
Home » Preclinical drugs
Category Archives: Preclinical drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
LINZAGOLIX
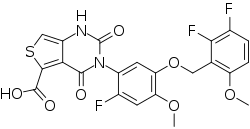
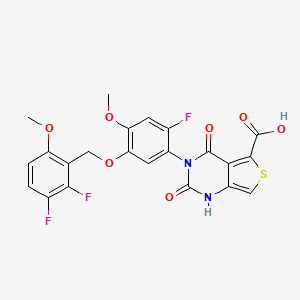
LINZAGOLIX
CAS 935283-04-8
C22H15F3N2O7S
- Hormone Antagonists
3-[5-[(2,3-difluoro-6-methoxyphenyl)methoxy]-2-fluoro-4-methoxyphenyl]-2,4-dioxo-1H-thieno[3,4-d]pyrimidine-5-carboxylic acid
- WHO 10711
- Treatment of Endometriosis Pain and Uterine Myoma-Associated Heavy Menstrual Bleeding
- OriginatorKissei Pharmaceutical
- DeveloperKissei Pharmaceutical; ObsEva
- Class2 ring heterocyclic compounds; Antihormones; Antineoplastics; Carboxylic acids; Fluorinated hydrocarbons; Ketones; Pyrimidines; Small molecules; Thiophenes
- Mechanism of ActionLHRH receptor antagonists
- PreregistrationUterine leiomyoma
- Phase IIIEndometriosis
- Phase IIAdenomyosis
- 22 Nov 2021FDA assigns PDUFA action date of (13/09/2022) for linzagolix for Uterine leiomyoma
- 22 Nov 2021The US FDA accepts NDA for linzagolix for Uterine leiomyoma for review
- 20 Oct 2021Efficacy and adverse events data from a phase II trial in Adenomyosis presented at the American Society for Reproductive Medicine (ASRM) 2021 Scientific Congress & Expo


Linzagolix choline
CAS#: 1321816-57-2 (choline)
Chemical Formula: C27H28F3N3O8S
Exact Mass: 611.1549
Molecular Weight: 611.58
Linzagolix is an orally bioavailable gonadotropin-releasing hormone (GnRH or LHRH) receptor antagonist, with potential hormone production inhibitory activity. Upon oral administration of linzagolix, this agent competes with GnRH for receptor binding and inhibits GnRH receptor signaling in the anterior pituitary gland, thereby inhibiting the secretion and release of luteinizing hormone (LH) and follicle stimulating hormone (FSH). In males, the inhibition of LH secretion prevents the release of testosterone. As a result, this may relieve symptoms associated with hormonally dependent disease states such as hormone-dependent prostate cancer. In women, this prevents the production of estrogen by the ovaries and may relieve symptoms from sex-hormone dependent diseases, such as pain associated with endometriosis, heavy menstrual bleeding or uterine fibroids.
Linzagolix (INN; developmental code names KLH-2109, OBE-2109; tentative brand name Yselty) is a small-molecule, non-peptide, orally active gonadotropin-releasing hormone antagonist (GnRH antagonist) which is under development by Kissei Pharmaceutical and ObsEva for the treatment of uterine fibroids, endometriosis, and adenomyosis.[1][3][2] As of December 2020, it is under review for approval for uterine fibroids, is in phase III clinical trials for endometriosis, and is in phase II clinical studies for adenomyosis.[1]
Estrogen-dependent disorders represent a challenging class of diseases that have a high incidence in the general population and are often associated with particularly severe symptomology. Uterine fibroids, for example, also referred to as leiomyomata, are among the most common benign tumors in women. Symptoms associated with uterine fibroids commonly include heavy or prolonged menstrual bleeding, pelvic pressure and pelvic organ compression, back pain, and adverse reproductive outcomes. Heavy menstrual bleeding may lead to iron deficiency anemia, a key symptom of uterine fibroids and the leading cause of surgical interventions that may include hysterectomy. Endometriosis is another estrogen-dependent gynecological condition, characterized by the presence of endometrial-like tissue outside the uterus.
Additional examples of estrogen-dependent diseases include adenomyosis and rectovaginal endometriosis, which are particularly severe endometrial growth disorders characterized by the invasion of endometrial tissue into the uterine myometrium and rectovaginal zones, respectively. The term adenomyosis or uterine adenomyosis is used to describe the presence of both endometrial glands and stroma deep within the myometrium. This condition is associated with hypertrophy and hyperplasia of the subjacent muscle cells, which may ultimately result in an altered size and globulous morphology of the uterus. Due to the severity of this disorder, one of the key symptoms is strong menstrual and even non-menstrual pelvic pain with abnormal uterine bleeding. Like adenomyosis, rectovaginal endometriosis patients present with a variety of pain symptoms including dysmenorrhea, dyspareunia, chronic pelvic pain, dysuria, and dyschezia. Treatment options for rectovaginal endometriosis are limited. Since medical therapies are either ineffective or have considerable side effects, rectovaginal endometriosis patients often undergo surgical procedures to reduce the endometrial node, and may even be subject to resection of the bowel if the node infiltrates the rectal or sigmoidal wall.

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Obseva Announces U.S. FDA Acceptance of New Drug Application for Linzagolix
November 22, 2021 01:05 ET | Source: ObsEva SA………. https://www.globenewswire.com/news-release/2021/11/22/2338610/0/en/Obseva-Announces-U-S-FDA-Acceptance-of-New-Drug-Application-for-Linzagolix.html
FDA Accepts NDA for Linzagolix for the Management of Heavy Menstrual Bleeding Associated with Uterine Fibroids
GENEVA, Switzerland November 22, 2021 – Obseva SA (NASDAQ: OBSV; SIX: OBSN), a biopharmaceutical company developing and commercializing novel therapies to improve women’s reproductive health, today announced that the New Drug Application (NDA) for linzagolix for the management of heavy menstrual bleeding associated with uterine fibroids in premenopausal women has been accepted for review by the United States Food and Drug Administration (FDA). The submission is based on data from the two Phase 3 PRIMROSE trials. Linzagolix has a differentiated profile and if approved, would be the first and only GnRH receptor antagonist with flexible dosing options for uterine fibroids, including a low dose option to address the needs of women who cannot or do not want to take hormones.1,4 The FDA set a target action date of September 13, 2022 for this NDA under the Prescription Drug User Fee Act (PDUFA).
“Today marks an important milestone not only in the linzagolix clinical development process, but for Obseva as a company, and most importantly, the millions of women living with uterine fibroids throughout the US. Linzagolix is a significant innovation in the field of women’s health – an area that is consistently underinvested in – and we are incredibly excited about the potential of bringing this important treatment to market” said Brian O’Callaghan, CEO of Obseva. “We are encouraged by our positive Phase 3 PRIMROSE results. If approved, we believe linzagolix will address a significant unmet need in offering a more individualized treatment option for a broader range of women.”
The Phase 3 PRIMROSE trials of linzagolix (PRIMROSE 1: US; n=574 and PRIMROSE 2: Europe and US; n=535) investigated the efficacy and safety of two dosing regimens, 100mg once daily and 200mg once daily, alone or in combination with hormonal ABT (1 mg estradiol and 0.5 mg norethisterone acetate) for the treatment of heavy menstrual bleeding associated with uterine fibroids. The NDA submission comprises positive 24-week treatment results from both studies, as well as supportive results from Week 52 and the 76-week post-treatment follow-up.
“Uterine fibroids can have a devastating impact on women’s day-to-day life. With its unique dosing options, linzagolix has the potential to significantly advance medical options for women,” stated Elizabeth Garner, MD, MPH, Chief Medical Officer of Obseva. “A dosing option without hormonal ABT would be welcomed by the significant number of women who either have contraindications to or a personal preference to avoid the use of estrogen-based therapies, while also providing a dosing option for women in whom hormonal ABT is indicated.”
The linzagolix marketing authorization application (MAA) was validated by the European Medicine Agency (EMA) with an approval recommendation from the Committee for Medicinal Products for Human Use (CHMP) expected in Q4 2021. Obseva announced previously that the company has entered into a partnership with Syneos Health to support commercialization of linzagolix in the US and EU.
About Linzagolix
Linzagolix is a novel, once daily, oral GnRH receptor antagonist with a potentially best-in-class profile1,2,3. Linzagolix is the subject of submitted marketing authorization applications for the treatment of heavy menstrual bleeding associated with uterine fibroids and is currently in late-stage clinical development for the treatment of pain associated with endometriosis. Obseva licensed linzagolix from Kissei in late 2015 and retains worldwide commercial rights, excluding Asia, for the product. Linzagolix is not currently approved anywhere in the world.
About the Phase 3 PRIMROSE Program in Uterine Fibroids
PRIMROSE 1 & 2 were prospective, randomized, parallel group, double-blind, placebo-controlled Phase 3 studies that investigated the efficacy and safety of two dosing regimens of linzagolix, 100 mg and 200 mg once daily, alone and in combination with hormonal ABT (1 mg estradiol and 0.5 mg norethisterone acetate) for the treatment of heavy menstrual bleeding associated with uterine fibroids. PRIMROSE 1 was conducted in the United States and enrolled 574 women. PRIMROSE 2 was conducted in Europe and the United States and enrolled 535 women. Both trials comprised a 52-week treatment period followed by a 6-month post treatment follow-up period. Additional information can be found here.
About Uterine Fibroids
Uterine fibroids are common benign tumors of the muscular tissue of the uterus which affect women of childbearing age and can vary in size from undetectable to large bulky masses. Few long-term medical treatments are available, and as a result, approximately 300,000 hysterectomies are performed for uterine fibroids every year in the US.
The symptoms of uterine fibroids are wide-ranging and include heavy menstrual bleeding, anemia, pelvic pressure and bloating, urinary frequency and pain that can be extremely debilitating with a significant impact on quality of life. These symptoms can also have an impact on mental health, creating the additional burden of anxiety and distress.
About Obseva
Obseva is a biopharmaceutical company built to address some of the most challenging unmet needs in women’s health – an under-researched, under-invested field of medicine. With deep expertise in clinical development, Obseva is passionate about the pursuit of advances that benefit women and their health and the importance of delivering truly meaningful innovation in this space. Through strategic in-licensing and disciplined drug development, Obseva has established a late-stage clinical pipeline with development programs focused on new therapies for the treatment of uterine fibroids, endometriosis, and preterm labor. Obseva is listed on the Nasdaq Global Select Market and is traded under the ticker symbol “OBSV” and on the SIX Swiss Exchange where it is traded under the ticker symbol “OBSN”. For more information, please visit http://www.ObsEva.com.
About Kissei
Kissei is a Japanese pharmaceutical company with approximately 70 years of history, specialized in the field of urology, kidney-dialysis and unmet medical needs. Silodosin is a Kissei product for the treatment of the signs and symptoms of benign prostatic hyperplasia which is sold worldwide through its licensees. KLH-2109/OBE2109 is a new chemical entity discovered by Kissei R&D.

……………………………

PATENT
WO 2007046392
https://patents.google.com/patent/WO2007046392A1/en
PATENT
WO 2014042176
https://patents.google.com/patent/WO2014042176A1/en

