
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TUCATINIB

Tucatinib
ツカチニブ;
N6-(4,4-dimethyl-4,5-dihydro-1,3-oxazol-2-yl)-N4-(3-methyl-4-{[1,2,4]triazolo[1,5-a]pyridin-7-yloxy}phenyl)quinazoline-4,6-diamine
| Formula | C26H24N8O2 |
|---|---|
| CAS | 937263-43-9 |
| Mol weight | 480.5212 |
To treat advanced unresectable or metastatic HER2-positive breast cancer
Drug Trials Snapshot
FDA APPROVED 4/17/2020 Tukysa
- ARRY 380
- ARRY-380
- ONT 380
- ONT-380
Tucatinib (INN),[1] sold under the brand name Tukysa, is a small molecule inhibitor of HER2 for the treatment of HER2-positive breast cancer.[2][3] It was developed by Array BioPharma and licensed to Cascadian Therapeutics (formerly Oncothyreon, subsequently part of Seattle Genetics).[4]
Common side effects are diarrhea, palmar-plantar erythrodysesthesia (burning or tingling discomfort in the hands and feet), nausea, fatigue, hepatotoxicity (liver damage), vomiting, stomatitis (inflammation of the mouth and lips), decreased appetite, abdominal pain, headache, anemia and rash.[5][6] Pregnant or breastfeeding women should not take Tucatinib because it may cause harm to a developing fetus or newborn baby.[5]
Tucatinib was approved for medical use in Australia in August 2020.[7]
Medical uses
Tucatinib is a kinase inhibitor indicated in combination with trastuzumab and capecitabine for treatment of adults with advanced unresectable or metastatic HER2-positive breast cancer, including those with brain metastases, who have received one or more prior anti-HER2-based regimens in the metastatic setting.[8]
Clinical trials
Two early stage clinical trials have reported encouraging results, both of which had options to enroll subjects with central nervous system (CNS) metastases.[2][9][10][11][12][10] HER2CLIMB is a Phase 2 randomized, double-blinded, placebo-controlled study of tucatinib in combination with trastuzumab and capecitabine in patients with pretreated, unresectable locally advanced or metastatic HER2-positive breast cancer.[13]
History
In April 2020, the U.S. Food and Drug Administration (FDA) approved tucatinib in combination with chemotherapy (trastuzumab and capecitabine) for the treatment of adults with advanced forms of HER2-positive breast cancer that can’t be removed with surgery, or has spread to other parts of the body, including the brain, and who have received one or more prior treatments.[5][6][14]
The FDA collaborated with the Australian Therapeutic Goods Administration (TGA), Health Canada, Health Sciences Authority (HSA, Singapore) and Swissmedic (SMC, Switzerland) on the review.[5] This was the first Project Orbis partnership between the FDA, HSA and Swissmedic.[5] As of 17 April 2020, the application is still under review at the other agencies.[5]
Tucatinib is a kinase inhibitor meaning it blocks a type of enzyme (kinase) and helps prevent the cancer cells from growing.[5] Tucatinib is approved for treatment after adults have taken one or more anti-HER2-based regimens in the metastatic setting.[5] The FDA approved tucatinib based on the results of the HER2CLIMB trial (NCT02614794) enrolling 612 subjects who had HER2-positive advanced unresectable or metastatic breast cancer and had prior treatment with trastuzumab, pertuzumab and ado-trastuzumab emtansine (T-DM1).[5][6] Subjects with previously treated and stable brain metastases, as well as those with previously treated and growing or untreated brain metastases, were eligible for the clinical trial, and 48% of enrolled subjects had brain metastases at the start of the trial.[5]
Subjects received either tucatinib 300 mg twice daily plus trastuzumab and capecitabine (tucatinib arm, n=410) or placebo plus trastuzumab and capecitabine (control arm, n=202).[6] The primary endpoint was progression-free survival (PFS), or the amount of time when there was no growth of the tumor, assessed by a blinded independent central review, evaluated in the initial 480 randomized patients.[5][6] The median PFS in subjects who received tucatinib, trastuzumab, and capecitabine was 7.8 months (95% CI: 7.5, 9.6) compared to 5.6 months (95% CI: 4.2, 7.1) in those subjects who received placebo, trastuzumab, and capecitabine (HR 0.54; 95% CI: 0.42, 0.71; p<0.00001).[5][6] Overall survival and PFS in subjects with brain metastases at baseline were key secondary endpoints.[5] The median overall survival in subjects who received tucatinib, trastuzumab, and capecitabine was 21.9 months (95% CI: 18.3, 31.0) compared to 17.4 months (95% CI: 13.6, 19.9) in subjects who received placebo, trastuzumab, and capecitabine (HR: 0.66; 95% CI: 0.50, 0.87; p=0.00480).[5][6] The median PFS in subjects with brain metastases at baseline who received tucatinib, trastuzumab and capecitabine was 7.6 months (95% CI: 6.2, 9.5) compared to 5.4 months (95% CI: 4.1, 5.7) in subjects who received placebo, trastuzumab and capecitabine (HR: 0.48; 0.34, 0.69; p<0.00001).[5][6]
The FDA granted the application for tucatinib priority review, breakthrough therapy, fast track, and orphan drug designations.[5][6][15] The FDA granted approval of Tukysa to Seattle Genetics, Inc.[5]
SYN
Recently, the Mao team reported a new route for the efficient synthesis of Tucatinib.
The results were published on Synthesis (DOI: 10.1055/s-0037-1610706).
Previously, the synthesis report route of Tucatinib was published by Array BioPharma in a patent document (WO 2007059257, 2007). The synthetic route reported in the patent is shown in the figure below:

Using 4-nitro-2-cyanoaniline as the raw material, the first step is to condense with DMF-DMA to prepare imine 3 (yield 87%); subsequent catalytic hydrogenation of palladium on carbon to reduce the nitro group to obtain the amine 4 (90% yield); followed by 1,1&39;-thiocarbonyldiimidazole (TCDI) and The amino alcohol undergoes condensation to prepare the thiourea derivative 5 (yield is only 34%); further with the intermediate 6 to undergo ring-closure reaction to obtain the key intermediate 7 (yield 62%) ; Finally, under the action of p-toluenesulfonic acid, intramolecular dehydration and ring closure to form oxazoline, complete the synthesis of the target compound tucatinib.
Reverse synthesis analysis
The author broke the bond of Tucatinib from two points a and b and split them into three fragments. : Thioether oxazoline 17, nitrobenzene 3 and the key fragment of the original research route 6.
Preparation of key fragment 6
4-nitro-3-methylphenol 8 as a starting point The material, with pyridine derivative 9, undergoes aromatic affinity substitution reaction to prepare aryl ether 10 (yield 64%); then it is condensed with DMF-DMA, and then treated with hydroxylamine hydrochloride. The step yield was 81% to obtain the oxime derivative 12; subsequently, the ring was closed under the treatment of trifluoroacetic anhydride, the mostAfter palladium-catalyzed hydrogenation to reduce the nitro group, the key aniline triazole 6 was successfully prepared, with a total yield of 32.8%.
aromatic ring skeleton construction
fragment 3 was synthesized according to the method reported in the literature. The estimated aromatic ring fragment was then constructed with the aniline triazole 6 prepared above:
Compound 6 and fragment 3 were cyclized in acetic acid , 14 was successfully prepared, and finally the nitro group was reduced by palladium-catalyzed hydrogenation to obtain the key arylamine 15 with a two-step yield of 76.4%.
Fragment 17 and Tucatinib synthesis
amino alcohol and 1,1&39;-thiocarbonyl diimidazole (TCDI) The ring is closed to obtain 16, which is then treated with methyl trifluoromethanesulfonate to obtain oxazoline 17, with a total yield of 67.23% in the two steps.
oxazoline17 and arylamine 15 in the presence of cesium carbonate, heated in DMF for 20 hours, and finally completed the synthesis of Tucatinib with a yield of 76%.
Comparison of the new route and the patent route
The yield of the last step of the patent is unknown, starting with key intermediates 3 and 6, total income The rate is less than 19%.
The overview of the new route is as follows:
Correspondingly, starting from the intermediate 3 and 6, the total yield of the new route There is a significant improvement to 39%. Moreover, the purity of the product and other aspects also meet the requirements of API.
Comment
Tucatinib (Tukysa) Tucatinib/Tucatinib as a small-molecule oral tyrosine kinase (TKI) inhibitor for HER2 Positive breast cancer has highly specific targeting selectivity. The study of the new synthetic route
effectively improves the production efficiency in terms of ensuring the purity of the compound, and the raw materials used are relatively simple and easy to obtain.
Medicinal chemists have completed the research and development and synthesis of compounds (from 0 to 1), while process chemists have optimized the synthetic routes and processes, so that the compounds can be prepared more simply, efficiently, economically and environmentally.
SYN PATENT
CN 111825604
PAPER
Synthesis (2019), 51(13), 2660-2664
Abstract
A new and improved synthetic route to tucatinib is described that involves three key intermediates. The first of these, 4-([1,2,4]triazolo[1,5-a]pyridin-7-yloxy)-3-methylaniline, was prepared on a 100 g scale in 33% yield over five steps and 99% purity. Next, N 4-(4-([1,2,4]triazolo[1,5-a]pyridin-7-yloxy)-3-methylphenyl)quinazoline-4,6-diamine was isolated in 67% yield over three steps and >99% purity. Then, 4,4-dimethyl-2-(methylthio)-4,5-dihydrooxazole trifluoromethanesulfonate was prepared under mild conditions in 67% yield over two steps. Finally, tucatinib was obtained in 17% yield over nine steps and in >99% purity (HPLC). Purification methods used to isolate the product and the intermediates involved in the route are also reported.

References
- ^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 75”. WHO Drug Information. 30 (1): 161. hdl:10665/331046.
- ^ Jump up to:a b “ONT-380 Active Against CNS Mets in HER2-Positive Breast Cancer”. Cancer Network. 15 December 2015. Retrieved 17 April 2020.
