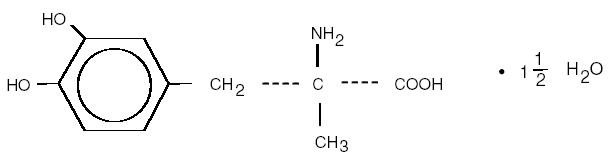
Desvenlafaxine

Desvenlafaxine Succinate Monohydrate
386750-22-7
93413-62-8 (free base, anhydrous)
448904-47-0 (anhydrous)
448904-48-1 (hemisuccinate)
DVS-233
PF-05212375
WY-45233 (free base)
| Name |
: |
Desvenlafaxine Succinate |
| Synonym |
: |
1-(2-(Dimethylamino)-1-(4- hydroxyphenyl)ethyl)cyclohexanol butanedioate; O-Desmethylvenlafaxine Succinate |
| Mol Formula |
: |
C20H31NO6 / C16H25NO2.C4H6O4 |
| CAS |
: |
448904-47-0 |
| Name |
: |
Desvenlafaxine Succinate Monohydrate |
| Synonym |
: |
1-((1RS)-2-(Dimethylamino)-1-(4- hydroxyphenyl)ethyl)cyclohexanol hydrogen butanedioate monohydrate ; O-Desmethylvenlafaxine Succinate monohydrate; Desvenlafaxine Succinate |
| Mol Formula |
: |
C20H33NO7 / C16H25NO2.C4H6O4.H2O |
| CAS |
: |
386750-22-7 |
Inventor/Developer – Wyeth Pharma Inc.
Status/Comment – FDA approved
Desvenlafaxine Succinate Hydrate
Research Code:DVS-233
Trade Name:Pristiq®
MOA:Serotonin and norepinephrine reuptake inhibitor (SNRI)
Indication:Major depressive disorder (MDD)
Status:Approved
Company:Pfizer (Originator)
Sales:$715 Million (Y2015); 
$737 Million (Y2014);;
$698 Million (Y2013);;
$630 Million (Y2012);;
$577 Million (Y2011);ATC Code:N06AX23
Desvenlafaxine succinate hydrate was approved by the U.S. Food and Drug Administration (FDA) on February 29, 2008.It was developed by Pfizer, then marketed as Pristiq® by Pfizer in the US.
The exact mechanism of the antidepressant action of Desvenlafaxine is unknown, but is thought to be related to the potentiation of serotonin and norepinephrine in the central nervous system, through inhibition of their reuptake. Non-clinical studies have shown that Desvenlafaxine is a potent and selective serotonin and norepinephrine reuptake inhibitor (SNRI). It is indicated for the treatment of major depressive disorder (MDD).
Pristiq® is available as extended release tablet for oral use, containing 50 mg or 100 mg of free Desvenlafaxine. The recommended dose is 50 mg once daily with or without food.
PRISTIQ®
(desvenlafaxine) Extended-release Tablets
WARNING
SUICIDAL THOUGHTS AND BEHAVIORS
Antidepressants increased the risk of suicidal thoughts and behavior in children, adolescents, and young adults in short-term studies. These studies did not show an increase in the risk of suicidal thoughts and behavior with antidepressant use in patients over age 24; there was a reduction in risk with antidepressant use in patients aged 65 and older [see WARNINGS AND PRECAUTIONS].
In patients of all ages who are started on antidepressant therapy, monitor closely for worsening, and for emergence of suicidal thoughts and behaviors. Advise families and caregivers of the need for close observation and communication with the prescriber [see WARNINGS AND PRECAUTIONS].
PRISTIQ is not approved for use in pediatric patients [ see Use in Specific Populations].
DESCRIPTION
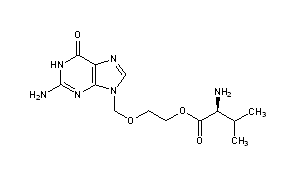
PRISTIQ is an extended-release tablet for oral administration that contains desvenlafaxine succinate, a structurally novel SNRI for the treatment of MDD. Desvenlafaxine (O-desmethylvenlafaxine) is the major active metabolite of the antidepressant venlafaxine, a medication used to treat major depressive disorder.
Desvenlafaxine is designated RS-4-[2-dimethylamino-1-(1-hydroxycyclohexyl)ethyl]phenol and has the empirical formula of C16H25NO2 (free base) and C16H25NO2 •C4H6O4•H2O (succinate monohydrate). Desvenlafaxine succinate monohydrate has a molecular weight of 399.48. The structural formula is shown below.
Desvenlafaxine succinate is a white to off-white powder that is soluble in water. The solubility of desvenlafaxine succinate is pH dependent. Its octanol:aqueous system (at pH 7.0) partition coefficient is 0.21.
