
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
SIBUTRAMINE
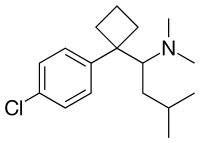
SIBUTRAMINE
- Molecular FormulaC17H26ClN
- Average mass279.848 Da
1-(4-Chlorophenyl)-N,N-dimethyl-a-(2-methylpropyl)cyclobutane methanamine
1-[1-(4-Chlorophenyl)cyclobutyl]-N,N,3-trimethyl-1-butanamine
106650-56-0[RN]
106650-56-0 (Sibutramine );
125494-59-9 (Sibutramine HCl Monohydrate);
84485-00-7 (Sibutramine HCl);
6124
UNII:WV5EC51866, WV5EC51866
сибутрамин[Russian]
سيبوترامين[Arabic]
西布曲明[Chinese]
Drug Name:Sibutramine Hydrochloride Hydrate
Research Code:BTS-54524
Trade Name:Meridia®
MOA:Serotonin-norepinephrine reuptake inhibitor
Indication:Obesity
Status:Withdrawn
Company:Abbott (Originator)
Sibutramine hydrochloride monohydrate, KES-524, BTS-54524, Meridia, Reductil
Sibutramine, formerly sold under the brand name Meridia among others, is an appetite suppressant which has been discontinued in many countries. Until 2010, it was widely marketed and prescribed as an adjunct in the treatment of obesity along with diet and exercise. It has been associated with increased cardiovascular events and strokes and has been withdrawn from the market in several countries and regions including Australia,[1] Canada,[2] China,[3] the European Union,[4] Hong Kong,[5] India,[6] Mexico, New Zealand,[7] the Philippines,[8] Thailand,[9] the United Kingdom,[10] and the United States.[11] However, the drug remains available in some countries.[12]
Sibutramine was originally developed in 1988 by Boots in Nottingham, UK,[13] and marketed by Knoll Pharmaceuticals after BASF/Knoll AG purchased the Boots Research Division in 1995, and was most recently manufactured and marketed by Abbott Laboratories before its withdrawal from most markets. It has been sold under a variety of brand names including Reductil, Meridia, Siredia, and Sibutrex. It is classified as a Schedule IV controlled substance in the United States.
Sibutramine hydrochloride hydrate was approved by the U.S. Food and Drug Administration (FDA) on Nov 16, 1997. It was developed and marketed as Meridia® by Abbott in the US.
Sibutramine hydrochloride hydrate is a serotonin-norepinephrine reuptake inhibitor, it produces its therapeutic effects by norepinephrine, serotonin and dopamine reuptake inhibition. Meridia® is indicated for the management of obesity, including weight loss and maintenance of weight loss, and should be used in conjunction with a reduced calorie diet.
Meridia® is available as capsule for oral use, containing 5, 10 or 15 mg of Sibutramine hydrochloride hydrate. The recommended dose is initiated at 10 mg once daily with or without food and may increase to 15 mg once daily.
Sibutramine has been withdrawn from the market in several countries and regions since 2010, owning to its side effect that associated with increased cardiovascular events and strokes.Route 1
Reference:1. US4746680A / US4806570A.
2. US4929629A.
SYN

SYN

SYN
PAT
https://patents.google.com/patent/KR20060019351A/en
Sibutramine hydrochloride (Sibutramine HCl: C 17 H 26 CIN HCl) is a chemical name {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl} -dimethylamine hydrochloride, and has the structure of Formula 1 .

Sibutramine was originally developed as a drug for the treatment of depression and was found to give weight to patients taking this drug. It was developed as an anti-obesity drug. Let your appetite decrease.
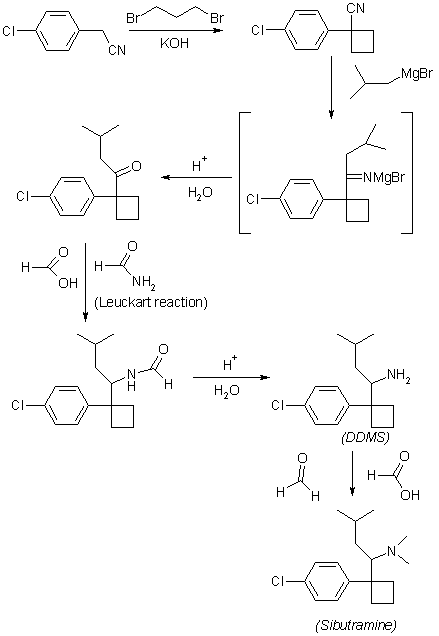
Korean Patent Publication No. 1990-274 (corresponding patent DE3212682 (Boots), filed Oct. 21, 1982; priority GB 1981.4.6.) For the preparation of 1- (1-arylcyclobutyl) alkylamine derivative comprising sibutramine It is described. The method for synthesizing sibutramine described in this document proceeds in a total of five steps as follows.
Step A

1- (4-chlorophenyl) -1-cyclobutyl cyanide is obtained from 4-Chlorobenzyl cyanide. Examples of the actual synthesis method described in this document are as follows.
After dissolving 25 g of 4-chlorobenzyl cyanide and 15 ml of 1,3-dibromopropane in 150 ml of DMSO, the solution was dissolved in nitrogen at room temperature (20-35 ° C.) and 7.5 g of NaH dispersed in mineral oil. And 200 ml of DMSO was added dropwise. The mixture was stirred at room temperature for 2 hours, 8 ml of IPA was added dropwise, and 110 ml of water was added dropwise. The mixture is filtered through CELITE ™ and the solid residue is washed with ether. The ether layer is separated, washed with water, dried, evaporated and vacuum distilled at high temperature to separate the desired 1- (4-chlorophenyl) -1-cyclobutyl cyanide. The total reaction time of this step is 5 hours and the yield from the starting material is 78%.
Step B

1- [1- (4-chlorophenyl) -cyanobutyl] -3-methyl-butan-1-one is obtained from 1- (4-chlorophenyl) -1-cyclobutyl cyanide. Specific synthesis examples are as follows. 35.2 g of 1- (4-chlorophenyl) -1-cyclobutyl cyanide are dissolved in 100 ml of ether and this solution is added to the product prepared by the reaction of 32 g of propylbromide and 6.36 g of magnesium. The ether is replaced with toluene and the mixture is heated under reflux for 1 hour. After adding water, concentrated hydrochloric acid is added, and the mixture is heated under reflux for 1 hour. The mixture obtained in the same manner as in the previous step was treated with ether, water, dried and evaporated and then vacuum distilled to give the desired 1- [1- (4-chlorophenyl) -cyanobutyl] -3-methyl-butan-1-one. To separate. The reaction time of this step is a total of 22 hours, the yield is 81%. The target product is bp 100-120 ° C / 0.2 mm / Hg.
Step C

N- {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl from 1- [1- (4-chlorophenyl) -cyanobutyl] -3-methyl-butan-1-one } Formamide is obtained. Specific synthesis examples are as follows. To 23.5 ml of formamide, 37 g (0.14 mol) of 1- [1- (4-chlorophenyl) -cyanobutyl] -3-methyl-butan-1-one and 9 ml of HCOOH were added dropwise at 160-170 ° C. The temperature is maintained at 175 ° C. to 180 ° C. for 24 hours. The mixture is extracted with ether and concentrated to afford an oil, which crystallizes the desired N- {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl} formamide from petroleum ether. The reaction time of this step is a total of 24 hours, the yield is 39%. The target is mp 110-112 ° C.
Step D

1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine hydrochloride from N- {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl} formamide Get Specific synthesis examples are as follows. 4 g of N- {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl} formamide, 25 ml of 2-methoxyethyl ether, 10 ml of water and 22 ml of concentrated hydrochloric acid were refluxed for 18 hours. Stir under. Dilute with water, wash with ether, and add 35 ml of 5M aqueous NaOH solution. After completion of the process by treatment with ether, water and brine, treated with magnesium sulfate, filtered and concentrated. The concentrated crude product is saturated with hydrochloric acid dissolved in 20 ml of ether. The resulting solid is filtered, concentrated and crystallized with petroleum ether to give the desired product 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine hydrochloride. The reaction time of this step is a total of 20 hours, the yield is 96% oil, 46% hydrochloride. The target product is mp 163-165 ° C.
Step E

The final target from 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine hydrochloride {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl} -dimethyl Amine hydrochloride, ie sibutramine hydrochloride, is obtained. Specific synthesis examples are as follows. 3.3 g of 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine hydrochloride, 2.99 g of HCOOH and 1 ml of water are mixed while cooling. 3.93 ml of 37% aqueous formaldehyde is added and heated at 85-95 ° C. for 18 hours. Excess hydrochloric acid is added and the solution is evaporated to dryness. 5N NaOH solution is added, extracted with ether and concentrated to give a pale yellow oil. This oil is dissolved in a mixture of 4 mL IPA, 20 mL ether and 2 mL hydrochloric acid is added dropwise. Concentrate, repeatedly dissolve in ethanol and concentrate again. Polishing with petroleum ether gives a yellow solid and recrystallizes with acetone to give the final target sibutramine hydrochloride. The reaction time of this step is a total of 18 hours, the yield is 80%. The target product is mp 195-197 ° C. The yield in 5 steps (A to E) is 18.9%.
As described above, the conventional sibutramine synthesis method has a total of five steps, which is complicated and takes a long time, and requires high temperature vacuum distillation (step A). Since the reaction proceeds at the high temperature of the raw material there was a problem that the yield is reduced. In fact, the synthesis was performed by applying the conventional sibutramine synthesis method, the total yield was very low as 18.9%.
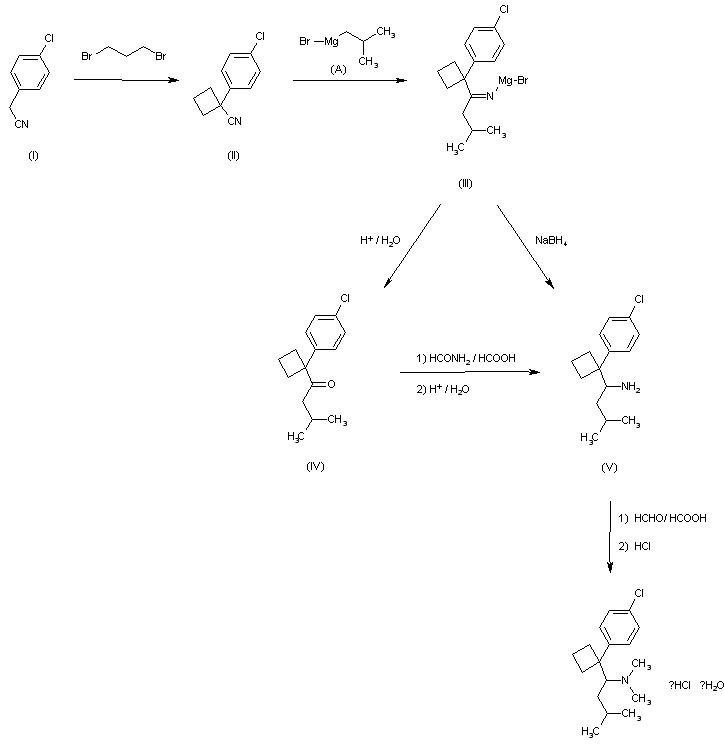
First step
4-Chlorobenzyl cyanide is reacted with 1,3-dibromopropane to give 1- (4-chlorophenyl) -1-cyclobutyl cyanide.

In a flask at room temperature (20-35 ° C.), 14.1 g (352 mmol) of NaH dispersed in mineral oil (200%) and 200 ml of DMSO were added. 25 g (160 mmol) of 4-chlorobenzyl cyanide and 1,3- A solution of 36 g (176 mmol) of dibromopropane dissolved in 200 ml of DMSO was added dropwise. The mixture is stirred at room temperature for 2 hours, 10 ml of IPA is added dropwise and 200 ml of water is added dropwise. The mixture is filtered through a CELITE ™ filter and the solid residue is washed with ether. The ether layer is separated, washed with water, filtered, concentrated and dried to give 34.72 g of crude product of 1- (4-chlorophenyl) -1-cyclobutyl cyanide. The yield of crude product is 109.8%.
2nd step
1- (4-chlorophenyl) -1-cyclobutyl cyanide isobutyl magnesium bromide is reacted to obtain 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine.

10 g (52 mmol) of 1- (4-chlorophenyl) -1-cyclobutyl cyanide was dissolved in 25 ml of toluene at room temperature, followed by addition of a 2.0 M solution of isobutyl magnesium bromide dissolved in 40 ml of diethyl ether. The mixture is heated at reflux at a temperature of at least 105 ° C. for 2 hours. After completion of the reaction at 0 ° C. with methanol, 2.4 g of NaBH 4 was added to the mixture at 0-25 ° C. and stirred for 1 hour or more. Concentrate, treat with ether, water, concentrate again, and vacuum dry. The yield of crude product is 91.6%.
3rd step
The amine group of 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine was dimethylated to obtain {1- [1- (4-chlorophenyl) -cyclobutyl]-which is the final object of the present invention. 3-methylbutyl} -dimethylamine hydrochloride, ie sibutramine hydrochloride, is obtained.

