
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Veterinary- Atipamezole
Atipamezole

4-(2-Ethyl-1,3-dihydroinden-2-yl)-3H-imidazole, Atipamezole, cas 104054-27-5
hydrochloride cas no 104075-48-1
- MPV 1248 (IS: FarmosGroupLt)
- UNII-03N9U5JAF6 (IS)
- UNII-2W4279571X (IS)
Atipamezole is a synthetic alpha2-adrenergic antagonist, indicated for the reversal of the sedative and analgesic effects of dexmedetomidine and medetomidine in dogs. It has also been researched in humans as a potential anti-Parkinsonian drug.Atipamezole is more potent than yohimbine; it is very selective for alpha2-adrenergic vs alpha1sites, but not selelctive for alpha2 – subtypes.
Atipamezole (brand name Antisedan, Pfizer) is a synthetic alpha2–adrenergic antagonist, indicated for the reversal of the sedative and analgesic effects of dexmedetomidine andmedetomidine in dogs.[1] It has also been researched in humans as a potential anti-Parkinsonian drug.[2]
- Pfizer Animal Health ANTISEDAN Product Overview
- Pertovaara A, Haapalinna A, Sirviö J, Virtanen R (2005). “Pharmacological properties, central nervous system effects, and potential therapeutic applications of atipamezole, a selective alpha2-adrenoceptor antagonist”. CNS Drug Reviews 11 (3): 273–88.doi:10.1111/j.1527-3458.2005.tb00047.x. PMID 16389294.

- ANTISEDAN product information page (Pfizer Animal Health)
- Drugs Future, vol. 15, no. 5, 1990, “Atipamezole“, pages 448-452, see page 449
Synonyms
1H-imidazole, 4-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-
1H-imidazole, 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-
5-(2-Ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole
Atipamezole
4-(2-Ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole
4-(2-Ethyl-2-indanyl)imidazole
4-(2-Ethyl-indan-2-yl)-1H-imidazole(Atipamezole)
4-(2-ethylindan-2-yl)imidazole
Antisedan
Antisedan
Atipamezol
Atipamezolum
Atipamezole Hydrochloride CAS 104075-48-1
………………………………
Atipamezole is a selective alpha2 – adrenoceptor antagonist which is currently marketed under the trademark Antisedan® for the reversal of sedative- analgesic veterinary drugs. Atipamezole has been disclosed e.g. in the European Patent EP 183492 as useful for the reversal of detomidine. European Patent EP 0589957 discloses the use of atipamezole for the treatment of male sexual impotence. In US 4698692 the use of atipamezole for the attenuation of ethyl alcohol intoxication is disclosed.
US Patent No. US6543389 discloses insecticidal pet collars for dogs comprising amitraz and atipamezole. Atipamezole in the collar provides amelioration of amitraz toxicosis in combination with the amitraz in case the dogs ingests the collar. The pet collar comprises 0.01 to 1%, preferably 0.1 to 1 %, by weight of atipamezole. Safe, effective ways to eliminate ectoparasites are desired for the companion animal’s well-being, for the well-being and comfort of its human associate and for the prevention of losses in livestock
A substantial amount of work has been devoted to identifying the neurotransmitters involved in the facilitation and inhibition of male sexual behaviour (see e.g. Bitran and Hull 1987, Neuroscience and Behavioral reviews 11 , 365-389). Noradrenergic neuro-transmission seems to have an important role.
Atipamezole is a selective and potent a2*-adrenoceptor antagonist which is currently marketed for the reversal of sedative-analgesic veterinary drugs. Atipamezole has been disclosed e.g. in the European Patent EP 183492 as useful for the reversal of detomidine.
We have now found that this compound is also very effective in increasing male sexual capacity in a monkey model. These findings suggest that atipamezole would be an effective therapy in male impotence in humans as well.
Another a2-adrenoreceptor antagonist, yohimbine, is currently used for the treatment of male impotence. Yohimbine increases noradrenergic neurotransmission and has been reported to facilitate the sexual capacity of male animals, although the results of different studies are conflicting.
Atipamezole is, however clearly advantageous over yohimbine for this use because of its excellent selectivity. The a2/a-|selectivity ratio of atipamezole is
200-300 times higher than that of yohimbine.
- EP 0310745 B (FARMOS OY) 1989.04.12. disclosed preparation of 5-(2-ethyl-2,3-dihydro-1 H-inden-2-yl)-1 H-imidazole salt by two synthetic routes.
-
First synthetic route as starting material was used 2-acetyl-1-indanone, which was alkylated with ethylbromide in acetone in the presence of sodium carbonate to 2-acetyl-2-ethyl-1-indanone. The acetyl group was brominated with bromine in methanol and to imidazole by heating in formamide. Then the intermediate was hydrogenated in 2N hydrochloric acid in the presence of 10% palladium on carbon.
-
Second synthetic route disclosed in the same patent is following, as starting material was used 2,3-dihydro-1H-indene-2-carboxylic acid methyl ester, which was prepared by methylation of 2,3-dihydro-1H-indene-2-carboxylic acid in the presence of sulphuric acid. The 2,3-dihydro-1H-indene-2-carboxylic acid methyl ester was reacted with N-isopropylcyclohexylamide and ethylbromide yielding 2,3-dihydro-2-ethyl-1H-indene-2-carboxylic acid, then thionyl chloride was added and 2,3-dihydro-2-ethyl-1H-indene-2-carboxylic acid chloride was obtained. In the next step ethoxymagnesiummalonic acid ethyl ester in dry ether was added to 2,3-dihydro-2-ethyl-1H-indene-2-carboxylic acid chloride and reaction mixture was treated with sulphuric acid, and 1-(2,3-dihydro-2-ethyl-1H-inden-2-yl)ethanone was obtained, then the intermediate was stirred in methylene chloride and bromine was added by giving a new intermediate 2-bromo-1-(2,3-dihydro-2-methyl-1H-inden-2-yl)ethanone, to which was thereafter added formamide and hydrochloric acid yielding crude product of 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole. The last step involved hydrogenation of the crude product of 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1 H-imidazole with 10% palladium on carbon.
-
EP 0247764 B (ORION-YHTYMÄ OY) 1987.02.12. disclosed the following process for preparation of 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole hydrochloride. The process starts by reaction of alpha, alpha-dibromo-o-xylene with 4-penten-2-one to obtain 1-(2,3-dihydro-2-vinyl-1H-inden-2-yl)ethanone. The obtained intermediate was brominated, e.g. with bromine, methylene chloride was used as solvent and 2-bromo-1-(2,3-dihydro-2-vinyl-1H-inden-2-yl)-ethanone was obtained, which is thereafter reacted with formamide in excess formamide to give a 4(5)-(2,3-dihydro-2-vinyl-1H-inden-2-ylimidazole hydrochloride. As the last step the vinyl group was catalytically hydrogenated to an ethyl group so as to form a product 4(5)-(2,3-dihydro-2-ethyl-1 H-inden-2-yl) imidazole.
-
Another synthetic route for obtaining 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole is disclosed in WAI, Wonf, et al. A Concise Synthesis of Atipamezole. Synthesis. 1995, no.2, p.139-140. The cyclization of alpha, alpha’-dibromo-o-xylene with acetylacetone by means of NaOH and tetrabutylammonium bromide in toluene/water at 80°C under phase-transfer conditions gives the unstable diacetyl derivative, which presumably undergoes cleavage to afford 2-acetylindane. The alkylation of 2-acetylindane with ethyl iodide and potassium tert-butoxide yields 2-acetyl-2-ethylindan, which is brominated with Br2 to give 2-bromoacetyl-2-ethylindan. Finally, this compound is cyclised with formamide at 160°C (some 2-ethyl-2-(4-oxazolyl)indane is also formed but easily eliminated); the cyclization can also be carried out with formamidine in liquid ammonia. Although the substitution of formamide by formamidine acetate eliminates the oxazole formation, it does not increase the yield of Atipamezole (<30%) WAI, Wonf, et al. A Concise Synthesis of Atipamezole. Synthesis. 1995, no.2, p.139-140 in the final step.
The preparation of atipamezole hydrochloride salt is described in U.S. Patent 4,689,339, wherein atipamezole obtained from the hydrogenation step is first recovered from alkaline solution as free base. After the evaporation of methylene chloride solvent to dryness the isolated crystalline product is converted into its hydrochloride salt by treatment with dry hydrogen chloride in ethyl acetate
Other compounds having alpha-2 adrenoceptor antagonist properties which may be useful in accordance with the present invention include idazoxan related compounds [Reckitt & Colman] Doxey, et al., Br. J. Parmacol., Vol. 78, p.489-505 (1983); imiloxan [Syntex] Michel, et al., Br. J. Pharmacol., Vol. 74, p.255-256 (1981); WY 26703 and related compounds [Wyeth] Latimer, et al., Naunvn Schmiedeberg’s Arch. Pharmacol., Vol. 327, p. 312-318 (1984); CH-38083 [Chinoin] Vizi, et a., J. Pharmacol. Exp. Ther., Vol. 238, p. 701-706 (1986); GR 50360A and related compounds [Glaxo] Halliday, et al., Br. J. Pharmacol., Vol. 95, p. 715 (1988); DG 5128 and related compounds of Daiichi Seiyaku Co., Ltd., Tokyo, Japan; and Yohimbine [Sigma].
………………………………….

