
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....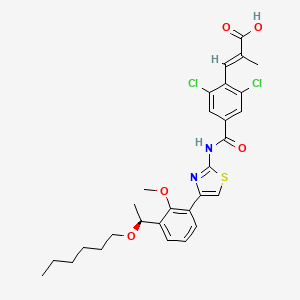
Lusutrombopag
Update…..FDA approved july2018
(E)-3-[2,6-dichloro-4-[[4-[3-[(1S)-1-hexoxyethyl]-2-methoxyphenyl]-1,3-thiazol-2-yl]carbamoyl]phenyl]-2-methylprop-2-enoic acid
(S)-(-)-(E)-3-(2,6-dichloro-4-{4-[3-(1-hexyloxyethyl)-2-methyloxyphenyl]thiazol-2-ylcarbamoyl}phenyl)-2-methylacrylic acid
(2E)-3-{2,6-Dichloro-4-[(4-{3-[(1S)-1-(hexyloxy)ethyl]-2-methoxyphenyl}-1,3-thiazol-2-yl)carbamoyl]phenyl}-2-methylacrylic acid
UNII 6LL5JFU42F, CAS 1110766-97-6,
D10476, MW591.546 , [US2010267783], MF C29H32Cl2N2O5S, S-888711
Shionogi & Co., Ltd., 塩野義製薬株式会社 INNOVATOR
Optically active compound (C-3B) Melting point: 142-145°C…………….EP2184279B1
NMR (DMSO-d6) δ ppm: 12.97 (brs, 1H), 8.29 (s, 2H), 7.90 (dd, 1H, J = 1.8 Hz, 7.5 Hz), 7.72 (s, 1H), 7.35 – 7.40 (m, 2H), 7.26 (t, 1H, J = 7.5 Hz), 4.82 (q, 1H, J = 6.3 Hz), 3.62 (s, 3H), 3.16 – 3.37 (m, 2H), 1.69 (s, 3H), 1.18 – 1.51 (m, 11H), 0.82-0.87 (m, 3H) Optical rotation -4.5 degrees (DMSO, c = 1.001, 25°C)………….EP2184279B1
Optical rotation: -7.0 ± 0.5 degrees (CHCl3, c = 1.040, 21°C), NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 – 1.4 (6H, m), 1.48 (3H, d, J = 6.4 Hz), 1.52 – 1.64 (2H, m), 1.86 (3H, d, J = 1.4Hz)), 3.35 (2H, t, J = 6.7Hz), 3.55 (3H, s), 4.87 (1H, q, J = 6.3 Hz), 7.25 (1H, t, J = 7.7 Hz), 7.41 (1H, s), 7.49 (1H, dd, J = 7.9 Hz, J = 1.6 Hz), 7.51 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.65 (1H, d, J = 1.4 Hz), 8.33 (2H, s), 13.4 (2H, brs)………EP2184279B1
Thrombopoietin receptor agonist, Oral thrombopoietin (TPO) mimetic
- 24 Mar 2015 Shionogi plans a phase III trial in Thrombocytopenia (in patients with chronic liver disease) in USA (NCT02389621)
- 31 Dec 2014 Preregistration for Thrombocytopenia in Japan (PO)
- 08 Nov 2013 Phase II development is ongoing in the US and the Europe
Process for preparing intermediates of an optically active 1,3-thiazole containing thrombopoietin receptor agonist Also claims crystalline forms of lusutrombopag intermediates and a process for preparing lusutrombopag. Shionogi is developing lusutrombopag, a small-molecule thrombopoietin mimetic, as an oral tablet formulation for treating thrombocytopenia.
In December 2014, an NDA was submitted in Japan. In May 2015, the drug was listed as being in phase III development for thrombocytopenia in the US and Europe.

The lusutrombopag, a low molecular-human thrombopoietin receptor agonist, its chemical formula, “(E) -3- [2,6-Dichloro-4- [4- [3 – [(S) -1-hexyloxyethyl] – 2-methoxyphenyl] -thiazol- 2-ylcarbamoyl] -phenyl] is a -2-methylacrylic acid “. lusutrombopag is represented by the following chemical structural formula.

Eltrombopag is represented by the following chemical structural formula.

Avatrombopag is represented by the following chemical structural formula.

Totrombopag choline is represented by the following chemical structural formula.



