
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Pegylated Interferon alpha-2b, (PegIFN), Virafin
DB00022 sequence CDLPQTHSLGSRRTLMLLAQMRRISLFSCLKDRHDFGFPQEEFGNQFQKAETIPVLHEMI QQIFNLFSTKDSSAAWDETLLDKFYTELYQQLNDLEACVIQGVGVTETPLMKEDSILAVR KYFQRITLYLKEKKYSPCAWEVVRAEIMRSFSLSTNLQESLRSKE
CDLPQTHSLG SRRTLMLLAQ MRRISLFSCL KDRHDFGFPQ EEFGNQFQKA ETIPVLHEMI
QQIFNLFSTK DSSAAWDETL LDKFYTELYQ QLNDLEACVI QGVGVTETPL MKEDSILAVR
KYFQRITLYL KEKKYSPCAW EVVRAEIMRS FSLSTNLQES LRSKE

Chemical structure of peginterferon α-2a and α-2b. Abbreviations: PeG-IFN, peginterferon; IFN, interferon; Lys, lysine; His, histidine; Cys, cysteine; Ser, serine.
Pegylated Interferon alpha-2b
(PegIFN), Virafin

| Formula | C860H1353N229O255S9 |
|---|---|
| CAS | 99210-65-8, 98530-12-2, 215647-85-1 |
| Mol weight | 19268.9111 |
- Interferon α2b, pegylated
- PegIFN a-2b
- PegIFN a-2b (biologics)
- PegIFN α-2b
- PegIntron
- Pegaferon
- PegiHep
- Peginterferon alfa-2b
- Peginterferon α-2b
- Pegylated interferon alfa-2b
- Pegylated interferon α-2b
- Pegylated interferons, PegIFN a-2b
- Proteinaceous biopharmaceuticals, PegIFN a-2b
- Sch 54031
- Sylatron
- ViraferonPeg
Active Moieties
| NAME | KIND | UNII | CAS | INCHI KEY |
|---|---|---|---|---|
| Interferon alfa-2b | unknown | 43K1W2T1M6 | 98530-12-2 | Not applicable |
| Clinical data | |
|---|---|
| Trade names | PegIntron, Sylatron, ViraferonPeg, others |
| AHFS/Drugs.com | Professional Drug Facts |
| MedlinePlus | a605030 |
| License data | EU EMA: by INN |
| Routes of administration | Subcutaneous injection |
| ATC code | L03AB10 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1][2]EU: Rx-only |
| Pharmacokinetic data | |
| Elimination half-life | 22–60 hrs |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 215647-85-1 |
| IUPHAR/BPS | 7462 |
| DrugBank | DB00022 |
| ChemSpider | none |
| UNII | G8RGG88B68 |
| KEGG | D02745 |
| ChEMBL | ChEMBL1201561 |
| ECHA InfoCard | 100.208.164 |
| Chemical and physical data | |
| Formula | C860H1353N229O255S9 |
| Molar mass | 19269.17 g·mol−1 |
NEW DRUG APPROVALS
ONE TIME
$10.00
New Delhi: ,,,,,,https://www.ndtv.com/india-news/zydus-virafin-gets-emergency-use-approval-for-treating-moderate-covid-19-cases-2420358
Zydus Cadila received emergency use approval from the Drugs Controller General of India (DGCI) on Friday for the use of “Virafin”, Pegylated Interferon alpha-2b (PegIFN) in treating moderate COVID-19 infection in adults.
A single-dose subcutaneous regimen of the antiviral Virafin will make the treatment more convenient for the patients. When administered early on during COVID-19, Virafin will help patients recover faster and avoid much of the complications, the company said.
In a release, Cadila Health highlighted that “the drug has also shown efficacy against other viral infections.”
Speaking on the development, Dr Sharvil Patel, Managing Director, Cadila Healthcare Limited said, “The fact that we are able to offer a therapy which significantly reduces the viral load when given early on can help in better disease management. It comes at a much-needed time for patients and we will continue to provide them access to critical therapies in this battle against COVID-19.”
In its Phase III clinical trials, the therapy had shown better clinical improvement in the patients suffering from COVID-19. During the trials, a higher proportion of patients administered with PegIFN arm were RT-PCR negative by day 7. The drug ensures faster viral clearance and has several add-on advantages compared to other anti-viral agents, the release further reads.
The development and the nod from DGCI come at a time when India is combating the second wave of coronavirus.
The central government in one of its major announcements decided to administer COVID-19 vaccines to all age above 18 years.
India recorded 3,32,730 new COVID-19 cases in the last 24 hours, the highest single-day spike since the pandemic broke out last year. India has crossed the mark of 3 lakh COVID-19 cases for two consecutive days now. This has taken the cumulative count of the COVID infection in the country to 1,62,63,695.
2CommentsThe country has recorded 2,263 new deaths due to COVID-19 in the last 24 hours. As many as 1,86,920 people have succumbed to the viral infection in India so far. There are 24,28,616 active COVID-19 cases in the country now.
PATENT
https://patents.google.com/patent/EP1562634B1/en
- Interferon alpha-2a plays an important role for the treatment of chronic hepatitis C, but it is limited in its efficacy by the short in vivo half-life. To improve the half-life and efficacy, interferon alpha-2a was conjugated with a polyethylene glycol moiety. Pegylation changes physicochemical and biological properties of the protein. One effect is the decrease of the proteolytic degradation and the renal clearance. This increases the half-life of the pegylated protein in blood. Another effect is the altered distribution in the body, depending on the size of the PEG moiety of the protein. Interferon alpha 2a pegylated with a large polyethylene glycol moiety (PEG moiety) such as a 40 kDa branched polyethylene moietywherein R and R’ are independently lower alkyl; n and n’ are integers having a sum of from 600 to 1500; and the average molecular weight of the polyethylene glycol units in said conjugate is from about 26,000 daltons to about 66,000 daltons;
has an improved biological activity and exhibits sustained adsorption and reduced renal clearance, resulting in a strong antiviral pressure throughout a once-weekly dosing schedule, see Perry M. C., et al. Drugs, 2001,15,2263-2288 and Lamb M. W., et al. The Annals of Pharmacotherapy, 2002, 36, 933-938. - [0003]See also Monkarsh et al. Analytical Biochemistry, 1997, 247, 434- 440 (Positional Isomers of Mono-pegylated Interferon α-2a) and Bailon et al. Bioconjugate Chemistry, 2001, 12, 195-202 (Rational Design of a Potent, Long-Lasting Form of interferon).
- [0004]The method for the pegylation of interferon alpha-2a is described in EP A 809 996. Since this pegylation is performed by reaction of PEG2-NHS of formulawith primary amino groups on for example lysine or to the N-terminus of the interferon alpha.one or more PEG moieties may be attached and form a mixture of unpegylated, mono- and multiple-pegylated interferon. Monopegylated interferon alpha can be isolated from the mixture by methods known in the art. Furthermore, since interferon alpha-2a molecule exhibits 12 sites for pegylation (11 lysines and the N-terminus) it is a mixture of positional isomers. From these possible twelve isomers, nine were isolated and characterized, each of these being conjugated to the branched polyethylene glycol chain at a specific lysine, namely,
at Lys(31) to form interferon alpha 2a pegylated at Lys(31) [referred to as PEG-Lys(31)],
at Lys(49) to form interferon.alpha 2a pegylated at Lys(49) [referred to as PEG-Lys(49)],
at Lys(70) to form interferon alpha 2a pegylated at Lys(70) [referred to as PEG-Lys(70)],
at Lys(83) to form interferon alpha 2a pegylated at Lys(83) [referred to as PEG-Lys(83)],
at Lys(112) to form interferon alpha 2a pegylated at Lys(112) [referred to as PEG-Lys(112)],
at Lys(121) to form interferon alpha 2a pegylated at Lys(121) [referred to as PEG-Lys(121)],
at Lys(131) to form interferon alpha 2a pegylated at Lys(131) [referred to as PEG-Lys(131)],
at Lys(134) to form interferon alpha 2a pegylated at Lys(134) [referred to as PEG-Lys(134)],
at Lys(164) to form interferon alpha 2a pegylated at Lys(164) [referred to as PEG-Lys(164)]. - [0005]It has been found that PEG-Lys(31) and PEG-Lys(134) have higher activities in an antiviral assay than the mixture, the activity of PEG-Lys(164) was equal to the mixture, whereas the activities of PEG-Lys(49), PEG-Lys(70), PEG-Lys(83), PEG-Lys(112), PEG-Lys(121) and PEG-Lys(131) were lower.
- The following examples will further illustrate the invention
Example 1A Separation of the positional isomers
- [0035]A two-step isolation and purification scheme was used to prepare the monopegylated isoforms of PEG-interferon alpha 2a.
- a) The first step was a separation of the positional isomers on a preparative low pressure liquid chromatography column with a weak-cation exchange matrix (TOSOH-BIOSEP, Toyopearl CM-650S, e.g. Resin Batch no. 82A the diameter of the column being 16 mm, the length 120 cm). A linear pH-gradient of increasing sodium acetate concentration (25 mM, pH 4.0 up 75 mM to pH 7.8) was applied at a flow rate of 0.7 mL/min. Detection was at 280 nm. With this chromatographic step species 1, 2, 5,6 and a mixture of 3, 4, 4a, 7 and 8 could be collected, see Table 1.
- b) The fractions were further separated and purified in the second preparation step. A preparative column with the same matrix as the analytical strong-cation exchange column (Resin Batch no. 82A having a ion exchange capacity of 123 mEq/ml) as described above but larger dimensions (30 mm i.d. and 70 mm length), further a higher flow rate and an extended run time was used. As for the analytical method the column was pre-equilibrated with 3.4 mM sodium acetate, 10% ethanol and 1% diethylene glycol, adjusted to pH 4.4 (buffer A). After loading the PEG-IFN samples, the column was washed with buffer A, followed by an ascending linear gradient to 10 mM dibasic potassium phosphate, 10% ethanol and 1% diethylene glycol, adjusted to pH 6.6 (buffer B). The flow rate was 1.0 mL/min and the detection at 218 nm.
- [0036]The protein concentration of the PEG-IFN alpha 2a isomer was determined by spectrophotometry, based on the 280 nm absorption of the.protein moiety of the PEG-IFN alpha 2a.
- [0037]An analytical elution profile of 180 µg of PEG-IFN alpha 2a is shown in Figure 1. The result of this method is a separation into 8 peaks, 2 peaks with baseline separation and 6 with partial separation. The decrease of the baseline absorption towards the end of the chromatogram suggests that there were no other monopegylated species of IFN alpha 2a eluting at higher retention time.
- [0038]In addition, looking carefully at the IEC-chromatogram a further peak close to the detection limit is visible between peaks 2 and 3 indicating the presence of additional positional isomers that should also contribute to the specific activity of the PEG-IFN alpha 2a mixture. Additional species were expected as the interferon alpha-2a molecule exhibits 12 sites for pegylation (11 lysines and the N-terminus). However, given the low abundance of the these species, they were not isolated and characterised.
- [0039]Isomer samples derived from IEC optimisation runs were investigated directly after the isolation (t = 0) and after 2 of weeks of storage at 5°C (data not shown). No significant differences were observed for the protein derived from IEC-peaks with regard to the protein content as determined by spectrometric methods; nor were any changes to be detected in the monopegylation site, the content of oligo-PEG-IFN alpha 2a, the amount of aggregates and the bioassay activity. Taking into account the relative abundance of the individual isomers – as determined by the IEC method – as well as the specific activities – as determined in the anti-viral assay – almost the total specific bioactivity of the PEG-IFN alpha 2a mixture used for their isolation is recovered (approximately 93%).
- [0040]The analytical IE-HPLC was used to check the purity of the individual isomers with respect to contamination with other positional isomers in the IEC fractions. The peaks 2, 3, 4, 4a, 5 and 7 had more than 98%, the peaks 1 and 8 had 93% and peak 6 had 88 % purity. Table 1:PEG-peptides identified by comparison of the Lys-C digest spectra of the isomers and the reference standard.Identified PEG Sites in the separated PEG-IFN SpeciesPeakmissing peaks in peptide mapPEG-IFNPEG siteMr (DA)SequencePeak 1K31A,E24-49Peak 2K134I, I’134-164Peak 3K131C122-131aPeak 4K121B, C113-131Peak 4aK164b134-164a,bPeak 5K70D, F50-83Peak 6K83D, H71-112Peak 7K49E, F32-70Peak 8K112B, H84-121a132-133 too small to detect.a,b RP-HPLC.
- [0041]The fractions were characterised by the methods described in examples 2 to 6.
Example 1B Analytical separation of positional isomers of mono-pegylated interferon alpha 2a
- [0042]HPLC Equipment:HP1100Column:SP-NPR, TosoH Bioscience, Particle size: 2.5µm, nonporous, Order#: 13076Injection:5-10 µg monopegylated IFNmobile Phase:Buffer A: 10% v/vEthanol 1% v/vDiethylenglycol 2.3 mMNa-Acetat 5.2 mMAcetic acid, in purified water, no pH adjustment Buffer B: 10% v/vEthanol 1% v/vDiethylenglycol 16.4 mMKH2PO4 4.4 mMK2HPO4, in purified water, no pH adjustmentGradient:0 Min40 %B 2 Min40 %B 2.1 Min48 %B 25 Min68 %B 27 Min75 %B 30 Min75 %B 34 Min40 %B 40 Min40 %BFlow:1.0 ml/min Column Temperature:25°C Detection:218 nm a typical Chromatogram is given in Figure 8.
Example 2 Analysis of the fractions by mass spectrometry peptide mapping
- [0043]Mass spectra were recorded on a MALDI-TOF MS instrument (PerSeptive Biosystems Voyager-DE STR with delayed extraction). Each IEC fraction (Ion Exchange Chromatography) was desalted by dialysis, reduced with 0.02 M 1,4-dithio-DL-threitol (DTT) and alkylated with 0.2 M 4-vinyl pyridine. Then the proteins were digested with endoproteinase Lys-C (Wako Biochemicals) in 0.25 M Tris (tris(hydroxymethyl)-aminoethane) at pH 8.5 with an approximate enzyme to protein ratio of 1:30. The reaction was carried out over night at 37 °C.
- [0044]A solution of 20 mg/ml α-cyano-4-hydroxycinnamic acid and 12 mg/ml nitrocellulose in acetone/isopropanol 40/60 (v/v) was used as matrix (thick-layer application). First, 0.5 µL of matrix was placed on the target and allowed to dry. Then, 1.0 µL of sample was added. The spectra were obtained in linear positive ionisation mode with an accelerating voltage of 20.000 V and a grid voltage of 95 %. At least 190 laser shots covering the complete spot were accumulated for each spectrum. Des-Arg1-bradykinin and bovine insulin were used for internal calibration.
Example 3 high-performance liquid chromatography (RP-HPLC) Peptide Mapping
- [0045]The peptides were characterized by reverse-phase high-performance liquid chromatography (RP-HPLC) Peptide Mapping. The IEC fractions were reduced, alkylated and digested with endoproteinase Lys-C as described for the MALDI-TOF MS peptide mapping. The analysis of the digested isomers was carried out on a Waters Alliance HPLC system with a Vydac RP-C18 analytical column (5 µm, 2.1 × 250 mm) and a precolumn with the same packing material. Elution was performed with an acetonitrile gradient from 1 % to 95 % for 105 min in water with a flow rate of 0.2 mL/min. Both solvents contained 0.1 % (v/v) TFA. 100 µL of each digested sample were injected and monitored at 215 nm.
Example 4 MALDI-TOF spectra of undigested protein
- [0046]An 18 mg/ml solution of trans-3-indoleacrylic acid in acetonitrile/0.1 % trifluoroacetic acid 70/30 (v/v) was premixed with the same volume of sample solution. Then 1.0 µL of the mixture was applied to the target surface. Typically 150 – 200 laser shots were averaged in linear positive ionisation mode. The accelerating voltage was set to 25.000 V and the grid voltage to 90 %. Bovine albumin M+ and M2+ were used for external calibration.
Example 5 SE-HPLC (size exclusion HPLC)
- [0047]SE-HPLC was performed with a Waters Alliance 2690 HPLC system equipped with a TosoHaas TSK gel G 4000 SWXL column (7.8 × 300 mm). Proteins were eluted using a mobile phase containing 0.02 M NaH2PO4, 0.15 M NaCl, 1% (v/v) diethylene glycol and 10 % (v/v) ethanol (pH 6.8) at a flow rate of 0.4 mL/min and detected at 210 nm. The injection amounts were 20 µg of each isomers.
- [0048]Size Exclusion HPLC and SDS-PAGE were used to determine the amount of oligo-PEG-IFN alpha 2a forms and aggregates in the different IEC fractions. The reference material contains 2.3 % aggregates and 2.2 % oligomers (Figure 4).
- [0049]Peaks 1, 4, 4a, 5, 6 and 8 contain < 0.7 % of the oligopegylated IFN alpha 2a forms, whereas in,peaks 2, 3, and 7 the percentage of the oligopegylated IFN alpha 2a forms are under the detection limit (< 0.2 %). In the case of the aggregates a different trend could be seen. In all peaks the amount of aggregates is below 0.9 %.
Example 6 SDS-PAGE
- [0050]SDS-PAGE was carried out both under non-reducing and under reducing conditions using Tris-Glycine gels of 16 % (1.5 mm, 10 well). Novex Mark 12 molecular weight markers with a mass range from 2.5 to 200 kDa were used for calibration, bovine serum albumin (BSA) was used as sensitivity standard (2 ng). Approximately 1 µg of all the samples and 0.5 µg of standard were applied to the gel. The running conditions were 125 V and 6 W for 120 min. The proteins were fixed and stained using the silver staining kit SilverXpress from Novex.
- [0051]The gels that were recorded under non-reducing conditions for the IEC fractions 1- 8 (Figure 2) show a pattern that is comparable to that of the PEG-IFN alpha 2a reference standard.
- [0052]Under reducing conditions, the gels show an increase in intensity of the minor bands at about 90 kDa as compared to the standard. Between 6 and 10 kDa protein fragments appear for peaks 6, 7 and 8 (Figure 3). Both bands together correspond to approximately 1 % of clipped material. In the lanes of isomer 1, 5, 6, 7, 8 additional bands with more than 100 kDa can be seen which are also present in the standard. These can be assigned to oligomers. Thus SDS-PAGE confirms the results of the SE-HPLC analysis.
- [0053]Overall, RP-HPLC and SDS-PAGE experiments indicate that the purity of the IEC fractions can be considered comparable to the PEG-IFN alpha 2a reference standard.
- [0054]The structure of the PEG-IFN alpha 2a species derived from the 9 IEC-fractions were identified based on the results of the methods described above using the strategy mentioned above.
Example 7 The antiviral activity (AVA)
- [0055]The antiviral activity was estimated by its protective effect on Madin-Darby bovine kidney (MDBK) cells against the infection by vesticular stomatitis virus (VSV) and compared with a PEG-IFN alpha 2a standard. Samples and reference standard were diluted in Eagle’s Minimum Essential Medium (MEM) containing 10 % fetal bovine serum to a final concentration of 10 ng/mL (assay starting concentration). Each sample was assayed in quadruplicate.
- [0056]The antiviral protection of Madin-Darby bovine kidney cells (MDBK) with vesicular stomatitis virus was tested according to the method described in Virol. 1981, 37, 755-758. All isomers induced an activity in the anti-viral assay as presented in Table 2. The activities range between 1061 and 339 U/µg, indicating that the difference in specific activities of the protein in the positional isomers is significant. The know-how and the results generated so far will allow the initiation of further investigations to establish this structure-function relationship between the positional isomers and the IFN alpha receptors. Table 2:In Vitro Antiviral Activities of PEG-IFN alpha 2a and individual PEG-IFN alpha 2a isomers. The Antiviral activity was determined in MDBK cells infected with vesicular stomatitis virus. The results present the averages of three assays performed independently.Antiviral Assay of PEG-IFNPeakU/µgPEG-IFN1061 ± 50Peak 11818 ± 127Peak 21358 ± 46Peak 3761197Peak 4339 ± 33Peak 4a966 ± 107Peak 5600 ± 27Peak 6463 ± 25Peak7513 ± 20Peak 8468 ± 23
- [0057]The results are further illustrated by the following figures
- Figure 1: Analytical IEC-HPLC of 180µg of PEG-IFN alpha 2a. An analytical strong-cation exchange column was used to check the purity of the separated positional isomers from each purification step (TOSOH-BIOSEP, SP-SPW,10 µm particle size, 7.5 mm diameter, 7.5 cm length).
- Figure 2: A/B: SDS-PAGE analysis with Tris-glycine (16%), the samples were electrophoresed under non-reduced conditions. The gels were stained for protein with Silver Stain. Lanes: M, molecular weight marker proteins/ 2, Peak 1/ 3, Peak 2/ 4, Peak 3/ 5, Peak 4/ 6, Peak 4a/ 7, Peak 5/ 8, Peak 6/ 9, Peak 7/10, Peak 8/ 11, Ix PEG-IFN standard/ 12, 1.5x PEG-IFN standard/ C1, IFN standard.
- Figure 3: A/B: SDS-PAGE analysis with Tris-glycine (16%), the samples were electrophoresed under reduced conditions. The gels were stained for protein with Silver Stain. Lanes: M, molecular weight marker proteins/ 2, Peak 1/ 3, Peak 2/ 4, Peak 3/ 5, Peak 4/ 6, Peak 4a/ 7, Peak 5/ 8, Peak 6/ 9, Peak 7/ 10, Peak 8/ 11, 1x PEG-IFN standard/ 12, 1.5x PEG-IFN standard/ C1, IFN standard.
- Figure 4: Size Exclusion (SE-) HPLC was used to determine the amount of oligo PEG-IFN forms and aggregates in the different IEC fractions. SE-HPLC was performed with a TosoHaas TSK gel G 4000 SWXL column (7.8 × 300 mm).
- Figure 5: MALDI-TOF spectrometry was used to determine the molecular weight of each isomer in order to ensure that the PEG-IFN molecules were still intact after IEC chromatography and to confirm the monopegylation.
- Figure 6: MALDI-TOF Lys-C peptide maps of the PEG-IFN reference standard and the peaks 1, 2, 3, 4, 4a, 5, 6, 7, 8. Missing peaks compared to the standard are indicated by arrows.
- Figure 7: RP-HPLC chromatograms of the Lys-C digests of the PEG-IFN reference and peak 4a
- Figure 8: Analytical HPLC of 5-10µg of PEG-IFN alpha 2a mixture of positional isomers on a column charged with SP-NPR, TosoH Bioscience, Particle size: 2.5µm, nonporous as described in Example 1B..
- Figure 9: Ribbon structure of interferon alpha-2a showing the pegylation sites. This is the high resolution structure of human interferon alpha-2a determined with NMR spectroscopy see J. Mol. Biol. 1997, 274, 661-675. The pegylation sites of pegylated interferon alpha-2a are coloured red and labelled with residue type and residue number.
Pegylated interferon alfa-2b, sold under the brand name PegIntron among others, is a medication used to treat hepatitis C and melanoma.[3] For hepatitis C it is typically used with ribavirin and cure rates are between 33 and 82%.[3][4] For melanoma it is used in addition to surgery.[3] It is given by injection under the skin.[3]
Side effects are common.[5] They may include headache, feeling tired, mood changes, trouble sleeping, hair loss, nausea, pain at the site of injection, and fever.[3] Severe side effects may include psychosis, liver problems, blood clots, infections, or an irregular heartbeat.[3] Use with ribavirin is not recommended during pregnancy.[3] Pegylated interferon alfa-2b is in the alpha interferon family of medications.[3] It is pegylated to protect the molecule from breakdown.[5]
Pegylated interferon alfa-2b was approved for medical use in the United States in 2001.[3] It is on the World Health Organization’s List of Essential Medicines.[6]
Peginterferon alfa-2b is a form of recombinant interferon used as part of combination therapy to treat chronic Hepatitis C, an infectious liver disease caused by infection with Hepatitis C Virus (HCV). HCV is a single-stranded RNA virus that is categorized into nine distinct genotypes, with genotype 1 being the most common in the United States, and affecting 72% of all chronic HCV patients 3. Treatment options for chronic Hepatitis C have advanced significantly since 2011, with the development of Direct Acting Antivirals (DAAs) resulting in less use of Peginterferon alfa-2b. Peginterferon alfa-2b is derived from the alfa-2b moeity of recombinant human interferon and acts by binding to human type 1 interferon receptors. Activation and dimerization of this receptor induces the body’s innate antiviral response by activating the janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway. Use of Peginterferon alfa-2b is associated with a wide range of severe adverse effects including the aggravation and development of endocrine and autoimmune disorders, retinopathies, cardiovascular and neuropsychiatric complications, and increased risk of hepatic decompensation in patients with cirrhosis. The use of Peginterferon alfa-2b has largely declined since newer interferon-free antiviral therapies have been developed.
In a joint recommendation published in 2016, the American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America (IDSA) no longer recommend Peginterferon alfa-2b for the treatment of Hepatitis C 2. Peginterferon alfa-2b was used alongside Ribavirin(https://go.drugbank.com/drugs/DB00811) with the intent to cure, or achieve a sustained virologic response (SVR), after 48 weeks of therapy. SVR and eradication of HCV infection is associated with significant long-term health benefits including reduced liver-related damage, improved quality of life, reduced incidence of Hepatocellular Carcinoma, and reduced all-cause mortality 1.
Peginterferon alfa-2b is available as a variable dose injectable product (tradename Pegintron) used for the treatment of chronic Hepatitis C. Approved in 2001 by the FDA, Pegintron is indicated for the treatment of HCV with Ribavirin or other antiviral drugs Label. When combined together, Peginterferon alfa-2b and Ribavirin have been shown to achieve a SVR between 41% for genotype 1 and 75% for genotypes 2-6 after 48 weeks of treatment.
Medical uses
It is used to treat hepatitis C and melanoma. For hepatitis C it is typically used with ribavirin. For melanoma it is used in addition to surgery.[3]
For hepatitis C it may also be used with boceprevir, telaprevir, simeprevir, or sofosbuvir.[5]
In India, in 2021, DGCI approved emergency use of Zydus Cadila‘s Virafin in treating moderate COVID-19 infection.[7]
Host genetic factors
For genotype 1 hepatitis C treated with pegylated interferon-alfa-2a or pegylated interferon-alfa-2b combined with ribavirin, it has been shown that genetic polymorphisms near the human IL28B gene, encoding interferon lambda 3, are associated with significant differences in response to the treatment. This finding, originally reported in Nature,[8] showed that genotype 1 hepatitis C patients carrying certain genetic variant alleles near the IL28B gene are more likely to achieve sustained virological response after the treatment than others. A later report from Nature[9] demonstrated that the same genetic variants are also associated with the natural clearance of the genotype 1 hepatitis C virus.
Side effects
Common side effects include headache, feeling tired, mood changes, trouble sleeping, hair loss, nausea, pain at the site of injection, and fever. Severe side effects may include psychosis, liver problems, blood clots, infections, or an irregular heartbeat.[3] Use with ribavirin is not recommended during pregnancy.[3]
Mechanism of action
One of the major mechanisms of PEG-interferon alpha-2b utilizes the JAK-STAT signaling pathway. The basic mechanism works such that PEG-interferon alpha-2b will bind to its receptor, interferon-alpha receptor 1 and 2 (IFNAR1/2). Upon ligand binding the Tyk2 protein associated with IFNAR1 is phosphorylated which in turn phosphorylates Jak1 associated with IFNAR2. This kinase continues its signal transduction by phosphorylation of signal transducer and activator of transcription (STAT) 1 and 2 via Jak 1 and Tyk2 respectively. The phosphorylated STATs then dissociate from the receptor heterodimer and form an interferon transcription factor with p48 and IRF9 to form the interferon stimulate transcription factor-3 (ISGF3). This transcription factor then translocates to the nucleus where it will transcribe several genes involved in cell cycle control, cell differentiation, apoptosis, and immune response.[10][11]
PEG-interferon alpha-2b acts as a multifunctional immunoregulatory cytokine by transcribing several genes, including interleukin 4 (IL4). This cytokine is responsible for inducing T helper cells to become type 2 helper T cells. This ultimately results in the stimulation of B cells to proliferate and increase their antibody production. This ultimately allows for an immune response, as the B cells will help to signal the immune system that a foreign antigen is present.[12]
Another major mechanism of type I interferon alpha (IFNα) is to stimulate apoptosis in malignant cell lines. Previous studies have shown that IFNα can cause cell cycle arrest in U266, Daudi, and Rhek-1 cell lines.[13]
A follow-up study researched to determine if the caspases were involved in the apoptosis seen in the previous study as well as to determine the role of mitochondrial cytochrome c release. The study confirmed that there was cleavage of caspase-3, -8, and -9. All three of these cysteine proteases play an important role in the initiation and activation of the apoptotic cascade. Furthermore, it was shown that IFNα induced a loss in the mitochondrial membrane potential which resulted in the release of cytochrome c from the mitochondria. Follow-up research is currently being conducted to determine the upstream activators of the apoptotic pathway that are induced by IFNα.[14]
History
It was developed by Schering-Plough. Merck studied it for melanoma under the brand name Sylatron. It was approved for this use in April 2011.
References
- ^ “PegIntron- peginterferon alfa-2b injection, powder, lyophilized, for solution PegIntron- peginterferon alfa-2b kit”. DailyMed. Retrieved 28 September 2020.
- ^ “Sylatron- peginterferon alfa-2b kit”. DailyMed. 28 August 2019. Retrieved 28 September 2020.
- ^ Jump up to:a b c d e f g h i j k l “Peginterferon Alfa-2b (Professional Patient Advice) – Drugs.com”. http://www.drugs.com. Archived from the original on 16 January 2017. Retrieved 12 January 2017.
- ^ “ViraferonPeg Pen 50, 80, 100, 120 or 150 micrograms powder and solvent for solution for injection in pre-filled pen CLEAR CLICK – Summary of Product Characteristics (SPC) – (eMC)”. http://www.medicines.org.uk. Archived from the original on 13 January 2017. Retrieved 12 January 2017.
- ^ Jump up to:a b c “Peginterferon alfa-2b (PegIntron)”. Hepatitis C Online. Archived from the original on 23 December 2016. Retrieved 12 January 2017.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ https://www.aninews.in/news/national/general-news/dgci-approves-emergency-use-of-zyduss-virafin-in-treating-moderate-covid-19-infection20210423163622/
- ^ Ge D, Fellay J, Thompson AJ, et al. (2009). “Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance”. Nature. 461 (7262): 399–401. Bibcode:2009Natur.461..399G. doi:10.1038/nature08309. PMID 19684573. S2CID 1707096.
- ^ Thomas DL, Thio CL, Martin MP, et al. (2009). “Genetic variation in IL28B and spontaneous clearance of hepatitis C virus”. Nature. 461 (7265): 798–801. Bibcode:2009Natur.461..798T. doi:10.1038/nature08463. PMC 3172006. PMID 19759533.
- ^ Ward AC, Touw I, Yoshimura A (January 2000). “The JAK-STAT pathway in normal and perturbed hematopoiesis”. Blood. 95 (1): 19–29. doi:10.1182/blood.V95.1.19. PMID 10607680. Archived from the original on 2014-04-26.
- ^ PATHWAYS :: IFN alpha[permanent dead link]
- ^ Thomas H, Foster G, Platis D (February 2004). “Corrigendum toMechanisms of action of interferon and nucleoside analogues J Hepatol 39 (2003) S93–8″. J Hepatol. 40 (2): 364. doi:10.1016/j.jhep.2003.12.003.
- ^ Sangfelt O, Erickson S, Castro J, Heiden T, Einhorn S, Grandér D (March 1997). “Induction of apoptosis and inhibition of cell growth are independent responses to interferon-alpha in hematopoietic cell lines”. Cell Growth Differ. 8 (3): 343–52. PMID 9056677. Archived from the original on 2014-04-26.
- ^ Thyrell L, Erickson S, Zhivotovsky B, et al. (February 2002). “Mechanisms of Interferon-alpha induced apoptosis in malignant cells”. Oncogene. 21 (8): 1251–62. doi:10.1038/sj.onc.1205179. PMID 11850845.
External links
- Peginterferon alfa-2b in the U.S. National Library of Medicine’s Drug Information Portal
- Medicines patent loophole ‘found’ at the BBC, 2007
- PEG-Intron (Peginterferon Alfa-2B) — Platelet Count Decreased from DrugLib.com
///////////Pegylated Interferon alpha-2b, PegIFN, Virafin, COVID 19, CORONA VIRUS, INDIA 2021, APPROVALS 2021
DEXMETHYLPHENIDATE
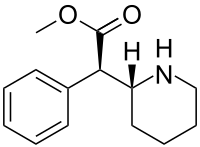
DEXMETHYLPHENIDATE
SynonymsDexmethylphenidate HCl, UNII1678OK0E08, CAS Number19262-68-1, WeightAverage: 269.77
Chemical FormulaC14H20ClNO2
methyl (2R)-2-phenyl-2-[(2R)-piperidin-2-yl]acetate hydrochloride