-
- 1. an essential process for obtaining 5-(2-ethyl-2,3-1H-inden-2-yl)-1H-imidazole, without bromination in any step of process, thus preventing the possibility of brominated by-products;
- 2. This process has given superior yields, compared to patents cited above;
- 3. This process is amenable to large scale production which does not require specialized equipment.
-
The condensing of commercially available 1-trityl-1H-imidazole-4-carboxaldehyde (I) with phtalide to form 2-(1-trityl-1H-imidazole-4-yl)indan-1,3-dione (II) is performed under the conditions that are similar to those used for synthesis of 4-(indane-1,3-dionyl) pyridine J. Org. Chem. 1971, vol.36, p.1563. surprisingly, the bulky 1-trityl-1H-imidazole-4-carboxaldehyde (I) reacted as expected and produced 2-(1-trityl-1H-imidazole-4-yl)indan-1,3-dione (II) in over 67% yield. Both ethyl acetate and dioxane can be used as reaction media.
-
The alkylation of (II) by ethyl iodide is performed in boiling acetone with potassium carbonate as basic agent. 2-Ethyl-2-(1-trityl-1H-imidazole-4-yl)indan-1,3-dione (III) is formed in over 67% yield and easily isolated from the acetone solution by concentrating it and diluting with water. A high purity (III) is obtained after crystallization from methanol or ethanol.
-
Removing the trityl group of 2-ethyl-2-(1-trityl-1H-imidazole-4-yl)indan-1,3-dione by acid hydrolysis to yield the deprotected 2-ethyl-2-(1H-imidazol-2-yl)indan-1,3-dione.
-
The reduction of (IV) to 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole hydrochloride (V) is performed in hydrogenation apparatus with Pd/C catalyst under hydrogen pressure in HCI solution. The reaction proceeds under variable pressure and temperature conditions, but a pressure of about 3 bar and the temperature of about 80-85°C is preferable. After removing the catalyst the product crystallizes on chilling in over 77% yield. It can be purified by additional crystallization.
-
EP 0310745 B (FARMOS OY) 1989.04.12. disclosed preparation of 5-(2-ethyl-2,3-dihydro-1 H-inden-2-yl)-1 H-imidazole salt by two synthetic routes.
-
First synthetic route as starting material was used 2-acetyl-1-indanone, which was alkylated with ethylbromide in acetone in the presence of sodium carbonate to 2-acetyl-2-ethyl-1-indanone. The acetyl group was brominated with bromine in methanol and to imidazole by heating in formamide. Then the intermediate was hydrogenated in 2N hydrochloric acid in the presence of 10% palladium on carbon.
-
Second synthetic route disclosed in the same patent is following, as starting material was used 2,3-dihydro-1H-indene-2-carboxylic acid methyl ester, which was prepared by methylation of 2,3-dihydro-1H-indene-2-carboxylic acid in the presence of sulphuric acid. The 2,3-dihydro-1H-indene-2-carboxylic acid methyl ester was reacted with N-isopropylcyclohexylamide and ethylbromide yielding 2,3-dihydro-2-ethyl-1H-indene-2-carboxylic acid, then thionyl chloride was added and 2,3-dihydro-2-ethyl-1H-indene-2-carboxylic acid chloride was obtained. In the next step ethoxymagnesiummalonic acid ethyl ester in dry ether was added to 2,3-dihydro-2-ethyl-1H-indene-2-carboxylic acid chloride and reaction mixture was treated with sulphuric acid, and 1-(2,3-dihydro-2-ethyl-1H-inden-2-yl)ethanone was obtained, then the intermediate was stirred in methylene chloride and bromine was added by giving a new intermediate 2-bromo-1-(2,3-dihydro-2-methyl-1H-inden-2-yl)ethanone, to which was thereafter added formamide and hydrochloric acid yielding crude product of 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole. The last step involved hydrogenation of the crude product of 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1 H-imidazole with 10% palladium on carbon.
-
EP 0247764 B (ORION-YHTYMÄ OY) 1987.02.12. disclosed the following process for preparation of 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole hydrochloride. The process starts by reaction of alpha, alpha-dibromo-o-xylene with 4-penten-2-one to obtain 1-(2,3-dihydro-2-vinyl-1H-inden-2-yl)ethanone. The obtained intermediate was brominated, e.g. with bromine, methylene chloride was used as solvent and 2-bromo-1-(2,3-dihydro-2-vinyl-1H-inden-2-yl)-ethanone was obtained, which is thereafter reacted with formamide in excess formamide to give a 4(5)-(2,3-dihydro-2-vinyl-1H-inden-2-ylimidazole hydrochloride. As the last step the vinyl group was catalytically hydrogenated to an ethyl group so as to form a product 4(5)-(2,3-dihydro-2-ethyl-1 H-inden-2-yl) imidazole.
-
Another synthetic route for obtaining 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole is disclosed in WAI, Wonf, et al. A Concise Synthesis of Atipamezole. Synthesis. 1995, no.2, p.139-140. The cyclization of alpha, alpha’-dibromo-o-xylene with acetylacetone by means of NaOH and tetrabutylammonium bromide in toluene/water at 80°C under phase-transfer conditions gives the unstable diacetyl derivative, which presumably undergoes cleavage to afford 2-acetylindane. The alkylation of 2-acetylindane with ethyl iodide and potassium tert-butoxide yields 2-acetyl-2-ethylindan, which is brominated with Br2 to give 2-bromoacetyl-2-ethylindan. Finally, this compound is cyclised with formamide at 160°C (some 2-ethyl-2-(4-oxazolyl)indane is also formed but easily eliminated); the cyclization can also be carried out with formamidine in liquid ammonia. Although the substitution of formamide by formamidine acetate eliminates the oxazole formation, it does not increase the yield of Atipamezole (<30%) WAI, Wonf, et al. A Concise Synthesis of Atipamezole. Synthesis. 1995, no.2, p.139-140 in the final step.
…………………………….
US Patent 8,431,717
Atipamezole [5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole, 1] is a veterinary drug that has been investigated for treating Parkinson’s disease in humans. V. Lusis and co-inventors summarize several ways to synthesize 1. Some routes give a low yield of 1 and produce large quantities of an oxazole byproduct. Other processes involve a sluggish bromination reaction that leads to many byproducts.
The inventors’ process is intended to overcome these problems. In particular, it does not use the bromination reaction and thus avoids forming brominated byproducts. The process, outlined in the figure, begins with the reaction of imidazole 2 with i-PrMgCl to form iodo Grignard reagent 3, which is treated with DMF to give 4. This intermediate is not isolated but is treated with aq NH4Cl to give aldehyde 5, isolated in 73.2% yield. The aldehyde is condensed with phthalide (6) in the presence of NaOMe to produce imidazolylindane 7, recovered in crude form in 67.2% yield.