5.02 g of 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine and 10 ml of HCOOH are mixed with cooling. 6 ml of 37% aqueous formaldehyde is added and heated at 85-95 ° C. for 18 hours. Excess 2M HCl is added and the solution is evaporated to dryness. 5N NaOH solution is added, extracted with ether and concentrated to give a pale yellow oil. After dissolving in a small amount of ether, ether saturated with HCl gas is slowly added dropwise at 0 ° C. The solid obtained was filtered and dried in vacuo to give 5.12 g of sibutramine hydrochloride as the final target. Yield of the product is 92%, mp 193.5-194.8 ° C. The total yield of the first to third stages is 52.7%. The H 1 NMR results of the final product are as follows: H 1 NMR (CDCl 3 ) 1.058 (6H, dd), 1.400 (2H, m), 1.508 (2H, m), 2.193 (3H, d), 2.316 (2H , m), 2.784 (2H, m), 2.910 (3H, d), 2.967 (1H, m), 3.568 (1H, m), 7.386 (2H, d), 7.638 (2H, d), 10.771 (1H, s)
The present invention is to shorten the process that was conventionally carried out in five steps to three steps to greatly shorten the process as well as to eliminate the difficult and time-consuming high-temperature vacuum distillation process to enable mass production In addition, it is possible to greatly reduce production time and production costs by improving the process step by step, and to reduce the production cost by showing a yield improvement effect nearly three times that of the conventional synthesis method in terms of overall yield.
Claims (3)
Hide Dependent
- In the method for synthesizing {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutyl} -dimethylamine from 4-chlorobenzyl cyanide,(a) reacting 4-chlorobenzyl cyanide with 1,3-dibromopropane to obtain 1- (4-chlorophenyl) -1-cyclobutyl cyanide;(b) To isobutyl magnesium bromide dissolved in diethyl ether is added to 1- (4-chlorophenyl) -1-cyclobutyl cyanide dissolved in toluene, and the mixture is refluxed at a temperature of 105 ° C. or higher at 1-3 ° C. Heating and cooling for a period of time, followed by addition of NaBH 4 at 0-25 ° C., followed by stirring for at least 1 hour to obtain 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine;(c) Dimethylating the amine group of 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine to yield {1- [1- (4-chlorophenyl) -cyclobutyl] -3-methyl Improved synthesis method of sibutramine consisting of a three-step reaction comprising the step of obtaining butyl} -dimethylamine.
- The method according to claim 1,In step (a), the solution of 4-chlorobenzyl cyanide and 1,3-dibromopropane dissolved in DMSO is added dropwise to the mixture of NaH and DMSO dispersed in mineral oil, followed by filtration. , Washing, concentrating and drying to obtain a crude product of 1- (4-chlorophenyl) -1-cyclobutyl cyanide, and proceeding to the next step (b) as it is without distillation at high temperature. Improved Synthesis of Sibutramine.
- The method according to claim 1,In step (c), 1- [1- (4-chlorophenyl) -cyclobutyl] -3-methylbutylamine is mixed with formaldehyde in a free base state, and 37% aqueous formaldehyde is added thereto, and 85 An improved method for synthesizing sibutramine, characterized in that sibutramine hydrochloride is obtained by heating at -95 ° C for 15-22 hours followed by addition of hydrochloric acid.
SYN
DE 3212682; GB 2098602; US 4806570
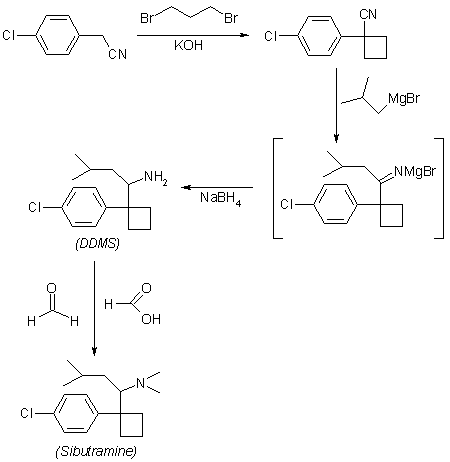
4-Chlorobenzyl cyanide (I) is cycloalkylated with 1,3-dibromopropane to yield 1-(4-chlorophenyl)cyclobutyl cyanide (II). The cyclobutyl cyanide (II) is treated with 2-methylpropyl magnesium bromide te give the imine salt (III), which may be either hydrolyzed to the ketone (IV), which is then formylaminated with formamide and formic acid and subsequently hydrolyzed, or reduced with sodium borohydride in ethanol to give 1-[1-(4-chlorophenyl)cyclobutyl]-3-methylbutylamine (V). Eschweiler-Clarke methylation and hydrochloride formation yield N-[1-[1-(4-chlorophenyl)cyclo butyl]-3-methylbutyl]-N,N-dimethylamine hydrochloride monohydrate
//////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
Sibutramine has been used to produce appetite suppression for the purpose of attaining weight loss in the treatment of patients with obesity.
Contraindications
Sibutramine is contraindicated in patients with:
- Psychiatric conditions as bulimia nervosa, anorexia nervosa, serious depression or pre-existing mania
- Patients with a history of or a predisposition to drug or alcohol abuse
- Hypersensitivity to the drug or any of the inactive ingredients
- Patients below 18 and above 65 years of age[14]
- Concomitant treatment with a MAO inhibitor, antidepressant or other centrally active drugs, particularly other anoretics
- History of peripheral arterial disease
- Hypertension that is not sufficiently controlled (e.g., >145/90 mmHg), caution in controlled hypertension
- Existing pulmonary hypertension
- Existing damage on heart valves, coronary heart disease, congestive heart failure, serious arrhythmias, previous myocardial infarction
- A history of coronary artery disease (e.g., angina, history of myocardial infarction), congestive heart failure, tachycardia, peripheral arterial occlusive disease, arrhythmia or cerebrovascular disease (stroke or transient ischemic attack (TIA))[14]
- Stroke or transient ischemic attack (TIA)
- Hyperthyroidism (overactive thyroid gland)
- Closed angle glaucoma
- Seizure disorders
- Enlargement of the prostate gland with urinary retention (relative contraindication)
- Pheochromocytoma
- Pregnant and lactating women (relative contraindication)
Side effects
A higher number of cardiovascular events has been observed in people taking sibutramine versus control (11.4% vs. 10.0%).[15] In 2010 the FDA noted the concerns that sibutramine increases the risk of heart attacks and strokes in patients with a history of cardiovascular disease.[15]
Frequently encountered side effects are: dry mouth, paradoxically increased appetite, nausea, strange taste in the mouth, upset stomach, constipation, trouble sleeping, dizziness, drowsiness, menstrual cramps/pain, headache, flushing, or joint/muscle pain.
In a 2016 Cochrane review sibutramine was found to substantially increase blood pressure and heart rate in some patients, in the updated review in 2021 sibutramine was not included since the drug had been withdrawn from the market.[16] When used, regular blood pressure monitoring needed to be performed.
The following side effects are infrequent but serious and require immediate medical attention: cardiac arrhythmias, paresthesia, mental/mood changes (e.g., excitement, restlessness, confusion, depression, rare thoughts of suicide).
Symptoms that require urgent medical attention are seizures, problems urinating, abnormal bruising or bleeding, melena, hematemesis, jaundice, fever and rigors, chest pain, hemiplegia, abnormal vision, dyspnea and edema.
Currently, no case of pulmonary hypertension has been noted. (Fenfluramine, of the 1990s “Fen-Phen” combo, forced excess release of neurotransmitters—a different action. Phentermine was uninvolved in the rare—but clinically significant—heart issues of fenfluramine.)
Interactions
Sibutramine has a number of clinically significant interactions. The concomitant use of sibutramine and monoamine oxidase inhibitors (MAOIs, such as selegiline) is not indicated, as it may increase the risk of serotonin syndrome, a somewhat rare but serious adverse drug reaction.[17] Sibutramine should not be taken within two weeks of stopping or starting an MAOI. Taking both sibutramine and certain medications used in the treatment of migraines—such as ergolines and triptans—as well as opioids, may also increase the risk for serotonin syndrome, as may the use of more than one serotonin reuptake inhibitor at the same time.[17]
The concomitant use of sibutramine and drugs which inhibit CYP3A4, such as ketoconazole and erythromycin, may increase plasma levels of sibutramine.[18] Sibutramine does not affect the efficacy of hormonal contraception.[17]
Pharmacology
Pharmacodynamics
| Compound | SERT | NET | DAT |
|---|---|---|---|
| Sibutramine | 298–2,800 | 350–5,451 | 943–1,200 |
| Desmethylsibutramine | 15 | 20 | 49 |
| (R)-Desmethylsibutramine | 44 | 4 | 12 |
| (S)-Desmethylsibutramine | 9,200 | 870 | 180 |
| Didesmethylsibutramine | 20 | 15 | 45 |
| (R)-Didesmethylsibutramine | 140 | 13 | 8.9 |
| (S)-Didesmethylsibutramine | 4,300 | 62 | 12 |
| Values are Ki (nM). |
Sibutramine is a serotonin–norepinephrine reuptake inhibitor (SNRI) that, in humans, reduces the reuptake of norepinephrine (by ~73%), serotonin (by ~54%), and dopamine (by ~16%),[21] thereby increasing the levels of these substances in synaptic clefts and helping enhance satiety; the serotonergic action, in particular, is thought to influence appetite. Older anorectic agents such as amphetamine and fenfluramine force the release of these neurotransmitters rather than affecting their reuptake.[22]
Despite having a mechanism of action similar to tricyclic antidepressants, sibutramine has failed to demonstrate antidepressant properties in animal studies. It was approved by the U.S. Food and Drug Administration (FDA) in November 1997[23] for the treatment of obesity.
Sibutramine is reported to be a prodrug to two active metabolites, desmethylsibutramine (M1; BTS-54354) and didesmethylsibutramine (M2; BTS-54505), with much greater potency as MRIs.[24][25]
Unlike other serotonergic appetite suppressants like fenfluramine, sibutramine and its metabolites have only low and likely inconsequential affinity for the 5-HT2B receptor.[21]
Pharmacokinetics
Sibutramine is well absorbed from the gastrointestinal tract (77%), but undergoes considerable first-pass metabolism, reducing its bioavailability. The drug itself reaches its peak plasma level after 1 hour and has also a half-life of 1 hour. Sibutramine is metabolized by cytochrome P450 isozyme CYP3A4 into two pharmacologically-active primary and secondary amines (called active metabolites 1 and 2) with half-lives of 14 and 16 hours, respectively. Peak plasma concentrations of active metabolites 1 and 2 are reached after three to four hours. The following metabolic pathway mainly results in two inactive conjugated and hydroxylated metabolites (called metabolites 5 and 6). Metabolites 5 and 6 are mainly excreted in the urine.
Chemistry
Sibutramine has usually been used in the form of the hydrochloride monohydrate salt.
Detection in body fluids
Sibutramine and its two active N-demethylated metabolites may be measured in biofluids by liquid chromatography–mass spectrometry. Plasma levels of these three species are usually in the 1–10 μg/L range in persons undergoing therapy with the drug. The parent compound and norsibutramine are often not detectable in urine, but dinorsibutramine is generally present at concentrations of >200 μg/L.[26][27][28]
Society and culture
Regulatory approval
Studies are ongoing into reports of sudden death, heart failure, renal failure and gastrointestinal problems. Despite a 2002 petition by Ralph Nader-founded NGO Public Citizen,[29] the FDA made no attempts to withdraw the drug, but was part of a Senate hearing in 2005.[30] Similarly, David Graham, FDA “whistleblower”, testified before a Senate Finance Committee hearing that sibutramine may be more dangerous than the conditions it is used for.[31]
Between January 2003 and November 2005, a large randomized-controlled “Sibutramine Cardiovascular OUTcomes” (SCOUT) study with 10,742 patients examined whether or not sibutramine administered within a weight management program reduces the risk for cardiovascular complications in people at high risk for heart disease and concluded that use of silbutramine had a RR 1.16 for the primary outcome (composit of nonfatal MI, nonfatal CVA, cardiac arrest, and CV death).[32]
In a dissenting article, “Sibutramine: gone, but not forgotten”, David Haslam (chairman of the National Obesity Forum) says that the SCOUT study is flawed as it only covered high-risk patients and did not consider obese patients who do not have cardiovascular complications or similar contraindications [33]
On January 21, 2010, the European Medicines Agency recommended suspension of marketing authorizations for sibutramine based on the SCOUT study results.[34]
In August 2010 the FDA added a new contraindication for patients over 65 years of age due to the fact that clinical studies of sibutramine did not include sufficient numbers of such patients.[14]
Abbott Laboratories announced on October 8, 2010 that it is withdrawing sibutramine from the US market under pressure from the FDA, citing concerns over minimal efficacy coupled with increased risk of adverse cardiovascular events.[35]
Counterfeit weight-loss products
On December 22, 2008, the United States Food and Drug Administration issued an alert to consumers naming 27 different products marketed as “dietary supplements” for weight loss, that illegally contain undisclosed amounts of sibutramine.[36][37] In March 2009, Dieter Müller et al. published a study of sibutramine poisoning cases from similar Chinese “herbal supplements” sold in Europe, containing as much as twice the dosage of the legally licensed drug.[38]
An additional 34 products were recalled by the FDA on April 22, 2009, further underscoring the risks associated with unregulated “herbal supplements” to unsuspecting persons. This concern is especially relevant to those with underlying medical conditions incompatible with undeclared pharmaceutical adulterants.[39] In January 2010, a similar alert was issued for counterfeit versions of the over-the-counter weight loss drug Alli sold over the Internet. Instead of the active ingredient orlistat, the counterfeit drugs contain sibutramine, and at concentrations at least twice the amount recommended for weight loss.[40]
In March 2010 Health Canada advised the public that illegal “Herbal Diet Natural” had been found on the market, containing sibutramine, which is a prescription drug in Canada, without listing sibutramine as an ingredient.[41] In October 2010 FDA notified consumers that “Slimming Beauty Bitter Orange Slimming Capsules contain the active pharmaceutical ingredient sibutramine, a prescription-only drug which is a stimulant. Sibutramine is not listed on the product label.”[42]
In October 2010 the MHRA in the UK issued a warning regarding “Payouji tea” and “Pai You Guo Slim Capsules” which were found to contain undeclared quantities of sibutramine.[43]
On December 30, 2010 the FDA released a warning regarding “Fruta Planta” dietary products, which were found to contain undeclared amounts of sibutramine. The recall stated that “there is NO SAFE formula on the US market and that all versions of Fruta Planta contain sibutramine. All versions of the formula are UNSAFE and should not be purchased from any source.”[44]
Some illegal weight loss products imported into Ireland have been found to contain sibutramine.[45][46] Similar concerns have been raised in Australia, where illegal imported supplements have been found to contain sibutramine, resulting in public alerts from Australia’s Therapeutic Goods Administration.[47]
In October 2011, the FDA warned that 20 brands of dietary supplements were tainted with sibutramine.[48] In a 2018 study FDA has found synthetic additives including sibutramine in over 700 diet supplements marketed as “natural”, “traditional” or “herbal remedies”.[49]
References
- ^ “Sibutramine (Reductil) – withdrawal in Australia”. Therapeutic Goods Administration (Tga). Therapeutic Goods Administration, Department of Health, Australian Government. 2010. Retrieved 2014-10-06.
- ^ Health Canada Endorsed Important Safety Information on MERIDIA (Sibutramine Hydrochloride Monohydrate): Subject: Voluntary withdrawal of Meridia (sibutramine) capsules from the Canadian market.
- ^ “Notification of Termination of Production, Sale, and Usage of Sibutramine Preparations and Their Active Pharmaceutical Ingredient”. sda.gov in People’s Republic of China. October 30, 2010. Retrieved 2011-05-21.
- ^ (in German) Sibutramin-Vertrieb in der Europäischen Union ausgesetzt [1]. Abbott Laboratories in Germany. Press Release 2010-01-21. Retrieved 2010-01-27
- ^ “De-registration of pharmaceutical products containing sibutramine” (Press release). info.gov in Hong Kong. November 2, 2010. Retrieved 2010-11-08.
- ^ “Banned Medicines” (Press release). Ministry of Health and Family Welfare. February 10, 2011. Retrieved 2011-03-15.
- ^ “Withdrawal of Sibutramine (Reductil) in New Zealand” (Press release). MedSafe in New Zealand. October 11, 2010. Retrieved 2012-11-06.
- ^ “FDA warns online sellers of banned slimming pills”. January 12, 2014. Retrieved February 20, 2014.
- ^ “Thai FDA reveals voluntary withdrawal of sibutramine from the Thai market” (PDF) (Press release). Food and Drug Administration of Thailand. October 20, 2010. Retrieved 2010-12-22.
- ^ “Top obesity drug sibutramine being suspended”. BBC News. 2010-01-22. Retrieved 2010-01-22.
- ^ Rockoff JD, Dooren JC (October 8, 2010). “Abbott Pulls Diet Drug Meridia Off US Shelves”. The Wall Street Journal. Retrieved 8 October 2010.
- ^ “Sibutramine – Drugs.com”. drugs.com.
- ^ Buckett WR, Thomas PC, Luscombe GP (1988). “The pharmacology of sibutramine hydrochloride (BTS 54 524), a new antidepressant which induces rapid noradrenergic down-regulation”. Progress in Neuro-Psychopharmacology & Biological Psychiatry. 12 (5): 575–84. doi:10.1016/0278-5846(88)90003-6. PMID 2851857. S2CID 24787523.
- ^ Jump up to:a b c “The FDA August 2010 drug safety update”. fda.gov.
- ^ Jump up to:a b “Early Communication about an Ongoing Safety Review of Meridia (sibutramine hydrochloride)”. United States Food and Drug Administration. 1 February 2010. Archived from the original on 6 January 2012.
- ^ Siebenhofer, Andrea; Winterholer, Sebastian; Jeitler, Klaus; Horvath, Karl; Berghold, Andrea; Krenn, Cornelia; Semlitsch, Thomas (2021-01-17). “Long-term effects of weight-reducing drugs in people with hypertension”. The Cochrane Database of Systematic Reviews. 1: CD007654. doi:10.1002/14651858.CD007654.pub5. ISSN 1469-493X. PMC 8094237. PMID 33454957.
- ^ Jump up to:a b c “Meridia Side Effects, and Drug Interactions”. RxList.com. 2007. Retrieved 2007-04-29.
- ^ (in Portuguese) Cloridrato de sibutramina monoidratado. Bula. [Sibutramine hydrochloride monohydrate—label information]. Medley (2007).
- ^ Nisoli E, Carruba MO (October 2000). “An assessment of the safety and efficacy of sibutramine, an anti-obesity drug with a novel mechanism of action”. Obesity Reviews. 1 (2): 127–39. doi:10.1046/j.1467-789x.2000.00020.x. PMID 12119986. S2CID 20553857.
- ^ Rothman RB, Baumann MH (May 2009). “Serotonergic drugs and valvular heart disease”. Expert Opinion on Drug Safety. 8 (3): 317–29. doi:10.1517/14740330902931524. PMC 2695569. PMID 19505264.
- ^ Jump up to:a b “Meridia (sibutramine hydrochloride monohydrate) Capsules CIV. Full Prescribing Information” (PDF). Abbott Laboratories, North Chicago, IL 60064, U.S.A. Retrieved 6 February 2016.
- ^ Heal DJ, Aspley S, Prow MR, Jackson HC, Martin KF, Cheetham SC (August 1998). “Sibutramine: a novel anti-obesity drug. A review of the pharmacological evidence to differentiate it from d-amphetamine and d-fenfluramine”. International Journal of Obesity and Related Metabolic Disorders. 22 Suppl 1: S18–28, discussion S29. PMID 9758240.
- ^ “FDA APPROVES SIBUTRAMINE TO TREAT OBESITY” (Press release). U.S. Food and Drug Administration. November 24, 1997. Retrieved 2007-04-29.
- ^ Kim KA, Song WK, Park JY (November 2009). “Association of CYP2B6, CYP3A5, and CYP2C19 genetic polymorphisms with sibutramine pharmacokinetics in healthy Korean subjects”. Clinical Pharmacology and Therapeutics. 86 (5): 511–8. doi:10.1038/clpt.2009.145. PMID 19693007. S2CID 24789264.
- ^ Hofbauer K (2004). Pharmacotherapy of obesity : options and alternatives. Boca Raton, Fla: CRC Press. ISBN 978-0-415-30321-7.
- ^ Jain DS, Subbaiah G, Sanyal M, et al. Liquid chromatography/electrospray ionization tandem mass spectrometry validated method for the simultaneous quantification of sibutramine and its primary and secondary amine metabolites in human plasma and its application to a bioequivalence study. Rapid Comm. Mass Spec. 20: 3509-3521, 2006.
- ^ Thevis M, Sigmund G, Schiffer AK, Schänzer W. Determination of N-desmethyl- and N-bisdesmethyl metabolites of Sibutramine in doping control analysis using liquid chromatography-tandem mass spectrometry. Eur. J. Mass Spec. 12: 129-136, 2006.
- ^ R. Baselt, Disposition of Toxic Drugs and Chemicals in Man, 8th edition, Biomedical Publications, Foster City, CA, 2008, pp. 1426–1427.
- ^ Wolfe SM, Sasich LD, Barbehenn E (March 19, 2002). “Petition to FDA to ban the diet drug sibutramine (MERIDIA) (HRG Publication #1613)”. Public Citizen. Retrieved 2007-04-29.
- ^ Japsen B (13 March 2005). “FDA weighs decision on Meridia; Health advisory likely for Abbott obesity drug”. Chicago Tribune. Chicago, Illinois. p. 1.
- ^ Hearing of 17 November 2004. Related CBS news item 19 November 2004.
- ^ James WP, Caterson ID, Coutinho W, Finer N, Van Gaal LF, Maggioni AP, Torp-Pedersen C, Sharma AM, Shepherd GM, Rode RA, Renz CL (September 2010). “Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects” (PDF). The New England Journal of Medicine. 363 (10): 905–17. doi:10.1056/NEJMoa1003114. hdl:2437/111825. PMID 20818901.
- ^ Haslam D (April 2010). “Sibutramine: gone, but not forgotten” (PDF). Pract Diab Int. 27 (3): 96–97. doi:10.1002/pdi.1453. Archived from the original (PDF) on 26 July 2015.
- ^ “European Medicines Agency recommends suspension of marketing authorisations for sibutramine” (PDF). European Medicines Agency. January 21, 2010. Archived from the original (PDF) on 2010-04-01.
- ^ Pollack A (October 8, 2010). “Abbott Labs Withdraws Meridia From Market”. The New York Times.
- ^ “FDA warns consumers about tainted weight loss pills” (Press release). U.S. Food and Drug Administration. 22 December 2008.
- ^ “Consumer directed questions and answers about FDA’s initiative against contaminated weight loss products”. U.S. Food and Drug Administration Center for Drug Evaluation and Research. 22 December 2008.
- ^ Müller D, Weinmann W, Hermanns-Clausen M (March 2009). “Chinese slimming capsules containing sibutramine sold over the Internet: a case series”. Deutsches Ärzteblatt International. 106 (13): 218–22. doi:10.3238/arztebl.2009.0218. PMC 2680571. PMID 19471631.
- ^ 34 weight loss products recalled, WebMD, 22 April 2009.
- ^ “Fake Alli diet pills can pose health risks”. CNN.com. January 23, 2010. Retrieved 2010-01-24.
- ^ “Herbal diet product poses heart risk”. CBC News. March 26, 2010.
- ^ “FDA Alert: Slimming Beauty Bitter Orange Slimming Capsules: Undeclared Drug Ingredient”. drugs.com.
- ^ “Press release: Warning over unlicensed herbal Payouji tea and Pai You Guo Slim Capsules”. United Kingdom Medicines & Healthcare Products Regulatory Agency. 20 October 2010. Archived from the original on 9 February 2012.
- ^ “PRock Marketing, LLC Issues a Voluntary Nationwide Recall of All weight loss formulas and variation of formulas of Reduce Weight Fruta Planta/Reduce Weight Dietary Supplement”. United States Food and Drug Administration. Archived from the original on 23 March 2012.
- ^ Pope C. “Seizures of illegal medicines rise”. The Irish Times.
- ^ “FDA Alert: Slim Xtreme Herbal Slimming Capsule: Undeclared Drug Ingredient”. drugs.com.
- ^ “Majestic slimming capsules: Safety advisory”. Therapeutic Goods Administration. Australian Government. 9 November 2012.
- ^ Carroll L (19 October 2011). “‘Natural’ diet pills tainted with banned prescription drug”. MSNBC. Archived from the original on 11 January 2012.
- ^ Cohen, Ronnie (12 October 2018). “No Wonder It Works So Well: There May Be Viagra In That Herbal Supplement”. NPR.org. Retrieved 2018-10-14.
External links
- Abbott Press Release on Meridia Withdrawal
- Sibutramine drug information from Merck Manual. Includes dosage information and a comprehensive list of international brand names
- U.S. Food and Drug Administration (FDA) product safety website
/////////////SIBUTRAMINE, UNII:WV5EC51866, WV5EC51866, сибутрамин , سيبوترامين , 西布曲明 , ABOTT, OBESITY

