
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Tebentafusp-tebn

Tebentafusp-tebn
- IMCGP100
UNIIN658GY6L3E
CAS number1874157-95-5
FDA APPROVED 1/25/2022, Kimmtrak, To treat unresectable or metastatic uveal melanoma
Immunocore Limited
- T cell receptor α chain (synthetic human) fusion protein with T cell receptor β chain (synthetic human) fusion protein with immunoglobulin, anti-(human CD3 antigen) (synthetic scFv fragment)
- Protein Sequence
- Sequence Length: 695, 500, 195
Sequence:
1AIQMTQSPSS LSASVGDRVT ITCRASQDIR NYLNWYQQKP GKAPKLLIYY51TSRLESGVPS RFSGSGSGTD YTLTISSLQP EDFATYYCQQ GNTLPWTFGQ101GTKVEIKGGG GSGGGGSGGG GSGGGGSGGG SEVQLVESGG GLVQPGGSLR151LSCAASGYSF TGYTMNWVRQ APGKGLEWVA LINPYKGVST YNQKFKDRFT201ISVDKSKNTA YLQMNSLRAE DTAVYYCARS GYYGDSDWYF DVWGQGTLVT251VSSGGGGSDG GITQSPKYLF RKEGQNVTLS CEQNLNHDAM YWYRQDPGQG301LRLIYYSWAQ GDFQKGDIAE GYSVSREKKE SFPLTVTSAQ KNPTAFYLCA351SSWGAPYEQY FGPGTRLTVT EDLKNVFPPE VAVFEPSEAE ISHTQKATLV401CLATGFYPDH VELSWWVNGK EVHSGVCTDP QPLKEQPALN DSRYALSSRL451RVSATFWQDP RNHFRCQVQF YGLSENDEWT QDRAKPVTQI VSAEAWGRAD
Sequence:
1AQQGEEDPQA LSIQEGENAT MNCSYKTSIN NLQWYRQNSG RGLVHLILIR51SNEREKHSGR LRVTLDTSKK SSSLLITASR AADTASYFCA TDGSTPMQFG101KGTRLSVIAN IQKPDPAVYQ LRDSKSSDKS VCLFTDFDSQ TNVSQSKDSD151VYITDKCVLD MRSMDFKSNS AVAWSNKSDF ACANAFNNSI IPEDT
Sequence Modifications
| Type | Location | Description |
|---|---|---|
| bridge | Cys-23 – Cys-88 | disulfide bridge |
| bridge | Cys-153 – Cys-227 | disulfide bridge |
| bridge | Cys-281 – Cys-349 | disulfide bridge |
| bridge | Cys-401 – Cys-466 | disulfide bridge |
| bridge | Cys-427 – Cys-157′ | disulfide bridge |
| bridge | Cys-23′ – Cys-89′ | disulfide bridge |
| bridge | Cys-132′ – Cys-182′ | disulfide bridge |
Tebentafusp, sold under the brand name Kimmtrak, is an anti-cancer medication used to treat uveal melanoma (eye cancer).[1][2]
The most common side effects include cytokine release syndrome, rash, pyrexia (fever), pruritus (itching), fatigue, nausea, chills, abdominal pain, edema, hypotension, dry skin, headache, and vomiting.[1][2]
Tebentafusp is a bispecific gp100 peptide-HLA-directed CD3 T cell engager.[1][2] It was approved for medical use in the United States in January 2022.[1][2]
Tebentafusp is a bispecific gp100 peptide-HLA-directed CD3 T cell engager used to treat unresectable or metastatic uveal melanoma.
Tebentafusp is a gp100 peptide-HLA-directed CD3 T cell engager.5 It is a bispecific, fusion protein and first-in-class drug of immune-mobilizing monoclonal T cell receptors against cancer (ImmTACs), a recently developed cancer immunotherapy with a novel mechanism of action. ImmTACs bind to target cancer cells that express a specific antigen of interest and recruit cytotoxic T cells to lyse the cells, such as melanocytes.1,2
Uveal melanoma is a rare ocular tumour with often poor prognosis and limited treatment options. Even after surgical ablation or removal of the ocular tumour, almost 50% of patients with uveal melanoma develop metastatic disease.1 On January 26, 2022, tebentafusp was first approved by the FDA for the treatment of HLA-A*02:01-positive adults with unresectable or metastatic uveal melanoma. This approval marks the first bispecific T cell engager to be approved by the FDA to treat a solid tumour and being the first and only therapy for the treatment of unresectable or metastatic uveal melanoma to be approved by the FDA.5
FDA approves tebentafusp-tebn for unresectable or metastatic uveal melanoma
On January 25, 2022, the Food and Drug Administration approved tebentafusp-tebn (Kimmtrak, Immunocore Limited), a bispecific gp100 peptide-HLA-directed CD3 T cell engager, for HLA-A*02:01-positive adult patients with unresectable or metastatic uveal melanoma.
Efficacy was evaluated in IMCgp100-202 (NCT03070392), a randomized, open-label, multicenter trial of 378 patients with metastatic uveal melanoma. Patients were required to be HLA-A*02:01 genotype positive identified by a central assay. Patients were excluded if prior systemic therapy or localized liver-directed therapy were administered. Prior surgical resection of oligometastatic disease was permitted. Patients with clinically significant cardiac disease or symptomatic, untreated brain metastases were excluded.
Patients were randomized (2:1) to receive tebentafusp-tebn (N=252) or investigator’s choice (N=126) of either pembrolizumab, ipilimumab, or dacarbazine. Tebentafusp-tebn was administered weekly by intravenous infusion at 20 mcg on day 1, 30 mcg on day 8, 68 mcg on day 15 and every subsequent week until disease progression or unacceptable toxicity. The main efficacy outcome measure was overall survival (OS). An additional efficacy outcome was investigator-assessed progression-free survival (PFS) per RECIST 1.1. Median OS was 21.7 months (95% CI: 18.6, 28.6) for patients treated with tebentafusp-tebn and 16 months (95% CI: 9.7, 18.4) in the investigator’s choice arm (HR=0.51, 95% CI: 0.37, 0.71, p<0.0001) PFS was 3.3 months (95% CI: 3, 5) for those receiving tebentafusp-tebn and 2.9 months (95% CI: 2.8, 3) in the investigator’s choice arm (HR=0.73, 95% CI: 0.58, 0.94, p=0.0139).
The most common adverse reactions (≥30%) were cytokine release syndrome, rash, pyrexia, pruritus, fatigue, nausea, chills, abdominal pain, edema, hypotension, dry skin, headache, and vomiting. The most common laboratory abnormalities (≥50%) were decreased lymphocyte count, increased creatinine, increased glucose, increased aspartate aminotransferase, increased alanine aminotransferase, decreased hemoglobin, and decreased phosphate.
The recommended tebentafusp-tebn dose administered intravenously is:
- 20 mcg on day 1,
- 30 mcg on day 8,
- 68 mcg on day 15, and
- 68 mcg once weekly thereafter.
View full prescribing information for Kimmtrak.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), Health Canada, and the United Kingdom’s Medicines and Healthcare product Regulatory Agency (MHRA). The application reviews may be ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted priority review, breakthrough designation and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
//////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Trade names | Kimmtrak |
| Other names | IMCgp100, tebentafusp-tebn |
| License data | US DailyMed: Tebentafusp |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 1874157-95-5 |
| DrugBank | DB15283 |
| UNII | N658GY6L3E |
Medical uses
Tebentafusp is indicated for HLA-A*02:01-positive adults with unresectable or metastatic uveal melanoma.[1][2]
History
Efficacy was evaluated in IMCgp100-202 (NCT03070392), a randomized, open-label, multicenter trial of 378 participants with metastatic uveal melanoma.[2] Participants were required to be HLA-A*02:01 genotype positive identified by a central assay.[2] Participants were excluded if prior systemic therapy or localized liver-directed therapy were administered.[2] Prior surgical resection of oligometastatic disease was permitted.[2] Participants with clinically significant cardiac disease or symptomatic, untreated brain metastases were excluded.[2]
The U.S. Food and Drug Administration (FDA) granted Immunocore‘s application for tebentafusp priority review, breakthrough therapy, and orphan drug designations.[2]
References
- ^ Jump up to:a b c d e f https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761228s000lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l “FDA approves tebentafusp-tebn for unresectable”. U.S. Food and Drug Administration (FDA). 25 January 2022. Retrieved 28 January 2022.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.
External links
- “Tebentafusp”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03070392 for “Safety and Efficacy of IMCgp100 Versus Investigator Choice in Advanced Uveal Melanoma” at ClinicalTrials.gov
/////////////////Tebentafusp-tebn, Kimmtrak, priority review, breakthrough designation, orphan drug designation, Immunocore Limited, IMCGP100, APPROVALS 2022, FDA 2022

NEW DRUG APPROVALS
ONE TIME
$10.00
VIP 152, BAY 1251152

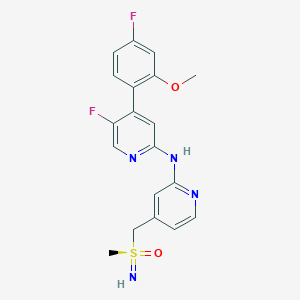
VIP 152, BAY 1251152
CAS RN.: 1610358-56-9
C19H18F2N4O2S
5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-[4-[(methylsulfonimidoyl)methyl]pyridin-2-yl]pyridin-2-amine
- 2-Pyridinamine, 5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-[4-[(S-methylsulfonimidoyl)methyl]-2-pyridinyl]-, (+)-
(+)-BAY-1251152 is a CDK9 inhibitor extracted from patent WO 2014076091 A1, example 1.
RN: 1610408-97-3
UNII: 1255AT22ZJ
UNII-1255AT22ZJ
2-Pyridinamine, 5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-[4-[[[S(S)]-S-methylsulfonimidoyl]methyl]-2-pyridinyl]-
Molecular Formula, C19-H18-F2-N4-O2-S, Molecular Weight, 404.4336
- OriginatorBayer
- DeveloperBayer; Vincerx Pharma
- ClassAntineoplastics; Fluorinated hydrocarbons; Organic sulfur compounds; Phenyl ethers; Pyridines; Small molecules
- Mechanism of ActionCyclin dependent kinase 9 inhibitors; Positive transcriptional elongation factor B inhibitors
- Orphan Drug StatusYes – Diffuse large B cell lymphoma
- Phase IChronic lymphocytic leukaemia; Haematological malignancies; Non-Hodgkin’s lymphoma; Richter’s syndrome; Solid tumours
- 17 Dec 2021Vincerx Pharma plans phase II trials for Cancer (IV, Infusion), in the second half of 2022
- 16 Dec 2021Phase-I clinical trials in Chronic lymphocytic leukaemia (Second-line therapy or greater) in USA (IV)
- 16 Dec 2021Phase-I clinical trials in Richter’s syndrome (Second-line therapy or greater) (IV) in USA

First-in-human dose escalation study of cyclin-dependent kinase-9 inhibitor VIP152 in patients with advanced malignancies shows early signs of clinical efficacyJennifer R. Diamond, Valentina Boni, Emerson Lim, Grzegorz Nowakowski, Raul Cordoba, Daniel Morillo, Ray Valencia, Isabelle Genvresse, Claudia Merz, Oliver Boix, Melanie M. Frigault, Joy M. Greer, Ahmed M. Hamdy, Xin Huang, Raquel Izumi, Harvey Wong and Victor Moreno
DOI: 10.1158/1078-0432.CCR-21-3617
Abstract
Purpose: To report on the first-in-human phase I study of VIP152 (NCT02635672), a potent and highly selective CDK9 inhibitor. Patients and Methods: Adults with solid tumors or aggressive non-Hodgkin lymphoma (NHL) who were refractory to or had exhausted all available therapies received VIP152 monotherapy as a 30-minute intravenous, once weekly infusion, as escalating doses (5, 10, 15, 22.5, or 30 mg in 21-day cycles) until the maximum tolerated dose (MTD) was determined. Results: Thirty-seven patients received {greater than or equal to} 1 VIP152 dose, with 30 mg identified as the MTD based on dose-limiting toxicity of grade 3/4 neutropenia. The most common adverse events were nausea and vomiting (75.7% and 56.8%, respectively), all of grade 1/2 severity. Of the most common events, Grade 3/4 events occurring in > 1 patient were neutropenia (22%), anemia (11%), abdominal pain (8%), increased alkaline phosphatase (8%), and hyponatremia (8%). Day 1 exposure for the MTD exceeded the predicted minimum therapeutic exposure and reproducibly achieved maximal pathway modulation; no accumulation occurred after multiple doses. Seven of 30 patients with solid tumors had stable disease (including 9.5 and 16.8 months in individual patients with pancreatic cancer and salivary gland cancer, respectively), and 2 of 7 patients with high-grade B-cell lymphoma with MYC and BCL2/BCL6 translocations (HGL) achieved durable complete metabolic remission (ongoing at study discontinuation, after 3.7 and 2.3 years of treatment). Conclusion: VIP152 monotherapy, administered intravenously once weekly, demonstrated a favorable safety profile and evidence of clinical benefit in patients with advanced HGL and solid tumors.
CLIP
Preclinical bioconjugation platform designed to overcome limitations of small–molecule and antibody–drug conjugates use to treat cancer
Vincera Pharma, Inc., a biopharmaceutical company aspiring to address the unmet medical needs of patients with cancer through paradigm-shifting therapeutics, today announced the signing of an exclusive license agreement with Bayer AG for the development and commercialization of an early development oncology portfolio. The license will become effective upon the closing of the transaction with LSAC (described below), and Vincera intends to use the funds it will receive upon closing of such transaction to initiate its clinical program.
Under the terms of the license agreement, Vincera will in-license VIP152 (formerly BAY 1251152 & CAS RN.: 1610358-56-9), a clinical-stage, highly selective, positive transcription elongation factor b (PTEFb)/cyclin-dependent kinase 9 (CDK9) inhibitor for the treatment of cancer. Additionally, Vincera will receive assets and license technology for a preclinical bioconjugation platform to address the limitations of small-molecule and antibody-drug conjugates in oncology. The preclinical assets include VIP236, a small molecule drug conjugate (SMDC) targeting advanced and metastatic cancer; as well as VIP943 (formerly BAY-943) and VIP924 (formerly BAY-924), two antibody-drug conjugates (ADC) targeting hematologic tumors; and VIP217, an oral PTEFb/CDK9 inhibitor in discovery. “This license agreement with Bayer creates the foundation of Vincera’s targeted clinical oncology pipeline, with a potentially best-in-class asset, while positioning us for long-term growth across two therapeutic platforms,” said Ahmed Hamdy M.D., Chief Executive Officer of Vincera. “Our lead asset, VIP152, is a small molecule PTEFb/CDK9 inhibitor with very encouraging data from monotherapy Phase 1 studies, including 2 of 7 patients with durable remissions of over 2 years in the very aggressive indication of relapsed/refractory double-hit DLBCL. In addition, preclinical data support our belief that VIP152 is the most selective CDK9 inhibitor in the clinic with on-target depletion of oncogenic MYC and MCL1 mRNA transcripts in patients. These results, combined with the acceptable safety profile seen to date, suggest that VIP152 could be an important new treatment option for patients with MYC- and MCL1-driven malignancies. Importantly, with proof-of-concept clinical data in hand, we are poised to execute on a strategic clinical development plan with the potential for multiple accelerated approvals in the U.S. Expansion of the current Phase 1b study to include these patient populations is expected to begin in 2021.”
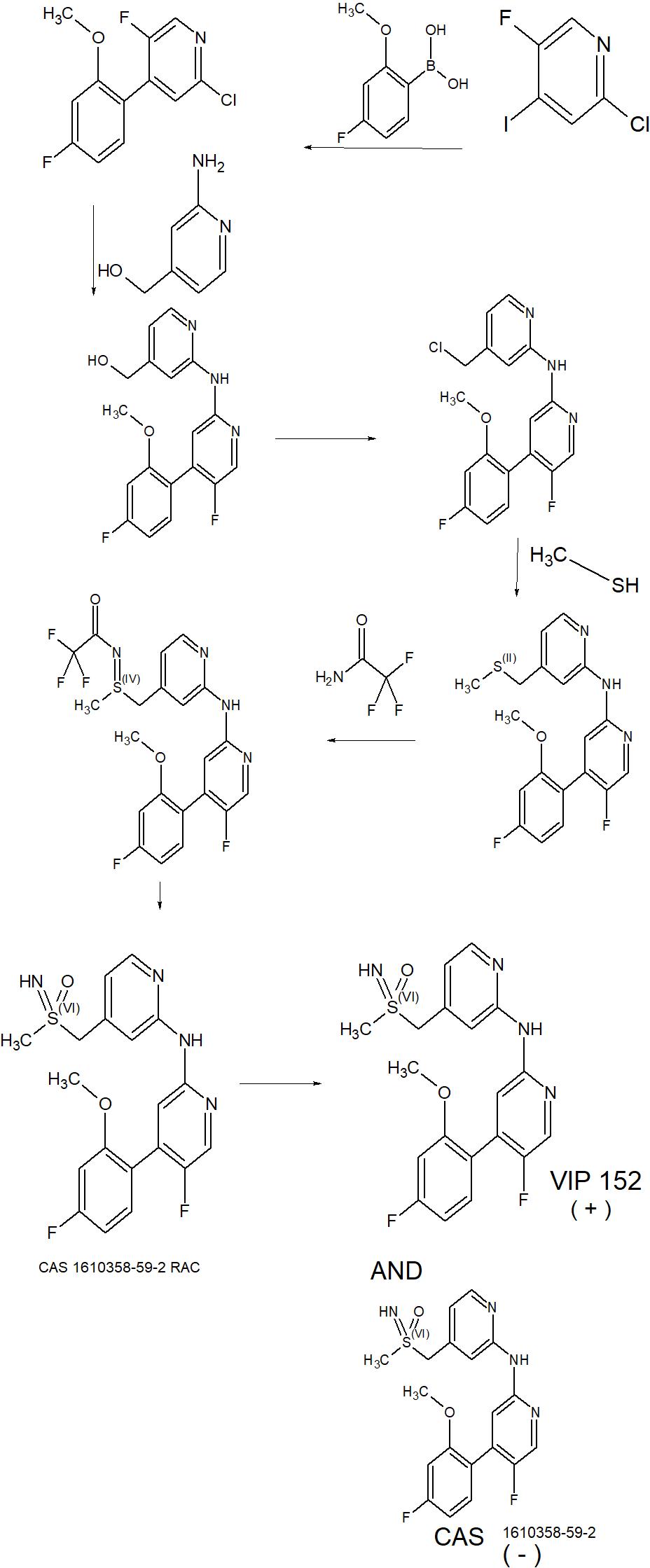
IP Information:
WO2014076091A1 (Product Patent)
Assignee: Bayer Pharma Aktiengesellschaft, Germany
Application Date: 2013-11-12
Family Equivalents:
AP3872A; AR093505A1; AU2013346939A1; AU2013346939B2; BR112015010707A2; BR112015010707A8; CA2891358A1; CL2015001304A1; CN105102444A; CN105102444B; CR20150256A; CU20150052A7; CY1118441T1; DK2928878T3; DOP2015000118A; EA027226B1; EA201590890A1; EP2928878B1; ES2612978T3; HK1213255A1; HRP20161547T1; HUE032868T2; IL238322A; JO3332B1; JP2015537015A; JP6263193B2; KR20150084968A; LT2928878T; MA38090A1; MA38090B1; ME02880B; MX2015006169A; NZ707084A; PE20151071A1; PH12015501003A1; PH12015501003B1; PL2928878T3; PT2928878T; RS55580B1; SG11201503079PA; SI2928878T1; SV2015004979A; TN2015000185A1; TW201420569A; TWI613193B; UA115254C2; US2015291528A1; US2017202815A1; US9650340B2; US9877954B2; UY35141A; WO2014076091A1
Title: 5-FLUORO-N-(PYRIDIN-2-YL)PYRIDIN-2-AMINE DERIVATIVES CONTAINING A SULFOXIMINE GROUP.
Abstract
The present invention relates to 5-fluoro-N-(pyridin-2-yl)pyridin-2-amine derivatives containing a sulfoximine group of general formula (I) as described and defined herein, and methods for their preparation, their use for the treatment and/or prophylaxis of disorders, in particular of hyper-proliferative disorders and/or virally induced infectious diseases and/or of cardiovascular diseases. The invention further relates to intermediate compounds useful in the preparation of said compounds of general formula (I).
“CDK9 represents a validated target for malignancies such as CLL where other less selective CDK inhibitors have shown clinical activity in high-risk patients,” says Dr. John C. Byrd, Chair of the Scientific Advisory Board of Vincera. “VIP-152 represents an exciting new therapy for this disease, particularly those with prior resistance to ibrutinib and venetoclax where a true unmet need exists for new treatments.”
Dr. Hamdy continued, “In addition to our planned clinical program, we intend to advance, in parallel, the development of our preclinical bioconjugation platform. We believe our next-generation platform has the potential to generate first-in-class and best-in-class opportunities in oncology, improving the specificity of drug targeting and release through a modular platform with innovative warhead design and linker-payload technologies. We are thrilled that the Bayer license will allow us to pursue the commercial potential of this promising oncology portfolio and look forward to providing updates as we execute across our pipeline in the coming quarters.”
In exchange for this license, Vincera will pay Bayer an upfront license fee and development and commercial sales milestone payments. In further consideration of the rights granted, we will also pay an annual royalty on the commercial sale of licensed products in the single- to low-double-digit percentage range on net commercial sales of licensed products.
On September 29, 2020, Vincera announced that it has entered into a merger agreement with LifeSci Acquisition Corp. (“LSAC”), a publicly-traded blank check company targeting biopharma, medical technology, digital health, and healthcare services sectors. Following the completion of the merger, the combined company is expected to have approximately $60 million in cash to fund its preclinical and clinical pipeline. Additional information about the merger and related transactions, including a copy of the merger agreement, are included in a Current Report on Form 8-K filed by LSAC with the SEC on September 29, 2020, and available at www.sec.gov.
About Vincera Pharma, Inc.
Vincera is a recently formed clinical-stage life sciences company focused on leveraging its extensive development and oncology expertise to advance new therapies intended to address unmet medical needs for the treatment of cancer. Vincera’s executive team has assembled a management team of biopharmaceutical experts with extensive experience in building and operating organizations that develop and deliver innovative medicines to patients. Vincera’s current pipeline is derived from an exclusive license agreement with Bayer and includes (i) a clinical-stage and follow-on small molecule drug program and (ii) a preclinical stage bioconjugation/next-generation antibody-drug conjugate platform. The company intends to develop multiple products through clinical proof-of-concept and potentially through Accelerated Approval in the United States. For more information, please visit www.vincerapharma.com.
//////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Patent
US 20150291528
Example 1
(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Preparation of Intermediate 1.12-Chloro-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridine
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.27 (m, 1H), 7.33 (m, 1H), 7.24 (m, 1H), 6.75 (m, 2H), 3.83 (s, 3H). |
Preparation of Intermediate 1.25-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.15 (m, 2H), 7.61 (m, 1H), 7.40 (s, 1H), 7.35 (br, 1H), 7.29 (m, 1H), 6.82 (m, 1H), 6.75 (m, 2H), 3.83 (s, 3H), 3.62 (s, 2H), 2.03 (s, 3H). |
Alternative Procedure for the Preparation of Intermediate 1.25-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine
Preparation of Intermediate 1.3(2-{[5-Fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methanol
Preparation of End Product (Alternative Preparation of Intermediate 1.2)
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.15 (m, 2H), 7.61 (m, 1H), 7.40 (br, 2H), 7.29 (m, 1H), 6.82 (m, 1H), 6.75 (m, 2H), 3.83 (s, 3H), 3.62 (s, 2H), 2.03 (s, 3H). |
Alternative Procedure for the Preparation of Example 1Preparation of Intermediate 1.4(rac)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.29 (m, 1H), 8.18 (m, 1H), 7.83 (s. 1H), 7.50 (br, 1H), 7.32 (m, 1H), 7.28 (m, 1H), 6.79 (m, 3H), 4.52 (d, 1H), 4.21 (d, 1H), 3.85 (s, 3H), 2.71 (s, 3H). |
Alternative Preparation of End Product (Example 1)
Example 2 and 3
Enantiomers of 5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
| (rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine (3.47 g) was separated into the single enantiomers by preparative chiral HPLC. |
Example 2(+)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Example 3(−)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Example 4(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Preparation of Intermediate 4.15-Fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine
| 1H NMR (400 MHz, CDCl 3, 300K) δ=7.95 (m, 1H), 7.20 (m, 1H), 6.72 (m, 2H), 6.46 (m, 1H), 4.33 (br, 2H), 3.61 (s, 3H). |
Preparation of Intermediate 4.2(2-Chloro-6-methylpyridin-4-yl)methanol
| 1H NMR (400 MHz, CDCl 3, 300K) δ=7.18 (s, 1H), 7.09 (s, 1H), 4.72 (d, 2H), 2.55 (s, 3H), 2.17 (tr, 1H). |
Preparation of Intermediate 4.32-Chloro-6-methyl-4-[(methylsulfanyl)methyl]pyridine
| 1H NMR (400 MHz, CDCl 3, 300K) δ=7.12 (s, 1H), 7.05 (s, 1H), 3.58 (s, 2H), 2.54 (s, 3H), 2.03 (s, 3H). |
Preparation of Intermediate 4.45-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine
Preparation of Intermediates 4.5 and 4.6(rac)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide and (rac)-N-{[(3-bromo-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-2-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}-2,2,2-trifluoroacetamide
Intermediate 4.5:
Intermediate 4.6:
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.18 (s, 1H), 7.84 (s, 1H), 7.33 (s, 1H), 7.29 (m, 1H), 7.23 (m, 1H), 6.78 (m, 2H), 4.77 (d, 1H), 4.36 (d, 1H), 3.86 (s, 3H), 2.80 (s, 3H), 2.63 (s, 3H). |
Preparation of End Product:
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.16 (s, 1H), 7.60 (s, 1H), 7.39 (m, 1H), 7.30 (m, 2H), 6.79 (m, 3H), 4.34 (d, 1H), 4.22 (d, 1H), 3.86 (s, 3H), 3.02 (s, 3H), 2.79 (br, 1H), 2.48 (s, 3H). |
Alternative Procedure for the Preparation of Example 4Preparation of Intermediate 4.15-Fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine
| 1H-NMR (300 MHz, DMSO-d 6, 300 K): δ [ppm]=7.85 (d, 1H), 7.25 (tr, 1H), 7.08-7.00 (m, 1H), 6.91-6.81 (m, 1H), 6.35 (d, 1H), 5.84 (s, 2H). |
Preparation of Intermediate 4.32-Chloro-6-methyl-4-[(methylsulfanyl)methyl]pyridine
| 1H-NMR (300 MHz, DMSO-d 6, 300 K): δ [ppm]=7.24 (s, 1H), 7.20 (s, 1H), 3.66 (s, 2H), 2.42 (s, 3H), 1.95 (s, 3H). |
Preparation of Intermediate 4.45-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine
| 1H-NMR (300 MHz, CDCl 3, 300 K): δ [ppm]=8.16 (d, 1H), 7.56 (d, 1H), 7.36-7.29 (m, 2H), 7.21 (s, 1H), 6.85-6.73 (m, 2H), 6.72 (s, 1H), 3.86 (s, 3H), 3.61 (s, 2H), 2.45 (s, 3H), 2.06 (s, 3H). |
Preparation of Intermediate 4.5(rac)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide
Preparation of Intermediate 4.6(rac)-N-{[(3-bromo-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-2-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}-2,2,2-trifluoroacetamide
Intermediate 4.5:
Intermediate 4.6(1H-NMR was Taken from a Different Batch)
Preparation of Intermediates 4.7 and 4.8
| 3.76 g of racemic 2,2,2-trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}acetamide were separated by chiral HPLC: |
Intermediate 4.7(+)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide
Intermediate 4.8(−)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide
Alternative Preparation of End Product (Example 4)(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Example 5
(rac)-5-Bromo-N-[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]-6-methyl-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-amine
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.14 (m, 1H), 7.80 (s, 1H), 7.32 (m, 2H), 7.29 (m, 1H), 6.78 (m, 2H), 4.87 (d, 1H), 4.59 (d, 1H), 3.85 (s, 3H), 3.07 (s, 3H), 2.99 (br, 1H), 2.62 (s, 3H). |
Example 6(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methoxy-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Preparation of Intermediate 6.1:2-Chloro-6-methoxy-4-[(methylsulfanyl)methyl]pyridine
| 1H NMR (400 MHz, CDCl 3, 300K) δ=6.92 (s, 1H), 6.61 (s, 1H), 3.96 (s, 3H), 3.56 (s, 2H), 2.03 (s, 3H). |
Preparation of Intermediate 6.25-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methoxy-4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.15 (m, 1H), 7.91 (m, 1H), 7.29 (m, 1H), 7.21 (s, 1H), 6.77 (m, 3H), 6.28 (s, 1H), 3.87 (s, 3H), 3.85 (s, 3H), 3.58 (s, 2H), 2.06 (s, 3H). |
Preparation of Intermediate 6.3(rac)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methoxypyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.18 (m, 1H), 7.56 (m, 1H), 7.29 (m, 2H), 7.12 (m, 1H), 6.78 (m, 2H), 6.25 (s, 1H), 4.52 (d, 1H), 4.07 (d, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 2.70 (s, 3H). |
Preparation of End Product:
Alternative Procedure for the Preparation of Example 6(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methoxy-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Example 7(rac)-N-{6-Chloro-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine
Preparation of Intermediate 7.12-Chloro-6-methoxy-4-[(methylsulfanyl)methyl]pyridine
| 1H NMR (400 MHz, d 6-DMSO, 300K) δ=10.06 (s, 1H), 8.25 (m, 1H), 7.71 (m, 1H), 7.56 (m, 1H), 7.35 (m, 1H), 7.10 (m, 1H), 6.93 (m, 1H), 6.85 (m, 1H), 5.47 (tr, 1H), 4.49 (d, 2H), 3.81 (s, 3H). |
Preparation of Intermediate 7.2N-{6-Chloro-4-[(methylsulfanyl)methyl]pyridin-2-yl}-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.17 (s, 1H), 7.50 (m, 3H), 7.32 (m, 1H), 6.90 (s, 1H), 6.79 (m, 2H), 3.87 (s, 3H), 3.62 (s, 2H), 2.07 (s, 3H). |
Preparation of Intermediate 7.3(rac)-N-{[(2-chloro-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}-2,2,2-trifluoroacetamide
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.18 (s, 1H), 8.12 (br, 1H), 7.84 (s, 1H), 7.37 (m, 1H), 7.31 (m, 1H), 6.80 (m, 3H), 4.46 (d, 1H), 4.24 (d, 1H), 3.87 (s, 3H), 2.75 (s, 3H). |
Preparation of End Product:
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014076091
Example 1:
(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Preparation of Intermediate 1.1:
2-Chloro-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridine
A batch with 2-chloro-5-fluoro-4-iodopyridine (1000 mg; 3.88 mmol; APAC Pharmaceutical, LLC), (4-fluoro-2-methoxyphenyl)boronic acid (660 mg; 3.88 mmol; Aldrich Chemical Company Inc.) and tetrakis(triphenylphosphin)palladium(0) (449 mg; 0.38 mmol) in 1,2-dimethoxyethane (10.0 mL) and 2 M aqueous solution of potassium carbonate (5.8 mL) was degassed using argon. The batch was stirred under an atmosphere of argon for 4 hours at 100 °C. After cooling, the batch was diluted with ethyl
acetate and THF and washed with a saturated aqueous solution of sodium chloride. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by column chromatography (hexane to hexane / ethyl acetate 50%) to give the desired product (947 mg; 3.70 mmol).
1H NMR (400MHz, CDCl3, 300K) δ = 8.27 (m, 1H), 7.33 (m, 1H), 7.24 (m, 1H), 6.75 (m, 2H), 3.83 (s, 3H).
Example 2: (+)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S- methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
1H-NMR (300 MHz, DMSO-d6, 300 K): δ [ppm] = 9.80 (s, 1H), 8.20 (m, 1H), 8.16 (m, 1H), 7.78 (m, 1H), 7.59 (s, 1H), 7.34 (m, 1H), 7.09 (m, 1H), 6.90 (m, 2H), 4.37 (d, 1H), 4.33 (d, 1H), 3.79 (s, 3H), 3.72 (s, 1H), 2.87 (s, 3H).
////////////VIP 152, BAY 1251152
COC1=C(C=CC(=C1)F)C2=CC(=NC=C2F)NC3=NC=CC(=C3)CS(=N)(=O)C

NEW DRUG APPROVALS
ONE TIME
$10.00
Gefapixant citrate

Gefapixant
- Molecular FormulaC14H19N5O4S
- Average mass353.397 Da
1015787-98-0[RN]
10642
AF 217
5-[(2,4-Diamino-5-pyrimidinyl)oxy]-4-isopropyl-2-methoxybenzenesulfonamide
5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzene- sulfonamide
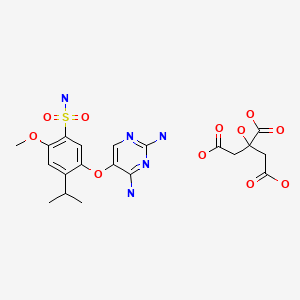
Gefapixant Citrate
| Formula | C14H19N5O4S. C6H8O7 |
|---|---|
| CAS | 2310299-91-1 |
| Mol weight | 545.5203 |
APPROVED JAPAN PMDA 2022/1/20, Lyfnua
ゲーファピキサントクエン酸塩
吉法匹生
| Efficacy | Analgesic, Anti-inflammatory, Antitussive, P2X3 receptor antagonist |
|---|---|
| Comment | Treatment of disorders associated with purinergic receptor activation |
Gefapixant (MK-7264) is a drug which acts as an antagonist of the P2RX3 receptor, and may be useful in the treatment of chronic cough.[1][2][3] It was named in honour of Geoff Burnstock.[4]
Gefapixant is under investigation in clinical trial NCT02397460 (Effect of Gefapixant (AF-219/MK-7264) on Cough Reflex Sensitivity).
PAPER
Organic Process Research & Development (2020), 24(11), 2445-2452.
https://pubs.acs.org/doi/10.1021/acs.oprd.0c00248
A robust, green, and sustainable manufacturing process has been developed for the synthesis of gefapixant citrate, a P2X3 receptor antagonist that is under investigation for the treatment of refractory and unexplained chronic cough. The newly developed commercial process features low process mass intensity (PMI), short synthetic sequence, high overall yield, minimal environmental impact, and significantly reduced API costs. The key innovations are the implementation of a highly efficient two-step methoxyphenol synthesis, an innovative pyrimidine synthesis in flow, a simplified sulfonamide synthesis, and a novel salt metathesis approach to consistently deliver the correct active pharmaceutical ingredient (API) salt form in high purity.

SYN
Organic Process Research & Development (2020), 24(11), 2478-2490.
https://pubs.acs.org/doi/10.1021/acs.oprd.0c00252
Gefapixant citrate (MK-7264) is a P2X3 antagonist for the treatment of chronic cough. The second generation manufacturing route developed for the Step 3A/3B formylation–cyclization reaction to generate the key intermediate diaminopyrimidine (1) (AF-072) required a significant excess of ethyl formate (EF), potassium tert-butoxide (KOt-Bu), and guanidine•HCl (G•HCl) when both steps were run as batch processes. It was imperative to develop an alternative process that required less of each reagent and generated less carbon monoxide byproducts, as the annual production of the final active pharmaceutical ingredient (API) is expected to be over 50 MT. In addition, the second generation process was misaligned with our company’s strategy of having the best science in place at the first regulatory filing. The final flow–batch process described herein, which features a flow-based formylation combined with a batch cyclization, has been performed on a 500 kg scale and now requires 35% less EF (leading to a 70% reduction in waste carbon monoxide), 38% less KOt-Bu, and 50% less G•HCl. These improvements, along with a twofold increase in concentration, have resulted in a 54% reduction in the step process mass intensity (step-PMI) from the second generation two-step batch–batch process (PMI of 17.16) to the flow–batch process (PMI of 7.86), without sacrificing reaction performance.

SYN
H. REN*, K. M. MALONEY* ET AL. (MERCK & CO., INC., RAHWAY USA) Development of a Green and Sustainable Manufacturing Process for Gefapixant Citrate (MK-7264) Part 1: Introduction and Process Overview Org. Process Res. Dev. 2020, 24, 2445–2452, DOI: 10.1021/acs.oprd.0c00248.