(Process 1)
Compound (D) can be produced by reacting compound (B) or a salt thereof with compound (C) in the presence of a base in a solvent. Examples of the solvent include halogen solvents such as dichloromethane, cyclic ethers such as tetrahydrofuran, 2-methyltetrahydrofuran, and tetrahydropyran, amide solvents such as N, N-dimethylformamide, aromatic hydrocarbon solvents such as toluene, A nitrile solvent such as acetonitrile, an ester solvent such as ethyl acetate, or a mixed solvent thereof and a mixed solvent thereof and water are preferable, and a mixed solvent of tetrahydrofuran and water is preferable. Examples of the base include organic bases such as triethylamine and pyridine, and inorganic bases such as sodium hydrogen carbonate, potassium hydrogen carbonate, cesium carbonate, sodium carbonate, and potassium carbonate, preferably triethylamine, sodium hydrogen carbonate, or potassium carbonate Is mentioned. The equivalent of the base may be an equivalent amount capable of neutralizing the salt and neutralizing the acid generated by the reaction. The equivalent of (C) can be used in an amount of 0.8 to 1.1 equivalents relative to (B), preferably 1.0 equivalent. The reaction temperature is usually 0 to 30 ° C., and the reaction time is usually 0.5 to 3 hours, although it varies depending on the raw material used, the solvent, the reaction temperature and the like. Examples of the salt of the compound (B) include a salt with an inorganic acid, a salt with an organic acid, a salt with an acidic amino acid, and the like. Examples of the salt with an inorganic acid include salts with hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid and the like. Examples of salts with organic acids include formic acid, acetic acid, trifluoroacetic acid, fumaric acid, oxalic acid, tartaric acid, maleic acid, citric acid, succinic acid, malic acid, methanesulfonic acid, benzenesulfonic acid, p-toluene And salts with sulfonic acid and the like. Examples of salts with acidic amino acids include salts with aspartic acid, glutamic acid and the like. Among these salts, salts with hydrochloric acid and methanesulfonic acid are preferable. Compound (C) used in Scheme 1 may be a commercially available product, or can be produced according to a known method or a method analogous thereto. Compound (D) may be isolated before the next step, but it can also be used in the next step without isolation.(Process 2)
Compound (F) can be produced by reacting compound (D) with compound (E) or a salt thereof in a solvent in the presence or absence of a base. Examples of the solvent include cyclic ethers such as tetrahydrofuran, 2-methyltetrahydrofuran, tetrahydropyran, amide solvents such as N, N-dimethylformamide, aromatic hydrocarbon solvents such as toluene, nitrile solvents such as acetonitrile, An ester solvent such as ethyl acetate or a mixed solvent thereof and a mixed solvent thereof with water, and the like are preferable, and a mixed solvent of tetrahydrofuran and water is preferable. Examples of the base include organic bases such as N, N-dimethylaminopyridine, triethylamine, N-methylpyrrolidine, N-methylmorpholine, diisopropylethylamine, and preferably N, N-dimethylaminopyridine, triethylamine and the like. . The equivalent of the base can be used in an amount of 0.1 to 2.0 equivalents relative to the compound (E), preferably 0.1 to 0.5 equivalents (provided that when a salt of the compound (E) is used, Further base necessary for neutralization is required). The reaction temperature is from room temperature to 60 ° C., and the reaction time is usually from 1 to 24 hours, although it varies depending on the raw material used, the solvent, the reaction temperature, and the like. Examples of the salt of compound (E) include a salt with an inorganic acid, a salt with an organic acid, a salt with an acidic amino acid, and the like. Examples of the salt with an inorganic acid include salts with hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid and the like. Examples of salts with organic acids include formic acid, acetic acid, trifluoroacetic acid, fumaric acid, oxalic acid, tartaric acid, maleic acid, citric acid, succinic acid, malic acid, methanesulfonic acid, benzenesulfonic acid, p-toluene And salts with sulfonic acid and the like. Examples of salts with acidic amino acids include salts with aspartic acid, glutamic acid and the like. Compound (F) may be isolated before the next step, but it can also be used in the next step without isolation.(Process 3)
The intramolecular cyclization and hydrolysis reaction in this step can be performed simultaneously or separately.
(Step 3-1)
Compound (A) can be produced by subjecting compound (F) to intramolecular cyclization and hydrolysis in the presence of a base in a solvent. Examples of the solvent include cyclic ethers such as tetrahydrofuran, 2-methyltetrahydrofuran and tetrahydropyran, lower alcohols such as methanol, ethanol and 2-propanol, amide solvents such as N, N-dimethylformamide, and nitriles such as acetonitrile. Examples thereof include a solvent and the like or a mixed solvent of a mixed solvent thereof and water, and a mixed solvent of tetrahydrofuran / methanol / water is preferable. Examples of the base include inorganic bases such as sodium hydroxide, potassium hydroxide, lithium hydroxide and sodium hydride, and metal alkoxides such as sodium methoxide and potassium tert-butoxide, preferably lithium hydroxide and sodium And methoxide. The base can be used in an amount of 3.0 to 6.0 equivalents, preferably 4.0 to 4.5 equivalents, relative to compound (F). The reaction temperature is usually from 0 to 20 ° C., and the reaction time is usually from 1 to 10 hours, although it varies depending on the raw material used, solvent, reaction temperature and the like.
(Step 3-2)
When isolating compound (G), compound (G) can be produced by subjecting compound (F) to an intramolecular cyclization reaction in a solvent in the presence of a base. Examples of the solvent include cyclic ethers such as tetrahydrofuran, 2-methyltetrahydrofuran and tetrahydropyran, lower alcohols such as methanol, ethanol and 2-propanol, amide solvents such as N, N-dimethylformamide, and nitriles such as acetonitrile. Examples thereof include a solvent and the like or a mixed solvent thereof, and a mixed solvent of tetrahydrofuran / methanol is preferable. Examples of the base include inorganic bases such as sodium hydroxide, potassium hydroxide, lithium hydroxide or sodium hydride, metal alkoxides such as sodium methoxide and potassium tert-butoxide, and lithium hydroxide, sodium methoxide and the like. preferable. The base can be used in an amount of 0.1 to 1.5 equivalents, preferably 1.0 to 1.1 equivalents, relative to compound (F). The reaction temperature is usually from 0 to 20 ° C., and the reaction time is usually from 1 to 10 hours, although it varies depending on the raw material used, solvent, reaction temperature and the like.
(Step 3-3)
The hydrolysis reaction in this step can be performed by the same method as in step 3-1 or a method analogous thereto.(Process 4)
Compound (A) can be converted to a salt thereof by a conventional method. Examples of such salts include inorganic salts such as sodium salt, potassium salt, calcium salt, magnesium salt, triethylamine, diisopropylamine, N, N′-dibenzylethylenediamine, ethanolamine, (2-hydroxyethyl) trimethylammonium. (Hereinafter referred to as choline), addition salts with organic bases such as N-methylglucamine, arginine, lysine and the like, and choline salts are preferred. Examples of the reagent used for conversion to the choline salt include choline hydroxide, choline bicarbonate, choline chloride and choline acetate.Here, the compound (B) and the salt thereof used in the above-mentioned scheme 1 are commercially available, or manufactured by the method described in a) to c), the method described in the reference examples, or a method analogous thereto. Can do.
a) JP-A 64-29373
b) Synthetic Communications, 32, 2565 (2002)
c) Synthesis, 200 (1977)Further, the compound (E) or a salt thereof used in the scheme 1 can be produced by the method described in Patent Document 1, the method described in Reference Examples, or a method analogous thereto.The compound obtained in the production process in the present specification includes hydrates or solvates thereof, and any of them can be used. Furthermore, the compound obtained in the production process in the present specification may have tautomers and / or geometric isomers, any of which can be used, and also a mixture thereof. be able to.By the production method of the present invention, the compound (A) useful as a pharmaceutical product or a salt thereof can be obtained in high yield and high purity through the compound (D) which is a production intermediate.The content of the present invention will be described in more detail by the following examples, but the present invention is not limited to the content.Reference example 1
Dimethyl 4-oxothiolane-2,3-dicarboxylate methylthioglycolate (15.0 g), tetrahydrofuran (45 g), piperidine (0.361 g) in a reaction mixture at room temperature with dimethyl maleate (21.4 g) in tetrahydrofuran (30 g) The solution was added. To the reaction mixture was added 20% sodium methoxide in methanol (43 g) at 55 ° C. under a nitrogen atmosphere. The reaction mixture was stirred at reflux for 3 hours. Diisopropyl ether (105 g) and acetic acid (0.85 g) were added to the reaction mixture at 45-50 ° C., and then cooled. The suspension was filtered to obtain wet crystals (43.3 g) of sodium salt of dimethyl 4-oxothiolane-2,3-dicarboxylate. The wet crystals were added to a mixture of 85% phosphoric acid (9.8 g), water (20 g) and ethyl acetate (150 g) at room temperature, and the aqueous layer was removed. The obtained organic layer was washed with 10% brine and then dried over anhydrous magnesium sulfate. The drying agent was removed by filtration, and the filtrate was concentrated under reduced pressure to obtain the title compound (22.7 g).Reference example 2
Dimethyl 4- (hydroxyimino) thiolane-2,3-dicarboxylate Dimethyl 4-oxothiolane-2,3-dicarboxylate (10.0 g), pyridine (5.44 g), hydroxylamine hydrochloride (3.34 g) Was stirred at 50 ° C. for 1 hour. Ethyl acetate and 7% aqueous phosphoric acid solution were added to the reaction mixture at room temperature, and the aqueous layer was removed. The obtained organic layer was washed with 5% sodium bicarbonate water and 10% brine. The organic layer was dried over anhydrous sodium sulfate. After removing the desiccant by filtration, the filtrate was concentrated under reduced pressure to obtain the title compound (10.4 g).Reference example 3
4-Aminothiophene-2,3-dicarboxylic acid dimethyl hydrochloride 4- (hydroxyimino) thiolane-2,3-dicarboxylate (10.4 g) in acetic acid (32 g) solution in 4N-hydrogen chloride / ethyl acetate solution ( 120 g) was added at room temperature. The reaction mixture was stirred at room temperature for 8 hours. After filtering the suspension, the obtained solid was dried to obtain the title compound (9.42 g).Reference example 4
4-Aminothiophene-2,3-dicarboxylic acid dimethyl methanesulfonate To a solution of methanesulfonic acid (80.0 g) in ethyl acetate (900 g), dimethyl 4- (hydroxyimino) thiolane-2,3-dicarboxylate (97. 1 g) of ethyl acetate (500 g) was added at 65-75 ° C. The reaction mixture was stirred at the same temperature for 2 hours. Methyl isobutyl ketone (100 g) was added at 45-50 ° C. and cooled to room temperature. After filtering the suspension, the obtained solid was dried to obtain the title compound (102 g).Reference Example 5
1,2-difluoro-3-[(4-fluoro-2-methoxyphenoxy) methyl] -4-methoxybenzene sodium borohydride in a solution of 2,3-difluoro-6-methoxybenzaldehyde (150 g) in toluene (900 g) (13.2 g) of 0.1N sodium hydroxide aqueous solution (180 g) was added at 35 to 39 ° C. The reaction mixture was stirred at the same temperature for 5 hours. After cooling the reaction mixture to room temperature, the aqueous layer was removed. The obtained organic layer was washed with 20% brine to obtain a toluene solution of 2,3-difluoro-6-methoxybenzyl alcohol. To this solution was added concentrated hydrochloric acid (610 g) at room temperature. The reaction mixture was stirred at 38-43 ° C. for 5 hours. After cooling the reaction mixture to room temperature, the aqueous layer was removed. The obtained organic layer was washed with water and 20% brine to obtain a toluene solution of 3- (chloromethyl) -1,2-difluoro-4-methoxybenzene. To this solution, 4-fluoro-2-methoxyphenol (125 g) and tetrabutylammonium bromide (56.2 g) were added at room temperature. A 25% aqueous sodium hydroxide solution (170 g) was added to the reaction mixture at 60 to 63 ° C., and the mixture was stirred at the same temperature for 4 hours. Water was added to the reaction mixture and the aqueous layer was removed. The obtained organic layer was washed with water and concentrated under reduced pressure. The residue was dissolved in 2-propanol and water was added. After filtering the suspension, the obtained solid was dried to obtain the title compound (232 g).Reference Example 6
1,2-difluoro-3-[(4-fluoro-2-methoxy-5-nitrophenoxy) methyl] -4-methoxybenzene 1,2-difluoro-3-[(4-fluoro-2-methoxyphenoxy) methyl ] To a solution of 4-methoxybenzene (158 g) in acetic acid (1200 g) was added 60% nitric acid (72.2 g) at 59-62 ° C., and the mixture was stirred at the same temperature for 2 hours. Water (1200 g) was added to the suspension at 15 to 19 ° C., and the mixture was stirred at the same temperature for 1 hour. After filtering the suspension, the obtained solid was washed with water to obtain wet crystals of the title compound (190 g, Net amount 168 g).Reference Example 7
2-Fluoro-5-[(2,3-difluoro-6-methoxyphenyl) methoxy] -4-methoxyaniline Raney nickel (2.5 g), ethyl acetate (180 g), 1,2-difluoro-3-[(4 -Fluoro-2-methoxy-5-nitrophenoxy) methyl] -4-methoxybenzene wet crystal (10.9 g, Net amount 10.0 g) was stirred at room temperature under a hydrogen atmosphere for 4 hours. The catalyst was removed by filtration, and the filtrate was concentrated under reduced pressure. The residue was dissolved with methanol and water was added. After filtering the suspension, the obtained solid was dried to obtain the title compound (7.97 g).Example 1
4- (phenoxycarbonylamino) thiophene-2,3-dicarboxylic acid dimethyl potassium carbonate (17.1 g), water (90 g), tetrahydrofuran (150 g) and 4-aminothiophene-2,3-dicarboxylic acid dimethyl hydrochloride (30 0.06) was added phenyl chloroformate (18.6 g) at 6-13 ° C. The reaction mixture was stirred at 12-13 ° C. for 30 minutes, and then the aqueous layer was removed. To the obtained organic layer, tert-butyl methyl ether was added and washed with 20% brine. The obtained organic layer was concentrated under reduced pressure. The residue was dissolved with diisopropyl ether and n-hexane was added. After filtering the suspension, the obtained solid was dried to obtain the title compound (37.0 g).
1 H-NMR (DMSO-d 6 ) δ ppm: 3.82 (3H, s), 3.82 (3H, s), 7.13-7.30 (3H, m), 7.40-7.46 (2H, m), 7.80 (1H, s ), 10.24 (1H, s)Example 2
4- {3- [2-Fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] ureido} dimethyl thiophene-2,3-dicarboxylate 2-fluoro-5-[( 2,3-difluoro-6-methoxyphenyl) methoxy] -4-methoxyaniline (7.70 g), dimethyl 4- (phenoxycarbonylamino) thiophene-2,3-dicarboxylate (8.65 g), triethylamine (0. 37 g) and tetrahydrofuran (80 mL) were stirred at room temperature for 24 hours. The reaction mixture was concentrated under reduced pressure. Ethyl acetate and methanol were added to the residue. After filtering the suspension, the obtained solid was dried to obtain the title compound (12.0 g).
1 H-NMR (DMSO-d 6 ) δ ppm: 3.71 (3H, s), 3.82 (3H, s), 3.83 (3H, s), 3.89 (3H, s), 5.00 (2H, d, J = 1.6 Hz), 6.87-6.93 (1H, m), 7.00 (1H, d, J = 12.8Hz), 7.41-7.50 (1H, m), 7.75 (1H, d, J = 8.0Hz), 7.94 (1H, s ), 8.82 (1H, s), 8.95 (1H, s)Example 3
3- [2-Fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] -2,4-dioxo-1,2,3,4-tetrahydrothieno [3,4 d] methyl pyrimidine-5-carboxylate 4- {3- [2-fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] ureido} thiophene-2,3-dicarboxylic acid A methanol solution (3.48 g) of 28% sodium methoxide was added to a suspension of dimethyl (10.0 g) in tetrahydrofuran (40 g), stirred at room temperature for 3 hours, and acetic acid (1.30 g) was added. The reaction mixture was concentrated under reduced pressure. Methanol was added to the residue, and water was further added. After filtering the suspension, the obtained solid was dried to obtain the title compound (8.58 g).
1 H-NMR (DMSO-d 6 ) δ ppm: 3.79 (3H, s), 3.81 (3H, s), 3.84 (3H, s), 4.95 (2H, s), 6.88-6.94 (1H, m), 7.08 (1H, d, J = 11.6Hz), 7.19-7.23 (2H, m), 7.44-7.53 (1H, m), 11.62 (1H, s)Example 4
4- (phenoxycarbonylamino) thiophene-2,3-dicarboxylate potassium carbonate (9.38 kg), water (49 kg), tetrahydrofuran (82 kg), dimethyl 4-aminothiophene-2,3-dicarboxylate hydrochloride (16 4 kg) of the reaction mixture was stirred for 40 minutes, and then phenyl chloroformate (10.1 kg) was added at 11-21 ° C. The reaction mixture was stirred for 30 minutes, and then the aqueous layer was removed to obtain a tetrahydrofuran solution of the title compound.Example 5
4- {3- [2-Fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] ureido} dimethyl thiophene-2,3-dicarboxylate 4-obtained in Example 4 To a tetrahydrofuran solution of dimethyl (phenoxycarbonylamino) thiophene-2,3-dicarboxylate, 2-fluoro-5-[(2,3-difluoro-6-methoxyphenyl) methoxy] -4-methoxyaniline (17.0 kg), Tetrahydrofuran (8.5 kg) and triethylamine (1.1 kg) were added, and the mixture was stirred at 50 ° C. for 3.5 hours to obtain a tetrahydrofuran solution of the title compound.Example 6
3- [2-Fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] -2,4-dioxo-1,2,3,4-tetrahydrothieno [3,4 d] pyrimidine-5-carboxylic acid tetrahydrofuranate 4- {3- [2-fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] ureido} obtained in Example 5 Methanol (41 kg) and water (47 kg) are added to a tetrahydrofuran solution of dimethyl thiophene-2,3-dicarboxylate, a 7.3% lithium hydroxide aqueous solution (80.1 kg) is added at 11 to 13 ° C., and 90 ° C. at 11 ° C. Stir for minutes. Acetic acid (11.4 kg) was added to the reaction mixture at 9 to 16 ° C., and acetic acid (13.0 kg) was further added at 29 to 31 ° C. Seed crystals were added to the reaction mixture, and the mixture was stirred at the same temperature for 30 minutes. Water (34 kg) was added to the suspension and stirred at 30 ° C. for 40 minutes. The suspension was stirred at 4-9 ° C. for 90 minutes. After the suspension was filtered, the obtained solid was washed with a mixed solution of methanol (54 kg) and water (68 kg) to give wet crystals of the title compound (31.64 kg, Net amount (compound (A) free form equivalent)) 26 0.7 kg) was obtained.
A part of the wet crystals of the title compound was dried under reduced pressure at an external temperature of 60 ° C., and 1 H-NMR, HPLC and powder X-ray diffraction were measured on the obtained dried crystals of the title compound.
1 H-NMR (DMSO-d 6 ) δ ppm: 1.68-1.82 (3H, m), 3.53-3.65 (3H, m), 3.80 (3H, s), 3.81 (3H, s), 4.94-4.98 (2H , m), 6.87-6.94 (1H, m), 7.13 (1H, d, J = 11.2Hz), 7.25 (1H, d, J = 7.2Hz), 7.39 (1H, s), 7.43-7.52 (1H, m), 11,99 (1H, s), 14.53 (1H, s)
PATENT
WO 2020089190
https://patents.google.com/patent/WO2020089190A2/enFor example, the GnRH antagonist may be 3-[2-fluoro-5-(2,3-difluoro-6-methoxybenzyloxy)4- methoxyphenyl]-2,4-dioxo-1 ,2,3,4- tetrahydrothieno [3,4d]pyrimidine-5-carboxylic acid, or a pharmaceutically acceptable salt thereof. The salt may be, for instance, the choline salt thereof, represented by formula (Via), below.
Compound (VI) and pharmaceutically acceptable salts thereof, such as the choline salt thereof (compound (Via)), can be synthesized, for example, using the methodology described in WO 2014/042176, the disclosure of which is incorporated herein by reference in its entirety. An exemplary synthetic scheme that may be used for the preparation of compound (VI) and the choline salt thereof is shown in Scheme 1 , below.Scheme 1 . Exemplary preparation of compound (VI) and the choline salt thereof




wherein Ri and R are each independently C alkoxy groups; LG is a nucleofugal leaving group, such as chlorine or bromine, among others; R represents an optional substituent, such as halogen, acyl group, C alkyl group, or a nitro substituent; DMAP denotes A/-dimethylaminopyridine; and TEA denotes trimethylamine.Crystalline compound (Via) has been characterized spectroscopically, for instance, in US Patent No. 9,169,266, the disclosure of which is incorporated herein by reference in its entirety. The foregoing crystalline form has been shown to exhibit characteristic X-ray powder diffraction peaks at about 7.10 2Q, about 11 .5° 2Q, about 19.4° 2Q, about 21 .5° 2Q, about 22.0° 2Q, about 22.6° 2Q, about 23.5° 2Q, and about 26.2° 2Q. Additionally, this crystalline form exhibits 13C solid-state nuclear magnetic resonance (NMR) peaks centered at about 55.5 ppm, about 57.1 ppm, about 58.7 ppm, about 69.8 ppm, about 98.1 ppm, about 110.3 ppm, about 1 1 1 .6 ppm, about 113.7 ppm, about 1 18.0 ppm, about 145.3 ppm, about 149.8 ppm, and about 155.8 ppm. This crystalline form further exhibits 19F solid-state NMR peaks centered at about -151.8 ppm, -145.2 ppm, and -131 .6 ppm.Compound (VI), as well as pharmaceutically acceptable salts thereof, such as the choline salt thereof, exhibit a high affinity for human GnRH receptor (27.4 nM). Using the compositions and methods described herein, a patient that is presenting with or has been diagnosed as having, adenomyosis or rectovaginal endometriosis may be administered a compound of formula (VI), or a pharmaceutically acceptable salt thereof, such as the choline salt thereof, to treat the disease or ameliorate one or more symptoms of the disease. Exemplary doses of compound (VI) and pharmaceutically acceptable salts thereof, such as the choline salt thereof, include doses of from 25 mg to 500 mg daily, such as doses of 100 mg per day and 200 mg per day. Additional dosing information is provided below.3-Aminoalkyl pyrimidine-2, 4(1 H,3H)-dionesAdditional GnRH antagonists that may be used in conjunction with the compositions and methods described herein include optionally substituted 3-aminoalkyl pyrimidine-2, 4(1 H,3H)-dione derivatives, such as compounds represented by formula (VII)

PATENTWO 2021023876https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021023876&_cid=P11-KWFRM2-91270-1
In some embodiments, the compound is the choline salt of the compound represented by formula (VI), choline 3- [2-fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) 4-methoxyphenyI] -2,4- dioxo-1,2,3,4-
tetrahydrothieno [3,4d] pyrimidine-5-carboxylate. It is to be understood that references herein to a compound represented by formula (VI) specifically include the choline salt of compound (VI), which is represented by formula (VIa), below.
In some embodiments, the choline 3- [2-fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) 4-methoxyphenyI] -2,4-dioxo-1,2,3,4- tetrahydrothieno [3,4d ] pyrimidine-5-carboxylate is in a crystalline state.
PATENT
WO 2021023877
References
- ^ Jump up to:a b c “Linzagolix – Kissei Pharmaceutical/ObsEva – AdisInsight”.
- ^ Jump up to:a b Ezzati M, Carr BR (2015). “Elagolix, a novel, orally bioavailable GnRH antagonist under investigation for the treatment of endometriosis-related pain”. Womens Health (Lond). 11 (1): 19–28. doi:10.2217/whe.14.68. PMID 25581052.
- ^ Chodankar, Rohan; Allison, Jennifer (2018). “New Horizons in Fibroid Management”. Current Obstetrics and Gynecology Reports. 7 (2): 106–115. doi:10.1007/s13669-018-0242-6. ISSN 2161-3303.
External links
| Clinical data | |
|---|---|
| Trade names | Yselty |
| Other names | KLH-2109; OBE-2109 |
| Routes of administration | By mouth[1][2] |
| Drug class | GnRH modulator; GnRH antagonist; Antigonadotropin |
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 935283-04-8 |
| PubChem CID | 16656889 |
| ChemSpider | 17590169 |
| UNII | 7CDW97HUEX |
| KEGG | D11608 |
| ChEMBL | ChEMBL3668014 |
| Chemical and physical data | |
| Formula | C22H15F3N2O7S |
| Molar mass | 508.42 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////LINZAGOLIX, Hormone Antagonists, WHO 10711, KLH-2109, KLH 2109, OBE-2109, OBE 2109

