
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
IIIM-290

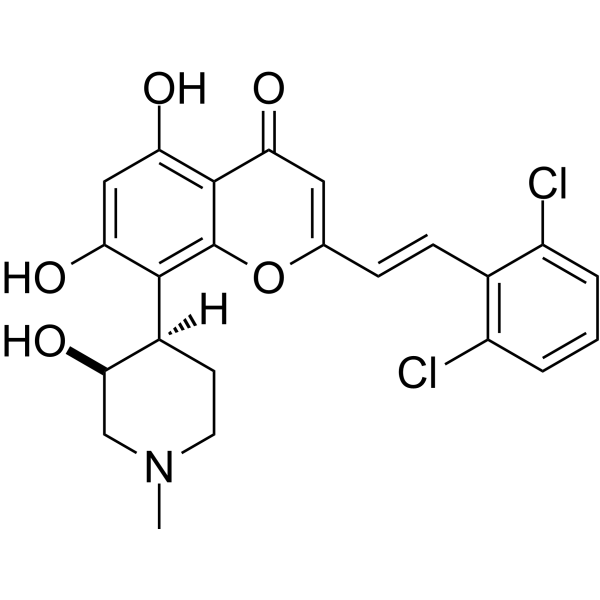
IIIM-290
4H-1-Benzopyran-4-one, 2-[2-(2,6-dichlorophenyl)ethenyl]-5,7-dihydroxy-8-[(3S,4R)-3-hydroxy-1-methyl-4-piperidinyl]-
| Molecular Weight |
462.32 |
|---|---|
| Formula |
C₂₃H₂₁Cl₂NO₅ |
| CAS No. |
2213468-64-3 |
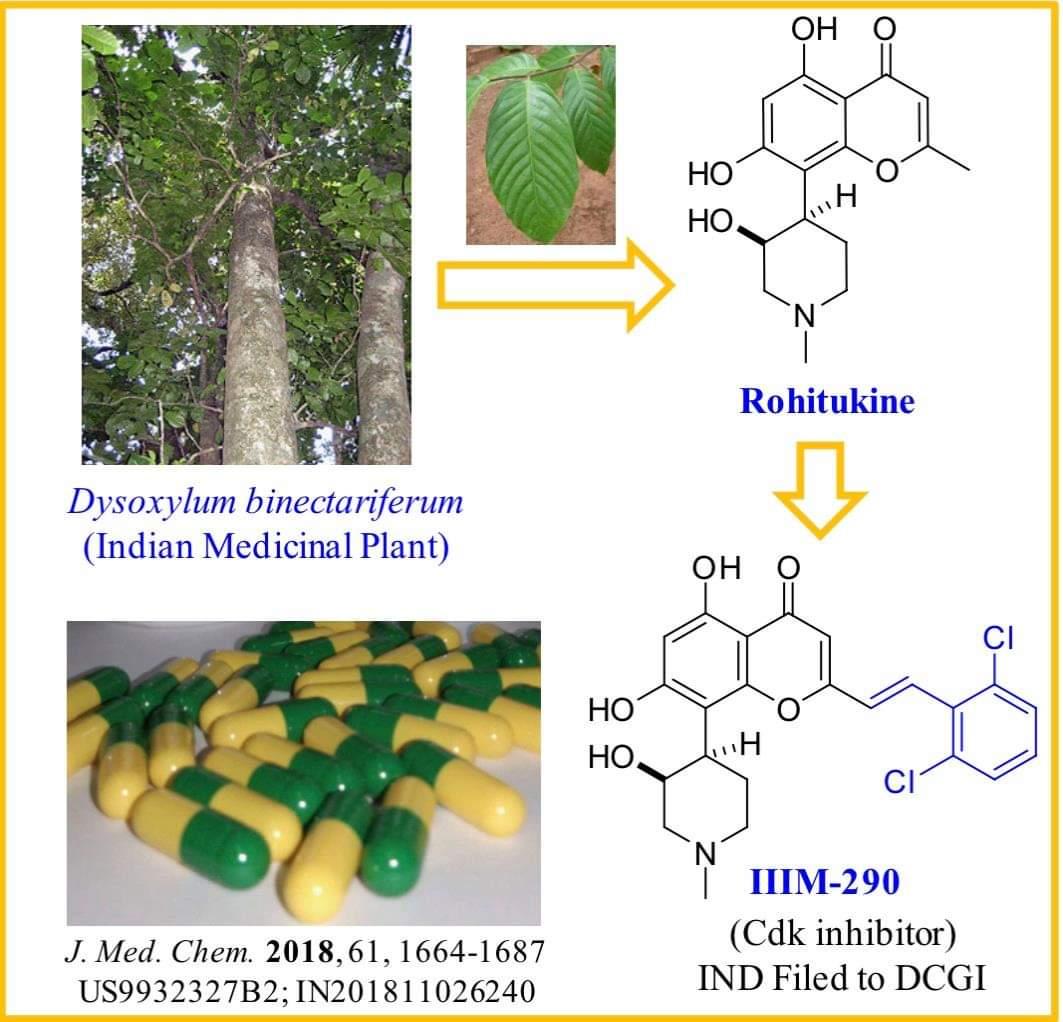
CSIR-IIIM Jammu has filed an IND Application of “IIIM-290” to Drug Controller General of India for conducting Phase I/Phase II clinical trial of its capsule formulation in patients with locally advanced or metastatic pancreatic cancer. This IND candidate has emerged from the eight years of medicinal chemistry/ preclinical efforts of IIIM Jammu in the area of small molecule kinase inhibitors. IIIM-290 (NCE) is an orally bioavailable CDK inhibitor, obtained via semisynthetic modification of a natural product rohitukine. Institute has already secured a patent on this small molecule as well as on its oral capsule formulation.

IIIM-290 is a potent and oral CDK inhibitor with IC50s of 90 and 94 nM for CDK2/A and CDK9/T1.


PAPER
https://pubs.acs.org/doi/pdf/10.1021/acs.jmedchem.7b01765
Discovery and Preclinical Development of IIIM-290, an Orally Active Potent Cyclin-Dependent Kinase Inhibitor
- Sandip B. Bharate
- Vikas Kumar
- Shreyans K. Jain
- Mubashir J. Mintoo
- Santosh K. Guru
- Vijay K. Nuthakki
- Mohit Sharma
- Sonali S. Bharate
- Sumit G. Gandhi
- Dilip M. Mondhe
- Shashi Bhushan
- Ram A. Vishwa
Abstract

Rohitukine (1), a chromone alkaloid isolated from Indian medicinal plant Dysoxylum binectariferum, has inspired the discovery of flavopiridol and riviciclib, both of which are bioavailable only via intravenous route. With the objective to address the oral bioavailability issue of this scaffold, four series of rohitukine derivatives were prepared and screened for Cdk inhibition and cellular antiproliferative activity. The 2,6-dichloro-styryl derivative IIIM-290 (11d) showed strong inhibition of Cdk-9/T1 (IC50 1.9 nM) kinase and Molt-4/MIAPaCa-2 cell growth (GI50 < 1.0 μM) and was found to be highly selective for cancer cells over normal fibroblast cells. It inhibited the cell growth of MIAPaCa-2 cells via caspase-dependent apoptosis. It achieved 71% oral bioavailability with in vivo efficacy in pancreatic, colon, and leukemia xenografts at 50 mg/kg, po. It did not have CYP/efflux-pump liability, was not mutagenic/genotoxic or cardiotoxic, and was metabolically stable. The preclinical data presented herein indicates the potential of 11d for advancement in clinical studies.


Patent
IN201811026240



Patent
InventorRam A. VishwakarmaSandip B. BharateShashi BhushanDilip M. MondheShreyans K. JainSamdarshi MeenaSantosh K. GuruAnup S. PathaniaSuresh KumarAkanksha BehlMubashir J. MintooSonali S. BharatePrashant Joshi Current Assignee Council of Scientific and Industrial Research (CSIR)
https://patents.google.com/patent/US9932327B2/en
The disruption of any internal and external regulation of cellular growth leads to tumorogenesis by uncontrolled proliferation. This loss of control occurs at multiple levels in most of the cancer cases. Cyclin-dependent kinases (CDKs) have been recognized as key regulators of cell cycle progression. Alteration and deregulation of CDK activity have pathogenic link to the cancer. Number of cancers are associated with hyper-activation of CDKs as a result of mutation of the CDK genes or CDK inhibitor genes. Therefore, CDK inhibitors or modulators are of great interest to explore as novel therapeutic agents against cancer (Senderowicz, A. M. Leukemia 2001, 15, 1). Several classes of chemical inhibitors of CDK activity have been described (Zhang, J. et. al. Nat Rev Cancer. 2009, 9, 28) and some of them have reached to clinical pipeline for cancer.
Because CDK inhibitors are ATP competitive ligands; hence earlier they were typically described as purine class of compounds for example dimethylaminopurine, a first substance to be known as a CDK inhibitor (Neant, I. et al. Exp. Cell Res. 1988, 176, 68), olomoucine (Vesely, J. et al. Eur. J. Biochem. 1994, 224, 771) and roscovitine (Meijer, L. et al. Eur. J. Biochem. 1997, 243, 527). The IC50values of these purine class of compounds for CDK1/cyclin B are 120, 7 and 0.2-0.8 μM respectively (Gray, N. et al. Curr. Med. Chem. 1999, 6, 859). Some of the more potent members of this series have been prepared by the Schultz group using combinatorial approaches (Gray, N. S. et al. Science 1998, 281, 533). Number of synthetic flavoalkaloids having potent CDK inhibitory activity has been reviewed recently (Jain, S. K. et al. Mini–Rev. Med. Chem. 2012, 12, 632).
Specific CDKs operate in distinct phases of the cell cycle. CDK complexes with their respective type cyclin partners such as, complex of CDK2 and cyclin A is responsible for the cell’s progression from G1 phase to S phase (Sherr, C. J. Science 1996, 274, 1672). DNA synthesis (S phase) begins with the CDK mediated phosphorylation of Rb (retinoblastoma) protein. Phosphorylated Rb is released from its complex with E2F. The released E2F then promotes the transcription of numerous genes required for the cell to progress through S phase, including thymidylate synthase and dihydrofolate reductase which are required for cell progression (Hatakeyama, M. et. al, Cell Cycle Res. 1995, 1, 9; Zhang, H. S. et. al. Cell 1999, 97, 53). Majority of human cancers have abnormalities in some component of the Rb pathway because of hyper-activation of CDKs resulting from the over-expression of positive cofactors (cyclins/CDKs) or a decrease in negative factors (endogenous CDK inhibitors) or Rb gene mutations (Sausville, E. A. et. al, Pharmacol. Ther. 1999, 82, 285).
The CDK-9 is a member of the Cdc2-like family of kinases. Its cyclin partners are members of the family of cyclin T (T1, T2a and T2b) and cyclin K. The CDK-9/cyclin T complexes appear to be involved in regulating several physiological processes. CDK9/cyclin T1 belongs to the P-TEFb complex, and is responsible for the phosphorylation of carboxyl terminal domain of the RNA Polymerase II, thus promoting general elongation. CDK-9 has also been described as the kinase of the TAK complex, which is homologous to the P-TEFb complex and is involved in HIV replication. CDK9 also appears to be involved in the differentiation program of several cell types, such as muscle cells, monocytes and neurons, suggesting that it may have a function in controlling specific differentiative pathways. In addition, CDK-9 seems to have an anti-apoptotic function in monocytes, that may be related to its control over differentiation of monocytes. This suggests the involvement of CDK-9 in several physiological processes in the cell, the deregulation of which may be related to the genesis of transforming events that may in turn lead to the onset of cancer. In addition, since the complex CDK-9/cyclin T1 is able to bind to the HIV-1 product Tat, the study of the functions of CDK-9/cyclin T may be of interest in understanding the basal mechanisms that regulate HIV replication (Falco, G. D. and Giordano A. Cancer Biol. Therapy 2002, 1, 337).
Rohitukine belongs to a class of chromone alkaloids and it was isolated by chemists at Hoechst India Ltd. in the early 1990’s from Dysoxylum binectariferum Hook. which is phylogenetically related to the Ayurvedic plant, D. malabaricum Bedd., used for rheumatoid arthritis. Rohitukine was isolated as the constituent responsible for anti-inflammatory and immunomodulatory activity (Naik, R. G. et. al. Tetrahedron 1988, 44, 2081; U.S. Pat. No. 4,900,727, 1990). Medicinal chemistry efforts around this nature-derived flavone alkaloid led to discovery of two promising clinical candidates for treatment of cancer viz. flavopiridol of Sanofi-Aventis and P-276-00 of Piramal life sciences. Recently FDA has granted the orphan drug status to flavopiridol for treatment of chronic lymphocytic leukemia (CLL).
The molecular formula of rohitukine is C16H19NO5 and the structure has a molecular weight of 305.32 g/mol. The chemical structure of rohitukine (1) is shown below. The present invention reports new semi-synthetic analogs of rohitukine as promising inhibitors of cyclin-dependent kinases such as CDK-2 and CDK-9.

