
Home » RADIOLABELLED
Category Archives: RADIOLABELLED
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Talogreptide mesaroxetan


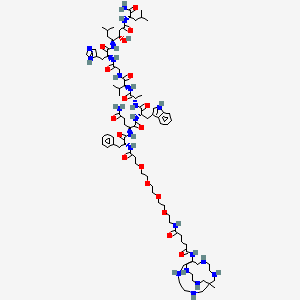
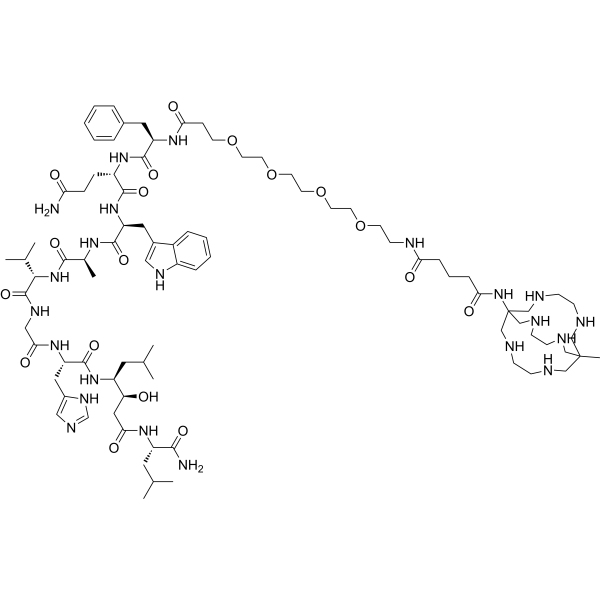
Talogreptide mesaroxetan
CAS 1801418-23-4
MF C86H140N22O18 MW1770.17
{MeCOSar}-PEG4-{d-Phe}-Gln-Trp-Ala-Val-Gly-His-{Sta}-Leu-NH2
(2S)-N-[(2S)-1-[[(2S)-1-[[(2S)-1-[[2-[[(2S)-1-[[(3S,4S)-1-[[(2S)-1-amino-4-methyl-1-oxopentan-2-yl]amino]-3-hydroxy-6-methyl-1-oxoheptan-4-yl]amino]-3-(1H-imidazol-5-yl)-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-methyl-1-oxobutan-2-yl]amino]-1-oxopropan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]-2-[[(2R)-2-[3-[2-[2-[2-[2-[[5-[(8-methyl-3,6,10,13,16,19-hexazabicyclo[6.6.6]icosan-1-yl)amino]-5-oxopentanoyl]amino]ethoxy]ethoxy]ethoxy]ethoxy]propanoylamino]-3-phenylpropanoyl]amino]pentanediamide
N-{21-[(8-methyl-3,6,10,13,16,19-hexaazabicyclo[6.6.6]icosan-1-yl)amino] -17,21-dioxo-4,7,10,13-tetraoxa-16-azahenicosan-1-oyl}-D-phenylalanyl-L-glutaminyl-L-tryptophyl-L-alanyl-Lvalylglycyl-L-histidyl-(3S,4S)-4-amino-3-hydroxy-6-methylheptanoyl-L-leucinamide
diagnostic imaging agent, antineoplastic, ZUN64K4H2X, SAR-BBN
Talogreptide mesaroxetan (CAS 1801418-23-4) is a synthetic peptide, a complex molecule used as a diagnostic imaging agent with potential antitumor effects, targeting G-protein coupled receptors (GRPr) often overexpressed in cancers, allowing for specific tumor visualization in PET scans, particularly for metastatic disease detection, known for its high specificity and contrast for imaging tumors like those expressing GRPr.
Key Characteristics:
- Type: A peptide-based diagnostic agent, often labeled with radioisotopes like Copper-64 ($^{64}$Cu) for Positron Emission Tomography (PET) imaging, notes Patsnap Synapse.
- Structure: It’s a modified peptide sequence incorporating elements like PEG4 and specific amino acids, MedchemExpress.com.
- Function: Binds strongly to GRPr, helping to highlight tumors and metastatic sites.
- Application: Used in research to create high-contrast PET scans for better tumor detection and monitoring, showing promise in visualizing lymph node metastasis.
In Simple Terms:
Imagine it as a “smart tracer” that seeks out specific cancer cells. When attached to a radioactive tag, it lights up tumors on a PET scan, helping doctors see cancer more clearly, notes Patsnap Synapse.
Syn
WO2024086891
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024086891&_cid=P10-MIZEJM-53111-1
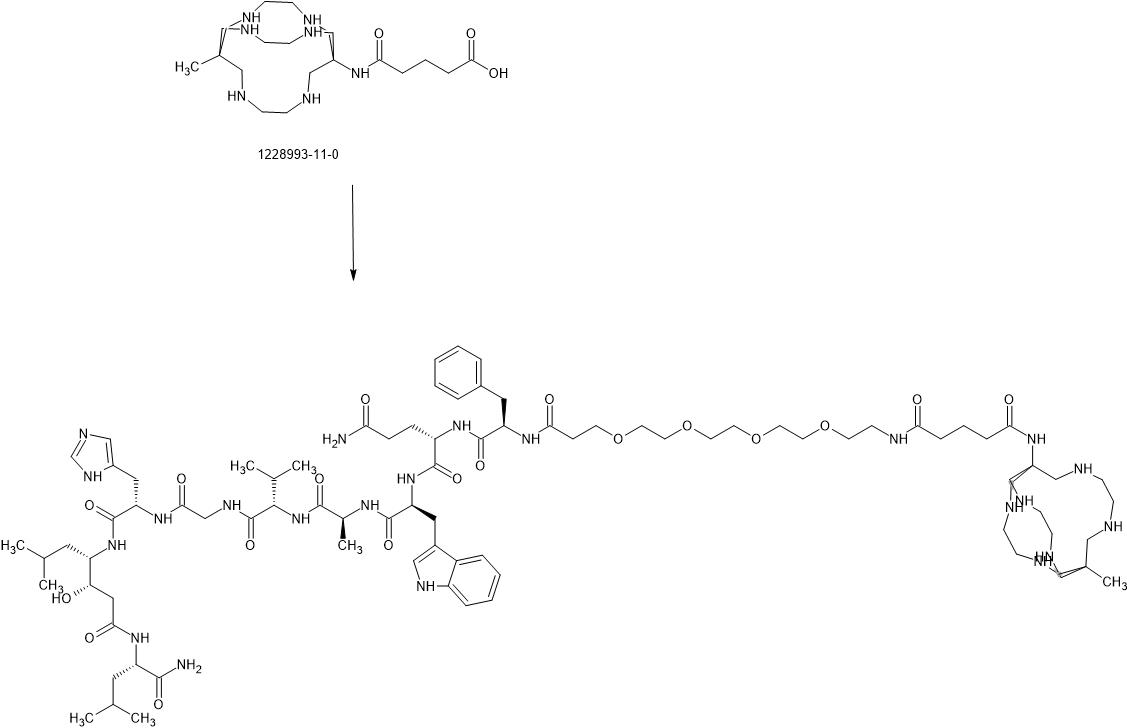
67Cu radioisotope
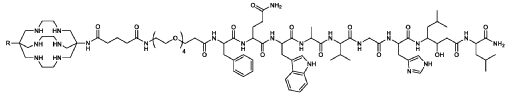
where R is CH3C(0)-;
(67CU-SAR-BBN)
Paper
Molecular Pharmaceutics (2015), 12(8), 2781-2790



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
/////////Talogreptide mesaroxetan, diagnostic imaging agent, antineoplastic, ZUN64K4H2X, SAR-BBN
Copper (64Cu) adarulatide tetraxetan
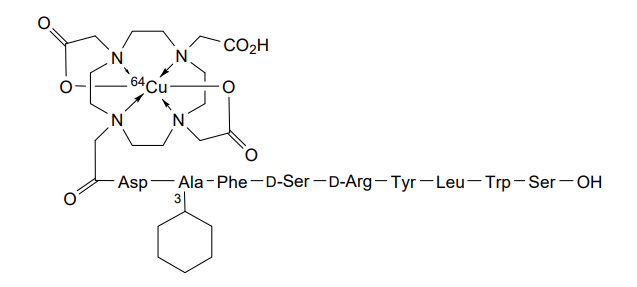
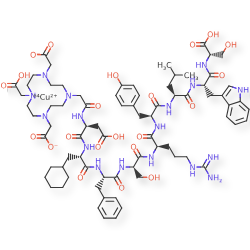
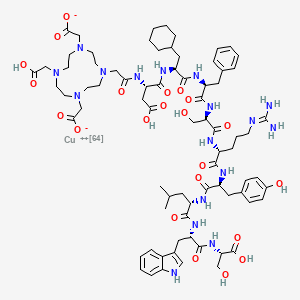
Copper (64Cu) adarulatide tetraxetan
| Adarulatide tetraxetan copper Cu-64 |
CAS 2841388-40-5
MF C76H10764CuN17O22, MF1,674.7
Cuprate(3-)-64Cu, [N-[2-[4,10-bis[(carboxy-κO)methyl]-7-(carboxymethyl)-1,4,7,10-tetrazacyclododec-1-yl-κN1,κN4,κN7,κN10 ]acetyl]-L-α-aspartyl-3-cyclohexyl-L-alanyl-L-phenylalanyl-D-seryl-D-arginyl-L-tyrosyl-L-leucyl-L-tryptophanyl-L-serinato(5-)]-, hydrogen (1:3)
2-[4-[2-[[(2S)-3-carboxy-1-[[(2S)-1-[[(2S)-1-[[(2R)-1-[[(2R)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(1S)-1-carboxy-2-hydroxyethyl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-cyclohexyl-1-oxopropan-2-yl]amino]-1-oxopropan-2-yl]amino]-2-oxoethyl]-7-(carboxylatomethyl)-10-(carboxymethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetate;copper-64(2+)
diagnostic imaging agent, QM8HMM6RJP

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Copper (64Cu) adarulatide tetraxetan. diagnostic imaging agent, QM8HMM6RJP
Borofalan (10B)

.png)
Borofalan (10B), ボロファラン (10B), 硼[10B]法仑
APPROVED JAPAN, 2020/3/25, Steboronine
Antineoplastic, Diagnostic aid, Radioactive agent
(2S)-2-amino-3-(4-(10B)dihydroxy(10B)phenyl)propanoic acid
| Formula | C9H12BNO4 |
|---|---|
| CAS | 80994-59-8 |
| Mol weight | 209.0069 |
- 4-(Borono-10B)-L-phenylalanine
- (10B)-4-Borono-L-phenylalanine
- Borofalan (10b)
- L-(p-[10B]Boronophenyl)alanine
- L-4-[10B]Boronophenylalanine
- p-[10B]Borono-L-phenylalanine
- L-Phenylalanine, 4-borono-10B-
Marketed Head and neck cancer - Originator Stella Pharma
- Developer Osaka University; Stella Pharma; Sumitomo Heavy Industries
- Class Antineoplastics; Borates; Propionic acids; Radiopharmaceuticals
- Mechanism of Action Ionising radiation emitters
- Phase IIGlioma
- Phase I Haemangiosarcoma; Malignant melanoma
- 20 May 2020 Launched for Head and neck cancer (Inoperable/Unresectable, Late-stage disease, Locally recurrent) in Japan (IV)
- 25 Mar 2020 Registered for Head and neck cancer (Inoperable/Unresectable, Late-stage disease, Locally recurrent) in Japan (IV) – First global approval
- 03 Mar 2020 Chemical structure information added
- Melting Point (Experimental)Value: 285-298 °C (decomp) Hattori, Yoshihide; Tetrahedron Letters 2008, VOL49(33), PG 4977-4980
- SP ROT -5.2 ° Conc: 0.50 g/100mL; Solv: hydrochloric acid 589.3 nm; Temp: 25 °C, Hattori, Yoshihide; Tetrahedron Letters 2008, VOL49(33), PG 4977-4980
Borofalan (10B)
4-[(10B)Borono]-L-phenylalanine
C9H1210BNO4 : 208.21
[80994-59-8]
With the development of atomic science, radiation therapy such as cobalt hexahydrate, linear accelerator, and electron beam has become one of the main methods of cancer treatment. However, traditional photon or electron therapy is limited by the physical conditions of the radiation itself. While killing the tumor cells, it also causes damage to a large number of normal tissues on the beam path. In addition, due to the sensitivity of tumor cells to radiation, traditional radiation therapy For the more radiation-resistant malignant tumors (such as: glioblastoma multiforme, melanoma), the treatment effect is often poor.
In order to reduce the radiation damage of normal tissues around the tumor, the concept of target treatment in chemotherapy has been applied to radiation therapy; and for tumor cells with high radiation resistance, it is currently actively developing with high relative biological effects (relative Biological effectiveness, RBE) radiation sources, such as proton therapy, heavy particle therapy, neutron capture therapy. Among them, neutron capture therapy combines the above two concepts, such as boron neutron capture therapy, by the specific agglomeration of boron-containing drugs in tumor cells, combined with precise neutron beam regulation, providing better radiation than traditional radiation. Cancer treatment options.
Boron Neutron Capture Therapy (BNCT) is a high-capture cross-section of thermal neutrons using boron-containing ( 10 B) drugs, with 10 B(n,α) 7 Li neutron capture and nuclear splitting reactions. Two heavy charged particles of 4 He and 7 Li are produced. The average energy of the two charged particles is about 2.33 MeV, which has high linear energy transfer (LET) and short range characteristics. The linear energy transfer and range of α particles are 150 keV/μm and 8 μm, respectively, while the 7 Li heavy particles are For 175 keV/μm, 5 μm, the total range of the two particles is equivalent to a cell size, so the radiation damage caused to the organism can be limited to the cell level, when the boron-containing drug is selectively aggregated in the tumor cells, with appropriate The sub-radiation source can achieve the purpose of locally killing tumor cells without causing too much damage to normal tissues.
Since the effectiveness of boron neutron capture therapy depends on the concentration of boron-containing drugs in the tumor cell position and the number of thermal neutrons, it is also called binary cancer therapy; thus, in addition to the development of neutron sources, The development of boron-containing drugs plays an important role in the study of boron neutron capture therapy.
4-( 10 B)dihydroxyboryl-L-phenylalanine (4-( 10 B)borono-L-phenylalanine, L- 10 BPA) is currently known to be able to utilize boron neutron capture therapy (boron neutron capture therapy) , BNCT) An important boron-containing drug for the treatment of cancer.
Therefore, various synthetic methods of L-BPA have been developed. As shown in the following formula (A), the prior art L-BPA synthesis method includes two methods of forming a bond (a) and a bond (b):