- ^ Martin M, López-Tarruella S (October 2018). “Emerging Therapeutic Options for HER2-Positive Breast Cancer”. American Society of Clinical Oncology Educational Book. American Society of Clinical Oncology. Annual Meeting. 35 (36): e64–70. doi:10.1200/EDBK_159167. PMID 27249772.
- ^ “Tucatinib” (PDF). Statement on a Nonproprietary Name Adopted by the USAN Council.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “FDA Approves First New Drug Under International Collaboration, A Treatment Option for Patients with HER2-Positive Metastatic Breast Cancer”. U.S. Food and Drug Administration (FDA) (Press release). 17 April 2020. Retrieved 17 April 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e f g h i “FDA approves tucatinib for patients with HER2-positive metastatic brea”. U.S. Food and Drug Administration (FDA). 17 April 2020. Retrieved 20 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Tukysa”. Therapeutic Goods Administration (TGA). 21 August 2020. Retrieved 22 September 2020.
- ^ “Tukysa (tucatinib) tablets, for oral use” (PDF). Seattle Genetics. Retrieved 17 April2020.
- ^ “Oncothyreon Inc. Announces Data For ONT-380 In HER2-Positive Breast Cancer Patients With And Without Brain Metastases At The San Antonio Breast Cancer Symposium”. BioSpace (Press release). 9 December 2015. Retrieved 18 April 2020.
- ^ Jump up to:a b Borges VF, Ferrario C, Aucoin N, Falkson CI, Khan QJ, Krop IE, et al. “Efficacy results of a phase 1b study of ONT-380, a CNS-penetrant TKI, in combination with T-DM1 in HER2+ metastatic breast cancer (MBC), including patients (pts) with brain metastases”. Journal of Clinical Oncology. 2016 ASCO Annual Meeting.
- ^ “SABCS15: Promising phase 1 results lead to phase 2 for ONT-380 in HER2+ breast cancer”. Colorado Cancer Blogs. Retrieved 10 June 2016.
- ^ “A Study of Tucatinib (ONT-380) Combined With Capecitabine and/or Trastuzumab in Patients With HER2+ Metastatic Breast Cancer”. ClinicalTrials.gov. 31 December 2013. Retrieved 18 April 2020.
- ^ “A Study of Tucatinib vs. Placebo in Combination With Capecitabine & Trastuzumab in Patients With Advanced HER2+ Breast Cancer (HER2CLIMB)”. ClinicalTrials.gov. Retrieved 18 April 2020.
- ^ “Tukysa: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 20 April 2020.
- ^ “Tucatinib Orphan Drug Designation and Approval”. U.S. Food and Drug Administration(FDA). 24 December 1999. Retrieved 20 April 2020.
External links
- “Tucatinib”. Drug Information Portal. U.S. National Library of Medicine.
- “Tucatinib”. National Cancer Institute.
- Clinical trial number NCT02614794 for “A Study of Tucatinib vs. Placebo in Combination With Capecitabine & Trastuzumab in Patients With Advanced HER2+ Breast Cancer (HER2CLIMB)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Tukysa |
| Other names | ONT-380, ARRY-380 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620032 |
| License data | US DailyMed: Tucatinib |
| Pregnancy category | AU: DUS: N (Not classified yet) |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | AU: S4 (Prescription only)US: ℞-only |
| Identifiers | |
| CAS Number | 937263-43-9 |
| PubChem CID | 51039094 |
| DrugBank | DB11652 |
| ChemSpider | 34995558 |
| UNII | 234248D0HH |
| KEGG | D11141 |
| ChEMBL | ChEMBL3989868 |
| Chemical and physical data | |
| Formula | C26H24N8O2 |
| Molar mass | 480.532 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]CC1=C(C=CC(=C1)NC2=NC=NC3=C2C=C(C=C3)NC4=NC(CO4)(C)C)OC5=CC6=NC=NN6C=C5 | |
| InChI[hide]InChI=1S/C26H24N8O2/c1-16-10-17(5-7-22(16)36-19-8-9-34-23(12-19)28-15-30-34)31-24-20-11-18(4-6-21(20)27-14-29-24)32-25-33-26(2,3)13-35-25/h4-12,14-15H,13H2,1-3H3,(H,32,33)(H,27,29,31)Key:SDEAXTCZPQIFQM-UHFFFAOYSA-N |
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Tukysa | Tablet | 150 mg/1 | Oral | Seattle Genetics, Inc. | 2020-04-17 | Not applicable |  | |
| Tukysa | Tablet | 150 mg | Oral | Seattle Genetics, Inc. | 2020-08-27 | Not applicable |  | |
| Tukysa | Tablet | 50 mg/1 | Oral | Seattle Genetics, Inc. | 2020-04-17 | Not applicable | | |
| Tukysa | Tablet | 50 mg | Oral | Seattle Genetics, Inc. | 2020-10-08 | Not applicable | |
Showing 1 to 4 of 4 entries
///////tucatinib, FDA 2020, TUKSYA, 2020 APROVALS, ARRY 380, ONT 380, ツカチニブ ,
Ripretinib

Ripretinib
リプレチニブ;
| Formula | C24H21BrFN5O2 |
|---|---|
| CAS | 1442472-39-0 |
| Mol weight | 510.3582 |
Antineoplastic, Receptor tyrosine kinase inhibitor
US FDA APPROVED 2020/5/15 QUINLOCK
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Qinlock | Tablet | 50 mg | Oral | Deciphera Pharmaceuticals. Llc | Not applicable | Not applicable | | |
| Qinlock | Tablet | 50 mg/1 | Oral | Deciphera Pharmaceuticals, LLC | 2020-05-15 | Not applicable | |
SYN

Ripretinib, sold under the brand name Qinlock, is a medication for the treatment of adults with advanced gastrointestinal stromal tumor (GIST), a type of tumor that originates in the gastrointestinal tract.[3] It is taken by mouth.[3] Ripretinib is a kinase inhibitor, meaning it works by blocking a type of enzyme called a kinase, which helps keep the cancer cells from growing.[3]
The most common side effects include alopecia (hair loss), fatigue, nausea, abdominal pain, constipation, myalgia (muscle pain), diarrhea, decreased appetite, palmar-plantar erythrodysesthesia syndrome (a skin reaction in the palms and soles) and vomiting.[3][4] Alopecia is a unique side effect to ripretinib, which is not seen with other tyrosine kinase inhibitors used to treat GISTs.
Ripretinib was approved for medical use in the United States in May 2020,[3] and in Australia in July 2020.[1] Ripretinib is the first new drug specifically approved in the United States as a fourth-line treatment for advanced gastrointestinal stromal tumor (GIST).
Medical uses
Ripretinib is indicated for the treatment of adults with advanced gastrointestinal stromal tumor (GIST), a type of tumor that originates in the gastrointestinal tract, who have received prior treatment with three or more kinase inhibitor therapies, including imatinib.[3] GIST is type of stomach, bowel, or esophagus tumor.[4]
Adverse effects
The most common side effects include alopecia (hair loss), fatigue, nausea, abdominal pain, constipation, myalgia (muscle pain), diarrhea, decreased appetite, palmar-plantar erythrodysesthesia syndrome (a skin reaction in the palms and soles) and vomiting.[3][4]
Ripretinib can also cause serious side effects including skin cancer, hypertension (high blood pressure) and cardiac dysfunction manifested as ejection fraction decrease (when the muscle of the left ventricle of the heart is not pumping as well as normal).[3][4]
Ripretinib may cause harm to a developing fetus or a newborn baby.[3][4]
History
Ripretinib was approved for medical use in the United States in May 2020.[3][5][6][4]
The approval of ripretinib was based on the results of an international, multi-center, randomized, double-blind, placebo-controlled clinical trial (INVICTUS/NCT03353753) that enrolled 129 participants with advanced gastrointestinal stromal tumor (GIST) who had received prior treatment with imatinib, sunitinib, and regorafenib.[3][7] The trial compared participants who were randomized to receive ripretinib to participants who were randomized to receive placebo, to determine whether progression free survival (PFS) – the time from initial treatment in the clinical trial to growth of the cancer or death – was longer in the ripretinib group compared to the placebo group.[3] During treatment in the trial, participants received ripretinib 150 mg or placebo once a day in 28-day cycles, repeated until tumor growth was found (disease progression), or the participant experienced intolerable side effects.[3][7] After disease progression, participants who were randomized to placebo were given the option of switching to ripretinib.[3][7] The trial was conducted at 29 sites in the United States, Australia, Belgium, Canada, France, Germany, Italy, the Netherlands, Poland, Singapore, Spain, and the United Kingdom.[4]
The major efficacy outcome measure was progression-free survival (PFS) based on assessment by blinded independent central review (BICR) using modified RECIST 1.1 in which lymph nodes and bone lesions were not target lesions and a progressively growing new tumor nodule within a pre-existing tumor mass must meet specific criteria to be considered unequivocal evidence of progression.[7] Additional efficacy outcome measures included overall response rate (ORR) by BICR and overall survival (OS).[7] The trial demonstrated a statistically significant improvement in PFS for participants in the ripretinib arm compared with those in the placebo arm (HR 0.15; 95% CI: 0.09, 0.25; p<0.0001).[7]
The U.S. Food and Drug Administration (FDA) granted the application for ripretinib priority review and fast track designations, as well as breakthrough therapy designation and orphan drug designation.[3][8] The FDA granted approval of Qinlock to Deciphera Pharmaceuticals, Inc.[3]
The FDA collaborated with the Australian Therapeutic Goods Administration (TGA) and Health Canada on the review of the application as part of Project Orbis.[3][7] The FDA approved ripretinib three months ahead of schedule.[3][7] As of May 2020, the review of the applications was ongoing for the Australian TGA and for Health Canada.[3][7]
Names
Ripretinib is the International nonproprietary name (INN) and the United States Adopted Name (USAN).[9][10]
| PATENT NUMBER | PEDIATRIC EXTENSION | APPROVED | EXPIRES (ESTIMATED) | |
|---|---|---|---|---|
| US8940756 | No | 2012-06-07 | 2032-06-07 | |
| US8461179 | No | 2012-06-07 | 2032-06-07 | |
| US8188113 | No | 2010-07-27 | 2030-07-27 | |
PATENT
US 8461179
PATENT
WO 2013184119
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2013184119
[0125] Example A13: A mixture of Example C5 (2.191 g, 7.94 mmol), Example Bl (1.538 g, 8.33 mmol) and KF on alumina (40 wt%) (9.22 g, 63.5 mmol) in DMA (40 mL) was sonicated for 2 h. The mixture was filtered through a shallow bed of silica gel and rinsed well with EtOAc. The filtrate was washed with satd. NaHC03 (lx), 5% LiCl (2x), then brine (lx), dried (MgS04), and concentrated to dryness to afford 3-(5-amino-2-bromo-4-fluorophenyl)-7-chloro-l -ethyl- l,6-naphthyridin-2(lH)-one (2.793 g, 89% yield) as a brown solid. 1H NMR (400 MHz, DMSO-<¾): δ 8.77 (s, 1 H), 8.00 (s, 1 H), 7.74 (s, 1 H), 7.37 (d, 1 H), 6.77 (d, 1 H), 5.45 (s, 2 H), 4.27 (q, 2 H), 1.20 (t, 3 H); MS (ESI) m z: 398.0 [M+H]+.
