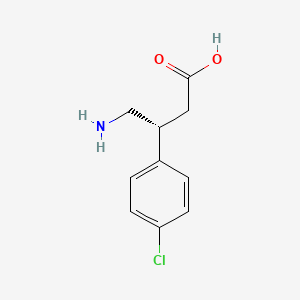
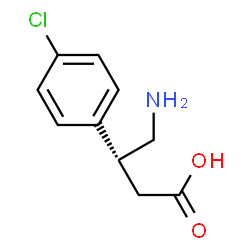
(R)-(–)-Baclofen, Arbaclofen, STX 209, AGI 006
| Chemical Names: |
(R)-Baclofen; Arbaclofen; 69308-37-8; (R)-4-Amino-3-(4-chlorophenyl)butanoic acid; (-)-Baclofen; D-Baclofen |
| Molecular Formula: |
C10H12ClNO2 |
| Molecular Weight: |
213.661 g/mol |
Spasticity, PREREGISTERD, OSMOTICA PHARMA
- Benzenepropanoic acid, β-(aminomethyl)-4-chloro-, (R)-
- (βR)-β-(Aminomethyl)-4-chlorobenzenepropanoic acid
- (-)-Baclofen
- (R)-(-)-Baclofen
- (R)-4-Amino-3-(4-chlorophenyl)butanoic acid
- (R)-4-Amino-3-(4-chlorophenyl)butyric acid
- (R)-Baclofen
- AGI 006
- Arbaclofen
- D-Baclofen
- R-(-)-Baclofen
- STX 209
- l-Baclofen
Optical Rotatory Power, -1.76 °, Conc: 0.5 g/100mL; Solv: water (7732-18-5); Wavlen: 589.3 nm; Temp: 25 °C, REF …..Paraskar, Abhimanyu S.; Tetrahedron 2006, VOL62(20), PG4907-4916

Arbaclofen, or STX209, is the R-enantiomer of baclofen. It is believed to be a selective gamma-amino butyric acid type B receptor agonist, and has been investigated as a treatment for autism spectrum disorder and fragile X syndrome in randomized, double blind, placebo controlled trials. It has also been investigated as a treatment for spasticity due to multiple sclerosis and spinal cord injury. Arbaclofen was investigated as a treatment for gastroesophageal reflux disease (GERD); however, with disappointing results.
AGI-006, a GABA(B) agonist, is currently in phase III clinical trials at Seaside Therapeutics for the treatment of social withdrawal in adolescents and adults with Fragile X Syndrome and for the treatment of autism spectrum disorders. AGI Therapeutics had been conducting clinical trials for the treatment of dyspepsia and for the treatment of delayed gastric emptying in diabetic patients; however, no recent development has been reported for this research. In 2015, Osmotica Pharmaceutical filed a NDA seeking approval of an extended-release formulation for the alleviation of spasticity due to multiple sclerosis.
AGI-006 is an oral formulation of arbaclofen, the R-isomer of baclofen. In 2012, a license option agreement was signed between Seaside and Roche by which the latter may commercialize the product upon completion of certain clinical development phases in fragile X syndrome and in autism spectrum disorders.
 Baclofen [USAN:USP:INN:BAN:JAN]
Baclofen [USAN:USP:INN:BAN:JAN]
1134-47-0
 Baclofen hydrochloride
Baclofen hydrochloride
28311-31-1
 Arbaclofen hydrochloride
Arbaclofen hydrochloride
63701-55-3
 (S)-Baclofen hydrochloride
(S)-Baclofen hydrochloride
63701-56-4
 (S)-Baclofen
(S)-Baclofen
66514-99-6
 Acamprosate mixture with baclofen
Acamprosate mixture with baclofen
1395997-58-6
CLIP1

Strategy for asymmetric synthesis of (R)-(-)-Baclofen is as represented in the Scheme 14. Herein, we made use of asymmetric Michael addition of nitromethane to 4- Chlorochalcone in the presence of Cu(acac)2 and (-)-Sparteine as a catalyst in DCM for 8 h to provide γ-nitro ketone as colorless solid, mp 105-109°C, in 87% yield with 82% ee. The Michael adduct 3d on Baeyer-Villiger reaction using m-CPBA to produce corresponding nitro ester 6a. The reduction of 6a containing nitro group can be reduced with sodium borohydride in presence of NiCl2. It resulted to generate 7 cyclic pyrrolidine moiety in 65% yield. Which upon hydrolysis with HCl will lead to (R)-(-)- Baclofen 8 as a neurotransmitter inhibitor drug molecule


