
Home » FDA 2018
Category Archives: FDA 2018
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
ENCORAFENIB, エンコラフェニブ

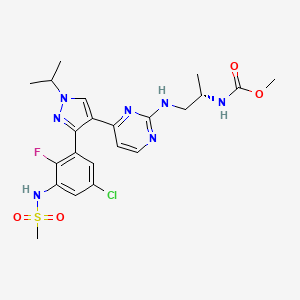
ENCORAFENIB, エンコラフェニブ
UNII:8L7891MRB6
Formula:C22H27ClFN7O4S, Average: 540.01
1269440-17-6
- BRAFTOVI
- NVP-LGX818
- NVP-LGX-818-NXA
- NVP-LGX818-NXA
- ENCORAFENIB [USAN]
- ENCORAFENIB [WHO-DD]
- ENCORAFENIB
- ENCORAFENIB [INN]
- METHYL N-((2S)-1-((4-(3-(5-CHLORO-2-FLUORO-3-(METHANESULFONAMIDO)PHENYL)(-1-(PROPAN-2-YL)-1H-PYRAZOL-4-YL(PYRIMIDIN-2-YL)AMINO)PROPAN-2-YL)CARBAMATE
- CARBAMIC ACID, N-((1S)-2-((4-(3-(5-CHLORO-2-FLUORO-3-((METHYLSULFONYL)AMINO)PHENYL)-1-(1-METHYLETHYL)-1H-PYRAZOL-4-YL)-2-PYRIMIDINYL)AMINO)-1-METHYLETHYL)-, METHYL ESTER
- LGX818
- LGX-818
Encorafenib, also known as BRAFTOVI, is a kinase inhibitor. Encorafenib inhibits BRAF gene, which encodes for B-raf protein, which is a proto-oncogene involved in various genetic mutations Label. This protein plays a role in regulating the MAP kinase/ERK signaling pathway, which impacts cell division, differentiation, and secretion. Mutations in this gene, most frequently the V600E mutation, are the most commonly identified cancer-causing mutations in melanoma, and have been isolated in various other cancers as well, including non-Hodgkin lymphoma, colorectal cancer, thyroid carcinoma, non-small cell lung carcinoma, hairy cell leukemia and adenocarcinoma of the lung 6.
On June 27, 2018, the Food and Drug Administration approved encorafenib and Binimetinib(BRAFTOVI and MEKTOVI, Array BioPharma Inc.) in combination for patients with unresectable or metastatic melanoma with a BRAF V600E or V600K mutation, as detected by an FDA-approved test Label.
Array Biopharma (a wholly owned subsidiary of Pfizer ), under license from Novartis , and licensees Pierre Fabre and Ono Pharmaceutical have developed and launched the B-Raf kinase inhibitor encorafenib . In January 2020, the US FDA’s Orange Book was seen to list encorafenib patents such as US8946250 , US8501758 , US9314464 and US9763941 , expiring in the range of 2029-2032. At that time Orange Book also reported that encorafenib as having NCE exclusivity expiring on July 27, 2023.
Encorafenib (trade name Braftovi) is a drug for the treatment of certain melanomas. It is a small molecule BRAF inhibitor [1] that targets key enzymes in the MAPK signaling pathway. This pathway occurs in many different cancers including melanoma and colorectal cancers.[2] The substance was being developed by Novartis and then by Array BioPharma. In June 2018, it was approved by the FDA in combination with binimetinib for the treatment of patients with unresectable or metastatic BRAF V600E or V600K mutation-positive melanoma.[3][4]
The most common (≥25%) adverse reactions in patients receiving the drug combination were fatigue, nausea, diarrhea, vomiting, abdominal pain, and arthralgia.[3]
Indication
Used in combination with Binimetinib in metastatic melanoma with a BRAF V600E or V600K mutation, as detected by an FDA-approved test 5.
Associated Conditions
Pharmacodynamics
Encorafenib has shown improved efficacy in the treatment of metastatic melanoma 3.
Encorafenib, a selective BRAF inhibitor (BRAFi), has a pharmacologic profile that is distinct from that of other clinically active BRAFis 7.
Once-daily dosing of single-agent encorafenib has a distinct tolerability profile and shows varying antitumor activity across BRAFi-pretreated and BRAFi-naïve patients with advanced/metastatic stage melanoma 7.
Mechanism of action
Encorafenib is a kinase inhibitor that specifically targets BRAF V600E, as well as wild-type BRAF and CRAF while tested with in vitro cell-free assays with IC50 values of 0.35, 0.47, and 0.3 nM, respectively. Mutations in the BRAF gene, including BRAF V600E, result in activated BRAF kinases that mahy stimulate tumor cell growth. Encorafenib is able to bind to other kinases in vitro including JNK1, JNK2, JNK3, LIMK1, LIMK2, MEK4, and STK36 and significantly reduce ligand binding to these kinases at clinically achievable concentrations (≤ 0.9 μM) Label.
In efficacy studies, encorafenib inhibited the in vitro cell growth of tumor cell lines that express BRAF V600 E, D, and K mutations. In mice implanted with tumor cells expressing the BRAF V600E mutation, encorafenib induced tumor regressions associated with RAF/MEK/ERK pathway suppression Label.
Encorafenib and binimetinib target two different kinases in the RAS/RAF/MEK/ERK pathway. Compared with either drug alone, co-administration of encorafenib and binimetinib result in greater anti-proliferative activity in vitro in BRAF mutation-positive cell lines and greater anti-tumor activity with respect to tumor growth inhibition in BRAF V600E mutant human melanoma xenograft studies in mice. In addition to the above, the combination of encorafenib and binimetinib acted to delay the emergence of resistance in BRAF V600E mutant human melanoma xenografts in mice compared with the administration of either drug alone Label.

Pharmacology
Encorafenib acts as an ATP-competitive RAF kinase inhibitor, decreasing ERK phosphorylation and down-regulation of CyclinD1.[5]This arrests the cell cycle in G1 phase, inducing senescence without apoptosis.[5] Therefore it is only effective in melanomas with a BRAF mutation, which make up 50% of all melanomas.[6] The plasma elimination half-life of encorafenib is approximately 6 hours, occurring mainly through metabolism via cytochrome P450 enzymes.[7]
Clinical trials
Several clinical trials of LGX818, either alone or in combinations with the MEK inhibitor MEK162,[8] are being run. As a result of a successful Phase Ib/II trials, Phase III trials are currently being initiated.[9]
History
Approval of encorafenib in the United States was based on a randomized, active-controlled, open-label, multicenter trial (COLUMBUS; NCT01909453) in 577 patients with BRAF V600E or V600K mutation-positive unresectable or metastatic melanoma.[3] Patients were randomized (1:1:1) to receive binimetinib 45 mg twice daily plus encorafenib 450 mg once daily, encorafenib 300 mg once daily, or vemurafenib 960 mg twice daily.[3] Treatment continued until disease progression or unacceptable toxicity.[3]
The major efficacy measure was progression-free survival (PFS) using RECIST 1.1 response criteria and assessed by blinded independent central review.[3] The median PFS was 14.9 months for patients receiving binimetinib plus encorafenib, and 7.3 months for the vemurafenib monotherapy arm (hazard ratio 0.54, 95% CI: 0.41, 0.71, p<0.0001).[3] The trial was conducted at 162 sites in Europe, North America and various countries around the world.[4]
SYN


PATENT
WO2010010154 , expiry , EU states, 2029, US in 2030 with US154 extension.
WO 2011025927
WO 2016089208
Patent
WO-2020011141
Novel deuterated analogs of diarylpyrazole compounds, particularly encorafenib are B-RAF and C-RAF kinase inhibitors, useful for treating proliferative diseases such as melanoma and colorectal cancer. Family members of the product case, WO2010010154 , expire in most of the EU states until 2029 and will expire in the US in 2030 with US154 extension. In January 2020, the US FDA’s Orange Book was seen to list encorafenib patents such as US8946250 , US8501758 , US9314464 and US9763941 , expiring in the range of 2029-2032. At that time Orange Book also reported that encorafenib as having NCE exclusivity expiring on July 27, 2023.
PAPER
European journal of cancer (Oxford, England : 1990) (2018), 88, 67-76.
References
- ^ Koelblinger P, Thuerigen O, Dummer R (March 2018). “Development of encorafenib for BRAF-mutated advanced melanoma”. Current Opinion in Oncology. 30 (2): 125–133. doi:10.1097/CCO.0000000000000426. PMC 5815646. PMID 29356698.
- ^ Burotto M, Chiou VL, Lee JM, Kohn EC (November 2014). “The MAPK pathway across different malignancies: a new perspective”. Cancer. 120 (22): 3446–56. doi:10.1002/cncr.28864. PMC 4221543. PMID 24948110.
- ^ Jump up to:a b c d e f g “FDA approves encorafenib and binimetinib in combination for unresectable or metastatic melanoma with BRAF mutations”. U.S. Food and Drug Administration (FDA)(Press release). 27 June 2018. Archived from the original on 18 December 2019. Retrieved 28 June 2018.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b “Drug Trial Snapshot: Braftovi”. U.S. Food and Drug Administration (FDA). 16 July 2018. Archived from the original on 19 December 2019. Retrieved 18 December 2019. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b Li Z, Jiang K, Zhu X, Lin G, Song F, Zhao Y, Piao Y, Liu J, Cheng W, Bi X, Gong P, Song Z, Meng S (January 2016). “Encorafenib (LGX818), a potent BRAF inhibitor, induces senescence accompanied by autophagy in BRAFV600E melanoma cells”. Cancer Letters. 370 (2): 332–44. doi:10.1016/j.canlet.2015.11.015. PMID 26586345.
- ^ Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. (July 2012). “A landscape of driver mutations in melanoma”. Cell. 150 (2): 251–63. doi:10.1016/j.cell.2012.06.024. PMC 3600117. PMID 22817889.
- ^ Koelblinger P, Thuerigen O, Dummer R (March 2018). “Development of encorafenib for BRAF-mutated advanced melanoma”. Current Opinion in Oncology. 30 (2): 125–133. doi:10.1097/CCO.0000000000000426. PMC 5815646. PMID 29356698.
- ^ “18 Studies found for: LGX818”. Clinicaltrials.gove.
- ^ Clinical trial number NCT01909453 for “Study Comparing Combination of LGX818 Plus MEK162 and LGX818 Monotherapy Versus Vemurafenib in BRAF Mutant Melanoma (COLUMBUS)” at ClinicalTrials.gov
External links
- “Encorafenib”. Drug Information Portal. U.S. National Library of Medicine.
- Li Z, Jiang K, Zhu X, Lin G, Song F, Zhao Y, Piao Y, Liu J, Cheng W, Bi X, Gong P, Song Z, Meng S: Encorafenib (LGX818), a potent BRAF inhibitor, induces senescence accompanied by autophagy in BRAFV600E melanoma cells. Cancer Lett. 2016 Jan 28;370(2):332-44. doi: 10.1016/j.canlet.2015.11.015. Epub 2015 Nov 14. [PubMed:26586345]
- Koelblinger P, Thuerigen O, Dummer R: Development of encorafenib for BRAF-mutated advanced melanoma. Curr Opin Oncol. 2018 Mar;30(2):125-133. doi: 10.1097/CCO.0000000000000426. [PubMed:29356698]
- Moschos SJ, Pinnamaneni R: Targeted therapies in melanoma. Surg Oncol Clin N Am. 2015 Apr;24(2):347-58. doi: 10.1016/j.soc.2014.12.011. Epub 2015 Jan 24. [PubMed:25769717]
- Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, Garbe C, Schadendorf D, Krajsova I, Gutzmer R, Chiarion-Sileni V, Dutriaux C, de Groot JWB, Yamazaki N, Loquai C, Moutouh-de Parseval LA, Pickard MD, Sandor V, Robert C, Flaherty KT: Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018 May;19(5):603-615. doi: 10.1016/S1470-2045(18)30142-6. Epub 2018 Mar 21. [PubMed:29573941]
- FDA approves encorafenib and binimetinib in combination for unresectable or metastatic melanoma with BRAF mutations [Link]
- BRAF B-Raf proto-oncogene, serine/threonine kinase [ Homo sapiens (human) ] [Link]
- Phase I Dose-Escalation and -Expansion Study of the BRAF Inhibitor Encorafenib (LGX818) in Metastatic BRAF-Mutant Melanoma [Link]
- Encorafenib FDA label [File]
- Encorafenib review [File]
 |
|
| Clinical data | |
|---|---|
| Trade names | Braftovi |
| Other names | LGX818 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a618040 |
| License data |
|
| Routes of administration |
Oral |
| Drug class | Antineoplastic Agents |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C22H27ClFN7O4S |
| Molar mass | 540.011 g/mol g·mol−1 |
| 3D model (JSmol) | |
///////////ENCORAFENIB, 1269440-17-6, BRAFTOVI, NVP-LGX818, LGX818, LGX 818, エンコラフェニブ ,
COC(=O)N[C@@H](C)CNc1nccc(n1)c2cn(nc2c3cc(Cl)cc(NS(=O)(=O)C)c3F)C(C)C
patent
[TABLE 0001]
| APCI | Atmospheric pressure chemical dissociation |
| HPLC | High performance liquid chromatography |
| TLC | TLC |
| h | hour |
| DMF | N, N-dimethylformamide |
| K 2 CO 3 | Potassium carbonate |
| DCM | Dichloromethane |
| THF | Tetrahydrofuran |
| CH 3 MgBr | Methyl magnesium bromide |
| PTSA | p-Toluenesulfonic acid |
| TFA | Trifluoroacetate |
| NMP | N-methylpyrrolidone |
| Diguanidinium carbonate | Guanidine carbonate |
| MTBE | Methyl tert-butyl ether |
| POCl 3 | Phosphorus oxychloride |
| DMSO | Dimethyl sulfoxide |
| Pd (dppf) Cl 2 | [1,1′-Bis (diphenylphosphino) ferrocene] Palladium dichloride |
| Dioxane | Dioxane |
| TsCl | 4-toluenesulfonyl chloride |
| Boc | Tert-butoxy carbon |
| DIPEA | N, N-diisopropylethylamine |
| CDCl 3 | Deuterated chloroform |
| TEA | Triethylamine |
| DMAP | 4-dimethylaminopyridine |
| Na 2 CO 3 | Sodium carbonate |
| HCl | hydrochloric acid |
[表 0002]
| MsCl | Methanesulfonyl chloride |
| Tol | Toluene |
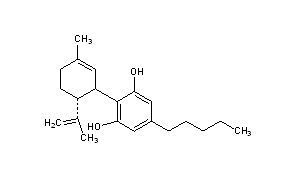
Cannabidiol, カンナビジオール;
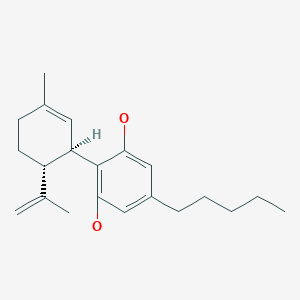
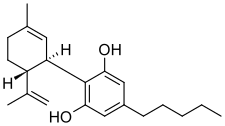
Cannabidiol
カンナビジオール;
| Formula |
C21H30O2
|
|---|---|
| CAS |
13956-29-1
|
| Mol weight |
314.4617
|
FDA APPROVED, 2018/6/25, Epidiolex
| Efficacy |
Anticonvulsant, Antiepileptic, Cannabinoid receptor agonist
|
|---|---|
| Comment |
Treatment of seizures
|
BRCX-014
BTX-1204
BTX-1503
CBD
GW-42003
GWP-42003
GWP-42003-P
PLT-101
PTL-101
ZYN-002
Cannabidiol
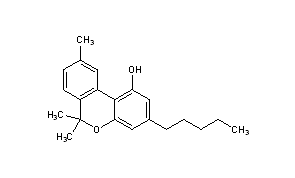
Cannabidiol (CBD) is a phytocannabinoid discovered in 1940. It is one of some 113 identified cannabinoids in Cannabis plants, accounting for up to 40% of the plant’s extract.[6] As of 2018, preliminary clinical research on cannabidiol included studies of anxiety, cognition, movement disorders, and pain.[7]
Cannabidiol can be taken into the body in multiple different ways, including by inhalation of cannabis smoke or vapor, as an aerosol spray into the cheek, and by mouth. It may be supplied as CBD oil containing only CBD as the active ingredient (no added THC or terpenes), a full-plant CBD-dominant hemp extract oil, capsules, dried cannabis, or as a prescription liquid solution.[2] CBD does not have the same psychoactivity as THC,[8][9][10] and may affect the actions of THC.[6][7][8][11] Although in vitro studies indicate CBD may interact with different biological targets, including cannabinoid receptors and other neurotransmitter receptors,[8][12] the mechanism of action for its possible biological effects has not been determined, as of 2018.[7][8]
In the United States, the cannabidiol drug Epidiolex has been approved by the Food and Drug Administration for treatment of two epilepsy disorders.[13] Side effects of long-term use listed on the Epidiolex label include somnolence, decreased appetite, diarrhea, fatigue, malaise, weakness, sleeping problems, and others.[2]
The U.S. Drug Enforcement Administration has assigned Epidiolex a Schedule V classification while non-Epidiolex CBD remains a Schedule I drug prohibited for any use.[14] CBD is not scheduled under any United Nations drug control treaties, and in 2018 the World Health Organization recommended that it remain unscheduled.[15]
Medical uses
Epilepsy
Medical reviews published in 2017 and 2018 incorporating numerous clinical trials concluded that cannabidiol is an effective treatment for certain types of childhood epilepsy.[16][17]
An orally administered cannabidiol solution (brand name Epidiolex) was approved by the US Food and Drug Administration in June 2018 as a treatment for two rare forms of childhood epilepsy, Lennox-Gastaut syndrome and Dravet syndrome.[13]
Other uses
Preliminary research on other possible therapeutic uses for cannabidiol include several neurological disorders, but the findings have not been confirmed by sufficient high-quality clinical research to establish such uses in clinical practice.[5][8][18][19][20][21]
Side effects
Preliminary research indicates that cannabidiol may reduce adverse effects of THC, particularly those causing intoxication and sedation, but only at high doses.[22] Safety studies of cannabidiol showed it is well-tolerated, but may cause tiredness, diarrhea, or changes in appetite as common adverse effects.[23] Epidiolex documentation lists sleepiness, insomnia and poor quality sleep, decreased appetite, diarrhea, and fatigue.[2]
Potential interactions
Laboratory evidence indicated that cannabidiol may reduce THC clearance, increasing plasma concentrations which may raise THC availability to receptors and enhance its effect in a dose-dependent manner.[24][25] In vitro, cannabidiol inhibited receptors affecting the activity of voltage-dependent sodium and potassium channels, which may affect neural activity.[26] A small clinical trial reported that CBD partially inhibited the CYP2C-catalyzed hydroxylation of THC to 11-OH-THC.[27]
Pharmacology
Pharmacodynamics
Cannabidiol has very low affinity for the cannabinoid CB1 and CB2 receptors but is said to act as an indirect antagonist of these receptors.[28][29] At the same time, it may potentiate the effects of THC by increasing CB1 receptor density or through another CB1receptor-related mechanism.[30]
Cannabidiol has been found to act as an antagonist of GPR55, a G protein-coupled receptor and putative cannabinoid receptor that is expressed in the caudate nucleus and putamen in the brain.[31] It has also been found to act as an inverse agonist of GPR3, GPR6, and GPR12.[12] Although currently classified as orphan receptors, these receptors are most closely related phylogenetically to the cannabinoid receptors.[12] In addition to orphan receptors, CBD has been shown to act as a serotonin 5-HT1A receptor partial agonist,[32] and this action may be involved in its antidepressant,[33][34] anxiolytic,[34][35] and neuroprotective effects.[36][37] It is an allosteric modulator of the μ- and δ-opioid receptorsas well.[38] The pharmacological effects of CBD have additionally been attributed to PPARγ agonism and intracellular calcium release.[6]
Research suggests that CBD may exert some of its pharmacological action through its inhibition of fatty acid amide hydrolase (FAAH), which may in turn increase the levels of endocannabinoids, such as anandamide, produced by the body.[6] It has also been speculated that some of the metabolites of CBD have pharmacological effects that contribute to the biological activity of CBD.[39]
Pharmacokinetics
The oral bioavailability of CBD is 13 to 19%, while its bioavailability via inhalation is 11 to 45% (mean 31%).[3][4] The elimination half-life of CBD is 18–32 hours.[5]
Cannabidiol is metabolized in the liver as well as in the intestines by CYP2C19 and CYP3A4 enzymes, and UGT1A7, UGT1A9, and UGT2B7 isoforms.[2]
Pharmaceutical preparations
Nabiximols (brand name Sativex) is a patented medicine containing CBD and THC in equal proportions. The drug was approved by Health Canada in 2005 for prescription to treat central neuropathic pain in multiple sclerosis, and in 2007 for cancer related pain.[40][41]
Chemistry
Cannabidiol is insoluble in water but soluble in organic solvents such as pentane. At room temperature, it is a colorless crystalline solid.[42] In strongly basic media and the presence of air, it is oxidized to a quinone.[43] Under acidic conditions it cyclizes to THC,[44] which also occurs during pyrolysis (smoking).[45] The synthesis of cannabidiol has been accomplished by several research groups.[46][47][48]
Biosynthesis

