
Home » INDIA 2022
Category Archives: INDIA 2022
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
OTERACIL POTTASIUM

OTERACIL
UNII4R7FFA00RX, CAS Number2207-75-2, WeightAverage: 195.175, Monoisotopic: 194.96823705, Chemical FormulaC4H2KN3O4
[K+].OC1=NC(=NC(=O)N1)C([O-])=O
1,3,5-Triazine-2-carboxylic acid, 1,4,5,6-tetrahydro-4,6-dioxo-, potassium salt (1:1)
218-627-5[EINECS]
2207-75-2[RN]
4,6-Dihydroxy-1,3,5-triazine-2-carboxylic acid potassium salt
- KOX
- NSC 28841
- Oxonate
- Oxonate, potassium
CDSCO APPROVED,01.02.2022

Gimeracil bulk & Oteracil potassium bulk and Tegafur 15mg/20mg, Gimeracil 4.35mg/5.8mg and Oteracil 11.8mg/15.8mg capsules
indicated in adults for the treatment of advanced gastric cancer when given in combination with cisplatin.
Oteracil Potassium is the potassium salt of oxonate, an enzyme inhibitor that modulates 5- fluorouracil (5-FU) toxicity. Potassium oxonate inhibits orotate phosphoribosyltransferase, which catalyzes the conversion of 5-FU to its active or phosphorylated form, FUMP. Upon oral administration, Oxonate is selectively distributed to the intracellular sites of tissues lining the small intestines, producing localized inhibitory effects within the gastrointestinal tract. As a result, 5-FU associated gastrointestinal toxic effects are reduced and the incidence of diarrhea or mucositis is decreased in 5-FU related therapy.
Oteracil is an adjunct to antineoplastic therapy, used to reduce the toxic side effects associated with chemotherapy. Approved by the European Medicines Agency (EMA) in March 2011, Oteracil is available in combination with Gimeracil and Tegafur within the commercially available product “Teysuno”. The main active ingredient in Teysuno is Tegafur, a pro-drug of Fluorouracil (5-FU), which is a cytotoxic anti-metabolite drug that acts on rapidly dividing cancer cells. By mimicking a class of compounds called “pyrimidines” that are essential components of RNA and DNA, 5-FU is able to insert itself into strands of DNA and RNA, thereby halting the replication process necessary for continued cancer growth.
Oteracil’s main role within Teysuno is to reduce the activity of 5-FU within normal gastrointestinal mucosa, and therefore reduce’s gastrointestinal toxicity 1. It functions by blocking the enzyme orotate phosphoribosyltransferase (OPRT), which is involved in the production of 5-FU.
/////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
SYNTHESIS
https://patents.google.com/patent/CN103435566A/zh


SYN
https://europepmc.org/article/pmc/pmc7717319
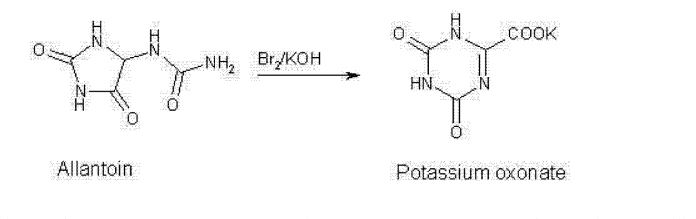
Poje et al. reported a two-step, gram-scale preparation of the TS-1 additive oteracil 21 (Scheme 16).226 Iodine-mediated-oxidation of uric acid 116 produced dehydroallantoin 117 as the major product, and subsequent treatment with potassium hydroxide resulted in the rearranged product oteracil 21.227