NEW DRUG APPROVALS
ONE TIME TO SUSTAIN THE BLOG SUBSCRIPTION
$10.00
Olipudase alfa
| HPLSPQGHPA RLHRIVPRLR DVFGWGNLTC PICKGLFTAI NLGLKKEPNV ARVGSVAIKL CNLLKIAPPA VCQSIVHLFE DDMVEVWRRS VLSPSEACGL LLGSTCGHWD IFSSWNISLP TVPKPPPKPP SPPAPGAPVS RILFLTDLHW DHDYLEGTDP DCADPLCCRR GSGLPPASRP GAGYWGEYSK CDLPLRTLES LLSGLGPAGP FDMVYWTGDI PAHDVWHQTR QDQLRALTTV TALVRKFLGP VPVYPAVGNH ESTPVNSFPP PFIEGNHSSR WLYEAMAKAW EPWLPAEALR TLRIGGFYAL SPYPGLRLIS LNMNFCSREN FWLLINSTDP AGQLQWLVGE LQAAEDRGDK VHIIGHIPPG HCLKSWSWNY YRIVARYENT LAAQFFGHTH VDEFEVFYDE ETLSRPLAVA FLAPSATTYI GLNPGYRVYQ IDGNYSGSSH VVLDHETYIL NLTQANIPGA IPHWQLLYRA RETYGLPNTL PTAWHNLVYR MRGDMQLFQT FWFLYHKGHP PSEPCGTPCR LATLCAQLSA RADSPALCRH LMPDGSLPEA QSLWPRPLFC (Disulfide bridge: 43-119, 46-111, 74-85, 175-180, 181-204, 339-385, 538-542, 548-561) |
Olipudase alfa
Xenpozyme, Japan 2022, APPROVALS 2022, 2022/3/28
PEPTIDE, オリプダーゼアルファ (遺伝子組換え)
Alternative Names: Acid sphingomyelinase Niemann Pick disease type B – Sanofi; Acid-sphingomyelinase – Sanofi; GZ-402665; Recombinant human acid sphingomyelinase – Sanofi; rhASM – Sanofi; Sphingomyelinase-C (synthetic human) – Sanofi; Synthetic human sphingomyelinase-C – Sanofi; Xenpozyme
| Formula | C2900H4373N783O791S24 |
|---|---|
| CAS | 927883-84-9 |
| Mol weight | 63631.0831 |
| Efficacy | Lysosomal storage disease treatment, Enzyme replacement (acid sphingomyelinase) |
|---|---|
| Comment | Enzyme replacement therapy product Treatment of Niemann-Pick disease type A/B |
- OriginatorGenzyme Corporation
- DeveloperSanofi
- ClassRecombinant proteins; Sphingomyelin phosphodiesterases
- Mechanism of ActionSphingomyelin-phosphodiesterase replacements
- Orphan Drug StatusYes – Niemann-Pick diseases
- RegisteredNiemann-Pick diseases
- 28 Mar 2022Registered for Niemann-Pick diseases (In adolescents, In children, In adults) in Japan (IV) – First global approval
- 09 Feb 2022FDA assigns PDUFA action date of (03/07/2022) for Olipudase alfa (In children, In adults) for Niemann-Pick diseases
- 09 Feb 2022Adverse e
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
CLIP
Olipudase Alfa Improves Lung Function, Spleen Volume in ASMD
Olipudase Alfa Improves Lung Function, Spleen Volume in ASMD
Olipudase alfa was associated with significant improvements in clinically relevant disease end points among patients with chronic visceral acid sphingomyelinase (ASM) deficiency (ASMD), according to results from the phase 2/3 ASCEND trial presented at the 17th Annual WORLDSymposium.
ASMD is a rare, debilitating lysosomal storage disease characterized by a deficiency of the enzyme acid sphingomyelinase, which results in the accumulation of sphingomyelin in various tissues of the body. Olipudase alfa is an investigational enzyme replacement therapy designed to replace deficient or defective ASM.
The multicenter, randomized, double-blind, placebo-controlled ASCEND trial evaluated the efficacy and safety of olipudase alfa in 36 adults with chronic visceral ASMD. Patients were randomly assigned 1:1 to receive olipudase alfa 3mg/kg intravenously every 2 weeks or placebo for 52 weeks. The coprimary end points were the percent change in spleen volume and percent-predicted diffusing capacity of the lung for carbon monoxide (DLCO).
At week 52, treatment with olipudase alfa resulted in a 39.45% reduction in spleen volume, compared with a 0.5% increase for placebo (P <.0001). A decrease in spleen volume of at least 30% was observed in 17 patients (94%) treated with olipudase afla compared with no patients treated with placebo. Additionally, olipudase alfa significantly improved lung function by 22% from baseline compared with 3% for patients receiving placebo (P =.0004), as measured by percent predicted DLCO.
Olipudase alfa also met key secondary end points including a 31.7% reduction in liver volume (vs a 1.4% reduction for placebo; P <.0001) and a 16.8% improvement in mean platelet counts (vs 2.5% with placebo; P =.019) at week 52. Significant improvements in HDL, LDL, AST, ALT, chitotriosidase (54% vs 12% with placebo; P =.0003), and lyso-sphingomyelin (78% vs 6% with placebo) were also observed in the olipudase alfa group at week 52.
With regard to Splenomegaly Related Score, a patient-reported outcome measurement that evaluates patient symptoms associated with an enlarged spleen, findings showed no meaningful difference between olipudase alfa and placebo (-8 point vs -9.3 points, respectively).
As for safety, olipudase alfa was well tolerated with most adverse events being mild to moderate in severity. There were no treatment-related serious adverse events and no adverse event-related discontinuations.
Disclosure: Some authors have declared affiliations with or received funding from the pharmaceutical industry. Please refer to the original study for a full list of disclosures.
Reference
Wasserstein M, Arash-Kaps L, Barbato A, et al. Adults with chronic acid sphingomyelinase deficiency show significant visceral, pulmonary, and hematologic improvements after enzyme replacement therapy with olipudase-alfa: 1-year results of the ASCEND placebo-controlled trial. Presented at: 17th Annual WORLDSymposium; February 8-12, 2021. Abstract 265.
CLIP
https://www.sanofi.com/en/media-room/press-releases/2021/2021-12-06-14-00-00-2346501
EMA accepts regulatory submission for olipudase alfa, the first potential therapy for ASMD
- Olipudase alfa has been granted PRIority MEdicines (PRIME) designation in Europe, Breakthrough Therapy designation in the United States, and SAKIGAKE designation in Japan
- European regulatory decision anticipated second half of 2022
DECEMBER 6, 2021
The European Medicines Agency (EMA) has accepted for review under an accelerated assessment procedure the Marketing Authorization Application (MAA) for olipudase alfa, Sanofi’s investigational enzyme replacement therapy which is being evaluated for the treatment of acid sphingomyelinase deficiency (ASMD). Historically referred to as Niemann-Pick disease (NPD) type A and type B, ASMD is a rare, progressive, and potentially life-threatening disease for which no treatments are currently approved. The estimated prevalence of ASMD is approximately 2,000 patients in the U.S., Europe (EU5 Countries) and Japan. If approved, olipudase alfa will become the first and only therapy for the treatment of ASMD.
“Today’s milestone has been decades in the making and our gratitude goes to the ASMD community who has stood by us with endless patience while olipudase alfa advanced through clinical development,” said Alaa Hamed, MD, MPH, MBA, Global Head of Medical Affairs, Rare Diseases, Sanofi. “Olipudase alfa represents the kind of potentially life-changing innovation that is possible when industry, medical professionals and the patient community work together toward a common goal.”
The MAA is based on positive results from two separate clinical trials (ASCEND and ASCEND-Peds) evaluating olipudase alfa in adult and pediatric patients with non-central nervous system (CNS) manifestations of ASMD type A/B and ASMD type B.
Olipudase alfa has received special designations from regulatory agencies worldwide, recognizing the innovation potential of the investigational therapy.
“Scientific innovation is the greatest source of hope for people living with diseases like ASMD where there are no approved treatments and is a critical component for ensuring a viable healthcare ecosystem,” said Bill Sibold, Executive Vice President of Sanofi Genzyme. “At Sanofi, we have a long history of pioneering scientific innovation, and we remain committed to finding solutions to address unmet medical needs, including those of the rare disease community.”
The EMA awarded olipudase alfa the PRIority MEdicines designation, also known as PRIME, intended to aid and expedite the regulatory process for investigational medicines that may offer a major therapeutic advantage over existing treatments, or benefit patients without treatment options.
The U.S. Food and Drug Administration (FDA) has granted Breakthrough Therapy designation to olipudase alfa. This designation is intended to expedite the development and review of drugs intended to treat serious or life-threatening diseases and conditions. The criteria for granting Breakthrough Therapy designation include preliminary clinical evidence indicating that the molecule may demonstrate substantial improvement on a clinically significant endpoint over available therapies.
In Japan, olipudase alfa was awarded the SAKIGAKE designation, which is intended to promote research and development in Japan for innovative new medical products that satisfy certain criteria, such as the severity of the intended indication. In September, Sanofi filed the J-NDA submission for olipudase alfa.
About ASMD
ASMD results from a deficient activity of the enzyme acid sphingomyelinase (ASM), which is found in special compartments within cells called lysosomes and is required to breakdown lipids called sphingomyelin. If ASM is absent or not functioning as it should, sphingomyelin cannot be metabolized properly and accumulates within cells, eventually causing cell death and the malfunction of major organ systems. The deficiency of the lysosomal enzyme ASM is due to disease-causing variants in the sphingomyelin phosphodiesterase 1 gene (SMPD1). The estimated prevalence of ASMD is approximately 2,000 patients in the U.S., Europe (EU5 Countries) and Japan.
ASMD represents a spectrum of disease caused by the same enzymatic deficiency, with two types that may represent opposite ends of a continuum sometimes referred to as ASMD type A and ASMD type B. ASMD type A is a rapidly progressive neurological form of the disease resulting in death in early childhood due to central nervous system complications. ASMD type B is a serious and potentially life-threatening disease that predominantly impacts the lungs, liver, and spleen, as well as other organs. ASMD type A/B represents an intermediate form that includes varying degrees of neurologic involvement. Patients with ASMD type A/B or ASMD type B were studied in the ASCEND trial program. Another type of NPD is NPD type C, which is unrelated to ASMD.
About olipudase alfa
Olipudase alfa is an investigational enzyme replacement therapy designed to replace deficient or defective ASM, allowing for the breakdown of sphingomyelin. Olipudase alfa is currently being investigated to treat non-CNS manifestations of ASMD. Olipudase alfa has not been studied in ASMD type A patients. Olipudase alfa is an investigational agent and the safety and efficacy have not been evaluated by the FDA, EMA, or any other regulatory authority worldwide.
About Sanofi
Sanofi is dedicated to supporting people through their health challenges. We are a global biopharmaceutical company focused on human health. We prevent illness with vaccines, provide innovative treatments to fight pain and ease suffering. We stand by the few who suffer from rare diseases and the millions with long-term chronic conditions.
With more than 100,000 people in 100 countries, Sanofi is transforming scientific innovation into healthcare solutions around the globe.
///////Olipudase alfa, japan 2022, APPROVALS 2022, Xenpozyme, PEPTIDE, オリプダーゼアルファ (遺伝子組換え) , ORPHAN DRUG, GZ-402665 , GZ 402665