Cannabidiol and THC biosynthesis[49]
Cannabis produces CBD-carboxylic acid through the same metabolic pathway as THC, until the next to last step, where CBDA synthase performs catalysis instead of THCA synthase.[50]
Isomerism

History
CBD was isolated from the cannabis plant in 1940, and its chemical structure was established in 1963.[7]
Society and culture
Names
Cannabidiol is the generic name of the drug and its INN.[51]
Food and beverage

An example of CBD-infused cold brew coffee & tea on a grocery store shelf.
Food and beverage products containing CBD were introduced in the United States in 2017.[52] Similar to energy drinks and protein barswhich may contain vitamin or herbal additives, food and beverage items can be infused with CBD as an alternative means of ingesting the substance.[53] In the United States, numerous products are marketed as containing CBD, but in reality contain little or none.[54] Some companies marketing CBD-infused food products with claims that are similar to the effects of prescription drugs have received warning lettersfrom the Food and Drug Administration for making unsubstantiated health claims.[55]
Plant sources
Selective breeding of cannabis plants has expanded and diversified as commercial and therapeutic markets develop. Some growers in the U.S. succeeded in lowering the proportion of CBD-to-THC to accommodate customers who preferred varietals that were more mind-altering due to the higher THC and lower CBD content.[56] Hemp is classified as any part of the cannabis plant containing no more than 0.3% THC in dry weight form (not liquid or extracted form).[57]
Legal status
Non-psychoactivity
CBD does not appear to have any psychotropic (“high”) effects such as those caused by ∆9-THC in marijuana, but may have anti-anxiety and anti-psychotic effects.[9] As the legal landscape and understanding about the differences in medical cannabinoids unfolds, it will be increasingly important to distinguish “medical marijuana” (with varying degrees of psychotropic effects and deficits in executive function) – from “medical CBD therapies” which would commonly present as having a reduced or non-psychoactive side-effect profile.[9][58]
Various strains of “medical marijuana” are found to have a significant variation in the ratios of CBD-to-THC, and are known to contain other non-psychotropic cannabinoids.[59] Any psychoactive marijuana, regardless of its CBD content, is derived from the flower (or bud) of the genus Cannabis. Non-psychoactive hemp (also commonly-termed industrial hemp), regardless of its CBD content, is any part of the cannabis plant, whether growing or not, containing a ∆-9 tetrahydrocannabinol concentration of no more than 0.3% on a dry-weight basis.[60] Certain standards are required for legal growing, cultivating, and producing the hemp plant. The Colorado Industrial Hemp Program registers growers of industrial hemp and samples crops to verify that the dry-weight THC concentration does not exceed 0.3%.[60]
United Nations
Cannabidiol is not scheduled under the Convention on Psychotropic Substances or any other UN drug treaty. In 2018, the World Health Organization recommended that CBD remain unscheduled.[15]
United States
In the United States, non-FDA approved CBD products are classified as Schedule I drugs under the Controlled Substances Act.[61] This means that production, distribution, and possession of non-FDA approved CBD products is illegal under federal law. In addition, in 2016 the Drug Enforcement Administration added “marijuana extracts” to the list of Schedule I drugs, which it defined as “an extract containing one or more cannabinoids that has been derived from any plant of the genus Cannabis, other than the separated resin (whether crude or purified) obtained from the plant.”[62] Previously, CBD had simply been considered “marijuana”, which is a Schedule I drug.[61][63]
In September 2018, following its approval by the FDA for rare types of childhood epilepsy,[13] Epidiolex was rescheduled (by the Drug Enforcement Administration) as a Schedule V drug to allow for its prescription use.[14] This change applies only to FDA-approved products containing no more than 0.1 percent THC.[14] This allows GW Pharmaceuticals to sell Epidiolex, but it does not apply broadly and all other CBD-containing products remain Schedule I drugs.[14] Epidiolex still requires rescheduling in some states before it can be prescribed in those states.[64][65]
A CNN program that featured Charlotte’s Web cannabis in 2013 brought increased attention to the use of CBD in the treatment of seizure disorders.[66][67] Since then, 16 states have passed laws to allow the use of CBD products with a doctor’s recommendation (instead of a prescription) for treatment of certain medical conditions.[68] This is in addition to the 30 states that have passed comprehensive medical cannabis laws, which allow for the use of cannabis products with no restrictions on THC content.[68] Of these 30 states, eight have legalized the use and sale of cannabis products without requirement for a doctor’s recommendation.[68]
Some manufacturers ship CBD products nationally, an illegal action which the FDA has not enforced in 2018, with CBD remaining the subject of an FDA investigational new drugevaluation, and is not considered legal as a dietary supplement or food ingredient as of December 2018.[69][70] Federal illegality has made it difficult historically to conduct research on CBD.[71] CBD is openly sold in head shops and health food stores in some states where such sales have not been explicitly legalized.[72][73]
The 2014 Farm Bill[74] legalized the sale of “non-viable hemp material” grown within states participating in the Hemp Pilot Program.[75] This legislation defined hemp as cannabis containing less than 0.3% of THC delta-9, grown within the regulatory framework of the Hemp Pilot Program.[76] The 2018 Farm Bill allowed for interstate commerce of hemp derived products, though these products still fall under the purview of the FDA.[77][78]
Australia
Prescription medicine (Schedule 4) for therapeutic use containing 2 per cent (2.0%) or less of other cannabinoids commonly found in cannabis (such as ∆9-THC). A schedule 4 drug under the SUSMP is Prescription Only Medicine, or Prescription Animal Remedy – Substances, the use or supply of which should be by or on the order of persons permitted by State or Territory legislation to prescribe and should be available from a pharmacist on prescription.[79]
New Zealand
Cannabidiol is currently a class B1 controlled drug in New Zealand under the Misuse of Drugs Act. It is also a prescription medicine under the Medicines Act. In 2017 the rules were changed so that anyone wanting to use it could go to the Health Ministry for approval. Prior to this, the only way to obtain a prescription was to seek the personal approval of the Minister of Health.
Associate Health Minister Peter Dunne said restrictions would be removed, which means a doctor will now be able to prescribe cannabidiol to patients.[80]
Canada
On October 17, 2018, cannabidiol became legal for recreational and medical use.[81][82]
Europe
In 2019, the European Food Safety Authority (EFSA) announced that CBD and other cannabinoids would be classified as “novel foods“,[83] meaning that CBD products would require authorization under the EU Novel Food Regulation stating: because “this product was not used as a food or food ingredient before 15 May 1997, before it may be placed on the market in the EU as a food or food ingredient, a safety assessment under the Novel Food Regulation is required.”[84] The recommendation – applying to CBD extracts, synthesized CBD, and all CBD products, including CBD oil – was scheduled for a final ruling by the European Commission in March 2019.[83] If approved, manufacturers of CBD products would be required to conduct safety tests and prove safe consumption, indicating that CBD products would not be eligible for legal commerce until at least 2021.[83]
Cannabidiol is listed in the EU Cosmetics Ingredient Database (CosIng).[85] However, the listing of an ingredient, assigned with an INCI name, in CosIng does not mean it is to be used in cosmetic products or is approved for such use.[85]
Several industrial hemp varieties can be legally cultivated in Western Europe. A variety such as “Fedora 17” has a cannabinoid profile consistently around 1%, with THC less than 0.1%.[86]
Sweden
CBD is classified as a medical product in Sweden.[87]
United Kingdom
Cannabidiol, in an oral-mucosal spray formulation combined with delta-9-tetrahydrocannabinol, is a product available (by prescription only until 2017) for relief of severe spasticity due to multiple sclerosis (where other anti-spasmodics have not been effective).[88]
Until 2017, products containing cannabidiol marketed for medical purposes were classed as medicines by the UK regulatory body, the Medicines and Healthcare products Regulatory Agency (MHRA) and could not be marketed without regulatory approval for the medical claims.[89][90] Cannabis oil is illegal to possess, buy, and sell.[91] In January 2019, the UK Food Standards Agency indicated it would regard CBD products, including CBD oil, as a novel food in the UK, having no history of use before May 1997, and indicating they must have authorization and proven safety before being marketed.[83][92]
Switzerland
While THC remains illegal, CBD is not subject to the Swiss Narcotic Acts because this substance does not produce a comparable psychoactive effect.[93] Cannabis products containing less than 1% THC can be sold and purchased legally.[94]
Research
A 2016 literature review indicated that cannabidiol was under basic research to identify its possible neurological effects,[10] although as of 2016, there was limited high-quality evidence for such effects in people.[20][95][96] A 2018 meta-analysis compared the potential therapeutic properties of “purified CBD” with full-plant, CBD-rich cannabis extracts with regard to treating refractory (treatment-resistant) epilepsy, noting several differences.[97] The daily average dose of people using full-plant extracts was more than four times lower than of those using purified CBD, a possible entourage effect of CBD interacting with THC.[97]