(R)-4-amino-3-(4-chlorophenyl)butanoic acid hydrochloride (8) The solution of 7 (100 mg, 0.51 mmol) in 6N HCl (2.7 mL) was refluxed at 100 °C. After 24 h, the reaction mixture was concentrated in vacuo to afford (R)-(–)- Baclofen 8 as colorless solid 93 mg, in 73% yield. Yield : 73% State : Solid. M.P. : 188-189 °C [a]D 25 : –3.4o (c = 0.65, H2O), lit.7 –3.79o (c = 0.65, H2O, 99 % ee) 1 H-NMR (300MHz, D2O) : δ. 7.36-7.49 (m, 4H) 3.50-3.37 (m, 2H), 2.30-3.22 (m, 1H), 2.71-2.92 (dd, 2H,) J = 9.5, 16.5 Hz).ppm 13C-NMR (75MHz, D2O) : δ. 175.46, 138.28, 136.95, 133.32, 129.32, 128.25, 127.81, 43.75, 39.91, 38.18.
7. Corey, E. J; Zhang, F. Y. Org. Lett. 2000, 2, 4257-4259
16. a) Thakur, V. V.; Nikalje, M. D.; Sudalai, A. Tetrahedron Asymmetry 2003, 14, 581. b) Chenevert R.; Desjardins, M.; Tetrahedron Lett. 1991, 32, 4249. c) Herdeis, C.; Hubmann, H. P. Tetrahedron Asymmetry 1992, 3, 1213. d) Meyers, A. I.; Snyder, L. J. Org. Chem. 1993, 58, 36.
clip 2

Yoshiji Takemoto (2005)6 Yoshiji Takemoto et al. have developed chiral thiourea catalyst 15 which was found to be highly efficient for the asymmetric Michael addition of 1,3-dicarbonyl compound to nitroolefins. Furthermore, a new synthetic route for (R)-(-)-Baclofen 14 and the generation of a chiral quaternary carbon center with high enantioselectivity by Michael reaction were developed (Scheme 6)
6. Okino, T.; Hoashi, Y.; Xuenong Xu,; Takemoto, Y.. J. Am. Chem. Soc. 2005, 127, 119.
CLIP3
Enantio- and Diastereoselective Michael Reaction of 1,3-Dicarbonyl Compounds to Nitroolefins Catalyzed by a Bifunctional Thiourea
Contribution from the Graduate School of Pharmaceutical Sciences, Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan
J. Am. Chem. Soc., 2005, 127 (1), pp 119–125
DOI: 10.1021/ja044370p
Publication Date (Web): December 3, 2004
Copyright © 2005 American Chemical Society
Abstract
We synthesized a new class of bifunctional catalysts bearing a thiourea moiety and an amino group on a chiral scaffold. Among them, thiourea 1e bearing 3,5-bis(trifluoromethyl)benzene and dimethylamino groups was revealed to be highly efficient for the asymmetric Michael reaction of 1,3-dicarbonyl compounds to nitroolefins. Furthermore, we have developed a new synthetic route for (R)-(−)-baclofen and a chiral quaternary carbon center with high enantioselectivity by Michael reaction. In these reactions, we assumed that a thiourea moiety and an amino group of the catalyst activates a nitroolefin and a 1,3-dicarbonyl compound, respectively, to afford the Michael adduct with high enantio- and diastereoselectivity.
http://pubs.acs.org/doi/full/10.1021/ja044370p
http://pubs.acs.org/doi/suppl/10.1021/ja044370p/suppl_file/ja044370psi20040916_090526.pdf
Synthesis of (R)–(−)-Baclofen. γ-Amino butylic acid (GABA) plays an important role as an inhibitory neurotransmitter in the central nervous system (CNS) of mammalians,20,21 and the deficiency of GABA is associated with diseases that exhibit neuromuscular dysfunctions such as epilespy, Huntington’s and Parkinson’s diseases, etc.22 Baclofen is a lipophilic analogue of GABA, and it is widely used as an antispastic agent. Although baclofen is commercialized in its racemic form, it has been reported that its biological activity resides exlusively in the (R)-enantiomer.23 We next applied our enantioselective Michael reaction for the synthesis of (R)-(−)-baclofen (Scheme 1). The reaction of 4-chlorobenzaldehyde with nitromethane and subsequent dehydration of the resultant alcohol 8 provided nitroolefin 9, which was reacted with diethyl malonate 3a in the presence of 10 mol % of 1e to afford the adduct 10 in 80% yield with 94% ee. Furthermore, enantiomerically pure 10 (>99% ee) was obtained after single recrystallization from Hexane/EtOAc. Reduction of the nitro group with nickel borite and in situ lactonization gave lactone 11 in 94%. The ester group of 11 was hydrolyzed and decarboxylated to afford 12. The specific rotation of 12 was compared with that of literature data24 ([α]30D −39.7° (c 1.00, EtOH), lit. [α]25D −39.0° (c 1, EtOH)), and, as expected, the absolute configuration of 12 was determined to be R. Lactam 12 was finally hydrolyzed with 6N HCl, affording enantiomerically pure (R)-(−)-baclofen as its hydrochloric salt with 38% overall yield in six steps from 4-chlorobenzaldehyde. Consequently, we succeeded in the synthesis of (R)-(−)-baclofen by the simple procedure with high enantioselctivity.