PRISTIQ is formulated as an extended-release tablet for once-a-day oral administration.
Each tablet contains 38 mg, 76 mg or 152 mg of desvenlafaxine succinate equivalent to 25 mg, 50 mg or 100 mg of desvenlafaxine, respectively.
Osmotica Pharmaceutical, Par Pharmaceutical and Pernix Therapeutics are marketing the product in the U.S. under the brand name Khedezla (TM) for the treatment of major depressive disorder.
In 2019, Pfizer and Mochida signed an agreement for joint development and commercialization of the product in Japan.
Desvenlafaxine, sold under the brand name Pristiq among others, is a medication used to treat major depressive disorder.[1] It is recommended that the need for further treatment be occasionally reassessed.[1] It appears less effective than its parent compound venlafaxine.[2] It is taken by mouth.[1]
Common side effects include dizziness, trouble sleeping, increased sweating, constipation, sleepiness, anxiety, and sexual problems.[1]Serious side effects may include suicide in those under the age of 25, serotonin syndrome, bleeding, mania, and high blood pressure.[1]A withdrawal syndrome may occur if the dose is rapidly decreased.[1] It is unclear if use during pregnancy or breastfeeding is safe.[3] It is an antidepressant of the serotonin-norepinephrine reuptake inhibitor (SNRI) class.[1]
Desvenlafaxine was approved for medical use in the United States in 2008.[1] Use in Europe was declined in 2009.[2] In the United States the wholesale cost is about 25.20 USD per month.[4] In 2016, it was the 272nd most prescribed medication in the United States, with more than a million prescriptions.[5]
Medical uses
Desvenlafaxine is primarily used as a treatment for major depressive disorder.[6] Use has only been studied up to 8 weeks.[1] It, however, appears less effective than venlafaxine.[2]
Doses of 50-400 mg/day appear effective for major depressive disorder, although no additional benefit was demonstrated at doses greater than 50 mg/day, and adverse events and discontinuations were more frequent at higher doses.[7]
Desvenlafaxine improves the HAM-D17 score[8] and measures of well being such as the Sheehan Disability Scale (SDS) and 5-item World Health Organization Well-Being Index (WHO-5).[9]
Adverse effects[edit]
Frequency of adverse effects:[6][10][11]
Very common adverse effects include:
- Nausea
- Headache
- Dizziness
- Dry mouth
- Hyperhidrosis
- Diarrhea
- Insomnia
- Constipation
- Fatigue
Common adverse effects include:
- Tremor
- Blurred vision
- Mydriasis
- Decreased appetite
- Sexual dysfunction
- Insomnia
- Anxiety
- Elevated cholesterol and triglycerides
- Proteinuria
- Vertigo
- Feeling jittery
- Asthenia
- Nervousness
- Hot flush
- Irritability
- Abnormal dreams
- Urinary hesitation
- Yawning
- Rash
Uncommon adverse effects include:
Rare adverse effects include:
Common however unknown intensity of adverse effects include:
Pharmacology
Desvenlafaxine is a synthetic form of the isolated major active metabolite of venlafaxine, and is categorized as a serotonin-norepinephrine reuptake inhibitor (SNRI). When most normal metabolizers take venlafaxine, approximately 70% of the dose is metabolized into desvenlafaxine, so the effects of the two drugs are expected to be very similar.[12] It works by blocking the “reuptake” transporters for key neurotransmitters affecting mood, thereby leaving more active neurotransmitters in the synapse. The neurotransmitters affected are serotonin (5-hydroxytryptamine) and norepinephrine (noradrenaline). It is approximately 10 times more potent at inhibiting serotonin uptake than norepinephrine uptake.[13]
Approval status
United States

Pristiq 50 mg tablets (US)
Wyeth announced on 23 January 2007 that it received an approvable letter from the Food and Drug Administration for desvenlafaxine. Final approval to sell the drug was contingent on a number of things, including:
- A satisfactory FDA inspection of Wyeth’s Guayama, Puerto Rico facility, where the drug is to be manufactured;
- Several postmarketing surveillance commitments, and follow-up studies on low-dose use, relapse, and use in children;
- Clarity by Wyeth around the company’s product education plan for physicians and patients;
- Approval of desvenlafaxine’s proprietary name, Pristiq.[15]
The FDA approved the drug for antidepressant use in February 2008, and was to be available in US pharmacies in May 2008.[16]
In March 2017, the generic form of the drug was made available in the US.
Canada
On February 4, 2009, Health Canada approved use of desvenlafaxine for treatment of depression.[17][18]
European Union
In 2009, an application to market desvenlafaxine for major depressive disorder in the European Union was declined.[2] In 2012, Pfizer received authorization in Spain to market desvenlafaxine for the disorder but it is not being sold.[19][20]
Australia
Desvenlafaxine is classified as a schedule 4 (prescription only) drug in Australia. It was listed on the PBS (Pharmaceutical Benefits Scheme) in 2008 for the treatment of major depressive disorders.
