PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Mechanism of ActionType 4 cyclic nucleotide phosphodiesterase inhibitors
RegisteredAtopic dermatitis
27 Sep 2021Registered for Atopic dermatitis (In adolescents, In children, In adults) in Japan (Topical)
11 Nov 2020Otsuka Pharmaceutical completes a phase III trial in Atopic dermatitis (In children, In adolescents, In adults) in Japan (Topical) (NCT03961529)
28 Sep 2020Preregistration for Atopic dermatitis in Japan (In children, In adolescents, In adults) (Topical)
Difamilast is under investigation in clinical trial NCT01702181 (A Safety Study to Evaluate the Use and Effectiveness of a Topical Ointment to Treat Adults With Atopic Dermatitis).
Synthesis of Oxazole Compound (Type A Crystal)
Compound (5) (white powder) was prepared in accordance with the method disclosed in Example 352 of PTL 1 (WO2007/058338).
Synthesis of difamilast commenced with the monobenzylated protocatechuic acid ethyl ester 15.1. Phenol 15.1 was first converted into the corresponding isopropyl ether, which was subsequently debenzylated under palladium-catalyzed hydrogenation conditions to generate the phenolic intermediate 15.3. Difluoromethylation of 15.3 was accomplished by introducing sodium chlorodifluoroacetate 15.4 in the presence of potassium carbonate at an elevated temperature. The decarboxylative C− O bond-forming reaction presumably proceeded via a difluorocarbene species. The difluoromethylated product was treated with acid followed by ester hydrolysis under a basic medium to furnish benzoic acid derivative 15.5. Benzoic acid 15.5 was subsequently transformed into benzamide 15.6 via a benzoyl imidazole intermediate. Condensation of benzamide 15.6 with 1-acetoxy-3-chloroacetone 15.7 produced an oxazole derivative, which was subsequently saponified and recrystallized from 50% aqueous MeOH to generate alcohol 15.8.
First, an activation−displacement process transformed alcohol 15.8 into bromide 15.9 via a mesylate intermediate. Alkyl bromide 15.9 was then treated with potassium phthalimide to incorporate the nitrogen center via an SN2-type displacement. Methylamine-mediated phthalimide deprotection and subsequent salt formation produced amine 15.11 as a hydrochloride salt in 69% yield over 3 steps. Finally, hydrochloride salt 15.11 was treated with aqueous sodium bicarbonate to generate a free amine, which was subjected to amide bond formation with 2-ethoxybenzoic acid 15.12 to deliver difamilast after recrystallization from aqueous EtOH.
Patent Documents 1 and 2 report an oxazole compound having a specific inhibitory action on phosphodiesterase 4 (PDE4) and a method for producing the same. PDE4 is the predominant PDE in inflammatory cells, inhibition of PDE4 increases intracellular cAMP concentration, and the increase in this concentration downregulates the inflammatory response through regulation of the expression of TNF-α, IL-23, and other inflammatory cytokines. .. Elevated cAMP levels also increase anti-inflammatory cytokines such as IL-10. Therefore, it is considered that the oxazole compound is suitable for use as an anti-inflammatory agent. For example, it may be useful for controlling skin eczema and dermatitis, including atopic dermatitis. Patent Document 3 describes an ointment that stably contains an oxazole compound having a specific inhibitory effect on PDE4 and can be efficiently absorbed into the skin. The contents of Patent Documents 1 to 3 are incorporated in the present specification by reference.
[Synthesis of Oxazole Compound (Type A Crystal)]
Compound (5) (white powder) was prepared by the method described in Example 352 of Patent Document 1 (International Publication No. 2007/088383).
[Preparation of B-type crystal 2]
Using the obtained B-type crystal as a seed crystal, it was examined to further prepare a B-type crystal. Specifically,
B-type crystals were prepared as follows according to the method described in Patent Document 3 (International Publication No. 2017/115780).
[0072]
[Chem. 6]
[0073]
Compound (1) 20.00 g (66.8 mmol) and 17.28 g (134 mmol) of diisopropylethylamine were added to 300 mL of ethyl acetate to cool the mixture, and 11.48 g (100 mmol) of methanesulfonyl chloride was introduced into the compound (1) at 10 to 30 ° C. Stir for hours. Subsequently, 17.41 g (200 mmol) of lithium bromide was added, and the mixture was stirred at 20 to 35 ° C. for 1 hour. 100 mL of water was added to the reaction solution to separate the layers, and the organic layer was concentrated under reduced pressure. 300 mL of ethyl acetate was added to the concentrated residue to dissolve it, and the mixture was concentrated again under reduced pressure. 200 mL of N, N-dimethylformamide and 17.33 g (93.6 mmol) of phthalimide potassium were added to the concentrated residue, and the mixture was reacted at 75 to 85 ° C. for 1 hour. 200 mL of water was added to the reaction solution to precipitate crystals, and the precipitated crystals were collected by filtration and dried at 80 ° C. to obtain 27.20 g (yield 95.01%) of compound (3).
[0074]
[Chem. 7]
[0075]
Compound (3) 20.00 g (46.7 mmol), 40 mL of a 40% aqueous methylamine solution, 40 mL of methanol, and 100 mL of water were mixed and reacted under reflux for 30 minutes. 200 mL of cyclopentyl methyl ether (CPME) and 20 mL of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the temperature was adjusted to 65 to 75 ° C. to separate the liquids. A mixed solution of 100 mL of water and 20.00 g of sodium chloride was added to the organic layer, and the temperature was adjusted again to 65 to 75 ° C. to separate the liquids. 5 mL of concentrated hydrochloric acid was added to the organic layer to precipitate crystals. Precipitated crystals were collected by filtration to obtain 27.58 g of wet crystals of compound (4).
[0076]
Wet crystals (46.7 mmol) of compound (4) were mixed with 120 mL of ethyl acetate and 7.1 mL (51.4 mmol) of triethylamine, and the mixture was stirred at 20 to 30 ° C. for 1 hour. To the reaction solution, 10.09 g (60.7 mmol) of 2-ethoxybenzoic acid and 11.63 g (60.7 mmol) of 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (WSC) were added, and 20 to 30 were added. The reaction was carried out at ° C. for 1 hour. 60 mL of water and 6 mL of concentrated hydrochloric acid were added to the reaction solution, and the temperature was adjusted to 40 to 50 ° C. to separate the solutions. 60 mL of water and 6 mL of a 25%
aqueous sodium hydroxide solution were added to the organic layer, the temperature was adjusted again to 40 to 50 ° C., the liquid was separated, and the organic layer was concentrated under reduced pressure. 50 mL of ethanol, 20 mL of water, 6 mL of a 25% aqueous sodium hydroxide solution, and 0.6 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. Activated carbon was removed by filtration, washed with 12 mL of ethanol, the filtrate was cooled, and 10 mg of B-type crystals (seed crystals) were added to precipitate crystals. Precipitated crystals were collected by filtration and dried at 60 ° C. to obtain 18.38 g (yield 88.18%) of crystals of compound (5).
Using the compound obtained in Example 347 and 2-bromopropane, white powdery N-[2-(4-difluoromethoxy-3-isopropoxyphenyl)oxazol-4-ylmethyl]-2-ethoxybenzamide was obtained following the procedure of Example 348.
Production Example 1: Production 1 of Compound (3)
Compound (3) was produced in accordance with the following reaction scheme.
[0146]
[Chem. 11]
[0147]
10.00 g (55.5 mmol) of compound (1a) and 9.20 g (66.6 mmol) of potassium carbonate were added to 40 ml of N,N-dimethylformamide and 6 ml of water, and the mixture was stirred until exotherm subsided. 16.92 g (111 mmol) of sodium chlorodifluoroacetate was added thereto, and the mixture was reacted at 95 to 110°C for 3 hours. 80 ml of butyl acetate and 80 ml of water were added to the reaction solution, and the solution was partitioned. 80 ml of water was added again to the organic layer, followed by partitioning. 3 ml of concentrated hydrochloric acid was added to the organic layer, and the mixture was stirred at 60 to 70°C for 30 minutes. 40 ml of water and 10 ml of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the mixture was partitioned. 5.93 g (61.1 mmol) of sulfamic acid and 10 ml of water were added to the organic layer, and 22.08 g (61.0 mmol) of a 25% sodium chlorite aqueous solution was added dropwise thereto at a temperature of 20°C or below. The mixture was reacted at 20°C or below for 15 minutes, and 10 ml of a 25% sodium hydroxide aqueous solution was added dropwise thereto at a temperature of 20°C or below, followed by pouring in 83.95 g (66.6 mmol) of a 10% sodium sulfite aqueous solution. Additionally, 2 ml of concentrated hydrochloric acid was added and the mixture was partitioned, followed by concentration of the organic layer under reduced pressure. 40 ml of methanol, 80 ml of water, and 10 ml of a 25% sodium hydroxide aqueous solution were added to the concentrated residue to dissolve the residue, and 5 ml of concentrated hydrochloric acid was added dropwise thereto to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 11.81 g (yield: 86.4%) of compound (3) as a white powder.
Production Example 2: Production 2 of Compound (3)
Compound (3) was produced in accordance with the following reaction scheme.
[0149]
[Chem. 12]
[0150]
10.00 g (53.2 mmol) of compound (1b), 9.55 g (69.1 mmol) of potassium carbonate, and 8.50 g (69.1 mmol) of isopropyl bromide were added to 40 ml of N,N-dimethylformamide, and the mixture was reacted at 75 to 85°C for 2 hours. 80 ml of butyl acetate and 80 ml of water were added to the reaction solution, and the mixture was partitioned. 5.68 g (58.5 mmol) of sulfamic acid and 10 ml of water were added to the organic layer, and 21.15 g (58.5 mmol) of a 25% sodium chlorite aqueous solution was added dropwise thereto at 20°C or below, followed by reaction for 15 minutes. 10 ml of a 25% sodium hydroxide aqueous solution was added thereto at 20°C or below, and subsequently 80.41 g (63.8 mmol) of a 10% sodium sulfite aqueous solution was poured in. Additionally, 2 ml of concentrated hydrochloric acid was added, and the mixture was partitioned, followed by concentration of the organic layer under reduced pressure. 40 ml of methanol, 80 ml of water, and 10 ml of a 25% sodium hydroxide aqueous solution were added to the concentrated residue, and the residue was dissolved, followed by dropwise addition of 5 ml of concentrated hydrochloric acid to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 12.09 g (yield: 92.4%) of compound (3) as a white powder.
[0151]
Production Example 3: Production of Compound (7)
Compound (7) was produced in accordance with the following reaction scheme.
[0152]
[Chem. 13]
Production Example 4: Production of Compound (11)
Compound (11) was produced in accordance with the following reaction scheme.
[0160]
[Chem. 14]
[0161]
Synthesis of Compound (9)
20.00 g (66.8 mmol) of compound (7) and 17.28 g (134 mmol) of N,N-diisopropylethylamine were added to 300 ml of ethyl acetate, and the mixture was cooled. 11.48 g (100 mmol) of methanesulfonyl chloride was poured in and stirred at 10 to 30°C for 1 hour. 17.41 g (200 mmol) of lithium bromide was added thereto and reacted at 20 to 35°C for 1 hour. 100 ml of water was added to the reaction solution, and the mixture was partitioned, followed by concentration of the organic layer under reduced pressure. 300 ml of ethyl acetate was added to the concentrated residue to dissolve the residue, and the solution was again concentrated under reduced pressure. 200 ml of N,N-dimethylformamide and 17.33 g (93.6 mmol) of potassium phthalimide were added to the concentrated residue and reacted at 75 to 85°C for 1 hour. 200 ml of water was added to the reaction solution to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 25.90 g (yield: 90.5%) of compound (9) as a white powder.
Synthesis of Compound (10)
15.00 g (35.0 mmol) of compound (9) was mixed with 30 ml of a 40% methylamine aqueous solution, 30 ml of methanol, and 75 ml of water, and reacted under reflux for 30 minutes. 150 ml of cyclopentyl methyl ether (CPME) and 15 ml of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the temperature was adjusted to 65 to 75°C, followed by partitioning. A mixture of 150 ml of water and 7.50 g of sodium chloride was added to the organic layer, and the temperature was adjusted to 65 to 75°C again, followed by partitioning. 3.75 ml of concentrated hydrochloric acid was added to the organic layer to precipitate crystals. The precipitated crystals were collected by filtration and dried at 60°C, thereby obtaining 11.95 g (yield: quant.) of compound (10) as a white powder.
Synthesis of Compound (11)
13.30 g (39.7 mmol) of compound (10) was mixed with 3.83 g (37.8 mmol) of triethylamine and 108 ml of ethyl acetate, and stirred at 20 to 30°C for 1 hour. 9.78 g (58.9 mmol) of 2-ethoxybenzoic acid and 11.28 g (58.8 mmol) of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (WSC) were added to the reaction solution, and reacted at 20 to 30°C for 1 hour. 54 ml of water and 5.4 ml of concentrated hydrochloric acid were added to the reaction solution, and the temperature was adjusted to 40 to 50°C, followed by partitioning. 54 ml of water and 5.4 ml of a 25% sodium hydroxide aqueous solution were added to the organic layer, and the temperature was adjusted to 40 to 50°C again. The mixture was partitioned, and the organic layer was concentrated under reduced pressure. 45 ml of ethanol, 18 ml of water, 5.4 ml of a 25% sodium hydroxide aqueous solution, and 0.54 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. The activated carbon was removed by filtration, and the filtrate was washed with 11 ml of ethanol. The filtrate was cooled, and a seed crystal was added thereto to precipitate crystals. The precipitated crystals were collected by filtration and dried at 35°C, thereby obtaining 12.88 g (72.6%) of compound (11) as a white powder.
Type B Crystal Preparation 2
Analysis was conducted to further prepare the type B crystal using the obtained type B crystal as a seed crystal. More specifically, the type B crystal was prepared as follows, in accordance with the method disclosed in PTL 3 (WO2017/115780).
[0072]
[0073]
20.00 g (66.8 mmol) of compound (1) and 17.28 g (134 mmol) of diisopropylethylamine were added to 300 mL of ethyl acetate, and the mixture was cooled. 11.48 g (100 mmol) of methanesulfonyl chloride was poured in and stirred at 10 to 30°C for 1 hour. 17.41 g (200 mmol) of lithium bromide was added thereto, and the mixture was stirred at 20 to 35°C for 1 hour. 100 mL of water was added to the reaction solution, and the mixture was separated, followed by concentration of the organic layer under reduced pressure. 300 mL of ethyl acetate was added to the concentrated residue to dissolve the residue, and the solution was again concentrated under reduced pressure. 200 mL of N,N-dimethylformamide and 17.33 g (93.6 mmol) of potassium phthalimide were added to the concentrated residue, and reacted at 75 to 85°C for 1 hour. 200 mL of water was added to the reaction solution to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 27.20 g (yield: 95.01%) of compound (3).
[0074]
[0075]
20.00 g (46.7 mmol) of compound (3), 40 mL of a 40% methylamine aqueous solution, 40 mL of methanol, and 100 mL of water were mixed and reacted for 30 minutes under reflux. 200 mL of cyclopentyl methyl ether (CPME) and 20 mL of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the temperature was adjusted to 65 to 75°C, followed by separation. A mixture of 100 mL of water and 20.00 g of sodium chloride was added to the organic layer, and the temperature was adjusted to 65 to 75°C again, followed by separation. 5 mL of concentrated hydrochloric acid was added to the organic layer to precipitate crystals. The precipitated crystals were collected by filtration, thereby obtaining 27.58 g of compound (4) as a wet crystal.
[0076]
The wet crystal (46.7 mmol) of compound (4) was mixed with 120 mL of ethyl acetate and 7.1 mL (51.4 mmol) of triethylamine, and stirred at 20 to 30°C for 1 hour. 10.09 g (60.7 mmol) of 2-ethoxybenzoic acid and 11.63 g (60.7 mmol) of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (WSC) were added to the reaction solution, and reacted at 20 to 30°C for 1 hour. 60 mL of water and 6 mL of concentrated hydrochloric acid were added to the reaction solution, and the temperature was adjusted to 40 to 50°C, followed by separation. 60 mL of water and 6 mL of a 25% sodium hydroxide aqueous solution were added to the organic layer, and the temperature was adjusted to 40 to 50°C again. The mixture was separated, and the organic layer was concentrated under reduced pressure. 50 mL of ethanol, 20 mL of water, 6 mL of a 25% sodium hydroxide aqueous solution, and 0.6 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. The activated carbon was removed by filtration, and the filtrate was washed with 12 mL of ethanol. The filtrate was cooled, and 10 mg of the type B crystal (a seed crystal) was added thereto to precipitate crystals. The precipitated crystals were collected by filtration and dried at 60°C, thereby obtaining 18.38 g (88.18%) of compound (5).
PATENT
WO2014034958A1
WO2007058338A2
WO2007058338A9
WO2007058338A3
US9181205B2
US2015239855A1
USRE46792E
US2020078340A1
US2017216260A1
US2019070151A1
US2009221586A1
US8637559B2
US2014100226A1
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Avacopan wasunder investigation in clinical trial NCT02994927 (A Phase 3 Clinical Trial of CCX168 (Avacopan) in Patients With ANCA-Associated Vasculitis).
VFMCRP announces approval for TAVNEOS® (avacopan) for the treatment of ANCA-associated vasculitis in Japan
First orally administered therapy for the treatment of two types of ANCA-associated vasculitis approved in Japan
Partner Kissei to market TAVNEOS® in Japan, with launch expected as soon as possible following National Health Insurance (NHI) price listing
September 27, 2021 02:02 AM Eastern Daylight Time
ST. GALLEN, Switzerland–(BUSINESS WIRE)–Vifor Fresenius Medical Care Renal Pharma (VFMCRP) today announced that Japan’s Ministry of Health and Labor Welfare (MHLW) has granted its partner, Kissei Pharmaceutical Co., Ltd., marketing authorization approval for TAVNEOS® for the treatment of patients with granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA), the two main types of ANCA-associated vasculitis, a rare and severe autoimmune renal disease with high unmet medical need.
“We are delighted that TAVNEOS® has been approved in Japan, the first market worldwide, and congratulate our partner Kissei for this significant milestone”
“We are delighted that TAVNEOS® has been approved in Japan, the first market worldwide, and congratulate our partner Kissei for this significant milestone,” said Abbas Hussain, CEO of Vifor Pharma Group. “ANCA-associated vasculitis is officially designated an intractable disease in Japan, indicating a rare disease without any effective treatment but for which long-term treatment is required. There is significant unmet medical need of over 10,000 patients in Japan, and we believe in the potential of TAVNEOS® for treating it. We are confident that Kissei will fully focus on bringing this breakthrough treatment to this patient population, helping them lead better, healthier lives.”
The approval is based on the marketing authorization application filing by Kissei which was supported by positive clinical data from the pivotal phase-III trial ADVOCATE in a total of 331 patients with MPA and GPA in 18 countries and regions, including Japan. TAVNEOS® demonstrated superiority over standard of care at week 52 based on Birmingham Vasculitis Activity Score (BVAS).
VFMCRP holds the rights to commercialize TAVNEOS® outside the U.S.. In June 2017, VFMCRP granted Kissei the exclusive right to develop and commercialize TAVNEOS® in Japan. Kissei expects to begin to market TAVNEOS® as soon as possible following NHI price listing. Outside Japan, TAVNEOS is currently in regulatory review with various agencies, including the U.S. Food and Drug Administration and the European Medicines Agency.
About Vifor Pharma Group
Vifor Pharma Group is a global pharmaceuticals company. It aims to become the global leader in iron deficiency, nephrology and cardio-renal therapies. The company is a partner of choice for pharmaceuticals and innovative patient-focused solutions. Vifor Pharma Group strives to help patients around the world with severe and chronic diseases lead better, healthier lives. The company develops, manufactures and markets pharmaceutical products for precision patient care. Vifor Pharma Group holds a leading position in all its core business activities and consists of the following companies: Vifor Pharma and Vifor Fresenius Medical Care Renal Pharma (a joint company with Fresenius Medical Care). Vifor Pharma Group is headquartered in Switzerland, and listed on the Swiss Stock Exchange (SIX Swiss Exchange, VIFN, ISIN: CH0364749348).
Kissei Pharmaceutical Co., Ltd. is a Japanese pharmaceutical company with approximately 70 years of history. Based on its management philosophy, “contributing to society through high-quality, innovative pharmaceutical products” and “serving society through our employees”, Kissei is concentrating on providing innovative pharmaceuticals to patients worldwide as a strongly R&D-oriented corporation. Kissei is engaged in R&D and licensing activities in the field of nephrology/dialysis, urology, and unmet medical needs in other disease areas. Kissei has an established collaboration with VFMCRP for sucroferric oxyhydroxide which Kissei fully developed in Japan as P-TOL® (known as Velphoro® in Europe/US) for the treatment of hyperphosphatemia. Since the launch in 2015, the market share of P-TOL® has been steadily expanding in Japan. For more information about Kissei Pharmaceutical, please visit www.kissei.co.jp.
About ChemoCentryx Inc.
ChemoCentryx is a biopharmaceutical company developing new medications for inflammatory and autoimmune diseases and cancer. ChemoCentryx targets the chemokine and chemoattractant systems to discover, develop and commercialize orally-administered therapies. Besides ChemoCentryx’s lead drug candidate, avacopan, ChemoCentryx also has early stage drug candidates that target chemoattractant receptors in other inflammatory and autoimmunediseases and in cancer.
About ANCA-associated vasculitis
ANCA-associated vasculitis is a systemic disease in which over-activation of the complement pathway further activates neutrophils, leading to inflammation and destruction of small blood vessels. This results in organ damage and failure, with the kidney as the major target, and is fatal if not treated. Currently, treatment for ANCA-associated vasculitis consists of courses of non-specific immuno-suppressants (cyclophosphamide or rituximab), combined with the administration of daily glucocorticoids (steroids) for prolonged periods of time, which can be associated with significant clinical risk including death from infection.
About TAVNEOS® (avacopan)
Avacopan is an orally-administered small molecule that is a selective inhibitor of the complement C5a receptor C5aR1. By precisely blocking the receptor (the C5aR) for the pro-inflammatory complement system fragment, C5a on destructive inflammatory cells such as blood neutrophils, avacopan arrests the ability of those cells to do damage in response to C5a activation, which is known to be the driver of inflammation. Moreover, avacopan’s selective inhibition of only the C5aR1 leaves the beneficial C5a l pathway through the C5L2 receptor functioning normally.
