
Home » New drugs china
Category Archives: New drugs china
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |

Peramivir, a Flu Treatment

PERAMIVIR
CAS 330600-85-6
- Molecular FormulaC15H28N4O4
- Average mass328.407 Da
(1S,2S,3R,4R)-4-carbamimidamido-3-[(1S)-1-acetamido-2-ethylbutyl]-2-hydroxycyclopentane-1-carboxylic acid
- RWJ 270201
- RWJ-270201
- RWJ270201
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Peramivir hydrate | QW7Y7ZR15U | 1041434-82-5 | RFUCJKFZFXNIGB-ZBBHRWOZSA-N |
| Molecular Weight | 382.45 |
| Formula | C15H28N4O4 • 3H2O |
| CAS No. | 330600-85-6 (Peramivir); 1041434-82-5 (Peramivir Trihydrate); |
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Rapivab | Solution | 600 mg/60mL | Intravenous | BioCryst Pharmaceuticals, Inc. | 2014-12-20 | Not applicable |  |
|
| Rapivab | Solution | 600 mg/60mL | Intravenous | Seqirus USA Inc. | 2014-12-20 | Not applicable | |
|
| Rapivab | Solution | 10 mg | Intravenous | Biocryst Pharmaceuticals Inc | Not applicable | Not applicable |  |
- Drug Name:
- Peramivir Hydrate
- Research Code:
- S-021812
- Trade Name:
- Rapiacta® / Peramiflu®
- MOA:
- Neuraminidase inhibitor
- Indication:
- Influenza infection
- Status:
- Approved
- Company:
- BioCryst (Originator) , Shionogi
- Sales:
- $21.6 Million (Y2015);

$23.7 Million (Y2014);
$20 Million (Y2013);
$24.1 Million (Y2012);
$17.7 Million (Y2011); - ATC Code:
Approved Countries or Area
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-12-19 | Marketing approval | Rapivab | Influenza infection | Solution | 200 mg/20 ml | BioCryst | Standard |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2010-01-13 | Marketing approval | Rapiacta | Influenza infection | Injection, Solution | Eq. 150 mg/300 mg Peramivir | BioCryst, Shionogi |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2013-04-05 | Marketing approval | 力纬 | Influenza infection | Injection, Solution | 100ml | 广州南新制药 | 1.1 |
Shionogi , GC Pharma and Seqirus , under license from BioCryst Pharmaceuticals (which licensed the program from the University of Alabama ), have developed and launched an iv formulation of the influenza neuraminidase inhibitor peramivir.
Peramivir hydrate was approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on January 13, 2010, and approved by the U.S. Food and Drug Administration (FDA) on December 19, 2014. It was co-developed and co-marketed as Rapiacta® by BioCryst and Shionogi in Japan.
Peramivir hydrate is a neuraminidase inhibitor, acting as a transition-state analogue inhibitor of influenza neuraminidase and thereby preventing new viruses from emerging from infected cells. It is indicated for the treatment of influenza A and B viral infections.
Rapiacta® is available as injection solution for intravenous, containing 150 mg/300 mg Peramivir hydrate. The recommended dose is once a day by an intravenous drip infusion over a period of 15 minutes or longer.
Peramivir (trade name Rapivab) is an antiviral drug developed by BioCryst Pharmaceuticals for the treatment of influenza. Peramivir is a neuraminidase inhibitor, acting as a transition-state analogue inhibitor of influenza neuraminidase and thereby preventing new viruses from emerging from infected cells. It is approved for intravenous administration.[1]
In October 2009, the U.S. Food and Drug Administration (FDA) issued an emergency use authorization (EUA) for the use of peramivir based on safety data from phase I, phase II, and limited phase III trial data. The emergency use authorization for peramivir expired in June 2010.[2][3] On 19 December 2014, the FDA approved peramivir to treat influenza infection in adults.[1]
Peramivir has also been approved in Japan and South Korea and is available in Japan as Rapiacta and in South Korea as Peramiflu.[ As of 2015, it is the only intravenous option for treating swine flu.
Peramivir is an antiviral agent developed by Biocryst Pharmaceuticals to treat influenza A/B. The development of peramivir has been supported by the US Department of Health and Human Services as part of the government’s effort to prepare for a flu pandemic. Being an influenza virus neuraminidase inhibitor, peramivir works by preventing new viruses from emerging from infected cells. Due to the poor oral bioavailability, the oral formulation of the drug was previously abandoned by Johnson and Johnson Company. The injectable intravenous formulation of peramivir was approved by the FDA in September 2017 for the treatment of acute uncomplicated influenza to pediatric patients 2 years and older who have been symptomatic for no more than two days.
History
An intramuscular (IM) peramivir phase II study for seasonal influenza in 2008–2009 found no effect for the primary endpoint of improvement in the median time to alleviation of symptoms in subjects with confirmed, acute, uncomplicated influenza infection versus placebo.
In October 2009, it was reported that the experimental antiviral drug peramivir had been “life-saving” effective in intravenous treating 8 serious cases of swine flu.[4] On October 23, the U.S. Food and Drug Administration (FDA) issued an Emergency Use Authorization for peramivir, allowing the use of the drug in intravenous form for hospitalized patients only in cases where the other available methods of treatment are ineffective or unavailable;[5] for instance, if oseltamivir resistance develops and a person is unable to take zanamivir via the inhaled route. The U.S. government (department of Health and Human Services) gave BioCryst Pharmaceuticals more than $77 million to finish the Phase III clinical development of peramivir. In 2009 the department of Health and Human Services had already given about $180 million to the program.[6] Biocryst also donated 1200 courses of treatment to the US department of Health and Human Services.[7] The Emergency Use Authorization expired on June 23, 2010. In 2011 a phase III trial found the median durations of influenza symptoms were the same with 1 intravenous injection of peramivir against 5 days of oral oseltamivir for people with seasonal influenza virus infection.[8]
In 2012 BioCryst reported that it should halt enrollment on its study for intravenous peramivir in potentially life-threatened people after an interim analysis led trial monitors to conclude that it would be futile to continue and the trial should be terminated. The difference between peramivir and control group (oral oseltamivir) for the primary endpoint, clinical or virologic, was small.[9] In 2013 the Biomedical Advanced Research and Development Authority (BARDA/HHS) released new funding under the current $234.8 million contract to enable completion of a New Drug Application filing for intravenous (IV) peramivir.[10]
According to a research report published in June 2011, a new variant of swine flu had emerged in Asia with a genetic adaptation (a S247N neuraminidase mutation) giving some resistance to oseltamivir and zanamivir, but no significant reduction in sensitivity to peramivir.[11][12] But a H274Y virus mutation showed resistance to oseltamivir and peramivir, but not to zanamivir, and only in N1 neuraminidases.[13] Ultimately 3.2% (19/599) of A(H1N1)pdm09 viruses collected between 2009 and 2012 had highly reduced peramivir inhibition due to the H275Y NA mutation.[14]
BioCryst Pharmaceuticals submitted a new drug application (NDA) to the U.S. Food and Drug Administration (FDA) for intravenous peramivir in December 2013.[15] Peramivir (Rapivab) was approved for intravenous administration in December 2014.[1][16]

Reference:1. WO9933781A1 / US6562861B1.

Reference:1. CN102372657A.

Reference:1. CN102633686A.

Reference:1. J. Med. Chem. 2001, 44, 4379-4392.