| CAS Number | 40431-64-9 as HCl: 19262-68-1 |
|---|---|
| PubChem CID | 154101as HCl: 154100 |
| IUPHAR/BPS | 7554 |
| DrugBank | DB06701 as HCl: DBSALT001458 |
| ChemSpider | 135807 as HCl: 135806 |
| UNII | M32RH9MFGPas HCl: 1678OK0E08 |
Trade Name:Focalin® / Attenade®MOA:Norepinephrine-dopamine reuptake inhibitorIndication:Attention deficit hyperactivity disorder (ADHD)Status:ApprovedCompany:Novartis (Originator) , CelgeneSales:$365 Million (Y2015); 
$492 Million (Y2014);
$594 Million (Y2013);
$554 Million (Y2012);
$550 Million (Y2011);ATC Code:N06BA11
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2005-05-26 | New dosage form | Focalin XR | Attention deficit hyperactivity disorder (ADHD) | Capsule, Extended release | 5 mg/10 mg/15 mg/20 mg/25 mg/30 mg/35 mg/40 mg | Novartis | |
| 2001-11-13 | Marketing approval | Focalin | Attention deficit hyperactivity disorder (ADHD) | Tablet | 2.5 mg/5 mg/10 mg | Novartis |
Dexmethylphenidate hydrochloride was approved by the U.S. Food and Drug Administration (FDA) on Nov 13, 2001. It was developed and marketed as Focalin® by Novartis in the US.
Dexmethylphenidate hydrochloride is a norepinephrine-dopamine reuptake inhibitor (NDRI). It is indicated for the treatment of attention deficit hyperactivity disorder (ADHD).
Focalin® is available as tablet for oral use, containing 2.5 mg, 5 mg or 10 mg of Dexmethylphenidate hydrochloride. The recommended dose is 10 mg twice daily, at least 4 hours apart.
NEW DRUG APPROVALS
ONE TIME
$10.00
NDA 212994, AZSTARYS
FDA APPROVE 2021

Drug Product Name Serdexmethylphenidate and Dexmethylphenidate (SDX/d-MPH)
Dosage Form capsule Strength 26.1/5.2 mg SDX/d-MPH 39.2/7.8 mg SDX/d-MPH 52.3/10.4 mg SDX/d-MPH
Route of Administration oral
Rx/OTC Dispensed Rx
Maximum Daily Dose 52.3 mg serdexmethylphenidate /10.4 mg dmethylphenidate as free base or 56 mg serdexmethylphenidate Chlorid
Dexmethylphenidate, sold under the brand name Focalin among others, is a medication used to treat attention deficit hyperactivity disorder (ADHD) in those over the age of five years.[3] If no benefit is seen after four weeks it is reasonable to discontinue its use.[3] It is taken by mouth.[3] The immediate release formulation lasts up to five hours while the extended release formulation lasts up to twelve hours.[4]
Common side effects include abdominal pain, loss of appetite, and fever.[3] Serious side effects may include abuse, psychosis, sudden cardiac death, mania, anaphylaxis, seizures, and dangerously prolonged erection.[3] Safety during pregnancy and breastfeeding is unclear.[5] Dexmethylphenidate is a central nervous system (CNS) stimulant.[6][3] How it works in ADHD is unclear.[3] It is the more active enantiomer of methylphenidate.[3]
Dexmethylphenidate was approved for medical use in the United States in 2001.[1] It is available as a generic medication.[3] In 2018, it was the 156th most commonly prescribed medication in the United States, with more than 3 million prescriptions.[7][8] It is also available in Switzerland.[9]
Reference:1. US6528530B2.
2. J. Org. Chem. 1998, 63, 9628-9629.Route 2
Reference:1. J. Am. Chem. Soc. 1999, 121,6509-6510.Route 3
Reference:1. Org. Process Res. Dev. 2010, 14, 1473–1475.Route 4
Reference:1. J. Med. Chem. 1998, 41,591-601.Route 5
Reference:1. Org. Lett. 1999, 1, 175-178.
2. Organic Syntheses 1985, 63, 206-212.
Four isomers of methylphenidate are possible, since the molecule has two chiral centers. One pair of threo isomers and one pair of erythro are distinguished, from which primarily d-threo-methylphenidate exhibits the pharmacologically desired effects.[102][124] The erythro diastereomers are pressor amines, a property not shared with the threo diastereomers. When the drug was first introduced it was sold as a 4:1 mixture of erythro:threo diastereomers, but it was later reformulated to contain only the threo diastereomers. “TMP” refers to a threo product that does not contain any erythro diastereomers, i.e. (±)-threo-methylphenidate. Since the threo isomers are energetically favored, it is easy to epimerize out any of the undesired erythro isomers. The drug that contains only dextrorotatory methylphenidate is sometimes called d-TMP, although this name is only rarely used and it is much more commonly referred to as dexmethylphenidate, d-MPH, or d-threo-methylphenidate. A review on the synthesis of enantiomerically pure (2R,2′R)-(+)-threo-methylphenidate hydrochloride has been published.[125]Methylphenidate synthesis

Method 1: Methylphenidate preparation elucidated by Axten et al. (1998)[126] via Bamford-Stevens reaction.

Method 2: Classic methylphenidate synthesis[127]

Method 3: Another synthesis route of methylphenidate which applies Darzens reaction to obtain aldehyde as an intermediate. This route is significant for its selectivity.SYNhttps://onlinelibrary.wiley.com/doi/abs/10.1002/jhet.2705SUN
1.9 Synthesis of (R, R), (R, S), (S, S) and (S, R) methyl 2-piperidin-2-yl-phenylacetate hydrochloride (1a, 1b, 1c and 1d)
Compound 8a, 8b, 8c or 8d (400 mg, 1.3 mmol) was dissolved into methanol solution (15 mL), and then thionyl chloride (1 mL) was added drop-wise. The reaction mixture was stirred for 12 hours and concentrated in vacuum; a white solid was precipitated and filtered to afford the final product. (1a. 0.28 g, 82% yield; 1b. 0.30 g, 84% yield; 1c. 0.31 g, 85% yield; 1d. 0.30 g, 84% yield). The characterization data of the four final products had been reported [2] by us in 2016.
SYN
https://patents.google.com/patent/US20040180928A1/en
- Dexmethylphenidate, also known as d-threo-methylphenidate, (R,R)-methylphenidate or (R,R)-α-phenyl-2-piperidineacetic acid methyl ester, having the formula:
- [0029]
is CNS (central nervous system) stimulant that is chemically and pharmacologically similar to the amphetamines. Dexmethylphenidate’s CNS actions is milder than those of the amphetamines and have more noticeable effects on mental activities than on motor activities. - [0030]
It has been reported by Sporzny (1961) that among racemic mixtures of threo and erythro diastereomers of methylphenidate, only threo-isomer displays stimulant properties. Dexmethylphenidate hydrochloride (i.e. the d-threo enantiomer of methylphenidate hydrochloride) has been reported to be 5 to 38 times more active than the corresponding (S,S)-methylphenidate hydrochloride (Prashad 2000). - [0031]
A commercially available drug is sold under the name Focalin™ (Novartis) and it consists of dexmethylphenidate in the form of the hydrochloride salt. This product is orally administered and clinically used in the treatment of narcolepsy and as adjunctive treatment in children with attention deficit disorder (ADD) and attention-deficit hyperactivity disorder (ADHD). - [0032]
A synthesis of dexmethylphenidate hydrochloride was firstly described in U.S. Pat. No. 2,838,519 and include resolution of erythro-α-phenyl-2-piperidineacetamide to obtain enantiopure (2R,2′S)-α-phenyl-2-piperidineacetamide, which was subjected to epimerization, hydrolysis, and esterification as shown in Scheme 1: - [0033]
Related example of preparation of dexmethylphenidate from erythro-α-phenyl-2-piperidineacetamide was described in U.S. Pat. No. 5,936,091. - [0034]
Preparation of dexmethylphenidate through optical resolution of threo-α-phenyl-2-piperidineacetamide was described in U.S. Pat. No. 5,965,734, as shown in Scheme 2: - [0035]
Synthetic methods for the preparation of racemic mixture of threo- and erythro-α-phenyl-2-piperidineacetamides as raw materials for the preparation of dexmethylphenidate were described by Panizzon (1944) and Patric (1982) and in U.S. Pat. Nos. 2,507,631, 2,838,519, 2,957,880 and 5,936,091, and in WO 01/27070. These methods include using sodium amide as base in the nucleophilic substitution of chlorine in 2-chloropyridine with phenylacetonitrile followed by hydrolysis of the formed nitrile and reduction of a pyridine ring to a piperidine one by hydrogenation on PtO 2 catalyst, as shown in Scheme 3: - [0036]
Alternatively, 2-bromopyridine was used instead of 2-chloropyridine by Deutsch (1996). - [0037]
In some other methods threo-methylphenidate was used as the raw material for the preparation of dexmethylphenidate. Threo-methylphenidate may be prepared by a several routes, inter alia by the following two processes: - [0038]
i) by esterification of threo-ritalinic acid which may be prepared from erythro-enriched and threo-α-phenyl-2-piperidineacetamides as shown in Scheme 4: - [0039]
ii) by cyclization of easily available 1-(phenylglyoxylyl)piperidine arenesulfonylhydrazone to (R*,R*)-enriched 7-phenyl-1-azabicyclo[4.2.0]octan-8-one and further converting the β-lactam to threo-methylphenidate hydrochloride, as described by Axten (1998), Corey (1965) and Earle (1969) and in WO 99/36403 and shown in Scheme 5: - [0040]
The resolution of threo-methylphenidate to afford dexmethylphenidate was first reported by Patric (1987) which used (R)-(−)-binaphthyl-2,2′-diyl hydrogen phosphate as the resolving agent. Several new resolutions of threo-methylphenidate have been reported recently by Prashad (1999) and in U.S. Pat. Nos. 6,100,401, 6,121,453, 6,162,919 and 6,242,464 as described in Scheme 6: - [0041]
wherein the chiral acid is one of the following: (R)-(−)-binaphthyl-2,2′-diyl hydrogen phosphate, (−)-menthoxyacetic acid, ditoluoyl-D-tartaric acid or dibenzoyl-D-tartaric acid. - [0042]
Resolution of threo-methylphenidate may be also achieved by enzymatic hydrolysis methods as proposed by Prashad (1998) and in WO 98/25902. Such resolution is described in Scheme 7: - [0043]
Resolution of threo-ritalinic acid hydrochloride with (S)-1-phenylethylamine give complex salt (R,R)-enriched threo-ritalinic acid.HCl.(S)-1-phenylethylamine with 77% ee optical purity of ritalinic acid (U.S. Ser. No. 2002/0019535), Scheme 8:
- [0119]
- [0120]
Gaseous hydrogen chloride was passed through a boiling solution of (R,R)-N-Boc-ritalinic acid (95.4 g, 299 mmol) in methanol (1.5 L). The mixture was stirred for 12 hours under reflux conditions and concentrated to the volume of 250 mL. Toluene (750 mL) was added to the stirred residue, then methanol lo was removed from boiling suspension under normal pressure. The obtained mixture was stirred overnight at 0-5° C. The precipitated solids were filtered off, washed on the filter with toluene (3×50 mL) and dried under reduced pressure to give 78.4 g (97.2% yield) of dexmethylphenidate hydrochloride as white crystals with mp 222-224° C. and [α]D 25 87.0° (c=1, MeOH).
- [0117]
- [0118]
A mixture of crystalline salt of (R,R)-N-Boc-ritalinic acid and (S)-1-phenylamine with [α]D 20 −28.6° (c=1, MeOH) (133.0 g, 302 mmol), ethyl acetate (1.3 L) and solution of citric acid (164.0 g, 845 mmol) in water (1.3 L) was stirred at 15-25° C. for 1.5 hours. The organic layer was separated, washed lo with brine (20 mL), dried over sodium sulfate, filtered and evaporated under reduced pressure to give 95.4 g (99%) of (R,R)-N-B
- [0115]
- [0116]
(S)-1-Phenylethylamine (113.8 g, 0.94 mol, 0.6 eq) was added dropwise to a stirred solution of N-Boc-threo-ritalinic acid (500 g, 1.57 mol, 1 eq) in ethyl acetate (5 L) for 1 hour at 20-40° C. The mixture was stirred for 1 hour at 40° C. and overnight at 5° C. The precipitated solids were filtered off, washed on the lo filter with cold ethyl acetate (2×500 mL) and dried under reduced pressure to give 380 g of white crystals with [α]D 20−23.3° (c=1, MeOH). The salt was twice recrystallized from aqueous methanol. The precipitated crystals were filtered off, washed on the filter with cold aqueous methanol and dried under reduced pressure to a constant weight to give 265 g (33.5% yield) of salt of (R,R)-N-Boc-ritalinic acid and (S)
- [0113]
- [0114]
A mixture of solution of N-Boc-threo-ritalinic acid sodium salt (1700 g, 4.98 mmol), citric acid (1150 g, 5.98 mmol) and water (5 mL) was stirred at 15-25° C. for 0.5 hour and extracted with ethyl acetate (3×4 L). Combined organic extracts were washed with brine (2×3 L), dried over sodium sulfate, filtered and evaporated under reduced pressure to constant weight to give 1560 g (98.1% yield) of N-Boc-threo-ritalinic acid with mp 133-134° C. (EtOAc/hexane) and 99.8% purity by HPLC.
Medical uses
Dexmethylphenidate is used as a treatment for ADHD, usually along with psychological, educational, behavioral or other forms of treatment. It is proposed that stimulants help ameliorate the symptoms of ADHD by making it easier for the user to concentrate, avoid distraction, and control behavior. Placebo-controlled trials have shown that once-daily dexmethylphenidate XR was effective and generally well tolerated.[6]
Improvements in ADHD symptoms in children were significantly greater for dexmethylphenidate XR versus placebo.[6] It also showed greater efficacy than osmotic controlled-release oral delivery system (OROS) methylphenidate over the first half of the laboratory classroom day but assessments late in the day favoured OROS methylphenidate.[6]
CLIP
An Improved and Efficient Process for the Production of Highly Pure Dexmethylphenidate Hydrochloride
Long-Xuan Xing, Cheng-Wu Shen, Yuan-Yuan Sun, Lei Huang, Yong-Yong Zheng,* Jian-Qi Li*
https://onlinelibrary.wiley.com/doi/abs/10.1002/jhet.2705
The present work describes an efficient and commercially viable process for the synthesis of dexmethylphenidate hydrochloride (1), a mild nervous system stimulant. The overall yield is 23% with ~99.9% purity (including seven chemical steps). Formation and control of possible impurities are also described in this report.

(R)-methyl 2-phenyl-2-((R)-piperidin-2-yl)acetate hydrochloride (1). ………… afford 1 as a white solid (107.6 g, 87.3% yield) with 99.50% purity and 99.70% ee. The crude product (107.6 g, 0.4 mol) was further purified by recrystallization from pure water (100 mL) to obtain the qualified product 1 (98.3 g, 91.4% yield) with 99.92 purity and 99.98% ee.
[α] 25 D +85.6 (MeOH, c 1) (lit [4b]. [α] 25 D +84 (MeOH, c 1));
Mp 222-223 C (lit [4b]. Mp 222– 224°C); MS m/z 234 [M + H]+ .
1 H NMR (400Hz, DMSO-d6) δ 1 H NMR (400 MHz, DMSO-d6) δ 9.64 (br, 1H), 8.97 (br, 1H), 7.41-7.26 (m, 5H), 4.18-4.16 (d, J = 9.2Hz, 1H), 3.77-3.75 (m, 1H), 3.66 (s, 3H), 3.25 (m, 1H), 2.94 (m, 1H), 1.67-1.64 (m, 3H), 1.41-1.25 (m, 3H).
13C NMR (100.6 MHz, DMSO-d6) δ 171.3, 134.2, 129.1, 128.6, 128.2, 56.8, 53.3, 52.6, 44.5, 25.7, 21.5, 21.4.
1H-NMR, and 13C-NMR of compound 1………………………………….. 10-11


DEPT,

COSY, NOESY, GHMBC, and HMQC of compound 1……………… 12-14

COSY

NOESY

GHMBC

HMQC
Contraindications
This section is transcluded from Methylphenidate. (edit | history)
Methylphenidate is contraindicated for individuals using monoamine oxidase inhibitors (e.g., phenelzine, and tranylcypromine), or individuals with agitation, tics, glaucoma, or a hypersensitivity to any ingredients contained in methylphenidate pharmaceuticals.[10]
The US Food and Drug Administration (FDA) gives methylphenidate a pregnancy category of C, and women are advised to only use the drug if the benefits outweigh the potential risks.[11] Not enough human studies have been conducted to conclusively demonstrate an effect of methylphenidate on fetal development.[12] In 2018, a review concluded that it has not been teratogenic in rats and rabbits, and that it “is not a major human teratogen”.[13]
Adverse effects
Part of this section is transcluded from Methylphenidate. (edit | history)
Products containing dexmethylphenidate have a side effect profile comparable to those containing methylphenidate.[14]