CLIP
https://cen.acs.org/pharmaceuticals/CBD-Medicine-marijuana/96/i30
CLIP
Cannabidiol: An overview of some chemical and pharmacological aspects. Part I: Chemical aspects

CLIP
https://www.sciencedirect.com/science/article/pii/S0076687917301490

CLIP

CLIP
Discovery of KLS-13019, a Cannabidiol-Derived Neuroprotective Agent, with Improved Potency, Safety, and Permeability

Cannabidiol is the nonpsychoactive natural component of C. sativa that has been shown to be neuroprotective in multiple animal models. Our interest is to advance a therapeutic candidate for the orphan indication hepatic encephalopathy (HE). HE is a serious neurological disorder that occurs in patients with cirrhosis or liver failure. Although cannabidiol is effective in models of HE, it has limitations in terms of safety and oral bioavailability. Herein, we describe a series of side chain modified resorcinols that were designed for greater hydrophilicity and “drug likeness”, while varying hydrogen bond donors, acceptors, architecture, basicity, neutrality, acidity, and polar surface area within the pendent group. Our primary screen evaluated the ability of the test agents to prevent damage to hippocampal neurons induced by ammonium acetate and ethanol at clinically relevant concentrations. Notably, KLS-13019 was 50-fold more potent and >400-fold safer than cannabidiol and exhibited an in vitro profile consistent with improved oral bioavailability.

Discovery of KLS-13019, a cannabidiol-derived neuroprotective agent, with improved potency, safety, and permeability
ACS Med Chem Lett 2016, 7(4): 424
Synthesis of cannabidiol by condensation of olivetol with 4(R)-isopropenyl-1(S)-methyl-2-cyclohexen-1-ol is described.
Cannabidiol is prepared by the condensation of olivetol with 4(R)-isopropenyl-1(S)-methyl-2-cyclohexen-1-ol in the presence of p-TsOH in toluene .
https://pubs.acs.org/doi/suppl/10.1021/acsmedchemlett.6b00009/suppl_file/ml6b00009_si_001.pdf
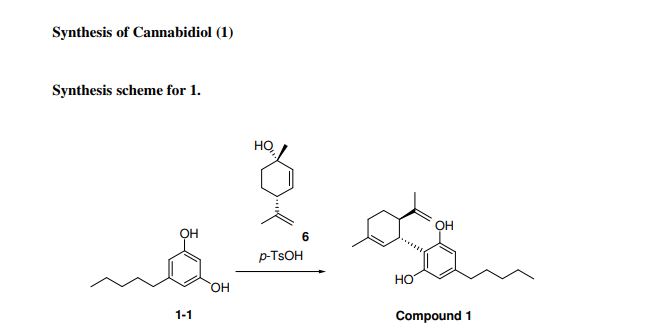
A solution of olivetol (1-1) (0.40 g, 2.2 mol, 1 equiv.), p-TsOH (40 mg, 0.21 mmol, 0.1 equiv.) and compound 6 (0.47 g, 3.1 mmol, 1.4 equiv.) in toluene (28 mL) was stirred at RT for 1.5 hours. TLC analysis indicated ~70% conversion of the starting olivetol. The reaction was stopped at this point and EtOAc (30 mL) was added to dilute the reaction mixture, which was then washed by saturated NaHCO3 aqueous solution (3 x 50 mL). The organic layer was dried over Na2SO4, filtered and concentrated to give crude compound 1 (0.9 g). It was purified by column chromatography to give compound 1 (140 mg, yield 20%). HPLC purity: 97%. LC/MS (ESI): m/z 315 (M+1). 1H-NMR (300 MHz, CDCl3) δ 6.40-6.20 (br s, 2H), 6.10-5.90 (br s, 1H), 5.59 (s, 1H), 4.68 (s, 2H), 4.58 (s, 1H), 3.90-3.80 (m, 1H), 2.50-2.40 (m, 3H), 2.30-2.00 (m, 2H), 1.90-1.70 (m, 5H), 1.67 (s, 3H), 1.65-1.50 (m, 2H), 1.40-1.20 (m, 4H), 0.90 (t, J = 6.6 Hz, 3H). The analytical data are attached below. Optical Rotation of 1: [α]D 22= -121.4 (c 1.00, EtOH), the average of two measurements: -121.7 and -121.1 Literature: [α]D 22= -125 (Ben-Shabat, 2006).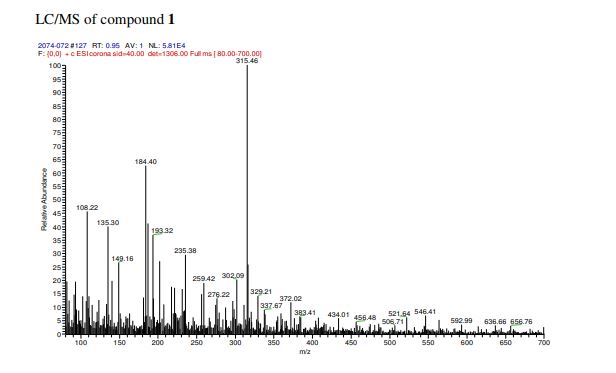


CLIP
https://onlinelibrary.wiley.com/doi/pdf/10.1002/pca.787




CLIP
J Am Chem Soc 1940, 62(1): 196

The red oil ethanolic extract from Minnesota wild hemp containing the carboxylated compound is submitted to a fractionated distillation with simultaneous thermal decarboxylation.
The fraction distilling at 190-210º C (2 mmHg) contains the desired compound as an intermediate oil, which is purified by treatment with 3,5-dinitrobenzoyl chloride in pyridine to yield the crystalline bis(3,5-dinitrobenzoate) .
Finally this compound is treated with liq ammonia at room temperature in a high pressure bomb to obtain the FINAL cannabidiol.
CLIP


