Lorlatinib, PF-6463922
For Cancer; Non-small-cell lung cancer
- Molecular Formula C21H19FN6O2
- Average mass 406.413 Da
Phase 2
WO 2013132376
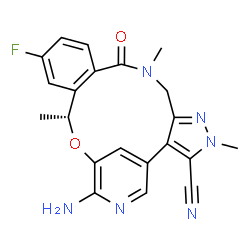
(10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-4,8- methenopyrazolo[4,3-h][2,5,11]benzoxadiazacyclotetradecine-3-carbonitrile
(16R)-19-Amino-13-fluoro-4,8,16-trimethyl-9-oxo-17-oxa-4,5,8,20-tetraazatetracyclo[16.3.1.02,6.010,15]docosa-1(22),2,5,10,12,14,18,20-octaene-3-carbonitrile
(10R)-7-Amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]benzoxadiazacyclotetradecine-3-carbonitrile
UNII:OSP71S83EU
лорлатиниб [Russian]
لورلاتينيب [Arabic]
洛拉替尼 [Chinese]
Ros1 tyrosine kinase receptor inhibitor; Anaplastic lymphoma kinase receptor inhibitor
useful for treating cancer mediated by anaplastic lymphoma kinase (ALK) or c-ros oncogene 1 (ROS1) receptor tyrosine kinase, particularly NSCLC. an ATP-competitive inhibitor of ROS1/ALK, for treating NSCLC. In February 2017, lorlatinib was reported to be in phase 2 clinical development.
- Originator Pfizer
- Developer Pfizer; The Childrens Hospital of Philadelphia; Yale University
- Class Antineoplastics; Aza compounds; Benzoxazines; Pyrazoles; Pyrazolones; Small molecules
- Mechanism of Action Anaplastic lymphoma kinase inhibitors; ROS1-protein-inhibitors
- Orphan Drug Status Yes – Non-small cell lung cancer
Lorlatinib (PF-6463922) is an experimental anti-neoplastic drug in development by Pfizer. It is a orally-administered small molecule inhibitor of ROS1 and ALK.
In 2015, FDA granted Pfizer orphan drug status for lorlatinib for the treatment of non-small cell lung cancer.[1]
- 05 Oct 2016 Massachusetts General Hospital plans a phase II trial for Non-small cell lung cancer (Late-stage disease, Metastatic disease) in USA (PO, unspecified formulation) (NCT02927340)
- 01 Oct 2016 Pfizer completes a phase I trial in pharmacokinetic trial in Healthy volunteers in USA (NCT02804399)
- 01 Aug 2016 Pfizer initiates a phase I drug-drug interaction trial in Healthy volunteers in Belgium (PO, unspecified formulation) (NCT02838264)

Structures of ALK inhibitors marketed or currently in the clinic
Synthesis

NEED COLOUR

Clinical studies
Several clinical trials are ongoing. A phase II trial comparing avelumab alone and in combination with lorlatinib or crizotinib for non-small cell lung cancer is expected to be complete in late 2017. A phase II trial comparing lorlatinib with crizotinib is expected to be complete in mid-2018.[2] A phase II trial for treatment of ALK-positive or ROS1-positive non-small cell lung cancer with CNA metastases is not expected to be complete until 2023.[3] Preclinical studies are investigating lorlatinib for treatment of neuroblastoma.
Lorlatinib is an investigational medicine that inhibits the anaplastic lymphoma kinase (ALK) and ROS1 proto-oncogene. Due to tumor complexity and development of resistance to treatment, disease progression is a challenge in patients with ALK-positive metastatic non-small cell lung cancer (NSCLC). A common site for progression in metastatic NSCLC is the brain. Lorlatinib was specifically designed to inhibit tumor mutations that drive resistance to other ALK inhibitors and to penetrate the blood brain barrier.
ABOUT LORLATINIB
ALK in NSCLC ROS1 in NSCLC PRECLINICAL DATA CLINICAL STUDIES Originally discovered as an oncogenic driver in a type of lymphoma, ALK gene alterations were also found to be among key drivers of tumor development in cancers, such as NSCLC.1 In ALK-positive lung cancer, a normally inactive gene called ALK is fused with another gene. This genetic alteration creates the ALK fusion gene and ultimately, the production of an ALK fusion protein, which is responsible for tumor growth.1,2 This genetic alteration is present in 3-5% of NSCLC patients.3,4,5 Another gene that can fuse with other genes is called ROS1. Sometimes a ROS1 fusion protein can contribute to cancer-cell growth and tumor survival. This genetic alteration is present in approximately 1% of NSCLC patients.5 Preclinical data showed lorlatinib is capable of overcoming resistance to existing ALK inhibitors and penetrated the blood brain barrier in ALK-driven tumor models.2 Specifically, in these preclinical models, lorlatinib had activity against all tested clinical resistance mutations in ALK.
A Phase 1/2 clinical trial of lorlatinib in patients with ALK-positive or ROS1-positive advanced NSCLC is currently ongoing. • The primary objective of the Phase 1 portion was to assess safety and tolerability of single-agent lorlatinib at increasing dose levels in patients with ALK-positive or ROS1-positive advanced NSCLC.6 • Data from the Phase 1 study showed that lorlatinib had promising clinical activity in patients with ALK-positive or ROS1- positive advanced NSCLC. Most of these patients had developed CNS metastases and had received ≥1 prior tyrosine kinase inhibitor.7 o The most common treatment-related adverse events (AEs) were hypercholesterolemia (69%) and peripheral edema (37%). Hypercholesterolemia was the most common (11%) grade 3 or higher treatment-related AE and the most frequent reason for dose delay or reduction. No patients discontinued due to treatment-related AEs. At the recommended Phase 2 dose, 4 out of 17 patients (24%) experienced a treatment-related AE of any grade that led to a dose delay or hold.
PATENT
WO2014207606
This invention relates to crystalline forms of the macrocyclic kinase inhibitor, (10R)-7-amino-12-fluoro-2, 10,16-trimethyl-15-OXO-10,15, 16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4, 3-?][2,5,1 1 ]benzoxadiazacyclotetradecine-3-carbonitrile, including crystalline solvates thereof, that may be useful in the treatment of abnormal cell growth, such as cancer, in mammals. The invention also relates to compositions including such crystalline forms, and to methods of using such compositions in the treatment of abnormal cell growth in mammals, especially humans.
Background of the Invention
The compound (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2/-/-8,4-(metheno)pyrazolo[4,3- ?][2,5,1 1 ]benzoxadiazacyclotetradecine-3-carbonitrile, represented by the formula (I):