Addiction experts in psychiatry, chemistry, pharmacology, forensic science, epidemiology, and the police and legal services engaged in delphic analysis regarding 20 popular recreational drugs. Methylphenidate was ranked 13th in dependence, 12th in physical harm, and 18th in social harm.[15]
The most common adverse effects include appetite loss, dry mouth, anxiety/nervousness, nausea, and insomnia. Gastrointestinal adverse effects may include abdominal pain and weight loss. Nervous system adverse effects may include akathisia (agitation/restlessness), irritability, dyskinesia (tics), lethargy (drowsiness/fatigue), and dizziness. Cardiac adverse effects may include palpitations, changes in blood pressure and heart rate (typically mild), and tachycardia (rapid heart rate).[16] Smokers with ADHD who take methylphenidate may increase their nicotine dependence, and smoke more often than before they began using methylphenidate, with increased nicotine cravings and an average increase of 1.3 cigarettes per day.[17] Ophthalmologic adverse effects may include blurred vision and dry eyes, with less frequent reports of diplopia and mydriasis.[18]
There is some evidence of mild reductions in height with prolonged treatment in children.[19] This has been estimated at 1 centimetre (0.4 in) or less per year during the first three years with a total decrease of 3 centimetres (1.2 in) over 10 years.[20][21]
Hypersensitivity (including skin rash, urticaria, and fever) is sometimes reported when using transdermal methylphenidate. The Daytrana patch has a much higher rate of skin reactions than oral methylphenidate.[22]
Methylphenidate can worsen psychosis in people who are psychotic, and in very rare cases it has been associated with the emergence of new psychotic symptoms.[23] It should be used with extreme caution in people with bipolar disorder due to the potential induction of mania or hypomania.[24] There have been very rare reports of suicidal ideation, but some authors claim that evidence does not support a link.[19] Logorrhea is occasionally reported. Libido disorders, disorientation, and hallucinations are very rarely reported. Priapism is a very rare adverse event that can be potentially serious.[25]
USFDA-commissioned studies from 2011 indicate that in children, young adults, and adults there is no association between serious adverse cardiovascular events (sudden death, heart attack, and stroke) and the medical use of methylphenidate or other ADHD stimulants.[26]
Because some adverse effects may only emerge during chronic use of methylphenidate, a constant watch for adverse effects is recommended.[27]
A 2018 Cochrane review found that methylphenidate might be associated with serious side effects such as heart problems, psychosis, and death; the certainty of the evidence was stated as very low and the actual risk might be higher.[28]
Overdose
The symptoms of a moderate acute overdose on methylphenidate primarily arise from central nervous system overstimulation; these symptoms include: vomiting, nausea, agitation, tremors, hyperreflexia, muscle twitching, euphoria, confusion, hallucinations, delirium, hyperthermia, sweating, flushing, headache, tachycardia, heart palpitations, cardiac arrhythmias, hypertension, mydriasis, and dryness of mucous membranes.[29][30] A severe overdose may involve symptoms such as hyperpyrexia, sympathomimetic toxidrome, convulsions, paranoia, stereotypy (a repetitive movement disorder), rapid muscle breakdown, coma, and circulatory collapse.[29][30][31] A methylphenidate overdose is rarely fatal with appropriate care.[31] Following injection of methylphenidate tablets into an artery, severe toxic reactions involving abscess formation and necrosis have been reported.[32]
Treatment of a methylphenidate overdose typically involves the administration of benzodiazepines, with antipsychotics, α-adrenoceptor agonists and propofol serving as second-line therapies.[31]
Addiction and dependence[edit]
| ΔFosB accumulation from excessive drug use Top: this depicts the initial effects of high dose exposure to an addictive drug on gene expression in the nucleus accumbens for various Fos family proteins (i.e., c-Fos, FosB, ΔFosB, Fra1, and Fra2). Bottom: this illustrates the progressive increase in ΔFosB expression in the nucleus accumbens following repeated twice daily drug binges, where these phosphorylated (35–37 kilodalton) ΔFosB isoforms persist in the D1-type medium spiny neurons of the nucleus accumbens for up to 2 months.[33][34] |
Methylphenidate is a stimulant with an addiction liability and dependence liability similar to amphetamine. It has moderate liability among addictive drugs;[35][36] accordingly, addiction and psychological dependence are possible and likely when methylphenidate is used at high doses as a recreational drug.[36][37] When used above the medical dose range, stimulants are associated with the development of stimulant psychosis.[38] As with all addictive drugs, the overexpression of ΔFosB in D1-type medium spiny neurons in the nucleus accumbens is implicated in methylphenidate addiction.[37][39]
Methylphenidate has shown some benefits as a replacement therapy for individuals who are addicted to and dependent upon methamphetamine.[40] Methylphenidate and amphetamine have been investigated as a chemical replacement for the treatment of cocaine addiction[41][42][43][44] in the same way that methadone is used as a replacement drug for physical dependence upon heroin. Its effectiveness in treatment of cocaine or psychostimulant addiction, or psychological dependence has not been proven and further research is needed.[45]
Biomolecular mechanisms
Further information: Addiction § Biomolecular mechanisms
Methylphenidate has the potential to induce euphoria due to its pharmacodynamic effect (i.e., dopamine reuptake inhibition) in the brain’s reward system.[39] At therapeutic doses, ADHD stimulants do not sufficiently activate the reward system, or the reward pathway in particular, to the extent necessary to cause persistent increases in ΔFosB gene expression in the D1-type medium spiny neurons of the nucleus accumbens;[36][39][46] consequently, when taken as directed in doses that are commonly prescribed for the treatment of ADHD, methylphenidate use lacks the capacity to cause an addiction.[36][39][46] However, when methylphenidate is used at sufficiently high recreational doses through a bioavailable route of administration (e.g., insufflation or intravenous administration), particularly for use of the drug as a euphoriant, ΔFosB accumulates in the nucleus accumbens.[36][39] Hence, like any other addictive drug, regular recreational use of methylphenidate at high doses eventually gives rise to ΔFosB overexpression in D1-type neurons which subsequently triggers a series of gene transcription-mediated signaling cascades that induce an addiction.[39][46][47]
Overdose
This section is transcluded from Methylphenidate. (edit | history)
The symptoms of a moderate acute overdose on methylphenidate primarily arise from central nervous system overstimulation; these symptoms include: vomiting, nausea, agitation, tremors, hyperreflexia, muscle twitching, euphoria, confusion, hallucinations, delirium, hyperthermia, sweating, flushing, headache, tachycardia, heart palpitations, cardiac arrhythmias, hypertension, mydriasis, and dryness of mucous membranes.[29][30] A severe overdose may involve symptoms such as hyperpyrexia, sympathomimetic toxidrome, convulsions, paranoia, stereotypy (a repetitive movement disorder), rapid muscle breakdown, coma, and circulatory collapse.[29][30][31] A methylphenidate overdose is rarely fatal with appropriate care.[31] Following injection of methylphenidate tablets into an artery, severe toxic reactions involving abscess formation and necrosis have been reported.[32]
Treatment of a methylphenidate overdose typically involves the administration of benzodiazepines, with antipsychotics, α-adrenoceptor agonists and propofol serving as second-line therapies.[31]
Interactions
This section is transcluded from Methylphenidate. (edit | history)
Methylphenidate may inhibit the metabolism of vitamin K anticoagulants, certain anticonvulsants, and some antidepressants (tricyclic antidepressants, and selective serotonin reuptake inhibitors). Concomitant administration may require dose adjustments, possibly assisted by monitoring of plasma drug concentrations.[48] There are several case reports of methylphenidate inducing serotonin syndrome with concomitant administration of antidepressants.[49][50][51][52]
When methylphenidate is coingested with ethanol, a metabolite called ethylphenidate is formed via hepatic transesterification,[53][54] not unlike the hepatic formation of cocaethylene from cocaine and ethanol. The reduced potency of ethylphenidate and its minor formation means it does not contribute to the pharmacological profile at therapeutic doses and even in overdose cases ethylphenidate concentrations remain negligible.[55][54]
Coingestion of alcohol (ethanol) also increases the blood plasma levels of d-methylphenidate by up to 40%.[56]
Liver toxicity from methylphenidate is extremely rare, but limited evidence suggests that intake of β-adrenergic agonists with methylphenidate may increase the risk of liver toxicity.[57]
Mode of activity
Methylphenidate is a catecholamine reuptake inhibitor that indirectly increases catecholaminergic neurotransmission by inhibiting the dopamine transporter (DAT) and norepinephrine transporter (NET),[58] which are responsible for clearing catecholamines from the synapse, particularly in the striatum and meso-limbic system.[59] Moreover, it is thought to “increase the release of these monoamines into the extraneuronal space.”[2]
Although four stereoisomers of methylphenidate (MPH) are possible, only the threo diastereoisomers are used in modern practice. There is a high eudysmic ratio between the SS and RR enantiomers of MPH. Dexmethylphenidate (d-threo-methylphenidate) is a preparation of the RR enantiomer of methylphenidate.[60][61] In theory, D-TMP (d-threo-methylphenidate) can be anticipated to be twice the strength of the racemic product.[58][62]
| Compd[63] | DAT (Ki) | DA (IC50) | NET (Ki) | NE (IC50) |
|---|---|---|---|---|
| D-TMP | 161 | 23 | 206 | 39 |
| L-TMP | 2250 | 1600 | >10K | 980 |
| DL-TMP | 121 | 20 | 788 | 51 |
Pharmacology
Main article: Methylphenidate § Pharmacology
Dexmethylphenidate has a 4–6 hour duration of effect (a long-acting formulation, Focalin XR, which spans 12 hours is also available and has been shown to be as effective as DL (dextro-, levo-)-TMP (threo-methylphenidate) XR (extended release) (Concerta, Ritalin LA), with flexible dosing and good tolerability.[64][65]) It has also been demonstrated to reduce ADHD symptoms in both children[66] and adults.[67] d-MPH has a similar side-effect profile to MPH[14] and can be administered without regard to food intake.[68]
References
- ^ Jump up to:a b “Focalin- dexmethylphenidate hydrochloride tablet”. DailyMed. 24 June 2020. Retrieved 15 November 2020.
- ^ Jump up to:a b “Focalin XR- dexmethylphenidate hydrochloride capsule, extended release”. DailyMed. 27 June 2020. Retrieved 15 November 2020.
- ^ Jump up to:a b c d e f g h i “Dexmethylphenidate Hydrochloride Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 15 April 2019.
- ^ Mosby’s Drug Reference for Health Professions – E-Book. Elsevier Health Sciences. 2013. p. 455. ISBN 9780323187602.
- ^ “Dexmethylphenidate Use During Pregnancy”. Drugs.com. Retrieved 15 April 2019.
- ^ Jump up to:a b c d Moen MD, Keam SJ (December 2009). “Dexmethylphenidate extended release: a review of its use in the treatment of attention-deficit hyperactivity disorder”. CNS Drugs. 23(12): 1057–83. doi:10.2165/11201140-000000000-00000. PMID 19958043. S2CID 24975170.
- ^ “The Top 300 of 2021”. ClinCalc. Retrieved 18 February 2021.
- ^ “Dexmethylphenidate Hydrochloride – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ “Focalin XR”. Drugs.com. Retrieved 15 April 2019.
- ^ “DAYTRANA” (PDF). United States Food and Drug Administration. Noven Pharmaceuticals, Inc. October 2013. Archived (PDF) from the original on 14 July 2014. Retrieved 13 June 2014.
- ^ “Methylphenidate: Use During Pregnancy and Breastfeeding”. Drugs.com. Archived from the original on 2 January 2018.
- ^ Humphreys C, Garcia-Bournissen F, Ito S, Koren G (2007). “Exposure to attention deficit hyperactivity disorder medications during pregnancy”. Canadian Family Physician. 53 (7): 1153–5. PMC 1949295. PMID 17872810.
- ^ Ornoy, Asher (6 February 2018). “Pharmacological Treatment of Attention Deficit Hyperactivity Disorder During Pregnancy and Lactation”. Pharmaceutical Research. 35 (3): 46. doi:10.1007/s11095-017-2323-z. PMID 29411149. S2CID 3663423.
- ^ Jump up to:a b Keating GM, Figgitt DP (2002). “Dexmethylphenidate”. Drugs. 62 (13): 1899–904, discussion 1905–8. doi:10.2165/00003495-200262130-00009. PMID 12215063.
- ^ Nutt D, King LA, Saulsbury W, Blakemore C (March 2007). “Development of a rational scale to assess the harm of drugs of potential misuse”. Lancet. 369 (9566): 1047–53. doi:10.1016/S0140-6736(07)60464-4. PMID 17382831. S2CID 5903121.
- ^ “Ritalin LA® (methylphenidate hydrochloride) extended-release capsules” (PDF). Novartis. Archived from the original (PDF)on 20 July 2011.
- ^ Bron TI, Bijlenga D, Kasander MV, Spuijbroek AT, Beekman AT, Kooij JJ (2013). “Long-term relationship between methylphenidate and tobacco consumption and nicotine craving in adults with ADHD in a prospective cohort study”. European Neuropsychopharmacology. 23 (6): 542–554. doi:10.1016/j.euroneuro.2012.06.004. PMID 22809706. S2CID 23148548.
- ^ Jaanus SD (1992). “Ocular side effects of selected systemic drugs”. Optometry Clinics. 2 (4): 73–96. PMID 1363080.
- ^ Jump up to:a b Cortese S, Holtmann M, Banaschewski T, Buitelaar J, Coghill D, Danckaerts M, et al. (March 2013). “Practitioner review: current best practice in the management of adverse events during treatment with ADHD medications in children and adolescents”. Journal of Child Psychology and Psychiatry, and Allied Disciplines. 54 (3): 227–46. doi:10.1111/jcpp.12036. PMID 23294014.
- ^ Poulton A (August 2005). “Growth on stimulant medication; clarifying the confusion: a review”. Archives of Disease in Childhood. 90 (8): 801–6. doi:10.1136/adc.2004.056952. PMC 1720538. PMID 16040876.
- ^ Hinshaw SP, Arnold LE (January 2015). “ADHD, Multimodal Treatment, and Longitudinal Outcome: Evidence, Paradox, and Challenge”. Wiley Interdisciplinary Reviews. Cognitive Science. 6(1): 39–52. doi:10.1002/wcs.1324. PMC 4280855. PMID 25558298.
- ^ Findling RL, Dinh S (March 2014). “Transdermal therapy for attention-deficit hyperactivity disorder with the methylphenidate patch (MTS)”. CNS Drugs. 28 (3): 217–28. doi:10.1007/s40263-014-0141-y. PMC 3933749. PMID 24532028.
- ^ Kraemer M, Uekermann J, Wiltfang J, Kis B (July 2010). “Methylphenidate-induced psychosis in adult attention-deficit/hyperactivity disorder: report of 3 new cases and review of the literature”. Clinical Neuropharmacology. 33 (4): 204–6. doi:10.1097/WNF.0b013e3181e29174. PMID 20571380. S2CID 34956456.
- ^ Wingo AP, Ghaemi SN (2008). “Frequency of stimulant treatment and of stimulant-associated mania/hypomania in bipolar disorder patients”. Psychopharmacology Bulletin. 41 (4): 37–47. PMID 19015628.
- ^ “Methylphenidate ADHD Medications: Drug Safety Communication – Risk of Long-lasting Erections”. U.S. Food and Drug Administration. 17 December 2013. Archived from the original on 17 December 2013. Retrieved 17 December 2013.
- ^ “FDA Drug Safety Communication: Safety Review Update of Medications used to treat Attention-Deficit/Hyperactivity Disorder (ADHD) in children and young adults”. United States Food and Drug Administration. 20 December 2011. Archived from the original on 30 October 2013. Retrieved 4 November 2013.
• Cooper WO, Habel LA, Sox CM, Chan KA, Arbogast PG, Cheetham TC, Murray KT, Quinn VP, Stein CM, Callahan ST, Fireman BH, Fish FA, Kirshner HS, O’Duffy A, Connell FA, Ray WA (November 2011). “ADHD drugs and serious cardiovascular events in children and young adults”. N. Engl. J. Med. 365 (20): 1896–1904. doi:10.1056/NEJMoa1110212. PMC 4943074. PMID 22043968.
• “FDA Drug Safety Communication: Safety Review Update of Medications used to treat Attention-Deficit/Hyperactivity Disorder (ADHD) in adults”. United States Food and Drug Administration. 15 December 2011. Archived from the original on 30 October 2013. Retrieved 4 November 2013.
• Habel LA, Cooper WO, Sox CM, Chan KA, Fireman BH, Arbogast PG, Cheetham TC, Quinn VP, Dublin S, Boudreau DM, Andrade SE, Pawloski PA, Raebel MA, Smith DH, Achacoso N, Uratsu C, Go AS, Sidney S, Nguyen-Huynh MN, Ray WA, Selby JV (December 2011). “ADHD medications and risk of serious cardiovascular events in young and middle-aged adults”. JAMA. 306 (24): 2673–2683. doi:10.1001/jama.2011.1830. PMC 3350308. PMID 22161946. - ^ Gordon N (1999). “Attention deficit hyperactivity disorder: possible causes and treatment”. International Journal of Clinical Practice. 53(7): 524–8. PMID 10692738.
- ^ Storebø OJ, Pedersen N, Ramstad E, Kielsholm ML, Nielsen SS, Krogh HB, et al. (May 2018). “Methylphenidate for attention deficit hyperactivity disorder (ADHD) in children and adolescents – assessment of adverse events in non-randomised studies”. The Cochrane Database of Systematic Reviews. 5: CD012069. doi:10.1002/14651858.CD012069.pub2. PMC 6494554. PMID 29744873.
- ^ Jump up to:a b c d Noven Pharmaceuticals, Inc. (17 April 2015). “Daytrana Prescribing Information” (PDF). United States Food and Drug Administration. pp. 1–33. Archived (PDF) from the original on 23 June 2015. Retrieved 23 June 2015.
- ^ Jump up to:a b c d Heedes G, Ailakis J. “Methylphenidate hydrochloride (PIM 344)”. INCHEM. International Programme on Chemical Safety. Archived from the original on 23 June 2015. Retrieved 23 June2015.
- ^ Jump up to:a b c d e f Spiller HA, Hays HL, Aleguas A (June 2013). “Overdose of drugs for attention-deficit hyperactivity disorder: clinical presentation, mechanisms of toxicity, and management”. CNS Drugs. 27 (7): 531–543. doi:10.1007/s40263-013-0084-8. PMID 23757186. S2CID 40931380.
The management of amphetamine, dextroamphetamine, and methylphenidate overdose is largely supportive, with a focus on interruption of the sympathomimetic syndrome with judicious use of benzodiazepines. In cases where agitation, delirium, and movement disorders are unresponsive to benzodiazepines, second-line therapies include antipsychotics such as ziprasidone or haloperidol, central alpha-adrenoreceptor agonists such as dexmedetomidine, or propofol. … However, fatalities are rare with appropriate care
- ^ Jump up to:a b Bruggisser M, Bodmer M, Liechti ME (2011). “Severe toxicity due to injected but not oral or nasal abuse of methylphenidate tablets”. Swiss Med Wkly. 141: w13267. doi:10.4414/smw.2011.13267. PMID 21984207.
- ^ Nestler EJ, Barrot M, Self DW (September 2001). “DeltaFosB: a sustained molecular switch for addiction”. Proceedings of the National Academy of Sciences of the United States of America. 98(20): 11042–6. Bibcode:2001PNAS…9811042N. doi:10.1073/pnas.191352698. PMC 58680. PMID 11572966.
Although the ΔFosB signal is relatively long-lived, it is not permanent. ΔFosB degrades gradually and can no longer be detected in brain after 1–2 months of drug withdrawal … Indeed, ΔFosB is the longest-lived adaptation known to occur in adult brain, not only in response to drugs of abuse, but to any other perturbation (that does not involve lesions) as well.
- ^ Nestler EJ (December 2012). “Transcriptional mechanisms of drug addiction”. Clinical Psychopharmacology and Neuroscience. 10 (3): 136–43. doi:10.9758/cpn.2012.10.3.136. PMC 3569166. PMID 23430970.
The 35–37 kD ΔFosB isoforms accumulate with chronic drug exposure due to their extraordinarily long half-lives. … As a result of its stability, the ΔFosB protein persists in neurons for at least several weeks after cessation of drug exposure. … ΔFosB overexpression in nucleus accumbens induces NFκB
- ^ Morton WA, Stockton GG (2000). “Methylphenidate Abuse and Psychiatric Side Effects”. Prim Care Companion J Clin Psychiatry. 2 (5): 159–164. doi:10.4088/PCC.v02n0502. PMC 181133. PMID 15014637.
- ^ Jump up to:a b c d e Malenka RC, Nestler EJ, Hyman SE (2009). “Chapter 15: Reinforcement and Addictive Disorders”. In Sydor A, Brown RY (eds.). Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York: McGraw-Hill Medical. p. 368. ISBN 9780071481274.
Cocaine, [amphetamine], and methamphetamine are the major psychostimulants of abuse. The related drug methylphenidate is also abused, although it is far less potent. These drugs elicit similar initial subjective effects ; differences generally reflect the route of administration and other pharmacokinetic factors. Such agents also have important therapeutic uses; cocaine, for example, is used as a local anesthetic (Chapter 2), and amphetamines and methylphenidate are used in low doses to treat attention deficit hyperactivity disorder and in higher doses to treat narcolepsy (Chapter 12). Despite their clinical uses, these drugs are strongly reinforcing, and their long-term use at high doses is linked with potential addiction, especially when they are rapidly administered or when high-potency forms are given.
- ^ Jump up to:a b Steiner H, Van Waes V (January 2013). “Addiction-related gene regulation: risks of exposure to cognitive enhancers vs. other psychostimulants”. Prog. Neurobiol. 100: 60–80. doi:10.1016/j.pneurobio.2012.10.001. PMC 3525776. PMID 23085425.
- ^ Auger RR, Goodman SH, Silber MH, Krahn LE, Pankratz VS, Slocumb NL (2005). “Risks of high-dose stimulants in the treatment of disorders of excessive somnolence: a case-control study”. Sleep. 28 (6): 667–72. doi:10.1093/sleep/28.6.667. PMID 16477952.
- ^ Jump up to:a b c d e f Kim Y, Teylan MA, Baron M, Sands A, Nairn AC, Greengard P (2009). “Methylphenidate-induced dendritic spine formation and DeltaFosB expression in nucleus accumbens”. Proc. Natl. Acad. Sci. U.S.A. 106 (8): 2915–20. Bibcode:2009PNAS..106.2915K. doi:10.1073/pnas.0813179106. PMC 2650365. PMID 19202072.
Despite decades of clinical use of methylphenidate for ADHD, concerns have been raised that long-term treatment of children with this medication may result in subsequent drug abuse and addiction. However, meta analysis of available data suggests that treatment of ADHD with stimulant drugs may have a significant protective effect, reducing the risk for addictive substance use (36, 37). Studies with juvenile rats have also indicated that repeated exposure to methylphenidate does not necessarily lead to enhanced drug-seeking behavior in adulthood (38). However, the recent increase of methylphenidate use as a cognitive enhancer by the general public has again raised concerns because of its potential for abuse and addiction (3, 6–10). Thus, although oral administration of clinical doses of methylphenidate is not associated with euphoria or with abuse problems, nontherapeutic use of high doses or i.v. administration may lead to addiction (39, 40).
- ^ Elkashef A, Vocci F, Hanson G, White J, Wickes W, Tiihonen J (2008). “Pharmacotherapy of methamphetamine addiction: an update”. Substance Abuse. 29 (3): 31–49. doi:10.1080/08897070802218554. PMC 2597382. PMID 19042205.
- ^ Grabowski J, Roache JD, Schmitz JM, Rhoades H, Creson D, Korszun A (December 1997). “Replacement medication for cocaine dependence: methylphenidate”. Journal of Clinical Psychopharmacology. 17 (6): 485–8. doi:10.1097/00004714-199712000-00008. PMID 9408812.
- ^ Gorelick DA, Gardner EL, Xi ZX (2004). “Agents in development for the management of cocaine abuse”. Drugs. 64 (14): 1547–73. doi:10.2165/00003495-200464140-00004. PMID 15233592. S2CID 5421657. Archived from the original on 1 July 2019. Retrieved 1 July 2019.
- ^ Karila L, Gorelick D, Weinstein A, Noble F, Benyamina A, Coscas S, et al. (May 2008). “New treatments for cocaine dependence: a focused review”. The International Journal of Neuropsychopharmacology. 11 (3): 425–38. doi:10.1017/S1461145707008097. PMID 17927843.
- ^ “NIDA InfoFacts: Understanding Drug Abuse and Addiction”(PDF). 2008. Archived from the original (PDF) on 15 December 2010.
- ^ Shearer J (May 2008). “The principles of agonist pharmacotherapy for psychostimulant dependence”. Drug and Alcohol Review. 27 (3): 301–8. doi:10.1080/09595230801927372. PMID 18368612.
- ^ Jump up to:a b c Nestler EJ (December 2013). “Cellular basis of memory for addiction”. Dialogues in Clinical Neuroscience. 15 (4): 431–43. doi:10.31887/DCNS.2013.15.4/enestler. PMC 3898681. PMID 24459410.
Despite the importance of numerous psychosocial factors, at its core, drug addiction involves a biological process: the ability of repeated exposure to a drug of abuse to induce changes in a vulnerable brain that drive the compulsive seeking and taking of drugs, and loss of control over drug use, that define a state of addiction. … A large body of literature has demonstrated that such ΔFosB induction in D1-type NAc neurons increases an animal’s sensitivity to drug as well as natural rewards and promotes drug self-administration, presumably through a process of positive reinforcement … Another ΔFosB target is cFos: as ΔFosB accumulates with repeated drug exposure it represses c-Fos and contributes to the molecular switch whereby ΔFosB is selectively induced in the chronic drug-treated state.41. … Moreover, there is increasing evidence that, despite a range of genetic risks for addiction across the population, exposure to sufficiently high doses of a drug for long periods of time can transform someone who has relatively lower genetic loading into an addict.4
- ^ Ruffle JK (November 2014). “Molecular neurobiology of addiction: what’s all the (Δ)FosB about?”. The American Journal of Drug and Alcohol Abuse. 40 (6): 428–37. doi:10.3109/00952990.2014.933840. PMID 25083822. S2CID 19157711.
The strong correlation between chronic drug exposure and ΔFosB provides novel opportunities for targeted therapies in addiction (118), and suggests methods to analyze their efficacy (119). Over the past two decades, research has progressed from identifying ΔFosB induction to investigating its subsequent action (38). It is likely that ΔFosB research will now progress into a new era – the use of ΔFosB as a biomarker. …
Conclusions
ΔFosB is an essential transcription factor implicated in the molecular and behavioral pathways of addiction following repeated drug exposure. The formation of ΔFosB in multiple brain regions, and the molecular pathway leading to the formation of AP-1 complexes is well understood. The establishment of a functional purpose for ΔFosB has allowed further determination as to some of the key aspects of its molecular cascades, involving effectors such as GluR2 (87,88), Cdk5 (93) and NFkB (100). Moreover, many of these molecular changes identified are now directly linked to the structural, physiological and behavioral changes observed following chronic drug exposure (60,95,97,102). New frontiers of research investigating the molecular roles of ΔFosB have been opened by epigenetic studies, and recent advances have illustrated the role of ΔFosB acting on DNA and histones, truly as a molecular switch(34). As a consequence of our improved understanding of ΔFosB in addiction, it is possible to evaluate the addictive potential of current medications (119), as well as use it as a biomarker for assessing the efficacy of therapeutic interventions (121,122,124). Some of these proposed interventions have limitations (125) or are in their infancy (75). However, it is hoped that some of these preliminary findings may lead to innovative treatments, which are much needed in addiction.
• Biliński P, Wojtyła A, Kapka-Skrzypczak L, Chwedorowicz R, Cyranka M, Studziński T (2012). “Epigenetic regulation in drug addiction”. Annals of Agricultural and Environmental Medicine. 19(3): 491–6. PMID 23020045.For these reasons, ΔFosB is considered a primary and causative transcription factor in creating new neural connections in the reward centre, prefrontal cortex, and other regions of the limbic system. This is reflected in the increased, stable and long-lasting level of sensitivity to cocaine and other drugs, and tendency to relapse even after long periods of abstinence. These newly constructed networks function very efficiently via new pathways as soon as drugs of abuse are further taken … In this way, the induction of CDK5 gene expression occurs together with suppression of the G9A gene coding for dimethyltransferase acting on the histone H3. A feedback mechanism can be observed in the regulation of these 2 crucial factors that determine the adaptive epigenetic response to cocaine. This depends on ΔFosB inhibiting G9a gene expression, i.e. H3K9me2 synthesis which in turn inhibits transcription factors for ΔFosB. For this reason, the observed hyper-expression of G9a, which ensures high levels of the dimethylated form of histone H3, eliminates the neuronal structural and plasticity effects caused by cocaine by means of this feedback which blocks ΔFosB transcription
• Robison AJ, Nestler EJ (October 2011). “Transcriptional and epigenetic mechanisms of addiction”. Nature Reviews. Neuroscience. 12 (11): 623–37. doi:10.1038/nrn3111. PMC 3272277. PMID 21989194.ΔFosB has been linked directly to several addiction-related behaviors … Importantly, genetic or viral overexpression of ΔJunD, a dominant negative mutant of JunD which antagonizes ΔFosB- and other AP-1-mediated transcriptional activity, in the NAc or OFC blocks these key effects of drug exposure14,22–24. This indicates that ΔFosB is both necessary and sufficient for many of the changes wrought in the brain by chronic drug exposure. ΔFosB is also induced in D1-type NAc MSNs by chronic consumption of several natural rewards, including sucrose, high fat food, sex, wheel running, where it promotes that consumption14,26–30. This implicates ΔFosB in the regulation of natural rewards under normal conditions and perhaps during pathological addictive-like states.
- ^ “Concerta product monograph” (PDF). Janssen Pharmaceuticals. Archived (PDF) from the original on 28 January 2017. Retrieved 4 December 2016.
- ^ Ishii M, Tatsuzawa Y, Yoshino A, Nomura S (April 2008). “Serotonin syndrome induced by augmentation of SSRI with methylphenidate”. Psychiatry and Clinical Neurosciences. 62 (2): 246. doi:10.1111/j.1440-1819.2008.01767.x. PMID 18412855. S2CID 5659107.
- ^ Türkoğlu S (2015). “Serotonin syndrome with sertraline and methylphenidate in an adolescent”. Clinical Neuropharmacology. 38(2): 65–6. doi:10.1097/WNF.0000000000000075. PMID 25768857.
- ^ Park YM, Jung YK (May 2010). “Manic switch and serotonin syndrome induced by augmentation of paroxetine with methylphenidate in a patient with major depression”. Progress in Neuro-Psychopharmacology & Biological Psychiatry. 34 (4): 719–20. doi:10.1016/j.pnpbp.2010.03.016. PMID 20298736. S2CID 31984813.
- ^ Bodner RA, Lynch T, Lewis L, Kahn D (February 1995). “Serotonin syndrome”. Neurology. 45 (2): 219–23. doi:10.1212/wnl.45.2.219. PMID 7854515. S2CID 35190429.
- ^ Patrick KS, González MA, Straughn AB, Markowitz JS (2005). “New methylphenidate formulations for the treatment of attention-deficit/hyperactivity disorder”. Expert Opinion on Drug Delivery. 2(1): 121–43. doi:10.1517/17425247.2.1.121. PMID 16296740. S2CID 25026467.
- ^ Jump up to:a b Markowitz JS, DeVane CL, Boulton DW, Nahas Z, Risch SC, Diamond F, Patrick KS (2000). “Ethylphenidate formation in human subjects after the administration of a single dose of methylphenidate and ethanol”. Drug Metabolism and Disposition. 28 (6): 620–4. PMID 10820132.
- ^ Markowitz JS, Logan BK, Diamond F, Patrick KS (1999). “Detection of the novel metabolite ethylphenidate after methylphenidate overdose with alcohol coingestion”. Journal of Clinical Psychopharmacology. 19 (4): 362–6. doi:10.1097/00004714-199908000-00013. PMID 10440465.
- ^ Patrick KS, Straughn AB, Minhinnett RR, Yeatts SD, Herrin AE, DeVane CL, Malcolm R, Janis GC, Markowitz JS (March 2007). “Influence of ethanol and gender on methylphenidate pharmacokinetics and pharmacodynamics”. Clinical Pharmacology and Therapeutics. 81 (3): 346–53. doi:10.1038/sj.clpt.6100082. PMC 3188424. PMID 17339864.
- ^ Roberts SM, DeMott RP, James RC (1997). “Adrenergic modulation of hepatotoxicity”. Drug Metab. Rev. 29 (1–2): 329–53. doi:10.3109/03602539709037587. PMID 9187524.
- ^ Jump up to:a b Markowitz JS, Patrick KS (June 2008). “Differential pharmacokinetics and pharmacodynamics of methylphenidate enantiomers: does chirality matter?”. Journal of Clinical Psychopharmacology. 28 (3 Suppl 2): S54-61. doi:10.1097/JCP.0b013e3181733560. PMID 18480678.
- ^ Schweri MM, Skolnick P, Rafferty MF, Rice KC, Janowsky AJ, Paul SM (October 1985). “[3H]Threo-(+/-)-methylphenidate binding to 3,4-dihydroxyphenylethylamine uptake sites in corpus striatum: correlation with the stimulant properties of ritalinic acid esters”. Journal of Neurochemistry. 45 (4): 1062–70. doi:10.1111/j.1471-4159.1985.tb05524.x. PMID 4031878. S2CID 28720285.
- ^ Ding YS, Fowler JS, Volkow ND, Dewey SL, Wang GJ, Logan J, et al. (May 1997). “Chiral drugs: comparison of the pharmacokinetics of [11C]d-threo and L-threo-methylphenidate in the human and baboon brain”. Psychopharmacology. 131 (1): 71–8. doi:10.1007/s002130050267. PMID 9181638. S2CID 26046917.
- ^ Ding YS, Gatley SJ, Thanos PK, Shea C, Garza V, Xu Y, et al. (September 2004). “Brain kinetics of methylphenidate (Ritalin) enantiomers after oral administration”. Synapse. 53 (3): 168–75. CiteSeerX 10.1.1.514.7833. doi:10.1002/syn.20046. PMID 15236349. S2CID 11664668.
- ^ Davids E, Zhang K, Tarazi FI, Baldessarini RJ (February 2002). “Stereoselective effects of methylphenidate on motor hyperactivity in juvenile rats induced by neonatal 6-hydroxydopamine lesioning”. Psychopharmacology. 160 (1): 92–8. doi:10.1007/s00213-001-0962-5. PMID 11862378. S2CID 8037050.
- ^ Williard RL, Middaugh LD, Zhu HJ, Patrick KS (February 2007). “Methylphenidate and its ethanol transesterification metabolite ethylphenidate: brain disposition, monoamine transporters and motor activity”. Behavioural Pharmacology. 18 (1): 39–51. doi:10.1097/FBP.0b013e3280143226. PMID 17218796. S2CID 20232871.
- ^ McGough JJ, Pataki CS, Suddath R (July 2005). “Dexmethylphenidate extended-release capsules for attention deficit hyperactivity disorder”. Expert Review of Neurotherapeutics. 5 (4): 437–41. doi:10.1586/14737175.5.4.437. PMID 16026226. S2CID 6561452.
- ^ Silva R, Tilker HA, Cecil JT, Kowalik S, Khetani V, Faleck H, Patin J (2004). “Open-label study of dexmethylphenidate hydrochloride in children and adolescents with attention deficit hyperactivity disorder”. Journal of Child and Adolescent Psychopharmacology. 14(4): 555–63. doi:10.1089/cap.2004.14.555. PMID 15662147.
- ^ Arnold LE, Lindsay RL, Conners CK, Wigal SB, Levine AJ, Johnson DE, et al. (Winter 2004). “A double-blind, placebo-controlled withdrawal trial of dexmethylphenidate hydrochloride in children with attention deficit hyperactivity disorder”. Journal of Child and Adolescent Psychopharmacology. 14 (4): 542–54. doi:10.1089/cap.2004.14.542. PMID 15662146.
- ^ Spencer TJ, Adler LA, McGough JJ, Muniz R, Jiang H, Pestreich L (June 2007). “Efficacy and safety of dexmethylphenidate extended-release capsules in adults with attention-deficit/hyperactivity disorder”. Biological Psychiatry. 61 (12): 1380–7. doi:10.1016/j.biopsych.2006.07.032. PMID 17137560. S2CID 45976373.
- ^ Teo SK, Scheffler MR, Wu A, Stirling DI, Thomas SD, Stypinski D, Khetani VD (February 2004). “A single-dose, two-way crossover, bioequivalence study of dexmethylphenidate HCl with and without food in healthy subjects”. Journal of Clinical Pharmacology. 44 (2): 173–8. doi:10.1177/0091270003261899. PMID 14747426. S2CID 20694072.
External links
- “Dexmethylphenidate”. Drug Information Portal. U.S. National Library of Medicine.
- “Dexmethylphenidate hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
///////////DEXMETHYLPHENIDATE, FDA 2021, APPROVALS 2021
Cl.[H][C@@](C(=O)OC)(C1=CC=CC=C1)[C@@]1([H])CCCCN1
NEW DRUG APPROVALS
ONE TIME
$10.00
Serdexmethylphenidate

