WO-2015136473
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015136473&redirectedID=true
WOCKHARDT LIMITED [IN/IN]; D-4, MIDC Area, Chikalthana, Aurangabad 431006 (IN)
Our New Drug Discovery team has developed a number of lead molecules, mainly in the area of anti-infectives; these are currently at various stages of development.
Of these molecules, the most advanced of the New Chemical Entities (NCE) is WCK 771, which has commenced Phase II human clinical trials.
WCK 771 is a broad-spectrum antibiotic, which has proven effective in treating diverse staphylococcal infections like MRSA and VISA.
Other lead molecules at various stages of pre-clinical trials are: WCK 2349, WCK 4873 and WCK 4086.
http://www.wockhardt.com/how-we-touch-lives/new-drug-discover.aspx
WO-2015136473
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015136473&redirectedID=true
Process for the synthesis of sodium (2S, 5R)-6-(benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboxylate (disclosed in WO2014135929) is claimed. Used as an intermediate in the synthesis of several antibacterial compounds. For a concurrent filing see WO2015136387, claiming the combination of an antibacterial agent with sulbactam.
In September 2015, Wockhardt’s pipeline lists several antibacterial programs, including WCK-771 and WCK-2349 (both in phase II), WCK-5107 (phase I), and also investigating iv and oral second generation oxazolidinones, WCK-4873, and iv and oral formulation of WCK-4086 (in preclinical stage) for treating the bacterial infection.
For a prior filing see WO2015125031, claiming the combination of an antibacterial agent (eg cefepime or cefpirome) and nitrogen containing bicyclic compound, useful for treating bacterial infection.

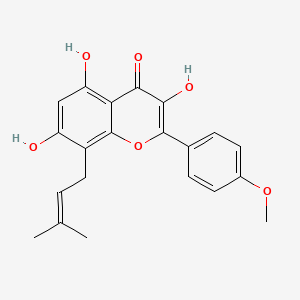
A compound of Formula (I), chemically known as sodium (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate, can be used as an intermediate in the synthesis of several antibacterial compounds and is disclosed in PCT International Patent Application No. PCT/IB2013/059264. The present invention discloses a process for preparation of a compound of Formula (I).

Scheme 1
Example 1
Synthesis of sodium (25, 5R)-6-(benzyloxy)-7-oxo-l,6-diazabicvclor3.2.11octane-2- carboxylate
Step 1; Preparation of -Γl-Γ(feΓt-butyldimethylsilyl -oxymethyll-5-Γdimethyl(oxido -λ-4-sulfanylidenel-4-oxo-pentyll-carbamic acid tert-butyl ester (III):
To a suspension of trimethylsulfoxonium iodide (180.36 gm, 0.819 mol) in tetrahydrofuran (900 ml), sodium hydride (32.89 g, 0.819 mol, 60% in mineral oil) was charged in one portion at 30°C temperature. The reaction mixture was stirred for 15 minutes and then dropwise addition of dimethylsulphoxide (1.125 ml) was done over a period of 3 hours at room temperature to provide a white suspension. The white suspension was added to a pre-cooled a solution of 2-(feri-butyldimethylsilyl-oxymethyl)-5-oxo-pyrrolidine-l-carboxylic acid tert-buty\ ester (II) (225 g, 0.683 mol, prepared as per J. Org Chem.; 2011, 76, 5574 and WO2009067600) in tetrahydrofuran (675 ml) and triethylamine (123.48 ml, 0.887 mol) mixture at -13°C by maintaining the reaction mixture temperature below -10°C. The resulting suspension was stirred for additional 1 hour at -10°C. The reaction mixture was carefully quenched by addition of saturated aqueous ammonium chloride (1.0 L) at -10°C to 10°C. The reaction was extracted by adding ethyl acetate (1.5 L). The layers were separated and aqueous layer was re-extracted with ethyl acetate (500 ml x 3). The combined organic layer was washed successively with saturated aqueous sodium bicarbonate (1.0 L), water (2.0 L) followed by saturated aqueous sodium chloride solution (1.0 L). Organic layer was dried over sodium sulfate and evaporated under vacuum to provide 265 g of 5-[l-[(ieri-butyldimethylsilyl)-oxymethyl]-5-[dimethyl(oxido)- -4-sulfanylidene]-4-oxo-pentyl]-carbamic acid tert-buty\ ester (III) as an yellow oily mass.
