
Home » EU SUBMISSION
Category Archives: EU SUBMISSION
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
EMA grants orphan drug designations to Alnylam’s ALN-AT3 for haemophilia treatment

EMA grants orphan drug designations to Alnylam’s ALN-AT3 for haemophilia treatment
Biopharmaceutical company Alnylam Pharmaceuticals has received orphan drug designations for ALN-AT3 from the European Medicines Agency (EMA) Committee to treat haemophilia A and B
SEE

May 13,2014
Alnylam Pharmaceuticals, Inc., a leading RNAi therapeutics company, announced today positive top-line results from its ongoing Phase 1 trial of ALN-AT3, a subcutaneously administered RNAi therapeutic targeting antithrombin (AT) in development for the treatment of hemophilia and rare bleeding disorders (RBD). These top-line results are being presented at the World Federation of Hemophilia (WFH) 2014 World Congress being held May 11 – 15, 2014 in Melbourne, Australia. In Part A of the Phase 1 study, human volunteer subjects received a single subcutaneous dose of ALN-AT3 and, per protocol, the maximum allowable level of AT knockdown was set at 40%. Initial results show that a single, low subcutaneous dose of ALN-AT3 at 0.03 mg/kg resulted in an up to 28-32% knockdown of AT at nadir that was statistically significant relative to placebo (p < 0.01 by ANOVA). This led to a statistically significant (p < 0.01) increase in peak thrombin generation, that was temporally associated and consistent with the degree of AT knockdown. ALN-AT3 was found to be well tolerated with no significant adverse events reported. With these data, the company has transitioned to the Multiple Ascending Dose (MAD) Part B of the study in moderate-to-severe hemophilia subjects. Consistent with previous guidance, the company plans to present initial clinical results from the Phase 1 study, including results in hemophilia subjects, by the end of the year. These human study results are the first to be reported for Alnylam’s Enhanced Stabilization Chemistry (ESC)-GalNAc conjugate technology, which enables subcutaneous dosing with increased potency, durability, and a wide therapeutic index. Further, these initial clinical results demonstrate a greater than 50-fold potency improvement with ESC-GalNAc conjugates relative to standard template chemistry conjugates.
“We are excited by these initial positive results for ALN-AT3 in the human volunteer ‘Part A’ of our Phase 1 study. Indeed, within the protocol-defined boundaries of single doses that provide no more than a 40% knockdown of AT in normal subjects, we were able to demonstrate a statistically-significant knockdown of AT of up to 28-32% and an associated increase in thrombin generation. Remarkably, this result was achieved at the lowest dose tested of 0.03 mg/kg, demonstrating a high and better than expected level of potency for ALN-AT3, our first ESC-GalNAc conjugate to enter clinical development,” said Akshay Vaishnaw, M.D., Ph.D., Executive Vice President and Chief Medical Officer of Alnylam. “With these results in hand, we are now proceeding to ‘Part B’ of the study, where we will administer multiple ascending doses to up to 18 patients with moderate-to-severe hemophilia A or B. Patients will receive three weekly doses, and we fully expect to achieve robust levels of AT knockdown as we dose escalate. In addition, we will aim to evaluate a once-monthly dosing regimen in future clinical studies, as we believe this could provide a highly attractive prophylactic regimen for patients. We look forward to sharing our detailed Phase 1 results, including data in hemophilia subjects, later this year, consistent with our original guidance.”
“There are several notable implications of these exciting initial results with ALN-AT3. First, ALN-AT3 now becomes the fourth program in our ‘Alnylam 5×15’ pipeline to demonstrate clinical activity. As such, these results increase our confidence level yet further across the entirety of our pipeline efforts, where we remain focused on genetically defined, liver-expressed disease targets with a modular and reproducible delivery platform. Moreover, these results with ALN-AT3 establish human proof of concept for our ESC-GalNAc conjugate technology, extending and broadening the human results we have previously shown with ALN-TTRsc which employs our standard template chemistry. Our ESC-GalNAc conjugate technology enables subcutaneous dosing with increased potency and durability and a wide therapeutic index, and has now become our primary approach for the delivery of RNAi therapeutics,” said John Maraganore, Ph.D., Chief Executive Officer of Alnylam. “Finally, the achievement of target knockdown at such a low dose of 0.03 mg/kg is unprecedented. Based on our evaluation of datasets from non-human primate (NHP) and human studies, these results demonstrate a 10-fold improved potency for ALN-AT3 as compared with NHP and a 50-fold improved potency in humans as compared with ALN-TTRsc. Based on data we announced earlier this week at TIDES, we believe that this increased potency is the combined result of enhanced stability for ESC-GalNAc conjugates and an attenuated nuclease environment in human tissue compared with other species. If these results extend to other ESC-GalNAc-siRNA conjugates, such as those in our complement C5 and PCSK9 programs, we believe we can expect highly potent clinical activities with very durable target knockdown effects.”
The ongoing Phase 1 trial of ALN-AT3 is being conducted in the U.K. as a single- and multi-dose, dose-escalation study comprised of two parts. Part A – which has now been completed – was a randomized, single-blind, placebo-controlled, single-dose, dose-escalation study, intended to enroll up to 24 healthy volunteer subjects. The primary objective of this part of the study was to evaluate the safety and tolerability of a single dose of ALN-AT3, with the potential secondarily to show changes in AT plasma levels at sub-pharmacologic doses. This part of the study evaluated only low doses of ALN-AT3, with a dose-escalation stopping rule at no more than a 40% level of AT knockdown. Based on the pharmacologic response achieved in this part of the study, only the lowest dose cohort (n=4; 3:1 randomization of ALN-AT3:placebo) was enrolled. Part B of the study is an open-label, multi-dose, dose-escalation study enrolling up to 18 people with moderate-to-severe hemophilia A or B. The primary objective of this part of the study is to evaluate the safety and tolerability of multiple doses, specifically three doses, of subcutaneously administered ALN-AT3 in hemophilia subjects. Secondary objectives include assessment of clinical activity as determined by knockdown of circulating AT levels and increase in thrombin generation at pharmacologic doses of ALN-AT3; thrombin generation is known to be a biomarker for bleeding frequency and severity in people with hemophilia (Dargaud, et al., Thromb Haemost; 93, 475-480 (2005)). In this part of the study, dose-escalation will be allowed to proceed beyond the 40% AT knockdown level.
In addition to reporting positive top-line results from the Phase 1 trial with ALN-AT3, Alnylam presented new pre-clinical data with ALN-AT3. First, in a saphenous vein bleeding model performed in hemophilia A (HA) mice, a single subcutaneous dose of ALN-AT3 that resulted in an approximately 70% AT knockdown led to a statistically significant (p < 0.0001) improvement in hemostasis compared to saline-treated HA mice. The improved hemostasis was comparable to that observed in HA mice receiving recombinant factor VIII. These are the first results in what can be considered a genuine bleeding model showing that AT knockdown with ALN-AT3 can control bleeding. Second, a number of in vitro studies were performed in plasma from hemophilia donors. Stepwise AT depletion in these plasma samples was shown to achieve stepwise increases in thrombin generation. Furthermore, it was shown that a 40-60% reduction of AT resulted in peak thrombin levels equivalent to those achieved with 10-15% levels of factor VIII in HA plasma and factor IX in hemophilia B (HB) plasma. These levels of factor VIII or IX are known to significantly reduce bleeding in hemophilia subjects. As such, these results support the hypothesis that a 40-60% knockdown of AT with ALN-AT3 could be fully prophylactic. Finally, a modified Activated Partial Thromboplastin Time (APTT) assay – an ex vivomeasure of blood coagulation that is significantly prolonged in hemophilia – was developed, demonstrating sensitivity to AT levels. Specifically, depletion of AT in HA plasma led to a shortening of modified APTT. This modified APTT assay can be used to routinely and simply monitor functional activity of AT knockdown in further ALN-AT3 clinical studies.
“The unmet need for new therapeutic options to treat hemophilia patients remains very high, particularly in those patients who experience multiple annual bleeds such as patients receiving replacement factor ‘on demand’ or patients who have developed inhibitory antibodies. Indeed, I believe the availability of a safe and effective subcutaneously administered therapeutic with a long duration of action would represent a marked improvement over currently available approaches for prophylaxis,” said Claude Negrier, M.D., head of the Hematology Department and director of the Haemophilia Comprehensive Care Centre at Edouard Herriot University Hospital in Lyon. “I continue to be encouraged by Alnylam’s progress to date with ALN-AT3, including these initial data reported from the Phase 1 trial showing statistically significant knockdown of antithrombin and increased thrombin generation, which has been shown to correlate with bleeding frequency and severity in hemophilia. I look forward to the advancement of this innovative therapeutic candidate in hemophilia subjects.”
About Hemophilia and Rare Bleeding Disorders
Hemophilias are hereditary disorders caused by genetic deficiencies of various blood clotting factors, resulting in recurrent bleeds into joints, muscles, and other major internal organs. Hemophilia A is defined by loss-of-function mutations in Factor VIII, and there are greater than 40,000 registered patients in the U.S. and E.U. Hemophilia B, defined by loss-of-function mutations in Factor IX, affects greater than 9,500 registered patients in the U.S. and E.U. Other Rare Bleeding Disorders (RBD) are defined by congenital deficiencies of other blood coagulation factors, including Factors II, V, VII, X, and XI, and there are about 1,000 patients worldwide with a severe bleeding phenotype. Standard treatment for hemophilia patients involves replacement of the missing clotting factor either as prophylaxis or on-demand therapy. However, as many as one third of people with severe hemophilia A will develop an antibody to their replacement factor – a very serious complication; these ‘inhibitor’ patients become refractory to standard replacement therapy. There exists a small subset of hemophilia patients who have co-inherited a prothrombotic mutation, such as Factor V Leiden, antithrombin deficiency, protein C deficiency, and prothrombin G20210A. Hemophilia patients that have co-inherited these prothrombotic mutations are characterized as having a later onset of disease, lower risk of bleeding, and reduced requirements for Factor VIII or Factor IX treatment as part of their disease management. There exists a significant need for novel therapeutics to treat hemophilia patients.
About Antithrombin (AT)
Antithrombin (AT, also known as “antithrombin III” and “SERPINC1″) is a liver expressed plasma protein and member of the “serpin” family of proteins that acts as an important endogenous anticoagulant by inactivating Factor Xa and thrombin. AT plays a key role in normal hemostasis, which has evolved to balance the need to control blood loss through clotting with the need to prevent pathologic thrombosis through anticoagulation. In hemophilia, the loss of certain procoagulant factors (Factor VIII and Factor IX, in the case of hemophilia A and B, respectively) results in an imbalance of the hemostatic system toward a bleeding phenotype. In contrast, in thrombophilia (e.g., Factor V Leiden, protein C deficiency, antithrombin deficiency, amongst others), certain mutations result in an imbalance in the hemostatic system toward a thrombotic phenotype. Since co-inheritance of prothrombotic mutations may ameliorate the clinical phenotype in hemophilia, inhibition of AT defines a novel strategy for improving hemostasis.
About GalNAc Conjugates and Enhanced Stabilization Chemistry (ESC)-GalNAc Conjugates
GalNAc-siRNA conjugates are a proprietary Alnylam delivery platform and are designed to achieve targeted delivery of RNAi therapeutics to hepatocytes through uptake by the asialoglycoprotein receptor. Alnylam’s Enhanced Stabilization Chemistry (ESC)-GalNAc-conjugate technology enables subcutaneous dosing with increased potency and durability, and a wide therapeutic index. This delivery platform is being employed in several of Alnylam’s genetic medicine programs, including programs in clinical development.
About RNAi
RNAi (RNA interference) is a revolution in biology, representing a breakthrough in understanding how genes are turned on and off in cells, and a completely new approach to drug discovery and development. Its discovery has been heralded as “a major scientific breakthrough that happens once every decade or so,” and represents one of the most promising and rapidly advancing frontiers in biology and drug discovery today which was awarded the 2006 Nobel Prize for Physiology or Medicine. RNAi is a natural process of gene silencing that occurs in organisms ranging from plants to mammals. By harnessing the natural biological process of RNAi occurring in our cells, the creation of a major new class of medicines, known as RNAi therapeutics, is on the horizon. Small interfering RNA (siRNA), the molecules that mediate RNAi and comprise Alnylam’s RNAi therapeutic platform, target the cause of diseases by potently silencing specific mRNAs, thereby preventing disease-causing proteins from being made. RNAi therapeutics have the potential to treat disease and help patients in a fundamentally new way.
About Alnylam Pharmaceuticals
Alnylam is a biopharmaceutical company developing novel therapeutics based on RNA interference, or RNAi. The company is leading the translation of RNAi as a new class of innovative medicines with a core focus on RNAi therapeutics as genetic medicines, including programs as part of the company’s “Alnylam 5x15TM” product strategy. Alnylam’s genetic medicine programs are RNAi therapeutics directed toward genetically defined targets for the treatment of serious, life-threatening diseases with limited treatment options for patients and their caregivers. These include: patisiran (ALN-TTR02), an intravenously delivered RNAi therapeutic targeting transthyretin (TTR) for the treatment of TTR-mediated amyloidosis (ATTR) in patients with familial amyloidotic polyneuropathy (FAP); ALN-TTRsc, a subcutaneously delivered RNAi therapeutic targeting TTR for the treatment of ATTR in patients with TTR cardiac amyloidosis, including familial amyloidotic cardiomyopathy (FAC) and senile systemic amyloidosis (SSA); ALN-AT3, an RNAi therapeutic targeting antithrombin (AT) for the treatment of hemophilia and rare bleeding disorders (RBD); ALN-CC5, an RNAi therapeutic targeting complement component C5 for the treatment of complement-mediated diseases; ALN-AS1, an RNAi therapeutic targeting aminolevulinate synthase-1 (ALAS-1) for the treatment of hepatic porphyrias including acute intermittent porphyria (AIP); ALN-PCS, an RNAi therapeutic targeting PCSK9 for the treatment of hypercholesterolemia; ALN-AAT, an RNAi therapeutic targeting alpha-1 antitrypsin (AAT) for the treatment of AAT deficiency-associated liver disease; ALN-TMP, an RNAi therapeutic targeting TMPRSS6 for the treatment of beta-thalassemia and iron-overload disorders; ALN-ANG, an RNAi therapeutic targeting angiopoietin-like 3 (ANGPTL3) for the treatment of genetic forms of mixed hyperlipidemia and severe hypertriglyceridemia; ALN-AC3, an RNAi therapeutic targeting apolipoprotein C-III (apoCIII) for the treatment of hypertriglyceridemia; and other programs yet to be disclosed. As part of its “Alnylam 5×15” strategy, as updated in early 2014, the company expects to have six to seven genetic medicine product candidates in clinical development – including at least two programs in Phase 3 and five to six programs with human proof of concept – by the end of 2015. Alnylam is also developing ALN-HBV, an RNAi therapeutic targeting the hepatitis B virus (HBV) genome for the treatment of HBV infection. The company’s demonstrated commitment to RNAi therapeutics has enabled it to form major alliances with leading companies including Merck, Medtronic, Novartis, Biogen Idec, Roche, Takeda, Kyowa Hakko Kirin, Cubist, GlaxoSmithKline, Ascletis, Monsanto, The Medicines Company, and Genzyme, a Sanofi company. In March 2014, Alnylam acquired Sirna Therapeutics, a wholly owned subsidiary of Merck. In addition, Alnylam holds an equity position in Regulus Therapeutics Inc., a company focused on discovery, development, and commercialization of microRNA therapeutics. Alnylam scientists and collaborators have published their research on RNAi therapeutics in over 200 peer-reviewed papers, including many in the world’s top scientific journals such as Nature, Nature Medicine, Nature Biotechnology, Cell, the New England Journal of Medicine, and The Lancet. Founded in 2002, Alnylam maintains headquarters in Cambridge, Massachusetts. For more information, please visit www.alnylam.com.
Eisai’s lenvatinib 兰伐替尼 レンバチニブ to get speedy review in Europe

Lenvatinib
For the treatment of patients with progressive radioiodine-refractory, differentiated thyroid cancer (RR-DTC).
European regulators have agreed to undertake an accelerated assessment of Eisai’s lenvatinib as a treatment for progressive radioiodine-refractory, differentiated thyroid cancer.
The drug, which carries Orphan Status in the EU, is to be filed “imminently” and could become the first in a new class of tyrosine kinase inhibitors, the drugmaker said.