(I)
is a potent, macrocyclic inhibitor of both wild type and resistance mutant forms of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) receptor tyrosine kinase. Preparation of the free base compound of formula (I) as an amorphous solid is disclosed in International Patent Publication No. WO 2013/132376 and in United States Patent Publication No. 2013/0252961 , the contents of which are incorporated herein by reference in their entirety.
Human cancers comprise a diverse array of diseases that collectively are one of the leading causes of death in developed countries throughout the world (American Cancer Society, Cancer Facts and Figures 2005. Atlanta: American Cancer Society; 2005). The progression of cancers is caused by a complex series of multiple genetic and molecular events including gene mutations, chromosomal translocations, and karyotypic abnormalities (Hanahan & Weinberg, The hallmarks of cancer. Cell 2000; 100: 57-70). Although the underlying genetic causes of
cancer are both diverse and complex, each cancer type has been observed to exhibit common traits and acquired capabilities that facilitate its progression. These acquired capabilities include dysregulated cell growth, sustained ability to recruit blood vessels (i.e., angiogenesis), and ability of tumor cells to spread locally as well as metastasize to secondary organ sites (Hanahan & Weinberg 2000). Therefore, the ability to identify novel therapeutic agents that inhibit molecular targets that are altered during cancer progression or target multiple processes that are common to cancer progression in a variety of tumors presents a significant unmet need.
Receptor tyrosine kinases (RTKs) play fundamental roles in cellular processes, including cell proliferation, migration, metabolism, differentiation, and survival. RTK activity is tightly controlled in normal cells. The constitutively enhanced RTK activities from point mutation, amplification, and rearrangement of the corresponding genes have been implicated in the development and progression of many types of cancer. (Gschwind et al., The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat. Rev. Cancer 2004; 4, 361-370; Krause & Van Etten, Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med. 2005; 353: 172-187.)
Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase, grouped together with leukocyte tyrosine kinase (LTK) to a subfamily within the insulin receptor (IR) superfamily. ALK was first discovered as a fusion protein with nucleophosmin (NPM) in anaplastic large cell lymphoma (ALCL) cell lines in 1994. (Morris et al., Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994; 263:1281-1284.) NPM-ALK, which results from a chromosomal translocation, is implicated in the pathogenesis of human anaplastic large cell lymphoma (ALCL) (Pulford et al., Anaplastic lymphoma kinase proteins in growth control and cancer. J. Cell Physiol., 2004; 199: 330-58). The roles of aberrant expression of constitutively active ALK chimeric proteins in the pathogenesis of ALCL have been defined (Wan et. al., Anaplastic lymphoma kinase activity is essential for the proliferation and survival of anaplastic large cell lymphoma cells. Blood, 2006; 107:1617-1623). Other chromosomal rearrangements resulting in ALK fusions have been subsequently detected in ALCL (50-60%), inflammatory myofibroblastic tumors (27%), and non-small-cell lung cancer (NSCLC) (2-7%). (Palmer et al., Anaplastic lymphoma kinase: signaling in development and disease. Biochem. J. 2009; 420:345-361 .)
The EML4-ALK fusion gene, comprising portions of the echinoderm microtubule associated protein-like 4 (EML4) gene and the ALK gene, was first discovered in NSCLC archived clinical specimens and cell lines. (Soda et al., Identification of the transforming EML4-ALK fusion gene in non-small cell lung cancer. Nature 2007; 448:561-566; Rikova et al., Cell 2007; 131 :1 190-1203.) EML4-ALK fusion variants were demonstrated to transform NIH-3T3 fibroblasts and cause lung adenocarcinoma when expressed in transgenic mice, confirming the
potent oncogenic activity of the EML4-ALK fusion kinase. (Soda et al., A mouse model for EML4-ALK-positive lung cancer. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:19893-19897.) Oncogenic mutations of ALK in both familial and sporadic cases of neuroblastoma have also been reported. (Caren et al., High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumors. Biochem. J. 2008; 416:153-159.)
ROS1 is a proto-oncogene receptor tyrosine kinase that belongs to the insulin receptor subfamily, and is involved in cell proliferation and differentiation processes. (Nagarajan et al. Proc Natl Acad Sci 1986; 83:6568-6572). ROS is expressed, in humans, in epithelial cells of a variety of different tissues. Defects in ROS expression and/or activation have been found in glioblastoma, as well as tumors of the central nervous system (Charest et al., Genes Chromos. Can. 2003; 37(1): 58-71). Genetic alterations involving ROS that result in aberrant fusion proteins of ROS kinase have been described, including the FIG-ROS deletion translocation in glioblastoma (Charest et al. (2003); Birchmeier et al. Proc Natl Acad Sci 1987; 84:9270-9274; and NSCLC (Rimkunas et al., Analysis of Receptor Tyrosine Kinase ROS1 -Positive Tumors in Non-Small Cell Lung Cancer: Identification of FIG-ROS1 Fusion, Clin Cancer Res 2012; 18:4449-4457), the SLC34A2-ROS translocation in NSCLC (Rikova et al. Cell 2007;131 :1 190-1203), the CD74-ROS translocation in NSCLC (Rikova et al. (2007)) and cholangiocarcinoma (Gu et al. PLoS ONE 201 1 ; 6(1 ): e15640), and a truncated, active form of ROS known to drive tumor growth in mice (Birchmeier et al. Mol. Cell. Bio. 1986; 6(9):3109-31 15). Additional fusions, including TPM3-ROS1 , SDC4-ROS1 , EZR-ROS1 and LRIG3-ROS1 , have been reported in lung cancer patient tumor samples (Takeuchi et al., RET, ROS1 and ALK fusions in lung cancer, Nature Medicine 2012; 18(3):378-381).
The dual ALK/c-MET inhibitor crizotinib was approved in 201 1 for the treatment of patients with locally advanced or metastatic NSCLC that is ALK-positive as detected by an FDA-approved test. Crizotinib has also shown efficacy in treatment of NSCLC with ROS1 translocations. (Shaw et al. Clinical activity of crizotinib in advanced rson-smali cell lung cancer (NSCLC) harboring ROS1 gene rearrangement. Presented at the Annual Meeting of the American Society of Clinical Oncology, Chicago, June 1-5, 2012.) As observed clinically for other tyrosine kinase inhibitors, mutations in ALK and ROS1 that confer resistance to ALK inhibitors have been described (Choi et ai., EML4-ALK Mutations in Lung Cancer than Confer Resistance to ALK Inhibitors, N Engl J Med 2010; 363:1734-1739; Awad et ai., Acquired Resistance to Crizotinib from a Mutation in CD74-ROS1, Engl J Med 2013; 368:2395-2401 ).
Thus, ALK and ROS1 are attractive molecular targets for cancer therapeutic intervention. There remains a need to identify compounds having novel activity profiles against wild-type and mutant forms of ALK and ROS1 .
The present invention provides crystalline forms of the free base of (10R)-7-amino-12-fluoro-2, 10,16-trimethyl-15-OXO-10,15, 16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3- ?][2, 5,1 1 ]-benzoxadiazacyclotetradecine-3-carbonitrile having improved properties, such as improved crystallinity, dissolution properties, decreased hygroscopicity, improved mechanical properties, improved purity, and/or improved stability, while maintaining chemical and enantiomeric stability.
Comparative Example 1A
Preparation of (10f?)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3- ?l[2,5,1 Hbenzoxadiazacyclo-tetradecine-3-carbonitrile (amorphous)