PATENT
peramivir’s product case, WO9933781 ,
hold SPC protection in the EU states until December 2023, and contain one of the Orange Book (OB) listed patents, US6562861 , that was due to expire in the US in December 2023. In January 2021, the US FDA’s OB was seen to list patents describing peramivir that expire in 2027.
PATENT
WO-2021002689
Process for preparing an inhibitor of neuraminidase infection ie peramivir trihydrate crystal from the free form of the drug ie peramivir. Discloses the use of peramivir trihydrate in treating influenza infection. Represents the first PCT filing from CKD Bio Corp that focuses on peramivir; however, this case was first seen as a Korean national filing that is published in February 2020.
PATENT
https://patents.google.com/patent/WO2012145932A1/en
Peramivir has the chemical name of
(1^,2^,3^, 4R)-3-[(llS)-l-acetamido-2-ethyl-butyl] -4-(diaminomethylideneamino)- 2-hydroxy-cyclopentane-l-carboxylicacid, and has the following structure:

(I)
[0003] Peramivir is currently being developed as an antiviral drug, and in particular, for treatment of influenza. Acting as a neuraminidase inhibitor, peramivir can efficiently inhibit the replication of all type of influenza viruses. Peramivir can be administered via injection, and is known to be well-tolerated and cause only mild adverse effect.
[0004] Several processes relating to the preparation of peramivir are disclosed in CN1227466, CN1282316, and WO01/00577A1.
[0005] As shown in Scheme 1, CN 1227466 discloses a process comprising ring-opening of chiral 2-azabicyclo [2.2.1] hept-5-en-3-one, followed by
amino-protecting reaction, Diel- Alder conjugate cycloaddition, reduction, acetylation, guanidylation and finally hydrolyzation to yield peramivir. The major drawback of this process is the use of highly expensive starting material 1. In addition, this process is not suitable for scale-up.
Scheme 1

[0006] WO2009021404 discloses a method comprising reacting
N-Boc-protected chiral 2-azabicyclo [2.2.1] hept-5-en-3-one and
2-ethylbutylaldehyde as starting material to prepare peramivir as illustrated in Scheme 2.
Scheme 2

eram v r
EXAMPLES
Example 1
1 (liS’,4J/?)-methyl-4-(2,3-bis(ieri-butoxycarbonyl)guanidino)cyclopent-2-ene- carboxylate (13)

11 12 13 [0063] To a mixture of (lS^R^methyl 4-aminocyclopent-2-enecarboxylate tartaric acid salt 11 ( 7.29 g, 25 mmol) in dichloromethane (150 mL), was added Et3N (9 mL, 65 mmol) at 0 °C, and the resulting mixture was stirred for 15 min. To this, tert-butyl (lH-pyrazol-l-yl)methylenedicarbamate 12 was added. After addition, the reaction was monitored for completion by TLC ( PE: EtOAc=5 : 1 ) . The organic layers were washed with water and brine and dried over anhydrous Na2S04. The mixture was filtered and concentrated to give 13 as a white solid, which was used in the next step without further purification.
[0064] MS (M+l ) : 384.
[0065] ‘H NMR (400 MHz, CDC13) δ 11.49 (s, 1H), 8.53 (d, J= 8.4 Hz, 1H), 5.94 – 5.83 (m, 2H), 5.38 – 5.31 (m, 1H), 3.73 (s, 3H), 3.56 – 3.44 (m, 1H), 2.60 (dt, J= 14.0, 8.5 Hz, 1H), 1.94 (dt, J= 13.9, 4.7 Hz, 1H), 1.50 (d, J= 7.4 Hz, 18H) (See attached Chart 1)
Example 2
2 (3aJ/?,4J/?,6iS’,6aiS -methyl-4-(2,3-bis(ieri-butoxycarbonyl)guanidino)-3-(pentan -3-yl)-4,5,6,6a-tetrahydro-3aH-cyclopenta[d]isoxazole-6-carboxylate (5)

14 85% a) Preparation of 2-Ethyl-N-hydroxybutanimidoyl chloride (14):
[0066] Hydroxylamine hydrochloride (7.2g, 0.1 mol) was dissolved in water (7 mL). Toluene (27 mL) was added, followed by addition of 2-ethylbutylraldehyde (lOg, 0.1 mol). The two-phase mixture was stirred vigorously while cooling.
Sodium hydroxide solution (ca.30%, 14.6g, O. l lmol) was added slowly (addition is very exothermic) to maintain a temperature between 15-25 °C. The mixture was stirred for 60 min, then allowed to stand to separate the layers. The organic extract was washed with water and brine, dried over Na2S04, and directly used in the next step.
[0067] N-Chlorosuccinimide (NCS) (13.3g, 0.1 mol) was suspended in dimethylformamide (DMF) (17ml) and cooled to about 10 °C. The toluene solution prepared above (3 .15 mol) was added dropwise with sufficient cooling to maintain the reaction temperature betweenlO-25°C. After addition, the reaction was monitored by TLC until completion of the reaction. Water (100ml) was added slowly (slightly exothermic) while maintaining the temperature at 15-25 °C. The two-phase mixture was stirred for 15 min at 15-25 °C and the layers were separated. The water layer was extracted with toluene (10ml) and the organic layer washed with water (3 X 20ml) and brine, dried over Na2S04, and directly used in the next step. b)
Preparation of (3aJ/?,4J/?,6iS’,6aiS -methyl-4-(2,3-bis(ieri-butoxycarbonyl)-guan idine)-3 -(pentan-3 -yl)-4,5,6,6a-tetrahy dro-3 aH-cy clopenta [d] is oxazole-6- carboxylate (15)
[0068] 13 (from example 1, 9.2g, 0.024 mol) was dissolved in toluene (100 mL) and triethylamine (10. Og, 0.099 mol) and the reaction mixture was heated to 60-70 °C. 2- Ethyl-N-hydroxylbutanimidoyl chloride 14 (from example 2a, 14.8 g, 0.099 mol) in toluene (40 mL) was added to the above solution. A white solid
(triethylammonium chloride) was formed. After addition, the reaction was
monitored for completion by TLC ( PE: EtOAc=3 : 1. The reaction mixture was cooled to 20-25 °C, the precipitate was removed by filtration and the filter cake was washed with toluene (50 g). The organic filtrate was washed with water, brine, and dried over anhydrous Na2S04. The mixture was filtered and concentrated by rotary evaporation. The resulting residue was purified by silica gel flash column
chromatography using PE/EtOAc (30 : 1-4: 1, v/v) to give 15 as a white solid.
[0069] Yield : 10.0 g (85%).
[0070] MS (M+l ) :497.
[0071] ‘H NMR (400 MHz, CDC13) δ 11.29 (s, 1H), 8.55 (d, J= 6.4 Hz, 1H), 5.30 (dd, J= 9.1, 1.5 Hz, 1H), 4.53 (d, J= 4.8 Hz, 1H), 3.78 (s, 3H), 3.70 (d, J = 9.1 Hz, 1H), 3.25 (t, J= 5.4 Hz, 1H), 2.93 – 2.84 (m, 1H), 2.20 (dd, J= 7.6, 3.7 Hz, 2H), 1.87 – 1.60 (m, 4H), 1.49 (d, J= 5.0 Hz, 18H), 0.95 (t, J= 7.4 Hz, 3H), 0.87 (t, J= 7.5 Hz, 3H). (See attached Chart 2)
Example 3 3. (1^,2^,3^,4^)-ιη6ίΗγ1-3-(1-306ίαιηΐάο-2-6ίΗγ1 υίγ1)-4-(2,3- Ϊ8(ί^ί- butoxycarb -onyl) guanidino)-2-hydroxycyclopentanecarboxylate (16):