Read more at: http://www.pharmatimes.com/Article/14-07-31/Eisai_s_lenvatinib_to_get_speedy_review_in_Europe.aspx#ixzz39OGhRHas
Lenvatinib was granted Orphan Drug Designation for thyroid cancer by the health authorities in Japan in 2012, and in Europe and the U.S in 2013. The first application for marketing authorization of lenvatinib in the world was submitted in Japan on June 2014. Eisai is planning to submit applications for marketing authorization in Europe and the U.S. in the second quarter of fiscal 2014.
Lenvatinib is an oral multiple receptor tyrosine kinase (RTK) inhibitor with a novel binding mode that selectively inhibits the kinase activities of vascular endothelial growth factor receptors (VEGFR), in addition to other proangiogenic and oncogenic pathway-related RTKs including fibroblast growth factor receptors (FGFR), the platelet-derived growth factor (PDGF) receptor PDGFRalpha, KIT and RET that are involved in tumor proliferation. This potentially makes lenvatinib a first-in-class treatment, especially given that it simultaneously inhibits the kinase activities of FGFR as well as VEGFR.
LENVATINIB BASE
COSY PREDICT
| Systematic (IUPAC) name | |
|---|---|
| 4-[3-chloro-4-(cyclopropylcarbamoylamino)phenoxy]-7-methoxy-quinoline-6-carboxamide | |
| Clinical data | |
| Legal status | ℞ Prescription only |
| Identifiers | |
| CAS number | |
| ATC code | None |
| PubChem | CID 9823820 |
| ChemSpider | 7999567 |
| UNII | EE083865G2 |
| Chemical data | |
| Formula | C21H19ClN4O4 |
| Mol. mass | 426.853 g/mol |
Lenvatinib (E7080) is a multi-kinase inhibitor that is being investigated for the treatment of various types of cancer by Eisai Co. It inhibits both VEGFR2 and VEGFR3 kinases.[1]
The substence was granted orphan drug status for the treatment of various types of thyroid cancer that do not respond toradioiodine; in the US and Japan in 2012 and in Europe in 2013[2] and is now approved for this use.

Clinical trials
Lenvatinib has had promising results from a phase I clinical trial in 2006[3] and is being tested in several phase II trials as of October 2011, for example against hepatocellular carcinoma.[4] After a phase II trial testing the treatment of thyroid cancer has been completed with modestly encouraging results,[5] the manufacturer launched a phase III trial in March 2011.[6]

Lenvatinib Mesilate
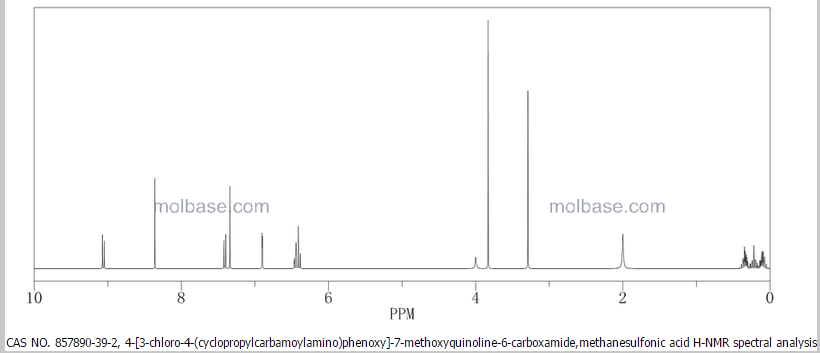
Molecular formula: C21H19ClN4O4,CH4O3S =523.0.
CAS: 857890-39-2.
UNII code: 3J78384F61.
About the Lenvatinib (E7080) Phase II Study
The open-label, global, single-arm Phase II study of multi-targeted kinase inhibitor lenvatinib (E7080) in advanced radioiodine (RAI)-refractory differentiated thyroid cancer involved 58 patients with advanced RAI refractory DTC (papillary, follicular or Hurthle Cell) whose disease had progressed during the prior 12 months. (Disease progression was measured using Response Evaluation Criteria in Solid Tumors (RECIST).) The starting dose of lenvatinib was 24 mg once daily in repeated 28 day cycles until disease progression or development of unmanageable toxicities.
2. About Thyroid Cancer
Thyroid cancer refers to cancer that forms in the tissues of the thyroid gland, located at the base of the throat or near the trachea. It affects more women than men and usually occurs between the ages of 25 and 65.
The most common types of thyroid cancer, papillary and follicular (including Hurthle Cell), are classified as differentiated thyroid cancer and account for 95 percent of all cases. While most of these are curable with surgery and radioactive iodine treatment, a small percentage of patients do not respond to therapy.
3. About Lenvatinib (E7080)
Lenvatinib is multi-targeted kinase inhibitor with a unique receptor tyrosine kinase inhibitory profile that was discovered and developed by the Discovery Research team of Eisai’s Oncology Unit using medicinal chemistry technology. As an anti-angiogenic agent, it inhibits tyrosine kinase of the VEGF (Vascular Endothelial Growth Factor) receptor, VEGFR2, and a number of other types of kinase involved in angiogenesis and tumor proliferation in balanced manner. It is a small molecular targeting drug that is currently being studied in a wide array of cancer types.
4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (additional name: 4-[3-chloro-4-(N′-cyclopropylureido)phenoxy]-7-methoxyquinoline-6-carboxamide) is known to exhibit an excellent angiogenesis inhibition as a free-form product, as described in Example 368 of Patent Document 1. 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide is also known to exhibit a strong inhibitory action for c-Kit kinase (Non-Patent Document 1, Patent Document 2).
However, there has been a long-felt need for the provision of a c-Kit kinase inhibitor or angiogenesis inhibitor that has high usability as a medicament and superior characteristics in terms of physical properties and pharmacokinetics in comparison with the free-form product of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide.
[Patent Document 1] WO 02/32872
[Patent Document 2] WO 2004/080462
[Non-Patent Document 1] 95th Annual Meeting Proceedings, AACR (American Association for Cancer Research), Volume 45, Page 1070-1071, 2004
………………………..
PATENT
http://www.google.co.in/patents/US8058474
EXAMPLES
Examples will now be described to facilitate understanding of the invention, but the invention is not limited to these examples.
Example 1Phenyl N-(2-chloro-4-hydroxyphenyl)carbamate

After suspending 4-amino-3-chlorophenol (23.7 g) in N,N-dimethylformamide (100 mL) and adding pyridine (23.4 mL) while cooling on ice, phenyl chloroformate (23.2 ml) was added dropwise below 20° C. Stirring was performed at room temperature for 30 minutes, and then water (400 mL), ethyl acetate (300 mL) and 6N HCl (48 mL) were added, the mixture was stirred and the organic layer was separated. The organic layer was washed twice with 10% brine (200 mL), and dried over magnesium sulfate. The solvent was removed to give 46 g of the title compound as a solid.
1H-NMR (CDCl3): 5.12 (1h, br s), 6.75 (1H, dd, J=9.2, 2.8 Hz), 6.92 (1H, d, J=2.8 Hz), 7.18-7.28 (4H, m), 7.37-7.43 (2H, m), 7.94 (1H, br s)
Example 21-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea

After dissolving phenyl N-(2-chloro-4-hydroxyphenyl)carbamate in N,N-dimethylformamide (100 mL), cyclopropylamine (22.7 mL) was added while cooling on ice and the mixture was stirred overnight at room temperature. Water (400 mL), ethyl acetate (300 mL) and 6N HCl (55 mL) were then added, the mixture was stirred and the organic layer was separated. The organic layer was washed twice with 10% brine (200 mL), and dried over magnesium sulfate. Prism crystals obtained by concentrating the solvent were filtered and washed with heptane to give 22.8 g of the title compound (77% yield from 4-amino-3-chlorophenol).
1H-NMR (CDCl3): 0.72-0.77 (2H, m), 0.87-0.95 (2H, m), 2.60-2.65 (1H, m), 4.89 (1H, br s), 5.60 (1H, br s), 6.71 (1H, dd, J=8.8, 2.8 Hz), 6.88 (1H, d, J=2.8 Hz), 7.24-7.30 (1H, br s), 7.90 (1H, d, J=8.8H)
Example 34-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide

To dimethylsulfoxide (20 mL) were added 7-methoxy-4-chloro-quinoline-6-carboxamide (0.983 g), 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea (1.13 g) and cesium carbonate (2.71 g), followed by heating and stirring at 70° C. for 23 hours. After the reaction mixture was allowed to cool down to room temperature, water (50 mL) was added, and the produced crystals were collected by filtration to give 1.56 g of the title compound (88% yield).
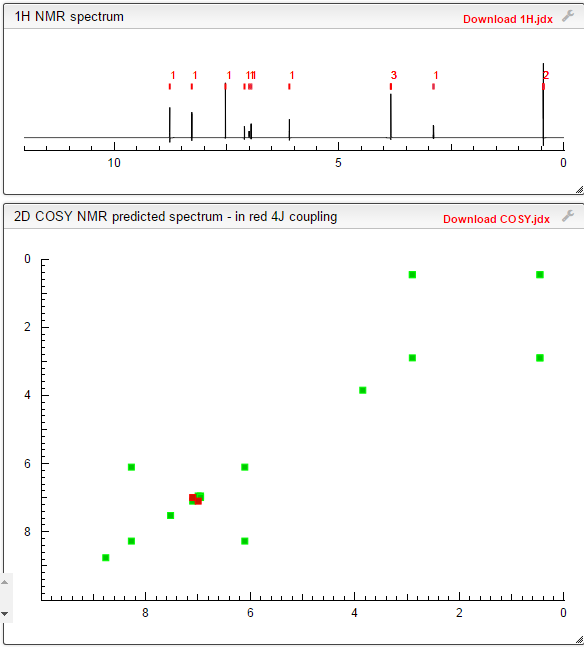
1H-NMR (d6-DMSO): 0.41 (2H, m), 0.66 (2H, m), 2.56 (1H, m), 4.01 (3H, s), 6.51 (1H, d, J=5.6 Hz), 7.18 (1H, d, J=2.8 Hz), 7.23 (1H, dd, J=2.8, 8.8 Hz), 7.48 (1H, d, J=2.8 Hz), 7.50 (1H, s), 7.72 (1H, s), 7.84 (1H, s), 7.97 (1H, s), 8.25 (1H, d, J=8.8 Hz), 8.64 (1H, s), 8.65 (1H, d, J=5.6 Hz)
Example 44-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide
In a reaction vessel were placed 7-methoxy-4-chloro-quinoline-6-carboxamide (5.00 kg, 21.13 mol), dimethylsulfoxide (55.05 kg), 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea (5.75 kg, 25.35 mol) and potassium t-butoxide (2.85 kg, 25.35 mol) in that order, under a nitrogen atmosphere. After stirring at 20° C. for 30 minutes, the temperature was raised to 65° C. over a period of 2.5 hours. After stirring at the same temperature for 19 hours, 33% (v/v) acetone water (5.0 L) and water (10.0 L) were added dropwise over a period of 3.5 hours. Upon completion of the dropwise addition, the mixture was stirred at 60° C. for 2 hours, and 33% (v/v) acetone water (20.0 L) and water (40.0 L) were added dropwise at 55° C. or higher over a period of 1 hour. After then stirring at 40° C. for 16 hours, the precipitated crystals were collected by filtration using a nitrogen pressure filter, and the crystals were washed with 33% (v/v) acetone water (33.3 L), water (66.7 L) and acetone (50.0 L) in that order. The obtained crystals were dried at 60° C. for 22 hours using a conical vacuum drier to give 7.78 kg of the title compound (96.3% yield).
…………………………
SYNTHESIS
1H NMR PREDICT

13 C NMR PREDICT


…………………..
PATENT
http://www.google.co.in/patents/US7253286
EX 368

Example 368
4-(3-Chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide
The title compound (22.4 mg, 0.052 mmol, 34.8%) was obtained as white crystals from phenyl N-(4-(6-carbamoyl-7-methoxy-4-quinolyl)oxy-2-chlorophenyl)carbamate (70 mg, 0.15 mmol) and cyclopropylamine, by the same procedure as in Example 11.
1H-NMR Spectrum (DMSO-d6) δ (ppm): 0.41 (2H, m), 0.66 (2H, m), 2.56 (1H, m), 4.01 (3H, s), 6.51 (1H, d, J=5.6 Hz), 7.18 (1H, d, J=2.8 Hz), 7.23 (1H, dd, J=2.8, 8.8 Hz), 7.48 (1H, d, J=2.8 Hz), 7.50 (1H, s), 7.72 (1H, s), 7.84 (1H, s), 7.97 (1H, s), 8.25 (1H, d, J=8.8 Hz), 8.64 (1H, s), 8.65 (1H, d, J=5.6 Hz).
The starting material was synthesized in the following manner.
Production Example 368-1Phenyl N-(4-(6-carbamoyl-7-methoxy-4-quinolyl)oxy-2-chlorophenyl)carbamate
The title compound (708 mg, 1.526 mmol, 87.4%) was obtained as light brown crystals from 4-(4-amino-3-chlorophenoxy)-7-methoxy-6-quinolinecarboxamide (600 mg, 1.745 mmol), by the same procedure as in Production Example 17.
1H-NMR Spectrum (CDCl3) δ (ppm): 4.14 (3H, s), 5.89 (1H, br), 6.50 (1H, d, J=5.6 Hz), 7.16 (2H, dd, J=2.4, 8.8 Hz), 7.22–7.30 (4H, m), 7.44 (2H, m), 7.55 (1H, s), 7.81 (1H, br), 8.31 (1H, d, J=8.8 Hz), 8.68 (1H, d, J=5.6 Hz), 9.27 (1H, s).
……………………
CRYSTALLINE FORM
http://www.google.co.in/patents/US7612208
Preparation Example 1
Preparation of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (1)
Phenyl N-(4-(6-carbamoyl-7-methoxy-4-quinolyl)oxy-2-chlorophenyl)carbamate (17.5 g, 37.7 mmol) disclosed in WO 02/32872 was dissolved in N,N-dimethylformamide (350 mL), and then cyclopropylamine (6.53 mL, 94.25 mmol) was added to the reaction mixture under a nitrogen atmosphere, followed by stirring overnight at room temperature. To the mixture was added water (1.75 L), and the mixture was stirred. Precipitated crude crystals were filtered off, washed with water, and dried at 70° C. for 50 min. To the obtained crude crystals was added ethanol (300 mL), and then the mixture was heated under reflux for 30 min to dissolve, followed by stirring overnight to cool slowly down to room temperature. Precipitated crystals was filtered off and dried under vacuum, and then further dried at 70° C. for 8 hours to give the titled crystals (12.91 g; 80.2%).
Preparation Example 2Preparation of 4-(3-cloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (2)
(1) Preparation of phenyl N-(2-chloro-4-hydroxyphenyl)carbamate

To a suspension of 4-amino-3-chlorophenol (23.7 g) in N,N-dimethylformamide (100 mL) was added pyridine (23.4 mL) while cooling in an ice bath, and phenyl chloroformate (23.2 mL) was added dropwise below 20° C. After stirring at room temperature for 30 min, water (400 mL), ethyl acetate (300 mL), and 6N-HCl (48 mL) were added and stirred. The organic layer was separated off, washed twice with a 10% aqueous sodium chloride solution (200 mL), and dried over magnesium sulfate. The solvent was evaporated to give 46 g of the titled compound as a solid.
- 1H-NMR Spectrum (CDCl3) δ(ppm): 5.12 (1H, br s), 6.75 (1H, dd, J=9.2, 2.8 Hz), 6.92 (1H, d, J=2.8 Hz), 7.18-7.28 (4H, m), 7.37-7.43 (2H, m), 7.94 (1H, br s).
(2) Preparation of 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea

To a solution of phenyl N-(2-chloro-4-hydroxyphenyl)carbamate in N,N-dimethylformamide (100 mL) was added cyclopropylamine (22.7 mL) while cooling in an ice bath, and the stirring was continued at room temperature overnight. Water (400 mL), ethyl acetate (300 mL), and 6N-HCl (55 mL) were added thereto, and the mixture was stirred. The organic layer was then separated off, washed twice with a 10% aqueous sodium chloride solution (200 mL), and dried over magnesium sulfate. The solvent was evaporated to give prism crystals, which were filtered off and washed with heptane to give 22.8 g of the titled compound (yield from 4-amino-3-chlorophenol: 77%).
- 1H-NMR Spectrum (CDCl3) δ(ppm): 0.72-0.77 (2H, m), 0.87-0.95 (2H, m), 2.60-2.65 (1H, m), 4.89 (1H, br s), 5.60 (1H, br s), 6.71 (1H, dd, J=8.8, 2.8 Hz), 6.88 (1H, d, J=2.8 Hz), 7.24-7.30 (1H, br s), 7.90 (1H, d, J=8.8 Hz)
(3) Preparation of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide
To dimethyl sulfoxide (20 mL) were added 7-methoxy-4-chloroquinoline-6-carboxamide (0.983 g), 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea (1.13 g) and cesium carbonate (2.71 g), and the mixture was heated and stirred at 70° C. for 23 hours. The reaction mixture was cooled to room temperature, and water (50 mL) was added, and the resultant crystals were then filtered off to give 1.56 g of the titled compound (yield: 88%).
Preparation Example 3Preparation of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (3)
7-Methoxy-4-chloroquinoline-6-carboxamide (5.00 kg, 21.13 mol), dimethyl sulfoxide (55.05 kg), 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea 5.75 kg, 25.35 mol) and potassium t-butoxide (2.85 kg, 25.35 mol) were introduced in this order into a reaction vessel under a nitrogen atmosphere. The mixture was stirred for 30 min at 20° C., and the temperature was raised to 65° C. over 2.5 hours. The mixture was stirred at the same temperature for 19 hours. 33% (v/v) acetone-water (5.0 L) and water (10.0 L) were added dropwise over 3.5 hours. After the addition was completed, the mixture was stirred at 60° C. for 2 hours. 33% (v/v) acetone-water (20.0 L) and water (40.0 L) were added dropwise at 55° C. or more over 1 hour. After stirring at 40° C. for 16 hours, precipitated crystals were filtered off using a nitrogen pressure filter, and was washed with 33% (v/v) acetone-water (33.3 L), water (66.7 L), and acetone (50.0 L) in that order. The obtained crystals were dried at 60° C. for 22 hours using a conical vacuum dryer to give 7.78 kg of the titled compound (yield: 96.3%).
1H-NMR chemical, shift values for 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamides obtained in Preparation Examples 1 to 3 corresponded to those for 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide disclosed in WO 02/32872.
Example 5
A Crystalline Form of the Methanesulfonate of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (Form A)
(Preparation Method 1)
In a mixed solution of methanol (14 mL) and methanesulfonic acid (143 μL, 1.97 mmol) was dissolved 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (700 mg, 1.64 mmol) at 70° C. After confirming the dissolution of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide, the reaction mixture was cooled to room temperature over 5.5 hours, further stirred at room temperature for 18.5 hours, and crystals were filtered off. The resultant crystals were dried at 60° C. to give the titled crystals (647 mg).
(Preparation Method 2)
In a mixed solution of acetic acid (6 mL) and methanesulfonic acid (200 μL, 3.08 mmol) was dissolved 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (600 mg, 1.41 mmol) at 50° C. After confirming the dissolution of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide, ethanol (7.2 mL) and seed crystals of a crystalline form of the methanesulfonate of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (Form A) (12 mg) were added in this order to the reaction mixture, and ethanol (4.8 mL) was further added dropwise over 2 hours. After the addition was completed, the reaction mixture was stirred at 40° C. for 1 hour then at room temperature for 9 hours, and crystals were filtered off. The resultant crystals were dried at 60° C. to give the titled crystals (545 mg).
Example 6A Crystalline Form of the Methanesulfonate of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (Form B)
A crystalline form of the acetic acid solvate of the methanesulfonate of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (Form I) (250 mg) obtained in Example 10 was dried under aeration at 30° C. for 3 hours and at 40° C. for 16 hours to give the titled crystals (240 mg)…………MORE IN PATENT
……………………………..
PATENT
https://www.google.com/patents/WO2014098176A1?cl=en
According to the present invention 4- (3-chloro-4- (cyclopropylamino-carbonyl) aminophenoxy) -7-methoxy-6-quinolinecarboxamide amorphous is excellent in solubility in water.
Example 1 4- (3-chloro-4- (cyclopropylamino-carbonyl) aminophenoxy) -7-methoxy-6-quinolinecarboxamide manufacture of amorphous amide
4- (3-chloro-4- (cyclopropylamino-carbonyl) amino phenoxy) -7-methoxy-6-quinolinecarboxamide B-type crystals (Patent Document 2) were weighed to 300mg, is placed in a beaker of 200mL volume, it was added tert- butyl alcohol (tBA) 40mL. This was heated to boiling on a hot plate, an appropriate amount of tBA to Compound A is dissolved, water was added 10mL. Then, the weakened heated to the extent that the solution does not boil, to obtain a sample solution. It should be noted, finally the solvent amount I was 60mL. 200mL capacity eggplant type flask (egg-plant shaped flask), and rotated in a state of being immersed in ethanol which had been cooled with dry ice. It was added dropwise a sample solution into the interior of the flask and frozen. After freezing the sample solution total volume, to cover the opening of the flask in wiping cloth, and freeze-dried. We got an amorphous A of 290mg.
Patent Document 2: US Patent Application Publication No. 2007/0117842 Patent specification 
………………………..
Paper
ACS Medicinal Chemistry Letters (2015), 6(1), 89-94
http://pubs.acs.org/doi/full/10.1021/ml500394m
……………..
Paper
Journal of Pharmaceutical and Biomedical Analysis (2015), 114, 82-87
http://www.sciencedirect.com/science/article/pii/S0731708515002940
KEEP WATCHING WILL BE UPDATED………….most of my posts are updated regularly
References
- Matsui, J.; Funahashi, Y.; Uenaka, T.; Watanabe, T.; Tsuruoka, A.; Asada, M. (2008). “Multi-Kinase Inhibitor E7080 Suppresses Lymph Node and Lung Metastases of Human Mammary Breast Tumor MDA-MB-231 via Inhibition of Vascular Endothelial Growth Factor-Receptor (VEGF-R) 2 and VEGF-R3 Kinase”. Clinical Cancer Research 14 (17): 5459–65.doi:10.1158/1078-0432.CCR-07-5270. PMID 18765537.
- “Phae III trial shows lenvatinib meets primary endpoint of progression free surival benefit in treatment of radioiodine-refactory differentiated thyroid cancer”. Eisai. 3 February 2014.
- Glen, H; D. Boss; T. R. Evans; M. Roelvink; J. M. Saro; P. Bezodis; W. Copalu; A. Das; G. Crosswell; J. H. Schellens (2007). “A phase I dose finding study of E7080 in patients (pts) with advanced malignancies”. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings Part I 25 (18S): 14073.
- ClinicalTrials.gov NCT00946153 Study of E7080 in Patients With Advanced Hepatocellular Carcinoma (HCC)
- Gild, M. L.; Bullock, M.; Robinson, B. G.; Clifton-Bligh, R. (2011). “Multikinase inhibitors: A new option for the treatment of thyroid cancer”. Nature Reviews Endocrinology 7 (10): 617–624.doi:10.1038/nrendo.2011.141. PMID 21862995.
- ClinicalTrials.gov NCT01321554 A Trial of E7080 in 131I-Refractory Differentiated Thyroid Cancer
UPDATES
EXTRAS……………
Martin Schlumberger et al. A phase 3, multicenter, double-blind, placebo-controlled trial of lenvatinib(E7080) in patients with 131I-refractory differentiated thyroid cancer (SELECT). 2014 ASCO Annual Meeting. Abstract Number:LBA6008. Presented June 2, 2014. Citation: J Clin Oncol 32:5s, 2014 (suppl; abstr LBA6008). Clinical trial information: NCT01321554.
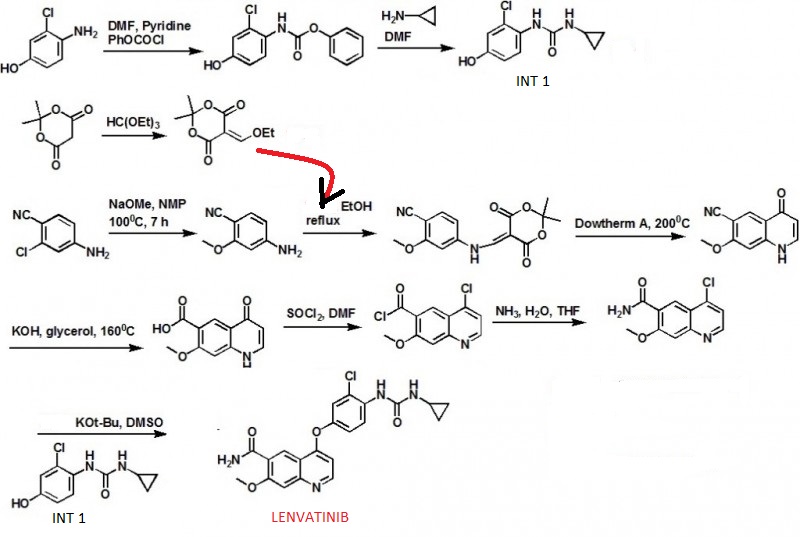
Bando, Masashi. Quinoline derivative-containing pharmaceutical composition. PCT Int. Appl. (2011), WO 2011021597 A1
Tomohiro Matsushima, four Nakamura, Kazuhiro Murakami, Atsushi Hoteido, Yusuke Ayat, Naoko Suzuki, Itaru Arimoto, Pinche Hirose, Masaharu Gotoda.Has excellent characteristics in terms of physical properties (particularly, dissolution rate) and pharmacokinetics (particularly, bioavailability), and is extremely useful as an angiogenesis inhibitor or c-Kit kinase inhibitor. US patent number US7612208 Also published as: CA2426461A1, CA2426461C, CN1308310C, CN1478078A, CN101024627A, DE60126997D1, DE60126997T2, DE60134679D1, DE60137273D1, EP1415987A1, EP1415987A4, EP1415987B1, EP1506962A2, EP1506962A3, EP1506962B1, EP1777218A1, EP1777218B1 , US7612092, US7973160, US8372981, US20040053908, US20060160832, US20060247259, US20100197911, US20110118470, WO2002032872A1, WO2002032872A8.Publication date: Aug 7, 2007 Original Assignee: Eisai Co., Ltd
Funahashi, Yasuhiro et al.Preparation of urea derivatives containing nitrogenous aromatic ring compounds as inhibitors of angiogenesis. US patent number US7253286, Also published as:CA2426461A1, CA2426461C, CN1308310C, CN1478078A, CN101024627A, DE60126997D1, DE60126997T2, DE60134679D1, DE60137273D1, EP1415987A1, EP1415987A4, EP1415987B1, EP1506962A2, EP1506962A3, EP1506962B1, EP1777218A1, EP1777218B1, US7612092, US7973160, US8372981, US20040053908, US20060160832, US20060247259, US20100197911, US20110118470, WO2002032872A1, WO2002032872A8.Publication date:Aug 7, 2007. Original Assignee:Eisai Co., Ltd
Sakaguchi, Takahisa; Tsuruoka, Akihiko. Preparation of amorphous salts of 4-[3-chloro-4-[(cyclopropylaminocarbonyl)amino]phenoxy]-7-methoxy-6-quinolinecarboxamide as antitumor agents. PCT Int. Appl. (2006), WO2006137474 A1 20061228.
Naito, Toshihiko and Yoshizawa, Kazuhiro. Preparation of urea moiety-containing quinolinecarboxamide derivatives. PCT Int. Appl., WO2005044788, 19 May 2005
Itaru Arimoto et al. Crystal of salt of 4-[3-chloro-4-(cyclopropylaminocarbonyl)amino-phenoxy]-7-methoxy-6-quinolinecarboxamide or solvate thereof and processes for producing these. PCT Int. Appl. (2005), WO2005063713 A1 20050714.
|
10-23-2009
|
ANTITUMOR AGENT FOR UNDIFFERENTIATED GASTRIC CANCER
|
|
|
10-2-2009
|
ANTI-TUMOR AGENT FOR MULTIPLE MYELOMA
|
|
|
8-21-2009
|
ANTITUMOR AGENT FOR THYROID CANCER
|
|
|
8-14-2009
|
THERAPEUTIC AGENT FOR LIVER FIBROSIS
|
|
|
2-27-2009
|
USE OF COMBINATION OF ANTI-ANGIOGENIC SUBSTANCE AND c-kit KINASE INHIBITOR
|
|
|
9-5-2008
|
Medicinal Composition
|
|
|
8-8-2007
|
Nitrogen-containing aromatic derivatives
|
|
|
5-25-2007
|
Polymorph of 4-[3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy]-7-methoxy-6- quinolinecarboxamide and a process for the preparation of the same
|
|
|
7-21-2006
|
Nitrogen-containing aromatic derivatives
|
|
|
6-23-2006
|
Use of sulfonamide-including compounds in combination with angiogenesis inhibitors
|
|
11-16-2011
|
UREA DERIVATIVE AND PROCESS FOR PREPARING THE SAME
|
|
|
8-10-2011
|
c-Kit kinase inhibitor
|
|
|
7-6-2011
|
Nitrogen-Containing Aromatic Derivatives
|
|
|
12-24-2010
|
COMBINED USE OF ANGIOGENESIS INHIBITOR AND TAXANE
|
|
|
9-24-2010
|
COMBINATION OF ANTI-ANGIOGENIC SUBSTANCE AND ANTI-TUMOR PLATINUM COMPLEX
|
|
|
4-30-2010
|
METHOD FOR PREDICTION OF THE EFFICACY OF VASCULARIZATION INHIBITOR
|
|
|
4-16-2010
|
METHOD FOR ASSAY ON THE EFFECT OF VASCULARIZATION INHIBITOR
|
|
|
3-24-2010
|
Urea derivative and process for preparing the same
|
|
|
2-26-2010
|
COMPOSITION FOR TREATMENT OF PANCREATIC CANCER
|
|
|
2-26-2010
|
COMPOSITION FOR TREATMENT OF UNDIFFERENTIATED GASTRIC CANCER
|
| US7253286 * | 18 Apr 2003 | 7 Aug 2007 | Eisai Co., Ltd | Nitrogen-containing aromatic derivatives |
| US20040053908 | 18 Apr 2003 | 18 Mar 2004 | Yasuhiro Funahashi | Nitrogen-containing aromatic derivatives |
| US20040242506 | 9 Aug 2002 | 2 Dec 2004 | Barges Causeret Nathalie Claude Marianne | Formed from paroxetine hydrochloride and ammonium glycyrrhyzinate by precipitation, spray, vacuum or freeze drying, or evaporation to glass; solid or oil; masks the bitter taste of paroxetine and has a distinctive licorice flavor; antidepressants; Parkinson’s disease |
| US20040253205 | 10 Mar 2004 | 16 Dec 2004 | Yuji Yamamoto | c-Kit kinase inhibitor |
| US20070004773 * | 22 Jun 2006 | 4 Jan 2007 | Eisai R&D Management Co., Ltd. | Amorphous salt of 4-(3-chiloro-4-(cycloproplylaminocarbonyl)aminophenoxy)-7-method-6-quinolinecarboxamide and process for preparing the same |
| US20070078159 | 22 Dec 2004 | 5 Apr 2007 | Tomohiro Matsushima | Has excellent characteristics in terms of physical properties (particularly, dissolution rate) and pharmacokinetics (particularly, bioavailability), and is extremely useful as an angiogenesis inhibitor or c-Kit kinase inhibitor |
| US20070117842 * | 22 Apr 2004 | 24 May 2007 | Itaru Arimoto | Polymorph of 4-[3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy]-7-methoxy-6- quinolinecarboxamide and a process for the preparation of the same |
| EP0297580A1 | 30 Jun 1988 | 4 Jan 1989 | E.R. SQUIBB & SONS, INC. | Amorphous form of aztreonam |
| JP2001131071A | Title not available | |||
| JP2005501074A | Title not available | |||
| JPS6422874U | Title not available | |||
| WO2002032872A1 | 19 Oct 2001 | 25 Apr 2002 | Itaru Arimoto | Nitrogenous aromatic ring compounds |
| WO2003013529A1 | 9 Aug 2002 | 20 Feb 2003 | Barges Causeret Nathalie Claud | Paroxetine glycyrrhizinate |
| WO2004039782A1 | 29 Oct 2003 | 13 May 2004 | Hirai Naoko | QUINOLINE DERIVATIVES AND QUINAZOLINE DERIVATIVES INHIBITING AUTOPHOSPHORYLATION OF Flt3 AND MEDICINAL COMPOSITIONS CONTAINING THE SAME |
| WO2004080462A1 | 10 Mar 2004 | 23 Sep 2004 | Eisai Co Ltd | c-Kit KINASE INHIBITOR |
| WO2004101526A1 | 22 Apr 2004 | 25 Nov 2004 | Itaru Arimoto | Polymorphous crystal of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-qunolinecarboxamide and method for preparation thereof |
| WO2005044788A1 | 8 Nov 2004 | 19 May 2005 | Eisai Co Ltd | Urea derivative and process for producing the same |
| WO2005063713A1 | 22 Dec 2004 | 14 Jul 2005 | Itaru Arimoto | Crystal of salt of 4-(3-chloro-4-(cyclopropylaminocarbonyl)amino-phenoxy)-7-methoxy-6-quinolinecarboxamide or of solvate thereof and processes for producing these |
| WO2006030826A1 | 14 Sep 2005 | 23 Mar 2006 | Eisai Co Ltd | Medicinal composition |
UPDATE………….
1H NMR PREDICT OF LENVATINIB BASE




