
Home » IND
Category Archives: IND
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
XL 114, AUR 104 and XL 102, AUR 102 (NO CONCLUSIONS, ONLY PREDICTIONS)

XL 114
FOR BOTH, JUST PREDICTION
PREDICTIONS
or


N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)C(C)O)CC(N)=O
![(2S)-2-[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=117943691&t=l)

(2S)-2-[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid
CAS 2305027-62-5
C12 H20 N6 O7, 360.32Threonine, N-[[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]carbonyl]-, (2S,3ξ)-N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)C(C)O)CC(N)=O
ALSO SEE


![(2S,3R)-2-[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](http://drugapprovalsint.com/wp-content/uploads/2021/10/str1-10.jpg)
1673534-76-3C12 H20 N6 O7, 360.32
L-Threonine, N-[[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]
(2S,3R)-2-[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acidN-[[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]carbonyl]-L-threonine
CAS 1673534-76-3
PD-1-IN-1 free base, EX-A1918, CS-6240, NSC-799645, CA-170 (AUPM-170)|PDL1 inhibitor, HY-101093, PD-1-IN-1
N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)[C@@H](C)O)CC(N)=O
XL 114, AUR 104
A novel covalent inhibitor of FABP5 for cancer therapy
XL 102, AUR 102
A potent, selective and orally bioavailable inhibitor of cyclin-dependent kinase 7 (CDK7)
NO CONCLUSIONS, ONLY PREDICTIONS
PREDICTIONS MORE
![(2R,3R)-2-[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=155529077&t=l)

(2R,3R)-2-[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid
C12H20N6O7, 360.32
![(2S,3S)-2-[[(1S)-3-Amino-1-[3-[(1S)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=155339943&t=l)

(2S,3S)-2-[[(1S)-3-amino-1-[3-[(1S)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid
XL102, AUR 102
XL102 is a potent, selective and orally bioavailable covalent inhibitor of CDK7, which is an important regulator of the cellular transcriptional and cell cycle machinery. CDK7 helps regulate cell cycle progression, with overexpression observed in multiple cancers, such as breast, prostate and ovarian cancers. In preclinical studies, XL102 revealed potent anti-proliferative activity, induced cell death in a large panel of cancer cell lines and caused tumor growth inhibition and regression in xenograft models, demonstrating its potential as a targeted antitumor agent.
In late 2020, Exelixis exercised its option to in-license XL102 (formerly AUR102) from Aurigene per the companies’ July 2019 collaboration, option and license agreement. Exelixis has assumed responsibility for the future clinical development, manufacturing and commercialization of XL102. Aurigene retains limited development and commercial rights for India and Russia.
SYN