ChemoCentryx is also developing avacopan for the treatment of patients with C3 Glomerulopathy (C3G) and hidradenitis suppurativa (HS). The U.S. Food and Drug Administration has granted avacopan orphan-drug designation for ANCA-associated vasculitis, C3G and atypical hemolytic uremic syndrome. The European Commission has granted orphan medicinal product designation for avacopan for the treatment of two forms of ANCA vasculitis: microscopic polyangiitis and granulomatosis with polyangiitis (formerly known as Wegener’s granulomatosis), as well as for C3G. In October 2020, European Medicines Agency (EMA) accepted to review the Marketing Authorization Application (MAA) for avacopan for the treatment of patients with ANCA-associated vasculitis (granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA)).
On May 6, 2021 the U.S. Food & Drug Administration’s (FDA’s) Arthritis Advisory Committee narrowly voted in support of avacopan, a C5a receptor inhibitor, for the treatment of adult patients with anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitis. Although the panelists were excited about the possibility of a steroid-sparing therapy, some raised questions about whether results from the single phase 3 trial could adequately inform the risk/benefit assessment.1 The FDA will weigh the panel’s recommendation as it considers possible approval.
Treatment Needs for ANCA-Associated Vasculitis
ANCA-associated vasculitis is a rare, severe and sometimes fatal form of vasculitis characterized by inflammation of small vessels, often including those in the kidney. One factor that distinguishes it from other forms of vasculitis is the dominant role of neutrophils in its pathogenesis. From work in both animal and mouse models, we know activation of the alternative complement pathway plays a role in the disease pathogenesis, triggering attraction and activation of neutrophils in a complex feedback loop.1-3
Morbidity and mortality from ANCA-associated vasculitis has improved in recent decades, partly due to the introduction of new treatment regimens. The FDA approved rituximab for ANCA-associated vasculitis in 2011, and, in 2018, its label was extended to include maintenance therapy. Most patients with newly diagnosed ANCA-associated vasculitis are now started on a tapering dose of glucocorticoids, paired either with cyclophosphamide or rituximab, with a later follow-up maintenance dose of rituximab at around six months.
High doses of glucocorticoids are often used for remission induction, and they may also be employed as part of maintenance therapy, flare management and relapsing disease. This is a concern for practitioners, who hope to reduce the toxicity that results from glucocorticoid use, especially when given at high doses for prolonged periods.
Avacopan is the first drug to be specifically developed for a vasculitis indication. Other vasculitis therapies—such as tocilizumab for giant cell arteritis or rituximab for ANCA-associated vasculitis—were first approved for other diseases. Avacopan is an oral C5a receptor antagonist that selectively blocks the effects of C5a, thus dampening neutrophil attraction and activation. It does not have FDA approval for other indications, but has orphan drug status for ANCA-associated vasculitis (specifically for microscopic polyangiitis and granulomatosis with polyangiitis) and for C3 glomerulopathy, a rare kidney disease.
Arthritis Advisory Panel Meeting
The FDA generally requires evidence from at least two adequate and well-controlled phase 3 trials to establish effectiveness of a drug. However, it exercises regulatory flexibility in certain circumstances, such as for some rare diseases. In this case, it may consider the results of a well-designed single study if the evidence is statistically persuasive and clinically meaningful.4
Study design is a challenge for any manufacturer attempting to develop a product to potentially decrease steroid use because the FDA does not accept steroid sparing as an assessable outcome for clinical trials. For example, in the GiACTA trial, the phase 3 trial used as evidence for approval of tocilizumab for patients with giant cell arteritis, the biotechnology company Genentech wanted to give tocilizumab and demonstrate patients could then be safely taken off glucocorticoids. But the FDA required a more complicated multi-arm design.5
Other issues come up because of the way glucocorticoids have been used historically. Although they have been used for vasculitis since before drug licensing was introduced, glucocorticoids are not themselves licensed for ANCA-associated vasculitis, which brings up certain regulatory barriers in study design. Additionally, the efficacy of glucocorticoids in vasculitis to control disease activity or prevent relapse has never been officially quantified in a placebo-controlled trial.
ADVOCATE Design
For avacopan, ChemoCentryx based its application on a single phase 3 trial and two phase 2 trials.1-3 In pre-meeting documents and during the meeting itself, the company drew comparisons to the RAVE trial, used to establish the non-inferiority of rituximab to standard cyclophosphamide therapy in patients with ANCA-associated vasculitis.6 In this case, a single phase 3 trial (with supporting phase 2 data) was used as evidence for approval of rituximab.
The phase 3 trial of avacopan, ADVOCATE, used a similar, double-blind, double-dummy design.1 ADVOCATE included 331 patients with either new or relapsing ANCA-associated vasculitis. Half the participants received 30 mg of avacopan twice a day orally, as well as a prednisone placebo, out to the study’s end at 12 months. The other half received oral prednisone (tapered to 0 mg at five months) plus an avacopan placebo.
Additionally, patients received immunosuppressive treatment, either cyclophosphamide (35%) or rituximab (65%), at the discretion of the prescribing physician. Patients who had received cyclophosphamide also received follow-up azathioprine at week 15. But after initial treatment, no patients received maintenance rituximab, as would now be common practice.
Prior to enrollment, many participants were already receiving glucocorticoids as part of their treatment, to help get their disease under control. Thus, open-label prednisone treatment continued to be tapered for the early part of the trial in both groups up to the end of week 4. This had to be tapered to 20 mg or less of prednisone daily before beginning the trial, in both treatment groups.
As reported by the investigators, at week 26, the avacopan group was non-inferior to the prednisone group in terms of sustained remission. At the study’s conclusion at week 52, 66% of patients in the avacopan group were in sustained remission, as were 55% of those in the prednisone group. Thus, in terms of remission, avacopan was superior to glucocorticoids at week 52 (P=0.007).
The researchers also provided encouraging secondary endpoints related to a number of other parameters, including reduced glucocorticoid-related toxicities, fewer relapses, better quality of life measures and improvements in kidney functioning (e.g., glomerular filtration rate changes).
David R.W. Jayne, MD, a professor of clinical autoimmunity at the University of Cambridge and director of the Vasculitis and Lupus Service at Addenbrooke’s Hospital, Cambridge, England, was one of the ADVOCATE investigators and says that in the context of previous vasculitis trials, which have only rarely displayed positive effects from interventions, the ADVOCATE results are impressive.
“We’ve never seen quality-of-life benefits or [glomerular filtration rate] recovery benefits in other vasculitis trials, but we saw them consistently in this one,” says Dr. Jayne.
Example 1: Preparation of Free Base Crystalline Form of Compound 1
Crude Compound 1 was prepared essentially as described in WO 2016/053890.
A free base crystalline form of Compound 1 was prepared by dissolving 18 g of crude Compound 1 in 50 mL acetone with heating at 40° C. (a concentration of about ˜0.36 g/mL). The warm solution was passed through a 10 μm polyethylene filter. The solution was then loaded into rotary evaporator at 30° C. bath temperature and 180 rpm rotational speed. The solid collected was dried further in a 45° C. oven for 1 hour. The XRPD data of the crystalline form is shown in Error! Reference source not found., and the table of peaks measured are listed in Table 1, below.
Example 2: Preparing an Amorphous Form of Compound 1
Method 1
Crude Compound 1 was prepared essentially as described in WO 2016/053890.
Crude Compound 1 (15 grams) was dissolved into 40 mL of acetone at 40° C. temperature. The solution was spray dried using a Buchi B290 Spray Dryer, equipped with a peristaltic pump. The spray drying process was completed by using target inlet temperature of 80° C., target spray rate of 5 mL/min, and process gas flow rate of 20.60 CFM. The spray dried powder collected in the sample collection chamber was the amorphous form of Compound 1 as assessed by XRFD, shown in Error! Reference source not found.
Method 2 An amorphous form of Compound 1 was prepared by dissolving 1 g of the free base crystalline form of Compound 1 in 9 mL of acetone without any heating (a concentration of about ˜0.11 g/mL). The solution was passed through a 10 μm polyethylene filter by gravity. The solution was then loaded into rotary evaporator at 45° C. bath temperature and 220 rpm rotational speed. The solid collected was dried further in a 45° C. oven for 30 hour. The XRPD data of the starting material (in crystalline form) and the amorphous form produced from Method 2 are shown in Error! Reference source not found.A & FIG. 3B. The DSC data of the starting material (in crystalline form) and the amorphous form produced from Method 2 are shown in Error! Reference source not found. Experimental details related to DSC data collection are described in Example 3.
A 3-L round bottom flask equipped with a magnetic stirrer was charged with (2R,3S)-2-(4-(cyclopentylamino)phenyl)-1-(2-fluoro-6-methylbenzoyl)-N-(4-methyl-3-(trifluoromethyl)phenyl)piperidine-3-carboxamide (Compound 1, 250 g, 430 mmol) and MeCN (1.84 L, 8 vol). The resulting mixture was stirred and heated to 75° C. (internal temperature) for 30 min to form a clear solution, and filtered through polyethylene frit filter and rinsed with MeCN (230 mL). To this solution at 60° C. was slowly added a pre-filtered solution of benzenesulfonic acid hydrate (77.9 g, 442 mmol (based on monohydrate), 1.03 eq) in MeCN (276 mL, 3 vol) over 10 min and rinsed with MeCN (92 mL) (internal temperature dropped to 55° C.). The resulting solution was cooled to 50° C., seeded with besylate crystals of Compound 1 (˜100 mg) and slowly cooled to 45° C. over 1 h. The resulting mixture was slowly cooled to RT and stirred for 42 h. The solid was collected by filtration, washed with MeCN (230 mL×2), air-dried and then dried in an oven under vacuum at 50° C. overnight (48 h) to afford N-cyclopentyl-4-((2R,3S)-1-(2-fluoro-6-methylbenzoyl)-3-((4-methyl-3-(trifluoromethyl)phenyl)-carbamoyl)piperidin-2-yl)benzenaminium benzenesulfonate as off-white crystals, with a recovery yield of 266.5 g (84%). 1H NMR (400 MHz, DMSO-d 6) (RT) δ 10.44 (s, 1H), 7.90-7.83 (m, 1H), 7.65-6.95 (m, 14H), 6.42-6.34 (m, 1H), 6.05-5.00 (br, 1H), 3.85-3.70 (m, 1H), 3.22-3.00 (m, 3H), 2.38-2.28 (m, 4H), 2.20-1.40 (m, 15H); (65° C.) δ 10.22 (d, J=8.4 Hz, 1H), 7.85 (d, J=8.4 Hz, 1H), 7.68-6.70 (m, 15H), 6.44-6.35 (m, 1H), 3.72-3.65 (m, 1H), 3.25-2.98 (m, 3H), 2.40-2.28 (m, 4H), 2.22-1.40 (m, 15H). MS: (ES) m/z calculated for C 33H 36F 4N 3O 2 [M+H] + 582.3, found 582.2. A plot of the XRPD is shown in FIG. 1, and Table 1, below, summarizes significant peaks observed in the XRPD plot. HPLC (both achiral analytical and chiral): >99%. Elemental Analysis consistent with formula of C 39H 41F 4N 3O 5S, KF: 0.66%.
Example 11[0147] The following are representative compounds prepared and evaluated using methods similar to the examples herein. Characterization data is provided for the compounds below. Biological evaluation is shown in Figure 1 for these compounds and others prepared as described herein.(2R,3S)-2-(4-Cyclopentylaminophenyl)-l-(2-fluoro-6-methylbenzoyl)piperidine-3- carboxylic acid (4-methyl-3-trifluoromethylphenyl)amide
[0097]This example illustrates the preparation of (2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methyl-benzoyl)-N-[4-methyl-3-(trifluoromethyl)phenyl]piperidine-3-carboxamide by the method provided more generally in FIG. 1 (Scheme 1) using the reagents provided below:
[0098]Step 1:
[0099]An oven-dried 12 L, 3-necked flask equipped with a mechanical stirrer, condenser, and thermometer was charged with acrolein diethyl acetal (1127 g, 8.666 mole, 1.05 equiv.) and warmed up to 40° C. A mixture of solid ethyl 3-(4-nitrophenyl)-3-oxo-propanoate (1956 g, 8.253 mole) and (R)-(−)-2-phenylglycinol (>99.5% e.e., 1187 g, 8.666 mole, 1.05 equiv.) was added in portions over 40 min. to maintain a stirrable mixture at an internal temperature of approximately 40° C. After all solids were added, the mixture was stirred at 40° C. for 10 minutes. 4M HCl in dioxane (206.2 mL, 0.825 mole, 10 mol. %) was subsequently added through the condenser within 2 minutes and the internal temperature was increased to 70 OC. The reaction was stirred for 22 h whereupon LC-MS showed consumption of starting materials and enamine intermediate. The heating was turned off and ethanol (6.6 L) was added. The solution was then seeded with 4 g of ethyl (3R,8aR)-5-(4-nitrophenyl)-3-phenyl-3,7,8,8a-tetrahydro-2H-oxazolo[3,2-a]pyridine-6-carboxylate and stirred at room temperature for 18 h. The solid was subsequently filtered off and 0.1 L of ethanol was used to rinse the flask and equipment onto the filter. The isolated solid was then washed three times on the filter with ethanol (250 mL each) and dried under vacuum to generate 1253 g of ethyl (3R,8aR)-5-(4-nitrophenyl)-3-phenyl-3,7,8,8a-tetrahydro-2H-oxazolo[3,2-a]pyridine-6-carboxylate as a bright yellow solid (38% yield, 98.5% HPLC wt/wt purity, 0.15 wt % of EtOH).
[0100]Step 2:
[0101]260 g of ethyl (3R,8aR)-5-(4-nitrophenyl)-3-phenyl-3,7,8,8a-tetrahydro-2H-oxazolo[3,2-a]pyridine-6-carboxylate (0.659 mol), 0.66 L of ethanol, and 56 g of palladium catalyst (10% Pd/C, Degussa type E101 NE/W, 50% wet, 21.5 wt. % of powder, 4.0 mol % Pd) were placed in a 2.2 L Parr bottle and purged with nitrogen. The bottle was mounted on a Parr shaker apparatus and hydrogen was added at a rate to keep the external temperature of the bottle below 30° C. After 4 hours, the consumption of hydrogen slowed down. The bottle was then shaken under 50 psi of hydrogen for 2 hours. 94 mL of glacial acetic acid (1.65 mol, 2.5 equiv.) was subsequently added to the bottle and the bottle was purged three times with hydrogen at 50 psi. The bottle was then shaken under 35-55 psi of hydrogen for 48 hours, keeping the temperature below 30° C. The bottle was removed from the apparatus and 55 mL of 12M HCl aq. was added (0.659 mol, 1 equiv.) followed by 87 mL of cyclopentanone (0.989 mol, 1.5 equiv.). The bottle was purged three times with hydrogen at 50 psi and then shaken under 50 psi of hydrogen for 16-20 hours. The mixture was removed from the apparatus and filtered through a fritted funnel containing celite (80 g) and then washed three times with 0.125 L of ethanol. 54.1 g of anhydrous sodium acetate (0.659 mol, 1 equiv.) was added and the mixture was concentrated in vacuo at 40-55° C. to remove 0.9 L of the volatile components. 2.0 L of acetonitrile was added and 2.0 L of volatile components were removed in vacuo. The crude material was diluted with 1.0 L of acetonitrile and mechanically stirred at r.t. for 30 minutes. The mixture was filtered through Celite (40 g) and the cake was washed with 0.28 L of acetonitrile. The combined filtrates gave a solution of the crude amine acetate (Solution A, e.e. =78%). Solutions A of two independent runs were combined for further processing.
[0102]In a 12-L 3-neck flask equipped with a mechanical stirrer, internal thermometer, and reflux condenser (−)-O,O′-di-p-toluoyl-L-tartaric acid (1.019 kg, 2.64 mol, 2 equiv.) was dissolved in 5.8 L of acetonitrile. The mixture was heated to 60° C. with stirring, followed by a quick addition of 1 L of Solution A. The resultant solution was seeded with 4 g of the crystalline ethyl (2R,3S)-2-[4-(cyclopentylamino)phenyl]piperidine-3-carboxylate (−)-O,O′-di-p-toluoyl-L-tartaric acid salt (1:2) and stirred at 60° C. for 15 minutes. After 15 minutes at 60 OC the seed bed has formed. The remaining amount of Solution A was added over a period of 2.5 hours, maintaining an internal temperature at 60° C. When the addition was complete, the heat source was turned off and the mixture was stirred for 17 hours, reaching a final temperature of 22.5° C. The suspension was filtered and the solids were washed with 0.50 L of acetonitrile to rinse the equipment and transfer all solids onto the filter. The resultant wet solids were washed on the funnel with 3.0 L of acetonitrile and dried in a vacuum oven at 45° C. for 48 hours to provide 1.005 kg of ethyl (2R,3S)-2-[4-(cyclopentylamino)phenyl]piperidine-3-carboxylate (−)-O,O′-di-p-toluoyl-L-tartaric acid salt (1:2) as an off-white solid (70% yield, contains 1 wt. % of acetonitrile). The enantiomeric ratio of the product was 99.4:0.6.
[0103]Step 3:
[0104]In a 5 L 3-necked flask equipped with a mechanical stirrer and an addition funnel, solid anhydrous potassium carbonate (K2CO3, 226 g, 1.64 mol, 4.1 equiv.) was dissolved in H2O (0.82 L) and cooled to ambient temperature. MTBE (0.82 L) was added, followed by solid ethyl (2R,3S)-2-[4-(cyclopentylamino)phenyl]piperidine-3-carboxylate (−)-O,O′-di-p-toluoyl-L-tartaric acid salt (1:2) (436 g, 0.400 mol). The mixture was vigorously stirred at r.t. for 1 hour, then 2-fluoro-6-methylbenzoyl chloride (72.5 g, 0.420 mmol, 1.05 equiv.) in MTBE (0.14 L) was added dropwise over 1 hour. The product started precipitating from the reaction before addition of the acid chloride was completed. The reaction was vigorously stirred at r.t. for 30 minutes and monitored by LC-MS for the disappearance of starting material. The mixture was subsequently transferred to a 5 L evaporation flask using 0.3 L of MTBE to rinse the equipment and remove all solids. The mixture was concentrated in vacuo to remove the MTBE, then 0.3 L of heptane was added and the mixture was evaporated again to leave only the product suspended in aqueous solution. The flask was removed from the rotavap and water (0.82 L) and heptane (0.82 L) were added. The suspension was vigorously stirred for 16 hours using a mechanical stirrer. The contents were then filtered and the solid was washed with water (2×0.42 L) and heptane (0.42 L). The solid was dried in a vacuum oven at 45° C. to provide 172 g of ethyl (2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methyl-benzoyl)piperidine-3-carboxylate as an off-white powder (95% yield).
[0105]Step 4:
[0106]A 0.5 L 3-necked round-bottom flask was dried overnight in an oven at 200° C. and then cooled under a stream of nitrogen. The flask was equipped with a magnetic stir bar, nitrogen inlet, and a thermometer. The flask was charged with 30.2 g of ethyl (2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methyl-benzoyl)piperidine-3-carboxylate (66.7 mmol), 11.5 mL of 4-methyl-5-trifluoromethylaniline (80 mmol, 1.2 equiv.) and 141 mL of dry toluene under an atmosphere of nitrogen. Nitrogen was bubbled through the resultant solution for 10 minutes and then the solution was warmed to 30° C. The oil bath was removed and 100 mL of a 2 M solution of AlMe3 in toluene (Aldrich, 200 mmol, 3 equiv.) was cannulated into the reaction mixture at a rate maintaining the reaction temperature between 35-40° C., a process that took approximately 45 minutes. The temperature of the reaction mixture was then increased to 55° C. over a period of 1 hour and the reaction mixture was stirred at 55° C. for 8 hours, whereupon all of the starting ester was consumed (monitored by LC-MS). The reaction was subsequently cooled overnight to ambient temperature and the solution was then cannulated into a mechanically stirred 1 L flask containing a solution of 67.8 g of sodium potassium tartrate tetrahydrate (240 mmol, 3.6 equiv.) in 237 mL of water, pre-cooled to 10 OC in an ice bath. The addition process took approximately 30 minutes, during which the reaction mixture self-heated to 57° C. The empty reaction flask was subsequently rinsed with 20 mL of dry toluene and the solution was combined with the quench mixture. The mixture was then cooled to r.t. with stirring, 91 mL of ethyl acetate was added, and the mixture was stirred an additional 15 minutes. The mixture was subsequently filtered through a pad of Celite and the filtrate was allowed to separate into two layers. The organic layer was then separated and washed with a solution of 5.7 g of sodium potassium tartrate tetrahydrate (20 mmol) in 120 mL of water and then with two 120 mL portions of water. The wet organic solution was concentrated in vacuo to a weight of ˜150 g and a solvent exchange with ethanol was performed maintaining a total volume of 0.2-0.3 L, until <1 mol. % toluene with respect to ethanol was observed by 1H NMR. The solution was then evaporated at elevated temperature to a weight of 223 g and heated to reflux. Mechanical stirring was initiated and 41 mL of water was added. The resulting solution was seeded with (2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methyl-benzoyl)-N-[4-methyl-3-(trifluoromethyl)phenyl]piperidine-3-carboxamide crystals at 60 OC and then slowly cooled to r.t. over 2 hours. The slurry was subsequently stirred for 18 hours and the solids were filtered off. The solids were then washed with two 30 mL portions of 7:3 ethanol/water and dried in a vacuum oven for 24 hours at 50 OC to afford 31.0 g of (2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methyl-benzoyl)-N-[4-methyl-3-(trifluoromethyl)phenyl]piperidine-3-carboxamide as off-white crystals (80% yield). Analytical data: HPLC purity: 99.59%; >99.8% d.e. and e.e. by HPLC; ICP-OES Pd: <1 ppm; Al: δ ppm; residual toluene by headspace GC-MS: 15 ppm; microash<0.1%; K—F 0.1%. 1H NMR (400 MHz, TFA-d) δ 7.91 (d, J=8.6 Hz, 1H), 7.84 (d, J=8.6 Hz, 1H), 7.58-6.82 (m, 8H), 6.75 (t, J=8.6 Hz, 1H), 4.10-4.00 (m, 1H), 3.60-3.47 (m, 1H), 3.45-3.41 (m, 1H), 3.33-3.25 (m, 1H), 2.44-2.22 (m, 7H), 2.04-1.92 (m, 4H), 1.82-1.69 (m, 7H), MS: (ES) m/z 582 (M+H+).
The present disclosure is directed to, inter alia, methods of treating ANCA-associated vasculitis (AAV) in a human in need thereof, the method comprising administering to the human a therapeutically effective amount of avacopan, having the structure shown below:
Announcement of Marketing Authorization Approval in Japan and Co-promotion Agreement of UPASITA® IV Injection Syringe for the Treatment of Secondary Hyperparathyroidism in Dialysis Patients
SANWA KAGAKU KENKYUSHO Co., Ltd. (Head Office: Nagoya, President and CEO : Shusaku Isono, Suzuken Group, ; “SANWA KAGAKU”) has received Marketing Authorization approval today for UPASITA® IV Injection Syringes (generic name: Upacicalcet Sodium Hydrate; “UPASITA®”) for the treatment of secondary hyperparathyroidism in patients on hemodialysis.
