
Home » Preclinical china
Category Archives: Preclinical china
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
JBI-802 BY JUBILANT
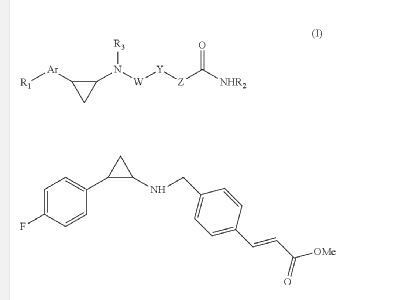
EXAMPLE
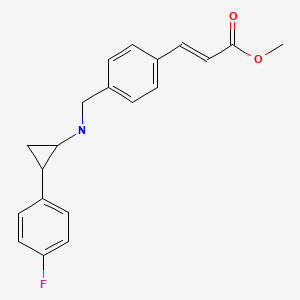
O=C(OC)/C=C/c1ccc(CNC2CC2c2ccc(F)cc2)cc1
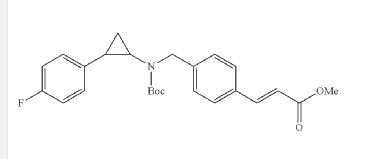
EXAMPLE ONLY NOT CONFIRMED
JBI-802
- Myeloid Leukemia Therapy
- Solid Tumors Therapy
Epigenetic Modifier Modulators
- Histone Deacetylase 6 (HDAC6) Inhibitors
- Lysine-Specific Histone Demethylase 1A (KDM1A; LSD1) Inhibitors
Jubilant Therapeutics Announces Successful Completion of Pre-IND Meeting with FDA for its Novel Dual LSD1 and HDAC6 Inhibitor JB1-802
https://markets.businessinsider.com/news/stocks/jubilant-therapeutics-announces-successful-completion-of-pre-ind-meeting-with-fda-for-its-novel-dual-lsd1-and-hdac6-inhibitor-jb1-802-1030834551
PRESS RELEASE PR Newswire
Sep. 30, 2021, 10:23 AM
BEDMINSTER, NJ, Sept. 30, 2021 /PRNewswire/ — Jubilant Therapeutics Inc., a biopharmaceutical company advancing small molecule precision therapeutics to address unmet medical needs in oncology and autoimmune diseases, today announced the successful completion of a pre-IND (Investigational New Drug) meeting with the U.S. Food and Drug Administration (FDA) regarding the development plan, clinical study design and dosing strategy for the Phase I/II trial of JB1-802, a dual inhibitor of LSD1 and HDAC6, for the treatment of small cell lung cancer, treatment-induced neuro-endocrine prostate cancer and other mutation-defined neuroendocrine tumors.
![]() A pre-IND meeting provides the drug development sponsor an opportunity for an open communication with the FDA to discuss the IND development plan and to obtain the agency’s guidance regarding planned clinical evaluation of the sponsor’s new drug candidate. After reviewing the preclinical data provided, plans for additional data generation and the Phase I/II clinical trial protocol, the FDA addressed Jubilant Therapeutics’ questions, provided guidance and aligned with the sponsor on the proposed development plan for JBI-802.
A pre-IND meeting provides the drug development sponsor an opportunity for an open communication with the FDA to discuss the IND development plan and to obtain the agency’s guidance regarding planned clinical evaluation of the sponsor’s new drug candidate. After reviewing the preclinical data provided, plans for additional data generation and the Phase I/II clinical trial protocol, the FDA addressed Jubilant Therapeutics’ questions, provided guidance and aligned with the sponsor on the proposed development plan for JBI-802.
“We appreciate the FDA’s guidance as we endeavor to find an innovative new treatment for high unmet-need tumors with devastatingly low survival rates,” said Hari S Bhartia, Chairman, Jubilant Therapeutics Inc.
“We are pleased with the outcome of the pre-IND meeting with the FDA and plan to submit the IND application by the end of 2021,” said Syed Kazmi, Chief Executive Officer, Jubilant Therapeutics Inc.
About Jubilant TherapeuticsJubilant Therapeutics Inc. is a patient-centric biopharmaceutical company advancing potent and selective small molecule modulators to address unmet medical needs in oncology and autoimmune diseases. Its advanced discovery engine integrates structure-based design and computational algorithms to discover and develop novel, precision therapeutics against both first-in-class and validated but intractable targets in genetically defined patient populations. The Company plans to file an IND later this year for the first in class dual inhibitor of LSD1/HDAC6, followed by two additional INDs in 2022 with novel modulators of PRMT5 and PAD4 in oncology and inflammatory indications. Jubilant Therapeutics is headquartered in Bedminster NJ and guided by globally renowned key opinion leaders and scientific advisory board members. For more information, please visit www.jubilanttx.com or follow us on Twitter @JubilantTx and LinkedIn.
View original content:https://www.prnewswire.com/news-releases/jubilant-therapeutics-announces-successful-completion-of-pre-ind-meeting-with-fda-for-its-novel-dual-lsd1-and-hdac6-inhibitor-jb1-802-301388983.html
SOURCE Jubilant Therapeutics Inc.

Mohd Zainuddin
Director at Jubilant Therapeutics Inc
PATENT
IN 201641016129
PATENT
US20200308110 – CYCLOPROPYL-AMIDE COMPOUNDS AS DUAL LSD1/HDAC INHIBITORS
https://patentscope.wipo.int/search/en/detail.jsf?docId=US306969204&tab=NATIONALBIBLIO&_cid=P21-KUANET-85789-2ApplicantsJubilant Epicore LLC
Inventors
Sridharan RAJAGOPAL
Mahanandeesha S. HALLUR
Purushottam DEWANG
Kannan MURUGAN
Durga Prasanna KUMAR C.H.
Pravin IYER
Chandrika MULAKALA
Dhanalakshmi SIVANANDHAN
Sreekala NAIR
Mohd ZAINUDDIN
Subramanyam Janardhan TANTRY
Chandru GAJENDRAN
Sriram RAJAGOPAL
Priority Data201641016129 09.05.2016 IN

Sridharan Rajagopal
Vice President-Head of Medicinal Chemistry at Jubilant Therapeutics Inc

Dhanalakshmi Sivanandhan
Vice President at Jubilant Therapeutics Inc

Mahanandeesha Hallur
Associate Director at Jubilant Biosys

Sreekala Nair

Chandrika Mulakala

Pravin Iyer
 Dewang")
Purushottam (M.) Dewang
ERRORS CALL ME , +919321316780
AND TO ADD TOO
EXAMPLE
C20 H20 F N O22-Propenoic acid, 3-[4-[[[2-(4-fluorophenyl)cyclopropyl]amino]methyl]phenyl]-, methyl ester, (2E)-Molecular Weight, 325.38
Patent
WO2017195216
I-3methyl (E)-3-(4-(((tert-butoxycarbonyl)(2-(4-((4-fluorobenzyl)oxy)phenyl) cyclopropyl)amino)methyl)phenyl)acrylate

The compound was synthesized using amine B6 and (E)-3-(4-Formyl-phenyl)-acrylic acid methyl esterfoUowing the procedure for the synthesis of 1-2. LC-MS m/z calcd for C32H34FN05, 531.2; found 532.2 [M+H]+.

| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| EP-3455204-A1 | Cyclopropyl-amide compounds as dual lsd1/hdac inhibitors | 2016-05-09 | |
| WO-2017195216-A1 | Cyclopropyl-amide compounds as dual lsd1/hdac inhibitors | 2016-05-09 | |
| US-2020308110-A1 | Cyclopropyl-amide compounds as dual lsd1/hdac inhibitors | 2016-05-09 |