Syn
https://doi.org/10.1021/acs.jmedchem.3c02374
J. Med. Chem. 2024, 67, 4376−4418
Gefapixant (Lyfnua). Gefapixant (34), also known as MK-7264, prior to that AF-219 and RO-4926219, is a P2 × 3antagonist for the treatment of chronic cough that was recently approved by the Japan Ministry of Health.243 Chronic cough is one of the most frequent reasons for patients to request medical consultation and is defined as cough ≥8 weeks in the past 12 months for those aged 18 years or older.244 The prevalence of chronic cough among US adults is 5% and can be associated with a deterioration of quality of life.244 The commercial manufacturing process of gefapixant has been described by Merck & Co., Inc., Rahway, NJ, USA, and is outlined in Scheme 59. Synthesis of 34 began with the regioselective bromination of isopropyl phenol 34.1. 245−247 The choice of polar MeCN solvent was found to play a critical role in the bromination regioselectivity providing the parabromophenol 34.2 in high yield. Interestingly, when toluene was used as the solvent the undesired ortho-substituted brominated phenol was the major product. In trial experiments it was discovered that a small amount of dibrominated product was formed which was alleviated using 1 mol % of methanesulfonic acid. Copper-mediated C−O bond formation proceeded with the use of NaOMe and CuBr in DMF to provide 34.3 in 92% yield. The authors describe in detail the screening conditions employed and the dimerization biproducts initially observed when obtaining 34.3. Ultimately, the use of DABCO in the first step allowed for the crystallization of the brominated phenol 34.2 as a DABCO
adduct. This enabled the Cu-catalyzed methoxylation to proceed without the need for phenol protection as well as the suppression of undesired dimerization products.247 Alkylation of phenol 34.3 with chloroacetonitrile in the presence of aqueous sodium hydroxide provided cyanomethyl intermediate 34.4.248 The diaminopyrimidine heterocycle was formed by formylation using ethyl formate and KOtBu
followed by reaction with guanidine HCl to complete the cyclization and obtain 34.5 in 81% yield.249 This was performed in a hybrid flow-batch telescoped process. Treat ment of 34.5 with chlorosulfonic acid in MeCN followed by ammonium hydroxide provided sulfonamide 34.6 in high yield.250,251 The final step in the manufacturing process was the isolation of gefapixant as a mono citrate salt.252,253 The free base of gefapixant was converted to a highly soluble glycolate salt which enabled complete dissolution in MeOH. Citric acid was added to crystallize final API as a mono citrate salt in 93%
yield.
(243) Merck & Co. Inc. Merck provides U.S. and Japan regulatory
update for gefapixant. https://www.merck.com/news/merck-providesu-s-and-japan-regulatory-update-for-gefapixant/ (accessed 2023-06).
(244) Yang, X.; Chung, K. F.; Huang, K. Worldwide prevalence, risk
factors and burden of chronic cough in the general population: a
narrative review. J. Thorac. Dis. 2023, 15, 2300−2313.
(245) Kocienski, P. Synthesis of gefapixant. Synfacts 2021, 17,
No. 0123.
(246) Ren, H.; Maloney, K. M.; Basu, K.; Di Maso, M. J.;
Humphrey, G. R.; Peng, F.; Desmond, R.; Otte, D. A. L.; Alwedi, E.;
Liu, W. J.; et al. Development of a green and sustainable
manufacturing process for gefapixant citrate (MK-7264). Part 1:
Introduction and process overview. Org. Process Res. Dev. 2020, 24,
2445−2452.
(247) Peng, F.; Humphrey, G. R.; Maloney, K. M.; Lehnherr, D.;
Weisel, M.; Levesque, F.; Naber, J. R.; Brunskill, A. P. J.; Larpent, P.;
Zhang, S. W.; et al. Development of a green and sustainable
manufacturing process for gefapixant citrate (MK-7264). Part 2:
Development of a robust process for phenol synthesis. Org. Process
Res. Dev. 2020, 24, 2453−2461.
(248) Basu, K.; Lehnherr, D.; Martin, G. E.; Desmond, R. A.; Lam,
Y.-h.; Peng, F.; Chung, J. Y. L.; Arvary, R. A.; Zompa, M. A.; Zhang,
S.-W.; et al. Development of a green and sustainable manufacturing
process for gefapixant citrate (MK-7264). Part 3: development of a
one-pot formylation−cyclization sequence to the diaminopyrimidine
core. Org. Process Res. Dev. 2020, 24, 2462−2477.
(249) Otte, D. A. L.; Basu, K.; Jellett, L.; Whittington, M.; Spencer,
G.; Burris, M.; Corcoran, E. B.; Stone, K.; Nappi, J.; Arvary, R. A.;
et al. Development of a green and sustainable manufacturing process
for gefapixant citrate (MK-7264). Part 4: Formylation−cyclization as
a flow−batch process leads to significant improvements in process
mass intensity (PMI) and CO generated versus the batch−batch
process. Org. Process Res. Dev. 2020, 24, 2478−2490.
(250) Di Maso, M. J.; Ren, H.; Zhang, S.-W.; Liu, W.; Desmond, R.;
Alwedi, E.; Narsimhan, K.; Kalinin, A.; Larpent, P.; Lee, A. Y.; et al.
Development of a green and sustainable manufacturing process for
gefapixant citrate (MK-7264). Part 5: Completion of the API free
base via a direct chlorosulfonylation process. Org. Process Res. Dev.
2020, 24, 2491−2497.
(251) Rivera, N. R.; Cohen, R. D.; Zhang, S.-W.; Dance, Z. E. X.;
Halsey, H. M.; Song, S.; Bu, X.; Reibarkh, M.; Ren, H.; Lee, A. Y.;
et al. Gefapixant citrate (MK-7264) sulfonamide step speciation
study: Investigation into precipitation−dissolution events during
addition of chlorosulfonic acid. Org. Process Res. Dev. 2023, 27,
286−294.
(252) Maloney, K. M.; Zhang, S.-W.; Mohan, A. E.; Lee, A. Y.;
Larpent, P.; Ren, H.; Humphrey, G. R.; Desmond, R.; DiBenedetto,
M.; Liu, W.; et al. Development of a green and sustainable
manufacturing process for gefapixant citrate (MK-7264). Part 6:
Development of an improved commercial salt formation process. Org.
Process Res. Dev. 2020, 24, 2498−2504.
(253) Mohan, A. E.; DiBenedetto, M.; Alwedi, E.; Ang, Y. S.; Asi
Sihombing, M. S. B.; Chang, H. Y. D.; Cote, A.; Desmond, R.; DiazSantana, A.; Khong, E.; et al. Development and Demonstration of a
Co-feed Process to Address Form and Physical Attribute Control of
the Gefapixant (MK-7264) Citrate Active Pharmaceutical Ingredient.
Org. Process Res. Dev. 2021, 25, 541−551.


AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
SYN
https://pubs.acs.org/doi/abs/10.1021/acs.oprd.0c00247

A scalable two-pot sulfonamidation through the process has been developed for the synthesis of gefapixant citrate, a P2X3 receptor antagonist that is under investigation for the treatment of refractory and unexplained chronic cough. Direct conversion of the diaryl ether precursor to a sulfonyl chloride intermediate using chlorosulfonic acid, followed by treatment with aqueous ammonia hydroxide, provided the desired sulfonamide in high yield. A pH-swing crystallization allowed for the formation of a transient acetonitrile solvate that enables the rejection of two impurities. After drying, the desired anhydrous free base form was isolated in high yield and purity.
SYN
https://www.sciencedirect.com/science/article/abs/pii/S1566070221000898
Gefapixant is the approved generic name for a compound also known as MK-7264, and prior to that AF-219 and RO-4926219. It is the first-in-class clinically developed antagonist for the P2X3 subtype of trimeric ionotropic purinergic receptors, a family of ATP-gated excitatory ion channels, showing nanomolar potency for the human P2X3 homotrimeric channel and essentially no activity at related channels devoid of P2X3 subunits. As the first P2X3 antagonist to have progressed into clinical studies it has now progressed to the point of successful completion of Phase 3 investigations for the treatment of cough, and the NDA application is under review with US FDA for treatment of refractory chronic cough or unexplained chronic cough. The molecule was discovered in the laboratories of Roche Pharmaceuticals in Palo Alto, California, but clinical development then continued with the formation of Afferent Pharmaceuticals for the purpose of identifying the optimal therapeutic indication for this novel mechanism and establishing a clinical plan for development in the optimal patient populations selected. Geoff Burnstock was a close collaborator and advisor to the P2X3 program for close to two decades of discovery and development. Progression of gefapixant through later stage clinical studies has been conducted by the research laboratories of Merck & Co., Inc., Kenilworth, NJ, USA (MRL; following acquisition of Afferent in 2016), who may commercialize the product once authorization has been granted by regulatory authorities.
PATENT
WO 2008040652
https://patents.google.com/patent/WO2008040652A1/en

SCHEME AExample 1: 5-(2,4-Diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonamideThe synthetic procedure used in this Example is outlined in Scheme B.


not isolated


SCHEME BStep 1 2-Isopropyl-4-methoxy-phenolTo a cooled solution of l-(2-hydroxy-5-methoxy-phenyl)-ethanone (10.0 kg) in 79.0 kg of THF was gradually added 46.4 kg of 3M solution of MeMgCl in THF at a rate such that the reaction mixture temperature did not exceed 25°C. Following addition of the MeMgCl solution, the reaction mixture was stirred at ambient temperature for 18 hours, at which point HPLC (high pressure liquid chromatography) analysis showed more than 98% conversion of l-(2-hydroxy-5-methoxy-phenyl)-ethanone to 2- (1 -hydroxy- 1- methyl-ethyl)-4-methoxy-phenol (not shown in Scheme D). To the stirred solution was then added 10% palladium on carbon (1.02 kg, 50% water wet) suspended in 3.5 kg of THF. The reaction mixture was cooled and placed under a hydrogen atmosphere at 0.34 atmosphere pressure, and concentrated HCl (19.5 kg) was added while maintaining the reaction temperature at 25°C. The resultant mixture was stirred at ambient temperature for 18 hours, then treated with 44.4 kg water and filtered through a bed of Celite to remove suspended catalyst. The filter cake was rinsed with EtOAc and the combined filtrate was separated. The organic phase was washed with water, then concentrated by distillation to provide an oil. This oil was dissolved in 2-butanone (20.4 kg) and the crude solution was employed directly in the next step. A 161.8 g aliquot of the solution was concentrated under vacuum to provide 49.5 g of 2-isopropyl-4-methoxyphenol as an oil, projecting to 10.4 kg crude contained product in the bulk 2-butanone solution. 1H NMR (DMSO) delta: 1.14 (d, 6H, J = 6.9 Hz), 3.18 (septet, IH, J = 6.9 Hz), 3.65 (s, 3H), 6.56, (dd, IH, J = 8.6 Hz, 3.1 Hz), 6.67 (d, IH, J = 3.1 Hz), 6.69 (d, IH, 8.6 Hz).Step 2 (2-Isopropyl-4-methoxy-phenoxy)-acetonitrileA stirred slurry of toluene-4-sulfonic acid cyanomethyl ester (13.0 kg), potassium carbonate (13.0 kg) and 2-isopropyl-4-methoxyphenol (9.57 kg) in 79.7 kg of 2-butanone was heated to 55-600C for 4 days, then heated to reflux for 18 hours. The resultant slurry was cooled and filtered to remove solids. The filtrate was concentrated under reduced pressure and the residue was redissolved in toluene. The toluene solution was extracted with IN KOH, and the organic phase was concentrated by distillation to give 20.6 g of a 1:1 (by weight) solution of (2-isopropyl-4-methoxy-phenoxy)-acetonitrile in toluene, which was used directly in the next step. A aliquot (96.7 g) of this solution was concentrated to dryness to give 50.9 g of crude (2-isopropyl-4-methoxy-phenoxy)- acetonitrile, projecting to a yield of 10.9 kg in the bulk solution: MS (M+H) = 206; 1H NMR (CDCl3) delta: 1.25 (d, J = 6.9 Hz), 3.31 (septet, IH, J = 6.9 Hz), 3.82 (s, 3H), 4.76 (s, 2H), 6.73 (dd. IH, J = 8.8 Hz, 3.1 Hz), 6.87 (d, IH, J = 3.1 Hz), 6.91 (d, IH, J = 8.8 Hz).Step 3 5-(2-Isopropyl-4-methoxy-phenoxy)-pyrimidine-2,4-diamine An approximately 1:1 (by weight) solution of 10.6 kg of (2-isopropyl-4-methoxy-phen- oxy) -acetonitrile in toluene was concentrated under reduced pressure and the residue was treated with 10.8 kg of tert-butoxybis(dimethylamino)methane (Brederick’s Reagent). The resulting mixture was dissolved in 20.2 kg of DMF and the solution was heated to 1100C for 2 hours, at which point HPLC analysis showed essentially complete conversion to 3,3-bis-dimethylamino-2-(2-isopropyl-4-methoxy-phenoxy)-propionitrile (not isolated, 1H NMR (CDCl3) delta: 1.21 (d, 3H, J = 7.2 Hz), 1.23 (d, 3H, J = 7.1 Hz), 2.46 (s, 6H), 2.48 (s, 6H), 3.43 (d, IH, J = 5.0 Hz), 3.31 (septet, IH, J = 6.9 Hz), 3.79 (s, 3H), 4.93 (d, IH, J = 5.0 Hz), 6.70 (dd, IH, J = 8.8 Hz, 3.0 Hz), 6.82 (d, IH, J = 3.0 Hz), 6.98 (d, IH, J = 8.8 Hz). The DMF solution was cooled and transferred onto 14.7 kg of aniline hydrochloride. The resulting mixture was heated to 1200C for 22 hours, at which point HPLC analysis showed greater than 97% conversion to 2-(2-isopropyl-4-methoxy-phenoxy)-3- phenylamino-acrylonitrile (not isolated, 1H nmr (CDCl3) delta: 1.31 (d, 6H, J = 6.9 Hz), 3.39 (septet, IH, J = 6.9 Hz), 3.82 (s, 3H), 6.61 (d (br), IH, J = 12.7 Hz), 6.73 (dd, IH, J = 8.9 Hz, 3.1 Hz), 6.88 (d, IH, J = 3.0 Hz), 6.93 (m, 2H), 6.97 (d, IH, J = 8.9 Hz), 7.05 (m, IH), 7.17 (d, IH, J = 12.6 Hz), 7.35 (m. 2H)).The mixture was cooled, diluted with 21.5 kg toluene, then with 72.2 L of water. The organic layer was separated, washed with water, and concentrated by distillation. The concentrate was transferred into 23.8 kg DMF, and the DMF solution was transferred onto 6.01 kg of guanidine carbonate. The resulting mixture was heated to 1200C for 3 days, at which point HPLC analysis showed greater than 95% conversion of 2-(2- isopropyl-4-methoxy-phenoxy)-3-phenylamino-acrylonitrile into 5-(2-Isopropyl-4- methoxy-phenoxy)-pyrimidine-2,4-diamine. The reaction mixture was cooled, diluted with 7.8 kg of EtOAc, then reheated to 600C. Water (75.1 L) was added and the resultant mixture was allowed to cool to ambient temperature. The precipitated solid was collected by filtration, rinsed with isopropanol and dried under vacuum at 50 degrees to give 9.62 kg of 5-(2-isopropyl-4-methoxy- phenoxy)-pyrimidine-2,4-diamine: m.p. 170-171 degrees C; MS (M+H) = 275; H nmr (chloroform) delta: 1.25 (d, 6H, J = 6.9 Hz), 3.30 (septet, IH, J = 6.9 Hz), 3.79 (s, 3H), 4.68 (br, 2H), 4.96 (br, 2H), 6.64 (dd, IH, J = 8.9 Hz, 3.0 Hz), 6.73, d, J = 8.9 Hz), 6.85 (d, IH, J = 3 Hz), 7.47 (s, IH).Step 4 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfon- amide, sulfolane solvate Chlorosulfonic acid (13.82 kg) was added to a slurry of 5-(2-isopropyl-4-methoxy-phen- oxy)-pyrimidine-2,4-diamine (10.07 kg) in sulfolane (50.0 kg) at a rate to maintain an internal pot temperature below 65°C. The reaction mixture was aged at 60-650C for 12 hours, at which point HPCL showed that all 5-(2-isopropyl-4-methoxy-phenoxy)- pyrimidine-2,4-diamine starting material had been converted to 5-(2,4-diamino- pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonic acid. MS (M+H) = 355. Phosphorus oxychloride (3.41 kg) was then added to the reaction mixture at 600C. The reaction mixture was heated to 75°C and aged for 12 hours, at which point HPLC showed that approximately 99% of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonic acid had been converted to 5-(2,4-diamino-pyrimidin-5-yloxy)-4-iso- propyl-2-methoxy-benzenesulfonyl chloride. MS (M+H) = 373. The solution of 5-(2,4- diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonyl chloride was then cooled to around 2°C).To a cooled (ca. 2°C) solution of ammonia (7N) in MeOH (74.1 kg) was added the cooled sulfolane solution of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonyl chloride (a homogeneous syrup) at a rate such that the internal temperature did not exceed 23°C. The resultant slurry was stirred for 18 hours at ambient temperature, then filtered on a coarse porosity frit filter. The collected solids were rinsed with MeOH (15.9 kg), then dried under reduced pressure at 700C to a constant weight of 23.90 kg. HPLC showed 97.5% conversion of 5-(2,4-diamino- pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonyl chloride to 5-(2,4-diamino- pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonamide sulfolane solvate. H nmr (DMSOd6) delta: 1.26 (d, 6H, J = 6.9 Hz), 2.07 (sym. m, 8H), 2.99 (sym. m, 8H), 3.41 (septet, IH, J = 6.9 Hz), 3.89 (s, 3H), 6.03 (s (br), 2H), 6.58 (s (br), 2H), 7.00 (s, IH), 7.04 (s (br), 2H), 7.08 (s, IH), 7.35 (s, IH).
Step 5 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzene- sulfonamideA slurry of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfon- amide sulfolane solvate (23.86 kg) in a mixture of ethanol (74.3 kg) and 0.44 N HCl (109.4 kg) was heated to reflux to provide a homogeneous solution of the monohydrochloride salt of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonamide. This solution was filterd while hot, then treated with concentrated ammonium hydroxide (3.4 L) to liberate the free base of 5-(2,4-diamino-pyrimidin-5- yloxy)-4-isopropyl-2-methoxy-benzenesulfonamide. The resultant mixture was cooled slowly to 200C and the crystalline product isolated by filtration. The filter cake was washed with water (20.1 kg) and dried under reduced pressure at 700C to a constant weight of 8.17 kg (57.7% yield based on di-solvate of sulfolane).MP = 281-282 0C.1H nmr (DMSOd6) delta: 1.27 (d, 6H, J = 6.9 Hz), 3.41 (septet, IH, J = 6.9 Hz), 3.89 (s, 3H), 5.87 (s (br), 2H), 6.40 (s (br), 2H), 6.98 (s, IH), 7.01 (s (br), 2H), 7.07 (s, IH), 7.36 (s, IH).
PATENT
US 20080207655https://patents.google.com/patent/US20080207655
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016004358
xample 20
5-(2,4-Diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-N-methyl-benzenemethylsulfonamide Step 1. 5-(2,4-Diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonyl chloride
[211] A mixture of pyrimidine (0.400 g, 1.5 mmol) in 2 ml chlorosulfonic acid was allowed to stir 20 min. The mixture was poured over ice. The precipitate was filtered, washed by cold H2O and dried under vacuum to afford 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonyl chloride (0.515 g, 95%) as a white solid; [MH]+= 373.
PATENT
WO 2017058645
https://patents.google.com/patent/WO2017058645A1/en
PATENTDisclosed herein is a novel process for preparing Compound A, a phenoxy diaminopyrimidine compound of the following formula, or a pharmaceutically acceptable salt thereof:

Compound A.Also disclosed herein are various salts and solvates of Compound A.
Scheme 1


Step 1. Preparation of 4-Bromo-2-isopropylphenol DABCO Co-crystalStep 1. Preparation of 4-Bromo-2-isopropylphenol DABCO Co-crystalThe following 4-bromo-2-isopropylphenol hemi-DABCO co-crystal is obtained in greater than 99% purity and at about 85-92% yield by the following process:

To a solution of 2-isopropyl phenol (75.0 g, 550 mmol) in acetonitrile (225 mL) was added MSA (0.520 g, 5.41 mmol). The mixture was cooled to -10 °C and NBS (98.01 g, 550 mmol) was added in portions while maintaining the internal temperature below 10 °C. The reaction was aged for 30 min to 1 h and then warmed to 20 °C, diluted with water (450 mL), and extracted with toluene (225 mL). The organic layer was sequentially washed with 9 wt% phosphoric acid (150 mL) and 5 wt% NaCl (150 mL). The organic layers were concentrated to roughly 150 mL and filtered into a clean reactor. The mixture was heated to 30-40 °C and n- heptane (28.5 mL) was added followed by DABCO (30.89 g, 275 mmol). The mixture was seeded (a seed can be synthesized from a previous batch of this procedure preformed without seeding) with 4-bromo-2-isopropylphenol hemi-DABCO co-crystal (75 mg, 0.277 mmol), diluted with 52.5 mL of n-heptane, and stirred for 1 h. The slurry was cooled to 20 °C over 1 h and 370 mL of n-heptane is added over 2 h. The slurry was cooled to 5 °C over 2 h, aged for 2 h, filtered, and washed with n-heptane (2 x 75 mL). The solid was dried at 20-25 °C under vacuum to yield 4-bromo-2-isopropylphenol hemi-DABCO co-crystal (134.8 g, 90 %) as a solid. 1H NMR (400 MHz, DMSO-76) d 7.20 (d, J= 2.5 Hz, 1H), 7.13 (dd, J= 8.5, 2.6 Hz, 2H), 6.73 (d, J = 8.5 Hz, 2H), 3.16 (hept, J= 6.9 Hz, 2H), 2.60 (s, 12H), 1.14 (d, J= 6.9 Hz, 12H).The crystallization of step 1 generates 4-bromo-2-isopropylphenol hemi-DABCO co-crystal, bromophenol mono-DABCO co-crystal, or a mixture of bromophenol hemi-DABCO co-crystal and bromophenol mono-DABCO co-crystal. An XRPD pattern of bromophenol hemi- DABCO co-crystal is shown in Figure 1.
The bromo-phenol mono-DABCO co-crystal can be generated in the following procedure:

bromophenol DABCO co-crystalTo a vial with a stir bar was charged DABCO (1.7 g, 15 mmol), phenol (2.5 g, 15 mmol), and 2 mL of n-heptane. The resulting slurry was stirred at 23 °C overnight. The slurry was then filtered and the resulting wet cake was washed with 2 mL of 5 °C n-heptane. The cake was dried under vacuum with nitrogen sweep to afford 4-bromo-2-isopropylphenol mono- DABCO co-crystal (2.9 g, 70% yield) as a solid. 1H NMR (500 MHz, DMSO-76) d 9.65 (s, 1H), 7.20 (s, 1H), 7.14 (d, J= 8.5 Hz, 1H), 6.74 (d, J= 8.5 Hz, 1H), 3.17 (hept, J= 6.8 Hz, 1H), 2.61(s, 12H), 1.15 (d, 7 = 6.9 Hz, 6H).An XRPD pattern of bromophenol mono-DABCO co-crystal is shown in Figure 2.Step 2a. Preparation of 2-Isopropyl-4-Methoxyphenol
The 2-isopropyl-4-Methoxyphenol shown below is obtained at about 92% yield by the following process:

bromophenol DABCO co-crystal methoxy phenolTo a solution of 4-bromo-2-isopropylphenol hemi-DABCO co-crystal (120 g, 442 mmol) in 25 wt% sodium methoxide in methanol (430 g) was added 60 mL of DMF. The solution was pressure purged with nitrogen, copper (I) bromide (3.23 g, 22.5 mmol) was added to the mixture, and the reaction was heated to reflux for 12-16 h. The reaction is cooled to 0-5 °C and quenched with 6M HC1 until the pH of the solution is less than 5. The slurry is diluted with 492 mL of toluene and 720 mL of water to provide a homogeneous solution with a rag between the layers. The aqueous layer is cut to waste. The organic layer is filtered to remove the rag and washed with 240 mL of water to provide 2-isopropyl-4-methoxylphenol (491 g, 13.3 wt%, 89% assay yield) as a solution in toluene. 1H NMR (500 MHz, DMSO-76) d 8.73 (s, 1H), 6.68 (d, J = 8.6 Hz, 1H), 6.66 (d, 7= 3.0 Hz, 1H), 6.55 (dd, 7= 8.6, 3.1 Hz, 1H), 3.65 (s, 3H), 3.17 (hept, j = 6.9 Hz, 1H), 1.14 (d, 7= 6.9 Hz, 6H).Step 2b. Preparation of 2-Isopropyl-4-Methoxyphenol
Alternatively, the methoxy phenol is obtained by the following process:

To a high-pressure vessel were charged 400 mL of anhydrous toluene, Re2(CO)io (3.16 g, 4.84 mmol) and mequinol (100 g, 806 mmol) at RT. The vessel was then degassed with propylene, and charged with propylene (85.0 g, 2.02 mol). The vessel was sealed and heated to 170 °C. Internal pressure was measured near 250 psi. The reaction was stirred at this condition for 72 h. The vessel was then allowed to cool down to 23 °C. The internal pressure was carefully released to 1 atmospheric pressure, and the toluene solution was assayed as 91% and used directly in the next step or isolated as a solid.Step 2a/2b results in anhydrous 2-isopropyl-4-methoxyphenol form 1. An XRPD pattern of the methoxy phenol form 1 is shown in Figure 3.In another embodiment, the product is isolated as a DMAP co-crystal:

To a vial with a stir bar was charged DMAP (3.67 g, 30.1 mmol), 2.5 ml of toluene, and 2-isopropyl-4-methoxylphenol (5.00 g, 30.1 mmol). The reaction mixture was stirred at RT for 5 min, and a homogeneous solution was formed. The reaction mixture was then cooled to 5 °C. Ten mL of n-heptane was slowly charged over 20 min. The resulting slurry was stirred at 5 °C overnight. The slurry was filtered and the resulting wet cake was washed with 3 mL of 5 °C n-heptane. The cake was dried under vacuum with a nitrogen sweep to provide 2- isopropyl-4-methoxylphenol DMAP co-crystal (7.01 g, 81%) as a solid. 1H NMR (500 MHz, DMSO-76) d 8.78 (s, 1H), 8.10 (d, J= 6.1 Hz, 2H), 6.71 – 6.65 (m, 2H), 6.57 (dd, J= 11.3, 6.0 Hz, 3H), 3.66 (s, 3H), 3.17 (hept, J= 6.8 Hz, 1H), 2.95 (s, 6H), 1.14 (d, J= 6.9 Hz, 6H).The crystallization generates anhydrous 2-isopropyl -4-methoxyphenol DMAP co crystal. An XRPD pattern of the 2-isopropyl-4-methoxyphenol DMAP co-crystal is shown in Figure 4.Step 3a. Preparation of the Cvanoether. 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile
The cyanoether is obtained at about 95 % yield by the following process:

A 12-15 wt% solution of 2-isopropyl-4-methoxylphenol (314.3 g, 12 wt%, 226.8 mmol) was concentrated to greater than 50 wt% 2-isopropyl-4-methoxyphenol in toluene under vacuum at 40-50°C. To the solution was added 189 mL of NMP, and the mixture was cooled to 5 °C. Sodium hydroxide (27.2 g, 50 wt% in water, 340 mmol) and chloroacetonitrile (36 g, 340 mmol) were added sequentially to the mixture while maintaining the internal temperature below 10 °C. The reaction was aged for 2 h and then diluted with 150 mL of toluene and 226 mL of water while maintaining the temperature below 10 °C. The mixture was warmed to 20-25 °C, the layers were separated, and the organic layer was washed with 75 mL of 20 wt% NaCl (aq.). The organic layer was and filtered to provide 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile (56.8 g, 74.6 wt%) as a solution in toluene. The filter was washed with NMP to provide additional 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile (27.1 g, 5.0 wt%) as a solution in NMP. The combined yield was about 94 %. 1H NMR (500 MHz, DMSO-i¾) d 7.05 (d, J= 8.8 Hz, 1H), 6.81 (d, 7= 3.0 Hz, 1H), 6.78 (dd, j= 8.8, 3.1 Hz, 1H), 5.11 (s, 2H), 3.73 (s, 3H), 3.20 (hept, j = 6.9 Hz, 1H), 1.17 (d, 7= 6.9 Hz, 6H).Step 3b. Preparation of the Cvanoether. 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile
Alternatively, the cyanoether shown below is obtained at about 92% yield by the following process:

A solution of 2-isopropyl-4-methoxyphenol in toluene (491 g, 13.3 wt%, 393 mmol) was concentrated and solvent switched to acetonitrile under vacuum at 40-50 °C.Potassium carbonate (164.5 g, 1190 mmol) and tetrabutylammonium hydrogensulfate (1.5 g, 4.42 mmol) were added to a separate vessel, and the vessel was pressure purged with nitrogen gas.The solution of phenol in acetonitrile and chloroacetonitrile was added sequentially to the reaction vessel. The vessel was heated to 40 °C and aged for 4 h. The mixture was allowed to cool to 25 °C, and was diluted with 326 mL water. The layers were separated, and the organic layer was washed with 130 mL of 10 wt% NaCl. A solvent switch to toluene was performed under vacuum, and the organic layer was filtered through two 16D Cuno #5 cartridges. The organic layer was concentrated to provide 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile in toluene (128.2 g, 58 wt%, 92% yield).Step 4 Preparation of the Dia inopyrimidine 5-(2-isopropyl-4-methoxyphenoxy)pyrimidine-2.4-di amineThe diaminopyrimidine is obtained at about 90 % yield by the following process:

A solution of potassium tert-butoxide (44.8 g, 0399 mmol) in NMP (180 mL) was cooled to -10 °C. A solution of 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile, the cyanoether, (59.3 g, 61.4 wt%, 177 mmol) in toluene and ethyl formate (26.3 g, 355 mmol) was charged to the base solution while maintaining the internal temperature between -12 °C and -8 °C. After a 3 h age, guanidine hydrochloride (136 g, 1420 mmol) was added to the mixture and the reaction was heated to 115 °C for 6 h. The mixture was allowed to cool to 90 °C, diluted with 200 mL of water, and aged until the reaction mixture was homogeneous (about 30-45 min). After all solids dissolved, vacuum (400 mm Hg) was applied to the reactor to remove toluene. Vacuum was disconnected and the solution was allowed to cool to 85°C. 5-(2-Isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine seed (49.8 mg) (a seed can be synthesized by a route described in U.S. Patent 7,741,484) was charged, the solution was aged for 2 h, 200 mL of water was added, and the batch was allowed to cool to 20 °C over 6 h. The slurry was aged for 10 h at 20 °C, filtered, washed with 2: 1 water :NMP (3 x 100 mL) and water (3 x 100 mL), and dried under vacuum at 50 °C to provide the title compound (42.2 g, 88%) as a solid. 1H NMR (500 MHz, DMSO-r¾) d 7.23 (s, 1H), 6.83 (d, J= 3.0 Hz, 1H), 6.70 (dd, J= 8.9, 3.0 Hz, 1H), 6.63 (d, j= 8.8 Hz, 1H), 6.32 (s, 2H), 5.75 (s, 2H), 3.71 (s, 3H), 3.28 (hept, j= 6.9 Hz, 1H), 1.20 (d, j = 6.9 Hz, 6H); 13C NMR (126 MHz, DMSO-r¾) d 159.7, 157.2, 155.1, 148.4, 144.2, 139.0, 130.4,116.9, 112.5, 111.3, 55.4, 26.57, 22.83.The crystallization of step 4 generates an anhydrous 5-(2-isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine form 1. An XRPD pattern of the 5-(2-isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine form 1 is shown in Figure 5.In one embodiment, 5-(2-isopropyl-4-methoxyphenoxy)pyrimidine-2, 4-diamineNMP solvate 1 is obtained by adding excess amount of 5-(2-isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine form 1 into NMP in a closed vessel to form a suspension. The suspension is stirred at RT until the completion of form transition. The crystals of 5-(2 -isopropyl -4-methoxyphenoxy)pyrimidine-2, 4-diamine NMP solvate 1 can be collected by filtration and measured immediately by XRPD to prevent desolvation. An XRPD pattern of the 5-(2 -isopropyl -4-methoxyphenoxy)pyrimidine-2, 4-diamine NMP solvate 1 is shown in Figure 6.Step 5. Preparation of Compound A Free BaseCompound A free base is obtained at about 91% yield by a process comprising the steps:

To a suspension of 5-(2 -isopropyl -4-methoxyphenoxy)pyrimidine-2, 4-diamine, the diaminopyrimidine, (47.0 g, 171 mmol) in 141 mL of acetonitrile at -10 °C was added chlorosulfonic acid (63.1 mL, 942 mmol) while maintaining the internal temperature below 25 °C. The solution was aged for 1 h at 25 °C and then heated to 45 °C for 12 h. The solution was allowed to cool to 20 °C and added to a solution of 235 mL ammonium hydroxide and 71 mL of acetonitrile at -10 °C while maintaining the internal temperature below 15 °C. The slurry was aged at l0°C for 1 h, heated to 25 °C, and aged for 1 h. The slurry was diluted with 564 mL of water and 188 mL of 50 wt% sodium hydroxide to provide a homogeneous solution that was heated to 35 °C for 2 h. The solution was allowed to cool to 22 °C and the pH of the solution was adjusted to 12.9 with a 2M solution of citric acid. The solution was seeded with Compound A free base (470 mg, 1.19 mmol) (a seed can be synthesized by a route described in U.S. Patent 7,741,484), aged for 2 h, acidified to pH 10.5-11.3 with a 2M solution of citric acid over 5-10 h, and then aged for 2 h. The slurry was filtered, the resulting cake was washed with 90: 10 water: acetonitrile (2 x 118 mL) and water (2 x 235 mL), and dried at 55 °C under vacuum to provide Compound A free base (50.9 g, 91%) as a solid. 1H NMR (500 MHz, DMSO-i¾) d 7.36 (s, 1H), 7.07 (s, 1H), 7.05 – 6.89 (m, 3H), 6.37 (s, 2H), 5.85 (s, 2H), 3.89 (s, 3H), 3.41 (hept, J = 6.6 Hz, 1H), 1.27 (d, J= 6.8 Hz, 6H).The crystallization of step 5 generates anhydrous Compound A free base form 1. In one embodiment, Compound A free base acetonitrile solvate 1 can be prepared by adding excess amount of Compound A free base form 1 into acetonitrile in a closed vessel to form a suspension. The suspension is stirred at 50 °C until the completion of form transition.The crystals of Compound A free base acetonitrile solvate 1 can be collected by filtration and measured immediately by XRPD to prevent desolvation. An XRPD pattern of Compound A free base acetonitrile solvate 1 is shown in Figure 7.Step 6a. Preparation of Compound A Citrate SaltCompound A citrate salt is obtained by a process comprising the steps:

Compound A free base (30.0 g, 84.9 mmol) and glycolic acid (22.6 g, 297 mmol) were added to methanol (360 mL). The solution was heated to 60 °C, aged for 1 h, and filtered through a 0.6 pm filter into a clean vessel. A solution of citric acid (32.6 g, 170 mmol) in 2- propanol (180 mL) at RT was filtered through a 0.6 pm filter into the methanol solution over 30 min while the temperature of the methanol solution was maintained between 58-62 °C. The solution was seeded with Compound A citrate salt (450 mg, 0.825 mmol) (a seed can be synthesized by a route described in patent application number PCT/US17/66562), aged for 1 h, and diluted with 180 mL of 2-propanol over 3 h while the temperature was maintained between 58-62 °C. The slurry was cooled to 50 °C over 3 h. The slurry was filtered at 50 °C, washed with 1 : 1 methanol :2-propanol (120 mL) and 2-propanol (120 mL) at 50 °C, and dried under vacuum at 35 °C to provide Compound A citrate salt (45.1 g, 97%) as a solid. 1H NMR (400 MHz, DMSO-76) d 10.89 (s, 3H), 7.33 (s, 1H), 7.10 (s, 1H), 7.07 (s, 3H), 7.04 (s, 2H), 6.44 (s, 2H), 3.91 (s, 3H), 3.34 (hept, J= 6.7 Hz, 1H), 2.69 (d, 7= 15.3 Hz, 2H), 2.60 (d, 7= 15.3 Hz, 2H), 1.26 (d, 7= 6.9 Hz, 6H). Step 6b. Alternative preparation of Compound A Citrate SaltAlternatively, Compound A citrate salt is obtained by a process comprising the steps:

To a suspension of Compound A citrate salt (4.5 g, 8.25 mmol) in methanol (72 mL) and 2-propanol (36 mL) at 50 °C were added simultaneously through separate 0.6 pm filters a solution of Compound A free base (30.0 g, 84.9 mmol) and glycolic acid (22.6 g, 297 mmol) in 360 mL of methanol at 50 °C and a solution of citric acid (19.5 g, 101 mmol) in 180 mL of 2- propanol at 25 °C over 8 h while maintaining the seed solution temperature of 60 °C. After the simultaneous addition is complete, citric acid (13.2 g, 68.7 mmol) in 180 mL of 2-propanol was added to the slurry over 8 h while the temperature was maintained at 60 °C. The slurry was allowed to cool to 50 °C and aged for 1 h, filtered at 50 °C, washed with 1 : 1 methanol :2- propanol (2 x 120 mL) and 2-propanol (120 mL), and dried under vacuum at 35 °C to provide Compound A citrate salt (45.1 g, 88%) as a solid.The crystallization of step 6a/6b generates anhydrous Compound A citrate form 1. In another embodiment, Compound A citrate methanol solvate 1 can be prepared via a saturated solution of Compound A citrate form 1 in methanol at 50C. The solution is naturally cooled to ambient temperature or evaporated at ambient temperature until the crystals of Compound A citrate methanol solvate 1 can be acquired. An XRPD pattern of Compound A citrate methanol solvate 1 is shown in Figure 8.
PATENT
https://patents.google.com/patent/CN111635368B/enPreparation of the Compound Gefapixant of example 11Adding compound 7(16g) and dichloromethane (64mL) into a 250mL three-necked bottle, stirring for dissolving, cooling to below 5 ℃ in an ice bath, dropwise adding a mixed solution of chlorosulfonic acid (21.1g) and dichloromethane (16mL) into the reaction solution, and stirring for 1 hour at the temperature of not higher than 5 ℃; then heating to room temperature and continuing stirring for 10 hours, after the reaction is finished, pouring the reaction liquid into ice water, and quickly separating a water layer; the organic layer was washed once with ice water, dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give a crude product. Dissolving the crude product with 30ml of acetonitrile, and cooling to below 5 ℃; 16ml of ammonia water (25-28%) is dripped into the solution, and after the dripping is finished, the solution is heated to room temperature and stirred for 20 hours. After the reaction is completed, concentrating the reaction solution under reduced pressure to remove acetonitrile, and separating out a white solid; and filtering again, and drying the filter cake at 70 ℃ under reduced pressure for 24h to obtain Gefapixant: white powder (19.50g), yield 94.6%, purity: 97.2 percent.Example 12 purification of the Compound GefapixantAdding a compound Gefapixant (20.77g) into a 500mL reaction bottle, adding 0.44N hydrochloric acid (95.4mL), absolute ethyl alcohol (64.4g) and nitrogen protection, heating to 75 ℃, stirring for dissolving, then carrying out heat preservation and reflux for 1 hour, filtering while hot, after filtering, heating the filtrate again to 60 ℃, dropwise adding ammonia water (25-28 percent and 2.96mL), closing and heating after dropwise adding, slowly cooling to room temperature, and gradually precipitating white solids. And continuously cooling the reaction solution to 20 ℃, keeping the temperature and stirring for 4h, filtering, washing a filter cake with 15ml of water, and performing vacuum drying on the obtained wet product at 60 ℃ for 24h to obtain Gefapixant: white powder (6.58g), yield 53.2%, purity: 99.5 percent.1H NMR(400MHz,DMSO)δ7.37(s,1H),7.08(s,1H),7.02(s,2H),7.00(s,1H),6.43(brs,2H),5.89(s,2H),3.90(s,3H),3.42(m,1H),1.28(d,J=8.0Hz,6H);LC-MS:m/z=354.1[M+H]+。
//////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Muccino D, Green S (June 2019). “Update on the clinical development of gefapixant, a P2X3 receptor antagonist for the treatment of refractory chronic cough”. Pulmonary Pharmacology & Therapeutics. 56: 75–78. doi:10.1016/j.pupt.2019.03.006. PMID 30880151.
- ^ Richards D, Gever JR, Ford AP, Fountain SJ (July 2019). “Action of MK-7264 (gefapixant) at human P2X3 and P2X2/3 receptors and in vivo efficacy in models of sensitisation”. British Journal of Pharmacology. 176 (13): 2279–2291. doi:10.1111/bph.14677. PMC 6555852. PMID 30927255.
- ^ Marucci G, Dal Ben D, Buccioni M, Martí Navia A, Spinaci A, Volpini R, Lambertucci C (December 2019). “Update on novel purinergic P2X3 and P2X2/3 receptor antagonists and their potential therapeutic applications”. Expert Opinion on Therapeutic Patents. 29 (12): 943–963. doi:10.1080/13543776.2019.1693542. hdl:11581/435751. PMID 31726893. S2CID 208037373.
- ^ Ford, Anthony P.; Dillon, Michael P.; Kitt, Michael M.; Gever, Joel R. (November 2021). “The discovery and development of gefapixant”. Autonomic Neuroscience. 235: 102859. doi:10.1016/j.autneu.2021.102859.
| Clinical data | |
|---|---|
| ATC code | R05DB29 (WHO) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1015787-98-0 |
| PubChem CID | 24764487 |
| DrugBank | DB15097 |
| ChemSpider | 58828660 |
| UNII | 6K6L7E3F1L |
| KEGG | D11349 |
| ChEMBL | ChEMBL3716057 |
| Chemical and physical data | |
| Formula | C14H19N5O4S |
| Molar mass | 353.40 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////Gefapixant, Lyfnua, JAPAN 2022, APPROVALS 2022, ゲーファピキサントクエン酸塩 , MK 7264, 吉法匹生 , AF 217

NEW DRUG APPROVALS
ONE TIME
$10.00
UPDATE
.WO/2022/060945SOLID STATE FORMS OF GEFAPIXANT AND PROCESS FOR PREPARATION THEREOF
TEVA
Gefapixant, 5-(2, 4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonamide, has the following chemical structure:
[0003] Gefapixant is a purinergic P2X3 receptor antagonist, and it is developed for the treatment of chronic cough. Gefapixant is also under clinical investigation as a treatment for asthma, interstitial cystitis, musculoskeletal pain, pelvic pain, and sleep apnea syndrome.
[0004] The compound is described in International Publication No. WO 2005/95359.
International Publication No. WO 2008/040652 disclosed a sulfonate solvate of Gefapixant. International Publication Nos. WO 2018/118668 and WO 2019/209607 disclose crystalline forms of Gefapixant as well as Gefapixant salts.
[0005] Polymorphism, the occurrence of different crystalline forms, is a property of some molecules and molecular complexes. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties like melting point, thermal behaviors (e.g., measured by thermogravimetric analysis (“TGA”), or differential scanning calorimetry (“DSC”)), X-ray diffraction (XRD) pattern, infrared absorption fingerprint, and solid state (13C) NMR spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
[0006] Different salts and solid state forms (including solvated forms) of an active pharmaceutical ingredient may possess different properties. Such variations in the properties of different salts and solid state forms and solvates may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, changing the dissolution profile in a favorable direction, or improving stability (polymorph as well as chemical stability) and shelf-life. These variations in the properties of different salts and solid state forms may also offer improvements to the final dosage form, for instance, if they serve to improve bioavailability. Different salts and solid state forms and solvates of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms, which may in turn provide additional opportunities to assess variations in the properties and characteristics of a solid active pharmaceutical ingredient.
[0007] Discovering new solid state forms and solvates of a pharmaceutical product may yield materials having desirable processing properties, such as ease of handling, ease of processing, storage stability, and ease of purification or as desirable intermediate crystal forms that facilitate conversion to other polymorphic forms. New solid state forms of a pharmaceutically useful compound can also provide an opportunity to improve the performance characteristics of a pharmaceutical product. It enlarges the repertoire of materials that a formulation scientist has available for formulation optimization, for example by providing a product with different properties, including a different crystal habit, higher crystallinity, or polymorphic stability, which may offer better processing or handling characteristics, improved dissolution profile, or improved shelf-life (chemi cal/phy si cal stability). For at least these reasons, there is a need for additional solid state forms (including solvated forms) of Gefapixant or salts or co-crystals thereof.
FLUPHENAZINE
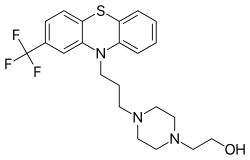
Fluphenazine
- Molecular FormulaC22H26F3N3OS
- Average mass437.522 Da
- SQ 10733
- Squibb 16144
UNIIS79426A41Z
CAS number69-23-8
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Fluphenazine decanoate | FMU62K1L3C | 5002-47-1 | VIQCGTZFEYDQMR-UHFFFAOYSA-N |
| Fluphenazine enanthate | QSB34YF0W9 | 2746-81-8 | LRWSFOSWNAQHHW-UHFFFAOYSA-N |
| Fluphenazine hydrochloride | ZOU145W1XL | 146-56-5 | MBHNWCYEGXQEIT-UHFFFAOYSA-N |
2-(Trifluoromethyl)-10-[3-[1-(b-hydroxyethyl)-4-piperazinyl]propyl]phenothiazine
200-702-9[EINECS]
4-(3-(2-(trifluoromethyl)phenothiazin-10-yl)propyl)-1-Piperazineethanol
4-[3-[2-(Trifluoromethyl)-10H-phenothiazin-10-yl]propyl]-1-piperazineethanol
69-23-8[RN]
فلوفينازين[Arabic][INN]
氟奋乃静[Chinese][INN]
1-(2-Hydroxyethyl)-4-[3-(trifluoromethyl-10-phenothiazinyl)propyl]piperazine
10-[3′-[4”-(b-Hydroxyethyl)-1”-piperazinyl]propyl]-3-trifluoromethylphenothiazine
1-Piperazineethanol, 4-(3-(2-(trifluoromethyl)-10H-phenothiazin-10-yl)propyl)-
1-Piperazineethanol, 4-[3-[2-(trifluoromethyl)-10H-phenothiazin-10-yl]propyl]-
read https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/071413s019lbl.pdfFluphenazineCAS Registry Number: 69-23-8
CAS Name: 4-[3-[2-(Trifluoromethyl)-10H-phenothiazin-10-yl]propyl]-1-piperazineethanol
Additional Names: 1-(2-hydroxyethyl)-4-[3-(trifluoromethyl-10-phenothiazinyl)propyl]piperazine; 10-[3¢-[4¢¢-(b-hydroxyethyl)-1¢¢-piperazinyl]propyl]-3-trifluoromethylphenothiazine; 2-(trifluoromethyl)-10-[3-[1-(b-hydroxyethyl)-4-piperazinyl]propyl]phenothiazine
Manufacturers’ Codes: S-94; SQ-4918
Molecular Formula: C22H26F3N3OS, Molecular Weight: 437.52
Percent Composition: C 60.39%, H 5.99%, F 13.03%, N 9.60%, O 3.66%, S 7.33%
Literature References: Prepn: H. L. Yale, F. Sowinski, J. Am. Chem. Soc.82, 2039 (1960); GB829246; G. E. Ullyot, US3058979 (1960, 1962 both to SKF); GB833474 (1960 to Scherico), C.A.54, 21143e (1960); E. L. Anderson et al.,Arzneim.-Forsch.12, 937 (1962); H. L. Yale, R. C. Merrill, US3194733 (1965 to Olin Mathieson). Metabolism: J. Dreyfuss, A. J. Cohen, J. Pharm. Sci.60, 826 (1971). Comprehensive description of the enanthate ester: K. Florey, Anal. Profiles Drug Subs.2, 245-262 (1973); of the dihydrochloride: idem,ibid. 263-294; of the decanoate ester: G. Clarke, ibid.9, 275-294 (1980).
Properties: Dark brown viscous oil, bp0.5 268-274°; bp0.3 250-252°.
Boiling point: bp0.5 268-274°; bp0.3 250-252°
Derivative Type: Dihydrochloride
CAS Registry Number: 146-56-5
Trademarks: Anatensol (BMS); Dapotum (BMS); Lyogen (Promonta Lundbeck); Moditen (Sanofi Winthrop); Omca (BMS); Pacinol (Schering); Permitil (Schering); Prolixin (Apothecon); Siqualone (BMS); Tensofin (BMS); Valamina (Schering)
Molecular Formula: C22H26F3N3OS.2HCl, Molecular Weight: 510.44
Percent Composition: C 51.77%, H 5.53%, F 11.17%, N 8.23%, O 3.13%, S 6.28%, Cl 13.89%
Properties: Crystals from abs ethanol, mp 235-237°. Also reported as mp 224.5-226°.
Melting point: mp 235-237°; Also reported as mp 224.5-226°
Derivative Type: Decanoate
CAS Registry Number: 5002-47-1
Manufacturers’ Codes: SQ-10733; QD-10733
Trademarks: Modecate (Sanofi Winthrop)
Molecular Formula: C32H44F3N3O2S, Molecular Weight: 591.77
Percent Composition: C 64.95%, H 7.49%, F 9.63%, N 7.10%, O 5.41%, S 5.42%
Properties: Pale yellow-orange, viscous liquid. Slowly crystallizes at room temp. mp 30-32°. Very sol in chloroform, ether, cyclohexane, methanol, ethanol. Insol in water.
Melting point: mp 30-32°
Derivative Type: Enanthate
CAS Registry Number: 2746-81-8
Manufacturers’ Codes: SQ-16144
Molecular Formula: C29H38F3N3O2S, Molecular Weight: 549.69Percent Composition: C 63.36%, H 6.97%, F 10.37%, N 7.64%, O 5.82%, S 5.83%
Properties: Pale yellow to yellow-orange viscous liquid or oily solid.
Therap-Cat: Antipsychotic.
Keywords: Antipsychotic; Phenothiazines.
Fluphenazine is a phenothiazine used to treat patients requiring long-term neuroleptic therapy.
A phenothiazine used in the treatment of psychoses. Its properties and uses are generally similar to those of chlorpromazine.
Fluphenazine, sold under the brand names Prolixin among others, is a high-potency typical antipsychotic medication.[1] It is used in the treatment of chronic psychoses such as schizophrenia,[1][2] and appears to be about equal in effectiveness to low-potency antipsychotics like chlorpromazine.[3] It is given by mouth, injection into a muscle, or just under the skin.[1] There is also a long acting injectable version that may last for up to four weeks.[1] Fluphenazine decanoate, the depot injection form of fluphenazine, should not be used by people with severe depression.[4]
Common side effects include movement problems, sleepiness, depression and increased weight.[1] Serious side effects may include neuroleptic malignant syndrome, low white blood cell levels, and the potentially permanent movement disorder tardive dyskinesia.[1] In older people with psychosis as a result of dementia it may increase the risk of dying.[1] It may also increase prolactin levels which may result in milk production, enlarged breasts in males, impotence, and the absence of menstrual periods.[1] It is unclear if it is safe for use in pregnancy.[1]
Fluphenazine is a typical antipsychotic of the phenothiazine class.[1] Its mechanism of action is not entirely clear but believed to be related to its ability to block dopamine receptors.[1] In up to 40% of those on long term phenothiazines, liver function tests become mildly abnormal.[5]
Fluphenazine came into use in 1959.[6] The injectable form is on the World Health Organization’s List of Essential Medicines.[7] It is available as a generic medication.[1] It was discontinued in Australia around mid 2017.[8]
Synthesis Reference
Ullyot, G.E.; U.S. Patent 3,058,979; October 16, 1962; assigned to Smith Kline & French Laboratories.
syn
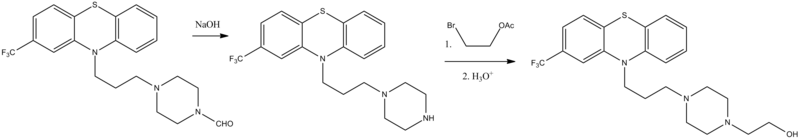
syn
Antipsychotics (Neuroleptics)
R.S. Vardanyan, V.J. Hruby, in Synthesis of Essential Drugs, 2006
Fluphenazine
Fluphenazine, 4-[3-[2-(trifluoromethyl)phenothiazin-10-yl]propyl]-1-piperazineethanol (6.1.8), is synthesized by any of the methods described above [21–27]. Alkylation of 2-trifluoromethylphenothiazine using 4-formyl-1-piperazineylpropylchlo-ride in the presence of sodium amide synthesizes 2-trifluoromethyl-10-[3-(4-formyl-1-piperazinyl)propyl]phenothizine (6.1.6). Further alkaline hydrolysis removes the N-formyl group, giving 2-trifluoromethyl-10-[3-(1-piperazinyl)propyl]phenothiazine (6.1.7). This is alkylated by 2-bromethanol-1 acetate, which upon further acidic hydrolysis removes the protecting acetyl group, yielding fluphenazine (6.1.8) [27,28].

Fluphenazine is an extremely strong antipsychotic drug. A stimulatory effect accompanies the neuroleptic effect. It is used in psychiatry for treating various forms of schizophrenia and other mental illnesses. The most common synonyms are fluorphenazine, moditen, dapotum, motival, permitil, and others.SYN
Manufacturing Process
A suspension of 69.0 grams of 2-trifluoromethylphenothiazine in 1 liter of toluene with 10.9 grams of sodium amide is heated at reflux with high speed stirring for 15 minutes. A solution of 54.1 grams of 1-formyl-4-(3’chloropropyl)-piperazine, [prepared by formylating 1-(3′-hydroxypropyl)piperazine by refluxing in an excess of methyl formate, purifying the 1-formyl4-(3′-hydroxypropyl)-piperazine by vacuum distillation, reacting this compound with an excess of thionyl chloride at reflux and isolating the desired 1-formyl-4(3′-chloropropyl)-piperazine by neutralization with sodium carbonate solution followed by distillation] in 200 ml of toluene is added. The reflux period is continued for 4 hours. The cooled reaction mixture is treated with 200 ml of water. The organic layer is extracted twice with dilute hydrochloric acid. The acid extracts are made basic with ammonia and extracted with benzene. The volatiles are taken off in vacuo at the steam bath to leave a dark brown oil which is 10-[3′-(N-formylpiperazinyl)-propyl]-2trifluoromethylphenothiazine. It can be distilled at 260°C at 10 microns, or used directly without distillation if desired.
A solution of 103.5 grams of 10-[3′-(N-formylpiperazinyl)-propyl]-2trifluoromethylphenothiazine in 400 ml of ethanol and 218 ml of water containing 26 ml of 40% sodium hydroxide solution is heated at reflux for 2 hours. The alcohol is taken off in vacuo on the steam bath. The residue is swirled with benzene and water. The dried benzene layer is evaporated in vacuo. The residue is vacuum distilled to give a viscous, yellow oil, 10(3’piperazinylpropyl)-2-trifluoromethylphenothiazine, distilling at 210° to235°C at 0.5 to 0.6 mm.
A suspension of 14.0 grams of 10-(3′-piperazinylpropyl)-2trifluoromethylphenothiazine, 6.4 grams of β-bromoethyl acetate and 2.6 grams of potassium carbonate in 100 ml of toluene is stirred at reflux for 16 hours. Water (50 ml) is added to the cooled mixture. The organic layer is extracted into dilute hydrochloric acid. After neutralizing the extracts and taking the separated base up in benzene, a viscous, yellow residue is obtained by evaporating the organic solvent in vacuo. This oil is chromatographed on alumina. The purified fraction of 7.7 grams of 10-[3′-(Nacetoxyethylpiperazinyl)-propyl] -2-trifluoromethylphenothiazine is taken up in ethyl acetate and mixed with 25 ml of alcoholic hydrogen chloride. Concentration in vacuo separates white crystals of the dihydrochloride salt, MP 225° to 227°C.
A solution of 1.0 gram of 10-[3′-(N-acetoxyethylpiperazinyl)-propyl]-2trifluoromethylphenothiazine in 25 ml of 1 N hydrochloric acid is heated at reflux briefly. Neutralization with dilute sodium carbonate solution and extraction with benzene gives the oily base, 10-[3′-(N-βhydroxyethylpiperazinyl)-propyl]-2-trifluoromethylphenothiazine. The base is reacted with an excess of an alcoholic hydrogen chloride solution. Trituration with ether separates crystals of the dihydrochloride salt, MP 224° to 226°C, (from US Patent 3,058,979).
Chemical Synthesis
Fluphenazine, 4-[3-[2-(trifluoromethyl)phenothiazin-10-yl]propyl]-1- piperazineethanol (6.1.8), is synthesized by any of the methods described above [21–27]. Alkylation of 2-trifluoromethylphenothiazine using 4-formyl-1-piperazineylpropylchloride in the presence of sodium amide synthesizes 2-trifluoromethyl-10-[3-(4-formyl- 1-piperazinyl)propyl]phenothizine (6.1.6). Further alkaline hydrolysis removes the N-formyl group, giving 2-trifluoromethyl-10-[3-(1-piperazinyl)propyl]phenothiazine (6.1.7). This is alkylated by 2-bromethanol-1 acetate, which upon further acidic hydrolysis removes the protecting acetyl group, yielding fluphenazine (6.1.8) [27,28].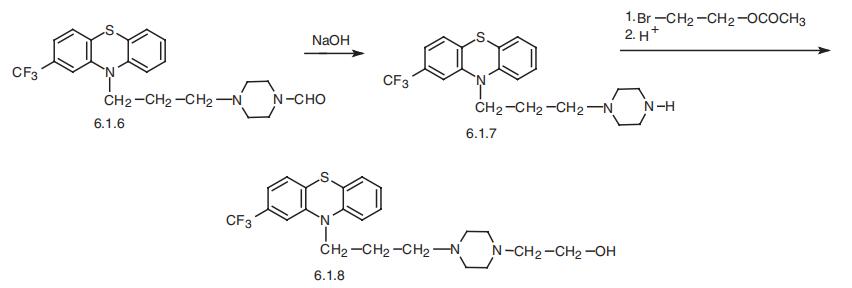
SYN
Indian Pat. Appl., 2014MU02033,
PATENT
CN 105153062
https://patents.google.com/patent/CN105153062A/en
Embodiment 1(1) preparation of 2-trifluoromethyl thiodiphenylamine: by 100g(0.356mol) Tecramine adds in reaction flask, be heated to 180-190 DEG C, open and stir, treat that it melts in backward reaction flask completely and add 10g(0.178mol) iron powder, stirring reaction about 2 hours at 180-190 DEG C of temperature, after reaction terminates, reaction solution is cooled to pour in beaker by reaction solution while hot when 100 DEG C, and iron powder stays (used water flushing) bottom reaction flask.Reaction solution is added underpressure distillation in clean reaction flask, collects 134-135 DEG C of (3mmHg) cut, obtain weak yellow liquid 3-trifluoromethyl pentanoic and be about 67.5g, yield about 80%.By 3-trifluoromethyl pentanoic 60g(0.253mol), sublimed sulphur 8g(0.253mol) add in reaction flask, whipped state is warming up to about 130 DEG C, after the complete melting of sulphur, in reaction flask, add 3g elemental iodine, continue to be warming up to 185-190 DEG C, react about 1 hour at this temperature.There is hydrogen sulfide to release in reaction process, note tail gas absorption.After reaction terminates, reaction solution is cooled to about 100 DEG C, adds 200g toluene in reaction flask, about raised temperature to 100 DEG C, in reaction flask, add 100g water, stir layering while hot after 5 minutes, water layer discarded, toluene layer returns reaction flask, and whipped state borehole cooling, to 15-18 DEG C, filters, filtrate retains (to be recycled apply mechanically 3-trifluoromethyl pentanoic), filter cake adopts 60 DEG C, vacuum to dry 10 hours, and obtain 29g intermediate 2-trifluoromethyl thiodiphenylamine, yield is about 85%(and calculates by sulphur)(2) preparation of 1-(3-chloropropyl)-4-(2-hydroxyethyl) piperazine: by 79g(0.5mol) 1,3-bromo-chloropropane, 320g toluene add in reaction flask, 130g(1.0mol is dripped under control 32-35 DEG C condition) 1-(2-hydroxyethyl) piperazine, time for adding about 2 hours.After dropwising, 32-35 DEG C of stirring reaction 10 hours, after reaction terminates, passes into hydrogen chloride gas to reaction system, regulate PH=8, solids removed by filtration, filtrate decompression distillation and concentration removing toluene solvant and unreacted complete 1,3-bromo-chloropropane, obtains viscous liquid product 95g, yield about 92%.(3) fluorine puts forth energy to be the preparation of nearly alkali: by 2-trifluoromethyl thiodiphenylamine 28g(0.105mol), toluene 140g, granular sodium hydroxide 28g(0.7mol) drop in reaction flask, whipped state is warming up to reflux state (110-112 DEG C), drip (the mixing solutions solution of 1-(3-chloropropyl)-4-(2-hydroxyethyl) piperazine and 50g toluene, the dropping process lasts about 1.5 hours of 26g (0.126mol) at reflux.After dropwising, reflux state reaction about 8 hours, whole reflux course notices that system moisture removes by timely water trap.After reaction terminates, be cooled to room temperature, solids removed by filtration insolubles, 150g purifying moisture three washing organic phases.Add the 10% concentration aqueous hydrochloric acid of 100g to organic phase, stir static layering after 10 minutes, discard upper toluene organic phase, retain lower floor’s aqueous phase, wash aqueous phase at twice with 150g toluene.In aqueous phase, add toluene 140g, drip the sodium hydroxide solution of 20% of 62g under whipped state, in process, hierarchy of control temperature is no more than 45 DEG C, after dropwising, stir 20 minutes, static layering, discard lower floor’s aqueous phase, retain upper organic phase, organic phase 15g anhydrous sodium sulfate drying, underpressure distillation removing toluene solvant, residue carries out underpressure distillation, collect 230 DEG C of (0.5mmHg) cuts, obtain 33g fluorine and put forth energy to be nearly alkali, yield 72%.(4) preparation of fluophenazine hydrochloride: 32g alkali is dissolved in 128g dehydrated alcohol, stirring is dissolved backward system completely and is led to hydrogen chloride gas, process temperature is no more than 20 DEG C, logical hydrogen chloride gas is stopped as PH=2, stir after 30 minutes and filter, filter cake 50g absolute ethanol washing, product puts into vacuum drying oven, dry after 10 hours for 45 DEG C and obtain fluophenazine hydrochloride 36g, yield about 95%.embodiment 2.(1) preparation of 2-trifluoromethyl thiodiphenylamine: by 500g(1.78mol) Tecramine adds in reaction flask, be heated to 180-190 DEG C, open and stir, treat that it melts in backward reaction flask completely and add 50g(0.89mol) iron powder, stirring reaction about 2 hours at 180-190 DEG C of temperature, after reaction terminates, reaction solution is cooled to pour in beaker by reaction solution while hot when 100 DEG C, and iron powder stays (used water flushing) bottom reaction flask.Reaction solution is added underpressure distillation in clean reaction flask, collects 134-135 DEG C of (3mmHg) cut, obtain weak yellow liquid 3-trifluoromethyl pentanoic and be about 346g, yield about 82%.By 3-trifluoromethyl pentanoic 300g(1.265mol), sublimed sulphur 40g(1.265mol) add in reaction flask, whipped state is warming up to about 130 DEG C, after the complete melting of sulphur, in reaction flask, add 15g elemental iodine, continue to be warming up to 185-190 DEG C, react about 1 hour at this temperature.There is hydrogen sulfide to release in reaction process, note tail gas absorption.After reaction terminates, reaction solution is cooled to about 100 DEG C, adds 1000g toluene in reaction flask, about raised temperature to 100 DEG C, in reaction flask, add 1000g water, stir layering while hot after 5 minutes, water layer discarded, toluene layer returns reaction flask, and whipped state borehole cooling, to 15-18 DEG C, filters, filtrate retains (to be recycled apply mechanically 3-trifluoromethyl pentanoic), filter cake adopts 60 DEG C, vacuum to dry 10 hours, and obtain 147g intermediate 2-trifluoromethyl thiodiphenylamine, yield is about 86%(and calculates by sulphur)(2) preparation of 1-(3-chloropropyl)-4-(2-hydroxyethyl) piperazine: by 395g(2.5mol) 1,3-bromo-chloropropane, 1600g toluene add in reaction flask, 650g(5.0mol is dripped under control 32-35 DEG C condition) 1-(2-hydroxyethyl) piperazine, time for adding about 2 hours.After dropwising, 32-35 DEG C of stirring reaction 10 hours, after reaction terminates, passes into hydrogen chloride gas to reaction system, regulate PH=8, solids removed by filtration, filtrate decompression distillation and concentration removing toluene solvant and unreacted complete 1,3-bromo-chloropropane, obtains viscous liquid product 470g, yield about 91%.(3) fluorine puts forth energy to be the preparation of nearly alkali: by 2-trifluoromethyl thiodiphenylamine 140g(0.525mol), toluene 700g, granular sodium hydroxide 140g(3.5mol) drop in reaction flask, whipped state is warming up to reflux state (110-112 DEG C), drip (the mixing solutions solution of 1-(3-chloropropyl)-4-(2-hydroxyethyl) piperazine and 300g toluene, the dropping process lasts about 1.5 hours of 130g (0.63mol) at reflux.After dropwising, reflux state reaction about 8 hours, whole reflux course notices that system moisture removes by timely water trap.After reaction terminates, be cooled to room temperature, solids removed by filtration insolubles, 750g purifying moisture three washing organic phases.Add the 10% concentration aqueous hydrochloric acid of 500g to organic phase, stir static layering after 10 minutes, discard upper toluene organic phase, retain lower floor’s aqueous phase, wash aqueous phase at twice with 750g toluene.In aqueous phase, add toluene 720g, drip the sodium hydroxide solution of 20% of 310g under whipped state, in process, hierarchy of control temperature is no more than 45 DEG C, after dropwising, stir 20 minutes, static layering, discard lower floor’s aqueous phase, retain upper organic phase, organic phase 75g anhydrous sodium sulfate drying, underpressure distillation removing toluene solvant, residue carries out underpressure distillation, collect 230 DEG C of (0.5mmHg) cuts, obtain 168g fluorine and put forth energy to be nearly alkali, yield 73%.(4) preparation of fluophenazine hydrochloride: 160g alkali is dissolved in 640g dehydrated alcohol, stirring is dissolved backward system completely and is led to hydrogen chloride gas, process temperature is no more than 20 DEG C, logical hydrogen chloride gas is stopped as PH=2, stir after 30 minutes and filter, filter cake 300g absolute ethanol washing, product puts into vacuum drying oven, dry after 10 hours for 45 DEG C and obtain fluophenazine hydrochloride 182g, yield about 96%.embodiment 3.(1) preparation of 2-trifluoromethyl thiodiphenylamine: by 1000g(3.56mol) Tecramine adds in reaction flask, be heated to 180-190 DEG C, open and stir, treat that it melts in backward reaction flask completely and add 100g(1.78mol) iron powder, stirring reaction about 2 hours at 180-190 DEG C of temperature, after reaction terminates, reaction solution is cooled to pour in beaker by reaction solution while hot when 100 DEG C, and iron powder stays (used water flushing) bottom reaction flask.Reaction solution is added underpressure distillation in clean reaction flask, collects 134-135 DEG C of (3mmHg) cut, obtain weak yellow liquid 3-trifluoromethyl pentanoic and be about 1029g, yield about 82%.By 3-trifluoromethyl pentanoic 600g(2.53mol), sublimed sulphur 80g(2.53mol) add in reaction flask, whipped state is warming up to about 130 DEG C, after the complete melting of sulphur, in reaction flask, add 30g elemental iodine, continue to be warming up to 185-190 DEG C, react about 1 hour at this temperature.There is hydrogen sulfide to release in reaction process, note tail gas absorption.After reaction terminates, reaction solution is cooled to about 100 DEG C, adds 2000g toluene in reaction flask, about raised temperature to 100 DEG C, in reaction flask, add 1000g water, stir layering while hot after 5 minutes, water layer discarded, toluene layer returns reaction flask, and whipped state borehole cooling, to 15-18 DEG C, filters, filtrate retains (to be recycled apply mechanically 3-trifluoromethyl pentanoic), filter cake adopts 60 DEG C, vacuum to dry 10 hours, and obtain 294g intermediate 2-trifluoromethyl thiodiphenylamine, yield is about 86%(and calculates by sulphur)(2) preparation of 1-(3-chloropropyl)-4-(2-hydroxyethyl) piperazine: by 790g(5mol) 1,3-bromo-chloropropane, 3200g toluene add in reaction flask, 1300g(10mol is dripped under control 32-35 DEG C condition) 1-(2-hydroxyethyl) piperazine, time for adding about 2 hours.After dropwising, 32-35 DEG C of stirring reaction 10 hours, after reaction terminates, passes into hydrogen chloride gas to reaction system, regulate PH=8, solids removed by filtration, filtrate decompression distillation and concentration removing toluene solvant and unreacted complete 1,3-bromo-chloropropane, obtains viscous liquid product 940g, yield about 91%.(3) fluorine puts forth energy to be the preparation of nearly alkali: by 2-trifluoromethyl thiodiphenylamine 280g(1.05mol), toluene 1400g, granular sodium hydroxide 280g(7mol) drop in reaction flask, whipped state is warming up to reflux state (110-112 DEG C), drip (the mixing solutions solution of 1-(3-chloropropyl)-4-(2-hydroxyethyl) piperazine and 500g toluene, the dropping process lasts about 1.5 hours of 260g (1.26mol) at reflux.After dropwising, reflux state reaction about 8 hours, whole reflux course notices that system moisture removes by timely water trap.After reaction terminates, be cooled to room temperature, solids removed by filtration insolubles, 1500g purifying moisture three washing organic phases.Add the 10% concentration aqueous hydrochloric acid of 1000g to organic phase, stir static layering after 10 minutes, discard upper toluene organic phase, retain lower floor’s aqueous phase, wash aqueous phase at twice with 1500g toluene.In aqueous phase, add toluene 1400g, drip the sodium hydroxide solution of 20% of 620g under whipped state, in process, hierarchy of control temperature is no more than 45 DEG C, after dropwising, stir 20 minutes, static layering, discard lower floor’s aqueous phase, retain upper organic phase, organic phase 150g anhydrous sodium sulfate drying, underpressure distillation removing toluene solvant, residue carries out underpressure distillation, collect 230 DEG C of (0.5mmHg) cuts, obtain 344g fluorine and put forth energy to be nearly alkali, yield 75%.(4) preparation of fluophenazine hydrochloride: 320g alkali is dissolved in 1280g dehydrated alcohol, stirring is dissolved backward system completely and is led to hydrogen chloride gas, process temperature is no more than 20 DEG C, logical hydrogen chloride gas is stopped as PH=2, stir after 30 minutes and filter, filter cake 500g absolute ethanol washing, product puts into vacuum drying oven, dry after 10 hours for 45 DEG C and obtain fluophenazine hydrochloride 364g, yield about 96%.
PATENT
WO 2015103587
https://patents.google.com/patent/WO2015103587A2/no
//////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical use
A 2018 Cochrane review found that fluphenazine was an imperfect treatment and other inexpensive drugs less associated with side effects may be an equally effective choice for people with schizophrenia.[9]
Side effects
Discontinuation
The British National Formulary recommends a gradual withdrawal when discontinuing antipsychotics to avoid acute withdrawal syndrome or rapid relapse.[10] Symptoms of withdrawal commonly include nausea, vomiting, and loss of appetite.[11] Other symptoms may include restlessness, increased sweating, and trouble sleeping.[11] Less commonly there may be a feeling of the world spinning, numbness, or muscle pains.[11] Symptoms generally resolve after a short period of time.[11]
There is tentative evidence that discontinuation of antipsychotics can result in psychosis.[12] It may also result in reoccurrence of the condition that is being treated.[13] Rarely tardive dyskinesia can occur when the medication is stopped.[11]
Pharmacology
Pharmacodynamics
See also: Antipsychotic § Pharmacodynamics, and Antipsychotic § Comparison of medications
Fluphenazine acts primarily by blocking post-synaptic D2 receptors in the basal ganglia, cortical and limbic system. It also blocks alpha-1 adrenergic receptors, muscarinic-1 receptors, and histamine-1 receptors.[14][15]
| Site | Ki (nM) | Action | Ref |
|---|---|---|---|
| 5-HT1A | 145-2829 | ND | [16] |
| 5-HT1B | 334 | ND | [16] |
| 5-HT1D | 334 | ND | [16] |
| 5-HT1E | 540 | ND | [16] |
| 5-HT2A | 3.8-98 | ND | [16] |
| 5-HT2B | ND | ND | [16] |
| 5-HT2C | 174–2,570 | ND | [16] |
| 5-HT3 | 4,265- > 10,000 | ND | [16] |
| 5-HT5A | 145 | ND | [16] |
| 5-HT6 | 7.9 – 38 | ND | [16] |
| 5-HT7 | 8 | ND | [16] |
| D1 | 14.45 | ND | [16] |
| D2 | 0.89 | ND | |
| D2L | ND | [16] | |
| D3 | 1.412 | ND | [16] |
| D4 | 89.12 | ND | [16] |
| D5 | 95–2,590 | ND | [16] |
| α1A | 6.4-9 | ND | [16] |
| α1B | 13 | ND | [16] |
| α2A | 304-314 | ND | [16] |
| α2B | 181.6-320 | ND | [16] |
| α2C | 28.8-122 | ND | [16] |
| β1 | > 10,000 | ND | [16] |
| β2 | > 10,000 | ND | [16] |
| H1 | 7.3-70 | ND | [16] |
| H2 | 560 | ND | [16] |
| H3 | 1,000 | ND | [16] |
| H4 | > 10,000 | ND | [16] |
| M1 | 1,095-3,235.93 | ND | [16] |
| M2 | 2,187.76-7,163 | ND | [16] |
| M3 | 1441–1445.4 | ND | [16] |
| M4 | 5,321 | ND | [16] |
| M5 | 357 | ND | [16] |
| SERT | ND | ND | [16] |
| NET | ND | ND | [16] |
| DAT | ND | ND | [16] |
| NMDA (PCP) | ND | ND | [16] |
| Values are Ki (nM). The smaller the value, the more strongly the drug binds to the site. All data are for human cloned proteins, except 5-HT3 (rat), D4 (human/rat), H3 (guinea pig), and NMDA/PCP (rat).[16] |
Pharmacokinetics
History
Fluphenazine came into use in 1959.[6]
Availability
The injectable form is on the World Health Organization’s List of Essential Medicines, the safest and most effective medicines needed in a health system.[7] It is available as a generic medication.[1] It was discontinued in Australia around mid 2017.[8]
Other animals
In horses, it is sometimes given by injection as an anxiety-relieving medication, though there are many negative common side effects and it is forbidden by many equestrian competition organizations.[27]
References
- ^ Jump up to:a b c d e f g h i j k l m n o “fluphenazine decanoate”. The American Society of Health-System Pharmacists. Archived from the original on 8 December 2015. Retrieved 1 December 2015.
- ^ “Product Information: Modecate (Fluphenazine Decanoate Oily Injection )” (PDF). TGA eBusiness Services. Bristol-Myers Squibb Australia Pty Ltd. 1 November 2012. Archived from the original on 2 August 2017. Retrieved 9 December 2013.
- ^ Tardy M, Huhn M, Engel RR, Leucht S (August 2014). “Fluphenazine versus low-potency first-generation antipsychotic drugs for schizophrenia”. The Cochrane Database of Systematic Reviews. 8 (8): CD009230. doi:10.1002/14651858.CD009230.pub2. PMID 25087165.
- ^ “Modecate Injection 25mg/ml – Patient Information Leaflet (PIL) – (eMC)”. http://www.medicines.org.uk. Retrieved 6 November 2017.
- ^ “Fluphenazine”. livertox.nih.gov. Retrieved 6 November 2017.
- ^ Jump up to:a b McPherson EM (2007). Pharmaceutical Manufacturing Encyclopedia (3rd ed.). Burlington: Elsevier. p. 1680. ISBN 9780815518563.
- ^ Jump up to:a b World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Jump up to:a b Rossi S, ed. (July 2017). “Fluphenazine – Australian Medicines Handbook”. Australian Medicines Handbook. Adelaide, Australia: Australian Medicines Handbook Pty Ltd. Retrieved 8 August 2017.
- ^ Matar HE, Almerie MQ, Sampson SJ (June 2018). “Fluphenazine (oral) versus placebo for schizophrenia”. The Cochrane Database of Systematic Reviews. 6: CD006352. doi:10.1002/14651858.CD006352.pub3. PMC 6513420. PMID 29893410.
- ^ Joint Formulary Committee, BMJ, ed. (March 2009). “4.2.1”. British National Formulary (57 ed.). United Kingdom: Royal Pharmaceutical Society of Great Britain. p. 192. ISBN 978-0-85369-845-6.
Withdrawal of antipsychotic drugs after long-term therapy should always be gradual and closely monitored to avoid the risk of acute withdrawal syndromes or rapid relapse.
- ^ Jump up to:a b c d e Haddad P, Haddad PM, Dursun S, Deakin B (2004). Adverse Syndromes and Psychiatric Drugs: A Clinical Guide. OUP Oxford. pp. 207–216. ISBN 9780198527480.
- ^ Moncrieff J (July 2006). “Does antipsychotic withdrawal provoke psychosis? Review of the literature on rapid onset psychosis (supersensitivity psychosis) and withdrawal-related relapse”. Acta Psychiatrica Scandinavica. 114 (1): 3–13. doi:10.1111/j.1600-0447.2006.00787.x. PMID 16774655. S2CID 6267180.
- ^ Sacchetti E, Vita A, Siracusano A, Fleischhacker W (2013). Adherence to Antipsychotics in Schizophrenia. Springer Science & Business Media. p. 85. ISBN 9788847026797.
- ^ Siragusa S, Saadabadi A (2020). “Fluphenazine”. StatPearls. PMID 29083807.
- ^ PubChem. “Fluphenazine”. pubchem.ncbi.nlm.nih.gov. Retrieved 30 September 2019.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai aj ak al Roth, BL; Driscol, J. “PDSP Ki Database”. Psychoactive Drug Screening Program (PDSP). University of North Carolina at Chapel Hill and the United States National Institute of Mental Health. Retrieved 14 August 2017.
- ^ Parent M, Toussaint C, Gilson H (1983). “Long-term treatment of chronic psychotics with bromperidol decanoate: clinical and pharmacokinetic evaluation”. Current Therapeutic Research. 34 (1): 1–6.
- ^ Jump up to:a b Jørgensen A, Overø KF (1980). “Clopenthixol and flupenthixol depot preparations in outpatient schizophrenics. III. Serum levels”. Acta Psychiatrica Scandinavica. Supplementum. 279: 41–54. doi:10.1111/j.1600-0447.1980.tb07082.x. PMID 6931472.
- ^ Jump up to:a b Reynolds JE (1993). “Anxiolytic sedatives, hypnotics and neuroleptics.”. Martindale: The Extra Pharmacopoeia (30th ed.). London: Pharmaceutical Press. pp. 364–623.
- ^ Ereshefsky L, Saklad SR, Jann MW, Davis CM, Richards A, Seidel DR (May 1984). “Future of depot neuroleptic therapy: pharmacokinetic and pharmacodynamic approaches”. The Journal of Clinical Psychiatry. 45 (5 Pt 2): 50–9. PMID 6143748.
- ^ Jump up to:a b Curry SH, Whelpton R, de Schepper PJ, Vranckx S, Schiff AA (April 1979). “Kinetics of fluphenazine after fluphenazine dihydrochloride, enanthate and decanoate administration to man”. British Journal of Clinical Pharmacology. 7 (4): 325–31. doi:10.1111/j.1365-2125.1979.tb00941.x. PMC 1429660. PMID 444352.
- ^ Young D, Ereshefsky L, Saklad SR, Jann MW, Garcia N (1984). Explaining the pharmacokinetics of fluphenazine through computer simulations. (Abstract.). 19th Annual Midyear Clinical Meeting of the American Society of Hospital Pharmacists. Dallas, Texas.
- ^ Janssen PA, Niemegeers CJ, Schellekens KH, Lenaerts FM, Verbruggen FJ, van Nueten JM, et al. (November 1970). “The pharmacology of fluspirilene (R 6218), a potent, long-acting and injectable neuroleptic drug”. Arzneimittel-Forschung. 20 (11): 1689–98. PMID 4992598.
- ^ Beresford R, Ward A (January 1987). “Haloperidol decanoate. A preliminary review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in psychosis”. Drugs. 33 (1): 31–49. doi:10.2165/00003495-198733010-00002. PMID 3545764.
- ^ Reyntigens AJ, Heykants JJ, Woestenborghs RJ, Gelders YG, Aerts TJ (1982). “Pharmacokinetics of haloperidol decanoate. A 2-year follow-up”. International Pharmacopsychiatry. 17 (4): 238–46. doi:10.1159/000468580. PMID 7185768.
- ^ Larsson M, Axelsson R, Forsman A (1984). “On the pharmacokinetics of perphenazine: a clinical study of perphenazine enanthate and decanoate”. Current Therapeutic Research. 36 (6): 1071–88.
- ^ Loving NS (31 March 2012). “Effects of Behavior-Modifying Drug Investigated (AAEP 2011)”. The Horse Media Group. Archived from the original on 6 January 2017. Retrieved 13 December 2016.
External links
- “Fluphenazine”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Prolixin, Modecate, Moditen others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682172 |
| License data | US DailyMed: Fluphenazine |
| Pregnancy category | AU: C |
| Routes of administration | By mouth, Intramuscular injection, depot injection (fluphenazine decanoate) |
| Drug class | Typical antipsychotic |
| ATC code | N05AB02 (WHO) |
| Legal status | |
| Legal status | AU: DiscontinuedCA: ℞-onlyUK: POM (Prescription only)US: ℞-only |
| Pharmacokinetic data | |
| Bioavailability | 2.7% (by mouth) |
| Metabolism | unclear[1] |
| Elimination half-life | IM 15 hours (HCL), 7–10 days (decanoate)[1] |
| Excretion | Urine, feces |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 69-23-8 |
| PubChem CID | 3372 |
| IUPHAR/BPS | 204 |
| DrugBank | DB00623 |
| ChemSpider | 3255 |
| UNII | S79426A41Z |
| KEGG | D07977 |
| ChEBI | CHEBI:5123 |
| ChEMBL | ChEMBL726 |
| CompTox Dashboard (EPA) | DTXSID2023068 |
| ECHA InfoCard | 100.000.639 |
| Chemical and physical data | |
| Formula | C22H26F3N3OS |
| Molar mass | 437.53 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
////////////Fluphenazine, فلوفينازين , 氟奋乃静 , SQ 10733, Squibb 16144
OCCN1CCN(CCCN2C3=CC=CC=C3SC3=C2C=C(C=C3)C(F)(F)F)CC1

