
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Zuclopenthixol

Zuclopenthixol
Clopenthixol
- Molecular FormulaC22H25ClN2OS
- Average mass400.965 Da
53772-83-1 [RN],
- N05AF05
1-Piperazineethanol, 4-[(3Z)-3-(2-chloro-9H-thioxanthen-9-ylidene)propyl]-
5443
2-{4-[(3Z)-3-(2-Chloro-9H-thioxanthen-9-ylidene)propyl]piperazin-1-yl}ethanol
258-758-5[EINECS]
Z)-Clopenthixol
1-piperazineethanol, 4-(3-(2-chloro-9h-thioxanthen-9-ylidene)propyl)-
1-Piperazineethanol, 4-(3-(2-chlorothioxanthen-9-ylidene)propyl)-
1-Piperazineethanol, 4-[(3Z)-3-(2-chloro-9H-thioxanthen-9-ylidene)propyl]-
- 1-Piperazineethanol, 4-[3-(2-chloro-9H-thioxanthen-9-ylidene)propyl]-, (Z)-
- 4-[(3Z)-3-(2-Chloro-9H-thioxanthen-9-ylidene)propyl]-1-piperazineethanol
- 9H-Thioxanthene, 1-piperazineethanol deriv.
- (Z)-Clopenthixol
- Acuphase
- Cisordinol
- Clopixol
- Clopixol depo
- Zuclopenthixol
- cis-(Z)-Clopenthixol
- cis-Clopenthixol
- α-Clopenthixol
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Zuclopenthixol acetate | 349S2ZHF05 | 85721-05-7 | OXAUOBQMCDIVPQ-IOXNKQMXSA-N |
| Zuclopenthixol decanoate | TSS9KIZ5OG | 64053-00-5 | QRUAPADZILXULG-WKIKZPBSSA-N |
| Zuclopenthixol dihydrochloride | 7042692VYN | 58045-23-1 | LPWNZMIBFHMYMX-MHKBYHAFSA-N |
Zuclopenthixol hydrochloride
58045-23-1, MW: 473.8933
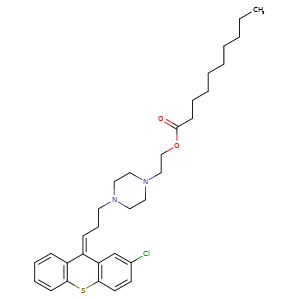
ZUCLOPENTHIXOL DECANOATE, CLOPENTHIXOL DECANOATE, CIS-
64053-00-5, Molecular Formula, C32-H43-Cl-N2-O2-S, Molecular Weight, 555.2227
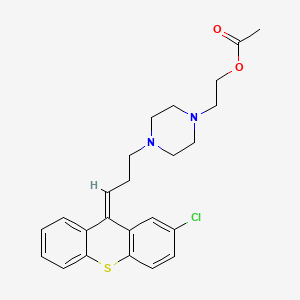
Zuclopenthixol acetate
85721-05-7, C24H27ClN2O2S, 443.0ClopenthixolCAS Registry Number: 982-24-1
CAS Name: 4-[3-(2-Chloro-9H-thioxanthen-9-ylidene)propyl]-1-piperazineethanol
Additional Names: 2-chloro-9-[3-[4-(2-hydroxyethyl)-1-piperazinyl]propylidene]thiaxanthene
Molecular Formula: C22H25ClN2OS
Molecular Weight: 400.96
Percent Composition: C 65.90%, H 6.28%, Cl 8.84%, N 6.99%, O 3.99%, S 8.00%
Literature References: Thioxanthene neuroleptic. Prepn (configuration not specified): BE585338; P. V. Petersen et al.,US3116291 (1960, 1963 both to Kefalas A/S). Prepn of the pharmacologically active cis-isomer: BE816855; N. Lassen, US3996211 (1974, 1976 both to Kefalas A/S). Pharmacology: Cazzullo, Andreola, Acta Neurol.20, 162 (1965); Weissman, Mod. Probl. Pharmacopsychiatry2, 15 (1969); Moeller Nielsen, ibid. 23. Metabolism: Khan, Acta Pharmacol. Toxicol.27, 202 (1969). HPLC determn of isomers in serum: T. Aaes-Jorgensen, J. Chromatogr.188, 239 (1980). Series of articles on pharmacology and clinical studies: Acta Psychiatr. Scand.64, Suppl. 294, 1-77 (1981).
Properties: Colorless syrup. Sparingly sol in ether. Readily sol in methanol.
Derivative Type: Dihydrochloride
CAS Registry Number: 633-59-0
Manufacturers’ Codes: AY-62021; N-746
Trademarks: Ciatyl (Troponwerke); Sordenac (Lundbeck); Sordinol (Ayerst)
Molecular Formula: C22H25ClN2OS.2HCl
Molecular Weight: 473.89
Percent Composition: C 55.76%, H 5.74%, Cl 22.44%, N 5.91%, O 3.38%, S 6.77%
Properties: Crystals from ethanol, mp 250-260° (dec). Freely sol in water; sparingly sol in alcohol. Practically insol in other organic solvents. LD50 in male mice (mg/kg): 111 i.v. (Lassen).
Melting point: mp 250-260° (dec)
Toxicity data: LD50 in male mice (mg/kg): 111 i.v. (Lassen)
Derivative Type:cis(Z)-Form
CAS Registry Number: 53772-83-1
Additional Names: a-Clopenthixol; zuclopenthixol
Properties: Crystals, mp 84-85°.
Melting point: mp 84-85°
Derivative Type:cis(Z)-Form dihydrochloride
CAS Registry Number: 58045-23-1
Trademarks: Cisordinol (Lundbeck); Clopixol (HMR)
Properties: Crystals, mp 250-260° (dec). LD50 in male mice (mg/kg): 105 i.v. (Lassen).
Melting point: mp 250-260° (dec)
Toxicity data: LD50 in male mice (mg/kg): 105 i.v. (Lassen)
Therap-Cat: Antipsychotic.
Keywords: Antipsychotic; Thioxanthenes.
Zuclopenthixol is an antipsychotic indicated for the management of schizophrenia. The acuphase formulation is indicated for initial treatment of acute psychosis or exacerbation of psychosis, while the depot formulation is best for maintenance.Zuclopenthixol, also known as Zuclopentixol or Zuclopenthixolum, is an antipsychotic agent. Zuclopenthixol is a thioxanthene-based neuroleptic with therapeutic actions similar to the phenothiazine antipsychotics. It is an antagonist at D1 and D2 dopamine receptors. Major brands of zuclopenthixol are Cisordinol, Acuphase, and Clopixol. This drug is a liquid. This compound belongs to the thioxanthenes. These are organic polycyclic compounds containing a thioxanthene moiety, which is an aromatic tricycle derived from xanthene by replacing the oxygen atom with a sulfur atom. Known drug targets of zuclopenthixol include 5-hydroxytryptamine receptor 2A, D(1B) dopamine receptor, D(2) dopamine receptor, D(1A) dopamine receptor, and alpha-1A adrenergic receptor. It is known that zuclopenthixol is metabolized by Cytochrome P450 2D6. Zuclopenthixol was approved for use in Canada in 2011, but is not approved for use in the United States.
Zuclopenthixol (brand names Cisordinol, Clopixol and others), also known as zuclopentixol, is a medication used to treat schizophrenia and other psychoses. It is classed, pharmacologically, as a typical antipsychotic. Chemically it is a thioxanthene. It is the cis–isomer of clopenthixol (Sordinol, Ciatyl).[1] Clopenthixol was introduced in 1961, while zuclopenthixol was introduced in 1978.
Zuclopenthixol is a D1 and D2 antagonist, α1-adrenergic and 5-HT2 antagonist.[2] While it is approved for use in Australia, Canada, Ireland, India, New Zealand, Singapore, South Africa and the UK it is not approved for use in the United States.[3][4]
Medical uses
Available forms
Zuclopenthixol is available in three major preparations:
- As zuclopenthixol decanoate (Clopixol Depot, Cisordinol Depot), it is a long-acting intramuscular injection. Its main use is as a long-acting injection given every two or three weeks to people with schizophrenia who have a poor compliance with medication and suffer frequent relapses of illness.[5] There is some evidence it may be more helpful in managing aggressive behaviour.[6]
- As zuclopenthixol acetate (Clopixol-Acuphase, Cisordinol-Acutard), it is a shorter-acting intramuscular injection used in the acute sedation of psychotic inpatients. The effect peaks at 48–72 hours providing 2–3 days of sedation.[7]
- As zuclopenthixol dihydrochloride (Clopixol, Cisordinol), it is a tablet used in the treatment of schizophrenia in those who are compliant with oral medication.[8]
It is also used in the treatment of acute bipolar mania.
Dosing
As a long-acting injection, zuclopenthixol decanoate comes in a 200 mg and 500 mg ampoule. Doses can vary from 50 mg weekly to the maximum licensed dose of 600 mg weekly. In general, the lowest effective dose to prevent relapse is preferred. The interval may be shorter as a patient starts on the medication before extending to 3 weekly intervals subsequently. The dose should be reviewed and reduced if side effects occur, though in the short-term an anticholinergic medication benztropine may be helpful for tremor and stiffness, while diazepam may be helpful for akathisia. 100 mg of zuclopenthixol decanoate is roughly equivalent to 20 mg of flupentixol decanoate or 12.5 mg of fluphenazine decanoate.
In acutely psychotic and agitated inpatients, 50 – 200 mg of zuclopenthixol acetate may be given for a calming effect over the subsequent three days, with a maximum dose of 400 mg in total to be given. As it is a long-acting medication, care must be taken not to give an excessive dose.
In oral form zuclopenthixol is available in 10, 25 and 40 mg tablets, with a dose range of 20–60 mg daily.
Side effects
Chronic administration of zuclopenthixol (30 mg/kg/day for two years) in rats resulted in small, but significant, increases in the incidence of thyroid parafollicular carcinomas and, in females, of mammary adenocarcinomas and of pancreatic islet cell adenomas and carcinomas. An increase in the incidence of mammary adenocarcinomas is a common finding for D2 antagonists which increase prolactin secretion when administered to rats. An increase in the incidence of pancreatic islet cell tumours has been observed for some other D2 antagonists. The physiological differences between rats and humans with regard to prolactin make the clinical significance of these findings unclear.
Withdrawal syndrome: Abrupt cessation of therapy may cause acute withdrawal symptoms (eg, nausea, vomiting, or insomnia). Symptoms usually begin in 1 to 4 days of withdrawal and subside within 1 to 2 weeks.[1][2]
Other permanent side effects are similar to many other typical antipsychotics, namely extrapyramidal symptoms as a result of dopamine blockade in subcortical areas of the brain. This may result in symptoms similar to those seen in Parkinson’s disease and include a restlessness and inability to sit still known as akathisia, a slow tremor and stiffness of the limbs.[8] Zuclopenthixol is thought to be more sedating than the related flupentixol, though possibly less likely to induce extrapyramidal symptoms than other typical depots.[5] As with other dopamine antagonists, zuclopenthixol may sometimes elevate prolactin levels; this may occasionally result in amenorrhoea or galactorrhoea in severe cases. Neuroleptic malignant syndrome is a rare but potentially fatal side effect. Any unexpected deterioration in mental state with confusion and muscle stiffness should be seen by a physician.
Zuclopenthixol decanoate induces a transient dose-dependent sedation. However, if the patient is switched to maintenance treatment with zuclopenthixol decanoate from oral zuclopenthixol or from i.m. zuclopenthixol acetate the sedation will be no problem. Tolerance to the unspecific sedative effect develops rapidly.[9]
SYN
Journal of the American Chemical Society (2019), 141(6), 2251-2256
https://pubs.acs.org/doi/10.1021/jacs.8b13907
Synthesis of Clopenthixol (4d)
Inside a nitrogen-filled glovebox, an oven-dried glass culture tube (Fischer Scientific part #14- 959-35A), equipped with a magnetic stirring bar, was charged with 2-chloro-9H-thioxanthen-9- one (245 mg, 1.0 mmol, 1 equiv), copper(II) acetate (0.91 mg, 0.0050 mmol, 0.0050 equiv), racBINAP (3.2 mg, 0.0050 mmol, 0.0050 equiv), and THF (1.0 mL). The tube was then fitted tightly with a Teflon-lined blow-out screw cap (Kimble-Chase part #73808-15425). The reaction tube was removed from the glovebox, and the mixture was stirred rapidly for 5 min. A balloon, connected to a 6 mL plastic syringe head, was filled with allene gas until its size was roughly 6 cm in diameter. A needle was attached to the head of the syringe. The reaction tube was evacuated by piercing the septum with a needle connected to a Schlenk line. Immediately after, the allene contained in the balloon was used to refill the reaction tube by piercing the septum with the needle. The balloon decreased to roughly half its original diameter during the refill process. The needle and balloon were left attached, and dimethoxy(methyl)silane (250 uL, 2.0 mmol, 2.0 equiv) was added to the reaction mixture using a 1 mL plastic syringe. The solution was then stirred overnight at rt. At this point, the flask was quickly evacuated by piercing the septum with a needle connected to a Schlenk line, and the headspace was refilled with dry nitrogen. This process was repeated a total of three times. THF (1 mL) solution containing 4e (367 mg, 1.2 mmol, 1.2 equiv), triphenylphosphine (11.5 mg, 0.044 mmol, 0.044 equiv), racDTBM-SEGPHOS (26.8 mg, 0.044 mmol, 0.044 equiv), and copper(II) acetate (3.6 mg, 0.040 mmol, 0.040 equiv) was added to the reaction mixture using a 1 mL plastic syringe. The reaction tube was heated to 40 °C by submersion in an oil bath overnight. After cooling to rt, the cap was removed and 4 M HCl in dioxane was slowly added to the reaction mixture (2.0 mL, WARNING: VIGOROUS HYDROGEN GAS EVOLUTION). The color of the reaction mixture turned to deep red, and after stirring for approximately 30 min, a tan precipitate evolved. After an additional 1 h, diethyl ether (10 mL) was added and the solids collected by filtration (950 mg). By LC/MS analysis, this solid contains mostly 4c (as the hydrochloride) with a trace amount of triphenylphosphine oxide. The entire solid was suspended in dry acetonitrile (1.0 mL) in another dry reaction tube, equipped with a magnetic stirring bar. Potassium carbonate (552 mg, 4 mmol) was added to the tube, which was then capped and placed under a nitrogen atmosphere using a needle connected to a Schlenk line. 2-bromoethanol (142 uL, 2 mmol) was added to the reaction mixture using a glass microsyringe, and the mixture was left to stir overnight at rt. After this time, the cap was removed, and the solution was diluted with water (10 mL). The mixture was extracted with dichloromethane (3 x 10 mL), and the combined organic phases was concentrated with the aid of a rotary evaporator. The mixture was purified by reverse phase preparative HPLC (C18 column, MeCN/water) to yield a 1.1:1 Z/E mixture of 4d as a yellow foamy solid (217 mg, 54% overall yield). The identity of 4d was confirmed by LC/MS analysis against a commercially available standard (Cayman Chemical) and by comparison of 1H NMR to the literature. 13 For further structural confirmation, a portion of 4d was repurified by HPLC to obtain pure (Z)-4d, the biologically active isomer, whose spectra have not been reported in the literature. 1H NMR (400 MHz, CDCl3) δ 7.43 (dd, J = 14.5, 6.2 Hz, 3H), 7.27 (q, J = 8.2 Hz, 4H), 7.17 (d, J = 8.3 Hz, 1H), 5.89 (t, J = 7.1 Hz, 1H), 3.60 (t, J = 5.4 Hz, 2H), 2.62 (t, J = 7.2 Hz, 2H), 2.58–2.35 (m, 12H); 13C NMR (101 MHz, CDCl3) δ 140.2, 135.7, 133.4, 133.2, 132.7, 130.9, 130.4, 128.6, 127.3, 126.9, 126.8, 126.7, 126.2, 125.6, 59.2, 58.3, 57.7, 53.1, 52.8, 27.3.
SYN
Chemical Engineering & Technology (2016), 39(10), 1821-1827.
https://onlinelibrary.wiley.com/doi/10.1002/ceat.201500673
SYN
European Journal of Pharmaceutics and Biopharmaceutics (2012), 82(2), 437-456.
https://www.sciencedirect.com/science/article/abs/pii/S0939641112002263?
SYN
Organic Process Research & Development (2013), 17(9), 1142-1148.
https://pubs.acs.org/doi/10.1021/op400069e
SUN
CN 103214453
https://patents.google.com/patent/CN103214453A/enDiuril ton (Clopenthixol), chemistry 2-chloro-9-[3 ‘ by name-(N ‘-the 2-hydroxyethyl piperazine-N)-allyl group]-thioxanthene, this product is a kind of Thiaxanthene derivative, has significant antipsycholic action and special sedative effect, is particularly useful for the schizophrenia patient.Its activeconstituents is its alpha-isomer, i.e. zuclopenthixol (structural formula is seen Fig. 1); Have the stereotypy effect that anti-Ritalin causes, and the effect of anti-Apomorphine is arranged, this product energy rejection condition avoiding reaction and catalepsy are stronger 10 times than chlorpromazine.A little less than the cholinolytic effect, and antihistamine effect is strong.Zuclopenthixol is applicable to that treatment has psychosis, class Paranoia-illusion type schizophrenia, hebephrenia, the manic and anxiety periodic psychosis of anxiety and illusion symptom; The uneasiness that mental element causes, excitement, psychiatric disorder, the encephalatrophy process, post-traumatic psychosis, the proverb of trembling are absurd etc.Be particularly useful for elderly patients.Recorded the quality standard of zuclopenthixol sheet, zuclopenthixol dihydrochloride, Ciatyl Depot and zuclopenthixol acetic ester etc. in the British Pharmacopoeia, wherein the zuclopenthixol quality standard has stipulated that its content should be 95%-105%.But in actual industrial production, guarantee that zuclopenthixol reaches pharmaceutically acceptable purity, and β-isomer (structural formula is seen Fig. 2) content being limited in 5%, is a very thing of difficulty.About the preparation method of zuclopenthixol, mainly containing of bibliographical information is following several:As if the general separation of having described diuril ton isomer can be undertaken by the fractional crystallization of dihydrochloride among the BE585338A of nineteen fifty-nine application, and still, this separation method yield is extremely low, and complicated operation does not also have actual industrial use.Described the preparation method of diuril ton isomer mixture among the US3116291, wherein alpha-isomer is that the content of zuclopenthixol is 30%-35%.Obtain purer zuclopenthixol by diuril ton alkali being carried out fractional separation in the literary composition with ether organic solvent, but, instructed crystallization to come the purifying zuclopenthixol can not obtain good result, especially in isomer mixture, had under the situation of a large amount of impurity existence by diuril ton alkali.Embodiment 1: the preparation of diuril ton base1) preparation of 2-chloro-9-allyl group-9-thioxanthene alcohol100.00g (0.405mol) 2-chloro-9-thioxanthone is dissolved in the 600mL tetrahydrofuran (THF), 20 ℃ of-30 ℃ of stirrings, add magnesium powder 26g then, iodine 1g, splash into chlorallylene 65g (0.855mol), 40 ℃-50 ℃ are reacted 2h down, and the cooling back drips 20% sodium chloride aqueous solution 1000ml in reaction solution, stir 10min, filter insolubles, use dichloromethane extraction then 2 times, each 500ml, merge organic phase, water 500ml washing is told organic layer, dry after-filtration, filtrate is concentrated except that desolvating, obtain 105.40g2-chloro-9-allyl group-9-thioxanthene alcohol.2) preparation of 2-chloro-9-(2-propenylidene) thioxanthene100.00g (0.346mol) 2-chloro-9-allyl group-9-thioxanthene alcohol is dissolved in the 100ml toluene, solution is heated to 40 ℃, the Acetyl Chloride 98Min. of 1.34g (0.017mol) is dissolved in the diacetyl oxide of 41.19g (0.403mol) and drops in the above-mentioned solution, temperature is controlled at about 40 ℃, dropwise, it is complete until the TLC monitoring reaction that heating makes temperature of reaction rise to 50 ℃ of-55 ℃ of reactions, and concentrating under reduced pressure steams solvent, obtains 94.01g2-chloro-9-(2-propenylidene) thioxanthene.3) preparation of clopenthixol baseN-(2-hydroxyethyl) piperazine of getting 90.00g (0.332mol) 2-chloro-9-(2-propenylidene) thioxanthene and 215.21g (1.65mol) adds in the 1L four-hole boiling flask, stirs and is warming up to 100 ℃ of reactions, and TLC monitors to reacting completely.Vacuum oil pump concentrating under reduced pressure excessive N-(2-hydroxyethyl) piperazine, temperature is controlled at 100 ℃-135 ℃, and oil pump vacuum tightness is at 0.2-1mmHg.Distillation finishes, and adds the benzene of 400ml and the water of 100ml in gained oily matter, and 70 ℃ are stirred 15min, and separatory is used the water washing organic phase of 100ml again, and simultaneous temperature is controlled at 60 ℃-70 ℃, separatory; With organic phase concentrate resistates.This resistates is dissolved in the 300ml methylene dichloride, after add 10% hydrochloric acid soln and transfer pH to 2-3, stirring 10min, separatory, water discard dichloromethane extraction liquid with the dichloromethane extraction of 150ml; Above-mentioned water adds ammoniacal liquor and regulates pH=9-10, extract with methylene dichloride (300ml * 2) after stirring 10min, merge organic phase, use anhydrous sodium sulfate drying, suction filtration, filtrate decompression concentrate 109.32g diuril ton base, isomer proportion α/β is that 45/55 (the HPLC area normalization method: analytical column is 4.6 * 250mmAgilent C18 post, moving phase is acetonitrile: methyl alcohol: phosphoric acid buffer=20: 30: 50, flow velocity are 1mL/min; With this understanding, the retention time of zuclopenthixol is 12min, and the retention time of β-isomer is 16min).Embodiment 2: the preparation of zuclopenthixol Chlorodracylic acid ester 2HCl100.00g (0.250mol) α/β-diuril ton is dissolved in the ethyl acetate of 500ml, under 40 ℃ of conditions, drips the 100ml ethyl acetate that is dissolved with 52.48g (0.30mol) parachlorobenzoyl chloride, dropwise the back back flow reaction, complete until the TLC monitoring reaction.Remove the 300ml solvent under reduced pressure, be cooled to 4 ℃, remove by filter precipitation.Mother liquor is heated to 40 ℃, drips the concentrated hydrochloric acid aqueous solution of 12.50g (0.125mol) 37%, react about 1h after, cool off, have solid to separate out, filter 56.27g zuclopenthixol Chlorodracylic acid ester 2HCl.Purity (HPLC is the same) is 97.11%, and productive rate is 36.82%.Embodiment 3: the preparation of zuclopenthixol2HCl is dissolved in the methanol aqueous solution of 300ml80% with 45.25g (0.074mol) zuclopenthixol Chlorodracylic acid ester, adds the potassium hydroxide of 16.83mol (0.30mol) then.With mixture heating up to 50 ℃, insulation reaction 1h.Underpressure distillation removes and desolvates, and with toluene (200ml * 2) and water extraction, merges organic phase, and concentrating under reduced pressure is removed toluene; Residue obtainedly carry out recrystallization with hexanaphthene, the 25.51g dried crystals.Purity is 99.7%, and productive rate is 86.17%. 1H?NMR(CDCl 3,400MHz),δ:7.10-7.46(7H,m),5.98(1H,t),3.41(2H,t),2.46-2.52(14H,m)。Embodiment 4: the preparation of Ciatyl Depot 2HClThe zuclopenthixol of 50.00g (0.125mol) is dissolved in the methylene dichloride of 500ml, and to wherein dripping 28.60g (0.150mol) decanoyl chloride, back flow reaction is complete to the TLC monitoring reaction after dropwising under the room temperature.Underpressure distillation removes and desolvates, and adds the ethyl acetate of 300ml in residue, drips the ethyl acetate solution that contains hydrogenchloride again, is transferred to 3-4 until pH.After the cooling, filter, vacuum-drying gets 70.63g Ciatyl Depot 2HCl.Productive rate is 90.12%.Embodiment 5: the preparation of Ciatyl DepotThe Ciatyl Depot 2HCl of 60g (0.0957mol) is suspended in the t-butyl methyl ether of 400ml, drips water (250ml) solution of 13.22g (0.0957mol) salt of wormwood, stirring reaction 0.5h.Two are separated, and use 100ml water washing organic phase again, use the anhydrous sodium sulfate drying organic phase, filter, and organic solvent is removed in underpressure distillation, get the 51.29g Ciatyl Depot.Productive rate is 96.58%. 1H?NMR(CDCl 3,400MHz),δ:7.12-7.50(7H,m),5.90(1H,t),4.35(2H,t),3.41(2H,t),2.97(2H,t),2.32-2.57(10H,m),2.06(2H,t),1.64(2H,m),1.30(12H,m),0.88(3H,t)。
SYN
https://patents.google.com/patent/WO2017121755A1/enPreparation of ZU3:9-(3-(4-(2-hydroxyethyl)piperazinyl)propylidene)-thioxanthene

ZU3To a solution of 9-oxothioxanthene (1.0 equiv.) in THF at reflux were added a solution of cyclopropylmagnesium bromide in THF (1.0 equiv.) and stirred during 2 hours. The mixture was cooled down at room temperature and a solution of hydrogen bromide in acetic acid (4 eq.) was added and stirred at room temperature. The reaction mixture was concentrated in vacuo and purified by silica gel column chromatography to obtain 9- (3bromopropylidene)thioxanthene in 30% yield.Then, to a solution of 9-(3bromopropylidene)thioxanthene in acetonitrile at reflux was added N-(2-hydroxyethyl)piperazine (1,5 eq.), potassium iodide (0.1 eq.) and potassium carbonate (3 eq.). The mixture was stirred at reflux then concentrated in vacuo and purified by silica gel column chromatography to afford ZU3 with 98% purity (HPLC). HPLC analysis (BEH C18 type, mobile phase: H20/ acetonitrile (HCOOH 0.1%)) : tR = 1.68 min. Preparation of ZUf:l-(3-(9H-thioxanthen-9-ylidene)propyl)piperidine-4-carboxylic acid), ZU4 (9-(3-(4- (ethylacetate) iperidine)propylidene)-thioxanthene

To a solution of 9-(3-bromopropylidene)thioxanthene in acetonitrile at reflux was added N-ethylacetate piperidine (1,5 eq.), potassium iodide (0.1 eq.) and potassium carbonate (3 eq.). The mixture was stirred at reflux then concentrated in vacuo and purified by silica gel column chromatography to afford ZU4 as a brown oil with a purity of 97% in HPLC analysis HPLC analysis (BEH C18 type, mobile phase: H20/acetonitrile (HCOOH 0.1%)) : tR = 2.07 min.The compound ZU4 was stirred during 2 hours at reflux in a mixture of THF and a solution of NaOH in water. After phase separation, the aqueous layer was extracted twice by diethyl ether. The global organic layer was, then, washed by a saturated solution of NaCl, dried over MgSC^, filtered and concentrated in vacuo to afford ZUf HPLC analysis (BEH CI 8 type, mobile phase: H20/acetonitrile (HCOOH 0.1%)) : tR = 2.24 min.Preparation of ZU5:(Z)-2-(4-(3-(2-chloro-9H-thioxanthen-9-ylidene)propyl)piperazin-l-yl)ethylacetate

To a solution of ZU (4-[3-(2-chloro-9H-thioxanthen-9-ylidene)propyl]-l- piperazineethanol) (leq.) in dichloromethane was added acetic anhydride (1.5 eq.), 4- dimethylaminopyridine (0,1 eq.) and trimethylamine (1 eq.). The mixture was stirred at room temperature and then concentrated in vacuo to afford ZU5 as a yellow oil with 97% of purity (HPLC). HPLC analysis {BEH C18 type, mobile phase: H20/acetonitrile (HCOOH 0.1%)): tR = 2.44 minPreparation of ZUe and ZU6:l-(3-(9H-xanthen-9-ylidene)propyl)piperidine-4-carboxylic acidand eth -(3-(9H-xanthen-9-ylidene)propyl)piperidine-4-carboxylate

To a solution of 9-oxoxanthene (1.0 equiv.) in THF at reflux were added a solution of cyclopropylmagnesium bromide in THF (1.0 equiv.) and stirred during 2 hours. The mixture was cooled down at room temperature and a solution of hydrogen bromide in acetic acid (4 eq.) was added and stirred at room temperature. The reaction mixture was concentrated in vacuo and purified by silica gel column chromatography to obtain 9- (3bromopropylidene)-oxoxanthene.Then, to a solution of 9-(3bromopropylidene)-oxoxanthene in acetonitrile at reflux was added Ethyl 4-piperidinecarboxylate (1,5 eq.), potassium iodide (0.1 eq.) and potassium carbonate (3 eq.). The mixture was stirred at reflux then concentrated in vacuo and purified by silica gel column chromatography to obtain ZU6 ZU6 is then dissolved in a mixture of THF and a solution of NaOH in water. After phase separation, the aqueous layer was extracted twice by diethyl ether. The global organic layer was, then, washed by a saturated solution of NaCl, dried over MgSC^, filtered and concentrated in vacuo to afford ZUe as a white solid with a purity up to 97% in HPLC. Preparation of ZUc:(Z)-2-(4-(3-(2-(trifluoromethyl)-9H-thioxanthen-9-ylidene)propyl)piperazin-l-yl) ethanamine

EtOH, reflux, 2hPurification withHCI buffer

To a solution of ZU1 (2-[4-[3-[2-(trifiuoromethyl)thioxanthen-9- ylidene]propyl]piperidin-l-yl] ethanol) (leq.) in THF was added diethylazodicarboxylate, phtalimide and triphenylphosphine. The solution was stirred at room temperature during 3 hours and then concentrated in vacuo. The crude oil was then dissolved in ethanol, hydrazine was added and the mixture was stirred at reflux during 2 hours. The crude product obtained after concentration was purified via a reversed phase chromatography using HCI as buffer to afford the compound ZUc as an hydrochloride salt (orange solid). [M+H]+ (ESI+) : 434. HPLC analysis (BEH C18 type, mobile phase: H20/acetonitrile (HCOOH 0.1%)): tR = 2.04 min Preparation of ZUd:(Z)-l-(2-fluoroethyl)-4-(3-(2-(trifluoromethyl)-9H-thioxanthen-9-ylidene)propyl) iperazine

To a solution of flupenthixol in dichloromethane was added at -10°C diethylamino sulfur trifluoride. The mixture was then stirred at room temperature. The crude product was purified via a reversed phase chromatography using HC1 as buffer to afford the compound ZUd as a hydrochloride salt (orange solid) with 97% purity in HPLC. HPLC analysis (BEH C18 type, mobile phase: H20/acetonitrile (HCOOH 0.1%)): tR = 3.59 min.Compounds ZU, ZUa, ZUb, ZU1, ZU2The following compounds can be easily found in commerce:

Example 2: Ebselen oxide derivatives

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Pharmacology
Pharmacodynamics