PATENT
https://patents.google.com/patent/CN104326923A/en
Sign succinic acid desvenlafaxine (Desvenlafaxinesuccinate, tradename Pristiq), chemical name RS-4- [2- dimethylamino-1- (1-hydroxycyclohexyl) ethyl] phenol succinic acid salt monohydrate; English name RS-4- (2 – (dimethylamino) -I- (1-hydroxycyclohexyl) ethyl) phenolsuccinatehydrate; formula C16H25NO2 · C4H6O · 4H20; relative molecular mass:. 399 48; CAS Registry number: 386750-22-7; of formula of formula I:
[0003]
[0004] The drug is produced by the US company Wyethphainsinc, February 29, 2008 listed by the US FDA approved, a serotonin – norepinephrine reuptake inhibitor is venlafaxine main active metabolite primarily for the treatment of major depressive disorder (MDD).
[0005] desvenlafaxine succinate is generally made with 0- desvenlafaxine succinate, aqueous salt synthesized, synthesized 0- desvenlafaxine earlier reports found in US Patent US4535186 discloses a method to 4-oxo-acetonitrile was synthesized from benzyl phenyl 0- desvenlafaxine, wherein the methylation step due to the use of formaldehyde as a methylating agent such that the reaction yield is very low, only 39 %, thereby affecting the overall yield of the overall reaction.
[0006] Currently many reported synthesis method of desvenlafaxine succinate is mostly based on the prior art (US4535186) Synthesis of venlafaxine, venlafaxine as a raw material to another, for which demethylation step reaction process improvement to synthesize 0- desmethylvenlafaxine, succinic acid and finally with water to a salt of desvenlafaxine succinate synthesis. The patent CN 1319934C, W0 0059851, CN101823969A venlafaxine are disclosed in as raw materials, the thiolate anion, lithium diphenylphosphide, HBr / HOAc as demethylating agent norepinephrine synthesis 0- venlafaxine oct, yields were> 73%. The reaction equation is as follows:
[0007]
[0008] In this class synthesis process, the raw material venlafaxine is synthesized by known techniques, the thiol compound used in the step of methylation away easily air pollution toxic, flammable irritant diphenyl compound and using a phosphine compound for corrosive HBr, increasing the difficulty of the operation and the post-treatment process such that the reaction unsuitable for industrial production.
[0009] Patent US7026508, US6689912 and US2005 / 0197392 each discloses a hydroxyphenyl acetonitrile as raw material, hydroxymethylated, α- ketone containing active hydrogen compound condensation, reducing the cyano group, an amino group and removal of methyl synthesis of methyl 〇- reaction desvenlafaxine, which synthetic route is shown below:
[0011] In such processes, the raw materials used are expensive cyano reagent, wherein the reagent lithium tri-secondary butyl borohydride risk patents US7026508 and method disclosed in patent US 6689912 to use, the patent US2005 / 0197392 use flammable irritating to diphenylphosphine compound, in addition, such methods involve harsh reaction conditions the reduction step cyano.Therefore, this method does not meet the economy, and is not suitable for industrial production.
[0012] Chinese Patent CN101781221Α discloses a synthetic method for the synthesis of 0- hydroxyphenylacetic acid desvenlafaxine, the acid-halo – aminolysis cyclohexanone condensation, amide reduction Synthesis 〇- desvenlafaxine, which scheme is as follows:
[0014] World Patent WO2008 / 093142 discloses a method similar to the above kind of oxygen acid as a raw material by benzyl, and finally debenzylation by synthesis 0- desvenlafaxine.
[0015] In such methods, the former are in a condensation reaction step with reduction of the amide using an unstable compound n-butyllithium, lithium tetrahydroaluminate; then using a compound unstable to hexamethyldisilazide amide, borane, this two-step reaction be carried out under an inert gas, dangerous reagents used twice, is not suitable for industrial operation.

Example 1
Synthesis [0041] The compound of formula III
The [0042] room temperature, was added the compound of formula II (81.69g, 0. 60mol), 1200mL acetone 3L reaction flask, stirred and dissolved. To this was added 1〇) 3 (265.368,1.92111〇1) was slowly added dropwise (112.888,0.65111〇1) of benzyl bromide was heated at reflux for LH, starting material after the reaction was cooled to room temperature, filtered off with suction, the filter cake was washed with a suitable amount of acetone , the filtrate by rotary evaporation, 50 ° C to give a compound of formula blast drying ΠI134 7g, yield 99% ZHNMR (300MHz, DMS0):… δ7 50-7 22 (m, 7H), δ6.93 (d, 2H) , S5.03 (s, 2H), S2.53 (s, 3H); 13C-NMR:… S197.02, δ163 · 42, δ136 · 51, δ129 · 83, δ129 02, δ128 91, δ127 64 , δ127. 10, δ114. 29, δ70. 13, δ26. 6.
