
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Avacopan
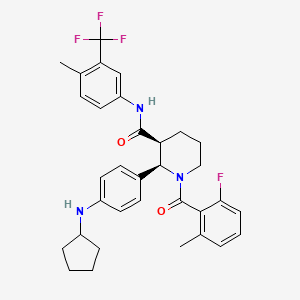

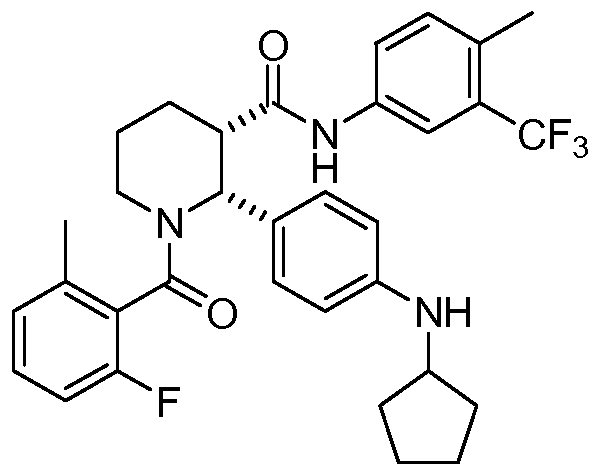
Avacopan
アバコパン
авакопан [Russian] [INN]
أفاكوبان [Arabic] [INN]
阿伐可泮 [Chinese] [INN]
| Formula | C33H35F4N3O2 |
|---|---|
| CAS | 1346623-17-3 |
| Mol weight | 581.6435 |
(2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methylbenzoyl)-N-[4-methyl-3-(trifluoromethyl)phenyl]piperidine-3-carboxamide
(2R,3S)-2-(4-Cyclopentylaminophenyl)-l-(2-fluoro-6-methylbenzoyl)piperidine-3- carboxylic acid (4-methyl-3-trifluoromethylphenyl)amide
3-Piperidinecarboxamide, 2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methylbenzoyl)-N-[4-methyl-3-(trifluoromethyl)phenyl]-, (2R,3S)-
- (2R,3S)-2-[4-(Cyclopentylamino)phenyl]-1-(2-fluoro-6-methylbenzoyl)-N-[4-methyl-3-(trifluoromethyl)phenyl]-3-piperidinecarboxamide
- (2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methylbenzoyl)-N-[4-methyl3-(trifluoromethyl)phenyl]piperidine-3-carboxamide
APPROVED PMDA JAPAN 2021/9/27, Tavneos

Anti-inflammatory, Complement C5a receptor antagonist
Treatment of anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis
Avacopan
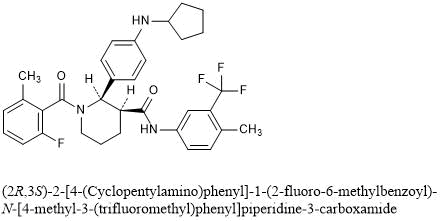
(2R,3S)-2-[4-(Cyclopentylamino)phenyl]-1-(2-fluoro-6-methylbenzoyl)-N-[4-methyl-3-(trifluoromethyl)phenyl]piperidine-3-carboxamide
C33H35F4N3O2 : 581.64
[1346623-17-3]
CCX 168
Avacopan wasunder investigation in clinical trial NCT02994927 (A Phase 3 Clinical Trial of CCX168 (Avacopan) in Patients With ANCA-Associated Vasculitis).
VFMCRP announces approval for TAVNEOS® (avacopan) for the treatment of ANCA-associated vasculitis in Japan
- First orally administered therapy for the treatment of two types of ANCA-associated vasculitis approved in Japan
- Partner Kissei to market TAVNEOS® in Japan, with launch expected as soon as possible following National Health Insurance (NHI) price listing
September 27, 2021 02:02 AM Eastern Daylight Time
ST. GALLEN, Switzerland–(BUSINESS WIRE)–Vifor Fresenius Medical Care Renal Pharma (VFMCRP) today announced that Japan’s Ministry of Health and Labor Welfare (MHLW) has granted its partner, Kissei Pharmaceutical Co., Ltd., marketing authorization approval for TAVNEOS® for the treatment of patients with granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA), the two main types of ANCA-associated vasculitis, a rare and severe autoimmune renal disease with high unmet medical need.
“We are delighted that TAVNEOS® has been approved in Japan, the first market worldwide, and congratulate our partner Kissei for this significant milestone”
“We are delighted that TAVNEOS® has been approved in Japan, the first market worldwide, and congratulate our partner Kissei for this significant milestone,” said Abbas Hussain, CEO of Vifor Pharma Group. “ANCA-associated vasculitis is officially designated an intractable disease in Japan, indicating a rare disease without any effective treatment but for which long-term treatment is required. There is significant unmet medical need of over 10,000 patients in Japan, and we believe in the potential of TAVNEOS® for treating it. We are confident that Kissei will fully focus on bringing this breakthrough treatment to this patient population, helping them lead better, healthier lives.”
The approval is based on the marketing authorization application filing by Kissei which was supported by positive clinical data from the pivotal phase-III trial ADVOCATE in a total of 331 patients with MPA and GPA in 18 countries and regions, including Japan. TAVNEOS® demonstrated superiority over standard of care at week 52 based on Birmingham Vasculitis Activity Score (BVAS).
VFMCRP holds the rights to commercialize TAVNEOS® outside the U.S.. In June 2017, VFMCRP granted Kissei the exclusive right to develop and commercialize TAVNEOS® in Japan. Kissei expects to begin to market TAVNEOS® as soon as possible following NHI price listing. Outside Japan, TAVNEOS is currently in regulatory review with various agencies, including the U.S. Food and Drug Administration and the European Medicines Agency.
About Vifor Pharma Group
Vifor Pharma Group is a global pharmaceuticals company. It aims to become the global leader in iron deficiency, nephrology and cardio-renal therapies. The company is a partner of choice for pharmaceuticals and innovative patient-focused solutions. Vifor Pharma Group strives to help patients around the world with severe and chronic diseases lead better, healthier lives. The company develops, manufactures and markets pharmaceutical products for precision patient care. Vifor Pharma Group holds a leading position in all its core business activities and consists of the following companies: Vifor Pharma and Vifor Fresenius Medical Care Renal Pharma (a joint company with Fresenius Medical Care). Vifor Pharma Group is headquartered in Switzerland, and listed on the Swiss Stock Exchange (SIX Swiss Exchange, VIFN, ISIN: CH0364749348).
For more information, please visit viforpharma.com.
About Kissei Pharmaceutical Co., Ltd.
Kissei Pharmaceutical Co., Ltd. is a Japanese pharmaceutical company with approximately 70 years of history. Based on its management philosophy, “contributing to society through high-quality, innovative pharmaceutical products” and “serving society through our employees”, Kissei is concentrating on providing innovative pharmaceuticals to patients worldwide as a strongly R&D-oriented corporation. Kissei is engaged in R&D and licensing activities in the field of nephrology/dialysis, urology, and unmet medical needs in other disease areas. Kissei has an established collaboration with VFMCRP for sucroferric oxyhydroxide which Kissei fully developed in Japan as P-TOL® (known as Velphoro® in Europe/US) for the treatment of hyperphosphatemia. Since the launch in 2015, the market share of P-TOL® has been steadily expanding in Japan. For more information about Kissei Pharmaceutical, please visit www.kissei.co.jp.
About ChemoCentryx Inc.
ChemoCentryx is a biopharmaceutical company developing new medications for inflammatory and autoimmune diseases and cancer. ChemoCentryx targets the chemokine and chemoattractant systems to discover, develop and commercialize orally-administered therapies. Besides ChemoCentryx’s lead drug candidate, avacopan, ChemoCentryx also has early stage drug candidates that target chemoattractant receptors in other inflammatory and autoimmunediseases and in cancer.
About ANCA-associated vasculitis
ANCA-associated vasculitis is a systemic disease in which over-activation of the complement pathway further activates neutrophils, leading to inflammation and destruction of small blood vessels. This results in organ damage and failure, with the kidney as the major target, and is fatal if not treated. Currently, treatment for ANCA-associated vasculitis consists of courses of non-specific immuno-suppressants (cyclophosphamide or rituximab), combined with the administration of daily glucocorticoids (steroids) for prolonged periods of time, which can be associated with significant clinical risk including death from infection.
About TAVNEOS® (avacopan)
Avacopan is an orally-administered small molecule that is a selective inhibitor of the complement C5a receptor C5aR1. By precisely blocking the receptor (the C5aR) for the pro-inflammatory complement system fragment, C5a on destructive inflammatory cells such as blood neutrophils, avacopan arrests the ability of those cells to do damage in response to C5a activation, which is known to be the driver of inflammation. Moreover, avacopan’s selective inhibition of only the C5aR1 leaves the beneficial C5a l pathway through the C5L2 receptor functioning normally.
ChemoCentryx is also developing avacopan for the treatment of patients with C3 Glomerulopathy (C3G) and hidradenitis suppurativa (HS). The U.S. Food and Drug Administration has granted avacopan orphan-drug designation for ANCA-associated vasculitis, C3G and atypical hemolytic uremic syndrome. The European Commission has granted orphan medicinal product designation for avacopan for the treatment of two forms of ANCA vasculitis: microscopic polyangiitis and granulomatosis with polyangiitis (formerly known as Wegener’s granulomatosis), as well as for C3G. In October 2020, European Medicines Agency (EMA) accepted to review the Marketing Authorization Application (MAA) for avacopan for the treatment of patients with ANCA-associated vasculitis (granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA)).
On May 6, 2021 the U.S. Food & Drug Administration’s (FDA’s) Arthritis Advisory Committee narrowly voted in support of avacopan, a C5a receptor inhibitor, for the treatment of adult patients with anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitis. Although the panelists were excited about the possibility of a steroid-sparing therapy, some raised questions about whether results from the single phase 3 trial could adequately inform the risk/benefit assessment.1 The FDA will weigh the panel’s recommendation as it considers possible approval.
Treatment Needs for ANCA-Associated Vasculitis
ANCA-associated vasculitis is a rare, severe and sometimes fatal form of vasculitis characterized by inflammation of small vessels, often including those in the kidney. One factor that distinguishes it from other forms of vasculitis is the dominant role of neutrophils in its pathogenesis. From work in both animal and mouse models, we know activation of the alternative complement pathway plays a role in the disease pathogenesis, triggering attraction and activation of neutrophils in a complex feedback loop.1-3
Morbidity and mortality from ANCA-associated vasculitis has improved in recent decades, partly due to the introduction of new treatment regimens. The FDA approved rituximab for ANCA-associated vasculitis in 2011, and, in 2018, its label was extended to include maintenance therapy. Most patients with newly diagnosed ANCA-associated vasculitis are now started on a tapering dose of glucocorticoids, paired either with cyclophosphamide or rituximab, with a later follow-up maintenance dose of rituximab at around six months.
High doses of glucocorticoids are often used for remission induction, and they may also be employed as part of maintenance therapy, flare management and relapsing disease. This is a concern for practitioners, who hope to reduce the toxicity that results from glucocorticoid use, especially when given at high doses for prolonged periods.
Avacopan is the first drug to be specifically developed for a vasculitis indication. Other vasculitis therapies—such as tocilizumab for giant cell arteritis or rituximab for ANCA-associated vasculitis—were first approved for other diseases. Avacopan is an oral C5a receptor antagonist that selectively blocks the effects of C5a, thus dampening neutrophil attraction and activation. It does not have FDA approval for other indications, but has orphan drug status for ANCA-associated vasculitis (specifically for microscopic polyangiitis and granulomatosis with polyangiitis) and for C3 glomerulopathy, a rare kidney disease.
Arthritis Advisory Panel Meeting
The FDA generally requires evidence from at least two adequate and well-controlled phase 3 trials to establish effectiveness of a drug. However, it exercises regulatory flexibility in certain circumstances, such as for some rare diseases. In this case, it may consider the results of a well-designed single study if the evidence is statistically persuasive and clinically meaningful.4
Study design is a challenge for any manufacturer attempting to develop a product to potentially decrease steroid use because the FDA does not accept steroid sparing as an assessable outcome for clinical trials. For example, in the GiACTA trial, the phase 3 trial used as evidence for approval of tocilizumab for patients with giant cell arteritis, the biotechnology company Genentech wanted to give tocilizumab and demonstrate patients could then be safely taken off glucocorticoids. But the FDA required a more complicated multi-arm design.5
Other issues come up because of the way glucocorticoids have been used historically. Although they have been used for vasculitis since before drug licensing was introduced, glucocorticoids are not themselves licensed for ANCA-associated vasculitis, which brings up certain regulatory barriers in study design. Additionally, the efficacy of glucocorticoids in vasculitis to control disease activity or prevent relapse has never been officially quantified in a placebo-controlled trial.
ADVOCATE Design
For avacopan, ChemoCentryx based its application on a single phase 3 trial and two phase 2 trials.1-3 In pre-meeting documents and during the meeting itself, the company drew comparisons to the RAVE trial, used to establish the non-inferiority of rituximab to standard cyclophosphamide therapy in patients with ANCA-associated vasculitis.6 In this case, a single phase 3 trial (with supporting phase 2 data) was used as evidence for approval of rituximab.
The phase 3 trial of avacopan, ADVOCATE, used a similar, double-blind, double-dummy design.1 ADVOCATE included 331 patients with either new or relapsing ANCA-associated vasculitis. Half the participants received 30 mg of avacopan twice a day orally, as well as a prednisone placebo, out to the study’s end at 12 months. The other half received oral prednisone (tapered to 0 mg at five months) plus an avacopan placebo.
Additionally, patients received immunosuppressive treatment, either cyclophosphamide (35%) or rituximab (65%), at the discretion of the prescribing physician. Patients who had received cyclophosphamide also received follow-up azathioprine at week 15. But after initial treatment, no patients received maintenance rituximab, as would now be common practice.
Prior to enrollment, many participants were already receiving glucocorticoids as part of their treatment, to help get their disease under control. Thus, open-label prednisone treatment continued to be tapered for the early part of the trial in both groups up to the end of week 4. This had to be tapered to 20 mg or less of prednisone daily before beginning the trial, in both treatment groups.
As reported by the investigators, at week 26, the avacopan group was non-inferior to the prednisone group in terms of sustained remission. At the study’s conclusion at week 52, 66% of patients in the avacopan group were in sustained remission, as were 55% of those in the prednisone group. Thus, in terms of remission, avacopan was superior to glucocorticoids at week 52 (P=0.007).
The researchers also provided encouraging secondary endpoints related to a number of other parameters, including reduced glucocorticoid-related toxicities, fewer relapses, better quality of life measures and improvements in kidney functioning (e.g., glomerular filtration rate changes).
David R.W. Jayne, MD, a professor of clinical autoimmunity at the University of Cambridge and director of the Vasculitis and Lupus Service at Addenbrooke’s Hospital, Cambridge, England, was one of the ADVOCATE investigators and says that in the context of previous vasculitis trials, which have only rarely displayed positive effects from interventions, the ADVOCATE results are impressive.
“We’ve never seen quality-of-life benefits or [glomerular filtration rate] recovery benefits in other vasculitis trials, but we saw them consistently in this one,” says Dr. Jayne.
………………………………………………………………………………………………………………………….

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……………………………………………………………………………………………………………………………
PATENT
Example 1: Preparation of Free Base Crystalline Form of Compound 1
Example 2: Preparing an Amorphous Form of Compound 1
PATENT
WO 2021163329
https://patents.google.com/patent/WO2021163329A1/en
PATENT
https://patents.google.com/patent/US20170283446A1/en
PATENT
Example 1: A Besylate Salt of Compound 1 (Form I)
PATENT
WO 2011163640
https://patents.google.com/patent/WO2011163640A1

Example 11[0147] The following are representative compounds prepared and evaluated using methods similar to the examples herein. Characterization data is provided for the compounds below. Biological evaluation is shown in Figure 1 for these compounds and others prepared as described herein.(2R,3S)-2-(4-Cyclopentylaminophenyl)-l-(2-fluoro-6-methylbenzoyl)piperidine-3- carboxylic acid (4-methyl-3-trifluoromethylphenyl)amide
[0148] 1H NMR (400 MHz, TFA-d) δ 7.91 (d, J= 8.6 Hz, 1 H), 7.84 (d, J= 8.6 Hz, 1 H), 7.58-6.82 (m, 8 H), 6.75 (t, J= 8.6 Hz, 1 H), 4.10-4.00 (m, 1H), 3.60-3.47 (m, 1H), 3.45-3.41 (m, 1H), 3.33-3.25 (m, 1H), 2.44-2.22 (m, 7H), 2.04-1.92 (m, 4H), 1.82-.169 (m, 7H)
PATENTUS 20110275639https://patents.google.com/patent/US20110275639PATENT
https://patents.google.com/patent/US20160090357A1/en
- [0097]This example illustrates the preparation of (2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methyl-benzoyl)-N-[4-methyl-3-(trifluoromethyl)phenyl]piperidine-3-carboxamide by the method provided more generally in FIG. 1 (Scheme 1) using the reagents provided below:
- [0098]Step 1:
- [0099]An oven-dried 12 L, 3-necked flask equipped with a mechanical stirrer, condenser, and thermometer was charged with acrolein diethyl acetal (1127 g, 8.666 mole, 1.05 equiv.) and warmed up to 40° C. A mixture of solid ethyl 3-(4-nitrophenyl)-3-oxo-propanoate (1956 g, 8.253 mole) and (R)-(−)-2-phenylglycinol (>99.5% e.e., 1187 g, 8.666 mole, 1.05 equiv.) was added in portions over 40 min. to maintain a stirrable mixture at an internal temperature of approximately 40° C. After all solids were added, the mixture was stirred at 40° C. for 10 minutes. 4M HCl in dioxane (206.2 mL, 0.825 mole, 10 mol. %) was subsequently added through the condenser within 2 minutes and the internal temperature was increased to 70 OC. The reaction was stirred for 22 h whereupon LC-MS showed consumption of starting materials and enamine intermediate. The heating was turned off and ethanol (6.6 L) was added. The solution was then seeded with 4 g of ethyl (3R,8aR)-5-(4-nitrophenyl)-3-phenyl-3,7,8,8a-tetrahydro-2H-oxazolo[3,2-a]pyridine-6-carboxylate and stirred at room temperature for 18 h. The solid was subsequently filtered off and 0.1 L of ethanol was used to rinse the flask and equipment onto the filter. The isolated solid was then washed three times on the filter with ethanol (250 mL each) and dried under vacuum to generate 1253 g of ethyl (3R,8aR)-5-(4-nitrophenyl)-3-phenyl-3,7,8,8a-tetrahydro-2H-oxazolo[3,2-a]pyridine-6-carboxylate as a bright yellow solid (38% yield, 98.5% HPLC wt/wt purity, 0.15 wt % of EtOH).
- [0100]Step 2:
- [0101]260 g of ethyl (3R,8aR)-5-(4-nitrophenyl)-3-phenyl-3,7,8,8a-tetrahydro-2H-oxazolo[3,2-a]pyridine-6-carboxylate (0.659 mol), 0.66 L of ethanol, and 56 g of palladium catalyst (10% Pd/C, Degussa type E101 NE/W, 50% wet, 21.5 wt. % of powder, 4.0 mol % Pd) were placed in a 2.2 L Parr bottle and purged with nitrogen. The bottle was mounted on a Parr shaker apparatus and hydrogen was added at a rate to keep the external temperature of the bottle below 30° C. After 4 hours, the consumption of hydrogen slowed down. The bottle was then shaken under 50 psi of hydrogen for 2 hours. 94 mL of glacial acetic acid (1.65 mol, 2.5 equiv.) was subsequently added to the bottle and the bottle was purged three times with hydrogen at 50 psi. The bottle was then shaken under 35-55 psi of hydrogen for 48 hours, keeping the temperature below 30° C. The bottle was removed from the apparatus and 55 mL of 12M HCl aq. was added (0.659 mol, 1 equiv.) followed by 87 mL of cyclopentanone (0.989 mol, 1.5 equiv.). The bottle was purged three times with hydrogen at 50 psi and then shaken under 50 psi of hydrogen for 16-20 hours. The mixture was removed from the apparatus and filtered through a fritted funnel containing celite (80 g) and then washed three times with 0.125 L of ethanol. 54.1 g of anhydrous sodium acetate (0.659 mol, 1 equiv.) was added and the mixture was concentrated in vacuo at 40-55° C. to remove 0.9 L of the volatile components. 2.0 L of acetonitrile was added and 2.0 L of volatile components were removed in vacuo. The crude material was diluted with 1.0 L of acetonitrile and mechanically stirred at r.t. for 30 minutes. The mixture was filtered through Celite (40 g) and the cake was washed with 0.28 L of acetonitrile. The combined filtrates gave a solution of the crude amine acetate (Solution A, e.e. =78%). Solutions A of two independent runs were combined for further processing.
- [0102]In a 12-L 3-neck flask equipped with a mechanical stirrer, internal thermometer, and reflux condenser (−)-O,O′-di-p-toluoyl-L-tartaric acid (1.019 kg, 2.64 mol, 2 equiv.) was dissolved in 5.8 L of acetonitrile. The mixture was heated to 60° C. with stirring, followed by a quick addition of 1 L of Solution A. The resultant solution was seeded with 4 g of the crystalline ethyl (2R,3S)-2-[4-(cyclopentylamino)phenyl]piperidine-3-carboxylate (−)-O,O′-di-p-toluoyl-L-tartaric acid salt (1:2) and stirred at 60° C. for 15 minutes. After 15 minutes at 60 OC the seed bed has formed. The remaining amount of Solution A was added over a period of 2.5 hours, maintaining an internal temperature at 60° C. When the addition was complete, the heat source was turned off and the mixture was stirred for 17 hours, reaching a final temperature of 22.5° C. The suspension was filtered and the solids were washed with 0.50 L of acetonitrile to rinse the equipment and transfer all solids onto the filter. The resultant wet solids were washed on the funnel with 3.0 L of acetonitrile and dried in a vacuum oven at 45° C. for 48 hours to provide 1.005 kg of ethyl (2R,3S)-2-[4-(cyclopentylamino)phenyl]piperidine-3-carboxylate (−)-O,O′-di-p-toluoyl-L-tartaric acid salt (1:2) as an off-white solid (70% yield, contains 1 wt. % of acetonitrile). The enantiomeric ratio of the product was 99.4:0.6.
- [0103]Step 3:
- [0104]In a 5 L 3-necked flask equipped with a mechanical stirrer and an addition funnel, solid anhydrous potassium carbonate (K2CO3, 226 g, 1.64 mol, 4.1 equiv.) was dissolved in H2O (0.82 L) and cooled to ambient temperature. MTBE (0.82 L) was added, followed by solid ethyl (2R,3S)-2-[4-(cyclopentylamino)phenyl]piperidine-3-carboxylate (−)-O,O′-di-p-toluoyl-L-tartaric acid salt (1:2) (436 g, 0.400 mol). The mixture was vigorously stirred at r.t. for 1 hour, then 2-fluoro-6-methylbenzoyl chloride (72.5 g, 0.420 mmol, 1.05 equiv.) in MTBE (0.14 L) was added dropwise over 1 hour. The product started precipitating from the reaction before addition of the acid chloride was completed. The reaction was vigorously stirred at r.t. for 30 minutes and monitored by LC-MS for the disappearance of starting material. The mixture was subsequently transferred to a 5 L evaporation flask using 0.3 L of MTBE to rinse the equipment and remove all solids. The mixture was concentrated in vacuo to remove the MTBE, then 0.3 L of heptane was added and the mixture was evaporated again to leave only the product suspended in aqueous solution. The flask was removed from the rotavap and water (0.82 L) and heptane (0.82 L) were added. The suspension was vigorously stirred for 16 hours using a mechanical stirrer. The contents were then filtered and the solid was washed with water (2×0.42 L) and heptane (0.42 L). The solid was dried in a vacuum oven at 45° C. to provide 172 g of ethyl (2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methyl-benzoyl)piperidine-3-carboxylate as an off-white powder (95% yield).
- [0105]Step 4:
- [0106]A 0.5 L 3-necked round-bottom flask was dried overnight in an oven at 200° C. and then cooled under a stream of nitrogen. The flask was equipped with a magnetic stir bar, nitrogen inlet, and a thermometer. The flask was charged with 30.2 g of ethyl (2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methyl-benzoyl)piperidine-3-carboxylate (66.7 mmol), 11.5 mL of 4-methyl-5-trifluoromethylaniline (80 mmol, 1.2 equiv.) and 141 mL of dry toluene under an atmosphere of nitrogen. Nitrogen was bubbled through the resultant solution for 10 minutes and then the solution was warmed to 30° C. The oil bath was removed and 100 mL of a 2 M solution of AlMe3 in toluene (Aldrich, 200 mmol, 3 equiv.) was cannulated into the reaction mixture at a rate maintaining the reaction temperature between 35-40° C., a process that took approximately 45 minutes. The temperature of the reaction mixture was then increased to 55° C. over a period of 1 hour and the reaction mixture was stirred at 55° C. for 8 hours, whereupon all of the starting ester was consumed (monitored by LC-MS). The reaction was subsequently cooled overnight to ambient temperature and the solution was then cannulated into a mechanically stirred 1 L flask containing a solution of 67.8 g of sodium potassium tartrate tetrahydrate (240 mmol, 3.6 equiv.) in 237 mL of water, pre-cooled to 10 OC in an ice bath. The addition process took approximately 30 minutes, during which the reaction mixture self-heated to 57° C. The empty reaction flask was subsequently rinsed with 20 mL of dry toluene and the solution was combined with the quench mixture. The mixture was then cooled to r.t. with stirring, 91 mL of ethyl acetate was added, and the mixture was stirred an additional 15 minutes. The mixture was subsequently filtered through a pad of Celite and the filtrate was allowed to separate into two layers. The organic layer was then separated and washed with a solution of 5.7 g of sodium potassium tartrate tetrahydrate (20 mmol) in 120 mL of water and then with two 120 mL portions of water. The wet organic solution was concentrated in vacuo to a weight of ˜150 g and a solvent exchange with ethanol was performed maintaining a total volume of 0.2-0.3 L, until <1 mol. % toluene with respect to ethanol was observed by 1H NMR. The solution was then evaporated at elevated temperature to a weight of 223 g and heated to reflux. Mechanical stirring was initiated and 41 mL of water was added. The resulting solution was seeded with (2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methyl-benzoyl)-N-[4-methyl-3-(trifluoromethyl)phenyl]piperidine-3-carboxamide crystals at 60 OC and then slowly cooled to r.t. over 2 hours. The slurry was subsequently stirred for 18 hours and the solids were filtered off. The solids were then washed with two 30 mL portions of 7:3 ethanol/water and dried in a vacuum oven for 24 hours at 50 OC to afford 31.0 g of (2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methyl-benzoyl)-N-[4-methyl-3-(trifluoromethyl)phenyl]piperidine-3-carboxamide as off-white crystals (80% yield). Analytical data: HPLC purity: 99.59%; >99.8% d.e. and e.e. by HPLC; ICP-OES Pd: <1 ppm; Al: δ ppm; residual toluene by headspace GC-MS: 15 ppm; microash<0.1%; K—F 0.1%. 1H NMR (400 MHz, TFA-d) δ 7.91 (d, J=8.6 Hz, 1H), 7.84 (d, J=8.6 Hz, 1H), 7.58-6.82 (m, 8H), 6.75 (t, J=8.6 Hz, 1H), 4.10-4.00 (m, 1H), 3.60-3.47 (m, 1H), 3.45-3.41 (m, 1H), 3.33-3.25 (m, 1H), 2.44-2.22 (m, 7H), 2.04-1.92 (m, 4H), 1.82-1.69 (m, 7H), MS: (ES) m/z 582 (M+H+).
PATENT
WO2019236820
The present disclosure is directed to, inter alia, methods of treating ANCA-associated vasculitis (AAV) in a human in need thereof, the method comprising administering to the human a therapeutically effective amount of avacopan, having the structure shown below:
References
- Jayne DRW, Merkel PA, Schall TJ, et al. Avacopan for the treatment of ANCA-associated vasculitis. N Engl J Med. 2021 Feb 18;384(7):599–609.
- Merkel PA, Niles J, Jimenez R, et al. Adjunctive treatment with avacopan, an oral C5a receptor inhibitor, in patients with antineutrophil cytoplasmic antibody-associated vasculitis. ACR Open Rheumatol. 2020;2(11):662–671.
- Jayne DRW, Bruchfeld AN, Harper L, et al. Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J Am Soc Nephrol. 2017 Sep;28(9):2756–2767.
- U.S. Department of Health and Human Services. Food and Drug Administration. Demonstrative substantial evidence of effectiveness for human drug and biological products: Guidance for industry. 2019.
- Stone JH, Tuckwell K, Dimonaco S, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med. 2017 Jul 27;377(4):317–328.
- Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010 Jul 15;363(3):221–232.
- Warrington KJ. Avacopan—time to replace glucocorticoids? N Engl J Med. 2021 Feb 18;384(7):664–665.
////////////Avacopan, アバコパン , JAPAN 2021, APPROVALS 2021, CCX 168, авакопан , أفاكوبان , 阿伐可泮 ,
CC1=C(C(=CC=C1)F)C(=O)N2CCCC(C2C3=CC=C(C=C3)NC4CCCC4)C(=O)NC5=CC(=C(C=C5)C)C(F)(F)F