ABOUT Fatty acid-binding proteins (FABPs)
Fatty acid-binding proteins (FABPs) are involved in binding and storing hydrophobic ligands such as long-chain fatty acids, as well as transporting them to the appropriate compartments in the cell. Epidermal fatty acid-binding protein (FABP5) is an intracellular lipid-binding protein that is abundantly expressed in adipocytes and macrophages. Previous studies have revealed that the FABP5 expression level is closely related to malignancy in various types of cancer. However, its precise functions in the metabolisms of cancer cells remain unclear. Here, we revealed that FABP5 knockdown significantly induced downregulation of the genes expression, such as hormone-sensitive lipase (HSL), monoacylglycerol lipase (MAGL), elongation of long-chain fatty acid member 6 (Elovl6), and acyl-CoA synthetase long-chain family member 1 (ACSL1), which are involved in altered lipid metabolism, lipolysis, and de novo FA synthesis in highly aggressive prostate and breast cancer cells. Moreover, we demonstrated that FABP5 induced inflammation and cytokine production through the nuclear factor-kappa B signaling pathway activated by reactive oxygen species and protein kinase C in PC-3 and MDA-MB-231 cells. Thus, FABP5 might regulate lipid quality and/or quantity to promote aggressiveness such as cell growth, invasiveness, survival, and inflammation in prostate and breast cancer cells. In the present study, we have revealed for the first time that high expression of FABP5 plays a critical role in alterations of lipid metabolism, leading to cancer development and metastasis in highly aggressive prostate and breast cancer cells.
Fatty acid-binding protein, epidermal is a protein that in humans is encoded by the FABP5 gene
Function
This gene encodes the fatty acid binding protein found in epidermal cells, and was first identified as being upregulated in psoriasis tissue. Fatty acid binding proteins are a family of small, highly conserved, cytoplasmic proteins that bind long-chain fatty acids and other hydrophobic ligands. It is thought that FABPs roles include fatty acid uptake, transport, and metabolism.[6]
The phytocannabinoids (THC and CBD) inhibit endocannabinoid anandamide (AEA) uptake by targeting FABP5, and competition for FABPs may in part or wholly explain the increased circulating levels of endocannabinoids reported after consumption of cannabinoids.[7] Results show that cannabinoids inhibit keratinocyte proliferation, and therefore support a potential role for cannabinoids in the treatment of psoriasis.[8]
Interactions
FABP5 has been shown to interact with S100A7.[
ABOUT CD47/SIRPa axis
CD47/SIRPa axis is established as a critical regulator of myeloid cell activation and serves as an immune checkpoint for macrophage mediated phagocytosis. Because of its frequent upregulation in several cancers, CD47 contributes to immune evasion and cancer progression. CD47 regulates phagocytosis primarily through interactions with SIRPla expressed on macrophages. Blockade of SIRPla/CD47 has been shown to dramatically enhance tumor cell phagocytosis and dendritic cells maturation for better antigen presentation leading to substantially improved antitumor responses in preclinical models of cancer (M. P. Chao et al. Curr Opin Immunol. 2012 (2): 225-232). Disruption of CD47-SIRPa interaction is now being evaluated as a therapeutic strategy for cancer with the use of monoclonal antibodies targeting CD47 or SIRPa and engineered receptor decoys.
CD47 is expressed on virtually all non-malignant cells, and blocking the CD47 or the loss of CD47 expression or changes in membrane distribution can serve as markers of aged or damaged cells, particularly on red blood cells (RBC). Alternatively, blocking SIRPa also allows engulfment of targets that are not normally phagocytosed, for those cells where pre-phagocytic signals are also present. CD47 is a broadly expressed transmembrane glycoprotein with a single Ig-like domain and five membrane- spanning regions, which functions as a cellular ligand for SIRPa with binding mediated through the NH2-terminal V-like domain of SIRPa. SIRPa is expressed primarily on myeloid cells, including macrophages, granulocytes, myeloid dendritic cells (DCs), mast cells, and their precursors, including hematopoietic stem cells.
CD47 is also constitutively upregulated on a number of cancers such as Non-Hodgkin Lymphoma (NHL), Acute myeloid leukemia (AML), breast, colon, glioblastoma, glioma, ovarian, bladder and prostate cancers, etc. Overexpression of CD47 by tumor cells, which efficiently helps them to escape immune surveillance and killing by innate immune cells. However, in most of the tumor types, blockade of the CD47-SIRPa interaction as a single agent may not be capable of inducing significant phagocytosis and antitumor immunity, necessitating the need to combine with other therapeutic agents. The concomitant engagement of activating receptors such as Fc-receptors (FcRs) or other prophagocytic receptors (collectively known as “eat-me” signals) may be necessary for exploiting the maximum potential of the CD-47-SIPRa pathway blockade.
The role of engagement of prophagocytic receptors is proved by inefficiency to trigger phagocytosis either by anti-CD47 F(ab) fragments, single chain variable fragments of CD-47 or non-Fc portion- containing SIRPa proteins in blocking of the CD47-SIRPa interaction. When activating prophagocytic receptors are engaged, as evident in the case of using Fc portion-containing blocking anti-CD47 antibodies, CD47- SIRPa blockade is able to trigger more efficient phagocytosis. Combining CD47-SIRPa blocking agents with therapeutic antibodies (Fc-containing) targeting tumor antigens stimulate activating Fc receptors (FcRs) leading to efficient phagocytosis. The Fc portion of therapeutic antibody targeting tumor antigen also induces antibody-dependent cellular cytotoxicity (ADCC), which also adds to the therapeutic efficacy. Hence antibodies selected from the group consisting of rituximab, herceptin, trastuzumab, alemtuzumab, bevacizumab, cetuximab and panitumumab, daratumumab due to its tumor targeting nature and ADCC, can trigger more efficient phagocytosis.
Earlier approaches to disrupt CD47- SIRPa interaction utilized monoclonal antibodies targeting CD47 or SIRPa and engineered receptor decoys fused to Fc fragment. However, a concern with this approach is that CD47 is highly expressed on both hematopoietic and non-hematopoietic normal cells. Hence along with tumor cells CD47-SIRPa blocking agents containing Fc-portion may also target many normal cells potentially leading to their elimination by macrophages. The interaction of blocking antibodies with normal cells is considered as a major safety issue resulting in anemia, thrombocytopenia, and leukopenia. These agents may also affect solid tissues rich in macrophages such as liver, lung, and brain. Hence it may be ideal to block the CD47- SIRPa interaction by agents devoid of Fc portion, such as small
molecules, peptides, Fab fragments etc. while activating prophagocytic receptors in tumor cells by appropriate combinations to induce efficient phagocytosis of tumor cells.
Apart from Fc Receptors, a number of other prophagocytic receptors are also reported to promote engulfment of tumor cells in response to CD47-SIRPa blockade by triggering the phagocytosis. These include receptors for SLAMF7, Mac-l, calreticulin and possibly yet to identified receptors. B cell tumor lines such as Raji and other diffuse large B cell lymphoma express SLAMF7 and are implicated in triggering prophagocytic signals during CD47-SIRPa blockade.
Therapeutic agents known to activate prophagocytic receptors are also therefore ideal partners for use in combination with CD47-SIRPa blocking agents to achieve efficient phagocytosis. These agents include proteasome inhibitors (bortezomib, ixazomib and carfilzomib), Anthracyclines (Doxorubicin, Epirubicin, Daunorubicin, Idarubicin, Mitoxantrone) Oxaliplatin, Cyclophosphamide, Bleomycin, Vorinostat, Paclitaxel, 5-Fluorouracil, Cytarabine, BRAF inhibitory drugs (Dabrafenib, Vemurafenib), PI3K inhibitor, Docetaxel, Mitomycin C, Sorafenib, Tamoxifen and oncolytic viruses.
Apart from the specific agents known to have effect on‘eat me’ signals other agents including Abiraterone acetate, Afatinib, Aldesleukin, Aldesleukin, Alemtuzumab, Anastrozole, Axitinib, Belinostat, Bendamustine, Bicalutamide, Blinatumomab, Bosutinib, Brentuximab, Busulfan, Cabazitaxel, Capecitabine, Carboplatin, Carfilzomib, Carmustine, Ceritinib, Clofarabine, Crizotinib, Dacarbazine, Dactinomycin, Dasatinib, Degarelix, Denileukin, Denosumab, Enzalutamide, Eribulin, Erlotinib, Everolimus, Exemestane, Exemestane, Fludarabine, Fulvestrant, Gefitinib, Goserelin, Ibritumomab, Imatinib, Ipilimumab, Irinotecan, Ixabepilone, Lapatinib, Lenalidomide, Letrozole, Leucovorin, Leuprolide, Lomustine, Mechlorethamine, Megestrol, Nelarabine, Nilotinib, Nivolumab, Olaparib, Omacetaxine, Palbociclib, Pamidronate, Panitumumab, Panobinostat, Pazopanib, Pegaspargase, Pembrolizumab, Pemetrexed Disodium, Pertuzumab, Plerixafor, Pomalidomide, Ponatinib, Pralatrexate, Procarbazine, Radium 223, Ramucirumab, Regorafenib, rIFNa-2b, Romidepsin, Sunitinib, Temozolomide, Temsirolimus, Thiotepa, Tositumomab, Trametinib, Vinorelbine, Methotrexate, Ibrutinib, Aflibercept, Toremifene, Vinblastine, Vincristine, Idelalisib, Mercaptopurine and Thalidomide could potentially have effect on‘eat me’ signal pathway on combining with CD-47-SIRPa blocking agents.