Analysis:
Mass: 422.3 (M+l); for Molecular weight: 421.68 and Molecular Formula: 
1H NMR (CDC13): δ 4.77 (br d, 1H), 4.38 (br s, 1H), 3.58 (br s, 3H), 3.39 (s, 3H), 3.38 (s, 3H), 2.17-2.27 (m, 2H), 1.73-1.82 (m, 2H), 1.43 (s, 9H), 0.88 (s, 9H), 0.01 (s, 3H), 0.04 (s, 3H).
Step 2: Preparation of 5-r4-benzyloxyimino-l-(fert-butyldimethylsilyl-oxymethyl)-5-chloro-pentyll-carbamic acid tert- butyl ester (IV):
To a suspension of 5-[l-[(ieri-butyldimethylsilyl)-oxymethyl]-5-[dimethyl(oxido)- -4-sulfanylidene]-4-oxo-pentyl]-carbamic acid tert-butyl ester (III) (440.0 g, 1.045 mol) in tetrahydrofuran (6.6 L), O-benzhydroxylamine hydrochloride (200.0 g, 1.254 mol) was charged. The reaction mixture was heated to 50°C for 2.5 hours. The reaction mixture was filtered through pad of celite and filtrate was concentrated to provide a residue. The residue was dissolved in ethyl acetate (5.0 L) and washed successively with saturated aqueous sodium bicarbonate (1.5 L), water (1.5 L) and saturated aqueous sodium chloride (1.5 L). Organic layer was dried over sodium sulfate. Solvent was evaporated under vacuum to yield 463.0 g of 5-[4-benzyloxyimino-l-(tert-butyldimethylsilyl-oxymethyl)-5-chloro-pentyl]-carbamic acid tert-butyl ester (IV) as an oily mass.
Analysis:
Mass: 486.1 (M+l); for Molecular weight: 485.4 and Molecular Formula: 
1H NMR (CDCI3): δ 7.26-1 6 (m, 5H), 5.10 (s, 2H), 4.66 (br d, 1H), 3.58-4.27 (m, 2H), 3.56-3.58 (m, 3H), 2.40-2.57 (m, 2H), 1.68-1.89 (m, 2H), 1.44 (s, 9H), 0.89 (s, 9H), 0.02 (s, 3H), 0.04 (s, 3H).
Step 3: Preparation of 5-5-benzyloxyimino-2-(fert-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (V):
To a solution of 5-[4-benzyloxyimino-l-(tert-butyldimethylsilyl-oxymethyl)-5-chloro-pentyl]-carbamic acid tert-butyl ester (IV) (463.0 g 0.954 mol) in tetrahydrofuran (6.9 L), was charged potassium feri-butoxide (139.2 g, 1.241 mol) in portions over a period of 30 minutes by maintaining temperature -10°C. The resulting suspension was stirred for additional 1.5 hours at -10°C to -5°C. The reaction mixture was quenched by addition of saturated aqueous ammonium chloride (2.0 L) at -5°C to 10°C. The organic layer was separated and aqueous layer was extracted with ethyl acetate (1.0 L x 2). The combined organic layer was washed with saturated aqueous sodium chloride solution (2.0 L). Organic layer was dried over sodium sulfate, and then evaporated under vacuum to yield 394.0 g of 5-5-benzyloxyimino-2-(ieri-butyldimethylsilyl-oxymethyl)-piperidine- 1 -carboxylic acid tert-butyl ester (V) as an yellow oily mass.
Analysis:
Mass: 449.4 (M+l) for Molecular weight: 448.68 and Molecular Formula: C24H4oN204Si;
1H NMR (CDC13): δ 7.25-1 3 (m, 5H), 5.04-5.14 (m, 2H), 4.35 (br s, 1H), 3.95 (br s, 1H), 3.63-3.74 (br d, 2H), 3.60-3.63 (m, 1H), 2.70-2.77 (m, 1H), 2.33-2.41 (m, 1H), 1.79-1.95 (m, 2H), 1.44 (s, 9H), 0.88 (s, 9H), 0.03 (s, 3H), 0.04 (s, 3H).