Synthesis of styryl analog 2-(2,6-dichlorostyryl)-5,7-dihydroxy-8-(3-hydroxy-1-methylpiperidin-4-yl)-4H-chromen-4-one (33)
This compound was synthesized using the procedure as described in example 4. Yellow solid; 1H NMR (DMSO-d6, 400 MHz): δ 7.68 (m, 2H), 7.61 (d, J=16 Hz, 1H), 7.49 (t, J=8 Hz, 1H), 7.14 (d, J=16 Hz, 1H), 6.41 (s, 1H), 5.85 (s, 1H), 4.53 (brs, 1H), 3.10-2.50 (m, 6H of piperidine), 2.65 (s, 3H), 1.62 (m, 1H); 13C NMR (DMSO-d6, 125 MHz): δ 179.68. 171.27, 159.20, 158.02, 154.03, 133.12, 131.49, 129.75, 128.35 (2C), 128.20, 127.90, 108.81, 106.79, 100.88, 100.52, 66.35, 59.82, 54.45, 43.15, 35.79, 22.01, 20.33, ESI-MS: m/z 462.01 [M+H]+; IR (CHCl3): νmax 3400, 2921, 1652, 1577, 1550, 1417, 1380, 1191, 1085 cm−1.
///////////IIIM-290, nda, india, phase 1, dcgi, CSIR, ROHITUKINE
|
OC1=C2C(OC(/C=C/C3=C(Cl)C=CC=C3Cl)=CC2=O)=C([C@]4([H])[C@H](O)CN(C)CC4)C(O)=C1 |
Zanubrutinib, ザヌブルチニブ , занубрутиниб , زانوبروتينيب ,
Zanubrutinib, BGB-3111
| Formula |
C27H29N5O3
|
|---|---|
| CAS |
1691249-45-2
|
| Mol weight |
471.5509
|
FDA , 2019/11/14, Brukinsa
ザヌブルチニブ ,
Antineoplastic, Bruton’s tyrosine kinase inhibitor, Mantle cell lymphoma
NEW PA
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023062504&_gid=202316
Zanubrutinib, sold under the brand name Brukinsa, is for the treatment of adult patients with mantle cell lymphoma (MCL) who have received at least one prior therapy.[3]
It was approved for medical use in the United States in November 2019.[4][3][5][6]
Zanubrutinib is classified as a Bruton’s tyrosine kinase (BTK) inhibitor. It is administered orally.
History
Efficacy was evaluated in BGB-3111-206 (NCT03206970), a phase II open-label, multicenter, single-arm trial of 86 patients with mantle cell lymphoma (MCL) who received at least one prior therapy.[5] Zanubrutinib was given orally at 160 mg twice daily until disease progression or unacceptable toxicity.[5] Efficacy was also assessed in BGB-3111-AU-003 (NCT 02343120), a phase I/II, open-label, dose-escalation, global, multicenter, single-arm trial of B‑cell malignancies, including 32 previously treated MCL patients treated with zanubrutinib administered orally at 160 mg twice daily or 320 mg once daily.[5][6]
The primary efficacy outcome measure in both trials was overall response rate (ORR), as assessed by an independent review committee.[5] In trial BGB-3111-206, FDG-PET scans were required and the ORR was 84% (95% CI: 74, 91), with a complete response rate of 59% (95% CI 48, 70) and a median response duration of 19.5 months (95% CI: 16.6, not estimable).[5] In trial BGB-3111-AU-003, FDG-PET scans were not required and the ORR was 84% (95% CI: 67, 95), with a complete response rate of 22% (95% CI: 9, 40) and a median response duration of 18.5 months (95% CI: 12.6, not estimable).[5] Trial 1 was conducted at 13 sites in China, and Trial 2 was conducted at 25 sites in the United States, United Kingdom, Australia, New Zealand, Italy, and South Korea.[6]
The U.S. Food and Drug Administration (FDA) granted zanubrutinib priority review, accelerated approval, breakthrough therapydesignation, and orphan drug designation.[3][5][7]
The FDA approved zanubrutinib in November 2019, and granted the application for Brukinsa to BeiGene USA Inc.[3][5][8]
PAPER
https://www.x-mol.com/paper/5799457
Discovery of Zanubrutinib (BGB-3111), a Novel, Potent, and Selective Covalent Inhibitor of Bruton’s Tyrosine Kinase Journal of Medicinal Chemistry ( IF 6.054 ) Pub Date: 2019-08-19 , DOI: 10.1021 / acs.jmedchem.9b00687
Yunhang Guo, Ye Liu, Nan Hu, Desheng Yu, Changyou Zhou, Gongyin Shi, Bo Zhang, Min Wei, Junhua Liu, Lusong Luo, Zhiyu Tang, Huipeng Song, Yin Guo, Xuesong Liu, Dan Su, Shuo Zhang, Xiaomin Song , Xing Zhou, Yuan Hong, Shuaishuai Chen, Zhenzhen Cheng, Steve Young, Qiang Wei, Haisheng Wang, Qiuwen Wang, Lei Lv, Fan Wang, Haipeng Xu, Hanzi Sun, Haimei Xing, Na Li, Wei Zhang, Zhongbo Wang, Guodong Liu, Zhijian Sun, Dongping Zhou, Wei Li, Libin Liu, Lai Wang, Zhiwei Wang
 |
Bruton’s tyrosine kinase (Btk) belongs to the Tec tyrosine kinase family (Vetrie et al., Nature 361: 226-233, 1993; Bradshaw, Cell Signal. 22: 1175-84, 2010). Btk is primarily expressed in most hematopoietic cells such as B cells, mast cells and macrophages (Smith et al., J. Immunol. 152: 557-565, 1994) and is localized in bone marrow, spleen and lymph node tissue. Btk plays important roles in B-cell receptor (BCR) and FcR signaling pathways, which involve in B-cell development, differentiation (Khan, Immunol. Res. 23: 147, 2001). Btk is activated by upstream Src-family kinases. Once activated, Btk in turn phosphorylates PLC gamma, leading to effects on B-cell function and survival (Humphries et al., J. Biol.Chem. 279: 37651, 2004).
[0003] These signaling pathways must be precisely regulated. Mutations in the gene encoding Btk cause an inherited B-cell specific immunodeficiency disease in humans, known as X-linked agammaglobulinemia (XLA) (Conley et al., Annu. Rev. Immunol. 27: 199-227, 2009). Aberrant BCR-mediated signaling may result in dysregulated B-cell activation leading to a number of autoimmune and inflammatory diseases. Preclinical studies show that Btk deficient mice are resistant to developing collagen- induced arthritis. Moreover, clinical studies of Rituxan, a CD20 antibody to deplete mature B-cells, reveal the key role of B-cells in a number of inflammatory diseases such as rheumatoid arthritis, systemic lupus erythematosus and multiple sclerosis (Gurcan et al, Int. Immunopharmacol. 9: 10-25, 2009). Therefore, Btk inhibitors can be used to treat autoimmune and/or inflammatory diseases.
[0004] In addition, aberrant activation of Btk plays an important role in pathogenesis of B-cell lymphomas indicating that inhibition of Btk is useful in the treatment of hematological malignancies (Davis et al, Nature 463: 88-92, 2010). Preliminary clinical trial results showed that the Btk inhibitor PCI-32765 was effective in treatment of several types of B-cell lymphoma (for example, 54thAmerican Society of Hematology (ASH) annual meeting abstract, Dec. 2012: 686 The Bruton’s Tyrosine Kinase (Btk) Inhibitor, Ibrutinib (PCI- 32765), Has Preferential Activity in the ABC Subtype of Relapsed/Refractory De Novo Diffuse Large B-Cell Lymphoma (DLBCL): Interim Results of a Multic enter, Open-Label, Phase I Study). Because Btk plays a central role as a mediator in multiple signal transduction pathways, inhibitors of Btk are of great interest as anti-inflammatory and/or anti-cancer agents {Mohamed et al., Immunol. Rev. 228: 58-73, 2009; Pan, Drug News perspect 21: 357-362, 200%; Rokosz et al., Expert Opin. Ther. Targets 12: 883-903, 2008; Uckun et al., Anti-cancer Agents Med. Chem. 7: 624-632, 2007; Lou et al, J. Med. Chem. 55(10): 4539-4550, 2012).
[0005] International application WO2014173289A disclosed a series of fused heterocyclic compounds as Btk inhibitors. In particular, WO2014173289A disclosed
(S)-7-(l-acryloylpiperidin-4-yl)-2-(4-phenoxyphenyl)-4,5,6,7-tetra-hydropyrazolo[l,5-a]pyrimi dine-3-carboxamide (hereinafter C
Compound 1
[0006] Compound 1 is a potent, specific and irreversible BTK kinase inhibitor. The data generated in preclinical studies using biochemical, cell based and animal studies suggested that Compound 1 could offer significant benefit in inhibiting tumor growth in B-cell malignancies. As Compound 1 was shown to be more selective than ibrutinib for inhibition of BTK vs. EGFR, FGR, FRK, HER2, HER4, ITK, JAK3, LCK, and TEC, it is expected to give rise to less side-effects than ibrutinib in clinic. In addition, Compound 1 showed significantly less inhibition of rituximab-induced antigen-dependent cell-mediated cytotoxicity (ADCC) than ibrutinib due to weaker ITK inhibition, and therefore may provide better efficacy when combined with rituximab or other ADCC-dependent antibody in treating B-cell malignancies.
[0007] Preclinical safety evaluation has demonstrated that Compound 1 was safer than ibrutinib in terms of the overall tolerance and severe toxicities in both rat and dog single and repeat dose toxicity studies up to 28 days. Additionally, Compound 1 had better bioavailability without accumulation issues observed for ibrutinib. These unique characteristics warrant further evaluation of Compound 1 in clinical studies.
[0008] However, Compound 1 was found to be an amorphous form according to the preparation method for Compound 27 in WO 2014173289A, which was further confirmed by the X-Ray Powder Diffraction pattern of FIG. 7A. The amorphous form was shown to have a low glass transition temperature as shown in FIG. 7B, indicating some difficulties in the drug formulation with the amorphous form, such as low stability and hard to purify. Therefore, it’s necessary to develop a new form of Compound 1 which possesses characteristics such as high melting point and better stability, suitable for drug formulation.
Scheme 1: Preparation of Compound 1 and deuterium-labeled Compound 1
Deuterium-Labeled Compound 1
Step 15: Synthesis of
(S)-7-(l-acryloylpiperidin-4-yl)-2-(4-phenoxyphenyl)-4,5,6,7-tetrahydropyrazolori,5-a1pyrimi dine-3-carboxamide (Compound 1
[0105] Under N2 atmosphere, ACN (12.0 v), water (12.5 v), BG-13 (8.0 Kg, 1.0 eq), and NaHC03 (2.5 eq.) were added to a reactor. The mixture was then cooled to -5-0 °C. To the mixture, the solution of acryloyl chloride (1.1 eq.) in MeCN (0.5 v) was added dropwise and
stirred until the reaction was completed. EA (6.0 v) was then added to the reactor, and stirred. The organic phase was collected. The aqueous layer was further extracted with EA (3.0 v). The organic phases were combined and washed with brine. The organic layer was collected and concentrated.
[0106] The residue was purified by silica gel (2 wt) column, eluted with 3% w/w methanol in DCM (21.0 v). The Compound 1 solution was collected and concentrated under vacuum. The residue was precipitated from EA/MTBE (2.0 v). The cake was collected by centrifugation as the product.
Step 15: Synthesis of (S)-7-(l-acryloylpiperidin-4-yl)-2-(4-phenoxyphenyl)
-4,5,6,7-tetrahydropyrazolori,5-a1pyrimidine-3-carboxamide (Compound 1, alternative method)
[0107] A mixture of CHsCN (10.0 v), purified water (5.0 v), NaOH (1.5 eq.) and BG-13 (1.0 eq.) was stirred to get a clear solution. EtOAc (6.0 v) was then charged to the reaction and separated. The organic phase was collected and washed with 15% brine (3.0 v) twice. The organic phase prepared above was concentrated and the solvent was swapped to CH3CN (residue volume: NMT 5.0 v). CH3CN (7.5 v) and purified water (12.5 v) were charged and cooled to 15-20°C. L-(+)-tartaric acid (0.5 eq) and NaHCCb (2.5 eq.) were charged to the reaction mixture. A solution of acryloyl chloride (1.1 eq.) in CH3CN (0.5 v) was charged drop-wise to the reaction mixture. After the reaction was completed, EtOAc (6.0 v) was charged to the reaction mixture and organic layer was collected. Aqueous phase was further extracted with EA (3.0 v). The organic layers were combined, washed with 15% brine (5.0 v) and concentrated. The solvent was swapped to DCM (volume of residue: 1.5-2.0 v) and purified by silica gel column (silica gel: 100-200 mush, 2.0 w/ w; eluent: 3%> w/ w MeOH in DCM (about 50 v). The collected solution was concentrated and swapped to EtOAc (4.0 v). MTBE (6.4 v) was charged drop-wise to residue at 50°C. The mixture was then cooled to 5°C and the cake was collected centrifugation.
Step 16: Preparation of Crystalline Form A of Compound 1
[0108] The above cake of Compound 1 was dissolved in 7.0 volumes of DCM, and then swapped to solvent EA. After recrystallization from EA/MTBE, the cakes was collected by centrifugation, and was dried under vacuum. This gave 4.44 Kg product (Yield: 70.2%).
[0109] The product was then characterized by X-ray powder diffraction (XRPD) pattern method, which was generated on a PANalytical Empyrean X-ray powder diffractometer with the XRPD parameters as follows: X-Ray wavelength (Cu, ka, Kal (A): 1.540598, Ka2(A): 1.544426; Ka2/Kal intensity ratio: 0.50); X-Ray tube setting (45 Kv, 40mA); divergence slit (automatic); scan mode (Continuous); scan range (°2TH) (3°-40); step size (°2TH) (0.0131); scan speed (°/min) (about 10). The XRPD result found the resultant product as a crystalline shown in FIG. 1.
[0110] The differential scanning calorimetry (DSC) curves shown as in FIG. 2 was generated on a TA Q2000 DSC from TA Instruments. The DSC parameters used includes: temperature (25°C-desired temperature); heating rate (10°C/min) ; method (ramp); sample pan (aluminum, crimped); purge gas (N2). DSC result showed a sharp melting point at 139.4°C (onset temperature).
[0111] The thermo-gravimetric analysis (TGA) curves shown as in FIG. 3 was generated on a TA Q5000 TGA from TA Instruments. The TGA parameters used includes: temperature
(RT-desired temperature); heating rate (10°C/min); method (ramp); sample pan (platinum, open); purge gas (N2). TGA result showed is anhydrous with no weight loss even up to 110 °C.
[0112] The proton nuclear magnetic resonance ^H-NMR) shown as in FIG. 4 was collected on a Bruker 400M NMR Spectrometer in DMSO-de. ¾-NMR (DMSO-de) δ 7.50 (d, J= 8.6 Hz, 2H), 7.46-7.38 (m, 2H), 7.17 (t, J = 7.6 Hz, 1H), 7.08 (d, J= 7.6 Hz, 2H), 7.05 (d, J= 8.8 Hz, 2H), 6.85-6.72 (m, 1H), 6.67 (s, 1H), 6.07 (dd, J= 16.8, 2.2 Hz, 1H), 5.64 (dd, J= 10.4 Hz, 2.2 Hz, 1H), 4.55-4.38 (m, 1H), 4.17-3.94 (m, 2H), 3.33-3.22 (m, 2H), 3.08-2.88 (m, 1H), 2.67-2.51 (m, 1H), 2.36-2.15 (m, 1H), 2.12-1.82 (m, 2H), 1.79-1.65 (m, 1H), 1.63-1.49 (m, 1H), 1.38-1.08 (m, 2H).
[0113] The carbon nuclear magnetic resonance (13C-NMR) shown as in FIG. 5 was collected on a Bruker 400M NMR Spectrometer in DMSO-de. 13C-NMR spectra for Crystalline Form A of Compound 1.
Step 15: Synthesis of (S)-7-(1-acrvlovlpiperidin-4-vl)-2-(4-phenoxvphenyl)-4.5.6.7-tetrahvdropvrazolo[1.5-a1pvrimidine-3-carboxamide (Compound 1)
[0119] Under N2 atmosphere, ACN (12.0 v), water (12.5 v), BG-13 (8.0 Kg, 1.0 eq), and NaHCO3 (2.5 eq.) were added to a reactor. The mixture was then cooled to -5-0 °C. To the mixture, the solution of acryloyl chloride (1.1 eq.) in MeCN (0.5 v) was added dropwise and stirred until the reaction was completed. EA (6.0 v) was then added to the reactor, and stirred. The organic phase was collected. The aqueous layer was further extracted with EA (3.0 v). The organic phases were combined and washed with brine. The organic layer was collected and concentrated.
[0120] The residue was purified by silica gel (2 wt) column, eluted with 3% w/w methanol in DCM (21.0 v). The Compound 1 solution was collected and concentrated under vacuum. The residue was precipitated from EA/MTBE (2.0 v). The cake was collected by centrifugation as the product.
Step 15: Synthesis of (S)-7-(l-acryloylpiperidin-4-yl)-2-(4-phenoxyphenyl) -4.5.6.7-tetrahvdropvrazolori.5-a1pvrimidine-3-carboxamide (Compound 1. alternative method)
[0121] A mixture of CH3CN (10.0 v), purified water (5.0 v), NaOH (1.5 eq.) and BG-13 (1.0 eq.) was stirred to get a clear solution. EtOAc (6.0 v) was then charged to the reaction and separated. The organic phase was collected and washed with 15% brine (3.0 v) twice. The organic phase prepared above was concentrated and the solvent was swapped to CH3CN (residue volume: NMT 5.0 v). CH3CN (7.5 v) and purified water (12.5 v) were charged and cooled to 15-20°C. L-(+)-tartaric acid (0.5 eq) and NaHCO3 (2.5 eq.) were charged to the reaction mixture. A solution of acryloyl chloride (1.1 eq.) in CH3CN (0.5 v) was charged drop-wise to the reaction mixture. After the reaction was completed, EtOAc (6.0 v) was charged to the reaction mixture and organic layer was collected. Aqueous phase was further extracted with EA (3.0 v). The organic layers were combined, washed with 15% brine (5.0 v) and concentrated. The solvent was swapped to DCM (volume of residue: 1.5-2.0 v) and purified by silica gel column (silica gel: 100-200 mush, 2.0 w/ w; eluent: 3% w/ w MeOH in DCM (about 50 v). The collected solution was concentrated and swapped to EtOAc (4.0 v). MTBE (6.4 v) was charged drop-wise to residue at 50°C. The mixture was then cooled to 5°C and the cake was collected centrifugation.
References
- ^ “Zanubrutinib (Brukinsa) Use During Pregnancy”. Drugs.com. 3 January 2020. Retrieved 26 January 2020.
- ^ “Zanubrutinib”. DrugBank. Retrieved 15 November 2019.
- ^ Jump up to:a b c d “FDA approves therapy to treat patients with relapsed and refractory mantle cell lymphoma supported by clinical trial results showing high response rate of tumor shrinkage”. U.S. Food and Drug Administration (FDA) (Press release). 14 November 2019. Retrieved 15 November 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Brukinsa (zanubrutinib) FDA Approval History”. Drugs.com. 14 November 2019. Archived from the original on 15 November 2019. Retrieved 15 November 2019.
- ^ Jump up to:a b c d e f g h i “FDA grants accelerated approval to zanubrutinib for mantle cell lymphoma”. U.S. Food and Drug Administration (FDA)(Press release). 15 November 2019. Archived from the original on 28 November 2019. Retrieved 27 November 2019. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c “Drug Trials Snapshots Brukinsa”. U.S. Food and Drug Administration (FDA). 14 November 2019. Retrieved 26 January 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Zanubrutinib Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 28 November 2019. Archived from the original on 28 November 2019. Retrieved 27 November 2019. This article incorporates text from this source, which is in the public domain.
- ^ “Drug Approval Package: Brukinsa”. U.S. Food and Drug Administration (FDA). 27 November 2019. Archived from the original on 28 November 2019. Retrieved 27 November 2019. This article incorporates text from this source, which is in the public domain.
External links
- “Zanubrutinib”. Drug Information Portal. U.S. National Library of Medicine.
 |
|
| Clinical data | |
|---|---|
| Trade names | Brukinsa |
| Other names | BGB-3111 |
| AHFS/Drugs.com | Monograph |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth |
| Drug class | Bruton’s tyrosine kinase(BTK) inhibitor |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| PubChem SID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C27H29N5O3 |
| Molar mass | 471.5509 g·mol−1 |
| 3D model (JSmol) | |
/////////////////Zanubrutinib, FDA 2019, ザヌブルチニブ , занубрутиниб , زانوبروتينيب , BGB-3111
Teprotumumab-trbw