In the next stage, compound 7 is alkylated with EtI in the presence of K2CO3. Product 8 is isolated in 50.9% yield after being recrystallized from EtOH. Product1 can be produced directly from 8 by making its HCl salt and hydrogenating the salt over Pd/C. Crude atipamezole is isolated as its HCl salt in 26.6% yield.
Alternatively, acid hydrolysis of 8 removes the trityl group to form dione 9, recovered as a white crystalline solid in 76.2% yield. The HCl salt of 9 is then hydrogenated to 1·HCl.
The patent’s claims cover the process to make 1 and new compounds 7 and 8. The overall yield of compound 1 is poor, partly because of the low yield from the hydrogenation step. The inventors claim, however, that the yield is higher than from earlier methods. They point out that the process is amenable to large-scale production without the use of specialized equipment. (JSC Grindeks [Riga, Latvia]. US Patent 8,431,717, April 30, 2013; Keith Turner), View the full-text here.
………………………………
nmr

Atipamezole Hydrochloride CAS 104075-48-1 HNMR
……………………………………………………….
GADODIAMIDE, OMNISCAN Drug Patent Expiration, 1 st oct 2013

GADODIAMIDE
GE HEALTHCARE, OMNISCAN
Drug Patent Expiration
1 st oct 2013, US5560903, CAS 122795-43-1
| GADODIAMIDE | INJECTABLE; INJECTION | 287MG/ML | RX | NDA 020123 |
Gadodiamide is a gadolinium-based MRI contrast agent, used in MR imaging procedures to assist in the visualization of blood vessels. It is commonly marketed under the trade name Omniscan.
For intravenous use in MRI to visualize lesions with abnormal vascularity (or those thought to cause abnormalities in the blood-brain barrier) in the brain (intracranial lesions), spine, and associated tissues.

Gadodiamide is a contrast medium for cranial and spinal magnetic resonance imaging (MRI) and for general MRI of the body after intravenous administration. The product provides contrast enhancement and facilitates visualisation of abnormal structures or lesions in various parts of the body including the central nervous system (CNS). It does not cross an intactblood brain barrier but might give enhancement in pathological conditions.
Based on the behavior of protons when placed in a strong magnetic field, which is interpreted and transformed into images by magnetic resonance (MR) instruments. Paramagnetic agents have unpaired electrons that generate a magnetic field about 700 times larger than the proton’s field, thus disturbing the proton’s local magnetic field. When the local magnetic field around a proton is disturbed, its relaxation process is altered. MR images are based on proton density and proton relaxation dynamics. MR instruments can record 2 different relaxation processes, the T1 (spin-lattice or longitudinal relaxation time) and the T2 (spin-spin or transverse relaxation time). In magnetic resonance imaging (MRI), visualization of normal and pathological brain tissue depends in part on variations in the radiofrequency signal intensity that occur with changes in proton density, alteration of the T1, and variation in the T2. When placed in a magnetic field, gadodiamide shortens both the T1 and the T2 relaxation times in tissues where it accumulates. At clinical doses, gadodiamide primarily affects the T1 relaxation time, thus producing an increase in signal intensity. Gadodiamide does not cross the intact blood-brain barrier; therefore, it does not accumulate in normal brain tissue or in central nervous system (CNS) lesions that have not caused an abnormal blood-brain barrier (e.g., cysts, mature post-operative scars). Abnormal vascularity or disruption of the blood-brain barrier allows accumulation of gadodiamide in lesions such as neoplasms, abscesses, and subacute infarcts.