European Medicines Agency recommends 39 medicines for human use for marketing authorisation in first half of 2014
![]()

10/07/2014
European Medicines Agency recommends 39 medicines for human use for marketing authorisation in first half of 2014
Thirty-nine medicines for human use were recommended for marketing authorisationby the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) in the first half of 2014, compared with 44 in first half of 2013 and 33 in first half of 2012.
This figure includes a number of new innovative medicines with the potential to meet unmet medical needs, treat diseases for which no treatments were previously available or bring significant added benefit to patients over existing therapies. Among these medicines are the anticancer medicines Mekinist (trametinib) and Gazyvaro (obinutuzumab), the anti-inflammatory* Entyvio (vedolizumab), the anti-infective Daklinza (daclatasvir), as well as Translarna (ataluren) and Sylvant (siltuximab), which are both intended for the treatment of rare conditions.
In parallel, the number of medicines recommended for approval via the European Union centralised procedure based on generic or informed consent applications has decreased compared with the first half of 2013 (6 versus 13).
More than two in three applicants received scientific advice from the CHMP during the development phase of their medicine, and for innovative medicines four in five applicants received such advice. This is a significant increase compared with the first half of 2013 (when one in two applicants received scientific advice), and mirrors the growing number of requests for scientific advice received by the Agency.
Confirming the trend observed in the past few years, the number of new medicines intended for the treatment of rare diseases is steadily increasing, providing treatments for patients who often have only few or no options. In the first half of 2014, eight medicines were recommended for the treatment of rare diseases. This number includes three medicines for which the CHMP recommended conditional approval but whose applications were withdrawn by the sponsor prior to a final decision by the European Commission![]() **.
**.
Conditional approval is one of the Agency’s mechanisms to provide early patient access to medicines that fulfill unmet medical needs or address life-threatening diseases. The CHMP also used this mechanism for the recommendation of the first treatment for Duchenne muscular dystrophy (Translarna), a life-threatening condition.
The CHMP granted two positive opinions after an accelerated assessment for the medicines Sylvant and Daklinza; this mechanism aims to speed up the assessment of medicines that are expected to be of major public health interest particularly from the point of view of therapeutic innovation.
The CHMP also gave an opinion on the use of a new combination product in the treatment of hepatitis C virus (HCV) infection in a compassionate use programme (ledipasvir and sofosbuvir). These programmes are intended to give patients with a life-threatening, long-lasting or seriously disabling disease access to treatments that are still under development. The treatment paradigm of hepatitis C is currently shifting rapidly, with the development of several new classes of direct-acting antivirals. By recommending the conduct of three compassionate use programmes and the marketing authorisation of three new medicines for HCV infection over the past eight months, the Agency is actively supporting this shift which is expected to bring significant added benefit to patients.
![]()

Notes
* On Friday 11 July 2014 at 11:00 the statement, ‘the anti-infectives Entyvio (vedolizumab) and Daklinza (daclatasvir)’ was corrected to ‘the anti-inflammatory Entyvio (vedolizumab), the anti-infective Daklinza (daclatasvir)’.
** The CHMP had recommended a conditional approval for Vynfinit (vintafolide) and its companion diagnostics Folcepri (etarfolatide) and Neocepri (folic acid). After authorisation, the company was to provide confirmatory data from an ongoing study with Vynfinit. However, before the authorisation process could be completed by the European Commission, preliminary data from this study became available which showed that the study could not confirm the benefit of Vynfinit in ovarian cancer patients. Therefore, the company terminated the study and decided to withdraw the applications.

Newly approved drugs: EMA presents figures
In the EU, the number of approved drugs is rising with a new active ingredient; however, stagnated, the number of newly approved generics.
read at
Teva Pharmaceutical has been given a green light by the European Commission (EC) for Lonquex, a rival to Amgen’s blockbuster Neulasta.

lipegfilgrastim
C 864 H 1369 N 225 O 258 S 9 [C 2 H 4 O] N
pegylated granulocyte colony stimulating factor; O3.133-[N5-(N-{[ω-methoxypoly (oxyethylene)] carbonyl} glycyl)-α-neuraminyl-(2 → 6)-α-D-galactopyranosyl]-L-methionyl -des-1-L-alanine-des-37-L-valine-des-38-L-serine-des-39-L-glutamic acid-human granulocyte colony-stimulating factor (G-CSF, pluripoietin)
Lonquex (lipegfilgrastim) has been approved to reduce the duration of neutropaenia (low white blood cell counts) and febrile neutropaenia in patients undergoing cytotoxic chemotherapy for cancer, and is given as a single subcutaneous dose per cycle of chemotherapy.
Like Neulasta (pegfilgrastim), Lonquex is a long-acting recombinant granulocyte colony-stimulating factor (G-CSF) and is dosed at the same frequency as Amgen’s drug.
http://www.pmlive.com/pharma_news/neulasta_rival_from_teva_cleared_in_eu_495953
Dolutegravir approved by the EU Commission (synthesis included in this post)

Dolutegravir
2H-Pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide, N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-, (4R,12aS)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide
(4R,12aS)-N-(2,4-difluorobenzyl)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide
Trade Name:Tivicay
Synonym:GSK1349572, S-349572, GSK572
Date of Approval: August 12, 2013 (US)
Indication:HIV infection
Drug class: Integrase strand transfer inhibitor
Company: ViiV Healthcare,GlaxoSmithKline
INNOVATOR …ViiV Healthcare
CAS number: 1051375-16-6
MF:C20H19F2N3O5
MW:419.4
Chemical Name: (4R,12aS)-N-[(2,4-difluorophenyl)methyl]-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a- hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide
Patent: US8129385
Patent expiration date: Oct 5, 2027
PCT patent application: W02006116764
Dolutegravir (DTG, GSK1349572) is an integrase inhibitor being developed for the treatment of human immunodeficiency virus (HIV)-1 infection by GlaxoSmithKline (GSK) on behalf of Shionogi-ViiV Healthcare LLC. DTG is metabolized primarily by uridine diphosphate glucuronyltransferase (UGT)1A1, with a minor role of cytochrome P450 (CYP)3A, and with renal elimination of unchanged drug being extremely low (< 1% of the dose).
The European Commission has on 21 January 2014 Dolutegravir (Tivicay, ViiV) permit as part of combination therapy for the treatment of HIV-infected persons over the age of 12 years.Dolutegravir (Tivicay, ViiV) is an integrase inhibitor, in combination with other antiretroviral drugs in adults and adolescents can be used from 12 years for the treatment of HIV infection.
Source: Communication from the European Commission
Dolutegravir[1] is a FDA-approved drug[2] for the treatment of HIV infection. Dolutegravir is an integrase inhibitor. Known as S/GSK1349572 or just “572” the drug is marketed as Tivicay[3] by GlaxoSmithKline (GSK). In February, 2013 the Food and Drug Administration announced that it would fast track dolutegravir’s approval process.[4] On August 13, 2013, dolutegravir was approved by the FDA. On November 4, 2013, dolutegravir was approved by Health Canada.[5]
The oral HIV integrase inhibitor S-349572 was originated by Shionogi-GlaxoSmithKline and Shionogi-ViiV Healthcare. In 2013, the product was approved and launched in the U.S. for the treatment of HIV-1 in adults and children aged 12 years and older, in combination with other antiretroviral agents. A positive opinion was received in the E.U for this indication and, in 2014, approval was attained in Europe for this indication. Registration is pending in Japan.
In 2013, orphan drug designation in Japan was assigned to the compound.
Dolutegravir is approved for use in a broad population of HIV-infected patients. It can be used to treat HIV-infected adults who have never taken HIV therapy (treatment-naïve) and HIV-infected adults who have previously taken HIV therapy (treatment-experienced), including those who have been treated with other integrase strand transfer inhibitors. Tivicay is also approved for children ages 12 years and older weighing at least 40 kilograms (kg) who are treatment-naïve or treatment-experienced but have not previously taken other integrase strand transfer inhibitors.[6]
Dolutegravir has also been compared head-to-head with a preferred regimen from the DHHS guidelines in each of the three classes (i.e. 1.) nuc + non-nuc, 2.) nuc + boosted PI, and 3.) nuc + integrase inhibitor).
SPRING-2 compared dolutegravir to another integrase inhibitor, raltegravir, with both coformulated with a choice of TDF/FTC orABC/3TC. After 48 weeks of treatment 88% of those on dolutegravir had less than 50 copies of HIV per mL compared to 85% in the raltegravir group, thus demonstrating non-inferiority.[9]
The FLAMINGO study has been presented at scientific meetings but as of early 2014 has not yet been published. It is an open-label trial of dolutegravir versus darunavir boosted with ritonavir. In this trial 90% of those on dolutegravir based regimens had viral loads < 50 at 48 weeks compared to 83% in the darunavir/r.[10] This 7% difference was statistically significant for superiority of the dolutegravir based regimens.
Another trial comparing dolutegravir to efavirenz, SINGLE, was the first trial to show statistical superiority to an efavirenz/FTC/TDF coformulated regimen for treatment naive patients.[11] After 48 weeks of treatment, 88% of the dolutegravir group had HIV RNA levels < 50 copies / mL versus 81% of the efavirenz group. This has led one commentator to predict that it may replace efavirenz as the first line choice for initial therapy as it can also be formulated in one pill, once-a-day regimens.[12]
Doultegravir has also been studied in patients who have been on previous antiretroviral medications. The VIKING trial looked at patients who had known resistance to the first generation integrase inhibitor raltegravir. After 24 weeks 41% of patients on 50mg dolutegravir once daily and 75% of patients on 50mg twice daily (both along with an optimized background regimen) achieved an HIV RNA viral load of < 50 copies per mL. This demonstrated that there was little clinical cross-resistance between the two integrase inhibitors. [13]
Dolutegravir (also known as S/GSK1349572), a second-generation integrase inhibitor under development by GlaxoSmithKline and its Japanese partner Shionogi for the treatment of HIV infection, was given priority review status from the US Food and Drug Administration (FDA) in February, 2013.
GlaxoSmithKline marketed the first HIV drug Retrovir in 1987 before losing out to Gilead Sciences Inc. (GILD) as the world’s biggest maker of AIDS medicines. The virus became resistant to Retrovir when given on its own, leading to the development of therapeutic cocktails.
The new once-daily drug Dolutegravir, which belongs to a novel class known as integrase inhibitors that block the virus causing AIDS from entering cells, is owned by ViiV Healthcare, a joint venture focused on HIV in which GSK is the largest shareholder.
Raltegravir (brand name Isentress) received approval by the U.S. Food and Drug Administration (FDA) on 12 October 2007, the first of a new class of HIV drugs, the integrase inhibitors, to receive such approval. it is a potent and well tolerated antiviral agent. However, it has the limitations of twice-daily dosing and a relatively modest genetic barrier to the development of resistance, prompting the search for agents with once-daily dosing.
Elvitegravir, approved by the FDA on August 27, 2012 as part of theelvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine fixed-dose combination pill (Quad pill, brand name Stribild) has the benefit of being part of a one-pill, once-daily regimen, but suffers from extensive cross-resistance with raltegravir.
Gilead’s Atripla (Emtricitabine/Tenofovir/efavirenz), approved in 2006 with loss of patent protection in 20121, is the top-selling HIV treatment. The $3.2 billion medicine combines three drugs in one pill, two compounds that make up Gilead’s Truvada (Emtricitabine/Tenofovir) and Bristol- Myers Squibb Co.’s Sustiva (Efavirenz).
A three-drug combination containing dolutegravir and ViiV’s older two-in-one treatment Epzicom(Abacavir/Lamivudine, marketed outside US as Kivexa) proved better than Gilead’s market-leading Atripla in a clinical trial released in July, 2012 (See the Full Conference Report Here), suggesting it may supplant the world’s best-selling AIDS medicine as the preferred front-line therapy. In the latest Phase III study, after 48 weeks of treatment, 88% of patients taking the dolutegravir-based regimen had reduced viral levels to the goal compared with 81% of patients taking Atripla. More patients taking Atripla dropped out of the study because of adverse events compared with those taking dolutegravir — 10% versus just 2% — which was the main driver of the difference in efficacy. The result was the second positive final-stage clinical read-out for dolutegravir, following encouraging results against U.S. company Merck & Co’s rival Isentress in April, 2012 (See the Conference Abstract Here)..
Dolutegravir is viewed by analysts as a potential multibillion-dollar-a-year seller, as its once-daily dosing is likely to be attractive to patients. The FDA is scheduled to issue a decision on the drug’s approval by August 17。
TIVICAY contains dolutegravir, as dolutegravir sodium, an HIV INSTI. The chemical name of dolutegravir sodium is sodium (4R,12aS)-9-{[(2,4-difluorophenyl)methyl]carbamoyl}-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazin-7-olate. The empirical formula is C20H18F2N3NaO5 and the molecular weight is 441.36 g/mol. It has the following structural formula:
 |
Dolutegravir sodium is a white to light yellow powder and is slightly soluble in water.
Each film-coated tablet of TIVICAY for oral administration contains 52.6 mg of dolutegravir sodium, which is equivalent to 50 mg dolutegravir free acid, and the following inactive ingredients: D-mannitol, microcrystalline cellulose, povidone K29/32, sodium starch glycolate, and sodium stearyl fumarate. The tablet film-coating contains the inactive ingredients iron oxide yellow, macrogol/PEG, polyvinyl alcohol-part hydrolyzed, talc, and titanium dioxide.
……………………………………
INTRODUCTION
Among viruses, human immunodeficiency virus (HIV), a kind of retrovirus, is known to cause acquired immunodeficiency syndrome (AIDS). The therapeutic agent for AIDS is mainly selected from a group of reverse transcriptase inhibitors (e.g., AZT, 3TC) and protease inhibitors (e.g., Indinavir), but they are proved to be accompanied by side effects such as nephropathy and the emergence of resistant viruses. Thus, the development of anti-HIV agents having the other mechanism of action has been desired.
On the other hand, a combination therapy is reported to be efficient in treatment for AIDS because of the frequent emergence of the resistant mutant. Reverse transcriptase inhibitors and protease inhibitors are clinically used as an anti-HIV agent, however agents having the same mechanism of action often exhibit cross-resistance or only an additional activity. Therefore, anti-HIV agents having the other mechanism of action are desired.
Under the circumstances above, an HIV integrase inhibitor has been focused on as an anti-HIV agent having a novel mechanism of action (Ref: Patent Documents 1 and 2). As an anti-HIV agent having such a mechanism of action, known are carbamoyl-substituted hydroxypyrimidinone derivative (Ref: Patent Documents 3 and 4) and carbamoyl-substituted hydroxypyrrolidione derivative (Ref: Patent Document 5). Further, a patent application concerning carbamoyl-substituted hydroxypyridone derivative has been filed (Ref: Patent Document 6, Example 8).
Other known carbamoylpyridone derivatives include 5-alkoxypyridine-3-carboxamide derivatives and γ-pyrone-3-carboxamide derivatives, which are a plant growth inhibitor or herbicide (Ref: Patent Documents 7-9).
Other HIV integrase inhibitors include N-containing condensed cyclic compounds (Ref: Patent Document 10).
- [Patent Document 1] WO03/0166275
- [Patent Document 2] WO2004/024693
- [Patent Document 3] WO03/035076
- [Patent Document 4] WO03/035076
- [Patent Document 5] WO2004/004657
- [Patent Document 6] JP Patent Application 2003-32772
- [Patent Document 7] JP Patent Publication 1990-108668
- [Patent Document 8] JP Patent Publication 1990-108683
- [Patent Document 9] JP Patent Publication 1990-96506
- [Patent Document 10] WO2005/016927
-
Patent Document 1 describes compounds (I) and (II), which are useful as anti-HIV drugs and shown by formulae:

-
This document describes the following reaction formula as a method of producing compound (I).