In addition to the therapeutic agents mentioned above, other treatment modalities that are in use in cancer therapy also activate prophagocytic receptors, and thus can be combined with CD47-SIRPa blocking agents to achieve efficient phagocytosis. These include Hypericin-based photodynamic therapy (Hyp-PDT), radiotherapy, High-hydrostatic pressure, Photofrin-based PDT and Rose Bengal acetate -based PDT.
However, there is an unmet need for combining small molecule CD-47-SIRPa pathway inhibitors with agents capable of stimulating activating receptors such as Fc-receptors (FcRs) or other prophagocytic receptors, or combining with other treatment modalities that are in use in cancer therapy to activate prophagocytic receptors for exploiting the maximum potential of the CD-47- SIRPa pathway blockade.
CLIP
Exelixis In-Licenses Second Anti-Cancer Compound from Aurigene Following FDA Acceptance of Investigational New Drug Application for Phase 1 Clinical Trial in Non-Hodgkin’s Lymphoma
– Robust preclinical data support Exelixis’ clinical development of XL114, with phase 1 trial in Non-Hodgkin’s lymphoma expected to begin in the coming months –
– Exelixis will make an option exercise payment of $10 million to Aurigene –
https://www.businesswire.com/news/home/20211014005549/en/Exelixis-In-Licenses-Second-Anti-Cancer-Compound-from-Aurigene-Following-FDA-Acceptance-of-Investigational-New-Drug-Application-for-Phase-1-Clinical-Trial-in-Non-Hodgkin%E2%80%99s-LymphomaOctober 14, 2021 08:00 AM Eastern Daylight Time
ALAMEDA, Calif.–(BUSINESS WIRE)–Exelixis, Inc. (Nasdaq: EXEL) and Aurigene Discovery Technologies Limited (Aurigene) today announced that Exelixis has exercised its exclusive option under the companies’ July 2019 agreement to in-license XL114 (formerly AUR104), a novel anti-cancer compound that inhibits the CARD11-BCL10-MALT1 (CBM) signaling pathway, which promotes lymphocyte survival and proliferation. Exelixis has now assumed responsibility for the future clinical development, commercialization and global manufacturing of XL114. Following the U.S. Food and Drug Administration’s (FDA) recent acceptance of its Investigational New Drug (IND) application, Exelixis will soon initiate a phase 1 clinical trial evaluating XL114 monotherapy in patients with Non-Hodgkin’s lymphoma (NHL). At the American Association of Cancer Research Annual Meeting in April of this year, Aurigene presented preclinical data (Abstract 1266) demonstrating that XL114 exhibited potent anti-proliferative activity in a large panel of cancer cell lines ranging from hematological cancers to solid tumors with excellent selectivity over normal cells. In addition, oral dosing of XL114 resulted in significant dose-dependent tumor growth inhibition in diffuse large B-cell lymphoma (DLBCL) and colon carcinoma models.
“We are pleased that our agreement with Aurigene has generated a second promising compound that warrants advancement into clinical development and believe the collaboration will continue to play an important role in expanding our pipeline”
XL114 is the second molecule that Exelixis in-licensed from Aurigene under the companies’ July 2019 collaboration, option and license agreement. Exelixis previously exercised its option to in-license XL102, a potent, selective and orally bioavailable inhibitor of cyclin-dependent kinase 7 (CDK7), from Aurigene in December 2020 and initiated a phase 1 trial of XL102 as a single agent and in combination with other anti-cancer agents in patients with advanced or metastatic solid tumors in January 2021.
“We are pleased that our agreement with Aurigene has generated a second promising compound that warrants advancement into clinical development and believe the collaboration will continue to play an important role in expanding our pipeline,” said Peter Lamb, Ph.D., Executive Vice President, Scientific Strategy and Chief Scientific Officer, Exelixis. “XL114 has shown potent anti-proliferative activity in lymphoma cell lines that have aberrant activation of the CBM signaling pathway and may have a differentiated profile and potential as a best-in-class molecule that could improve outcomes for patients with Non-Hodgkin’s lymphoma and other hematologic cancers.”
XL114 was identified to have anti-proliferative activity in cell lines with constitutive activation of CBM signaling, including activated B-cell-like DLBCL (ABC-DLBCL), mantle cell lymphoma and follicular lymphoma cell lines. Further characterization of XL114 in cell-based assays demonstrated a functional role in B-cell (BCR) signaling pathways. Additionally, XL114 showed dose-dependent tumor growth inhibition in an ABC-DLBCL mouse xenograft tumor model. In preclinical development, XL114 also demonstrated a high degree of selectivity against a broad safety pharmacology panel of enzymes and receptors. While the precise molecular mechanism underlying XL114’s function in repressing BCR signaling and MALT1 activation has yet to be characterized, the fatty acid-binding protein 5 (FABP5) has been identified as a prominent XL114-binding target.
“XL114 is the second molecule that Exelixis has opted to in-license under our July 2019 agreement, underscoring the significant potential of our approach to the discovery and preclinical development of innovative cancer therapies that target novel mechanisms of action,” said Murali Ramachandra, Ph.D., Chief Executive Officer, Aurigene. “Exelixis has a track record of success in the clinical development and commercialization of anti-cancer therapies that provide patients with important new treatment options, and we are pleased that the continued advancement of XL114 will be supported by the company’s extensive clinical, regulatory and commercialization infrastructure.”
Under the terms of the July 2019 agreement, Exelixis made an upfront payment of $10 million for exclusive options to obtain an exclusive license from Aurigene to three preexisting programs, including the compounds now known as XL102 and XL114. In addition, Exelixis and Aurigene initiated three Aurigene-led drug discovery programs on mutually agreed upon targets, in exchange for an additional upfront payment of $2.5 million per program. The collaboration was expanded in 2021 to include three additional early discovery programs. Exelixis is also contributing research funding to Aurigene to facilitate discovery and preclinical development work on all nine programs. Exelixis may exercise its option for a program at any time up until the first IND for the program becomes effective. Having exercised options on two programs thus far (XL102 and XL114), if and when Exelixis exercises a future option, it will make an option exercise payment to Aurigene and assume responsibility for that program’s future clinical development and commercialization including global manufacturing. To exercise its option for XL114, Exelixis will make an option exercise payment to Aurigene of $10 million. Once Exelixis exercises its option for a program, Aurigene will be eligible for clinical development, regulatory and sales milestones, as well as royalties on future potential sales of the compound. Under the terms of the agreement, Aurigene retains limited development and commercial rights for India and Russia.
About Aurigene
Aurigene Discovery Technologies Limited is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY, NYSE: RDY, NSEIFSC: DRREDDY). Aurigene is focused on precision-oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene’s programs currently in clinical development include an oral ROR-gamma inhibitor AUR101 for moderate to severe psoriasis in phase 2 under a U.S. FDA IND and a PD-L1/VISTA antagonist CA-170 for non-squamous non-small cell lung cancer in phase 2b/3 in India. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has also partnered with several large and mid-pharma companies in the U.S. and Europe and has multiple programs in clinical development. For more information, please visit Aurigene’s website at www.aurigene.com.
About Exelixis
Founded in 1994, Exelixis, Inc. (Nasdaq: EXEL) is a commercially successful, oncology-focused biotechnology company that strives to accelerate the discovery, development and commercialization of new medicines for difficult-to-treat cancers. Following early work in model system genetics, we established a broad drug discovery and development platform that has served as the foundation for our continued efforts to bring new cancer therapies to patients in need. Our discovery efforts have resulted in four commercially available products, CABOMETYX® (cabozantinib), COMETRIQ® (cabozantinib), COTELLIC® (cobimetinib) and MINNEBRO® (esaxerenone), and we have entered into partnerships with leading pharmaceutical companies to bring these important medicines to patients worldwide. Supported by revenues from our marketed products and collaborations, we are committed to prudently reinvesting in our business to maximize the potential of our pipeline. We are supplementing our existing therapeutic assets with targeted business development activities and internal drug discovery – all to deliver the next generation of Exelixis medicines and help patients recover stronger and live longer. Exelixis is a member of the Standard & Poor’s (S&P) MidCap 400 index, which measures the performance of profitable mid-sized companies. In November 2020, the company was named to Fortune’s 100 Fastest-Growing Companies list for the first time, ranking 17th overall and the third-highest biopharmaceutical company. For more information about Exelixis, please visit www.exelixis.com, follow @ExelixisInc on Twitter or like Exelixis, Inc. on Facebook.