1.Schenker MP, Solomon JA, Roberts DA. (2001). Gadolinium Arteriography Complicated by Acute Pancreatitis and Acute Renal Failure, Journal of vascular and interventional radiology 12(3):393.[1]
2 Unal O, Arslan H. (1999). Cardiac arrest caused by IV gadopentetate dimeglumine. AJR Am J Roentgenol 172:1141.[2]
3 Cacheris WP, Quay SC, Rocklage SM. (1990). The relationship between thermodynamics and the toxicity of gadolinium complexes, Magn Reson Imaging 8(6):467-81. doi:10.1016/0730-725X(90)90055-7
4 Canavese, C; Mereu, MC; Aime, S; Lazzarich, E; Fenoglio, R; Quaglia, M; Stratta, P (2008). “Gadolinium-associated nephrogenic systemic fibrosis: the need for nephrologists’ awareness”. Journal of nephrology 21 (3): 324–36. PMID 18587720.
COUNTRY PATENT APPROVED, EXPIRY
| United States | 5560903 | 1993-10-01 | 2013-10-01 |
| Canada | 1335819 | 1995-06-06 | 2012-06-06 |
| United States | 5362475 | 1994-11-08 | 2011-11-08 |
| Canada | 1335819 | 1995-06-06 | 2012-06-06 |
| United States | 5560903 | 1993-10-01 | 2013-10-01 |
Gadolinium contrast agents are used as contrast media to enhance magnetic resonance imaging as they are paramagnetic. This compound has a low incidence of adverse side effects, although there is a rare association with nephrogenic systemic fibrosis (NSF) when given to people with severe renal impairment (ie, GFRglomerular filtration rate <30mL/min/1·73m2).It seems to be related to the liberation of free gadolinium ions, and UK CHM advice is against using the least stable of the agents – Omniscan (gadodiamide) – in patients with severe renal impairment, and carefully considering whether to use others where renal function is impaired.
OMNISCAN (gadodiamide) Injection is the formulation of the gadolinium complex of diethylenetriamine pentaacetic acid bismethylamide, and is an injectable, nonionic extracellular enhancing agent for magnetic resonance imaging. OMNISCAN is administered by intravenous injection. OMNISCAN is provided as a sterile, clear, colorless to slightly yellow, aqueous solution. Each 1 mL contains 287 mg gadodiamide and 12 mg caldiamide sodium in Water for Injection.
The pH is adjusted between 5.5 and 7.0 with hydrochloric acid and/or sodium hydroxide. OMNISCAN contains no antimicrobial preservative. OMNISCAN is a 0.5 mol/L solution of aqua[5,8-bis(carboxymethyl)11-[2-(methylamino)-2-oxoethyl]-3-oxo-2,5,8,11-tetraazatridecan-13-oato (3-)-N5, N8, N11, O3, O5, O8, O11, O13] gadolinium hydrate, with a molecular weight of 573.66 (anhydrous), an empirical formula of C16H28GdN5O9•xH2O, and the following structural formula:
 |
Pertinent physicochemical data for OMNISCAN are noted below:
PARAMETER
| Osmolality (mOsmol/kg water) | @ 37°C | 789 |
| Viscosity (cP) | @ 20°C | 2 |
| @ 37°C | 1.4 | |
| Density (g/mL) | @ 25°C | 1.14 |
| Specific gravity | @ 25°C | 1.15 |
OMNISCAN has an osmolality approximately 2.8 times that of plasma at 37°C and is hypertonic under conditions of use.
gadodiamide, chemical name: [5,8 _ bis (carboxymethyl) -11 – [2_ (methylamino)-2_ ethyl] -3 – O 2 ,5,8, 11 – tetraazacyclododecane-decane -13 – oxo-(3 -)] gadolinium trihydrate. Its structure is shown in formula one.
[0003] Structural Formula:
[0004]

[0005] Magnetic resonance contrast agent gadodiamide resonance than ionic contrast agents safer generation of products, it is non-ionic structure significantly reduces the number of particles in solution, osmotic balance of body fluids is very small.Meanwhile, gadodiamide relatively low viscosity to bring the convenience of nursing staff, making it easier to bolus. In addition, gadodiamide pioneered the use of amide-substituted carboxyl part, not only reduces the toxicity of carboxyl groups and ensure the non-ionic nature of the product solution.
[0006] reported in the literature and their intermediates gadodiamide synthetic route is as follows:
[0007] 1. Compound III synthetic routes for its preparation in U.S. Patent No. US5508388 described as: In the synthesis process, the inventors using acetonitrile as solvent, acetic anhydride as dehydrating agent, pyridine as acid-binding agent, at 55 ~ 60 ° C, the reaction 18h. Anti-
See the reaction should be a process. The disadvantage of this synthesis are acetonitrile toxicity, not widely used.
[0008]

[0009] Reaction a
[0010] (2) Synthesis of Compound III in many articles are reported in the patent and its implementation method similar to the patent US5508388.
[0011] In US3660388, the diethylenetriamine pentaacetic acid (Compound II), pyridine, acetic anhydride, the mixture was reacted at 65 ° C or 20h at 125 ° C the reaction 5min, to give compound III.
[0012] In US4822594, the compounds II, pyridine, acetic anhydride mixture was reacted at 65 ° C 20h, to give compound III.
[0013] In US4698263, the compounds II, pyridine, acetic anhydride heated in a nitrogen or argon atmosphere under reflux for 18h, to give compound III. [0014] In the EPO183760B1, the compounds II, pyridine, acetic anhydride mixture was reacted at 55 ° C 24h, to give compound III.
[0015] In CN1894223A, the compounds II, pyridine, acetic anhydride, the mixture above 65 ° C the reaction mixture, and the pyridine of DTPA feed ratio is: 1: (0.5 to 3).
[0016] The above patents do not provide for the compound III is post-processing method.
[0017] 3 Synthesis of Compound IV.
[0018] In U.S. Patent US4859451, the diethylenetriamine pentaacetic acid dianhydride (compound III) and ammonia, methanol and the reaction of compounds IV, see Reaction Scheme II.
[0019]

[0020] Reaction two
[0021] In the patent US5087439, the compound III with methylamine in aqueous solution for several hours, or overnight reactions, see reaction formula III.
[0022]

[0023] Reactive three
[0024] These two patents using ammonia and methylamine, which can form explosive mixtures with air, in case of fire or high pressure can cause an explosion in the production process of great insecurity. Although raw material prices are lower, but higher production conditions (such as requiring sealed, low temperature, etc.). Compared to this synthesis process,
[0025] 4, gadodiamide (Compound I) synthesis.
[0026] In the patent US4859451, the use of gadolinium chloride with the compound IV is carried out under acidic conditions, complexing. Finally, tune
Section PH neutral, see reaction IV.
[0027]

[0028] Reaction formula tetrakis [0029] in the patent US5087439, the chlorides are used as reactants, and details of the post-processing method of Compound I.
[0030] In the patent US5508388, the use of gadolinium oxide with compound IV in acetonitrile, water with stirring, the resulting compound I.
[0032] The synthetic route is as follows:
[0033]

[0034] 1) Compound II (diethylenetriamine pentaacetic acid) in pyridine, acetic anhydride in the presence of a dehydration reaction into the acid anhydride, and the product was stirred with cold DMF, leaving the solid filtered, washed with ether reagents, drying , to obtain a white powdery solid compound III (diethylenetriamine pentaacetic acid anhydride);
[0035] 2) Compound III in DMF with methylamine hydrochloride, the reaction of the compound IV (5,8 _ bis carboxymethyl methyl-11 – [2 – (dimethylamino) -2 – oxoethyl] – 3 – oxo -2,5,8,11 – tetraazacyclododecane _13_ tridecyl acid); and the control compound III: MeNH2 · HCl molar ratio = 1: (1 to 4), control the temperature between 20 ~ 80 ° C, the reaction time is 4 ~ 6h, after the treatment, the method of distillation under reduced pressure to remove DMF, the product is dissolved in a polar solvent, methanol, and then adding a solvent polarity modulation, so that the target Compound IV from system completely precipitated;
[0036] 3) Compound IV with gadolinium oxide formed in the presence of hydrochloric acid of the complex, after the reaction, filtration and drying, to obtain a white powdery compound I, i.e. gadodiamide.
[0037] Existing gadodiamide Synthesis basically from the synthesis of Compound IV as a starting material, the present invention is first introduced to the compound II as a starting material to synthesize gadodiamide. Synthesis of the conventional method of gadodiamide, the present invention has the advantage of inexpensive starting materials, convenient and easy to get. In addition, the synthetic pathway intermediates are involved in the post-processing is simple, enabling continuous reaction, saving time and cost savings, the reaction becomes controlled step by step, and try to avoid the use of toxic reagents, reducing the possibility of operator injury , while also greatly reducing damage to the environment.
Covidien sells Confluent product line for $235 million
October 30, 2013 | By Anabela Farrica
![]()
Covidien announced yesterday that it has reached an agreement with Integra LifeSciences Corporation to sell its Confluent Surgical product line for $235 million in up-front cash, plus an additional $30 million in milestones. The transaction depends on a number of regulatory approvals, but it is expected to be finished by March 31, 2014.
View original post 152 more words
Sales of Biogen Idec’s multiple sclerosis drug Tecfidera soar
October 29 ,2013 | By Márcio Barra