Serdexmethylphenidate
- Molecular FormulaC25H30ClN3O8
- Average mass535.974 Da
CAS 1996626-29-9 base
| 1996626-30-2 chloride |
L-Serine, N-[[1-[[[[(2R)-2-[(1R)-2-methoxy-2-oxo-1-phenylethyl]-1-piperidinyl]carbonyl]oxy]methyl]-3-pyridiniumyl]carbonyl]-, chloride (1:1)
N-[(1-{[({(2R)-2-[(1R)-2-Methoxy-2-oxo-1-phenylethyl]-1-piperidinyl}carbonyl)oxy]methyl}-3-pyridiniumyl)carbonyl]-L-serine chloride
Azstarys, FDA APPROVED, 3/2/2021, Products on NDA 212994, Type 1 – New Molecular Entity and Type 4 – New Combination
Serdexmethylphenidate Chloride (SDX), SDX or KP145


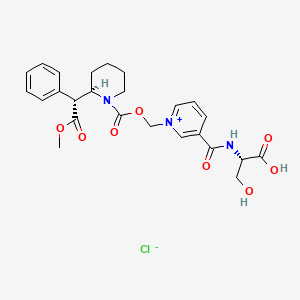
| Molecular Formula | C25H30ClN3O8 |
|---|---|
| Synonyms | UNII-FN54BT298YKP415 ClSerdexmethylphenidate chlorideFN54BT298YSerdexmethylphenidate chloride (USAN) |
| Molecular Weight | 536 g/mol |
CAS 1996626-30-2 chloride
(2S)-3-hydroxy-2-[[1-[[(2R)-2-[(1R)-2-methoxy-2-oxo-1-phenylethyl]piperidine-1-carbonyl]oxymethyl]pyridin-1-ium-3-carbonyl]amino]propanoic acid;chloride
Serdexmethylphenidate is a derivative of dexmethylphenidate created by pharmaceutical company KemPharm. The compound is under investigation for the treatment of ADHD in children, adolescents, and adults as of 2020.[2] The drug was approved for medical use by the FDA in March, 2021. Serdexmethylphenidate is a prodrug which has a delayed onset of action and a prolonged duration of effects compared to dexmethylphenidate, its parent compound.
SCHEME

WO2021173533
US20200237742
WO2019241019
US20190381017
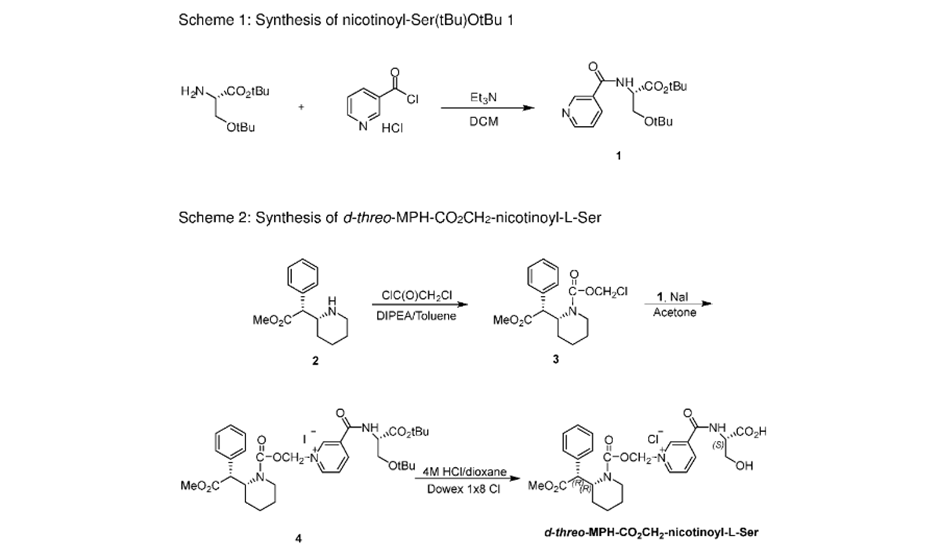
NEW DRUG APPROVALS
ONE TIME
$10.00
Formulations
Serdexmethylphenidate/dexmethylphenidate (Azstarys), a co-formulation of serdexmethylphenidate and dexmethylphenidate, was approved by the Food and Drug Administration (FDA) in March 2021, for the treatment of ADHD in those above six years of age. Co-formulation of serdexmethylphenidate with dexmethylphenidate allows for a more rapid onset of action while still retaining up to 13 hours of therapeutic efficacy.[3][4]
Due to serdexmethylphenidate’s delayed onset and prolonged duration of effects, several dosage forms containing serdexmethylphenidate have been investigated for use as long-acting psychostimulants in the treatment of ADHD. Under the developmental codename KP484, serdexmethylphenidate has been investigated as a “super-extended duration” psychostimulant, with therapeutic efficacy lasting up to 16 hours following oral administration. In 2011, MonoSol Rx entered into a partnership with KenPharm to develop oral films containing KP415.[5]
Abuse potential
The abuse potential of serdexmethylphenidate is theorized to be lower than other psychostimulants because serdexmethylphenidate is an inactive prodrug of dexmethylphenidate, and must undergo enzymatic metabolism prior to exerting any stimulant effects.[6] Common routes of administration used during the abuse of psychostimulants such as insufflation and intravenous injection have little impact on the pharmacokinetics and metabolism of serdexmethylphenidate and do not result in a faster onset of action.[7]
SYN

SYN
US 20200237742
Title(EN) Serdexmethylphenidate Conjugates, Compositions And Methods Of Use Thereof
Abstract
(EN)
The present technology is directed to one or more compositions comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof. The present technology also relates to one or more compositions and oral formulations comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof. The present technology also relates to one or more methods of using compositions comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof. The present technology additionally relates to one or more pharmaceutical kits containing a composition comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof.
| Synthetic Process for Making Serdexmethylphenidate |
| In one embodiment, the protected serdexmethylphenidate intermediate can be prepared as shown in Scheme 4. |
| In an alternate embodiment, the protected serdexmethylphenidate intermediate can be prepared according to Scheme 5. |
PATENT
US 20190381017
Title(EN) Compositions Comprising Serdexmethylphenidate Conjugates And Methods Of Use Thereof
Abstract
(EN)
The present technology is directed to one or more compositions comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof. The present technology also relates to one or more compositions and oral formulations comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof. The present technology also relates to one or more methods of using compositions comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof. The present technology additionally relates to one or more pharmaceutical kits containing a composition comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof.
PATENT
WO 2019241019
PAT
WO 2018107131
WO 2018107132
References
- ^ “Azstarys Prescribing Information” (PDF). United States Food and Drug Administration. Retrieved 18 March 2021.
- ^ “KemPharm’s KP415 and Serdexmethylphenidate (SDX) Prodrug to be Featured in Multiple Sessions at the AACAP 2020 Virtual Meeting”. http://www.globenewswire.com.
- ^ Mickle T. “Prodrugs for ADHD Treatments: Opportunities & Potential to Fill Unmet Medical Needs” (PDF). Retrieved 15 November 2020.
- ^ Eric Bastings, MD (2 March 2021). “NDA 212994 Approval” (PDF). United States Food and Drug Administration. Retrieved 6 March 2021.
- ^ Van Arnum P (1 March 2012). “Meeting Solubility Challenges”. Pharmaceutical Technology. 2012 (2): S6–S8. Retrieved 15 November 2020.
- ^ Mickle T. “Prodrugs for ADHD Treatments: Opportunities & Potential to Fill Unmet Medical Needs” (PDF). Retrieved 15 November 2020.
- ^ Braeckman R (1 October 2018). “Human Abuse Potential of Intravenous Serdexmethylphenidate (SDX), A Novel Prodrug of D-Methylphenidate, in Recreational Stimulant Abusers”. Journal of the American Academy of Child & Adolescent Psychiatry. 57 (10): 176. doi:10.1016/j.jaac.2018.09.141. Retrieved 15 November 2020.
External links
- “Serdexmethylphenidate”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Other names | KP484 |
| License data | US DailyMed: Serdexmethylphenidate |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only |
| Pharmacokinetic data | |
| Bioavailability | 3%[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1996626-30-2 |
| PubChem CID | 134823897 |
| ChemSpider | 81368035 |
| UNII | FN54BT298Y |
| KEGG | D11401 |
| ChEMBL | ChEMBL4298139 |
| Chemical and physical data | |
| Formula | C25H30ClN3O8 |
| Molar mass | 535.98 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
//////////Serdexmethylphenidate, Azstarys, FDA 2021 APPROVALS 2021, SDX, KP 145,



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
NEW DRUG APPROVALS
ONW TIME
$10.00
Sacituzumab govitecan-hziy


Sacituzumab govitecan-hziy
1601.8 g/mol
(2R)-2-amino-3-[1-[[4-[[1-[2-[2-[2-[2-[2-[2-[2-[2-[2-[[2-[2-[[(2S)-6-amino-1-[4-[[(19S)-10,19-diethyl-7-hydroxy-14,18-dioxo-17-oxa-3,13-diazapentacyclo[11.8.0.02,11.04,9.015,20]henicosa-1(21),2,4(9),5,7,10,15(20)-heptaen-19-yl]oxycarbonyloxymethyl]anilino]-1-oxohexan-2-yl]amino]-2-oxoethoxy]acetyl]amino]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethyl]triazol-4-yl]methylcarbamoyl]cyclohexyl]methyl]-2,5-dioxopyrrolidin-3-yl]sulfanylpropanoic acid
Trodelvy
- hRS 7SN38
- hRS7-SN38
- IMMU 132
- IMMU-132
CAS: 1491917-83-9
UNII-DA64T2C2IO component ULRUOUDIQPERIJ-PQURJYPBSA-N
UNII-SZB83O1W42 component ULRUOUDIQPERIJ-PQURJYPBSA-N
| Efficacy | Antineoplastic, Topoisomerase I inhibitor |
|---|---|
| Disease | Breast cancer (triple negative) |

Sacituzumab Govitecan is an antibody drug conjugate containing the humanized monoclonal antibody, hRS7, against tumor-associated calcium signal transducer 2 (TACSTD2 or TROP2) and linked to the active metabolite of irinotecan, 7-ethyl-10-hydroxycamptothecin (SN-38), with potential antineoplastic activity. The antibody moiety of sacituzumab govitecan selectively binds to TROP2. After internalization and proteolytic cleavage, SN-38 selectively stabilizes topoisomerase I-DNA covalent complexes, resulting in DNA breaks that inhibit DNA replication and trigger apoptosis. TROP2, also known as epithelial glycoprotein-1 (EGP-1), is a transmembrane calcium signal transducer that is overexpressed by a variety of human epithelial carcinomas; this antigen is involved in the regulation of cell-cell adhesion and its expression is associated with increased cancer growth, aggressiveness and metastasis.
NEW DRUG APPROVALS
one time
$10.00
CLIP
FDA Approves Trodelvy®, the First Treatment for Metastatic Triple-Negative Breast Cancer Shown to Improve Progression-Free Survival and Overall Survival
– Trodelvy Significantly Reduced the Risk of Death by 49% Compared with Single-Agent Chemotherapy in the Phase 3 ASCENT Study –
– Trodelvy is Under Regulatory Review in the EU and in the United Kingdom, Canada, Switzerland and Australia as Part of Project Orbis –April 07, 2021 07:53 PM Eastern Daylight Time
FOSTER CITY, Calif.–(BUSINESS WIRE)–Gilead Sciences, Inc. (Nasdaq: GILD) today announced that the U.S. Food and Drug Administration (FDA) has granted full approval to Trodelvy® (sacituzumab govitecan-hziy) for adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (TNBC) who have received two or more prior systemic therapies, at least one of them for metastatic disease. The approval is supported by data from the Phase 3 ASCENT study, in which Trodelvy demonstrated a statistically significant and clinically meaningful 57% reduction in the risk of disease worsening or death (progression-free survival (PFS)), extending median PFS to 4.8 months from 1.7 months with chemotherapy (HR: 0.43; 95% CI: 0.35-0.54; p<0.0001). Trodelvy also extended median overall survival (OS) to 11.8 months vs. 6.9 months (HR: 0.51; 95% CI: 0.41-0.62; p<0.0001), representing a 49% reduction in the risk of death.
Trodelvy is directed to the Trop-2 receptor, a protein frequently expressed in multiple types of epithelial tumors, including TNBC, where high expression is associated with poor survival and relapse. Prior to the FDA approval of Trodelvy, patients with previously treated metastatic TNBC had few treatment options in this high unmet-need setting. The FDA granted accelerated approval to Trodelvy in April 2020 based on objective response rate and duration of response results in a Phase 1/2 study. Today’s approval expands the previous Trodelvy indication to include treatment in adult patients with unresectable locally advanced or metastatic TNBC who have received two or more prior systemic therapies, at least one of them for metastatic disease.
“Women with triple-negative breast cancer have historically had very few effective treatment options and faced a poor prognosis,” said Aditya Bardia, MD, MPH, Director of Breast Cancer Research Program, Mass General Cancer Center and Assistant Professor of Medicine at Harvard Medical School, and global principal investigator of the ASCENT study. “Today’s FDA approval reflects the statistically significant survival benefit seen in the landmark ASCENT study and positions sacituzumab govitecan-hziy as a potential standard of care for pre-treated TNBC.”
“A metastatic TNBC diagnosis is frightening. As an aggressive and difficult-to-treat disease, it’s a significant advance to have an FDA-approved treatment option with a proven survival benefit for patients with metastatic disease that continues to progress,” said Ricki Fairley, Founder and CEO of Touch, the Black Breast Cancer Alliance. “For far too long, people with metastatic TNBC had very few treatment options. Today’s news continues the progress of bringing more options to treat this devastating disease.”
Among all patients evaluable for safety in the ASCENT study (n=482), Trodelvy had a safety profile consistent with the previously approved FDA label. The most frequent Grade ≥3 adverse reactions for Trodelvy compared to single-agent chemotherapy were neutropenia (52% vs. 34%), diarrhea (11% vs. 1%), leukopenia (11% vs. 6%) and anemia (9% vs. 6%). Adverse reactions leading to treatment discontinuation occurred in 5% of patients receiving Trodelvy.
“Today’s approval is the culmination of a multi-year development program and validates the clinical benefit of this important treatment in metastatic TNBC,” said Merdad Parsey, MD, PhD, Chief Medical Officer, Gilead Sciences. “Building upon this milestone, we are committed to advancing Trodelvy with worldwide regulatory authorities so that, pending their decision, Trodelvy may become available to many more people around the world who are facing this difficult-to-treat cancer.”
Regulatory submissions for Trodelvy in metastatic TNBC have been filed in the United Kingdom, Canada, Switzerland and Australia as part of Project Orbis, an initiative of the FDA Oncology Center of Excellence (OCE) that provides a framework for concurrent submission and review of oncology products among international partners, as well as in Singapore through our partner Everest Medicines.The European Medicines Agency has also validated a Marketing Authorization Application for Trodelvy in the European Union. All filings are based on data from the Phase 3 ASCENT study.
Trodelvy Boxed Warning
The Trodelvy U.S. Prescribing Information has a BOXED WARNING for severe or life-threatening neutropenia and severe diarrhea; see below for Important Safety Information.
About Trodelvy
Trodelvy (sacituzumab govitecan-hziy) is a first-in-class antibody and topoisomerase inhibitor conjugate directed to the Trop-2 receptor, a protein frequently expressed in multiple types of epithelial tumors, including metastatic triple-negative breast cancer (TNBC), where high expression is associated with poor survival and relapse.
Trodelvy is also being developed as an investigational treatment for metastatic urothelial cancer, hormone receptor-positive/human epidermal growth factor receptor 2-negative (HR+/HER 2-) metastatic breast cancer and metastatic non-small cell lung cancer. Additional evaluation across multiple solid tumors is also underway.
About Triple-Negative Breast Cancer (TNBC)
TNBC is an aggressive type of breast cancer, accounting for approximately 15% of all breast cancers. The disease is diagnosed more frequently in younger and premenopausal women and is more prevalent in African American and Hispanic women. TNBC cells do not have estrogen and progesterone receptors and have limited HER 2. Medicines targeting these receptors therefore are not typically effective in treating TNBC.
About the ASCENT Study
The Phase 3 ASCENT study, an open-label, active-controlled, randomized confirmatory trial, enrolled more than 500 patients with relapsed/refractory metastatic triple-negative breast cancer (TNBC) who had received two or more prior systemic therapies (including a taxane), at least one of them for metastatic disease. Patients were randomized to receive either Trodelvy or a chemotherapy chosen by the patients’ treating physicians. The primary efficacy outcome was progression-free survival (PFS) in patients without brain metastases at baseline, as measured by a blinded, independent, centralized review using RECIST v1.1 criteria. Additional efficacy measures included PFS for the full population (all patients with and without brain metastases) and overall survival (OS). More information about ASCENT is available at http://clinicaltrials.gov/show/NCT02574455.
Important Safety Information for Trodelvy
BOXED WARNING: NEUTROPENIA AND DIARRHEA
- Severe, life-threatening, or fatal neutropenia may occur. Withhold TRODELVY for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment. Consider G-CSF for secondary prophylaxis. Initiate anti-infective treatment in patient with febrile neutropenia without delay.
- Severe diarrhea may occur. Monitor patients with diarrhea and give fluid and electrolytes as needed. Administer atropine, if not contraindicated, for early diarrhea of any severity. At the onset of late diarrhea, evaluate for infectious causes and, if negative, promptly initiate loperamide. If severe diarrhea occurs, withhold TRODELVY until resolved to ≤ Grade 1 and reduce subsequent doses.
CONTRAINDICATIONS
- Severe hypersensitivity to TRODELVY
WARNINGS AND PRECAUTIONS
Neutropenia: Dose modifications may be required due to neutropenia. Neutropenia occurred in 62% of patients treated with TRODELVY, leading to permanent discontinuation in 0.5% of patients. Grade 3-4 neutropenia occurred in 47% of patients. Febrile neutropenia occurred in 6%.
Diarrhea: Diarrhea occurred in 64% of all patients treated with TRODELVY. Grade 3 diarrhea occurred in 12% of patients. Neutropenic colitis occurred in 0.5% of patients. Withhold TRODELVY for Grade 3-4 diarrhea and resume when resolved to ≤ Grade 1. At onset, evaluate for infectious causes and if negative, promptly initiate loperamide, 4 mg initially followed by 2 mg with every episode of diarrhea for a maximum of 16 mg daily. Discontinue loperamide 12 hours after diarrhea resolves. Additional supportive measures (e.g., fluid and electrolyte substitution) may also be employed as clinically indicated. Patients who exhibit an excessive cholinergic response to treatment can receive appropriate premedication (e.g., atropine) for subsequent treatments.
Hypersensitivity and Infusion-Related Reactions: TRODELVY can cause severe and life-threatening hypersensitivity and infusion-related reactions, including anaphylactic reactions. Hypersensitivity reactions within 24 hours of dosing occurred in 37% of patients. Grade 3-4 hypersensitivity occurred in 1% of patients. The incidence of hypersensitivity reactions leading to permanent discontinuation of TRODELVY was 0.4%. Pre-infusion medication is recommended. Observe patients closely for hypersensitivity and infusion-related reactions during each infusion and for at least 30 minutes after completion of each infusion. Medication to treat such reactions, as well as emergency equipment, should be available for immediate use.
Nausea and Vomiting: Nausea occurred in 67% of all patients treated with TRODELVY. Grade 3-4 nausea occurred in 5% of patients. Vomiting occurred in 40% of patients and Grade 3-4 vomiting occurred in 3% of these patients. Premedicate with a two or three drug combination regimen (e.g., dexamethasone with either a 5-HT3 receptor antagonist or an NK-1 receptor antagonist as well as other drugs as indicated) for prevention of chemotherapy-induced nausea and vomiting (CINV). Withhold TRODELVY doses for Grade 3 nausea or Grade 3-4 vomiting and resume with additional supportive measures when resolved to Grade ≤ 1. Additional antiemetics and other supportive measures may also be employed as clinically indicated. All patients should be given take-home medications with clear instructions for prevention and treatment of nausea and vomiting.
Increased Risk of Adverse Reactions in Patients with Reduced UGT1A1 Activity: Individuals who are homozygous for the uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1)*28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia and may be at increased risk for other adverse reactions with TRODELVY. The incidence of Grade 3-4 neutropenia in genotyped patients was 69% in patients homozygous for the UGT1A1*28, 48% in patients heterozygous for the UGT1A1*28 allele and 46% in patients homozygous for the wild-type allele. The incidence of Grade 3-4 anemia in genotyped patients was 24% in patients homozygous for the UGT1A1*28 allele, 8% in patients heterozygous for the UGT1A1*28 allele, and 10% in patients homozygous for the wild-type allele. Closely monitor patients with known reduced UGT1A1 activity for adverse reactions. Withhold or permanently discontinue TRODELVY based on severity of the observed adverse reactions in patients with evidence of acute early-onset or unusually severe adverse reactions, which may indicate reduced UGT1A1 function.
Embryo-Fetal Toxicity: Based on its mechanism of action, TRODELVY can cause teratogenicity and/or embryo-fetal lethality when administered to a pregnant woman. TRODELVY contains a genotoxic component, SN-38, and targets rapidly dividing cells. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TRODELVY and for 6 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRODELVY and for 3 months after the last dose.
ADVERSE REACTIONS
In the ASCENT study (IMMU-132-05), the most common adverse reactions (incidence ≥25%) were nausea, neutropenia, diarrhea, fatigue, alopecia, anemia, vomiting, constipation, rash, decreased appetite, and abdominal pain. The most frequent serious adverse reactions (SAR) (>1%) were neutropenia (7%), diarrhea (4%), and pneumonia (3%). SAR were reported in 27% of patients, and 5% discontinued therapy due to adverse reactions. The most common Grade 3-4 lab abnormalities (incidence ≥25%) in the ASCENT study were reduced hemoglobin, lymphocytes, leukocytes, and neutrophils.
DRUG INTERACTIONS
UGT1A1 Inhibitors: Concomitant administration of TRODELVY with inhibitors of UGT1A1 may increase the incidence of adverse reactions due to potential increase in systemic exposure to SN-38. Avoid administering UGT1A1 inhibitors with TRODELVY.
UGT1A1 Inducers: Exposure to SN-38 may be substantially reduced in patients concomitantly receiving UGT1A1 enzyme inducers. Avoid administering UGT1A1 inducers with TRODELVY
Please see full Prescribing Information, including BOXED WARNING.
About Gilead Sciences
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer. Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California.
Sacituzumab govitecan, sold under the brand name Trodelvy, is a Trop-2-directed antibody and topoisomerase inhibitor drug conjugate indicated for the treatment of metastatic triple-negative breast cancer (mTNBC) in adult patients that have received at least two prior therapies.[1][2]
The most common side effects are nausea, neutropenia, diarrhea, fatigue, anemia, vomiting, alopecia (hair loss), constipation, decreased appetite, rash and abdominal pain.[1][2] Sacituzumab govitecan has a boxed warning about the risk of severe neutropenia (abnormally low levels of white blood cells) and severe diarrhea.[1][2] Sacituzumab govitecan may cause harm to a developing fetus or newborn baby.[1] Women are advised not to breastfeed while on sacituzumab govitecan and 1 month after the last dose is administered.[3]
The U.S. Food and Drug Administration (FDA) considers it to be a first-in-class medication.[4]
Mechanism
Sacituzumab govitecan is a conjugate of the humanized anti-Trop-2 monoclonal antibody linked with SN-38, the active metabolite of irinotecan.[5] Each antibody having on average 7.6 molecules of SN-38 attached.[6] SN-38 is too toxic to administer directly to patients, but linkage to an antibody allows the drug to specifically target cells containing Trop-2.
Sacituzumab govitecan is a Trop-2-directed antibody and topoisomerase inhibitor drug conjugate, meaning that the drug targets the Trop-2 receptor that helps the cancer grow, divide and spread, and is linked to topoisomerase inhibitor, which is a chemical compound that is toxic to cancer cells.[1] Approximately two of every ten breast cancer diagnoses worldwide are triple-negative.[1] Triple-negative breast cancer is a type of breast cancer that tests negative for estrogen receptors, progesterone receptors and human epidermal growth factor receptor 2 (HER2) protein.[1] Therefore, triple-negative breast cancer does not respond to hormonal therapy medicines or medicines that target HER2.[1]
Development
Immunomedics announced in 2013, that it had received fast track designation from the US Food and Drug Administration (FDA) for the compound as a potential treatment for non-small cell lung cancer, small cell lung cancer, and metastatic triple-negative breast cancer. Orphan drug status was granted for small cell lung cancer and pancreatic cancer.[7][8] In February 2016, Immunomedics announced that sacituzumab govitecan had received an FDA breakthrough therapy designation (a classification designed to expedite the development and review of drugs that are intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition) for the treatment of patients with triple-negative breast cancer who have failed at least two other prior therapies for metastatic disease.[9][10]
History
Sacituzumab govitecan was added to the proposed INN list in 2015,[11] and to the recommended list in 2016.[12]
Sacituzumab govitecan-hziy was approved for use in the United States in April 2020.[1][13][14][2]
Sacituzumab govitecan-hziy was approved based on the results of IMMU-132-01, a multicenter, single-arm clinical trial (NCT01631552) of 108 subjects with metastatic triple-negative breast cancer who had received at least two prior treatments for metastatic disease.[1][14][2] Of the 108 patients involved within the study, 107 were female and 1 was male.[15] Subjects received sacituzumab govitecan-hziy at a dose of 10 milligrams per kilogram of body weight intravenously on days one and eight every 21 days.[14][15] Treatment with sacituzumab govitecan-hziy was continued until disease progression or unacceptable toxicity.[15] Tumor imaging was obtained every eight weeks.[14][2] The efficacy of sacituzumab govitecan-hziy was based on the overall response rate (ORR) – which reflects the percentage of subjects that had a certain amount of tumor shrinkage.[1][14] The ORR was 33.3% (95% confidence interval [CI], 24.6 to 43.1). [1][14][15] Additionally, with the 33.3% of study participants who achieved a response, 2.8% of patients experienced complete responses.[15] The median time to response in patients was 2.0 months (range, 1.6 to 13.5), the median duration of response was 7.7 months (95% confidence interval [CI], 4.9 to 10.8), the median progression free survival was 5.5 months, and the median overall survival was 13.0 months.[15] Of the subjects that achieved an objective response to sacituzumab govitecan-hziy, 55.6% maintained their response for six or more months and 16.7% maintained their response for twelve or more months.[1][14]
Sacituzumab govitecan-hziy was granted accelerated approval along with priority review, breakthrough therapy, and fast track designations.[1][14] The U.S. Food and Drug Administration (FDA) granted approval of Trodelvy to Immunomedics, Inc.[1]
References
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA Approves New Therapy for Triple Negative Breast Cancer That Has Spread, Not Responded to Other Treatments”. U.S. Food and Drug Administration (FDA). 22 April 2020. Retrieved 22 April 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e f “Drug Trial Snapshot: Trodelvy”. U.S. Food and Drug Administration (FDA). 22 April 2020. Retrieved 29 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ (PDF)https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761115s000lbl.pdf. Missing or empty
|title=(help) - ^ “New Drug Therapy Approvals 2020”. U.S. Food and Drug Administration (FDA). 31 December 2020. Retrieved 17 January2021. This article incorporates text from this source, which is in the public domain.
- ^ Sacituzumab Govitecan (IMMU-132), an Anti-Trop-2/SN-38 Antibody-Drug Conjugate: Characterization and Efficacy in Pancreatic, Gastric, and Other Cancers. 2015
- ^ “Novel Agents are Targeting Drivers of TNBC”. http://www.medpagetoday.com. 28 June 2016.
- ^ “Sacituzumab govitecan Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 22 April 2020.
- ^ “Sacituzumab govitecan Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 22 April 2020.
- ^ “New Therapy Shows Early Promise, Continues to Progress in Triple-Negative Breast Cancer”. Cure Today.
- ^ “U.S. Food and Drug Administration (FDA) Grants Breakthrough Therapy Designation to Immunomedics for Sacituzumab Govitecan for the Treatment of Patients With Triple-Negative Breast Cancer”(Press release). Immunomedics. 5 February 2016. Retrieved 25 April 2020 – via GlobeNewswire.
- ^ World Health Organization (2015). “International nonproprietary names for pharmaceutical substances (INN): proposed INN: list 113”. WHO Drug Information. 29 (2): 260–1. hdl:10665/331080.
- ^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 75”. WHO Drug Information. 30 (1): 151–3. hdl:10665/331046.
- ^ “Trodelvy: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 22 April 2020.
- ^ Jump up to:a b c d e f g h “FDA grants accelerated approval to sacituzumab govitecan-hziy for metastatic triple negative breast cancer”. U.S. Food and Drug Administration (FDA). 22 April 2020. Retrieved 23 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f “Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer”. The New England Journal of Medicine.
Further reading
- Bardia A, Mayer IA, Vahdat LT, et al. (February 2019). “Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer”. N. Engl. J. Med. 380 (8): 741–751. doi:10.1056/NEJMoa1814213. PMID 30786188.
- Weiss J, Glode A, Messersmith WA, et al. (August 2019). “Sacituzumab govitecan: breakthrough targeted therapy for triple-negative breast cancer”. Expert Rev Anticancer Ther. 19 (8): 673–679. doi:10.1080/14737140.2019.1654378. PMID 31398063. S2CID 199518147.
External links
- “Sacituzumab govitecan”. Drug Information Portal. U.S. National Library of Medicine.
- “Sacituzumab govitecan”. ADC Review.
- “Sacituzumab govitecan”. National Cancer Institute.
- Clinical trial number NCT01631552 for “Phase I/II Study of IMMU-132 in Patients With Epithelial Cancers” at ClinicalTrials.gov
- Sacituzumab govitecan at the US National Library of Medicine Medical Subject Headings (MeSH)
| Monoclonal antibody | |
|---|---|
| Type | ? |
| Source | Humanized (from mouse) |
| Target | Trop-2 |
| Clinical data | |
| Trade names | Trodelvy |
| Other names | IMMU-132, hRS7-SN-38, sacituzumab govitecan-hziy |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620034 |
| License data | US DailyMed: Sacituzumab_govitecan |
| Pregnancy category | Contraindicated |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only |
| Identifiers | |
| CAS Number | 1491917-83-9 |
| PubChem CID | 91668186 |
| DrugBank | DB12893 |
| ChemSpider | none |
| UNII | M9BYU8XDQ6 |
| KEGG | D10985 |
| Chemical and physical data | |
| Formula | C76H104N12O24S |
| Molar mass | 1601.79 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| show |
//////////sacituzumab govitecan-hziy, fda 2021, approvals 2021, Trodelvy , hRS 7SN38, hRS7-SN38, IMMU 132, IMMU-132, MONOCLONAL ANTIBODY, Sacituzumab govitecan, sacituzumab govitecan-hziy, CANCER, MONOCLONAL ANTIBODIES
#sacituzumab govitecan-hziy, #fda 2021, #approvals 2021, #Trodelvy , #hRS 7SN38, #hRS7-SN38, #IMMU 132, #IMMU-132, #MONOCLONAL ANTIBODY, #Sacituzumab govitecan, #sacituzumab govitecan-hziy, #CANCER, #MONOCLONAL ANTIBODIES
CCC1=C2CN3C(=CC4=C(C3=O)COC(=O)C4(CC)OC(=O)OCC5=CC=C(C=C5)NC(=O)C(CCCCN)NC(=O)COCC(=O)NCCOCCOCCOCCOCCOCCOCCOCCOCCN6C=C(N=N6)CNC(=O)C7CCC(CC7)CN8C(=O)CC(C8=O)SCC(C(=O)O)N)C2=NC9=C1C=C(C=C9)O
NEW DRUG APPROVALS
ONE TIME
$10.00
PF-07321332, Nirmatrelvir
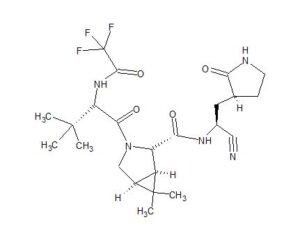