NEW DRUG APPROVALS
ONE TIME
$10.00
JBI-802 BY JUBILANT
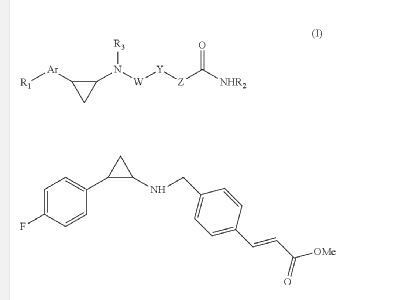
EXAMPLE
O=C(OC)/C=C/c1ccc(CNC2CC2c2ccc(F)cc2)cc1
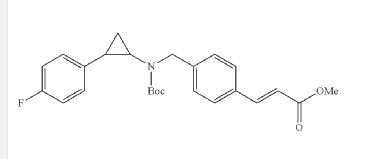
EXAMPLE ONLY NOT CONFIRMED
JBI-802
- Myeloid Leukemia Therapy
- Solid Tumors Therapy
Epigenetic Modifier Modulators
- Histone Deacetylase 6 (HDAC6) Inhibitors
- Lysine-Specific Histone Demethylase 1A (KDM1A; LSD1) Inhibitors
Jubilant Therapeutics Announces Successful Completion of Pre-IND Meeting with FDA for its Novel Dual LSD1 and HDAC6 Inhibitor JB1-802
https://markets.businessinsider.com/news/stocks/jubilant-therapeutics-announces-successful-completion-of-pre-ind-meeting-with-fda-for-its-novel-dual-lsd1-and-hdac6-inhibitor-jb1-802-1030834551
PRESS RELEASE PR Newswire
Sep. 30, 2021, 10:23 AM
BEDMINSTER, NJ, Sept. 30, 2021 /PRNewswire/ — Jubilant Therapeutics Inc., a biopharmaceutical company advancing small molecule precision therapeutics to address unmet medical needs in oncology and autoimmune diseases, today announced the successful completion of a pre-IND (Investigational New Drug) meeting with the U.S. Food and Drug Administration (FDA) regarding the development plan, clinical study design and dosing strategy for the Phase I/II trial of JB1-802, a dual inhibitor of LSD1 and HDAC6, for the treatment of small cell lung cancer, treatment-induced neuro-endocrine prostate cancer and other mutation-defined neuroendocrine tumors.
![]() A pre-IND meeting provides the drug development sponsor an opportunity for an open communication with the FDA to discuss the IND development plan and to obtain the agency’s guidance regarding planned clinical evaluation of the sponsor’s new drug candidate. After reviewing the preclinical data provided, plans for additional data generation and the Phase I/II clinical trial protocol, the FDA addressed Jubilant Therapeutics’ questions, provided guidance and aligned with the sponsor on the proposed development plan for JBI-802.
A pre-IND meeting provides the drug development sponsor an opportunity for an open communication with the FDA to discuss the IND development plan and to obtain the agency’s guidance regarding planned clinical evaluation of the sponsor’s new drug candidate. After reviewing the preclinical data provided, plans for additional data generation and the Phase I/II clinical trial protocol, the FDA addressed Jubilant Therapeutics’ questions, provided guidance and aligned with the sponsor on the proposed development plan for JBI-802.
“We appreciate the FDA’s guidance as we endeavor to find an innovative new treatment for high unmet-need tumors with devastatingly low survival rates,” said Hari S Bhartia, Chairman, Jubilant Therapeutics Inc.
“We are pleased with the outcome of the pre-IND meeting with the FDA and plan to submit the IND application by the end of 2021,” said Syed Kazmi, Chief Executive Officer, Jubilant Therapeutics Inc.
About Jubilant TherapeuticsJubilant Therapeutics Inc. is a patient-centric biopharmaceutical company advancing potent and selective small molecule modulators to address unmet medical needs in oncology and autoimmune diseases. Its advanced discovery engine integrates structure-based design and computational algorithms to discover and develop novel, precision therapeutics against both first-in-class and validated but intractable targets in genetically defined patient populations. The Company plans to file an IND later this year for the first in class dual inhibitor of LSD1/HDAC6, followed by two additional INDs in 2022 with novel modulators of PRMT5 and PAD4 in oncology and inflammatory indications. Jubilant Therapeutics is headquartered in Bedminster NJ and guided by globally renowned key opinion leaders and scientific advisory board members. For more information, please visit www.jubilanttx.com or follow us on Twitter @JubilantTx and LinkedIn.
View original content:https://www.prnewswire.com/news-releases/jubilant-therapeutics-announces-successful-completion-of-pre-ind-meeting-with-fda-for-its-novel-dual-lsd1-and-hdac6-inhibitor-jb1-802-301388983.html
SOURCE Jubilant Therapeutics Inc.

Mohd Zainuddin
Director at Jubilant Therapeutics Inc
PATENT
IN 201641016129
PATENT
US20200308110 – CYCLOPROPYL-AMIDE COMPOUNDS AS DUAL LSD1/HDAC INHIBITORS
https://patentscope.wipo.int/search/en/detail.jsf?docId=US306969204&tab=NATIONALBIBLIO&_cid=P21-KUANET-85789-2ApplicantsJubilant Epicore LLC
Inventors
Sridharan RAJAGOPAL
Mahanandeesha S. HALLUR
Purushottam DEWANG
Kannan MURUGAN
Durga Prasanna KUMAR C.H.
Pravin IYER
Chandrika MULAKALA
Dhanalakshmi SIVANANDHAN
Sreekala NAIR
Mohd ZAINUDDIN
Subramanyam Janardhan TANTRY
Chandru GAJENDRAN
Sriram RAJAGOPAL
Priority Data201641016129 09.05.2016 IN

Sridharan Rajagopal
Vice President-Head of Medicinal Chemistry at Jubilant Therapeutics Inc

Dhanalakshmi Sivanandhan
Vice President at Jubilant Therapeutics Inc

Mahanandeesha Hallur
Associate Director at Jubilant Biosys

Sreekala Nair

Chandrika Mulakala

Pravin Iyer
 Dewang")
Purushottam (M.) Dewang
ERRORS CALL ME , +919321316780
AND TO ADD TOO
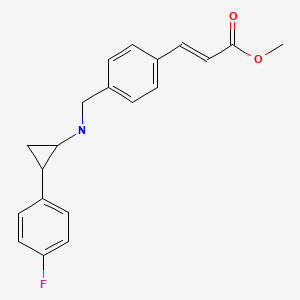
EXAMPLE
C20 H20 F N O22-Propenoic acid, 3-[4-[[[2-(4-fluorophenyl)cyclopropyl]amino]methyl]phenyl]-, methyl ester, (2E)-Molecular Weight, 325.38
Patent
WO2017195216
I-3methyl (E)-3-(4-(((tert-butoxycarbonyl)(2-(4-((4-fluorobenzyl)oxy)phenyl) cyclopropyl)amino)methyl)phenyl)acrylate

The compound was synthesized using amine B6 and (E)-3-(4-Formyl-phenyl)-acrylic acid methyl esterfoUowing the procedure for the synthesis of 1-2. LC-MS m/z calcd for C32H34FN05, 531.2; found 532.2 [M+H]+.

| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| EP-3455204-A1 | Cyclopropyl-amide compounds as dual lsd1/hdac inhibitors | 2016-05-09 | |
| WO-2017195216-A1 | Cyclopropyl-amide compounds as dual lsd1/hdac inhibitors | 2016-05-09 | |
| US-2020308110-A1 | Cyclopropyl-amide compounds as dual lsd1/hdac inhibitors | 2016-05-09 |
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
Step 2: (E)-3-[4-({tert-Butoxycarbonyl-[2-(4-fluoro-phenyl)-cyclopropyl]-amino}-methyl)-phenyl]-acrylic acid methyl ester (I-2)
| To a stirred solution of (E)-3-(4-{[2-(4-fluoro-phenyl)-cyclopropylamino]-methyl}-phenyl)-acrylic acid methyl ester (XLVI, 0.25 g, 0.76 mmol) in tetrahydrofuran and water mixture (6 mL, 1:1) was added sodium bicarbonate (0.087 g, 2.3 mmol) and Boc anhydride (0.22 mL, 0.92 mmol) at room temperature and the resulting mixture was stirred at that temperature for 2 h. The progress of the reaction was monitored by TLC. The reaction mixture was diluted with ethylacetate and the organic portion was washed with water and brine solution, dried over sodium sulphate and concentrated under reduced pressure to get the crude product which was purified by column chromatography using ethylacetate-hexane gradient to afford the titled product as sticky oil (I-2, 0.19 g, 58%). LC-MS m/z calcd for C 25H 28FNO 4, 425.2; found 326.3 [M-Boc+1] +. |
| The following compounds were synthesized using procedure for the synthesize of I-2 |
REFJBI-802, novel dual inhibitor of LSD1-HDAC6 for treatment of cancerSivanandhan, D.; Rajagopal, S.; Nair, S.; et al.Annu Meet Am Assoc Cancer Res (AACR) · 2020-06-22 / 2020-06-24 · Virtual, N/A · Abst 1756Synthesis and optimization of a novel series of LSD1-HDAC dual inhibitors led to the discovery of JBI-802 as the lead compound, with IC50 of 0.05 mcM against LSD1 and isoform selective HDAC6/8 activity, with IC50 of 0.011 and 0.098 mcM for HDAC6 and HDAC8, respectively. The candidate also showed excellent selectivity against other HDACs, with approximately 77-fold selectivity for HDAC6. In vitro, JBI-802 showed strong antiproliferative activity on selected cell lines, including acute myeloid leukemia, chronic lymphocytic leukemia, lymphoma and certain solid tumors, such as small cell lung cancer and sarcoma. In vivo, JBI-802 demonstrated strong efficacy in erythroleukemia xenograft model, leading to prolonged survival of mice bearing HEL92.1.7 tumors. The candidate showed excellent dose-response and superior efficacy compared to single agents in this model, with ED50 of approximately 6.25 mg/kg twice-daily by oral administration. When evaluated in CT-26 syngeneic model, JBI-802 showed promising activity as single agent and in the combination of JBI-802 plus anti-programmed cell death protein 1 (PD-1) monoclonal antibody (MAb), with approximately 80% tumor growth inhibition observed for the combination. Exploratory toxicology studies showed that JBI-802 was well tolerated at efficacious doses. Further preclinical IND-enabling studies are currently underway for this molecule, which is to be developed as a clinical candidate for the treatment of acute myeloid leukemia and other tumor types.
REFNovel dual inhibitor of LSD1-HDAC6/8 for treatment of cancerDhanalakshmi, S.; Rajagopal, S.; Sadhu, N.; et al.62nd Annu Meet Am Soc Hematol · 2020-12-05 / 2020-12-08 · Virtual, N/A · Abst 3378 Blood 2020, 136(Suppl. 1)
REFJubilant Therapeutics Presents Preclinical Data at the American Association for Cancer Research, Reveals Unique Dual-Action Anti-Cancer Mechanism Underscoring First-in-Class Pipeline Asset in Hematological Tumors
Jubilant Therapeutics Press Release 2020, June 22
////////////////JB1-802, JUBILANT, CANCER, PRECLINICAL
EXTRAS…………
PATENTWO2021062327 – FUSED PYRIMIDINE COMPOUNDS, COMPOSITIONS AND MEDICINAL APPLICATIONS THEREOFhttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021062327&_cid=P21-KUAMRR-83330-1PCT/US2020/052953
Priority Data
| 201941039277 | 27.09.2019 | IN |
Inventors
- VENKATESHAPPA, Chandregowda
- SIVANANDHAN, Dhanalakshmi
- RAJAGOPAL, Sridharan
- ROTH, Bruce
- PANDEY, Anjali
- SAXTON, Tracy
- HALLUR, Gurulingappa
- MADHYASTHA, Naveena
- SADHU M, Naveen
Lung cancer accounts for the greatest number of cancer deaths, and approximately 85% of lung cancer cases are non-small cell lung cancer (NSCLC). The development of targeted therapies for lung cancer has primarily focused on tumors displaying specific oncogenic drivers, namely mutations in epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK). Three generations of tyrosine kinase inhibitors (TKIs) have been developed for cancers with the most frequently observed EGFR mutations, however, other oncogenic drivers in the EGFR family of receptor tyrosine kinases have received less research and development focus and several oncogenic drivers, including insertions in the exon 20 gene of EGFR, have no currently approved therapeutics to treat their cancers.
[0003] The mutation, amplification and/or overexpression of human epidermal growth factor receptor 2 (HER2), another member of the human epidermal growth factor receptor family of receptor tyrosine kinases, has been implicated in the oncogenesis of several cancers, including lung, breast, ovarian, and gastric cancers. Although targeted therapies such as trastuzumab and lapatinib have shown clinical efficacy especially in breast tumors, their utility in lung cancer has been limited. It is likely that this variation is due to tissue-specific factors, including the low potency of kinase inhibitors like lapatinib for the mutagenic alterations in HER2 that are observed in the lung cancer patient population, including insertions in the exon 20 gene of HER2.
[0004] Given that many patients with mutations in EGFR and HER2 do not derive clinical benefit from currently available therapies against these targets, there remains a significant unmet need for the development of novel therapies for the treatment of cancers associated with EGFR and HER2 mutations.
Compound 49: (E)-N-(3-(3-benzyl-7-((1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4-(dimethylamino)but-2-enamide
Step 1: Synthesis of (E)-4-(dimethylamino)but-2-enoyl chloride
[0280] To a stirred mixture of acetonitrile (2 mL) and DMF (2 drop) under N2 atmosphere was added N,N-dimethylamino crotonic acid hydrochloride (0.1 g, 0.77 mmol). After 10 min, this solution was cooled to 0-5 °C. Oxalyl chloride (0.122 g, 0.968 mmol) was added and the reaction mixture was maintained at 0-5 °C for 30 min. It was allowed to warm to RT and stirring was continued for 2 h. It was then heated to 40 °C for 5 min and again brought to RT and stirred for 10 min. Formation of product was confirmed by TLC and the reaction mass was used as such to the next step without any workup.
Step-2: Synthesis of (E)-N-(3-(3-benzyl-7-((1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4-(dimethylamino)but-2-enamide (Compound 49)
[0281] 1-(3-Aminophenyl)-3-benzyl-7-((1-methyl-1H-pyrazol-4-yl)amino)-3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one (0.11g, 0.7 mmol) in DMP (2 mL) was cooled to -15 °C and then (E)-4-(dimethylamino)but-2-enoylchloride was added. The reaction mixture was stirred for 1 h at -15 °C to RT. After the completion of reaction, the reaction mass was quenched with ice water, sodium bicarbonate solution and extracted with DCM (100 mL x 2). The combined organic layer was washed with cold water (3 x 50 mL), brine solution (10 mL), dried over anhydrous sodium sulfate and evaporated under reduced pressure to obtain crude product. The crude product was purified by prep HPLC to get pure product (E)-N-(3-(3-benzyl-7-((1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4-(dimethylamino)but-2-enamide (Compound 49, 0.022 g, 16 % yield) as white solid.1H NMR (400 MHz, DMSO-d6): δ 10.21 (s, 1H), 9.32 (s, 1H), 8.06 (s, 1H), 7.76 (bs, 1H) 7.65 (s, 1H), 7.48 (bs, 1H), 7.39-7.29 (m, 5H), 7.03 (d, J = 7.2 Hz, 2H), 6.74-6.68 (m, 1H), 6.62 (s, 1H), 6.25 (d, J = 15.2 Hz, 1H), 4.62 (s, 2H), 4.37 (s, 2H), 3.47 (s, 3H), 3.03 (d, J = 5.6 Hz, 2H), 2.15 (s, 6H); LCMS Calcd for [M+H] + 538.2, found 538.5
Compound 50: (E)-N-(3-(3-benzyl-7-((1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-3-chloroacrylamide
Step-1: Synthesis of (Z)-3-chloroacrylic acid
[0282] To a stirred solution propiolic acid (2 g, 28.5 mmol) in DMF (15 mL) under N2 atmosphere was added thionyl chloride (4.07 g, 34.2 moles) slowly and the reaction mixture was maintained at 25 °C for 1 h. The reaction was monitored by TLC, after the completion of reaction, the residue was poured into ice and the resulting aqueous solution was extracted with ether (3 x100 mL). The organic layer was washed with brine (20 mL), dried over anhydrous sodium sulfate and evaporated under reduced pressure to obtain crude product. The crude product was purified to get pure product (Z)-3-chloroacrylic acid (1.9 g, 62.9 % yield). LCMS Calcd for [M-H] +, 104.98, found 105.1
Step-2: Synthesis of (Z)-3-chloroacryloyl chloride
[0283] To a stirred solution of acetonitrile (3 mL) and DMF (3 drop) under N2 atmosphere was added of (Z)-3-chloroacrylic acid (0.2 g, 1.87 mmol). After 10 min this solution was cooled 0-5 °C. Oxalyl chloride (0.122 g, 0.968 mmol) was added and the reaction mixture was maintained at 0-5 °C for 30 min. It was allowed to warm to RT and stirring was continued for 2 h to get (Z)-3-chloroacryloyl chloride. Formation of product was confirmed by TLC and the reaction mass was used as such to the next step without any workup.
Step-3: Synthesis of (E)-3-((3-(3-benzyl-7-((1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)amino)acryloyl chloride (Compound 50)
[0284] A solution of 1-(3-Aminophenyl)-3-benzyl-7-((1-methyl-1H-pyrazol-4-yl)amino)-3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one (0.11 g, 0.7 mmol) in DMP (2 mL) was cooled to -15 °C and then (Z)-3-chloroacryloyl chloride was added. The reaction mixture was stirred for 1 h at -15 °C to RT. The reaction was monitored by TLC. After the completion of reaction, reaction mass was quenched with ice water and sodium bicarbonate solution. The aqueous layer was e 0.028 g, 22% yield) as a white solid.1H NMR (400 MHz, DMSO-d6): δ 10.35 (s, 1H), 9.32 (s, 1H), 8.06 (s, 1H), 7.74 (s, 1H), 7.59 (s, 1H), 7.51 (s, 1H), 7.41-7.35 (m, 5H), 7.30-7.29 (m, 1H), 7.08-7.02 (m, 2H), 6.62-6.58 (m, 2H), 4.62 (s, 2H), 4.37 (s, 2H), 3.47 (s, 3H); LCMS Calcd for [M+H] + 515.1, LCMS found 515.2
Compound 51: (E)-N-(3-(7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-3-phenyl-2-thioxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4-(dimethylamino)but-2-enamide
Step-1: Synthesis of 2,4-dichloro-5-(chloromethyl)pyrimidine
[0285] Title compound was prepared in a similar manner to general procedure I.5-(hydroxymethyl)pyrimidine-2,4-diol (15 g, 106 mmol) gave 2,4-dichloro-5-(chloromethyl)pyrimidine (11.50 g, 55% yield) as a white solid.1H NMR (400 MHz, CDCl3): δ 8.66 (s, 1H), 4.65 (s, 2H).
Step-2: Synthesis of 2,4-dichloro-5-(iodomethyl)pyrimidine
[0286] Title compound was prepared in a similar manner to general procedure J.2,4-dichloro-5-(chloromethyl)pyrimidine (11.50 g, 58.20 mmol) on treatment with NaI (10.50 g, 69.0 mmol) in acetone (100 mL) resulted in 2,4-dichloro-5-(iodomethyl)pyrimidine (15.20 g, 91% yield). The solid was immediately taken up in toluene and stored under refrigeration.1H NMR (400 MHz, CDCl3): δ 8.60 (s, 1H), 4.39 (s, 2H).
Step-3: Synthesis of N-((2,4-dichloropyrimidin-5-yl)methyl)aniline
[0287] A solution of iodo compound (18, 7.0 g, 24.20 mmol) in toluene (50 mL) was cooled to 0 °C and aniline (2.20 g, 24.20 mmol) was added. The reaction mixture was stirred for 30 min at 0 °C. Then a solution of sodium hydroxide (1.30 g, 32.50 mmol) in water (5 ml) was added and reaction mixture was stirred for 16 h at RT. The reaction was monitored by TLC. After completion of the reaction, water (25 mL) was added and extracted with ethyl acetate (2 x 100 mL). The organic layer was washed with brine solution, dried over anhydrous sodium sulfate and evaporated under reduced pressure to obtain the crude residue. The crude compound was purified by silica gel column chromatography to afford the title compound as a white solid (10 g, 81% yield). LCMS Calcd for [M+H] + 254.11, found 254.09
Step-4: Synthesis of tert-butyl (3-((2-chloro-5-((phenylamino)methyl)pyrimidin-4-yl)amino)phenyl)carbamate
[0288] To a stirred solution of N-((2,4-dichloropyrimidin-5-yl)methyl)aniline (4.0 g, 15.08 mmol) in IPA (30 mL), tert-butyl (3-aminophenyl)carbamate (4.90 g, 23.0 mmol) and DIPEA (8.20 mL, 47 mmol) were added. The reaction mixture was heated at 100 °C for 16 h in a sealed tube. Solvent was then evaporated and the crude thus obtained was purified by flash column chromatography to afford the title compound as off white solid (2.50 g, 37% yield). LCMS Calcd for [M+H] + 425.92, found 426.35
Step-5: Synthesis of tert-butyl (3-(7-chloro-3-phenyl-2-thioxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate
[0289] To a solution of tert-butyl (3-((2-chloro-5-((phenylamino)methyl)pyrimidin-4-yl)amino)phenyl)carbamate (1.50 g, 3.50 mmol) in THF (35 mL) was added DIPEA (2.40 mL, 14.10 mmol) and thiophosgene (0.27 g, 3.50 mmol) at 0 °C. The reaction mixture was stirred at RT for 24 h with TLC monitoring. After completion of the reaction, sodium bicarbonate solution was added. The reaction mixture was partitioned between DCM (2 x 100 mL) and water (50 mL). The organic layer was washed with brine (10 mL), dried over anhydrous sodium sulfate and evaporated under reduced pressure to obtain crude product. The crude product was purified by silica gel column chromatography to afford the title compound as a yellow solid (1.36 g, 82% yield). LCMS Calcd for [M+H] + 467.97, found 468.27
Step-6: Synthesis of tert-butyl (3-(7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-3-phenyl-2-thioxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate
[0290] To a solution of tert-butyl (3-(7-chloro-3-phenyl-2-thioxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (1.30 g, 2.78 mmol) in IPA (15 mL) was added 3-
chloro-1-methyl-1H-pyrazol-4-amine (0.44 g, 3.34 mmol) and TFA (1 mL). The reaction mixture was heated for 16 h at 110 °C. Reaction was monitored by TLC. After the completion of reaction, the reaction mixture was concentrated, water (10 mL) and saturated sodium bicarbonate (20 mL) solution were added to the residue and extracted with DCM (3 x 200 mL). The combined organic layer was washed with brine solution, dried over anhydrous sodium sulfate and evaporated under reduced pressure to obtain the title compound (1.30 g) that was used as such for the next step without further purification. LCMS Calcd for [M+H] + 563.08, found 562.90
Step-7: Synthesis of 1-(3-aminophenyl)-7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidine-2(1H)-thione
[0291] To an ice-cold solution of tert-butyl (3-(7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-3-phenyl-2-thioxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (1.30 g, 2.30 mmol) in DCM (20 mL) and MeOH (10 mL) was added 4N HCl in dioxane (5 mL). The reaction mixture was stirred for 16 h at RT. The reaction was monitored by TLC. After completion of the reaction, the solvent was evaporated followed by addition of water (10 mL) and saturated sodium bicarbonate (20 mL) solution and extraction with DCM (3 x 200 mL). The combined organic layer was washed with brine solution, dried over anhydrous sodium sulfate and evaporated under reduced pressure to obtain crude product. The crude product was purified by silica gel column chromatography to afford the title compound as a brown solid (0.20 g). LCMS Calcd for [M+H] + 462.96, found 463.0. Purity: 68%
Step-8: Synthesis of (E)-N-(3-(7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-3-phenyl-2-thioxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4-(dimethylamino)but-2-enamide (Compound 51)
[0292] To an ice-cold solution of 1-(3-aminophenyl)-7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidine-2(1H)-thione (0.18 g, 0.39 mmol) and trans-N,N-dimethylaminocrotonic acid hydrochloride (0.077 g, 0.47 mmol) in dichloromethane (10 mL) was added triethyl amine (1.2 mmol) followed by drop wise addition of propylphosphonic anhydride (T3P) (0.26 g, 0.97 mmol). The mixture was stirred at RT for 6 h. Completion of the reaction was monitored by TLC. The reaction mixture was portioned between 5% methanol in dichloromethane and saturated bicarbonate solution. The organic phase was dried over anhydrous sodium sulfate, filtered and concentrated. The crude obtained was purified by silica gel chromatography to afford the title compound as off white solid (Compound 51, 0.010 g, 5% yield).1H NMR (400 MHz, DMSO-d6): δ 10.36 (bs, 1H), 8.97 (bs, 1H), 8.25 (s, 1H), 7.72 (bs, 2H), 7.48-7.42 (m, 5H), 7.36-7.32 (m, 1H), 7.03 (d, J = 7.6 Hz, 1H), 6.76-6.60 (m, 2H), 6.30 (d, J = 14.8 Hz, 1H), 4.95 (s, 2H), 3.50 (s, 3H), 3.12 (bs, 2H), 2.21 (s, 6H); LCMS Calcd for [M+H] + 574.10, found 574.41
Scheme 28: Preparation of (E)-N-(3-(3-benzyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4-(dimethylamino)but-2-enamide (Compound 52):
Step 1: Preparation of ethyl 4-((3-((tert-butoxycarbonyl) amino) phenyl) amino)-2-(methylthio) pyrimidine-5-carboxylate (106):
[0293] Title compound (106) was prepared as off-white solid (142 g; Yield: 74%) in a manner substantially similar to procedure mentioned in General procedure O.1H-NMR (400 MHz, CDCl3): ^ 10.36 (s, 1H), 8.77 (d, 1H), 7.89 (s, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.25-7.22 (m, 1H), 7.03 (d, J = 8.0 Hz, 1H), 6.51 (s, 1H), 4.35 (q, J = 7.2 Hz, 2H), 2.54 (s, 3H), 1.51 (s, 9H), 1.42-1.38 (m, 3H). LCMS: [M+H]+ 405.21, 89.28%.
Step 2: Preparation of tert-butyl (3-((5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)amino)phenyl)carbamate (107):
[0294] Title compound was prepared in a manner substantially similar to procedure mentioned in General procedure P. The crude was triturated with dichloromethane afforded 107 as off white solid (40.0 g; Yield: 31%).1H-NMR (400 MHz, CDCl3): ^ 8.09 (s, 1H), 7.86 (m, 2H),
7.36 (d, J = 8.0 Hz, 1H), 7.25-7.15 (m, 1H), 6.95 (d, J = 8.0 Hz, 1H), 6.55 (s, 1H), 4.59 (s, 2H), 2.50 (s, 3H), 1.51 (s, 9H). LCMS: [M+H]+ 363.05, 91.24%.
Step 3: Preparation of tert-butyl (3-((5-formyl-2-(methylthio)pyrimidin-4-yl)amino)phenyl)carbamate (108):
[0295] Title compound (108) was prepared as a pale yellow solid (31.0 g; Yield: 78%) in a manner substantially similar to procedure mentioned in General procedure Q.1H-NMR (400 MHz, CDCl3): ^ 10.59 (s, 1H), 9.75 (s, 1H), 8.42 (s, 1H), 7.97 (s, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.04 (d, J = 8.0 Hz, 1H), 6.59 (s, 1H), 3.48 (s, 1H), 2.58 (s, 3H), 1.52 (s, 9H). LCMS: [M+H]+ 361.30, 97.51%.
Step 4: Preparation of tert-butyl (E)-(3-((5-((benzylimino)methyl)-2(methylthio)pyrimidin-4-yl)amino)phenyl)carbamate (110):
[0296] Title compound (110) was prepared as a yellow solid (28 g; Yield: 72%) in a manner substantially similar to procedure mentioned in General procedure R.1H-NMR (400 MHz, CDCl3): ^ 12.15 (s, 1H), 8.31 (s, 1H), 8.16 (s, 1H), 7.91 (s, 1H), 7.41 (m, 4H), 7.35-7.33 (m, 1H), 7.32-7.29 (m, 1H), 7.26-7.22 (m, 1H), 7.03 (d, J = 8.0 Hz, 1H), 6.46 (s, 1H), 4.84 (s, 2H), 2.59 (s, 3H), 1.52 (s, 9H). LCMS: [M+H]+ 450.38; 99.66%.
Step 5: Preparation of tert-butyl (3-((5-((benzylamino)methyl)-2-(methylthio)pyrimidin-4-yl)amino)phenyl)carbamate (111):
[0297] Title compound (111) was prepared as a pale yellow solid (40 g; Yield: 80%) in a manner substantially similar to procedure mentioned in General procedure S. LCMS: [M+H]+ 452.44; 83.57%
Step 6: Preparation of tert-butyl (3-(3-benzyl-7-(methylthio)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (112):
[0298] Title compound was prepared in a manner substantially similar to procedure mentioned in General procedure T. The crude was triturated with diethyl ether afforded 112 as off white solid (12 g; Yield: 28%).1H-NMR (400 MHz, CDCl3): ^ 8.03 (s, 1H), 7.50 (s, 1H), 7.37 (m, 6H), 7.26 (m, 1H), 6.96 (m, 1H), 6.59 (s, 1H), 4.69 (s, 2H), 4.34 (s, 2H), 2.16 (s, 3H), 1.50 (s, 9H). LCMS: [M+H]+ 478.16; 95.62%.
Step 7: Preparation of tert-butyl (3-(3-benzyl-7-(methylsulfonyl)-2-oxo-3,4-dihydropyrimido [4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (113):
[0299] Title compound was prepared in a manner substantially similar to procedure mentioned in General procedure U. The crude was triturated with diethyl ether afforded 113 as an off white solid (8.0 g; Yield: 76%).1H-NMR (400 MHz, CDCl3): ^ 8.39 (s, 1H), 7.63 (s, 1H), 7.40 (m, 6H), 7.17 (d, J = 8.0 Hz, 1H), 6.95 (d, J = 8.0 Hz, 1H), 6.61 (s, 1H), 4.71 (s, 2H), 4.48 (s, 2H), 2.97 (s, 3H), 1.49 (s, 9H). LCMS: [M+H]+ 510.31, 93.69%.
Step 8: Preparation of tert-butyl (3-(3-benzyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (114):
[0300] Title compound was prepared in a manner substantially similar to General procedure V, tert-butyl (3-(3-benzyl-7-(methylsulfonyl)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (113) and 1-methyl-1H-pyrazol-3-amine (41) gave (tert-butyl (3-(3-benzyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (114) as a brown solid (Yield: 77%), which was used directly for the next step without any further purification. MS: [M+H]+ 527.46.
Step 9: Preparation of 1-(3-aminophenyl)-3-benzyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one (115):
[0301] Title compound was prepared in a manner substantially similar to General procedure W, tert-butyl (3-(3-benzyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (114) gave 1-(3-aminophenyl)-3-benzyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one (115) as a brown solid (Yield: 93%), which was used directly for the next step. MS: [M+H]+ 427.44.
Step 10: Preparation of (E)-N-(3-(3-benzyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4-(dimethylamino)but-2-enamide (Compound 52):
[0302] Title compound was prepared in a manner substantially similar General procedure X, 1-(3-aminophenyl)-3-benzyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one (115) and trans-N,N-dimethylaminocrotonic acid hydrochloride gave (E)-N-(3-(3-benzyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4-(dimethylamino)but-2-enamide Compound 52, as a white solid (48 mg; Yield: 13%), after prep-HPLC purification.1H-NMR (400 MHz, CDCl3): δ 10.17 (s, 1H), 9.51 (s, 1H), 8.08 (s, 1H), 7.72 (d, J = 8.4 Hz, 1H), 7.60 (s, 1H), 7.43-7.35 (m, 5H), 7.33-7.29 (m, 1H), 7.10 (s, 1H), 7.01 (d, J = 8.8 Hz, 1H), 6.75-6.69 (m, 1H), 6.27 (d, J = 15.3 Hz, 1H), 5.51 (s, 1H), 4.62 (s, 2H), 4.39 (s, 2H), 3.59 (s, 3H), 3.06 (d, J = 4.8 Hz, 2H), 2.17 (s, 6H). MS: [M+H]+ 538.32.
Scheme 30: Alternative Preparation of (E)-N-(3-(7-((3-chloro-1-methyl-1H-pyrazol-4- yl)amino)-2-oxo-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4- (dimethylamino)but-2-enamide (Compound 35):
Step 1: Preparation of 5-(hydroxymethyl)pyrimidine-2,4(1H,3H)-dione (119):
[0308] An ice-cold solution of pyrimidine-2,4(1H,3H)-dione (118) (10 g, 89.21 mmol) and paraformaldehyde (9.63 g, 107.05 mmol) in aqueous potassium hydroxide (132 mL, 0.5 M,
66.74 mmol) was heated at 55 °C for 14 hours. After completion of starting material (TLC), the reaction mixture was cooled to 0 °C and the pH was adjusted to 6 with 12N hydrochloric acid, the resulting white precipitate was filtered through sintered funnel and washed with diethyl ether afforded 119 as a white solid (6.3 g, Yield: 50%) which was used directly for the next step.1H-NMR (400 MHz, DMSO-d6): ^ 10.98 (bs, 1H), 10.64 (bs, 1H), 7.24 (s, 1H), 4.78 (m, 1H), 4.12 (d, J = 12.8 Hz, 2H). LCMS: [M+H]+ 143.04 (99.92% purity).
Step 2: Preparation of 2,4-dichloro-5-(chloromethyl)pyrimidine (120):
[0309] To an ice-cold solution of 5-(hydroxymethyl)pyrimidine-2,4(1H,3H)-dione (119) (10 g, 70.36 mmol) in toluene (25 mL) was added phosphoryl chloride (14 mL, 140.72 mmol) then N,N-diisopropylethylamine (37 mL, 211 mmol). The reaction mixture was heated at 120 °C for 16 hours. After the complete disappearance of starting material on TLC, the reaction mixture was quenched slowly with sodium bicarbonate solution and extracted with ethyl acetate (3 x 200 mL). The combined organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered and evaporated under reduced pressure afforded 120 as a brown solid (12 g, Yield: 86%) which was used directly for the next step.1H NMR (400 MHz, CDCl3): ^ 8.66 (s, 1H), 4.64 (s, 2H). MS: [M+H]+ 197.0
Step 3: Preparation of 2,4-dichloro-5-(iodomethyl)pyrimidine (121):
[0310] To a solution of 2,4-dichloro-5-(chloromethyl)pyrimidine (120) (8.0 g, 40.51 mmol in acetone (40 mL) was added sodium iodide (9.71 g, 64.82 mmol). The reaction mixture was stirred at room temperature for 30 min and heated to reflux for 2 hours. After completion of reaction (TLC monitoring), the reaction mixture cooled to room temperature. The resulting white precipitate was filtered through sintered funnel and washed with acetone. The filtrate was concentrated under reduced pressure afforded 121 as a brown solid (10 g, Yield: 85%) which was used directly for the next step.1H-NMR (400 MHz, CDCl3): ^ 8.60 (s, 1H), 4.39 (s, 2H). Step 4: Preparation of N-((2,4-dichloropyrimidin-5-yl)methyl)aniline (122):
[0311] To an ice-cold solution of 2, 4-dichloro-5-(iodomethyl)pyrimidine (121) (5.0 g, 17.30 mmol) in acetone (50 mL) was added potassium carbonate (5.26 g, 38.06 mmol) and aniline (1.93 g, 20.76 mmol). The resulting reaction mixture was stirred at room temperature for 16 hours. After completion the reaction (as per TLC monitoring), the resulting white precipitate was filtered through sintered funnel and washed with acetone. The filtrate was concentrated under reduced pressure and crude was purified by column chromatography on silica gel (100-200 mesh) using 15% ethyl acetate-hexane as an eluent afforded 122 as a brown solid (2.5 g, Yield: 57%).1H-NMR (400 MHz, CDCl3): ^ 8.61 (s, 1H), 7.07 (t, J = 7.6 Hz, 2H), 6.58 (m, 3H), 6.30 (bs, 1H), 4.33 (m, 2H). LCMS: [M+H]+ 254.03 (99.01% purity).
Step 5: Preparation of tert-butyl (3-(7-chloro-2-oxo-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (123):
[0312] To an ice-cold solution of N-((2,4-dichloropyrimidin-5-yl)methyl)aniline (122) (500 mg, 1.96 mmol), in isopropanol (5 mL) was added N,N-diisopropylethylamine (1.47 mL, 8.42 mmol) and tert-butyl (3-aminophenyl)carbamate (105) (409 mg, 1.96 mmol). The resulting reaction mixture was heated at 100 °C for 16 hours in a sealed tube. After completion of reaction (TLC monitoring), the solvent was then evaporated under reduced pressure and resulting crude was purified by column chromatography on silica gel (100-200 mesh) using 30% ethyl acetate-hexane as an eluent afforded 123 as a brown solid (500 mg, Yield: 60%).1H-NMR (400 MHz, DMSO-d6): δ 9.41 (s, 1H), 8.96 (s, 1H), 8.10 (s, 1H), 7.73 (s, 1H), 7.25 (m, 2H), 7.12 (m, 3H), 6.61 (m, 3H), 6.14 (t, J = 7.2 Hz, 1H), 4.26 (m, 2H) and 1.53 (s, 9H). LCMS: [M+H]+ 426.14 (93% purity).
Step 6: Preparation of tert-butyl (3-(7-chloro-2-oxo-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (124):
[0313] To an ice-cold solution of tert-butyl (3-(7-chloro-2-oxo-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (123) (500 mg, 1.17 mmol) in tetrahydrofuran (6 mL) was added N,N-diisopropylethylamine (0.81 ml, 4.68 mmol) and triphosgene (139 mg, 0.46 mmol). The reaction mixture was stirred at room temperature for 3 hours. After completion of the reaction (TLC monitoring), aqueous triethylamine solution was added and extracted with dichloromethane (3 times). The combined organic layer was washed with brine and dried over sodium sulfate and evaporated under reduced pressure to obtain the crude residue. The crude was purified by column chromatography on silica gel (100-200 mesh) using 30% ethyl acetate-hexane as an eluent afforded 124 as a brown solid (450 mg, Yield: 85%).1H-NMR (400 MHz, DMSO-d6): δ 9.54 (s, 1H), 8.43 (s, 1H), 7.58 (s, 1H), 7.44 (m, 4H), 7.29 (t, J = 7.2 Hz, 3H), 6.94 (s, 1H), 5.0 (s, 2H) and 1.47 (s, 9H). LCMS: [M+H]+ 452.27 (99% purity).
Step 7: Preparation of tert-butyl (3-(7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (125):
[0314] Title compound was prepared in a manner substantially similar to procedure mentioned in General procedure V, (tert-butyl(3-(7-chloro-2-oxo-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (124) and 3-chloro-1-methyl-1H-pyrazol-4-amine (44) gave tert-butyl (3-(7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (125) as a brown solid in 70% yield, which was used directly for the next step. MS: [M+H]+ 547.17.
Step 8: Preparation of 1-(3-aminophenyl)-7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one (126):
[0315] Title compound was prepared in a manner substantially similar to procedure mentioned in General procedure W, tert-butyl (3-(7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (125) gave 1-(3-aminophenyl)-7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one (126) as a brown solid (800 mg, Yield: 82%) which was used directly for the next step. MS: [M+H]+ 447.08.
Step 9: Preparation of (E)-N-(3-(7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4-(dimethylamino)but-2-enamide (Compound 35):
[0316] Title compound was prepared in a manner substantially similar to procedure mentioned in General procedure X, 1-(3-aminophenyl)-7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one (126) and trans-N,N-dimethylaminocrotonic acid hydrochloride gave the titled compound, which was purified by prep-HPLC purification to afforded the title compound Compound 35 as a white solid (285 mg, Yield: 23%).1H-NMR (400 MHz, DMSO-d6): δ 10.27 (bs, 1H), 8.86 (s, 1H), 8.21 (s, 1H), 7.73 (s, 2H), 7.51-7.40 (m, 5H), 7.30-7.25 (m, 1H), 7.09 (d, J = 7.6 Hz, 1H), 6.76-6.70 (m, 2H), 6.29 (d, J = 15.4 Hz, 1H), 4.88 (s, 2H), 3.50 (s, 3H), 3.05 (d, J = 4.8 Hz, 2H) and 2.16 (s, 6H). MS:
[M+H]+ 558.16.

