
Home » 2019
Yearly Archives: 2019
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
ADAFOSBUVIR, адафосбувир , أدافوسبوفير ,
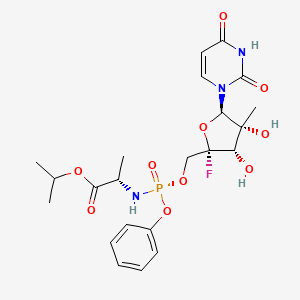


ADAFOSBUVIR
AL335; ALS-335; JNJ-64146212 , D11364
Propan-2-yl N-((P5’S)-4′-fluoro-2′-C-methyl-p-o-phenyl- 5′-uridylyl)-L-alaninate
propan-2-yl (2S)-2-{[(S)-{[(2S,3S,4R,5R)-5-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)-2-fluoro-3,4-dihydroxy-4-methyloxolan-2-yl]methoxy}(phenoxy)phosphoryl]amino}propanoate
Isopropyl (2S)-2-{[(S)-{[(2S,3S,4R,5R)-5-(2,4-dioxo-3,4-dihydro-1(2H)-pyrimidinyl)-2-fluoro-3,4-dihydroxy-4-methyltetrahydro-2-furanyl]methoxy}(phenoxy)phosphoryl]amino}propanoate (non-preferred name
Propan-2-yl N-((P5’S)-4′-fluoro-2′-C-methyl-p-o-phenyl- 5′-uridylyl)-L-alaninate
545.5 g/mol, C22H29FN3O10P
CAS Registry Number 1613589-09-5
Adafosbuvir is under investigation in clinical trial NCT02894905 (A Study to Evaluate the Effect of Renal Impairment on the Pharmacokinetics of AL-335).
- Originator Alios BioPharma
- Developer Alios BioPharma; Janssen
- Class Antivirals; Pyrimidine nucleotides; Uracil nucleotides
- Mechanism of Action Hepatitis C virus NS 5 protein inhibitors
- Phase II Hepatitis C
- 28 Oct 2019 No recent reports of development identified for phase-I development in Hepatitis-C(In volunteers) in USA (PO)
- 28 Sep 2018 No recent reports of development identified for phase-I development in Hepatitis-C in France (PO)
- 28 Sep 2018 No recent reports of development identified for phase-I development in Hepatitis-C in Georgia (PO)
Adafosbuvir (AL 335), a monophosphate prodrug, is being developed by Alios BioPharma (a subsidiary of Johnson & Johnson) for the treatment of hepatitis C virus (HCV) infections. Adafosbuvir acts a uridine-based nucleotide analogue polymerase inhibitor. Clinical development is underway in New Zealand, Japan, the UK, the US, France, Georgia, Mauritius and Moldova.
Adafosbuvir has emerged from the company’s research programme focused on developing anti-viral nucleotides for the treatment of HCV infections , In November 2014, Alios BioPharma was acquired by Johnson & Johnson As at September 2018, no recent reports of development had been identified for phase-I development in Hepatitis-C in France (PO), Georgia (PO).
As at October 2019, no recent reports of development had been identified for phase-I development in Hepatitis-C (In volunteers) in USA (PO).
useful for the treatment of hepatitis C viral infections, assignaed to Janssen Pharmaceuticals Inc and Achillion Pharmaceuticals Inc . Janssen Pharmaceuticals, following Johnson & Johnson’s acquisition of Alios , was developing adafosbuvir, a uridine (pyrimidine) nucleotide analog, from a series of back-up compounds, that acts by inhibiting HCV NS5B polymerase, for the potential treatment of HCV infection.
As of December 2019, AL-335 dose increased from 400 to 800 mg qd in the presence of reduced simeprevir and odalasvlr doses increased ALS-022227 less than dose proportionally. However, this effect was minimal in the absence of slmeprevir [1973148]. Also, the company was also developing JNJ-4178 , a triple combination of adafosbuvir, odalasvir and simeprevir for the same indication.

McGuigan phosphoramidate nucleotide prodrugs. (a) Sofosbuvir (GS-7977) (Sp isomer), (b) BMS-986094 (Rp and Sp isomer mixture), (c) Adafosbuvir (AL-335) (Sp isomer), (d) ACH-3422*, and (e) MIV-802* (Sp isomer)

Figure 3. Clinical and preclinical 30,50-CPO prodrug. (a) GS-0938 (Rp isomer) and (b) IDX19368 (Sp isomer).

PAPER
Journal of Medicinal Chemistry (2019), 62(9), 4555-4570.
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.9b00143
We report the synthesis and biological evaluation of a series of 4′-fluoro-2′-C-substituted uridines. Triphosphates of the uridine analogues exhibited a potent inhibition of hepatitis C virus (HCV) NS5B polymerase with IC50values as low as 27 nM. In an HCV subgenomic replicon assay, the phosphoramidate prodrugs of these uridine analogues demonstrated a very potent activity with EC50 values as low as 20 nM. A lead compound AL-335(53) demonstrated high levels of the nucleoside triphosphate in vitro in primary human hepatocytes and Huh-7 cells as well as in dog liver following a single oral dose. Compound 53 was selected for the clinical development where it showed promising results in phase 1 and 2 trials.

PATENT
WO 2014209979
WO2014100505
Family members of the product case of adafosbuvir, WO2014100505 , expire in the US in December 2033.
PATENT
US 20150368286
WO 2015054465
PATENT
WO2017059147 ( US20170087174 ), claiming combination comprising simeprevir , odalasvir and AL-335
PATENT
WO-2019237297
Process for preparing AL-335 (also known as adafosbuvir) and its intermediates. AL-355 is a nucleoside inhibitor of NS3B polymerase, which plays an important role in the replication of the hepatitis C virus.



Compound 4 may be prepared in accordance with the procedures described in international patent application WO 2015/200216. Compound 4 (1.0 equiv) was then dissolved in THF (10 L/kg) and cooled down to -25℃. iPrMgCl (2M in THF) was added slowly over one hour and the resulting mixture was stirred for one hour. The Compound 3 solution previously made (see above) was then added dropwise at -25℃ and the mixture was stirred for 5h at that temperature before being warmed to -5℃ and stirred for 10 additional hours at that temperature. Once the reaction was complete, the reaction was warmed up to 5℃ and an aqueous solution of NH 4Cl (5L/kg -9 w/w%) was added slowly over 30 minutes. After phase separation, the organic layer was washed with aqueous NaHCO 3 solution (5L/kg -10 w/w%) and twice with aqueous NaCl solution (5L/kg -10 w/w%) . After solvent switch to acetonitrile, the reaction was assayed and stored under nitrogen and used as such in the next step.
/////////////ADAFOSBUVIR, AL335, ALS-335, JNJ-64146212, Alios BioPharma, Janssen, hepatitis C viral infections, D11364, адафосбувир , أدافوسبوفير , PHASE 2
CC(C)OC(=O)[C@H](C)N[P@](=O)(OC[C@@]1(F)O[C@@H](N2C=CC(=O)NC2=O)[C@](C)(O)[C@@H]1O)OC1=CC=CC=C1
Cefiderocol, セフィデロコル , цефидерокол , سيفيديروكول , 头孢德罗 ,