Cisordinol 10 mg tablet
Zuclopenthixol antagonises both dopamine D1 and D2 receptors, α1-adrenoceptors and 5-HT2 receptors with a high affinity, but has no affinity for cholinergic muscarine receptors. It weakly antagonises the histamine (H1) receptor but has no α2-adrenoceptor blocking activity[citation needed].
Evidence from in vitro work and clinical sources (i.e. therapeutic drug monitoring databases) suggests that both CYP2D6 and CYP3A4 play important roles in zuclopenthixol metabolism.[11]
Pharmacokinetics
History
Zuclopenthixol was introduced by Lundbeck in 1978.[22]
References
- ^ Sneader, Walter (2005). Drug discovery: a history. New York: Wiley. p. 410. ISBN 0-471-89980-1.
- ^ Pharmacological effects of a specific dopamine D-1 antagonist SCH 23390 in comparison with neuroleptics Life sciences 1984 Apr 16;34(16):1529-40.
- ^ Green, Alan I.; Noordsy, Douglas L.; Brunette, Mary F.; O’Keefe, Christopher (2008). “Substance abuse and schizophrenia: Pharmacotherapeutic intervention”. Journal of Substance Abuse Treatment. 34 (1): 61–71. doi:10.1016/j.jsat.2007.01.008. ISSN 0740-5472. PMC 2930488. PMID 17574793.
- ^ Sweetman, Sean C., ed. (2009). “Anxiolytic Sedatives Hypnotics and Antipsychotics”. Martindale: The complete drug reference (36th ed.). London: Pharmaceutical Press. pp. 1040–1. ISBN 978-0-85369-840-1.
- ^ Jump up to:a b da Silva Freire Coutinho E, Fenton M, Quraishi SN (1999). “Zuclopenthixol decanoate for schizophrenia”. The Cochrane Database of Systematic Reviews. John Wiley and Sons, Ltd. (2): CD001164. doi:10.1002/14651858.CD001164. PMC 7032616. PMID 10796607. Retrieved 2007-06-12.
- ^ Haessler F, Glaser T, Beneke M, Pap AF, Bodenschatz R, Reis O (2007). “Zuclopenthixol in adults with intellectual disabilities and aggressive behaviours”. British Journal of Psychiatry. 190 (5): 447–448. doi:10.1192/bjp.bp.105.016535. PMID 17470962.
- ^ Lundbeck P/L (1991). “Clopixol Acuphase 50 mg/mL Injection Clopixol Acuphase 100 mg / 2 mL Injection”. Lundbeck P/L. Retrieved 2007-06-12.
- ^ Jump up to:a b Bryan, Edward J.; Purcell, Marie Ann; Kumar, Ajit (16 November 2017). “Zuclopenthixol dihydrochloride for schizophrenia”. The Cochrane Database of Systematic Reviews. 2017 (11): CD005474. doi:10.1002/14651858.CD005474.pub2. ISSN 1469-493X. PMC 6486001. PMID 29144549.
- ^ “Summary of Product Characteristics” (PDF).
- ^ Jump up to:a b c d e “TGA eBS – Product and Consumer Medicine Information Licence”.
- ^ Davies SJ, Westin AA, Castberg I, Lewis G, Lennard MS, Taylor S, Spigset O (2010). “Characterisation of zuclopenthixol metabolism by in vitro and therapeutic drug monitoring studies”. Acta Psychiatrica Scandinavica. 122 (6): 445–453. doi:10.1111/j.1600-0447.2010.01619.x. PMID 20946203. S2CID 41869401.
- ^ Parent M, Toussaint C, Gilson H (1983). “Long-term treatment of chronic psychotics with bromperidol decanoate: clinical and pharmacokinetic evaluation”. Current Therapeutic Research. 34 (1): 1–6.
- ^ Jump up to:a b Jørgensen A, Overø KF (1980). “Clopenthixol and flupenthixol depot preparations in outpatient schizophrenics. III. Serum levels”. Acta Psychiatrica Scandinavica. Supplementum. 279: 41–54. doi:10.1111/j.1600-0447.1980.tb07082.x. PMID 6931472.
- ^ Jump up to:a b Reynolds JE (1993). “Anxiolytic sedatives, hypnotics and neuroleptics.”. Martindale: The Extra Pharmacopoeia (30th ed.). London: Pharmaceutical Press. pp. 364–623.
- ^ Ereshefsky L, Saklad SR, Jann MW, Davis CM, Richards A, Seidel DR (May 1984). “Future of depot neuroleptic therapy: pharmacokinetic and pharmacodynamic approaches”. The Journal of Clinical Psychiatry. 45 (5 Pt 2): 50–9. PMID 6143748.
- ^ Jump up to:a b Curry SH, Whelpton R, de Schepper PJ, Vranckx S, Schiff AA (April 1979). “Kinetics of fluphenazine after fluphenazine dihydrochloride, enanthate and decanoate administration to man”. British Journal of Clinical Pharmacology. 7 (4): 325–31. doi:10.1111/j.1365-2125.1979.tb00941.x. PMC 1429660. PMID 444352.
- ^ Young D, Ereshefsky L, Saklad SR, Jann MW, Garcia N (1984). Explaining the pharmacokinetics of fluphenazine through computer simulations. (Abstract.). 19th Annual Midyear Clinical Meeting of the American Society of Hospital Pharmacists. Dallas, Texas.
- ^ Janssen PA, Niemegeers CJ, Schellekens KH, Lenaerts FM, Verbruggen FJ, van Nueten JM, et al. (November 1970). “The pharmacology of fluspirilene (R 6218), a potent, long-acting and injectable neuroleptic drug”. Arzneimittel-Forschung. 20 (11): 1689–98. PMID 4992598.
- ^ Beresford R, Ward A (January 1987). “Haloperidol decanoate. A preliminary review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in psychosis”. Drugs. 33 (1): 31–49. doi:10.2165/00003495-198733010-00002. PMID 3545764.
- ^ Reyntigens AJ, Heykants JJ, Woestenborghs RJ, Gelders YG, Aerts TJ (1982). “Pharmacokinetics of haloperidol decanoate. A 2-year follow-up”. International Pharmacopsychiatry. 17 (4): 238–46. doi:10.1159/000468580. PMID 7185768.
- ^ Larsson M, Axelsson R, Forsman A (1984). “On the pharmacokinetics of perphenazine: a clinical study of perphenazine enanthate and decanoate”. Current Therapeutic Research. 36 (6): 1071–88.
- ^ William Andrew Publishing (22 October 2013). Pharmaceutical Manufacturing Encyclopedia. Elsevier. pp. 1102–. ISBN 978-0-8155-1856-3.
External links
- Product information for Zuclopenthixol (CLOPIXOL), provided by the Therapeutic Goods Administration — https://www.ebs.tga.gov.au/ebs/picmi/picmirepository.nsf/pdf?OpenAgent&id=CP-2010-PI-05705-3
| Clinical data | |
|---|---|
| Trade names | Clopixol |
| AHFS/Drugs.com | International Drug Names |
| Pregnancy category | AU: C |
| Routes of administration | Oral, IM |
| Drug class | Typical antipsychotic |
| ATC code | N05AF05 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)UK: POM (Prescription only)In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | 49% (oral) |
| Protein binding | 98% |
| Metabolism | Hepatic (CYP2D6 and CYP3A4-mediated) |
| Elimination half-life | 20 hours (oral), 19 days (IM) |
| Excretion | Feces |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 53772-83-1 85721-05-7 (acetate) 64053-00-5 (decanoate) |
| PubChem CID | 5311507 |
| DrugBank | DB01624 |
| ChemSpider | 4470984 |
| UNII | 47ISU063SG |
| KEGG | D03556 |
| ChEBI | CHEBI:51364 |
| ChEMBL | ChEMBL53904 |
| CompTox Dashboard (EPA) | DTXSID3048233 |
| ECHA InfoCard | 100.053.398 |
| Chemical and physical data | |
| Formula | C22H25ClN2OS |
| Molar mass | 400.97 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
/////////zuclopenthixol, N05AF05, Clopenthixol, Cisordinol, Clopixol
OCCN1CCN(CC\C=C2\C3=C(SC4=C2C=C(Cl)C=C4)C=CC=C3)CC1

NEW DRUG APPROVALS
ONE TIME
$10.00
Aztreonam

Aztreonam
- Molecular FormulaC13H17N5O8S2
- Average mass435.433 Da
(2S,3S)-3-{[(2Z)-2-(2-Ammonio-1,3-thiazol-4-yl)-2-{[(2-carboxy-2-propanyl)oxy]imino}acetyl]amino}-2-methyl-4-oxo-1-azetidinesulfonate2-[[(Z)-[1-(2-amino-4-thiazolyl)-2-[[(2S,3S)-2-methyl-4-oxo-1-sulfo-3-azetidinyl]amino]-2-oxoethylidene]amino]oxy]-2-methyl-propanoic acid
278-839-9[EINECS]
5159
78110-38-0[RN]
UA2451400
азтреонам [Russian] [INN]
أزتريونام [Arabic] [INN]
氨曲南 [Chinese] [INN]
AztreonamCAS Registry Number: 78110-38-0
CAS Name: [2S-[2a,3b(Z)]]-2-[[[1-(2-Amino-4-thiazolyl)-2-[(2-methyl-4-oxo-1-sulfo-3-azetidinyl)amino]-2-oxoethylidene]amino]oxy]-2-methylpropanoic acid
Additional Names: azthreonam
Manufacturers’ Codes: SQ-26776
Trademarks: Azactam (BMS); Primbactam (Menarini)
Molecular Formula: C13H17N5O8S2, Molecular Weight: 435.43
Percent Composition: C 35.86%, H 3.94%, N 16.08%, O 29.40%, S 14.73%
Literature References: The first totally synthetic monocyclic b-lactam (monobactam) antibiotic. It has a high degree of resistance to b-lactamases and shows specific activity vs aerobic gram-negative rods.
Prepn: R. B. Sykes et al.,NL8100571 (1981 to Squibb), C.A.96, 181062x (1982).
Fast-atom-bombardment mass spectra: A. I. Cohen et al.,J. Pharm. Sci.71, 1065 (1982). Activity vs gram-negative bacteria: R. B. Sykes et al.,Antimicrob. Agents Chemother.21, 85 (1982). Series of articles on structure-activity, in vitro and in vivo properties, pharmacokinetics: J. Antimicrob. Chemother.8, Suppl. E, 1-148 (1981).
Toxicology: G. R. Keim et al.,ibid. 141. Mechanism of action study: A. D. Russell, J. R. Furr, ibid.9, 329 (1982). Comparative stability to renal dipeptidase: H. Mikami et al.,Antimicrob. Agents Chemother.22, 693 (1982). Human pharmacokinetics: E. A. Swabb et al.,ibid.21, 944 (1982).
Clinical evaluation in urinary tract infection: C. Donadio et al.,Drugs Exp. Clin. Res.13, 167 (1987). Clinical efficacy in neonatal sepsis: S. Sklavunu-Tsurutsoglu et al.,Rev. Infect. Dis.13, Suppl. 7, S591 (1991). Comprehensive description: K. Florey, Anal. Profiles Drug Subs.17, 1-39 (1988).
Properties: White crystalline, odorless powder, dec 227°. Very slightly sol in ethanol, slightly sol in methanol, sol in DMF, DMSO. Practically insol in toluene, chloroform, ethyl acetate.
Derivative Type: Disodium salt
Molecular Formula: C13H15N5Na2O8S2, Molecular Weight: 479.40
Percent Composition: C 32.57%, H 3.15%, N 14.61%, Na 9.59%, O 26.70%, S 13.38%
Properties: LD50 (mg/kg): 3300 i.v. in mice; 6600 i.p. in rats (Keim).
Toxicity data: LD50 (mg/kg): 3300 i.v. in mice; 6600 i.p. in rats (Keim)
Therap-Cat: Antibacterial.
Keywords: Antibacterial (Antibiotics); ?Lactams; Monobactams.
Aztreonam is a beta-lactam antibiotic used to treat select aztreonam sensitive gram negative bacteria.
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Aztreonam lysine | XNM7LT65NP | 827611-49-4 | KPPBAEVZLDHCOK-JHBYREIPSA-N |
A monocyclic beta-lactam antibiotic originally isolated from Chromobacterium violaceum. It is resistant to beta-lactamases and is used in gram-negative infections, especially of the meninges, bladder, and kidneys. It may cause a superinfection with gram-positive organisms.
Aztreonam, sold under the brand name Azactam among others, is an antibiotic used primarily to treat infections caused by gram-negative bacteria such as Pseudomonas aeruginosa.[1][2] This may include bone infections, endometritis, intra abdominal infections, pneumonia, urinary tract infections, and sepsis.[1] It is given by intravenous or intramuscular injection or by inhalation.[1]
Common side effects when given by injection include pain at the site of injection, vomiting, and rash.[1] Common side effects when inhaled include wheezing, cough, and vomiting.[1] Serious side effects include Clostridium difficile infection and allergic reactions including anaphylaxis.[1] Those who are allergic to other β-lactam have a low rate of allergy to aztreonam.[1] Use in pregnancy appears to be safe.[1] It is in the monobactam family of medications.[1] Aztreonam inhibits cell wall synthesis by blocking peptidoglycan crosslinking to cause bacterial death.[1]
Aztreonam was approved for medical use in the United States in 1986.[1] It was removed from the World Health Organization’s List of Essential Medicines in 2019.[3][4] It is available as a generic medication.[1] It is a manufactured version of a chemical from the bacterium Chromobacterium violaceum.[5]
Medical uses
Nebulized forms of aztreonam are used to treat infections that are complications of cystic fibrosis and are approved for such use in Europe and the US; they are also used off-label for non-CF bronchiectasis, ventilator-associated pneumonia, chronic obstructive pulmonary disease, mycobacterial disease, and to treat infections in people who have received lung transplants.[6]
Aztreonam has strong activity against susceptible Gram-negative bacteria, including Pseudomonas aeruginosa. It is resistant to some beta-lactamases, but is inactivated by extended-spectrum beta-lactamases.
It has no useful activity against Gram-positive bacteria or anaerobes. It is known to be effective against a wide range of bacteria including Citrobacter, Enterobacter, E. coli, Haemophilus, Klebsiella, Proteus, and Serratia species.[7] The following represents minimum inhibitory concentration (MIC) susceptibility data for a few medically significant microorganisms.
- Staphylococcus aureus 8 – >128 μg/ml
- Staphylococcus epidermidis 8 – 32 μg/ml
- Streptococcus pyogenes 8 – ≥128 μg/ml
Synergism between aztreonam and arbekacin or tobramycin against P. aeruginosa has been suggested.[9]
SYN
ACS Medicinal Chemistry Letters, 11(2), 162-165; 2020
https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00534

Aztreonam, first discovered in 1980, is an FDA approved, intravenous, monocyclic beta-lactam antibiotic. Aztreonam is active against Gram-negative bacteria and is still used today. The oral bioavailability of aztreonam in humans is less than 1%. Herein we describe the design and synthesis of potential oral prodrugs of aztreonam.
A stirring mixture of CES1 (20 mg, 2200 Units) in a 15 mM solution of sodium phosphate monobasic (enzyme grade) in acetonitrile-d3 / D2O (1 mL; ratio of 2.5 : 97.5) was heated at 37 °C for 5 min. 2-(((Z)-(1-(2-aminothiazol-4-yl)-2-(((2S,3S)-1-((2,2- dimethyl-4-(pivaloyloxy)butoxy)sulfonyl)-2-methyl-4-oxoazetidin-3-yl)amino)-2- oxoethylidene)amino)oxy)-2-methylpropanoic acid TFA salt 28c (10 mg, 14 µmol) was added, and the suspension was stirred for 70 min at 37 °C. Over the course of the reaction a fine precipitate formed. The mixture was filtered through a 25 mm, 0.45 µM glass fiber syringe filter (Pall Corporation Acrodisc). The filtrate was analyzed by 1H-NMR spectroscopy to reveal that 3,3-dimethyltetrahydrofuran was released as one of the products. The presence of 3,3-dimethyltetrahydrofuran was confirmed by 1H-NMR analysis of the same sample spiked with 2 µL of authentic materialSYN Faming Zhuanli Shenqing, 106520857,
SYN
Synthesis Reference
Neal G. Anderson, Carl F. Anderson, “Delta form of aztreonam and preparation thereof.” U.S. Patent US4826973, issued January, 1983.
SYN
https://patents.google.com/patent/US7145017B2/enAztreonam is a monobactam antibiotic disclosed in U.S. Pat. No. 4,775,670, which is incorporated by reference herein in its entirety. Aztreonam has the chemical name (Z)-2-[[[(2-amino-4-thiazolyl)[[(2S,-3S)-2-methyl-4-oxo-1-sulfo-3-azetidinyl]carbamoyl]methylene]amino]oxy]-2-methylpropionic acid. Aztreonam is also known as [3S-[3α(Z),4β]]-3-[[(2-amino-4-thiazolyl)[(1-carboxy-1-methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-azetidinesulfonic acid and (2S, 3S)-3-[[2-[2-amino-4-thiazolyl]-(Z)-2[(1-carboxy-1-methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-azetidine-1-sulfonic acid.Aztreonam has the structure:

Aztreonam is known to exist in various polymorphic forms including the α, β, δ, and γ forms.U.S. Pat. No. 4,775,670 discloses a process for making Aztreonam, a compound of formula I:

The process includes acylating a compound of formula IV:

The acylation entails reacting a compound of formula IV with a carboxylic acid or the corresponding carboxylic acid halide or carboxylic acid anhydride (R1—OH) in the presence of a carbodiimide such as dicyclohexylcarbodiimide and a substance capable of forming an active ester in situ such as N-hydroxybenzotriazole. U.S. Pat. No. 4,775,670 discloses that when the acyl group (R1) contains reactive functional groups, such as amino or carboxyl groups, it may be necessary to first protect those functional groups, then carry out the acylation reaction, and finally deprotect the resulting product. The deprotection is carried out by reaction of the acylation product with trifluoroacetic acid in the presence of anisole under anhydrous conditions.Similarly, U.S. Pat. No. 4,946,838 discloses a process for making crystalline anhydrous Aztreonam comprising reacting the diphenylmethyl ester of Aztreonam ([3S-[3β(Z),4α]]-3-[[(2-amino-4-thiazolyl)[(1-diphenylmethoxycarbonyl-1-methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-azetidinesulfonic acid) with trifluoroacetic acid in the presence of anisole under anhydrous conditions to produce the α-form of Aztreonam. The α-form is recrystallized from an anhydrous organic solvent to produce the β-form of Aztreonam. The β-form is anhydrous, substantially non-hygroscopic and more stable than the α-form.U.S. Pat. No. 5,254,681 discloses a process for preparing monobactams of formula (I):

wherein R is acyl. The process comprises acylating azetidin with 2-(2-amino-4-thiazolyl)-2-(Z)-(alkoxyimino) acetic acid in the presence of 1-hydroxy-benzotriazole and dicyclohexylcarbodiimide.U.S. Pat. No. 5,194,604 discloses a process and intermediates for making beta-lactams having aminothiazole(iminooxyacetic acid)acetic acid sidechains of formula (I), such as Aztreonam. The process comprises acylating a compound of formula III:

with a compound of formula (II):

in which R7 is

wherein

is a 4, 5, 6 or 7 membered heterocyclic ring having at least one nitrogen atom in the ring or such a group fused to a phenyl or substituted phenyl ring, to form a compound of formula (I):

wherein R1–R6 are as defined in U.S. Pat. No. 5,194,604.U.S. Pat. No. 4,652,651, which is incorporated by reference herein in its entirety, discloses a process for making 1-sulpho-2-oxoazetidine derivatives of the formula (I):

in which Het is an optionally amino-substituted, 5- or 6-membered, aromatic heterocycle containing 1 or 2 nitrogen atoms and optionally also an oxygen or sulphur atom, R1 may be lower alkoxycarbonyl-lower alkyl and R2 may be lower alkyl. The process entails acylating a compound of formula (II):

in which R20 equals R2 and R3 is hydrogen or sulpho, with a thioester of the formula (III):

in which Het is as above and R10 has any of the values of R1. U.S. Pat. No. 4,652,651 discloses that where R10 is a lower alkoxycarbonyl-lower alkyl group, for example the t-butoxycarbonylmethyl group, this can be converted, if desired, into the corresponding carboxylower alkyl group by treatment with a strong acid such as trifluoroacetic acid (optionally in the presence of anisole), hydrochloric acid or p-toluenesulphonic acid at a low temperature such as −10° C. to room temperature.There remains a need in the art for a process of making Aztreonam which does not require anhydrous reaction conditions and which also enables high yield and high purity. The present invention answers this need.
SUMMARY OF THE INVENTIONThe invention is based on the discovery that Aztreonam can be produced by reacting [3S-[3α(Z),4β]]-3-[[(2-amino-4-thiazolyl)[(1-t-butoxycarbonyl-1-methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-azetidinesulfonic acid with aqueous acid. The process of the invention, enables yields of between 70–75% and purities above 98%, preferably above 99%. The inventive aqueous process is advantageous over the prior art anhydrous processes in that the reaction conditions are more mild, there is no need to clean the final product and there is no need to keep the system dry. Thus, the aqueous process is less expensive than the anhydrous processes.The present invention is directed to a process for preparing [3S-[3α(Z),4β]]-3-[[(2-amino-4-thiazolyl)[(1-carboxy-1-methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-azetidinesulfonic acid by hydrolyzing the ester group of [3S-[3α(Z),4β]]-3-[[(2-amino-4-thiazolyl)[(1-t-butoxycarbonyl-1-methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-azetidinesulfonic acid. The hydrolysis may be effected by reacting the ester with aqueous acid, at elevated temperatures.One reaction scheme for carrying out the process is shown below:

EXAMPLE 15.4 g Azetidin is dissolved in 20 ml acetonitrile (or dimethyl formamide) with the assistance of 5 ml of triethylamine at room temperature. The solution is cooled to 0° C. A solution of 4 g TAEM in 25 ml THF is added with magnetic stirring. If the color disappears, 8 g TAEM in 50 ml THF is added. After 10 minutes, another 4.1 g TAEM in 25 ml THF is added. The solution is stirred at 0° C. for an additional hour. The pH is adjusted to about 4–5 with a freshly prepared TFA solution (TFA-THF 1:4, V/V). Being careful not to evaporate the acetonitrile, the THF is evaporated (weight loss is about 90 g) at 30° C. under vacuum. The remaining residue is diluted with 200 ml ethylacetate and then extracted with 100 ml and then 50 ml of distilled water. The aqueous extracts are combined and washed twice with 50 ml ethylacetate after readjustment of the pH to about 4–5. The dissolved ethylacetate is removed from the aqueous phase by vacuum at 30° C. 10–15 g KCl (or NaCl) is dissolved. The solution is acidified with HCl solution (cc. HCl-distilled water 1:4, V/V) with stirring (approx. 10 ml). The solution is cooled to 0° C. with slow stirring and crystallization occurs. The resulting suspension is refrigerated overnight (at about 5° C.). The suspension is filtered on a glass filter, and the crystals are washed with chilled water. The washed crystals are dried at room temperature. The product, Aztreonam t-butyl ester, is about 12.5–13 g white solid, which is sufficiently pure for the next step.
EXAMPLE 265 g Azetidine is dissolved in a mixture of 240 ml acetonitrile and 60 ml triethylamine. When dissolution is complete, TAEM is added in four portions. The suspension is stirred for 20–30 min, then diluted with 500 ml EtOAc and 500 ml water and stirred for 5–10 min. The pH of the emulsion is set to 5 with 2.4 M HCl solution. After the phases separate, the pH of the aqueous phase is checked. If the pH is between 4.20 and 5.30, the two phases are filtered and separated, otherwise more HCl is added. The upper phase is diluted with 900 ml ethylacetate and extracted with 2×500 ml water (faster phase separation). The combined aqueous phase is diluted with 500 ml water and washed with 2×500 ml ethylacetate. The dissolved ethylacetate is removed from the aqueous phase by vacuum. The aqueous phase is acidified further to pH 2 with 2.4 M HCl solution. The solution is stirred and cooled. Crystallization starts soon. The suspension is stirred and cooled to 0° C., stirring at this temperature overnight. The suspension is filtered, washed with chilled water, dried at 38° C. in air-circulated oven for 3 h. The yield is approx. 116–120 g of Aztreonam t-butyl ester.
EXAMPLE 3Aztreonam t-butyl ester (113.6 g, 0.231 mol) is suspended in 975 ml water at 60° C. with stirring and 325 ml trifluoroacetic acid is added. The solution is stirred for 60 min., then it is cooled slowly using an ice-water bath. After the product precipitates, the suspension is refrigerated overnight. The product is filtered on a glass-filter, suspended in 240 ml chilled water and filtered again. The filtrate is re-suspended in 360 ml cold acetone and filtered. The latter step is repeated and the product is dried at room temperature to yield 61.6 g Aztreonam (water content: 15–16%).
EXAMPLE 4Aztreonam t-butyl ester (18.0 g, 0.0366 mol) is suspended in 144 ml water at 60° C. with stirring and 40 ml aqueous hydrochloric acid (1:1, V/V) is added. The solution is stirred for 60 min, then 37 ml 5.4 M NaOH solution is added. The solution is cooled slowly using an ice-water bath. After the product precipitates, the suspension is refrigerated overnight. The product is filtered on a glass-filter, suspended in 50 ml chilled water and filtered again. The filtrate is re-suspended in 70 ml cold acetone and filtered. The latter step is repeated and the product is dried at room temperature to yield 8.3 g Aztreonam (water content: 15–16%). The crude Aztreonam is crystallized.
EXAMPLE 5Aztreonam t-butyl ester (100.00 g, Assay as is: 97.2%, 0.19796 mol)) is suspended in a mixture of 450 ml water and 5 ml trifluoroacetic acid. The suspension, which slowly becomes clear, is heated to 58° C. with stirring and 100 ml trifluoroacetic acid is added. The solution is stirred for 105 min at 60–63° C. The solution is added to chilled water (450 ml) with efficient stirring and the resulting slurry is cooled further to 25° C. After two hours it is cooled to 0° C. and stirred for 18 hours. The product is filtered on a glass-filter and washed with 300 ml chilled water. The product is suspended in 650 ml chilled water, then filtered and washed with 300 ml cold acetone. The product is suspended in 400 ml cold acetone and filtered and dried in an air-ventilation oven at 30° C. for 30 min. Yield: 66.6 g (63%, according to assays) Aztreonam (Assay: 100.5%, water content: 18.0%).HPLC Impurity Profile:
- Aztreonam: 99.22%
- Aztreonam t-butyl ester: 0.44%
HPLC Impurity Profile of Sample from Reaction Mixture: - Aztreonam: 82.20%
- Aztreonam t-butyl ester: 0.43%
- Aztreonam, open-chained: 7.22%
- Other main degradation product (RRT=0.56): 5.24%
EXAMPLE 6Aztreonam t-butyl ester (27.11 g, Assay as is: 96.5%, 0.05328 mol) is suspended in a mixture of 122 ml water and 1.35 ml cc. HCl. The suspension is heated to 62° C. with stirring and 30 ml cc. HCl is added. The suspension, which becomes clear after approx. 15 min, (then the product starts to precipitate), is stirred for 30 min at 63–65° C. Chilled water (162 ml) is added with efficient stirring and the resulting slurry is cooled further to 25° C. After two hours it is cooled to 0° C. and stirred for 2 hours. The product is filtered on a glass-filter, washed twice with 120 ml chilled water, twice with 125 ml cold acetone and filtered. The product is dried at room temperature overnight. Yield: 19.44 g (72%, according to assays) Aztreonam (Assay: 100.1%, water content: 14.4%).HPLC Impurity Profile:
- Aztreonam: 99.65%
- Aztreonam t-butyl ester: 0.21%
HPLC Impurity Profile of Sample from Reaction Mixture: - Aztreonam: 89.43%
- Aztreonam t-butyl ester: 0.26%
- Aztreonam, open-chained: 4.70%
- Other main degradation product (RRT=0.56): 1.47%
SYN
Manufacturing Process
This mixture was sterilized for 15 minutes at 121°C at 15 lbs/inch2 steam pressure prior to use. The fermentation flasks were incubated at 25°C for 40 to 45 hours on a of rotary shaker. A 250 liter batch of Agrobacterium radiobacter A.T.C.C. No. 31700 is fermented in a 100 gallon steel vessel with a media and operating conditions described below. Culture of Agrobacterium radiobacter grown out on agar slants, pH 7.3 consisted of yeast extract (1 g), beef extract (1 g), NZ amine A (2 g), glucose (10 g), agar (15 g) in 1000 ml distilled water. Loopful of surface growth from agar slant was used as the source of incolumn. Medium of oatmeal (20 g), tomato paste (20 g) tapped water to 1000 ml, pH 7, was sterilized for 15 min at 121°C at 15 lbs/inch2 steam pressure prior to use. 100 ml of the medium, containing incolumn is incubated at 25°C for about 24 hours on a rotary shaker. It was added to a mixture of yeast extract (5 g), glucose (10 g) in 1 L distilled water and incubated for about 42 hours at 25°C in 100 gallon stainless steel fermentation vessel.
During incubation, the broth is agitated at 155 r.p.m. and aerated at rate of 10.0 cubic feet per minute. An antifoam agent (Ucon LB625, Union Carbide) was added as needed. The fermentation beer was adjusted to pH 4 with aqueous HCl and calls separated by centrifugation. The supernatante (200 L) was extracted with 40 L of 0.05 m cetyldimethylbenzyl ammonium chloride in dichloromethane and extract concentrated in vacuo to 5.5 L. The concentrate was then extracted with solution of 177 g of sodium thiocyanate in 2 L of water, adjusting the mixture of pH 4.35 with phosphoric acid. The aqueous extract was concentrated in vacuo to 465 ml and added to 1840 ml of methanol. Solids are filtrated yielded 194 g of crude solid product. It was dissolved and chromatographed on a 5×106.5 cm column of Sephadex G-10 three times and after concentrating in vacuo gave 3.5 g of crude antibiotic M53 (azetreonam) which was chromatographed at first on QAE Sephadex A- 25 (liner gradient, prepared from 2.5 L of water and 2.5 L of 0.25 M sodium nitrate). Then the residue (fractions 26-75) gave M53 (natrium salt) after evaporation. It was triturated with methanol and the souble fraction, 0.40 g was chromatographed on a 2.5×20 cm column of Diaion HP20AG, eluting at 2 ml per minute with water and collecting 20 ml fractions. Fractions 26-75 gave 51.9 mg of antibiotic M53 (sodium salt).
Chemical Synthesis
Aztreonam, (Z)-2[[[(2-amino-4-thiazolyl)[[(2S,3S)-2-methyl-4-oxo-1-sulfo-3-azetidinyl]cabamoyl]methylen]amino]oxy]-2-methylpropionoic acid (32.1.4.9), is synthesized from tert-butyloxycarbonylthreonine, which is reacted with O-benzylhydroxylamine in the presence of dicyclohexylcarbodimide and 1-hydroxybenzotriazole, to form the benzyl hydroxamide derivative (32.1.4.1). This product undergoes a reaction with triphenylphosphine and ethyl azodicarboxylate, which results in the cyclodehydration of the product to (3S-trans)-N-benzyloxy-3-tert-butyloxycarbonylamino-4-methyl-azetidinone (32.1.4.2). Debenzylating this by hydrogen reduction using a palladium on carbon catalyst forms (3S-trans)-N-hydroxy-3-tertbutyloxycarbonyl-amino-4-methyl-azetidinone (32.1.4.3). The hydroxyl group in this compound is removed by reducing it with titanium trichloride, which forms azetidinone (32.1.4.4). Removing the tert-butyloxycarbonyl protection using trifluoroacetic acid and subsequent acylation of the resulting product with the benzyl chloroformate gives (3S-trans)-benzyloxycarbonylamino-4-methylazetidinone (32.1.4.5). Sulfonating this product with a mixture of sulfur trioxide and dimethylformamide gives the corresponding N-sulfonic acid. Turning the resulting Nsulfonic acid into a potassium salt by reacting it with potassium hydrophosphate, followed by replacing the potassium cation with a tetrabutylammonium cation by reacting it with tetrabutylammonium sulfate gives the product (32.1.4.6). Reducing this with hydrogen using a palladium on carbon catalyst gives 3-amino-4-methyl-monobactamic acid (32.1.4.7). Acylating this with (Z) 2-amino-α-[[2-(diphenylmethoxy)-1,1-dimethyl-2-oxoethoxy]imino] 4-thiazoleacetic acid in the presence of dicyclohexylcarbodiimide and 1-hydroxy-benzotriazole gives the diphenylmethyl ester of the desired aztreonam (32.1.4.8), which is hydrolyzed to aztreonam (32.1.4.9) using trifluoroacetic acid.
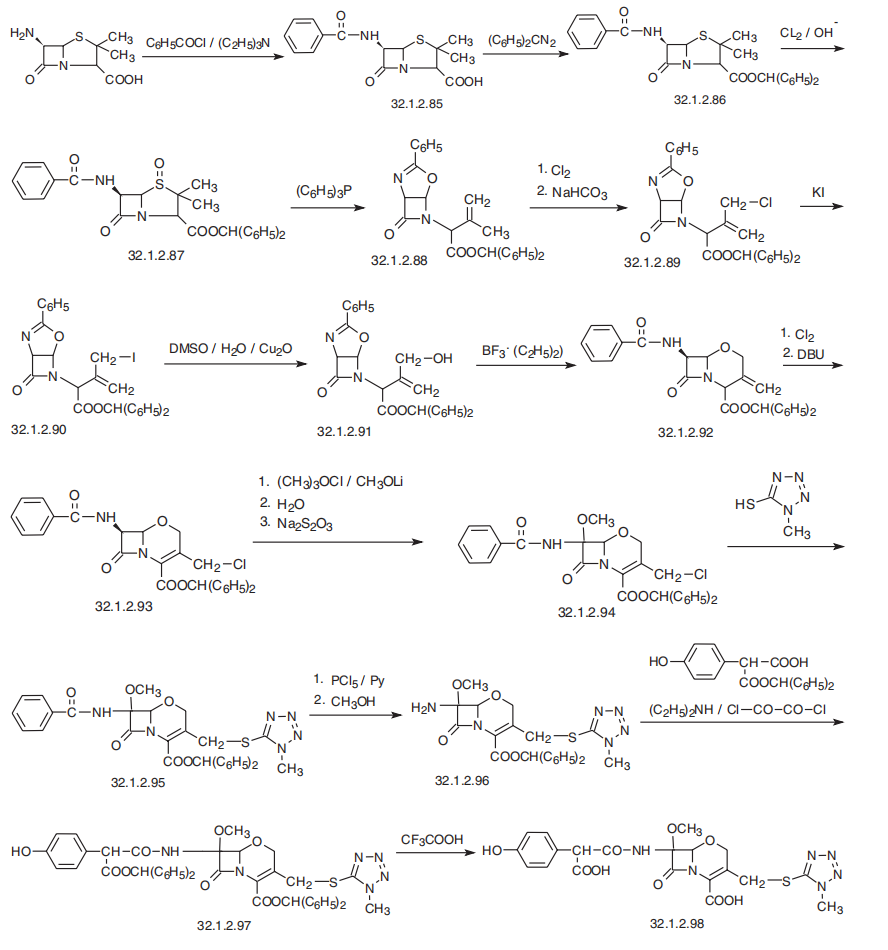
It is believed that the methyl group at position 4 increases the stability of the beta-lactam ring with respect to most beta-lactamases, and at the same time it does not induce formation of beta-lactamase as cephalosporins and imipenems do.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Spectrum of activity
Acinetobacter anitratus, Escherichia coli, Pseudomonas aeruginosa, and Proteus mirabilis are generally susceptible to aztreonam, while some staphylococci, Staphylococcus aureus, Staphylococcus haemolyticus and Xanthomonas maltophilia are resistant to it. Furthermore, Aeromonas hydrophila, Citrobacter koseri (Citrobacter diversus), Pantoea agglomerans (Enterobacter agglomerans), Haemophilus spp. and Streptococcus pyogenes have developed resistance to aztreonam to varying degrees.[10]
Aztreonam is often used in people who are penicillin allergic or who cannot tolerate aminoglycosides.[medical citation needed]
Administration[edit]
Aztreonam is poorly absorbed when given orally, so it must be administered as an intravenous or intramuscular injection (trade name Azactam ), or inhaled (trade name Cayston) using an ultrasonic nebulizer. In the United States, the Food and Drug Administration (FDA) approved the inhalation form on 22 February 2010, for the suppression of P. aeruginosa infections in patients with cystic fibrosis.[11] It received conditional approval for administration in Canada and the European Union in September 2009,[11] and has been fully approved in Australia.[12]
Side effects
Reported side effects include injection site reactions, rash, and rarely toxic epidermal necrolysis. Gastrointestinal side effects generally include diarrhea and nausea and vomiting. There may be drug-induced eosinophilia. Because of the unfused beta-lactam ring there is somewhat lower cross-reactivity between aztreonam and many other beta-lactam antibiotics, and it may be safe to administer aztreonam to many patients with hypersensitivity (allergies) to penicillins and nearly all cephalosporins.[13] There is a much lower risk of cross-sensitivity between aztreonam and other beta-lactam antibiotics than within other beta-lactam antibiotics. However, there is a higher chance of cross-sensitivity if a person is specifically allergic to ceftazidime, a cephalosporin. Aztreonam exhibits cross-sensitivity with ceftazidime due to a similar side chain.[14]
Mechanism of action
Aztreonam is similar in action to penicillin. It inhibits synthesis of the bacterial cell wall, by blocking peptidoglycan crosslinking. It has a very high affinity for penicillin-binding protein-3 and mild affinity for penicillin-binding protein-1a. Aztreonam binds the penicillin-binding proteins of Gram-positive and anaerobic bacteria very poorly and is largely ineffective against them.[13] Aztreonam is bactericidal, but less so than some of the cephalosporins.[medical citation needed]
References
- ^ Jump up to:a b c d e f g h i j k l “Aztreonam”. The American Society of Health-System Pharmacists. Retrieved 8 December 2017.
- ^ British national formulary : BNF 69 (69 ed.). British Medical Association. 2015. p. 381. ISBN 9780857111562.
- ^ World Health Organization (2019). Executive summary: the selection and use of essential medicines 2019: report of the 22nd WHO Expert Committee on the selection and use of essential medicines. Geneva: World Health Organization. hdl:10665/325773. WHO/MVP/EMP/IAU/2019.05. License: CC BY-NC-SA 3.0 IGO.
- ^ World Health Organization (2019). The selection and use of essential medicines: report of the WHO Expert Committee on Selection and Use of Essential Medicines, 2019 (including the 21st WHO Model List of Essential Medicines and the 7th WHO Model List of Essential Medicines for Children). Geneva: World Health Organization. hdl:10665/330668. ISBN 9789241210300. ISSN 0512-3054. WHO technical report series;1021.
- ^ Yaffe SJ, Aranda JV (2010). Neonatal and Pediatric Pharmacology: Therapeutic Principles in Practice. Lippincott Williams & Wilkins. p. 438. ISBN 9780781795388.
- ^ Quon BS, Goss CH, Ramsey BW (March 2014). “Inhaled antibiotics for lower airway infections”. Annals of the American Thoracic Society. 11 (3): 425–34. doi:10.1513/annalsats.201311-395fr. PMC 4028738. PMID 24673698.
- ^ Mosby’s Drug Consult 2006 (16th ed.). Mosby, Inc. 2006.
- ^ “Aztreonam Susceptibility and Minimum Inhibitory Concentration (MIC) Data” (PDF). toku-e.com. 3 February 2020.
- ^ Kobayashi Y, Uchida H, Kawakami Y (December 1992). “Synergy with aztreonam and arbekacin or tobramycin against Pseudomonas aeruginosa isolated from blood”. The Journal of Antimicrobial Chemotherapy. 30 (6): 871–2. doi:10.1093/jac/30.6.871. PMID 1289363.
- ^ “Aztreonam spectrum of bacterial susceptibility and Resistance” (PDF). Retrieved 15 May 2012.
- ^ Jump up to:a b Larkin C (22 February 2010). “Gilead’s Inhaled Antibiotic for Lungs Wins Approval”. BusinessWeek. Archived from the original on 2 March 2010. Retrieved 5 March 2010.
- ^ “FDA approves Gilead cystic fibrosis drug Cayston”. BusinessWeek. 23 February 2010. Retrieved 5 March 2010.
- ^ Jump up to:a b AHFS Drug Information 2006 (2006 ed.). American Society of Health-System Pharmacists. 2006.
- ^ Terico, AT; Gallagher, JC (December 2014). “Beta-lactam hypersensitivity and cross-reactivity”. Journal of Pharmacy Practice. 27 (6): 530–44. doi:10.1177/0897190014546109. PMID 25124380. S2CID 19275020.
External links
- “Aztreonam”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Azactam, Cayston, others |
| AHFS/Drugs.com | Monograph |
| License data | EU EMA: by INN |
| Pregnancy category | AU: B1 |
| Routes of administration | Intravenous, intramuscular, inhalation |
| ATC code | J01DF01 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only)US: ℞-onlyEU: Rx-only |
| Pharmacokinetic data | |
| Bioavailability | 100% (IM) 0.1% (by mouth in rats) Unknown (by mouth in humans) |
| Protein binding | 56% |
| Metabolism | Liver (minor %) |
| Elimination half-life | 1.7 hours |
| Excretion | Kidney |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 78110-38-0 |
| PubChem CID | 5742832 |
| DrugBank | DB00355 |
| ChemSpider | 4674940 |
| UNII | G2B4VE5GH8 |
| KEGG | D00240 |
| ChEBI | CHEBI:161680 |
| ChEMBL | ChEMBL158 |
| CompTox Dashboard (EPA) | DTXSID0022640 |
| ECHA InfoCard | 100.071.652 |
| Chemical and physical data | |
| Formula | C13H17N5O8S2 |
| Molar mass | 435.43 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 227 °C (441 °F) (dec.) |
| showSMILES | |
| showInChI | |
| (verify) |
Patent
Publication numberPriority datePublication dateAssigneeTitleEP0070024A11981-07-131983-01-19E.R. Squibb & Sons, Inc.The crystalline anhydrous form of (3S-(3 alpha(z),4 beta))-3-(((2-amino-4-thiazolyl)(1-carboxy-1-methylethoxy)-imino)-acetyl)-amino)-4-methyl-2-oxo-1-azetidinesulfonic acid, method for its preparation, mixture and pharmaceutical composition containing itUS4529698A1981-01-191985-07-16E. R. Squibb & Sons, Inc.Process for preparing a 2-oxo-1-azetidinesulfonic acid saltUS4652651A1983-05-311987-03-24Hoffmann-La Roche Inc.Process for the manufacture of 1-sulpho-2-oxoazetidine carboxylic acid intermediates via catalytic ester cleavageUS4775670A1980-09-291988-10-04E. R. Squibb & Sons, Inc.2-oxo-1-azetidinesulfonic acid saltsEP0297580A11987-07-011989-01-04E.R. Squibb & Sons, Inc.Amorphous form of aztreonamUS4826973A1984-07-201989-05-02E. R. Squibb & Sons, Inc.Delta form of aztreonam and preparation thereofUS4923998A1977-03-141990-05-08Fujisawa Pharmaceutical Company, Ltd.Cephem and cepham compounds and processes for preparation thereofUS4946838A1981-07-131990-08-07E. R. Squibb & Sons, Inc.Crystalline anhydrous aztreonamUS5194604A1990-06-291993-03-16E. R. Squibb & Sons, Inc.Process and intermediates for beta-lactams having aminothiazole(iminooxyacetic acid)acetic acid sidechainsUS5254681A1989-08-021993-10-19Consiglio Nazionale Delle RicercheProcess for preparing monobactames and their intermediate productPL165700B11991-10-151995-01-31PanMethod of obtaining z2/2-aminothiazolyl-4/-2/-t-butoxycarbonyl-1-methylethoxyimine/ acetic acidWO2002051356A22000-12-272002-07-04Salus Pharma, Inc.Inhalable aztreonam for treatment and prevention of pulmonary bacterial infectionsWO2003018578A12001-08-272003-03-06Aurobindo Pharma Ltd.Method for producing beta form of crystalline anhydrous aztreonamUS20040062721A12000-12-272004-04-01Montgomery Alan BruceInhalable aztreonam lysinate formulation for treatment and prevention of pulmonary bacterial infectionsWO2004052333A12002-12-112004-06-24Pari GmbhPharmaceutical compositions for the pulmonary delivery of aztreonamUS20050014739A12003-05-152005-01-20Viktor GyollaiAztreonam beta polymorph with very low residual solvent contentUS20050032775A12003-07-022005-02-10Viktor GyollaiAztreonam L-lysine and methods for the preparation thereof
Publication numberPriority datePublication dateAssigneeTitle
Family To Family CitationsCN1802371A2003-05-152006-07-12特瓦药厂有限公司Aztreonam beta-polymorph with very low residual solvent contentAU2004256124B2 *2003-07-022011-04-28Corus Pharma, Inc.Aztreonam L-lysine and methods for the preparation thereofWO2006122253A1 *2005-05-092006-11-16Sicor, Inc.Process for making aztreonamWO2007083187A2 *2006-01-162007-07-26Orchid Chemicals & Pharmaceuticals LimitedAn improved process for the preparation of monobactam antibioticCN101412715B *2008-12-162010-04-14海南百那医药发展有限公司Aztreonam compound and preparation thereofCN102127068B *2010-12-312012-08-29山西普德药业股份有限公司Method for synthesizing aztreonam compoundCN102311431B *2011-08-302014-12-10海南海药股份有限公司Method for preparing anhydrous beta-aztreonamCN105017241B *2015-06-242018-03-06山东罗欣药业集团股份有限公司A kind of aztreonam compound and its preparation
////////////////Aztreonam, SQ 26776, antibacterial, lactam, monobactam, UA2451400, азтреонам , أزتريونام , 氨曲南 ,
C[C@H]1[C@H](NC(=O)C(=N/OC(C)(C)C(=O)O)\C2=CSC([NH3+])=N2)C(=O)N1S([O-])(=O)=O

