
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
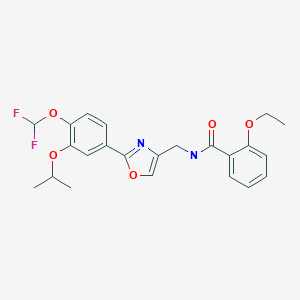
Difamilast
FDA 2026, APPROVALS 2026, difamilast, Adquey, 2/12/2026, To treat mild to moderate atopic dermatitis
PMDA Moizerto, JAPAN APPROVED 2021/9/27
ジファミラスト
ディファミラスト;
地法米司特
N-({2-[4-(difluoromethoxy)-3-(propan-2-yloxy)phenyl]-1,3- oxazol-4-yl}methyl)-2-ethoxybenzamide
OPA-15406
| Formula |
C23H24F2N2O5
|
|---|---|
| CAS |
937782-05-3
|
| Mol weight |
446.4439
|
MM 36; MM-36-Medimetriks-Pharmaceuticals; Moizerto; OPA-15406
| Efficacy |
Anti-inflammatory, Phosphodiesterase IV inhibitor
|
|---|---|
| Comment |
Treatment of atopic dermatitis
|
OriginatorOtsuka Pharmaceutical Development & Commercialization- DeveloperMedimetriks Pharmaceuticals; Otsuka Pharmaceutical Development & Commercialization
- ClassBenzamides; Nonsteroidal anti-inflammatories; Oxazoles; Skin disorder therapies
- Mechanism of ActionType 4 cyclic nucleotide phosphodiesterase inhibitors
- RegisteredAtopic dermatitis
- 27 Sep 2021Registered for Atopic dermatitis (In adolescents, In children, In adults) in Japan (Topical)
- 11 Nov 2020Otsuka Pharmaceutical completes a phase III trial in Atopic dermatitis (In children, In adolescents, In adults) in Japan (Topical) (NCT03961529)
- 28 Sep 2020Preregistration for Atopic dermatitis in Japan (In children, In adolescents, In adults) (Topical)