Scheme 1. Total Synthesis of (R)-(−)-Baclofena
a Conditions: (a) MeNO2, NaOMe, MeOH, room temperature, 15 h; (b) MsCl, TEA, THF, room temperature, 1 h; (c) diethyl malonate, 1e, toluene, room temperature, 24 h; (d) NiCl2·6H2O, NaBH4, MeOH, room temperature, 7.5 h; (e) NaOH, EtOH, room temperature, 45 h; (f) toluene, reflux, 6.5 h; (g) 6N HCl, reflux, 24 h.
Total synthesis of (R)-(–)-baclofen. 9: The mixture of 4-chlorobenzaldehyde (1.41 g, 10 mmol), nitromethane (10 equiv, 5.4 ml) and NaOMe (0.10 equiv, 54.0 mg) in MeOH (10 ml) was stirred overnight. Saturated ammonium chloride was added to the mixture and aqueous phase was extracted with AcOEt. The extract was washed with brine, dried over MgSO4, filtrated and concentrated in vacuo. The residue was purified by by column chromatography on silica gel (Hexane/AcOEt = 3/1 as eluent) to afford desired nitroalcohol 8 (1.82 g, 90%). To the stirred solution of the obtained nitroalcohol 8 and MsCl (1.2 equiv, 0.84 ml) in THF (9.0 ml) was added TEA (2.1 equiv, 2.7 ml) dropwise at 0 °C. After 1 h, saturated ammonium chloride was added to the reaction mixture and aqueous phase was extracted with AcOEt. The extract was washed with 1N HCl (two times), saturated NaHCO3 and brine, dried over MgSO4, filtrated and concentrated in vacuo. The residual solid was purified by recrystallization from AcOEt/Hexane to afford the desired nitroolefin 9 (1.20 g, 72%). yellow needle; m.p. 112 °C (AcOEt/Hexane); 1 H NMR (500 MHz, CDCl3) δ 7.97 (d, J = 13.7 Hz, 1H), 7.57 (d, J = 13.7 Hz, 1H), 7.50 (d, J = 8.6 Hz, 2H), 7.44 (d, J = 8.6 Hz, 2H) ppm; 13 C NMR (126 MHz, CDCl3) δ 138.4, 137.7, 137.5, 130.3, 129.8, 128.6 ppm; IR (CHCl3) ν 3113, 3029, 1637, 1594, 1525, 1494 cm-1 ; MS (EI + ) 183 (M+ , 51), 101 (100); Anal. Calcd. for C8H6ClNO2: C 52.34; H, 3.29; N, 7.63; Cl, 19.31. Found: C, 52.35; H, 3.40; N, 7.67; Cl, 19.24. 10: Under argon atmosphere, to the stirred solution of p-chloro-β-nitrostylene 9 (36.7 mg, 0.20 mmol) and thiourea (0.10 equiv, 8.3 mg) in toluene (0.40 ml) was added diethylmalonate (2 equiv, 0.060 ml) at rt. After 24 h, the reaction mixture was concentrated in vacuo. The residue was purified by column chromatography on silica gel (AcOEt/hexane = 1/5 as eluent) to afford desired product 10 (55.3 mg, 80%) as colorless solid. Enantiomerically pure 10 (>99% ee) was obtained after single recrystallization from Hexane/AcOEt. m.p. 56-57 °C (Hexane/AcOEt); [α]D 25 –8.56 (c 1.02, CHCl3, >99% ee); 1 H NMR (500 MHz, CDCl3) δ 7.30 (d, J = 8.2 Hz, 2H), 7.19 (d, J = 8.6 Hz, 2H), 4.91 (dd, J = 4.6, 13.1 Hz, 1H), 4.83 (dd, J = 9.5, 13.1 Hz, 1H), 4.23 (m, 3H), 4.04 (q, J = 7.22 Hz, 2H), 3.78 (d, J = 9.5 Hz, 1H), 1.27 (t, J = 7.2 Hz, 3H), 1.09 (t, J = 7.0 Hz, 3H); 13 C NMR (126 MHz, CDCl3) δ 167.4, 166.8, 134.9, 134.5, 129.6, 129.3, 77.5, 62.3, 62.1, 54.8, 42.4, 14.0, 13.8 ppm; IR (CHCl3) ν 3031, 2994, 1733, 1558, 1494, 1374 cm-1 ; MS (FAB+ ) 344 (MH+ , 100); Anal. Calcd for C15H18ClNO6: C, 52.42, H, 5.28, N, 4.07, Cl, 10.31; Found: C, 52.52, H, 5.21, N, 4.07, Cl, 10.25; HPLC [Chiralcel OD-H, hexane/2-propannol = 90/10, 0.5 mL/min, λ = 210 nm, retention times: (major) 28.3 min, (minor) 25.1 min]. 