NEW DRUG APPROVALS
ONE TIME
$10.00
Tezepelumab-ekko

(Heavy chain)
QMQLVESGGG VVQPGRSLRL SCAASGFTFR TYGMHWVRQA PGKGLEWVAV IWYDGSNKHY
ADSVKGRFTI TRDNSKNTLN LQMNSLRAED TAVYYCARAP QWELVHEAFD IWGQGTMVTV
SSASTKGPSV FPLAPCSRST SESTAALGCL VKDYFPEPVT VSWNSGALTS GVHTFPAVLQ
SSGLYSLSSV VTVPSSNFGT QTYTCNVDHK PSNTKVDKTV ERKCCVECPP CPAPPVAGPS
VFLFPPKPKD TLMISRTPEV TCVVVDVSHE DPEVQFNWYV DGVEVHNAKT KPREEQFNST
FRVVSVLTVV HQDWLNGKEY KCKVSNKGLP APIEKTISKT KGQPREPQVY TLPPSREEMT
KNQVSLTCLV KGFYPSDIAV EWESNGQPEN NYKTTPPMLD SDGSFFLYSK LTVDKSRWQQ
GNVFSCSVMH EALHNHYTQK SLSLSPGK
(Light chain)
SYVLTQPPSV SVAPGQTARI TCGGNNLGSK SVHWYQQKPG QAPVLVVYDD SDRPSWIPER
FSGSNSGNTA TLTISRGEAG DEADYYCQVW DSSSDHVVFG GGTKLTVLGQ PKAAPSVTLF
PPSSEELQAN KATLVCLISD FYPGAVTVAW KADSSPVKAG VETTTPSKQS NNKYAASSYL
SLTPEQWKSH RSYSCQVTHE GSTVEKTVAP TECS
(Disulfide bridge: H22-H96, H136-L213, H149-H205, H224-H’224, H225-H’225, H228-H’228, H231-H’231, H262-H322, H368-H426, H’22-H’96, H’136-L’213, H’149-H’205, H’262-H’322, H’368-H’426, L22-L87, L136-L195, L’22-L’87, L’136-L’195)
Tezepelumab-ekko
テゼペルマブ (遺伝子組換え)
| Formula | C6400H9844N1732O1992S52 |
|---|---|
| CAS | 1572943-04-4 |
| Mol weight | 144588.4306 |
PEPTIDE
UD FDA APPROVED, 12/17/2021, To treat severe asthma as an add-on maintenance therapy , Tezspire
Monoclonal antibody
Treatment of asthma and atopic dermatitis
Tezepelumab, sold under the brand name Tezspire, is a human monoclonal antibody used for the treatment of asthma.[4][5]
It blocks thymic stromal lymphopoietin (TSLP),[2] an epithelial cytokine that has been suggested to be critical in the initiation and persistence of airway inflammation.[6]
It was approved for medical use in the United States in December 2021.[2][3]
Medical uses
Tezepelumab is indicated for the add-on maintenance treatment of people aged twelve years and older with severe asthma.[2]
Research
In Phase III trials, tezepelumab demonstrated efficacy compared to placebo for patients with severe, uncontrolled asthma.[7][8]
Structural studies by X-ray crystallography showed that Tezepelumab competes against a critical part of the TSLPR binding site on TSLP.[1]
It is being studied for the treatment of chronic obstructive pulmonary disease, chronic rhinosinusitis with nasal polyps, chronic spontaneous urticaria and eosinophilic esophagitis (EoE).[3]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Jump up to:a b Verstraete K, Peelman F, Braun H, Lopez J, Van Rompaey D, Dansercoer A, et al. (April 2017). “Structure and antagonism of the receptor complex mediated by human TSLP in allergy and asthma”. Nature Communications. 8 (1): 14937. Bibcode:2017NatCo…814937V. doi:10.1038/ncomms14937. PMC 5382266. PMID 28368013.
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761224s000lbl.pdf
- ^ Jump up to:a b c “Tezspire (tezepelumab) approved in the US for severe asthma”. AstraZeneca (Press release). 17 December 2021. Retrieved 17 December 2021.
- ^ Marone G, Spadaro G, Braile M, Poto R, Criscuolo G, Pahima H, et al. (November 2019). “Tezepelumab: a novel biological therapy for the treatment of severe uncontrolled asthma”. Expert Opinion on Investigational Drugs. 28 (11): 931–940. doi:10.1080/13543784.2019.1672657. PMID 31549891. S2CID 202746054.
- ^ Matera MG, Rogliani P, Calzetta L, Cazzola M (February 2020). “TSLP Inhibitors for Asthma: Current Status and Future Prospects”. Drugs. 80 (5): 449–458. doi:10.1007/s40265-020-01273-4. PMID 32078149. S2CID 211194472.
- ^ “Tezepelumab granted Breakthrough Therapy Designation by US FDA”. AstraZeneca (Press release). 7 September 2018.
- ^ “Studies found for: Tezepelumab”. ClinicalTrials.Gov. National Library of Medicine, National Institutes of Health, U.S. Department of Health and Human Services.
- ^ Menzies-Gow A, Corren J, Bourdin A, Chupp G, Israel E, Wechsler ME, et al. (May 2021). “Tezepelumab in Adults and Adolescents with Severe, Uncontrolled Asthma”. New England Journal of Medicine. 384 (19): 1800–09. doi:10.1056/NEJMoa2034975. PMID 33979488. S2CID 234484931.
External links
- “Tezepelumab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02054130 for “Study to Evaluate the Efficacy and Safety of MEDI9929 (AMG 157) in Adult Subjects With Inadequately Controlled, Severe Asthma” at ClinicalTrials.gov
- Clinical trial number NCT03347279 for “Study to Evaluate Tezepelumab in Adults & Adolescents With Severe Uncontrolled Asthma (NAVIGATOR)” at ClinicalTrials.gov
| Structural basis for inhibition of TSLP-signaling by Tezepelumab (PDB 5J13)[1] | |
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | thymic stromal lymphopoietin (TSLP) |
| Clinical data | |
| Trade names | Tezspire |
| Other names | MEDI9929, AMG 157, tezepelumab-ekko |
| License data | US DailyMed: Tezepelumab |
| Routes of administration | Subcutaneous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [2][3] |
| Identifiers | |
| CAS Number | 1572943-04-4 |
| DrugBank | DB15090 |
| ChemSpider | None |
| UNII | RJ1IW3B4QX |
| KEGG | D11771 |
| Chemical and physical data | |
| Formula | C6400H9844N1732O1992S52 |
| Molar mass | 144590.40 g·mol−1 |
////////////Tezepelumab-ekko, Tezspire, PEPTIDE, APPROVALS 2021, FDA 2021, Monoclonal antibody
, asthma, atopic dermatitis, ANTI INFLAMATORY, テゼペルマブ (遺伝子組換え)

NEW DRUG APPROVALS
ONE TIME
$10.00
Efgartigimod alfa-fcab
DKTHTCPPCP APELLGGPSV FLFPPKPKDT LYITREPEVT CVVVDVSHED PEVKFNWYVD
GVEVHNAKTK PREEQYNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK
GQPREPQVYT LPPSRDELTK NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS
DGSFFLYSKL TVDKSRWQQG NVFSCSVMHE ALKFHYTQKS LSLSPGK
(Disulfide bridge: 6-6′, 9-9′, 41-101, 147-205, 41′-101′, 147′-205′)
Efgartigimod alfa-fcab
| Formula | C2310H3554N602O692S14 |
|---|---|
| CAS | 1821402-21-4 |
| Mol weight | 51279.464 |
US FDA APPROVED 12/17/2021, To treat generalized myasthenia gravis
Press Release, Vyvgart, BLA 761195
| エフガルチギモドアルファ (遺伝子組換え) |
PEPTIDE
Treatment of IgG-driven autoimmune diseases
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-myasthenia-gravis
FDA Approves New Treatment for Myasthenia Gravis
Approval is the First of a New Class of Medication for this Rare, Chronic, Autoimmune, Neuromuscular DiseaseFor Immediate Release:December 17, 2021
The U.S. Food and Drug Administration today approved Vyvgart (efgartigimod) for the treatment of generalized myasthenia gravis (gMG) in adults who test positive for the anti-acetylcholine receptor (AChR) antibody.
Myasthenia gravis is a chronic autoimmune, neuromuscular disease that causes weakness in the skeletal muscles (also called voluntary muscles) that worsens after periods of activity and improves after periods of rest. Myasthenia gravis affects voluntary muscles, especially those that are responsible for controlling the eyes, face, mouth, throat, and limbs. In myasthenia gravis, the immune system produces AChR antibodies that interfere with communication between nerves and muscles, resulting in weakness. Severe attacks of weakness can cause breathing and swallowing problems that can be life-threatening.
“There are significant unmet medical needs for people living with myasthenia gravis, as with many other rare diseases,” said Billy Dunn, M.D., director of the Office of Neuroscience in the FDA’s Center for Drug Evaluation and Research. “Today’s approval is an important step in providing a novel therapy option for patients and underscores the agency’s commitment to help make new treatment options available for people living with rare diseases.”
Vyvgart is the first approval of a new class of medication. It is an antibody fragment that binds to the neonatal Fc receptor (FcRn), preventing FcRn from recycling immunoglobulin G (IgG) back into the blood. The medication causes a reduction in overall levels of IgG, including the abnormal AChR antibodies that are present in myasthenia gravis.
The safety and efficacy of Vyvgart were evaluated in a 26-week clinical study of 167 patients with myasthenia gravis who were randomized to receive either Vyvgart or placebo. The study showed that more patients with myasthenia gravis with antibodies responded to treatment during the first cycle of Vyvgart (68%) compared to those who received placebo (30%) on a measure that assesses the impact of myasthenia gravis on daily function. More patients receiving Vyvgart also demonstrated response on a measure of muscle weakness compared to placebo.
The most common side effects associated with the use of Vyvgart include respiratory tract infections, headache, and urinary tract infections. As Vyvgart causes a reduction in IgG levels, the risk of infections may increase. Hypersensitivity reactions such as eyelid swelling, shortness of breath, and rash have occurred. If a hypersensitivity reaction occurs, discontinue the infusion and institute appropriate therapy. Patients using Vyvgart should monitor for signs and symptoms of infections during treatment. Health care professionals should administer appropriate treatment and consider delaying administration of Vyvgart to patients with an active infection until the infection is resolved.
The FDA granted this application Fast Track and Orphan Drug designations. The FDA granted the approval of Vyvgart to argenx BV.
///////////efgartigimod alfa-fcab, Vyvgart, FDA 2021,APPROVALS 2021, myasthenia gravis, argenx BV, Fast Track, Orphan Drug, PEPTIDE,
| エフガルチギモドアルファ (遺伝子組換え) |

NEW DRUG APPROVALS
one time
$10.00
CLOBETASOL
Clobetasol propionate
- Molecular FormulaC25H32ClFO5
- Average mass466.970 Da
CCI 4725, CCI-4725, GR 2/925, GR-2/925,(8S,9R,10S,11S,13S,14S,16S,17R)-17-(chloroacetyl)-9-fluoro-11-hydroxy-10,13,16-trimethyl-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthren-17-yl propanoate
246-634-3[EINECS], 25122-46-7[RN]
(11β,16β)-21-chloro-9-fluoro-11-hydroxy-16-methyl-3,20-dioxopregna-1,4-dien-17-yl propanoate
Active Moieties
| NAME | KIND | UNII | CAS | INCHI KEY |
|---|---|---|---|---|
| Clobetasol | prodrug | ADN79D536H | 25122-41-2 | FCSHDIVRCWTZOX-DVTGEIKXSA-N |
Clobetasol
CAS Registry Number: 25122-41-2
CAS Name: (11b,16b)-21-Chloro-9-fluoro-11,17-dihydroxy-16-methylpregna-1,4-diene-3,20-dione
Molecular Formula: C22H28ClFO4, Molecular Weight: 410.91
Percent Composition: C 64.30%, H 6.87%, Cl 8.63%, F 4.62%, O 15.57%
Literature References: Topical corticosteroid. Prepn: Elks et al.,DE1902340; eidem,US3721687 (1969, 1973 both to Glaxo). Review of pharmacology and clinical efficacy in skin disorders: E. A. Olsen, R. C. Cornell, J. Am. Acad. Dermatol.15, 246-255 (1986).
Derivative Type: 17-Propionate
CAS Registry Number: 25122-46-7
Manufacturers’ Codes: GR-2/925
Trademarks: Clobesol (GSK); Dermovate (GSK); Olux (Connetics); Psorex (GSK); Temovate (GSK)
Molecular Formula: C25H32ClFO5
Molecular Weight: 466.97
Percent Composition: C 64.30%, H 6.91%, Cl 7.59%, F 4.07%, O 17.13%
Properties: White or almost white colorless, crystalline powder, mp 195.5-197°. [a]D +103.8° (c = 1.04 in dioxane). uv max (ethanol): 237 nm (e 15000). Insol in water.
Melting point: mp 195.5-197°
Optical Rotation: [a]D +103.8° (c = 1.04 in dioxane)
Absorption maximum: uv max (ethanol): 237 nm (e 15000)
Therap-Cat: Glucocorticoid; anti-inflammatory.
Keywords: Glucocorticoid.
Clobetasol propionate is a corticosteroid used to treat corticosteroid-responsive dermatoses and plaque psoriasis.
Clobetasol propionate is a corticosteroid used to treat skin conditions such as eczema, contact dermatitis, seborrheic dermatitis, and psoriasis.[2] It is applied to the skin as a cream, ointment, or shampoo.[2][3] Use should be short term and only if other weaker corticosteroids are not effective.[3] Use is not recommended in rosacea or perioral dermatitis.[2]
Common side effects include skin irritation, dry skin, redness, pimples, and telangiectasia.[2] Serious side effects may include adrenal suppression, allergic reactions, cellulitis, and Cushing’s syndrome.[2] Use in pregnancy and breastfeeding is of unclear safety.[4] Clobetasol is believed to work by activating steroid receptors.[2] It is a US Class I (Europe: class IV) corticosteroid, making it one of the strongest available.
Clobetasol propionate was patented in 1968 and came into medical use in 1978.[5] It is available as a generic medication.[3] In 2019, it was the 180th most commonly prescribed medication in the United States, with more than 3 million prescriptions.[6][7]
SYNTHESIS OF KEY INTERMEDIATE
SYN

DE 1902340
US 3992422
DE 2613875
EP 72200
WO 2012122452
CN 112110972
PATENT
IN 201821008147
Clobetasol propionate (C25H32ClFO5); CAS Registry No.[25112-46-7]; IUPAC name: 17-(2′- Chloroacetyl)-9-fluoro-l l-hydroxy-10,13,16-trimethyl-3-oxo-6,7,8,l 1,12,14,15,16-octahydrocyclopenta[a]phenanthren-17-yl] propionate is a potent halogen adrenal corticosteroid of the gluco-corticoid class used to treat various skin disorders including eczema and psoriasis. It is also highly effective for contact dermatitis caused by exposure to poison ivy/oak.In the US 3721687Apatentshow use of methanesulfonyl chloride and Pyridine as base to protect alcohol and at the time of LiCI reaction results with 10-15%ene impurity and less yield.In the methanesulfonyl chloride step used with pyridine as base which is a hazardous.Mesyl compound converted to Clobetasol propionate by using LiCl in Dimethylformamide reaction at IOO-IlO0C forms 10-15% with ene impurity. The synthesis of Clobetasol propionate results in small quantities of the eneimpurity. Clobetasol propionate desired compound to be with impurities which must be minimized. Ene impurity can be reduced to very low levels by reaction itself. However, if used recrystallization reduce ene impurity it is time consuming and very expensive. Further, because recrystallizations have high losses, unacceptably low yields.


Example I: Betamethasone to betamethasone 17- propionate To a 100 ml 4-neck round bottom flask (RBF) equipped with halfmoon stirrer, thermowelland addition funnel, mounted in a tub bath, was charged betamethasone (5.0g, 0.0127mole), Dimethylformamide (20ml). Cooled the reaction mass to 10-15°C. Slowly added trimethyl ortho propionate (3.42g, 0.0255mole) and p-toluenesulfonic acid (PTSA)(0.30g, 0.00174 mole) to the reaction mass at 10-15°C. Stirred the contents 10-15°C for 4 hr. The reaction was monitored for completion by TLC. Further continued stirring at the same temperature for Ihr till reaction complies by TLC. After reaction completion, added H2SO4UP to pH=1.0-2.0 in to reaction mass.Reaction mass was quenched in Purified water (25ml) at 25-30°C. Cooled reaction mass temperature to 0-5°C. Stirred for I hr and filtered and washed with Purified water (10mlX2). Suck dried under vacuum completely to get cream coloured solid. Dried in tray drier at 50-55°C.Dry weight-5.40g(94.50%); HPLC: 98.5%;mp215-218°C. IR (KBr, on’):3454.90, 3370.99 (-OH); 1719.86, 1659.10 (C=O)iC25H33FO6;
MS 448.52m/z 449.2255 [M+H]; 1HNMR (300MHz, CDCl3S ppm): Spectrum is recorded on Varian, and Tetra Methyl Silane (TMS) as internal standard. 1H-NMR Spectrum shows Aromatic-HK-7.17-7.22(d,lH); Hj-6.37-6.38 (d,lH);Hr6.14 (s,lH); HF-4.04-4.06(s,2H);HE2.23-2.28 (q,2H);Hc-l.39-1.43 (d,3H); HB-1.14 (t,3H);HA-0.96-2.96 (m,20H). 13CMR (300MHz, CDCl3Sppm): 8.692 (CH2-CH3); 16.693; 19.664; 21.353; 23.042; 27.568; 30.387; 36.456; 43.400; 46.547; 47.422; 71.760; 93.547; 124.307; 125.728; 127.903; 129.222; 130.443; 132.472; 145.632; 153.044; 167.424; 175.031 (O-C=O); 185.772 (Cyclic C=O); 196.732 (CH-CO-CH2-OH).
Example 2: Betamethasone 17- propionateto betamethasone 21-tosylate 100ml 4-neck RBF equipped with halfmoon stirrer, thermowell, reflux condenser mountained in water bath, was charged Stage-1 (5.0g, O.Olllmole), Dimethylformamide (20ml). Added 4-Dimethylaminopyridine as base (4.10g, 0.0335mole) and p-toluenesulfonyl chloride (4.24.Og, 0.0222mole)slowly, Stirredfor2-3 hr at 25-30°C. Stirred reaction mass at 25-30°C till reaction complies by TLC.As such reaction mass used insitue for next step. Reaction mass aliquot taken (2ml) and quenched in DM water (20ml), precipited material fdtered and washed with DM water (20ml). Suck dried well. Dried in tray drier at 50-55°C to get dry white solid. Dry weight-0.598g, (89.0%); HPLC: 98.5%; mp-170-175°C (dec). IR (KBr,cm”1):3291.91, 2980.39 (-OH); 1739.15, 1661.99 (C=O); C32H39FO8S; MS 602.71mA 603.2317 [M+H]; 1HNMR (300MHz, CDCl3Sppm): Spectrum is recorded on Varian, and Tetra Methyl Silane (TMS) as internal standard. 1H-NMR Spectrum shows Aromatic-Ηκ7.17-7.22(d,lH); Hj-6.37-6.38 (d,lH);Hr6.14 (s,lH); HG-4.334-4.393 (m,lH);HF-3.846- 4.007(d,2H);HE-2.273-2.349 (q,2H);HD-l.671-1.688 (s,lH); Hc-I-306-1.331 (d,3H); Hb1.055-1.105 (t,3H);HA-0.941 -2.634 (m,18H).13CMR (300MHz, CDCl3Sppm): 9.055 (CH2- CH3); 17.244; 20.002; 21.353; 23.168; 27.901; 30.622; 33.508; 34.881; 36.783; 43.642; 46.637; 47.330; 48.113; 48.417; 66.613; 70.902; 93.801; 102.732; 124.415; 129.324; 130.443; 132.472; 145.632; 153.583; 168.042; 174.853 (O-C=O); 186.208 (Cyclic C=O); 205.491 (CH-CO-CH2-OAr).
Example 3:Betamethasone 21-tosylateto Clobetasol propionate As such reaction mass used insitue for next step. Added. lithium chloride (LiCl)1.04 gm (0.0245mole). Stirred the reaction mass at 60-65°C for 5-6 hr.Reaction completion checked by TLC.After reaction completion, Added DM water (200ml). Stirred the reaction mass at 10-15°C for Ihr and Filtered washed with DM water (30mlx2).Dried in oven at 50-55°C to get white crystalline powder. Dry weight-4.42gm, (85.0%); HPLC:99.70%;mp-158-161°C. IR (KBr, cm_1):3299.62, 2976.53 (-OH); 1734.32, (C=0);1662.95 (C=C);C25H32C1F05; MSΑβ6.9Ίτη/ζ 467 [M+H];’HNMR (300MHz, CDCl35ppm): Spectrum is recorded on Varian, and Tetra Methyl Silane (TMS) as internal standard. 1H-NMR Spectrum shows AromaticHk-7.094-7. 128(d,IH); Hj-6.267-6.307 (d,lH); Hr6.066-6.076 (s,IH); H0-4.334-4.393 (m,lH); Hf-3.846-4.007 (d,2H); HE-2.273-2.349 (q,2H); Hd-I .671-1.688 (s,lH); Hc-1.306- 1.331 (d,3H); Hb-I .055-1.105 (t,3H); HA-0.941-2.634 (m,17H).13CMR (300MHz, CDCl35ppm): 8.692 (CH2-CH3); 16.693; 19.664; 21.353; 23.042; 27.568; 30.387; 36.456; 41.104; 46.547; 47.422; 71.760; 93.547; 124.307; 125.728; 127.903; 129.222; 130.443; 132.472; 145.632; 153.044; 168.312; 173.101 (O-C=O); 185.802 (Cyclic C=O); 204.602(CH-CO-CH2-C1)
SYN
Ruben Vardanyan, Victor Hruby, in Synthesis of Best-Seller Drugs, 2016
Synthesis of clobetasol propionate (27.1.13) starts from the known betamethasone 17-propionate (27.1.26), a potent glucocorticoid steroid with antiinflammatory and immunosuppressive properties, which was mesylated with methanesulfonyl chloride in pyridine to produce 9α-fluoro-11β-hydroxy-21-methylsulfonyloxy-16β-methyl 17-propionyloxypregna-1,4-diene-3,20-dione (27.1.27). The obtained product was refluxed in acetone, DMF, and dry LiCl mixture to produce the desired clobetasol propionate (27.1.13) [34] (Scheme 27.2.).