Difamilast is under investigation in clinical trial NCT01702181 (A Safety Study to Evaluate the Use and Effectiveness of a Topical Ointment to Treat Adults With Atopic Dermatitis).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019194211&_cid=P10-MLOK7B-25338-2
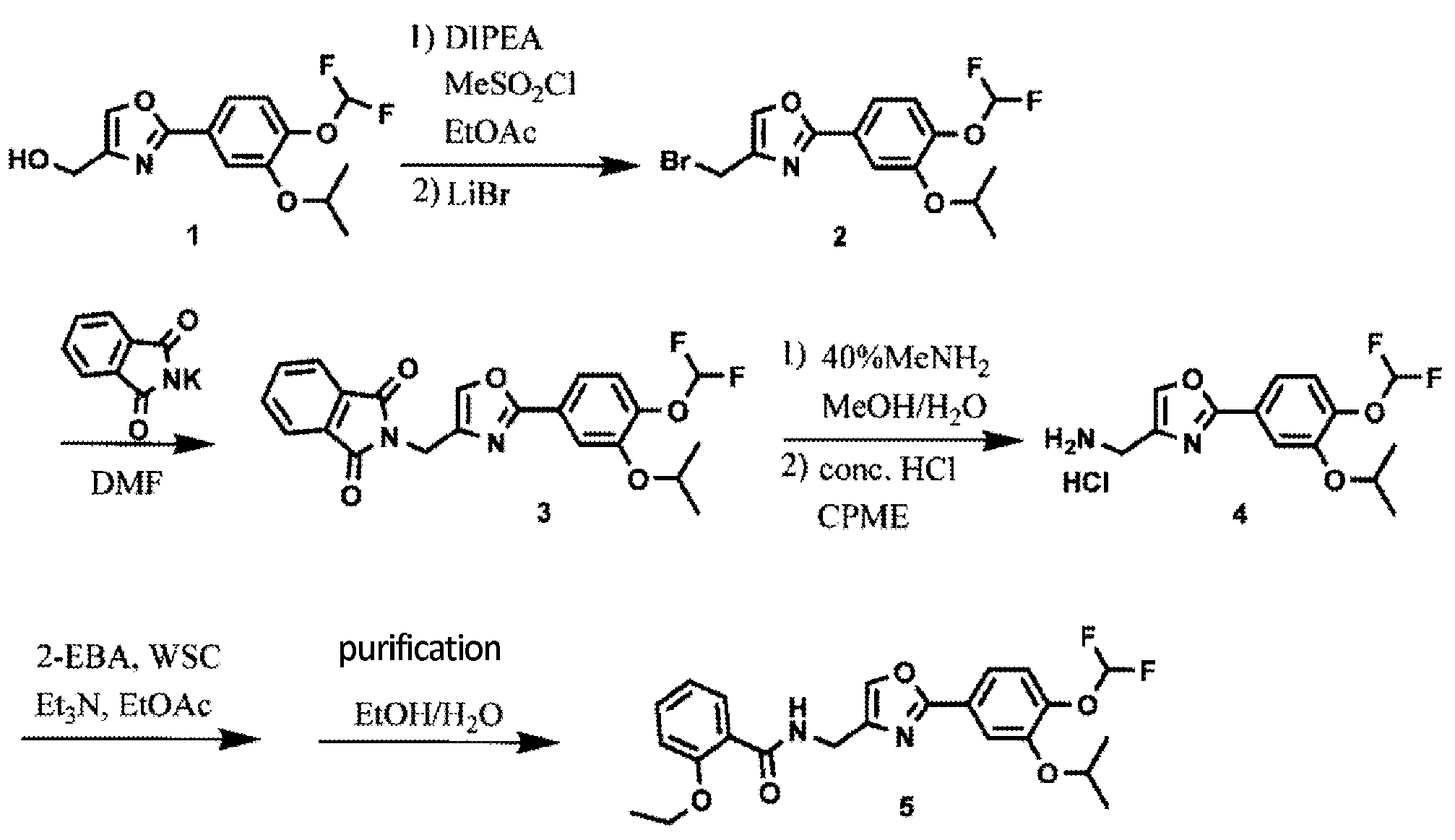
Synthesis of Oxazole Compound (Type A Crystal)
Compound (5) (white powder) was prepared in accordance with the method disclosed in Example 352 of PTL 1 (WO2007/058338).
SYN
https://www.chemicalbook.com/article/how-is-difamilast-synthesised.htm
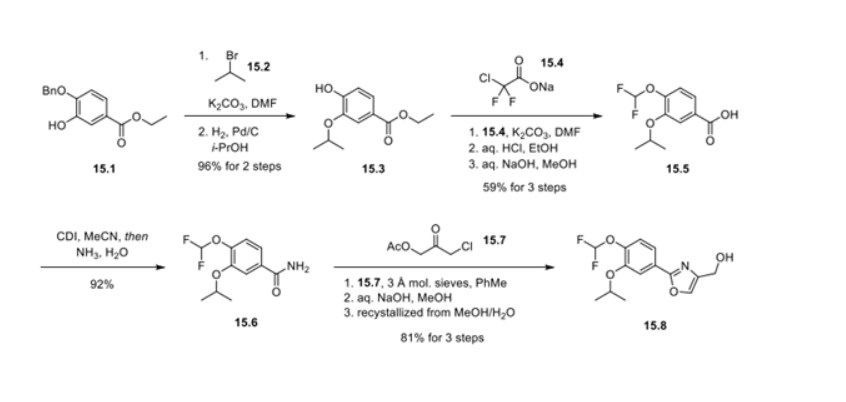
Synthesis of difamilast commenced with the monobenzylated protocatechuic acid ethyl ester 15.1. Phenol 15.1 was first converted into the corresponding isopropyl ether, which was subsequently debenzylated under palladium-catalyzed hydrogenation conditions to generate the phenolic intermediate 15.3. Difluoromethylation of 15.3 was accomplished by introducing sodium chlorodifluoroacetate 15.4 in the presence of potassium carbonate at an elevated temperature. The decarboxylative C− O bond-forming reaction presumably proceeded via a difluorocarbene species. The difluoromethylated product was treated with acid followed by ester hydrolysis under a basic medium to furnish benzoic acid derivative 15.5. Benzoic acid 15.5 was subsequently transformed into benzamide 15.6 via a benzoyl imidazole intermediate. Condensation of benzamide 15.6 with 1-acetoxy-3-chloroacetone 15.7 produced an oxazole derivative, which was subsequently saponified and recrystallized from 50% aqueous MeOH to generate alcohol 15.8.