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
Andexanet alfa
(heavy chain)
IVGGQECKDG ECPWQALLIN EENEGFCGGT ILSEFYILTA AHCLYQAKRF KVRVGDRNTE
QEEGGEAVHE VEVVIKHNRF TKETYDFDIA VLRLKTPITF RMNVAPACLP ERDWAESTLM
TQKTGIVSGF GRTHEKGRQS TRLKMLEVPY VDRNSCKLSS SFIITQNMFC AGYDTKQEDA
CQGDAGGPHV TRFKDTYFVT GIVSWGEGCA RKGKYGIYTK VTAFLKWIDR SMKTRGLPKA
KSHAPEVITS SPLK
(light chan)
ANSFLFWNKY KDGDQCETSP CQNQGKCKDG LGEYTCTCLE GFEGKNCELF TRKLCSLDNG
DCDQFCHEEQ NSVVCSCARG YTLADNGKAC IPTGPYPCGK QTLER
(Disulfide bridge: H7-H12, H27-H43, H108-L98, H156-H170, H181-H209, L16-L27, L21-L36, L38-L47, L55-L66, L62-L75, L77-L90)
Andexanet alfa
JAPAN 2022, PEPTIDE
| Ondexxya |
| 2022/3/28 |
| Anticoagulant reversal (factor Xa inhibitors) |
CAS: 1262449-58-0
アンデキサネットアルファ (遺伝子組換え)
- Andexanet alfa
- r-Antidote
- rfXa Inhibitor Antidote
- PRT-4445
- PRT064445
Andexanet alfa, sold under the trade name Andexxa among others, is an antidote for the medications rivaroxaban and apixaban, when reversal of anticoagulation is needed due to uncontrolled bleeding.[1] It has not been found to be useful for other factor Xa inhibitors.[2] It is given by injection into a vein.[2]
Common side effects include pneumonia and urinary tract infections.[2] Severe side effects may include blood clots, heart attacks, strokes, or cardiac arrest.[2] It works by binding to rivaroxaban and apixaban.[2]
It was approved for medical use in the United States in May 2018.[1] It was developed by Portola Pharmaceuticals.[3]
ndexanet alfa is a recombinant human coagulation Factor Xa that promotes blood coagulation. It was developed by Portola Pharmaceuticals and was approved in in May 2018. It is marketed as Andexxa for intravenous injection or infusion and is indicated for the reversal of anticoagulation in combination with rivaroxaban and apixaban in cases of life-threatening or uncontrolled bleeding. Rivaroxaban and apixaban are Factor Xa inhibitors that promote anticoagulation in situations where blood clotting is unfavourable, such as in deep vein thrombosis and pulmonary embolism. However, the use of these agents is associated with a risk for uncontrollable bleeding episodes that can lead to can cause serious or fatal bleeding. Andexanet alfa is currently under regulatory review by the European Union and is undergoing clinical development in Japan 1.
Andexanet alfa works by binding to Factor Xa inhibitors and prevent them from interacting with endogenous Factor Xa. It displayed high affinity (0.53–1.53 nmol/L) to apixaban, betrixaban, edoxaban and rivaroxaban 1. However, the effectiveness of andexanet alfa on treating bleeding related to any FXa inhibitors other than apixaban and rivaroxaban was not demonstrated, thus such use is limited 7. Its pharmacokinetic properties are not reported to be affected by factor Xa inhibitors 1. Andexanet alfa retains the structural similarity to that of endogenous human factor Xa, but exists in its mature functional form without the need for activation via the intrinsic or extrinsic coagulation pathways 5 and remains catalytically inactive due to structural modification 1. The procoagulation potential of andexanet alfa is eliminated through the removal of a 34-residue fragment containing Gla: via this truncation, andexanet alfa is unable to bind to membrane surfaces and assemble the prothrombinase complex 5. It also prevents andexanet alfa from taking up space on phospholipid surface membranes, so that native FXa may bind and assemble the prothrominase complex 5. The amino acid residue modification from serine to alanine in the binding site of the catalytic domain allows more effective binding to FXa inhibitors and deters the andexanet alfa from converting prothrombin to thrombin 5.
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////