1H NMR spectrum of C21H30O2 in CDCL3 at 400 MHz.
R.J. Abraham, M. Mobli Modelling 1H NMR Spectra of Organic Compounds: Theory, Applications and NMR Prediction Software, Wiley, Chichester, 2008.
CLIP
References
- ^ “Sativex (Cannabidiol/Tetrahydrocannabinol) Bayer Label” (PDF). bayer.ca. Retrieved 28 June 2018.
- ^ Jump up to:a b c d e “Epidiolex (Cannabidiol) FDA Label” (PDF). fda.gov. Retrieved 28 June 2018.For label updates see FDA index page for NDA 210365
- ^ Jump up to:a b Mechoulam R, Parker LA, Gallily R (November 2002). “Cannabidiol: an overview of some pharmacological aspects”. Journal of Clinical Pharmacology. 42 (11 Suppl): 11S–19S. doi:10.1002/j.1552-4604.2002.tb05998.x. PMID 12412831.
- ^ Jump up to:a b Scuderi C, Filippis DD, Iuvone T, Blasio A, Steardo A, Esposito G (May 2009). “Cannabidiol in medicine: a review of its therapeutic potential in CNS disorders”. Phytotherapy Research (Review). 23 (5): 597–602. doi:10.1002/ptr.2625. PMID 18844286.
- ^ Jump up to:a b c Devinsky, Orrin; Cilio, Maria Roberta; Cross, Helen; Fernandez-Ruiz, Javier; French, Jacqueline; Hill, Charlotte; Katz, Russell; Di Marzo, Vincenzo; Jutras-Aswad, Didier; Notcutt, William George; Martinez-Orgado, Jose; Robson, Philip J.; Rohrback, Brian G.; Thiele, Elizabeth; Whalley, Benjamin; Friedman, Daniel (22 May 2014). “Cannabidiol: Pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders”. Epilepsia. 55 (6): 791–802. doi:10.1111/epi.12631. PMC 4707667. PMID 24854329.
- ^ Jump up to:a b c d Campos AC, Moreira FA, Gomes FV, Del Bel EA, Guimarães FS (December 2012). “Multiple mechanisms involved in the large-spectrum therapeutic potential of cannabidiol in psychiatric disorders”. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences (Review). 367 (1607): 3364–78. doi:10.1098/rstb.2011.0389. PMC 3481531. PMID 23108553.
- ^ Jump up to:a b c d Boggs, Douglas L; Nguyen, Jacques D; Morgenson, Daralyn; Taffe, Michael A; Ranganathan, Mohini (6 September 2017). “Clinical and preclinical evidence for functional interactions of cannabidiol and Δ9-tetrahydrocannabinol”. Neuropsychopharmacology. 43(1): 142–154. doi:10.1038/npp.2017.209. ISSN 0893-133X. PMC 5719112. PMID 28875990.
- ^ Jump up to:a b c d e Pisanti S, Malfitano AM, Ciaglia E, Lamberti A, Ranieri R, Cuomo G, Abate M, Faggiana G, Proto MC, Fiore D, Laezza C, Bifulco M (July 2017). “Cannabidiol: State of the art and new challenges for therapeutic applications”. Pharmacol. Ther. 175: 133–150. doi:10.1016/j.pharmthera.2017.02.041. PMID 28232276.
- ^ Jump up to:a b c Iseger TA, Bossong MG (March 2015). “A systematic review of the antipsychotic properties of cannabidiol in humans”. Schizophrenia Research. 162 (1–3): 153–61. doi:10.1016/j.schres.2015.01.033. PMID 25667194.
- ^ Jump up to:a b Jurkus R, Day HL, Guimarães FS, Lee JL, Bertoglio LJ, Stevenson CW (2016). “Cannabidiol Regulation of Learned Fear: Implications for Treating Anxiety-Related Disorders”. Frontiers in Pharmacology. 7: 454. doi:10.3389/fphar.2016.00454. PMC 5121237. PMID 27932983.
- ^ Aizpurua-Olaizola O, Soydaner U, Öztürk E, Schibano D, Simsir Y, Navarro P, Etxebarria N, Usobiaga A (February 2016). “Evolution of the Cannabinoid and Terpene Content during the Growth of Cannabis sativa Plants from Different Chemotypes”. Journal of Natural Products. 79 (2): 324–31. doi:10.1021/acs.jnatprod.5b00949. PMID 26836472.
- ^ Jump up to:a b c Laun AS, Shrader SH, Brown KJ, Song ZH (June 2018). “GPR3, GPR6, and GPR12 as novel molecular targets: their biological functions and interaction with cannabidiol”. Acta Pharmacol. Sin. doi:10.1038/s41401-018-0031-9. PMID 29941868.
- ^ Jump up to:a b c “FDA approves first drug comprised of an active ingredient derived from marijuana to treat rare, severe forms of epilepsy”. US Food and Drug Administration. 25 June 2018. Retrieved 25 June 2018.
- ^ Jump up to:a b c d “DEA reschedules Epidiolex, marijuana-derived drug, paving the way for it to hit the market”. CNBC. September 27, 2018.
- ^ Jump up to:a b Angell T (13 August 2018). “UN Launches First-Ever Full Review Of Marijuana’s Status Under International Law”. Marijuana Moment. Retrieved 1 November 2018.
- ^ Stockings E, Zagic D, Campbell G, Weier M, Hall WD, Nielsen S, Herkes GK, Farrell M, Degenhardt L (July 2018). “Evidence for cannabis and cannabinoids for epilepsy: a systematic review of controlled and observational evidence”. J. Neurol. Neurosurg. Psychiatry. 89 (7): 741–753. doi:10.1136/jnnp-2017-317168. PMID 29511052.
- ^ Perucca E (December 2017). “Cannabinoids in the Treatment of Epilepsy: Hard Evidence at Last?”. J Epilepsy Res. 7 (2): 61–76. doi:10.14581/jer.17012. PMC 5767492. PMID 29344464.
- ^ Silva TB, Balbino CQ, Weiber AF (1 May 2015). “The relationship between cannabidiol and psychosis: A review”. Annals of Clinical Psychiatry. 27 (2): 134–41. PMID 25954940.
- ^ Blessing EM, Steenkamp MM, Manzanares J, Marmar CR (October 2015). “Cannabidiol as a Potential Treatment for Anxiety Disorders”. Neurotherapeutics. 12 (4): 825–36. doi:10.1007/s13311-015-0387-1. PMC 4604171. PMID 26341731.
- ^ Jump up to:a b Prud’homme M, Cata R, Jutras-Aswad D (2015). “Cannabidiol as an Intervention for Addictive Behaviors: A Systematic Review of the Evidence”. Substance Abuse. 9: 33–8. doi:10.4137/SART.S25081. PMC 4444130. PMID 26056464.
- ^ Fernández-Ruiz J, Sagredo O, Pazos MR, García C, Pertwee R, Mechoulam R, Martínez-Orgado J (February 2013). “Cannabidiol for neurodegenerative disorders: important new clinical applications for this phytocannabinoid?”. British Journal of Clinical Pharmacology. 75 (2): 323–33. doi:10.1111/j.1365-2125.2012.04341.x. PMC 3579248. PMID 22625422.
- ^ Fischer B, Russell C, Sabioni P, van den Brink W, Le Foll B, Hall W, Rehm J, Room R (August 2017). “Lower-Risk Cannabis Use Guidelines: A Comprehensive Update of Evidence and Recommendations”. American Journal of Public Health. 107 (8): e1–e12. doi:10.2105/AJPH.2017.303818. PMID 28644037.
- ^ Iffland K, Grotenhermen F (2017). “An Update on Safety and Side Effects of Cannabidiol: A Review of Clinical Data and Relevant Animal Studies”. Cannabis and Cannabinoid Research. 2 (1): 139–154. doi:10.1089/can.2016.0034. PMC 5569602. PMID 28861514.
- ^ Bornheim LM, Kim KY, Li J, Perotti BY, Benet LZ (August 1995). “Effect of cannabidiol pretreatment on the kinetics of tetrahydrocannabinol metabolites in mouse brain”. Drug Metabolism and Disposition. 23 (8): 825–831. PMID 7493549.
- ^ Klein C, Karanges E, Spiro A, Wong A, Spencer J, Huynh T, Gunasekaran N, Karl T, Long LE, Huang XF, Liu K, Arnold JC, McGregor IS (November 2011). “Cannabidiol potentiates Δ⁹-tetrahydrocannabinol (THC) behavioural effects and alters THC pharmacokinetics during acute and chronic treatment in adolescent rats”. Psychopharmacology. 218 (2): 443–457. doi:10.1007/s00213-011-2342-0. PMID 21667074.
- ^ Ghovanloo MR, Shuart NG, Mezeyova M, Dean RA, Ruben PC, Goodchild SJ (September 2018). “Inhibitory effects of cannabidiol on voltage-dependent sodium currents”. Journal of Biological Chemistry. doi:10.1074/jbc.RA118.004929.
- ^ Nadulski T, Pragst F, Weinberg G, Roser P, Schnelle M, Fronk EM, Stadelmann AM (December 2005). “Randomized, double-blind, placebo-controlled study about the effects of cannabidiol (CBD) on the pharmacokinetics of Delta9-tetrahydrocannabinol (THC) after oral application of THC verses standardized cannabis extract”. Ther Drug Monit. 27 (6): 799–810. PMID 16306858.
- ^ Mechoulam R, Peters M, Murillo-Rodriguez E, Hanus LO (August 2007). “Cannabidiol–recent advances”. Chemistry & Biodiversity (Review). 4 (8): 1678–92. doi:10.1002/cbdv.200790147. PMID 17712814.
- ^ Pertwee RG (January 2008). “The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin”. British Journal of Pharmacology. 153 (2): 199–215. doi:10.1038/sj.bjp.0707442. PMC 2219532. PMID 17828291.
- ^ Hayakawa K, Mishima K, Hazekawa M, Sano K, Irie K, Orito K, Egawa T, Kitamura Y, Uchida N, Nishimura R, Egashira N, Iwasaki K, Fujiwara M (January 2008). “Cannabidiol potentiates pharmacological effects of Delta(9)-tetrahydrocannabinol via CB(1) receptor-dependent mechanism”. Brain Research. 1188: 157–64. doi:10.1016/j.brainres.2007.09.090. PMID 18021759.
- ^ Ryberg E, Larsson N, Sjögren S, Hjorth S, Hermansson NO, Leonova J, Elebring T, Nilsson K, Drmota T, Greasley PJ (December 2007). “The orphan receptor GPR55 is a novel cannabinoid receptor”. British Journal of Pharmacology. 152 (7): 1092–101. doi:10.1038/sj.bjp.0707460. PMC 2095107. PMID 17876302.
- ^ Russo EB, Burnett A, Hall B, Parker KK (August 2005). “Agonistic properties of cannabidiol at 5-HT1a receptors”. Neurochemical Research. 30 (8): 1037–43. doi:10.1007/s11064-005-6978-1. PMID 16258853.
- ^ Zanelati TV, Biojone C, Moreira FA, Guimarães FS, Joca SR (January 2010). “Antidepressant-like effects of cannabidiol in mice: possible involvement of 5-HT1A receptors”. British Journal of Pharmacology. 159 (1): 122–8. doi:10.1111/j.1476-5381.2009.00521.x. PMC 2823358. PMID 20002102.
- ^ Jump up to:a b Resstel LB, Tavares RF, Lisboa SF, Joca SR, Corrêa FM, Guimarães FS (January 2009). “5-HT1A receptors are involved in the cannabidiol-induced attenuation of behavioural and cardiovascular responses to acute restraint stress in rats”. British Journal of Pharmacology. 156 (1): 181–8. doi:10.1111/j.1476-5381.2008.00046.x. PMC 2697769. PMID 19133999.
- ^ Campos AC, Guimarães FS (August 2008). “Involvement of 5HT1A receptors in the anxiolytic-like effects of cannabidiol injected into the dorsolateral periaqueductal gray of rats”. Psychopharmacology. 199 (2): 223–30. doi:10.1007/s00213-008-1168-x. PMID 18446323.
- ^ Mishima K, Hayakawa K, Abe K, Ikeda T, Egashira N, Iwasaki K, Fujiwara M (May 2005). “Cannabidiol prevents cerebral infarction via a serotonergic 5-hydroxytryptamine1A receptor-dependent mechanism”. Stroke. 36 (5): 1077–82. doi:10.1161/01.STR.0000163083.59201.34. PMID 15845890.
- ^ Hayakawa K, Mishima K, Nozako M, Ogata A, Hazekawa M, Liu AX, Fujioka M, Abe K, Hasebe N, Egashira N, Iwasaki K, Fujiwara M (March 2007). “Repeated treatment with cannabidiol but not Delta9-tetrahydrocannabinol has a neuroprotective effect without the development of tolerance”. Neuropharmacology. 52 (4): 1079–87. doi:10.1016/j.neuropharm.2006.11.005. PMID 17320118.
- ^ Kathmann M, Flau K, Redmer A, Tränkle C, Schlicker E (February 2006). “Cannabidiol is an allosteric modulator at mu- and delta-opioid receptors”. Naunyn-Schmiedeberg’s Archives of Pharmacology. 372 (5): 354–61. doi:10.1007/s00210-006-0033-x. PMID 16489449.
- ^ Ujváry I, Hanuš L (2014). “Human Metabolites of Cannabidiol: A Review on Their Formation, Biological Activity, and Relevance in Therapy”. Cannabis and Cannabinoid Research. 1 (1): 90–101. doi:10.1089/can.2015.0012. PMC 5576600. PMID 28861484.
- ^ Russo, E. B. (2008). “Cannabinoids in the management of difficult to treat pain”. Therapeutics and Clinical Risk Management. 4 (1): 245–259. PMC 2503660. PMID 18728714.
- ^ Russo, E. B. (2008). “Cannabinoids in the management of difficult to treat pain”. Therapeutics and Clinical Risk Management. 4 (1): 245–259. PMC 2503660. PMID 18728714.
- ^ Jones PG, Falvello L, Kennard O, Sheldrick GM, Mechoulam R (1977). “Cannabidiol”. Acta Crystallogr. B. 33 (10): 3211–3214. doi:10.1107/S0567740877010577.
- ^ Mechoulam R, Ben-Zvi Z, Gaoni Y (August 1968). “Hashish–13. On the nature of the Beam test”. Tetrahedron. 24 (16): 5615–24. doi:10.1016/0040-4020(68)88159-1. PMID 5732891.
- ^ Gaoni Y, Mechoulam R (1966). “Hashish—VII The isomerization of cannabidiol to tetrahydrocannabinols”. Tetrahedron. 22 (4): 1481–1488. doi:10.1016/S0040-4020(01)99446-3.
- ^ Cannabis—XV: Pyrolysis of cannabidiol. Structure elucidation of four pyrolytic products, doi:10.1016/0040-4020(75)87002-5
- ^ Petrzilka T, Haefliger W, Sikemeier C, Ohloff G, Eschenmoser A (March 1967). “[Synthesis and optical rotation of the (-)-cannabidiols]”. Helvetica Chimica Acta. 50 (2): 719–23. doi:10.1002/hlca.19670500235. PMID 5587099.
- ^ Gaoni Y, Mechoulam R (1985). “Boron trifluoride etherate on alumuna — a modified Lewis acid reagent. An improved synthesis of cannabidiol”. Tetrahedron Letters. 26 (8): 1083–1086. doi:10.1016/S0040-4039(00)98518-6.
- ^ Kobayashi Y, Takeuchi A, Wang YG (June 2006). “Synthesis of cannabidiols via alkenylation of cyclohexenyl monoacetate”. Organic Letters. 8 (13): 2699–702. doi:10.1021/ol060692h. PMID 16774235.
- ^ Taura F, Sirikantaramas S, Shoyama Y, Yoshikai K, Shoyama Y, Morimoto S (June 2007). “Cannabidiolic-acid synthase, the chemotype-determining enzyme in the fiber-type Cannabis sativa”. FEBS Letters. 581 (16): 2929–34. doi:10.1016/j.febslet.2007.05.043. PMID 17544411.
- ^ Marks MD, Tian L, Wenger JP, Omburo SN, Soto-Fuentes W, He J, Gang DR, Weiblen GD, Dixon RA (2009). “Identification of candidate genes affecting Delta9-tetrahydrocannabinol biosynthesis in Cannabis sativa”. Journal of Experimental Botany. 60 (13): 3715–26. doi:10.1093/jxb/erp210. PMC 2736886. PMID 19581347.
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN)” (PDF). WHO Drug Information. 30 (2): 241. 2016.
- ^ “Billboard featuring hemp leaf raises questions about new beverage for sale in Cincinnati | WLWT5”. WLWT5. 2017-09-29. Retrieved 2017-09-29.
- ^ “CBD-Infused Foods Becoming a New Health Trend and Penetrating the Market”. Retrieved 2017-12-14.
- ^ “Warning Letters and Test Results for Cannabidiol-Related Products”. Food and Drug Administration. November 2, 2017. Retrieved January 2, 2018.
- ^ Fox A, Ravitz JR, Leongini EM, Brian J M. “Companies Marketing CBD Products Be Warned: FDA Is Watching”. Lexology. Retrieved 2017-12-14.
- ^ Romney L (September 13, 2012). “On the frontier of medical pot to treat boy’s epilepsy”. Los Angeles Times.
- ^ “How are CBD Extracts & Isolates Made?”. IntelliCBD. June 22, 2018.
- ^ Sachs J, McGlade E, Yurgelun-Todd D (October 2015). “Safety and Toxicology of Cannabinoids”. Neurotherapeutics. 12 (4): 735–46. doi:10.1007/s13311-015-0380-8. PMC 4604177. PMID 26269228.
- ^ Izzo AA, Borrelli F, Capasso R, Di Marzo V, Mechoulam R (October 2009). “Non-psychotropic plant cannabinoids: new therapeutic opportunities from an ancient herb”. Trends in Pharmacological Sciences. 30 (10): 515–27. doi:10.1016/j.tips.2009.07.006. PMID 19729208.
- ^ Jump up to:a b “Industrial hemp”. Department of Agriculture, State of Colorado. 2018. Retrieved 14 September 2018.
- ^ Jump up to:a b Hudak J, Stenglein C (February 6, 2017). “DEA guidance is clear: Cannabidiol is illegal and always has been”. FixGov. Brookings Institution. Retrieved December 10, 2017.
- ^ “Establishment of a New Drug Code for Marijuana Extract”. Federal Register. 81 (240): 90194–90196. December 14, 2016. 81 FR 90195
- ^ “Clarification of the New Drug Code (7350) for Marijuana Extract”. U.S. Department of Justice. Retrieved December 10, 2017.
- ^ “Epilepsy Foundation Statement on DEA’s Scheduling of Epidiolex” (Press release). Landover, MD: Epilepsy Foundation. 27 September 2018. Retrieved 1 November 2018.
- ^ “State Rescheduling for FDA-approved Therapies Derived from CBD”. Epilepsy Foundation. Retrieved 1 November 2018.
- ^ Maa E, Figi P (June 2014). “The case for medical marijuana in epilepsy”. Epilepsia. 55 (6): 783–6. doi:10.1111/epi.12610. PMID 24854149.
- ^ Young S. “Marijuana stops child’s severe seizures”. CNN. CNN. Retrieved 14 May 2018.
- ^ Jump up to:a b c “State Medical Marijuana Laws”. National Conference of State Legislatures. 27 April 2018. Retrieved 14 May 2018.
- ^ “FDA and Marijuana: Questions and Answers. No. 12 – Can products that contain THC or cannabidiol (CBD) be sold as dietary supplements?”. US Food and Drug Administration. 20 December 2018. Retrieved 13 January 2019.
- ^ Stephen Daniells (6 November 2018). “Top FDA official: ‘Anyone who thinks CBD is lawful is mistaken‘“. NutraIngredients-USA, William Reed Business Media Ltd. Retrieved 6 November 2018.
- ^ Corba, Jacqueline. “Super Bowl Champ: CBD Can Solve NFL’s Opioid Problem‘“. Cheddar. Retrieved 2019-01-29.
- ^ Summers DJ (March 22, 2017). “Is CBD Oil Legal? Depends on Where You Are and Who You Ask”. Leafly. Retrieved January 3, 2018.
- ^ Gaines LV (March 23, 2017). “Why are CBD products sold over the counter some places and tightly regulated in others?”. Chicago Reader. Retrieved January 3, 2018.
- ^ “the-2014-farm-bill | THE BILL”. The 2014 Farm Bill. Retrieved 2018-11-27.
- ^ “7 U.S. Code § 5940 – Legitimacy of industrial hemp research”. LII / Legal Information Institute. Retrieved 2018-11-27.
- ^ Zhang, Mona. “No, CBD Is Not ‘Legal in All 50 States‘“. Forbes. Retrieved 2018-11-27.
- ^ “Trump just signed a law that could spark a boom for the $1 billion marijuana-linked CBD industry”. Business Insider. December 20, 2018. Retrieved January 29, 2019.
- ^ Estevez, Lauren. “Guide to CBD Laws”. LME Law. Retrieved 2019-01-29.
- ^ “Poisons Standard June 2017”. Legislation.gov.au. Retrieved December 4, 2016.
- ^ “Doctors now able to prescribe cannabidiol”. radionz.co.nz. Retrieved June 2, 2017.
- ^ “Health products containing cannabis or for use with cannabis: Guidance for the Cannabis Act, the Food and Drugs Act, and related regulations”. Government of Canada. 11 July 2018. Retrieved 19 October 2018.
- ^ Communications, Government of Canada, Department of Justice, Electronic. “Cannabis Legalization and Regulation”. http://www.justice.gc.ca.
- ^ Jump up to:a b c d Will Chu (31 January 2019). “Updated EFSA ruling for CBD classes supplement ingredient as Novel Food”. NutraIngredients.com, William Reed Business Media Ltd. Retrieved 1 January 2019.
- ^ “Cannabinoids, searched in the EU Novel food catalogue (v.1.1)”. European Commission. 1 January 2019. Retrieved 1 February 2019.
- ^ Jump up to:a b “CosIng – Cosmetics – Cannabidiol”. European Commission. Retrieved December 4,2016.
- ^ Fournier G, Beherec O, Bertucelli S (2003). “Intérêt du rapport Δ-9-THC / CBD dans le contrôle des cultures de chanvre industriel” [The advantage of the Δ-9-THC / CBD ratio in the control of industrial hemp crops]. Annales de Toxicologie Analytique (in French). 15 (4): 250–259. doi:10.1051/ata/2003003.
- ^ “CBD products should follow the drug laws”. Swedish Medical Products Agency. 4 April 2018. Retrieved 31 July 2018.
- ^ “Sativex Oromucosal Spray – Summary of Product Characteristics (SPC) – (eMC)”. Medicines.org.uk. Retrieved December 4, 2016.
- ^ “MHRA statement on products containing Cannabidiol (CBD)”. Gov.uk. December 14, 2016.
- ^ “Is CBD Legal in the UK?”. The CBD Blog. October 31, 2018.
- ^ “CBD oil UK law: The latest news”. Business Matters. 23 July 2018. Retrieved 6 October 2018.
- ^ Gunn L, Haigh L (29 January 2019). “British watchdog deems CBD a novel food, seeks to curtail sale on UK market”. Nutrition Insight, CNS Media BV. Retrieved 1 January 2019.
- ^ “Products Containing Cannabidiol (CBD) – Overview”. SwissMedic.ch. Retrieved May 20, 2017.
- ^ “Cannabis à faible teneur en THC et CBD” (in French). BAG.Admin.ch. Retrieved May 20, 2017.
- ^ Zlebnik NE, Cheer JF (July 2016). “Beyond the CB1 Receptor: Is Cannabidiol the Answer for Disorders of Motivation?”. Annual Review of Neuroscience. 39: 1–17. doi:10.1146/annurev-neuro-070815-014038. PMC 5818147. PMID 27023732.
- ^ Hurd YL, Yoon M, Manini AF, Hernandez S, Olmedo R, Ostman M, Jutras-Aswad D (October 2015). “Early Phase in the Development of Cannabidiol as a Treatment for Addiction: Opioid Relapse Takes Initial Center Stage”. Neurotherapeutics. 12 (4): 807–15. doi:10.1007/s13311-015-0373-7. PMC 4604178. PMID 26269227.
- ^ Jump up to:a b Pamplona, Fabricio A.; da Silva, Lorenzo Rolim; Coan, Ana Carolina (12 September 2018). “Potential Clinical Benefits of CBD-Rich Cannabis Extracts Over Purified CBD in Treatment-Resistant Epilepsy: Observational Data Meta-analysis”. Frontiers in Neurology. 9. doi:10.3389/fneur.2018.00759. ISSN 1664-2295. PMC 6143706. PMID 30258398.
Further reading
- Williams, Alex (October 27, 2018). “Why Is CBD Everywhere?”. The New York Times. ISSN 0362-4331.
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Sativex (with THC), Epidiolex |
| Synonyms | CBD |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
Inhalation (smoking, vaping), buccal (aerosol spray), oral (solution)[1][2] |
| Drug class | Cannabinoid |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | • Oral: 13–19%[3] • Inhaled: 31% (11–45%)[4] |
| Elimination half-life | 18–32 hours[5] |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ECHA InfoCard | 100.215.986 |
| Chemical and physical data | |
| Formula | C21H30O2 |
| Molar mass | 314.464 g/mol |
| 3D model (JSmol) | |
| Melting point | 66 °C (151 °F) |
| (verify) | |
| Part of a series on |
| Cannabis |
|---|
 |
/////////////////////Cannabidiol, カンナビジオール , FDA 2018, GW Research Ltd , APH-1501, BRCX-014, BTX-1204, BTX-1503, CBD, GW-42003, GWP-42003, GWP-42003-P, PLT-101, PTL-101, ZYN-002
Voretigene neparvovec , ボレチジーンネパルボベック;
Voretigene neparvovec
Voretigene neparvovec-rzyl;
Luxturna (TN)
ボレチジーンネパルボベック;
DNA (synthetic adeno-associated virus 2 vector AAV2-hRPE65v2)
CAS: 1646819-03-5
2017/12/19, FDA Luxturna, SPARK THERAPEUTICS
Vision loss treatment, Retinal dystrophy
AAV2-hRPE65v2
AAV2.RPE65
LTW-888
SPK-RPE65
rAAV.hRPE65v2
rAAV2-CBSB-hRPE65
2SPI046IKD (UNII code)
| melting point (°C) | 72-90ºC | Rayaprolu V. et al. J. Virol. vol. 87. no. 24. (2013) |
FDA
LUXTURNA
STN: 125610
Proper Name: voretigene neparvovec-rzyl
Trade Name: LUXTURNA
Manufacturer: Spark Therapeutics, Inc.
Indication:
- Is an adeno-associated virus vector-based gene therapy indicated for the treatment of patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy. Patients must have viable retinal cells as determined by the treating physician(s).
Product Information
- Package Insert – LUXTURNA (PDF – 602KB)
- Demographic Subgroup Information – voretigene neparvovec [LUXTURNA] (PDF – 2.7MB)
Refer to Section 1.1 of the clinical reviewer memo for information about participation in the clinical trials and any analysis of demographic subgroup outcomes that is notable.
Supporting Documents
Related Information
Voretigene neparvovec (Luxturna) is a novel gene therapy for the treatment of Leber’s congenital amaurosis.[1] It was developed by Spark Therapeutics and Children’s Hospital of Philadelphia.[2][3] It is the first in vivo gene therapy approved by the FDA.[4]
Leber’s congenital amaurosis, or biallelic RPE65-mediated inherited retinal disease, is an inherited disorder causing progressive blindness. Voretigene is the first treatment available for this condition.[5] The gene therapy is not a cure for the condition, but substantially improves vision in those treated.[6] It is given as an subretinal injection.
It was developed by collaboration between the University of Pennsylvania, Yale University, the University of Florida and Cornell University. In 2018, the product was launched in the U.S. by Spark Therapeutics for the treatment of children and adult patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy. The same year, Spark Therapeutics received approval for the product in the E.U. for the same indication.
Chemistry and production
Voretigene neparvovec is an AAV2 vector containing human RPE65 cDNA with a modified Kozak sequence. The virus is grown in HEK 293 cells and purified for administration.[7]
History
Married researchers Jean Bennett and Albert Maguire, among others, worked for decades on studies of congenital blindness, culminating in approval of a novel therapy, Luxturna.[8]
It was granted orphan drug status for Leber congenital amaurosis and retinitis pigmentosa.[9][10] A biologics license application was submitted to the FDA in July 2017 with Priority Review.[5] Phase III clinical trial results were published in August 2017.[11] On 12 October 2017, a key advisory panel to the Food and Drug Administration (FDA), composed of 16 experts, unanimously recommended approval of the treatment.[12] The US FDA approved the drug on December 19, 2017. With the approval, Spark Therapeutics received a pediatric disease priority review voucher.[13]
The first commercial sale of voretigene neparvovec — the first for any gene therapy product in the US — occurred in March 2018.[14][14][4] The price of the treatment has been announced at $425,000 per eye.[15]
INDICATION
LUXTURNA (voretigene neparvovec-rzyl) is an adeno-associated virus vector-based gene therapy indicated for the treatment of patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy.
Patients must have viable retinal cells as determined by the treating physicians.
IMPORTANT SAFETY INFORMATION FOR LUXTURNA
Warnings and Precautions
-
Endophthalmitis may occur following any intraocular surgical procedure or injection. Use proper aseptic injection technique when administering LUXTURNA, and monitor for and advise patients to report any signs or symptoms of infection or inflammation to permit early treatment of any infection.
-
Permanent decline in visual acuity may occur following subretinal injection of LUXTURNA. Monitor patients for visual disturbances.
-
Retinal abnormalities may occur during or following the subretinal injection of LUXTURNA, including macular holes, foveal thinning, loss of foveal function, foveal dehiscence, and retinal hemorrhage. Monitor and manage these retinal abnormalities appropriately. Do not administer LUXTURNA in the immediate vicinity of the fovea. Retinal abnormalities may occur during or following vitrectomy, including retinal tears, epiretinal membrane, or retinal detachment. Monitor patients during and following the injection to permit early treatment of these retinal abnormalities. Advise patients to report any signs or symptoms of retinal tears and/or detachment without delay.
-
Increased intraocular pressure may occur after subretinal injection of LUXTURNA. Monitor and manage intraocular pressure appropriately.
-
Expansion of intraocular air bubbles Instruct patients to avoid air travel, travel to high elevations or scuba diving until the air bubble formed following administration of LUXTURNA has completely dissipated from the eye. It may take one week or more following injection for the air bubble to dissipate. A change in altitude while the air bubble is still present can result in irreversible vision loss. Verify the dissipation of the air bubble through ophthalmic examination.
-
Cataract Subretinal injection of LUXTURNA, especially vitrectomy surgery, is associated with an increased incidence of cataract development and/or progression.
Adverse Reactions
-
In clinical studies, ocular adverse reactions occurred in 66% of study participants (57% of injected eyes), and may have been related to LUXTURNA, the subretinal injection procedure, the concomitant use of corticosteroids, or a combination of these procedures and products.
-
The most common adverse reactions (incidence ≥5% of study participants) were conjunctival hyperemia (22%), cataract (20%), increased intraocular pressure (15%), retinal tear (10%), dellen (thinning of the corneal stroma) (7%), macular hole (7%), subretinal deposits (7%), eye inflammation (5%), eye irritation (5%), eye pain (5%), and maculopathy (wrinkling on the surface of the macula) (5%).
Immunogenicity
Immune reactions and extra-ocular exposure to LUXTURNA in clinical studies were mild. No clinically significant cytotoxic T-cell response to either AAV2 or RPE65 has been observed.
In clinical studies, the interval between the subretinal injections into the two eyes ranged from 7 to 14 days and 1.7 to 4.6 years. Study participants received systemic corticosteroids before and after subretinal injection of LUXTURNA to each eye, which may have decreased the potential immune reaction to either AAV2 or RPE65.
Pediatric Use
Treatment with LUXTURNA is not recommended for patients younger than 12 months of age, because the retinal cells are still undergoing cell proliferation, and LUXTURNA would potentially be diluted or lost during the cell proliferation. The safety and efficacy of LUXTURNA have been established in pediatric patients. There were no significant differences in safety between the different age subgroups.
Please see US Full Prescribing Information for LUXTURNA.
References:
1. LUXTURNA [package insert]. Philadelphia, PA: Spark Therapeutics, Inc; 2017. 2. Gupta PR, Huckfeldt RM. Gene therapy for inherited retinal degenerations: initial successes and future challenges. J Neural Eng. 2017;14(5):051002. 3. Kay C. Gene therapy: the new frontier for inherited retinal disease. Retina Specialist. March 2017. http://www.retina-specialist.com/CMSDocuments/2017/03/RS/rs0317I.pdf. Accessed November 14, 2017 4. Polinski NK, Gombash SE, Manfredsson FP, et al. Recombinant adeno-associated virus 2/5-mediated gene transfer is reduced in the aged rat midbrain. Neurobiol Aging. 2015;36(2):1110-1120. 5. Moore T. Restoring retinal function in a mouse model of hereditary blindness. PLoS Med. 2005;2(11):e399. 6. McBee JK, Van Hooser JP, Jang GF, Palczewski K. Isomerization of 11-cis-retinoids to all-trans-retinoids in vitro and in vivo. J Biol Chem. 2001;276(51):48483-48493. 7. Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4(5):346-358. 8. Trapani I, Puppo A, Auricchio A. Vector platforms for gene therapy of inherited retinopathies. Prog Retin Eye Res. 2014;43:108-128. 9. Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390(10097):849-860.