NEW DRUG APPROVALS
ONE TIME
$10.00
ADAPALENE

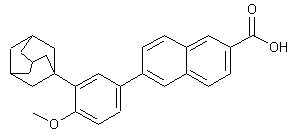
ADAPALENE
- Molecular FormulaC28H28O3
- Average mass412.520 Da
- CD 271
- CD-271
CD-271, Differin, Differine106685-40-9[RN]
2-Naphthalenecarboxylic acid, 6-(4-methoxy-3-tricyclo[3.3.1.13,7]dec-1-ylphenyl)-
6-[3-(Adamantan-1-yl)-4-methoxyphenyl]-2-naphthoic acid
6-[4-methoxy-3-(tricyclo[3.3.1.13,7]dec-1-yl)phenyl]naphthalene-2-carboxylic acid AdapaleneCAS Registry Number: 106685-40-9
CAS Name: 6-(4-Methoxy-3-tricyclo[3.3.1.13,7]dec-1-ylphenyl)-2-naphthalenecarboxylic acid
Additional Names: 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid
Manufacturers’ Codes: CD-271
Trademarks: Differin (Galderma)
Molecular Formula: C28H28O3
Molecular Weight: 412.52
Percent Composition: C 81.52%, H 6.84%, O 11.64%
Literature References: Retinoid selective for retinoic acid receptor (RAR) subtypes b and g. Prepn: B. Shroot et al.,EP199636; eidem,US4717720 (1986, 1988 both to Cent. Int. Recher. Dermatol.); and structure-activity study: B. Charpentier et al.,J. Med. Chem.38, 4993 (1995). Pilot-scale synthesis: Z. Liu, J. Xiang, Org. Process Res. Dev.10, 285 (2006). HPLC determn in plasma and tissue: R. Ruhl, H. Nau, Chromatographia45, 269 (1997). Clinical pharmacology: C. E. M. Griffiths et al.,J. Invest. Dermatol.101, 325 (1993). Clinical trial in acne: A. Shalita et al.,J. Am. Acad. Dermatol.34, 482 (1996). Reviews of pharmacology and clinical potential: B. A. Bernard, Skin Pharmacol.6, Suppl. 1, 61-69 (1993); R. N. Brogden, K. L. Goa, Drugs53, 511-519 (1997); of clinical use in acne vulgaris: J. Waugh et al.,Drugs64, 1465-1478 (2004).
Properties: White crystals from THF and ethyl acetate, mp 319-322°. pK 4.2. Stable to light.
Melting point: mp 319-322°
pKa: pK 4.2
Therap-Cat: Antiacne.
Keywords: Antiacne.
Adapalene is a third-generation topical retinoid primarily used in the treatment of mild-moderate acne, and is also used off-label to treat keratosis pilaris as well as other skin conditions.[1] Studies have found adapalene is as effective as other retinoids, while causing less irritation.[2] It also has several advantages over other retinoids. The adapalene molecule is more stable compared to tretinoin and tazarotene, which leads to less concern for photodegradation.[2] It is also chemically more stable compared to the other two retinoids, allowing it to be used in combination with benzoyl peroxide.[2] Due to its effects on keratinocyte proliferation and differentiation, adapalene is superior to tretinoin for the treatment of comedonal acne and is often used as a first-line agent. [3]
Adapalene is a third-generation topical retinoid with anti-comedogenic, comedolytic, and anti-inflammatory properties used to treat acne vulgaris in adolescents and adults.

SYN
AU 9047961; EP 0199636; US 4717720; US 5098895; US 5183889
J Med Chem 1995,38(26),4993
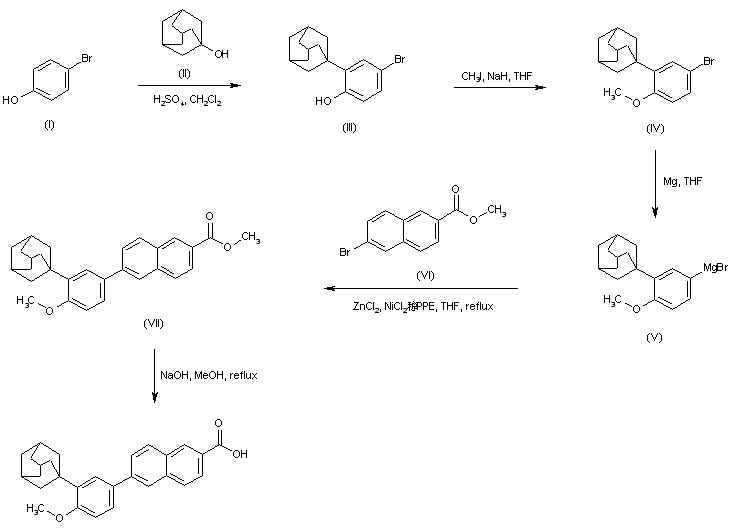
Friedel-Crafts condensation of 4-bromophenol (I) with 1-adamantanol (II) in the presence of H2SO4 yielded the adamantyl phenol (III). Subsequent alkylation of the sodium phenoxide of (III) with iodomethane produced the methyl ether (IV). The Grignard reagent (V), prepared from aryl bromide (IV), was converted to the organozincate derivative, and then subjected to a nickel-catalyzed cross-coupling with methyl 6-bromo-2-naphthoate (VI) to furnish adduct (VII). The target carboxylic acid was finally obtained by saponification of the methyl ester (VII).
SYN
CA 2021550; EP 0409740; FR 2649976; JP 1991063246; US 5073361; US 5149631
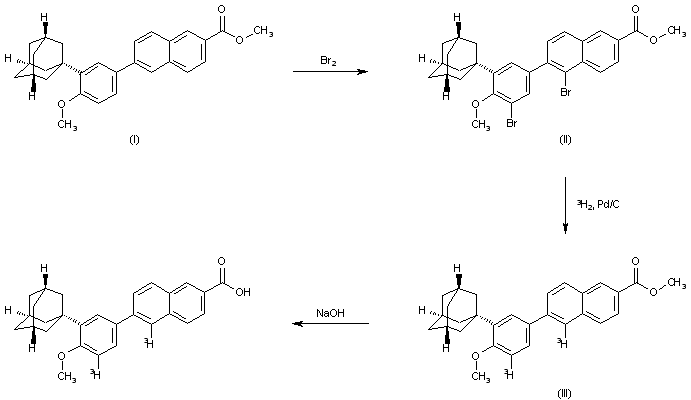
The bromination of 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid methyl ester (I) with Br2 in dichloromethane gives the dibromo derivative (II), which is hydrogenated with tritium gas over Pd/C in THF containing TEA to yield the bis tritiated ester (III). Finally, ester (III) is hydrolyzed with NaOH in refluxing methanol to afford the target tritiated naphthoic acid.
SYN
doi:10.1071/CH9732303c US4717720
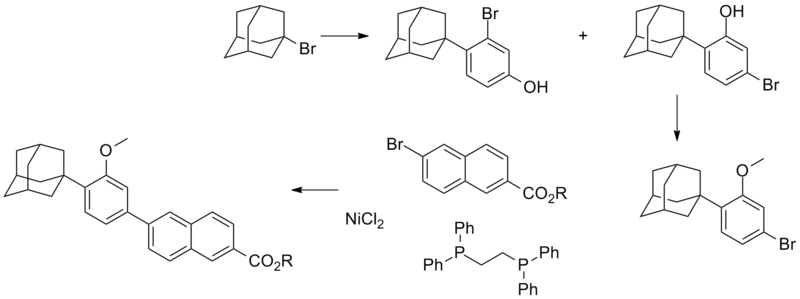
SYN
Adapalene (CAS NO.: 106685-40-9), with its systematic name of 2-Naphthalenecarboxylic acid, 6-(4-methoxy-3-tricyclo(3.3.1.1(sup 3,7))dec-1-ylphenyl)-, could be produced through many synthetic methods.
Following is one of the synthesis routes:
Firstly, Friedel-Crafts condensation of 4-bromophenol (I) with 1-adamantanol (II) in the presence of H2SO4 yields the adamantyl phenol (III). Next, subsequent alkylation of the sodium phenoxide of (III) with iodomethane produces the methyl ether (IV). The Grignard reagent (V), prepared from aryl bromide (IV), is converted to the organozincate derivative, and then subjects to a nickel-catalyzed cross-coupling with methyl 6-bromo-2-naphthoate (VI) to furnish adduct (VII). Finally, the target carboxylic acid is obtained by saponification of the methyl ester (VII).

Synthesis Reference
Graziano Castaldi, Pietro Allegrini, Gabriele Razzetti, Mauro Ercoli, “Process for the preparation of adapalene.” U.S. Patent US20060229465, issued October 12, 2006.
PATENT
https://patents.google.com/patent/US8119834B2/enThe chemical name for adapalene is 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid, which is represented by Compound I (below):

Adapalene has been approved by the FDA as a cream, a gel, a solution and pledgets for the topical treatment of acne vulgaris and is marketed under the tradename of DIFFERIN®.U.S. Pat. No. 4,717,720 (“the ‘720 patent”) discloses benzonaphthalene derivatives, including adapalene. The ‘720 patent describes a process for preparing adapalene (i.e., according to example 9c followed by example 10) that involves two reaction steps.The first step for preparing adapalene according to the ‘720 patent involves the preparation of the methyl ester of 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid. According to example 9c of the ‘720 patent, 2-(1-adamantyl)-4-bromoanisole (also known as 1-(5-bromo-2-methoxyphenyl)adamantane) is converted to its organomagnesium derivative and then into its organozinc derivative. The organozinc derivative is next coupled to methyl 6-bromo-2-naphthoate by adding a catalytic amount of NiCl2/DPPE complex (also known as [bis(diphenylphosphino) ethane]dichloronickel(II)). Upon completion of the reaction, the mixture is poured into water, extracted with dichloromethane, and then dried. The product is next isolated by column chromatography by eluting with a mixture of heptane (70%) and dichloromethane (30%). The resulting product is then recrystallized in ethyl acetate (yield: 78%).The second step for preparing adapalene according to the ‘720 patent involves hydrolyzing the product of step 1 (above). According to example 10 of the ‘720 patent, the ester obtained in Example 9c can be treated with a solution of soda in methanol followed by heating at reflux for 48 hours. The solvents are then evaporated and the resulting residue is taken up in water and acidified with concentrated HCl to neutralize the resulting adapalene sodium salt. The resulting solid is next filtered and dried under vacuum over phosphoric anhydride and then recrystallized in a mixture of tetrahydrofuran and ethyl acetate to yield adapalene (yield: 81%).The process of preparing adapalene according to the ‘720 patent is both difficult and uneconomical to conduct on an industrial scale. Regarding step 1, the use of dichloromethane is both toxic and hazardous for the environment. Additionally, purification of the intermediate product by column chromatography, followed by recrystallization, in order to obtain a crystalline product of acceptable purity is both expensive and laborious. Moreover, the step 1 process produces as a biaryllic C—C bond, and the catalytic coupling is noticeably exothermic. Regarding step 2, the synthesis of adapalene and/or its sodium salt requires a long reaction time (i.e., 48 hours) at methanol reflux and further requires a high ratio of solvent (volume) to product (mass).Additionally, according to the prior art, the manufacture of adapalene is not satisfactory for industrial implementation because the presence of high amounts of undesired by-products makes it necessary to use uneconomical purification procedures to isolate the product according to quality specifications. One significant undesired by-product produced during the Grignard reaction of step 1 in the synthesis of adapalene is 3,3′-diadamantyl-4,4′-dimethoxybiphenyl, which has not been previously described in the literature and which is represented by Compound VI (below):

The level of the by-product in a sample of adapalene, adapalene methyl ester and/or an adapalene salt can be determined using standard analytical techniques known to those of ordinary skill in the art. For example, the level can be determined by HPLC. A specific method for determining the level of this impurity is provided herein.Since the solubility of the dimeric by-product is very low in most solvents, the design of an economical industrial process that yields pure adapalene without the use of expensive chromatographic methods requires the selection of the proper solvents and conditions to inhibit formation of the by-product during the manufacturing process.Additionally, adapalene has been described as being white (see, e.g., Merck Index, 13th ed., p. 29). It has been observed that adapalene has a tendency to yellow under certain synthetic conditions or due to the quality of the starting materials used in its preparation. In this regard, color must be attributed to the presence of some specific impurities that may or may not be detectable by conventional methods such as HPLC.

ExampleStep 2: Preparation of 6-[3-(1-adamantyl)-4-methoxy phenyl]-2-naphthoic acid-potassium Salt (i.e., Adapalene Potassium Salt)In a 2 L, five necked cylindrical reaction vessel equipped with reflux condenser, distillation kit, heat-transfer jacket, anchor impeller and purged with nitrogen, were added 48.38 g (dry equivalent amount) of methyl 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoate (1.134×10−1 mol), wet with methanol, 2.73 g of tetrabutylammonium bromide (8.47×10−3 mol), 18.39 g of potassium hydroxide (85% alkali content, freshly titrated. 2.79×10−1 mol) and 581 mL of toluene. The mixture was heated to reflux temperature, and the methanol/water was removed by distillation. The distilled mixture was replaced by pure toluene and the mixture was stirred at reflux for approximately three hours (including the time required for the distillation). The solution was then cooled to approximately 20-25° C., filtered and the resulting solid was washed with toluene.The solid was next suspended in 187 mL of tetrahydrofuran and stirred for approximately 30 minutes. Then, 375 mL of toluene was added, and the mixture was heated to reflux and maintained at that temperature for approximately 1 hour. The solution was then cooled to approximately 20-25° C., filtered, and the resulting solid washed with toluene. The toluene-wet product was then suspended in 256 mL of methanol, heated to reflux for approximately 30 minutes and cooled to 50-60° C. After cooling, 409 mL of water was added dropwise. The mixture was then again heated to reflux for approximately 15 additional minutes, cooled to room temperature and filtered. The resulting solid was washed with water to yield 50.69 g (wet) of adapalene potassium salt (1.12×10−1 mol, dry equivalent amount calculated from loss on drying; yield: 99.18%). Analytical data: HPLC Purity (HPLC at 272 nm): 99.86%; Impurity (i.e., 3,3′-diadamantyl-4,4′-dimethoxybiphenyl) area percent (HPLC at 272 nm): not detected; 1H-NMR (300 MHz, CD3OD): δ 1.83 (broad s, 6H), 2.08 (broad s, 3H), 2.21 (broad s, 6H), 3.88 (s, 3H), 7.04 (d, 1H, J=8.4 Hz), 7.56 (overlapped, 1H, J=2.4, 9.6 Hz), 7.57 (overlapped s, 1H), 7.74 (dd, 1H, J=8.7, 1.8 Hz), 7.87 (d, 1H, J=9.0 Hz), 7.97 (d, 1H, J=8.7 Hz), 8.00 (broad d, 1H, J=0.9 Hz), 8.06 (dd, 1H, 8.4, J=1.8 Hz), 8.47 (broad d, 1H, J=0.9 Hz); 13C-NMR (75.4 MHz, CD3OD): δ 30.6, 38.3, 41.8, 55.5, 113.3, 125.3, 126.4, 126.6, 127.8, 128.3, 130.0, 130.4, 133.0, 134.2, 136.1, 136.3, 139.7, 141.1, 159.9, 175.4.
ExampleStep 3: Preparation of 6-[3-(1-adamantyl)-4-methoxy phenyl]-2-naphthoic Acid (i.e., Adapalene)In 500 mL of methanol was added 49.59 g (1.10×10−1 mol, dry equivalent amount) of the wet solid obtained in Example/Step 2, and the mixture was heated to reflux for 30 minutes and cooled to approximately 40° C. Next, 33.17 g of concentrated HCl was slowly added over approximately 1 hour with gentle stirring in order to ensure homogeneity, followed by the slow addition of 248 mL of water. The resulting mixture was stirred for approximately 30 additional minutes at approximately 40° C. and then cooled to room temperature, filtered and washed with methanol. The wet solid was then suspended with 1020 mL of tetrahydrofuran and heated to reflux for approximately 10 minutes or until complete dissolution. The solution was then cooled to approximately 35° C., the solid particles were removed by filtration, and the filter was washed with tetrahydrofuran.The collected mother liquors were heated to reflux, and 654 g of tetrahydrofuran was removed by distillation. The mixture was then cooled to approximately 55-60° C. Thereafter, 650 mL of methanol was added over approximately 10 minutes, and the mixture heated to reflux for approximately 30 minutes, cooled, and filtered. The resulting solid was filtered with methanol and dried at 80° C. in a vacuum oven to yield 40.54 g of adapalene (9.83×10−2 mol; yield: 89.29% (from adapalene potassium salt); 88.56% (from adapalene methyl ester); and 78.67% (from methyl 6-bromo-2-naphthoate)). Analytical data: HPLC Purity (HPLC at 272 nm): 100.00%; Assay: 99.99%; Residue on Ignition: 0.02%; IR: matches reference.Table 1 (below) lists the peak assignments of the X-ray powder diffractogram of the adapalene obtained and are illustrated in FIG. 1.
| TABLE 1 | |||
| peak | peak_position | peak_intensity | background |
| 1 | 9.94547 | 175.32198 | 42.94638 |
| 2 | 13.18338 | 239.32156 | 48.88440 |
| 3 | 14.87487 | 234.32591 | 47.91444 |
| 4 | 15.28319 | 573.40082 | 53.73505 |
| 5 | 16.37472 | 1207.21631 | 69.64595 |
| 6 | 16.54000 | 882.00000 | 68.42529 |
| 7 | 17.39657 | 110.88804 | 58.39248 |
| 8 | 17.93203 | 114.02068 | 55.36037 |
| 9 | 19.44575 | 285.34473 | 113.52401 |
| 10 | 19.94692 | 569.60516 | 153.63921 |
| 11 | 22.43198 | 2846.14307 | 110.81189 |
| 12 | 24.02238 | 140.20882 | 85.37505 |
| 13 | 25.04586 | 925.64282 | 121.97974 |
| 14 | 25.41035 | 240.42351 | 102.81077 |
| 15 | 26.68556 | 362.45480 | 68.05973 |
| 16 | 27.71646 | 141.77916 | 72.53469 |
| 17 | 40.51307 | 133.00453 | 43.44914 |
| 18 | 46.52728 | 130.31587 | 50.16773 |
ExampleStep 4: Preparation of 3,3′-diadamantyl-4,4′-dimethoxybiphenylTo a 100 mL rounded bottom reaction vessel equipped with a magnetic stirrer, thermometer, reflux condenser, pressure compensated addition funnel, were added 0.15 g of 1-(5-bromo-2-methoxyphenyl)adamantane, 0.47 g of magnesium turnings and 7 mL of tetrahydrofuran. The mixture was heated to approximately 35° C., and 0.13 mL of 1,2-dibromoethane were added to the mixture. Reaction exothermy self-heated the mixture. Next, a solution of 4.85 g of 1-(5-bromo-2-methoxyphenyl)adamantane and 28 mL of tetrahydrofuran was added to the mixture dropwise. During this addition, the temperature of the mixture dropped from reflux temperature to approximately 45° C. The reaction was then stirred for approximately 45 additional minutes at approximately 45° C. and was permitted to cool to approximately 22° C. Next, 2.3 g of ZnCl2 was added to the mixture, resulting in an exothermic reaction that raised the temperature of the mixture to approximately 38° C. The mixture was then permitted to cool to approximately 22° C. and was stirred for approximately 1 hour at this temperature.Next, 0.03 g of Pd(OAc)2 and 3.5 g of 1-(5-bromo-2-methoxyphenyl) adamantane were added to the mixture, followed by 25 mL of tetrahydrofuran in order to improve agitation, and the mixture was heated at reflux for approximately 24 hours. The resulting mixture was then evaporated to dryness and poured into 103 mL of 0.015 N HCl. Next, 150 mL of dichloromethane and 100 mL of water were added to yield a mixture consisting of a solid, an aqueous layer and an organic layer. The mixture was then filtered to separate the solid, the aqueous layer was discarded, and the organic layer was washed with 200 mL of water and decanted again. This process was repeated twice on the filtered solid. The three collected organic layers were evaporated to dryness, washed in methanol, and dried to yield 2.1 g of 3,3′-diadamantyl-4,4′-dimethoxybiphenyl (yield: 39.9%). Analytical data: Melting point: 288.1-289.1° C.; Elemental analysis: C 83.63%, H 8.73%; 1H-NMR (300 MHz, CDCl3): δ 1.78 (broad s, 12H), 2.08 (broad s, 6H), 2.15 (broad s, 12H), 3.86 (s, 6H), 6.92 (dm, 2H, J=8.1 Hz), 7.34 (dd, 2H, J=2.4, 8.1 Hz), 7.39 (d, 2H, J=2.4 Hz); 13C-NMR (75.4 MHz, CDCl3): δ 29.2, 37.1, 37.2, 40.6, 55.1, 111.9, 125.0, 125.5, 134.0, 138.5, 157.8; MS (EI, 70 eV): m/z=484 (6), 483 (36), 412 (1,100), 410 (5), 347 (8), 135 (22), 107 (7), 93 (14), 79 (17), 67 (9), 55 (6); IR (Selected absorption bands): 2992, 2964, 2898, 2850, 1603 cm−1.
PATENT
https://patents.google.com/patent/WO2008126104A2/en
The compound 6-[3-(l – Adamantyl) – 4 – methoxy phenyl] – 2 – naphthoic acid of Formula – I known as Adapalene is used in dermatology, particularly in the treatment of acne vulgaris and psoriasis.

Formula – 1Adapalene was first time disclosed in the US patent No. 4,717,720 (herein after referred as ‘720) describe the preparation of compound of Formula – I using Negishi cross Coupling. In this reaction, 2-(l-adamantyl)-4-bromoanisole is converted to its organomagnesium compound followed by conversion to organozinc compound using zinc chloride and reacted with 6-bromo-2-methylnaphthoate employing transition metal as reaction catalyst such as palladium or nickel or one of its complexes with various phosphines. The reaction sequence is as shown in scheme – 1 below:

Scheme – 1 Another US patent No. 5,015,758 describe the process for preparation of 6[3-(l- Adamantyl) – 4 – methoxyphenyl] – 2 – naphthoate a penultimate step for preparation of Adapalene using Friedel – Crafts alkylation by reacting 1 – acetoxy adamantane with methyl – 6 – (4 – hydroxyphenyl) – 2 – naphthoate in presence of cone. Sulfuric acid in solvent n – heptane.Another improved process was published in the journal, Organic Process Research & Development, 2006, 10, 285 – 288 for the preparation of Adapalene. The process involves the preparation of intermediates followed by Negishi cross Coupling, where in 2-(l-adamantyl)-4-bromophenol was prepared using 1 – adamentol and 4- bromo phenol in presence of 98% sulphuric acid and acetic acid, which on methylation with dimethyl sulfate and potassium carbonate in dry acetone yields 2-(l -adamantyl)-4-bromoanisole. The compound is reacted with magnesium to form Grignard reagent and then coupled with 6-bromo-2-methylnaphthoate in presence of novel Pd – Zn double metal catalyst to yield ester, which on saponification followed by treatment with acid yields Adapalene.The recent published application WO 2006/108717 describes the use of Suzuki coupling for the synthesis of adapalene the compound of formula – 1. The application describes the preparation of 3-adamantyl-4-methoxyphenyl boronic acid from 2-(l-adamantyl)-4- bromoanisole using n-Butyl Lithium and triisopropyl borate in solvent tetrahydrofuran. Finally 3-adamantyl-4-methoxyphenyl boronic acid is reacted with 6-bromo-2-naphthoic acid involving Suzuki coupling in presence of Palladium acetate catalyst, a ligand 2 – (dicyclohexyl – phosphino) biphenyl, an inorganic base in solvent to get the compound adapalene.Some of the drawbacks of the prior art processes include:- The reported process in US patent 4717720, using Negishi cross coupling involves Grignard reaction. This requires anhydrous condition and a possibility of runaway reaction during Grignard reagent formation. Also the reaction involves the addition of fused ZnC12 and the preparation of the catalyst NiC12 (DPPE) complex, which needs to be freshly prepared increases the reaction step and has to be thoroughly dried before its use for coupling. Further the coupling reaction, results in the formation of dimer impurities during the organozinc compound reaction, with 2-(I -adamantyl)-4-bromoanisole and 6-bromo-2-methylnaphthoate respectively, which are difficult to remove. All these operations make the entire synthesis extremely sensitive and difficult to handle.Some of the above drawbacks were addressed by the authors in the article published in Organic Process Research & Development, 2006, 10, 285 – 288 for the preparation of Adapalene. But the use of Pd catalyst with the ligand like PdCl2 (PPh3)2 for the direct conversion of Grignard reagent employing ZnCYl in catalytic amount has its own limitations. The use of Grignard reagent, palladium catalyst with ligand and hygroscopic ZnCl2 demerits this process for industrial application.The recent published application WO 2006/108717; describes the use of Suzuki coupling for the synthesis of adapalene the compound of formula – I. The use of organo boronic acids for the Suzuki reaction has some limitations because of the indeterminate stoichiometry associated with the use of boronic acid, and its difficulty in purification and the byproducts formed during the reaction.Therefore there remains a need for an improved process for preparing adapalene that eliminates or substantially reduces the impurities, decreases the number of steps, and employs a more robust process which is convenient and cost efficient.