Tecfidera (dymethil Fumarate), Biogen Idec’s prized multiple sclerosis drug, is fast approaching blockbuster status according to the recently released third-quarter numbers of Biogen Idec’s, beating along the way analyst expectation and pushing Biogen Idec’s net profit up 22%.
For Q3, Tecfidera sales garnered $286.4 million, far higher than the $217.2 million that analysts expected. It is now expected that Tecfidera will reach $3.5 billion in annual revenue by 2016, a no doubt impressive number for a drug that, in its first quarter in the market (the drug was launched in March 2013 in the US), posted $192.1 million in sales. This revenue mark pushed Biogen’s net profit up 22% to $488 million, up from $398 million last year.
View original post 302 more words
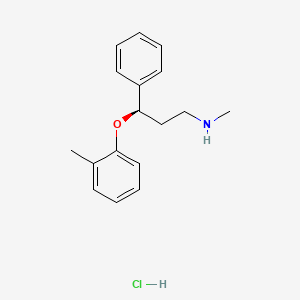
Atomoxetine

Atomoxetine
Atomoxetine hydrochloride (CAS NO.: 82248-59-7)
(R)-(-)-N-Methyl-gamma-(2-methylphenoxy)benzenepropanamine hydrochloride
| Patent No | PatentExpiry Date | |
|---|---|---|
| 5658590 | Nov 26, 2016 | |
| 5658590*PED | May 26, 2017 |
nda 021411 app 2002-11-26
TREATMENT OF ATTENTION-DEFICIT HYPERACTIVITY DISORDER
label
|
Country
|
Patent Number
|
Approved
|
Expires (estimated)
|
|---|---|---|---|
| United States | 5658590 | 1997-05-26 | 2017-05-26 |
The HCl salt of atomoxetine , with the (R)-configuration], which is marketed under the trade name Strattera, is used for treating attention-deficit hyperactivity disorder (ADHD
Atomoxetine is a drug approved for the treatment of attention-deficit hyperactivity disorder(ADHD).[1] It is a selective norepinephrine reuptake inhibitor (NRI),[1] not to be confused with serotonin norepinephrine reuptake inhibitors (SNRIs) or selective serotonin reuptake inhibitors (SSRIs), both of which are currently the most prescribed form of antidepressants.
his compound is manufactured, marketed and sold in theUnited States under the brand name Strattera by Eli Lilly and Company as a hydrochloride salt (atomoxetine HCl), the original patent filing company, and current U.S. patent owner. Generics of atomoxetine are sold in all other countries; they are manufactured by Torrent Pharmaceuticals using the label Tomoxetin, Ranbaxy Laboratories (through its Division: Solus) using the label Attentin, Sun Pharmaceuticals(through its Division: Milmet Pharmaceuticals), and Intas Biopharmaceuticals There is currently no generic manufactured directly in the United States since it is under patent until 2017.[2]
On August 12, 2010, Lilly lost a lawsuit that challenged Lilly’s patent on Strattera, increasing the likelihood of an earlier entry of a generic into the US market.[3] On September 1, 2010, Sun Pharmaceuticals announced it would begin manufacturing a generic in the United States.[4] In a July 29, 2011 conference call, however, Sun Pharmaceutical’s Chairman stated “Lilly won that litigation on appeal so I think [generic Strattera]’s deferred.”[5]
Atomoxetine is designated chemically as (−)-N-methyl-3-phenyl-3-(o-tolyloxy)-propylamine hydrochloride, and has a molecular mass of 291.82.[1] It has a solubility of 27.8 mg/mL in water.[1] Atomoxetine is a white solid that exists as a granular powder inside the capsule, along with pre-gelatinized starch and dimethicone.[1] The capsule shells contain gelatin, sodium lauryl sulfate, FD&C Blue No. 2, synthetic yellow iron oxide, titanium dioxide, red iron oxide, edible black ink, and trace amounts of other inactive ingredients.[1]
The compound (-)-N-methyl-3-(2-methylphenoxy)-3-phenylpropylamine, or (-)-Λ/-methyl-3-phenyl-3-(o-tolyloxy)-propylamine hydrochloride, is usually known by its adopted name “atomoxetine hydrochloride.” It is represented as shown in Formula 1 and is a selective norepinephrine reuptake inhibitor. A commercialatomoxetine hydrochloride product is sold as STRATTERA™ in the form of capsules containing 10, 18, 25, 40, 60, 80, or 100 mg of atomoxetine, for treating attention-deficit/hyperactivity disorder.
- “STRATTERA® (atomoxetine hydrochloride) CAPSULES for Oral Use. Full Prescribing Information.” Eli Lilly and Company, 2002, 2013. Revised August 5, 2013. [1]
- “Patent and Exclusivity Search Results”. Electronic Orange Book. US Food and Drug Administration. Retrieved 26 April 2009.
- “Drugmaker Eli Lilly loses patent case over ADHD drug, lowers revenue outlook”. Chicago Tribune.
- “Sun Pharma receives USFDA approval for generic Strattera capsules”. International Business Times.
- “Sun Pharma Q1 2011-12 Earnings Call Transcript 10.00 am, July 29, 2011”.
- Strattera by Eli Lilly and Company
- RxList.com – Strattera
- Detailed Strattera Consumer Information: Uses, Precautions, Side Effects
- All disclosed Lilly trials
- MSDS for Atomoxetine HCl
- Strattera Related Published Studies
Synthesis
Also known as: Atomoxetine hydrochloride, Strattera, Atomoxetine HCL, (R)-Tomoxetine hydrochloride, TOMOXETINE HYDROCHLORIDE, Tomoxetine, 82248-59-7
First step appears to be a Mannich reaction between acetophenone, paraformaldehyde and dimethylamine, although not formally written in the scheme.

Foster, B. J.; Lavagnino, E. R.; European Patent, 1982, EP 0052492.

Eli Lilly’s Strattera capsules.


Atomoxetine, designated chemically as (-)-N-methyl-3-phenyl-3-(0-tolyloxy)- propylamine hydrochloride, is structurally represented by the compound of Formula-I and is indicated for the potential treatment of attention-deficit hyperactivity disorder (ADHD). This compound is manufactured, marketed and sold in the United States under the brand name Strattera.

Formula-I Atomoxetine was first disclosed in US Patent No 4314081. The said patent disclosed Atomoxetine, its pharmaceutically acceptable salts and composition containing them.
4-hydroxy Atomoxetine, chemically known as R-(-)-N-methyl-3-(2-methyl-4- hydroxyphenyl)oxy)-3 -phenyl- 1-aminopropane, structurally represented by Formula-II, is a metabolite of Atomoxetine.