[0126] Example A14: A suspension of Example A13 (1.50 g, 3.78 mmol) in dioxane (15 mL) was treated with methylamine (40% in water) (26.4 mL, 303 mmol) in a pressure tube and heated to 100°C overnight. The mixture was cooled to RT, treated with a large amount of brine, then diluted with EtOAc until all of the solids dissolved. The layers were separated, the aqueous layer extracted with additional EtOAc (lx) and the combined organics were washed with satd. NaHC03 (lx), dried (MgS04) and concentrated to dryness. The resulting solid was suspended in MeCN/H20, frozen and lyophilized to afford 3-(5-amino-2-bromo-4-fluorophenyl)-l-ethyl-7-(methylamino)-l,6-naphthyridin-2(lH)-one (1.32g, 89% yield) as a light brown solid. 1H NMR (400 MHz, DMSO-<¾): δ 8.37 (s, 1 H), 7.62 (s, 1 H), 7.30 (d, 1 H), 6.99 (q, 1 H), 6.73 (d, 1 H), 6.21 (s, 1 H), 5.33 (s, 2 H), 4.11 (q, 2 H), 2.84 (d, 3 H), 1.19 (t, 3 H); MS (ESI) m/z: 393.0 [M+H]+.
[0263] Example 31: A mixture of Example A14 (0.120 g, 0.307 mmol) and TEA (0.043 mL, 0.307 mmol) in THF (3.0 mL) was treated with phenyl isocyanate (0.040 g, 0.337 mmol) and stirred at RT for 4 h. Over the course of the next 4 days the mixture was treated with additional phenyl isocyanate (0.056 mL) and stirred at RT. The resulting solid was filtered, rinsed with THF, then triturated with MeOH to afford l-(4-bromo-5-(l-ethyl-7-(methylamino)-2-oxo- 1 ,2-dihydro- 1 ,6-naphthyridin-3 -yl)-2-fluorophenyl)-3 -phenylurea (101 mg, 64.5% yield) as a bright white solid. 1H NMR (400 MHz, DMSO-<¾): δ 9.09 (s, 1 H), 8.68 (s, 1 H), 8.41 (s, 1 H), 8.17 (d, 1 H), 7.70 (s, 1 H), 7.65 (d, 1 H), 7.41 (d, 2 H), 7.27 (m, 2 H), 7.03 (m, 1 H), 6.96 (t, 1 H), 6.23 (s, 1 H), 4.13 (q, 2 H), 2.86 (d, 3 H), 1.20 (t, 3 H); MS (ESI) m/z: 510.1 [M+H]+.
References
- ^ Jump up to:a b c “Qinlock Australian Prescription Medicine Decision Summary”. Therapeutic Goods Administration (TGA). 21 July 2020. Retrieved 17 August 2020.
- ^ “Ripretinib (Qinlock) Use During Pregnancy”. Drugs.com. 10 August 2020. Retrieved 17 August 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t “FDA Approves First Drug for Fourth-Line Treatment of Advanced Gastrointestinal Stromal Tumors”. U.S. Food and Drug Administration (FDA) (Press release). 15 May 2020. Retrieved 15 May 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f g “Drug Trial Snapshot: Qinlock”. U.S. Food and Drug Administration (FDA). 15 May 2020. Retrieved 2 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ “FDA Grants Full Approval of Deciphera Pharmaceuticals’ Qinlock (ripretinib) for the Treatment of Fourth-Line Gastrointestinal Stromal Tumor”. Deciphera Pharmaceuticals, Inc. (Press release). 15 May 2020. Retrieved 15 May 2020.
- ^ “Qinlock: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 15 May 2020.
- ^ Jump up to:a b c d e f g h i “FDA approves ripretinib for advanced gastrointestinal stromal tumor”. U.S. Food and Drug Administration (FDA). 15 May 2020. Retrieved 18 May 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Ripretinib Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 2 October 2014. Retrieved 15 May 2020.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 81”. WHO Drug Information. 33 (1): 106. hdl:10665/330896. License: CC BY-NC-SA 3.0 IGO.
- ^ “Ripretinib” (PDF). United States Adopted Name (USAN) Drug Finder. Retrieved 17 May 2020.
Further reading
- Schneeweiss M, Peter B, Bibi S, et al. (May 2018). “The KIT and PDGFRA switch-control inhibitor DCC-2618 blocks growth and survival of multiple neoplastic cell types in advanced mastocytosis”. Haematologica. 103 (5): 799–809. doi:10.3324/haematol.2017.179895. PMC 5927976. PMID 29439183.
External links
- “Ripretinib”. Drug Information Portal. U.S. National Library of Medicine.
- “Ripretinib”. NCI Drug Dictionary. National Cancer Institute.
- “Ripretinib”. National Cancer Institute.
- Clinical trial number NCT03353753 for “Phase 3 Study of DCC-2618 vs Placebo in Advanced GIST Patients Who Have Been Treated With Prior Anticancer Therapies (invictus)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Pronunciation | rip re’ ti nib |
| Trade names | Qinlock |
| Other names | DCC-2618 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620035 |
| License data | US DailyMed: Ripretinib |
| Pregnancy category | AU: D[1]US: N (Not classified yet)[2]Use should be avoided |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | AU: S4 (Prescription only) [1]US: ℞-only [3] |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 1442472-39-0 |
| PubChem CID | 71584930 |
| DrugBank | DB14840 |
| ChemSpider | 67886378 |
| UNII | 9XW757O13D |
| KEGG | D11353 |
| ChEMBL | ChEMBL4216467 |
| Chemical and physical data | |
| Formula | C24H21BrFN5O2 |
| Molar mass | 510.367 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]CCN1C(=O)C(=CC2=C1C=C(NC)N=C2)C1=C(Br)C=C(F)C(NC(=O)NC2=CC=CC=C2)=C1 | |
| InChI[hide]InChI=1S/C24H21BrFN5O2/c1-3-31-21-12-22(27-2)28-13-14(21)9-17(23(31)32)16-10-20(19(26)11-18(16)25)30-24(33)29-15-7-5-4-6-8-15/h4-13H,3H2,1-2H3,(H,27,28)(H2,29,30,33)Key:CEFJVGZHQAGLHS-UHFFFAOYSA-N |
////////////Ripretinib, QINLOCK, リプレチニブ , 2020 APPROVALS, FDA 2020
REPROXALAP
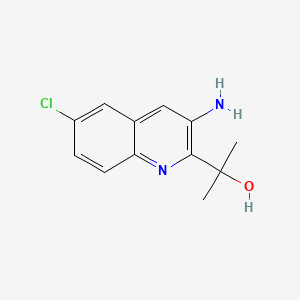
REPROXALAP
レプロキサラップ;
ADX-102
2-(3-amino-6-chloroquinolin-2-yl)propan-2-ol
C12H13ClN2O, 236.7 g/mol
CAS 916056-79-6
UNII-F0GIZ22IJH
2-(3-amino-6-chloroquinolin-2-yl)propan-2-ol
Phase 3 Clinical
Aldeyra Therapeutics is developing reproxalap, which binds and traps free aldehydes, formulated using Captisol technology licensed from Ligand Pharmaceuticals as an eye drop formulation, for treating acute noninfectious anterior uveitis, allergic conjunctivitis and dry eye syndrome.

PATENT
product case, WO2006127945 ,
EU states until 2026
expire US in 2029 with US154 extension.
PATENTS
WO2018170476
United States patent application serial number US 13/709,802, filed December 10, 2012 and published as US 2013/0190500 on July 25, 2013 (“the ‘500 publication,” the entirety of which is hereby incorporated herein by reference), describes certain aldehyde scavenging compounds. Such compounds include com ound A:
[0036] Compound A, (6-chloro-3-amino-2-(2-hydroxypropyl)-l-azanaphthalene), is designated as compound A in the ‘500 publication and the synthesis of compound A is described in detail at Example 5 of the ‘500 publication, and is reproduced herein for ease of reference.
Example A – General Preparation of Compound A
Compound A
[00436] The title compound was prepared according to the steps and intermediates (e.g., Scheme 1) described below and in the ‘500 publication, the entirety of which is incorporated herein by reference.
Step 1: Synthesis of Intermediate A- 1
[00437] To a 2 L round bottom flask was charged ethanol (220 mL), and pyridine (31 g, 392 mmol) and the resulting solution stirred at a moderate rate of agitation under nitrogen. To this solution was added ethyl bromopyruvate (76.6 g, 354 mmol) in a slow, steady stream. The reaction mixture was allowed to stir at 65±5° C. for 2 hours.
