
Home » New drugs EU
Category Archives: New drugs EU
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Europe grants conditional OK to Pfizer’s Bosulif
Good News For Pfizer’s Orphan Drug Bosulif (Bosutinib) in Europe

mar 28,2013
Regulators in Europe have given a partial green light to Pfizer ‘s leukaemia drug Bosulif.
The European Commission has granted conditional marketing authorisation for Bosulif (bosutinib) for the treatment of adults with chronic, accelerated or blast phase Philadelphia chromosome-positive (Ph+) chronic myelogenous leukaemia (CML). The drug can be given to patients previously treated with one or more tyrosine kinase inhibitors ie Novartis’ Gleevec (imatinib) and Tasigna (nilotinib) or Bristol-Myers Squibb’s Sprycel (dasatinib).
The European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) on January 17, 2013, adopts a positive opinion, recommending a conditional marketing authhorization for Pfizer’s orphan drug Bosulif (Bosutinib) for Chronic Leukemia (CML). Bosutinib receives orphan designation from the European Commission (EC) on August 4, 2010, for CML.
Pfizer receives FDA approval on September 4, 2012, for orphan drug Bosulif (Bosutinib) for CML. Pfizer receives on February 24, 2009, FDA Orphan Drug Designation (ODD) for Bosutinib for CML.

Bosutinib (rINN/USAN; codenamed SKI-606, marketed under the trade name Bosulif) is atyrosine kinase inhibitor undergoing research for use in the treatment of cancer. [1] [2]Originally synthesized by Wyeth, it is being developed by Pfizer.
Some commercial stocks of bosutinib (from sources other than the Pfizer material used for clinical trials) have recently been found to have the incorrect chemical structure, calling the biological results obtained with them into doubt.[3]
Bosutinib received US FDA approval on September 5, 2012 for the treatment of adult patients with chronic, accelerated, or blast phase Philadelphia chromosome-positive (Ph+)chronic myelogenous leukemia (CML) with resistance, or intolerance to prior therapy.[4][5][6]
- Puttini M, Coluccia AM, Boschelli F, Cleris L, Marchesi E, Donella-Deana A, Ahmed S, Redaelli S, Piazza R, Magistroni V, Andreoni F, Scapozza L, Formelli F, Gambacorti-Passerini C. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res. 2006 Dec 1;66(23):11314-22. Epub 2006 Nov 17.
- Vultur A, Buettner R, Kowolik C, et al. (May 2008). “SKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cells”.Mol. Cancer Ther. 7 (5): 1185–94. doi:10.1158/1535-7163.MCT-08-0126.PMC 2794837. PMID 18483306.
- Derek Lowe, In The Pipeline (blog), “Bosutinib: Don’t Believe the Label!”
- Cortes JE, Kantarjian HM, Brümmendorf TH, Kim DW, Turkina AG, Shen ZX, Pasquini R, Khoury HJ, Arkin S, Volkert A, Besson N, Abbas R, Wang J, Leip E, Gambacorti-Passerini C. Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood. 2011 Oct 27;118(17):4567-76. Epub 2011 Aug 24.
- Cortes JE, Kim DW, Kantarjian HM, Brümmendorf TH, Dyagil I, Griskevicus L, Malhotra H, Powell C, Gogat K, Countouriotis AM, Gambacorti-Passerini C. Bosutinib Versus Imatinib in Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia: Results From the BELA Trial. J Clin Oncol. 2012 Sep 4. [Epub ahead of print]
- “Bosulif Approved for Previously Treated Philadelphia Chromosome-Positive Chronic Myelogenous Leukemia”. 05 Sep 2012.
ThromboGenics NV, European Commission has approved JETREA® (ocriplasmin) in the European Union
Leuven, March 15, 2013
ThromboGenics NV an integrated biopharmaceutical company focused on developing and commercializing innovative ophthalmic medicines, today announces that the European Commission has approved JETREA® (ocriplasmin) in the European Union. JETREA® is approved for the treatment of vitreomacular traction (VMT), including when associated with macular hole of diameter less than or equal to 400 microns. The EU approval triggers a €45 million milestone payment to ThromboGenics from its partner Alcon. The first sale of JETREA® in the EU by Alcon will trigger a further €45 million milestone payment to ThromboGenics.
Alcon, a division of Novartis, acquired the rights to commercialize JETREA® outside the United States in March 2012. ThromboGenics retains the right to commercialize the drug in the US. ThromboGenics launched JETREA® in the US in mid-January 2013 where it is approved for the treatment of patients with symptomatic vitreomacular adhesion (VMA).
Ocriplasmin (trade name Jetrea) is a recombinant protease with activity against fibronectin and laminin, components of the vitreoretinal interface. It is used for treatment of symptomatic vitreomacular adhesion, for which it received FDA approval on 17 October 2012. It works by dissolving the proteins that link the vitreous to the macula, resulting in posterior detachment of the vitreous from the retina.[1]
Novartis’ Ilaris canakizumab has become the first biologic drug to be approved in the EU to treat the symptoms of gouty arthritis
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | IL-1β |

Novartis’ Ilaris has become the first biologic drug to be approved in the EU to treat the symptoms of gouty arthritis in another gain for the interleukin-1 beta inhibitor.
march01,2013
First biologic drug approved for condition in Europe
The European Commission (EC) cleared llaris (canakizumab) for the treatment of adult patients who have suffered at least three gouty arthritis attacks in the previous 12 months, but who are unsuitable for treatment with non-steroidal anti-inflammatory drugs (NSAIDs) and colchicine or repeated courses of corticosteroids.
Gouty arthritis – commonly known as gout – is an “excruciating condition”, according to Novartis division head David Epstein, who noted that Ilaris offers new hope to patients who do not currently have treatment options.
Data from two phase III trials of Ilaris in acute gouty arthritis attacks showed that patients treated with the drug experienced significantly greater pain relief compared to the injectable steroid triamcinolone acetonide, while most adverse events were mild to moderate in severity.
The most frequent side effects were infections, and particularly upper respiratory tract infections and nasopharyngitis.
Ilaris was launched in the US and EU in 2009 as a treatment for an auto-inflammatory condition called cryopyrin-associated periodic syndrome (CAPS). The rarity of that condition has meant sales have been relatively small, coming in at $72m last year, albeit a 56 per cent gain over 2011.
Gouty arthritis is a much bigger market for the drug and, along with a juvenile arthritis indication Novartis is pursuing, could push Ilaris towards blockbuster status with sales in excess of $1bn a year.
“Our vision is to realise the potential of Ilaris wherever IL-1 beta plays a key role and available treatment options don’t give patients the help they need,” said Epstein.
EU approval comes after the US FDA knocked back Ilaris for gouty arthritis, saying in 2011 that Novartis needed to provide more data on the drug’s risk-benefit profile, specifically its potential to leave patients vulnerable to infections.
Gout has been a tricky indication for drug developers to crack, with the FDA turning down another CAPS treatment – Regeneron’s IL-1 inhibitor Arcalyst (rilonacept) – in 2012 on the grounds of inadequate safety data and concern about a risk of malignancy.
One success came in 2010 when Savient secured approval for its Krystexxa (pegloticase) drug as a second-line treatment after oral xanthine oxidase inhibitors in patients with severe debilitating chronic tophaceous gout.
However, the drug has failed to make significant inroads because of a high price and tendency to stimulate neutralising antibodies that limit its therapeutic effect, according to Decision Resources.
There is still a great demand for safer and more effective therapies with the phase III pipeline featuring another potential blockbuster in the form of AstraZeneca/Ardea Biosciences URAT1 inhibitor lesinurad.
Canakinumab (INN, trade name Ilaris, previously ACZ885)[1] is a human monoclonal antibody targeted at interleukin-1 beta. It has no cross-reactivity with other members of the interleukin-1 family, including interleukin-1 alpha.[2]
Canakinumab was approved for the treatment of cryopyrin-associated periodic syndromes (CAPS) by the US FDA on June 2009[3] and by the European Medicines Agency in October 2009.[4] CAPS is a spectrum of autoinflammatory syndromes including familial cold autoinflammatory syndrome, Muckle–Wells syndrome, and neonatal-onset multisystem inflammatory disease.
Canakinumab was being developed by Novartis for the treatment of rheumatoid arthritis but this trial has been discontinued.[5] Canakinumab is also in phase I clinical trials as a possible treatment for chronic obstructive pulmonary disease.[6]
References
- Dhimolea, Eugen (2010). “Canakinumab”. MAbs 2 (1): 3–13. doi:10.4161/mabs.2.1.10328. PMC 2828573. PMID 20065636.
- Lachmann, HJ; Kone-Paut I, Kuemmerle-Deschner JB et al. (4 June 2009). “Use of canakinumab in the cryopyrin-associated periodic syndrome”. New Engl J Med 360 (23): 2416–25. doi:10.1056/NEJMoa0810787. PMID 19494217.
- “New biological therapy Ilaris approved in US to treat children and adults with CAPS, a serious life-long auto-inflammatory disease” (Press release). Novartis. 18 June 2009. Retrieved 28 July 2009.
- Wan, Yuet (29 October 2009). “Canakinumab (Ilaris) and rilonacept (Arcalyst) approved in EU for treatment of cryopyrin-associated periodic syndrome”. National electronic Library for Medicines. Retrieved 14 April 2010.
- “clinicaltrials.gov, Identifier NCT00784628: Safety, Tolerability and Efficacy of ACZ885 (Canakinumab) in Patients With Active Rheumatoid Arthritis”. Retrieved 2010-08-21.
- Yasothan U, Kar S (2008). “Therapies for COPD”. Nat Rev Drug Discov 7 (4): 285. doi:10.1038/nrd2533.