Among them, the method for synthesizing L-BPA by forming the bond (a) is to try to introduce a substituent containing a dihydroxylboryl group or a borono group into the skeleton of the phenylalanine, thereby the pair of the amide substituent. The position forms a carbon-boron bond to produce L-BPA.
J. Org. Chem. 1998, 63, 8019 discloses a method for the cross-coupling reaction of (S)-4-iodophenylalanine with a diboron compound by palladium-catalyzed amine end treatment. Amine-protected (S)-4-iodophenylalanine (eg (S)-N-tert-butoxycarbonyl-4-iodophenylalanine ((S)-N-Boc-4-) Iodophenylalanine)) is prepared by cross-coupling with a diboron compound such as bis(pinacolato diboron) to give (S)-N-tert-butoxycarbonyl-4-pentanoylboryl phenylalanine The amine-terminated (S)-4-boranyl ester phenylalanine of the acid ((S)-N-Boc-4-pinacolatoborono phenylalanine); afterwards, the protecting group on the amine end and the boronic end are removed. The above substituents complete the preparation of L-BPA.
However, since the selected 10 B-doped divaleryl diboron is not a commercially available compound, this method requires additional pretreatment of the preparation of the borating agent, resulting in a high process complexity and a long time consuming process. It is impossible to prepare a high yield of L-BPA. In addition, the carboxylic acid group of the protected (S)-4-iodophenylalanine at the amine end needs to be protected by a substituent to form a benzyl ester group to increase the process yield to 88%; however, The preparation of L-BPA in this manner also requires an additional step of deprotecting the carboxylic acid group, which in turn increases the process complexity of L-BPA.
Accordingly, the method provided in this document not only involves pre-treatment of the preparation of the borating agent, but also requires a large amount of process time and synthesis steps to complete the steps of protecting and deprotecting the carboxylic acid group, and is not advantageous as an industry. The main method of synthesizing L-BPA.
On the other hand, a method for synthesizing L-BPA by forming a bond (b) is a coupling reaction of an amino acid with a boron-containing benzyl fragment or a boron-containing benzaldehyde fragment. To synthesize L-BPA. Biosci. Biotech. Biochem. 1996, 60, 683 discloses an enantioselective synthesis of L-BPA which gives the hands of a cyclic ethers of boronic acid and L-proline The chiral derivatives from L-valine are subjected to a coupling reaction to produce L-BPA. However, this method requires the formation of a cyclic ether compound of boric acid from 4-boronobenzylbromide, followed by a coupling reaction with a chiral derivative of L-proline, and in the latter stage. The amino acid undergoes an undesired racemization in the synthesis step, so that the method requires an enzymatic resolution step to reduce the yield to obtain L-BPA having a certain optical purity.
Accordingly, the method provided in the literature still includes the steps of pretreatment of the preparation of the borating agent and post-treatment of the enzymatic resolution, so that the process involved in the method is complicated and takes a long time, and cannot be obtained. High yield of L-BPA.
In addition, L- 10 BPA (4-( 10 B)borono-L-phenylalanine, 4-( 10 B)dihydroxyboryl-L-phenylalanine) containing 10 boron is currently known to accumulate in tumor cells. The key factor is to use the thermal neutron beam to irradiate the boron element accumulated in the tumor cells to kill the tumor cells by capturing the high-energy particles generated by the reaction, thereby achieving the purpose of treating cancer. Therefore, 10 boron can promote the treatment of L- 10 BPA by boron neutron capture treatment.
However, the boron element present in nature contains about 19.9% of 10 boron and about 80.1% of 11 boron. Therefore, many researchers are still actively developing methods that can be applied to the synthesis of L-BPA, especially for the synthesis of 10- boron-rich L-BPA.
J.Org.Chem.1998,63,8019 additionally provides a method of synthesizing 10 boronated agents, since the method involves multiple steps, it is easy to greatly reduce the boron content of 10 10 boron enriched material in the manufacturing process. Therefore, the method provided in this document is not suitable for the synthesis of 10- boron-rich L-BPA.
Another example is the Biosci.Biotech.Biochem.1996,60,683, before the enzymatic resolution step is not performed, the method provided by the articles could not be obtained with a certain L-BPA optical purity; 10 and the method for preparing boronated agents when also relates to multi-step, resulting in conversion of boron-rich material 10 occurs during the manufacturing process. Therefore, the method provided in this document is also not suitable for the synthesis of 10- boron-rich L-BPA.
Furthermore, Bull. Chem. Soc. Jpn. 2000, 73, 231 discloses the use of palladium to catalyze 4-iodo-L-phenylalanine with 4,4,5,5-tetramethyl-1,3,2 A method in which a dioxonium pentoxide (common name: pinacolborane) is subjected to a coupling reaction. However, this document does not mention how to prepare articles 10 boron enriched L-BPA using this method, and 4,4,5,5-tetramethyl-1,3,2-dioxaborolane not a commercial 10 The compounds available in the literature are not suitable for the synthesis of 10- boron-rich L-BPA.
In addition, Synlett. 1996, 167 discloses a method for coupling a iodophenylborate with a zinc derivative of L-serine zinc derivatives, which involves first preparing phenyl iodoborate. The ester and the preparation of a zinc derivative of L-type serine acid, etc., result in a lower yield of the produced L-BPA. In addition, since the 10- boron-rich triiodide 10 boron and 1,3-diphenylpropane-1,3-diol selected for this method are not commercially available compounds, the methods provided in this document are also provided. Still not suitable for the synthesis of 10- boron-rich L-BPA.
SYN

Repub. Korean Kongkae Taeho Kongbo, 2018060319,
PAPER
Research and Development in Neutron Capture Therapy, Proceedings of the International Congress on Neutron Capture Therapy, 10th, Essen, Germany, Sept. 8-13, 2002 (2002), 1-8.
PAPER
European Journal of Pharmaceutical Sciences (2003), 18(2), 155-163
https://www.sciencedirect.com/science/article/abs/pii/S0928098702002567


PAPER
Tetrahedron Letters (2008), 49(33), 4977-4980
PATENT
WO 2004009135
PATENT
US 20130331599
PATENT
WO 2017028751
https://patents.google.com/patent/WO2017028751A1/en
Example 1
Before preparing (S)-N-tert-butoxycarbonyl-4-dihydroxyborylphenylalanine from (S)-N-tert-butoxycarbonyl-4-iodophenylalanine, it is necessary to reveal Process for preparing (S)-N-tert-butoxycarbonyl-4-iodophenylalanine by using (S)-4-iodophenylalanine as a starting material and a process for preparing 10 tributyl borate with 10 boric acid.
1. Preparation of (S)-N-tert-butoxycarbonyl-4-iodophenylalanine from (S)-4-iodophenylalanine
Please refer to the following reaction formula I, which is (S)-4-iodophenylalanine in a solvent of 1,4-dioxane (1,4-dioxane) and water (H 2 O) with hydrogen peroxide. Sodium (NaOH) and di-tert-butyl dicarbonate (Boc 2 O) are reacted to obtain a chemical reaction formula of (S)-N-tert-butoxycarbonyl-4-iodophenylalanine.

In the preparation process, two reaction vessels were selected for the reaction.
The specific operation process is as follows:
1. Set up a reaction using a 3L three-neck bottle.
2. (S)-4-iodo-L-phenylalanine (200.00 g, 687.10 mmol, 1.00 eq) was added to the reaction system.
3. Add 1,4-dioxane (1.00 L) and water (1.00 L) to the reaction system, respectively.
4. Sodium hydroxide (68.71 g, 1.72 mol, 2.50 eq) was added to the reaction system, the solution gradually became clear, and the temperature rose slightly to 19 °C.
5. When the system is cooled to 0-10 ° C, di-tert-butyl dicarbonate (254.93 g, 1.17 mol, 268.35 mL, 1.70 eq) is added to the reaction system, and the temperature of the reaction system is naturally raised to 10 to 30 ° C and Stir at room temperature (about 30 ° C) for 8 hours.
6. The reaction was detected using high performance liquid chromatography (HPLC) until the starting of the reaction.
7. The temperature of the control system is less than 40 ° C, and the 1,4-dioxane in the reaction solution is concentrated.
8. The reaction system was lowered to room temperature (about 25 ° C), 100 mL of water was added, and the pH was adjusted to 1.8-2 with hydrochloric acid (2M (ie, molarity, M)).
9. Extract three times with ethyl acetate (2 L).
10. Combine the organic phases and wash twice with saturated brine (1 L).
11. The organic phase was dried over sodium sulfate (200 g).
12. Continue drying in an oven (40-45 ° C) to give (S)-N-tert-butoxycarbonyl-4-iodo-L-phenylalanine (250.00 g, 626.28 mmol, HPLC analysis, yield 93.00 %, purity 98%).
The prepared (S) -N- tert-butoxycarbonyl-4-iodo-phenylalanine was -L- Hydrogen 1 nuclear magnetic resonance spectrum analysis (1 HNMR) as follows:
1 H NMR: (400 MHz DMSO-d 6 )
δ 7.49 (d, J = 7.8 Hz, 2H), 6.88 (d, J = 7.8 Hz, 2H), 5.80 (d, J = 5.9 Hz, 1H), 3.68 (d, J = 5.5 Hz, 1H), 3.00-2.90 (m, 1H), 2.87-2.75 (m, 1H), 1.35-1.15 (m, 9H).
Second, tributyl borate 10 was prepared from boronic acid 10
See the following reaction formulas II, 10 as boric acid (H 2 SO 4) is reacted with sulfuric acid in a solvent (butan-1-ol), and toluene (Toluene) in n-butanol, to obtain 10 tributyl borate (10 The chemical reaction formula of B(OBu) 3 ).