Dinesh Chikkanna
Director, Medicinal Chemistry Aurigene Discovery Technologies

Murali Ramachandra
CEO at Aurigene Discovery Technologies

CLIP
https://cancerres.aacrjournals.org/content/81/13_Supplement/1266
Abstract 1266: Discovery and preclinical evaluation of a novel covalent inhibitor of FABP5 for cancer therapyDinesh Chikkanna, Leena Khare Satyam, Sunil Kumar Pnaigrahi, Vinayak Khairnar, Manoj Pothuganti, Lakshmi Narayan Kaza, Narasimha Raju Kalidindi, Vijaya Shankar Nataraj, Aditya Kiran Gatta, Narasimha Rao Krishnamurthy, Sandeep Patil, DS Samiulla, Kiran Aithal, Vijay Kamal Ahuja, Nirbhay Kumar Tiwari, KB Charamannna, Pravin Pise, Thomas Anthony, Kavitha Nellore, Sanjeev Giri, Shekar Chelur, Susanta Samajdar and Murali Ramachandra
DOI: 10.1158/1538-7445.AM2021-1266 Published July 2021
Proceedings: AACR Annual Meeting 2021; April 10-15, 2021 and May 17-21, 2021; Philadelphia, PA
Abstract
Dysregulated fatty acid metabolism is thought to be a hallmark of cancer, wherein fatty acids function both as an energy source and as signals for enzymatic and transcriptional networks contributing to malignancy. Fatty acid-binding protein 5 (FABP5) is an intracellular protein that facilitates transport of fatty acids and plays a role in regulating the expression of genes associated with cancer progression such as cell growth, survival, and metastasis. Overexpression of FABP5 has been reported to contribute to an aggressive phenotype and a poor survival correlation in several cancers. Therefore, inhibition of FABP5 is considered as a therapeutic approach for cancers. Phenotypic screening of a library of covalent compounds for selective sensitivity of cancer cells followed by medicinal chemistry optimization resulted in the identification of AUR104 with desirable properties. Chemoproteomic-based target deconvolution revealed FABP5 as the cellular target of AUR104. Covalent adduct formation with Cys43 of FABP5 by AUR104 was confirmed by mass spectrometry. Target occupancy studies using a biotin-tagged AUR104 demonstrated potent covalent binding to FABP5 in both cell-free and cellular conditions. Ligand displacement assay with a fluorescent fatty acid probe confirmed the competitive binding mode of AUR104 with fatty acids. Binding at the fatty acid site and covalent bond formation with Cys43 were also demonstrated by crystallography. Furthermore, AUR104 showed a high degree of selectivity against a broad safety pharmacology panel of enzymes and receptors. AUR104 exhibited potent anti-proliferative activity in a large panel of cell lines derived from both hematological and solid cancers with a high degree of selectivity over normal cells. Anti-proliferative activity in lymphoma cell lines correlated with inhibition of MALT1 pathway activity, cleavage of RelB/Bcl10 and secretion of cytokines, IL-10 and IL-6. AUR104 displayed desirable drug-like properties and dose-dependent oral exposure in pharmacokinetic studies. Oral dosing with AUR104 resulted in dose-dependent anti-tumor activity in DLBCL (OCI-LY10) and NSCLC (NCI-H1975) xenograft models. In a repeated dose MTD studies in rodents and non-rodents, AUR104 showed good tolerability with an exposure multiple of >500 over cellular EC50 for up to 8 hours. In summary, we have identified a novel covalent FABP5 inhibitor with optimized properties that showed anti-tumor activity in in vitro and in vivo models with acceptable safety profile. The data presented here strongly support clinical development of AUR104.
Citation Format: Dinesh Chikkanna, Leena Khare Satyam, Sunil Kumar Pnaigrahi, Vinayak Khairnar, Manoj Pothuganti, Lakshmi Narayan Kaza, Narasimha Raju Kalidindi, Vijaya Shankar Nataraj, Aditya Kiran Gatta, Narasimha Rao Krishnamurthy, Sandeep Patil, DS Samiulla, Kiran Aithal, Vijay Kamal Ahuja, Nirbhay Kumar Tiwari, KB Charamannna, Pravin Pise, Thomas Anthony, Kavitha Nellore, Sanjeev Giri, Shekar Chelur, Susanta Samajdar, Murali Ramachandra. Discovery and preclinical evaluation of a novel covalent inhibitor of FABP5 for cancer therapy [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2021; 2021 Apr 10-15 and May 17-21. Philadelphia (PA): AACR; Cancer Res 2021;81(13_Suppl):Abstract nr 1266.
Patent
US20200147054 – COMBINATION OF SMALL MOLECULE CD-47 INHIBITORS WITH OTHER ANTI-CANCER AGENTS
Muralidhara Ramachandra
Pottayil Govindan Nair Sasikumar
Girish Chandrappa Daginakatte
Kiran Aithal Balkudru
PATENT
WO 2020095256
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020095256
Example- 1: The synthetic procedures for the preparation of compounds described in the present invention were described in co-pending Indian provisional patent application 201841001438 dated 12* Jan 2018, which is converted as PCT application
PCT/IB2019/050219, the contents of which are hereby incorporated by reference in their entirety.
PATENT
WO 2018178947https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018178947&tab=PCTDESCRIPTION
PATENT
WO 2019138367
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019138367
PATENT
WO 2019073399
https://patents.google.com/patent/WO2019073399A1/en
Priority to IN201741036169
Example 4 of WO 2015/033299
PATENT
https://patents.google.com/patent/BR112020014202A2/en

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
PATENT
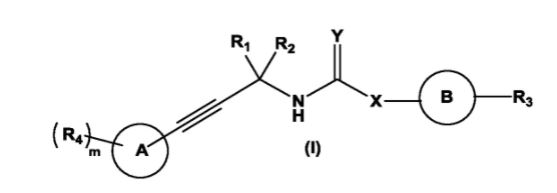
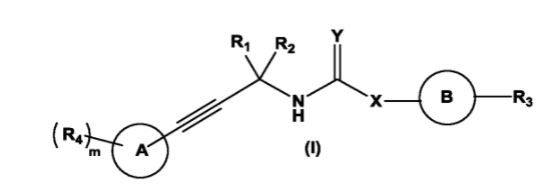
The present invention relates to substituted alkynylene compounds represented by compound of formula (I) pharmaceutically acceptable salts and stereoisomers thereof. The present invention further provides the methods of preparation of compound of formula (I) and therapeutic uses thereof as anti-cancer agents.
Patent
Example 1
(((S)-4-amino-1-(3-((S)-1,5-diaminopentyl)-1,2,4-oxadiazol-5-yl)-4-oxobutyl)carbamoyl)-L-proline (Compound 1)
Synthesis of Compound 1 b
Synthesis of Compound 1C
Synthesis of Compound 1d
Synthesis of Compound 1f
Synthesis of Compound 1g
Synthesis of Compound 1h
Synthesis of Compound 1i
Synthesis of compound 1j
Synthesis of Compound 1
Example 2
(S)-4-(3-((S)-1-amino-4-guanidinobutyl)-1,2,4-oxadiazol-5-yl)-4-(3-((S)-1-carboxy-2-phenylethyl) ureido)butanoic acid (Compound 7)
Synthesis of Compound 2b
Synthesis of Compound 2c
Synthesis of Compound 2d
Synthesis of Compound 2f
Synthesis of Compound 2g
Synthesis of Compound 2h
Synthesis of Compound 2i
Synthesis of Compound 2j
Synthesis of Compound 7
PATENT
WO 2015/033299
https://patents.google.com/patent/WO2015033299A1/en?oq=WO+2015%2f033299
Pottayil Govindan Nair SasikumarMuralidhara RamachandraSeetharamaiah Setty Sudarshan Naremaddepalli
Example 1: Synthesis of Compound 1

Step la:

Ethylchloroformate (1.5 g, 13.78 mniol) and N-Methylmorpholine ( 1.4 g, 13.78 mmol) were added to a solution of compound la (3 g, 11.48 mmol) in THE (30 mL) arid stirred at -20 °C. After 20 min. Liquid ammonia (0.77 g, 45.92 mmol) was added to the active mixed anhydride formed in- situ and stirred at 0-5 °C for 20 min. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with NaHCOs, citric acid, brine solution, dried over Na2S04 and evaporated under reduced pressure to get 2.9 g of compound lb (Yield: 96.3%). LCMS: 261.0 ( Vi+H ; .
Step lb:

1 b 1cTrifluroacetic anhydride (9.7 g, 46.0 mmol) was added to a solution of compound lb (8 g, 30.7 mmol) in pyridine (24.3 g, 307.0 mmol) and stirred at room temperature for 3 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with NaHCO?,, citric acid, brine solution, dried over Na2-S04 and evaporated under reduced pressure to afford 7 g of compound lc (Yield: 94.0%). LCMS: 187.2 (M-¾u )+.
Step lc:

1 c 1dHydroxylamine hydrochloride (3 g, 43.37 mmol) and potassium carbonate (6 g, 43.37 mmol) were added to a solution of compound lc (7 g, 28.91 mmol) in EtOH (70 m L) and stirred at 90 °C for 2 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with brine solution, dried over Na2S04 and evaporated under reduced pressure. The crude compound was purified by silica gel column chromatography (Eluent: 0-5% ethyl acetate in hexane) to get 4.2 g of compound Id (Yield: 52.8%). LCMS: 276.4 (M+H)+.Step Id:

Deoxo-Fluor® (1.83 g, 8.3 mmol) was added to a solution of Fmoc-Asn(Trt)-OH (4.5 g, 7.5 mmol) in CH2Q2 (50 mL) and stirred at 0 °C for 3 h. Then CH2CI2 was evaporated and triturated with hexane, decanted and evaporated under vacuum to get the corresponding acid fluoride. NMM (1.17 g, 1 1.6 mmol) and compound Id (1.6 g, 5.8 mmol) in THF were added to the acid fluoride and stirred at room temperature for 12 h. Then THF was evaporated and sodium acetate (0.72 g, 8.7 mmol) was added followed by EtOH (50 mL). The reaction mixture was stirred at 90 °C for 2 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with NaHCOa, citric acid, brine solution, dried over Na2S04 and evaporated under reduced pressure, which was further purified by silica gel column chromatography (Eluent: 0-5% ethyl acetate in hexane) to afford 2.8 g of compound le (Yield: 44.4%). LCMS: 836.4 (M+Hf .Step le:
Ph3

To compound le (2.3 g, 2.7 mmol) in CH2CI2 (10 mL) diethyiarnine (10 mL) was added and the reaction mixture was stirred at room temperature for 30 min. The resulting solution was concentrated in vacuum to get gummy residue. The crude compound was purified by neutral alumina column chromatography (Eluent: 0-50% ethyl acetate in hexane then 0-5% methanol in chloroform) to get 1.4 g of If (Yield: 90 %). LCMS: 636.5 (M+Na)+.

1f 1To a solution of compound If (0.45 g) in CH2CI2 (5 mL), trifluoroacetic acid (5 mL) and catalytic amount of triisopropylsilane were added and stirred for 3 h at room temperature to remove the acid sensitive protecting groups. The resulting solution was concentrated in vacuum to afford 0.29 g of crude compound 1 which was purified using prep-HPLC method described under experimental conditions. \H NMR (DMSQ-d6, 400 MHz): δ 2.58 (m, 2H), 3.53 (m, 3H), 3.91 (t, 1H), 4.36 (t, 1H), 6.91 (s, 1H), 7.45 (s, 1H); 1 C NMR (DMSO-de, 400 MHz): δ 20.85, 45.71 , 50.23, 65.55, 171.03, 171 .41, 181.66. LCMS: 216.2 (Μ+ΗΓ; HPLC: tR = 13.1 min.Example 2: Synthesis of Co

Step 2a:

1f2a
The urea linkage was carried out by the coupling compound If (2.7 g, 4.39 mmoi) in THF (30 mL) at room temperature with compound 2b (1.67 g, 4.39 mmoi). The coupling was initiated by the addition of TEA (0.9 g, 8.78 mmoi) in THF (10 m L) and the resultant mixture was stirred at room temperature. After completion of 20 h, THF was evaporated from the reaction mass, and partitioned between water and ethyl acetate. Organic layer was washed with water, brine, dried over Na2S04 and evaporated under reduced pressure to get compound 2a, which was further purified by silica gel column chromatography (Fluent: 0-50% ethyl acetate in hexane) to afford 3.46 g of compound 2a (Yield: 92.10%). LCMS 857.4 (M+H)+.

2aTo a solution of compound 2a (0.22 g, 0.25 mmol) in 0¾ί¾ (5 m L), trifluoroaeetic acid (5 mL) and catalytic amount of triisopropyisilane were added and stirred for 3h at room, temperature. The resulting solution was concentrated under reduced pressure to obtain 0.35 g of crude compound. The crude solid material was purified using preparative- HPLC method described under experimental conditions. LCMS: 347.1 (M+H)+; HPLC: tR = 12.9 min.
Synthesis of

2bTo the compound H-Ser(tBu)-OiBu (2 g, 9.2 mmol) in C I I■■(.‘{■ (20 mL), triethylamine (1.39 g, 13.8 mmol) was added and the solution was stirred at room temperature for 5-10 min. To this mixture, solution of 4-Nitrophenyl chioro formate (2.22 g, 11.04 mmol) in CH2CI2 was added and the resultant mixture was stirred at room temperature for 30 min. The completion of the reaction was confirmed by TLC analysis. After completion of reaction, reaction mixture was diluted with CH2CI2 and washed with water and 5.0 M citric acid solution, dried over Na2SC>4 and evaporated under reduced pressure to get crude compound 2b, which was further purified by silica gel column chromatography (Eiuent: 0-20% ethyl acetate in hexane) to yield 2.1 g (58.9%) of 2b.Example 3: Synthesis of Compound 3

The compound was synthesised using similar procedure as depicted in Example 1 (compound 1) and D-amino acids are linked up in reverse order. Boc-D-Thr(‘Bu)-OH was used in place of Boc-Ser(‘Bu)-OH (compound la, Example 1) and Fmoc-D- Asn(trt)-OH in place of Fmoc-Asn(trt)-OH to yield 0.15 g crude material of the title compound 3. LCMS: 230.1 (M+H)+.Example 4: Synthesis of Co
The compound was synthesised using similar procedure as depicted in Example 2 for synthesising compound 2 using

instead of H-Ser(‘Bu)-0’Bu (in synthesis of compound 2b) to yield 0.35 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 361.2 (M+H)+, HPLC: tR = 12.19 min.Example 5: Synthesis of

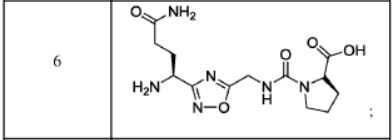
The compound was synthesised using similar procedure as depicted in Example 4 (compound 4) using D-amino acids are linked up in reverse order. Boc-D-Thr(‘Bu)-OH was used in place of Boc-Ser(‘Bu)-OH, Fmoc-D-Asn(trt)-OH in place of Fmoc-Asn(trt)- OH and H-D-Ser(‘Bu)-0’Bu was used in place of H-Thr^Bu^O’Bu to yield 0.3 g crude material of the title compound. The cmde solid material was purified using preparative HPLC described under experimental conditions. LCMS: 361.3 (M+H)+. HPLC: tR = 13.58 min.Example 6: Synthesis of Compound 6

The compound was synthesised using similar procedure as depicted in Example 2 by using H-Thr(‘Bu)-OMe instead of H-Ser(‘Bu)-0’Bu (in synthesis of compound 2b) to yield 0.2 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 375.1 (M+H)+, HPLC: tR = 1.84 min.Example 7: Synthesis of Compound 7