Monoclonal Antibody Therapeutic Uses
EU OKs Lundbeck’s Selincro, Nalmefene to cut alcoholic urges

Nalmefene
17-cyclopropylmethyl-4,5α-epoxy-6-methylenemorphinan-3,14-diol
march 1 2013
Lundbeck will be celebrating news that European regulators have issued a green light for Selincro, making it the first therapy approved for the reduction of alcohol consumption in dependent adults.
Selincro (nalmefene) is a unique dual-acting opioid system modulator that acts on the brain’s motivational system, which is dysregulated in patients with alcohol dependence.
The once daily pill has been developed to be taken on days when an alcoholic feels at greater risk of having a drink, in a strategy that aims to reduce – rather than stop – alcohol consumption, which some experts believe is a more realistic goal.
Clinical trials of the drug have shown that it can reduce alcohol consumption by approximately 60% after six months treatment, equating to an average reduction of nearly one bottle of wine per day.
In March last year, data was published from two Phase III trials, ESENSE 1 and ESENSE 2, showing that the mean number of heavy drinking days decreased from 19 to 7 days/month and 20 to 7 days/month, while TAC fell from 85 to 43g/day and from 93 to 30g/day at month six. However, the placebo effect was also strong in the studies.
According to Anders Gersel Pedersen, Executive Vice President and Head of Research & Development at Lundbeck, Selincro “represents the first major innovation in the treatment of alcohol dependence in many years,” and he added that its approval “is exciting news for the many patients with alcohol dependence who otherwise may not seek treatment”.
Alcohol dependence is considered a major public health concern, and yet it is both underdiagnosed and undertreated, highlighting the urgent need for better management of the condition.
In Europe, more than 90% of the 14 million patients with alcohol dependence are not receiving treatment, but research suggests that treating just 40% of these would save 11,700 lives each year.
The Danish firm said it expects to launch Selincro in its first markets in mid-2013, and that it will provide the drug as part of “a new treatment concept that includes continuous psychosocial support focused on the reduction of alcohol consumption and treatment adherence”.
Nalmefene (Revex), originally known as nalmetrene, is an opioid receptor antagonistdeveloped in the early 1970s, and used primarily in the management of alcoholdependence, and also has been investigated for the treatment of other addictions such aspathological gambling and addiction to shopping.
Nalmefene is an opiate derivative similar in both structure and activity to the opiate antagonist naltrexone. Advantages of nalmefene relative to naltrexone include longer half-life, greater oral bioavailability and no observed dose-dependent liver toxicity. As with other drugs of this type, nalmefene can precipitate acute withdrawal symptoms in patients who are dependent on opioid drugs, or more rarely when used post-operatively to counteract the effects of strong opioids used in surgery.
Nalmefene differs from naltrexone by substitution of the ketone group at the 6-position of naltrexone with a methylene group (CH2), which considerably increases binding affinity to the μ-opioid receptor. Nalmefene also has high affinity for the other opioid receptors, and is known as a “universal antagonist” for its ability to block all three.
- US patent 3814768, Jack Fishman et al, “6-METHYLENE-6-DESOXY DIHYDRO MORPHINE AND CODEINE DERIVATIVES AND PHARMACEUTICALLY ACCEPTABLE SALTS”, published 1971-11-26, issued 1974-06-04
- Barbara J. Mason, Fernando R. Salvato, Lauren D. Williams, Eva C. Ritvo, Robert B. Cutler (August 1999). “A Double-blind, Placebo-Controlled Study of Oral Nalmefene for Alcohol Dependence”. Arch Gen Psychiatry 56 (8): 719.
- Clinical Trial Of Nalmefene In The Treatment Of Pathological Gambling
- http://www.fda.gov/cder/foi/label/2000/20459S2lbl.pdf
- “Efficacy of Nalmefene in Patients With Alcohol Dependence (ESENSE1)”. “Lundbeck submits Selincro in EU; Novo Nordisk files Degludec in Japan”. thepharmaletter. 22 December 2011.
- Nalmefene Hydrochloride Drug Information, Professional
Alexza gets EU thumbs-up for Adasuve, Loxapine

LOXAPINE
2-Chloro-11-(4-methylpiperazin-1-yl)dibenzo[b,f][1,4]oxazepine
FEBRUARY 22, 2013
Alexza Pharmaceuticals of the USA and partnerFerrer are celebrating after getting the green light for their inhaled antipsychotic Adasuve.
The European Commission has granted marketing authorisation to Adasuve (loxapine) for the rapid control of mild-to-moderate agitation in adults with schizophrenia or bipolar disorder. The approval is based on two Phase III studies involving over 650 patients which showed that the drug demonstrated statistically significant reductions in agitation from baseline compared to placebo.
The approval requires that patients receive regular treatment immediately after control of acute agitation symptoms, and that Adasuve is administered only in a hospital setting. Also, short-acting beta-agonist bronchodilator treatment should be available for treatment of possible severe respiratory side-effects.
Alexza chief executive Thomas King noted that Adasuve is the first authorised non-injectable therapy for these indications, noting that it will be launched on both sides of the Atlantic in the third quarter – US approval was granted in December.
Spain’s Ferrer has Adasuve rights in Europe, Latin America, Russia and the Commonwealth of Independent States countries. Chief executive Jordi Ramental said the focus for the initial EU launch will be Germany and Austria in 2013, adding that “at the same time, we are compiling the registration dossiers for the non-EU countries in our licensed territory”.
Links
Loxapine (Loxapac, Loxitane) is a typical antipsychotic medication, used primarily in the treatment of schizophrenia. It is a member of the dibenzoxazepine class and structurally related to clozapine (which belongs to the chemically akin class of dibenzodiazepines). Several researchers have argued that Loxapine may behave as an atypical antipsychotic.[1]
Loxapine may be metabolized by N-demethylation to amoxapine, a tetracyclic antidepressant.[2]
Dosage
The typical starting dosage is 10 mg twice daily; usual dose range 30–50 mg twice daily; maximum recommended dosage is 250 mg per day. The US Food and Drug Administration (FDA) has approved loxapine ( Adasuve , Alexza Pharmaceuticals) inhalation powder 10 mg for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults. [4]

Schmutz, J.; Kunzle, F.; Hunziker, F.; Gauch, R.; Helv. Chim. Acta 1967, 50, 245.
- Glazer WM (1999). “Does loxapine have “atypical” properties? Clinical evidence”. The Journal of Clinical Psychiatry 60 (Suppl 10): 42–6. PMID 10340686.
- Cheung SW, Tang SW, Remington G (March 1991). “Simultaneous quantitation of loxapine, amoxapine and their 7- and 8-hydroxy metabolites in plasma by high-performance liquid chromatography”. Journal of Chromatography 564 (1): 213–21. doi:10.1016/0378-4347(91)80083-O. PMID 1860915.
- Sperry L, Hudson B, Chan CH (March 1984). “Loxapine abuse”. The New England Journal of Medicine 310 (9): 598. doi:10.1056/NEJM198403013100920. PMID 6694719.
- Harrison, Pam: Inhalant Approved for Agitation in Bipolar I, Schizophrenia. Medscape. Dec 24, 2012. “Clozapine and loxapine for schizophrenia”. Drug and Therapeutics Bulletin 29 (11): 41–2. May 1991. PMID 1747161.
- Chakrabarti A, Bagnall A, Chue P, et al. (2007). Chakrabarti, Abhijit. ed. “Loxapine for schizophrenia”. Cochrane Database of Systematic Reviews (Online) (4): CD001943. doi:10.1002/14651858.CD001943.pub2. PMID 17943763.
- Brennand, Kristen; Anthony Simone, Jessica Jou, Chelsea Gelboin-Burkhart, Ngoc Tran, Sarah Sangar, Yan Li, Yangling Mu, Gong Chen, Diana Yu, Shane McCarthy, Jonathan Sebat & Fred H. Gage (13 April 2011). “Modelling schizophrenia using human induced pluripotent stem cells”. Nature 473 (7346): 221–5. doi:10.1038/nature09915. PMID 21490598.
Launch of Ipsen/Braintree’s Eziclen/Izinova in Europe expected by end 2013
Eziclen® / Izinova® a new bowel cleansing preparation, is a sulphate-based formulation (sodium, magnesium and potassium sulphates).
The dose for cleansing the colon will require two administrations of about 0.5 litre / 16 ounces of the product diluted in water, each followed by about 1 litre / 32 ounces, of clear liquids, each.
The drug will be registered under the trade name Eziclen® in the large majority of the concerned EU countries and under the name Izinova® in the other few countries including France & UK
7 feb 2013
French drugmaker Ipsen and US partner Braintree Laboratories yesterday announced today that Eziclen/Izinova (BLI-800) has successfully completed its European decentralized registration procedure involving 16 countries, paving the way for a launch of the product before the end of 2013.
The product will be indicated in adults for bowel cleansing prior to any procedure requiring a clean bowel (eg, bowel visualization including bowel endoscopy and radiology or surgical procedure). Each European Union member states (France [reference member state], Belgium, Czech Republic, Estonia, Germany, Greece, Italy, Latvia, Lithuania, Luxemburg, the Netherlands, Poland, Portugal, Romania, Spain and UK) should now adopt a national decision within 30 days. In practice, the grant of the national marketing authorization may vary from one to several months, Ipsen noted.
Jean Fabre, senior vice president Intercontinental Operations and Franchise “Primary Care” at Ipsen, stated: “The completion of the decentralized procedure for Eziclen/Izinova (BLI-800) is an important step forward to national marketing authorizations in Europe. The availability of Eziclen/Izinova (BLI-800) will provide physicians and patients with a valuable agent for pre-colonoscopy colonic cleansing, particularly in the screening of colorectal cancer. This decision gives also perspectives for our plant in Dreux where Eziclen/Izinova will be manufactured”.
In 2009, Ipsen acquired the exclusive manufacturing, marketing and distribution rights of Braintree’s proprietary formulation BLI-800. The agreement covers countries within the EU, Commonwealth of Independent States, selected Asian countries (including China) and some North African countries. This product was approved by US Food and Drug Administrtion in 2010 and is marketed as Suprep Bowel Prep Kit.
The European Medicines Agency (EMA) Approves Otsuka’s Aripiprazole (ABILIFY®) for the Treatment of Moderate to Severe Manic Episodes in Bipolar I Disorder in Adolescents

Aripiprazole (OPC-14597, OPC-31, BMS-337039) cas no 129722-12-9
Aripiprazole is a psychotropic drug that is available as ABILIFY® (aripiprazole) Tablets, ABILIFY DISCMELT® (aripiprazole) Orally Disintegrating Tablets, ABILIFY® (aripiprazole) Oral Solution, and ABILIFY® (aripiprazole) Injection, a solution for intramuscular injection.