NEW DRUG APPROVALS
ONE TIME
$10.00
BIAPENEM

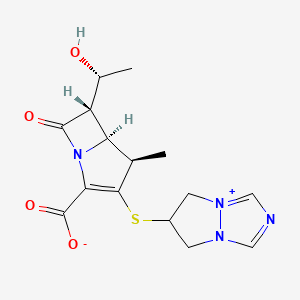
Biapenem
RPX7009
- Molecular FormulaC15H18N4O4S
- Average mass350.393 Da
Biapenern
CL 186-815LJ
C10,627LJ
C10627LJC 10627
omegacin
YR5U3L9ZH1
(4R,5S,6S)-3-((6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium-6-yl)thio)-6-((R)-1-hydroxyethyl)-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylate
[4R-[4a,5b,6b(R*)]]-6-[[2-Carboxy-6-(1-hydroxyethyl)-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-5 H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt
120410-24-4[RN]
5H-Pyrazolo[1,2-a][1,2,4]triazol-4-ium, 6-[[(4R,5S,6S)-2-carboxy-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-, inner salt [ACD/Index Name]
6-[[(4R,5S,6S)-2-Carboxy-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt
7074
(4R,5S,6S)-3-(6,7-Dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium-6-ylsulfanyl)-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylate
TL8000539UNII:YR5U3L9ZH1UNII-YR5U3L9ZH1биапенем
بيابينام比
阿培南
INDIA CDSCO APPROVED 25 SEPT 2021, BDR PHARMA, 
https://www.cdsco.gov.in/opencms/resources/UploadCDSCOWeb/2018/UploadCTApprovals/BDR.pdfhttps://medicaldialogues.in/news/industry/pharma/bdr-pharma-gets-dcgi-nod-for-generic-antibiotic-drug-biapenem-82384
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter a
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
Biapenem (INN) is a carbapenem antibiotic. It has in vitro activity against anaerobes.[1] 1-β-methyl-carbapenem antibiotic. Approved in Japan in 2001.
PATENT
EP 168707
EP 289801
JP 02088578
ZA 9100014
EP 533149
CN 1995040
IN 2006DE01555
CN 101121716
IN 2008CH00177
CN 101805359
CN 101851206
CN 101935321
CN 111875622
WO 2018074916
WO 2016059622
US 20150328323
WO 2015151081
WO 2015155753
WO 2015151078
US 20150284416
WO 2015151080
US 20150038726
WO 2014104488
IN 2013MU00181
WO 2014111957
CN 103570750
WO 2014097221
IN 2012CH01371
WO 2013150550
PAPERS
Journal of Organic Chemistry (1992), 57(15), 4243-9.
Heterocycles (1993), 36(8), 1729-34.
Journal of Antibiotics (1993), 46(12), 1866-82.
e-EROS Encyclopedia of Reagents for Organic Synthesis (2008), 1-3.
Bioorganic & medicinal chemistry letters (2009), 19(17), 5162-5.
IP.com Journal (2014), 14(12A), 1-3
IP.com Journal (2014), 14(10A), 1-2.
Bioorganic & medicinal chemistry (2013), 21(18), 5841-50.

NEW DRUG APPROVALS
one time
$10.00
PATENT
https://patents.google.com/patent/WO2014097221A1/esBiapenem is chemically known as 6-[[2(4R,5S,6S)-carboxy-6-[(lR)- hydroxy ethyl] -4-methyl-7-oxo- 1 -azabicyclo [3.2.0]hept-2-en-3 -yljthio] 6,7-dihydro-5H- pyrazolo[l,2-a][l,2,4]triazol-4-ium inner salt, and is represented by Formula 1. It is indicated for the treatment of bacterial infection and sepsis.

Formula 1U.S. Patent No. 4,866,171, in Example 6, discloses the purification of biapenem using chromatography and/or lyophilization techniques. This patent also describes a process for the conversion of amorphous biapenem into a crystalline form by dissolving the amorphous biapenem in water while heating, followed by cooling, then washing the obtained crystals with a 50% aqueous ethanol solution.U.S. Patent No. 5,241,073 describes a process for the purification of biapenem involving column chromatography and crystallization with ethanol.U.S. Patent No. 5,286,856 describes a process for the crystallization of biapenem from an aqueous solution, comprising maintaining the temperature of the aqueous solution from eutectic temperature (-10°C to -2°C) to a temperature lower than 0°C, followed by lyophilization.The Journal of Organic Chemistry, 63(23):8145-8149 (1998) describes the purification of biapenem involving resin chromatography.The present invention provides an alternate process for the purification of biapenem that avoids making use of tedious techniques like chromatography and lyophilization. At the same time, it results in a high yield and high purity of the final product. Advantageously, the crystalline biapenem of this invention can be directly isolated from the reaction mixture. Further, the process of the present invention involves fewer steps, is easily scalable, and industrially advantageous.EXAMPLESExample 1 : Purification of BiapenemBiapenem (12 g) was added into water (300 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.6 g) was added to the reaction mixture and stirred for 10 minutes to 15 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (36 mL). The filtrate obtained was passed through a 0.45 micron filter, and its pH was adjusted to 5.5 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (336 mL) was added to the reaction mixture at 5°C to 10°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (60 mL). The solid was dried under reduced pressure (720 mmHg) at 30°C to 35°C to obtain the title product as white crystals.Yield: 84%HPLC Purity: 99.87% Example 2: Purification of BiapenemBiapenem (18 g) was added into water (450 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.9 g) was added to the reaction mixture and stirred for 30 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (54 mL). The filtrate obtained was passed through a 0.45 micron filter and its pH was adjusted to 4.9 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (504 mL) was added to the reaction mixture at 10°C to 15°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (90 mL). The solid was dried under reduced pressure (720 mmHg) at 35°C to 40°C to obtain the title product as white crystals.Yield: 81.77%HPLC Purity: 99.80%
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2013150550
The present invention relates to an improved process for the preparation of carbapenem antibiotic; more particularly relates to the preparation of Ertapenem monosodium salt of formula (I) having purity greater than 98.5% and having pharmaceutically acceptable level of residual solvent and palladium content.
The US patents namely US 5,478,820 and US 5,856,321 disclose various processes for preparing Ertapenem and its sodium salt. Example 12 of US 5,478,820 discloses a process in which the Ertapenem was isolated using column purification followed by freeze-drying technique. According to Example-4 of this patent disodium salt of Ertapenem was prepared by dissolving crude product in water using NaHCO3, followed by purification using column chromatography and subsequent lyophilization.
US 6,504,027 provides a process for preparing Ertapenem in crystalline form which comprises deprotecting and extracting a polar organic solution containing a crude mono-protected Ertapenem of formula
wherein P represents protecting group and X represents charge balancing group like sodium
with C4.10 alcohol in the presence of ion-pairing reagent followed by adjusting the pH of the aqueous layer to 5.5 and crystallizing using methanol and 1-propanol to produce a crystalline compound; this patent process involves operations like
multiple extractions which is cumbersome in plant and said operation affects the overall yield.
US 7,145,002 provides a process for producing Ertapenem or its sodium salt and/or its solvate in crystalline form. This patent states (refer para 3, lines 31-41) that contact of Ertapenem sodium with water and alcoholic solvents results in the formation of crystalline solvates. The processes reported in examples- 1 & 2 provide crystalline Ertapenem monosodium which is isolated from a mixture of methanol, 1-propanol and water followed by washing with aqueous isopropyl alcohol which results in the formation of crystalline solvate of Ertapenem sodium. Applicant found the Ertapenem monosodium obtained according to this process contain higher amount of residual solvent and palladium content.
US 7,022,841 provide a process for reducing the levels of organic solvents in Ertapenem to pharmaceutically acceptable levels. This patent discloses (Refer para 1, lines 52-60) that Ertapenem sodium obtained from water/alcohol mixture according to US 7, 145,002 becomes amorphous when water content of the solid is reduced and further the organic solvent present in the solid is not readily removed. In view of this drawback, this patent provides a process wherein the water content of Ertapenem sodium is maintained between 13-25% during the washing and drying process. This patent further discloses that (Refer para 9, lines 6-14) the washing of Ertapenem sodium can be carried out using anhydrous solvents which results in the formation of amorphous solid, which is then dried using hydrated nitrogen by increasing the water content of the solid. Due to the hygroscopic and unstable nature of Ertapenem sodium when in contact with water, the above processes result in more degradation of Ertapenem. The patent further discloses in example 5 that the degradation of Ertapenem sodium is more when it takes more time for drying.
Further this patent requires repetitive washing and control of moisture content to get the desired results.
For isolation of Ertapenem sodium from the reaction mass, all the above discussed prior art patents utilize methanol and 1-propanol as crystallization solvent. The filtration of Ertapenem sodium formed by using these solvents or their mixture takes longer time duration and subsequent drying for the removal of residual solvent also takes several hours due to occlusion of solvent into Ertapenem sodium. During these operations the Ertapenem sodium degrades an results in the formation of many impurities such as several dimers, methanolysis impurity etc., and hence the reported processes is not suitable to manufacture Ertapenem sodium on commercial scale with purity greater than 98.5% and with pharmaceutically acceptable level of residual solvent content.
Methanolysis impurity Dimer-I
Dimer-II
Further the applicant found that Ertapenem monosodium isolated by following the process reported in prior art was having palladium content above the pharmaceutically acceptable level. Hence the process reported in prior art is not suitable on manufacturing scale where maintaining stringent technological condition is cumbersome and involves higher operating cost.
Thus all the reported processes suffer in terms of one or more of the following facts:
■ Filtration time of Ertapenem sodium takes several hours.
■ Drying time takes several hours due to occlusion of solvent and nature of the solid.
■ Stringent technological condition is required for maintenance of moisture content during washing & drying operation.
■ Palladium content is found to be higher (greater than 25 ppm) which is not acceptable for pharmaceutical products.
■ The isolated Ertapenem sodium is having higher amount of residual solvents.
■ The purity is reduced over to several hours of filtration & drying.
With our continued research for developing a process for the preparation of Ertapenem monosodium of formula (I) to overcome the above mentioned drawbacks, we surprisingly found that when esters of organic acid were used as solvents in place of 1-propanol, the solid obtained was easily filterable with less cycle time. Further the washing with hydrocarbon solvents containing 0-75% alcoholic solvent followed by drying results in Ertapenem having residual solvent content well below the pharmaceutically acceptable levels. The use of thiourea, thiosemicarbazide or their N-substituted derivatives in the presence of organic solvents during isolation brings down the palladium content to pharmaceutically acceptable level.
The Ertapenem or its sodium salt can be prepared according the processes provi
(I)
P’ and P” represent carboxylic protecting groups and X is H or Na
Scheme-1
The present invention is illustrated with the following examples, which should not be construed to limit the scope of the invention.
Example- I
Preparation of Ertapenem monosodium of formula (I)
Step-I:
To a stirred solution of p-nitrobenzyl (4R,5S,6S)-3-(diphenyloxy)phosphoryloxy-6-[(lR)-l-hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3,2,0]hept-2-ene-2-carboxylate (compound II) (100 g) and (2S,4S)-2-[[(3-carboxyphenyl) amino]carbonyl]-4-mercapto-l-(4-nitrobenzyl)pyrrolidinecarboxylate (compound III) (75 g) in N,N-dimethylformamide was added Ν,Ν-diisopropylethylamine at -30 to -40° C and stirred. The reaction mass, after completion of the reaction, was quenched with a mixture of phosphate buffer solution-ethyl acetate and the pH was adjusted to 5 – 6 with phosphoric acid. The organic layer was separated, washed with water and subjected to carbon treatment. To the organic layer containing the compound of formula (IV) (wherein P’ and P” refers to p-nitrobenzyl), a solution of sodium 2-ethylhexanoate (42 g in 500 mL methanol) was added and taken to next step as such. (If required the compound of formula (IV) is isolated either as sodium salt or as free acid by following the process reported in prior art and taken further)
Step-II:
To the Step-I organic layer containing the compound of formula (IV) (wherein P’ and P” refers to p-nitrobenzyl & X is Na), 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8- 10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered to remove palladium on carbon. To the filtrate, thiourea (5 g) and tetrahydrofuran were added and stirred. The aqueous layer was separated and treated with carbon and neutral alumina at 10-15° C while degassing and filtered. The filtrate was added to methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol (200 ml) and dried under vacuum. Yield: 46 g; Purity by HPLC: 98.93%; Palladium content: 1.8 ppm by ICP MS
The HPLC purity of Ertapenem monosodium was checked using the following parameters
Column : Zorbax Eclipse plus C8, (50 mm x 4.6 mm), 1.8μ).
Mobile phase : Ammoniam acetate buffer: Acetonitile: water
Detector : UV at 250 nm
Flow rate : 0.5 mL/min
Run time : 45 min.
Example- II
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8-10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and 2-methyltetrahydrofuran and the layers separated. The aqueous layer was treated with carbon & neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with cyclohexane (200 ml) and
dried under vacuum. Yield: 44 g; Purity by HPLC: 98.84%; Palladium content: 0.93 ppm by ICP MS
The term ICP MS method refers to the inductively coupled plasma mass spectrometry. The following parameter was used to determine the content of palladium.
The carbapenem was digested in a closed vessel system in presence of reagents Nitric acid, Hydrogen peroxide and Hydrochloric acid by using Microwave reaction system with microwave radiation power 1200 Watts. The digested sample was introduced into inductively coupled plasma mass spectrometer by help of Peltier cooled spray chamber. The sample aerosol is getting atomized then ionized in the argon plasma. The ionized Palladium was estimated by using Quadrupole mass detector. The sample was quantified against NIST traceable reference standards at mass number ! 05.
Example- III
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and tetrahydrofuran and the layers separated. The aqueous layer was separated and treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of toluene: ethanol (200 ml) and dried under vacuum. Yield: 42 g; Purity by HPLC: 99.03%
Example- IV
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction was filtered and the filtrate was treated with thiosemicarbazide and tetrahydrofuran and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C followed by the addition of ethyl acetate and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol (200 ml) and dried under vacuum. Yield: 41 g; Purity by HPLC: 99.13%; Palladium content: 1.71 ppm by ICP MS
Example- V
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8-10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and 2-methyltetrahydrofuran and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, a mixture of ethyl acetate containing 10% methyl acetate was added and stirred. The solid obtained was
filtered, washed with cyclohexane:ethanol and dried under vacuum. Yield: 40.5 g; Purity by HPLC: 98.77%; Palladium content: 1.43 ppm by ICP MS
Example-VI
(V ) (V I )
The diprotected Meropenem of formula (V) (where P and P’ were p-nitrobenzyl) was dissolved in tetrahydrofuran and 3-(N-morpholino)propanesulfonic acid buffer and hydrogenated using palladium on carbon at 9-10 kg hydrogen pressure. The mass was filtered and the filtrate was washed with ethyl acetate. The aqueous layer was treated with thiourea and 2-methyltetrahydrofuran. The aqueous layer was separated, treated with carbon and degassed. The carbon was filtered off and acetone was added to the filtrate to crystallize Meropenem trihydrate of formula (VI). The product was filtered and washed with aq. acetone and dried under vacuum to get Meropenem trihydrate. Purity: 99.8%; Pd content: 0.08 ppm
Reference example-I:
Preparation of Ertapenem monosodium of formula (I)
To Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered. The filtrate was treated with thiourea and tetrahydrofuran and the layers separated. The aqueous layer was treated with carbon and neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass ethyl acetate was added and stirred. The solid obtained was filtered, washed with ethanol (5 * 100 ml) and dried under vacuum. Yield: 31 g; Purity by HPLC: 96.76%
Reference example-II:
Preparation of Ertapenem monosodium of formula (I)
To the Step-I reaction mass , as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction was filtered and the layers separated. The aqueous layer was treated with carbon and neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol and dried under vacuum. Yield: 43 g; Purity by HPLC: 98.6%; Palladium content: 35.8 ppm by ICP MS.
Reference example-HI:
Preparation of Ertapenem monosodium of formula (I)
To the Step-I reaction mass as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with 1-propanol at -5° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass methanol and 1-propanol were added and stirred. The solid obtained was filtered, washed with ethanol and dried under nitrogen atmosphere in vacuum. Yield: 25 g; Purity by HPLC: 97 %.: palladium content: 38.2 ppm
The following tables illustrate the advantages of the present invention over prior art process:
Table-I: Comparison of present process with prior art process
The crystallization and washing method disclosed in US 7,022,841 was followed.
The above table indicates that the use of ethyl acetate as crystallization solvent results with improved yield and high purity with less filtration and drying time thereby increasing the productivity significantly on manufacturing scale. Further the use of thiourea or thiosemicarbazide as reagents in the present process results in the pharmaceutically acceptable level of palladium content.
Table-II: Comparison of solvents for washing Ertapenem monosodium
The above table indicates that the use of hydrocarbon solvents containing 0-75% of alcoholic solvent helps in washing to remove the residual solvent content in shorter duration and with single run wash. On the other hands the use of ethanol alone results in Ertapenem monosodium having less yield and purity requiring repetitive washing.
Table-IH: Effect of different reagent in reduction of palladium content
Reagent : thiourea, thiosemicarbazide or its N-substituted derivatives
Advantages of the process of the present invention:
> The use of ester of an organic acid for the crystallization of Ertapenem sodium results in fast filtration and reduced cycle time, thereby increasing the productivity.
> Washing of Ertapenem sodium with hydrocarbon solvent optionally containing alcohol results in improved physical nature of Ertapenem sodium resulting in reduced washing and drying time thereby avoid the degradation of Ertapenem and providing Ertapenem sodium with purity greater than 98.5% by HPLC.
Use of thiourea, thiosemicarbazide or their N-substituted derivatives in the process results in Ertapenem sodium having pharmaceutically acceptable level of palladium content.
PATENT
https://patents.google.com/patent/WO2002057266A1/enEXAMPLE