The specific operation process is as follows:
1. Set up a reaction device R1 using a 3L three-necked bottle, and configure a water separator on the device.
2. 10 boric acid (150.00 g, 2.46 mol, 1.00 eq) was added to the reaction R1 at room temperature (about 25 ° C).
3. Add n-butanol (1.00 L) to the reaction R1 at room temperature (about 25 ° C) and stir, and most of the boric acid cannot be dissolved.
4. Toluene (1.00 L) was added to the reaction R1 at room temperature (about 25 ° C) and stirred.
5. Concentrated sulfuric acid (4.82 g, 49.16 mmol, 2.62 mL, 0.02 eq) was added dropwise to the reaction at room temperature (about 25 ° C), at which time a large amount of solid remained undissolved.
6. The reaction system was heated to 130 ° C, and the water was continuously removed, stirred for 3.5 hours, and water (about 140 g) was formed in the water separator. The solids were all dissolved, and the solution changed from colorless to brown. .
7. TLC (DCM: MeOH = 5:1, Rf = 0.43, bromocresol green).
8. Distill off most of the toluene at atmospheric pressure.
9. After most of the toluene is distilled off, the temperature of the system is lowered to 20 to 30 ° C, and the reaction liquids of the two reactions are combined, and the apparatus is changed for distillation.
10. Oil bath external temperature 108-110 ° C pump distillation under reduced pressure, Kelvin thermometer 45 ° C, distilled n-butanol.
11. Oil bath external temperature 108-110 ° C oil pump distillation under reduced pressure, the residual butanol was distilled off.
12. Oil bath external temperature 118-120 ° C oil pump vacuum distillation, Kelvin thermometer 55 ° C, began to produce products.
13. The temperature is raised to 135-140 ° C oil pump vacuum distillation, the product is completely distilled.
14. The product is obtained as a colorless liquid 10 tributyl borate (830.00g, 3.62mol, yield 73.58%).
The results of the 1 H NMR analysis of the obtained tributyl 10 borate were as follows:
1 H NMR: (400 MHz CDCl 3 )
δ 3.82-3.68 (m, 6H), 1.57-1.42 (m, 6H), 1.34 (qd, J = 7.4, 14.9 Hz, 6H), 0.95-0.80 (m, 9H).
Three, -N- tert-butoxycarbonyl-4-iodo-phenylalanine was prepared (S) of (S) -N- tert-butoxycarbonyl-4-hydroxy-10-yl -L- phenylalanine boron
Please refer to the following reaction formula III, which is (S)-N-tert-butoxycarbonyl-4-iodophenylalanine with tributyl 10 borate, t-butyl magnesium chloride (t-BuMgCl) and bis (2-A) yl aminoethyl) ether (BDMAEE) reaction, to produce (S) -N- tert-butoxycarbonyl group -4- (10 B) dihydroxyboryl -L- phenylalanine chemical reaction.

In the preparation process, two reaction vessels were selected for the reaction.
The specific operation process is as follows:
1. Set up a reaction using a 3L three-neck bottle.
2. Tributyl 10 borate (187.60 g, 87.98 mmol, 3.20 eq) was placed in the reaction system at room temperature (about 22 ° C).
3. Sodium hydride (20.45 g, 511.24 mmol, purity 60%, 2.00 eq) was added to the reaction system at room temperature (about 22 ° C). The reaction solution was a suspension and stirred at room temperature (about 22 ° C). 5 minutes.
4. Bis(2-methylaminoethyl)ether (327.73 g, 2.04 mol, 8.00 eq) was added to the reaction at room temperature (about 22 ° C).
5. N-tert-Butoxycarbonyl-4-iodo-L-phenylalanine (100.00 g, 255.62 mmol, 1.00 eq) was added to the reaction system at room temperature (about 22 ° C), and a large amount of solid was not dissolved.
6. Lower the temperature of the reaction system to 0-5 ° C, add t-butyl magnesium chloride (1.7 M, 1.20 L, 2.04 mol, 8.00 eq) to the reaction, control the temperature between 0-10 ° C, the dropping time is about It is 1.5 hours.
7. After the completion of the charging, the temperature of the reaction system was naturally raised to room temperature (20 to 30 ° C) and stirred at this temperature for 12 hours.
8. Using high performance liquid chromatography (HPLC) to detect about 9.00% of the remaining material.
9. When the temperature of the reaction system was lowered to -5 to 0 ° C, it was quenched by dropwise addition of 500 mL of water.
10. Lower the temperature of the system to 0-5 ° C, add methyl tert-butyl ether (500 mL) to the reaction system and adjust the pH to 2.9-3.1 (using a pH meter) with 37% HCl (about 500 mL). Exothermic, the temperature of the control system is between 0-15 °C.
11. The aqueous phase obtained by liquid separation was extracted once with methyl tert-butyl ether (500 mL), and the obtained organic phases were combined to give an organic phase of about 1.1 L.
12. Slowly add a sodium hydroxide aqueous solution (1 M, 400 mL) to the obtained organic phase, exotherm during the dropwise addition, and control the system temperature between 0-15 °C.
13. After the completion of the dropwise addition, the pH of the system was about 10, and the pH was adjusted to between 12.10 and 12.6 with an aqueous sodium hydroxide solution (4M). (measured with a pH meter)
14. Dispensing.
15. The aqueous phase 1 obtained after liquid separation was extracted once with n-butanol (500 ml) to obtain aqueous phase 2.
16. Combine the aqueous phase 2 of the two reaction vessels.
17. Adjust the pH of the aqueous phase to 2.9-3.1 with 37% HCl, stir for about 40 minutes, and precipitate a large amount of solid.
18. Filtration gave a white solid which was washed once with dichloromethane (50 mL).
19. At 25 ° C, the precipitated solid was slurried with dichloromethane (150 mL) and stirred for 10 min.
20. A white solid was filtered to give (S) -N- tert-butoxycarbonyl group -4- (10 B) dihydroxyboryl -L- phenylalanine (75.00g, 240.82mmol, by HPLC analysis, a yield of 47.11% , purity 99%).
The prepared (S) -N- tert-butoxycarbonyl group -4- (10 B) results dihydroxyboryl -L- phenylalanine 1 HNMR was as follows:
1 H NMR: (400 MHz DMSO-d 6 )
Δ12.55 (br.s., 1H), 7.91 (s, 2H), 7.66 (d, J = 7.5 Hz, 2H), 7.17 (d, J = 7.5 Hz, 2H), 4.08-4.01 (m, 1H) ), 3.61-3.53 (m, 1H), 2.98 (dd, J = 4.2, 13.9 Hz, 1H), 2.79 (dd, J = 10.4, 13.5 Hz, 1H), 1.79-1.67 (m, 1H), 1.35- 1.17 (m, 9H).
Preparation of L- 10 BPA from (S)-N-tert-Butoxycarbonyl-4-dihydroxyboryl-L-phenylalanine
See the following reaction scheme IV, which is (S) -N- tert-butoxycarbonyl group -4- (10 B) of amine end dihydroxyboryl -L- phenylalanine deprotection of the chemical reaction, to obtain L- 10 BPA.

The specific operation process is as follows:
1. Set up a reaction using a 1L three-neck bottle.
2. room temperature (20-30 deg.] C) to (S) -N- tert-butoxycarbonyl group -4- (10 B) dihydroxyboryl -L- phenylalanine (67.00g, 217.31mmol, 1.00eq) was added the reaction In the system.
3. room temperature (20-30 deg.] C) water (23.75mL) and acetone (Acetone, 420.00mL) were added dropwise to the reaction flask, stirred (S) -N- tert-butoxycarbonyl group -4- (10 B) dihydroxy Boronyl-L-phenylalanine.
4. Concentrated hydrochloric acid (23.93 g, 656.28 mmol, 23.46 mL, 3.02 eq) was added dropwise to the reaction system at room temperature (20-30 ° C). After the addition was completed, the reaction system was heated to 55-60 ° C and stirred for 4.5 hours.
5. HPLC detection until the reaction of the starting material is completed.
6. The temperature is controlled below 40 ° C, and the acetone in the reaction system is concentrated.
7. Lower the concentrated system to below 15 °C, adjust the pH of the system to about 1.5 with sodium hydroxide solution (4M) (pH meter detection), stir for 40 minutes and continue to adjust the pH of the system to 6.15 using sodium hydroxide solution (4M). ~6.25, a large amount of white solid precipitated, which was filtered to give a white solid, and rinsed with acetone (200mL).
8. Obtained as a white solid L- 10 BPA (36.00 g, 171.17 mmol, HPLC, yield 78.77%, purity 99%).
The analytical results obtained by the L- 10 BPA 1 HNMR are as follows:
1 H NMR: (400 MHz D 2 O, CF 3 COOH)
δ 7.44 (d, J = 7.9 Hz, 1H), 7.03 (d, J = 7.9 Hz, 1H), 4.06 (dd, J = 5.7, 7.5 Hz, 1H), 3.11-3.01 (m, 1H), 2.98 -2.87 (m, 1H).
xample 6
Preparation of (S)-N-tert-butoxycarbonyl-4-dihydroxyboryl-L-phenylalanine from (S)-N-tert-butoxycarbonyl-4-iodophenylalanine
Please refer to the following reaction formula VII, which is a reaction of (S)-N-tert-butoxycarbonyl-4-iodophenylalanine with tributyl borate and t-butylmagnesium chloride (t-BuMgCl) to obtain (S The chemical reaction formula of -N-tert-butoxycarbonyl-4-dihydroxyboryl-L-phenylalanine.

The specific operation process is as follows:
1. Construct a reaction unit with a 250 mL three-neck bottle.
2. Tributyl borate (17.65 g, 76.68 mmol, 3.00 eq) was placed in a 250 mL reaction flask at 20-30 °C.
3. Sodium hydride (1.02 g, 25.56 mmol, 1.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
4. (S)-N-tert-Butoxycarbonyl-4-iodo-L-phenylalanine (10.00 g, 25.56 mmol, 1.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
5. Reduce the temperature of the reaction system to 0 ° C under nitrogen atmosphere, slowly add t-butyl magnesium chloride (1.7 M in THF, 120 mL, 8.00 eq) to the reaction, the dropping time is about 30 minutes, and the control temperature is 0. Between °C and 10 °C.
Stir at 20.20 ~ 30 ° C for 20 hours.
7. HPLC detection of the basic reaction of the raw materials, leaving only about 0.7% of the raw materials.
8. At a temperature of 0 ° C, 5 mL of water was added dropwise to the reaction to quench it. After complete quenching, stirring was continued for 10 minutes.
9. Cool down to 0 ° C, add methyl tert-butyl ether (50 mL) to the reaction and adjust the pH to 3 with 37% HCl (about 50 mL) (detected with a pH meter), adjust the pH during the process to exotherm, control the temperature at 0 Between °C and 15 °C.
12. The aqueous phase obtained by liquid separation was extracted once with methyl t-butyl ether (50 mL) and the organic phases were combined.
12. Add NaOH solution (1M, 55mL) to the obtained organic phase to adjust the pH to between 12.10-12.6. The process is exothermic and the temperature is controlled between 0 °C and 15 °C.
13. Liquid separation, the obtained aqueous phase was extracted once with n-butanol (50 mL), and most of the impurities were extracted and removed.
14. The aqueous phase obtained by liquid separation was adjusted to pH 3 with 37% HCl and stirred for about 30 minutes to precipitate a white solid.
15. Filtration gave a white solid which was washed once with dichloromethane (50 mL).
16. The precipitated solid was slurried with 25 mL of dichloromethane at 25 ° C and stirred for 10 minutes.
17. Filtration of (S)-N-tert-butoxycarbonyl-4-dihydroxyboryl-L-phenylalanine (6.8 g, HPLC, yield: 83.15%, purity 98%).
Example 7
Please continue to refer to Reaction Scheme VII. The specific operation process is as follows:
1. Construct a reaction unit with a 250 mL three-neck bottle.
2. Tributyl borate (8.82 g, 38.34 mmol, 3.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
3. Sodium hydride (511.25 mg, 12.78 mmol, 1.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
4. (S)-N-tert-Butoxycarbonyl-4-iodo-L-phenylalanine (5.00 g, 12.78 mmol, 1.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
5. The temperature of the reaction system was lowered to 0 ° C under nitrogen atmosphere, and t-butyl magnesium chloride (1.7 M in THF, 60 mL, 8.00 eq) was added dropwise to the reaction, the dropwise addition time was about 30 minutes, and the control temperature was 0 ° C. -10 ° C between.
Stir at 6.20 ~ 30 ° C for 22 hours.
7. HPLC detection of the raw material reaction is completed.
8. At a temperature of 0 ° C, 2.5 mL of water was added dropwise to the reaction to quench it. After complete quenching, stirring was continued for 10 minutes.
9. Cool down to 0 ° C, add methyl tert-butyl ether (25 mL) to the reaction and adjust the pH to 3 with 37% HCl (about 25 mL) (detected with a pH meter), adjust the pH during the process to exotherm, control the temperature at 0 Between °C and 15 °C.
12. The aqueous phase obtained by liquid separation was extracted once with methyl t-butyl ether (25 mL) and the organic phases were combined.
12. Add NaOH solution (1M, 30mL) to the obtained organic phase to adjust the pH to between 12.10-12.6. The process is exothermic and the temperature is controlled between 0 °C and 15 °C.
13. Liquid separation, the obtained aqueous phase was extracted once with n-butanol (25 ml), and most of the impurities were extracted and removed.
14. The aqueous phase obtained by liquid separation was adjusted to pH 3 with 37% HCl and stirred for about 30 minutes to precipitate a white solid.
15. Filtration gave a white solid which was washed once with dichloromethane (25 mL).
16. The precipitated solid was slurried with 15 mL of dichloromethane at 25 ° C and stirred for 10 minutes.
17. Filtration gave (S)-N-tert-butoxycarbonyl-4-dihydroxyboryl-L-phenylalanine (3.4 g, obtained by HPLC, yield: 85.26%, purity 98%).
Bis(2-methylaminoethyl)ether is a complexing agent for Mg, which can reduce the occurrence of side reactions in the reaction. The reactions of Examples 6 and 7 were carried out without adding bis(2-methylaminoethyl)ether. The analysis showed that the iodine impurity in the reaction of Example 6 was about 17%, and the iodine impurity in the reaction of Example 7 was observed. About 28%. Therefore, it has been proved from the side that the addition of bis(2-methylaminoethyl)ether can protect the reaction from reducing iodine.
The BPA or 10 BPA obtained in the above examples were analyzed by chiral HPLC, and the ratio of the L-enantiomer to the D-enantiomer was 100:0.
The boron-containing drug L-BPA for neutron capture therapy disclosed in the present invention is not limited to the contents described in the above examples. The above-mentioned embodiments are only examples for convenience of description, and the scope of the claims should be determined by the claims.
PATENT
KR 2018060319
PATENT
WO 2019163790
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019163790
///////////Borofalan (10B), Borofalan, Steboronine, JAPAN 2020, 2020 APPROVALS, ボロファラン (10B), ボロファラン , 硼[10B]法仑 ,
B(C1=CC=C(C=C1)CC(C(=O)O)N)(O)O
FDA approves new diagnostic imaging agent FLUCICLOVINE F-18 to detect recurrent prostate cancer