11: Under argon atmosphere, to the suspension of 10 (550 mg, 1.60 mmol, >99% ee) and NiCl2· 6H2O (1.0 equiv, 380 mg) in MeOH (8.0 ml) was added NaBH4 (12 equiv, 726 mg) at 0 °C. After the reaction mixture was stirred 7.5 h at rt, the reaction mixture was quenched with NH4Cl and diluted with CHCl3. The organic layer was separated and dried over MgSO4, filtrated and concentrated in vacuo. The residue was purified by column chromatography on silica gel (MeOH/CHCl3 = 1/20 as eluent) to afford desired product (402 mg, 94%) as colorless powder. m.p. 126-128 °C (Hexane/AcOEt); [α]D 26 –123.4 (c 0.96, CHCl3); 1 H NMR (500 MHz, CDCl3) δ 7.31 (m, 2H), 7.20 (d, J = 8.2 Hz, 2H), 7.12 (s, 1H), 4.24 (q, J = 7.0 Hz, 1H), 4.09 (m, 1H), 3.81 (m, 2H), 3.54 (m, 1H), 3.41 (m, 1H), 1.28 (t, J = 6.9 Hz, 3H); 13 C NMR (126 MHz, CDCl3) δ 172.5, 169.0, 138.3, 133.5, 129.2, 128.4, 61.9, 55.2, 47.5, 43.7, 14.1 ppm; IR (CHCl3) ν 3435, 3229, 3017, 2360, 1710, 1493 cm-1 ; MS (FAB+ ) 268 (MH+ , 100); Anal. Calcd for C13H14ClNO3: C, 58.32, H, 5.27, N, 5.23, Cl, 13.24; Found: C, 58.10, H, 5.15, N, 5.43, Cl, 13.13. 12 : To the solution of 11 (240mg, 0.90 mmol) in EtOH (3.6 ml) was added 1N NaOH (1.1 ml) at rt. After 30 min, the reaction mixture was concerned in vacuo. To the residue was added H2O and 5N HCl, and the aqueous phase was extracted with CHCl3. The extract was dried over MgSO4, filtrated andconcentrated in vacuo to afford corresponding carboxylic acid (194 mg, 90%). The solution of carboxylic acid (194 mg, 0.81 mmol) in toluene (11 ml) was refluxed at 140 °C. After 6 h, the mixture was concentrated in vacuo. The residue was purified by column chromatography on silica gel (MeOH/ CHCl3 = 1/7) to afford desired product 12 (148 mg, 93%) as colorless needle. m.p. 109-111 °C (Hexane/AcOEt); [α]D 30 –39.7 (c 1.00, CHCl3); 1 H NMR (500 MHz, CDCl3) δ 7.32 (d, J = 7.9 Hz, 2H), 7.19 (t, J = 8.2 Hz, 2H), 6.15 (s, 1H), 3.79 (t, J = 8.9 Hz, 1H), 3.68 (m, 1H), 3.38 (t, J = 8.4 Hz, 1H), 2.74 (dd, J = 9.0, 16.9 Hz, 1H), 2.45 (dd, J = 8.6, 16.8 Hz, 1H); 13 C NMR (126 MHz, CDCl3) δ 177.5, 140.7, 132.9, 129.0, 128.1, 49.3, 39.6, 37.8 ppm; IR (CHCl3) ν 3439, 3006, 2361, 1699, 1494 cm-1 ; MS (FAB+ ) 196 (MH+ , 100); Anal. Calcd for C10H10ClNO: C, 61.39, H, 5.15, N, 7.16, Cl, 18.12; Found: C, 61.50, H, 5.21, N, 7.25, Cl, 17.98. (R)-(–)-baclofen : The solution of 12 (107 mg, 0.55 mmol) in 6N HCl (2.7 ml) was refluxed at 100 °C. After 24 h, the reaction mixture was concentrated in vacuo to afford (R)-(–)-baclofen (129 mg, 94%) as colorless solid. m.p. 188-189 °C (exane/i-PrOH); [α]D 25 –3.79 (c 0.65, H2O); 1 H NMR (500 MHz, DMSO-d6) δ 12.26 (s, 1H), 8.13 (s, 3H), 7.35 (m, 4H), 3.09 (m, 1H), 2.94 (m, 1H), 2.85 (dd, J = 5.5, 16.2 Hz, 1H), 2.56 (dd, J = 9.5, 16.5 Hz, 1H); 13 C NMR (126 MHz, DMSO-d6) δ 172.5, 139.5, 131.9, 130.0, 128.7, 128.6, 128.0, 43.1, 39.1, 37.8 ppm; MS (FAB+ ) 214 (MH+ , 100); HRMS (FAB+ ) Calcd for [C10H13ClNO2] + : 214.0635; Found: 214.0637.