Cefiderocol
セフィデロコル;
| Formula |
C30H34ClN7O10S2
|
|---|---|
| CAS |
1225208-94-5
|
| Mol weight |
752.2149
|
Antibacterial, Cell wall biosynthesis inhibitor, enicillin binding protein, Siderophore cephalosporin
Fetroja (TN)
FDA, Cefiderocol, APPROVED, 2019/11/14
(6R,7R)-7-{[(2Z)-2-(2-Amino-1,3-thiazol-4-yl)-2-{[(2-carboxy-2-propanyl)oxy]imino}acetyl]amino}-3-[(1-{2-[(2-chloro-3,4-dihydroxybenzoyl)amino]ethyl}-1-pyrrolidiniumyl)methyl]-8-oxo-5-thia-1-azabicycl o[4.2.0]oct-2-ene-2-carboxylate
S-649266, GSK 2696266D
Cefiderocol, sold under the brand name Fetroja, is an antibiotic used to treat complicated urinary tract infections when no other options are available.[2] It is indicated for the treatment of multi-drug-resistant Gram-negative bacteria including Pseudomonas aeruginosa.[3][4][5] It is given by injection into a vein.[6]
It is in the cephalosporin family of medications.[2][7] Cefiderocol was approved for medical use in the United States on November 14, 2019.[2][8]
Cefiderocol, also known as S-649266, is a potent siderophore cephalosporin antibiotic with a catechol moiety on the 3-position side chain. S-649266 shows potent in vitro activity against the non-fermenting Gram-negative bacteria Acinetobacter baumannii, Pseudomonas aeruginosa and Stenotrophomonas maltophilia, including MDR strains such as carbapenem-resistant A. baumannii and metallo-β-lactamase-producing P. aeruginosa. S-649266 showed potent in vitro activities against A. baumannii producing carbapenemases such as OXA-type β-lactamases, and P. aeruginosa producing metallo-β-lactamases such as IMP type and VIM type. FDA approved this drug in 11/14/2019 To treat patients with complicated urinary tract infections who have limited or no alternative treatment options
Medical uses
Cefiderocol is used to treat adults with complicated urinary tract infections, including kidney infections caused by susceptible Gram-negative microorganisms, who have limited or no alternative treatment options.[2][7]
Mechanism of action
Its mechanism of entry into bacterial cells is by binding to iron, which is actively transported into the bacterial cells along with the cefiderocol.[6][9][10][11][12] It is in a medication class known as siderophores,[6][7] and was the first siderophore antibiotic to be approved by the U.S. Food and Drug Administration (FDA).[13] It bypasses the bacterial porin channels by using the bacteria’s own iron-transport system for being transported in.[14]
History
In 2019, cefiderocol was approved in the United States as an antibacterial drug for treatment of adults 18 years of age or older with complicated urinary tract infections (cUTI), including kidney infections caused by susceptible Gram-negative microorganisms, who have limited or no alternative treatment options.[2][8]
The safety and effectiveness of cefiderocol was demonstrated in a study of 448 patients with cUTIs.[2] Of the patients who were administered cefiderocol, 72.6% had resolution of symptoms and eradication of the bacteria approximately seven days after completing treatment, compared with 54.6% in patients who received an alternative antibiotic.[2] The clinical response rates were similar between the two treatment groups.[2]
Labeling for cefiderocol includes a warning regarding the higher all-cause mortality rate observed in cefiderocol-treated patients compared to those treated with other antibiotics in a trial in critically ill patients with multidrug-resistant Gram-negative bacterial infections.[2] The cause of the increase in mortality has not been established.[2] Some of the deaths were a result of worsening or complications of infection, or underlying co-morbidities.[2] The higher all-cause mortality rate was observed in patients treated for hospital-acquired/ventilator-associated pneumonia (i.e.nosocomial pneumonia), bloodstream infections, or sepsis.[2] The safety and efficacy of cefiderocol has not been established for the treatment of these types of infections.[2]
Cefiderocol received a Qualified Infectious Disease Product designation from the U.S. Food and Drug Administration (FDA) and was granted priority review.[2] The FDA granted approval of Fetroja, on November 14, 2019, to Shionogi & Co., Ltd.[2]
PATENT
WO 2010050468
WO 2016035845
WO 2016035847
PATENT
WO 2017216765,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017216765&tab=PCTDESCRIPTION
Bacterial infections continue to remain one of the major causes contributing towards human diseases. One of the key challenges in treatment of bacterial infections is the ability of bacteria to develop resistance to one or more antibacterial agents over time. Examples of such bacteria that have developed resistance to typical antibacterial agents include: Penicillin-resistant Streptococcus pneumoniae, Vancomycin-resistant Enterococci, and Methicillin-resistant Staphylococcus aureus. The problem of emerging drug-resistance in bacteria is often tackled by switching to newer antibacterial agents, which can be more expensive and sometimes more toxic. Additionally, this may not be a permanent solution as the bacteria often develop resistance to the newer antibacterial agents as well in due course. In general, bacteria are particularly efficient in developing resistance, because of their ability to multiply very rapidly and pass on the resistance genes as they replicate. Therefore, there is a need for development of newer ways to treat infections that are becoming resistant to known therapies and methods.
Surprisingly, it has been found that the compositions comprising a compound of Formula (I) or a pharmaceutically acceptable salt thereof and at least one beta-lactamase inhibitor or a pharmaceutically acceptable salt thereof, exhibit synergistic antibacterial activity, even against resistant bacterial strains.
Formula (I)
Example 1
Synthesis of Compound of formula (I)
Step-1: Preparation of intermediate (1):
To the clear solution of (Z)-2[(2-tert-butoxycarbonyl amino-thiazol-4-yl)-carboxy-methyleneaminooxy]2-methyl-propionic acid tert-butyl ester (30 gm, 69.93 mmol) in N,N-dimethyl acetamide (300 ml) was charged triethylamine (17.68 ml, 125.87 mmol) under stirring. The reaction mixture was cooled to -15°C. Methane sulfonyl chloride (12.01 gm, 104. 89 mmol) was charged to this cooled reaction mixture via addition funnel while maintaining temperature at about -15°C. The reaction mixture was stirred for 30 minutes at -15°C after the addition. To the reaction mixture was charged (6 ?,75)-4-methoxybenzyl-7-amino-3-chloromethyl-8-oxo-5-thia-l-aza-bicyclo[4.2.0]oct-2-ene-2-carboxylate hydrochloride salt (28.25 gm, 69.93 mmol) along with N-methyl morpholine (15.5 ml, 139.86 mmol). The reaction mixture was stirred further for 1 hour at -15°C and the reaction progress was monitored using TLC. After completion of reaction, ethyl acetate (1.2 L) was charged followed by IN aqueous hydrochloric acid (1.2 L) under stirring and cooling was removed to warm up reaction mixture to room temperature. Layers were separated and organic layer was washed with saturated aqueous sodium bicarbonate solution (500 ml) followed by brine (500 ml). Organic layer was dried over sodium sulphate and was evaporated under vacuum to provide a crude mass. It was purified using silica gel column chromatography (60-120 mesh, 30% ethyl acetate in hexane) to provide 38 gm of intermediate (1).
Analysis:
1H NMR (CDCls) δ ppm: 8.29 (br s, 1H), 8.17 (d, 1H), 7.35 (d, 2H), 7.31 (s, 1H), 6.91 (d, 2H), 6.21 (dd, 1H), 5.23 (dd, 2H), 5.05 (d, 1H), 4.55 (d, 1H), 4.46 (d, 1H), 3.82 (s, 3H), 3.65 (d, 1H), 3.48 (d, 1H), 1.62 (s, 3H), 1.59 (s, 3H), 1.53 (s, 9H), 1.45 (s, 9H).
Step-2: Preparation of intermediate (2):
The solution of intermediate 1 (45 gm, 57.76 mmol) in dichloro methane (450 ml) was cooled to about -40°C and m-chloroperbenzoic acid (18 gm, 57.76 mmol) was added in three lots at -40°C under stirring. The mixture was stirred for 30 minutes and allowed to warm at -20°C. As TLC showed complete conversion, 5% aqueous sodium thiosulfate solution (1.2 L) was added at -15°C under stirring. The mixture was allowed to warm at room temperature and was charged with ethyl acetate (1.5 L) and stirred for 30 minutes and layers were separated. Organic layer was washed with saturated aqueous sodium bicarbonate solution (1 L) followed by brine (500 ml).
Organic layer was dried over sodium sulphate and evaporated under vacuum to provide 46 gm of intermediate (2).
Analysis:
1H NMR (CDC13) δ ppm: 8.48 (br s, 1H), 7.89 (d, 1H), 7.34 (d, 2H), 7.29 (s, 1H), 6.92 (d, 2H), 6.21 (dd, 1H), 5.27 (dd, 2H), 5.04 (br d, 1H), 4.58 (d, 1H), 4.23 (d, 1H), 3.83 (s, 3H), 3.82 (d, 1H), 3.43 (d, 1H), 1.60 (s, 3H), 1.58 (s, 3H), 1.53 (9H)1.42 (s, 9H).
Step-3: Preparation of intermediate (3):
Part-1: To the clear solution of intermediate 2 (35 gm, 44.02 mmol) in tetrahydrofuran (350 ml) was charged potassium iodide (14.61 gm, 88.05 mmol) under stirring at 25°C. The suspension was stirred for 5 hours at the same temperature and the reaction was monitored using mass spectroscopy. After completion of the reaction ethyl acetate (600 ml) was added to the reaction mixture followed by 5% aqueous sodium thiosulphate (600 ml). The reaction mixture was stirred for 15 minutes and layers were separated. Organic layer was washed with demineralised water (500 ml) followed by brine (500 ml). Organic layer was dried over sodium sulphate and evaporated to dryness under vacuum to provide 38 gm of corresponding iodo-methyl intermediate.
Part-2: To the iodo-methyl intermediate obtained (37.24 gm, 41.98 mmol) in N,N-dimethylformamide (35 ml) was added 2-chloro-3,4-di-(4-methoxybenzyloxy)-N-(pyrrolidin-l-ylethyl)-benzamide (22 gm, 42.98 mmol). The thick mass was stirred at 25°C for 15 hours and the reaction was monitored using mass spectroscopy. Potassium iodide (48.78 gm, 293.8 mmol) was charged to the reaction mass under stirring at 25 °C. The reaction mixture was cooled to -40°C and acetyl chloride (12 ml, 167.9 mmol) was added. After completion of the reaction ethyl acetate (1.2 L) followed by demineralised water (1.2 L) was added to the reaction mass at 0°C. Layers were separated and organic layer was washed with demineralised water (500 ml) followed by brine (500 ml). Organic layer was dried over sodium sulphate and was evaporated to dryness under vacuum to obtain quaternary intermediate (3) as iodide salt.
Step-4: Preparation compound of Formula (I):
Compound (3) (30 gm, 21.5 mmol) was dissolved in dichloro methane (300 ml) and anisole (30 gm, mmol) was added under stirring at 25°C. The mixture was cooled to -40° C and 2M aluminium chloride solution in nitromethane (150 ml) was added over 45 minutes at -40°C. As addition was completed reaction mixture was stirred for 1 hour at 0°C. To the reaction mixture 2M aqueous hydrochloric acid (750 ml) and acetonitrile (750 ml) were added and the stirring was
continued for 15 minutes. Di-isopropyl ether (1.5 L) was charged to the reaction mixture and the reaction mass was stirred for 15 minutes at 25°C, and the layers were separated. Aqueous layer was washed with additional di-isopropyl ether (500 ml). HP-21 resin (150 gm) was charged to the aqueous layer. The aqueous layer along with resin was loaded on a resin HP-21 column. The column was eluted with demineralised water till pH of eluent became neutral. Then the column was eluted with 10% acetonitrile in water mixture. Finally the column was eluted with 20% acetonitrile in water mixture. Evaporation of required fractions below 40°C under vacuum provided 5.5 gm of crude compound (I). The crude compound (I) was purified by dissolving in acetonitrile (200 ml) and demineralised water (200 ml) mixture followed by addition of HP-21 resin (200 gm).The slurry thus obtained was loaded on HP-21 resin column. The column was eluted first with demineralised water (3 L) followed by 10% acetonitrile in water mixture (2 L) then followed by 20% acetonitrile in water mixture till complete pure compound from the column is eluted. Pure fractions were collected and lyophilized under vacuum to provide titled compound (I) in pure form.
Analysis:
1H NMR (DMSO d6) δ ppm: 12.5 (br s, 2H), 9.42 (br s, 1H), 8.41 (br t, 1H), 7.28 (br s, 3H), 6.78 (s, 2H), 6.73 (s, 1H), 5.73 (dd, 1H), 5.15 (d, 1H), 5.08 (br d, 1H), 3.71-3.91 (m, 4H), 3.21-3.60 (m, 7H), 1.95-2.19 (m, 4H)1.76 (s, 3H), 1.44 (s, 3H).
HPLC purity: 90.80%
PATENT
WO 2019093450
Prior art documents
Non-patent literature
Non-patent Document 2: The Lancet Infrction diseases, 13 (9), 785-796,2013
Non-patent Document 3: Antimicrobial Agents and Chemotherapy, 61 (3), 1-11, 2017
PAPER
European Journal of Medicinal Chemistry (2018), 155, 847-868
References
- ^ Katsube, T.; Echols, R.; Arjona Ferreira, J. C.; et al. (2017). “Cefiderocol, a Siderophore Cephalosporin for Gram‐Negative Bacterial Infections: Pharmacokinetics and Safety in Subjects With Renal Impairment”. Journal of Clinical Pharmacology. 57 (5): 584–591. doi:10.1002/jcph.841. PMC 5412848. PMID 27874971.
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA approves new antibacterial drug to treat complicated urinary tract infections as part of ongoing efforts to address antimicrobial resistance”. U.S. Food and Drug Administration (FDA) (Press release). 14 November 2019. Archived from the original on 16 November 2019. Retrieved 15 November 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Choi, Justin J; McCarthy, Matthew W. (24 January 2018). “Cefiderocol: a novel siderophore cephalosporin”. Expert Opinion on Investigational Drugs. 27 (2): 193–197. doi:10.1080/13543784.2018.1426745. PMID 29318906.
- ^ Aoki, Toshiaki; Yoshizawa, Hidenori; Yamawaki, Kenji; et al. (15 July 2018). “Cefiderocol (S-649266), A new siderophore cephalosporin exhibiting potent activities against Pseudomonas aeruginosa and other gram-negative pathogens including multi-drug resistant bacteria: Structure activity relationship”. European Journal of Medicinal Chemistry. 155: 847–868. doi:10.1016/j.ejmech.2018.06.014. ISSN 1768-3254. PMID 29960205.
- ^ Portsmouth, Simon; van Veenhuyzen, David; Echols, Roger; et al. (25 October 2018). “Cefiderocol versus imipenem-cilastatin for the treatment of complicated urinary tract infections caused by Gram-negative uropathogens: a phase 2, randomised, double-blind, non-inferiority trial”. The Lancet Infectious Diseases. 0 (12): 1319–1328. doi:10.1016/S1473-3099(18)30554-1. ISSN 1473-3099. PMID 30509675.
- ^ Jump up to:a b c “Fetroja (cefiderocol) for injection, for intravenous use full prescribing information”(PDF). November 2019. Retrieved 17 November 2019. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c Zhanel GG, Golden AR, Zelenitsky S, et al. (February 2019). “Cefiderocol: A Siderophore Cephalosporin with Activity Against Carbapenem-Resistant and Multidrug-Resistant Gram-Negative Bacilli”. Drugs. 79 (3): 271–289. doi:10.1007/s40265-019-1055-2. PMID 30712199.
- ^ Jump up to:a b “Cefiderocol New Drug Application”. U.S. Food and Drug Administration (FDA). Archived from the original on 4 December 2019. Retrieved 22 November 2019. This article incorporates text from this source, which is in the public domain.
- ^ Sato T, Yamawaki K (November 2019). “Cefiderocol: Discovery, Chemistry, and In Vivo Profiles of a Novel Siderophore Cephalosporin”. Clin. Infect. Dis. 69 (Supplement_7): S538–S543. doi:10.1093/cid/ciz826. PMC 6853759. PMID 31724047.
- ^ Matthews-King A (26 October 2018). “Antibiotic ‘Trojan horse’ could defeat superbugs causing global medical crisis, trial finds”. The Independent. Retrieved 26 October 2018.
- ^ Newey S (26 October 2018). “New ‘Trojan horse’ drug proves effective against antibiotic resistant bacteria”. The Telegraph. ISSN 0307-1235. Retrieved 26 October 2018.
- ^ Simpson DH, Scott P (2017). “Antimicrobial Metallodrugs”. In Lo K (ed.). Inorganic and Organometallic Transition Metal Complexes with Biological Molecules and Living Cells. Elsevier. ISBN 9780128038871.
- ^ Saisho, Yutaka; Katsube, Takayuki; White, Scott; et al. (March 2018). “Pharmacokinetics, Safety, and Tolerability of Cefiderocol, a Novel Siderophore Cephalosporin for Gram-Negative Bacteria, in Healthy Subjects” (PDF). Antimicrobial Agents and Chemotherapy. 62 (3): 1–12. doi:10.1128/AAC.02163-17. PMC 5826143. PMID 29311072. Retrieved 22 November 2019.
- ^ Ito A, Nishikawa T, Matsumoto S, et al. (December 2016). “Siderophore Cephalosporin Cefiderocol Utilizes Ferric Iron Transporter Systems for Antibacterial Activity against Pseudomonas aeruginosa”. Antimicrobial Agents and Chemotherapy. 60 (12): 7396–7401. doi:10.1128/AAC.01405-16. PMC 5119021. PMID 27736756.
External links
- “Cefiderocol”. Drug Information Portal. U.S. National Library of Medicine.
ADDITIONAL INFORMATION
S-649266 shows potent in vitro activity against the non-fermenting Gram-negative bacteria Acinetobacter baumannii, Pseudomonas aeruginosa and Stenotrophomonas maltophilia, including MDR strains such as carbapenem-resistant A. baumannii and metallo-β-lactamase-producing P. aeruginosa. MIC90s of S-649266 for A. baumannii, P. aeruginosa and S. maltophilia were 2, 1 and 0.5 mg/L, respectively, whereas MIC90s of meropenem were >16 mg/L. S-649266 showed potent in vitro activities against A. baumannii producing carbapenemases such as OXA-type β-lactamases, and P. aeruginosa producing metallo-β-lactamases such as IMP type and VIM type. MIC90 values for these A. baumannii strains and P. aeruginosa strains were 8 and 4 mg/L, respectively.
REFERENCES
1: Yamano Y. In Vitro Activity of Cefiderocol Against a Broad Range of Clinically Important Gram-negative Bacteria. Clin Infect Dis. 2019 Nov 13;69(Supplement_7):S544-S551. doi: 10.1093/cid/ciz827. PubMed PMID: 31724049; PubMed Central PMCID: PMC6853761.
2: Echols R, Ariyasu M, Nagata TD. Pathogen-focused Clinical Development to Address Unmet Medical Need: Cefiderocol Targeting Carbapenem Resistance. Clin Infect Dis. 2019 Nov 13;69(Supplement_7):S559-S564. doi: 10.1093/cid/ciz829. PubMed PMID: 31724048; PubMed Central PMCID: PMC6853756.
3: Sato T, Yamawaki K. Cefiderocol: Discovery, Chemistry, and In Vivo Profiles of a Novel Siderophore Cephalosporin. Clin Infect Dis. 2019 Nov 13;69(Supplement_7):S538-S543. doi: 10.1093/cid/ciz826. PubMed PMID: 31724047; PubMed Central PMCID: PMC6853759.
4: Bonomo RA. Cefiderocol: A Novel Siderophore Cephalosporin Defeating Carbapenem-resistant Pathogens. Clin Infect Dis. 2019 Nov 13;69(Supplement_7):S519-S520. doi: 10.1093/cid/ciz823. PubMed PMID: 31724046; PubMed Central PMCID: PMC6853757.
5: Katsube T, Echols R, Wajima T. Pharmacokinetic and Pharmacodynamic Profiles of Cefiderocol, a Novel Siderophore Cephalosporin. Clin Infect Dis. 2019 Nov 13;69(Supplement_7):S552-S558. doi: 10.1093/cid/ciz828. PubMed PMID: 31724042; PubMed Central PMCID: PMC6853762.
6: Kidd JM, Abdelraouf K, Nicolau DP. Efficacy of Humanized Cefiderocol Exposure is Unaltered by Host Iron Overload in the Thigh Infection Model. Antimicrob Agents Chemother. 2019 Oct 28. pii: AAC.01767-19. doi: 10.1128/AAC.01767-19. [Epub ahead of print] PubMed PMID: 31658966.
7: Chen IH, Kidd JM, Abdelraouf K, Nicolau DP. Comparative In Vivo Antibacterial Activity of Human-Simulated Exposures of Cefiderocol and Ceftazidime against Stenotrophomonas maltophilia in the Murine Thigh Model. Antimicrob Agents Chemother. 2019 Oct 7. pii: AAC.01558-19. doi: 10.1128/AAC.01558-19. [Epub ahead of print] PubMed PMID: 31591126.
8: Stevens RW, Clancy M. Compassionate Use of Cefiderocol in the Treatment of an Intraabdominal Infection Due to Multidrug-Resistant Pseudomonas aeruginosa: A Case Report. Pharmacotherapy. 2019 Nov;39(11):1113-1118. doi: 10.1002/phar.2334. Epub 2019 Oct 22. PubMed PMID: 31550054.
9: Sanabria C, Migoya E, Mason JW, Stanworth SH, Katsube T, Machida M, Narukawa Y, Den Nagata T. Effect of Cefiderocol, a Siderophore Cephalosporin, on QT/QTc Interval in Healthy Adult Subjects. Clin Ther. 2019 Sep;41(9):1724-1736.e4. doi: 10.1016/j.clinthera.2019.07.006. Epub 2019 Aug 1. PubMed PMID: 31378318.
10: Trecarichi EM, Quirino A, Scaglione V, Longhini F, Garofalo E, Bruni A, Biamonte E, Lionello R, Serapide F, Mazzitelli M, Marascio N, Matera G, Liberto MC, Navalesi P, Torti C; IMAGES Group . Successful treatment with cefiderocol for compassionate use in a critically ill patient with XDR Acinetobacter baumannii and KPC-producing Klebsiella pneumoniae: a case report. J Antimicrob Chemother. 2019 Nov 1;74(11):3399-3401. doi: 10.1093/jac/dkz318. PubMed PMID: 31369095.
11: Nakamura R, Ito-Horiyama T, Takemura M, Toba S, Matsumoto S, Ikehara T, Tsuji M, Sato T, Yamano Y. In Vivo Pharmacodynamic Study of Cefiderocol, a Novel Parenteral Siderophore Cephalosporin, in Murine Thigh and Lung Infection Models. Antimicrob Agents Chemother. 2019 Aug 23;63(9). pii: e02031-18. doi: 10.1128/AAC.02031-18. Print 2019 Sep. PubMed PMID: 31262762; PubMed Central PMCID: PMC6709502.
12: Katsube T, Saisho Y, Shimada J, Furuie H. Intrapulmonary pharmacokinetics of cefiderocol, a novel siderophore cephalosporin, in healthy adult subjects. J Antimicrob Chemother. 2019 Jul 1;74(7):1971-1974. doi: 10.1093/jac/dkz123. PubMed PMID: 31220260; PubMed Central PMCID: PMC6587409.
13: Jean SS, Hsueh SC, Lee WS, Hsueh PR. Cefiderocol: a promising antibiotic against multidrug-resistant Gram-negative bacteria. Expert Rev Anti Infect Ther. 2019 May;17(5):307-309. doi: 10.1080/14787210.2019.1612240. Epub 2019 May 6. PubMed PMID: 31055983.
14: Hackel MA, Tsuji M, Yamano Y, Echols R, Karlowsky JA, Sahm DF. Reproducibility of broth microdilution MICs for the novel siderophore cephalosporin, cefiderocol, determined using iron-depleted cation-adjusted Mueller-Hinton broth. Diagn Microbiol Infect Dis. 2019 Aug;94(4):321-325. doi: 10.1016/j.diagmicrobio.2019.03.003. Epub 2019 Mar 23. PubMed PMID: 31029489.
15: Miyazaki S, Katsube T, Shen H, Tomek C, Narukawa Y. Metabolism, Excretion, and Pharmacokinetics of [(14) C]-Cefiderocol (S-649266), a Siderophore Cephalosporin, in Healthy Subjects Following Intravenous Administration. J Clin Pharmacol. 2019 Jul;59(7):958-967. doi: 10.1002/jcph.1386. Epub 2019 Feb 7. PubMed PMID: 30730562; PubMed Central PMCID: PMC6593826.
16: Zhanel GG, Golden AR, Zelenitsky S, Wiebe K, Lawrence CK, Adam HJ, Idowu T, Domalaon R, Schweizer F, Zhanel MA, Lagacé-Wiens PRS, Walkty AJ, Noreddin A, Lynch Iii JP, Karlowsky JA. Cefiderocol: A Siderophore Cephalosporin with Activity Against Carbapenem-Resistant and Multidrug-Resistant Gram-Negative Bacilli. Drugs. 2019 Feb;79(3):271-289. doi: 10.1007/s40265-019-1055-2. Review. PubMed PMID: 30712199.
17: Huttner A. Cefiderocol for treatment of complicated urinary tract infections – Author’s reply. Lancet Infect Dis. 2019 Jan;19(1):24-25. doi: 10.1016/S1473-3099(18)30728-X. PubMed PMID: 30587291.
18: Portsmouth S, Echols R, Den Nagata T. Cefiderocol for treatment of complicated urinary tract infections. Lancet Infect Dis. 2019 Jan;19(1):23-24. doi: 10.1016/S1473-3099(18)30721-7. PubMed PMID: 30587290.
19: Wagenlehner FME, Naber KG. Cefiderocol for treatment of complicated urinary tract infections. Lancet Infect Dis. 2019 Jan;19(1):22-23. doi: 10.1016/S1473-3099(18)30722-9. PubMed PMID: 30587289.
20: Portsmouth S, van Veenhuyzen D, Echols R, Machida M, Ferreira JCA, Ariyasu M, Tenke P, Nagata TD. Cefiderocol versus imipenem-cilastatin for the treatment of complicated urinary tract infections caused by Gram-negative uropathogens: a phase 2, randomised, double-blind, non-inferiority trial. Lancet Infect Dis. 2018 Dec;18(12):1319-1328. doi: 10.1016/S1473-3099(18)30554-1. Epub 2018 Oct 25. PubMed PMID: 30509675.
 |
|
| Clinical data | |
|---|---|
| Trade names | Fetroja |
| Routes of administration |
Intravenous infusion |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | 56–58%[1] |
| Elimination half-life | 2.8 hours |
| Excretion | mainly renal (60–70% unchanged) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C30H34ClN7O10S2 |
| Molar mass | 752.21 g·mol−1 |
| 3D model (JSmol) | |
////////////Cefiderocol, セフィデロコル , FDA 2019, цефидерокол , سيفيديروكول , 头孢德罗 , S-649266, GSK 2696266D
Givosiran, ギボシラン ,
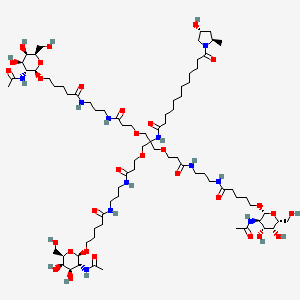