Step 7a:

1f7aThe compound 7a was synthesised using similar procedure as for compound 2a (Example 2, step 2a) using H-Thr(‘Bu)-OMe instead of H-Ser(‘Bu)-OtBu to get crude material which was further purified by silica gel column chromatography (Eluent: 0-50% ethyl acetate in he ane) to get 2.0 g of compound 7a (Yield: 74 %). LCMS: 829.2 (M+H)+.Step 7b:

7a 7bTo a solution of compound 7a (0.35 g, 4.0 mmol) in THF (5 mL) was added lithium hydroxide (0.026 g, 0.63 mmol) at 0 °C and the mixture was stirred for 2 h at room temperature. The completion of the reaction was confirmed by TLC analysis. THF was evaporated from the reaction mass, and partitioned between water and ethyl acetate. Organic layer was washed with citric acid, brine solution, dried over Na2S04 and evaporated under reduced pressure to afford 7b, which was further purified by silica gel column chromatography (Eluent: 0-5% methanol in DCM) to get 0.3 g of product 7b (Yield: 86.7%). LCMS 815.2 (M+H)+.
Step 7c:

7b 7Compound 7b (0.295 g, 0.39 mmol) was anchored to Rink amide resin (0.7 g, 0.55 mmol/g) using HOBT (0.072 g, 0.54 mmol) and DIC (0.068 g, 0.54 mmol) method in DMF (10 mL). The resin was stirred for 12 h at room temperature. The resin was washed with DCM, DMF and DCM and dried. The target compound was cleaved from the rink amide resin using TFA (5 mL) and catalytic amount of TIPS. The resin was allowed to remain at room temperature for 2 h with occasional stirring. After 2 h, TFA and TIPS were evaporated under nitrogen atmosphere and the resulting residue was washed with diethyl ether to yield 0.1 g crude material of the title compound 7. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 360.0 (M+H)+, HPLC: tR = 13.88 min.Example 8: Synthesis of

The compound was synthesised using similar procedure as depicted in Example 2 (compound 2) using Fmoc-Glu(0’Bu)-OH instead of Fmoc-Asn(Trt)-OH to get 0.4 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 362.1 (M+H)+. HPLC: tR = 13.27 min.
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019061324&tab=FULLTEXT
Patenthttps://patents.google.com/patent/WO2019067678A1/enPATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019061324
PATENThttps://patents.google.com/patent/WO2018073754A1/en
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019087087
PAPERSScientific Reports (2019), 9(1), 1-19. https://www.nature.com/articles/s41598-019-48826-6
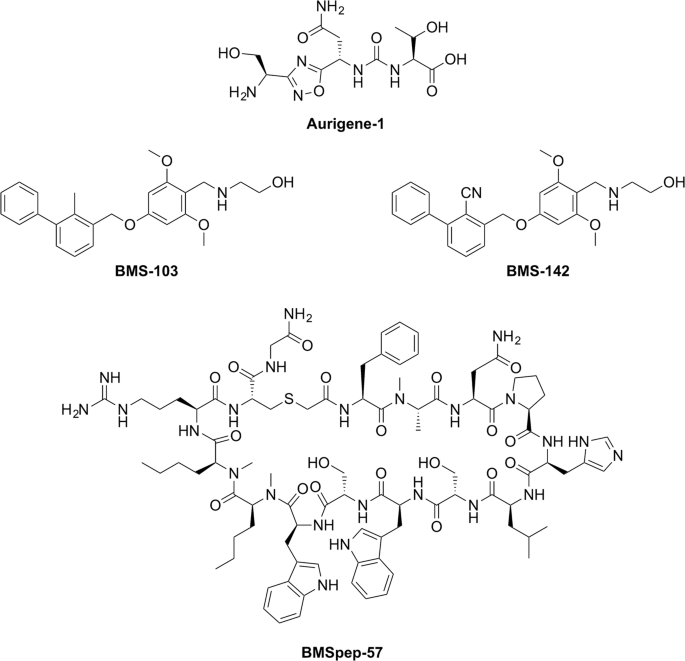
Chemical structures of PD-L1 inhibitors developed by Aurigene (Aurigene-1) and Bristol-Meyers Squibb (BMSpep-57, BMS-103, and BMS-142). Chemical structures were generated using ChemDraw Professional 15. PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019087087
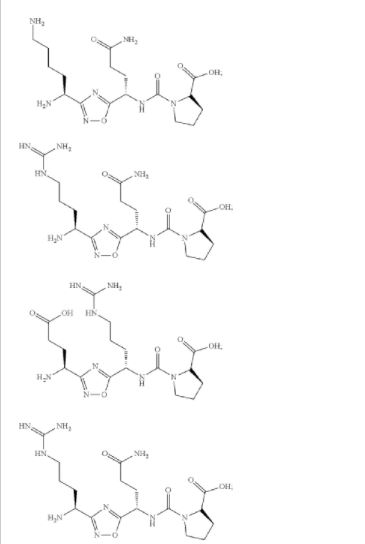
L-threonine’ mentioned in compound of formula (I) thereof can be represented by any one of the following formulae:
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2020289477-A1 | Conjoint therapies for immunomodulation | 2017-11-06 | |
| WO-2019073399-A1 | CRYSTALLINE FORMS OF 1,2,4-OXADIAZOLE SUBSTITUTED IN POSITION 3 | 2017-10-11 | |
| AU-2018341583-A1 | Crystal forms of immunomodulators | 2017-09-29 | |
| WO-2019061324-A1 | CRYSTALLINE FORMS OF IMMUNOMODULATORS | 2017-09-29 | |
| WO-2019067678-A1 | CRYSTALLINE FORMS OF IMMUNOMODULATORS | 2017-09-29 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2020247766-A1 | Crystal forms of immunomodulators | 2017-09-29 | |
| US-2020061030-A1 | Dual inhibitors of vista and pd-1 pathways | 2016-10-20 | |
| WO-2018073754-A1 | Dual inhibitors of vista and pd-1 pathways | 2016-10-20 | |
| US-2020361880-A1 | 1,2,4-Oxadiazole and Thiadiazole Compounds as Immunomodulators | 2015-03-10 | |
| EP-3041827-B1 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2018-04-18 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| EP-3363790-B1 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2020-02-19 |
| US-10173989-B2 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2019-01-08 |
| US-10590093-B2 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2020-03-17 |
| US-2015073024-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-2017101386-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2018072689-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-2019144402-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-2020199086-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-9771338-B2 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2017-09-26 |
| WO-2015033299-A1 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 |
////////////Investigational New Drug Application, Phase 1, Clinical Trial, Non-Hodgkin’s Lymphoma, XL 114, AUR 104, aurigene, Exelixis
N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)C(C)O)CC(N)=O
https://patentscope.wipo.int/search/en/result.jsf?inchikey=HFOBENSCBRZVSP-WHFCDURNSA-N