[0072] Compound 15 ( from example 2, 5.0 g, 10.08 mmol) and nickel chloride hexahydrate (2.5g, 10.5 mmol) were dissolved in methanol (40 mL). The green solution was cooled to -15 °C, while a suspension formed. Sodium borohydride
( 0.456 g, 12 mmol) was added to the reaction mixture at -10 to -5 °C (reaction is highly exothermic). A black suspension was formed along with gas evolution.
After complete addition of the sodium borohydride solution, the reaction mixture was stirred until TLC showed 15 was fully consumed. A solution of acetic
anhydride ( 15g, 0.13 mol) was added slowly and maintained the reaction temperature at 0-5 °C, the reaction mixture was stirred for 2-12 h at 0 °C (The black solution change into green solution ), The pH of the mixture was adjusted to ~9 by addition of 25% aq. ammonium hydroxide. The mixture was concentrated by rotary evaporator. The resulting residue was diluted with water (30 mL) and extracted with EtOAc (50 mL><3). The combined organic extracts were washed with water and brine and dried over anhydrous Na2S04. The mixture was filtered and concentrated by rotary evaporation. The residue was purified by flash chromatography using DCM/Methanol (100:0 to 100:2, v/v) to give 16 as a white solid. [0073] Yield: 3.8 g (71%).
[0074] MS (M+l ) : 543.
[0075] ‘H NMR (400 MHz, CDC13) δ 11.39 (s, 1H), 8.72 (d, J= 9.9 Hz, 1H), 8.59 (d, J= 8.5 Hz, 1H), 4.53 – 4.39 (m, 1H), 4.26 (d, J= 16.4 Hz, 2H), 3.96 (t, J = 10.2 Hz, 1H), 3.71 (s, 3H), 2.90 – 2.75 (m, 1H), 2.53 (dt, J= 13.6, 8.8 Hz, 1H), 2.10 (s, 3H), 2.03 (d, J= 6.3 Hz, 1H), 1.90 – 1.76 (m, 1H), 1.38 (dd, J= 73.9, 7.9 Hz, 18H), 1.25 (ddd, J= 15.2, 13.1, 7.3 Hz, 4H), 0.79 (t, J= 7.3 Hz, 3H), 0.75 (dd, J = 14.1, 6.9 Hz, 3H). (See attached Chart 3)
Example 4
4. (1^,2^,3^,4^)-3-(1-306ίαιηΐάο-2-6ίΗγ1 υίγ1)-4-(2,3- Ϊ8(ί^ί- butoxycarbonyl)gu -anidino)-2-hydroxycyclopentanecarboxylic acid (17)

[0076] To a mixture of compound 16 ( from example 3, 2.0 g, 3.69 mmol) in MeOH/THF (1 : 1, v/v, 12 mL), was added aq. NaOH (IN, 7 mL) at room
temperature. After completion of the reaction (monitored by TLC,
DCM:MeOH=10: l, the mixture was concentrated by rotary evaporation. The resulting solution was neutralized to pH 7 using ice-cold 1 N HCl aq. solution and quickly extracted with EtOAc twice. The combined organic extracts were washed with water, brine, and dried over anhydrous Na2S04. The mixture was filtered and the filtrate was concentrated by rotary evaporation. The resulting white foam was washed triturated by hexane, filtered, dried to give 17 as a white solid
[0077] Yield: 1.6 g (84%).
[0078] MS (M+l ) : 529 o
[0079] ‘H NMR (400 MHz, CDC13) δ 11.41 (s, 1H), 8.80 (d, J= 9.8 Hz, 1H), 8.62 (d, J= 8.3 Hz, 1H), 4.43 (dd, J= 23.3, 14.3 Hz, 2H), 4.00 (t, J= 9.8 Hz, 1H), 2.83 (s, 1H), 2.53 (dt, J= 16.9, 8.4 Hz, 1H), 2.14 (s, 3H), 1.91 (dd, J= 12.5, 6.0 Hz, 1H), 1.46 (dd, J = 30.1, 9.5 Hz, 18H), 1.47 – 1.14 (m, 6H), 0.97 – 0.69 (m, 6H). (See attached Chart 4)
Example 5
5. (1^,2^,3^,4^)-3-(1-306ίαιηΐάο-2-6ίΗγ1 υίγ1)-4^υαηΐ(1ΐηο-2- hydroxycyclopent -anecarboxylic acid (Peramivir I)

[0080] Compound 17 ( from example 4, 1.1 g, 2 mmol) was dissolved in aq. HC1 ( 6N, 6 mL, 36 mmol) at 0 °C. The mixture was stirred at room temperature overnight. The resulting solution was neutralized to pH 6-7 using ice-cold 1 N NaOH aq. solution. The mixture was concentrated to 1.5 ml by rotary evaporation. To this, methanol (20 mL) was added. The precipitate was filtered, and the filtrate was concentrated. The resulting white solid was recrystallized from
methanol/water (1 : 1, v/v) to give Peramivir I as a white solid.
[0081] Yield: 500 mg (73%).
[0082] MS (M+l ) : 329.
[0083] H NMR (400 MHz, D20) δ 4.21 (d, J= 10.6 Hz, 2H), 3.70 (dd, J= 14.6, 9.0 Hz, 1H), 2.57 (d, J= 4.8 Hz, 1H), 2.40 (dt, J= 17.7, 8.9 Hz, 1H), 2.14 – 2.01 (m, 1H), 1.81 (s, 3H), 1.75 – 1.58 (m, 1H), 1.31 (s, 3H), 0.78 (ddd, J= 21.6, 18.6, 6.8 Hz, 8H). (See attached Chart 5)
SYN

Method of synthesis
i. Lactan is dissolved I alcoholic solution, esterification by ring opening under HCl and acid catalysis.
ii. Compound formed is reacted with hydrochloride and tert-butyl dicarbonate.
iii. Reaction with oxammonium hydrochloride reactant salt in alcohol-carbonate system.
iv. Cycloaddition reaction takes place under base catalysis.
v. Hydrogenation in alcoholic solution to generate intermediate compound.
vi. Taking off BOC protect hydrochloride
vii. Two guanidine radicals are put in the reaction vessel, system made of alcohol, organic solvent, aqueous sodium hydroxide solution.
viii. Taking off BOC, product is obtained under organic acid.
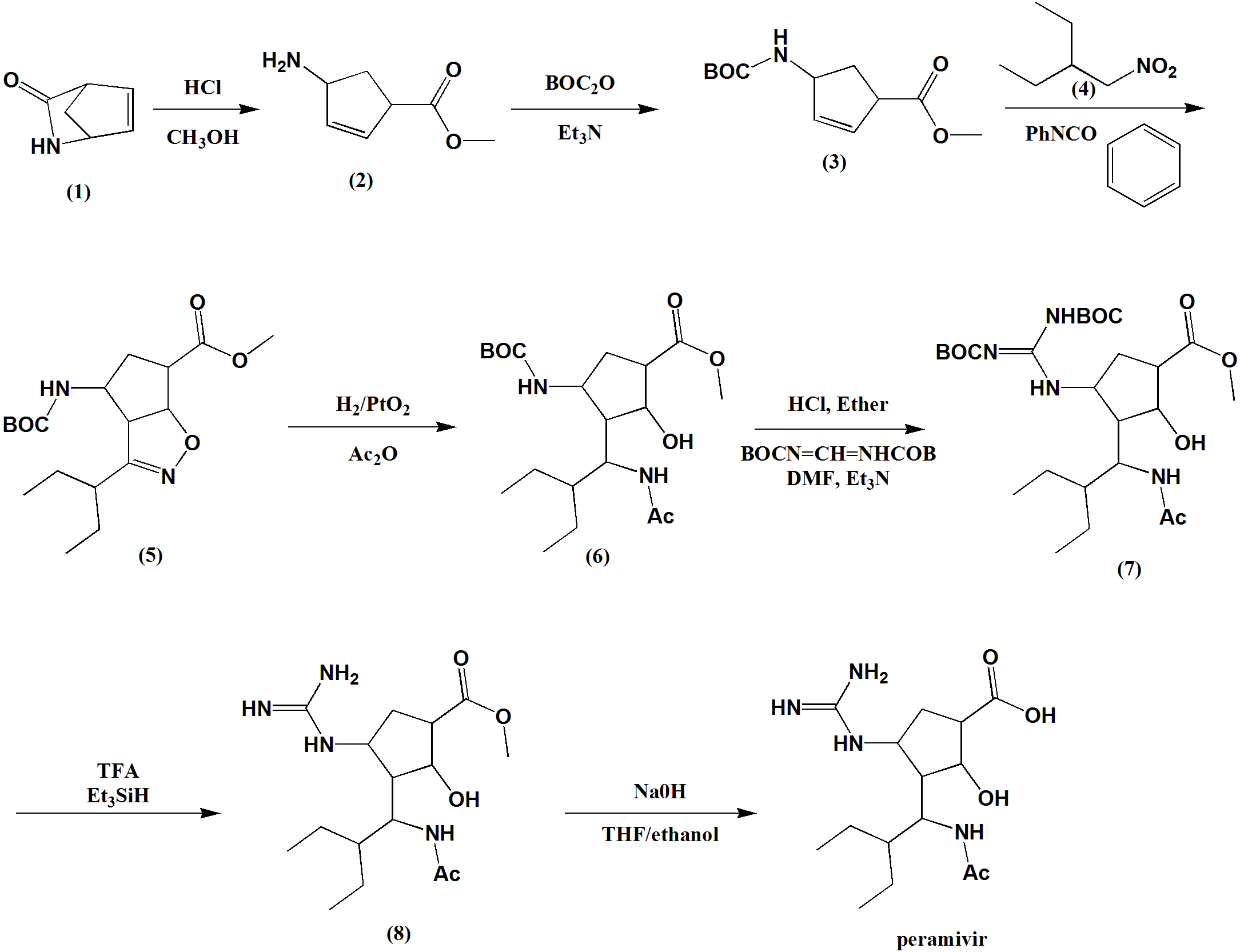
SYN
https://www.tandfonline.com/doi/abs/10.1080/00397911.2012.729279?journalCode=lsyc20
Abstract
An improved and convenient synthetic route for the synthesis of peramivir has been developed with a total 34% yield. The process was improved from previous methods in three key reaction steps including 1,3-dipolar cycloaddition, reductive ring cleavage of the isoxazoline, and incorporation of the peripheral guanidino group. First, an activated sodium hypochlorite (Cl% = 10%) was employed for the catalytic 1,3-dipolar cycloaddition, and 61–68% yields were obtained. Second, the NaBH4-NiCl2 was used as a new reducing reagent instead of the expensive catalyst PtO2. Most important, an innovative and environmentally friendly method of guanylation in the final step was developed using chloroformamidine hydrochloride as the amidino reagent, which avoided the use of the highlytoxic reagent of HgCl2 and made the process greener.
Supplemental materials are available for this article. Go to the publisher’s online edition of Synthetic Communications® to view the free supplemental file.
GRAPHICAL ABSTRACT