Step 4: Preparation of (25,5R5)-5-benzyloxyamino-2-(tert-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (VI):
To a solution of 5-5-benzyloxyimino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (V) (394.0 g, 0.879 mol) in dichloromethane (5.0 L) and glacial acetic acid (788 ml), was charged sodium cyanoborohydride (70.88 g, 1.14 mol) one portion. The resulting reaction mixture was stirred at temperature of about 25 °C to 30°C for 2 hours. The mixture was quenched with adding aqueous solution of sodium bicarbonate (1.3 kg) in water (5.0 L). The organic layer was separated and aqueous layer was extracted with dichloromethane (2.0 L). The combined organic layer washed successively with water (2.0 L), saturated aqueous
sodium chloride (2.0 L) and dried over sodium sulfate. Solvent was evaporated under vacuum to provide a residue. The residue was purified by silica gel column chromatography to yield 208 g of (25,5i?5)-5-benzyloxyamino-2-(ieri-butyldimethylsilyl-oxymethyl)-piperidine- 1 -carboxylic acid tert-buty\ ester (VI) as pale yellow liquid.
Analysis:
Mass: 451.4 (M+l); for Molecular weight: 450.70 and Molecular Formula: C24H42N204Si;
1H NMR (CDC13): δ 7..26-7.36 (m, 5H), 4.90-5.50 (br s, 1H), 4.70 (dd, 2H), 4.09-4.25 (m, 2H), 3.56-3.72 (m, 2H), 2.55-3.14 (m, 2H), 1.21-1.94 (m, 4H), 1.45 (s, 9H), 0.89 (s, 9H), 0.05 (s, 6H).
Step 5: Preparation of (25,5R5)-5-benzyloxyamino-2-(tert-butyldimethylsilyl-oxymethyl)-piperidine (VII):
To a solution of 5-5-benzyloxyamino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (VI) (208 g, 0.462 mol) in dichloromethane (3.0 L), boron trifluoride diethyletherate complex (114.15 ml, 0.924 mol) was charged in one portion. The resulting reaction mixture was stirred at temperature of about 25°C to 35°C temperature for 2 hours. The reaction mixture was quenched with saturated aqueous sodium bicarbonate (2.0 L). The organic layer was separated and aqueous layer was extracted with dichloromethane (1.5 L x 2). The combined organic layer was washed with saturated aqueous sodium chloride (1.0 L) and dried over sodium sulfate. Solvent was evaporated under vacuum to yield 159 g of (25,5i?5)-5-benzyloxyamino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine (VII) as a yellowish syrup.
Analysis:
Mass: 351.3 (M+l); for Molecular weight: 350.58 and Molecular Formula: C19H34N202Si.
Step-6: Preparation of (25,5R)-6-benzyloxy-2-(fert-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicyclo-r3.2.11octane (VIII):
Part 1; Preparation of (2S,5RS)-6-benzyloxy-2-(fert-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicvclo-r3.2.11octane:
To a solution of (25,5i?5)-5-benzyloxyamino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine (VII) (159.0 g, 0.454 mol) in a mixture of acetonitrile (2.38 L) and diisopropylethylamine (316.5 ml, 1.81 mol) was added triphosgene (59.27 gm, 0.199 mol) dissolved in acetonitrile (760 ml) at -15°C over 30 minutes under stirring. The resulting reaction mixture was stirred for additional 1 hour at -10°C. The reaction mixture was quenched by addition of saturated aqueous sodium bicarbonate (2.0 L) at -5°C to 10°C. Acetonitrile was evaporated from the reaction mixture under vacuum and to the left over aqueous phase, dichloromethane (2.5 L) was added. The organic layer was separated and aqueous layer extracted with dichloromethane (1.5 L x 2). The combined organic layer was washed successively with water (2.0 L), saturated aqueous sodium chloride (2.0 L) and dried over sodium sulfate. Solvent was evaporated under vacuum and the residue was passed through a silica gel bed to yield 83.0 g of diastereomeric mixture (25, 5i?5)-6-benzyloxy-2-(feri-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicyclo-[3.2.1]octane in 50:50 ratio as a yellow liquid.