Example 1A
Step 1 :
Palladium (II) acetate (53 mg, 0.24 mmol) and cataCXium® A (180 mg, 0.5 mmol) were mixed together in toluene (1 .5 mL, de-gassed) and the resulting solution was added via pipette to a stirred solution of compound 7 (0.9 g, 2.4 mmol), compound 15 (1 .0 g, 3.0 mmol) bis-pinacolato diboron (0.9 g, 3.6 mmol) and CsF (1 .9 g, 12.6 mmol) in MeOH/H20 (9:1 , 12 mL, degassed) at 60 °C. The resulting mixture was then stirred at reflux for 3 hrs. A further portion of Palladium (II) acetate (26 mg, 0.12 mmol) and cataCXium® A (90 mg, 0.25 mmol) in toluene (1 .5 mL, de-gassed) was added, and the yellow reaction mixture stirred at 60 °C overnight. After cooling to room temperature, the mixture was diluted with EtOAc (150 mL) and filtered through CELITE®. The filtrate was washed with water (100 mL), then brine (100 mL), dried (Na2S04) and evaporated. The residue was purified by flash chromatography over silica gel, which was eluted with 1 :1 EtOAc/cyclohexane, to give compound 22 as a yellow oil (570 mg, 43% yield). TLC (Rf = 0.40, 1 :1 EtOAc/cyclohexane). 1H NMR (400 MHz, CDCI3) δ 8.03 (m, 1 H), 7.65 (s, 1 H), 7.27 (dd,1 H, J = 9.9, 2.7 Hz), 7.01 (m, 1 H), 6.68 (m, 1 H), 6.40 (m, 1 H), 4.90 (br s, 2 H), 4.20 – 4.30 (m, 2 H), 3.96 (s, 3 H), 3.94 (s, 3 H), 2.55 – 2.85 (m, 3 H), 1 .68 (d, 3 H, J = 6.6 Hz), 1 .24 (s, 9 H). LCMS ES m/z 539 [M+H]+.
Step 2:
To a solution of compound 22 (69% purity, 0.95 g, assumed 1 .05 mmol) in MeOH (20 mL) was added a solution NaOH (1 .0 g, 25 mmol) in water (2 mL). The mixture was stirred at 40 °C for 3.5 hours. The reaction was diluted with water (80 mL), concentrated by 20 mL to remove MeOH on the rotary evaporator, and washed with MTBE (100 mL). The aqueous layer was then acidified carefully with 1 M aq HCI to approx. pH 2 (pH paper). Sodium chloride (15 g) was added to the mixture and the mixture was extracted with EtOAc (100 mL). The organic layer was separated, dried (Na2S04) and evaporated to give compound 23 as a pale yellow solid (480 mg, 87% yield). 1H NMR (400 MHz, CD3OD) δ 8.05 (m, 1 H), 7.45 (s, 1 H), 7.37 (dd,1 H, J = 10.4, 2.8 Hz), 7.10 (dt, 1 H, J = 8.5, 2.4 Hz), 6.50 – 6.60 (m, 2 H), 4.05 – 4.30 (m, 2 H), 3.99 (s, 3 H), 2.60 – 2.80 (m, 3 H), 1 .72 (d, 3 H, J = 6.5 Hz). LCMS ES m/z 525 [M+H]+.
Step 3:
A solution of HCI in dioxane (4 M, 6.0 mL) was added to a solution of compound 23
(480 mg, 0.91 mmol) in MeOH (methanol) (6 mL) and the reaction was stirred at 40 °C for 2.5 hours. The reaction mixture was then concentrated to dryness under reduced pressure. The residue was taken-up in MeOH (50 mL) and acetonitrile (100 mL) was added and the mixture was then again evaporated to dryness, to give compound 24 as an off white solid (400 mg, 87% yield). 1H NMR (400 MHz, CD3OD) δ 8.07 (dd, 1 H, J = 8.9. 5.9 Hz), 7.51 (d, 1 H, J = 1 .7 Hz), 7.42 (dd, 1 H, J = 9.8, 2.6 Hz), 7.23 (d, 1 H, J = 1 .6 Hz), 7.16 (dt, 1 H, J = 8.5, 2.7 Hz), 6.73 (dd, 1 H, J = 1 1 .9, 6.9 Hz), 4.22 (d, 1 H, J = 14.7 Hz), 4.14 (d, 1 H, J = 14.7 Hz), 4.07 (s, 3 H), 2.75 (s, 3 H), 1 .75 (d, 3 H, J = 5.5 Hz). LCMS ES m/z 425 [M+H]+.
Step 4:
A solution of compound 24 (400 mg, assumed 0.91 mmol) as the HCI salt and DIPEA
(diisopropylethylamine) (1 .17 g, 9.1 mmol) in DMF (dimethylformamide) (5.0 mL) and THF (0.5 mL) was added drop-wise to a solution of HATU (2-(1 H-7-azabenzotriazol-1 -yl)-1 ,1 ,3,3-tetramethyl uronium hexafluorophosphate methanaminium) (482 mg, 1 .27 mmol) in DMF (10.0 mL) at 0 °C over 30 minutes. After complete addition, the mixture was stirred at 0 °C for a further 30 mins. Water (70 mL) was added and the mixture was extracted into EtOAc (2 x 60 mL). The combined organics were washed with saturated aqueous NaHC03 (2 x 100 mL), brine (100 mL), dried over Na2S04, and evaporated. The residue was purified by column chromatography over silica gel, which was eluted with 70% EtOAc/cyclohexane giving 205 mg of a pale yellow residue (semi-solid). The solids were dissolved in MTBE (7 mL) and cyclohexane (20 mL) was added slowly with good stirring to precipitate the product. After stirring for 30 minutes, the mixture was filtered, and Example 1A was collected as an
amorphous white solid (1 10 mg, 29% yield). TLC (Rf = 0.40, 70% EtOAc in cyclohexane). 1H NMR (400 MHz, CDCI3) δ 7.83 (d, 1 H, J = 2.0 Hz), 7.30 (dd, 1 H, J = 9.6, 2.4 Hz), 7.21 (dd, 1 H, J = 8.4, 5.6 Hz), 6.99 (dt, 1 H, J = 8.0, 2.8 Hz), 6.86 (d, 1 H, J = 1 .2 Hz), 5.75 – 5.71 (m, 1 H), 4.84 (s, 2 H), 4.45 (d, 1 H, J = 14.4 Hz), 4.35 (d ,1 H, J = 14.4 Hz), 4.07 (s, 3 H), 3.13 (s, 3 H), 1 .79 (d, 3 H, J = 6.4Hz). LCMS ES m/z 407 [M+H]+.
Example 1
Preparation of crystalline hydrate of (10 ?)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo- 10,15,16,17-tetrahvdro-2/-/-8,4-(metheno)pyrazolo[4,3- ?l[2,5,1 Hbenzoxa-diazacyclo-tetradecine-3-carbonitrile (Form 1)