Optically active compound (C-3B)Melting point: 142-145°C DESIRED
NMR (DMSO-d6) δ ppm: 12.97 (brs, 1H), 8.29 (s, 2H), 7.90 (dd, 1H, J = 1.8 Hz, 7.5 Hz), 7.72 (s, 1H), 7.35 – 7.40 (m, 2H), 7.26 (t, 1H, J = 7.5 Hz), 4.82 (q, 1H, J = 6.3 Hz), 3.62 (s, 3H), 3.16 – 3.37 (m, 2H), 1.69 (s, 3H), 1.18 – 1.51 (m, 11H), 0.82-0.87 (m, 3H) Optical rotation -4.5 degrees (DMSO, c = 1.001, 25°C)
Example 4: Synthesis of (C-3B)

First step: Synthesis of (S)-1-(3-bromo-2-methyloxyphenyl)ethane-1-ol (17)
Using the same method as that of the first step of Example 3, the compound (17) was obtained from the compound (16) at a yield 77%.
-
-
Optical rotation: -23.5 ± 0.6 degrees (CHCl3, c = 1.050, 21°C)
NMR (CDCl3) θ ppm: 1.49 (3H, d, J = 6.6 Hz), 2.33 (1H, brs), 3.88 (3H, s), 5.19 (1H, q, J = 6.4 Hz), 7.01 (1H, t, J = 7.9 Hz), 7.40 (1H, dd, J = 7.7 Hz, J = 1.1 Hz), 7.46 (1H, dd, J = 8.0 Hz, J = 1.4 Hz)
-
Second step: Synthesis of (S)-1-bromo-3-(1-hexyloxyethyl)-2-methyloxybenzene (18)
-
-
Using the same method as that of the second step of Example 3, the compound (18) was obtained from the compound (17) at a yield of 96%.
Optical rotation: -29.8 ± 0.6 degrees (CHCl3, c = 1.055, 21°C)
NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 – 1.4 (6H, m), 1.42 (3H, d, J = 6.5 Hz), 1.54 (2H, m), 3.29 (2H, m), 3.85 (3H, s), 4.78 (1H, q, J = 6.4 Hz), 7.02 (1H, t, J = 7.9 Hz), 7.39 (1H, dd, J = 7.8 Hz, J = 1.7 Hz), 7.45 (1H, dd, J = 7.9 Hz, J = 1.7 Hz)
-
Third step and fourth step: Synthesis of (S)-4-(3-(1-hexyloxyethyl)-2-methyloxyphenyl)thiazole-2-amine (20)
-
-
Using the same method as that of the fourth step of Example 3, the compound (19) was obtained from the compound (18), subsequently according to the same method as that of the fourth step, the compound (20) was obtained.
-
Compound (19)
-
-
NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.9 Hz), 1.2-1.4 (6H, m), 1.45 (3H, d, J = 6.6 Hz), 1.55 (2H, m), 3.29 (2H, m), 3.78 (3H, s), 4.73 (2H, m), 4.80 (1H, q, J = 6.4 Hz), 7.24 (1H, t, J = 7.8Hz), 7.52 (1H, dd, J = 7.7 Hz, J = 1.8 Hz), 7.65 (1H, dd, J = 7.7 Hz, J = 1.8 Hz)
-
Compound (20)
-
Optical rotation: -4.2 ± 0.4 degrees (DMSO, c = 1.025, 21°C)
NMR (CDCl3) δ ppm: 0.84 (3H, t, J = 7.0 Hz), 1.2 – 1.3 (6H, m), 1.35 (3H, d, J = 6.5 Hz), 1.48 (2H, m), 3.25 (2H, m), 3.61 (3H, s), 4.78 (1H, q, J = 6.4 Hz), 6.99 (2H, brs), 7.05 (1H, s), 7.16 (1H, t, J = 7.7 Hz), 7.27 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.81 (1H, dd, J = 7.6 Hz, J = 1.9 Hz) -
Fifth step: Synthesis of ethyl (S)-(E)-3-(2,6-dichloro-4-(4-(3-(1-hexyloxyethyl)-2-metyloxyphenyl)thiazol-2-ylcarbamoyl)phenyl)-2-methylacrylate (21)
-
-
Using the same method as that of the fifth step of Example 3, the compound (21) was obtained from the compound (20) at a yield of 94%.