UPASITA® was created by Ajinomoto Pharmaceuticals Co., Ltd. (currently EA Phama Co., Ltd.) and developed by SANWA KAGAKU for the treatment of secondary hyperparathyroidism under a licensing agreement with EA Pharma. UPASITA® acts on calcium sensing receptor in the parathyroid and suppresses excessive secretions of parathyroid hormones (PTH). UPASITA® is administered by intravenous injection to dialysis patients through dialysis circuit by physicians or medical staffs upon completion of dialysis and such administration is expected to reduce the burden of patients with many oral medications whose drinking water volume is severely restricted.
Regarding provision of medical and drug information, SANWA KAGAKU entered into a co-promotion agreement in Japan with Kissei Pharmaceutical Co., Ltd. (Head Office: Matsumoto, Nagano; Chairman and CEO: Mutsuo Kanzawa ; “Kissei”). SANWA KAGAKU will handle the production, marketing, and distribution of the Product while SANWA KAGAKU and Kissei collaboratively promote it to medical institutions in the field in accordance with the agreement. Through the co-promotion activity in the field, SANWA KAGAKU and Kissei will contribute to the treatment of dialysis patients suffering from secondary hyperparathyroidism.
《Reference》
About secondary hyperparathyroidism (SHPT) SHTP is one of complications that occur as chronic kidney disease (chronic kidney failure) progresses and is a pathological condition where excessive PTH is secreted by the parathyroid gland. It has been reported that excessive secretion of parathyroid hormone promotes efflux of phosphorus and calcium from the bone into the blood, thereby increasing the risk of developing bone fractures and arteriosclerosis due to calcification of the cardiovascular system and affecting the vital prognosis.
Product Summary of UPASITA® IV Injection Syringe for Dialysis Brand name: UPASITA® IV Injection Syringe for Dialysis 25μg UPASITA® IV Injection Syringe for Dialysis 50μg UPASITA® IV Injection Syringe for Dialysis 100μg UPASITA® IV Injection Syringe for Dialysis 150μg UPASITA® IV Injection Syringe for Dialysis 200μg UPASITA® IV Injection Syringe for Dialysis 250μg UPASITA® IV Injection Syringe for Dialysis 300μg
Generic Name (JAN): Upacicalcet Sodium Hydrate
Date of Marketing Approval: June 23, 2021
Indications: Secondary hyperparathyroidism in patients on hemodialysis
Dosage and Administration: In adults, UPASITA® is usually administered into venous line of the dialysis circuit at the end of dialysis session during rinse back at a dose of 25 μg sodium upacicalcet 3 times a week as a starting dose. The starting dose can be 50 μg depending on the concentration of serum calcium. Thereafter, the dose may be adjusted in a range from 25 to 300 μg while parathyroid hormone (PTH) and serum calcium level should be carefully monitored in patients.
SYN
WO 2020204117
PATENT
WO 2011108724
WO 2011108690
JP 2013063971
WO 2016194881
JP 6510136
PATENT
WO 2016194881
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016194881&tab=FULLTEXT(Example 1) Synthesis of (2S) -2-amino-3-{[(5-chloro-2-hydroxy-3-sulfophenyl) carbamoyl] amino} propanoic acid (Compound 1 ) [Chemical formula 14] CDI 150. 2 g (926.6Mmol, 1.1 eq. vs Boc-DAP-O t Bu) to and stirred at 5 ° C. acetone was added 750mL (3.0L / kg). 250 g (842.6 mmol) of Boc-DAP-OtBu was added in two portions, and the mixture was washed with 125 mL (0.5 L / kg) of acetone. After stirring for 30 minutes, completion of the IC (imidazolylcarbonylation) reaction was confirmed by HPLC. 282.6 g (1263.8 mmol, 1.5 eq.) Of ACHB was added in 3 portions, and the mixture was washed with 125 mL (0.5 L / kg) of acetone. After raising the temperature to 30 ° C. and stirring for 18 hours, the completion of the urea conversion reaction was confirmed by HPLC. After cooling to 5 ° C., 124.5 mL (1432.4 mmol, 1.7 eq.) Of concentrated hydrochloric acid was added, and the mixture was stirred for 1 hour. The precipitated unwanted material was filtered and washed with 1000 mL (4.0 L / kg) of acetone. The filtrate was concentrated to 1018 g (4.1 kg / kg), the temperature was raised to 50 ° C., and 625.0 mL (7187 mmol, 8.5 eq.) Of concentrated hydrochloric acid was added dropwise. After stirring for 30 minutes and confirming the completion of deprotection by HPLC, 750 mL of water was added (3.0 L / kg). This liquid was concentrated under reduced pressure to 1730 g (6.9 kg / kg) to precipitate a solid. After stirring at 20 ° C. for 14 hours, vacuum filtration was performed. The filtered solid was washed with 500 mL (2.0 L / kg) of acetone and then dried under reduced pressure at 60 ° C. for 6 hours to obtain 201.4 g of the target product (64.5%). 1H-NMR (400MHz, DMSO-d6): δ 8.3 (s, 1H), 8.2 (bs, 3H), 8.1 (d, 1H, J = 2.6Hz), 7.3 (t, 1H, J = 6.0Hz), 7.0 (d, 1H, J = 2.6Hz), 4.0-4.1 (m, 1H), 3.6-3.7 (m, 1H), 3.4-3.5 (m, 1H)[0026](Example 2) Synthesis of (2S) -2-amino-3-{[(3-sulfophenyl) carbamoyl] amino} propanoic acid (Compound 2 ) [Chemical formula 15] CDI 120.2 g (741.2 mmol, 1. 600 mL (3.0 L / kg) of acetone was added to 1 eq. Vs Boc-DAP-OtBu), and the mixture was stirred at 5 ° C. 200 g (673.9 mmol) of Boc-DAP-OtBu was added in two portions, and the mixture was washed with 100 mL (0.5 L / kg) of acetone. After stirring for 30 minutes, the completion of the IC reaction was confirmed by HPLC. 175.0 g (1010.8 mmol, 1.5 eq.) Of ABS was added in 3 portions and washed with 100 mL (0.5 L / kg) of acetone. After raising the temperature to 30 ° C. and stirring for 18 hours, the completion of the urea conversion reaction was confirmed by HPLC. After cooling to 5 ° C., 99.6 mL (1145.4 mmol, 1.7 eq.) Of concentrated hydrochloric acid was added, and the mixture was stirred for 1 hour. The precipitated unwanted material was filtered and washed with 1400 mL (7.0 L / kg) of acetone. The filtrate was concentrated to 800.1 g (4.0 kg / kg), heated to 50 ° C., and then 500.0 mL (5750.0 mmol, 8.5 eq.) Of concentrated hydrochloric acid was added dropwise. After stirring for 30 minutes and confirming the completion of deprotection by HPLC, 600 mL of water was added (3.0 L / kg). This liquid was concentrated under reduced pressure to 1653.7 g to precipitate a solid. After aging at 20 ° C. for 15 hours, vacuum filtration was performed. The filtered solid was washed with 400 mL (2.0 L / kg) of acetone and then dried under reduced pressure at room temperature for 6 hours to obtain 140.3 g of the desired product (net 132.2 g, 64.7%). 1H-NMR (400MHz, DMSO-d6): δ 8.8 (s, 1H), 8.2 (bs, 3H), 7.7 (s, 1H), 7.3-7.4 (m, 1H), 7.1-7.2 (m, 2H) , 6.3-6.4 (bs, 1H), 4.0-4.1 (bs, 1H), 3.6-3.7 (bs, 1H), 3.5-3.6 (bs, 1H)[0027](Example 3) Synthesis of (2S) -2-amino-3-{[(3-chloro-2-methyl-5-sulfophenyl) carbamoyl] amino} propanoic acid (Compound 3 ) [Chemical formula 16] CDI 14. To 4 g (88.8 mmol, 1.05 eq. Vs Boc-DAP-OtBu), 75 mL (3.0 L / kg vs DAP-OtBu) of acetone was added and stirred at 5 ° C. After adding 25 g (84.3 mmol) of Boc-DAP-OtBu in two portions and stirring for 30 minutes, the completion of the IC reaction was confirmed by HPLC. 26.1 g (118.0 mmol, 1.4 eq.) Of ACTS was added in 3 portions and washed with 25 mL (1.0 L / kg) of acetone. After the temperature was raised to 30 ° C., the mixture was stirred overnight, and the completion of the urea conversion reaction was confirmed by HPLC. After concentrating under reduced pressure at 10 kPa and 40 ° C. until the solvent was completely removed, 37.5 mL (1.5 L / kg) of water and 22.8 mL (257.6 mmol) of concentrated hydrochloric acid were added to perform deprotection for 2 hours. After confirming the completion of the reaction by HPLC, the mixture was cooled to 5 ° C., 60 mL (2.4 L / kg) of MeCN was added, and the mixture was stirred overnight. Further, when 120 mL (4.8 L / kg) of MeCN was added, stratification occurred, so 10 mL (0.4 L / kg) of water and 2.5 mL (0.1 L / kg) of MeCN were added. The precipitated solid was filtered under reduced pressure, washed with 60 mL of MeCN / water (1/2), and then dried under reduced pressure at 60 ° C. for 14 hours to obtain 20.1 g of the desired product as a white solid (net18.3 g, yield 61). 0.8%). 1H-NMR (400MHz, DMSO-d6): δ 14.70-13.30 (bs, 1H), 8.27 (bs, 3H), 8.15 (s, 1H), 7.98 (d, 1H, J = 1.6Hz), 7.27 (d , 1H, J = 1.6Hz), 6.82 (t, 1H, J = 6.0Hz), 4.04 (bs, 1H), 3.70-3.60 (m, 1H), 3.60-3.50 (m, 1H), 2.22 (s, 3H)[0028](Example 4) Synthesis of compound 3 using phenylchloroformate as a carbonyl group-introducing reagent (Step 1) [Chemical formula 17] MeCN 375 mL (7.5 L / kg vs ACTS), Py for 50 g (225.6 mmol) of ACTS. 38.1 mL (473.7 mmol, 2.1 eq.) Was added and stirred at 25 ° C. 29.9 mL (236.8 mmol, 1.05 eq.) Of ClCO 2 Ph (phenyl chloroformate) was added dropwise, and after stirring for 30 minutes, completion of the CM (carbamate) reaction was confirmed by HPLC. 68.9 g (232.4 mmol) of Boc-DAP-OtBu was added, 97.5 mL (699.3 mmol, 3.1 eq.) Of TEA was added dropwise, and the mixture was stirred at 25 ° C. for 3 hours. The completion of the urea conversion reaction was confirmed by HPLC. Here, 103.5 g of the total amount of 517.43 g was used to move to the next step (down to ACTS 10 g scale). 30 mL of water was added and concentrated to 77.0 g at 40 ° C. and 5 kPa. After 100 mL (10 L / kg) of AcOEt was added and the liquid separation operation was performed, 30 mL of water was added to the organic layer and the liquid separation operation was performed again. The organic layer was concentrated to 47.6 g at 40 ° C. and 10 kPa, and then 15 mL (1.5 L / kg) of AcOEt and 100 mL (10 L / kg) of THF were added. Again, it was concentrated to 50.7 g and THF was added up to 146 g. When it was concentrated again to 35.5 g and added to AcOEt 30 mL (3 L / kg) and THF 100 mL (10 L / kg), a solid was precipitated. It was cooled to 5 ° C. and aged overnight. The precipitated solid was filtered under reduced pressure, washed with 20 mL (2.0 L / kg) of THF, and then dried under reduced pressure at 40 ° C. for 3 hours overnight at 30 ° C. to obtain 24.9 g of the desired product as a white solid (net). 23.0 g, 83.6%). 1 H-NMR (400MHz, DMSO-d6): δ 8.86 (bs, 1H), 8.09 (s, 1H), 7.88 (s, 1H), 7.25 (d, 1H, J = 1.6Hz), 7.14 (d, 1H, J = 7.6Hz), 6.60 (t, 1H, J = 5.6Hz), 4.00-3.90 (m, 1H), 3.60-3.50 (m, 1H), 3.30-3.20 (m, 1H), 3.15-3.05 (m, 6H), 2.19 (s, 3H), 1.50-1.30 (m, 18H), 1.20-1.10 (m, 9H)
(Step 2) [Chemical
formula 18] Compound 4 21.64 g (net. 20.0 g, 68 mL of water (3.4 L / kg vs. compound 4) vs. 32.8 mmol) ) Was added, the mixture was stirred at 50 ° C., and 12 mL (135.6 mmol, 4.1 eq.) Of concentrated hydrochloric acid was added dropwise. After stirring for 1 hour, the temperature was raised to 70 ° C. to dissolve the precipitated solid. After confirming the completion of the reaction by HPLC, the mixture was cooled to 50 ° C. and aged for 1 hour, and then cooled to 5 ° C. over 4 hours. The precipitated solid was filtered under reduced pressure, washed with 40 mL (2.0 L / kg) of MeCN / water (2/1), and then dried under reduced pressure at 60 ° C. for 3 hours to obtain 11.2 g of the desired product as a white solid (11.2 g). net 10.5 g, 91.1%).[0029](Example 5) [Chemical formula 19] MeCN 10.0 mL (10.0 L / kg vs ACSS), Py 0.75 mL (9.25 mmol, 2.05 eq.) For 1.00 g (4.51 mmol) of ACTS. , And stirred at 8 ° C. After dropping 0.59 mL (4.74 mmol, 1.05 eq.) Of ClCO 2 Ph, raising the temperature to room temperature and stirring for 1 hour, completion of the CM conversion reaction was confirmed by HPLC. 1.33 g (4.51 mmol, 1.0 eq.) Of Boc-DAP-OtBu was added, 1.92 mL (13.76 mmol, 3.05 eq.) Of TEA was added dropwise, and the mixture was stirred at 40 ° C. for 1 hour. After confirming the completion of the urea conversion reaction by HPLC, the mixture was concentrated until the solvent was completely removed. 1.0 mL of water and 2.0 mL of concentrated hydrochloric acid (22.6 mmol, 5.0 eq.) Were added, and the mixture was stirred at 50 ° C. for 4 hours. After confirming the completion of deprotection by HPLC, MeCN 7.5 mL (7.5 L / kg), 1 M HCl aq. After adding 4.5 mL, the mixture was stirred at 5 ° C. overnight. The precipitated solid was filtered under reduced pressure, washed with 3.0 mL (3.0 L / kg) of MeCN, and then dried at 60 ° C. overnight to obtain 1.28 g of the desired product as a white solid (net 1.18 g, 77). .0%).[0030](Example 6) (Step 1) 3-({[(2S) -2-amino-3-methoxy-3-oxopropyl] carbamoyl} amino) -5-chloro-4-methylbenzene-1-sulfonic acid ( Synthesis of Compound 5 ) [Chemical formula 20] To 5 g (22.56 mmol) of ACTS, 37.5 mL (7.5 L / kg vs ACTS) of MeCN and 3.81 mL (47.38 mmol, 2.1 eq.) Of Py were added. The mixture was stirred at 25 ° C. 2.99 mL (23.68 mmol, 1.05 eq.) Of ClCO 2 Ph was added dropwise, and after stirring for 30 minutes, the completion of the CM reaction was confirmed by HPLC. 5.92 g (23.23 mmol, 1.03 eq.) Of Boc-DAP-OMe was added, 9.75 mL (69.93 mmol, 3.1 eq.) Of TEA was added dropwise, and the mixture was stirred at 25 ° C. for 3 hours. 0.4 g (1.58 mmol, 0.07 eq.) Of Boc-DAP-OMe and 0.22 mL (1.58 mmol, 0.07 eq.) Of TEA were added, and the completion of the ureaization reaction was confirmed by HPLC. 7.32 mL (112.8 mmol, 5.0 eq.) Of MsOH was added, the temperature was raised to 50 ° C., and the mixture was stirred for 4 hours. After confirming the completion of deprotection by HPLC, the mixture was cooled to 25 ° C. and 37.5 mL (7.5 L / kg) of MeCN and 7.5 mL (1.5 L / kg) of water were added to precipitate a solid. It was cooled to 5 ° C. and aged for 16 hours. The precipitated solid was filtered under reduced pressure, washed with 20 mL (4.0 L / kg) of water / MeCN (1/2), and then dried under reduced pressure at 40 ° C. for 5 hours to obtain 7.72 g of the target product as a white solid (772 g of the target product). net 7.20 g, 87.3%). 1H-NMR (400MHz, DMSO-d6): δ 8.39 (bs, 3H), 8.16 (d, 1H, J = 1.2Hz), 7.90 (d, 1H, J = 1.6Hz), 7.28 (d, 1H, J = 1.6Hz), 6.78 (t, 1H, J = 5.6Hz), 4.20-4.10 (m, 1H), 3.77 (s, 3H), 3.70-3.60 (m, 1H), 3.55-3.45 (m, 1H) , 2.21 (s, 3H) HRMS (FAB – ): calcd for m / z 364.0369 (MH), found The m / z 364.0395 (MH)
(step 2) [Formula 21]
compound 5 10.64 g (net Non 10.0 g, To 27.34 mmol), 18 mL of water (1.8 L / kg vs. compound 5 ) was added and stirred at 8 ° C. 3.42 mL (57.41 mmol, 2.1 eq.) Of a 48% aqueous sodium hydroxide solution was added dropwise, and the mixture was washed with 1.0 mL (1.0 L / kg) of water and then stirred at 8 ° C. for 15 minutes. After confirming the completion of hydrolysis by HPLC, the temperature was raised to 25 ° C. and 48% HBr aq. The pH was adjusted to 5.8 by adding about 3.55 mL. After confirming the precipitation of the target product by dropping 65 mL (6.5 L / kg) of IPA, the mixture was aged for 1 hour. 81 mL (8.1 L / kg) of IPA was added dropwise and aged at 8 ° C. overnight. The precipitated solid was filtered under reduced pressure, washed with 20 mL (2.0 L / kg) of IPA, and then dried under reduced pressure at 40 ° C. for 4 hours to obtain 10.7 g of the desired product as a white solid (net 9.46 g, 92. 6%). 1 H-NMR (400MHz, DMSO-d6): δ8.76 (s, 1H), 7.91 (d, 1H, J = 1.6Hz), 8.00-7.50 (bs, 2H), 7.24 (d, 1H, J = 1.6Hz), 7.20 (t, 1H, J = 5.6Hz), 3.58-3.54 (m, 1H), 3.47-3.43 (m, 1H), 3.42-3.37 (m, 1H), 2.23 (s, 3H)[0031](Example 7) (Step 1) [Chemical formula 22] For 10.0 g (45.1 mmol) of ACTS, 50 mL (5.0 L / kg vs ACTS) of MeCN, 7.46 mL (92.5 mmol, 2.05 eq. ) Was added, and the mixture was stirred at 8 ° C. 5.98 mL (47.4 mmol, 1.05 eq.) Of ClCO 2 Ph was added dropwise, the temperature was raised to 25 ° C., and the mixture was stirred for 1 hour, and then the completion of the CM reaction was confirmed by HPLC. 100 ml of acetone (10.0 L / kg vs ACTS) was added, the mixture was cooled to 8 ° C., and aged for 1 hour. The precipitated solid was filtered under reduced pressure, washed with 30 mL of acetone (3.0 L / kg vs ACTS), and then dried under reduced pressure at 60 ° C. for 2 hours to obtain 17.8 g of the target product (net 14.4 g as a free form). Quant). 1 H-NMR (400MHz, DMSO-d6): δ 9.76 (bs, 1H), 8.93-8.90 (m, 2H), 8.60-8.50 (m, 1H), 8.10-8.00 (m, 2H), 7.60 (s , 1H), 7.50-7.40 (m, 3H), 7.30-7.20 (m, 3H), 2.30 (s, 3H)
(Step 2) [Chemical 23]
Compound 6 To 5.0 g (11.9 mmol), 50 ml of acetonitrile and 3.53 g (11.9 mmol) of Boc-DAP-OtBu were added, and the mixture was stirred at 8 ° C. 3.5 ml (25 mmol) of triethylamine was added dropwise, and the mixture was stirred overnight at room temperature. The solvent was distilled off under reduced pressure, and 25 ml of ethyl acetate and 5 ml of water were added for extraction. The organic layer was washed with 5 ml of water, the solvent was distilled off, 50 ml of tetrahydrofuran was added, the mixture was cooled to 8 ° C., and aged for 1 hour. The precipitated solid was filtered under reduced pressure, washed with 10 ml of tetrahydrofuran, and dried under reduced pressure at 60 ° C. overnight to obtain 6.3 g of the desired product as a white solid.[0032](Example 8) [Chemical formula 24] For 1.08 g (4.89 mmol) of ACTS, 8.1 mL (7.5 L / kg vs ACTS) of MeCN and 827 μL (10.27 mmol, 2.1 eq.) Of Py were added. In addition, it was stirred at room temperature. ClCO 2 Ph 649 μL (5.14 mmol, 1.05 eq.) Was added dropwise, and the mixture was stirred for 30 minutes, and then the completion of the CM conversion reaction was confirmed by HPLC. 1.48 g (5.04 mmol, 1.03 eq.) Of Cbz-DAP-OMe HCl was added, 2.1 mL (15.17 mmol, 3.1 eq.) Of TEA was added dropwise, and the mixture was stirred at room temperature for about 5 hours. After confirming the completion of the urea conversion reaction by HPLC, the mixture was concentrated until the solvent was completely removed. 15.0 mL of 30% HBr / AcOH was added, and the mixture was stirred at room temperature for 70 minutes, and the completion of deprotection was confirmed by HPLC. After concentration to dryness, 10 mL of water and 4 mL of AcOEt were added to carry out an extraction operation, and then the aqueous layer was stirred at room temperature overnight. The precipitated solid was filtered under reduced pressure, washed with 15 mL of water and 10 mL of AcOEt, and then dried at 40 ° C. for 3 hours to obtain 1.45 g of the desired product as a white solid (58.8%).[0033](Example 9) Synthesis of compound 7 ( methyl ester of compound 1 ) using phenyl chloroformate as a carbonyl group introduction reagent [Chemical formula 25] MeCN 73 mL (14.6 L) with respect to 5.00 g (22.4 mmol) of ACHB. / Kg vs ACHB), Py 3.8 mL (47 mmol, 2.1 eq.), Was added and stirred at 40 ° C. After adding 3.0 mL (24 mmol, 1.05 eq.) Of ClCO 2 Ph and stirring for 30 minutes, the completion of the CM conversion reaction was confirmed by HPLC. 5.87 g (23 mmol, 1.0 eq.) Of Boc-DAP-OMe was added, washed with a small amount of MeCN, 9.7 mL (70 mmol, 3.1 eq.) Of TEA was added dropwise, and the mixture was stirred at 40 ° C. for 3 hours. After confirming the completion of the urea conversion reaction by HPLC, the mixture was cooled to room temperature. 7.3 mL (112 mmol, 5.0 eq.) Of MsOH was added, the temperature was raised to 50 ° C., and the mixture was stirred for 7 hours. Further, 1.5 mL (23 mmol, 1.0 eq.) Of MsOH was added, and the reaction was carried out at 50 ° C. overnight. After confirming the completion of deprotection by HPLC, 90 mL of acetone was added to the reaction solution, and the mixture was cooled to room temperature. The precipitated solid was obtained and dried under reduced pressure at 60 ° C. to obtain the desired product. 1 H-NMR (400MHz, DMSO-d6): δ 7.22 (m, 1H), 7.14 (m, 1H), 4.36 (m, 1H), 3.80 (s, 3H), 3.20-3.40 (m, 2H).[0034](Example 10) Synthesis of compound 5 using 4-chlorophenylchloroformate as a carbonyl group-introducing reagent [Chemical formula 26] For 5.00 g (22.6 mmol) of ACTS, 73 mL (14.6 L / kg vs ACTS) of MeCN, 3.8 mL (47 mmol, 2.1 eq.) Of Py was added and stirred at 40 ° C. After adding 3.25 mL (23.7 mmol, 1.05 eq.) Of 4-chloroformic acid 4-chlorophenylate and stirring at 40 ° C. for 1.5 hours, completion of the CM conversion reaction was confirmed by HPLC. Add 5.92 g (23.2 mol, 1.0 eq.) Of Boc-DAP-OMe, wash with a small amount of MeCN, add 9.7 mL (70 mmol, 3.1 eq.) Of TEA, and stir at 40 ° C. for 2 hours. did. After confirming the completion of the urea conversion reaction by HPLC, the mixture was cooled to room temperature. 7.3 mL (113 mmol, 5.0 eq.) Of MsOH was added, the temperature was raised to 50 ° C., and the mixture was stirred for 3.5 hours. After confirming the completion of deprotection by HPLC, the reaction solution was cooled to room temperature, 7.5 mL of water was added, the mixture was cooled to 8 ° C., and the mixture was stirred overnight. The precipitated solid was filtered, washed with a small amount of MeCN water, and dried at 60 ° C. overnight to obtain 6.94 g of the desired product as a white solid (84.1%).[0035](Example 11) Synthesis of compound 5 using 4-nitrophenyl chloroformate as a carbonyl group-introducing reagent [Chemical formula 27] 73 mL (14.6 L / kg vs. ACTS) of MeCN with respect to 5.00 g (22.6 mmol) of ACTS. , Py 3.8 mL (47 mmol, 2.1 eq.), And stirred at 40 ° C. 4.77 mL (23.7 mmol, 1.05 eq.) Of 4-nitrophenyl chloroformate was added dropwise, and the mixture was stirred at 40 ° C. for 3.5 hours, and then the completion of the CM reaction was confirmed by HPLC. Add 5.92 g (23.2 mmol, 1.0 eq.) Of Boc-DAP-OMe, wash with a small amount of MeCN, add 9.7 mL (70 mmol, 3.1 eq.) Of TEA, and stir at 40 ° C. for 2 hours. did. After confirming the completion of the urea conversion reaction by HPLC, the mixture was cooled to room temperature. 7.3 mL (113 mmol, 5.0 eq.) Of MsOH was added, the temperature was raised to 50 ° C., and the mixture was stirred for 3.5 hours. After confirming the completion of deprotection by HPLC, the reaction solution was cooled to room temperature, 7.5 mL of water was added, the mixture was cooled to 8 ° C., and the mixture was stirred overnight. The precipitated solid was filtered, washed with a small amount of MeCN water, and dried at 60 ° C. overnight to obtain 5.96 g of the desired product as a white solid (72.2%).[0036](Example 12) Synthesis of compound 3 using Boc-DAP-OH [Chemical 28] MeCN 73 mL (14.6 L / kg vs ACTS), Py 3.8 mL, relative to 5.00 g (22.6 mmol) of ACTS. (47 mmol, 2.1 eq.) Was added and stirred at 40 ° C. After adding 3.00 mL (23.8 mmol, 1.05 eq.) Of phenylchloroformate and stirring at 40 ° C. for 0.5 hours, the completion of the CM conversion reaction was confirmed by HPLC (CM conversion reaction product: 4.37 minutes). , ACTS: N.D.). Add 4.75 g (23.2 mmol, 1.0 eq.) Of Boc-DAP-OH, wash with a small amount of MeCN, add 9.7 mL (70 mmol, 3.1 eq.) Of TEA, and stir at 40 ° C. for 2 hours. did. After confirming the completion of the urea-forming reaction by HPLC (urea-forming reaction product: 3.81 minutes, CM-forming reaction product: 0.02 area% vs. urea-forming reaction product), the mixture was cooled to room temperature. By adding 7.3 mL (113 mmol, 5.0 eq.) Of MsOH, raising the temperature to 50 ° C., stirring for 4.5 hours, and further adding 1.5 mL (23 mmol, 1.0 eq.) Of MsOH, stirring for 1 hour. , The formation of the target product was confirmed by HPLC (Compound 3: 2.49 minutes, urea conversion reaction product: 0.50 area vs. compound 3, area of compound 3 with respect to the total area excluding pyridine: 71.0 area).