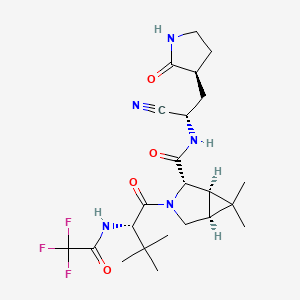
PF-07321332
Nirmatrelvir
UNII-7R9A5P7H32
7R9A5P7H32
PF07321332
CAS 2628280-40-8
C23H32F3N5O4, 499.5
(1R,2S,5S)-N-[(1S)-1-cyano-2-[(3S)-2-oxopyrrolidin-3-yl]ethyl]-3-[(2S)-3,3-dimethyl-2-[(2,2,2-trifluoroacetyl)amino]butanoyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxamide
https://clinicaltrials.gov/ct2/show/NCT04756531
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
SYN
https://pubmed.ncbi.nlm.nih.gov/34726479/
https://www.science.org/doi/10.1126/science.abl4784
Science. 2021 Dec 24;374(6575):1586-1593. doi: 10.1126/science.abl4784. Epub 2021 Nov 2.
An oral SARS-CoV-2 M pro inhibitor clinical candidate for the treatment of COVID-19
file:///C:/Users/Inspiron/Downloads/science.abl4784_sm.pdf
The worldwide outbreak of COVID-19 caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has become a global pandemic. Alongside vaccines, antiviral therapeutics are an important part of the healthcare response to countering the ongoing threat presented by COVID-19. Here, we report the discovery and characterization of PF-07321332, an orally bioavailable SARS-CoV-2 main protease inhibitor with in vitro pan-human coronavirus antiviral activity and excellent off-target selectivity and in vivo safety profiles. PF-07321332 has demonstrated oral activity in a mouse-adapted SARS-CoV-2 model and has achieved oral plasma concentrations exceeding the in vitro antiviral cell potency in a phase 1 clinical trial in healthy human participants.
Synthesis of PF-07321332 (Compound 6): Anhydrous, MTBE solvate form
(1R,2S,5S)-N-{(1S)-1-Cyano-2-[(3S)-2-oxopyrrolidin-3-yl]ethyl}-6,6-dimethyl-3-[3-methyl-N- (trifluoroacetyl)-L-valyl]-3-azabicyclo[3.1.0]hexane-2-carboxamide (1 eq tert-butyl methyl ether solvate) (6, MTBE solvate). This experiment was carried out in 2 parallel batches. Methyl N- (triethylammoniosulfonyl)carbamate, inner salt (Burgess reagent; 69.3 g, 276 mmol) was added to a solution of T18 (61 g, 111 mmol) in dichloromethane (550 ml). After the reaction mixture had been stirred at 25 °C for 1 h. The reaction mixture was quenched by a mixture of saturated aqueous sodium bicarbonate solution (200 ml) and saturated aqueous sodium chloride solution (100 ml). The separated organic phase was concentrated. The resulting residue was dissolved in 50% ethyl acetate/ tert-butyl methyl ether (600 ml), washed by a mixture of saturated aqueous sodium bicarbonate solution (200 ml) and saturated aqueous sodium chloride solution (100 ml) twice, saturated aqueous sodium chloride solution (200 ml), a mixture of HCl (1 M; 200 ml) and saturated aqueous sodium chloride solution (100 ml) twice. The organic layer was then dried over magnesium sulfate, filtered, and concentrated. The residue was treated with a mixture of ethyl acetate and tert-butyl methyl ether (1:10, 400 ml) and heated to 50 °C; after stirring for 1 hour at 50 °C, it was cooled to 25 °C and stirred overnight. The solid was collected via filtration, dissolved in dichloromethane (100 ml) and filtered through silica gel (200 g); the silica gel was then washed with ethyl acetate (1 Liter), 10% methanol in ethyl acetate (2 Liters). The combined eluates were concentrated. The 2 batches were combined, taken up in a mixture of ethyl acetate and tert-butyl methyl ether (5:95, 550 ml). This mixture was heated to 50 °C for 1 h, cooled to 25 °C, and stirred overnight. Filtration afforded 6, MTBE solvate, as a white solid. Yield: 104 g, 75 %. 1H NMR (600 MHz, DMSO-d6) δ 9.43 (d, J = 8.4 Hz, 1H), 9.03 (d, J = 8.6 Hz, 1H), 7.68 (s, 1H), 4.97 (ddd, J = 10.9, 8.6, 5.1 Hz, 1H), 4.41 (d, J = 8.4 Hz, 1H), 4.15 (s, 1H), 3.91 (dd, J = 10.4, 5.5 Hz, 1H), 3.69 (d, J = 10.4 Hz, 1H), 3.17 – 3.11 (m, 1H), 3.07 (s, 3H, MTBE), 3.04 (td, J = 9.4, 7.1 Hz, 1H), 2.40 (tdd, J = 10.4, 8.4, 4.4 Hz, 1H), 2.14 (ddd, J = 13.4, 10.9, 4.4 Hz,
1H), 2.11 – 2.03 (m, 1H), 1.76 – 1.65 (m, 2H), 1.57 (dd, J = 7.6, 5.5 Hz, 1H), 1.32 (d, J = 7.6 Hz, 1H), 1.10 (s, 9H, MTBE), 1.03 (s, 3H), 0.98 (s, 9H), 0.85 (s, 3H). Anal. Calcd for C23H32F3N5O4 .C5H12O: C, 57.23; H, 7.55; N, 11.92. Found: C, 57.08; H, 7.55; N, 11.85. mp = 118.8 oC
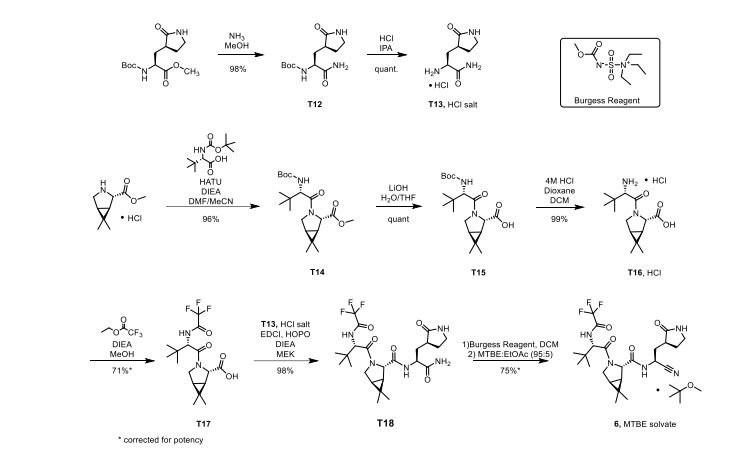
cry
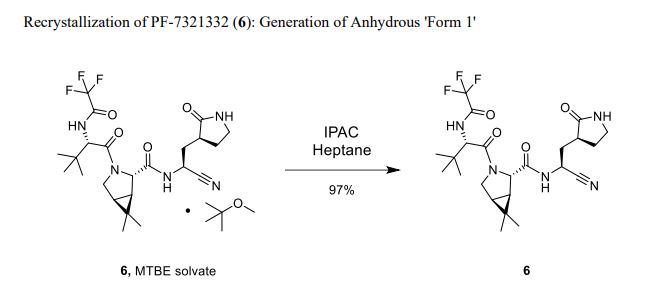
Compound 6 (anhydrous MTBE solvate, 200 g, 332.8 mmol, 83.11 mass%) was charged into a reactor with overhead half-moon stirring at 350 rpm. Heptane (1000 ml) was charged, followed by isopropyl acetate (1000 ml) and the stirring was continued at 20 oC overnight. Additional heptane (1000 ml) was charged over 120 minutes. The reaction vessel was then cooled to 10 oC over 30 min and stirred at that temp for 3 days. The solid was filtered, washing with a mixture of isopropyl acetate (80 ml) and heptane (320 ml). It was then dried under vacuum at 50 °C to provide 6, anhydrous ‘Form 1’, as a white crystalline solid. Yield: 160.93 g, 322 mmol, 97%. 1H NMR (600 MHz, DMSO-d6) δ 9.43 (d, J = 8.4 Hz, 1H), 9.03 (d, J = 8.6 Hz, 1H), 7.68 (s, 1H), 4.97 (ddd, J = 10.9, 8.6, 5.1 Hz, 1H), 4.41 (d, J = 8.4 Hz, 1H), 4.15 (s, 1H), 3.91 (dd, J = 10.4, 5.5 Hz, 1H), 3.69 (d, J = 10.4 Hz, 1H), 3.17 – 3.11 (m, 1H), 3.04 (td, J = 9.4, 7.1 Hz, 1H), 2.40 (tdd, J = 10.4, 8.4, 4.4 Hz, 1H), 2.14 (ddd, J = 13.4, 10.9, 4.4 Hz, 1H), 2.11 – 2.03 (m, 1H), 1.76 – 1.65 (m, 2H), 1.57 (dd, J = 7.6, 5.5 Hz, 1H), 1.32 (d, J = 7.6 Hz, 1H), 1.03 (s, 3H), 0.98 (s, 9H), 0.85 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 177.50, 170.72, 167.45, 156.95 (q, J = 37.0 Hz), 119.65, 115.84 (q, J = 286.9 Hz), 60.08, 58.19, 47.63, 37.77, 36.72, 34.60, 34.15, 30.28, 27.34, 26.86, 26.26, 25.72, 18.86, 12.34. 19F NMR (376 MHz, DMSO-d6) δ -72.94. HRMS (ESI-TOF) m/z calcd. for C23H33F3N5O4 [M + H]+ 500.2474, found 500.2472. Anal. Calcd for C23H32F3N5O4: C, 55.30; H, 6.46; N, 14.02. Found: C, 55.30; H, 6.49; N, 13.96. mp = 192.9 oC
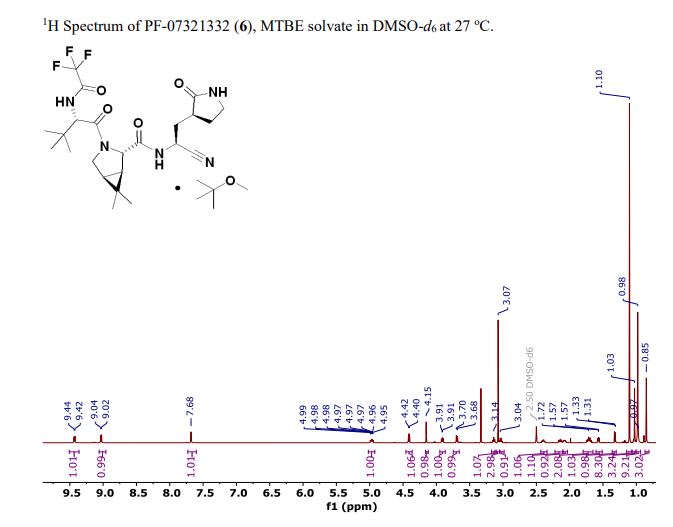



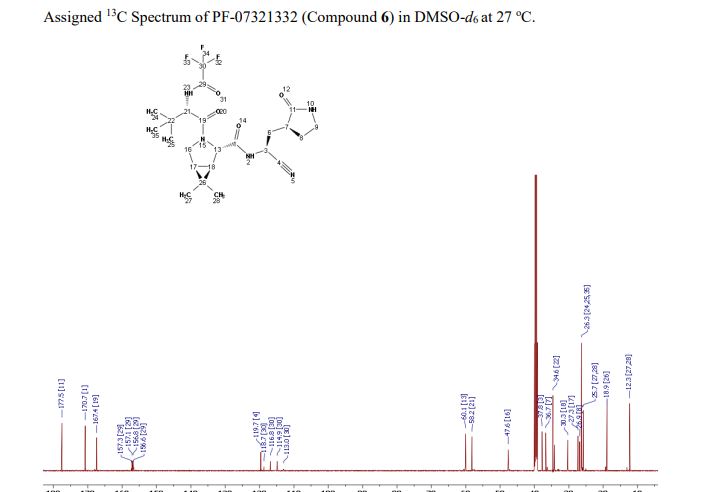




SYN
Bioorganic & medicinal chemistry letters (2021), 50, 128333.
https://www.sciencedirect.com/science/article/pii/S0960894X21005606
https://pubmed.ncbi.nlm.nih.gov/34418570/
pecific anti-coronaviral drugs complementing available vaccines are urgently needed to fight the COVID-19 pandemic. Given its high conservation across the betacoronavirus genus and dissimilarity to human proteases, the SARS-CoV-2 main protease (Mpro) is an attractive drug target. SARS-CoV-2 Mpro inhibitors have been developed at unprecedented speed, most of them being substrate-derived peptidomimetics with cysteine-modifying warheads. In this study, Mpro has proven resistant towards the identification of high-affinity short substrate-derived peptides and peptidomimetics without warheads. 20 cyclic and linear substrate analogues bearing natural and unnatural residues, which were predicted by computational modelling to bind with high affinity and designed to establish structure-activity relationships, displayed no inhibitory activity at concentrations as high as 100 μM. Only a long linear peptide covering residues P6 to P5‘ displayed moderate inhibition (Ki = 57 µM). Our detailed findings will inform current and future drug discovery campaigns targeting Mpro.
SYN
https://pubmed.ncbi.nlm.nih.gov/34498651/
Chemical communications (Cambridge, England) (2021), 57(72), 9096-9099We present a detailed computational analysis of the binding mode and reactivity of the novel oral inhibitor PF-07321332 developed against the SARS-CoV-2 3CL protease. Alchemical free energy calculations suggest that positions P3 and P4 could be susceptible to improvement in order to get a larger binding strength. QM/MM simulations unveil the reaction mechanism for covalent inhibition, showing that the nitrile warhead facilitates the recruitment of a water molecule for the proton transfer step.
PATENT
WO 2021234668
SYNOral inhibitors of the SARS-CoV-2 main protease for the treatment of COVID-19Owen, D., 261st Am Chem Soc (ACS) Natl Meet · 2021-04-05 Abst 243Synthesis of intermediate : Aminolysis of methyl N-Boc-3-[2-oxopyrrolidin-3(S)-yl]-L-alaninate in the presence of NH3 and subsequent N-deprotection using HCl leads to 2(S)-amino-3-[2-oxopyrrolidin-3(S)-yl]propenamide hydrochloride