NEW DRUG APPROVALS
ONE TIME
$10.00
PATENT


The present invention relates to substituted alkynylene compounds represented by compound of formula (I) pharmaceutically acceptable salts and stereoisomers thereof. The present invention further provides the methods of preparation of compound of formula (I) and therapeutic uses thereof as anti-cancer agents.
XL 102
EXELIXIS AND AURIGENE ANNOUNCE THAT PROMISING PRECLINICAL DATA TO BE PRESENTED AT THE ENA SYMPOSIUM SUPPORT THE CLINICAL DEVELOPMENT OF A NOVEL CDK7 INHIBITOR
Exelixis and Aurigene Announce That Promising Preclinical Data to Be Presented at the ENA Symposium Support the Clinical Development of a Novel CDK7 Inhibitor
– Detailed characterization of an oral inhibitor of CDK7 demonstrates potent activity against multiple hematologic and solid tumor cell lines, as monotherapy and in combination with chemotherapies –
October 09, 2020 03:02 AM Eastern Daylight Time
ALAMEDA, Calif.–(BUSINESS WIRE)–Exelixis, Inc. (Nasdaq: EXEL) and Aurigene Discovery Technologies Limited (Aurigene) today disclosed new preclinical data showing that AUR102 has potent anti-tumor activity in a large panel of cancer cell lines. AUR102 is a potent, selective, and orally bioavailable covalent inhibitor of cyclin-dependent kinase 7 (CDK7), which is an important regulator of the cellular transcriptional and cell cycle machinery. Exelixis has an exclusive option for AUR102 under its July 2019 exclusive collaboration, option and license agreement with Aurigene. The new data will be presented in a poster (Abstract 170) at the 32nd EORTC-NCI-AACR (ENA) Symposium, which is being held virtually on October 24-25, 2020.
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy”
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy,” said Murali Ramachandra, Ph.D., Chief Executive Officer of Aurigene. “The data to be presented at ENA 2020 demonstrate that AUR102 effectively engages CDK7 and inhibits a key mediator of the cell cycle and transcription. The ability to inhibit CDK7 activity with an orally available therapeutic such as AUR102 holds great potential to improve care and outcomes for patients with diverse cancer indications, including breast cancer, prostate cancer, leukemia and lymphoma.”
The abstract provides a summary of results from a detailed characterization of AUR102 in cancer cell lines and animal tumor models. Additional data will be presented in the poster. Key findings included in the abstract are:
• AUR102 exhibited potent anti-proliferative activity in a large panel of cell lines with induction of cell death in cell lines derived from multiple cancer types.
• The observed anti-proliferative activity correlated with cellular CDK7 target engagement and decreased levels of P-Ser5 RNAPII, a key mediator of transcription.
• AUR102 studies showed synergy when used in combination with multiple chemotherapies.
• Oral dosing with AUR102 resulted in dose-dependent anti-tumor activity, including complete tumor regression in diffuse large B-cell lymphoma, acute myeloid leukemia, and triple-negative breast cancer xenograft models.
• Inhibition of tumor growth was accompanied by complete target engagement as demonstrated in a parallel PK-PD study.
• AUR102 significantly impacts several pathways and key cancer driver and immune-response genes.
The study authors conclude that the data support clinical evaluation of AUR102 as a single agent and in combination with chemotherapies for the treatment of cancer.
“The exciting AUR102 data to be presented at ENA 2020 provide further validation of our partnering strategy, which gives us multiple opportunities to build a pipeline of best-in-class cancer therapies,” said Peter Lamb, Ph.D., Executive Vice President of Scientific Strategy and Chief Scientific Officer of Exelixis. “AUR102 could be the subject of an Investigational New Drug filing later this year, which would be an important value driver for the program itself and for our collaboration with Aurigene. We commend the Aurigene team on their ongoing success in building a robust body of data supporting the broad clinical potential of AUR102.”
Under the terms of the July 2019 agreement, Exelixis made an upfront payment of $10 million for exclusive options to license three preexisting programs from Aurigene. In addition, Exelixis and Aurigene initiated three Aurigene-led drug discovery programs on mutually agreed upon targets, in exchange for additional upfront option payments of $2.5 million per program. Exelixis is also contributing research funding to Aurigene to facilitate discovery and preclinical development work on all six programs. As the programs mature, Exelixis will have the opportunity to exercise an exclusive option for each program up until the time of Investigational New Drug (IND) filing acceptance. If Exelixis decides to exercise an option, it will make an option exercise payment to Aurigene and assume responsibility for that program’s future clinical development and commercialization including global manufacturing. Aurigene will be eligible for clinical development, regulatory, and sales milestones, as well as royalties on sales. Under the terms of the agreement, Aurigene retains limited development and commercial rights for India and Russia.
About Aurigene
Aurigene is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY, NYSE: RDY). Aurigene is focused on precision-oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene’s programs currently in clinical development include an oral ROR-gamma inhibitor AUR101 for moderate to severe psoriasis in phase 2 under a U.S. FDA IND and a PD-L1/ VISTA antagonist CA-170 for non-squamous non-small cell lung cancer in phase 2b/3 in India. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has also partnered with several large and mid-pharma companies in the United States and Europe and has multiple programs in clinical development. For more information, please visit Aurigene’s website at http://www.aurigene.com.
About Exelixis
Founded in 1994, Exelixis, Inc. (Nasdaq: EXEL) is a commercially successful, oncology-focused biotechnology company that strives to accelerate the discovery, development and commercialization of new medicines for difficult-to-treat cancers. Following early work in model system genetics, we established a broad drug discovery and development platform that has served as the foundation for our continued efforts to bring new cancer therapies to patients in need. Our discovery efforts have resulted in four commercially available products, CABOMETYX® (cabozantinib), COMETRIQ® (cabozantinib), COTELLIC® (cobimetinib) and MINNEBRO® (esaxerenone), and we have entered into partnerships with leading pharmaceutical companies to bring these important medicines to patients worldwide. Supported by revenues from our marketed products and collaborations, we are committed to prudently reinvesting in our business to maximize the potential of our pipeline. We are supplementing our existing therapeutic assets with targeted business development activities and internal drug discovery – all to deliver the next generation of Exelixis medicines and help patients recover stronger and live longer. Exelixis is a member of Standard & Poor’s (S&P) MidCap 400 index, which measures the performance of profitable mid-sized companies. For more information about Exelixis, please visit http://www.exelixis.com, follow @ExelixisInc on Twitter or like Exelixis, Inc. on Facebook.
EXELIXIS AND AURIGENE ANNOUNCE THAT PROMISING PRECLINICAL DATA TO BE PRESENTED AT THE ENA SYMPOSIUM SUPPORT THE CLINICAL DEVELOPMENT OF A NOVEL CDK7 INHIBITOR
Exelixis and Aurigene Announce That Promising Preclinical Data to Be Presented at the ENA Symposium Support the Clinical Development of a Novel CDK7 Inhibitor
– Detailed characterization of an oral inhibitor of CDK7 demonstrates potent activity against multiple hematologic and solid tumor cell lines, as monotherapy and in combination with chemotherapies –
October 09, 2020 03:02 AM Eastern Daylight Time
ALAMEDA, Calif.–(BUSINESS WIRE)–Exelixis, Inc. (Nasdaq: EXEL) and Aurigene Discovery Technologies Limited (Aurigene) today disclosed new preclinical data showing that AUR102 has potent anti-tumor activity in a large panel of cancer cell lines. AUR102 is a potent, selective, and orally bioavailable covalent inhibitor of cyclin-dependent kinase 7 (CDK7), which is an important regulator of the cellular transcriptional and cell cycle machinery. Exelixis has an exclusive option for AUR102 under its July 2019 exclusive collaboration, option and license agreement with Aurigene. The new data will be presented in a poster (Abstract 170) at the 32nd EORTC-NCI-AACR (ENA) Symposium, which is being held virtually on October 24-25, 2020.
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy”