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
Step 2: (E)-3-[4-({tert-Butoxycarbonyl-[2-(4-fluoro-phenyl)-cyclopropyl]-amino}-methyl)-phenyl]-acrylic acid methyl ester (I-2)
| To a stirred solution of (E)-3-(4-{[2-(4-fluoro-phenyl)-cyclopropylamino]-methyl}-phenyl)-acrylic acid methyl ester (XLVI, 0.25 g, 0.76 mmol) in tetrahydrofuran and water mixture (6 mL, 1:1) was added sodium bicarbonate (0.087 g, 2.3 mmol) and Boc anhydride (0.22 mL, 0.92 mmol) at room temperature and the resulting mixture was stirred at that temperature for 2 h. The progress of the reaction was monitored by TLC. The reaction mixture was diluted with ethylacetate and the organic portion was washed with water and brine solution, dried over sodium sulphate and concentrated under reduced pressure to get the crude product which was purified by column chromatography using ethylacetate-hexane gradient to afford the titled product as sticky oil (I-2, 0.19 g, 58%). LC-MS m/z calcd for C 25H 28FNO 4, 425.2; found 326.3 [M-Boc+1] +. |
| The following compounds were synthesized using procedure for the synthesize of I-2 |
REFJBI-802, novel dual inhibitor of LSD1-HDAC6 for treatment of cancerSivanandhan, D.; Rajagopal, S.; Nair, S.; et al.Annu Meet Am Assoc Cancer Res (AACR) · 2020-06-22 / 2020-06-24 · Virtual, N/A · Abst 1756Synthesis and optimization of a novel series of LSD1-HDAC dual inhibitors led to the discovery of JBI-802 as the lead compound, with IC50 of 0.05 mcM against LSD1 and isoform selective HDAC6/8 activity, with IC50 of 0.011 and 0.098 mcM for HDAC6 and HDAC8, respectively. The candidate also showed excellent selectivity against other HDACs, with approximately 77-fold selectivity for HDAC6. In vitro, JBI-802 showed strong antiproliferative activity on selected cell lines, including acute myeloid leukemia, chronic lymphocytic leukemia, lymphoma and certain solid tumors, such as small cell lung cancer and sarcoma. In vivo, JBI-802 demonstrated strong efficacy in erythroleukemia xenograft model, leading to prolonged survival of mice bearing HEL92.1.7 tumors. The candidate showed excellent dose-response and superior efficacy compared to single agents in this model, with ED50 of approximately 6.25 mg/kg twice-daily by oral administration. When evaluated in CT-26 syngeneic model, JBI-802 showed promising activity as single agent and in the combination of JBI-802 plus anti-programmed cell death protein 1 (PD-1) monoclonal antibody (MAb), with approximately 80% tumor growth inhibition observed for the combination. Exploratory toxicology studies showed that JBI-802 was well tolerated at efficacious doses. Further preclinical IND-enabling studies are currently underway for this molecule, which is to be developed as a clinical candidate for the treatment of acute myeloid leukemia and other tumor types.
REFNovel dual inhibitor of LSD1-HDAC6/8 for treatment of cancerDhanalakshmi, S.; Rajagopal, S.; Sadhu, N.; et al.62nd Annu Meet Am Soc Hematol · 2020-12-05 / 2020-12-08 · Virtual, N/A · Abst 3378 Blood 2020, 136(Suppl. 1)
REFJubilant Therapeutics Presents Preclinical Data at the American Association for Cancer Research, Reveals Unique Dual-Action Anti-Cancer Mechanism Underscoring First-in-Class Pipeline Asset in Hematological Tumors
Jubilant Therapeutics Press Release 2020, June 22
////////////////JB1-802, JUBILANT, CANCER, PRECLINICAL
EXTRAS…………
PATENTWO2021062327 – FUSED PYRIMIDINE COMPOUNDS, COMPOSITIONS AND MEDICINAL APPLICATIONS THEREOFhttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021062327&_cid=P21-KUAMRR-83330-1PCT/US2020/052953
Priority Data
| 201941039277 | 27.09.2019 | IN |
Inventors
- VENKATESHAPPA, Chandregowda
- SIVANANDHAN, Dhanalakshmi
- RAJAGOPAL, Sridharan
- ROTH, Bruce
- PANDEY, Anjali
- SAXTON, Tracy
- HALLUR, Gurulingappa
- MADHYASTHA, Naveena
- SADHU M, Naveen
Lung cancer accounts for the greatest number of cancer deaths, and approximately 85% of lung cancer cases are non-small cell lung cancer (NSCLC). The development of targeted therapies for lung cancer has primarily focused on tumors displaying specific oncogenic drivers, namely mutations in epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK). Three generations of tyrosine kinase inhibitors (TKIs) have been developed for cancers with the most frequently observed EGFR mutations, however, other oncogenic drivers in the EGFR family of receptor tyrosine kinases have received less research and development focus and several oncogenic drivers, including insertions in the exon 20 gene of EGFR, have no currently approved therapeutics to treat their cancers.
[0003] The mutation, amplification and/or overexpression of human epidermal growth factor receptor 2 (HER2), another member of the human epidermal growth factor receptor family of receptor tyrosine kinases, has been implicated in the oncogenesis of several cancers, including lung, breast, ovarian, and gastric cancers. Although targeted therapies such as trastuzumab and lapatinib have shown clinical efficacy especially in breast tumors, their utility in lung cancer has been limited. It is likely that this variation is due to tissue-specific factors, including the low potency of kinase inhibitors like lapatinib for the mutagenic alterations in HER2 that are observed in the lung cancer patient population, including insertions in the exon 20 gene of HER2.
[0004] Given that many patients with mutations in EGFR and HER2 do not derive clinical benefit from currently available therapies against these targets, there remains a significant unmet need for the development of novel therapies for the treatment of cancers associated with EGFR and HER2 mutations.
Compound 49: (E)-N-(3-(3-benzyl-7-((1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4-(dimethylamino)but-2-enamide
Step 1: Synthesis of (E)-4-(dimethylamino)but-2-enoyl chloride
[0280] To a stirred mixture of acetonitrile (2 mL) and DMF (2 drop) under N2 atmosphere was added N,N-dimethylamino crotonic acid hydrochloride (0.1 g, 0.77 mmol). After 10 min, this solution was cooled to 0-5 °C. Oxalyl chloride (0.122 g, 0.968 mmol) was added and the reaction mixture was maintained at 0-5 °C for 30 min. It was allowed to warm to RT and stirring was continued for 2 h. It was then heated to 40 °C for 5 min and again brought to RT and stirred for 10 min. Formation of product was confirmed by TLC and the reaction mass was used as such to the next step without any workup.
Step-2: Synthesis of (E)-N-(3-(3-benzyl-7-((1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4-(dimethylamino)but-2-enamide (Compound 49)
[0281] 1-(3-Aminophenyl)-3-benzyl-7-((1-methyl-1H-pyrazol-4-yl)amino)-3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one (0.11g, 0.7 mmol) in DMP (2 mL) was cooled to -15 °C and then (E)-4-(dimethylamino)but-2-enoylchloride was added. The reaction mixture was stirred for 1 h at -15 °C to RT. After the completion of reaction, the reaction mass was quenched with ice water, sodium bicarbonate solution and extracted with DCM (100 mL x 2). The combined organic layer was washed with cold water (3 x 50 mL), brine solution (10 mL), dried over anhydrous sodium sulfate and evaporated under reduced pressure to obtain crude product. The crude product was purified by prep HPLC to get pure product (E)-N-(3-(3-benzyl-7-((1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4-(dimethylamino)but-2-enamide (Compound 49, 0.022 g, 16 % yield) as white solid.1H NMR (400 MHz, DMSO-d6): δ 10.21 (s, 1H), 9.32 (s, 1H), 8.06 (s, 1H), 7.76 (bs, 1H) 7.65 (s, 1H), 7.48 (bs, 1H), 7.39-7.29 (m, 5H), 7.03 (d, J = 7.2 Hz, 2H), 6.74-6.68 (m, 1H), 6.62 (s, 1H), 6.25 (d, J = 15.2 Hz, 1H), 4.62 (s, 2H), 4.37 (s, 2H), 3.47 (s, 3H), 3.03 (d, J = 5.6 Hz, 2H), 2.15 (s, 6H); LCMS Calcd for [M+H] + 538.2, found 538.5
Compound 50: (E)-N-(3-(3-benzyl-7-((1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-3-chloroacrylamide
Step-1: Synthesis of (Z)-3-chloroacrylic acid
[0282] To a stirred solution propiolic acid (2 g, 28.5 mmol) in DMF (15 mL) under N2 atmosphere was added thionyl chloride (4.07 g, 34.2 moles) slowly and the reaction mixture was maintained at 25 °C for 1 h. The reaction was monitored by TLC, after the completion of reaction, the residue was poured into ice and the resulting aqueous solution was extracted with ether (3 x100 mL). The organic layer was washed with brine (20 mL), dried over anhydrous sodium sulfate and evaporated under reduced pressure to obtain crude product. The crude product was purified to get pure product (Z)-3-chloroacrylic acid (1.9 g, 62.9 % yield). LCMS Calcd for [M-H] +, 104.98, found 105.1
Step-2: Synthesis of (Z)-3-chloroacryloyl chloride
[0283] To a stirred solution of acetonitrile (3 mL) and DMF (3 drop) under N2 atmosphere was added of (Z)-3-chloroacrylic acid (0.2 g, 1.87 mmol). After 10 min this solution was cooled 0-5 °C. Oxalyl chloride (0.122 g, 0.968 mmol) was added and the reaction mixture was maintained at 0-5 °C for 30 min. It was allowed to warm to RT and stirring was continued for 2 h to get (Z)-3-chloroacryloyl chloride. Formation of product was confirmed by TLC and the reaction mass was used as such to the next step without any workup.
Step-3: Synthesis of (E)-3-((3-(3-benzyl-7-((1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)amino)acryloyl chloride (Compound 50)
[0284] A solution of 1-(3-Aminophenyl)-3-benzyl-7-((1-methyl-1H-pyrazol-4-yl)amino)-3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one (0.11 g, 0.7 mmol) in DMP (2 mL) was cooled to -15 °C and then (Z)-3-chloroacryloyl chloride was added. The reaction mixture was stirred for 1 h at -15 °C to RT. The reaction was monitored by TLC. After the completion of reaction, reaction mass was quenched with ice water and sodium bicarbonate solution. The aqueous layer was e 0.028 g, 22% yield) as a white solid.1H NMR (400 MHz, DMSO-d6): δ 10.35 (s, 1H), 9.32 (s, 1H), 8.06 (s, 1H), 7.74 (s, 1H), 7.59 (s, 1H), 7.51 (s, 1H), 7.41-7.35 (m, 5H), 7.30-7.29 (m, 1H), 7.08-7.02 (m, 2H), 6.62-6.58 (m, 2H), 4.62 (s, 2H), 4.37 (s, 2H), 3.47 (s, 3H); LCMS Calcd for [M+H] + 515.1, LCMS found 515.2
Compound 51: (E)-N-(3-(7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-3-phenyl-2-thioxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4-(dimethylamino)but-2-enamide
Step-1: Synthesis of 2,4-dichloro-5-(chloromethyl)pyrimidine
[0285] Title compound was prepared in a similar manner to general procedure I.5-(hydroxymethyl)pyrimidine-2,4-diol (15 g, 106 mmol) gave 2,4-dichloro-5-(chloromethyl)pyrimidine (11.50 g, 55% yield) as a white solid.1H NMR (400 MHz, CDCl3): δ 8.66 (s, 1H), 4.65 (s, 2H).
Step-2: Synthesis of 2,4-dichloro-5-(iodomethyl)pyrimidine
[0286] Title compound was prepared in a similar manner to general procedure J.2,4-dichloro-5-(chloromethyl)pyrimidine (11.50 g, 58.20 mmol) on treatment with NaI (10.50 g, 69.0 mmol) in acetone (100 mL) resulted in 2,4-dichloro-5-(iodomethyl)pyrimidine (15.20 g, 91% yield). The solid was immediately taken up in toluene and stored under refrigeration.1H NMR (400 MHz, CDCl3): δ 8.60 (s, 1H), 4.39 (s, 2H).
Step-3: Synthesis of N-((2,4-dichloropyrimidin-5-yl)methyl)aniline
[0287] A solution of iodo compound (18, 7.0 g, 24.20 mmol) in toluene (50 mL) was cooled to 0 °C and aniline (2.20 g, 24.20 mmol) was added. The reaction mixture was stirred for 30 min at 0 °C. Then a solution of sodium hydroxide (1.30 g, 32.50 mmol) in water (5 ml) was added and reaction mixture was stirred for 16 h at RT. The reaction was monitored by TLC. After completion of the reaction, water (25 mL) was added and extracted with ethyl acetate (2 x 100 mL). The organic layer was washed with brine solution, dried over anhydrous sodium sulfate and evaporated under reduced pressure to obtain the crude residue. The crude compound was purified by silica gel column chromatography to afford the title compound as a white solid (10 g, 81% yield). LCMS Calcd for [M+H] + 254.11, found 254.09
Step-4: Synthesis of tert-butyl (3-((2-chloro-5-((phenylamino)methyl)pyrimidin-4-yl)amino)phenyl)carbamate
[0288] To a stirred solution of N-((2,4-dichloropyrimidin-5-yl)methyl)aniline (4.0 g, 15.08 mmol) in IPA (30 mL), tert-butyl (3-aminophenyl)carbamate (4.90 g, 23.0 mmol) and DIPEA (8.20 mL, 47 mmol) were added. The reaction mixture was heated at 100 °C for 16 h in a sealed tube. Solvent was then evaporated and the crude thus obtained was purified by flash column chromatography to afford the title compound as off white solid (2.50 g, 37% yield). LCMS Calcd for [M+H] + 425.92, found 426.35
Step-5: Synthesis of tert-butyl (3-(7-chloro-3-phenyl-2-thioxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate
[0289] To a solution of tert-butyl (3-((2-chloro-5-((phenylamino)methyl)pyrimidin-4-yl)amino)phenyl)carbamate (1.50 g, 3.50 mmol) in THF (35 mL) was added DIPEA (2.40 mL, 14.10 mmol) and thiophosgene (0.27 g, 3.50 mmol) at 0 °C. The reaction mixture was stirred at RT for 24 h with TLC monitoring. After completion of the reaction, sodium bicarbonate solution was added. The reaction mixture was partitioned between DCM (2 x 100 mL) and water (50 mL). The organic layer was washed with brine (10 mL), dried over anhydrous sodium sulfate and evaporated under reduced pressure to obtain crude product. The crude product was purified by silica gel column chromatography to afford the title compound as a yellow solid (1.36 g, 82% yield). LCMS Calcd for [M+H] + 467.97, found 468.27
Step-6: Synthesis of tert-butyl (3-(7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-3-phenyl-2-thioxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate
[0290] To a solution of tert-butyl (3-(7-chloro-3-phenyl-2-thioxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (1.30 g, 2.78 mmol) in IPA (15 mL) was added 3-
chloro-1-methyl-1H-pyrazol-4-amine (0.44 g, 3.34 mmol) and TFA (1 mL). The reaction mixture was heated for 16 h at 110 °C. Reaction was monitored by TLC. After the completion of reaction, the reaction mixture was concentrated, water (10 mL) and saturated sodium bicarbonate (20 mL) solution were added to the residue and extracted with DCM (3 x 200 mL). The combined organic layer was washed with brine solution, dried over anhydrous sodium sulfate and evaporated under reduced pressure to obtain the title compound (1.30 g) that was used as such for the next step without further purification. LCMS Calcd for [M+H] + 563.08, found 562.90
Step-7: Synthesis of 1-(3-aminophenyl)-7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidine-2(1H)-thione
[0291] To an ice-cold solution of tert-butyl (3-(7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-3-phenyl-2-thioxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (1.30 g, 2.30 mmol) in DCM (20 mL) and MeOH (10 mL) was added 4N HCl in dioxane (5 mL). The reaction mixture was stirred for 16 h at RT. The reaction was monitored by TLC. After completion of the reaction, the solvent was evaporated followed by addition of water (10 mL) and saturated sodium bicarbonate (20 mL) solution and extraction with DCM (3 x 200 mL). The combined organic layer was washed with brine solution, dried over anhydrous sodium sulfate and evaporated under reduced pressure to obtain crude product. The crude product was purified by silica gel column chromatography to afford the title compound as a brown solid (0.20 g). LCMS Calcd for [M+H] + 462.96, found 463.0. Purity: 68%
Step-8: Synthesis of (E)-N-(3-(7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-3-phenyl-2-thioxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4-(dimethylamino)but-2-enamide (Compound 51)
[0292] To an ice-cold solution of 1-(3-aminophenyl)-7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidine-2(1H)-thione (0.18 g, 0.39 mmol) and trans-N,N-dimethylaminocrotonic acid hydrochloride (0.077 g, 0.47 mmol) in dichloromethane (10 mL) was added triethyl amine (1.2 mmol) followed by drop wise addition of propylphosphonic anhydride (T3P) (0.26 g, 0.97 mmol). The mixture was stirred at RT for 6 h. Completion of the reaction was monitored by TLC. The reaction mixture was portioned between 5% methanol in dichloromethane and saturated bicarbonate solution. The organic phase was dried over anhydrous sodium sulfate, filtered and concentrated. The crude obtained was purified by silica gel chromatography to afford the title compound as off white solid (Compound 51, 0.010 g, 5% yield).1H NMR (400 MHz, DMSO-d6): δ 10.36 (bs, 1H), 8.97 (bs, 1H), 8.25 (s, 1H), 7.72 (bs, 2H), 7.48-7.42 (m, 5H), 7.36-7.32 (m, 1H), 7.03 (d, J = 7.6 Hz, 1H), 6.76-6.60 (m, 2H), 6.30 (d, J = 14.8 Hz, 1H), 4.95 (s, 2H), 3.50 (s, 3H), 3.12 (bs, 2H), 2.21 (s, 6H); LCMS Calcd for [M+H] + 574.10, found 574.41
Scheme 28: Preparation of (E)-N-(3-(3-benzyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4-(dimethylamino)but-2-enamide (Compound 52):
Step 1: Preparation of ethyl 4-((3-((tert-butoxycarbonyl) amino) phenyl) amino)-2-(methylthio) pyrimidine-5-carboxylate (106):
[0293] Title compound (106) was prepared as off-white solid (142 g; Yield: 74%) in a manner substantially similar to procedure mentioned in General procedure O.1H-NMR (400 MHz, CDCl3): ^ 10.36 (s, 1H), 8.77 (d, 1H), 7.89 (s, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.25-7.22 (m, 1H), 7.03 (d, J = 8.0 Hz, 1H), 6.51 (s, 1H), 4.35 (q, J = 7.2 Hz, 2H), 2.54 (s, 3H), 1.51 (s, 9H), 1.42-1.38 (m, 3H). LCMS: [M+H]+ 405.21, 89.28%.
Step 2: Preparation of tert-butyl (3-((5-(hydroxymethyl)-2-(methylthio)pyrimidin-4-yl)amino)phenyl)carbamate (107):
[0294] Title compound was prepared in a manner substantially similar to procedure mentioned in General procedure P. The crude was triturated with dichloromethane afforded 107 as off white solid (40.0 g; Yield: 31%).1H-NMR (400 MHz, CDCl3): ^ 8.09 (s, 1H), 7.86 (m, 2H),
7.36 (d, J = 8.0 Hz, 1H), 7.25-7.15 (m, 1H), 6.95 (d, J = 8.0 Hz, 1H), 6.55 (s, 1H), 4.59 (s, 2H), 2.50 (s, 3H), 1.51 (s, 9H). LCMS: [M+H]+ 363.05, 91.24%.
Step 3: Preparation of tert-butyl (3-((5-formyl-2-(methylthio)pyrimidin-4-yl)amino)phenyl)carbamate (108):
[0295] Title compound (108) was prepared as a pale yellow solid (31.0 g; Yield: 78%) in a manner substantially similar to procedure mentioned in General procedure Q.1H-NMR (400 MHz, CDCl3): ^ 10.59 (s, 1H), 9.75 (s, 1H), 8.42 (s, 1H), 7.97 (s, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.04 (d, J = 8.0 Hz, 1H), 6.59 (s, 1H), 3.48 (s, 1H), 2.58 (s, 3H), 1.52 (s, 9H). LCMS: [M+H]+ 361.30, 97.51%.
Step 4: Preparation of tert-butyl (E)-(3-((5-((benzylimino)methyl)-2(methylthio)pyrimidin-4-yl)amino)phenyl)carbamate (110):
[0296] Title compound (110) was prepared as a yellow solid (28 g; Yield: 72%) in a manner substantially similar to procedure mentioned in General procedure R.1H-NMR (400 MHz, CDCl3): ^ 12.15 (s, 1H), 8.31 (s, 1H), 8.16 (s, 1H), 7.91 (s, 1H), 7.41 (m, 4H), 7.35-7.33 (m, 1H), 7.32-7.29 (m, 1H), 7.26-7.22 (m, 1H), 7.03 (d, J = 8.0 Hz, 1H), 6.46 (s, 1H), 4.84 (s, 2H), 2.59 (s, 3H), 1.52 (s, 9H). LCMS: [M+H]+ 450.38; 99.66%.
Step 5: Preparation of tert-butyl (3-((5-((benzylamino)methyl)-2-(methylthio)pyrimidin-4-yl)amino)phenyl)carbamate (111):
[0297] Title compound (111) was prepared as a pale yellow solid (40 g; Yield: 80%) in a manner substantially similar to procedure mentioned in General procedure S. LCMS: [M+H]+ 452.44; 83.57%
Step 6: Preparation of tert-butyl (3-(3-benzyl-7-(methylthio)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (112):
[0298] Title compound was prepared in a manner substantially similar to procedure mentioned in General procedure T. The crude was triturated with diethyl ether afforded 112 as off white solid (12 g; Yield: 28%).1H-NMR (400 MHz, CDCl3): ^ 8.03 (s, 1H), 7.50 (s, 1H), 7.37 (m, 6H), 7.26 (m, 1H), 6.96 (m, 1H), 6.59 (s, 1H), 4.69 (s, 2H), 4.34 (s, 2H), 2.16 (s, 3H), 1.50 (s, 9H). LCMS: [M+H]+ 478.16; 95.62%.
Step 7: Preparation of tert-butyl (3-(3-benzyl-7-(methylsulfonyl)-2-oxo-3,4-dihydropyrimido [4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (113):
[0299] Title compound was prepared in a manner substantially similar to procedure mentioned in General procedure U. The crude was triturated with diethyl ether afforded 113 as an off white solid (8.0 g; Yield: 76%).1H-NMR (400 MHz, CDCl3): ^ 8.39 (s, 1H), 7.63 (s, 1H), 7.40 (m, 6H), 7.17 (d, J = 8.0 Hz, 1H), 6.95 (d, J = 8.0 Hz, 1H), 6.61 (s, 1H), 4.71 (s, 2H), 4.48 (s, 2H), 2.97 (s, 3H), 1.49 (s, 9H). LCMS: [M+H]+ 510.31, 93.69%.
Step 8: Preparation of tert-butyl (3-(3-benzyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (114):
[0300] Title compound was prepared in a manner substantially similar to General procedure V, tert-butyl (3-(3-benzyl-7-(methylsulfonyl)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (113) and 1-methyl-1H-pyrazol-3-amine (41) gave (tert-butyl (3-(3-benzyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (114) as a brown solid (Yield: 77%), which was used directly for the next step without any further purification. MS: [M+H]+ 527.46.
Step 9: Preparation of 1-(3-aminophenyl)-3-benzyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one (115):
[0301] Title compound was prepared in a manner substantially similar to General procedure W, tert-butyl (3-(3-benzyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (114) gave 1-(3-aminophenyl)-3-benzyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one (115) as a brown solid (Yield: 93%), which was used directly for the next step. MS: [M+H]+ 427.44.
Step 10: Preparation of (E)-N-(3-(3-benzyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4-(dimethylamino)but-2-enamide (Compound 52):
[0302] Title compound was prepared in a manner substantially similar General procedure X, 1-(3-aminophenyl)-3-benzyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one (115) and trans-N,N-dimethylaminocrotonic acid hydrochloride gave (E)-N-(3-(3-benzyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4-(dimethylamino)but-2-enamide Compound 52, as a white solid (48 mg; Yield: 13%), after prep-HPLC purification.1H-NMR (400 MHz, CDCl3): δ 10.17 (s, 1H), 9.51 (s, 1H), 8.08 (s, 1H), 7.72 (d, J = 8.4 Hz, 1H), 7.60 (s, 1H), 7.43-7.35 (m, 5H), 7.33-7.29 (m, 1H), 7.10 (s, 1H), 7.01 (d, J = 8.8 Hz, 1H), 6.75-6.69 (m, 1H), 6.27 (d, J = 15.3 Hz, 1H), 5.51 (s, 1H), 4.62 (s, 2H), 4.39 (s, 2H), 3.59 (s, 3H), 3.06 (d, J = 4.8 Hz, 2H), 2.17 (s, 6H). MS: [M+H]+ 538.32.
Scheme 30: Alternative Preparation of (E)-N-(3-(7-((3-chloro-1-methyl-1H-pyrazol-4- yl)amino)-2-oxo-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4- (dimethylamino)but-2-enamide (Compound 35):
Step 1: Preparation of 5-(hydroxymethyl)pyrimidine-2,4(1H,3H)-dione (119):
[0308] An ice-cold solution of pyrimidine-2,4(1H,3H)-dione (118) (10 g, 89.21 mmol) and paraformaldehyde (9.63 g, 107.05 mmol) in aqueous potassium hydroxide (132 mL, 0.5 M,
66.74 mmol) was heated at 55 °C for 14 hours. After completion of starting material (TLC), the reaction mixture was cooled to 0 °C and the pH was adjusted to 6 with 12N hydrochloric acid, the resulting white precipitate was filtered through sintered funnel and washed with diethyl ether afforded 119 as a white solid (6.3 g, Yield: 50%) which was used directly for the next step.1H-NMR (400 MHz, DMSO-d6): ^ 10.98 (bs, 1H), 10.64 (bs, 1H), 7.24 (s, 1H), 4.78 (m, 1H), 4.12 (d, J = 12.8 Hz, 2H). LCMS: [M+H]+ 143.04 (99.92% purity).
Step 2: Preparation of 2,4-dichloro-5-(chloromethyl)pyrimidine (120):
[0309] To an ice-cold solution of 5-(hydroxymethyl)pyrimidine-2,4(1H,3H)-dione (119) (10 g, 70.36 mmol) in toluene (25 mL) was added phosphoryl chloride (14 mL, 140.72 mmol) then N,N-diisopropylethylamine (37 mL, 211 mmol). The reaction mixture was heated at 120 °C for 16 hours. After the complete disappearance of starting material on TLC, the reaction mixture was quenched slowly with sodium bicarbonate solution and extracted with ethyl acetate (3 x 200 mL). The combined organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered and evaporated under reduced pressure afforded 120 as a brown solid (12 g, Yield: 86%) which was used directly for the next step.1H NMR (400 MHz, CDCl3): ^ 8.66 (s, 1H), 4.64 (s, 2H). MS: [M+H]+ 197.0
Step 3: Preparation of 2,4-dichloro-5-(iodomethyl)pyrimidine (121):
[0310] To a solution of 2,4-dichloro-5-(chloromethyl)pyrimidine (120) (8.0 g, 40.51 mmol in acetone (40 mL) was added sodium iodide (9.71 g, 64.82 mmol). The reaction mixture was stirred at room temperature for 30 min and heated to reflux for 2 hours. After completion of reaction (TLC monitoring), the reaction mixture cooled to room temperature. The resulting white precipitate was filtered through sintered funnel and washed with acetone. The filtrate was concentrated under reduced pressure afforded 121 as a brown solid (10 g, Yield: 85%) which was used directly for the next step.1H-NMR (400 MHz, CDCl3): ^ 8.60 (s, 1H), 4.39 (s, 2H). Step 4: Preparation of N-((2,4-dichloropyrimidin-5-yl)methyl)aniline (122):
[0311] To an ice-cold solution of 2, 4-dichloro-5-(iodomethyl)pyrimidine (121) (5.0 g, 17.30 mmol) in acetone (50 mL) was added potassium carbonate (5.26 g, 38.06 mmol) and aniline (1.93 g, 20.76 mmol). The resulting reaction mixture was stirred at room temperature for 16 hours. After completion the reaction (as per TLC monitoring), the resulting white precipitate was filtered through sintered funnel and washed with acetone. The filtrate was concentrated under reduced pressure and crude was purified by column chromatography on silica gel (100-200 mesh) using 15% ethyl acetate-hexane as an eluent afforded 122 as a brown solid (2.5 g, Yield: 57%).1H-NMR (400 MHz, CDCl3): ^ 8.61 (s, 1H), 7.07 (t, J = 7.6 Hz, 2H), 6.58 (m, 3H), 6.30 (bs, 1H), 4.33 (m, 2H). LCMS: [M+H]+ 254.03 (99.01% purity).
Step 5: Preparation of tert-butyl (3-(7-chloro-2-oxo-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (123):
[0312] To an ice-cold solution of N-((2,4-dichloropyrimidin-5-yl)methyl)aniline (122) (500 mg, 1.96 mmol), in isopropanol (5 mL) was added N,N-diisopropylethylamine (1.47 mL, 8.42 mmol) and tert-butyl (3-aminophenyl)carbamate (105) (409 mg, 1.96 mmol). The resulting reaction mixture was heated at 100 °C for 16 hours in a sealed tube. After completion of reaction (TLC monitoring), the solvent was then evaporated under reduced pressure and resulting crude was purified by column chromatography on silica gel (100-200 mesh) using 30% ethyl acetate-hexane as an eluent afforded 123 as a brown solid (500 mg, Yield: 60%).1H-NMR (400 MHz, DMSO-d6): δ 9.41 (s, 1H), 8.96 (s, 1H), 8.10 (s, 1H), 7.73 (s, 1H), 7.25 (m, 2H), 7.12 (m, 3H), 6.61 (m, 3H), 6.14 (t, J = 7.2 Hz, 1H), 4.26 (m, 2H) and 1.53 (s, 9H). LCMS: [M+H]+ 426.14 (93% purity).
Step 6: Preparation of tert-butyl (3-(7-chloro-2-oxo-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (124):
[0313] To an ice-cold solution of tert-butyl (3-(7-chloro-2-oxo-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (123) (500 mg, 1.17 mmol) in tetrahydrofuran (6 mL) was added N,N-diisopropylethylamine (0.81 ml, 4.68 mmol) and triphosgene (139 mg, 0.46 mmol). The reaction mixture was stirred at room temperature for 3 hours. After completion of the reaction (TLC monitoring), aqueous triethylamine solution was added and extracted with dichloromethane (3 times). The combined organic layer was washed with brine and dried over sodium sulfate and evaporated under reduced pressure to obtain the crude residue. The crude was purified by column chromatography on silica gel (100-200 mesh) using 30% ethyl acetate-hexane as an eluent afforded 124 as a brown solid (450 mg, Yield: 85%).1H-NMR (400 MHz, DMSO-d6): δ 9.54 (s, 1H), 8.43 (s, 1H), 7.58 (s, 1H), 7.44 (m, 4H), 7.29 (t, J = 7.2 Hz, 3H), 6.94 (s, 1H), 5.0 (s, 2H) and 1.47 (s, 9H). LCMS: [M+H]+ 452.27 (99% purity).
Step 7: Preparation of tert-butyl (3-(7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (125):
[0314] Title compound was prepared in a manner substantially similar to procedure mentioned in General procedure V, (tert-butyl(3-(7-chloro-2-oxo-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (124) and 3-chloro-1-methyl-1H-pyrazol-4-amine (44) gave tert-butyl (3-(7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (125) as a brown solid in 70% yield, which was used directly for the next step. MS: [M+H]+ 547.17.
Step 8: Preparation of 1-(3-aminophenyl)-7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one (126):
[0315] Title compound was prepared in a manner substantially similar to procedure mentioned in General procedure W, tert-butyl (3-(7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)carbamate (125) gave 1-(3-aminophenyl)-7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one (126) as a brown solid (800 mg, Yield: 82%) which was used directly for the next step. MS: [M+H]+ 447.08.
Step 9: Preparation of (E)-N-(3-(7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-1(2H)-yl)phenyl)-4-(dimethylamino)but-2-enamide (Compound 35):
[0316] Title compound was prepared in a manner substantially similar to procedure mentioned in General procedure X, 1-(3-aminophenyl)-7-((3-chloro-1-methyl-1H-pyrazol-4-yl)amino)-3-phenyl-3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one (126) and trans-N,N-dimethylaminocrotonic acid hydrochloride gave the titled compound, which was purified by prep-HPLC purification to afforded the title compound Compound 35 as a white solid (285 mg, Yield: 23%).1H-NMR (400 MHz, DMSO-d6): δ 10.27 (bs, 1H), 8.86 (s, 1H), 8.21 (s, 1H), 7.73 (s, 2H), 7.51-7.40 (m, 5H), 7.30-7.25 (m, 1H), 7.09 (d, J = 7.6 Hz, 1H), 6.76-6.70 (m, 2H), 6.29 (d, J = 15.4 Hz, 1H), 4.88 (s, 2H), 3.50 (s, 3H), 3.05 (d, J = 4.8 Hz, 2H) and 2.16 (s, 6H). MS:
[M+H]+ 558.16.