Clobetasol propionate, its structural formula (formula (I)), is a potent halogen-containing adrenocorticoid drug, has strong anti-inflammatory, anti-pruritic and vasoconstrictive effects, and its anti-inflammatory effect is approximately hydrogenated It is 112 times that of cortisone, and it is also used to treat neurodermatitis, contact dermatitis, eczema, discoid lupus erythematosus and other symptoms. It is currently widely used in clinical practice. It has been very popular in the international market and ranks among the top hormones. At present, there are only a few domestic companies in normal production, and the total yield is about 88%.[0003]

[0004] Formula (I).[0005] The process route for the production of synthetic clobetasol propionate is complex, technically difficult, and product quality requirements are strict. This is due to the complex structure of corticosteroids. The chemical structure of this type of drug is composed of three six-membered rings and one five-membered ring fused together to form a special molecular structure composed of 21 carbon atoms, with special molecular configuration steric effects and steric barriers. Group role. The functional groups on the drug structure interfere with each other, which makes the chemical reaction very complicated. It is manifested in many synthetic process steps, low raw material utilization rate, large amount of auxiliary materials, long production cycle, and many side reactions. The reaction process has various problems such as a large amount of solvents, a large amount of waste water and waste gas, and difficulty in recycling. Low technical indicators, low cost and other aspects.[0006] US patent, patent number 3721687, discloses two synthetic processes.[0007] Process method (1) adopts 9a-fluoro-113-hydroxy-16a-methyl-17 oxopropyl-1,4-diene-3,20-dione to synthesize clobetasol propionate, 9a -Fluoro-11-hydroxy-16 a -methyl-17oxopropyl-1,4-diene-3,20-dione and lithium chloride mixture, mixed with dimethylformamide (DMF) in acetone The solution is refluxed for four days, the solution is moved to a vacuum, ethanol, methanol, and acetone are added, and the mixture is refluxed for another 4 days. Most of the solution is moved to a vacuum, water is added to the residue, the crude product is put into the ether solution, and the mixture is passed through with chloroform. The aluminum is purified by filtration and recrystallized with ethanol to produce clobetasol propionate as a raw material. Method I uses too much acetone, and there is a certain risk of operation.[0008] Process method 2 adopts 21-chloro-9a-fluoro-1I@ -hydroxy-16a-methyl-17_oxopropyl-4ene-3,20-dione to synthesize clobetasol propionate Cable. Dissolve 21-chloro-9 a -fluoro-11 P -hydroxy-16 a -methyl-17oxopropyl_4ene-3,20-dione in acetone, cool in an ice bath, and add slowly while stirring Chromic acid (prepared by chromic acid: add 53.3ml of concentrated sulfuric acid to 250ml of water and add 66.7g of chromium trioxide); 4 hours later, the mixture reaches room temperature, ether is added, and it is left for another 20 minutes. The mixture is washed with water, and then the solution is moved to a vacuum ; The residue is recrystallized with acetone-petroleum ether, which pollutes the environment. In the past, organic solvents were not safe for production operations. [0009] Chinese patent, application number 200610053511.5, provides a method of mixing betamethasone 17-propionate sulfonate and anhydrous lithium chloride in a ratio of 1:1 to 2 and dissolving in dimethylformamide ( DMF), the chlorination reaction is carried out; second, after the chlorination reaction is complete, it is separated by ice water, and then centrifuged to dry, after drying, the crude clobetasol propionate is obtained; third, the clobetasol propionate is crude The crude tasol is dissolved in methanol or ethanol, activated carbon is added, decolorized, filtered, and the activated carbon is recovered; fourth, the filtrate is concentrated under reduced pressure, crystallized, dehydrated, and dried to obtain the raw material of clobetasol propionate. It has the characteristics of easy availability of starting materials, simple reaction steps, less dangerous and harmful solvents, mature technology, and convenient industrial production.[0010] The process route is as follows:[0011]

[0012] Clobetasol propionate uses betamethasone as the starting material, goes through the steps of cyclic ester-hydrolysis-sulfonation-chlorination, and then undergoes rough refinement to obtain clobetasol propionate-a refined substance, and then undergoes dissolution , Filtration, concentration, cooling, centrifugation, and drying to obtain clobetasol propionate. But its process route is longer, there are many influencing factors, and there are many side reactions. Moreover, the solvents used are very polluting and difficult to recycle.

Example 1[0034] 20g of Betamethasone 17-ester obtained by cyclic ester hydrolysis reaction was dissolved in 150ml of acetone, and after fully stirring and dissolving, 6g of ZnCl2 was added, and the temperature was raised to 35°C, and then 30g of BTC was introduced, After the BTC is passed, the reaction is kept warm for 3 hours. After the reaction is completed, the temperature is 40°C, and the concentration is reduced under reduced pressure until the solution contains 30ml of acetone. Then 300ml of drinking water is added for water separation and filtration. After drying for 16 hours at °C, 19.64 g of crude clobetasol propionate was obtained. The yield was 98.2%, and the crude clobetasol propionate content was 96.9% after analysis.Example 2[0036] 20g of betamethasone 17-ester compound obtained by cyclic ester hydrolysis reaction was dissolved in 150ml of acetone, and after fully stirring and dissolving, 7.2g of FeCl3 was added, heated to 30°C, and then 24g of BTC was introduced After the BTC is passed, the reaction is kept warm for 2 hours. After the reaction is completed, the solution is concentrated under reduced pressure at a temperature of 35°C until the solution contains 20ml of acetone, and then 300ml of drinking water is added for water precipitation, filtered, and finally at the temperature After drying for 10 hours at 85°C, 19.5 g of crude clobetasol propionate was obtained. The yield was 97.5%, and the crude clobetasol propionate content was 95.6% after analysis.Example 3[0038] 20g of Betamethasone 17-ester obtained by the cyclic ester hydrolysis reaction was dissolved in 150ml of acetone, and after fully stirring and dissolving, 4g of AlCl3 was added and the temperature was raised to 35°C, and then 28g of BTC was introduced, After the BTC is passed through, the reaction is kept warm for 4 hours. After the reaction is completed, the temperature is 30°C, and concentrated under reduced pressure until the solution contains 20ml of acetone. Then 300ml of drinking water is added for water precipitation, filtered, and finally at a temperature of 75 After drying for 18 hours at °C, 19.62g crude clobetasol propionate was obtained. The yield was 98.1%, and the crude clobetasol propionate content was 95.8% after analysis.Example 4[0040] The betamethasone 17-ester 20g obtained by the cyclic ester hydrolysis reaction was dissolved in 100ml of acetone, and after being fully stirred to dissolve, 4g of ZnCl3 was added, heated to 40°C, and then passed into 25g of BTC, After the BTC is passed, the reaction is kept for 5 hours. After the reaction is completed, the temperature is 40°C, and the concentration is reduced under reduced pressure until the solution contains 20ml of acetone. Then 200ml of drinking water is added for water precipitation, filtered, and finally at a temperature of 80°C. After drying for 18 hours at °C, 19.54 g of crude clobetasol propionate was obtained. The yield was 97.7%, and the crude clobetasol propionate content was 96.2% after analysis.[0041] Example 5 [0042] 20g of Betamethasone 17-ester obtained by cyclic ester hydrolysis reaction was dissolved in 200ml of acetone, and after fully stirring and dissolving, 8g of ZnCl3 was added and the temperature was raised to 50°C. Then pass in 40g BTC. After passing the BTC, keep it warm and react for 3 hours. After the reaction is completed, perform vacuum concentration at a temperature of 40°C until the solution contains 40ml of acetone, and then add 400ml of drinking water for hydrolysis. Filter, and finally dry at 85°C for 18 hours to obtain 19.58 g of crude clobetasol propionate. The yield was 97.9%, and the crude clobetasol propionate content was 96.9% after analysis.Example 6[0044] 20g of betamethasone 17-ester compound obtained by cyclic ester hydrolysis reaction was dissolved in 80ml of acetone, and after fully stirring and dissolving, 6g of ZnCl3 was added, and after the temperature was raised to 40°C, 25g of BTC was introduced, After the BTC is passed, the reaction is kept for 3 hours. After the reaction is completed, it is concentrated under reduced pressure at a temperature of 40°C, and concentrated until the solution contains 10ml of acetone. Then 150ml of drinking water is added for water precipitation, filtered, and finally at a temperature of 85°C. After drying for 18 hours at °C, 19.3g crude clobetasol propionate was obtained. The yield was 96.5%, and the crude clobetasol propionate content was 96.5% after analysis.
Publication numberPriority datePublication dateAssigneeTitleUS3721687A *1968-01-191973-03-20Glaxo Lab Ltd3-keto-delta 4-9alpha-halo-11-oxygenated-16-methyl or methylene-17alpha-acyloxy-20-keto-21-halo pregnenesCN1923842A *2006-09-112007-03-07Zhejiang Dingtai Pharmaceutical Co., Ltd.Manufacturing method of clobetasol propionate
Publication numberPriority datePublication dateAssigneeTitleFamily To Family CitationsCN105646630A *2015-08-102016-06-08Shandong Taihua Biological Technology Co., Ltd.One-pot Preparation of Clobetasol Propionate IntermediateCN112110972A *2019-06-212020-12-22Henan Lihua Pharmaceutical Co., Ltd.A kind of preparation method of clobetasol propionateCN112028957A *2020-07-292020-12-04Henan Lihua Pharmaceutical Co., Ltd.A kind of clobetasol propionate intermediate and preparation method
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
Clobetasol propionate is used for the treatment of various skin disorders including eczema, herpes labialis,[8] psoriasis, and lichen sclerosus. It is also used to treat several auto-immune diseases including alopecia areata, lichen planus (auto immune skin nodules), and mycosis fungoides (T-cell skin lymphoma). It is used as first-line treatment for both acute and chronic GVHD of the skin.[9]
Clobetasol propionate is used cosmetically by dark-skinned women for skin whitening, although this use is controversial. The U.S. Food and Drug Administration has not approved it for that purpose, and sales without a prescription are illegal in the U.S. Nonetheless, skin-whitening creams containing this ingredient can sometimes be found in ethnic beauty supply stores in New York City and on the internet. It is also sold internationally, and does not require a prescription in some countries. Whitening creams with clobetasol propionate, such as Hyprogel, can make skin thin and easily bruised, with visible capillaries, and acne. It can also lead to hypertension, elevated blood sugar, suppression of the body’s natural steroids, and stretch marks, which may be permanent.[10]
Clobetasol propionate is, along with mercury and hydroquinone, “amongst the most toxic and most used agents in lightening products.” Many products sold illegally have higher concentrations of clobetasol propionate than is permitted for prescription drugs.[11]
Contraindications
According to the California Environmental Protection Agency, clobetasol propionate should not be used by pregnant women, or women expecting to become pregnant soon, as studies with rats shows a risk of birth defects:[12]
“Studies in the rat following oral administration at dosage levels up to 50 mcg/kg per day revealed that the females exhibited an increase in the number of resorbed embryos and a decrease in the number of living fetuses at the highest dose. Pregnancy: Teratogenic Effects (i.e., possibility of causing abnormalities in fetuses): Pregnancy Category C: Clobetasol propionate has not been tested for teratogenicity when applied topically; however, it is absorbed percutaneously, and when administered subcutaneously it was a significant teratogen in both the rabbit and mouse. Clobetasol propionate has greater teratogenic potential than steroids that are less potent. There are no adequate and well-controlled studies of the teratogenic effects of clobetasol propionate in pregnant women. Temovate Cream and Ointment should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.”
Forms
Clobetasol propionate is marketed and sold worldwide under numerous names, including Clobex, Clob-x (Colombia), Clovate, Clobet (Biolab Thailand) Clonovate (T.O. Chemicals, Thailand), Cormax (Watson, US), Haloderm (Switzerland, by ELKO Org), Pentasol (Colombia), Cosvate, Clop (Cadila Healthcare, India), Propysalic (India), Temovate (US), Dermovate[13] (GlaxoSmithKline, Canada, Estonia, Pakistan, Switzerland, Portugal, Romania, Israel), Olux, ClobaDerm, Tenovate, Dermatovate, Butavate, Movate, Novate, Salac (Argentina), and Powercort, Lotasbat and Kloderma (Indonesia), Lemonvate (Italy), Delor (Ethiopia), Psovate (Turkey).
References
- ^ “Clobetasol Propionate Topical Ointment 0.05% Information – Drug Encyclopedia”. Kaiser Permanente.
- ^ Jump up to:a b c d e f “Clobetasol Propionate Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 13 April 2019.
- ^ Jump up to:a b c British national formulary : BNF 76 (76 ed.). Pharmaceutical Press. 2018. p. 1210. ISBN 9780857113382.
- ^ “Clobetasol topical Use During Pregnancy”. Drugs.com. Retrieved 13 April 2019.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 487. ISBN 9783527607495.
- ^ “The Top 300 of 2019”. ClinCalc. Retrieved 16 October 2021.
- ^ “Clobetasol – Drug Usage Statistics”. ClinCalc. Retrieved 16 October 2021.
- ^ Hull C, McKeough M, Sebastian K, Kriesel J, Spruance S (March 2009). “Valacyclovir and topical clobetasol gel for the episodic treatment of herpes labialis: a patient-initiated, double-blind, placebo-controlled pilot trial”. Journal of the European Academy of Dermatology and Venereology. 23 (3): 263–7. doi:10.1111/j.1468-3083.2008.03047.x. PMID 19143902. S2CID 205588376.
- ^ E. Fougera and Co. “CLOBETASOL PROPIONATE CREAM USP, 0.05% CLOBETASOL PROPIONATE OINTMENT USP, 0.05%<“. NIH Daily Med.
- ^ Saint Louis C (January 15, 2010). “Creams Offering Lighter Skin May Bring Risks”. New York Times.
- ^ Gbetoh MH, Amyot M (October 2016). “Mercury, hydroquinone and clobetasol propionate in skin lightening products in West Africa and Canada”. Environmental Research. 150: 403–410. Bibcode:2016ER….150..403G. doi:10.1016/j.envres.2016.06.030. hdl:1866/19006. PMID 27372064.
- ^ Office of Environmental Health Hazard Assessment (August 22, 1997). “Chemicals Under Consideration For Possible Listing Via The “Formally Required To Be Labeled Or Identified” Mechanism”. California Environmental Protection Agency. Archived from the original on 2001-07-20. Retrieved 2007-05-06.
- ^ “DERMOVATE 0.05% W/V OINTMENT – Clobetasol Topical(0.05% w/v) Glaxo SmithKline Pharmaceuticals Ltd”. GNH. Retrieved 2021-07-16.
External links
- “Clobetasol propionate”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Pronunciation | /kloʊˈbeɪtəsɒl/[1] |
| Trade names | Temovate, Clobex, Cormax, others |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Clobetasol_propionate |
| Pregnancy category | AU: B3 |
| Routes of administration | Topical |
| ATC code | D07AD01 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 25122-46-7 |
| PubChem CID | 32798 |
| IUPHAR/BPS | 7062 |
| DrugBank | DB01013 |
| ChemSpider | 30399 |
| UNII | 779619577M |
| KEGG | D01272 |
| ChEBI | CHEBI:31414 |
| ChEMBL | ChEMBL1159650 |
| CompTox Dashboard (EPA) | DTXSID6045907 |
| ECHA InfoCard | 100.042.380 |
| Chemical and physical data | |
| Formula | C25H32ClFO5 |
| Molar mass | 466.97 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
//////////////////Clobetasol propionate, CCI 4725, CCI-4725, GR 2/925, GR-2/925, Glucocorticoid, anti-inflammatory
[H][C@@]12C[C@H](C)[C@](OC(=O)CC)(C(=O)CCl)[C@@]1(C)C[C@H](O)[C@@]1(F)[C@@]2([H])CCC2=CC(=O)C=C[C@]12C

NEW DRUG APPROVALS
ONE TIME
$10.00
SEVELAMER
SEVELAMER
2-(chloromethyl)oxirane;prop-2-en-1-amine
CAS 52757-95-6, Molecular Formula, (C3-H7-N.C3-H5-Cl-O)x, Molecular Weight, 149.6198, HSDB 7608
Sevelamer hydrochloride [USAN]
152751-57-0, (C3-H7-N.C3-H5-Cl-O.Cl-H)x-, 186.0807, GT 16-026A
- A crosslinked polymeric amine that binds PHOSPHATES and BILE ACIDS; it is nonabsorbed; used for hyperphosphatemia during HEMODIALYSIS and in END-STAGE RENAL DISEASE; used like calcium acetate.
Sevelamer carbonate [USAN]
845273-93-0, (C3-H7-N.C3-H5-Cl-O)x-.x-C-H2-O3, 211.6436, GT335-012
- A polymeric amine that binds phosphate and is used to treat HYPERPHOSPHATEMIA in patients with kidney disease.
Drug Name:Sevelamer Carbonate,
Trade Name:Renvela®
MOA:Phosphate binder
Indication:Hyperphosphatemia
Company:Genzyme (Originator)
Sales:$1,037.9 Million (Y2015); 
$902.9 Million (Y2014);
$997.5 Million (Y2013);
$842.4 Million (Y2012);
$581 Million (Y2011);ATC Code:
Sevelamer Carbonate was first approved by the U.S. Food and Drug Administration (FDA) on Jul 19, 2007, then approved by European Medicine Agency (EMA) on Jun 21, 2009. However, till 2013, China Food and Drug Administration approved this drug. It was developed by Genzyme, and the trade name is Renvela®. On the other hand, US FDA firstly approved Sevelamer HCl (Renagel®) on Oct 30, 1998.
Renvela® is a non-absorbed phosphate binding crosslinked polymer, containing multiple amines separated by one carbon from the polymer backbone. These amines exist in a protonated form in the intestine and interact with phosphate molecules through ionic and hydrogen bonding. By binding phosphate in the gastrointestinal tract and decreasing absorption, sevelamer carbonate lowers the phosphate concentration in the serum (serum phosphorus).
Renvela® is available as film-coated tablet for oral use, containing 800 mg of free sevelamer carbonate on an anhydrous basis. The initial dose is 0.8 or 1.6 grams orally three times per day with meals.SevelamerCAS Registry Number: 52757-95-6
CAS Name: 2-Propen-1-amine polymer with (chloromethyl)oxirane
Additional Names: allylamine polymer with 1-chloro-2,3-epoxypropane; allylamine-epichlorohydrin copolymer; poly(allylamin-co-N,N¢-diallyl-1,3-diamino-2-hydroxypropane)
Literature References: Polymeric non-absorbed phosphate binder consisting of polyallylamine crosslinked with epichlorohydrin to form a hydrogel where 40% of the amines are protonated. Follows the general formula of (C3H7N.C3H5ClO)n. Binds dietary phosphate leading to increased fecal excretion, decreased absorption and decreased serum phosphorous levels. Prepd not claimed: S. R. Holmes-Farley et al.,WO9505184; eidem,US5496545 (1995, 1996 both to GelTex). Mechanism of action: S. R. Holmes-Farley et al.,J. Macromol. Sci. Pure Appl. Chem.A36, 1085 (1999). Determn of phosphate binding capacity: J. R. Mazzeo et al.,J. Pharm. Biomed. Anal.19, 911 (1999). Clinical studies in end stage renal disease: E. A. Slatopolsky et al.,Kidney Int.55, 299 (1999).
Derivative Type: Hydrochloride
CAS Registry Number: 152751-57-0
Manufacturers’ Codes: PB-94; GT16-026A
Trademarks: Renagel (Genzyme)
Properties: Insol in water. Hydrophilic.
Therap-Cat: Antihyperphosphatemic.
Keywords: Antihyperphosphatemic.
Sevelamer (rINN) is a phosphate binding medication used to treat hyperphosphatemia in patients with chronic kidney disease. When taken with meals, it binds to dietary phosphate and prevents its absorption. Sevelamer was invented and developed by GelTex Pharmaceuticals. Sevelamer is marketed by Sanofi under the brand names Renagel (sevelamer hydrochloride) and Renvela (sevelamer carbonate).
Chemistry and pharmacology
Sevelamer consists of polyallylamine that is crosslinked with epichlorohydrin.[1] The marketed form sevelamer hydrochloride is a partial hydrochloride salt being present as approximately 40% amine hydrochloride and 60% sevelamer base. The amine groups of sevelamer become partially protonated in the intestine and interact with phosphate ions through ionic and hydrogen bonding.
Medical uses
Sevelamer is used in the management of hyperphosphatemia in adult patients with stage 4 and 5 chronic kidney disease (CKD) on hemodialysis. Its efficacy at lowering phosphate levels is similar to that of calcium acetate, but without the accompanying risk of hypercalcemia and arterial calcification.[2][3] In patients with CKD, it has also been shown to reduce triglycerides and LDL, and increase HDL.[4]
This is a phosphate binder.
Contraindications
Sevelamer therapy is contraindicated in hypophosphatemia or bowel obstruction. In hypophosphatemia, sevelamer could exacerbate the condition by further lowering phosphate levels in the blood, which could be fatal.[5]
Adverse effects
Common adverse drug reactions (ADRs) associated with the use of sevelamer include: hypotension, hypertension, nausea and vomiting, dyspepsia, diarrhea, flatulence, and/or constipation.
Other effects
Sevelamer can significantly reduce serum uric acid.[6] This reduction has no known detrimental effect and several beneficial effects, including reducing hyperuricemia, uric acid nephrolithiasis, and gout.
Sevelamer is able to sequester advanced glycation end products (AGEs) in the gut, preventing their absorption into the blood. AGEs contribute to oxidative stress, which can damage cells (like beta cells, which produce insulin in the pancreas). As Vlassara and Uribarri explain in a 2014 review on AGEs, this may explain why sevelamer, but not calcium carbonate (a phosphate binder that does not sequester AGEs), has been shown to lower AGEs in the blood, as well as oxidative stress and inflammatory markers.[7]
SYN
https://www.oatext.com/polymer-and-heterocyclic-compounds-their-utility-and-application-as-drug.php
Sevelamer hydrochloride (Renagel VR ) was the first polymeric phosphate control and removal of excess phosphate is of benefit to patients with chronic kidney disease CKD where dialysis is unable to maintain safe phosphorus levels. Sevelamer limits the absorption of dietary phosphorus by binding phosphate in the intestine through ionic interaction with the polyamine polymer. Sevelamer is across linked form of poly(allylamine) containing primary and secondary aliphatic amine residues and was approved for the treatment of hyperphosphatemia by the FDA in 1998.The following Scheme 5 shows the synthetic lines for Sevelamer hydrochloride.
Scheme 5. Represent the formation of poly allylamine as a residue of sevelamer structure
Synthesis of Sevelamer Hydrochloride. Approximately 40 percent of the amine moieties are in the HCl form. Crosslinking degree is 10%.
SYN
- The synthesis of Sevelamer consists of crosslinkung poly(allylamine hydrochloride) with epichlorohydrin. The product is washed, dried and ground to the desired particle size to give the active substance.
Reference:1. US2009155368A1.Route 2
Reference:1. WO2011099038A2.
Reference:1. WO2010146603A1.Route 4
Reference:1. CN102796262A.
SYN
https://patents.google.com/patent/US20100331516A1/en
- The present invention relates to the process for preparation of carbonate salt of amine polymers, preferably Poly(allylamine-co-N,N′-diallyl-1,3-diamino-2-hydroxypropane)carbonate Formula-I, an antihyperphosphatemic agent.
- a, b=number of primary amine groups a+b=9
c=number of crosslinking groups c=1
m=large number to indicate extended polymer network
- Sevelamer carbonate is non-absorbable polymer marketed as Renvela™ by Genzyme Corporation. It is known chemically as poly(allylamine-co-N,N′-diallyl-1,3-diamino-2-hydroxypropane) carbonate salt. It was developed as a pharmaceutical alternative to Sevelamer hydrochloride (Renagel®). Renvela™ contains Sevelamer carbonate, a non-absorbed phosphate binding crosslinked polymer, free of metal and calcium. It contains multiple amines separated by one carbon from the polymer backbone. These amines exist in a protonated form in the intestine and interact with phosphate molecules through ionic and hydrogen bonding. By binding phosphate in the dietary tract and decreasing absorption, Sevelamer carbonate lowers the phosphate concentration in the serum.
- [0004]
Sevelamer carbonate is an anion exchange resin with the same polymeric structure as Sevelamer hydrochloride in which carbonate replaces chloride as the counterion. While the counterions differ for the two salts, the polymer itself, the active moiety, is the same. The protonated amines can be indirectly measured as carbonate content in meq/gm. Renvela™ is used in End Stage Renal Disease (ESRD) which leads to hyperphosphatemia due to retention of phosphorous. This condition can lead to ectopic calcification. Renvela™ binds dietary phosphate in GI tract and thus controls the serum phosphate levels. The potency of Renvela™ is measured in terms of its Phosphate Binding Capacity (PBC) by Phosphate Assay (PA). Treatment of hyperphosphatemia includes reduction in dietary intake of phosphate, inhibition of intestinal phosphate absorption with phosphate binders, and removal of phosphate with dialysis. Sevelamer carbonate taken with meals has been shown to control serum phosphorus concentrations in patients with CKD who are on dialysis. Currently Sevelamer hydrochloride is used to cure hyperphosphatemia. As a consequence ESRD patients still need a high dosage of Renagel® to meet clinical end-points, leading to adverse effect such as gastrointestinal discomfort and problems with patient compliance. But systemic acidosis development or worsening of pre-existing acidosis has been reported in many patients on long term dialysis who are given Sevelamer hydrochloride (Perit Dial Int. 2002, 22, 737-738, Nephron 2002, 92, 499-500, Kidney Int. 2004, 66, S39-S45, Ren. Fail 2005, 27,143-147). - [0005]
Administration of Sevelamer hydrochloride adds to metabolic acid load because the resin removes some bicarbonate or bicarbonate precursor (mainly short chain fatty acid anions) from the body and replaces it with chloride. Each molecule of chloride contributed to the body in exchange for carbonate or bicarbonate precussor is equivalent to a molecule of hydrochloric acid added to the body, so the tendency of patients on long term haemodialysis to acidosis is inevitably increased when they take Sevelamer hydrochloride. (Kidney Int., 2005; 67: 776-777) - [0006]
This problem can be countered by an increase in the dialysate concentration of bicarbonate used in each dialysis session. A more fundamental solution, suitable for both dialyzed and non-dialyzed patients, would be the administration of Sevelamer free base, or any other suitable resin, not as the chloride but as body suitable counterion such as bicarbonate. Anion exchange resins have traditionally been synthesized in the chloride form, but the chloride in the current Sevelamer preparation is of no benefit to patients with renal failure. A change in the formulation of Sevelamer from its current chloride form to Sevelamer attached to bicarbonate would convert an acid load into a mild alkali load. (Cli. Sci. 1963; 24:187-200) - [0007]
U.S. Pat. No. 6,858,203 relates to phosphate-binding polymers provided for removing phosphate from the gastrointestinal tract. These polymers are useful for the treatment of hyperphosphatemia. - [0008]
WO 2006/050315 describes pharmaceutical compositions comprising a carbonate salt of an aliphatic amine polymer wherein the monovalent anion can prevent or ameliorate acidosis, in particular acidosis in patients with renal disease. - [0009]
HPLC Ion Chromatography PA method is used for the determination of PBC of Sevelamer HCl which can be adopted for determining the carbonate content from Sevelamer carbonate (J R Mazzeo et al, J. Pharm. Biomed. Anal. 19 (1999) 911-915). - [0010]
Our co-pending application number 1402/MUM/2006 dated 1 Sep. 2006 discloses process for preparation of Sevelamer HCl having phosphate binding capacity in the range of about 5.0 meq/gm to about 6.0 meq/gm and chloride content in the range of about 3.74 to about 5.60 meq/gm. - [0011]
The prior art mentioned above discussed advantages of Sevelamer carbonate over Sevelamer hydrochloride thus there remains need for commercially viable and industrially useful process for the preparation of Sevelamer carbonate having consistency in phosphate binding capacity, degree of cross linking, chloride content and carbonate content.
- The reaction is represented by the following reaction scheme:
- [0078]
100 gm Sevelamer hydrochloride was dispersed in 500 ml purified water and sodium hydroxide solution [20 gm sodium hydroxide dissolved in 500 ml purified water] was added to the obtained suspension followed by stirring at 25-35° C. for 30 minutes. The obtained material was filtered and wet cake was stirred in 1.0 L purified water for an hour. The material was filtered and cake was washed twice. Wet cake was dried at 50-90° C. for 5-6 hrs to get Sevelamer base (70 gm). LOD: 0.4% Chloride content: Nil.
Example 2
- [0079]
10 gm Sevelamer was suspended in 200 ml water and stirred. Carbon dioxide gas was purged into the obtained suspension at 25-35° C. for 8 hrs using dry ice. The obtained material was filtered and washed with 100 ml water [3×100] and the wet cake was dried on rotavapor at 90-95° C. to get Sevelamer carbonate (11.5 gm). Yield—115% w/w [Chloride content: 0.3%, Phosphate binding: 5.75 mMole/g, Carbonate content: 4.78 meq/g and Degree of crosslinking—16.4%], Solid state 13C NMR shows prominent peak at 164 ppm which is for carbon of carbonate.
Example 3
- [0080]
10 gm Sevelamer was added to 200 ml water and reacted with carbon dioxide gas under pressure at 25-35° C. for 7-8 hrs with stirring. The obtained material was filtered and washed with 100 ml water thrice [3×100]. The wet cake thus obtained was dried on rotavapor at 90-95° C. to get Sevelamer carbonate (11.3 gm). Yield—113% w/w Degree of crosslinking—16.4%, Solid state 13C NMR shows prominent peak at 164 ppm which is for carbon of carbonate.
Example 4
- [0081]
Sevelamer (7 gm) was added to 150 ml water and reacted with carbon dioxide gas under pressure at 60-65° C. for 7-8 hrs with stirring. The material obtained was filtered and washed with 100 ml purified water thrice [3×100]. The wet cake thus obtained was dried on rotavapor at 90-95° C. to get Sevelamer carbonate (9.3 gm). - [0082]
Yield—120% w/w Degree of crosslinking—16.4%, Solid state 13C NMR shows prominent peak at 164 ppm which is for carbon of carbonate.
Example 5
- [0083]
Sevelamer (7 gm) was added to 150 ml water and reacted with carbon dioxide gas by purging under pressure at 60-65° C. for 7-8 hrs with stirring. The material obtained was filtered and washed with 100 ml purified water thrice [3×100]. The wet cake thus obtained was dried on rotavapor at 90-95° C. to get Sevelamer carbonate (9.0 gm). - [0084]
[Degree of crosslinking—16.4%, Chloride content: 0.5%, Phosphate binding: 5.56 mMole/g and Carbonate content: 4.46 meq/g] Yield—110% w/w Solid state 13C NMR shows prominent peak at 164 ppm which is for carbon of carbonate.
Example 6
- [0085]
Sevelamer hydrochloride (10 gm) was treated Sodium hydroxide solution (2M) for 1 hr at temperature 25 to 35° C. to get Sevelamer base. Filter the free base and was added to 150 ml water and reacted with carbon dioxide gas by purging under pressure at 60-65° C. for 7-8 hrs with stirring. The material obtained was filtered and washed with 100 ml purified water thrice [3×100]. The wet cake thus obtained was dried on rotavapor under vacuum at 90-95° C. to get Sevelamer carbonate (9.3 gm). Yield—120% w/w. - [0086]
[Degree of crosslinking—16.4%, Chloride content: 0.2%, Phosphate binding: 5.45 mMole/g and Carbonate content: 4.36 meq/g]. - [0087]
Solid state 13C NMR shows prominent peak at 164 ppm which is for carbon of carbonate.
Example 7
- [0088]
Sevelamer hydrochloride (10 gm) was treated sodium hydroxide solution (2M) for 1 hr at temperature 25 to 35° C. to get Sevelamer base. Filter the free base and was added to 100 ml water. Sodium bicarbonate (10 gm dissolved in 1000 ml purified water) solution was added at temperature 60-65° C. for 4 hrs with stirring. Sevelamer Carbonate thus obtained was filtered and again subjected to for treatment of sodium bicarbonate solution (10 gm in 1000 ml). Reaction mixture was heated for 4 hrs at 60-65° C. with stirring. The material obtained was filtered and washed with 100 ml purified water thrice [3×100]. The wet cake thus obtained was dried under vacuum tray dryer at 80-90° C. for 24 hrs and further dried in atmospheric tray dryer at 100° C. for 36 hrs to get Sevelamer carbonate (9.0 gm). The loss of drying of material was about 5-7% achieved as per requirement. Yield—120% w/w, [Degree of crosslinking—16.4%, Chloride content: 0.01%, Phosphate binding: 5.68 mMole/g and Carbonate content: 4.85 meq/g]
Example 8
- [0089]
Sodium hydroxide pellets (41 gm) is dissolved in 600 ml methanol at 25-35° C. and polyallylamine hydrochloride (100 gm) is added to it followed by stirring for 5-6 hrs at temperature 25-35° C. The obtained reaction mass is filtered through hyflobed and filtrate is concentrated to reduce to half volume and the separated inorganic salt is filtered off over hyflobed. The obtained filtrate is concentrated completely under vacuum to get sticky mass (61 gm) of polyallylamine. Yield—61% w/w
Example 9
- [0090]
Polyallylamine (27.5 gm) dissolved in 100 ml water is charged into 1 L SS 316 autoclave and interacted with carbon dioxide gas under pressure (5.0 Kg/cm2). Initially 2-3 Kg/cm2 gas is consumed by the reaction mass and exotherm is observed from 28 C to 35° C. Then 5 Kg/cm2 pressure is maintained for 5-6 hours. After completion of the reaction the reaction mass is slowly added to 700 methanol and stirred for 3-4 hours. The separated solid (31 gm) is filtered, washed with 50 ml methanol and dried at 40-50° C. in vacuum oven. Yield—112% w/w
Example 10
- [0091]
Polyallylamine carbonate (20 gm) is dissolved in 30 ml water and cooled at 5-15° C. under stirring. The aqueous sodium hydroxide solution [dissolving 4.23 gm sodium hydroxide pellets into 4.2 ml of water] is added to reaction mass dropwise at 10-15° C. with continued stirring for 30 minutes. 101 ml toluene and 0.6 ml SPAN-85 is added to it and heated at 55-60° C. Epichlorohydrin (1.06 gm) is added to the reaction mass followed by stirring and heating for 3 hrs. The reaction mass is cooled at 25-35° C. and filtered through Buchner funnel. The obtained wet cake is added to 1 L acetone followed by stirring for 1 hour to get solid which was filtered through Buchner funnel. The aqueous organic washings are repeated for 7-10 times till polymer is free from excess alkalinity and the obtained wet cake is dried at 40-50° C. on rotavapor and then at 90-95° C. till constant weight of polymer is obtained (9 gm). Yield—45% w/w, Solid state 13C NMR shows prominent peak at 164 ppm which is for carbon of carbonate.
Example 11
- [0092]
Polyallylamine carbonate (20 gm) is dissolved in 30 ml water and cooled at 5-15° C. under stirring. The aqueous sodium hydroxide solution [dissolving 4.23 gm sodium hydroxide pellets into 4.2 ml of purified water] is added to obtained reaction mass dropwise at 10-15° C. with continued stirring for 30 minutes. 150 ml water and 0.6 ml SPAN-85 is added to it and heated at 60-80° C. Epichlorohydrin (1.06 gm) is added followed by stirring and heating is continued for 3 hours. The reaction mass is cooled at 25-35° C. and filtered through Buchner funnel. The obtained wet cake is added to 1 L acetone followed by stirring for 1 hour to get solid which is filtered through Buchner funnel. This aqueous organic washings are repeated for 7-10 times till the polymer is free from excess alkalinity and the obtained material is dried at 40-50° C. on rotavapor and/or Fluidised bed dryer then at 90-95° C. till constant weight of polymer is obtained (9 gm).
Example 12
- [0093]
Polyallylamine carbonate (20 gm) is dissolved in 30 ml water and cooled at 5-15° C. under stirring. The aqueous sodium hydroxide solution [dissolving 4.23 gm sodium hydroxide pellets into 4.2 ml of purified water] is added to the obtained reaction mass dropwise at 10-15° C. with continued stirring for 30 minutes. 150 ml water and 0.6 ml SPAN-85 is added to it and heated at 60-80° C. Epichlorohydrin (1.06 gm) is added followed by stirring and heating is continued for 3 hours. The reaction mass is cooled at 25-35° C. and filtered through Buchner funnel. The obtained wet cake is added to 1 L isopropyl alcohol (IPA) followed by stirring for 1 hour to get solid which is filtered through Buchner funnel. The obtained material is washed with water and organic solvents for 4-5 times till the polymer is free from excess alkalinity. The obtained wet cake is dried under vacuum tray dryer at 80-90° C. for 24 hrs and further dried in atmospheric tray dryer at 100° C. for 36 hrs till constant weight of dried polymer is obtained (15 gm). The loss on drying of material is around 6% as per requirement.
Example 13
- [0094]
In 1 L SS 316 autoclave, 75 gm allylamine and 200 ml water is charged and carbon dioxide gas under pressure (5 Kg/cm2) is purged into autoclave for 3-4 hours followed by stirring. Nitrogen gas is purged for 15 minutes. 9.8 gm VA-086 is added to the reaction mass and stirred at 70-80° C. for 12 hours and this solution is added to 1 L methanol under stirring. The separated material is filtered and washed with 100 ml methanol, suck dried and dried in vacuum oven at 50-60° C. to get 90 gm of polyallylamine carbonate. Yield—120% w/w
Example 14
- [0095]
Polyallylamine carbonate (20 gm) dissolved in 30 ml water is cooled at 5-15° C. under stirring and sodium hydroxide solution [dissolving 4.23 gm sodium hydroxide pellets into 4.2 ml of purified water] is added to the obtained reaction mass dropwise at 10-15° C. followed by continued stirring for 30 minutes. 101 ml toluene and 0.6 ml SPAN-85 is added to it and heated at 55-60° C. Epichlorohydrin (1.06 gm) is added and reaction mass is stirred and heated for 3 hours. Then it is cooled to 25-35° C. and filtered through Buchner funnel. The wet cake obtained is added to 1 to 1.5 L acetone followed by stirring for 1 hour to get solid which is filtered through Buchner funnel. The washings are repeated for 7-10 times till polymer is free from excess alkalinity. Wet cake (9 gm) is dried at 40-50° C. on rotavapor and then at 90-95° C. till constant weight of polymer is obtained. Yield—45% w/w
Example 15
- [0096]
Sevelamer hydrochloride (10 gm) was added to 10% aqueous sodium bicarbonate solution at 25-35° C. and stirred for 7-8 hrs. The material obtained was filtered and washed with 100 ml purified water thrice and the wet cake was dried on rotavapor at 90-95° C. to get Sevelamer carbonate (7.5 gm). Yield—75% w/w - [0097]
Solid state 13C NMR shows prominent peak at 164 ppm which is for carbon of carbonate. - [0098]
[Chloride content: 0.4%, Phosphate binding: 5.45 mMole/g and Carbonate content: 4.85 meq/g]
Example 16
- [0099]
Sevelamer hydrochloride (10 gm) was added to 10% aqueous sodium bicarbonate solution. The mixture was stirred at 60-65° C. for 4 hrs. The material obtained was filtered and the obtained wet cake was again subjected to the treatment of 10% sodium bicarbonate solution. Reaction mixture was heated for 4 hrs at 60-65° C. with stirring. The material obtained was filtered and washed with 100 ml purified water four times and the wet cake was dried on rotavapor under vacuum at 90-95° C. to get Sevelamer carbonate (7.5 gm). Yield—75% w/w, Solid state 13C NMR shows prominent peak at 164 ppm which is for carbon of carbonate, [Chloride content: 0.03%, Phosphate binding: 5.25 mMole/g and Carbonate content: 4.65 meq/g].
Example 17
- [0100]
Sevelamer hydrochloride (10 gm) was added into 130 ml solution of sodium bicarbonate (10 gm NaHCO3 in 130 ml water) and the mixture was stirred at 60-65° C. for 4 hrs. The material was filtered using Buckner funnel assembly. The obtained wet cake was added into 130 ml solution of sodium bicarbonate (10 gm NaHCO3 in 130 ml water) and stirred at 60-65° C. for 4 hrs. The material was filtered using Buckner funnel assembly and the wet cake was washed by stirring it in 100 ml water for 1 hr at 60-65° C. The material was filtered using Buckner funnel assembly. The wet cake was washed twice at 60-65° C. and dried on rotavapor at 90-95° C. to get Sevelamer carbonate (8.5 gm). Yield—75% w/w, Chloride content: 0.03%
Example 18
- [0101]
Sevelamer hydrochloride (1.1 Kg) was added into 15.5 L solution of sodium bicarbonate (1.1 Kg NaHCO3 in 14.3 L water). The obtained mixture was stirred at 60-65° C. for 4 hrs. The obtained material was filtered by centrifuge filter. The obtained wet cake was added into 15.5 L solution of sodium bicarbonate (1.1 Kg NaHCO3 in 14.3 L water) and maintained stirring at 60-65° C. for 4 hrs. The material was filtered by centrifuge filter assembly and obtained wet cake was stirred in 11 L water for 1 hr at 60-65° C. The material was filtered by centrifuge filter and the washing of wet cake was repeated at 60-65° C. for two more times. The obtained wet cake was dried in air tray dryer (ATD) at 90-100° C. for 30-36 hrs and LOD was checked after every five hours till LOD was in the range of 5 to 10%. to get Sevelamer carbonate (0.995 Kg), [Chloride content: 0.03%, Phosphate binding capacity: 5.5 mmole/gm, Carbonate content: 5.1 meq/gm]
Example 19
- [0102]
Sevelamer hydrochloride (10 gm) was added to sodium bicarbonate solution (10 gm in 200 ml) at 25-35° C. The reaction mixture was heated for 4 hrs at 60-65° C. with stirring. Sevelamer Carbonate thus obtained was filtered and again subjected to treatment of Sodium bicarbonate solution (10 gm in 200 ml). Reaction mixture was heated for 4 hrs at 60-65° C. with stirring. The material was filtered off and washed with 100 ml purified water four times (4×100 ml) and the wet cake was dried under vacuum tray dryer at 80-90° C. for 24 hrs and further dried in atmospheric tray dryer at 100° C. for 36 hrs till constant weight of dried polymer was obtained. The loss on drying of material was around 6% (Limit: 4-10%), achieved as per requirement. Sevelamer carbonate (7.5 gm) was obtained which can be sieved through 30 mesh for uniformity of the sample. Yield—75% w/w. Solid state 13C NMR shows prominent peak at 164 ppm which is for carbon of carbonate. [Chloride content: 0.02%, Phosphate binding: 5.56 mMole/g and Carbonate content: 4.74 meq/g].
Example 20
- [0103]
10 g wet cake of Sevelamer carbonate was subjected to drying in air tray dryer at 80-100° C. at atmospheric pressure for 36 hours and LOD was measured after every five hours. LOD: 7.5% Yield: 3.1 gm
Example 21
- [0104]
100 g wet cake of Sevelamer carbonate was subjected to drying in air tray dryer at 80-100° C. at atmospheric pressure for 37 hours and LOD was measured. LOD: 8.4% Yield: 30 gm
Example 22
- [0105]
10 g wet cake of Sevelamer carbonate was subjected to drying in vacuum tray dryer at 50-100° C. at reduced pressure for 24 hours and LOD was measured. LOD: 8.5% Yield: 3.2 gm
Example 23
- [0106]
100 g wet cake of Sevelamer carbonate was subjected to drying in vacuum tray dryer at 50-100° C. at reduced pressure for 24 hours and LOD was measured. LOD: 8.9% Yield: 31 gm
Example 24
- [0107]
10 Kg wet cake of Sevelamer carbonate was subjected to drying in fluidized bed dryer at 80-100° C. for 16 hours and LOD was measured after every five hours. LOD: 7.9% Yield: 3.4 kg
Example 25
- [0108]
15 Kg wet cake of Sevelamer carbonate was subjected to drying in fluidised bed dryer at 80-110° C. for 16 hours and LOD was measured. LOD: 8.8% Yield: 4.9 kg.
Example 26
- [0109]
10 g wet cake of Sevelamer carbonate was subjected to drying in rotary evaporator at 50-100° C. at reduced pressure for 16 hours and LOD was measured after every five hours. - [0110]
LOD: 9.1% Yield: 3.1 gm
Example 27
- [0111]
100 g wet cake of polyallylamine carbonate is subjected to drying in rotary evaporator at 50-100° C. at reduced pressure for 16 hours and LOD is measured. LOD: 8.9% Yield: 33 gm.
SYN
https://patents.google.com/patent/CN102675510A/enHyperphosphatemia is a kind of patient’s disease on one’s body that often appears at renal tubal dysfunction, hypothyroidism, acute acromegaly or phosphoric acid salt drug overdose; Its treatment is normal adopts the pharmacotherapy of regimen or oral phosphorus adsorbent to carry out; But it has been generally acknowledged that the regimen effect is relatively poor, the use of phosphorus adsorbent is essential.In recent years; Discover that the compound that contains the polyallylamine structure has good phosphorus adsorptive power (as: USP 5496545,20040191212, Chinese patent 95193521.6 etc.), crosslinked polyallylamine class medicine is evident in efficacy especially; Wherein, SEVELAMER (Sevelamer) is because of its good clinical manifestation, and granted listing is used to treat hyperphosphatemia.The structural formula of SEVELAMER is following:
Synthesizing of crosslinked polyallylamine class medicine SEVELAMER (Sevelamer); Be to react by polyallylamine hydrochloride and linking agent; Different because of used linking agent, the phosphorus adsorptive power of product has than big-difference, and the most option table chloropharin of the present document of reporting is a linking agent.For example: in USP 5496545; Introduced the synthetic of a series of crosslinked allyl amine polymers oral, that the phosphorus adsorptive power is arranged, listed linking agent has: Epicholorohydrin, 1,4-butanediol diglycidyl ether, 1; 2-ethylene glycol bisthioglycolate glycidyl ether, 1; 3-propylene dichloride, 1,2-ethylene dichloride, 1,3-dibromopropane, 1; 2-ethylene dibromide, succinyl dichloride, dimethyl succinate salt, TDI, acrylate chloride and pyromellitic dianhydride, preferred cross-linking agents is an Epicholorohydrin.U.S. Pat 5496545 described SEVELAMER building-up processes are: in the alkaline aqueous solution, polyallylamine hydrochloride and Epicholorohydrin carry out crosslinking reaction under room temperature, react agglutination thing after 18 hours; Pour into and make its curing in the Virahol; Filter, repeatedly washing back redispersion is in water, after the filtration; Be scattered in a large amount of Virahols, obtain product through filtration, drying.The required reaction times of this technology is longer, and multiple times of filtration and washing in the operating process need be used a large amount of organic solvents, and in the series product, the highest phosphorus adsorptive power is 3.1mmol/g.Embodiment 1: sevelamer hydrochloride syntheticIn the 500mL flask; Add 46.2g (0.374mol) PAH hydrochloride, the 108.0mL deionized water dissolving adds sodium hydroxide and regulates pH=10-11; Drip the toluene solution of 11.0g (0.042mol) 1-chloro-3-p-toluenesulfonyl-2-propyl alcohol, heat up in 70-75 ℃ of reaction 4 hours.Reaction finishes, and adds hydrochloric acid and regulates pH=1-2, filters, and obtains the sevelamer hydrochloride bullion.The sevelamer hydrochloride bullion is scattered in the 300.0mL deionized water, and hydro-oxidation sodium is regulated pH=10.0-11.0, filters; Deionized water wash; The gained white solid through pulverizing, gets product sevelamer hydrochloride 38.8g again 70 ℃ of vacuum-dryings 8 hours; The phosphorus adsorptive value is 5.5mmol/g, and chloride ion content is 16.5%.Embodiment 2: carbonic acid SEVELAMER syntheticIn the 500mL flask; Add 150.0g (0.404mol; Weight concentration is 30%) the PAH hydrochloride aqueous solution; Add sodium hydroxide and regulate pH=10-11, drip the acetonitrile solution of 13.0g (0.042mol) 1-bromo-3-p-toluenesulfonyl-2-propyl alcohol, heat up in 70-75 ℃ of reaction 4 hours.Reaction finishes, and adds hydrochloric acid and regulates pH=1-2, filters, and obtains the sevelamer hydrochloride bullion.The sevelamer hydrochloride bullion is scattered in the 300.0mL deionized water, adds yellow soda ash and regulate pH=8.5-9.5, filter; Deionized water wash, gained white solid are 70 ℃ of vacuum-dryings 8 hours, again through pulverizing; Get product carbonic acid SEVELAMER 36.4g, the phosphorus adsorptive value is 5.4mmol/g.Embodiment 3: carbonic acid SEVELAMER syntheticIn the 500mL flask; Add 46.2g (0.374mol) PAH hydrochloride, the 108.0mL deionized water dissolving adds sodium hydroxide and regulates pH=10-11; Drip the toluene solution of 7.9g (0.042mol) 1-chloro-3-methylsulfonyl-2-propyl alcohol, heat up in 70-75 ℃ of reaction 4 hours.Reaction finishes, and adds hydrochloric acid and regulates pH=1-2, filters, and obtains the sevelamer hydrochloride bullion.The sevelamer hydrochloride bullion is scattered in the 300.0mL deionized water, and hydro-oxidation sodium is regulated pH=12.0-12.5, feeds dioxide gas to saturated; Filter; Deionized water wash, gained white solid are 70 ℃ of vacuum-dryings 8 hours, again through pulverizing; Get product carbonic acid SEVELAMER 40.5g, the phosphorus adsorptive value is 5.0mmol/g.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Ramsdell R (June 1999). “Renagel: a new and different phosphate binder”. review. ANNA Journal. 26 (3): 346–7. PMID 10633608.
- ^ Burke SK (September 2000). “Renagel: reducing serum phosphorus in haemodialysis patients”. review. Hospital Medicine. 61 (9): 622–7. doi:10.12968/hosp.2000.61.9.1419. PMID 11048603.
- ^ Habbous S, Przech S, Acedillo R, Sarma S, Garg AX, Martin J (January 2017). “The efficacy and safety of sevelamer and lanthanum versus calcium-containing and iron-based binders in treating hyperphosphatemia in patients with chronic kidney disease: a systematic review and meta-analysis”. review. Nephrology, Dialysis, Transplantation. 32 (1): 111–125. doi:10.1093/ndt/gfw312. PMID 27651467.
- ^ Patel L, Bernard LM, Elder GJ (February 2016). “Sevelamer Versus Calcium-Based Binders for Treatment of Hyperphosphatemia in CKD: A Meta-Analysis of Randomized Controlled Trials”. review. Clinical Journal of the American Society of Nephrology. 11 (2): 232–44. doi:10.2215/CJN.06800615. PMC 4741042. PMID 26668024.
- ^ Emmett M (September 2004). “A comparison of clinically useful phosphorus binders for patients with chronic kidney failure”. review. Kidney International Supplements. 66 (90): S25–32. doi:10.1111/j.1523-1755.2004.09005.x. PMID 15296504.
- ^ Locatelli F, Del Vecchio L (May 2015). “Cardiovascular mortality in chronic kidney disease patients: potential mechanisms and possibilities of inhibition by resin-based phosphate binders”. review. Expert Review of Cardiovascular Therapy. 13 (5): 489–99. doi:10.1586/14779072.2015.1029456. PMID 25804298. S2CID 32586527.
- ^ Vlassara H, Uribarri J (January 2014). “Advanced glycation end products (AGE) and diabetes: cause, effect, or both?”. review. Current Diabetes Reports. 14 (1): 453. doi:10.1007/s11892-013-0453-1. PMC 3903318. PMID 24292971.
External links
- “Sevelamer”. Drug Information Portal. U.S. National Library of Medicine.
- “Sevelamer hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
- “Sevelamer carbonate”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Pronunciation | (/sɛˈvɛləmər/ or /sɛˈvɛləmɪər/) |
| Trade names | Renagel, Renvela |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a601248 |
| License data | EU EMA: by INNUS DailyMed: Sevelamer |
| Pregnancy category | AU: B3 |
| Routes of administration | By mouth |
| ATC code | V03AE02 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)US: ℞-onlyEU: Rx only |
| Pharmacokinetic data | |
| Bioavailability | 0% |
| Excretion | Feces 100% |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 52757-95-6 |
| PubChem CID | 3085017 |
| DrugBank | DB00658 |
| ChemSpider | 2341997 |
| UNII | 941N5DUU5C |
| KEGG | D08512 as HCl: D01983 |
| ChEMBL | ChEMBL1201492 |
| CompTox Dashboard (EPA) | DTXSID80872282 |
| Chemical and physical data | |
| Formula | [(C3H7N)a+b.(C9H17N2O)c]m where a+b:c = 9:1 |
| Molar mass | variable |
| (what is this?) (verify) |
//////////////////////SEVELAMER, HSDB 7608, GT335-012, GT 16-026A, PB 94, Antihyperphosphatemic