Example 1A Example 1
(amorphous) (Form 1 }
Amorphous (10f?)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3- ?][2,5,11 ]benzoxa-diazacyclo-tetradecine-3-carbonitrile free base, prepared as described in Example 1A (and Example 2 of United States Patent Publication No. 2013/0252961), was dissolved in 1 .0 : 1 .1 (v:v) H20:MeOH at a concentration of 22 mg/mL at 50°C, then allowed to cool to room temperature . This slurry was granulated for approximately 72 hours. The solids were isolated by filtration and vacuum dried overnight at 60°C to produce crystalline hydrate Form 1 of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-/?][2,5,1 1 ]benzoxadiazacyclotetradecine-3-carbonitrile.
Example 4
Alternative preparation of crystalline acetic acid solvate of (10 ?)-7-amino-12-fluoro-2, 10,16-trimethyl-15-OXO-10,15, 16,17-tetrahvdro-2H-8,4-(metheno)pyrazolo[4,3- ?U2,5, 1 1 lbenzoxa-diazacyclotetradecine-3-carbonitrile (Form 3)

Step 1 :
To a reaction vessel under N2 were charged compound 9 (9.97 kg, 17.95 mol), compound 21 (3.52 kg, 18.85 mol) and 2-methyltetrahydrofuran (97 L). Triethylamine (7.45 kg, 73.6 mol) was added while keeping the internal temperature below 35°C. The reaction mixture was held for 30 min and n-propylphosphonic anhydride (T3P), 50% solution in ethyl acetate (22.85 kg, 35.9 mol) was charged slowly, maintaining the internal temperature below 25°C. The reaction mixture was held at 20°C for at least 2 h until reaction was deemed complete. Ethyl acetate (35 L) and water (66 L) were added followed by 0.5N Hydrochloric acid solution (80 L). The aqueous layer was removed and the organic layer was washed with brine solution (80 L). The organic layer was concentrated and solvent exchanged with 2-methyl-2-butanol (80 L) give compound 25 (23 wt/wt%) solution in 2-methyl-2-butanol . This solution was carried forward to the next step directly in three batches, assuming 12.00 kg (100% yield) from this step.
Step 2:
2-Methyl-2-butanol (100 L) was combined with potassium acetate (1 .8 kg, 18.34 mol), palladium(ll) acetate (0.10 kg, 0.46 mol) and water (0.10 kg, 5.73 mol). The resulting mixture was purged with nitrogen. Di(1 -adamantyl)n-butylphosphine (0.23 kg, 0.43 mol) was added. An amount of 20% of compound 25 (3.97 kg active or 17.3 L of step 1 solution in 2-methyl-2-butanol) was added, and the resulting reaction mixture was heated at reflux for 2 h. The remaining solution of compound 25 in 2-methyl-2-butanol was subsequently added to the reaction over a period of 5 h. The resulting mixture was heated until the reaction was deemed complete (typically 16 – 20 h). This reaction step was processed in three batches, and the isolation was done in one single batch. Thus, the combined three batches were filtered through CELITE® to remove insoluble materials. The filtrate was concentrated to a low volume (approximately 20 L). Acetonitrile (60 L) was added. The resulting mixture was heated to reflux for 2 – 4 h, then cooled to RT for granulation. The resulting slurry was filtered to give compound 26 as a crude product. The crude product was combined with ethyl acetate (80 L) and Silicycle thiol (5 kg). The resulting mixture was heated for 2 h, cooled to RT and filtered. The filtrate was concentrated to approx. 20 L, and the resulting slurry was granulated and filtered. The filter cake was rinsed with ethyl acetate (4 L) and dried in a vacuum oven to give compound 26 as a pure product (4.74 kg, 43.5% overall last two steps). 1H NMR (CDCI3) δ 8.25 – 8.23 (m, 1 H), 7.28 (1 H, dd, 2.76 and 9.79 Hz), 7.22 (1 H, dd, 5.52 and 8.53 Hz), 7.18 (1 H, d, J = 1 .76 Hz), 7.01 (1 H, dt, J = 2.50 and 8.03 Hz), 5.78 – 5.70 (m, 1 H), 4.76 (1 H, d, J = 14.3 Hz), 4.13 (s, 3H), 3.16 (s, 3H), 1 .78 (d, 3H, J = 6.02 Hz), 1 .45 (s, 18H); 13C NMR (CDCI3) δ 167.0, 162.9, 160.4, 148.7, 146.3, 143.0, 140.7, 139.9, 135.5, 129.9, 129.8, 126.1 , 123.8, 123.5, 1 19.7, 1 13.8, 1 13.5, 1 1 1 .6, 108.1 , 81 .1 , 70.1 , 45.5, 37.0, 29.7, 26.0, 20.7; LCMS (M+1)+ 607.3, 507.1 , 451 .2.
Step 3:
To a reactor under N2 was added compound 26 (4.74 kg, 7.82 mol) and ethyl acetate (54 L). Hydrochloric acid 37% (5.19 L, 63.2 mol) was charged slowly while keeping the internal temperature below 25°C. The reaction mixture was stirred for 24 – 48 h until the reaction was complete. Ethyl acetate (54L) and water (54 L) were added. The reaction mixture was then treated with triethylamine until pH 8 – 9 was reached. The aqueous layer was removed and then the organic layer was washed water (2 x 54 L). The organic layer was concentrated under reduced pressure to approx. 54 L to give compound 27 (unisolated).
Step 4:
Acetic acid (1 .0 kg, 16.6 mol) was added to the organic layer containing compound 27. The reaction mixture was concentrated and then held for at least 3 h with stirring at RT. The resulted slurry was filtered. The filter cake was washed with ethyl acetate (2 L) and dried under vacuum to give 3.20 kg (87.8% yield) of Example 4 acetic acid solvate (Form 3). The spectroscopic data of this material was identical to that of an authentic sample of the crystalline acetic acid Form 3 of (10R)-7-amino-12-fluoro-2, 10, 16-trimethyl-15-oxo-10, 15,16, 17-tetrahydro-2/-/-8,4-(metheno)pyrazolo[4,3- ?][2,5,1 1 ]-benzoxadiazacyclo-tetradecine-3-carbonitrile prepared according to Example 3.
Preparation of Synthetic Intermediates