PAPERS
Progress in Retinal and Eye Research (2018), 63, 107-131
Lancet (2017), 390(10097), 849-860.
References
- ^ “Luxturna (voretigene neparvovec-rzyl) label” (PDF). FDA. December 2017. Retrieved 31 December 2017. (for label updates, see FDA index page)
- ^ “Spark’s gene therapy for blindness is racing to a historic date with the FDA”. Statnews.com. 9 October 2017. Retrieved 9 October 2017.
- ^ Clarke,Reuters, Toni. “Gene Therapy for Blindness Appears Initially Effective, Says U.S. FDA”. Scientific American. Retrieved 2017-10-12.
- ^ Jump up to:a b “First Gene Therapy For Inherited Disease Gets FDA Approval”. NPR.org. 19 Dec 2017.
- ^ Jump up to:a b “Press Release – Investors & Media – Spark Therapeutics”. Ir.sparktx.com. Retrieved 9 October 2017.
- ^ McGinley, Laurie (19 December 2017). “FDA approves first gene therapy for an inherited disease”. Washington Post.
- ^ Russell, Stephen; Bennett, Jean; Wellman, Jennifer A.; Chung, Daniel C.; Yu, Zi-Fan; Tillman, Amy; Wittes, Janet; Pappas, Julie; Elci, Okan; McCague, Sarah; Cross, Dominique; Marshall, Kathleen A.; Walshire, Jean; Kehoe, Taylor L.; Reichert, Hannah; Davis, Maria; Raffini, Leslie; George, Lindsey A.; Hudson, F Parker; Dingfield, Laura; Zhu, Xiaosong; Haller, Julia A.; Sohn, Elliott H.; Mahajan, Vinit B.; Pfeifer, Wanda; Weckmann, Michelle; Johnson, Chris; Gewaily, Dina; Drack, Arlene; et al. (2017). “Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65 -mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial”. The Lancet. 390 (10097): 849–860. doi:10.1016/S0140-6736(17)31868-8. PMC 5726391. PMID 28712537.
- ^ “FDA approves Spark’s gene therapy for rare blindness pioneered at CHOP – Philly”. Philly.com. Retrieved 2018-03-24.
- ^ “Voretigene neparvovec – Spark Therapeutics – AdisInsight”. adisinsight.springer.com.
- ^ Ricki Lewis, PhD (October 13, 2017). “FDA Panel Backs Gene Therapy for Inherited Blindness”. Medscape.
- ^ Lee, Helena; Lotery, Andrew (2017). “Gene therapy for RPE65 -mediated inherited retinal dystrophy completes phase 3”. The Lancet. 390 (10097): 823–824. doi:10.1016/S0140-6736(17)31622-7. PMID 28712536.
- ^ “Landmark Therapy to Treat Blindness Gets One Step Closer to FDA Approval”. Bloomberg.com. 2017-10-12. Retrieved 2017-10-12.
- ^ “Spark grabs FDA nod for Luxturna, a breakthrough gene therapy likely bearing a pioneering price”. FiercePharma.
- ^ Jump up to:a b “The anxious launch of Luxturna, a gene therapy with a record sticker price”. STAT. 2018-03-21. Retrieved 2018-03-24.
- ^ Tirrell, Meg (3 January 2018). “A US drugmaker offers to cure rare blindness for $850,000”. CNBC. Retrieved 3 January 2018.
Further reading
- Ledford, Heidi (2017). “FDA advisers back gene therapy for rare form of blindness”. Nature. 550 (7676): 314. doi:10.1038/nature.2017.22819. PMID 29052639.
- Wilson, James M. (2018). “Interview with Jean Bennett, MD, PhD”. Human Gene Therapy Clinical Development. 29 (1): 7–9. doi:10.1089/humc.2018.29032.int. PMID 29641279.
- Ameri, Hossein (2018). “Prospect of retinal gene therapy following commercialization of voretigene neparvovec-rzyl for retinal dystrophy mediated by RPE65 mutation”. Journal of Current Ophthalmology. 30 (1): 1–2. doi:10.1016/j.joco.2018.01.006. PMC 5859497. PMID 29564403.
- Russell, Stephen; Bennett, Jean; Maguire, Albert M.; High, Katherine A. (2018). “Voretigene neparvovec-rzyl for the treatment of biallelic RPE65 mutation–associated retinal dystrophy”. Expert Opinion on Orphan Drugs. 6 (8): 457–464. doi:10.1080/21678707.2018.1508340.
- Bakall, Benjamin; Hariprasad, Seenu M.; Klein, Kendra A. (2018). “Emerging Gene Therapy Treatments for Inherited Retinal Diseases”. Ophthalmic Surgery, Lasers and Imaging Retina. 49 (7): 472–478. doi:10.3928/23258160-20180628-02. PMID 30021033.
- “Drug and Device News”. P & T. 43 (2): 74–104. 2018. PMC 5768294. PMID 29386862.
| Gene therapy | |
|---|---|
| Vector | Adeno-associated virusserotype 2 |
| Nucleic acid type | DNA |
| Editing method | RPE65 |
| Clinical data | |
| Trade names | Luxturna |
| Pregnancy category |
|
| Routes of administration |
subretinal injection |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| KEGG | |
//////////FDA 2017, Voretigene neparvovec , Voretigene neparvovec-rzyl, Luxturna, ボレチジーンネパルボベック, 1646819-03-5 , FDA Luxturna, SPARK THERAPEUTICS, Vision loss treatment, Retinal dystrophy., AAV2-hRPE65v2, LTW-888, SPK-RPE65, Orphan drug,
Elapegademase, エラペグアデマーゼ (遺伝子組換え)
AQTPAFNKPK VELHVHLDGA IKPETILYYG RKRGIALPAD TPEELQNIIG MDKPLSLPEF
LAKFDYYMPA IAGSREAVKR IAYEFVEMKA KDGVVYVEVR YSPHLLANSK VEPIPWNQAE
GDLTPDEVVS LVNQGLQEGE RDFGVKVRSI LCCMRHQPSW SSEVVELCKK YREQTVVAID
LAGDETIEGS SLFPGHVKAY AEAVKSGVHR TVHAGEVGSA NVVKEAVDTL KTERLGHGYH
TLEDTTLYNR LRQENMHFEV CPWSSYLTGA WKPDTEHPVV RFKNDQVNYS LNTDDPLIFK
STLDTDYQMT KNEMGFTEEE FKRLNINAAK SSFLPEDEKK ELLDLLYKAY GMPSPA