Tepezza (teprotumumab-trbw)
Company: Horizon Therapeutics plc
Date of Approval: January 21, 2020
Treatment for: Thyroid Eye Disease
UNIIY64GQ0KC0A
CAS number1036734-93-6
R-1507 / R1507 / RG-1507 / RG1507 / RO-4858696 / RO-4858696-000 / RO-4858696000 / RO4858696 / RO4858696-000 / RV-001 / RV001
Tepezza (teprotumumab-trbw) is a fully human monoclonal antibody (mAb) and a targeted inhibitor of the insulin-like growth factor 1 receptor (IGF-1R) for the treatment of active thyroid eye disease (TED).
FDA Approves Tepezza (teprotumumab-trbw) for the Treatment of Thyroid Eye Disease (TED) – January 21, 2020
Today, the U.S. Food and Drug Administration (FDA) approved Tepezza (teprotumumab-trbw) for the treatment of adults with thyroid eye disease, a rare condition where the muscles and fatty tissues behind the eye become inflamed, causing the eyes to be pushed forward and bulge outwards (proptosis). Today’s approval represents the first drug approved for the treatment of thyroid eye disease.
“Today’s approval marks an important milestone for the treatment of thyroid eye disease. Currently, there are very limited treatment options for this potentially debilitating disease. This treatment has the potential to alter the course of the disease, potentially sparing patients from needing multiple invasive surgeries by providing an alternative, non surgical treatment option,” said Wiley Chambers, M.D., deputy director of the Division of Transplant and Ophthalmology Products in the FDA’s Center for Drug Evaluation and Research. “Additionally, thyroid eye disease is a rare disease that impacts a small percentage of the population, and for a variety of reasons, treatments for rare diseases are often unavailable. This approval represents important progress in the approval of effective treatments for rare diseases, such as thyroid eye disease.”
Thyroid eye disease is associated with the outward bulging of the eye that can cause a variety of symptoms such as eye pain, double vision, light sensitivity or difficulty closing the eye. This disease impacts a relatively small number of Americans, with more women than men affected. Although this condition impacts relatively few individuals, thyroid eye disease can be incapacitating. For example, the troubling ocular symptoms can lead to the progressive inability of people with thyroid eye disease to perform important daily activities, such as driving or working.
Tepezza was approved based on the results of two studies (Study 1 and 2) consisting of a total of 170 patients with active thyroid eye disease who were randomized to either receive Tepezza or a placebo. Of the patients who were administered Tepezza, 71% in Study 1 and 83% in Study 2 demonstrated a greater than 2 millimeter reduction in proptosis (eye protrusion) as compared to 20% and 10% of subjects who received placebo, respectively.
The most common adverse reactions observed in patients treated with Tepezza are muscle spasm, nausea, alopecia (hair loss), diarrhea, fatigue, hyperglycemia (high blood sugar), hearing loss, dry skin, dysgeusia (altered sense of taste) and headache. Tepezza should not be used if pregnant, and women of child-bearing potential should have their pregnancy status verified prior to beginning treatment and should be counseled on pregnancy prevention during treatment and for 6 months following the last dose of Tepezza.
The FDA granted this application Priority Review, in addition to Fast Track and Breakthrough Therapy Designation. Additionally, Tepezza received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases or conditions. Development of this product was also in part supported by the FDA Orphan Products Grants Program, which provides grants for clinical studies on safety and efficacy of products for use in rare diseases or conditions.
The FDA granted the approval of Tepezza to Horizon Therapeutics Ireland DAC.
Teprotumumab (RG-1507), sold under the brand name Tepezza, is a medication used for the treatment of adults with thyroid eye disease, a rare condition where the muscles and fatty tissues behind the eye become inflamed, causing the eyes to be pushed forward and bulge outwards (proptosis).[1]
The most common adverse reactions observed in people treated with teprotumumab-trbw are muscle spasm, nausea, alopecia (hair loss), diarrhea, fatigue, hyperglycemia (high blood sugar), hearing loss, dry skin, dysgeusia (altered sense of taste) and headache.[1] Teprotumumab-trbw should not be used if pregnant, and women of child-bearing potential should have their pregnancy status verified prior to beginning treatment and should be counseled on pregnancy prevention during treatment and for six months following the last dose of teprotumumab-trbw.[1]
It is a human monoclonal antibody developed by Genmab and Roche. It binds to IGF-1R.
Teprotumumab was first investigated for the treatment of solid and hematologic tumors, including breast cancer, Hodgkin’s and non-Hodgkin’s lymphoma, non-small cell lung cancer and sarcoma.[2][3] Although results of phase I and early phase II trials showed promise, research for these indications were discontinued in 2009 by Roche. Phase II trials still in progress were allowed to complete, as the development was halted due to business prioritization rather than safety concerns.
Teprotumumab was subsequently licensed to River Vision Development Corporation in 2012 for research in the treatment of ophthalmic conditions. Horizon Pharma (now Horizon Therapeutics, from hereon Horizon) acquired RVDC in 2017, and will continue clinical trials.[4] It is in phase III trials for Graves’ ophthalmopathy (also known as thyroid eye disease (TED)) and phase I for diabetic macular edema.[5] It was granted Breakthrough Therapy, Orphan Drug Status and Fast Track designations by the FDA for Graves’ ophthalmopathy.[6]
In a multicenter randomized trial in patients with active Graves’ ophthalmopathy Teprotumumab was more effective than placebo in reducing the clinical activity score and proptosis.[7] In February 2019 Horizon announced results from a phase 3 confirmatory trial evaluating teprotumumab for the treatment of active thyroid eye disease (TED). The study met its primary endpoint, showing more patients treated with teprotumumab compared with placebo had a meaningful improvement in proptosis, or bulging of the eye: 82.9 percent of teprotumumab patients compared to 9.5 percent of placebo patients achieved the primary endpoint of a 2 mm or more reduction in proptosis (p<0.001). Proptosis is the main cause of morbidity in TED. All secondary endpoints were also met and the safety profile was consistent with the phase 2 study of teprotumumab in TED.[8] On 10th of July 2019 Horizon submitted a Biologics License Application (BLA) to the FDA for teprotumumab for the Treatment of Active Thyroid Eye Disease (TED). Horizon requested priority review for the application – if so granted (FDA has a 60-day review period to decide) it would result in a max. 6 month review process.[9]
History[edit]
Teprotumumab-trbw was approved for use in the United States in January 2020, for the treatment of adults with thyroid eye disease.[1]
Teprotumumab-trbw was approved based on the results of two studies (Study 1 and 2) consisting of a total of 170 patients with active thyroid eye disease who were randomized to either receive teprotumumab-trbw or a placebo.[1] Of the subjects who were administered Tepezza, 71% in Study 1 and 83% in Study 2 demonstrated a greater than two millimeter reduction in proptosis (eye protrusion) as compared to 20% and 10% of subjects who received placebo, respectively.[1]
The U.S. Food and Drug Administration (FDA) granted the application for teprotumumab-trbw fast track designation, breakthrough therapy designation, priority review designation, and orphan drug designation.[1] The FDA granted the approval of Tepezza to Horizon Therapeutics Ireland DAC.[1]
References
- ^ Jump up to:a b c d e f g h “FDA approves first treatment for thyroid eye disease”. U.S. Food and Drug Administration (FDA) (Press release). 21 January 2020. Retrieved 21 January 2020. This article incorporates text from this source, which is in the public domain.
- ^ https://clinicaltrials.gov/ct2/show/NCT01868997
- ^ http://adisinsight.springer.com/drugs/800015801
- ^ http://www.genmab.com/product-pipeline/products-in-development/teprotumumab
- ^ http://adisinsight.springer.com/drugs/800015801
- ^ http://www.genmab.com/product-pipeline/products-in-development/teprotumumab
- ^ Smith, TJ; Kahaly, GJ; Ezra, DG; Fleming, JC; Dailey, RA; Tang, RA; Harris, GJ; Antonelli, A; Salvi, M; Goldberg, RA; Gigantelli, JW; Couch, SM; Shriver, EM; Hayek, BR; Hink, EM; Woodward, RM; Gabriel, K; Magni, G; Douglas, RS (4 May 2017). “Teprotumumab for Thyroid-Associated Ophthalmopathy”. The New England Journal of Medicine. 376 (18): 1748–1761. doi:10.1056/NEJMoa1614949. PMC 5718164. PMID 28467880.
- ^ “Horizon Pharma plc Announces Phase 3 Confirmatory Trial Evaluating Teprotumumab (OPTIC) for the Treatment of Active Thyroid Eye Disease (TED) Met Primary and All Secondary Endpoints”. Horizon Pharma plc. Retrieved 22 March 2019.
- ^ “Horizon Therapeutics plc Submits Teprotumumab Biologics License Application (BLA) for the Treatment of Active Thyroid Eye Disease (TED)”. Horizon Therapeutics plc. Retrieved 27 August 2019.
External links
- “Teprotumumab”. Drug Information Portal. U.S. National Library of Medicine.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | IGF-1R |
| Clinical data | |
| Other names | teprotumumab-trbw, RG-1507 |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| DrugBank | |
| ChemSpider |
|
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.081.384 |
| Chemical and physical data | |
| Formula | C6476H10012N1748O2000S40 |
| Molar mass | 145.6 kg/mol g·mol−1 |
/////////Teprotumumab-trbw, APPROVALS 2020, FDA 2020, ORPHAN, BLA, fast track designation, breakthrough therapy designation, priority review designation, and orphan drug designation, Tepezza, Horizon Therapeutics, MONOCLONAL ANTIBODY, 2020 APPROVALS, active thyroid eye disease, Teprotumumab
https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-thyroid-eye-disease
ENCORAFENIB, エンコラフェニブ

ENCORAFENIB, エンコラフェニブ
UNII:8L7891MRB6
Formula:C22H27ClFN7O4S, Average: 540.01
1269440-17-6
- BRAFTOVI
- NVP-LGX818
- NVP-LGX-818-NXA
- NVP-LGX818-NXA
- ENCORAFENIB [USAN]
- ENCORAFENIB [WHO-DD]
- ENCORAFENIB
- ENCORAFENIB [INN]
- METHYL N-((2S)-1-((4-(3-(5-CHLORO-2-FLUORO-3-(METHANESULFONAMIDO)PHENYL)(-1-(PROPAN-2-YL)-1H-PYRAZOL-4-YL(PYRIMIDIN-2-YL)AMINO)PROPAN-2-YL)CARBAMATE
- CARBAMIC ACID, N-((1S)-2-((4-(3-(5-CHLORO-2-FLUORO-3-((METHYLSULFONYL)AMINO)PHENYL)-1-(1-METHYLETHYL)-1H-PYRAZOL-4-YL)-2-PYRIMIDINYL)AMINO)-1-METHYLETHYL)-, METHYL ESTER
- LGX818
- LGX-818
Encorafenib, also known as BRAFTOVI, is a kinase inhibitor. Encorafenib inhibits BRAF gene, which encodes for B-raf protein, which is a proto-oncogene involved in various genetic mutations Label. This protein plays a role in regulating the MAP kinase/ERK signaling pathway, which impacts cell division, differentiation, and secretion. Mutations in this gene, most frequently the V600E mutation, are the most commonly identified cancer-causing mutations in melanoma, and have been isolated in various other cancers as well, including non-Hodgkin lymphoma, colorectal cancer, thyroid carcinoma, non-small cell lung carcinoma, hairy cell leukemia and adenocarcinoma of the lung 6.
On June 27, 2018, the Food and Drug Administration approved encorafenib and Binimetinib(BRAFTOVI and MEKTOVI, Array BioPharma Inc.) in combination for patients with unresectable or metastatic melanoma with a BRAF V600E or V600K mutation, as detected by an FDA-approved test Label.
Array Biopharma (a wholly owned subsidiary of Pfizer ), under license from Novartis , and licensees Pierre Fabre and Ono Pharmaceutical have developed and launched the B-Raf kinase inhibitor encorafenib . In January 2020, the US FDA’s Orange Book was seen to list encorafenib patents such as US8946250 , US8501758 , US9314464 and US9763941 , expiring in the range of 2029-2032. At that time Orange Book also reported that encorafenib as having NCE exclusivity expiring on July 27, 2023.
Encorafenib (trade name Braftovi) is a drug for the treatment of certain melanomas. It is a small molecule BRAF inhibitor [1] that targets key enzymes in the MAPK signaling pathway. This pathway occurs in many different cancers including melanoma and colorectal cancers.[2] The substance was being developed by Novartis and then by Array BioPharma. In June 2018, it was approved by the FDA in combination with binimetinib for the treatment of patients with unresectable or metastatic BRAF V600E or V600K mutation-positive melanoma.[3][4]
The most common (≥25%) adverse reactions in patients receiving the drug combination were fatigue, nausea, diarrhea, vomiting, abdominal pain, and arthralgia.[3]
Indication
Used in combination with Binimetinib in metastatic melanoma with a BRAF V600E or V600K mutation, as detected by an FDA-approved test 5.
Associated Conditions
Pharmacodynamics
Encorafenib has shown improved efficacy in the treatment of metastatic melanoma 3.
Encorafenib, a selective BRAF inhibitor (BRAFi), has a pharmacologic profile that is distinct from that of other clinically active BRAFis 7.
Once-daily dosing of single-agent encorafenib has a distinct tolerability profile and shows varying antitumor activity across BRAFi-pretreated and BRAFi-naïve patients with advanced/metastatic stage melanoma 7.
Mechanism of action
Encorafenib is a kinase inhibitor that specifically targets BRAF V600E, as well as wild-type BRAF and CRAF while tested with in vitro cell-free assays with IC50 values of 0.35, 0.47, and 0.3 nM, respectively. Mutations in the BRAF gene, including BRAF V600E, result in activated BRAF kinases that mahy stimulate tumor cell growth. Encorafenib is able to bind to other kinases in vitro including JNK1, JNK2, JNK3, LIMK1, LIMK2, MEK4, and STK36 and significantly reduce ligand binding to these kinases at clinically achievable concentrations (≤ 0.9 μM) Label.
In efficacy studies, encorafenib inhibited the in vitro cell growth of tumor cell lines that express BRAF V600 E, D, and K mutations. In mice implanted with tumor cells expressing the BRAF V600E mutation, encorafenib induced tumor regressions associated with RAF/MEK/ERK pathway suppression Label.
Encorafenib and binimetinib target two different kinases in the RAS/RAF/MEK/ERK pathway. Compared with either drug alone, co-administration of encorafenib and binimetinib result in greater anti-proliferative activity in vitro in BRAF mutation-positive cell lines and greater anti-tumor activity with respect to tumor growth inhibition in BRAF V600E mutant human melanoma xenograft studies in mice. In addition to the above, the combination of encorafenib and binimetinib acted to delay the emergence of resistance in BRAF V600E mutant human melanoma xenografts in mice compared with the administration of either drug alone Label.