PATENT
JP 6510136
PATENT
WO 2020204117
Reference Example 1 Synthesis of 3-{[(2S) -2-amino-2-carboxyethyl] carbamoylamino} -5-chloro-4-methylbenzenesulfonate sodium (Compound A1) (Step 1) Synthesis of 3 -({[(2S) -2-amino-3-methoxy-3-oxopropyl] carbamoyl} amino) -5-chloro-4-methylbenzene-1-sulfonic acid 3-amino- 37.5 mL (7.5 L / kg vs ACTS) of acetonitrile and 3.81 mL (47.38 mmol, 2.1 eq.) Of pyridine against 5 g (22.56 mmol) of 5-chloro-4-methylbenzenesulfonic acid (ACTS). Was added and stirred at 25 ° C. 2.99 mL (23.68 mmol, 1.05 eq.) Of ClCO 2 Ph was added dropwise, and after stirring for 30 minutes, the completion of the carbamate reaction was confirmed by HPLC. Add 5.92 g (23.23 mmol, 1.03 eq.) Of 3-amino-N- (tert-butoxycarbonyl) -L-alanine methyl ester hydrochloride and 9.75 mL (69.93 mmol, 3.1 eq.) Triethylamine. Was added dropwise, and the mixture was stirred at 25 ° C. for 3 hours. Add 0.4 g (1.58 mmol, 0.07 eq.) Of 3-amino-N- (tert-butoxycarbonyl) -L-alanine methyl ester hydrochloride and 0.22 mL (1.58 mmol, 0.07 eq.) Of triethylamine. Then, the completion of the urea conversion reaction was confirmed by HPLC. 7.32 mL (112.8 mmol, 5.0 eq.) Of methanesulfonic acid was added, the temperature was raised to 50 ° C., and the mixture was stirred for 4 hours. After confirming the completion of deprotection by HPLC, the mixture was cooled to 25 ° C. and 37.5 mL (7.5 L / kg) of acetonitrile and 7.5 mL (1.5 L / kg) of water were added to precipitate a solid. It was cooled to 5 ° C. and aged for 16 hours. The precipitated solid was filtered under reduced pressure, washed with 20 mL (4.0 L / kg) of water / acetonitrile (1/2), and then dried under reduced pressure at 40 ° C. for 5 hours to obtain 7.72 g of the desired product as a white solid (. net 7.20 g, 87.3%).
1 H-NMR (400MHz, DMSO-d6): δ 8.39 (bs, 3H), 8.16 (d, 1H, J = 1.2Hz), 7.90 (d, 1H, J = 1.6Hz), 7.28 (d, 1H, J = 1.6Hz), 6.78 (t, 1H, J = 5.6Hz), 4.20-4.10 (m, 1H), 3.77 (s, 3H), 3.70-3.60 (m, 1H), 3.55-3.45 (m, 1H) ), 2.21 (S, 3H)HRMS (FAB – ): Calcd For M / Z 364.0369 (MH & lt;), Found M / Z 364.0395 (MH & lt;) (Step 2) (2) Compound obtained in step 1 of synthesis of 3-{[(2S) -2-amino-2-carboxyethyl] carbamoylamino} -5-chloro-4-methylbenzenesulfonate . To 64 g (net 10.0 g, 27.34 mmol), 18 mL of water (1.8 L / kg vs. the compound of Step 1) was added, and the mixture was stirred at 8 ° C. 3.42 mL (57.41 mmol, 2.1 eq.) Of a 48% aqueous sodium hydroxide solution was added dropwise, and the mixture was washed with 1.0 mL (1.0 L / kg) of water and then stirred at 8 ° C. for 15 minutes. After confirming the completion of hydrolysis by HPLC, the temperature was raised to 25 ° C. and 48% HBr aq. About 3.55 mL was added to adjust the pH to 5.8. After confirming the precipitation of the desired product by dropping 65 mL (6.5 L / kg) of isopropyl alcohol, the mixture was aged for 1 hour. 81 mL (8.1 L / kg) of isopropyl alcohol was added dropwise and the mixture was aged at 8 ° C. overnight. The precipitated solid was filtered under reduced pressure, washed with 20 mL (2.0 L / kg) of isopropyl alcohol, and then dried under reduced pressure at 40 ° C. for 4 hours to obtain 10.7 g of the desired product as a white solid (net 9.46 g, 92). .6%). 1 H-NMR (400MHz, DMSO-d6): δ8.76 (s, 1H), 7.91 (d, 1H, J = 1.6Hz), 8.00-7.50 (bs, 2H), 7.24 (d, 1H, J = 1.6Hz), 7.20 (t, 1H, J = 5.6Hz), 3.58-3.54 (m, 1H), 3.47-3.43 (m, 1H), 3.42-3.37 (m, 1H), 2.23 (s, 3H)
https://patents.google.com/patent/WO2012072663A1/enEXAMPLESExample 1 : Synthesis and isolation of (+)-2-amino-3,6-dihydro-4-dimethylamino-6- methyl-l,3,5-triazine hydrochloride by the process according to the invention
Preliminary step: Synthesis of racemic 2-amino-3,6-dihydro-4-dimethylamino- 6-methyl-l,3,5-triazine hydrochloride:
Metformin hydrochloride is suspended in 4 volumes of isobutanol. Acetaldehyde diethylacetal (1.2 eq.) and para-toluenesulfonic acid (PTSA) (0.05 eq) are added and the resulting suspension is heated to reflux until a clear solution is obtained. Then 2 volumes of the solvent are removed via distillation and the resulting suspension is cooled to 20°C. The formed crystals are isolated on a filter dryer and washed with isobutanol (0.55 volumes). Drying is not necessary and the wet product can be directly used for the next step.Acetaldehyde diethylacetal can be replaced with 2,4,6-trimethyl-l,3,5-trioxane (paraldehyde).- Steps 1 and 2: formation of the diastereoisomeric salt and isolation of the desired diastereoisomer
Racemic 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine hydrochloride wet with isobutanol (obtained as crude product from preliminary step without drying) and L-(+)-Tartaric acid (1 eq.) are dissolved in 2.2 volumes of methanol at 20-40°C. The obtained clear solution is filtered and then 1 equivalent of triethylamine (TEA) is added while keeping the temperature below 30°C. The suspension is heated to reflux, stirred at that temperature for 10 minutes and then cooled down to 55°C. The temperature is maintained at 55°C for 2 hours and the suspension is then cooled to 5- 10°C. After additional stirring for 2 hours at 5-10°C the white crystals are isolated on a filter dryer, washed with methanol (2 x 0.5 Vol) and dried under vacuum at 50°C. The yield after drying is typically in the range of 40-45%
– Steps 3 and 4: transformation of the isolated diastereoisomer of the tartrate salt into the hydrochloride salt and recovery of the salt
γ ethanol HN^NH(+) 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate salt is suspended in 2 volumes of ethanol and 1.02 equivalents of HCl-gas are added under vacuum (-500 mbar). The suspension is heated to reflux under atmospheric pressure (N2) and 5% of the solvent is removed via distillation. Subsequent filtration of the clear colourless solution into a second reactor is followed by a cooling crystallization, the temperature is lowered to 2°C. The obtained suspension is stirred at 2°C for 3 hours and afterwards the white crystals are isolated with a horizontal centrifuge. The crystal cake is washed with ethanol and dried under vacuum at 40°C. The typical yield is 50-55% and the mother liquors can be used for the recovery of about 25-30%) of (+)-2-amino- 3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate.Example 2: Modification of the solvent of steps 3 and 4
– Steps 3 and 4: transformation of the isolated diastereoisomer of the tartrate salt into the hydrochloride salt and recovery of the salt
HN^NH acetone HN^NH(+) 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate salt synthesized according to steps 1 and 2 of example 1 is suspended in 1 volume (based on total amount of (+) 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate salt) of acetone at 20°C. To this suspension 1.01 equivalents of 37% Hydrochloric acid are added. The suspension is heated to reflux under atmospheric pressure (N2) and water is added until a clear solution is obtained. 1.5 vol of acetone are added at reflux temperature. The compound starts crystallising and the obtained suspension is kept at reflux for 2 hours followed by a cooling crystallization to 0°C. The obtained suspension is stirred at 0°C for 2 hours and the white crystals are isolated by centrifugation. The crystal cake is washed with isopropanol and dried under vacuum at 40°C in a continuous drying oven.
Chidamide is being researched as a treatment for pancreatic cancer.[4][5][6] However, it is not US FDA approved for the treatment of pancreatic cancer.
Chidamide (Epidaza®), a class I HDAC inhibitor, was discovered and developed by ChipScreen and approved by the CFDA in December 2014 for the treatment of recurrent of refractory peripheral T-cell lymphoma. Chidamide, also known as CS055 and HBI- 8000, is an orally bioavailable benzamide type inhibitor of HDAC isoenzymes class I 1–3, as well as class IIb 10, with potential antineoplastic activity. It selectively binds to and inhibits HDAC, leading to an increase in acetylation levels of histone protein H3.74 This agent also inhibits the expression of signaling kinases in the PI3K/ Akt and MAPK/Ras pathways and may result in cell cycle arrest and the induction of tumor cell apoptosis. Currently, phases I and II clinical trials are underway for the treatment of non-small cell lung cancer and for the treatment of breast cancer, respectively.
Chemical Synthesis
The scalable synthetic approach to chidamide very closely follows the discovery route. The sequence began with the condensation of commercial nicotinaldehyde (52) and malonic acid (53) in a mixture of pyridine and piperidine. Next, activation of acid 54 with N,N0-carbonyldiimidazole (CDI) and subsequent reaction with 4-aminomethyl benzoic acid (55) under basic conditions afforded amide 56 in 82% yield. Finally, activation of 56 with CDI prior to treatment with 4-fluorobenzene- 1,2-diamine (57) and subsequent treatment with TFA and THF yielded chidamide (VIII) in 38% overall yield from 52. However, no publication reported that mono-N-Boc-protected bis-aniline was used to approach Chidamide.
^ Qiao Z, Ren S, Li W, Wang X, He M, Guo Y, et al. (April 2013). “Chidamide, a novel histone deacetylase inhibitor, synergistically enhances gemcitabine cytotoxicity in pancreatic cancer cells”. Biochemical and Biophysical Research Communications. 434 (1): 95–101. doi:10.1016/j.bbrc.2013.03.059. PMID23541946.
^ Guha M (April 2015). “HDAC inhibitors still need a home run, despite recent approval”. Nature Reviews. Drug Discovery. 14 (4): 225–6. doi:10.1038/nrd4583. PMID25829268. S2CID36758974.
Immunoglobulin G1, anti-(human transferrin receptor) (human-mus musculus monoclonal JR-141 gamma1-chain) fusion protein with peptide (synthetic 2-amino acid linker) fusion protein with human iduronate-2-sulfatase, disulfide with human-mus musculus mono
Immunoglobulin G1-kappa, anti-(human transferrin receptor 1, tfr1) humanized monoclonal antibody, fused with human iduronate-2-sulfatase, glycoform alfa:
Pabinafusp alfa is under investigation in clinical trial NCT03568175 (A Study of JR-141 in Patients With Mucopolysaccharidosis II).
JR-141
NEW DRUG APPROVALS
ONE TIME
$10.00
JCR Pharmaceuticals Announces Approval of IZCARGO® (Pabinafusp Alfa) for Treatment of MPS II (Hunter Syndrome) in Japan
– First Approved Enzyme Replacement Therapy for MPS II to Penetrate Blood-Brain Barrier via Intravenous Administration, Validating JCR’s J-Brain Cargo® Technology –March 23, 2021 07:30 AM Eastern Daylight Time
HYOGO, Japan–(BUSINESS WIRE)–JCR Pharmaceuticals Co., Ltd. (TSE 4552; “JCR”) today announced that the Ministry of Health, Labour and Welfare (MHLW) in Japan has approved IZCARGO® (pabinafusp alfa 10 mL, intravenous drip infusion) for the treatment of mucopolysaccharidosis type II (MPS II, or Hunter syndrome). IZCARGO® (formerly known as JR-141) is a recombinant iduronate-2-sulfatase enzyme replacement therapy (ERT) that relies on J-Brain Cargo®, a proprietary technology developed by JCR, to deliver therapeutics across the blood-brain barrier (BBB). It is the first-ever approved ERT that penetrates the BBB via intravenous administration, a potentially life-changing benefit for individuals with lysosomal storage disorders (LSDs) such as MPS II.
“Subsequent to this approval in Japan, I look forward to further accumulation of clinical evidence for pabinafusp alfa in Brazil, the US and EU”Tweet this
Many patients with MPS II show complications not only in somatic symptoms but also in the central nervous system (CNS), which are often severe, with significant effects on patients’ neurocognitive development, independence, and quality of life. By delivering the enzyme to both the body and the brain, IZCARGO® treats the neurological complications of Hunter syndrome that other available therapies have been unable or inadequate to address so far.
“Approval of IZCARGO® in Japan under SAKIGAKE designation is a key milestone in JCR Pharmaceuticals’ global expansion. It comes on the heels of Fast Track designation from the US FDA, orphan designation from the European Medicines Agency, and the FDA’s acceptance of the JR-141 Investigational New Drug application, enabling JCR to begin our Phase 3 trial in the US,” said Shin Ashida, chairman and president of JCR Pharmaceuticals. “These critical regulatory milestones in Japan, where we have such a strong record of success, and those in the US and Europe, provide important validation of the value of our J-Brain Cargo® technology to deliver therapies across the blood-brain barrier, which we believe is essential to addressing the central nervous system complications of lysosomal storage disorders. We will continue our uncompromising effort to take on the challenge of providing new treatment options for patients with lysosomal storage disorders around the world as soon as possible.”
The MHLW’s approval of IZCARGO® is based on totality of evidence from non-clinical and clinical studies1-4. In a phase 2/3 clinical trial conducted in Japan, all 28 patients experienced significant reductions in heparan sulfate (HS) concentrations in the cerebrospinal fluid (CSF) – a biomarker for effectiveness against CNS symptoms of MPS II – after 52 weeks of treatment, thus meeting the trial’s primary endpoint. IZCARGO® maintained somatic disease control in patients who switched from standard ERT to IZCARGO®. The study also confirmed an improvement in somatic symptoms in participants who had not previously received standard ERT prior to the start of the trial. Additionally, a neurocognitive development assessment demonstrated maintenance or improvement of age-equivalent function in 21 of the 28 patients. There were no reports of serious treatment-related adverse events in the trial, suggestive of a favorable safety and tolerability profile for IZCARGO®.4
“Subsequent to this approval in Japan, I look forward to further accumulation of clinical evidence for pabinafusp alfa in Brazil, the US and EU,” said Dr. Paul Harmatz of University of California – San Francisco (UCSF) Benioff Children’s Hospital Oakland, Oakland, CA, United States. “The availability of an enzyme replacement therapy that crosses the blood-brain barrier is expected to treat both CNS and somatic symptoms associated with this devastating and life-threatening disorder, including developmental and cognitive delays, bone deformities, and abnormal behavior, which have, historically, been unaddressed.”
JCR recently filed an application with the Brazilian Health Surveillance Agency (Agência Nacional de Vigilância Sanitária [ANVISA]) for marketing approval of IZCARGO® for the treatment of patients with MPS II. JCR is also preparing to launch a Phase 3 trial of IZCARGO® in the US, Brazil, the UK, Germany, and France.
About pabinafusp alfa
Pabinafusp alfa (10 mL, intravenous drip infusion) is a recombinant fusion protein of an antibody against the human transferrin receptor and idursulfase, the enzyme that is missing or malfunctioning in subjects with Hunter syndrome. It incorporates J-Brain Cargo®, JCR’s proprietary BBB-penetrating technology, to cross the BBB through transferrin receptor-mediated transcytosis, and its uptake into cells is mediated through the mannose-6-phosphate receptor. This novel mechanism of action is expected to make pabinafusp alfa effective against the CNS symptoms of Hunter syndrome.
In pre-clinical trials, JCR has confirmed both high-affinity binding of pabinafusp alfa to transferrin receptors, and passage across the BBB into neuronal cells, as evidenced by electron microscopy. In addition, JCR has confirmed enzyme uptake in various brain tissues. The company has also confirmed a reduction of substrate accumulation in the CNS and peripheral organs in an animal model of Hunter syndrome.1
In several clinical trials of pabinafusp alfa, JCR obtained evidence of reduced HS concentrations in the CSF, a biomarker for assessing effectiveness against CNS symptoms. The results were consistent with those obtained in pre-clinical studies. Clinical studies have also demonstrated positive effects of pabinafusp alfa on CNS symptoms.2
About J-Brain Cargo® Technology
JCR’s first-in-class proprietary technology, J-Brain Cargo®, enables the development of therapies that cross the BBB and penetrate the CNS. The CNS complications of diseases are often severe, resulting in developmental delays, an impact on cognition and, above all, poor prognosis, which affect patients’ independence as well as the quality of life of patients and their caregivers. With J-Brain Cargo®, JCR seeks to address the unresolved clinical challenges of LSDs by delivering the enzyme to both the body and the brain.
About Mucopolysaccharidosis II (Hunter Syndrome)
Mucopolysaccharidosis II (Hunter syndrome) is an X-linked recessive LSD caused by a deficiency of iduronate-2-sulfatase, an enzyme that breaks down complex carbohydrates called glycosaminoglycans (GAGs, also known as mucopolysaccharides) in the body. Hunter syndrome, which affects an estimated 7,800 individuals worldwide (according to JCR research), gives rise to a wide range of somatic and neurological symptoms. The current standard of care for Hunter syndrome is ERT. CNS symptoms related MPS II have been unmet medical needs so far.