NEW DRUG APPROVALS
ONE TIME
$10.00
GST-HG-121
GST-HG-121
mw 431.4
C23 H29 N07
Fujian Cosunter Pharmaceutical Co Ltd
Preclinical for the treatment of hepatitis B virus infection
This compound was originally claimed in WO2018214875 , and may provide the structure of GST-HG-121 , an HBsAg inhibitor which is being investigated by Fujian Cosunter for the treatment of hepatitis B virus infection; in June 2019, an IND application was planned in the US and clinical trials of the combination therapies were expected in 2020. Fujian Cosunter is also investigating GST-HG-131 , another HBsAg secretion inhibitor, although this appears to be being developed only as a part of drug combination.
WO2017013046A1
PATENT
WO2018214875
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018214875&_cid=P21-KB0QYA-12917-1
PATENT
WO-2020103924
Novel crystalline forms of 11-oxo-7,11-dihydro-6H-benzo[f]pyrido[1,2-d][1,4]azepine, a hepatitis B surface antigen and HBV replication inhibitor, useful for treating HBV infection.
Step H: Compound 9 (15.80 g, 35.95 mmol) was dissolved in dichloromethane (150.00 mL), and trifluoroacetic acid (43.91 mL, 593.12 mmol) was added. The reaction solution was stirred at 10 degrees Celsius for 3 hours. The reaction solution was concentrated under reduced pressure and spin-dried, sodium bicarbonate aqueous solution (100.00 mL) was added, and dichloromethane (100.00 mL) was extracted. The organic phase was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain compound 10.
Step J: Compound 12 (875.00 mg, 1.90 mmol) was dissolved in toluene (20.00 mL) and ethylene glycol dimethyl ether (20.00 mL), and tetrachlorobenzoquinone (1.40 g, 5.69 mmol) was added. The reaction solution was stirred at 120 degrees Celsius for 12 hours. The reaction solution was cooled to room temperature, and a saturated aqueous sodium carbonate solution (50.00 ml) and ethyl acetate (60.00 ml) were added. The mixed solution was stirred at 10-15 degrees Celsius for 20 minutes, and the liquid was separated to obtain an organic phase. Add 2.00 mol/L aqueous hydrochloric acid solution (60.00 mL) to the organic phase, stir at 10-15 degrees Celsius for 20 minutes, and separate the liquid. Wash the organic phase with 2 mol/L aqueous hydrochloric acid solution (60.00 mL×2), separate the liquid, and separate the water phase A 2 mol/L aqueous sodium hydroxide solution (200.00 ml) and dichloromethane (200.00 ml) were added. The layers were separated, and the organic phase was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain compound 13.
Step K: Compound 13 (600.00 mg, 1.31 mmol) was dissolved in methanol (6.00 mL), and 4.00 mol/L aqueous sodium hydroxide solution (2.00 mL, 6.39 equiv) was added. The reaction solution was stirred at 15 degrees Celsius for 0.25 hours. The reaction solution was adjusted to pH=3-4 with a 1.00 mol/L hydrochloric acid aqueous solution, and then extracted with dichloromethane (50.00 mL×3). The organic phases were combined, washed with saturated brine (50.00 mL), and dried over anhydrous sodium sulfate , Filtered and concentrated under reduced pressure to obtain the compound of formula (I). ee value (enantiomeric excess): 100%.
////////////GST-HG-121, Fujian Cosunter, Preclinical , hepatitis B, virus infection
O=C(O)C1=CN2C(=CC1=O)c3cc(OC)c(OCCCOC)cc3OC[C@H]2C(C)(C)C
O=C(O)C1=CN2C(=CC1=O)c3cc(OC)c(OCCCOC)cc3OC[C@H]2C(C)(C)C
HS 10340