Fomnula-ll
4-hydroxy Atomoxetine hydrochloride was first disclosed in US Patent No 7384983, wherein 4-hydroxy Atomoxetine free base was dissolved in ethylacetate, treated the solution with 0.1N HC1; followed by lyophilization yielded a yellow solid which was dissolved in methanol and passed through a short column of activated carbon; the solvent was removed and finally the hydrochloride salt was recrystallized from water to afford 4-hydroxy Atomoxetine hydrochloride. However, this patent does not mention about the nature of the polymorph obtained through this process.
The asymmetric epoxidation of (E)-3-phenyl-2-propen-1-ol (I) by means of titanium tetraisopropoxide, (+)-diethyl tartrate (+)-(DET) and tBu-OOH in dichloromethane gives the chiral epoxide (II), which is opened by means of bis(2-methoxyethoxy)aluminum hydride (Red-Al) in DME to yield the chiral diol (III). The regioselective reaction of (III) with Ms-Cl and TEA in ethyl ether affords the primary mesylate (IV), which is condensed with 2-methylphenol (V) by means of PPh3 and DEAD in ethyl ether to provide the adduct (VI). Finally this compound is treated with methylamine in hot aq. THF to give rise to the target (R)-tomoxetine.

The reduction of omega-chloropropiophenone (I) with NaBH4 in ethanol gives 3-chloro-1-phenyl-1-propanol (II), which is treated with butyric anhydride and pyridine in dichloromethane to yield the corresponding racemic ester (III). The optical resolution of (III) with immobilized lipase B from Candida antarctica (CALB) affords a mixture of unreacted (S)-ester and (R)-alcohol (IV) that are separated by column chromatography. Condensation of th (R)-alcohol (IV) with 2-methylphenol (V) by means of PPh3 and diethyl azodicarboxylate (DEAD) in THF gives the corresponding ether (VI), which is finally treated with methylamine in refluxing ethanol.
more info
U.S. Patent No. 4,314,081 describes 3-Aryloxy-3-phenyl polyamines, which possess central nervous system activity. Atomoxetine is a member of the above class of compounds, and is a useful drug for the treatment of depression.Atomoxetine was claimed in U. S. Patent No. 4,314,081 and the patent describes a process for the preparation of atomoxetine and related compounds in two different ways as depicted below as Scheme A and Scheme B, respectively.
Scheme A
Atomoxetinc
Scheme B

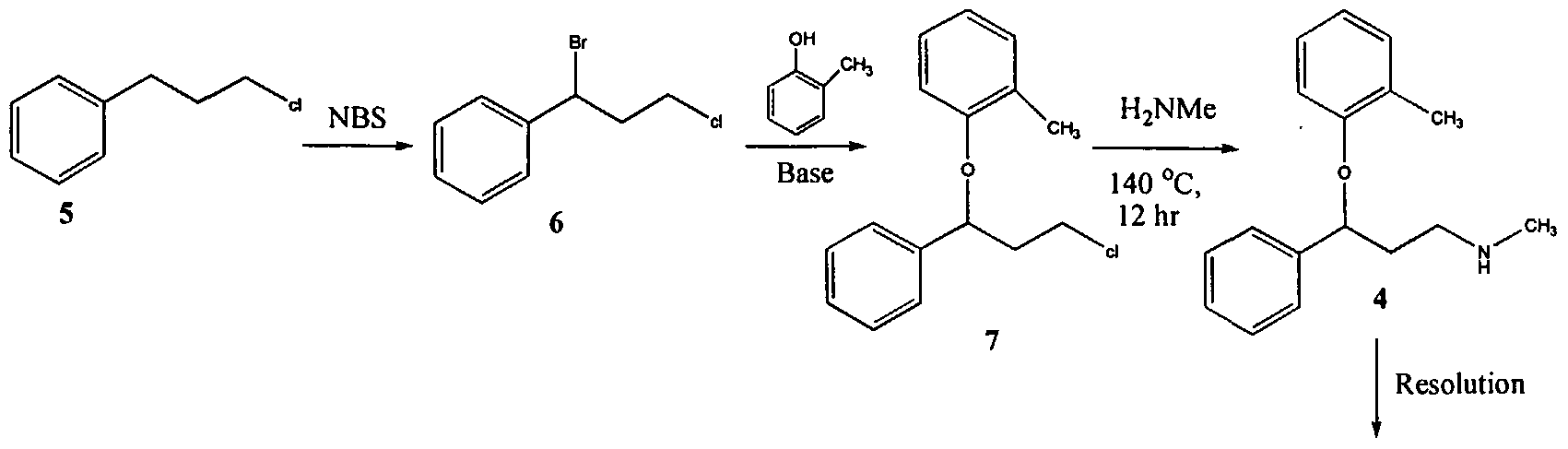
The process illustrated in Scheme A involves the preparation of the atomoxetineusing 3-phenyl chloropropyl amine (Formula 5) as a starting material. The process involves bromination of said starting compound (Formula 5) by using N-bromosuccinimide. Further the bromo derivative is condensed with o-cresol to result in a compound of Formula 7, which is then subjected to amination using methylamine. Though the process looks very simple, it involves the following disadvantages: i) N-bromosuccinimide being a corrosive and sensitive chemical, its usage demands special care; ii) the workup of the compound formula 7 involves high vacuum (0.03 torr) distillation at 135-1450C, which is a tedious and cumbersome process to carry out at the plant level; and iii) the reaction conditions involved in some of the steps are harsh, for example the amination reaction is conducted at 14O0C. at pressures of 10 kg/cm2 for 12 hours in an autoclave.
All the above points make the process not viable for practicing on a commercial scale. Further, as described in U.S. Patent 4,314,081 , the free base compounds exist as high boiling oils, but form white crystalline salts.
On the other hand, Scheme B describes the preparation of atomoxetine using β-dimethylaminopropiophenone produced by a Mannich reaction; which is reduced to the hydroxy derivative having Formula 9 using diborane; further the hydroxy compound (Formula 9) is converted to the corresponding chloro derivative of Formula 10 using dry HCI gas and thionyl chloride and is followed by condensation with o-cresol.
The said reaction is carried out in methanol at reflux for a duration of five days to achieve the compound of formula 11 and is followed by demethylation using cyanogen bromide to end up with atomoxetine. As can be clearly understood the process is associated with the following problems: i) the use of costly reagents such as diborane makes the process uneconomical; ii) the passage of dry HCI gas followed by thionyl chloride addition is ^ very cumbersome and is not advisable in the plant; iii) this is a time-consuming process, involving a reaction which requires five days for its completion; and iv) use of cyanogen bromide, which is highly toxic, is not desirable.
All of the above-quoted drawbacks make the process unfriendly to practice in a production plant as well as to the environment.
Further, M. Srebnik et al., Journal of Organic Chemistry, Vol. 53, pages2916-2920 (1988); E. Corey et al., Tetrahedron Letters, Vol. 30, pages 5207-5210 (1989);
U.S. Patent No. 4,868,344; Y. Gao et al., Journal of Organic Chemistry, Vol. 53, pages 4081-4084 (1988); J. Deeter et al.,
Tetrahedron Letters, Vol. 31, pages 7101-7104 (1990);
and U.S. Patent No. 4,950,791 disclose stereospecific methods for the preparation of 3-aryloxy-3-phenylpropylamines; the enantiomers of 3-hydroxy-3-phenylpropylamines are prepared by the stereospecific reduction of the corresponding ketones. The thus obtained (S)-3-hydroxy-3-phenyl propylamines are subjected to condensation with aryl alcohols using the Mitsunobo reaction. As can be seen in Scheme C, the reaction involves two critical steps.
Scheme C
Dusopinocampheny) chloroborane