>>Elapegademase<<< AQTPAFNKPKVELHVHLDGAIKPETILYYGRKRGIALPADTPEELQNIIGMDKPLSLPEF LAKFDYYMPAIAGSREAVKRIAYEFVEMKAKDGVVYVEVRYSPHLLANSKVEPIPWNQAE GDLTPDEVVSLVNQGLQEGERDFGVKVRSILCCMRHQPSWSSEVVELCKKYREQTVVAID LAGDETIEGSSLFPGHVKAYAEAVKSGVHRTVHAGEVGSANVVKEAVDTLKTERLGHGYH TLEDTTLYNRLRQENMHFEVCPWSSYLTGAWKPDTEHPVVRFKNDQVNYSLNTDDPLIFK STLDTDYQMTKNEMGFTEEEFKRLNINAAKSSFLPEDEKKELLDLLYKAYGMPSPA
Elapegademase, エラペグアデマーゼ (遺伝子組換え)
EZN-2279
Protein chemical formula C1797H2795N477O544S12
Protein average weight 115000.0 Da
Peptide
APPROVED, FDA, Revcovi, 2018/10/5
CAS: 1709806-75-6
Elapegademase-lvlr, Poly(oxy-1,2-ethanediyl), alpha-carboxy-omega-methoxy-, amide with adenosine deaminase (synthetic)
EZN-2279; PEG-rADA; Pegademase recombinant – Leadiant Biosciences; Pegylated recombinant adenosine deaminase; Polyethylene glycol recombinant adenosine deaminase; STM-279, UNII: 9R3D3Y0UHS
- Originator Sigma-Tau Pharmaceuticals
- Developer Leadiant Biosciences; Teijin Pharma
- Class Antivirals; Polyethylene glycols
- Mechanism of Action Adenosine deaminase stimulants
- Orphan Drug Status Yes – Immunodeficiency disorders; Adenosine deaminase deficiency
- Registered Adenosine deaminase deficiency; Immunodeficiency disorders
- 05 Oct 2018 Registered for Adenosine deaminase deficiency (In adults, In children) in USA (IM)
- 05 Oct 2018 Registered for Immunodeficiency disorders (In adults, In children) in USA (IM)
- 04 Oct 2018 Elapegademase receives priority review status for Immunodeficiency disorders and Adenosine deaminase deficiency in USA
検索キーワード:Elapegademase (Genetical Recombination)
検索件数:1
| エラペグアデマーゼ(遺伝子組換え) Elapegademase (Genetical Recombination)  [1709806-75-6] |
Elapegademase is a PEGylated recombinant adenosine deaminase. It can be defined molecularly as a genetically modified bovine adenosine deaminase with a modification in cysteine 74 for serine and with about 13 methoxy polyethylene glycol chains bound via carbonyl group in alanine and lysine residues.[4] Elapegademase is generated in E. coli, developed by Leadiant Biosciences and FDA approved on October 5, 2018.[1, 5]
Indication
Elapegademase is approved for the treatment of adenosine deaminase severe combined immune deficiency (ADA-SCID) in pediatric and adult patients.[1] This condition was previously treated by the use of pegamedase bovine as part of an enzyme replacement therapy.[2]
ADA-SCID is a genetically inherited disorder that is very rare and characterized by a deficiency in the adenosine deaminase enzyme. The patients suffering from this disease often present a compromised immune system. This condition is characterized by very low levels of white blood cells and immunoglobulin levels which results in severe and recurring infections.[3]
Pharmacodynamics
In clinical trials, elapegademase was shown to increase adenosine deaminase activity while reducing the concentrations of toxic metabolites which are the hallmark of ADA-SCID. As well, it was shown to improve the total lymphocyte count.[6]
Mechanism of action
The ADA-SCID is caused by the presence of mutations in the ADA gene which is responsible for the synthesis of adenosine deaminase. This enzyme is found throughout the body but it is mainly active in lymphocytes. The normal function of adenosine deaminase is to eliminate deoxyadenosine, created when DNA is degraded, by converting it into deoxyinosine. This degradation process is very important as deoxyadenosine is cytotoxic, especially for lymphocytes. Immature lymphocytes are particularly vulnerable as deoxyadenosine kills them before maturation making them unable to produce their immune function.[3]
Therefore, based on the causes of ADA-SCID, elapegademase works by supplementing the levels of adenosine deaminase. Being a recombinant and an E. coli-produced molecule, the use of this drug eliminates the need to source the enzyme from animals, as it was used previously.[1]
Absorption
Elapegademase is administered intramuscularly and the reported Tmax, Cmax and AUC are approximately 60 hours, 240 mmol.h/L and 33000 hr.mmol/L as reported during a week.[Label]
Volume of distribution
This pharmacokinetic property has not been fully studied.
Protein binding
This pharmacokinetic property is not significant as the main effect is in the blood cells.
Metabolism
Metabolism studies have not been performed but it is thought to be degraded by proteases to small peptides and individual amino acids.
Route of elimination
This pharmacokinetic property has not been fully studied.
Half life
This pharmacokinetic property has not been fully studied.
Clearance
This pharmacokinetic property has not been fully studied.
Toxicity
As elapegademase is a therapeutic protein, there is a potential risk of immunogenicity.
There are no studies related to overdose but the highest weekly prescribed dose in clinical trials was 0.4 mg/kg. In nonclinical studies, a dosage of 1.8 fold of the clinical dose produced a slight increase in the activated partial thromboplastin time.[Label]
FDA label. Download (145 KB)
General References
- Rare DR [Link]
- Globe News Wire [Link]
- NIH [Link]
- NIHS reports [File]
- WHO Drug Information 2017 [File]
- Revcovi information [File]
/////////////Elapegademase, Peptide, エラペグアデマーゼ (遺伝子組換え) , EZN-2279, Elapegademase-lvlr, Orphan Drug, STM 279, FDA 2018
COCCOC(=O)NCCCC[C@H](N)C(=O)O
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent

READ
ANTHONY MELVIN CRASTO

DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE



 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
CALL +919323115463 INDIA
//////////////
Calaspargase pegol, カラスパルガーゼペゴル
|
LPNITILATG GTIAGGGDSA TKSNYTAGKV GVENLVNAVP QLKDIANVKG EQVVNIGSQD
MNDDVWLTLA KKINTDCDKT DGFVITHGTD TMEETAYFLD LTVKCDKPVV MVGAMRPSTS MSADGPFNLY NAVVTAADKA SANRGVLVVM NDTVLDGRDV TKTNTTDVAT FKSVNYGPLG YIHNGKIDYQ RTPARKHTSD TPFDVSKLNE LPKVGIVYNY ANASDLPAKA LVDAGYDGIV SAGVGNGNLY KTVFDTLATA AKNGTAVVRS SRVPTGATTQ DAEVDDAKYG FVASGTLNPQ KARVLLQLAL TQTKDPQQIQ QIFNQY (tetramer; disulfide bridge 77-105, 77′-105′, 77”-105”, 77”’-105”’) |