NEW DRUG APPROVALS
ONE TIME
$10.00
GST-HG-121
GST-HG-121
mw 431.4
C23 H29 N07
Fujian Cosunter Pharmaceutical Co Ltd
Preclinical for the treatment of hepatitis B virus infection
This compound was originally claimed in WO2018214875 , and may provide the structure of GST-HG-121 , an HBsAg inhibitor which is being investigated by Fujian Cosunter for the treatment of hepatitis B virus infection; in June 2019, an IND application was planned in the US and clinical trials of the combination therapies were expected in 2020. Fujian Cosunter is also investigating GST-HG-131 , another HBsAg secretion inhibitor, although this appears to be being developed only as a part of drug combination.
WO2017013046A1
PATENT
WO2018214875
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018214875&_cid=P21-KB0QYA-12917-1
PATENT
WO-2020103924
Novel crystalline forms of 11-oxo-7,11-dihydro-6H-benzo[f]pyrido[1,2-d][1,4]azepine, a hepatitis B surface antigen and HBV replication inhibitor, useful for treating HBV infection.
Step H: Compound 9 (15.80 g, 35.95 mmol) was dissolved in dichloromethane (150.00 mL), and trifluoroacetic acid (43.91 mL, 593.12 mmol) was added. The reaction solution was stirred at 10 degrees Celsius for 3 hours. The reaction solution was concentrated under reduced pressure and spin-dried, sodium bicarbonate aqueous solution (100.00 mL) was added, and dichloromethane (100.00 mL) was extracted. The organic phase was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain compound 10.
Step J: Compound 12 (875.00 mg, 1.90 mmol) was dissolved in toluene (20.00 mL) and ethylene glycol dimethyl ether (20.00 mL), and tetrachlorobenzoquinone (1.40 g, 5.69 mmol) was added. The reaction solution was stirred at 120 degrees Celsius for 12 hours. The reaction solution was cooled to room temperature, and a saturated aqueous sodium carbonate solution (50.00 ml) and ethyl acetate (60.00 ml) were added. The mixed solution was stirred at 10-15 degrees Celsius for 20 minutes, and the liquid was separated to obtain an organic phase. Add 2.00 mol/L aqueous hydrochloric acid solution (60.00 mL) to the organic phase, stir at 10-15 degrees Celsius for 20 minutes, and separate the liquid. Wash the organic phase with 2 mol/L aqueous hydrochloric acid solution (60.00 mL×2), separate the liquid, and separate the water phase A 2 mol/L aqueous sodium hydroxide solution (200.00 ml) and dichloromethane (200.00 ml) were added. The layers were separated, and the organic phase was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain compound 13.
Step K: Compound 13 (600.00 mg, 1.31 mmol) was dissolved in methanol (6.00 mL), and 4.00 mol/L aqueous sodium hydroxide solution (2.00 mL, 6.39 equiv) was added. The reaction solution was stirred at 15 degrees Celsius for 0.25 hours. The reaction solution was adjusted to pH=3-4 with a 1.00 mol/L hydrochloric acid aqueous solution, and then extracted with dichloromethane (50.00 mL×3). The organic phases were combined, washed with saturated brine (50.00 mL), and dried over anhydrous sodium sulfate , Filtered and concentrated under reduced pressure to obtain the compound of formula (I). ee value (enantiomeric excess): 100%.
////////////GST-HG-121, Fujian Cosunter, Preclinical , hepatitis B, virus infection
O=C(O)C1=CN2C(=CC1=O)c3cc(OC)c(OCCCOC)cc3OC[C@H]2C(C)(C)C
O=C(O)C1=CN2C(=CC1=O)c3cc(OC)c(OCCCOC)cc3OC[C@H]2C(C)(C)C
ADX-103
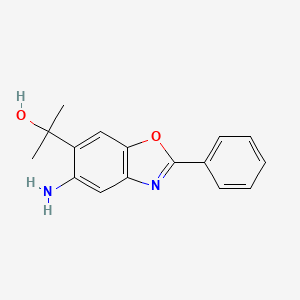
ADX-103
CAS 916056-81-0
Preclinical, Antiinflammatory Ophthalmic Agents, Diabetic Retinopathy,
Agents for Ophthalmic Drugs
MF C16 H16 N2 O2
5-Amino-α,α-dimethyl-2-phenyl-6-benzoxazolemethanol
Aldeyra Therapeutics Inc
ADX-103 , an aldehyde trap being investigated by Aldeyra for the treatment of dry eye syndrome; in May 2018, preclinical data were presented at 2018 ARVO Meeting in Honolulu, HI. Aldeyra, in collaboration with an undisclosed company, is also investigating an anti-inflammatory agent for treating ocular inflammation.
PATENT
WO-2020033344
Novel crystalline forms of a specific benzoxazole and it’s salts, process for their preparation, and compositions comprising them are claimed, useful for treating dry eye, inflammation and diabetes, through action as an aldehyde scavenger.
It has now been found that compounds of the present invention, and compositions thereof, are useful for treating, preventing, and/or reducing a risk of a disease, disorder, or condition in which aldehyde toxicity is implicated in the pathogenesis. In general, salt forms or freebase forms, and pharmaceutically acceptable compositions thereof, are useful for treating or lessening the severity of a variety of diseases or disorders as described in detail herein. Such compounds are represented by the chemical structure below, denoted as compound A:
or a pharmaceutically acceptable salt thereof.
[0008] Compounds of the present invention, and pharmaceutically acceptable compositions thereof, are useful for treating a variety of diseases, disorders or conditions, associated with toxic aldehydes. Such diseases, disorders, or conditions include those described herein.
[0009] Compounds provided by this invention are also useful for the study of certain aldehydes in biology and pathological phenomena.
Scheme 1 – Synthesis of Compound A
Step 1: Synthesis of Compound A2
[00549] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with methanol (10L). Compound A1 (2.0kg) was added, followed by further methanol to rinse (9L). The reaction mixture was warmed to Tjacket=40°C. Once temperature had stabilized, sulfuric acid (220 mL, 0.4eq.) was slowly added. Once addition was complete, agitation was maintained for 30 mins then the vessel was heated to Tjmt=62°C. Reaction progress was
monitored by LC-MS analysis of reaction mixture. The reaction does not go to completion but is deemed complete when no change is apparent in ratio of starting material : product.
[00550] The vessel contents were cooled to Tjmt=24°C and stirred 60 minutes before filtration under vacuum. The filter cake was air dried for 2 hours and the contents then dissolved in ethyl acetate (18L) which was then washed sequentially with saturated sodium bicarbonate (8L), water (8L) and brine (8L) before drying over sodium sulfate, filtration and evaporation in vacuo. Compound A2 (1.5kg, 68.1%) was obtained as a bright orange powder.
Step 2: Synthesis of Compound A3
[00551] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with /V,/V-dimethylformamide (16L). Compound A2 (1.5kg) was added and the brown reaction mixture set to cool to Tint<20oC. Once temperature had stabilized, A-bromosucci ni mi de (l.5kg, 1.1 eq.) was added portion wise, maintaining Tint<27°C. Once addition was complete, the reaction was allowed to stir until starting material content was <1% AUC (250nm) by LCMS analysis.
[00552] A secondary jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with ethyl acetate (16L) and deionized water (22L). The reaction mixture was vacuum transferred into this vessel and held at high agitation for not less than 30 minutes. The aqueous layer was discharged and the organic layer washed with saturated sodium chloride (2 x 8L) then dried over sodium sulfate before evaporation in vacuo to Compound A3 as a deep brown oil (2.lkg, 100.8%), suitable for use in following step without purification.
Step 3: Synthesis of Compound A4
[00553] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with dichloromethane (9L). Compound A3 (2.lkg) was added and the reaction mixture cooled to Tmt<l°C. A solution of Di-/er/-butyl dicarbonate (3.6kg, 2.2 eq.) in dichloromethane (0.5L) was added followed by a solution of A, A-di methyl ami nopyri di ne (92g, 0.1 eq.) in dichloromethane (0.5L). The resultant clear brown solution was stirred for 30 minutes whereupon pyridine (1.3L, 1.7 eq.) was dropwise added, maintaining Tint<5°C. Upon complete addition internal temperature was ramped from Tint=l°C to Tint=20°C over 18 hours.
[00554] The reaction mixture was sequentially washed with saturated sodium chloride (3 x 4.5L), 10 % w/v aqueous citric acid (2 x 4L), saturated sodium bicarbonate (4L), aqueous hydrochloric acid (1M, 4L), saturated sodium bicarbonate (4L) and saturated sodium chloride (4L) then dried over sodium sulfate and evaporated in vacuo with one azeotropic distillation with toluene (2L) to a very dark, heavy tar (3.4kg).
[00555] The isolated tar was mixed with absolute ethanol (3.1L) for 2 days whereupon it was filtered providing light cream colored, granular solids and a black mother liquor. The solids were washed with ice-cold ethanol (3 x 1L) and dried to constant mass. Compound A4 was obtained as off- white granules (1.7 kg, 50.2%).
Step 4: Synthesis of Compound AS
[00556] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with reagent alcohol (6.1 L) and Compound A4 (0.8kg), Tmt<20°C. Iron powder (0.5kg, 5.0 eq.) was added and the suspension stirred vigorously for 30 minutes. Acetic acid (glacial, 1.6L, 15.7 eq.) was added, maintaining Tint<30C.
[00557] Once LCMS confirmed complete consumption of starting material, ethyl acetate (10.2L) and water (10.2L) were added. Sodium bicarbonate (2.3kg, 15.9 eq.) was added portion wise and the layers separated once gas evolution had ceased. The aqueous layer was washed with ethyl acetate until LCMS indicated no further product was being extracted (8 x 2L) and the combined organic layers were sequentially washed with deionized water (6L) then saturated sodium chloride (6L) before drying over magnesium sulfate and evaporation in vacuo. Compound A5 was obtained as a light orange solid (0.7kg, 91.5%).
Step 5: Synthesis of Compound A6
[00558] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with dichloromethane (9L), Compound A5 (0.7kg), and the reaction mixture cooled to Tint 20°C. Benzoyl chloride (0.3L, 1.5 eq.) was added and the reaction stirred 15 minutes. N,N-dimethylaminopyridine (7g, 0.04 eq.) in dichloromethane (0.1L) was added and the reaction stirred 15 minutes. Pyridine (0.5L, 2.5 eq.) was dropwise added, maintaining Tint<20°C. Upon complete addition the reaction was stirred until LCMS indicated consumption of starting material.
[00559] The reaction mixture was washed with deionized water (11L) and the organic layer extracted sequentially with aqueous hydrochloric acid (1M, 3 x 5L), saturated aqueous sodium bicarbonate (11 L), saturated sodium chloride (11 L), dried over magnesium sulfate and evaporated in vacuo. Compound A6 was obtained as a cream colored solid, suitable for use without further purification (0.9kg, 100.7%).
Step 6: Synthesis of Compound A 7
[00560] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with l,2-dimethoxy ethane (16L) and temperature set to Tint=2l°C. Compound A6 (0.9kg) was added and stirred to dissolution. Copper iodide (0.3kg, 1.0 eq.) was added and the mixture stirred 15 minutes. l, lO-phenanthroline (0.3kg, 1.2 eq.) was added and the mixture stirred 15 minutes. Cesium carbonate (l .5kg, 3.0 eq.) was added and the reaction was stirred for 15 minutes. The reaction temperature was ramped to Tint=80-85oC and maintained for 23 hours whereupon it was cooled to Tmt=20°C.
[00561] The reaction mixture was filtered through a celite pad, washing sequentially with deionized water (8L) and ethyl acetate (8L). The organic layer was extracted sequentially with deionized water (2 x 5L), saturated sodium chloride (4L), dried over sodium sulfate and evaporated in vacuo. Compound A7 was obtained as a brown solid, suitable for use without further purification (0.8kg, 104.1%).
Step 7: Synthesis of Compound A8
[00562] A 12L 3 -neck round bottom flask with nitrogen bleed and mechanical stirring was charged with a solution of Compound A7 (0.8kg) in dichloromethane (3.6L) and cooled to Tmt<5°C in an ice bath. Hydrochloric acid in dioxane (4M, 1 2L, 3.1 eq.) was added dropwise with vigorous stirring, maintaining Tmt<25°C. Once addition was complete, the reaction mixture was allowed to stir for 18 hours at Tint=20-25oC.
[00563] The reaction mixture was filtered and the filter cake washed with dichloromethane (2 x 1L) and dried to constant mass. The hydrochloride salt of Compound A8 was isolated as an off-white solid (0.5kg, 88.7%).
Step 8: Synthesis of Compound A
[00564] A 12L 3 -neck round bottom flask with nitrogen bleed and mechanical stirring was charged with a solution of Compound A8 (0.5kg) in tetrahydrofuran (4.8L) and cooled to Tint<-30°C in a dry-ice / acetone bath. Methylmagnesium bromide (3.4M in 2-methyltetrahydrofuran, 2.4L, 5.0eq.) was added slowly, maintaining Tmt<-lO°C. Once addition was complete, the reaction was allowed to warm to room temperature overnight.
[00565] Saturated aqueous ammonium chloride (2L) and ethyl acetate (2L) were added and the reaction mixture stirred for 30 minutes. The aqueous layer was extracted with further ethyl acetate (2 x 2L) and the combined organic layers washed with saturated sodium chloride (2L), dried over sodium sulfate and evaporated in vacuo to a dark heavy oil. The heavy oil was purified by column chromatography on silica gel, eluting with ethyl acetate : heptane 1 : 19 to 1 : 1. Pure Compound A was obtained after evaporation and drying as a brown powder (99.8 g, 23.0%).
Example 1 – Preparation of Free Base Forms A, B and C of Compound A
Compound A
Primary Polymorph Screen
[00566] Based on solubility screen results, a primary polymorph screen using an initial set of 24 solvents, as shown in Table 18, was performed as follows: A) To 24 x 20 mL vials, approximately 50 mg of the received ADX-103 was added; B) The solids were then slurried in 2 mL of the solvents and left placed in an incubator/shaker to temperature cycle between ambient and 40 °C in 4 hour cycles; C) After 72 hours temperature cycling, the mother liquors were removed from the vials and split evenly between 4 x 2 mL vials. The vials were then split between evaporation, crash cooling to 2 °C and -18 °C and anti-solvent addition; and D) Any solids
recovered were analysed by XRPD, any new patterns identified were also analysed by TG/DTA and PLM.
Table 18. Solvents Selected for Initial Primary Polymorph Screen
PATENT
WO2018039197 , as compound I-8.
PATENT
WO 2006127945
WO 2011072141
WO 2014116593
US 20150344447
WO 2020028820
////////////ADX-103, Preclinical, Antiinflammatory, Ophthalmic Agents, Diabetic Retinopathy, Aldeyra Therapeutics Inc,
CC(C)(O)c1cc2oc(nc2cc1N)c3ccccc3
SK1-I , BML 258