Synthesis of Oteracil 21a
aReagents and conditions: (a) LiOH, I2, H2O, 5 °C, 5 min, then AcOH, 75%; (b) aq KOH, 20 min, rt, 82%.
(226) Poje M; Sokolić-Maravić L The mechanism for the conversion of uric acid into allantoin and dehydro-allantoin: A new look at an old problem. Tetrahedron 1986, 42 (2), 747–751. [Google Scholar]
(227) Sugi M; Igi M EP Patent 0957096, 1999.
EP0957096A1 *1998-05-111999-11-17SUMIKA FINE CHEMICALS Co., Ltd.Method for producing potassium oxonate
CN101475539A *2009-02-112009-07-08鲁南制药集团股份有限公司Refining method for preparing high-purity oteracil potassium
CN102250025A *2011-05-182011-11-23深圳万乐药业有限公司Preparation method suitable for industrially producing oteracil potassium
CN102746244A *2012-07-272012-10-24南京正大天晴制药有限公司Refining method of oteracil potassium
//////////OTERACIL POTTASIUM, KOX, NSC 28841, Oxonate, Oxonate potassium, INDIA 2022, APPROVALS 2022, CANCER
[K+].OC1=NC(=NC(=O)N1)C([O-])=O

NEW DRUG APPROVALS
ONE TIME
$10.00
GIMERACIL

GIMERACIL
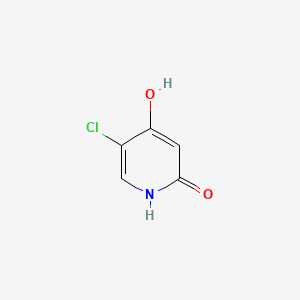
C5H4ClNO2, 145.54
5-chloro-4-hydroxy-1H-pyridin-2-one
5-Chloro-2,4-dihydroxypyridine
5-Chloro-4-hydroxy-2(1H)-pyridone
CDSCO APPROVED,01.02.2022
Gimeracil bulk & Oteracil potassium bulk and Tegafur 15mg/20mg, Gimeracil 4.35mg/5.8mg and Oteracil 11.8mg/15.8mg capsules
indicated in adults for the treatment of advanced gastric cancer when given in combination with cisplatin.
| Combination of | |
|---|---|
| Tegafur | Antineoplastic drug |
| Gimeracil | Enzyme inhibitor |
| Oteracil | Enzyme inhibitor |
| Clinical data | |
| Trade names | Teysuno, TS-1 |
| Other names | S-1[1] |
| AHFS/Drugs.com | UK Drug Information |
| License data | EU EMA: by Tegafur |
| Pregnancy category | Contraindicated |
| Routes of administration | By mouth |
| ATC code | L01BC53 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only) [2]EU: Rx-only [3]In general: ℞ (Prescription only) |
| Identifiers | |
| CAS Number | 150863-82-4 |
| PubChem CID | 54715158 |
Tegafur/gimeracil/oteracil, sold under the brand names Teysuno and TS-1,[3][4] is a fixed-dose combination medication used for the treatment of advanced gastric cancer when used in combination with cisplatin,[3] and also for the treatment of head and neck cancer, colorectal cancer, non–small-cell lung, breast, pancreatic, and biliary tract cancers.[5]: 213
The most common severe side effects when used in combination with cisplatin include neutropenia (low levels of neutrophils, a type of white blood cell), anaemia (low red blood cell counts) and fatigue (tiredness).[3]
Tegafur/gimeracil/oteracil (Teysuno) was approved for medical use in the European Union in March 2011.[3] It has not been approved by the U.S. Food and Drug Administration (FDA).[5]: 213
Medical uses
In the European Union tegafur/gimeracil/oteracil is indicated in adults for the treatment of advanced gastric cancer when given in combination with cisplatin.[3]
Contraindications
In the European Union, tegafur/gimeracil/oteracil must not be used in the following groups:
- people receiving another fluoropyrimidine (a group of anticancer medicines that includes tegafur/gimeracil/oteracil) or who have had severe and unexpected reactions to fluoropyrimidine therapy;[3]
- people known to have no DPD enzyme activity, as well as people who, within the previous four weeks, have been treated with a medicine that blocks this enzyme;[3]
- pregnant or breastfeeding women;[3]
- people with severe leucopenia, neutropenia, or thrombocytopenia (low levels of white cells or platelets in the blood);[3]
- people with severe kidney problems requiring dialysis;[3]
- people who should not be receiving cisplatin.[3]
Mechanism of action
Tegafur is the actual chemotherapeutic agent. It is a prodrug of the active substance fluorouracil (5-FU).[3] Tegafur, is a cytotoxic medicine (a medicine that kills rapidly dividing cells, such as cancer cells) that belongs to the ‘anti-metabolites’ group. Tegafur is converted to the medicine fluorouracil in the body, but more is converted in tumor cells than in normal tissues.[3] Fluorouracil is very similar to pyrimidine.[3] Pyrimidine is part of the genetic material of cells (DNA and RNA).[3] In the body, fluorouracil takes the place of pyrimidine and interferes with the enzymes involved in making new DNA.[3] As a result, it prevents the growth of tumor cells and eventually kills them.[3]