Condensation of Boc-L-tert-leucine with methyl (1R,2S,5S)-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxylate using HATU gives the corresponding amide, which upon hydrolysis of methyl ester moiety in the presence of LiOH and subsequent N-deprotection by means of HCl affords intermediate,. N-Acylation of amine with ethyl trifluoroacetate yields diamide derivative, which upon condensation with 2(S)-amino-3-[2-oxopyrrolidin-3(S)-yl]propenamide hydrochloride using EDC and HOPO generates compound N-1 STEP. Burgess dehydration of amide derivative furnishes PF-7321332 .
In about November to December 2019 a novel coronavirus was identified as the cause of pneumonia cases in Wuhan (China). It spread, resulting in an epidemic throughout China, and thereafter in other countries throughout the world. In February 2020, the World Health Organization designated the disease COVTD-19, which stands for coronavirus disease 2019. The virus is also known as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (1)
COVID-19 is a betacoronavirus in the same subgenus as the severe acute respiratory syndrome (SARS) virus (as well as several bat coronaviruses), but in a different clade. The structure of the receptor- binding gene region is very similar to that of the SARS coronavirus, and the virus has been shown to use the same receptor, the angiotensin converting enzyme 2 (ACE2), for cell entry (2).
In the situation of rapidly increasing cases, inappropriate management of mild cases could increase the burden of healthcare system and medical costs. Viral clearance is a major standard in the assessment of recovery and discharge from medical care, but early results illustrated that the persistence of viral RNA is heterogeneous despite the rapid remission of symptoms and can last over three weeks even in very mild cases. In addition, long hospitalization stays may increase the risk for hospital-associated mental health problems and unexpected hospital-acquired infections. (9)
At the beginning, the outbreak identified an initial association with a seafood market that sold live animals in Wuhan, China. However, as the outbreak progressed, person-to-person spread became the main mode of transmission.
Person to person transmission is thought to occur mainly via respiratory droplets, resembling the spread of influenza. With droplet transmission, the virus is released in respiratory secretions when an infected person breathes, coughs, sneezes, or talks, and can infect another person if such secretions make direct contact with the mucous membranes. Infection can also occur if a person touches an infected surface and then touches his or her eyes, nose, or mouth. Droplets typically do not travel more than six feet (about two meters) and do not linger in the air. There is still controversy about this topic.
Whether SARS-CoV-2 can be transmitted through the airborne route (through particles smaller than droplets that remain in the air over time and distance) under natural conditions has been controversial.
Reflecting the current uncertainty regarding transmission mechanisms, recommendations on airborne precautions in the health care setting vary by location; airborne precautions are universally recommended when aerosol-generating procedures are performed.
It appears that SARS-CoV-2 can be transmitted prior to the development of symptoms and throughout the course of illness. However, most data informing this issue is from studies evaluating viral RNA detection from respiratory and other specimens, and detection of viral RNA does not necessarily indicate the presence of infectious virus.
A study suggested infectiousness started 2.3 days prior to symptom onset, peaked 0.7 days before symptom onset, and declined within seven days; however, most patients were isolated following symptom onset, which would reduce the risk of transmission later in illness regardless of infectiousness. These findings raise the possibility that patients might be more infectious in the earlier stage of infection, but additional data is needed to confirm this hypothesis (3).
How long a person remains infectious is also uncertain. The duration of viral shedding is variable; there appears to be a wide range, which may depend on severity of the illness. In one study of 21 patients with mild illness (no hypoxia), 90 percent had repeated negative viral RNA tests on nasopharyngeal swabs by 10 days after the onset of symptoms; tests were positive for longer in patients with more severe illness (4). In contrast, in another study of 56 patients with mild to moderate illness (none required intensive care), the median duration of viral RNA shedding from nasal or oropharyngeal specimens was 24 days, and the longest was 42 days (5). However, as mentioned above, detectable viral RNA does not always correlate with isolation of infectious virus, and there may be a threshold of viral RNA level below which infectivity is unlikely. In the study of nine patients with mild COVID-19 described above, infectious virus was not detected from respiratory specimens when the viral RNA level was <106 copies/mL (6).
Risk of transmission from an individual with SARS-CoV-2 infection varies by the type and duration of exposure, use of preventive measures, and likely individual factors (e.g., the amount of virus in respiratory secretions).
Antibodies against the virus are induced in those who have become infected. Preliminary evidence suggests that some of these antibodies are protective, but this remains to be definitively established. It is unknown whether all infected patients develop a protective immune response and how long any protective effect will last.
Diagnosis of COVID-19 is made by detection of SARS-CoV-2 RNA by reverse transcription polymerase chain reaction (RT-PCR). Various RT-PCR assays are used around the world; different assays amplify and detect different regions of the SARSCoV-2 genome. Common gene targets include nucleocapsid (N), envelope (E), spike (S), and RNA-dependent RNA polymerase (RdRp), as well as regions in the first open reading frame (7).
Serologic tests detect antibodies to SARS-CoV-2 in the blood, and those that have been adequately validated can help identify patients who have had COVID-19. However, sensitivity and specificity are still not well defined. Detectable antibodies generally take several days to weeks to develop, for example, up to 12 days for IgM and 14 days for IgG(Si-
………………………………..
PF-07321332 (or nirmatrelvir) is an antiviral drug developed by Pfizer which acts as an orally active 3CLprotease inhibitor. The combination of PF-07321332 with ritonavir has been in phase III trials for the treatment of COVID-19 since September 2021[2][3][4] and is expected to be sold under the brand name Paxlovid.[5] After promising results preventing hospitalization and death if given within the first 3 days of symptoms, Pfizer submitted an application to the U.S. Food and Drug Administration (FDA) for emergency authorization for PF-07321332 in combination with ritonavir in November 2021.[6]
PF-07321332 is an azabicyclohexane that is (1R,5S)-3-azabicyclo[3.1.0]hexane substituted by {(1S)-1-cyano-2-[(3S)-2-oxopyrrolidin-3-yl]ethyl}aminoacyl, 3-methyl-N-(trifluoroacetyl)-L-valinamide, methyl and methyl groups at positions 2S, 3, 6 and 6, respectively. It is an inhibitor of SARS-CoV-2 main protease which is currently under clinical development for the treatment of COVID-19. It has a role as an EC 3.4.22.69 (SARS coronavirus main proteinase) inhibitor and an anticoronaviral agent. It is a nitrile, a member of pyrrolidin-2-ones, a secondary carboxamide, a pyrrolidinecarboxamide, a tertiary carboxamide, an organofluorine compound and an azabicyclohexane.
Development
Pharmaceutical
Coronaviral proteases cleave multiple sites in the viral polyprotein, usually after glutamine residues. Early work on related human rhinoviruses showed that the flexible glutamine side chain could be replaced by a rigid pyrrolidone.[7][8] These drugs had been further developed prior to the SARS CoV2 pandemic for other diseases including SARS.[9] The utility of targeting the 3CL protease in a real world setting was first demonstrated in 2018 when GC376 (a prodrug of GC373) was used to treat the previously 100% lethal cat coronavirus disease, feline infectious peritonitis, caused by Feline coronavirus.[10]
The Pfizer drug is an analog of GC373, where the aldehyde covalent cysteine acceptor has been replaced by a nitrile.[11][12]
PF-07321332 was developed by modification of an earlier clinical candidate lufotrelvir,[13][14] which is also a covalent inhibitor but its warhead is a phosphate prodrug of a hydroxyketone. However, lufotrelvir needs to be administered intravenously limiting its use to a hospital setting. Stepwise modification of the tripeptide protein mimetic led to PF-0732133, which is suitable for oral administration.[1] Key changes include a reduction in the number of hydrogen bond donors, and the number of rotatable bonds by introducing the rigid bicyclic non-canonical amino acid, which mimics the leucine residue found in earlier inhibitors. This residue had previously been used in the synthesis of boceprevir.[15]
Clinical
In April 2021, Pfizer began phase I trials.[16] In September 2021, Pfizer began a phase II/III trial.[17] In November 2021, Pfizer announced 89% reduction in hospitalizations of high risk patients studied when given within three days after symptom onset.[5]
On December 14, Pfizer announced that Paxlovid, when given within three days of symptom onset, reduced risk of hospitalization or death by 89% compared to placebo in 2,246 high risk patients studied.[18]
Chemistry and pharmacology
Full details of the synthesis of PF-07321332 were first published by scientists from Pfizer.[1]
In the penultimate step, a synthetic homochiral amino acid is coupled with a homochiral amino amide using the water-soluble carbodiimide EDCI as coupling agent. The resulting intermediate is then treated with Burgess reagent, which dehydrates the amide group to the nitrile of the product.
PF-07321332 is a covalent inhibitor, binding directly to the catalytic cysteine (Cys145) residue of the cysteine protease enzyme.[19]
In the drug combination, ritonavir serves to slow down metabolism of PF-07321332 by cytochrome enzymes to maintain higher circulating concentrations of the main drug.[20]
Public health system reactions to development
Despite not being approved yet in any country, the UK placed an order for 250,000 courses after Pfizer´s press release in October 2021,[21][22] and Australia pre-ordered 500,000 courses of the drug.[23]
As of November 2021, the US government was expected to sign a contract to buy around 10 million courses of the combination treatment.[24][25]
Legal status
In November 2021, Pfizer signed a license agreement with the United Nations–backed Medicines Patent Pool to allow PF-07321332 to be manufactured and sold in 95 countries.[26] Pfizer stated that the agreement will allow local medicine manufacturers to produce the pill “with the goal of facilitating greater access to the global population”. However, the deal excludes several countries with major COVID-19 outbreaks including Brazil, China, Russia, Argentina, and Thailand.[27][28]
On 16 November 2021, Pfizer submitted an application to the U.S. Food and Drug Administration (FDA) for emergency authorization for PF-07321332 in combination with ritonavir.[29][30][31]
Misleading comparison with ivermectin
Conspiracy theorists on the internet have claimed that Paxlovid is merely a “repackaged” version of the antiparasitic drug ivermectin, which has been erroneously promoted as a COVID-19 “miracle cure”. Their claims, sometimes using the nickname “Pfizermectin”,[32] are based on a narrative that Pfizer is suppressing the true benefits of ivermectin and rely on superficial correspondences between the drugs and a misunderstanding of their respective pharmacokinetics.[33] Paxlovid is not structurally related or similar to ivermectin, and while both are 3C-like protease inhibitors, Paxlovid is much more potent with an IC50 around 10,000 times lower, allowing for effective oral dosing within the therapeutic margin.[34]
References
- ^ Jump up to:a b c Owen DR, Allerton CM, Anderson AS, Aschenbrenner L, Avery M, Berritt S, et al. (November 2021). “An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19″. Science: eabl4784. doi:10.1126/science.abl4784. PMID 34726479. S2CID 240422219.
- ^ Vandyck K, Deval J (August 2021). “Considerations for the discovery and development of 3-chymotrypsin-like cysteine protease inhibitors targeting SARS-CoV-2 infection”. Current Opinion in Virology. 49: 36–40. doi:10.1016/j.coviro.2021.04.006. PMC 8075814. PMID 34029993.
- ^ Şimşek-Yavuz S, Komsuoğlu Çelikyurt FI (August 2021). “Antiviral treatment of COVID-19: An update”. Turkish Journal of Medical Sciences. doi:10.3906/sag-2106-250. PMID 34391321. S2CID 237054672.
- ^ Ahmad B, Batool M, Ain QU, Kim MS, Choi S (August 2021). “Exploring the Binding Mechanism of PF-07321332 SARS-CoV-2 Protease Inhibitor through Molecular Dynamics and Binding Free Energy Simulations”. International Journal of Molecular Sciences. 22 (17): 9124. doi:10.3390/ijms22179124. PMC 8430524. PMID 34502033.
- ^ Jump up to:a b “Pfizer’s Novel COVID-19 Oral Antiviral Treatment Candidate Reduced Risk Of Hospitalization Or Death By 89% In Interim Analysis Of Phase 2/3 EPIC-HR Study”. Pfizer Inc. 5 November 2021.
- ^ Mahase E (November 2021). “Covid-19: Pfizer’s paxlovid is 89% effective in patients at risk of serious illness, company reports”. BMJ. 375: n2713. doi:10.1136/bmj.n2713. PMID 34750163. S2CID 243834203.
- ^ Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R (June 2003). “Coronavirus Main Proteinase (3CLpro) Structure: Basis for Design of Anti-SARS Drugs”. Science. 300 (5626): 1763–1767. Bibcode:2003Sci…300.1763A. doi:10.1126/science.1085658. PMID 12746549. S2CID 13031405.
- ^ Dragovich PS, Prins TJ, Zhou R, Webber SE, Marakovits JT, Fuhrman SA, et al. (April 1999). “Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 4. Incorporation of P1 lactam moieties as L-glutamine replacements”. Journal of Medicinal Chemistry. 42 (7): 1213–1224. doi:10.1021/jm9805384. PMID 10197965.
- ^ Pillaiyar T, Manickam M, Namasivayam V, Hayashi Y, Jung SH (July 2016). “An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy”. Journal of Medicinal Chemistry. 59 (14): 6595–6628. doi:10.1021/acs.jmedchem.5b01461. PMC 7075650. PMID 26878082.
- ^ Pedersen NC, Kim Y, Liu H, Galasiti Kankanamalage AC, Eckstrand C, Groutas WC, et al. (April 2018). “Efficacy of a 3C-like protease inhibitor in treating various forms of acquired feline infectious peritonitis”. Journal of Feline Medicine and Surgery. 20 (4): 378–392. doi:10.1177/1098612X17729626. PMC 5871025. PMID 28901812.
- ^ Halford B (7 April 2021). “Pfizer unveils its oral SARS-CoV-2 inhibitor”. Chemical & Engineering News. 99 (13): 7. doi:10.47287/cen-09913-scicon3. S2CID 234887434.
- ^ Vuong W, Khan MB, Fischer C, Arutyunova E, Lamer T, Shields J, et al. (August 2020). “Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication”. Nature Communications. 11 (1): 4282. doi:10.1038/s41467-020-18096-2. PMC 7453019. PMID 32855413.
- ^ Clinical trial number NCT04535167 for “First-In-Human Study To Evaluate Safety, Tolerability, And Pharmacokinetics Following Single Ascending And Multiple Ascending Doses of PF-07304814 In Hospitalized Participants With COVID-19 ” at ClinicalTrials.gov
- ^ Boras B, Jones RM, Anson BJ, Arenson D, Aschenbrenner L, Bakowski MA, et al. (February 2021). “Discovery of a Novel Inhibitor of Coronavirus 3CL Protease for the Potential Treatment of COVID-19”. bioRxiv: 2020.09.12.293498. doi:10.1101/2020.09.12.293498. PMC 7491518. PMID 32935104.
- ^ Njoroge FG, Chen KX, Shih NY, Piwinski JJ (January 2008). “Challenges in modern drug discovery: a case study of boceprevir, an HCV protease inhibitor for the treatment of hepatitis C virus infection”. Accounts of Chemical Research. 41 (1): 50–59. doi:10.1021/ar700109k. PMID 18193821. S2CID 2629035.
- ^ Nuki P (26 April 2021). “Pfizer is testing a pill that, if successful, could become first-ever home cure for COVID-19”. National Post. Archived from the original on 27 April 2021.
- ^ “Pfizer begins dosing in Phase II/III trial of antiviral drug for Covid-19”. Clinical Trials Arena. 2 September 2021.
- ^ Press release (14 December 2021). “Pfizer Announces Additional Phase 2/3 Study Results Confirming Robust Efficacy of Novel COVID-19 Oral Antiviral Treatment Candidate in Reducing Risk of Hospitalization or Death”.
- ^ Pavan M, Bolcato G, Bassani D, Sturlese M, Moro S (December 2021). “Supervised Molecular Dynamics (SuMD) Insights into the mechanism of action of SARS-CoV-2 main protease inhibitor PF-07321332”. J Enzyme Inhib Med Chem. 36 (1): 1646–1650. doi:10.1080/14756366.2021.1954919. PMC 8300928. PMID 34289752.
- ^ Woodley M (19 October 2021). “What is Australia’s potential new COVID treatment?”. The Royal Australian College of General Practitioners (RACGP). Retrieved 6 November 2021.
- ^ “Pfizer Covid pill ‘can cut hospitalisations and deaths by nearly 90%'”. The Guardian. 5 November 2021. Retrieved 17 November 2021.
- ^ Mahase E (October 2021). “Covid-19: UK stockpiles two unapproved antiviral drugs for treatment at home”. BMJ. 375: n2602. doi:10.1136/bmj.n2602. PMID 34697079. S2CID 239770104.
- ^ “What are the two new COVID-19 treatments Australia has gained access to?”. ABC News (Australia). 17 October 2021. Retrieved 5 November 2021.
- ^ “U.S. to Buy Enough of Pfizer’s Covid Antiviral Pills for 10 Million People”. The New York Times. 17 November 2021. Retrieved 17 November 2021.
- ^ Pager T, McGinley L, Johnson CY, Taylor A, Parker C. “Biden administration to buy Pfizer antiviral pills for 10 million people, hoping to transform pandemic”. The Washington Post. Retrieved 16 November 2021.
- ^ “Pfizer and The Medicines Patent Pool (MPP) Sign Licensing Agreement for COVID-19 Oral Antiviral Treatment Candidate to Expand Access in Low- and Middle-Income Countries” (Press release). Pfizer. 16 November 2021. Retrieved 17 November 2021 – via Business Wire.
- ^ “Covid-19: Pfizer to allow developing nations to make its treatment pill”. BBC News. 16 November 2021. Archived from the original on 16 November 2021. Retrieved 17 November 2021.
- ^ “Pfizer Will Allow Its Covid Pill to Be Made and Sold Cheaply in Poor Countries”. The New York Times. 16 November 2021. Retrieved 17 November 2021.
- ^ “Pfizer Seeks Emergency Use Authorization for Novel COVID-19 Oral Antiviral Candidate”. Business Wire (Press release). 16 November 2021. Retrieved 17 November 2021.
- ^ Kimball S (16 November 2021). “Pfizer submits FDA application for emergency approval of Covid treatment pill”. CNBC. Retrieved 17 November 2021.
- ^ Robbins R (5 November 2021). “Pfizer Says Its Antiviral Pill Is Highly Effective in Treating Covid”. The New York Times. ISSN 0362-4331. Archived from the original on 8 November 2021. Retrieved 9 November 2021.
- ^ Bloom J (2 December 2021). “How Does Pfizer’s Pavloxid Compare With Ivermectin?”. American Council on Science and Health. Retrieved 12 December 2021.
- ^ Gorski D (15 November 2021). “Pfizer’s new COVID-19 protease inhibitor drug is not just ‘repackaged ivermectin'”. Science-Based Medicine.
- ^ von Csefalvay C (27 November 2021). “Why Paxlovid is not Pfizermectin”. Bits and Bugs. Chris von Csefalvay. Retrieved 28 November 2021.
External links
- “PF-07321332”. Drug Information Portal. U.S. National Library of Medicine.
- “Early Data Suggest Pfizer Pill May Prevent Severe COVID-19”. National Institutes of Health. 16 November 2021.
Pfizer to make COVID-19 pill available in low- and middle-income nations
If authorized by global health authorities, the drug promises to reduce deaths and hospitalizations linked to
the novel coronavirus.
By Brian Buntz | November 16, 2021FacebookTwitterLinkedInShare
In late October, Merck (NYSE:MRK) and its partner Ridgeback Biotherapeutics agreed to make the COVID-19 antiviral molnupiravir available in the developing world.
Now, Pfizer (NYSE:PFE) is taking a similar approach for its investigational antiviral cocktail Paxlovid, which contains PF-07321332 and ritonavir.
Pfizer, like Merck, struck an agreement with the Medicines Patent Pool (MPP) related to Paxlovid.
MPP’s mission is to expand low- and middle-income countries’ access to vital medicines. The United Nations supports the organization.
Pfizer announced earlier this month that Paxlovid was 89% effective in reducing the risk of hospitalization or death in an interim analysis of the Phase 2/3 EPIC-HR trial.
The collaboration with MPP will enable generic drug makers internationally with sub-licenses to produce Paxlovid for use in 95 countries, which comprise more than half of the world’s population.
“This license is so important because, if authorized or approved, this oral drug is particularly well-suited for low- and middle-income countries and could play a critical role in saving lives, contributing to global efforts to fight the current pandemic,” said Charles Gore, executive director of MPP, in a press release. “PF-07321332 is to be taken together with ritonavir, an HIV medicine we know well, as we have had a license on it for many years, and we will be working with generic companies to ensure there is enough supply for both COVID-19 and HIV.”
At present, MPP has signed agreements with ten patient holders for 13 HIV antiretrovirals and several other drugs.
Filed Under: clinical trials, Drug Discovery, Infectious Disease
Tagged With: Medicines Patent Pool, Merck, PF-07321332, Pfizer, Ridgeback Biotherapeutics, ritonavir
PFIZER INITIATES PHASE 1 STUDY OF NOVEL ORAL ANTIVIRAL THERAPEUTIC AGENT AGAINST SARS-COV-2
Tuesday, March 23, 2021 – 11:00am
- In-vitro studies conducted to date show that the clinical candidate PF-07321332 is a potent protease inhibitor with potent anti-viral activity against SARS-CoV-2
- This is the first orally administered coronavirus-specific investigational protease inhibitor to be evaluated in clinical studies, and follows Pfizer’s intravenously administered investigational protease inhibitor, which is currently being evaluated in a Phase 1b multi-dose study in hospitalized clinical trial participants with COVID-19
NEW YORK–(BUSINESS WIRE)– Pfizer Inc. (NYSE: PFE) announced today that it is progressing to multiple ascending doses after completing the dosing of single ascending doses in a Phase 1 study in healthy adults to evaluate the safety and tolerability of an investigational, novel oral antiviral therapeutic for SARS-CoV-2, the virus that causes COVID-19. This Phase 1 trial is being conducted in the United States. The oral antiviral clinical candidate PF-07321332, a SARS-CoV2-3CL protease inhibitor, has demonstrated potent in vitro anti-viral activity against SARS-CoV-2, as well as activity against other coronaviruses, suggesting potential for use in the treatment of COVID-19 as well as potential use to address future coronavirus threats.
“Tackling the COVID-19 pandemic requires both prevention via vaccine and targeted treatment for those who contract the virus. Given the way that SARS-CoV-2 is mutating and the continued global impact of COVID-19, it appears likely that it will be critical to have access to therapeutic options both now and beyond the pandemic,” said Mikael Dolsten, MD, PhD., Chief Scientific Officer and President, Worldwide Research, Development and Medical of Pfizer. “We have designed PF-07321332 as a potential oral therapy that could be prescribed at the first sign of infection, without requiring that patients are hospitalized or in critical care. At the same time, Pfizer’s intravenous antiviral candidate is a potential novel treatment option for hospitalized patients. Together, the two have the potential to create an end to end treatment paradigm that complements vaccination in cases where disease still occurs.”
Protease inhibitors bind to a viral enzyme (called a protease), preventing the virus from replicating in the cell. Protease inhibitors have been effective at treating other viral pathogens such as HIV and hepatitis C virus, both alone and in combination with other antivirals. Currently marketed therapeutics that target viral proteases are not generally associated with toxicity and as such, this class of molecules may potentially provide well-tolerated treatments against COVID-19.
The Phase 1 trial is a randomized, double-blind, sponsor-open, placebo-controlled, single- and multiple-dose escalation study in healthy adults evaluating the safety, tolerability and pharmacokinetics of PF-07321332.
Initiation of this study is supported by preclinical studies that demonstrated the antiviral activity of this potential first-in-class SARS-CoV-2 therapeutic designed specifically to inhibit replication of the SARS-CoV2 virus. The structure of PF-07321332, together with the pre-clinical data, will be shared in a COVID-19 session of the Spring American Chemical Society meeting on April 6.
Pfizer is also investigating an intravenously administered investigational protease inhibitor, PF-07304814, which is currently in a Phase 1b multi-dose trial in hospitalized clinical trial participants with COVID-19.
About Pfizer: Breakthroughs That Change Patients’ Lives
At Pfizer, we apply science and our global resources to bring therapies to people that extend and significantly improve their lives. We strive to set the standard for quality, safety and value in the discovery, development and manufacture of health care products, including innovative medicines and vaccines. Every day, Pfizer colleagues work across developed and emerging markets to advance wellness, prevention, treatments and cures that challenge the most feared diseases of our time. Consistent with our responsibility as one of the world’s premier innovative biopharmaceutical companies, we collaborate with health care providers, governments and local communities to support and expand access to reliable, affordable health care around the world. For more than 170 years, we have worked to make a difference for all who rely on us. We routinely post information that may be important to investors on our website at www.Pfizer.com. In addition, to learn more, please visit us on www.Pfizer.com and follow us on Twitter at @Pfizer and @Pfizer News, LinkedIn, YouTube and like us on Facebook at Facebook.com/Pfizer.
.CLIP
https://cen.acs.org/content/cen/articles/99/i13/Pfizer-unveils-oral-SARS-CoV.html

Drugmaker Pfizer revealed its oral COVID-19 antiviral clinical candidate PF-07321332 on Tuesday at the American Chemical Society Spring 2021 meeting. The compound, which is currently in Phase 1 clinical trials, is the first orally administered compound in the clinic that targets the main protease (also called the 3CL protease) of SARS-CoV-2, the virus that causes COVID-19. By inhibiting the main protease, PF-07321332 prevents the virus from cleaving long protein chains into the parts it needs to reproduce itself. Dafydd Owen, director of medicinal chemistry at Pfizer, presented the compound in a symposium of the Division of Medicinal Chemistry.
Last year, Pfizer reported PF-07304814, a different small molecule inhibitor of SARS-CoV-2’s main protease. The work to develop that compound began during the 2002-2003 outbreak of SARS-CoV, severe acute respiratory syndrome. But that molecule can only be given intravenously, which limits its use to hospital settings.
Because PF-07321332 can be taken orally, as a pill or capsule, it could be given outside of hospitals if it proves to be safe and effective. People who have been exposed to SARS-CoV-2 could take it as a preventative measure, for example.
“For the foreseeable future, we will expect to see continued outbreaks from COVID-19. And therefore, as with all viral pandemics, it’s important we have a full toolbox on how to address it,” Charlotte Allerton, Pfizer’s head of medicine design, told C&EN.
PF-07321332 was developed from scratch during the current pandemic. It’s a reversible covalent inhibitor that reacts with one of the main protease’s cysteine residues. Owen also discussed the chemistry involved in scaling up the compound. The first 7 mg of the compound were synthesized in late July 2020. Encouraged by the early biological data, the Pfizer team aimed to scale up the synthesis. By late October, they’d made 100 g of the compound. Just two weeks later, the chemists had scaled up the synthesis to more than 1 kg. Owen said 210 researchers had worked on the project. Ana Martinez, who studies COVID-19 treatments at the Spanish National Research Council CSIC and also presented during the symposium, told C&EN that having a COVID-19 antiviral is of critical importance. She eagerly anticipates the safety and efficacy data from the trials of PF-07321332. “Hopefully we will have a new drug to fight against COVID-19,” Martinez said. And because the molecule targets the main protease, she said that it might be useful for fighting other coronaviruses and preventing future pandemics.Chemical & Engineering News
| Clinical data | |
|---|---|
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2628280-40-8 |
| PubChem CID | 155903259 |
| UNII | 7R9A5P7H32 |
| KEGG | D12244 |
| ChEBI | CHEBI:170007 |
| Chemical and physical data | |
| Formula | C23H32F3N5O4 |
| Molar mass | 499.535 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 192.9[1] °C (379.2 °F) |
| showSMILES | |
| showInChI |
Xray crystal structure (PDB:7SI9 and 7VH8) of the SARS-CoV-2 protease inhibitor PF-07321332 bound to the viral 3CLpro (Mpro) protease enzyme. Ribbon diagram of the protein with the drug shown as sticks. The catalytic residues (His41, Cys145) are shown as yellow sticks.
./////////////////PF-07321332, PF 07321332, COVID 19, CORONA VIRUS, SARS-CoV-2 inhibitor, PHASE 1, nirmatrelvir, PAXLOVID, CORONA VIRUS, COVID 19
C1N(C([C@@H]2C1[C@]2(C)C)C(=O)N[C@@H](CC3C(NCC3)=O)C#N)C(C([C@@](C)(C)C)NC(=O)C(F)(F)F)=O
C1N(C(C2C1C2(C)C)C(=O)N[C@@H](CC3C(NCC3)=O)C#N)C(C([C@@](C)(C)C)NC(=O)C(F)(F)F)=O
NEW DRUG APPROVALS
ONE TIME
$10.00
Pabinafusp alfa
(Heavy chain)
EVQLVQSGAE VKKPGESLKI SCKGSGYSFT NYWLGWVRQM PGKGLEWMGD IYPGGDYPTY
SEKFKVQVTI SADKSISTAY LQWSSLKASD TAMYYCARSG NYDEVAYWGQ GTLVTVSSAS
TKGPSVFPLA PSSKSTSGGT AALGCLVKDY FPEPVTVSWN SGALTSGVHT FPAVLQSSGL
YSLSSVVTVP SSSLGTQTYI CNVNHKPSNT KVDKKVEPKS CDKTHTCPPC PAPELLGGPS
VFLFPPKPKD TLMISRTPEV TCVVVDVSHE DPEVKFNWYV DGVEVHNAKT KPREEQYNST
YRVVSVLTVL HQDWLNGKEY KCKVSNKALP APIEKTISKA KGQPREPQVY TLPPSRDELT
KNQVSLTCLV KGFYPSDIAV EWESNGQPEN NYKTTPPVLD SDGSFFLYSK LTVDKSRWQQ
GNVFSCSVMH EALHNHYTQK SLSLSPGKGS SETQANSTTD ALNVLLIIVD DLRPSLGCYG
DKLVRSPNID QLASHSLLFQ NAFAQQAVCA PSRVSFLTGR RPDTTRLYDF NSYWRVHAGN
FSTIPQYFKE NGYVTMSVGK VFHPGISSNH TDDSPYSWSF PPYHPSSEKY ENTKTCRGPD
GELHANLLCP VDVLDVPEGT LPDKQSTEQA IQLLEKMKTS ASPFFLAVGY HKPHIPFRYP
KEFQKLYPLE NITLAPDPEV PDGLPPVAYN PWMDIRQRED VQALNISVPY GPIPVDFQRK
IRQSYFASVS YLDTQVGRLL SALDDLQLAN STIIAFTSDH GWALGEHGEW AKYSNFDVAT
HVPLIFYVPG RTASLPEAGE KLFPYLDPFD SASQLMEPGR QSMDLVELVS LFPTLAGLAG
LQVPPRCPVP SFHVELCREG KNLLKHFRFR DLEEDPYLPG NPRELIAYSQ YPRPSDIPQW
NSDKPSLKDI KIMGYSIRTI DYRYTVWVGF NPDEFLANFS DIHAGELYFV DSDPLQDHNM
YNDSQGGDLF QLLMP
(Light chain)
DIVMTQTPLS LSVTPGQPAS ISCRSSQSLV HSNGNTYLHW YLQKPGQSPQ LLIYKVSNRF
SGVPDRFSGS GSGTDFTLKI SRVEAEDVGV YYCSQSTHVP WTFGQGTKVE IKRTVAAPSV
FIFPPSDEQL KSGTASVVCL LNNFYPREAK VQWKVDNALQ SGNSQESVTE QDSKDSTYSL
SSTLTLSKAD YEKHKVYACE VTHQGLSSPV TKSFNRGEC
(Disulfide bridge: H22-H96, H145-H201, H221-L219, H227-H’227, H230-H’230, H262-H322, H368-H426, H596-H609, H847-H857, H’22-H’96, H’145-H’201, H’221-L’219, H’262-H’322, H’368-H’426, H’596-H’609, H’847-H’857, L23-L93, L139-L199, L’23-L’93, L’139-L’199)
Pabinafusp alfa
CAS 2140211-48-7
PMDA 2021/3/23, JAPAN
Pabinafusp alfa (genetical recombination) (JAN)
2140211-48-7, UNII: TRF8S0U6ON
Immunoglobulin G1, anti-(human transferrin receptor) (human-mus musculus monoclonal JR-141 gamma1-chain) fusion protein with peptide (synthetic 2-amino acid linker) fusion protein with human iduronate-2-sulfatase, disulfide with human-mus musculus mono
Immunoglobulin G1-kappa, anti-(human transferrin receptor 1, tfr1) humanized monoclonal antibody, fused with human iduronate-2-sulfatase, glycoform alfa:
Pabinafusp alfa is under investigation in clinical trial NCT03568175 (A Study of JR-141 in Patients With Mucopolysaccharidosis II).
JR-141
NEW DRUG APPROVALS
ONE TIME
$10.00
JCR Pharmaceuticals Announces Approval of IZCARGO® (Pabinafusp Alfa) for Treatment of MPS II (Hunter Syndrome) in Japan
– First Approved Enzyme Replacement Therapy for MPS II to Penetrate Blood-Brain Barrier via Intravenous Administration, Validating JCR’s J-Brain Cargo® Technology –March 23, 2021 07:30 AM Eastern Daylight Time
HYOGO, Japan–(BUSINESS WIRE)–JCR Pharmaceuticals Co., Ltd. (TSE 4552; “JCR”) today announced that the Ministry of Health, Labour and Welfare (MHLW) in Japan has approved IZCARGO® (pabinafusp alfa 10 mL, intravenous drip infusion) for the treatment of mucopolysaccharidosis type II (MPS II, or Hunter syndrome). IZCARGO® (formerly known as JR-141) is a recombinant iduronate-2-sulfatase enzyme replacement therapy (ERT) that relies on J-Brain Cargo®, a proprietary technology developed by JCR, to deliver therapeutics across the blood-brain barrier (BBB). It is the first-ever approved ERT that penetrates the BBB via intravenous administration, a potentially life-changing benefit for individuals with lysosomal storage disorders (LSDs) such as MPS II.
“Subsequent to this approval in Japan, I look forward to further accumulation of clinical evidence for pabinafusp alfa in Brazil, the US and EU”Tweet this
Many patients with MPS II show complications not only in somatic symptoms but also in the central nervous system (CNS), which are often severe, with significant effects on patients’ neurocognitive development, independence, and quality of life. By delivering the enzyme to both the body and the brain, IZCARGO® treats the neurological complications of Hunter syndrome that other available therapies have been unable or inadequate to address so far.
“Approval of IZCARGO® in Japan under SAKIGAKE designation is a key milestone in JCR Pharmaceuticals’ global expansion. It comes on the heels of Fast Track designation from the US FDA, orphan designation from the European Medicines Agency, and the FDA’s acceptance of the JR-141 Investigational New Drug application, enabling JCR to begin our Phase 3 trial in the US,” said Shin Ashida, chairman and president of JCR Pharmaceuticals. “These critical regulatory milestones in Japan, where we have such a strong record of success, and those in the US and Europe, provide important validation of the value of our J-Brain Cargo® technology to deliver therapies across the blood-brain barrier, which we believe is essential to addressing the central nervous system complications of lysosomal storage disorders. We will continue our uncompromising effort to take on the challenge of providing new treatment options for patients with lysosomal storage disorders around the world as soon as possible.”
The MHLW’s approval of IZCARGO® is based on totality of evidence from non-clinical and clinical studies1-4. In a phase 2/3 clinical trial conducted in Japan, all 28 patients experienced significant reductions in heparan sulfate (HS) concentrations in the cerebrospinal fluid (CSF) – a biomarker for effectiveness against CNS symptoms of MPS II – after 52 weeks of treatment, thus meeting the trial’s primary endpoint. IZCARGO® maintained somatic disease control in patients who switched from standard ERT to IZCARGO®. The study also confirmed an improvement in somatic symptoms in participants who had not previously received standard ERT prior to the start of the trial. Additionally, a neurocognitive development assessment demonstrated maintenance or improvement of age-equivalent function in 21 of the 28 patients. There were no reports of serious treatment-related adverse events in the trial, suggestive of a favorable safety and tolerability profile for IZCARGO®.4
“Subsequent to this approval in Japan, I look forward to further accumulation of clinical evidence for pabinafusp alfa in Brazil, the US and EU,” said Dr. Paul Harmatz of University of California – San Francisco (UCSF) Benioff Children’s Hospital Oakland, Oakland, CA, United States. “The availability of an enzyme replacement therapy that crosses the blood-brain barrier is expected to treat both CNS and somatic symptoms associated with this devastating and life-threatening disorder, including developmental and cognitive delays, bone deformities, and abnormal behavior, which have, historically, been unaddressed.”
JCR recently filed an application with the Brazilian Health Surveillance Agency (Agência Nacional de Vigilância Sanitária [ANVISA]) for marketing approval of IZCARGO® for the treatment of patients with MPS II. JCR is also preparing to launch a Phase 3 trial of IZCARGO® in the US, Brazil, the UK, Germany, and France.
About pabinafusp alfa
Pabinafusp alfa (10 mL, intravenous drip infusion) is a recombinant fusion protein of an antibody against the human transferrin receptor and idursulfase, the enzyme that is missing or malfunctioning in subjects with Hunter syndrome. It incorporates J-Brain Cargo®, JCR’s proprietary BBB-penetrating technology, to cross the BBB through transferrin receptor-mediated transcytosis, and its uptake into cells is mediated through the mannose-6-phosphate receptor. This novel mechanism of action is expected to make pabinafusp alfa effective against the CNS symptoms of Hunter syndrome.
In pre-clinical trials, JCR has confirmed both high-affinity binding of pabinafusp alfa to transferrin receptors, and passage across the BBB into neuronal cells, as evidenced by electron microscopy. In addition, JCR has confirmed enzyme uptake in various brain tissues. The company has also confirmed a reduction of substrate accumulation in the CNS and peripheral organs in an animal model of Hunter syndrome.1
In several clinical trials of pabinafusp alfa, JCR obtained evidence of reduced HS concentrations in the CSF, a biomarker for assessing effectiveness against CNS symptoms. The results were consistent with those obtained in pre-clinical studies. Clinical studies have also demonstrated positive effects of pabinafusp alfa on CNS symptoms.2
About J-Brain Cargo® Technology
JCR’s first-in-class proprietary technology, J-Brain Cargo®, enables the development of therapies that cross the BBB and penetrate the CNS. The CNS complications of diseases are often severe, resulting in developmental delays, an impact on cognition and, above all, poor prognosis, which affect patients’ independence as well as the quality of life of patients and their caregivers. With J-Brain Cargo®, JCR seeks to address the unresolved clinical challenges of LSDs by delivering the enzyme to both the body and the brain.
About Mucopolysaccharidosis II (Hunter Syndrome)
Mucopolysaccharidosis II (Hunter syndrome) is an X-linked recessive LSD caused by a deficiency of iduronate-2-sulfatase, an enzyme that breaks down complex carbohydrates called glycosaminoglycans (GAGs, also known as mucopolysaccharides) in the body. Hunter syndrome, which affects an estimated 7,800 individuals worldwide (according to JCR research), gives rise to a wide range of somatic and neurological symptoms. The current standard of care for Hunter syndrome is ERT. CNS symptoms related MPS II have been unmet medical needs so far.
About JCR Pharmaceuticals Co., Ltd.
JCR Pharmaceuticals Co., Ltd. (TSE 4552) is a global specialty pharmaceuticals company that is redefining expectations and expanding possibilities for people with rare and genetic diseases worldwide. We continue to build upon our 45-year legacy in Japan while expanding our global footprint into the US, Europe, and Latin America. We improve patients’ lives by applying our scientific expertise and unique technologies to research, develop, and deliver next-generation therapies. Our approved products in Japan include therapies for the treatment of growth disorder, Fabry disease, acute graft-versus host disease, and renal anemia. Our investigational products in development worldwide are aimed at treating rare diseases including MPS I (Hurler syndrome, Hurler-Scheie, and Scheie syndrome), MPS II (Hunter syndrome), Pompe disease, and more. JCR strives to expand the possibilities for patients while accelerating medical advancement at a global level. Our core values – reliability, confidence, and persistence – benefit all our stakeholders, including employees, partners, and patients. Together we soar. For more information, please visit https://www.jcrpharm.co.jp/en/site/en/.
1 Sonoda H, Morimoto H, Yoden E, et al. A blood-brain-barrier-penetrating anti-human transferrin receptor antibody fusion protein for neuronopathic mucopolysaccharidosis II. Molecular Therapy. 2018;26(5):1366-1374.
2 Morimoto H, Kida K, Yoden E, et al. Clearance of heparan sulfate in the brain prevents neurodegeneration and neurocognitive impairment in MPS II mice. Molecular Therapy. 2021;S1525-0016(21)00027-7.
3 Okuyama T, Eto Y, Sakai N, et al. Iduronate-2-sulfatase with anti-human transferrin receptor antibody for neuropathic mucopolysaccharidosis II: a phase 1/2 trial. Molecular Therapy. 2019;27(2):456-464.
4 Okuyama T, Eto Y, Sakai N, et al. A phase 2/3 trial of pabinafusp alfa, IDS fused with anti-human transferrin receptor antibody, targeting neurodegeneration in MPS-II. Molecular Therapy. 2021;29(2):671-679.
//////////Pabinafusp alfa, JR-141, JR 141,APPROVALS 21, JAPAN 2021
#Pabinafusp alfa, #JR-141, #JR 141, #APPROVALS 21, #JAPAN 2021
Diclofenac etalhyaluronate sodium