FLUCICLOVINE F-18
Cyclobutanecarboxylic acid, 1-amino-3-(fluoro-18F)-, trans- [
- Molecular FormulaC5H818FNO2
- Average mass132.124 Da
May 27, 2016
Release
The U.S. Food and Drug Administration today approved Axumin, a radioactive diagnostic agent for injection. Axumin is indicated for positron emission tomography (PET) imaging in men with suspected prostate cancer recurrence based on elevated prostate specific antigen (PSA) levels following prior treatment.
Prostate cancer is the second leading cause of death from cancer in U.S. men. In patients with suspected cancer recurrence after primary treatment, accurate staging is an important objective in improving management and outcomes.
“Imaging tests are not able to determine the location of the recurrent prostate cancer when the PSA is at very low levels,” said Libero Marzella, M.D., Ph.D., director of the Division of Medical Imaging Products in the FDA’s Center for Drug Evaluation and Research. “Axumin is shown to provide another accurate imaging approach for these patients.”
Two studies evaluated the safety and efficacy of Axumin for imaging prostate cancer in patients with recurrent disease. The first compared 105 Axumin scans in men with suspected recurrence of prostate cancer to the histopathology (the study of tissue changes caused by disease) obtained by prostate biopsy and by biopsies of suspicious imaged lesions. Radiologists onsite read the scans initially; subsequently, three independent radiologists read the same scans in a blinded study.
The second study evaluated the agreement between 96 Axumin and C11 choline (an approved PET scan imaging test) scans in patients with median PSA values of 1.44 ng/mL. Radiologists on-site read the scans, and the same three independent radiologists who read the scans in the first study read the Axumin scans in this second blinded study. The results of the independent scan readings were generally consistent with one another, and confirmed the results of the onsite scan readings. Both studies supported the safety and efficacy of Axumin for imaging prostate cancer in men with elevated PSA levels following prior treatment.
Axumin is a radioactive drug and should be handled with appropriate safety measures to minimize radiation exposure to patients and healthcare providers during administration. Image interpretation errors can occur with Axumin PET imaging. A negative image does not rule out the presence of recurrent prostate cancer and a positive image does not confirm the presence of recurrent prostate cancer. Clinical correlation, which may include histopathological evaluation of the suspected recurrence site, is recommended.
The most commonly reported adverse reactions in patients are injection site pain, redness, and a metallic taste in the mouth.
Axumin is marketed by Blue Earth Diagnostics, Ltd., Oxford, United Kingdom
Patent
http://www.google.com/patents/WO2014023775A1?cl=en
The non-natural amino acid [ F]-l-amino-3-fluorocyclobutane-l-carboxylic acid
([18F]-FACBC, also known as [18F]-Fluciclovine) is taken up specifically by amino acid transporters and has shown promise for tumour imaging with positron emission tomography (PET).
A known synthesis of [18F]-FACBC begins with the provision of the protected precursor compound 1 -(N-(t-butoxycarbonyl)amino)-3 –
[((trifluoromethyl)sulfonyl)oxy]-cyclobutane-l-carboxylic acid ethyl ester. This precursor compound is first labelled with [18F]-fluoride:

II before removal of the two protecting groups:

IT III
EP2017258 (Al) teaches removal of the ethyl protecting group by trapping the [18F]- labelled precursor compound (II) onto a solid phase extraction (SPE) cartridge and incubating with 0.8 mL of a 4 mol/L solution of sodium hydroxide (NaOH). After 3 minutes incubation the NaOH solution was collected in a vial and a further 0.8 mL 4 mol/L NaOH added to the SPE cartridge to repeat the procedure. Thereafter the SPE cartridge was washed with 3 mL water and the wash solution combined with the collected NaOH solution. Then 2.2 mL of 6 mol/L HCl was then added with heating to 60°C for 5 minutes to remove the Boc protecting group. The resulting solution was purified by passing through (i) an ion retardation column to remove Na+ from excess NaOH and Cl~ from extra HCl needed to neutralise excess of NaOH to get a highly acidic solution before the acidic hydrolysis step, (ii) an alumina column, and (iii) a reverse-phase column. There is scope for the deprotection step(s) and/or the
purification step in the production of [18F]-FACBC to be simplified.
Example 1: Synthesis of f FIFACBC
No-carrier- added [18F]fluoride was produced via the 180(p,n)18F nuclear reaction on a GE PETtrace 6 cyclotron (Norwegian Cyclotron Centre, Oslo). Irradiations were performed using a dual-beam, 30μΑ current on two equal Ag targets with HAVAR foils using 16.5 MeV protons. Each target contained 1.6 ml of > 96% [180]water (Marshall Isotopes). Subsequent to irradiation and delivery to a hotcell, each target was washed with 1.6 ml of [160]water (Merck, water for GR analysis), giving approximately 2-5 Gbq in 3.2 ml of [160]water. All radiochemistry was performed on a commercially available GE FASTlab™ with single-use cassettes. Each cassette is built around a one-piece-moulded manifold with 25 three-way stopcocks, all made of polypropylene. Briefly, the cassette includes a 5 ml reactor (cyclic olefin copolymer), one 1 ml syringe and two 5 ml syringes, spikes for connection with five prefilled vials, one water bag (100 ml) as well as various SPE cartridges and filters. Fluid paths are controlled with nitrogen purging, vacuum and the three syringes. The fully automated system is designed for single-step fluorinations with cyclotron-produced [18F]fluoride. The FASTlab was programmed by the software package in a step-by-step time-dependent sequence of events such as moving the syringes, nitrogen purging, vacuum, and temperature regulation. Synthesis of
[18F]FACBC followed the three general steps: (a) [18F]fluorination, (b) hydrolysis of protection groups and (c) SPE purification.
Vial A contained K222 (58.8 mg, 156 μπιοΐ), K2C03 (8.1 mg, 60.8 μπιοΐ) in 79.5% (v/v)
MeCN(aq) (1105 μΐ). Vial B contained 4M HC1 (2.0 ml). Vial C contained MeCN
(4.1ml). Vial D contained the precursor (48.4 mg, 123.5 μιηοΐ) in its dry form (stored at -20 °C until cassette assembly). Vial E contained 2 M NaOH (4.1 ml). The 30 ml product collection glass vial was filled with 200 mM trisodium citrate (10 ml). Aqueous
[18F]fluoride (1-1.5 ml, 100-200 Mbq) was passed through the QMA and into the 180-
H20 recovery vial. The QMA was then flushed with MeCN and sent to waste. The trapped [18F]fluoride was eluted into the reactor using eluent from vial A (730 μΐ) and then concentrated to dryness by azeotropic distillation with acetonitrile (80 μΐ, vial C). Approximately 1.7 ml of MeCN was mixed with precursor in vial D from which 1.0 ml of the dissolved precursor (corresponds to 28.5 mg, 72.7 mmol precursor) was added to the reactor and heated for 3 min at 85°C. The reaction mixture was diluted with water and sent through the tC18 cartridge. Reactor was washed with water and sent through the tC18 cartridge. The labelled intermediate, fixed on the tC18 cartridge was washed with water, and then incubated with 2M NaOH (2.0 ml) for 5 min after which the 2M NaOH was sent to waste. The labelled intermediate (without the ester group) was then eluted off the tC18 cartridge into the reactor using water. The BOC group was hydrolysed by adding 4M HC1 (1.4 ml) and heating the reactor for 5 min at 60 °C. The reactor content with the crude [18F]FACBC was sent through the HLB and Alumina cartridges and into the 30 ml product vial. The HLB and Alumina cartridges were washed with water (9.1 ml total) and collected in the product vial. Finally, 2M NaOH (0.9 ml) and water (2.1 ml) was added to the product vial, giving a purified formulation of [18F]FACBC with a total volume of 26 ml. Radiochemical purity was measured by radio-TLC using a mixture of MeCN:MeOH:H20:CH3COOH (20:5:5: 1) as the mobile phase. The radiochemical yield (RCY) was expressed as the amount of radioactivity in the [18F]FACBC fraction divided by the total used [18F]fluoride activity (decay corrected). Total synthesis time was 43 min.
The RCY of [18F]FACBC was 62.5% ± 1.93 (SD), n=4.
/////FDA, diagnostic imaging agent, recurrent prostate cancer, fda 2016, Axumin, marketed, Blue Earth Diagnostics, Ltd., Oxford, United Kingdom, fluciclovine F 18
C1[C@@](C[C@H]1[18F])(N)C(=O)O
UPDATE
![]()
SEE EMA
| Axumin : EPAR – Summary for the public | EN = English | 06/07/2017 |
The active substance fluciclovine (18F) is prepared from the precursor AH113487 by nucleophilic substitution
of a triflate group by 18F-fluoride, followed by two deprotection steps. Due to the short half-life of the 18Ffluorine
radioisotope, each batch is prepared on the day of clinical use.
The active substance is prepared in a proprietary automated synthesiser unit. The synthesiser module is
computer-controlled. A fluid path for synthesis is provided in the form of a single use cassette (FASTlab). The
cassette contains 3 reagent vials and 3 solid phase cartridges. Two other reagent vials are supplied
separately as they have a recommended storage temperature of 2-8°C. These 2 vials are inserted into the
cassette on the day of production.
Assessment report
EMA/237809/2017 Page 13/90
Fluciclovine (18F) is produced in a continuous operation from the precursor AH113487. Due to the radioactive
nature of the process, and the short half-life of [18F] fluorine, intermediates are not isolated and there is no
opportunity for operator intervention or in-process testing. Control of the synthesis of fluciclovine (18F) from
the precursor is achieved through the automated synthesis platform, which is pre-programmed with
synthesis parameters optimised for the process. On-board detectors record transfers of radioactivity through
the fluid path at critical points and monitor temperature and pressure as appropriate so that the operator
may track the progress of the synthesis.
The active substance fluciclovine (18F) progressses immediately to purification, formulation and dispensing as
the finished product within a single, continuous operation. Validation of the manufacturing process for
fluciclovine (18F) is therefore described as part of finished product validation.
The characterisation of the active substance is in accordance with the EU guideline on chemistry of new
active substances.
As mentioned, the manufacture of the active substance and finished product takes place in a single,
continuous process. The active substance is not isolated at any point. Therefore, relevant information about
impurities is given only for the finished product.
For the same reason, information for the container closure system is provided only for the finished product.http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004197/WC500230836.pdf
The USFDA has approved Navidea Biopharmaceuticals’ Supplemental New Drug Application (sNDA) for the expanded use of Lymphoseek (technetium Tc 99m tilmanocept) Injection
 [99mTc]-DTPA-mannosyl-dextran [99mTc]-DTPA-mannosyl-dextran |
|||
| Systematic (IUPAC) name | |||
|---|---|---|---|
| Dextran 3-[(2-aminoethyl)thio]propyl 17-carboxy-10,13,16-tris(carboxymethyl)-8-oxo-4-thia-7,10,13,16-tetraazaheptadec-1-yl 3-[[2-[[1-imino-2-(D-mannopyranosylthio)ethyl]amino]ethyl]thio]propyl ether technetium-99m complexes…………………………………………………..………………..OTHER NAME ………………Dextran 3-[(2-aminoethyl)thio]propyl 17-carboxy-10,13,16-tris(carboxymethyl)-8-oxo-4-thia-7,10,13,16-tetraazaheptadec-1-yl 3-[[2-[[1-imino-2-(D-mannopyranosylthio)ethyl]amino]ethyl]thio]propyl ether technetium-99Tc complexes (1-6)-alpha-D-pyranoglucan partially etherified by 3-[(2-aminoethyl)sulfanyl]propyl 17-carboxy-10,13,16-tris(carboxymethyl)-8-oxo-4-thia-7,10,13,16-tetraazaheptadecyl and 3-[(2-{[2-(L-mannopyranosylsulfanyl)acetimidoyl]amino}ethyl)sulfanyl]propyl [99mTc]technetium coordination compound [99mTc]-DTPA-mannosyl-dextran composed of a dextran backbone linked to multiple units of mannose and DTPA (diethylenetriamine pentaacetic acid) with an average molecular weight of 35800………………..LAUNCHED………….Launched – 2013 |
|||
| Clinical data | |||
| Trade names | Lymphoseek | ||
| AHFS/Drugs.com | entry | ||
| Pregnancy cat. | C (US) | ||
| Legal status | ℞-only (US) | ||
| Routes | Intradermal, subcutaneous | ||
| Pharmacokinetic data | |||
| Half-life | 1.75 to 3.05 hours at injection site | ||
| Identifiers | |||
| ATC code | V09IA09 | ||
| Chemical data | |||
| Formula | (C6H10O5)n(C19H28N4O9S99mTc)3–8(C13H24N2O5S2)12–20(C5H11NS)0–17 | ||
| Mol. mass 15,281–23,454 g/mol[1]……………………..CODES1600 NEO3-06 TcDTPAmanDx Tilmanocepthttp://chem.sis.nlm.nih.gov/chemidplus/rn/1262984-82-6NDA N202207 APPROVED
|
|||
PATENT US 6409990, EXPMay 12, 2020
商品名:Lymphoseek 通用名:Technetium Tc 99m tilmanocept 中文名:未知
药企:Navidea Biopharmaceuticals, Inc.
FDA approves Navidea’s Lymphoseek for expanded use in head and neck cancer patients
The US Food and Drug Administration (FDA) has approved Navidea Biopharmaceuticals’ Supplemental New Drug Application (sNDA) for the expanded use of Lymphoseek (technetium Tc 99m tilmanocept) Injection indicated for guiding sentinel lymph node (SLN) biopsy in head and neck cancer patients with squamous cell carcinoma of the oral cavity.
NCI: 99mTc-DTPA-mannosyl-dextran A radiolabeled macromolecule consisting of the chelating agent diethylenetriamine pentaacetic acid (DTPA) and mannose each attached to a dextran backbone and labeled with metastable technetiumTc-99 (Tc-99m), with mannose binding and radioisotopic activities. Upon injection, the mannose moiety of 99mTc-DTPA-mannosyl-dextran binds to mannose-binding protein (MBP). As MBPs reside on the surface of dendritic cells and macrophages, this gamma-emitting macromolecule tends to accumulate in lymphatic tissue where it may be imaged using gamma scintigraphy. This agent exhibits rapid clearance from the injection site, rapid uptake and high retention within the first draining lymph node, and low uptake by the remaining lymph nodes. MBP is a C-type lectin that binds mannose or fucose carbohydrate residues, such as those found on the surfaces of many pathiogens, and once bound activates the complement system.
The active ingredient in technetium Tc 99m tilmanocept is technetium Tc 99m tilmanocept. The active ingredient is formed when Technetium Tc 99m pertechnetate, sodium injection is added to the tilmanocept powder vial.
Technetium Tc 99m binds to the diethylenetriaminepentaacetic acid (DTPA) moieties of the tilmanocept molecule.
Chemically, technetium Tc 99m tilmanocept consists of technetium Tc 99m, dextran 3-[(2- aminoethyl)thio]propyl 17-carboxy-10,13,16- tris(carboxymethyl)-8-oxo-4-thia-7,10,13,16- tetraazaheptadec-1-yl 3-[[2-[[1-imino-2-(D- mannopyranosylthio) ethyl]amino]ethyl]thio]propyl ether complexes. Technetium Tc 99m tilmanocept has the following structural formula:

Empirical formula: [C6H10O5]n.(C19H28N4O9S99mTc)b.(C13H24N2O5S2)c.(C5H11NS)a
Calculated average molecular weight: 15,281 to 23,454 g/mol
It contains 3-8 conjugated DTPA (diethylenetriamine pentaacetic acid) molecules (b); 12-20 conjugated mannose molecules (c) with 0-17 amine side chains (a) remaining free.
The tilmanocept powder vial contains a sterile, non-pyrogenic, white to off-white powder that consists of a mixture of 250 mcg tilmanocept, 20 mg trehalose dihydrate, 0.5 mg glycine, 0.5 mg sodium ascorbate, and 0.075 mg stannous chloride dihydrate. The contents of the vial are lyophilized and are under nitrogen.
Technetium Tc 99m tilmanocept injection is supplied as a Kit. The Kit includes tilmanocept powder vials which contain the necessary non-radioactive ingredients needed to produce technetium Tc 99m tilmanocept. The Kit also contains DILUENT for technetium Tc 99m tilmanocept. The diluent contains a preservative and is specifically formulated for technetium Tc 99m tilmanocept. No other diluent should be used.
The DILUENT for technetium Tc 99m tilmanocept contains 4.5 mL sterile buffered saline consisting of 0.04% (w/v) potassium phosphate, 0.11% (w/v) sodium phosphate (heptahydrate), 0.5% (w/v) sodium chloride, and 0.4% (w/v) phenol. The pH is 6.8 – 7.2.http://www.druginformation.com/RxDrugs/T/Technetium%20Tc%2099m%20Tilmanocept%20Injection.html

Lymphoseek(TM) is a lymphatic tissue-targeting agent which was first launched in 2013 in the U.S. by Navidea Biopharmaceuticals (formerly known as Neoprobe) for lymphatic mapping with a hand-held gamma counter to assist in the localization of lymph nodes draining a primary tumor site in patients with breast cancer or melanoma. In 2014, a supplemental NDA was approved in the U.S. for its use as a sentinel lymph node tracing agent in patients with head and neck squamous cell carcinoma of the oral cavity. Although several tracing agents exist that are used in “off-label” capacities, Lymphoseek is the first tracing agent specifically labeled for lymph node detection.
In 2012, an MAA was filed in the E.U. for the detection of lymphatic tissue in patients with solid tumors, and in 2013, a supplemental MAA was filed in the E.U. for sentinel lymph node detection in patients with head and neck cancer. The products is also awaiting registration to support broader and more flexible use in imaging and lymphatic mapping procedures, including lymphoscintigraphy and other optimization capabilities.
Navidea holds an exclusive worldwide license of Lymphoseek(TM) through the University of California at San Diego (UCSD), and, in 2007, Lymphoseek(TM) was licensed to Cardinal Health by Navidea for marketing and distribution in the U.S.
Lymphoseek(TM), also known as [99mTc]DTPA-mannosyl-dextran, is a receptor-binding radiopharmaceutical designed specifically for the mapping of sentinel lymph nodes in connection with gamma detection devices in a surgical procedure known as intraoperative lymphatic mapping (ILM). It is made up of multiple DTPA and mannose units, each attached by a 5-carbon thioether spacer to a dextran backbone. The compound features subnanomolar affinity for the mannose binding protein receptor, and consequently shows low distal node accumulation. Additionally, its small molecular diameter of 7 nanometers allows for enhanced diffusion into lymphatic channels and capillaries.

1600
99mTc-tilmanocept
Tc-DTPA-mannosyl-dextran
Technetium Tc 99m Tilmanocept
Tilmanocept
UNII-8IHI69PQTC
Chemical structure of [99mTc]tilmanocept. [99mTc]Tilmanocept is composed of a dextran backbone (black) to which are attached multiple units of mannose (green) and DTPA (blue). The mannose units provide a molecular mechanism by which [99mTc]tilmanocept avidly binds to a receptor specific to reticuloendothelial cells (CD206), and the DTPA units provide a highly stable means to radiolabel tilmanocept with 99mtechnetium (red). The molecular weight of [99mTc]tilmanocept is approximately 19,000 g/mol; the molecular diameter is 7.1 nm
[(99m)Tc]Tilmanocept is a CD206 receptor-targeted radiopharmaceutical designed for sentinel lymph node (SLN) identification. Two nearly identical nonrandomized phase III trials compared [(99m)Tc]tilmanocept to vital blue dye.
Technetium (99mTc) tilmanocept, trade name Lymphoseek, is a radiopharmaceutical diagnostic imaging agent approved by the U.S. Food and Drug Administration (FDA) for the imaging of lymph nodes.[1][2] It is used to locate those lymph nodes which may be draining from tumors, and assist doctors in locating those lymph nodes for removal during surgery.[3]

http://blog.sina.com.cn/u/1242475203
…………………….
WO 2000069473
http://www.google.com/patents/EP1178838A2?cl=en
…………………………………………..
US 6409990
http://www.google.co.in/patents/US6409990
References
- FDA Professional Drug Information
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm343525.htm
- Marcinow, A. M.; Hall, N.; Byrum, E.; Teknos, T. N.; Old, M. O.; Agrawal, A. (2013). “Use of a novel receptor-targeted (CD206) radiotracer, 99mTc-tilmanocept, and SPECT/CT for sentinel lymph node detection in oral cavity squamous cell carcinoma: Initial institutional report in an ongoing phase 3 study”. JAMA otolaryngology– head & neck surgery 139 (9): 895–902. doi:10.1001/jamaoto.2013.4239. PMID 24051744.
http://www.google.com/patents/US8247538
Radiopharmaceuticals for use in therapy employ radionuclides which are generally longer in half-life and weaker in penetration capability, but emit stronger radiation, sufficient to kill cells, in relation to that for use in diagnosis. Alpha ray-emitting radionuclides are excluded from radiopharmaceuticals for the reason that they are highly radioactive and difficult to purchase and to attach to other compounds. All of the radionuclides currently used in pharmaceuticals are species that emit beta rays.
As mentioned above, radiopharmaceuticals, whether for use in therapy or diagnosis, are prepared by labeling pharmaceuticals with specific radionuclides. Technetium-99m (99mTc) is known as the radioisotope most widely used to label radiopharmaceuticals. Technetium-99m has a half life of as short as 6 hours and emits gamma rays at 140 KeV, and thus it is not so toxic to the body. In addition, gamma radiation from the radioisotope is highly penetrative enough to obtain images. Thanks to these advantages, technetium-99m finds a broad spectrum of therapeutic and diagnostic applications in the nuclear medicine field (Sivia, S. J., John, D. L., Potential technetium small molecule radiopharmaceuticals. Chem. Rev. 99, 2205-2218, 1999; Shuang, L., Edwards, D. S., 99mTc-Labeled small peptides as diagnostic radiopharmaceuticals. Chem. Rev. 99, 2235-2268, 1999).
Methods of labeling 99mTc-2,6-diisopropylacetanilidoiminodiacetic acid are well known in the art (Callery, P. S., Faith, W. C., et al., 1976. Tissue distribution of technetium-99m and carbon-labeled N-(2,6)-dimetylphenylcarbamoylmethyl iminodiacetic acid. J. Med. Chem. 19, 962-964; Motter, M. and Kloss, G., 1981. Properties of various IDA derivatives. J. Label. Compounds Padiopharm. 18, 56-58; Cao, Y. and Suresh, M. R. 1998. A Simple And Efficient Method For Radiolabeling Of Preformed Liposomes. J Pharm Pharmaceut Sci. 1 (1), 31-37).
Basically, the conventional methods are based on the following reaction formula. In practice, a solution of SnCl2.2H2O, serving as a reducing agent of technetium-99m, in 0.1 N HCl and 0.1 ml (10 mCi) of sodium pertechnetium were added to lyophilized 2,6-diisopropylacetanilidoiminodiacetic acid in a vial, followed by stirring at room temperature for 30 min to prepare 99mTc-2,6-diisopropylacetanilidoiminodiacetic acid. The preparation of 99mTc-2,6-diisopropylacetanilidoiminodiacetic acid may be realized according to the following reaction formula.