Abilify 2mg tablets (US)
Aripiprazole is 7-[4-[4-(2,3-dichlorophenyl)-1- piperazinyl]butoxy]-3,4-dihydrocarbostyril. The empirical formula is C23H27Cl2N3O2 and its molecular weight is 448.38.
 |
ABILIFY Tablets are available in 2 mg, 5 mg, 10 mg, 15 mg, 20 mg, and 30 mg strengths. Inactive ingredients include cornstarch, hydroxypropyl cellulose, lactose monohydrate, magnesium stearate, and microcrystalline cellulose. Colorants include ferric oxide (yellow or red) and FD&C Blue No. 2 Aluminum Lake.
ABILIFY DISCMELT Orally Disintegrating Tablets are available in 10 mg and 15 mg strengths. Inactive ingredients include acesulfame potassium, aspartame, calcium silicate, croscarmellose sodium, crospovidone, crème de vanilla (natural and artificial flavors), magnesium stearate, microcrystalline cellulose, silicon dioxide, tartaric acid, and xylitol. Colorants include ferric oxide (yellow or red) and FD&C Blue No. 2 Aluminum Lake.

ABILIFY Oral Solution is a clear, colorless to light yellow solution available in a concentration of 1 mg/mL. The inactive ingredients for this solution include disodium edetate, fructose, glycerin, dl-lactic acid, methylparaben, propylene glycol, propylparaben, sodium hydroxide, sucrose, and purified water. The oral solution is flavored with natural orange cream and other natural flavors.
ABILIFY Injection is available in single-dose vials as a ready-to-use, 9.75 mg/1.3 Ml (7.5 mg/mL) clear, colorless, sterile, aqueous solution for intramuscular use only. Inactive ingredients for this solution include 150 mg/mL of sulfobutylether β-cyclodextrin (SBECD), tartaric acid, sodium hydroxide, and water for injection.
Wednesday, February 6, 2013
Otsuka Pharmaceutical Co. Ltd. announced today that the European Medicines Agency (EMA) has approved a label extension for aripiprazole for the treatment up to 12 weeks of moderate to severe manic episodes in Bipolar I Disorder in adolescents aged 13 and older.
| United States | 5006528 | 1994-10-20 | 2014-10-20 |
| United States | 7115587 | 2005-01-21 | 2025-01-21 |

Aripiprazole, 7-{4-[4-(2,3-dichlorophenyl)-1-piperazinyl]-butoxy}-3,4-dihydro carbostyril or 7-{4-[4-(2,3-dichlorophenyl)-1-piperazinyl]-butoxy}-3,4-dihydro-2 (1H)-quinolinone, is an atypical antipsychotic agent useful for the treatment of schizophrenia (U.S. Pat. No. 4,74,416 and U.S. Pat. No. 5,006,528). Schizophrenia is a common type of psychosis characterized by delusions, hallucinations and extensive withdrawal from others. Onset of schizophrenia typically occurs between the age of 16 and 25 and affects 1 in 100 individuals worldwide. It is more prevalent, than Alzheimer’s disease, multiple sclerosis, insulin-dependent diabetes and muscular dystrophy. Early diagnosis and treatment can lead to significantly improved recovery and outcome. Moreover, early therapeutic intervention can avert costly hospitalization.
Aripiprazole (Aripiprazole) is an atypical antipsychotic, on 15 November 2002 by the U.S. FDA clearance to market, its efficacy is through the dopamine D2 receptor and serotonin 5HT1A receptor partial agonist activity and serotonin 5HT2A receptor antagonism activity mediated common. With its unique mechanism of action and safety assessment, aripiprazole known as third-generation antipsychotic drugs.
[0003] Aripiprazole is a quinolinone derivative, developed by the Japanese company Otsuka Pharmaceutical, the chemical name
Is: 7 – {4 – [4 – (2,3 – dichlorophenyl)-1_ piperazinyl] butoxy} -3,4 – dihydro-quinolone, the following structural formula:
[0004]

[0005] For the preparation of aripiprazole, Japanese OtsukaPharmaceutical’s patent EP 0367141A2, and related patents US4234585, CN89108934 preparation methods described in 5. In addition, the patent CN1450056A, CN1562973A, CN1784385A, CN1680328A, CN1576273A, etc. describe some of these five Preparation
Method is very similar way. These preparation methods are direct or indirect use of 7 – hydroxy -3,4 – dihydro – quinolin-2 – one (HCS) that the key to higher prices of raw materials, and some methods involve harsh reaction conditions, poor selectivity, low yield, but also increases the cost of industrial production of the product.
[0006] Chinese patent CN1304373C preparation method is not described in the 7 – hydroxy-3 ,4 _ dihydro-2_ (1H) – quinoline
Quinolone intermediates for their preparation of the core reaction is as follows:
[0007]

[0008] This reaction is Friedel-Crafts alkylation reaction, there is a harsh reaction conditions, the yield is low, the reaction selectivity is poor, the shortcomings of high emissions, is not conducive to industrial mass production. SUMMARY OF THE INVENTION
[0009] In order to solve the above problems, the present invention provides a simple, high selectivity, high yield, low cost, environmentally friendly, easy to prepare industrialization aripiprazole and intermediates thereof.
[0010] The technical solution of the present invention, the present invention provides in one aspect a process for preparingaripiprazole novel intermediates.
[0011] The present invention, on the other hand provides a method for the preparation of intermediates.
[0012] The present invention provides the use of the other intermediates for preparing aripiprazole two new preparation methods.
[0013] Specifically, the present invention relates to novel intermediates, compounds of formula ⑴:
[0014]