Examples:Example 1: Preparation of 3 – Adamantyl – 4 – methoxy phenyl potassium trifluoroborate:In a 2.0 L round bottom flask equipped with stirring and under nitrogen atmosphere 100.0 gm of 2-(l- adamantyl) 4-bromo anisole was charged in 600 ml tetrahydrofuran. The reaction mixture was cooled to -55 ± 50C and 302 ml of 1.6 M n – butyl Lithium was slowly added and stirred. 87 ml of tri isopropyl borate was then charged and stirring was continued for 30 minutes at -55 ± 5°C. Cooling was removed and the temperature raised slowly to 25 – 300C. 1.0 L of 1.2N hydrochloric acid was then charged and reaction mass was stirred for 30 minutes and separated the organic layer. The organic layer was charged in 1.0 L round bottom flask and freshly prepared aqueous solution of potassium hydrogen difluoride (230 gm, in 700 ml water) was added at 25 – 300C and stirring was maintained till white precipitate is obtained. The mixture was continued under stirring and cooled to 0 – 50C. The product, 3 – adamantyl – 4 – methoxyphenyl potassium trifluoroborate obtained was filtered, washed with 100 ml of ethyl acetate. The product was dried at 60 – 65°C till constant weight. Yield: 90.5 gm (83%), Purity: 99.0 % by HPLC.Example 2: Preparation of 6 – [3-(l- Adamantyl) – 4 – methoxyphenyl] – 2 – naphthoic acid:In a 1.0 L round bottom flask equipped with stirring and under nitrogen atmosphere 50.0 gm of 3 – Adamantyl – 4 – methoxyphenyl potassium trifluoroborate, 23 gm of 6- bromo -2-methyl napthoate in 300 ml tetrahydrofuran (THF) was charged. Stirred for 15 min and charged 3.0 gm of 5% Pd / C was and aqueous potassium hydroxide solution (50.0 gm in 300 ml water). Stirring was continued and the temperature was raised to reflux. The reaction mass was maintained for 10 hours at reflux and after the completion of the reaction, 200 ml of tetrahydrofuran: water (1 : 1) mixture was added and then filtered through hyflow bed at 45-500C. The hyflow bed was washed with tetrahydrofuran: water (1 : 1) mixture at 45-500C. 500 ml water was charged and the reaction mass was stirred. The aqueous layer was acidified with 1.2N hydrochloric acid. The precipitated mass was filtered, washed with water till neutral pH. The solid product obtained was dried at 70 – 75°C till constant weight to get 6 – [3-(l- adamantyl) – 4 – methoxyphenyl] – 2 – naphthoic acid.The dried product was taken in 300 ml of tetrahydrofuran and stirred. The temperature was raised to reflux and was maintained for 30 minutes. The heating was stopped and cooled the reaction mass to 25 – 300C. 500 ml of n – heptane was charged to the reaction mass and stirred for 30 minutes. The reaction mass was then chilled to 0 – 5°C and maintained stirring at 0 – 5°C temperature for 2.0 hours. The precipitated solid was filtered and washed with n – heptane. The pure crystalline 6 – [3-(l- adamantyl) – 4 methoxyphenyl] – 2 – naphthoic acid thus obtained was then dried till constant weight. Yield = 40 – 42 gms (68 – 72 %)
PATENT
https://pubs.acs.org/doi/10.1021/op050223f
Strategies that were adopted during the process development of adapalene to achieve a cost-effective commercial-scale synthesis are described herein. These included (1) the use of AcOH/H2SO4 to afford 2-(1-adamantyl)-4-bromophenol in quantitative yield; (2) the dimethyl sulfate methylation to enhance the yield of methylation to 95%; (3) direct conversion of the Grignard reagent into methyl 6-(3-(1-adamantyl)-4-methoxyphenyl)-2-naphthoate by the catalysis of both PdCl2(PPh3)2 and ZnCl2 in high yield; (4) the use of EDTA-disodium salt dihydrate to ensure the heavy metal’s content within acceptable limits; (5) the use of toluene to simplify the original chromatographic purification to recrystallization. The pilot-scale synthesis of adapalene is described in detail in the Experimental Section.

6-(3-(1-Adamantyl)-4-methoxyphenyl)-2-naphthoic Acid (Adapalene, 1). Compound 7 (213 g, 0.5 mol) was treated with 2 N NaOH solution (8 L) in methanol under reflux for 8 h. After evaporation of methanol (7 L) and addition of water (1.5 L), the mixture was acidified until pH 1 with 6 N HCl and filtrated through Celite. The residue was washed with water (3 × 5 L), and recrystallized twice in THF (194 g/2 L/time) to give pure (99% HPLC) 1 (177 g, 85%), mp 320-322 °C.1 H NMR (400 MHz, DMSO-d6) δ 1.77 (6 H,s, H on 1-adamantyl), 2.07 (3 H, s, H on 1-adamantyl), 2.14 (6 H, s, H on 1-adamantyl), 3.87 (3 H, s, H on ArOCH3), 7.12 (1 H, d, J ) 8.4 Hz, 5-phenyl H), 7.58 (1 H, d, J ) 2.0 Hz, 2-phenyl H), 7.65 (1 H, dd, J ) 8.4 Hz, J ) 2.0 Hz, 6-phenyl H), 7.89 (1 H, d, J ) 8.8 Hz, 7-naphthyl H), 7.98 (1 H, d, J ) 8.8 Hz, 4-naphthyl H), 8.08 (1 H, d, J ) 8.8 Hz, 8-naphthyl H), 8.15 (1 H, d, J ) 8.8 Hz, 3-naphthyl H), 8.22 (1 H, s, 5-naphthyl H), 8.60 (1 H, s, 1-naphthyl H), 13.05 (1 H, s, -COOH); 13C NMR (100 MHz, DMSO-d6) δ 28.32, 36.47, 40.09, 55.28, 112.68, 123.99, 124.99, 125.38, 125.68, 125.85, 127.55, 128.25, 129.72, 130.13, 130.83, 131.46, 135.38, 138.00, 140.13, 158.53, 167.34.
PATENT
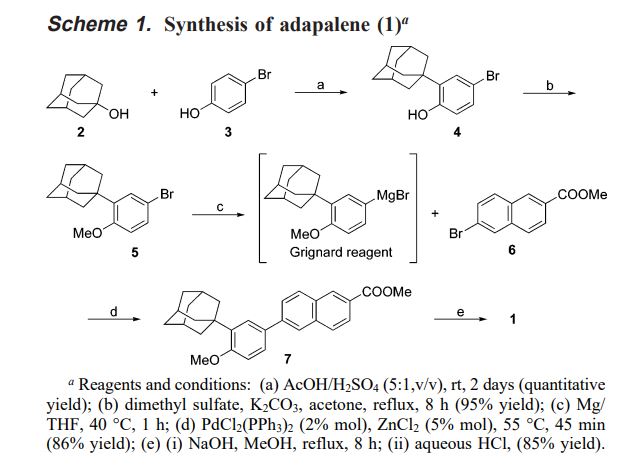
https://patents.google.com/patent/US7345189B2/en
Adapalene, namely 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid, having the following chemical formula:

is disclosed in U.S. Pat. No. 4,717,720 and used in dermatology, in particular for the treatment of acne vulgaris and psoriasis.According to U.S. Pat. No. 4,717,720 the synthesis is carried out by a coupling reaction between a magnesium, lithium or zinc derivative of a compound of formula (A) and a compound of formula (B), wherein X and Y are Cl, Br, F or I; R is hydrogen or alkyl; and Ad is 1-adamantyl

in an anhydrous solvent, in the presence of a metal transition or a complex thereof as a catalyst.A number of alternative synthetic approaches have been suggested in order to reduce the preparation costs. Surprisingly, particularly advantageous proved the alternative synthesis of the invention, which makes use of easily-available, low-cost 6-hydroxy-2-naphthoic acid alkyl esters as intermediates, and provides good yields.EXAMPLE 1Synthesis of 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid methyl ester [adapalene methyl ester]A round-bottom flask is loaded with nickel (II) chloride (0.158 g; 1.2 mmol) and THF (20 ml), and tris(hydroxypropyl)phosphine (1.53 g; 7.3 mmol) is added to the mixture, which is refluxed for an hour, then cooled to a temperature of 50° C. and added in succession with methyl 6-tosyl-naphthalene-2-carboxylate (8.7 g; 24.4 mmol), potassium phosphate (10.38 g; 48.8 mmol), 4-methoxy-3-adamantyl-phenylboronic acid (7-g; 24.4 mmol), water (0.88 g; 48.8 mmol) and THF (50 ml). The mixture is heated under reflux for 24 hours, then cooled to a temperature ranging from 50 to 55° C. and added with water, adjusting pH to a value below 7 with acetic acid. After cooling to a temperature of 15° C., the resulting product is filtered, thereby obtaining crystalline adapalene methyl ester (8.5 g; 20.08 mmol) in 82% yield.1H NMR: (300 MHz, DMSO), δ 8.6 (s, 1H), δ 8.3-7.8 (m, 6H), δ 7.7-7.5 (m, 2H), δ 7.1 (d, 1H), δ 3.9 (s, 3H), δ 3.85 (s, 3H), δ 2 (m, 9H), δ 1.7 (m, 6H).EXAMPLE 2Synthesis of 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid sodium salt [adapalene sodium salt]A round-bottom flask is loaded with adapalene methyl ester (7 g; 16.41 mmol), THF (42 ml), water (7 ml) and a 50% w/w sodium hydroxide aqueous solution (1.44 g; 18.05 mmol). The mixture is refluxed for 6 hours, then added with water (133 ml) and THF is distilled off to a residual content of approx. 5% w/w, heated to a temperature of about 80° C. until complete dissolution of the solid, then cooled to 15° C. The crystallized product is filtered and dried under vacuum in a static dryer at a temperature of 50° C., thereby obtaining adapalene sodium salt (6.7 g; 15.42 mmol) in 94% yield.EXAMPLE 3Synthesis of 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid [adapalene]A round-bottom flask is loaded with adapalene sodium salt (6.7 g; 15.42 mmol), THF (40 ml) and water (7 ml) and the mixture is refluxed until complete dissolution of the solid. The resulting solution is dropped into a 3% w/w acetic acid aqueous solution, keeping the temperature above 60-70° C., to precipitate adapalene acid (6.3 g; 15.27 mmol), which is filtered and dried under vacuum at a temperature of 50-60° C. The yield is 95%.EXAMPLE 4Synthesis of adapalene methyl esterA round-bottom flask is loaded with nickel (II) chloride (0.158 g; 1.2 mmol) and THF (20 ml), and tris(hydroxypropyl)phosphine (1.53 g; 7.3 mmol) is added. The mixture is refluxed for an hour, then cooled to a temperature of 50° C. and added in succession with methyl 6-tosyl-naphthalene-2-carboxylate (8.7 g; 24.4 mmol), potassium phosphate (10.38 g; 48.8 mmol), 4-methoxy-3-adamantyl-phenylboronic acid (9.1 g; 31.8 mmol), water (10.53 g; 585.3 mmol) and THF (50 ml). The mixture is refluxed for 24 hours, then cooled to a temperature ranging from 50 to 55° C., added with water, and adjusted to pH lower than 7 with acetic acid. After cooling to 15° C., the resulting product is filtered, thereby obtaining adapalene methyl ester (9 g; 21.2 mmol) in 86% yield.EXAMPLE 5Synthesis of adapalene methyl esterA round-bottom flask is loaded with nickel (II) chloride (0.158 g; 1.2 mmol) and THF (15 ml), and tris(hydroxypropyl)phosphine (1.53 g; 7.3 mmol) is added. The mixture is refluxed for an hour, then cooled to a temperature of 50° C. and added in succession with methyl 6-tosyl-naphthalene-2-carboxylate (8.7 g; 24.4 mmol), potassium carbonate (6.75 g; 48.8 mmol), 4-methoxy-3-adamantyl-phenylboronic acid (9.1 g; 31.8 mmol), water (8.11 g; 450.5 mmol) and THF (30 ml). The mixture is refluxed for 24 hours, then cooled to a temperature ranging from 50 to 55° C., added with water, and adjusted to pH lower than 7 with acetic acid. After cooling to 15° C., the resulting product is filtered, thereby obtaining adapalene methyl ester (9.37 g; 21.96 mmol) in 90% yield.EXAMPLE 6Synthesis of adapalene methyl esterA round-bottom flask is loaded with methyl 6-tosyl-naphthalene-2-carboxylate (8.7 g; 24.4 mmol), THF (70 ml), potassium phosphate (10.38 g; 48.8 mmol), 4-methoxy-3-adamantyl-phenylboronic acid (7 g; 24.4 mmol), nickel chloride complexed with tri(cyclohexyl)phosphine (0.83 g; 1.2 mmol) and tri(cyclohexyl)phosphine (1.37 g; 4.88 mmol). The mixture is refluxed for 24 hours, then cooled to a temperature ranging from 50 to 55° C. and added with water, then cooled to 15° C. The resulting product is filtered, thereby obtaining adapalene methyl ester (8.1 g; 19.0 mmol) in 78% yield.
PATENThttps://patents.google.com/patent/WO2008126104A2/en
The compound 6-[3-(l – Adamantyl) – 4 – methoxy phenyl] – 2 – naphthoic acid of Formula – I known as Adapalene is used in dermatology, particularly in the treatment of acne vulgaris and psoriasis.
Formula – 1Adapalene was first time disclosed in the US patent No. 4,717,720 (herein after referred as ‘720) describe the preparation of compound of Formula – I using Negishi cross Coupling. In this reaction, 2-(l-adamantyl)-4-bromoanisole is converted to its organomagnesium compound followed by conversion to organozinc compound using zinc chloride and reacted with 6-bromo-2-methylnaphthoate employing transition metal as reaction catalyst such as palladium or nickel or one of its complexes with various phosphines. The reaction sequence is as shown in scheme – 1 below:
Scheme – 1 Another US patent No. 5,015,758 describe the process for preparation of 6[3-(l- Adamantyl) – 4 – methoxyphenyl] – 2 – naphthoate a penultimate step for preparation of Adapalene using Friedel – Crafts alkylation by reacting 1 – acetoxy adamantane with methyl – 6 – (4 – hydroxyphenyl) – 2 – naphthoate in presence of cone. Sulfuric acid in solvent n – heptane.Another improved process was published in the journal, Organic Process Research & Development, 2006, 10, 285 – 288 for the preparation of Adapalene. The process involves the preparation of intermediates followed by Negishi cross Coupling, where in 2-(l-adamantyl)-4-bromophenol was prepared using 1 – adamentol and 4- bromo phenol in presence of 98% sulphuric acid and acetic acid, which on methylation with dimethyl sulfate and potassium carbonate in dry acetone yields 2-(l -adamantyl)-4-bromoanisole. The compound is reacted with magnesium to form Grignard reagent and then coupled with 6-bromo-2-methylnaphthoate in presence of novel Pd – Zn double metal catalyst to yield ester, which on saponification followed by treatment with acid yields Adapalene.The recent published application WO 2006/108717 describes the use of Suzuki coupling for the synthesis of adapalene the compound of formula – 1. The application describes the preparation of 3-adamantyl-4-methoxyphenyl boronic acid from 2-(l-adamantyl)-4- bromoanisole using n-Butyl Lithium and triisopropyl borate in solvent tetrahydrofuran. Finally 3-adamantyl-4-methoxyphenyl boronic acid is reacted with 6-bromo-2-naphthoic acid involving Suzuki coupling in presence of Palladium acetate catalyst, a ligand 2 – (dicyclohexyl – phosphino) biphenyl, an inorganic base in solvent to get the compound adapalene.Some of the drawbacks of the prior art processes include:- The reported process in US patent 4717720, using Negishi cross coupling involves Grignard reaction. This requires anhydrous condition and a possibility of runaway reaction during Grignard reagent formation. Also the reaction involves the addition of fused ZnC12 and the preparation of the catalyst NiC12 (DPPE) complex, which needs to be freshly prepared increases the reaction step and has to be thoroughly dried before its use for coupling. Further the coupling reaction, results in the formation of dimer impurities during the organozinc compound reaction, with 2-(I -adamantyl)-4-bromoanisole and 6-bromo-2-methylnaphthoate respectively, which are difficult to remove. All these operations make the entire synthesis extremely sensitive and difficult to handle.Some of the above drawbacks were addressed by the authors in the article published in Organic Process Research & Development, 2006, 10, 285 – 288 for the preparation of Adapalene. But the use of Pd catalyst with the ligand like PdCl2 (PPh3)2 for the direct conversion of Grignard reagent employing ZnCYl in catalytic amount has its own limitations. The use of Grignard reagent, palladium catalyst with ligand and hygroscopic ZnCl2 demerits this process for industrial application.The recent published application WO 2006/108717; describes the use of Suzuki coupling for the synthesis of adapalene the compound of formula – I. The use of organo boronic acids for the Suzuki reaction has some limitations because of the indeterminate stoichiometry associated with the use of boronic acid, and its difficulty in purification and the byproducts formed during the reaction.Therefore there remains a need for an improved process for preparing adapalene that eliminates or substantially reduces the impurities, decreases the number of steps, and employs a more robust process which is convenient and cost efficient.The present inventors have come out with a novel process which ameliorates the problems in the prior art with a one – pot process for the preparation of adapalene by employing Suzuki – Miyaura coupling involving the use of novel reactant 3-adamantyl-4- methoxyphenyl potassium trifiuoroborate.The novel compound 3 – Adamantyl – 4 – methoxy phenyl potassium trifiuoroborate, exhibit superb behavior in the Suzuki-Miyaura reaction and provides a powerful method for the preparation of 6 – [3-(I – Adamantyl) – 4 – methoxy phenyl] – 2 – naphthoic acid, the compound of Formula – I.

Formula – 1Potassium organotrifluoroborates are air and moisture-stable crystalline solids which can be stored for extended periods of time making it more industrial friendly to use on large scale production.The other advantage of the present invention is in the use of methyl ester of 6 – Bromo – 2 -naphthoic acid and isolating adapalane directly from the reaction instead of its methyl ester, the above process becomes more robust and eliminates the saponification step as reported in prior art. Also the use of readily and cheaply available Pd catalyst on carbon over the conventional and costlier Pd-catalyst with ligands offers further advantage to the current process.Examples:Example 1: Preparation of 3 – Adamantyl – 4 – methoxy phenyl potassium trifluoroborate:In a 2.0 L round bottom flask equipped with stirring and under nitrogen atmosphere 100.0 gm of 2-(l- adamantyl) 4-bromo anisole was charged in 600 ml tetrahydrofuran. The reaction mixture was cooled to -55 ± 50C and 302 ml of 1.6 M n – butyl Lithium was slowly added and stirred. 87 ml of tri isopropyl borate was then charged and stirring was continued for 30 minutes at -55 ± 5°C. Cooling was removed and the temperature raised slowly to 25 – 300C. 1.0 L of 1.2N hydrochloric acid was then charged and reaction mass was stirred for 30 minutes and separated the organic layer. The organic layer was charged in 1.0 L round bottom flask and freshly prepared aqueous solution of potassium hydrogen difluoride (230 gm, in 700 ml water) was added at 25 – 300C and stirring was maintained till white precipitate is obtained. The mixture was continued under stirring and cooled to 0 – 50C. The product, 3 – adamantyl – 4 – methoxyphenyl potassium trifluoroborate obtained was filtered, washed with 100 ml of ethyl acetate. The product was dried at 60 – 65°C till constant weight. Yield: 90.5 gm (83%), Purity: 99.0 % by HPLC.Example 2: Preparation of 6 – [3-(l- Adamantyl) – 4 – methoxyphenyl] – 2 – naphthoic acid:In a 1.0 L round bottom flask equipped with stirring and under nitrogen atmosphere 50.0 gm of 3 – Adamantyl – 4 – methoxyphenyl potassium trifluoroborate, 23 gm of 6- bromo -2-methyl napthoate in 300 ml tetrahydrofuran (THF) was charged. Stirred for 15 min and charged 3.0 gm of 5% Pd / C was and aqueous potassium hydroxide solution (50.0 gm in 300 ml water). Stirring was continued and the temperature was raised to reflux. The reaction mass was maintained for 10 hours at reflux and after the completion of the reaction, 200 ml of tetrahydrofuran: water (1 : 1) mixture was added and then filtered through hyflow bed at 45-500C. The hyflow bed was washed with tetrahydrofuran: water (1 : 1) mixture at 45-500C. 500 ml water was charged and the reaction mass was stirred. The aqueous layer was acidified with 1.2N hydrochloric acid. The precipitated mass was filtered, washed with water till neutral pH. The solid product obtained was dried at 70 – 75°C till constant weight to get 6 – [3-(l- adamantyl) – 4 – methoxyphenyl] – 2 – naphthoic acid.The dried product was taken in 300 ml of tetrahydrofuran and stirred. The temperature was raised to reflux and was maintained for 30 minutes. The heating was stopped and cooled the reaction mass to 25 – 300C. 500 ml of n – heptane was charged to the reaction mass and stirred for 30 minutes. The reaction mass was then chilled to 0 – 5°C and maintained stirring at 0 – 5°C temperature for 2.0 hours. The precipitated solid was filtered and washed with n – heptane. The pure crystalline 6 – [3-(l- adamantyl) – 4 methoxyphenyl] – 2 – naphthoic acid thus obtained was then dried till constant weight. Yield = 40 – 42 gms (68 – 72 %)
/////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
Per the recommendations of the Global Alliance on Improving Outcomes of Acne, retinoids such as adapalene are considered first-line therapy in acne treatment and are to be used either independently or in conjunction with benzoyl peroxide and/or an antimicrobial agent, like clindamycin, for maximum efficacy.[4][5] Furthermore, adapalene, like other retinoids, increases the efficacy and penetration of other topical acne medications that are used in conjunction with topical retinoids as well as hastens the improvement of the post-inflammatory hyperpigmentation caused by acne.[4] In the long term, it can be used as maintenance therapy.[4]
Off-label uses
Adapalene has the unique ability to inhibit keratinocyte differentiation and decrease keratin deposition. This property makes adapalene an effective treatment for keratosis pilaris and callus. It may be used by men undergoing foreskin restoration to reduce excess keratin that forms a layer on the exterior of the human penis after circumcision. Other non-FDA approved indications that have been reported in the literature include treatment of warts, molluscum contagiosum, Darier disease, photoaging, pigmentary disorders, actinic keratoses and alopecia areata.[6]
Side effects
Adapalene is known to cause mild adverse effects such as photosensitivity, irritation, redness, dryness, itching, and burning.[2] It is common (between 1% and 10% of users)[7] to experience a brief sensation of warmth or stinging, as well as dry skin, peeling and redness during the first 2–4 weeks of using the medication.[4][8] These effects are considered mild and generally decrease over time.[4][8] Any serious allergic reaction is rare.[8] Furthermore, of the three topical retinoids, adapalene is often regarded as the most tolerable.[6]
In pregnancy
Use of topical adapalene in pregnancy has not been well studied, but has a theoretical risk of retinoid embryopathy.[9] Thus far, there is no evidence that the cream causes problems in the baby if used during pregnancy. Use is at the consumer’s own risk.[10]
According to the Drugs and Lactation Database, topical adapalene has poor systemic absorption and results in low blood levels (less than 0.025 mcg/L) despite long term use, suggesting that there is low risk of harm for a nursing infant.[11] However, it is recommended that the topical medication should not be applied to the nipple or any other area that may come into direct contact with the infant’s skin.[11]
Interactions
Adapalene has been shown to enhance the efficacy of topical clindamycin, although adverse effects are also increased.[12][13] Application of adapalene gel to the skin 3–5 minutes before application of clindamycin enhances penetration of clindamycin into the skin, which may enhance the overall efficacy of the treatment as compared to clindamycin alone.[14]
Pharmacology
Unlike the retinoid tretinoin (Retin-A), adapalene has also been shown to retain its efficacy when applied at the same time as benzoyl peroxide due to its more stable chemical structure.[15] Furthermore, photodegradation of the molecule is less of a concern in comparison to tretinoin and tazarotene.[6]
Pharmacokinetics
Absorption of adapalene through the skin is low. A study with six acne patients treated once daily for five days with two grams of adapalene cream applied to 1,000 cm2 (160 sq in) of skin found no quantifiable amounts, or less than 0.35 ng/mL of the drug, in the patients’ blood plasma.[16] Controlled trials of chronic users of adapalene have found drug levels in the patients’ plasma to be 0.25 ng/mL.[9]
Pharmacodynamics
Adapalene is highly lipophilic. When applied topically, it readily penetrates hair follicles and absorption occurs 5 minutes after topical application.[2] After penetration into the follicle, adapalene binds to nuclear retinoic acid receptors (namely retinoic acid receptor beta and gamma).[5][9] These complexes then bind to the retinoid X receptor which induces gene transcription by binding to specific DNA sites, thus modulating downstream keratinocyte proliferation and differentiation.[2][9] This results in normalization of keratinocyte differentiation, allowing for decreased microcomedone formation, decreased clogging of pores, and increased exfoliation by increasing cell turnover.[6][9][17] Adapalene is also regarded as an anti-inflammatory agent, as it suppresses the inflammatory response stimulated by the presence of Cutibacterium acnes,[6] and inhibits both lipoxygenase activity and the oxidative metabolism of arachidonic acid into prostaglandins.[9]
Adapalene selectively targets retinoic acid receptor beta and retinoic acid receptor gamma when applied to epithelial cells such as those found in the skin.[18] Its agonism of the gamma subtype is largely responsible for adapalene’s observed effects. In fact, when adapalene is applied in conjunction with a retinoic acid receptor gamma antagonist, adapalene loses clinical efficacy.[19]
Retinization is a common temporary phenomenon reported by patients when initiating use of retinols.[20] Within the initial period of treatment, skin can become red, irritated, dry and may burn or itch from retinol application; however, this tends to resolve within four weeks with once a day use.[20]
History
Adapalene is a research product of Galderma Laboratories, France.[21] Adapalene was approved in 1996 by the U.S. Food and Drug Administration (FDA) for use in the treatment of acne.[22]
Research
A study has concluded that adapalene can be used to treat plantar warts and may help clear lesions faster than cryotherapy.[23]
References
- ^ Rolewski SL (October 2003). “Clinical review: topical retinoids”. Dermatology Nursing. 15 (5): 447–50, 459–65. PMID 14619325.
- ^ Jump up to:a b c d e f Tolaymat, L; Zito, PM (January 2021). “Adapalene”. PMID 29494115.
- ^ Asai, Yuka; Baibergenova, Akerke; Dutil, Maha; Humphrey, Shannon; Hull, Peter; Lynde, Charles; Poulin, Yves; Shear, Neil H.; Tan, Jerry; Toole, John; Zip, Catherine (2 February 2016). “Management of acne: Canadian clinical practice guideline”. Canadian Medical Association Journal. 188 (2): 118–126. doi:10.1503/cmaj.140665. PMC 4732962. PMID 26573753.
- ^ Jump up to:a b c d e Kolli, Sree S.; Pecone, Danielle; Pona, Adrian; Cline, Abigail; Feldman, Steven R. (2019-01-23). “Topical Retinoids in Acne Vulgaris: A Systematic Review”. American Journal of Clinical Dermatology. 20 (3): 345–365. doi:10.1007/s40257-019-00423-z. ISSN 1179-1888. PMID 30674002. S2CID 59225325.
- ^ Jump up to:a b Xiang, Leihong Flora; Troielli, Patricia; Lozada, Vicente Torres; Tan, Jerry; Suh, Dae Hun; See, Jo-Ann; Piquero-Martin, Jaime; Perez, Montserrat; Orozco, Beatriz (2018-02-01). “Practical management of acne for clinicians: An international consensus from the Global Alliance to Improve Outcomes in Acne”. Journal of the American Academy of Dermatology. 78 (2): S1–S23.e1. doi:10.1016/j.jaad.2017.09.078. hdl:10067/1492720151162165141. ISSN 0190-9622. PMID 29127053. S2CID 31654121.
- ^ Jump up to:a b c d e Tolaymat, Leila; Zito, Patrick M. (2018), “Adapalene”, StatPearls, StatPearls Publishing, PMID 29494115, retrieved 2019-03-13
- ^ “Differin”. Swedish Drug Formulary. Retrieved 2017-12-11.
- ^ Jump up to:a b c “Adapalene Gel”. WebMD. Retrieved 2017-12-11.
- ^ Jump up to:a b c d e f Piskin, Suleyman; Uzunali, Erol (August 2007). “A review of the use of adapalene for the treatment of acne vulgaris”. Therapeutics and Clinical Risk Management. 3 (4): 621–624. ISSN 1176-6336. PMC 2374937. PMID 18472984.
- ^ “FDA approves Differin Gel 0.1% for over-the-counter use to treat acne”. July 8, 2016. Retrieved 14 July 2016.
- ^ Jump up to:a b “Adapalene”, Drugs and Lactation Database (LactMed), National Library of Medicine (US), 2006, PMID 30000483, retrieved 2019-03-13
- ^ Wolf JE, Kaplan D, Kraus SJ, Loven KH, Rist T, Swinyer LJ, Baker MD, Liu YS, Czernielewski J (September 2003). “Efficacy and tolerability of combined topical treatment of acne vulgaris with adapalene and clindamycin: a multicenter, randomized, investigator-blinded study”. Journal of the American Academy of Dermatology. 49 (3 Suppl): S211-7. doi:10.1067/S0190-9622(03)01152-6. PMID 12963897.
- ^ Jain, GauravK; Ahmed, FarhanJ (2007). “Adapalene pretreatment increases follicular penetration of clindamycin: In vitro and in vivo studies”. Indian Journal of Dermatology, Venereology and Leprology. 73 (5): 326–9. doi:10.4103/0378-6323.34010. ISSN 0378-6323. PMID 17921613.
- ^ Jain GK, Ahmed FJ (2007). “Adapalene pretreatment increases follicular penetration of clindamycin: in vitro and in vivo studies” (PDF). Indian Journal of Dermatology, Venereology and Leprology. 73 (5): 326–9. doi:10.4103/0378-6323.34010. PMID 17921613.
- ^ Martin B, Meunier C, Montels D, Watts O (October 1998). “Chemical stability of adapalene and tretinoin when combined with benzoyl peroxide in presence and in absence of visible light and ultraviolet radiation”. The British Journal of Dermatology. 139 Suppl 52: 8–11. doi:10.1046/j.1365-2133.1998.1390s2008.x. PMID 9990414. S2CID 43287596.
- ^ “DIFFERIN® (adapalene) Cream, 0.1% Label” (PDF). FDA. May 25, 2000. Retrieved 4 Oct 2011.
- ^ “DIFFERIN® (adapalene) Gel, 0.3%” (PDF). Retrieved March 12, 2019.
- ^ Mukherjee S, Date A, Patravale V, Korting HC, Roeder A, Weindl G (2006). “Retinoids in the treatment of skin aging: an overview of clinical efficacy and safety”. Clinical Interventions in Aging. 1 (4): 327–48. doi:10.2147/ciia.2006.1.4.327. PMC 2699641. PMID 18046911.
- ^ Michel S, Jomard A, Démarchez M (October 1998). “Pharmacology of adapalene”. The British Journal of Dermatology. 139 Suppl 52: 3–7. doi:10.1046/j.1365-2133.1998.1390s2003.x. PMID 9990413. S2CID 23084886.
- ^ Jump up to:a b “Differin Gel: An Over-the-Counter Retinoid for Acne”. http://www.differin.com. Retrieved 2019-03-25.
- ^ US Patent 4717720A, Shroot B, Eustache J, Bernardon J-M, “Benzonaphthalene derivatives and compositions”, published 1988-01-05, issued 1988-01-05, assigned to Galderma Research and Development SNC
- ^ “FDA approval of DIFFERIN® (adapalene) Solution, 0.1%”. FDA. May 31, 1996. Retrieved 29 May 2017.
- ^ Gupta, Ramji; Gupta, Sarthak (2015). “Topical Adapalene in the Treatment of Plantar Warts; Randomized Comparative Open Trial in Comparison with Cryo-Therapy”. Indian Journal of Dermatology. 60 (1): 102. doi:10.4103/0019-5154.147835. ISSN 0019-5154. PMC 4318023. PMID 25657417.
External links
- “Adapalene”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Differin, Pimpal, Gallet, Adelene, Adeferin |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a604001 |
| License data | US DailyMed: Adapalene |
| Pregnancy category | AU: D |
| Routes of administration | Topical |
| Drug class | Retinoids |
| ATC code | D10AD03 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only) / S3CA: ℞-onlyUK: POM (Prescription only)US: OTC / Rx-only |
| Pharmacokinetic data | |
| Bioavailability | Very low[medical citation needed] |
| Excretion | Bile |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 106685-40-9 |
| PubChem CID | 60164 |
| IUPHAR/BPS | 5429 |
| DrugBank | DB00210 |
| ChemSpider | 54244 |
| UNII | 1L4806J2QF |
| KEGG | D01112 |
| ChEBI | CHEBI:31174 |
| ChEMBL | ChEMBL1265 |
| CompTox Dashboard (EPA) | DTXSID5046481 |
| ECHA InfoCard | 100.149.379 |
| Chemical and physical data | |
| Formula | C28H28O3 |
| Molar mass | 412.529 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
/////////////////ADAPALENE, CD 271, CD-271, ANTIACNE, Differin, Differine
COC1=C(C=C(C=C1)C1=CC2=C(C=C1)C=C(C=C2)C(O)=O)C12CC3CC(CC(C3)C1)C2

NEW DRUG APPROVALS
ONE TIME
$10.00
Fam-trastuzumab deruxtecan-nxki

Fam-trastuzumab deruxtecan-nxki
| Formula | C6460H9972N1724O2014S44. (C52H57FN9O13)8 |
|---|---|
| CAS | 1826843-81-5 |
| Mol weight | 153701.9811 |
| Antineoplastic | |
| Disease | Breast cancer (HER2 positive) |
|---|
DS-8201a
Trastuzumab deruxtecan, sold under the brand name Enhertu, is an antibody-drug conjugate consisting of the humanized monoclonal antibody trastuzumab (Herceptin) covalently linked to the topoisomerase I inhibitor deruxtecan (a derivative of exatecan).[5][6] It is licensed for the treatment of breast cancer or gastric or gastroesophageal adenocarcinoma.[6][7] Trastuzumab binds to and blocks signaling through epidermal growth factor receptor 2 (HER2/neu) on cancers that rely on it for growth. Additionally, once bound to HER2 receptors, the antibody is internalized by the cell, carrying the bound deruxtecan along with it, where it interferes with the cell’s ability to make DNA structural changes and replicate its DNA during cell division, leading to DNA damage when the cell attempts to replicate itself, destroying the cell.[7]
It was approved for medical use in the United States in December 2019,[6] in Japan in March 2020,[8] in the European Union in January 2021,[3][4] and in Australia in October 2021.[1]

Trastuzumab deruxtecan (DS-8201a) is a HER2-targeting antibody-drug conjugate or ADC), structurally composed of a humanized anti-human HER2 (anti-hHER2) antibody, an enzymatically cleavable peptide-linker, and a proprietary topoisomerase I inhibitor payload (exatecan derivative or DX-8951 / DXd).

CLIP
Trastuzumab deruxtecan active substance, also referred to as DS-8201a, results from the conjugation of the following intermediates: – Trastuzumab monoclonal antibody (MAAL-9001); – A drug-linker (MAAA-1162a) comprised of a Topoisomerase I inhibitor derivative of exatecan (MAAA1181a) and a tetrapeptide based cleavable linker (MFAH). MAAL-9001 is covalently conjugated to approximately 8 molecules of MAAA-1162a. The linker is designed to be stable in plasma to reduce systemic exposure to the released MAAA-1181a drug. After cell internalisation, the released MAAA-1181a drug leads to apoptosis of the target tumour cells via the inhibition of topoisomerase I. The released MAAA-1181a drug is cell-membrane permeable, giving the ability to penetrate and act in surrounding cells. The effect of the ADC derives primarily from the released MAAA-1181a drug and to a lesser extent to the antibody-dependent cellular cytotoxic (ADCC) effector function of the conjugated antibody. The quality of MAAL-9001 antibody, MAAA-1162a drug-linker and the conjugated antibody is described in separate sections. The structures of DS-8201a, MAAA-1162a, MAAA-1181a, and MAAL-9001 are provided in Figure 1.