PNB = p-nitrobenzyl

Ia’A hydrogenator is charged with 63 g of 10% Pd on carbon catalyst (dry weight) in 1.8 L of water. The vessel is placed under hydrogen then vented and placed under nitrogen. Sodium hydroxide (68 g, 50%) is charged adjusting the pH to about 7.5 with carbon dioxide.The enol phosphate (170 g) and the thiol (86 g) are dissolved in 1.3‘L of N- ethylpyrrolidinone (NEP). The mixture is cooled to below -40°C and 1,1,3,3- tetramethylguanidine (109 g) is added. After 3 hours, the reaction mixture is quenched into the hydrogenator at below 15°C adjusting the pH to about 8 with carbon dioxide. The vessel is placed under hydrogen. When the reaction is complete, the hydrogen is vented and the reaction mixture is treated with activated carbon and filtered. The filtrate is extracted with iso-amyl alcohol containing diphenylphosphoric acid (240 g) and 50% NaOH (44 g). The resulting aqueous solution is further extracted with iso-amyl alcohol to give an aqueous solution containing at least 90 mg/mL of the product. Both extractions are performed using two CINC centrifugal separators set in series for countercurrent extraction. The pH is adjusted to 5.5 with acetic acid. The product is crystallized by adding equal volumes of methanol and 1- propanol at below -5°C and isolated by filtration. The solid is washed with a mixture of 2-propanol and water (85: 15 v/v) then dried to yield a compound of formula la’.While certain preferred embodiments of the invention have been described herein in detail, numerous alternative embodiments are contemplated as falling within the scope of the appended claims. Consequently the invention is not to be limited thereby.
Patent Citations
Publication numberPriority datePublication dateAssigneeTitleUS4866171A1987-04-111989-09-12Lederle (Japan), Ltd.(1R,5S,6S)-2-[(6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazolium-6-yl)]thio-6-[R-1-hydroxyethyl]-1-methyl-carbapenum-3-carboxylateUS5241073A1990-10-121993-08-31Lederle (Japan)Process for preparing (1R,5S,6S)-2-[(6,7-dihydro-5H-pyrazolo [1,2-a][1,2,4]triazolium-6-yl)]thio-6-[(R)-1-hydroxyethyl]-1-methyl-carbapenem-3-carboxylate and starting materials thereofUS5286856A1991-09-201994-02-15Takeda Chemical Industries, Ltd.Production of crystalline penemWO2002057266A1 *2001-01-162002-07-25Merck & Co., Inc.Improved process for carbapenem synthesisWO2009047604A1 *2007-10-082009-04-16Orchid Chemicals & Pharmaceuticals LimitedProcess for the preparation of carbapenem antibioticCN102268025A *2011-07-152011-12-07海南美兰史克制药有限公司一种比阿培南化合物及其制法
References
- ^ Aldridge KE, Morice N, Schiro DD (April 1994). “In vitro activity of biapenem (L-627), a new carbapenem, against anaerobes”. Antimicrob. Agents Chemother. 38 (4): 889–93. doi:10.1128/aac.38.4.889. PMC 284564. PMID 8031067.
External links
- (in Japanese) Omegacin
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | IV |
| ATC code | J01DH05 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 120410-24-4 |
| PubChem CID | 71339 |
| ChemSpider | 64442 |
| UNII | YR5U3L9ZH1 |
| ChEBI | CHEBI:3089 |
| ChEMBL | ChEMBL285347 |
| CompTox Dashboard (EPA) | DTXSID5046435 |
| Chemical and physical data | |
| Formula | C15H18N4O4S |
| Molar mass | 350.39 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
ClinicalTrials.gov
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT04552444 | Clinical Efficacy of Combination Therapy Based on High-dose Biapenem in CRKP Infections | Recruiting | 2020-09-17 | |
| NCT01772836 | Safety Study of Intravenous Biapenem (RPX2003) and RPX7009 Given Alone and in Combination | Phase 1 | Completed | 2013-07-11 |
| NCT01702649 | Safety, Tolerability, Pharmacokinetics of Intravenous RPX2003 (Biapenem) in Healthy Adult Subjects | Phase 1 | Completed | 2012-12-03 |
NIPH Clinical Trials Search of Japan
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| UMIN000017219 | Feasibility and efficacy of the de-escalation therapy by Biapenem for postoperative bacterial pneumonia. | None | Recruiting | 2015-04-22 |
| UMIN000003964 | Clinical evaluation of Biapenem 0.3g, three times daily dosing in eldery patients with pneumonia (moderate and severe infection) | Not applicable | Complete: follow-up complete | 2010-07-29 |
/////////BIAPENEM, TL8000539, UNII:YR5U3L9ZH1, UNII-YR5U3L9ZH1, биапенем, بيابينام ,比阿培南 , Biapenern, CL 186-815, CL 186815, L 627, LJC 10627, Omegacin, Antibacterial, Antibiotics, Lactams, Carbapenems, ind 2021, india 2021, approvals 2021
CC1C2C(C(=O)N2C(=C1SC3CN4C=NC=[N+]4C3)C(=O)[O-])C(C)O
https://clinicaltrials.gov/search/intervention=Biapenem

updated
Biapenem is chemically known as 6-[[2(4R,5S,6S)-carboxy-6-[(lR)-hydroxy ethyl] -4-methyl-7-oxo- 1 -azabicyclo [3.2.0]hept-2-en-3-yljthio] 6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt, and is represented by Formula 1. It is indicated for the treatment of bacterial infection and sepsis.
Formula 1
U.S. Patent No. 4,866,171, in Example 6, discloses the purification of biapenem using chromatography and/or lyophilization techniques. This patent also describes a process for the conversion of amorphous biapenem into a crystalline form by dissolving the amorphous biapenem in water while heating, followed by cooling, then washing the obtained crystals with a 50% aqueous ethanol solution.
U.S. Patent No. 5,241,073 describes a process for the purification of biapenem involving column chromatography and crystallization with ethanol.
U.S. Patent No. 5,286,856 describes a process for the crystallization of biapenem from an aqueous solution, comprising maintaining the temperature of the aqueous solution from eutectic temperature (-10°C to -2°C) to a temperature lower than 0°C, followed by lyophilization.
The Journal of Organic Chemistry, 63(23):8145-8149 (1998) describes the purification of biapenem involving resin chromatography.
The present invention provides an alternate process for the purification of biapenem that avoids making use of tedious techniques like chromatography and lyophilization. At the same time, it results in a high yield and high purity of the final product. Advantageously, the crystalline biapenem of this invention can be directly isolated from the reaction mixture. Further, the process of the present invention involves fewer steps, is easily scalable, and industrially advantageous.
EXAMPLES
Example 1 : Purification of Biapenem
Biapenem (12 g) was added into water (300 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.6 g) was added to the reaction mixture and stirred for 10 minutes to 15 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (36 mL). The filtrate obtained was passed through a 0.45 micron filter, and its pH was adjusted to 5.5 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (336 mL) was added to the reaction mixture at 5°C to 10°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (60 mL). The solid was dried under reduced pressure (720 mmHg) at 30°C to 35°C to obtain the title product as white crystals.
Yield: 84%
HPLC Purity: 99.87%
Example 2: Purification of Biapenem
Biapenem (18 g) was added into water (450 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.9 g) was added to the reaction mixture and stirred for 30 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (54 mL). The filtrate obtained was passed through a 0.45 micron filter and its pH was adjusted to 4.9 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (504 mL) was added to the reaction mixture at 10°C to 15°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (90 mL). The solid was dried under reduced pressure (720 mmHg) at 35°C to 40°C to obtain the title product as white crystals.
Yield: 81.77%
HPLC Purity: 99.80%
PATENT
Background of the Invention Biapenem is a synthetic broad-spectrum carbapenem antibiotic which suppresses bacterial growth by inhibiting the enzymes responsible for bacterial cell wall synthesis, and shows broad-spectrum antibacterial activity both against gram-positive bacteria and gram-negative bacteria. Biapenem is chemically known as (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][ 1,2,4] triazol-8-ium-6-ylsulfanyl)-6-( 1 -hydroxyethyl)-4-methyl-7-oxo-1 -azabicyclo [3.2.0]hept-2-ene-2-carboxylate and marketed in Japan as OMEGACIN®.Various methods are reported in the prior art for the preparation of Biapenem of formula (I) which includes the condensation of compound of formula (II) with compound of formula (III) and subsequent deprotection of the protecting group as shown in scheme-1. wherein R1 is hydrogen or hydroxy protecting group such as tert-butyl dimethyl silyl and the like, R2 is hydrogen or carboxyl protecting group such as p-nitrobenzyl, p-methoxy benzyl, allyl and the like, A is an activating group such as P(0)(OR)2, SO2R and the like wherein R is selected from substituted or unsubstituted C1-6 alkyl, aralkyl or aryl to form the compound of formula (II). The X” in compound of formula (III) is halogen selected from Br or CI.Biapenem was first disclosed in US 4,866,171 and the said patent also discloses a process for the preparation of the same. US 5,241,073 disclosed the method for the preparation of compound of formula (III) followed by condensation with compound of general formula (II) using base such as N-ethyldiisopropylamine and subsequent deprotection yields Biapenem which was isolated by column chromatography followed by crystallization from ethanol.EP 0289801 discloses a process for the preparation of crystalline Biapenem wherein Biapenem was dissolved in water and lyophilized to get amorphous compound. The amorphous compound was dissolved in water at 40° C followed by cooling to get crystalline product. This patent further provides the PXRD values of the crystalline Biapenem. The Biapenem obtained according to the process provided in this patent takes longer time for reconstitution and hence not suitable.US 5,286,856 and US 5,424,069 provide a process for the crystallization of Biapenem which utilizes freeze-drying technique and vial lyophillisation method respectively. These patents disclose (refer para 1, lines 10-33 of US’ 856) that the process provided in EP 0289801 results with Biapenem crystals which take relatively longer time for dissolution during use. To overcome the above issues, these patents utilize the freeze-drying and vial lyophillisation methods. The said methods involve freezing of the solution containing Biapenem followed by raising the temperature and repeating the cooling and heating process followed by lyophillisation to get the crystalline product. Lyophillisation and related process are capital intensive techniques and uneconomical in commercial scale operations.All the above said prior arts utilize either the lyophillisation technique or preparing the amorphous material and crystallizing it from water to get crystalline Biapenem.Biapenem is available as powder for injection which needs to be reconstituted with water or saline solution before injection. The process of preparing a solution having an appropriate concentration of an active ingredient for the administration is called “reconstitution”. The reconstitution time (RCT) plays a critical role in injectable powders. Short reconstitution time is preferable for both a member of medical center and patients. If the reconstitution time is too long, it will increase the preparation time thus making it difficult to administrate it to many patients at the same, which will eventually lower the competitiveness of the drug. The problem before the applicants is to find economic and robust process for the preparation of Biapenem with high purity and yield which should dissolve in water in less than 25 seconds (reconstitution time). With our continued intensive and diligent research for developing a process for the preparation of Biapenem having high purity and yield with reconstitution time of less than 25 seconds, we have identified an improved process which is commercially viable and eliminates the issues associated with reconstitution time. The process of this invention is simple and obviates the use of freeze crystallization. Further the present invention fulfils the need for a process for the manufacture of Biapenem which is convenient to operate in commercial scale
Objectives of the inventionThe main objective of the present invention is to provide a simple and commercially viable, industrially scalable process for the crystallization of Biapenem of formula (I) with high purity and good yield.Yet another objective of the present invention is to provide a simple and commercially suitable process for the preparation of Biapenem of formula (I) with reconstitution time less than 25 seconds. The reconstitution time is calculated by the time taken to dissolve 300 mg of Biapenem in 100 ml of water or saline solution.Summary of the inventionAccordingly the primary aspect of the present invention is to provide an improved process for the preparation of Biapenem of formula (I) the said process comprises;(i) obtaining a solution of Biapenem in water containing co-solvent; and(ii) adding anti-solvent in to the solution of step (i) or vice-versa to crystallize Biapenem followed by filtration. Detailed Description In an embodiment of the present invention, the co-solvent used in step (i) is selected from alcoholic solvents consisting of methanol, ethanol, isopropyl alcohol, n-propanol, n-butanol and iso-butanol or mixtures thereof; preferably methanol, ethanol and isopropyl alcohol; more preferably methanol.In another embodiment of the present invention the anti-solvent used in step (ii) is selected from acetone, methyl ethyl ketone, methyl isobutyl ketone, ethyl acetate, methyl acetate, butyl acetate, tetrahydrofuran or mixtures thereof; preferably acetone. In yet another embodiment of the present invention, the solution of Biapenem in step (i) can be obtained by (a) dissolving Biapenem in water followed by addition of co-solvent (b) dissolving Biapenem in water containing the co-solvent (c) the aqueous solution containing Biapenem can be obtained directly from the reaction mass followed by addition of co-solvent (d) the aqueous solution of Biapenem containing co-solvent can be obtained directly from the reaction mass. The said solutions, if necessary can be subjected to sterile filtration before the addition of anti-solvent. Thus the present invention provided a process for the preparation of sterile Biapenem having reconstitution time less than 25 seconds, more preferably less than 15 seconds.The prior art lyophillisation process for the preparation of Biapenem requires capital investment and high operating cost due to the involvement of repetitive heating and cooling process which is tedious technology in commercial scale operations. The reported prior art process for the crystallization of Biapenem of formula (I) from water results in the formation of crystalline powder which takes longer time for dissolution in water or saline solution (reconstitution time). Surprisingly, applicant found that the use of co-solvents during the crystallization of Biapenem results with Biapenem having reconstitution time of less than 25 seconds. This constitutes the novelty of the present invention.In this present invention the Biapenem of formula (I) is obtained as crystalline solid with purity above 99.0 % by HPLC with good stability and further can be easily filled in vials.
The following examples are provided by way of illustration only and should not be construed to limit the scope of the invention.
Crystallization of (4R,5S,6S)-3-(6,7-dihvdro-5H-pyrazolo[l,2-al 11,2,41 triazol-8-ium-6-vlsulfanvl)-6-(l-hydroxvethvl)-4-methvl-7-oxo-l-azabicyclo [3.2.01hept-2-ene-2-carboxvlate [Biapenem of formula (1)1:Example -1:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. Activated carbon and EDTA were added to the clear solution and filtered through hi-flow bed, washed with water followed by filtration through micron filters in sterile area. To the filtrate, methanol (600 mL) was added followed by acetone under stirring. To the reaction mass, Biapenem seed material was added and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 85 g Purity by HPLC: 99.5% Reconstitution time (RCT): < 15 seconds
Example -2:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. To the filtrate, isopropyl alcohol (500 ml) was added followed by acetone under stirring. The mass was cooled and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 83 g Purity by HPLC: 99.6% Reconstitution time: < 15 seconds
Example -3;To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. The solution was filtered through micron filters. To the filtrate, ethanol (600 ml) was added followed by acetone and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 84 g Purity by HPLC: 99.5% Reconstitution time : < 15 seconds
Example -4:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. The solution was filtered through hi-flow bed, washed with water followed by filtration through micron filters. To the filtrate, methanol (450 ml) was added followed by acetone and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem. Yield: 87 g Purity by HPLC: 99.4% Reconstitution time (RCT): < 15 seconds
Reference example -1:Preparation of Biapenem (Non-Sterile)Step-I: Preparation of p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pvrazolofl,2-al[l,2,41triazol-8-ium-6-vlsulfanvn-6-(l-hvdroxvethyl)-4-methvI-7-oxo-l-azabicvclo[3.2.01hept-2-ene-2-carboxylate [Compound of formula (IV)1To a mixture of acetonitrile and DMF, P-Nitrobenzyl (4R,5S,6S)-3-(dipheny loxy)phosphory loxy-6- [(1R)-1 -hydroxyethy 1] -4-methy 1-7-oxo-1 -azabicyclo[3,2,0]hept-2-ene-2-carboxylate (compound of formula II) and 6,7-dihydro-6-mercapto-5H-pyrazolo[l,2-a] [1,2,4] triazole chloride (compound of formula III) were added and cooled to 0-5° C. To this mixture, N-ethyldiisopropyl amine was added and stirred till the completion of the reaction, followed by the addition of dichloromethane to crystallize the p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][l,2,4]triazol-8-ium-6-ylsulfanyl)-6-(l -hydroxyethyl)-4-methyl-7-oxo-1 -azabicyclo[3.2.0] hept-2-ene-2-carboxylate which was filtered and dried under nitrogen.
Step-II: Preparation of BiapenemTo a solution of MOPS buffer and THF, p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][l,2,4]triazol-8-ium-6-ylsulfanyl)-6-(l-hydroxy ethyl)-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (Compound of formula-IV) was added at pH 7-8 and cooled to 5-10° C. The mixture was hydrogenated using palladium on carbon as catalyst. The catalyst was filtered and the filtrate was treated with activated carbon and filtered. The filtrate was extracted with dichloromethane and the layers separated. The aqueous layer was degassed. To the aqueous layer, acetone was added to crystallize Biapenem at 20-25° C. The product was filtered, washed with aqueous acetone and dried under vacuum to get Biapenem (Non-Sterile).
Reference example -2: Crystallization of Biapenem
Example -1 was repeated without the addition of methanol.Yield: 84 g Purity by HPLC: 99.5%Reconstitution time : > 90 secondsThe reconstitution time is calculated by the time taken to dissolve 300 mg of Biapenem in 100 ml of water or saline solution.Table-1: Comparative Data:The comparative data provided in the table-1 clearly indicates that the addition of co-solvent during crystallization provides Biapenem with reconstitution time less than 25 seconds.
VX 759
VX 759
CAS#: 478025-29-5
Chemical Formula: C22H27NO3S
Molecular Weight: 385.522
478025-29-5
Drug Name:VCH-759Research Code:VX-759; BCH-27759; VCH-759
VX-759; BCH-27759; VCH-759; VX759; BCH27759; VCH759; VX 759; BCH 27759; VCH 759; NNI-1
3-(N-isopropyl-4-methylcyclohexane-1-carboxamido)-5-phenylthiophene-2-carboxylic acid
- 3-[[(trans-4-Methylcyclohexyl)carbonyl](1-methylethyl)amino]-5-phenyl-2-thiophenecarboxylic acid
- 3-[Isopropyl(trans-4-methylcyclohexylcarbonyl)amino]-5-phenylthiophene-2-carboxylic acid
- NNI 1
MOA:NS5B inhibitorIndication:HCV infectionStatus:Phase Ⅱ (Discontinued)Company:Vertex (Originator)
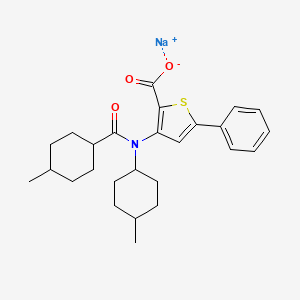
| Molecular Formula | C26H32NNaO3S |
|---|---|
| Synonyms | VX-759 sodium saltBCP17193 |
| Molecular Weight | 461.6 |
VCH-759 had been in phase II clinical trials by ViroChem Pharma (acquired by Vertex in 2009) for the treatment of HCV infection. However, this research has been discontinued.
Infection with HCV is a major cause of human liver disease throughout the world. In the US, an estimated 4.5 million Americans are chronically infected with HCV. Although only 30% of acute infections are symptomatic, greater than 85% of infected individuals develop chronic, persistent infection. Treatment costs for HCV infection have been estimated at $5.46 billion for the US in 1997. Worldwide over 200 million people are estimated to be infected chronically. HCV infection is responsible for 40-60% of all chronic liver disease and 30% of all liver transplants. Chronic HCV infection accounts for 30% of all cirrhosis, end-stage liver disease, and liver cancer in the U.S. The CDC estimates that the number of deaths due to HCV will minimally increase to 38,000/year by the year 2010.
Due to the high degree of variability in the viral surface antigens, existence of multiple viral genotypes, and demonstrated specificity of immunity, the development of a successful vaccine in the near future is unlikely. Alpha-interferon (alone or in combination with ribavirin) has been widely used since its approval for treatment of chronic HCV infection. However, adverse side effects are commonly associated with this treatment: flu-like symptoms, leukopenia, thrombocytopenia, depression from interferon, as well as anemia induced by ribavirin (Lindsay, K. L. (1997) Hepatology 26 (suppl 1 ): 71 S-77S). This therapy remains less effective against infections caused by HCV genotype 1 (which constitutes -75% of all HCV infections in the developed markets) compared to infections caused by the other 5 major HCV genotypes. Unfortunately, only -50-80% of the patients respond to this treatment (measured by a reduction in serum HCV RNA levels and normalization of liver enzymes) and, of responders, 50-70% relapse within 6 months of cessation of treatment. Recently, with the introduction of pegylated interferon (Peg-IFN), both initial and sustained response rates have improved substantially, and combination treatment of Peg-IFN with ribavirin constitutes the gold standard for therapy. However, the side effects associated with combination therapy and the impaired response in patients with genotype 1 present opportunities for improvement in the management of this disease.
First identified by molecular cloning in 1989 (Choo, Q-L et al (1989) Science 244:359-362), HCV is now widely accepted as the most common causative agent of post-transfusion non A, non-B hepatitis (NANBH) (Kuo, G et al (1989) Science 244:362-364). Due to its genome structure and sequence homology, this virus was assigned as a new genus in the Flaviviridae family. Like the other members of the Flaviviridae, such as flaviviruses (e.g. yellow fever virus and Dengue virus types 1-4) and pestiviruses (e.g. bovine viral diarrhea virus, border disease virus, and classic swine fever virus) (Choo, Q-L et al (1989) Science 244:359-362; Miller, R.H. and R.H. Purcell (1990) Proc. Natl. Acad. Sci. USA 87:2057-2061 ), HCV is an enveloped virus containing a single strand RNA molecule of positive polarity. The HCV genome is approximately 9.6 kilobases (kb) with a long, highly conserved, noncapped 5′ nontranslated region (NTR) of approximately 340 bases which functions as an internal ribosome entry site (IRES) (Wang CY et al ‘An RNA pseudoknot is an essential structural element of the internal ribosome entry site located within the hepatitis C virus 5′ noncoding region’ RNA- A Publication of the RNA Society. 1 (5): 526-537, 1995 JuL). This element is followed by a region which encodes a single long open reading frame (ORF) encoding a polypeptide of -3000 amino acids comprising both the structural and nonstructural viral proteins.
Upon entry into the cytoplasm of the cell, this RNA is directly translated into a polypeptide of -3000 amino acids comprising both the structural and nonstructural viral proteins. This large polypeptide is subsequently processed into the individual structural and nonstructural proteins by a combination of host and virally-encoded proteinases (Rice, CM. (1996) in B.N. Fields, D.M.Knipe and P.M. Howley (eds) Virology 2nd Edition, p931-960; Raven Press, N.Y.). Following the termination codon at the end of the long ORF, there is a 3′ NTR which roughly consists of three regions: an – 40 base region which is poorly conserved among various genotypes, a variable length poly(U)/polypyrimidine tract, and a highly conserved 98 base element also called the “3′ X-tail” (Kolykhalov, A. et al (1996) J. Virology 70:3363-3371 ; Tanaka, T. et al (1995) Biochem Biophys. Res. Commun. 215:744-749; Tanaka, T. et al (1996) J. Virology 70:3307-3312; Yamada, N. et al (1996) Virology 223:255-261 ). The 3′ NTR is predicted to form a stable secondary structure which is essential for HCV growth in chimps and is believed to function in the initiation and regulation of viral RNA replication.
The NS5B protein (591 amino acids, 65 kDa) of HCV (Behrens, S. E. et al (1996) EMBO J. 15:12-22), encodes an RNA-dependent RNA polymerase (RdRp) activity and contains canonical motifs present in other RNA viral polymerases. The NS5B protein is fairly well conserved both intra-typically (-95-98% amino acid (aa) identity across 1 b isolates) and inter-typically (-85% aa identity between genotype 1 a and 1 b isolates). The essentiality of the HCV NS5B RdRp activity for the generation of infectious progeny virions has been formally proven in chimpanzees (A. A. Kolykhalov et al.. (2000) Journal of Virology, 74(4): 2046-2051 ). Thus, inhibition of NS5B RdRp activity (inhibition of RNA replication) is predicted to be useful to treat HCV infection.
Although the predominant HCV genotype worldwide is genotype 1, this itself has two main subtypes, denoted 1a and 1 b. As seen from entries into the Los Alamos HCV database
(www.hcv.lanl.gov) (Table 1 ) there are regional differences in the distribution of these subtypes: while genotype 1 a is most abundant in the United States, the majority of sequences in Europe and Japan are from genotype 1 b.
Table 1
Based on the foregoing, there exists a significant need to identify synthetic or biological compounds for their ability to inhibit replication of both genotype 1 a and genotype 1 b of HCV.
PATENT
WO 2002100851
WO 2007071434
PATENT
WO 2009000818
Compound A
5-Phenyl-3-[[(frans-4-methylcyclohexyl)carbonyl](1-methylethyl)amino]-2-thiophenecarboxylic acid
To a mixture of methyl S-^trans^-methylcyclohexyOcarbonylKI-methylethy^aminol-S-phenyl-2-thiophenecarboxylate (Intermediate 31 ) (390 mg) in THF/MeOH/water (3:2:1, vol/vol, 40 ml. total) was added lithium hydroxide monohydrate (246 mg). The mixture was stirred at room temperature for 20 hours, the solvents removed in vacuo, and the residue partitioned between water (40 ml.) and ethyl acetate (40 ml_). The organic layer was dried
(Na2SC>4), evaporated and triturated with ether to give the title compound.
MS calcd for (C22H27NO3S+ H)+: 356
MS found (electrospray): (M+H)+ =356
Compounds A, B, C and D may be made according to the processes described in WO2002/100851 or as described hereinabove.
Structures of Compounds A, B, C and D are shown below for the avoidance of doubt.
The compounds of Formula (I) which have been tested demonstrate a surprisingly superior potency as HCV polymerase inhibitors, as shown by the IC5O values in the cell-based assays across both of the 1 a and 1 b genotypes of HCV, compared to Compounds A, B, C and D. Accordingly, the compounds of Formula (I) are of great potential therapeutic benefit in the treatment and prophylaxis of HCV.
PAPER
Bioorganic & Medicinal Chemistry Letters (2016), 26(18), 4536-4541.
https://www.sciencedirect.com/science/article/abs/pii/S0960894X16300427
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter a
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
| Application Id | Application Number | Application Date | Country | Title |
| US333830037 | 16610899 | 04.05.2018 | US | IDENTIFICATION AND TARGETED MODULATION OF GENE SIGNALING NETWORKS |
| US77274496 | 13661508 | 26.10.2012 | US | COMPOUNDS AND METHODS FOR THE TREATMENT OR PREVENTION OF FLAVIVIRUS INFECTIONS |
| US73506396 | 13172477 | 29.06.2011 | US | Thiophene analogues for the treatment or prevention of flavivirus infections |
| EP29858255 | 10185737 | 11.06.2002 | EP | Thiophene derivatives as antiviral agents for flavivirus infection |
| US73326585 | 13081780 | 07.04.2011 | US | Compounds and methods for the treatment or prevention of <i>Flavivirus </i>infections |
| JP272602128 | 2010103706 | 28.04.2010 | JP | COMPOUND AND METHOD FOR TREATMENT OR PREVENTION OF FLAVIVIRUS INFECTION |
| EP11167270 | 09167203 | 11.06.2002 | EP | Thiophene derivatives as antiviral agents for flavivirus infection |
| CN83819886 | 200910139375.5 | 11.06.2002 | CN | Thiophene derivatives as antiviral agents for flavivirus infection |
| US42846304 | 12097840 | 20.12.2006 | US | Antiviral 2-Carboxy-Thiophene Compounds |
| JP272180287 | 2008546276 | 20.12.2006 | JP | 抗ウイルス性2-カルボキシ-チオフェン化合物 |
| WO2007071434 | PCT/EP2006/012442 | 20.12.2006 | WO | ANTIVIRAL 2-CARBOXY-THIOPHENE COMPOUNDS |
| US41429134 | 11042442 | 26.01.2005 | US | Compounds and methods for the treatment or prevention of <i>Flavivirus </i>infections |
| CN82792692 | 02815768.0 | 11.06.2002 | CN | Thiophene derivatives used as antiviral agent against flavivirus infections |
| JP270324690 | 2003503618 | 11.06.2002 | JP | FLAVIVIRUS感染の治療または予防のための化合物および方法 |
| EA95398282 | 200400022 | 11.06.2002 | EA | COMPOUNDS AND METHODS FOR THE TREATMENT OR PREVENTION OF FLAVIVIRUS INFECTIONS |
| US40367952 | 10166031 | 11.06.2002 | US | Compounds and methods for the treatment or prevention of Flavivirus infections |
| KR588271 | 1020037016240 | 11.12.2003 | KR | THIOPHENE DERIVATIVES AS ANTIVIRAL AGENTS FOR FLAVIVIRUS INFECTION |
| EP14092312 | 02742563 | 11.06.2002 | EP | THIOPHENE DERIVATIVES AS ANTIVIRAL AGENTS FOR FLAVIVIRUS INFECTION |
| WO2002100851 | PCT/CA2002/000876 | 11.06.2002 | WO | THIOPHENE DERIVATIVES AS ANTIVIRAL AGENTS FOR FLAVIVIRUS INFECTION |
////////////////VX-759, BCH-27759, VCH-759, VX759, BCH27759, VCH759, VX 759, BCH 27759, VCH 759, NNI-1
O=C(C1=C(N(C(C2CCC(C)CC2)=O)C(C)C)C=C(C3=CC=CC=C3)S1)O