SK1-I , BML 258
Sphingosine kinase 1 (SphK1) inhibitor; antiproliferative
- (1E)-1,2,4-Trideoxy-4-(methylamino)-1-(4-pentylphenyl)-D-erythro-pent-1-enitol
- (E,2R,3S)-2-(Methylamino)-5-(4-pentylphenyl)pent-4-ene-1,3-diol
- D-erythro-Pent-1-enitol, 1,2,4-trideoxy-4-(methylamino)-1-(4-pentylphenyl)-, (1E)-
| Name: | (2R,3S,4E)-N-methyl-5-(4′-pentylphenyl)-2-aminopent-4-ene-1,3-diol . HCl |
| Formula: | C17H27NO2 . HCl |
| MW: | 313.9 |
| CAS: | 1072443-89-0
|
- Originator Enzo Biochem; Virginia Commonwealth University
- Developer Enzo Biochem
- Class Antineoplastics; Small molecules
- Mechanism of Action Sphingosine kinase inhibitors
- Preclinical Autoimmune hepatitis; Haematological malignancies; Liver cancer; Solid tumours
- 07 May 2019 Preclinical trials in Liver cancer in USA (unspecified route)
- 03 Dec 2018 SK1 I is available for licensing as of 03 Dec 2018. http://www.enzo.com/
- 03 Dec 2018 Enzo Biochem has patent pending for SK1 I worldwide
SK1 I, a small molecule that specifically inhibits sphingosine kinase 1, is being developed by Enzo Biochem for the treatment of cancer and autoimmune diseases. Preclinical development is underway for the treatment of solid tumours, liver cancer, haematological malignancies and autoimmune hepatitis in the US.
As at December 2018, Enzo Biochem seeks partners for the development of SK1
SK1-I is a sphingosine analog and a sphingosine competitive inhibitor specific for sphingosine kinase 1 (SK1), with ki~10µM and excellent water solubility. It is not to be confused with SKI-I, 5-naphthalen-2-yl-2H-pyrazole-3-carboxylic acid (2-hydroxy-naphthalen-1-ylmethylene)-hydrazide, CAS 306301-68-8, a noncompetitive inhibitor of both SK1 and SK2 with poor water solubility (K.J. French, et al., 2006; N.J. Pyne and S. Pyne, 2010). SK1-I does not inhibit SK2, PKCα, PKCδ, PKA, AKT1, ERK1, EGFR, CDK2, IKKβ or CamK2β. Not only does it decrease sphingosine-1-phosphate levels, it also causes an accumulation of its proapoptotic precursor ceremide. Inhibits tumor cell growth in vitro and in vivo.
PATENTS
US 20100035959
WO 2010127093
US 20100278741
WO 2011025545
Patent
US-10364211
This patent was granted in July 30, 2019 and set to expire on October 24, 2038. Claims methods for synthesizing the compound (2R,3S,4E)-N-methyl-5-(4′-pentylphenyl)-2-aminopent-4-ene-1,3-diol (also known as SK1-I and BML-258 (as HCl salt)) and its intermediates.
(2R,3S,4E)-N-methyl-5-(4′-pentylphenyl)-2-aminopent-4-ene-1,3-diol, also known as SK1-I and BML-258 (as HCl salt), is a pharmaceutical inhibitor of sphingosine kinase 1 initially described in Paugh et al., Blood. 2008 Aug. 15; 112(4): 1382-1391. An existing method for synthesizing SK1-I is disclosed in U.S. Pat. No. 8,314,151.
|
and |
|
|
N-Boc-(D)-Serine Methyl Ester
Protection of N-Boc-(D)-Serine Methyl Ester
(R)—Garner Aldehyde
Addition of 4-Pentylphenyl Acetylene to the Above Aldehyde
Deprotection of the Above Oxazolidine
Reduction of the Above Alcohol
Deprotection to SK1-I (BML-258)
PATENT
WO2018237379 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018237379
claiming sphingosine pathway modulating compounds for the treatment of cancers, assigned to Enzo Biochem Inc , naming different team
Sphingosine- 1 -phosphate (SIP) was discovered to be a bioactive signaling molecule over 20 years ago. Studies have since identified two related kinases, sphingosine kinase 1 and 2 (a/k/a sphingosine kinase “type I” and “type II” respectively, and SphKl and SphK2 respectively), which catalyze the phosphorylation of sphingosine to SIP. Extracellular SIP can bind to and activate each of five S IP-specific, G protein-coupled receptors (designated S IPR1-5) to regulate cellular and physiological processes in an autocrine or paracrine manner. Selective inhibitors of each of sphingosine kinase 1 and 2, as well as both nonselective and selective agonists of SlPRs, have been developed and are known in the art.
Product Literature References
General Literature References
/////////SK1-I , SK1I , SK1 I , BML 258, Enzo Biochem, Virginia Commonwealth, Preclinical, solid tumours, liver cancer, haematological malignancies, autoimmune hepatitis,
CCCCCC1=CC=C(/C=C/[C@H](O)[C@H](NC)CO)C=C1.Cl
SYN 01

SYN-01, SYN-510
Synthena AG
Preclinical
Synthena , presumed to be under license from University of Bern , is investigating (presumably SYN-01 ), a lead from the tricyclo(tc)-DNA based antisense oligonucleotides (AON) developed using its proprietary tricyclo-DNA technology platform, for the treatment of Duchenne muscular dystrophy. In January 2017, the drug was listed as being in preclinical development.
Patent
WO-2019142135
Process for preparing tricyclo-deoxyribonucleic acid (tc-DNA) which may be used as building blocks for tc-DNA containing antisense oligonucleotide-based therapies.
Antisense technology is an effective means for reducing the expression of specific gene products and can therefore be useful in therapeutic, diagnostic, and research applications.
Generally, the principle behind antisense technology is that an antisense oligomeric compound (a sequence of nucleotides or analogues thereof) hybridizes to a target nucleic acid and modulates gene expression activities or function, such as transcription and/or translation.
[003] Antisense oligomeric compounds may be prepared from chemically-modified antisense oligonucleotides, which may include a variety of different structural variations depending upon the therapeutic strategy. For example, tricyclo-deoxyribonucleic acids (tc-DNA) are conformationally constrained DNA analogs.
[004] There is a need in the field for processes that allow for the bulk preparation of tc-DNA nucleoside precursors that may be used as building blocks for tc-DNA containing antisense oligonucleotide-based therapies.
Example 4 – Cvclopropanation of Compound 17 with Carbenoid Prepared from CH2I2 and Et2Zn in the Absence of Additives
[00127] According to the following scheme, compound 17 was converted to tc-DNA Nucleoside Precursor 18 using the cyclopropanation conditions set forth in Examples 4 to 7 :
[00128] 1.07 g purified a-anomer (3.736 mmol) 17 was dissolved in 37 ml of dry CH2C12 and cooled to 0 °C (ice). Subsequently, 22.3 ml (22.3 mmol, 6 eq.) Et2Zn 1.0 M in hexane (Aldrich) were added dropwise and stirred under Ar for 30 min at 0 °C. Then, 3.02 ml (37.2 mmol, 10 eq.) of CH2I2 were added dropwise over 15 min at the same temperature and stirred for further 2 h at 0 °C. Afterwards the cooling bath was removed and the mixture was stirred for additional 21 h at ambient temperature. TLC showed substantial amount of unreacted a- 17. It was diluted by addition of EtOAc and quenched with 50 mL of sat. aqueous NH4Cl. Extractive work-up provided 1.79 g of crude which was purified by chromatography on silica-gel giving 0.43 g (39%) of 18 and 0.49 g of mixture of compound 17 and 18 (approximately 20:80).
PATENT
WO2018193428
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018193428
claiming a composition comprising an oligomeric compound having tricyclo-deoxyribonucleic acid (tc-DNA) nucleosides and a lipid moiety.
EXAMPLE 1
Inventive compositions for the treatment of Duchenne muscular dystrophy
Evaluation of efficacy
[00464] Adult mdx mice were treated weekly over 4 weeks with intravenous injections of different 13-mer AONs targeting the donor splice site of exon 23 of the dystrophin pre-mRNA (M23D: +2-11), namely with either SY-0308, SY-0210 and the inventive SY-0299, SY-0343, SY-0442 and SY-0455. SY-0308 (also named “tcDNA-PO M23D” interchangeably herein) corresponds to p-CCTCGGCTTACCT-OH of SEQ ID NO: l, with all nucleotides being tc-DNAs and all internucleosidic linkage groups being phosphorodiester linkage groups, and p being a phosphate moiety at the 5′ end. SY-0210 (also named “tcDNA-PS M23D” interchangeably herein) corresponds to p-CCTCGGCTTACCT-OH of SEQ ID NO: 1, with all nucleotides being tc-DNAs and all internucleosidic linkage groups being phosphorothioate linkage groups, and p being a phosphate moiety at the 5′ end. The inventive composition SY-0343 is herein interchangeably referred to as “Palm-2PS-tcDNA-PO M23D” which is depicted in the following:
[00465] The inventive composition SY-0442 is herein interchangeably referred to as “Palm-lPS-tcDNA-PO M23D” which is depicted in the following:
[00466] The inventive composition SY-0299 is herein interchangeably referred to as “Palm-2PO-tcDNA-PO M23D” which is depicted in the following:
//////////////////SYN-01, SYN 01, SYN01, preclinical , Duchenne muscular dystrophy, University of Bern,
HS 10340

HS-10340
CAS 2156639-66-4

CAS 2307670-65-9
Jiangsu Hansoh Pharmaceutical Group Co Ltd
Being investigated by Jiangsu Hansoh, Shanghai Hansoh Biomedical and Changzhou Hengbang Pharmaceutical ; in June 2018, the product was being developed as a class 1 chemical drug in China.
Useful for treating liver cancer, gastric cancer and prostate cancer.
Use for treating cancers, liver cancer, gastric cancer, prostate cancer, skin cancer, ovary cancer, lung cancer, breast cancer, colon cancer, glioma and rhabdomyosarcoma


PATENT
WO2017198149
where it is claimed to be an FGFR-4 inhibitor for treating liver and prostate cancers, assigned to Jiangsu Hansoh Pharmaceutical Group Co Ltd and Shanghai Hansoh Biomedical Co Ltd .
PATENT
WO2019085860
PATENT
WO-2019085927
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019085927&tab=FULLTEXT
Novel crystalline salt (such as hydrochloride, sulfate, methane sulfonate, mesylate, besylate, ethanesulfonate, oxalate, maleate, p-toluenesulfonate) forms of FGFR4 inhibitor, particularly N-[5-cyano-4-[[(1R)-2-methoxy-1-methyl-ethyl]amino]-2-pyridyl]-7-formyl-6-[(2-oxo-1,3-oxazepan-3-yl)methyl]-3,4-dihydro-2H-1,8-naphthyridine-1-carboxamide (designated as Forms I- IX), compositions comprising them and their use as an FGFR4 inhibitor for the treatment of cancer such as liver cancer, gastric cancer, prostate cancer, skin cancer, ovarian cancer, lung cancer, breast cancer, colon cancer and glioma or rhabdomyosarcoma are claimed.
///////////HS-10340 , HS 10340 , HS10340, CANCER, Jiangsu Hansoh, Shanghai Hansoh Biomedical, Changzhou Hengbang, CHINA, liver cancer, gastric cancer, prostate cancer, skin cancer, ovary cancer, lung cancer, breast cancer, colon cancer, glioma, rhabdomyosarcoma
C[C@H](COC)Nc1cc(ncc1C#N)NC(=O)N4CCCc3cc(CN2CCCCOC2=O)c(C=O)nc34
CCS(=O)(=O)O.C[C@H](COC)Nc1cc(ncc1C#N)NC(=O)N4CCCc3cc(CN2CCCCOC2=O)c(C=O)nc34
CS 3001
CS-3001
BB 7, VX 033
- Molecular Weight, 478.37
C17 H18 Br F2 N3 O2 S2
CStone Pharmaceuticals Co Ltd, JUNE 2018 IND FILED CHINA
URAT1 inhibitor – useful for treating hyperuricemia and gout.
The compound was originally claimed in WO2017202291 , covering thiophene derivative URAT1 inhibitors, useful for treating hyperuricemia and gouty arthritis, assigned to Medshine Discovery Inc , but naming the inventors.and has been reported in some instances to be a URAT1 modulator. In June 2018, an IND application was filed in



WO-2019101058
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019101058&tab=FULLTEXT&maxRec=1000
Novel crystalline forms of URAT1 inhibitor (designated as Forms A and B) are claimed. The compounds are disclosed to be useful for treating hyperuricemia and gouty arthritis.
Novel crystalline forms of a URAT1 inhibitor, designated as Forms A and B, and their preparation.
For example, remove 2.0 mL of phosphoric acid into 2000 mL of water, sonicate for 10 minutes, mix, and let cool to room temperature as mobile phase A.
////////////CS-3001, BB 7, VX 033, CHINA, PRECLINICAL, CStone Pharmaceuticals, URAT1 inhibitor, hyperuricemia, gout
O=C(O)C(C)(C)Sc4nnc(Br)n4c2sc(c1CC(F)(F)CCc12)C3CC3
TL 487
TL-487
- Molecular Weight, 507.58, MF C30 H29 N5 O3
Teligene Inc(2E)-N-[3-Cyano-7-ethoxy-4-[(4-phenoxyphenyl)amino]-6-quinolinyl]-4-(dimethylamino)-2-butenamide
(E)-N-(3-cyano-7-ethoxy-4-((4-phenoxyphenyl)amino)quinolin-6-yl)-4-(dimethylamino)but-2-enamide
Maleate in anhydrous or monohydrate CAS, 2326561-36-6, AND 2326561-38-8 form are BTK and HER-2 kinase inhibitor useful for treating cancer
Useful for treating breast cancer, ovary cancer and colon cancer. are BTK and HER-2 kinase inhibitor useful for treating cancer.
Anticancer protein kinase inhibitor
The compound was originally claimed in WO2013152135 , and may provide the structure of TL-487 , a small molecule inhibitor to HERs, being investigated by Teligene for the treatment of breast cancer; in July 2016, the company intended to develop the product as a class 1.1 chemical drug in China.
PATENT
US 20150057312
PATENT
WO2013152135

PATENT
WO-2019096327
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019096327&redirectedID=true
Novel crystalline maleate salt of (E)-N-(3-cyano-7-ethoxy-4-((4-phenoxyphenyl)amino)quinolin-6-yl)-4-(dimethylamino)but-2-enamide (first disclosed in WO2013152135) and its hydrates (monohydrate) and anhydrates, process for its preparation, composition comprising it and its use for treating cancers such as breast cancer, ovary cancer, colon cancer, prostate cancer, kidney cancer, bladder cancer, stomach cancer, lung cancer, mantle cell lymphoma and multiple myeloma are claimed. The compound is disclosed to be an irreversible inhibitor to BTK and Her-2 (also known as Erb-2 or neu).
///////////////TL-487, PRECLINICAL, CHINA, breast cancer, ovary cancer, olon cancer, BTK, HER-2 kinase inhibitor,
CN(C)C\C=C\C(=O)Nc3cc4c(Nc2ccc(Oc1ccccc1)cc2)c(cnc4cc3OCC)C#N
HM04 or H0900
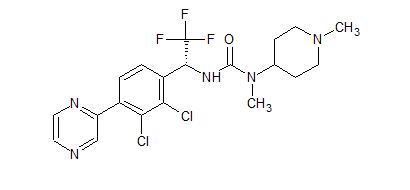
![3-[(1R)-1-(2,3-Dichloro-4-pyrazin-2-ylphenyl)-2,2,2-trifluoroethyl]-1-methyl-1-(1-methylpiperidin-4-yl)urea.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=91884613&t=l)
HM04 or H0900
Cas 1808913-24-7
(R)-3-(1-(2,3-dichloro-4-(pyrazin-2-yl)phenyl)-2,2,2-trifluoroethyl)-1-methyl-1-(1-methylpiperidin-4-yl) urea
The compound was disclosed in WO2015134839 . Helsinn under license from Novo Nordisk , is investigating ghrelin antagonists for treating obesity, Prader-Willi syndrome and other metabolic disorders; in May 2015, the program was listed as being in preclinical development
Helps reducing ghrelin signaling activity and treating disorder associated with an increase in ghrelin level (eg food abuse, alcohol addiction, and Prader-Willi syndrome).
Ghrelin, a growth hormone-releasing peptide produced by ghrelinergic cells in the gastrointestinal tract, is understood to function as a neuropeptide that regulates energy metabolism by stimulating appetite. The modulation, for example inhibition, of ghrelin signaling, through the ghrelin/growth hormone secretagogue receptor (GHS-Rla), is an attractive target for pharmacological treatment of disorders associated with high ghrelin level. Potential disorders for treatment using ghrelin modulators include food abuse (such as binge eating, obesity, hyperphagia (or uncontrollable appetite), post-dieting body weight rebound (including post-dieting hyperphagia), alcohol addiction, and genetic diseases associated with increased ghrelin level (e.g., Prader-Willi syndrome (PWS)).
PATENT
US 20150252021