Peramivir – Synthetic Route 1

Synthetic Reference
Chand, Pooran; Kotian, Pravin L.; Dehghani, Ali; El-Kattan, Yahya; Lin, Tsu-Hsing; Hutchison, Tracy L.; Babu, Y. Sudhakar; Bantia, Shanta; Elliott, Arthur J.; Montgomery, John A. Systematic Structure-Based Design and Stereoselective Synthesis of Novel Multi-Substituted Cyclopentane Derivatives with Potent Anti-influenza Activity. Journal of Medicinal Chemistry. Volume 44. Issue 25. Pages 4379-4392. Journal. (2001).
Synthetic Reference
Jia, Fei; Hong, Juan; Sun, Ping-Hua; Chen, Jian-Xin; Chen, Wei-Min. Facile synthesis of the neuraminidase inhibitor peramivir. Synthetic Communications. Volume 43. Issue 19. Pages 2641-2647. Journal; Online Computer File. (2013).
Peramivir – Synthetic Route 3

Peramivir – Synthetic Route 4

Synthetic Reference
Li, Mingjie; Zhu, Quanming; Wang, Chunyan. A process for preparing peramivir. Assignee Shandong Luoxin Pharmacy Stock Co., Ltd., Peop. Rep. China. CN 103524383. (2014).
Synthetic Reference
Ge, Min; Li, Liang; Hu, Chunchen; Wang, Huaiqiu; Fu, Mingwei; Li, Lingchao. Synthesis of Peramivir via 3+2 ring closure. Assignee Acesys Pharmatech Ltd., Peop. Rep. China. CN 108997171. (2018).
SYN 1
WO 9933781
Ring opening of cis-2-azabicyclo[2.2.1]hept-5-en-3-one (I) with HCl in refluxing methanol gives cis-4-amino-2-cyclopentene-1-carboxylic acid (II), which is esterified with HCl/methanol, yielding the methyl ester (III). The protection of (III) with tert-butoxycarbonyl anhydride and triethylamine in dichloromethane affords carbamate (IV), which is cyclized with 2-ethyl-1-nitrobutane (V) by means of phenyl isocyanate in refluxing benzene to give the cyclopenta-oxazoline (VI). Ring opening of (VI) by hydrogenation with H2 over PtO2 in HCl/methanol yields amine (VII), which is acylated with acetic anhydride and triethylamine in dichloromethane to afford acetamide (VIII). The deprotection of (VIII) with HCl in ethyl ether gives the cyclopentylamine (IX), which is condensed with N,N’-bis(tert-butoxycarbonyl)-O-methylisourea (X) by means of HgCl2 in DMF to yield the protected guanidino compound (XI). Deprotection of the guanidino group of (XI) with trifluoroacetic acid in dichloromethane affords the methyl ester (XII), which is finally hydrolyzed with NaOH in THF/methanol.
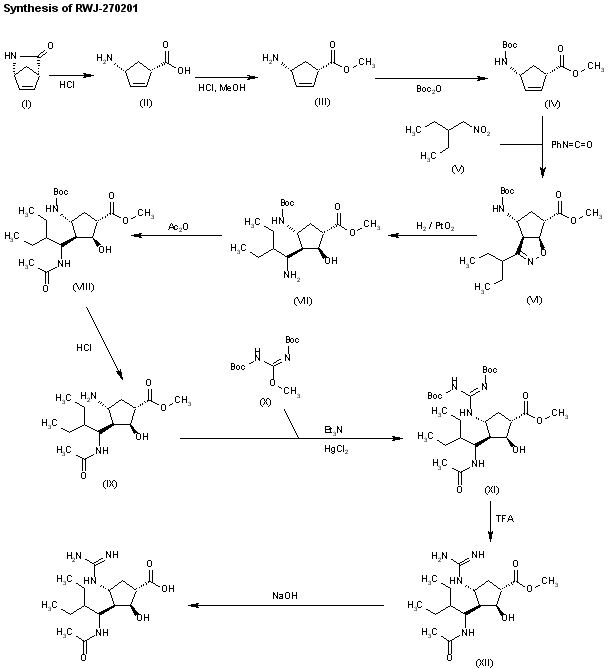
SYN 2
J Med Chem 2000,43(19),3482
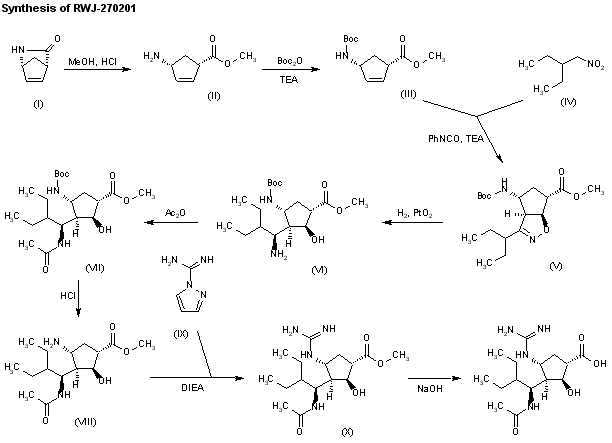
The stereospecific synthesis of BCX-1812(RWJ-270201) has been reported: Ring opening of (-)-2-azabicyclo[2.2.1]hept-5-en-3-one (I) with methanolic HCl gives the methyl ester (II), which is N-protected with Boc2O and TEA yielding the carbamate (III). Cyclization of (III) with 2-ethyl-1-nitrobutane (IV) by means of phenyl isocyanate and TEA affords the bicyclic compound (V) and other isomers. Compound (V) is isolated from the mixture and then hydrogenated with H2 over PtO2 in MeOH and a catalytic amount of HCl to provide amine (VI). Reaction of (VI) with acetic anhydride gives the acetamide (VII), which is Boc-deprotected with ethereal HCl yielding amine (VIII). The guanylation of (VIII) with pyrazolecarboxamidine hydrochloride (IX) and DIEA affords the guanidino derivative (X), which is finally hydrolyzed with NaOH to the desired (1S,2S,3R,4R,1’S)-diastereomer.
SYN 3
J Med Chem 2001,44(25),4379
The ring opening of (-)-2-azabicyclo [2,2,1]hept-5-en-3-one (I) with methanolic HCl gives the methyl ester (II), which is N-protected with Boc2O and TEA, yielding the carbamate (III). The cyclization of (III) with 2-ethyl-1-nitrobutane (IV) by means of phenyl isocyanate and TEA affords the bicyclic compound (V) along with other isomers that are separated by column chromatography. Compound (V) is hydrogenated with H2 over PtO2 in methanol to provide amine (VI), which is acylated with Ac2O, giving the acetamide (VII). N-Boc deprotection of (VII) by means of HCl in ethyl ether yields the amine (VIII), which is condensed with protected isothiourea (IX) by means of HgCl2 to afford the guanidine derivative (X). The hydrolysis of (X) with NaOH provides the carboxylic acid (XI), which is finally deprotected with trifluoroacetic acid to yield the target cyclopentanecarboxylic acid derivative.