[0015] wherein, R is selected from methyl, ethyl, propyl, isopropyl, butyl, t-butyl, benzyl and other common alkyl groups in any one, and preferably is ethyl.
[0016] Compound of formula ⑴: 3 – (4 – (4 – (4 – (2,3 _-dichlorophenyl)-piperazinyl) butoxy) _2_ nitrophenyl) propionate, is the following prepared by the procedure:
[0017] Step one, the acylation reaction: with 4 – methyl – 3 – nitro-phenol (VIII) and acetic anhydride as the raw material, DMAP as catalyst, to give 4 – methyl – 3 – nitrophenyl acetate ( VII).
[0018] wherein 4 – methyl – 3 – nitro-phenol (VIII), acetic anhydride, DMAP molar ratio is preferably 1: 1.0 to 1.4: 0.05, at room temperature, the reaction time is preferably 0.5 to 3 hours.
[0019] Step two, the bromination reaction: The resulting product, 4 to Step one – methyl – 3 – nitrophenyl acetate (VII), N-bromosuccinimide and benzoyl peroxide as a raw material , carbon tetrachloride solvent reflux, to give 4 – bromomethyl-3 – nitrophenyl acetate (VI).
[0020] wherein 4 – methyl – 3 – nitrophenyl acetate (VII), N-bromosuccinimide, benzoyl peroxide molar ratio is preferably 1: 1 to 1.2: 0.05, reaction time is preferably 4-18 hours.
[0021] Step three, instead of the reaction: in an appropriate solvent, adding an alkaline agent and diethyl malonate was stirred in an ice bath, was added dropwise step two the resulting product, 4 – bromomethyl-3 – nitrophenyl yl acetate (VI) solution after completion of the addition reaction of 1 to 3 hours to obtain a brown liquid product, 2 – (4_ acetoxy-2 – nitrobenzyl) malonate (V).
[0022], wherein the alkali agent is a common organic or inorganic base selected from sodium methoxide, sodium ethoxide, sodium hydride, sodium tert-butoxide or potassium tert-butoxide, preferably sodium tert-butoxide; the solvent is selected from tetrahydrofuran, methanol, ethanol, butanol, tert-butanol, toluene or N, N-dimethylformamide; 4 – bromomethyl-3 – nitrophenyl acetate (VI), alkaline agent and lipid diethyl molar ratio is preferably 1: 1.0 to 1.8: 1.0 to 1.4.
[0023] Step 4 Hydrolysis decarboxylation: the product obtained in Step Three 2 – (4_ acetoxy-2 – nitro-benzyl)-malonic acid diethyl ester (V) was added concentrated hydrochloric acid and a suitable solvent, heating and stirring reflux, to give a yellow solid product 3 – (4_ hydroxy-2 – nitrophenyl) propionic acid (IV).
[0024] wherein the solvent is selected from water, methanol, ethanol or acetic acid, water soluble solvent, was heated with stirring under reflux time is preferably 3 to 18 hours. [0025] Step five, the esterification reaction: the product obtained in step 4, 3 – (4 – hydroxy-2 – nitrophenyl) propionic acid (IV) was added to an appropriate solvent, the mixture was stirred in an ice bath, was added dropwise thionyl sulfone, after completion of the addition reaction of 1 to 3 hours, to give a pale brown liquid product 3 – (4 – hydroxy-2 – nitrophenyl) propionate (III).
[0026] wherein the solvent is selected from anhydrous methanol, ethanol, propanol, isopropanol, butanol, t-butanol, benzyl alcohol, alcohol and other common solvents.
[0027] Step VI substitution reaction: 1,4 – dibromobutane was added to an appropriate solvent and an alkaline reagent, heated to 50 ~ 100 ° C, the product obtained was added dropwise Step Five 3 – (4_ hydroxy – nitrophenyl) propionate (III) solution, after the addition was complete the reaction was kept 2 to 4 hours to obtain a brown liquid product 3 – (4 – (4 – bromo-butoxy)-2 – nitrophenyl) propionate (II).
[0028] wherein the solvent is selected from methanol, 95% ethanol, ethanol, acetonitrile and N, N-dimethylformamide, and the like; said alkaline agent is a common organic or inorganic weak base, such as triethylamine, pyridine, potassium carbonate, sodium carbonate, etc..
[0029] Step 7 condensation reaction: the product obtained in Step Six 3 – (4 – (4 – bromo-butoxy)-2 – nitrophenyl) propionate (II) adding a suitable solvent, (2,3 – dichlorophenyl)-piperazine hydrochloride 1_, alkaline reagents and catalysts, to obtain
The intermediate product 3 – (4 – (4 – (4 – (2,3 – dichlorophenyl)-piperazin-1 – yl) butoxy)-2 – nitrophenyl) propionate ⑴.
[0030] Among them, 3 – (4 – (4 – (4 – (2,3 _-dichlorophenyl)-piperazinyl) butoxy) _2_ nitrophenyl) propionate (I), (2, 3 – dichloro-phenyl)-piperazine hydrochloride 1_, alkaline reagents and catalysts, the four molar ratio is preferably 1: 0.9 to 1.0: 2.0 to 2.2: 0.05 to 0.5. The solvent is selected from methanol, ethanol and N, N-dimethylformamide, acetonitrile and the like. Step six of the alkaline reagent and alkaline reagent used in the same, said catalyst is a common low-iodine salts, such as sodium iodide, potassium iodide.
[0031] The present invention provides two other hand, the use of a compound of formula ⑴ preparing aripiprazole new method.
[0032] Method one: ⑴ intermediate compound of formula in an appropriate solvent in the acid or salt or a base in the presence of a reducing agent under the action of restoring ring closure reaction to obtain aripiprazole.
[0033] Method one reductive cyclization of the reducing agent used is iron, zinc, sodium sulfide, stannous chloride, and preferably iron; reaction solvent is selected from water, methanol, ethanol, ethyl acetate or in one or more of the mixed solvent; said acid is a common organic or inorganic acid, preferably acetic acid or hydrochloric acid; said salt is a common inorganic or organic salts selected from chloride, ferrous chloride, , ammonium sulfate, calcium chloride, zinc chloride, sodium chloride, sodium bromide or sodium acetate and the like; common said base is an inorganic base selected from sodium hydroxide, potassium hydroxide or sodium bicarbonate; the reduction ring-closing reaction temperature range of 30 ~ 140 ° C, preferably about 80 ° C; reaction time ranges from about 0.5 to 8 hours, preferably 2 hours.
[0034] Method two: ⑴ intermediate compound of formula in an appropriate solvent in the first catalyst, the reduction reaction, and then carried out in a suitable solvent can be prepared by cyclization of aripiprazole.
[0035] The reduction reaction of the second approach, the reducing agent is hydrogen or a carboxylic acid; the catalyst is selected from molybdenum, molybdenum dioxide or Raney nickel, preferably Raney nickel; the solvent is selected from methanol, ethanol, ethyl acetate or acetic acid, preferably ethanol; said ring-closing reaction of the solvent is selected from N, N-dimethylformamide, trichlorobenzene or xylene; reaction temperature range of 50 ~ 180 ° C, preferably about 70 ~ 150 ° C; reaction time the range of about 1 to 8 hours.
[0036] In summary, the present invention is described for preparing aripiprazole method in 4– methyl – 3 – nitro-phenol (VIII) as a starting material, by acetylation protected hydroxy, radical instead of 4 – bromomethyl-3 – nitrophenyl acetate (VI), the diethyl malonate and a nucleophilic substitution reaction to obtain 2 – (4_ acetoxy-2 – nitrobenzyl ) malonic acid diethyl ester (V), which is decarboxylated by hydrolysis, esterification, to give 3 – (4 – hydroxy-2 – nitrophenyl) propionate (III), the reaction product with dibromobutane an ether compounds, and with (2,3 – dichlorophenyl)-piperazine hydrochloride 1_ condensation, to give 3 – (4 – (4 – (4 – (2,3 – dichlorophenyl) piperazine -1 – yl) butoxy) -2 – nitrophenyl) propionate (I), and then by reductive cyclization step, or first reduced and then ring-closing reaction of aripiprazole. The synthetic route of the present invention is as follows: [0037]

According to Example 1 of Japanese Unexamined Patent Publication No. 191256/1990, anhydrous aripiprazole crystals are manufactured for example by reacting 7-(4-bromobutoxy)-3,4-dihydrocarbostyril with 1-(2,3-dichlorophenylpiperadine and recrystallizing the resulting raw anhydrousaripiprazole with ethanol. Also, according to the Proceedings of the 4th Japanese-Korean Symposium on Separation Technology (Oct. 6-8, 1996), anhydrousaripiprazole crystals are manufactured by heating aripiprazole hydrate at 80° C. However, the anhydrous aripiprazole crystals obtained by the aforementioned methods have the disadvantage of being significantly hygroscopic.
The hygroscopicity of these crystals makes them difficult to handle since costly and burdensome measures must be taken in order ensure they are not exposed to moisture during process and formulation. Exposed to moisture, the anhydrous form can take on water and convert to a hydrous form. This presents several disadvantages. First, the hydrous forms of aripiprazole have the disadvantage of being less bioavailable and less dissoluble than the anhydrous forms ofaripiprazole. Second, the variation in the amount of hydrous versus anhydrousaripiprazole drug substance from batch to batch could fail to meet specifications set by drug regulatory agencies. Third, the milling may cause the drug substance, Conventional Anhydrous Aripiprazole, to adhere so manufacturing equipment which may further result in processing delay, increased operator involvement, increased cost, increased maintenance, and lower production yield. Fourth, in addition to problems caused by introduction of moisture during the processing of these hygroscopic crystals, the potential for absorbance of moisture during storage and handling would adversely affect the dissolubility of aripiprazole drug substance. Thus shelf-life of the product could be significantly decreased and/or packaging costs could be significantly increased. It would be highly desirable to discover a form of aripiprazole that possessed low hygroscopicity thereby facilitating pharmaceutical processing and formulation operations required for producing dosage units of an aripiprazole medicinal product having improved shelf-life, suitable dissolubility and suitable bioavailability.
Also, Proceedings of the 4 the Japanese-Korean Symposium on Separation Technology (Oct. 6-8, 1996) state that, anhydrous aripiprazole crystals exist as type-I crystals and type-II crystals; the type-I crystals of anhydrous aripiprazolecan be prepared by recrystallizing from an ethanol solution of aripiprazole, or by heating aripiprazole hydrate at 80° C.; and the type-II crystals of anhydrousaripiprazole can be prepared by heating the type-I crystals of anhydrousaripiprazole at 130 to 140° C. for 15 hours.
By the aforementioned methods, anhydrous aripiprazole type-II crystals having high purity can not be easily prepared in an industrial scale with good repeatability.
Chemical Synthesis of Aripiprazole (active ingredient for Abilify)