Optical rotation: +4.7 ± 0.4 degrees (CHCl3, c = 1.07, 21°C)
NMR (CDCl3 ) δ ppm: 0.87 (3H, t, J = 6.9 Hz), 1.2 – 1.35 (6H, m), 1.38 (3H, t, J = 7.1
Hz), 1.44 (3H, d, J = 6.4 Hz), 1.57 (2H, m), 1.77 (3H, d, J = 1.4 Hz), 3.30 (2H, m), 3.59 (3H, s), 4.31 (2H, q, J = 7.1 Hz), 4.83 (1H, q, J = 6.4 Hz), 7.17 (1H, t, J = 7.7 Hz), 7.42 (1H, d, J = 1.7 Hz), 7.42 (1H, dd, J = 7.7 Hz, J = 1.8 Hz), 7.51 (1H, s), 7.67 (1H, dd, J = 7.6 Hz, J = 1.7 Hz), 7.89 (2H, s), 10.30 (1H, brs)
-
Sixth step: Synthesis of (S)-(E)-3-(2,6-dichloro-4-(4-(3-(1-hexyloxyethyl)-2-metyloxyphenyl)thiazol-2-ylcarbamoyl)phenyl)-2-methylacrylic acid (C-3B)
-
Using the same method as that of the sixth step of Example 3, the compound (C-3B) was obtained from the compound (21) at a yield of 80%.Optical rotation: -7.0 ± 0.5 degrees (CHCl3, c = 1.040, 21°C)
NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 – 1.4 (6H, m), 1.48 (3H, d, J = 6.4 Hz), 1.52 – 1.64 (2H, m), 1.86 (3H, d, J = 1.4Hz)), 3.35 (2H, t, J = 6.7Hz), 3.55 (3H, s), 4.87 (1H, q, J = 6.3 Hz), 7.25 (1H, t, J = 7.7 Hz), 7.41 (1H, s), 7.49 (1H, dd, J = 7.9 Hz, J = 1.6 Hz), 7.51 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.65 (1H, d, J = 1.4 Hz), 8.33 (2H, s), 13.4 (2H, brs) -
Results of powder X-ray deffraction are shown in Fig. 5.
-
Diffraction angle of main peak: 2θ = 17.8, 21.1, 22.5, 23.3, 24.1, and 24.4 degrees
WO2005014561/EP1655291A1
https://www.google.co.in/patents/EP1655291A1?cl=en
WO2014003155, claiming a composition comprising lusutrombopag, useful for treating thrombocytopenia.
https://www.google.co.in/patents/US20150148385?cl=en
.
Methods respectively for producing optically active compound having agonistic activity on thrombopoietin receptors and intermediate of said compound
(Step 1) Synthesis of compound (VII ‘) under a nitrogen atmosphere, it was dissolved compound 1 (2.00kg) in 1,2-dimethoxyethane (28.0kg). 25% LDA tetrahydrofuran – heptane – ethyl benzene solution (13.20kg) was added dropwise over 1 hour at -55 ℃, and stirred for 30 minutes. It was added dropwise over 40 minutes to 1,2-dimethoxyethane (3.0kg) solution of N- formyl morpholine (3.74kg) at -55 ℃, and stirred for 1 hour. 1,2-dimethoxyethane (3.0kg) solution of 2-phosphono-propanoic acid triethyl (3.74kg) was added dropwise over 45 minutes at 0 ℃, and stirred for 2 hours. 35% aqueous solution of sulfuric acid (15.8kg) was added dropwise over 40 minutes to the reaction solution. Water (16.0kg) was added and extracted. The resulting organic layer was washed with water (8.0kg), and the solvent was evaporated under reduced pressure. Acetonitrile (16.0kg) was added, and the mixture was stirred for 1 hour at 25 ℃, and the mixture was stirred and cooled to 0 ℃ 5 hours and 30 minutes. The precipitated crystals were collected by filtration, and washed with 5 ℃ acetonitrile (3.2kg). The resulting crystals it was dissolved in acetonitrile (16.0kg) at 75 ℃. It was cooled to 60 ℃, and the mixture was stirred for 30 minutes. Over 1 hour and then cooled to 30 ℃, and the mixture was stirred for 45 minutes. Over 40 minutes and then cooled to 5 ℃, and the mixture was stirred for 3 hours.The precipitated crystals were collected by filtration, and washed with 5 ℃ acetonitrile (3.2kg). The resulting crystals it was dissolved in acetonitrile (13.0kg) at 75 ℃. It was cooled to 60 ℃, and the mixture was stirred for 30 minutes. Furthermore, up to 30 ℃ over 1 hour and then cooled and stirred for 70 minutes. Over 30 minutes and then cooled to 5 ℃, and the mixture was stirred for 4 hours. I precipitated crystals were collected by filtration. Washed with 5 ℃ acetonitrile (3.2kg), and dried to give the compound (VII ‘) (1.63kg, 51.2% yield). NMR (CDCl 3 ) delta ppm: 8.07 (s, 2H), 7.47 (s, 1H), 4.32 (Q, 2H, J = 7.0 Hz), 1.79 (s, 3H), 1.38 (t, 3H, J = 7.0 Hz) Results of powder X-ray diffraction and I shown in Figure 1 and Table 3. [Table 3] In the powder X-ray diffraction spectrum, diffraction angle (2θ): 8.1 ± 0.2 °, 16.3 ± 0.2 °, 19.2 ± 0.2 °, 20.0 ± 0. 2 °, the peak was observed at 24.8 ± 0.2 °, and 39.0 ± 0.2 ° degrees.
(Synthesis of Compound (XI ‘))
(Step 2) Synthesis of Compound 4 under a nitrogen atmosphere over Compound 3 (3.00kg) and 1mol / L isopropylmagnesium chloride in tetrahydrofuran (11.40kg) 1 hour at 25 ℃ in The dropped, and stirred for 2 hours. 1mol / L isopropylmagnesium chloride in tetrahydrofuran solution (0.56kg) was added at 25 ℃, and stirred for 2 hours. To the reaction mixture N- methoxymethyl -N- methylacetamide the (1.45kg) was added dropwise over at 25 ℃ 40 minutes, and stirred for 80 minutes. 7% hydrochloric acid (9.7kg) was added to the reaction mixture, and the mixture was extracted with toluene (11.0kg). The resulting organic layer twice with water (each 7.5kg) washed, the solvent was evaporated under reduced pressure to give Compound 4 (2.63kg). NMR (CDCl 3 ) delta ppm: 7.69 (dd, 1H, J = 7.7 Hz, J = 1.5 Hz), 7.55 (dd, 1H, J = 7.7 Hz, J = 1.5 Hz), 7.05 (t, 1H, J = 7.7 Hz), 3.88 (s, 3H), 2.64 (s, 3H) ppm:
(Step 3) Synthesis of Compound 5 Under a nitrogen atmosphere, chloro [(1S Compound 4 (2.63kg), 2S) -N- ( p- toluenesulfonyl) -1,2-diphenyl-ethane diamine] (p- cymene) ruthenium (II) (28.6g), it was added to tetrahydrofuran (1.3kg) and triethylamine (880.0g). Formic acid (570.0g) was added dropwise over 6 hours at 40 ℃, and stirred for 1 hour. In addition 3.5% hydrochloric acid (14.4kg) to the reaction mixture, and the mixture was extracted with toluene (13.0kg).The organic layer was washed with 3.5% hydrochloric acid (14.4kg) and water (7.5kg), the solvent was concentrated under reduced pressure to obtain a toluene solution of Compound 5 (4.44kg).
(Step 4) Synthesis of Compound 6 under a nitrogen atmosphere, it was a potassium hydroxide (6.03kg) was dissolved in water (6.0kg). To the solution, it added tetrabutylammonium bromide (182.0g) and toluene solution of Compound 5 (4.44kg). 1-bromo-hexane (2.79kg) was added dropwise over 1 hour at 60 ℃, and the mixture was stirred for 4 hours. And extracted by adding water (4.4kg) to the reaction solution. The resulting organic layer was filtered through powdered cellulose and extracted with toluene (3.0kg) and water (7.6kg) to the filtrate. The solvent it was evaporated under reduced pressure from the organic layer. Toluene operation of evaporated under reduced pressure and the solvent by the addition of a (7.8kg) was repeated five times to obtain a toluene solution of Compound 6 (10.0kg).