OH
,CH,
DEAD/ tn phenyl phosphine

The first critical step is an asymmetric reduction of the ketone to its corresponding alcohol. The second critical step involves the condensation of the obtained enantiomeric alcohol with the corresponding aryl alcohol. The process suffers from the following disadvantages:
1) the reagent used for the asymmetric reduction of the ketone is highly expensive;
2) the reagent diethyl azodicarboxylate (“DEAD”) is expensive;
3) the DEAD reagent is known to be highly carcinogenic, thus creating problems in handling; and
4) the reaction involves the use of triphenylphosphine and DEAD and the resulting byproducts formed in the reaction, phoshineoxide and a hydrazine derivative, are very difficult to remove.
Therefore, commercial applicability of the said process is limited owing to the above noted disadvantages.
International Patent Publication No. WO 00/58262 relates to a stereo- specific process for the preparation of atomoxetineusing nucleophilic aromatic displacement of an aromatic ring having a functional group, which can be converted to a methyl group. As can be seen, the process is very lengthy and involves many steps and is thus not commercially desirable.
U.S. Patent No. 5,847,214 describes the nucleophilic aromatic displacement reaction of 3-hydroxy-3-arylpropylamines with activated aryl halides, for example the reaction of N-methyl-3-phenyl-3-hydroxypropylamine with 4- triflouromethyl-1-cholro benzene has been reported; the success of this reaction is mainly due to electron withdrawing group on benzene ring of the aryl halides.
U.S. Patent No. 6,541 ,668 describes a process for the preparation of atomoxetine and its pharmaceutically acceptable addition salts which comprises reacting an alkoxide of N-methyl-3-phenyl-3-hydroxy propyl amine or an N protected derivative thereof, with 2-flouro toluene in the presence of 1 ,3-Dimethyl – 2-imidazolidinone (“DMI”) or N-Methyl-3-pyrrolidinone (“NMP”) as the solvent. The process disclosed in the said patent can be shown as Scheme D. Further, the process disclosed in the said patent restricts itself to the solvents DMI and NMP.
Scheme D

Nevertheless, a new crystalline form of N-methyl-3-phenyl-3-(o- tolyloxy)propylamine oxalate and an isolation technique of (±)-atmoxetine free base in a solid form, an intermediate useful in the synthesis of atomoxetine hydrochloride, is desirable.

http://www.sciencedirect.com/science/article/pii/S0040403906025068
There have been several methods reported for preparing (R)-(−)-N-methyl-3-(2-methylphenoxy)-3-phenylpropylamine (Atomoxetine®). For example, U.S. Pat. No. 4,868,344 discloses a process as shown in the following scheme:

In this example, 3-chloropropiophenone is used as the starting material to be asymmetrically reduced with (−)-diisopinocamphenylchloroborane ((−)-IPc2BC1) to give the corresponding chiral alcohol. The resulting chiral alcohol is then reacted with o-cresol via Mitsunobu reaction to form the chiral ether compound. Subsequently, amination of the chiral ether compound with methylamine provided atomoxetine. In this process, the materials such as chiral-borane ((−)-IPc2BC1) and diethyl azodicarboxylate (DEAD) are expensive, and result in high manufacturing cost.
Further, WO 2006/009884 discloses another method for preparing atomoxetine, including the step of reacting N-methyl-3-phenyl-3-hydroxypropylamine with 2-fluorotoluene which is followed by resolution of the resulting product to provide optically pure atomoxetine as shown in the following scheme:

This process involving a chiral resolution step is inefficient due to low product yield, complicated and long time process that renders this process economically less competitive.
………………………………………………………………………………………………

see below
B.-F. Chen and co-inventors describe a synthesis of 5 that avoids costly reagents. It includes the preparation of the chiral amino alcohol 3 as a key intermediate. The route for preparing 5 starts with a Mannich reaction between benzophenone, N,O-dimethylhydroxylamine, and paraformaldehyde to give compound 1, isolated in 87.6% yield.
Ketone 1 is asymmetrically reduced to form alcohol 2 by using the chiral ruthenium catalyst RuCl2-[(S)-DMSEGPHOS)][(S)-DAIPEN]. The hydrogen pressure is described as “predetermined”, but no value is given. Product 2 is recovered as an oily product in 98.7% yield with 98.8% purity and 99% ee. It appears that the catalyst is not removed before the next step in which the oil is hydrogenated over a Raney nickel catalyst to form amino alcohol 3.
Intermediate 3 is also isolated as an oil in 96.4% yield, 96.5% % purity, and 99% ee. After recrystallization from toluene–heptane, the solid product is recovered with 100% ee. In the last step, 3 is treated with fluorotoluene 4 in the presence of t-BuOK to form atomoxetine, isolated as an oil in 91% yield with 97% ee. The purification of 5 and its conversion to the HCl salt are not described.
The inventors provide basic 1H-NMR data for all compounds except 5. The example describing the preparation of 1 lists one of the reactants as 2-acetylthiophene, which is clearly incorrect; and another reagent is called “32% hydrochloride”. These errors should have been spotted by anyone with a fundamental knowledge of chemistry who was involved in writing the patent—perhaps none were. (Sci Pharmtech [Taiwan]. US Patent 8,299,305, Oct. 30, 2012; Keith Turner)
View the full-text patent here.
| Patent Number: | US 8299305 |
| Title: | Process for preparation of atomoxetine |
| Inventor(s): | Chen, Bo-Fong; Li, Yan-Wei; Yeh, Jinun-Ban; Wong, Wei-Chyun |
| Patent Assignee(s): | SCI Pharmtech, Inc., Taiwan |
Bristol-Myers Squibb announced promising results from an expanded phase 1 dose-ranging study of its lung cancer drug nivolumab