HS-10340
CAS 2156639-66-4

CAS 2307670-65-9
Jiangsu Hansoh Pharmaceutical Group Co Ltd
Being investigated by Jiangsu Hansoh, Shanghai Hansoh Biomedical and Changzhou Hengbang Pharmaceutical ; in June 2018, the product was being developed as a class 1 chemical drug in China.
Useful for treating liver cancer, gastric cancer and prostate cancer.
Use for treating cancers, liver cancer, gastric cancer, prostate cancer, skin cancer, ovary cancer, lung cancer, breast cancer, colon cancer, glioma and rhabdomyosarcoma


PATENT
WO2017198149
where it is claimed to be an FGFR-4 inhibitor for treating liver and prostate cancers, assigned to Jiangsu Hansoh Pharmaceutical Group Co Ltd and Shanghai Hansoh Biomedical Co Ltd .
PATENT
WO2019085860
PATENT
WO-2019085927
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019085927&tab=FULLTEXT
Novel crystalline salt (such as hydrochloride, sulfate, methane sulfonate, mesylate, besylate, ethanesulfonate, oxalate, maleate, p-toluenesulfonate) forms of FGFR4 inhibitor, particularly N-[5-cyano-4-[[(1R)-2-methoxy-1-methyl-ethyl]amino]-2-pyridyl]-7-formyl-6-[(2-oxo-1,3-oxazepan-3-yl)methyl]-3,4-dihydro-2H-1,8-naphthyridine-1-carboxamide (designated as Forms I- IX), compositions comprising them and their use as an FGFR4 inhibitor for the treatment of cancer such as liver cancer, gastric cancer, prostate cancer, skin cancer, ovarian cancer, lung cancer, breast cancer, colon cancer and glioma or rhabdomyosarcoma are claimed.
///////////HS-10340 , HS 10340 , HS10340, CANCER, Jiangsu Hansoh, Shanghai Hansoh Biomedical, Changzhou Hengbang, CHINA, liver cancer, gastric cancer, prostate cancer, skin cancer, ovary cancer, lung cancer, breast cancer, colon cancer, glioma, rhabdomyosarcoma
C[C@H](COC)Nc1cc(ncc1C#N)NC(=O)N4CCCc3cc(CN2CCCCOC2=O)c(C=O)nc34
CCS(=O)(=O)O.C[C@H](COC)Nc1cc(ncc1C#N)NC(=O)N4CCCc3cc(CN2CCCCOC2=O)c(C=O)nc34
CS 3001
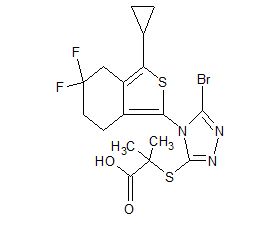
CS-3001
BB 7, VX 033
- Molecular Weight, 478.37
C17 H18 Br F2 N3 O2 S2
CStone Pharmaceuticals Co Ltd, JUNE 2018 IND FILED CHINA
URAT1 inhibitor – useful for treating hyperuricemia and gout.
The compound was originally claimed in WO2017202291 , covering thiophene derivative URAT1 inhibitors, useful for treating hyperuricemia and gouty arthritis, assigned to Medshine Discovery Inc , but naming the inventors.and has been reported in some instances to be a URAT1 modulator. In June 2018, an IND application was filed in



WO-2019101058
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019101058&tab=FULLTEXT&maxRec=1000
Novel crystalline forms of URAT1 inhibitor (designated as Forms A and B) are claimed. The compounds are disclosed to be useful for treating hyperuricemia and gouty arthritis.
Novel crystalline forms of a URAT1 inhibitor, designated as Forms A and B, and their preparation.
For example, remove 2.0 mL of phosphoric acid into 2000 mL of water, sonicate for 10 minutes, mix, and let cool to room temperature as mobile phase A.
////////////CS-3001, BB 7, VX 033, CHINA, PRECLINICAL, CStone Pharmaceuticals, URAT1 inhibitor, hyperuricemia, gout
O=C(O)C(C)(C)Sc4nnc(Br)n4c2sc(c1CC(F)(F)CCc12)C3CC3
TL 487
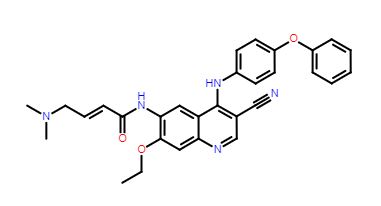
TL-487
- Molecular Weight, 507.58, MF C30 H29 N5 O3
Teligene Inc(2E)-N-[3-Cyano-7-ethoxy-4-[(4-phenoxyphenyl)amino]-6-quinolinyl]-4-(dimethylamino)-2-butenamide
(E)-N-(3-cyano-7-ethoxy-4-((4-phenoxyphenyl)amino)quinolin-6-yl)-4-(dimethylamino)but-2-enamide
Maleate in anhydrous or monohydrate CAS, 2326561-36-6, AND 2326561-38-8 form are BTK and HER-2 kinase inhibitor useful for treating cancer
Useful for treating breast cancer, ovary cancer and colon cancer. are BTK and HER-2 kinase inhibitor useful for treating cancer.
Anticancer protein kinase inhibitor
The compound was originally claimed in WO2013152135 , and may provide the structure of TL-487 , a small molecule inhibitor to HERs, being investigated by Teligene for the treatment of breast cancer; in July 2016, the company intended to develop the product as a class 1.1 chemical drug in China.
PATENT
US 20150057312
PATENT
WO2013152135

PATENT
WO-2019096327
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019096327&redirectedID=true
Novel crystalline maleate salt of (E)-N-(3-cyano-7-ethoxy-4-((4-phenoxyphenyl)amino)quinolin-6-yl)-4-(dimethylamino)but-2-enamide (first disclosed in WO2013152135) and its hydrates (monohydrate) and anhydrates, process for its preparation, composition comprising it and its use for treating cancers such as breast cancer, ovary cancer, colon cancer, prostate cancer, kidney cancer, bladder cancer, stomach cancer, lung cancer, mantle cell lymphoma and multiple myeloma are claimed. The compound is disclosed to be an irreversible inhibitor to BTK and Her-2 (also known as Erb-2 or neu).
///////////////TL-487, PRECLINICAL, CHINA, breast cancer, ovary cancer, olon cancer, BTK, HER-2 kinase inhibitor,
CN(C)C\C=C\C(=O)Nc3cc4c(Nc2ccc(Oc1ccccc1)cc2)c(cnc4cc3OCC)C#N
Quisapride Hydrochloride
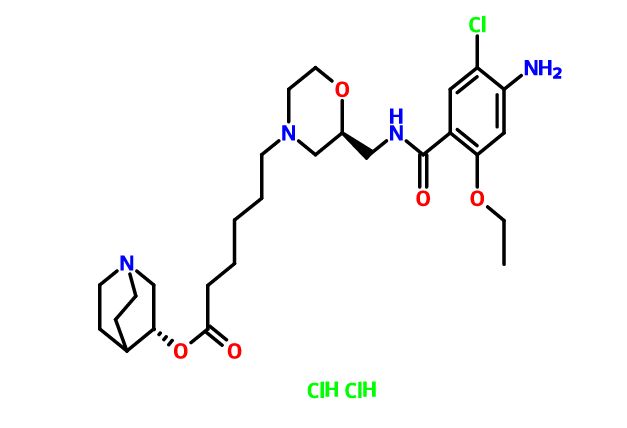
Quisapride Hydrochloride
(R) – quinuclidine-3-5 – ((S) -2 – (( 4 – amino-5-chloro-2-ethoxy benzoylamino) methyl) morpholino) hexanoate
IND Filed china
A 5-HT4 agonist potentially for the treatment of gastrointestinal motility disorders.
![]()
SHR-116 958, SHR 116958
CAS 1132682-83-7 (Free)
| Shanghai Hengrui Pharmaceutical Co., Ltd. |

CAS 1274633-87-2 (dihcl)
- (3R)-1-Azabicyclo[2.2.2]oct-3-yl (2S)-2-[[(4-amino-5-chloro-2-ethoxybenzoyl)amino]methyl]-4-morpholinehexanoate hydrochloride (1:2)
- SHR 116958
-
C27 H41 Cl N4 O5 . 2 Cl H,4-Morpholinehexanoic acid, 2-[[(4-amino-5-chloro-2-ethoxybenzoyl)amino]methyl]-, (3R)-1-azabicyclo[2.2.2]oct-3-yl ester, hydrochloride (1:2), (2S)-
5-HT is a neurotransmitter Chong, widely distributed in the central nervous system and peripheral tissues, 5-HT receptor subtypes at least seven, and a wide variety of physiological functions of 5-HT receptor with different interactions related. Thus, the 5-HT receptor subtypes research is very necessary.
The study found that the HT-5 4 receptor agonists useful for treating a variety of diseases, such as gastroesophageal reflux disease, gastrointestinal disease, gastric motility disorder, non-ulcer dyspepsia, functional dyspepsia, irritable bowel syndrome, constipation, dyspepsia, esophagitis, gastroesophageal disease, nausea, postoperative intestinal infarction, central nervous system disorders, Alzheimer’s disease, cognitive disorder, emesis, migraine, neurological disease, pain, cardiovascular disease, heart failure , arrhythmias, intestinal pseudo-obstruction, gastroparesis, diabetes and apnea syndrome.
The HT-5 4 receptor agonists into benzamides, benzimidazole class and indole alkylamines three kinds, which benzamides derivatives act on the neurotransmitter serotonin in the central nervous system by modulation, It showed significant pharmacological effect. The role of serotonin and benzamides derivatives and pharmacologically related to many diseases. Therefore, more and more people will focus on the human body produce serotonin, a storage position and the position of serotonin receptors, and to explore the relationship between these positions with a variety of diseases.
Commonly used in clinical cisapride (cisapride) and Mosapride (Tony network satisfied) is one of the novel benzamides drugs.