-
Furthermore, Patent Documents 2 to 6 describe the following reaction formula as an improved method of producing compound (I).


-
- [Patent Document 1] International publication No.2006/116764 pamphlet
- [Patent Document 2] International publication No.2010/011812 pamphlet
- [Patent Document 3] International publication No.2010/011819 pamphlet
- [Patent Document 4] International publication No.2010/068262 pamphlet
- [Patent Document 5] International publication No.2010/067176 pamphlet
- [Patent Document 6] International publication No.2010/068253 pamphlet
- [Patent Document 7] US Patent 4769380A
- [Patent Document 8] International applicationPCT/JP2010/055316
- [PATENT DOCUMENTS]
[NON-PATENT DOCUMENTS]
-
- [Non-Patent Document 1] Journal of Organic Chemistry, 1991, 56(16), 4963-4967
- [Non-Patent Document 2] Science of Synthesis, 2005, 15, 285-387
- [Non-Patent Document 3] Journal of Chemical Society Parkin Transaction. 1, 1997, Issue. 2, 163-169
-



…………………………………………
Dolutegravir synthesis (EP2602260, 2013). LiHMDS as the non-nucleophilic strong base pulling compound 1 carbonyl group proton alpha position with an acid chloride after 2 and ring closure reaction to obtain 3 , 3 via primary amine 4 ring opening ring closure to obtain 5 , NBS the bromine under acidic conditions to obtain aldehyde acetal becomes 6 , 6 of the aldehyde and amino alcohols 7 and turn off the condensation reaction obtained by the ring 8 , alkaline hydrolysis 8 of bromine into a hydroxyl group and hydrolyzable ester obtained 9 after the 10 occurred acid condensation Dolutegravir.
………………………………………………………
Synthesis of Dolutegravir (S/GSK1349572, GSK1349572)

………………………
SYNTHESIS

2H-Pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide, N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-, (4R,12aS) ………..dolutegravir
PATENT US8129385

Desired isomer
Example Z-1
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt

a)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide. To a solution of 16a (409 mg, 0.87 mmol) in dichloroethane (20 mL) was added (2R)-2-amino-1-propanol (0.14 mL, 1.74 mmol) and 10 drops of glacial acetic acid. The resultant solution was heated at reflux for 2 h. Upon cooling, Celite was added to the mixture and the solvents removed in vacuo and the material was purified via silica gel chromatography (2% CH3OH/CH2Cl2 gradient elution) to give (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (396 mg, 92%) as a glass. 1H NMR (CDCl3) δ 10.38 (m, 1H), 8.42 (s, 1H), 7.54-7.53 (m, 2H), 7.37-7.24 (m, 4H), 6.83-6.76 (m, 2H), 5.40 (d, J=10.0 Hz, 1H), 5.22 (d, J=10.0 Hz, 1H), 5.16 (dd, J=9.6, 6.0 Hz, 1H), 4.62 (m, 2H), 4.41 (m, 1H), 4.33-4.30 (m, 2H), 3.84 (dd, J=12.0, 10.0 Hz, 1H), 3.63 (dd, J=8.4, 7.2 Hz, 1H), 1.37 (d, J=6.0 Hz, 3H); ES+MS: 496 (M+1).
b)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt. To a solution of (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (396 mg, 0.80 mmol) in methanol (30 mL) was added 10% Pd/C (25 mg). Hydrogen was bubbled through the reaction mixture via a balloon for 2 h. The resultant mixture was filtered through Celite with methanol and dichloromethane.
The filtrate was concentrated in vacuo to give (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide , DOLUTEGRAVIR as a pink tinted white solid (278 mg, 86%).
1H NMR (CDCl3) δ 11.47 (m, 1H), 10.29 (m, 1H), 8.32 (s, 1H), 7.36 (m, 1H), 6.82 (m, 2H), 5.31 (dd, J=9.6, 3.6 Hz, 1H), 4.65 (m, 2H), 4.47-4.38 (m, 3H), 3.93 (dd, J=12.0, 10.0 Hz, 1H), 3.75 (m, 1H), 1.49 (d, J=5.6 Hz, 3H); ES+ MS: 406 (M+1).
DOLUTEGRAVIR NA SALT
The above material (278 mg, 0.66 mmol) was taken up in ethanol (10 mL) and treated with 1 N sodium hydroxide (aq) (0.66 ml, 0.66 mmol). The resulting suspension was stirred at room temperature for 30 min. Ether was added and the liquids were collected to provide the sodium salt of the title compound as a white powder (291 mg, 99%). 1H NMR (DMSO-d6) δ 10.68 (m, 1H), 7.90 (s, 1H), 7.35 (m, 1H), 7.20 (m, 1H), 7.01 (m, 1H), 5.20 (m, 1H), 4.58 (m, 1H), 4.49 (m, 2H), 4.22 (m, 2H), 3.74 (dd, J=11.2, 10.4 Hz, 1H), 3.58 (m, 1H), 1.25 (d, J=4.4 Hz, 3H).
UNDESIRED ISOMER
Example Z-9
(3S,11aR)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt

The title compound was made in two steps using a similar process to that described in example Z-1. 16a (510 mg, 1.08 mmol) and (25)-2-amino-1-propanol (0.17 mL, 2.17 mmol) were reacted in 1,2-dichloroethane (20 mL) with acetic acid to give (3S,11aR)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (500 mg, 93%). This material was hydrogenated in a second step as described in example Z-1 to give (3S,11aR)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (386 mg, 94%) as a tinted white solid. 1H NMR (CDCl3) δ 11.46 (m, 1H), 10.28 (m, 1H), 8.32 (s, 1H), 7.35 (m, 1H), 6.80 (m, 2H), 5.30 (dd, J=10.0, 4.0 Hz, 1H), 4.63 (m, 2H), 4.48-4.37 (m, 3H), 3.91 (dd, J=12.0, 10.0 Hz, 1H), 3.73 (m, 1H), 1.48 (d, J=6.0 Hz, 3H); ES+ MS: 406 (M+1). This material (385 mg, 0.95 mmol) was treated with sodium hydroxide (0.95 mL, 1.0 M, 0.95 mmol) in ethanol (15 mL) as described in example Z-1 to provide its corresponding sodium salt (381 mg, 94%) as a white solid. 1H NMR (DMSO-d6) δ 10.66 (m, 1H), 7.93 (s, 1H), 7.33 (m, 1H), 7.20 (m, 1H), 7.01 (m, 1H), 5.19 (m, 1H), 4.59 (m, 1H), 4.48 (m, 2H), 4.22 (m, 2H), 3.75 (m, 1 H), 3.57 (m, 1H), 1.24 (d, J=5.6 Hz, 3H).
SYNTHESIS OF INTERMEDIATES

IN ABOVE SCHEME SYNTHESIS UPTO COMPD 9 MAY BE USEFUL IN SYNTHESIS BUT READERS DISCRETION IS SOUGHT IN THIS ?????????????????
1) Maltol 1 (189 g, 1.5 mol) was dissolved in dimethylformamide (1890 ml), and benzyl bromide (184 ml, 1.5 mol) was added. After the solution was stirred at 80° C. for 15 minutes, potassium carbonate (228 g, 1.65 mol) was added, and the mixture was stirred for 1 hour. After the reaction solution was cooled to room temperature, an inorganic salt was filtered, and the filtrate was distilled off under reduced pressure. To the again precipitated inorganic salt was added tetrahydrofuran (1000 ml), this was filtered, and the filtrate was distilled off under reduced pressure to obtain the crude product (329 g, >100%) of 3-benzyloxy-2-methyl-pyran-4-one 2 as a brown oil.
NMR (CDCl3) δ: 2.09 (3H, s), 5.15 (2H, s), 6.36 (1H, d, J=5.6 Hz), 7.29-7.41 (5H, m), 7.60 (1H, d, J=5.6 Hz).
2) The compound 2 (162.2 g, 750 mmol) was dissolved in ethanol (487 ml), and aqueous ammonia (28%, 974 ml) and a 6N aqueous sodium hydroxide solution (150 ml, 900 mmol) were added. After the reaction solution was stirred at 90° C. for 1 hour, this was cooled to under ice-cooling, and ammonium chloride (58 g, 1080 mmol) was added. To the reaction solution was added chloroform, this was extracted, and the organic layer was washed with an aqueous saturated sodium bicarbonate solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, isopropyl alcohol and diethyl ether were added to the residue, and precipitated crystals were filtered to obtain 3-benzyloxy-2-methyl-1H-pyridine-4-one 3 (69.1 g, 43%) as a pale yellow crystal.
NMR (DMSO-d6) δ: 2.05 (3H, s), 5.04 (2H, s), 6.14 (1H, d, J=7.0 Hz), 7.31-7.42 (5H, m), 7.46 (1H, d, J=7.2 Hz), 11.29 (1H, brs).
3) The above compound 3 (129 g, 699 mmol) was suspended in acetonitrile (1300 ml), and N-bromosuccinic acid imide (117 g, 659 mmol) was added, followed by stirring at room temperature for 90 minutes. Precipitated crystals were filtered, and washed with acetonitrile and diethyl ether to obtain 3-benzyloxy-5-bromo-2-methyl-pyridine-4-ol 4 (154 g, 88%) as a colorless crystal.
NMR (DMSO-d6) δ: 2.06 (3H, s), 5.04 (2H, s), 7.32-7.42 (5H, m), 8.03 (1H, d, J=5.5 Hz), 11.82 (1H, brs).
4) To a solution of the compound 4 (88 g, 300 mmol), palladium acetate (13.4 g, 60 mmol) and 1,3-bis(diphenylphosphino)propane (30.8 g, 516 mmol) in dimethylformamide (660 ml) were added methanol (264 ml) and triethylamine (210 ml, 1.5 mol) at room temperature. The interior of a reaction vessel was replaced with carbon monoxide, and the material was stirred at room temperature for 30 minutes, and stirred at 80 degree for 18 hours. A vessel to which ethyl acetate (1500 ml), an aqueous saturated ammonium chloride solution (1500 ml) and water (1500 ml) had been added was stirred under ice-cooling, and the reaction solution was added thereto. Precipitates were filtered, and washed with water (300 ml), ethyl acetate (300 ml) and diethyl ether (300 ml) to obtain 5-benzyloxy-4-hydroxy-6-methyl-nicotinic acid methyl ester 5 (44.9 g, 55%) as a colorless crystal.
NMR (DMSO-d6) δ: 2.06 (3H, s), 3.72 (3H, s), 5.02 (2H, s), 7.33-7.42 (5H, m), 8.07 (1H, s).
5) After a solution of the compound 5 (19.1 g, 70 mmol) in acetic anhydride (134 ml) was stirred at 130° C. for 40 minutes, the solvent was distilled off under reduced pressure to obtain 4-acetoxy-5-benzyloxy-6-methyl-nicotinic acid methyl ester 6 (19.9 g, 90%) as a flesh colored crystal.
NMR (CDCl3) δ: 2.29 (3H, s), 2.52 (3H, s), 3.89 (3H, s), 4.98 (2H, s), 7.36-7.41 (5H, m), 8.85 (1H, s).
6) To a solution of the compound 6 (46.2 g, 147 mmol) in chloroform (370 ml) was added metachloroperbenzoic acid (65%) (42.8 g, 161 mmol) in portions under ice-cooling, and this was stirred at room temperature for 90 minutes. To the reaction solution was added a 10% aqueous potassium carbonate solution, and this was stirred for 10 minutes, followed by extraction with chloroform. The organic layer was washed with successively with a 10% aqueous potassium carbonate solution, an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under induced pressure, and the residue was washed with diisopropyl ether to obtain 4-acetoxy-5-benzyloxy-6-methyl-1-oxy-nicotinic acid methyl ester 7 (42.6 g, 87%) as a colorless crystal.
NMR (CDCl3) δ: 2.30 (3H, s), 2.41 (3H, s), 3.90 (3H, s), 5.02 (2H, s), 7.37-7.39 (5H, m), 8.70 (1H, s).
7) To acetic anhydride (500 ml) which had been heated to stir at 130° C. was added the compound 7 (42.6 g, 129 mmol) over 2 minutes, and this was stirred for 20 minutes. The solvent was distilled off under reduced pressure to obtain 4-acetoxy-6-acetoxymethyl-5-benzyloxy-nicotinic acid methyl ester 8 (49.6 g, >100%) as a black oil.
NMR (CDCl3) δ: 2.10 (3H, s), 2.28 (3H, s), 3.91 (3H, s), 5.07 (2H, s), 5.20 (2H, s), 7.35-7.41 (5H, m), 8.94 (1H, s).
8) To a solution of the compound 8 (46.8 g, 125 mmol) in methanol (140 ml) was added a 2N aqueous sodium hydroxide solution (376 ml) under ice-cooling, and this was stirred at 50° C. for 40 minutes. To the reaction solution were added diethyl ether and 2N hydrochloric acid under ice-cooling, and precipitated crystals were filtered. Resulting crystals were washed with water and diethyl ether to obtain 5-benzyloxy-4-hydroxy-6-hydroxymethyl-nicotinic acid 9 (23.3 g, 68%) as a colorless crystal.
NMR (DMSO-d6) δ: 4.49 (2H, s), 5.19 (2H, s), 5.85 (1H, brs), 7.14-7.20 (2H, m), 7.33-7.43 (7H, m), 8.30 (1H, s), 10.73 (1H, t, J=5.8 Hz), 11.96 (1H, brs).
9) To a solution of the compound 9 (131 g, 475 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (219 g, 1140 mmol) and 1-hydroxybenzotriazole (128 g, 950 mmol) in dimethylformamide (1300 ml) was added 4-fluorobenzylamine (109 ml, 950 mmol), and this was stirred at 80° C. for 1.5 hours. After the reaction solution was cooled to room temperature, hydrochloric acid was added, followed by extraction with ethyl acetate. The extract was washed with a 5% aqueous potassium carbonate solution, an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under reduced pressure to obtain a mixture (175 g) of 10 and 11. the resulting mixture was dissolved in acetic acid (1050 ml) and water (1050 ml), and zinc (31.1 g, 475 mmol) was added, followed by heating to reflux for 1 hour. After the reaction solution was cooled to room temperature, a 10% aqueous potassium carbonate solution was added, followed by extraction with ethyl acetate. The extract was washed with an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. After the solvent was distilled off under reduced pressure, this was washed with diethyl ether to obtain 5-benzyloxy-N-(4-fluoro-benzyl)-4-hydroxy-6-hydroxymethyl-nicotinic acid amide 10 (107 g, 59%) as a colorless crystal.
NMR (DMSO-d6) δ: 4.45 (2H, d, J=4.3 Hz), 4.52 (2H, d, J=5.8 Hz), 5.09 (2H, s), 6.01 (1H, brs), 7.36-7.43 (5H, m), 8.31 (1H, s), 12.63 (1H, brs).
………………..
SYNTHESIS
- Example 3