Gimeracil inhibits the degradation of fluorouracil by reversibly blocking the dehydrogenase enzyme dihydropyrimidine dehydrogenase (DPD). This results in higher 5-FU levels and a prolonged half-life of the substance.[6]
Oteracil mainly stays in the gut because of its low permeability, where it reduces the production of 5-FU by blocking the enzyme orotate phosphoribosyltransferase. Lower 5-FU levels in the gut result in a lower gastrointestinal toxicity.[6]
Within the medication, the molar ratio of the three components (tegafur:gimeracil:oteracil) is 1:1:0.4.[7]
The maximum tolerated dose differed between Asian and Caucasian populations (80 mg/m2 and 25 mg/m2 respectively), perhaps due to differences in CYP2A6 genotype.[5]: 213
Research
It is being developed for the treatment of hepatocellular carcinoma.[8] and has activity in esophageal,(Perry Chapter 33) breast,[citation needed] cervical,[citation needed] and colorectal cancer.[9]
References
- ^ Liu TW, Chen LT (201). “S-1 with leucovorin for gastric cancer: how far can it go?”. Lancet Oncol. 17 (1): 12–4. doi:10.1016/S1470-2045(15)00478-7. PMID 26640038.
- ^ “Teysuno 20mg/5.8mg/15.8mg hard capsules – Summary of Product Characteristics (SmPC)”. (emc). Retrieved 30 July 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r “Teysuno EPAR”. European Medicines Agency (EMA). Retrieved 30 July 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “ティーエスワン 患者さん・ご家族向け総合情報サイト | 大鵬薬品工業株式会社”.
- ^ Jump up to:a b c DeVita, DeVita; Lawrence, TS; Rosenberg, SA (2015). DeVita, Hellman, and Rosenberg’s Cancer: Principles and Practice of Oncology (10th ed.). LWW. ISBN 978-1451192940.
- ^ Jump up to:a b A. Klement (22 July 2013). “Dreier-Kombination gegen Magenkrebs: Teysuno”. Österreichische Apothekerzeitung (in German) (15/2013): 23.
- ^ Peters GJ, Noordhuis P, Van Kuilenburg AB et al. (2003). “Pharmacokinetics of S-1, an oral formulation of ftorafur, oxonic acid and 5-chloro-2,4-dihydroxypyridine (molar ratio 1:0.4:1) in patients with solid tumors”. Cancer Chemother. Pharmacol. 52 (1): 1–12. doi:10.1007/s00280-003-0617-9. PMID 12739060. S2CID 10858817.
- ^ “BCIQ”.
- ^ Miyamoto Y, Sakamoto Y, Yoshida N, Baba H (2014). “Efficacy of S-1 in colorectal cancer”. Expert Opin Pharmacother. 15 (12): 1761–70. doi:10.1517/14656566.2014.937706. PMID 25032886. S2CID 23637808.
External links
- “Tegafur”. Drug Information Portal. U.S. National Library of Medicine.
- “Gimeracil”. Drug Information Portal. U.S. National Library of Medicine.
- “Oteracil”. Drug Information Portal. U.S. National Library of Medicine.
Gimeracil is an adjunct to antineoplastic therapy, used to increase the concentration and effect of the main active componets within chemotherapy regimens. Approved by the European Medicines Agency (EMA) in March 2011, Gimeracil is available in combination with Oteracil and Tegafur within the commercially available product “Teysuno”. The main active ingredient in Teysuno is Tegafur, a pro-drug of Fluorouracil (5-FU), which is a cytotoxic anti-metabolite drug that acts on rapidly dividing cancer cells. By mimicking a class of compounds called “pyrimidines” that are essential components of RNA and DNA, 5-FU is able to insert itself into strands of DNA and RNA, thereby halting the replication process necessary for continued cancer growth.
Gimeracil’s main role within Teysuno is to prevent the breakdown of Fluorouracil (5-FU), which helps to maintin high enough concentrations for sustained effect against cancer cells 2. It functions by reversibly and selectively blocking the enzyme dihydropyrimidine dehydrogenase (DPD), which is involved in the degradation of 5-FU 1. This allows higher concentrations of 5-FU to be achieved with a lower dose of tegafur, thereby also reducing toxic side effects.
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////