Step 2: Synthesis of Intermediate A-2
[00438] Upon completion of the 2-hour stir time in example 1, the reaction mixture was slowly cooled to 18-22° C. The flask was vacuum-purged three times at which time 2-amino-5-chloro-benzaldehyde (ACB) (50.0 g, 321 mmol) was added directly to the reaction flask as a solid using a long plastic funnel. Pyridine (64.0 g, 809 mmol) was added followed by an EtOH rinse (10 mL) and the reaction mixture was heated at 80±3° C. under nitrogen for about 16 hours (overnight) at which time HPLC analysis indicated that the reaction was effectively complete.
Step 3: Synthesis of Intermediate A-3
[00439] The reaction mixture from example 2 was cooled to about 70° C. and morpholine (76.0 g, 873 mmol)) was added to the 2 L reaction flask using an addition funnel. The reaction mixture was heated at 80±2° C. for about 2.5 hours at which time the reaction was considered complete by HPLC analysis (area % of A-3 stops increasing). The reaction mixture was cooled to 10-15° C. for the quench, work up, and isolation.
Step 4: Isolation of Intermediate A-3
[00440] To the 2 L reaction flask was charged water (600 g) using the addition funnel over 30-60 minutes, keeping the temperature below 15° C. by adjusting the rate of addition and using a cooling bath. The reaction mixture was stirred for an additional 45 minutes at 10-15° C. then the crude A-3 isolated by filtration using a Buchner funnel. The cake was washed with water (100 mLx4) each time allowing the water to percolate through the cake before applying a vacuum. The cake was air dried to provide crude A-3 as a nearly dry brown solid. The cake was returned to the 2 L reaction flask and heptane (350 mL) and EtOH (170 mL) were added and the mixture heated to 70±3° C. for 30-60 minutes. The slurry was cooled to 0-5° C. and isolated by filtration under vacuum. The A-3 was dried in a vacuum drying oven under vacuum and 35±3° C. overnight (16-18 hours) to provide A-3 as a dark green solid.
Step 5: Synthesis of Compound A
[00441] To a 2 L round bottom flask was charged methylmagnesium chloride (200 mL of 3.0 M solution in THF, 600 mmol). The solution was cooled to 0-5° C. using an ice bath.
[00442] A 500 mL flask (magnetic stirring) was charged with 22.8 grams A-3 from example 4 and THF (365 mL), stirred to dissolve then transferred to an addition funnel on the 2 L Reaction Flask. The A-3 solution was added drop-wise to the reaction flask over 5.75 hours, keeping the temperature of the reaction flask between 0-5° C throughout the addition. At the end of the addition the contents of the flask were stirred for an additional 15 minutes at 0-5° C. then the cooling bath was removed and the reaction was allowed to stir overnight at ambient temperature.
[00443] The flask was cooled in an ice bath and the reaction mixture was carefully quenched by adding EtOH (39.5 g, 857 mmol) drop-wise to the reaction mixture, keeping the temperature of the reaction mixture below 15° C. during the course of the addition. An aqueous solution of H4C1 (84.7 g H4C1 in 415 mL water) was then carefully added and the mixture stirred under moderate agitation for about 30 minutes then transferred to a separately funnel to allow the layers to separate. Solids were present in the aqueous phase so HO Ac (12.5 g) was added and the contents swirled gently to obtain a nearly homogeneous lower aqueous phase. The lower aqueous layer was transferred back to the 2 L reaction flask and stirred under moderate agitation with 2-methylTHF (50 mL) for about 15 minutes. The original upper organic layer was reduced in volume to approximately 40 mL using a rotary evaporator at≤40° C. and vacuum as needed. The phases in the separatory funnel were separated and the upper 2-MeTHF phase combined with the product residue, transferred to a 500 mL flask and vacuum distilled to an approximate volume of 25 mL. To this residue was added 2-MeTHF (50 mL) and distilled to an approximate volume of 50 mL. The crude compound A solution was diluted with 2-MeTHF (125 mL), cooled to 5-10° C. and 2M H2S04 (aq) (250 mL) was slowly added and the mixture stirred for 30 minutes as the temperature was allowed to return to ambient. Heptane (40 mL) was charged and the reaction mixture stirred for an additional 15 minutes then transferred to a separatory funnel and the layers were allowed to separate. The lower aqueous product layer was extracted with additional heptane (35 mL) then the lower aqueous phase was transferred to a 1 L reaction flask equipped with a mechanical stirrer and the mixture was cooled to 5-10° C. The combined organic layers were discarded. A solution of 25% NaOH(aq) was prepared (NaOH, 47 g, water, 200 mL) and slowly added to the 1 L reaction flask to bring the pH to a range of 6.5-8.5.
[00444] EtOAc (250 mL) was added and the mixture was stirred overnight. The mixture was transferred to a separatory funnel and the lower phase discarded. The upper organic layer was washed with brine (25 mL) then the upper organic product layer was reduced in volume on a rotary evaporator to obtain the crude compound A as a dark oil that solidified within a few minutes. The crude compound A was dissolved in EtOAc (20 mL) and filtered through a plug of silica gel (23 g) eluting with 3/1 heptane/EtOAc until all compound A was eluted (approximately 420 mL required) to remove most of the dark color of compound A. The solvent was removed in vacuo to provide 14.7 g of compound A as a tan solid. Compound A was taken up in EtOAc (25 mL) and eluted through a column of silica gel (72 g) using a mobile phase gradient of 7/1 heptane/EtOAc to 3/lheptane/EtOAc (1400 mL total). The solvent fractions containing compound A were stripped, compound A diluted with EtOAc (120 mL) and stirred in a flask with Darco G-60 decolorizing carbon (4.0 g) for about 1 hour. The mixture was filtered through celite using a fitted funnel, rinsing the cake with EtOAc (3 x 15 mL). The combined filtrates were stripped on a rotary evaporator and compound A dissolved in heptane (160 mL)/EtOAc(16 mL) at 76° C. The
homogeneous solution was slowly cooled to 0-5° C, held for 2 hours then compound A was isolated by filtration. After drying in a vacuum oven for 5 hours at 35° C. under best vacuum, compound A was obtained as a white solid. HPLC purity: 100% (AUC).
Example 1 – Preparation of Free Base Forms A and B of Compound A
Compound A
[00445] Compound A is prepared according to the method described in detail in Examples 1-5 of the ‘500 publication, the entirety of which is hereby incorporated herein by reference.
PATENT
example 5 [WO2018039197A1]
https://patents.google.com/patent/WO2018039197A1/en
Exam le 5: Synthesis of NS2

NS2
[00190] 2-(3-amino-6-chloroquinolin-2-yl)propan-2-ol. To a 2 L round bottom flask was charged methylmagnesium chloride (200 mL of 3.0 M solution in THF, 600 mmol). The solution was cooled to 0-5 °C using an ice bath.
[00191] A 500 mL flask (magnetic stirring) was charged with 22.8 grams A-3a from Example 4 and THF (365 mL), stirred to dissolve, and then transferred to an addition funnel on the 2 L reaction flask. The A-3a solution was added drop-wise to the reaction flask over 5.75 hours, keeping the temperature of the reaction flask between 0-5 °C throughout the addition. At the end of the addition the contents of the flask were stirred for an additional 15 minutes at 0-5 °C, then the cooling bath was removed and the reaction was allowed to stir overnight at ambient temperature.
[00192] The flask was cooled in an ice bath and the reaction mixture was carefully quenched by adding EtOH (39.5 g, 857 mmol) drop-wise to the reaction mixture, keeping the temperature of the reaction mixture below 15 °C during the course of the addition. An aqueous solution of H4CI (84.7 g H4CI in 415 mL water) was then carefully added and the mixture stirred under moderate agitation for about 30 minutes then transferred to a separatory funnel to allow the layers to separate. Solids were present in the aqueous phase so HOAc (12.5 g) was added and the contents swirled gently to obtain a nearly homogeneous lower aqueous phase. The lower aqueous layer was transferred back to the 2 L reaction flask and stirred under moderate agitation with 2-methyl-tetrahydrofuran (2-MeTHF) (50 mL) for about 15 minutes. The original upper organic layer was reduced in volume to approximately 40 mL using a rotary evaporator at < 40 °C under vacuum as needed. The phases in the separatory funnel were separated and the upper 2-MeTHF phase combined with the product residue was transferred to a 500 mL flask and vacuum distilled to an approximate volume of 25 mL. To this residue was added 2-MeTHF (50 mL) and the mixture again distilled to an approximate volume of 50 mL. The crude compound NS2 solution was diluted with 2-MeTHF (125 mL), cooled to 5-10 °C, and 2 M H2S04 (aq) (250 mL) was slowly added and the mixture stirred for 30 minutes as the temperature was allowed to return to ambient. Heptane (40 mL) was charged and the reaction mixture stirred for an additional 15 minutes then transferred to a separatory funnel, and the layers were allowed to separate. The lower aqueous product layer was extracted with additional heptane (35 mL), then the lower aqueous phase was transferred to a 1 L reaction flask equipped with a mechanical stirrer, and the mixture was cooled to 5-10 °C. The combined organic layers were discarded. A solution of 25% NaOH (aq) was prepared (NaOH, 47 g, water, 200 mL) and slowly added to the 1 L reaction flask to bring the pH to a range of 6.5 – 8.5.
[00193] EtOAc (250 mL) was added and the mixture was stirred overnight. The mixture was transferred to a separatory funnel and the lower phase discarded. The upper organic layer was washed with brine (25 mL), then the upper organic product layer was reduced in volume on a rotary evaporator to obtain a obtain the crude compound NS2 as a dark oil that solidified within a few minutes. The crude compound NS2 was dissolved in EtOAc (20 mL) and filtered through a plug of silica gel (23 g) eluting with 3/1 heptane/EtOAc until all compound NS2 was eluted (approximately 420 mL required) to remove most of the dark color of compound NS2. The solvent was removed in vacuo to provide 14.7 g of compound NS2 as a tan solid. Compound NS2 was taken up in EtOAc (25 mL) and eluted through a column of silica gel (72g) using a mobile phase gradient of 7/1 heptane/EtOAc to 3/1 heptane/EtOAc (1400 mL total). The solvent fractions containing compound NS2 were evaporated. Compound NS2 was diluted with EtOAc (120 mL) and stirred in a flask with Darco G-60 decolorizing carbon (4.0 g) for about 1 hour. The mixture was filtered through celite using a firtted funnel, rinsing the cake with EtOAc (3 x 15 mL). The combined filtrates were evaporated on a rotary evaporator and compound NS2 dissolved in heptane (160 mL)/EtOAc (16 mL) at 76 °C. The homogeneous solution was slowly cooled to 0-5 °C, held for 2 hours, then compound NS2 was isolated by filtration. After drying in a vacuum oven for 5 hours at 35 °C under best vacuum, compound NS2 was obtained as a white solid. HPLC purity: 100% (AUC); HPLC (using standard conditions): A-2: 7.2 minutes; A-3 : 11.6 minutes.