About JCR Pharmaceuticals Co., Ltd.
JCR Pharmaceuticals Co., Ltd. (TSE 4552) is a global specialty pharmaceuticals company that is redefining expectations and expanding possibilities for people with rare and genetic diseases worldwide. We continue to build upon our 45-year legacy in Japan while expanding our global footprint into the US, Europe, and Latin America. We improve patients’ lives by applying our scientific expertise and unique technologies to research, develop, and deliver next-generation therapies. Our approved products in Japan include therapies for the treatment of growth disorder, Fabry disease, acute graft-versus host disease, and renal anemia. Our investigational products in development worldwide are aimed at treating rare diseases including MPS I (Hurler syndrome, Hurler-Scheie, and Scheie syndrome), MPS II (Hunter syndrome), Pompe disease, and more. JCR strives to expand the possibilities for patients while accelerating medical advancement at a global level. Our core values – reliability, confidence, and persistence – benefit all our stakeholders, including employees, partners, and patients. Together we soar. For more information, please visit https://www.jcrpharm.co.jp/en/site/en/.
1 Sonoda H, Morimoto H, Yoden E, et al. A blood-brain-barrier-penetrating anti-human transferrin receptor antibody fusion protein for neuronopathic mucopolysaccharidosis II. Molecular Therapy. 2018;26(5):1366-1374.
2 Morimoto H, Kida K, Yoden E, et al. Clearance of heparan sulfate in the brain prevents neurodegeneration and neurocognitive impairment in MPS II mice. Molecular Therapy. 2021;S1525-0016(21)00027-7.
3 Okuyama T, Eto Y, Sakai N, et al. Iduronate-2-sulfatase with anti-human transferrin receptor antibody for neuropathic mucopolysaccharidosis II: a phase 1/2 trial. Molecular Therapy. 2019;27(2):456-464.
4 Okuyama T, Eto Y, Sakai N, et al. A phase 2/3 trial of pabinafusp alfa, IDS fused with anti-human transferrin receptor antibody, targeting neurodegeneration in MPS-II. Molecular Therapy. 2021;29(2):671-679.
//////////Pabinafusp alfa, JR-141, JR 141,APPROVALS 21, JAPAN 2021
ClassAmides; Analgesics; Antirheumatics; Drug conjugates; Glycosaminoglycans; Nonsteroidal anti-inflammatories
Mechanism of ActionCyclooxygenase inhibitors
RegisteredOsteoarthritis
Phase IITendinitis
23 Mar 2021Registered for Osteoarthritis in Japan (Intra-articular)
25 Sep 2020Phase II for Osteoarthritis is still ongoing in USA (Seikagaku Corporation pipeline, September 2020)
25 Sep 2020Phase II for Tendinitis is still ongoing in Japan (Seikagaku Corporation pipeline, September 2020)
In today’s aging society, osteoarthritis (hereinafter also referred to as “OA” in the present specification), which is a dysfunction caused by joint pain and joint degeneration, is the most common joint disease in the world. It is one of the major causes of physical disorders that interfere with daily life in the elderly. Further, as a disease accompanied by swelling and pain in joints, rheumatoid arthropathy (hereinafter, also referred to as “RA” in the present specification), which is polyarthritis, is known. In RA as well, when the condition progresses over a long period of time, cartilage and bones are destroyed and degeneration or deformation occurs, resulting in physical disorders that interfere with daily life, such as narrowing the range in which joints can be moved.
Currently, preparations using hyaluronic acid and its derivatives are used as medicines for arthropathy such as osteoarthritis and rheumatoid arthropathy. Hyaluronic acid preparations are usually formulated as injections, and for the purpose of improving dysfunction due to arthropathy and suppressing pain through the lubricating action, shock absorption action, cartilage metabolism improving action, etc. of hyaluronic acid, the affected knee, It is administered directly to joints such as the shoulders. Commercialized hyaluronic acid preparations include, for example, those containing purified sodium hyaluronate as an active ingredient (for example, Alz (registered trademark) and Svenir (registered trademark)). The preparation requires continuous administration of 3 to 5 times at a frequency of once a week. In addition, preparations containing crosslinked hyaluronan as an active ingredient require three consecutive doses once a week (for example, Synvisc®), or treatment is completed with a single dose. For single dose administration (eg, Synvisc-One®, Gel-One®, MONOVISC®) are known.On the other hand, steroids and non-steroidal anti-inflammatory compounds are known as quick-acting drugs, and are also used for treatments aimed at relieving joint pain caused by OA and RA. For example, the steroid triamcinolone acetonide has been used as a therapeutic target for joint diseases such as rheumatoid arthritis. Triamcinolone acetonide is commercially available as a drug that is injected intra-articularly and requires administration every 1 to 2 weeks for treatment. Further, as non-steroidal anti-inflammatory compounds, for example, ointments containing diclofenac sodium as an active ingredient and oral administration agents are known.It is also known that a mixture or a conjugate of hyaluronic acid or a derivative thereof and a steroid or a non-steroidal anti-inflammatory compound is used as an active ingredient. For example, a mixture of crosslinked hyaluronic acid and triamcinolone hexaacetonide (CINGAL®) has been commercialized as a single-dose drug. Further, a compound in which hyaluronic acid or a derivative thereof is linked to a steroid or a non-steroidal anti-inflammatory compound is also known. For example, Patent Documents 1 and 2 describe derivatives in which an anti-inflammatory compound is introduced into hyaluronic acid via a spacer. These aim to achieve both fast-acting pain relief and long-term pain relief through improvement of dysfunction. However, it has not yet reached the stage where it can be said that sufficient treatment methods for OA and RA have been established and provided.
<Synthesis Example> Aminoethanol-diclofenac-introduced sodium hyaluronate (test substance) was synthesized according to the method described in Examples of International Publication No. 2005/066241 (hyaluronic acid weight average molecular weight: 800,000, introduction rate). : 18 mol%). More specifically, it was synthesized by the following method. 2.155 g (10.5 mmol) of 2-bromoethylamine hydrobromide is dissolved in 20 mL of dichloromethane, 1.436 mL (10.5 mmol) of triethylamine is added under ice-cooling, and di-tert-butyl-dicarbonate (Boc) is added. 2 O) 2.299 g (10.5 mmol) of a dichloromethane solution of 5 mL was added and stirred. After stirring at room temperature for 90 minutes, ethyl acetate was added, and the mixture was washed successively with 5 wt% citric acid aqueous solution, water and saturated brine. After dehydration with sodium sulfate, the solvent was distilled off under reduced pressure to obtain Boc-aminoethyl bromide. 5 mL of a dimethylformamide (DMF) solution of 2.287 g (10.2 mmol) of Boc-aminoethyl bromide obtained above is ice-cooled, 6 mL of a DMF solution of 3.255 g (10.2 mmol) of diclofenac sodium is added, and the mixture is added at room temperature. Stirred overnight. The mixture was stirred at 60 ° C. for 11 hours and at room temperature overnight. Ethyl acetate was added, and the mixture was sequentially separated and washed with a 5 wt% aqueous sodium hydrogen carbonate solution, water, and saturated brine. After dehydration with sodium sulfate, ethyl acetate was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (toluene: ethyl acetate = 20: 1 (v / v), 0.5% by volume triethylamine) to obtain Boc-aminoethanol-diclofenac. 2.108 g (4.80 mmol) of Boc-aminoethanol-diclofenac obtained above was dissolved in 5 mL of dichloromethane, 20 mL of 4M hydrochloric acid / ethyl acetate was added under ice-cooling, and the mixture was stirred for 2.5 hours. Diethyl ether and hexane were added and precipitated, and the precipitate was dried under reduced pressure. As a result, aminoethanol-diclofenac hydrochloride was obtained. Structure 1 was identified by-NMR H: 1 H-NMR (500 MHz, CDCl 3 ) [delta] (ppm) = 3.18 (2H,t, NH 2 CH 2 CH 2 O-), 3.94 (2H, s, Ph-CH 2 -CO), 4.37 (2H, t, NH 2 CH 2 CH 2 O-), 6.47-7.31 (8H, m, Aromatic H, NH). After dissolving 500 mg (1.25 mmol / disaccharide unit) of hyaluronic acid having a weight average molecular weight of 800,000 in 56.3 mL of water / 56.3 mL of dioxane, imide hydroxysuccinate (1 mmol) / 0.5 mL of water, water-soluble carbodiimide Hydrochloride (WSCI / HCl) (0.5 mmol) / water 0.5 mL, aminoethanol-diclofenac hydrochloride (0.5 mmol) / (water: dioxane = 1: 1 (v / v), 5 mL obtained above ) Was added in sequence, and the mixture was stirred all day and night. 7.5 mL of a 5 wt% sodium hydrogen carbonate aqueous solution was added to the reaction mixture, and the mixture was stirred for about 4 hours. 215 μL of a 50% (v / v) acetic acid aqueous solution was added to the reaction solution for neutralization, and then 2.5 g of sodium chloride was added and the mixture was stirred. 400 ml of ethanol was added to precipitate, and the precipitate was washed twice with an 85% (v / v) aqueous ethanol solution, twice with ethanol, and twice with diethyl ether, dried under reduced pressure overnight at room temperature, and aminoethanol-diclophenac. Introduction Sodium hyaluronate (test substance) was obtained. The introduction rate of diclofenac measured by a spectrophotometer was 18 mol%.
Anamorelin is a non-peptidic ghrelin mimetic Treatment of cancer anorexia and cancer cachexia
Anamorelin hydrochloride has been submitted New Drug Application (NDA) for the treatment of cachexia in non-small cell lung cancer (NSCLC) patients.
It was originally developed by Novo Nordisk, then it was licensed to Ono and Helsinn Therapeutics for the treatment of cachexia and anorexia in cancer patients.
Anamorelin hydrochloride has been submitted New Drug Application (NDA) for the treatment of cachexia in non-small cell lung cancer (NSCLC) patients.
It was originally developed by Novo Nordisk, then it was licensed to Ono and Helsinn Therapeutics for the treatment of cachexia and anorexia in cancer patients.
On 18 May 2017, the European Medicines Agency recommended the refusal of the marketing authorisation for the medicinal product, intended for the treatment of anorexia, cachexia or unintended weight loss in patients with non-small cell lung cancer. Helsinn requested a re-examination of the initial opinion. After considering the grounds for this request, the European Medicines Agency re-examined the opinion, and confirmed the refusal of the marketing authorisation on 14 September 2017.[8] The European Medicines Agency concluded that the studies show a marginal effect of anamorelin on lean body mass and no proven effect on hand grip strength or patients’ quality of life. In addition, following an inspection at clinical study sites, the agency considered that the safety data on the medicine had not been recorded adequately. Therefore, the agency was of the opinion that the benefits of anamorelin did not outweigh its risks.[9]
The chemical name of anamorelin hydrochloride is 2-Amino-N-((R)-1-((R)-3-benzyl-3-(1,2,2-trimethylhydrazine-1-carbonyl)piperidin-1-yl)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-2-methylpropanamide hydrochloride corresponding to the molecular formula C31H42N6O3•HCl and has a relative molecular mass 583.16 g/mol and has the following structure:
The structure of the active substance was elucidated by a combination of 1 H-NMR, 13C-NMR, elemental analysis, FT-IR, UV and and mass spectrometry. Anamorelin HCl appears as a white to off-white hygroscopic solid, freely soluble in water, methanol and ethanol, sparingly soluble in acetonitrile and practically insoluble in ethyl acetate, isopropyl acetate and n-heptane. Its pka was found to be 7.79 and the partition coefficient 2.98. It has two chiral centres with the R,R absolute configuration, which is controlled in the active substance specification by chiral HPLC. Based on the presented data, neither anamorelin hydrochloride, nor any of its salts have been previously authorised in medicinal products in the European Union. Anamorelin is therefore considered as a new active substance.
SYN
OPRD
PATENT
WO 9958501
PATENT
WO 2001034593
https://patents.google.com/patent/WO2001034593A1/enExample 1A procedure for the preparation of the compound which is either 2-Amino-N-[(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1- (1 H-indol-3-ylmethyl)-2-oxoethyl]-2-methylpropionamide
A one-necked round-bottom flask (1 I) equipped with a magnetic stirrer and addition funnel was charged with NaOH-pellets (15,6 g), tetrahydrofuran (400 ml) and ethylnipecotate (50 ml, 324 mmol). To the stirred mixture at room temperature was added dropwise a solution of Boc2O (84,9 g, 389 mmol) dissolved in tetrahydrofuran (150 ml) (1 hour, precipitation of white solid, NaOH-pellets dissolved, exoterm). The mixture was stirred overnight at room temperature. The mixture was added to EtOAc (500 ml) and H2O (2000 ml), and the aqueous layer was re-extracted with EtOAc (2 X 500 ml) and the combined organic layers were washed with brine (100 ml), dried over MgSO4, filtered and concentrated in vacuo to afford piperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester 3-ethyl ester (82,5 g) as a thin yellow oil.1H-NMR (300 MHz, CDCI3): δ 1,25 (t, 3H, CH3); 1 ,45 (s, 9H, 3 X CH3); 2,05 (m, 1H); 2,45 (m, 1H); 2,85 (m, 1 H); 3,95 (d (broad), 1 H); 4,15 (q, 2H, CH2)Step b3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tetf-butyl ester 3-ethyl ester (racemic mixture)
A three-necked round-bottom flask (2 I) equipped with a magnetic stirrer, thermometer, nitrogen bubbler and addition funnel was evacuated, flushed with nitrogen, charged with anhydrous tetrahydrofuran (500 ml) and cooled to -70 °C. Then lithium diisopropylamine (164 ml of a 2,0 M solution in tetrahydrofuran, 327 mmol) was added. To the stirred solution at -70 °C was added dropwise over 45 min. a solution of piperidine-1 ,3-dicarboxylic acid 1- tert-butyl ester 3-ethyl ester (80 g, 311 mmol) in anhydrous tetrahydrofuran (50 ml) (temperature between -70 °C and -60 °C, clear red solution). The mixture was stirred for 20 min. and followed by dropwise addition over 40 min. of a solution of benzylbromide (37 ml, 311 mmol) in anhydrous tetrahydrofuran (250 ml) (temperature between -70 °C and -60 °C). The mixture was stirred for 1 hour at -70 °C, and then left overnight at room temperature (pale orange).The reaction mixture was concentrated in vacuo to approx. 300 ml, transferred to a separating funnel, diluted with CH2CI2 (900 ml) and washed with H2O (900 ml). Due to poor separation the aqueous layer was re-extracted with CH2CI2 (200 ml), the combined organic layers were washed with aqueous NaHSO4 (200 ml, 10%), aqueous NaHCO3 (200 ml, saturated), H2O (200 ml), brine (100 ml), dried over MgSO4> filtered and concentrated in vacuo to afford an oil, which was dissolved in EtOAc(1):heptane(10) and aged overnight. The solids formed was removed by filtration, washed with heptane and dried in vacuo to give a racemic mixture of 3-benzylpiperidine-1 ,3-dicarboxylic acid 1-ter–butyl ester 3-ethyl ester (81 ,4 g). ■ HPLC (h8): Rt = 15,79 min.LC-MS: Rt = 7,67 min. (m+1) = 348,0Step c 3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester (racemic mixture)
3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester 3-ethyl ester (81 g, 233 mmol) was dissolved in EtOH (400 ml) and NaOH (400 ml, 16% aqueous solution) in a one neck round- bottom flask (1 L) equipped with a condenser and a magnetic stirrer. The mixture was refluxed for 10 h under nitrogen, and cooled to room temperature, concentrated in vacuo to approx. 600 ml (precipitation of a solid), diluted with H2O (400 ml), cooled in an icebath, and under vigorous stirring acidified with 4 M H2SO4 until pH = 3 (final temperature: 28 °C). The mixture was extracted with EtOAc (2 X 700 ml), and the combined organic layers were washed with brine (200 ml), dried over MgSO4, filtered and concentrated in vacuo to afford an oil, which was dissolved in EtOAc(1):heptane(10) and aged overnight. The crystals formed were removed by filtration, washed with heptane and dried in vacuo to give a racemic mixture of 3-benzylpiperidine-1 ,3-dicarboxylic acid 1-tetf-butyl ester (66,0 g)HPLC (h8): Rt = 12,85 min.LC-MS: Rt = 5,97 min. (m+1) = 320,0Chirale HPLC (Chiracel OJ, heptane(92):iPrOH(8):TFA(0,1)): Rt = 8,29 min. 46,5 % Rt = 13,69 min. 53,5 %Step d(3R)-3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester or (3S)-3-Benzylpiperidine-1,3-dicarboxylic acid 1-tert-butyl ester
(Resolution of 3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester)
3-Benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester (76 g, 238 mmol) was dissolved in EtOAc (3,0 L) in a one neck flask (5L) equipped with magnetic stirring. Then H2O (30 ml), R(+)-1-phenethylamine (18,2 ml, 143 mmol) and Et3N (13,2 ml, 95 mmol) were added and the mixture was stirred overnight at room temperature resulting in precipitation of white crystals (41 ,9 g), which were removed by filtration, washed with EtOAc and dried in vacuo. The precipitate was dissolved in a mixture of aqueous NaHSO4 (300 ml, 10%) and EtOAc (600 ml), layers were separated and the aqueous layer re-extracted with EtOAc (100 ml). The combined organic layers were washed with brine (100 ml), dried over MgSO4 and filtered. The solvent was removed in vacuo to afford a colourless oil, which was dissolved in EtOAc(1):heptane(10) and aged overnight. The crystals that had been formed were removed by filtration, washed with heptane and dried in vacuo to give one compound which is either (3R)-3-benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester or (3S)-3-benzylpiperidine- 1,3-dicarboxylic acid 1-tert-butyl ester (27,8 g).Chirale HPLC (Chiracel OJ, heptane(92):iPrOH(8):TFA(0,1)):Rt = 7,96 min. 95,8 % eeStep e(3R)-3-Benzyl-3-(N,N’1N’-trimethylhvdrazinocarbonyl)piperidine-1-carboxylic acid tert-butyl ester or (3S)-3-Benzyl-3-(N,N’,N’-trimethylhvdrazinocarbonyl)piperidine-1-carboxylic acid tert-butyl ester
Trimethylhydrazine dihydrochloride (15,3 g, 104 mmol) was suspended in tetrahydrofuran (250 ml) in a one-neck round-bottom flask (1 I) equipped with a large magnetic stirrer, and an addition funnel/nitrogen bubbler. The flask was then placed in a water-bath (temp: 10- 20°C), bromo-rrts-pyrrolydino-phosphonium-hexafluorophosphate (40,4 g, 86,7 mmol) was added, and under vigorous stirring dropwise addition of diisopropylethylamine (59 ml, 347 mmol). The mixture (with heavy precipitation) was stirred for 5 min., and a solution of the product from step d which is either (3R)-3-benzylpiperidine-1 ,3-dicarboxylic acid 1-tert-butyl ester or (3S)-3-benzylpiperidine-1,3-dicarboxylic acid 1-tert-butyl ester (27,7 g, 86,7 mmol) in tetrahydrofuran (250 ml) was added slowly over 1 ,5 hour. The mixture was stirred overnight at room temperature. The reaction was diluted with EtOAc (1000 ml), washed with H2O (500 ml), aqueous NaHSO4, (200 ml, 10%), aqueous NaHCO3 (200 ml, saturated), brine (200 ml), dried over MgSO4, filtered and concentrated in vacuo to afford a thin orange oil. The mixture was dissolved in EtOAc (300 ml), added to SiO2 (150 g) and concentrated in vacuo to a dry powder which was applied onto a filter packed with SiO2 (150 g), washed with heptan (1 I) and the desired compound was liberated with EtOAc (2,5 I). After concentration in vacuo, the product which is either (3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)-piperidine-1- carboxylic acid tert-butyl ester or (3S)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)- piperidine-1-carboxylic acid tert-butyl ester (49 g) as an orange oil was obtained.HPLC (h8): Rt = 14,33 min.Ste f(3R)-3-Benzyl-piperidine-3-carboxylic acid trimethylhydrazide or (3S)-3-Benzyl-piperidine-3- carboxylic acid trimethylhydrazide
The product from step e which is either (3R)-3-Benzyl-3-(N,N’,N’- trimethylhydrazinocarbonyl)-piperidine-1 -carboxylic acid tert-butyl ester or (3S)-3-Benzyl-3- (N,N’,N’-trimethylhydrazinocarbonyl)-piperidine-1 -carboxylic acid tert-butyl ester (56,7 g, 100,9 mmol) was dissolved in EtOAc (500 ml) (clear colourless solution) in a one-neck roundbottom flask (2L) equipped with magnetic stirring. The flask was then placed in a waterbath (temp: 10-20 °C), and HCI-gas was passed through the solution for 5 min. (dust- like precipitation). After stirring for 1 hour (precipitation of large amount of white crystals), the solution was flushed with N2 to remove excess of HCI. The precipitate was removed by gentle filtration, washed with EtOAc (2 X 100 ml), and dried under vacuum at 40 °C overnight to give the product which is either (3R)-3-benzyl-piperidine-3-carboxylic acid trimethylhydrazide or (3S)-3-benzyl-piperidine-3-carboxylic acid trimethylhydrazide (37,0 g).HPLC (h8): Rt = 7,84 min.Step q r(1 R)-2-r(3R)-3-Benzyl-3-(N,N’,N’-trimethylhvdrazinocarbonyl)piperidin-1-vn-1-((1 H-indol-3- yl)methyl)-2-oxoethvncarbamic acid tert-butyl ester or .(1 R)-2-..3S)-3-Benzyl-3-(N,N’,N’- trimethylhvdrazinocarbonyl)piperidin-1-vn-1-((1 H-indol-3-yl)methyl)-2-oxoethyllcarbamic acid tert-butyl ester
Boc-D-Trp-OH (32,3 g, 106 mmol) was dissolved in dimethylacetamide (250 ml) in a one- neck roundbottom flask (500 ml) equipped with a magnetic stirrer and a nitrogen bubbler. The solution was cooled to 0-5 °C and 1-hydroxy-7-azabenzotriazole (14,4 g, 106 mmol), 1- ethyl-3-(3-dimethylaminopropyl)carbodiimid hydrochloride (20,3 g, 106 mmol), N- methylmorpholine (11 ,6 ml, 106 mmol) were added. After stirring for 20 min. at 0-5 °C the product from step f which is either (3R)-3-benzyl-piperidine-3-carboxylic acid trimethylhydrazide or (3S)-3-benzyl-piperidine-3-carboxylic acid trimethylhydrazide (37,0 g, 106 mmol) and N-methylmorpholine (24,4 ml, 223 mmol) were added. The reaction was stirred overnight at room temperature. The mixture was then added to EtOAc (750 ml) and washed with aqueous NaHSO4 (300 ml, 10 %). The layers were allowed to separate, and the aqueous layer was re-extracted with EtOAc (500 ml). The combined organic layers were washed with H2O (100 ml), aqueous NaHCO3 (300 ml, saturated), H2O (100 ml), brine (300 ml), dried over MgSO4, filtered and concentrated in vacuo to afford the product which is either [(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1-((1H- indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester or [(1 R)-2-[(3S)-3-benzyl-3- (N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1-((1 H-indol-3-yl)methyl)-2- oxoethyljcarbamic acid tert-butyl ester (56,7g) as an orange oil.HPLC (h8): Rt = 14,61 min.LC-MS: Rt = 7,35 min. (m+1 ) = 562,6Step h1 -f(2R)-2-Amino-3-(1 H-indol-3-yl)propionylH3R)-3-benzylpiperidine-3-carboxylic acid trimethylhydrazide or 1-f(2R)-2-Amino-3-(1 H-indol-3-yl)propionvn-(3S)-3-benzylpiperidine-3- carboxylic acid trimethylhydrazide