7 6 5
Step 1 :
A solution of (-)-DIPCI ((-)-B-chlorodiisopinocampheylborane) (57.1 g, 178 mmol) in THF
(tetrahydrofuran) (100 ml) was cooled to -20 to -30 °C. A solution of compound 1 (31 .3 g, 1 19 mmol) in THF (100 ml) was then added dropwise, via addition funnel (30 min addition). The reaction was left to warm up to room temperature (RT). After 2 h, the reaction was cooled to -30 °C and another portion of (-)-DIPCI (38.0 g, 1 19 mmol) was added. After 30 min, the reaction was allowed to warm to RT and after 1 h, the solvents were removed in vacuo and the residue re-dissolved in MTBE (methyl tertiary-butyl ether) (200 ml). A solution of diethanolamine (31 g, 296 mmol) in ethanol/THF (15 ml/30 ml) was added via addition funnel, to the reaction mixture under an ice bath. The formation of a white precipitate was observed. The suspension was heated at reflux for 2 hours then cooled to room temperature, filtered and the mother liquids concentrated in vacuo. The residue was suspended in heptane/EtOAc (7:3, 200 ml) and again
filtered. This procedure was repeated until no more solids could be observed after the liquids were concentrated. The final yellow oil was purified by column chromatography (eluent: cyclohexane/EtOAc 99:1 to 96:4). The resulting colorless oil was further purified by recrystallization from heptanes, to give alcohol compound 2 (25 g, 80% yield, 99% purity and 96% ee) as white crystals. 1H NMR (400 MHz, CDCI3) δ 7.73 (dd, 1 H), 7.32 (dd, 1 H), 6.74 (ddd, 1 H), 4.99 – 5.04 (m, 1 H), 2.01 (d, 1 H), 1 .44 (d, 3 H). LCMS-ES: No ionization, Purity 99%. Chiral GC (column CP-Chirasil-DexnCB): 96% ee; Rt (minor) 17.7 minutes and Rt (major) 19.4 minutes.
Step 2:
A solution of compound 2 (22 g, 83 mmol) in MTBE (350 mL) was cooled under an ice bath and triethylamine (23 mL, 166 mmol) followed by mesyl chloride (9.6 mL, 124 mmol) were added drop-wise. The reaction was then warmed to RT and stirred for 3 h. The reaction mixture was filtered and the solids washed with EtOAc. The mother liquids were concentrated in vacuo to give compound 3 (35 g, 80% yield) as a pale yellow oil. This material was taken into the following step without further purification. 1H NMR (400 MHz, CDCI3) δ 7.78 (dd, 1 H), 7.24 (dd, 1 H), 6.82 (ddd, 1 H), 2.92 (s, 3 H), 1 .64 (d, 3 H). LCMS-ES no ionization.
Step 3:
A suspension of Cs2C03 (65 g, 201 mmol) and compound 4 (13.3 g, 121 mmol) in 2-CH3-THF (2-methyitetrahydrofuran) (600 mL) and acetone (300 mL) was stirred at RT for 30 minutes then heated at 40 °C before drop-wise addition of a solution of compound 3 (34.4 g, 80 mmol) in 2-CH3-THF (300 mL) via addition funnel. The resulting mixture was left stirring at 75 -80 °C for 24 h. The reaction was then filtered through CELITE® with MTBE, the solvents removed in vacuo and the residue purified by column chromatography over silica gel which was eluted with cyclohexane/EtOAc (9:1 to 1 :1) to give compound 5 (14.3 g, 39 % yield, 90% ee) as a white solid. The solids were then re crystallized from heptane/EtOAc to give compound 5 (10.8 g, 37% yield, 95% ee). 1H NMR (400 MHz, CDCI3) 5 7.38 (dd, 1 H), 7.62 (dd, 1 H), 7.10 (dd, 1 H), 6.75 (ddd, 1 H), 6.44 – 6.51 (m, 2 H), 5.34 – 5.39 (m, 1 H), 4.73 (br s, 2 H), 1 .61 (d, 3 H). LCMS-ES m/z 359 [M+H]+. HPLC (Chiralpak IC 4.6 x 250 mm): 95% ee; Rt (minor) 10.4 minutes; Rt (major) 14.7 minutes; eluent: Heptane 80%/IPA 20% with 0.2% DEA, 0.7 mL/min. Step 4:
Compound 5 (20 g, 57 mmol) was dissolved in methanol (300 mL), and sequentially treated with triethylamine (TEA) (15.4 mL, 1 13 mmol) and PdCI2(dppf) (1 ,1 -bis(diphenylphosphino)ferrocene]dichloropalladium(ll) ) (4.1 g, 5.7 mmol). This mixture was heated at 100 °C for 16 hours, under a 100 psi carbon monoxide atmosphere. LCMS indicated consumption of starting material. The reaction mixture was filtered through a pad of CELITE®, and the filtrate evaporated to a brown oil. The crude product was purified by flash
chromatography over silica gel which was eluted with 50% to 75% ethyl acetate in cyclohexane, affording the pure product 6 as a brick-red solid (13.0 g, 79% yield). 1H NMR (400 MHz, CDCI3) δ 1 .65 (d, 3 H), 3.94 (s, 3 H), 4.75 (br s, 2 H), 6.32 (q, 1 H), 6.42 (dd, 1 H), 6.61 (dd, 1 H), 7.00 (ddd, 1 H), 7.28 (dd, 1 H), 7.60 (dd, 1 H), 8.03 (dd, 1 H). LCMS ES m/z 291 for [M+H]+.
Step 5:
Compound 6 (13.0 g, 45 mmol) was dissolved in acetonitrile (195 mL), and cooled to <10 °C in an ice water bath. NBS (N-bromosuccinimide) (7.9 g, 45 mmol) was added drop-wise to the cooled reaction mixture as a solution in acetonitrile (195 mL), monitoring the internal temperature to ensure it did not rise above 10 °C. After addition was complete, the mixture was stirred for 15 minutes. Thin layer chromatography (TLC) (1 :1 cyclohexane/ethyl acetate) showed consumption of starting material. The reaction mixture was evaporated, and the residue redissolved in ethyl acetate (400 mL), and washed with 2M aqueous NaOH (2 x 300 mL), and 10% aqueous sodium thiosulfate solution (300 mL). The organic extracts were dried over MgS04, and evaporated to a red oil (17.6 g). The crude product was purified over silica gel, which was eluted with 10% to 50% ethyl acetate in cyclohexane, which gave compound 7 (12.0 g, 73% yield). 1H NMR (400 MHz, CDCI3) δ 1 .65 (d, 3 H), 3.96 (s, 3 H), 4.74 – 4.81 (br s, 2 H), 6.33 (q, 1 H), 6.75 (d, 1 H), 7.03 (ddd, 1 H), 7.25 (dd, 1 H), 7.66 (d, 1 H), 8.06 (dd, 1 H). LCMS ES m/z 369/371 [M+H]+. A Chiralpak AD-H (4.6 x 100 mm, 5 micron) column was eluted with 10% MeOH (0.1 % DEA) in C02 at 120 bar. A flow rate of 5.0 mL/min gave the minor isomer Rt 0.6 minutes and the major isomer Rt 0.8 minutes (99% ee). Optical rotation: [ ]d20 = -92.4 deg (c=1 .5, MeOH).
Preparation of (/?)-methyl 2-(1 -((N,N-di-Boc-2-amino-5-bromopyridin-3-yl)oxy)ethyl)-4-fluorobenzoic acid (9)