References
- ^ Jump up to:a b c “Drug Approval Package: Rapivab (peramivir) Injection NDA #206426”. U.S. Food and Drug Administration (FDA). 16 January 2015. Retrieved 11 February 2020.
- ^ Thorlund K, Awad T, Boivin G, Thabane L (May 2011). “Systematic review of influenza resistance to the neuraminidase inhibitors”. BMC Infectious Diseases. 11 (1): 134. doi:10.1186/1471-2334-11-134. PMC 3123567. PMID 21592407.
- ^ “Peramivir authorized for Emergency use”. LifeHugger. 2009-12-04. Archived from the original on 2011-07-13. Retrieved 2009-12-04.
- ^ “Life-Saving H1N1 Drug Unavailable to Most”. CBS Evening News. Atlanta, GA, USA: CBS Interactive. 2009-10-19. Retrieved 2009-10-20.
- ^ “Emergency Use Authorization Granted For BioCryst’s Peramivir”. Reuters. 2009-10-24.
- ^ “Feds hand BioCryst $77M for anti-viral trial”. Fierce biotech. September 21, 2009.
- ^ “FDA Authorizes Emergency Use of Intravenous Antiviral Peramivir for 2009 H1N1 Influenza for Certain Patients, Settings”. Reuters. 2009-10-24.
- ^ Kohno S, Yen MY, Cheong HJ, Hirotsu N, Ishida T, Kadota J, et al. (November 2011). “Phase III randomized, double-blind study comparing single-dose intravenous peramivir with oral oseltamivir in patients with seasonal influenza virus infection”. Antimicrobial Agents and Chemotherapy. 55 (11): 5267–76. doi:10.1128/AAC.00360-11. PMC 3195028. PMID 21825298.
- ^ “BioCryst scraps $235M late-stage flu drug program backed by feds”. Fierce Biotech. November 8, 2012.
- ^ “BioCryst to File Peramivir NDA Supported by BARDA/HHS Funding”. Fierce Biotech. July 11, 2013.
- ^ Hurt, A.C. (9 June 2011). “Increased detection in Australia and Singapore of a novel influenza A(H1N1)2009 variant with reduced oseltamivir and zanamivir sensitivity due to a S247N neuraminidase mutation”. Eurosurveillance.
- ^ Hirschler, Ben (2011-06-10). “Swine flu starting to show resistance to drugs”. Reuters.
- ^ McKimm-Breschkin JL (January 2013). “Influenza neuraminidase inhibitors: antiviral action and mechanisms of resistance”. Influenza and Other Respiratory Viruses. 7 Suppl 1: 25–36. doi:10.1111/irv.12047. PMC 4942987. PMID 23279894.
- ^ Leang SK, Kwok S, Sullivan SG, Maurer-Stroh S, Kelso A, Barr IG, Hurt AC (March 2014). “Peramivir and laninamivir susceptibility of circulating influenza A and B viruses”. Influenza and Other Respiratory Viruses. 8 (2): 135–9. doi:10.1111/irv.12187. PMC 4186459. PMID 24734292.
- ^ “BioCryst Files Peramivir NDA for the Treatment of Influenza”(Press release). BioCryst Pharmaceuticals. 2013-12-20.
- ^ “Rapivab: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 11 February 2020.
External links
- “Peramivir”. Drug Information Portal. U.S. National Library of Medicine.
- “Peramivir: Requirements for Administration under EUA”. Lifehugger. Archived from the original on 2011-07-13.
- “CDC H1N1 Flu – Termination of the Emergency Use Authorization (EUA) of Medical Products and Devices”. U.S. Centers for Disease Control and Prevention (CDC).
- Clinical trial number NCT00957996 for “Safety Study of IV Peramivir in Hospitalized Subjects With Confirmed or Suspected Influenza” at ClinicalTrials.gov
- “A Novel Anti-viral Drug for Influenza, RAPIACTA Launch” (PDF). Shionogi & Co., Ltd. January 26, 2010. Archived from the original (PDF) on 2013-05-22.
 |
|
| Clinical data | |
|---|---|
| Trade names | Rapivab |
| AHFS/Drugs.com | Monograph |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Intravenous |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 100% (IV) |
| Elimination half-life | 7.7 to 20.8 hours (in patients with normal renal function) |
| Excretion | Kidney |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII |
|
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C15H28N4O4 |
| Molar mass | 328.413 g·mol−1 |
| 3D model (JSmol) | |
| |
|
| Influenza (Flu) |
|---|
 |
////////////////PERAMIVIR, RWJ 270201, RWJ-270201, RWJ270201M, BCX 1812, RWJ 270201
/////////////////