Calaspargase pegol
Molecular Formula, C1516-H2423-N415-O492-S8 (peptide monomer), Molecular Weight, 10261.2163
APPROVED, Asparlas, FDA 2018/12/20
CAS 941577-06-6
UNII T9FVH03HMZ
カラスパルガーゼペゴル;
(27-Alanine,64-aspartic acid,252-threonine,263-asparagine)-L-asparaginase 2 (EC 3.5.1.1, L-asparagineamidohydrolase II) Escherichia coli (strain K12) tetramer alpha4, carbamates with alpha-carboxy-omega-methoxypoly(oxyethylene)
Asparaginase (Escherichia coli isoenzyme II), conjugate with alpha-(((2,5-dioxo-1-pyrrolidinyl)oxy)carbonyl)-omega-methoxypoly(oxy-1,2-ethanediyl)
|
Peptide
|
- Calaspargase pegol
- calaspargase pegol-mknl
- EZN-2285
- Used to treat acute lymphoblastic leukemia., Antineoplastic
- BAX-2303
SC-PEG E. Coli L-asparaginase
SHP-663
Calaspargase pegol-mknl (trade name Asparlas) is a drug for the treatment of acute lymphoblastic leukemia (ALL). It is approved by the Food and Drug Administration for use in the United States as a component of a multi-agent chemotherapeutic regimen for ALL in pediatric and young adult patients aged 1 month to 21 years.[1]
Calaspargase pegol was first approved in 2018 in the U.S. as part of a multi-agent chemotherapeutic regimen for the treatment of patients with acute lymphoblastic leukemia.
In 2008, orphan drug designation was assigned in the E.U.
Calaspargase pegol is an engineered protein consisting of the E. coli-derived enzyme L-asparaginase II conjugated with succinimidyl carbonate monomethoxypolyethylene glycol (pegol).[2] The L-asparaginase portion hydrolyzes L-asparagine to L-aspartic acid depriving the tumor cell of the L-asparagine it needs for survival.[2] The conjugation with the pegol group increases the half-life of the drug making it longer acting.
Asparaginase is an important agent used to treat acute lymphoblastic leukemia (ALL) [1]. Asparagine is incorporated into most proteins, and the synthesis of proteins is stopped when asparagine is absent, which inhibits RNA and DNA synthesis, resulting in a halt in cellular proliferation. This forms the basis of asparaginase treatment in ALL [1], [2], [6].
Calaspargase pegol, also known as asparlas, is an asparagine specific enzyme which is indicated as a part of a multi-agent chemotherapy regimen for the treatment of ALL [3]. The asparagine specific enzyme is derived from Escherichia coli, as a conjugate of L-asparaginase (L-asparagine amidohydrolase) and monomethoxypolyethylene glycol (mPEG) with a succinimidyl carbonate (SC) linker to create a stable molecule which increases the half-life and decreases the dosing frequency [Label], [1].
Calaspargase pegol, by Shire pharmaceuticals, was approved by the FDA on December 20, 2018 for acute lymphoblastic anemia (ALL) [3].
Indication
This drug is is an asparagine specific enzyme indicated as a component of a multi-agent chemotherapeutic regimen for the treatment of acute lymphoblastic leukemia in pediatric and young adult patients age 1 month to 21 years [Label].
The pharmacokinetics of calaspargase pegol were examined when given in combination with multiagent chemotherapy in 124 patients with B-cell lineage ALL [3]. The FDA approval of this drug was based on the achievement and maintenance of nadir serum asparaginase activity above the level of 0.1 U/mL when administering calaspargase, 2500 U/m2 intravenously, at 3-week intervals.
Associated Conditions
Pharmacodynamics
The effect of this drug is believed to occur by selective killing of leukemic cells due to depletion of plasma L-asparagine. Leukemic cells with low expression of asparagine synthetase are less capable of producing L-asparagine, and therefore rely on exogenous L-asparagine for survival [Label]. When asparagine is depleted, tumor cells cannot proliferate [6].
During remission induction, one dose of SC-PEG (2500 IU/m2) results in a sustained therapeutic serum asparaginase activity (SAA) without excessive toxicity or marked differences in the proportion of patients with low end-induction minimum residual disease (MRD) [5].
Pharmacodynamic (PD) response was studied through measurement of plasma and cerebrospinal fluid (CSF) asparagine concentrations with an LC-MS/MS assay (liquid chromatography–mass spectrometry). Asparagine concentration in plasma was sustained below the assay limit of quantification for more than 18 days after one dose of calaspargase pegol, 2,500 U/m2, during the induction phase of treatment. Average cerebrospinal asparagine concentrations decreased from a pretreatment concentration of 0.8 μg/mL (N=10) to 0.2 μg/mL on Day 4 (N=37) and stayed decreased at 0.2 μg/mL (N=35) 25 days after the administration of one of 2,500 U/m2 in the induction phase [Label].
Mechanism of action
L-asparaginase (the main component of this drug) is an enzyme that catalyzes the conversion of the amino acid L-asparagine into both aspartic acid and ammonia [Label], [2]. This process depletes malignant cells of their required asparagine. The depletion of asparagine then blocks protein synthesis and tumor cell proliferation, especially in the G1 phase of the cell cycle. As a result, tumor cell death occurs. Asparagine is important in protein synthesis in acute lymphoblastic leukemia (ALL) cells which, unlike normal cells, cannot produce this amino acid due to lack of the enzyme asparagine synthase [2], [Label].
Pegylation decreases enzyme antigenicity and increases its half-life. Succinimidyl carbamate (SC) is used as a PEG linker to facilitate attachment to asparaginase and enhances the stability of the formulation [4], [1]. SC-PEG urethane linkages formed with lysine groups are more hydrolytically stable [2].
Toxicity
Pancreatitis, hepatotoxicity, hemorrhage, and thrombosis have been observed with calaspargase pegol use [Label].
Pancreatitis: Discontinue this drug in patients with pancreatitis, and monitor blood glucose.
Hepatotoxicity: Hepatic function should be tested regularly, and trough levels of this drug should be measured during the recovery phase of the drug cycle [Label].
Hemorrhage or Thrombosis: Discontinue this drug in serious or life-threatening hemorrhage or thrombosis. In cases of hemorrhage, identify the cause of hemorrhage and treat appropriately. Administer anticoagulant therapy as indicated in thrombotic events [Label].
A note on hypersensitivity:
Observe the patient for 1 hour after administration of calaspargase pegol for possible hypersensitivity [Label]. In cases of previous hypersensitivity to this drug, discontinue this drug immediately.
Lactation: Advise women not to breastfeed while taking this drug [Label].
Pregnancy: There are no available data on the use of calaspargase pegol in pregnant women to confirm a risk of drug-associated major birth defects and miscarriage. Published literature studies in pregnant animals suggest asparagine depletion can cause harm to the animal offspring. It is therefore advisable to inform women of childbearing age of this risk. The background risk of major birth defects and miscarriage for humans is unknown at this time [Label].
Pregnancy testing should occur before initiating treatment. Advise females of reproductive potential to avoid becoming pregnant while taking this drug. Females should use effective contraceptive methods, including a barrier methods, during treatment and for at least 3 months after the last dose. There is a risk for an interaction between calaspargase pegol and oral contraceptives. The concurrent use of this drug with oral contraceptives should be avoided. Other non-oral contraceptive methods should be used in women of childbearing potential [Label].
References
- Angiolillo AL, Schore RJ, Devidas M, Borowitz MJ, Carroll AJ, Gastier-Foster JM, Heerema NA, Keilani T, Lane AR, Loh ML, Reaman GH, Adamson PC, Wood B, Wood C, Zheng HW, Raetz EA, Winick NJ, Carroll WL, Hunger SP: Pharmacokinetic and pharmacodynamic properties of calaspargase pegol Escherichia coli L-asparaginase in the treatment of patients with acute lymphoblastic leukemia: results from Children’s Oncology Group Study AALL07P4. J Clin Oncol. 2014 Dec 1;32(34):3874-82. doi: 10.1200/JCO.2014.55.5763. Epub 2014 Oct 27. [PubMed:25348002]
- Appel IM, Kazemier KM, Boos J, Lanvers C, Huijmans J, Veerman AJ, van Wering E, den Boer ML, Pieters R: Pharmacokinetic, pharmacodynamic and intracellular effects of PEG-asparaginase in newly diagnosed childhood acute lymphoblastic leukemia: results from a single agent window study. Leukemia. 2008 Sep;22(9):1665-79. doi: 10.1038/leu.2008.165. Epub 2008 Jun 26. [PubMed:18580955]
- Blood Journal: Randomized Study of Pegaspargase (SS-PEG) and Calaspargase Pegol (SPC-PEG) in Pediatric Patients with Newly Diagnosed Acute Lymphoblastic Leukemia or Lymphoblastic Lymphoma: Results of DFCI ALL Consortium Protocol 11-001 [Link]
References
- ^ “FDA approves longer-acting calaspargase pegol-mknl for ALL” (Press release). Food and Drug Administration. December 20, 2018.
- ^ Jump up to:a b “Calaspargase pegol-mknl”. NCI Drug Dictionary. National Cancer Institute.
FDA label, Download(300 KB)
General References
- Angiolillo AL, Schore RJ, Devidas M, Borowitz MJ, Carroll AJ, Gastier-Foster JM, Heerema NA, Keilani T, Lane AR, Loh ML, Reaman GH, Adamson PC, Wood B, Wood C, Zheng HW, Raetz EA, Winick NJ, Carroll WL, Hunger SP: Pharmacokinetic and pharmacodynamic properties of calaspargase pegol Escherichia coli L-asparaginase in the treatment of patients with acute lymphoblastic leukemia: results from Children’s Oncology Group Study AALL07P4. J Clin Oncol. 2014 Dec 1;32(34):3874-82. doi: 10.1200/JCO.2014.55.5763. Epub 2014 Oct 27. [PubMed:25348002]
- Appel IM, Kazemier KM, Boos J, Lanvers C, Huijmans J, Veerman AJ, van Wering E, den Boer ML, Pieters R: Pharmacokinetic, pharmacodynamic and intracellular effects of PEG-asparaginase in newly diagnosed childhood acute lymphoblastic leukemia: results from a single agent window study. Leukemia. 2008 Sep;22(9):1665-79. doi: 10.1038/leu.2008.165. Epub 2008 Jun 26. [PubMed:18580955]
- Asparlas Approval History [Link]
- NCI: Calaspargase Pegol [Link]
- Blood Journal: Randomized Study of Pegaspargase (SS-PEG) and Calaspargase Pegol (SPC-PEG) in Pediatric Patients with Newly Diagnosed Acute Lymphoblastic Leukemia or Lymphoblastic Lymphoma: Results of DFCI ALL Consortium Protocol 11-001 [Link]
- Medsafe NZ: Erwinaze inj [File]
| Clinical data | |
|---|---|
| Trade names | Asparlas |
| Synonyms | EZN-2285 |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| DrugBank | |
| UNII | |
| KEGG | |
| ChEMBL | |
/////////////Calaspargase pegol, Peptide, FDA 2018, EZN-2285, カラスパルガーゼペゴル , BAX-2303, SC-PEG E. Coli L-asparaginase , SHP-663, orphan drug
CC(C)C[C@@H](C(=O)O)NC(=O)OCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOC.COCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOC(=O)NCCCC[C@@H](C(=O)O)N
Omadacycline tosylate
Omadacycline tosylate
728.8521, C29H40N4O7. C7H8O3S
CAS: 1075240-43-5
389139-89-3 FREE FORM
FDA 2018/10/3, Nuzyra
オマダサイクリントシル酸塩;
UNII-5658Y89YCD

Omadacycline has been used in trials studying the treatment of Bacterial Pneumonia, Bacterial Infections, Community-Acquired Infections, and Skin Structures and Soft Tissue Infections. Omadacycline represents a significant advance over the well-known tetracycline family, and has been shown to be highly effective in animal models at treating increasingly problematic, clinically prevalent infections caused by gram-positive bacteria, such as methicillin-resistant Staphylococcus aureus (MRSA), and by gram-negative, atypical and anaerobic bacteria, including those resistant to currently available classes of antibiotics and known to cause diseases such as pneumonias, urinary tract infections, skin diseases and blood-borne infections in both the hospital and community settings.
Omadacycline (formerly known as PTK-0796)[1] is a broad spectrum antibiotic belonging to the aminomethylcycline subclass[2] of tetracycline antibiotics. In the United States, it was approved in October 2018 for the treatment of community-acquired bacterial pneumonia and acute skin and skin structure infections.
In vitro studies
In vitro studies have shown that omadacycline has activity against a broad range of Gram-positive and select Gram-negativepathogens.[3] Omadacycline has potent in vitro activity against Gram-positive aerobic bacteria including methicillin-resistant Staphylococcus aureus (MRSA), pencillin-resistant and multi-drug resistant Streptococcus pneumoniae, and vancomycin-resistant Enterococcus. Omadacycline also has antimicrobial activity against common Gram-negative aerobes, some anaerobes, and atypical bacteria such as Legionella and Chlamydia.[4] This activity translated to potent efficacy for omadacycline in an in vivo systemic infection model in mice.[5]
Additional in vitro and in vivo studies of omadacycline metabolism, disposition, and drug interactions show that omadacycline is metabolically stable (i.e., it does not undergo significant biotransformation) and neither inhibits nor interacts with metabolizing enzymes or transporters.[6]
Mechanism of action
The mechanism of action of omadacycline is similar to that of other tetracyclines – inhibition of bacterial protein synthesis. Omadacycline has activity against bacterial strains expressing the two main forms of tetracycline resistance (efflux and ribosomal protection).[7]
Clinical trials
A phase 2 study was conducted comparing the safety and efficacy of omadacycline to linezolid for the treatment of complicated skin and skin structure infections. Patients were randomized at 11 sites in the US to receive either omadacycline 100 mg intravenously once daily with an option to transition to 200 mg orally once daily or linezolid 600 mg intravenously twice daily with an option to transition to 600 mg orally twice daily. The results indicated that omadacycline is well-tolerated and has the potential to be an effective treatment in patients with complicated skin and skin structure infections.[8]
In June 2013, the US Food and Drug Administration (FDA) designated the intravenous and oral formulations of omadacycline as a qualified infectious disease product in the treatment of acute bacterial skin and skin structure infections and community-acquired bacterial pneumonia.[9]
A 650 patient phase 3 registration study comparing omadacycline to linezolid for the treatment of acute bacterial skin and skin structure infections began in June 2015.[10][11]Omadacycline met the primary efficacy endpoint of early clinical response with statistical non-inferiority (10% margin) compared to linezolid, and was generally safe and well-tolerated. The most common treatment-emergent adverse events were gastrointestinal side effects (18.0% for omadacycline vs. 15.8% for linezolid).[12]
A 750 patient phase 3 study comparing omadacycline to moxifloxacin for the treatment of community-acquired bacterial pneumonia began in November 2015.[13] Omadacycline was statistically non-inferior to moxifloxacin at the early clinical response, 72 to 120 hours after therapy was initiated.[14]
In May 2016, a phase 1b study of omadacycline in urinary tract infection was initiated.[15]
In August 2016, a second phase 3 study of omadacycline was initiated in patients with acute bacterial skin and skin structure infections, comparing the efficacy and safety of once-daily, oral omadacycline to that of twice-daily, oral linezolid.[16] In July 2017, analysis of the data showed that all of the primary and secondary endpoints required for submission to the FDA and EMA were met. This was the third phase 3 registration study of omadacycline with favorable results.[17]
Discovery
Omadacycline was invented at Tufts University School of Medicine by a research team led by Mark L. Nelson with Mohamed Ismail while at Tufts and Kwasi Ohemeng and Laura Honeyman at Paratek Pharmaceuticals, Boston. The team applying their chemistry methods to the tetracycline scaffolds created over 3000 new derivatives, leading to the novel third generation compounds omadacycline and sarecycline. 18[18]
PAPERS
Tetrahedron Letters (2008), 49(42), 6095-6100