3H IS DOLUTEGRAVIR
Step 1
-
N,N-dimethylformamide dimethyl acetal (4.9 ml, 36.5 mmol) was added dropwise to compound 3A (5.0 g, 30.4 mmol) under cooling at 0°C. After stirring at 0°C for 1 hour, 100 ml of ethyl acetate was added to the reaction solution, and the organic layer was washed with a 0.5 N aqueous hydrochloric acid solution (50 ml). The aqueous layer was separated, followed by extraction with ethyl acetate (50 ml). The organic layers were combined, washed with a saturated aqueous solution of sodium bicarbonate and saturated saline in this order, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (n-hexane-ethyl acetate: 1:1 (v/v) → ethyl acetate) to obtain 4.49 g (yield: 67%) of compound 3B as an oil.
1H-NMR (CDCl3)δ:1.32 (3H, t, J = 7.1 Hz), 2.90 (3H, br s), 3.29 (3H, br s), 4.23 (2H, q, J = 7.1 Hz), 4.54 (2H, s), 7.81 (1H, s).
Step 2
-
Lithium hexamethyldisilazide (1.0 M solution in toluene, 49 ml, 49.0 mmol) was diluted with tetrahydrofuran (44 ml). A tetrahydrofuran (10 ml) solution of compound 3B (4.49 g, 20.4 mmol) was added dropwise thereto under cooling at -78°C, and a tetrahydrofuran (10 ml) solution of ethyl oxalyl chloride (3.35 g, 24.5 mmol) was then added dropwise to the mixture. The mixture was stirred at -78°C for 2 hours and then heated to 0°C. 2 N hydrochloric acid was added to the reaction solution, and the mixture was stirred for 20 minutes, followed by extraction with ethyl acetate (200 ml x 2). The organic layer was washed with a saturated aqueous solution of sodium bicarbonate and saturated saline and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (n-hexane-ethyl acetate: 7:3 → 5:5 → 0:10 (v/v)) to obtain 1.77 g (yield: 31%) of compound 3C as a white solid.
1H-NMR (CDCl3)δ:1.36-1.46 (6H, m), 4.35-4.52 (8H, m), 8.53 (1H, s).
Step 3
-
Aminoacetaldehyde dimethyl acetal (0.13 ml, 1.20 mmol) was added to an ethanol (6 ml) solution of compound 3C (300 mg, 1.09 mmol) at 0°C, and the mixture was stirred at 0°C for 1.5 hours, then at room temperature for 18 hours, and at 60°C for 4 hours. The solvent in the reaction solution was distilled off under reduced pressure, and the obtained residue was then purified by silica gel column chromatography (n-hexane-ethyl acetate: 5:5 → 0:10 (v/v)) to obtain 252 mg (yield: 64%) of compound 3D as an oil.
1H-NMR (CDCl3)δ:1.36-1.47 (6H, m), 3.42 (6H, s), 3.90 (2H, d, J = 5.2 Hz), 4.37 (3H, q, J = 7.2 Hz), 4.50 (2H, q, J = 7.2 Hz), 8.16 (1H, s).
Step 4
-
62% H2SO4 (892 mg, 5.64 mmol) was added to a formic acid (10 ml) solution of compound 3D (1.02 g, 2.82 mmol), and the mixture was stirred at room temperature for 16 hours. The formic acid was distilled off under reduced pressure. To the residue, methylene chloride was added, and the mixture was pH-adjusted to 6.6 by the addition of a saturated aqueous solution of sodium bicarbonate. The methylene chloride layer was separated, while the aqueous layer was subjected to extraction with methylene chloride. The methylene chloride layers were combined and dried over anhydrous sodium sulfate. The solvent was distilled off to obtain 531.8 mg of compound 3E as a yellow oil.
1H-NMR (CDCl3) δ: 1.28-1.49 (6H, m), 4.27-4.56 (4H, m), 4.84 (2H, s), 8.10 (1H, s), 9.72 (1H, s).
Step 5
-
Methanol (0.20 ml, 5.0 mmol), (R)-3-amino-butan-1-ol (179 mg, 2.0 mmol), and acetic acid (0.096 ml, 1.70 mmol) were added to a toluene (5 ml) solution of compound 3E (531 mg, 1.68 mmol), and the mixture was heated to reflux for 4 hours. The reaction solution was cooled to room temperature, then diluted with chloroform, and then washed with a saturated aqueous solution of sodium bicarbonate. The aqueous layer was subjected to extraction with chloroform. The chloroform layers were combined, washed with saturated saline, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (chloroform-methanol: 100:0 → 90:10) to obtain 309.4 mg of compound 3F as a brown oil.
1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.1 Hz), 1.40 (3H, d, J = 7.1 Hz), 1.55-1.61 (1H, m), 2.19-2.27 (1H, m), 4.00 (1H, d, J = 1.5 Hz), 4.03 (1H, d, J = 2.5 Hz), 4.10 (1H, dd, J = 13.2, 6.3 Hz), 4.26 (1H, dd, J = 13.2, 3.8 Hz), 4.38 (2H, q, J = 7.1 Hz), 5.00-5.05 (1H, m), 5.31 (1H, dd, J = 6.4, 3.9 Hz), 8.10 (1H, s).
Step 6
-
Potassium trimethylsilanolate (333 mg, 2.34 mmol) was added to a 1,2-dimethoxyethane (2 ml) solution of compound 3F (159 mg, 0.47 mmol), and the mixture was stirred at room temperature for 7 hours. 1 N hydrochloric acid and saturated saline were added to the reaction solution, followed by extraction with chloroform. The chloroform layers were combined and dried over anhydrous sodium sulfate. The solvent was distilled off to obtain 34.4 mg (yield: 25%) of compound 3G as an orange powder.
1H-NMR (CDCl3) δ: 1.46 (3H, d, J = 3.5 Hz), 1.58-1.65 (1H, m), 2.26-2.30 (1H,m), 4.06-4.10 (2H, m), 4.31 (1H, dd, J = 13.8, 5.6 Hz), 4.48 (1H, dd, J = 13.6, 3.9 Hz), 5.03 (1H, t, J = 6.4 Hz), 5.36 (1H, dd, J = 5.5, 4.0 Hz), 8.44 (1H, s), 12.80 (1H, s), 14.90 (1H, s).
Step 7
-
Compound 3G (16 mg, 0.054 mmol) and 2,4-difluorobenzylamine (17 mg, 0.12 mmol) were dissolved in N,N-dimethylformamide (1 ml). To the solution, N,N,N’,N’-tetramethyl-O-(7-aza-benzotriazol-1-yl)uronium hexafluorophosphate (HATU) (53 mg, 0.14 mmol) and N-methylmorpholine (0.031 ml, 0.28 mmol) were added, and the mixture was stirred at room temperature for 16 hours. 2,4-difluorobenzylamine (17 mg, 0.12 mmol), HATU (64 mg, 0.17 mmol), and N-methylmorpholine (0.037 ml, 0.34 mmol) were further added thereto, and the mixture was stirred at room temperature for additional 16 hours. 0.5 N hydrochloric acid was added to the reaction solution, followed by extraction with ethyl acetate. The ethyl acetate layers were combined, washed with 0.5 N hydrochloric acid and then with saturated saline, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by preparative high-performance liquid chromatography to obtain 12.5 mg (yield: 55%) of compound 3H as an orange solid.
- DOLUTEGRAVIR
-
1H-NMR (DMSO-d6) δ: 1.36 (3H, d, J = 6.9 Hz), 1.55-1.60 (1H, m), 2.01-2.05 (1H, m), 3.92-3.94 (1H, m), 4.04 (1H, t, J = 12.6 Hz), 4.38-4.41 (1H, m), 4.57-4.60 (1H, m), 4.81-4.83 (1H, m), 5.46-5.49 (1H, m), 7.08-7.11 (1H, m), 7.25-7.30 (1H, m), 7.41 (1H, dd, J = 15.3, 8.7 Hz), 8.53 (1H, s), 10.38 (1H, s), 12.53 (1H, s).
ISOMERS OF DOLUTEGRAVIR
- Reference Example 1

Step 1
-
Acetic acid (180 mg, 3.00 mmol) was added to a toluene (90 ml) solution of compound A-1 (4.39 g, 9.33 mmol) and (R)-3-aminobutan-1-ol (998 mg, 11.2 mmol), and the mixture was stirred at 50°C for 90 minutes. The reaction solution was allowed to cool to room temperature and then poured to a saturated aqueous solution of sodium bicarbonate. The organic layer was separated, while the aqueous layer was subjected to extraction three times with ethyl acetate. The combined extracts were washed with saturated saline and then dried over sodium sulfate. The solvent was distilled off to obtain 4.29 g of crude product A-2.
Step 2
-
The crude product A-2 obtained in the preceding step was dissolved in ethanol (40 ml). To the solution, a 2 N aqueous sodium hydroxide solution (20 ml) was added at room temperature, and the mixture was stirred at the same temperature for 2 hours. The reaction solution was neutralized to pH 7 using a 2 N aqueous hydrochloric acid solution. The solvent was directly distilled off. The obtained crude product A-3 was subjected to azeotropy with toluene (100 ml) and used in the next step without being purified.
Step 3
-
HOBt (1.65 g, 12.2 mmol) and WSC HCl (2.34 g, 12.2 mmol) were added at room temperature to a DMF (100 ml) solution of the crude product A-3 obtained in the preceding step, and the mixture was stirred at the same temperature for 15 hours. Water was added to the reaction solution, followed by extraction three times with ethyl acetate. The combined extracts were washed with water three times and then dried over sodium sulfate. The solvent was distilled off, and the obtained oil was subjected to silica gel column chromatography for purification. Elution was performed first with n-hexane-ethyl acetate (3:7, v/v) and then with only ethyl acetate. The fraction of interest was concentrated, and the obtained oil was then dissolved in ethyl acetate. The solution was crystallized with diisopropyl ether as a poor solvent. The obtained crystals were collected by filtration and dissolved again in ethyl acetate. The solution was recrystallized to obtain 1.84 g of compound A-4.
1HNMR (CDCl3) δ: 1.49 (3H, d, J = 6.6 Hz), 1.88-1.96 (1H, m), 2.13-2.26 (1H, m), 3.90-4.17 (4H, m), 4.42-4.47 (1H, m), 4.63 (2H, d, J = 6.0 Hz), 5.12-5.17 (1H, m), 5.17 (1H, d, J = 9.9 Hz), 5.33 (1H, d, J = 9.9 Hz), 6.77-6.87 (2H, m), 7.27-7.42 (4H, m), 7.59-7.62 (2H, m), 8.35 (1H, s), 10.41 (1H, t, J = 5.7 Hz).
Step 4
-
The compound A-4 was subjected to the hydroxy deprotection reaction described in Step F of the paragraph [0088] to obtain compound A-5.
1HNMR (DMSO-d6) δ:1.41 (3H, d, J = 6.3 Hz), 1.85-1.92 (1H, m), 1.50-1.75 (1H, m), 4.02-4.09 (3H, m), 4.28-4.34 (1H, m), 4.53 (2H, d, J = 5.7 Hz), 4.64 (1H, dd, J = 3.9 Hz, 12.6 Hz), 5.45 (1H, dd, J = 3.6 Hz, 9.3 Hz), 7.06 (1H, ddd, J = 2.7 Hz, 8.4 Hz, 8.4 Hz), 7.20-7.28 (1H, m), 7.35-7.42 (1H, m), 8.43 (1H, s),10.37 (1H, t, J = 6.0 Hz),12.37 (1H, brs).
- Reference Example 2
-

-
Compound A-1 was reacted with (S)-3-aminobutan-1-ol in Step 1. Compound B-5 was obtained in the same way as in Reference Example 1.
1HNMR (DMSO-d6) δ:1.41 (3H, d, J = 6.3 Hz), 1.85-1.92 (1H, m), 1.50-1.75 (1H, m), 4.02-4.09 (3H, m), 4.28-4.34 (1H, m), 4.53 (2H, d, J = 5.7 Hz), 4.64 (1H, dd, J = 3.9 Hz, 12.6 Hz), 5.45 (1H, dd, J = 3.6 Hz, 9.3 Hz), 7.06 (1H, ddd, J = 2.7 Hz, 8.4 Hz, 8.4 Hz), 7.20-7.28 (1H, m), 7.35-7.42 (1H, m), 8.43 (1H, s),10.37 (1H, t, J = 6.0 Hz),12.37 (1H, brs).

……………..

ENTRY 68
………………………….
WO 2010068262
…………………………
WO 2010068253
…………………………………
WO 2011119566
…………………………..
Synthesis
Example 3