http://www.sciencedirect.com/science/article/pii/S0957416604003672

http://www.sciencedirect.com/science/article/pii/S0957416699002359
 The thiourea catalyst L7 bearing 3,5-bis(trifluoromethyl) benzene and dimethylamino groups has been revealed to be efficient for the asymmetric Michael reaction of 1,3-dicarbonyl compounds to nitroolefins (Scheme 8). This methodology has been applied for the total synthesis of (R)-(−)-baclofen. Reaction of 4-chloronitrostyrene and 1,3-dicarbonyl compound generates quaternary carbon center with 94% ee. Reduction of the nitro gruop to amine and subsequent cyclization, esterification and ring opening provides ( R )-(−)-baclofen in 38% yield.
The thiourea catalyst L7 bearing 3,5-bis(trifluoromethyl) benzene and dimethylamino groups has been revealed to be efficient for the asymmetric Michael reaction of 1,3-dicarbonyl compounds to nitroolefins (Scheme 8). This methodology has been applied for the total synthesis of (R)-(−)-baclofen. Reaction of 4-chloronitrostyrene and 1,3-dicarbonyl compound generates quaternary carbon center with 94% ee. Reduction of the nitro gruop to amine and subsequent cyclization, esterification and ring opening provides ( R )-(−)-baclofen in 38% yield.

http://pubs.rsc.org/en/content/articlelanding/2010/np/b924964h/unauth#!divAbstract

http://pubs.rsc.org/en/content/articlelanding/2010/np/b924964h/unauth#!divAbstract