[0043] Example 2
[0044] following the experimental procedure of Example 1, benzyl bromide (112. 88g, 0. 65mol) is replaced with benzyl chloride (82. 28g, 0. 65mol), heated at reflux overnight, cooled to room temperature after completion of the reaction evacuated filter cake washed with a suitable amount of acetone, the filtrate by rotary evaporation, 50 ° C to give a compound of formula blast drying III103. 17g, yield 76%.
[0045] Example 3
Synthesis [0046] The compound of Formula IV
[0047] The compound of formula III is added to the 3L reaction flask (67. 9g, 0 · 30mol), Cufc2 (147. 4g, 0 · 65mol), 0 · 9L methylene chloride, I. 35LEA, heated to reflux, after completion of the reaction cooling suction filtered, the filter cake was washed with 200mL dichloromethane. The filtrates were combined, and the filtrate was washed with hydrochloric acid and then washed twice with water, dried, rotary evaporation, 50 ° C overnight blast drying. A compound of formula IV to give the crude 89.438, yield 98%.
[0048] 89. 43g was added the compound of formula IV (crude) was added to IL-neck round bottom flask, recrystallized from isopropanol, filtered off with suction, the filter cake was dried by blowing 45 ° C. Refined products 84. 9g, 95% yield. .. 1Hnmr (SoomhzJMSO-CI6): δ7 53-7 20 (m, 7H), 5 6. 95 (d, 2H), δ5 09 (s, 2H), δ4 69 (s, 2H); 13C-.. NMR:. δ190 83, 5 164.12, 5 136.71, δ129 96, δ128 91, δ128 31, δ127 00, δ126 73, δ114 37, δ70 82, δ32 45………
[0049] Example 4
Synthesis [0050] The compound of formula V
[0051] Add the compound of formula IV (36. 6g, 0. 12mol) A 250mL round bottom flask, 120mL of ethanol was stirred. Aqueous solution was slowly added dropwise thereto 39mL of dimethylamine (33%), dropwise, with stirring until completion of the reaction starting material. Rotary evaporation and water, to which ethanol was added, under ice-water bath, thereto was slowly added dropwise 12mL hydrobromic acid. After stirring 30min 25 ° C incubation rotary evaporation and water was added thereto and dissolved with 70mL of methylene chloride over anhydrous sodium sulfate, after rotary evaporation, and thereto was added 60mLEA refluxed 30min, 30min stirring ice-water bath. Filtered off with suction, 45 ° C blast drying to give the purified product compound of formula V41.lg, yield 98%.1H NMR (300MHz, DMS0-d6): δ7 · 55-7 · 24 (πι, 7Η), S6.97 (d, 2H), S5.13 (s, 2H), S3.79 (s, 2H), 5 2. 23 (s, 6H); 13C-NMR:….. δ195 45, 5 163.12, 5 136.53, 5 129.85, 5 128.96, 5 127.61, δ127 27, δ127 06, δ114 65, δ75 31, δ70 . 72, δ46. 55.
[0052] Example 5
Synthesis [0053] The compound of formula VI
[0054] Compound of formula V was added to 500mL round bottom flask (41g, 0. 12mol), 205mL of ethanol under ice-water bath, to which was added a likelihood of 0! 1 (9.368,0.23111〇1), after stirring for 11 ^ 301, to which was added portionwise like 8! 14 (8.888,0.23111〇1). The reaction was stirred for 4h at End material to room temperature. Rotary evaporation, and thereto was added 60mL of water, 120mL of methylene chloride, stirred, separated, the aqueous phase was extracted twice with 120mL dichloromethane. The organic phases were combined, spin dry.The crude 31. 0g, 98% yield.
[0055] The crude product 31g was dissolved in 180mL of ethanol, under ice-water bath, was slowly added dropwise thereto 16mL of concentrated hydrochloric acid to pH = 1-2, stirred at room temperature for 30 min. Rotary evaporation, 45 ° C blast drying. A compound of formula VI to give crude product 31. 2g, 89% yield.