Givosiran
N-[1,3-bis[3-[3-[5-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxypentanoylamino]propylamino]-3-oxopropoxy]-2-[[3-[3-[5-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxypentanoylamino]propylamino]-3-oxopropoxy]methyl]propan-2-yl]-12-[(2R,4R)-4-hydroxy-2-methylpyrrolidin-1-yl]-12-oxododecanamide
Treatment of Acute Hepatic Porphyria (AHP)
| Formula |
C524H694F16N173O316P43S6
|
|---|---|
| CAS |
1639325-43-1
|
| Mol weight |
16300.3229
|
Treatment of acute hepatic porphyria, RNA interference (RNAi) drug
FDA APPROVED, Givlaari, 2019/11/20
ギボシラン;
Givosiran, sold under the brand name Givlaari, is for the treatment of adults with acute hepatic porphyria, a genetic disorder resulting in the buildup of toxic porphyrin molecules which are formed during the production of heme (which helps bind oxygen in the blood).[1][2]
History
The U.S. Food and Drug Administration (FDA) granted the application for givosiran breakthrough therapy designation, priority reviewdesignation, and orphan drug designation.[1] The FDA granted the approval of Givlaari to Alnylam Pharmaceuticals.[1]
The full ENVISION results demonstrated a 74 percent mean and 90 percent median reduction in the primary endpoint measure of annualized rate of composite attacks in patients on givosiran relative to placebo during the six-month double-blind period. In addition, givosiran achieved statistically significant positive results for five of nine secondary endpoints, with an overall safety and tolerability profile that the Company believes is encouraging, especially in this high unmet need disease. Adverse events (AEs) were reported in 89.6 percent of givosiran patients and 80.4 percent of placebo patients; serious adverse events (SAEs) were reported in 20.8 percent of givosiran patients and 8.7 percent of placebo patients. Ninety-three of 94 patients, or 99 percent, enrolled in the open-label extension (OLE) period of the study. Based on the ENVISION results, the Company plans to complete its rolling submission of a New Drug Application (NDA) and file a Marketing Authorisation Application (MAA) in mid-2019.
“Given the high unmet need in this disease setting, we are very pleased for the patients and families living with acute hepatic porphyria for whom these results signal hope for a potential new therapeutic option,” said Akshay Vaishnaw, M.D., Ph.D., President of R&D at Alnylam. “Givosiran substantially reduced the frequency of attacks, providing strong support for a treatment benefit, with a consistent effect across all components of the primary endpoint and all subgroups analyzed. In this disease with high burden and associated comorbidities, we’re encouraged by the overall tolerability profile. We firmly believe givosiran has the potential to be a transformative medicine for patients living with AHP.”
“Currently, there are no approved therapies aimed at preventing the painful, often incapacitating attacks and chronic symptoms associated with AHP,” said Manisha Balwani, M.D., M.S, Associate Professor of the Department of Genetics and Genomic Sciences and Department of Medicine at the Icahn School of Medicine at Mount Sinai and principal investigator of the ENVISION study. “The results from ENVISION are promising and demonstrate a strong treatment effect for givosiran, with reduction of attacks and improvement in patient-reported measures of overall health status and quality of life. Thus, givosiran represents a novel and targeted treatment approach that has the potential to make a significant impact on the lives of patients who are struggling with the disabling symptoms of this disease.”
Efficacy Results
Givosiran met the primary efficacy endpoint with a 74 percent mean reduction relative to placebo in the annualized rate of composite porphyria attacks, defined as those requiring hospitalization, urgent healthcare visit, or hemin administration, in patients with acute intermittent porphyria (AIP) over six months (p equal to 6.04×10-9). There was a corresponding 90 percent median reduction in composite annualized attack rate (AAR), with a median AAR of 1.0 in givosiran patients compared with a median AAR of 10.7 in placebo patients. Fifty percent of givosiran-treated patients were attack-free during the six-month treatment period as compared to 16.3 percent of placebo-treated patients. The reductions in attack rates were observed across all components of the primary endpoint. The treatment benefit for givosiran compared to placebo was maintained across all pre-specified patient subgroups, including age, race, geography, historical attack rates, prior hemin prophylaxis status, disease severity, and other baseline characteristics.
Givosiran also demonstrated statistically significant differences in five of nine hierarchically tested secondary endpoints relative to placebo. These included mean reductions of:
- 91 percent in urinary aminolevulinic acid (ALA) in patients with AIP at three months (p equal to 8.74×10-14).
- 83 percent in urinary ALA in patients with AIP at six months (p equal to 6.24×10-7).
- 73 percent in urinary levels of porphobilinogen (PBG) in patients with AIP at six months (p equal to 8.80×10-7).
- 77 percent in the number of annualized days on hemin in patients with AIP (p equal to 2.35×10-5).
- 73 percent in composite AAR for patients with any AHP (p equal to 1.35×10-8).
The remaining four secondary endpoints did not meet the prespecified criteria for statistical significance in hierarchical testing.

About Acute Hepatic Porphyria
Acute hepatic porphyria (AHP) refers to a family of rare, genetic diseases characterized by potentially life-threatening attacks and for some patients chronic debilitating symptoms that negatively impact daily functioning and quality of life. AHP is comprised of four subtypes, each resulting from a genetic defect leading to deficiency in one of the enzymes of the heme biosynthesis pathway in the liver: acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), variegate porphyria (VP), and ALAD-deficiency porphyria (ADP). These defects cause the accumulation of neurotoxic heme intermediates aminolevulinic acid (ALA) and porphobilinogen (PBG), with ALA believed to be the primary neurotoxic intermediate responsible for causing both attacks and ongoing symptoms between attacks. Common symptoms of AHP include severe, diffuse abdominal pain, weakness, nausea, and fatigue. The nonspecific nature of AHP signs and symptoms can often lead to misdiagnoses of other more common conditions such as irritable bowel syndrome, appendicitis, fibromyalgia, and endometriosis, and consequently, patients afflicted by AHP often remain without a proper diagnosis for up to 15 years. In addition, long-term complications of AHP and its treatment can include chronic neuropathic pain, hypertension, chronic kidney disease and liver disease, including iron overload, fibrosis, cirrhosis and hepatocellular carcinoma. Currently, there are no treatments approved to prevent debilitating attacks or to treat the chronic manifestations of the disease.
About Givosiran
Givosiran is an investigational, subcutaneously administered RNAi therapeutic targeting aminolevulinic acid synthase 1 (ALAS1) in development for the treatment of acute hepatic porphyria (AHP). Monthly administration of givosiran has the potential to significantly lower induced liver ALAS1 levels in a sustained manner and thereby decrease neurotoxic heme intermediates, aminolevulinic acid (ALA) and porphobilinogen (PBG), to near normal levels. By reducing accumulation of these intermediates, givosiran has the potential to prevent or reduce the occurrence of severe and life-threatening attacks, control chronic symptoms, and decrease the burden of the disease. Givosiran utilizes Alnylam’s Enhanced Stabilization Chemistry ESC-GalNAc conjugate technology, which enables subcutaneous dosing with increased potency and durability and a wide therapeutic index. Givosiran has been granted Breakthrough Therapy Designation by the U.S. Food and Drug Administration (FDA) and PRIME Designation by the European Medicines Agency (EMA). Givosiran has also been granted Orphan Drug Designations in both the U.S. and the EU for the treatment of AHP. The safety and efficacy of givosiran were evaluated in the ENVISION Phase 3 trial with positive results; these results have not been evaluated by the FDA, the EMA or any other health authority.
About RNAi
RNAi (RNA interference) is a natural cellular process of gene silencing that represents one of the most promising and rapidly advancing frontiers in biology and drug development today. Its discovery has been heralded as “a major scientific breakthrough that happens once every decade or so,” and was recognized with the award of the 2006 Nobel Prize for Physiology or Medicine. By harnessing the natural biological process of RNAi occurring in our cells, a new class of medicines, known as RNAi therapeutics, is now a reality. Small interfering RNA (siRNA), the molecules that mediate RNAi and comprise Alnylam’s RNAi therapeutic platform, function upstream of today’s medicines by potently silencing messenger RNA (mRNA) – the genetic precursors – that encode for disease-causing proteins, thus preventing them from being made. This is a revolutionary approach with the potential to transform the care of patients with genetic and other diseases.
References
- ^ Jump up to:a b c “FDA approves first treatment for inherited rare disease”. U.S. Food and Drug Administration (FDA) (Press release). 20 November 2019. Archived from the original on 21 November 2019. Retrieved 20 November 2019. This article incorporates text from this source, which is in the public domain.
- ^ “FDA approves givosiran for acute hepatic porphyria”. U.S. Food and Drug Administration (FDA) (Press release). 20 November 2019. Archived from the original on 21 November 2019. Retrieved 20 November 2019. This article incorporates text from this source, which is in the public domain.
- The New England journal of medicine (2019), 380(6), 549-558.
- New England Journal of Medicine (2019), 380(6), 549-558.
- Toxicologic Pathology (2018), 46(7), 735-745.
External links
- “Givosiran”. Drug Information Portal. U.S. National Library of Medicine (NLM).
-
GIVLAARI
(givosiran) Injection, for Subcutaneous UseDESCRIPTION
GIVLAARI is an aminolevulinate synthase 1-directed small interfering RNA (siRNA), covalently linked to a ligand containing three N-acetylgalactosamine (GalNAc) residues to enable delivery of the siRNA to hepatocytes.
The structural formulas of the givosiran drug substance in its sodium form, and the ligand (L96), are presented below.
Abbreviations: Af = adenine 2′-F ribonucleoside; Cf = cytosine 2′-F ribonucleoside; Uf = uracil 2′-F ribonucleoside; Am = adenine 2′-OMe ribonucleoside; Cm = cytosine 2′-OMe ribonucleoside; Gf = guanine 2′-F ribonucleoside; Gm = guanine 2′-OMe ribonucleoside; Um = uracil 2′-OMe ribonucleoside; L96 = triantennary GalNAc (N-acetylgalactosamine)
GIVLAARI is supplied as a sterile, preservative-free, 1-mL colorless-to-yellow solution for subcutaneous injection containing 189 mg givosiran in a single-dose, 2-mL Type 1 glass vial with a TEFLON®-coated stopper and a flip-off aluminum seal. GIVLAARI is available in cartons containing one single-dose vial each. Water for injection is the only excipient used in the manufacture of GIVLAARI.
The molecular formula of givosiran sodium is C524 H651 F16 N173 Na43 O316 P43 S6 with a molecular weight of 17,245.56 Da.
The molecular formula of givosiran (free acid) is C524 H694 F16 N173 O316 P43 S6 with a molecular weight of 16,300.34 Da.
| Clinical data | |
|---|---|
| Trade names | Givlaari |
| Routes of administration |
Subcutaneous injection |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C524H694F16N173O316P43S6 |
| Molar mass | 16300.42 g·mol−1 |
| 3D model (JSmol) | |
/////////Givosiran, ギボシラン , FDA 2019, Acute Hepatic Porphyria,
Golodirsen, ゴロジルセン;

