



| Patent ID
|
Patent Title
|
Submitted Date
|
Granted Date
|
|---|---|---|---|
| US2016108123 | ANTIBODY MOLECULES TO PD-L1 AND USES THEREOF |
2015-10-13
|
2016-04-21
|
| US2014343086 | COMPOUNDS AND COMPOSITIONS FOR INHIBITING THE ACTIVITY OF ABL1, ABL2 AND BCR-ABL1 |
2014-07-31
|
2014-11-20
|
| US8829195 | Compounds and compositions for inhibiting the activity of ABL1, ABL2 and BCR-ABL1 |
2013-05-13
|
2014-09-09
|
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA approves novel device Zephyr Endobronchial Valve (Zephyr Valve) for treating breathing difficulty from severe emphysema

Depiction of the Zephyr ® endobronchial valve. Image courtesy of Pulmonx, Inc.
FDA approves novel device for treating breathing difficulty from severe emphysema
The U.S. Food and Drug Administration today approved a new device, the Zephyr Endobronchial Valve (Zephyr Valve), intended to treat breathing difficulty associated with severe emphysema.
The U.S. Food and Drug Administration today approved a new device, the Zephyr Endobronchial Valve (Zephyr Valve), intended to treat breathing difficulty associated with severe emphysema.
“Treatment options are limited for people with emphysema who have severe symptoms that have not improved from taking medicines. These have included lung surgery, such as lung volume reduction or lung transplants, which may not be suitable or appropriate for all patients,” said Tina Kiang, Ph.D., acting director, Division of Anesthesiology, General Hospital, Respiratory, Infection Control and Dental Devices, in the FDA’s Center for Devices and Radiological Health. “This novel device is a less invasive treatment that expands the options available to patients.”
June 29, 2018
Release
The U.S. Food and Drug Administration today approved a new device, the Zephyr Endobronchial Valve (Zephyr Valve), intended to treat breathing difficulty associated with severe emphysema.
“Treatment options are limited for people with emphysema who have severe symptoms that have not improved from taking medicines. These have included lung surgery, such as lung volume reduction or lung transplants, which may not be suitable or appropriate for all patients,” said Tina Kiang, Ph.D., acting director, Division of Anesthesiology, General Hospital, Respiratory, Infection Control and Dental Devices, in the FDA’s Center for Devices and Radiological Health. “This novel device is a less invasive treatment that expands the options available to patients.”
The Centers for Disease Control and Prevention estimates that 3.5 million American adults have been diagnosed with emphysema. Emphysema, including severe emphysema, is a type of chronic obstructive pulmonary disease (COPD) due to damage to the air sacs (alveoli) in the lungs. Lung damage from emphysema is irreversible. The damaged alveoli can cause used air to become trapped in the lungs during exhalation. This can cause the diseased parts of the lung to get larger and put pressure on the healthy part of the lung, which makes it difficult to breathe. As a result, the body may not get the oxygen it needs.
Using a flexible bronchoscope, a doctor places Zephyr Valves, similar in size to pencil erasers, into the diseased areas of the lung airways during a procedure in a hospital setting. Design of the device is intended to prevent air from entering the damaged parts of the lung and allow trapped air and fluids to escape. During inhalation, the valves close, preventing air from entering the damaged part of the lung and during exhalation, the valves open, letting out trapped air, which is intended to relieve pressure.
The FDA reviewed data from a multi-center study of 190 patients with severe emphysema. In this study, 128 patients were treated with Zephyr Valves and medical management according to current clinical guidelines, including medications (bronchodilators, corticosteroids, antibiotics or anti-inflammatory maintenance medications) and pulmonary rehabilitation, while 62 patients (the control group) received medical management only. Results of treatment were measured by how many patients in each arm of the study had at least a 15 percent improvement in pulmonary function scores (the volume of air that can forcibly be blown out in one second after full inhalation). At one year, 47.7 percent of patients treated with Zephyr Valves experienced at least a 15 percent improvement in their pulmonary function scores, compared with 16.8 percent of patients in the control group. Adverse events observed in the study include death, air leak (pneumothorax), pneumonia, worsening of emphysema, coughing up blood, shortness of breath and chest pain.
The Zephyr Valve device is contraindicated for patients with active lung infections; those who are allergic to nitinol, nickel, titanium or silicone; active smokers and those who are not able to tolerate the bronchoscopic procedure. Patients who have had major lung procedures, heart disease, large bubbles of air trapped in the lung or who have not responded to other treatments should talk with their providers to determine if the Zephyr Valve device is appropriate for them.
The Zephyr Valve was granted Breakthrough Device designation, meaning the FDA provided intensive interaction and guidance to the company on efficient device development, to expedite evidence generation and the agency’s review of the device. To qualify for such designation, a device must provide for more effective treatment or diagnosis of a life-threatening or irreversibly debilitating disease or condition, and meet one of the following criteria: the device must represent a breakthrough technology; there must be no approved or cleared alternatives; the device must offer significant advantages over existing approved or cleared alternatives; or the availability of the device is in the best interest of patients.
The FDA reviewed the Zephyr Valve device through the premarket approval review pathway, a regulatory pathway for the highest risk class of devices.
The FDA granted approval of the Zephyr Valve device to Pulmonx Inc.
////////////fda 2018, medical devices, Zephyr Valve device, Pulmonx Inc, Breakthrough Device designation, Zephyr Endobronchial Valve, Zephyr Valve,
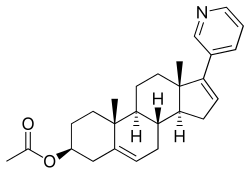
Abiraterone acetate, アビラテロン酢酸エステル
Abiraterone acetate
- Molecular FormulaC26H33NO2
- Average mass391.546 Da
Abiraterone, CB-7598, アビラテロン酢酸エステル
(3β)-17-(pyridin-3-yl)androsta-5,16-dien-3-yl acetate
CAS 154229-18-2
(1S,2R,5S,10R,11S,15S)-2,15-dimethyl-14-(pyridin-3-yl)tetracyclo[8.7.0.02,7.011,15]heptadeca-7,13-dien-5-yl acetate
CB-7630;CB7630;CB 7630
MFCD00934213 [MDL number]
UNII:EM5OCB9YJ6
(1S,2R,5S,10R,11S,15S)-2,15-dimethyl-14-(pyridin-3-yl)tetracyclo[8.7.0.0²,⁷.0¹¹,¹⁵]heptadeca-7,13-dien-5-ol
- (3β)-17-(pyridin-3-yl)androsta-5,16-dien-3-ol
- 17-(3-Pyridyl)androsta-5,16-dien-3beta-ol
Centocor Ortho Biotech
Abiraterone is a derivative of steroidal progesterone and is an innovative drug that offers clinical benefit to patients with hormone refractory prostate cancer. Abiraterone is administered as an acetate salt prodrug because it has a higher bioavailability and less susceptible to hydrolysis than abiraterone itself. FDA approved on April 28, 2011.
Used in combination with prednisone for the treatment of metastatic, castration-resistant prostate cancer.
- Originator The Institute of Cancer Research
- Developer All Ireland Cooperative Oncology Research Group; Cancer Research UK; Cougar Biotechnology; Janssen Research & Development; Johnson & Johnson; UNICANCER
- Class Androstenols; Antiandrogens; Antineoplastics; Small molecules
- Mechanism of Action CYP17A1 protein inhibitors
Highest Development Phases
- Marketed Prostate cancer
- Phase II Breast cancer; Ovarian cancer
- No development reported Congenital adrenal hyperplasia
Most Recent Events
- 06 Jun 2018 The National Institute for Health and Clinical Excellence does not recommend abiraterone for Prostate cancer (Combination therapy, First-line therapy, Hormone refractory, Metastatic disease)
- 06 Mar 2018 Janssen initiates a phase II trial for Prostate cancer (Combination therapy, Hormone refractory, Metastatic disease, Second-line therapy or greater) in USA (PO) (NCT03360721)
- 01 Mar 2018 Janssen plans the phase II OPTIMABI trial in Prostate cancer (Hormone refractory, Metastatic disease) in France (PO, Tablet) (NCT03458247)
- Abiraterone is associated with decreases in PSA levels, tumor shrinkage (as evaluated by RECIST criteria), radiographic regression of bone metastases and improvement in pain. Levels of adrenocorticotropic hormones increased up to 6-fold but this can be suppressed by dexamethasone.


FDA
NDA 202379, ZYTIGA (abiaterone acetate)
(3β)-17-(3-pyridinyl)androsta-5,16-dien-3-yl acetate
OND Division: NDA: Applicant: Stamp Date: PDUFA Goal Date: Established Name: Trade Name Dosage Form and Strength: Route of Administration: Indication: eCTD Reference for CMC Regulatory Filing Related IND Assessed by: Division of Drug Oncology Products 202-379 Centocor Ortho Biotech, Inc. 20 December, 2010 20 June, 2011 (Priority) Abiraterone Acetate ZYTIGA (proposed) Tablet – 250 mg Oral Indicated with prednisone for the treatment of metastatic (castrationresistant prostate cancer) in patients who have received prior chemotherapy containing a
https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202379Orig1s000ChemR.pdf
Abiraterone acetate, the drug substance, is an acetyl ester of abiraterone. It is a pro-drug of the active metabolite abiraterone. Abiraterone acetate is converted in vivo to abiraterone which selectively inhibits the enzyme CYP17. Abiraterone acetate is designated chemically as (3β)-17- (3-pyridinyl)androsta-5,16-dien-3-yl acetate. It is a white to off-white, non-hygroscpic, crystalline powder. It is freely soluble in organic solvents like tetrahydrofuran and dichloromethane but practically insoluble in water. It shows some solubility in 0.1N HCl. It should be noted that abiraterone acetate contains a . The dissociation constant (pKa) of abiraterone acetate is 5.19. It indicates that most of the abiraterone acetate will be soluble in stomach pH and most of the drug will be absorbed in the unionized form in the intestine at higher pH. The partition coefficient (log P) value of abiraterone acetate is 5.12 indicating high lipophilicity. Based on low aqueous solubility and low permeability thru the cells in GI tract, the drug substance is considered BCS Class IV.
Abiraterone acetate, sold under the brand name Zytiga among others, is an antiandrogen medication which is used in the treatment of prostate cancer.[1] It is specifically indicated for use in conjunction with castration and prednisone for the treatment of metastaticcastration-resistant prostate cancer (mCRPC) and in the treatment of metastatic high-risk castration-sensitive prostate cancer (mCSPC).[1] It is taken by mouth once per day with food.[1]
Side effects of abiraterone acetate include fatigue, arthralgia, hypertension, nausea, edema, hypokalemia, hot flashes, diarrhea, vomiting, cough, headache, glucocorticoid deficiency, mineralocorticoid excess, and hepatotoxicity among others.[1] The drug is an androgen synthesis inhibitor – specifically, a CYP17A1 inhibitor – and thereby inhibits the production of androgens like testosterone and dihydrotestosterone in the body.[1] In doing so, it prevents the effects of these hormones in the prostate gland and elsewhere in the body.[1] Abiraterone acetate is a prodrug of abiraterone.[1]Abiraterone acetate, sold under the brand name Zytiga among others, is an antiandrogen medication which is used in the treatment of prostate cancer.[1] It is specifically indicated for use in conjunction with castration and prednisone for the treatment of metastaticcastration-resistant prostate cancer (mCRPC) and in the treatment of metastatic high-risk castration-sensitive prostate cancer (mCSPC).[1] It is taken by mouth once per day with food.[1]
Abiraterone acetate was first described in 1993 and was introduced for medical use in 2011.[5][6][7] It was approved for the treatment of mCRPC in 2011 and was subsequently approved for the treatment of mCSPC in 2018.[8] The medication is marketed widely throughout the world.[9] It is not available as a generic medication.[10]
Medical uses
Prostate cancer
Abiraterone acetate is indicated for use in combination with prednisone, a corticosteroid, as a treatment for mCRPC (previously called hormone-resistant or hormone-refractory prostate cancer).[11][12][13][14] This is a form of prostate cancer that is not responding to first-line androgen deprivation therapy or treatment with androgen receptor antagonists. Abiraterone acetate has received FDA (28 April 2011), EMA (23 September 2011), MHRA (5 September 2011) and TGA (1 March 2012) approval for this indication.[11][12][13][14] In Australia it is covered by the Pharmaceutical Benefits Scheme when being used to treat castration-resistant prostate cancer and given in combination with prednisone/prednisolone (subject to the conditions that the patient is not currently receiving chemotherapy, is either resistant or intolerant of docetaxel, has a WHO performance status of <2, and his disease has not since become progressive since treatment with PBS-subsidised abiraterone acetate has commenced).[15]
Clinical effectiveness
A phase III study in subjects previously treated with docetaxel started in 2008.[16] In September 2010, an independent panel found that the interim results in patients previously treated with docetaxel were so much better compared to those treated with placebo that it was unethical to keep half the study participants on placebo, and all patients began receiving the drug. Overall survival was increased by 3.9 months in to this study (14.8 months versus 10.9 months for placebo).[17]
A placebo-controlled double-blind randomized phase III study in patients with castration-refractory prostate cancer but who had not received chemotherapy opened to accrual in April 2009.[18][19] 1,088 men received either abiraterone acetate (1000 mg daily) plus prednisone (5 mg twice daily), or placebo plus prednisone. The median radiographic progression-free survival was 16.5 months with abiraterone acetate–prednisone and 8.3 months with prednisone alone (hazard ratio (HR) = 0.53; 95% confidence interval (CI), 0.45 to 0.62; P<0.001). After a median follow-up period of 22.2 months, overall survival was better with abiraterone acetate plus prednisone (median not reached) compared to placebo plus prednisone (27.2 months); HR = 0.75; 95% CI, 0.61 to 0.93; P=0.01).[20]
Available forms
Abiraterone acetate is available in the form of 250 mg and 500 mg film-coated oral tablets and 250 mg uncoated oral tablets.[1] It is used at a dosage of 1,000 mg orally once per day with food in conjunction with castration (via GnRH analogue therapy or orchiectomy) and in combination with 5 mg prednisone orally twice per day.[1]
Contraindications
Contraindications include hypersensitivity to abiraterone acetate. Although documents state that it should not be taken by women who are or who may become pregnant,[12][21] there is no medical reason that any woman should take it. Women who are pregnant should not even touch the pills unless they are wearing gloves.[21] Other cautions include severe baseline hepatic impairment, mineralocorticoid excess, cardiovascular disease including heart failure and hypertension, uncorrected hypokalemia, and adrenocorticoid insufficiency.[22]
Side effects
Side effects by frequency:[11][12][13][14][22]
Very common (>10% frequency):
Common (1-10% frequency):
- Hypertriglyceridaemia
- Sepsis
- Cardiac failure
- Angina pectoris
- Arrhythmia
- Atrial fibrillation
- Tachycardia
- Dyspepsia (indigestion)
- Rash
- Alanine aminotransferase increased
- Aspartate aminotransferase increased
- Fractures
- Hematuria
Uncommon (0.1-1% frequency):
Rare (<0.1% frequency):
- Allergic alveolitis
Overdose
Clinical experience with overdose of abiraterone acetate is limited.[1] There is no specific antidote for abiraterone acetate overdose, and treatment should consist of general supportive measures, including monitoring of cardiac and liver function.[1]
Interactions
Abiraterone acetate is a CYP3A4 substrate and hence should not be administered concurrently with strong CYP3A4 inhibitors such as ketoconazole, itraconazole, clarithromycin, atazanavir, nefazodone, saquinavir, telithromycin, ritonavir, indinavir, nelfinavir, voriconazole) or inducers such as phenytoin, carbamazepine, rifampin, rifabutin, rifapentine, phenobarbital.[22][21] It also inhibits CYP1A2, CYP2C9, and CYP3A4 and likewise should not be taken concurrently with substrates of any of these enzymes that have a narrow therapeutic index.[22][21]
Pharmacology
Pharmacodynamics

Abiraterone, the active metaboliteof abiraterone acetate.
Antiandrogenic activity
Abiraterone, the active metabolite of abiraterone acetate, inhibits CYP17A1, which manifests as two enzymes, 17α-hydroxylase (IC50 = 2.5 nM) and 17,20-lyase (IC50 = 15 nM) (approximately 6-fold more selective for inhibition of 17α-hydroxylase over 17,20-lyase)[23][24] that are expressed in testicular, adrenal, and prostatic tumor tissues. CYP17A1 catalyzes two sequential reactions: (a) the conversion of pregnenolone and progesterone to their 17α-hydroxy derivatives by its 17α-hydroxylase activity, and (b) the subsequent formation of dehydroepiandrosterone (DHEA) and androstenedione, respectively, by its 17,20-lyase activity.[25] DHEA and androstenedione are androgens and precursors of testosterone. Inhibition of CYP17A1 activity by abiraterone thus decreases circulating levels of androgens such as DHEA, testosterone, and dihydrotestosterone (DHT). Abiraterone acetate, via its metabolite abiraterone, has the capacity to lower circulating testosterone levels to less than 1 ng/dL (i.e., undetectable) when added to castration.[23][26] These concentrations are considerably lower than those achieved by castration alone (~20 ng/dL).[26] The addition of abiraterone acetate to castration was found to reduce levels of DHT by 85%, DHEA by 97 to 98%, and androstenedione by 77 to 78% relative to castration alone.[26] In accordance with its antiandrogenic action, abiraterone acetate decreases the weights of the prostate gland, seminal vesicles, and testes.[27]
Abiraterone also acts as a partial antagonist of the androgen receptor (AR), and as an inhibitor of the enzymes 3β-hydroxysteroid dehydrogenase (3β-HSD), CYP11B1 (steroid 11β-hydroxylase), CYP21A2 (Steroid 21-hydroxylase), and other CYP450s (e.g., CYP1A2, CYP2C9, and CYP3A4).[22][28][29][30] In addition to abiraterone itself, part of the activity of the drug has been found to be due to a more potent active metabolite, δ4-abiraterone (D4A), which is formed from abiraterone by 3β-HSD.[31] D4A is an inhibitor of CYP17A1, 3β-hydroxysteroid dehydrogenase/Δ5-4 isomerase, and 5α-reductase, and has also been found to act as a competitive antagonist of the AR reportedly comparable to the potent antagonist enzalutamide.[31] However, the initial 5α-reduced metabolite of D4A, 3-keto-5α-abiraterone, is an agonist of the AR, and promotes prostate cancer progression.[32] Its formation can be blocked by the coadministration of dutasteride, a potent and selective 5α-reductase inhibitor.[32]
Estrogenic activity
There has been interest in the use of abiraterone acetate for the treatment of breast cancer due to its ability to lower estrogen levels.[33] However, abiraterone has been found to act as a direct agonist of the estrogen receptor, and induces proliferation of human breast cancer cells in vitro.[33] If abiraterone acetate is used in the treatment of breast cancer, it should be combined with an estrogen receptor antagonist like fulvestrant.[33] In spite of its antiandrogenic and estrogenic properties, abiraterone acetate does not appear to produce gynecomastia as a side effect.[34]
Other activities
Due to inhibition of glucocorticoid biosynthesis, abiraterone acetate can cause glucocorticoid deficiency, mineralocorticoid excess, and associated adverse effects.[35] This is why the medication is combined with prednisone, a corticosteroid, which serves as a means of glucocorticoid replacement and prevents mineralocorticoid excess.[36]
Abiraterone acetate, along with galeterone, has been identified as an inhibitor of sulfotransferases (SULT2A1, SULT2B1b, SULT1E1), which are involved in the sulfation of DHEA and other endogenous steroids and compounds, with Ki values in the sub-micrmolar range.[37]
Pharmacokinetics
After oral administration, abiraterone acetate, the prodrug form in the commercial preparation, is converted into the active form, abiraterone. This conversion is likely to be esterase-mediated and not CYP-mediated. Administration with food increases absorption of the drug and thus has the potential to result in increased and highly variable exposures; the drug should be consumed on an empty stomach at least one hour before or two hours after food. The drug is highly protein bound (>99%), and is metabolised in the liver by CYP3A4 and SULT2A1 to inactive metabolites. The drug is excreted in feces (~88%) and urine (~5%), and has a terminal half-life of 12 ± 5 hours.[21]
Chemistry
Abiraterone acetate, also known as 17-(3-pyridinyl)androsta-5,16-dien-3β-ol acetate, is a synthetic androstane steroid and a derivative of androstadienol (androsta-5,16-dien-3β-ol), an endogenous androstane pheromone. It is specifically a derivative of androstadienol with a pyridine ring attached at the C17 position and an acetate ester attached to the C3β hydroxyl group. Abiraterone acetate is the C3β acetate ester of abiraterone.
History
In the early 1990s, Mike Jarman, Elaine Barrie, and Gerry Potter of the Cancer Research UK Centre for Cancer Therapeutics in the Institute of Cancer Research in London set out to develop drug treatments for prostate cancer. With the nonsteroidal androgen synthesis inhibitor ketoconazole as a model, they developed abiraterone, filing a patent in 1993 and publishing the first paper describing it the following year.[5][38] Rights for commercialization of the drug were assigned to BTG, a UK-based specialist healthcare company. BTG then licensed the product to Cougar Biotechnology, which began development of the commercial product.[39] In 2009, Cougar was acquired by Johnson & Johnson, which developed and sells the commercial product, and is conducting ongoing clinical trials to expand its clinical uses.[40]
Abiraterone acetate was approved by the United States Food and Drug Administration on April 28, 2011.[6][7] The FDA press release made reference to a phase III clinical trial in which abiraterone use was associated with a median survival of 14.8 months versus 10.9 months with placebo; the study was stopped early because of the successful outcome.[41]Abiraterone acetate was also licensed by the European Medicines Agency.[42] Until May 2012 the National Institute for Health and Clinical Excellence (NICE) did not recommend use of the drug within the NHS on cost-effectiveness grounds. This position was reversed when the manufacturer submitted revised costs.[43] The use is currently limited to men who have already received one docetaxel-containing chemotherapy regimen.[44][45]
Society and culture
Generic names
Abiraterone acetate is the generic name of the drug and its USAN, BANM, and JAN, while abiraterone is the INN and BAN of abiraterone, its deacetylated form.[9] Abiraterone acetate is also known by its developmental code names CB-7630 and JNJ-212082, while CB-7598 was the developmental code name of abiraterone.[9][46]
Brand names
Abiraterone acetate is marketed by Janssen Biotech (a subsidiary of Johnson & Johnson) under the brand name Zytiga.[9] In addition, Intas Pharmaceuticals markets the drug under the brand name Abiratas, Cadila Pharmaceuticals markets the drug as Abretone, and Glenmark Pharmaceuticals as Abirapro.[citation needed]
Availability
Abiraterone acetate is marketed widely throughout the world, including in the United States, Canada, the United Kingdom, Ireland, elsewhere in Europe, Australia, New Zealand, Latin America, Asia, and Israel.[9]
Research
Abiraterone acetate is under development for the treatment of breast cancer and ovarian cancer and as of March 2018 is in phase II clinical trials for these indications.[46] It was also under investigation for the treatment of congenital adrenal hyperplasia, but no further development has been reported for this potential use.[46] An oral ultramicrosize tablet formulation of abiraterone acetate (also known as abiraterone acetate fine particle (AAFP) or submicron abiraterone acetate) with improved bioavailability is in pre-registration in the United States for the treatment of prostate cancer as of April 2018 and has the tentative brand name Yonza.[47]
PAPER
https://pubs.acs.org/doi/abs/10.1021/op500044p
Improved Procedure for Preparation of Abiraterone Acetate
Chemical Research Division, Ranbaxy Research Laboratory, Gurgaon, Haryana 122001, India
Org. Process Res. Dev., 2014, 18 (4), pp 555–558
DOI: 10.1021/op500044p
*E-mail: Mukesh.madhra@ranbaxy.com. Tel: (91-124)4011832.

An improved procedure for the preparation of abiraterone acetate is described. The present process highlights reduced reaction time, isolation with acid–base treatment without involving column chromatography, multiple crystallization and is amenable to large-scale synthesis.
Abiraterone Acetate (1)
1 in 81% yield (1.8 kg). HPLC Purity: 99.72%, Assay: 98.8% (HPLC, w/w). MS: m/z = 392.7 [M + H]+. IR (KBr) (cm–1): 3047, 2936, 1735, 1244, 1035, 801, 714. 1H NMR (400 MHz, DMSO-d6): δ 8.58 (s, 1 H), 8.43–8.42 (d, 1 H), 7.76–7.74 (d, 1 H), 7.34–7.31 (dd, 1 H), 6.11 (s, 1 H), 5.38 (s, 1 H), 4.44 (m, 1H), 2.19–2.50 (m, 3H), 1.98–2.08 (m, 6H), 1.39–1.85 (m, 9H), 1.03–1.11 (m, 8H). 13C NMR (CDCl3): δ 170.4, 151.6, 147.9, 147.8, 140.0, 133.6, 132.9, 129.1, 122.9, 122.2, 73.8, 57.4, 50.2, 47.3, 38.1, 36.9, 36.7, 35.1, 31.7, 31.4, 30.3, 27.7, 21.4, 20.8, 19.2, 16.5.
https://pubs.acs.org/doi/suppl/10.1021/op500044p/suppl_file/op500044p_si_001.pdf




Abiraterone (2)
2 (2.88 kg, 72%) as a white solid. HPLC Purity: 99.87%. MS: m/z = 350.3 [M + H]+. IR (KBr) (cm–1): 3236, 3062, 3031, 2931,1596, 1065, 803. 1H NMR (400 MHz, CDCI3): δ 8.61 (s, 1 H), 8.44–8.46 (d, 1 H), 7.63–7.65 (d, 1 H), 7.20–7.23 (dd, 1 H), 5.993–5.996 (d, 1 H), 5.38–5.99 (d, 1 H), 3.48–3.54 (m, 1 H), 2.24–2.32 (m, 3H), 1.97–2.10 (m, 3H), 1.47–1.86 (m, 10H), 1.04–1.10 (s, 8H). 13C NMR (CDCl3): δ 16.58, 19.34, 20.88, 30.45, 31.52, 31.63, 31.81, 35.27, 36.71, 37.20, 42.32, 47.34, 50.37, 57.56, 71.62, 121.28, 123.03, 129.24, 132.99, 133.70, 141.21, 147.79, 147.88, 151.68
see supp info
PATENT
PATENT
The abiraterone acetate was the ester of formula (Abiraterone acetate) structure.
[0004]

[0005] So far, the search route may abiraterone acetate ester (Abiraterone acetate) are two.
[0006] Patent W09509178, CN 102030798, WO 2006021777, 2006021777, WO2006021776, J.Med.Chem.38,2463-2471,1995, synthetic route reported in the literature like the following formula WO.
[0007]

[0008] The route is DHEA as raw material, with an acetyl group protecting the hydroxyl group, the product obtained is then reacted with trifluoromethanesulfonic anhydride to give triflate product was finally reagent under palladium catalysis, Suzuki coupling reaction with 3-pyridyl diethyl borane, to give an ester of abiraterone acetate.
[0009] Patent GB 2282377,0PPI, 29 (I), 123-134,1997 the reported another method of synthesis.
The synthetic procedure the following formula.
[0010]

[0011] The route is DHEA as raw material, the reaction with hydrazine hydrate, and then reacted with iodine to give the 17-iodo – androsta-5,16-diene–3beta- alcohol, and catalytic agent in the button with Li-yl-pyrazol-diethyl _3_ boron burning Suzuki coupling reaction to give abiraterone, and finally acetylated abiraterone acetate to give abiraterone acetate.
[0012] By comparing the two lines, a synthetic routes can be found with a reagent such as trifluoromethanesulfonic anhydride, 2,6-di-t-butyl-4-methylpyridine and the like are expensive, relatively high chemical costs. In comparison, two synthetic route mild reaction conditions, the reagents are cheap, and therefore have more industrialized prospects. However, according to the synthesis process reported in the literature, the route to industrial production, there are still some technical problems.
[0013] More specifically, to 17- iodo – androsta-5,16-diene–3beta_ when alcohol (2) Synthesis of abiraterone (3) as a raw material for the Suzuki coupling reaction, the solvent is tetrahydrofuran, the solvent high cost; shall reaction refluxed for 4 days, the reaction time is too long. More importantly, when the Suzuki coupling reaction, starting material 17- iodo – male left diene-5,16-ol _3beta_
(2) will react with the impurities abiraterone (3) 4, 4 impurities not removed by recrystallization, can only be purified by column chromatography. If the compound is not 4 Ex, abiraterone prepared by acetylation reaction of abiraterone acetate ester, the impurities will be converted to 4 5 impurities, the impurities by recrystallization 5 likewise not removed, only purified by column chromatography.
[0014]

[0015] The abiraterone acetate ester synthesis, synthesis is reported abiraterone 24h the reaction with acetic anhydride and pyridine at room temperature, the reaction time is too long. The mixture was then evaporated under reduced pressure to be excess acetic anhydride and pyridine, and then crystallized from diethyl ether again, to give the final acetate abiraterone acetate was purified by column chromatography.
[0016] In summary, two synthetic routes reported in the literature of the last two long reaction time and complicated operation, product purification difficult. All this has seriously hampered the industrialization prospects abiraterone acetate esters.It is essential to two synthetic routes to optimize the improvement, in order to achieve the industrial production of abiraterone acetate ester.