SYNTHESIS

SYN
https://europepmc.org/article/pmc/pmc7717319
Synthesis of Gimeracil 20a
aReagents and conditions: (a) CH3C(OCH3)3, MeOH, then (CH3)2NHCH(OCH3)2, reflux, 92%; (b) aq AcOH, 130 °C, 2 h, 95%; (c) SO2Cl2, HOAc, 50 °C, 0.5 h, 91%; (d) 40% H2SO4, 130 °C, 4 h, 91%; (e) SO2Cl2, HOAc, 50 °C, 45 min, 86%; (f) 75% H2 SO4, 140 °C, 3 h, then NaOH, then pH 4–4.5, 89%


In 1953, Kolder and Hertog reported a synthesis of the TS-1 additive gimeracil 20, which was completed in seven steps using 4-nitropyridine N-oxide as starting material.222 Later, Yano et al. reported an alternative gram-scale synthesis (Scheme 15).223 The one-pot, three component condensation of malononitrile 111, 1,1,1-trimethoxyethane, and 1,1-dimethyoxytrimethylamine generated the dicyano intermediate 112, which was into 2(1H)-pyridinone 113.224 Selective chlorination of 113 was followed by acid-mediated demethylation, hydrolysis, and decarboxylation, to afford gimeracil 20. Interestingly, Xu et al. found that treatment of intermediate 113 with sulfuryl chloride resulted in dichloro 115 formation, which could still be converted to gimeracil 20 by treatment with sulfuric acid.225
(222) Kolder CR; den Hertog HJ Synthesis and reactivity of 5-chloro-2,4-dihydroxypyridine. Rec. Trav. Chim 1953, 72, 285–295. [Google Scholar]
(223) Yano S; Ohno T; Ogawa K Convenient and practical synthesis of 5-chloro-4-hydroxy-2(1H)-pyridinone. Heterocycles 1993, 36, 145–148. [Google Scholar]
(224) Mittelbach M; Kastner G; Junek H Synthesen mit Nitrilen, 71. Mitt. Zur Synthese von 4-Hydroxynicotinsaure aus Butadiendicarbonitrilen. Arch. Pharm 1985, 318 (6), 481–486. [Google Scholar]
(225) Xu Y; Mao D; Zhang F CN Patent 1915976, 2007.