(Step 5) Synthesis of Compound 7 under a nitrogen atmosphere, magnesium powder (301.0g), in tetrahydrofuran (1.3kg), the compound in toluene (6.4kg) and 1mol / L isopropylmagnesium chloride in tetrahydrofuran (432.0g) 6 In addition of the toluene solution (0.50kg) at 30 ℃, and the mixture was stirred for 2 hours. Toluene solution of Compound 6 (9.50kg) was added dropwise over 3 hours at 50 ℃, and stirred for 2 hours. 1-bromo-hexane (746.0g) was added at 50 ℃, and the mixture was stirred for 1 hour. It was added dropwise over 1 hour at 5 ℃ toluene (5.3kg) solution of 2-chloro -N- methoxy -N- methyl-acetamide (1.78kg), and stirred for 1 hour. 3.7% hydrochloric acid (16.7kg) was added to the reaction mixture, and the mixture was extracted. The obtained organic layer was washed with water (15.0kg), and concentrated under reduced pressure to give a toluene solution of Compound 7 (8.25kg).
(Step 6) Synthesis of Compound (II ‘) under a nitrogen atmosphere, thiourea (1.03kg), in ethanol (1.2kg) and 65 ℃ toluene solution of compound 7 (8.25kg) in toluene (6.3kg) over 3 hours was added dropwise and stirred for 2 hours. The reaction solution was extracted by adding 0.7% hydrochloric acid (30.6kg), and washed twice with water (30.0kg). Ethanol in the organic layer (9.5kg), and extracted by addition of heptane (10.0kg) and 3.5% hydrochloric acid (5.9kg). The resulting aqueous layer with 4% hydrochloric acid (1.5kg) and ethanol (3.5kg) merged the aqueous layer was extracted from the organic layer, the ethanol was washed with heptane (10.0kg) (3.1kg) It was added. 8% aqueous sodium hydroxide (6.0kg) was added dropwise over at 5 ℃ 30 minutes, and stirred for 20 minutes. 8% aqueous sodium hydroxide (5.8kg) was added dropwise over a period at 5 ℃ 15 minutes.The precipitated crystals were collected by filtration, washed with 45% aqueous ethanol (10.9kg) and water (15.0kg) (crude crystals of Compound (II ‘)). The resulting crude crystals were dissolved in 50 ℃ in ethanol (8.1kg), over a period of 1 hour and then cooled to 10 ℃, and the mixture was stirred for 30 minutes. Water (10.0kg) over 2 hours was added dropwise and stirred for 30 minutes. The precipitated crystals were collected by filtration, washed with 50% aqueous ethanol (7.5kg) and water (10.0kg) (crystals of the compound after recrystallization from ethanol / water system (II ‘)). The resulting crystals were dissolved at 55 ℃ in toluene (1.6kg) and heptane (1.3kg), over 1 hour and cooled to 20 ℃, and stirred for 30 minutes. Heptane (6.3kg) over a period of 30 minutes was added dropwise and stirred for 15 minutes. The obtained crystals precipitated were collected by filtration, washed with a mixed solvent of toluene (0.3kg) and heptane (2.3kg), and dried to give compound (II ‘) (1.67kg, 44.5% yield) a (crystalline compound after recrystallization from toluene / heptane system (II ‘)).
NMR (CDCl 3 ) delta ppm: 0.84 (3H, t, J = 7.0 Hz), 1.2 – 1.3 (6H, M), 1.35 (3H, D, J = 6.5 Hz), 1.48 (2H, M), 3.25 ( 2H, m), 3.61 (3H, s), 4.78 (1H, q, J = 6.4 Hz), 6.99 (2H, brs), 7.05 (1H, s), 7.16 (1H, t, J = 7.7 Hz), 7.27 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.81 (1H, dd, J = 7.6 Hz, J = 1.9 Hz) it is shown in Figure 2 and Table 4 the results of powder X-ray diffraction. [Table 4] In the powder X-ray diffraction spectrum, diffraction angle (2θ): 12.5 ± 0.2 °, 13.0 ± 0.2 °, 13.6 ± 0.2 °, 16.4 ± 0. 2 °, 23.0 ± 0.2 °, a peak was observed at 24.3 ± 0.2 ° degrees. Above, each of the compounds (II ‘) of the crude crystals, the ethanol / compound after recrystallization from water (II’) crystals and toluene / heptane compound after recrystallization from (II ‘) crystallographic purity of the results of the , Fig. 3, I 4 and 5 as well as Table 5. [Table 5](HPLC was measured by the above method A.) As shown in the results of the above table, as compared to recrystallization from ethanol / water, recrystallized with toluene / heptane system, compounds having a high optical purity it is possible to manufacture a crystal of (II ‘). Next, the above-mentioned compound (II ‘) of the crude crystals, the ethanol / compound after recrystallization from water (II’) crystals and toluene / heptane compound after recrystallization from (II ‘) results of crystals of HPLC of the respectively, Fig. 6, I 7 and 8 and Table 6. [Table 6] (units, .N.D shows the peak area of the (%). is, .HPLC to indicate not detected was measured by the above method B.) As shown in the results of Table, with ethanol / water system Compared to recrystallization, recrystallization from toluene / heptane system is found to be efficiently remove organic impurities A and organic impurities B.