Experimental Procedures for the preparation of Aripiprazole (Abilify, aripiprazole)
US 5,006,528 discloses process for the preparation of Aripiprazole in two steps The first step comprises synthesis of 7 -. (4-bromobutoxy) -3,4-dihydrocarbostyril (7-BBQ) by alkylating the hydroxy group of 7-hydroxy-3, 4 -dihydrocarbostyril (7-HQ) with 1 ,4-dibromobutane using potassium carbonate in water at reflux temperature for 3 hours to obtain 7-BBQ in 68% yield The resulting 7-BBQ is further reacted with 1 -. (2,3 – dichlorophenyl)-piperazine to obtain Aripiprazole.
Preparation of 7 – (4-Bromobutoxy) 3 ,4-dihydro-2 (1H) quinolinon ( 7 – (4-Bromobutoxy) 3 ,4-dihydrocarbostyril; 7-BBQ)
7-Hydroxy-3 ,4-dihydro-2 (1H)-quinolinone (aka 7-Hydroxy-3 ,4-dihydrocarbostyril, 60gm) and potassium carbonate (76.3 gm) were taken in acetonitrile (1200ml) at room temperature. To this tetra butyl ammonium iodide (13.7 gm) and 1 ,4-dibromobutane (238.5gm) were added and heated at 40 – 45 ° C for 24 hours Reaction mass was cooled upto room temperature and was filtered off The resulting filtrate was distilled off.. under vacuum. The resultant mass was cooled to 25-30 ° C and cyclohexane (300 ml) was added under stirring. The resulting solid was filtered off and was dried. The resulting solid was taken in water and was stirred for few minutes. The . solid was filtered and dried under vacuum at 55-60 ° C for 20 hours to obtain title compound mp 110.5-111 ° C; 1H NMR (DMSO-d6) ä 1.81 (2H, m,-CH2-), 1.95 (2H , m,-CH2-), 2.41 (2H, t, J) 7 Hz,-CH2CO-), 2.78 (2H, t, J) 7 Hz,-CH2-C-CO-), 3.60 (2H, t, J) 6 Hz,-CH2Br), 3.93 (2H, t, J) 6 Hz, O-CH2-), 6.43 (1H, d, J) 2.5 Hz), 6.49 (1H, dd, J) 2.5, 8 Hz ), 7.04 (1H, d, J) 8 Hz), 9.98 (1H, s, NHCO). Anal. (C13H16NO2Br) C, H, N.
Yield: 73-75%; Purity: 93-95%
Preparation of Aripiprazole (7 – {4 – [4 – (2,3-Dichlorophenyl) piperazin-1-yl] butoxy} 3 ,4-dihydroquinolin-2 (1H)-One)
7 – (4-Bromobutoxy)-l ,2,3,4-tetrahydroquinolin-2-one (50 gm) was taken in acetonitrile (500 ml) at 25-30 ° C. To this potassium carbonate (67.2 gm) and l – (2,3 – dichlorophenyl). piperazine hydrochloride (44.9gm) were added under stirring The reaction mixture was refluxed at 80-85 ° C for 8 hours The reaction mass was cooled to room temperature, filtered and the resulting solid was washed. with acetonitrile. To the resulting solid, water was added and was stirred. The solid was filtered off, washed with water and dried under vacuum at 75-80 ° C for 15 hrs. The resulting crude aripiprazole was crystallized from isopropyl alcohol and water to . obtain title compound Yield: 75-80%; Dimer Impurity: <0.1% 1H NMR:. DMSO-d6 d 9.96 [1H, s, NH]; 7.29 [2H, m, Ar]; 7.13 [1H, q, Ar ]; 7.04 [1H, d, Ar]; 6.49 [1H, dd, Ar]; 6.45 [1H, d, Ar]; 3.92 [2H, t,-CH2-O-]; 2.97 [4H, bb, 2 ( -CH2-)]; 2.78 [2H, t,-CH2-N2-)]; 2.39 [4H, m, 2 (-CH2-)]; 1.73 [2H, m, – CH2-]; 1.58 [2H, m .,-CH2-] IR: cm-1 3193; 2939; 2804; 1680; 1627; 1579; 1520; 1449; 1375; 1270; 1245; 1192; 1169; 1045; 965; 649; 869; 780; 712; 588 .
For the Process of references Aripiprazole (Abilify, Japanese: Oh, Bldg re phi, Ann reピplastic AKZO have suitable; Chinese: Ann-law who, aripiprazole)
Yasuo Oshiro, Seiji Sato, Nobuyuki Kurahashi, Tatsuyoshi Tanaka, Tetsuro Kikuchi, Katsura Tottori, Yasufumi Uwahodo, and Takao Nishi; Novel Antipsychotic Agents with Dopamine autoreceptor Agonist Properties: Synthesis and Pharmacology of 7 – [4 – (4-Phenyl-1- piperazinyl) butoxy] – 3,4-dihydro-2 (1H)-quinolinone Derivatives ; J. Med Chem. 1998, 41, 658-667.
Yasuo Oshiro, Seiji Sato, Nobuyuki Kurahashi; Carbostyril Derivatives , Otsuka Pharmaceutical Co., Ltd.;. U.S. Patent 5006528 ; Issue Date: Apr 9, 1991
BANDO, Takuji, YANO, Katsuhiko, FUKANA, Makoto, AOKI, Satoshi; Method for producing fine particles of aripiprazole anhydride crystals b; OTSUKA PHARMACEUTICAL CO, LTD, WO 2013002420 A1..
Yuanqiu Hui, Chen Hongwen, Qian Wen, firewood rain column, Xu Dan, Yang Zhimin, Tian Zhoushan; method for preparing high purity of aripiprazole; NJCTT Pharmaceutical Co., Ltd.; application number: 201210292382.0; Publication Number: CN102863377A; Publication date: 2013.01.09 After (The invention relates to the field of medicine and chemical industry, in particular to a method for preparing high purity of aripiprazole would join aripiprazole A solvent is heated, filtered, and the filtrate was added to a solvent B, low temperature mixing, filtration, the filter cake is suspended in water, adjusted to alkaline pH of the aqueous solution, filtration, high temperature vacuum dried to obtain a high-purity refined product Aripiprazole This method is simple, high purity, suitable for the industrial the large-scale application)
ZHENG Siji, LIU Xiaoyi, FU Linyong, TAN Bo, ZHOU Min:.. ARIPIPRAZOLE MEDICAMENT FORMULATION AND PREPARATION METHOD THEREFOR / FORMULATION DE MÉDICAMENT ARIPIPRAZOLE ET SON PROCÉDÉ DE PRÉPARATION / a aripiprazole pharmaceutical formulation and preparation method SHANGHAI ZHONGXI. PHARMACEUTICAL January 2013: WO 2013/000391
Zheng Si Ji, Liu Xiaoyi, Fulin Yong, Tan Bo, Zhou Min: A aripiprazole pharmaceutical formulation and preparation method; Shanghai Pharmaceutical Co., Ltd. and Western; Publication date: 2013.01.02: Application Number: CN 201210235157.3; Publication Number: CN102846543A (the invention provides a method for preparing aripiprazole pharmaceutical formulation, comprising the steps of: an acidic solution containing aripiprazole is dissolved in the acidulant, to obtain an acidic solution containing the drug; Thereafter, the resulting drug-containing acidic solution alkalizing agents and materials prepared by wet granulation or suspension to give aripiprazole pharmaceutical formulation; said excipients include antioxidants)
Zheng Si Ji; Tan wave; Fulin Yong; Liu Xiaoyi; Yuanshao Qing; Cao Zhihui; aripiprazole Ⅰ type microcrystalline, aripiprazole solid preparation and preparation methods; application number: 201110180032.0; Publication Number: CN102850268A; Publication Date: 2013.01.02
Cai Fu Bo, Qin Xinrong, Du Xiaochun, Li Ling; kind of aripiprazole improved method of synthesis; Chengdu Nakasone Pharmaceutical Group Co., Ltd.; Application Number: 200910058148.X; Publication Number: CN101781246A; Publication date: 2010.07.21 (the invention provides a method of synthesis of aripiprazole improved method according to the modified method of the present invention, aripiprazole into the etherification reaction and condensation reaction of two-step synthesis, by an etherification reaction in the quinolone compound and at least 6-fold molar equivalents of 1,4 – dihalo-butane reacted with a non-polar solvent ether aripiprazole precipitate, and recovering 1,4 – dihalo-butane recycling; azeotropic condensation reaction of a ketone to be / water mixture as solvent, aripiprazole etherified with a piperazine compound or a salt thereof in the presence of a base under reflux and alkaline metal iodide compound conditions, the amount of water added to the end of the reaction, cooling crystallization, filtration, and dried to give aripiprazole. improved high yield synthesis of high purity, step simple, low cost, suitable for industrial production.)
GUPTA, Vijay Shankar, KUMAR, Pramod, VIR, Dharam; Process for producing aripiprazole in anhydrous type i crystals; JUBILANT LIFE SCIENCES LIMITED; WO 2012131451 A1
SRIVASTAVA JAYANT GUPTA Vijay Shankar;. Improved process for the preparation of 7 (4-bromobutoxy) 3,4-dihydrocarbostyril, a precursor of aripiprazole; wo2011030213 A1
No Generic Abilify in the US until April 2015
On May 7, 2012, The US Court of Appeals for the Federal Circuit ruled in favor of Otsuka Pharmaceutical Co., Ltd. In its patent litigation against several companies including Israel-based Teva and Weston, Ontario-based Apotex seeking FDA approval to market generic copies of Abilify ®.. The Federal Circuit Affirmed a Decision of the U.S. District Court for the District of New Jersey Holding that the asserted claims ofU.S. Patent No. 5,006,528 Covering aripiprazole, the active Ingredient in Abilify ®, are Valid, THUS Maintaining Patent and Regulatory Protection for Abilify ® in the U.S. until at least April 20, 2015 . The Case is Otsuka Pharma Co.. V. sand Inc.., 2011-1126 and 2011-1127, US Court of Appeals for the Federal Circuit (Washington). The lower court case is Otsuka Pharmaceutical Co. v. Sandoz Inc., 07cv1000, US District Court for the District of New Jersey (Trenton).
Chemical Name for Aripiprazole (Abilify for active Ingredient): 7 – {4 – [4 – (2,3-Dichlorophenyl) piperazin-1-yl] butoxy} 3 ,4-dihydroquinolin-2 (1H)-One
CAS Number 129722 -12-9
aripiprazole chemical name 7 – [4 – [4 – (2,3 – dichlorophenyl) -1 – piperazinyl] butoxy] -3,4 – dihydro-2 ( 1H) – quinolinone
Aripiprazole (, Aripiprazole, Abilify) is an atypical antipsychotic medication for the quinoline derivatives, aripiprazole is a dopamine system stabilizer first, positive and schizophrenia negative symptoms have a significant effect. For the treatment of schizophrenia, the development of Otsuka Pharmaceutical Co., Ltd., in November 15, 2002 by the U.S. Food and Drug Administration (FDA) approval in the U.S., domestic aripiprazole has (Booz clear (brisking, manufacturers : Chengdu Nakasone Pharmaceutical), Austrian (Manufacturer: Shanghai Pharmaceutical Co., Ltd. and Western)) have been approved by the listing in China. On sale in the United States where the law by Bristol-Myers Squibb is responsible. An law where the main patent protection in the United States, and more than three-quarters of its sales from the U.S., patent will expire in April 2015.
Aripiprazole synthetic route
7 – hydroxy-3 ,4. Dihydro -2 (1H) – quinolinone as a starting material, 1,4. Dibromobutane ether to give 7 – (4 – Bromo-butoxy) -3,4 – dihydro – 2 (1H) quinolinone, and then with 1 – (2,3 – dichlorophenyl) piperazine acid condensation aripiprazole (7 – [4 – [4 – (2,3 – dichlorophenyl) -1 – piperazinyl] butoxy] -3,4 – dihydro -2 (1H) – quinolinone)
Aripiprazole preparation method
7 – (4 – Bromo-butoxy) -3,4 – dihydro -2 (1H) – quinolone
A reaction flask was added 7 – hydroxy – 3,4 – dihydro -2 (1H) – quinolone 32.6 g (0.2mol), 1,4 – dibromo butane 129.5g (0.6mol), 11.2% KOH solution 250ml (0.5mol) and DMF975ml, was heated to 60 º C for 2h diluted with 1L water, the aqueous layer with ethyl acetate. acetate (300ml × 2) and the combined organic layers were washed with water, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to recover the solvent, the residue was recrystallized from isopropanol, to give 7 – (4 – Bromo-butoxy) – 3,4 – dihydro -2 (1H) – quinolone 38.7g, yield 68%, mp108 ~ 110 º C.
Synthesis of aripiprazole
in the reaction flask was added 7 – (4 – Bromo-butoxy) -3,4 – dihydro -2 (1H) – quinolone, 29.8g (0.1mol), KI25g (0.15mol) 95% Ethanol 596ml, stirred and heated to 60 º C, was added N-2 30min after 3 – dichlorophenyl piperazine 23.1g (0.1mol) and triethylamine 20ml (0.15mol), stirred for 8h at 60 º C the mixture is filtered. crystallization filtrate was cooled, filtered and the filter cake was recrystallized twice from ethanol and dried to obtain aripiprazole 25.6g, yield 57%, mp138.9 ~ 139.6 º C.
emea
Aripiprazole is a new antipsychotic belonging to the class of atypical antipsychotic drugs. It has been proposed that aripiprazole antipsychotic action could be mediated through a combination of partial agonist action at dopamine D2 and serotonin 5-HT1A receptors and antagonism at serotonin 5-HT2A receptors
Aripiprazole is a quinolinone derivative with the chemical name 7-[4-[4-(2,3-dichlorophenyl)-1- piperazinyl]butoxy]- 3,4-dihydro-2(1H)-quinolinone.
The active substance does not contain any chiral centres and does not exhibit any optical isomerism.
Aripiprazole active substance is a white crystalline powder and is practically insoluble in water and its solubility is pH dependent. Therefore, a particle size effect on dissolution of the tablets can be expected. In order to ensure batch-to-batch consistency of the product, and to ensure adequate bioavailability, aripiprazole is subject to milling.
Aripiprazole can exist in several crystalline forms, Form I was chosen for the development and commercialisation.. Aripiprazole is synthesised by a 2-step process. In the first step, 7-hydroxy-3,4-dihydro-2(H)- quinolinone is transformed into an intermediate, which is reacted with 1-(2,3-dichlorophenyl) piperazine hydrochloride to obtain aripiprazole. The process, specifications and control methods are adequately described in the restricted section of the EDMF.
Schizophrenia is a major psychotic disorder. Its essential features consist of a mixture of characteristic
signs and symptoms that have been present for a significant length of time during a 1-month period (or
for a shorter time if successfully treated), with some signs of the disorder persisting for at least 6
months.
The characteristic symptoms of schizophrenia have often been conceptualized as falling into two
broad categories positive and negative (or deficit) symptoms with a third category, disorganized,
recently added because statistical analyses have revealed that it is a dimension independent of the
positive symptom category, where it was previously included. The positive symptoms include
delusions and hallucinations. Disorganized symptoms include disorganized speech (thought disorder)
and disorganized behaviour and poor attention. Negative symptoms include restricted range and
intensity of emotional expression (affective flattening), reduced thought and speech productivity
(alogia), anhedonia, and decreased initiation of goal-directed behaviour (avolition).
The onset of schizophrenia typically occurs during adolescence or early adulthood. It affects men and
women with equal frequency. The peak age at onset for males, however, is the early 20s, and for
women it is the late 20s and early 30s. The majority of patients alternate between acute psychotic
episodes and stable phases with full or partial remission. Inter-episode residual symptoms are
common. This often-chronic illness can be characterized by three phases that merge into one another
without absolute, clear boundaries between them. These phases, which form the structure for
integrating treatment approaches, are described below:
Acute phase. During this florid psychotic phase, patients exhibit severe psychotic symptoms, such as
delusions and/or hallucinations and severely disorganized thinking, and are usually unable to care for
themselves appropriately. Negative symptoms often become more severe as well.
Stabilization phase. During this phase, acute psychotic symptoms decrease in severity. This phase may
last for 6 or more months after the onset of an acute episode.
Stable phase. Symptoms are relatively stable and, if present at all, are almost always less severe than
in the acute phase. Patients can be asymptomatic; others may manifest non-psychotic symptoms, such
as tension, anxiety, depression, or insomnia. When negative (deficit) symptoms and/or positive
symptoms, such as delusions, hallucinations, or thought disorder, persist, they are often present in
attenuated, non-psychotic forms (e.g., circumstantiality rather than looseness, illusions rather than hallucinations, overvalued ideas rather than delusions).
There are a number of antipsychotics in use but none is ideal in particular because their safety profile is complex. The in vitro affinity profile of aripiprazole for dopamine and serotonin receptors is similar to the one of so-called atypical antipsychotics. It is postulated that aripiprazole’s mechanism of action is novel as it involves a combination of partial agonist action (agonist/antagonism) at dopamine D2 and serotonin 5-HT1A receptors and antagonism at serotonin 5-HT2A receptors.
Aripiprazole is a new antipsychotic belonging to the class of atypical antipsychotic drugs. It has been
proposed that aripiprazole antipsychotic action could be mediated through a combination of partial
agonist at dopamine D2 and serotonin 5-HT1A receptors and antagonism at serotonin 5-HT2A receptors.
The non-clinical characterization of aripiprazole as a dopamine D2 receptor partial agonist at pituitary
lactotrophs predicts a low potential to induce hyperprolactinemia in humans but it may be dose related.
Studies on the sedative liability of aripiprazole suggest a reduced sedative potential compared to
typical antipsychotics. The safety pharmacology profile of aripiprazole r
ZALTRAP Approved in the EU for Patients with Previously Treated Metastatic Colorectal Cancer