The product from step g which is either [(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’- trimethylhydrazinocarbonyl)piperidin-1 -yl]-1 -((1 H-indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester or [(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1- yl]-1-((1 H-indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester (56,7 g, 100,9 mmol) was dissolved in EtOAc (500 ml) (clear colourless solution) in a one-neck round-bottom flask (2L) equipped with magnetic stirring. The flask was then placed in a water-bath (temp: 10-20 °C), and HCI-gas was passed through the solution for 10 min. (heavy precipitation of oil). The mixture was flushed with N2 to remove excess of HCI and then separated into an oil and an EtOAc-layer. The EtOAc-layer was discarded. The oil was dissolved in H2O (500 ml), CH2CI2 (1000 ml), and solid Na2CO3 was added until pH > 7. The layers were separated, and the organic layer was washed with H2O (100 ml), brine (100 ml), dried over MgSO4, filtered and concentrated in vacuo to afford the product which is either 1-[(2R)-2-amino-3-(1 H-indol- 3-yl)propionyl]-(3R)-3-benzylpiperidine-3-carboxylic acid trimethylhydrazide or 1-[(2R)-2- amino-3-(1H-indol-3-yl)propionyl]-(3S)-3-benzylpiperidine-3-carboxylic acid trimethylhydrazide (27 g) as an orange foam.HPLC (h8): Rt = 10,03 min.Step i(1-r(1 R)-2-r(3R)-3-Benzyl-3-(N,N’,N’-trimethylhvdrazinocarbonyl)piperidin-1-vn-1-(1H-indol-3- ylmethyl)-2-oxo-ethylcarbamovπ-1 -methylethyl fcarbamic acid tert-butyl ester or1-r(1 R)-2-r(3S)-3-Benzyl-3-(N,N’.N’-trimethylhvdrazinocarbonyl)piperidin-1-vn-1-(1 H-indol-3- ylmethyl)-2-oxo-ethylcarbamovπ-1-methylethyl)carbamic acid tert-butyl ester
Boc-Aib-OH (11 ,9 g, 58,4 mmol) was dissolved in dimethylacetamide (125 ml) in a one-neck roundbottom flask (500 ml) equipped with a magnetic stirrer and nitrogen bubbler. To the stirred solution at room temperature were added 1-hydroxy-7-azabenzotriazole (7,95 g, 58,4 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimid hydrochloride (11 ,2 g, 58,4 mmol), and diisopropylethylamine (13,0 ml, 75,8 mmol). After 20 min. (yellow with precipitation) a solution of the product from step h which is either 1-[(2R)-2-amino-3-(1 H-indol-3- yl)propionyl]-(3R)-3-benzylpiperidine-3:carboxylic acid trimethylhydrazide or 1-[(2R)-2- amino-3-(1 H-indol-3-yl)propionyl]-(3S)-3-benzylpiperidine-3-carboxylic acid trimethylhydrazide (27,0 g, 58,4 mmol) in dimethylacetamide (125 ml) was added. The reaction was stirred at room temperature for 3 h. The mixture was added to EtOAc (750 ml) and washed with aqueous NaHSO4 (300 ml, 10 %). The layers were allowed to separate, and the aqueous layer was re-extracted with EtOAc (500 ml). The combined organic layers were washed with H2O (100 ml), aqueous NaHCO3 (300 ml, saturated), H2O (100 ml), brine (300 ml), dried over MgSO4, filtered and concentrated in vacuo to approx. 500 ml. Then SiO2 (150 g) was added and the remaining EtOAc removed in vacuo to give a dry powder which was applied onto a filter packed with SiO2 (150 g), washed with heptan (1 L), and the desired compound was liberated with EtOAc (2,5 L). After concentration in vacuo, the product which is either {1-[(1 R)-2-[(3R)-3-benzyl-3-(N, N’, N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1- (1H-indol-3-ylmethyl)-2-oxo-ethylcarbamoyl]-1-methylethyl}carbamic acid tert-butyl ester or {1-[(1R)-2-[(3S)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1-(1 H-indol-3- ylmethyl)-2-oxo-ethylcarbamoyl]-1-methylethyl}carbamic acid tert-butyl ester 33,9 g as an orange foam was obtained.HPLC (h8): Rt = 14,05 min.Step j2-Amino-N-r(1 R)-2-f(3R)-3-benzyl-3-(N,N’,N’-trimethylhvdrazinocarbonyl)piperidin-1-vπ-1- (1 H-indol-3-ylmethyl)-2-oxoethyll-2-methylpropionamide, fumarate or2-Amino-N-r(1 R)-2-r(3S)-3-benzyl-3-(N1N’1N’-trimethylhvdrazinocarbonyl)piperidin-1-yll-1- (1H-indol-3-ylmethyl)-2-oxoethvπ-2-methylpropionamide, fumarate
The product from step i which is either {1-[(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’- trimethylhydrazinocarbonyl)piperidin-1-yl]-1-(1H-indol-3-ylmethyl)-2-oxo-ethylcarbamoyl]-1- methylethyl}carbamic acid tert-butyl ester or {1-[(1 R)-2-[(3S)-3-benzyl-3-(N,N’,N’- trimethylhydrazinocarbonyl)piperidin-1 -yl]-1 -(1 H-indol-3-ylmethyl)-2-oxo-ethylcarbamoyl]-1 – methylethyljcarbamic acid tert-butyl ester (23,8 g, 36,8 mmol) was dissolved in of EtOAc (800 ml) (clear yellow solution) in a one neck round-bottom flask (1L) equipped with magnetic stirring. The flask was then placed in a water-bath (temp: 10-20 °C), and HCI-gas was passed through the solution for 5 min. (dust-like precipitation). After stirring for 1 hour (precipitation of large amount of yellow powder), the solution was flushed with N2 to remove excess of HCI. The precipitate was removed by gentle filtration and dried under vacuum at 40 °C overnight.The non-crystallinic precipitate was dissolved in H2O (500 ml) and washed with EtOAc (100 ml). Then CH2CI2 (1000 ml) and solid Na2CO3 was added until pH > 7. The 2 layers were separated, and the aqueous layer was e-extracted with CH2CI2 (200 ml). The combined organic layers were washed with brine (100 ml), dried over MgSO4 and filtered. The solvent was evaporated under reduced pressure and redissolved in EtOAc (500 ml) in a one neck round-bottom flask (1 L) equipped with magnetic stirring. A suspension of fumaric acid (3,67 g) in isopropanol (20 ml) and EtOAc (50 ml) was slowly added (5 min.), which resulted in precipitation of a white crystallinic salt. After 1 hour the precipitation was isolated by filtration and dried overnight in vacuum at 40 °C to give the fumarate salt of the compound which is either 2-amino-N-[(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1- yl]-1-(1 H-indol-3-ylmethyl)-2-oxoethyl]-2-methylpropionamide or 2-amino-N-[(1 R)-2-[(3S)-3- benzyl-3-(N,N,,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1-(1 H-indol-3-ylmethyl)-2- oxoethyl]-2-methylpropionamide (13,9 g) as a white powder.HPLC (A1): Rt = 33,61 min.HPLC (B1): Rt = 34,62 min. LC-MS: Rt = 5,09 min. (m+1) = 547,4 ClaimsHide Dependent 1. The compound obtainable by the procedure as described in example 1 , or a pharmaceutically acceptable salt thereof.2. The compound obtainable by the procedure as described in example 1 , and which compound is2-Amino-N-[(1 R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylhydrazinocarbonyl)piperidin-1-yl]-1- (1 H-indol-3-ylmethyl)-2-oxoethyl]-2-methylpropionamide
or a pharmaceutically acceptable salt thereof.3. A pharmaceutical composition comprising, as an active ingredient, a compound according to any one of claims 1-2 or a pharmaceutically acceptable salt thereof together with a pharmaceutically acceptable carrier or diluent.4. A pharmaceutical composition according to claim 3 for stimulating the release of growth hormone from the pituitary.5. A pharmaceutical composition according to claim 3 or claim 4 for administration to animals to increase their rate and extent of growth, to increase their milk and wool production, or for the treatment of ailments.6. A method of stimulating the release of growth hormone from the pituitary of a mammal, the method comprising administering to said mammal an effective amount of a compound according to any one of claims 1 or 2 or a pharmaceutically acceptable salt thereof, or of a composition according to any one of claims 3 – 5.7. A method of increasing the rate and extent of growth, the milk and wool production, or for the treatment of ailments, the method comprising administering to a subject in need thereof an effective amount of a compound according to any one of claims 1-2 or a pharmaceutically acceptable salt thereof, or of a composition according to any one of claims 3-5.8. Use of a compound according to any one of claims 1-2 or a pharmaceutically acceptable salt thereof for the preparation of a medicament.9. Use according to claim 8 wherein the medicament is for stimulating the release of growth hormone from the pituitary of a mammal.
Growth hormone is a major participant in the control of several complex physiologic processes, including growth and metabolism. Growth hormone is known to have a number of effects on metabolic processes, e.g., stimulation of protein synthesis and free fatty acid mobilization and to cause a switch in energy metabolism from carbohydrate to fatty acid metabolism. Deficiency in growth hormone can result in a number of severe medical disorders, e.g., dwarfism.
The release of growth hormone from the pituitary is controlled, directly or indirectly, by number of hormones and neurotransmitters. Growth hormone release can be stimulated by growth hormone releasing hormone (GHRH) and inhibited by somatostatin. In both cases the hormones are released from the hypothalamus but their action is mediated primarily via specific receptors located in the pituitary. Other compounds which stimulate the release of growth hormone from the pituitary have also been described. For example, arginine, L-3,4-dihydroxyphenylalanine (1-Dopa), glucagon, vasopressin, PACAP (pituitary adenylyl cyclase activating peptide), muscarinic receptor agonists and a synthetic hexapeptide, GHRP (growth hormone releasing peptide) release endogenous growth hormone either by a direct effect on the pituitary or by affecting the release of GHRH and/or somatostatin from the hypothalamus.
The use of certain compounds for increasing the levels of growth hormone in mammals has previously been proposed. For example, U.S. Pat. Nos. 6,303,620 and 6,576,648 (the entire contents of which are incorporated herein by reference), disclose a compound: (3R)-1-(2-methylalanyl-D-tryptophyl)-3-(phenylmethyl)-3-piperidinecarboxylic acid 1,2,2-trimethylhydrazide, having the following chemical structure:
(MOL)(CDX) which acts directly on the pituitary cells under normal experimental conditions in vitro to release growth hormone therefrom. This compound is also known under the generic name “anamorelin.” This growth hormone releasing compound can be utilized in vitro as a unique research tool for understanding, inter alia, how growth hormone secretion is regulated at the pituitary level. Moreover, this growth hormone releasing compound can also be administered in vivo to a mammal to increase endogenous growth hormone release.
Example 1
Crystallization of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-(phenylmethyl)-3-piperidinecarboxylic acid 1,2,2-trimethylhydrazide form A
0.0103 g of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-(phenylmethyl)-3-piperidinecarboxylic acid 1,2,2-trimethylhydrazide was dissolved in methanol (0.1 mL) in a glass vial. The glass vial was then covered with PARAFILM® (thermoplastic film) which was perforated with a single hole. The solvent was then allowed to evaporate under ambient conditions. An X-ray diffraction pattern showed the compound was crystalline ( FIG. 1).
PATENT
WO 2017067438
https://patents.google.com/patent/WO2017067438A1/enAnamorelin, whose chemical name is: (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2- Trimethylformylhydrazide is a compound that increases mammalian growth hormone levels and has a compound structure as shown in Formula I:
Cancer cachexia is a state of consumption in which patients lose a lot of weight and muscle mass. It is necessary for the treatment of cachexia because it weakens the patient, affects the quality of life and interferes with the patient’s treatment plan. The drug alamorelin produces the same effect as the so-called “starved hormone” ghrelin, which stimulates hunger. Alamolin is a mimetic of ghrelin, which is secreted by the stomach and is a ligand for growth hormone receptors. . Alamolin binds to this receptor, causing the release of growth hormone, causing a metabolic cascade that affects a variety of different factors, including fat-removing body weight, as well as blood sugar metabolism. Therefore, alamorelin can also enhance the appetite of patients and help patients stay healthy. The 2014 European Society of Medical Oncology (ESMO) in Madrid, Spain, announced that Alamolin is expected to be the first drug in history to effectively improve cancer cachexia.Alamolin is a drug developed by Helsinn Therapeutics (Switzerland) from Novo Nordisk for the development of a cachexia and anorexia for patients with cancer, including non-small cell lung cancer. It can also be used to treat hip fractures and preventive diseases. The strength of the elderly and the elderly has continued to decline. In two key, 12-week Phase III clinical trials (ROMANA 1, ROMANA 2), alamorelin can significantly increase the body fat loss, and is generally tolerated; the incidence of serious adverse drug reactions is less than 3%, mainly related to hyperglycemia and diabetes. Compared with the placebo group, alamorelin continued to increase body weight and improve cancer anorexia-cachexia-related symptoms and concerns; however, there was no significant difference in the improvement of grip strength between the alamolin group and the placebo group. Therefore, this product has excellent clinical value and market value.The polymorphic form of the drug free base and its preparation are reported as follows:Synthesis of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylmethyl is disclosed in the patent ZL99806010.0 A method for synthesizing hydrazide, and using [(1R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylmethylcarbonyl)piperidin-1-yl tert-Butyl ester of 1-((1H-indol-3-yl)methyl)-2-oxoethyl]carbamate is dissolved in dichloromethane, then trifluoroacetic acid is added to remove tert-butyl formate After the base, the mixture was concentrated to remove the solvent, and then the product was extracted with dichloromethane, and the obtained extract was concentrated to dryness to give (3R)-1-(2-methylalanyl-D-color ammonia as an amorphous powder. Acyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide.Patent ZL00815145.8 discloses the synthesis of alamorelin and its compounds as pharmaceutically acceptable salts, relating to novel diastereomeric compounds, pharmaceutically acceptable salts thereof, compositions containing them and their use in therapy Lack of use of medical conditions caused by growth hormone. Synthesis of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformyl is disclosed in this patent. The synthesis method of hydrazine, and using [(1R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylmethylcarbonylcarbonyl)piperidin-1-yl] 1-((1H-Indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester was dissolved in ethyl acetate, and then hydrogen chloride gas was passed to remove the tert-butyl formate protection group. , the solid is dissolved in water, and then the pH is adjusted to about 7 with sodium carbonate, and the product is extracted with dichloromethane; the extract phase is concentrated to obtain (3R)-1-(2-methylalanyl-D-tryptophan). -3-Benzyl-3-piperidine 1,2,2-trimethylformylhydrazide.Patent WO2006016995 discloses (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylmethyl as a medicament Crystalline polymorphs of hydrazides, methods of producing and separating these polymorphs, and pharmaceutical compositions and drug therapies containing these polymorphs, the crystalline polymorphs for direct application to the pituitary Gland cells release the growth hormone. This patent discloses (4R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazone 4 Crystal form: Form A, Form B, Form C and Form D. The patent also provides the preparation of 3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazone. The method of crystal form, especially the preparation method of Form C, in which the method of removing the tert-butyl formate protecting group of methanesulfonic acid in methanol is utilized without exception. As a well-known cause in the art, clinical studies have found that mesylate is genotoxic, and its DNA alkylation leads to mutagenic effects, in which methyl methanesulfonate and ethyl methanesulfonate have been reported. (eg document EMEA/44714/2008). The invention adopts hydrochloric acid or hydrogen chloride gas to remove the tert-butyl formate protecting group, avoids the method of removing methanesulfonic acid, thereby avoiding the risk of the genotoxic impurities in the process, and increasing the risk. The safety of the drug.(3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-three prepared by the patent ZL99806010.0 and the patent ZL00815145.8 Methyl formyl hydrazide, no data on the purity of its compounds, we found that (3R)-1-(2-methylalanyl-D-tryptophan)-3 was prepared by this method. -Benzyl-3-piperidine 1,2,2-trimethylformylhydrazide does not help to remove the impurities produced, and the purity of the obtained product is not high, and it is difficult to meet the medicinal requirements. And (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethyl obtained by the preparation method of the present invention. The crystal form of the formyl hydrazide has a purity of 99.8% and a single impurity of less than 0.1%, which fully meets the requirements for medicinal purity. Moreover, the crystal form is stable to conditions such as pressure, temperature, humidity and illumination, and the preparation method is simple in operation and suitable for industrial production.Example 1:300 g of [(1R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylcarbamidocarbonyl)piperidin-1-yl]-1-(( 1H-Indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester was added to the reaction flask, and then 4 L of dichloromethane was added to the reaction flask, and the raw material was completely dissolved by stirring.Then, the reaction system is cooled to 10 ° C or lower in an ice bath, hydrogen chloride gas is continuously supplied to the reaction liquid, and solids are gradually precipitated, and the reaction is further maintained at about 10 ° C for 3 to 5 hours, and the sample is detected. After the reaction of the raw materials is completed, the reaction system is completed. 1.5 L of water was added thereto, the solid was completely dissolved, and then the pH was adjusted to about 8 with a 20% aqueous sodium hydroxide solution, and the layers were separated; the aqueous phase was extracted once more with dichloromethane, and the organic phases were combined.The organic phase was dried over anhydrous sodium sulfate for 3 hrs, filtered, and then evaporated to ethylamine 3-Benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude 246 g, yield 97.2%. HPLC content (area normalization method) was 96.1%.Example 2:300 g of [(1R)-2-[(3R)-3-benzyl-3-(N,N’,N’-trimethylcarbamidocarbonyl)piperidin-1-yl]-1-(( 1H-Indol-3-yl)methyl)-2-oxoethyl]carbamic acid tert-butyl ester was added to the reaction flask, 36% concentrated hydrochloric acid was added to the reaction flask, and the reaction system was heated to 40 with stirring. The reaction was carried out at ° C to 50 for 3 hours.Then, the sample is detected. After the reaction of the raw material is completed, the reaction system is cooled to 10 or less, and 2.0 L of dichloromethane is added to the reaction system, and then the pH is adjusted to about 8 with a 20% aqueous sodium hydroxide solution, and the aqueous phase is further separated. It was extracted once with dichloromethane and the organic phases were combined.The organic phase was dried over anhydrous sodium sulfate for 3 hrs, filtered, and then evaporated to ethylamine 3-Benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude 248 g, yield 98%. HPLC content (area normalization method) was 96.2%.Preparation of (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazone E crystal formExample 3Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10g was added to the reaction flask and 30 ml of N- was added.Methylpyrrolidone, stirred and dissolved completely. Then, 60 ml of water was added dropwise to the reaction flask at room temperature, and the reaction liquid was heated to 60 ° C. The solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 hours.Slowly cooled to below 20 ° C, filtered, and the filter cake was washed with a mixture of N-methylpyrrolidone / H 2 O; the cake was vacuum dried at about 55 ° C to obtain (3R)-1-(2-methylalanyl) -D-tryptophan)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 9.5 g), HPLC content (area normalization) 99.72%. The XRD pattern is shown in Fig. 1, the DSC chart is shown in Fig. 2, and the TGA pattern is shown in Fig. 3, where the crystal form is defined as the E crystal form. The DSC of the crystal form has an endotherm at 120.05, the TGA is heated at 60A, and the crystal loss of 5 is about 3.1%. Combined with the Karl Fischer method, the moisture content of the product is determined. 3.1% and 3.2% indicate that the sample is present as a monohydrate.Example 4:Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 30 ml of N,N-dimethylformamide was added, stirred, and dissolved completely. Then, 30 ml of water was added dropwise to the reaction flask at room temperature, and the reaction solution was heated to 50 ° C. The solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 h.Slowly cool to below 10 ° C, filter, filter cake washed with N, N-dimethylformamide / H 2 O mixture; vacuum cake dried at around 55 ° C to obtain (3R)-1-(2-A Alanyl-D-tryptophanyl-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 8.5 g), HPLC content (area normalization) ) 99.87%. Upon comparison, it was confirmed that the solid was in the E crystal form.Example 5:Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 30 ml of dimethyl sulfoxide was added, stirred, and dissolved completely. Then, 40 ml of water was added dropwise to the reaction flask at room temperature, and the reaction liquid was heated to 60 ° C, the solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 hours.Slowly cooled to below 10 ° C, filtered, and the filter cake was washed with a mixture of dimethyl sulfoxide / H 2 O; the cake was vacuum dried at about 50 ° C to obtain (3R)-1-(2-methylalanyl) -D-tryptophanyl-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 9.1 g), HPLC content (area normalization) 99.61%. Upon comparison, it was confirmed that the solid was in the E crystal form.Example 6Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 40 ml of 1,4-dioxane was added, stirred, and dissolved completely. Then, 50 ml of water was added dropwise to the reaction flask at room temperature, and the reaction solution was heated to 70 ° C. The solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 hours.Slowly cooled to below 10 ° C, filtered, and the filter cake was washed with a mixture of 1,4-dioxane/H 2 O; the cake was vacuum dried at about 50 ° C to obtain (3R)-1-(2-methyl alanyl-D-tryptophan-3-Benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 8.7 g), HPLC content (area normalization) 99.11%. Upon comparison, it was confirmed that the solid was in the E crystal form.Example 7Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 40 ml of N,N-dimethylacetamide was added, stirred, and dissolved completely. Then, 40 ml of water was added dropwise to the reaction flask at room temperature, and the reaction solution was heated to 70 ° C. The solution became cloudy, and was slowly cooled to about 50 ° C. Seed crystals were added thereto, and cooling was continued to gradually precipitate a solid.The reaction system was cooled to about 10 ° C, filtered, and the filter cake was washed with a mixture of N,N-dimethylacetamide/H 2 O; the cake was vacuum dried at about 50 ° C to obtain (3R)-1-(2- Methylalanyl-D-tryptophanyl-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 8.1 g), HPLC content (area normalized) Law) 99.78%. Upon comparison, it was confirmed that the solid was in the E crystal form.Example 7Taking the above amorphous (3R)-1-(2-methylalanyl-D-tryptophyl)-3-benzyl-3-piperidine 1,2,2-trimethylformylhydrazine crude product 10 g was added to the reaction flask, 50 ml of acetone was added, stirred, and dissolved completely. Then, 70 ml of water was added dropwise to the reaction flask at room temperature, and the reaction liquid was heated to 45 ° C. The solution became cloudy, and a white solid was gradually precipitated, and stirring was continued for 2 hours.Slowly cool to below 10 ° C, filter, filter cake washed with acetone / H 2 O mixture; filter cake vacuum dried at around 50 ° C to obtain (3R)-1-(2-methylalanyl-D-color Aminoacyl-3-phenylmethyl-3-piperidine 1,2,2-trimethylformylhydrazide (white solid, 9.3 g), HPLC content (area normalization) 98.9%. Upon comparison, it was confirmed that the solid was in the E crystal form.