Pharmacology
Encorafenib acts as an ATP-competitive RAF kinase inhibitor, decreasing ERK phosphorylation and down-regulation of CyclinD1.[5]This arrests the cell cycle in G1 phase, inducing senescence without apoptosis.[5] Therefore it is only effective in melanomas with a BRAF mutation, which make up 50% of all melanomas.[6] The plasma elimination half-life of encorafenib is approximately 6 hours, occurring mainly through metabolism via cytochrome P450 enzymes.[7]
Clinical trials
Several clinical trials of LGX818, either alone or in combinations with the MEK inhibitor MEK162,[8] are being run. As a result of a successful Phase Ib/II trials, Phase III trials are currently being initiated.[9]
History
Approval of encorafenib in the United States was based on a randomized, active-controlled, open-label, multicenter trial (COLUMBUS; NCT01909453) in 577 patients with BRAF V600E or V600K mutation-positive unresectable or metastatic melanoma.[3] Patients were randomized (1:1:1) to receive binimetinib 45 mg twice daily plus encorafenib 450 mg once daily, encorafenib 300 mg once daily, or vemurafenib 960 mg twice daily.[3] Treatment continued until disease progression or unacceptable toxicity.[3]
The major efficacy measure was progression-free survival (PFS) using RECIST 1.1 response criteria and assessed by blinded independent central review.[3] The median PFS was 14.9 months for patients receiving binimetinib plus encorafenib, and 7.3 months for the vemurafenib monotherapy arm (hazard ratio 0.54, 95% CI: 0.41, 0.71, p<0.0001).[3] The trial was conducted at 162 sites in Europe, North America and various countries around the world.[4]
SYN


PATENT
WO2010010154 , expiry , EU states, 2029, US in 2030 with US154 extension.
WO 2011025927
WO 2016089208
Patent
WO-2020011141
Novel deuterated analogs of diarylpyrazole compounds, particularly encorafenib are B-RAF and C-RAF kinase inhibitors, useful for treating proliferative diseases such as melanoma and colorectal cancer. Family members of the product case, WO2010010154 , expire in most of the EU states until 2029 and will expire in the US in 2030 with US154 extension. In January 2020, the US FDA’s Orange Book was seen to list encorafenib patents such as US8946250 , US8501758 , US9314464 and US9763941 , expiring in the range of 2029-2032. At that time Orange Book also reported that encorafenib as having NCE exclusivity expiring on July 27, 2023.
PAPER
European journal of cancer (Oxford, England : 1990) (2018), 88, 67-76.
References
- ^ Koelblinger P, Thuerigen O, Dummer R (March 2018). “Development of encorafenib for BRAF-mutated advanced melanoma”. Current Opinion in Oncology. 30 (2): 125–133. doi:10.1097/CCO.0000000000000426. PMC 5815646. PMID 29356698.
- ^ Burotto M, Chiou VL, Lee JM, Kohn EC (November 2014). “The MAPK pathway across different malignancies: a new perspective”. Cancer. 120 (22): 3446–56. doi:10.1002/cncr.28864. PMC 4221543. PMID 24948110.
- ^ Jump up to:a b c d e f g “FDA approves encorafenib and binimetinib in combination for unresectable or metastatic melanoma with BRAF mutations”. U.S. Food and Drug Administration (FDA)(Press release). 27 June 2018. Archived from the original on 18 December 2019. Retrieved 28 June 2018. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “Drug Trial Snapshot: Braftovi”. U.S. Food and Drug Administration (FDA). 16 July 2018. Archived from the original on 19 December 2019. Retrieved 18 December 2019. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b Li Z, Jiang K, Zhu X, Lin G, Song F, Zhao Y, Piao Y, Liu J, Cheng W, Bi X, Gong P, Song Z, Meng S (January 2016). “Encorafenib (LGX818), a potent BRAF inhibitor, induces senescence accompanied by autophagy in BRAFV600E melanoma cells”. Cancer Letters. 370 (2): 332–44. doi:10.1016/j.canlet.2015.11.015. PMID 26586345.
- ^ Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. (July 2012). “A landscape of driver mutations in melanoma”. Cell. 150 (2): 251–63. doi:10.1016/j.cell.2012.06.024. PMC 3600117. PMID 22817889.
- ^ Koelblinger P, Thuerigen O, Dummer R (March 2018). “Development of encorafenib for BRAF-mutated advanced melanoma”. Current Opinion in Oncology. 30 (2): 125–133. doi:10.1097/CCO.0000000000000426. PMC 5815646. PMID 29356698.
- ^ “18 Studies found for: LGX818”. Clinicaltrials.gove.
- ^ Clinical trial number NCT01909453 for “Study Comparing Combination of LGX818 Plus MEK162 and LGX818 Monotherapy Versus Vemurafenib in BRAF Mutant Melanoma (COLUMBUS)” at ClinicalTrials.gov
External links
- “Encorafenib”. Drug Information Portal. U.S. National Library of Medicine.
- Li Z, Jiang K, Zhu X, Lin G, Song F, Zhao Y, Piao Y, Liu J, Cheng W, Bi X, Gong P, Song Z, Meng S: Encorafenib (LGX818), a potent BRAF inhibitor, induces senescence accompanied by autophagy in BRAFV600E melanoma cells. Cancer Lett. 2016 Jan 28;370(2):332-44. doi: 10.1016/j.canlet.2015.11.015. Epub 2015 Nov 14. [PubMed:26586345]
- Koelblinger P, Thuerigen O, Dummer R: Development of encorafenib for BRAF-mutated advanced melanoma. Curr Opin Oncol. 2018 Mar;30(2):125-133. doi: 10.1097/CCO.0000000000000426. [PubMed:29356698]
- Moschos SJ, Pinnamaneni R: Targeted therapies in melanoma. Surg Oncol Clin N Am. 2015 Apr;24(2):347-58. doi: 10.1016/j.soc.2014.12.011. Epub 2015 Jan 24. [PubMed:25769717]
- Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, Garbe C, Schadendorf D, Krajsova I, Gutzmer R, Chiarion-Sileni V, Dutriaux C, de Groot JWB, Yamazaki N, Loquai C, Moutouh-de Parseval LA, Pickard MD, Sandor V, Robert C, Flaherty KT: Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018 May;19(5):603-615. doi: 10.1016/S1470-2045(18)30142-6. Epub 2018 Mar 21. [PubMed:29573941]
- FDA approves encorafenib and binimetinib in combination for unresectable or metastatic melanoma with BRAF mutations [Link]
- BRAF B-Raf proto-oncogene, serine/threonine kinase [ Homo sapiens (human) ] [Link]
- Phase I Dose-Escalation and -Expansion Study of the BRAF Inhibitor Encorafenib (LGX818) in Metastatic BRAF-Mutant Melanoma [Link]
- Encorafenib FDA label [File]
- Encorafenib review [File]
 |
|
| Clinical data | |
|---|---|
| Trade names | Braftovi |
| Other names | LGX818 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a618040 |
| License data |
|
| Routes of administration |
Oral |
| Drug class | Antineoplastic Agents |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C22H27ClFN7O4S |
| Molar mass | 540.011 g/mol g·mol−1 |
| 3D model (JSmol) | |
///////////ENCORAFENIB, 1269440-17-6, BRAFTOVI, NVP-LGX818, LGX818, LGX 818, エンコラフェニブ ,
COC(=O)N[C@@H](C)CNc1nccc(n1)c2cn(nc2c3cc(Cl)cc(NS(=O)(=O)C)c3F)C(C)C
patent
[TABLE 0001]
| APCI | Atmospheric pressure chemical dissociation |
| HPLC | High performance liquid chromatography |
| TLC | TLC |
| h | hour |
| DMF | N, N-dimethylformamide |
| K 2 CO 3 | Potassium carbonate |
| DCM | Dichloromethane |
| THF | Tetrahydrofuran |
| CH 3 MgBr | Methyl magnesium bromide |
| PTSA | p-Toluenesulfonic acid |
| TFA | Trifluoroacetate |
| NMP | N-methylpyrrolidone |
| Diguanidinium carbonate | Guanidine carbonate |
| MTBE | Methyl tert-butyl ether |
| POCl 3 | Phosphorus oxychloride |
| DMSO | Dimethyl sulfoxide |
| Pd (dppf) Cl 2 | [1,1′-Bis (diphenylphosphino) ferrocene] Palladium dichloride |
| Dioxane | Dioxane |
| TsCl | 4-toluenesulfonyl chloride |
| Boc | Tert-butoxy carbon |
| DIPEA | N, N-diisopropylethylamine |
| CDCl 3 | Deuterated chloroform |
| TEA | Triethylamine |
| DMAP | 4-dimethylaminopyridine |
| Na 2 CO 3 | Sodium carbonate |
| HCl | hydrochloric acid |
[表 0002]
| MsCl | Methanesulfonyl chloride |
| Tol | Toluene |
GRAPIPRANT


GRAPIPRANT
- Molecular FormulaC26H29N5O3S
- Average mass491.605 Da
CAS 415903-37-6
UNII-J9F5ZPH7NB, CJ 023423, CJ-023423,
Phase II, Arrys Therapeutics, CANCER,
PAIN, AskAt Phase II,
- N-[[[2-[4-(2-Ethyl-4,6-dimethyl-1H-imidazo[4,5-c]pyridin-1-yl)phenyl]ethyl]amino]carbonyl]-4-methylbenzenesulfonamide
- 1-[2-[4-(2-Ethyl-4,6-dimethylimidazo[4,5-c]pyridin-1-yl)phenyl]ethyl]-3-(4-methylphenyl)sulfonylurea
- 2-Ethyl-4,6-dimethyl-1-[4-[2-[[[[(4-methylphenyl)sulfonyl]amino]carbonyl]amino]ethyl]phenyl]-1H-imidazo[4,5-c]pyridine
- AAT 007
- CJ 023423
- Grapiprant
- MR 10A7
- RQ 00000007
- RQ 7
Synonyms and Mappings
- 415903-37-6
- GRAPIPRANT [GREEN BOOK]
- CJ-023
- GRAPIPRANT [INN]
- GRAPIPRANT [WHO-DD]
- MR-10A7
- AAT-007
- MR10A7
- RQ-00000007
- RQ-7
- GRAPIPRANT [USAN]
- GRAPIPRANT
- 2-ETHYL-4,6-DIMETHYL-1-(4-(2-(((((4-METHYLPHENYL)SULFONYL)AMINO)CARBONYL)AMINO)ETHYL)PHENYL)-1H-IMIDAZO(4,5-C)PYRIDINE
- N-(((2-(4-(2-ETHYL-4,6-DIMETHYL-1H-IMIDAZO(4,5-C)PYRIDIN-1-YL)PHENYL)ETHYL)AMINO)CARBONYL)-4-METHYLBENZENESULFONAMIDE
- CJ 023423
- BENZENESULFONAMIDE, N-(((2-(4-(2-ETHYL-4,6-DIMETHYL-1H-IMIDAZO(4,5-C)PYRIDIN-1-YL)PHENYL)ETHYL)AMINO)CARBONYL)-4-METHYL-
- CJ-023,423
- N-(((2-(4-(2-ETHYL-4,6-DIMETHYL-1H-IMIDAZO(4,5-C)PYRIDIN-1-YL)PHENYL)ETHYL)AMINO)CARBONYL)-4-METHYL-BENZENESULFONAMIDE
- CJ-023423
SYN

Arrys Therapeutics (under license from AskAt ) and affiliate Ikena Oncology (formerly known as Kyn Therapeutics ) are developing ARY-007 , an oral formulation of grapiprant, for treating cancers; in December 2019, preliminary data were expected in 2020
Grapiprant (trade name Galliprant) is a small molecule drug that belongs in the piprant class. This analgesic and anti-inflammatory drug is primarily used as a pain relief for mild to moderate inflammation related to osteoarthritis in dogs. Grapiprant has been approved by the FDA’s Center for Veterinary Medicine and was categorized as a non-cyclooxygenase inhibiting non-steroidal anti-inflammatory drug (NSAID) in March 2016.[1]
Preclinical studies also indicate that grapiprant is not only efficacious as a acute pain but also in chronic pain relief and inflammation drug. The effect of the drug is directly proportional to the dosage and its effects were comparable to human medication such as rofecoxib and piroxicam.[2]
Grapiprant, a prostanoid EP4 receptor antagonist, is in phase II clinical trials at AskAt for the treatment of chronic pain. Phase I/II clinical trials are ongoing at Arrys Therapeutics in combination with pembrolizumab for the treatment of patients with microsatellite stable colorectal cancer and in patients with advanced or metastatic PD-1/L1 refractory non-small cell lung cancer (NSCLC).
Grapiprant is also used in humans, and was researched to be used as a pain control and inflammation associated with osteoarthritis. The effect of grapiprant could be explained through the function of prostaglandin E2, in which acts as a pro-inflammatory mediator of redness of the skin, edema and pain which are the typical signs of inflammation. The effect of PGE2 stems from its action through the four prostaglandin receptor subgroups EP1, EP2, EP3 and EP4, in which the prostaglandin EP4 receptor acts as the main intermediary of the prostaglandin-E2-driven inflammation. Grapiprant is widely accepted in veterinary medicine due to its specific and targeted approach to pain management in dogs. The serum concentration of grapiprant is increased when used in conjunction with other drugs such as acetaminophen, albendazole, and alitretinoin.
Common side effects are intestinal related effects such as mild diarrhea, appetite loss, and vomiting.[3] Additionally, it is found that it might lead to reduced tear production due to it being a sulfa-based medication and also reduced albumin levels.
Grapiprant, a prostanoid EP4 receptor antagonist, is in phase II clinical trials at AskAt for the treatment of chronic pain. Phase I/II clinical trials are ongoing at Arrys Therapeutics in combination with pembrolizumab for the treatment of patients with microsatellite stable colorectal cancer and in patients with advanced or metastatic PD-1/L1 refractory non-small cell lung cancer (NSCLC).
Medical uses
Grapiprant is used once a day as an oral pain relief for dogs with inflammation-related osteoarthritis. It is a non-steroidal anti-inflammatory (NSAID) that functions as a targeted action to treat osteoarthritis pain and inflammation in dogs.
Mechanism of action
Grapiprant acts as a specific antagonist that binds and blocks the prostaglandin EP4 receptor, one out of the four prostaglandin E2 (PGE2) receptor subgroups. The EP4 receptor then mediates the prostaglandin-E2-elicited response to pain, and hence grapiprant was proven to be effective in the decrease of pain in several inflammatory pain models of rats. It was also proven to be effective in reducing osteoarthritis-related pain in humans, which serves as a proof for its mechanism of action. The approximate calculation for canine efficacy dose is between the range of 1.3 and 1.7 mg/kg, in conjunction with a methylcellulose suspending agent. Based on the calculations from the comparisons of binding affinity of grapiprant to the EP4 receptors of dogs, rats, and humans, the study of plasma and serum protein binding determinations, the effective doses determined in inflammation pain models of rats, and human-related clinical studies, it is evaluated that Grapiprant should be administered just once a day. The approved dose of the commercial Grapiprant tablet by the FDA for the pain relief and inflammation associated with osteoarthritis to dogs is reported to be 2 mg/kg a day.[4]
Absorption
Studies in animals such as horses have shown the presence of Grapiprant in serum 72 hours with a concentration >0.005 ng/ml after the initial administration of a dose of 2 mg/kg. Grapiprant is expeditiously absorbed and the reported serum concentration was reported to be 31.9 ng/ml in an amount of time of 1.5 hours. The actual body exposure to grapiprant after administration of one dose was shown to be 2000 ng.hr/ml. The degree and rate at which grapiprant is absorbed into the body, presents a mean bioavailability of 39%. A significant reduction in the bioavailability, concentration time and maximal concentration were reported to have occurred after food intake.[1] And thus, grapiprant is usually not administered with food as it will not be as efficient.[5]
Distribution
The volume of distribution in cat studies was reported to be 918 ml/kg.[1]
Route of elimination
Following an oral administration, the majority of the dose was metabolized within the first 72 hours. Equine studies have shown that grapiprant is present in urine 96 hours after the first administration of a dose of 2 mg/kg and has a concentration >0.005 ng/ml. From the excreted dose conducted in horses, it is found that 55%, 15% and 19% of the orally-administered dose was excreted in bile, urine, and faeces respectively.[1]
Toxicity
Safety studies conducted on grapiprant have demonstrated that it generally possesses an exceptional safety profile and a wide safety margin in veterinary studies.[6] In animal studies, a research on 2.5-12 times overdose was conducted for grapiprant and the study resulted in soft-blobs and mucous-filled faeces, occasional bloody stools and emesis.
PATENT
WO-2020014465
Novel crystalline forms of grapiprant and their salts eg HCl (designated as Form A), useful for inhibiting prostaglandin EP4 receptor activity and treating cancers.
Prostaglandins are mediators of pain, fever and other symptoms associated with inflammation. Prostaglandin E2 (PGE2) is the predominant eicosanoid detected in inflammation conditions. In addition, it is also involved in various physiological and/or pathological conditions such as hyperalgesia, uterine contraction, digestive peristalsis, awakeness, suppression of gastric acid secretion, blood pressure, platelet function, bone metabolism, angiogenesis or the like.
[0003] Four PGE2 receptor subtypes (EP1, EP2, EP3 and EP4) displaying different pharmacological properties exist. The EP4 subtype, a Gs-coupled receptor, stimulates cAMP production as well as PI3K and GSK3P signaling, and is distributed in a wide variety of tissue suggesting a major role in PGE2-mediated biological events. Various EP4 inhibitors have been described previously, for example, in WO 2002/032900, WO 2005/021508, EiS 6,710,054, and US 7,238,714, the contents of which are incorporated herein by reference in their entireties.
[0004] Accordingly, there is a need for treating, preventing, and/or reducing severity of a proliferative disorder associated with prostaglandin EP4 receptor activity. The present invention addresses such a need.
It has now been found that compounds of the present invention, and compositions thereof, are useful for treating, preventing, and/or reducing severity of a proliferative disorder associated with prostaglandin EP4 receptor activity. In general, salt forms and co-crystal forms, and pharmaceutically acceptable compositions thereof, are useful for treating or lessening the severity of proliferative disorders associated with prostaglandin EP4 receptor activity, as described in detail herein. Such compounds are represented by the chemical structure below, denoted as compound A (also known as grapiprant):
A
or a pharmaceutically acceptable salt thereof.
United States Patent 7,960,407, filed March 1, 2006 and issued June 14, 2011 (“the ‘407 patent,” the entirety of which is hereby incorporated herein by reference), describes certain EP4 inhibitor compounds. Such compounds include compound A:
or a pharmaceutically acceptable salt thereof.
[0037] Compound A, N-[({2-[4-(2-Ethyl-4,6-dimethyl-lH-imidazo[4,5-c]pyridin-l-yl) phenyl]ethyl}amino)carbonyl]-4-methylbenzenesulfonamide, is described in detail in the ‘407
patent, including its synthetic route. The ‘407 patent also discloses a variety of physical forms of compound A.
[0038] It would be desirable to provide a solid form of compound A (e.g., as a co-crystal thereof or salt thereof) that imparts characteristics such as improved aqueous solubility, stability and ease of formulation. Accordingly, the present invention provides both co-crystal forms and salt forms of compound A:
A.
PATENT
WO 2002032900
PATENT
WO 2002032422
Family members of the product case ( WO0232422 ) of grapiprant have protection in most of the EU states until October 2021 and expire in the US in October 15, 2021.
PATENT
WO 2003086371
PATENT
WO2020014445 covering combinations of grapiprant and an immuno-oncology agent.
WO 2005102389
WO 2006095268
US 7960407
US 20190314390
References
- ^ Jump up to:a b c d “Grapiprant”. http://www.drugbank.ca. Retrieved 2019-05-15.
- ^ PubChem. “Grapiprant”. pubchem.ncbi.nlm.nih.gov. Retrieved 2019-05-15.
- ^ Paul Pion, D. V. M.; Spadafori, Gina (2017-08-08). “Veterinary Partner”. VIN.com.
- ^ Nagahisa, A.; Okumura, T. (2017). “Pharmacology of grapiprant, a novel EP4 antagonist: receptor binding, efficacy in a rodent postoperative pain model, and a dose estimation for controlling pain in dogs”. Journal of Veterinary Pharmacology and Therapeutics. 40 (3): 285–292. doi:10.1111/jvp.12349. ISSN 1365-2885. PMID 27597397.
- ^ Paul Pion, D. V. M.; Spadafori, Gina (2017-08-08). “Veterinary Partner”. VIN.com.
- ^ Kirkby Shaw, Kristin; Rausch-Derra, Lesley C.; Rhodes, Linda (February 2016). “Grapiprant: an EP4 prostaglandin receptor antagonist and novel therapy for pain and inflammation”. Veterinary Medicine and Science. 2 (1): 3–9. doi:10.1002/vms3.13. ISSN 2053-1095. PMC 5645826. PMID 29067176.
 |
|
| Clinical data | |
|---|---|
| Trade names | Galliprant |
| Routes of administration |
Oral |
| ATCvet code | |
| Pharmacokinetic data | |
| Bioavailability | 6.6 L/kg, high volume of distribution |
| Elimination half-life | 5.86 hours in horses |
| Excretion | Urine |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C26H29N5O3S |
| Molar mass | 491.61 g·mol−1 |
| 3D model (JSmol) | |
//////////////GRAPIPRANT, 415903-37-6, UNII-J9F5ZPH7NB, CJ 023423, CJ-023423, RQ-00000007, MR10A7, Galliprant, Phase II, Arrys Therapeutics, CANCER, PAIN, AskAt
CCC1=NC2=C(N1C3=CC=C(C=C3)CCNC(=O)NS(=O)(=O)C4=CC=C(C=C4)C)C=C(N=C2C)C
Brilliant blue G , ブリリアントブルーG ,
Brilliant blue G
FDA 2019, 12/20/2019, TISSUEBLUE, New Drug Application (NDA): 209569
Company: DUTCH OPHTHALMIC, PRIORITY; Orphan
OPQ recommends APPROVAL of NDA 209569 for commercialization of TissueBlue (Brilliant Blue G Ophthalmic Solution), 0.025%
Neuroprotectant
sodium;3-[[4-[[4-(4-ethoxyanilino)phenyl]-[4-[ethyl-[(3-sulfonatophenyl)methyl]azaniumylidene]-2-methylcyclohexa-2,5-dien-1-ylidene]methyl]-N-ethyl-3-methylanilino]methyl]benzenesulfonate
| Formula |
C47H48N3O7S2. Na
|
|---|---|
| CAS |
6104-58-1
|
| Mol weight |
854.0197
|
ブリリアントブルーG, C.I. Acid Blue 90
UNII-M1ZRX790SI
M1ZRX790SI
6104-58-1
Brilliant Blue G
Derma Cyanine G
SYN