7
Step 1 :
To a solution of compound 7 (2000 g, 5.4 mol) in dry DCM (dichloromethane) (32000 mL) was added DIPEA (N.N-dsisopropyleibylamine) (2100 g, 16.28 mol) and DMAP (4-dimethylaminopyridine) (132 g, 1 .08 mol). Then Boc20 (di-tert-butyl-dicarbonate) (3552 g, 16.28 mol) was added to the mixture in portions. The reaction was stirred at RT for overnight. TLC (petroleum ether/EtOAc =5:1) show the reaction was complete, the mixture was washed with sat. NH4CI (15 L) two times, then dried over Na2S04and concentrated to give a crude product which was purified by column (silica gel, petroleum ether/EtOAc from 20:1 to 10:1) to give compound 8 (2300 g, 75%) as a white solid.
Step 2:
Compound 8 (50 g, 87.81 mmol, 100 mass%) was charged to a round bottom flask (RBF) containing tetrahydrofuran (12.25 mol/L) in Water (5 mL/g, 3060 mmol, 12.25 mol/L) and sodium hydroxide (1 mol/L) in Water (1 .5 equiv., 131 .7 mmol, 1 mol/L). The biphasic mixture was stirred at RT for 14 hours. 1 N HCI was added to adjust pH to < 2. THF was then removed by vacuum distillation. The product precipitated out was collected by filtration. The filter cake was rinsed with water, pulled dried then dried in vacuum oven to constant weight (48 h, 55°C, 25 mbar). 48.3g isolated, 99% yield. 1H NMR (CDCI3, 400MHz) δ 8.24 (1 H, dd, 1 H, J = 5.76 and 3.0 Hz), 8.16 (1 H, d, J = 2.0 Hz), 7.37 (1 H, dd, J = 2.5 and 9.8 Hz), 7.19 (1 H, d, J = 2 Hz), 7.14 – 7.06 (1 H, m), 6.50 (1 H, q, J = 6.3 Hz), 1 .67 (3H, d, J = 8.4 Hz), 1 .48 (18H, s). 13C NMR (CDCI3, 100 MHz), δ 170.1 , 169.2, 167.6, 165.1 , 150.6, 149.2, 148.6, 141 .4, 140.7, 135.2, 135.1 , 124.2, 122.2,122.1 , 1 19.9, 1 15.4, 1 15.1 , 1 13.4, 1 13.2, 100.0, 83.4, 73.3, 27.9, 23.9. LCMS (M+ +1) 557.2, 555.3, 457.1 , 455.1 , 401 , 0, 399.0.

Step 1 :
Ethyl 1 ,3-dimethylpyrazole-5-carboxylate (5.0 g, 30 mmol) was dissolved in 1 ,2-dichloroethane (200 mL), followed by addition of NBS (5.3 g, 30 mmol) and dibenzoyi peroxide (727 mg, 3.0 mmol), in small portions and stirred at 85 °C for 2 hours. The mixture was allowed to cool, diluted to 400 mL with dichloromethane, and washed with water (2 x 200 mL). The organic layer was dried over MgS04, and evaporated to give compound 10 (4.1 g, 42% yield). TLC (EtOAc/Cyclohexane; 1 :10; KMn04): Rf~0.3. 1H NMR (400 MHz, CDCI3) δ 4.47 (s, 2 H), 4.41 (q, 2 H), 4.15 (s, 3 H), 1 .42 (t, 3 H). LCMS ES m/z 324/326/328 [M+H]+.
Step 2:
Compound 10 (3.0 g, 9.2 mmol) was dissolved in methylamine solution (33% solution in ethanol, 70 mL), and stirred at RT for 16 hours. The mixture was evaporated to give compound 11 (1 .8 g, 71 % yield). 1H NMR (400 MHz, CDCI3) δ 4.39 (q, 2 H), 4.14 (s, 3 H), 4.05 (s, 2 H), 2.62 (d, 3 H), 1 .41 (t, 3 H). LCMS ES m/z 276/278 [M+H]+.
Step 3:
Compound 11 (1 .8 g, 6.5 mmol) was dissolved in dichloromethane (20 mL), and the mixture cooled to 0 °C. A solution of di(fe/?-butyl) dicarbonate (1 .75 g, 8 mmol) in dichloromethane (17.5 mL) was added dropwise. The ice bath was removed and the mixture stirred for 18 hours at room temperature. The mixture was diluted to 100 mL with dichloromethane, and washed with water (2 x 50 mL). Organic extracts were dried over magnesium sulfate, and evaporated to give compound 12 (1 .8 g, 72% yield). 1H NMR (400 MHz, CDCI3) δ 4.48 – 4.44 (m, 2 H), 4.41 (q, 2 H), 4.12 (s, 3 H), 2.82 – 2.79 (m, 3 H), 1 .47 (s, 9 H), 1 .41 (t, 3 H). LCMS ES m/z 376/378 [M+H]+ and 276/278 [M-BOC]+.
Step 4:
Compound 12 (4 g, 1 1 mmol) was dissolved in dioxane (43 mL). Sodium amide (1 g, 27 mmol) was added in one portion. The reaction mixture was stirred at 100 °C for 24 h. After this time, the solvent was removed under reduced pressure to give a white solid. The material was suspended in EtOAc (100 mL) and washed with 5% citric acid solution (100 mL). The organic phase was separated and washed with water (100 mL), dried over MgS04, filtered and the solvent removed in vacuo to give compound 13 as a yellow gum (3.1 g, 84% yield). 1H NMR (400 MHz, DMSO-c/6) δ 4.27 (s, 2 H), 3.92 (s, 3 H), 2.70 (s, 3 H), 1 .40 (s, 9 H). LCMS ES m/z 348/350 [M+H]+ and 248/250 [M-BOC]+.
Step 5:
Compound 13 (3 g, 8.6 mmol) was dissolved in DMF (43 mL, 0.2 M). HOBt (1 .2 g, 8.6 mmol) was added, followed by ammonium chloride (0.9 g, 17.2 mmol). EDCI (2.5 g, 13 mmol) was then added, followed by TEA (2.4 mL, 17 mmol). The reaction mixture was stirred at room temperature. After 18h, the solvent was removed under reduced pressure to give a yellow oil
(8.0 g). The residue was dissolved in EtOAc (75ml_). The organic phase was washed with NaHC03 (sat. solution, 70 ml_) and then brine (100 ml_). The combined organic layers were dried over MgS04 and the solvent removed in vacuo to give compound 14 as a dark yellow oil (2.7 g, 91 % yield). This material was used directly in the next step without further purification. 1H NMR (400 MHz, CDCI3) δ 6.74 (br s, 1 H), 5.95 (br s, 1 H), 4.49 (br s, 2 H), 4.16 (s, 3 H), 2.81 (br s, 3 H), 1 .47 (s, 9 H). LCMS ES m/z 347/349 [M+H]+ and 247/249 [M-BOC]+.
Step 6:
Compound 14 (2.7 g, 7.9 mmol) was dissolved in DCM (80 ml_, 0.1 M). TEA (3.3 ml_, 23.8 mmol) was then added and the reaction mixture cooled down to -5 °C. Trifluoroacetic anhydride (2.2 ml_, 15.8 mmol) in DCM (15 ml_) was added dropwise over 30 min. After addition, the reaction mixture was stirred at 0 °C for 1 h. After this time, the solvents were removed under reduced pressure to give a dark yellow oil. This residue was diluted in DCM (100 ml_), washed with 5% citric acid, sat. NaHC03and brine, dried over MgS04, filtered and the solvents removed in vacuo to give a dark yellow oil (2.6 g). The crude product was purified by reverse phase chromatography to give compound 15 as a yellow oil (2.3 g, 87% yield). 1H NMR (400 MHz, CDCI3) δ 4.46 (br s, 2 H), 4.01 (s, 3 H), 2.83 (br s, 3 H), 1 .47 (s, 9 H). LCMS ES m/z 331 /329 [M+H]+ and 229/231 [M-BOC]+ as the base ion.
Preparation o/: 1 -methyl-3-((methylamino)methyl)-1 H-pyrazole-5-carbonitrile (21)