NEW DRUG APPROVALS
THIS MAY NOT RUN WITHOUT SUBSCRIPTION HELP. AVOID CLOSURE OF THIS BLOG
$10.00
//////////GIMERACIL, APPROVALS 2022, INDIA 2022
OC1=CC(=O)NC=C1Cl
Desidustat


Ranjit Desai
DESIDUSTAT
2-(1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxamido)acetic acid
desidustat
Glycine, N-((1-(cyclopropylmethoxy)-1,2-dihydro-4-hydroxy-2-oxo-3-quinolinyl)carbonyl)-
N-(1-(Cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycine
ZYAN1 compound
(1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl) glycine in 98% yield, as a solid. MS (ESI-MS): m/z 333.05 (M+H) +. 1H NMR (DMSO-d 6): 0.44-0.38 (m, 2H), 0.62-0.53 (m, 2H), 1.34-1.24 (m, 1H), 4.06-4.04 (d, 2H), 4.14-4.13 (d, 2H), 7.43-7.39 (t, 1H), 7.72-7.70 (d, 1H), 7.89-7.85 (m, 1H), 8.11-8.09 (dd, 1H), 10.27-10.24 (t, 1H), 12.97 (bs, 1H), 16.99 (s, 1H). HPLC Purity: 99.85%
Oxemia (Desidustat) has received approval from the Drug Controller General of India. This was an incredible team effort by Zydans across the organization and I am so proud of what we have accomplished. Oxemia is a breakthrough treatment for Anemia associated with Chronic Kidney Disease in Patients either on Dialysis or Not on Dialysis, and will help improve quality of life for CKD patients. Team #zydus , on to our next effort!