These drugs mainly through the intestinal muscle between the excited 5-HT neurofilament preganglionic and postganglionic neurons 4 receptor to promote the release of acetylcholine and enhancing cholinergic role in strengthening the peristalsis and contraction of gastrointestinal smooth muscle. In large doses, it can antagonize the HT-53 receptors play a central antiemetic effect, when typical doses, through the promotion of gastrointestinal motility and antiemetic effect. These drugs can increase the lower esophageal smooth muscle tension and promote esophageal peristalsis, improving the antrum and duodenum coordinated motion, and promote gastric emptying, but also promote the intestinal movement and enhanced features, increase the role of the proximal colon emptying, It is seen as the whole digestive tract smooth muscle prokinetic effect of the whole gastrointestinal drugs.
Mainly used for reflux esophagitis, functional dyspepsia, gastroparesis, postoperative gastrointestinal paralysis, functional constipation and intestinal pseudo-obstruction patients. Since there is slight antagonism cisapride the HT-5 3 and anti-D2 receptor, can cause cardiac adverse reactions, prolonged QT occurs, and therefore, patients with severe heart disease, ECG QT prolonged, low potassium, and low blood magnesium prohibited drug. Liver and kidney dysfunction, lactating women and children is not recommended. Due to increase between drug diazepam, ethanol, acenocoumarol, cimetidine and ranitidine the absorption of anticholinergic drugs may also antagonize the effect of this product to promote peristalsis of the stomach, should be aware of when using these, such as when diarrhea should reduce, anticoagulant therapy should pay attention to monitoring the clotting time. Mosapride selective gastrointestinal tract the HT-5 4 receptor agonists, there is no antagonism of D2 receptors, does not cause QT prolonged, reduce adverse reactions, mainly fatigue, dizziness, loose stools, mild abdominal pain , the efficacy of cisapride equivalent clinical effect broader Puka cisapride (prucalopride, Pru) of benzimidazole drugs, with high selectivity and specificity of the HT-5 4 receptor, increasing cholinergic neurotransmitters quality release, stimulate peristalsis reflex, enhance colon contraction, and accelerate gastric emptying, gastrointestinal motility to promote good effect, can effectively relieve the patient’s symptoms of constipation, constipation and for treatment of various gastrointestinal surgery peristalsis slow and weak, and intestinal pseudo-obstruction.
WO2005068461 discloses as the HT-5 4 receptor agonists benzamides compounds, particularly discloses compounds represented by the formula:

ATI-7505
ATI-7505 is stereoisomeric esterified. Cisapride analogs, safe and effective treatment of various gastrointestinal disorders, including gastroparesis, gastroesophageal reflux disease and related disorders. The drug can also be used to treat a variety of central nervous system disorders. ATI-7505 for the treatment or prevention of gastroesophageal reflux disease, also taking cisapride significantly reduced side effects. These side effects include diarrhea, abdominal cramps and blood pressure and heart rate rise.
Further, the compounds and compositions of this patent disclosure also useful in treating emesis and other diseases. Such as indigestion, gastroesophageal reflux, constipation, postoperative ileus, and intestinal pseudo-obstruction. In the course of treatment, but also taking cisapride reduce the side effects.
ΑΉ-7505 as the HT-5 4 receptor ligands may be mediated by receptors to treat the disease. These receptors are located in several parts of the central nervous system, modulate the receptor can be used to affect the CNS desired modulation.
ATI-7505 contained in the ester moiety does not detract from the ability of the compounds to provide treatment, but to make the compound easier to serum and / or cytosolic esterases degraded, so you can avoid the drug cytochrome P450 detoxification system, and this system with cisapride cause side effects related, thus reducing side effects.
The HT-Good 5 4 receptor agonists and should the HT-5 4 receptor binding powerful, while the other hardly shows affinity for the receptor, and show functional activity as agonists. They should be well absorbed from the gastrointestinal tract, metabolically stable and possess desirable pharmacokinetic properties. When targeting the receptor in the central nervous system, they should cross the blood-free, selectively targeting peripheral nervous system receptors, they should not pass through the blood-brain barrier. They should be non-toxic, and there is little proof of side effects. Furthermore, the ideal drug candidate will be a stable, non-hygroscopic and easily formulated in the form of physical presence.
Based on the HT-5 4 receptor agonists current developments, the present invention relates to a series of efficacy better, safer, less side effects of the benzamide derivatives.

Synthesis
PATENT
Example 3
(R) – quinuclidine-3-5 – ((S) -2 – (( 4 – amino-5-chloro-2-ethoxy benzoylamino) methyl) morpholino) hexanoate


REFERENCES
China Pharmaceuticals: Asia Insight: China Has R&D
Nov 6, 2012 – levofolinate, sevoflurane inhalation, ambroxol hydrochloride, ioversol, etc ….. dextromethorphan hydrochloride 复方沙芬那敏. 3.2 …… quisapride.
//////SHR-116 958, SHR 116958, Quisapride Hydrochloride, preclinical
Cl.Cl.Clc1cc(c(OCC)cc1N)C(=O)NC[C@H]4CN(CCCCCC(=O)O[C@H]3CN2CCC3CC2)CCO4
Tianagliflozin IND filed by Tianjin Institute of Pharmaceutical research


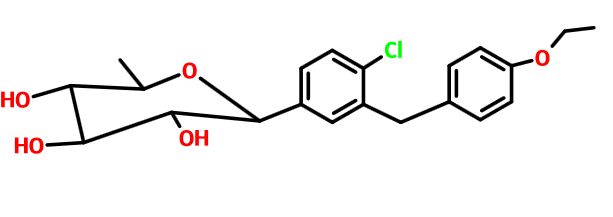
Tianagliflozin,
taigeliejing, 6-deoxydapagliflozin
| Molecular Formula: | C21H25ClO5 |
|---|---|
| Molecular Weight: | 392.8732 g/mol |
IND Filing…Tianjin Institute of Pharmaceutical research
Tianjin Institute Of Pharmaceutical Research,
(3R,4S,5S,6R)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-methyloxane-3,4,5-triol
1–[4–Chloro–3–(4–ethoxybenzyl)phenyl]–1,6–dideoxy–β–d–glucopyranose
![]()
CAS N. 1461750-27-5
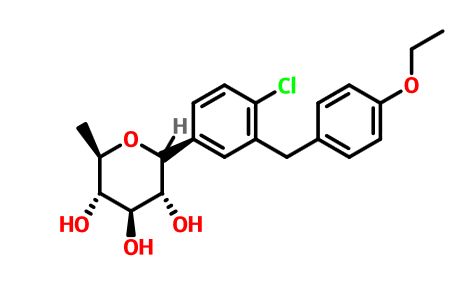

The structures of dapagliflozin and 6-deoxydapagliflozin (1)
,deletion of the 6-OH in the sugar moiety of dapagliflozin led to the discovery of a more potent SGLT2 inhibitor, 6-deoxydapagliflozin (1, ). In an in vitro assay, 1 was a more active SGLT2 inhibitor, with IC 50 = 0.67 nM against human SGLT2 (hSGLT2), as compared with 1.1 nM for dapagliflozin, leading to the identification of 1 as the most active SGLT2 inhibitor discovered so far in this field. Also in an in vivo assay, 1 also introduced more urinary glucose in a rat urinary glucose excretion test (UGE) and exhibited more potent blood glucose inhibitory activity in a rat oral glucose tolerance test (OGTT) than dapagliflozin.
Tianjin Institute Of Pharmaceutical Research,天津药物研究院

SPECTRAL DATA of Tianagliflozin
1 as a white solid (3.65 g, 93 %). R f = 0.35 (EtOAc);
m.p.: 148–149 °C;
1H NMR (400 MHz, DMSO-d 6): δ = 7.35 (d, 1H, J = 8.4 Hz), 7.25 (s, 1H), 7.18 (d, 1H, J = 8.0 Hz), 7.08 (d, 2H, J = 8.4 Hz), 6.81 (d, 2H, J = 8.4 Hz), 4.95 (d, 1H, J = 5.2 Hz, OH), 4.90 (d, 1H, J = 4.4 Hz, OH), 4.79 (d, 1H, J = 5.6 Hz, OH), 3.92–4.01 (m, 5H), 3.24–3.29 (m, 1H), 3.18–3.22 (m, 1H), 3.09–3.15 (m, 1H), 2.89–2.95 (m, 1H), 1.29 (t, 3H, J = 7.0 Hz, CH2 CH 3 ), 1.15 (d, 3H, J = 6.0 Hz, CHCH 3 ) ppm;
13C NMR (100 MHz, DMSO-d 6): δ = 156.85, 139.65, 137.82, 131.83, 131.16, 130.58, 129.52, 128.65, 127.14, 114.26, 80.71, 77.98, 75.77, 75.51, 74.81, 62.84, 37.55, 18.19, 14.62 ppm;
IR (KBr): v¯¯¯ = 3,564 (w), 3,385 (s), 2,981 (s), 2,899 (s), 2,861 (s), 1,613 (m), 1,512 (s), 1,477 (m), 1,247 (s), 1,102 (s), 1,045 (s), 1,012 (s) cm−1;
HR–MS: calcd for C21H29ClNO5 ([M + NH4]+) 410.1729, found 410.1724.
PATENT
CN 103864737
http://www.google.com/patents/CN103864737A?cl=en
PATENT
WO 2014094544
http://www.google.com/patents/WO2014094544A1?cl=en


-27-


1 D1 -6 Optionally, the step (7 ‘) is the step (7’) in place:
LS l- [4 – D (I- Dl- 6)

A.

(DMSO-d 6, 400 MHz), δ 7.35 (d, 1H, J = 8.0 Hz), 7.28 (d, 1H, J ‘. 2.0 Hz), 7.17 (dd, IH, / = 2.0 Hz and 8.4 Hz), 7.05 (d, 2H, J: 8.8 Hz), 6.79 (d, 2H, 8.8 Hz): 4.924,95 (m, 2H), 4,81 (d, IH, 6,0 Hz), 3.93- 3.99 (m, 5H), 3,85 (d, 1H, J = 10,4 Hz), 3,66 (dd, IH, 5,2 Hz and 11,6 Hz), 3.17-3,28 (m, 3H), 3.02-3.08 (m: IH), 1.28 (t, 3H, J = 7,0 Hz), 0,80 (s, 9H), -0.05 (s, 3H), -0.09 (s, 3H) .
PATENT
[0066] The added 100mL dried over anhydrous methanol 0. 5g of sodium metal, nitrogen at room temperature with stirring, until the sodium metal disappeared. Followed by addition of 5. 2g (10mmol) of compound 6, stirring was continued at room temperature for 3 hours. To the reaction system was added 5g strong acid cation exchange resin, stirred at room temperature overnight, the reaction mixture until pH = 7. The resin was removed by suction, and the filtrate evaporated to dryness on a rotary evaporator, the residue was further dried on a vacuum pump to give the product I-D1-6, as a white foamy solid.
PATENT
WO 2014139447
PATENT related
http://www.google.com/patents/WO2013044608A1?cl=en
http://link.springer.com/article/10.1007%2Fs40242-014-4043-9#/page-1
Design of SGLT2 Inhibitors for the Treatment of Type 2 Diabetes: A History Driven by Biology to Chemistry.
Abstract
A brief history of the design of sodium-dependent glucose cotransporter 2 (SGLT2) inhibitors is reviewed. The design of O-glucoside SGLT2 inhibitors by structural modification of phlorizin, a naturally occurring O-glucoside, in the early stage was a process mainly driven by biology with anticipation of improving SGLT2/SGLT1 selectivity and increasing metabolic stability. Discovery of dapagliflozin, a pioneering C-glucoside SGLT2 inhibitor developed by Bristol-Myers Squibb, represents an important milestone in this history. In the second stage, the design of C-glycoside SGLT2 inhibitors by modifications of the aglycone and glucose moiety of dapagliflozin, an original structural template for almost all C-glycoside SGLT2 inhibitors, was mainly driven by synthetic organic chemistry due to the challenge of designing dapagliflozin derivatives that are patentable, biologically active and synthetically accessible. Structure-activity relationships (SAR) of the SGLT2 inhibitors are also discussed.
http://www.ncbi.nlm.nih.gov/pubmed/25557661
Paper

Discovery of 6-Deoxydapagliflozin as a Highly Potent Sodium-dependent Glucose Cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes
http://www.ingentaconnect.com/content/ben/mc/2014/00000010/00000003/art00009?crawler=true
CLIP