Step 1 :
To /V-benzylmethylamine (2.40 kg, 19.8 mol) and ethyldiisopropylamine (2.61 kg, 20.2 mol) in acetonitrile (6 L) at 16°C was added chloroacetone (1 .96 kg, 21 .2 mol) over 60 mins [exothermic, temp kept <30°C]. The mixture was stirred at 22°C for 18 hours then concentrated to an oily solid. The residue was triturated with MTBE (5 L), and then filtered through a pad of CELITE® (600 g, top) and silica (1 .5 kg, bottom), washing with MTBE (8 L). The filtrate was evaporated to afford compound 16 (3.35 kg, 18.9 mol, 95%) as a brown oil.
Step 2:
Compound 16 (1 .68 kg, 9.45 mol), Boc-anhydride (2.1 kg, 9.6 mol) and 20wt% Pd/C (50% H20, 56 g) in ethanol (5 L) were hydrogenated in an 1 1 -L autoclave at 50 psi [exotherm to 40°C with 20°C jacket]. The atmosphere became saturated with carbon dioxide during the reaction and so needed to be vented and de-gassed twice to ensure sufficient hydrogen uptake and completion of the reaction. The total reaction time was ~1 .5 hours. Two runs (for a total of 18.9 mol) were combined and filtered through a pad of SOLKA-FLOC®, washing with methanol. The filtrate was treated with DMAP (45 g, 0.37 mol) and stirred at room temperature overnight to destroy the excess Boc-anhydride. The mixture was then concentrated to dryness, dissolved in MTBE (6 L) and filtered through a pad of magnesol (1 kg), washing with MTBE (4 L). The filtrate was evaporated to afford compound 17 (3.68 kg, ~95 wt%, 18.7 mol, 99%) as an orange-brown oil.
Step 3:
To compound 17 (3.25 kg, -95 wt%, 16.5 mol) and diethyl oxalate (4.71 kg, 32.2 mol) in methanol (12 L) at 15°C was added 25 wt% sodium methoxide in methanol (6.94 kg, 32.1 mol) over 25 mins [temp kept <25°C]. The mixture was stirred at 20°C for 16 hours then cooled to -37°C and 37% hydrochloric acid (3.1 kg, 31 mol) was added over 5 mins [temp kept <-10°C]. The mixture was cooled to -40°C and methylhydrazine (1 .42 kg, 30.8 mol) was added over 7 mins [temp kept <-17°C]. The mixture was warmed to 5°C over 90 minutes, then re-cooled to 0°C and quenched by addition of 2.4M KHS04 (6.75 L, 16.2 mol) in one portion [exotherm to 27°C]. The mixture was diluted with water (25 L) and MTBE (15 L), and the layers separated. The organic layer was washed with brine (7 L) and the aqueous layers then sequentially re-extracted with MTBE (8 L). The combined organics were evaporated and azeotroped with toluene (2 L) to afford crude compound 18. Chromatography (20 kg silica, 10-40% EtOAc in hexane) afforded compound 18 (3.4 kg, ~95 wt%, 11 .4 mol, 69%) as an orange oil.
Step 4:
Ammonia (3 kg, 167 mol) was bubbled in to cooled methanol (24 L) [temp kept <18°C]. A solution of compound 18 (4.8 kg, ~95 wt%, 16.1 mol) in methanol (1 .5 L) was added over 30 minutes and the mixture stirred at 25°C for 68 hours and then at 30°C for 24 hours. Two runs (from a total of 9.68 kg of ~95 wt% Step 3) were combined and concentrated to ~13 L volume. Water (30 L) was slowly added over 80 minutes, keeping the temperature 30 to 40°C. The resulting slurry was cooled to 20°C, filtered, washed with water (12 L) and pulled dry on the filter overnight. The solids were triturated in MTBE (8 L) and hexane (8 L) at 45°C then re-cooled to 15°C, filtered, washed with hexane (4 L) and dried under vacuum to afford compound 19 (7.95 kg, 29.6 mol, 90%) as an off-white solid.
Step 5:
To compound 19 (7.0 kg, 26.1 mol) in DCM (30 L) at 0°C was added triethylamine (5.85 kg, 57.8 mol). The mixture was further cooled to -6°C then trifluoroacetic anhydride (5.85 kg, 27.8 mol) added over 90 minutes [temp kept 0 to 5°C]. TLC assay showed the reaction was incomplete. Additional triethylamine (4.1 kg, 40.5 mol) and trifluoroacetic acid (4.1 kg, 19.5 mol) were added over 2 hours until TLC showed complete reaction. The reaction mixture was quenched in to water (40 L) [temp to 23°C]. The layers were separated and the aqueous re-extracted with DCM (8 L). The organic layers were sequentially washed with brine (7 L), filtered through a pad of silica (3 kg) and eluted with DCM (10 L). The filtrate was evaporated and chromatographed (9 kg silica, eluent 10-30% EtOAc in hexane). Product fractions were evaporated and azeotroped with IPA to afford compound 20 (6.86 kg, -94 wt%, 25.8 mol, 99%) as an orange oil.
Step 6:
To compound 20 (6.86 kg, -94 wt%, 25.8 mol) in IPA (35 L) at 17°C was added 37% hydrochloric acid (6.4 L, 77.4 mol). The mixture was heated to 35°C overnight then concentrated to a moist solid and residual water azeotroped with additional IPA (8 L). The resulting moist solid was triturated with MTBE (12 L) at 45°C for 30 minutes then cooled to 20°C and filtered, washing with MTBE (5 L). The solids were dried under vacuum at 45°C to afford compound 21 (4.52 kg, 24.2 mol, 94%) as a white solid. 1H-NMR was consistent with desired product; mp 203-205°C; HPLC 99.3%. 1H NMR (CD3OD, 400 MHz) δ 7.12 (1 H, s), 4.28 (2H, s), 4.09 (3H, s), 2.77 (3H, s). 13C NMR (CD3OD, 100 MHz) δ 144.5, 177.8, 1 14.9, 110.9, 45.9, 39.0, 33.2. LCMS (M++1) 151 .1 , 138.0, 120.0.
PATENT
WO2013132376
PATENT
WO 2016089208
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017021823&redirectedID=true
Preparation of the free base of lorlatinib as an amorphous solid is disclosed in
International Patent Publication No. WO 2013/132376 and in United States Patent No. 8,680,1 1 1 . Solvated forms of lorlatinib free base are disclosed in International Patent Publication No. WO 2014/207606.
Example 1
Lab Scale Preparation of Form 7 of (10 ?‘)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2/-/-8,4-(metheno)pyrazolo[4,3- ?l[2,5,1 l lbenzoxadiazacyclotetra-decine- -carbonitrile (lorlatinib) Free Base
[AcOH solvate]
Form 7 of lorlatinib free base was prepared by de-solvation of the acetic acid solvate of lorlatinib (Form 3), prepared as described in International Patent Publication No. WO 2014/207606, via an intermediate methanol solvate hydrate form of lorlatinib (Form 2).
The acetic acid solvate of lorlatinib (Form 3) (5 g, 10.72 mmol) was slurried in methanol
(10 mL/g, 1235.9 mmol) at room temperature in an Easymax flask with magnetic stirring to which triethylamine (1 .2 equiv., 12.86 mmol) was added over 10 minutes. The resulting solution was heated to 60°C and water (12.5 mL/g, 3469.3 mmol) was added over 10 minutes, while maintaining a temperature of 60°C. Crystallization was initiated by scratching the inside of the glass vessel to form a rapidly precipitating suspension which was triturated to make the system mobile. The suspension was then cooled to 25°C over 1 hour, then cooled to 5°C and granulated for 4 hours. The white slurry was filtered and washed with 1 mL/g chilled
water/methanol (1 :1) then dried under vacuum at 50°C overnight to provide the methanol solvate hydrate Form 2 of lorlatinib.
Form 7 was then prepared via a re-slurry of the methanol solvate hydrate Form 2 of lorlatinib in heptane. 100 mg of lorlatinib Form 2 was weighed into a 4-dram vial and 3 mL of heptane was added. The mixture was slurried at room temperature on a roller mixer for 2 hours. Form conversion was confirmed by PXRD revealing complete form change to Form 7 of lorlatinib free base.
Paper
http://pubs.acs.org/doi/abs/10.1021/jm500261q
*E-mail: ted.w.johnson@pfizer.com. Phone: (858) 526-4683., *E-mail: paul.f.richardson@pfizer.com. Phone: (858) 526-4290.