[0056] To a 250mL round-bottom flask was added 31. 2g crude compound of formula VI, 78 mL ethyl acetate, heated to reflux to dissolve, cooled to room temperature naturally, filtered off with suction, the filter cake was washed with the amount of EA. Refined products 29. 3g, 94% yield. 1HNMR (300MHz, DMS0-d6):….. Δ7 45-7 24 (m, 7H), δ6 95 (d, 2H), δ5 07 (s, 2H), δ4 88 (br, 1H), δ4 . 60 (m, 1Η), δ2 48-2 27 (m, 2H), 52.19 (s, 6H); 13C-NMR:…. δ157 26, 5 137.21, 5 136.83, δ128 · 35, δ127 68, δ127. 52, δ127. 17, δ114. 19, δ69. 62, δ69. 13, δ67. 57,45.56.
[0057] Example 6
[0058] Following the procedure of Example 5 experiments embodiment, the shame 0! 1 (9.368,0.23111〇1) shame Alternatively 0! 1 (14.048,0.72111〇1), and NaBH4 (8. 88g, 0. 23mol) is replaced with NaBH4 (13. 32g, 0. 72mol), stirred Ih at room temperature the reaction was complete feed. Rotary evaporation, and thereto was added 60mL of water, burning 120mL dichloromethane, stirred, separated, the aqueous phase was extracted twice with 120mL dichloromethane. The organic phases were combined, spin dry. The crude 31. 33g, yield 99%.
[0059] Example 7
Synthesis [0060] The compound of formula VII
[0061] was added (24g, 0.078 mol) compound of formula VI to a 500mL round bottom flask, 240 mL of toluene, under ice-water bath, and thereto is added thionyl chloride (10. 2g, 0. 086mol), the reaction was heated to 60 ° C 4h, cooled, stirred for about 25 ° C 2h, filtered off with suction, the filter cake washed with 30mL toluene, and drying, the product compound of formula to give an off-white VII22. 4g, 88% yield. 1H NMR (300MHz, DMS〇-d6):… Δ7 44-7 29 (m, 7H), δ7 07 (d, 2Η), δ5 12 (s, 2Η), δ4 69 (m, 1Η).. , δ3 37-3 15 (m, 2H), δ2 80 (s, 6H); 13C-NMR:…. δ158 26, 5 137.33, 5 136.71, 5 128.31, δ127 72, δ127 47, δ127… 13, δ114. 21, δ69. 62, δ67. 57, δ59. 39, δ46. 74.
[0062] Example 8
Synthesis [0063] The compound of formula VIII
[0064] Add the compound of formula ¥ 11 (218,0.064 11〇1!) Was added to a round bottom flask, 2001 ^ toluene, triethylamine (7.21 8, 0.071mol), stirred for 3h, filtered off with suction, washed with a little toluene; the 500mL three-neck flask was added to the toluene solution, cooled to -80 ° C, n-butyllithium was slowly added dropwise 35mL (2. 5mol / L), dropwise with stirring incubated 0. 5h, 9. 48g was slowly added dropwise cyclohexanone, After dropping the reaction 4h, slowly warmed to room temperature, the reaction was quenched with saturated ammonium chloride, using lmol / L sodium hydroxide to adjust pH = about 9, EA extraction, rotary evaporation, the crude product was slurried with diethyl ether, dried to obtain a compound of formula VIII19. 56g, yield 86%. 1H bandit R (300MHz, DMS0-d6): δ7 · 45-7 · 23 (πι, 7Η), S6.87 (d, 2H), S5.09 (s, 2H), S3.05 (t, 1H) , S2.75 (t, lH), δ2 · 41-2 · 34 (πι, 1Η), S2.18 (s, 6H), Sl.58-0.92 (m, 10H); 13C-Mffi:. δ157 32 , δ136. 73, δ134. 73, δ129. 11, δ128. 90, δ127. 65, δ127. 16, δ114. 23, δ73. 42, δ70. 87, δ59. 85, δ48. 52, δ47. 35, δ38 . 95, δ26. 33, δ22. 30.
[0065] Example 9
After the [0066] following the experimental procedure of Example 8, the reaction temperature is added dropwise n-butyllithium was replaced _65 ° C, to give compound VIII crude, beaten with ether and drying to give pure 15. 33g, yield 67 %.
[0067] Example 10
Synthesis [0068] The compound of formula IX
[0069] The compound of formula ¥ 111 (1 (^, 0.028111〇1) was dissolved in ^ booklet 1,501,111, was added 18 10% wet palladium on carbon, into hydrogen, I.SMPa at room temperature for 5h, filtration, rotary evaporation as a white solid compound of formula 1X6 83g, yield 92% 1HNMR (300MHz, DMSO- (I6):… δ9 13 (br, 1H), 5 6.96 (d, 2H), 5 6.64 (d, 2H), 53.01 (t, 1H), S2.72 (t, lH), δ2 · 39-2 · 35 (πι, 1Η), S2.15 (s, 6H), Sl.57-0.90 (m, 10H); 13C, MR:. δ155 56, δ131 56, δ130 04, δ114 23, δ72 52, δ60 36, δ51 57, δ45 21, δ37 11, δ32 38, δ25 67, δ21 23〇………..