] Example 1
Preparation of [0031] 17- (3-pyrazol Li-yl) androsta-5,16-diene-_3beta_ alcohol (abiraterone) of
[0032] A 750ml NMP was added to a 3L three-necked flask, were added with stirring 50gl7_ iodo – androsta-5,16-diene–3beta- alcohol, 88 mg of bis (triphenylphosphine) palladium chloride and diethyl 19.74g yl – (3-pyridyl) borane, and finally adding 345ml 2mol / L Na2CO3 solution. Heating, holding temperature of about 70-80 ° C, TLC monitored the reaction was complete. The reaction was cooled to room temperature, the reaction solution was added 1500ml of water, stirred, filtered and washed with water. Blast drying, 26.3g abiraterone.
[0033] Example 2
[0034] Preparation and purification of abiraterone acetate ester
[0035] The abiraterone 26g 156ml dissolved in pyridine, 52 ml of acetic anhydride was added at room temperature, heating, holding temperature of about 70-80 ° C, the reaction for about 4 h, TLC monitoring of the reaction was complete. The reaction was cooled to room temperature, the ice bath, 560ml of ice water was added to the reaction mixture, the precipitated white solid was stirred 20min, filtered, washed with water. 55 ° C blast drying. The crude product was added to 26ml of ethanol was dissolved by heating to clarify. Water was added 26ml, stirred for lh. Cooled to room temperature and filtered. Blast drying. Abiraterone acetate to give the final acetate 22.lg, HPLC> 99.5%.
[0036] Example 3
Preparation of [0037] 17- (3-pyridyl) androsta-5,16-diene-_3beta_ alcohol (abiraterone) of
[0038] The IlOL NMP was added to a 3L three-necked flask, were added with stirring 7.5kgl7_ iodo – androsta-5,16-diene–3beta- alcohol, 132 g of bis (triphenylphosphine) palladium chloride and 29.6kg two ethyl – (3-pyridyl) borane and finally 500L2mol / L Na2CO3 solution. To maintain the internal temperature of about 70_80 ° C, TLC monitored the reaction was complete. The reaction was cooled to room temperature, 220L of water was added to the reaction mixture, stirred for 30min, filtered, washed with water. Blast drying, 39.2kg abiraterone.
[0039] Example 4
[0040] Preparation and purification of abiraterone acetate ester
[0041] The abiraterone 39kg dissolved in pyridine 230L, 78L of acetic anhydride was added at room temperature, heating, holding temperature of about 70-80 ° C, the reaction for about 4 h, TLC monitoring of the reaction was complete. The reaction was cooled to room temperature, the ice bath, ice water was added to the 840L reaction solution, stirred 30min, filtered, washed with water, 50_55 ° C blast drying. The crude product was added to 39L of ethanol was dissolved by heating to clarify. Water was added 39L, stirred for lh then cooled to room temperature and filtered. Blast drying. Abiraterone acetate to obtain the final ester 33.2kg, HPLC> 99.5% ο
PATENT
Abiraterone acetate [17-(3-pyridyl)-5,16-androstadien-33-acetate] is a steroid compound which inhibits selectively and efficiently the enzyme 17-ohydroxylase-C17- 20-lyase, which catalyzes the conversion of dehydroepiandrosterone and androstenedione to testosterone. The inhibition of said enzyme causes a strong decrease of testosterone levels in the patient and therefore this drug is used in the treatment of certain hormone-dependent tumors resistant to chemotherapy such as prostate cancer. This compound has the followin chemical formula:

This product was disclosed for the first time in WO 93/20097, which also provides a synthetic process for its preparation including as last step the reaction of an enol triflate with a pyridine borate by Suzuki coupling (see scheme below). However, this process is not viable in practice, mainly because of the difficulty in preparing the enol trifluorosulfonate at the 17-position 2: this step, apart from proceeding with a poor conversion and low yield, gives place to the impurity tri-unsaturated 3 in a 10% yield, which only may be removed by column chromatography. Further, the product obtained after the subsequent Suzuki coupling must be also purified by column chromatography according to the examples provided therein.

Abiraterone-acetate
The above-mentioned impurity was prevented in later processes (EP 1 781 683 y EP 1 789 432) thanks to the use of alternative bases to that previously employed (i.e. 2,5-ditert-butyl-4-methylpyridine) such as DABCO, DBU or tryethylamine. However, in the sole example described in said documents, whilst the final product is achieved without using any column chromatography, it is obtained in a global yield of scarcely 21 % and shows a purity of only 96.4%.

EP 0 721 461 proposes the use of a vinyl iodide or bromide intermediate instead of the enol triflate, as depicted in the following scheme:

However, the iodo-enol is much less reactive than the triflate in the coupling with the pyridine borane, resulting in long reaction times (48 hours – 4 days) with a part of the starting material unreacted and wherein until a 5% of a dimeric impurity is obtained, which can only be removed by purification by means of reverse phase column chromatograp

Therefore, there is still a need of developing new processes for obtaining 17-(3- pyridyl)-5,16-androstadien-33-ol and related compounds, some of which are of therapeutic interest (e.g. abiraterone acetate) which overcome all or part of the drawbacks associated to the known processes belonging to the state of the art.
PATENT
The chemical name of abiraterone acetate ⑽) -17- (3- pyridyl) – androsta-5,16-diene-3-acetate, by the oral US Centocor Ortho developed CYP17 inhibitor . As anti-cancer drugs on the market April 28, 2011 by the US Food and Drug Administration (FDA) approval, in combination with prednisone therapy with castration-resistant prostate cancer. Trade name Zyitga. Abiraterone acetate is an oral androgen synthesis inhibitors, capable of inhibiting 17a- hydroxylase / C17,20-lyase (CYP17). Clinical results show that abiraterone acetate can significantly prolong patients with advanced prostate cancer include the use of one or both docetaxel-containing chemotherapy but her condition is still deteriorating lives of patients, the risk of death by 35%, and the side effects of drugs is very small, good safety.
[0003] Currently, the synthesis of abiraterone acetate routes are mainly three: (1) dehydroepiandrosterone acetic acid as a starting material, first-butyl-4-methylpyridine 2,6_ di ( esterified by trifluoromethanesulfonic anhydride, then with diethyl under DTBMP) under catalytic bis-triphenylphosphine palladium chloride – acetate proceeded abiraterone acetate Suzuki coupling (2-pyridyl) borane the total yield of the reaction is 48.7%; short reaction step of the process, but after the first-stage reaction a lot of by-products, to be purified by column chromatography, and the double bonds can not be divided by-products, and therefore remains a need for second-stage reaction column chromatography Analysis and purified by recrystallization complicated operation; (2) DHEA as a starting material, a condensation reaction of a hydrazone with hydrazine hydrate, and the presence (TMG) in 1,1,3,3-tetramethylguanidine ene reaction with iodine to generate iodine compound 17- iodo – male left -5,16_ diene -30- alcohol, then in the catalytic bis-triphenylphosphine palladium dichloride and diethyl – (3-pyridyl ) borane was prepared by Suzuki coupling abiraterone abiraterone acetate to give finally acetylated hydroxyl prepared. The total yield of the reaction is 41.5%. This synthesis step is longer, lower yields, and since the active iodides easy to generate high polymer impurities that can not be removed by recrystallization or column chromatography, can only be purified by preparative chromatography to give A in the Suzuki coupling process Long bit acetate pure, can not meet the requirements of industrial production; (3) in the method (1) was treated with triethylamine instead of DTBMP, reduces the formation of byproducts double bond, then after the reaction the remaining starting material was recrystallized removed. This reaction increases the process steps and the purity of the final product was only 96.4%, the drug does not meet the quality standards.
Example 1
[0021] One method of synthesis of abiraterone acetate, comprising the steps of:
[0022] A, in a 100ml round bottom flask were sequentially added 0 • 95g (5mmol) of p-toluenesulfonyl chloride, 15 mL of toluene, sufficiently stirred to dissolve, to obtain X-solution, (solution cooled to 15 ° C X) at 15 ° C under a slow was added dropwise by molar ratio of 1: 2 was added 1.5mL (lOmmol) 80% hydrazine hydrate to the solution X, dropwise within 5min; 3〇min reaction was continued, white precipitate appears in the flask. TLC analysis of the reaction end point is determined. After completion of the reaction, cold water was added 3〇mllO ° C., Stirred, filtered off with suction, the filter cake was then washed with purified water 3-5 times, and dried to obtain a white crystalline p-toluene sulfonyl hydrazide billion • 82g, 86.3% yield ( literature values: yield 92%).
[0023] B, DHEA -17- Synthesis of p-toluenesulfonyl hydrazone
[0024] In a 100ml round bottom flask were sequentially added in square • 75g CMmo 1) dehydroepiandrosterone (DHEA), 25 mL of methanol, 0.81 g sufficiently stirred to dissolve the p-toluene sulfonyl hydrazide, rt (15_25 ° C), was added O.lmL 0.2 mol .L-1 sulfuric acid, 60 ° C in an oil bath at reflux for 2h, TLC analysis of the reaction end point is determined. After completion of the reaction, the solvent was largely removed by rotary evaporation, a heavy white precipitate appeared in the flask. Was added 30mL of water, filtered off with suction, the filter cake was then washed with purified water 3-5 times to remove water at one thousand bake 50 ° C, to give a white solid 1.27g i.e. -17- Dehydroepiandrosterone p-toluenesulfonyl hydrazone, yield 81.4%.
[0025] C, 17- (3- pyridyl) androsta-5,16-diene–30- _ Synthesis of alcohol
[0026] In a 100ml round bottom flask was added 0.91g (2mmol) -17- Dehydroepiandrosterone p-toluenesulfonyl hydrazone, 25mLl, 4- dioxane, 〇.27g (3 mmol of) lithium tert-butoxide, 0_012g (0.013mmol) Pd2 (dba) 3,0.023g stirred for five minutes to fully dissolve the (0_005mmol) Xphos, at room temperature, wherein Pd2 (dba) 3 were added under nitrogen, and then quickly added 0.39g (2.5mmol) 3- bromo pyridine, 95 ° C oil bath reactor 12h, TLC analysis of the reaction end point is determined. After the reaction, ice water was added 30mL0 ° C, thoroughly shaken, ethyl acetate was added 20mL of acetic acid, liquid separation, was extracted with ethyl acetate (1 〇ml each, extracted three times) and the combined organic phase was dried over anhydrous sodium sulfate (by lg / ml was added over anhydrous sodium ratio) sulfate, filtered, and the filtrate rotary evaporated to give a pale yellow solid which was recrystallized from n-hexane (20ml) to give a white solid that is 17- (3-pyridyl) – male stay -5, 16- -3P- diene alcohols, a yield of 42.6%.
[0027] D, Synthesis of abiraterone acetate
[0028] In 0.39gl7- successively added 100mL round bottom flask (3-pyridyl) – androsta-5,16-diene-fir -3 – ol, 3〇111 dagger diethyl ether, 0.15mL (0.25mmol) triethylamine amine, are hook stirring, was slowly added dropwise 0.3 mL (2mm〇l) of acetyl chloride, the reaction stirred at room temperature Jiao 3h, TLC analysis of the reaction end point is determined. The mixture was then suction filtered, the filtrate was decolorized with charcoal, rotary evaporation, to give a white solid, i.e. abiraterone acetate product yield of 80.6%.
[0029] Example 2
[0030] A, in a 100ml round bottom flask were added 1. (^ (5.311111101) p-toluenesulfonyl chloride, 15 mL of toluene, sufficiently stirred to dissolve to give the solution X, at 15 ° C was slowly added dropwise 1 • 7mL (12mmol) X 80% hydrazine hydrate to the solution in dropwise within 5min; reaction was continued for 30min, a white precipitate appeared .TLC analysis to determine the end of the reaction after the completion of the reaction flask was added 30ml 10 ° C water with stirring, suction filtered, then the filter cake. washed 3-5 times purified water, was dry, i.e., p-toluenesulfonyl hydrazide to give white crystals 0.93 g, 86.7% yield (literature: yield 92%).
[0031] B, DHEA -17- Synthesis of p-toluenesulfonyl hydrazone
[0032] successively added 0 • 97g (3mmo 1) dehydroepiandrosterone (DHEA) in a 100ml round bottom flask, 25 mL of methanol, 1 • 08g p-toluene sulfonyl hydrazide, fully dissolved with stirring at room temperature, was added O.lmL 0.2mol • L_1 sulfuric acid, 60 ° C in an oil bath at reflux for 2h, TLC analysis of the reaction end point is determined. After completion of the reaction, the solvent was largely removed by rotary evaporation, a heavy white precipitate appeared in the flask. Was added 30mL of water, filtered off with suction, the filter cake was then washed with purified water 3-5 times, 5 (TC drying under removal of water, to give a white solid 1.43g i.e. Dehydroepiandrosterone p-toluenesulfonyl hydrazone -17- yield 80.9%.
[0033] C, 17- (3- pyridyl) -30- _ male left diene -5,16-ol Synthesis
[0034] In a 100ml round bottom flask was added in 1.32g (3 mmol of) -17- Dehydroepiandrosterone p-toluenesulfonyl hydrazone, 25mLl, 4- dioxane, 0.35g (4mmol) of lithium t-butoxide, 0.012g (0.013_ol) Pd2 (dba) 3,0.023g stirring for five minutes under fully dissolved (0.005mmol) Xphos, at room temperature, wherein Pd2 (dba) 3 were added under nitrogen, and then quickly added 0.48g (3mmol) 3- bromo pyridine, 80 ° C oil bath and the reaction 19h, TLC analysis of the reaction end point is determined. After the reaction, 30mL of ice water was added, shaken well, 2〇mL ethyl acetate was added, liquid separation, was extracted with 30ml ethyl acetate (10ml each, extracted three times) and the combined organic phases were scaled lg / ml was added anhydrous dried over sodium sulfate, filtered, and the filtrate was rotary evaporated to give a pale yellow solid which was recrystallized from 20ml of n-hexane to give a white solid that is 17- (3-pyridyl) – androsta-5,16-diene–3P- alcohol, 42 • 6% yield.
[0035] D, Synthesis of abiraterone acetate
[0036] In 0.41gl7- successively added 100mL round bottom flask (3-pyridyl) -! -33- androst-5,16-diene-ol, ^ 3〇 diethyl ether, 0.2mL (0 • 3 round 〇1 ) of triethylamine, stir, slowly added dropwise 0.3mL (2mmol) of acetyl chloride, the reaction was stirred at room temperature for 3h, TLC analysis of the reaction end point is determined. The mixture was then suction filtered, the filtrate was decolorized with charcoal, rotary evaporation, to give a white solid, i.e. abiraterone acetate product yield of 81 • 2%.
[0037] Example 3
[0038] A, were added in a 100ml round-bottomed flask 1.08g (5.7mmo 1) p-toluenesulfonyl chloride, 15 mL of toluene, sufficiently stirred to dissolve to give the solution X, at 15 ° C was slowly added dropwise 1 • 8mL (13mmo 1 ) of 80% hydrazine hydrate to the solution X, dropwise within 5min; reaction was continued for 30min, a white precipitate appeared in the flask. TLC analysis of the reaction end point is determined. After completion of the reaction, water 30ml 10 ° C with stirring, filtered off with suction, the filter cake was then washed with purified water 3-5 times, and dried to obtain a white crystalline p-toluene sulfonyl hydrazide 0.94g, 85.6% (Yield literature values: yield 92%).
[0039] B, DHEA -17- Synthesis of p-toluenesulfonyl hydrazone
[0040] 1 • 18g were added in a 100ml round bottom flask (3.3 mmol) Dehydroepiandrosterone, 25 mL of methanol, 1.28 g of p-toluene sulfonyl hydrazide, fully dissolved with stirring at room temperature, was added O.lmL 0.2mol • I / a sulfate, an oil bath at reflux for 2h, TLC analysis of the reaction end point is determined. After completion of the reaction, the solvent was largely removed by rotary evaporation, a heavy white precipitate appeared in the flask. Was added 30mL of water, filtered off with suction, the filter cake was then washed with purified water 3-5 times, 5 (TC drying under removal of water, to give a white solid 1.51g i.e. Dehydroepiandrosterone p-toluenesulfonyl hydrazone -17- yield 80.9%.
[0041] C, 17- (3- pyridyl) – androst _5,16- diene synthetic alcohols -3P-
[0042] Add l_45g (3.2mmol) -17- Dehydroepiandrosterone p-toluenesulfonyl hydrazone, 25mLl, 4- dioxane, 0 • 35g in 100ml round bottom flask (4mmol) of lithium tert-butoxide, 0.012 g (0.013mmol) Pd2 (dba) 3,0.023g (0.005ramol) Xphos, fully dissolved after stirred at room temperature for five minutes, wherein Pd2 (dba) 3 were added under nitrogen, then added rapidly 0 • 60g (4mmol) 3 – bromopyridine, 120 ° C oil bath and the reaction 9h, TLC analysis of the reaction end point is determined. After the reaction, 30mL of ice water was added, shaken well, was added 20mL of ethyl acetate, separated, extracted with 30ml ethyl acetate (10ml each, extracted three times) and the combined organic phases were scaled lg / ml was added over anhydrous sodium to intervene sulfate, filtered, and the filtrate rotary evaporated to give a pale yellow solid which was recrystallized from burning 2〇ml n-hexyl, i.e. 17_ to give a white solid (3-Jie ratio piperidyl) – androst -5,16_ diene -30- alcohols, a yield of 43.1%.
[0043] D, Synthesis of abiraterone acetate
[0044] successively added in a round bottom flask i〇〇mL 0.52gl7- (3- pyridyl) -! -30- androst-5,16-diene-ol, ^ 3〇 diethyl ether, 0.25mL (0.36mmol) triethylamine, stir, slowly added dropwise 0.35 mL (2.2 mmol) of acetyl chloride, the reaction was stirred at room temperature for 3h, TLC analysis of the reaction end point is determined. The mixture was then suction filtered, the filtrate was decolorized with charcoal, rotary evaporation, to give a white solid, i.e. abiraterone acetate product yield of 81.8%.
[0045] In each of the above embodiments, the improved synthetic route abiraterone acetate to DHEA as raw materials by the condensation of p-toluenesulfonyl hydrazide, and then reacted with 3-bromopyridine coupling occurs, acetylation, etc. 3 target product was synthesized from abiraterone acetate, 43.4% overall yield.Route and the mild reaction conditions, readily available and inexpensive raw materials, low production cost.
PAPER
Four-Step Synthesis of Abiraterone Acetate from Dehydroepiandrosterone
https://link.springer.com/article/10.1007/s11094-016-1459-1
Balaev, A.N., Gromyko, A.V. & Fedorov, V.E. Pharm Chem J (2016) 50: 404. https://doi.org/10.1007/s11094-016-1459-1
Syn 1
J Med Chem 1995,38(13),2463

Treatment of dehydroepiandrosterone 3-acetate (I) with triflic anhydride and 2,6-di-tert-butyl-4-methylpyridine provided the desired enol triflate (III) along with some 3,5-diene (II), which were separated by column chromatography. Subsequent coupling of triflate (III) with pyridylborane (IV) using bis(triphenylphosphine)- palladium(II) chloride as the catalyst afforded the (3-pyridyl)androstadiene (V), which after hydrolysis of the acetate ester with NaOH provided the target compound.
Abiraterone
-
- Synonyms:CB-7598
- ATC:L02BX03
- Use:androgen biosynthesis inhibitor for treating prostate cancer
- Chemical name:(3β)-17-(3-pyridinyl)androsta-5,16-dien-3-ol
- Formula:C24H31NO
- MW:349.51 g/mol
- CAS-RN:154229-19-3
Substance Classes
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 853-23-6 | C21H30O3 | dehydroepiandrosterone-3-acetate | Androst-5-en-17-one, 3-(acetyloxy)-, (3β)- |
| 89878-14-8 | C9H14BN | diethyl (3-pyridyl)borane | Pyridine, 3-(diethylboryl)- |
| C26H33NO2 | (3β)-acetoxy-17-(3-pyridyl)androsta-5,16-diene |
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| EU | Zytiga | Janssen Cilag, 2011 | |
| USA | Zytiga | Johnson & Johnson, 2011 |
Formulations
- tabs. 250 mg
References
-
- Potter, G. A. et al., J. Med. Chem., (1995) 38, 2463.
- US 5 604 213 (British Technology Group; 18.2.1997; appl. 30.9.1994; GB-prior. 31.3.1992).
- EP 633 893 (Brit. Technology Group; 18.1.1995; appl. 15.3.1993; GB-prior. 31.3.1992).
- WO 9 320 097 (Brit. Technology Group; 14.10.1993; appl. 15.3.1993; GB-prior. 31.3.1992).
-
large scale synthesis of acetate:
- Potter, G. A. et al., Org. Prep. Proced. Int., (1997) 29, 123.
CLIP
Abiraterone acetate, the active ingredient of ZYTIGA is the acetyl ester of abiraterone. Abiraterone is an inhibitor of CYP17 (17α-hydroxylase/C17,20-lyase). Each ZYTIGA tablet contains either 250 mg or 500 mg of abiraterone acetate. Abiraterone acetate is designated chemically as (3β)-17-(3-pyridinyl) androsta-5,16-dien-3-yl acetate and its structure is:
 |
Abiraterone acetate is a white to off-white, non-hygroscopic, crystalline powder. Its molecular formula is C26H33NO2 and it has a molecular weight of 391.55. Abiraterone acetate is a lipophilic compound with an octanol-water partition coefficient of 5.12 (Log P) and is practically insoluble in water. The pKa of the aromatic nitrogen is 5.19.
ZYTIGA tablets are available in 500 mg film-coated tablets, 250 mg film-coated tablets and 250 mg uncoated tablets with the following inactive ingredients:
- 500 mg film-coated tablets: colloidal silicon dioxide, croscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, silicified microcrystalline cellulose, and sodium lauryl sulfate. The coating, Opadry® II Purple, contains iron oxide black, iron oxide red, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
- 250 mg film-coated tablets: colloidal silicon dioxide, croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone, and sodium lauryl sulfate. The coating, Opadry® II Beige, contains iron oxide red, iron oxide yellow, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
- 250 mg uncoated tablets: colloidal silicon dioxide, croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone, and sodium lauryl sulfate.
PAPER
A CONVENIENT, LARGE-SCALE SYNTHESIS OF ABIRATERONE ACETATE [3β-ACETOXY-17-(3-PYRIDYL)ANDROSTA-5,16-DIENE], A POTENTIAL NEW DRUG FOR THE TREATMENT OF PROSTATE CANCER
Organic Preparations and Procedures International , The New Journal for Organic Synthesis , Volume 29, 1997 – Issue 1
https://www.tandfonline.com/doi/abs/10.1080/00304949709355175
- 10.1080/00304949709355175
- 3P-Acetoxy- 17-(3-pyridyl)androsta-5,16-diene (abiraterone acetate, 5) is a pro-drug for 17- (3-pyridyl)androsta-5,16-dien-3P-ol (abiraterone, 4), a potent inhibitor of human cytochrome P450,,, (steroidal 17a-hydroxylase-C,,,,,-lyase).’ This enzyme is a potential target for a drug designed to treat hormone-dependent prostatic carcinoma, and 5 has been approved for clinical trial in patients with this cancer. In connection with this trial, a method amenable to the synthesis of 5 on a large scale (ca. 100 g or more) was required. The present paper reports such a synthesis.