NEW DRUG APPROVALS
ONE TIME
$10.00
Mobocertinib

Mobocertinib
1847461-43-1
MF C32H39N7O4
MW 585.70
propan-2-yl 2-[4-[2-(dimethylamino)ethyl-methylamino]-2-methoxy-5-(prop-2-enoylamino)anilino]-4-(1-methylindol-3-yl)pyrimidine-5-carboxylate
TAK-788, AP32788, TAK788, UNII-39HBQ4A67L, AP-32788, 39HBQ4A67L
US10227342, Example 10, MFCD32669806, NSC825519, s6813, TAK-788;AP32788, WHO 11183
NSC-825519, example 94 [WO2015195228A1], GTPL10468, BDBM368374, BCP31045, EX-A3392
US FDA APPROVED 9/15/2021, Exkivity, To treat locally advanced or metastatic non-small cell lung cancer with epidermal growth factor receptor exon 20 insertion mutation

Mobocertinib succinate Chemical Structure
CAS No. : 2389149-74-8
| Molecular Weight | 703.78 |
|---|---|
| Formula | C₃₆H₄₅N₇O₈ |
Mobocertinib mesylateCAS# 2389149-85-1 (mesylate)C33H43N7O7S
Molecular Weight: 681.809
CAS #: 2389149-85-1 (mesylate) 1847461-43-1 (free base) 2389149-74-8 (succinate) 2389149-76-0 (HBr) 2389149-79-3 (HCl) 2389149-81-7 (sulfate) 2389149-83-9 (tosylate) 2389149-87-3 (oxalate) 2389149-89-5 (fumarate)
JAPANESE ACCEPTED NAME
Mobocertinib Succinate
Propan-2-yl 2-[4-{[2-(dimethylamino)ethyl](methyl)amino}-2-methoxy-5-(prop-2-enamido)anilino]-4-(1-methyl-1H-indol-3-yl)pyrimidine-5-carboxylate monosuccinate
C32H39N7O4▪C4H6O4 : 703.78
[2389149-74-8]
FDA grants accelerated approval to mobocertinib for metastatic non-small cell lung cancer with EGFR exon 20 insertion mutations……. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-mobocertinib-metastatic-non-small-cell-lung-cancer-egfr-exon-20
On September 15, 2021, the Food and Drug Administration granted accelerated approval to mobocertinib (Exkivity, Takeda Pharmaceuticals, Inc.) for adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy.
Today, the FDA also approved the Oncomine Dx Target Test (Life Technologies Corporation) as a companion diagnostic device to select patients with the above mutations for mobocertinib treatment.
Approval was based on Study 101, an international, non-randomized, open-label, multicohort clinical trial (NCT02716116) which included patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations. Efficacy was evaluated in 114 patients whose disease had progressed on or after platinum-based chemotherapy. Patients received mobocertinib 160 mg orally daily until disease progression or intolerable toxicity.
The main efficacy outcome measures were overall response rate (ORR) according to RECIST 1.1 as evaluated by blinded independent central review (BICR) and response duration. The ORR was 28% (95% CI: 20%, 37%) with a median response duration of 17.5 months (95% CI: 7.4, 20.3).
The most common adverse reactions (>20%) were diarrhea, rash, nausea, stomatitis, vomiting, decreased appetite, paronychia, fatigue, dry skin, and musculoskeletal pain. Product labeling includes a boxed warning for QTc prolongation and Torsades de Pointes, and warnings for interstitial lung disease/pneumonitis, cardiac toxicity, and diarrhea.
The recommended mobocertinib dose is 160 mg orally once daily until disease progression or unacceptable toxicity.
View full prescribing information for mobocertinib.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), and United Kingdom’s Medicines & Healthcare products Regulatory Agency (MHRA). The application reviews are ongoing at the other regulatory agencies.
This review used the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment. The FDA approved this application approximately 6 weeks ahead of the FDA goal date.
This application was granted priority review, breakthrough therapy designation and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.Takeda’s EXKIVITY™ (mobocertinib) Approved by U.S. FDA as the First Oral Therapy Specifically Designed for Patients with EGFR Exon20 Insertion+ NSCLC…….. https://www.takeda.com/newsroom/newsreleases/2021/takeda-exkivity-mobocertinib-approved-by-us-fda/September 15, 2021
- Approval based on Phase 1/2 trial results, which demonstrated clinically meaningful responses with a median duration of response (DoR) of approximately 1.5 years
- Next-generation sequencing (NGS) companion diagnostic test approved simultaneously to support identification of patients with EGFR Exon20 insertion mutations
OSAKA, Japan, and CAMBRIDGE, Mass. September 15, 2021 – Takeda Pharmaceutical Company Limited (TSE:4502/NYSE:TAK) (“Takeda”) today announced that the U.S. Food and Drug Administration (FDA) has approved EXKIVITY (mobocertinib) for the treatment of adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 20 insertion mutations as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy. EXKIVITY, which was granted priority review and received Breakthrough Therapy Designation, Fast Track Designation and Orphan Drug Designation from the FDA, is the first and only approved oral therapy specifically designed to target EGFR Exon20 insertion mutations. This indication is approved under Accelerated Approval based on overall response rate (ORR) and DoR. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
“The approval of EXKIVITY introduces a new and effective treatment option for patients with EGFR Exon20 insertion+ NSCLC, fulfilling an urgent need for this difficult-to-treat cancer,” said Teresa Bitetti, president, Global Oncology Business Unit, Takeda. “EXKIVITY is the first and only oral therapy specifically designed to target EGFR Exon20 insertions, and we are particularly encouraged by the duration of the responses observed with a median of approximately 1.5 years. This approval milestone reinforces our commitment to meeting the needs of underserved patient populations within the oncology community.”
The FDA simultaneously approved Thermo Fisher Scientific’s Oncomine Dx Target Test as an NGS companion diagnostic for EXKIVITY to identify NSCLC patients with EGFR Exon20 insertions. NGS testing is critical for these patients, as it can enable more accurate diagnoses compared to polymerase chain reaction (PCR) testing, which detects less than 50% of EGFR Exon20 insertions.
“EGFR Exon20 insertion+ NSCLC is an underserved cancer that we have been unable to target effectively with traditional EGFR TKIs,” said Pasi A. Jänne, MD, PhD, Dana Farber Cancer Institute. “The approval of EXKIVITY (mobocertinib) marks another important step forward that provides physicians and their patients with a new targeted oral therapy specifically designed for this patient population that has shown clinically meaningful and sustained responses.”
“Patients with EGFR Exon20 insertion+ NSCLC have historically faced a unique set of challenges living with a very rare lung cancer that is not only underdiagnosed, but also lacking targeted treatment options that can improve response rates,” said Marcia Horn, executive director, Exon 20 Group at ICAN, International Cancer Advocacy Network. “As a patient advocate working with EGFR Exon20 insertion+ NSCLC patients and their families every day for nearly five years, I am thrilled to witness continued progress in the fight against this devastating disease and am grateful for the patients, families, healthcare professionals and scientists across the globe who contributed to the approval of this promising targeted therapy.”
The FDA approval is based on results from the platinum-pretreated population in the Phase 1/2 trial of EXKIVITY, which consisted of 114 patients with EGFR Exon20 insertion+ NSCLC who received prior platinum-based therapy and were treated at the 160 mg dose. Results were presented at the 2021 American Society of Clinical Oncology (ASCO) Annual Meeting from the Phase 1/2 trial and demonstrated a confirmed ORR of 28% per independent review committee (IRC) (35% per investigator) as well as a median DoR of 17.5 months per IRC, a median overall survival (OS) of 24 months and a median progression-free survival (PFS) of 7.3 months per IRC.
The most common adverse reactions (>20%) were diarrhea, rash, nausea, stomatitis, vomiting, decreased appetite, paronychia, fatigue, dry skin, and musculoskeletal pain. The EXKIVITY Prescribing Information includes a boxed warning for QTc prolongation and Torsades de Pointes, and warnings and precautions for interstitial lung disease/pneumonitis, cardiac toxicity, and diarrhea.
The FDA review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence (OCE), which provides a framework for concurrent submission and review of oncology products among international partners. We look forward to continuing our work with regulatory agencies across the globe to bring mobocertinib to patients.
About EXKIVITY (mobocertinib)
EXKIVITY is a first-in-class, oral tyrosine kinase inhibitor (TKI) specifically designed to selectively target epidermal growth factor receptor (EGFR) Exon20 insertion mutations.
EXKIVITY is approved in the U.S. for the treatment of adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with EGFR exon 20 insertion mutations as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy.
Results from the Phase 1/2 trial of mobocertinib have also been accepted for review by the Center for Drug Evaluation (CDE) in China for locally advanced or metastatic NSCLC patients with EGFR Exon20 insertion mutations who have been previously treated with at least one prior systemic chemotherapy.
For more information about EXKIVITY, visit http://www.EXKIVITY.com. For the Prescribing Information, including the Boxed Warning, please visit https://takeda.info/Exkivity-Prescribing-Information.
About EGFR Exon20 Insertion+ NSCLC
Non-small cell lung cancer (NSCLC) is the most common form of lung cancer, accounting for approximately 85% of the estimated 2.2 million new cases of lung cancer diagnosed each year worldwide, according to the World Health Organization.1,2 Patients with epidermal growth factor receptor (EGFR) Exon20 insertion+ NSCLC make up approximately 1-2% of patients with NSCLC, and the disease is more common in Asian populations compared to Western populations.3-7 This disease carries a worse prognosis than other EGFR mutations, as EGFR TKIs – which do not specifically target EGFR Exon20 insertions – and chemotherapy provide limited benefit for these patients.
Takeda is committed to continuing research and development to meet the needs of the lung cancer community through the discovery and delivery of transformative medicines.
EXKIVITY IMPORTANT SAFETY INFORMATION
QTc Interval Prolongation and Torsades de Pointes: EXKIVITY can cause life-threatening heart rate-corrected QT (QTc) prolongation, including Torsades de Pointes, which can be fatal, and requires monitoring of QTc and electrolytes at baseline and periodically during treatment. Increase monitoring frequency in patients with risk factors for QTc prolongation. Avoid use of concomitant drugs which are known to prolong the QTc interval and use of strong or moderate CYP3A inhibitors with EXKIVITY, which may further prolong the QTc. Withhold, reduce the dose, or permanently discontinue EXKIVITY based on the severity of QTc prolongation.
Interstitial Lung Disease (ILD)/Pneumonitis: Monitor patients for new or worsening pulmonary symptoms indicative of ILD/pneumonitis. Immediately withhold EXKIVITY in patients with suspected ILD/pneumonitis and permanently discontinue EXKIVITY if ILD/pneumonitis is confirmed.
Cardiac Toxicity: Monitor cardiac function, including left ventricular ejection fraction, at baseline and during treatment. Withhold, resume at reduced dose or permanently discontinue based on severity.
Diarrhea: Diarrhea may lead to dehydration or electrolyte imbalance, with or without renal impairment. Monitor electrolytes and advise patients to start an antidiarrheal agent at first episode of diarrhea and to increase fluid and electrolyte intake. Withhold, reduce the dose, or permanently discontinue EXKIVITY based on the severity.
Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective non-hormonal contraception.
Mobocertinib, sold under the brand name Exkivity, is used for the treatment of non-small cell lung cancer.[2][3]
The most common side effects include diarrhea, rash, nausea, stomatitis, vomiting, decreased appetite, paronychia, fatigue, dry skin, and musculoskeletal pain.[2]
Mobocertinib is a small molecule tyrosine kinase inhibitor. Its molecular target is epidermal growth factor receptor (EGFR) bearing mutations in the exon 20 region.[4][5]
Mobocertinib was approved for medical use in the United States in September 2021.[2][3] It is a first-in-class oral treatment to target EGFR Exon20 insertion mutations.[3]
Medical uses
Mobocertinib is indicated for adults with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy.[2]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter a
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
PATENT
WO 2019222093
https://patents.google.com/patent/WO2019222093A1

Scheme I



Example 1 Procedure for the preparation of isopropyl 2-((5-acrylamido-4-((2- (dimethylamino)ethyl) (methyl)amino)-2-methoxyphenyl)amino)-4-(l -methyl- lH-indol-3- yl)pyrimidine-5-carboxylate (Compound (A)).

[00351] Step 1 : Preparation of isopropyl 2-chloro-4-(l -methyl- lH-indo 1-3 -yl)pyrimidine-5- carboxylate.

[00352] To a 2 L Radley reactor equipped with a mechanical stirrer, a thermometer, and a refluxing condenser was charged isopropyl 2,4-dichloropyrimidine-5-carboxylate (100 g, 42.5 mmol, 1.00 eq.) andl,2-dimethoxyethane (DME, 1.2 L, 12 vol) at RT. The mixture was cooled to 3 °C, and granular AlCb (65.5 g, 49.1 mmol, 1.15 eq.) was added in 2 portions (IT 3-12 °C, jacket set 0 °C). The white slurry was stirred 15-25 °C for 60 minutes. 1 -Methylindole (59 g, 44.9 mmol, 1.06 eq.) was added in one portion (IT 20-21°C). DME (100 mL) was used to aid 1- Methylindole transfer. The reaction mixture was aged for at 35 °C for 24 h. Samples (1 mL) were removed at 5 h and 24 h for HPLC analysis (TM1195).[00353] At 5 h the reaction had 70 % conversion, while after 24 h the desired conversion was attained (< 98%).[00354] The reaction mixture was cooled to 0 °C to 5 °C and stirred for 1 h. The solids were collected via filtration and washed with DME (100 mL). The solids (AlCb complex) were charged back to reactor followed by 2-MeTHF (1 L, 10 vol), and water (400 mL, 4 vol). The mixture was stirred for 10 minutes. The stirring was stopped to allow the layers to separate.The organic phase was washed with water (200 mL, 2 vol). The combined aqueous phase was re-extracted with 2-MeTHF (100 mL, 1 vol).[00355] During workup a small amount of product title compound started to crystallize.Temperature during workup should be at about 25-40 °C.[00356] The combined organic phase was concentrated under mild vacuum to 300-350 mL (IT 40-61 °C). Heptane (550 mL) was charged while maintaining the internal temperature between 50 °C and 60 °C. The resulting slurry was cooled at 25 °C over 15 minutes, aged for 1 h (19-25 °C) and the resulting solids isolated by filtration.[00357] The product was dried at 50 °C under vacuum for 3 days to yield 108.1 g (77 % yield) of the title compound, in 100% purity (AUC) as a yellow solid.‘H NMR (400 MHz, DMSO-i/e) d ppm 1.24 (d, J= 6.53 Hz, 6 H) 3.92 (s, 3 H) 5.19 (spt, J=6.27 Hz, 1 H) 7.25 – 7.35 (m, 2 H) 7.59 (d, J=8.03 Hz, 1 H) 8.07 (s, 1 H) 8.16 (d, J= 7.53 Hz, 1 H) 8.82 (s, 1 H).[00358] Step 2: Preparation of isopropyl 2-((4-fhioro-2-methoxy-5-nitrophenyl)amino)-4-(l- methyl-lH-indol-3-yl)pyrimidine-5-carboxylate.

[00359] A mixture of the product of step 1 (85.0 g, 258 mmol, 1.0 eq.), 4-fluoro-2-methoxy- 5nitroaniline (57.0 g, 306 mmol, 1.2 eq.) and PTSA monohydrate (13.3 g, 70.0 mmol, 0.27 eq.) in acetonitrile (1.4 L, 16.5 v) was heated to 76-81 °C under nitrogen in a 2 L Radley reactor. IPC at 19 h indicated that the reaction was complete.[00360] The reaction mixture was cooled to 25 °C and water (80 mL) was charged in one portion (IT during charge dropped from 25 °C to 19 °C). The reaction mixture was aged for 1 h at 21 °C and then the resulting solids were isolated by filtration. The product was washed with EtOAc (2 x 150 mL) and dried in high vacuum at 50 °C to 60 °C for 44 h to give 121.5 g of the title compound (98% yield), HPLC purity 100 % a/a; NMR indicated that PTSA was purged.¾ NMR (400 MHz, DMSO-7,) d ppm 1.21 (d, 7=6.02 Hz, 6 H) 3.91 (s, 3 H) 4.02 (s, 3 H) 5.09 (spt, 7=6.27 Hz, 1 H) 7.10 (t, 7=7.53 Hz, 1 H) 7.26 (t, 7=7.58 Hz, 1 H) 7.42 (d, 7=13.05 Hz, 1 H) 7.55 (d, 7=8.53 Hz, 1 H) 7.90 (br d, 7=7.53 Hz, 1 H) 7.98 (s, 1 H) 8.75 (s, 1 H) 8.88 (d, 7=8.03 Hz, 1 H) 9.03 (s, 1 H).[00361] Step 3: Preparation of isopropyl 2-((4-((2-(dimethylamino)ethyl(methyl)amino)-2- methoxy-5-nitrophenyl)amino)-4-(l-methyl-lH-indol-3-yl)pyrimidine-5-carboxylate.

[00362] A 50 L flask was charged 1.500 kg of the product of step 2 (3.1 moles, l.O equiv.), 639.0 g A,A,A-trimethylethylenediamine (6.3 mol, 2 equiv.), and 21 L MeCN. The resulting slurry was mixed for 7 hours at reflux. The reaction was cooled overnight. Water (16.5 L) was added before the solids were isolated. After isolation of the solids, a wash of 2.25 L MeCN in 2.25 L water was conducted to provide the title compound. The solids were dried, under vacuum, at 75 °C. HPLC purity a/a % of the dry solid was 99.3%.¾ NMR (400 MHz, DMSO-7,) d ppm 1.22 (d, 7=6.02 Hz, 6 H) 2.09 – 2.13 (m, 1 H) 2.19 (s, 6 H) 2.49 – 2.52 (m, 1 H) 2.89 (s, 3 H) 3.29 – 3.35 (m, 2 H) 3.89 (s, 3 H) 3.94 (s, 3 H) 5.10 (spt, 7=6.19 Hz, 1 H) 6.86 (s, 1 H) 7.07 (br t, 7=7.53 Hz, 1 H) 7.24 (t, 7=7.28 Hz, 1 H) 7.53 (d, 7=8.53Hz, 1 H) 7.86 – 8.02 (m, 2 H) 8.36 (s, 1 H) 8.69 (s, 1 H) 8.85 (s, 1 H).[00363] Step 4: Preparation of isopropyl 2-((5-amino-4-((2-(dimethylamino)ethyl)(methyl)- amino)-2-methoxyphenyl)amino)-4-(l -methyl- lH-indo 1-3 -yl)pyrimidine-5-carboxy late.