Preparation of ACB

[00194] After a N2 atmosphere had been established and a slight stream of N2 was flowing through the vessel, platinum, sulfided, 5 wt. % on carbon, reduced, dry (9.04 g, 3.0 wt. % vs the nitro substrate) was added to a 5 L heavy walled pressure vessel equipped with a large magnetic stir-bar and a thermocouple. MeOH (1.50 L), 5-chloro-2-nitrobenzaldehyde (302.1 g, 1.63 mol), further MeOH (1.50 L) and Na2C03 (2.42 g, 22.8 mmol, 0.014 equiv) were added. The flask was sealed and stirring was initiated at 450 rpm. The solution was evacuated and repressurized with N2 (35 psi), 2x. The flask was evacuated and repressurized with H2 to 35 psi. The temperature of the solution reached 30 °C w/in 20 min. The solution was then cooled with a water bath. Ice was added to the water bath to maintain a temperature below 35 °C. Every 2h, the reaction was monitored by evacuating and repressurizing with N2 (5 psi), 2x prior to opening. The progress of the reaction could be followed by TLC: 5-Chloro-2-nitrobenzaldehyde (Rf = 0.60, CH2CI2, UV) and the intermediates (Rf = 0.51, CH2CI2, UV and Rf = 0.14, CH2CI2, UV) were consumed to give ACB (Rf = 0.43, CH2CI2, UV). At 5 h, the reaction had gone to 98% completion (GC), and was considered complete. To a 3 L medium fritted funnel was added celite (ca. 80 g). This was settled with MeOH (ca. 200 mL) and pulled dry with vacuum. The reduced solution was transferred via cannula into the funnel while gentle vacuum was used to pull the solution through the celite plug. This was chased with MeOH (4 x 150 mL). The solution was transferred to a 5 L three-necked round-bottom flask. At 30 °C on a rotavap, solvent (ca. 2 L) was removed under reduced pressure. An N2 blanket was applied. The solution was transferred to a 5L four-necked round-bottomed flask equipped with mechanical stirring and an addition funnel. Water (2.5 L) was added dropwise into the vigorously stirring solution over 4 h. The slurry was filtered with a minimal amount of vacuum. The collected solid was washed with water (2 x 1.5 L), 2-propanol (160 mL) then hexanes (2 x 450 mL). The collected solid (a canary yellow, granular solid) was transferred to a 150 x 75 recrystallizing dish. The solid was then dried under reduced pressure (26-28 in Hg) at 40°C overnight in a vacuum-oven. ACB (> 99% by HPLC) was stored under a N2 atmosphere at 5°C.
PATENT
WO-2020223717
Process for preparing reproxalap as acetaldehyde dehydrogenase inhibitor useful for treating ocular diseases and cancer.
PATENT
WO-2020223685
Novel crystalline forms of reproxalap (compound 1; designated as Forms A and B) as acetaldehyde dehydrogenase inhibitor useful for treating ocular diseases and cancer.
PATENT
WO 2020123730
//////////REPROXALAP, レプロキサラップ , ADX-102, Phase 3 Clinical
CC(C)(C1=C(C=C2C=C(C=CC2=N1)Cl)N)O
Lercanidipine
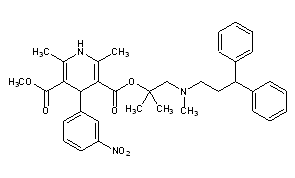
LercanidipineCAS Registry Number: 100427-26-7CAS Name: 1,4-Dihydro-2,6-dimethyl-4-(3-nitrophenyl)-3,5-pyridinedicarboxylic acid 2-[(3,3-diphenylpropyl)methylamino]-1,1-dimethylethyl methyl esterAdditional Names: methyl 1,1,N-trimethyl-N-(3,3-diphenylpropyl)-2-aminoethyl 1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylate; methyl 1,1-dimethyl-2-[N-(3,3-diphenylpropyl)-N-methylamino]ethyl 2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate; masnidipineMolecular Formula: C36H41N3O6Molecular Weight: 611.73Percent Composition: C 70.68%, H 6.76%, N 6.87%, O 15.69%Literature References: Dihydropyridine calcium channel blocker. Prepn: D. Nardi et al.,EP153016; eidem,US4705797 (1985, 1987 both to Recordati). Pharmacology: G. Bianchi et al.,Pharmacol. Res.21, 193 (1989). Clinical evaluation in hypertension: E. Rimoldi et al.,Acta Ther.20, 23 (1994).
Derivative Type: HydrochlorideCAS Registry Number: 132866-11-6Manufacturers’ Codes: Rec-15-2375; R-75Trademarks: Lerdip (Recordati); Zanidip (Napp)Molecular Formula: C36H41N3O6.HClMolecular Weight: 648.19Percent Composition: C 66.71%, H 6.53%, N 6.48%, O 14.81%, Cl 5.47%Properties: Prepd as the hemihydrate, mp 119-123°. LD50 in mice (mg/kg): 83 i.p.; 657 orally (Nardi).Melting point: mp 119-123°Toxicity data: LD50 in mice (mg/kg): 83 i.p.; 657 orally (Nardi)
Therap-Cat: Antihypertensive.Keywords: Antihypertensive; Dihydropyridine Derivatives; Calcium Channel Blocker; Dihydropyridine Derivatives.
Masnidipine hydrochloride, Lercanidipine hydrochloride, TJN-324, Rec-15/2375, Lercan, Cardiovasc, Lerzam, Zanidip, Lerdip, Lercadip, Zanedip
Syn 1
EP 0153016; JP 60199874; US 4772621; US 4968832
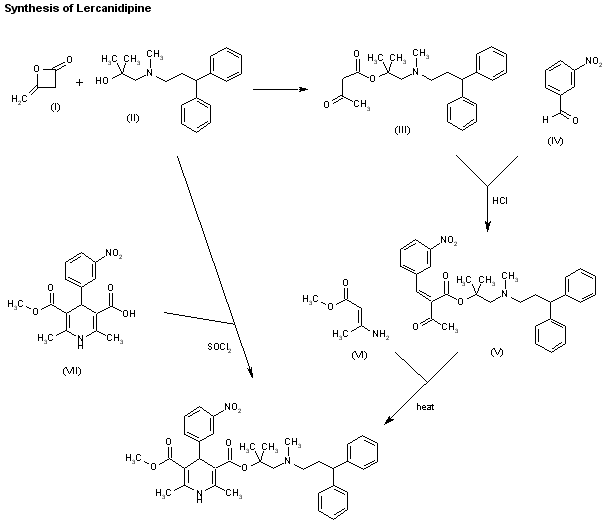
Two new related ways for the synthesis of lercanidipine have been reported: 1) The condensation of diketene (I) with the aminoalcohol (II) gives the corresponding acetoacetate ester (III), which is allowed to react with 3-nitrobenzaldehyde (IV) by means of HCl in chloroform yielding the expected benzylidene derivative (V). Finally, this compound is cyclized with methyl 3-aminocrotonate (VI) in refluxing isopropanol. 2) By esterification of 2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylic acid monomethyl ester (VIII) with alcohol (II) by means of SOCl2 in DMF/dichloromethane.
PATENT
https://patents.google.com/patent/WO2007054969A2/en

PATENT
https://patents.google.com/patent/EP1860102A1/en

PATENT

SPINOSAD
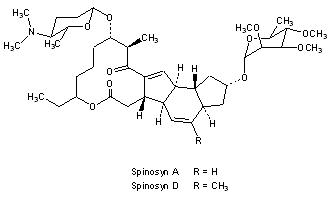

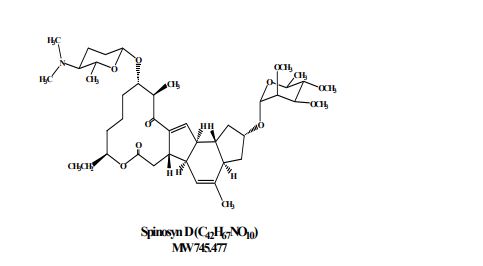
Spinosad
Spinosyn A: The chemical name is: 1H-as-Indaceno[3,2- d]oxacyclododecin-7,a5-dione, 2-[(6-deoxy-2,3,4-tri-O-methyl-alphaL-mannopyranosyl)oxy]-13-[[2R,5S,6R)-5-(dimethylamino) tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-9-ethyl2,3,3a,5a,5b,6,9,10,11,12,13,14,16a,16b-tetradecahydro-14-metyl-, (2R,3aS,5aR,5bS,9S,13S,14R,16aS,16bR)-
Spinosyn D: The chemical name is: 1H-as-Indaceno[3,2- d]oxacyclododecin-7,15-dione, 2-[(6-deoxy-2,3,4-tri-O-methyl-alphaL-mannopyranosyl)oxy]-13-[[2R,5S,6R)-5-(dimethylamino) tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-9-ethyl2,3,3a,5a,5b,6,9,10,11,12,13,14,16a,16b-tetradecahydro-4,14-dimetyl-, (2S,3aSR,5aS,5bS,9S,13S,14R,16aS,16bS)-
168316-95-8
- Molecular FormulaC83H132N2O20
- Average mass1477.938 Da
- Comfortis
- Conserve
- EC 434-300-1
- Natroba
- NaturaLyte
- Spinosad
- Tracer
- Tracer Naturalyte
- UNII-XPA88EAP6V
- XDE 105
Natroba (Spinosad) Suspension 0.9% ParaPro Pharma
New Drug Application (NDA): 022408 appr 01/18/2011
spinosad, is a new molecular entity, and a fermentation product produced by the actinomycete, Saccharopolyspora spinosa. Spinosad contains two components, spinosyn A and D. T

Figure 1. Structure of spinosyn A and DTitle: SpinosynsCAS Registry Number: 131929-60-7Literature References: Class of fermentation derived 12 membered macrocyclic lactones in a unique tetracyclic ring. At least 20 spinosyns have been isolated from Saccharopolyspora spinosa; variations in the two sugars account for most of the structural and insecticidal activity differences. Isolation and biological activity: L. D. Boeck et al.,EP375316 (1990 to Lilly); eidem,US5496931 (1996 to DowElanco); and structure determn: H. A. Kirst et al.,Tetrahedron Lett.32, 4839 (1991). Soil degradation: K. A. Hale, D. E. Portwood, J. Environ. Sci. HealthB31, 477 (1996). HPLC determn in vegetables: L.-T. Yeh et al.,J. Agric. Food Chem.45, 1746 (1997); in soil and water: S. D. West, ibid. 3107. Uptake and metabolism in larvae: T. C. Sparks et al.,Proc. Beltwide Cotton Conf.2, 1259 (1997). Mode of action study: V. L. Salgado et al.,Pestic. Biochem. Physiol.60, 103 (1998). Review of physical and biological properties: C. V. DeAmicis et al.,ACS Symp. Ser.658, 144-154 (1997). Review: G. D. Crouse, T. C. Sparks, Rev. Toxicol.2, 133-146 (1998).