(Step 7) Compound ‘Synthesis of DMSO adduct of (VIII) Under a nitrogen atmosphere, the compound (II ‘) (1.50kg) and compound (VII’) (1.43kg) in ethyl acetate (17.6kg) and triethylamine (1.09kg) were sequentially added, was dissolved.Diphenyl phosphorochloridate the (1.46kg) was added dropwise over 1 hour at 50 ℃, and the mixture was stirred for 3 hours. The reaction mixture was cooled to 25 ℃, after the addition of 2.6% hydrochloric acid (8.1kg), and extracted. The resulting organic layer to 6.3% aqueous solution of sodium hydroxide (3.2kg) and 14% aqueous sodium carbonate (5.2kg) was added and stirred for 20 minutes. Adjusted to pH7.5 with 8.3% hydrochloric acid and extracted. The organic layer it was washed with 4.8% sodium chloride aqueous solution (11.0kg). DMSO and (16.5kg) was added, and the mixture was concentrated under reduced pressure.DMSO and (5.8kg) was added, over a period at 40 ℃ 30 minutes was added dropwise water (0.9kg), and stirred for 1 hour. Over a period of 30 minutes, cooled to 25 ℃, and the mixture was stirred for 30 minutes. Over at 25 ℃ 30 minutes was added dropwise water (1.4kg), and the precipitated crystals were collected by filtration. After washing with 90% DMSO solution (10.0kg) and water (27.0kg), to obtain crystals of DMSO adduct and dried to Compound (VIII ‘) (2.98kg, 95.2% yield).
1H-NMR (CDCl 3 ) delta: 0.87 (t, J = 6.8 Hz, 3H), 1.20-1.34 (M, 6H), 1.37 (t, J = 7.1 Hz, 3H), 1.44 (D, J = 6.5 Hz , 3H), 1.52-1.59 (m, 2H), 1.77 (d, J = 1.3Hz, 3H), 2.62 (s, 6H), 3.28-3.34 (m, 2H), 3.59 (s, 3H), 4.31 ( q, J = 7.1Hz, 2H), 4.83 (q, J = 6.5Hz, 1H), 7.16 (t, J = 7.7Hz, 1H), 7.40-7.43 (m, 2H), 7.51 (s, 1H), 7.68 (dd, J = 7.7, 1.8Hz, 1H), 7.92 (d, J = 1.3Hz, 2H), 10.58 (s, 1H). The results of the powder X-ray diffraction and I are shown in Figure 9 and Table 7. [Table 7]
In the powder X-ray diffraction spectrum, diffraction angle (2θ): 5.2 ° ± 0.2 °, 7.0 ° ± 0.2 °, 8.7 ° ± 0.2 °, 10.5 ° ± 0.2 °, 12.3 ° ± 0.2 °, 14.0 ° ± 0.2 °, 15.8 ° ± 0.2 °, 19.3 ° ± 0.2 °, 22.5 ° peak was observed to ± 0.2 ° and 24.1 ° ± 0.2 °. TG / DTA analysis result it is shown in Figure 10. Then, each result of HPLC of concentrated dry solid and the above DMSO adduct crystals described in the following Reference Examples 1, 11 and 12, 13 and 14, and I are shown in Table 8. [Table 8] (unit, .HPLC showing peak areas of (%) was measured by the above methods C.) As shown in the results of the above Table, when compared with the extract, DMSO adduct of the compound (VIII ‘) The in the crystal, less residual organic impurities D, and it found to be about 56% removal.