Structure of andexant alfa. Andexanet alfa is a modified activated human factor Xa (FXa) that binds FXa with high affinity and a 1:1 stoichiometric ratio but does not have intrinsic catalytic activity (the amino acid serine at position 419 is replaced by alanine) and lacks the membrane-binding-carboxyglutamic acid domain (Gla domain) of native FX. The Gla domains are responsible for the binding of FXa to phospholipids
Medical uses
Andexanet alfa is used to stop life threatening or uncontrollable bleeding in people who are taking rivaroxaban or apixaban.[1]
There are no randomised clinical trials as of 2019. Studies in healthy volunteers show that the molecule binds factor Xa inhibitors and counters their anti-Xa-activity.[4] The only published clinical trial is a prospective, open label, single group study.[5] This study reports results on 352 people and demonstrates a reduction of anti-Xa-activity while also showing an excellent or good hemostatic efficacy in 82%. While people who were expected to die in 30 days were excluded from the study, 14% of participants died. There was no relationship between hemostatic efficacy and reduced anti-Xa-activity.[6] The FDA has demanded a randomised clinical trial: the first results are not expected before 2023.[7]
Adverse effects
Common side effects include pneumonia and urinary tract infections.[2] Severe side effects may include blood clots or cardiac arrest.[2]
Andexanet alfa has a boxed warning that it is associated with arterial and venous blood clots, ischemic events, cardiac arrest, and sudden deaths.[1]
Pharmacology
Mechanism of action
Andexanet alfa is a biologic agent, a recombinant modified version of human activated factor X (FXa).[8] Andexanet alfa differs from native FXa due to the removal of a 34 residue fragment that contains the Gla domain. This modification reduces andexanet alfa’s procoagulant potential. Additionally, a serine to alanine (S419A) mutation in the active site eliminates its activity as a prothrombin to thrombin catalyst, but still allows the molecule to bind to FXa inhibitors.[9] FXa inhibitors bind to andexanet alfa with the same affinity as to natural FXa. As a consequence in the presence of andexanet alfa natural FXa is partially freed, which can lead to effective hemostasis.[3][10] In other words, it acts as a decoy receptor. Andexanet alfa reverses effect of all anticoagulants that act directly through FXa or by binding antithrombin III. The drug is not effective against factor IIa inhibitor dabigatran.[11]
History[edit]
It was approved in the United States in 2018 based on data from two phase III studies on reversing the anticoagulant activity of FXa inhibitors rivaroxaban and apixaban in healthy volunteers.[4] As a condition of its accelerated approval there is a study being conducted comparing it to other currently used reversal agents (“usual care”).[5][12]
Society and culture
Economics
Initial pricing (AWP) is $58,000 per reversal (800 mg bolus + 960 mg infusion, $3,300 per 100 mg vial) which is higher than reversal agents for other DOAC agents (idarucizumab for use in dabigatran reversal is $4,200 per reversal).[13]
References
- ^ Jump up to:a b c d e “Andexxa- andexanet alfa injection, powder, lyophilized, for solution”. DailyMed. 21 September 2020. Retrieved 12 November 2020.
- ^ Jump up to:a b c d e f g “Andexxa Monograph for Professionals”. Drugs.com. Retrieved 19 December 2018.
- ^ Jump up to:a b Dolgin E (March 2013). “Antidotes edge closer to reversing effects of new blood thinners”. Nature Medicine. 19 (3): 251. doi:10.1038/nm0313-251. PMID 23467222. S2CID 13340319.
- ^ Jump up to:a b Siegal DM, Curnutte JT, Connolly SJ, Lu G, Conley PB, Wiens BL, Mathur VS, Castillo J, Bronson MD, Leeds JM, Mar FA, Gold A, Crowther MA (December 2015). “Andexanet Alfa for the Reversal of Factor Xa Inhibitor Activity”. New England Journal of Medicine. 373 (25): 2413–24. doi:10.1056/NEJMoa1510991. PMID 26559317.
- ^ Jump up to:a b Connolly SJ, Crowther M, Eikelboom JW, Gibson CM, Curnutte JT, Lawrence JH, et al. (April 2019). “Full Study Report of Andexanet Alfa for Bleeding Associated with Factor Xa Inhibitors”. New England Journal of Medicine. 380 (14): 1326–1335. doi:10.1056/NEJMoa1814051. PMC 6699827. PMID 30730782.
- ^ Justin Morgenstern, “Andexanet Alfa: More garbage science in the New England Journal of Medicine”, First10EM blog, February 11, 2019. Available at: https://first10em.com/andexanet-alfa/.
- ^ “A Randomized Clinical Trial of Andexanet Alfa in Acute Intracranial Hemorrhage in Patients Receiving an Oral Factor Xa Inhibitor”. 11 January 2022.
- ^ Lu, Genmin; DeGuzman, Francis R.; Lakhotia, Sanjay; Hollenbach, Stanley J.; Phillips, David R.; Sinha, Uma (2008-11-16). “Recombinant Antidote for Reversal of Anticoagulation by Factor Xa Inhibitors”. Blood. 112 (11): 983. doi:10.1182/blood.V112.11.983.983. ISSN 0006-4971.
- ^ Kaatz, Scott; Bhansali, Hardik; Gibbs, Joseph; Lavender, Robert; Mahan, Charles E.; Paje, David G. (2017-09-13). “Reversing factor Xa inhibitors – clinical utility of andexanet alfa”. Journal of Blood Medicine. 8: 141–149. doi:10.2147/JBM.S121550. PMC 5602457. PMID 28979172.
- ^ Lu G, Deguzman FR, Hollenbach SJ, et al. (March 2013). “A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa”. Nature Medicine. 19 (4): 446–51. doi:10.1038/nm.3102. PMID 23455714. S2CID 11235887.
- ^ H. Spreitzer (23 December 2013). “Neue Wirkstoffe – Andexanet Alfa”. Österreichische Apothekerzeitung (in German) (26/2013): 40.
- ^ “Trial of Andexanet in ICH Patients Receiving an Oral FXa Inhibitor”. ClinicalTrials.gov. 11 January 2022.
- ^ “Lexi Comp Drug Information Online”. 24 May 2018.
Further reading
- Connolly SJ, Milling TJ, Eikelboom JW, Gibson CM, Curnutte JT, Gold A, et al. (September 2016). “Andexanet Alfa for Acute Major Bleeding Associated with Factor Xa Inhibitors”. New England Journal of Medicine. 375 (12): 1131–41. doi:10.1056/NEJMoa1607887. PMC 5568772. PMID 27573206.
External links
- “Andexanet alfa”. Drug Information Portal. U.S. National Library of Medicine.
- “Ondexxya (andexanet alfa): Avoid use of andexanet prior to heparinization”. European
| Clinical data | |
|---|---|
| Trade names | Andexxa, Ondexxya, others |
| Other names | Coagulation factor Xa (recombinant), inactivated-zhzo, PRT06445, r-Antidote, PRT4445 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Andexanet_alfa |
| Routes of administration | Intravenous injection |
| ATC code | V03AB38 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only)US: ℞-only [1]EU: Rx-only |
| Pharmacokinetic data | |
| Metabolism | Not studied |
| Elimination half-life | 5 h to 7 h |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1262449-58-0 |
| IUPHAR/BPS | 7576 |
| DrugBank | DB14562 |
| ChemSpider | none |
| UNII | BI009E452R |
| KEGG | D11029 |
| ChEMBL | ChEMBL3301583 |
//////////Andexanet alfa, JAPAN 2022, APPROVALS 2022, アンデキサネットアルファ (遺伝子組換え) , Ondexxya , PRT-4445, PRT064445