Derivative Type: Spinosyn Acas 131929-60-7CAS Name: (2R,3aS,5aR,5bS,9S,13S,14R,16aS,16bR)-2-[(6-Deoxy-2,3,4-tri-O-methyl-a-L-mannopyranosyl)oxy]-13-[[(2R,5S,6R)-5-(dimethylamino)tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-9-ethyl-2,3,3a,5a,5b,6,9,10,11,12,13,14,16a,16b-tetradecahydro-14-methyl-1H-as-indaceno[3,2-d]oxacyclododecin-7,15-dioneAdditional Names: lepicidin AManufacturers’ Codes: A-83543A; LY-232105Molecular Formula: C41H65NO10Molecular Weight: 731.96Percent Composition: C 67.28%, H 8.95%, N 1.91%, O 21.86%Literature References: Total synthesis: L. A. Paquette et al.,J. Am. Chem. Soc.120, 2553 (1998).Properties: White, odorless crystalline solid, mp 118°. pKa 8.1. uv max (methanol): 243 nm (e 11000). [a]27436 -262.7° (methanol). Vapor pressure: 2.4 ´ 10-10. Soly in water (ppm): 290 (pH 5), 235 (pH 7), 16 (pH 9), distilled 20. Soly (w/v%): methanol 19, acetone 17, dichloromethane >50, hexane 0.45%. LD50 in rats (mg/kg): 3783-5000 orally (Crouse).Melting point: mp 118°pKa: pKa 8.1Optical Rotation: [a]27436 -262.7° (methanol)Absorption maximum: uv max (methanol): 243 nm (e 11000)Toxicity data: LD50 in rats (mg/kg): 3783-5000 orally (Crouse)
Derivative Type: Spinosyn DCAS Registry Number: 131929-63-0Manufacturers’ Codes: A-83543DMolecular Formula: C42H67NO10Molecular Weight: 745.98Percent Composition: C 67.62%, H 9.05%, N 1.88%, O 21.45%Properties: Odorless, white crystalline solid. mp 169°. pKa 7.8. uv max (methanol): 243 nm (e 11000). [a]27436 -297.5° (methanol). Vapor pressure: 2.0 ´ 10-10. Soly in water (ppm): 28 (pH 5), 0.329 (pH 7), 0.04 (pH 9), distilled 1.3. Soly (w/v%): methanol 0.25, acetone 1.0, dichloromethane 45, hexane 0.07%.Melting point: mp 169°pKa: pKa 7.8Optical Rotation: [a]27436 -297.5° (methanol)Absorption maximum: uv max (methanol): 243 nm (e 11000)
Derivative Type: SpinosadCAS Registry Number: 168316-95-8Manufacturers’ Codes: XDE-105; DE-105Trademarks: Conserve (Dow AgroSci.); Justice (Dow AgroSci.); Naturalyte (Dow AgroSci.); SpinTor (Dow AgroSci.); Success (Dow AgroSci.); Tracer (Dow AgroSci.)Literature References: Mixture of spinosyns A and D. Effect on beneficial insects: D. Murray, R. Lloyd, Australian Cottongrower18, 62 (1997).Properties: Light grey to white crystals (tech). LD50 in rats, mallard ducks, quail (mg/kg): >3600, >2000, >2000 orally (Crouse).Toxicity data: LD50 in rats, mallard ducks, quail (mg/kg): >3600, >2000, >2000 orally (Crouse)
Use: Insecticide.(2R,3aS,5aR,5bS,9S,13S,14R,16aS,16bR)-13-{[(2R,5S,6R)-5-(Dimethylamino)-6-methyltetrahydro-2H-pyran-2-yl]oxy}-9-ethyl-14-methyl-7,15-dioxo-2,3,3a,5a,5b,6,7,9,10,11,12,13,14,15,16a,16b-hexadecahydro-1H ;-as-indaceno[3,2-d]oxacyclododecin-2-yl 6-deoxy-2,3,4-tri-O-methyl-α-L-mannopyranoside – (2S,3aR,5aS,5bS,9S,13S,14R,16aS,16bS)-13-{[(2R,5S,6R)-5-(dimethylamino)-6-methyltetrahydro-2H-pyran-2-yl]ox y}-9-ethyl-4,14-dimethyl-7,15-dioxo-2,3,3a,5
1H-as-Indaceno[3,2-d]oxacyclododecin-7,15-dione, 2-[(6-deoxy-2,3,4-tri-O-methyl-α-L-mannopyranosyl)oxy]-13-[[(2R,5S,6R)-5-(dimethylamino)tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-9-ethyl-2,3,3a,5a,5b ,6,9,10,11,12,13,14,16a,16b-tetradecahydro-14-methyl-, (2R,3aS,5aR,5bS,9S,13S,14R,16aS,16bR)-, compd. with (2S,3aR,5aS,5bS,9S,13S,14R,16aS,16bS)-2-[(6-deoxy-2,3,4-tri-O-methyl-α-L-mannopyranosyl)o xy]-13-[[(2R,5S,6R)-5-(dimethylamino)tetrahySpinosad[USAN] [Wiki]168316-95-8 [RN]1H-as-Indaceno[3,2-d]oxacyclododecin-7,15-dione, 2-[(6-deoxy-2,3,4-tri-O-methyl-α-L-mannopyranosyl)oxy]-13-[[2R,5S,6R)-5-(dimethylamino) tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-9-ethyl-2,3,3a,5a,5b,6,9,10,11,12,13,14,16a,16b-tetradecahydro-4,14-dimetyl-,(2S,3aSR,5aS,5bS,9S,13S,14R,16aS,16bS)-1H-as-Indaceno[3,2-d]oxacyclododecin-7,a5-dione, 2-[(6-deoxy-2,3,4-tri-O-methyl-α-L-mannopyranosyl)oxy]-13-[[2R,5S,6R)-5-(dimethylamino) tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-9-ethyl-2,3,3a,5a,5b,6,9,10,11,12,13,14,16a,16b-tetradecahydro-14-metyl-, (2R,3aS,5aR,5bS,9S,13S,14R,16aS,16bR)-NAF-144Spinosad|spinosyn A and D (mixture)spinosyn A and D (mixture)
Spinosad is an insecticide based on chemical compounds found in the bacterial species Saccharopolyspora spinosa. The genus Saccharopolyspora was discovered in 1985 in isolates from crushed sugarcane. The bacteria produce yellowish-pink aerial hyphae, with bead-like chains of spores enclosed in a characteristic hairy sheath.[1] This genus is defined as aerobic, Gram-positive, nonacid-fast actinomycetes with fragmenting substrate mycelium. S. spinosa was isolated from soil collected inside a nonoperational sugar mill rum still in the Virgin Islands. Spinosad is a mixture of chemical compounds in the spinosyn family that has a generalized structure consisting of a unique tetracyclic ring system attached to an amino sugar (D-forosamine) and a neutral sugar (tri-Ο-methyl-L-rhamnose).[2] Spinosad is relatively nonpolar and not easily dissolved in water.[3]
Spinosad is a novel mode-of-action insecticide derived from a family of natural products obtained by fermentation of S. spinosa. Spinosyns occur in over 20 natural forms, and over 200 synthetic forms (spinosoids) have been produced in the lab.[4] Spinosad contains a mix of two spinosoids, spinosyn A, the major component, and spinosyn D (the minor component), in a roughly 17:3 ratio.[1
Mode of action
Spinosad is highly active, by both contact and ingestion, in numerous insect species.[5] Its overall protective effect varies with insect species and life stage. It affects certain species only in the adult stage, but can affect other species at more than one life stage. The species subject to very high rates of mortality as larvae, but not as adults, may gradually be controlled through sustained larval mortality.[5] The mode of action of spinosoid insecticides is by a neural mechanism.[6] The spinosyns and spinosoids have a novel mode of action, primarily targeting binding sites on nicotinic acetylcholine receptors (nAChRs) of the insect nervous system that are distinct from those at which other insecticides have their activity. Spinosoid binding leads to disruption of acetylcholine neurotransmission.[2] Spinosad also has secondary effects as a γ-amino-butyric acid (GABA) neurotransmitter agonist.[2] It kills insects by hyperexcitation of the insect nervous system.[2] Spinosad so far has proven not to cause cross-resistance to any other known insecticide.[7]
Use
Spinosad has been used around the world for the control of a variety of insect pests, including Lepidoptera, Diptera, Thysanoptera, Coleoptera, Orthoptera, and Hymenoptera, and many others.[8] It was first registered as a pesticide in the United States for use on crops in 1997.[8] Its labeled use rate is set at 1 ppm (1 mg a.i./kg of grain) and its maximum residue limit (MRL) or tolerance is set at 1.5 ppm. Spinosad’s widespread commercial launch was deferred, awaiting final MRL or tolerance approvals in a few remaining grain-importing countries. It is considered a natural product, thus is approved for use in organic agriculture by numerous nations.[5] Two other uses for spinosad are for pets and humans. Spinosad has recently been used in oral preparations (as Comfortis) to treat C. felis, the cat flea, in canines and felines; the optimal dose set for canines is reported to be 30 mg/kg.[2]
Spinosad is sold under the trade names, Comfortis, Trifexis, and Natroba.[9][10] Trifexis also includes milbemycin oxime. Comfortis and Trifexis brands treat adult fleas on pets; the latter also prevents heartworm disease. Natroba is sold for treatment of human head lice. Spinosad is also commonly used to kill thrips.[11][12][13]
Spinosyn A