A facile synthesis of 6-deoxydapagliflozin


The synthetic route to the target compound 1 is shown in Scheme 3. The starting material methyl 2,3,4-tri-O-benzyl-6-deoxy-6-iodo-α–d-glucopyranoside (3) was prepared from commercially available methyl α–d-glucopyranoside (2) according to a known method [5, 6].
Iodide 3 was reductively deiodinated to give 4 in 91 % yield under hydrogenolytic conditions using 10 % Pd/C as catalyst in the presence of Et3N as base in THF/MeOH at room temperature.
when the iodide 3 was treated with Barton–McCombie reagent (n-Bu3SnH/AIBN) [7] in toluene at room temperature no reaction occurred; however, when the reaction was carried out at elevated temperatures, such as reflux, a complex mixture formed with only a trace amount (3 %, entry 1) of the desired product 4.
When the iodide 3 was treated with LiAlH4 in THF at 0 °C to room temperature, another complex mixture was produced with only a trace amount (2 %, entry 2) of 4.
When Pd(OH)2 was used as the hydrogenolysis catalyst instead of 10 % Pd/C, the desired 4 was indeed formed (14 %, entry 4), but most of the starting material was converted to a few more polar byproducts, which were believed to result from the cleavage of at least one of the benzyl groups.
pdf available
Monatshefte für Chemie – Chemical Monthly
December 2013, Volume 144, Issue 12, pp 1903-1910
////////IND Filing, SGLT-2 inhibitor, type 2 diabetes, Tianagliflozin, taigeliejing, 6-deoxydapagliflozin, 1461750-27-5
Clc1c(cc(cc1)C2[C@@H]([C@H]([C@@H]([C@H](O2)C)O)O)O)Cc3ccc(cc3)OCC
Shanghai Hengrui’s potent inhibitors of Human Uric Acid Transporter 1 (hURAT1)

| MF | C 1 4 H 1 2 BrNO 2 S |
|---|---|
| MW | 338.21958 g / mol |
1- (6-bromoquinolin-4-yl) sulfanylcyclobutane-1-carboxylic acid
CAS…….1638327-48-6
Cyclobutanecarboxylic acid, 1-[(6-bromo-4-quinolinyl)thio]-
COMING ………….
MS m / z (ESI): 338.0 [M + l]
1H NMR (400 MHz, DMSO) δ 13.17 (s, 1H), 8.75-8.79 (m, 1H), 8.24 (s, 1H), 7.87-7.98 (m, 2H), 7.21-7.25 (m, 1H), 2.83-2.95 (m, 2H), 2.30-2.41 (m, 2H), 2.16-2.27 (m, 1H), 1.97-2.08 (m, 1H)
WO-2014183555-A1 / 2014-11-20
http://www.google.co.in/patents/WO2014183555A1?cl=en
PROCEDURE
6-bromo-quinoline-4-thiol
A mixture of 6-bromo-4-chloro-quinoline 3a (260 mg, 1.1 mmol, using known methods “Bioorganic &
Medicinal Chemistry Letters, 2012, 22 (4), 1569-1574 “prepared to give) and sodium sulfide (100 mg, 1.3 mmol) was added to 4 mL of N, N- dimethyl formamide, plus complete, heated 80 ° C, the reaction was stirred for 2 hours. To the reaction mixture was added 50 mL of water, 1 M hydrochloric acid was added dropwise to the reaction solution to pH 5-6, extracted with ethyl acetate (50 mL X 3), the combined organic phases, with no over anhydrous sodium sulfate, filtered, and the filtrate concentrated under reduced pressure to give the title product 6-bromo-quinolin-4-thiol 3b (257 mg, yellow oil), it was used directly in the next reaction.
The second step
L – ((6-bromo-quinolin-4-yl) thio) cyclobutyl carboxylate
Under an argon atmosphere, 6-bromo-quinolin-4-thiol 3b (257 mg, 1.1 mmol), 1- bromo-cyclobutyloxy embankment carboxylate (266 mg, 1.3 mmol) and cesium carbonate (371 mg, 1.1 mmol) were sequentially added to 5 mL of N, N- dimethylformamide and heated to 60 ° C, the reaction was stirred for 2 hours. The reaction solution was filtered, the filter cake washed with ethyl acetate (10 mL X 3) and the filtrate was concentrated under reduced pressure to give the title product l – ((6-bromo-quinolin-4-yl) thio) ethyl cyclobutyl 3c ( 300 mg, brown oil). Yield: 77%.
MS m / z (ESI): 368.2 [M + l]
1H MR (400 MHz, CDCl 3 ) [delta] 8.67 (d, = 4.77 Hz, IH), 8.31 (d, = 2.13 Hz, IH), 7.94 (d, = 8.91Hz, IH), 7.78 (dd, = 9.03, 2.13Hz, IH), 7.15 (d, = 4.89Hz, IH), 4.16 (q, = 7.15Hz, 2H), 2.86-3.04 (m, 2H), 2.39-2.51 (m, 2H), 2.25-2.37 ( m, IH), 2.00-2.15 (m, IH), 1.16 (t, = 7.09Hz, 3H)
third step
L – ((6-bromo-quinolin-4-yl) thio) cyclobutyl acid
L – ((6-bromo-quinolin-4-yl) thio) ethyl cyclobutyl 3c (100 mg, 0.27 mmol) and lithium hydroxide monohydrate (23 mg, 0.55 mmol) was dissolved in 6 mL of tetrahydrofuran, ethanol and water (^ = 4: 1: 1) mixed solvent, the reaction was stirred for 3 hours. 1M hydrochloric acid was added dropwise to the reaction solution pH of 5 to 6, liquid separation, the aqueous phase was extracted (10 mL X 3) with dichloromethane, the combined organic phases, the organic phase was washed with a saturated sodium chloride solution (10 mL XI), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure, the resulting A by thin layer chromatography in a developing solvent system, and the residue was purified to give the title product l – ((6-bromo-quinolin-4-yl) thio) cyclobutyl acid 3 (20 mg, white solid), yield: 22%.
MS m / z (ESI): 338.0 [M + l]
1H NMR (400 MHz, DMSO) δ 13.17 (s, 1H), 8.75-8.79 (m, 1H), 8.24 (s, 1H), 7.87-7.98 (m, 2H), 7.21-7.25 (m, 1H), 2.83-2.95 (m, 2H), 2.30-2.41 (m, 2H), 2.16-2.27 (m, 1H), 1.97-2.08 (m, 1H)
L – ((6-bromo-quinolin-4-yl) thio) cyclobutyl acid
First step
6-bromo-quinoline-4-thiol
A mixture of 6-bromo-4-chloro-quinoline 3a (260 mg, 1.1 mmol, a known method of “Bioorganic &
Medicinal Chemistry Letters, 2012, 22 (4), 1569-1574 “prepared to give) and sodium sulfide (100 mg, 1.3 mmol) was added to 4 mL of N, N- dimethyl formamide, plus complete, heated 80 ° C, the reaction was stirred for 2 hours. To the reaction mixture was added 50 mL of water, 1 M hydrochloric acid was added dropwise to the reaction solution to pH 5-6, extracted with ethyl acetate (50 mL X 3), the combined organic phases, with no over anhydrous sodium sulfate, filtered, and the filtrate concentrated under reduced pressure to give the title product 6-bromo-quinolin-4-thiol 3b (257 mg, yellow oil), it was used directly in the next reaction.
The second step
L – ((6-bromo-quinolin-4-yl) thio) ethyl cyclobutyl
Under an argon atmosphere, 6-bromo-quinolin-4-thiol 3b (257 mg, 1.1 mmol), 1- bromo-cyclobutyloxy embankment carboxylate (266 mg, 1.3 mmol) and cesium carbonate (371 mg, 1.1 mmol) were added to 5 mL of N, N- dimethylformamide and heated to 60 ° C, the reaction was stirred for 2 hours. The reaction mixture was filtered, the filter cake washed with ethyl acetate (10 mL X 3) and the filtrate was concentrated under reduced pressure to give the title product l – ((6-bromo-quinolin-4-yl) thio) ethyl cyclobutyl 3c ( 300 mg, brown oil). Yield: 77%.
MS m / z (ESI): 368.2 [M + l]
1H MR (400 MHz, CDC1 3) δ 8.67 (d, = 4.77Hz, IH), 8.31 (d, = 2.13Hz, IH), 7.94 (d, = 8.91Hz, IH), 7.78 (dd, = 9.03, 2.13Hz, IH), 7.15 (d, = 4.89Hz, IH), 4.16 (q, = 7.15Hz, 2H), 2.86-3.04 (m, 2H), 2.39-2.51 (m, 2H), 2.25-2.37 ( m, IH), 2.00-2.15 (m, IH), 1.16 (t, = 7.09Hz, 3H) Step
L – ((6-bromo-quinolin-4-yl) thio) cyclobutyl acid
L – ((6-bromo-quinolin-4-yl) thio) ethyl cyclobutyl 3c (100 mg, 0.27 mmol) and lithium hydroxide monohydrate (23 mg, 0.55 mmol) was dissolved in 6 mL of tetrahydrofuran, ethanol and water (^ = 4: 1: 1) mixed solvent, the reaction was stirred for 3 hours. 1M hydrochloric acid was added dropwise to the reaction solution pH of 5 to 6, liquid separation, the aqueous phase was extracted (10 mL X 3) with dichloromethane, the combined organic phases, the organic phase was washed with a saturated sodium chloride solution (10 mL XI), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure, to the resulting thin layer chromatography using a developing solvent system A and the residue was purified to give the title product l – ((6-bromo-quinolin-4-yl) thio) cyclobutyl acid 3 (20 mg, white solid), yield: 22%. MS m / z (ESI): 338.0 [M + l]
1H NMR (400 MHz, DMSO) δ 13.17 (s, 1H), 8.75-8.79 (m, 1H), 8.24 (s, 1H), 7.87-7.98 (m, 2H), 7.21-7.25 (m, 1H), 2.83-2.95 (m, 2H), 2.30-2.41 (m, 2H), 2.16-2.27 (m, 1H), 1.97-2.08 (m, 1H)
Discovery of potent and orally bioavailable inhibitors of Human Uric Acid Transporter 1 (hURAT1) and binding mode prediction using homology model
- Shanghai Hengrui Pharmaceutical Co. Ltd, 279 Wenjing Rd., Shanghai 200245, China
This Letter describes the Discovery of a series of potent inhibitors of Human Uric Acid Transporter 1 (hURATl). Lead generation via 3D pharmacophore Analysis and Optimization resulted in compound 41 . With an IC 50 of 33.7 nM, 41 Also Demonstrated good Oral Bioavailability in RAT (74.8%) and displayed a consistent PK profile across all species tested (rat, dog and monkey).

http://www.sciencedirect.com/science/article/pii/S0960894X1530353X
Volume 26, Issue 2 , 15 January 2016, Pages 277-282

//////// Shanghai Hengrui, inhibitors of Human Uric Acid Transporter 1 (hURAT1), 1- (6-bromoquinolin-4-yl) sulfanylcyclobutane-1-carboxylic acid
c13cc (ccc3nccc1SC2 (C (= O) O) CCC2) Br
Lefucoxib (乐福昔布)

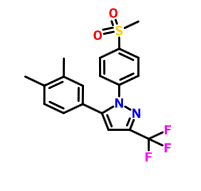
Lefucoxib (乐福昔布)
5-(3,4-dimethyl-phenyl)-1-methanesulfonyl-3-trifluoromethol-pyrazole
1 [4- (methylsulfonyl) phenyl] -3-trifluoromethyl-5- (3,4-dimethylphenyl) – pyrazole
CAS 849048-84-6
![]()
| Molecular Formula: | C19H17F3N2O2S |
|---|---|
| Molecular Weight: | 394.41069 g/mol |
IND FILED
Prostaglandin G/H Synthase 2 (PTGS2; COX-2) Inhibitors
A COX-2 inhibitor potentially for the treatment of rheumatoid arthritis.
cyclooxygenase-2 (COX-2) inhibitor
National Center of Biomedical Analysis
![]()
 CHINA FLAG
CHINA FLAG

PATENT
CN 1468854
http://www.google.com/patents/CN1468854A?cl=en
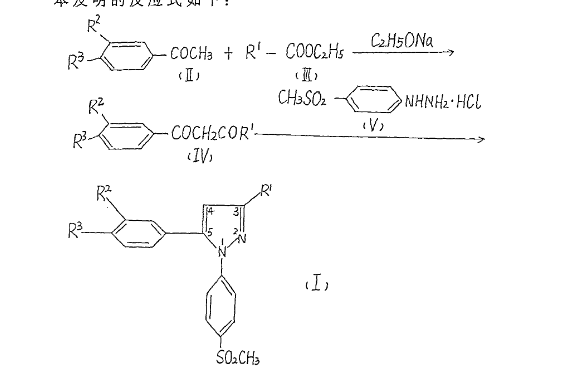
Example 1
1 [4- (methylsulfonyl) phenyl] -3-trifluoromethyl-5- (3,4-dimethylphenyl) – pyrazole (I1)
1- (3,4- two toluene-yl) -4,4,4-trifluoro-methyl – D-1,3-dione (IV1) of sodium metal was weighed 2.3g (0.1mol) was added 50ml of anhydrous toluene to prepare a sodium sand. After cooling, ethanol was added dropwise 12ml, and then heated at 60 ℃, complete reaction of sodium metal. After cooling to room temperature, was added 3,4-dimethylphenyl ethanone 23.8g (0.1mol) and trifluoroacetic ethyl acetate 20ml (0.2mol), reacted at 100 ℃ 5 hours. Toluene was distilled off under reduced pressure, a 10% aqueous hydrochloric acid was added, the pH was adjusted to 2-3, extracted with ethyl acetate, washed with water, dried over anhydrous MgSO4, ethyl acetate was distilled off under reduced pressure. Then under reduced pressure, distillation, collecting fractions 105-107 ℃ / 0.7mmHg, was 14.6g, 60% yield.
1- [4- (methylsulfonyl) phenyl] -3-trifluoromethyl-5- (3,4-dimethylphenyl) – pyrazole (I1) take the above-prepared substituted (IV1) 2.38g (0.01mol ), 15ml of ethanol, then added p-methanesulfonyl phenyl hydrazine salt alkoxide 2.3g (0.01ml). Was refluxed for 15 hours. Place the refrigerator overnight, the crystals were collected by filtration, recrystallized from ethanol, mp 129-31 ℃, to give 3.1 g.
Elemental analysis: C19H17F3N2O2S Calculated: C, 57.86; H, 4.34; N, 7.10 Found: C, 57.97; H, 4.29; N, 7.20MS (m / z): 395 (M + 1)