[0070] Example 11
Synthesis [0071] The compounds of formula I
Under [0072] nitrogen, the compound of formula IX is added to the three-necked flask (4g, 0. 015mol), succinic acid (1.85g, 0. 015mol), IOOmL acetone / water mixed solvent = 71/19 was heated at reflux for 3h proceeds down to room temperature to crystallization under ice-cooling, filtered off with suction, 40 ° C dried to give 5. 13g as a white solid compound of formula I, yield 85%.
PATENT
https://patents.google.com/patent/WO2008090465A2/en
Desvenlafaxine (Formula I, below) is an active pharmaceutical substance with an empirical formula of C16H25NO2 and a molecular weight of 263.38. Desvenlafaxine, which can also be referred to as desmethylvenlafaxine and/or O-desmethylvenlafaxine, is the major active metabolite of venlafaxine, an active pharmaceutical ingredient indicated for the treatment of major depressive disorder.
U.S. Patent No. 4,535, 186 discloses the first process for preparing desvenlafaxine. In U.S. Patent No.4,535,186, desvenlafaxine is synthesized by the process illustrated in Scheme 1:

Scheme 1 Additional alternative processes for preparing desvenlafaxine are described in the literature. These alternative processes generally proceed via the demethylation of venlafaxine, see, for example, U.S. Patent Application Publication No. 2005/197392, U.S. Patent Nos. 7,026,508, and 6,689,912, and International Patent Publication No. WO07/071404. These processes make use of different demethylating agents, such as lithium diphenylphosphide, alkali metal salts of trialkylborohydrides, high molecular weight thiolate anions, and metal sulfides. However, the use of the aforementioned demethylating agents presents several drawbacks, i.e., requires extensive purification procedures aimed to isolate desvenlafaxine from said demethylating agents and/or corresponding by-products, and involve odor workups, which make these processes unsuitable for industrial implementation.
In view of the foregoing, there is a need for an alternative process for preparing desvenlafaxine from venlafaxine including, for instance, an alternative process which avoids the drawbacks of current state of the art processes (e.g., makes use of simpler and shorter purification procedures, allows an essentially odorless workup, and which is well suited for industrial implementation).
Salts of O-desmethylvenlafaxine, including the fumarate, succinate and formate salts, have been described in the literature. For example, U.S. Patent No. 4,535,186 reports the preparation of O-desmethylvenlafaxine fumarate salt. More recently, the preparation of several polymorphic forms of the succinate salt have been reported in U.S. Patent No. 6,673,838 B2. Additionally, U.S. Patent Application Publication No. 2006/0058552 discloses the preparation of the formate salt.

HPLC Method
In the examples described below, the following analytical chromatographic HPLC method was used:
The chromatographic separation was carried out in a Kromasil C8, 5 μm, 25 cm x 4.6 mm. I. D column at room temperature.
The mobile phase was prepared by mixing 1,600 g of (NH4)H2P(It buffer solution pH = 4.4 and 313.2 g of acetonitrile HPLC grade. The pH of the mixture should be 4.9, adjust if necessary.
A (NH4)H2PO4 buffer solution (pH = 4.4) was prepared by dissolving 17 g of (NH4)H2PO4 in 1600 mL of water and adjusting the pH = 4.4 with HaPO4 or ammonium hydroxide.
The chromatograph was equipped with a 225 nm detector, and the flow rate was 1.2 mL per minute at room temperature. Test samples (20 μl) were prepared by dissolving the appropriate amount of sample to obtain 1 mg per mL concentration in the mobile phase.
In those conditions the retention time of desvanlafaxine, compound (I), is about 7 minutes, and the retention time of venlafaxine, compound (IV), is about 22 minutes.
EXAMPLE 1: Preparation of Desvenlafaxine (Le., Compound I).
This example illustrates a process for converting Compound IV into desvenlafaxine {i.e., Compound I) according to one aspect of the invention.
In a 100 mL flask 8 g (0.027 mol) of Venlafaxine free base, 13 mL of polyethylene glycol 400 (PEG400) and 6.9 g (0.041 mol) of 2-(diethylamino)ethanothiol were charged. 13.2 g (13.6 mL, 0.073 mol) of 30 % w/w solution of sodium methanolate in methanol were slowly added. The resulting suspension was heated to about 195° C and methanol was distilled off in the meantime. The stirring was continued for four hours at that temperature and then was cooled down to 20-25° C. 30 mL of 1 M hydrochloric acid were added to adjust pH to approx. 9.5. The resulting suspension was filtered at 20-25° C and the solid was dried at 50° C. The solid corresponded to desvenlafaxine (3.4 g; yield: 45 %; purity HPLC: 96.8 %).