Ziv-aflibercept is a recombinant fusion protein consisting of Vascular Endothelial Growth Factor (VEGF)-binding portions from the extracellular domains of human VEGF Receptors 1 and 2 fused to the Fc portion of the human IgG1. Ziv-aflibercept is produced by recombinant DNA technology in a Chinese hamster ovary (CHO) K-1 mammalian expression system. Ziv-aflibercept is a dimeric glycoprotein with a protein molecular weight of 97 kilodaltons (kDa) and contains glycosylation, constituting an additional 15% of the total molecular mass, resulting in a total molecular weight of 115 kDa.
ZALTRAP is a sterile, clear, colorless to pale yellow, non-pyrogenic, preservative-free, solution for administration by intravenous infusion. ZALTRAP is supplied in single-use vials of 100 mg per 4 ml and 200 mg per 8 ml formulated as 25 mg/mL ziv-aflibercept in polysorbate 20 (0.1%), sodium chloride (100 mM), sodium citrate (5 mM), sodium phosphate (5 mM), and sucrose (20%), in Water for Injection USP, at a pH of 6.2.
5th feb 2013
Sanofi and Regeneron Pharmaceuticals, Inc. today announced that the European Commission (EC) has granted marketing authorization in the European Union for ZALTRAP 25mg/ml concentrate for solution for infusion in combination with irinotecan/5-fluorouracil/folinic acid (FOLFIRI) chemotherapy in adults with metastatic colorectal cancer (mCRC) that is resistant to or has progressed after an oxaliplatin-containing regimen. This decision was based on the efficacy and safety results of the VELOUR Phase 3 trial
“ZALTRAP is an important addition to the metastatic colorectal cancer treatment landscape and helps to fill a critical treatment gap,” said Eric Van Cutsem, M.D., Ph.D., University Hospitals Leuven, Belgium and lead investigator of the VELOUR study. “ZALTRAP is the first and only agent to demonstrate a survival improvement in a Phase 3 trial in patients previously treated with an oxaliplatin-based regimen who are being treated with FOLFIRI for their metastatic disease.” “I would like to thank the physicians, patients, and their families for their support in moving ZALTRAP through the clinical trial process leading to approval in Europe,” said Debasish Roychowdhury, M.D., Senior Vice President and Head, Sanofi Oncology. “We are thrilled to provide a new therapy that further extends the lives of patients with metastatic colorectal cancer and look forward to working with European health authorities to ensure patients have access to ZALTRAP.” In Europe, colorectal cancer is the most common cancer in both men and women and is the second leading cause of cancer death. In 2008, there were 436,000 new cases diagnosed and 212,000 deaths from colorectal cancer.[1] Commenting on the marketing authorization, George D. Yancopoulos, M.D., Ph.D., Chief Scientific Officer of Regeneron and President of Regeneron Laboratories, added: “The European approval of ZALTRAP provides a new option to address the unmet medical need in this patient population. There continues to be a need to develop new cancer therapies and Regeneron and Sanofi are committed to finding novel investigational treatments and combinations.” ZALTRAP received approval from the U.S. Food and Drug Administration (FDA) in August 2012 after a Priority Review, and marketing authorization applications for ZALTRAP are under review with other regulatory agencies worldwide
EU approves Lyxumia (Lixisenatide), SANOFI, for the treatment of type 2 diabetes
Sun feb3, 2013, Sanofi today announced its novel once daily GLP-1 – Lyxumia has been approved by EU authorities.Lyxumia is the fourth GLP-1 and the second once daily GLP-1 to reach the market. Lyxumia is distinguished from other GLP-1’s on the market by virtue of its superior post prandial glucoselowering impact. Like other GLP-1’s it has a positive impact on hypoglycemia rates and weight lowering.
READ OLD ARTICLE AT