SYN
Reference:
1. Org. Process Res. Dev.2006, 10, 339–345.
Abstract
The rapid process development of a scaleable synthesis of the pseudotripeptide RC-1291 for preclinical and clinical evaluation is described. By employing a nontraditional N-to-C coupling strategy, the peptide chain of RC-1291 was assembled in high yield, with minimal racemization and in an economical manner by introducing the most expensive component last. A one-pot deprotection/crystallization procedure was developed for the isolation of RC-1291 free base, which afforded the target compound in excellent yield and with a purity of >99.5% without chromatographic purification.
(R,R)-2-Amino-N-[2-[3-benzyl-3-(N,N′,N′-trimethyl-hydrazinocarbonyl)piperidin-1-yl]-1-(1H-indol-3-ylmethyl)- 2-oxo-ethyl]-2-methyl-propionamide (1). Crude 7 (911 g; 1.28 mol theoretical)10 was dissolved in methanol (4.12 L) in a 22-L round-bottom flask equipped with a mechanical stirrer, a temperature probe, a reflux condenser, a gas (N2) inlet, and an addition funnel. The solution was heated to 55 °C; then methanesulfonic acid (269.5 g, 2.805 mol) was added over a period of 15 min. (Caution: gas evolution!) The solution was then heated to 60 °C for a period of 1 h, after which HPLC analysis showed that no 7 remained. The temperature of the reaction mixture was increased to reflux (68-72 °C) over a period of 35 min, while simultaneously adding a solution of KOH (85%, 210.4 g, 3.187 mol) in water (4.12 L). The clear, slightly yellow solution was then allowed to cool to 20 °C at a rate of 5 °C/h. The free base of RC1291 (1) crystallized as a pale-yellow solid, which was isolated by filtration. The filter cake was washed with two portions of 50% aqueous methanol (500 mL each) and then dried under high vacuum at 20 ( 5 °C to afford 1 as an off-white, crystalline solid (595 g, 85% yield for two steps, >99.5% AUC by HPLC).
HRMS (ESI) calcd for C31H43N6O3 [M + H]+ 547.3397, found 547.3432.
Cachexia and muscle wasting are very common among patients suffering from cancer, chronic obstructive pulmonary disease, and other chronic diseases. Ghrelin stimulates growth hormone secretion via the ghrelin receptor, which subsequently leads to increase of IGF-1 plasma levels. The activation of the GH/IGF-1 axis leads to an increase of muscle mass and functional capacity. Ghrelin further acts on inflammation, appetite, and adipogenesis and for this reason was considered an important target to address catabolic conditions. We report the synthesis and properties of an indane based series of ghrelin receptor full agonists; they have been shown to generate a sustained increase of IGF-1 levels in dog and have been thoroughly investigated with respect to their functional activity.
Patent
https://patents.google.com/patent/EP2838892A1/enGrowth hormone is a major participant in the control of several complex physiologic processes including growth and metabolism. Growth hormone is known to have a number of effects on metabolic processes such as stimulating protein synthesis and mobilizing free fatty acids, and causing a switch in energy metabolism from carbohydrate to fatty acid metabolism. Deficiencies in growth hormone can result in dwarfism and other severe medical disorders.The release of growth hormone from the pituitary gland is controlled directly and indirectly by a number of hormones and neurotransmitters. Growth hormone release can be stimulated by growth hormone releasing hormone (GHRH) and inhibited by somatostatin.The use of certain compounds to increase levels of growth hormone in mammals has previously been proposed. Anamorelin is one such compound. Anamorelin is a synthetic orally active compound originally synthesized in the 1990s as a growth hormone secretogogue for the treatment of cancer related cachexia. The free base of anamorelin is chemically defined as:® (3R) 1 -(2-methylaIanyl~D ryptophyl)~3-(phenylraethyl)~3~piperidineearboxylie acid 1 ,2,2trimethyihydrazide,* 3-{(2R)-3-{(3R)-3-benzyi-3-| (trimethylhydrazino)carbonyi]piperidin-l»yl}-2-[(2»met hylaianyl)amino]-3-ox.opropyi}-IH-indole, or• 2-Amino-N-[(lR)-2-[(3R)-3~benzyWcarbony piperidin- 1 -yl] – 1-( 1 H-indol-3 -yl^^and has the below chemical structure; U.S. Patent No. 6,576,648 to Artkerson reports a process of preparing anamorelin as the fumarate salt, with the hydrochloride salt produced as an intermediate in Step (j) of Example 1 . U.S. Patent No. 7,825, 138 to Lorimer describes a process for preparing crystal forms of the free base of anamorelin.There is a need to develop anamorelin monohydrochloride as an active pharmaceutical ingredient with reduced impurities and improved stability over prior art forms of anamorelin hydrochloride, such as those described in U.S. Patent No, 6,576,648, having good solubility, bioavailability and processabi!ity. There is also a need to develop methods of producing pharmaceutically acceptable forms of anamorelin monohydrochloride thai have improved yield over prior art processes, reduced residual solvents, and controlled distribution of chloride content,it has unexpectedly been discovered that the process of making the hydrochloride salt of anamorelin described in Step (j) of U.S. Patent No. 6.576,648 can result in excessive levels of chloride in the final product, and that this excess chloride leads to the long-term instability of the final product due at least, partially to an increase in the amount of the less stable dihydrochloride salt of anamorelin. Conversely, because anamorelin free base is less soluble in water than the hydrochloride salt, deficient chloride content in the final product can lead to decreased solubility of the molecule. The process described in U.S. Patent No, 6,576,648 also yields a final product that contains more than 5000 ppm (0.5%) of residual solvents, which renders the product less desirable from a pharmaceutical standpoint, as described in CH Harmonized Tripartite Guideline. See Impurities; Guideline for residual solvents Q3C(R3). in order to overcome these problems, methods have been developed which, for the first time, allow for the efficient and precise control of the reaction between anarnorehn tree base and hydrochloric acid in situ, thereby increasing the yield of anarnorehn monohydrochioride from the reaction and reducing the incidence of unwanted anamorelin dihydroeh ride. According to the method, the free base of anamorelin is dissolved in an organic solvent and combined with water and hydrochloric acid, with the molar ratio of anarnorehn and chloride tightly controlled to prevent an excess of chloride in the final product. The water and hydrochloric acid can be added either sequentially or at the same time as long as two separate phases are formed. Without wishing to be bound by any theory, it is believed thai as the anamorelin free base in the organic phase is protonated by the hydrochloric acid it migrates into the aqueous phase. The controlled ratio of anamorelin free base and hydrochloric acid and homogenous distribution in the aqueous phase allows for the controlled formation of the monohydrochioride salt over the dihydrochloride, and the controlled distribution of the resulting chloride levels within individual batches and among multiple batches of anamorelin monohydrochioride.Thus, in a fust embodiment the invention provides methods for preparing anamorelin monohydrochioride or a composition comprising anamorelin monohydrochioride comprising: (a) dissolving anamorelin free base in an organic solvent to form a solution; (b) mixing said solution with water and hydrochloric acid for a time sufficient to: (i) react said anamorelin free base with said hydrochloric acid, and (ii) form an organic phase and an aqueous phase; (c) separating the aqueous phase from the organic phase; and (d) isolating anamorelin monohydrochioride from the aqueous phase.In a particularly preferred embodiment, the molar ratio of anamorelin to hydrochloric acid used in the process is less than or equal to 1 : 1 , so as to reduce the production of anamorelin dihydrochloride and other unwanted chemical species. Thus, for example, hydrochloric acid can be added at a molar ratio of from 0,90 to 1 ,0 relative to said anamorelin, from 0.90 to 0.99, or from 0.93 to 0.97.n another particularly preferred embodiment, the anamorelin monohydrochioride or a composition comprising anamorelin monohydrochioride is isolated from the aqueous phase via spray drying, preferably preceded by distillation. This technique has proven especially useful in the manufacture of anamorelin monohydrochioride or a composition comprising anamorelin monohydrochioride because of the excellent reduction in solvent levels observed, and the production of a stable amorphous form of anamorelin monohydrochioride or a composition comprising anamorelin monohydrochioride. In other embodiments, the invention relates to the various forms of anamorelin monohvdrochloride and compositions comprising anamorelin monohvdrochloride produced by the methods of the present invention. In a first embodiment, which derives from the controlled chloride content among batches accomplished by the present methods, the invention provides anamorelin monohvdrochloride or a composition comprising anamorelin monohydrochloride having an inter-batch chloride content of from 5.8 to 6.2%, preferably from 5.8 to less than 6.2%. Alternatively, the invention provides anamorelin monohydrochloride or a composition comprising anamorelin monohydrochloride having a molar ratio of chloride to anamorelin less than or equal to 1 : 1 , such as from 0.9 to 1.0 or 0.99, in yet another embodiment the invention provides an amorphous form of anamorelin monohydrochloride or a composition comprising anamorelin monohydrochloride. Further descriptions of the anamorelin monohydrochloride and compositions comprising the anamorelin monohydrochloride are given in the detailed description which follows.EXAMPLE 1 . PREPARATION OF ANAMOREUN HYDROCHLORIDEVarious methods have been developed to prepare the hydrochloric acid salt of anarnorelin, with differing results.In a first method, which is the preferred method of the present invention, anarnorelin free base was carefully measured and dissolved in isopropyl acetate. Anarnorelin free base was prepared according to known method (e.g., U.S. Patent No, 6,576,648). A fixed volume of HCl in water containing various molar ratios (0.80, 0,95, 1.00 or 1.05) of HCl relative to the anarnorelin free base was then combined with the anamorelin/isopropyl acetate solution, to form a mixture having an organic and an aqueous phase, The aqueous phase of the mixture was separated from the organic phase and the resulting aqueous phase was concentrated by spray drying to obtain the batches of anarnorelin monohydrochloride (or a composition comprising anarnorelin monohydrochloride ) shown in Table 1 A.Approximately 150mg of the resulting spray dried sample of anarnorelin monohydrochloride (or composition comprising anarnorelin monohydrochloride) was accurately weighed out and dissolved in methanol (50mL). Acetic acid (5mL) and distilled water (5mL) were added to the mixture. The resulting mixture was potentiometricaJ ly titrated using 0,0 IN silver nitrate and the e dpoint was determined. A blank determination was also performed and correction was made, if necessary. The chloride content in the sample was calculated by the following formula. This measurement method of chloride content was performed without any cations other than proton (! ! ‘ ).Chloride content (%) = VxNx35.453x l 00x l 00/{Wx[1 00-(water content (%))-(residual solvent (%))]}V: volume at the endpoint (ml.)N; actual normality of 0.01 mol/L silver nitrate35.453 : atomic weight of ChlorineW: weight of sample (mg)TABLE 1 AHCl Chloride ContentThis data showed that anamorelin monohydrochlonde produced by a fixed volume of HCl in water containing 0.80 or 1 .05 molar equivalents of HC1 relative to anamorelin free base had levels of chloride thai were undesirable, and associated with product instability as shown in Example 3.Alternatively, a fixed volume of HCl in water containing 0.95 moles of HCl relative to anamorelin free base was used to prepare anamorelin monohydrochlonde (or composition comprising anamorelin monohydrochloride) as follows. Anamorelin free base (18.8g, 34.4mmoi) and isopropyl acetate (341.8g) were mixed in a 1000 mL flask. The mixture was heated at 40±5°C to confirm dissolution of the crystals and then cooled at 25±5°C. Distilled water (22.3g) and 3.6% diluted hydrochloric acid (33. Ig, 32.7mmoL 0.95 equivalents) were added into the flask and washed with distilled water. After 30 minutes stirring, the reaction was static for more than 15 minutes and the lower layer (aqueous layer) was transferred into a separate 250mL flask. Distilled water was added to the flask and concentrated under pressure at 50i5cC. The resulting aqueous solution was then filtered and product isolated by spray drying to afford anamorelin monohydrochlonde A (the present invention).The physical properties of anamorelin monohydrochloride A were compared to anamorelin monohydrochloride produced by a traditional comparative method (“anamorelin monohydrochloride B”) (comparative example). Anamorelin mono hydrochloride B in the comparative example was produced by bubbling HCl gas into isopropyl acetate to produce a 2M solution of HCl, and reacting 0.95 molar equivalents of the 2M HCl in isopropyl acetate with anamorelin free base. The physical properties of anamorelin monohydrochloride B are reported in Table IB. This data shows that when 0.95 equivalents of HCl is added to anamorelin free base, the chloride content (or amount of anamorelin dihydrochloride) is increased, even when a stoichiometric ratio of hydrochloride to anamorelin of less than 1 ,0 is used, possibly due to uncontrolled precipitation. In addition, this data shows that the concentration of residual solvents in anamorelin monohydrochloride B was greater than the concentration in anamorelin monohydrochloride A, TABLE I B A similar decrease in residual solvent concentration was observed when 2-methyltetrahydrofuran was used as the dissolving solvent for anamorelin free base instead of isopropvi acetate in the process for preparing spray dried anamorelin monohydrochloride A (data not reported).The residual solvent (organic volatile impurities) concentration (specifically isopropyl acetate) of anamorelin monohydrochloride in TABLE IB was measured using gas chromatography (GC-2010, Shimadzu Corporation) according to the conditions shown in TABLE 1 C,
^ Currow DC, Abernethy AP (April 2014). “Anamorelin hydrochloride in the treatment of cancer anorexia-cachexia syndrome”. Future Oncology. 10 (5): 789–802. doi:10.2217/fon.14.14. PMID24472001.
^ Jump up to:abc Garcia JM, Polvino WJ (June 2009). “Pharmacodynamic hormonal effects of anamorelin, a novel oral ghrelin mimetic and growth hormone secretagogue in healthy volunteers”. Growth Hormone & IGF Research. 19 (3): 267–73. doi:10.1016/j.ghir.2008.12.003. PMID19196529.
^ Jump up to:ab Garcia JM, Boccia RV, Graham CD, Yan Y, Duus EM, Allen S, Friend J (January 2015). “Anamorelin for patients with cancer cachexia: an integrated analysis of two phase 2, randomised, placebo-controlled, double-blind trials”. The Lancet. Oncology. 16 (1): 108–16. doi:10.1016/S1470-2045(14)71154-4. PMID25524795.
^ Jump up to:ab Garcia JM, Friend J, Allen S (January 2013). “Therapeutic potential of anamorelin, a novel, oral ghrelin mimetic, in patients with cancer-related cachexia: a multicenter, randomized, double-blind, crossover, pilot study”. Supportive Care in Cancer. 21 (1): 129–37. doi:10.1007/s00520-012-1500-1. PMID22699302. S2CID22853697.
^ Temel JS, Abernethy AP, Currow DC, Friend J, Duus EM, Yan Y, Fearon KC (April 2016). “Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): results from two randomised, double-blind, phase 3 trials”. The Lancet. Oncology. 17 (4): 519–531. doi:10.1016/S1470-2045(15)00558-6. PMID26906526.
The U.S. Food and Drug Administration today approved Azedra (iobenguane I 131) injection for intravenous use for the treatment of adults and adolescents age 12 and older with rare tumors of the adrenal gland (pheochromocytoma or paraganglioma) that cannot be surgically removed (unresectable), have spread beyond the original tumor site and require systemic anticancer therapy. This is the first FDA-approved drug for this use.
update………APPROVED JAPAN 2021, 2021/9/27, Raiatt MIBG-I 131
July 30, 2018
Release
The U.S. Food and Drug Administration today approved Azedra (iobenguane I 131) injection for intravenous use for the treatment of adults and adolescents age 12 and older with rare tumors of the adrenal gland (pheochromocytoma or paraganglioma) that cannot be surgically removed (unresectable), have spread beyond the original tumor site and require systemic anticancer therapy. This is the first FDA-approved drug for this use.
“Many patients with these ultra-rare cancers can be treated with surgery or local therapies, but there are no effective systemic treatments for patients who experience tumor-related symptoms such as high blood pressure,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Patients will now have an approved therapy that has been shown to decrease the need for blood pressure medication and reduce tumor size in some patients.”
Pheochromocytomas are rare tumors of the adrenal glands. These glands are located right above the kidneys and make hormones including stress hormones called epinephrines and norepinephrines. Pheochromocytomas increase the production of these hormones, leading to hypertension (high blood pressure) and symptoms such as headaches, irritability, sweating, rapid heart rate, nausea, vomiting, weight loss, weakness, chest pain or anxiety. When this type of tumor occurs outside the adrenal gland, it is called a paraganglioma.
The efficacy of Azedra was shown in a single-arm, open-label, clinical trial in 68 patients that measured the number of patients who experienced a 50 percent or greater reduction of all antihypertensive medications lasting for at least six months. This endpoint was supported by the secondary endpoint, overall tumor response measured by traditional imaging criteria. The study met the primary endpoint, with 17 (25 percent) of the 68 evaluable patients experiencing a 50 percent or greater reduction of all antihypertensive medication for at least six months. Overall tumor response was achieved in 15 (22 percent) of the patients studied.
The most common severe side effects reported by patients receiving Azedra in clinical trials included low levels of white blood cells (lymphopenia), abnormally low count of a type of white blood cells (neutropenia), low blood platelet count (thrombocytopenia), fatigue, anemia, increased international normalized ratio (a laboratory test which measures blood clotting), nausea, dizziness, hypertension and vomiting.
As it is a radioactive therapeutic agent, Azedra includes a warning about radiation exposure to patients and family members, which should be minimized while the patient is receiving Azedra. The risk of radiation exposure is greater in pediatric patients. Other warnings and precautions include a risk of lower levels of blood cells (myelosuppression), underactive thyroid, elevations in blood pressure, renal failure or kidney injury and inflammation of lung tissue (pneumonitis). Myelodysplastic syndrome and acute leukemias, which are cancers of the blood and bone marrow, were observed in patients who received Azedra, and the magnitude of this risk will continue to be studied. Azedra can cause harm to a developing fetus; women should be advised of the potential risk to the fetus and to use effective contraception after receiving Azedra. Radiation exposure associated with Azedra may cause infertility in males and females.
The FDA granted this application Fast Track, Breakthrough Therapy and Priority Review designations. Azedra also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Azedra to Progenics Pharmaceuticals, Inc.
AdreView (iobenguane I 123 Injection) is a sterile, pyrogen-free radiopharmaceutical for intravenous injection. Each mL contains 0.08 mg iobenguane sulfate, 74 MBq (2 mCi) of I 123 (as iobenguane sulfate I 123) at calibration date and time on the label, 23 mg sodium dihydrogen phosphate dihydrate, 2.8 mg disodium hydrogen phosphate dihydrate and 10.3 mg (1% v/v) benzyl alcohol with a pH of 5.0 – 6.5. Iobenguane sulfate I 123 is also known as I 123 meta-iodobenzlyguanidine sulfate and has the following structural formula:
Physical Characteristics
Iodine 123 is a cyclotron-produced radionuclide that decays to Te 123 by electron capture and has a physical half-life of 13.2 hours.
Iobenguane I-131 is a guanidine analog with specific affinity for tissues of the sympathetic nervous system and related tumors. The radiolabeled forms are used as antineoplastic agents and radioactive imaging agents. (Merck Index, 12th ed) MIBG serves as a neuron-blocking agent which has a strong affinity for, and retention in, the adrenal medulla and also inhibits ADP-ribosyltransferase.
Iobenguane i-131 is a Radioactive Diagnostic Agent. The mechanism of action of iobenguane i-131 is as a Radiopharmaceutical Activity.
Iobenguane I-131 is an I 131 radioiodinated synthetic analogue of the neurotransmitter norepinephrine. Iobenguane localizes to adrenergic tissue and, in radioiodinated forms, may be used to image or eradicate tumor cells that take up and metabolize norepinephrine.
The radioisotope of iodine used for the label can be iodine-123 (for imaging purposes only) or iodine-131 (which must be used when tissue destruction is desired, but is sometimes used for imaging also).
Pheochromocytoma seen as dark sphere in center of the body (it is in the left adrenal gland). Image is by MIBG scintigraphy, with radiation from radioiodine in the MIBG. Two images are seen of the same patient from front and back. Note dark image of the thyroid due to unwanted uptake of iodide radioiodine from breakdown of the pharmaceutical, by the thyroid gland in the neck. Uptake at the side of the head are from the salivary glands. Radioactivity is also seen in the bladder, from normal renal excretion of iodide.
It localizes to adrenergic tissue and thus can be used to identify the location of tumors[2] such as pheochromocytomas and neuroblastomas. With I-131 it can also be used to eradicate tumor cells that take up and metabolize norepinephrine.
Thyroid precautions
Thyroid blockade with (nonradioactive) potassium iodide is indicated for nuclear medicine scintigraphy with iobenguane/mIBG. This competitively inhibits radioiodine uptake, preventing excessive radioiodine levels in the thyroid and minimizing the risk of thyroid ablation ( in the case of I-131). The minimal risk of thyroid carcinogenesis is also reduced as a result.
The FDA-approved dosing of potassium iodide for this purpose are as follows: infants less than 1 month old, 16 mg; children 1 month to 3 years, 32 mg; children 3 years to 18 years, 65 mg; adults 130 mg.[3] However, some sources recommend alternative dosing regimens.[4]
Not all sources are in agreement on the necessary duration of thyroid blockade, although agreement appears to have been reached about the necessity of blockade for both scintigraphic and therapeutic applications of iobenguane. Commercially available iobenguane is labeled with iodine-123, and product labeling recommends administration of potassium iodide 1 hour prior to administration of the radiopharmaceutical for all age groups,[5] while the European Associated of Nuclear Medicine recommends (for iobenguane labeled with either I-131 or I-123,) that potassium iodide administration begin one day prior to radiopharmaceutical administration, and continue until the day following the injection, with the exception of newborns, who do not require potassium iodide doses following radiopharmaceutical injection.[4]
Product labeling for diagnostic iodine-131 iobenguane recommends potassium iodide administration one day before injection and continuing 5 to 7 days following.[6] Iodine-131 iobenguane used for therapeutic purposes requires a different pre-medication duration, beginning 24–48 hours prior to iobenguane injection and continuing 10–15 days following injection.[7]
Alternative imaging modality for pheochromocytoma
The FDOPAPET/CT scan has proven to be nearly 100% sensitive for detection of pheochromocytomas, vs. 90% for MIBG scans.[8][9][10] Centers which offer FDOPA PET/CT, however, are rare.