NEW DRUG APPROVALS
ONE TIME
$10.00
METHOCARBAMOL

Methocarbamol
- Molecular FormulaC11H15NO5
- Average mass241.240 Da
- метокарбамол , ميثوكاربامول , 美索巴莫
1,2-Propanediol, 3-(2-methoxyphenoxy)-, 1-carbamate
208-524-3[EINECS]
2-Hydroxy-3-(2-methoxyphenoxy)propyl carbamate
532-03-6[RN]
MethocarbamolCAS Registry Number: 532-03-6
CAS Name: 3-(2-Methoxyphenoxy)-1,2-propanediol 1-carbamate
Additional Names: 3-(o-methoxyphenoxy)-2-hydroxypropyl 1-carbamate; 2-hydroxy-3-(o-methoxyphenoxy)propyl 1-carbamate; guaiacol glyceryl ether carbamate
Manufacturers’ Codes: AHR-85Trademarks: Neuraxin; Miolaxene (Lepetit); Lumirelax; Etroflex; Delaxin (Ferndale); Robamol (Cenci); Traumacut (Brenner-Efeka); Tresortil; Relestrid; Robaxin (Robins)
Molecular Formula: C11H15NO5, Molecular Weight: 241.24Percent Composition: C 54.77%, H 6.27%, N 5.81%, O 33.16%
Literature References: Prepn from 3-(o-methoxyphenoxy)-2-hydroxypropyl chlorocarbonate: Murphey, US2770649 (1956 to A. H. Robins). Comprehensive description: S. Alessi-Severini et al.,Anal. Profiles Drug Subs. Excip.23, 371-399 (1994).
Properties: Crystals from benzene, mp 92-94°. uv max (water): 222, 274 nm (E1%1cm 298, 94). 1og P -0.06. Soly in water at 20°: 2.5 g/100 ml. Sol in alcohol, propylene glycol. Sparingly sol in chloroform. Practically insol in n-hexane.
Melting point: mp 92-94°
Absorption maximum: uv max (water): 222, 274 nm (E1%1cm 298, 94)
Therap-Cat: Muscle relaxant (skeletal).
Therap-Cat-Vet: Muscle relaxant (skeletal).
Keywords: Muscle Relaxant (Skeletal).
Methocarbamol, sold under the brand name Robaxin among others, is a medication used for short-term musculoskeletal pain.[3][4] It may be used together with rest, physical therapy, and pain medication.[3][5][6] It is less preferred in low back pain.[3] It has limited use for rheumatoid arthritis and cerebral palsy.[3][7] Effects generally begin within half an hour.[3] It is taken by mouth or injection into a vein.[3]
Methocarbamol is a CNS depressant indicated with rest, physical therapy and other treatments to control the discomfort associated with various acute musculoskeletal conditions.
Methocarbamol was developed in the early 1950s as a treatment for muscle spasticity and the associated pain.6,7 It is a guaiacol glyceryl ether.7
Methocarbamol tablets and intramuscular injections are prescription medicines indicated in the United States as an adjunct to rest, physical therapy, and other measures for the relief of discomforts associated with acute, painful musculoskeletal conditions.Label,9 In Canada, methocarbamol can be sold as an over the counter oral medicine at a lower dose that may be combined with acetaminophen or ibuprofen.10 A combination product with acetylsalicylic acid and codeine is available in Canada by prescription.10
Methocarbamol was FDA approved on 16 July 1957.8
Common side effect include headaches, sleepiness, and dizziness.[3][8] Serious side effects may include anaphylaxis, liver problems, confusion, and seizures.[4] Use is not recommended in pregnancy and breastfeeding.[3][4] Because of risk of injury, skeletal muscle relaxants should generally be avoided in geriatric patients.[3] Methocarbamol is a centrally acting muscle relaxant.[3] How it works is unclear, but it does not appear to affect muscles directly.[3]
Methocarbamol was approved for medical use in the United States in 1957.[3] It is available as a generic medication.[3][4] It is relatively inexpensive as of 2016.[9] In 2019, it was the 136th most commonly prescribed medication in the United States, with more than 4 million prescriptions.[10][11]
SYN
CN 109970606

SYN
Synthesis of methocarbamol from guaifenesin. (a) methocarbamol and (b) β-isomer of methocarbamol.
SYN
https://www.sciencedirect.com/science/article/abs/pii/S0957416607003801
The muscle relaxant methocarbamol 2 and tranquilizer mephenoxalone 3, as well as intermediate cyclic carbonate 4, have been prepared in enantiopure form by starting from enantiopure guaifenesin 1 easily available by an entrainment resolution procedure. Thermal investigations reveal that 2 is probably a conglomerate forming substance, 3 forms a stable racemic compound, and 4 occupies an intermediate position. The enantiomeric excess of a binary phase eutectic point for these substances comprises 0%, 85%, and 10%, respectively.
Graphical abstract

PATENT
US 2770649
https://patents.google.com/patent/US2770649A/en
PAPER
Journal of pharmaceutical sciences (1970), 59(7), 1043-4
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical use
Methocarbamol is a muscle relaxant used to treat acute, painful musculoskeletal spasms in a variety of musculoskeletal conditions.[12] However, there is limited and inconsistent published research on the medication’s efficacy and safety in treating musculoskeletal conditions, primarily neck and back pain.[12]
Methocarbamol injection may have a beneficial effect in the control of the neuromuscular spasms of tetanus.[6] It does not, however, replace the current treatment regimen.[6]
It is not useful in chronic neurological disorders, such as cerebral palsy or other dyskinesias.[3]
Currently, there is some suggestion that muscle relaxants may improve the symptoms of rheumatoid arthritis; however, there is insufficient data to prove its effectiveness as well as answer concerns regarding optimal dosing, choice of muscle relaxant, adverse effects, and functional status.[7]
Comparison to similar agents
The clinical effectiveness of methocarbamol compared to other muscle relaxants is not well-known.[12] One trial of methocarbamol versus cyclobenzaprine, a well-studied muscle relaxant, in those with localized muscle spasm found there was no significant differences in their effects on improved muscle spasm, limitation of motion, or limitation of daily activities.[12]
Contraindications
There are few contraindications to methocarbamol. They include:
- Hypersensitivity to methocarbamol or to any of the injection components.[6]
- For the injectable form, suspected kidney failure or renal pathology, due to large content of polyethylene glycol 300 that can increase pre-existing acidosis and urea retention.[6]
Side effects
Methocarbamol is a centrally acting skeletal muscle relaxant that has significant adverse effects, especially on the central nervous system.[3]
Potential side effects of methocarbamol include:
- Most commonly drowsiness, blurred vision, headache, nausea, and skin rash.[8]
- Possible clumsiness (ataxia), upset stomach, flushing, mood changes, trouble urinating, itchiness, and fever.[13][14]
- Both tachycardia (fast heart rate) and bradycardia (slow heart rate) have been reported.[14]
- Hypersensitivity reactions and anaphylatic reactions are also reported.[5][6]
- May cause respiratory depression when combined with benzodiazepines, barbiturates, codeine, or other muscle relaxants.[15]
- May cause urine to turn black, blue or green.[13]
While the product label states that methocarbamol can cause jaundice, there is minimal evidence to suggest that methocarbamol causes liver damage.[8] During clinical trials of methocarbamol, there were no laboratory measurements of liver damage indicators, such as serum aminotransferase (AST/ALT) levels, to confirm hepatotoxicity.[8] Although unlikely, it is impossible to rule out that methocarbamol may cause mild liver injury with use.[8]
Elderly
Skeletal muscle relaxants are associated with an increased risk of injury among older adults.[16] Methocarbamol appeared to be less sedating than other muscle relaxants, most notably cyclobenzaprine, but had similar increased risk of injury.[15][16] Methocarbamol is cited along with “most muscle relaxants” in the 2012 Beers Criteria as being “poorly tolerated by older adults, because of anticholinergic adverse effects, sedation, increased risk of fractures,” noting that “effectiveness dosages tolerated by older adults is questionable.”[17]
Pregnancy
Methocarbamol is labeled by the FDA as a pregnancy category C medication.[6] The teratogenic effects of the medication are not known and should be given to pregnant women only when clearly indicated.[6]
Overdose
There is limited information available on the acute toxicity of methocarbamol.[5][6] Overdose is used frequently in conjunction with CNS depressants such as alcohol or benzodiazepines and will have symptoms of nausea, drowsiness, blurred vision, hypotension, seizures, and coma.[6] There are reported deaths with an overdose of methocarbamol alone or in the presence of other CNS depressants.[5][6]
Abuse
Unlike other carbamates such as meprobamate and its prodrug carisoprodol, methocarbamol has greatly reduced abuse potential.[18] Studies comparing it to the benzodiazepine lorazepam and the antihistamine diphenhydramine, along with placebo, find that methocarbamol produces increased “liking” responses and some sedative-like effects; however, at higher doses dysphoria is reported.[18] It is considered to have an abuse profile similar to, but weaker than, lorazepam.[18]
Interactions
Methocarbamol may inhibit the effects of pyridostigmine bromide.[5][6] Therefore, methocarbamol should be used with caution in those with myasthenia gravis taking anticholinesterase medications.[6]
Methocarbamol may disrupt certain screening tests as it can cause color interference in laboratory tests for 5-hydroxy-indoleacetic acid (5-HIAA) and in urinary testing for vanillylmandelic acid (VMA) using the Gitlow method.[6]
Pharmacology
Mechanism of action
The mechanism of action of methocarbamol has not currently been established.[3] Its effect is thought to be localized to the central nervous system rather than a direct effect on skeletal muscles.[3] It has no effect on the motor end plate or the peripheral nerve fiber.[6] The efficacy of the medication is likely related to its sedative effect.[3] Alternatively, methocarbamol may act via inhibition of acetylcholinesterase, similarly to carbamate.[19]
Pharmacokinetics
In healthy individuals, the plasma clearance of methocarbamol ranges between 0.20 and 0.80 L/h/kg.[6] The mean plasma elimination half-life ranges between 1 and 2 hours, and the plasma protein binding ranges between 46% and 50%.[6] The elimination half-life was longer in the elderly, those with kidney problems, and those with liver problems.[6]
Metabolism
Methocarbamol is the carbamate derivative of guaifenesin, but does not produce guaifenesin as a metabolite, because the carbamate bond is not hydrolyzed metabolically;[8][6] its metabolism is by Phase I ring hydroxylation and O-demethylation, followed by Phase II conjugation.[6] All the major metabolites are unhydrolyzed carbamates.[20][21] Small amounts of unchanged methocarbamol are also excreted in the urine.[5][6]
Society and culture
Methocarbamol was approved as a muscle relaxant for acute, painful musculoskeletal conditions in the United States in 1957.[8] Muscle relaxants are widely used to treat low back pain, one of the most frequent health problems in industrialized countries. Currently, there are more than 3 million prescriptions filled yearly.[8] Methocarbamol and orphenadrine are each used in more than 250,000 U.S. emergency department visits for lower back pain each year.[22] In the United States, low back pain is the fifth most common reason for all physician visits and the second most common symptomatic reason.[23] In 80% of primary care visits for low back pain, at least one medication was prescribed at the initial office visit and more than one third were prescribed two or more medications.[24] The most commonly prescribed drugs for low back pain included skeletal muscle relaxants.[25] Cyclobenzaprine and methocarbamol are on the U.S. Medicare formulary, which may account for the higher use of these products.[16]
Economics
The generic formulation of the medication is relatively inexpensive, costing less than the alternative metaxalone in 2016.[26][9]
Marketing