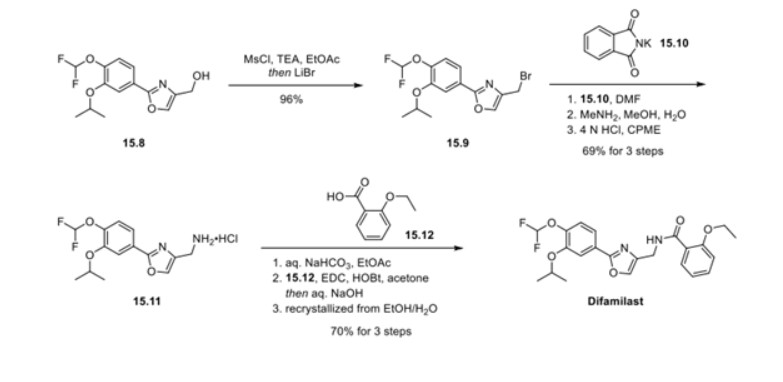
First, an activation−displacement process transformed alcohol 15.8 into bromide 15.9 via a mesylate intermediate. Alkyl bromide 15.9 was then treated with potassium phthalimide to incorporate the nitrogen center via an SN2-type displacement. Methylamine-mediated phthalimide deprotection and subsequent salt formation produced amine 15.11 as a hydrochloride salt in 69% yield over 3 steps. Finally, hydrochloride salt 15.11 was treated with aqueous sodium bicarbonate to generate a free amine, which was subjected to amide bond formation with 2-ethoxybenzoic acid 15.12 to deliver difamilast after recrystallization from aqueous EtOH.
PATENT
JP 2021059538
https://patentscope.wipo.int/search/en/detail.jsf?docId=JP322244172&_cid=P20-L1WXG6-04592-1
patcit 2 : International Publication No. 2014/034958 (Japanese Publication No. 2015-528433 )
patcit 3 : International Publication No. 2017/115780
Compound (5) (white powder) was prepared by the method described in Example 352 of Patent Document 1 (International Publication No. 2007/088383).
N−({2−[4−(difluoromethoxy)−3−isopropoxyphenyl]oxazol−4−yl}methyl)−2−ethoxybenzamide
: white powder.
1H NMR (400 MHz, CDCl3): δ = 8.56 (br s,
1H, NH), 8.23 (dd, J = 7.6 Hz, 1.6 Hz, 1H, ArH), 7.66 (s, 1H, ArH), 7.63 (d, J = 2.0 Hz, 1H, ArH), 7.58 (dd, J = 8.4 Hz, 2.0 Hz, 1H, ArH), 7.44−7.39 (m, 1H, ArH), 7.21 (d, J = 8.0 Hz, 1H, ArH), 7.08−7.04 (m, 1H, ArH), 6.94 (d, J = 8.0 Hz, 1H, ArH), 6.61 (t, J = 75.2 Hz, 1H, CHF 2), 4.68 (sept, J = 6.0 Hz, 1H, CH), 4.62
(d, J = 6.0 Hz, 2H, CH 2), 4.17 (q, J = 6.93, 2H, CH 2), 1.48 (t, J = 7.2 Hz, 3H,
CH 3), 1.39 (d, J = 5.6 Hz, 6H, 2CH 3).
Using the obtained B-type crystal as a seed crystal, it was examined to further prepare a B-type crystal. Specifically,
B-type crystals were prepared as follows according to the method described in Patent Document 3 (International Publication No. 2017/115780).
aqueous sodium hydroxide solution were added to the organic layer, the temperature was adjusted again to 40 to 50 ° C., the liquid was separated, and the organic layer was concentrated under reduced pressure. 50 mL of ethanol, 20 mL of water, 6 mL of a 25% aqueous sodium hydroxide solution, and 0.6 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. Activated carbon was removed by filtration, washed with 12 mL of ethanol, the filtrate was cooled, and 10 mg of B-type crystals (seed crystals) were added to precipitate crystals. Precipitated crystals were collected by filtration and dried at 60 ° C. to obtain 18.38 g (yield 88.18%) of crystals of compound (5).
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007058338&_cid=P10-MLOK7P-25609-1
Example 352
Using the compound obtained in Example 347 and 2-bromopropane, white powdery N-[2-(4-difluoromethoxy-3-isopropoxyphenyl)oxazol-4-ylmethyl]-2-ethoxybenzamide was obtained following the procedure of Example 348.
1H-NMR (CDCl3) δ: 8.57 (1H, br s), 8.24 (1H, dd, J = 7.5, 1.8 Hz), 7.67 (1H, s), 7.65-7.57 (2H, m), 7.46-7.40 (1H, m), 7.26-7.21 (1H, m), 7.08 (1H, t, J = 7.5 Hz), 6.95 (1H, d, J = 8.4 Hz), 6.63 (1H, t, J = 75 Hz), 4.74-4.62 (3H, m), 4.19 (2H, q, J = 6.9 Hz), 1.49 (3H, t, J = 6.9 Hz), 1.40 (6H, d, J = 6.3 Hz)
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017115780
Compound (3) was produced in accordance with the following reaction scheme.
1H-NMR (CDCl 3) δ: 7.70 (2H,dd,J = 6.4 Hz,2.0 Hz),7.22 (1H,d,J = 9.2 Hz),6.66 (1H,t,J = 74.8 Hz),4.66(1H,sept,J = 6.0 Hz),1.39 (6H,d,J = 6.0 Hz).
Production Example 2: Production 2 of Compound (3)
Compound (3) was produced in accordance with the following reaction scheme.