http://pubs.rsc.org/en/Content/ArticleHtml/2016/SC/c5sc02913a

REF
Highly enantioselective biotransformations of 2-aryl-4-pentenenitriles, a novel chemoenzymatic approach to (R)-(-)-baclofen
Tetrahedron Lett 2002, 43(37): 6617
Enantioselective Michael addition of nitromethane to alpha,beta-enones catalyzed by chiral quaternary ammoniun salts. A simple synthesis of (R)-baclofen
Org Lett 2000, 2(26): 4257
Stereospecific synthesis of (R)- and (S)-baclofen and (R)- and (S)-PCPGABA [4-amino-2-(4chlorophenyl)butyric Acid] via (R)- and (S)-3-(4-Chlorophenyl)pyrrolidines
Chem Pharm Bull 1995, 43(8): 1302
Enantioselective syntheses of (-)-(R)-rolipram, (-)-(R)-baclofen and other GABA analogues via rhodium-catalyzed conjugate addition of arylboronic acids
Synthesis (Stuttgart) 2003, (18): 2805
Palladium-catalyzed, asymmetric Baeyer-Villiger oxidation of prochiral cyclobutanones with PHOX ligands
Tetrahedron 2011, 67(24): 4352
An efficient synthesis of (R)- and (S)-baclofen via desymmetrization
Tetrahedron Lett 2009, 50(45): 6166
Recoverable resin-supported pyridylamide ligand for microwave-accelerated molybdenum-catalyzed asymmetric allylic alkylations: Enantioselective synthesis of baclofen
Org Lett 2003, 5(13): 2275
Asymmetric synthesis of ß-substituted ?-lactams via rhodium/diene-catalyzed 1,4-additions: Application to the synthesis of (R)-baclofen and (R)-rolipram
Org Lett 2011, 13(4): 788
Multisite organic-inorganic hybrid catalysts for the direct sustainable synthesis of GABAergic drugs
Angew Chem Int Ed 2014, 53(33): 8687
///////////////
http://www.jocpr.com/articles/a-facile-synthesis-of-baclofean-via-feacac3-catalyzed-michael-addition-and-pinner-reaction.pdf
http://shodhganga.inflibnet.ac.in/bitstream/10603/93509/10/10_chapter1.pdf





(±)-Baclofen, hydrochloride (2)
A mixture of 4-(4-Chlorophenyl) pyrrolidin-2-one 15 (0.070 g, 0.35 mmol) in HCl aqueous solution (6 mol L-1, 1.5 cm3) was heated at 100 °C for 6 h. The solvent was removed under reduced pressure and the residue was triturated in isopropanol yielding a crystalline (±)-baclofen hydrochloride 2 (0.071 g, 82%).; IR nmax/cm -1: 3415, 3006, 1713, 1562, 1492, 1407, 1251, 1186, 815 cm-1 (KBr, neat); 1H NMR (300 MHz, CDCl3) d 2.55 (dd, J 16.5 and 8.7 Hz, 1 H); 2.82 (dd, J 16.5 and 5.7 Hz, 1 H); 2.93-3.50 (m, 3 H); 7.34 (d, J 8.7 Hz, 2 H), 7.40 (d, J 8.7 Hz, 2 H), 7.94 (bs, 3H, NH3+), 12.23 (bs, 1 H, COOH), 13C NMR (CDCl3, 75 MHz) d 37.94, 39.70, 43.28, 128.89, 130.27, 132.20, 139.56, 172.71.
http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0103-50532001000500011
Title: Baclofen
CAS Registry Number: 1134-47-0
CAS Name: b-(Aminomethyl)-4-chlorobenzenepropanoic acid
Additional Names: b-(aminomethyl)-p-chlorohydrocinnamic acid; g-amino-b-(p-chlorophenyl)butyric acid; b-(4-chlorophenyl)GABA
Manufacturers’ Codes: Ba-34647
Trademarks: Baclon (Leiras); Clofen (Alphapharm); Lioresal (Novartis)
Molecular Formula: C10H12ClNO2
Molecular Weight: 213.66
Percent Composition: C 56.21%, H 5.66%, Cl 16.59%, N 6.56%, O 14.98%
Literature References: Specific GABA-B receptor agonist. Prepn: NL 6407755; H. Keberle et al., US 3471548 (1965, 1969 both to Ciba). Toxicity study: T. Tadokoro et al., Osaka Daigaku Igaku Zasshi 28, 265 (1976), C.A. 88, 183016u (1978). Comprehensive description: S. Ahuja, Anal. Profiles Drug Subs. 14, 527-548 (1985). Review of pharmacology and therapeutic efficacy in spasticity: R. N. Brogden et al., Drugs 8, 1-14 (1974); of intrathecal use in spinal cord injury: K. S. Lewis, W. M. Mueller, Ann. Pharmacother.27, 767-774 (1993). Clinical evaluation in reflex sympathetic dystrophy: B. J. van Hilten et al., N. Engl. J. Med. 343, 625 (2000).
Properties: Crystals from water, mp 206-208° (Keberle); 189-191°, (Uchimaru). LD50 in male mice, rats (mg/kg): 45, 78 i.v.; 103, 115 s.c.; 200, 145 orally (Tadokoro).
Melting point: mp 206-208° (Keberle); 189-191°, (Uchimaru)
Toxicity data: LD50 in male mice, rats (mg/kg): 45, 78 i.v.; 103, 115 s.c.; 200, 145 orally (Tadokoro)
Derivative Type: Hydrochloride
Molecular Formula: C10H13Cl2NO2
Molecular Weight: 250.12
Percent Composition: C 48.02%, H 5.24%, Cl 28.35%, N 5.60%, O 12.79%
Properties: mp 179-181°.
Melting point: mp 179-181°
Therap-Cat: Muscle relaxant (skeletal).
Keywords: Muscle Relaxant (Skeletal).
/////////////////(R)-(–)-Baclofen, Arbaclofen, STX 209, AGI 006, Spasticity, PREREGISTERD, OSMOTICA PHARMA
c1cc(ccc1[C@@H](CC(=O)O)CN)Cl