Diclofenac etalhyaluronate sodium
RN: 1398396-25-2
UNII: LG1II3835L
Molecular Formula, [(C30-H35-Cl2-N3-O12)a-(C14-H20-N-Na-O11)b]n-H2-O
Molecular Weight, 1101.8195
HYALURONIC ACID PARTLY AMIDIFIED WITH 2-(2-(2-((2,6-DICHLOROPHENYL)AMINO)PHENYL)ACETYLOXY)ETHANAMINE, SODIUM SALT
HYALURONAMIDE, N-(2-((2-(2-((2,6-DICHLOROPHENYL)AMINO)PHENYL)ACETYL)OXY)ETHYL), SODIUM SALT
SI 613
APPROVED PMDA JAPAN 2021/3/23, Joycle
Anti-inflammatory, Joint function improving agent

NEW DRUG APPROVALS
One time
$10.00
Treatment of Signs and Symptoms of Osteoarthritis of the Knee
![Chemical structure of N-[2-[[2-[2-[(2,6-dichlorophenyl)amino]phenyl]acetyl]oxy]ethyl]hyaluronamide (diclofenac etalhyaluronate, SI-613)](https://www.researchgate.net/publication/325304331/figure/fig1/AS:669091362766859@1536535220444/Chemical-structure-of.png)
Diclofenac Etalhyaluronate Sodium
Sodium hyaluronate partially amidated with 2- (2- {2-[(2,6-dichlorophenyl) amino] phenyl} acetyloxy) ethaneamine
Hyaluronic acid sodium salt partly amidified with 2- (2- {2-[(2,6-dichlorophenyl) amino] phenyl} acetyloxy) ethanamine
[(C 30 H 35 Cl 2 N 3 O 12 ) a (C 14 H 20 NNaO 11 ) b ] n
[ 1398396-25-2 ]
Hyaluronic acid/non-steroidal anti-inflammatory drug; Hyaluronic acid/NSAID; JOYCLU; ONO 5704; ONO-5704/SI-613; SI-613
- OriginatorSeikagaku Corporation
- DeveloperOno Pharmaceutical; Seikagaku Corporation
- ClassAmides; Analgesics; Antirheumatics; Drug conjugates; Glycosaminoglycans; Nonsteroidal anti-inflammatories
- Mechanism of ActionCyclooxygenase inhibitors
- RegisteredOsteoarthritis
- Phase IITendinitis
- 23 Mar 2021Registered for Osteoarthritis in Japan (Intra-articular)
- 25 Sep 2020Phase II for Osteoarthritis is still ongoing in USA (Seikagaku Corporation pipeline, September 2020)
- 25 Sep 2020Phase II for Tendinitis is still ongoing in Japan (Seikagaku Corporation pipeline, September 2020)
In today’s aging society, osteoarthritis (hereinafter also referred to as “OA” in the present specification), which is a dysfunction caused by joint pain and joint degeneration, is the most common joint disease in the world. It is one of the major causes of physical disorders that interfere with daily life in the elderly. Further, as a disease accompanied by swelling and pain in joints, rheumatoid arthropathy (hereinafter, also referred to as “RA” in the present specification), which is polyarthritis, is known. In RA as well, when the condition progresses over a long period of time, cartilage and bones are destroyed and degeneration or deformation occurs, resulting in physical disorders that interfere with daily life, such as narrowing the range in which joints can be moved.
Currently, preparations using hyaluronic acid and its derivatives are used as medicines for arthropathy such as osteoarthritis and rheumatoid arthropathy. Hyaluronic acid preparations are usually formulated as injections, and for the purpose of improving dysfunction due to arthropathy and suppressing pain through the lubricating action, shock absorption action, cartilage metabolism improving action, etc. of hyaluronic acid, the affected knee, It is administered directly to joints such as the shoulders. Commercialized hyaluronic acid preparations include, for example, those containing purified sodium hyaluronate as an active ingredient (for example, Alz (registered trademark) and Svenir (registered trademark)). The preparation requires continuous administration of 3 to 5 times at a frequency of once a week.
In addition, preparations containing crosslinked hyaluronan as an active ingredient require three consecutive doses once a week (for example, Synvisc®), or treatment is completed with a single dose. For single dose administration (eg, Synvisc-One®, Gel-One®, MONOVISC®) are known.On the other hand, steroids and non-steroidal anti-inflammatory compounds are known as quick-acting drugs, and are also used for treatments aimed at relieving joint pain caused by OA and RA. For example, the steroid triamcinolone acetonide has been used as a therapeutic target for joint diseases such as rheumatoid arthritis. Triamcinolone acetonide is commercially available as a drug that is injected intra-articularly and requires administration every 1 to 2 weeks for treatment. Further, as non-steroidal anti-inflammatory compounds, for example, ointments containing diclofenac sodium as an active ingredient and oral administration agents are known.It is also known that a mixture or a conjugate of hyaluronic acid or a derivative thereof and a steroid or a non-steroidal anti-inflammatory compound is used as an active ingredient. For example, a mixture of crosslinked hyaluronic acid and triamcinolone hexaacetonide (CINGAL®) has been commercialized as a single-dose drug. Further, a compound in which hyaluronic acid or a derivative thereof is linked to a steroid or a non-steroidal anti-inflammatory compound is also known. For example, Patent Documents 1 and 2 describe derivatives in which an anti-inflammatory compound is introduced into hyaluronic acid via a spacer. These aim to achieve both fast-acting pain relief and long-term pain relief through improvement of dysfunction. However, it has not yet reached the stage where it can be said that sufficient treatment methods for OA and RA have been established and provided.
PATENT
WO 2018168920
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018168920
<Synthesis Example>
Aminoethanol-diclofenac-introduced sodium hyaluronate (test substance) was synthesized according to the method described in Examples of International Publication No. 2005/066241 (hyaluronic acid weight average molecular weight: 800,000, introduction rate). : 18 mol%).
More specifically, it was synthesized by the following method.
2.155 g (10.5 mmol) of 2-bromoethylamine hydrobromide is dissolved in 20 mL of dichloromethane, 1.436 mL (10.5 mmol) of triethylamine is added under ice-cooling, and di-tert-butyl-dicarbonate (Boc) is added. 2 O) 2.299 g (10.5 mmol) of a dichloromethane solution of 5 mL was added and stirred. After stirring at room temperature for 90 minutes, ethyl acetate was added, and the mixture was washed successively with 5 wt% citric acid aqueous solution, water and saturated brine. After dehydration with sodium sulfate, the solvent was distilled off under reduced pressure to obtain Boc-aminoethyl bromide.
5 mL of a dimethylformamide (DMF) solution of 2.287 g (10.2 mmol) of Boc-aminoethyl bromide obtained above is ice-cooled, 6 mL of a DMF solution of 3.255 g (10.2 mmol) of diclofenac sodium is added, and the mixture is added at room temperature. Stirred overnight. The mixture was stirred at 60 ° C. for 11 hours and at room temperature overnight. Ethyl acetate was added, and the mixture was sequentially separated and washed with a 5 wt% aqueous sodium hydrogen carbonate solution, water, and saturated brine. After dehydration with sodium sulfate, ethyl acetate was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (toluene: ethyl acetate = 20: 1 (v / v), 0.5% by volume triethylamine) to obtain Boc-aminoethanol-diclofenac.
2.108 g (4.80 mmol) of Boc-aminoethanol-diclofenac obtained above was dissolved in 5 mL of dichloromethane, 20 mL of 4M hydrochloric acid / ethyl acetate was added under ice-cooling, and the mixture was stirred for 2.5 hours. Diethyl ether and hexane were added and precipitated, and the precipitate was dried under reduced pressure. As a result, aminoethanol-diclofenac hydrochloride was obtained. Structure 1 was identified by-NMR
H: 1 H-NMR (500 MHz, CDCl 3 ) [delta] (ppm) = 3.18 (2H, t, NH 2 CH 2 CH 2 O-), 3.94 (2H, s, Ph-CH 2 -CO), 4.37 (2H, t, NH 2 CH 2 CH 2 O-), 6.47-7.31 (8H, m, Aromatic H, NH).
After dissolving 500 mg (1.25 mmol / disaccharide unit) of hyaluronic acid having a weight average molecular weight of 800,000 in 56.3 mL of water / 56.3 mL of dioxane, imide hydroxysuccinate (1 mmol) / 0.5 mL of water, water-soluble carbodiimide Hydrochloride (WSCI / HCl) (0.5 mmol) / water 0.5 mL, aminoethanol-diclofenac hydrochloride (0.5 mmol) / (water: dioxane = 1: 1 (v / v), 5 mL obtained above ) Was added in sequence, and the mixture was stirred all day and night. 7.5 mL of a 5 wt% sodium hydrogen carbonate aqueous solution was added to the reaction mixture, and the mixture was stirred for about 4 hours. 215 μL of a 50% (v / v) acetic acid aqueous solution was added to the reaction solution for neutralization, and then 2.5 g of sodium chloride was added and the mixture was stirred. 400 ml of ethanol was added to precipitate, and the precipitate was washed twice with an 85% (v / v) aqueous ethanol solution, twice with ethanol, and twice with diethyl ether, dried under reduced pressure overnight at room temperature, and aminoethanol-diclophenac. Introduction Sodium hyaluronate (test substance) was obtained. The introduction rate of diclofenac measured by a spectrophotometer was 18 mol%.
PATENT
WO 2018168921
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018168921
//////////Diclofenac etalhyaluronate sodium, JOYCLU, ONO 5704, ONO-5704/SI-613, SI 613, JAPAN 2021, Joycle, APPROVALS 2021
#Diclofenac etalhyaluronate sodium, #JOYCLU, #ONO 5704, #ONO-5704/SI-613, #SI 613, #JAPAN 2021, #Joycle, #APPROVALS 2021
Dasiglucagon




Dasiglucagon
Treatment of Hypoglycemia in Type 1 and Type 2 Diabetes Patients
| Formula | C152H222N38O50 |
|---|---|
| CAS | 1544300-84-6 |
| Mol weight | 3381.6137 |
FDA APPROVED, 2021/3/22, Zegalogue
Zealand Pharma A/S
UNIIAD4J2O47FQ
HypoPal rescue pen
(4S)-4-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-2-[[(2S,3R)-2-[[2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-amino-3-(1H-imidazol-4-yl)propanoyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]acetyl]amino]-3-hydroxybutanoyl]amino]-3-phenylpropanoyl]amino]-3-hydroxybutanoyl]amino]-3-hydroxypropanoyl]amino]-3-carboxypropanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-hydroxypropanoyl]amino]hexanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-methylpentanoyl]amino]-3-carboxypropanoyl]amino]-2-methylpropanoyl]amino]propanoyl]amino]-5-carbamimidamidopentanoyl]amino]propanoyl]amino]-5-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-4-carboxy-1-[[(2S)-1-[[(1S,2R)-1-carboxy-2-hydroxypropyl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-1-oxobutan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-1-oxohexan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-5-oxopentanoic acid
. [16-(2-methylalanine)(S>X),17-L-alanine(R>A),20-L-α-glutamyl(Q>E),21-L-αglutamyl(D>E),24-L-lysyl(Q>K),27-L-α-glutamyl(M>E),28-L-serine(N>S)]human glucagon
L-Threonine, L-histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L- phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L- lysyl-L-tyrosyl-L-leucyl-L-α-aspartyl-2-methylalanyl-L-alanyl-L- arginyl-L-alanyl-L-α-glutamyl-L-α-glutamyl-L-phenylalanyl-L- valyl-L-lysyl-L-tryptophyl-L-leucyl-L-α-glutamyl-L-seryl
L-Threonine, L-histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-alpha-aspartyl-L-tyrosyl-L-seryl-L-lysyl-L-tyrosyl-L-leucyl-L-alpha-aspartyl-2-methylalanyl-L-alanyl-L-arginyl-L-alanyl-L-alpha-glutamyl-L-alphaC152 H222 N38 O50L-Threonine, L-histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L-lysyl-L-tyrosyl-L-leucyl-L-α-aspartyl-2-methylalanyl-L-alanyl-L-arginyl-L-alanyl-L-α-glutamyl-L-α-glutamyl-L-phenylalanyl-L-valyl-L-lysyl-L-tryptophyl-L-leucyl-L-α-glutamyl-L-seryl-Molecular Weight3381.61
Other Names
- L-Histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L-lysyl-L-tyrosyl-L-leucyl-L-α-aspartyl-2-methylalanyl-L-alanyl-L-arginyl-L-alanyl-L-α-glutamyl-L-α-glutamyl-L-phenylalanyl-L-valyl-L-lysyl-L-tryptophyl-L-leucyl-L-α-glutamyl-L-seryl-L-threonine
- Developer Beta Bionics; Zealand Pharma
- ClassAntihyperglycaemics; Antihypoglycaemics; Peptides
- Mechanism of ActionGlucagon receptor agonists
- Orphan Drug StatusYes – Hypoglycaemia; Congenital hyperinsulinism
- RegisteredHypoglycaemia
- Phase IIICongenital hyperinsulinism
- Phase II/IIIType 1 diabetes mellitus
- 22 Mar 2021Registered for Hypoglycaemia (In children, In adolescents, In adults, In the elderly) in USA (SC) – First global approval
- 22 Mar 2021Zealand Pharma anticipates the launch of dasiglucagon in USA (SC, Injection) in June 2021
- 22 Mar 2021Pooled efficacy and safety data from three phase III trials in Hypoglycaemia released by Zealand Pharma
NEW DRUG APPROVALS
one time
$10.00
PATENTS
WO 2014016300
US 20150210744
PAPER
Pharmaceutical Research (2018), 35(12), 1-13
Dasiglucagon, sold under the brand name Zegalogue, is a medication used to treat severe hypoglycemia in people with diabetes.[1]
The most common side effects include nausea, vomiting, headache, diarrhea, and injection site pain.[1]
Dasiglucagon was approved for medical use in the United States in March 2021.[1][2][3] It was designated an orphan drug in August 2017.[4]
Dasiglucagon is under investigation in clinical trial NCT03735225 (Evaluation of the Safety, Tolerability and Bioavailability of Dasiglucagon Following Subcutaneous (SC) Compared to IV Administration).
Medical uses
Dasiglucagon is indicated for the treatment of severe hypoglycemia in people aged six years of age and older with diabetes.[1][2]
Contraindications
Dasiglucagon is contraindicated in people with pheochromocytoma or insulinoma.[1]
References
- ^ Jump up to:a b c d e f https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214231s000lbl.pdf
- ^ Jump up to:a b “Dasiglucagon: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 22 March 2021.
- ^ “Zealand Pharma Announces FDA Approval of Zegalogue (dasiglucagon) injection, for the Treatment of Severe Hypoglycemia in People with Diabetes” (Press release). Zealand Pharma. 22 March 2021. Retrieved 22 March 2021 – via GlobeNewswire.
- ^ “Dasiglucagon Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 10 August 2017. Retrieved 22 March 2021.
External links
- “Dasiglucagon”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03378635 for “A Trial to Confirm the Efficacy and Safety of Dasiglucagon in the Treatment of Hypoglycemia in Type 1 Diabetes Subjects” at ClinicalTrials.gov
- Clinical trial number NCT03688711 for “Trial to Confirm the Clinical Efficacy and Safety of Dasiglucagon in the Treatment of Hypoglycemia in Subjects With T1DM” at ClinicalTrials.gov
- Clinical trial number NCT03667053 for “Trial to Confirm the Efficacy and Safety of Dasiglucagon in the Treatment of Hypoglycemia in T1DM Children” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Zegalogue |
| AHFS/Drugs.com | Zegalogue |
| License data | US DailyMed: Dasiglucagon |
| Routes of administration | Subcutaneous |
| Drug class | Glucagon receptor agonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1544300-84-6 |
| PubChem CID | 126961379 |
| DrugBank | DB15226 |
| UNII | AD4J2O47FQ |
| KEGG | D11359 |
| Chemical and physical data | |
| Formula | C152H222N38O50 |
| Molar mass | 3381.664 g·mol−1 |
| 3D model (JSmol) | Interactive image |
///////////Dasiglucagon, FDA 2021, APPROVALS 2021, Zegalogue, ダシグルカゴン, ZP 4207, ZP-GA-1, Hypoglycemia, Type 1, Type 2 , Diabetes Patients, Zealand Pharma A/S, Orphan Drug Status, Hypoglycaemia, Congenital hyperinsulinism, HypoPal rescue pen, DIABETES
#Dasiglucagon, #FDA 2021, #APPROVALS 2021, #Zegalogue, #ダシグルカゴン, #ZP 4207, ZP-GA-1, #Hypoglycemia, #Type 1, #Type 2 , #Diabetes Patients, #Zealand Pharma A/S, #Orphan Drug Status, #Hypoglycaemia, #Congenital hyperinsulinism, #HypoPal rescue pen, #DIABETESSMILES
- C[C@H]([C@@H](C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(=O)O)C(=O)N[C@@H](CC2=CC=C(C=C2)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC3=CC=C(C=C3)O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(=O)O)C(=O)NC(C)(C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CC4=CC=CC=C4)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC5=CNC6=CC=CC=C65)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)O)NC(=O)CNC(=O)[C@H](CCC(=O)N)NC(=O)[C@H](CO)NC(=O)[C@H](CC7=CNC=N7)N)O
CLARITHROMYCIN