NIVOLUMAB
Anti-PD-1;BMS-936558; ONO-4538
PRONUNCIATION nye vol’ ue mab
THERAPEUTIC CLAIM Treatment of cancer
CHEMICAL DESCRIPTION
A fully human IgG4 antibody blocking the programmed cell death-1 receptor (Medarex/Ono Pharmaceuticals/Bristol-Myers Squibb)
MOLECULAR FORMULA C6362H9862N1712O1995S42
MOLECULAR WEIGHT 143.6 kDa
SPONSOR Bristol-Myers Squibb
CODE DESIGNATION MDX-1106, BMS-936558
CAS REGISTRY NUMBER 946414-94-4
Bristol-Myers Squibb announced promising results from an expanded phase 1 dose-ranging study of its lung cancer drug nivolumab
Nivolumab (nye vol’ ue mab) is a fully human IgG4 monoclonal antibody designed for the treatment of cancer. Nivolumab was developed by Bristol-Myers Squibb and is also known as BMS-936558 and MDX1106.[1] Nivolumab acts as an immunomodulator by blocking ligand activation of the Programmed cell death 1 receptor.
A Phase 1 clinical trial [2] tested nivolumab at doses ranging from 0.1 to 10.0 mg per kilogram of body weight, every 2 weeks. Response was assessed after each 8-week treatment cycle, and were evaluable for 236 of 296 patients. Study authors concluded that:”Anti-PD-1 antibody produced objective responses in approximately one in four to one in five patients with non–small-cell lung cancer, melanoma, or renal-cell cancer; the adverse-event profile does not appear to preclude its use.”[3]
Phase III clinical trials of nivolumab are recruiting in the US and EU.[4]
- Statement On A Nonproprietary Name Adopted By The USAN Council – Nivolumab, American Medical Association.
- A Phase 1b Study of MDX-1106 in Subjects With Advanced or Recurrent Malignancies (MDX1106-03), NIH.
- Topalian SL, et al. (June 2012). “Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer”. New England Journal of Medicine 366. doi:10.1056/NEJMoa1200690. Lay summary – New York Times.
- Nivolumab at ClinicalTrials.gov, A service of the U.S. National Institutes of Health.
The PD-1 blocking antibody nivolumab continues to demonstrate sustained clinical activity in previously treated patients with advanced non-small cell lung cancer (NSCLC), according to updated long-term survival data from a phase I trial.
Survival rates at one year with nivolumab were 42% and reached 24% at two years, according to the median 20.3-month follow up. Additionally, the objective response rate (ORR) with nivolumab, defined as complete or partial responses by standard RECIST criteria, was 17% for patients with NSCLC. Results from the updated analysis will be presented during the 2013 World Conference on Lung Cancer on October 29.
“Lung cancer is very difficult to treat and there continues to be a high unmet medical need for these patients, especially those who have received multiple treatments,” David R. Spigel, MD, the program director of Lung Cancer Research at the Sarah Cannon Research Institute and one of the authors of the updated analysis, said in a statement.
“With nivolumab, we are investigating an approach to treating lung cancer that is designed to work with the body’s own immune system, and these are encouraging phase I results that support further investigation in larger scale trials.”
In the phase I trial, 306 patients received intravenous nivolumab at 0.1–10 mg/kg every-other-week for ≤12 cycles (4 doses/8 week cycle). In all, the trial enrolled patients with NSCLC, melanoma, renal cell carcinoma, colorectal cancer, and prostate cancer.
The long-term follow up focused specifically on the 129 patients with NSCLC. In this subgroup, patients treated with nivolumab showed encouraging clinical activity. The participants had a median age of 65 years and good performance status scores, and more than half had received three or more prior therapies. Across all doses of nivolumab, the median overall survival was 9.9 months, based on Kaplan-Meier estimates.
In a previous update of the full trial results presented at the 2013 ASCO Annual Meeting, drug-related adverse events of all grades occurred in 72% of patients and grade 3/4 events occurred in 15%. Grade 3/4 pneumonitis related to treatment with nivolumab emerged early in the trial, resulting in 3 deaths. As a result, a treatment algorithm for early detection and management was developed to prevent this serious side effect.
Nivolumab is a fully human monoclonal antibody that blocks the PD-1 receptor from binding to both of its known ligands, PD-L1 and PD-L2. This mechanism, along with early data, suggested an associated between PD-L1 expression and response to treatment.
In separate analysis presented at the 2013 World Conference on Lung Cancer, the association of tumor PD-L1 expression and clinical activity in patients with NSCLC treated with nivolumab was further explored. Of the 129 patients with NSCLC treated with nivolumab in the phase I trial, 63 with NSCLC were tested for PD-L1 expression by immunohistochemistry (29 squamous; 34 non-squamous).
Bristol-Myers Squibb announced promising results from phase 2b study of its rheumatoid arthritis drug clazakizumab
NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL
CLAZAKIZUMAB
PRONUNCIATION klaz” a kiz’ ue mab
THERAPEUTIC CLAIM Autoimmune diseases, rheumatoid arthritis
CHEMICAL NAMES
1. Immunoglobulin G1, anti-(human interleukin 6) (human-Oryctolagus cuniculus monoclonal BMS-945429/ALD518 heavy chain), disulfide with human-Oryctolagus cuniculus monoclonal BMS-945429/ALD518 κ-chain, dimer
2. Immunoglobulin G1, anti-(human interleukin-6 (B-cell stimulatory factor 2, CTL differentiation factor, hybridoma growth factor, interferon beta-2)); humanized rabbit monoclonal BMS-945429/ALD518 [300-alanine(CH2-N67>A67)]1 heavy chain (223-217′)-disulfide with humanized rabbit monoclonal BMS-945429/ALD518 light chain dimer (229-229”:232-232”)-bisdisulfide, O-glycosylated
MOLECULAR FORMULA C6426H9972N1724O2032S42
MOLECULAR WEIGHT 145.2 kDa
SPONSOR Bristol-Myers Squibb
CODE DESIGNATION BMS-945429, ALD518
CAS REGISTRY NUMBER 1236278-28-6
Monoclonal antibody
Type Whole antibody
Source Humanized
Target IL6
CAS number 1236278-28-6
Clazakizumab is a humanized monoclonal antibody designed for the treatment of rheumatoid arthritis.[1]
Clazakizumab was developed by Alder Biopharmaceuticals and Bristol-Myers Squibb.
gamma1 heavy chain (1-450) [humanized VH (Homo sapiens IGHV3-66*01 (83.50%) -(IGHD)-IGHJ3*02 M123>L (115)) [8.8.14] (1-120) -Homo sapiens IGHG1*03 CH

FDA approves GE’s imaging drug Vizamyl for Alzheimer’s
October 28, 2013 | By Anabela Farrica

Last Friday, FDA approved Vizamyl (flutemetamol F 18), a radioactive agent to be used with PET to help evaluate the brain of patients for Alzheimer’s disease or dementia. Vyzamil works by binding to beta-amyloid plaques, which can be found in the brain of people with Alzheimer’s disease or other dementias, as well as in the brain of elderly people who do not have neurological problems.
View original post 206 more words
Orphan drugs in Portugal
October 28 ,2013 | By Márcio Barra
What follows is a list of Orphan Drugs available in Portugal. This data has been compiled from two different databases, the INFOMED database, managed by the Portuguese National Competent Authrority on Medicines, INFARMED, and the OrphaNet database, from which sales numbers from Portugal were obtained, when available (note, sales numbers were, according to the Orphanet Website, last updated in September 28, 2013). The Portuguese Marketing approval date was also provided,
View original post 1,584 more words
Biosimilars-in-India
http://www.ibef.org/download/Biosimilars-in-India-30312.pdf
-
Biosimilars – India Brand Equity Foundation
www.ibef.org/download/Biosimilars-in-India-30312.pdfpatented/registered biotech products, but are manufactured by new companies after the patent expiry of the originator product. The global. Biosimilars market is …