[00364] To a mixture of the product of step 3 (1.501 kg, 2.67 mol, 1.00 eq.) and 10% Pd/C (64 % wet, 125.0 g, 0.01 1 eq.) was added 2-MeTHF (17.7 L) in a 20 L pressure reactor. The mixture was hydrogenated at 6- 10 psi ¾ and at 40 °C until IPC indicated complete conversion (1 1 h, the reaction product 99.0%). The reaction mixture was filtered (Celite), and the pad rinsed with MeTHF (2.5 L total). The filtrate was stored under N2 in a refrigerator until crystallization.[00365] Approximately 74% of 2-MeTHF was evaporated under reduced pressure while maintaining IT 23-34 °C (residual volume in the reactor was approximately 4.8 L). To the mixture was added n-heptane (6 L) over 15 min via dropping funnel. The resulting slurry was aged at room temperature overnight. The next day the solids on the walls were scraped to incorporate them into the slurry and the solids were isolated by filtration. The isolated solids were washed with n-heptane containing 5% MeTFlF (2 x 750 mL), and dried (75 °C, 30 inch Flg) to yield 1287 g (91 % yield) of the title compound as a yellow solid. F1PLC purity: 99.7% pure.[00366] ¾ NMR (400 MHz, DMSO- ) d ppm 1.20 (d, .7=6.02 Hz, 6 H) 2.21 (s, 6 H) 2.37 -2.44 (m, 2 H) 2.68 (s, 3 H) 2.93 (t, .7=6.78 Hz, 2 H) 3.74 (s, 3 H) 3.90 (s, 3 H) 4.60 (s, 2 H) 5.08 (spt, 7=6.19 Hz, 1 H) 6.80 (s, 1 H) 7.08 – 7.15 (m, 1 H) 7.19 – 7.26 (m, 2 H) 7.52 (d, .7=8.03 Hz, 1 H) 7.94 – 8.01 (m, 2 H) 8.56 (s, 1 H) 8.66 (s, 1 H).[00367] Step 5: Preparation of isopropyl 2-((4-((2-(dimethylamino)ethyl)(methyl)amino)-2- methoxy-5 -(3 -(phenylsulfonyl)propanamido)phenyl)amino)-4-(l -methyl- lH-indol-3- yl)pyrimidine-5-carboxylate.

lnt-5[00368] A mixture of the product of step 4 (1.284 kg, 2.415 mol, 1.0 eq.) and 3- (phenylsulfonyl)propionic acid (0.5528 kg, 2.580 mol, 1.07 eq.) in anhydrous DCM (8.5 L) was cooled to 2 °C, and treated with DIEA (0.310 kg, 2.399 mol, 1.0 eq.). To the reaction mixture was charged over 40 min, 50 % w/w T3P in MeTHF (1.756 kg, 2.759 mol, 1.14 eq.) while maintaining the internal temperature between 0 °C and 8 °C. The mixture was stirred at 0 °C to 5 °C for 15 minutes and then warmed over 30 min to 15 °C then held at 15 °C to 30 °C for 60 min.[00369] The reaction was quenched with water (179 mL). The reaction mixture was stirred at ambient temperature for 30 min then DIEA (439 g) was charged in one portion. The resulting mixture was aged for 15 min, and then treated with 5% aqueous K2CO3 (7.3 L) at 22-25 °C. The organic layer was separated and the aqueous layer back extracted with DCM (6.142 L). The combined organic extract was washed with brine (2 x 5.5 L).[00370] The organic extract was concentrated to 6.5 L, diluted with EtOFl, 200 Proof (14.3 kg), and the mixture concentrated under vacuum (23-25 inch Flg/IT40-60 °C) to a residual volume of 12.8 L.[00371] The residual slurry was treated with EtOFl, 200 Proof (28.8 Kg), and heated to 69 °C to obtain a thin slurry. The reaction mixture was cooled to 15 °C over 2 h, and stored overnight at 15 °C under nitrogen.[00372] The next day, the mixture was cooled to 5 °C, and aged for 30 minutes. The resulting solid was isolated by filtration, washed with EtOFl (2 x 2.16 kg) and dried to give 1.769 kg (100% yield) of the title compound. F1PLC purity 99.85%.‘H NMR (400 MHz, DMSO-i¾ d ppm 1.08 – 1.19 (m, 8 H) 2.15 (s, 6 H) 2.32 (t, J= 5.77 Hz, 2 H) 2.66 – 2.76 (m, 5 H) 2.88 (br t, J= 5.52 Hz, 2 H) 3.48 (qd, J= 7.03, 5.02 Hz, 1 H) 3.60 – 3.69 (m, 2 H) 3.83 (s, 3 H) 3.89 (s, 3 H) 4.40 (t, J=5.02 Hz, 1 H) 5.04 (quin, J=6.27 Hz, 1 H) 7.01 – 7.09 (m, 2 H) 7.22 (t, J= 7.53 Hz, 1 H) 7.52 (d, J= 8.53 Hz, 1 H) 7.67 – 7.82 (m, 4 H) 7.97 (s, 1 H) 7.98 – 8.00 (m, 1 H) 8.14 (s, 1 H) 8.61 – 8.70 (m, 3 H) 10.09 (s, 1 H).[00373] Step 6: Preparation of isopropyl 2-((5-acrylamido-4-((2-(dimethylamino)ethyl) (methyl)amino)-2-methoxyphenyl)amino)-4-(l -methyl- lH-indol-3-yl)pyrimidine-5-carboxylate (Compound (A)).

compound (A)[00374] The product of step 5 (1.600 kg, 2.198 mol, 1.0 equiv.) was dissolved in anhydrous THF (19.5 kg) and was treated at -1 °C to 1 °C with 2M KOSi(CH3)3 in THF (2.72 L, 5.44 mol, 2.47 equiv.). KOSi(CFb)3 was added over 5 minutes, reactor jacket set at -5 °C to 10 °C. 2 M KOSi(CFh)3 solution was prepared by dissolving 871 g of KOSi(CFh)3 technical grade (90%) in 3.056 L of anhydrous TF1F.[00375] The reaction mixture was aged for 60 minutes. Potable water (22 L) was charged to the reaction mixture over 1 10 minutes, while maintaining temperature at 2-7 °C. The resulting suspension was aged at 3-7 °C for 60 minutes; the product was isolated by filtration (the filtration rate during crude product isolation was (1.25 L/min), washed with potable water (2 x 1.6 L) and air dried overnight and then in high vacuum for 12 h at 45 °C to give 1.186 kg of crude title compound (92% yield).‘H NMR (500 MHz, DMSO-i¾ d ppm 1.05 (t, J= 7.09 Hz, 2 H) 1.1 1 (d, J= 6.36 Hz, 6 H) 2.1 1 (s, 6 H) 2.28 (br t, .7=5.38 Hz, 3 H) 2.55 – 2.67 (m, 3 H) 2.69 (s, 3 H) 2.83 (br t, .7=5.38 Hz, 3 H) 3.31 (s, 3 H) 3.36 – 3.51 (m, 2 H) 3.54 – 3.70 (m, 3 H) 3.75 – 3.82 (m, 3 H) 4.33 (t, .7=5.14 Hz, 1 H) 4.99 (dt, 7=12.35, 6.30 Hz, 2 H) 5.75 (s, 1 H) 6.95 – 7.07 (m, 2 H) 7.17 (br t, .7=7.58 Hz, 2 H) 7.48 (d, 7=8.31 Hz, 2 H) 7.62 – 7.71 (m, 3 H) 7.71 – 7.83 (m, 2 H) 7.93 (d, .7=7.83 Hz, 3 H) 8.09 (s, 2 H) 8.53 – 8.67 (m, 3 H) 10.03 (s, 2 H).[00376] Step 7: Preparation of polymorphic Form-I of isopropyl 2-((5-acrylamido-4-((2- (dimethylamino)ethyl) (methyl)amino)-2-methoxyphenyl)amino)-4-(l -methyl- lH-indol-3- yl)pyrimidine-5-carboxylate (Free base Compound (A)).[00377] Method 1 : The crude product of step 6 (1.130 kg) was recrystallized by dissolving it in EtOAc (30.1 kg) at 75 °C, polish filtered (1.2 pm in-line filter), followed by concentration of the filtrate to 14 L of residue (IT during concentration is 58-70 °C). The residual slurry was cooled to 0 °C over 70 minutes and then aged at 0-2 °C for 30 minutes. Upon isolation the product was dried to a constant weight to give 1.007 kg (89% recovery) of the title compound as polymorphic Form-I. Purity (HPLC, a/a %, 99.80%).
PATENT
WO 2015195228
https://patents.google.com/patent/WO2015195228A1/en
PATENT
https://patents.google.com/patent/US10227342
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215310s000lbl.pdf
- ^ Jump up to:a b c d e “FDA grants accelerated approval to mobocertinib for metastatic non-sma”. U.S. Food and Drug Administration (FDA). 16 September 2021. Retrieved 16 September 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c “Takeda’s Exkivity (mobocertinib) Approved by U.S. FDA as the First Oral Therapy Specifically Designed for Patients with EGFR Exon20 Insertion+ NSCLC” (Press release). Takeda Pharmaceutical Company. 15 September 2021. Retrieved 16 September 2021 – via Business Wire.
- ^ “TAK-788 as First-line Treatment Versus Platinum-Based Chemotherapy for Non-Small Cell Lung Cancer (NSCLC) With EGFR Exon 20 Insertion Mutations”. Clinicaltrials.gov. Retrieved 17 February 2021.
- ^ Zhang SS, Zhu VW (2021). “Spotlight on Mobocertinib (TAK-788) in NSCLC with EGFR Exon 20 Insertion Mutations”. Lung Cancer. Auckland, N.Z. 12: 61–65. doi:10.2147/LCTT.S307321. PMC 8286072. PMID 34285620.
External links
- “Mobocertinib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02716116 for “A Study of TAK-788 in Adults With Non-Small Cell Lung Cancer” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Exkivity |
| Other names | TAK-788 |
| License data | US DailyMed: Mobocertinib |
| Pregnancy category | Contraindicated[1] |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1847461-43-12389149-74-8 |
| PubChem CID | 118607832 |
| DrugBank | DB16390DBSALT003192 |
| ChemSpider | 84455481 |
| UNII | 39HBQ4A67L |
| KEGG | D12001D11969 |
| ChEMBL | ChEMBL4650319 |
| Chemical and physical data | |
| Formula | C32H39N7O4 |
| Molar mass | 585.709 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////mobocertinib, Exkivity, TAK 788, AP32788, fda 2021, approvals 2021, cancer
CC(C)OC(=O)C1=CN=C(N=C1C2=CN(C3=CC=CC=C32)C)NC4=C(C=C(C(=C4)NC(=O)C=C)N(C)CCN(C)C)OC

NEW DRUG APPROVALS
one time to maintain this blog
$10.00
Verdiperstat
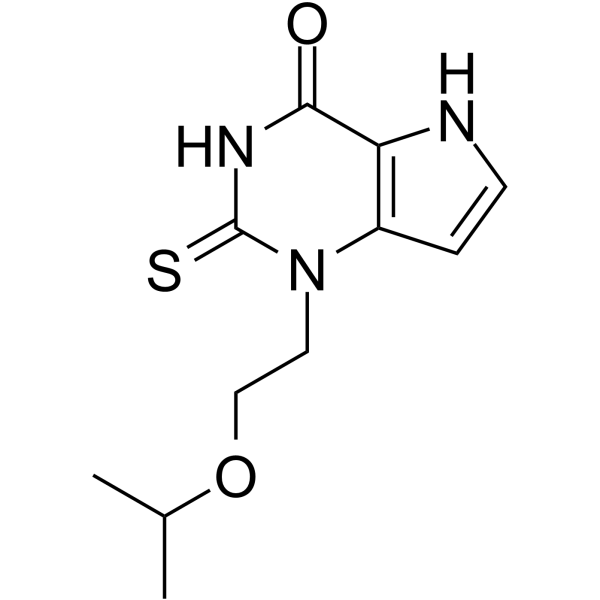
Verdiperstat
AZD 3241; BHV-3241
CAS No. : 890655-80-8
1-(2-propan-2-yloxyethyl)-2-sulfanylidene-5H-pyrrolo[3,2-d]pyrimidin-4-one
4H-Pyrrolo[3,2-d]pyrimidin-4-one, 1,2,3,5-tetrahydro-1-[2-(1-methylethoxy)ethyl]-2-thioxo-
1-(2-isopropoxyethyl)-2-thioxo-1,2,3,5-tetrahydro-pyrrolo[3,2-d] pyrimidin-4-one
l-(2-Isopropoxyethyl)-2-thioxo-l,2,3,5-tetrahydro-pyrrolo[3,2-d]pyrimidin-4-one
- Molecular FormulaC11H15N3O2S
- Average mass253.321 Da
AZD-3241, BHV-3421, UNII-TT3345YXVR, TT3345YXVR, BHV-3241, WHO 10251вердиперстат [Russian] [INN]فيرديبيرستات [Arabic] [INN]维地泊司他 [Chinese] [INN]
- OriginatorAstraZeneca
- DeveloperAstraZeneca; Biohaven Pharmaceuticals
- ClassAntiparkinsonians; Ethers; Organic sulfur compounds; Pyrimidinones; Small molecules
- Mechanism of ActionPeroxidase inhibitors
- Orphan Drug StatusYes – Multiple system atrophy
- Phase IIIMultiple system atrophy
- Phase II/IIIAmyotrophic lateral sclerosis
- DiscontinuedParkinson’s disease
- 23 Jun 20213574186: Added patent info and HE
- 23 Jun 2021Biohaven Pharmaceuticals has patents pending for the composition of matter of verdiperstat, pharmaceutical compositions and various neurological diseases in Europe, Japan and other countries
- 01 Nov 2020Brigham and Women’s Hospital plans a phase I trial for Multiple System Atrophy in USA , (NCT04616456)
EU/3/14/1404: Orphan designation for the treatment of multiple system atrophy
This medicine is now known as verdiperstat.
On 16 December 2014, orphan designation (EU/3/14/1404) was granted by the European Commission to Astra Zeneca AB, Sweden, for 1-(2-isopropoxyethyl)-2-thioxo-1,2,3,5-tetrahydro-pyrrolo[3,2-d] pyrimidin-4-one for the treatment of multiple system atrophy.
The sponsorship was transferred to Richardson Associates Regulatory Affairs Limited, Ireland, in March 2019.
The sponsorship was transferred to Biohaven Pharmaceutical Ireland DAC, Ireland, in September 2021.
Key facts
| Active substance | 1-(2-isopropoxyethyl)-2-thioxo-1,2,3,5-tetrahydro-pyrrolo[3,2-d] pyrimidin-4-one (verdiperstat) |
| Intented use | Treatment of multiple system atrophy |
| Orphan designation status | Positive |
| EU designation number | EU/3/14/1404 |
| Date of designation | 16/12/2014 |
| Sponsor | Biohaven Pharmaceutical Ireland DAC |
VERDIPERSTAT
For Initial Indications in Multiple System Atrophy (MSA) and Amyotrophic Lateral Sclerosis (ALS)
Verdiperstat is a first-in-class, potent, selective, brain-penetrant, irreversible myeloperoxidase (MPO) enzyme inhibitor. Verdiperstat was progressed through Phase 2 clinical trials by AstraZeneca. Seven clinical studies were completed by AstraZeneca, including four Phase 1 studies in healthy subjects, two Phase 2a studies in subjects with Parkinson’s Disease, and one Phase 2b study in subjects with MSA. These Phase 2 clinical studies provide evidence that verdiperstat achieves peripheral target engagement (i.e., reduces MPO specific activity in plasma) and central target engagement in the brain and offer proof of its mechanism of action (i.e., reduce microglial activation and neuroinflamation).
A Phase 3 clinical trial to evaluate the efficacy of verdiperstat in MSA is currently ongoing. A Phase 2/3 trial to evaluate the efficacy of verdiperstat in ALS is currently ongoing as part of the HEALEY ALS Platform Trial.
Verdiperstat has received Fast Track and Orphan Drug designations by the U.S. Food and Drug Administration (FDA) and the European Medicine Agency due to the unmet medical needs in MSA.
Verdiperstat Overview
DESCRIPTIONClick to expendFirst-in-class, brain-penetrant, irreversible inhibitor of MPO
CLINICAL STATUSClick to expendOver 250 healthy volunteers and patients have been treated with verdiperstat in Phase 1 and Phase 2 studies. A Phase 3 study in MSA is currently underway and a Phase 2/3 study in ALS is currently enrolling.
Verdiperstat (AZD3241) is a selective, irreversible and orally active myeloperoxidase (MPO) inhibitor, with an IC50 of 630 nM, and can be used in the research of neurodegenerative brain disorders.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter a
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp

PATENTWO 2006062465https://patents.google.com/patent/WO2006062465A1/enExample 9 l-(2-Isopropoxyethyl)-2-thioxo-l,2,3,5-tetrahydro-pyrrolo[3,2-d]pyrimidin-4-one (a) 3-[(2-Isopropoxyethyl)ωnino]-lH-pyrwle-2-carboxylic acid ethyl ester Trichlorocyanuric acid (1.84 g, 7.93 mmol) was added to a solution of 2- isopropoxyethanol (0.75 g, 7.21 mmol) in CH2Cl2 (3 mL). The reaction mixture was cooled to 0 °C and TEMPO (0.022 g, 0.14 mmol) was carefully added in small portions. The mixture was stirred at r.t. for 20 minutes then filtered through Celite and washed with CH2Cl2. The filtrate was kept cold, 0 °C, during filtration. The aldehyde solution was added to a stirred mixture of 3-amino-lH-pyrrole-2-carboxylic acid ester (0.83 g, 5.41 mmol) and HOAc (0.62 mL, 10.8 mmol) at 0 °C in methanol (5 mL). The mixture was stirred for 20 minutes, then NaCNBH3 (0.34 g, 5.41 mmol) was added. After stirring at r.t for 2 h, the solution was evaporated onto silica and purified by flash column chromatography (heptane/ethyl acetate gradient; 0 to 100% ethyl acetate) to yield the title compound (0.75 g, 58%) as an oil. 1H NMR (DMSO-d6) δ ppm 10.72 (IH, br s), 6.76-6.74 (IH, m), 5.66-5.65 (IH, m), 5.34(1H, br s), 4.17 (2H, q, J=7.0 Hz), 3.59-3.49 (3H, m), 3.15 (2H, q, J=5.6 Hz), 1.26 (3H, t, J=7.0 Hz), 1.10 (3H, s), 1.08 (3H, s); MS (ESI) m/z 241 (M +1).(b) l-(2-Isopropoxyethyl)-2-thioxo-l,2,3,5-tetrahydro-pyrrolo[3,2-d]pyrimidin-4-one The title compound (0.17 g, 23%) was prepared in accordance with the general method B using 3-[(2-isopropoxyethyl)amino]-lH-pyrrole-2-carboxylic acid ethyl ester (0.7 g, 2.91 mmol) and ethoxycarbonyl isothiocyanate (0.40 mL, 3.50 mmol).1H NMR (DMSO-d6) δ ppm 12.74 (2H, br s), 7.35 (IH, d, J=2.8 Hz), 6.29 (IH, d, J=3.0Hz), 4.49 (2H, t, J=6.3 Hz), 3.72 (2H, t, J=6.3 Hz), 3.60-3.58 (IH, m), 1.02 (3H, s), 1.01 (3H, s);MS (ESI) m/z 254 (M +1).
/////////verdiperstat, вердиперстат , فيرديبيرستات , 维地泊司他 , WHO 10251, AZD-3241, BHV-3421, UNII-TT3345YXVR, TT3345YXVR, BHV-3241, AZD 3241, BHV 3241, BHV 3421
CC(C)OCCN1C2=C(C(=O)NC1=S)NC=C2

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
Vosoritide
| PGQEHPNARK YKGANKKGLS KGCFGLKLDR IGSMSGLGC (Disulfide bridge: 23-39) |

H-Pro-Gly-Gln-Glu-His-Pro-Asn-Ala-Arg-Lys-Tyr-Lys-Gly-Ala-Asn-Lys-Lys-Gly-Leu-Ser-Lys-Gly-Cys(1)-Phe-Gly-Leu-Lys-Leu-Asp-Arg-Ile-Gly-Ser-Met-Ser-Gly-Leu-Gly-Cys(1)-OH
PGQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC
H-PGQEHPNARKYKGANKKGLSKGC(1)FGLKLDRIGSMSGLGC(1)-OH
PEPTIDE1{P.G.Q.E.H.P.N.A.R.K.Y.K.G.A.N.K.K.G.L.S.K.G.C.F.G.L.K.L.D.R.I.G.S.M.S.G.L.G.C}$PEPTIDE1,PEPTIDE1,23:R3-39:R3$$$
L-prolyl-glycyl-L-glutaminyl-L-alpha-glutamyl-L-histidyl-L-prolyl-L-asparagyl-L-alanyl-L-arginyl-L-lysyl-L-tyrosyl-L-lysyl-glycyl-L-alanyl-L-asparagyl-L-lysyl-L-lysyl-glycyl-L-leucyl-L-seryl-L-lysyl-glycyl-L-cysteinyl-L-phenylalanyl-glycyl-L-leucyl-L-lysyl-L-leucyl-L-alpha-aspartyl-L-arginyl-L-isoleucyl-glycyl-L-seryl-L-methionyl-L-seryl-glycyl-L-leucyl-glycyl-L-cysteine (23->39)-disulfide
(4R,10S,16S,19S,22S,28S,31S,34S,37S,40S,43S,49S,52R)-52-[[2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-6-amino-2-[[(2S)-6-amino-2-[[(2S)-4-amino-2-[[(2S)-2-[[2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-5-amino-5-oxo-2-[[2-[[(2S)-pyrrolidine-2-carbonyl]amino]acetyl]amino]pentanoyl]amino]-4-carboxybutanoyl]amino]-3-(1H-imidazol-5-yl)propanoyl]pyrrolidine-2-carbonyl]amino]-4-oxobutanoyl]amino]propanoyl]amino]-5-carbamimidamidopentanoyl]amino]hexanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]hexanoyl]amino]acetyl]amino]propanoyl]amino]-4-oxobutanoyl]amino]hexanoyl]amino]hexanoyl]amino]acetyl]amino]-4-methylpentanoyl]amino]-3-hydroxypropanoyl]amino]hexanoyl]amino]acetyl]amino]-40-(4-aminobutyl)-49-benzyl-28-[(2S)-butan-2-yl]-31-(3-carbamimidamidopropyl)-34-(carboxymethyl)-16,22-bis(hydroxymethyl)-10,37,43-tris(2-methylpropyl)-19-(2-methylsulfanylethyl)-6,9,12,15,18,21,24,27,30,33,36,39,42,45,48,51-hexadecaoxo-1,2-dithia-5,8,11,14,17,20,23,26,29,32,35,38,41,44,47,50-hexadecazacyclotripentacontane-4-carboxylic acid
Vosoritide
| Formula | C176H290N56O51S3 |
|---|---|
| CAS | 1480724-61-5 |
| Mol weight | 4102.7254 |
1480724-61-5[RN]BMN 111L-Cysteine, L-prolylglycyl-L-glutaminyl-L-α-glutamyl-L-histidyl-L-prolyl-L-asparaginyl-L-alanyl-L-arginyl-L-lysyl-L-tyrosyl-L-lysylglycyl-L-alanyl-L-asparaginyl-L-lysyl-L-lysylglycyl-L-leucyl-L-seryl-L-lysylglycyl-L-cysteinyl-L-phenylalanylglycyl-L-leucyl-L-lysyl-L-leucyl-L-α-aspartyl-L-arginyl-L-isoleucylglycyl-L-seryl-L-methionyl-L-serylglycyl-L-leucylglycyl-, cyclic (23→39)-disulfideL-prolylglycyl-(human C-type natriuretic peptide-(17-53)-peptide (CNP-37)), cyclic-(23-39)-disulfideUNII:7SE5582Q2Pвосоритид [Russian] [INN]فوسوريتيد [Arabic] [INN]伏索利肽 [Chinese] [INN]
Voxzogo, 2021/8/26 EU APPROVED
| Product details | |
|---|---|
| Name | Voxzogo |
| Agency product number | EMEA/H/C/005475 |
| Active substance | Vosoritide |
| International non-proprietary name (INN) or common name | vosoritide |
| Therapeutic area (MeSH) | Achondroplasia |
| Anatomical therapeutic chemical (ATC) code | M05BX |
| Orphan |
This medicine was designated an orphan medicine. This means that it was developed for use against a rare, life-threatening or chronically debilitating condition or, for economic reasons, it would be unlikely to have been developed without incentives. For more information, see Orphan designation. |
| Publication details | |
|---|---|
| Marketing-authorisation holder | BioMarin International Limited |
| Date of issue of marketing authorisation valid throughout the European Union | 26/08/2021 |
On 24 January 2013, orphan designation (EU/3/12/1094) was granted by the European Commission to BioMarin Europe Ltd, United Kingdom, for modified recombinant human C-type natriuretic peptide for the treatment of achondroplasia.
The sponsorship was transferred to BioMarin International Limited, Ireland, in February 2019.
This medicine is now known as Vosoritide.
The medicinal product has been authorised in the EU as Voxzogo since 26 August 2021.
PEPTIDE
| Treatment of Achondroplasia modified recombinant human C-type natriuretic peptide (CNP) |
Vosoritide, sold under the brand name Voxzogo, is a medication used for the treatment of achondroplasia.[1]
The most common side effects include injection site reactions (such as swelling, redness, itching or pain), vomiting and decreased blood pressure.[1]
Vosoritide was approved for medical use in the European Union in August 2021.[1][2]
Voxzogo is a medicine for treating achondroplasia in patients aged 2 years and older whose bones are still growing.
Achondroplasia is an inherited disease caused by a mutation (change) in a gene called fibroblast growth-factor receptor 3 (FGFR3). The mutation affects growth of almost all bones in the body including the skull, spine, arms and legs resulting in very short stature with a characteristic appearance.
Achondroplasia is rare, and Voxzogo was designated an ‘orphan medicine’ (a medicine used in rare diseases) on 24 January 2013. Further information on the orphan designation can be found here: ema.europa.eu/medicines/human/orphan-designations/EU3121094.
Voxzogo contains the active substance vosoritide.
Medical uses
Vosoritide is indicated for the treatment of achondroplasia in people two years of age and older whose epiphyses are not closed.[1]
Mechanism of action