Generic methocarbamol 750mg tablet.
Methocarbamol without other ingredients is sold under the brand name Robaxin in the U.K., U.S., Canada[27] and South Africa; it is marketed as Lumirelax in France, Ortoton in Germany and many other names worldwide.[28] In combination with other active ingredients it is sold under other names: with acetaminophen (paracetamol), under trade names Robaxacet and Tylenol Body Pain Night; with ibuprofen as Robax Platinum; with acetylsalicylic acid as Robaxisal in the U.S. and Canada.[29][30] However, in Spain the tradename Robaxisal is used for the paracetamol combination instead of Robaxacet.[citation needed] These combinations are also available from independent manufacturers under generic names.[citation needed]
Research
Although opioids are a typically first line in treatment of severe pain, several trials suggest that methocarbamol may improve recovery and decrease hospital length of stay in those with muscles spasms associated with rib fractures.[31][32][33] However, methocarbamol was less useful in the treatment of acute traumatic pain in general.[34]
Long-term studies evaluating the risk of development of cancer in using methocarbamol have not been performed.[5][6] There are currently no studies evaluating the effect of methocarbamol on mutagenesis or fertility.[5][6]
The safety and efficacy of methocarbamol has not been established in pediatric individuals below the age of 16 except in tetanus.[5][6]
References
- ^ “Robaxin-750 – Summary of Product Characteristics (SmPC)”. (emc). 8 August 2017. Retrieved 19 April 2020.
- ^ Sica DA, Comstock TJ, Davis J, Manning L, Powell R, Melikian A, Wright G (1990). “Pharmacokinetics and protein binding of methocarbamol in renal insufficiency and normals”. European Journal of Clinical Pharmacology. 39 (2): 193–4. doi:10.1007/BF00280060. PMID 2253675. S2CID 626920.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r “Methocarbamol Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists.
- ^ Jump up to:a b c d British national formulary : BNF 76 (76 ed.). Pharmaceutical Press. 2018. p. 1093. ISBN 9780857113382.
- ^ Jump up to:a b c d e f g h i “Robaxin- methocarbamol tablet, film coated”. DailyMed. 18 July 2019. Retrieved 19 April 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w x “Robaxin- methocarbamol injection”. DailyMed. 10 December 2018. Retrieved 19 April 2020.
- ^ Jump up to:a b Richards, Bethan L.; Whittle, Samuel L.; Buchbinder, Rachelle (18 January 2012). “Muscle relaxants for pain management in rheumatoid arthritis”. The Cochrane Database of Systematic Reviews. 1: CD008922. doi:10.1002/14651858.CD008922.pub2. ISSN 1469-493X. PMID 22258993.
- ^ Jump up to:a b c d e f g h “Methocarbamol”. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. National Institute of Diabetes and Digestive and Kidney Diseases. 30 January 2017. PMID 31643609.
- ^ Jump up to:a b Fine, Perry G. (2016). The Hospice Companion: Best Practices for Interdisciplinary Care of Advanced Illness. Oxford University Press. p. PT146. ISBN 978-0-19-045692-4.
- ^ “The Top 300 of 2019”. ClinCalc. Retrieved 16 October 2021.
- ^ “Methocarbamol – Drug Usage Statistics”. ClinCalc. Retrieved 16 October 2021.
- ^ Jump up to:a b c d Chou, Roger; Peterson, Kim; Helfand, Mark (August 2004). “Comparative efficacy and safety of skeletal muscle relaxants for spasticity and musculoskeletal conditions: a systematic review”. Journal of Pain and Symptom Management. 28 (2): 140–175. doi:10.1016/j.jpainsymman.2004.05.002. ISSN 0885-3924. PMID 15276195.
- ^ Jump up to:a b “Methocarbamol”. MedlinePlus. Retrieved 18 April 2020.
- ^ Jump up to:a b “Methocarbamol Side Effects: Common, Severe, Long Term”. Drugs.com. Retrieved 18 April 2020.
- ^ Jump up to:a b See, Sharon; Ginzburg, Regina (1 August 2008). “Choosing a skeletal muscle relaxant”. American Family Physician. 78 (3): 365–70. ISSN 0002-838X. PMID 18711953.
- ^ Jump up to:a b c Spence, Michele M.; Shin, Patrick J.; Lee, Eric A.; Gibbs, Nancy E. (July 2013). “Risk of injury associated with skeletal muscle relaxant use in older adults”. The Annals of Pharmacotherapy. 47 (7–8): 993–8. doi:10.1345/aph.1R735. ISSN 1542-6270. PMID 23821610. S2CID 9037478.
- ^ “Beers Criteria Medication List”. DCRI. Retrieved 18 October 2020.
- ^ Jump up to:a b c Preston KL, Wolf B, Guarino JJ, Griffiths RR (1992). “Subjective and behavioral effects of diphenhydramine, lorazepam and methocarbamol: evaluation of abuse liability”. Journal of Pharmacology and Experimental Therapeutics. 262 (2): 707–20. PMID 1501118.
- ^ PubChem. “Methocarbamol”. pubchem.ncbi.nlm.nih.gov. Retrieved 6 July 2020.
- ^ Methocarbamol. In: DRUGDEX System [intranet database]. Greenwood Village, Colorado: Thomson Healthcare; c1974–2009 [cited 2009 Feb 10].
- ^ Bruce RB, Turnbull LB, Newman JH (January 1971). “Metabolism of methocarbamol in the rat, dog, and human”. J Pharm Sci. 60 (1): 104–6. doi:10.1002/jps.2600600120. PMID 5548215.
- ^ Friedman BW, Cisewski D, Irizarry E, Davitt M, Solorzano C, Nassery A, et al. (March 2018). “A Randomized, Double-Blind, Placebo-Controlled Trial of Naproxen With or Without Orphenadrine or Methocarbamol for Acute Low Back Pain”. Annals of Emergency Medicine. 71 (3): 348–356.e5. doi:10.1016/j.annemergmed.2017.09.031. ISSN 1097-6760. PMC 5820149. PMID 29089169.
- ^ Chou, Roger; Huffman, Laurie Hoyt (2 October 2007). “Medications for Acute and Chronic Low Back Pain: A Review of the Evidence for an American Pain Society/American College of Physicians Clinical Practice Guideline”. Annals of Internal Medicine. 147 (7): 505–14. doi:10.7326/0003-4819-147-7-200710020-00008. ISSN 0003-4819. PMID 17909211. S2CID 32719708.
- ^ Cherkin, D. C.; Wheeler, K. J.; Barlow, W.; Deyo, R. A. (1 March 1998). “Medication use for low back pain in primary care”. Spine. 23 (5): 607–14. doi:10.1097/00007632-199803010-00015. ISSN 0362-2436. PMID 9530793. S2CID 23664539.
- ^ Luo, Xuemei; Pietrobon, Ricardo; Curtis, Lesley H.; Hey, Lloyd A. (1 December 2004). “Prescription of nonsteroidal anti-inflammatory drugs and muscle relaxants for back pain in the United States”. Spine. 29 (23): E531–7. doi:10.1097/01.brs.0000146453.76528.7c. ISSN 1528-1159. PMID 15564901. S2CID 72742439.
- ^ Robbins, Lawrence D. (2013). Management of Headache and Headache Medications. Springer Science & Business Media. p. PT147. ISBN 978-1-4612-2124-1.
- ^ “ROBAXIN product appearance in Canada”. ctchealth.ca. Retrieved 13 December 2021.
- ^ “Methocarbamol”. Drugs.com. Retrieved 12 May 2018.
- ^ “New Drugs and Indications Reviewed at the May 2003 DEC Meeting” (PDF). ESI Canada. Archived from the original (PDF) on 10 July 2011. Retrieved 14 November 2008.
- ^ “Tylenol Body Pain Night Overview and Dosage”. Tylenol Canada. Archived from the original (website) on 31 March 2012. Retrieved 23 April 2012.
- ^ Patanwala, Asad E.; Aljuhani, Ohoud; Kopp, Brian J.; Erstad, Brian L. (October 2017). “Methocarbamol use is associated with decreased hospital length of stay in trauma patients with closed rib fractures”. The American Journal of Surgery. 214 (4): 738–42. doi:10.1016/j.amjsurg.2017.01.003. ISSN 0002-9610. PMID 28088301.
- ^ Deloney, Lindsay; Smith, Melanie; Carter, Cassandra; Privette, Alicia; Leon, Stuart; Eriksson, Evert (January 2020). “946: Methocarbamol reduces opioid use and length of stay in young adults with traumatic rib fractures”. Critical Care Medicine. 48 (1): 452. doi:10.1097/01.ccm.0000633320.62811.06. ISSN 0090-3493.
- ^ Smith, Melanie; Deloney, Lindsay; Carter, Cassandra; Leon, Stuart; Privette, Alicia; Eriksson, Evert (January 2020). “1759: Use of methocarbamol in geriatric patients with rib fractures is associated with improved outcomes”. Critical Care Medicine. 48 (1): 854. doi:10.1097/01.ccm.0000649332.10326.98. ISSN 0090-3493.
- ^ Aljuhani, Ohoud; Kopp, Brian J.; Patanwala, Asad E. (2017). “Effect of Methocarbamol on Acute Pain After Traumatic Injury”. American Journal of Therapeutics. 24 (2): e202–6. doi:10.1097/mjt.0000000000000364. ISSN 1075-2765. PMID 26469684. S2CID 24284482.
External links
- “Methocarbamol”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Robaxin, Marbaxin, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682579 |
| License data | US DailyMed: Methocarbamol |
| Pregnancy category | AU: B2 |
| Routes of administration | By mouth, intravenous |
| ATC code | M03BA03 (WHO) M03BA53 (WHO) M03BA73 (WHO) |
| Legal status | |
| Legal status | CA: OTCUK: POM (Prescription only) [1]US: ℞-only |
| Pharmacokinetic data | |
| Metabolism | Liver |
| Elimination half-life | 1.14–1.24 hours[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 532-03-6 |
| PubChem CID | 4107 |
| IUPHAR/BPS | 6829 |
| DrugBank | DB00423 |
| ChemSpider | 3964 |
| UNII | 125OD7737X |
| KEGG | D00402 |
| ChEBI | CHEBI:6832 |
| ChEMBL | ChEMBL1201117 |
| CompTox Dashboard (EPA) | DTXSID6023286 |
| ECHA InfoCard | 100.007.751 |
| Chemical and physical data | |
| Formula | C11H15NO5 |
| Molar mass | 241.243 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
- Sica DA, Comstock TJ, Davis J, Manning L, Powell R, Melikian A, Wright G: Pharmacokinetics and protein binding of methocarbamol in renal insufficiency and normals. Eur J Clin Pharmacol. 1990;39(2):193-4. [Article]
- Bruce RB, Turnbull LB, Newman JH: Metabolism of methocarbamol in the rat, dog, and human. J Pharm Sci. 1971 Jan;60(1):104-6. [Article]
- Witenko C, Moorman-Li R, Motycka C, Duane K, Hincapie-Castillo J, Leonard P, Valaer C: Considerations for the appropriate use of skeletal muscle relaxants for the management of acute low back pain. P T. 2014 Jun;39(6):427-35. [Article]
- Crankshaw DP, Raper C: Mephenesin, methocarbamol, chlordiazepoxide and diazepam: actions on spinal reflexes and ventral root potentials. Br J Pharmacol. 1970 Jan;38(1):148-56. doi: 10.1111/j.1476-5381.1970.tb10343.x. [Article]
- Muir WW 3rd, Sams RA, Ashcraft S: The pharmacology and pharmacokinetics of high-dose methocarbamol in horses. Equine Vet J Suppl. 1992 Feb;(11):41-4. [Article]
- Authors unspecified: Methocarbamol-A New Lissive Agent. Can Med Assoc J. 1958 Dec 15;79(12):1008-9. [Article]
- O’DOHERTY DS, SHIELDS CD: Methocarbamol; new agent in treatment of neurological and neuromuscular diseases. J Am Med Assoc. 1958 May 10;167(2):160-3. [Article]
- FDA Approved Drug Products: Robaxin [Link]
- FDA Approved Drug Products: Robaxin Intramuscular Injection [Link]
- Pfizer Canada: Robax [Link]
////////////////Methocarbamol, метокарбамол , ميثوكاربامول , 美索巴莫, AHS 85, Muscle Relaxant
COC1=C(OCC(O)COC(N)=O)C=CC=C1

NEW DRUG APPROVALS
ONE TIME
$10.00
L-CARNOSINE

L-CARNOSINE
- Molecular FormulaC9H14N4O3
- Average mass226.232 Da
(2S)-2-(3-aminopropanamido)-3-(1H-imidazol-5-yl)propanoic acid
(E)-N-(3-Amino-1-hydroxypropylidene)-L-histidine [ACD/IUPAC Name]
206-169-9[EINECS], 305-84-0[RN]
8HO6PVN24Wカルノシン , Dragosine, Ignotin, Ignotine, Karnozin, L-Carnosine, N-(β-Alanyl)-L-histidine, NSC 524045, Sevitin, β-Alanylhistidine
CarnosineCAS Registry Number: 305-84-0CAS Name: b-Alanyl-L-histidine
Additional Names: ignotine
Molecular Formula: C9H14N4O3, Molecular Weight: 226.23
Percent Composition: C 47.78%, H 6.24%, N 24.77%, O 21.22%
Literature References: Naturally occurring dipeptide found in large amounts in skeletal muscle. Also present in other tissues such as brain, cardiac muscle, kidney. Water soluble antioxidant; functions as a free-radical scavenger. Isoln: Gulewitsch, Amiradzibi, Ber.33, 1902 (1900); Wolff, Wilson, J. Biol. Chem.95, 495 (1932); 109, 565 (1935). Synthesis from histidine and b-iodo- or b-nitropropionyl chloride: Baumann, Ingvaldsen, ibid.35, 271 (1918); Barger, Tutin, Biochem. J.12, 406 (1918). Later syntheses: Sifford, du Vigneaud, J. Biol. Chem.108, 753 (1935); R. A. Turner, J. Am. Chem. Soc.75, 2388 (1953); F. J. Vinick, S. Jung, J. Org. Chem.48, 392 (1983). Crystal structure: H. Itoh et al.,Acta Crystallogr.33B, 2959 (1977). Possible role in wound healing: D. E. Fischer et al.,Proc. Soc. Exp. Biol. Med.158, 402 (1978). Review of physiological properties and therapeutic potential: S. E. Gariballa, A. J. Sinclair, Age Ageing29, 207-210 (2000).
Properties: Crystals from aqueous ethanol, mp 262° (dec) (Vinick, Jung); also reported as mp 260° (capillary tube) and as mp 308-309° (Dennis bar) (Sifford, du Vigneaud). [a]D25 +21.0° (c = 1.5 in water). pK1 2.64; pK2 6.83; pK3 9.51. Alkaline reaction. One gram dissolves in 3.1 ml water at 25°.
Melting point: mp 262° (dec) (Vinick, Jung); mp 260° (capillary tube) and as mp 308-309° (Dennis bar) (Sifford, du Vigneaud)
pKa: pK1 2.64; pK2 6.83; pK3 9.51
Optical Rotation: [a]D25 +21.0° (c = 1.5 in water)
Derivative Type: Nitrate
CAS Registry Number: 5852-98-2
Molecular Formula: C9H15N5O6, Molecular Weight: 289.25
Percent Composition: C 37.37%, H 5.23%, N 24.21%, O 33.19%
Properties: Crystals, dec 222°. [a]D20 +24.1° (c = 1.5 in water). Very sol in water.
Optical Rotation: [a]D20 +24.1° (c = 1.5 in water)
Derivative Type: Hydrochloride
CAS Registry Number: 5852-99-3
Molecular Formula: C9H15ClN4O3, Molecular Weight: 262.69
Percent Composition: C 41.15%, H 5.76%, Cl 13.50%, N 21.33%, O 18.27%
Properties: Crystals, dec 245°. Very sol in water.
Derivative Type: D-Form
CAS Registry Number: 5853-00-9
Properties: Crystals, mp 260°. [a]D28 -20.4° (c = 1.5).
Melting point: mp 260°
Optical Rotation: [a]D28 -20.4° (c = 1.5)
Carnosine (beta-alanyl-L-histidine) is a dipeptide molecule, made up of the amino acids beta-alanine and histidine. It is highly concentrated in muscle and brain tissues.[citation needed] Carnosine was discovered by Russian chemist Vladimir Gulevich.[2]
Carnosine is naturally produced by the body in the liver[3] from beta-alanine and histidine. Like carnitine, carnosine is composed of the root word carn, meaning “flesh”, alluding to its prevalence in meat.[4] There are no plant-based sources of carnosine,[5] however synthetic supplements do exist.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
SYN
WO2009033754 PAGE: 98 claimed protein
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2009033754
SYN
Showa Igakkai Zasshi 1974, V34(3), P271-83
Russian Journal of General Chemistry 2007, V77(9), P1576-1579
Chemische Berichte 1961, V94, P2768-78
Farmaco, Edizione Scientifica 1968, V23(9), P859-69
Paper
Journal of the American Chemical Society 1953, V75, P2388-90
| +21.9 ° |
Conc: 3.0 g/100mL;water ; Wavlenght: 589.3 nm; Temp: 20 °C
Annali di Chimica (Rome, Italy) 1968, V58(11), P1431-4
Z. physiol. Chem. 1914, V87, P1-11
PAPER
Chemistry – A European Journal (2003), 9, (8), 1714-1723.
PAPER
Journal of Magnetic Resonance (2003), 164, (2), 256-269.

SYN
WO 2001064638
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2001064638
Example 1
(S) -2- (Cyanoacetylamino) -3- (l_ * H-imidazol-4-yl) propionic acid, sodium salt
To a solution of sodium ethoxide obtained by dissolving 5.57 g (0.24 mol) of sodium in 800 ml of ethanol was added 40.0 g (0.26 mol) of L-histidine at room temperature. After 15 minutes, 44.12 g (0.39 mol) of ethyl cyanoacetate were added and the suspension was refluxed for 16 hours. After cooling to room temperature, the mixture was filtered. The yellowish filtrate was concentrated in vacuo, the residue was slurried in ethyl acetate, filtered, washed with ethyl acetate and purified by flash chromatography on silica gel (eluent: gradient ethyl acetate → methanol / ethyl acetate 3: 1).
Yield: 28.42 g (46%)
1HNMR (DMSO- ^ 6, 00 MHz): δ = 8,28 (d, 1H); 7,45 (s, 1H); 6,7 (s, 1H); 5,5 (br. s, 1H); 4,12-4,20 (m, 1H); 3,65 (s, 2H); 2,95-3,05 (m, 1H); 2,8-2,9 (m, 1H).
13C NMR (DMSO- 6, 100 MHz): δ = 174,05; 161,09; 134,25; 131,97; 119,66; 116,43; 54,83; 29,13; 25,20.
Example 2
(• S) -2- (Cyanoacetylamino) -3- (1-δ-imidazol-4-yl) propionic acid, sodium salt
9.80 g of sodium hydride (60% in mineral oil) and 50.6 g
(0.51 mol) were added at room temperature to a suspension of 40.0 g (0.26 mol) of L-histidine in 750 ml of N, N-dimethylformamide Given methyl cyanoacetate. The mixture was heated to 155 ° C. for 2 h in an open flask and the solution thus obtained was analyzed by means of HPLC.
Histidine (8 area%) and (S) -2- (cyanoacetylamino) -3- (1H-imidazol-4-yl) propionic acid sodium salt (38 area%) were identified.
Example 3
(S) -2- (Cyanoacetylamino) -3- (l-ö r -imidazol-4-yl) propionic acid
To a solution of sodium ethoxide obtained by dissolving 4.02 g (0.175 mol) of sodium in 280 ml of ethanol, 28.27 g (0.18 mol) of L-Ηistidine were added at room temperature. The mixture was heated slowly and 30.92 g (0.27 mol) of ethyl cyanoacetate were added dropwise at a temperature of 60.degree. The mixture was heated further and the ethanol was distilled off, the amount of ethanol distilled off being continuously replaced in portions by N, N-dimethylformamide. At the end of the reaction, the temperature of the solution was 130 ° C. The mixture was stirred at this temperature for a further 2 hours. The brown reaction mixture (200 g) was cooled to 50 ° C. and 30 g of concentrated hydrochloric acid were metered in. About 70 g of solvent (Η 2O / N, N-dimethylformamide mixture) distilled off. The viscous suspension was mixed with 200 g of acetone, cooled to -10 ° C. and filtered. For recrystallization, the residue was dissolved in water and the pH was adjusted to 5.0. On cooling (<5 ° C.) a white solid precipitated out, which was filtered off, washed with ethanol and dried at 40 ° C./20 mbar.
Yield: 26.39 g (66%).
IR (KBr): v = 3421, 3240, 3149, 3059, 2970, 2255, 1653, 1551, 1396, 1107, 1088, 979, 965, 826, 786, 638 cm is “1 .
1HΝMR (DMSO-c 6 , 400 MHz): δ = 11.0 (br., 2H); 8.50 (d, 1H); 7.68 (s, 1H); 6.85 (s, 1H); 4.35-4.48 ( m, 1H); 3.68 (s, 2H); 2.92-3.03 (, 1H); 2.82-2.91 (m, 1H).
13 C NMR (DMSO- 6 , 100 MHz): δ = 172.23; 161.92; 134.55; 132.70; 116.73; 115.87; 52.80; 28.68; 25.06.
LC-MS: mlz = 223 ([M + H]), 205, 177, 156, 110.
The optical purity was determined to be> 99.8% on a sample obtained according to the above procedure. The determination was carried out by hydrolysis of the amide bond (6 N hydrochloric acid, 110 ° C., 24 h), followed by derivatization of the released histidine with trifluoroacetic anhydride and isobutyl chloroformate. A D-histidine content of <0.1% was detected by gas chromatography on a chiral stationary phase.
Example 4
L-Carnosine
To a solution of 1.90 g (7.8 mmol) of (<S) -2- (cyanoacetylamino) -3- (1H-imidazol-4-yl) propionic acid sodium salt (prepared according to Example 1) in 50 ml of ethanol / conc.
Ammonia solution (V: V- 4: 1) were given 0.3 g of rhodium / activated charcoal (5% Rh). The
The mixture was hydrogenated at 110 ° C. and 45 bar for 1 hour. The catalyst was then filtered off and the filtrate was adjusted to pH 8.2 with formic acid. After the solution had been concentrated in vacuo, the residue was suspended in 200 ml of ethanol and heated to 60 ° C. for 30 minutes. The product was filtered off, washed successively with ethanol, ethyl acetate and diethyl ether and finally dried.
Yield: 1.33 g (76%)
1H NMR (D 2 O, 400 MΗz): δ = 7.70 (s, 1Η); 6.93 (s, 1Η); 4.43-4.50 (m, 1Η); 3.20-3.28 (m, 2Η); 3.11-3.19 (m, 1H); 2.95-3.03 (m, 1H); 2.61-2.71 (m, 2H).
The optical purity was determined by the method described in Example 3 to be 99.5%.
Example 5
(S) -2- (Cyanoacetylamino) -3- (1-O-imidazol-4-yl) propionic acid methyl ester
To a solution of sodium methoxide obtained by dissolving 0.94 g (40.7 mmol; 1.95 equiv.) Of sodium in 100 ml of methanol, 5.0 g (20.4 mmol) were added at room temperature
L-histidine methyl ester dihydrochloride added. After 30 minutes, 3.03 g
(30.6 mmol) of methyl cyanoacetate were added and the mixture was left on for 16 hours
Boiled under reflux. After cooling to room temperature, the mixture was filtered.
The yellowish filtrate was concentrated in vacuo and the residue was purified by means of flash chromatography on silica gel (eluent: gradient ethyl acetate – »ethyl acetate / methanol 3: 1).
Yield: 1.51 g (31%)
1H MR (OMSO-de, 400 MHz): δ = 8.65 (d, 1H); 7.52 (s. 1H); 6.8 (s, 1H); 4.45 ^ 1.55 (m,
1H); 3,69 (s, 2H); 3,62 (s, 3H); 3,3 (br., 1H); 2,82-2,98 (m, 2H).
Example 6
L-Carnosine
1.76 g of Rh / C (0.4 mol% of pure Rh based on the starting material used) in a mixture of 94.2 g of ammonia solution (25% in H 2 O) and 62.8 g of methanol were placed in a 1 liter pressure autoclave . The autoclave was closed, the contents were heated to 90 ° C. and 40 bar hydrogen was injected. A solution of 20.0 g (0.09 mol) (* S) -2- (cyanoacetylamino) -3- (1H-imidazol-4-yl) propionic acid (prepared according to Example 3) was then within one hour Mixture 94.2 g ammonia solution (25% in Η 2O) and 62.8 g of methanol are metered in. After a one hour post-reaction at 90 ° C., the reaction mixture was cooled to room temperature. The pressure in the autoclave was released and the catalyst was filtered off over activated charcoal. An HPLC in-process analysis showed that the clear greenish reaction solution (326.2 g) contained 5.74% (m / m) carnosine, which corresponds to a selectivity of 92% with complete conversion. The reaction mixture was then concentrated to approx. 60 g on a rotary evaporator. As a result of the dropwise addition of 174 g of ethanol, a white solid precipitated out, which was filtered off and dried at 50 ° C./20 mbar.
Ausbeute: 13,0 g (64%)
1H NMR (D2O, 400 MHz): δ = 7,70 (s, 1H); 6,93 (s, 1H); 4,43-4,50 (m, 1H); 3,20-3,28 (m, 2H); 3,11-3,19 (m, 1H); 2,95-3,03 (m, 1H); 2,61-2,71 (m, 2H).
I3C NMR (D20, 100 MHz): δ = 178,58; 172,39; 136,46; 133,90; 118,37; 55,99; 36,65; 33,09; 29,74.
LC-MS: m/z = 227 ([M+H]+), 210, 192, 164, 146, 136, 110.
Example 7
L-Carnosine
In a 1 liter pressure autoclave, a solution of 10.00 g (45.0 mmol) (S) -2- (cyanoacetylamino) -3 was added to 0.88 g of Rh / C (0.4 mol% of pure Rh based on the starting material used) – (1H-imidazol-4-yl) propionic acid (prepared according to Example 3) in a mixture of 157 g conc. NΗ 3/ Methanol (m / m = 3: 2) was added. The autoclave was closed and flushed twice with 40 bar nitrogen and once with hydrogen. The mixture was heated to 90 ° C. and 40 bar hydrogen was injected. After 3 h at 90 ° C., the reaction mixture was cooled to room temperature, the autoclave was depressurized and the catalyst was separated off by filtration. An in-process analysis (HPLC) showed that the reaction solution (147.2 g) contained 6.38% (m / m) carnosine, which corresponds to a selectivity of 92% when the conversion is complete. The reaction mixture was then concentrated to 41.2 g on a rotary evaporator. 124 g of ethanol were added dropwise at room temperature and the flask was placed in a refrigerator overnight. The next day the precipitate was filtered off, washed with ethanol and dried in a drying cabinet at 40 ° C./20 mbar. 7.96 g (78%) of a slightly greenish solid with a content (HPLC) of 98.0% (m / m) were obtained.
Example 8
L-Carnosine
The procedure was as described in Example 7, with the difference that 5% Rh on aluminum oxide was used as the catalyst. Under these conditions, L-carnosine was formed with 83% selectivity.
Example 9
L-Carnosine
4.5 g of Raney cobalt (doped with 0.3% iron) in 195 g of methanol were placed in a 1 liter pressure autoclave. A solution of 30.0 g (0.135 mol) (S) -2- (cyanoacetylamino) -3- (1H-imidazol-4-yl) propionic acid (prepared according to Example 3) in 375 g ammonia solution (25% in Η O) was admitted. The autoclave was closed and flushed twice with 40 bar nitrogen. Then 45 bar of hydrogen were injected and the contents were heated to 100 ° C. within half an hour. After an after-reaction of 3 hours at 100 ° C., the reaction mixture was cooled to room temperature and the pressure in the autoclave was released. An HPLC in-process analysis showed that the reaction solution (590.8 g) contained 4.68% (mim) carnosine, which corresponds to a selectivity of 91% with complete conversion.
Example 10
L-Carnosine
In a 100 ml
pressure autoclave were to a solution of 2.0 g (9.0 mmol) (ιS) -2- (cyanoacetylamino) -3- (lH-imidazol-4-yl) propionic acid (prepared according to Example 3) in a Mixture of 25 g of ammonia solution (25% in Η 2 O) and 13 g of methanol, 1.1 g of Raney nickel (doped with 1.8% molybdenum) were added. The autoclave was closed and placed in an oil bath preheated to 100.degree. After 10 minutes, 50 bar of hydrogen were injected. After 2.5 hours at 100 ° C., the reaction mixture was
cooled to room temperature and the pressure on the autoclave was released. An HPLC in-process analysis showed that the reaction solution (39.4 g) contained 4.54% (m / m) carnosine, which, with a conversion of 99%, corresponds to a selectivity of 89%.
Example 11
L-Carnosine
In a 1 liter pressure autoclave, 4.50 g of Raney cobalt (doped with 0.3% iron) in a mixture of 285 g of conc. Ammonia / methanol (mim = 1.9: 1) submitted. The autoclave was closed and flushed twice with 40 bar nitrogen. Then 45 bar of hydrogen were injected and the mixture was heated to 100.degree. A solution of 30.0 g (0.135 mol) of (S) -2- (cyanoacetylamino) -3- (1H-imidazol-4-yl) propionic acid (prepared according to Example 3) in a mixture of 285 g was then obtained within one hour conc. Ammonia / methanol (m / m = 1.9: 1) metered in. After a one hour post-reaction at 100 ° C., the reaction mixture was cooled to room temperature. The pressure in the autoclave was released and the catalyst was filtered off. A ΗPLC in-process analysis showed that the reddish brown reaction solution (310.5 g) contained 9.57% (m / m) carnosine,
Example 12
(S) -2- (Cyanoacetylamino) -3- (3-methyl-3-ö r -imidazol-4-yl) propionic acid, sodium salt
0.50 g (2.95 mmol) of 3-methyl-L-histidine were added at 40 ° C. to a solution of 0.20 g (2.94 mmol) of sodium ethoxide in 5.60 g of ethanol. The clear solution was heated to 60 ° C. and 0.50 g (4.43 mmol) ethyl cyanoacetate was added dropwise. The mixture was refluxed for 1 hour. Then 10 mg (0.15 mmol) of imidazole were added. The ethanol was then slowly distilled off and the amount of ethanol distilled off was continuously replaced in portions by N, N-dimethylformamide. After a subsequent reaction time of 2 h at 125 ° C., the reaction mixture was carefully concentrated and the residue was purified by means of flash column chromatography on silica gel (eluent: gradient ethyl acetate → ethyl acetate / methanol 2: 1). 0.49 g (64%) of a slightly yellowish solid were obtained.
DC: Rf •= 0,46 (Ethanol/H2O 3:7).
1H NMR (DMSO-öfe, 400 MHz): δ = 7,91 (d, 1H); 7,38 (s, 1H); 6,58 (s, 1H); 3,97 (q, 1H);
3,68 (s, 2H); 3,50 (s, 3H); 3,01 (dd, 1H); 2,85 (dd, 1H).
13C NMR (DMSO-^6, 100 MHz): δ = 171,54; 160,80; 136,95; 128,68; 126,91; 116,40;
54,26; 30,65; 25,97; 25,11.
LC-MS: m/z = 237 ([M+H]+), 219, 193, 191, 176, 166, 164, 150, 109.
Example 13
(S) -2- (3-aminopropionylamino) -3- (3-methyl-3Jϊ-imidazol-4-yl) propionic acid
(= anserine)
To a solution of 0.20 g (0.77 mmol) (5) -2- (cyanoacetylamino) -3- (3-methyl-3H-imidazol-4-yl) propionic acid sodium salt (prepared according to Example 12) in 2 , 4 g of methanol and 1.6 g of ammonia solution (25% in Η 2 O), 16 mg of rhodium / Al 2 O 3 (5% Rh) were added. The mixture was hydrogenated at 85 ° C. and 50 bar for 1 hour. The catalyst was then filtered off. Anserine could be clearly detected in the filtrate by means of thin-layer chromatography, HPLC (by co-injection with a commercial reference substance) and LC-MS.
Gross yield: approx. 45%.
TLC: R f = 0.25 (ethyl acetate / methanol / Ammom ‘ ak H 2 O 43: 35: 8: 10).
LC-MS: m / z = 241 ([M + H] +), 224, 206, 180, 170, 126, 109.

SYN

Synthesis of L-carnosine from two amino acids β -alanine-amide and L-histidine
SYN
https://pubs.rsc.org/en/content/articlelanding/2019/cy/c9cy01622h
L-Carnosine (L-Car, β-alanyl-L-histidine) is a bioactive dipeptide with important physiological functions. Direct coupling of unprotected β-Ala (β-alanine) with L-His (L-histidine) mediated by an enzyme is a promising method for L-Car synthesis. In this study, a new recombinant dipeptidase (SmPepD) from Serratia marcescens with a high synthetic activity toward L-Car was identified by a genome mining approach and successfully expressed in Escherichia coli. Divalent metal ions strongly promoted the synthetic activity of SmPepD, with up to 21.7-fold increase of activity in the presence of 0.1 mM MnCl2. Higher temperature, lower pH and increasing substrate loadings facilitated the L-Car synthesis. Pilot biocatalytic syntheses of L-Car were performed comparatively in batch and continuous modes. In the continuous process, an ultra-filtration membrane reactor with a working volume of 5 L was employed for catalyst retention. The dipeptidase, SmPepD, showed excellent operational stability without a significant decrease in space–time yield after 4 days. The specific yield of L-Car achieved was 105 gCar gcatalyst−1 by the continuous process and 30.1 gCar gcatalyst−1 by the batch process. A nanofiltration membrane was used to isolate the desired product L-Car from the reaction mixture by selectively removing the excess substrates, β-Ala and L-His. As a result, the final L-Car content was effectively enriched from 2.3% to above 95%, which gave L-Car in 99% purity after ethanol precipitation with a total yield of 60.2%. The recovered substrate mixture of β-Ala and L-His can be easily reused, which will enable the economically attractive and environmentally benign production of the dipeptide L-Car.