str from chemblink Online Database of Chemicals from Around the World

OLD ARTICLE
Sanofi Receives Positive CHMP Opinion in the European Union for Once-Daily Lyxumia® (lixisenatide)
 Sanofi announced today that the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) has issued a positive opinion recommending the approval of once-daily Lyxumia® (lixisenatide) for the treatment of adults with type 2 diabetes mellitus to achieve glycemic control in combination with oral glucose-lowering medicinal products and/or basal insulin when these, together with diet and exercise, do not provide adequate glycemic control.
Sanofi announced today that the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) has issued a positive opinion recommending the approval of once-daily Lyxumia® (lixisenatide) for the treatment of adults with type 2 diabetes mellitus to achieve glycemic control in combination with oral glucose-lowering medicinal products and/or basal insulin when these, together with diet and exercise, do not provide adequate glycemic control.
 http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002445/smops/Positive/human_smop_000448.jsp&mid=WC0b01ac058001d127
http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002445/smops/Positive/human_smop_000448.jsp&mid=WC0b01ac058001d127Lixisenatide is a once-daily injectable GLP-1 receptor agonist that has been developed by Sanofi.[1] As of September 2010 it is in clinical trials for diabetes.[2]
Lixisenatide, a glucagon-like peptide-1 agonist (GLP-1), is in development for the treatment of patients with type 2 diabetes mellitus. Lixisenatide was discovered by Zealand Pharma A/S and the global rights are licensed to Sanofi. Lyxumia® is the intended trademark for lixisenatide. Lixisenatide is not currently approved or licensed anywhere in the world.
GLP-1 is a naturally-occurring peptide that is released within minutes of eating a meal. It is known to suppress glucagon secretion from pancreatic alpha cells and stimulate insulin secretion by pancreatic beta cells. GLP-1 receptor agonists are in development as an add-on treatment for type 2 diabetes and their use is endorsed by the European Association for the Study of Diabetes, the American Diabetes Association, the American Association of Clinical Endocrinologists and the American College of Endocrinology.
The GetGoal phase III clinical program will provide data for lixisenatide in adults with type 2 diabetes treated with various oral anti-diabetic agents or insulin. With ten trials in the program, GetGoal started in May 2008 and has enrolled more than 4,300 patients. To date, GetGoal-X, GetGoal-L, GetGoal-L Asia, GetGoal-Mono, GetGoal-S and GetGoal-F1 have reported positive top-line results supporting efficacy and safety for lixisenatide
- Christensen, M; Knop, FK; Holst, JJ; Vilsboll, T (2009). “Lixisenatide, a novel GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus”. IDrugs : the investigational drugs journal 12 (8): 503–13. PMID 19629885.
- http://www.genengnews.com/gen-news-highlights/positive-phase-iii-data-for-two-separate-type-2-diabetes-therapies-reported-at-esad/81243945/
Sanofi Submits Application for Regulatory Approval for Lyxumia
®
(lixisenatide) for the Treatment of Type 2 Diabetes in Japan
 http://en.sanofi.com/Images/30529_20120611_LYXUMIA-JAPAN_en.pdf
http://en.sanofi.com/Images/30529_20120611_LYXUMIA-JAPAN_en.pdf

| CAS No. | 320367-13-3 | ||
| Chemical Name: | Lixisenatide | ||
| Synonyms: | lixisenatide | ||
| CBNumber: | CB01518519 | ||
Molecular Formula:
|
C215H347N61O65S |
伯舒替尼 Bosutinib


Bosutinib Monohydrate (伯舒替尼)
(Bosulif®)
Approved sept4 2012 by FDA
PMDA SEPT26 2014
EMA MAR 27 2013
A kinase inhibitor indicated for the treatment of adult patients with Ph+ chronic myelogenous leukemia (CML).
WYETH INNOVATOR
PFIZER DEVELOPER

SKI-606; SK-606
CAS No.380843-75-4 (Free form)
CAS 918639-08-4(Bosutinib Monohydrate)
Bosutinib (rINN/USAN; codenamed SKI-606, marketed under the trade name Bosulif) is atyrosine kinase inhibitor undergoing research for use in the treatment of cancer. [1] [2]Originally synthesized by Wyeth, it is being developed by Pfizer.
。

Some commercial stocks of bosutinib (from sources other than the Pfizer material used for clinical trials) have recently been found to have the incorrect chemical structure, calling the biological results obtained with them into doubt.[3]
Bosutinib received US FDA approval on September 5, 2012 for the treatment of adult patients with chronic, accelerated, or blast phase Philadelphia chromosome-positive (Ph+)chronic myelogenous leukemia (CML) with resistance, or intolerance to prior therapy.[4][5][6]

Article
The European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) on January 17, 2013, adopts a positive opinion, recommending a conditional marketing authhorization for Pfizer’s orphan drug Bosulif (Bosutinib) for Chronic Leukemia (CML). Bosutinib receives orphan designation from the European Commission (EC) on August 4, 2010, for CML.
Pfizer receives FDA approval on September 4, 2012, for orphan drug Bosulif (Bosutinib) for CML. Pfizer receives on February 24, 2009, FDA Orphan Drug Designation (ODD) for Bosutinib for CML.
Per a September 2012 article in FierceBioTech.com, a Pfizer spokesperson says that “the drug will cost less than $8,200/month”/patient in the US. In other words, treatment will cost approximately $98,400/patient/year. Also per FierceBiotech,“Bosulif is the 3rd new medicine from Pfizer Oncology’s pipeline to be approved by the FDA in just 13 months ….”.
ARTICLE
Pfizer’s response to compound fraud spotlights quality issues
read ………http://www.rsc.org/chemistryworld/2015/12/pfizer-bogus-bosutinib-isomer-fraud-leukaemia-drug

Synthesis
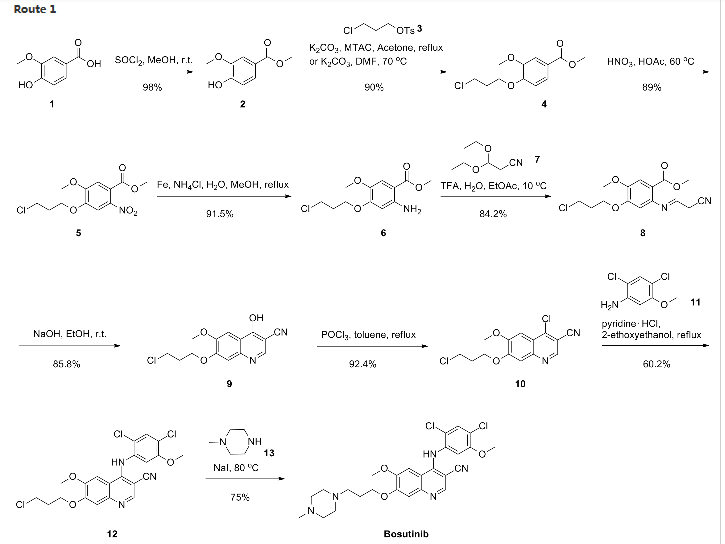

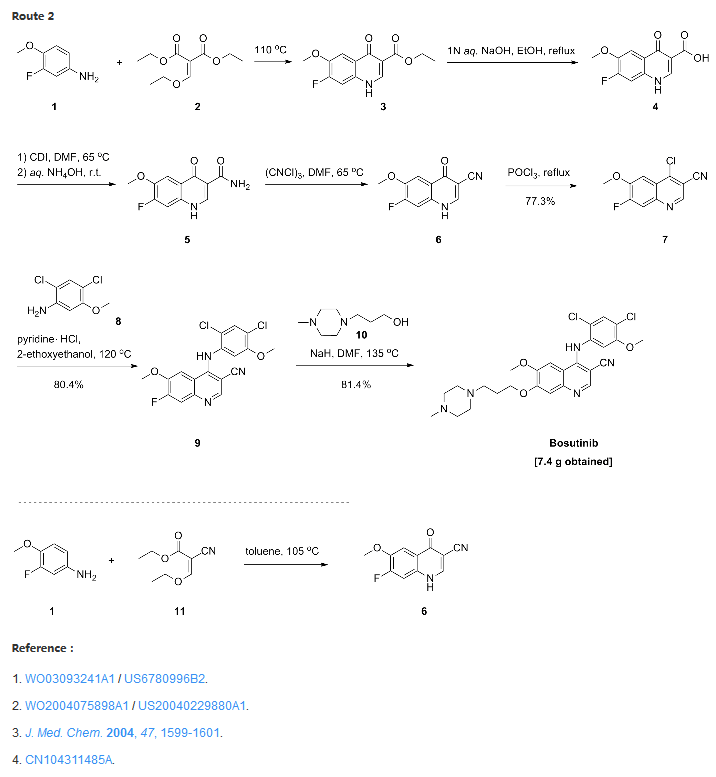
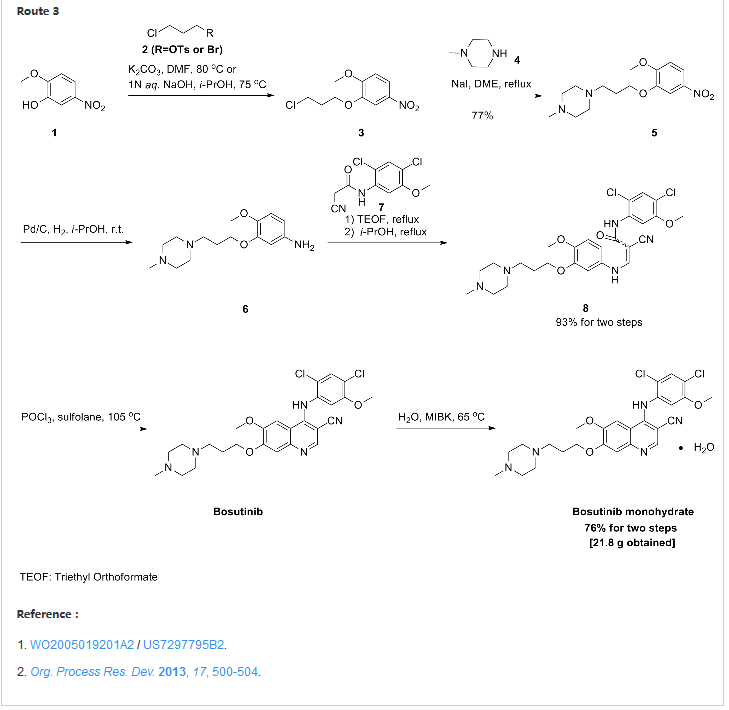