Part-2: Separation of diastereomers to prepare (25,5R)-6-benzyloxy-2-(fert-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicvclo-r3.2.11octane:
A mixture of diastereomers (2S,5Z?S)-6-benzyloxy-2-(teri-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicyclo-[3.2.1]octane in 50:50 ratio (47.0 gm, 0.125 mol), was dissolved in n-hexane (141 ml) and stirred at temperature of about 10°C to 15°C for 1 hour. Precipitated solid was filtered and washed with n-hexane (47 ml) to provide 12.0 g of diastereomerically pure (25,5i?)-6-benzyloxy-2-(tert-butyl-dimethylsilyl-oxymethyl)-7-oxo- 1,6-diaza-bicyclo-[3.2.1] octane (VIII) as a white crystalline material.
Analysis:
Mass: 377.3 (M+l); for Molecular weight: 376.58 and Molecular Formula: 
1H NMR (CDCI3): δ Ί -Ί.ΑΑ (m, 5H), 4.95 (dd, 2H), 3.76-3.85 (ddd, 2H), 3.37-3.40 (m, 1H), 3.28-3.31 (m, 2H), 2.89 (brd, 1H), 1.90-2.02 (m, 2H), 1.62- 1.74 (m, 2H), 1.56 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H).
Diastereomeric purity as determined by HPLC: 99.85%
Step-7: Preparation of (25,5R)-6-benzyloxy-2-hvdroxymethyl)-7-oxo-l,6-diaza-bicvclo-r3.2.11octane (IX):
To a solution of (25,5i?)-6-benzyloxy-2-(ieri-butyl-dimethylsilyl-oxymethyl)-7-oxo- l,6-diaza-bicyclo-[3.2.1]octane (VIII) ( 12.0 g, 31.9 rnmol) in tetrahydrofuran (180 ml) was charged tetra 7? -butyl ammonium fluoride (38.0 ml, 38 mmol, 1 M in tetrahydrofuran) at room temperature. The reaction mixture was stirred for 2 hours. It was quenched with saturated aqueous ammonium chloride ( 100 ml). The organic layer was separated and aqueous layer extracted with dichloromethane (150 ml x 3). The combined organic layer was washed with saturated aqueous sodium chloride (150 ml), dried over sodium sulfate and evaporated under vacuum to yield 24.0 g of (25,5i?)-6-benzyloxy-2-hydroxymethyl)-7-oxo-l ,6-diaza-bicyclo-[3.2.1]octane (IX) as a yellow liquid. The compound of Formula (IX) was purified by silica gel (60-120 mesh) column chromatography using a mixture of ethyl acetate and hexane as an eluent.
Analysis:
Mass: 263.1 (M+l); for Molecular weight: 262.31 and Molecular Formula: C14H18N203
1H NMR (CDCb): δ 7.34-7.42 (m, 5H), 4.95 (dd, 2H), 3.67-3.73 (m, 1H), 3.53-3.60 (m, 2H), 3.32-3.34 (m, 1H), 2.88-3.01 (m, 2H), 2.09 (brs, 1H), 1.57-2.03 (m, 2H), 1.53- 1.57 (m, 1H), 1.37- 1.40 (m, 1H).