NEW DRUG APPROVALS
$10.00
Ferric derisomaltose

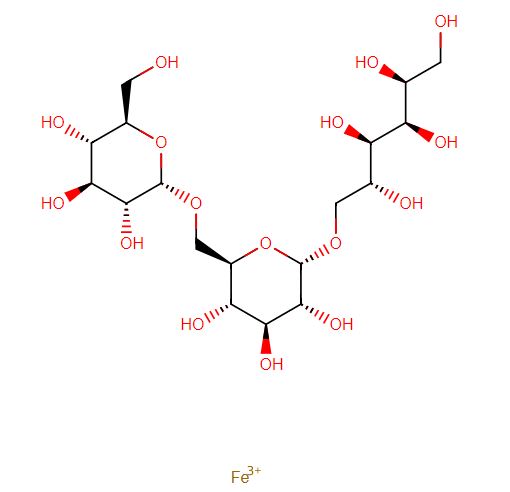
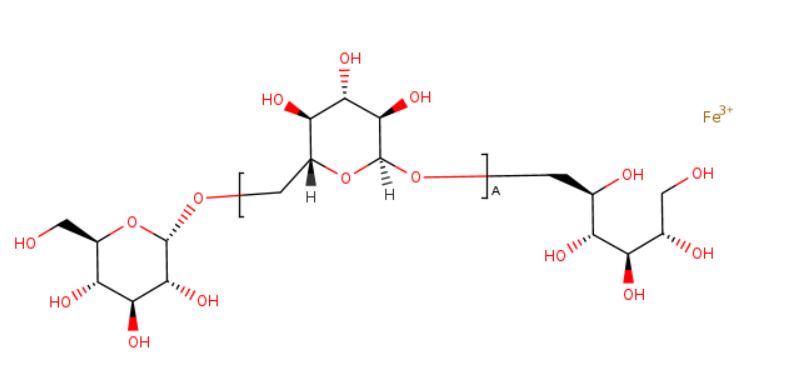
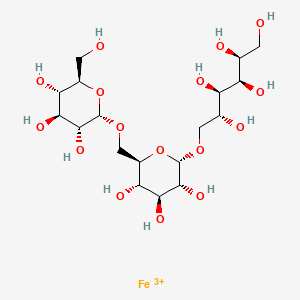
Ferric derisomaltose
WeightAverage: 562.297
Monoisotopic: 562.117975Chemical FormulaC18H34FeO16
Monover, JAPAN 2022, 2022/3/28
Monoferric (TN);
Monover (TN)
Anti-anemic, Hematinic, Supplement (iron)
CAS 1345510-43-1
デルイソマルトース第二鉄
- NS32
- WHO 9712
- UNII-AHU547PI9H
| Originator Company | Pharmacosmos |
| Active Companies | Nippon Shinyaku Co Ltd;Pharmacosmos A/S;Wasserburger Arzneimittelwerk Gmbh;Zealand University Hospital |
iron(3+) (2S,3R,4R,5R)-6-{[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-({[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy}methyl)oxan-2-yl]oxy}hexane-1,2,3,4,5-pentol
- alpha-D-Glucan, (1-6)-, reduced, reaction products with iron hydroxide (Fe(OH)3)
Ferric derisomaltose is an iron injection used in the treatment of iron deficiency anemia.
Ferric derisomaltose, sold under the brand name Monoferric, is a medication for the treatment of iron deficiency anemia (IDA) in adults who have intolerance to oral iron or have had unsatisfactory response to oral iron or who have non-hemodialysis dependent chronic kidney disease (NDD-CKD).[1] It was approved for use in the United States in January 2020.[1][2][3] It is given intravenously.[1]
Iron deficiency is an extremely common condition and is the most frequent cause of anemia worldwide. Iron deficiency results when iron intake, iron stores, and loss of iron from the body do not adequately support production of erythrocytes, also known as red blood cells. Though it is generally considered non life-threatening, iron deficiency may considerably affect quality of life.3
Ferric derisomaltose is a form of iron used in the treatment of iron deficiency. This drug is a complex of iron (III) hydroxide and derisomaltose. The latter is an iron carbohydrate oligosaccharide that works to release iron. Ferric derisomaltose was developed by Pharmacosmos Therapeutics ad was granted FDA approval in January 2020.8,9 Clinical trials show that it is non-inferior to iron sucrose, another form of iron that is often administered in iron deficiency, and less likely to cause serious hypersensitivity that is associated with other forms of injectable iron.1,4
This drug is indicated for the treatment of iron deficiency anemia in adult patients who have experienced intolerance to oral iron preparations or insufficient clinical response to orally administered iron. Ferric derisomaltase is also indicated for patients with non-hemodialysis dependent chronic kidney disease.8 In Australia and United Kingdom, ferric derisomaltase is indicated for cases in which rapid delivery of iron is required.10,11
Iron deficiency is an extremely common condition and is the most frequent cause of anemia worldwide. Iron deficiency results when iron intake, iron stores, and loss of iron from the body do not adequately support production of erythrocytes, also known as red blood cells. Though it is generally considered non life-threatening, iron deficiency may considerably affect quality of life. Ferric derisomaltose is a form of iron used in the treatment of iron deficiency. This drug is a complex of iron (III) hydroxide and derisomaltose. The latter is an iron carbohydrate oligosaccharide that works to release iron. Ferric derisomaltose was developed by Pharmacosmos Therapeutics ad was granted FDA approval in January 2020. Clinical trials show that it is non-inferior to [iron sucrose], another form of iron that is often administered in iron deficiency, and less likely to cause serious hypersensitivity that is associated with other forms of injectable iron.
Monoferric is an iron replacement product containing ferric derisomaltose for intravenous infusion. Ferric derisomaltose is an iron carbohydrate complex with a matrix structure composed of interchanging layers of ferric hydroxide and the carbohydrate derisomaltose. Derisomaltose consists of linear, hydrogenated isomaltooligosaccharides with an average molecular weight of 1000 Da and a narrow molecular weight distribution that is almost devoid of mono-and disaccharides.
Ferric derisomaltose has an average molecular weight of 155,000 Da and has the following empirical formula:
{FeO(1-3X) (OH)(1+3X) (C6H5O73-)X}, (H20)T, –
(C6H10O6)R(-C6H10O5-)Z(C6H13O5)R, (NaCl)Y
X= 0.0311; T = 0.25; R = 0.14; Z = 0.49; Y = 0.14
Iron atoms placed in the electronegative cavities of the 3-D structure between and within the derisomaltose molecules. A schematic representation is presented below
 |
Monoferric is a sterile, dark brown, non-transparent aqueous solution with pH 5.0-7.0, containing ferric derisomaltose dissolved in water for injections and filled into Type I glass vials.
Each 1 mL of solution contains 100 mg of elemental iron as ferric derisomaltose in water for injection.
Each 1 mL of solution contains 100 mg of elemental iron as ferric derisomaltose in water for injection.
| Mkt. Status | Active Ingredient | Proprietary Name | Appl. No. | Dosage Form | Route | Strength | TE Code | RLD | RS | Applicant Holder |
|---|---|---|---|---|---|---|---|---|---|---|
| RX | FERRIC DERISOMALTOSE | MONOFERRIC | N208171 | SOLUTION | INTRAVENOUS | 1GM/10ML (100MG/ML) | RLD | RS | PHARMACOSMOS AS | |
| DISCN | FERRIC DERISOMALTOSE | MONOFERRIC | N208171 | SOLUTION | INTRAVENOUS | 100MG/ML (100MG/ML) | RLD | PHARMACOSMOS AS | ||
| DISCN | FERRIC DERISOMALTOSE | MONOFERRIC | N208171 | SOLUTION | INTRAVENOUS | 500MG/5ML (100MG/ML) | RLD | PHARMACOSMOS AS | ||
| Mkt. Status | Active Ingredient | Proprietary Name | Appl. No. | Dosage Form | Route | Strength | TE Code | >RLD | RS | Applicant Holder |
MONOFERRIC (FERRIC DERISOMALTOSE)
1GM/10ML (100MG/ML)
Marketing Status: Prescription
Active Ingredient: FERRIC DERISOMALTOSE
Proprietary Name: MONOFERRIC
Dosage Form; Route of Administration: SOLUTION; INTRAVENOUS
Strength: 1GM/10ML (100MG/ML)
Reference Listed Drug: Yes
Reference Standard: Yes
TE Code:
Application Number: N208171
Product Number: 003
Approval Date: Jan 16, 2020
Applicant Holder Full Name: PHARMACOSMOS AS
Marketing Status: Prescription
Patent and Exclusivity Information
Patent and Exclusivity for: N208171
Product 003
FERRIC DERISOMALTOSE (MONOFERRIC) SOLUTION 1GM/10ML (100MG/ML)
Patent Data
| Product No | Patent No | Patent Expiration | Drug Substance | Drug Product | Patent Use Code | Delist Requested | Submission Date |
|---|---|---|---|---|---|---|---|
| 003 | 8815301 | 08/14/2029 | DS | DP | U-2734 | 02/14/2020 | |
| 003 | 10414831 | 03/25/2029 | DS | DP | 02/14/2020 |
PATENT
AU2009342799B2
US10414831B2
US2012010166A1
US2014303364A1
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Jump up to:a b c d “Monoferric- ferric derisomaltose solution”. DailyMed. 24 January 2020. Retrieved 16 February 2020.
- ^ “Monoferric approval letter” (PDF). U.S. Food and Drug Administration (FDA). 16 January 2020. Retrieved 16 February 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Drug Approval Package: Monoferric Injection”. U.S. Food and Drug Administration (FDA). 7 May 2020. Retrieved 13 August 2020.
External links
- “Ferric derisomaltose”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Monoferric |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Ferric_derisomaltose |
| Routes of administration | Intravenous (IV) |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1345510-43-1 |
| PubChem CID | 86278348 |
| DrugBank | DB15617 |
| UNII | AHU547PI9H |
| KEGG | D11808 |
| Chemical and physical data | |
| Formula | C18H34FeO16+3 |
| Molar mass | 562.299 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////////////Ferric derisomaltose, デルイソマルトース第二鉄 , APPROVALS 2022, JAPAN 2022, NS32, WHO 9712
[Fe+3].OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO[C@H]1O[C@H](CO[C@H]2O[C@H](CO)[C@@H](O)[C@H](O)[C@H]2O)[C@@H](O)[C@H](O)[C@H]1O

NEW DRUG APPROVALS
one time
$10.00
Difamilast
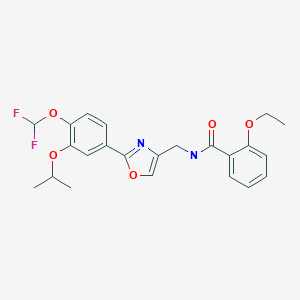
Difamilast
FDA 2026, APPROVALS 2026, difamilast, Adquey, 2/12/2026, To treat mild to moderate atopic dermatitis
PMDA Moizerto, JAPAN APPROVED 2021/9/27
ジファミラスト
ディファミラスト;
地法米司特
N-({2-[4-(difluoromethoxy)-3-(propan-2-yloxy)phenyl]-1,3- oxazol-4-yl}methyl)-2-ethoxybenzamide
OPA-15406
| Formula |
C23H24F2N2O5
|
|---|---|
| CAS |
937782-05-3
|
| Mol weight |
446.4439
|
MM 36; MM-36-Medimetriks-Pharmaceuticals; Moizerto; OPA-15406
| Efficacy |
Anti-inflammatory, Phosphodiesterase IV inhibitor
|
|---|---|
| Comment |
Treatment of atopic dermatitis
|
OriginatorOtsuka Pharmaceutical Development & Commercialization- DeveloperMedimetriks Pharmaceuticals; Otsuka Pharmaceutical Development & Commercialization
- ClassBenzamides; Nonsteroidal anti-inflammatories; Oxazoles; Skin disorder therapies
- Mechanism of ActionType 4 cyclic nucleotide phosphodiesterase inhibitors
- RegisteredAtopic dermatitis
- 27 Sep 2021Registered for Atopic dermatitis (In adolescents, In children, In adults) in Japan (Topical)
- 11 Nov 2020Otsuka Pharmaceutical completes a phase III trial in Atopic dermatitis (In children, In adolescents, In adults) in Japan (Topical) (NCT03961529)
- 28 Sep 2020Preregistration for Atopic dermatitis in Japan (In children, In adolescents, In adults) (Topical)