Golodirsen
- RNA, [P-deoxy-P-(dimethylamino)](2′,3′-dideoxy-2′,3′-imino-2′,3′-seco)(2’a→5′)(G-m5U-m5U-G-C-C-m5U-C-C-G-G-m5U-m5U-C-m5U-G-A-A-G-G-m5U-G-m5U-m5U-C), 5′-[P-[4-[[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]carbonyl]-1-piperazinyl]-N,N-dimethylphosphonamidate]
- Nucleic Acid Sequence
- Sequence Length: 25
| Formula |
C305H481N138O112P25
|
|---|---|
| CAS |
1422959-91-8
|
| Mol weight |
8647.2841
|
- Exon 53: NG-12-0163
- Golodirsen
- SRP 4053
Nucleic Acid Sequence
Sequence Length: 252 a 6 c 8 g 9 umodified
FDA APPROVED, Vyondys 53, 019/12/12
Antisense oligonucleotide
|
ゴロジルセン;
|
Duchenne muscular dystrophy (DMD variant amenable to exon 53 skipping)
VYONDYS 53 (golodirsen) injection is a sterile, aqueous, preservative-free, concentrated solution for dilution prior to intravenous administration. VYONDYS 53 is a clear to slightly opalescent, colorless liquid. VYONDYS 53 is supplied in single-dose vials containing 100 mg golodirsen (50 mg/mL). VYONDYS 53 is formulated as an isotonic phosphate buffered saline solution with an osmolality of 260 to 320 mOSM and a pH of 7.5. Each milliliter of VYONDYS 53 contains: 50 mg golodirsen; 0.2 mg potassium chloride; 0.2 mg potassium phosphate monobasic; 8 mg sodium chloride; and 1.14 mg sodium phosphate dibasic, anhydrous, in water for injection. The product may contain hydrochloric acid or sodium hydroxide to adjust pH.
Golodirsen is an antisense oligonucleotide of the phosphorodiamidate morpholino oligomer (PMO) subclass. PMOs are synthetic molecules in which the five-membered ribofuranosyl rings found in natural DNA and RNA are replaced by a six-membered morpholino ring. Each morpholino ring is linked through an uncharged phosphorodiamidate moiety rather than the negatively charged phosphate linkage that is present in natural DNA and RNA. Each phosphorodiamidate morpholino subunit contains one of the heterocyclic bases found in DNA (adenine, cytosine, guanine, or thymine). Golodirsen contains 25 linked subunits. The sequence of bases from the 5′ end to 3′ end is GTTGCCTCCGGTTCTGAAGGTGTTC. The molecular formula of golodirsen is C305H481N138O112P25 and the molecular weight is 8647.28 daltons. The structure of golodirsen is:
|
SIDE EFFECTS
- Hypersensitivity Reactions [see WARNINGS AND PRECAUTIONS]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In the VYONDYS 53 clinical development program, 58 patients received at least one intravenous dose of VYONDYS 53, ranging between 4 mg/kg (0.13 times the recommended dosage) and 30 mg/kg (the recommended dosage). All patients were male and had genetically confirmed Duchenne muscular dystrophy. Age at study entry was 6 to 13 years. Most (86%) patients were Caucasian.
VYONDYS 53 was studied in 2 double-blind, placebo-controlled studies.
In Study 1 Part 1, patients were randomized to receive once-weekly intravenous infusions of VYONDYS 53 (n=8) in four increasing dose levels from 4 mg/kg to 30 mg/kg or placebo (n=4), for at least 2 weeks at each level. All patients who participated in Study 1 Part 1 (n=12) were continued into Study 1 Part 2, an open-label extension, during which they received VYONDYS 53 at a dose of 30 mg/kg IV once weekly [see Clinical Studies].
In Study 2, patients received VYONDYS 53 (n=33) 30 mg/kg or placebo (n=17) IV once weekly for up to 96 weeks, after which all patients received VYONDYS 53 at a dose of 30 mg/kg.
Adverse reactions observed in at least 20% of treated patients in the placebo-controlled sections of Studies 1 and 2 are shown in Table 1.
Table 1: Adverse Reactions That Occurred in At Least 20% of VYONDYS 53-Treated Patients and at a Rate Greaterthan Placebo in Studies 1 and 2
| Adverse Reaction | VYONDYS 53 (N = 41) % |
Placebo (N = 21) % |
| Headache | 41 | 10 |
| Pyrexia | 41 | 14 |
| Fall | 29 | 19 |
| Abdominal pain | 27 | 10 |
| Nasopharyngitis | 27 | 14 |
| Cough | 27 | 19 |
| Vomiting | 27 | 19 |
| Nausea | 20 | 10 |
Other adverse reactions that occurred at a frequency greater than 5% of VYONDYS 53-treated patients and at a greater frequency than placebo were: administration site pain, back pain, pain, diarrhea, dizziness, ligament sprain, contusion, influenza, oropharyngeal pain, rhinitis, skin abrasion, ear infection, seasonal allergy, tachycardia, catheter site related reaction, constipation, and fracture.
Hypersensitivity reactions have occurred in patients treated with VYONDYS 53 [see WARNINGS AND PRECAUTIONS].
Antisense technology provides a means for modulating the expression of one or more specific gene products, including alternative splice products, and is uniquely useful in a number of therapeutic, diagnostic, and research applications. The principle behind antisense technology is that an antisense compound, e.g., an oligonucleotide, which hybridizes to a target nucleic acid, modulates gene expression activities such as transcription, splicing or translation through any one of a number of antisense mechanisms. The sequence specificity of antisense compounds makes them attractive as tools for target validation and gene functionalization, as well as therapeutics to selectively modulate the expression of genes involved in disease.
Duchenne muscular dystrophy (DMD) is caused by a defect in the expression of the protein dystrophin. The gene encoding the protein contains 79 exons spread out over more than 2 million nucleotides of DNA. Any exonic mutation that changes the reading frame of the exon, or introduces a stop codon, or is characterized by removal of an entire out of frame exon or exons, or duplications of one or more exons, has the potential to disrupt production of functional dystrophin, resulting in DMD.
Recent clinical trials testing the safety and efficacy of splice switching
oligonucleotides (SSOs) for the treatment of DMD are based on SSO technology to induce alternative splicing of pre-mRNAs by steric blockade of the spliceosome (Cirak et al., 2Q\ \; Goemans et al., 2011; Kinali et al., 2009; van Deutekom et al., 2007). However, despite these successes, the pharmacological options available for treating DMD are limited. Golodirsen is a phosphorodiamidate morpholino oligomer (PMO) designed to skip exon 53 of the human dystrophin gene in patients with DMD who are amendable to exon 53 skipping to restore the read frame and produce a functional shorter form of the dystrophin protein.
Although significant progress has been made in the field of antisense technology, there remains a need in the art for methods of preparing phosphorodiamidate morpholino oligomers with improved antisense or antigene performance.
PATENT
https://patents.google.com/patent/WO2017205880A1/en
Provided herein are processes for preparing phosphorodiamidate morpholino oligomers (PMOs). The synthetic processes described herein allow for a scaled-up PMO synthesis while maintaining overall yield and purity of a synthesized PMO.
Accordingly, in one aspect, provided herein is a process for preparing an oligomeric compound of Formula A):

(A).
In certain embodiments, provided herein is a process for preparing an oligomeric compound of Formula (G):

In yet another embodiment, the oligomeric compound of the disclosure including, for example, some embodiments of an oligomeric compound of Formula (G), is an oligomeric compound of Formula (XII):

(XII).
For clarity, the structural formulas including, for example, oligomeric compound of Formula (C) and Golodirsen depicted by Formula (XII), are a continuous structural formula from 5′ to 3′, and, for the convenience of depicting the entire formula in a compact form in the above structural formulas, Applicants have included various illustration breaks labeled “BREAK A” and “BREAK B.” As would be understood by the skilled artisan, for example, each indication of “BREAK A” shows a continuation of the illustration of the structural formula at these points. The skilled artisan understands that the same is true for each instance of “BREAK B” in the structural formulas above including Golodirsen. None of the illustration breaks, however, are intended to indicate, nor would the skilled artisan understand them to mean, an actual discontinuation of the structural formulas above including
Example 1: NCP2 Anchor Synthesis
1. Preparation of Meth l 4-Fluoro-3-Nitrobenzoate (1)

To a 100L flask was charged 12.7kg of 4-fluoro-3-nitrobenzoic acid was added 40kg of methanol and 2.82kg concentrated sulfuric acid. The mixture was stirred at reflux (65° C) for 36 hours. The reaction mixture was cooled to 0° C. Crystals formed at 38° C. The mixture was held at 0° C for 4 hrs then filtered under nitrogen. The 100L flask was washed and filter cake was washed with 10kg of methanol that had been cooled to 0° C. The solid filter cake was dried on the funnel for 1 hour, transferred to trays, and dried in a vacuum oven at room temperature to a constant weight of 13.695kg methyl 4-fluoro-3-nitrobenzoate (100% yield; HPLC 99%).
2. Preparation of 3-Nitro-4-(2-oxopropyl)benzoic Acid
A. (Z)-Methyl 4-(3 -Hydroxy- l-Methoxy-l-Oxobut-2-en-2-yl)-3-Nitrobenzoate (2)

To a 100L flask was charged 3.98kg of methyl 4-fluoro-3-nitrobenzoate (1) from the previous step 9.8kg DMF, 2.81kg methyl acetoacetate. The mixture was stirred and cooled to 0° C. To this was added 3.66kg DBU over about 4 hours while the temperature was maintained at or below 5° C. The mixture was stirred an additional 1 hour. To the reaction flask was added a solution of 8.15kg of citric acid in 37.5kg of purified water while the reaction temperature was maintained at or below 15° C. After the addition, the reaction mixture was stirred an addition 30 minutes then filtered under nitrogen. The wet filter cake was returned to the 100L flask along with 14.8kg of purified water. The slurry was stirred for 10 minutes then filtered. The wet cake was again returned to the 100L flask, slurried with 14.8kg of purified water for 10 minutes, and filtered to crude (Z)-methyl 4-(3 -hydroxy- 1 – methoxy-l-oxobut-2-en-2-yl)-3-nitrobenzoate.
B. 3-Nitro-4-(2-oxopropyl)benzoic Acid

2 3
The crude (Z)-m ethyl 4-(3 -hydroxy- 1-methoxy-l -ox obut-2-en-2-yl)-3-nitrobenzoate was charged to a 100L reaction flask under nitrogen. To this was added 14.2kg 1,4-dioxane and the stirred. To the mixture was added a solution of 16.655kg concentrated HC1 and 13.33kg purified water (6M HC1) over 2 hours while the temperature of the reaction mixture was maintained below 15° C. When the addition was complete, the reaction mixture was heated at reflux (80° C) for 24 hours, cooled to room temperature, and filtered under nitrogen. The solid filter cake was triturated with 14.8kg of purified water, filtered, triturated again with 14.8kg of purified water, and filtered. The solid was returned to the 100L flask with 39.9kg of DCM and refluxed with stirring for 1 hour. 1.5kg of purified water was added to dissolve the remaining solids. The bottom organic layer was split to a pre-warmed 72L flask, then returned to a clean dry 100L flask. The solution was cooled to 0° C, held for 1 hour, then filtered. The solid filter cake was washed twice each with a solution of 9.8kg DCM and 5kg heptane, then dried on the funnel. The solid was transferred to trays and dried to a constant weight of 1.855kg 3-Nitro-4-(2-oxopropyl)benzoic Acid. Overall yield 42% from compound 1. HPLC 99.45%.
3. Preparation of N-Tritylpiperazine Succinate (NTP)

To a 72L jacketed flask was charged under nitrogen 1.805kg triphenylmethyl chloride and 8.3kg of toluene (TPC solution). The mixture was stirred until the solids dissolved. To a 100L jacketed reaction flask was added under nitrogen 5.61kg piperazine, 19.9kg toluene, and 3.72kg methanol. The mixture was stirred and cooled to 0° C. To this was slowly added in portions the TPC solution over 4 hours while the reaction temperature was maintained at or below 10° C. The mixture was stirred for 1.5 hours at 10° C, then allowed to warm to 14° C. 32.6kg of purified water was charged to the 72L flask, then transferred to the 100L flask while the internal batch temperature was maintained at 20+/-50 C. The layers were allowed to split and the bottom aqueous layer was separated and stored. The organic layer was extracted three times with 32kg of purified water each, and the aqueous layers were separated and combined with the stored aqueous solution.
The remaining organic layer was cooled to 18° C and a solution of 847g of succinic acid in 10.87kg of purified water was added slowly in portions to the organic layer. The mixture was stirred for 1.75 hours at 20+/-50 C. The mixture was filtered, and the solids were washed with 2kg TBME and 2kg of acetone then dried on the funnel. The filter cake was triturated twice with 5.7kg each of acetone and filtered and washed with 1kg of acetone between triturations. The solid was dried on the funnel, then transferred to trays and dried in a vacuum oven at room temperature to a constant weight of 2.32kg of NTP. Yield 80%. 4. Preparation of (4-(2-Hydroxypropyl)-3-NitrophenyI)(4-Tritylpiperazin-l-yl)Methanone A. Preparation of l-(2-Nitro-4(4-Tritylpiperazine-l-Carbonyl)Phenyl)Propan-2-one

3 4
To a 100L jacketed flask was charged under nitrogen 2kg of 3-Nitro-4-(2- oxopropyl)benzoic Acid (3), 18.3 kg DCM, 1.845kg N-(3-dimethylaminopropyl)-N’- ethylcarbodiimide hydrochloride (EDC.HC1). The solution was stirred until a homogenous mixture was formed. 3.048kg of NTP was added over 30 minutes at room temperature and stirred for 8 hours. 5.44kg of purified water was added to the reaction mixture and stirred for 30 minutes. The layers were allowed to separate and the bottom organic layer containing the product was drained and stored. The aqueous layer was extracted twice with 5.65kg of DCM. The combined organic layers were washed with a solution of 1.08kg sodium chloride in 4.08kg purified water. The organic layers were dried over 1.068kg of sodium sulfate and filtered. The sodium sulfate was washed with 1.3kg of DCM. The combined organic layers were slurried with 252g of silica gel and filtered through a filter funnel containing a bed of 252g of silica gel. The silica gel bed was washed with 2kg of DCM. The combined organic layers were evaporated on a rotovap. 4.8kg of THF was added to the residue and then evaporated on the rotovap until 2.5 volumes of the crude l-(2-nitro-4(4-tritylpiperazine-l- carbonyl)phenyl)propan-2-one in THF was reached.
B. Preparation of (4-(2-Hydroxypropyl)-3-NitrophenyI)(4-Tritylpiperazin-l- yl)Methano

To a 100L jacketed flask was charged under nitrogen 3600g of 4 from the previous step and 9800g THF. The stirred solution was cooled to <5° C. The solution was diluted with 11525g ethanol and 194g of sodium borohydride was added over about 2 hours at <5° C. The reaction mixture was stirred an additional 2 hours at <5° C. The reaction was quenched with a solution of about 1.1kg ammonium chloride in about 3kg of water by slow addition to maintain the temperature at <10° C. The reaction mixture was stirred an additional 30 minutes, filtered to remove inorganics, and recharged to a 100L jacketed flask and extracted with 23kg of DCM. The organic layer was separated and the aqueous was twice more extracted with 4.7kg of DCM each. The combined organic layers were washed with a solution of about 800g of sodium chloride in about 3kg of water, then dried over 2.7kg of sodium sulfate. The suspension was filtered and the filter cake was washed with 2kg of DCM. The combined filtrates were concentrated to 2.0 volumes, diluted with about 360g of ethyl acetate, and evaporated. The crude product was loaded onto a silica gel column of 4kg of silica packed with DCM under nitrogen and eluted with 2.3kg ethyl acetate in 7.2kg of DCM. The combined fractions were evaporated and the residue was taken up in 11.7kg of toluene. The toluene solution was filtered and the filter cake was washed twice with 2kg of toluene each. The filter cake was dried to a constant weight of 2.275kg of compound 5 (46% yield from compound 3) HPLC 96.99%. 5. Preparation of 2,5-Dioxopyrrolidin-l-yl(l-(2-Nitro-4-(4-triphenylmethylpiperazine-l Carbon l)Phenyl)Propan-2-yl) Carbonate (NCP2 Anchor)