Full information for the active substance intermediate MAAA-1162a (C52H56FN9O13, MW 1034.05) was provided in the dossier. MAAA-1162a is composed of DX-8951·MsOH (drug intermediate) and MFAH (linker intermediate with maleimide functionality). The maleimide moiety reacts with the antibody (MAAL-9001) in the conjugation reaction to yield trastuzumab deruxtecan (DS-8201a). MAAA-1162a contains 3 stereogenic centres. General information was provided for solid state form, melting point, moisture sorption, UV-Vis absorption, optical rotation and solubility.
/////////////////////////////////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
CLIP
Fam-trastuzumab deruxtecan-nxki is an ADC that is comprised of an anti-HER2 antibody and the potent topoisomerase inhibitor exatecan [206]. These two entities are connected via a liner consisting of a maleimide conjugation handle that includes a protease-cleavable Gly-Gly-Phe-Gly (GGFG) tetrapeptide linker. This conjugation handle could release deruxtecan after internalization of the conjugate by the cancer cells that recognize the antibody. Fam-trastuzumabderuxtecan-nxki was developed by Daiichi Sankyo and AstraZeneca, and granted approval by the FDA in December 2019 [207]. There are approximately eight payload molecules/antibodies. Fam-trastuzumab deruxtecan-nxki has been approved for the treatment of adult patients with HER2 positive breast cancer that is unresectable or metastatic [208]. The synthesis of GGFG linker is described in Scheme 38 [209,210]. First, commercial tert-butyl 2-aminoacetate 274 was treated with Fmoc-L-Phe-OH 275 in the presence of HOBt and DIC, giving the corresponding product 276. Further deprotection of Fmoc group and amide formation gave the intermediate 278, which then underwent removal of Fmoc group to give GGFG linker 279. Lastly, treatment of 279 with the activated ester 280 provided 281.
Preparation of the payload exatecan derivative is described in Scheme 39 [211]. Aluminum-catalyzed Friedel-Crafts acylation of o-fluorotoluene 282 with succinic andydride 283 gave intermediate 284 in 90% yield. Next, hydrogenation reduction ofthe carbonyl group of 284, followed by reaction with SOCl2 in MeOH, furnished 285, which then underwent nitration with H2SO4 and KNO3 to give compound 286 in 48% overall yield. Hydrolysis of 286 followed by treatment with polyphosphoric acid (PPA) gave the cyclization product 287 in only 27% yield. The transformation of 287 into 288 was realized following the four-step sequence: carbonyl reduction with NaBH4, acid-mediated elimination reaction, PtO2-catalyzed hydrogenation reduction, and acetylation with Ac2O. Regioselective benzylic oxidation of 288 in acetone with KMnO4 gave 289 in 65% yield, further functionalization with butyl nitrile and Zn-mediated acylation gave compound 290 in 66% yield over 2 steps. Treatment of 290 with aqueous HCl provided hydrolysis product 291 in 50% yield, which then coupled with ethyl trifluoroacetate to provide intermediate 292. Polycyclic compound 294 was prepared from 292 and 293 through a [4+2] cycloaddition reaction in refluxing toluene. The key intermediate 294 next underwent acidic hydrolysis and chiral resolution to provide the chiral product 295. Further condensation reaction with 296 in the presence of T3P and Et3N in DCM and TFA-promoted removal of the Boc group formed 297. The synthesis of fam-trastuzumab deruxtecan-nxki is described in Scheme 40 [212]. The linker 281 was coupled to 297 in the presence of T3P and Et3N to give the linker-payload 298. Through transformation of the disulfide bonds into free sulfhydryl groups for linkage (DTT in pH 8.0 buffer), followed by re-oxidation of the remaining disulfide bonds with cysteine, the linker-payload 298 was conjugated to the anti-HER2 mAb to give fam-trastuzumab deruxtecan-nxki (XXIX) based on the amount of protein with approximately eight linker/payloads per antibody.



[206] T.N. Iwata, K. Sugihara, T. Wada, T. Agatsuma, [Fam-] trastuzumab deruxtecan (DS-8201a)-induced antitumor immunity is facilitated by the anti-CTLA-4 antibody in a mouse model, PLoS One 14 (2019) 0222280.
[207] R. Voelker, Another targeted therapy for ERBB2-positive breast cancer, JAMA 323 (2020) 408.
[208] S. Modi, C. Saura, T. Yamashita, Y.H. Park, S.B. Kim, K. Tamura, F. Andre, H. Iwata, Y. Ito, J. Tsurutani, J. Sohn, N. Denduluri, C. Perrin, K. Aogi, E. Tokunaga, S.A. Im, K.S. Lee, S.A. Hurvitz, J. Cortes, C. Lee, S. Chen, L. Zhang, J. Shahidi, A. Yver, I. Krop, Trastuzumab deruxtecan in previously treated HER2-positive breast cancer, N. Engl. J. Med. 382 (2020) 610-621.
[209] C.L. Law, K. Klussman, A.F. Wahl, P. Senter, S. Doronina, B. Toki, Treatment of immunological disorders using anti-CD30 antibodies, 2003.WO2003043583.
[210] S. Doronina, P.D. Senter, B.E. Toki, Pentapeptide compounds and uses related thereto, 2002. WO2002088172. [211] H. Terasawa, A. Ejima, S. Ohsuki, K. Uoto, Hexa-cyclic compound, 1998. US5834476.
[212] G.M. Dubowchik, R.A. Firestone, L. Padilla, D. Willner, S.J. Hofstead, K. Mosure, J.O. Knipe, S.J. Lasch, P.A. Trail, Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: model studies of enzymatic drug release and antigen-specific in vitro anticancer activity, Bioconjugate. Chem 13 (2002) 855-869.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized |
| Target | HER2 |
| Clinical data | |
| Trade names | Enhertu |
| Other names | DS-8201a, fam-trastuzumab deruxtecan-nxki |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Trastuzumab_deruxtecanUS FDA: Enhertu |
| Pregnancy category | AU: D[1] |
| Routes of administration | Intravenous |
| ATC code | L01FD04 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only) [1]US: ℞-only [2]EU: Rx-only [3]Rx-only[4] |
| Identifiers | |
| CAS Number | 1826843-81-5 |
| PubChem SID | 384585505 |
| DrugBank | DB14962 |
| UNII | 5384HK7574 |
| KEGG | D11529 |
| ChEMBL | ChEMBL4297844 |
| Chemical and physical data | |
| Formula | C6460H9972N1724O2014S44.(C52H57F1N9O13)8 |
Medical uses
Trastuzumab deruxtecan-nxki is indicated for the treatment of adults with unresectable (unable to be removed with surgery) or metastatic (when cancer cells spread to other parts of the body) HER2-positive breast cancer who have received two or more prior anti-HER2-based regimens in the metastatic setting and for adults with locally advanced or metastatic HER2-positive gastric or gastroesophageal junction adenocarcinoma who have received a prior trastuzumab-based regimen.[6][7]
Side effects and label warnings
The most common side effects are nausea, fatigue, vomiting, alopecia (hair loss), constipation, decreased appetite, anemia (hemoglobin in blood is below the reference range), decreased neutrophil count (white blood cells that help lead your body’s immune system response to fight infection), diarrhea, leukopenia (other white blood cells that help the immune system), cough and decreased platelet count (component of blood whose function is to react to bleeding from blood vessel injury by clumping, thereby initiating a blood clot).[6]
The prescribing information for fam-trastuzumab deruxtecan-nxki includes a boxed warning to advise health care professionals and patients about the risk of interstitial lung disease (a group of lung conditions that causes scarring of lung tissues) and embryo-fetal toxicity.[6] Interstitial lung disease and pneumonitis, including cases resulting in death, have been reported with fam-trastuzumab deruxtecan-nxki.[6]
History
The U.S. Food and Drug Administration (FDA) approved fam-trastuzumab deruxtecan-nxki in December 2019.[6][9] The application for fam-trastuzumab deruxtecan-nxki was granted accelerated approval, fast track designation, and breakthrough therapy designation.[6]
The FDA approved fam-trastuzumab deruxtecan-nxki based on the results of one clinical trial enrolling 184 female patients with HER2-positive, unresectable and/or metastatic breast cancer who had received two or more prior anti-HER2 therapies in the metastatic setting.[6] These patients were heavily pretreated in the metastatic setting, receiving between two and 17 therapies prior to receiving fam-trastuzumab deruxtecan-nxki.[6] Patients in the clinical trial received fam-trastuzumab deruxtecan-nxki every three weeks and tumor imaging was obtained every six weeks.[6] The overall response rate was 60.3%, which reflects the percentage of patients who had a certain amount of tumor shrinkage with a median duration of response of 14.8 months.[6]
The FDA granted the approval of Enhertu to Daiichi Sankyo.[6]
On 10 December 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a conditional marketing authorization for the medicinal product Enhertu, intended for the treatment of metastatic HER2-positive breast cancer.[10][11] Enhertu was reviewed under EMA’s accelerated assessment program. The applicant for this medicinal product is Daiichi Sankyo Europe GmbH. Trastuzumab deruxtecan was approved for medical use in the European Union in January 2021.[3][4]
In January 2021, the U.S. Food and Drug Administration (FDA) granted accelerated approval to fam-trastuzumab deruxtecan-nxki for the treatment of adults with locally advanced or metastatic HER2-positive gastric or gastroesophageal (GEJ) adenocarcinoma who have received a prior trastuzumab-based regimen.[7][12]
Efficacy was evaluated in a multicenter, open-label, randomized trial (DESTINY-Gastric01, NCT03329690) in participants with HER2-positive locally advanced or metastatic gastric or GEJ adenocarcinoma who had progressed on at least two prior regimens, including trastuzumab, a fluoropyrimidine- and a platinum-containing chemotherapy.[7] A total of 188 participants were randomized (2:1) to receive fam-trastuzumab deruxtecan-nxki 6.4 mg/kg intravenously every three weeks or physician’s choice of either irinotecan or paclitaxel monotherapy.[7]
References
- ^ Jump up to:a b c “Enhertu”. Therapeutic Goods Administration (TGA). 18 October 2021. Retrieved 22 October 2021.
- ^ “Enhertu- fam-trastuzumab deruxtecan-nxki injection, powder, lyophilized, for solution”. DailyMed. Retrieved 15 January 2021.
- ^ Jump up to:a b c “Enhertu EPAR”. European Medicines Agency (EMA). 9 December 2020. Retrieved 31 March 2021.
- ^ Jump up to:a b c “Enhertu approved in the EU for the treatment of HER2-positive metastatic breast cancer” (Press release). AstraZeneca. 20 January 2021. Retrieved 21 January 2021.
- ^ A HER2-Targeting Antibody–Drug Conjugate, Trastuzumab Deruxtecan (DS-8201a), Enhances Antitumor Immunity in a Mouse Model
- ^ Jump up to:a b c d e f g h i j k l m n “FDA approves new treatment option for patients with HER2-positive breast cancer who have progressed on available therapies”. U.S.Food and Drug Administration (FDA) (Press release). 20 December 2019. Archived from the original on 20 December 2019. Retrieved 20 December 2019. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f “FDA approves fam-trastuzumab deruxtecan-nxki for HER2-positive gastric adenocarcinomas”. U.S. Food and Drug Administration (FDA). 15 January 2021. Retrieved 15 January 2021. This article incorporates text from this source, which is in the public domain.
- ^ “Enhertu Approved in Japan for Treatment of Patients with HER2 Positive Unresectable or Metastatic Breast Cancer” (Press release). Daiichi Sankyo. 25 March 2020. Retrieved 21 January 2021 – via Business Wire.
- ^ “Drug Trials Snapshot: Enhertu”. U.S. Food and Drug Administration (FDA). 20 December 2019. Retrieved 24 January 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Enhertu: Pending EC decision”. European Medicines Agency (EMA). 10 December 2020. Retrieved 11 December 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Trastuzumab deruxtecan recommended for approval in the EU by CHMP for HER2-positive metastatic breast cancer” (Press release). AstraZeneca. 14 December 2020. Retrieved 21 January 2021.
- ^ “Enhertu approved in the US for the treatment of patients with previously treated HER2-positive advanced gastric cancer” (Press release). AstraZeneca. 18 January 2021. Retrieved 22 January 2021.
Further reading
- Iwata TN, Sugihara K, Wada T, et al. (October 2019). “[Fam-] trastuzumab deruxtecan (DS-8201a)-induced antitumor immunity is facilitated by the anti-CTLA-4 antibody in a mouse model”. PLOS ONE. 14 (10): e0222280. Bibcode:2019PLoSO..1422280I. doi:10.1371/journal.pone.0222280. PMC 6772042. PMID 31574081.
- Modi S, Saura C, Yamashita T, et al. (February 2020). “Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer”. N. Engl. J. Med. 382 (7): 610–621. doi:10.1056/NEJMoa1914510. PMC 7458671. PMID 31825192.
External links
- “Trastuzumab_deruxtecan”. Drug Information Portal. U.S. National Library of Medicine.
- Deruxtecan shows structure
- Clinical trial number NCT03329690 for “DS-8201a in Human Epidermal Growth Factor Receptor 2 (HER2)-Expressing Gastric Cancer [DESTINY-Gastric01]” at ClinicalTrials.gov
////////////Fam-trastuzumab deruxtecan-nxki ,, FDA 2019, APROVALS 2019, DS-8201a

NEW DRUG APPROVALS
ONE TIME
$10.00
Tixagevimab
| (Heavy chain) QMQLVQSGPE VKKPGTSVKV SCKASGFTFM SSAVQWVRQA RGQRLEWIGW IVIGSGNTNY AQKFQERVTI TRDMSTSTAY MELSSLRSED TAVYYCAAPY CSSISCNDGF DIWGQGTMVT VSSASTKGPS VFPLAPSSKS TSGGTAALGC LVKDYFPEPV TVSWNSGALT SGVHTFPAVL QSSGLYSLSS VVTVPSSSLG TQTYICNVNH KPSNTKVDKR VEPKSCDKTH TCPPCPAPEF EGGPSVFLFP PKPKDTLYIT REPEVTCVVV DVSHEDPEVK FNWYVDGVEV HNAKTKPREE QYNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKALPASIEK TISKAKGQPR EPQVYTLPPS REEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT PPVLDSDGSF FLYSKLTVDK SRWQQGNVFS CSVMHEALHN HYTQKSLSLS PGK (Light chain) EIVLTQSPGT LSLSPGERAT LSCRASQSVS SSYLAWYQQK PGQAPRLLIY GASSRATGIP DRFSGSGSGT DFTLTISRLE PEDFAVYYCQ HYGSSRGWTF GQGTKVEIKR TVAAPSVFIF PPSDEQLKSG TASVVCLLNN FYPREAKVQW KVDNALQSGN SQESVTEQDS KDSTYSLSST LTLSKADYEK HKVYACEVTH QGLSSPVTKS FNRGEC (Disulfide bridge: H22-H96, H101-H106, H150-H206, H216-L216, H232-H’232, H235-H’235, H267-H327, H373-H431, H’22-H’96, H’101-H’106, H’150-H’206, H’226-L’216, H’267-H’327, H’373-H’431, L23-L89, L136-L196, L’23-L’89, L’136-L’196) |
Tixagevimab
FDA 2021, 2021/12/8
ANTI VIRAL, CORONA VIRUS, PEPTIDE
Monoclonal antibody
Treatment and prevention of SARS-CoV-2 infection
| Formula | C6488H10034N1746O2038S50 |
|---|---|
| CAS | 2420564-02-7 |
| Mol weight | 146704.817 |
- 2196
- AZD-8895
- AZD8895
- COV2-2196
- Tixagevimab
- Tixagevimab [INN]
- UNII-F0LZ415Z3B
- WHO 11776
- OriginatorVanderbilt University
- DeveloperAstraZeneca; INSERM; National Institute of Allergy and Infectious Diseases
- ClassAntivirals; Monoclonal antibodies
- Mechanism of ActionVirus internalisation inhibitors
- RegisteredCOVID 2019 infections
- 24 Dec 2021Pharmacodynamics data from a preclinical trial in COVID-2019 infections released by AstraZeneca
- 16 Dec 2021Pharmacodynamics data from a preclinical trial in COVID-2019 infections released by AstraZeneca
- 10 Dec 2021Registered for COVID-2019 infections (In the elderly, Prevention, In adults) in USA (IM) – Emergency Use Authorization
Tixagevimab/cilgavimab is a combination of two human monoclonal antibodies, tixagevimab (AZD8895) and cilgavimab (AZD1061) targeted against the surface spike protein of SARS-CoV-2[4][5] used to prevent COVID-19. It is being developed by British-Swedish multinational pharmaceutical and biotechnology company AstraZeneca.[6][7] It is co-packaged and given as two separate consecutive intramuscular injections (one injection per monoclonal antibody, given in immediate succession).[2]
/////////////////////////////////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Development
In 2020, researchers at Vanderbilt University Medical Center discovered particularly potent monoclonal antibodies, isolated from COVID-19 patients infected with a SARS-CoV-2 circulating at that time. Initially designated COV2-2196 and COV2-2130, antibody engineering was used to transfer their SARS-CoV-2 binding specificity to IgG scaffolds that would last longer in the body, and these engineered antibodies were named AZD8895 and AZD1061, respectively (and the combination was called AZD7442).[8]
To evaluate the antibodies’ potential as monoclonal antibody based prophylaxis (prevention), the ‘Provent’ clinical trial enrolled 5,000 high risk but not yet infected individuals and monitored them for 15 months.[9][10] The trial reported that those receiving the cocktail showed a 77% reduction in symptomatic COVID-19 and that there were no severe cases or deaths. AstraZeneca also found that the antibody cocktail “neutralizes recent emergent SARS-CoV-2 viral variants, including the Delta variant“.[7]
In contrast to pre-exposure prophylaxis, the Storm Chaser study of already-exposed people (post-exposure prophylaxis) did not meet its primary endpoint, which was prevention of symptomatic COVID-19 in people already exposed. AZD7442 was administered to 1,000 volunteers who had recently been exposed to COVID.[9]
Regulatory review
In October 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) started a rolling review of tixagevimab/cilgavimab, which is being developed by AstraZeneca AB, for the prevention of COVID-19 in adults.[11]
Also in October 2021, AstraZeneca requested Emergency Use Authorization for tixagevimab/cilgavimab to prevent COVID-19 from the U.S. Food and Drug Administration (FDA).[12][13]
Emergency use authorization
On 14 November 2021, Bahrain granted emergency use authorization.[14]
On 8 December 2021, the U.S. Food and Drug Administration (FDA) granted emergency use authorization of this combination to prevent COVID-19 (before exposure) in people with weakened immunity or who cannot be fully vaccinated due to a history of severe reaction to coronavirus vaccines.[15] The FDA issued an emergency use authorization (EUA) for AstraZeneca’s Evusheld (tixagevimab co-packaged with cilgavimab and administered together) for the pre-exposure prophylaxis (prevention) of COVID-19 in certain people aged 12 years of age and older weighing at least 40 kilograms (88 lb).[2] The product is only authorized for those individuals who are not currently infected with the SARS-CoV-2 virus and who have not recently been exposed to an individual infected with SARS-CoV-2.[2]
References
- ^ “Evusheld- azd7442 kit”. DailyMed. Retrieved 4 January 2022.
- ^ Jump up to:a b c d “Coronavirus (COVID-19) Update: FDA Authorizes New Long-Acting Monoclonal Antibodies for Pre-exposure Prevention of COVID-19 in Certain Individuals”. U.S. Food and Drug Administration (FDA) (Press release). 8 December 2021. Retrieved 9 December 2021. This article incorporates text from this source, which is in the public domain.
- ^ O’Shaughnessy, Jacqueline A. (20 December 2021). “Re: Emergency Use Authorization 104” (PDF). Food and Drug Administration. Letter to AstraZeneca Pharmaceuticals LP | Attention: Stacey Cromer Berman, PhD. Archived from the original on 29 December 2021. Retrieved 18 January 2022.
- ^ “IUPHAR/BPS Guide to PHARMACOLOGY”. IUPHAR. 27 December 2021. Retrieved 27 December 2021.
- ^ “IUPHAR/BPS Guide to PHARMACOLOGY”. IUPHAR. 27 December 2021. Retrieved 27 December 2021.
- ^ Ray, Siladitya (21 August 2021). “AstraZeneca’s Covid-19 Antibody Therapy Effective In Preventing Symptoms Among High-Risk Groups, Trial Finds”. Forbes. ISSN 0015-6914. Archived from the original on 21 August 2021. Retrieved 18 January 2022.
- ^ Jump up to:a b Goriainoff, Anthony O. (20 August 2021). “AstraZeneca Says AZD7442 Antibody Phase 3 Trial Met Primary Endpoint in Preventing Covid-19”. MarketWatch. Archived from the original on 21 August 2021. Retrieved 18 January 2022.
- ^ Dong J, Zost SJ, Greaney AJ, Starr TN, Dingens AS, Chen EC, et al. (October 2021). “Genetic and structural basis for SARS-CoV-2 variant neutralization by a two-antibody cocktail”. Nature Microbiology. 6 (10): 1233–1244. doi:10.1038/s41564-021-00972-2. ISSN 2058-5276. PMC 8543371. PMID 34548634.
- ^ Jump up to:a b Haridy, Rich (23 August 2021). “”Game-changing” antibody cocktail prevents COVID-19 in the chronically ill”. New Atlas. Retrieved 23 August 2021.
- ^ “AZD7442 PROVENT Phase III prophylaxis trial met primary endpoint in preventing COVID-19”. AstraZeneca (Press release). 20 August 2021. Retrieved 15 October 2021.
- ^ “EMA starts rolling review of Evusheld (tixagevimab and cilgavimab)”. European Medicines Agency. 14 October 2021. Retrieved 15 October 2021.
- ^ “AZD7442 request for Emergency Use Authorization for COVID-19 prophylaxis filed in US”. AstraZeneca US (Press release). 5 October 2021. Retrieved 15 October 2021.
- ^ “AZD7442 request for Emergency Use Authorization for COVID-19 prophylaxis filed in US”. AstraZeneca (Press release). 5 October 2021. Retrieved 15 October 2021.
- ^ Abd-Alaziz, Moaz; Elhamy, Ahmad (14 November 2021). Macfie, Nick (ed.). “Bahrain authorizes AstraZeneca’s anti-COVID drug for emergency use”. Reuters. Archived from the original on 23 November 2021. Retrieved 18 January 2022.
- ^ Mishra, Manas; Satija, Bhanvi (8 December 2021). Dasgupta, Shounak (ed.). “U.S. FDA authorizes use of AstraZeneca COVID-19 antibody cocktail”. Reuters. Archived from the original on 13 January 2022. Retrieved 18 January 2022.
External links
“Tixagevimab”. Drug Information Portal. U.S. National Library of Medicine.
- “Cilgavimab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT04625972 for “Phase III Double-blind, Placebo-controlled Study of AZD7442 for Post-exposure Prophylaxis of COVID-19 in Adults (STORM CHASER)” at ClinicalTrials.gov
- Clinical trial number NCT04625725 for “Phase III Double-blind, Placebo-controlled Study of AZD7442 for Pre-exposure Prophylaxis of COVID-19 in Adult. (PROVENT)” at ClinicalTrials.gov
| Tixagevimab (teal, right) and cilgavimab (purple, left) binding the spike protein RBD. From PDB: 7L7E. | |
| Combination of | |
|---|---|
| Tixagevimab | Monoclonal antibody |
| Cilgavimab | Monoclonal antibody |
| Clinical data | |
| Trade names | Evusheld |
| Other names | AZD7442 |
| License data | US DailyMed: Tixagevimab |
| Routes of administration | Intramuscular |
| ATC code | J06BD03 (WHO) |
| Legal status | |
| Legal status | US: ℞-only via emergency use authorization[1][2][3] |
| Identifiers | |
| KEGG | D12262 |
| Clinical data | |
|---|---|
| Drug class | Antiviral |
| ATC code | None |
| Identifiers | |
| CAS Number | 2420564-02-7 |
| DrugBank | DB16394 |
| UNII | F0LZ415Z3B |
| KEGG | D11993 |
| Chemical and physical data | |
| Formula | C6488H10034N1746O2038S50 |
| Molar mass | 146706.82 g·mol−1 |
| Clinical data | |
|---|---|
| Drug class | Antiviral |
| ATC code | None |
| Identifiers | |
| CAS Number | 2420563-99-9 |
| DrugBank | DB16393 |
| UNII | 1KUR4BN70F |
| KEGG | D11994 |
| Chemical and physical data | |
| Formula | C6626H10218N1750O2078S44 |
| Molar mass | 149053.44 g·mol−1 |
/////////////////Tixagevimab, ANTI VIRAL, CORONA VIRUS, PEPTIDE, Monoclonal antibody, SARS-CoV-2 , WHO 11776, 2196, AZD-8895, AZD 8895, COV2-2196, COVID 19
NEW DRUG APPROVALS
ONE TIME
$10.00
Daridorexant

Daridorexant
- Molecular FormulaC23H23ClN6O2
- Average mass450.921 Da
[(2S)-2-(5-Chloro-4-methyl-1H-benzimidazol-2-yl)-2-methyl-1-pyrrolidinyl][5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl]methanone
1505484-82-1[RN]
Methanone, [(2S)-2-(5-chloro-4-methyl-1H-benzimidazol-2-yl)-2-methyl-1-pyrrolidinyl][5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl]-
ACT-541468, , Nemorexant
FDA APPROVED 2022, 1/7/2022, To treat insomnia,

Daridorexant HCl
CAS#: 1792993-84-0 (HCl)
Chemical Formula: C23H24Cl2N6O2
Molecular Weight: 487.39
Elemental Analysis: C, 56.68; H, 4.96; Cl, 14.55; N, 17.24; O, 6.57
Methanone, ((2S)-2-(6-chloro-7-methyl-1H-benzimidazol-2-yl)-2-methyl-1-pyrrolidinyl)(5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl)-, hydrochloride (1:1)
Daridorexant HCl; Daridorexant hydrochloride; ACT541468A; ACT 541468A; ACT-541468A; ACT541468 hydrochloride; ACT 541468 hydrochloride; ACT-541468 hydrochloride
Daridorexant HCl is used in the treat of Insomnia Disorder in Adult Patients
Daridorexant, sold under the brand name Quviviq, is a medication used for the treatment of insomnia.[1] Daridorexant is a dual orexin receptor antagonist (DORA) which was originated by Actelion Pharmaceuticals and is under development by Idorsia Pharmaceuticals.[3][4] It acts as a selective dual antagonist of the orexin receptors OX1 and OX2.[3][4] The medication has a relatively short elimination half-life of 6 to 10 hours.[2] As of April 2020, daridorexant has passed its first phase III clinical trial for the treatment of insomnia.[3]Daridorexant was approved for medical use in the United States in January 2022.[1][5][6]
Daridorexant, formerly known as nemorexant, is a selective dual orexin receptor antagonist used to treat insomnia. Insomnia is characterized by difficulties with sleep onset and/or sleep maintenance and impairment of daytime functioning. It chronically affects the person’s daily functioning and long-term health effects, as insomnia is often associated with comorbidities such as hypertension, diabetes, and depression. Conventional treatments for insomnia include drugs targeting gamma-aminobutyric acid type-A (GABA-A), serotonin, histamine, or melatonin receptors; however, undesirable side effects are frequently reported, such as next-morning residual sleepiness, motor incoordination, falls, memory and cognitive impairment. Novel drugs that target orexin receptors gained increasing attention after discovering the role of orexin signalling pathway in wakefulness and almorexant, an orexin receptor antagonist that improved sleep. Daridorexant was designed via an intensive drug discovery program to improve the potency and maximize the duration of action while minimizing next-morning residual activity.1
Daridorexant works on orexin receptors OX1R and OX2R to block the binding of orexins, which are wake-promoting neuropeptides and endogenous ligands to these receptors. Daridorexant reduces overactive wakefulness: in the investigational trials, daridorexant reportedly improved sleep and daytime functioning in patients with insomnia.1 It was approved by the FDA on January 10, 2022, under the name QUVIVIQ.6 as the second orexin receptor antagonist approved to treat insomnia following suvorexant.2
QUVIVIQ
- Generic Name: daridorexant tablets
- Brand Name: Quviviq
QUVIVIQ contains daridorexant, an orexin receptor antagonist. The chemical name of daridorexant hydrochloride is (S)-(2-(5-chloro-4-methyl-1H-benzo[d]imidazol-2-yl)-2-methylpyrrolidin-1-yl)(5- methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone hydrochloride. The molecular formula is C23H23N6O2Cl * HCl. The molecular weight is 487.38 g/mol.
The structural formula is:
 |
Daridorexant hydrochloride is a white to light yellowish powder that is very slightly soluble in water.
QUVIVIQ tablets are intended for oral administration. Each film-coated tablet contains 27 mg or 54 mg of daridorexant hydrochloride equivalent to 25 mg or 50 mg of daridorexant, respectively. The inactive ingredients are croscarmellose sodium, magnesium stearate, mannitol, microcrystalline cellulose, povidone, and silicon dioxide.
In addition, the film coating contains the following inactive ingredients: glycerin, hypromellose, iron oxide black, iron oxide red, microcrystalline cellulose, talc, titanium dioxide, and, in the 50 mg tablet only, iron oxide yellow.
Dosage Forms And Strengths
QUVIVIQ (daridorexant) tablets are available as:
25 mg: light purple, arc-triangle shaped, film-coated tablet debossed with “25” on one side and “i” (Idorsia logo) on the other side, containing 25 mg daridorexant.
50 mg: light orange, arc-triangle shaped, film-coated tablet debossed with “50” on one side and “i” (Idorsia logo) on the other side, containing 50 mg daridorexant.
QUVIVIQ tablets are available as:
25 mg, light purple, arc-triangle shaped film-coated tablets debossed with “25” on one side, and “i” on the other side. NDC 80491-7825-3, bottle of 30 with child-resistant closure
50 mg: light orange, arc-triangle shaped film-coated tablets debossed with “50” on one side, and “i” on the other side. NDC 80491-7850-3, bottle of 30 with child-resistant closure
SYN
https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cmdc.202000453
Since its discovery in 1998, the orexin system has been of interest to the research community as a potential therapeutic target for the treatment of sleep/wake disorders. Herein we describe our efforts leading to the identification of daridorexant, which successfully finished two pivotal phase 3 clinical trials for the treatment of insomnia disorders.

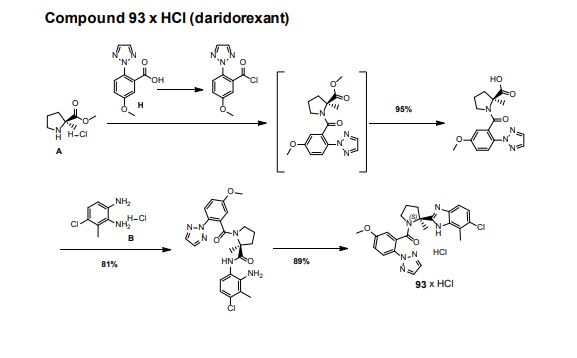
Step 3. Amide (S7) (1000 g, 2.13 mmol) was dissolved in EtOH (5 L) and 32% aqueous HCl (500 mL) was added at 23 °C. The solution was filtered through a Whatman filter (5 µm). The filtrate was heated to 75 °C for 4h. The resulting suspension was cooled to 0 °C and filtered. The product was dried under reduced pressure to yield 93 x HCl (922 g, 89%) as a white solid.
LC-MS B: tR = 0.78 min; [M+H]+ = 451.19, mp 280 °C.
1H NMR (500 MHz, D6-DMSO) δ: 15.05- 15.65 (m, 1 H), 8.06 (s, 2 H), 7.79 (s, 1 H), 7.75 (d, J = 8.9 Hz, 2 H), 7.66 (m, 1 H), 7.57 (d, J = 8.7 Hz, 1 H), 7.15 (dd, J1 = 2.9 Hz, J2 = 8.9 Hz, 1 H), 4.06-4.10 (m, 1 H), 3.92 (s, 3 H), 3.35 (s, 1 H), 2.78 (s, 3 H), 2.54-2.67 (m, 1 H), 2.23-2.31 (m, 1 H), 2.06-2.20 (m, 2 H), 1.97 (s, 3 H),
13C NMR (125 MHz, D6-DMSO) δ: 166.2, 159.3, 158.6, 136.5, 132.7, 131.9, 130.4, 130.3, 129.4, 126.8, 124.5, 123.4, 116.4, 113.7, 113.0, 61.6, 56.8, 49.7, 41.1, 23.9, 20.2, 15.7.

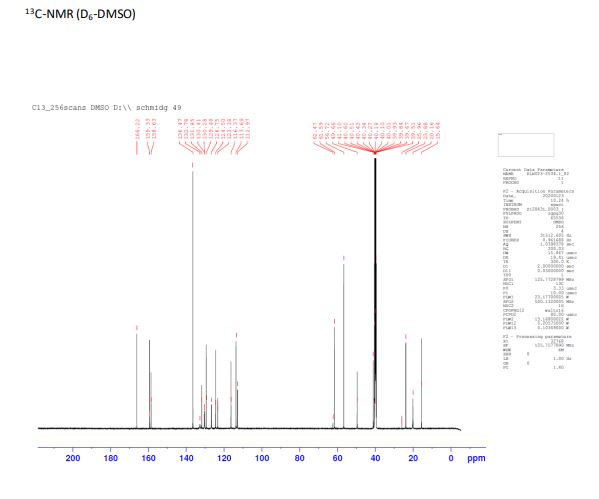
SYN
https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cmdc.201900618
Abstract
DORA explorers: The orexin system plays an important role in regulating the sleep-wake cycle. Herein we report our optimization efforts toward a novel dual orexin receptor antagonist (DORA) with improved properties over compound 6. Replacing the oxadiazole by a triazole resulted in compounds (e. g. compound 33) with improved properties, such as higher intrinsic metabolic stability, lower plasma protein binding, higher brain free fraction, and increased solubility. Further optimization was needed to decrease the compounds P-glycoprotein susceptibility. Our work led to the identification of compound 42, a potent, brain-penetrating DORA with improved in vivo efficacy in dogs compared with compound 6.