////////////Brilliant blue G , ブリリアントブルーG , C.I. Acid Blue 90, FDA 2019, PRIORITY, Orphan
CCN(CC1=CC(=CC=C1)S(=O)(=O)[O-])C2=CC(=C(C=C2)C(=C3C=CC(=[N+](CC)CC4=CC(=CC=C4)S(=O)(=O)[O-])C=C3C)C5=CC=C(C=C5)NC6=CC=C(C=C6)OCC)C.[Na+]
- Benzenemethanaminium, N-[4-[[4-[(4-ethoxyphenyl)amino]phenyl][4-[ethyl[(3-sulfophenyl)methyl]amino]-2-methylphenyl]methylene]-3-methyl-2,5-cyclohexadien-1-ylidene]-N-ethyl-3-sulfo-, hydroxide, inner salt, monosodium salt
- Benzenemethanaminium, N-[4-[[4-[(4-ethoxyphenyl)amino]phenyl][4-[ethyl[(3-sulfophenyl)methyl]amino]-2-methylphenyl]methylene]-3-methyl-2,5-cyclohexadien-1-ylidene]-N-ethyl-3-sulfo-, inner salt, monosodium salt (9CI)
- Brilliant Indocyanine G (6CI)
- C.I. Acid Blue 90 (7CI)
- C.I. Acid Blue 90, monosodium salt (8CI)
- Acid Blue 90
- Acid Blue G 4061
- Acid Blue PG
- Acid Bright Blue G
- Acid Brilliant Blue G
- Acid Brilliant Cyanine G
- Acidine Sky Blue G
- Amacid Brilliant Cyanine G
- Anadurm Cyanine A-G
- BBG
- Benzyl Cyanine G
- Biosafe Coomassie Stain
- Boomassie blue silver
- Brilliant Acid Blue G
- Brilliant Acid Blue GI
- Brilliant Acid Blue J
- Brilliant Acid Cyanine G
- Brilliant Blue G
- Brilliant Blue G 250
- Brilliant Blue J
- Brilliant Indocyanine GA-CF
- Bucacid Brilliant Indocyanine G
- C.I. 42655
- CBB-G 250
- Colocid Brilliant Blue EG
- Coomassie Blue G
- Coomassie Blue G 250
- Coomassie Brilliant Blue G
- Coomassie Brilliant Blue G 250
- Coomassie G 250
- Cyanine G
- Daiwa Acid Blue 300
- Derma Cyanine G
- Derma Cyanine GN 360
- Dycosweak Acid Brilliant Blue G
- Eriosin Brilliant Cyanine G
- Fenazo Blue XXFG
- Impero Azure G
- Kayanol Cyanine G
- Lerui Acid Brilliant Blue G
- Milling Brilliant Blue 2J
- NSC 328382
- Optanol Cyanine G
- Orient Water Blue 105
- Orient Water Blue 105S
- Polar Blue G
- Polar Blue G 01
- Polycor Blue G
- Sandolan Cyanine N-G
- Sellaset Blue B
- Serva Blue G
- Serva Blue G 250
- Silk Fast Cyanine G
- Simacid Blue G 350
- Sumitomo Brilliant Indocyanine G
- Supranol Cyanin G
- Supranol Cyanine G
- TissueBlue
- Triacid Fast Cyanine G
- Water Blue 105
- Water Blue 105S
- Water Blue 150
- Xylene Brilliant Cyanine G
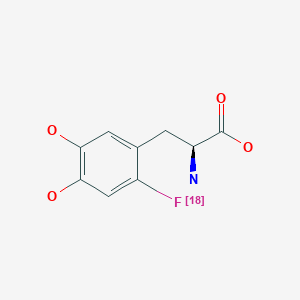
Fluorodopa F 18, フルオロドパ (18F), флуородопа (18F) , فلورودوبا (18F) , 氟[18F]多巴 ,
Fluorodopa F 18
2019/10/10, fda 2019,
| Formula |
C9H10FNO4
|
|---|---|
| Cas |
92812-82-3
|
| Mol weight |
215.1784
|
Diagnostic aid (brain imaging), Radioactive agent, for use in positron emission tomography (PET)
CAS 92812-82-3
フルオロドパ (18F)
Fluorodopa, also known as FDOPA, is a fluorinated form of L-DOPA primarily synthesized as its fluorine-18isotopologue for use as a radiotracer in positron emission tomography (PET).[1] Fluorodopa PET scanning is a valid method for assessing the functional state of the nigrostriatal dopaminergic pathway. It is particularly useful for studies requiring repeated measures such as examinations of the course of a disease and the effect of treatment
In October 2019, Fluorodopa was approved in the United States for the visual detection of certain nerve cells in adult patients with suspected Parkinsonian Syndromes (PS).[2][3]
The U.S. Food and Drug Administration (FDA) approved Fluorodopa F 18 based on evidence from one clinical trial of 56 patients with suspected PS.[2] The trial was conducted at one clinical site in the United States.[2]
PAPER
A one-pot two-step synthesis of 6-[18F]fluoro-L-DOPA ([18F]FDOPA) has been developed involving Cu-mediated radiofluorination of a pinacol boronate ester precursor. The method is fully automated, provides [18F]FDOPA in good activity yield (104 ± 16 mCi, 6 ± 1%), excellent radiochemical purity (>99%) and high molar activity (3799 ± 2087 Ci mmol−1), n = 3, and has been validated to produce the radiotracer for human use.
![Graphical abstract: One-pot synthesis of high molar activity 6-[18F]fluoro-l-DOPA by Cu-mediated fluorination of a BPin precursor](https://pubs.rsc.org/en/Image/Get?imageInfo.ImageType=GA&imageInfo.ImageIdentifier.ManuscriptID=C9OB01758E&imageInfo.ImageIdentifier.Year=2019 "Graphical abstract")

PATENT
KR 2019061368

The present invention relates to an L-dopa precursor compd., a method for producing the same, and a method for producing 18F-labeled L-dopa using the same. The method of prepg. 18F-labeled L-dopa I using the L-dopa precursor II [A = halogen-(un)substituted alkyl; W, X, Y = independently protecting group] can improve the labeling efficiency of 18F. After the labeling reaction, sepn. and purifn. steps of the product can be carried out continuously and it can be performed with on-column labeling (a method of labeling through the column). The final product I, 18 F-labeled L-dopa, can be obtained at a high yield relative to conventional methods. Further, it has an advantage that it is easy to apply various methods such as bead labeling.
PAPER
Science (Washington, DC, United States) (2019), 364(6446), 1170-1174.

PAPER
European Journal of Organic Chemistry (2018), 2018(48), 7058-7065.
PATENT
WO 2018115353
CN 107311877
References
- ^ Deng WP, Wong KA, Kirk KL (June 2002). “Convenient syntheses of 2-, 5- and 6-fluoro- and 2,6-difluoro-L-DOPA”. Tetrahedron: Asymmetry. 13 (11): 1135–1140. doi:10.1016/S0957-4166(02)00321-X.
- ^ Jump up to:a b c “Drug Trials Snapshots: Fluorodopa F 18”. U.S. Food and Drug Administration (FDA). 27 November 2019. Archived from the original on 27 November 2019. Retrieved 27 November 2019. This article incorporates text from this source, which is in the public domain.
- ^ “Drug Approval Package: Fluorodopa F18”. U.S. Food and Drug Administration (FDA). 20 November 2019. Archived from the original on 27 November 2019. Retrieved 26 November 2019. This article incorporates text from this source, which is in the public domain.
 |
|
| Clinical data | |
|---|---|
| Other names | 6-fluoro-L-DOPA, FDOPA |
| License data |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| ChemSpider | |
| UNII | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C9H10FNO4 |
| Molar mass | 215.18 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////////////Fluorodopa F 18, フルオロドパ (18F), FDA 2019, флуородопа (18F) , فلورودوبا (18F) , 氟[18F]多巴 , radio labelled
N[C@@H](CC1=CC(O)=C(O)C=C1[18F])C(O)=O
Enfortumab vedotin