References
Cheng, Feixiong, Edited by Lee, Philip W, From Handbook of Metabolic Pathways of Xenobiotics (2014), 4, 1655-1656
Bi, X.; Meng, Z.; Chen, H.; Zhu, X.; Dou, G.
In vivo and in vitro metabolism of lefucoxib in rats, J Pharm Biomed Anal. 2008 Sep 10;48(1):134-9. doi: 10.1016/j.jpba.2008.04.024. Epub 2008 Apr 30.
Bi, X.; Meng, Z.; Dou, G. Determination of lefucoxib in rat plasma, urine, and feces by high-performance liquid chromatography with fluorescence detection: Application in pharmacokinetic studies
J Chromatogr B Anal Technol Biomed Life Sci 2007, 850(1-2): 199
Talanta (2011), 85(1), 8-27
Jiefangjun Yaoxue Xuebao (2009), 25(6), 496-498.
Yaowu Fenxi Zazhi (2006), 26(9), 1222-1224.
Zhongguo Yaolixue Yu Dulixue Zazhi (2007), 21(2), 147-151.
| CN101497585B | Jan 31, 2008 | Jan 12, 2011 | 中国科学院理化技术研究所 | Method for photocatalytic synthesis of 1,3,5-trisubstituted-2-pyrazole derivative |

 .
.
//////////c1c(ccc(c1C)C)c2n(nc(c2)C(F)(F)F)c3ccc(cc3)S(=O)(=O)C
CC1=C(C=C(C=C1)C2=CC(=NN2C3=CC=C(C=C3)S(=O)(=O)C)C(F)(F)F)C
What is SMU-B?

cas 1253286-89-3
Spiro[3H-indole-3,4′-piperidin]-2(1H)-one, 5-[6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridinyl]-1′-methyl-
SMU-B
or is it
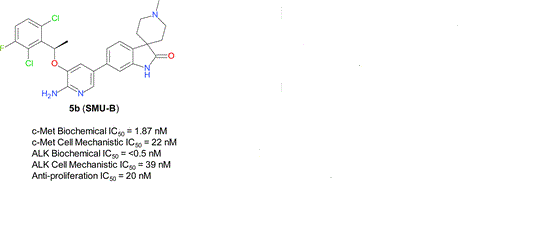
1253286-90-6
Spiro[3H-indole-3,4′-piperidin]-2(1H)-one, 6-[6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridinyl]-1′-methyl-
SMU-B

A series of novel aminopyridyl/pyrazinyl-substituted spiro[indoline-3,4′-piperidine]-2-ones were designed, synthesized, and tested in various in vitro/in vivo pharmacological and antitumor assays. 6-[6-Amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridyl]-1′-methylspiro[indoline-3,4′-piperidine]-2-one (compound 5b or SMU-B) was identified as a potent, highly selective, well-tolerated, and orally efficacious c-Met/ALK dual inhibitor, which showed pharmacodynamics effect by inhibiting c-Met phosphorylation in vivo and significant tumor growth inhibitions (>50%) in GTL-16 human gastric carcinoma xenograft models.
see..http://pubs.acs.org/doi/abs/10.1021/ml400203d
cas 1253286-90-6
6-[6-Amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridyl]-1′-methylspiro[indoline-3,4′-piperidine]-2-one (compound 5b or SMU-B)
SEE

1_4,3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] -2-nitro-approved P set

obtained in Step 1-3 (IS) -I- (2,6- dichloro-3-fluorophenyl) ethanol (2. 09g, IOmmol) was dissolved in dry THF (80 ml). Then, at room temperature under a nitrogen atmosphere, a solution of 3-hydroxy-2-nitro-pyridine (1.54g, llmmol) and triphenylphosphine (3. 409g, 13mmol), and so is completely dissolved, cooled to 0 ° C, was added Diisopropyl azodicarboxylate (DIAD, 2.63g, 13mmol), After the addition, the mixture was stirred at 0 ° C for 16 hours, the solvent was removed by rotary evaporation and the oily residue was purified by silica gel column chromatography (petroleum ether / ethyl acetate : 4/1) to give the desired product as a white solid (3. 046g, yield: 92%) o 1H-NMR (CDClySOOMHz): 8 (ppm) I. 86 (d, J = 6. 4Hz, 3H), 6 . 10 (q, J = 6. 4Hz, 1H), 7. 09 (dd, J = 7. 6,8. 8Hz, 1H), 7. 21 (dd, J = 8. 4, I. 2Hz, 1H ), 7. 31 (dd, J = 4. 8,8. 8Hz, 1H),
7. 37 (dd, J = 4. 8,8. OHz, 1H), 8. 04 (dd, J = L 6,4. 4Hz, 1H). Mass spectrum m / z:. 330 94 [M + H, 35C1,35Cl], 332. 92 [M + H, 35Cl, 37Cl].
1_5,3_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -2_ atmosphere based grant given P

to take steps 1-4 to get the 3 – [(lR) _l- (2,6_ dichloro-3-fluorophenyl) ethoxy] -2_ nitro than Li Jie (2. 649g, 8mmol) was dissolved in ethanol (15mL) was added iron powder (3. 575g, 64mmol) were mixed under nitrogen with vigorous stirring at 90 ° C oil bath, was added via syringe 0.8mL IM HCl (aq), after 10 minutes, was added 0. 8mL IMHCl (aq). Stirring was continued for 30 minutes, TLC showed the reaction. Cooled to room temperature, filtered through Celite, the filter residue washed with ethanol (3X IOmL). The combined organic phase was removed by rotary evaporation of the solvent gave the desired product as a light brown solid (2. 41g, yield: 100%) o 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 81 (d, J = 6. 8Hz, 3H ), 5. 03 (s, br, 2H), 6. 01 (q, J = 6. 8Hz, 1H), 6. 47 (dd, J = 4. 8,7. 6Hz, 1H), 6. 70 (d, J = 8. OHz, 1H), 7. 05 (t, J = 8. 8Hz, 1H), 7. 28 (dd, J = 4. 0,8. 0Hz, 1H), 7. 57 ( d, J = 5.2Hz, lH). Mass spectrum m / z:. 301 00 [M + H, 35Cl, 35Cl], 302. 77 [M + H, 35Cl, 37Cl].
l-6,5_ desert _3_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -2_ atmosphere base than Li Jie

The steps 1-5 obtained 3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] -2-yl atmosphere than Li Jie (1.506g, 5mmol) dissolved in acetonitrile (20mL) in. Then, at 0 ° C and the degree of stirring in added portionwise N- bromosuccinimide (0.908g, 5. Lmmol), After the addition, stirring was continued for 30 minutes. The solvent was removed by rotary evaporation, the crude product was obtained as a white solid was the desired product (1.045g, yield: 55%) was purified by column chromatography on silica gel. 1H-NMR (⑶Cl3,500MHz): 8 (ppm) I. 81 (d, J = 6. 8Hz, 3H), 4 85 (s, br, 2H), 6 98 (q, J = 6. 8Hz.. , 1H), 6. 82 (d, J =
2. 0Hz, 1H), 7. 08 (t, J = 8. 4Hz, 1H), 7. 31 (dd, J = 4. 8,8. 8Hz, 1H), 7. 65 (d, J = 2 . OHz, 1H). Mass spectrum m / z:… 378 84 [M + H, 35Cl, 35Cl, 79Br], 380 82 [M + H, 35Cl, 35Cl, 81Br or 35Cl, 37Cl, 79Br], 382 80 [M + H, 35Cl , 37Cl, 81Bror 37Cl, 37Cl, 79Br].
Step 2, I ‘- methyl-5- (4,4,5,5-tetramethyl -I, 3,2- dioxolane boron-2-yl) spiro [indoline Spray – 3,4 ‘- piperidin] -2_ one

2-1,5- bromo -I ‘- methyl-spiro [indoline-3,4’ – piperidin] _2_ one

[0300] 5-bromo – indol-2-one (I. 272g, 6mmol) was suspended THF (15mL) at, and cooled to -78 ° C, added dropwise with stirring IM NaN (SiMe3) THF solution of 2 (30mL, 30mmol). After the addition was stirred at _78 ° C 30 min, then 2-chloro -N- (2- chloro-ethyl) -N- methyl-ethylamine hydrochloride solid (I. 155g, 6mmol). After the addition stirring was continued for 30 minutes, then warmed to room temperature and stirred for two days. TLC showed the reaction was completed, to the pink suspension was carefully added aqueous 4M hydrochloric acid (IOmL), and then adjusted with concentrated aqueous ammonia to pH ^ 9, and extracted with DCM (3 X 80mL). The organic phases were combined, dried (Na2SO4), and concentrated to give the crude product was purified by silica gel column chromatography (7M NH3 in methanol solution / DCM: 5/95) to give the desired product (I. 38g, yield: 78%) was purified. 1H-NMR (CD3ODjOOMHz):. 8 (ppm) I. 86-1 92 (m, 2H), I 94-1 98 (m, 2H), 2 44 (s, 3H), 2 62-…. 2. 68 (m, 2H), 2. 86-2. 91 (m, 2H), 6. 76 (d, J = 7. 6Hz, 1H), 7. 33 (dd, J = I. 2,7 . 6Hz, 1H), 7. 44 (d, J = I. 6Hz, 1H), 7. 81 (s, br, 1H). Mass spectrum m / z:. 294 99 [M + H, 79Br], 296 82 [M + H, 81Br]..
2-2, V – methyl-5- (4,4,5,5-tetramethyl–1,3,2_ dioxolane Borane _2_ yl) spiro [indoline – 3,4 ‘- piperidin] -2_ one

Under nitrogen, obtained in Step 2-1 to 5-bromo -I ‘- methyl-spiro [indoline-_3,4’ – piperidin] _2_ one (147. 6mg, 0. 5mmol) , the United pinacols drop acid unitary purpose (140mg, 0. 55mmol) and acetic acid Bell (147mg, I. 5mmol) in DMSO (0. 2ml) was added in PdCl2 (dppf) • CH2Cl2 (20. 4mg, 0. 025mmol ), to the resulting solution was bubbled with nitrogen for 2 minutes, and then stirred at 80 ° C of 16 hours. LC-MS showed completion of the reaction, after cooling to room temperature, water (2mL), extracted with DCM (3X5mL). The organic phases were combined, dried (Na2SO4), and concentrated to give the desired product (170mg, yield: 100%) o MS m / z:. 342 07 [M + H], 343. 08 [M + H, 100%], 344. 11 [M + H].
Step 3,5_ [6_ atmosphere base _5_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -3_ batch P fixed base] -I ‘- A group spiro [indoline-3,4 ‘- piperidin] -2_ one
The steps 1-6 5_ desert obtained _3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] -2-yl batch atmosphere pyridine (75. 8mg , 0. 2mmol), I’- step 2_2 obtained methyl 5- (4,4,5,5-tetramethyl-l, 3,2-dioxolane Borane 2-yl) spiro [ indoline-3,4′-piperidin] -2-one (82mg, 0. 24mmol) and potassium carbonate (82. 9mg, 0. 6mmol) was dissolved in DME / water mixture solution (4 / 1,2. Oml ). Then, under nitrogen, was added Pd (PPh3) 4 (II. 6mg, 0. Olmmol), to the resulting mixture was bubbled with nitrogen for 2 minutes, and then stirred at 80 ° C of 18 hours. LC-MS showed completion of the reaction, after cooling to room temperature, water (5mL), extracted (3 X IOmL) with DCM. The organic phases were combined, dried (Na2SO4), and concentrated to give the crude product was purified by silica gel column chromatography (7M NH3 in methanol solution / DCM: 5/95) to give the desired product (88. 6mg, yield: 86%) was purified. 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 86 (d, J = 6. 4Hz, 3H), I 93-2 02 (m, 4H), 2 44 (s, 3H),…
2. 66-2. 72 (m, 2H), 2. 89-2. 93 (m, 2H), 4. 87 (s, br, 2H), 6. ll (q, J = 6. 4Hz, 1H ), 6. 88 (d, J =
8. OHz, 1H), 6. 94 (d, J = I. 2Hz, 1H), 7. 06 (t, J = 8. 4Hz, 1H), 7. 19 (dd, J = I. 2,8 . OHz, 1H),
7. 31 (m, 1H), 7. 36 (s, 1H), 7. 66 (s, br, 1H), 7. 80 (d, J = 2. OHz, 1H). Mass spectrum m / z:.. 515 05 [M + H, 35Cl, 35Cl], 517 03 [M + H, 35Cl, 37Cl].
Example 2: 6_ [6_ atmosphere base _5_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -3_ than Li Jie base] -I ‘ – methyl-spiro [indoline-3,4 ‘- piperidin] -2_ one

Step I, I ‘- methyl-6- (4,4,5,5-tetramethyl–I, 3,2- dioxolane boron-2-yl) spiro [indoline Spray – 3,4 ‘- piperidin] -2_ one
1-1,6- bromo -I ‘- methyl-spiro [indoline-3,4’ – piperidin] -2_ one

As described in Example I steps 2-1 of the method from the commercially available 6-bromo – indol-2-one was prepared, Yield: 82%. Analysis of the data obtained the desired product are = 1H-Nmr (Cd3OdJOOmHz): 8 (ppm) 1.90-1.98 (m, 4H),
2. 44 (s, 3H), 2. 64-2. 68 (m, 2H), 2. 86-2. 92 (m, 2H), 7. 05 (d, J = 2. 0Hz, 1H), 7. 16-7. 21 (m, 2H), 7. 91 (s, br, 1H). Mass spectrum m / z: 295 00 [M + H, 79Br], 296 78 [M + H, 81Br]… [0312] 1-2, 1 ‘- methyl-6- (4,4,5,5-tetramethyl-_1,3,2_ dioxolane Borane _2_ yl) spiro [indoline – 3,4 ‘- piperidin] -2_ one