PATENT
WO2015134839
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015134839
Example 1
nthesis of Intermediate lk
Intermediate k
Step 1:
To a solution of la (100 g, 0.62 mol) in DMF (1.2 L) was added N-bromosuccinimide (110 g, 0.62 mol) at 0 °C. The mixture was stirred at room temperature for 4 h, then water (800 mL) was added and the resulting mixture was extracted with EtOAc (3 x 500 mL). The combined organic layers were dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was triturated with petroleum ether to provide lb (133.7 g, 89% yield) as a brown solid. !H-NMR (CDC13, 300 MHz): δ= 7.30 (d, 1 H), 6.59 (d, 1 H), 4.22 (br, 2 H). LC-MS: 241 [M+l]+.
Step 2:
To a solution of lb (133.7 g, 0.55 mol) in dry CH2C12 (1.5 L) was added acetic anhydride (110 g, 0.62 mol) dropwise over a period of 20 minutes at room temperature. The mixture was stirred at room temperature overnight, then diluted with CH2C12 (300 mL) and washed with water (150 mL) and brine (200 mL). The organic layer was separated, dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was triturated with petroleum ether (300 mL) to provide compound lc (143.0 g, 91% yield) as a white solid. ¾-NMR (CDC13, 400 MHz): δ= 8.26 (d, 1 H), 7.63 (br, 1 H), 7.54 (d, 1 H), 2.26 (s, 3 H). LC-MS: 280 [M-l]“.
Step 3:
A mixture of compound lc (50.0 g, 0.18 mol), butyl vinyl ether (Id, 89.0 g, 0.89 mol), bis(l,3-diphenylphosphino)propane (DPPP, 22.0 g, 0.053 mol), TEA (100 mL, 0.71 mol) and Pd(OAc)2 (6.4 g, 0.027 mol) in DMSO (1.2 L) was heated at 130 °C under N2 overnight. After the reaction was completed, the mixture was cooled to 0 °C and 2N HC1 (480 mL) was added dropwise over a period of 30 minutes. Then, the mixture was extracted with EtOAc (3 x 100 mL). The combined organic layers were dried over anhydrous a2S04 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 : 10) to provide le (19.5 g, 45% yield) as a yellow solid. 1H-NMR (CDC13, 400 MHz): 3= 8.46 (d, 1 H), 7.82 (br, 1 H), 7.51 (d, 1 H), 2.63 (s, 3 H), 2.29 (s, 3 H). LC-MS: 244 [M-l]“.
Step 4:
To a solution of le (21.9 g, 89.4 mmol) in MeOH (350 mL) was added 2N NaOH solution (350 mL) at room temperature. The mixture was heated at 50 °C overnight, then cooled and concentrated under reduced pressure. The resulting solid was triturated with water (100 mL) for 30 min and filtered to provide If (18.0 g, 98% yield) as a brown solid. ¾-NMR (CDC13, 400 MHz): 3= 7.48 (d, 1 H), 6.68 (d, 1 H), 4.56 (br, 2 H), 2.62 (s, 3 H). LC-MS: 202[M-1]\
Step 5:
To a mixture of compound If (18.0 g, 89.2 mmol) and ice (360 g) in cone. HC1 (180 mL) was added a solution of NaN02 (9.2 g, 133.7 mmol) in water (20 mL) dropwise over a period of 30 minutes, and the resulting mixture stirred in an ice bath for 30 min. A solution of KI (74.0 g, 446 mmol) in water (360 mL) was added dropwise over 45 min at 0 °C. The mixture was stirred for 30 min and then extracted with EtOAc (3 x 100 mL). The combined organic layers were dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 :40) to provide lg (23.9 g, 86% yield) as a yellow solid. 1H-NMR (CDC13, 400 MHz): 3= 7.6 (d, 1 H), 7.06 (d, 1 H), 2.62 (s, 3 H).
Step 6:
To a solution of lg (23.9 g, 76.1 mmol) in MeOH (100 mL)/THF (100 mL) was slowly added NaB¾ (2.9 g, 76.1 mmol) at 0 °C. The mixture was stirred at room temperature for 5 min, and then quenched with water (100 mL). The mixture was extracted with EtOAc (3 x 100 mL). The combined organic layers were dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 : 10) to provide lh (22.4 g, 93% yield) as a white solid. 1H-NMR (CDC13, 400 MHz): 3= 7.81 (d, 1 H), 7.26 (d, 1 H), 5.23 (q, 1 H), 2.17 (br, 1 H), 1.47 (d, 3 H).
Step 7:
To a mixture of lh (22.4 g, 70.9 mmol), phthalimide (12.5 g, 85.0 mmol) and PPh3 (22.3 g, 85.0 mmol) in dry THF (450 mL) was added DIAD (21.5 g, 106.3 mmol) at room temperature under N2 protection. The mixture was stirred at room temperature overnight and then concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 : 15) to provide li (18.5 g, 58% yield) as a white solid. 1H-NMR (CDC13, 400 MHz): 3= 7.78-7.84 (m, 3 H), 7.70-7.73 (m, 2 H), 7.41-7.43 (d, 1 H), 5.76-5.81 (q, 1 H), 1.84 (d, 3 H).
Step 8:
A solution of li (7.2 g, 16.2 mmol) and hydrazine hydrate (98%, 4.0 g, 80.9 mmol) in MeOH (150 mL) was heated under reflux for 2 h, then cooled and concentrated under reduced pressure. The residue was diluted with water (100 mL) and extracted with CH2C12 (3 x 100 mL). The combined organic layers were dried over anhydrous Na2SC>4 and concentrated under reduced pressure to give lj (3.8 g, 75% yield) as a white solid. 1H-NMR (CDC13, 400 MHz): 3= 7.81 (d, 1 H), 7.25 (d, 1 H), 4.55 (q, 1 H), 1.36-1.38 (d, 3 H). LC-MS: 316 [M+l]+.
Step 9:
To a solution of lj (41. Og, 0.13 mol) in methyl tert-butyl ether (750 mL) was added slowly a solution of D-mandelic acid (7.8 g, 0.052 mol) in methyl tert-butyl ether (1 10 mL) at 45°C. The mixture was stirred at this temperature for 30 min then cooled and filtered. White solid obtained was partitioned between 5% NaOH solution (300 mL) and methyl tert-butyl ether (300 mL). The bi -phases were separated and the aqueous phase was extracted with methyl tert-butyl ether (300 mL). The combined organic layer was concentrated to provide Intermediate lk (12 g, 58.5% yield) as a white solid (ee%=98.0%, Chiralpak AD-H, 5 μπι, 4.6*250mm, mobile phase: Hex: EtOH : DEA=80 : 20 : 0.2), retention time = 6.408 min).
Example 2
Synthesis of Compoun
A suspension of N-methyl-4-piperidone 2a (13.3 g, 58.6 mmol), NH2Me (30% in MeOH, 100 mL) and Pd/C (0.66 g) in MeOH (200 mL) was heated at 60 °C under H2 atmosphere (50 psi) overnight, then cooled and filtered. The filtrate was concentrated under reduced pressure and the residue was dissolved in HC1 in dioxane (3N, 100 mL) and stirred for 30 min. The precipitate was filtered and washed with EtOAc (50 mL) to provide 2b (7.7g, 54% yield) as white powder. 1H-NMR (DMSO, 400 MHz): δ= 9.50 (br, 2 H), 3.48 (d, 2 H), 3.15-3.16 (m, 1 H), 2.96-3.01 (m, 2 H), 2.70 (s, 3 H), 2.51 (s, 3 H), 2.22-2.28 (m, 2 H), 1.94-2.02 (m, 2 H), LC-MS: 129 [M+l]+ .
Example 20
Synthesis of H0900
Step 1:
To a mixture of 16d (32 g, 120 mmol) in dry CH2CI2 (800 mL) was added Dess-Martin peroxide reagent (76 g, 180 mmol) portion- wise at 0 °C. The mixture was stirred at room temperature for 1 h, then diluted with DCM (800 mL), washed with aqueous NaHC03 solution (300 mL) and brine (300 mL). The organic phase was separated, dried over anhydrous Na2S04 and
concentrated under reduced pressure to afford crude 18a (31.4 g) which was used directly in the next step without further purification.
Step 2:
To a solution of 18a (12 g, 40 mmol) and 3b (22.2 g, 60 mmol) in DME (560 mL) were added Pd(PPh3)4 (9.25 g, 8 mmol) and Cul (1.52 g, 8 mmol) at room temperature. The mixture was stirred at 90 °C overnight, then concentrated under reduced pressure. The residue was purified with silica gel column chromatography (silica, EA : PE = 1 :5) to provide 18b (8.0 g, 79.3%) as a white solid. LC-MS: 253 [M+l]+.
Step 3:
To a solution of 18b (7 g, 27.7 mmol) and (¾)-tert-butylsulfinamide (7.27 g, 30.56 mmol) in dry THF (200 mL) was added Ti(i-OPr)4 (15.7 g, 55.4 mmol) dropwise at room temperature. The mixture was stirred at 80 °C overnight, and then cooled. Ethyl acetate (40 mL) was added, the resulting mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified with silica gel column chromatography (silica, EA:PE =1 :5) to provide 18c (6.8 g, 69%) as a yellow solid. 1H-NMR (CDC13, 400 MHz): 3= 9.10 (s, 1H), 8.97 (s, 1H), 8.72 (s, 1H), 8.64 (d, 1H),8.12 (d, 1H), 7.59 (d, 1H), 1.30 (s, 9H).LC-MS: 356 [M+l]+.
Step 4:
To a stirred solution of 18c (6.8 g, 19 mmol) and Tetrabutylammonium difluorotnphenylsilicate (15.8 g, 29 mmol) in dry THF (250 mL) was added a solution of TMSCF3 (11 g, 77 mmol) in anhydrous THF (50 mL) at -65 °C. The mixture was then stirred at -65 °C for 2 h, and at that point aqueous NH4CI solution (250 mL) was added. The mixture was diluted with ethyl acetate (250 mL), washed with brine (250 mL), dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was purified with silica gel column chromatography (silica, EA : PE=1 :2) to provide 18d (4.3 g, 52%) as a yellow solid. LC-MS: 426 [M+l]+.
Step 5:
To a stirred solution of 18d (4.3 g, 10.1 mmol) in MeOH (40 mL) was added a solution of HCl/MeOH (4N, 40 mL) at room temperature. The mixture was stirred for 1 h, then concentrated under reduced pressure. The residue was triturated with ethyl acetate (40 mL) to afford crude 18e (4.3g) which was directly in the next step without further purification. LC-MS: 322 [M+l]+.
Step 6:
To a solution of 18e (2.7 g, 7.1 mmol), 2b (3.4 g, 21.3 mmol) and TEA (80 mL) in DCM (220 mL) was added thiphosgene (3.15 g, 10.6 mmol) in DCM (40 mL) dropwise at 0 °C. The solution was warmed to ambient temperature and stirred for 1 h, then diluted with DCM ( 100 mL) and washed with aqueous Na2C03 solution (100 mL) and brine (100 mL). The organic layer was separated, dried over anhydrous Na2SC>4 and concentrated. The residue was purified with silica gel column chromatography (silica, DCM : CH3OH=10 : 1) to provide crude H0900 (2.13 g, ee%=92.5%) which was further purified through chiral separation to afford H0900 (1.6 g, 49% yield) as a white solid. (ee%=98.5%, Chiralpak IC 5um, 4.6*250mm, Phase: Hex: EtOH:
DEA=90: 10:0.2), retention tine =12.829 min. 1H-NMR (CDC13, 400 MHz): δ= 8.86 (d, 1H), 8.63 (dd, 1H), 8.55 (d, 1H), 7.47 (d, 1H), 7.40 (d, 1H), 6.28 (m, 1H), 5.18 (d, 1H), 4.12 (m, 1H), 2.88 (t, 2H), 2.77 (s, 3H), 2.22 (s, 3H), 2.05 (m, 2H), 2.48 (m, 2H), 1.52 (m, 2H), 1.73-1.49 (m, 4H). LC-MS: 476 [M+l]+.
PATENT
WO-2019118298
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019118298&tab=PCTDESCRIPTION&maxRec=1000
Novel crystalline fumarate salt forms of (R)-3-(1-(2,3-dichloro-4-(pyrazin-2-yl)phenyl)-2,2,2-trifluoroethyl)-1-methyl-1-(1-methylpiperidin-4-yl) urea (also referred to as HM04 or H0900; designated as Forms 1-4), process for their preparation and compositions comprising them are claimed.
PWS occurs in approximately 1 in 10,000 births and is associated with deletion or lack of expression of region 15ql 1.2 of the paternal chromosome 15.
Characteristics of PWS include short stature, low muscle tone, and hyperphagia. Growth hormone replacement is frequently used to treat growth deficiencies and hypotonia. However, treatment for the insatiable appetite is lacking and PWS children can mature into adults suffering from obesity and type 2 diabetes. Levels of ghrelin are generally elevated in PWS; however, the relationship with ghrelin signaling and food intake in PWS remains unclear. See Purtell L., et ah, In adults with Prader-Willi syndrome, elevated ghrelin levels are more consistent with hyperphagia than high PYY and GLP-l levels. Neuropeptides. 201 l;45(4):30l-7; Cummings D.E., et ah, Elevated plasma ghrelin levels in Prader Willi syndrome. Nature Medicine . 2002;8(7):643-4; DelParigi A., et ah, High circulating ghrelin: a potential cause for hyperphagia and obesity in Prader-Willi syndrome. The Journal of Clinical Endocrinology and Metabolism. 2002;87(l2):546l-4.
[005] Accordingly, it is desirable to find treatments that effectively inhibit GHSRla, that are tolerable to the patient, and that do not interfere with other functions of the growth hormones. GHSRla modulators, including inhibitors such as (R)-3-(l-(2,3-dichloro-4-(pyrazin-2-yl)phenyl)-2, 2, 2-trifluoroethyl)-l -methyl- l-(l-m ethylpiperidin-4-yl) urea (HM04, H0900) depicted below, are reported in LT.S. Patent No. 9,546,157.
Step 1 : Synthesis of compound 2A
[00106] 2,2,6,6-tetramethylpiperidine (7.20 kg, 51.1 mol, 3.0 eq.,
KF=0.30%) was added into a 100 L reactor equipped with a temperature probe and overhead stirrer and mixed at RT under nitrogen protection. THF (50 L) was added into the reactor and stirred. The vessel was purged with nitrogen three times and cooled to 0 °C. n-BuLi (20.4 L, 3.0 eq.; 2.5 M hexane solution) was added to the mixture dropwise while keeping the temperature at about 0 °C to about 5 °C for over one hour. The color of the solution turned yellow. The mixture was stirred at about 0 °C to about 5 °C for 30 minutes. The mixture was cooled to about -78 °C to about -70 °C to form Solution A.
[00107] Compound 1 (3.25 kg, 17.0 mol. 1.0 eq., KF=0.03%) was dissolved in 15 L of THF to form Solution B.
[00108] Solution B was added to solution A dropwise at a temperature of about -70 °C to about -78 °C over one hour and then stirred for 30 minutes to form solution C. Tri-isopropyl borate ((i-PrO)3B) (3.52 kg, 18.7 mol., 1.1 eq.) was added dropwise into solution C over 10 minutes. The reaction mixture was stirred at a
temperature of about -70 °C to about -78 °C for one hour. HC1 (40 L, 3M, 7.0 eq.) was added over 30 minutes to quench the reaction. A 10 degree rise in temperature was noted.
[00109] The resulting aqueous layer was separated and extracted with EtOAc (40 L). The aqueous layer was separated and extracted twice again with EtOAc (35 L, 30 L). The organic layers were combined resulting in about 160 L of liquid. The combined organic layer was washed twice with 50 L of a 1M aqueous HC1 solution saturated with NaCl. The organic layer was concentrated to about 5 L in a 50 L rotavapor at a temperature of about 50 °C to about 55 °C under 30-40 mmHg for about 8 hours.
[00110] The residual EtOAc was swapped with DME for 3 times (10 L x 3). The organic layer was concentrated in the 50 L rotavapor at a temperature of about 50 °C to about 55 °C under 30-40 mmHg for about 6 hours. Each time about 5 L of residual remained. DME (20 L) was added to the residual to obtain a deep brown solution of 14.2% compound 2A (3.55 kg in 25 kg of solution; 88.8% yield; 97.4% purity (AETC by HPLC, retention time = 1.6 minutes); 0.24% residual ethyl acetate). 1H-NMR (400 MHz, DMSO): 5=8.55 (s, 2H), 7.36 (d, 1H), 7.69 (d, 1H). A second batch of compound 2A was prepared by the same method to produce 3.29 kg (95.4% purity, 82.3% yield, 0.11% residual ethyl acetate).
[00111] Step 2: Synthesis of Compound 3A
C! , N
M
K2CO3 (I .O equiv)
2A OH
DME/H20 3:1 (20 vol), 50 e C 3A N
[00112] Compound 2 A (2.91 kg in 20.5 kg solution) was added into a 100
L reactor at room temperature under nitrogen. DME (45 mL), 2-chloropyrazin (1.42 kg,
12.4 mol., 1.0 eq.), and Pd(dppf)Cl2 (10% w/w, 291 g) were added sequentially, and each
mixed at room temperature under nitrogen. Nitrogen was bubbled into the mixture for 20
minutes and the resulting mixture was purged and filled with nitrogen (3 times). The
mixture was heated to 48-52°C over 60 minutes. K2CO3 (2.57 kg, 18.6 mol, 1.5 eq.) was
added to 22 L of water in another reactor at room temperature and then added dropwise to
the compound 2 A mixture over 10 minutes. The mixture was stirred at 48-52°C for 16
hours and then cooled to room temperature. This procedure was repeated twice and all
three batches were combined.
[00113] An aqueous solution of K2CO3 (1.0 kg) was dissolved in 22 L of
water and added to the combined mixture to adjust the pH to 9. TBME (50 L) was added
into the mixture and filtered (PET filter, 3-5 pm, 205g/m2) to remove about 50 g of
sticky, brown solid material (catalyst analog). The aqueous layer was twice separated and
extracted with TBME (40 L, 40L).
[00114] The aqueous layer was combined with the aqueous layer of a
fourth batch prepared according to the above method. The pH of the combined aqueous layers was adjusted to pH<3 with HC1 (2N, 48 L). The solid precipitated out slowly as
the mixture was stirred at room temperature for 1 hour. The mixture was filtered (PET
filter, 3-5 pm, 205g/m2) over 30 minutes to obtain 20 kg of wet product. ACN (40 L) was
added into a 100 L reactor equipped with an overhead stirrer at room temperature. The 20
kg of wet product was added into the reactor and the reaction mixture heated to reflux
and stirred at reflux for 4 hours. The reaction mixture was cooled to room temperature
over 3 hours (around 15 °C/hour) and filtered to obtain 8.5 kg of wet solid. The wet solid
was dried under vacuum (20-30 mmHg) at 50-55 °C for 15 hours to obtain compound 3 A
as a pale white solid (6.1 kg; 97.4% purity (AUC by HPLC, retention time = 3.7
minutes); 83.8% yield). 1H-NMR (400 MHz, DMSO): 5=7.67 (d, 1H), 7.82 (d, 1H), 8.75
(d, 1H), 8.82 (t, 1H), 8.98 (d, 1H), 13.89 (bs, 1H).
[00115] Step 3: Synthesis of compound 6A
3A 6A N
N
[00116] Compound 3 A (6.1 kg, 22.7 mol, 1.0 eq.) was added into a 100 L
reactor equipped with a temperature probe, overhead stirrer, and condenser. Methanol
(92 L) was added into the reactor at room temperature. The mixture was cooled to
0-10 °C and added with SOCk (5.4 kg, 45.3 mol, 2.0 eq.) dropwise at 0-10 °C over 30
minutes. The reaction mixture was heated to reflux (65 °C) and stirred at reflux for 15
hours. A suspension was formed. Most of the solvent and SOCk was removed under
vacuum distillation until about 30 L remained. The mixture was concentrated under
vacuum (30-40 mmHg) at 50-55 °C for about 6 hours. Water (10 L) was added to the residual at -5 to 15 °C. The pH was adjusted to 8-9 with an aqueous solution of K2CO3 (200 g, dissolved in 2L of water) at -5 to 15 °C. The resulting aqueous layer was extracted twice with isopropyl acetate (25 L, 25 L). The combination of organic layers (about 50 kg) was washed with 20 L of NaHCCb aqueous layer. The organic layer was separated and washed with 10 L of of an aqueous solution of NaHCCb. All the aqueous layers were combined (55.8 kg). The organic layer was filtered through a silica pad (30 cm) and the pad washed with extra isopropyl acetate until the compound 6 A was filtered from the silica gel (about 3 hours). The organic layer was concentrated to about 5 L. THF (10 L) was added to the residual and concentrated to about 5 L (3 times) under vacuum (30-40 mmHg) at 50-55 °C for about 3 hours. Another 10 L of THF was added to the residual concentrate, giving a concentrated solution of compound 6A (15.8 kg; 32.83%,
5.19 kg compound 6A in solution; 97.9% purity (AUC by HPLC, retention time = 8.5 min); 80.8% yield). 1H-NMR (400 MHz, DMSO): 5=3.98 (s, 3H), 7.54 (d, 1H), 7.78 (d, 1H), 8.63 (d, 1H), 8.72 (t, 1H), 8.94 (d, 1H).
[00117] Step 4: Synthesis of compound 6B