The key step in the previously reported’ synthesis of 5 was the palladium-catalysed crosscoupling reaction between diethyl(3-pyridy1)borane and the 17-en01 triflate derived from the 3-acetate of dehydroepiandrosterone 1. The procedure has potential drawbacks as a method for large-scale synthesis. Aside from the use of the expensive and noxious triflic anhydride, the formation of the enol triflate requires the expensive hindered base 2,6-di-tert-b~tyl-4-methylpyridine.~ Further, it was accompanied by some elimination of acetic acid to give androsta-3,5,16-trien- 17-yl triflate which required chromatographic separation from the desired product, and contributed to reducing its isolated yield from the acetate of 1 to a moderate 58%. These problems prompted consideration of an altemative steroidal precursor suitable for the cross-coupling reaction. It occurred to us that the vinyl iodide 3 might provide a viable alternative to an enol triflate in the palladium catalyzed cross-coupling step. Such steroidal vinyl iodides are easily and cheaply obtained via the corresponding 17-h~drazones.’-~ The synthesis of 3 iself from the hydrazone3 2 by oxidation with iodine in the presence of a hindered guanidine base has been optimi~ed~.~ to obtain the product on a small scale (0.13 g) in 95% yield. We were able to repeat this reaction on a large scale and obtain a similar yield of 3. The palladium catalysed cross-coupling reaction of 3 with diethyl(3-pyridy1)borane proceeded without the need to protect the 3-hydroxyl function to give 4, whereas the use of an enol triflate in the coupling reaction does not conveniently allow this option. However, coupling with the iodide was much slower, requiring 4 days at 80″ as compared with the 1 hr required when an enol triflate precursor was used.’ RO A ‘ OR Recrystallization of the crude 4 obtained by the foregoing procedure gave a product with melting point lower than that previously reported,’ and TLC revealed a less mobile contaminant that was not removed by further recrystallization. The crude product was therefore acetylated to give the crude target compound 5 contaminated with a by-product. This by-product was 6, formed from a precursor 7 present as a contaminant in crude 4. The prolonged reaction time required for the cross-coupling reaction using the vinyl iodide 3 had enabled a Heck-type reaction7 to occur between the initial product 4 and the bis(tripheny1phosphine)- palladium derivative of 3 to form 7. The very recently reported8 palladium-catalysed dimerisation of 17-i0dod’~-steroids to give 16,17′-coupled products provides a precedent for this side-reaction. Whereas column chromatography on silica-gel of crude 5 afforded pure 6, which was eluted first, compound 6 contaminated later fractions and could not be completely removed from 5 by recrystallization. However subsequent reverse phase chromatography allowed the complete separation of the now faster eluting 5 from 6, and recovery of >lo0 g of pure 5 by batchwise chromatography of the crude product. The by-product 6 was deacetylated to give 7, the contaminant present in 4 prepared by the present route. Neither of the new compounds 6 and 7 was appreciably inhibitory towards the human cytochrome P450,,, (S. E. Barrie, personal communication). The availability of pure 7 affords the option of exploring the purification of 4 prior to acetylation. However, for chromatographic purification, the greater solubilities of 5 and 6 in suitable organic solvents compared with their non-acetylated counterparts favor the present choice of purification after acetylation.
3P-Acetoxy-17-(3-pyridyl)androsta-5,16-diene ( 5) and 3~-acetoxy-16-(3~-acetoxyandrosta-5,16- dien-17-yl)-17-(3-pyridyl)androsta-5,16-diene (6).- To a stirred suspension of the product from the foregoing reaction (36.5 g, 0.104 mol) in dry pyridine (200 mL) in a 500 mL round-bottomed flask was added acetic anhydride (75 mL) and the mixture stirred at room temperature for 24 hrs. The pyridine and excess acetic anhydride was removed on a rotary evaporator, initially at water pump pressure with the water bath at 70 “, and finally under high vacuum at 80″ for 30 min. The resulting oil was dissolved in Et,O (500 mL), washed with saturated aqueous NaHCO, (2 x 200 mL), dried (N%CO,), and concentrated to an oil which crystallised on standing. The crude 5 was partly purified by preparative flash chromatography on silica gel using a 9 cm diameter column, eluting with dichloromethane. A by-product (6) eluted first and was followed by fractions variously enriched in 5. The foregoing reaction and purification procedure was carried out a total of four times, thus using a total of 146 g (0.41 8 mol) of crude 4. The dichloromethane fractions containing the least by-product were combined and concentrated. Recrystallisation from hexane afforded product (1 08 g) consisting of 5 containing 6.8% w/wof 6 as impurity as determined by analytical HPLC. The more contaminated fractions similarly afforded product (25 g) containing 21 3% w/w of 6 (we thank Dr C. P. Quarterman, Aston Molecules Ltd, Birmingham U.K. for these analyses). A pure sample of 6 (4 g) was isolated from the combined initial fractions as pale yellow crystals, mp. 269-270” (from hexane); IR 1732 cm-‘ (GO str); ‘H-NMR: 6 0.85 (s, 3, H-18′), 1.02, 1.04 (2s, 6, H-19,19′), 1.06 (s, 3, H-18), 2.034, 2.039 (2s, 6, CH,CO), 4.59 (2m, 2, H-3,3’), 5.13 (s, 1, H-16), 5.39 (dd, 2, H-6,6), 7.62 (dd, 1, Js,4 = 8.0 Hz, Js,6 = Anal. Calcd for C,,H,,NO,: C, 80.18; H, 8.73; N, 1.99. Found: C, 80.19; H, 8.78; N, 1.95
The crude 5 was purified by reverse-phase column chromatography. A solution of material from the 108 g batch (10 g) in a hot mixture of acetonitrile (200 mL) and methanol (40 mL) was allowed to cool and filtered. The filtrate was applied to a 10 cm diameter column (500 g) of LiChroprep@ RP-8 reverse-phase C, packing Art. No. 9324. The column was eluted with acetonitrile-0.05 M ammonium acetate (20: 1) with a flow rate of 25 amin and 500 mL fractions were collected and analysed by analytical HPLC (see below). Fractions 4-10 contained pure 5. After a further two fractions, the eluant was changed to acetonitrile-acetic acid (20:l) and pure 6 was completely eluted in fractions 16-19. In 3 subsequent runs using the same column, 25 g portions of the same batch of crude 5 were each dissolved in a mixture of hot acetonitrile (350 mL) and methanol (100 mL) and processed as before. Fractions 2-7 contained pure 5 and, following the change of solvent, fractions 8-12 contained 6. The four eluates containing 5 were combined and recrystallised from acetonitrile (1200 mL) to give pure 5 (57.5 g), mp. 146-148″, lit.’ mp. 144-145′, in which 6 was not detected by analytical HPLC (for procedure, see below) at the limit of detection (<0,05% w/w 6). Anal. Calcd for C,,H,,NO,: C, 79.75; H, 8.50; N, 3.58. Found: C, 79.73; H, 8.48; N, 3.62
Further material (14 g) from the 108 g batch was combined with a portion (22 g) of the more impure 25 g batch and the total of 36 g was chromatographed in one batch as above, again giving complete separation of 5 from 6. Recrystallisation from acetonitrile (600 mL) gave a further 28.5 g of 5 of purity equal to the foregoing crop of 57.5 g 5. Concentration of the combined mother liquors from these crops followed by addition of water (MeCN:&O, 12:l v/v) gave further pure 5 (17.5 g). The total recovery of pure 5 was therefore 103.5 g (36% based on 3). The spectroscopic data (NMR, IR, and MS) of the final products from this procedure were identical with those reported for the product obtained by the route previously described.’
Procedure for Analysis of Purity of Batches of 5 Using Analytical HPLC.- The eluant was acetonitrile-0.05M ammonium acetate and the flow rate 1.5 mumin. Components were monitored either by fluorescence detection (excitation wavelength hex 262 nm, emission wavelength hem 353 nm) or by UV detection (254 nm). Typical retention times were: for 5,225 sec; for 6, 1162 sec. For analysis of crystalline products, a solution (5 mg/mL) in acetonitrile was diluted 50 fold to 100 pg/mL and 100 pl of this solution was injected onto the column.
1 G. A. Potter, S. E. Barrie, M. Jarman and M. G. Rowlands, J. Med. Chem., 38,2463 (1995).
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q Janssen Biotech, Inc. “ZYTIGA Prescribing Information”(PDF). Horsham, PA: U.S. Food and Drug Administration.
- ^ Jump up to:a b c d “Meeting Library – Meeting Library”. meetinglibrary.asco.org.
- ^ Jump up to:a b c d Benoist GE, Hendriks RJ, Mulders PF, Gerritsen WR, Somford DM, Schalken JA, van Oort IM, Burger DM, van Erp NP (November 2016). “Pharmacokinetic Aspects of the Two Novel Oral Drugs Used for Metastatic Castration-Resistant Prostate Cancer: Abiraterone Acetate and Enzalutamide”. Clin Pharmacokinet. 55 (11): 1369–1380. doi:10.1007/s40262-016-0403-6. PMC 5069300
 . PMID 27106175.
. PMID 27106175. - Jump up^ Potter et al. J. Med. Chem., 1995, 38 (13), pp 2463–2471
- ^ Jump up to:a b Scowcroft H (2011-09-21). “Where did abiraterone come from?”. Cancer Research UK. Retrieved 2011-09-28.
- ^ Jump up to:a b “Drugs@FDA – FDA Approved Drug Products – Zytiga”. http://www.fda.gov. United States Food and Drug Administration. Retrieved 4 March 2016.
- ^ Jump up to:a b “FDA approves Zytiga for late-stage prostate cancer” (Press release). United States Food and Drug Administration. 2011-04-28.
- Jump up^ NCI Staff. “Abiraterone Approved for Earlier Use in Men with Metastatic Prostate Cancer”. U.S. Department of Health and Human Services, National Institutes of Health, National Cancer Institute.
- ^ Jump up to:a b c d e “Abiraterone”. Drugs.com.
- Jump up^ “Generic Zytiga Availability”. Drugs.com.
- ^ Jump up to:a b c “Zytiga (abiraterone acetate) tablet [Janssen Biotech, Inc.]”. DailyMed. Janssen Biotech, Inc. September 2013. Retrieved 24 January 2014.
- ^ Jump up to:a b c d “Zytiga: EPAR – Product Information” (PDF). European Medicines Agency. Janssen-Cilag International N.V. 29 October 2013. Retrieved 24 January 2014.
- ^ Jump up to:a b c “Zytiga 250 mg tablets – Summary of Product Characteristics”. electronic Medicines Compendium. Janssen-Cilag Ltd. 21 January 2014. Retrieved 24 January 2014.
- ^ Jump up to:a b c “Zytiga abiraterone acetate product information” (PDF). TGA eBusiness Services. Janssen-Cilag Pty Ltd. 1 March 2012. Retrieved 24 January 2014.
- Jump up^ “Pharmaceutical Benefits Scheme – Abiraterone”. Pharmaceutical Benefits Scheme. Retrieved 24 January 2014.
- Jump up^ Clinical trial number NCT00638690 for “Abiraterone Acetate in Castration-Resistant Prostate Cancer Previously Treated With Docetaxel-Based Chemotherapy” at ClinicalTrials.gov
- Jump up^ de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Saad F, Staffurth JN, Mainwaring P, Harland S, Flaig TW, Hutson TE, Cheng T, Patterson H, Hainsworth JD, Ryan CJ, Sternberg CN, Ellard SL, Fléchon A, Saleh M, Scholz M, Efstathiou E, Zivi A, Bianchini D, Loriot Y, Chieffo N, Kheoh T, Haqq CM, Scher HI (May 2011). “Abiraterone and increased survival in metastatic prostate cancer”. The New England Journal of Medicine. 364 (21): 1995–2005. doi:10.1056/NEJMoa1014618. PMC 3471149 . PMID 21612468.
- Jump up^ Clinical trial number NCT00887198 for “Abiraterone Acetate in Asymptomatic or Mildly Symptomatic Patients With Metastatic Castration-Resistant Prostate Cancer” at ClinicalTrials.gov
- Jump up^ “BTG and Ortho Biotech’s Prostate Cancer Trial Unblinded”. Genetic Engineering & Biotechnology News. 2010-09-10. Retrieved 2011-05-26.
- Jump up^ Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, Fizazi K, Mainwaring P, Piulats JM, Ng S, Carles J, Mulders PF, Basch E, Small EJ, Saad F, Schrijvers D, Van Poppel H, Mukherjee SD, Suttmann H, Gerritsen WR, Flaig TW, George DJ, Yu EY, Efstathiou E, Pantuck A, Winquist E, Higano CS, Taplin ME, Park Y, Kheoh T, Griffin T, Scher HI, Rathkopf DE (January 2013). “Abiraterone in metastatic prostate cancer without previous chemotherapy”. The New England Journal of Medicine. 368 (2): 138–48. doi:10.1056/NEJMoa1209096. PMC 3683570 . PMID 23228172.
- ^ Jump up to:a b c d e “Zytiga prescribing information” (pdf). Janssen Biotech. May 2012. Retrieved 4 March 2016.
- ^ Jump up to:a b c d e “Zytiga (abiraterone) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 24 January 2014.
- ^ Jump up to:a b Neidle S (30 September 2013). Cancer Drug Design and Discovery. Academic Press. pp. 341–342. ISBN 978-0-12-397228-6.
- Jump up^ Fernández-Cancio, Mónica; Camats, Núria; Flück, Christa E.; Zalewski, Adam; Dick, Bernhard; Frey, Brigitte M.; Monné, Raquel; Torán, Núria; Audí, Laura (2018-04-29). “Mechanism of the Dual Activities of Human CYP17A1 and Binding to Anti-Prostate Cancer Drug Abiraterone Revealed by a Novel V366M Mutation Causing 17,20 Lyase Deficiency”. Pharmaceuticals. 11 (2): 37. doi:10.3390/ph11020037.
- Jump up^ Attard G, Belldegrun AS, de Bono JS (December 2005). “Selective blockade of androgenic steroid synthesis by novel lyase inhibitors as a therapeutic strategy for treating metastatic prostate cancer”. BJU International. 96 (9): 1241–6. doi:10.1111/j.1464-410X.2005.05821.x. PMID 16287438.
- ^ Jump up to:a b c Small EJ (November 2014). “Can targeting the androgen receptor in localized prostate cancer provide insights into why men with metastatic castration-resistant prostate cancer die?”. Journal of Clinical Oncology. 32 (33): 3689–91. doi:10.1200/JCO.2014.57.8534. PMID 25311216.
Abiraterone acetate is a prodrug for abiraterone, a CYP17 inhibitor, which has the capacity to lower serum testosterone levels to less than 1 ng/dL (compared with levels closer to 20 ng/dL that are achieved with conventional ADT).19 […] Relative to LHRHa alone, the addition of abiraterone resulted in an 85% decline in dihydrotestosterone (DHT) levels, a 97% to 98% decline in dehydroepiandrosterone (DHEA) levels, and a 77% to 78% decline in androstenedione levels.
- Jump up^ Tindall DJ, James M (20 April 2009). Androgen Action in Prostate Cancer. Springer Science & Business Media. pp. 748–. ISBN 978-0-387-69179-4.
- Jump up^ Yin L, Hu Q (January 2014). “CYP17 inhibitors–abiraterone, C17,20-lyase inhibitors and multi-targeting agents”. Nature Reviews. Urology. 11 (1): 32–42. doi:10.1038/nrurol.2013.274. PMID 24276076.
- Jump up^ Malikova, Jana; Brixius-Anderko, Simone; Udhane, Sameer S.; Parween, Shaheena; Dick, Bernhard; Bernhardt, Rita; Pandey, Amit V. (2017). “CYP17A1 inhibitor abiraterone, an anti-prostate cancer drug, also inhibits the 21-hydroxylase activity of CYP21A2”. The Journal of Steroid Biochemistry and Molecular Biology. 174: 192–200. doi:10.1016/j.jsbmb.2017.09.007. ISSN 1879-1220. PMID 28893623.
- Jump up^ Udhane, Sameer S.; Dick, Bernhard; Hu, Qingzhong; Hartmann, Rolf W.; Pandey, Amit V. (2016). “Specificity of anti-prostate cancer CYP17A1 inhibitors on androgen biosynthesis”. Biochemical and Biophysical Research Communications. 477 (4): 1005–1010. doi:10.1016/j.bbrc.2016.07.019. ISSN 1090-2104. PMID 27395338.
- ^ Jump up to:a b Li Z, Bishop AC, Alyamani M, Garcia JA, Dreicer R, Bunch D, Liu J, Upadhyay SK, Auchus RJ, Sharifi N (July 2015). “Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer”. Nature. 523 (7560): 347–51. doi:10.1038/nature14406. PMC 4506215 . PMID 26030522.
- ^ Jump up to:a b Li Z, Alyamani M, Li J, Rogacki K, Abazeed M, Upadhyay SK, Balk SP, Taplin ME, Auchus RJ, Sharifi N (May 2016). “Redirecting abiraterone metabolism to fine-tune prostate cancer anti-androgen therapy”. Nature. 533 (7604): 547–51. doi:10.1038/nature17954. PMID 27225130.
- ^ Jump up to:a b c Capper CP, Larios JM, Sikora MJ, Johnson MD, Rae JM (May 2016). “The CYP17A1 inhibitor abiraterone exhibits estrogen receptor agonist activity in breast cancer”. Breast Cancer Research and Treatment. 157 (1): 23–30. doi:10.1007/s10549-016-3774-3. PMID 27083183.
- Jump up^ Alesini D, Iacovelli R, Palazzo A, Altavilla A, Risi E, Urbano F, Manai C, Passaro A, Magri V, Cortesi E (2013). “Multimodality treatment of gynecomastia in patients receiving antiandrogen therapy for prostate cancer in the era of abiraterone acetate and new antiandrogen molecules”. Oncology. 84 (2): 92–9. doi:10.1159/000343821. PMID 23128186.
- Jump up^ Figg WD, Chau CH, Small EJ (14 September 2010). Drug Management of Prostate Cancer. Springer Science & Business Media. p. 97. ISBN 978-1-60327-829-4.
- Jump up^ Rosenthal L, Burchum J (17 February 2017). Lehne’s Pharmacotherapeutics for Advanced Practice Providers – E-Book. Elsevier Health Sciences. p. 936. ISBN 978-0-323-44779-9.
- Jump up^ Yip CK, Bansal S, Wong SY, Lau AJ (April 2018). “Identification of Galeterone and Abiraterone as Inhibitors of Dehydroepiandrosterone Sulfonation Catalyzed by Human Hepatic Cytosol, SULT2A1, SULT2B1b, and SULT1E1”. Drug Metabolism and Disposition. 46 (4): 470–482. doi:10.1124/dmd.117.078980. PMID 29436390.
- Jump up^ “A new way to treat prostate cancer: The story of abiraterone”. The Institute of Cancer Research. 2012-09-10. Retrieved 2012-11-12.
- Jump up^ “Abiraterone Acetate (CB7630)”. Cougar Biotechnology. Archived from the original on 7 September 2008. Retrieved 2008-08-20.
- Jump up^ “Johnson & Johnson Announces Definitive Agreement to Acquire Cougar Biotechnology, Inc” (Press release). Cougar Biotechnology. 2009-05-11. Archived from the original on 29 May 2009. Retrieved 2009-06-03.
- Jump up^ “FDA Approval for Abiraterone Acetate”. U.S. Department of Health and Human Services, National Institutes of Health, National Cancer Institute.
- Jump up^ “EMA assessment of Zytiga (abiraterone)”. European Medicines Agency.
- Jump up^ “Prostate cancer (metastatic, castration resistant) – abiraterone (following cytoxic therapy): final appraisal determination guidance” (PDF). NICE guidance. 15 May 2012. Archived from the original (PDF) on February 19, 2013.
- Jump up^ “NICE technology appraisal guidance [TA259]”. NICE guidance. June 2012.
- Jump up^ “NICE appraisal of earlier treatment with abiraterone for prostate cancer”. NICE press release. 14 August 2014.
- ^ Jump up to:a b c “Abiraterone acetate – Johnson & Johnson”. Adis Insight.
- Jump up^ “Abiraterone acetate – Churchill Pharmaceuticals”. Adis Insight.
External links
 |
|
| Clinical data | |
|---|---|
| Trade names | Zytiga, others |
| Synonyms | CB-7630; JNJ-212082; 17-(3-Pyridinyl)androsta-5,16-dien-3β-ol acetate |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a611046 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth (tablets)[1] |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | Unknown, but may be 50% at most on empty stomach[3] |
| Protein binding | Abiraterone: ~99.8% (to albumin and α1-AGp)[3][1][2] |
| Metabolism | Esterases, CYP3A4, SULT2A1[2] |
| Metabolites | Abiraterone, others[1][3] |
| Elimination half-life | Abiraterone: 12–24 hours[1][3] |
| Excretion | Feces: 88%[1][2] Urine: 5%[1][2] |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C26H33NO2 |
| Molar mass | 391.555 g/mol |
| 3D model (JSmol) | |
| Melting point | 144 to 145 °C (291 to 293 °F) [4] |
Publication number/Priority date/Publication date/AssigneeTitle
WO1993020097A11992-03-31/1993-10-14/British Technology Group Ltd./17-substituted steroids useful in cancer treatment
US5618807A1992-03-311997-04-08British Technology Group LimitedMethod for preparing 17-substituted steroids useful in cancer treatment
EP1781683A12004-08-242007-05-09Btg International LimitedProcess fot the preparation of 17-0-vinyl- triflates as intermediates
EP1789432A12004-08-242007-05-30Btg International LimitedMethanesulfonate salts of abiraterone-3-esters and recovery of salts of abiraterone-3-esters from solution in methyl tert-butyl ether
US20130252930A1 *2010-12-162013-09-26Biomarin Pharmaceutical Inc.Cyp11b, cyp17, and/or cyp21 inhibitors
Non-Patent
Title
A. TAKEMIYA; J.F. HARTWIG J. AM.CHEM.SOC. vol. 128, 2006, page 14800
GREEN TW ET AL.: ‘Protective Groups in Organic Synthesis’, 1999, JOHN WILEY & SONS
J. BARLUENGA ET AL. ANGEW. CHEM. INT. ED. vol. 46, 2007, pages 5587 – 90
J. BARLUENGA ET AL. CHEM. EUR. J. vol. 14, 2008, pages 4792 – 5
J. ORG. CHEM. vol. 50, 1985, pages 2438 – 43
J.AM.CHEM.SOC. vol. 130, 2008, pages 8481 – 8490
ORGANIC LETTERS vol. 12, no. 18, 2010, pages 4042 – 4045
ROBERT H. CRABTREE: ‘The Organometallic Chemistry of the Transition Metals’, 2005, WILEY-INTERSCIENCE
Publication numberPriority datePublication dateAssigneeTitle
CN104558090A *2013-10-282015-04-29重庆医药工业研究院有限责任公司Abiraterone acetate impurity and determination method thereof
WO2015200837A1 *2014-06-272015-12-30Fl Therapeutics LlcAbiraterone derivatives and non-covalent complexes with albumin
CN105223282A *2014-06-262016-01-06深圳海王药业有限公司High performance liquid chromatography gradient method for determining related substances of abiraterone acetate
WO2016004910A12014-07-092016-01-14Zentiva, K.S.Method of preparing abiraterone acetate of high purity applicable on industrial scale
CN105693809A *2016-01-132016-06-22华中农业大学Compound with anti-tumor activity and application of compound
US9556218B22013-06-282017-01-31Scinopharm Taiwan, Ltd.Process for the preparation of abiraterone and intermediates thereof
Family To Family Citations
New ICH Guidelines: ICH Q13 on Conti Manufacturing and ICH Q14 on AQbD
DRUG REGULATORY AFFAIRS INTERNATIONAL
![]()
New ICH Guidelines:
*ICH Q13* on Continuous Manufacturing &![]() 🎛
🎛![]() 🎚
🎚
*ICH Q14* on ATP – QbD (Analytical target profile and quality by design)![]() ⚗
⚗![]() ⏱
⏱
New ICH Guidelines: ICH Q13 on Conti Manufacturing and ICH Q14 on AQbD
In a press release from 22 June the International Council for Harmonisation (ICH) has announced that they will prepare new topics for the future. The Assembly agreed to begin working on two new topics for ICH harmonisation:
Analytical Procedure Development and Revision of Q2(R1) Analytical Validation (Q2(R2)/Q14)
and
Continuous Manufacturing (Q13)
The long anticipated revision of ICH Q2(R1) “Guideline on Validation of Analytical Procedures: Text and Methodology” has been approved and the work plan is scheduled to commence in Q3 2018. It is intended that the new guidelines will be consistent with ICH Q8(R2), Q9, Q10, Q11 and Q12 .
The AQbD approach is very important to collect information in order…
View original post 154 more words
Rebamipide, ребамипид , ريباميبيد ,瑞巴派特 ,