Such conventional processes of preparing radiopharmaceuticals labeled with technetium-99m can be divided into reactions between the radioisotope and a physiologically active material to be labeled and the separation of labeled compounds from unlabeled compounds.
| M. Molter, et al., Properties of Various IDA Derivatives, J. Label. Compounds Padiopharm., vol. 18, pp. 56-58, 1981. | ||
| 2 | Patrick S. Callery, et al., Tissue Distribution of Technetium-99m and Carbon . . . , J. Med. Chem., vol. 19, pp. 962-964, 1976. | |
| 3 | * | Sang Hyun Park et al. Synthesis and Radiochemical Labeling of N-(2,6-diisopropylacetanilido)-Iminodiacetic acid and it s analogues under microwave irradiation: A hepatobiliary imaging agent, QSAR Comb. Sci. 2004, 23, 868-874. |
| 4 | Shuang Liu, et al., 99mTc-Labeled Small Peptides as Diagnostic . . . , Chem. Rev., vol. 99, pp. 2235-2268, 1999. | |
| 5 | Shuang Liu, et al., 99mTc—Labeled Small Peptides as Diagnostic . . . , Chem. Rev., vol. 99, pp. 2235-2268, 1999. | |
| 6 | Silvia S. Jurisson, et al., Potential Technetium Small Molecule . . . , Chem. Rev., vol. 99, pp. 2205-2218, 1999. | |
| 7 | Y. Cao, et al., A Simple and Efficient Method for Radiolabeling . . . , J. Phar,. Pharmaceut. Sci., pp. 31-37, 1998. |
|
Diagnostic radiopharmaceuticals (V09)
|
|
|---|---|
| Central nervous system | |
| Skeletal system | |
| Renal | |
| Hepatic/reticuloendothelial | |
| Respiratory system | |
| Cardiovascular system | |
| Inflammation/infection |
|
| Tumor | |
| Adrenal cortex | |
| Radionuclides (including tracers) |
|
FDA Approves Neuraceq (florbetaben F18 injection) for PET Imaging of Beta-Amyloid Plaques
FLORBETABEN F18
Diagnostic radiopharmaceutical
1. Benzenamine, 4-[(1E)-2-[4-[2-[2-[2-(fluoro-18F)ethoxy]ethoxy]ethoxy]phenyl]
ethenyl]-N-methyl-
2. 4-{(1E)-2-(4-{2-[2-(2-[18F]fluoroethoxy)ethoxy]ethoxy}phenyl)eth- 1-en-1-yl}-N-methylaniline
C21H26[18F]NO3
358.5
Bayer Healthcare
UNII-TLA7312TOI
CAS REGISTRY NUMBER 902143-01-5
https://www.ama-assn.org/resources/doc/usan/florbetaben-f18.pdf


Berlin/Boston, March 20, 2014‒ Piramal Imaging today announced that the U.S. Food and Drug Administration (FDA) has approved Neuraceq. This approval comes only four weeks after receiving marketing authorization for Neuraceq from the European Commission.
Neuraceq is indicated for Positron Emission Tomography (PET) imaging of the brain to estimate beta-amyloid neuritic plaque density in adult patients with cognitive impairment who are being evaluated for Alzheimer’s disease (AD) and other causes of cognitive decline.
read at
4-[(E)-2-(4-{2-[2-(2-fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline has been labeled with [F-18]fluoride and is claimed by patent application WO2006066104 and members of the corresponding patent family.

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
The usefulness of this radiotracer for the detection of Αβ plaques have been reported in the literature (W. Zhang et al., Nuclear Medicine and Biology 32 (2005) 799-809; C. Rowe et al., Lancet Neurology 7 (2008) 1 -7).
The synthesis of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)- vinyl]-N-methylaniline has been described before:
a) W. Zhang et al., Nuclear Medicine and Biology 32 (2005) 799-809.

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
4 mg precursor 2a (2-[2-(2-{4-[(E)-2-{4-[(tert-butoxycarbonyl)(methyl)amino]- phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl methanesulfonate) in 0.2 mL
DMSO were reacted with [F-18]fluoride/kryptofix/potassium carbonate complex. The intermediate was deprotected with HCI and neutralized with
NaOH. The mixture was extracted with ethyl acetate. The solvent was dried and evaporated, the residue was dissolved in acetonitrile and purified by semi-preparative HPLC. 20% (decay corrected), 1 1 % (not corrected for decay) 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N- methylaniline were obtained in 90 min.
WO2006066104
4 mg precursor 2-[2-(2-{4-[(E)-2-{4-[(tert-butoxycarbonyl)(methyl)amino]- phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl methanesulfonate in 0.2 mL DMSO were reacted with [F-18]fluoride/kryptofix/potassium carbonate complex. The intermediates was deprotected with HCI and neutralized with NaOH. The mixture was extracted with ethyl acetate. The solvent was dried and evaporated, the residue was dissolved in acetonitrile and purified by semi- preparative HPLC. 30% (decay corrected), 17% (not corrected for decay) 4- [(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N- methylaniline were obtained in 90 min. to yield N-Boc protected 4-[(E)-2-(4-{2-[2-(2-[F- 18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline. The unreacted perfluorinated precursor was removed using a fluorous phase cartridge.
Deprotection, final purification and formulation to obtain a product, suitable for injection into human is not disclosed. Furthermore, the usefulness (e.g. regarding unwanted F-19/F-18 exchange) of this approach at a higher radioactivity level is not demonstrated. Finally, this method would demand a two-pot setup (first reaction vessel: fluorination, followed by solid-phase- extraction, and deprotection in the second reaction vessel).
However, the focus of the present invention are compounds and methods for an improved “one-pot process” for the manufacturing of 4-[(E)-2-(4-{2-[2-(2- [F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline.
Very recently, further methods have been described:
d) US201001 13763
The mesylate precursor 2a was reacted with [F-18]fluoride species in a solvent mixture consisting of 100 μΙ_ acetonitrile and 500 μΙ_ tertiary alcohol. After fluorination for 10 min at 100-150 °C, the solvent was evaporated. After deprotection (1 N HCI, 5 min, 100-120 °C), the crude product was purified by HPLC (C18 silica, acetonitrile / 0.1 M ammonium formate).
e) H. Wang et al., Nuclear Medicine and Biology 38 (201 1 ) 121 -127
5 mg precursor 2a (2-[2-(2-{4-[(E)-2-{4-[(tert-butoxycarbonyl)(methyl)amino]- phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl methanesulfonate) in 0.5 ml_
DMSO were reacted with [F-18]fluoride/kryptofix/potassium carbonate complex. The intermediate was deprotected with HCI and neutralized with NaOH. The crude product was diluted with acetonitrile / 0.1 M ammonium dformate (6/4) and purified by semi-preparative HPLC. The product fraction was collected, diluted with water, passed through a C18 cartridge and eluted with ethanol, yielding 17% (not corrected for decay) 4-[(E)-2-(4-{2-[2-(2-[F- 18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline within 50 min. In the paper, the conversion of an unprotected mesylate precursor (is described:
5 mg unprotected mesylate precursor (2-{2-[2-(4-{(E)-2-[4- (methylamino)phenyl]vinyl}phenoxy)ethoxy]-ethoxy}ethyl 4- methanesulfonate) in 0.5 ml_ DMSO were reacted with [F- 18]fluoride/kryptofix/potassium carbonate complex. The crude product was diluted with acetonitrile / 0.1 M ammonium formate (6/4) and purified by semi- preparative HPLC. The product fraction was collected, diluted with water, passed through a C18 cartridge and eluted with ethanol, yielding 23% (not corrected for decay) 4-[(E)-2-(4-{2-[2-(2-[F-
18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline within 30 min. Beside the purification by HPLC, a process based on solid-phase-extraction was investigated, but the purity was inferior to that with HPLC purification. So far, one-pot radiolabelings have been performed using a mesylate precursor. It is know, that for F-18 labeling of stilbenes, mesylates have advantages over corresponding tosylates by providing more clean reactions with less amount of by-products (W. Zhang et al. Journal of Medicinal Chemistry 48 (2005) 5980- 5988), whereas the purification starting from the tosylate precursor was tedious and time consuming resulting in a low yield.
In contrast to this teaching of the prior art, we found advantages of tosylate and further arylsulfonate precursors for 4-[(E)-2-(4-{2-[2-(2-[F- 18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline compared to the corresponding mesylate. Less non-radioactive by-products that eluted close to the retention time of 4-[(E)-2-(4-{2-[2-(2-[F-
18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline were found in the crude products if arylsulfonate precursors were used (Example 2 – Example 6) compared to the crude mixture that was obtained after conversion of the mesylate precursor (Example 1 ).
The favorable by-product profile after radiolabeling of tosylate precursor 2b (Figure 10) compared to the radiolabeling of mesylate precursor 2a (Figure 7) supported an improved cartridge based purification (Example 8, Example 9).
…………………
The term “F-18” means fluorine isotope 18F. The term”F-19″ means fluorine isotope 19F. EXAMPLES
Example 1 Radiolabeling of mesylate precursor 2a

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2a (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 1 ). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 21 % (corrected for decay).
Example 2 Synthesis and radiolabeling of tosylate precursor 2b


4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
4-Dimethylaminopyridine (26.7 mg) and triethylamine (225 μΙ_) were added to a solution of 1 .0 g terf-butyl {4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate (4) in dichloromethane (12 mL) at 0 °C. A solution of p- toluenesulfonyl chloride (417 mg) in dichloromethane (13.5 mL) was added at 0 °C. The resulting mixture was stirred at room temperature over night. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography (silica, 0- 80% ethyl acetate in hexane). 850 mg 2b were obtained as colorless solid.
1 H NMR (300 MHz, CDCI3) δ ppm 1 .46 (s, 9 H), 2.43 (s, 3 H), 3.27 (s, 3 H), 3.59-3.73 (m, 6 H), 3.80- 3.86 (m, 2 H), 4.05-4.19 (m, 2 H), 6.88-7.05 (m, 4 H), 7.21 (d, J = 8.3 Hz, 2 H), 7.32 (d, J = 8.3 Hz, 2 H), 7.39-7-47 (m, 4 H), 7.80 (d, J = 8.3 Hz, 2 H). MS (ESIpos): m/z = 612 (M+H)+
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2b (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 2). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 25% (corrected for decay).
Example 3 Synthesis and radiolabeling of 2c (2-[2-(2-{4-[(E)-2-{4-[(tert- butoxycarbonyl)(methyl)amino]phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl
4-bromobenzenesulfonate)