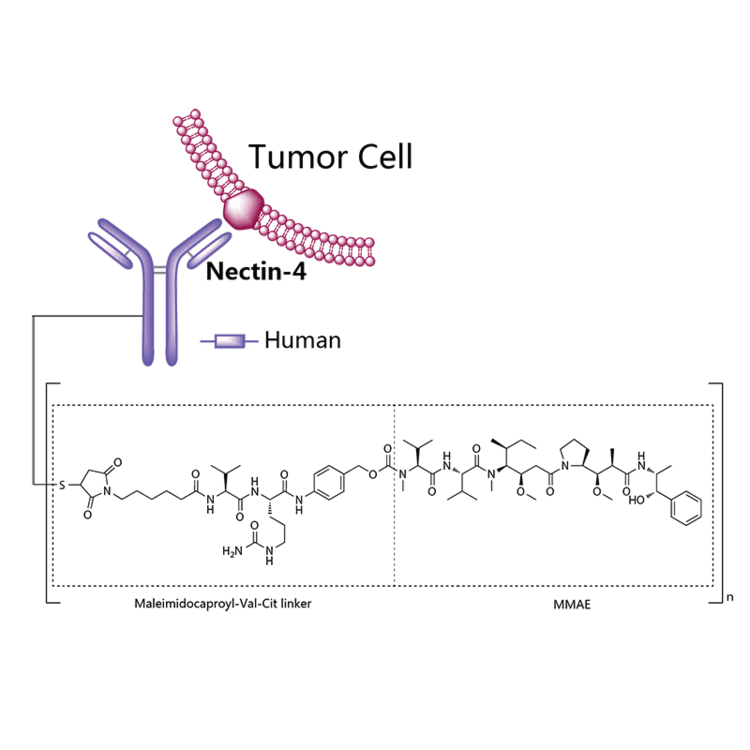

Enfortumab vedotin
| Formula |
C6642H10284N1742O2063S46
|
|---|---|
| Cas |
1346452-25-2
|
| Mol weight |
149022.148
|
AGS-22M6E, enfortumab vedotin-ejfv
Fda approved 2019/12/18, Padcev
Antineoplastic, Nectin-4 antibody, Tubulin polymerization inhibitor, Urothelial cancer
エンホルツマブベドチン (遺伝子組換え);
protein Based Therapies, Monoclonal antibody, mAb,
UNII DLE8519RWM
Other Names
- AGS 22CE
- AGS 22M6E
- AGS 22ME
- Enfortumab vedotin
- Enfortumab vedotin-ejfv
- Immunoglobulin G1 (human monoclonal AGS-22M6 γ1-chain), disulfide with human monoclonal AGS-22M6 κ-chain, dimer, tetrakis(thioether) with N-[[[4-[[N-[6-(3-mercapto-2,5-dioxo-1-pyrrolidinyl)-1-oxohexyl]-L-valyl-N5-(aminocarbonyl)-L-ornithyl]amino]phenyl]methoxy]carbonyl]-N-methyl-L-valyl-N-[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-hydroxy-1-methyl-2-phenylethyl]amino]-1-methoxy-2-methyl-3-oxopropyl]-1-pyrrolidinyl]-2-methoxy-1-[(1S)-1-methylpropyl]-4-oxobutyl]-N-methyl-L-valinamide
- Padcev
Protein Sequence
Sequence Length: 1322, 447, 447, 214, 214multichain; modified (modifications unspecified)
Enfortumab vedotin is an antibody-drug conjugate used in the treatment of patients with advanced, treatment-resistant urothelial cancers.3 It is comprised of a fully human monoclonal antibody targeted against Nectin-4 and a microtubule-disrupting chemotherapeutic agent, monomethyl auristatin E (MMAE), joined by a protease-cleavable link.3 It is similar to brentuximab vedotin, another antibody conjugated with MMAE that targets CD-30 instead of Nectin-4.
The clinical development of enfortumab vedotin was the result of a collaboration between Astellas Pharma and Seattle Genetics2 and it was first approved for use in the United States in December 2019 under the brand name PadcevTM.3
The most common side effects for patients taking enfortumab vedotin were fatigue, peripheral neuropathy (nerve damage resulting in tingling or numbness), decreased appetite, rash, alopecia (hair loss), nausea, altered taste, diarrhea, dry eye, pruritis (itching) and dry skin. [4]Enfortumab vedotin[1] (AGS-22M6E) is an antibody-drug conjugate[2] designed for the treatment of cancer expressing Nectin-4.[3]Enfortumab refers to the monoclonal antibody part, and vedotin refers to the payload drug (MMAE) and the linker.
The fully humanized antibody was created by scientists at Agensys (part of Astellas) using Xenomice from Amgen; the linker technology holding the antibody and the toxin together was provided by and licensed from Seattle Genetics.[5]
Results of a phase I clinical trial were reported in 2016.[2]
In December 2019, enfortumab vedotin-ejfv was approved in the United States for the treatment of adult patients with locally advanced or metastatic urothelial cancer who have previously received a programmed death receptor-1 (PD-1) or programmed death ligand 1 (PD-L1) inhibitor and a platinum-containing chemotherapy.[4]
Enfortumab vedotin was approved based on the results of a clinical trial that enrolled 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy.[4] The overall response rate, reflecting the percentage of patients who had a certain amount of tumor shrinkage, was 44%, with 12% having a complete response and 32% having a partial response.[4] The median duration of response was 7.6 months.[4]
The application for enfortumab vedotin-ejfv was granted accelerated approval, priority review designation, and breakthrough therapydesignation.[4] The U.S. Food and Drug Administration (FDA) granted the approval of Padcev to Astellas Pharma US Inc.[4]
Indication
Enfortumab vedotin is indicated for the treatment of adult patients with locally advanced or metastatic urothelial cancer who have previously received a programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor, and a platinum-containing chemotherapy in the neoadjuvant/adjuvant, locally advanced, or metastatic setting.3
Associated Conditions
Pharmacodynamics
Enfortumab vedotin is an anti-cancer agent that destroys tumor cells by inhibiting their ability to replicate.3 Patients with moderate to severe hepatic impairment should not use enfortumab vedotin – though it has not been studied in this population, other MMAE-containing antibody-drug conjugates have demonstrated increased rates of adverse effects in patients with moderate-severe hepatic impairment.3 Enfortumab vedotin may also cause significant hyperglycemia leading, in some cases, to diabetic ketoacidosis, and should not be administered to patients with a blood glucose level >250 mg/dl.3
Mechanism of action
Enfortumab vedotin is an antibody-drug conjugate comprised of multiple components.3 It contains a fully human monoclonal antibody directed against Nectin-4, an extracellular adhesion protein which is highly expressed in urothelial cancers,1 attached to a chemotherapeutic microtubule-disrupting agent, monomethyl auristatin E (MMAE). These two components are joined via a protease-cleavable linker. Enfortumab vedotin binds to cells expressing Nectin-4 and the resulting enfortumab-Nectin-4 complex is internalized into the cell. Once inside the cell, MMAE is released from enfortumab vedotin via proteolytic cleavage and goes on to disrupt the microtubule network within the cell, arresting the cell cycle and ultimately inducing apoptosis.3
PATENT
WO 2016176089
WO 2016138034
WO 2017186928
WO 2017180587
WO 2017200492
US 20170056504
PAPER
Cancer Research (2016), 76(10), 3003-3013.
General References
- Hanna KS: Clinical Overview of Enfortumab Vedotin in the Management of Locally Advanced or Metastatic Urothelial Carcinoma. Drugs. 2019 Dec 10. pii: 10.1007/s40265-019-01241-7. doi: 10.1007/s40265-019-01241-7. [PubMed:31823332]
- McGregor BA, Sonpavde G: Enfortumab Vedotin, a fully human monoclonal antibody against Nectin 4 conjugated to monomethyl auristatin E for metastatic urothelial Carcinoma. Expert Opin Investig Drugs. 2019 Oct;28(10):821-826. doi: 10.1080/13543784.2019.1667332. Epub 2019 Sep 17. [PubMed:31526130]
- FDA Approved Drug Products: Padcev (enfortumab vedotin-ejfv) for IV injection [Link]
References
- ^ World Health Organization (2013). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 109”(PDF). WHO Drug Information. 27 (2).
- ^ Jump up to:a b Seattle Genetics and Agensys, an Affiliate of Astellas, Highlight Promising Enfortumab Vedotin (ASG-22ME) and ASG-15ME Phase 1 Data in Metastatic Urothelial Cancer at 2016 ESMO Congress. Oct 2016
- ^ Statement On A Nonproprietary Name Adopted By The USAN Council – Enfortumab Vedotin, American Medical Association.
- ^ Jump up to:a b c d e f g “FDA approves new type of therapy to treat advanced urothelial cancer”. U.S. Food and Drug Administration (FDA) (Press release). 18 December 2019. Archived from the original on 19 December 2019. Retrieved 18 December 2019. This article incorporates text from this source, which is in the public domain.
- ^ Challita-Eid PM, Satpayev D, Yang P, et al. (May 2016). “Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models”. Cancer Research. 76 (10): 3003–13. doi:10.1158/0008-5472.can-15-1313. PMID 27013195.
External links
- “Enfortumab vedotin”. Drug Information Portal. U.S. National Library of Medicine.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Nectin-4 |
| Clinical data | |
| Trade names | Padcev |
| Other names | AGS-22M6E, AGS-22CE, enfortumab vedotin-ejfv |
| License data | |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChemSID | |
| DrugBank | |
| ChemSpider |
|
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C6642H10284N1742O2063S46 |
| Molar mass | 149.0 kg/mol g·mol−1 |
PADCEV™
(enfortumab vedotin-ejfv) for Injection, for Intravenous Use
DESCRIPTION
Enfortumab vedotin-ejfv is a Nectin-4 directed antibody-drug conjugate (ADC) comprised of a fully human anti-Nectin-4 IgG1 kappa monoclonal antibody (AGS-22C3) conjugated to the small molecule microtubule disrupting agent, monomethyl auristatin E (MMAE) via a protease-cleavable maleimidocaproyl valine-citrulline (vc) linker (SGD-1006). Conjugation takes place on cysteine residues that comprise the interchain disulfide bonds of the antibody to yield a product with a drug-to-antibody ratio of approximately 3.8:1. The molecular weight is approximately 152 kDa.
Figure 1: Structural Formula
|
Approximately 4 molecules of MMAE are attached to each antibody molecule. Enfortumab vedotin-ejfv is produced by chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells and the small molecule components are produced by chemical synthesis.
PADCEV (enfortumab vedotin-ejfv) for injection is provided as a sterile, preservative-free, white to off-white lyophilized powder in single-dose vials for intravenous use. PADCEV is supplied as a 20 mg per vial and a 30 mg per vial and requires reconstitution with Sterile Water for Injection, USP, (2.3 mL and 3.3 mL, respectively) resulting in a clear to slightly opalescent, colorless to slightly yellow solution with a final concentration of 10 mg/mL [see DOSAGE AND ADMINISTRATION]. After reconstitution, each vial allows the withdrawal of 2 mL (20 mg) and 3 mL (30 mg). Each mL of reconstituted solution contains 10 mg of enfortumab vedotin-ejfv, histidine (1.4 mg), histidine hydrochloride monohydrate (2.31 mg), polysorbate 20 (0.2 mg) and trehalose dihydrate (55 mg) with a pH of 6.0.
///////////////Enfortumab vedotin, AGS-22M6E, エンホルツマブベドチン (遺伝子組換え) , protein Based Therapies, Monoclonal antibody, mAb, FDA 2019
[*]SC1CC(=O)N(CCCCCC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(=O)N)C(=O)Nc2ccc(COC(=O)N(C)[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@@H](CC(=O)N3CCC[C@H]3[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)c4ccccc4)OC)cc2)C1=O
RESMETIROM
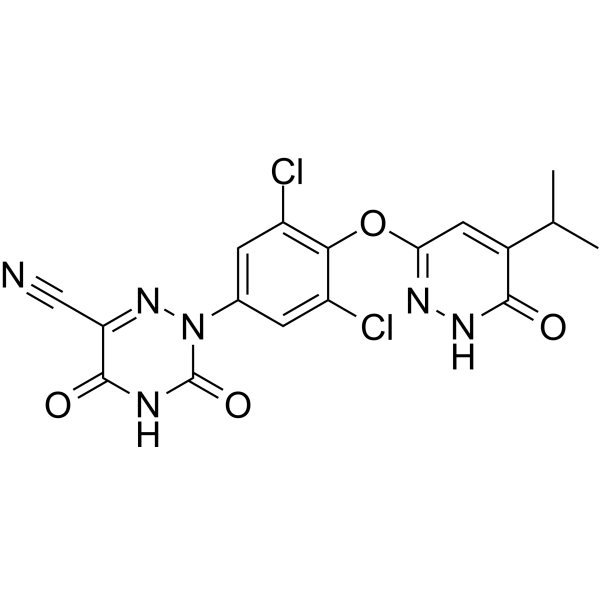
RESMETIROM
| C17H12Cl2N6O4 |
435.2 g/mol
MGL-3196
CAS 920509-32-6, Resmetirom, VIA-3196, UNII-RE0V0T1ES0
FDA APPROVED 3/14/2024, To treat noncirrhotic non-alcoholic steatohepatitis with moderate to advanced liver scarring
Press Release
Phase III, Non-alcoholic fatty liver disease (NAFLD)
2-[3,5-dichloro-4-[(6-oxo-5-propan-2-yl-1H-pyridazin-3-yl)oxy]phenyl]-3,5-dioxo-1,2,4-triazine-6-carbonitrile
2-(3,5-DICHLORO-4-((5-ISOPROPYL-6-OXO-1,6-DIHYDROPYRIDAZIN-3-YL)OXY)PHENYL)-3,5-DIOXO-2,3,4,5-TETRAHYDRO-(1,2,4)TRIAZINE-6-CARBONITRILE
1,2,4-TRIAZINE-6-CARBONITRILE, 2-(3,5-DICHLORO-4-((1,6-DIHYDRO-5-(1-METHYLETHYL)-6-OXO-3-PYRIDAZINYL)OXY)PHENYL)-2,3,4,5-TETRAHYDRO-3,5-DIOXO-
Madrigal Pharmaceuticals , following the merger between Synta and Madrigal Pharmaceuticals (pre-merger) (following the acquisition of VIA Pharmaceuticals ‘ assets (originally under license from Roche )), is developing resmetirom (MGL-3196, VIA-3196), the lead from oral capsule formulation thyroid hormone receptor (THR) beta agonists, cholesterol and triglyceride modulators, for the use in the treatment of metabolic disorders including hypercholesterolemia and other dyslipidemias, and non-alcoholic steatohepatitis.
MGL-3196 is a first-in-class, orally administered, small-molecule, liver-directed, THR β-selective agonist. Preclinical, toxicology and Phase 1 clinical data suggest MGL-3196 has an attractive, differentiated profile as a potential treatment for non-alcoholic steatohepatitis (NASH) and dyslipidemias. THR-β selectivity also enhances the safety profile of MGL-3196, compared to non-selective agents. MGL-3196 has shown no suppression of the central thyroid axis, no THR-α effects on heart rate or bone, and no elevation of liver enzymes. These characteristics make MGL-3196 among the most promising molecules in development in this therapeutic area. MGL-3196 is in a Phase 2 clinical trial for the treatment of non-alcoholic steatohepatitis (NASH).
PATENT
WO-2020010068
Novel crystalline salt of resmetirom as thyroid hormone receptor agonists useful for treating obesity, hyperlipidemia, hypercholesterolemia and diabetes. Appears to be the first filing from the assignee and the inventors on this compound,
Thyroid hormones are critical for normal growth and development and for maintaining metabolic homeostasis (Paul M. Yen, Physiological reviews, Vol. 81(3): pp. 1097-1126 (2001)). Circulating levels of thyroid hormones are tightly regulated by feedback mechanisms in the hypothalamus/pituitary/thyroid (HPT) axis. Thyroid dysfunction leading to hypothyroidism or hyperthyroidism clearly demonstrates that thyroid hormones exert profound effects on cardiac function, body weight, metabolism, metabolic rate, body temperature, cholesterol, bone, muscle and behavior.
[0005] The biological activity of thyroid hormones is mediated by thyroid hormone receptors (TRs or THRs) (M. A. Lazar, Endocrine Reviews, Vol. 14: pp. 348-399 (1993)). TRs belong to the superfamily known as nuclear receptors. TRs form heterodimers with the retinoid receptor that act as ligand-inducible transcription factors. TRs have a ligand binding domain, a DNA binding domain, and an amino terminal domain, and regulate gene expression through interactions with DNA response elements and with various nuclear co-activators and co repressors. The thyroid hormone receptors are derived from two separate genes, a and b. These distinct gene products produce multiple forms of their respective receptors through differential RNA processing. The major thyroid receptor isoforms are aΐ, a2, bΐ, and b2. Thyroid hormone receptors aΐ, bΐ, and b2 bind thyroid hormone. It has been shown that the thyroid hormone receptor subtypes can differ in their contribution to particular biological responses. Recent studies suggest that TIIb 1 plays an important role in regulating TRH (thyrotropin releasing hormone) and on regulating thyroid hormone actions in the liver. T11b2 plays an important role in the regulation of TSH (thyroid stimulating hormone) (Abel et. al, J. Clin. Invest., Vol 104: pp. 291-300 (1999)). TIIb 1 plays an important role in regulating heart rate (B. Gloss et. al. Endocrinology, Vol. 142: pp. 544-550 (2001); C. Johansson et. al, Am. J. Physiol., Vol. 275: pp. R640-R646 (1998)).
[0006] Efforts have been made to synthesize thyroid hormone analogs which exhibit increased thyroid hormone receptor beta selectivity and/or tissue selective action. Such thyroid hormone mimetics may yield desirable reductions in body weight, lipids, cholesterol, and lipoproteins, with reduced impact on cardiovascular function or normal function of the hypothalamus/pituitary/thyroid axis (see, e.g., Joharapurkar et al, J. Med. Chem, 2012, 55 (12), pp 5649-5675). The development of thyroid hormone analogs which avoid the undesirable effects of hyperthyroidism and hypothyroidism while maintaining the beneficial effects of thyroid hormones would open new avenues of treatment for patients with metabolic disease such as obesity, hyperlipidemia, hypercholesterolemia, diabetes and other disorders and diseases such as liver steatosis and NASH, atherosclerosis, cardiovascular diseases, hypothyroidism, thyroid cancer, thyroid diseases, a resistance to thyroid hormone (RTH) syndrome, and related disorders and diseases.
PATENT
WO2018075650
In one embodiment, the metabolite of Compound A comprises a compound
having the following structure:
(“Ml”).
PATENT
WO 2007009913
PATENT
WO 2014043706
https://patents.google.com/patent/WO2014043706A1/en
Example 3: Preparation of (Z)-ethyl (2-cyano-2-(2-(3,5-dichloro-4-((5-isopropyl-6- oxo- l,6-dihydropyridazin-3-yl)oxy)phenyl)hydrazono)acetyl)carbamate (Int. 8)
A 2 L, three-neck, round-bottom flask equipped with overhead stirring, a thermocouple, N2 inlet/outlet was charged with Int. 7 (75.0 g, 0.239 mol, 1 wt), acetic acid (600 mL, 8 vol), water (150 mL, 2 vol), and concentrated HC1 (71.3 mL, 0.95 vol). The resulting thin slurry was cooled to 6 °C and a solution of NaN02 (16.8 g, 0.243 mol, 1.02 equiv) in water (37.5 mL, 0.5 vol) was added over a period of 10 min while maintaining a batch temperature below 10 °C. After an additional 10 min of agitation between 5-10 °C, HPLC analysis showed complete conversion of Int. 7 to the diazonium intermediate. A solution of NaOAc (54.5 g, 0.664 mol, 2.78 equiv) in water (225 mL, 3 vol) was added over a period of 6 min while maintaining a batch temperature below 10 °C. N-cyanoacetylurethane (37.9 g, 0.243 mol, 1.02 equiv) was immediately added, the cooling was removed, and the batch naturally warmed to 8 °C over 35 min. HPLC analysis showed complete consumption of the diazonium intermediate and the reaction was deemed complete. The batch warmed naturally to 21 °C and was filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with water (375 mL, 5 vol) twice. The collected orange solid was dried in a 35 °C vacuum oven for 64 h to provide crude Int. 8 (104.8 g, 91%).
A I L, three-neck, round-bottom flask equipped with overhead stirring, a
thermocouple, and N2 inlet/outlet was charged with crude Int. 8 (104.4 g, 1 wt) and acetic acid (522 mL, 5 vol). The resulting slurry was heated to 50 °C and held at that temperature for 1.5 h. The batch cooled naturally to 25 °C over 2 h and was filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with water (522 mL, 5 vol) and the cake conditioned under vacuum for 1.75 h. The light orange solid was dried to constant weight in a 40 °C vacuum oven to provide 89.9 g (78% from Int. 7) of the desired product. 1H NMR (DMSO) was consistent with the assigned structure.
Example 4: Preparation of 2-(3,5-dichloro-4-((5-isopropyl-6-oxo-l,6- dihydropyridazin-3-yl)oxy)phenyl)-3,5-dioxo-2,3,4,5-tetrahydro-l,2,4-triazine-6-carbonitrile (Compound A)
A 2 L, three-neck, round-bottom flask equipped with overhead stirring, a
thermocouple, N2 inlet/outlet, and reflux condenser was charged with Int. 8 (89.3 g, 0.185 mol, 1 wt), DMAC (446 mL, 5 vol), and KOAc (20.0 g, 0.204 mol, 1.1 equiv). The mixture was heated to 120 °C and held at that temperature for 2 h. HPLC analysis showed complete conversion to Compound A. The batch temperature was adjusted to 18 °C over 1 h, and acetic acid (22.3 mL, 0.25 vol) was added. The batch temperature was adjusted to 8 °C, and water (714 mL, 8 vol) was added over 1 h; an orange slurry formed. The batch was filtered through Sharkskin filter paper and the cake was allowed to condition overnight under N2 without vacuum for convenience. A premixed solution of 1 : 1 acetone/water (445 mL, 5 vol) was charged to the flask and added to the cake as a rinse with vacuum applied. After 2 h of conditioning the cake under vacuum, it was transferred to a clean 1 L, three-neck, round- bottom flask equipped with overhead stirring, a thermocouple, and N2inlet/outlet. Ethanol (357 mL, 4 vol) and acetone (357 mL, 4 vol) were charged and the resulting slurry was heated to 60 °C; dissolution occurred. Water (890 mL, 10 vol) was added over a period of 90 min while maintaining a batch temperature between 55-60 °C. The resulting slurry was allowed to cool to 25 °C and filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with a solution of 1:1 EtOH/water (446 mL, 5 vol). The cake was conditioned overnight under N2 without vacuum for convenience. The cracks in the cake were smoothed and vacuum applied. The cake was washed with water (179 mL, 2 vol) and dried in a 45 °C vacuum oven to a constant weight of 70.5 g (87%, crude Compound A). HPLC analysis showed a purity of 94.8%.
A 500 mL, three-neck, round-bottom flask equipped with overhead stirring, a thermocouple, N2 inlet/outlet, and reflux condenser was charged with crude Compound A (70.0 g) and MIBK (350 mL, 5 vol). The orange slurry was heated to 50 °C and held at that temperature for 2 h. The batch cooled naturally to 23 °C and was filtered through Sharkskin filter paper. The reactor and cake were washed sequentially with MIBK (35 mL, 0.5 vol) twice. The collected solids were dried in a 45 °C vacuum oven to a constant weight of 58.5 g (84%). This solid was charged to a 500 mL, three-neck, round-bottom flask equipped with overhead stirring, a thermocouple, N2 inlet/outlet, and reflux condenser. Ethanol (290 mL, 5 vol) was added and the slurry was heated to reflux. After 3.5 h at reflux, XRPD showed the solid was consistent with Form I, and heating was removed. Upon reaching 25 °C, the batch was filtered through filter paper, and the reactor and cake were washed sequentially with EtOH (174 mL, 3 vol). The tan solid Compound A was dried in a 40 °C vacuum oven to a constant weight of 50.4 g (87%, 64% from Int. 8). HPLC analysis showed a purity of 99.1%. 1H NMR (DMSO) was consistent with the assigned structure.
Example 5: Scaled up preparation of 2-(3,5-dichloro-4-((5-isopropyl-6-oxo-l,6- dihydropyridazin-3-yl)oxy)phenyl)-3,5-dioxo-2,3,4,5-tetrahydro-l,2,4-triazine-6-carbonitrile (Compound A)
A larger scale batch of Compound A was synthesized according to the scheme below. The conditions in the scheme below are similar to those described in Examples 1-4 above.