Rebamipide
- Molecular FormulaC19H15ClN2O4
- Average mass370.786 Da
- Monoisotopic mass370.072021 Da
OPC-12759
OPC-12759E
OPC-759
(±)-a-(p-Chlorobenzamido)-1,2-dihydro-2-oxo-4-quinolinepropionic acid
2-(4-Chlorobenzoylamino)-3-[2(1H)-quinolinon-4-yl]propionic acid
4-Quinolinepropanoic acid, α-[(4-chlorobenzoyl)amino]-1,2-dihydro-2-oxo- [ACD/Index Name]
4-quinolinepropanoic acid, α-[(4-chlorobenzoyl)amino]-2-hydroxy-
6454
CAS 90098-04-7 [RN]
a-[(4-Chlorobenzoyl)amino]-1,2-dihydro-2-oxo-4-quinolinepropanoic acid
LR583V32ZR
UNII:LR583V32ZR
ребамипид [Russian] [INN]
ريباميبيد [Arabic] [INN]
瑞巴派特 [Chinese] [INN]
(±)-2-(4-CHLOROBENZOYLAMINO)-3-(2(1H)-QUINOLINON-4-YL)-PROPIONIC ACID
obtain the white powder from dimethylformamide-water with its hemihydrate m.p. being 288-290°C (decomposition).
(-)-Configuration: from dimethylformamide to give colorless needles, mp 305~306 °C (decomposition). [α] D20-116.7 ° (C = 1.0, dimethylformamide).
(+)-Configuration: from dimethylformamide to give colorless needles, mp 305~306 °C (decomposition). [α] D20 + 116.9 ° (C = 1.0, dimethylformamide).
(-)-Configuration: from dimethylformamide to give colorless needles, mp 305~306 °C (decomposition). [α] D20-116.7 ° (C = 1.0, dimethylformamide).
(+)-Configuration: from dimethylformamide to give colorless needles, mp 305~306 °C (decomposition). [α] D20 + 116.9 ° (C = 1.0, dimethylformamide).
Rebamipide is a quinolone derivative that was launched in 1990 by Otsuka in Japan for the oral treatment of Helicobacter pylori-induced gastric inflammation after eradication therapy and peptic ulcer
Title: Rebamipide
CAS Registry Number: 90098-04-7
CAS Name: a-[(4-Chlorobenzoyl)amino]-1,2-dihydro-2-oxo-4-quinolinepropanoic acid
Additional Names: (±)-a-(p-chlorobenzamido)-1,2-dihydro-2-oxo-4-quinolinepropionic acid; 2-(4-chlorobenzoylamino)-3-[2(1H)-quinolinon-4-yl]propionic acid; proamipide
Manufacturers’ Codes: OPC-12759
Trademarks: Mucosta (Otsuka)
Molecular Formula: C19H15ClN2O4
Molecular Weight: 370.79
Percent Composition: C 61.55%, H 4.08%, Cl 9.56%, N 7.56%, O 17.26%
Literature References: Gastric cytoprotectant. Prepn: M. Uchida et al., DE 3324034; eidem, US 4578381; (1984, 1986 both to Otsuka). Synthesis and pharmacology: M. Uchida et al., Chem. Pharm. Bull. 33, 3775 (1985); of enantiomers: eidem, ibid. 35, 853 (1987). Antiulcer activity in rats: K. Yamasaki et al., Eur. J. Pharmacol. 142, 23 (1987); K. Yamasaki et al., Jpn. J. Pharmacol. 49,441 (1989). HPLC determn in plasma and urine: Y. Shioya, T. Shimizu, J. Chromatogr. 434, 283 (1988).
Properties: White powder from DMF-water, mp 288-290° (dec) as hemihydrate.
Melting point: mp 288-290° (dec) as hemihydrate
Derivative Type: (-)-Form
Properties: Colorless needles from DMF, mp 305-306° (dec). [a]D20 -116.7° (c = 1.0 in DMF).
Melting point: mp 305-306° (dec)
Optical Rotation: [a]D20 -116.7° (c = 1.0 in DMF)
Derivative Type: (+)-Form
Properties: Colorless needles from DMF, mp 305-306° (dec). [a]D20 +116.9° (c = 1.0 in DMF).
Melting point: mp 305-306° (dec)
Optical Rotation: [a]D20 +116.9° (c = 1.0 in DMF)
Therap-Cat: Antiulcerative.
Keywords: Antiulcerative; Cytoprotectant (Gastric).
Rebamipide has been investigated for the treatment of Stomach Ulcer, Keratoconjunctivitis Sicca, and Gastric Adenoma and Early Gastric Cancer.
Rebamipide is a quinolinone derivative that stimulates endogenous PGE2 generation in gastric mucosa, enhancing gastric mucosal defense in a COX-2-dependent manner.
Rebamipide has been shown to inhibit the production of reactive oxygen species and to decrease cytokine release induced by H. pylori infection.
A daily oral dose of 100 mg/kg was found to be protective against the development of pyloric channel ulcers in Mongolian gerbils infected with H. pylori.
In addition to the stomach, rebamipide can also enhance secretion of mucin covering the conjunctiva and cornea, which is important for tear film adhesion.
Rebamipide, a gastroprotective drug, was developed in Japan and was proven to be superior to cetraxate, the former most prescribed drug of the same category, in 1989 in the treatment for gastric ulcers. The initially discovered basic mechanisms of action of rebamipide included its action as a prostaglandin inducer and oxygen free-radical scavenger. In the last 5 years, several basic and clinical studies have been performed for functional dyspepsia, chronic gastritis, NSAID-induced gastrointestinal injuries, gastric ulcer following eradication therapy for Helicobacter pylori, gastric ulcer after endoscopic surgery and ulcerative colitis. In addition, several molecules have been identified as therapeutic targets of rebamipide to explain its pleiotropic pharmacological actions.
Rebamipide has been shown to inhibit the production of reactive oxygen species and to decrease cytokine release induced by H. pylori infection.
A daily oral dose of 100 mg/kg was found to be protective against the development of pyloric channel ulcers in Mongolian gerbils infected with H. pylori.
In addition to the stomach, rebamipide can also enhance secretion of mucin covering the conjunctiva and cornea, which is important for tear film adhesion.
Rebamipide, a gastroprotective drug, was developed in Japan and was proven to be superior to cetraxate, the former most prescribed drug of the same category, in 1989 in the treatment for gastric ulcers. The initially discovered basic mechanisms of action of rebamipide included its action as a prostaglandin inducer and oxygen free-radical scavenger. In the last 5 years, several basic and clinical studies have been performed for functional dyspepsia, chronic gastritis, NSAID-induced gastrointestinal injuries, gastric ulcer following eradication therapy for Helicobacter pylori, gastric ulcer after endoscopic surgery and ulcerative colitis. In addition, several molecules have been identified as therapeutic targets of rebamipide to explain its pleiotropic pharmacological actions.
Rebamipide, an amino acid derivative of 2-(1H)-quinolinone, is used for mucosal protection, healing of gastroduodenal ulcers, and treatment of gastritis. It works by enhancing mucosal defense, scavenging free radicals, and temporarily activating genes encoding cyclooxygenase-2.
Rebamipide is used in a number of Asian countries including Japan (marketed as Mucosta), South Korea, China[1] and India (where it is marketed under the trade name Rebagen). It is also approved in Russia under the brand name Rebagit.[2] It is not approved by the Food and Drug Administration for use in the United States.
Studies have shown that rebamipide can fight the damaging effects of NSAIDs on the GIT mucosa, and more recently, the small intestine.[citation needed] It has also been studied for the treatment of Behçet’s disease.[3] It was shown to successfully treat pouchitis in a single-N study after first-line therapies for the condition were unsuccessful.[4] Some studies have shown effectiveness in presbyacusis(age-related hearing loss).[citation needed]
It has also been shown to alleviate signs and symptoms of dry eyes in a randomised controlled trial although this is not yet widely available clinically.[5]
SYN
Rebamipide (CAS NO.: 111911-87-6), with its systematic name of 4-Quinolinepropanic acid, alpha-((4-chlorobenzoyl)amino)-1,2-dihydro-2-oxo-, (+-)-, could be produced through many synthetic methods.
Following is one of the reaction routes:

4-(Bromomethyl)quinolin-2(1H)-one (I) could react with hot phosphorus oxychloride to produce a mixture of 4-(bromomethyl)-2-chloroquinoline (II) and 2-chloro-4-(chloromethyl)quinoline (III), and then the mixture without separation is ondensed with 2(S)-isopropyl-3,6-dimethoxy-2,5-dihydropyrazine (IVs) in the presence of butyllithium in hexane, affording (-)-2-chloro-4-[6(S)-isopropyl-2,5-dimethoxy-3,6-dihydropyrazin-3(R)-yl methyl]quinoline (Vr). The hydrolysis of (Vr) with HCl produces 3-(2-chloroquinolin-4-yl)-(R)-alanine methyl ester (VIr), which is treated with HCl and propylene oxide to afford 3-(2-oxo-2,3-dihydroquinolin-4-yl)-(R)-alanine (VIIr). At last, this compound is acylated with 4-chlorobenzoyl chloride (VIII) by means of K2CO3in acetone, affording (R)-OPC-12759.

Figure 2 The synthetic route of Rebamipide.

DE 3324034; US 4578381 ABOVE
The condensation of 4-(bromomethyl)quinolin-2(1H)-one (I) with diethyl acetamidomalonate (II) by means of sodium ethoxide in refluxing ethanol gives ethyl 2-acetamido-2-(ethoxycarbonyl)-3-(2-oxo-1,2-dihydroquinolin-4yl)propionate (III), which is submitted to a decarboxylative hydrolysis with refluxing 20% HCl yielding 3-(2-oxo-1,2-dihydroquinolin-4yl)alanine (IV). Finaily this compound is acylated with 4-chlorobenzoyl chloride by means of K2CO3 in acetone water.
SYN

Chem Pharm Bull 1991,39(11),2906 ABOVE
The synthesis of (R)- and (S)-isomers of OPC-12759 has been described: These optical isomers can be obtained in three different ways: 1) The reaction of 4-(bromomethyl)quinolin-2(1H)-one (I) with hot phosphorus oxychloride gives a mixture of 4-(bromomethyl)-2-chloroquinoline (II) and 2-chloro-4-(chloromethyl)quinoline (III), which, without separation, is condensed with 2(S)-isopropyl-3,6-dimethoxy-2,5-dihydropyrazine (IVs) by means of butyllithium in hexane, yielding (-)-2-chloro-4-[6(S)-isopropyl-2,5-dimethoxy-3,6-dihydropyrazin-3(R)-yl methyl]quinoline (Vr). The hydrolysis of (Vr) with HCl affords 3-(2-chloroquinolin-4-yl)-(R)-alanine methyl ester (VIr), which is treated with HCl and propylene oxide to give 3-(2-oxo-2,3-dihydroquinolin-4-yl)-(R)-alanine (VIIr). Finally, this compound is acylated with 4-chlorobenzoyl chloride (VIII) by means of K2CO3 in acetone, affording (R)-OPC-12759.
SYN

 3) The methylation of 3-(2-oxo-1,2-dihydroquinolin-4-yl)-(R,S)-alanine (IX) with SOCl2 and methanol yields the corresponding methyl ester (X), which is submitted to optical resolution with D-(-)-mandelic acid, affording adducts (XII) and (XIII). The hydrolytic treatment of (XII) and (XIII) with HCl and propylene oxide finally yields isomers (VIIr) and (VIIs), already obtained. Racemic OPC-12759 can also be resolved into its optical isomers by treatment with brucine and fractionated crystallization.
3) The methylation of 3-(2-oxo-1,2-dihydroquinolin-4-yl)-(R,S)-alanine (IX) with SOCl2 and methanol yields the corresponding methyl ester (X), which is submitted to optical resolution with D-(-)-mandelic acid, affording adducts (XII) and (XIII). The hydrolytic treatment of (XII) and (XIII) with HCl and propylene oxide finally yields isomers (VIIr) and (VIIs), already obtained. Racemic OPC-12759 can also be resolved into its optical isomers by treatment with brucine and fractionated crystallization.
Rebamipide
-
- Synonyms:Proamipide
- ATC:A02BX
- Use:ulcer therapeutic
- Chemical name:α-[(4-chlorobenzoyl)amino]-1,2-dihydro-2-oxo-4-quinolinepropanoic acid
- Formula:C19H15ClN2O4
- MW:370.79 g/mol
- CAS-RN:90098-04-7
- LD50:572 mg/kg (M, i.v.);
700 mg/kg (R, i.v.);
>2 g/kg (dog, p.o.)
Substance Classes
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 39098-85-6 | C4H5ClO2 | acetoacetyl chloride | Butanoyl chloride, 3-oxo- |
| 62-53-3 | C6H7N | aniline | Benzenamine |
| 4876-10-2 | C10H8BrNO | 4-(bromomethyl)-2(1H)-quinolinone | 2(1H)-Quinolinone, 4-(bromomethyl)- |
| 128-08-5 | C4H4BrNO2 | N-bromosuccinimide | 2,5-Pyrrolidinedione, 1-bromo- |
| 122-01-0 | C7H4Cl2O | 4-chlorobenzoyl chloride | Benzoyl chloride, 4-chloro- |
| 1068-90-2 | C9H15NO5 | diethyl acetamidomalonate | Propanedioic acid, (acetylamino)-, diethyl ester |
| 4900-38-3 | C19H22N2O6 | ethyl 2-acetamido-2-(ethoxycarbonyl)-3-(2-oxo-1,2-dihydroquinolin-4-yl)propionate | Propanedioic acid, (acetylamino)[(1,2-dihydro-2-oxo-4-quinolinyl)methyl]-, diethyl ester |
| 5162-90-3 | C12H12N2O3 | 3-(2-oxo-1,2-dihydroquinolin-4-yl)alanine | 4-Quinolinepropanoic acid, α-amino-1,2-dihydro-2-oxo- |
| 102-01-2 | C10H11NO2 | 3-oxo-N-phenylbutanamide | Butanamide, 3-oxo-N-phenyl- |
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| J | Mucosta | Otsuka |
Formulations
- tabl. 100 mg
References
-
- Uchida, M. et al.: Chem. Pharm. Bull. (CPBTAL) 33, 3775 (1985).
- DOS 3 324 034 (Otsuka; appl. 7.4.1983; J-prior. 7.5.1982).
- GB 2 123 825 (Otsuka; appl. 7.5.1983; J-prior. 7.5.1982).
-
oral and parenteral formulations:
- JP 60 019 767 (Otsuka; appl. 7.11.1983).
PAPER
Magic Bullet! Rebamipide, a Superior Anti-ulcer and Ophthalmic Drug and Its Large-Scale Synthesis in a Single Organic Solvent via Process Intensification Using Krapcho Decarboxylation
https://pubs.acs.org/doi/10.1021/acs.oprd.7b00382#
Prashanth Kumar Babu, Mohan Reddy Bodireddy, Reshma Choudlu Puttaraju, Dnyaneshwar Vagare, Raghu Nimmakayala, Naresh Surineni, Madhusudana Rao Gajula*, and Pramod Kumar* 
Chemical Research Division, API R&D Centre, Micro Labs Ltd., Plot No.43-45, KIADB Industrial Area, fourth phase, Bommasandra-Jigani Link Road, Bommasandra, Bangalore 560 105, Karnataka, India
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.7b00382
Publication Date (Web): May 31, 2018
Copyright © 2018 American Chemical Society
*E-mail: pramodkumar@microlabs.in., *E-mail: gmadhusudanrao@yahoo.com.

Rebamipide (1) is a superior drug compared to existing drugs for use in healing of peptic ulcers, gastrointestinal bleeding, and dyspepsia. It is also useful as an ophthalmic drug for the treatment of dry eye syndrome. Process intensification for its synthesis was achieved by (i) averting uncontrollable frothing using Krapcho decarboxylation instead of conventional acid hydrolysis, where uncontrollable frothing became chaotic, (ii) minimizing organic waste generation by using a single organic solvent, and (iii) avoiding anti-foaming agents (n-octanol, acetophenone) and acetic acid. With these trifling modifications, the overall yield of active pharmaceutical ingredient (API) was ≥83% with excellent purity (≥99.89%), and the process meets the metrics of “green” chemistry with an E-factor = 11.5. The developed hassle-free commercial process is viable for multi-kilogram synthesis of Rebamipide (1) as the key step, Krapcho decarboxylation is safe to run at 130–140 °C in DMSO, and it was proved to be effective by differential scanning calorimetry thermal screening studies. The characterization data of intermediates, process-related impurities, and API are reported. The carryover and process-related impurities were controlled efficiently. The present work can enhance the scope and worldwide adoptability of Rebamipide (1), which is currently limited to Asian countries.
https://pubs.acs.org/doi/suppl/10.1021/acs.oprd.7b00382/suppl_file/op7b00382_si_001.pdf
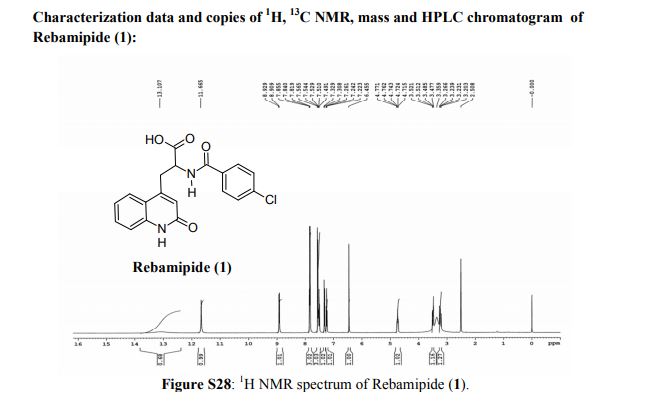
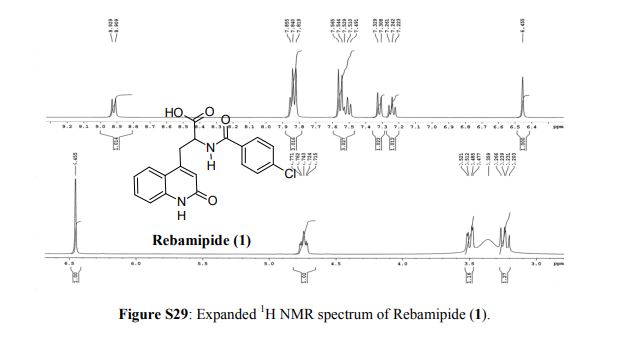
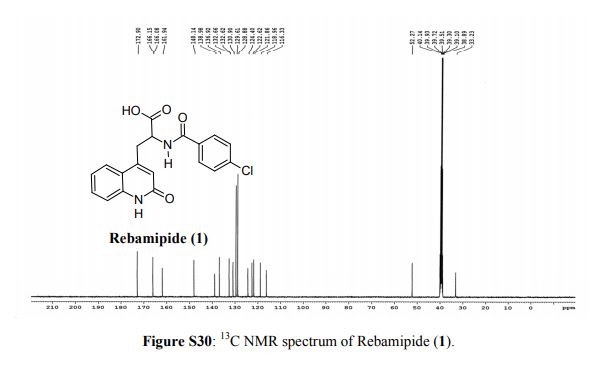
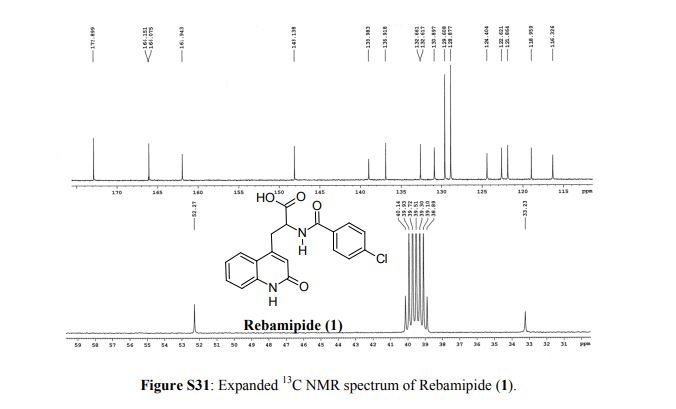
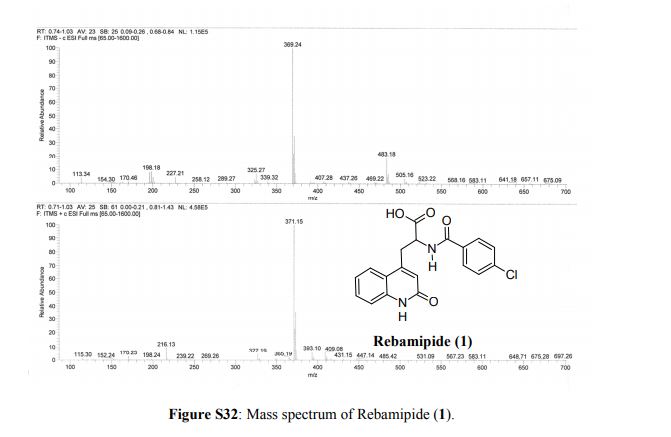
Articles
- Arakawa T, Watanabe T, Fukuda T, Yamasaki K, Kobayashi K (1995). “Rebamipide, novel prostaglandin-inducer accelerates healing and reduces relapse of acetic acid-induced rat gastric ulcer. Comparison with cimetidine”. Dig Dis Sci. 40 (11): 2469–72. doi:10.1007/BF02063257. PMID 7587834.
- Arakawa T, Kobayashi K, Yoshikawa T, Tarnawski A (1998). “Rebamipide: overview of its mechanisms of action and efficacy in mucosal protection and ulcer healing”. Dig Dis Sci. 43 (9 Suppl): 5S–13S. PMID 9753220.
- Tarnawski AS, Chai J, Pai R, Chiou SK (2004). “Rebamipide activates genes encoding angiogenic growth factors and Cox2 and stimulates angiogenesis: a key to its ulcer healing action?”. Dig Dis Sci. 49 (2): 202–9. doi:10.1023/B:DDAS.0000017439.60943.5c. PMID 15104358.
- Takumida M, Anniko M (2009). “Radical scavengers for elderly patients with age-related hearing loss”. Acta Otolaryngol. 129 (1): 36–44. doi:10.1080/00016480802008215. PMID 18607930.
References
- Jump up^ drugs.com
- Jump up^ “Russian State Register of Medicines. Registration Sertificate: Rebagit (rebamipide) Film-Coated Tablets” (in Russian). Retrieved 10 June 2017.
- Jump up^ Matsuda T, Ohno S, Hirohata S, Miyanaga Y, Ujihara H, Inaba G, Nakamura S, Tanaka S, Kogure M, Mizushima Y (2003). “Efficacy of rebamipide as adjunctive therapy in the treatment of recurrent oral aphthous ulcers in patients with Behcet’s disease: a randomised, double-blind, placebo-controlled study”. Drugs R D. 4 (1): 19–28. doi:10.2165/00126839-200304010-00002. PMID 12568631.
- Jump up^ http://www.wjgnet.com/1007-9327/12/656.pdf Archived October 20, 2013, at the Wayback Machine.
- Jump up^ Kinoshita, S.; K. Oshiden; S. Awamura; H. Suzuki; N. Nakamichi (2013). “A randomized, multicenter phase 3 study comparing 2% rebamipide (OPC-12759) with 0.1% sodium hyaluronate in the treatment of dry eye”. Ophthalmology. 120 (6): 1158–65. doi:10.1016/j.ophtha.2012.12.022. PMID 23490326.
 |
|
| Clinical data | |
|---|---|
| Trade names | Mucosta (JP), Rebagen (KR,CN, IN), Rebagit (RU) |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
Oral (tablets) |
| ATC code | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C19H15ClN2O4 |
| Molar mass | 370.786 g/mol |
| 3D model (JSmol) | |
/////////Rebamipide, UNII:LR583V32ZR, ребамипид , ريباميبيد ,瑞巴派特 , OPC-12759 , OPC-12759E , OPC-759 , OPC 12759 , OPC 12759E , OPC 759 , OTSUKA, JAPAN 1990
OC(=O)C(CC1=CC(O)=NC2=CC=CC=C12)NC(=O)C1=CC=C(Cl)C=C1
Alfuzosin, 塩酸アルフゾシン
Alfuzosin
- Molecular FormulaC19H27N5O4
- Average mass389.449 Da
N-{3-[(4-Amino-6,7-dimethoxy-2-quinazolinyl)(methyl)amino]propyl}tetrahydro-2-furancarboxamide
N-{3-[(4-amino-6,7-dimethoxyquinazolin-2-yl)(methyl)amino]propyl}tetrahydrofuran-2-carboxamide
SL 77499-10
UNII:90347YTW5F
Urion
Xatral
2-furancarboxamide, N-[3-[(4-amino-6,7-dimethoxy-2-quinazolinyl)methylamino]propyl]tetrahydro-
5357
cas 81403-80-7 [RN]
| CAS: | 81403-68-1 HCL SALT |
90347YTW5F
塩酸アルフゾシン
Title: Alfuzosin
CAS Registry Number: 81403-80-7
CAS Name: N-[3-[(4-Amino-6,7-dimethoxy-2-quinazolinyl)methylamino]propyl]tetrahydro-2-furancarboxamide
Additional Names: N1-(4-amino-6,7-dimethoxyquinazol-2-yl)-N1-methyl-N2-(tetrahydrofuroyl-2)-propylenediamine
Manufacturers’ Codes: SL-77.499
Molecular Formula: C19H27N5O4
Molecular Weight: 389.45
Percent Composition: C 58.60%, H 6.99%, N 17.98%, O 16.43%
Literature References: a1-Adrenoceptor antagonist structurally similar to prazosin, q.v. Prepn: P. M. J. Manoury, DE 2904445; idem, US 4315007 (1979, 1982 both to Synthelabo); and antihypertensive activity in rats: P. M. Manoury et al., J. Med. Chem. 29,19 (1986). Pharmacology: A. G. Ramage, Eur. J. Pharmacol. 129, 307 (1986). HPLC determn in biological fluids: P. Guinebault et al., J. Chromatogr. 353, 361 (1986). Pharmacology in humans: A. H. Deering, Br. J. Clin. Pharmacol. 25, 417 (1988). Clinical evaluation in essential hypertension: S. Leto Di Priolo et al., Eur. J. Clin. Pharmacol. 35, 25 (1988); A. K. Ghosh, S. Ghosh, Ger. Cardiovasc. Med. 1, 81 (1988). Clinical trial in benign prostatic hyperplasia (BPH): C. G. Roehrborn et al., BJU Int. 92, 257 (2003). Review of clinical experience in BPH: D. M. Weiner, F. C. Lowe, Expert Opin. Pharmacother. 4, 2057-2063 (2003).
Alfuzosin hydrochloride (CAS 81403-68-1)
Derivative Type: Hydrochloride
CAS Registry Number: 81403-68-1
Manufacturers’ Codes: SL-77.499-10
Trademarks: Mittoval (Schering AG); Urion (Zambon); UroXatral (Sanofi-Synthelabo); Xatral (Sanofi-Synthelabo)
Molecular Formula: C19H27N5O4.HCl
Molecular Weight: 425.91
Percent Composition: C 53.58%, H 6.63%, N 16.44%, O 15.03%, Cl 8.32%
Properties: Crystals from ethanol + ether, mp 225° (Manoury, 1986), also reported earlier as mp 235° (dec) (Manoury, 1982). pKa 8.13.
Melting point: mp 225° (Manoury, 1986); mp 235° (dec) (Manoury, 1982)
pKa: pKa 8.13
Therap-Cat: Antihypertensive. In treatment of benign prostatic hypertrophy.
Keywords: Antihypertensive; Quinazoline Derivatives; Antiprostatic Hypertrophy; a-Adrenergic Blocker.
Alfuzosin (INN, provided as the hydrochloride salt) is a pharmaceutical drug of the α1 blocker class. As an antagonist of the α1adrenergic receptor, it works by relaxing the muscles in the prostate and bladder neck, making it easier to urinate. It is thus used to treat benign prostatic hyperplasia (BPH).[1]
Alfuzosin is marketed in the United States by Sanofi Aventis under the brand name Uroxatral and elsewhere under the tradenames Xat, Xatral, Prostetrol and Alfural. Alfuzosin was approved by the U.S. FDA for treatment of BPH in June 2003.
Side effects
The most common side effects are dizziness (due to postural hypotension), upper respiratory tract infection, headache, fatigue, and abdominal disturbances. Side effects include stomach pain, heartburn, and congested nose.[2] Adverse effects of alfuzosin are similar to that of tamsulosin with the exception of retrograde ejaculation.[3]
Contraindications
Alfuzosin should be used with caution in patients with severe renal insufficiency, and should not be prescribed to patients with a known history of QT prolongation who are taking medications known to prolong the QT interval.
Chemistry
Alfuzosin contains a stereocenter and is therefore chiral. There are two enantiomeric forms, (R)-alfuzosin and (S)-alfuzosin. The drug is used as a racemate, (RS)-alfuzosin, a 1: 1 mixture of the (R)- and (S)-forms.[4]
| Enantiomers of alfuzosin | |
|---|---|
-Alfuzosin_Structural_Formula_V2.svg) CAS number: 123739-69-5 |
-Alfuzosin_Structural_Formula_V2.svg) CAS number.: 123739-70-8 |
Alfuzosin
-
- ATC:G04CA01
- Use:antihypertensive, α1-adrenoceptor antagonist, α-blocker, treatment of benign prostatic hypertrophy (BPH)
- Chemical name:(±)-N-[3-[(4-amino-6,7-dimethoxy-2-quinazolinyl)methylamino]propyl]tetrahydro-2-furancarboxamide
- Formula:C19H27N5O4
- MW:389.46 g/mol
- CAS-RN:81403-80-7
Derivatives
monohydrochloride
- Formula:C19H27N5O4 • HCl
- MW:425.92 g/mol
- CAS-RN:81403-68-1
Substance Classes
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 23680-84-4 | C10H10ClN3O2 | 4-amino-2-chloro-6,7-dimethoxyquinazoline | 4-Quinazolinamine, 2-chloro-6,7-dimethoxy- |
| 5004-88-6 | C9H12N2O3 | 2-amino-4,5-dimethoxybenzamide | Benzamide, 2-amino-4,5-dimethoxy- |
| 541-41-3 | C3H5ClO2 | chloroformic acid ethyl ester | Carbonochloridic acid, ethyl ester |
| 72104-44-0 | C9H14N2O2 | 2-cyano-N-methyl-N-tetrahydrofuroylethylamine | 2-Furancarboxamide, N-(2-cyanoethyl)tetrahydro-N-methyl- |
| 27631-29-4 | C10H8Cl2N2O2 | 2,4-dichloro-6,7-dimethoxyquinazoline | Quinazoline, 2,4-dichloro-6,7-dimethoxy- |
| 28888-44-0 | C10H10N2O4 | 2,4-dihydroxy-6,7-dimethoxyquinazoline | 2,4(1H,3H)-Quinazolinedione, 6,7-dimethoxy- |
| 20357-25-9 | C9H9NO5 | 4,5-dimethoxy-2-nitrobenzaldehyde | Benzaldehyde, 4,5-dimethoxy-2-nitro- |
| 4959-60-8 | C9H10N2O5 | 4,5-dimethoxy-2-nitrobenzamide | Benzamide, 4,5-dimethoxy-2-nitro- |
| 28888-44-0 | C10H10N2O4 | 6,7-dimethoxyquinazoline-2,4-dione | 2,4(1H,3H)-Quinazolinedione, 6,7-dimethoxy- |
| 541-41-3 | C3H5ClO2 | ethyl chloroformate | Carbonochloridic acid, ethyl ester |
| 693-05-0 | C4H8N2 | 3-(methylamino)propanenitrile | Propanenitrile, 3-(methylamino)- |
| 81403-67-0 | C9H18N2O2 | N1-methyl-N2-tetrahydrofuroyltrimethylenediamine | 2-Furancarboxamide, tetrahydro-N-[3-(methylamino)propyl]- |
| 16874-33-2 | C5H8O3 | (±)-tetrahydrofuran-2-carboxylic acid | 2-Furancarboxylic acid, tetrahydro- |
| 167391-50-6 | C8H12O5 | tetrahydro-2-furancarboxylic acid anhydride with ethyl hydrogen carbonate | 2-Furancarboxylic acid, tetrahydro-, anhydride with ethyl hydrogen carbonate |
| 57-13-6 | CH4N2O | urea | Urea |
| 120-14-9 | C9H10O3 | veratraldehyde | Benzaldehyde, 3,4-dimethoxy- |
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| D | Alfunar | Apogepha | |
| Alfusin | TAD Pharma | ||
| Urion | Sanofi-Aventis | ||
| Uroxatral | Sanofi-Aventis | ||
| F | Urion | Zambon | |
| Xatral | Sanofi-Aventis | ||
| GB | Xatral | Sanofi-Aventis | |
| I | Mittoval | Sanofi-Aventis | |
| Xatral | Sanofi-Aventis |
Formulations
- film tabl. 2.5 mg; retard tabl. 10 mg (hydrochloride)
References
-
- Manoury, P.M. et al.: J. Med. Chem. (JMCMAR) 29, 19 (1986).
- US 4 315 007 (Synthelabo; 9.2.1982; F-prior. 6.2.1978, 29.12.1978).
- DE 2 904 445 (Synthelabo; appl. 16.8.1979; F-prior. 6.2.1978, 29.12.1978).
-
synthesis of 6,7-dimethoxyquinazoline-2,4-dione:
- Althuis, T.H.; Hess, H.J.: J. Med. Chem. (JMCMAR) 20, 146 (1977).
SYN
Mathias Scheer, “Alfuzosin tablets and synthesis.” U.S. Patent US20060062845, issued March 23, 2006.
Syn, DOI: 10.1021/jm00151a003 NB: (WO2009001369)