Difamilast is under investigation in clinical trial NCT01702181 (A Safety Study to Evaluate the Use and Effectiveness of a Topical Ointment to Treat Adults With Atopic Dermatitis).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019194211&_cid=P10-MLOK7B-25338-2
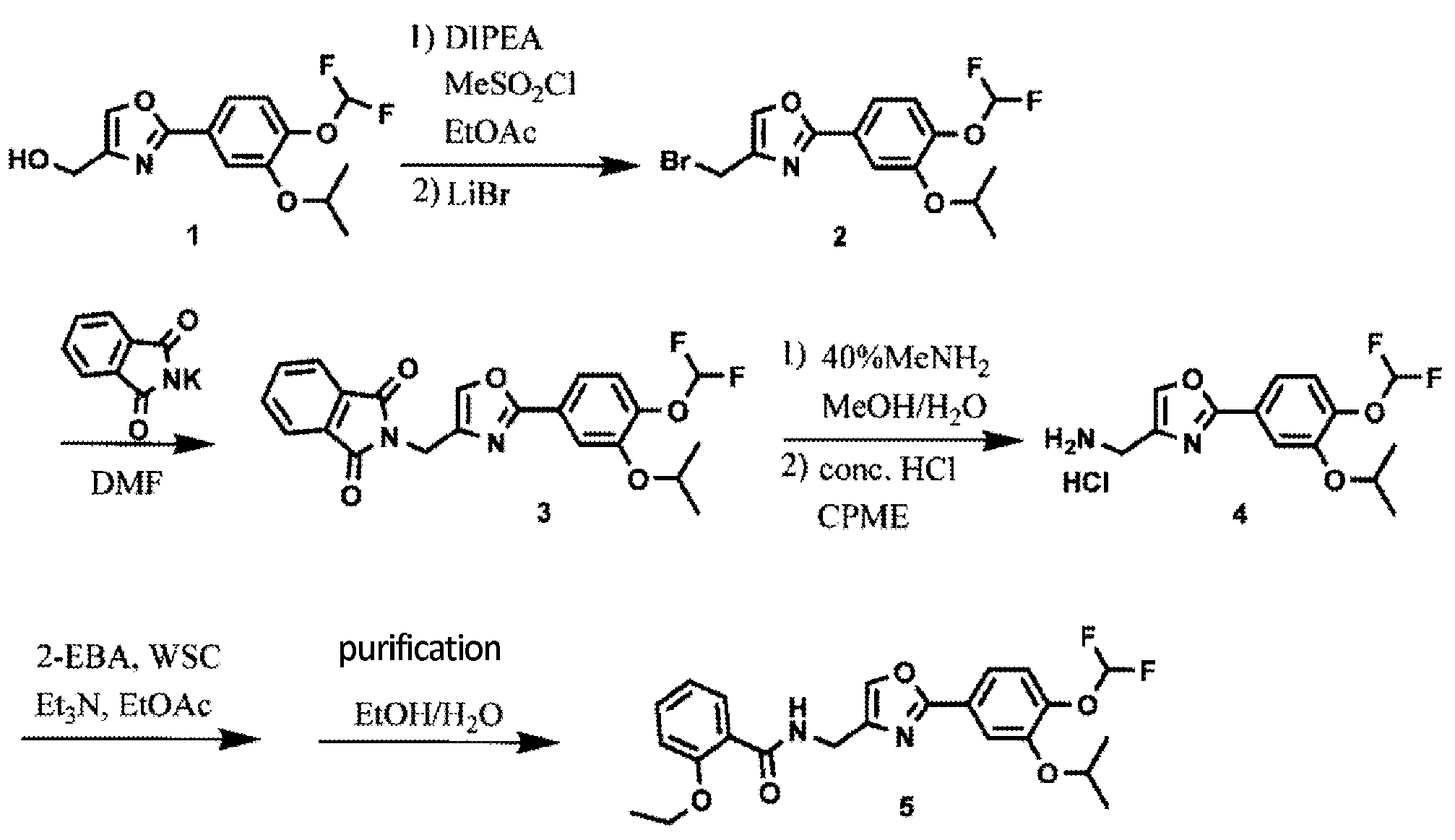
Synthesis of Oxazole Compound (Type A Crystal)
Compound (5) (white powder) was prepared in accordance with the method disclosed in Example 352 of PTL 1 (WO2007/058338).
SYN
https://www.chemicalbook.com/article/how-is-difamilast-synthesised.htm
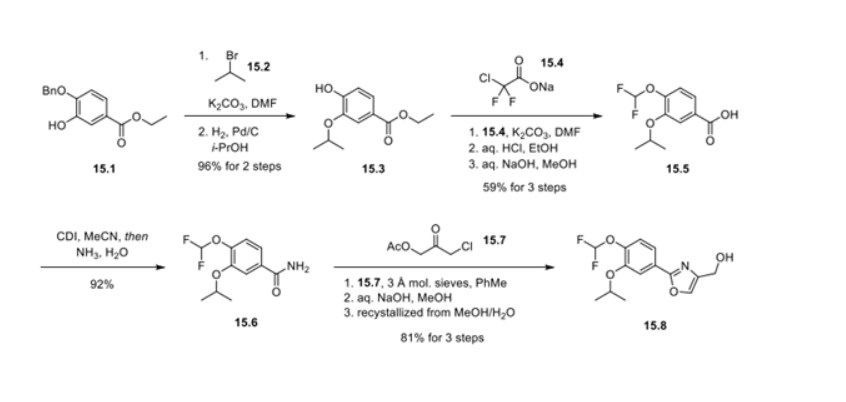
Synthesis of difamilast commenced with the monobenzylated protocatechuic acid ethyl ester 15.1. Phenol 15.1 was first converted into the corresponding isopropyl ether, which was subsequently debenzylated under palladium-catalyzed hydrogenation conditions to generate the phenolic intermediate 15.3. Difluoromethylation of 15.3 was accomplished by introducing sodium chlorodifluoroacetate 15.4 in the presence of potassium carbonate at an elevated temperature. The decarboxylative C− O bond-forming reaction presumably proceeded via a difluorocarbene species. The difluoromethylated product was treated with acid followed by ester hydrolysis under a basic medium to furnish benzoic acid derivative 15.5. Benzoic acid 15.5 was subsequently transformed into benzamide 15.6 via a benzoyl imidazole intermediate. Condensation of benzamide 15.6 with 1-acetoxy-3-chloroacetone 15.7 produced an oxazole derivative, which was subsequently saponified and recrystallized from 50% aqueous MeOH to generate alcohol 15.8.
First, an activation−displacement process transformed alcohol 15.8 into bromide 15.9 via a mesylate intermediate. Alkyl bromide 15.9 was then treated with potassium phthalimide to incorporate the nitrogen center via an SN2-type displacement. Methylamine-mediated phthalimide deprotection and subsequent salt formation produced amine 15.11 as a hydrochloride salt in 69% yield over 3 steps. Finally, hydrochloride salt 15.11 was treated with aqueous sodium bicarbonate to generate a free amine, which was subjected to amide bond formation with 2-ethoxybenzoic acid 15.12 to deliver difamilast after recrystallization from aqueous EtOH.
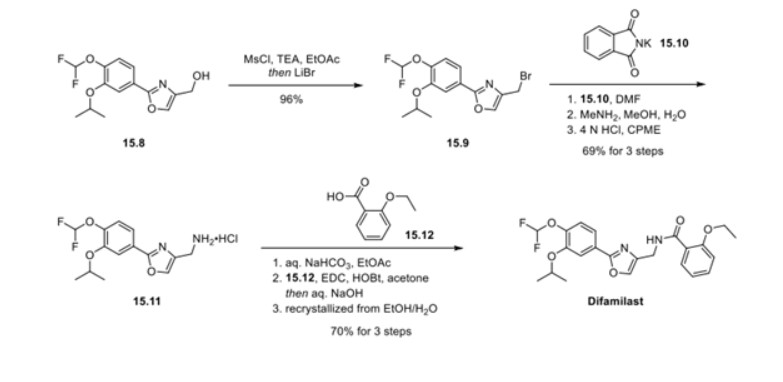
PATENT
JP 2021059538
https://patentscope.wipo.int/search/en/detail.jsf?docId=JP322244172&_cid=P20-L1WXG6-04592-1
patcit 2 : International Publication No. 2014/034958 (Japanese Publication No. 2015-528433 )
patcit 3 : International Publication No. 2017/115780
Compound (5) (white powder) was prepared by the method described in Example 352 of Patent Document 1 (International Publication No. 2007/088383).
N−({2−[4−(difluoromethoxy)−3−isopropoxyphenyl]oxazol−4−yl}methyl)−2−ethoxybenzamide
: white powder.
1H NMR (400 MHz, CDCl3): δ = 8.56 (br s,
1H, NH), 8.23 (dd, J = 7.6 Hz, 1.6 Hz, 1H, ArH), 7.66 (s, 1H, ArH), 7.63 (d, J = 2.0 Hz, 1H, ArH), 7.58 (dd, J = 8.4 Hz, 2.0 Hz, 1H, ArH), 7.44−7.39 (m, 1H, ArH), 7.21 (d, J = 8.0 Hz, 1H, ArH), 7.08−7.04 (m, 1H, ArH), 6.94 (d, J = 8.0 Hz, 1H, ArH), 6.61 (t, J = 75.2 Hz, 1H, CHF 2), 4.68 (sept, J = 6.0 Hz, 1H, CH), 4.62
(d, J = 6.0 Hz, 2H, CH 2), 4.17 (q, J = 6.93, 2H, CH 2), 1.48 (t, J = 7.2 Hz, 3H,
CH 3), 1.39 (d, J = 5.6 Hz, 6H, 2CH 3).
Using the obtained B-type crystal as a seed crystal, it was examined to further prepare a B-type crystal. Specifically,
B-type crystals were prepared as follows according to the method described in Patent Document 3 (International Publication No. 2017/115780).
aqueous sodium hydroxide solution were added to the organic layer, the temperature was adjusted again to 40 to 50 ° C., the liquid was separated, and the organic layer was concentrated under reduced pressure. 50 mL of ethanol, 20 mL of water, 6 mL of a 25% aqueous sodium hydroxide solution, and 0.6 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. Activated carbon was removed by filtration, washed with 12 mL of ethanol, the filtrate was cooled, and 10 mg of B-type crystals (seed crystals) were added to precipitate crystals. Precipitated crystals were collected by filtration and dried at 60 ° C. to obtain 18.38 g (yield 88.18%) of crystals of compound (5).
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007058338&_cid=P10-MLOK7P-25609-1
Example 352
Using the compound obtained in Example 347 and 2-bromopropane, white powdery N-[2-(4-difluoromethoxy-3-isopropoxyphenyl)oxazol-4-ylmethyl]-2-ethoxybenzamide was obtained following the procedure of Example 348.
1H-NMR (CDCl3) δ: 8.57 (1H, br s), 8.24 (1H, dd, J = 7.5, 1.8 Hz), 7.67 (1H, s), 7.65-7.57 (2H, m), 7.46-7.40 (1H, m), 7.26-7.21 (1H, m), 7.08 (1H, t, J = 7.5 Hz), 6.95 (1H, d, J = 8.4 Hz), 6.63 (1H, t, J = 75 Hz), 4.74-4.62 (3H, m), 4.19 (2H, q, J = 6.9 Hz), 1.49 (3H, t, J = 6.9 Hz), 1.40 (6H, d, J = 6.3 Hz)
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017115780
Compound (3) was produced in accordance with the following reaction scheme.
1H-NMR (CDCl 3) δ: 7.70 (2H,dd,J = 6.4 Hz,2.0 Hz),7.22 (1H,d,J = 9.2 Hz),6.66 (1H,t,J = 74.8 Hz),4.66(1H,sept,J = 6.0 Hz),1.39 (6H,d,J = 6.0 Hz).
Production Example 2: Production 2 of Compound (3)
Compound (3) was produced in accordance with the following reaction scheme.
Compound (7) was produced in accordance with the following reaction scheme.
Compound (11) was produced in accordance with the following reaction scheme.
20.00 g (66.8 mmol) of compound (7) and 17.28 g (134 mmol) of N,N-diisopropylethylamine were added to 300 ml of ethyl acetate, and the mixture was cooled. 11.48 g (100 mmol) of methanesulfonyl chloride was poured in and stirred at 10 to 30°C for 1 hour. 17.41 g (200 mmol) of lithium bromide was added thereto and reacted at 20 to 35°C for 1 hour. 100 ml of water was added to the reaction solution, and the mixture was partitioned, followed by concentration of the organic layer under reduced pressure. 300 ml of ethyl acetate was added to the concentrated residue to dissolve the residue, and the solution was again concentrated under reduced pressure. 200 ml of N,N-dimethylformamide and 17.33 g (93.6 mmol) of potassium phthalimide were added to the concentrated residue and reacted at 75 to 85°C for 1 hour. 200 ml of water was added to the reaction solution to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 25.90 g (yield: 90.5%) of compound (9) as a white powder.
15.00 g (35.0 mmol) of compound (9) was mixed with 30 ml of a 40% methylamine aqueous solution, 30 ml of methanol, and 75 ml of water, and reacted under reflux for 30 minutes. 150 ml of cyclopentyl methyl ether (CPME) and 15 ml of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the temperature was adjusted to 65 to 75°C, followed by partitioning. A mixture of 150 ml of water and 7.50 g of sodium chloride was added to the organic layer, and the temperature was adjusted to 65 to 75°C again, followed by partitioning. 3.75 ml of concentrated hydrochloric acid was added to the organic layer to precipitate crystals. The precipitated crystals were collected by filtration and dried at 60°C, thereby obtaining 11.95 g (yield: quant.) of compound (10) as a white powder.
13.30 g (39.7 mmol) of compound (10) was mixed with 3.83 g (37.8 mmol) of triethylamine and 108 ml of ethyl acetate, and stirred at 20 to 30°C for 1 hour. 9.78 g (58.9 mmol) of 2-ethoxybenzoic acid and 11.28 g (58.8 mmol) of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (WSC) were added to the reaction solution, and reacted at 20 to 30°C for 1 hour. 54 ml of water and 5.4 ml of concentrated hydrochloric acid were added to the reaction solution, and the temperature was adjusted to 40 to 50°C, followed by partitioning. 54 ml of water and 5.4 ml of a 25% sodium hydroxide aqueous solution were added to the organic layer, and the temperature was adjusted to 40 to 50°C again. The mixture was partitioned, and the organic layer was concentrated under reduced pressure. 45 ml of ethanol, 18 ml of water, 5.4 ml of a 25% sodium hydroxide aqueous solution, and 0.54 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. The activated carbon was removed by filtration, and the filtrate was washed with 11 ml of ethanol. The filtrate was cooled, and a seed crystal was added thereto to precipitate crystals. The precipitated crystals were collected by filtration and dried at 35°C, thereby obtaining 12.88 g (72.6%) of compound (11) as a white powder.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019194211
*DIPEA: Diisopropylethylamine, CPME: Cyclopentyl methyl ether,
DMF: N,N-dimethylformamide, 2-EBA: 2-Ethoxybenzoic acid,
WSC: 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
Analysis was conducted to further prepare the type B crystal using the obtained type B crystal as a seed crystal. More specifically, the type B crystal was prepared as follows, in accordance with the method disclosed in PTL 3 (WO2017/115780).
PATENT
WO2014034958A1
WO2007058338A2
WO2007058338A9
WO2007058338A3
US9181205B2
US2015239855A1
USRE46792E
US2020078340A1
US2017216260A1
US2019070151A1
US2009221586A1
US8637559B2
US2014100226A1
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
/////////////Difamilast, JAPAN 2021, APPROVALS 2021, ジファミラスト , MM 36, MM-36-Medimetriks-Pharmaceuticals, Moizerto, OPA-15406, OPA 15406, 地法米司特
O=C(NCC1=COC(C2=CC=C(OC(F)F)C(OC(C)C)=C2)=N1)C3=CC=CC=C3OCC
INTrmediate No.CAS No.DIFAM-001177429-27-5DIFAM-00293652-48-3DIFAM-0031574285-26-9DIFAM-00470-23-5DIFAM-0051574285-28-1DIFAM-0061574285-30-5DIFAM-0071574285-32-7DIFAM-0081574285-36-1DIFAM-0091574285-38-3DIFAM-010DIFAM-0111574285-40-7DIFAM-0121574285-43-0DIFAM-013134-11-2Difamilast937782-05-3
TOLDIMFOS SODIUM