Confirmation of Bosutinib Structure; Demonstration of Controls To Ensure Product Quality

Nonbranded/unauthorized vendors had been manufacturing/selling what they described as bosutinib, while the material supplied was actually an isomer of bosutinib. This raised concerns within the worldwide research community around the established control strategies for bosutinib. This manuscript summarizes that the appropriate testing was in place to ensure product quality, along with additional experimentation that was performed to confirm that testing (methods) can differentiate the potential isomeric compounds. Testing includes the use of IR for identity confirmation of raw materials, material characterization by NMR, single crystal X-ray to confirm structure, and evaluation of several potential isomers by HPLC, melting point, and IR, thus demonstrating the control strategy needed to ensure the product controls.




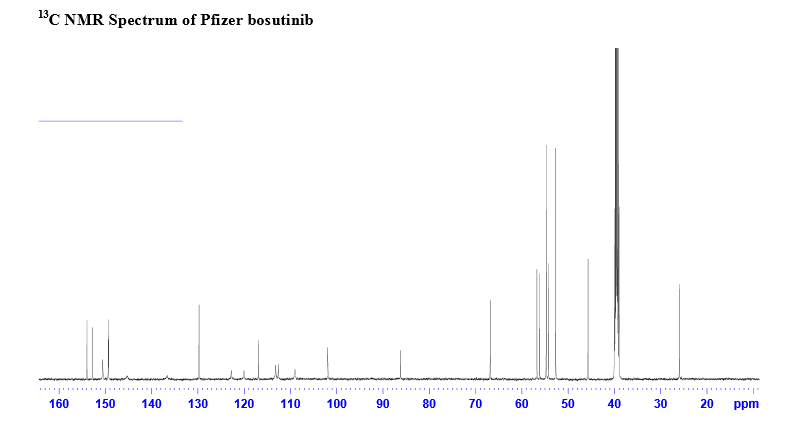
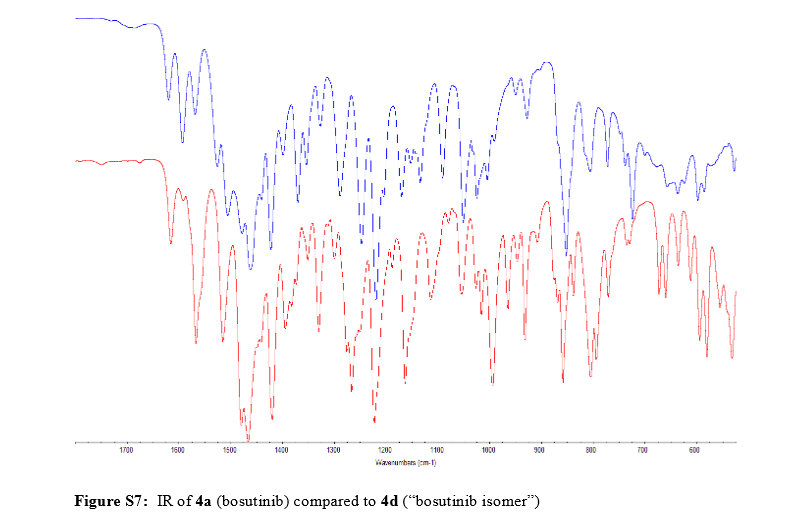
REFERENCES
- Puttini M, Coluccia AM, Boschelli F, Cleris L, Marchesi E, Donella-Deana A, Ahmed S, Redaelli S, Piazza R, Magistroni V, Andreoni F, Scapozza L, Formelli F, Gambacorti-Passerini C. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res. 2006 Dec 1;66(23):11314-22. Epub 2006 Nov 17.
- Vultur A, Buettner R, Kowolik C, et al. (May 2008). “SKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cells”.Mol. Cancer Ther. 7 (5): 1185–94. doi:10.1158/1535-7163.MCT-08-0126.PMC 2794837. PMID 18483306.
- Derek Lowe, In The Pipeline (blog), “Bosutinib: Don’t Believe the Label!”
- Cortes JE, Kantarjian HM, Brümmendorf TH, Kim DW, Turkina AG, Shen ZX, Pasquini R, Khoury HJ, Arkin S, Volkert A, Besson N, Abbas R, Wang J, Leip E, Gambacorti-Passerini C. Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood. 2011 Oct 27;118(17):4567-76. Epub 2011 Aug 24.
- Cortes JE, Kim DW, Kantarjian HM, Brümmendorf TH, Dyagil I, Griskevicus L, Malhotra H, Powell C, Gogat K, Countouriotis AM, Gambacorti-Passerini C. Bosutinib Versus Imatinib in Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia: Results From the BELA Trial. J Clin Oncol. 2012 Sep 4. [Epub ahead of print]
- “Bosulif Approved for Previously Treated Philadelphia Chromosome-Positive Chronic Myelogenous Leukemia”. 05 Sep 2012.
-
P Bowles et al, Org. Process Res. Dev., 2015, DOI: 10.1021/acs.oprd.5b00244
8 N M Levinson and S G Boxer, PLoS One, 2012, 7, e29828 (DOI: 10.1371/journal.pone.0029828)
9 N Beeharry et al, Cell Cycle, 2014, 13, 2172 (DOI: 10.4161/cc.29214)
update…………….
file:///C:/Users/Inspiron/Downloads/molecules-15-04261%20(1).pdf
Synthesis of Bosutinib from 3-Methoxy-4-hydroxybenzoic Acid
Jun 11, 2010 – Abstract: This paper reports a novel synthesis of bosutinib starting from … C-NMR, MS and the purities of all the compounds were determined …
4-(2,4-Dichloro-5-methoxyphenylamino]-6-methoxy-7-[3-(4-methylpiperazin-1-yl)propoxy]quinoline- 3-carbonitrile (10). A mixture of 7-(3-chloropropoxy)-4-(2,4-dichloro-5-methoxyphenylamino)-6-methoxyquinoline-3-carbonitrile (9, 0.328 g, 0.7 mmol) and sodium iodide (0.11 g, 0.70 mmol) in N-methylpiperazine (4 mL) was heated at 80 ºC for 12 h. The reaction mixture was concentrated in vacuo and partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The organic layer was washed with brine, dried over sodium sulfate, filtered, and concentrated in vacuo. The residue was purified by column chromatography, eluting with 30% methanol in dichloromethane. The fractions containing product were collected and concentrated in vacuo. Diethyl ether was added to the residue, and the light pink solid was collected by filtration (0.28 g, 75% yield, 98.7% HPLC purity): m.p. 116–120 ºC;
1H-NMR (DMSO-d6): 1.92–1.97 (m, 2H), 2.15 (s, 3H), 2.32–2.46 (m, 10H), 3.84 (s, 3H), 3.93 (s, 3H), 4.19 (t, J = 6.3 Hz, 2H), 7.31 (br s, 2H), 7.43 (s, 1H), 7.64 (s, 1H), 8.52 (s, 1H), 9.51 (s, 1H);
13C-NMR (CDCl3): 25.96, 45.68, 52.67, 52.67, 54.24, 54.72, 54.72, 56.01, 60.71, 66.87, 89.10, 101.66, 101.66, 109.12, 113.95, 117.17, 122.99, 122.99, 128.27, 137.88, 146.15, 148.13, 148.51, 149.50, 150.43, 153.03;
MS (ES) m/z 530.2, 532.2 (M+1).
…………………..
Structural and spectroscopic analysis of the kinase inhibitor …
files.figshare.com/337292/Figure_S2.doc
by NM Levinson – Cited by 39 – Related articles
NMR spectroscopy on bosutinib and the bosutinib isomer. As described in the main text, the 1H NMR spectra of the compounds we purchased from LC Labs and …
Figure S2. NMR experiments on bosutinib and the bosutinib isomer. A) The structure of bosutinib and a putative structure for the bosutinib isomer are shown. The blue numbers on the bosutinib structure represent the five aromatic proton-carbon pairs. The numbers on the aniline ring of the bosutinib isomer are 13C chemical shifts. B) NMR spectra. In the top left panel, 1H-13C HSQC spectra of bosutinib and the bosutinib isomer are shown. The thick black lines connect the peaks that arise from the equivalent proton-carbon pairs in the two compounds. The thin gray lines are intended to guide the eye to the corresponding peaks in the 1-dimensional spectra. The peaks for the five aromatic proton-carbon pairs in authentic bosutinib are indicated with large blue numbers. These putative assignments are based on 13C chemical shift predictions. The bottom panel shows the 1H NMR spectra of both compounds. The peak located at 7.34 ppm in the bosutinib isomer sample, which integrates to 2, is indicated. The colored numbers directly next to the peaks are the peak integrations. The panel on the upper right shows the aromatic region of the 13C NMR spectrum of the bosutinib isomer. The peak located at 123 ppm, which displays an integrated intensity of 2, is indicated.
//////////////Clc1c(OC)cc(c(c1)Cl)Nc4c(C#N)cnc3cc(OCCCN2CCN(CC2)C)c(OC)cc34