PATENTS
WO 2009120389
WO 2009111064
WO 2017165729
WO 2018026987
WO 2018085216
SYNTHESIS BY PHARMACODIA WEBSITE
Omadacycline, www.pharmacodia.com
REF Omadacycline, www.pharmacodia.com
References
- Jump up^ Boggs, Jennifer. “Antibiotic Firm Paratek Joins IPO Queue; Aiming for $92M”. bioworld.com. Clarivate Analytics. Retrieved October 17, 2017.
- Jump up^ Honeyman, Laura; Ismail, Mohamed; Nelson, Mark L.; Bhatia, Beena; Bowser, Todd E.; Chen, Jackson; Mechiche, Rachid; Ohemeng, Kwasi; Verma, Atul K.; Cannon, E. Pat; MacOne, Ann; Tanaka, S. Ken; Levy, Stuart (2015). “Structure-Activity Relationship of the Aminomethylcyclines and the Discovery of Omadacycline”. Antimicrobial Agents and Chemotherapy. 59 (11): 7044–7053. doi:10.1128/AAC.01536-15. PMC 4604364. PMID 26349824.
- Jump up^ Tanaka, S. Ken (20 June 2016). “In Vitro and In Vivo Assessment of Cardiovascular Effects with Omadacycline”. Antimicrobial Agents and Chemotherapy. 60 (9): 5247–53. doi:10.1128/AAC.00320-16. PMC 4997885. PMID 27324778.
- Jump up^ Villano, Stephen (19 August 2016). “Omadacycline: development of a novel aminomethylcycline antibiotic for treating drug-resistant bacterial infections”. Future Microbiology. 11: 1421–1434. doi:10.2217/fmb-2016-0100. Retrieved 24 August 2016.
- Jump up^ MacOne, A. B.; Caruso, B. K.; Leahy, R. G.; Donatelli, J.; Weir, S.; Draper, M. P.; Tanaka, S. K.; Levy, S. B. (February 2014). “In Vitro and in Vivo Antibacterial Activities of Omadacycline, a Novel Aminomethylcycline”. Antimicrobial Agents and Chemotherapy. 58 (2): 1127–1135. doi:10.1128/AAC.01242-13. PMC 3910882. PMID 24295985.
- Jump up^ Flarakos, Jimmy (8 August 2016). “Clinical disposition, metabolism and in vitro drug–drug interaction properties of omadacycline”. Xenobiotica: 1–15. doi:10.1080/00498254.2016.1213465.
- Jump up^ Draper, M. P.; Weir, S.; MacOne, A.; Donatelli, J.; Trieber, C. A.; Tanaka, S. K.; Levy, S. B. (March 2014). “Mechanism of Action of the Novel Aminomethylcycline Antibiotic Omadacycline”. Antimicrobial Agents and Chemotherapy. 58 (3): 1279–1283. doi:10.1128/AAC.01066-13. PMC 3957880. PMID 24041885.
- Jump up^ Noel, G. J.; Draper, M. P.; Hait, H.; Tanaka, S. K.; Arbeit, R. D. (November 2012). “A Randomized, Evaluator-Blind, Phase 2 Study Comparing the Safety and Efficacy of Omadacycline to Those of Linezolid for Treatment of Complicated Skin and Skin Structure Infections”. Antimicrobial Agents and Chemotherapy. 56 (11): 5650–5654. doi:10.1128/AAC.00948-12. PMC 3486554. PMID 22908151.
- Jump up^ “Paratek Pharmaceuticals Announces FDA Grant of Qualified Infectious Disease Product (QIDP) Designation for Its Lead Product Candidate, Omadacycline”. prnewsire.com. PR Newswire. January 3, 2013. Retrieved October 17, 2017.
- Jump up^ Seiffert, Don (2015). “Paratek presents new trial data for antibiotic as late-stage trials continue”. bizjournals.com. American City Business Journals. Retrieved October 17,2017.
- Jump up^ “Omadacycline Versus Linezolid for the Treatment of ABSSSI (EudraCT #2013-003644-23)”. clinicaltrials.gov. Retrieved 2015-10-13.
- Jump up^ “Paratek Announces that Omadacycline Met All Primary and Secondary Efficacy Outcomes Designated by FDA and EMA in a Phase 3 Study in Acute Bacterial Skin Infections; Omadacycline was Generally Safe and Well-Tolerated”. finance.yahoo.com. Retrieved 3 July 2016.
- Jump up^ “Omadacycline vs Moxifloxacin for the Treatment of CABP (EudraCT #2013-004071-13)”. clinicaltrials.gov. Retrieved 2015-10-13.
- Jump up^ “Paratek Announces Positive Phase 3 Study of Omadacycline in Community-Acquired Bacterial Pneumonia”. http://www.globenewswire.com. April 3, 2017. Retrieved 16 May 2017.
- Jump up^ “Paratek Initiates Phase 1b Study of Omadacycline in Urinary Tract Infection”. globenewswire.com. May 2, 2016. Retrieved 3 July 2016.
- Jump up^ “Paratek Initiates Phase 3 Study of Oral-only Omadacycline in ABSSSI”. globenewswire.com. August 15, 2016. Retrieved 15 August 2016.
- Jump up^ “Paratek Announces Phase 3 Study of Oral-Only Dosing of Omadacycline Met All Primary and Secondary FDA and EMA Efficacy Endpoints in Acute Bacterial Skin Infections”. http://www.globenewswire.com. July 17, 2017. Retrieved 19 July 2017.
- Jump up^ Ref: Mark L. Nelson and Kwasi Ohemeng: 4-dedimethylamino tetracycline compounds, United States (US) patent number 7,056,902 (2006)
|
|
| Clinical data | |
|---|---|
| Trade names | Nuzyra |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C29H40N4O7 |
| Molar mass | 556.66 g·mol−1 |
| 3D model (JSmol) | |
/////////////FDA 2018, Nuzyra, Omadacycline tosylate, Omadacycline, オマダサイクリントシル酸塩 ,PTK-0796, PTK 0796
CC1=CC=C(C=C1)S(O)(=O)=O.[H][C@@]12CC3=C(C=C(CNCC(C)(C)C)C(O)=C3C(=O)C1=C(O)[C@]1(O)C(=O)C(C(N)=O)=C(O)[C@@H](N(C)C)[C@]1([H])C2)N(C)C
FDA approves first treatment Firdapse (amifampridine) for Lambert-Eaton myasthenic syndrome, a rare autoimmune disorder
FDA approves first treatment Firdapse (amifampridine) for Lambert-Eaton myasthenic syndrome, a rare autoimmune disorder
The U.S. Food and Drug Administration today approved Firdapse (amifampridine) tablets for the treatment of Lambert-Eaton myasthenic syndrome (LEMS) in adults. LEMS is a rare autoimmune disorder that affects the connection between nerves and muscles and causes weakness and other symptoms in affected patients. This is the first FDA approval of a treatment for LEMS.
November 28, 2018
Release
The U.S. Food and Drug Administration today approved Firdapse (amifampridine) tablets for the treatment of Lambert-Eaton myasthenic syndrome (LEMS) in adults. LEMS is a rare autoimmune disorder that affects the connection between nerves and muscles and causes weakness and other symptoms in affected patients. This is the first FDA approval of a treatment for LEMS.
“There has been a long-standing need for a treatment for this rare disorder,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “Patients with LEMS have significant weakness and fatigue that can often cause great difficulties with daily activities.”
In people with LEMS, the body’s own immune system attacks the neuromuscular junction (the connection between nerves and muscles) and disrupts the ability of nerve cells to send signals to muscle cells. LEMS may be associated with other autoimmune diseases, but more commonly occurs in patients with cancer such as small cell lung cancer, where its onset precedes or coincides with the diagnosis of cancer. The prevalence of LEMS is estimated to be three per million individuals worldwide.
The efficacy of Firdapse was studied in two clinical trials that together included 64 adult patients who received Firdapse or placebo. The studies measured the Quantitative Myasthenia Gravis score (a 13-item physician-rated categorical scale assessing muscle weakness) and the Subject Global Impression (a seven-point scale on which patients rated their overall impression of the effects of the study treatment on their physical well-being). For both measures, the patients receiving Firdapse experienced a greater benefit than those on placebo.
The most common side effects experienced by patients in the clinical trials were burning or prickling sensation (paresthesia), upper respiratory tract infection, abdominal pain, nausea, diarrhea, headache, elevated liver enzymes, back pain, hypertension and muscle spasms. Seizures have been observed in patients without a history of seizures. Patients should inform their health care provider immediately if they have signs of hypersensitivity reactions such as rash, hives, itching, fever, swelling or trouble breathing.
The FDA granted this application Priority Review and Breakthrough Therapydesignations. Firdapse also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Firdapse to Catalyst Pharmaceuticals, Inc.
///////////Priority Review, Breakthrough Therapy, Firdapse, Orphan Drug designation, fda 2018, amifampridine
FDA approves new treatment for patients with acute myeloid leukemia
|
||
November 21, 2018
Release
The U.S. Food and Drug Administration today approved Daurismo (glasdegib) tablets to be used in combination with low-dose cytarabine (LDAC), a type of chemotherapy, for the treatment of newly-diagnosed acute myeloid leukemia (AML) in adults who are 75 years of age or older or who have other chronic health conditions or diseases (comorbidities) that may preclude the use of intensive chemotherapy.
“Intensive chemotherapy is usually used to control AML, but many adults with AML are unable to have intensive chemotherapy because of its toxicities. Today’s approval gives health care providers another tool to use in the treatment of AML patients with various, unique needs. Clinical trials showed that overall survival was improved using Daurismo in combination with LDAC compared to LDAC alone for patients who would not tolerate intensive chemotherapy,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research.
AML is a rapidly progressing cancer that forms in the bone marrow and results in an increased number of abnormal white blood cells in the bloodstream and bone marrow. The National Cancer Institute at the National Institutes of Health estimates that in 2018, approximately 19,520 people will be diagnosed with AML and approximately 10,670 patients with AML will die of the disease. Almost half of the adults diagnosed with AML are not treated with intensive chemotherapy because of comorbidities and chemotherapy related toxicities.
The efficacy of Daurismo was studied in a randomized clinical trial in which 111 adult patients with newly diagnosed AML were treated with either Daurismo in combination with LDAC or LDAC alone. The trial measured overall survival (OS) from the date of randomization to death from any cause. Results demonstrated a significant improvement in OS in patients treated with Daurismo. The median OS was 8.3 months for patients treated with Daurismo plus LDAC compared with 4.3 months for patients treated with LDAC only.
Common side effects reported by patients receiving Daurismo in clinical trials include low red blood cell count (anemia), tiredness (fatigue), bleeding (hemorrhage), fever with low white blood cell count (febrile neutropenia), muscle pain, nausea, swelling of the arms or legs (edema), low platelet counts (thrombocytopenia), shortness of breath (dyspnea), decreased appetite, distorted taste (dysgeusia), pain or sores in the mouth or throat (mucositis), constipation and rash.
The prescribing information for Daurismo includes a Boxed Warning to advise health care professionals and patients about the risk of embryo-fetal death or severe birth defects. Daurismo should not be used during pregnancy or while breastfeeding. Pregnancy testing should be conducted in females of reproductive age prior to initiation of Daurismo treatment and effective contraception should be used during treatment and for at least 30 days after the last dose. The Boxed Warning also advises male patients of the potential risk of drug exposure through semen and to use condoms with a pregnant partner or a female partner that could become pregnant both during treatment and for at least 30 days after the last dose. Daurismo must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks. Patients should also be advised not to donate blood or blood products during treatment. Health care providers should also monitor patients for changes in the electrical activity of the heart, called QT prolongation.
The FDA granted this application Priority Review designation. Daurismo also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Daurismo to Pfizer.
//////////////Daurismo, glasdegib, fda 2018, Priority Review, Orphan Drug
FDA approves first treatment Gamifant (emapalumab) specifically for patients with rare and life-threatening type of immune disease
FDA approves first treatment Gamifant (emapalumab) specifically for patients with rare and life-threatening type of immune disease
The U.S. Food and Drug Administration today approved Gamifant (emapalumab) for the treatment of pediatric (newborn and above) and adult patients with primary hemophagocytic lymphohistiocytosis (HLH) who have refractory, recurrent or progressive disease or intolerance with conventional HLH therapy. This FDA approval is the first for a drug specifically for HLH.
“Primary HLH is a rare and life-threatening condition typically affecting children and this approval fills an unmet medical need for these patients,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “We are committed to continuing to expedite the development and review of therapies that offer meaningful treatment options for …
November 20, 2018
Release
The U.S. Food and Drug Administration today approved Gamifant (emapalumab-lzsg) for the treatment of pediatric (newborn and above) and adult patients with primary hemophagocytic lymphohistiocytosis (HLH) who have refractory, recurrent or progressive disease or intolerance with conventional HLH therapy. This FDA approval is the first for a drug specifically for HLH.
“Primary HLH is a rare and life-threatening condition typically affecting children and this approval fills an unmet medical need for these patients,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “We are committed to continuing to expedite the development and review of therapies that offer meaningful treatment options for patients with rare conditions.”
HLH is a condition in which the body’s immune cells do not work properly. The cells become overactive releasing molecules, which leads to inflammation. The immune cells start to damage the body’s own organs, including the liver, brain and bone marrow. It can be inherited, which is known as primary or “familial” HLH. It can also have non-inherited causes. People with primary HLH usually develop symptoms within the first months or years of life. Symptoms may include fever, enlarged liver or spleen and decreased number of blood cells.
The efficacy of Gamifant was studied in a clinical trial of 27 pediatric patients with suspected or confirmed primary HLH with either refractory, recurrent or progressive disease during conventional HLH therapy or who were intolerant of conventional HLH therapy. The median age of the patients in the trial was 1 year old. The study showed that 63 percent of patients experienced a response and 70 percent were able to proceed to stem cell transplant.
Common side effects reported by patients receiving Gamifant in clinical trials included infections, hypertension, infusion-related reactions, low potassium and fever. Patients receiving Gamifant should not receive any live vaccines and should be tested for latent tuberculosis. Patients should be closely monitored and treated promptly for infections while receiving Gamifant.
The FDA granted this application Priority Review and Breakthrough Therapydesignation. Gamifant also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Gamifant to Novimmune SA.
////////////Gamifant, emapalumab, FDA 2018
FDA approves new drug Aemcolo (rifamycin), to treat travelers’ diarrhea
November 16, 2018
Release
The U.S. Food and Drug Administration today approved Aemcolo (rifamycin), an antibacterial drug indicated for the treatment of adult patients with travelers’ diarrhea caused by noninvasive strains of Escherichia coli (E. coli), not complicated by fever or blood in the stool.
“Travelers’ diarrhea affects millions of people each year and having treatment options for this condition can help reduce symptoms of the condition,” said Edward Cox, M.D., M.P.H., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Travelers’ diarrhea is the most common travel-related illness, affecting an estimated 10 to 40 percent of travelers worldwide each year. Travelers’ diarrhea is defined by having three or more unformed stools in 24 hours, in a person who is traveling. It is caused by a variety of pathogens, but most commonly bacteria found in food and water. The highest-risk destinations are in most of Asia as well as the Middle East, Africa, Mexico, and Central and South America.
The efficacy of Aemcolo was demonstrated in a randomized, placebo-controlled clinical trial in 264 adults with travelers’ diarrhea in Guatemala and Mexico. It showed that Aemcolo significantly reduced symptoms of travelers’ diarrhea compared to the placebo.
The safety of Aemcolo, taken orally over three or four days, was evaluated in 619 adults with travelers’ diarrhea in two controlled clinical trials. The most common adverse reactions with Aemcolo were headache and constipation.
Aemcolo was not shown to be effective in patients with diarrhea complicated by fever and/or bloody stool or diarrhea due to pathogens other than noninvasive strains of E. coli and is not recommended for use in such patients. Aemcolo should not be used in patients with a known hypersensitivity to rifamycin, any of the other rifamycin class antimicrobial agents (e.g. rifaximin), or any of the components in Aemcolo.
The FDA granted Aemcolo a Qualified Infectious Disease Product (QIDP)designation. QIDP designation is given to antibacterial and antifungal drug products that treat serious or life-threatening infections under the Generating Antibiotic Incentives Now (GAIN) title of the FDA Safety and Innovation Act. As part of QIDP designation, the Aemcolo marketing application was granted Priority Review under which the FDA’s goal is to take action on an application within an expedited time frame.
The FDA granted approval of Aemcolo to Cosmo Technologies, Ltd.