TOLDIMFOS SODIUM
C9H12NNaO2P+ , 220.16
Toldimfos sodium
575-75-7
Sodium (4-(dimethylamino)-2-methylphenyl)phosphinate
UNII-6139240O1E
sodium;[4-(dimethylamino)-2-methylphenyl]-oxido-oxophosphanium
Sodium (4-(dimethylamino)-2-methylphenyl)phosphinate
sodium;[4-(dimethylamino)-2-methylphenyl]-oxido-oxophosphanium
Phosphinic acid, [4-(dimethylamino)-2-methylphenyl]-, sodium salt
Phosphinic acid, (4-(dimethylamino)-2-methylphenyl)-, sodium salt
Phosphinic acid, P-(4-(dimethylamino)-2-methylphenyl)-, sodium salt (1:1)
Toldimfos is an aromatic phosphorus compound which falls between phosphorous itself and phosphoric acid in the stages of oxidation. Toldimfos sodium is the sodium salt of 2- methyl-4-(dimethylamino)phenylphosphinic acid. It is used to treat and prevent diseases associated with parturition and peri-partum period, developmental and nutritional disorders in young animals, and bone growth disorders and tetany or paresis caused by calcium, magnesium, and phosphorus metabolism disorders. Toldimfos has been used as a human medicine since 1920. While it is no longer indicated for human use, it is used in horses, cattle, sheep, pigs, and goats, and administered by intravenous, intramuscular, or subcutaneous injection. No specific data on the pharmacodynamic action of toldimfos was submitted. The precise mode of action of toldimfos is unknown and it is questionable whether the effect of toldimfos is simply a matter of the substitution of deficient phosphorus. It appears more likely that the effect of toldimfos arises due to multiple stimulation of metabolism with the body.
Toldimfos sodium trihydrate
5787-63-3
Toldimfos [INN:BAN]
57808-64-7
Toldimfos Sodium
CAS Registry Number: 575-75-7
CAS Name: (4-Dimethylamino-o-tolyl)phosphonous acid sodium salt
Additional Names: sodium (4-dimethylamino-o-tolyl)phosphonate; p-dimethylamino-o-toluenephosphonous acid sodium salt
Trademarks: Foston (Hoechst); Tonofosfan (Hoechst)
Molecular Formula: C9H13NNaO2P, Molecular Weight: 221.17
Percent Composition: C 48.87%, H 5.92%, N 6.33%, Na 10.39%, O 14.47%, P 14.00%
Literature References: Prepd from N,N-dimethyl-m-toluidine and phosphorus trichloride: Benda, Schmidt, DE397813 (1924 to Cassella), Frdl.14, 1409.
Derivative Type: Trihydrate
CAS Registry Number: 5787-63-3
Properties: Scales, needles, or prisms from alc. Freely sol in cold water, hot alcohol.
Therap-Cat-Vet: Phosphorus source.
SYN

PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011026571
PATENT
https://patents.google.com/patent/CN103830261A/en
China’s animal husbandry fast development is the important motivity that promotes China’s agricultural and rural economy development, improves farmers’ income.The disease relevant with Nutrition and Metabolism of serious harm animal health is in rising trend in recent years, the direct economic loss that raising poultry nutritive metabolic disease causes over ten billion to China’s animal husbandry every year, and indirect economic loss is difficult to estimate.
Due to the life-time service of chemicals, will cause some poultrys, poultry product drug residue is serious, this is harm humans healthy not only, also affecting the export of farm produce earns foreign exchange, therefore, tackle this problem, research and development are efficient, the new Nutrition and Metabolism medicine of low toxicity, wide spectrum will have huge market.
Toldimfos (Toldimfos Sodium) belongs to the nutritional supplementation medicine of phosphorus supplement, can be used as benzenephosphonic acid (Phenylphosphinicacid, BPA) succedaneum uses, can be used for treating the disease relevant with childbirth and perinatal stage of the food animals such as horse, cattle, pig, sheep, the diseases such as the bone lengthening obstacle being caused by calcium, magnesium, phosphorus metabolism obstacle.
Toldimfos has higher water solublity, mainly excretes through urine rapidly with the former medicine form of not metabolism in animal body, and the half-life is short, can in tissue, not accumulate.
Toldimfos is developed by German Hoechst company, the symptom such as since nineteen twenty, once physical weakness, chronic stress, depression, mental muscle power postoperative for human treatment, that catch was overtired.Now be not used in the mankind, be mainly used in animal.Its commodity are called onofosfan.
In sum, toldimfos, as a kind of nutritional supplementation medicine of new and effective noresidue, has wide market prospect in China.The development of this product will be made outstanding contributions to the sound development of China’s animal husbandry, remarkable economic and social benefits with application.
PATENT
https://patents.google.com/patent/WO2004003198A1/ja
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
//////////TOLDIMFOS SODIUM, HOECHST, Foston, Tonofosfan,
O.O.O.[Na+].CN(C)c1ccc(c(C)c1)P(=O)[O-]
NEW DRUG APPROVALS
ONE TIME
$10.00
Carotegrast methyl
Carotegrast methyl
| Formula | C28H26Cl2N4O5 |
|---|---|
| CAS | 401905-67-7 |
| Mol weight | 569.4358 |
PMDA APROVED, CAROGRA, カロテグラストメチル
| ON 2022/3/28 |
Antiasthmatic, Integrin alpha 4 inhibitor
- An alpha4 integrin antagonist.
401905-67-7[RN]
L-Phenylalanine, N-(2,6-dichlorobenzoyl)-4-[6-(dimethylamino)-1,4-dihydro-1-methyl-2,4-dioxo-3(2H)-quinazolinyl]-, methyl ester
methyl (2S)-2-[(2,6-dichlorophenyl)formamido]-3-{4-[6-(dimethylamino)-1-methyl-2,4-dioxo-1,2,3,4-tetrahydroquinazolin-3-yl]phenyl}propanoate
Methyl N-(2,6-dichlorobenzoyl)-4-[6-(dimethylamino)-1-methyl-2,4-dioxo-1,4-dihydro-3(2H)-quinazolinyl]-L-phenylalaninate
Carotegrast Methyl
Methyl (2S)-2-(2,6-dichlorobenzamido)-3-{4-[6-(dimethylamino)-1-methyl-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl]phenyl}propanoate
C28H26Cl2N4O5 : 569.44
[401905-67-7]
PATENT
WO 2008062859
https://patents.google.com/patent/WO2008062859A1/en



Step 1
(Method 2): The title compound was prepared starting from 2-amino-5-dimethylamino- benzoic acid methyl ester dihydrochloride through the hydrolysis under basic condition To 5.0 g of 2-amino-5-dimethylamino-benzoic acid methyl ester di-hydrochloride, there were added 15 mL of water and 15.6 mL of a 6M aqueous solution of sodium hydroxide and the resulting mixture was heated to 40°C for 2 hours. After the confirmation of the progress of the reaction according to HPLC, the reaction system was cooled to room temperature, a 6M hydrochloric acid aqueous solution was dropwise added to the reaction system to thus neutralize the same and to separate out crystals (pH 4.9) and then the reaction system was stirred at 10°C for 2 hours. The solid thus obtained was isolated through the filtration under reduced pressure, washed with 30 mL of water and then dried under reduced pressure at 60°C for 14 hours. Title compound 3.14 g was obtained as gray-colored solid. The physical properties determined were almost identical to those observed for the same compound prepared in the above-mentioned synthesis example. H-NMR (400MHz, DMSO-d6): δ 8.21 (bs, 3H), 7.10 (d, 1H, J=2.8Hz), 6.97 (dd, 1H, J=9.1, 2.8Hz), 6.70 (d, 1H, J=9.1 Hz), 2.72 (s, 6H); 13C-NMR (100MHz, DMSO-d6): δ168.89, 144.55, 141.61, 123.29, 117.90, 114.78, 110.11,41.95; MS (ESI+): m/z 181.3 (MH+), (ESI-): m/z 179.2 (M-H–).
Step 2
Step 1: Synthesis of Nα-(2,6-dichlorobenzoyl) -4-{2-ethoxycarbonylamino-5-dimethyl- amino-benzoylamino}-L-phenylalanine methyl ester To 1.96 g of 2-amino-5-dimethylaminobenzoic acid, there were added 12 mL of acetonitrile and 5.29 mL of pyridine to form a suspension and then the resulting suspension was cooled to 4°C. To this suspension there was dropwise added 4.17 mL of ethyl chloroformate over 5 minutes and then the mixture was stirred at 25°C for one hour. After confirming the disappearance of the starting material by HPLC, 0.7 mL of ethanol was added to the mixture to thus decompose the excess ethyl chloroformate and the mixture was further stirred for additional one hour. To this reaction solution there were added 4.0 g of 4-amino-Nα-(2,6-dichlorobenzoyl)-L-phenylalanine methyl ester and 12 mL of N,Ndimethylformamide, and the resulting mixture was stirred overnight. Subsequently, 48 mL of methanol was drop-wise added, the resulting mixture was stirred at 10°C overnight and then the solid separated from the mixture was isolated through filtration under reduced pressure. The solid was then washed with 8 mL of methanol and dried at 70°C for 5 hours under reduced pressure. Title compound 5.50 g was obtained as pale yellow solid. 1H-NMR (400MHz, DMSO-d6): δ 10.29 (s, 1H), 9.42 (bs, 1H), 9.24 (d, 1H, J=7.9Hz), 7.73 (bs, 1H), 7.62 (d, 2H, J=8.4Hz), 7.48-7.44 (m, 2H), 7.41 (dd, 1H, J=9.5, 6.2Hz), 7.27 (d, 2H,J=8.4Hz), 7.01 (d, 1H, J=2.7Hz), 6.93 (dd, 1H, J=9.1, 2.9Hz), 4.71 (ddd, 1H, J=9.2, 8.1, 5.7Hz), 4.05 (q, 2H, J=7.0Hz), 3.66 (s, 3H), 3.10 (dd, 1H, J=14.0, 5.6Hz), 2.96 (dd, 1H, J=14.0, 9.2Hz), 2.93 (s, 6H), 1.18 (t, 3H, J=7.2Hz); MS (ESI+): m/z 601.2 (MH+) and 623.2 (M+Na), (ESI–): m/z 599.1 (M-H–).
Step 3
Step2: Synthesis of Na-(2,6-dichlorobenzoyl)-4-{6-dimethylamino-1-methylquinazoline-2,4[1H,3H]-dion-3-yl}-L-phenylalanine methyl ester To 2.0 g of Na-(2,6-dichlorobenzoyl)-4-{2-ethoxycarbonylamino -5-dimethyl- amino-benzoylamino}-L-phenylalanine methyl ester prepared in above-mentioned step 1, were added 16 mL of N,N-dimethylfbrmamide, 0.8 mL of methanol and 0.91 g of potassium carbonate, followed by the stirring of the resulting mixture at 25°C overnight. To this reaction solution, there was added 0.75 mL of methyl p-toluenesulfonate for subjecting the methyl ester to alkylation at 25~40°C. After confirming the disappearance of the starting material by HPLC, 0.75 mL of acetic acid was added to quench the reaction, 16 mL of water was dropped and the solid was separated. Further, 8 mLof N,N-dimethylformamide/water = 1/1 mixed liquid was added to the resulting mixture, followed by the stirring of the mixture at 25°C. Then the solid thus separated was isolated through filtration under reduced pressure and then washed with 8 mL of water. Thereafter, the isolated solid was dried at 70°C for 4 hours under reduced pressure. Desired compound 1.77 g was obtained as pale yellow solid. 1H-NMR (400MHz, DMSO-d6): δ 9.28 (d, 1H, J=8.1 Hz), 7.48-7.36 (m, 6H), 7.31 (dd, 1H, J=3.0, 9.0Hz), 7.24 (d, 1H, J=3.0Hz), 7.20-7.15 (m, 2H), 4.18 (ddd, 1H, J=10.2, 8.1,4.8Hz), 3.69 (s, 3H), 3.49 (s, 3H), 3.22 (dd, 1H, J=14.1, 4.8Hz), 3.02 (dd, 1H, J=14.2, 10.5Hz), 2.94 (s, 6H); MS (ESI+): m/z 569.2 (MH+) and 591.1 (M+Na), (ESI-): m/z 567.2 (M-H–).
PATENT
https://patents.google.com/patent/WO2004074264A1/en
PATENT’ WO 2003070709
https://patents.google.com/patent/WO2003070709A1/en
PATENT
WO 2002016329
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
/////////////Carotegrast methyl, CAROGRA, カロテグラストメチル , JAPAN 2022, APPROVALS 2022,
COC(=O)[C@H](Cc1ccc(cc1)N2C(=O)N(C)c3ccc(cc3C2=O)N(C)C)NC(=O)c4c(Cl)cccc4Cl

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}