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....





















 Chemical Structure of Clomifene
Chemical Structure of Clomifene























































 The Grignard condensation of 5-cyanophthalide (I) with 4-fluorophenylmagnesium bromide (II) in THF gives 1-(4-fluorophenyl)-1-hydroxy-1,3-dihydroisobenzofuran-5-carbonitrile bromomagnesium salt (III), which slowly rearranges to the benzophenone (IV). A new Grignard condensation of (IV) with 3-(dimethylamino)propylmagnesium chloride (V) in THF affords the expected bis(magnesium) salt (VI), which is hydrolyzed with acetic acid to provide the diol (VII) as a racemic mixture. Selective esterification of the primary alcohol of (VII) with (+)-3,3,3-trifluoro-2-methoxy-2-phenylacetyl chloride (VIII) gives the monoester (IX) as a mixture of diastereomers. This mixture is separated by HPLC and the desired diastereomer (X) is treated with potassium tert-butoxide in toluene.
The Grignard condensation of 5-cyanophthalide (I) with 4-fluorophenylmagnesium bromide (II) in THF gives 1-(4-fluorophenyl)-1-hydroxy-1,3-dihydroisobenzofuran-5-carbonitrile bromomagnesium salt (III), which slowly rearranges to the benzophenone (IV). A new Grignard condensation of (IV) with 3-(dimethylamino)propylmagnesium chloride (V) in THF affords the expected bis(magnesium) salt (VI), which is hydrolyzed with acetic acid to provide the diol (VII) as a racemic mixture. Selective esterification of the primary alcohol of (VII) with (+)-3,3,3-trifluoro-2-methoxy-2-phenylacetyl chloride (VIII) gives the monoester (IX) as a mixture of diastereomers. This mixture is separated by HPLC and the desired diastereomer (X) is treated with potassium tert-butoxide in toluene. A new method for the preparation of citalopram has been developed: The chlorination of 1-oxo-1,3-dihydroisobenzofuran-5-carboxylic acid (I) with refluxing SOCl2 gives the acyl chloride (II), which is condensed with 2-amino-2-methyl-1-propanol (III) in THF yielding the corresponding amide (IV). The cyclization of (IV) by means of SOCl2 affords the oxazoline (V), which is treated with 4-fluorophenylmagnesium bromide (VI) in THF giving the benzophenone (VII). This compound (VII), without isolation, is treated with 3-(dimethylamino)propylmagnesium chloride (VIII) in the same solvent, providing the cabinol (IX), which is cyclized by means of methanesulfonyl chloride and Et3N in CH2Cl2 yielding the isobenzofuran (X). Finally, this compound is treated with POCl3 in refluxing pyridine to generate the 5-cyano substituent of citalopram.
A new method for the preparation of citalopram has been developed: The chlorination of 1-oxo-1,3-dihydroisobenzofuran-5-carboxylic acid (I) with refluxing SOCl2 gives the acyl chloride (II), which is condensed with 2-amino-2-methyl-1-propanol (III) in THF yielding the corresponding amide (IV). The cyclization of (IV) by means of SOCl2 affords the oxazoline (V), which is treated with 4-fluorophenylmagnesium bromide (VI) in THF giving the benzophenone (VII). This compound (VII), without isolation, is treated with 3-(dimethylamino)propylmagnesium chloride (VIII) in the same solvent, providing the cabinol (IX), which is cyclized by means of methanesulfonyl chloride and Et3N in CH2Cl2 yielding the isobenzofuran (X). Finally, this compound is treated with POCl3 in refluxing pyridine to generate the 5-cyano