Spinosyn A does not appear to interact directly with known insecticidal-relevant target sites, but rather acts via a novel mechanism.[6] Spinosyn A resembles a GABA antagonist and is comparable to the effect of avermectin on insect neurons.[4] Spinosyn A is highly active against neonate larvae of the tobacco budworm, Heliothis virescens, and is slightly more biologically active than spinosyn D. In general, spinosyns possessing a methyl group at C6 (spinosyn D-related analogs) tend to be more active and less affected by changes in the rest of the molecule.[7] Spinosyn A is slow to penetrate to the internal fluids of larvae; it is also poorly metabolized once it enters the insect.[7] The apparent lack of spinosyn A metabolism may contribute to its high level of activity, and may compensate for the slow rate of penetration.[7]
Safety and ecotoxicology
Spinosad has high efficacy, a broad insect pest spectrum, low mammalian toxicity, and a good environmental profile, a unique feature of the insecticide compared to others currently used for the protection of grain products.[5] It is regarded as natural product-based, and approved for use in organic agriculture by numerous national and international certifications.[8] Spinosad residues are highly stable on grains stored in bins, with protection ranging from 6 months to 2 years.[5][clarification needed] Ecotoxicology parameters have been reported for spinosad, and are:[14]
- in rat (Rattus norvegicus Bergenhout, 1769), acute oral: LD50 >5000 mg/kg (nontoxic)
- in rat (R. norvegicus), acute dermal: LD50 >2000 mg/kg (nontoxic)
- in California quail (Callipepla californica Shaw, 1798), oral toxicity: LD50 >2000 mg/kg (nontoxic)
- in duck (Anas platyrhynchos domestica Linnaeus, 1758), dietary toxicity: LC50 >5000 mg/kg (nontoxic)
- in rainbow trout (Oncorhynchus mykiss Walbaum, 1792), LC50-96h = 30.0 mg/l (slightly toxic)
- in Honeybee (Apis mellifera Linnaeus, 1758), LD50 = 0.0025 mg/bee (highly toxic if directly sprayed on and of dried residues).
Chronic exposure studies failed to induce tumor formation in rats and mice; mice given up to 51 mg/kg/day for 18 months resulted in no tumor formation.[15] Similarly, administration of 25 mg/kg/day to rats for 24 months did not result in tumor formation.[16]
syn
EP 0375,316 (1994, to DowElanco)
US 5496931 (1996 to DowElanco)
PATENT
CN 102190694
https://patents.google.com/patent/CN102190694A/en
Pleocidin compounds (spinosyns) is soil actinomycete thorn many armfuls of bacterium Saccharopolysporaspinosa of sugar secondary metabolites behind aerobic fermentation under developing medium.Pleocidin belongs to macrolides compound, it comprises one a plurality of chiral carbon tetracyclic ring systems (Macrolide tetracycle), big ring is gone up the 9-hydroxyl and is being linked two different hexa-atomic sugar respectively with the 17-hydroxyl, wherein that 17 connections is an aminosugar (Forosamine sugar), and that connect on the 9-position is a rhamnosyl (Rhamnose sugar).Tetracyclic ring system is by one 5,, 6,5-is suitable-and anti–anti–three-loop system condenses one 12 membered macrolide to be formed, and wherein contains an alpha, beta-unsaturated ketone and an independently two key.When 6 on ring is pleocidin A when being substituted by hydrogen, in mixture, account for 85-90%, when ring 6 bit substituents when connecing methyl, be pleocidin D then, in mixture, account for about 10-15%.Up to the present B, C, D, E, F, G, K, L, M, N, O, P, Q, R, S, T, U, more than 20 derivative such as V, W etc. have been found and have isolated it to comprise Spinosyn A.
The commercialization kind has pleocidin Spinosyns (mixture of pleocidin A and pleocidin D) at present, the s-generation pleocidin insecticides Spinetoram. latter is got through semisynthesis by the thick product pleocidin L of biological method preparation and the mixture of J, promptly by 5 of pleocidin J, 6 two key selective reductions, reach 3 ‘ O-ethylization of rhamnosyl and obtain its major ingredient, ethylizing by 3 ‘ O-of pleocidin L rhamnosyl obtains its minor consistuent.
The pleocidin compound can be controlled lepidopteran, Diptera and Thysanoptera insect effectively.It can prevent and treat the pest species of some blade of eating in a large number in Coleoptera and the Orthoptera well.Pleocidin has very high activity to lepidopterous larvaes such as Heliothis virescens, bollworm, beet armyworm, prodenia litura, cabbage looper, small cabbage moth and rice-stem borers, and they are suitable environmental protection, have interesting toxicology character.
U.S. Patent No. 5362634 discloses the derivative that natural pleocidin is replaced by methyl or ethyl on C-21, U.S. Patent application No.60/153513 has disclosed the natural butenyl pleocidin derivative that the 3-4 carbochain replaces on C-21.Pleocidin derivative (John Daeuble, ThomasC.Sparks, Peter Johnson, Paul R.Graupner, the Bioorganic ﹠amp that can prepare C-21 position different substituents by replacement(metathesis)reaction; Medicinal Chemistry17 (2009) 4197-4205).U.S. Patent No. 6001981A, WO 9700265A have openly opened the chemosynthesis of pleocidin compound and have modified, and comprise aminosugar and rhamnosyl and the big chemically modified that encircles in the structure.
PATENT
https://patents.google.com/patent/WO2002077005A1/en
Spinosyns (A83543) are produced by derivatives of Saccharopolyspora spinosa NRRL18395 including strains NRRL 18537, 18538, 18539, 18719, 18720, 18743 and 18823 and derivatives thereof. A more preferred nomenclature for spinosyns is to refer to the pseudoaglycones as spinosyn A 17-Psa, spinosyn D 17-Psa, etc., and to the reverse pseudoaglycones as spinosyn A 9-Psa, spinosyn D 9-Psa, etc. (see Kirst et al., 1991). The known members of this family have been referred to as factors or components, and each has been given an identifying letter designation. These compounds are hereinafter referred to as spinosyn A, B, etc. The spinosyn compounds are useful for the control of arachnids, nematodes and insects, in particular Lepidoptera and Diptera species, and they are quite environmentally friendly and have an appealing toxicological profile. [0004] U.S. Patent No. 5,362,634 and corresponding European Patent Application No. 375316 Al disclose spinosyns A, B, C, D, E, F, G, H, and J. WO 93/09126 discloses spinosyns L, M, N, Q, R, S, and T. WO 94/20518 and US 5,6704,486 disclose spinosyns K,
O, P, U, V, W, and Y, and derivatives thereof. A large number of synthetic modifications to spinosyn compounds have been made, as disclosed in U.S. Patent No. 6,001,981 and WO
97/00265.
PAPER
J. Am. Chem. Soc. 120, 2553 (1998).
Further reading
- The non‐target impact of spinosyns on beneficial arthropods
- Spinosad toxicity to pollinators and associated risk
References
- ^ Jump up to:a b Mertz, Frederick; Raymond C. Yao (Jan 1990). “Saccharopolyspora spinosa sp. nov. Isolated from soil Collected in a Sugar Mill Rum Still”. International Journal of Systematic Bacteriology. 40 (1): 34–39. doi:10.1099/00207713-40-1-34.
- ^ Jump up to:a b c d e Qiao, Meihua; Daniel E. Snyder; Jeffery Meyer; Alan G. Zimmerman; Meihau Qiao; Sonya J. Gissendanner; Larry R. Cruthers; Robyn L. Slone; Davide R. Young (12 September 2007). “Preliminary Studies on the effectiveness of the novel pulicide, spinosad, for the treatment and control of fleas on dogs”. Veterinary Parasitology. 150 (4): 345–351. doi:10.1016/j.vetpar.2007.09.011. PMID 17980490.
- ^ Crouse, Gary; Thomas C Sparks; Joseph Schoonover; James Gifford; James Dripps; Tim Brue; Larry L Larson; Joseph Garlich; Chris Hatton; Rober L Hill; Thomas V Worden; Jacek G Martynow (27 September 2000). “Recent advances in the chemistry of spinosyns”. Pest Manag Sci. 57 (2): 177–185. doi:10.1002/1526-4998(200102)57:2<177::AID-PS281>3.0.CO;2-Z. PMID 11455648.
- ^ Jump up to:a b Watson, Gerald (31 May 2001). “Actions of Insecticidal Spinosyns on gama-Aminobutyric Acid Responses for Small-Diameter Cockroach Neurons”. Pesticide Biochemistry and Physiology. 71: 20–28. doi:10.1006/pest.2001.2559.
- ^ Jump up to:a b c d e Hertlein, Mark; Gary D. Thompson; Bhadriraju Subramanyam; Christos G. Athanassiou (12 January 2011). “Spinosad: A new natural product for stored grain protection”. Stored Products. 47 (3): 131–146. doi:10.1016/j.jspr.2011.01.004. Retrieved 3 May 2012.