Desidustat (INN, also known as ZYAN1) is a drug for the treatment of anemia of chronic kidney disease. This drug with the brand name Oxemia is discovered and developed by Zydus Life Sciences.[1] The subject expert committee of CDSCO has recommended the grant of permission for manufacturing and marketing of Desidustat 25 mg and 50 mg tablets in India,based on some conditions related to package insert, phase 4 protocols, prescription details, and GCP.[2] Clinical trials on desidustat have been done in India and Australia.[3] In a Phase 2, randomized, double-blind, 6-week, placebo-controlled, dose-ranging, safety and efficacy study, a mean hemoglobin increase of 1.57, 2.22, and 2.92 g/dL in desidustat 100, 150, and 200 mg arms, respectively, was observed.[4] The Phase 3 clinical trials were conducted at additional lower doses as of 2019.[5] Desidustat is developed for the treatment of anemia as an oral tablet, where currently injections of erythropoietin and its analogues are drugs of choice. Desidustat is a HIF prolyl-hydroxylase inhibitor. In preclinical studies, effects of desidustat was assessed in normal and nephrectomized rats, and in chemotherapy-induced anemia. Desidustat demonstrated hematinic potential by combined effects on endogenous erythropoietin release and efficient iron utilization.[6][7] Desidustat can also be useful in treatment of anemia of inflammation since it causes efficient erythropoiesis and hepcidin downregulation.[8] In January 2020, Zydus entered into licensing agreement with China Medical System (CMS) Holdings for development and commercialization of desidustat in Greater China. Under the license agreement, CMS will pay Zydus an initial upfront payment, regulatory milestones, sales milestones and royalties on net sales of the product. CMS will be responsible for development, registration and commercialization of desidustat in Greater China.[9] It has been observed that desidustat protects against acute and chronic kidney injury by reducing inflammatory cytokines like IL-6 and oxidative stress [10] A clinical trial to evaluate the efficacy and safety of desidustat tablet for the management of Covid-19 patients is ongoing in Mexico, wherein desidustat has shown to prevent acute respiratory distress syndrome (ARDS) by inhibiting IL-6.[11] Zydus has also received approval from the US FDA to initiate clinical trials of desidustat in chemotherapy Induced anemia (CIA).[12]. Desidustat has met the primary endpoints in the phase 3 clinical trials and Zydus had filed the New Drug Application (NDA) to DCGI in November, 2021.[13]\
CLIP
Zydus receives DCGI approval for new drug Oxemia; what you need to know
The new drug is an oral, small molecule hypoxia-inducible factor-prolyl hydroxylase (HIF-PH) inhibitor, Zydus said in a statement.
Gujarat-based pharma company Zydus Lifesciences on Monday received the Drugs Controller General of India (DCGI) approval for its new drug application for a first-of-its-kind oral treatment for anemia associated with Chronic Kidney Disease (CKD) – Oxemia (Desidustat).
The new drug is an oral, small molecule hypoxia-inducible factor-prolyl hydroxylase (HIF-PH) inhibitor, the drug firm said in a statement.
Desidustat showed good safety profile, improved iron mobilization and LDL-C reduction in CKD patients in DREAM-D and DREAM-ND Phase III clinical trials, conducted in approximately 1,200 subjects. Desidustat provides CKD patients with an oral convenient therapeutic option for the treatment of anemia. The pharma major did not, however, declare the cost per dose if the drug is available in the market.
“After more than a decade of research and development into the science of HIF-PH inhibitors, results have demonstrated that Oxemia addresses this unmet need and additionally reduces hepcidin, inflammation and enables better iron mobilization. This advancement offers ease of convenience for the patient and will also reduce the disease burden by providing treatment at an affordable cost, thereby improving the quality of life for patients suffering from Chronic Kidney Disease,” Chairman of Zydus Lifesciences Pankaj Patel said.
Chronic Kidney Disease (CKD) is a progressive medical condition characterised by a gradual loss of kidney function and is accompanied by comorbidities like anemia, cardiovascular diseases (hypertension, heart failure and stroke), diabetes mellitus, eventually leading to kidney failure.
PATENT
|
Scheme 3:
|
Step 1′a Process for Preparation of ethyl 2-iodobenzoate (XI-a)
Step-2 Process for the Preparation of ethyl 2-((tert-butoxycarbonyl)(cyclopropylmethoxy)aminolbenzoate (XII-a)
Step 3 Process for the Preparation of ethyl 2-((cyclopropylmethoxy)amino)benzoate (XIII-a)
Step 4 Process for the Preparation of ethyl 24N-(cyclopropylinethoxy)-3-ethoxy-3-oxopropanamido)benzoate (XIV-a)
Step 5: Process for the Preparation of ethyl 1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2 dihydroquinolline-3-carboxylate (XY-a)