FTIR spectrum of alfuzosin hydrochloride
CLIP


Add the following:

C19H27N5O4·HCl ![]()
![]()
![]()
![]()
![]() 425.91
425.91
2-Furancarboxamide, N-[3-[(4-amino-6,7-dimethoxy-2-quinazolinyl)methylamino]propyl]tetrahydro-, monohydrochloride (±).
(±)-N-[3-[(4-Amino-6,7-dimethoxy-2-quinazolinyl)methylamino]propyl]tetrahydro-2-furamide monohydrochloride [81403-68-1].
[81403-68-1].
(±)-N-[3-[(4-Amino-6,7-dimethoxy-2-quinazolinyl)methylamino]propyl]tetrahydro-2-furamide monohydrochloride
» Alfuzosin Hydrochloride contains not less than 99.0 percent and not more than 101.0 percent of C19H27N5O4·HCl, calculated on the anhydrous basis.
Packaging and storage— Preserve in tight, well-closed containers, protected from light and humidity. Store at room temperature.
USP Reference standards  11
11 —
—
USP Alfuzosin Hydrochloride RS.
USP Alfuzosin System Suitability Mixture RS.
USP Alfuzosin Hydrochloride RS.
USP Alfuzosin System Suitability Mixture RS.
Identification—
B: It meets the requirements of the test for Chloride 191.
pH ![]() 791
791![]() : between 4.0 and 5.5
: between 4.0 and 5.5
Test solution: 20 mg per mL, in carbon dioxide-free water.
Optical rotation ![]() 781
781![]() :
: ![]() 0.10
0.10![]() to +0.10
to +0.10![]()
Test solution: 20 mg per mL, in carbon dioxide-free water.
Water, Method I 921: not more than 0.5%.
Residue on ignition 281: not more than 0.1%.
Related compounds—
Solution A— Dilute 5.0 mL of perchloric acid in 900 mL of water, adjust with 2 M sodium hydroxide solution to a pH of 3.5, and dilute with water to 1000 mL.
Mobile phase— Prepare a filtered and degassed mixture of Solution A, acetonitrile, and tetrahydrofuran (80:20:1). Make adjustments if necessary (see System Suitability under Chromatography 621).
System suitability solution— Dissolve an accurately weighed quantity of USP Alfuzosin System Suitability Mixture RS in Mobile phase, and dilute quantitatively with Mobile phase to obtain a solution containing about 0.4 mg per mL.
Test solution— Dissolve 40.0 mg of Alfuzosin Hydrochloride in Mobile phase, and dilute with Mobile phase to 100.0 mL.
Reference solution— Quantitatively dilute an accurately measured volume of the Test solution by a factor of 1000 with Mobile phase.
Chromatographic system (see Chromatography ![]() 621
621![]() )— The liquid chromatograph is equipped with a detector set at 254 nm and a 4.6-mm × 15-cm column that contains 5-µm packing L1. The flow rate is about 1.5 mL per minute. Chromatograph the System suitability solution, and record the peak responses as directed for Procedure: the peak-to-valley ratio is at least 5. [NOTE—The peak-to-valley ratio is determined as the ratio of the height above the baseline of the impurity A peak to the height above the baseline of the lowest point of the curve separating this impurity peak from the peak due to alfuzosin.]
)— The liquid chromatograph is equipped with a detector set at 254 nm and a 4.6-mm × 15-cm column that contains 5-µm packing L1. The flow rate is about 1.5 mL per minute. Chromatograph the System suitability solution, and record the peak responses as directed for Procedure: the peak-to-valley ratio is at least 5. [NOTE—The peak-to-valley ratio is determined as the ratio of the height above the baseline of the impurity A peak to the height above the baseline of the lowest point of the curve separating this impurity peak from the peak due to alfuzosin.]
Procedure— Separately inject equal volumes (about 10 µL) of the Reference solution and the Test solution, record the chromatograms, and measure the peak responses. Calculate the percentage of each impurity in the portion of Alfuzosin Hydrochloride taken by the formula:
100[rU / (1000 rS)]
in which 100 is the percentage conversion factor; rU is the peak response for any impurity obtained from the Test solution; 1000 is the dilution factor; and rS is the peak response for alfuzosin obtained from the Reference solution: the limits are as shown in the accompanying table. Disregard any peak with an area less than 0.05%.
| Compound | Relative Retention Time |
Limit (%) |
| Alfuzosin | 1.0 | — |
| Impurity A1 | 1.2 | * |
| Impurity D2 | 0.5 | 0.20 |
| Any individual unspecified impurity | — | 0.10 |
| Total impurities | — | 0.30 |
|
1 N-[3-[(4-Amino-6,7-dimethoxyquinazolin-2-yl)(methyl)amino]propyl]furan-2-carboxamide.
2 N-(4-Amino-6,7-dimethoxyquinazolin-2-yl)-N-methylpropane-1,3-diamine.
* Impurity A, a component of USP Alfuzosin System Suitability Mixture RS, is not a specified impurity.
|
||
Assay— Dissolve about 300 mg of Alfuzosin Hydrochloride, accurately weighed, in a mixture of 40 mL of anhydrous acetic acid and 40 mL of acetic anhydride. Titrate with 0.1 M perchloric acid, determining the endpoint potentiometrically. Each mL of 0.1 M perchloric acid is equivalent to 42.59 mg of C19H27N5O4·HCl. USP32
USP32
Auxiliary Information— Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
| Monograph | Daniel K. Bempong, Ph.D. Senior Scientist 1-301-816-8143 |
(MDPS05) Monograph Development-Pulmonary and Steroids |
| Reference Standards | Lili Wang, Technical Services Scientist 1-301-816-8129 RSTech@usp.org |
USP32–NF27 Page 1449
Pharmacopeial Forum: Volume No. 34(1) Page 69
Chromatographic Column—
Chromatographic columns text is not derived from, and not part of, USP 32 or NF 27.
References
- Jump up^ Lepor, Herbert (2016). “Alpha-blockers for the Treatment of Benign Prostatic Hyperplasia”. Urologic Clinics of North America. 43 (3): 311–23. doi:10.1016/j.ucl.2016.04.009. PMC 2213889 . PMID 27476124.
- Jump up^ “Alfuzosin”. MedlinePlus. United States National Library of Medicine. April 15, 2016.
- Jump up^ Hills, Robert K; Liu, Chenli; Zeng, Guohua; Kang, Ran; Wu, Wenqi; Li, Jiasheng; Chen, Kang; Wan, Show P. (2015). “Efficacy and Safety of Alfuzosin as Medical Expulsive Therapy for Ureteral Stones: A Systematic Review and Meta-Analysis”. PLOS ONE. 10 (8): e0134589. doi:10.1371/journal.pone.0134589. ISSN 1932-6203. PMC 4526635 . PMID 26244843.
 This article incorporates text available under the CC BY 4.0 license.
This article incorporates text available under the CC BY 4.0 license. - Jump up^ Rote Liste Service GmbH (Hrsg.): Rote Liste 2017 – Arzneimittelverzeichnis für Deutschland (einschließlich EU-Zulassungen und bestimmter Medizinprodukte). Rote Liste Service GmbH, Frankfurt/Main, 2017, Aufl. 57, S. 159, ISBN 978-3-946057-10-9.
External links
- Uroxatral (alfuzosin HCl) Extended-Release Tablets Prescribing Information
- Alfuzosin (General information from the NIH)
 |
|
| Clinical data | |
|---|---|
| Pronunciation | /ælˈfjuːzoʊsɪn/ al-FEW-zoh-sin |
| Trade names | Uroxatral, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a64002 |
| Pregnancy category |
|
| Routes of administration |
By mouth (tablets) |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 49% |
| Protein binding | 82–90% |
| Metabolism | Liver (CYP3A4-mediated) |
| Elimination half-life | 10 hours |
| Excretion | Feces (69%) and Urine (24%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.108.671 |
| Chemical and physical data | |
| Formula | C19H27N5O4 |
| Molar mass | 389.46 g·mol−1 |
| 3D model (JSmol) | |
/////////////////塩酸アルフゾシン, Uroxatral, alfuzosin
COC1=C(OC)C=C2C(N)=NC(=NC2=C1)N(C)CCCNC(=O)C1CCCO1
Afloqualone, アフロクアロン
Afloqualone
| Molecular Formula: | C16H14FN3O |
|---|---|
| Molecular Weight: | 283.306 g/mol |
6-amino-2-(fluoromethyl)-3-(2-methylphenyl)quinazolin-4-one
HQ 495, C033541, QA-3735, UNII:CO4U2C8ORZ
4(3H)-Quinazolinone, 6-amino-2-(fluoromethyl)-3-(2-methylphenyl)- [ACD/Index Name]
4831
56287-74-2 [RN], アフロクアロン
Afloqualone; 56287-74-2; Arofuto; Aroft; Afloqualon; Afloqualone [INN:JAN]
Afloqualone (Arofuto) is a quinazolinone family GABAergic drug and is an analogue of methaqualone developed in the 1970s by a team at Tanabe Seiyaku.[1] It has sedative and muscle-relaxant effects resulting from its agonist activity at the β subtype of the GABAareceptor ,[2] and has had some clinical use, although it causes photosensitization as a side-effect that can cause skin problems such as dermatitis.[3]

PATENT
CN 106496145
PATENT
CN 106496144
https://patents.google.com/patent/CN106496144A/en
Example 1:
[0027] A-fluoro-quinolin-one process, comprising the steps of:
[0028] A. To the hydrogenation apparatus 10g 6- nitro-2- (fluoromethyl) -3- (2-methylphenyl) -4- (3H) -1,3- phthalazinone , 150ml acid content of 0.1 ~ 0.4N n-butanol solution of acetic acid, lg palladium ruthenium bimetallic catalyst, hydrogen pressure 0.02 ~ 0.4MPa, reaction temperature of 25-50 ° C, after 1 hour, filtered to give the filtrate ;
[0029] B. washed catalyst with ethanol, at normal temperature, under reduced pressure to obtain a solution ⑴;
[0030] C. was added to the filtrate and the solution ⑴ water, 0.1N sodium hydroxide solution was added, the pH adjusted to 10.2 to 11.0, and stirred at 50-60 ° C 0.5 h, cooled to room temperature and filtered to give the crude fluoro-quinolin-one;
[0031] D.-fluoro-quinolin per gram of the recrystallization solvent was added 5 ~ 15ml crude ketone, wherein the recrystallization solvent is a volume ratio of 1: 1: 0.2 in a solution of isopropanol (m), a solution of acid butyl ester (II ) and water mixture; crystallized at room temperature, filtered to give a fluorine methaqualone.
[0032] Example 2:
[0033] A-fluoro-quinolin-one process, comprising the steps of:
[0034] A. hydrogenation apparatus added to 20g 6- nitro _2_ (fluoromethyl) -3- (2_-methylphenyl) -4- (3-1,3-phthalazinone buckle, acid content of the acid-containing 240ml 0.1 ~ 0.4N ethanol solution of hydrochloric acid, lg palladium ruthenium bimetallic catalyst, hydrogen pressure 0.02 ~ 0.4MPa, reaction temperature of 25-50 ° C. after 0.5 hours the reaction was filtered to obtain filtrate;
[0035] B. washed catalyst with ethanol, at normal temperature, under reduced pressure to obtain a solution ⑴;
[0036] C. was added to the filtrate and the solution ⑴ water, 0.1N sodium hydroxide solution was added, the pH adjusted to 10.2 to 11.0, and stirred at 50-60 ° C 1 hour, cooled to room temperature and filtered to give the crude fluoro-quinolin-one;
[0037] D.-fluoro-quinolin added per gram of crude ketone was recrystallized from 5 ~ 15ml of the solvent, wherein the recrystallization solvent is a volume ratio of 1: o.2: methanol solution of i (m), an ethyl acetate solution (II ) and water mixture; crystallized at room temperature, filtered to give a fluorine methaqualone.
[0038] Example 3:
[0039] – quinolin-fluoro-one kind of process, comprising the steps of:
[0040] A. Add 5g 6- nitro apparatus _2_ hydride (fluoromethyl) -3- (2-methylphenyl) -4- (3-1,3-Perot phthalazinone, 80ml methanol containing an acid in an amount of 0.1 ~ 0.4N solution of sulfuric acid, lg palladium ruthenium bimetallic catalyst, hydrogen pressure 0.02 ~ 0.4MPa, reaction temperature of 25-50 ° C. after 1.5 hours the reaction was filtered to obtain filtrate;
[0041] B. the catalyst was washed with ethanol, normal temperature under reduced pressure to obtain a solution (the I);
[0042] C. was added to the filtrate and the solution (I) water, 0.1N sodium hydroxide solution was added, the pH adjusted to 10.2 to 11.0, and stirred at 50-60 ° C 1 hour, cooled to room temperature and filtered to give fluoro-quinolin-one Crude;
[0043] D. methaqualone fluorine per gram of crude product were added 5 ~ 15ml recrystallization solvent, wherein the recrystallization solvent is a volume ratio of 1: 0.2: 0.2 ethanol solution (m), carboxylic acid butyl ester (II) and water mixture; crystallization at room temperature, and filtered to give fluoro-quinolin-one.
PAPER
6-Amino-2-(fluoromethyl)-3-(2-methylphenyl)quinazolin-4(3H)-one
Acta Crystallographica, Section E: Structure Reports Online (2007), 63, (7), o3109
http://scripts.iucr.org/cgi-bin/paper?S1600536807026670
PAPER
Synthesis of the metabolites of afloqualone and related compounds
Chemical & pharmaceutical bulletin (1983), 31, (4), 1158-65.
Seven main metabolites (3-9) of afloqualone (1, 6-amino-2-fluoromethyl-3-(o-tolyl)-4 (3H)-quinazolinone and related 4 (3H)-quinazolinone derivatives were synthesized. The metabolites 4 and 5 containing a sulfur atom were prepared by the reaction of 6-acetamido-2-chloromethyl-3-(o-tolyl)-4 (3H)-quinazolinone (11) with NaSCH3 followed by oxidation with H2O2. Reaction of 11 and N-acetyl-L-cysteine gave the mercapturic acid-conjugated metabolite 6. Condensation of 2-fluoroacetamido-5-nitrobenzoic acid (19) and 2-amino-benzyl alcohol (20) with dicyclohexylcarbodiimide (DCC) in the presence of 1-hydroxy-benzotriazole afforded 2-fluoromethyl-3-(o-hydroxymethylphenyl)-6-nitro-4 (3H)-quinazolinone (21), which was converted to the metabolites 7 and 8. Treatment of the 2-bromomethyl-4 (3H)-quinazolinone (24) with AgBF4-H2O in dimethylsulfoxide (DMSO) gave the 2-hydroxymethyl metabolite 9. None of the main metabolites (2-9) showed significant central nervous system depressant activity
https://www.jstage.jst.go.jp/article/cpb1958/31/4/31_4_1158/_article
References
- Jump up^ US Patent 3966731 – 2-Fluoromethyl-3-o-tolyl-6-amino-4(3H)-quinazolinone
- Jump up^ Ochiai T, Ishida R. Pharmacological studies on 6-amino- 2-fluoromethyl- 3-(O-tolyl)- 4(3H)- quinazolinone (afloqualone), a new centrally acting muscle relaxant. (II) Effects on the spinal reflex potential and the rigidity. Japanese Journal of Pharmacology. 1982 Jun;32(3):427-38.
- Jump up^ Ishikawa T, Kamide R, Niimura M. Photoleukomelanodermatitis (Kobori) induced by afloqualone. Journal of Dermatology. 1994 Jun;21(6):430-3.
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C16H14FN3O |
| Molar mass | 283.3 |
| 3D model (JSmol) | |
//////////////Afloqualone, HQ 495, アフロクアロン , C033541, QA-3735, UNII:CO4U2C8ORZ, 4831
CC1=CC=CC=C1N2C(=NC3=C(C2=O)C=C(C=C3)N)CF
Afloqualone
-
- ATC:M03A
- Use:muscle relaxant
- Chemical name:6-amino-2-(fluoromethyl)-3-(2-methylphenyl)-4(3H)-quinazolinone
- Formula:C16H14FN3O
- MW:283.31 g/mol
- CAS-RN:56287-74-2
- LD50:397 mg/kg (M, p.o.);
249 mg/kg (R, p.o.)
Derivatives
hydrochloride
- Formula:C16H14FN3O • xHCl
- MW:unspecified
- CAS-RN:56287-75-3
Substance Classes
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 108-24-7 | C4H6O3 | acetic anhydride | Acetic acid, anhydride |
| 69123-71-3 | C7H5ClN2O3 | 2-amino-5-nitrobenzoyl chloride | Benzoyl chloride, 2-amino-5-nitro- |
| 23076-31-5 | C14H13N3O3 | N-(2-amino-5-nitrobenzoyl)-o-toluidine | Benzamide, 2-amino-N-(2-methylphenyl)-5-nitro- |
| 56287-72-0 | C16H14FN3O4 | 2-[(fluoroacetyl)amino]-N-(2-methylphenyl)-5-nitrobenzamide | Benzamide, 2-[(fluoroacetyl)amino]-N-(2-methylphenyl)-5-nitro- |
| 359-06-8 | C2H2ClFO | fluoroacetyl chloride | Acetyl chloride, fluoro- |
| 56287-73-1 | C16H12FN3O3 | 2-(fluoromethyl)-3-(2-methylphenyl)-6-nitro-4(3H)-quinazolinone | 4(3H)-Quinazolinone, 2-(fluoromethyl)-3-(2-methylphenyl)-6-nitro- |
| 616-79-5 | C7H6N2O4 | 5-nitroanthranilic acid | Benzoic acid, 2-amino-5-nitro- |
| 95-53-4 | C7H9N | o-toluidine | Benzenamine, 2-methyl- |
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| J | Aflomus | Hishiyama | |
| Airomate | SawaiNippon Chemiphar | ||
| Arofuto | Tanabe |
Formulations
- tabl. 20 mg
References
-
- Tani, J. et al.: J. Med. Chem. (JMCMAR) 22, 95 (1979).
- DOS 2 449 113 (Tanabe; appl. 15.10.1974; J-prior. 15.10.1973).
- US 3 966 731 (Tanabe; 29.6.1976; J-prior. 15.10.1973)
Title: Afloqualone
CAS Registry Number: 56287-74-2
CAS Name: 6-Amino-2-(fluoromethyl)-3-(2-methylphenyl)-4(3H)-quinazolinone
Additional Names: 6-amino-2-fluoromethyl-3-(o-tolyl)-4(3H)-quinazolinone
Manufacturers’ Codes: HQ-495
Trademarks: Arofuto (Tanabe)
Molecular Formula: C16H14FN3O
Molecular Weight: 283.30
Percent Composition: C 67.83%, H 4.98%, F 6.71%, N 14.83%, O 5.65%
Literature References: A centrally acting muscle relaxant. Prepn: I. Inoue et al., DE 2449113; eidem, US 3966731 (1975, 1976 to Tanabe); J. Tani et al., J. Med. Chem. 22, 95 (1979). Pharmacology: T. Ochiai, R. Ishida, Jpn. J. Pharmacol. 31, 491 (1981); 32,427 (1982). Metabolism: N. Otsuka et al., J. Pharmacobio-Dyn. 5, S-59 (1982); S. Furuuchi et al., Drug Metab. Dispos. 11, 371 (1983).
Properties: Pale yellow prisms from 2-propanol, mp 195-196°. LD50 in mice (mg/kg): 315.1 i.p. (Tani).
Melting point: mp 195-196°
Toxicity data: LD50 in mice (mg/kg): 315.1 i.p. (Tani)
Therap-Cat: Muscle relaxant (skeletal).
Keywords: Muscle Relaxant (Skeletal).
FDA approves first drug Epidiolex (cannabidiol) comprised of an active ingredient derived from marijuana to treat rare, severe forms of epilepsy
The U.S. Food and Drug Administration today approved Epidiolex (cannabidiol) [CBD] oral solution for the treatment of seizures associated with two rare and severe forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome, in patients two years of age and older. This is the first FDA-approved drug that contains a purified drug substance derived from marijuana. It is also the first FDA approval of a drug for the treatment of patients with Dravet syndrome.
June 25, 2018
Release
The U.S. Food and Drug Administration today approved Epidiolex (cannabidiol) [CBD] oral solution for the treatment of seizures associated with two rare and severe forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome, in patients two years of age and older. This is the first FDA-approved drug that contains a purified drug substance derived from marijuana. It is also the first FDA approval of a drug for the treatment of patients with Dravet syndrome.
CBD is a chemical component of the Cannabis sativa plant, more commonly known as marijuana. However, CBD does not cause intoxication or euphoria (the “high”) that comes from tetrahydrocannabinol (THC).
It is THC (and not CBD) that is the primary psychoactive component of marijuana.
“This approval serves as a reminder that advancing sound development programs that properly evaluate active ingredients contained in marijuana can lead to important medical therapies. And, the FDA is committed to this kind of careful scientific research and drug development,” said FDA Commissioner Scott Gottlieb, M.D. “Controlled clinical trials testing the safety and efficacy of a drug, along with careful review through the FDA’s drug approval process, is the most appropriate way to bring marijuana-derived treatments to patients. Because of the adequate and well-controlled clinical studies that supported this approval, prescribers can have confidence in the drug’s uniform strength and consistent delivery that support appropriate dosing needed for treating patients with these complex and serious epilepsy syndromes. We’ll continue to support rigorous scientific research on the potential medical uses of marijuana-derived products and work with product developers who are interested in bringing patients safe and effective, high quality products. But, at the same time, we are prepared to take action when we see the illegal marketing of CBD-containing products with serious, unproven medical claims. Marketing unapproved products, with uncertain dosages and formulations can keep patients from accessing appropriate, recognized therapies to treat serious and even fatal diseases.”
Dravet syndrome is a rare genetic condition that appears during the first year of life with frequent fever-related seizures (febrile seizures). Later, other types of seizures typically arise, including myoclonic seizures (involuntary muscle spasms). Additionally, status epilepticus, a potentially life-threatening state of continuous seizure activity requiring emergency medical care, may occur. Children with Dravet syndrome typically experience poor development of language and motor skills, hyperactivity and difficulty relating to others.
Lennox-Gastaut syndrome begins in childhood. It is characterized by multiple types of seizures. People with Lennox-Gastaut syndrome begin having frequent seizures in early childhood, usually between ages 3 and 5. More than three-quarters of affected individuals have tonic seizures, which cause the muscles to contract uncontrollably. Almost all children with Lennox-Gastaut syndrome develop learning problems and intellectual disability. Many also have delayed development of motor skills such as sitting and crawling. Most people with Lennox-Gastaut syndrome require help with usual activities of daily living.
“The difficult-to-control seizures that patients with Dravet syndrome and Lennox-Gastaut syndrome experience have a profound impact on these patients’ quality of life,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “In addition to another important treatment option for Lennox-Gastaut patients, this first-ever approval of a drug specifically for Dravet patients will provide a significant and needed improvement in the therapeutic approach to caring for people with this condition.”
Epidiolex’s effectiveness was studied in three randomized, double-blind, placebo-controlled clinical trials involving 516 patients with either Lennox-Gastaut syndrome or Dravet syndrome. Epidiolex, taken along with other medications, was shown to be effective in reducing the frequency of seizures when compared with placebo.
The most common side effects that occurred in Epidiolex-treated patients in the clinical trials were: sleepiness, sedation and lethargy; elevated liver enzymes; decreased appetite; diarrhea; rash; fatigue, malaise and weakness; insomnia, sleep disorder and poor quality sleep; and infections.
Epidiolex must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks. As is true for all drugs that treat epilepsy, the most serious risks include thoughts about suicide, attempts to commit suicide, feelings of agitation, new or worsening depression, aggression and panic attacks. Epidiolex also caused liver injury, generally mild, but raising the possibility of rare, but more severe injury. More severe liver injury can cause nausea, vomiting, abdominal pain, fatigue, anorexia, jaundice and/or dark urine.
Under the Controlled Substances Act (CSA), CBD is currently a Schedule I substance because it is a chemical component of the cannabis plant. In support of this application, the company conducted nonclinical and clinical studies to assess the abuse potential of CBD.
The FDA prepares and transmits, through the U.S. Department of Health and Human Services, a medical and scientific analysis of substances subject to scheduling, like CBD, and provides recommendations to the Drug Enforcement Administration (DEA) regarding controls under the CSA. DEA is required to make a scheduling determination.
The FDA granted Priority Review designation for this application. Fast-Track designation was granted for Dravet syndrome. Orphan Drug designation was granted for both the Dravet syndrome and Lennox-Gastaut syndrome indications.
The FDA granted approval of Epidiolex to GW Research Ltd.