3 NCP2 Anchor
To a 100L jacketed flask was charged under nitrogen 4.3kg of compound 5 (weight adjusted based on residual toluene by 1H MR; all reagents here after were scaled accordingly) and 12.7kg pyridine. To this was charged 3.160 kg of DSC (78.91 weight % by 1H NMR) while the internal temperature was maintained at <35° C. The reaction mixture was aged for about 22 hours at ambience then filtered. The filter cake was washed with 200g of pyridine. In two batches each comprising ½ the filtrate volume, filtrate wash charged slowly to a 100L jacketed flask containing a solution of about 11kg of citric acid in about 50 kg of water and stirred for 30 minutes to allow for solid precipitation. The solid was collected with a filter funnel, washed twice with 4.3kg of water per wash, and dried on the filter funnel under vacuum.
The combined solids were charged to a 100L jacketed flask and dissolved in 28kg of DCM and washed with a solution of 900g of potassium carbonate in 4.3kg of water. After 1 hour, the layers were allowed to separate and the aqueous layer was removed. The organic layer was washed with 10kg of water, separated, and dried over 3.5kg of sodium sulfate. The DCM was filtered, evaporated, and dried under vacuum to 6.16kg of NCP2 Anchor (114% yield).
Example 2: Anchor Loaded Resin Synthesis
To a 75L solid phase synthesis reactor was charged about 52L of NMP and 2600g of aminomethyl polystyrene resin. The resin was stirred in the NMP to swell for about 2 hours then drained. The resin was washed twice with about 39L DCM per wash, then twice with 39L Neutralization Solution per wash, then twice with 39L of DCM per wash. The NCP2 Anchor Solution was slowly added to the stirring resin solution, stirred for 24 hours at room temperature, and drained. The resin was washed four times with 39L of NMP per wash, and six times with 39L of DCM per wash. The resin was treated and stirred with ½ the DEDC Capping Solution for 30 minutes, drained, and was treated and stirred with the 2nd ½ of the DEDC Capping Solution for 30 minutes and drained. The resin was washed six times with 39L of DCM per wash then dried in an oven to constant weight of 3573.71g of Anchor Loaded Resin.
Example 3: Preparation of Activated EG3 Tail
1. Preparation of Trityl Piperazine Phenyl Carbamate 35

To a cooled suspension of NTP in dichloromethane (6 mL/g NTP) was added a solution of potassium carbonate (3.2 eq) in water (4 mL/g potassium carbonate). To this two- phase mixture was slowly added a solution of phenyl chloroformate (1.03 eq) in
dichloromethane (2 g/g phenyl chloroformate). The reaction mixture was warmed to 20° C. Upon reaction completion (1-2 hr), the layers were separated. The organic layer was washed with water, and dried over anhydrous potassium carbonate. The product 35 was isolated by crystallization from acetonitrile. Yield=80%
2. Preparation of Carbamate Alcohol (36)

Sodium hydride (1.2 eq) was suspended in l-methyl-2-pyrrolidinone (32 mL/g sodium hydride). To this suspension were added triethylene glycol (10.0 eq) and compound 35 (1.0 eq). The resulting slurry was heated to 95° C. Upon reaction completion (1-2 hr), the mixture was cooled to 20° C. To this mixture was added 30% dichloromethane/methyl tert- butyl ether (v:v) and water. The product-containing organic layer was washed successively with aqueous NaOH, aqueous succinic acid, and saturated aqueous sodium chloride. The product 36 was isolated by crystallization from dichloromethane/methyl tert-butyl ether/heptane. Yield=90%.
3. Preparation of EG3 Tail Acid (37)

To a solution of compound 36 in tetrahydrofuran (7 mL/g 36) was added succinic anhydride (2.0 eq) and DMAP (0.5 eq). The mixture was heated to 50° C. Upon reaction completion (5 hr), the mixture was cooled to 20° C and adjusted to pH 8.5 with aqueous NaHC03. Methyl tert-butyl ether was added, and the product was extracted into the aqueous layer. Dichloromethane was added, and the mixture was adjusted to pH 3 with aqueous citric acid. The product-containing organic layer was washed with a mixture of pH=3 citrate buffer and saturated aqueous sodium chloride. This dichloromethane solution of 37 was used without isolation in the preparation of compound 38. 4. Preparation of Activated EG3 Tail (38)

To the solution of compound 37 was added N-hydroxy-5-norbornene-2,3-dicarboxylic acid imide (HONB) (1.02 eq), 4-dimethylaminopyridine (DMAP) (0.34 eq), and then l-(3- dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDC) (1.1 eq). The mixture was heated to 55° C. Upon reaction completion (4-5 hr), the mixture was cooled to 20° C and washed successively with 1 : 1 0.2 M citric acid/brine and brine. The dichloromethane solution underwent solvent exchange to acetone and then to Ν,Ν-dimethylformamide, and the product was isolated by precipitation from acetone/N,N-dimethylformamide into saturated aqueous sodium chloride. The crude product was reslurried several times in water to remove residual Ν,Ν-dimethylformamide and salts. Yield=70% of Activated EG3 Tail 38 from compound 36.
Example 4: 50L Solid-phase Synthesis of
Golodirsen [Oligomeric Compound (XII)] Crude Drug Substance
1. Materials
Table 2: Starting Materials

Activated Phosphoramidochloridic acid, 1155373-31-1 C37H37CIN5O5P 698.2 C Subunit N,N-dimethyl-,[6-[4-
(benzoylamino)-2-oxo-l(2H)- pyrimidinyl]-4-
(triphenylmethyl)-2- morpholinyljmethyl ester
Activated Propanoic Acid, 2,2-dimethyl- 1155309-89-9 C5iH53ClN707P 942.2
DPG ,4-[[[9-[6-
Subunit [[[chloro(dimethylamino)phosp
hinyl]oxy]methyl]-4-
(triphenylmethyl)-2- morpholinyl]-2-[(2- phenylacetyl)amino]-9H-purin-
6-yl]oxy]methyl]phenyl ester
Activated Phosphoramidochloridic acid, 1155373-34-4 C3iH34ClN405P 609.1 T Subunit N,N-dimethyl-,[6-(3,4-dihydro- 5-methyl-2,4-dioxo- 1 (2H)- pyrimidinyl)]-4- (triphenylmethyl)-2- morpholinyljmethyl ester
Activated Butanedioic acid, 1- 1380600-06-5 C43H47N3Oio 765.9 EG3 Tail [3aR,4S,7R,7aS)-l,3,3a,4,7,7a- hexahydro- 1 ,3 -dioxo-4,7- methano-2H-isoindol-2-yl] 4- [2-[2-[2-[[[4-(triphenylmethyl)- 1- piperazinyl ] carb onyl ] oxy] ethox
y]ethoxy] ethyl] ester
Golodirsen.
Example 5: Purification of Golodirsen Crude Drug Substance
The deprotection solution from Example 4, part E, containing the Golodirsen crude drug substance was loaded onto a column of ToyoPearl Super-Q 650S anion exchange resin (Tosoh Bioscience) and eluted with a gradient of 0-35% B over 17 column volume (Buffer A: 10 mM sodium hydroxide; Buffer B: 1 M sodium chloride in 10 mM sodium hydroxide) and fractions of acceptable purity (CI 8 and SCX HPLC) were pooled to a purified drug product solution. HPLC: 93.571% (C18; Fig. 3) 88.270% (SCX; Fig. 4).
The purified drug substance solution was desalted and lyophilized to 1450.72 g purified Golodirsen drug substance. Yield 54.56 %; HPLC: 93.531% (Fig. 5; C18) 88.354% (Fig. 6; SCX).
PATENT
WO 2019067979
Duchenne Muscular Dystrophy (DMD) is a serious, progressively debilitating, and ultimately fatal inherited X-linked neuromuscular disease. DMD is caused by mutations in the dystrophin gene characterized by the absence, or near absence, of functional dystrophin protein that disrupt the mRNA reading frame, resulting in a lack of dystrophin, a critically important part of the protein complex that connects the cytoskeletal actin of a muscle fiber to the extracellular matrix. In the absence of dystrophin, patients with DMD follow a predictable disease course. Affected patients, typically boys, develop muscle weakness in the first few years of life, lose the ability to walk during childhood, and usually require respiratory support by their late teens. Loss of functional abilities leads to loss of independence and increasing caregiver burden. Once lost, these abilities cannot be recovered. Despite improvements in the standard of care, such as the use of glucocorticoids, DMD remains an ultimately fatal disease, with patients usually dying of respiratory or cardiac failure in their mid to late 20s.
Progressive loss of muscle tissue and function in DMD is caused by the absence or near absence of functional dystrophin; a protein that plays a vital role in the structure and function of muscle cells. A potential therapeutic approach to the treatment of DMD is suggested by Becker muscular dystrophy (BMD), a milder dystrophinopathy. Both dystrophinopathies are caused by mutations in the DMD gene. In DMD, mutations that disrupt the pre-mRNA reading frame,
referred to as “out-of-frame” mutations, prevent the production of functional dystrophin. In BMD, “in-frame” mutations do not disrupt the reading frame and result in the production of internally shortened, functional dystrophin protein.
An important approach for restoring these “out-of-frame” mutations is to utilize an antisense oligonucleotide to exclude or skip the molecular mutation of the DMD gene
(dystrophin gene). The DMD or dystrophin gene is one of the largest genes in the human body and consists of 79 exons. Antisense oligonucleotides (AONs) have been specifically designed to target specific regions of the pre-mRNA, typically exons to induce the skipping of a mutation of the DMD gene thereby restoring these out-of-frame mutations in-frame to enable the production of internally shortened, yet functional dystrophin protein.
The skipping of exon 53 in the dystrophin gene has been an area of interest for certain research groups due to it being the most prevalent set of mutations in this disease area, representing 8% of all DMD mutations. A prominent AON being developed by Sarepta
Therapeutics, Inc., for DMD patients that are amenable to exon 53 skipping is golodirsen.
Golodirsen is a phosphorodiamidate morpholino oligomer, or PMO. Another AON being developed by Nippon Shinyaku CO., LTD., for DMD patients that are amenable to exon 53 skipping is viltolarsen (NS-065 which is a PMO.
Exondys 51 ® (eteplirsen), is another PMO that was approved in 2016 by the United States Food and Drug Administration (FDA) for the treatment of Duchenne muscular dystrophy (DMD) in patients who have a confirmed mutation of the DMD gene that is amenable to exon 51 skipping. However, the current standard of care guidelines for the treatment of DMD in patients that are not amenable to exon 51 skipping include the administration of glucocorticoids in conjunction with palliative interventions. While glucocorticoids may delay the loss of ambulation, they do not sufficiently ameliorate symptoms, modify the underlying genetic defect or address the absence of functional dystrophin characteristic of DMD.
Previous studies have tested the efficacy of an antisense oligonucleotides (AON) for exon skipping to generate at least partially functional dystrophin in combination with a steroid for reducing inflammation in a DMD patient (see WO 2009/054725 and van Deutekom, et al., N. Engl. J. Med. 2007; 357:2677-86, the contents of which are hereby incorporated herein by reference for all purposes). However, treatment with steroids can result in serious complications, including compromise of the immune system, reduction in bone strength, and growth
suppression. Notably, none of the previous studies suggest administering an antisense
oligonucleotide for exon skipping with a non-steroidal anti-inflammatory compound to a patient for the treatment of DMD.
Thus, there remains a need for improved methods for treating muscular dystrophy, such as DMD and BMD in patients.
EXAMPLE 1
CAT- 1004 in Combination with M23D PMO Reduces Inflammation and Fibrosis in Mdx Mice.
To assess the effectiveness of a combination treatment of an exon skipping antisense oligonucleotide and an F-Kb inhibitor in Duchenne muscular dystrophy, M23D PMO and
CAT-1004 were utilized in the Mdx mouse model. The effect on inflammation and fibrosis was determined by analyzing samples of muscle taken from the quadriceps, of (1) wild-type mice treated with saline, (2) mdx mice treated with saline, (3) mdx mice treated with CAT-1004, (4) mdx mice treated with the M23D PMO, and (5) mdx mice treated with the M23D PMO in combination with CAT-1004. The tissue sections were analyzed for fibrosis by picrosirius red staining and for inflammation and fibrosis by Hematoxylin and Eosin (H&E) staining, as described in the Materials and Methods section above.
Treatment of Mdx mice with either M23D PMO or CAT-1004 as monotherapies resulted in a reduction of inflammation and fibrosis as compared to Mdx mice treated with saline.
Surprisingly, treatment of Mdx mice with the M23D PMO in combination with CAT-1004 resulted in reduced inflammation and fibrosis as compared with mice treated with CAT-1004
alone or M23D alone (Fig. 9). These results indicate the combination treatment enhances muscle fiber integrity.
EXAMPLE 2
Exon Skipping and Dystrophin Production in Mdx Mice Treated with the M23D
PMO and the M23D PMO in Combination with CAT- 1004
To analyze the extent of exon skipping and dystrophin production in mice treated with the M23D PMO in combination with CAT- 1004, samples of muscle were taken from the quadriceps, diaphragm, and heart of (1) wild-type mice treated with saline, (2) mdx mice treated with saline, (3) mdx mice treated with CAT- 1004, (4) mdx mice treated with the M23D PMO, and (5) mdx mice treated with the M23D PMO in combination with CAT- 1004. RT-PCR analysis for exon 23 skipping was performed as well as Western blot analysis to determine dystrophin protein levels.
Exon skipping was observed in the muscle of the quadriceps, diaphragm, and heart of the Mdx mice treated with the M23D PMO as well as mice treated with the M23D PMO in combination with CAT-1004 (Fig. 10). Surprisingly, enhanced dystrophin production was observed in the muscle of the quadriceps, diaphragm, and heart of the mice treated with the M23D PMO in combination with CAT-1004 as compared to treatment with M23D PMO monotherapy (Fig. 11). These results indicated the increase in dystrophin levels extended to the heart, a tissue known to have low efficiency of dystrophin upregulation by these agents when used alone. Notably, neither exon skipping nor dystrophin production were observed in mdx mice treated with CAT-1004 monotherapy (Figs. 10 and 11).
PATENT
WO 2019046755
PAPER
Methods in Molecular Biology (New York, NY, United States) (2018), 1828(Exon Skipping and Inclusion Therapies), 31-55.
PAPER
Human Molecular Genetics (2018), 27(R2), R163-R172.
///////////Golodirsen, ゴロジルセン , FDA 2019, ANTISENSE, Exon 53: NG-12-0163, SRP 4053, OLIGONUCLEOTIDE, Duchenne Muscular Dystrophy
FDA approves novel treatment Oxbryta (voxelotor) to target abnormality in sickle cell disease
Sickle cell disease is a lifelong, inherited blood disorder in which red blood cells are abnormally shaped (in a crescent, or “sickle” shape), which restricts the flow in blood vessels and limits oxygen delivery to the body’s tissues, leading to severe pain and organ damage. It is also characterized by severe and chronic inflammation that worsens vaso-occlusive crises during which patients experience episodes of extreme pain and organ damage. Nonclinical studies have demonstrated that Oxbryta inhibits red blood cell sickling, improves red blood cell deformability (ability of a red blood cell to change shape) and improves the blood’s ability to flow.
“Oxbryta is an inhibitor of deoxygenated sickle hemoglobin polymerization, which is the central abnormality in sickle cell disease,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research. “With Oxbryta, sickle cells are less likely to bind together and form the sickle shape, which can cause low hemoglobin levels due to red blood cell destruction. This therapy provides a new treatment option for patients with this serious and life-threatening condition.”
Oxbryta’s approval was based on the results of a clinical trial with 274 patients with sickle cell disease. In the study, 90 patients received 1500 mg of Oxbryta, 92 patients received 900 mg of Oxbryta and 92 patients received a placebo. Effectiveness was based on an increase in hemoglobin response rate in patients who received 1500 mg of Oxbryta, which was 51.1% for these patients compared to 6.5% in the placebo group.
/////////fda 2019, Fast Track designation, Oxbryta, Orphan Drug designation, voxelotor, Global Blood Therapeutics, sickle cell disease
FDA approves new treatment XCOPRI (cenobamate tablets) for adults with partial-onset seizures
FDA approves first treatment Givlaari (givosiran) for inherited rare disease
///////////Givlaari, givosiran, fda 2019, Breakthrough Therapy designation, Priority Review, Orphan Drug
Blarcamesine, ブラルカメシン ,