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline To a stirred solution of 100 mg (0,219 mmol) tert-butyl-{4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate
(WO2006/066104) in 2 mL THF was added a solution of 140 mg (0.548 mmol) 4-brombenzene sulfonylchlorid in 3 mL THF drop by drop. The reaction mixture was cooled to 0°C. 156.8 mg (1 .1 mmol) potassium trimethylsilanolat was added. The milky suspension was stirred at 0°C for 2 hours and at 80°C for another 2 hours. The reaction mixture was poured onto ice-cooled water. The aqueous solution was extracted with dichloromethane several times. The combined organic phases were dried with sodium sulphate and concentrated in vacuum. The crude product was purified using silica gel with ethyl acetate/hexane-gradient as mobile phase. The desired product 2c was obtained with 77 mg (0.1 14 mmol, 52.0 % yield).
1 H NMR (300 MHz, CDCI3) δ ppm 1 .39 (s, 10 H) 3.20 (s, 3 H) 3.50 – 3.57 (m, 2 H) 3.57 – 3.61 (m, 2 H) 3.61 – 3.66 (m, 2 H) 3.72 – 3.80 (m, 2 H) 4.02 – 4.10 (m, 2 H) 4.10 – 4.17 (m, 2 H) 6.79 – 6.85 (m, 2 H) 6.91 (d, J=8.10 Hz, 2 H) 7.10 – 7.17 (m, 2 H) 7.32 – 7.41 (m, 5 H) 7.57 – 7.65 (m, 2 H) 7.67 – 7.74 (m, 2 H)
MS (ESIpos): m/z = 676/678 (M+H)+
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2c (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 3). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 43% (corrected for decay). Example 4 Synthesis and radiolabeling of 2d (2-[2-(2-{4-[(E)-2-{4-[(tert- butoxycarbonyl)(methyl)amino]phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl
4-(adamantan-1 -yl)benzenesulfonate)

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
To a stirred solution of 151 mg (0,330 mmol) tert-butyl-{4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate
(WO2006/066104), 4.03 mg (0,033 mmol) DMAP und 36.7 mg (363 mmol) triethylamine in 4 mL dichlormethane was added a solution of 97,4 mg (0,313 mmol) 4-(adamantan-1 -yl)benzene sulfonylchloride in 1 mL dichlormethane at 0°C. The reaction mixture was stirred at 0°C for 1 hour and over night at room temperature. 7.3 mg (0,072 mmol) triethylamin und 19.5 mg (0.062 mmol) 4- (adamantan-l -yl)benzenesulfonyl chloride were added to the reaction mixture. The reaction mixture was stirred at room temperature for 3 days. It was concentrated in vacuum. The crude product was purified using silica gel with ethyl acetate/hexane-gradient as mobile phase. The desired product 2d was obtained with 104 mg (0.142 mmol, 43.4% yield).
1 H NMR (300 MHz, CDCI3) δ ppm 1 .51 (s, 9 H), 1 .62 (s, 1 H), 1 .74 – 1 .91 (m, 6 H), 1 .94 (d, J=3.20 Hz, 6 H), 2.16 (br. s., 3 H), 3.31 (s, 3 H), 3.63 – 3.69 (m, 2 H), 3.69 – 3.73 (m, 2 H), 3.76 (dd, J=5.27, 4.52 Hz, 2 H), 3.89 (d, J=4.90 Hz, 2 H), 4.13 – 4.26 (m, 4 H), 6.95 (d, J=8.85 Hz, 2 H), 7.02 (d, J=8.29 Hz, 2 H), 7.25 (d, J=8.48 Hz, 2 H), 7.40 – 7.52 (m, 4 H), 7.55 (m, J=8.67 Hz, 2 H), 7.89 (m, J=8.67 Hz, 2 H)
MS (ESIpos): m/z = 732 (M+H)+
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2d (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 4). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 27% (corrected for decay).
Example 5 Synthesis and radiolabeling of 2e (2-[2-(2-{4-[(E)-2-{4-[(tert- butoxycarbonyl)(methyl)amino]phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl
4-cyanobenzenesulfonate)


4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
To a stirred solution of 151 mg (0.330 mmol) tert-butyl-{4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate
(WO2006/066104), 4.03 mg (0.033 mmol) DMAP und 36.7 mg (0.363 mmol) triethylamine in 4 mL dichlormethane was added a solution of 63.2 mg (0.313 mmol) 4-cyanobenzenesulfonyl chloride in 1 mL dichlormethane at 0°C. The reaction mixture was stirred over night and concentrated in vacuum. The crude product was purified using silica gel with ethyl acetate/hexane-gradient as mobile phase. The desired product 2e was obtained with 118 mg (0.190 mmol, 57.6 % yield).
1 H NMR (400 MHz, CDCI3) δ ppm 1 .47 (s, 9 H) 3.28 (s, 3 H) 3.58 – 3.63 (m, 2 H) 3.63 – 3.68 (m, 2 H) 3.70 – 3.75 (m, 2 H) 3.81 – 3.87 (m, 2 H) 4.1 1 – 4.18 (m, 2 H) 4.24 – 4.30 (m, 2 H) 6.91 (d, J=8.59 Hz, 2 H) 6.99 (dt, 2 H) 7.22 (d, J=8.34 Hz, 2 H) 7.39 – 7.50 (m, 4 H) 7.83 (m, J=8.59 Hz, 2 H) 7.98 – 8.1 1 (m, 2 H)
MS (ESIpos): m/z = 623 (M+H)+
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2e (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 5). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 31 % (corrected for decay).
Example 6 Synthesis and radiolabeling of 2f (2-[2-(2-{4-[(E)-2-{4-[(tert- butoxycarbonyl)(methyl)amino]phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl
2-nitrobenzenesulfonate)


4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- eth oxy} phe nyl )vi ny I] -N -methyla n i I i ne
To a stirred solution of 200 mg (0.437 mmol) tert-butyl-{4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate
(WO2006/066104), 5.34 mg (0.044 mmol) DMAP und 47.5 mg (0.470 mmol) triethylamine in 4 mL dichlormethane was added a solution of 92 mg (0,415 mmol) 2-nitrobenzenesulfonyl chloride in 1 mL dichlormethane at 0°C. The reaction mixture was stirred over night and concentrated in vacuum. The crude product was purified with ethyl acetate/hexane-gradient as mobile phase using silica gel. The desired product 2f was obtained with 77 mg (0.1 19 mmol, 59.5 % yield). 1 H NMR (400 MHz, CDCI3) δ ppm 1 .39 (s, 9 H) 3.20 (s, 3 H) 3.55 – 3.63 (m, 4 H) 3.59 (m, 4 H) 3.69 – 3.74 (m, 2 H) 3.75 – 3.80 (m, 2 H) 4.06 (dd, J=5.68, 3.92 Hz,
2 H) 4.32 – 4.37 (m, 2 H) 6.80 – 6.84 (m, 2 H) 6.84 – 6.98 (dt, 2 H) 7.14 (d, J=8.59 Hz, 2 H) 7.35 (d, J=3.03 Hz, 2 H) 7.37 (d, J=2.78 Hz, 2 H) 7.62 – 7.74 (m,
3 H) 8.06 – 8.1 1 (m, 1 H)
FDA approves second brain imaging drug Vizamyl (flutemetamol F 18 injection)to help evaluate patients for Alzheimer’s disease, dementia
 is the structure on right
is the structure on right
Vizamyl (flutemetamol F 18 injection)
http://jnm.snmjournals.org/content/50/8/1251/F1.expansion.html get structure

| Chemical name: | 2-{3-[18F]fluoro-4-(methylamino)phenyl}-1,3-benzothiazol-6-ol diagnostic aid |
| Cas Number | 765922-62-1 |
| INN name | flutemetamol |
| Molecular Formula: |  |
GE Healthcare
FDA PRESS RELEASE
For Immediate Release: Oct. 25, 2013
http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm372261.htm
The U.S. Food and Drug Administration today approved Vizamyl (flutemetamol F 18 injection), a radioactive diagnostic drug for use with positron emission tomography (PET) imaging of the brain in adults being evaluated for Alzheimer’s disease (AD) and dementia.
Dementia is associated with diminishing brain functions such as memory, judgment, language and complex motor skills. The dementia caused by AD is associated with the accumulation in the brain of an abnormal protein called beta amyloid and damage or death of brain cells. However, beta amyloid can also be found in the brain of patients with other dementias and in elderly people without neurologic disease.
Vizamyl works by attaching to beta amyloid and producing a PET image of the brain that is used to evaluate the presence of beta amyloid. A negative Vizamyl scan means that there is little or no beta amyloid accumulation in the brain and the cause of the dementia is probably not due to AD. A positive scan means that there is probably a moderate or greater amount of amyloid in the brain, but it does not establish a diagnosis of AD or other dementia. Vizamyl does not replace other diagnostic tests used in the evaluation of AD and dementia.
http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm372261.htm
About GE Healthcare
GE Healthcare provides transformational medical technologies and services to meet the demand for increased access, enhanced quality and more affordable healthcare around the world. GE GE -0.23% works on things that matter – great people and technologies taking on tough challenges. From medical imaging, software & IT, patient monitoring and diagnostics to drug discovery, biopharmaceutical manufacturing technologies and performance improvement solutions, GE Healthcare helps medical professionals deliver great healthcare to their patients.
For our latest news, please visit http://newsroom.gehealthcare.com
Flutemetamol [ 18 F] Injection is a diagnostic positron emission tomography (PET) agent for the imaging of β-amyloid plaques in the brain. The synthesis of the agent can be performed using automated synthesis platforms with or without using specially-tailored cassettes. For example, the synthesis can be performed using either the TRACERlab FX F-N platform or the FASTlab™ platform, commercially available from GE Healthcare a division of General Electric Company in conjunction with auxiliary preparative high pressure liquid
chromatography equipment. After synthesis, the bulk agent is transferred to high pressure liquid chromatography (HPLC) equipment to separate the physico-chemically similar compounds [ 18 F] flutemetamol from its deprotected precursor, AHl 11832 (6-hydroxy-2-(4′-(N- methyl)amino-3′-nitro)phenylbenzothiazole) and hence obtain purified [ 18 F] flutemetamol.
However there still exists a need in the art for alternative purification methods for the preparation of [ 18 F] flutemetamol. The invention as described below answers such a need. Specifically, Applicants have now found a process that eliminates the use of preparative HPLC equipment. SUMMARY OF THE INVENTION
As [ 18 F]flutemetamol and its deprotected precursor, AH111832 (6-hydroxy-2-(4′-(N- methyl)amino-3′-nitro)phenylbenzothiazole) are physico-chemically very similar, preparative HPLC is required to separate them. However, Applicants have now found that it is possible to replace the preparative HPLC equipment in previous purification processes with low cost, single-use solid phase extraction (SPE) cartridges for purification of [ 18 F]flutemetamol.
Accordingly, the present invention provides a purification process comprising the following steps:
(a) passing a diluted crude product reaction mixture comprising flutemetamol through a first reverse phase SPE cartridge;
(b) washing said first reverse phase SPE cartridge with a water/acetonitrile,
tetrahydrofuran(THF)/water, methanol(MeOH)/water or isopropanol/water mixture; preferably, a water/acetonitrile mixture;
(c) rinsing said first reverse phase SPE cartridge with water once step (b) is completed; (d) eluting said first reverse phase SPE cartridge with acetonitrile or tetrahydrofuran; preferably, acetonitrile;
(e) directly passing the mixture from said eluting step (d) through a normal phase SPE cartridge to give an acetonitrile or tetrahydrofuran solution; preferably, an acetonitrile solution, comprising purified flutemetamol; (f) diluting said acetonitrile or tetrahydrofuran solution; preferably, an acetonitrile solution, comprising purified flutemetamol, with water to form a diluted water/acetonitrile or a diluted water/tetrahydrofuran solution; preferably, a diluted water/acetonitrile solution, comprising purified flutemetamol, wherein said water/acetonitrile solution contains about 40- 70% (v/v) water; preferably at least about 40% (v/v) water; more preferably at least about 50% (v/v) water;
(g) passing the diluted water/acetonitrile or diluted water/tetrahydrofuran solution; preferably, diluted water/acetonitrile solution, comprising purified flutemetamol of step (f) through a second reverse phase SPE cartridge and trapping the flutemetamol on said cartridge second reverse phase SPE cartridge;
(h) rinsing said second reverse phase SPE cartridge with water; and
(i) eluting the trapped purified flutemetamol from second reverse phase SPE cartridge with an injectable organic solvent; preferably, ethanol or DMSO; preferably with ethanol.
According to the invention, the purified flutemetamol can be collected after step (i).
The present invention also provides a purification process of the present invention, wherein the process is automated.
yr 2011 clip
Alzheimer’s disease (AD) is defined histologically by the presence of extracellular β-amyloid (Aβ) plaques and intraneuronal neurofibrillary tangles in the cerebral cortex. The diagnosis of dementia, along with the prediction of who will develop dementia, has been assisted by magnetic resonance imaging and positron emission tomography (PET) by using [18F]fluorodeoxyglucose (FDG). These techniques, however, are not specific for AD. Based on the chemistry of histologic staining dyes, several Aβ-specific positron-emitting radiotracers have been developed to image neuropathology of AD. Among these, [11C]PiB is the most studied Aβ-binding PET radiopharmaceutical in the world. The histologic and biochemical specificity of PiB binding across different regions of the AD brain was demonstrated by showing a direct correlation between Aβ-containing amyloid plaques and in vivo [11C]PiB retention measured by PET imaging. Because 11C is not ideal for commercialization, several 18F-labeled tracers have been developed. At this time, [18F]3′-F-PiB (Flutemetamol), 18F-AV-45 (Florbetapir), and 18F-AV-1 (Florbetaben) are undergoing extensive phase II and III clinical trials. This article provides a brief review of the amyloid biology and chemistry of Aβ-specific 11C and 18F-PET radiopharmaceuticals. Clinical trials have clearly documented that PET radiopharmaceuticals capable of assessing Aβ content in vivo in the brains of AD subjects and subjects with mild cognitive impairment will be important as diagnostic agents to detect in vivo amyloid brain pathology. In addition, PET amyloid imaging will also help test the amyloid cascade hypothesis of AD and as an aid to assess the efficacy of antiamyloid therapeutics currently under development in clinical trials.
Xofigo Injection Recommended for Approval in EU