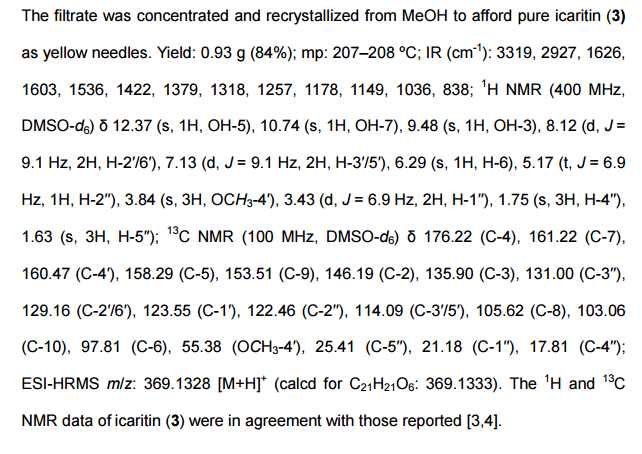
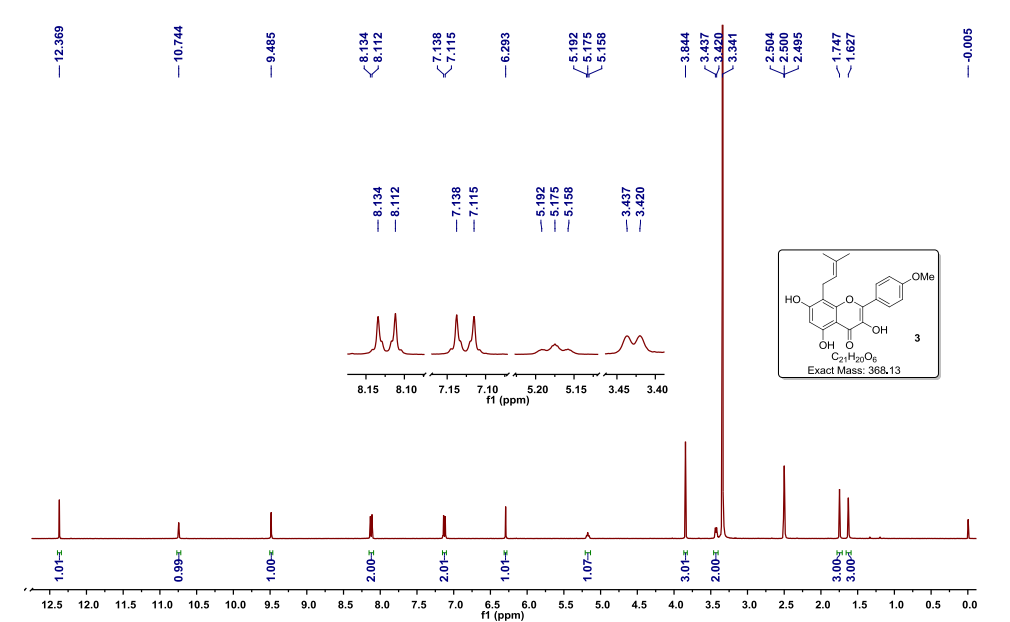
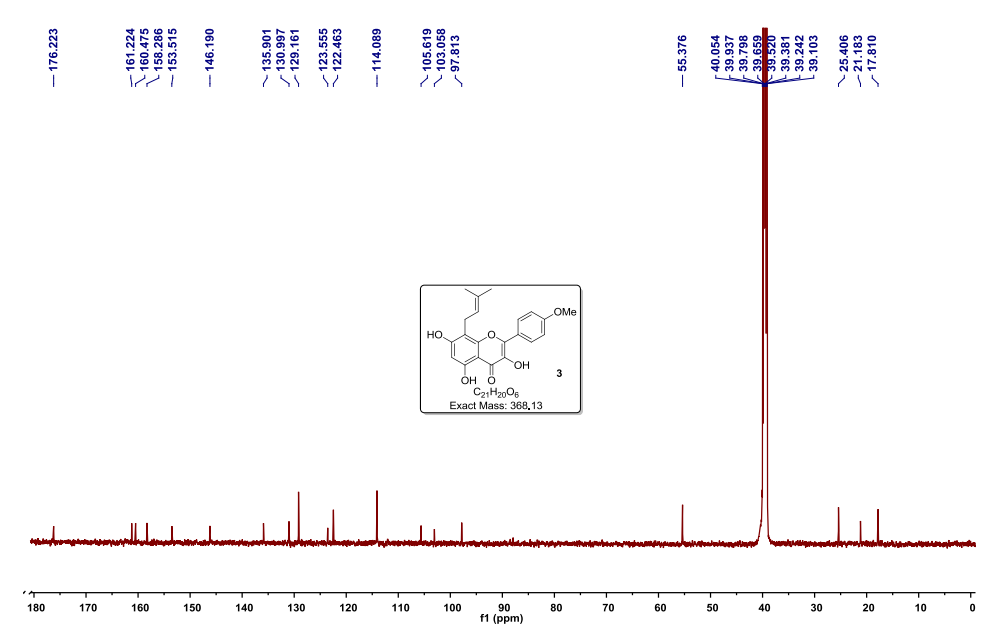
Step 8: Preparation of sodium salt of (25, 5R)-6-benzyloxy-7-oxo-l,6-diaza-bicvclor3.2.11-octane-2-carboxylic acid (I):
Step I:
Compound of Formula (IX) obtained in step 8 above was used without any further purification. To the clear solution of (25,5i?)-6-benzyloxy-2-hydroxymethyl)-7-oxo-l,6-diaza-bicyclo-[3.2.1]octane (IX) (24.0 g, 31.8 mmol) (quantities added based upon theoretical basis i.e 8.3 g ) in dichloromethane (160 ml), was added Dess-Martin reagent (24.1 g, 57.24 mmol) in portions over 15 minutes. The resulting suspension was stirred for 2 hours at 25°C. The reaction was quenched by adding a solution, prepared from saturated aqueous sodium hydrogen carbonate solution (160 ml) and 72.0 g of sodium thiosulfate. Diethyl ether (160 ml) was added to the reaction mixture and it was stirred for 5-10 minutes and filtered through celite. Biphasic layer from filtrate was separated. Organic layer was washed with saturated aqueous sodium hydrogen carbonate solution (160 ml) followed by saturated aqueous sodium chloride solution (160 ml). Organic layer was dried over sodium sulfate and evaporated to dryness at 30°C to obtain 20.0 g of intermediate aldehyde, which was used immediately for the next reaction.