I was under cooling added dropwise at 0 ℃ (4.9 ml, 36.5 mmol) and N, N-dimethylformamide dimethyl acetal (5.0 g, 30.4 mmol) in the first step compound 3A. After stirring for 1 hour at 0 ℃, ethyl acetate was added to 100ml, the reaction mixture was washed with 0.5N aqueous hydrochloric acid (50 ml). Was extracted with ethyl acetate (50ml) and solution was separated and the aqueous layer. The organic layers were combined, washed successively with saturated aqueous sodium bicarbonate solution and saturated brine, and then dried over anhydrous sodium sulfate. After the solvent was distilled off, silica gel column chromatography and the residue obtained was – and purified by (n-hexane (v / v) → ethyl acetate 1:1) to an oil (67% yield) of Compound 3B 4.49 g I got a thing.
1 H-NMR (CDCl 3) δ: 1.32 (3H, t, J = 7.1 Hz), 2.90 (3H, br s), 3.29 (3H, br s), 4.23 (2H, q, J = 7.1 Hz), 4.54 (2H, s), 7.81 (1H, s).
Diluted with tetrahydrofuran (44 ml) (1.0M toluene solution, 49 ml, 49.0 mmol) the second step lithium hexamethyldisilazide, under cooling at -78 ℃, compound 3B (4.49 g, 20.4 mmol) in this After dropwise tetrahydrofuran (10 ml) was added dropwise tetrahydrofuran (3.35 g, 24.5 mmol) of ethyl oxalyl chloride and (10 ml) solution. After stirring for 2 hours at -78 ℃, I was warmed to 0 ℃. After washing (200 ml x 2), saturated aqueous sodium bicarbonate solution and the organic layer with saturated brine After stirring for 20 minutes, extracted with ethyl acetate by adding 2N hydrochloric acid, the reaction solution was dried over anhydrous sodium sulfate. After removal of the solvent, silica gel column chromatography and the residue obtained – was purified (n-hexane (v / v) ethyl acetate 7:3 → 5:5 → 0:10), compound 3C 1.77 g (yield I as a white solid 31%).
1 H-NMR (CDCl 3) δ :1.36-1 .46 (6H, m), 4.35-4.52 (8H, m), 8.53 (1H, s).
Was added at 0 ℃ (0.13 ml, 1.20 mmol) the aminoacetaldehyde dimethyl acetal ethanol (300 mg, 1.09 mmol) of the third step compound 3C to (6 ml) solution, 1 hour and 30 minutes at 0 ℃, 18 hours at room temperature , then I was stirred for 4 hours at 60 ℃. After the solvent was evaporated under reduced pressure and the reaction mixture by silica gel column chromatography and the residue obtained was – and purified by (n-hexane (v / v) ethyl acetate 5:5 → 0:10), compound 3D 252 mg (yield: I got as an oil 64%) rate.
1 H-NMR (CDCl 3) δ :1.36-1 .47 (6H, m), 3.42 (6H, s), 3.90 (2H, d, J = 5.2 Hz), 4.37 (3H, q, J = 7.2 Hz), 4.50 (2H, q, J = 7.2 Hz), 8.16 (1H, s).
Was added (892 mg, 5.64 mmol) and 2 SO 4 62-H% formic acid (1.02 g, 2.82 mmol) in a fourth step the compound for 3D (10 ml) solution was stirred at room temperature for 16 hours. Methylene chloride was added to the residue Shi distilled off under reduced pressure and formic acid was adjusted to pH = 6.6 by addition of saturated aqueous sodium bicarbonate. The solution was separated methylene chloride layer was extracted with methylene chloride and the aqueous layer. I was dried over anhydrous sodium sulfate combined methylene chloride layers. The solvent was then distilled off and was obtained as a yellow oil 531.8 mg compound 3E.
1H-NMR (CDCl3) δ: 1.28-1.49 (6H, m), 4.27-4.56 (4H, m), 4.84 (2H, s), 8.10 (1H, s), 9.72 (1H, s).
Amino – – butane – 1 – ol (179 mg, 2.0 mmol), methanol (0.20 ml, 5.0 mmol), (R) -3 toluene (531 mg, 1.68 mmol) in the fifth step to compound 3E (5 ml) solution was added (0.096 ml, 1.70 mmol) acetic acid was heated under reflux for 4 hours. After dilution with chloroform, cooled to room temperature, the reaction mixture was washed with a saturated aqueous sodium bicarbonate solution, and the aqueous layer was extracted with chloroform. After washing with saturated brine combined chloroform layer was dried over anhydrous sodium sulfate. The solvent was then distilled off, silica gel column chromatography and the residue obtained – and (chloroform methanol 100:0 → 90:10), was obtained as a brown oil 309.4 mg compound 3F.
1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.1 Hz), 1.40 (3H, d, J = 7.1 Hz), 1.55-1.61 (1H, m), 2.19-2.27 (1H, m), 4.00 (1H, d, J = 1.5 Hz), 4.03 (1H, d, J = 2.5 Hz), 4.10 (1H, dd, J = 13.2, 6.3 Hz), 4.26 (1H, dd, J = 13.2, 3.8 Hz ), 4.38 (2H, q, J = 7.1 Hz), 5.00-5.05 (1H, m), 5.31 (1H, dd, J = 6.4, 3.9 Hz), 8.10 (1H, s).
1,2 (159 mg, 0.47 mmol) in the sixth step compound 3F – was added (333 mg, 2.34 mmol) and potassium trimethylsilanolate dimethoxyethane (2 ml) solution was stirred for 7 hours at room temperature. Brine was added to the 1N-hydrochloric acid to the reaction mixture, followed by extraction with chloroform. The combined chloroform layer was dried over anhydrous sodium sulfate. The solvent was removed by distillation, and I as an orange powder (25% yield) of compound 3G 34.4 mg.
1H-NMR (CDCl3) δ: 1.46 (3H, d, J = 3.5 Hz), 1.58-1.65 (1H, m), 2.26-2.30 (1H, m), 4.06-4.10 (2H, m), 4.31 (1H , dd, J = 13.8, 5.6 Hz), 4.48 (1H, dd, J = 13.6, 3.9 Hz), 5.03 (1H, t, J = 6.4 Hz), 5.36 (1H, dd, J = 5.5, 4.0 Hz) , 8.44 (1H, s), 12.80 (1H, s), 14.90 (1H, s).
2,4 (16 mg, 0.054 mmol) and the seventh step compound 3G – was dissolved in N, N-dimethylformamide (1 ml) (17 mg, 0.12 mmol) difluorobenzyl amine, N, N, N ‘, N was added (0.031 ml, 0.28 mmol) and N-methylmorpholine uronium hexafluorophosphate (HATU) (53 mg, 0.14 mmol), and ‘- tetramethyl-O-(yl 7 – aza – – benzo triazolopyrimidine -1) I was stirred at room temperature for 16 h. 2,4 – was added (0.037 ml, 0.34 mmol) and N-methylmorpholine (64 mg, 0.17 mmol) and (17 mg, 0.12 mmol), HATU difluorobenzylamine, and the mixture was stirred for 16 hours at room temperature. I was extracted with ethyl acetate addition of 0.5N-hydrochloric acid to the reaction mixture. 0.5N-hydrochloric acid and then was washed with saturated brine, and dried over anhydrous sodium sulfate and combined ethyl acetate layer. The solvent was then distilled off, and purified by preparative high performance liquid chromatography residue was obtained as an orange solid (55% yield) of compound 3H 12.5 mg.
1H-NMR (DMSO-d6) δ: 1.36 (3H, d, J = 6.9 Hz), 1.55-1.60 (1H, m), 2.01-2.05 (1H, m), 3.92-3.94 (1H, m), 4.04 (1H, t, J = 12.6 Hz), 4.38-4.41 (1H, m), 4.57-4.60 (1H, m), 4.81-4.83 (1H, m), 5.46-5.49 (1H, m), 7.08-7.11 (1H, m), 7.25-7.30 (1H, m), 7.41 (1H, dd, J = 15.3, 8.7 Hz), 8.53 (1H, s), 10.38 (1H, s), 12.53 (1H, s)
References
- [1] American Medical Association (AMA), STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL (Dolutegravir) Accessed 3 December 2011.
- FDA approves new drug to treat HIV infection http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm364744.htm Aug. 12, 2013
- “U.S. FDA approves GlaxoSmithKline’s HIV drug Tivicay”. Reuters. 12 August 2013. Retrieved 13 February 2013.
- “GSK wins priority status for new HIV drug in U.S”. Reuters. 16 February 2013. Retrieved 18 February 2013.
- “ViiV Healthcare receives approval for Tivicay™ (dolutegravir) in Canada for the treatment of HIV”. Retrieved 11 November 2013.
- FDA approves new drug to treat HIV infection http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm364744.htm Aug. 12, 2013
- U.S. FDA approves GlaxoSmithKline’s HIV drug Tivicay http://www.reuters.com/article/2013/08/12/us-glaxosmithkline-hivdrug-idUSBRE97B0WU20130812 Mon Aug 12, 2013 6:40pm EDT
- “Dolutegravir Prescribing Information”. Retrieved 3 January 2014.
- Raffi, F; Jaeger, H; Quiros-Roldan, E; Albrecht, H; Belonosova, E; Gatell, JM; Baril, JG; Domingo, P; Brennan, C; Almond, S; Min, S; extended SPRING-2 Study, Group (Nov 2013). “Once-daily dolutegravir versus twice-daily raltegravir in antiretroviral-naive adults with HIV-1 infection (SPRING-2 study): 96 week results from a randomised, double-blind, non-inferiority trial.”. The Lancet infectious diseases 13 (11): 927–35. PMID 24074642.
- http://www.natap.org/2013/ICAAC/ICAAC_24.htm
- Walmsley, Sharon L.; Antela, Antonio; Clumeck, Nathan; Duiculescu, Dan; Eberhard, Andrea; Gutiérrez, Felix; Hocqueloux, Laurent; Maggiolo, Franco; Sandkovsky, Uriel; Granier, Catherine; Pappa, Keith; Wynne, Brian; Min, Sherene; Nichols, Garrett (7 November 2013). “Dolutegravir plus Abacavir–Lamivudine for the Treatment of HIV-1 Infection”. New England Journal of Medicine 369 (19): 1807–1818. doi:10.1056/NEJMoa1215541.
- Sax, Paul. “SINGLE Study Underscores Waning of the Efavirenz Era — But Probably Just in the USA – See more at:http://blogs.jwatch.org/hiv-id-observations/index.php/single-study-underscores-waning-of-the-efavirenz-era-but-probably-just-in-the-usa/2013/11/06/#sthash.A39SderN.dpuf”. Retrieved 19 December 2013.
- Eron, JJ; Clotet, B; Durant, J; Katlama, C; Kumar, P; Lazzarin, A; Poizot-Martin, I; Richmond, G; Soriano, V; Ait-Khaled, M; Fujiwara, T; Huang, J; Min, S; Vavro, C; Yeo, J; VIKING Study, Group (2013 Mar 1). “Safety and efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant HIV type 1 infection: 24-week results of the VIKING Study.”. The Journal of infectious diseases 207 (5): 740–8. PMID 23225901.
-
WO2010011812A1 * Jul 23, 2009 Jan 28, 2010 Smithkline Beecham Corporation Chemical compounds WO2010011819A1 * Jul 23, 2009 Jan 28, 2010 Smithkline Beecham Corporation Chemical compounds -
-
- [Patent Document 1] International publication No.2006/116764 pamphlet
- [Patent Document 2] International publication No.2010/011812 pamphlet
- [Patent Document 3] International publication No.2010/011819 pamphlet
- [Patent Document 4] International publication No.2010/068262 pamphlet
- [Patent Document 5] International publication No.2010/067176 pamphlet
- [Patent Document 6] International publication No.2010/068253 pamphlet
- [Patent Document 7] US Patent 4769380A
- [Patent Document 8] International applicationPCT/JP2010/055316
[NON-PATENT DOCUMENTS]
-
- [Non-Patent Document 1] Journal of Organic Chemistry, 1991, 56(16), 4963-4967
- [Non-Patent Document 2] Science of Synthesis, 2005, 15, 285-387
- [Non-Patent Document 3] Journal of Chemical Society Parkin Transaction. 1, 1997, Issue. 2, 163-169
-
…………………
Sources:
Johns, Brian Alvin; Kawasuji, Takashi; Taishi, Teruhiko; Taoda, Yoshiyuki ; Polycyclic carbamoylpyridone derivative having HIV integrase inhibitory activity and their preparation; PCT Int. Appl., WO2006116764, 02 Nov 2006
Johns, Brian Alvin; Weatherhead, Jason Gordon;Tricyclic heterocyclic compounds as antiviral agents and their preparation and use in the treatment of HIV infection; PCT Int. Appl., WO2010011812, 28 Jan 2010
Johns, Brian Alvin; Weatherhead, Jason Gordon; Tricyclic heterocyclic compounds as antiviral agents and their preparation and use in the treatment of HIV infection;PCT Int. Appl., WO2010011819, 28 Jan 2010
Yoshida, Hiroshi; Taoda, Yoshiyuki; Johns, Brian Alvin; Synthesis of fused tricyclic carbamoylpyridone HIV integrase inhibitors and intermediates;PCT Int. Appl.,WO2010068253, 17 Jun 2010
Johns, Brian Alvin; Duan, Maosheng; Hakogi, Toshikazu;Processes and intermediates for fused tricyclic carbamoylpyridone HIV integrase inhibitors;PCT Int. Appl., WO2010068262, 17 Jun 2010
Sumino, Yukihito; Okamoto, Kazuya; Masui, Moriyasu; Yamada, Daisuke; Ikarashi, Fumiya;Preparation of compounds having HIV integrase inhibitory activity; PCT Int. Appl.,WO2012018065, 09 Feb 2012
Kawasuji, Takashi; Johns, Brian A.;Discovery of dolutegravir and S/GSK1265744: Carbamoyl pyridone HIV-1 integrase inhibitors;Abstracts, 64th Southeast Regional Meeting of the American Chemical Society, Raleigh, NC, United States, November 14-17 (2012), SERM-176.
Kawasuji, Takashi; Johns, Brian A.; Yoshida, Hiroshi; Weatherhead, Jason G.; Akiyama, Toshiyuki; Taishi, Teruhiko; Taoda, Yoshiyuki; Mikamiyama-Iwata, Minako; Murai, Hitoshi; Kiyama, Ryuichi; Fuji, Masahiro; Tanimoto, Norihiko; Yoshinaga, Tomokazu; Seki, Takahiro; Kobayashi, Masanori; Sato, Akihiko; Garvey, Edward P.; Fujiwara, Tamio; Carbamoyl Pyridone HIV-1 Integrase Inhibitors. 2. Bi- and Tricyclic Derivatives Result in Superior Antiviral and Pharmacokinetic Profiles;Journal of Medicinal Chemistry (2013), 56(3), 1124-1135
Walmsley S et al. Dolutegravir (DTG; S/GSK1349572) + abacavir/lamivudine once daily statistically superior to tenofovir/emtricitabine/efavirenz: 48-week results – SINGLE (ING114467). 52nd ICAAC, 9-12 September 2012, San Francisco. Abstract H-556b.
http://www.abstractsonline.com/Plan/ViewAbstract.aspx?sKey=e1c18d5b-830f-4b4e-8671-35bcfb20eed5&cKey=af219b7d-2171-46b2-91ef-b8049552c9e5&mKey=%7b6B114A1D-85A4-4054-A83B-04D8B9B8749F%7d
http://www.natap.org/2012/ICAAC/ICAAC_06.htm
http://i-base.info/htb/20381
Raffi F et al. Once-daily dolutegravir (DTG; S/GSK1349572) is non-inferior to raltegravir (RAL) in antiretroviral-naive adults: 48 week results from SPRING-2 (ING113086). 19th International AIDS Conference. 22-27 July 2012, Washington. Late breaker oral presentation THLBB04.
http://pag.aids2012.org/abstracts.aspx?aid=20990
National Institutes of Health (U.S.). A trial comparing GSK1349572 50 mg plus abacavir/lamivudine once daily to Atripla (also called the SINGLE trial). Available from:http://clinicaltrials.gov/ct2/show/NCT01263015.
Stellbrink HJ, Reynes J, Lazzarin A, et al. Dolutegravir in combination therapy exhibits rapid and sustained antiviral response in ARV-naïve adults: 96-week results from SPRING-1 (ING112276) (Abstract 102LB). Paper presented at: 19th Conference on Retroviruses and Opportunistic Infections; 2012 March 5–8; Seattle, WA. Available from:http://www.retroconference.org/2012b/Abstracts/45432.html
Merck’s New Drug Application for an Investigational Intravenous (IV) Formulation of NOXAFIL® (posaconazole) Receives FDA Priority Review

Posaconazole, SCH 56592, Noxafil (Schering-Plough)
Posaconazole is a triazole antifungal drug that is used to treat invasive infections by Candida species and Aspergillus species in severely immunocompromised patients.
For prophylaxis of invasive Aspergillus and Candida infections in patients, 13 years of age and older, who are at high risk of developing these infections due to being severely immunocompromised as a result of procedures such as hematopoietic stem cell transplant (HSCT) recipients with graft-versus-host disease (GVHD), or due to hematologic malignancies with prolonged neutropenia from chemotherapy. Also for the treatment of oropharyngeal candidiasis, including oropharyngeal candidiasis refractory to itraconazole and/or fluconazole. Posaconazole is used as an alternative treatment for invasive aspergillosis, Fusarium infections, and zygomycosis in patients who are intolerant of, or whose disease is refractory to, other antifungals
Posaconazole is designated chemically as 4-[4-[4-[4-[[ (3R,5R)-5- (2,4-difluorophenyl)tetrahydro-5-(1H-1,2,4-triazol-1 -ylmethyl)-3-furanyl]methoxy]phenyl]-1 -piperazinyl]phenyl]-2-[ (1S,2S)-1 -ethyl-2- hydroxypropyl]-2,4-dihydro-3H-1,2,4-triazol-3-one with an empirical formula of C37H42F2N8O4 and a molecular weight of 700.8.
Posaconazole is used, for example, to prevent and/or treat invasive fungal infections caused by Candida species, Mucor species, Aspergillus species,Fusarium species, or Coccidioides species in immunocompromised patients and/or in patients where the disease is refractory to other antifungal agents such as amphothericin B, fluconazole, or itraconazole, and/or in patients who do not tolerate these antifungal agents.
CAS No. 171228-49-2
Posaconazole compounds have been described inU.S. Pat. Appl. No. 2003/0055067 for “Antifungal Composition with Enhanced Bioavailability,” U.S. Pat. Appl. No. 2004/0058974 for “Treating Fungal Infections,” and European Patent Publication1372394 (A1 ) for “Liquid Suspensions of Posaconazole (SCH 56592) with Enhanced Bioavailability for Treating Fungal Infections.”
| Synonyms: | Pcz;Pos;Noxafil;Sch 56592;Aids058495;Aids-058495;Posconazole;Posaconazole;Posaconazole for research;HYDROXYPROPYL]-2,4-DIHYDRO-3H-1,2,4-TRIAZOL-3-ONE |
| Molecular Formula: | C37H42F2N8O4 |
| Formula Weight: | 700.78 |
Merck’s New Drug Application for an Investigational Intravenous (IV) Formulation of NOXAFIL® (posaconazole) Receives FDA Priority Review
Marketing Authorization Application also Filed with the European Medicines Agency
WHITEHOUSE STATION, N.J., Nov. 18, 2013–(BUSINESS WIRE)–Merck (NYSE:MRK), known as MSD outside the United States and Canada, today announced that its New Drug Application for an investigational intravenous (IV) solution formulation of the company’s antifungal agent, NOXAFIL® (posaconazole), has been accepted for priority review by the U.S. Food and Drug Administration (FDA).http://www.pharmalive.com/mercks-noxafil-nda-gets-fda-priority-review
Posaconazole (CAS Registry Number 171228-49-2; CAS Name: 2,5-anhydro-1 ,3,4-trideoxy-2- C-(2,4-difluorophenyl)-4-[[4-[4-[4-[1-[(1S,2S)-1-ethyl-2-hydroxypropyl]-1 ,5-dihydro-5-oxo-4H- 1 ,2,4-triazol-4-yl]phenyl]-1-piperazinyl]phenoxy]methyl]-1-(1 H-1 ,2,4-triazol-1-yl)-D-threo-pentitol) which is represented by the following general formula (I)

(I)
is known as an antifungal agent. It is available as an oral suspension (40 mg/ml) under the trademark NOXAFIL® from Schering Corporation, Kenilworth, NJ. WO95/17407 and WO 96/38443 disclose the compound having the general formula (I) and its use in treating fungal infections. Various pharmaceutical compositions comprising posaconazole and being adapted for oral, topical or parenteral use are described e.g. in WO 02/80678, U.S. Patent No. 5,972,381 , U.S. Patent No. 5,834,472, U.S. Patent No. 4,957,730 and WO 2005/117831. As was mentioned above, WO 95/17407 and WO 96/38443 disclose the compound having the general formula (I). However, during prosecution of the subsequently filed European patent application no. 98951994.7, now European patent EP 1 021 439 B1 , the applicant declared that the methods disclosed in these publications only lead to the compound of formula (I) as an amorphous solid.
Polymorphism is a phenomenon relating to the occurrence of different crystal forms for one molecule. There may be several different crystalline forms for the same molecule with distinct crystal structures and distinct and varying physical properties like melting point, XRPD pattern, IR-spectrum and solubility profile. These polymorphs are thus distinct solid forms which share the molecular formula of the compound from which the crystals are made up, however, they may have distinct advantageous physical properties which can have a direct effect on the ability to process and/or manufacture the drug product, like flowability, as well as physical properties such as solubility, stability and dissolution properties which can have a direct effect on drug product stability, solubility, dissolution, and bioavailability.
Three polymorphic forms of posaconazole designated as forms I, Il and III are described and characterized in WO 99/18097 (US-B-6,713,481 , US-B-6,958,337). Crystalline forms Il and III were found to be unstable under the conditions investigated, so that crystalline form I was considered to be useful in the development of a pharmaceutical product.
A. K. Saksena et al., WO 9517407; eidem, US 5661151 (1995, 1997 both to Schering);
eidem, Tetrahedron Lett. 37, 5657 (1996).
SCH-56592, a novel orally active broad spectrum antifungal agent35th Intersci Conf Antimicrob Agents Chemother (Sept 17-20, San Francisco) 1995,Abst F61
seeSaksena, A.K.; Girijavallabhan, V.M.; Lovey, R.G.; Pike, R.E.; Wang, H.; Liu, Y.-T.; Ganguly, A.K.; Bennett, F. (Schering Corp.) EP 0736030; JP 1997500658; US 5661151; US 5703079; WO 9517407
Process for the preparation of triazolonesWO 9633178
Mono N-arylation of piperazine(III): Metal-catalyzed N-arylation and its application to the novel preparations of the antifungal posaconazole and its advanced intermediateTetrahedron Lett 2002,43(18),3359
Comparative antifungal spectrum: A. Cacciapuoti et al., Antimicrob. Agents Chemother. 44, 2017 (2000).
Pharmacokinetics, safety and tolerability: R. Courtney et al., ibid. 47, 2788 (2003).
HPLC determn in serum: H. Kim et al., J. Chromatogr. B 738, 93 (2000).
Review of development: A. K. Saksena et al. inAnti-Infectives: Recent Advances in Chemistry and Structure Activity Relationships (Royal Soc. Chem., Cambridge, 1997) pp 180-199; and clinical efficacy in fungal infections: R. Herbrecht, Int. J. Clin. Pract. 58, 612-624 (2004).
synthesis 1

……………..