PERAMIVIR
(1S,2S,3S,4R)-3-[(1S)-1-acetamido-2-ethyl-butyl]-4- (diaminomethylideneamino)-2-hydroxy-cyclopentane- 1-carboxylic acid
8th, APRIL 2013
In the wake of the growing bird flu outbreak, China’s SFDA will fast-track approvals of a flu treatment – not a vaccine – known as peramivir (peramivir sodium chloride injection). The drug is a neuraminidase inhibitor, which works by preventing the virus from moving from an infected cell to infect other cells. Guangzhou Nanxin Pharma has been approved to produce the medicine, and it expects to begin distributing the drug in 30 days. Other China pharmas have also filed for approval to make the drug or conduct human clinical trials
Peramivir is an experimental antiviral drug developed by BioCryst Pharmaceuticals for the treatment of influenza. It has been authorized for the emergency use of treatment of certain hospitalized patients with known or suspected 2009 H1N1 influenza.[1]
Peramivir is a neuraminidase inhibitor, acting as a transition-state analogue inhibitor of influenza neuraminidase and thereby preventing new viruses from emerging from infected cells.
The development of peramivir is supported by the US Department of Health and Human Services as part of the US government’s effort to prepare against the threat of an influenza pandemic.[2]
Peramivir is already available in Japan as RAPIACTA (R) and also available in South Korea as PERAMIFLU. Peramivir is currently the only intravenous option for treating swine flu. The drug is in Phase III studies in US.[3][4]
Use in treating Influenza A (H1N1) “Swine Flu”
In October 2009, it was reported that the experimental antiviral drug peramivir had been effective in treating serious cases of swine flu.[5] On October 23, the U.S. Food and Drug Administration (FDA) issued an Emergency Use Authorization for Peramivir, allowing the use of the drug in intravenous form for hospitalized patients only in cases where the other available methods of treatment are ineffective or unavailable;[6] for instance, if oseltamivir (Tamiflu) resistance develops and a person is unable to take Relenza via the inhaled route. The Emergency Use Authorization expired on June 23, 2010.
Biocryst also donated 1200 courses of treatment to the US department of Health and Human Services.[7]
According to a research report published in June 2011, a novel variant of swine flu has emerged in Asia with a genetic adaptation giving some resistance to Roche’s (ROG.VX) Tamiflu and GlaxoSmithKline’s (GSK.L) Relenza, the two mainstay drugs used to tackle the disease. There was no significant reduction in sensitivity to Peramivir. [8]
Initial treatment courses are for 5 to 10 days duration. Treatment beyond 10 days is permitted depending on clinical presentation such as critical illness (e.g., respiratory failure or intensive care unit admission), continued viral shedding or unresolved clinical influenza illness.[9]
- “Peramivir authorized for Emergency use”. LifeHugger. 2009-12-04. Retrieved 2009-12-04.
- “HHS Pursues Advance Development of New Influenza Antiviral Drug” (Press release). USDepartment of Health and Human Services. 2007-01-04. Retrieved 2007-05-25.
- “Evaluation of the Efficacy and Safety of Peramivir in Subjects With Uncomplicated Acute Influenza”.National Institutes of Health. 2007-03-16. Retrieved 2007-05-25.
- “Evaluation of the Efficacy and Safety of Peramivir in Adults With Acute Serious or Potentially Life-Threatening Influenza”. National Institutes of Health. 2007-03-28. Retrieved 2007-05-25.
- “Life-Saving H1N1 Drug Unavailable to Most”. CBS Evening News (Atlanta, GA, USA: CBS Interactive). 2009-10-19. Retrieved 2009-10-20.
- “Emergency Use Authorization Granted For BioCryst’s Peramivir”. Reuters. 2009-10-24.
- “FDA Authorizes Emergency Use of Intravenous Antiviral Peramivir for 2009 H1N1 Influenza for Certain Patients, Settings”. Reuters. 2009-10-24.
- Hirschler, Ben (2011-06-10). “Swine flu starting to show resistance to drugs”. Reuters.
- “Peramivir: Dosage and Administration”. LifeHugger. 2009-12-04. Retrieved 2009-12-04.
TLC388 (Lipotecan®) Taiwan Liposome Company Hepatic cancer drug candidate gets fast track approval status from SFDA
TLC388 (Lipotecan®) structure can be figured out from a link below of a poster
http://www.tlcbio.com/files/news/2011111701580783.pdf
IT IS A CAMPOTHECIN ANALOGUE
The str can be concluded from above picture from a poster by TLC BIO
TLC388 (Lipotecan) is a potent Topoisomerase-1 inhibitor and it can disrupt both Sonic Hedgehog and HIF1-α pathways to overcome cancer drug resistance and inhibit angiogenesis induced by tumor hypoxia. This phase I first-in-human study of Lipotecan examined the MTD, safety, anti-tumor activity and pharmacokinetic profiles of TLC388 in patients with advanced incurable solid tumors.
Methods: Lipotecan was administered intravenously on day 1, 8 and 15 of a 28-day cycle. Patients underwent safety assessments regularly and tumor assessments every other cycle. Pharmacokinetic samples were drawn on days 1, 8 and 15 of cycles 1 and 2 for all treated patients.
http://mct.aacrjournals.org/cgi/content/meeting_abstract/10/11_MeetingAbstracts/A89
http://clinicaltrials.gov/show/NCT00747474
MAR19 2013
China SFDA has granted fast track approval status to Taiwan Liposome company hepatic cancer drug Lipotecan, shortening the review period. The drug will enter Phase 2 clinical trials in China in the second half of this year. Lipotecan has been granted orphan drug status by US FDA and EU EMEA for the treatment of hepatocellular carcinoma (HCC)
Nexavar is the standard of care in first line advanced liver cancer patients. Lipotecan as a second-line treatment allows patients who have failed prior treatment with Nexavar to maintain a six month course of the disease without progressing
Pfizer Gains China Approval of Kinase-Specific Lung Cancer Drug, Xalkori (crizotinib)

Xalkori, crizotinib,
(PF-02341066)
3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine
Crizotinib; 877399-52-5; Xalkori; PF-2341066; PF-02341066; (R)-crizotinib; 877399-52-5
| Molecular Formula: | C21H22Cl2FN5O |
|---|---|
| Molecular Weight: | 450.336683 g/mol |
Crizotinib an inhibitor of receptor tyrosine kinase for the treatment of non-small cell lung cancer (NSCLC). Verification of the presence of ALK fusion gene is done by Abbott Molecular’s Vysis ALK Break Apart FISH Probe Kit. This verification is used to select for patients suitable for treatment. FDA approved in August 26, 2011.
Crizotinib (1), an anaplastic lymphoma kinase (ALK) receptor tyrosine kinase inhibitor approved by the U.S. Food and Drug Administration in 2011, is efficacious in ALK and ROS positive patients
Feb 25, 2013
Pfizer has been granted China approval for Xalkori (crizotinib), an innovative treatment for patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) that is anaplastic lymphoma kinase (ALK) positive. The ALK-positive variation, which comprises between 3% and 5% of all NSCLC tumors, must be proved by a biomarker test. Pfizer said China’s approval came just eleven months after it submitted a new drug application to the SFDA for Xalkori
Crizotinib (trade name Xalkori,[1] Pfizer), is an anti-cancer drug acting as an ALK (anaplastic lymphoma kinase) and ROS1 (c-ros oncogene 1) inhibitor, approved for treatment of some non-small cell lung carcinoma (NSCLC) in the US and some other countries, and undergoing clinical trials testing its safety and efficacy in anaplastic large cell lymphoma, neuroblastoma, and other advanced solid tumors in both adults and children.[2]
- FDA approves Xalkori with companion diagnostic for a type of late-stage lung cancer. U.S. Food and Drug Administration.http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm269856.htm
- ClinicalTrials.gov NCT00932451 An Investigational Drug, PF-02341066, Is Being Studied In Patients With Advanced Non-Small Cell Lung Cancer With A Specific Gene Profile Involving The Anaplastic Lymphoma Kinase (ALK) Gene
Crizotinib the core structure is a substituted pyridine, the 3 – position of the ether as a chiral center adjacent, so with Mitsunobu reaction to complete, as is a typical Mitsunobu SN2 reaction, the reaction chiral center occurs in reverse, so easy to control, no racemization occurs. Pyridine substituted at position 5 by Suzuki reaction constructed.
Compound 1 The activation of the hydroxyl groups of methanesulfonyl chloride, and then with a 4 – iodopyrazole reaction 2 , 2 to 4 Suzuki reaction conversion can be used, but will generate a large quantity of the reaction product of their coupling, the first 2 converted to a Grignard reagent, and then with a boronic acid ester of 3 reaction 4 .

……………………
http://www.specchemonline.com/articles/view/biocatalyst-breakthroughs#.VTcW9yxabEs
http://www.google.com/patents/WO2014020467A2?cl=en
(R)-3-[l-(2,6-Dichloro-3-fluoro-phenyl)-ethoxy]-5-(l-piperidin-4-yl-lH-py- razol-4-yl)-pyridin-2-ylamine, also known as Crizotinib, is represented by the Formula (I):

Formula (I)
Crizotinib is a potent small-molecule inhibitor of c-Met/HGFR (hepatocyte growth factor receptor) kinase and ALK (anaplastic lymphoma kinase) activity. Enantiomerically pure compound of formula I was first disclosed in US Patent No. 7,858,643. Additionally, the racemate of compound of formula I was disclosed in U.S. patent application 2006/0128724, both of these references discloses similar methods for the synthesis of Compound of Formula I.
Conventionally, the compounds of formula I are prepared by reacting Bis(pinacolato)diboron with protected 5-bromo-3-[l-(2,6-dichloro-3-fluoro-phenyl)-ethoxy]-pyridin-2-ylamine in the presence of Pd catalyst. The obtained product after deprotection is reacted with N- protected 4-(4-bromo-pyrazol-l-yl)-piperidine in the presence of Pd Catalyst. The obtained product is filtered through celite pad and purified by Column Chromatography. The final product of formula I was obtained by deprotection of the purified compound by using HCl/dioxane. US Patent No. 7,858,643 provides enantiomerically pure aminoheteroaryl compounds, particularly aminopyridines and aminopyrazines, having protein tyrosine kinase activity. More particularly, US 7,858,643 describes process for the preparation of 3-[(lR)-l-(2,6- dichloro-3-fluorophenyl)ethoxy]-5-(l-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine. The Scheme is summarized below in Scheme- 1 :