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy,” said Murali Ramachandra, Ph.D., Chief Executive Officer of Aurigene. “The data to be presented at ENA 2020 demonstrate that AUR102 effectively engages CDK7 and inhibits a key mediator of the cell cycle and transcription. The ability to inhibit CDK7 activity with an orally available therapeutic such as AUR102 holds great potential to improve care and outcomes for patients with diverse cancer indications, including breast cancer, prostate cancer, leukemia and lymphoma.”
The abstract provides a summary of results from a detailed characterization of AUR102 in cancer cell lines and animal tumor models. Additional data will be presented in the poster. Key findings included in the abstract are:
• AUR102 exhibited potent anti-proliferative activity in a large panel of cell lines with induction of cell death in cell lines derived from multiple cancer types.
• The observed anti-proliferative activity correlated with cellular CDK7 target engagement and decreased levels of P-Ser5 RNAPII, a key mediator of transcription.
• AUR102 studies showed synergy when used in combination with multiple chemotherapies.
• Oral dosing with AUR102 resulted in dose-dependent anti-tumor activity, including complete tumor regression in diffuse large B-cell lymphoma, acute myeloid leukemia, and triple-negative breast cancer xenograft models.
• Inhibition of tumor growth was accompanied by complete target engagement as demonstrated in a parallel PK-PD study.
• AUR102 significantly impacts several pathways and key cancer driver and immune-response genes.
The study authors conclude that the data support clinical evaluation of AUR102 as a single agent and in combination with chemotherapies for the treatment of cancer.
“The exciting AUR102 data to be presented at ENA 2020 provide further validation of our partnering strategy, which gives us multiple opportunities to build a pipeline of best-in-class cancer therapies,” said Peter Lamb, Ph.D., Executive Vice President of Scientific Strategy and Chief Scientific Officer of Exelixis. “AUR102 could be the subject of an Investigational New Drug filing later this year, which would be an important value driver for the program itself and for our collaboration with Aurigene. We commend the Aurigene team on their ongoing success in building a robust body of data supporting the broad clinical potential of AUR102.”
Under the terms of the July 2019 agreement, Exelixis made an upfront payment of $10 million for exclusive options to license three preexisting programs from Aurigene. In addition, Exelixis and Aurigene initiated three Aurigene-led drug discovery programs on mutually agreed upon targets, in exchange for additional upfront option payments of $2.5 million per program. Exelixis is also contributing research funding to Aurigene to facilitate discovery and preclinical development work on all six programs. As the programs mature, Exelixis will have the opportunity to exercise an exclusive option for each program up until the time of Investigational New Drug (IND) filing acceptance. If Exelixis decides to exercise an option, it will make an option exercise payment to Aurigene and assume responsibility for that program’s future clinical development and commercialization including global manufacturing. Aurigene will be eligible for clinical development, regulatory, and sales milestones, as well as royalties on sales. Under the terms of the agreement, Aurigene retains limited development and commercial rights for India and Russia.
About Aurigene
Aurigene is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY, NYSE: RDY). Aurigene is focused on precision-oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene’s programs currently in clinical development include an oral ROR-gamma inhibitor AUR101 for moderate to severe psoriasis in phase 2 under a U.S. FDA IND and a PD-L1/ VISTA antagonist CA-170 for non-squamous non-small cell lung cancer in phase 2b/3 in India. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has also partnered with several large and mid-pharma companies in the United States and Europe and has multiple programs in clinical development. For more information, please visit Aurigene’s website at http://www.aurigene.com.
About Exelixis
Founded in 1994, Exelixis, Inc. (Nasdaq: EXEL) is a commercially successful, oncology-focused biotechnology company that strives to accelerate the discovery, development and commercialization of new medicines for difficult-to-treat cancers. Following early work in model system genetics, we established a broad drug discovery and development platform that has served as the foundation for our continued efforts to bring new cancer therapies to patients in need. Our discovery efforts have resulted in four commercially available products, CABOMETYX® (cabozantinib), COMETRIQ® (cabozantinib), COTELLIC® (cobimetinib) and MINNEBRO® (esaxerenone), and we have entered into partnerships with leading pharmaceutical companies to bring these important medicines to patients worldwide. Supported by revenues from our marketed products and collaborations, we are committed to prudently reinvesting in our business to maximize the potential of our pipeline. We are supplementing our existing therapeutic assets with targeted business development activities and internal drug discovery – all to deliver the next generation of Exelixis medicines and help patients recover stronger and live longer. Exelixis is a member of Standard & Poor’s (S&P) MidCap 400 index, which measures the performance of profitable mid-sized companies. For more information about Exelixis, please visit http://www.exelixis.com, follow @ExelixisInc on Twitter or like Exelixis, Inc. on Facebook.
Exelixis Forward-Looking Statements
This press release contains forward-looking statements, including, without limitation, statements related to: Exelixis’ and Aurigene’s plans to present preclinical data in support of the continued development of AUR102 in a poster as part of the 32nd ENA Symposium; the potential for AUR102 to improve care and outcomes for patients with diverse cancer indications, including breast cancer, prostate cancer, leukemia and lymphoma; the potential for AUR102 to be the subject of an Investigational New Drug filing later in 2020; Exelixis’ potential future financial and other obligations under the exclusive collaboration, option and license agreement with Aurigene; and Exelixis’ plans to reinvest in its business to maximize the potential of the company’s pipeline, including through targeted business development activities and internal drug discovery. Any statements that refer to expectations, projections or other characterizations of future events or circumstances are forward-looking statements and are based upon Exelixis’ current plans, assumptions, beliefs, expectations, estimates and projections. Forward-looking statements involve risks and uncertainties. Actual results and the timing of events could differ materially from those anticipated in the forward-looking statements as a result of these risks and uncertainties, which include, without limitation: the availability of data at the referenced times; the level of costs associated with Exelixis’ commercialization, research and development, in-licensing or acquisition of product candidates, and other activities; uncertainties inherent in the drug discovery and product development process; Exelixis’ dependence on its relationship with Aurigene, including Aurigene’s adherence to its obligations under the exclusive collaboration, option and license agreement and the level of Aurigene’s assistance to Exelixis in completing clinical trials, pursuing regulatory approvals or successfully commercializing partnered compounds in the territories where they may be approved; the continuing COVID-19 pandemic and its impact on Exelixis’ research and development operations; complexities and the unpredictability of the regulatory review and approval processes in the U.S. and elsewhere; Exelixis’ and Aurigene’s continuing compliance with applicable legal and regulatory requirements; Exelixis’ and Aurigene’s ability to protect their respective intellectual property rights; market competition; changes in economic and business conditions; and other factors affecting Exelixis and its product pipeline discussed under the caption “Risk Factors” in Exelixis’ Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission (SEC) on August 6, 2020, and in Exelixis’ future filings with the SEC. All forward-looking statements in this press release are based on information available to Exelixis as of the date of this press release, and Exelixis undertakes no obligation to update or revise any forward-looking statements contained herein, except as required by law.
Exelixis, the Exelixis logo, CABOMETYX, COMETRIQ and COTELLIC are registered U.S. trademarks. MINNEBRO is a registered Japanese trademark.