[00118] THF (26 L) was added into a 100 L reactor equipped with a temperature probe and overhead stirrer under nitrogen. DIBAL-H (26 kg, 46 mol, 5.0 eq.) was added and the system purged and filled with nitrogen three times. The mixture was cooled to -78 to -70 °C to form solution A. A room temperature solution of compound 6A (2.6 kg, 9.2 mol, 1.0 eq.) in 52 L of THF was added dropwise at -78 to -70 °C over 30 minutes under nitrogen. The mixture was warmed to -30 °C over about 5-6 hours. The reaction mixture was stirred at -40 to -30 °C for 30 minutes. The mixture was slowly added to 42 L of 2N HCL over 1 hour reaching a maximum temperature of 35 °C. The mixture was extracted with 26 L of isopropyl acetate. The organic layer was separated and washed with 30 L of brine. This procedure was repeated and both batches of organic layer were combined and concentrated from about 100 L to about 5-10 L under vacuum.
A solid slowly formed during concentration. The mixture was cooled to 5-15 °C and stirred for 1 hour. The mixture was filtered (30-50 pm) over 30 minutes. The solid was dried under vacuum at 50 °C for 6 hours to obtain compound 6B as a brown solid (2.1 kg; 97.5% purity (AUC by HPLC, retention time = 8.6 min); 45.7% yield). 1H-NMR (400 MHz, DMSO): d = 4.65 (d, 2H), 5.68 (t, 1H), 7.62 (d, 1H), 7.68 (d, 1H), 8.72 (d, 1H),
8.80 (t, 1H), 8.94 (d, 1H).
[00119] Step 5: Synthesis of compound 7
[00120] DMSO (10 L) was added to a 50 L flask equipped with a temperature probe and overhead stirrer under nitrogen at room temperature. Compound 6B (2.05 kg, 8.04 mol, 1.0 eq.) was added under nitrogen at room temperature. Et3N (8 L) was added under nitrogen at RT and the mixture was then cooled to 15-20 °C.
SCb. pyridine (5.1 kg, 32.08 mol, 4.0 eq.) was dissolved into 10 L of DMSO at 5-15 °C in a separate flask and added to the mixture dropwise over 3.5 hours at about 20 °C. The reaction mixture was transferred to 70 L of ice-water. The suspension mixture was stirred at 0-10 °C for 1 hour and filtered (PET, 3-5 pm, 205 g/m2) by centrifuge over 1.5 hours to obtain compound 7 as a brown solid. The solid was dissolved in 35 L of DCM at room temperature. The resulting DCM layer was washed with 5 L of brine. The organic layer was separated and concentrated under vacuum at 40-45 °C to dryness to obtain compound 7 as a brown solid (2.33 kg; 96.3% purity (AEiC by HPLC, retention time = 9.2 minutes); 93.5% yield). 1H-NMR (400 MHz, DMSO): d = 7.67 (d, 1H), 7.99 (d, 1H), 8.67 (d, 1H), 8.75 (s, 1H), 8.99 (d, 1H), 10.56 (s, 1H).
[00121] Step 6: Synthesis of compound 8
[00122] THF (23 L) was added to a 50 L flask equipped with a temperature probe and overhead stirrer under nitrogen at room temperature. Compound 7 (2.3 kg, 9.1 mol, 1.0 eq.) and (S)-2-methylpropane-2-sulfmamide (1.21 kg, 10 mol, 1.1 eq.) were added sequentially to the flask under nitrogen. Ti(OEt)4 (6.22 kg, 27.3 mol, 3.0 eq.) was added dropwise to the flask over 1 hour at 30-35 °C under nitrogen. The system was purged with nitrogen three times and then the mixture was stirred at room temperature for 2 hours. Isopropyl acetate (40 L) was added to the reaction mixture. The entire reaction mixture was then charged to 20 L of brine while stirring slowly at RT. A lot of solid was formed and no heat release was observed. The solid (about 18 kg) was filtered using centrifuge, and then the solid was slurried with 20 L of isopropyl acetate again for 20 minutes, and filtered again, resulting is slightly less solid (17.3 kg). The filtrates were then combined and washed with 20 L of brine. The organic layer was separated and concentrated in a rotavapor under vacuum (30-40 mmHg) at 40-50 °C for about 4 hours to remove the solvents and obtain a brown oil (compound 8). The oil was dissolved in DMF to obtain a black solution (7.36 kg; 40.1%; 3.0 kg compound 8 in solution; 92.1% purity (AUC by HPLC, retention time = 9.7 minutes); >100% yield). 1H-NMR (400 MHz, CDCb): d = 1.30 (s, 9H), 7.59 (d, 1H), 8.11 (d, 1H), 8.64 (s, 1H), 8.73 (m, 1H), 8.97 (s, 1H), 9.10 (s, 1H).
[00123] Step 7: Synthesis of compound 11
O
S
10 s C
8
11 N
[00124] DMF (26 L, 10 v/w) was added to a 50 L flask equipped with a temperature probe and overhead stirrer under nitrogen at 15 °C. Compound 8 (7.3 kg of
DMF solution, containing 2.9 kg, 8.1 mol, 1.0 eq.) and TBAA (2.44 kg, 8.1 mol, 1.0 eq.) were added sequentially to the flask under nitrogen. The mixture was cooled to 0-10 °C.
TMSCF3 (2.88 kg, 20.3 mol, 2.5 eq.) was then added to the flask over 60 min at 0-10 °C.
The reaction mixture was stirred at 0-5 °C under nitrogen protection for 3 hours.
Isopropyl acetate (60 L) was added to the mixture, followed by the addition of 45 L of
NaHCCb under stirring at 5-25 °C. The organic layer was separated, washed three times with NaHC03 (30 L x 3), and concentrated from 60 kg to 2.5 kg of brown oil. The oil product was dissolved in 20 L of TBME and filtered through a pad of silica gel (about 40 cm high, 30 cm diameter) over 2 hours to obtain 2.14 kg of compound 1 1 in TBME solution. The solution was concentrated at 45-50 °C to dryness to obtain compound 1 1 as a black oil (1.85 kg; 85.2% purity (AETC by HPLC, retention time = 9.1 minutes, 9.6 minutes for diastereoisomer); 53.6% yield). 1H-NMR (400 MHz, CDCh): d = 1.33 (s, 9H), 3.82-3.85 (d, 1H), 5.61-5.66 (m, 1H), 7.53-7.60 (m, 2H), 8.63-8.64 (d, 1H), 8.71-8.72 (m, 1H), 8.95 (s, 1H).
[00125] Step 8: Synthesis of compound 12 (free base)
[00126] Compound 1 1 (1.8 kg, 4.23 mol, 1.0 eq., crude) was added to a 50 L reactor equipped with a temperature probe and overhead stirrer under nitrogen at 25 °C. Anhydrous MeOH (18 L) was added to dissolve compound 1 1. Then MeOH/HCl (18 L, 1 N) was added dropwise at 25-30 °C over 10 minutes and the mixture was stirred at 25-30 °C for 1 hour. Water (15 L) was added to the reaction and the mixture concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 4 hours to remove the solvent. The pH of the mixture was adjusted to 10 with 5 L of K2CO3 solution. 20 L of EtOAc was then added to the mixture and the organic layer was separated and the aqueous layer extracted twice with EtOAc (15 L x 2). The organic layers were combined and washed with 10 L of brine. The combined organic layers contained 996 g of
compound 12 in 40 kg of EtOAc solution (84% purity (AUC by HPLC, retention time =
2.8 minutes). The organic layers were concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 3 hours to a 7.5 kg volume of compound 12 in EtOAc solution (83% purity (AETC by HPLC, retention time = 2.7 minutes).
[00127] In a separate 50 L reactor equipped with a temperature probe and overhead stirrer, D-CSA was added (930 g, 4.0 mol, 1.0 eq. to 1.26 kg compound 12) and stirred at room temperature under nitrogen. EtOAc (10 L) and then the EtOAc solution of compound 12 (1.26 kg, 3.9 mol, 1.0 eq.) were each sequentially added to the reactor. The mixture was stirred at room temperature for 1 hour and slowly became a suspension. The mixture was filtered by centrifuge and washed with EtOAc to produce 2.3 kg of compound 12 as an off-white solid (96.0% purity).
[00128] The solid product, 20 L of EtOAc, and 10 L of 10% aqueous K2CO3 were added sequentially to a 50 L flask and stirred at room temperature until no solid remained (pH = 9-10). The organic layer was separated and the aqueous layer extracted twice with EtOAc (10 L x 2). The organic layers were combined (about 32 kg) and washed with 10 L of brine. The organic layer contained 716 g of compound 12 in
31.8 kg of solution.
[00129] The organic layer was concentrated under vacuum at 45-50 °C to about 8 L. Activated carbon (200 g) was added to the organic layer and the mixture stirred at 60-70 °C for 1 hour, cooled to room temperature, and filtered using a Buchner funnel and filter paper (pore size: 30-50 pm) over 30 minutes to remove the activated carbon. The mixture was concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 3 hours to yield 710 g of compound 12 as a yellow solid (99.4% purity). [00130] D-CSA (410 g, 1.77 mol, 1.0 eq. to 680 g compound 12), 3.4 L iPrOH, and 68 mL of water were added sequentially to a 10 L reactor equipped with a temperature probe and overhead stirrer and stirred at room temperature under nitrogen. The mixture was heated to reflux (84 °C) to form solution A after 1 hour. Compound 12 (680 g) was dissolved in 3.4 L of iPrOH and added into solution A for one partition. A clear solution was formed and the temperature decreased to 65 °C. The mixture was stirred at 65 °C for about 15 minutes after which a solid appeared. The mixture was cooled to 10 °C over 2 hours, stirred at 10 °C for an additional 30 minutes, and filtered through a Buchner funnel and filter paper (pore size: 30-50 pm) over 30 minutes to collect the 1.1 kg of white solid.
[00131] EtOAc (10 L), 1.1 kg of white solid product, and 5 L of 10% K2CO3 were added sequentially to a 20 L flask and mixed for 5 minutes. The solid dissolved (pH = 9-10). The EtOAc layer was separated and the aqueous layer extracted twice with EtOAc (5 L each). The organic layers were combined (about 20 L), washed with 5 L of brine, and concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-55 °C for about 3 hours to remove most of the solution and until the residue weight reached 1 kg. Heptanes (1 L) was added to the mixture and stirred at room temperature for 30 minutes. The mixture was filtered using a Buchner funnel and filter paper (pore size: 30-50 pm) over 30 minutes to obtain 419 g of compound 12 base as a white solid (99.7% purity). The filtrate was concentrated to 135 g of compound 12 as a yellow solid (98.7% purity). 1H-NMR (400 MHz, CDCh): d = 1.85 (bs, 2H), 5.17 (m, 1H), 7.56 (d, 1H), 7.68 (d, 1H), 8.62 (d, 1H), 8.70-8.71 (m, 1H), 8.93 (s, 1H). Combined, the products resulted in a 40.7% yield of compound 12.
[00132] Step 9: Synthesis of compound 10
10A 10
[00133] Pd/C (40 g, 5% w/w) was added into a 10 L autoclave reactor at room temperature under nitrogen. THF (2 L), 2 L of methylamine (27%-30% alcoholic solution, 2.1 eq.), and 800 g of compound 10A (7 mol, 1.0 eq.) were sequentially added into the reactor. The system was purged with hydrogen three times. The mixture was stirred at hydrogen pressure (50 psi) at 70-75 °C overnight and was then filtered using a Biichner funnel and filter paper (pore size: 30-50 pm) over 10 minutes to remove the Pd/C. The filtrate was concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 3 hours to obtain 933 g of yellow oil. The mixture was distilled without a column at atmospheric pressure and the 140-170 °C portion was collected to obtain 763 g of compound 10 as a colorless oil (98.6% purity (AUC by HPLC, retention time = 4.8 minutes); 84.2% yield; 8000 ppm residual ethanol). A portion of the oil (563 g) was distilled using a 3 cm column at atmospheric pressure and the 140-170 °C portion was collected to obtain 510 g of compound 10 (75.8% yield; 134 ppm residual ethanol). 1H-NMR (400 MHz, CDCb): d = 0.82 (bs, 1H), 1.10-1.12 (q, 2H), 1.66 (d, 2H), 1.73-1.81 (t, 2H), 2.05 (s, 3H), 2.08-2.19 (m, 1H), 2.22 (s, 3H), 2.60 (d, 2H).
[00134] Step 10: Synthesis of HM04 fumarate salt
[00135] DCM (1L), 200 g CDI (1.23 mol, 2.0 eq.), and 35 g DABCO (0.31 mol, 0.5 eq.) were sequentially added into a 3 L reactor equipped with a temperature probe and overhead stirrer, and stirred at room temperature under nitrogen. The mixture was cooled to -10 to -5 °C. Compound 12 (200 g) was dissolved in 1 L of DCM and added into the mixture dropwise over 1 hour, followed by stirring for 16 hours at -10 to -5 °C. Compound 10 (159 g, 1.24 mol, 2.0 eq.) was added at -10 to 0 °C over 10 minutes. The mixture was then warmed to 0 to 5 °C and held for 2 hours. The mixture was concentrated under vacuum at 40-45 °C to about 1 L. HC1 (1 L of 1 N) was added to the residual and concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 2 hours to remove the DCM. Another 3 L of 1N HC1 was added to the residual and extracted three times with TBME (4 L, 2 L, 2 L). The aqueous layer was slowly adjusted to pH = 9-10 with 20% aqueous K2CO3 (about 1.5 L) and extracted with DCM (2 L x 3). The organic layers were combined (about 4 L) and washed three times with 0.25 N KH2PO4 (1.2 L x 3). The organic layer was washed with 2 L of brine to bring the pH to neutral and concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 2 hours to 450 g (335 mL). MTBE (1.5 L) was added to the residual and distilled until 500 mL of liquid was collected. This step was repeated four times with the addition of 500 mL of TBME and collection of 500 mL of distillate, with the exception that 330 mL of liquid was collected at the final distillation. About 1 to 1.2 L of residual remained in the flask. The residual was slowly cooled to room temperature and stirred at room temperature overnight. The mixture was filtered, washed twice with TBME (400 mL x 2), and dried to obtain 192 g of HM04 free base a light yellow solid (99.3% purity (AUC by HPLC, retention time = 11.0 minutes). The product on the wall was dissolved in DCM and concentrated under vacuum to obtain 22 g of HM04 free base as a brown sticky oil (97.6% purity). The filtrate was concentrated under vacuum to obtain 22.5 g of yellow solid (94.0% purity).
[00136] HM04 free base (187 g, 0.39 mol, 1.0 eq., 99.3% purity) and 1.9 L of ACN were sequentially added to a 3 L flask equipped with a temperature probe and overhead stirrer and stirred at 15 °C under nitrogen to obtain a light-yellow suspension. Fumaric acid (45.6 g, 0.39 mol, 1.0 eq.) was added to the flask and generated a white suspension after 1 minute. The reaction suspension was stirred overnight at room temperature, filtered (15-20 pm, ash<0.l5), washed twice with ACN (50 mL x 2), and dried under vacuum at 50 °C for 6 hours to obtain 207 g of HM04 fumarate salt as a light yellow solid (99.4% purity (AUC by HPLC, retention time = 11.1 minutes); 57.8% yield; 3100 ppm residual ACN). The filtrate was concentrated under vacuum to obtain 20.1 g of HM04 fumarate salt as a light yellow solid (97.3% purity).
[00137] A portion of the product (117 g) was further dried in a vacuum oven (20-40 mmHg) to lower the residual acetonitrile content. After drying at 60 °C for 6 hours, 15 hours, and 72 hours; and at 65 °C for 18 hours, the residual acetonitrile content was measured as 3100 ppm, 2570 ppm, 1300 ppm, and 256 ppm, respectively. After the drying process, 98 g of HM04 fumarate salt was isolated (99.4% purity (AUC by HPLC, retention time = 11.0 minutes); 1H-NMR (400 MHz, DMSO): d = 1.49-1.58 (m, 2H),
1.81-1.92 (m, 2H), 2.44-2.53 (m, 5H), 2.78 (s, 3H), 3.12 (m, 2H), 4.06-4.13 (m, 1H), 6.36-6.41 (m, 1H), 6.55 (s, 2H), 7.47 (d, 1H), 7.73 (d, 1H), 8.11 (d, 1H), 8.75 (d, 1H),
8.81-8.82 (m, 1H), 8.99 (d, 1H). The yield of 98g of HM04 fumarate salt isolated after drying the partial batch was extrapolated over the whole batch to calculate an
approximate yield of 48% for step 10.
[00138] XRPD analysis of HM04 fumarate salt products obtained after drying at 60 °C for 6 hours, 15 hours, and 72 hours; and at 65 °C for 18 hours was performed (see Figures 6-9, respectively). The XRPD profile showed that the HM04 fumarate salt product was consistent with Form 1.
Example 6. Streamlined Synthesis of HM04 Fumarate Salt Form 1
[00139] The overall yield of HM04 fumarate salt produced using Step 10 of Example 5 was calculated as approximately 48%. In order to increase the overall yield, a streamlined synthesis was investigated that eliminated the step of isolating HM04 free base. In particular, step 10 of the method of Example 5 shown in Figure 5 was changed. An overview of the streamlined synthesis beginning after step 9 of Example 5 is shown in Figure 10.
[00140] Streamlined HM04 Fumarate Salt Trial 1 : PCM (121.4 g). CPI (20.0 g, 123 mmol, 2 eq.) and DABCO (3.5 g, 31 mmol) were sequentially added into an inertized 1 L reactor. The mixture was cooled to -10 °C. Separately, a solution of DCM (132.5 g) and compound 12 (20.0 g, 62.1 mmol) were charged into a vessel and stirred until a solution was obtained. This solution was dropped into the 1 L reactor over 33 minutes by keeping the internal temperature at -10 to -5 °C. At the end of the addition, the vessel was rinsed with DCM (7.0 g), which was then added to the reaction mixture.
After stirring overnight (19 hours) and positive IPC, compound 10 (15.9 g, 124 mmol, 2 eq.) was added over 15 minutes and the vessel rinsed with DCM (9.0 g). After heating at 0 °C, 1 hour of stirring, positive IPC, and a further 1.5 hours of stirring, the mixture was heated at room temperature and charged with water (200.1 g). The aqueous layer was separated and the organic layer extracted twice with 1 N HC1 (201, 200 g). The combined aqueous layers containing the product were washed with TBME (148 g). After removal of the organic layer, the aqueous layer was charged with DCM (265.0 g) and 50% K2CO3 solution (about 240 ml) until reaching pH 9.61.
[00141] Meanwhile, a solution of KH2PO4 (8.2 g) in water (240 g) was prepared. The organic layer containing the product was charged with the KH2PO4 solution until reaching pH 7.12 (142.2 g). After separation of the aqueous layer, the organic layer was washed with water (200 g). After separation of the aqueous layer, the organic layer was evaporated at 50 °C. ACN (314.4 g) was added and the solvent distilled again at 70-75 °C under vacuum. ACN (235.8 g) was added and the solvent distilled again under vacuum. ACN (141.5 g) was added, the resulting solution polish filtered and the filter washed with ACN (16 g). After heating at 60 °C, fumaric acid (7.2 g, 62 mmol) was added to the solution, causing a white precipitate. After cooling to 20 °C over 1 hour, the suspension was filtered and washed twice with TBME (2 x 30 g). After drying on the filter with nitrogen flow, 70.7 g of wet raw product was obtained. This was slurried with TBME (177.0 g) for 1 hour, filtered, and washed with TBME (70 g). After drying on the filter under nitrogen flow, 33.0 g of wet product was obtained. Heating at 50 °C under vacuum afforded the dry product as a white powder of HM04 fumarate salt (21.1 g;
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9926337 | SUBSTITUTED ASYMMETRIC UREAS AND MEDICAL USES THEREOF | 2016-12-02 | |
| US9546157 | p-Substituted Asymmetric Ureas and Medical Uses Thereof | 2015-03-06 | 2015-09-10 |
////////////HM04, H0900, Helsinn, Novo Nordisk, PRECLINICAL, obesity, Prader-Willi syndrome, ghrelin
CN(C1CCN(C)CC1)C(=O)N[C@H](c3ccc(c2cnccn2)c(Cl)c3Cl)C(F)(F)F

{kind=link}
{kind=link}