Abstract
The orexin system is responsible for regulating the sleep-wake cycle. Suvorexant, a dual orexin receptor antagonist (DORA) is approved by the FDA for the treatment of insomnia disorders. Herein, we report the optimization efforts toward a DORA, where our starting point was (5-methoxy-4-methyl-2-[1,2,3]triazol-2-yl-phenyl)-{(S)-2-[5-(2-trifluoromethoxy-phenyl)-[1,2,4]oxadiazol-3-yl]-pyrrolidin-1-yl}methanone (6), a compound which emerged from our in-house research program. Compound 6 was shown to be a potent, brain-penetrating DORA with in vivo efficacy similar to suvorexant in rats. However, shortcomings from low metabolic stability, high plasma protein binding (PPB), low brain free fraction (fu brain), and low aqueous solubility, were identified and hence, compound 6 was not an ideal candidate for further development. Our optimization efforts addressing the above-mentioned shortcomings resulted in the identification of (4-chloro-2-[1,2,3]triazol-2-yl-phenyl)-{(S)-2-methyl-2-[5-(2-trifluoromethoxy-phenyl)-4H-[1,2,4]triazol-3-yl]-pyrrolidin-1-yl}l-methanone (42), a DORA with improved in vivo efficacy compared to 6.
PAT
WO 2015083071
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015083071
Reference Example 1
1) Synthesis of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-lodo-5-methoxy benzoic acid (15.0 g; 53.9 mmol) is dissolved in anhydrous DMF (45 ml) followed by the addition of 1 H-1 ,2,3-triazole (7.452 g; 108 mmol) and cesium carbonate (35.155 g; 108 mmol). By the addition of cesium carbonate the temperature of the reaction mixture increases to 40°C and gas evolved from the reaction mixture. Copper(l)iodide (514 mg; 2.7 mmol) is added. This triggers a strongly exothermic reaction and the temperature of the reaction mixture reaches 70°C within a few seconds. Stirring is continued for 30 minutes. Then the DMF is evaporated under reduced pressure followed by the addition of water (170 ml) and EtOAc (90 ml). The mixture is vigorously stirred and by the addition of citric acid monohydrate the pH is adjusted to 3-4. The precipitate is filtered off and washed with water and EtOAc and discarded. The filtrate is poured into a separation funnel and the phases are separated. The water phase is extracted again with EtOAc. The combined organic layers are dried over MgS04, filtered and the solvent is evaporated to give 7.1 g of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid as a white powder of 94% purity (6 % impurity is the regioisomerically N1-linked triazolo-derivative); tR [min] = 0.60; [M+H]+ = 220.21
2) Synthesis of (S)-1 -(tert-butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic acid
2-Methyl-L-proline hydrochloride (99.7 g; 602 mmol) is dissolved in a 1/1-mixture of MeCN and water (800 ml) and triethylamine (254 ml; 1810 mmol) is added. The temperature of the reaction mixture slightly rises. The reaction mixture is cooled to 10°C to 15°C followed by careful addition of a solution of Boc20 (145 g; 662 mmol) in MeCN (200 ml) over 10 minutes.
Stirring at RT is continued for 2 hours. The MeCN is evaporated under reduced pressure and aq. NaOH solution (2M; 250 ml) is added to the residual aq. part of the reaction mixture. The water layer is washed with Et20 (2x 300 ml) then cooled to 0°C followed by slow and careful addition of aq. HCI (25%) to adjust the pH to 2. During this procedure a suspension forms.
The precipitate is filtered off and dried at HV to give 1 10.9 g of the title compound as a beige powder; tR [min] = 0.68; [M+H]+ = 230.14
3) Synthesis of (S)-tert-butyl 2-((2-amino-4-chloro-3-methylphenyl)carbamoyl)-2-
(S)-1-(tert-butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic acid (60 g; 262 mmol) and HATU (100 g; 264 mmol) is suspended in DCM (600 ml) followed by the addition of DIPEA (84.6 g; 654 mmol) and 6-chloro-2,3-diaminotoluene (41 g; 262 mmol). The reaction mixture is stirred at rt for 14 hours then concentrated under reduced pressure and to the residue is added water followed by the extraction of the product with EtOAc (3x). The combined organic layers are washed with brine, dried over MgS04, filtered and the solvent is evaporated under
reduced pressure to give 185 g of the title compound as a dark brownish oil, which is used in the next step without further purification; tR [min] = 0.89; [M+H]+ = 368.01
4) Synthesis of (S)-tert-butyl 2-(5-chloro-4-methyl-1 H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1 -carboxylate
(S)-tert-butyl 2-((2-amino-4-chloro-3-methylphenyl)carbamoyl)-2-methylpyrrolidine-1-carboxylate (185 g; 427 mmol) are dissolved in AcOH (100%; 611 ml), heated to 100°C and stirring continued for 90 minutes. The AcOH is evaporated under reduced pressure and the residue is dissolved in DCM followed by careful addition of saturated sodium bicarbonate solution. The phases are separated, the aq. phase is extracted once more with DCM, the combined aq. phases are dried over MgS04, filtered and the solvent is evaporated under reduced pressure to give 142.92 g of the title compound as a dark brown oil which is used in the next step without further purification; tR [min] = 0.69; [M+H]+ = 350.04
5) Synthesis of (S)-5-chloro-4-methyl-2-(2-methylpyrrolidin-2-yl)-1 H-benzo[d]imidazole hydrochloride
(S)-tert-butyl 2-(5-chloro-4-methyl-1 H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1-carboxylate (355.53 g; 1.02 mol) are dissolved in dioxane (750 ml) followed by careful addition of HCI solution in dioxane (4M; 750 ml; 3.05 mol). The reaction mixture is stirred for 3 hours followed by the addition of Et20 (800 ml) which triggered precipitation of the product. The solid is filtered off and dried at high vacuum to give 298.84 g of the title compound as a redish powder; tR [min] = 0.59; [M+H]+ = 250.23
6) Synthesis of [(S)-2-(5-chloro-4-methyl-1 H-benzoimidazol-2-yl)-2-methyl-pyrrolidin-1- -(5-methoxy-2-[1,2,3]triazol-2-yl-phenyl)-methanone
(S)-5-chloro-4-methyl-2-(2-methylpyrrolidin-2-yl)-1 H-benzo[d]imidazole hydrochloride (62.8 g; 121 mmol) is dissolved in DCM (750 ml) followed by the addition of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid (62.8 g; 121 mmol) and DIPEA (103 ml; 603 mmol). Stirring is continued for 10 minutes followed by the addition of HATU (47 g; 124 mmol). The reaction mixture is stirred for 16 hours at RT. The solvents are evaporated under reduced pressure and the residue is dissolved in EtOAc (1000 ml) and washed with water (3x 750 ml). The organic phase is dried over MgS04, filtered and the solvent is evaporated under reduced pressure. The residue is purified by CC with EtOAc / hexane = 2 / 1to give 36.68 g of the title compound as an amorphous white powder. tR [min] = 0.73; [M+H]+ = 450.96
Table 1 : Characterisation data for COMPOUND as free base in amorphous form
II. Preparation of crystalline forms of COMPOUND
Example 1 :
Preparation of seeding material of COMPOUND hydrochloride in crystalline Form 1
10 mg COMPOUND is mixed with 0.2 mL 0.1 M aq. HCI and 0.8 mL EtOH. The solvent is fully evaporated and 0.05 mL isopropanol is added. Alternatively 0.05 mL methyl-isobutylketone can be added. The sample is stored closed at room temperature for 4 days and crystalline material of COMPOUND hydrochloride in crystalline Form 1 is obtained. This material can be used as seeding material for further crystallization of COMPOUND hydrochloride in crystalline Form 1.
Example 2: Preparation and characterization of COMPOUND hydrochloride in crystalline form 1
5g COMPOUND is mixed with 0.9 mL 1 M aq. HCI and 20 mL EtOH. The solvent is evaporated and 25 mL isopropanol is added. Seeds of COMPOUND hydrochloride are added and the sample is allowed to stand at room temperature. After about 2 days the suspension is filtered and the solid residue is dried at reduced pressure (2 mbar for 1 hour) and allowed to equilibrate open for 2 hours at 24°C/46% relative humidity. The obtained solid is COMPOUND hydrochloride in crystalline Form 1
Table 2: Characterisation data for COMPOUND hydrochloride in crystalline form 1
PAT
WO 2018202689
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018202689
Examples
Reference Example 1
Synthesis of 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid
4,5-dibromo-2-(4-methoxy-2-nitrophenyl)-2H-1,2,3-triazole
4- Fluoro-3-nitroanisole (3.44 g, 1 eq.), 4,5-dibromo-2/-/-1 ,2,3-triazole (4.56 g, 1 eq.)1, K2C03 (2.78 g, 1 eq.) and DMF (30 mL) are heated to 1 10 °C for 32 h. The reaction mixture is cooled to 22 °C and treated with water (70 mL). The resulting suspension is filtered, washed with water (15 mL). The product is slurried in isopropanol (40 mL), filtered and dried under reduced pressure to yield a white solid. Yield: 6.42 g, 84%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, CDCI3) δ: 7.71 (d, J = 8.9 Hz, 1 H), 7.47 (d, J = 2.8 Hz, 1 H), 7.25 (dd, Ji = 2.8 Hz, J2 = 8.9 Hz, 1 H), 3.97 (s, 3 H).
1 X. Wang, L. Zhang, D. Krishnamurthy, C. H. Senanayake, P. Wipf Organic Letters 2010 12 (20), 4632-4635.
5- methoxy-2-(2H-1 ,2,3-triazol-2-yl)aniline
4, 5-Dibromo-2-(4-methoxy-2-nitrophenyl)-2/-/-1 ,2,3-triazole (2 g, 1 eq.), sodium acetate (1.3 g, 3 eq.), and 10% Pd/C 50% water wet (0.3 g) is suspended in EtOAc (10 mL). The mixture is heated to 50 °C and set under hydrogen until conversion is complete. The reaction mixture is filtered over Celite. The filtrate is washed with 1 N NaOH (10 mL) and water (15 mL). The organic layer is concentrated under reduced pressure to yield an oil. Yield: 0.95 g, 94%. Purity: 96% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.05 (s, 2 H), 7.53 (d, J = 8.9 Hz, 1 H), 6.49 (d, J = 2.7 Hz, 1 H), 6.30 (dd, Ji = 2.7 Hz, J2 = 8.9 Hz, 1 H), 5.94 (s, 2 H), 3.74 (s, 3 H).
5-methoxy-2-(2H-1,2,3-triazol-2-yl)aniline monosulfate
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)aniline (455 g, 1 eq ) is dissolved in isopropanol (3 L). To the solution is added cone. H2SO4 (235 g, 1 eq.) below 40 °C. The suspension is cooled to
20 °C and filtered. The cake is washed with isopropanol (700 mL) and TBME (1.5 L). The product is dried to obtain a white solid. Yield: 627 g, 91 %. Purity: 100% a/a (LC-MS method 2).
2-(2-iodo-4-methoxyphenyl)-2H-1,2,3-triazole
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)aniline monosulfate (200 g, 1 eq.) is dissolved in 2 M aq. H2SO4 soln. (1.4 L) and cooled to -5 °C. To the solution is added a solution of sodium nitrite (62 g, 1.3 eq.) in water (600 mL) at -5 to 0 °C. The mixture is stirred at 0 °C for 30 min and then added to a preheated mixture of Kl (161 g, 1.4 eq.) in water (700 mL) at 65 °C. The resulting solution is stirred at 60 °C for 20 min, cooled to 20 °C and treated with a soln. of sulfamic acid (27 g, 0.4 eq.) in water (120 mL). The mixture is extracted with isopropyl acetate (2 L). The organic layer is washed with a mixture of 2 N NaOH (500 mL) and 40% NaHS03 soln. (100 mL), and a mixture of 1 N HCI (50 mL) and water (500 mL). The organic layer is concentrated to dryness. The residue is dissolved in isopropanol (700 mL) and cooled to 0 °C. The resulting suspension is filtered. The solid is dried under reduced pressure. Yield: 164 g, 79%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.08 (s, 2 H), 7.57 (d, J = 2.8 Hz, 1 H), 7.43 (d, J = 8.8 Hz, 1 H), 7.13 (dd, Ji = 2.8 Hz, J2 = 8.8 Hz, 1 H), 3.85 (s, 3 H).
5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-(2-lodo-4-methoxyphenyl)-2/-/-1 ,2,3-triazole (200 g, 1 eq.) is dissolved in THF (2 L) and cooled to 0 °C. 2 M iPrMgCI soln. in THF (350 mL, 1.05 eq.) is added at 0 °C. The mixture is cooled to -20 °C and C02 (gas) is bubbled into the solution over 30 min until the exothermicity is ceased. To the mixture is added 2 N HCI (600 mL) at 8 °C and concentrated under reduced pressure to remove 2.4 L solvent. The residue is extracted with TBME (1.6 L). The organic layer is washed with 1 N HCI (200 mL) and extracted with 1 N NaOH (600 mL and 200 mL). The aq. layer is filtered over charcoal (15 g), diluted with water (200 mL) and treated with 32% HCI (160 mL). The resulting suspension is filtered and washed with water (200 mL). Yield: 127 g, 87%. Purity: 100% a/a (LC-MS method 2); MP: 130 °C (DSC goldpan). The obtained product may be re-crystallized from toluene (MP: 130.9 °C) or water (MP: 130 °C).
Table Ref 1 : Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid in crystalline form 2 (recrystallization from toluene)
Technique Data Summary Remarks
XRPD Crystalline see Fig. 8
Reference Example 2
Synthesis of 4-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
4,5-Dibromo-2-(5-methyl-2-nitrophenyl)-2H-1 ,2,3-triazole
3- Fluoro-4-nitrotoluene (1367 g, 1 eq.), 4,5-dibromo-2/-/-1 ,2,3-triazole (1999 g, 1 eq.), K2C03 (1340 g, 1.1 eq.) and DMF (1 1 L) is heated to 75 °C for 15 h. The reaction mixture is cooled to 22 °C and treated with water (18 L). The resulting suspension is filtered, washed with water (4 L). The product is washed with isopropanol (5 L), and dried under reduced pressure to yield a white solid. Yield: 281 1 g, 88%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.10 (d, J = 8.3 Hz, 1 H), 7.86 (d, J = 1.0 Hz, 1 H), 7.66 (dd, J1 = 0.9 Hz, J2 = 8.3 Hz, 1 H), 2.51 (s, 3 H).
4- Methyl-2-(2H-1 ,2,3-triazol-2-yl)aniline
4, 5-Dibromo-2-(5-methyl-2-nitrophenyl)-2/-/-1 ,2,3-triazole (205 g, 1 eq.), sodium acetate (149 g, 3.2 eq.), and 5% Pd/C 50% water wet (37.8 g) is suspended in EtOAc (0.8 L). The mixture is heated to 40-50 °C and set under hydrogen (2 bar) until conversion is complete. The reaction mixture is filtered over Celite. The filtrate is washed with water (300 mL), 2N NaOH (300 ml_+250 mL) and water (300 mL). The organic layer is concentrated under reduced pressure to yield a yellow oil. Yield: 132 g, 90%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.09 (s, 2 H), 7.48 (d, J = 1.3 Hz, 1 H), 6.98 (dd, J1 = 1.8 Hz, J2 = 8.3 Hz, 1 H), 6.85 (d, J = 8.2 Hz, 1 H), 5.79 (s, 2 H), 2.23 (s, 3 H).
4-Methyl-2-(2H-1,2,3-triazol-2-yl)aniline monosulfate
4-Methyl-2-(2/-/-1 ,2,3-triazol-2-yl) aniline (199 g, 1 eq ) is dissolved in isopropanol (1.7 L). To the solution is added cone. H2SO4 (118 g, 1.05 eq.) below 40 °C. The suspension is cooled to 20 °C and filtered. The cake is washed with isopropanol (500 mL). The product is dried to obtain a white solid. Yield: 278 g, 89%. Purity: 100% a/a (LC-MS method 2). 1H NMR (400 MHz, DMSO) <5: 8.21 (s, 2 H), 7.70 (s, 1 H), 7.23 (s, 2 H), 2.35 (s, 3 H).
2-(2-iodo-5-methylphenyl)-2H-1 ,2,3-triazole
4-Methyl-2-(2/-/-1 ,2,3-triazol-2-yl)aniline monosulfate (1553 g, 1 eq.) is dissolved in 1 M aq. H2S04 Soln. (1 1 L) and cooled to -5 °C. To the solution is added a solution of sodium nitrite (433 g, 1.1 eq.) in water (4 L) at -5 to 0 °C. The mixture is stirred at 0 °C for 30 min and then added to a preheated mixture of potassium iodide (1325 g, 1.4 eq.) in water (4 L) at 55-70 °C. The resulting solution is stirred at 60 °C for 20 min, cooled to 20 °C and treated with a soln. of sulfamic acid (220 g, 0.4 eq.) in water (900 mL). The mixture is extracted with isopropyl acetate (13 L). The organic layer is washed with a mixture of 2 N NaOH (3.5 L) and 40% NaHSOs soln. (330 g), and a mixture of 1 N HCI (280 mL) and water (3.5 L). The
organic layer is concentrated to dryness. Yield: 1580 g, 97%. Purity: 91 % a/a (LC-MS method 2). 1 H NMR (400 MHz, CDCI3) <5: 7.90 (s, 2 H), 7.87 (d, J = 8.1 Hz, 1 H), 7.34 (d, J = 1 .6 Hz, 1 H), 7.03-7.06 (m, 1 H), 2.40 (s, 3 H).
The crude product, together with a second batch (141 1 g) is purified by distillation on a short path distillation equipment at 120 °C jacket temperature, feeding tank (70 °C), cooling finger (20 °C) and at a pressure of 0.004 mbar. Yield: 2544 g (78%), Purity: 100 % a/a ()LC-MS method 2).
4-Methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-(2-lodo-5-methylphenyl)-2/-/-1 ,2,3-triazole (1250 g, 1 eq.) is dissolved in THF (13 L) and cooled to 0 °C. 2 M iPrMgCI soln. in THF (2.2 L, 1 eq.) is added at 0 °C. The mixture is cooled to -25 °C and CO2 (gas) is bubbled into the solution over 60 min until the exothermicity is ceased. To the mixture is added 2 N HCI (5 L) at 4 °C and concentrated under reduced pressure to remove 14.5 L solvent. The residue is extracted with TBME (10 L). The organic layer is extracted with 1 N NaOH (6 L and 3 L). The aq. layer is filtered over charcoal (15 g), diluted with water (200 mL) and treated with 32% HCI (1 .23 L). The resulting suspension is filtered and washed with water (5 L). Yield: 796 g, 89%. Purity: 100% a/a (LC-MS method 2); MP: 125 °C (DSC goldpan).
The following examples illustrate the invention.
Example 1 :
Example 1.1: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt (potassium 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoate)
2-Bromo-5-methoxybenzoic acid (21 .5 g, 0.093 mol, 1 eq.) copper (I) iodide (0.886 g, 0.05 eq.), and K2CO3 powder (32.2 g, 2.5 eq.) were suspended in dioxane (600 mL) and water (8.4 mL). To the mixture were added 1 H-1 ,2,3-triazole (10.8 mL, 2 eq.) and trans-/V,/V-dimethylcyclohexane-1 ,2-diamine (1 .32 g, 0.1 eq.). The mixture was heated at reflux for 3.5 h. IPC showed full conversion. The ratio of the desired N(2) to the regioisomeric Λ/(1 ) isomer was 84: 16. The mixture was cooled to 40 °C and filtered. The cake was washed with dioxane (100 mL). The solid was dried to obtain 50.6 g of a blue solid. The ratio of N{2) to Λ/(1 ) isomer of was 98.6: 1 .4.
Table 1 : Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt in crystalline form 1
Example 1.2: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid
The solid of Example 1.1 was dissolved in water (300 mL). TBME (200 mL) and 32% aq. HCI (35 mL) was added. The aq. layer was separated and discarded. The organic layer was washed with a mixture of 2N aq. HCI (100 mL) and 32% aq. HCI (20 mL). The organic layer was washed with 1 N aq. HCI (50 mL). The organic layer was extracted with 1 N aq. NaOH (200 mL). The aq. layer was heated to 45 °C and traces of TBME were removed under reduced pressure. To the aq. layer was added at 45 °C 32% aq. HCI (20 mL). At a pH of 6 optionally seed crystals were added. The resulting suspension was filtered at 40 °C. The cake was washed with water (30 mL). The product was dried at 60 °C and 5 mbar. Yield: 12.4 g, 61 %. Purity: 100% a/a, tR 0.63 min. Seed crystals may be obtained by careful crystallization according to the above procedure.
MP: 80 °C (DSC).
1H NMR (400 MHz, DMSO) & 3.87 (s, 3 H), 7.26 (m, 2 H), 7.64 (d, J = 8.7 Hz, 1 H), 8.02 (s, 2 H), 13.01-13.22 (br, 1 H).
Table 2: Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid in crystalline form 1
Example 1.3: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)benzoic acid, e.g. obtained according to the procedure of Reference Example 1 (5 g, 0.0228 mol) and KHCO3 (1.61 g, 0.7 eq) were suspended in dioxane (100 mL) and water (1 mL). The mixture was heated at reflux for 40 min. The mixture was cooled to 20 °C and filtered. Yield: 2.56 g, 44%. 1H NMR (400 MHz, D20) & 3.80 (s, 3 H), 7.04 (m, 2 H), 7.46 (d, J = 8.7 Hz, 1 H), 7.82 (s, 2 H). MP: 279.5°C (DSC shows additionally a broad endothermic event at about 153 °C to 203 °C which may be attributed to endothermic desolvations; melting is immediately followed by exothermic degradation).
Table 3: Characterisation data for 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt in crystalline form 2
Example 1.4: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
In an alternative procedure, 2-Bromo-5-methoxybenzoic acid (20 g, 0.086 mol, 1 eq.) copper (I) iodide (0.824 g, 0.05 eq.), and K2C03 powder (26.9 g, 2.25 eq.) were suspended in dioxane (494 mL). To the mixture was added 1 H-1 ,2,3-triazole (12 g, 2 eq.). The mixture was heated at reflux for 1 h. To the mixture was added water (12.5 g, 8 eq.). The mixture was heated at reflux for 2 h. Solvent (100 mL) was removed by distillation. The residue was cooled to 45 °C in 8 min, filtered and washed with dioxane (50 mL).
XRPD corresponds to crystalline form 1 (see Fig. 1 , Example 1.1 ).
Example 1.5: Crystalline 5-methoxy-2-(2H-1,2,3-triazol-2-yl)benzoic acid
The solid of Example 1.4 was dissolved in water (200 mL). The mixture was heated to 50 °C and 20% aq. H2SO4 (40 mL) was added to adjust the pH to 5. The mixture was filtered over Celite. The filtrate was treated at 45 °C with 20% aq. H2S04 (40 mL). At pH 3 seeds (obtained for example using the procedure of reference example 1 ) were added. The suspension was stirred at 45 °C and filtered. The product was washed with water (20 mL) and dried at 60 °C and 10 mbar to yield a white solid. Yield: 10.8 g, 57%. Purity: 100% a/a, tR 0.63 min.
Characterisation of 5-methoxy-2-(2/-/-1 ,2,3-triazol-2-yl)benzoic acid obtained according to Example 1.5:
XRPD corresponds to crystalline form 1 (see Fig. 2, Example 1.2).
Example 2:
Example 2.1: Crystalline 4-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt (potassium 4-methyl-2-(2H-1,2,3-triazol-2-yl)benzoate)
2-Bromo-4-methylbenzoic acid (20 g, 0.093 mol, 1 eq.) copper (I) iodide (0.886 g, 0.05 eq.), and K2CO3 powder (32.2 g, 2.5 eq.) were suspended in dioxane (300 mL) and water (10.1 mL). To the mixture was added 1 A7-1 ,2,3-triazole (10.8 mL, 2 eq.) and trans-Λ/,ΛΑ-
dimethylcyclohexane-1 ,2-diamine (1 .32 g, 0.1 eq.). The mixture was heated at reflux for 4 h. IPC showed a conversion of 98.5%. The ratio of the desired N(2) to the regioisomeric Λ/(1 ) isomer was 75:25. The mixture was concentrated at normal pressure and external temperature of 130 °C. Solvent (100 mL) was removed. To the residue was added dioxane (100 mL) and the mixture was cooled to 45 °C and filtered. The cake was washed with dioxane (80 mL). The solid was dried to obtain 48.8 g of a blue solid. The ratio of N(2) to Λ/(1 ) isomer was 98.7: 1 .3.
Table 4: Characterisation data for 4-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid potassium salt in crystalline form 1
Example 2.2: Crystalline 4-methyl-2-(2H-1,2,3-triazol-2-yl) benzoic acid
The solid of Example 2.1 was dissolved in water (300 mL) and filtered. To the filtrate were added TBME (200 mL) and 32% aq. HCI (30 mL). The aq. layer was separated and discarded. The organic layer was washed with a mixture of 2N aq. HCI (100 mL) and 32% aq. HCI (10 mL). The organic layer was washed with 1 N aq. HCI (50 mL). The organic layer was extracted with 1 N aq. NaOH (200 mL). The aq. layer was heated to 45 °C and traces of TBME were removed under reduced pressure. To the aq. layer was added at 45 °C 32% aq. HCI (20 mL). At a pH of 6 seed crystals (obtained for example using the procedure of reference example 2) were added. The resulting suspension was filtered at 40 °C. The cake was washed with water (30 mL). The product was dried at 60 °C and 5 mbar. Yield: 1 1 .7 g, 62%. Purity: 100% a/a. tR 0.66 min.
MP: 125 °C (DSC).
1H NMR (400 MHz, DMSO) & 2.44 (s, 3 H), 7.41 (d, J = 7.9 Hz, 1 H), 7.56 (s, 1 H), 7.68 (d, J = 7.9 Hz, 1 H), 8.06 (s, 2 H), 12.53-13.26 (br, 1 H)
Table 5: Characterisation data for 4-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid in crystalline form 1
Technique Data Summary Remarks
XRPD Crystalline see Fig. 5
Example 2.3: Crystalline 4-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
4-Methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid (5 g, 0.0246 mol) and KHC03 (1 .74 g , 0.7 eq) were suspended in dioxane ( 100 mL) and water (1 mL). The mixture was heated at reflux for 40 min. The mixture was cooled to 20 °C and filtered. Yield: 2.47 g, 42% . MP: 277 °C (DSC Alupan) 1 H NMR (400 MHz, D20) & 2.32 (s, 3 H), 7.28 (d, J = 7.9 Hz, 1 H), 7.39 (m, 2 H), 7.84 (s, 2 H).
MP: 276.8 °C (DSC shows additionally a broad endothermic event at about 140 °C to 208 °C which may be attributed to endothermic desolvations; melting is immediately followed by exothermic degradation).
XRPD corresponds to crystalline form 1 (see Fig. 4, Example 2.1 ).
Reference Example 3:
Reference Example 3.1: Crystalline 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid sodium salt (sodium 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoate)
2-Bromo-5-methylbenzoic acid (20 g, 0.093 mol, 1 eq. ) copper (I) iodide (0.886 g, 0.05 eq.), Na2CC>3 powder (24.6 g, 2.5 eq.) were suspended in dioxane (300 mL) and water (10.1 mL). To the mixture was added 1 /-/-1 ,2,3-triazole ( 10.8 mL, 2 eq.) and 8-hydroxy quinoline ( 1 .35 g, 0.1 eq.). The mixture was heated at reflux for 5 h. IPC showed a conversion of >99%. The ratio of the desired N(2) to the regioisomeric Λ/(1 ) isomer was 78:22. The mixture was concentrated at normal pressure and external temperature of 135 °C. Solvent (100 mL) was removed. To the residue was added dioxane (100 mL) and the mixture was cooled to 45 °C and filtered. The cake was washed with dioxane (80 mL). The solid was dried to obtain 36.2 g of a yellow solid. The ratio of N(2) to Λ/( 1 ) isomer of was 99: 1 .
Table 6: Characterisation data for 5-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid sodium salt in crystalline form 1
Reference Example 3.2: Crystalline 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid
The solid obtaind in Reference Example 3.1 was dissolved in water (300 mL) and filtered. To the filtrate was added TBME (200 mL) and 32% aq. HCI (30 mL) was added. The aq. layer was separated and discarded. The organic layer was washed with 1 N aq. HCI ( 100 mL). The organic layer was washed with 1 N aq. HCI (50 mL). The organic layer was extracted with 1 N aq. NaOH (200 mL). The aq. layer was heated to 45 °C and traces of TBME were removed
under reduced pressure. To the aq. layer was added at 45 °C 32% aq. HCI (20 mL). At a pH of 6 seed crystals (obtained for example using the procedure of Reference example 2) were added. The resulting suspension was filtered at 40 °C. The cake was washed with water (30 mL). The product was dried at 60 °C and 5 mbar. Yield: 12.1 g, 64%. Purity: 100% a/a. tR 0.67 min.
MP: 173 °C (DSC)
1 H NMR (400 MHz, DMSO) & 2.42 (s, 3 H), 7.50-7.52 (m, 1 H), 7.58 (s, 1 H), 7.63 (m, 1 H), 8.05 (s, 2 H), 13.01 (s, 1 H).
Table 7: Characterisation data for 5-methyl-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid in crystalline form 1
Reference Example 3.3: Crystalline 5-methyl-2-(2H-1,2,3-triazol-2-yl) benzoic acid sodium salt
5-Methyl-2-(2/-/-1 ,2,3-triazol-2-yl)benzoic acid (5 g, 0.0246 mol) and Na2C03 (1 .05 g, 0.4 eq) were suspended in dioxane ( 100 mL) and water (1 mL). The mixture was heated at reflux for 40 min. The mixture was cooled to 20 °C and filtered. Yield: 2.79 g, 50%. MP: 341 °C (DSC Alupan) 1 H NMR (400 MHz, D20) & 2.32 (s, 3 H), 7.30 (m, 2 H), 7.43 (m, 1 H), 7.83 (s, 2 H).
XRPD corresponds to crystalline form 1 (see Fig. 6, Reference Example 3.1 ).
Reference Example 3.4: 5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoic acid potassium salt
2-Bromo-5-methylbenzoic acid (20 g, 0.093 mol, 1 eq. ) copper (I) iodide (0.886 g, 0.05 eq.), and K2CO3 powder (32.1 g, 2.5 eq.) were suspended in dioxane (600 mL). To the mixture was added 1 /-/-1 ,2,3-triazole ( 10.8 mL, 2 eq.) and 8-hydroxy quinoline ( 1 .35 g, 0.1 eq.). The mixture was heated at reflux for 4 h. IPC showed a conversion of >94%. The ratio of the desired N(2) to the regioisomeric Λ/( 1 ) isomer was 78:22. The mixture was cooled to 35 °C and filtered. The cake was washed with dioxane (100 mL). The products were dissolved in water and a LC-MS was recorded. The ratio of N(2) to Λ/(1 ) isomer of was 83: 17.
Reference Example 4.1: Methyl (S)-1-(5-methoxy-2-(2H-1,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylate
5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl) benzoic acid (100 g, 0.46 mol) was suspended in DCM (650 mL) and DMF (10 mL) at 20 °C. To this suspension was added oxalyl chloride (51 mL, 0.59 mol) over a period of 30 min. LC-MS showed 60% conversion to acid chloride intermediate. Oxalyl chloride (17.6 mL, 0.45 eq.) was added dropwise. LC-MS showed full conversion to acid chloride intermediate.
Methyl (S)-2-methylpyrrolidine-2-carboxylate hydrochloride (84 g, 0.47 mol) was suspended in DCM (800 mL) in a second flask. The suspension was cooled to 10 °C. Triethylamine (200 mL, 1.41 mol) was added over 15 min. The acid chloride solution was added to the reaction mixture at 10-20 °C over at least 15 min. The reaction mixture was washed with 1 M HCI (500 mL), 1 N NaOH (500 mL) and water (500 mL). The organic layer was concentrated to dryness to give a light-yellow solid as product. Yield: 157 g, 100%, 99% a/a (LC-MS), M+1 =345. 1H NMR (400 MHz, DMSO) δ: 8.06 (s, 2 H), 7.79 (d, J = 8.9 Hz, 1 H), 7.21 (dd, J1 = 2.9 Hz, J2 = 8.9 Hz, 1 H), 6.85 (d, J = 1.9 Hz, 1 H), 3.89 (s, 3 H), 3.66 (s, 3 H), 3.29 (m, 1 H), 3.03 (m, 1 H), 2.08 (m, 1 H), 1.82 (m, 3 H), 1.50 (s, 3 H).
Reference Example 4.2: (S)-1-(5-methoxy-2-(2H-1,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylic acid
Methyl (S)-1-(5-methoxy-2-(2/-/-1 ,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylate (157 g, 0.46 mol) was dissolved in MeOH (750 mL) at 20 °C. To this solution was added 16% NaOH (300 mL). The resulting solution was heated up to 80 °C and stirred for 60 min. Solvent was distilled off under reduced pressure (850 mL). The residue was taken up in DCM (1500 mL) and water (450 ml) at 20 °C. 32% HCI (200 mL) was added. Layers were separated and the organic layer was washed with water (450 mL). The organic layer was concentrated to the minimum stirring volume under reduced pressure. Toluene (750 mL) was added and solvent was further distilled under vacuum (150 mL distilled). The mixture was cooled to 20 °C and stirred for 15 min. The suspension was filtered at 20 °C. The cake was rinsed with toluene (150 mL) and then dried under reduced pressure at 50 °C to give a white solid as product. Yield: 128 g, 85%, 94% a/a (LC-MS), M+1 =331. Melting point: 178 °C (DSC). 1H NMR (400 MHz, DMSO) δ: 12.3 (s, 1 H), 8.04 (s, 2 H), 7.79 (d, 1 H), 7.20 (dd, J1 = 2.8 Hz, J2 = 8.9 Hz, 1 H), 6.84 (m, 1 H), 3.88 (s, 3 H), 3.29 (m, 1 H), 2.99 (m, 1 H), 2.1 1 (m, 1 H), 1.81 (m, 3 H), 1.47 (s, 3 H).
Reference Example 4.3: (S)-N-(2-amino-4-chloro-3-methylphenyl)-1-(5-methoxy-2-(2H-1,2,3-triazol-2-yl) benzoyl)-2 methylpyrrolidine-2-carboxamide
(S)-1-(5-Methoxy-2-(2/-/-1 ,2,3-triazol-2-yl) benzoyl)-2-methylpyrrolidine-2-carboxylic acid (128 g, 0.39 mol) was suspended in DCM (850 mL) and DMF (6 mL) at 20 °C. To this suspension was added oxalyl chloride (39 mL, 0.45 mol) over a period of 30 min. 4-Chloro-3-methylbenzene-1 ,2-diamine hydrochloride (75 g, 0.39 mol) was suspended in DCM (1300 mL) in a second flask. The suspension was cooled down to 10 °C. Triethylamine (180 mL, 1.27 mol) was added. The acid chloride solution was added to the reaction mixture at 10-20 °C over at least 15 min. Water (650 mL) was added to the reaction mixture. Layers were separated and the organic phase was concentrated under reduced pressure (1900 mL distilled out). TBME (1000 mL) was added and solvent was further distilled under vacuum (400 mL distilled). The mixture was finally cooled down to 20 °C and stirred for 15 min. The resulting suspension was filtered off at 20 °C. The cake was rinsed with TBME (250 mL) and then dried under reduced pressure at 50 °C to give a white solid as product. Yield: 145 g, 80%, 97% a/a (LC-MS), M+1=469. Melting point: 185 °C (DSC). 1H NMR (400 MHz, DMSO) δ: 9.10-9.14 (m, 1 H), 7.88-8.12 (m, 2 H), 7.81-7.82 (m, 1 H), 7.38-7.44 (m, 1 H), 7.21 (dd, J1 = 2.7 Hz, J2 = 8.9 Hz, 1 H), 6.84 (d, J = 7.8 Hz, 1 H), 6.64 (d, J = 8.3 Hz, 1 H), 5.01 (brs, 2 H), 3.88 (s, 3 H), 3.61-3.73 (m, 1 H), 3.14-3.26 (m, 1 H), 2.25-2.30 (m, 1 H), 2.13 (s, 3 H), 1.97 (m, 3 H), 1.47-1.61 (m, 3 H).
Reference Example 4.4: (S)-(2-(5-chloro-4-methyl-1H-benzo[d]imidazol-2-yl)-2-methylpyrrolidin-1-yl) (5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone hydrochloride
(S)-/V-(2-amino-4-chloro-3-methylphenyl)-1-(5-methoxy-2-(2H-1 ,2,3-triazol-2-yl) benzoyl)-2 methylpyrrolidine-2-carboxamide (145 g, 0.31 mol) was dissolved in isopropanol (870 mL) at 20 °C. To this solution was added carefully 5-6 N HCI in isopropanol (260 mL) over 10 min. the reaction mixture was then heated up to 90 °C and stirred for 4 hours. Water (28 mL) was added and the reaction mixture was stirred for an additional one hour. The reaction mixture was cooled to 20 °C. A light brown suspension was obtained which was filtered. The cake was rinsed with isopropanol (220 mL). The solid was finally dried under reduced pressure at 60 °C to give a beige solid. Yield: 133 g, 88%, 100% a/a (LC-MS), M+1 =451. Melting point: 277 °C (DSC). Ή NMR (400 MHz, DMSO) δ: 8.06 (s, 2 H), 7.76 (d, J = 8.9 Hz, 1 H), 7.63 (d, J = 8.8 Hz, 2 H), 7.55 (m, 1 H), 7.16 (dd, J1 = 2.7 Hz, J2 = 8.9 Hz, 1 H), 3.98 (m, 1 H), 3.90 (s, 3 H), 3.33 (m, 2H), 3.32 (m, 1 H), 2.74 (s, 3 H), 2.55 (m, 1 H), 2.23 (m, 1 H), 2.10 (m, 2 H), 1.95 (s, 3 H).
/////////////////////////////////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Trade names | Quviviq |
| Other names | Nemorexant; ACT-541468 |
| License data | US DailyMed: Daridorexant |
| Routes of administration | By mouth |
| Drug class | Orexin antagonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Pharmacokinetic data | |
| Elimination half-life | 6–10 hours[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1505484-82-1 |
| PubChem CID | 91801202 |
| DrugBank | DB15031 |
| ChemSpider | 64854514 |
| UNII | LMQ24G57E9 |
| KEGG | D11886 |
| PDB ligand | NS2 (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C23H23ClN6O2 |
| Molar mass | 450.93 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
REF
References
- ^ Jump up to:a b c https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/214985s000lbl.pdf
- ^ Jump up to:a b Muehlan C, Vaillant C, Zenklusen I, Kraehenbuehl S, Dingemanse J (November 2020). “Clinical pharmacology, efficacy, and safety of orexin receptor antagonists for the treatment of insomnia disorders”. Expert Opin Drug Metab Toxicol. 16 (11): 1063–1078. doi:10.1080/17425255.2020.1817380. PMID 32901578.
- ^ Jump up to:a b c “Daridorexant – Idorsia Pharmaceuticals – AdisInsight”.
- ^ Jump up to:a b Equihua-Benítez AC, Guzmán-Vásquez K, Drucker-Colín R (July 2017). “Understanding sleep-wake mechanisms and drug discovery”. Expert Opin Drug Discov. 12 (7): 643–657. doi:10.1080/17460441.2017.1329818. PMID 28511597.
- ^ “Daridorexant: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 11 January 2022.
- ^ “Idorsia receives US FDA approval of Quviviq (daridorexant)” (Press release). Idorsia Pharmaceuticals. 10 January 2022. Retrieved 11 January 2022 – via GlobeNewswire.
External links
- “Daridorexant”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03545191 for “Study to Assess the Efficacy and Safety of ACT-541468 in Adult and Elderly Subjects With Insomnia Disorder” at ClinicalTrials.gov
- Clinical trial number NCT03575104 for “Study to Assess the Efficacy and Safety of ACT-541468 in Adult and Elderly Subjects Suffering From Difficulties to Sleep” at ClinicalTrials.gov
- Clinical trial number NCT03679884 for “Study to Assess the Long Term Safety and Tolerability of ACT-541468 in Adult and Elderly Subjects Suffering From Difficulties to Sleep” at ClinicalTrials.gov
///////////////Daridorexant, Quviviq, FDA 2022, APPROVALS 2022, INSOMNIA, ACT541468A, ACT 541468A. ACT-541468A, ACT541468, FDA 2022, APPROVALS 2022
O=C(N1[C@](C)(C2=NC3=CC=C(Cl)C(C)=C3N2)CCC1)C4=CC(OC)=CC=C4N5N=CC=N5.[H]Cl

NEW DRUG APPROVALS
ONE TIME
$10.00
TAUROLIDINE

TAUROLIDINE
- Molecular FormulaC7H16N4O4S2
- Average mass284.356 Da
19388-87-5[RN]
243-016-5[EINECS]
2H-1,2,4-Thiadiazine, 4,4′-methylenebis[tetrahydro-, 1,1,1′,1′-tetraoxide
4,4′-methanediylbis(1,2,4-thiadiazinane) 1,1,1′,1′-tetraoxide
UNII-8OBZ1M4V3V
тауролидин
توروليدين
牛磺利定
NMR https://www.apexbt.com/downloader/document/C4559/NMR-2.pdf
MS https://www.apexbt.com/downloader/document/C4559/MS-2.pdf
Taurolidine
CAS Registry Number: 19388-87-5
CAS Name: 4,4¢-Methylenebis(tetrahydro-1,2,4-thiadiazine) 1,1,1¢,1¢-tetraoxide
Additional Names: 4,4¢-methylenebis(perhydro-1,2,4-thiadiazine 1,1-dioxide); bis(1,1-dioxoperhydro-1,2,4-thiadiazin-4-yl)methane
Trademarks: Drainasept (Geistlich); Taurolin (HMR); Tauroflex (Geistlich)
Molecular Formula: C7H16N4O4S2, Molecular Weight: 284.36
Percent Composition: C 29.57%, H 5.67%, N 19.70%, O 22.51%, S 22.55%
Literature References: Broad spectrum, synthetic formaldehyde carrier formed by the condensation of two molecules of taurine and three molecules of formaldehyde. Prepn: FR1458701; R. W. Pfirrmann, US3423408 (1966, 1969 both to Ed. Geistlich Söhne). Antibacterial activity in mice: M. K. Browne et al.,J. Appl. Bacteriol.41, 363 (1976). Anti-endotoxin activity in lab animals: R. W. Pfirrmann, G. B. Leslie, ibid.46, 97 (1979). Mechanism of action: E. Myers et al.,ibid.48, 89 (1980). HPLC determn of metabolites in plasma: A. D. Woolfson et al.,Int. J. Pharm.49, 135 (1989). Pharmacokinetics: C. Steinbach-Lebbin et al.,Arzneim.-Forsch.32, 1542 (1982). Metabolism in humans: B. I. Knight et al.,Br. J. Clin. Pharmacol.12, 695 (1981). Clinical trials in peritonitis: M. K. Browne et al.,Surg. Gynecol. Obstet.146, 721 (1978); G. Wesch et al.,Fortschr. Med.101, 545 (1983); in wound sepsis: A. K. Halsall et al.,Pharmatherapeutica2, 673 (1981); in pleural infection: A. A. Conlan et al.,S. Afr. Med. J.64, 653 (1983).
Properties: White crystals, mp 154-158°. Sol in water.
Melting point: mp 154-158°
Therap-Cat: Antibacterial.
Keywords: Antibacterial (Synthetic).
Taurolidine is an antimicrobial that is used to try to prevent infections in catheters.[1] Side effects and the induction of bacterial resistance is uncommon.[1] It is also being studied as a treatment for cancer.[2]
It is derived from the endogenous amino acid taurine. Taurolidine’s putative mechanism of action is based on a chemical reaction. During the metabolism of taurolidine to taurinamide and ultimately taurine and water, methylol groups are liberated that chemically react with the mureins in the bacterial cell wall and with the amino and hydroxyl groups of endotoxins and exotoxins. This results in denaturing of the complex polysaccharide and lipopolysaccharide components of the bacterial cell wall and of the endotoxin and in the inactivation of susceptible exotoxins.[3]
PATENT
https://patents.google.com/patent/WO2012070066A1/en
Taurolidine is an antibacterial drug and also has antiendotoxic substance, which is used as an antiseptic solution in surgery for washing out the abdominal cavity and it also prevents septic shock. It is commercially sold as Taurolidine (Formula I). The present invention relates to a process for the preparation of Taurolidine which provides significant advantages over the existing processes.