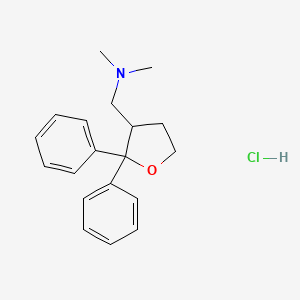
Blarcamesine
ブラルカメシン;
[(2,2-diphenyloxolan-3-yl)methyl]dimethylamine
- Anavex 2-73
- Tetrahydro-N,N-dimethyl-2,2-diphenyl-3-furanemethanamine
- THD-DP-FM
- AE-37 / AE37 / ANAVEX 2-73 FREE BASE
- UNII 9T210MMZ3F
| Formula |
C19H23NO
|
|---|---|
| Cas |
195615-83-9
195615-84-0 HCL
|
| Mol weight |
281.392
|
Treatment of Rett syndrome, Investigated for use/treatment in breast cancer.
Anti-amnesic, Muscarinic/sigma receptor agonist
- Originator Anavex Life Sciences
- Developer ABX-CRO; Anavex Life Sciences; The Michael J. Fox Foundation for Parkinsons Research
- Class Antidementias; Antidepressants; Antiepileptic drugs; Antiparkinsonians; Anxiolytics; Behavioural disorder therapies; Dimethylamines; Furans; Neuroprotectants; Neuropsychotherapeutics; Nootropics; Small molecules
- Mechanism of Action Muscarinic receptor modulators; Sigma-1 receptor agonists
- Orphan Drug Status Yes – Epilepsy; Rett syndrome
- Phase II/III Alzheimer’s disease
- Phase II Parkinson’s disease; Rett syndrome
- Preclinical Amyotrophic lateral sclerosis; Angelman syndrome; Anxiety disorders; Autistic disorder; Fragile X syndrome; Multiple sclerosis
- No development reported Cognition disorders; Epilepsy; Stroke
- 28 Oct 2019 No recent reports of development identified for phase-I development in Cognition-disorders in USA
- 09 Oct 2019 Anavex Life Sciences initiates enrolment in the long term extension ATTENTION-AD trial for Alzheimer’s disease in (country/ies)
- 02 Oct 2019 Anavex Life Sciences has patent protection covering compositions of matter and methods of treating Alzheimer’s disease for blarcamesine in USA
- Anavex Life Sciences is developing ANAVEX-2-73 and its active metabolite ANAVEX-19-144, for treating Alzheimer’s disease, epilepsy, stroke and Rett syndrome.
ANAVEX2-73 is an experimental drug is in Phase II trials for Alzheimer’s disease, phase I trials for epilepsy, and in preclinical trials for amyotrophic lateral sclerosis, Parkinson’s disease, Rett syndrome, stroke.[1][2] ANAVEX2-73 acts as a muscarinic receptor and a moderate sigma1 receptor agonist.[1] ANAVEX2-73 may function as a pro-drug for ANAVEX19-144 as well as a drug itself. ANAVEX19-144 is the active metabolite of ANAVEX 1-41, which is similar to ANAVEX2-73 but it is not as selective for sigma receptor.[2]
Properties and uses
ANAVEX2-73 has an inhibitory constant (ki) lower than 500 nM for all M1–M4 muscarinic acetylcholine receptor subtypes, demonstrating that it acts as a powerful antimuscarinic compound.[2] ANAVEX2-73 was originally tested in mice against the effect of the muscarinic receptor antagonist scopolamine, which induces learning impairment.[1] M1 receptor agonists are known to reverse the amnesia caused by scopolamine.[3] Scopolamine is used in the treatment of Parkinson’s disease and motion sickness by reducing the secretions of the stomach and intestines and can also decreases nerve signals to the stomach.[3] This is via competitive inhibition of muscarinic receptors.[3] Muscarinic receptors are involved in the formation of both short term and long term memories.[1] Experiments in mice have found that M1 and M3 receptor agonists inhibit the formation of amyloid-beta and target GSK-3B.[clarification needed]Furthermore, stimulation of the M1 receptor activates AF267B, which in turn blocks β-secretase, which cleaves the amyloid precursor protein to produce the amyloid-beta peptide. These amyloid-beta peptides aggregate together to form plaques. This enzyme[clarification needed] is involved in the formation of Tau plaques, which are common in Alzheimer’s disease.[clarification needed][4]Therefore. M1 receptor activation appears to decreases tau hyperphosphorylation and amyloid-beta accumulation.[4]
Sigma1 activation appears to be only involved in long-term memory processes. This partly explains why ANAVEX2-73 seems to be more effective in reversing scopolamine-induced long-term memory problems compared to short-term memory deficits.[1] The sigma-1 receptor is located on mitochondria-associated endoplasmic reticulum membranes and modulates the ER stress response and local calcium exchanges with the mitochondria. ANAVEX2-73 prevented Aβ25-35-induced increases in lipid peroxidation levels, Bax/Bcl-2ratio and cytochrome c release into the cytosol, which are indicative of elevated toxicity.[clarification needed] ANAVEX2-73 inhibits mitochondrial respiratory dysfunction and therefore prevents against oxidative stress and apoptosis. This drug prevented the appearance of oxidative stress. ANAVEX2-73 also exhibits anti-apoptotic and anti-oxidant activity. This is due in part because sigma-1 agonists stimulate the anti-apoptoic factor Bcl-2 due to reactive oxygen species dependent transcriptional activation of nuclear factor kB.[5] Results from Marice (2016) demonstrate that sigma1 compounds offer a protective potential, both alone and possibly with other agents like donepezil, an acetylcholinesterase inhibitor, or the memantine, a NMDA receptor antagonist.[6]
PATENT
WO9730983
PATENT
Novel crystalline forms of A2-73 (blarcamesine hydrochloride, ANAVEX2-73, AV2-73), a mixed muscarinic receptor ligand and Sig-1 R agonist useful for treating Alzheimer’s disease.
PATENT
WO2017013498
SYN
By Foscolos, George B. et alFrom Farmaco, 51(1), 19-26; 1996

References
- ^ Jump up to:a b c “ANAVEX 2-73 – AdisInsight”. Adisinsight.springer.com. Retrieved 2016-05-25.
- ^ Jump up to:a b c Malviya, M; Kumar, YC; Asha, D; Chandra, JN; Subhash, MN; Rangappa, KS (2008). “Muscarinic receptor 1 agonist activity of novel N-arylthioureas substituted 3-morpholino arecoline derivatives in Alzheimer’s presenile dementia models”. Bioorg Med Chem. 16: 7095–7101. doi:10.1016/j.bmc.2008.06.053.
- ^ Jump up to:a b Leal, NS; Schreiner, B; Pinho, CM; Filadi, R; Wiehager, B; Karlström, H; Pizzo, P; Ankarcrona, M (2016). “Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid β-peptide production”. J Cell Mol Med. 20: 1686–1695. doi:10.1111/jcmm.12863. PMC 4988279. PMID 27203684.
- ^ Lahmy, V; Long, R; Morin, D; Villard, V; Maurice, T (2015-09-28). “Mitochondrial protection by the mixed muscarinic/σ1 ligand ANAVEX2-73, a tetrahydrofuran derivative, in Aβ25-35 peptide-injected mice, a nontransgenic Alzheimer’s disease model”. Front Cell Neurosci. 8: 463. doi:10.3389/fncel.2014.00463. PMC 4299448. PMID 25653589.
- ^ Maurice, T (2015-09-28). “Protection by sigma-1 receptor agonists is synergic with donepezil, but not with memantine, in a mouse model of amyloid-induced memory impairments”. Behav. Brain Res. 296: 270–8. doi:10.1016/j.bbr.2015.09.020. PMID 26386305.
//////////Blarcamesine, ブラルカメシン , Orphan Drug Status, PHASE 2
CN(C)CC1CCOC1(C1=CC=CC=C1)C1=CC=CC=C1
LEUPRORELIN, リュープロレリン;