/////////// Epidiolex, cannabidiol, fda 2018, Dravet syndrome, epilepsy, Priority Review , Fast-Track designation, Orphan Drug designation
RG7440, Ipatasertib, アイパタセルチブ;
Ipatasertib
GDC-0068 , RG7440
CAS 1001264-89-6, C24H32ClN5O2, 457.9962
アイパタセルチブ;
イパタセルチブ;
Antineoplastic, AKT serine/threonine kinase inhibitor
2(S)-(4-Chlorophenyl)-1-[4-[7(R)-hydroxy-5(R)-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl]piperazin-1-yl]-3-(isopropylamino)propan-1-one
(2S)-2-(4-Chlorophenyl)-1-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta(d)pyrimidin-4-yl)piperazin-1-yl(-3-((propan-2-yl)amino)propan-1-one
1-Propanone, 2-(4-chlorophenyl)-1-(4-((5R,7R)-6,7-dihydro-7-hydroxy-5-methyl-5H-cyclopentapyrimidin-4-yl)-1-piperazinyl)-3-((1-methylethyl)amino)-, (2S)-
Ipatasertib dihydrochloride
1396257-94-5
Ipatasertib (RG7440) is an experimental cancer drug in development by Roche. It is a small molecule inhibitor of Akt. It was discovered by Array Biopharma and is currently in phase II trials for treatment of breast cancer.[1]
In vitro, ipatasertib showed activity against all three isoforms of Akt.[2]
Ipatasertib is an orally-available protein kinase B (PKB/Akt) inhibitor in phase III clinical development at Genentech for the treatment of metastatic castration-resistant prostate cancer in combination with abiraterone and prednisone.
In 2014, orphan drug designation was assigned in the U.S. for the treatment of gastric cancer including cancer of the gastro-esophageal junction.
Ipatasertib. An orally bioavailable inhibitor of the serine/threonine protein kinase Akt (protein kinase B) with potential antineoplastic activity. Ipatasertib binds to and inhibits the activity of Akt in a non-ATP-competitive manner, which may result in the inhibition of the PI3K/Akt signaling pathway and tumor cell proliferation and the induction of tumor cell apoptosis. Activation of the PI3K/Akt signaling pathway is frequently associated with tumorigenesis and dysregulated PI3K/Akt signaling may contribute to tumor resistance to a variety of antineoplastic agents. Check for active clinical trials using this agent.
PROBLEM
It has been found that ipatasertib exhibits a very high solubility (>1 g/g water; >2 g/g water/ethanol 1:1) and a very high hygroscopicity (˜6% at 50% RH, >35% at 95% RH). Whereas poor solubility is often a limiting factor in the development of galenical formulations of other API’s (active pharmaceutical ingredient), a high solubility can equally be problematic for the process performance. Due to this very high intrinsic hygroscopicity of the API, ipatasertib drug substance tends to auto-dissolve to a honey-like viscous liquid at increased humidity. Such high solubility and hygroscopicity may pose serious problems for processing as well as for stability and shelf-life of the final product. Therefore, conventional pharmaceutical compositions comprising ipatasertib and processes for the manufacture of pharmaceutical compositions comprising wetting (e.g. wet granulation) are difficult due to the high solubility and high hygroscopicity of the API.
SYN

Bromination of (+)-(R)-pulegone (I) with Br2 in the presence of NaHCO3 in Et2O, followed by ring contraction via Favorskii rearrangement with NaOEt in EtOH, and treatment with semicarbazide hydrochloride and NaOAc in refluxing EtOH/H2O gives rise to cyclopentanecarboxylate (II) (1). Subsequent ozonolysis of olefin (II) by means of O3 in EtOAc at -78 °C, and reductive treatment with Zn in AcOH provides beta-ketoester (III). Reaction of ketoester (III) with ammonium acetate (IVa) in MeOH/CH2Cl2 yields enamine (V), which upon cyclization with ammonium formate (IVb) and formamide (VI) at 150 °C provides cyclopentapyrimidinol (VII). Chlorination of pyrimidinol (VII) using POCl3 in refluxing CH2Cl2 results in 4-chloro-5(R)-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidine (VIII), which is condensed with N-Boc-piperazine (IX) in the presence of DIEA in refluxing BuOH to produce piperazinyl cyclopentapyrimidine (X). Oxidation of compound (X) using mCPBA and NaHCO3 in CHCl3 furnishes N-oxide (XI). Subsequent rearrangement of N-oxide (XI) using Ac2O in CH2Cl2 at 100 °C yields acetate (XII). This compound (XII) is hydrolyzed with LiOH in H2O/THF to give alcohol (XIII), which upon Swern oxidation with (COCl)2, DMSO and Et3N in CH2Cl2 at -78 °C affords ketone (XIV) (1-6). Asymmetric transfer hydrogenation of ketone (XIV) in the presence of RuCl[(R,R)-TsDPEN(p-cymene)], HCOOH and Et3N in CH2Cl2, followed by protection with PNBCl in the presence of Et3N in CH2Cl2, and hydrolysis with LiOH in H2O/THF gives rise to alcohol (XV) (1-6). Also, intermediate (XV) can be produced by enzymatic reduction of ketone (XI) using KRED-101 in the presence of GDH, NADP, KOH and PEG-400, KRED-X1.1-P1F01 in the presence of glucose and NAD in DMSO/i-PrOH or KRED-X1.1-P1B06, KRED-X1.1-P1F01 or KRED-X1.1-P1H10 in the presence of NADP in DMSO/i-PrOH or i-PrOH (11,12). In an alternative method, asymmetric transfer hydrogenation of ketone (XIV) in the presence of RuCl[(R,R)-MsDPEN(p-cymene)], HCOOH and Et3N in CH2Cl2, followed by O-protection of the resultant cis/trans mixture of alcohols with PNBCl and Et3N or protection with pivaloyl chloride in the presence of DIEA in CH2Cl2, followed by separation of the resulting cis/trans mixture of esters by means of HPLC. Hydrolysis of trans ester with LiOH in THF yields alcohol (XV) (11). N-Deprotection of piperazine derivative (XV) by means of HCl in CH2Cl2, i-PrOH or toluene at 62 °C provides amine dihydrochloride (XVI) (1-7,11,12), which is then coupled with aminoacid derivative (XVIIa) (1-7,11) or its sodium salt (XVIIb) (12,13) in the presence of DIEA and HBTU in CH2Cl2 or NMM and T3P in i-PrOH or toluene to produce amide (XVIII) (1-7,11-13). Finally, Boc-deprotection of precursor (XVIII) by means of HCl in MeOH/Et2O, PrOH, i-PrOH or toluene at 57 °C furnishes the target GDC-0068

Synthesis of intermediate (XVII): Condensation of methyl (4-chlorophenyl)acetate (XIX) with formaldehyde (XX) in the presence of NaOMe in DMSO gives beta-hydroxyester (XXI). Subsequent dehydration of alcohol (XXI) using MsCl and Et3N in CH2Cl2 provides arylacrylate (XXII), which upon conjugate addition with isopropylamine (XXIII) in the presence of Boc2O in THF yields N-Boc beta-aminoester (XXIV). Basic hydrolysis of ester (XXIV) using KOSiMe3 in THF generates the potassium carboxylate (XXV), which upon condensation with 4(R)-benzyl-2-oxazolidinone (XXVI) via activation with pivaloyl chloride and BuLi in THF at -78 °C affords the N-acyl oxazolidinone (XXVII) (2-6). Finally, removal of the chiral auxiliary group of (XXVII) using LiOH and H2O2 in THF/H2O furnishes the key intermediate (XVII) (1-6,11). Alternative synthesis of intermediate (XXVII): Protection of isopropylamine (XXIII) with Boc2O in toluene affords tert-butyl isopropylcarbamate (XXVIII), which upon N-alkylation with bromomethyl methyl ether (XXIX) in the presence of NaHMDS in 2-MeTHF gives tert-butyl isopropyl(methoxymethyl)carbamate (XXX) (11). Condensation of 4(R)-benzyl-2-oxazolidinone (XXVI) with 2-(4-chlorophenyl)acetyl chloride (XXXIIa) using BuLi in THF at -50 °C (1) or with 2-(4-chlorophenyl)acetic acid (XXXIIb) via activation with pivaloyl chloride and Et3N in refluxing toluene (11) affords N-acyl oxazolidinone(XXXI). After conversion of intermediate (XXXI) to its titanium enolate with TiCl4 and DIEA in CH2Cl2 at -50 °C, diastereoselective Mannich reaction with formaldehyde hemiaminal (XXX) affords adduct (XXVII)
PAPER
Synthesis of Akt inhibitor ipatasertib. Part 2. Total synthesis and first kilogram scale-up
Org Process Res Dev 2014, 18(12): 1652
https://pubs.acs.org/doi/full/10.1021/op500270z
https://pubs.acs.org/doi/suppl/10.1021/op500270z/suppl_file/op500270z_si_001.pdf
Synthesis of Akt Inhibitor Ipatasertib. Part 2. Total Synthesis and First Kilogram Scale-up
† Small Molecule Process Chemistry, Genentech, Inc., a member of the Roche Group, 1 DNA Way, South San Francisco, California 94080-4990, United States
‡ Array BioPharma Inc., 3200 Walnut Street, Boulder, Colorado 80301, United States
Org. Process Res. Dev., 2014, 18 (12), pp 1652–1666
DOI: 10.1021/op500270z
*E-mail: travisr@gene.com.

Herein, the first-generation process to manufacture Akt inhibitor Ipatasertib through a late-stage convergent coupling of two challenging chiral components on multikilogram scale is described. The first of the two key components is a trans-substituted cyclopentylpyrimidine compound that contains both a methyl stereocenter, which is ultimately derived from the enzymatic resolution of a simple triester starting material, and an adjacent hydroxyl group, which is installed through an asymmetric reduction of the corresponding cyclopentylpyrimidine ketone substrate. A carbonylative esterification and subsequent Dieckmann cyclization sequence was developed to forge the cyclopentane ring in the target. The second key chiral component, a β2-amino acid, is produced using an asymmetric aminomethylation (Mannich) reaction. The two chiral intermediates are then coupled in a three-stage endgame process to complete the assembly of Ipatasertib, which is isolated as a stable mono-HCl salt.
(S)-2-(4-Chlorophenyl)-1-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-(isopropylamino)propan-1-one, Ipatasertib Mono-HCl
Ipatasertib mono-HCl (3.23 kg, 80% yield) as an off-white solid. Analytical results: 99.7 A% [0.26% S,R,S-diastereomer observed)]; impurity 23 (M399) was not detected (<0.02 A%) [Method 2.2]; ruthenium content by IPC-AES = 5 ppm; analysis for PF6 anion by CAD-HPLC resulted in not detected [Method 2.3]; residual solvent = 0.4% EtOAc; ion chromatography (IC) = 8.5% chloride (1.14 salt equivalent); DSC = 141 °C; FTIR (neat) 3269 (br OH), 2961–2865 (N–H stretch), 1637 (C═O stretch); 1H NMR (600 MHz, DMSO-d6) 9.39 (s, 1H), 8.64 (s, 1H), 8.49 (s, 1H), 7.49 (q, J = 2.9 Hz, 2H), 7.41 (q, J = 2.9 Hz, 2H), 5.58 (s, 1H), 4.91 (t, J = 6.9 Hz, 1H), 4.78 (dd, J = 8.9, 4.5 Hz, 1H), 3.81 (m, J = 3.3 Hz, 1H), 3.68 (m, J = 3.3 Hz, 1H), 3.67 (m, J = 3.1 Hz, 1H), 3.65 (m, J = 3.2 Hz, 1H), 3.63 (m, J = 3.6 Hz, 1H), 3.59 (m, J = 4.3 Hz, 1H), 3.51 (m, J = 3.5 Hz, 1H), 3.46 (m, J = 3.5 Hz, 1H), 3.36 (m, J = 3.2 Hz, 1H), 3.30 (m, J = 5.7 Hz, 1H), 3.21 (m, J = 3.4 Hz, 1H), 2.98 (m, J = 5.8 Hz, 1H), 1.97 (m, J = 4.8 Hz, 2H), 1.26 (d, J = 6.6 Hz, 3H), 1.25 (d, J = 7.0 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) 170.2, 168.2, 159.4, 155.2, 135.3, 132.5, 129.7 (2C), 129.1 (2C), 120.8, 71.7, 50.4, 47.0, 44.8, 44.5, 44.1, 41.4, 40.8, 34.5, 19.8, 18.4, 18.1; HRMS calcd for C24H32ClN5O2 457.2245; found [M+H]+ 458.2306.
Ipatasertib freebase (3.9 kg, 98.2 A% containing ~1.2% impurity 23 (M399) and impurity M416 at 0.2 A% [Method 2.2]) as tan solid. By CAD-HPLC (see Figure S1-2), the PF6 anion was present in ~0.86 A% [Method 2.3]; Ion chromatography (IC) = 4.0% chloride (0.56 salt equivalent); 1 H NMR (600 MHz, DMSO-d6) 8.44 (s, 1H), 7.45 (d, J = 8.5 Hz, 2H), 7.40 (d, J = 8.5 Hz, 2H), 5.48 (br s, 1H), 4.86 (t, J = 6.9 Hz, 1H), 4.58 (dd, J = 7.3, 4.6 Hz, 1H), 3.74 (m, 1H), 3.40 (m, 1H), 3.63 (m, 2H), 3.61 (m, 1H), 3.42 (m, 1H), 3.57 (m, 1H), 3.18 (m, 1H), 3.50 (m, J = 2.9 Hz, 1H), 3.09 (m, J = 3.1 Hz, 1H), 3.42 (m, 1H), 2.87 (m, J = 4.7 Hz, 1H), 2.00 (m, 1H), 1.92 (m, J = 3.1 Hz, 1H), 1.15 (d, J = 6.4 Hz, 6H), 1.03 (d, J = 6.9 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) 172.0, 169.0, 159.6, 156.3, 136.3, 132.1, 129.7 (2C), 128.9 (2C), 120.9, 72.0, 49.4, 48.7, 45.4, 44.9, 44.8, 44.6, 41.4, 40.9, 34.3, 20.1, 19.9, 19.7; HRMS calcd for C24H32ClN5O2 [M+H]+ 458.2317; found 458.2312. See supporting information (S2) for the NMR spectra (DMSO-d6) of Ipatasertib freebase: ( 1 H) S2, Figure S2-5.12 and ( 13C) Figure S2-5.13.
https://pubs.acs.org/doi/suppl/10.1021/op500270z/suppl_file/op500270z_si_002.pdf
Table S2-1 1 H NMR Assignments of Ipatasertib mono-HCl. S2-52 Figure S2-5.10. 13C NMR (DMSO-d6) spectrum of Ipatasertib mono-HCl. S2-53 Table S2-2 13C NMR Assignments of Ipatasertib mono-HCl. S2-54 Table S2-3 Characteristic Ipatasertib mono-HCl Infrared Signals. S2-55 Figure S2-5.11. FTIR Spectrum of Ipatasertib mono-HCl. S2-56 Figure S2-5.12. XRPD Pattern of Ipatasertib mono-HCl. S2-57
PAPER
https://pubs.acs.org/doi/abs/10.1021/op500271w
https://pubs.acs.org/doi/suppl/10.1021/op500271w/suppl_file/op500271w_si_001.pdf
Synthesis of Akt Inhibitor Ipatasertib. Part 1. Route Scouting and Early Process Development of a Challenging Cyclopentylpyrimidine Intermediate
† Array BioPharma Inc., 3200 Walnut Street, Boulder, Colorado 80301, United States
‡ Genentech Inc., a member of the Roche Group, 1 DNA Way, South San Francisco, California 94080-4990, United States
Org. Process Res. Dev., 2014, 18 (12), pp 1641–1651
DOI: 10.1021/op500271w
*E-mail: jlane@arraybiopharma.com., *E-mail: travisr@gene.com.

Herein, the route scouting and early process development of a key cyclopentylpyrimidine ketone intermediate toward the synthesis of Akt inhibitor Ipatasertib are described. Initial supplies of the intermediate were prepared through a method that commenced with the natural product (R)-(+)-pulegone and relied on the early construction of a methyl-substituted cyclopentyl ring system. The first process chemistry route, detailed herein, enabled the synthesis of the ketone on a hundred-gram scale, but it was not feasible for the requisite production of multikilogram quantities of this compound and necessitated the exploration of alternative strategies. Several new synthetic approaches were investigated towards the preparation of the cyclopentylpyrimidine ketone, in either racemic or chiral form, which resulted in the discovery of a more practical route that hinged on the initial preparation of a highly substituted dihydroxypyrimidine compound. The cyclopentane ring in the target was then constructed through a key carbonylative esterification and subsequent tandem Dieckmann cyclization–decarboxylation sequence that was demonstrated in a racemic synthesis. This proof-of-concept was later developed into an asymmetric synthesis of the cyclopentylpyrimidine ketone, which will be described in a subsequent paper, along with the synthesis of Ipatasertib.
PAPER
Discovery and preclinical pharmacology of a selective ATP-Competitive akt inhibitor (GDC-0068) for the treatment of human tumors
J Med Chem 2012, 55(18): 8110
PAPER
Asymmetric synthesis of akt kinase inhibitor ipatasertib
Org Lett 2017, 19(18): 4806
PATENT
WO 2008006040
PATENT
WO 2012135753
PATENT
WO 2012135759
PATENT
WO 2012135781
PATENT
WO 2013173784
PATENT
WO 2015073739
PATENT
WO 2012135779
PATENT
WO 2013173768
References
- Jump up^ https://www.clinicaltrials.gov/ct2/show/NCT02301988
- Jump up^ Lin K, Friedman L, Gloor S, Gross S, Liederer BM, Mitchell I, et al. Preclinical characterization of GDC-0068, a novel selective ATP competitive inhibitor of Akt. 22nd-EORTC-NCI-AACR-2010 2010; abstr. 79
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
PO |
| ATC code |
|
| Identifiers | |
| ChemSpider | |
| KEGG | |
| Chemical and physical data | |
| Formula | C24H32ClN5O2 |
| Molar mass | 458.00 g·mol−1 |
| 3D model (JSmol) | |
////////////// ipatasertib, orphan drug designation, GDC-0068 , RG7440, PHASE 3
CC(C)NC[C@@H](C(=O)N1CCN(CC1)c2ncnc3[C@H](O)C[C@@H](C)c23)c4ccc(Cl)cc4
It has been found that ipatasertib exhibits a very high solubility (>1 g/g water; >2 g/g water/ethanol 1:1) and a very high hygroscopicity (˜6% at 50% RH, >35% at 95% RH). Whereas poor solubility is often a limiting factor in the development of galenical formulations of other API’s (active pharmaceutical ingredient), a high solubility can equally be problematic for the process performance. Due to this very high intrinsic hygroscopicity of the API, ipatasertib drug substance tends to auto-dissolve to a honey-like viscous liquid at increased humidity. Such high solubility and hygroscopicity may pose serious problems for processing as well as for stability and shelf-life of the final product. Therefore, conventional pharmaceutical compositions comprising ipatasertib and processes for the manufacture of pharmaceutical compositions comprising wetting (e.g. wet granulation) are difficult due to the high solubility and high hygroscopicity of the API.
Linrodostat BMS 986205, ONO 7701
cas 2221034-29-1
- Linrodostat
- (2R)-N-(4-chlorophenyl)-2-(cis-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
- Linrodostat mesylate
- Linrodostat [USAN]
- UNII-OS7OBU191R
- OS7OBU191R
- Linrodostat mesylate [USAN]
- BMS-986205-04
- 2221034-29-1
- Cyclohexaneacetamide, N-(4-chlorophenyl)-4-(6-fluoro-4-quinolinyl)-alpha- methyl-, (alphaR,1alpha,4alpha)-, methanesulfonate (1:1)
Linrodostat; (2R)-N-(4-chlorophenyl)-2-(cis-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide; Linrodostat mesylate; Linrodostat [USAN]; UNII-OS7OBU191R; OS7OBU191R


BMS 986205
(2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoro-4-quinolinyl)cyclohexyl]propanamide
Cyclohexaneacetamide, N-(4-chlorophenyl)-4-(6-fluoro-4-quinolinyl)-α-methyl-, cis-
Cyclohexaneacetamide, N-(4-chlorophenyl)-4-(6-fluoro-4-quinolinyl)-α-methyl-, cis-(αR)-
(i?)-N-(4-chlorophenyl)-2- c 5-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
CAS: 1923833-60-6
Phase III Head and neck cancer; Malignant melanoma
BMS-986205, ONO-7701, F- 001287
- Molecular Formula C24H24ClFN2O
- Average mass 410.912 Da
BMS986205, BMS 986205, ONO-7701
Cyclohexaneacetamide, N-(4-chlorophenyl)-4-(6-fluoro-4-quinolinyl)-α-methyl-, cis-(αR)-
A potent and selective IDO1 (indoleamine 2,3-dioxygenase 1) inhibitor.
| Alternate Name | (R)-N-(4-chlorophenyl)-2-((1s,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propenamide |
|---|---|
| Appearance | Crystalline solid |
| CAS # | 1923833-60-6 |
| Molecular Formula | C₂₄H₂₄ClFN₂O |
| Molecular Weight | 410.92
|
- Originator Bristol-Myers Squibb
- Developer Bristol-Myers Squibb; Ono Pharmaceutical
- Class Antineoplastics; Cyclohexanes; Quinolines; Small molecules
- Mechanism of Action Indoleamine-pyrrole 2,3-dioxygenase inhibitors
Highest Development Phases
- Phase II IHead and neck cancer; Malignant melanoma
- Phase I/II Cancer
- Phase I Solid tumours
Most Recent Events
- 01 Jun 2018Efficacy and adverse events data from a phase I/IIa trial in Bladder cancer (Combination therapy, Late-stage disease) presented at the 54th Annual Meeting of the American Society of Clinical Oncology (ASCO- 2018)
- 08 May 2018Bristol-Myers Squibb plans the CheckMate 9UT phase II trial for Bladder Cancer in USA, Canada, Italy, Mexico, Netherlands, Spain and United Kingdom , (NCT03519256)
- 30 Apr 2018Bristol-Myers Squibb withdraws a phase III trial for Non-small cell lung cancer (First-line therapy, Combination therapy, Late-stage disease) in USA, Austria, Australia, Brazil, Canada, Czech Republic, France, Germany, Greece, Italy, Japan, South Korea, Mexico, Spain, Switzerland, Taiwan and Turkey prior to enrolment (NCT03417037)
BMS , following its acquisition of Flexus Biosciences , and licensee Ono Pharmaceutical are developing linrodostat, a once-daily, indoleamine 2,3-dioxygenase 1 inhibitor for the potential oral treatment of cancer including renal cell carcinoma, muscle-invasive bladder cancer and melanoma. In October 2018, the trial was initiated in the US, Europe, Israel and Brazil.
WO2015031295 product pat
WO2016073770 first disclosed
WO2018209049
- WO 2016073770