Step II:
To the crude intermediate aldehyde (20.0 g, 31.6 mmol) (quantities added based upon theoretical yield i.e. 8.2 g) obtained as above, was charged i-butyl alcohol (160 ml) and cyclohexene (10.8 ml, 110.6 mmol). The reaction mixture was cooled to temperature of about 10°C to 15°C. To this mixture was added clear solution prepared from sodium hypophosphate (14.8 g, 94.8 mmol) and sodium chlorite (5.7 g, 63.2 mmol) in water (82.0 ml) over a period of 30 minutes by maintaining temperature between 10°C to 15°C. The reaction mixture was further stirred for 1 hour and was quenched with saturated aqueous ammonium chloride solution. The reaction mixture was subjected to evaporation under vacuum at 40°C to remove i-butyl alcohol. Resulting mixture was extracted with dichloromethane (3 x 150 ml). Layers were separated. Combined organic layer was washed with aqueous brine solution, dried over sodium sulfate and evaporated to dryness under vacuum to obtain 16.0 g of crude residue. To this residue was added acetone (83 ml) to provide a clear solution and to it was added dropwise a solution of sodium 2-ethyl hexanoate (4.5 g) in acetone (24 ml). The reaction mixture was stirred for 15 hours at 25°C to 30°C to provide a suspension. To the suspension was added diethyl ether (215 ml) and stirred for 30 minutes. Resulting solid was filtered over suction, and wet cake was washed with cold acetone (83 ml) followed by diethyl ether (83 ml). The solid was dried under vacuum at 40°C to provide 3.6 g of off-white colored, non-hygroscopic sodium salt of (25, 5i?)-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]-octane-2-carboxylic acid (I).
Analysis:
Mass: 275.2 as M-1 (for free acid) for Molecular Weight: 298 and Molecular Formula: 
NMR (DMSO-d6): δ 7.43-7.32 (m, 5H), 4.88 (q, 2H), 3.48 (s, IH), 3.21 (d, IH), 2.73 (d, IH), 2.04-2.09 (m, IH), 1.77-1.74 (m, IH), 1.65-1.72 (m, IH), 1.55-1.59 (m, IH);
Purity as determined by HPLC: 97.47%;
[a]D25: -42.34° (c 0.5, water).