LEUPRORELIN
- Molecular FormulaC59H84N16O12
- Average mass1209.398 Da
| INGREDIENT | UNII | CAS | |
|---|---|---|---|
| Leuprolide acetate | 37JNS02E7V | 74381-53-6 |
Synthesis Reference, Daniel Kadzimirzs, Gerhard Jas, Volker Autze, “Solution-Phase Synthesis of Leuprolide and Its Intermediates.” U.S. Patent US20090005535, issued January 01, 2009.US20090005535
Jitsubo , a subsidiary of Sosei , was investigating JIT-1007 , presumed to be a biosimilar version of an undisclosed peptide therapeutic, generated using its proprietary Molecular Hiving, for the treatment of an unidentified indication, however no development has been reported for some time, this program is assumed to be discontinued.
Leuprorelin, also known as leuprolide, is a manufactured version of a hormone used to treat prostate cancer, breast cancer, endometriosis, uterine fibroids, and early puberty.[1][2] It is given by injection into a muscle or under the skin.[1]
Common side effects include hot flashes, unstable mood, trouble sleeping, headaches, and pain at the site of injection.[1] Other side effects may include high blood sugar, allergic reactions, and problems with the pituitary gland.[1] Use during pregnancy may harm the baby.[1] Leuprorelin is in the gonadotropin-releasing hormone (GnRH) analogue family of medications.[1] It works by decreasing gonadotropin and therefore decreasing testosterone and estradiol.[1]
Leuprorelin was patented in 1973 and approved for medical use in the United States in 1985.[1][3] It is on the World Health Organization’s List of Essential Medicines, the most effective and safe medicines needed in a health system.[4] In the United Kingdom a monthly dose costs the NHS about GB£75.24.[5] In the United States the equivalent dose has a wholesale cost of US$1,011.93.[6] It is sold under the brand name Lupron among others.[1]
Medical use
Leuprorelin may be used in the treatment of hormone-responsive cancers such as prostate cancer and breast cancer. It may also be used for estrogen-dependent conditions such as endometriosis[7] or uterine fibroids.
It may be used for precocious puberty in both males and females,[8] and to prevent premature ovulation in cycles of controlled ovarian stimulation for in vitro fertilization (IVF).
It may be used to reduce the risk of premature ovarian failure in women receiving cyclophosphamide for chemotherapy.[9]
Along with triptorelin and goserelin, it is has been used to delay puberty in transgender youth until they are old enough to begin hormone replacement therapy.[10] Researchers have recommended puberty blockers after age 12, when the person has developed to Tanner stages 2-3, and then cross-sex hormones treatment at age 16. This use of the drug is off-label, however, not having been approved by the Food and Drug Administration and without data on long-term effects of this use.[11]
They are also sometimes used as alternatives to antiandrogens like spironolactone and cyproterone acetate for suppressing testosterone production in transgender women.[citation needed]
It is considered a possible treatment for paraphilias.[12] Leuprorelin has been tested as a treatment for reducing sexual urges in pedophiles and other cases of paraphilia.[13][14]
Side effects
Common side effects of Lupron Injection include redness/burning/stinging/pain/bruising at the injection site, hot flashes (flushing), increased sweating, night sweats, tiredness, headache, upset stomach, nausea, diarrhea, constipation, stomach pain, breast swelling or tenderness, acne, joint/muscle aches or pain, trouble sleeping (insomnia), reduced sexual interest, vaginal discomfort/dryness/itching/discharge, vaginal bleeding, swelling of the ankles/feet, increased urination at night, dizziness, breakthrough bleeding in a female child during the first 2 months of leuprorelin treatment, weakness, chills, clammy skin, skin redness, itching, or scaling, testicle pain, impotence, depression, or memory problems.[15] The rates of gynecomastia with leuprorelin have been found to range from 3 to 16%.[16]
Mechanism of action
Leuprorelin is a gonadotropin-releasing hormone (GnRH) analogue acting as an agonist at pituitary GnRH receptors. Agonism of GnRH receptors initially results in the stimulation of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) secretion by the anterior pituitary ultimately leading to increased serum estradiol and testosterone levels via the normal physiology of the hypothalamic–pituitary–gonadal axis (HPG axis); however, because propagation of the HPG axis is incumbent upon pulsatile hypothalamic GnRH secretion, pituitary GnRH receptors become desensitised after several weeks of continuous leuprorelin therapy. This protracted downregulation of GnRH receptor activity is the targeted objective of leuprorelin therapy and ultimately results in decreased LH and FSH secretion, leading to hypogonadism and thus a dramatic reduction in estradiol and testosterone levels regardless of sex.[17][18]
In the treatment of prostate cancer, the initial increase in testosterone levels associated with the initiation of leuprorelin therapy is counterproductive to treatment goals. This effect is avoided with concurrent utilisation of 5α-reductase inhibitors, such as finasteride, which function to block the downstream effects of testosterone.
Chemistry
The peptide sequence is Pyr-His-Trp-Ser-Tyr-D-Leu-Leu-Arg-Pro-NHEt (Pyr = L–Pyroglutamyl).
History
Leuprorelin was discovered and first patented in 1973 and was introduced for medical use in 1985.[19][20] It was initially marketed only for daily injection, but a depot injectionformulation was introduced in 1989.[20]
Society and culture
Names
Leuprorelin is the generic name of the drug and its INN and BAN, while leuprorelin acetate is its BANM and JAN, leuprolide acetate is its USAN and USP, leuprorelina is its DCIT, and leuproréline is its DCF.[21][22][23][24] It is also known by its developmental code names A-43818, Abbott-43818, DC-2-269, and TAP-144.[21][22][23][24]
Leuprorelin is marketed by Bayer AG under the brand name Viadur, by Tolmar under the brand name Eligard, and by TAP Pharmaceuticals (1985–2008), by Varian Darou Pajooh under the brand name Leupromer and Abbott Laboratories (2008–present) under the brand name Lupron. It is available as a slow-release implant or subcutaneous/intramuscular injection.
In the UK and Ireland, leuprorelin is marketed by Takeda UK as Prostap SR (one-month injection) and Prostap 3 (three-month injection).
Approvals
Available formsLupron injection was first approved by the FDA for treatment of advanced prostate cancer on April 9, 1985.
- Lupron depot for monthly intramuscular injection was first approved by the FDA for palliative treatment of advanced prostate cancer on January 26, 1989, and subsequently in 22.5 mg/vial and 30 mg/vial for intramuscular depot injection every 3 and 4 months, respectively. 3.75 mg/vial and 11.25 mg/vial dosage forms were subsequently approved for subcutaneous depot injection every month and every 3 months, respectively for treatment of endometriosis or fibroids. 7.5 mg/vial, 11.25 mg/vial, and 15 mg/vial dosage forms were subsequently approved for subcutaneous depot injection for treatment of children with central precocious puberty.
- Viadur (72 mg yearly subcutaneous implant) was first approved by the FDA for palliative treatment of advanced prostate cancer on March 6, 2000. Bayer will fulfill orders until current supplies are depleted, expected by the end of April 2008
- Eligard (7.5 mg for monthly subcutaneous depot injection) was first approved by the FDA for palliative treatment of advanced prostate cancer on January 24, 2002, and subsequently in 22.5 mg, 30 mg, and 45 mg doses for subcutaneous depot injection every 3, 4, and 6 months, respectively.
- Leupromer 7.5 (7.5 mg, one month depot for subcutaneous injection) is the second in situ-forming injectable drug in the world. It is used for palliative treatment of advanced prostate cancer, endometriosis, and uterine fibroids. It was approved by The Ministry of Health and Medical Education Of Iran.
Leuprorelin is available in the following forms, among others:[25][26][27]
- Short-acting daily intramuscular injection (Lupron): 5 mg/mL (2.8 mL) used as 1 mg every day.
- Long-acting depot intramuscular injection (Lupron Depot): 7.5 mg once a month, 22.5 mg every 3 months, or 30 mg every 4 months.
- Long-acting depot subcutaneous injection (Eligard): 7.5 mg once a month, 22.5 mg every 3 months, 30 mg every 4 months, or 45 mg every 6 months.
- Long-acting subcutaneous implant (Viadur): 65 mg pellet once every 12 months.
“Lupron protocol”
A 2005 paper in the controversial and non-peer reviewed journal Medical Hypotheses suggested leuprorelin as a possible treatment for autism,[28] the hypothetical method of action being the now defunct hypothesis that autism is caused by mercury, with the additional unfounded assumption that mercury binds irreversibly to testosterone and therefore leuprorelin can help cure autism by lowering the testosterone levels and thereby mercury levels.[29] However, there is no scientifically valid or reliable research to show its effectiveness in treating autism.[30] This use has been termed the “Lupron protocol”[31] and Mark Geier, the proponent of the hypothesis, has frequently been barred from testifying in vaccine-autism related cases on the grounds of not being sufficiently expert in that particular issue[32][33][34] and has had his medical license revoked.[31] Medical experts have referred to Geier’s claims as “junk science”.[35]
Veterinary use
Leuprorelin is frequently used in ferrets for the treatment of adrenal disease. Its use has been reported in a ferret with concurrent primary hyperaldosteronism,[36] and one with concurrent diabetes mellitus.[37]
Research
As of 2006 leuprorelin was under investigation for possible use in the treatment of mild to moderate Alzheimer’s disease.[38]
A by mouth formulation of leuprorelin is under development for the treatment of endometriosis.[39] It was also under development for the treatment of precocious puberty, prostate cancer, and uterine fibroids, but development for these uses was discontinued.[39] The formulation has the tentative brand name Ovarest.[39] As of July 2018, it is in phase II clinical trials for endometriosis.[39]
Patent
WO-2019198834
Process for producing leuprorelin as LH-RH (GnRH) agonist useful for treating endometriosis, uterine fibroids, premenopausal breast cancer and prostate cancer.
PATENT
WO2019198833
WO2016140232
References
- ^ Jump up to:a b c d e f g h i “Leuprolide Acetate”. The American Society of Health-System Pharmacists. Archived from the original on 23 December 2016. Retrieved 8 December2016.
- ^ “19th WHO Model List of Essential Medicines (April 2015)” (PDF). WHO. April 2015. Archived (PDF) from the original on May 13, 2015. Retrieved May 10, 2015.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 514. ISBN 9783527607495.
- ^ “WHO Model List of Essential Medicines (19th List)” (PDF). World Health Organization. April 2015. Archived (PDF) from the original on 13 December 2016. Retrieved 8 December 2016.
- ^ British national formulary : BNF 69 (69 ed.). British Medical Association. 2015. p. 655. ISBN 9780857111562.
- ^ “NADAC as of 2016-12-07 | Data.Medicaid.gov”. Centers for Medicare and Medicaid Services. Archived from the original on 21 December 2016. Retrieved 23 December 2016.
- ^ Crosignani PG, Luciano A, Ray A, Bergqvist A (January 2006). “Subcutaneous depot medroxyprogesterone acetate versus leuprolide acetate in the treatment of endometriosis-associated pain”. Human Reproduction. 21 (1): 248–56. doi:10.1093/humrep/dei290. PMID 16176939.
- ^ Badaru A, Wilson DM, Bachrach LK, et al. (May 2006). “Sequential comparisons of one-month and three-month depot leuprolide regimens in central precocious puberty”. The Journal of Clinical Endocrinology and Metabolism. 91 (5): 1862–7. doi:10.1210/jc.2005-1500. PMID 16449344.
- ^ Clowse ME, Behera MA, Anders CK, Copland S, Coffman CJ, Leppert PC, Bastian LA (March 2009). “Ovarian preservation by GnRH agonists during chemotherapy: a meta-analysis”. Journal of Women’s Health. 18 (3): 311–9. doi:10.1089/jwh.2008.0857. PMC 2858300. PMID 19281314.
- ^ David A. Wolfe; Eric J. Mash (9 October 2008). Behavioral and Emotional Disorders in Adolescents: Nature, Assessment, and Treatment. Guilford Press. pp. 556–. ISBN 978-1-60623-115-9. Archived from the original on 2 July 2014. Retrieved 24 March 2012.
- ^ Dreger, A. (2009, Jan.-Feb.). Gender Identity Disorder in childhood: Inconclusive advice to parents. Hastings Center Report, pp. 26-29.
- ^ Saleh FM, Niel T, Fishman MJ (2004). “Treatment of paraphilia in young adults with leuprolide acetate: a preliminary case report series”. Journal of Forensic Sciences. 49 (6): 1343–8. doi:10.1520/JFS2003035. PMID 15568711.
- ^ Schober JM, Byrne PM, Kuhn PJ (2006). “Leuprolide acetate is a familiar drug that may modify sex-offender behaviour: the urologist’s role”. BJU International. 97 (4): 684–6. doi:10.1111/j.1464-410X.2006.05975.x. PMID 16536753.
- ^ Schober JM, Kuhn PJ, Kovacs PG, Earle JH, Byrne PM, Fries RA (2005). “Leuprolide acetate suppresses pedophilic urges and arousability”. Archives of Sexual Behavior. 34 (6): 691–705. doi:10.1007/s10508-005-7929-2. PMID 16362253.
- ^ “Common Side Effects of Lupron (Leuprolide Acetate Injection) Drug Center”. Archived from the original on 2015-07-29. Retrieved 2015-07-26.[full citation needed]
- ^ Di Lorenzo G, Autorino R, Perdonà S, De Placido S (December 2005). “Management of gynaecomastia in patients with prostate cancer: a systematic review”. Lancet Oncol. 6 (12): 972–9. doi:10.1016/S1470-2045(05)70464-2. PMID 16321765.
- ^ Mutschler E, Schäfer-Korting M (2001). Arzneimittelwirkungen (in German) (8 ed.). Stuttgart: Wissenschaftliche Verlagsgesellschaft. pp. 372–3. ISBN 978-3-8047-1763-3.
- ^ Wuttke W, Jarry H, Feleder C, Moguilevsky J, Leonhardt S, Seong JY, Kim K (1996). “The neurochemistry of the GnRH pulse generator”. Acta Neurobiologiae Experimentalis. 56(3): 707–13. PMID 8917899. Archived from the original on 2015-12-08.
- ^ Jamil, George Leal (30 September 2013). Rethinking the Conceptual Base for New Practical Applications in Information Value and Quality. IGI Global. pp. 111–. ISBN 978-1-4666-4563-9.
- ^ Jump up to:a b Hara T (1 January 2003). Innovation in the Pharmaceutical Industry: The Process of Drug Discovery and Development. Edward Elgar Publishing. pp. 106–107. ISBN 978-1-84376-566-0.
- ^ Jump up to:a b J. Elks (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 730–. ISBN 978-1-4757-2085-3.
- ^ Jump up to:a b Index Nominum 2000: International Drug Directory. Taylor & Francis. 2000. pp. 599–. ISBN 978-3-88763-075-1.
- ^ Jump up to:a b I.K. Morton; Judith M. Hall (6 December 2012). Concise Dictionary of Pharmacological Agents: Properties and Synonyms. Springer Science & Business Media. pp. 164–. ISBN 978-94-011-4439-1.
- ^ Jump up to:a b “Leuprorelin”.
- ^ Sara K. Butler; Ramaswamy Govindan (25 October 2010). Essential Cancer Pharmacology: The Prescriber’s Guide. Lippincott Williams & Wilkins. pp. 262–. ISBN 978-1-60913-704-5.
- ^ Richard A. Lehne; Laura Rosenthal (25 June 2014). Pharmacology for Nursing Care – E-Book. Elsevier Health Sciences. pp. 1296–. ISBN 978-0-323-29354-9.
- ^ Prostate Cancer. Demos Medical Publishing. 20 December 2011. pp. 503–. ISBN 978-1-935281-91-7.
- ^ Geier M, Geier D (2005). “The potential importance of steroids in the treatment of autistic spectrum disorders and other disorders involving mercury toxicity”. Med Hypotheses. 64 (5): 946–54. doi:10.1016/j.mehy.2004.11.018. PMID 15780490.
- ^ Allen A (2007-05-28). “Thiomersal on trial: the theory that vaccines cause autism goes to court”. Slate. Archived from the original on 2008-02-03. Retrieved 2008-01-30.
- ^ “Testosterone regulation”. Research Autism. 2007-05-07. Archived from the original on 2015-04-18. Retrieved 2015-04-09.
- ^ Jump up to:a b “Maryland medical board upholds autism doctor’s suspension”. Chicago Tribune. May 11, 2011. Archived from the original on October 21, 2011.
- ^ “John and Jane Doe v. Ortho-Clinical Diagnostics, Inc Archived 2008-03-06 at the Wayback Machine“, US District Court for the Middle District of North Carolina, July 6, 2006
- ^ “Dr. Mark Geier Severely Criticized Archived 2016-12-02 at the Wayback Machine“, Stephen Barrett, M.D., Casewatch.org
- ^ Mills S, Jones T (2009-05-21). “Physician team’s crusade shows cracks”. Chicago Tribune. Archived from the original on 2009-05-25. Retrieved 2009-05-21.
- ^ ‘Miracle drug’ called junk science: Powerful castration drug pushed for autistic children, but medical experts denounce unproven claims Archived 2013-12-03 at the Wayback Machine, Chicago Tribune, May 21, 2009
- ^ Desmarchelier M, Lair S, Dunn M, Langlois I (2008). “Primary hyperaldosteronism in a domestic ferret with an adrenocortical adenoma”. Journal of the American Veterinary Medical Association. 233 (8): 1297–301. doi:10.2460/javma.233.8.1297. PMID 19180717.
- ^ Boari A, Papa V, Di Silverio F, Aste G, Olivero D, Rocconi F (2010). “Type 1 diabetes mellitus and hyperadrenocorticism in a ferret”. Veterinary Research Communications. 34(Suppl 1): S107–10. doi:10.1007/s11259-010-9369-2. PMID 20446034.
- ^ Doraiswamy PM, Xiong GL (2006). “Pharmacological strategies for the prevention of Alzheimer’s disease”. Expert Opinion on Pharmacotherapy. 7 (1): 1–10. doi:10.1517/14656566.7.1.S1. PMID 16370917.
- ^ Jump up to:a b c d “Leuprorelin oral – Enteris BioPharma – AdisInsight”. adisinsight.springer.com. Retrieved 16 July 2018.
External links
- Lupron Injection (package insert from abbott)
- Reforming (purportedly) Non-Punitive Responses to Sexual Offending (journal article discussing use of Lupron as a form of reforming sex offender law)
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Lupron, Eligard, Lucrin, others |
| Synonyms | Leuprolide; Leuprolidine; A-43818; Abbott-43818; DC-2-269; TAP-144 |
| AHFS/Drugs.com | Consumer Drug Information |
| MedlinePlus | a685040 |
| Pregnancy category |
|
| Routes of administration |
implant, injection |
| Drug class | GnRH analogue; GnRH agonist; Antigonadotropin |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Elimination half-life | 3 hours |
| Excretion | Kidney |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.161.466 |
| Chemical and physical data | |
| Formula | C59H84N16O12 |
| Molar mass | 1209.421 g·mol−1 |
| 3D model (JSmol) | |
|
|
//////////LEUPRORELIN, リュープロレリン ,
Penfluridol
- Molecular FormulaC28H27ClF5NO
- Average mass523.965 Da
| Summary |
|---|
| Although there are shortcomings and gaps in the data, there appears to be enough overall consistency for different outcomes. The effectiveness and adverse effects profile of penfluridol are similar to other typical antipsychotics; both oral and depot. Furthermore, penfluridol is shown to be an adequate treatment option for people with schizophrenia, especially those who do not respond to oral medication on a daily basis and do not adapt well to depot drugs. One of the results favouring penfluridol was a lower drop out rate in medium term when compared to depot medications. It is also an option for people with long-term schizophrenia with residual psychotic symptoms who nevertheless need continuous use of antipsychotic medication. An additional benefit of penfluridol is that it is a low-cost intervention.[3] |
Penfluridol
-
- ATC:N05AG03
- Use:neuroleptic
- Chemical name:1-[4,4-bis(4-fluorophenyl)butyl]-4-[4-chloro-3-(trifluoromethyl)phenyl]-4-piperidinol
- Formula:C28H27ClF5NO
- MW:523.97 g/mol
- CAS-RN:26864-56-2
- EINECS:248-074-5
- LD50:87 mg/kg (M, p.o.);
160 mg/kg (R, p.o.)
Synthesis
PAPER
Late stage functionalization of secondary amines via a cobalt-catalyzed electrophilic amination of organozinc reagents
Org Lett 2019, 21(2): 494
https://pubs.acs.org/doi/10.1021/acs.orglett.8b03787
Scheme 6

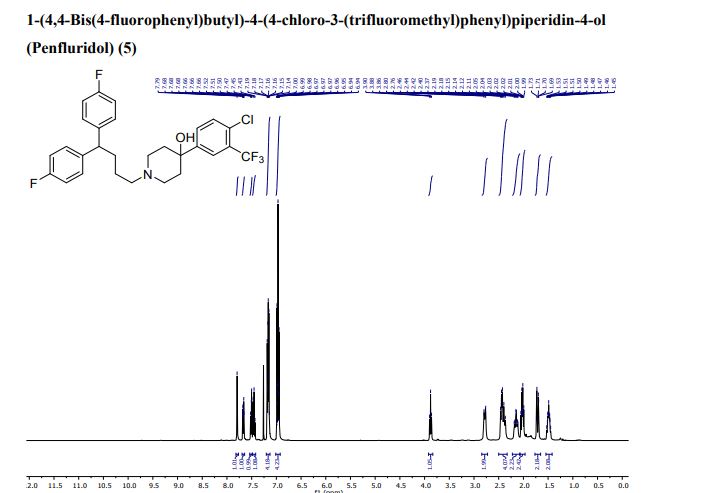
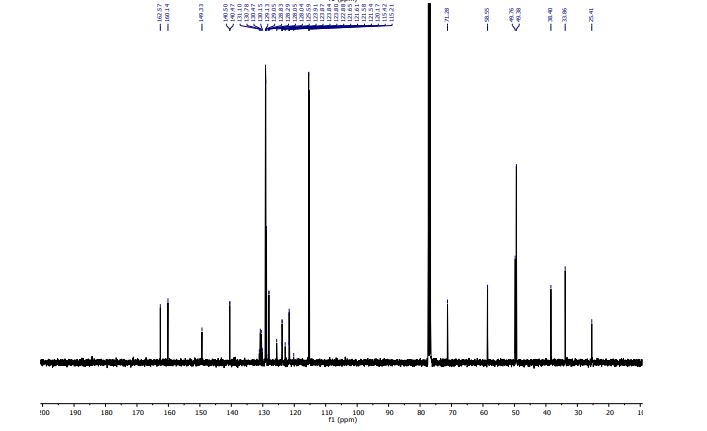