EXAMPLE 2: Preparation of Desvenlafaxine Succinate Monohydrate.
This example illustrates a process for converting desvenlafaxine (i.e., Compound I) into desvenlafaxine succinate monohydrate according to one aspect of the invention.
Desvenlafaxine base (18.1 g, 0.069 mol) was charged into a 500 mL round bottomed flask under nitrogen atmosphere with 9.75 g (0.083 mol) of succinic acid, 135 g (170 mL) of acetone and 54 g of deionized water. The suspension was heated to reflux temperature and maintained at this temperature 30 minutes. The resulting solution was cooled to 50-55° C and filtered.
The filtered solution was cooled to 30-35° C in approximately 1 hour. In the interim, seeding was performed at approximately 40-45° C. The suspension was maintained for 3 hours at 30-35° C. Thereafter, the suspension was cooled to 20-25° C in approximately 1 hour, and maintained at this temperature for 2 hours. Then, the suspension was cooled to 10 ± 3° C in approximately 30 minutes and maintained at this temperature for 1 hour. Finally, the suspension was filtered and washed twice with 2 x 7.5 g (2 x 9.4 mL) of acetone. The wet solid was dried under vacuum at 60 ± 5° C to yield 22.96 g of desvenlafaxine succinate (yield: 83.6 %). Analytical data: HPLC Purity: 99.9 %; assay: 99.6 %.
Desvenlafaxine
-
- Synonyms:metabolite of Venlafaxine, O-desmethylvenlafaxine, WY-45233, DVS-233
- ATC:N06AX23
- Use:antidepressant
- Chemical name:4-[2-(dimethylamino)-1-(1-hydroxycyclohexyl)ethyl]phenol
- Formula:C16H25NO2
- MW:263.38 g/mol
- CAS-RN:93413-62-8
- InChI Key:KYYIDSXMWOZKMP-UHFFFAOYSA-N
- InChI:InChI=1S/C16H25NO2/c1-17(2)12-15(13-6-8-14(18)9-7-13)16(19)10-4-3-5-11-16/h6-9,15,18-19H,3-5,10-12H2,1-2H3
Derivatives
succinate monohydrate
- Formula:C20H33NO7
- MW:399.48 g/mol
- CAS-RN:386750-22-7
| Country |
Trade Name |
Vendor |
Annotation |
| USA |
Pristiq |
Wyeth ,2008 |
|
Formulations
- tabl. 50 mg, 100 mg (as succinate)
References
-
- a EP 1 973 866 (Synthon; 1.10.2008; appl. 19.12.2006; USA-prior. 20.12.2005).
- b WO 2 008 090 465 (Medichem SA; 31.7.2008; appl. 22.1.2008; USA-prior. 22.1.2007).
- c US 7 026 508 (Wyeth; 5.5.2005; appl. 10.11.2004; USA-prior. 12.2.2001).
- d US 4 535 186 (American Home Products; 13.8.1985; appl. 26.10.1983; USA-prior. 19.4.1983).
-
new polymorph:
- WO 2 008 110 338 (Synthon; 18.9.2008; appl. 6.3.2008; USA-prior. 9.3.2007).
-
crystalline polymorphs of Desvenlafaxine succinate:
- CN 101 274 897 (Mai DE Ltd.; 1.10.2008; appl. 4.1.2008; USA-prior. 8.1.2007).
- US 20 080 188 567 (Mai DE Ltd.; 8.7.2008; USA-prior. 8.1.2006).
-
enantiomers of Desvenlafaxine:
- US 2 002 022 662 (American Home Products; 21.2.2002; appl. 21.9.2001; USA-prior. 15.6.1999).
References
- ^ Jump up to:a b c d e f g h i “Desvenlafaxine Succinate Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 18 March 2019.
- ^ Jump up to:a b c d “Withdrawal Assessment Report for Dessvenlafaxime” (PDF). EMA. p. 3. Retrieved 22 March 2019.
- ^ “Desvenlafaxine Pregnancy and Breastfeeding Warnings”. Drugs.com. Retrieved 19 March 2019.
- ^ “NADAC as of 2019-02-27”. Centers for Medicare and Medicaid Services. Retrieved 3 March 2019.
- ^ “The Top 300 of 2019”. clincalc.com. Retrieved 22 December 2018.
- ^ Jump up to:a b “PRODUCT INFORMATION PRISTIQ® desvenlafaxine (as succinate)” (PDF). TGA eBusiness Services. Pfizer Australia Pty Ltd. 10 December 2012. Retrieved 8 November2013.