Scheme-1
wherein, “Boc” means tert-butoxycarbonyl; and a) (Boc)2, DMF, Dimethylaminopyridine b) Pd(dppf)Cl2, KOAc, Dichloromethane; c) HC1, Dioxane, Dichloromethane; d) Pd(PPh3)2Cl2, Na2C03, DME/H20; e) 4M HCl/Dioxane, Dichloromethane
A similar process has been disclosed in the U.S. patent application 2006/0128724 for the preparation of Crizotinib. J. Jean Cui et. al. in J. Med. Chem. 2011, 54, 6342-6363, also provides a similar process for the preparation of Crizotinib and its derivatives.
However, above mentioned synthetic process requires stringent operational conditions such as filtration at several steps through celite pad. Also column chromatography is required at various steps which is not only tedious but also results in significant yield loss. Another disadvantage of above process involves extensive use of palladium catalysts, hence metal scavengers are required to remove palladium content from the desired product at various steps which makes this process inefficient for commercial scale.
Yet another disadvantage of above process is the cost of Bis(pinacolato)diboron. This reagent is used in excess in the reaction mixture resulting in considerable cost, especially during large-scale syntheses.
US Patent No. 7,825,137 also discloses a process for the preparation of Crizotinib where Boc protected 4-(4-iodo-pyrazol-l-yl)-piperidine is first reacted with Bis(pinacolato)diboron in the presence of Pd catalyst. The reaction mixture is filtered through a bed of celite and the obtained filtrate is concentrated and purified by silica gel chromatography to give to form tert-butyl-4-[4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl]piperidine-l- carboxylate. To this compound, 5-bromo-3-[l-(2,6-dichloro-3-fluoro-phenyl)-ethoxy]- pyridin-2-ylamine is added in the presence of a Pd catalyst. The reaction mixture is stirred for 16h at 87°C. The reaction mixture is filtered through celite pad and the concentrated filtrate is purified on silica gel column to obtain (4-{6-amino-5-[(R)-l-(2,6-dichloro-3-fluoro- phenyl)-ethoxy]-pyri- din-3-yl}-pyrazol-l-yl)-piperidine-l-carboxylic acid tert-butyl ester of 95% purity. To the solution of resulting compound in dichloromethane 4N HCl/Dioxane is added and thereby getting the reaction suspension is filtered in Buchner funnel lined with filter paper. The obtained solid is dissolved in HPLC water and pH is adjusted to 10 with the addition of Na2C03 Compound is extracted using dichloroform and is purified on a silica gel column by eluting with CH2Cl2 MeOH/NEt3 system to obtain Crizotinib. The scheme is summarized below in scheme 2:

Formula (i) Formula (ii)

Formula (iii) Formula (ii) ula (iv)

Formula (v) Formula (I)
Scheme-2
Preparation of Crizotinib:
To a stirred solution of Tert-butyl 4-(4-{ 6-amino-5-[(li?)-l-(2,6-dichloro-3- fluorophenyl)ethoxy]pyridin-3 -yl } – lH-pyrazol- 1 -yl)piperidine- 1 -carboxylate (material obtained in Example 3) (l.Og, 0.00181 moles) in dichloromethane (-13 ml) at 0°C was added 4.0 M dioxane HQ (6.7 ml, 0.0272 moles). Reaction mixture was stirred at room temperature for 4h. After the completion of reaction monitored by TLC, solid was filtered and washed with dichloromethane (10 ml). The obtained solid was dissolved in water (20 ml); aqueous layer was extracted with dichloromethane (10×2). The pH of aqueous layer was adjusted to 9-10 with Na2C03 and compound was extracted with dichloromethane (10 x 3), combined organic layers were washed with water (20 ml), evaporated under vacuum to get solid product. The solid was stirred with ether (10 ml), filtered off, washed well with ether, dried under vacuum to get Crizotinib.
Yield: 0.45g (55 %)
HPLC Purity: 99.35 %
1HNMR (400 MHz, CDC13) δ: 7.76 (d, J = 1.6 Hz, 1H), 7.56 (s, 1H), 7.49 (s, 1H), 7.30 (dd, J = 9.2 Hz), 7.0 (m, 1H), 6.86 (d, J = 1.6 Hz, 1H), 6.09 ( q, J= 6.8 Hz, 1H), 4.75 (brs, 1H), 4.19 (m, 1H), 3.25 (m, 2H), 2.76 (m, 2H), 2.16 (m, 2H), 1.92 (m, 2H), 1.85 (d, J= 6.8 Hz, 3H), 1.67 (brs, 1H)
…………………………
http://www.sciencedirect.com/science/article/pii/S0040403914000872
Abstract
A novel approach for the synthesis of Crizotinib (1) is described. In addition, new efficient procedures have been developed for the preparation of (S)-1-(2,6-dichloro-3-fluorophenyl)ethanol (2) and tert-butyl 4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)piperidine-1-carboxylate (4), the key intermediates required for the synthesis of Crizotinib.
Graphical abstract

- …………………
- http://www.sciencedirect.com/science/article/pii/S0040403911021745
-
Abstract
4-(4-Iodo-1H-pyrazol-1-yl)piperidine is a key intermediate in the synthesis of Crizotinib. We report a robust three-step synthesis that has successfully delivered multi-kilogram quantities of the key intermediate. The process includes nucleophilic aromatic substitution of 4-chloropyridine with pyrazole, followed by hydrogenation of the pyridine moiety and subsequent iodination of the pyrazole which all required optimization to ensure successful scale-up.
Graphical abstract

……………………

A robust six-step process for the synthesis of crizotinib, a novel c-Met/ALK inhibitor currently in phase III clinical trials, has been developed and used to deliver over 100 kg of API. The process includes a Mitsunobu reaction, a chemoselective reduction of an arylnitro group, and a Suzuki coupling, all of which required optimization to ensure successful scale-up. Conducting the Mitsunobu reaction in toluene and then crystallizing the product from ethanol efficiently purged the reaction byproduct. A chemoselective arylnitro reduction and subsequent bromination reaction afforded the key intermediate 6. A highly selective Suzuki reaction between 6 and pinacol boronate 8, followed by Boc deprotection, completed the synthesis of crizotinib 1.
3-[(1R)-1-(2,6-Dichloro-3-fluorophenyl)ethoxy]-5-[1-(piperidin-4-yl)-1H-pyrazol-4-yl]pyridin-2-amine 1
crizotinib1 (20.7 kg, 80%) as a white solid.
Mp 192 °C;
1H NMR (400 MHz, CDCl3) δ: 7.78 (d, J = 1.8 Hz, 1H), 7.58 (s, 1H), 7.52 (s, 1H), 7.31 (dd, J = 9.0, 4.9 Hz, 1H), 7.06 (m, 1H), 6.89 (d, J = 1.7 Hz, 1H), 6.09 (q, 1H), 4.79 (br s, 2H), 4.21 (m, 1H), 3.26 (m, 2H), 2.78 (m, 2H), 2.17 (m, 2H), 1.90 (m, 2H), 1.87 (d, J = 6.7 Hz, 3H), 1.63 (br s, 1H).
13C NMR (100.6 MHz, CDCl3) δ: 157.5 (d, J = 250.7 Hz), 148.9, 139.8, 137.0, 135.7, 135.6, 129.9, 129.0 (d, J = 3.7 Hz), 122.4, 122.1 (d, J = 19.0 Hz), 119.9, 119.3, 116.7 (d, J = 23.3 Hz), 115.0, 72.4, 59.9, 45.7, 34.0, 18.9.
LC-MS: found m/z 450.0, 451.0, 452.0, 453.0, 454.0, 455.0.
Anal. Calcd for C21H22Cl2FN5O: C, 56.01; H, 4.92; N, 15.55. Found: C, 56.08; H, 4.94; N, 15.80.
Cui, J. J.; Botrous, I.; Shen, H.; Tran-Dube, M. B.; Nambu, M. D.; Kung, P.-P.; Funk, L. A.; Jia, L.; Meng, J. J.; Pairish, M. A.; McTigue, M.; Grodsky, N.; Ryan, K.; Alton, G.; Yamazaki, S.; Zou, H.; Christensen, J. G.; Mroczkowski, B.Abstracts of Papers; 235th ACS National Meeting, New Orleans, LA, United States, April 6–10, 2008.