(Step 8) under nitrogen atmosphere, DMSO adduct of the compound (VIII ‘) and (2.50kg) it was dissolved in ethanol (15.8kg). 24% sodium hydroxide aqueous solution (1.97kg) was added dropwise over a period at 45 ℃ 30 minutes to the solution and stirred for 3 hours. The reaction mixture was cooled to 25 ℃, water was added (20.0kg) and ethanol (7.8kg). 18% hydrochloric acid (2.61kg) was added dropwise over at 25 ℃ 30 minutes, followed by addition of seed crystals prepared according to the method described in Patent Document 23. After stirring for 3 hours and allowed to stand overnight. Thereafter, the precipitated crystals were collected by filtration, to give after washing with 50% aqueous ethanol solution (14.2kg), and dried to a compound (XI ‘) (1.99kg, 93.9% yield).
NMR (CDCl 3 ) delta ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 – 1.4 (6H, M), 1.48 (3H, D, J = 6.4 Hz), 1.52 – 1.64 (2H, M), 1.86 (3H, d, J = 1.4Hz), 3.35 (2H, t, J = 6.7Hz), 3.55 (3H, s), 4.87 (1H, q, J = 6.3 Hz), 7.25 (1H, t, J = 7.7 Hz), 7.41 (1H, s), 7.49 (1H, dd, J = 7.9 Hz, J = 1.6 Hz), 7.51 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.65 (1H, d, J = 1.4 Hz), 8.33 (2H, s), 13.4 (2H, brs) I is shown in Figure 15 the results of powder X-ray diffraction.
Patent Document 1: JP-A-10-72492 JP
Patent Document 2: WO 96/40750 pamphlet
Patent Document 3: JP-A-11-1477 JP
Patent Document 4: Japanese Unexamined Patent Publication No. 11-152276
Patent Document 5: International Publication No. 00/35446 pamphlet
Patent Document 6: JP-A-10-287634 JP
Patent Document 7: WO 01/07423 pamphlet
Patent Document 8: International Publication WO 01/53267 pamphlet
Patent Document 9: International Publication No. 02 / 059 099 pamphlet
Patent Document 10: International Publication No. 02/059100 pamphlet
Patent Document 11: International Publication No. 02/059100 pamphlet
Patent Document 12: International Publication No. 02/062775 pamphlet
Patent Document 13: International Publication No. 2003/062233 pamphlet
Patent Document 14: International Publication No. 2004/029049 pamphlet
Patent Document 15: International Publication No. 2005/007651 pamphlet
Patent Document 16: International Publication No. 2005/014561 pamphlet
Patent Document 17: JP 2005-47905 Japanese
patent Document 18: Japanese Patent Publication No. 2006-219480
Patent Document 19: Japanese Patent Publication No. 2006-219481
Patent Document 20: International Publication No. 2007/004038 pamphlet
Patent Document 21: International Publication No. 2007/036709 pamphlet
Patent Document 22: International Publication No. 2007/054783 pamphlet
Patent Document 23: International Publication No. 2009/017098 pamphlet
Non-Patent Document 1: Proceedings of the National Akademyi of Science of the United State of America (…. Proc Natl Acad Sci USA) 1992, Vol. 89, p 5640-5644.
Non-Patent Document 2: Journal of Organic (.. J. Org Chem) Chemistry 1984, Vol. 49, p 3856-3857.