A: Chondrocyte with constitutionally active FGFR3 that down-regulates its development via the MAPK/ERK pathway
B: Vosoritide (BMN 111) blocks this mechanism by binding to the atrial natriuretic peptide receptor B (NPR-B), which subsequently inhibits the MAPK/ERK pathway at the RAF-1 protein.[3]
Vosoritide works by binding to a receptor (target) called natriuretic peptide receptor type B (NPR-B), which reduces the activity of fibroblast growth factor receptor 3 (FGFR3).[1] FGFR3 is a receptor that normally down-regulates cartilage and bone growth when activated by one of the proteins known as acidic and basic fibroblast growth factor. It does so by inhibiting the development (cell proliferation and differentiation) of chondrocytes, the cells that produce and maintain the cartilaginous matrix which is also necessary for bone growth. Children with achondroplasia have one of several possible FGFR3 mutations resulting in constitutive (permanent) activity of this receptor, resulting in overall reduced chondrocyte activity and thus bone growth.[3]
The protein C-type natriuretic peptide (CNP), naturally found in humans, reduces the effects of over-active FGFR3. Vosoritide is a CNP analogue with the same effect but prolonged half-life,[3] allowing for once-daily administration.[4]
Chemistry
Vosoritide is an analogue of CNP. It is a peptide consisting of the amino acids proline and glycine plus the 37 C-terminal amino acids from natural human CNP. The complete peptide sequence isPGQEHPNARKYKGANKKGLS KGCFGLKLDR IGSMSGLGC
with a disulfide bridge between positions 23 and 39 (underlined).[5] The drug must be administered by injection as it would be rendered ineffective by the digestive system if taken by mouth.
History
Vosoritide is being developed by BioMarin Pharmaceutical and, being the only available causal treatment for this condition, has orphan drug status in the US as well as the European Union.[1][2][6] As of September 2015, it is in Phase II clinical trials.[7][4]
Society and culture
Controversy
Some people with achondroplasia, as well as parents of children with this condition, have reacted to vosoritide’s study results by saying that dwarfism is not a disease and consequently does not need treatment.[8]
Research
Vosoritide has resulted in increased growth in a clinical trial with 26 children. The ten children receiving the highest dose grew 6.1 centimetres (2.4 in) in six months, compared to 4.0 centimetres (1.6 in) in the six months before the treatment (p=0.01).[9] The body proportions, more specifically the ratio of leg length to upper body length – which is lower in achondroplasia patients than in the average population – was not improved by vosoritide, but not worsened either.[7][10]
As of September 2015, it is not known whether the effect of the drug will last long enough to result in normal body heights,[10] or whether it will reduce the occurrence of achondroplasia associated problems such as ear infections, sleep apnea or hydrocephalus. This, together with the safety of higher doses, is to be determined in further studies.[4]
References
- ^ Jump up to:a b c d e f g “Voxzogo EPAR”. European Medicines Agency. 23 June 2021. Retrieved 9 September 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “European Commission Approves BioMarin’s Voxzogo (vosoritide) for the Treatment of Children with Achondroplasia from Age 2 Until Growth Plates Close”. BioMarin Pharmaceutical Inc. (Press release). 27 August 2021. Retrieved 9 September 2021.
- ^ Jump up to:a b c Lorget F, Kaci N, Peng J, Benoist-Lasselin C, Mugniery E, Oppeneer T, et al. (December 2012). “Evaluation of the therapeutic potential of a CNP analog in a Fgfr3 mouse model recapitulating achondroplasia”. American Journal of Human Genetics. 91 (6): 1108–14. doi:10.1016/j.ajhg.2012.10.014. PMC 3516592. PMID 23200862.
- ^ Jump up to:a b c Clinical trial number NCT02055157 for “A Phase 2 Study of BMN 111 to Evaluate Safety, Tolerability, and Efficacy in Children With Achondroplasia (ACH)” at ClinicalTrials.gov
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN): List 112” (PDF). WHO Drug Information. 28 (4): 539. 2014.
- ^ “Food and Drug Administration Accepts BioMarin’s New Drug Application for Vosoritide to Treat Children with Achondroplasia” (Press release). BioMarin Pharmaceutical. 2 November 2020. Retrieved 9 September 2021 – via PR Newswire.
- ^ Jump up to:a b Spreitzer H (6 July 2015). “Neue Wirkstoffe – Vosoritid”. Österreichische Apothekerzeitung (in German) (14/2015): 28.
- ^ Pollack A (17 June 2015). “Drug Accelerated Growth in Children With Dwarfism, Pharmaceutical Firm Says”. The New York Times.
- ^ “BMN 111 (vosoritide) Improves Growth Velocity in Children With Achondroplasia in Phase 2 Study”. BioMarin. 17 June 2015.
- ^ Jump up to:a b “Vosoritid” (in German). Arznei-News.de. 20 June 2015.
External links
- “Vosoritide”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Voxzogo |
| Other names | BMN-111 |
| Routes of administration |
Subcutaneous injection |
| ATC code | None |
| Legal status | |
| Legal status | EU: Rx-only [1] |
| Identifiers | |
| CAS Number | 1480724-61-5 |
| DrugBank | DB11928 |
| ChemSpider | 44210446 |
| UNII | 7SE5582Q2P |
| KEGG | D11190 |
| Chemical and physical data | |
| Formula | C176H290N56O51S3 |
| Molar mass | 4102.78 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////////Vosoritide, Voxzogo, PEPTIDE, ボソリチド (遺伝子組換え) , восоритид , فوسوريتيد , 伏索利肽 , APPROVALS 2021, EU 2021, BMN 111, ORPHAN DRUG
CCC(C)C1C(=O)NCC(=O)NC(C(=O)NC(C(=O)NC(C(=O)NCC(=O)NC(C(=O)NCC(=O)NC(CSSCC(C(=O)NC(C(=O)NCC(=O)NC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O)N1)CCCNC(=N)N)CC(=O)O)CC(C)C)CCCCN)CC(C)C)CC2=CC=CC=C2)NC(=O)CNC(=O)C(CCCCN)NC(=O)C(CO)NC(=O)C(CC(C)C)NC(=O)CNC(=O)C(CCCCN)NC(=O)C(CCCCN)NC(=O)C(CC(=O)N)NC(=O)C(C)NC(=O)CNC(=O)C(CCCCN)NC(=O)C(CC3=CC=C(C=C3)O)NC(=O)C(CCCCN)NC(=O)C(CCCNC(=N)N)NC(=O)C(C)NC(=O)C(CC(=O)N)NC(=O)C4CCCN4C(=O)C(CC5=CN=CN5)NC(=O)C(CCC(=O)O)NC(=O)C(CCC(=O)N)NC(=O)CNC(=O)C6CCCN6)C(=O)O)CC(C)C)CO)CCSC)CO
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn

join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter a
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
MEVOCICLIB, SY 1365
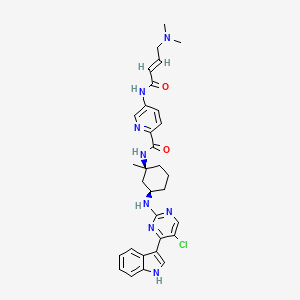
MEVOCICLIB,
CAS 1816989-16-8
SY 1365
N-[(1S,3R)-3-[[5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl]amino]-1-methylcyclohexyl]-5-[[(E)-4-(dimethylamino)but-2-enoyl]amino]pyridine-2-carboxamide
N-((lS,3R)-3-(5-chloro-4-(lH-indol-3-yl)pyrimidin-2-ylamino)-l-methylcvclohexyl)-5-((E)-4-(dimethylamino)but-2-enamido)picolinamide
HS Tariff Code: 2934.99.9001
Syros
| Molecular Weight | 587.12 |
|---|---|
| Formula | C₃₁H₃₅ClN₈O₂ |
- OriginatorSyros Pharmaceuticals
- ClassAmides; Amines; Antineoplastics; Chlorinated hydrocarbons; Cyclohexanes; Indoles; Pyridines; Pyrimidines; Small molecules
- Mechanism of ActionCyclin-dependent kinase-activating kinase inhibitors
- DiscontinuedAcute myeloid leukaemia; Breast cancer; Haematological malignancies; Ovarian cancer; Solid tumours
- 23 Oct 2019Discontinued – Preclinical for Haematological malignancies and Acute myeloid leukaemia in the USA (Parenteral); Phase-I for Solid tumours, Ovarian cancer and Breast cancer in the USA (IV) because data obtained did not support an optimal profile for patients and indicated higher or frequent dosing
- 07 Dec 2018Pharmacodynamics data from preclinical trials in Breast cancer presented at the 41st Annual San Antonio Breast Cancer Symposium (SABCS-2018)
- 15 Nov 2018Adverse events, efficacy and pharmacokinetics data from a phase I trial in Solid tumours presented at the 30th EORTC-NCI-AACR Molecular Targets and Cancer Therapeutics Symposium (EORTC-NCI-AACR-2018)
| Clinical Trial | NCT NumberSponsorConditionStart DatePhaseNCT03134638Syros PharmaceuticalsAdvanced Solid Tumors|Ovarian Cancer|Breast CancerMay 12, 2017Phase 1 |
|---|
Mevociclib (SY-1365) is a potent and first-in-class selective CDK7 inhibitor, with a Ki of 17.4 nM. Mevociclib exhibits anti-proliferative and apoptotic effects in solid tumor cell lines. Mevociclib possesses anti-tumor activity in hematological and multiple aggressive solid tumors.
Mevociclib, also known as SY-1365, is a CDK7 inhibitor. In vitro, SY-1365 inhibited cell growth of many different cancer types at nanomolar concentrations. SY-1365 treatment decreased MCL1 protein levels, and cancer cells with low BCL-XL expression were found to be more sensitive to SY-1365. Transcriptional changes in acute myeloid leukemia (AML) cell lines were distinct from those following treatment with other transcriptional inhibitors. SY-1365 demonstrated substantial anti-tumor effects in multiple AML xenograft models as a single agent; SY-1365-induced growth inhibition was enhanced in combination with the BCL2 inhibitor venetoclax.
Syn
WO2015154038
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015154038
Example 16. Synthesis of N-((lS,3R)-3-(5-chloro-4-(lH-indol-3-yl)pyrimidin-2-ylamino)-l-methylcvclohexyl)-5-((E)-4-(dimethylamino)but-2-enamido)picolinamide (Compound 267).
[251] (+/-) Benzyl tert-butyl ((lS,3R)-l-methylcvclohexane-l,3-diyl)dicarbamate
(+/-)
[252] A solution of (+/-)-(lS,3R) -3-((ieit-¾itoxycarbonyl)amino)–l -raethylcyclohexanecarboxylic acid prepared as in WO2010/148197 (4.00 g, 15.5 mmol) in toluene (Ϊ 55 mL) was treated with Et3N (2.4 mL, 17.1 mmol) and DPPA (3.68 mL, Ϊ7.1 mmol) and heated at reflux for lh. Benzyl alcohol (8.0 mL, 77.7 mmol) and Et3N (4.4 mL , 31 .4 mmol) were added to the reaction mixture and the solution was heated at 100 °C for 72h. The mixture was cooled to room temperature and then diluted with EtOAc (300 mL) and H20 (300 mL). The layers were separated and the aqueous layer was extracted with EtOAc (3 x 200 mL). The combined organics layers were washed with brine (100 mL), filtered and evaporated to dryness. The residue was purified by Si02 chromatography (EtOAc in hexanes, 0 to 50% gradient) and afforded the title compound (3.40 g, 9.38 mmol, 60%) as a white solid.
[253] Benzyl tert-butyl ((lS,3R)-l-methylcvclohexane-l,3-diyl)dicarbamate and benzyl tert- -l-methylcvclohexane-l,3-diyl)dicarbamate
(+/-)
[254] Both enantiomers of (+/-)-Benzyl tert-butyl ((lS,3R)-l-methylcyclohexane-l,3-diyl)dicarbamate (3.40 g, 9.38 mmol) were separated using preparative chiral HPLC (Chiralpak IA, 5 urn, 20×250 mm; hex/MeOH/DCM = 90/5/5) to yield both compounds benzyl tert-butyl ((lS,3R)-l-methylcyclohexane-l,3-diyl)dicarbamate (1.20 g, 3.31 mmol) and benzyl iert-butyl ((lR,3S)-l-methylcyclohexane-l,3-diyl)dicarbamate (1.15 g, 3.17 mmol) as white solids.
255 Benzyl ((lS,3R)-3-amino-l-methylcvclohexyl)carbamate hydrochloride
[256] A solution of benzyl tert-butyl ((lS,3R)-l-methylcyclohexane-l,3-diyl)dicarbamate (700 mg, 1.93 mmol) in DCM (19 mL) was treated with a 4M solution of HCI in dioxane (9.66 mL, 38.6 mmol) and stirred 16h at rt. The mixture was evaporated to dryness and afforded the title compound (577 mg, 1.93 mmol, 100%) as a white solid which was used in the next step without further purification.
[257] (lS,3R)-Benzyl-3-(5-chloro-4-(l-(phenylsulfonyl)-lH ndol-3-yl)pyrimidin-2-ylamino)-1-methylcyclohexylcarbamate
[258] A solution of 3-(2,5-dichloropyrimidin-4-yl)-l-(phenylsulfonyl)-lH-indole (1.02 g, 2.53 mmol), benzyl (( iS,3 )-3- amino- 1 -methylcyclohexyljcarbaniaie hydrochloride (577 mg, 1.93 mmol) and DIPEA (1.15 mL, 6.60 mmol) in NMP (11 mL) was heated at 135 °C (microwave) for 60 min. The cooled mixture was diluted with EtOAc (250 mL), washed with H20 (100 mL), brine (100 mL), dried over MgS04, filtered and evaporated to dryness. The residue was purified by Si02 chromatography (EtOAc in DCM, 0 to 50% gradient) and afforded the title compound (747 mg, 1.19 mmol, 54%) as a yellow foam.
[259] (lS,3R)-N-(5-chloro-4-(l-(phenylsulfonyl)-lH ndol-3-yl)pyrimidin-2-yl)-3-methylcvclohexane-l,3-diamine
[260] A cooled (-78°C) solution of (lS,3R)-benzyl-3-(5-chloro-4-(l-(phenylsulfonyl)-lH-indol-3-yl)pyrimidin-2-ylamino)-l-methylcyclohexylcarbamate (747 mg, 1.19 mmol) in DCM (39 mL) was treated with a 1M solution of BBr3 in DCM (2.83 mL, 2.83 mmol) and was slowly warmed up to rt. MeOH (10 mL) was added to the mixture was the resulting solution was stirred lh at rt. The resulting mixture was evaporated to dryness. The residue was purified by reverse phase chromatography (C18, H20/ACN +0.1% HC02H, 0 to 60% gradient) and afforded the title compound (485 mg, 0.978 mmol, 83%) as a yellow solid.
[261] 5-amino-N-( ( lS,3R)-3-( 5-chloro-4-(l-(phenylsulfonyl)-lH-indol-3-yl)pyrimidin-2-ylamino)-l-methylcvclohexyl)picolinamide
[262] A solution of (lR,3S)-N-(5-chloro-4-(l-(phenylsulfonyl)-lH-indol-3-yl)pyrimidin-2-yl)-3-methylcyclohexane-l,3-diamine (75.0 mg, 0.150 mmol) and 5-aminopicolinic acid (25.0 mg, 0.180 mmol) in DMF (5.0 mL) was treated with HBTU (86.0 mg, 0.230 mmol) and DIPEA (79 μί, 0.45 mmol). The resulting mixture was stirred 5h at rt and diluted with MeTHF (50 mL) and saturated NaHC03 (50 mL). The layers were separated and the aqueous layer was extracted with MeTHF (2 x 50 mL). The combined organic layers were dried over MgS04, filtered and evaporated to dryness. The residue was purified by Si02 chromatography (EtOAc in DCM, 0 to 50% gradient) and afforded the title compound (74.0 mg, 0.120 mmol, 79%) as a light yellow oil.
[263] 5-amino-N-((lS,3R)-3-(5-chloro-4-(lH ndol-3-yl)pyrimidin-2-ylamino)-l-methylcyclohexyDpicolinamide
[264] A solution of 5-amino-N-((lS,3R)-3-(5-chloro-4-(l-(phenylsulfonyl)-lH-indol-3-yl)pyrimidin-2-ylamino)-l-methylcyclohexyl)picolinamide (74.0 mg, 0.120 mmol) in 1,4-dioxane (4.0 mL) was treated with a 2M solution of NaOH in H20 (960 μί, 4.78 mmol) and heated at 60°C for lh. The cooled mixture was diluted with MeTHF (30 mL) and H20 (30 mL). The layers were separated and the aqueous layer was extracted with MeTHF (3 x 30 mL). The combined organic layers were dried over MgS04, filtered and evaporated to dryness affording the title compound (57.0 mg, 0.120 mmol, 100%) as a light yellow oil which was used in the next step without further purification.
[265] N-((lS,3R)-3-(5-chloro-4-(lH ndol-3-yl)pyrimidin-2-ylamino)-l-methylcvd^
( ( E)-4-(dimethylamino)but-2-enamido )picolinamide ( Compound 267)
[266] A cooled (-78°C) solution of 5-amino-N-((lS,3R)-3-(5-chloro-4-(lH-indol-3-yl)pyrimidin-2-ylamino)-l-methylcyclohexyl)picolinamide (57.0 mg, 0.120 mmol) and DIPEA (104 0.598 mmol) in THF/NMP (4.0 mL/1.0 mL) was treated with a 54.2 mg/mL solution of (E)-4-bromobut-2-enoyl chloride in DCM (104 μί, 0.598 mmol). The resulting mixture was stirred 4h at -78°C before addition of a 2M solution of dimethylamine in THF (359 μί, 0.717 mmol). The resulting mixture was warmed up to rt and stirred 45min at this temperature before being evaporated to dryness. The residue was purified by reverse phase chromatography (C18, H20/ACN +0.1% HC02H, 0 to 50% gradient) and afforded the title compound (15.0 mg, 0.026 mmol, 22%) as a white solid after lyophilization. LCMS: Calculated: 587.12; Found (M+H+): 587.39. 1H NMR (500 MHz, DMSO) δ 11.84 (s, 1H), 10.54 (s, 1H), 8.82 (d, J = 2.3 Hz, 1H), 8.64 (s, 1H), 8.47 (s, 1H), 8.25 (dd, J = 8.6, 2.4 Hz, 2H), 7.98 (d, J = 8.9 Hz, 2H), 7.50 (d, J = 7.7 Hz, 1H), 7.25 – 7.07 (m, 3H), 6.81 (dt, J = 15.5, 5.8 Hz, 1H), 6.29 (d, J = 15.4 Hz, 1H), 4.23 – 4.08 (m, 1H), 3.08 (dd, J = 5.7, 1.1 Hz, 2H), 2.46 – 2.37 (m, 1H), 2.18 (s, 6H), 2.04 – 1.95 (m, 2H), 1.87 – 1.70 (m, 3H), 1.63 – 1.46 (m, 4H), 1.39 – 1.26 (m, 1H).
Ref
- [1]. Hu S, et al. Discovery and characterization of SY-1365, a selective, covalent inhibitor of CDK7. Cancer Res. 2019 May 7.[2]. Shanhu Hu, et al. SY-1365, a potent and selective CDK7 inhibitor, exhibits promising anti-tumor activity in multiple preclinical models of aggressive solid tumors.
///////////////
CN(C)C\C=C\C(=O)Nc1ccc(nc1)C(=O)N[C@]1(C)C[C@@H](CCC1)Nc1ncc(Cl)c(n1)c1c[NH]c2ccccc21


NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
SY 5609
[ Fig. 0001] [ Fig. 0002]
[ Fig. 0003]
[ Fig. 0004]
SY 5609
CAS 2519828-12-5
Cancer, solid tumor
PHASE 1
A highly selective and potent oral inhibitor of cyclin-dependent kinase 7 (CDK7) for potential treatment of advanced solid tumors that harbor the Rb pa thway alterations (Syros Pharmaceuticals, Inc., Cambridge, Massachusetts, USA)
SY-5609 is an oral non-covalent CDK7 inhibitor in early clinical development at Syros Pharmaceuticals for the treatment of patients with advanced breast, colorectal, lung or ovarian cancer, or with solid tumors of any histology that harbor Rb pathway alterations.
- OriginatorSyros Pharmaceuticals
- ClassAntineoplastics; Small molecules
- Mechanism of ActionCyclin-dependent kinase-activating kinase inhibitors
- Phase IBreast cancer; Solid tumours
- 05 Aug 2021Roche plans the phase I/Ib INTRINSIC trial in Colorectal cancer (Combination therapy, Metastatic disease) in USA, Canada, Italy, South Korea, Spain and United Kingdom (NCT04929223)
- 05 Aug 2021Roche and Syros Pharmaceuticals enters into a clinical trial collaboration to evaluate atezolizumab in combination with SY 5609 in a clinical trial
- 05 Aug 2021Syros Pharmaceuticals plans a phase I trial in Cancer in second half of 2021
- NCT04247126
- https://clinicaltrials.gov/ct2/show/NCT04247126
At #ESMO21, we will be presenting new preclinical and clinical data on SY-5609, our highly selective and potent oral CDK7 inhibitor. #oncology #biotech Learn more: https://lnkd.in/gqYmWYhb
A Promising Approach for Difficult-to-Treat Cancers
SY-5609 is a highly selective and potent oral inhibitor of the cyclin-dependent kinase 7 (CDK7) in a Phase 1 dose-escalation trial in patients with advanced breast, colorectal, lung, ovarian or pancreatic cancer, or with solid tumors of any histology that harbor Rb pathway alterations.
SY-5609 represents a new approach to treating cancer that we believe has potential in a range of difficult-to-treat cancers. It has shown robust anti-tumor activity, including complete regressions, in preclinical models of breast, colorectal, lung and ovarian cancers at doses below the maximum tolerated dose. In preclinical studies of breast, lung and ovarian cancers, deeper and more sustained responses were associated with the presence of Rb pathway alterations. SY-5609 has also shown substantial anti-tumor activity in combination with fulvestrant in treatment-resistant models of estrogen receptor-positive breast cancer, including those resistant to both fulvestrant and a CDK4/6 inhibitor. Early dose-escalation data demonstrated proof-of-mechanism at tolerable doses.
Syros to Present New Data from Phase 1 Clinical Trial of SY-5609 in Oral Presentation at ESMO Congress 2021SEPTEMBER 13, 2021
Management to Host Conference Call on Monday, September 20, 2021 at 4:00 p.m. ET
CAMBRIDGE, Mass.–(BUSINESS WIRE)– Syros Pharmaceuticals (NASDAQ:SYRS), a leader in the development of medicines that control the expression of genes, today announced that it will present new data from the dose-escalation portion of the Phase 1 clinical trial of SY-5609, its highly selective and potent oral cyclin-dependent kinase 7 (CDK7) inhibitor, at the ESMO Congress 2021, taking place virtually September 16-21, 2021. The oral presentation will include safety, tolerability, and initial clinical activity data for SY-5609 in patients with breast, colorectal, lung, ovarian and pancreatic cancers, as well as in patients with solid tumors of any histology harboring Rb pathway alterations.
In separate poster presentations, Syros will present new preclinical data evaluating the antitumor and pharmacodynamic activity of intermittent dosing regimens for SY-5609 in ovarian cancer models, as well as new preclinical data evaluating antitumor activity of SY-5609 as a single agent and in combination with chemotherapy in KRAS-mutant models.
The abstracts for the two poster presentations are now available online on the ESMO conference website at: https://www.esmo.org/meetings/esmo-congress-2021/abstracts, and the presentations will become available for on-demand viewing starting September 16 at 08:30 CEST (September 16 at 2:30 a.m. ET). The abstract for the oral presentation on the Phase 1 dose-escalation data will remain embargoed until September 17 at 00:05 CEST (September 16 at 6:05 p.m. ET).
Details of the oral presentation are as follows:
Presentation Title: Tolerability and Preliminary Clinical Activity of SY-5609, a Highly Potent and Selective Oral CDK7 Inhibitor, in Patients with Advanced Solid Tumors
Session Date & Time: Monday, September 20, 17:30-18:30 CEST (11:30-12:30 p.m. ET)
Presentation Time: 17:55-18:00 CEST (11:55-12:00 p.m. ET)
Session Title: Mini Oral Session: Developmental Therapeutics
Presenter: Manish Sharma, M.D., START Midwest
Abstract Number: 518MO
Details of the poster presentations are as follows:
Presentation Title: Preclinical Evaluation of Intermittent Dosing Regimens on Antitumor and PD Activity of SY-5609, a Potent and Selective Oral CDK7 Inhibitor, in Ovarian Cancer Xenografts
Abstract Number: 14P
Presentation Title: SY-5609, a Highly Potent and Selective Oral CDK7 inhibitor, Exhibits Robust Antitumor Activity in Preclinical Models of KRAS Mutant Cancers as a Single Agent and in Combination with Chemotherapy
Abstract Number: 13P
Conference Call Information
Syros will host a conference call on Monday, September 20, 2021 at 4:00 p.m. ET to discuss the new clinical and preclinical data for SY-5609, which will be presented at the ESMO Congress 2021.
To access the live conference call, please dial 866-595-4538 (domestic) or 636-812-6496 (international) and refer to conference ID 4648345. A webcast of the call will also be available on the Investors & Media section of the Syros website at www.syros.com. An archived replay of the webcast will be available for approximately 30 days following the conference call.
About Syros Pharmaceuticals
Syros is redefining the power of small molecules to control the expression of genes. Based on its unique ability to elucidate regulatory regions of the genome, Syros aims to develop medicines that provide a profound benefit for patients with diseases that have eluded other genomics-based approaches. Syros is advancing a robust clinical-stage pipeline, including: tamibarotene, a first-in-class oral selective RARα agonist in RARA-positive patients with higher-risk myelodysplastic syndrome and acute myeloid leukemia; SY-2101, a novel oral form of arsenic trioxide in patients with acute promyelocytic leukemia; and SY-5609, a highly selective and potent oral CDK7 inhibitor in patients with select solid tumors. Syros also has multiple preclinical and discovery programs in oncology and monogenic diseases.
View source version on businesswire.com: https://www.businesswire.com/news/home/20210913005090/en/
PATENT
CN(C)C\C=C\C(=O)Nc1ccc(cc1)C(=O)Nc1cccc(c1)Nc1ncc(Cl)c(n1)c1c[NH]c2ccccc21
THZ1; 1604810-83-4; THZ-1; HY-80013
CLIP
SY 1365 MEVOCICLIB, CAS 1816989-16-8
CN(C)C\C=C\C(=O)Nc1ccc(nc1)C(=O)N[C@]1(C)C[C@@H](CCC1)Nc1ncc(Cl)c(n1)c1c[NH]c2ccccc21
PATENT
PATENT
3-fluoro-4-(methylamino)-N-[(1S,3R)-1-methyl-3-[[4-(7-methyl-1H-indol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]cyclohexyl]benzamide (Compound 130)
3-chloro-4-[[4-(dimethylamino)-3-hydroxy-butanoyl]amino]-N-[(1S,3R)-3-[[4-(1H-indazol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-1-methyl-cyclohexyl]benzamide (Compound 129)
4-amino-N-((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)-1-methylcyclohexyl)benzamide (Compound 128)
4-amino-3-fluoro-N-[(1S,3R)-3-[[4-(1H-indazol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-1-methyl-cyclohexyl]benzamide (Compound 127)
4-amino-N-((1S,3R)-3-((5-chloro-4-(2-methyl-1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)benzamide (Compound 126)
4-amino-N-((1S,3R)-3-((5-chloro-4-(1H-indazol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)benzamide (Compound 124)
Example 25 Synthesis of N1-(4-(((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)carbamoyl)phenyl)oxalamide (Compound 113)
Example 24 Synthesis of N-((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)-4-(4-(dimethylamino)butanamido)benzamide (Compound 105)
PATENT
4-amino-N-(3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)tricyclo[3.3.1.13,7]decanyl)benzamide (Compound 100).
+/−)-4-amino-N-(3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)-5 hydroxycyclohexyl)benzamide (Compound 101)
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide (Compound 102)
(1S,3R)-N-(4-aminophenyl)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexanecarboxamide (Compound 106)
4-amino-N-((1S,3R)-3-(5-cyclopropyl-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide.HCl (Compound 103)
4-amino-N-((1S,3R)-3-(5-chloro-4-(pyridin-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide (Compound 108)
4-amino-N-((1S,3R)-3-(5-cyano-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide (Compound 107)
(+/−)-4-amino-N-(3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)-5-fluorocyclohexyl)benzamide (Compound 110)
4-amino-N-(5-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)bicyclo[3.1.1]heptan-1-yl)benzamide (Compound 104)
4-amino-N4(1R,5S)-5-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)-3,3-difluorocyclohexyl)benzamide (Compound 115)
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzenesulfonamide (Compound 109).
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)-2-fluorobenzamide (Compound 112)
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)-3-fluorobenzamide (Compound 111).
(+/−)-4-amino-N-(3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)-1-methylcyclohexyl)benzamide (Compound 116).
N-((1S,3R)-3-(4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)-4-aminobenzamide (Compound 114).
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)-2-morpholinobenzamide(Compound 117).
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyridin-2-ylamino)cyclohexyl)benzamide (Compound 118).
3-amino-N-(trans-4-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide.HCl (Compound 119).
(1S,3R)-N1-(R)-1-(4-aminophenyl)-2,2,2-trifluoroethyl)-N3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)cyclohexane-1,3-diamine (Compound 120).
(1S,3R)-N1-(4-aminobenzyl)-N3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)-N1-methylcyclohexane-1,3-diamine.HCl (Compound 122).
4-amino-N-((1S,3R)-3-(5-chloro-4-(pyrazolo[1,5-a]pyridin-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide.HCl (Compound 123).
Synthesis of 5-amino-N-((1S,3R)-3-(5-chloro-4-(1-methyl-1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)picolinamide (Compound 125)
Synthesis of N-((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)-4-(4-(dimethylamino)butanamido)benzamide (Compound 105)
Synthesis of N1-(4-(((1S,3R)-3-)(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)carbamoyl)phenyl)oxalamide (Compound 113)
Synthesis of 4-amino-N-((1S,3R)-3-((5-chloro-4-(1H-indazol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)benzamide (Compound 124)
Synthesis of 4-amino-N-((1S,3R)-3-((5-chloro-4-(2-methyl-1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)benzamide (Compound 126)
Synthesis of 4-amino-3-fluoro-N-[(1S,3R)-3-[[4-(1H-indazol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-1-methyl-cyclohexyl]benzamide (Compound 127).
Synthesis of 4-amino-N-((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl) pyrimidin-2-yl)amino)-1-methylcyclohexyl)benzamide (Compound 128)
Synthesis of 3-chloro-4-[[4-(dimethylamino)-3 hydroxy-butanoyl]amino]-N-[(1S,3R)-3-[[4-(1H-indazol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-1-methyl-cyclohexyl]benzamide (Compound 129).
Synthesis of 3-fluoro-4-(methylamino)-N-[(1S,3R)-1-methyl-3-[[4-(7-methyl-1H-indol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]cyclohexyl]benzamide (Compound 130)
//////////////SY 5609, 2519828-12-5, Cancer, solid tumor, PHASE 1, SYROS

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
Ferric pyrophosphate citrate



Ferric pyrophosphate citrate
1802359-96-1
tetrairon(3+) bis((phosphonooxy)phosphonic acid) tris(2-hydroxypropane-1,2,3-tricarboxylate) (hydrogen phosphonooxy)phosphonate
Iron(3+) diphosphate (4:3)
Proper name: ferric pyrophosphate citrate Chemical names: Iron (3+) cation; 2-oxidopropane-1,2,3-tricarboxylate; diphosphate 1,2,3-propanetricarboxylic acid, 2-hydroxy-, iron (3+), diphosphate Molecular formula: [Fe4 3+(C6H5O7)3(P2O7)3] Molecular mass: 1313
Physicochemical properties: TRIFERIC AVNU (ferric pyrophosphate citrate) contains no asymmetric centers. Ferric pyrophosphate citrate is a yellow to green amorphous powder. The drug substance does not melt, or change state, below 300 °C. Thermal decomposition was observed at 263 ± 3ºC. Ferric pyrophosphate citrate is freely soluble in water (>100 g/L). Ferric pyrophosphate citrate is completely insoluble in most organic solvents (MeOH, Acetone, THF, DMF, DMSO). A 5% solution in water exhibits a solution pH of about 6. … https://pdf.hres.ca/dpd_pm/00060816.PDF
- Ferric pyrophosphate citrate
- FPC
- SFP
- Tetraferric nonahydrogen citrate pyrophosphate
- Triferic
Active Moieties
| NAME | KIND | UNII | CAS | INCHI KEY |
|---|---|---|---|---|
| Ferric cation | ionic | 91O4LML611 | 20074-52-6 | VTLYFUHAOXGGBS-UHFFFAOYSA-N |
CANADA