6A

Compound A
Synthesis of 4: A 50 L jacketed glass vessel (purged with N2) was charged with 3,6- dichloropyridazine (2.00 kg), 4-amino-2,6-dichlorophenol (2.44 kg) and N,N- dimethylacetamide (10.0 L). The batch was vacuum (26 inHg) / nitrogen (1 PSIG) purged 3 times. Cesium carbonate (5.03 kg) was added and the batch temperature was adjusted from 22.3 °C to 65.0 °C over 3.5 hours. The batch was held at 65.0 °C for 20 hours. At this point,
NMR analysis indicated 3.34% 3.6-dichloropyridazine relative to 2. The batch temperature was adjusted to 21.5 °C and ethyl acetate (4.00 L) was added to the batch. The batch was agitated for 10 minutes and then filtered through a 18″ Nutsche filter equipped with polypropylene filter cloth. The filtration took 15 minutes. Ethyl acetate (5.34 L) was charged to the vessel and transferred to the filter as a rinse. The batch was then manually re- suspended in the filter before re-applying vacuum. This process was repeated 2 more times and the filter cake was conditioned for 10 minutes. The filtrate was charged to a 100-L vessel that contained (16.0 L) of a previously prepared 15% sodium chloride in H20. The batch was agitated for 5 minutes and then allowed to separate for 35 minutes. The interface was not visible, so the calculated 23 L of the lower aqueous phase was removed. 16.0 L of 15% Sodium chloride in H20 was added to the batch. The batch was agitated for 6 minutes and then allowed to separate for 7 minutes. The interface was visible at -19 L and the lower aqueous phase was removed. 17.0 L of 15% Sodium chloride in H20 was added to the batch. The batch was agitated for 7 minutes and then allowed to separate for 11 minutes. The lower aqueous phase was removed. The vessel was set up for vacuum distillation and the batch was concentrated from 17.0 L to 8.0 L over 2 hours 20 minutes with the batch temperature kept around 21 °C. Benzoic anhydride (3.19 kg) and acetic acid (18.0 L) were charged to the vessel. The vessel was set up for vacuum distillation and the batch was concentrated from 28.0 L to 12.0 L over 2 days (overnight hold at 20 °C) with the batch temperature kept between 20 and 55 °C. At this point, JH NMR analysis indicated a mol ratio of acetic acid to ethyl acetate of 1.0:0.015. Acetic acid (4.0 L) was charged to the batch and the batch was distilled to 12 L. JH NMR analysis indicated a mol ratio of acetic acid to ethyl acetate of 1.0:0.0036. Acetic acid (20.0 L) was charged to the batch and the batch temperature was adjusted to 70.0 °C. The batch was sampled for HPLC analysis and 2 was 0.16%. Sodium acetate (2,20 kg) was added to the batch and the batch temperature was adjusted from 72.4 °C to 110.0 °C. After 18.5 hours, HPLC analysis indicated no Int. B detected. The batch temperature was adjusted from 111.3 to 74.7 °C and DI water (30.0 L) was added to the batch over 2 hours. The batch temperature was adjusted to 20 .5 °C and then filtered using a 24″ Haselloy Nutsche filter equipped with polypropylene filter cloth. A previously prepared solution of 1:1 acetic acid in DI H20 (10.0 L) was charged to the vessel and agitated for 5 minutes. The wash was transferred to the filter and the batch was then manually re- suspended in the filter before re-applying vacuum. DI H20 (10.0 L) was charged to the vessel and then transferred to the filter. The batch was manually re-suspended in the filter before re-applying vacuum. DI H20 (10.0 L) was charged directly to the filter and the batch was then manually re-suspended in the filter before re-applying vacuum. The filter cake was allowed to condition for 18 hours to give 14.4 kg of 4. HPLC analysis indicated a purity of 93.7%. This wet cake was carried forward into the purification. A 100 L jacketed glass vessel (purged with N2) was charged with crude 4 (wet cake 14.42 kg), acetic acid (48.8 L) and the agitator was started. DI H20 (1.74 L) was charged. The batch (a slurry) temperature was adjusted from 18.1 to 100.1 °C over 4.25 hours. The batch was held at 100.1 to 106.1 °C for 1 hour and then adjusted to 73.1 °C. DI H20 (28.0 L) was added to the batch over 1 hour keeping the batch temperature between 73.1 and 70.3 °C. The batch temperature was adjusted further from 70.3 °C to 25.0 °C overnight. The batch was filtered using a 24″ Hastelloy Nutsche filter equipped with polypropylene filter cloth. The filtration took 13 minutes. A solution of DI H20 (9.00 L) and acetic acid (11.0 L) was prepared and added to the 100 L vessel. The mixture was agitated for 5 minutes and then transferred to the filter cake. DI H20 (20.0 L) was charged to the vessel, agitated for 6 minutes and then transferred to the filter cake. DI H20 (20.0 L) was charged to the vessel, agitated for 9 minutes and then transferred to the filter cake. The batch was allowed to condition for 3 days and then transferred to drying trays for vacuum oven drying. After 3 days at 50 °C and 28’7Hg, the batch gave a 74% yield (3.7 kg) of4 as an off-white solid. The JH NMR spectrum was consistent with the assigned structure, HPLC analysis indicated a purity of 98.87% and KF analysis indicated 0.14% H20. Synthesis of Int. 7: A 100-L jacketed glass vessel (purged with N2) was charged with tetrahydrofuran (44.4 L). The agitator was started (125 RPM) and 4 (3.67 kg) was charged followed by lithium chloride (1.26 kg). The batch temperature was observed to be 26.7 ° C and was an amber solution. Isopropenylmagnesium bromide 1.64 molar solution in 2-methyl THF (21.29 kg) was added over 2 ½ hours keeping the batch between 24.3 and 33.6 °C. The batch was agitated at 24.5 °C for 17 hours at which point HPLC analysis indicated 9% 4. A 2nd 100-L jacketed glass vessel (purged with N2) was charged with 3N hydrogen chloride (18.3 L). The batch was transferred to the vessel containing the 3N HC1 over 25 minutes keeping the batch temperature between 20 and 46 °C. A bi-phasic solution was observed. The quenched batch was transferred back to the 1st 100-L vessel to quench the small amount of residue left behind. THF (2.00 L) was used as a rinse. The batch temperature was observed to be 40.9 ° C and was agitated at 318 RPM for 45 minutes. The batch temperature was adjusted to 21.8 ° C and the layers were allowed to separate. The separation took 10 minutes. The lower aqueous phase was removed (-26.0 L). A solution of sodium chloride (1.56 kg) in DI water (14.0 L) was prepared and added to the batch. This was agitated at 318 RPM for 10 minutes and agitator was stopped. The separation took 3 minutes. The lower aqueous phase was removed (-16.0 L). The batch was vacuum distilled from 58.0 L to 18.4 L using ~24’7Hg and a jacket temperature of 50 to 55 °C. A solution of potassium hydroxide (2.30 kg) in DI water (20.7 L) was prepared in a 72-L round bottom flask. The vessel was set up for atmospheric distillation using 2 distillation heads and the batch was transferred to the 72-L vessel. THF (0.75 L) was used as a rinse. The batch volume was -41.0 L, the temperature was adjusted to 64.1 °C and distillation started with the aid of a N2 sweep. Heating was continued to drive the batch temperature to 85.4 °C while distilling at which point the 72-L vessel was set up for reflux (batch volume was about 28.0 L at the end of the distillation). The batch was held at 85 °C for 13 hours at which point HPLC analysis indicated 0.3% compound 6A. Heating was stopped and the batch was transferred to a 100-L jacketed glass vessel. Solids were observed. The batch temperature was adjusted from 70.6 °C to 56.7 °C. A previously prepared solution of sodium hydrogen carbonate (2.82 kg) in DI water (35.0 L) was added over 80 minutes keeping the batch temperature between 56.7 and 46.7 °C. The batch pH at the end of the addition was 9.8. The batch was held at
46.7 to 49.0 °C for 40 minutes and then cooled to 25.0 °C. The batch was filtered using a 18″ stainless steel Nutsche filter. DI water (18.4 L) was charged to the vessel and transferred to the filter. The filter cake was manually re-suspended in the filter and then the liquors were removed. This process was repeated once more and the filter cake was 3″ thick. The filter cake was conditioned on the filter for 3 days, was transferred to drying trays and dried in a vacuum oven at 45 °C to provide 2.93 kg Int. 7 (95% yield) with an HPLC purity of 87.6%.
Synthesis of Int. 8: A 100 L jacketed glass vessel (purged with N2 and plumbed to a caustic scrubber) was charged with acidic acid (13.0 L). Int. 7 (2.85 kg) was charged to the vessel and the agitator was started. N-Cyanoacetylurethane (1.56 kg) and DI water (5.70 L) were charged to the vessel. The batch temperature was adjusted from 17.0 °C to 5.5 °C and a thin slurry was observed. At this point 37% hydrogen chloride (2.70 L) was added over 10 minutes keeping the batch temperature between 4.8 °C and 8.8 °C. A previously prepared solution of sodium nitrite (638 g) in DI water (1.42 L) was added over 26 minutes keeping the batch temperature between 5.8 °C and 8.7 °C. A brown gas was observed in the vessel head space during the addition. HPLC analysis indicated no Int. 7 detected. At this point a previously prepared solution of sodium acetate (2.07 kg) in DI water (8.50 L) was added over 47 minutes keeping the batch temperature between 5.5 °C and 9.5 °C. After the addition, a thin layer of orange residue was observed on the vessel wall just above the level of the batch. The batch temperature was adjusted from 9.4 °C to 24.5 °C and held at 25 °C (+ 5 °C) for 12 hours. The batch was filtered using a 24″ Hastelloy Nutsche filter equipped with tight-weave polypropylene filter cloth. The filtration took 30 minutes. The vessel was rinsed with 14.3 L of a 1 : 1 acidic acid / DI water. The orange residue on the reactor washed away with the rinse. The rinse was transferred to the filter where the batch was manually re-suspended. Vacuum was re-applied to remove the wash. A 2nd 1 : 1 acidic acid / DI water wash was performed as above and the batch was conditioned on the filter for 26 hours. HPLC analysis of the wet filter cake indicated purity was 90.4%. The batch was dried to a constant weight of 3.97 kg (91% yield) in a vacuum oven at 45 °C and 287Hg. Preparation of Compound A DMAC Solvate
A 100 L, jacketed, glass vessel purged with N2 was charged with Int. 8 (3.90 kg) and potassium acetate (875 g). N,N-dimethylacetamide (DMAC, 18.3 L) was charged to the vessel and the agitator was started. The batch temperature was adjusted to 115 °C over 2 h. After 2 h at 115 °C, the batch was sampled and HPLC analysis indicated 0.27% Int. 8 remained. The batch temperature was adjusted to 25.0 °C overnight. Acetic acid (975 mL) was added to the batch and the batch was agitated further for 3 h. The batch was transferred to a carboy and the vessel was rinsed clean with 800 mL of DMAC. The batch was transferred back to the 100 L vessel using vacuum through a 10 μιη in-line filter and a DMAC rinse (1.15 L) was used. The filtration was fast at the beginning but slow at the end, plugging up the filter. The batch temperature was adjusted to 11.1 °C and DI water (35.1 L) was added over 2 h 20 min, keeping the batch temperature between 5-15 °C. The batch was held for 1 h and filtered, using an 18″ Nutsche filter equipped with tight-weave
polypropylene cloth. The filtration took 15 h. A 1: 1 ethanol/DI water wash (19.5 L) was charged to the vessel, cooled to 10 °C, and transferred to the filter cake. The cake was allowed to condition under N2 and vacuum for 8 h and transferred to drying trays. The batch was dried in a vacuum oven at 45 °C and 28’7Hg to give 89% yield (3.77 kg) of Compound A DMAC solvate as an orange/tan solid. The 1H NMR spectrum was consistent with the assigned structure and Karl Fischer analysis indicated 0.49% H20. XRPD indicated the expected form, i.e., Compound A DMAC solvate. Thermogravimetric analysis (TGA) indicated 16% weight loss. HPLC analysis indicated a purity of 93.67%.
Preparation of Crude Compound A
A 100 L, jacketed, glass vessel purged with N2 was charged with Compound A
DMAC solvate (3.75 kg) and ethanol (15.0 L). The agitator was started and acetone (15.0 L) was added. The batch temperature was adjusted from 10.6 °C to 60.0 °C over 1 h. At this point, the batch was in solution. DI water was added to the batch over 1.5 h, keeping the batch temperature at 60 + 5 °C. The batch was held at 60 + 5 °C for 1 h and cooled to 23.5 °C. An 18″ Nutsche filter equipped with tight-weave (0.67 CFM) polypropylene cloth was set up and the batch was filtered. The filtration took 15 h. A 1: 1 ethanol/DI water wash (19.5 L) was charged to the vessel and transferred to the filter cake. The cake was allowed to condition under N2 and vacuum for 8 h and transferred to drying trays. The batch was dried in a vacuum oven at 45 °C and 28’7Hg for five days to give a 94% yield (2.90 kg) of Compound A as a powdery tan solid. The NMR spectrum is consistent with the assigned structure and Karl Fischer analysis indicated 6.6% H20. XRPD indicated the expected form of dihydrate. TGA indicated 6.7% weight loss. HPLC analysis indicated a purity of 96.4% (AUC).
Purification of Crude Compound A
A 50 L, jacketed, glass vessel purged with N2 was charged with Compound A crude
(2.90 kg) and methyl isobutyl ketone (14.5 L). The agitator was started and the batch temperature was adjusted from 20.2 °C to 50.4 °C over 1.5 h. The batch was held at 50 °C (+ 5 °C) for 1 h and cooled to 20-25 °C. The batch was held at 20-25 °C for 2.5 h. An 18″ Nutsche filter equipped with tight- weave (0.67 CFM) polypropylene cloth was set up and the batch was filtered. The filtration took 20 min. Methyl isobutyl ketone (MIBK, 1.45 L) was charged to the vessel and transferred to the filter cake. The cake was manually resuspended and the liquors were pulled through with vacuum. Methyl isobutyl ketone (2.90 L) was charged to the filter cake and the cake was manually resuspended. The liquors were pulled through with vacuum and the cake was conditioned with vacuum and nitrogen for 15 h. The filter cake dried into a tan, hard 18″ x 1 ½” disc. This was manually broken up and run through coffee grinders to give a 76% yield (2.72 kg) of MGL-3196 MIBK solvate as a tan, powdery solid. No oven drying was necessary. The NMR spectrum was consistent with the assigned structure and Karl Fischer analysis indicated <0.1 % H20. XRPD indicated the expected form MIBK solvate. TGA indicated 17.3% weight loss. HPLC analysis indicated a purity of 98.5%.
Example 6: Conversion of Compound A to Form I
Purified Compound A (4802 g) as a 1:1 MIBK solvate which was obtained from Int. 8 as described in Example 5 above was added into a jacketed, 100 L reactor along with 24 liters of ethanol. The resulting slurry was heated to 80 + 5 °C (reflux) over 1 h 25 min; the mixture was stirred at that temperature for 4 h 25 min. Analysis of the filtered solids at 2 h 55 min indicated that the form conversion was complete, with the XRPD spectra conforming to Form I. The mixture was cooled to 20 + 5 °C over 45 min and stirred at that temperature for 15 min. The slurry was filtered and the filter cake was washed twice with prefiltered ethanol (2 x 4.8 L). The wet cake (4.28 kg) was dried under vacuum at 40 + 5 °C for 118 h to afford 3390 g of Compound A form I.
PAPER
Journal of Medicinal Chemistry (2014), 57(10), 3912-3923
https://pubs.acs.org/doi/abs/10.1021/jm4019299
The beneficial effects of thyroid hormone (TH) on lipid levels are primarily due to its action at the thyroid hormone receptor β (THR-β) in the liver, while adverse effects, including cardiac effects, are mediated by thyroid hormone receptor α (THR-α). A pyridazinone series has been identified that is significantly more THR-β selective than earlier analogues. Optimization of this series by the addition of a cyanoazauracil substituent improved both the potency and selectivity and led to MGL-3196 (53), which is 28-fold selective for THR-β over THR-α in a functional assay. Compound 53 showed outstanding safety in a rat heart model and was efficacious in a preclinical model at doses that showed no impact on the central thyroid axis. In reported studies in healthy volunteers, 53 exhibited an excellent safety profile and decreased LDL cholesterol (LDL-C) and triglycerides (TG) at once daily oral doses of 50 mg or higher given for 2 weeks.