Non-Patent Document 3: (.. J. Org Chem). Journal of Organic Chemistry, 1992, Vol. 57, p 6667-6669
Non-Patent Document 4:. Shinretto (Synlett) 2004 year Vol. 6, p 1092-1094
| 101 | Discovery and biological evaluation of Lusutrombopag (S-888711) as a novel nonpeptide drug candidate for thrombocytopenia Masami Takayama, Hajime Yamada, Hiroshi Takemoto, Takeshi Shiota, Yoshikazu Tanaka, Noriko Yamane, Kouji Takahashi, Naoki Oyabu, Kenji Kuwabara, Itsuki Oshima, Kenzo Koizumi, Hiroshi Yoshida, Ayumu Nogami, Tomomi Yamada, Yutaka Yoshida, Takami Murashi, Shinichiro Hara. |
| 101 – Discovery and biological evaluation of Lusutrombopag (S-888711) as a novel nonpeptide drug candidate for thrombocytopenia
Masami Takayama1, masami.takayama@shionogi.co.jp, Hajime Yamada3, Hiroshi Takemoto2, Takeshi Shiota2, Yoshikazu Tanaka2, Noriko Yamane2, Kouji Takahashi2, Naoki Oyabu3, Kenji Kuwabara3, Itsuki Oshima2, Kenzo Koizumi3, Hiroshi Yoshida3, Ayumu Nogami3, Tomomi Yamada3, Yutaka Yoshida3, Takami Murashi3, Shinichiro Hara2. (1) Department of Strategic Research Planning Offices, Shionogi & CO., LTD, Toyonaka, Osaka 561-0825, Japan, (2) Department of Innovative Drug Discovery Research Laboratories, Shionogi & CO.,LTD, Toyonaka, Osaka 561-0825, Japan, (3) Department of Medicinal Research Laboratories, Shionogi & CO., LTD, Toyonaka, Osaka 561-0825, Japan As a drug candidate of thrombocytopenia, Lusutrombopag (S-888711) is in Phase III clinical trial stage right now. It is been proven that Lusutrombopag (S-888711) is excellent property in safety and efficacy by clinical trials. In this meeting, we will present in detail about the history of drug discovery of Lusutrombopag.Because Lusutrombopag (S-888711) acts specifically to human TPO receptor, we prepared TPOR-Ki/Shi mice expressing a mouse-human chimeric TPOR for evaluating the efficacy. This TPOR-Ki/Shi mice worked very well as an evaluation model of drug efficacy, so we were able to select Lusutrombopag from many candidate compounds. In this meeting, we will present the results of the efficacy in TPOR-Ki/Shi mice of Lusutrombopag and the similar drug (Eltrombopag). |
update………..
FDA approves lusutrombopag for thrombocytopenia in adults with chronic liver disease
https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm615348.htm
synthesis………..https://newdrugapprovals.org/2015/08/20/lusutrombopag-oral-thrombopoietin-tpo-mimetic/
On July 31, 2018, the Food and Drug Administration approved lusutrombopag (Mulpleta, Shionogi Inc.) for thrombocytopenia in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure.
Approval was based on two randomized, double-blind, placebo-controlled trials (L-PLUS 1 and L-PLUS 2, NCT02389621) involving 312 patients with chronic liver disease and severe thrombocytopenia who were undergoing an invasive procedure and had a platelet count less than 50 x 109/L. Patients were randomized 1:1 to receive 3 mg of lusutrombopag or placebo once daily for up to 7 days.
In L-PLUS 1, 78% of patients (38/49) receiving lusutrombopag required no platelet transfusion prior to the primary invasive procedure, compared with 13% (6/48) who received placebo (95% CI for treatment difference: 49%, 79%; p<0.0001). In L-PLUS 2, 65% (70/108) of patients who received lusutrombopag required no platelet transfusion prior to the primary invasive procedure or rescue therapy for bleeding from randomization through 7 days after the procedure, compared with 29% (31/107) receiving placebo (95% CI for treatment difference: 25%, 49%; p<0.0001).
The most common adverse reaction in ≥ 3% of patients was headache.
The recommended lusutrombopag dosage is 3 mg orally once daily with or without food for 7 days.
View full prescribing information for Mulpleta.
FDA granted this application priority review and fast track designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Healthcare professionals should report all serious adverse events suspected to be associated with the use of any medicine and device to FDA’s MedWatch Reporting System or by calling 1-800-FDA-1088.
Follow the Oncology Center of Excellence on Twitter @FDAOncology.
Check out recent approvals at the OCE’s podcast, Drug Information Soundcast in Clinical Oncology.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


Join me on twitter

 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL 
//////
phase 3, shionogi, japan, lusutrombopag, S 888711
CCCCCCOC(C)C1=CC=CC(=C1OC)C2=CSC(=N2)NC(=O)C3=CC(=C(C(=C3)Cl)C=C(C)C(=O)O)Cl


[…] synthesis………..https://newdrugapprovals.org/2015/08/20/lusutrombopag-oral-thrombopoietin-tpo-mimetic/ […]