Summary Basis of Decision – Triferic AVNU – Health Canada
Date SBD issued:2021-07-29
The following information relates to the new drug submission for Triferic AVNU.
Iron (supplied as ferric pyrophosphate citrate)
Drug Identification Number (DIN):
DIN 02515334 – 1.5 mg/mL iron (supplied as ferric pyrophosphate citrate), solution, intravenous administration
Rockwell Medical Inc.
New Drug Submission Control Number: 239850
On April 22, 2021, Health Canada issued a Notice of Compliance to Rockwell Medical Inc. for the drug product Triferic AVNU.
The market authorization was based on quality (chemistry and manufacturing), non-clinical (pharmacology and toxicology), and clinical (pharmacology, safety, and efficacy) information submitted. Based on Health Canada’s review, the benefit-harm-uncertainty profile of Triferic AVNU is favourable for the replacement of iron to maintain hemoglobin in adult patients with hemodialysis-dependent chronic kidney disease (CKD-HD). Triferic AVNU is not intended for use in patients receiving peritoneal dialysis and has not been studied in patients receiving home hemodialysis.
Triferic AVNU, an iron preparation, was authorized for the replacement of iron to maintain hemoglobin in adult patients with hemodialysis-dependent chronic kidney disease (CKD-HD). Triferic AVNU is not intended for use in patients receiving peritoneal dialysis and has not been studied in patients receiving home hemodialysis.
Triferic AVNU is not authorized for use in pediatric patients (<18 years of age), as its safety and effectiveness have not been established in this population. No overall differences in efficacy or safety were observed in geriatric patients (≥65 years of age) compared to younger patients in clinical trials.
Triferic AVNU is contraindicated for patients who are hypersensitive to this drug or to any ingredient in the formulation, or component of the container.
Triferic AVNU was approved for use under the conditions stated in its Product Monograph taking into consideration the potential risks associated with the administration of this drug product.
Triferic AVNU (1.5 mg/mL iron [supplied as ferric pyrophosphate citrate]) is presented as a solution. In addition to the medicinal ingredient, the solution contains water for injection.
For more information, refer to the Clinical, Non-clinical, and Quality (Chemistry and Manufacturing) Basis for Decision sections.
Additional information may be found in the Triferic AVNU Product Monograph, approved by Health Canada and available through the Drug Product Database.
Health Canada considers that the benefit-harm-uncertainty profile of Triferic AVNU is favourable for the replacement of iron to maintain hemoglobin in adult patients with hemodialysis-dependent chronic kidney disease (CKD-HD). Triferic AVNU is not intended for use in patients receiving peritoneal dialysis and has not been studied in patients receiving home hemodialysis.
Chronic kidney disease (CKD) is a worldwide public health concern. One of the most common comorbidities of CKD-HD patients is anemia, which may be due to low body iron stores (as a result of blood loss during dialysis) and impaired utilization of iron. Consequently, there is an ongoing need to replenish body iron in CKD-HD patients.
Iron deficiency anemia in CKD-HD patients is generally treated using parenteral (intravenous) iron administration used in conjunction with erythropoiesis stimulating agents (ESAs). Intravenous administration is preferred, as oral iron is not well absorbed and gastrointestinal intolerance is common. At the time of authorization of Triferic AVNU, there were four other intravenous iron products marketed in Canada: Dexiron, an iron dextran (≥1,000 mg/dose); Ferrlecit (sodium ferric gluconate; 125 mg/dose); Venofer (iron sucrose; 200 mg/dose); and the more recently approved Monoferric (Iron Isomaltoside 1,000; up to 500 mg/bolus injection and up to 1,500 mg/infusion). Each of these intravenous iron products are indicated for the treatment of iron deficiency anemia and are associated with safety concerns for hypersensitivity reactions. Serious hypersensitivity reactions have been reported, including life threatening and fatal anaphylactic/anaphylactoid reactions.
Triferic AVNU is an iron replacement product delivered via intravenous infusion into the blood lines pre- and post-dialyzer in CKD-HD patients at each hemodialysis treatment. It is a preservative-free sterile solution containing 1.5 mg elemental iron/mL in water for injection.
Triferic AVNU has been shown to be efficacious in maintaining hemoglobin (Hb) during the treatment period in CKD-HD patients. The market authorization was primarily based on the results of two pivotal, randomized, placebo-controlled, single blind, Phase III clinical studies (Studies SFP-4 and SFP-5). Both studies were identical in design and enrolled a combined total of 599 adult patients with CKD-HD who were iron-replete. Patients were randomized to receive either Triferic AVNU added to bicarbonate concentrate with a final concentration of 110 μg of iron/L in dialysate or placebo (standard dialysate) administered 3 to 4 times per week during hemodialysis. All patients were to remain randomized in their treatment group until pre-specified Hb or ferritin criteria were met, indicating the need for a change in anemia management, or until they had completed 48 weeks of treatment. After randomization, patients’ ESA product, doses, or route of administration were not to be changed and oral or intravenous iron administration were not allowed.
The primary efficacy endpoint (mean change in Hb level from baseline to the end-of-treatment period) was met in both pivotal studies. In Study SFP-4, the mean Hb decreased 0.04 g/dL in the Triferic AVNU group compared to 0.39 g/dL in the placebo group. In Study SFP-5, the mean Hb decreased 0.09 g/dL in the Triferic AVNU group compared to 0.45 g/dL in the placebo group. In both studies, the treatment difference in mean hemoglobin change was 0.36 g/dL (p = 0.011) between the Triferic AVNU and the placebo groups. This value was statistically significant for both studies. The treatment difference of 0.35 g/dL was also statistically significant (p = 0.010) for both studies in the analysis using the intent-to-treat population. A high proportion of patients did not complete the planned 48 weeks of study treatment mainly due to protocol-mandated changes in anemia management (ESA dose changes). However, the proportion was similar for both arms and the analysis of Hb change in this subgroup was consistent with that of the primary efficacy analysis. Secondary endpoints which included changes in reticulocyte Hb content, serum ferritin, and pre-dialysis serum iron panel to the end of treatment, were consistent with the primary efficacy results.
The safety of Triferic AVNU was evaluated in seven controlled and uncontrolled Phase II/III studies, which included the two pivotal studies. In total, 1,411 CKD-HD patients were exposed to Triferic AVNU in the clinical program. In the pivotal studies, 78% of patients in the Triferic AVNU group and 75% of patients in the placebo group had at least one treatment-emergent adverse event (TEAE). The most common TEAEs in the Triferic AVNU group (which were higher than the placebo group) were procedural hypotension (21.6%), muscle spasms (9.6%), headache (9.2%), pain in extremity (6.8%), edema peripheral (6.8%) and dyspnoea (5.8%). Serious TEAEs were reported at similar rates for the two groups at 27.7% for the Triferic AVNU group and 27.4% for the placebo group. The most common serious TEAEs occurring in the Triferic AVNU group (which were higher than the placebo group) were cardiac arrest (1.7%), arteriovenous fistula thrombosis (1.7%), and pulmonary edema (1.4%). Few patients discontinued study treatment due to TEAEs (4.5% in the Triferic AVNU group and 2.4% in the placebo group).
In the overall clinical program, there were two cases (0.1%) of hypersensitivity reactions related to treatment out of the 1,411 patients treated with Triferic AVNU. There were no cases of serious hypersensitivity reaction and no cases of anaphylaxis related to Triferic AVNU treatment. A Serious Warnings and Precautions box describing a warning for hypersensitivity reaction has been included in the Product Monograph for Triferic AVNU.
A Risk Management Plan (RMP) for Triferic AVNU was submitted by Rockwell Medical Inc. to Health Canada. The RMP is designed to describe known and potential safety issues, to present the monitoring scheme and when needed, to describe measures that will be put in place to minimize risks associated with the product. In the RMP, the sponsor included ‘hypersensitivity reactions’ as an important identified risk; ‘systemic/serious infections’ as an important potential risk; and ‘use in pregnant and breastfeeding women’, ‘use in children’ and ‘concomitant use with other intravenous iron product’ as missing information. Labelling for these safety concerns has been included in the Product Monograph and the sponsor has committed to systemically review clinical and post-marketing safety data as part of routine pharmacovigilance activities. Upon review, the RMP was considered to be acceptable.
The submitted inner and outer labels, package insert and Patient Medication Information section of the Triferic AVNU Product Monograph meet the necessary regulatory labelling, plain language and design element requirements.
A review of the submitted brand name assessment, including testing for look-alike sound-alike attributes, was conducted and the proposed name Triferic AVNU was accepted.
Overall, the therapeutic benefits of Triferic AVNU therapy seen in the pivotal studies are positive and are considered to outweigh the potential risks. Triferic AVNU has an acceptable safety profile based on the non-clinical data and clinical studies. The identified safety issues can be managed through labelling and adequate monitoring. Appropriate warnings and precautions are in place in the Triferic AVNU Product Monograph to address the identified safety concerns.
This New Drug Submission complies with the requirements of sections C.08.002 and C.08.005.1 and therefore Health Canada has granted the Notice of Compliance pursuant to section C.08.004 of the Food and Drug Regulations. For more information, refer to the Clinical, Non-clinical, and Quality (Chemistry and Manufacturing) Basis for Decision sections.
- See MedEffect Canada for the latest advisories, warnings and recalls for marketed products.
- See the Notice of Compliance (NOC) Database for a listing of the authorization dates for all drugs that have been issued an NOC since 1994.
- See the Drug Product Database (DPD) for the most recent Product Monograph. The DPD contains product-specific information on drugs that have been approved for use in Canada.
- See the Notice of Compliance with Conditions (NOC/c)-related documents for the latest fact sheets and notices for products which were issued an NOC under the Notice of Compliance with Conditions (NOC/c) Guidance Document, if applicable. Clicking on a product name links to (as applicable) the Fact Sheet, Qualifying Notice, and Dear Health Care Professional Letter.
- See the Patent Register for patents associated with medicinal ingredients, if applicable.
- See the Register of Innovative Drugs for a list of drugs that are eligible for data protection under C.08.004.1 of the Food and Drug Regulations, if applicable.
The Chemistry and Manufacturing information submitted for Triferic AVNU has demonstrated that the drug substance and drug product can be consistently manufactured to meet the approved specifications. Proper development and validation studies were conducted, and adequate controls are in place for the commercial processes. Changes to the manufacturing process and formulation made throughout the pharmaceutical development are considered acceptable upon review. Based on the stability data submitted, the proposed shelf life of 36 months is acceptable when the drug product is stored protected from light in the aluminum pouch at room temperature (15 ºC to 30 ºC).
Proposed limits of drug-related impurities are considered adequately qualified (i.e. within International Council for Harmonisation [ICH] limits and/or qualified from toxicological studies).
All sites involved in production are compliant with Good Manufacturing Practices.
None of the excipients used in the formulation of Triferic AVNU are of human or animal origin. All non-medicinal ingredients (described earlier) found in the drug product are acceptable for use in drugs according to the Food and Drug Regulations.
DIN:
02515334
Product Monograph/Veterinary Labelling:
Date: 2021-04-21 Product monograph/Veterinary Labelling (PDF version ~ 175K)
Company:
ROCKWELL MEDICAL INC
30142 S Wixom Rd
Wixom
Michigan
United States 48393
Class:
Human
Dosage form(s):
Solution
Route(s) of administration:
Intravenous
Number of active ingredient(s):
1
Schedule(s):
Prescription
Biosimilar Biologic Drug:
No
American Hospital Formulary Service (AHFS):See footnote3
20:04.04 IRON PREPARATIONS
Anatomical Therapeutic Chemical (ATC):See footnote4
B03AC IRON, PARENTERAL PREPARATIONS
Active ingredient group (AIG) number:See footnote5
0108536041
| Active ingredient(s) | Strength |
|---|---|
| IRON (FERRIC PYROPHOSPHATE CITRATE) | 1.5 MG / ML |
RXLIST
TRIFERIC®
(ferric pyrophosphate citrate) Solution, for Hemodialysis Use
TRIFERIC®
(ferric pyrophosphate citrate) powder packet for hemodialysis use
DESCRIPTION
Triferic (ferric pyrophosphate citrate) solution, an iron replacement product, is a mixed-ligand iron complex in which iron (III) is bound to pyrophosphate and citrate. It has a molecular formula of Fe4(C6H4O7)3(H2P2O7)2(P2O7) and a relative molecular weight of approximately 1313 daltons. Ferric pyrophosphate citrate has the following structure:
|
Triferic Solution
Triferic (ferric pyrophosphate citrate) solution–is a clear, slightly yellow-green color sterile solution containing 27.2 mg of elemental iron (III) per 5 mL (5.44 mg iron (III) per mL) filled in a 5 mL or 272 mg of elemental iron (III) per 50 mL (5.44 mg iron (III) per mL) filled in a 50 Ml low density polyethylene (LDPE) ampule. Each Triferic ampule contains iron (7.5-9.0% w/w), citrate (15-22% w/w), pyrophosphate (15-22% w/w), phosphate (< 2% w/w), sodium (18-25% w/w) and sulfate (20-35%). One Triferic 5 mL ampule is added to 2.5 gallons (9.46 L) of bicarbonate concentrate. One Triferic 50 mL ampule is added to 25 gallons (94.6 L) of master bicarbonate mix.
Triferic Powder Packets
Triferic (ferric pyrophosphate citrate) powder is a slightly yellow-green powder, packaged in single use paper, polyethylene and aluminum foil packets, each containing 272.0 mg of elemental iron (III). Each Triferic packet contains iron (7.5-9.0% w/w), citrate (15-22% w/w), pyrophosphate (15-22% w/w), phosphate (< 2% w/w), sodium (18-25% w/w) and sulfate (20- 35%). One Triferic powder packet is added to 25 (94.6 L) gallons of master bicarbonate mix.
Ferric pyrophosphate citrate (FPC), a novel iron-replacement agent, was approved by the US Food and Drug Administration in January 2015 for use in adult patients receiving chronic hemodialysis (HD). This iron product is administered to patients on HD via the dialysate.
Ferric pyrophosphate citrate is a soluble iron replacement product. Free iron presents several side effects as it can catalyze free radical formation and lipid peroxidation as well as the presence of interactions of iron in plasma. The ferric ion is strongly complexed by pyrophosphate and citrate.1 FPC is categorized in Japan as a second class OTC drug.6 This category is given to drugs with ingredients that in rare cases may cause health problems requiring hospitalization or worst.7 It is also FDA approved since 2015.Label
Iron(III) pyrophosphate is an inorganic chemical compound with the formula Fe4(P2O7)3.
Synthesis
Anhydrous iron(III) pyrophosphate can be prepared by heating the mixture of iron(III) metaphosphate and iron(III) phosphate under oxygen with the stoichiometric ratio 1:3. The reactants can be prepared by reacting iron(III) nitrate nonahydrate with phosphoric acid.[2]
It can be also prepared via the following reaction:[3]3 Na4P2O7(aq) + 4 FeCl3(aq) → Fe4(P2O7)3(s) + 12 NaCl(aq)
References
- ^ W.M.Haynes. CRC Handbook of Chemistry and Physics (97th edition). New York: CRC Press, 2016. pp 4-68
- ^ Elbouaanani, L.K; Malaman, B; Gérardin, R; Ijjaali, M (2002). “Crystal Structure Refinement and Magnetic Properties of Fe4(P2O7)3 Studied by Neutron Diffraction and Mössbauer Techniques”. Journal of Solid State Chemistry. Elsevier BV. 163 (2): 412–420. doi:10.1006/jssc.2001.9415. ISSN 0022-4596.
- ^ Rossi L, Velikov KP, Philipse AP (May 2014). “Colloidal iron(III) pyrophosphate particles”. Food Chem. 151: 243–7. doi:10.1016/j.foodchem.2013.11.050. PMID 24423528.
- Gupta A, Amin NB, Besarab A, Vogel SE, Divine GW, Yee J, Anandan JV: Dialysate iron therapy: infusion of soluble ferric pyrophosphate via the dialysate during hemodialysis. Kidney Int. 1999 May;55(5):1891-8. doi: 10.1046/j.1523-1755.1999.00436.x. [Article]
- Naigamwalla DZ, Webb JA, Giger U: Iron deficiency anemia. Can Vet J. 2012 Mar;53(3):250-6. [Article]
- Fidler MC, Walczyk T, Davidsson L, Zeder C, Sakaguchi N, Juneja LR, Hurrell RF: A micronised, dispersible ferric pyrophosphate with high relative bioavailability in man. Br J Nutr. 2004 Jan;91(1):107-12. [Article]
- Pratt RD, Swinkels DW, Ikizler TA, Gupta A: Pharmacokinetics of Ferric Pyrophosphate Citrate, a Novel Iron Salt, Administered Intravenously to Healthy Volunteers. J Clin Pharmacol. 2017 Mar;57(3):312-320. doi: 10.1002/jcph.819. Epub 2016 Oct 3. [Article]
- Underwood E. (1977). Trace elements in human and animal nutrition (4th ed.). Academic press.
- KEGG [Link]
- Nippon [Link]
- FDA Reports [Link]
| Names | |
|---|---|
| Other namesFerric pyrophosphate | |
| Identifiers | |
| CAS Number | 10058-44-3 (anhydrous) 10049-18-0 (nonahydrate) |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:132767 |
| ChemSpider | 23258 |
| DrugBank | DB09147 |
| ECHA InfoCard | 100.030.160 |
| EC Number | 233-190-0 |
| PubChem CID | 24877 |
| UNII | QK8899250F 1ZJR117WBQ (nonahydrate) |
| CompTox Dashboard (EPA) | DTXSID6047600 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | Fe4(P2O7)3 |
| Molar mass | 745.224 (anhydrate) 907.348 (nonahydrate) |
| Appearance | yellow solid (nonahydrate)[1] |
| Solubility in water | insoluble |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
CLIP
https://link.springer.com/article/10.1007/s10534-018-0151-1
Iron deficiency is a significant health problem across the world. While many patients benefit from oral iron supplements, some, including those on hemodialysis require intravenous iron therapy to maintain adequate iron levels. Until recently, all iron compounds suitable for parenteral administration were colloidal iron–carbohydrate conjugates that require uptake and processing by macrophages. These compounds are associated with variable risk of anaphylaxis, oxidative stress, and inflammation, depending on their physicochemical characteristics. Ferric pyrophosphate citrate (FPC) is a novel iron compound that was approved for parenteral administration by US Food and Drug Administration in 2015. Here we report the physicochemical characteristics of FPC. FPC is a noncolloidal, highly water soluble, complex iron salt that does not contain a carbohydrate moiety. X-ray absorption spectroscopy data indicate that FPC consists of iron (III) complexed with one pyrophosphate and two citrate molecules in the solid state. This structure is preserved in solution and stable for several months, rendering it suitable for pharmaceutical applications in solid or solution state.
Iron deficiency with or without associated anemia represents a significant health problem worldwide. While many patients can restore iron levels with the use of oral iron supplements, oral supplementation is not suitable in some patients, including those undergoing chronic hemodialysis for chronic kidney disease (CKD) (Fudin et al. 1998; Macdougall et al. 1996; Markowitz et al. 1997). The limitations of oral iron replacement in patients undergoing hemodialysis likely arise from excessive ongoing losses and insufficient absorption, thus intravenous (IV) iron has become the primary route of administration in such patients (Shah et al. 2016). Multiple IV iron formulations are available, including iron dextran, iron sucrose, sodium ferric gluconate, iron carboxymaltose, ferrumoxytol, and iron isomaltoside (Macdougall et al. 1996). All such formulations are iron–carbohydrate macromolecular complexes, and the majority consist of an iron oxide core surrounded by a carbohydrate moiety (Macdougall et al. 1996; Markowitz et al. 1997).
Intravenous iron products have been used extensively for over 30 years for the treatment of iron-deficiency anemia and to maintain iron balance in hemodialysis patients since these patients have obligatory excessive losses. While these agents are generally well tolerated, they have been associated with risk of anaphylaxis (Wang et al. 2015). Compared to oral iron agents, there may be an increased risk of cardiovascular complications and infections in nondialysis patients with CKD (Macdougall et al. 1996). Additionally, higher mortality rates have been reported with use of high-dose IV iron in hemodialysis patients (Bailie et al. 2015).
Iron possesses oxidizing properties that may cause injury to cells and tissues (Koskenkorva-Frank et al. 2013; Vaziri 2013). Iron loading in general is associated with endocrinological, gastrointestinal, infectious, neoplastic, neurodegenerative, obstetric, ophthalmic, orthopedic, pulmonary, and vascular complications. In addition, excessive or misplaced tissue iron also can contribute to aging and mortality (Weinberg 2010). Normally, the body is able to protect tissues from the damaging effects of iron by regulating iron absorption in the intestine and sequestering iron with iron-binding proteins. However, the concentrations of iron introduced into the bloodstream with IV iron therapy can be as much as 100 times more than that absorbed normally through the intestine. Combined with the fact that IV iron is administered over a period of minutes compared to the slow, regulated absorption in the gut, it is possible that the increased iron load may damage cells and tissues.
A novel parenteral iron formulation, ferric pyrophosphate citrate (FPC), potentially offers a more physiologic delivery of iron. Unlike previous forms of IV iron, FPC contains no carbohydrate shell. Soluble ferric pyrophosphate-citrate complexes, generally referred to as soluble ferric pyrophosphate (SFP) were first described in the mid-1800s by Robiquet and Chapman (Chapman 1862; Robiquet 1857). This class of food-grade iron salts has been available for over 100 years as oral iron supplements and for fortification of food. In the late-1990s, Gupta et al. demonstrated that food-grade SFP could be administered to hemodialysis patients via the dialysate (Gupta et al. 1999). However, the commercially available compounds are poorly characterized and not suitable for further development as a parenteral iron supplement. Therefore, a pharmaceutical-grade SFP was developed. This product had a higher solubility than food-grade SFP and was granted a new USAN name—FPC. In 2015 FPC was approved by the US Food and Drug Administration (FDA) for parenteral delivery by hemodialysis to replace iron losses and thereby maintain hemoglobin levels in hemodialysis-dependent patients with CKD (Rockwell Medical Inc 2018). FPC is currently marketed under the trade name Triferic® (Rockwell Medical Inc., Wixom, Michigan, USA). FPC is the first carbohydrate-free, noncolloidal, water-soluble iron salt suitable for parenteral administration.
Infrared spectroscopy
Infrared (IR) spectroscopy was used to determine the main functional groups present in FPC. Figure 1 shows a representative IR spectrum of FPC. Peak assignments and positions for FPC as well as for sodium citrate, sodium pyrophosphate, and ferric sulfate, which were used to confirm the peak assignments, are shown in Table 1.
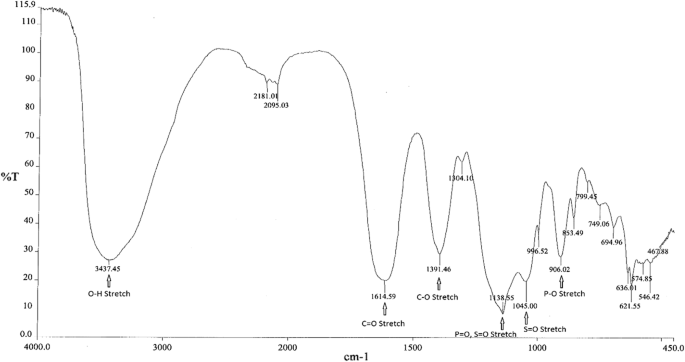
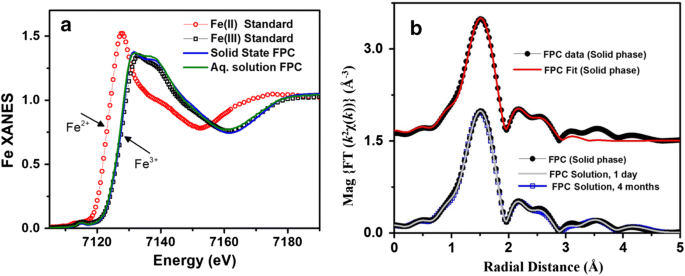
X-ray spectra of solid and aqueous iron standards and FPC. a XANES spectra of iron (II) and iron (III) standards as well as FPC in the solid and solution phases show that FPC consists exclusively of iron (III) and that the solid-phase structure is maintained in solution. b EXFAS modeling of FPC in the solid phase (top) and in solution (bottom) at Day 1 and Month 4
Chemical composition of ferric pyrophosphate citrate
From: Physicochemical characterization of ferric pyrophosphate citrate
| Ion | Percentage |
|---|---|
| Iron | 8 |
| Citrate | 19 |
| Pyrophosphate | 18 |
| Phosphate | < 1 |
| Sulfate | 25–28 |
PATENT
https://patents.google.com/patent/WO2017040937A1/enProperties of Conventional SFP

Another example of SFP is the composition is the chelate composition described in US Patent Nos. 7,816,404 and 8,178,709. The SFP may be a ferric pyrophosphate citrate (FPC) comprising a mixed-ligand iron compound comprising iron chelated with citrate andpyrophosphate, optionally FPC has the following formula: Fe4(C6H407)3(H2P207)2(P207) (relative MW 1313 daltons), e.g., structure (I):
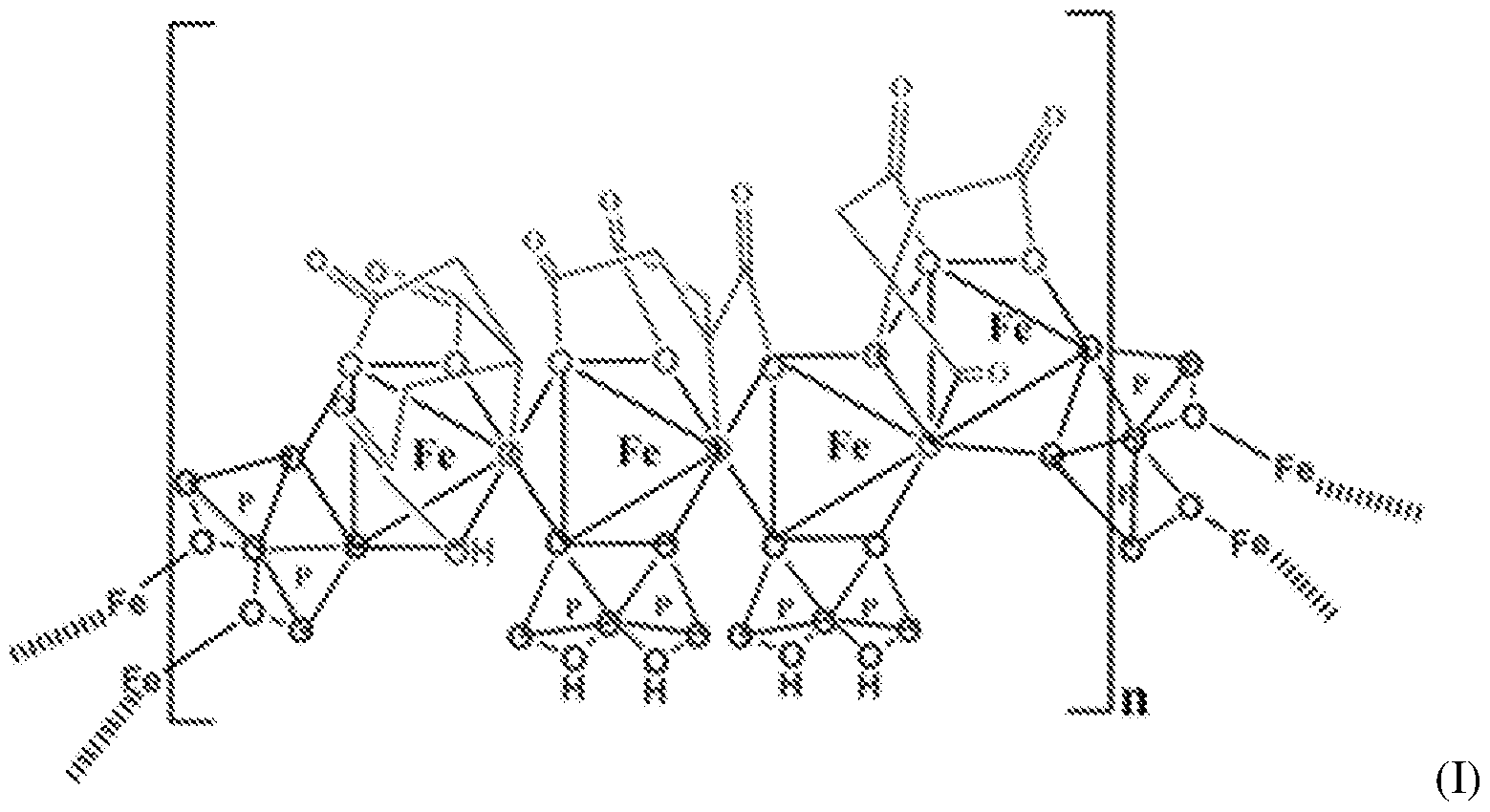
[0036] An exemplary SFP according to the present disclosure is known to have the properties described in Table 3.Table 3 – Properties of SFP according to the present disclosure


NEW DRUG APPROVALS
one time
$10.00
////////////ferric pyrophosphate citrate, Triferic AVNU, , Ferric pyrophosphate citrate, FPC, SFP, Tetraferric nonahydrogen citrate pyrophosphate, Triferic, FDA 2015, APPROVALS 2021, CANADA 2021, hemodialysis-dependent chronic kidney disease
[Fe+3].[Fe+3].[Fe+3].[Fe+3].OP(O)(=O)OP(O)(O)=O.OP(O)(=O)OP(O)(O)=O.OP([O-])(=O)OP([O-])([O-])=O.OC(CC([O-])=O)(CC([O-])=O)C([O-])=O.OC(CC([O-])=O)(CC([O-])=O)C([O-])=O.OC(CC([O-])=O)(CC([O-])=O)C([O-])=O
Pegvaliase
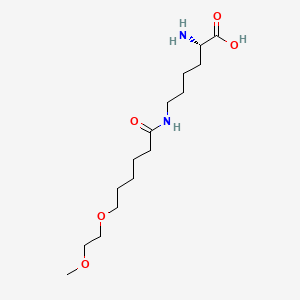

Pegvaliase
(2S)-2-amino-6-[6-(2-methoxyethoxy)hexanoylamino]hexanoic acid
CAS 1585984-95-7
- Molecular FormulaC15H30N2O5
- Average mass318.409 Da
BMN-165
Palynziq
pegvaliase
pegvaliase-pqpz
L-Lysine, N6-[6-(2-methoxyethoxy)-1-oxohexyl]-
N6-[6-(2-Methoxyethoxy)hexanoyl]-L-lysine
AUSTRALIA APPROVAL 2021
PALYNZIQ![]()
Evaluation commenced: 30 Sep 2020
Registration decision: 6 Jul 2021
Date registered: 14 Jul 2021
Approval time: 166 (175 working days)
pegvaliase
BioMarin Pharmaceutical Australia Pty Ltd
PALYNZIQ (solution for injection, pre-filled syringe) is indicated for the treatment of patients with phenylketonuria (PKU) aged 16 years and older who have inadequate blood phenylalanine control despite prior management with available treatment options.
Pegvaliase, sold under the brand name Palynziq, is a medication for the treatment of the genetic disease phenylketonuria.[2][3] Chemically, it is a pegylated derivative of the enzyme phenylalanine ammonia-lyase that metabolizes phenylalanine to reduce its blood levels.[4]
It was approved by the Food and Drug Administration for use in the United States in 2018.[2] The U.S. Food and Drug Administration (FDA) considers it to be a first-in-class medication.[5]
Pegvaliase is a recombinant phenylalanine ammonia lyase (PAL) enzyme derived from Anabaena variabilis that converts phenylalanine to ammonia and trans-cinnamic acid. Both the U.S. Food and Drug Administration and European Medicines Agency approved pegvaliase-pqpz in May 2018 for the treatment of adult patients with phenylketonuria (PKU). Phenylketonuria is a rare autosomal recessive disorder that is characterized by deficiency of the enzyme phenylalanine hydroxylase (PAH) and affects about 1 in 10,000 to 15,000 people in the United States. PAH deficiency and inability to break down an amino acid phenylalanine (Phe) leads to elevated blood phenylalanine concentrations and accumulation of neurotoxic Phe in the brain, causing chronic intellectual, neurodevelopmental and psychiatric disabilities if untreated. Individuals with PKU also need to be under a strictly restricted diet as Phe is present in foods and products with high-intensity sweeteners. The primary goal of lifelong treatment of PKU, as recommended by the American College of Medical Genetics and Genomics (ACMG) guidelines, is to maintain blood Phe concentration in the range of 120 µmol/L to 3690 µmol/L. Pegvaliase-pqpz, or PEGylated pegvaliase, is used as a novel enzyme substitution therapy and is marketed as Palynziq for subcutanoues injection. It is advantageous over currently available management therapies for PKU, such as [DB00360], that are ineffective to many patients due to long-term adherence issues or inadequate Phe-lowering effects. The presence of a PEG moiety in pegvaliase-pqpz allows a reduced immune response and improved pharmacodynamic stability.
References
- ^ Jump up to:a b “Palynziq”. Therapeutic Goods Administration (TGA). 23 July 2021. Retrieved 5 September 2021.
- ^ Jump up to:a b “FDA approves a new treatment for PKU, a rare and serious genetic disease” (Press release). Food and Drug Administration. May 24, 2018.
- ^ Mahan KC, Gandhi MA, Anand S (April 2019). “Pegvaliase: a novel treatment option for adults with phenylketonuria”. Current Medical Research and Opinion. 35 (4): 647–651. doi:10.1080/03007995.2018.1528215. PMID 30247930.
- ^ “Palynziq”. BioMarin Pharmaceutica.
- ^ New Drug Therapy Approvals 2018 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2019. Retrieved 16 September 2020.
External links
- “Pegvaliase”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Pronunciation | peg val’ i ase |
| Trade names | Palynziq |
| Other names | Pegvaliase-pqpz; PEG-PAL; RAvPAL-PEG |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a618057 |
| License data | US DailyMed: Pegvaliase |
| Pregnancy category | AU: D[1] |
| Routes of administration | Subcutaneous |
| ATC code | A16AB19 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only) [1]US: ℞-onlyEU: Rx-only |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1585984-95-7 |
| PubChem CID | 86278362 |
| DrugBank | DB12839 |
| ChemSpider | 58172730 |
| UNII | N6UAH27EUV |
| KEGG | D11077 |
| Chemical and physical data | |
| Formula | C15H30N2O5 |
| Molar mass | 318.414 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////pegvaliase, PALYNZIQ, AUSTRALIA 2021, APPROVALS 2021, BioMarin, BMN 165, Palynziq, pegvaliase, pegvaliase-pqpz
COCCOCCCCCC(=O)NCCCCC(C(=O)O)N

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
_pyrophosphate.svg){kind=link}
{kind=link}