///////

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

































































![[1860-5397-11-135-1]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-11-135-1.png?scale=2.0&max-width=1024&background=FFFFFF)

![[1860-5397-11-135-i1]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-135-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-11-135-i2]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-135-i2.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-11-135-i3]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-135-i3.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-11-135-i4]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-135-i4.png?scale=2.0&max-width=1024&background=FFFFFF)
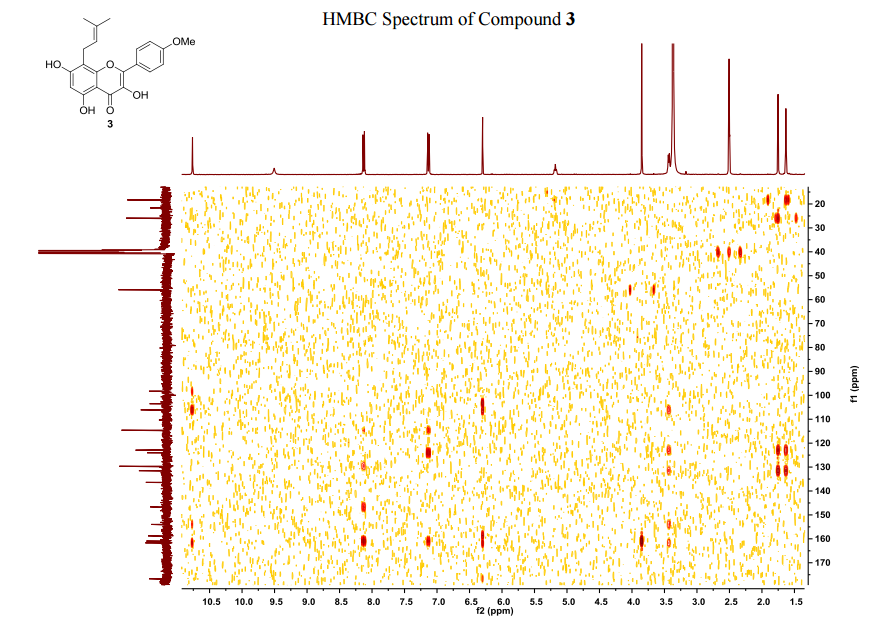
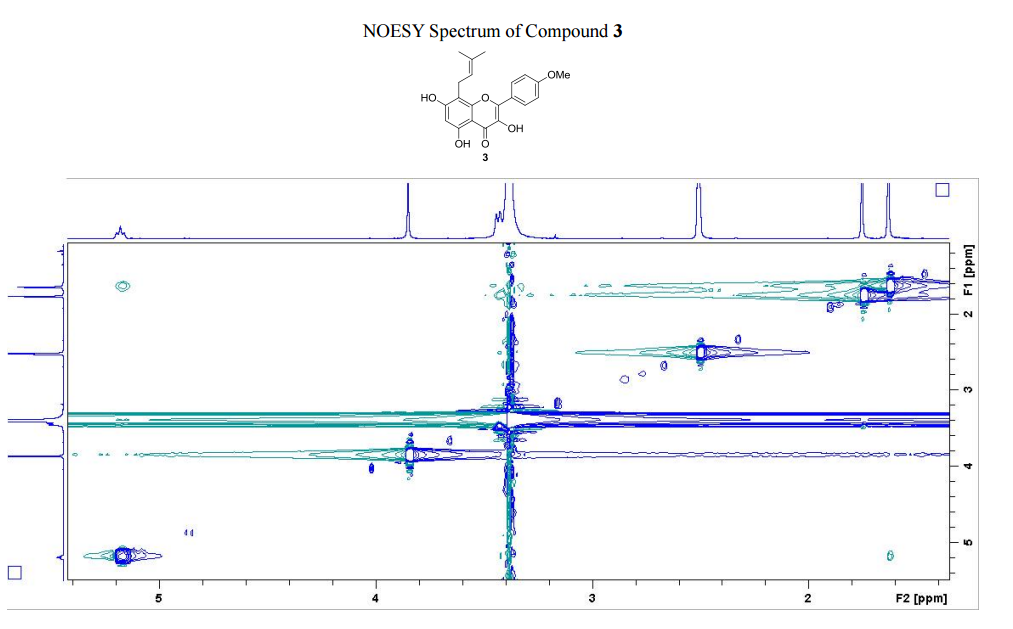












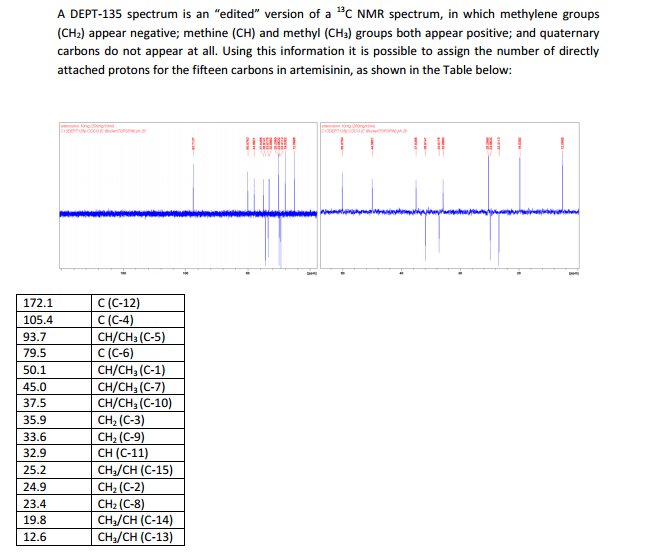

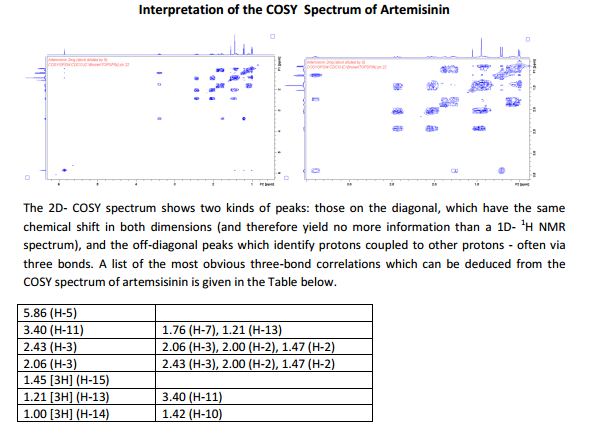
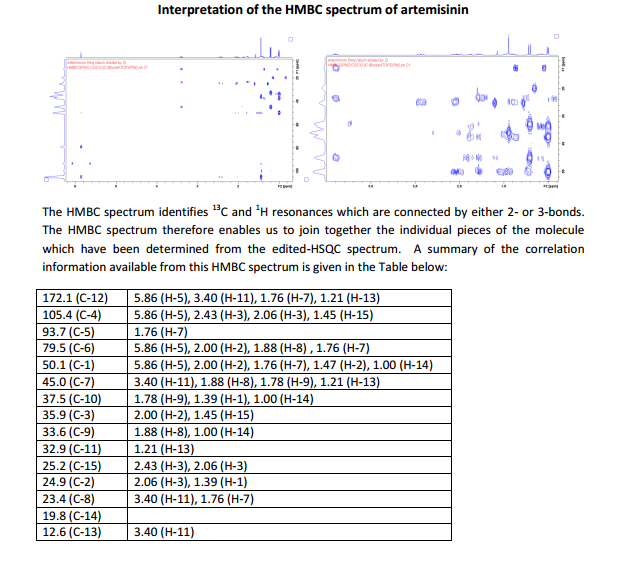