Cl 223Ra Cl
is the structure
http://www.ama-assn.org/resources/doc/usan/radium-ra-223-dichloride.pdf check out yourself
Xofigo® (radium Ra 223 dichloride) Injection Recommended for Approval in the European Union
Oslo, Norway, 20 September 2013 – Algeta ASA (OSE: ALGETA), announced today that Bayer has received a positive opinion from the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) recommending approval of Xofigo® (radium Ra 223 dichloride) in Europe. The proposed indication is for the treatment of adults with castration-resistant prostate cancer, symptomatic bone metastases and no known visceral metastases. The decision of the European Commission (EC) on the approval is expected in the fourth quarter of 2013.
Xofigo® (radium Ra 223 dichloride) injection was approved by the US Food and Drug Administration (FDA) in May 2013 for the treatment of patients with CRPC, symptomatic bone metastases and no known visceral metastatic disease and is now available in the United States at licensed facilities. read all at
http://www.pharmalive.com/xofigo-injection-recommended-for-approval-in-eu
old article
FDA Approves Xofigo for Advanced Prostate Cancer
May 15, 2013 — The U.S. Food and Drug Administration today approved Xofigo (radium Ra 223 dichloride) to treat men with symptomatic late-stage (metastatic) castration-resistant prostate cancer that has spread to bones but not to other organs. It is intended for men whose cancer has spread after receiving medical or surgical therapy to lower testosterone.
Prostate cancer forms in a gland in the male reproductive system found below the bladder and in front of the rectum. The male sex hormone testosterone stimulates the prostate tumors to grow. According to the National Cancer Institute, an estimated 238,590 men will be diagnosed with prostate cancer and 29,720 will die from the disease in 2013.
Xofigo is being approved more than three months ahead of the product’s prescription drug user fee goal date of Aug. 14, 2013, the date the agency was scheduled to complete review of the drug application. The FDA reviewed Xofigo under the agency’s priority review program, which provides for an expedited review of drugs that appear to provide safe and effective therapy when no satisfactory alternative therapy exists, or offer significant improvement compared to marketed products.
“Xofigo binds with minerals in the bone to deliver radiation directly to bone tumors, limiting the damage to the surrounding normal tissues,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Xofigo is the second prostate cancer drug approved by the FDA in the past year that demonstrates an ability to extend the survival of men with metastatic prostate cancer.”
In August 2012, the FDA approved Xtandi to treat men with metastatic castration-resistant prostate cancer that has spread or recurred, even with medical or surgical therapy to minimize testosterone. Xtandi is approved for patients who have previously been treated the chemotherapy drug docetaxel.
Xofigo’s safety and effectiveness were evaluated in a single clinical trial of 809 men with symptomatic castration-resistant prostate cancer that spread to bones but not to other organs. Patients were randomly assigned to receive Xofigo or a placebo plus best standard of care.
The study was designed to measure overall survival. Results from a pre-planned interim analysis showed men receiving Xofigo lived a median of 14 months compared to a median of 11.2 months for men receiving placebo. An exploratory updated analysis conducted later in the trial confirmed Xofigo’s ability to extend overall survival.
The most common side effects reported during clinical trials in men receiving Xofigo were nausea, diarrhea, vomiting and swelling of the leg, ankle or foot. The most common abnormalities detected during blood testing included low levels of red blood cells (anemia), lymphocytes (lymphocytopenia), white blood cells (leukopenia), platelets (thrombocytopenia) and infection-fighting white blood cells (neutropenia).
Xofigo is marketed by Wayne, N.J.-based Bayer Pharmaceuticals. Xtandi is co-marketed by Astellas Pharma U.S., Inc. of Northbrook, Ill., and Medivation, Inc. of San Francisco, Calif.
Navidea starts clinical trial for Alzheimer’s diagnostic drug


AZD4694, NAV4694 STRUCTURE
Navidea starts clinical trial for Alzheimer’s diagnostic drug
Business First of Columbus
The Phase 3 trial for the Alzheimer’s agent, at the moment named NAV4694, will compare how well the drugdisplays the buildup of a damaging protein in the brain of patients believed to have Alzheimer’s compared with what’s found in the autopsy. There …
read all at
http://www.bizjournals.com/columbus/news/2013/06/27/navidea-starts-clinical-trial-for.html
http://jnm.snmjournals.org/content/54/6/880.abstract
Navidea Biopharmaceuticals, a Dublin, Ohio biopharmaceutical company focused on precision diagnostics, earlier this week announced the completion of a study of its novel radiopharmaceutical NAV4694 as a biomarker for Alzheimer’s disease (AD).
 NAV4694 is designed to aid visual detection and quantification of cerebral beta amyloid in diagnosing Alzheimer’s disease (AD). One hallmark of AD is the accumulation of beta amyloid plaques between nerve cells in the brain.
NAV4694 is designed to aid visual detection and quantification of cerebral beta amyloid in diagnosing Alzheimer’s disease (AD). One hallmark of AD is the accumulation of beta amyloid plaques between nerve cells in the brain.
The study was designed and conducted by Navidea’s partner, AstraZeneca, to assess the safety and of the biomarker during PET scanning in subjects with AD and in healthy volunteers. Efficacy measures included binding parameters and overall image quality. The 16-patient trial was completed at Karolinska Institutet sites in Stockholm, Sweden.
PHASE1,Progenics Pharmaceuticals’ Novel Small Molecule Drugs Targeting PSMA Successfully Visualize Prostate Cancer, 123-I-MIP-1095

Name: 123-I-MIP-1095
Synonym: 123-I-MIP-1095; [123I]-MIP-1095; iodine I 123 IMP-1095; 2-(3-{l-carboxy-5-[3-(4-iodo-phenyl)-ureido]-pentyl}-ureido)-pentanedioic acid.; [123I]-(S)-2-(3-((S)-1-carboxy-5-(3-(4-iodophenyl)ureido)pentyl)ureido)pentanedioic acid
IUPAC/Chemical name:
2-(3-(1-carboxy-5-(3-(4-iodophenyl)ureido)pentyl)ureido)pentanedioic acid
Chemical Formula: C19H25123IN4O8
Exact Mass: 560.07284
Molecular Weight: 560.33
123-I-MIP-1095
An iodine 123-radiolabled small molecule that exhibits high affinity for prostate-specific membrane antigen (PSMA) with potential use in molecular imaging. 123-I-MIP-1095, a radiolabeled glutamate-urea-lysine analogue, selectively binds PSMA, which allows imaging of PSMA-expressing prostate cancer cells with gamma scintigraph. PSMA is a transmembrane glycoprotein highly expressed by malignant prostate epithelial cells and vascular endothelial cells of various solid tumors.
![]()
| Synonym: | iodine I 123 IMP-1095 | ||
| Chemical structure: | 2-(3-{l-carboxy-5-[3-(4-iodo-phenyl)-ureido]-pentyl}-ureido)-pentanedioic acid | ||
March 5, 2013
Progenics Pharmaceuticals, Inc. (Nasdaq:PGNX) reported positive clinical data from a study of two novel radiolabeled small molecules targeting prostate-specific membrane antigen (PSMA). The imaging agents — 123I-MIP-1072 and 123I-MIP-1095 — had a high sensitivity of lesion detection in bone, tissue and the prostate gland with minimal retention in non-target tissue. The research was published as the cover article in the March issue of The Journal of Nuclear Medicine.
“Existing imaging techniques are limited in their ability to diagnose and stage prostate cancer,” said John J. Babich, Ph.D., senior author of the article “First-in-Man Evaluation of Two High-Affinity PSMA-Avid Small Molecules for Imaging Prostate Cancer.” “The approach described in this paper has the potential to assess disease status more accurately. It could help clinicians select optimal treatments and lead to better patient outcomes.”
Separate phase 1 studies were conducted under an exploratory investigational new drug (IND) application to measure the potential effectiveness of the small molecules in diagnosing and staging prostate cancer. In the first study, seven patients with documented prostate cancer were administered doses of 123I-MIP-1072 and 123I-MIP-1095, two weeks apart. In the second study, six healthy volunteers received 123I-MIP-1072 only. Whole body planar imaging and single photon emission computed tomography (SPECT)/computed tomography (CT) were performed for each group, and pharmacokinetics, tissue distribution, excretion, safety and organ radiation dose were analyzed.
Based on the data reported, Progenics is conductinga global, multi-center phase 2 trial investigating a next generation radiolabeled small molecule targeting PSMA, MIP-1404.
Mark R. Baker, chief executive officer of Progenics, said, “We recently acquired all of the rights to the compounds described in this Journal of Nuclear Medicine paper, as well as to the phase 2 stage imaging agent MIP-1404, through Progenics’ acquisition of Molecular Insight Pharmaceuticals. It is gratifying to see this expansion of our oncology pipeline demonstrating progress so soon.”
Robert J. Israel, M.D., Progenics’ senior vice president of medical affairs and clinical research, said, “We believe that MIP-1404 has excellent potential as a diagnostic radiopharmaceutical. Results to date from the study compounds and MIP-1404 show PSMA as a robust target for prostate cancer molecular imaging, and that a radiolabeled small molecule, which binds PSMA with high affinity, has the potential to detect prostate cancer throughout the body. Cancer treatment guidelines call for imaging prostate cancer with conventional bone scans or MRI. A more accurate method of imaging prostate cancer could be of great value.”
Mr. Baker further added, “Thought leaders in prostate cancer care are focused on avoiding unnecessary surgery and other invasive procedures due to the complications associated with them. Clinicians generally prefer “watchful waiting” when the cancer appears to be indolent. At the same time, some therapeutics to treat aggressive prostate cancer have recently been approved or are under development, such as Progenics’ own PSMA ADC, which currently is in phase 2 testing. Patients and their physicians would benefit from feedback on how therapeutic agents are impacting the course of cancer, and guidance on how and when to use therapeutic agents. It is clear that an improved way to visualize prostate cancer, with a high degree of specificity and sensitivity, would better inform both “watchful waiting” and the treatment of aggressive disease. We believe that data from the ongoing phase 2 trial of MIP-1404 will demonstrate its capabilities to assist prostate cancer patients and their physicians in making these critical decisions.”
About Prostate Cancer
Prostate cancer is the most common form of cancer affecting men in the United States and is the second leading cause of cancer deaths among men each year. The American Cancer Society estimates that in 2013, 238,590 new cases of prostate cancer will be diagnosed and approximately 29,720 American men will die from the disease. Accurate diagnosis and staging of prostate cancer is critical to determining appropriate patient management.
About Progenics
Progenics Pharmaceuticals, Inc. is discovering and developing innovative medicines for oncology, with a pipeline that includes product candidates in preclinical through late-stage development. Progenics’ first commercial product, Relistor® (methylnaltrexone bromide) for opioid-induced constipation, is marketed and in further development by Salix Pharmaceuticals, Ltd. for markets worldwide other than Japan, where Ono Pharmaceutical Co., Ltd. holds an exclusive license for the subcutaneous formulation. For additional information, please visit http://www.progenics.com.