English: DE patent 2040231
US patent 3575990
doi:10.1135/cccc19733879

SYN
References
-
- US 3 575 990 (Janssen; 20.4.1971; appl. 3.9.1969).
- DOS 2 040 231 (Janssen; appl. 13.8.1970; USA-prior. 3.9.1969).
-
alternative synthesis:
- FR-appl. 2 161 007 (Janssen; appl. 23.11.1972; J-prior. 25.11.1971).
PATENT
https://patents.google.com/patent/CN106187863A/en
Although Penfluridol listed for many years, but its chemical preparation technology abroad little studied in the earlier literature, there are several prepared as follows:
[0013] Process (a): 1971 Document Ger.0ffen [P], 2040231, (1971) Hermans.HKF first reported Penfluridol chemical synthesis, which process is as follows:
[0014]

[0015] The process of cyclopropyl methanol (ΙΠ) by 4,4, _-difluorophenyl-one ([pi) as a starting material, the reaction of cyclopropyl magnesium bromide-bis 4- (fluorophenyl), then the reaction with thionyl chloride to give 1,1_-bis (4-fluorophenyl) -4-chloro-butene (IV), obtained by catalytic hydrogenation 1,1_-bis (4-phenyl gas) burning chlorobutanol _4_ (V), and finally with 4-chloro-3-methylphenyl gas-4-piperidinol (X VH) in methyl isobutyl ketone was refluxed for three days the reaction to produce Penfluridol (the I), Document: Sindelar.K.et al, Collect Czech.Chem.Commun [J], 38 (12): 3879-3901, (1973).
[0016] In the above process, starting material and documentation of cyclopropyl magnesium bromide hardly prepared each reaction were not reported preparation yield, and therefore Document Sindelar · K · et al, Collect Czech · Chem · Commun [ J], 38 (12):. 3879-3901, (1973) that this technology is not very good.
[0017] Process (b): 1973, Sindelar.K successful research and the following other technology, which process is as follows:
[0018]

[0019] The process consists of 4,4_-bis (4-fluorophenyl) butoxy alkyl iodide as a starting material, 4,4_ ethylenedioxythiophene condensing piperidone removal of generated hydrogen iodide in N-pentanone – [4,4-bis (4-fluorophenyl) butoxy group] -4,4-dioxo-condensing vinyl piperidone, N-then obtained by acid hydrolysis [4,4-bis (4-fluorophenyl ) azetidinyl] -4-piperidone (W), the compound ![]() with 4-chloro-3-trifluoromethyl phenyl magnesium bromide reacted Penfluridol (I).
with 4-chloro-3-trifluoromethyl phenyl magnesium bromide reacted Penfluridol (I).
[0020] This process route may seem simple, but there are more desired to prepare intermediates, the process is more complex, with low yields reported in the literature.
[0021] Process (c): as follows:
[0022]

[0023] In this process, 4-chloro – (4-fluorophenyl) butyryl-one (Shan) starts, 4-fluorophenyl magnesium bromide reacts with 4-chloro – bis (4-fluorophenyl) butanol ( IX), and then boiling the reaction hydroiodic acid to give 4-iodo-in, red phosphorus catalyst – bis (4-fluorophenyl) butoxy left foot and finally burning ^^ – ^ – methyl ^ two gas – chlorophenyl Bu ‘piperidinol prepared products San ^ top five gas profitable ⑴.
[0024] This synthesis has the characteristics of high yield, but the intermediate (IX), (X) quality is not purified, many by-products, difficult to control the quality of products, and hydroiodic acid to be used, the source of raw material is difficult, therefore, not ideal technology.
[0025] Process (d), as follows:
[0026]

[0027] The process begins by Stobber reaction with 4,4 – fluorophenyl ketone reaction product diethyl succinate and compound (XI), and then generates bis (4-fluorophenyl) methine acid or base hydrolysis after succinic acid (M), by catalytic hydrogenation to give 4,4_-bis (4-fluorophenyl) butanoic acid after, the reaction with thionyl chloride without isolating the compound (XIV) with the compound directly (XW), by reduction after obtain the final product – Penfluridol. The disadvantage of this process is that, in the above reaction step, Stobber the reaction yield is low; hydrogenation catalyst manufacturing operation more difficult and unsafe; reaction with thionyl chloride, large air pollution, and other refractory.
[0028] The various preparation techniques Penfluridol other drug earlier British Patent Brit. 1141664 and German patent Ger. Off en. 2040231 has been reported, but no other foreign patent reports. In neither country has patent coverage, and no magazine reported.
The reaction formula is as follows:
[0058]

[0059] Step (5), the preparation of compounds of formula (XW) as shown, may be employed a method reported in the literature, or prepared using a method specifically includes the following steps:
[0060]

0124] (6) Penfluridol drug (I) were prepared:
[0125]

[0126] In three 500ml reaction flask equipped with a mechanical stirrer, a condenser, a thermometer, a calcium chloride tube, was added 250ml of anhydrous diethyl ether, 2 · 4g (0 · 0631mol) tetrahydro lithium aluminum hydride, stirring was started, was added 20g (0 · 0372mol) amide (6), the addition was completed, 38 ° C for 6 hours.
[0127] completion of the reaction, water was added 4.2ml decomposition for 25 minutes, followed by addition of 5.4ml of 20% by weight concentration of sodium hydroxide solution decomposition for 20 minutes, 14.2ml decomposed with water for 15 minutes;
[0128] The decomposition was filtered, the filtrate (ethyl ether) and dried over anhydrous potassium carbonate. Filtered, the filter cake was washed with a little ether. The filtrate and the washings added to a distillation flask, recovery ether atmospheric distillation, vacuum drained, was added a mixed solvent l〇〇ml [chloroform: petroleum ether (60-90 ° C) = 1: 4, weight ratio, stirred and heated to reflux dissolution, filtered while hot, the filtrate was allowed to stand for crystallization at about 10 ° C, to be naturally deposited crystal after freezing -5 ° C overnight, filtered, the cake was washed with a mixed solvent, drain, ventilation pressure at 70 ° C dried to constant weight to give white crystalline product Penfluridol drug (I), mp 105-107 ° C, yield 81.5%.
[0129] Intermediate 4_ (3-trifluoromethyl-4-chlorophenyl) -4-piperidinol (XW) (referred piperidinol) Preparation:
[0130] (1) benzylamine (Beta) Preparation:
[0131]

[0132] equipped with a mechanical stirrer, a condenser, a thermometer 2000ml three reaction bottle, were added ammonium bicarbonate 240g (3.04mol), aqueous ammonia at a concentration of 20 wt %% of 15148 (17.812111〇1,
[0133] 1640ml), benzyl chloride 80g (0.632mol), reaction was stirred for 6 hours.Reaction to complete rested stratification. Aqueous layer was separated, and aqueous ammonia recovery bicarbonate atmospheric heating to 100 ° c, the water was distilled off under reduced pressure, with 50% sodium hydroxide PH12 above, extraction with benzene and dried solid sodium hydroxide. Recovery of benzene atmospheric distillation, vacuum distillation, collecting 33.4 g of the product obtained, yield 50.7%, content 99%,
[0134]

[0135] (3) N_ benzyl – bis ([beta] methoxycarbonyl-ethyl) amine (C) (referred to as diester thereof):
[0136]

[0137] The reaction flask equipped with a mechanical stirrer, a condenser, a thermometer three 250ml, 43g methyl acrylate (0.5111〇1) methanol 328 (401111), was added with stirring 21.48 benzylamine (0.2111〇1), The reaction was stirred for 7 hours. Completion of the reaction, recovery of excess methyl acrylate and methanol, water chestnut vacuum distillation until the internal temperature l〇〇-ll〇 ° C, to give the crude product as a yellow oil (C) 54g, yield 97%, content 94.3%.
[0138] (3) 1 – benzyl-4-piperidone (E) (referred to as the hydrolyzate) is prepared:
[0139]

[0140] In a reaction flask equipped with a 500ml three mechanical stirrer, thermometer, fractional distillation apparatus, was added 27% sodium methoxide 27g, crude diester was 33.4g (0.12mol), toluene 300ml, stirred and heated, the temperature reached 90 when ° C or more, additional 50ml toluene was reacted for 3 hours. Cooled to room temperature, and neutralized with acetic acid to PH6, standing layer. The toluene layer was separated and extracted with 150ml of 22% hydrochloric acid three times. Hydrochloric acid extracts were combined, heated with stirring for 4 hours. Recovered by distillation under reduced pressure and hydrochloric acid (about 120ml distilled dilute hydrochloric acid) was cooled to distillation l〇 ° C below, with 40% sodium hydroxide PH12 above. With 80ml ethyl acetate 3 times extracted with ethyl acetate extracts were combined, sub-net water, dried over anhydrous sodium sulfate. Sodium sulfate was removed by filtration, recovering ethyl acetate atmospheric distillation, vacuum drained hydrolyzed to give (E) and the crude product 19g, yield 84%.

[0141] (4) 1-ethoxycarbonyl-4-piperidone (F) (referred to as a carbonyl group-piperidone) Preparation:
[0142]

[0143] equipped with a mechanical stirrer, a condenser, 250ml three reaction flask thermometer, was added ethyl chloroformate 23.9g (0 · 22mo 1), benzene 100ml, stirring slowly added dropwise [The crude hydrolyzate (E ) 37 · 8g (0 · 2mo 1) + 20ml phenyl] solution dropwise, the reaction was heated with stirring for 5 hours.Water chestnut evaporated under reduced pressure and ethyl benzene chlorine, Li mechanical change stream distilled off under reduced pressure, low boiling point evaporated to give the product 268 was collected, yield 76%.
[0144] (5) 1 – ethoxycarbonyl-4- (3-trifluoromethyl-4-chlorophenyl) -4-piperidinol (G) (referred to as a carbonyl group-piperidinol) is:
[0145]

[0146] In three 500ml reaction flask equipped with a mechanical stirrer, a condenser, a thermometer, a dropping funnel and a calcium chloride drying tube over anhydrous anhydrous absolute, at room temperature was added magnesium metal shoulder 2.5g (0.103mol) 20ml of anhydrous ethyl ether and slowly stirring was started.
[0147] 2-chloro-5-bromo – trifluorotoluene (referred bromide) was dissolved under 27g (0.104mol) at room temperature in 130ml anhydrous diethyl ether and stirred to obtain a uniform liquid mass (W is);
[0148] When the liquid material taken ![]() 15ml was added to the above reaction, a solution of iodine 0.13g, 1,2- dibromoethane 0.2g, initiated Grignard reaction was heated until the iodine color disappeared, the reaction slowed down, slow slow dropping liquid material (W). The addition was completed, refluxing was continued for 1 hour. Completion of the reaction, cooled to room temperature, slowly added dropwise at room temperature carbonyl piperidone (F) water solution was cooled at normal [carbonyl-piperidone 13.6g (0.0795mol) + 40ml dry ether], dropwise, the reaction was heated with stirring 1.5 hour. L〇〇ml ammonium chloride solution concentration of 20% by weight was added, refluxed for 15 minutes and allowed to stand 30 minutes at room temperature stratification. Discharged aqueous layer (lower layer), the residual liquid was distilled (upper layer) at an external temperature of 55 ° C atmospheric distillation recovery ether, discharge hot, refrigerated overnight, the precipitated solid. Filtered, washed with a small amount of time, drained, and dried to give the product (G) 24.1g, yield 85.7%, mp 118-126Γ.
15ml was added to the above reaction, a solution of iodine 0.13g, 1,2- dibromoethane 0.2g, initiated Grignard reaction was heated until the iodine color disappeared, the reaction slowed down, slow slow dropping liquid material (W). The addition was completed, refluxing was continued for 1 hour. Completion of the reaction, cooled to room temperature, slowly added dropwise at room temperature carbonyl piperidone (F) water solution was cooled at normal [carbonyl-piperidone 13.6g (0.0795mol) + 40ml dry ether], dropwise, the reaction was heated with stirring 1.5 hour. L〇〇ml ammonium chloride solution concentration of 20% by weight was added, refluxed for 15 minutes and allowed to stand 30 minutes at room temperature stratification. Discharged aqueous layer (lower layer), the residual liquid was distilled (upper layer) at an external temperature of 55 ° C atmospheric distillation recovery ether, discharge hot, refrigerated overnight, the precipitated solid. Filtered, washed with a small amount of time, drained, and dried to give the product (G) 24.1g, yield 85.7%, mp 118-126Γ.
[0149] (6) 4- (3-trifluoromethyl-4-chlorophenyl) -4-piperidinol (X VH) (referred piperidinol) Preparation:
[0150]

[0151] equipped with a mechanical stirrer, a condenser, 250ml three reaction flask thermometer, were added ethanol 40ml, 158 of sodium hydroxide (0.375111〇1), carbonyl piperidinol (6) 2 (^ (0.0569111〇1 ), heated to reflux, and the reaction stirred for 3.5 hours. the reaction was completed, 50ml of water was added, the reaction was refluxed for 10 minutes, the hot reaction solution was placed in 300g of crushed ice, stirred well, and the precipitated solid, -5 ° C frozen standing for 2 hours the above.
[0152] filtered, washed with water to pH 8-9, drained, and dried to give piperidinol (XVH) 15g, yield 94%, mp 137-144 ° C, ash content <5%.
[0153] Example 2
[0154] (a) 3- (4-fluorobenzoyl) propionic acid (2) (the acid) is prepared:
[0155]

[0156] The reaction flask equipped with a mechanical stirrer, a condenser, a thermometer three 500ml, was added 17.1g (0.171mol) of succinic anhydride, l〇5g (1 · 09mol) fluorobenzene, stirred and dissolved. Added in one portion 60g (0 · 306mol) in dry wrong trichloride, stirring, the reaction was stirred at 100 ° C for 2 hours, at a concentration of 10% by weight hydrochloric acid 165ml exploded 30 minutes;
[0157] Other embodiments with Example 1, the product, 111.? 105-107 ° (:, this step a yield of 81.5%, 46.7% overall yield.
References
- ^ van Praag HM, Schut T, Dols L, van Schilfgaarden R., Controlled trial of penfluridol in acute psychosis, Br Med J. 1971 December 18;4(5789):710-3
- ^ Janssen PA, Niemegeers CJ, Schellekens KH, Lenaerts FM, Verbruggen FJ, Van Nueten JM, Schaper WK., The pharmacology of penfluridol (R 16341) a new potent and orally long-acting neuroleptic drug, Eur J Pharmacol. 1970 July 15;11(2):139-54
- ^ Jump up to:a b Soares, B; Silva de Lima, M (2006). “Penfluridol for schizophrenia”. Cochrane Database of Systematic Reviews. 2: CD002923.pub2. doi:10.1002/14651858.CD002923.pub2.
Further reading
- Benkert O, Hippius H.: Psychiatrische Pharmakotherapie, Springer-Verlag, 1976, 2. Auflage. ISBN3-540-07916-5
- R Bhattacharyya, R Bhadra U Roy, S Bhattacharyya, J Pal S Sh Saha – Resurgence of Penfluridol:Merits and Demerits, Eastern Journal of Psychiatry, January-June 2015 vol 18, Issue 1 p 23 –29
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| ATC code | |
| Identifiers | |
| CAS Number | |
| PubChemCID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard(EPA) | |
| ECHA InfoCard | 100.043.689 |
| Chemical and physical data | |
| Formula | C28H27ClF5NO |
| Molar mass | 523.965 g·mol−1 |
| 3D model (JSmol) | |
/////////Penfluridol, Antipsychotic, Semap, Micefal, Longoperidol, MCN-JR-16,341, R 16,341, MCN-JR-16,341 / R 16,341,