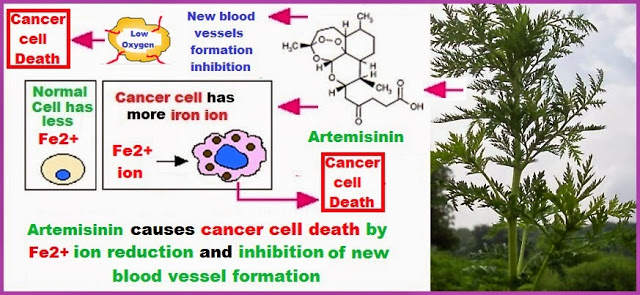

 The number of sequential operations required in traditional approaches to making molecules can make synthesis time-consuming. In particular, downstream processes such as purification of the desired compound from waste products can take much longer than the actual reaction. Importantly, flow chemistry can also offer significant improvements to work health and safety as hazardous chemicals can be manipulated in a closed system and therefore, risks associated with exposure are reduced.
The number of sequential operations required in traditional approaches to making molecules can make synthesis time-consuming. In particular, downstream processes such as purification of the desired compound from waste products can take much longer than the actual reaction. Importantly, flow chemistry can also offer significant improvements to work health and safety as hazardous chemicals can be manipulated in a closed system and therefore, risks associated with exposure are reduced.