Purification
Step 6 Process for the Preparation of ethyl (1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycinate (XVI-a)
Purification
Step 7: Process for the Preparation of (1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycine (I-a)
Polymorphic Data (XRPD):
References[edit]
- ^ “Zydus receives DCGI approval for new drug Oxemia; what you need to know”.
- ^ CDSCO, SEC Committee. “SEC meeting to examine IND proposals, dated 29.12.2021”. CDSCO website Govt of India. CDSCO. Retrieved 19 January 2022.
- ^ Kansagra KA, Parmar D, Jani RH, Srinivas NR, Lickliter J, Patel HV, et al. (January 2018). “Phase I Clinical Study of ZYAN1, A Novel Prolyl-Hydroxylase (PHD) Inhibitor to Evaluate the Safety, Tolerability, and Pharmacokinetics Following Oral Administration in Healthy Volunteers”. Clinical Pharmacokinetics. 57 (1): 87–102. doi:10.1007/s40262-017-0551-3. PMC 5766731. PMID 28508936.
- ^ Parmar DV, Kansagra KA, Patel JC, Joshi SN, Sharma NS, Shelat AD, Patel NB, Nakrani VB, Shaikh FA, Patel HV; on behalf of the ZYAN1 Trial Investigators. Outcomes of Desidustat Treatment in People with Anemia and Chronic Kidney Disease: A Phase 2 Study. Am J Nephrol. 2019 May 21;49(6):470-478. doi: 10.1159/000500232.
- ^ “Zydus Cadila announces phase III clinical trials of Desidustat”. 17 April 2019. Retrieved 20 April 2019 – via The Hindu BusinessLine.
- ^ Jain MR, Joharapurkar AA, Pandya V, Patel V, Joshi J, Kshirsagar S, et al. (February 2016). “Pharmacological Characterization of ZYAN1, a Novel Prolyl Hydroxylase Inhibitor for the Treatment of Anemia”. Drug Research. 66 (2): 107–12. doi:10.1055/s-0035-1554630. PMID 26367279.
- ^ Joharapurkar AA, Pandya VB, Patel VJ, Desai RC, Jain MR (August 2018). “Prolyl Hydroxylase Inhibitors: A Breakthrough in the Therapy of Anemia Associated with Chronic Diseases”. Journal of Medicinal Chemistry. 61 (16): 6964–6982. doi:10.1021/acs.jmedchem.7b01686. PMID 29712435.
- ^ Jain M, Joharapurkar A, Patel V, Kshirsagar S, Sutariya B, Patel M, et al. (January 2019). “Pharmacological inhibition of prolyl hydroxylase protects against inflammation-induced anemia via efficient erythropoiesis and hepcidin downregulation”. European Journal of Pharmacology. 843: 113–120. doi:10.1016/j.ejphar.2018.11.023. PMID 30458168. S2CID 53943666.
- ^ Market, Capital (20 January 2020). “Zydus enters into licensing agreement with China Medical System Holdings”. Business Standard India. Retrieved 20 January 2020 – via Business Standard.
- ^ Joharapurkar, Amit; Patel, Vishal; Kshirsagar, Samadhan; Patel, Maulik; Savsani, Hardikkumar; Jain, Mukul (22 January 2021). “Prolyl hydroxylase inhibitor desidustat protects against acute and chronic kidney injury by reducing inflammatory cytokines and oxidative stress”. Drug Development Research. 82 (6): 852–860. doi:10.1002/ddr.21792. PMID 33480036. S2CID 231680317.
- ^ “Zydus’ trials of Desidustat shows positive results for Covid-19 management”. The Hindu Business Line. The Hindu. Retrieved 25 January 2021.
- ^ “Zydus receives approval from USFDA to initiate clinical trials of Desidustat in cancer patients receiving chemotherapy”. PipelineReview.com. La Merie Publishing. Retrieved 22 January 2021.
- ^ “Stock Share Price | Get Quote | BSE”.
|
WO – 30.04.2020
|
|
2.WO/2020/058882METHODS OF PRODUCING VENOUS ANGIOBLASTS AND SINUSOIDAL ENDOTHELIAL CELL-LIKE CELLS AND COMPOSITIONS THEREOF
WO – 26.03.2020
|
|
3.110876806APPLICATION OF HIF2ALPHA AGONIST AND ACER2 AGONIST IN PREPARATION OF MEDICINE FOR TREATING ATHEROSCLEROSIS
CN – 13.03.2020
|
|
US – 28.11.2019
|
|
WO – 06.09.2019
|
|
EA – 30.10.2015
Настоящее изобретение относится к новым соединениям общей формулы (I), фармацевтическим композициям, содержащим указанные соединения, применению этих соединений для лечения состояний, опосредованных пролилгидроксилазой HIF, и к способу лечения анемии, включающему введение заявленных соединений |
|
EP – 28.10.2015
|
|
US – 22.10.2015
The present invention relates to novel compounds of the general formula (I), their tautomeric forms, their stereoisomers, their pharmaceutically acceptable salts, pharmaceutical compositions containing them, methods for their preparation, use of these compounds in medicine and the intermediates involved in their preparation. |
|
WO – 03.07.2014
|
 |
|
| Clinical data | |
|---|---|
| Other names | ZYAN1 |
| Identifiers | |
| CAS Number | |
| UNII | |
| Chemical and physical data | |
| Formula | C16H16N2O6 |
| Molar mass | 332.312 g·mol−1 |
| 3D model (JSmol) | |
Date
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT04215120 | Desidustat in the Treatment of Anemia in CKD on Dialysis Patients | Phase 3 | Recruiting | 2020-01-02 |
| NCT04012957 | Desidustat in the Treatment of Anemia in CKD | Phase 3 | Recruiting | 2019-12-24 |
////////// DESIDUSTAT, ZYDUS CADILA, COVID 19, CORONA VIRUS, PHASE 3, ZYAN 1, OXEMIA, APPROVALS 2022, INDIA 2022


{kind=link}
{kind=link}
{kind=link}