SYNhttps://patents.google.com/patent/US20170211105A1/en
- Carnosine is a dipeptide of the amino acids beta-alanine and histidine. It is highly concentrated in muscle and brain tissues.
- [0005]
β-Alanine (or beta-alanine) is a naturally occurring beta amino acid, which is an amino acid in which the amino group is at the β-position from the carboxylate group (i.e., two atoms away). - [0006]
β-Alanine is not used in the biosynthesis of any major proteins or enzymes. It is formed in vivo by the degradation of dihydrouracil and carnosine. It is a component of the naturally occurring peptides carnosine and anserine and also of pantothenic acid (vitamin B5), which itself is a component of coenzyme A. Under normal conditions, β-alanine is metabolized into acetic acid. - [0007]
β-Alanine is the rate-limiting precursor of carnosine, which is to say carnosine levels are limited by the amount of available β-alanine, not histidine. Supplementation with β-alanine has been shown to increase the concentration of carnosine in muscles, decrease fatigue in athletes and increase total muscular work done. - [0008]
Carnosine and beta-alanine are popular dietary supplements currently produced using chemical methods. Beta-alanine is also a synthetic precursor to pantothenic acid, the essential vitamin B5. Beta-alanine can also be used as a monomer for the production of a polymeric resin (U.S. Pat. No. 4,082,730). - [0009]
Naturally, carnosine is produced exclusively in animals from beta-alanine (via uracil) and histidine. In yeasts and animals, beta-alanine is typically produced by degradation of uracil. Chemically, carnosine can be synthesized from histidine and beta-alanine derivatives. For example, the coupling of an N-(thiocarboxy) anhydride of beta-alanine with histidine has been described (Vinick et al. A simple and efficient synthesis of L-carnosine. J. Org. Chem, 1983, 48(3), pp. 392-393). - [0010]
Beta-alanine can be produced synthetically by Michael addition of ammonia to ethyl- or methyl-acrylate. This requires the use of the caustic agent ammonia and high pressures. It is also natively produced in bacteria and yeasts in small quantities. In bacteria, beta-alanine is produced by decarboxylation of aspartate. Lysates of bacteria have been used in biocatalytic production from aspartate (Patent CN104531796A). - [0011]
There remains a need in the industry for a safer, more economical system for the production of carnosine and beta-alanine.
- [0105]
The present disclosure provides methods for the biosynthetic production of beta-alanine and carnosine using engineered microorganisms of the present invention. - [0106]
In one embodiment, a method of producing beta-alanine is provided. The method comprises providing a fermentation media comprising a carbon substrate, contacting said media with a recombinant yeast microorganism expressing an engineered beta-alanine biosynthetic pathway wherein said pathway comprises an aspartate to beta-alanine conversion (pathway step a), and culturing the yeast in conditions whereby beta-alanine is produced. - [0107]
In another embodiment of the present invention, a method of producing carnosine is provided. The method comprises providing a fermentation media comprising a carbon substrate, contacting said media with a recombinant yeast microorganism expressing an engineered carnosine biosynthetic pathway wherein said pathway comprises (i) an aspartate to beta-alanine conversion (pathway step a) and (ii) a beta-alanine to carnosine conversion (pathway step b), and culturing the yeast in conditions whereby carnosine is produced. - [0108]
In another embodiment of the present invention, a method of producing carnosine via biotransformation is provided. The method comprises providing a media comprising a carbon substrate and exogenously added beta-alanine, contacting said media with a recombinant yeast microorganism expressing an engineered carnosine biosynthetic pathway wherein said pathway comprises (i) a beta-alanine to carnosine conversion (pathway step b), and culturing the yeast in conditions whereby carnosine is produced. - [0109]
Some embodiments of the present invention comprise yeast strains designated ca1 and ca2 and are derived from S. cerevisiae strain S288C. Each encodes at least 2 foreign genes under inducible Gal promoters. Strain ca1 also contains an additional gene, panM. The specific proteins encoded by each strain and their sequences, source, and accession numbers are provided in Table 1. The genes for these proteins are synthesized with yeast-optimized codon usage, assembled into singular genetic cassettes, and then inserted into the HO locus of S288C under URA2 selection. Strains ca1 and ca2 served as parent strains to derivatives comprising various heterologous genes. Ca2 served as a parent strain for ca7, ca8, ca9, ca10, ca11, ca12, ca14, ca15 in which the carnosine synthase is a different ortholog. Strain ca1 served as the parent strain to strains ca19, ca20, ca21, ca22, ca23, ca24, ca27, and ca28 in which the aspartate decarboxylase is a different ortholog. The specific proteins encoded by each strain and their sequences, source, and accession numbers are provided in Table 2. - [0110]
Aspartate, histidine, and the cofactors involved in the carnosine and beta-alanine pathway are universal to all organisms, and thus the host organism could be any genetically tractable organism (plants, animals, bacteria, or fungi). Amongst yeasts, other species such as S. pombe or P. pastoris are plausible alternatives. Within the S. cerevisiae species, other strains more amenable to large-scale productions, such as CENPalpha, may be utilized. - [0111]
The Gal promoter used in embodiments of the present invention could be replaced with constitutive promoters, or other chemically-inducible, growth phase-dependent, or stress-induced promoters. Heterologous genes of the present invention may be genomically encoded or alternatively encoded on plasmids or yeast artificial chromosomes (YACs). All genes introduced could be encoded with alternate codon usage without altering the biochemical composition of the system. All enzymes used in embodiments of the present invention have extensive orthologs in the biosphere that could be encoded as alternatives. - [0112]
Aspartate, histidine, and the cofactors involved in this pathway are universal to all organisms, and thus the host organism could be any genetically tractable organism (plants, animals, bacteria, or fungi). Among yeast, other species such as S. pombe or P. pastoris are plausible alternatives. Within the S. cerevisiae species, other strains more amenable to large scale productions, such as CENPalpha, may be preferable. The panD gene can replaced with orthologs from other bacteria. Examples include Corynebacterium glutamicum Escherichia coli, Helicobacter pylori, Tribolium castaneum, Pectobacterium carotovorum, Actinoplanes sp. SE50/110, Taoultella ornithinolytica, Methanocaldococcus jannaschii DSM 2661 and Methanocaldococcus bathoardescens. This is shown in Table 2. Carnosine synthase is natively found in mammals, birds, and reptiles. Therefore, the chicken enzyme used in ca1 and ca2 could be replaced by various orthologs. Examples include Gorilla gorilla, Falco perefrinus, Allpiucator mississsippiensis, Ailuoropoda melanoleuca, Ursus maritimus, Python bivittatus, and Orcinus orca. This is shown in Table 2.
Culture Conditions
- [0113]
The growth medium used to test for production of carnosine by the engineered strains was Teknova SC Minimal Broth with Raffinose supplemented with 1% galactose. - [0114]
A variety of purification protocols including solid phase extraction and cation exchange chromatography may be employed to purify the desired products from the culture supernatant or the yeast cell pellet fraction.
SYN


| Names | |
|---|---|
| Preferred IUPAC name(2S)-2-(3-Aminopropanamido)-3-(3H-imidazol-4-yl)propanoic acid | |
| Other namesβ-Alanyl-L-histidine | |
| Identifiers | |
| CAS Number | 305-84-0 |
| 3D model (JSmol) | Interactive imageInteractive image |
| ChEBI | CHEBI:15727 |
| ChEMBL | ChEMBL242948 |
| ChemSpider | 388363 |
| ECHA InfoCard | 100.005.610 |
| IUPHAR/BPS | 4559 |
| KEGG | C00386 |
| PubChem CID | 439224 |
| UNII | 8HO6PVN24W |
| CompTox Dashboard (EPA) | DTXSID80879594 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C9H14N4O3 |
| Molar mass | 226.236 g·mol−1 |
| Appearance | Crystalline solid |
| Melting point | 253 °C (487 °F; 526 K) (decomposition) |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Biosynthesis
Carnosine is synthesized within the body from beta-alanine and histidine. Beta-alanine is a product of pyrimidine catabolism[6] and histidine is an essential amino acid. Since beta-alanine is the limiting substrate, supplementing just beta-alanine effectively increases the intramuscular concentration of carnosine.[7][8]
Physiological effects
pH buffer
Carnosine has a pKa value of 6.83, making it a good buffer for the pH range of animal muscles.[9] Since beta-alanine is not incorporated into proteins, carnosine can be stored at relatively high concentrations (millimolar). Occurring at 17–25 mmol/kg (dry muscle),[10] carnosine (β-alanyl-L-histidine) is an important intramuscular buffer, constituting 10-20% of the total buffering capacity in type I and II muscle fibres.
Anti-oxidant
Carnosine has been proven to scavenge reactive oxygen species (ROS) as well as alpha-beta unsaturated aldehydes formed from peroxidation of cell membrane fatty acids during oxidative stress. It also buffers pH in muscle cells, and acts as a neurotransmitter in the brain. It is also a zwitterion, a neutral molecule with a positive and negative end.[citation needed]
Antiglycating
Carnosine acts as an antiglycating agent, reducing the rate of formation of advanced glycation end-products (substances that can be a factor in the development or worsening of many degenerative diseases, such as diabetes, atherosclerosis, chronic kidney failure, and Alzheimer’s disease[11]), and ultimately reducing development of atherosclerotic plaque build-up.[12][13][14]
Geroprotective
Carnosine is considered as a geroprotector.[15] Carnosine can increase the Hayflick limit in human fibroblasts,[16] as well as appearing to reduce the telomere shortening rate.[17] Carnosine may also slow aging through its anti-glycating properties (chronic glycolysis is speculated to accelerate aging).[18]
Other
Carnosine can chelate divalent metal ions.[12]
Carnosine administration has been shown to have cardioprotective properties, protecting against ischaemia-reperfusion injury, and doxorubicin-induced cardiomyopathy.[19]
Carnosine demonstrated neuroprotective effects in multiple animal studies.[20][21][22]
Research has demonstrated a positive association between muscle tissue carnosine concentration and exercise performance.[23][24][25] β-Alanine supplementation is thought to increase exercise performance by promoting carnosine production in muscle. Exercise has conversely been found to increase muscle carnosine concentrations, and muscle carnosine content is higher in athletes engaging in anaerobic exercise.[23]
Carnosine appears to protect in experimental ischemic stroke by influencing a number of mechanisms that are activated during stroke. It is a potent pH buffer and has anti matrix metalloproteinase activity, antioxidant and antiexcitotoxic properties and protects the blood brain barrier [26], [27], [28], [29], [30], [31], [32]. [33], [34], [35].
References
- ^ “C9625 L-Carnosine ~99%, crystalline”. Sigma-Aldrich.
- ^ Gulewitsch, Wl.; Amiradžibi, S. (1900). “Ueber das Carnosin, eine neue organische Base des Fleischextractes”. Berichte der Deutschen Chemischen Gesellschaft. 33 (2): 1902–1903. doi:10.1002/cber.19000330275.
- ^ Trexler, Eric T.; Smith-Ryan, Abbie E.; Stout, Jeffrey R.; Hoffman, Jay R.; Wilborn, Colin D.; Sale, Craig; Kreider, Richard B.; Jäger, Ralf; Earnest, Conrad P.; Bannock, Laurent; Campbell, Bill (2015-07-15). “International society of sports nutrition position stand: Beta-Alanine”. Journal of the International Society of Sports Nutrition. 12: 30. doi:10.1186/s12970-015-0090-y. ISSN 1550-2783. PMC 4501114. PMID 26175657.
- ^ Hipkiss, A. R. (2006). “Does chronic glycolysis accelerate aging? Could this explain how dietary restriction works?”. Annals of the New York Academy of Sciences. 1067 (1): 361–8. Bibcode:2006NYASA1067..361H. doi:10.1196/annals.1354.051. PMID 16804012. S2CID 41175541.
- ^ Alan R. Hipkiss (2009). “Chapter 3: Carnosine and Its Possible Roles in Nutrition and Health”. Advances in Food and Nutrition Research.
- ^ “beta-ureidopropionate + H2O => beta-alanine + NH4+ + CO2”. reactome. Retrieved 2020-02-08.
Cytosolic 3-ureidopropionase catalyzes the reaction of 3-ureidopropionate and water to form beta-alanine, CO2, and NH3 (van Kuilenberg et al. 2004).
- ^ Derave W, Ozdemir MS, Harris R, Pottier A, Reyngoudt H, Koppo K, Wise JA, Achten E (August 9, 2007). “Beta-alanine supplementation augments muscle carnosine content and attenuates fatigue during repeated isokinetic contraction bouts in trained sprinters”. J Appl Physiol. 103 (5): 1736–43. doi:10.1152/japplphysiol.00397.2007. PMID 17690198. S2CID 6990201.
- ^ Hill CA, Harris RC, Kim HJ, Harris BD, Sale C, Boobis LH, Kim CK, Wise JA (2007). “Influence of beta-alanine supplementation on skeletal muscle carnosine concentrations and high intensity cycling capacity”. Amino Acids. 32 (2): 225–33. doi:10.1007/s00726-006-0364-4. PMID 16868650. S2CID 23988054.
- ^ Bate-Smith, EC (1938). “The buffering of muscle in rigor: protein, phosphate and carnosine”. Journal of Physiology. 92 (3): 336–343. doi:10.1113/jphysiol.1938.sp003605. PMC 1395289. PMID 16994977.
- ^ Mannion, AF; Jakeman, PM; Dunnett, M; Harris, RC; Willan, PLT (1992). “Carnosine and anserine concentrations in the quadriceps femoris muscle of healthy humans”. Eur. J. Appl. Physiol. 64 (1): 47–50. doi:10.1007/BF00376439. PMID 1735411. S2CID 24590951.
- ^ Vistoli, G; De Maddis, D; Cipak, A; Zarkovic, N; Carini, M; Aldini, G (Aug 2013). “Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): an overview of their mechanisms of formation”. Free Radic. Res. 47: Suppl 1:3–27. doi:10.3109/10715762.2013.815348. PMID 23767955. S2CID 207517855.
- ^ Jump up to:a b Reddy, V. P.; Garrett, MR; Perry, G; Smith, MA (2005). “Carnosine: A Versatile Antioxidant and Antiglycating Agent”. Science of Aging Knowledge Environment. 2005 (18): pe12. doi:10.1126/sageke.2005.18.pe12. PMID 15872311.
- ^ Rashid, Imran; Van Reyk, David M.; Davies, Michael J. (2007). “Carnosine and its constituents inhibit glycation of low-density lipoproteins that promotes foam cell formation in vitro”. FEBS Letters. 581 (5): 1067–70. doi:10.1016/j.febslet.2007.01.082. PMID 17316626. S2CID 46535145.
- ^ Hipkiss, A. R. (2005). “Glycation, ageing and carnosine: Are carnivorous diets beneficial?”. Mechanisms of Ageing and Development. 126 (10): 1034–9. doi:10.1016/j.mad.2005.05.002. PMID 15955546. S2CID 19979631.
- ^ Boldyrev, A. A.; Stvolinsky, S. L.; Fedorova, T. N.; Suslina, Z. A. (2010). “Carnosine as a natural antioxidant and geroprotector: From molecular mechanisms to clinical trials”. Rejuvenation Research. 13 (2–3): 156–8. doi:10.1089/rej.2009.0923. PMID 20017611.
- ^ McFarland, G; Holliday, R (1994). “Retardation of the Senescence of Cultured Human Diploid Fibroblasts by Carnosine”. Experimental Cell Research. 212 (2): 167–75. doi:10.1006/excr.1994.1132. PMID 8187813.
- ^ Shao, Lan; Li, Qing-Huan; Tan, Zheng (2004). “L-Carnosine reduces telomere damage and shortening rate in cultured normal fibroblasts”. Biochemical and Biophysical Research Communications. 324 (2): 931–6. doi:10.1016/j.bbrc.2004.09.136. PMID 15474517.
- ^ Hipkiss, A. R. (2006). “Does Chronic Glycolysis Accelerate Aging? Could This Explain How Dietary Restriction Works?”. Annals of the New York Academy of Sciences. 1067 (1): 361–8. Bibcode:2006NYASA1067..361H. doi:10.1196/annals.1354.051. PMID 16804012. S2CID 41175541.
- ^ McCarty, Mark F; DiNicolantonio, James J (2014-08-04). “β-Alanine and orotate as supplements for cardiac protection”. Open Heart. 1 (1): e000119. doi:10.1136/openhrt-2014-000119. ISSN 2053-3624. PMC 4189254. PMID 25332822.
- ^ Virdi, Jasleen Kaur; Bhanot, Amritansh; Jaggi, Amteshwar Singh; Agarwal, Neha (2020-10-02). “Investigation on beneficial role of l -carnosine in neuroprotective mechanism of ischemic postconditioning in mice: possible role of histidine histamine pathway”. International Journal of Neuroscience. 130 (10): 983–998. doi:10.1080/00207454.2020.1715393. ISSN 0020-7454. PMID 31951767. S2CID 210710039.
- ^ Rajanikant, G.K.; Zemke, Daniel; Senut, Marie-Claude; Frenkel, Mark B.; Chen, Alex F.; Gupta, Rishi; Majid, Arshad (November 2007). “Carnosine Is Neuroprotective Against Permanent Focal Cerebral Ischemia in Mice”. Stroke. 38 (11): 3023–3031. doi:10.1161/STROKEAHA.107.488502. ISSN 0039-2499. PMID 17916766.
- ^ Min, Jiangyong; Senut, Marie-Claude; Rajanikant, Krishnamurthy; Greenberg, Eric; Bandagi, Ram; Zemke, Daniel; Mousa, Ahmad; Kassab, Mounzer; Farooq, Muhammad U.; Gupta, Rishi; Majid, Arshad (October 2008). “Differential neuroprotective effects of carnosine, anserine, and N -acetyl carnosine against permanent focal ischemia”. Journal of Neuroscience Research. 86 (13): 2984–2991. doi:10.1002/jnr.21744. PMC 2805719. PMID 18543335.
- ^ Jump up to:a b Culbertson, Julie Y.; Kreider, Richard B.; Greenwood, Mike; Cooke, Matthew (2010-01-25). “Effects of Beta-Alanine on Muscle Carnosine and Exercise Performance:A Review of the Current Literature”. Nutrients. 2 (1): 75–98. doi:10.3390/nu2010075. ISSN 2072-6643. PMC 3257613. PMID 22253993.
- ^ Baguet, Audrey; Bourgois, Jan; Vanhee, Lander; Achten, Eric; Derave, Wim (2010-07-29). “Important role of muscle carnosine in rowing performance”. Journal of Applied Physiology. 109 (4): 1096–1101. doi:10.1152/japplphysiol.00141.2010. ISSN 8750-7587. PMID 20671038.
- ^ Varanoske, Alyssa N.; Hoffman, Jay R.; Church, David D.; Wang, Ran; Baker, Kayla M.; Dodd, Sarah J.; Coker, Nicholas A.; Oliveira, Leonardo P.; Dawson, Virgil L.; Fukuda, David H.; Stout, Jeffrey R. (2017-09-07). “Influence of Skeletal Muscle Carnosine Content on Fatigue during Repeated Resistance Exercise in Recreationally Active Women”. Nutrients. 9 (9): 988. doi:10.3390/nu9090988. ISSN 2072-6643. PMC 5622748. PMID 28880219.
26. Kim EH, Kim ES, Shin D, Kim D, Choi S, Shin YJ, Kim KA, Noh D, Caglayan AB, Rajanikant GK, Majid A, Bae ON. Carnosine Protects against Cerebral Ischemic Injury by Inhibiting Matrix-Metalloproteinases. Int J Mol Sci. 2021 Jul 13;22(14):7495. doi: 10.3390/ijms22147495. PMID: 34299128; PMCID: PMC8306548.
27. Jain S, Kim ES, Kim D, Burrows D, De Felice M, Kim M, Baek SH, Ali A, Redgrave J, Doeppner TR, Gardner I, Bae ON, Majid A. Comparative Cerebroprotective Potential of d- and l-Carnosine Following Ischemic Stroke in Mice. Int J Mol Sci. 2020 Apr 26;21(9):3053. doi: 10.3390/ijms21093053. PMID: 32357505; PMCID: PMC7246848.
28. Kim ES, Kim D, Nyberg S, Poma A, Cecchin D, Jain SA, Kim KA, Shin YJ, Kim EH, Kim M, Baek SH, Kim JK, Doeppner TR, Ali A, Redgrave J, Battaglia G, Majid A, Bae ON. LRP-1 functionalized polymersomes enhance the efficacy of carnosine in experimental stroke. Sci Rep. 2020 Jan 20;10(1):699. doi: 10.1038/s41598-020-57685-5. PMID: 31959846; PMCID: PMC6971073.
29. Schön M, Mousa A, Berk M, Chia WL, Ukropec J, Majid A, Ukropcová B, de Courten B. The Potential of Carnosine in Brain-Related Disorders: A Comprehensive Review of Current Evidence. Nutrients. 2019 May 28;11(6):1196. doi: 10.3390/nu11061196. PMID: 31141890; PMCID: PMC6627134.
30. Davis CK, Laud PJ, Bahor Z, Rajanikant GK, Majid A. Systematic review and stratified meta-analysis of the efficacy of carnosine in animal models of ischemic stroke. J Cereb Blood Flow Metab. 2016 Oct;36(10):1686-1694. doi: 10.1177/0271678X16658302. Epub 2016 Jul 8. PMID: 27401803; PMCID: PMC5046161.
31. Baek SH, Noh AR, Kim KA, Akram M, Shin YJ, Kim ES, Yu SW, Majid A, Bae ON. Modulation of mitochondrial function and autophagy mediates carnosine neuroprotection against ischemic brain damage. Stroke. 2014 Aug;45(8):2438-2443. doi: 10.1161/STROKEAHA.114.005183. Epub 2014 Jun 17. PMID: 24938837; PMCID: PMC4211270.
32. Bae ON, Majid A. Role of histidine/histamine in carnosine-induced neuroprotection during ischemic brain damage. Brain Res. 2013 Aug 21;1527:246-54. doi: 10.1016/j.brainres.2013.07.004. Epub 2013 Jul 11. PMID: 23850642.
33. Bae ON, Serfozo K, Baek SH, Lee KY, Dorrance A, Rumbeiha W, Fitzgerald SD, Farooq MU, Naravelta B, Bhatt A, Majid A. Safety and efficacy evaluation of carnosine, an endogenous neuroprotective agent for ischemic stroke. Stroke. 2013 Jan;44(1):205-12. doi: 10.1161/STROKEAHA.112.673954. Epub 2012 Dec 18. PMID: 23250994; PMCID: PMC3678096.
34. Min J, Senut MC, Rajanikant K, Greenberg E, Bandagi R, Zemke D, Mousa A, Kassab M, Farooq MU, Gupta R, Majid A. Differential neuroprotective effects of carnosine, anserine, and N-acetyl carnosine against permanent focal ischemia. J Neurosci Res. 2008 Oct;86(13):2984-91. doi: 10.1002/jnr.21744. PMID: 18543335; PMCID: PMC2805719.
35. Rajanikant GK, Zemke D, Senut MC, Frenkel MB, Chen AF, Gupta R, Majid A. Carnosine is neuroprotective against permanent focal cerebral ischemia in mice. Stroke. 2007 Nov;38(11):3023-31. doi: 10.1161/STROKEAHA.107.488502. Epub 2007 Oct 4. PMID: 17916766.
////////L-CARNOSINE, カルノシン , b-Alanyl-L-histidine, ignotine, 8HO6PVN24W, カルノシン , Dragosine, Ignotin, Ignotine, Karnozin, L-Carnosine, N-(β-Alanyl)-L-histidine, NSC 524045, Sevitin, β-Alanylhistidine

NEW DRUG APPROVALS
one time
$10.00
FOMEPIZOLE

FOMEPIZOLE
- Molecular FormulaC4H6N2
- Average mass82.104 Da
4-Methylpyrazole, 4-MP
7554-65-6[RN]
105204[Beilstein]
1H-Pyrazole, 4-methyl-
231-445-0[EINECS]фомепизол , فوميبيزول
甲吡唑
Launched – 1998 EUSA PHARMA
Fomepizole, also known as 4-methylpyrazole, is a medication used to treat methanol and ethylene glycol poisoning.[2] It may be used alone or together with hemodialysis.[2] It is given by injection into a vein.[2]
Common side effects include headache, nausea, sleepiness, and unsteadiness.[2] It is unclear if use during pregnancy is safe for the baby.[2] Fomepizole works by blocking the enzyme that converts methanol and ethylene glycol to their toxic breakdown products.[2]
Fomepizole was approved for medical use in the United States in 1997.[2] It is on the World Health Organization’s List of Essential Medicines.[3]FomepizoleCAS Registry Number: 7554-65-6
CAS Name: 4-Methyl-1H-pyrazole
Additional Names: 4-MP
Trademarks: Antizol (Orphan Med.)
Molecular Formula: C4H6N2, Molecular Weight: 82.10
Percent Composition: C 58.52%, H 7.37%, N 34.12%
Literature References: Alcohol dehydrogenase inhibitor. Prepn: H. Pechmann, E. Burkard, Ber.33, 3590 (1900); D. S. Noyce et al.,J. Org. Chem.20, 1681 (1955); T. Momose et al.,Heterocycles30, 789 (1990). Inhibition of human liver alcohol dehydrogenase: T.-K. Li, H. Theorell, Acta Chem. Scand.23, 892 (1969). Toxicity study: G. Magnusson et al.,Experientia28, 1198 (1972). GC determn in plasma and urine: R. Achari, M. Mayersohn, J. Pharm. Sci.73, 690 (1984). Clinical pharmacology: D. Jacobsen et al.,Alcohol. Clin. Exp. Res.12, 516 (1988). Pharmacokinetics: eidem,Eur. J. Clin. Pharmacol.37, 599 (1989). Clinical trial in ethylene glycol poisoning: J. Brent et al.,N. Engl. J. Med.340, 832 (1999); in methanol poisoning: idem et al., ibid.344, 424 (2001). Review: J. Likforman et al.,J. Toxicol. Clin. Exp.7, 373-382 (1987). Review of use in methanol poisoning: M. B. Mycyk, J. B. Leikin, Am. J. Therapeut.10, 68-70 (2003).
Properties: mp 15.5-18.5°. bp18mm 98.5-99.5°; bp730 204-205°. nD22 1.4913. uv max in 95% ethanol: 220 nm (log e 3.47); in 6N HCl: 226 nm (log e 3.65). Sol in water, alcohol. LD50 (7 days) in mice, rats (mmol/kg): 3.8, 3.8 i.v.; 7.8, 6.5 orally (Magnusson).
Melting point: mp 15.5-18.5°
Boiling point: bp18mm 98.5-99.5°; bp730 204-205°
Index of refraction:nD22 1.4913
Absorption maximum: uv max in 95% ethanol: 220 nm (log e 3.47); in 6N HCl: 226 nm (log e 3.65)
Toxicity data: LD50 (7 days) in mice, rats (mmol/kg): 3.8, 3.8 i.v.; 7.8, 6.5 orally (Magnusson)
Therap-Cat: Antidote to methanol and ethylene glycol poisoning.
Therap-Cat-Vet: Antidote to ethylene glycol poisoning in dogs.
Keywords: Antidote (Methanol and Ethylene Glycol Poisoning).
Fomepizole was approved by the U.S. Food and Drug Administration (FDA) on Dec 4, 1997. It was developed and marketed as Antizol® by Paladin in the US.
Fomepizole is a competitive alcohol dehydrogenase inhibitor, Alcohol dehydrogenase catalyzes the oxidation of ethanol to acetaldehyde, and it also catalyzes the initial steps in the metabolism of ethylene glycol and methanol to their toxic metabolites. Antizol® is indicated as an antidote for ethylene glycol (such as antifreeze) or methanol poisoning, or for use in suspected ethylene glycol or methanol ingestion, either alone or in combination with hemodialysis.
Antizol® is available as injection solution for intravenous use, containing 1 g/ml of free Fomepizole. The recommended dose is 15 mg/kg should be administered, followed by doses of 10 mg/kg every 12 hours for 4 doses, then 15 mg/kg every 12 hours thereafter until ethylene glycol or methanol concentrations are undetectable or have been reduced below 20 mg/dL.
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 1997-12-04 | First approval | Antizol | Methanol or ethylene glycol poisoning | Injection | 1 g/mL | Paladin | Orphan |
SYN

SYN
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 5920-30-9 | C4H8N2 | 4,5-dihydro-4-methylpyrazole | |
| 7803-57-8 | H6N2O | hydrazine hydrate | Hydrazine, monohydrate |
| 78-85-3 | C4H6O | methacrolein | 2-propenal, 2-methyl- |
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
SYN
Reference:
US7553863B2.
https://patents.google.com/patent/US7553863B2/enEthylene glycol is commonly available as automobile radiator antifreeze. Because of its sweet taste, improperly stored antifreeze is a common source of ethylene glycol poisoning, particularly in children. Ethylene glycol is rapidly absorbed from the gastrointestinal tract. Toxicity can be divided into three stages:
- Stage 1—Neurological (0.5-12 hours post-ingestion)
- Stage 2—Cardiopulmonary (12-24 hours post-ingestion)
- Stage 3—Renal (24-72 hours post-ingestion)
4-Methylpyrazole, marketed as Antizol® (fomepizole) by Orphan Medical, Inc. is a specific antidote for the treatment of ethylene glycol poisoning. It works by inhibiting the enzyme alcohol dehydrogenase which is responsible for the conversion of ethylene glycol, which itself is relatively non-toxic, into its toxic metabolites that in turn cause the renal injury and metabolic acidosis. Antizol® is currently approved by the FDA as an antidote for ethylene glycol poisoning or suspected ethylene glycol poisoning and is recommended by poison control centers as first line therapy. See Antizol® (fomepizole) Injection, Product Monograph, Orphan Medical, Inc., 2001, the entire contents of which are hereby incorporated by reference.Methanol is commonly available in the home in automobile windshield washer fluid and as a gas line anti-icing additive. Methanol has a minor degree of direct toxicity. Its major toxicity follows its metabolism to formic acid. Antizol® is also a specific antidote for the treatment of methanol toxicity. It works by inhibiting the enzyme alcohol dehydrogenase which is responsible for the conversion of methanol into its toxic metabolites, formaldehyde and formic acid. Again, Antizol® is approved by the FDA for use in treating methanol poisoning or suspected methanol poisoning and is recommended by poison control centers as first line therapy.Known methods of preparing 4-methylpyrazole include the reaction of alpha, beta-unsaturated carbonyl compounds or diketones with hydrazine or hydrazine derivatives or the dehydrogenation of the corresponding 2-pyrazoline. See U.S. Pat. Nos. 3,200,128, 4,996,327, and 5,569,769. Other processes for preparing 4-methylpyrazole are disclosed in U.S. Pat. Nos. 6,229,022, 5,569,769, and 4,996,327.4-methylpyrazole prepared by synthetic routes employed heretofore may contain impurities and toxic by-products, including pyrazole, hydrazine, and nitrobenzaldehyde. Pyrazole, like 4-methylpyrazole, is also an inhibitor of alcohol dehydrogenase, but is more toxic than 4-methylpyrazole. Pyrazole is a known teratogen (Eisses, 1995) with 10 fold less potency against alcohol dehydrogenase (T. Li et al., Acta Chem. Scan. 1969, 23, 892-902). In addition, Ewen MacDonald published a paper in 1976 that showed pyrazole in contrast to 4-methylpyrazole has a detrimental effect on brain levels of noradrenaline (E. MacDonald, Acta Pharmacol. et Toxicol. 1976, 39, 513-524). Hydrazine and nitrobenzaldehyde are known mutagens and carcinogens (H. Kohno et al., Cancer Sci. 2005, 96, 69-76).These impurities and toxic by-products have been tolerated heretofore because methods of making ultrapure 4-methylpyrazole have not been available. The FDA has previously approved up to 0.5% pyrazole in Antizol®, but recently is requesting a higher level of purity of less than 0.1% pyrazole to qualify such high levels with animal and other studies. Therefore, while the purity of Antizol® is sufficiently high for its antidotal use in emergency medicine, such toxic impurities are not ideal. For example a pregnant woman who needs antidote therapy would risk exposure of a fetus to potentially toxic pyrazole of known teratogenicity and potentially high levels of known carcinogens. Therefore, a need exists for a 4-methylpyrzaole with even lower amounts of pyrazole and other impurities and for a synthesis of such an ultrapure 4-methylpyrazole.The process of the present invention is set forth in the following exemplary scheme:

EXAMPLE 1Preparation of 1,1-diethoxypropane 1Into a 2-liter flask under nitrogen were added 586 g (3.96 moles) of triethyl orthoformate, 46 g (56 ml, 1 mole) of ethanol, and 16 g of ammonium nitrate. Over the course of one hour 232 g (4 moles) of propionaldehyde were added with stirring. An ice bath was used as necessary to keep maintain the mixture at 30-36° C. The mixture turned yellow orange after one-third of the propionaldehyde had been added. The mixture was stirred overnight at room temperature and then brought to pH 7.5±0.2 with 10% aqueous sodium carbonate (about 30 ml). The aqueous layer was decanted, and the organic layer was distilled over sodium carbonate at atmospheric pressure to produce 124 g (81.6%) of 1.
EXAMPLE 2Preparation of 1-ethoxy-1-propene 2Into a 500 ml flask equipped with a 12″×¾″ packed column were added 0.25 g (0.0013 moles) of p-toluene sulfonic acid, followed by 241 g (1.82 moles) of 1. Nitrogen was bubbled into the mixture while 0.157 g (0.00065 moles) of bis(2-ethylhexyl)amine were added. The nitrogen flow was reduced, and the mixture was distilled to 160° C. to partially remove ethyl alcohol and 1-ethoxy-1-propene. The reaction mixture washed with 320 ml of water and then with 70 ml of water. The organic layer was dried over magnesium sulfate and filtered to produce 121 g (77.5%) of 2, bp 67-76° C., as a clear, colorless liquid. Gas chromatographic analysis showed less than 0.01% ethylvinyl ether.
EXAMPLE 3Preparation of 1,1,3,3-tetraethoxy-2-methylpropane 3Into a 5 liter flask equipped with a mechanical stirrer were added 790 g (5.34 moles) of triethyl orthoformate and 4.28 ml of boron trifluoride-diethyl etherate under a nitrogen atmosphere. Temperature was maintained at 25° C. with cooling as needed. To this mixture were added 230 g (2.67 moles) of 1-ethoxy-1-propene were added slowly and dropwise. The reaction mixture was exothermic; the temperature rose to about 35-38° C. The pot was cooled to 25° C. and stirring was continued for one hour. Solid anhydrous sodium carbonate (32.1 g, 0.3 moles) was added in one portion to the flask and stirring was continued for one hour. The mixture was filtered and the filtrate was fractionally distilled under reduced pressure. The light fraction was removed at a pot temperature of 55-60° C. at 10 mm pressure. The vacuum was improved to 3 mm and the pot temperature was permitted to rise to about 100-140° C. to produce 500 g (80%) of 3, bp 80-81° C. at 3 mm, as a clear, colorless to yellow-brown liquid.
EXAMPLE 4Preparation of 4-methylpyrazoleInto a 5 liter flask equipped with a mechanical stirrer were added 1750 ml of sterile USP water to which 266.7 g (2.05 moles) of hydrazine hydrosulfate were added gradually over one hour with stirring. To the above mixture was added dropwise 481 g (2.053 moles) of 3 and the reaction mixture was warmed to 80° C. Heating and stirring were maintained for 3 hours, the flask was cooled to 40° C., and the volatile components were distilled off under a reduced pressure of about 125 mm. The resulting mixture was cooled to 10° C. first with water and then with glycol; 20 ml of water were added to the flask, and cooling was continued to a temperature of 3° C. Thereafter 50% sodium hydroxide solution was added with cooling so as to maintain the temperature below 30° C. The pH of the reaction mixture should be between 4 and 6. A solution of sodium bicarbonate containing 4.9 g of sodium bicarbonate to 55 ml of water was added to the flask. Additional sodium bicarbonate solution was added until the pH reached 7.0. The flask temperature was allowed to rise to 27° C. with continued stirring. The contents of the flask were extracted with ethyl acetate and the aqueous layer was separated. The organic layer was dried over magnesium sulfate, filtered, and the extract was distilled under vacuum. The light fraction was removed at a pot temperature of 55-60° C. at 125 mm pressure. The vacuum was improved to 5 mm for the remainder of the distillation; pot temperatures were permitted to rise to 100-110° C. to produce 134.8 g (84% based on 3) of 4-methylpyrazole, bp 77-80° C. at 5 mm, as a clear, colorless to yellow liquid. Gas chromatographic analysis showed less than 0.1% pyrazole and less than 10 ppm hydrazine.
SYN
Syn
Journal of the American Chemical Society (1949), 71, 3994-4000.
SYN
Journal of Organic Chemistry (1962), 27, 2415-19.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Fomepizole is an alcohol dehydrogenase inhibitor originally commercialized in 1998 by Orphan Medical as an antidote for ethylene glycol (such as antifreeze) or methanol poisoning, or for use in suspected ethylene glycol or methanol ingestion, either alone or in combination with hemodialysis. In January 2015, Takeda launched the product for the treatment of ethylene glycol and methanol poisoning in Japan. Raptor Pharmaceuticals (currently Horizon Therapeutics) was evaluating the compound in phase II clinical studies for the treatment of the symptoms associated with alcohol intolerance due to ALDH2 deficiency; however, no recent developments have been reported. The compound has been licensed to Paladin and Swedish Orphan Biovitrum (formerly Swedish Orphan). Prior to being acquired by Alliance Pharma in 2010, Cambridge Laboratories obtained a license to fomepizole. In 2005, Orphan Medical was acquired by Jazz Pharmaceuticals. In 2011, Takeda licensed the product from Paladin for development and commercialization rights in Japan. In 2015, orphan drug designation in Australia was assigned to the compound for the treatment of ethylene glycol and methanol poisonings. In 2015, the product was acquired by EUSA Pharma from Jazz Pharmaceuticals for the treatment of poisoning. In 2021, the compound was granted orphan drug designation in the U.S. for the treatment of acetaminophen overdose.
NMR