Clarithromycin
Synonyms:A-56268, TE-031, 6-O-methylerythromycin, ATC:J01FA09Use:macrolide antibioticChemical name:6-O-methylerythromycinFormula:C38H69NO13
- MW:747.96 g/mol
- CAS-RN:81103-11-9
- 81103-11-9
klacid XL / Klaricid XL / Macladin / Naxy / Veclam / Zeclar
(3R,4S,5S,6R,7R,9R,11R,12R,13S,14R)-6-{[(2S,3R,4S,6R)-4-(dimethylamino)-3-hydroxy-6-methyloxan-2-yl]oxy}-14-ethyl-12,13-dihydroxy-4-{[(2R,4R,5S,6S)-5-hydroxy-4-methoxy-4,6-dimethyloxan-2-yl]oxy}-7-methoxy-3,5,7,9,11,13-hexamethyl-1-oxacyclotetradecane-2,10-dione
Synthesis Reference
Jih-Hua Liu, David A. Riley, “Preparation of crystal form II of clarithromycin.” U.S. Patent US5844105, issued May, 1997. US5844105
NEW DRUG APPROVALS
ONE TIME
$10.00
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Clarithromycin citrate | 16K08R7NG0 | 848130-51-8 | MDRWXDRMSKEMRE-AZFLODHXSA-N |
ClarithromycinCAS Registry Number: 81103-11-9CAS Name: 6-O-MethylerythromycinManufacturers’ Codes: A-56268; TE-031Trademarks: Biaxin (Abbott); Clarosip (Grñenthal); Clathromycin (Taisho); Cyllind (Abbott); Klacid (Abbott); Klaricid (Abbott); Macladin (Guidotti); Naxy (Sanofi Winthrop); Veclam (Zambon); Zeclar (Abbott)Molecular Formula: C38H69NO13Molecular Weight: 747.95Percent Composition: C 61.02%, H 9.30%, N 1.87%, O 27.81%Literature References: Semisynthetic macrolide antibiotic; derivative of erythromycin, q.v. Prepn: Y. Watanabe et al.,EP41355; eidem,US4331803 (1981, 1982 both to Taisho); and in vitro antibacterial activity: S. Morimoto et al.,J. Antibiot.37, 187 (1984). In vitro and in vivo antibacterial activity: P. B. Fernandes et al.,Antimicrob. Agents Chemother.30, 865 (1986). Comparative antibacterial spectrum in vitro: C. Benson et al.,Eur. J. Clin. Microbiol.6, 173 (1987); H. M. Wexler, S. M. Finegold, ibid. 492. HPLC determn in biological fluids: D. Croteau et al.,J. Chromatogr.419, 205 (1987); in plasma: H. Amini, A. Ahmadiani, J. Chromatogr. B817, 193 (2005). Acute toxicity study: S. Abe et al.,Chemotherapy (Tokyo)36, Suppl. 3, 274 (1988). Symposium on pharmacology and comparative clinical studies: J. Antimicrob. Chemother.27, Suppl. A, 1-124 (1991). Comprehensive description: I. I. Salem, Anal. Profiles Drug Subs. Excip.24, 45-85, (1996).Properties: Colorless needles from chloroform + diisopropyl ether (1:2), mp 217-220° (dec). Also reported as crystals from ethanol, mp 222-225° (Morimoto). uv max (CHCl3): 288 nm (e 27.9). uv max (CHCl3): 240, 288 nm; (methanol): 211, 288 nm. [a]D24 -90.4° (c = 1 in CHCl3). Stable at acidic pH. LD50 in male, female mice, male, female rats (mg/kg): 2740, 2700, 3470, 2700 orally, 1030, 850, 669, 753 i.p., >5000 all s.c. (Abe).Melting point: mp 217-220° (dec); mp 222-225° (Morimoto)Optical Rotation: [a]D24 -90.4° (c = 1 in CHCl3)Absorption maximum: uv max (CHCl3): 288 nm (e 27.9). uv max (CHCl3): 240, 288 nmToxicity data: LD50 in male, female mice, male, female rats (mg/kg): 2740, 2700, 3470, 2700 orally, 1030, 850, 669, 753 i.p., >5000 all s.c. (Abe)Therap-Cat: Antibacterial.Keywords: Antibacterial (Antibiotics); Macrolides.
Clarithromycin, a semisynthetic macrolide antibiotic derived from erythromycin, inhibits bacterial protein synthesis by binding to the bacterial 50S ribosomal subunit. Binding inhibits peptidyl transferase activity and interferes with amino acid translocation during the translation and protein assembly process. Clarithromycin may be bacteriostatic or bactericidal depending on the organism and drug concentration.
Clarithromycin, sold under the brand name Biaxin among others, is an antibiotic used to treat various bacterial infections.[2] This includes strep throat, pneumonia, skin infections, H. pylori infection, and Lyme disease, among others.[2] Clarithromycin can be taken by mouth as a pill or liquid.[2]
Common side effects include nausea, vomiting, headaches, and diarrhea.[2] Severe allergic reactions are rare.[2] Liver problems have been reported.[2] It may cause harm if taken during pregnancy.[2] It is in the macrolide class and works by decreasing protein production of some bacteria.[2]
Clarithromycin was developed in 1980 and approved for medical use in 1990.[3][4] It is on the World Health Organization’s List of Essential Medicines, the safest and most effective medicines needed in a health system.[5] Clarithromycin is available as a generic medication.[2] It is made from erythromycin and is chemically known as 6-O-methylerythromycin.[6]
Medical uses
Clarithromycin is primarily used to treat a number of bacterial infections including pneumonia, Helicobacter pylori, and as an alternative to penicillin in strep throat.[2] Other uses include cat scratch disease and other infections due to bartonella, cryptosporidiosis, as a second line agent in Lyme disease and toxoplasmosis.[2] It may also be used to prevent bacterial endocarditis in those who cannot take penicillin.[2] It is effective against upper and lower respiratory tract infections, skin and soft tissue infections and helicobacter pylori infections associated with duodenal ulcers.
Spectrum of bacterial susceptibility
Staphylococcus aureusAerobic Gram-positive bacteria
Aerobic Gram-negative bacteria
- Haemophilus parainfluenzae
- Haemophilus influenzae
- Moraxella catarrhalis
Helicobacter
Mycobacteria
Mycobacterium avium complex consisting of:
Other bacteria
Safety and effectiveness of clarithromycin in treating clinical infections due to the following bacteria have not been established in adequate and well-controlled clinical trials:[7]
Aerobic Gram-positive bacteria
- Streptococcus agalactiae
- Streptococcus (Groups C, F, G)
- Viridans group streptococci
Aerobic Gram-negative bacteria
Anaerobic Gram-positive bacteria
- Clostridium perfringens
- Peptococcus Niger
- Cutibacterium acnes
Anaerobic Gram-negative bacteria
- Prevotella melaninogenica (formerly Bacteroides melaninogenicus)
Contraindications
- Clarithromycin should not be taken by people who are allergic to other macrolides or inactive ingredients in the tablets, including microcrystalline cellulose, croscarmelose sodium, magnesium stearate, and povidone
- Clarithromycin should not be used by people with a history of cholestatic jaundice and/or liver dysfunction associated with prior clarithromycin use.[7]
- Clarithromycin should not be used in the setting of hypokalaemia (low blood potassium)
- Use of clarithromycin with the following medications: cisapride, pimozide, astemizole, terfenadine, ergotamine, ticagrelor, ranolazine or dihydroergotamine is not recommended.[7]
- It should not be used with colchicine in people with kidney or liver impairment.[7]
- Concomitant use with cholesterol medications such as lovastatin or simvastatin.[7]
- Hypersensitivity to clarithromycin or any component of the product, erythromycin, or any macrolide antibiotics.[7]
- QT prolongation or ventricular cardiac arrhythmias, including torsade de pointes.[7]
Side effects
The most common side effects are gastrointestinal: diarrhea (3%), nausea (3%), abdominal pain (3%), and vomiting (6%). It also can cause headaches, insomnia, and abnormal liver function tests. Allergic reactions include rashes and anaphylaxis. Less common side effects (<1%) include extreme irritability, hallucinations (auditory and visual), dizziness/motion sickness, and alteration in senses of smell and taste, including a metallic taste. Dry mouth, panic attacks, and nightmares have also been reported, albeit less frequently.[8]
Cardiac
In February 2018, the FDA issued a Safety Communication warning with respect to an increased risk for heart problems or death with the use of clarithromycin, and has recommended that alternative antibiotics be considered in those with heart disease.[9]
Clarithromycin can lead to a prolonged QT interval. In patients with long QT syndrome, cardiac disease, or patients taking other QT-prolonging medications, this can increase risk for life-threatening arrhythmias.[10]
In one trial, the use of short-term clarithromycin treatment was correlated with an increased incidence of deaths classified as sudden cardiac deaths in stable coronary heart disease patients not using statins.[11] Some case reports suspect it of causing liver disease.[12]
Liver and kidney
Clarithromycin has been known to cause jaundice, cirrhosis, and kidney problems, including kidney failure.[citation needed]
Central nervous system
Common adverse effects of clarithromycin in the central nervous system include dizziness, headaches. Rarely, it can cause ototoxicity, delirium and mania.
Infection
A risk of oral candidiasis and vaginal candidiasis, due to the elimination of the yeast’s natural bacterial competitors by the antibiotic, has also been noted.
Pregnancy and breastfeeding
Clarithromycin should not be used in pregnant women except in situations where no alternative therapy is appropriate.[7] Clarithromycin can cause potential hazard to the fetus hence should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.[7] For lactating mothers it is not known whether clarithromycin is excreted in human milk.[7]
Interactions
Clarithromycin inhibits a liver enzyme, CYP3A4, involved in the metabolism of many other commonly prescribed drugs. Taking clarithromycin with other medications that are metabolized by CYP3A4 may lead to unexpected increases or decreases in drug levels.
A few of the common interactions are listed below.
Colchicine
Clarithromycin has been observed to have a dangerous interaction with colchicine as the result of inhibition of CYP3A4 metabolism and P-glycoprotein transport. Combining these two drugs may lead to fatal colchicine toxicity, particularly in people with chronic kidney disease.[7]
Statins
Taking clarithromycin concurrently with certain statins (a class of drugs used to reduce blood serum cholesterol levels) increases the risk of side effects, such as muscle aches and muscle break down (rhabdomyolysis).[13]
Calcium channel blockers
Concurrent therapy with calcium channel blocker may increase risk of low blood pressure, kidney failure, and death, compared to pairing calcium channel blockers with azithromycin, a drug similar to clarithromycin but without CYP3A4 inhibition.[14] Administration of clarithromycin in combination with verapamil have been observed to cause low blood pressure, low heart rate, and lactic acidosis.[7]
Carbamazepine
Clarithromycin may double the level of carbamazepine in the body by reducing its clearance, which may lead to toxic symptoms of carbamazepine, such as double vision, loss of voluntary body movement, nausea, as well as hyponatremia.[15]
HIV medications
Depending on the combination of medications, clarithromycin therapy could be contraindicated, require changing doses of some medications, or be acceptable without dose adjustments.[16] For example, clarithromycin may lead to decreased zidovudine concentrations.[17]
Mechanism of action
Clarithromycin prevents bacteria from multiplying by acting as a protein synthesis inhibitor. It binds to 23S rRNA, a component of the 50S subunit of the bacterial ribosome, thus inhibiting the translation of peptides.[citation needed]
Pharmacokinetics
MetabolismUnlike erythromycin, clarithromycin is acid-stable, so can be taken orally without having to be protected from gastric acids. It is readily absorbed, and diffuses into most tissues and phagocytes. Due to the high concentration in phagocytes, clarithromycin is actively transported to the site of infection. During active phagocytosis, large concentrations of clarithromycin are released; its concentration in the tissues can be over 10 times higher than in plasma. Highest concentrations are found in liver, lung tissue, and stool.
Clarithromycin has a fairly rapid first-pass metabolism in the liver. Its major metabolites include an inactive metabolite, N-desmethylclarithromycin, and an active metabolite, 14-(R)-hydroxyclarithromycin. Compared to clarithromycin, 14-(R)-hydroxyclarithromycin is less potent against mycobacterial tuberculosis and the Mycobacterium avium complex. Clarithromycin (20%-40%) and its active metabolite (10%-15%) are excreted in urine. Of all the drugs in its class, clarithromycin has the best bioavailability at 50%, which makes it amenable to oral administration. Its elimination half-life is about 3 to 4 hours with 250 mg administered every 12 h, but increased to 5 to 7 h with 500 mg administered every 8 to 12 h. With any of these dosing regimens, the steady-state concentration of this metabolite is generally attained within 3 to 4 days.[18]
History
Clarithromycin was invented by researchers at the Japanese drug company Taisho Pharmaceutical in 1980.[3] The product emerged through efforts to develop a version of the antibiotic erythromycin that did not experience acid instability in the digestive tract, causing side effects, such as nausea and stomachache. Taisho filed for patent protection for the drug around 1980 and subsequently introduced a branded version of its drug, called Clarith, to the Japanese market in 1991. In 1985, Taisho partnered with the American company Abbott Laboratories for the international rights, and Abbott also gained FDA approval for Biaxin in October 1991. The drug went generic in Europe in 2004 and in the US in mid-2005.
Society and culture

A pack of Clarithromycin tablets manufactured by Taisho Pharmaceutical
Available forms
Clarithromycin is available as a generic medication.[2] In the United States, clarithromycin is available as immediate release tablets, extended release tablets, and granules for oral suspension.[2]
Brand names
Clarithromycin is available under several brand names in many different countries, for example Biaxin, Crixan, Claritron, Clarihexal, Clacid, Claritt, Clacee, Clarac, Clariwin, Claripen, Clarem, Claridar, Cloff, Fromilid, Infex, Kalixocin, Karicin, Klaricid, Klaridex, Klacid, Klaram, Klabax, MegaKlar, Monoclar, Resclar, Rithmo, Truclar, Vikrol and Zeclar.
Manufacturers
In the UK the drug product is manufactured in generic form by a number of manufacturers including Somex Pharma, Ranbaxy, Aptil and Sandoz.
SYN
CN 109705180

SYN
Indian Pat. Appl., 2014DE00731, 31 Aug 2016

SYN
Heterocycles, 31(12), 2121-4; 1990

SYN
https://patents.google.com/patent/WO2006064299A1/enErythromycin A is known to be a useful macrolide antibiotic having a strong activity against Gram-positive bacteria, this compound has an undesirable property that it loses rapidly the antibacterial activity by the acid in stomach when administered orally, where- upon its blood concentration remains at a low level. 6-0-Alkyl derivatives of Erythromycin- A are well known as an useful antibacterial agents. 6-O-Methyl-Erythromycin-A (Clarithromycin) and a pharmaceutically acceptable salt is a potent macrolide antibiotic as reported in US Patent No. 4,331 ,803. Clarithromycin is stable in acidic medium and also remarkable in vivo activity and has a strong antibacterial property against Gram-positive bacteria compared to Erythromycin- A. This compound shows excellent effect for the treatment of infections by oral administration.A number of synthetic processes have been reported for preparing 6-O-alkyl erythromycin. US Patent No. 4,331 ,803 discloses a method for the preparation of Clarithromycin by methylating 6-OH group of 2′-O-3′-N-benzyloxycarbonyl erythromycinFormula (III)

21,3′-O-Protected ErythromycinMethylation of 6-OH group of the 2′,3′-benzyloxycarbonyl erythromycin was carried out using methyl iodide in the presence of a suitable base in a solvent. Clarithromycin was obtained from the compound after removing benzyloxycarbonyl group by hydrogenolysis and then subjecting to the reductive methylation in the presence of excess amount of farmaldehyde. Clarithromycin can also be synthesized by the methylation of 6-OH position of Erythromycin-A-9-OximeFormula (II)

Erythromycin-9-OximeSynthesis of Clarithromycin using 9-oxime or its derivatives are well reported in US Patent Nos. 5,274,085; 4,680,386; 4,668,776; 4,670,549 and 4,672,109. In case of Erythromycin-9-Oxime derivatives, the oxime is protected before methylation step with 2- alkenyl group (US Patent Nos. 4,670,549; 4,668,776) or benzyl group (US Patent Nos. 4,680,386 and 4,670,549). However, it has been reported (Ref. Journal of Antibiotics 46, No. 6, Page No. 647, year 1993) that when the Erythromycin-A-9-Oxime is protected by trimethylsilyl group, which is very unstable under basic condition pose potential impurities formation during methylation. There are some methods reported in US Patent Nos., e.g. , 4,680,386; 4,670,549 and US Patent No. 4,311,803 for the synthesis of Clarithromycin by using chlorobenzyloxycarbonyl group for protection at 2′ and 3′ function of of Erythromycin-A-9-Oxime derivatives.For the protection of 2′-OH group (US Patent No. 4,311 ,803) requires large amounts of benzyl chloroformate which poses problems in handling because of its severe irritating and toxic properties. This protection step also leads to the formation of 3′ -N- demethylation, which requires an additional re-methylation step. The de-protection of chlorobenzyloxy carbonyl group leads to the formation of undesired side products. In earlier reported processes, e.g. , US Patent No. 4,990,602; EP 0,272,110 Al where the methylation has been done on Erythromycin-A-9-Oxime derivatives by the protection of 2′ and 4″ hydroxyl groups using DMSO and THF as a solvent at 0° to 50C or at room temperature, smooth methylation takes place with less side product formation. However, by using the above methylation processes the formation of 6, 11-O-dimethyl erythromycin- A (Compound- A) is always more than 1.0 % in Clarithromycin. Hence, there is a need for an efficient methylation process for the production of Clarithromycin with lesser amount of 6,11-O-dimethyl erythromycin-A than reported previously.




EXAMPLE 1Erythromycin-A-9-OximeTo a solution of 201 Ltr water in 561 Kg isopropyl alcohol is added 282 Kg (4057 mol) of hydroxyl amine hydrochloride under stirring and the reaction mixture is brought to 10 to 200C. Caustic flakes (162 Kg, 4050 mol) is added slowly to the reaction mixture by keeping temperature between 10° to 200C. After 15 minutes of completion of addition, pH of reaction mixture is adjusted to 6.5 to 7.0 by the slow addition of glacial acetic acid (96 Ltr, 100.8 Kg, 1678.6 mole). To the stirred reaction mass is added 300 Kg (408.8 mole) of Erythromycin-A base and reaction mixture is stirred at 55° C for 28 hours. After completion of the reaction, mixture is brought to ambient temperature and to it a mixture of ammonia solution (270 Kg) and water (600 Ltr) is added within 1 hour followed by 3000 Ltr of fresh water in next two hours and stirred the reaction mass for further 1 hour. White solid product obtained is centrifuged, wet cake is washed with water and dried at 6O0C for 12 hours to give 270 Kg of erythromycin-A Oxime. Melting point = 156° to 158°C.EXAMPLE 22′,4″-O-Bis(trimethylsilyl)-erythro?nycin-A-9[O-(l-methoxy-l-methyl ethyl)oximeTo a solution of 80 Kg (106.8 mole) of Erythromycin-A-9-Oxime in 400 Ltr of dichloromethane is added 38.50 Kg (534 mole) of 2-methoxy propene at 100C temperature 19.25 Kg (166.6 mole) of pyridine hydrochloride is added under stirring and the reaction mixture is stirred at 8 to 12° C for 6 hours then to it is added 19.30 Kg (119.5 mole) of HMDS and stirring is continued for 12 to 15 hours at 15° to 18°C temperature. After completion of reaction, 400 Ltr of saturated aqueous sodium carbonate solution is added and the mixture is stirred thoroughly at room temperature. Aqueous layer is further extracted with fresh DCM (100 Ltr). Both DCM extracts are mixed together and washed with water (200 Ltr) followed by brine solution (200 Ltr). The solvent is evaporated under reduced pressure. To the obtained crude solid mass is charged isopropyl alcohol (240 Ltr) and distilled out 80 Ltr of isopropyl alcohol. To the reaction mixture 160 Ltr of water is charged and stirring continued at room temperature for 1 hour. Solid crystalline product obtained is centrifuged and dried at 60° to 650C for 8 hours under vacuum to give 85 Kg of title compound. Melting point = 125° to 126°C. HPLC Purity = More than 90 % .EXAMPLE 3Clarithromycin-9- OximeTo a solution of 80 Kg (82.98 mole) of 2′,4″-O-bis(trimethylsilyl)-erythromycin-A- 9-[O-(l-methoxy methyl ethyl)Oxime] in 1200 Ltr of a mixture of dimethyl sulfoxide and diethylether (1 : 1) are added methyl iodide (20.62 Kg, 145.2 mole) and 6.48 Kg (98.35 mole) of 85 % potassium hydroxide powder and the reaction mixture is stirred for 90 minutes at room temperature. To the reaction mass is added 53 Kg of 40 % dimethylamine solution and stirring is continued for 1 hour diethylether layer is separated and DMSO layer is further extracted with fresh diethylether (200 Ltr). Combined ether layer is washed with water and concentrated in vacuum. To the obtained semi solid mass 330 Ltr of isopropyl alcohol is charged and then distilled out 165 Ltr of isopropyl alcohol. To the obtained slurry 165 Ltr of water and 21.71 Kg formic acid (99%) are added and the mixture is stirred at room temperature for 30 minutes. 622 Ltr of water is added to the reaction mixture and pH is adjusted between 10.5 and 11.5 with 25 % aqueous sodium hydroxide solution. Solid compound obtained is centrifuged and wet cake is kept as such for further reaction on the basis of moisture content. Wet weight = 95 Kg, Moisture Content = 33 %, Dried weight = 62 KgEXAMPLE 46-O-Methyl erythromycin- A (Clarithromycin)62 Kg of 6-O-Methyl erythromycin-9-Oxime is charged into a mixture of 434 Ltr of isopropyl alcohol and water (1: 1) and to it is added 38.80 Kg of sodium metabisulphite (203 mole) and then the mixture is heated to reflux for 6 to 8 hours. To the reaction mixture is charged water (620 Ltr) at ambient temperature and then the mixture is adjusted to pH about 10.5 to 11.5 by adding 25% aqueous sodium hydroxide solution and stirred for further 1 hour. White solid crude product is centrifuged, washed with water (300 Ltr), dried at 65° to 750C for 8 hours to give 40 Kg of crude Clarithromycin which on re- crystallization with chloroform isopropyl alcohol mixture provided 20 Kg of Clarithromycin (Form II).
SYNEP 0041355; US 4331803J Antibiot 1984,37(2),187-189
| EP 0147062 |
The methylation of 2′-O,N-bis(benzyloxycarbonyl)-N-demethylerythromycin A (I) with methyl iodide and KOH or NaHI in DMSO-dimethoxyethane gives the 6-O-methyl derivative (II), which is deprotected by hydrogenation with H2 over Pd/C in ethanol acetic acid affording 6-O-methyl-N-demethylerythromycin A (III). Finally, this compound is methylated with formaldehyde under reductive conditions (H2-Pd/C) in ethanol/acetic acid.
CLIP


2 Clarithromycin. Initial attempts of making clarithromycin (2) from erythromycin (1) by methylation of 8 gave approximately equal amounts of 2 and 10 by methylation at O-6 and O-11, respectively (Scheme 2, route A).[28–30] This allowed 2 to be obtained in approximately 39% yield, but it contained a small impurity of di-O-methylated 9. To improve the yields and obtain 2 in pure form, other alternatives were explored. During methylation of analogues of 8 it was observed that the conformation of the macrocyclic core plays an important role for the regioselectivity of the O-methylation.[31] As oximes are readilyhydrolysed and may have different conformations than ketone 8, oximes 11 and 13 were subjected to methylation. Interestingly, methylation of 13, but not of 11, proved to be highly selective for O-6 and provided 14 in 86% yield (Scheme2 route B); an observation which supports that 13 populates different conformations compared to 8 and 11 under the methylation conditions.[31] Compound 14 was then hydrogenated with Pd/C to deprotect the two benzyloxycarbonyl groups and the 2-chlorobenzyl group. The N-methylamine was then methylated by reductive amination and the oxime was deprotected by hydrolysis to provide clarithromycin (2). This procedure was further modified for process-scale synthesis so that clarithromycin (2) could be obtained in 70% yield starting from oxime 11 without the isolation of any intermediate.[32][28] M. Shigeo, T. Yoko, W. Yoshiaki, O. Sadafumi, J. Antibiot. 1984, 37, 187 – 189. [29] Y. Watanabe, T. Adachi, T. Asaka, M. Kashimura, S. Morimoto, Heterocycles 1990, 31, 2121 – 2124. [30] E. H. Flynn, H. W. Murphy, R. E. McMahon, J. Am. Chem. Soc. 1955, 77, 3104 – 3106. [31] Y. Watanabe, S. Morimoto, T. Adachi, M. Kashimura, T. Asaka, J. Antibiot. 1993, 46, 647 – 660.32] R. A. Dominguez, M. D. C. C. Rodriguez, L. . D. Tejo, R. N. Rib, J. S. Cebrin, J. I. B. Bilbao, 2003, US6642364B2.
References
- ^ https://www.ema.europa.eu/documents/psusa/clarithromycin-list-nationally-authorised-medicinal-products-psusa/00000788/202004_en.pdf
- ^ Jump up to:a b c d e f g h i j k l m n “Clarithromycin”. The American Society of Health-System Pharmacists. Archivedfrom the original on September 3, 2015. Retrieved September 4, 2015.
- ^ Jump up to:a b Greenwood D (2008). Antimicrobial drugs : chronicle of a twentieth century medical triumph (1 ed.). Oxford: Oxford University Press. p. 239. ISBN 9780199534845. Archived from the original on 2016-03-05.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 498. ISBN 9783527607495.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Kirst HA (2012). Macrolide Antibiotics (2 ed.). Basel: Birkhäuser Basel. p. 53. ISBN 9783034881050. Archived from the original on 2016-03-05.
- ^ Jump up to:a b c d e f g h i j k l “BIAXIN® Filmtab® (clarithromycin tablets, USP) BIAXIN® XL Filmtab® (clarithromycin extended-release tablets) BIAXIN® Granules (clarithromycin for oral suspension, USP)” (PDF). November 2, 2015. Archived (PDF) from the original on August 24, 2015. Retrieved November 2, 2015.
- ^ “Clarithromycin Side Effects in Detail – Drugs.com”. Drugs.com. Archived from the original on 2017-08-19. Retrieved 2017-08-18.
- ^ “Safety Alerts for Human Medical Products – Clarithromycin (Biaxin): Drug Safety Communication – Potential Increased Risk of Heart Problems or Death in Patients With Heart Disease”. FDA. Retrieved 24 February 2018.
- ^ Yamaguchi S, Kaneko Y, Yamagishi T, et al. [Clarithromycin-induced torsades de pointes]. Nippon Naika Gakkai Zasshi. 2003;92(1):143–5.
- ^ Winkel P, Hilden J, Fischer Hansen J, Hildebrandt P, Kastrup J, Kolmos HJ, et al. (2011). “Excess sudden cardiac deaths after short-term clarithromycin administration in the CLARICOR trial: why is this so, and why are statins protective?”. Cardiology. 118 (1): 63–7. doi:10.1159/000324533. PMID 21447948. S2CID 11873791.
- ^ Tietz A, Heim MH, Eriksson U, Marsch S, Terracciano L, Krähenbühl S (January 2003). “Fulminant liver failure associated with clarithromycin”. The Annals of Pharmacotherapy. 37 (1): 57–60. doi:10.1345/1542-6270(2003)037<0057:flfawc>2.0.co;2. PMID 12503933.
- ^ Patel AM, Shariff S, Bailey DG, Juurlink DN, Gandhi S, Mamdani M, et al. (June 2013). “Statin toxicity from macrolide antibiotic coprescription: a population-based cohort study”. Annals of Internal Medicine. 158 (12): 869–76. doi:10.7326/0003-4819-158-12-201306180-00004. PMID 23778904. S2CID 21222679.
- ^ Gandhi S, Fleet JL, Bailey DG, McArthur E, Wald R, Rehman F, Garg AX (December 2013). “Calcium-channel blocker-clarithromycin drug interactions and acute kidney injury”. JAMA. 310 (23): 2544–53. doi:10.1001/jama.2013.282426. PMID 24346990.
- ^ Gélisse P, Hillaire-Buys D, Halaili E, Jean-Pastor MJ, Vespignan H, Coubes P, Crespel A (November 2007). “[Carbamazepine and clarithromycin: a clinically relevant drug interaction]”. Revue Neurologique. 163 (11): 1096–9. doi:10.1016/s0035-3787(07)74183-8. PMID 18033049.
- ^ Sekar VJ, Spinosa-Guzman S, De Paepe E, De Pauw M, Vangeneugden T, Lefebvre E, Hoetelmans RM (January 2008). “Darunavir/ritonavir pharmacokinetics following coadministration with clarithromycin in healthy volunteers”. Journal of Clinical Pharmacology. 48 (1): 60–5. doi:10.1177/0091270007309706. PMID 18094220. S2CID 38368595.
- ^ Polis MA, Piscitelli SC, Vogel S, Witebsky FG, Conville PS, Petty B, et al. (August 1997). “Clarithromycin lowers plasma zidovudine levels in persons with human immunodeficiency virus infection”. Antimicrobial Agents and Chemotherapy. 41 (8): 1709–14. doi:10.1128/AAC.41.8.1709. PMC 163990. PMID 9257746.
- ^ Ferrero JL, Bopp BA, Marsh KC, Quigley SC, Johnson MJ, Anderson DJ, et al. (1990). “Metabolism and disposition of clarithromycin in man”. Drug Metabolism and Disposition. 18 (4): 441–6. PMID 1976065.
- ReferencesAllevi, P. et al.: Bioorg. Med. Chem. (BMECEP) 7, 12, 2749 (1999)Watanabe, Y. et al.: Heterocycles (HTCYAM) 31, 12, 2121 (1990).EP 158 467 (Taisho Pharmaceutical Co.; 22.3.1985; J-prior. 6.4.1984).EP 272 110 (Taisho Pharmaceutical Co.; 16.12.1987; J-prior. 17.12.1986).US 2 001 037 015 (Teva Pharm.; 15.12.2000; USA-prior. 29.2.2000).KR 2 000 043 839 (Hanmi Pharm.; ROK-prior. 29.12.1998).EP 1 150 990 (Hanmi Pharm.; 7.11.2001; ROK-prior. 29.12.1998)EP 41 355 (Taisho Pharmaceutical Co.; 27.5.1981; J-prior. 4.6.1980).Preparation of O,N-dicarbobenzoxy-N-demethylerythromycin:Flynn, E. H. et al.: J. Am. Chem. Soc. (JACSAT) 77, 3104 (1955).Process for preparation of erythromycin A oxime:US 5 808 017 (Abbott; 15.9.1998; USA-prior. 10.4.1996).Alternative synthesis of clarithromycin:Liao, G.; Zhang, G.; He, T.: Zhongguo Kangshengsu Zazhi (ZKZAEY) 27, 3, 148 (2002) (in Chinese).EP 1 134 229 (Hanmi Pharmac. Co.; 19.9.2001; ROK-prior. 15.3.2000).Crystal form 0 of clarithromycin:The Merck Index, 13th Ed., 2362, p. 408.US 5 945 405 (Abbott; 31.8.1999; USA-prior. 17.1.1997).
External links
- “Clarithromycin”. Drug Information Portal. U.S. National Library of Medicine.
- US patent 4331803, Watanabe Y, Morimoto S, Omura S, “Novel erythromycin compounds”, issued 1981-05-19, assigned to Taisho Pharmaceutical
- “FDA review finds additional data supports the potential for increased long-term risks with antibiotic clarithromycin (Biaxin) in patients with heart disease” (PDF). FDA Drug Safety Communication.
| Clinical data | |
|---|---|
| Trade names | Biaxin, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a692005 |
| License data | EU EMA: by INNUS DailyMed: Clarithromycin |
| Pregnancy category | AU: B3 |
| Routes of administration | By mouth, intravenous |
| Drug class | Macrolides |
| ATC code | J01FA09 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)US: ℞-onlyEU: Rx-only [1]In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | 50% |
| Protein binding | low binding |
| Metabolism | hepatic |
| Elimination half-life | 3–4 h |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 81103-11-9 |
| PubChem CID | 84029 |
| DrugBank | DB01211 |
| ChemSpider | 10342604 |
| UNII | H1250JIK0A |
| KEGG | D00276 |
| ChEMBL | ChEMBL1741 |
| CompTox Dashboard (EPA) | DTXSID3022829 |
| ECHA InfoCard | 100.119.644 |
| Chemical and physical data | |
| Formula | C38H69NO13 |
| Molar mass | 747.964 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESCC[C@@H]1[C@@]([C@@H]([C@H](C(=O)[C@@H](C[C@@]([C@@H]([C@H]([C@@H]([C@H](C(=O)O1)C)O[C@H]2C[C@@]([C@H]([C@@H](O2)C)O)(C)OC)C)O[C@H]3[C@@H]([C@H](C[C@H](O3)C)N(C)C)O)(C)OC)C)C)O)(C)O | |
| hideInChIInChI=1S/C38H69NO13/c1-15-26-38(10,45)31(42)21(4)28(40)19(2)17-37(9,47-14)33(52-35-29(41)25(39(11)12)16-20(3)48-35)22(5)30(23(6)34(44)50-26)51-27-18-36(8,46-13)32(43)24(7)49-27/h19-27,29-33,35,41-43,45H,15-18H2,1-14H3/t19-,20-,21+,22+,23-,24+,25+,26-,27+,29-,30+,31-,32+,33-,35+,36-,37-,38-/m1/s1 Key:AGOYDEPGAOXOCK-KCBOHYOISA-N | |
| (what is this?) (verify) |
////////////////////CLARITHROMYCIN, Antibacterial, Antibiotics, Macrolides, A-56268, TE-031,
#CLARITHROMYCIN, #Antibacterial, #Antibiotics, #Macrolides, #A-56268, #TE-031,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}