- ^ Jump up to:a b Orr, Nailah; Andrew J. Shaffner; Kimberly Richey; Gary D. Crouse (30 April 2009). “Novel mode of action of spinosad: Receptor binding studies demonstrating lack of interaction with known insecticidal target sites”. Pesticide Biochemistry and Physiology. 95: 1–5. doi:10.1016/j.pestbp.2009.04.009.
- ^ Jump up to:a b c d Sparks, Thomas; Gary D crouse; Gregory Durst (30 March 2001). “Natural products as insecticides: the biology, biochemistry and quantitative structure-activity relationships of spinosyns and spinosoids”. Pest Manag Sci. 57 (10): 896–905. doi:10.1002/ps.358. PMID 11695182.
- ^ Jump up to:a b c Sparks, Thomas; James E. Dripps; Gerald B Watson; Doris Paroonagian (6 November 2012). “Resistance and cross-resistance to the spinosyns- A review and analysis”. Pesticide Biochemistry and Physiology. 102: 1–10. doi:10.1016/j.pestbp.2011.11.004. Retrieved 17 November 2011.
- ^ “Spinosad international brands”. Drugs.com. 3 January 2020. Retrieved 30 January2020.
- ^ “Spinosad US brands”. Drugs.com. 3 January 2020. Retrieved 30 January 2020.
- ^ “Spinosad – brand name list from”. Drugs.com. Retrieved 2012-10-20.
- ^ “UC Davis School of Vet Med”. Vetmed.ucdavis.edu. Retrieved 2012-10-20.
- ^ “Safer Flea Control | Insects in the City”. Citybugs.tamu.edu. Retrieved 2012-10-20.
- ^ “Codling Moth and Leafroller Control Using Chemicals” (PDF). Entomology.tfrec.wsu.edu. Retrieved 2012-10-20.
- ^ Stebbins, K. E. (2002). “Spinosad Insecticide: Subchronic and Chronic Toxicity and Lack of Carcinogenicity in CD-1 Mice”. Toxicological Sciences. 65 (2): 276–287. doi:10.1093/toxsci/65.2.276. PMID 11812932. Retrieved 2015-03-08.
- ^ Yano, B. L. (2002). “Spinosad Insecticide: Subchronic and Chronic Toxicity and Lack of Carcinogenicity in Fischer 344 Rats”. Toxicological Sciences. 65 (2): 288–298. doi:10.1093/toxsci/65.2.288. PMID 11812933. Retrieved 2015-03-08.
External links
- “Spinosad”. Drug Information Portal. U.S. National Library of Medicine.
- Monograph
| Spinosyn A | |
| Spinosyn D | |
| Identifiers | |
|---|---|
| CAS Number | 168316-95-8 (A)131929-60-7 (D) |
| ChEBI | CHEBI:9230 (A) CHEBI:9232 (D) |
| ChEMBL | ChEMBL1615373 |
| ChemSpider | 16736513 |
| ECHA InfoCard | 100.103.254 |
| PubChem CID | 183094 (A)443059 (D) |
| CompTox Dashboard (EPA) | DTXSID7032478 |
| InChI[show] | |
| Properties | |
| Chemical formula | C41H65NO10 (A) C42H67NO10 (D) |
| Pharmacology | |
| ATCvet code | QP53BX03 (WHO) |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). |
//////////Spinosad
Dr. Darrin Lew https://www.drdarrinlew.us/insect-control/production-of-spinosad.html
Production of Spinosad
Last Updated on Tue, 29 Oct 2019 | Insect Control
Spinosad is produced directly from the fermentation of a strain of Saccharo-polyspora spinosa. Production strains of S. spinosa have been selected for increased titers of spinosyns A and D, however, no genetic engineering techniques have been used in this process and no genetically-modified organisms are used in the production process. After fermentation, the spinosyn A and D mixture is extracted from the fermentation broth, precipitated and dried to create technical spinosad, which is then formulated into end-use products. Spinosad technical material is also produced under pharmaceutical manufacturing guidelines to be used as a flea control agent in companion animals.
5.9.2 Production of Spinetoram
Production of spinetoram begins with the fermentation of a mutant strain of Saccharopolyspora spinosa that produces primarily spinosyns J and L, rather than spinosyns A and D. This strain was generated through mutagenesis of S. spinosa. However, like the spinosad-producing strains, no genetic engineering techniques were used in this process and no genetically-modified organisms are used in the production process. After fermentation, the spinosyn J and L mixture is extracted from the fermentation broth and precipitated in preparation for the two chemical synthesis steps required to produce spinetoram. The solvents used in extracting and precipitating the spinosyn J and L mixture are recycled.
Spinosyns J and L, unlike spinosyns A and D, have a free hydroxyl group at the 30-position on the rhamnose sugar, which allows for chemical manipulation of this site (see Figure 5.10). In the first synthetic step, the free hydroxyl at the 30-position in spinosyn J and spinosyn L is ethylated to yield a mixture of 30-O-ethyl spinosyn J and 30-O-ethyl spinosyn L. This material is then hydrogenated to yield a mixture of spinetoram-J (30-O-ethyl-5,6-dihydro spinosyn J; see Figure 5.2, structure 5.5) and spinetoram-L (30-O-ethyl spinosyn L; see Figure 5.2, structure 5.6). The hydrogenation conditions are selective and reduce only the disubstituted double bond between C5 and C6 in the 30-O-ethyl spinosyn J intermediate, leaving the 30-O-ethyl spinosyn L unchanged. The material is crystallized from the reaction mixture and dried to create technical spinetoram, which is then formulated into end-use products.
5.9.3 Formulation Attributes of the Spinosyns
To meet a variety of market needs, spinosad and spinetoram products span a very wide range of formulation types (see Table 5.8).
The range of possible formulations for any pesticide is determined by the physical and chemical properties of the active ingredient. Three primary properties determine the formulation characteristics of the spinosyns: (1) both Figure 5.10 Chemical synthesis steps in spinetoram manufacturing.
Figure 5.10 Chemical synthesis steps in spinetoram manufacturing.
Table 5.8 Spinosyn product formulation types and associated uses.
Formulation type
Use pattern
Suspension concentrate
Emulsifiable concentrate Wettable granule Wettable powder Dustable powder Sprayable bait Granular bait Bait stations Granules Tablets
Chewable tablets Gel, paste Creme rinse
Crops, ornamentals, forestry, stored grain, animal health, public health, turf, home and garden Public health Crops
Crops, ornamentals, seed treatment
Stored grain, crops
Crops
Crops, animal health, urban pests
Urban pests
Public health
Public health
Animal health
Urban pests
Public health are fermentation-derived mixtures; (2) both are weak bases; and (3) both have significant solubility in organic solvents.
As fermentation-derived products, spinosad and spinetoram are mixtures composed primarily of two similar, but not identical molecules. In terms of physical properties, a significant difference between the major and minor components of both spinosad and spinetoram is the presence or absence of a methyl group at C6 on the tetracycle (see Table 5.9). With regard to components of spinosad, spinosyn D (methyl group at C6) has a melting point 71 °C higher than that of spinosyn A (hydrogen at C6), and the water solubility of spinosyn D (at pH 7) is almost 1000-fold lower than that of spinosyn A. With regard to the components of spinetoram, spinetoram-L (methyl group at C6) has a melting point 72 °C lower than that of spinetoram-J (hydrogen at C6), and the water solubility of spinetoram-L (at pH 7) is four-fold higher than that of spinetoram-J. The melting points and water solubilities of the mixtures that constitute technical spinosad and technical spinetoram are determined by the relative ratios of the major and minor components.
The predominant components of both spinosad and spinetoram all have pKa values of about 8 (see Table 5.9). As a weak base, the solubility of spinosyns in water increases as the pH is reduced. From a formulation perspective, at pH level above 5, the spinosyns behave like high-melting solids with little water solubility, which results in the predominant agricultural formulations being suspension concentrates and wettable granule formulations composed of milled crystalline particles. Acid salts of spinosyns can be produced and are used in animal health formulations. The basic nature of the spinosyns is also a consideration when combining multiple active ingredients into the same formulation.
The spinosyns have significant solubility in organic solvents (see Table 5.9). This property is relatively rare in high-melting solids with limited water solubility, and has proven to be useful in a number of formulations for
Table 5.9 Selected physical properties of spinosyn A, spinosyn D, spinetoram-
J, and spinetoram-L.
Table 5.9 Selected physical properties of spinosyn A, spinosyn D, spinetoram-
J, and spinetoram-L.
| Property | Spinosyn A133 | Spinosyn D133 | Spinetoram-J134 | Spinetoram-L134 |
| Melting point, °C | 84-99.5a | 161.6-170a | 143.4b | 70.8b |
| Water solubility, | 235 | 0.332 | 11.3 | 46.7 |
| mg/lc’d’e | ||||
| pKaf | 8.10e | 7.87e | 7.86g | 7.59g |
| Solubility in organic solvents, mg/Lc | ||||
| Acetone | 168 000 | 10100 | >250000 | >250000 |
| Ethyl acetate | 194000 | 19 000 | >250000 | >250000 |
| w-Heptane | 12 400 | 300 | 23 900 | >250000 |
| Methanol | 190000 | 2520 | 163 000 | >250000 |
| Xylene | > 250 000 | 64000 | >250000 | >250000 |
“Visual determination. bDiffential scanning calorimetry. cShake flask. ^Buffered to pH 7. eAt 20 °C.
fCapillary zone electrophoresis. gAt 25 °C.
“Visual determination. bDiffential scanning calorimetry. cShake flask. ^Buffered to pH 7. eAt 20 °C.
fCapillary zone electrophoresis. gAt 25 °C.
non-agricultural markets, such as mosquito control and animal health. It is also a consideration when combining the spinosyns with other active ingredients.
////////
https://aem.asm.org/content/82/18/5603


{kind=link}
{kind=link}
{kind=link}
{kind=link}