Bristol-Myers Squibb, following its acquisition of Flexus Biosciences, is developing BMS-986205 (previously F- 001287), the lead from an immunotherapy program of indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors for the potential treatment of cancer. In February 2016, a phase I/IIa trial was initiated .
BMS-986205 (ONO-7701) is being evaluated at Bristol-Myers Squibb in phase I/II clinical trials for the oral treatment of adult patients with advanced cancers in combination with nivolumab. Early clinical development is also ongoing at Ono in Japan for the treatment of hematologic cancer and for the treatment of solid tumors.
In April 2017, data from the trial were presented at the 108th AACR Annual Meeting in Washington DC. As of February 2017, the MTD had not been reached, but BMS-986205 plus nivolumab treatment was well tolerated, with only two patients discontinuing treatment due to DLTs. The most commonly reported treatment-related adverse events (TRAEs) were decreased appetite, fatigue, nausea, diarrhea, and vomiting. Grade 3 TRAEs were reported in three patients during the combination therapy; however, no grade 3 events were reported during BMS-986205 monotherapy lead-in. No grade 4 or 5 TRAEs were reported with BMS-986205 alone or in combination with nivolumab
Indoleamine 2,3-dioxygenase (IDO; also known as IDOl) is an IFN-γ target gene that plays a role in immunomodulation. IDO is an oxidoreductase and one of two enzymes that catalyze the first and rate-limiting step in the conversion of tryptophan to N-formyl-kynurenine. It exists as a 41kD monomer that is found in several cell populations, including immune cells, endothelial cells, and fibroblasts. IDO is relatively well-conserved between species, with mouse and human sharing 63% sequence identity at the amino acid level. Data derived from its crystal structure and site-directed mutagenesis show that both substrate binding and the relationship between the substrate and iron-bound dioxygenase are necessary for activity. A homolog to IDO (ID02) has been identified that shares 44% amino acid sequence homology with IDO, but its function is largely distinct from that of IDO. (See, e.g., Serafini P, et al, Semin. Cancer Biol, 16(l):53-65 (Feb. 2006) and Ball, H.J. et al, Gene, 396(1):203-213 (Jul. 2007)).
IDO plays a major role in immune regulation, and its immunosuppressive function manifests in several manners. Importantly, IDO regulates immunity at the T cell level, and a nexus exists between IDO and cytokine production. In addition, tumors frequently manipulate immune function by upregulation of IDO. Thus, modulation of IDO can have a therapeutic impact on a number of diseases, disorders and conditions.
A pathophysiological link exists between IDO and cancer. Disruption of immune homeostasis is intimately involved with tumor growth and progression, and the production of IDO in the tumor microenvironment appears to aid in tumor growth and metastasis. Moreover, increased levels of IDO activity are associated with a variety of different tumors (Brandacher, G. et al, Clin. Cancer Res., 12(4): 1144-1151 (Feb. 15, 2006)).
Treatment of cancer commonly entails surgical resection followed by chemotherapy and radiotherapy. The standard treatment regimens show highly variable degrees of long-term success because of the ability of tumor cells to essentially escape by regenerating primary tumor growth and, often more importantly, seeding distant metastasis. Recent advances in the treatment of cancer and cancer-related diseases, disorders and conditions comprise the use of combination therapy incorporating immunotherapy with more traditional chemotherapy and radiotherapy. Under most scenarios, immunotherapy is associated with less toxicity than traditional chemotherapy because it utilizes the patient’s own immune system to identify and eliminate tumor cells.
In addition to cancer, IDO has been implicated in, among other conditions, immunosuppression, chronic infections, and autoimmune diseases or disorders (e.g. , rheumatoid arthritis). Thus, suppression of tryptophan degradation by inhibition of IDO activity has tremendous therapeutic value. Moreover, inhibitors of IDO can be used to enhance T cell activation when the T cells are suppressed by pregnancy, malignancy, or a virus (e.g., HIV). Although their roles are not as well defined, IDO inhibitors may also find use in the treatment of patients with neurological or neuropsychiatric diseases or disorders (e.g., depression).
Small molecule inhibitors of IDO have been developed to treat or prevent IDO-related diseases. For example, the IDO inhibitors 1-methyl-DL-tryptophan; p-(3-benzofuranyl)-DL-alanine; p-[3-benzo(b)thienyl]-DL-alanine; and 6-nitro-L-tryptophan have been used to modulate T cell-mediated immunity by altering local extracellular concentrations of tryptophan and tryptophan metabolites (WO 99/29310). Compounds having IDO inhibitory activity are further reported in WO 2004/094409.
In view of the role played by indoleamine 2,3-dioxygenase in a diverse array of diseases, disorders and conditions, and the limitations (e.g., efficacy) of current IDO inhibitors, new IDO modulators, and compositions and methods associated therewith, are needed.
In April 2017, preclinical data were presented at the 108th AACR Annual Meeting in Washington DC. BMS-986205 inhibited kynurenine production with IC50 values of 1.7, 1.1 and > 2000 and 4.6, 6.3 and > 2000 nM in human (HeLa, HEK293 expressing human IDO-1 and tryptophan-2, 3-dioxygenase cell-based assays) and rat (M109, HEK293 expressing mouse ID0-1 and -2 cell-based assays) respectively. In human SKOV-3 xenografts (serum and tumor) AUC (0 to 24h; pharmacokinetic and pharmacodynamic [PK and PD])) was 0.8, 4.2 and 23 and 3.5, 11 and 40 microM h, respectively; area under the effect curve (PK and PD) was 39, 32 and 41 and 60, 63 and 76% kyn, at BMS-986205 (5, 25 and 125 mg/kg, qd×5), respectively
In April 2017, preclinical data were presented at the 253rd ACS National Meeting and Exhibition in San Francisco, CA. BMS-986205 showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. A good pharmacokinetic profile was seen at oral and iv doses in rats, dogs and monkeys. The compound showed good oral exposure and efficacy in in vivo assays
Preclinical studies were performed to evaluate the activity of BMS-986205, a potent and selective optimized indoleamine 2, 3-dioxygenase (IDO)- 1inhibitor, for the treatment of cancer. BMS-986205 inhibited kynurenine production with IC50 values of 1.7, 1.1 and > 2000 and 4.6, 6.3 and > 2000 nM in human (HeLa, HEK293 expressing human IDO-1 and tryptophan-2, 3-dioxygenase cell-based assays) and rat (M109, HEK293 expressing mouse ID0-1 and -2 cell-based assays) respectively. BMS-986205 was also found to be potent when compared with IDO-1from other species (human < dog equivalent monkey equivalent mouse > rat). In cell-free systems, incubation of inhibitor lead to loss of heme absorbance of IDO-1 which was observed in the presence of BMS-986205 (10 microM), while did not observed with epacadostat (10 microM). The check inhibitory activity and check reversibility (24 h after compound removal) of BMS-986205 was found to be < 1 and 18% in M109 (mouse) and < 1 and 12% SKOV3 (human) cells, respectively. In human whole blood IDO-1, human DC mixed lymphocyte reaction and human T cells cocultured with SKOV3 cells- cell based assays, BMS-986205 showed potent cellular effects (inhibition of kynurenine and T-cell proliferation 3H-thymidine) with IC50 values of 2 to 42 (median 9.4 months), 1 to 7 and 15 nM, respectively. In human SKOV-3 xenografts (serum and tumor) AUC (0 to 24h; pharmacokinetic and pharmacodynamic [PK and PD])) was 0.8, 4.2 and 23 and 3.5, 11 and 40 microM h, respectively; area under the effect curve (PK and PD) was 39, 32 and 41 and 60, 63 and 76% kyn, at BMS-986205 (5, 25 and 125 mg/kg, qd×5), respectively. In vivo human-SKOV3 and hWB-xenografts, IC50 values of BMS-986205 were 3.4 and 9.4 NM, respectively. The ADME of BMS-986205 at parameters iv/po dose was 0.5/2, 0.5/1.5 and 0.5/1.2 mg/kg, respectively; iv/clearance was 27, 25 and 19 ml, min/kg, respectively; iv Vss was 3.8, 5.7 and 4.1 l/kg, respectively; t1/2 (iv) was 3.9, 4.7 and 6.6 h, respectively; fraction (po) was 64, 39 and 10%, respectively. At the time of presentation, BMS-986205 was being evaluated in combination with nivolumab.
The chemical structure and preclinical profile was presented for BMS-986205 ((2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoroquinolin-4-yl)cyclohexyl]propanamide), a potent IDO-1 inhibitor in phase I for the treatment of cancer. This compound showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. The pharmacokinetic profile in rats dosed at 0.5 mg/kg iv and 2 mg/kg po, with clearance, Vss, half-life and bioavailability of 27 ml/min/kg, 3.8 l/kg, 3.9 h and 4%, respectively; in dogs at 0.5 iv and 1.5 po mg/kg dosing results were 25 ml/min/kg, 5.7 l/kg, 4.7 h and 39%; and, in cynomolgus monkeys with the same doses as dogs results were 19 ml/min/kg, 4.1 l/kg, 6.6 h and 10%, respectively. The compound showed good oral exposure and efficacy in in vivo assays.
BMS-986158: a BET inhibitor for cancerAshvinikumar Gavai of Bristol Myers Squibb (BMS) gave an overview of his company’s research into Bromodomian and extra-terminal domain (BET) as oncology target for transcriptional suppression of key oncogenes, such as MYC and BCL2. BET inhibition has been defined as strong rational strategy for the treatment of hematologic malignancies and solid tumors. From crystal-structure guided SAR studies, BMS-986158, 2-{3-(1,4-Dimethyl-1H-1,2,3-triazol-5-yl)-5-[(S)-(oxan-4-yl)(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl}propan-2-ol, was chosen as a potent BET inhibitor, showing IC50 values for BRD2, BRD3 and BRD4 activity of 1 nM; it also inhibited Myc oncogene (IC50 = 0.5 nM) and induced chlorogenic cancer cell death. In vitro the compound also displayed significant cytotoxicity against cancer cells. When administered at 0.25, 0.5 and 1 mg/kg po, qd to mice bearing human lung H187 SCLC cancer xenograft, BMS-986158 was robust and showed efficacy as a anticancer agent at low doses. In metabolic studies, it showed t1/2 of 36, 40 and 24 min in human, rat and mice, respectively, and it gave an efflux ratio of 3 in Caco-2 permeability assay. In phase 1/II studies, BMS-986158 was well tolerated at efficacious doses and regimens, and drug tolerable toxicity at efficacy doses and regimens. Selective Itk inhibitors for inflammatory disordersThe development of highly selective Itk inhibitors for the treatment of diseases related to T-cell function, such as inflammatory disorders, was described by Shigeyuki Takai (Ono Pharmaceutical). Inhibitory properties of a hit compound, ONO-8810443, were modified via X-ray structure and Molecular Dynamics stimulation to get ONO-212049 with significant kinase selectivity (140-fold) against Lck, a tyrosine kinase operating upstream of Itk in the TCR cascade. Further modifications identified final lead compound ONO-7790500 (N-[6-[3-amino-6-[2-(3-methoxyazetidin-1-yl)pyridin-4-yl]pyrazin-2-yl]pyridin-3-yl]-1-(3-methoxyphenyl)-2,3-dimethyl-5-oxopyrazole-4-carboxamide), which selectively inhibited Itk (IC50 = < 0.004 microM) over Lck (IC50 = 9.1 microM; SI 2000-fold) and suppressed Jurkat T-cell proliferation (IC50 = 0.014 microM). This compound suppressed alphaCD3/CDP28 CD4+T-cell stimulation (IC50 = 0.074 microM) with selectivity over PMA/Ionomycin (IC50 = > 10 microM). ONO-7790500 also exhibited in vivo IL-2 inhibitory properties (62% inhibition at 30 mg/kg po) in mice. In pharmacokinetic studies in balb/c mice, the compound administered orally (10 mg/kg) showed a Cmax of 1420 ng/ml, AUClast of 11,700 ng*h/ml, t1/2 of 5.3 h and oral bioavailability of 68%. Administration iv at 0.3 mg/kg gave an AUC last of 610 ng*h/ml, t1/2 of 3.8 h, Vss of 1260 ml/kg and Cl of 5.1 ml/min/kg. ADMET data showed ONO-7790500 did not have relevant activity in cytochromes and hERG channels (IC50 > 10 microM) in toxicological studies, and gave a PAMPA value of 5.0 x 10(-6) cm/s. Fused imidazole and pyrazole derivatives as TGF-beta inhibitorsDual growth and differentiation factor-8 (GDF-8; also known as myostatin) and TGF-beta inhibitors were described. Both targets belong to TGF-beta superfamily consisting of a large group of structurally related cell regulatory proteins involved in fundamental biological and pathological processes, such as cell proliferation or immunomodulation. Myostatin (GDF8) is a negative regulator negative regulator of skeletal muscle growth and has also been related to bone metabolism. Investigators at Rigel Pharmaceuticals found that compounds designed to be GDF-8 inhibitors were able to inhibit TGF-beta as well, this could be an advantage for the treatment of diseases associated with muscle and adipose tissue disorders, as well as potentially immunosuppressive disorders. Jiaxin Yu from the company described new fused imidazole derivatives, of which the best compound was 6-[2-(2,4,5-Trifluorophenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-3-yl]quinoxaline. This compound was very potent at TGF-beta Receptor Type-1 (ALK5) inhibition with an IC50 value of 1nM. In an in vivo mouse assay this compound showed good activity at 59.7 mg/kg, po, and good plasma exposure; inhibition of GDF-8 and TGFbeta growth factors was 90 and 81.6 %, respectively.Rigel’s Ihab Darwish described a series of fused pyrazole derivatives, with the best compound being 6-[2-(2,4-Difluorophenyl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl][1,2,4]triazolo[1,5-a]pyridine. This compound showed an IC50 of 0.06 and 0.23 microM for GDF-8 and TGFbeta, respectively, in the pSMAD (MPC-11) signaling inhibition test. The compound had a good pharmacokinetic profile, with 40% of bioavailability in mice after a 5-mg/kg po dose. An iv dose of 1 mg/kg showed t1/2 of 0.7 h and Vss of 1.0 l/h/kgDiscovery of selective inhibitor of IDO BMS-986205 for cancerIndoleamine-2,3-dioxygenase (IDO)-1 enzyme initiates and regulates the first step of the kynurenine pathway (KP) of tryptophan metabolism, and evidence has shown that overexpression of IDO-1 in cancer tumors is a crucial mechanism facilitating tumor immune evasion and persistence. The chemical structure and preclinical profile of BMS-986205 was presented by Aaron Balog from BMS. BMS-986205 ((2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoroquinolin-4-yl)cyclohexyl]propanamide), is a potent IDO-1 inhibitor in phase I for the treatment of cancer. This compound showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. The pharmacokinetic profile in rats dosed at 0.5 mg/kg iv and 2 mg/kg po, with clearance, Vss, half-life and bioavailability of 27 ml/min/kg, 3.8 l/kg, 3.9 h and 4%, respectively; in dogs at 0.5 iv and 1.5 po mg/kg dosing results were 25 ml/min/kg, 5.7 l/kg, 4.7 h and 39%; and, in cynomolgus monkeys with the same doses as dogs results were 19 ml/min/kg, 4.1 l/kg, 6.6 h and 10%, respectively. The compound showed good oral exposure and efficacy in in vivo assays.Three further reports have been published from this meeting .The website for this meeting can be found at https://www.acs.org/content/acs/en/meetings/spring-2017.html.
SYNTHESIS
1 Wittig NaH
2 REDUCTION H2, Pd, AcOEt, 4 h, rt, 50 psi
3 Hydrolysis HCl, H2O, Me2CO, 2 h, reflux
4 4-Me-2,6-(t-Bu)2-Py, CH2Cl2, overnight, rt
5 SUZUKI AcOK, 72287-26-4, Dioxane, 16 h, 80°C
6 Heck Reaction, Suzuki Coupling, Hydrogenolysis of Carboxylic Esters, Reduction of Bonds, HYDROGEN
7 Et3N, THF, rt – -78°C , Pivaloyl chloride, 15 min, -78°C; 1 h, 0°C ,THF, 0°C – -78°C, BuLi, Me(CH2)4Me, 15 min, -78°C, R:(Me3Si)2NH •Na, THF, 10 min, -50°C , HYDROLYSIS, (PrP(=O)O)3, C5H5N, AcOEt, 5 min, rt

Product Patent
WO2016073770
Example 19
(i?)-N-(4-chlorophenyl)-2- c 5-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
Example 19 : (i?)-N-(4-chlorophenyl)-2-(cz5-4-(6-fluoroquinolin-4- yl)cyclohexyl)propanamide
[0277] Prepared using General Procedures K, B, E, L, M, N, and O. General Procedure L employed 2-(4-(6-fluoroquinolin-4-yl)-cyclohexyl)acetic acid (mixture of
diastereomers), and ( ?)-2-phenyl-oxazolidinone. General Procedure M employed the cis product and iodomethane. The auxiliary was removed following General Procedure N and the desired product formed employing General Procedure O with 4-chloroaniline.
Purified using silica gel chromatography (0% to 100% ethyl acetate in hexanes) to afford Example 19. 1H NMR of czs-isomer (400 MHz; CDC13): δ 9.14 (s, 1H), 8.70 (d, J= 4.6 Hz, 1H), 8.06 (dd, J= 9.2 Hz, J= 5.6 Hz, 1H), 7.58-7.64 (m, 3H), 7.45 (ddd, J= 9.3 Hz, J= 7.8 Hz, J= 2.7 Hz, 1H), 7.19-7.24 (m, 2H), 7.15 (d, J= 4.6Hz, 1H), 3.16-3.26 (m, 1H), 2.59-2.69 (m, 1H), 2.08-2.16 (m, 1H), 1.66-1.86 (m, 7H), 1.31-1.42 (m, 1H), 1.21 (d, J= 6.8Hz, 3H) ppm. m/z 411.2 (M+H)+.
PAPER
Bioorganic & Medicinal Chemistry Letters (2018), 28(3), 319-329.
https://www.sciencedirect.com/science/article/pii/S0960894X17312180

PATENT
WO 2018022992
PATENT
WO 2018071500
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018071500&redirectedID=true
WO-2019006292
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019006292&tab=PCTDESCRIPTION&maxRec=1000
Improved methods for the preparation of substituted quinolinycyclohexylpropanamide compounds, such as linrodostat claiming substituted pyridine compounds as IDO1 inhibitors, useful for treating cancers.
Indoleamine 2,3 -di oxygenase (IDO; also known as IDOl) is an IFN-γ target gene that plays a role in immunomodulation. IDO plays a major role in immune regulation, and its immunosuppressive function manifests in several manners. A pathophysiological link exists between IDO and cancer. Disruption of immune homeostasis is intimately involved with tumor growth and progression, and the production of IDO in the tumor microenvironment appears to aid in tumor growth and metastasis. Moreover, increased levels of IDO activity are associated with a variety of different tumors (Brandacher, G. et al, Clin. Cancer Res. , 12(4): 1144-1151 (Feb. 15, 2006)). In addition to cancer, IDO has been implicated in, among other conditions, immunosuppression, chronic infections, and autoimmune diseases or disorders (e.g., rheumatoid arthritis).
Substituted quinolinylcyclohexylpropanamide pharmaceutical compounds that inhibit IDO and are useful for the treatment of cancer have been previously described. See, e.g., WO2016/073770. Improved methods of making such compounds, which reduce production costs and improve production safety, are, therefore, needed.
Scheme 4
[0076] The disclosure is also directed to methods of preparing intermediate compounds of formula IV. Methods to produce compounds of formula IV are depicted in Schemes 5 and 6.
Scheme 5
IX-A
Scheme 6
IX-B IV
Compounds of the disclosure that include one or more radioisotopes can be used in imaging. See, e.g., WO2018017529. For example, radiolabeled compounds of the disclosure can be used in Positron Emission Tomography (PET). Such methods are useful in the imaging of cancer in a subject. A preferred radiolabeled compound is
1
Pharmaceutically acceptable salts of [18F]-Compound 1 are also within the scope of the disclosure. An exemplary method for the preparation of [18F]-Compound 1 is depicted in Scheme below.
1 . reaction
[18F]-Compound 1
Example 9
(R)-N-(4-chlorophenyl)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
[00258] To a 10 L glass-lined reactor under a blanket of nitrogen was charged 349 g Ν,Ν,Ν’,Ν’-tetramethylchloroformamidinium hexafluorophosphate (TCFH) and 2 L acetonitrile. 245 g N-methylimidazole was added followed by 0.3 L acetonitrile. 300 g (R)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanoic acid was added followed by 0.3 L acetonitrile. The mixture was held for 0.5 h then 139 g 4-chloroaniline charged followed by 0.4 L acetonitrile. The mixture was maintained at 20 °C until the reaction was deemed complete by HPLC analysis. The solution was then heated to 60°C, and 1.2 L water was charged. The solution was then cooled to 40 °C, seeds (3 g) were charged, and the resulting slurry was maintained for 1 h. The slurry was then cooled to 20 °C and 2.7 L water was charged. The slurry was filtered and the cake was washed three times with 3 L of 2: 1 water: acetonitrile. The cake was dissolved with 5.1 L ethyl acetate and the solution was distilled to a volume of 4.2 L at 41 °C under vacuum. The slurry was cooled to 20 °C, 4.14 g seeds were charged, and a solution of 95.7 g methanesulfonic acid in 2.9 L ethyl acetate was added. The slurry was then filtered and washed two times with 1.65 L ethyl acetate and dried under vacuum at 50°C to yield 445 g of (R)-N-(4-chlorophenyl)-2-((l s,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide methanesulfonate as a white solid in 88% yield.
[00259] ¾ NMR (600 MHz, DMSO-de) δ 10.19 (s, IH), 9.24 (d, J=5.7 Hz, IH), 8.40 (dd, J=10.3, 2.6 Hz, IH), 8.33 (dd, J=9A, 5.3 Hz, IH), 8.09 (d, J=5.7 Hz, IH), 8.04 (t, J=8.6 Hz, IH), 7.71 – 7.64 (m, 2H), 7.37 – 7.30 (m, 2H), 3.64 (ddt, J=10.8, 7.3, 3.8 Hz, IH), 2.98 – 2.89 (m, IH), 2.43 (s, 3H), 2.05 – 1.60 (m, 9H), 1.14 (d, J=6.7 Hz, 3H); 13C NMR (126 MHz, DMSO-de) δ 175.0, 162.7, 161.1 , 145.4, 138.2, 136.8, 128.6, 128.1 , 126.7, 126.4, 123.3, 120.8, 119.8, 109.0, 39.8, 39.7, 38.6, 35.5, 28.3, 27.6, 27.2, 26.1 , 16.2 MS (ESI): calcd for C24H24CIFN2O
([M + H]+), 410.16; found, 410.15.
[00260] HPLC analysis: Column: Sigma-Aldrich Supelco Ascentis Express CI 8 2.7um, 150 x 4.6 mm ID; Solvent A: 0.05% TFA with MeCN:water (5/95 v/v); Solvent B: 0.05% TFA with MeCN: water (95/5 v/v); Gradient: %B: 0 Min. 15%; 1 Min. 15%; 13 Min. 55%; 19 Min. 65%; 24 Min. 100%; 24.1 15%; 28 Min. 15%; Stop Time: 24 Min; Flow Rate: 1.0 ml/min;
Column temperature: 30 °C; wavelength: 218 nm. The retention time (R)-N-(4-chlorophenyl)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide peak was 12.6 min.
Example 7
(R)-N-(4-chlorophenyl)-2-((ls, -4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
[00252] To a 50 L glass-lined reactor under a blanket of nitrogen was charged 13.75 kg acetonitrile, then 2.68 Kg Ν,Ν,Ν’,Ν’-tetramethylchloroformamidinium hexafluorophosphate (TCFH) and rinsed with 2.0 Kg acetonitrile. 2.03 Kg N-methylimidazole was added followed by 1.95 Kg acetonitrile. 2.48 Kg (R)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanoic acid was added followed by 1.05 Kg acetonitrile. The mixture was held for 0.5 h then 1.21 Kg 4-chloroaniline charged followed by 1.0 Kg acetonitrile. The mixture was maintained at 20 °C until the reaction was deemed complete by HPLC analysis. The solution was then heated to 60°C, and 9.25 Kg water was charged. The solution was then cooled to 40 °C, the mixture was aged
for 1 h, seeds (32 g) were charged and rinsed with 1.15 Kg 2: 1 water: acetonitrile, and the resulting slurry was maintained for 1 h. The slurry was then cooled to 20 °C and 25.75 Kg water was charged. The slurry was filtered and the cake was washed three times with 6.9 Kg of 2: 1 water: acetonitrile. The cake was dried under vacuum at 50°C to yield 3.33 Kg of (R)-N-(4-chlorophenyl)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide hydrate as a white solid in 94.1% yield.
[00253] ¾ NMR (600 MHz, DMSO-de) δ 10.09 (s, 1H), 8.86 (d, J=4.5 Hz, 1H), 8.08 (dd, J=9.0, 5.6 Hz, 1H), 7.95 (dd, J=10.9, 2.6 Hz, 1H), 7.70 – 7.60 (m, 3H), 7.54 (d, J=4.5 Hz, 1H), 7.33 (d, J=9.0 Hz, 2H), 3.43 – 3.31 (m, 3H), 2.90 – 2.80 (m, 1H), 1.99 – 1.55 (m, 9H), 1.13 (d, J=6.8 Hz, 3H); 13C NMR (151 MHz, DMSO-de) δ 175.0, 159.9, 152.4, 149.7, 145.2, 138.1, 132.7, 128.5, 127.2, 126.7, 120.8, 119.0, 118.6, 107.2, 40.2, 37.4, 35.6, 28.5, 27.6, 27.4, 26.3, 16.1 ; HRMS (ESI); calcd for C24H24CIFN2O ([M + H]+), 411.1619; found 411.1649.
WO-2019006283
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019006283&redirectedID=true
Novel crystalline forms of linrodostat , its salts and hydrates, designated as Forms 1, 2 and 4 (first disclosed in WO2016073770 ), processes for their preparation and compositions comprising them are claimed. Also claims are their use for treating prostate cancer, liver cancer, brain cancer, bladder cancer, ovary cancer and breast cancer.
(R)-N-(4-chlorophenyl)-2-((l S,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanami the below structure:
[0003] Compound 1 is a potent inhibitor of indoleamine 2,3-dioxygenase (IDO; also known as IDOl), which is an IFN-γ target gene that plays a role in immunomodulation.
Compound 1 is being investigated as a treatment for cancer and other diseases. Compound 1 has been previously described in WO2016/073770.
[0004] A compound, as a free base, hydrate, solvate, or salt, can exist in amorphous form and/or one or more crystalline forms, each having different physical properties, for example, different X-ray diffraction patterns (XRPD or PXRD) and different thermal behavior. The free base, hydrate, solvate, and salt forms of a compound can also differ with respect to their individual stabilities, processing, formulation, dissolution profile, bioavailability, and the like. [0005] New forms of (R)-N-(4-chlorophenyl)-2-((l S,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide, having desirable and beneficial chemical and physical properties are needed. There is also a need for reliable and reproducible methods for the manufacture, purification, and formulation of Compound 1 (and its hydrates, solvates, salt,, and hydrated salt forms) to facilitate commercialization. The present disclosure is directed to these, as well as other important aspects.
REFERENCES
23-Feb-2015
Bristol-Myers Squibb To Expand Its Immuno-Oncology Pipeline with Agreement to Acquire Flexus Biosciences, Inc
Bristol-Myers Squibb Co; Flexus Biosciences Inc
17-Dec-2014
Flexus Biosciences, a Cancer Immunotherapy Company Focused on Agents for the Reversal of Tumor Immunosuppression (ARTIS), Announces $38M Financing
Flexus Biosciences Inc
2015106thApril 21Abs 4290
Potent and selective next generation inhibitors of indoleamine-2,3-dioxygenase (IDO1) for the treatment of cancer
American Association for Cancer Research Annual Meeting
Jay P. Powers, Matthew J. Walters, Rajkumar Noubade, Stephen W. Young, Lisa Marshall, Jan Melom, Adam Park, Nick Shah, Pia Bjork, Jordan S. Fridman, Hilary P. Beck, David Chian, Jenny V. McKinnell, Maksim Osipov, Maureen K. Reilly, Hunter P. Shunatona, James R. Walker, Mikhail Zibinsky, Juan C. Jaen
2017108thApril 04Abs 4964
Structure, in vitro biology and in vivo pharmacodynamic characterization of a novel clinical IDO1 inhibitor
American Association for Cancer Research Annual Meeting
John T Hunt, Aaron Balog, Christine Huang, Tai-An Lin, Tai-An Lin, Derrick Maley, Johnni Gullo-Brown, Jesse Swanson, Jennifer Brown
2017253rdApril 05Abs MEDI 368
Discovery of a selective inhibitor of indoleamine-2,3-dioxygenase for use in the therapy of cancer
American Chemical Society National Meeting and Exposition
Aaron Balog
April 2-62017
American Chemical Society – 253rd National Meeting and Exhibition (Part IV) – OVERNIGHT REPORT, San Francisco, CA, USA
Casellas J, Carceller V
////////////////PHASE 1, BMS 986205, 1923833-60-6, BMS-986205, ONO-7701,Bristol-Myers Squibb, Antineoplastics, F- 001287
C[C@H]([C@H]1CC[C@@H](C2=CC=NC3=CC=C(F)C=C23)CC1)C(NC4=CC=C(Cl)C=C4)=O
Wrapping up #MEDI‘s 1st time disclosures is Aaron Balog of @bmsnews talking about an IOD-1 inhibitor to treat cancer #ACSSanFran
////////////////BMS986205, BMS 986205, BM-986205, ONO-7701, Phase III, Head and neck cancer, Malignant melanoma, 1923833-60-6, Linrodostat
CC(C1CCC(CC1)C2=C3C=C(C=CC3=NC=C2)F)C(=O)NC4=CC=C(C=C4)Cl
ABL 001, Asciminib
ABL001 / Asciminib
Cas 1492952-76-7
Chemical Formula: C20H18ClF2N5O3
Molecular Weight: 449.8428
Elemental Analysis: C, 53.40; H, 4.03; Cl, 7.88; F, 8.45; N, 15.57; O, 10.67
N-[4-[Chloro(difluoro)methoxy]phenyl]-6-[(3R)-3-hydroxypyrrolidin-1-yl]-5-(1H-pyrazol-5-yl)pyridine-3-carboxamide
3-Pyridinecarboxamide, N-[4-(chlorodifluoromethoxy)phenyl]-6-[(3R)-3-hydroxy-1-pyrrolidinyl]-5-(1H-pyrazol-3-yl)-
PHASE 3, Chronic Myeloid Leukemia, NOVARTIS
UPDATE FDA APPROVED 10/29/2021,
| Scemblix |
To treat Philadelphia chromosome-positive chronic myeloid leukemia with disease that meets certain criteria
Asciminib, sold under the brand name Scemblix, is a medication used to treat Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML).[1][2][3] Asciminib is a protein kinase inhibitor.[1]
The most common adverse reactions include upper respiratory tract infections, musculoskeletal pain, fatigue, nausea, rash, and diarrhea.[2]
Asciminib was approved for medical use in the United States in October 2021.[1][4][5]
The U.S. Food and Drug Administration (FDA) granted the application for asciminib priority review, fast track, orphan drug, and breakthrough therapy designations.[2][6][7]
Asciminib is an orally bioavailable, allosteric Bcr-Abl tyrosine kinase inhibitor with potential antineoplastic activity. Designed to overcome resistance, ABL001 binds to the Abl portion of the Bcr-Abl fusion protein at a location that is distinct from the ATP-binding domain. This binding results in the inhibition of Bcr-Abl-mediated proliferation and enhanced apoptosis of Philadelphia chromosome-positive (Ph+) hematological malignancies. The Bcr-Abl fusion protein tyrosine kinase is an abnormal enzyme produced by leukemia cells that contain the Philadelphia chromosome.
ABL001 has been used in trials studying the health services research of Chronic Myelogenous Leukemia and Philadelphia Chromosome-positive Acute Lymphoblastic Leukemia.