Synthesis of intermediate (XX): The reaction of 2-chloro-2′,4′-difluoroacetophenone (I) with sodium acetate and NaI in DMF gives 2-acetoxy-2′,4′-difluoroacetophenone (II), which by methylenation with methyltriphenylphosphonium bromide and sodium bis(trimethylsilyl)amide in THF yields 2-(2,4-difluorophenyl)-2-propen-1-ol acetate ester (III). The hydrolysis of (III) with KOH in dioxane/water affords the corresponding alcohol (IV), which is regioselectively epoxidized with titanium tetraisopropoxide and L-(+)-diethyl tartrate in dichloromethane to (S)-(-)-2-(2,4-difluorophenyl)oxirane-2-methanol (V). The reaction of (V) with 1,2,4-triazole (VI) in DMF affords (R)-2-(2,4-difluorophenyl)-3-(1,2,4-triazol-1-yl)propane-1,2-diol (VII), which is selectively mesylated with methanesulfonyl chloride and triethylamine to the monomesylate (VIII). The cyclization of (VIII) with NaH in DMF gives the oxirane (IX), which is condensed with diethyl malonate (X) by means of NaH in DMSO to yield a mixture of (5R-cis)- and (5R-trans)-5-(2,4-difluorophenyl)-2-oxo-5-(1,2,4-triazol-1-ylmethyl) tetrahydrofuran-3-carboxylic acid ethyl ester (XI). The reduction of (XI) with NaBH4 and LiCl in ethanol affords (R)-4-(2,4-difluorophenyl)-2-(hydroxymethyl)-5-(1,2,4-triazol-1-yl) pentane-1,4-diol (XII), which is selectively tosylated with tosyl chloride and triethylamine in THF to the bistosylate (XIII). The cyclization of (XIII) by means of NaH in refluxing toluene gives (5R-cis)-5-(2,4-difluorophenyl)-5-(1,2,4-triazol-1-ylmethyl) tetrahydrofuran-3-methanol tosylate ester (XIV). The reaction of (XIV) with 1-(4-hydroxyphenyl)-4-(4-nitrophenyl)piperazine (XV) to obtain compound (XVI), and the following reaction sequence (XVI) to (XVII) to (XVIII) to (XIX) to (5R-cis)-4-[4-[4-[4-[5-(2,4-difluorophenyl)-5-(1,2,4-triazol-1-ylmethyl)tetrahydrofuran-3-ylmethoxy]phenyl]piperazin-1-yl]phenyl-3,4-dihydro-2H-1,2,4-triazol-3-one (XX) has been performed according to J Med Chem 1984, 27: 894-900.
………………….pat approved expiry
| United States | 5661151 | 1999-07-19 | 2019-07-19 |
| Canada | 2305803 | 2009-12-22 | 2018-10-05 |
| Canada | 2179396 | 2001-04-17 | 2014-12-20 |
| United States | 5703079 | 1994-08-26 | 2014-08-26 |
MORE INFO
| US Patent No | Patent expiry | |
|---|---|---|
| 5661151 | Jul 19, 2019 | |
| 5703079 | Aug 26, 2014 | |
| 6958337 | Oct 5, 2018 | |
| 8263600 | Apr 1, 2022 |
- Cornely OA, Maertens J, Winston DJ, Perfect J, Ullmann AJ, Walsh TJ, Helfgott D, Holowiecki J, Stockelberg D, Goh YT, Petrini M, Hardalo C, Suresh R, Angulo-Gonzalez D: Posaconazole vs. fluconazole or itraconazole prophylaxis in patients with neutropenia. N Engl J Med. 2007 Jan 25;356(4):348-59. Pubmed
- Ullmann AJ, Lipton JH, Vesole DH, Chandrasekar P, Langston A, Tarantolo SR, Greinix H, Morais de Azevedo W, Reddy V, Boparai N, Pedicone L, Patino H, Durrant S: Posaconazole or fluconazole for prophylaxis in severe graft-versus-host disease. N Engl J Med. 2007 Jan 25;356(4):335-47. Pubmed
- Bhattacharya M, Rajeshwari K, Dhingra B: Posaconazole. J Postgrad Med. 2010 Apr-Jun;56(2):163-7. Pubmed
- Frampton JE, Scott LJ: Posaconazole : a review of its use in the prophylaxis of invasive fungal infections. Drugs. 2008;68(7):993-1016.Pubmed
- Schiller DS, Fung HB: Posaconazole: an extended-spectrum triazole antifungal agent. Clin Ther. 2007 Sep;29(9):1862-86. Pubmed
- Kwon DS, Mylonakis E: Posaconazole: a new broad-spectrum antifungal agent. Expert Opin Pharmacother. 2007 Jun;8(8):1167-78.Pubmed
- Groll AH, Walsh TJ: Posaconazole: clinical pharmacology and potential for management of fungal infections. Expert Rev Anti Infect Ther. 2005 Aug;3(4):467-87. Pubmed
- Rachwalski EJ, Wieczorkiewicz JT, Scheetz MH: Posaconazole: an oral triazole with an extended spectrum of activity. Ann Pharmacother. 2008 Oct;42(10):1429-38. Epub 2008 Aug 19. Pubmed
- Li Y, Theuretzbacher U, Clancy CJ, Nguyen MH, Derendorf H: Pharmacokinetic/pharmacodynamic profile of posaconazole. Clin Pharmacokinet. 2010 Jun;49(6):379-96. doi: 10.2165/11319340-000000000-00000. Pubmed
YERVOY®(ipilimumab) Receives Marketing Authorisation for First-Line Treatment of Adult Patients with Advanced Melanoma in Europe
PARIS, France, November 8, 2013 /PRNewswire/ —
Yervoy, an innovative immuno-oncology therapy that has demonstrated durable long-term survival in some patients, [1] , [2] is now approved for use in previously-untreated patients
Bristol-Myers Squibb today announced that the European Commission (EC) has approved YERVOY® (ipilimumab) for the first-line treatment of adult patients with advanced (unresectable or metastatic) melanoma.[3] When initially approved in Europe in July 2011 for the treatment of adult patients with previously-treated advanced melanoma, ipilimumab represented the first major treatment advance in this disease in more than 30 years, providing the first overall survival benefit ever seen in the treatment of metastatic melanoma in a phase III study.[ 1 ]
http://www.pharmalive.com/ec-approves-yervoy
![]()
Ipilimumab
by Todd Campbell, The Motley Fool Sep 28th 2013 1:00PM
Updated Sep 28th 2013 1:02PM
In early 2011, the Food and Drug Administration approved Bristol-Myers Squibb‘s drug Yervoy as a treatment for skin cancer melanoma. The drug marked the first approved treatment proven to extend the life of a person diagnosed with the disease. It marked a big leap forward in medicine as an early leader in immunotherapy, or the unleashing of the body’s immune system on cancer.
read all at
http://www.dailyfinance.com/2013/09/28/yervoy-battles-melanoma-but-can-it-become-a-blockb/

Ipilimumab’s molecular target is CTLA-4 (Uniprot: P16410; canSAR ; PFAM: P16410), a negative regulator of T-cell activation. Ipilimumab augments T-cell activation and proliferation by binding to CTLA-4 and preventing its interaction with its ligands (CD80 and CD86). CTLA-4 is a membrane-bound, 223 amino acid long, T-cell protein. It contains an immunoglobulin V-type domain (PFAM:PF07686). The structure of CTLA-4 is determined (see e.g. PDBe:3osk)
Ipilimumab (i pi lim′ ue mab; also known as MDX-010 and MDX-101), marketed asYervoy, is a drug used for the treatment of melanoma, a type of skin cancer. It is a U.S. Food and Drug Administration (FDA) approved human monoclonal antibody developed byBristol-Myers Squibb, and works by activating the immune system by targeting CTLA-4.
Cytotoxic T lymphocytes (CTLs) can recognize and destroy cancer cells. However, there is also an inhibitory mechanism that interrupts this destruction. Ipilimumab turns off this inhibitory mechanism and allows CTLs to continue to destroy cancer cells.
In addition to melanoma, ipilimumab is undergoing clinical trials for the treatment of non-small cell lung carcinoma (NSCLC), small cell lung cancer (SCLC) and metastatic hormone-refractory prostate cancer.
Yervoy is a monoclonal antibody drug indicated for treating metastatic melanoma. The drug was developed by Bristol-Myers Squibb.
In March 2011, The US Food and Drug Administration (FDA) approved Yervoy to treat patients with newly diagnosed or previously-treated unresectable or metastatic melanoma. Yervoy is the first drug approved vor the treatment of metastatic melanoma in the US.
Bristol-Myers Squibb submitted a marketing authorisation application to the European Medicines Agency in May 2010. The drug received approval from the European Commission in July 2011.
Approval from Australia’s Therapeutic Goods Association was received in July 2011. The drug is currently being reviewed by Health Canada.
Metastatic melanoma
Melanoma responsible for majority of skin cancer deaths in the US. In metastatic melanoma the cancer spreads to other parts of the body from its starting point. It becomes difficult to treat the disease once it spreads beyond the skin to other parts of the body. The disease is also known as stage IV melanoma.
If the melanoma spreads to the lungs then the patient faces breathing problems. The patients with metastatic melanoma may feel symptoms of fatigue, loss of weight, and appetite and bowel problems.
The incidence of the disease has increased steadily in the US after 1970s. The American Cancer Society (ACS) estimated that more than 68,000 new cases of melanoma were registered in the US in 2009. The ACS estimated that the number of deaths occurred due to melanoma in 2010 was more than 8,700.
Yervoy mechanism
Yervoy treats metastatic melanoma by activating the immune system. The drug works by binding or inhibiting cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), a molecule that plays vital role in relating natural immune responses. The presence or absence of CTLA-4 can curb or increase the immune system’s T-cell response in fighting disease.
The drug also works by blocking a complex set of interactions in the immune system. It is designed to inhibit the activity of CTLA-4, thereby sustaining an active immune response in its attack on cancer cells.
Approvals and indications
Ipilimumab was approved by the FDA in March 2011 to treat patients with late-stage melanoma that has spread or cannot be removed by surgery. On February 1, 2012, Health Canada approved ipilimumab for “treatment of unresectable or metastatic melanoma in patients who have failed or do not tolerate other systemic therapy for advanced disease.” Additionally Ipilimumab was approved in the European Union (EU), for second line treatment of metastatic melanoma, November 2012

Glaxo Plans to File for Malaria Vaccine Approval Next Year

Malaria vaccine candidate reduces disease over 18 months of follow-up in late-stage study of more than 15,000 infants and young children
Malaria is a significant public health burden, claiming 660,000 lives a year – mostly children in sub-Saharan Africa
-Data support plan to submit regulatory application in 2014
Multilateral Initiative on Malaria Pan African Conference, Durban, South Africa — Results from a large-scale Phase III trial, presented today in Durban, show that the most clinically advanced malaria vaccine candidate, RTS,S, continued to protect young children and infants from clinical malaria up to 18 months after vaccination. Based on these data, GSK now intends to submit, in 2014, a regulatory application to the European Medicines Agency (EMA). The World Health Organization (WHO) has indicated that a policy recommendation for the RTS,S malaria vaccine candidate is possible as early as 2015 if it is granted a positive scientific opinion by EMA.
READ ALL AT
http://www.pharmalive.com/glaxo-plans-to-file-for-malaria-vaccine-approval-next-year
![]()
GSK and Genmab seek alternative approval for leukaemia drug Arzerra

Arzerra
GlaxoSmithKline and Genmab A/S have announced the submission of leukaemia drug Arzerra to the European Medicines Agency (EMA) for a variation in marketing authorisation.
The companies are seeking authorisation for the drug to be used in combination with an alkylator-based therapy for treatment of Chronic Lymphocytic Leukemia (CLL) patients who have not received prior treatment and are inappropriate for fludarabine-based therapy.
READ ALL AT
Ofatumumab(trade name Arzerra, also known as HuMax-CD20) is a human monoclonal antibody (for the CD20 protein) which appears to inhibit early-stage B lymphocyte activation. It is FDA approved for treating chronic lymphocytic leukemia that is refractory to fludarabine and alemtuzumab (Campath) and has also shown potential in treating Follicular non-Hodgkin’s lymphoma, Diffuse large B cell lymphoma, rheumatoid arthritis and relapsing remitting multiple sclerosis. Ofatumumab has also received conditional approval in Europe for the treatment of refractory chronic lymphocytic leukemia. This makes ofatumumab the first marketing application for an antibody produced by Genmab, as well as the first human monoclonal antibody which targets the CD20 molecule that will be available for patients with refractory CLL.
Chronic lymphocytic leukemia (CLL) is a slowly progressing cancer of the blood and bone marrow. Arzerra (ofatumumab) has been approved by the FDA for treating CLL.
Patients with CLL whose cancer is no longer being controlled by other forms of chemotherapy can be prescribed Arzerra.
People older than fifty are mainly affected by CLL. A group of white blood cells known as B-cells that are part of the body’s immune system is the source of CLL. Every year, about ¼ of people diagnosed with CLL die from the disease.

Arzerra is an anti-CD20 monoclonal antibody that targets a membrane-proximal (which means close to the cell surface), small loop epitope, which is a portion of a molecule to which an antibody binds, on the CD20 molecule on B-cells. This epitope isn’t similar to binding sites that are targeted by other CD20 antibodies that are currently available. The CD20 molecule is highly expressed in most B-cell malignancies, making it a key target in CLL therapy.

MECHANISM OF ACTION:
Binding specifically to both the small and large extracellular loops of the CD20 molecule, Arzerra is an anti-CD20 monoclonal antibody. The CD20 molecule is expressed on normal B lymphocytes (pre-B- to mature B-lymphocyte) and on B-cell CLL. The CD20 molecule isn’t internalized following antibody binding and it isn’t shed from the cell surface. The Fc domain of ofatumumab mediates immune effector functions to result in B-cell lysis in vitro, while the Fab domain binds to the CD20 molecule. Complement-dependent cytotoxicity and antibody-dependent, cell-mediated cytotoxicity has been suggested as the possible mechanisms of cell lysis.
Products receive accelerated approval based on a surrogate endpoint, such as a reduction in the size of the tumor or decrease in the number of cancerous white cells or in an enlarged spleen or lymph nodes. These indirect measures for clinical outcomes are considered reasonably likely to predict that the drug will allow patients to live with fewer side effects of a disease or to live longer. Arzerra was approved under the FDA’s accelerated approval process, which allows earlier approval of drugs that meet unmet medical needs.

To confirm that the addition of Arzerra to standard chemotherapy delays the progression of the disease, the manufacturer of this medication is currently conducting a clinical trial in CLL patients. This is because the accelerated approval process requires further study of the drug.