substituent of citalopram. The chlorination of 1-oxo-1,3-dihydroisobenzofuran-5-carboxylic acid (XII) with refluxing SOCl2 gives the acyl chloride (XIII), which is condensed with 2-amino-2-methyl-1-propanol (XIV) in THF to yield the corresponding amide (XV). The cyclization of (XV) by means of SOCl2 affords the oxazoline (XVI), which is treated with 4-fluorophenylmagnesium bromide (XVII) in THF to give the benzophenone (XVIII). This compound (XVIII), without isolation, is treated with 3-(dimethylamino)propylmagnesium chloride (XIX) in the same solvent to provide the carbinol (XX), which is submitted to optical resolution with (+)- or (-)-tartaric acid, or (+)- or (-)-camphor-10-sulfonic acid (CSA) to give the desired (S)-enantiomer (XXI). Cyclization of (XXI) by means of methanesulfonyl chloride and TEA in dichloromethane yields the chiral isobenzofuran (XXII), which is finally treated with POCl3 in refluxing pyridine.
The chlorination of 1-oxo-1,3-dihydroisobenzofuran-5-carboxylic acid (XII) with refluxing SOCl2 gives the acyl chloride (XIII), which is condensed with 2-amino-2-methyl-1-propanol (XIV) in THF to yield the corresponding amide (XV). The cyclization of (XV) by means of SOCl2 affords the oxazoline (XVI), which is treated with 4-fluorophenylmagnesium bromide (XVII) in THF to give the benzophenone (XVIII). This compound (XVIII), without isolation, is treated with 3-(dimethylamino)propylmagnesium chloride (XIX) in the same solvent to provide the carbinol (XX), which is submitted to optical resolution with (+)- or (-)-tartaric acid, or (+)- or (-)-camphor-10-sulfonic acid (CSA) to give the desired (S)-enantiomer (XXI). Cyclization of (XXI) by means of methanesulfonyl chloride and TEA in dichloromethane yields the chiral isobenzofuran (XXII), which is finally treated with POCl3 in refluxing pyridine.
 The esterification of racemic 1-[4-bromo-2-(hydroxymethyl)phenyl]-4-(dimethylamino)-1-(4-fluorophenyl)-1-butanol (I) with (S)-2-(6-methoxynaphth-2-yl)propionyl chloride (II) by means of TEA and DMAP in THF gives the corresponding ester (III) as a diastereomeric mixture that is separated by chiral chromatography over Daicel AD, the desired diastereomer (IV) is easily isolated. Finally, this ester is hydrolyzed and simultaneously cyclized by means of NaH in DMF to provide the target intermediate (V). Other acyl chlorides such as (S)-2-(4-isobutylphenyl)propionyl chloride, (S)-O-acetylmandeloyl chloride, (S)-benzyloxycarbonylprolyl chloride, (S)-2-phenylbutyryl chloride, (S)-2-methoxy-2-phenylacetyl chloride or (S)-N-acetylalanine can also be used in the preceding sequence.
The esterification of racemic 1-[4-bromo-2-(hydroxymethyl)phenyl]-4-(dimethylamino)-1-(4-fluorophenyl)-1-butanol (I) with (S)-2-(6-methoxynaphth-2-yl)propionyl chloride (II) by means of TEA and DMAP in THF gives the corresponding ester (III) as a diastereomeric mixture that is separated by chiral chromatography over Daicel AD, the desired diastereomer (IV) is easily isolated. Finally, this ester is hydrolyzed and simultaneously cyclized by means of NaH in DMF to provide the target intermediate (V). Other acyl chlorides such as (S)-2-(4-isobutylphenyl)propionyl chloride, (S)-O-acetylmandeloyl chloride, (S)-benzyloxycarbonylprolyl chloride, (S)-2-phenylbutyryl chloride, (S)-2-methoxy-2-phenylacetyl chloride or (S)-N-acetylalanine can also be used in the preceding sequence.