Formula I
The current process for the preparation of Taurolidine is depicted in Scheme 1

Formula II

Formula IVFormula IThe present inventors thus propose an industrially viable procedure for isolation of Taurolidine in substantially pure form.Taurolidine is dissolved in a suitable solvent to obtain a clear solution. The product starts to precipitate and an anti solvent is added optionally to maximize the precipitation procedure. The solvents employed for the purification are non -aqueous aprotic solvents comprising DMSO, DMAc, DMF, Acetonitirle, DMSO being the most preferred solvent. The antisolvents employed are toluene, ethyl acetate, dichloromethane, ether; toluene being the most preferred.Taurolidine obtained by the instant procedure has purity greater than or equal to 99.5 %. The process of the invention is illustrated by the following examples to obtain Taurolidine. Example ICbz-Taurine sodium salt (Formula II)To 1000ml of water in the RBF charge 192gm of (3.0 eq) of sodium hydroxide under cooling followed by 200gm of Taurine and dissolve it until clear solution is obtained. Cool to 0°C to 5°C, and Charge 50% CBZ-C1 in toluene at 0°C to 5°C. After completion of addition, maintain at room temperature for 14h. Separate the toluene layer and wash the aqueous layer with 2x200ml of ethyl acetate. Add slowly 27gm of sodium hydroxide in 60ml of water to the aqueous layer and adjust pH to 12- 14. Cool to 0°C to 5°C and a white solid separates from the solution. Filter the solid and dry the solid at 60 -70 °C. Weight of the solid: 320 gExample 2Cbz-Taurinamide (Formula III)To a clean dry flask charge 1500ml of toluene and charge 320gm of Formula II and cool to 0°C to 5°C. Charge 308 gm of PC15 slowly at 0°C to 5°C for 2hrs. Maintain at 0°C to 5°C up to completion of reaction. Quench the RM into another flask containing 2 ltr of water at 0°C to 5°C. Separate the organic layer, wash and extract the aqueous layer with toluene. Dry the organic layer with sodium sulphate and cool to 0°C to 5°C. Purge ammonia gas into the reaction mass till the reaction is complete. Filter the solid and dissolve the solid in 21tr of water and extract the aqueous layer with 2x600ml of ethyl acetate. Dry the organic layer with sodium sulphate and concentrate it under reduced pressure to obtain a white solid. Weight of the solid: 150 gExample 3Taurinamide Succinate (Formula IV)Take a suspension of 100 g of Cbz-Taurinamide in 1000 ml methanol, and 10% Pd/C (1 .0 g) and subject to hydrogenation at 45-50 psi. Upon completion of the reaction filter the catalyst and add succinic acid (1 .0 eq) to the solvent and distill off the solvent under vacuum to provide the title compound in about 90% yield as a white solid.Example 4Taurolidine (Formula I)To a solution of 100 g Taurinamide succinate in water is added sat sodium bicarbonate solution and pH adjusted to 7-8. To the solution was added formaldehyde (50 ml) and allowed to stir for 4 h. The solid obtained was filtered and washed with water to give Taurolidine. The title compound was obtained in about 70% yield and about 98% purity.Example 5Purification of TaurolidineTaurolidine (100 g) was dissolved in DMSO (400 ml) and a clear solution is obtained and a precipitate is obtained immediately. The solid is filtered and washed with toluene and dried to give a white solid in 40 % yield. The product obtained was >99.5% pure.Example 6Purification of TaurolidineTaurolidine (100 g) was dissolved in DMSO (400 ml) and a clear solution is obtained and a precipitate is obtained immediately. To the solution, toluene (1000 ml) is added. The solid is filtered and washed with toluene and dried to give a white solid in 70 % yield. The product obtained was >99.5% pure by HPLC and passed elemental analysis within 0.4% of the theoretical values.Example 7Purification of TaurolidineTaurolidine (100 g) was dissolved in DMAc (800 ml) and to the solution, toluene (1000 ml) is added. The solid is filtered and washed with toluene and dried to give a white solid in 70% yield.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022007713&_cid=P12-KYJIDL-52672-1Taurolidine (English name Taurolidine, chemical name is 4,4′-methylenebis[tetrahydro-2H-1,2,4-thiadiazine] 1,1,1′,1′-tetraoxide , the molecular formula is C 7 H 16 N 4 O 4 S 2 ) is a derivative of the amino acid taurine, and its structure is as follows:
Taurolidine has anti-endotoxin, anti-bacterial and anti-adherent properties. In terms of bacteria, taurolidine can chemically react with cell walls, endotoxins and exotoxins to inhibit microbial adhesion and play an antibacterial role. In addition, in terms of anti-tumor, taurolidine can induce cytotoxicity of tumor cells by inducing apoptosis, autophagy and necrosis. The extent to which these processes are involved may vary with the type of tumor cell. Until July 2020, there were more than 260 foreign literature searches on taurolidine research reports, most of which focused on the exploration of the effect of taurolidine on tumor-related signaling pathways, while the application of taurolidine in antiviral activity was not yet available. See research reports.
PATENThttps://patents.google.com/patent/CN101274921B/en
Taurolidine synthetic operation step:1. the preparation of tauryl villaumite hydrochlorate
In being housed, ventpipe, escape pipe, thermometer and churned mechanically 300ml four-necked bottle add Mercaptamine 25g, 200ml methylene dichloride and 32ml dehydrated alcohol, under ice-water bath (below the 10 ℃) mechanical stirring, feed dry appropriate chlorine, reaction begins and heat release immediately, the thick solid of adularescent generates, and temperature remains on below 50 ℃ and stirs, reaction 5h.The whole process HCl gas and the monochloroethane gas of alkali lye absorption reaction process.Stop logical chlorine after reacting end, get yellow mercury oxide, suction filtration is used washed with dichloromethane four times, and vacuum-drying gets white solid 50g, 152~154 ℃ of fusing points.2. the preparation of tauryl azide salt hydrochlorate
The reaction flask ice-water bath of containing 45ml water is cooled to-15 ℃, adds NaN 3(2g), after stirring is molten entirely, add slightly pinkiness of tauryl villaumite hydrochlorate (9g) solution in batches, the water-bath of 20 minutes recession deicings, room temperature continues stirring 60 minutes.3. the preparation of tauryl amine hydrochlorate
Above-mentioned reaction solution is joined in the 500ml autoclave, add 0.5g 5%Pd/C, feed hydrogen, pressure is 7Mpa, stirring at room 6h.Turn off hydrogen, pour out reaction solution, elimination Pd/C gets colourless reaction solution.The reaction solution that takes a morsel adds in the nuclear-magnetism pipe, adds deuterated reagent D 2O, with 1HNMR determines the transformation efficiency of hydrogenation reaction.Two kinds of CH of tauryl amine hydrochlorate 2D 1The HNMR peak is 3.29~3.31 and 3.40~3.42ppm place, and two kinds of CH of reactant tauryl azide salt hydrochlorate 2D 1The HNMR peak is 3.37~3.38 and 3.82~3.84ppm place.Determine that with the peak height ratio of two kinds of compounds the 4th step added the amount of formaldehyde.4. the preparation of taurolidine
With the above-mentioned reaction solution that removes by filter Pd/C, add 5g NaHCO 3, be stirred to molten entirely, frozen water cooling, stir slowly splash into down formaldehyde solution (37%, 2ml), have milky white precipitate to produce after 30 minutes, continue to stir 1h, suction filtration, filter cake is washed 3 times with frozen water.Vacuum-drying gets white powdery solid 2.3g, 170~174 ℃ of fusing points.Embodiment 2:Making with extra care of taurolidine:The above-mentioned taurolidine white powder 5~10g that obtains adds 50~200ml acetonitrile, and heating for dissolving removes by filter a small amount of insolubles, concentrates, and cooling below 10 ℃ gets white powder 5~10g, 172~174 ℃ of fusing points.Embodiment 3:Proton nmr spectra ( 1H-NMR) data are as follows:1HNMR(DMSO-D6,TMS7.26-7.28(t,2H,NH),4.09-4.10(d,4H,N-CH 2),3.53(s,2H,N-CH 2-N),3.28-3.29(t,4H,N-CH 2-CH 2),2.96-2.97(t,4H,S-CH 2-CH 2)。The infrared absorption spectrum data are as follows:IR (KBr compressing tablet cm -1): 3425,3263,1633,1450,1404,1317,1278,1228,1160,1134,1073,1026,993,958,924,830,757,667,532,511.See Fig. 3.The ultimate analysis analytical value:C, 29.04%, N, 18.55%, H, 5.85%; Calculated value: C, 29.57%, N, 19.71%, H, 5.67%Embodiment 4:Taurolidine formulation optimizing injection type of the present invention, as: infusion solution, injection liquid, freeze-dried powder injection or powder ampoule agent for injection etc., more preferably infusion solution.The preparation of infusion solution[prescription 1] taurolidine 10.0~30.0gPVP 40.0~80.0gNaCl 2~5gAdd water to 1000ml[method for making] takes by weighing taurolidine, is dissolved in water, and stirs, and adds the PVP dissolving, and adjust pH to 7.0 is crossed the moisture film of 0.22 μ m, packing, and 121 ℃ of sterilizations 20 minutes, promptly.[prescription 2] taurolidine 10.0~30.0gCitric acid 0.1~1.0gLemon enzyme sodium 10.0~20.0gAdd water to 1000ml[method for making] takes by weighing taurolidine, is dissolved in water, stir, and the dissolving of adding citric acid sodium, adjust pH to 7.0, the moisture film of mistake 0.22 μ m, packing was sterilized 20 minutes for 121 ℃, promptly.
PATENThttps://patents.google.com/patent/US8952148B2/en

Example ICbz-Taurine Sodium Salt (Formula II)To 1000 ml of water in the RBF charge 192 gm of (3.0 eq) of sodium hydroxide under cooling followed by 200 gm of Taurine and dissolve it until clear solution is obtained. Cool to 0° C. to 5° C., and Charge 50% CBZ-Cl in toluene at 0° C. to 5° C. After completion of addition, maintain at room temperature for 14 h. Separate the toluene layer and wash the aqueous layer with 2×200 ml of ethyl acetate. Add slowly 27 gm of sodium hydroxide in 60 ml of water to the aqueous layer and adjust pH to 12-14. Cool to 0° C. to 5° C. and a white solid separates from the solution. Filter the solid and dry the solid at 60-70° C. Weight of the solid: 320 g
Example 2Cbz-Taurinamide (Formula III)To a clean dry flask charge 1500 ml of toluene and charge 320 gm of Formula II and cool to 0° C. to 5° C. Charge 308 gm of PCl5 slowly at 0° C. to 5° C. for 2 hrs. Maintain at 0° C. to 5° C. up to completion of reaction. Quench the RM into another flask containing 2 ltr of water at 0° C. to 5° C. Separate the organic layer, wash and extract the aqueous layer with toluene. Dry the organic layer with sodium sulphate and cool to 0° C. to 5° C. Purge ammonia gas into the reaction mass till the reaction is complete. Filter the solid and dissolve the solid in 2 ltr of water and extract the aqueous layer with 2×600 ml of ethyl acetate. Dry the organic layer with sodium sulphate and concentrate it under reduced pressure to obtain a white solid. Weight of the solid: 150 g
Example 3Taurinamide Succinate (Formula IV)Take a suspension of 100 g of Cbz-Taurinamide in 1000 ml methanol, and 10% Pd/C (1.0 g) and subject to hydrogenation at 45-50 psi. Upon completion of the reaction filter the catalyst and add succinic acid (1.0 eq) to the solvent and distill off the solvent under vacuum to provide the title compound in about 90% yield as a white solid.
Example 4Taurolidine (Formula I)To a solution of 100 g Taurinamide succinate in water is added sat sodium bicarbonate solution and pH adjusted to 7-8. To the solution was added formaldehyde (50 ml) and allowed to stir for 4 h. The solid obtained was filtered and washed with water to give Taurolidine. The title compound was obtained in about 70% yield and about 98% purity.
Example 5Purification of TaurolidineTaurolidine (100 g) was dissolved in DMSO (400 ml) and a clear solution is obtained and a precipitate is obtained immediately. The solid is filtered and washed with toluene and dried to give a white solid in 40% yield. The product obtained was >99.5% pure.
Example 6Purification of TaurolidineTaurolidine (100 g) was dissolved in DMSO (400 ml) and a clear solution is obtained and a precipitate is obtained immediately. To the solution, toluene (1000 ml) is added. The solid is filtered and washed with toluene and dried to give a white solid in 70% yield. The product obtained was >99.5% pure by HPLC and passed elemental analysis within 0.4% of the theoretical values.
Example 7Purification of TaurolidineTaurolidine (100 g) was dissolved in DMAc (800 ml) and to the solution, toluene (1000 ml) is added. The solid is filtered and washed with toluene and dried to give a white solid in 70% yield.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
SYN
European Journal of Medicinal Chemistry 265 (2024) 116124
Defencath, a product created by CorMedix, Inc., was granted FDA approval on November 15, 2023. Its purpose is to decrease the occur rence of catheter-related bloodstream infections (CRBSI) in a limited
population of adult individuals who have chronic renal failure and undergo hemodialysis (HD) using a central venous catheter (CVC) [11]. Defencath is the first and only FDA-approved antimicrobial catheter
locking solution (CLS) in the United States. It is composed of the anticoagulant Heparin and the broad-spectrum non-antibiotic antimicrobial and antifungal agent Taurolidine. Taurolidine has been proven to have activity against the following microorganisms: Staphylococcus aureus, Staphylococcus epidermidis, Escherichia coli, Enterococcus faecalis, Pseudomonas aeruginosa, Klebsiella pneumoniae, Serratia marcescens, Candida albicans, Candida glabrata [12,13].

The synthesis route of Taurolidine is showed in Scheme 4 [14].
Benzyl (2-sulfamoylethyl)carbamate (TAUR-001) was removed benzy
loxycarbonyl (Cbz) group under the action of palladium carbon to obtain
TAUR-002. After the reaction with formaldehyde, the product Taur
olidine was obtained.
[11] A.K. Agarwal, P. Roy-Chaudhury, P. Mounts, E. Hurlburt, A. Pfaffle, E.C. Poggio,
Taurolidine/Heparin lock solution and catheter-related bloodstream infection in
hemodialysis: a randomized, double-blind, active-control, phase 3 study, Clin. J.
Am. Soc. Nephrol. 18 (2023) 1446–1455.
[12] C.H. van den Bosch, B. Jeremiasse, J.T. van der Bruggen, F.N.J. Frakking, Y.G.
T. Loeffen, C.P. van de Ven, A.F.W. van der Steeg, M.F. Fiocco, M.D. van de
Wetering, M. Wijnen, The efficacy of taurolidine containing lock solutions for the
prevention of central-venous-catheter-related bloodstream infections: a
systematic review and meta-analysis, J. Hosp. Infect. 123 (2022) 143–155.
[13] Y. Wouters, G.R.H. Mennen, R.H.M. Te Morsche, H.M.J. Roelofs, G.J.A. Wanten,
The antiseptic and antineoplastic agent taurolidine modulates key leukocyte
functions, In Vivo 36 (2022) 2074–2082.
[14] R.M. Berrios, Synthesis of Taurolidine, Purity Profiles and Polymorphs, 2023.
US11738120B1
Medical uses
Taurolidine is an antimicrobial agent used in an effort to prevent catheter infections. It however is not approved for this use in the United States as of 2011.[4]
- Catheter lock solution in home parenteral nutrition (HPN) or total parenteral nutrition (TPN): catheter-related blood stream infections (CRBSI) remains the most common serious complication associated with long-term parenteral nutrition. The use of taurolidine as a catheter lock solution shows a reduction of CRBSI.[1][5] The overall quality of the evidence however is not strong enough to justify routine use.[1][5]
- Catheter lock solution: Taurolidine decreases the adherence of bacteria and fungi to host cells by destructing the fimbriae and flagella and thus prevent the biofilm formation.[6][7] Taurolidine is the active ingredient of antimicrobial catheter lock solutions for the prevention and treatment of catheter related bloodstream infections (CRBSIs) and is suitable for use in all catheter based vascular access devices.[8][1] Bacterial resistance against taurolidine has never been observed in various studies.[9][10]
- The use of a taurolidine lock solution may decrease the risk of catheter infection in children with cancer but the evidence is tentative.[11]
Side effects
No systemic side effects have been identified. The safety of taurolidine has also been confirmed in clinical studies with long-term intravenous administration of high doses (up to 20 grams daily). In the body, taurolidine is metabolized rapidly via the metabolites taurultam and methylol taurinamide, which also have a bactericidal action, to taurine, an endogenous aminosulphonic acid, carbon dioxide and water. Therefore, no toxic effects are known or expected in the event of accidental injection. Burning sensation while instilling, numbness, erythema, facial flushing, headache, epistaxis, and nausea have been reported.[12]
Toxicology
Taurolidine has a relatively low acute and subacute toxicity.[1] Intravenous injection of 5 grams taurolidine into humans over 0.5–2 hours produce only burning sensation while instilling, numbness, and erythema at the injection sites.[12] For treatment of peritonitis, taurolidine was administered by peritoneal lavage, intraperitoneal instillation or intravenous infusion, or by a combination thereof. The total daily dose ranged widely from 0.5 to 50 g. The total cumulative dose ranged from 0.5 to 721 g. In those patients who received intravenous taurolidine, the daily intravenous dose was usually 15 to 30 g but several patients received up to 40 g/day. Total daily doses of up to 40 g and total cumulative doses exceeding 300 g were safe and well tolerated.[12][13][14][15][16]
Pharmacology
- Metabolism: Taurolidine and taurultam are quickly metabolized to taurinamide, taurine, carbon dioxide and water. Taurolidine exists in equilibrium with taurultam and N-methylol-taurultam in aqueous solution.[17]
- Pharmacokinetic (elimination): The half-life of the terminal elimination phase of taurultam is about 1.5 hours, and of the taurinamide metabolite about 6 hours. 25% of the taurolidine dose applied is renally eliminated as taurinamide and/or taurine.[13][14][18]
Mechanism of action
Following administration of taurolidine, the antimicrobial and antiendotoxin activity of the taurolidine molecule is conferred by the release of three active methylol (hydroxymethyl) groups as taurolidine is rapidly metabolized by hydrolysis via methylol taurultam to methylol taurinamide and taurine. These labile N-methylol derivatives of taurultam and taurinamide react with the bacterial cell-wall resulting in lysis of the bacteria, and by inter- and intramolecular cross-linking of the lipopolysaccharide-protein complex, neutralization of the bacterial endotoxins which is enhanced by enzymatic activation. This mechanism of action is accelerated and maximised when taurolidine is pre-warmed to 37 °C (99 °F). Microbes are killed and the resulting toxins are inactivated; the destruction time in vitro is 30 minutes.[19]
The chemical mode of action of taurolidine via its reactive methylol groups confers greater potency in vivo than indicated by in vitro minimum inhibitory concentration (MIC) values, and also appears to preclude susceptibility to resistance mechanisms.[14]
Taurolidine binding to lipopolysaccharides (LPS) prevents microbial adherence to host epithelial cells, thereby prevents microbial invasion of uninfected host cells. Although the mechanism underlying its antineoplastic activity has not been fully elucidated, it may be related to this agent’s anti-adherence property.[6][7] Taurolidine has been shown to block interleukin 1 (IL-1) and tumour necrosis factor (TNF) in human peripheral blood mononuclear cells (PBMC).[20] In addition, taurolidine also promotes apoptosis by inducing various apoptotic factors and suppresses the production of vascular endothelial growth factor (VEGF), a protein that plays an important role in angiogenesis.[21]
Taurolidine is highly active against the common infecting pathogens associated with peritonitis and catheter sepsis, this activity extends across a wide-spectrum of aerobic and anaerobic bacteria and fungi (with no diminution of effect in the presence of biological fluids, e.g. blood, serum, pus).[15][16][22]
- Gram-positive bacteria (MIC of 1–2 mg/mL): Staphylococcus (including multiple-antibiotic resistant coagulase negative strains, methicillin-resistant S. aureus), Streptococcus, Enterococcus.
- Gram-negative bacteria (MIC of 0.5–5 mg/mL): Aerobacter species, Citrobacter species, Enterobacter species, Escherichia coli, Proteus species (indole negative), Proteus mirabilis, Pseudomonas species (including P. aeruginosa), Salmonella species, Serratia marcescans, Klebsiella species.
- Anaerobes (MIC 0.03–0.3 mg/mL): Bacteroides species (including B. fragilis), Fusobacteria, Clostridium species, Peptostreptococcus anaerobius.
- Fungi (MIC 0.3–5 mg/mL): Candida albicans, Cryptococcus neoformans, Aspergillus species, Trichophyton rubrum, Epidermophyton floccosum, Pityrosporum ovale.[10][17][22]
Chemical properties
The chemical name for taurolidine is 4,4′-Methylene-bis(1,2,4-thiadiazinane)-1,1,1’,1′-tetraoxide.
It is a white to off white odourless crystalline powder. It is practically insoluble in chloroform, slightly soluble in boiling acetone, ethanol, methanol, and ethyl acetate, sparingly soluble in water 8 at 20° and ethyl alcohol, soluble in dilute hydrochloric acid, and dilute sodium hydroxide, and freely soluble in N,N-dimethylformamide (at 60 °C).
History
Taurolidine was first synthesized in the laboratories of Geistlich Pharma AG, Switzerland in 1972. Clinical trials begun in 1975 in patients with severe peritonitis.
Research
Taurolidine demonstrates some anti-tumor properties, with positive results seen in early-stage clinical investigations using the drug to treat gastrointestinal malignancies and tumors of the central nervous system.[23] More recently, it has been found to exert antineoplastic activity. Taurolidine induces cancer cell death through a variety of mechanisms. Even now, all the antineoplastic pathways it employs are not completely elucidated. It has been shown to enhance apoptosis, inhibit angiogenesis, reduce tumor adherence, downregulate pro-inflammatory cytokine release, and stimulate anticancer immune regulation following surgical trauma. Apoptosis is activated through both a mitochondrial cytochrome-c-dependent mechanism and an extrinsic direct pathway. A lot of in vitro and animal data support taurolidine’s tumoricidal action.[24][25][26] Taurolidine has been used as an antimicrobial agent in the clinical setting since the 1970s and thus far appears nontoxic. The nontoxic nature of taurolidine makes it a favorable option compared with current chemotherapeutic regimens. Few published clinical studies exist evaluating the role of taurolidine as a chemotherapeutic agent. The literature lacks a gold-standard level 1 randomized clinical trial to evaluate taurolidine’s potential antineoplastic benefits. However, these trials are currently underway. Such randomized control studies are vital to clarify the role of taurolidine in modern cancer treatment.[21][2]
References
- ^ Jump up to:a b c d e f Liu Y, Zhang AQ, Cao L, Xia HT, Ma JJ (2013). “Taurolidine lock solutions for the prevention of catheter-related bloodstream infections: a systematic review and meta-analysis of randomized controlled trials”. PLOS ONE. 8 (11): e79417. Bibcode:2013PLoSO…879417L. doi:10.1371/journal.pone.0079417. PMC 3836857. PMID 24278133.
- ^ Jump up to:a b Neary PM, Hallihan P, Wang JH, Pfirrmann RW, Bouchier-Hayes DJ, Redmond HP (April 2010). “The evolving role of taurolidine in cancer therapy”. Annals of Surgical Oncology. 17 (4): 1135–43. doi:10.1245/s10434-009-0867-9. PMID 20039217. S2CID 23807182.
- ^ Waser PG, Sibler E (1986). “Taurolidine: A new concept in antimicrobial chemotherapy”. In Harms AF (ed.). Innovative Approaches in Drug Research. Elsevier Science Publishers. pp. 155–169.
- ^ O’Grady NP, Alexander M, Burns LA, Dellinger EP, Garland J, Heard SO, et al. (May 2011). “Guidelines for the prevention of intravascular catheter-related infections”. Clinical Infectious Diseases. 52 (9): e162-93. doi:10.1093/cid/cir257. PMC 3106269. PMID 21460264.
- ^ Jump up to:a b Bradshaw JH, Puntis JW (August 2008). “Taurolidine and catheter-related bloodstream infection: a systematic review of the literature”. Journal of Pediatric Gastroenterology and Nutrition. 47 (2): 179–86. doi:10.1097/MPG.0b013e318162c428. PMID 18664870. S2CID 19136945.
- ^ Jump up to:a b Gorman SP, McCafferty DF, Woolfson AD, Jones DS (April 1987). “Reduced adherence of micro-organisms to human mucosal epithelial cells following treatment with Taurolin, a novel antimicrobial agent”. The Journal of Applied Bacteriology. 62 (4): 315–20. doi:10.1111/j.1365-2672.1987.tb04926.x. PMID 3298185.
- ^ Jump up to:a b Blenkharn JI (July 1989). “Anti-adherence properties of taurolidine and noxythiolin”. Journal of Chemotherapy. 1 (4 Suppl): 233–4. PMID 16312382.
- ^ Liu H, Liu H, Deng J, Chen L, Yuan L, Wu Y (2014). “Preventing catheter-related bacteremia with taurolidine-citrate catheter locks: a systematic review and meta-analysis”. Blood Purification. 37 (3): 179–87. doi:10.1159/000360271. PMID 24777144.
- ^ Olthof ED, Rentenaar RJ, Rijs AJ, Wanten GJ (August 2013). “Absence of microbial adaptation to taurolidine in patients on home parenteral nutrition who develop catheter related bloodstream infections and use taurolidine locks”. Clinical Nutrition. 32 (4): 538–42. doi:10.1016/j.clnu.2012.11.014. PMID 23267744.
- ^ Jump up to:a b Torres-Viera C, Thauvin-Eliopoulos C, Souli M, DeGirolami P, Farris MG, Wennersten CB, et al. (June 2000). “Activities of taurolidine in vitro and in experimental enterococcal endocarditis”. Antimicrobial Agents and Chemotherapy. 44 (6): 1720–4. doi:10.1128/aac.44.6.1720-1724.2000. PMC 89943. PMID 10817739.
- ^ Simon A, Bode U, Beutel K (July 2006). “Diagnosis and treatment of catheter-related infections in paediatric oncology: an update”. Clinical Microbiology and Infection. 12 (7): 606–20. doi:10.1111/j.1469-0691.2006.01416.x. PMID 16774556.
- ^ Jump up to:a b c Gong L, Greenberg HE, Perhach JL, Waldman SA, Kraft WK (June 2007). “The pharmacokinetics of taurolidine metabolites in healthy volunteers”. Journal of Clinical Pharmacology. 47 (6): 697–703. doi:10.1177/0091270007299929. PMID 17395893. S2CID 31059736.
- ^ Jump up to:a b Knight BI, Skellern GG, Browne MK, Pfirrmann RW (November 1981). “Peritoneal absorption of the antibacterial and antiendotoxin taurolin in peritonitis”. British Journal of Clinical Pharmacology. 12 (5): 695–9. doi:10.1111/j.1365-2125.1981.tb01292.x. PMC 1401955. PMID 7332737.
- ^ Jump up to:a b c Stendel R, Scheurer L, Schlatterer K, Stalder U, Pfirrmann RW, Fiss I, et al. (2007). “Pharmacokinetics of taurolidine following repeated intravenous infusions measured by HPLC-ESI-MS/MS of the derivatives taurultame and taurinamide in glioblastoma patients”. Clinical Pharmacokinetics. 46 (6): 513–24. doi:10.2165/00003088-200746060-00005. PMID 17518510. S2CID 33321671.
- ^ Jump up to:a b Browne MK, MacKenzie M, Doyle PJ (May 1978). “C controlled trial of taurolin in established bacterial peritonitis”. Surgery, Gynecology & Obstetrics. 146 (5): 721–4. PMID 347606.
- ^ Jump up to:a b Browne MK (1981). “The treatment of peritonitis by an antiseptic – taurolin”. Pharmatherapeutica. 2 (8): 517–22. PMID 7255507.
- ^ Jump up to:a b Knight BI, Skellern GG, Browne MK, Pfirrmann RW (September 1981). “The characterisation and quantitation by high-performance liquid chromatography of the metabolites of taurolin”. British Journal of Clinical Pharmacology. 12 (3): 439–40. doi:10.1111/j.1365-2125.1981.tb01245.x. PMC 1401804. PMID 7295478.
- ^ Browne MK, Leslie GB, Pfirrmann RW (December 1976). “Taurolin, a new chemotherapeutic agent”. The Journal of Applied Bacteriology. 41 (3): 363–8. doi:10.1111/j.1365-2672.1976.tb00647.x. PMID 828157.
- ^ Braumann C, Pfirrman RW, et al. (2013). “Taurolidine, an Effective Multimodal Antimicrobial Drug Versus Traditional Antiseptics and Antibiotics”. In Willy C (ed.). Antiseptics in Surgery – Update 2013. Lindqvist Book Publishing. pp. 119–125.
- ^ Bedrosian I, Sofia RD, Wolff SM, Dinarello CA (November 1991). “Taurolidine, an analogue of the amino acid taurine, suppresses interleukin 1 and tumor necrosis factor synthesis in human peripheral blood mononuclear cells”. Cytokine. 3 (6): 568–75. doi:10.1016/1043-4666(91)90483-t. PMID 1790304.
- ^ Jump up to:a b Jacobi CA, Menenakos C, Braumann C (October 2005). “Taurolidine–a new drug with anti-tumor and anti-angiogenic effects”. Anti-Cancer Drugs. 16 (9): 917–21. doi:10.1097/01.cad.0000176502.40810.b0. PMID 16162968. S2CID 33876185.
- ^ Jump up to:a b Nösner K, Focht J (1994). “In-vitro Wirksamkeit von Taurolidin und 9 Antibiotika gegen klinische Isolate aus chirurgischem Einsendegut sowie gegen Pilze”. Chirurgische Gastroenterologie. 10 (Suppl 2): 10.
- ^ Stendel R, Picht T, Schilling A, Heidenreich J, Loddenkemper C, Jänisch W, Brock M (2004-04-01). “Treatment of glioblastoma with intravenous taurolidine. First clinical experience”. Anticancer Research. 24 (2C): 1143–7. PMID 15154639.
- ^ Calabresi P, Goulette FA, Darnowski JW (September 2001). “Taurolidine: cytotoxic and mechanistic evaluation of a novel antineoplastic agent”. Cancer Research. 61 (18): 6816–21. PMID 11559556.
- ^ Clarke NW, Wang JH, et al. (2005). “Taurolidine inhibits colorectal adenocarcinoma metastases in vivo and in vitro by inducing apoptosis”. Ir J Med Sci. 174 (Supplement 3): 1.
- ^ Stendel R, Scheurer L, Stoltenburg-Didinger G, Brock M, Möhler H (2003-06-01). “Enhancement of Fas-ligand-mediated programmed cell death by taurolidine”. Anticancer Research. 23 (3B): 2309–14. PMID 12894508.
| Clinical data | |
|---|---|
| ATC code | B05CA05 (WHO) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 19388-87-5 |
| ChemSpider | 27486 |
| UNII | 8OBZ1M4V3V |
| CompTox Dashboard (EPA) | DTXSID00173001 |
| ECHA InfoCard | 100.039.090 |
| Chemical and physical data | |
| Formula | C7H16N4O4S2 |
| Molar mass | 284.35 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////////////TAUROLIDINE, UNII-8OBZ1M4V3V, тауролидин ,توروليدين , 牛磺利定 ,
C1CS(=O)(=O)NCN1CN2CCS(=O)(=O)NC2

NEW DRUG APPROVALS
ONE TIME
$10.00
Anthony crasto is now Consultant Glenmark Lifesciences at Glenmark Life Sciences!

I’m happy to share that I’m starting a new position as Consultant Glenmark Lifesciences at Glenmark Life Sciences!
17th Jan 2022, A new innings
I retired 16th Jan 2022 at 58 yrs from Glenmark . completed 16 yrs 2 months
30 plus years in the field of Process research
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
///////////////

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}