- Originator Novartis
- Developer Novartis; Novartis Oncology
- Class Antineoplastics; Pyrazoles; Pyrrolidines; Small molecules
- Mechanism of Action Bcr-abl tyrosine kinase inhibitors
Highest Development Phases
- Phase III Chronic myeloid leukaemia
- No development reported Precursor cell lymphoblastic leukaemia-lymphoma
Most Recent Events
- 04 Nov 2017 No recent reports of development identified for phase-I development in Acute-lymphoblastic-leukaemia(Second-line therapy or greater) in Australia (PO)
- 04 Nov 2017 No recent reports of development identified for phase-I development in Acute-lymphoblastic-leukaemia(Second-line therapy or greater) in France (PO)
- 04 Nov 2017 No recent reports of development identified for phase-I development in Acute-lymphoblastic-leukaemia(Second-line therapy or greater) in Germany (PO)
- The tyrosine kinase activity of the ABLl protein is normally tightly regulated, with the N-terminal cap region of the SH3 domain playing an important role. One regulatory mechanism involves the N-terminal cap glycine-2 residue being myristoylated and then interacting with a myristate binding site within the SHI catalytic domain. A hallmark of chronic myeloid leukemia (CML) is the Philadelphia chromosome (Ph), formed by the t(9,22) reciprocal chromosome translocation in a haematopoietic stem cell. This chromosome carries the BCR-ABL1 oncogene which encodes the chimeric BCR-ABL1 protein, that lacks the N-terminal cap and has a constitutively active tyrosine kinase domain.Although drugs that inhibit the tyrosine kinase activity of BCR-ABL1 via an ATP-competitive mechanism, such as Gleevec® / Glivec® (imatinib), Tasigna® (nilotinib) and Sprycel® (dasatinib), are effective in the treatment of CML, some patients relapse due to the emergence of drug-resistant clones, in which mutations in the SHI domain compromise inhibitor binding. Although Tasigna® and Sprycel® maintain efficacy towards many Gleevec-resistant mutant forms of BCR-ABLl, the mutation in which the threonine-315 residue is replaced by an isoleucine (T315I) remains insensitive to all three drugs and can result in CML patients developing resistance to therapy. Therefore, inhibiting BCR-ABLl mutations, such as T315I, remains an unmet medical need. In addition to CML, BCR-ABLl fusion proteins are causative in a percentage of acute lymphocytic leukemias, and drugs targeting ABL kinase activity also have utility in this indication.Agents targeting the myristoyl binding site (so-called allosteric inhibitors) have potential for the treatment of BCR-ABLl disorders (J. Zhang, F. J. Adrian, W. Jahnke, S. W. Cowan- Jacob, A. G. Li, R. E. Iacob4, T. Sim, J. Powers, C. Dierks, F. Sun, G.-R. Guo, Q. Ding, B. Okram, Y. Choi, A. Wojciechowski, X. Deng, G. Liu, G. Fendrich, A. Strauss, N. Vajpai, S. Grzesiek, T. Tuntland, Y. Liu, B. Bursulaya, M. Azam, P. W. Manley, J. R. Engen, G. Q. Daley, M. Warmuth., N. S. Gray. Targeting BCR-ABL by combining allosteric with ATP -binding-site inhibitors. Nature 2010;463:501-6). To prevent the emergence of drug resistance from ATP inhibitor and/or allosteric inhibitor use, a combination treatment using both types of inhibitor can be developed for the treatment of BCR-ABLl related disorders. In particular, the need exists for small molecules, or combinations thereof, that inhibit the activity of BCR-ABLl and BCR-ABLl mutations via the ATP binding site, the myristoyl binding site or a combination of both sites.Further, inhibitors of ABL 1 kinase activity have the potential to be used as therapies for the treatment of metastatic invasive carcinomas and viral infections such as pox and Ebola viruses.The compounds from the present invention also have the potential to treat or prevent diseases or disorders associated with abnormally activated kinase activity of wild-type ABL1, including non-malignant diseases or disorders, such as CNS diseases in particular neurodegenerative diseases (for example Alzheimer’s, Parkinson’s diseases), motoneuroneuron diseases (amyotophic lateral sclerosis), muscular dystrophies, autoimmune and inflammatory diseases (diabetes and pulmonary fibrosis), viral infections, prion diseases.
Asciminib is an allosteric inhibitor of BCR-ABL kinase in phase III clinical development at Novartis for the treatment of patients with chronic myelogenous leukemia (CML) in chronic phase who have been previously treated with ATP-binding site tyrosine kinase inhibitors. Early clinical trials are also under way in patients with Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) and as first-line threapy of CML.
PATENT
To illustrate tautomerism with the following specific examples, (R)-N-(4- (chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin-l-yl)-5-(lH-pyrazol-5-yl)nicotinamide
(right structure, below) is a tautomer of (R)-N-(4-(chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin-l-yl)-5-(lH-pyrazol-3-yl)nicotinamide (left structure, below) and vice versa:

[0045] Where the plural form (e.g. compounds, salts) is used, this includes the singular
Example 9
(R)-N-(4-(Chlorodifluoromethoxy)phenyl)-6-(3-hvdroxypyrrolidin-l-yl)-5-(lH-pyrazol-5- vDnicotinamide

[00365] A mixture of (R)-5-Bromo-N-(4-(chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin-l-yl)nicotinamide (Stage 9.2, 100 mg, 0.216 mmol) and 5-(4 ,4,5,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -((2-(trimethylsilyl)ethoxy)methyl)- IH-pyrazole (215 mg, 0.663 mmol), Pd(PPh3)2Cl2 (17 mg, 0.024 mmol), Na2C03 (115 mg, 1.081 mmol), DME (917 μί), water (262 μΕ) and EtOH (131 μί) in a MW vial was sealed, evacuated / purged 3 times with argon and subjected to MW irradiation at 125°C for 20 min. The RM was diluted with 2 mL
of DME, stirred with Si-Thiol (Silicycle 1.44 mmol/g, 90 mg, 0.130 mmol) for 3 h. The mixture was centrifuged and the supernatant was filtered through a 0.45 μηι PTFE filter and the solvent was evaporated off under reduced pressure. The crude product was purified by flash
chromatography (RediSep® Silica gel column, 12 g, cyclohexane / EtOAc from 40% to 100% EtOAc) to afford the protected intermediate as a colorless oil. Ethylene diamine (96 μί, 1.428 mmol) and TBAF 1 M in THF (1.428 mL, 1.428 mmol) were then added and the RM was stirred at 80-85°C for 5 days. The solvent was evaporated off under reduced pressure and the residue was dissolved in EtOAc (40 mL), washed 3 times with sat. aq. NaHCC and brine, dried over Na2S04 and The solvent was evaporated off under reduced pressure to give a residue which was purified by preparative SFC (Column DEAP, from 25% to 30% in 6 min) to yield the title compound as a white solid.
[00366] Alternatively, Example 9 was prepared by adding TFA (168 mL, 2182 mmol) to a solution of N-(4-(chlorodifluoromethoxy)phenyl)-6-((R)-3-hydroxypyrrolidin-l-yl)-5-(l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazol-5-yl)nicotinamide (Stage 9.1, 31.3 g, 54.6 mmol) in DCM (600 mL). The mixture was stirred at RT for 2.5 h. The solvent was evaporated off under reduced pressure and the residue was dissolved in EtOAc (1.5 L),washed with a sat. solution of NaHC03 (3 x 500 mL) and brine (500 mL), dried over Na2S04 and the solvent was evaporated off under reduced pressure to give a residue which was suspended in DCM (300 mL), stirred at RT for 15 min, filtered, washed with DCM (200 mL), dried and purified by chromatography (Silica gel, 1 kg, DCM / MeOH 95:5). The residue was dissolved in MeOH (500 mL) and treated with Si-Thiol (Biotage, 5.0 g , 6.5 mmol) for 16 h at 25°C. The resin was filtered off, the solvent was evaporated off under reduced pressure and the residue was crystallized from MeCN to afford the title compound as a white crystalline solid.
[00367] Alternatively, Example 9 was prepared by the dropwise addition of aqueous HC1
(7.7 mL of 6M) to a solution of N-(4-(chlorodifluoromethoxy)phenyl)-6-((R)-3-hydroxypyrrolidin- 1 -yl)-5-( 1 -(tetrahydro-2H-pyran-2-yl)- 1 H-pyrazol-5-yl)nicotinamide (Stage 9.1, 3.8 g, 7.12 mmol) in MeOH (20 mL) and THF (10 mL) with cooling (below 35°C). The mixture was stirred at 22°C for 2 h and then added to cooled (10°C) 1.2 M NaOH (22 mL).
Throughout the addition the temperature was kept below 30°C and pH was kept in the range of 9-10. The RM was then stirred for 30 min at 30°C. The solvent was evaporated off under reduced pressure, until the desired compound precipitated. The precipitate was filtered and dried to give the title compound as a yellow solid.
[00368] Analytical data for Example 9: HPLC (Condition 5) tR = 5.54 min, HPLC Chiral
(CHIRALCEL® OD-H, 250 x 4.6 mm, eluent : n-heptane/EtOH/MeOH (85: 10:5), 1 mL/min, UV 210 nm) tR = 10.17 min, UPLC-MS (condition 3) tR = 0.93 min, m/z = 450.3 [M+H]+, m/z = 494.1 [M+formic acid-H]“; XH-NMR (400 MHz, DMSO-d6) δ ppm 1.65 – 1.76 (m, 1 H) 1.76 – 1.87 (m, 1 H) 2.93 (d, J=l 1.73 Hz, 1 H) 3.19 – 3.29 (m, 2 H) 3.35 – 3.51 (m, 1 H) 4.10 – 4.25 (m, 1 H) 4.89 (br. s, 1 H) 6.41 (br. s, 1 H) 7.33 (d, J=8.50 Hz, 2 H) 7.57/7.83 (br. s, 1 H) 7.90 (d, J=8.50 Hz, 2 H) 8.07 (br. s, 1 H) 8.77 (br. s, 1 H) 10.23 (s, 1 H) 12.97/13.15 (br. s, 1 H).
[00369] Stage 9.1 : N-(4-(Chlorodifluoromethoxy)phenyl)-6-((R)-3-hydroxypyrrolidin- 1 -yl)-5-( 1 -(tetrahydro-2H-pyran-2- l)- 1 H-pyrazol-5-yl)nicotinamide

[00370] l-(Tetrahydro-2H-pyran-2-yl)-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (29.6 g, 102 mmol), K3P04 (51.6 g, 236 mmol) and Pd(PPh3)4 (4.55 g, 3.93 mmol) were added to a suspension of (R)-5-bromo-N-(4-(chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin-l-yl)nicotinamide (Stage 9.2, 36.4 g, 79 mmol) in toluene (360 mL) under an argon atmosphere and the mixture was stirred at 110°C for 4 h. The RM was poured into brine (500 mL) and extracted with EtOAc (2 x 1 L). The combined extracts were washed with brine (500 mL), dried over Na2S04, and the solvent was evaporated off under reduced pressure to give a residue which was purified by chromatography (Silica gel column, 1.5 kg, DCM / MeOH 95:5) to afford a dark yellow foam, that was dissolved in MeOH / DCM (1 L of 3: l) and treated with Si-Thiol (Biotage, 35 g , 45.5 mmol) for 17 h at 30°C. The resin was filtered off, and solvent was evaporated off under reduced pressure, until the desired compound crystallized. The product was filtered washed with MeOH and dried to afford the title compound.
[00371] Alternatively, Stage 9.1 was prepared by adding 4-(chlorodifluoromethoxy)aniline
(16.6 g, 84.9 mmol), NMM (21.7 g, 212.1 mmol), hydroxybenzotriazole hydrate (HOBt H20, 11.9 g, 77.77 mmol) and l-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCIHCl, 20.9 g, 109.0 mmol) to a solution of 6-((R)-3-hydroxypyrrolidin-l-yl)-5-(l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazol-5-yl)nicotinic acid (Stage 9.4, 29.83 g, 70.7 mmol) in THF (271 mL). The mixture was stirred for 1.5 h at 25°C and then at 65°C for 16 h. After cooling the RM to 35 °C, further EDCIHCl (13.3 g, 69.4 mmol) was added and the RM was stirred for 1.5 h at 35°C then again at 65°C for 16 h. After cooling the RM to 35°C, water (150 mL) was added, the THF was removed under reduced pressure, EtOAc (180 mL) was added and the mixture was stirred for at 35 °C fori h. The two layers were separated and the aq. phase was then extracted with EtOAc (60 mL). The combined organic layers were washed with water (90 mL), brine (90 mL). The solvent was evaporated off under reduced pressure to give a brown solid which was purified by column chromatography (Silica gel, DCM / MeOH 40: 1 to 20: 1) to afford the title compound as a yellow solid.
[00372] Analytical data for Stage 9.1: HPLC (Condition 5) tR = 6.12 min, UPLC-MS
(Condition 3) tR = 1.06 min, m/z = 533.2 [M+H]+; XH-NMR (400 MHz, DMSO-d6) δ ppm 1.36 -2.02 (m, 7 H) 2.23 – 2.38 (m, 1 H) 3.08 – 3.29 (m, 2 H) 3.32 – 3.52 (m, 2 H) 3.73 – 3.93 (m, 1 H) 4.13 – 4.25 (m, 1 H) 4.80 – 4.90 (m, 1 H) 4.95 – 5.17 (m, 1 H) 6.33 – 6.50 (m, 1 H) 7.33 (d, J=8.99 Hz, 2 H) 7.61 (d, J=1.56 Hz, 1 H) 7.86 (d, J=8.99 Hz, 2 H) 7.97 – 8.11 (m, 1 H) 8.82 (s, 1 H) 10.13 – 10.25 (m, 1 H).
[00373] Stage 9.2: (R)-5-Bromo-N-(4-(chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin- 1 -yl)nicotinamide

[00374] (R)-Pyrrolidin-3-ol (9.55 g, 109.6 mmol) and DIPEA (35.1 ml, 201.3 mmol) were added to a suspension of 5-bromo-6-chloro-N-(4-(chlorodifluoromethoxy)phenyl)nicotinamide (Stage 9.3, 37.7 g, 91.5 mmol) in iPrOH (65 mL) and stirred at 140°C for 1 h. EtOAc (700 mL) was added and the solution was washed IN HC1 (2 x 200 mL), sat. NaHCC (200 mL) and brine (2 x 200 mL), dried over Na2S04, and the solution was concentrated under reduced pressure until crystallization commenced. n-Heptane (1 L) were added and the mixture was stirred at RT for 30 min, filtered and washed with ΪΡΓ20 (500 mL) to afford the title compound as a white crystalline solid. HPLC (Condition 5) tR = 6.68 min, UPLC-MS (Condition 3) tR = 1.10 min, m/z =
462.2/464.2 [M+H]+; XH-NMR (400 MHz, DMSO-d6) δ ppm 1.78 – 2.01 (m, 2 H) 3.55 (d, J=l 1.34 Hz, 1 H) 3.66 – 3.75 (m, 1 H) 3.79 – 3.93 (m, 2 H) 4.34 (br. s, 1 H) 4.98 (d, =3.13 Hz, 1 H) 7.32 (d, J=8.99 Hz, 2 H) 7.84 (d, J=8.99 Hz, 2 H) 8.33 (d, J=1.96 Hz, 1 H) 8.66 (d, J=1.96 Hz, 1 H) 10.21 (s, 1 H).
[00375] Stage 9.3: 5-Bromo-6-chloro-N- 4-(chlorodifluoromethoxy)phenyl)nicotinamide

[00376] DMF (2.55 mL, 33.0 mmol) and SOCl2 (24.08 ml, 330 mmol) were added to a suspension of 5-bromo-6-chloro-nicotinic acid (26 g, 110 mmol) in toluene (220 mL) and the RM was stirred at 80°C for 1 h. The solvent was evaporated off under reduced pressure and the residue was dissolved in THF (220 mL) and cooled to -16°C. DIPEA (38.4 mL, 220 mmol) was added, followed by dropwise addition of a solution of 4-(chlorodifluoromethoxy)aniline (22.35 g, 115 mmol) in THF (220 mL) over 15 min. The suspension was stirred for 1 h at RT. The solvent was evaporated off under reduced pressure and the residue was dissolved in TBME (700 mL), washed with IN HC1 (2 x 200 mL), sat. NaHC03 (200 mL) and brine (2 x 200 mL), dried over Na2S04, and the solvent was evaporated off under reduced pressure to give the product which was crystallized from EtOAc – n-heptane to afford the title compound as a white crystalline solid. HPLC (Condition 5) tR = 7.77 min, UPLC-MS (Condition 3) tR = 1.24 min, m/z =
409.1/411.1/413.1 [M+H]+; XH-NMR (400 MHz, DMSO-d6) δ ppm 7.38 (d, =8.99 Hz, 2 H) 7.85 (d, =8.99 Hz, 2 H) 8.72 (br. s, 1 H) 8.92 (br. s, 1 H) 10.68 (s, 1 H).
[00377] Stage 9.4: 6-((R)-3-Hydroxypyrrolidin-l-yl)-5-(l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazol-5-yl)nicotinic acid

[00378] Aq. NaOH (180 niL of 2.6 M) was added to a solution of methyl 6-((R)-3-hydroxypyrrolidin- 1 -yl)-5-(l -(tetrahydro-2H-pyran-2-yl)- 1 H-pyrazol-5-yl)nicotinate (Stage 9.5, 11 lg, 299 mmol) in MeOH (270 mL) and the RM was stirred at RT for 14 h. The MeOH was evaporated off under reduced pressure and the aq. residue was treated with brine (90 mL), extracted with MeTHF twice (540 mL + 360 mL) and the combined organic layers were washed with water (90 mL). MeTHF was added to the combined aq. layers, the biphasic mixture was cooled to 0 °C and acidified (pH = 4-4.5) with aq. HC1 solution (18%) and extracted with
MeTHF. The combined organic extracts were washed with brine and the solvent was evaporated off under reduced pressure to give a residue which was recrystallized from a EtOAc / TBME (1 : 1) to afford the title compound as a white solid. HPLC (Condition 7) tR = 4.74 min, LC-MS
(Condition 8) tR = 3.37 min, m/z = 359.0 [M+H]+; XH-NMR (400 MHz, DMSO-d6) δ ppm 1.44 (br. s, 2 H), 1.51 (d, J=11.54 Hz, 2 H), 1.64 – 1.86 (m, 4 H), 1.90 (br. s, 1 H), 2.31 (d, J=9.29 Hz, 1 H), 2.77 (br. s, 1 H), 3.10 (br. s, 1 H), 3.21 (d, J=8.78 Hz, 2 H), 3.27 – 3.51 (m, 4 H), 3.87 (d, J=11.54 Hz, 1 H), 4.16 (br. s, 1 H), 4.75 – 4.93 (m, 1 H), 5.04 (br. s, 1 H), 6.35 (d, J=17.32 Hz, 1 H), 7.51 – 7.64 (m, 1 H), 7.64 – 7.82 (m, 1 H), 8.67 (d, J=2.26 Hz, 1 H), 12.58 (br. s, 1 H).
[00379] Stage 9.5: Methyl 6-((R)-3-hydroxypyrrolidin-l-yl)-5-(l-(tetrahydro-2H-pyran-2-yl)- 1 H-pyrazol-5-yl)nicotinate

[00380] A mixture of (R)-methyl 5-bromo-6-(3-hydroxypyrrolidin-l-yl)nicotinate (Stage
9.6, 90 g, 299 mmol), l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazole-5-boronic acid pinacol ester (103.9 g, 373.6 mmol), K3P04 (126.9 g, 597.7 mmol), Pd(PPh3)2Cl2 (6.29 g, 8.97 mmol) in toluene (900 mL) was stirred at 92°C and for 16 h. After cooling the mixture to RT, the solution was washed with water (450 mL), 5% NaHCC solution (430 mL) and the solvent was evaporated off under reduced pressure to give a residue which was used without further purifications in the next step. HPLC (Condition 7) tR = 6.929 min, LC-MS (Condition 8) tR = 4.30 min, m/z = 373.0 [M+H ; XH-NMR (400 MHz, DMSO-d6) δ ppm 1.19 – 1.28 (m, 1 H), 1.35 – 1.63 (m, 4 H), 1.63 -1.86 (m, 3 H), 1.89 (br. s, 1 H), 2.12 – 2.39 (m, 1 H), 3.11 (br. s, 1 H), 3.18 – 3.48 (m, 4 H), 3.78 (s, 4 H), 3.88 (d, J=11.54 Hz, 1 H), 4.08 – 4.24 (m, 1 H), 4.86 (dd, J=18.20, 2.89 Hz, 1 H), 5.02 (d, J=8.28 Hz, 1 H), 6.39 (br. s, 1 H), 7.58 (d, J=1.25 Hz, 1 H), 7.78 (br. s, 1 H), 8.69 (t, J=2.01 Hz, 1 H).
[00381] Stage 9.6: (R)-methyl 5-bromo-6-(3-hydroxypyrrolidin-l-yl)nicotinate

[00382] DIPEA (105.3 g, 142.2 mL, 814.4 mmol) was added to a solution of methyl-5-bromo-6-chroronicotinate (85 g, 339.5 mmol) and (R)-pyrrolidin-3-ol (54.2 g, 441.2 mmol) in isopropyl acetate and the RM was stirred at 70°C for 14 h . The solvent was evaporated off under reduced pressure to give a the residue which was dissolved in toluene (850 mL), washed with water (127 mL) and brine (127 mL)and concentrated under reduced pressure until precipitation commenced. n-Heptane (340 mL) was slowly added to the stirred mixture at 22 °C, which was then cooled to 0 °C and the product was filtered, washed with a toluene / n-heptane mixture
(1 : 1.5) and dried to give the title compound as a yellow solid. HPLC (Condition 7) tR = 8.54 min, LC-MS (Condition 8) tR = 4.62 min, m/z = 300.9/302.9 [M+H]+; XH-NMR (400 MHz, DMSO-d6) δ ρριη 1.77 – 1.99 (m, 2 H), 3.57 (d, J=11.54 Hz, 1 H), 3.72 (ddd, J=l 1.11, 7.97, 3.26 Hz, 1 H), 3.78 (s, 3 H), 3.81 -3.90 (m, 2 H), 4.26 – 4.39 (m, 1 H), 4.99 (br. s, 1 H), 8.11 (d, J=2.01 Hz, 1 H), 8.56 (d, J=1.76 Hz, 1 H).
PAPER
- By Wylie, Andrew A.; Schoepfer, Joseph; Jahnke, Wolfgang; Cowan-Jacob, Sandra W.; Loo, Alice; Furet, Pascal; Marzinzik, Andreas L.; Pelle, Xavier; Donovan, Jerry; Zhu, Wenjing; et al
- From Nature (London, United Kingdom) (2017), 543(7647), 733-737.
By Wylie, Andrew A. et alFrom Nature (London, United Kingdom), 543(7647), 733-737; 2017
PAPER
- By Molica, Matteo; Massaro, Fulvio; Breccia, Massimo
- From Expert Opinion on Pharmacotherapy (2017), 18(1), 57-65.
PATENT
US 20170216289
PAPER
- By El Rashedy, Ahmed A.; Olotu, Fisayo A.; Soliman, Mahmoud E. S.
- From Chemistry & Biodiversity (2018), 15(3), n/a.

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
 |
|
| Clinical data | |
|---|---|
| Trade names | Scemblix |
| Other names | ABL001 |
| Routes of administration |
By mouth |
| Drug class | Tyrosine kinase inhibitor |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII |
|
| KEGG | |
| ChEMBL |
|
| PDB ligand | |
| Chemical and physical data | |
| Formula | C20H18ClF2N5O3 |
| Molar mass | 449.84 g·mol−1 |
| 3D model (JSmol) | |
References
- ^ Jump up to:a b c d “Scemblix- asciminib tablet, film coated”. DailyMed. Retrieved 4 November 2021.
- ^ Jump up to:a b c “FDA approves asciminib for Philadelphia chromosome-positive chronic myeloid leukemia”. U.S. Food and Drug Administration (FDA) (Press release). 29 October 2021. Retrieved 4 November 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Breccia M, Colafigli G, Scalzulli E, Martelli M (August 2021). “Asciminib: an investigational agent for the treatment of chronic myeloid leukemia”. Expert Opinion on Investigational Drugs. 30 (8): 803–811. doi:10.1080/13543784.2021.1941863. PMID 34130563.
- ^ “Scemblix: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 29 October 2021.
- ^ “FDA approves Novartis Scemblix (asciminib), with novel mechanism of action for the treatment of chronic myeloid leukemia”. Novartis (Press release). Retrieved 29 October 2021.
- ^ “Asciminib Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 27 February 2017. Retrieved 29 October 2021.
- ^ “Novartis receives FDA Breakthrough Therapy designations for investigational STAMP inhibitor asciminib (ABL001) in chronic myeloid leukemia”. Novartis (Press release). 8 February 2020. Retrieved 29 October 2021.
External links
- “Asciminib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02081378 for “A Phase I Study of Oral ABL001 in Patients With CML or Ph+ ALL” at ClinicalTrials.gov
- Clinical trial number NCT03106779 for “Study of Efficacy of CML-CP Patients Treated With ABL001 Versus Bosutinib, Previously Treated With 2 or More TKIs” at ClinicalTrials.gov
////////////////ABL001, Asciminib, ABL 001, ABL-001, PHASE 3, Chronic Myeloid Leukemia, NOVARTIS
O=C(NC1=CC=C(OC(F)(Cl)F)C=C1)C2=CN=C(N3C[C@H](O)CC3)C(C4=CC=NN4)=C2

NEW DRUG APPROVALS
one time
$10.00