//////////////RESMETIROM , MGL-3196, VIA-3196, UNII-RE0V0T1ES0, Phase III
CC(C)C1=CC(=NNC1=O)OC2=C(C=C(C=C2Cl)N3C(=O)NC(=O)C(=N3)C#N)Cl
Avapritinib, アバプリチニブ , авапритиниб , أفابريتينيب ,

Avapritinib
BLU-285, BLU285
Antineoplastic, Tyrosine kinase inhibitor
アバプリチニブ
(1S)-1-(4-fluorophenyl)-1-[2-[4-[6-(1-methylpyrazol-4-yl)pyrrolo[2,1-f][1,2,4]triazin-4-yl]piperazin-1-yl]pyrimidin-5-yl]ethanamine
| Formula |
C26H27FN10
|
|---|---|
| CAS |
1703793-34-3
|
| Mol weight |
498.558
|
| No. | Drug Name | Active Ingredient | Approval Date | FDA-approved use on approval date* |
|---|---|---|---|---|
| 1. | Ayvakit | avapritinib | 1/9/2020 | To treat adults with unresectable or metastatic gastrointestinal stromal tumor (GIST) |
PRIORITY; Orphan, NDA 212608
Avapritinib, sold under the brand name Ayvakit, is a medication used for the treatment of tumors due to one specific rare mutation: It is specifically intended for adults with unresectable or metastatic ( y) gastrointestinal stromal tumor (GIST) that harbor a platelet-derived growth factor receptor alpha (PDGFRA) exon 18 mutation.[1]
Common side effects are edema (swelling), nausea, fatigue/asthenia (abnormal physical weakness or lack of energy), cognitive impairment, vomiting, decreased appetite, diarrhea, hair color changes, increased lacrimation (secretion of tears), abdominal pain, constipation, rash. and dizziness.[1]
Ayvakit is a kinase inhibitor.[1]
History
The U.S. Food and Drug Administration (FDA) approved avapritinib in January 2020.[1] The application for avapritinib was granted fast track designation, breakthrough therapy designation, and orphan drug designation.[1] The FDA granted approval of Ayvakit to Blueprint Medicines Corporation.[1]
Avapritinib was approved based on the results from the Phase I NAVIGATOR[2][3] clinical trial involving 43 patients with GIST harboring a PDGFRA exon 18 mutation, including 38 subjects with PDGFRA D842V mutation.[1] Subjects received avapritinib 300 mg or 400 mg orally once daily until disease progression or they experienced unacceptable toxicity.[1] The recommended dose was determined to be 300 mg once daily.[1] The trial measured how many subjects experienced complete or partial shrinkage (by a certain amount) of their tumors during treatment (overall response rate).[1] For subjects harboring a PDGFRA exon 18 mutation, the overall response rate was 84%, with 7% having a complete response and 77% having a partial response.[1] For the subgroup of subjects with PDGFRA D842V mutations, the overall response rate was 89%, with 8% having a complete response and 82% having a partial response.[1] While the median duration of response was not reached, 61% of the responding subjects with exon 18 mutations had a response lasting six months or longer (31% of subjects with an ongoing response were followed for less than six months).[1]
PATENT
WO 2015057873
https://patents.google.com/patent/WO2015057873A1/en
Example 7: Synthesis of (R)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4- yl)pyrrolo[2, 1 -f\ [ 1 ,2,4] triazin-4-yl)piperazin- 1 -yl)pyrimidin-5-yl)ethanamine and (S)- 1 – (4- fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethanamine (Compounds 43 and 44)


Step 1 : Synthesis of (4-fluorophenyl)(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2,l- f] [ 1 ,2,4] triazin-4-yl)piperazin- 1 -yl)pyrimidin-5-yl)methanone:

4-Chloro-6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2,l-/] [l,2,4]triazine (180 mg, 0.770 mmol), (4-fluorophenyl)(2-(piperazin-l-yl)pyrimidin-5-yl)methanone, HC1 (265 mg, 0.821 mmol) and DIPEA (0.40 mL, 2.290 mmol) were stirred in 1,4-dioxane (4 mL) at room temperature for 18 hours. Saturated ammonium chloride was added and the products extracted into DCM (x2). The combined organic extracts were dried over Na2S04, filtered through Celite eluting with DCM, and the filtrate concentrated in vacuo. Purification of the residue by MPLC (25- 100% EtOAc-DCM) gave (4-fluorophenyl)(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2,l- ] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)methanone (160 mg, 0.331 mmol, 43 % yield) as an off-white solid. MS (ES+) C25H22FN90 requires: 483, found: 484 [M + H]+.
Step 2: Synthesis of (5,Z)-N-((4-fluorophenyl)(2-(4-(6-(l-methyl- lH-p razol-4-yl)p rrolo[2, l- ] [l,2,4]triazin-4- l)piperazin- l-yl)pyrimidin-5-yl)methylene)-2-methylpropane-2-sulfinamide:

(S)-2-Methylpropane-2-sulfinamide (110 mg, 0.908 mmol), (4-fluorophenyl)(2-(4-(6-(l- methyl- lH-pyrazol-4-yl)pyrrolo[2,l-/][l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5- yl)methanone (158 mg, 0.327 mmol) and ethyl orthotitanate (0.15 mL, 0.715 mmol) were stirred in THF (3.2 mL) at 70 °C for 18 hours. Room temperature was attained, water was added, and the products extracted into EtOAc (x2). The combined organic extracts were washed with brine, dried over Na2S04, filtered, and concentrated in vacuo while loading onto Celite. Purification of the residue by MPLC (0- 10% MeOH-EtOAc) gave (5,Z)-N-((4-fluorophenyl)(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)methylene)-2- methylpropane-2-sulfinamide (192 mg, 0.327 mmol, 100 % yield) as an orange solid. MS (ES+) C29H3iFN10OS requires: 586, found: 587 [M + H]+.
Step 3: Synthesis of (lS’)-N-(l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4- l)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethyl)-2-methylpropane-2-

(lS’,Z)-N-((4-Fluorophenyl)(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2,l- ] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)methylene)-2-methylpropane-2-sulfinamide (190 mg, 0.324 mmol) was taken up in THF (3 mL) and cooled to 0 °C. Methylmagnesium bromide (3 M solution in diethyl ether, 0.50 mL, 1.500 mmol) was added and the resulting mixture stirred at 0 °C for 45 minutes. Additional methylmagnesium bromide (3 M solution in diethyl ether, 0.10 mL, 0.300 mmol) was added and stirring at 0 °C continued for 20 minutes. Saturated ammonium chloride was added and the products extracted into EtOAc (x2). The combined organic extracts were washed with brine, dried over Na2S04, filtered, and concentrated in vacuo while loading onto Celite. Purification of the residue by MPLC (0-10% MeOH-EtOAc) gave (lS’)-N-(l-(4-fluorophenyl)-l-(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2, l- ] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethyl)-2-methylpropane-2-sulfinamide (120 mg, 0.199 mmol, 61.5 % yield) as a yellow solid (mixture of diastereoisomers). MS (ES+) C3oH35FN10OS requires: 602, found: 603 [M + H]+. Step 4: Synthesis of l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2,l- f\ [ 1 ,2,4] triazin-4- l)piperazin- 1 -yl)pyrimidin-5-yl)ethanamine:

(S)-N- ( 1 – (4-Fluorophenyl)- 1 -(2- (4- (6-( 1 -methyl- 1 H-pyrazol-4-yl)pyrrolo [2,1- /] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethyl)-2-methylpropane-2-sulfinamide (120 mg, 0.199 mmol) was stirred in 4 M HCl in 1,4-dioxane (1.5 mL)/MeOH (1.5 mL) at room temperature for 1 hour. The solvent was removed in vacuo and the residue triturated in EtOAc to give l-(4-fluorophenyl)- l-(2-(4-(6-(l -methyl- lH-pyrazol-4-yl)pyrrolo[2, l-/][l,2,4]triazin-4- yl)piperazin- l-yl)pyrimidin-5-yl)ethanamine, HCl (110 mg, 0.206 mmol, 103 % yield) as a pale yellow solid. MS (ES+) C26H27FN10requires: 498, found: 482 [M- 17 + H]+, 499 [M + H]+.
Step 5: Chiral separation of (R)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4- yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine and (5)-1-(4- fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin- 1 -yl)pyrimidin- -yl)ethanamine:

The enantiomers of racemic l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4- yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine (94 mg, 0.189 mmol) were separated by chiral SFC to give (R)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl-lH- pyrazol-4-yl)pyrrolo[2, l-/][l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethanamine (34.4 mg, 0.069 mmol, 73.2 % yield) and (lS,)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4- yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine (32.1 mg, 0.064 mmol, 68.3 % yield). The absolute stereochemistry was assigned randomly. MS (ES+)
C26H27FN10 requires: 498, found: 499 [M + H]+.
References
- ^ Jump up to:a b c d e f g h i j k l m “FDA approves the first targeted therapy to treat a rare mutation in patients with gastrointestinal stromal tumors”. U.S. Food and Drug Administration (FDA) (Press release). 9 January 2020. Archived from the original on 11 January 2020. Retrieved 9 January 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Blueprint Medicines Announces FDA Approval of AYVAKIT (avapritinib) for the Treatment of Adults with Unresectable or Metastatic PDGFRA Exon 18 Mutant Gastrointestinal Stromal Tumor”. Blueprint Medicines Corporation (Press release). 9 January 2020. Archived from the original on 11 January 2020. Retrieved 9 January 2020.
- ^ “Blueprint Medicines Announces Updated NAVIGATOR Trial Results in Patients with Advanced Gastrointestinal Stromal Tumors Supporting Development of Avapritinib Across All Lines of Therapy”. Blueprint Medicines Corporation (Press release). 15 November 2018. Archived from the original on 10 January 2020. Retrieved 9 January 2020.
Further reading
- Wu CP, Lusvarghi S, Wang JC, et al. (July 2019). “Avapritinib: A Selective Inhibitor of KIT and PDGFRα that Reverses ABCB1 and ABCG2-Mediated Multidrug Resistance in Cancer Cell Lines”. Mol. Pharm. 16 (7): 3040–3052. doi:10.1021/acs.molpharmaceut.9b00274. PMID 31117741.
- Gebreyohannes YK, Wozniak A, Zhai ME, et al. (January 2019). “Robust Activity of Avapritinib, Potent and Highly Selective Inhibitor of Mutated KIT, in Patient-derived Xenograft Models of Gastrointestinal Stromal Tumors”. Clin. Cancer Res. 25 (2): 609–618. doi:10.1158/1078-0432.CCR-18-1858. PMID 30274985.
External links
- “Avapritinib”. Drug Information Portal. U.S. National Library of Medicine (NLM).
| Clinical data | |
|---|---|
| Trade names | Ayvakit |
| Other names | BLU-285, BLU285 |
| License data | |
| Routes of administration |
By mouth |
| Drug class | Antineoplastic agents |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C26H27FN10 |
| Molar mass | 498.570 g·mol−1 |
| 3D model (JSmol) | |
///////Avapritinib, 2020 APPROVALS, PRIORITY, Orphan, BLU-285, BLU285, FDA 2020, Ayvakit, アバプリチニブ , авапритиниб , أفابريتينيب ,

{kind=link}