Clinical trials
Iobenguane I 131 for cancers
Iobenguane I 131 (as Azedra) has had a clinical trial as a treatment for malignant, recurrent or unresectable pheochromocytoma and paraganglioma, and the US FDA has granted it a Priority Review.[11]
Percent Composition: C 34.93%, H 3.66%, I 46.13%, N 15.28%
Literature References: Norepinephrine analog with specific affinity for tissues of sympathetic nervous system and related tumors; prepd as 123I and 131I labeled forms. Prepn and imaging studies: D. M. Wieland et al.,J. Nucl. Med.21, 349 (1980); eidem,US4584187 (1986). Improved synthesis: P. A. P. M. van Doremalen, A. G. M. Janssen, J. Radioanal. Nucl. Chem. Lett.96, 97 (1985). Metabolism in man: T. J. Mangner et al.,J. Nucl. Med.27, 37 (1986). HPLC determn in serum and urine: D. Schwabe et al.,J. Chromatogr.487, 177 (1989). Radiopharmacokinetics: S. Ertl et al.,Nucl. Med. Commun.8, 643 (1987). Clinical evaluation of myocardial imaging: D. Fagret et al.,Eur. J. Nucl. Med.15, 624 (1989). Diagnostic use in pheochromocytoma: B. Shapiro et al.,J. Nucl. Med.26, 576 (1985); therapeutic use: M. Krempf et al.,J. Clin. Endocrinol. Metab.72, 455 (1991). Symposia on therapeutic and diagnostic use in neuroblastoma: Advances in Neuroblastoma Research2, A. E. Evans et al., Eds. (Alan R. Liss, Inc., New York, 1988) p 643-726; Med. Pediatr. Oncol.15, 157-228 (1987). Review of pharmacology: J. C. Sisson, D. M. Weiland, Am. J. Physiol. Imaging1, 96-103 (1986); of biodistribution and clinical studies: A. R. Wafelman et al.,Eur. J. Nucl. Med.21, 545-559 (1994); of therapeutic use in neural crest tumors: L. Troncone, V. Rufini, Anticancer Res.17, 1823-1832 (1997).
Derivative Type: Sulfate
Molecular Formula: (C8H10IN3)2.H2SO4
Molecular Weight: 648.26
Percent Composition: C 29.64%, H 3.42%, I 39.15%, N 12.96%, S 4.95%, O 9.87%
Properties: Colorless crystals from water + ethanol, mp 166-167°.
Melting point: mp 166-167°
Therap-Cat: Radiolabeled forms as antineoplastic; diagnostic aid (radioactive imaging agent).
Jump up^Scarsbrook AF, Ganeshan A, Statham J, et al. (2007). “Anatomic and functional imaging of metastatic carcinoid tumors”. Radiographics. 27 (2): 455–77. doi:10.1148/rg.272065058. PMID17374863.
Jump up^Kowalsky RJ, Falen, SW. Radiopharmaceuticals in Nuclear Pharmacy and Nuclear Medicine. 2nd ed. Washington DC: American Pharmacists Association; 2004.
Jump up^6-[18FFluorodopamine Positron Emission Tomographic (PET) Scanning for Diagnostic Localization of Pheochromocytoma. Pacek et al. 2001] full text
Jump up^Luster M, Karges W, Zeich K, Pauls S, Verburg FA, Dralle H; et al. (2010). “Clinical value of (18)F-fluorodihydroxyphenylalanine positron emission tomography/computed tomography ((18)F-DOPA PET/CT) for detecting pheochromocytoma”. European journal of nuclear medicine and molecular imaging. 37 (3): 484–93. doi:10.1007/s00259-009-1294-7. PMID19862519.
BIM-44058 is a 34 amino acid analog of native human PTHrP currently in phase III clinical trials at Radius Health for the treatment of postmenopausal osteoporosis. Radius is also developing a microneedle transdermal patch using a 3M drug delivery system in phase II clinical trials. The drug candidate was originally developed at Biomeasure (a subsidiary of Ipsen), and was subsequently licensed to Radius and Teijin Pharma.
Abaloparatide is indicated to treat postmenopausal women with osteoporosis who are more susceptible to bone fractures.[2]
Dosage
The dose recommended is 80mcg subcutaneous injection once a day, administered in the periumbilical area using a prefilled pen device containing 30 doses.[4]
Warnings and Precautions
Preclinical studies revealed that abaloparatide systemic daily administration leads to a dose- and time-dependent increase in the incidence of osteosarcoma in rodents.[5] However, whether abaloparatide-SC will cause osteosarcoma in humans is unknown. Thus, the use of abaloparatide is not recommended for individuals at increased risk of osteosarcoma. Additionally, its use is not advised for more than 2 years during a patient’s lifetime.[4][6]
Side Effects
The most common side effects reported by more than 2% of clinical trials subjects are hypercalciuria, dizziness, nausea, headache, palpitations, fatigue, upper abdominal pain and vertigo.[4]
Pharmacology
Abaloparatide is 34 amino acid synthetic analog of PTHrP. It has 41% homology to parathyroid hormone (PTH) (1-34) and 76% homology to parathyroid hormone-related protein (PTHrP) (1-34).[7] It works as an anabolic agent for the bone, through selective activation of the parathyroid hormone 1 receptor (PTH1R), a G protein-coupled receptor (GPCR) expressed in the osteoblasts and osteocytes. Abaloparatide preferentially binds the RG conformational state of the PTH1R, which in turn elicits a transient downstream cyclic AMP signaling response towards to a more anabolic signaling pathway.[8][9]
History
Preclinical studies
Abaloropatide was previously known as BA058 and BIM-44058 while under development. The anabolic effects of abaloparatide on bone were demonstrated in two preclinical studies conducted in ovarectomized rats. Both studies showed increased cortical and trabecular bone volume and density, and trabecular microarchitecture improvement in vertebral and nonvertebral bones after short-term[10] and long-term[11] daily subcutaneous injection of abaloparatide compared to controls. Recent studies indicated a dose-dependent increased in bone mass and strength in long-term abalorapatide treatment.[12] However, it was also indicated that prolonged abalorapatide-SC treatment leads to increased incidence of osteosarcoma.[5] To date, there is no yet evidence for increased risk of bone tumors due to prolonged abalorapatide systemic administration in humans. Based on this preclinical data, the FDA does not advised the use of abaloparatide-SC for more than 2 years, or in patients with history of Paget disease and/or other conditions that exacerbates the risk of developing osteosarcoma.[4]
Clinical Trials
Phase II trials were initiated in 2008. A 24-week randomized trial was conducted in postmenopausal women with osteoporosis (n=222) assessing bone mass density (BMD) changes as the primary endpoint.[13] Significant BMD increase at doses of 40 and 80 mcg were found in the lumbar spine, femur and hips of abaloparatide-treated participants compared to placebo. Additionally, abaloparatide showed superior anabolic effects on the hips compared to teriparatide.[14]
In the phase III (2011-2014) Abaloparatide Comparator Trial in Vertebral Endpoints (ACTIVE) trial, a 18-months randomized, multicenter, double-blinded, placebo-controlled study evaluated the long-term efficacy of abaloparatide compared to placebo and teriparatide in 2,463 postmenopausal women (± 69 years old).[2] Women who received daily injections of abaloparatide experienced substantial reduction in the incidence of fractures compared to placebo. Additionally, greater BMD increase at 6, 12 and 18 months in spinal, hips and femoral bones was observed in abaloparatide compared to placebo and teriparatide-treated subjects.[3]
Participants who completed 18 months of abaloparatide or placebo in the ACTIVE study were invited to participate in an extended open-labeled study – ACTIVExtend study (2012-2016).[15] Subjects (n=1139) received additional 2 years of 70 mg of alendronate, Vitamin D (400 to 800 IU), and calcium (500–1000 mg) supplementation daily. Combined abaloparatide and alendronate therapy reduced significantly the incidence of vertebral and nonvertebral fractures.[16]
A clinical trial assessing the effectiveness of abaloparatide in altering spinal bone mineral density (BMD) in male subjects is expected to start in the first quarter of 2018. If successful, Radius Health aims to submit a sNDA to expand the use of abaloparatide-SC to treat men with osteoporosis.[17]
In addition to the injectable form of abaloparatide, a transdermal patch is also in development.[1]
Commercialization
As previously noted, abaloparatide-SC is manufactured by Radius Health, Inc. (Nasdaq: RDUS), a biomedical company based in Waltham, Massachusetts. This company is focused on the development of new therapeutics for osteoporosis, cancer and endocrine diseases. Abaloparatide is the only drug currently marketed by Radius Health. RDUS reported that sales for abaloparatide were $3.5million for the third quarter of 2017.[17] The company announced a net loss of $57.8 million, or $1.31 per share for the third quarter of 2017, compared to $19.2 million for the same quarter of 2016.[18] The net loss most likely reflects the substantial expenses associated with the preparation and launching of abaloparatide into the US market in May 2017.
In July 2017, Radius Health licensed rights to Teijin Limited for abaloparatide-SC manufacture and commercialization in Japan. Teijin is developing abaloparatide-SC under agreement with Ipsen Pharma S.A.S., and is conducting a phase III clinical trial in Japanese patients with osteoporosis.[19]
Regulatory Information
Radius Health filed a Marketing Authorization Application (MAA) in November 2015,[20] which was validated in December, 2015, and still under regulatory assessment by the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA). As in July 2017, the CHMP issued a second Day-180 List of Outstanding Issues, which Radius is addressing with the CHMP.[17]
In February 2016 a NDA was filed to the FDA, Radius NDA for abaloparatide-SC was accepted in May, 2016.[21] A Prescription Drug User Fee Act (PDUFA) date was initially granted in March 30, 2016, but then extended to June 30, 2017.[22] As previously stated, abaloparatide injection was approved for use in postmenopausal osteoporosis on April 28, 2017.[6]
Intellectual Property
Radius Health currently holds three patents on abaloparatide-SC, with expiration dates from 2027-2028.[23] The patents relate to the drug composition (US 8148333), and the drug delivery methods (US 7803770 B2 and US 8748382-B2).
As previously mentioned, Teijin Limited was granted use of Radius Health intellectual property in July 2017, for the development, manufacture and commercialization of abaloparatide-sc in Japan.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











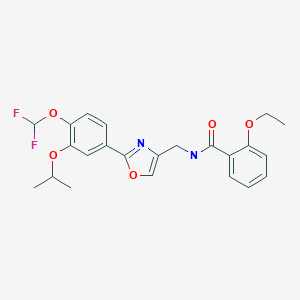

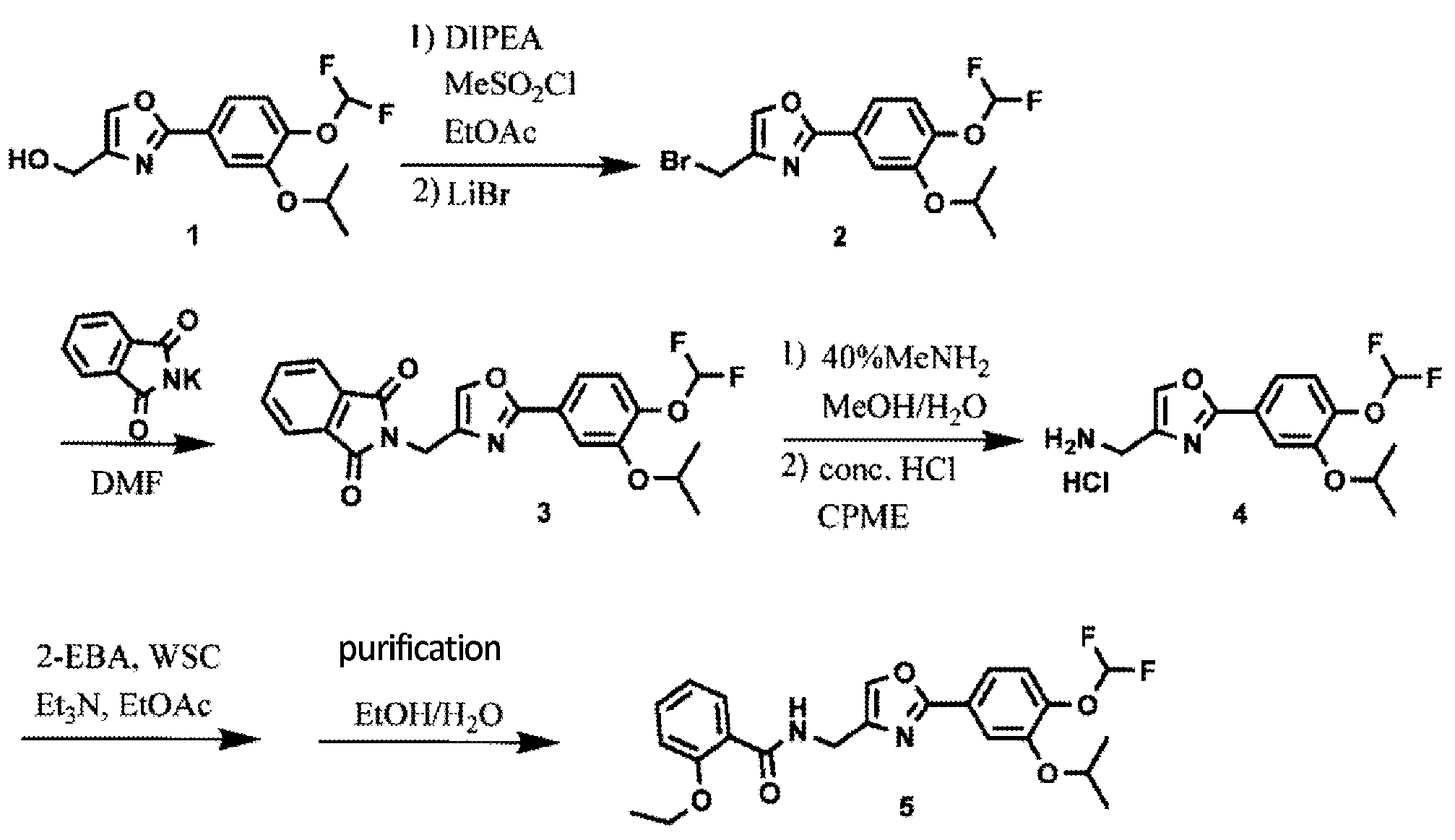
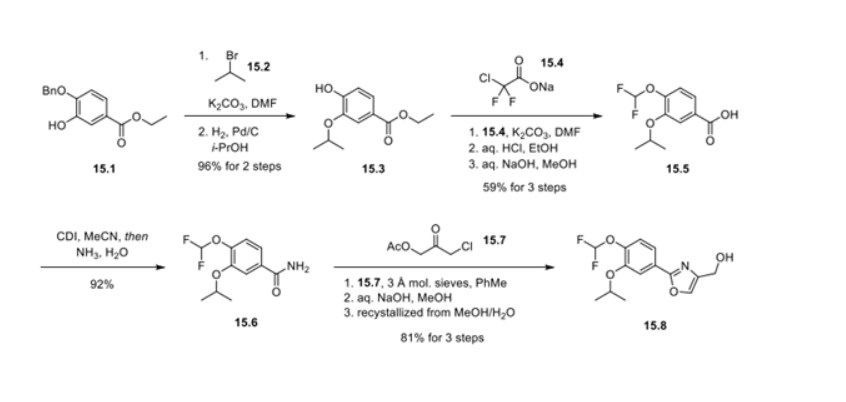
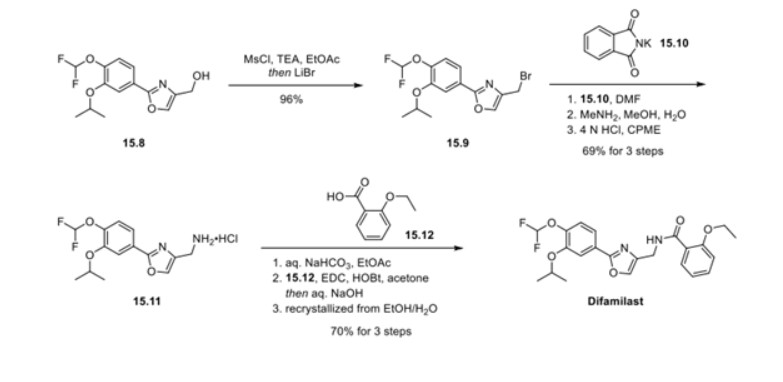



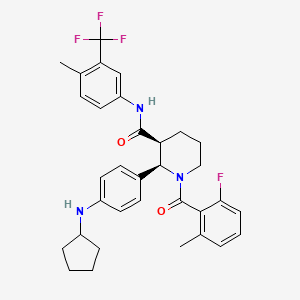

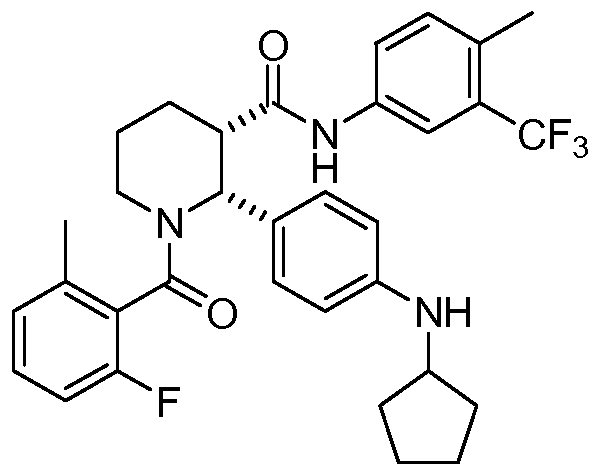

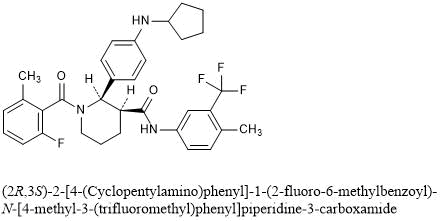





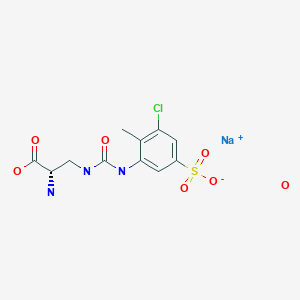

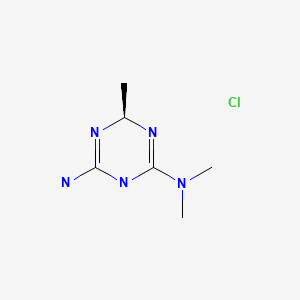




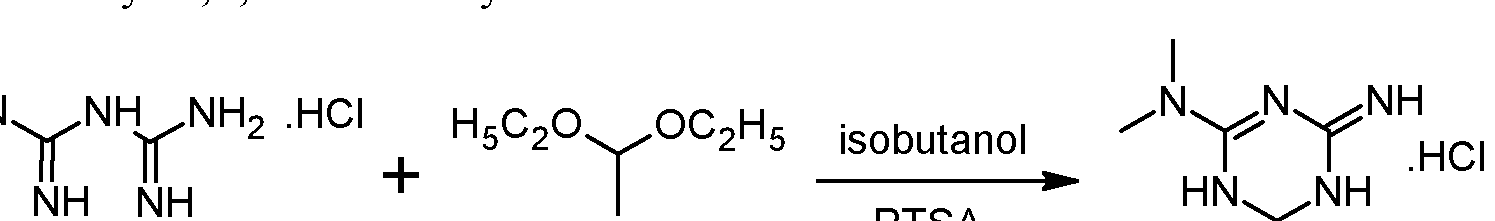


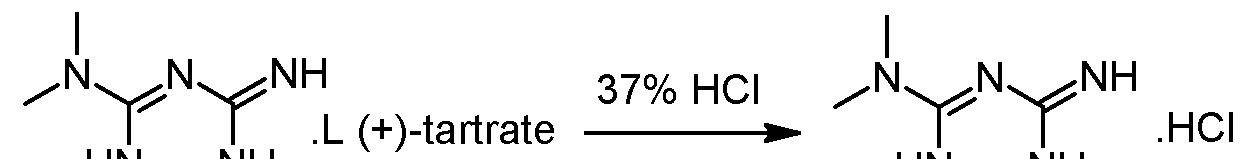
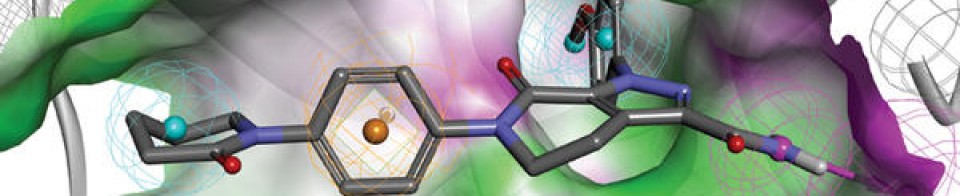

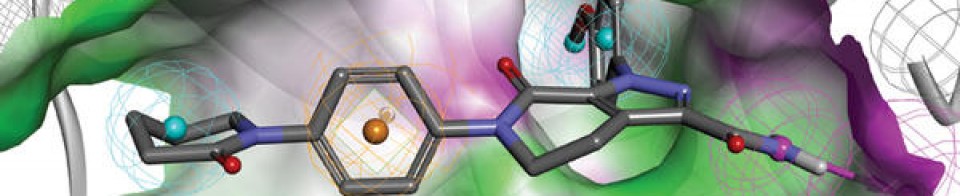



![Chemical structure of N-[2-[[2-[2-[(2,6-dichlorophenyl)amino]phenyl]acetyl]oxy]ethyl]hyaluronamide (diclofenac etalhyaluronate, SI-613)](https://www.researchgate.net/publication/325304331/figure/fig1/AS:669091362766859@1536535220444/Chemical-structure-of.png)

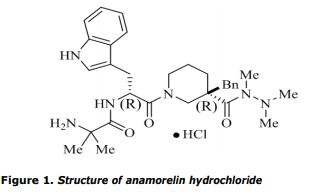

































{kind=link}
{kind=link}
{kind=link}
{kind=link}