| Chemical structure of fomepizole | |
| Clinical data | |
|---|---|
| Pronunciation | /ˌfoʊˈmɛpɪzoʊl/ |
| Trade names | Antizol, others |
| Other names | 4-Methylpyrazole |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Fomepizole |
| Routes of administration | Intravenous |
| ATC code | V03AB34 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 7554-65-6 |
| PubChem CID | 3406 |
| DrugBank | DB01213 |
| ChemSpider | 3289 |
| UNII | 83LCM6L2BY |
| KEGG | D00707 |
| ChEBI | CHEBI:5141 |
| ChEMBL | ChEMBL1308 |
| CompTox Dashboard (EPA) | DTXSID3040649 |
| ECHA InfoCard | 100.028.587 |
| Chemical and physical data | |
| Formula | C4H6N2 |
| Molar mass | 82.106 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Density | 0.99 g/cm3 |
| Boiling point | 204 to 207 °C (399 to 405 °F) (at 97,3 kPa) |
| showSMILES | |
| show |
Medical use
Fomepizole is used to treat ethylene glycol and methanol poisoning. It acts to inhibit the breakdown of these toxins into their active toxic metabolites. Fomepizole is a competitive inhibitor of the enzyme alcohol dehydrogenase,[4] found in the liver. This enzyme plays a key role in the metabolism of ethylene glycol, and of methanol.
- Ethylene glycol is first metabolized to glycolaldehyde by alcohol dehydrogenase. Glycolaldehyde then undergoes further oxidation to glycolate, glyoxylate, and oxalate. Glycolate and oxalate are the primary toxins responsible for the metabolic acidosis, and for the renal damage, seen in ethylene glycol poisoning.
- Methanol is first metabolized to formaldehyde by alcohol dehydrogenase. Formaldehyde then undergoes further oxidation, via formaldehyde dehydrogenase, to become formic acid.[5] Formic acid is the primary toxin responsible for the metabolic acidosis, and for the visual disturbances, associated with methanol poisoning.
By competitively inhibiting the first enzyme, alcohol dehydrogenase, in the metabolism of ethylene glycol and methanol, fomepizole slows the production of the toxic metabolites. The slower rate of metabolite production allows the liver to process and excrete the metabolites as they are produced, limiting the accumulation in tissues such as the kidney and eye. As a result, much of the organ damage is avoided.[6]
Fomepizole is most effective when given soon after ingestion of ethylene glycol or methanol. Delaying its administration allows for the generation of harmful metabolites.[6]
Interaction with alcohol
Concurrent use with ethanol is contraindicated because fomepizole is known to prolong the half-life of ethanol via inhibiting its metabolism. Extending the half-life of ethanol may increase and extend the intoxicating effects of ethanol, allowing for greater (potentially dangerous) levels of intoxication at lower doses. Fomepizole slows the production of acetaldehyde by inhibiting alcohol dehydrogenase, which in turn allows more time to further convert acetaldehyde into acetic acid by acetaldehyde dehydrogenase. The result is a patient with a prolonged and deeper level of intoxication for any given dose of ethanol, and reduced “hangover” symptoms (since these adverse symptoms are largely mediated by acetaldehyde build up).
In a chronic alcoholic who has built up a tolerance to ethanol, this removes some of the disincentives to ethanol consumption (“negative reinforcement“) while allowing them to become intoxicated with a lower dose of ethanol. The danger is that the alcoholic will then overdose on ethanol (possibly fatally). If alcoholics instead very carefully reduce their doses to reflect the now slower metabolism, they may get the “rewarding” stimulus of intoxication at lower doses with less adverse “hangover” effects – leading potentially to increased psychological dependency. However, these lower doses may therefore produce less chronic toxicity and provide a harm minimization approach to chronic alcoholism.
It is, in essence, the antithesis of a disulfiram approach which tries to increase the buildup of acetaldehyde resulting in positive punishment for the patient. Compliance, and adherence, is a substantial problem in disulfiram-based approaches. Disulfiram also has a considerably longer half-life than that of fomepizole, requiring the person to not drink ethanol in order to avoid severe effects. If the person is not adequately managed on a benzodiazepine, barbiturate, acamprosate, or another GABAA receptor agonist, the alcohol withdrawal syndrome, and its attendant, life-threatening risk of delirium tremens “DT”, may occur. Disulfiram treatment should never be initiated until the risk of DT has been evaluated, and mitigated appropriately. Fomepizole treatment may be initiated while the DT de-titration sequence is still being calibrated based upon the person’s withdrawal symptoms and psychological health.[citation needed]
Adverse effects
Common side effects associated with fomepizole use include headache and nausea.[7]
Kinetics
Absorption and distribution
Fomepizole distributes rapidly into total body water. The volume of distribution is between 0.6 and 1.02 L/kg. The therapeutic concentration is from 8.2 to 24.6 mg (100 to 300 micromoles) per liter. Peak concentration following single oral doses of 7 to 50 mg/kg of body weight occurred in 1 to 2 hours. The half-life varies with dose, so has not been calculated.
Metabolism and elimination
Hepatic; the primary metabolite is 4-carboxypyrazole (about 80 to 85% of an administered dose). Other metabolites include the pyrazoles 4-hydroxymethylpyrazole and the N -glucuronide conjugates of 4-carboxypyrazole and 4-hydroxymethylpyrazole.
Following multiple doses, fomepizole rapidly induces its own metabolism via the cytochrome P450 mixed-function oxidase system.
In healthy volunteers, 1.0 to 3.5% of an administered dose was excreted unchanged in the urine. The metabolites also are excreted unchanged in the urine.
Fomepizole is dialyzable.
Other uses
Apart from medical uses, the role of 4-methylpyrazole in coordination chemistry has been studied.[8]
References
- ^ “Antizol- fomepizole injection”. DailyMed. Retrieved 24 December 2020.
- ^ Jump up to:a b c d e f g “Fomepizole”. The American Society of Health-System Pharmacists. Archived from the original on 21 December 2016. Retrieved 8 December 2016.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Casavant MJ (January 2001). “Fomepizole in the treatment of poisoning”. Pediatrics. 107 (1): 170–171. doi:10.1542/peds.107.1.170. PMID 11134450.
- ^ “Forensic Pathology”. Archived from the original on 2008-09-17.
- ^ Jump up to:a b Brent, J (May 2009). “Fomepizole for Ethylene Glycol and Methanol Poisoning”. N. Engl. J. Med. 360 (21): 2216–23. doi:10.1056/NEJMct0806112. PMID 19458366.
- ^ Lepik, KJ; Levy, AR; Sobolev, BG; Purssell, RA; DeWitt, CR; Erhardt, GD; Kennedy, JR; Daws, DE; Brignall, JL (April 2009). “Adverse drug events associated with the antidotes for methanol and ethylene glycol poisoning: a comparison of ethanol and fomepizole”. Annals of Emergency Medicine. 53 (4): 439–450.e10. doi:10.1016/j.annemergmed.2008.05.008. PMID 18639955.
- ^ Vos, Johannes G.; Groeneveld, Willem L. (1979). “Pyrazolato and related anions. Part V. Transition metal salts of 4-methylpyrazole”. Transition Metal Chemistry. 4 (3): 137–141. doi:10.1007/BF00619054. S2CID 93580021.
External links
- “Fomepizole”. Drug Information Portal. U.S. National Library of Medicine.
/////////////FOMEPIZOLE, фомепизол , فوميبيزول ,甲吡唑 , 4 MP

NEW DRUG APPROVALS
ONE TIME
$10.00
ALLOPURINOL

ALLUPURINOL
- Molecular FormulaC5H4N4O
- Average mass136.111 Da
- аллопуринол [Russian]ألوبيرينول [Arabic]别嘌醇 [Chinese]
1H-Pyrazolo(3,4-d)pyrimidin-4-ol
2,5-Dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one
206-250-9[EINECS], 315-30-0[RN]
4H-Pyrazolo[3,4-d]pyrimidin-4-one, 1,5-dihydro-, radical ion(1+)
4H-Pyrazolo[3,4-d]pyrimidin-4-one, 1,7-dihydro-
691008-24-9[RN]
7H-Pyrazolo[3,4-d]pyrimidin-4-ol
Allopurinol is a medication used to decrease high blood uric acid levels.[2] It is specifically used to prevent gout, prevent specific types of kidney stones and for the high uric acid levels that can occur with chemotherapy.[3][4] It is taken by mouth or injected into a vein.[4]
Common side effects when used by mouth include itchiness and rash.[4] Common side effects when used by injection include vomiting and kidney problems.[4] While not recommended historically, starting allopurinol during an attack of gout appears to be safe.[5][6] In those already on the medication, it should be continued even during an acute gout attack.[5][3] While use during pregnancy does not appear to result in harm, this use has not been well studied.[1] Allopurinol is in the xanthine oxidase inhibitor family of medications.[4]
Allopurinol was approved for medical use in the United States in 1966.[4] It is on the World Health Organization’s List of Essential Medicines, the safest and most effective medicines needed in a health system.[7] Allopurinol is available as a generic medication.[4] In 2019, it was the 43rd most commonly prescribed medication in the United States, with more than 15 million prescriptions.[8][9]
ALLUPRINOLCAS Registry Number: 315-30-0
CAS Name: 1,5-Dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one
Additional Names: 1H-pyrazolo[3,4-d]pyrimidin-4-ol; 4-hydroxypyrazolo[3,4-d]pyrimidine; HPP
Manufacturers’ Codes: BW-56158
Trademarks: Adenock (Mitsubishi); Allurit (Aventis); Aloral (Lagap); Alositol (Tanabe); Allo-Puren (Isis); Allozym (Sawai); Allural (Rovi); Anoprolin (Azwell); Anzief (Nippon Chemiphar); Apulonga (Dorsch); Apurol (Siegfried); Apurin (GEA); Bleminol (Gepepharm); Caplenal (Teva); Cellidrin (Hennig); Cosuric (DDSA); Dabroson (Hoyer); Embarin (Merckle); Epidropal (Teofarma); Foligan (DESMA); Gichtex (Gerot); Hamarin (Roche); Hexanurat (Durascan); Ketanrift (Ohta); Lopurin (Abbott); Lysuron (Roche); Miniplanor (Galen); Monarch (SS Pharm.); Remid (TAD); Riball (Schering AG); Sigapurol (Siegfried); Suspendol (Merckle); Takanarumin (Takata); Uricemil (Molteni); Uripurinol (Azupharma); Urosin (Roche); Urtias (Novartis); Zyloprim (GSK); Zyloric (GSK)
Molecular Formula: C5H4N4O, Molecular Weight: 136.11
Percent Composition: C 44.12%, H 2.96%, N 41.16%, O 11.75%
Literature References: Xanthine oxidase inhibitor; decreases uric acid production. Prepn: Robins, J. Am. Chem. Soc.78, 784 (1956); Schmidt, Druey, Helv. Chim. Acta39, 986 (1956); Druey, Schmidt, US2868803 (1959 to Ciba); GB798646 (1958 to Wellcome Found.); Hitchings, Falco, US3474098 (1969 to Burroughs Wellcome). Physiological and biochemical studies: Hitchings, in Biochem. Aspects Antimetab. Drug Hydroxylation, D. Shugar, Ed. (Academic Press, London, 1969) pp 11-22, C.A.75, 3531h (1971). Clinical trial in treatment of renal calculi: M. J. V. Smith, J. Urol.117, 690 (1977); B. Ettinger et al.,N. Engl. J. Med.315, 1386 (1986). Use in hyperuricemia and gout: G. R. Boss, J. E. Seegmiller, ibid.300, 1459 (1977). Effect on renal function in treatment of gout: T. Gibson, Ann. Rheum. Dis.41, 59 (1982). Comprehensive description: S. A. Benezra, T. R. Bennett, Anal. Profiles Drug Subs.7, 1-17 (1978).
Properties: Crystals, mp above 350°. uv max (0.1N NaOH): 257 nm (e 7200); (0.1N HCl): 250 nm (e 7600); (methanol): 252 nm (e 7600). Soly in mg/ml at 25°: water 0.48; n-octanol <0.01; chloroform 0.60; ethanol 0.30; DMSO 4.6. pKa 10.2.
Melting point: mp above 350°
pKa: pKa 10.2
Absorption maximum: uv max (0.1N NaOH): 257 nm (e 7200); (0.1N HCl): 250 nm (e 7600); (methanol): 252 nm (e 7600)
Derivative Type: Sodium salt
CAS Registry Number: 17795-21-0
Trademarks: Aloprim (Nabi)
Molecular Formula: C5H3N4NaO, Molecular Weight: 158.09Percent Composition: C 37.99%, H 1.91%, N 35.44%, Na 14.54%, O 10.12%
Properties: White amorphous mass. pKa 9.31.
pKa: pKa 9.31
Therap-Cat: Treatment of hyperuricemia and chronic gout. Antiurolithic.
Keywords: Antigout; Antiurolithic; Xanthine Oxidase Inhibitor.
Synthesis ReferenceDruey, J. and Schmidt, P.; US. Patent 2868,803; January 13,1959; assigned to Ciba Pharmaceutical Products Inc. Hitchings, G.H. and Falco, EA.; U.S. Patent 3,474,098; October 21,1969; assigned to Bur- roughs Wellcome & Co. Cresswell, R.M.and Mentha, J.W.; US.Patent4,146,713; March27,1979; assigned to Bur- roughs Wellcome & Co.
SYN

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
SYN

http://drugsynthesis.blogspot.co.uk/2011/11/laboratory-synthesis-of-allopurinol.html
Reference(s):
- US 2 868 803 (Ciba; 13.1.1959; CH-prior. 10.2.1956).
- DAS 1 720 024 (Wellcome Found; appl. 12.7.1967; GB-prior. 14.7.1966).
Similar process:
- DAS 1 904 894 (Wellcome Found; appl. 31.1.1969; GB-prior. 2.2.1968).
- US 4 146 713 (Burroughs Wellcome; 27.3.1979; GB-prior. 2.2.1968).
Alternative syntheses:
- US 3 474 098 (Burroughs Wellcome; 21.10.1969; prior. 29.3.1956).
- DAS 2 224 382 (Henning Berlin; appl. 18.5.1972).
- DE 1 118 221 (Wellcome Found; appl. 4.8.1956; GB-prior. 10.8.1955).
- DAS 1 814 082 (Wellcome Found; appl. 11.12.1968).
- DAS 1 950 075 (Henning Berlin; appl. 3.10.1969).
SYNCondensation of hydrazine with ethoxymethylenemalononitrile (I) leads to 3-amino-4-cyanopyrazole (II), which, by hydrolysis with sulphuric acid, gives the corresponding amide (III); heating III with formamide in excess results in allopurinol (IV). The synthesis of allopurinol can be illustrated as below:

SYN
Synthesis

IR
https://www.sciencedirect.com/science/article/abs/pii/S0099542808600878
Infrared Spectrum The infrared spectrum of allopurinol is shown in Figure 1 . in KBr with a Perkin Elmer model 457 infrared spectrophotometer. with the structure of allopurinol . It was taken as a 0.2% dispersion of allopurinol Table I gives the infrareg assignments consistent Table I Infrared Spectral Assignments for Allopurinol Frequency (cm-l) Assignment
3060 CH stretching vibrations of the pyrimidine ring
1700 CO stretching vibration of the keto form of the 4-hydroxy tautomer 1
590 ring vibrations
1245 CH in-plane deformation




NMR

| Clinical data | |
|---|---|
| Trade names | Zyloprim, Caplenal, Zyloric, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682673 |
| License data | US DailyMed: Allopurinol |
| Pregnancy category | AU: B2[1] |
| Routes of administration | By mouth (tablet), intravenous |
| ATC code | M04AA01 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)UK: POM (Prescription only)US: ℞-only |
| Pharmacokinetic data | |
| Bioavailability | 78±20% |
| Protein binding | Negligible |
| Metabolism | liver (80% oxipurinol, 10% allopurinol ribosides) |
| Elimination half-life | 2 h (oxipurinol 18–30 h) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 315-30-0 |
| PubChem CID | 135401907 |
| IUPHAR/BPS | 6795 |
| DrugBank | DB00437 |
| ChemSpider | 2010 |
| UNII | 63CZ7GJN5I |
| KEGG | D00224 |
| ChEBI | CHEBI:40279 |
| ChEMBL | ChEMBL1467 |
| CompTox Dashboard (EPA) | DTXSID4022573 |
| ECHA InfoCard | 100.005.684 |
| Chemical and physical data | |
| Formula | C5H4N4O |
| Molar mass | 136.114 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
Gout
Allopurinol is used to reduce urate formation in conditions where urate deposition has already occurred or is predictable. The specific diseases and conditions where it is used include gouty arthritis, skin tophi, kidney stones, idiopathic gout; uric acid lithiasis; acute uric acid nephropathy; neoplastic disease and myeloproliferative disease with high cell turnover rates, in which high urate levels occur either spontaneously, or after cytotoxic therapy; certain enzyme disorders which lead to overproduction of urate, for example: hypoxanthine-guanine phosphoribosyltransferase, including Lesch–Nyhan syndrome; glucose 6-phosphatase including glycogen storage disease; phosphoribosyl pyrophosphate synthetase, phosphoribosyl pyrophosphate amidotransferase; adenine phosphoribosyltransferase.
It is also used to treat kidney stones caused by deficient activity of adenine phosphoribosyltransferase.
Tumor lysis syndrome
Allopurinol was also commonly used to treat tumor lysis syndrome in chemotherapeutic treatments, as these regimens can rapidly produce severe acute hyperuricemia;[10] however, it has gradually been replaced by urate oxidase therapy.[11] Intravenous formulations are used in this indication when people cannot take medicine by mouth.[12]
Inflammatory bowel disease
Allopurinol cotherapy is used to improve outcomes for people with inflammatory bowel disease and Crohn’s disease who do not respond to thiopurine monotherapy.[13][14] Cotherapy has also been shown to greatly improve hepatoxicity side effects in treatment of IBD.[15] Cotherapy invariably requires dose reduction of the thiopurine, usually to one-third of the standard dose depending upon the patient’s genetic status for thiopurine methyltransferase.[16]
Psychiatric disorders
Allopurinol has been tested as an augmentation strategy for the treatment of mania in bipolar disorder. Meta-analytic evidence showed that adjunctive allopurinol was superior to placebo for acute mania (both with and without mixed features).[17] Its efficacy was not influenced by dosage, follow-up duration, or concurrent standard treatment.[17]
Side effects
Because allopurinol is not a uricosuric, it can be used in people with poor kidney function. However, for people with impaired kidney function, allopurinol has two disadvantages. First, its dosing is complex.[18] Second, some people are hypersensitive to the drug; therefore, its use requires careful monitoring.[19][20]
Allopurinol has rare but potentially fatal adverse effects involving the skin. The most serious adverse effect is a hypersensitivity syndrome consisting of fever, skin rash, eosinophilia, hepatitis, and worsened renal function, collectively referred to as DRESS syndrome.[19] Allopurinol is one of the drugs commonly known to cause Stevens–Johnson syndrome and toxic epidermal necrolysis, two life-threatening dermatological conditions.[19] More common is a less-serious rash that leads to discontinuing this drug.[19]
More rarely, allopurinol can also result in the depression of bone marrow elements, leading to cytopenias, as well as aplastic anemia. Moreover, allopurinol can also cause peripheral neuritis in some patients, although this is a rare side effect. Another side effect of allopurinol is interstitial nephritis.[21]
Allopurinol should not be given to people who are allergic to it.[10]
Drug interactions
Drug interactions are extensive, and are as follows:[10]
- Azathioprine and 6-mercaptopurine: Azathioprine is metabolised to 6-mercaptopurine which in turn is inactivated by the action of xanthine oxidase – the target of allopurinol. Giving allopurinol with either of these drugs at their normal dose will lead to overdose of either drug; only one-quarter of the usual dose of 6-mercaptopurine or azathioprine should be given;
- Didanosine: plasma didanosine Cmax and AUC values were approximately doubled with concomitant allopurinol treatment; it should not be co-administered with allopuroinol and if it must be, the dose of should be reduced and the person should be closely monitored.
Allopurinol may also increase the activity or half-life of the following drugs, in order of seriousness and certainty of the interaction:[10]
- Ciclosporin
- Coumarin anticoagulants, such as warfarin (reported rarely, but is serious when it occurs)
- Vidarabine
- Chlorpropamide
- Phenytoin
- Theophylline
- Cyclophosphamide, doxorubicin, bleomycin, procarbazine, mechlorethamine
Co-administration of the following drugs may make allopurinol less active or decrease its half-life:[10]
- Salicylates and medicines that increase the secretion of uric acid
- furosemide (see more on diuretics below)
Co-administration of the following drugs may cause hypersensitivity or skin rash:[10]
- Ampicillin and amoxicillin
- Diuretics, in particular thiazides, especially in renal impairment
- Angiotensin-converting-enzyme inhibitors (ACE inhibitors)
Pharmacology
A common misconception is that allopurinol is metabolized by its target, xanthine oxidase, but this action is principally carried out by aldehyde oxidase.[22] The active metabolite of allopurinol is oxipurinol, which is also an inhibitor of xanthine oxidase. Allopurinol is almost completely metabolized to oxipurinol within two hours of oral administration, whereas oxipurinol is slowly excreted by the kidneys over 18–30 hours. For this reason, oxipurinol is believed responsible for the majority of allopurinol’s effect.[23]
Mechanism of action
Allopurinol is a purine analog; it is a structural isomer of hypoxanthine (a naturally occurring purine in the body) and is an inhibitor of the enzyme xanthine oxidase.[2] Xanthine oxidase is responsible for the successive oxidation of hypoxanthine and xanthine, resulting in the production of uric acid, the product of human purine metabolism.[2] In addition to blocking uric acid production, inhibition of xanthine oxidase causes an increase in hypoxanthine and xanthine. While xanthine cannot be converted to purine ribotides, hypoxanthine can be salvaged to the purine ribotides adenosine and guanosine monophosphates. Increased levels of these ribotides may cause feedback inhibition of amidophosphoribosyl transferase, the first and rate-limiting enzyme of purine biosynthesis. Allopurinol, therefore, decreases uric acid formation and may also inhibit purine synthesis.[24]
Pharmacogenetics
The HLA-B*5801 allele is a genetic marker for allopurinol-induced severe cutaneous adverse reactions, including Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN).[25][26] The frequency of the HLA-B*5801 allele varies between ethnicities: Han Chinese and Thai populations have HLA-B*5801 allele frequencies of around 8%, as compared to European and Japanese populations, who have allele frequencies of around 1.0% and 0.5%, respectively.[27] The increase in risk for developing allopurinol-induced SJS or TEN in individuals with the HLA-B*5801 allele (as compared to those who do not have this allele) is very high, ranging from a 40-fold to a 580-fold increase in risk, depending on ethnicity.[25][26] As of 2011 the FDA-approved drug label for allopurinol did not contain any information regarding the HLA-B*5801 allele, though FDA scientists did publish a study in 2011 which reported a strong, reproducible and consistent association between the allele and allopurinol-induced SJS and TEN.[28] However, the American College of Rheumatology recommends screening for HLA-B*5801 in high-risk populations (e.g. Koreans with stage 3 or worse chronic kidney disease and those of Han Chinese and Thai descent), and prescribing patients who are positive for the allele an alternative drug.[29] The Clinical Pharmacogenetics Implementation Consortium guidelines state that allopurinol is contraindicated in known carriers of the HLA-B*5801 allele.[30][31]
History
Allopurinol was first synthesized and reported in 1956 by Roland K. Robins (1926-1992), in a search for antineoplastic agents.[2][32] Because allopurinol inhibits the breakdown (catabolism) of the thiopurine drug mercaptopurine, and it was later tested by Wayne Rundles, in collaboration with Gertrude Elion‘s lab at Wellcome Research Laboratories to see if it could improve treatment of acute lymphoblastic leukemia by enhancing the action of mercaptopurine.[2][33] However, no improvement in leukemia response was noted with mercaptopurine-allopurinol co-therapy, so that work turned to other compounds and the team then started testing allopurinol as a potential for gout.[34] Allopurinol was first marketed as a treatment for gout in 1966.[33]
Society and culture

Pure allopurinol is a white powder.
Formulations
Allopurinol is sold as an injection for intravenous use[12] and as a tablet.[10]
Brands
Allopurinol has been marketed in the United States since 19 August 1966, when it was first approved by FDA under the trade name Zyloprim.[35] Allopurinol was marketed at the time by Burroughs-Wellcome. Allopurinol is a generic drug sold under a variety of brand names, including Allohexal, Allosig, Milurit, Alloril, Progout, Ürikoliz, Zyloprim, Zyloric, Zyrik, and Aluron.[36]
See also
- Lesinurad/allopurinol, a fixed-dose combination drug
- Hydroxychavicol, potent xanthine oxidase inhibitor
References
- ^ Jump up to:a b “Allopurinol Use During Pregnancy”. Drugs.com. Archived from the original on 20 August 2016. Retrieved 20 December 2016.
- ^ Jump up to:a b c d e Pacher P, Nivorozhkin A, Szabó C (March 2006). “Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol”. Pharmacological Reviews. 58 (1): 87–114. doi:10.1124/pr.58.1.6. PMC 2233605. PMID 16507884.
- ^ Jump up to:a b World Health Organization (2009). Stuart MC, Kouimtzi M, Hill SR (eds.). WHO Model Formulary 2008. World Health Organization. p. 39. hdl:10665/44053. ISBN 9789241547659.
- ^ Jump up to:a b c d e f g “Allopurinol”. The American Society of Health-System Pharmacists. Archived from the original on 29 April 2016. Retrieved 8 December 2016.
- ^ Jump up to:a b Robinson PC, Stamp LK (May 2016). “The management of gout: Much has changed”. Australian Family Physician. 45 (5): 299–302. PMID 27166465.
- ^ Satpanich, P; Pongsittisak, W; Manavathongchai, S (18 August 2021). “Early versus Late Allopurinol Initiation in Acute Gout Flare (ELAG): a randomized controlled trial”. Clinical Rheumatology. doi:10.1007/s10067-021-05872-8. PMID 34406530. S2CID 237156638.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ “The Top 300 of 2019”. ClinCalc. Retrieved 16 October 2021.
- ^ “Allopurinol – Drug Usage Statistics”. ClinCalc. Retrieved 16 October 2021.
- ^ Jump up to:a b c d e f g “300 mg Allopurinol tables”. UK Electronic Medicines Compendium. 7 April 2016. Archived from the original on 11 September 2016.
- ^ Jeha S (October 2001). “Tumor lysis syndrome”. Seminars in Hematology. 38 (4 Suppl 10): 4–8. doi:10.1016/S0037-1963(01)90037-X. PMID 11694945.
- ^ Jump up to:a b “Label for injectable Allopurinol”. DailyMed. June 2014. Archived from the original on 13 September 2016.
- ^ Bradford K, Shih DQ (October 2011). “Optimizing 6-mercaptopurine and azathioprine therapy in the management of inflammatory bowel disease”. World Journal of Gastroenterology. 17 (37): 4166–73. doi:10.3748/wjg.v17.i37.4166. PMC 3208360. PMID 22072847.
- ^ Sparrow MP, Hande SA, Friedman S, Cao D, Hanauer SB (February 2007). “Effect of allopurinol on clinical outcomes in inflammatory bowel disease nonresponders to azathioprine or 6-mercaptopurine”. Clinical Gastroenterology and Hepatology. 5 (2): 209–14. doi:10.1016/j.cgh.2006.11.020. PMID 17296529.
- ^ Ansari A, Patel N, Sanderson J, O’Donohue J, Duley JA, Florin TH (March 2010). “Low-dose azathioprine or mercaptopurine in combination with allopurinol can bypass many adverse drug reactions in patients with inflammatory bowel disease”. Alimentary Pharmacology & Therapeutics. 31 (6): 640–7. doi:10.1111/j.1365-2036.2009.04221.x. PMID 20015102. S2CID 6000856.
- ^ Ansari AR, Duley JA (March 2012). “Azathioprine co-therapy with allopurinol for inflammatory bowel disease: trials and tribulations” (PDF). Rev Assoc Med Bras. 58 (Suppl.1): S28–33.
- ^ Jump up to:a b Bartoli F, Cavaleri D, Bachi B, Moretti F, Riboldi I, Crocamo C, Carrà G (September 2021). “Repurposed drugs as adjunctive treatments for mania and bipolar depression: A meta-review and critical appraisal of meta-analyses of randomized placebo-controlled trials”. Journal of Psychiatric Research. 143: 230–238. doi:10.1016/j.jpsychires.2021.09.018. PMID 34509090. S2CID 237485915.
- ^ Dalbeth N, Stamp L (2007). “Allopurinol dosing in renal impairment: walking the tightrope between adequate urate lowering and adverse events”. Seminars in Dialysis. 20 (5): 391–5. doi:10.1111/j.1525-139X.2007.00270.x. PMID 17897242. S2CID 1150852.
- ^ Jump up to:a b c d Chung WH, Wang CW, Dao RL (July 2016). “Severe cutaneous adverse drug reactions”. The Journal of Dermatology. 43 (7): 758–66. doi:10.1111/1346-8138.13430. PMID 27154258. S2CID 45524211.
- ^ Tsai TF, Yeh TY (2010). “Allopurinol in dermatology”. American Journal of Clinical Dermatology. 11 (4): 225–32. doi:10.2165/11533190-000000000-00000. PMID 20509717. S2CID 36847530.
- ^ De Broe ME, Bennett WM, Porter GA (2003). Clinical Nephrotoxins: Renal Injury from Drugs and Chemicals. Springer Science+Business Media. ISBN 9781402012778.
Acute interstitial nephritis has also been reported associated with by the administration of allopurinol.
- ^ Reiter S, Simmonds HA, Zöllner N, Braun SL, Knedel M (March 1990). “Demonstration of a combined deficiency of xanthine oxidase and aldehyde oxidase in xanthinuric patients not forming oxipurinol”. Clinica Chimica Acta; International Journal of Clinical Chemistry. 187 (3): 221–34. doi:10.1016/0009-8981(90)90107-4. PMID 2323062.
- ^ Day RO, Graham GG, Hicks M, McLachlan AJ, Stocker SL, Williams KM (2007). “Clinical pharmacokinetics and pharmacodynamics of allopurinol and oxypurinol”. Clinical Pharmacokinetics. 46 (8): 623–44. doi:10.2165/00003088-200746080-00001. PMID 17655371. S2CID 20369375.
- ^ Cameron JS, Moro F, Simmonds HA (February 1993). “Gout, uric acid and purine metabolism in paediatric nephrology”. Pediatric Nephrology. 7 (1): 105–18. doi:10.1007/BF00861588. PMID 8439471. S2CID 34815040.
- ^ Jump up to:a b “Uric Acid-Lowering Drugs Pathway, Pharmacodynamics”. PharmGKB. Archived from the original on 8 August 2014.
- ^ Jump up to:a b “PharmGKB”. Archived from the original on 8 August 2014. Retrieved 1 August 2014.
- ^ “Allele Frequency Net Database”. Archived from the original on 28 August 2009.
- ^ Zineh I, Mummaneni P, Lyndly J, Amur S, La Grenade LA, Chang SH, et al. (December 2011). “Allopurinol pharmacogenetics: assessment of potential clinical usefulness”. Pharmacogenomics. 12 (12): 1741–9. doi:10.2217/pgs.11.131. PMID 22118056.
- ^ Khanna D, Fitzgerald JD, Khanna PP, Bae S, Singh MK, Neogi T, et al. (October 2012). “2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia”. Arthritis Care & Research. 64 (10): 1431–46. doi:10.1002/acr.21772. PMC 3683400. PMID 23024028.
- ^ “Annotation of CPIC Guideline for allopurinol and HLA-B”. PharmGKB. Archived from the original on 8 August 2014. Retrieved 1 August 2014.
- ^ Hershfield MS, Callaghan JT, Tassaneeyakul W, Mushiroda T, Thorn CF, Klein TE, Lee MT (February 2013). “Clinical Pharmacogenetics Implementation Consortium guidelines for human leukocyte antigen-B genotype and allopurinol dosing”. Clinical Pharmacology and Therapeutics. 93 (2): 153–8. doi:10.1038/clpt.2012.209. PMC 3564416. PMID 23232549.
- ^ Robins RK (1956). “Potential Purine Antagonists. I. Synthesis of Some 4,6-Substituted Pyrazolo \3,4-d] pyrimidines1”. J. Am. Chem. Soc. 78 (4): 784–790. doi:10.1021/ja01585a023.
- ^ Jump up to:a b Sneader W (2005). Drug Discovery: A History. John Wiley & Sons. p. 254. ISBN 9780471899792.
- ^ Elion GB (April 1989). “The purine path to chemotherapy”. Science. 244 (4900): 41–7. Bibcode:1989Sci…244…41E. doi:10.1126/science.2649979. PMID 2649979.
- ^ “FDA Approved Drug Products”. Drugs@FDA. Archived from the original on 14 August 2012. Retrieved 8 November 2013.
- ^ “Search Results for Allopurinol”. DailyMed. Archived from the original on 25 March 2012. Retrieved 27 July 2011.
Further reading
- Dean L (March 2016). “Allopurinol Therapy and HLA-B*58:01 Genotype”. In Pratt VM, McLeod HL, Rubinstein WS, et al. (eds.). Medical Genetics Summaries. National Center for Biotechnology Information (NCBI). PMID 28520356.
External links
- “Allopurinol”. Drug Information Portal. U.S. National Library of Medicine.
- “PRODUCT INFORMATION Allopurinol Tablets USP”. U.S. National Library of Medicine. Medication handout sheet (Revised: 07/2019 0603-2115)
- Allopurinol pathway on PharmGKB
/////////////////////ALLUPURINOL, BW-56158, аллопуринол , ألوبيرينول , 别嘌醇 ,

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}