Compound (7) was produced in accordance with the following reaction scheme.
Compound (11) was produced in accordance with the following reaction scheme.
20.00 g (66.8 mmol) of compound (7) and 17.28 g (134 mmol) of N,N-diisopropylethylamine were added to 300 ml of ethyl acetate, and the mixture was cooled. 11.48 g (100 mmol) of methanesulfonyl chloride was poured in and stirred at 10 to 30°C for 1 hour. 17.41 g (200 mmol) of lithium bromide was added thereto and reacted at 20 to 35°C for 1 hour. 100 ml of water was added to the reaction solution, and the mixture was partitioned, followed by concentration of the organic layer under reduced pressure. 300 ml of ethyl acetate was added to the concentrated residue to dissolve the residue, and the solution was again concentrated under reduced pressure. 200 ml of N,N-dimethylformamide and 17.33 g (93.6 mmol) of potassium phthalimide were added to the concentrated residue and reacted at 75 to 85°C for 1 hour. 200 ml of water was added to the reaction solution to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 25.90 g (yield: 90.5%) of compound (9) as a white powder.
15.00 g (35.0 mmol) of compound (9) was mixed with 30 ml of a 40% methylamine aqueous solution, 30 ml of methanol, and 75 ml of water, and reacted under reflux for 30 minutes. 150 ml of cyclopentyl methyl ether (CPME) and 15 ml of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the temperature was adjusted to 65 to 75°C, followed by partitioning. A mixture of 150 ml of water and 7.50 g of sodium chloride was added to the organic layer, and the temperature was adjusted to 65 to 75°C again, followed by partitioning. 3.75 ml of concentrated hydrochloric acid was added to the organic layer to precipitate crystals. The precipitated crystals were collected by filtration and dried at 60°C, thereby obtaining 11.95 g (yield: quant.) of compound (10) as a white powder.
13.30 g (39.7 mmol) of compound (10) was mixed with 3.83 g (37.8 mmol) of triethylamine and 108 ml of ethyl acetate, and stirred at 20 to 30°C for 1 hour. 9.78 g (58.9 mmol) of 2-ethoxybenzoic acid and 11.28 g (58.8 mmol) of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (WSC) were added to the reaction solution, and reacted at 20 to 30°C for 1 hour. 54 ml of water and 5.4 ml of concentrated hydrochloric acid were added to the reaction solution, and the temperature was adjusted to 40 to 50°C, followed by partitioning. 54 ml of water and 5.4 ml of a 25% sodium hydroxide aqueous solution were added to the organic layer, and the temperature was adjusted to 40 to 50°C again. The mixture was partitioned, and the organic layer was concentrated under reduced pressure. 45 ml of ethanol, 18 ml of water, 5.4 ml of a 25% sodium hydroxide aqueous solution, and 0.54 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. The activated carbon was removed by filtration, and the filtrate was washed with 11 ml of ethanol. The filtrate was cooled, and a seed crystal was added thereto to precipitate crystals. The precipitated crystals were collected by filtration and dried at 35°C, thereby obtaining 12.88 g (72.6%) of compound (11) as a white powder.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019194211
*DIPEA: Diisopropylethylamine, CPME: Cyclopentyl methyl ether,
DMF: N,N-dimethylformamide, 2-EBA: 2-Ethoxybenzoic acid,
WSC: 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
Analysis was conducted to further prepare the type B crystal using the obtained type B crystal as a seed crystal. More specifically, the type B crystal was prepared as follows, in accordance with the method disclosed in PTL 3 (WO2017/115780).
PATENT
WO2014034958A1
WO2007058338A2
WO2007058338A9
WO2007058338A3
US9181205B2
US2015239855A1
USRE46792E
US2020078340A1
US2017216260A1
US2019070151A1
US2009221586A1
US8637559B2
US2014100226A1
///////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
/////////////Difamilast, JAPAN 2021, APPROVALS 2021, ジファミラスト , MM 36, MM-36-Medimetriks-Pharmaceuticals, Moizerto, OPA-15406, OPA 15406, 地法米司特
O=C(NCC1=COC(C2=CC=C(OC(F)F)C(OC(C)C)=C2)=N1)C3=CC=CC=C3OCC
INTrmediate No.CAS No.DIFAM-001177429-27-5DIFAM-00293652-48-3DIFAM-0031574285-26-9DIFAM-00470-23-5DIFAM-0051574285-28-1DIFAM-0061574285-30-5DIFAM-0071574285-32-7DIFAM-0081574285-36-1DIFAM-0091574285-38-3DIFAM-010DIFAM-0111574285-40-7DIFAM-0121574285-43-0DIFAM-013134-11-2Difamilast937782-05-3