Although crizotinib demonstrates robust efficacy in anaplastic lymphoma kinase (ALK)-positive non-small-cell lung carcinoma patients, progression during treatment eventually develops. Resistant patient samples revealed a variety of point mutations in the kinase domain of ALK, including the L1196M gatekeeper mutation. In addition, some patients progress due to cancer metastasis in the brain. Using structure-based drug design, lipophilic efficiency, and physical-property-based optimization, highly potent macrocyclic ALK inhibitors were prepared with good absorption, distribution, metabolism, and excretion (ADME), low propensity for p-glycoprotein 1-mediated efflux, and good passive permeability. These structurally unusual macrocyclic inhibitors were potent against wild-type ALK and clinically reported ALK kinase domain mutations. Significant synthetic challenges were overcome, utilizing novel transformations to enable the use of these macrocycles in drug discovery paradigms. This work led to the discovery of 8k (PF-06463922), combining broad-spectrum potency, central nervous system ADME, and a high degree of kinase selectivity.
Discovery of (10R)-7-Amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a Macrocyclic Inhibitor of Anaplastic Lymphoma Kinase (ALK) and c-ros Oncogene 1 (ROS1) with Preclinical Brain Exposure and Broad-Spectrum Potency against ALK-Resistant Mutations
Ted W. Johnson*, Paul F. Richardson*, Simon Bailey, Alexei Brooun, Benjamin J. Burke, Michael R. Collins, J. Jean Cui, Judith G. Deal, Ya-Li Deng, Dac Dinh, Lars D. Engstrom, Mingying He, Jacqui Hoffman, Robert L. Hoffman, Qinhua Huang, Robert S. Kania, John C. Kath, Hieu Lam, Justine L. Lam, Phuong T. Le, Laura Lingardo, Wei Liu, Michele McTigue, Cynthia L. Palmer, Neal W. Sach, Tod Smeal, Graham L. Smith, Albert E. Stewart, Sergei Timofeevski, Huichun Zhu, Jinjiang Zhu, Helen Y. Zou, and Martin P. Edwards
La Jolla Laboratories, Pfizer Worldwide Research and Development, 10770 Science Center Drive, San Diego, California 92121, United States
J. Med. Chem., 2014, 57 (11), pp 4720–4744
DOI: 10.1021/jm500261q
(10R)-7-Amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]benzoxadiazacyclotetradecine-3-carbonitrile (8k)
white solid:
TLC Rf = 0.40 (70% EtOAc in cyclohexane);
LC–MS (ESI), m/z 407.1 [M + H]+;
1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 2.0 Hz, 1 H), 7.30 (dd, J = 9.6, 2.4 Hz, 1 H), 7.21 (dd, J = 8.4, 5.6 Hz, 1 H), 6.99 (dt, J = 8.0, 2.8 Hz, 1 H), 6.86 (d, J = 1.2 Hz, 1 H), 5.75–5.71 (m, 1 H), 4.84 (s, 2 H), 4.45 (d, J = 14.4 Hz, 1 H), 4.35 (d, J = 14.4 Hz, 1 H), 4.07 (s, 3 H), 3.13 (s, 3 H), 1.79 (d, J = 6.4 Hz, 3 H).
References
1H NMR PREDICT


13C NMR PREDICT


///////////////////Lorlatinib, PF-6463922, anti-neoplastic, Pfizer, ROS1, ALK, phase 2, UNII:OSP71S83EU, лорлатиниб , لورلاتينيب , 洛拉替尼 , Orphan Drug, PF 6463922
Fc2ccc3C(=O)N(C)Cc1nn(C)c(C#N)c1c4cc(O[C@H](C)c3c2)c(N)nc4

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....








































 Deregulation of the HER receptor family, comprising four related receptor tyrosine kinases (EGFR, HER2, HER3, and HER4), promotes proliferation, invasion, and tumor cell survival.Such deregulation has been observed in many human cancers, including lung, head and neck, and breast. Numerous small molecules have been investigated for inhibition of tyrosine kinases with the aminoquinazoline motif coming to the forefront as a privileged scaffold. Three of the clinically available treatments, gefitinib (1),lapatinib (2), and erlotinib (3),as well as the candidate drug dacomitinib (4), contain this arrangement
Deregulation of the HER receptor family, comprising four related receptor tyrosine kinases (EGFR, HER2, HER3, and HER4), promotes proliferation, invasion, and tumor cell survival.Such deregulation has been observed in many human cancers, including lung, head and neck, and breast. Numerous small molecules have been investigated for inhibition of tyrosine kinases with the aminoquinazoline motif coming to the forefront as a privileged scaffold. Three of the clinically available treatments, gefitinib (1),lapatinib (2), and erlotinib (3),as well as the candidate drug dacomitinib (4), contain this arrangement