![3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine NMR spectra analysis, Chemical CAS NO. 877399-52-5 NMR spectral analysis, 3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-29/000/437/336/877399-52-5-1h.png)
![3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine NMR spectra analysis, Chemical CAS NO. 877399-52-5 NMR spectral analysis, 3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-29/000/437/336/877399-52-5-13c.png)
| WO2006021881A2 * | 15 Aug 2005 | 2 Mar 2006 | Pfizer | Pyrazole-substituted aminoheteroaryl compounds as protein kinase inhibitors |
| WO2006021884A2 * | 15 Aug 2005 | 2 Mar 2006 | Pfizer | Enantiomerically pure aminoheteroaryl compounds as protein kinase inhibitors |
| WO2013181251A1 * | 29 May 2013 | 5 Dec 2013 | Ratiopharm Gmbh | Crizotinib hydrochloride salt in crystalline |
| EP2620140A1 * | 26 Jan 2012 | 31 Jul 2013 | ratiopharm GmbH | Crizotinib containing compositions |
-
WO2010048131A1 * Oct 20, 2009 Apr 29, 2010 Vertex Pharmaceuticals Incorporated C-met protein kinase inhibitors WO2011042389A2 * Oct 4, 2010 Apr 14, 2011 Bayer Cropscience Ag Phenylpyri(mi)dinylazoles US7825137 Nov 23, 2006 Nov 2, 2010 Pfizer Inc. Method of treating abnormal cell growth US7858643 Aug 26, 2005 Dec 28, 2010 Agouron Pharmaceuticals, Inc. Crizotinib, a c-Met protein kinase inhibitor anticancer agent; 3-[(R)-1-(2,6-dichloro-3-fluoro-phenyl)-ethoxy]-5-(1-piperidin-4-yl-1H-pyrazol-4-yl)-pyridin-2-ylamine is crizotinib US20060128724 Aug 26, 2005 Jun 15, 2006 Agouron Pharmaceuticals, Inc. Pyrazole-substituted aminoheteroaryl compounds as protein kinase inhibitors 1 J. JEAN CUI J. MED. CHEM. vol. 54, 2011, pages 6342 – 6363 2 ORG. PROCESS RES. DEV. vol. 15, 2011, pages 1018 – 1026 3 * PIETER D. DE KONING ET AL: “Fit-for-Purpose Development of the Enabling Route to Crizotinib (PF-02341066)“, ORGANIC PROCESS RESEARCH & DEVELOPMENT, vol. 15, no. 5, 16 September 2011 (2011-09-16), pages 1018-1026, XP055078841, ISSN: 1083-6160, DOI: 10.1021/op200131n
State Food and Drug Administration, China Grants Approval to Sihuan Pharmaceutical for Clinical Trial of Innovative Drug — Pinoxacin Hydrochloride
HONG KONG, Feb. 22, 2013, Sihuan Pharmaceutical Holdings Group Ltd. a leading pharmaceutical company with the largest cardio-cerebral vascular (“CCV”) drug franchise in China’s prescription market, today announced that Pinoxacin Hydrochloride, a Category 1.1 new drug developed by the Company’s innovative drug research and development team, received Approval for Clinical Studies from the State Food and Drug Administration. Phase I of clinical studies are set to begin in the first half of this year. It is the fourth Category 1 innovative drug for which the Company has received Approval for Clinical Studies.
Pinoxacin Hydrochloride is DPP-4 inhibitor class of oral hypoglycemic agents, a drug with a brand new structure for treating type II diabetes. It is clinically used to enhance the function of endogenous insulin for improving glycemic control, and long-term use can improve islet beta-cells function. Pre-clinical research has shown that DPP-4 inhibitors have potent in vitro and in vivo activities, a good selection profile, great stability and controllable quality, as well as better tolerance, with long-term administration showing a protective effect on pancreatic beta-cells. In addition, the DPP-4 inhibitor will not cause serious side effects such as weight gain and hypoglycaemia seen in traditional diabetes drugs. Pinoxacin Hydrochloride has good pharmacokinetic characteristics, high oral bioavailability, quick absorption, rapid onset and a longer duration. A once daily dosage is expected to keep the patients’ symptoms under control. The advantages of Pinoxacin Hydrochloride have proven the drug’s growth potential present in the market.
Celgene gains SFDA China, approval for marketing Revlimid (lenalidomide) to treat multiple myeloma

(RS)-3-(4-amino-1-oxo 1,3-dihydro-2H-isoindol- 2-yl)piperidine-2,6-dione
Lenalidomide
REVLIMID® is an oral immunomodulatory drug marketed in the United States and many international markets, in combination with dexamethasone, for treatment of patients with multiple myeloma who have received at least one prior therapy. It is also marketed in the United States and certain international markets for the treatment of transfusion-dependent anemia due to low- or intermediate-1-risk myelodysplastic syndromes, or MDS, associated with a deletion 5q cytogenetic abnormality with or without additional cytogenetic abnormalitie.Revlimid Worldwide annual sales in 2011 was $3.2bLenalidomide (Revlimid) is a derivative of thalidomideintroduced in 2004.It was initially intended as a treatment for multiple myeloma, for which thalidomide is an accepted therapeutic treatment. Lenalidomide has also shown efficacy in the class of hematological disorders known as myelodysplastic syndromes (MDS). Lenalidomide has significantly improved overall survival in myeloma (which generally carries a poor prognosis), although toxicity remains an issue for users. [1]It costs $163,381 per year for the average patient.[2]
Mechanism of action
Lenalidomide has been used to successfully treat both inflammatory disorders and cancers in the past 10 years. There are multiple mechanisms of action, and they can be simplified by organizing them as mechanisms of action in vitro and in vivo.[3] In vitro, lenalidomide has three main activities: direct anti-tumor effect, inhibition of the microenvironment support for tumor cells, and immunomodulatory role. In vivo, lenalidomide induces tumor cell apoptosis directly and indirectly by inhibition of bone marrow stromal cell support, by anti-angiogenic and anti-osteoclastogenic effects, and by immunomodulatory activity. Lenalidomide has a broad range of activities that can be exploited to treat many hematologic and solid cancers.
- McCarthy; Philip L. McCarthy, Kouros Owzar, Craig C. Hofmeister, et al. (May 10, 2012). “Lenalidomide after Stem-Cell Transplantation for Multiple Myeloma”. N Engl J Med 366 (19): 1770–1781. doi:10.1056/NEJMoa1114083. PMID 22571201.
- Badros, Ashraf Z. Badros (May 10, 2012). “Lenalidomide in Myeloma — A High-Maintenance Friend”. N Engl J Med 366 (19): 1836–1838. doi:10.1056/NEJMe1202819. PMID 22571206.
- Vallet S, Palumbo A, Raje N, Boccadoro M, Anderson KC (July 2008). “Thalidomide and lenalidomide: Mechanism-based potential drug combinations”. Leukemia & Lymphoma 49 (7): 1238–45. doi:10.1080/10428190802005191. PMID 18452080.