- ^ Perry, Richard; Cassagnol, Manouchkathe (2009). “Desvenlafaxine: a new serotonin-norepinephrine reuptake inhibitor for the treatment of adults with major depressive disorder”. Clinical Therapeutics. 31 Pt 1: 1374–1404. doi:10.1016/j.clinthera.2009.07.012. ISSN 1879-114X. PMID 19698900.
- ^ Thase ME, Kornstein SG, Germain JM, Jiang Q, Guico-Pabia C, Ninan PT (March 2009). “An integrated analysis of the efficacy of desvenlafaxine compared with placebo in patients with major depressive disorder”. CNS Spectr. 14 (3): 144–54. PMID 19407711.
- ^ Soares CN, Kornstein SG, Thase ME, Jiang Q, Guico-Pabia CJ (October 2009). “Assessing the efficacy of desvenlafaxine for improving functioning and well-being outcome measures in patients with major depressive disorder: a pooled analysis of 9 double-blind, placebo-controlled, 8-week clinical trials”. J Clin Psychiatry. 70 (10): 1365–71. doi:10.4088/JCP.09m05133blu. PMID 19906341.
- ^ “DESVENLAFAXINE tablet, extended release [Ranbaxy Pharmaceuticals Inc.]”. DailyMed. Ranbaxy Pharmaceuticals Inc. March 2013. Retrieved 9 November 2013.
- ^ “desvenlafaxine (Rx) – Pristiq, Khedezla”. Medscape Reference. WebMD. Retrieved 9 November 2013.
- ^ Lemke, Thomas L.; Williams, David A. (2012). Foye’s Principles of Medicinal Chemistry. Lippincott Williams & Wilkins. p. 609. ISBN 978-1-60913-345-0.
- ^ Jump up to:a b Deecher, DC; Beyer, CE; Johnston, G; Bray, J; Shah, S; Abou-Gharbia, M; Andree, TH (August 2006). “Desvenlafaxine succinate: A new serotonin and norepinephrine reuptake inhibitor” (PDF). The Journal of Pharmacology and Experimental Therapeutics. 318 (2): 657–665. doi:10.1124/jpet.106.103382. PMID 16675639.
- ^ Roth, BL; Driscol, J (Dec 2012). “PDSP Ki Database”. Psychoactive Drug Screening Program (PDSP). University of North Carolina at Chapel Hill and the United States National Institute of Mental Health. Retrieved 7 July 2018.
- ^ “Wyeth Receives Approvable Letter From FDA For Pristiq (Desvenlafaxine Succinate) For The Treatment Of Major Depressive Disorder” (Press release). 2007-01-23. Retrieved 2007-04-04.
- ^ “FDA Approves Pristiq” (Press release). Wyeth. 2008-02-29. Archived from the original on 2008-03-05. Retrieved 2008-02-29.
- ^ Health Canada Notice of Compliance – Pristiq[permanent dead link]. February 4, 2009, retrieved on March 9, 2009.
- ^ “Summary Basis of Decision (SBD) PrPristiq™”. Health Canada. 2009-05-29. Retrieved 2016-12-30.
- ^ “Pristiq 100 mg Comprimidos de Liberacion Prolongada”. AEMPS Medicines Online Information Center – CIMA. Retrieved 2016-12-30.
- ^ “Pristiq 50 mg Comprimidos de Liberacion Prolongada”. AEMPS Medicines Online Information Center – CIMA. Retrieved 2016-12-30.
External links
Route 1

Route 2

Route 3

Route 4

Route 5

Route 6

Publication numberPriority datePublication dateAssigneeTitle
WO2008093142A1 *2007-01-312008-08-07Generics [Uk] LimitedProcess for the preparation of o-desmethyl venlafaxine
WO2010028130A2 *2008-09-032010-03-11Concert Pharmaceuticals, Inc.Antidepressant compounds
CN101781221A *2010-02-112010-07-21上海凯米侬医药科技有限公司Preparation method of O-desmethylvenlafaxine
CN102249936A *2010-05-192011-11-23江苏豪森医药研究院有限公司Hydrate of O-desmethylvenlafaxine hydrochloride and preparation method thereof
FUJII, TOZO等: “Quinolizidines. XXII. An extension of the “3-acetylpyridine route” to the syntheses of 9-hydroxy-10-methoxy- and 10-hydroxy-9-methoxybenzo[a]quinolizidine-type Alangium alkaloids”, 《CHEMICAL & PHARMACEUTICAL BULLETIN》, vol. 35, no. 9, 31 December 1987 (1987-12-31), XP002311646 *
周金培等: “抗抑郁药文拉法辛的合成研究”, 《中国药科大学学报》, vol. 30, no. 4, 31 December 1999 (1999-12-31) *
///////DVS-233 , PF-05212375 , WY-45233, DESVENLAFAXINE SUCCINATE, WYETH

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....