In the step 1-1 of the obtained 6-bromo -I ‘- methyl-spiro [indoline-_3,4’ – piperidin] -2_ ketone and commercially available linking pinacol boronic ester material, the method of Example I was prepared in accordance with steps 2-2, Yield: 95%. Analysis of the data obtained of the target product are as follows: Mass spectrum m / z:. 342 06 [M + H], 343 04 [M + H, 100%], 344. 12 [M + H]..
Step 2,6_ [6_ atmosphere base _5_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -3 ratio Li Jie base] -I ‘- methyl-spiro [indoline-3,4 ‘- piperidin] -2_ one
Example I steps 1-6 to obtain 5-bromo -3 – [(IR) -I- (2,6- dichloro-3-fluorophenyl) ethoxy] -2-amino- pyridine, I obtained in Example 1-2 of the present embodiment in step ‘- methyl-6- (4,4,5,5-tetramethyl-l, 3,2-dioxolane-2-yl borane) spiro [indoline-_3,4 ‘- piperidin] -2-one, prepared as in Example I Step 3. Yield: 82%. 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 86 (d, J = 6. 4Hz, 3H), I. 91-1 95 (m, 2H), I 97-2 03 (m, 2H… ), 2. 45 (s, 3H), 2. 65-2. 72 (m, 2H), 2. 89-2. 95 (m, 2H), 5. 12 (s, hr, 2H),
6. 12 (q, J = 6. 4Hz, 1H), 6. 94-7. 00 (m, 3H), 7. 06 (t, J = 8. 4Hz, 1H), 7. 31 (m, 1H ), 7. 35 (d, J = 7. 2Hz, 1H), 7. 90 (d, J = 2. 0Hz, 1H), 9. 28 (s, br, 1H). Mass spectrum m / z:.. 515 05 [M + H, 35Cl, 35Cl], 517 03 [M + H, 35Cl, 37Cl].
5- [6-amino-5 – [(2,6-dichloro-3-fluorophenyl) methoxy] _3_ pyridinyl] -I’–methyl-spiro [indole: 3 [0317] Example morpholine-3,4 ‘- piperidin] -2-one
H2N N


Step I, 5_ desert _3_ (2,6_ two gas -3- integrity oxy) _2_ atmosphere based grant given P
1-1,2,6_ two gas acid gas _3_
Cl OF
Sodium hydroxide (13g, 325mmol) in water (IlOmL) was cooled to _5 ° C was added dropwise under vigorous stirring of liquid bromine (12. 5g, 78. 2mmol), added after the addition of pre-cooled to 10 ° C dioxane (75mL). The above mixture under vigorous stirring was added dropwise a pre-cooled to 5 ° C of I- (2,6- dichloro-3-fluorophenyl) ethanone (5g, 21. 2mmol) in dioxane (330mL) and water (90mL) was added. After the addition, at room temperature for 2 hours Lan Xiang, Xiang Lan then 90 C for 30 minutes. TLC was not shown with the S starting material disappeared, and was acidified with concentrated hydrochloric acid to PH~9. The resulting mixture was rotary evaporated to dryness, added water (20mL), and extracted with diethyl ether (2X80mL), the organic phases were combined, dried (Na2SO4), and concentrated to give an oily product solidified after cooling to a transparent, slightly yellow solid (3. 4g, Yield: 67%). 1H-Nmr (Cdci3AOOmHz):. 8 (ppm) 7. 21 (. Dd, J = 8. 0,8 8Hz, 1H), 7 35 (. Dd, J = 4. 4,9 2Hz, 1H), 9 . 79 (s, br, 1H). Mass spectrum m / z (ES “:. 207 11 [M_H, 35Clj35Cl], 209 10 [MH, 35Cl, 37Cl]..
1-2,2,6–dichloro-3-fluoro-benzyl alcohol
^ Coh
F
[0325] To be filled with 2,6-dichloro-3-fluoro benzoic acid (3g, 14. 35mmol) added dropwise to the flask IM BH3. THF (43mL, 43mmol), added after the mixture was stirred under reflux for 24 hours. TLC showed the reaction was complete, methanol (50mL) to destroy excess borane, and the solvent was distilled off under reduced pressure and the resulting trimethyl borate, the process is repeated twice more to give a viscous product 2. I g, yield: 75% . 1H-Nmr (Cdci3JOOmHz): 8 (ppm) 2. 09 (t, J = 6. 4Hz, 1H), 4. 97 (d, J = 6. 4Hz, 2H), 7 09 (t, J = 8. . 8Hz, 1H), 7. 32 (dd, J = 4. 8,9. 1Hz, 1H). Mass spectrum m / z (ES-):.. 193 08 [M_H, 35Cl, 35Cl], 195 12 [MH, 35Cl, 37Cl].
1-3,3_ (2,6-gas _3_ integrity oxy) _2_ nitro grant given P

Following the procedure of steps 1-4 of Example I, was prepared from 2,6-dichloro-3-fluoro-benzyl alcohol and 3-hydroxy-2-nitropyridine prepared in yield (in this example embodiment steps 1_2) : 90%. 1H-Nmr (Cdci3AOOmHz): 8 (ppm) 5. 45 (s, 2H), 7 20, 7 37 (dd, J = 4. 8. (Dd, J = 8. 0,9 2Hz, 1H.). , 9. 2Hz, 1H), 7. 59 (dd, J = 4. 4,8. 4Hz, 1H),
7. 74 (dd, J = L 2,8. 4Hz, 1H), 8. 17 (dd, J = L 6,4. 4Hz, 1H). Mass spectrum m / z:. 316 89 [M + H, 35Cl, 35Cl], 318. 89 [M + H, 35Cl, 37Cl].
1_4,3_ (2,6-gas _3_ integrity oxy) _2_ atmosphere based grant given P

The method according to Example I step 1_5 from 3- (2,6-gas -3- integrity oxy) _2_ nitro Jie ratio 唳 preparation (in this case, steps 1-3), that Yield: 95% o 1H-Nmr (Cdci3JOOmHz):. 8 (ppm) 4 65 (s, br, 2H), 5 31 (s, 2H), 6 66 (dd, J = 5. 2,8.. . 0Hz, 1H), 7. 14 (dd, J = I. 2,8. 0Hz, 1H), 7. 18 (dd, J =
8. 4,9. 2Hz, 1H), 7. 37 (dd, J = 4. 8,8. 8Hz, 1H), 7. 73 (dd, J = I. 6,5. 6Hz, 1H). Mass spectrum m / z:. 286 95 [M + H, 35Cl, 35Cl], 288 85 [M + H, 35Cl, 37Cl]..
1-5,5_ desert -3- (2,6-gas -3_ integrity oxy) ~ 2 ~ atmosphere based grant given P

Following the procedure of Example I step 1_6 embodiment, starting from 3- (2,6-gas _3_ integrity yloxy) _2_ atmosphere group given the preparation of the batch P (in the example of the present embodiment in step 1-4), Yield: 60% o 1H-Nmr (Cdci3JOOmHz):. 8 (ppm) 4 68 (s, br, 2H), 5 28 (s, 2H), 7 21 (dd, J = 8. 0,8.. . 8Hz, lH), 7. 24 (dd, J = 2. OHz, 1H), 7. 39 (dd, J = 4. 8,
9. 2Hz, 1H), 7. 78 (d, J = 2. OHz, 1H). Mass spectrum m / z:. 364 83 [M + H, 35Cl, 36Cl, 79Br], 366 77 [M + H], 368 69 [M + H]…
Step 2,5_ [6_ atmosphere base _5_ [(2,6_ two gas -3- gas) methoxy] -3_ than Li Jie base] -I-methyl-spiro [indoline _ 3,4 ‘- piperidin] -2-one
The present embodiment 5_ desert steps 1_5 obtained _3_ (2,6_ two gas _3_ integrity yloxy) pyridine ~ 2 ~ atmosphere, Examples 2-2 obtained in step I I ‘- methyl-5- (4,4,5,5-tetramethyl-borane _1,3,2- dioxolane-2-yl) spiro [indoline-_3,4’ – piperidine ] -2-one, prepared as in Example I Step 3. Yield: 85 V0o 1H-Nmr (Cdci3JOOmHz):.. 8 (ppm) I. 92-2 02 (m, 4H), 2. 43 (s, 3H), 2. 65-2 71 (m, 2H) , 2. 90-2. 91 (m, 2H), 4. 92 (s, br, 2H), 5. 52 (s, 2H), 6. 89 (d, J = 8. 4Hz, 1H), 6 . 90 (d, J = L 2Hz, 1H), 7. 06 (t, J = 8. OHz, 1H), 7. 21 (dd, J = L 2,8. OHz, 1H), 7. 31 ( m, 1H),
7. 37 (s, 1H), 7. 79 (s, br, 1H), 7. 80 (d, J = 2.0Hz, lH). MS m / z:. 501 06 [M + H, 35Cl, 35Cl], 503 04 [M + H, 35Cl, 37Cl]..
6- [6-amino-5 – [(2,6-dichloro-3-fluorophenyl) methoxy] _3_ pyridinyl] -I’- methyl-spiro [indole: 4 [0337] Example morpholine _3,4 ‘- piperidin] -2-one



H2N N
Following the procedure in Example I step of Example 3, the procedure of Example 3 to give 5-bromo-1-5 _3_ (2,6-dichloro-3-fluoro-benzyloxy) -2-amino-pyridine and Step 2 in Example I to give the embodiment 1-2 ‘- methyl-6- (4,4,5,5-tetramethyl-1,3,2-dioxolane Borane 2-yl) spiro [ indoline-3,4 ‘- piperidine] _2_ ester -one, yield:. 78 V0o 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 96-2 00 (m, 2H), 2. 01 -2. 12 (m, 2H), 2. 46 (s, 3H), 2. 66-2. 73 (m, 2H), 2. 90-2. 96 (m, 2H), 5. 30 (s , hr, 2H), 6. 94-7. 01 (m, 3H), 7. 07 (t, J =
8. 4Hz, 1H), 7. 30 (m, 1H), 7. 34 (d, J = 7. 2Hz, 1H), 7. 89 (d, J = 2. OHz, 1H), 8. 56 ( s, br, 1H). MS m / z:. 501 06 [M + H, 35Cl, 35Cl], 503 04 [M + H, 35Cl, 37Cl]..
Example 5: 5_ [5_ atmosphere base -6- [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] Batch-2-yl] -I ‘ – methyl-spiro [indoline-3,4 ‘- piperidin] -2-one
J0A = o
. | J: too
[0342] Step 1,5_ desert _2_ atmosphere base _3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] Jie than exposure
Cl 6, / ISL / Br
xy
H2N N
In at 0 ° C, NaH (80mg of NaH in mineral oil, 2mmol) force the mouth (1R) -1_ (2,6- dichloro-3-fluorophenyl) ethanol (418mg, 2mmol. See example Example I Step 1_3) in anhydrous THF (6mL) and stirred for half an hour, a solution of 2-amino-3,5-dibromo-pyrazine (506mg, 2mmol) in THF (6mL) was added. The resulting mixture was warmed to room temperature, heated under reflux for 20 hours. TLC showed the reaction was substantially complete. After cooling to room temperature, water was added (IOmL), the mixture was extracted three times with ethyl acetate (3x20mL), the organic phases were combined, dried, concentrated, and the residue to give 594mg product was purified by column chromatography (l-3Me0H inhexanes), yield: 78%. 1H-NMR (O) Cl3, 500MHz):. 8 (ppm) I. 83 (d, J = 7. 2Hz, 3H), 5. 12 (s, br, 2H), 6 73 (q, J = 6 . 8Hz, 1H), 7. 05 (t, J = 8. OHz, 1H), 7. 28 (dd, J = 4. 8,
8. 8Hz, 1H), 7. 58 (s, 1H). Mass spectrum m / z:. 379 83 [M + H, 35Cl, 35Cl, 79Br], 381. 81 [M + H, 35Cl, 35Cl, 81Br], 383 79 [M + H, 35Cl, 37Cl, 81Br]..
Step 2,5_ [5_ atmosphere base _6_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] Batch-2-yl] -I ‘- A group spiro [indoline-3,4 ‘- piperidin] -2-one
5_ bromide present embodiment obtained in step I _2_ amino _3_ [(IR) -I- (2,6_ dichloro _3_ fluorophenyl) ethoxy] pyrazine, implemented I’- methyl step 2-2 obtained in Example I-5 (4,4,5,5-tetramethyl -I, 3,2- dioxolane boron
2-yl) spiro [indoline-3,4 ‘- piperidin] -2-one, prepared as in Example I Step 3. Yield: 54%. 1H-NMR (CD3ODjOOMHz): 8 (ppm) I. 85 (d, J = 6. 8Hz, 3H), I 85-1 88 (m, 2H), I 97-2 04 (m, 2H…. ), 2. 46 (s, 3H), 2. 76-2. 82 (m, 2H), 2. 97-3. 02 (m, 2H), 6. 74 (q, J = 6. 4Hz, 1H ), 6. 85 (d, J = 8. OHz, 1H), 7. 15 (t, J = 8. 4Hz, 1H), 7. 41 (dd, J = 4. 8,9. 2Hz, lH) , 7. 54 (dd, J = I. 6,
8. OHz, 1H), 7. 69 (d, J = I. 8Hz, 1H), 7. 81 (dt, J = 2. 0,8. 0Hz, 1H), 7. 87 (s, 1H). Mass spectrum m / z:. 515 92 [M + H, 35Cl, 35Cl], 517. 90 [M + H, 35Cl, 37Cl].
Example 6: 6- [5-amino -6 – [(lR) -l_ (2,6- dichloro _3_ fluorophenyl) ethoxy] pyrazin-2-yl] -I ‘ – methyl-spiro [indoline-3,4 ‘- piperidin] -2-one

The embodiment of Example 5, 5_ bromo obtained in step I _2_ amino _3_ [(IR) -I- (2,6_ dichloro _3_ fluorophenyl) ethoxy] pyrazine, Example I’- methyl-2 obtained in steps 1-2 6- (4,4,5,5-tetramethyl–I, 3,2- dioxolane boron-2-yl) spiro [indole morpholine -3,4’_ piperidin] -2-one, prepared as in Example I Step 3. Yield: 67% 0
1H-NMR (CD3ODjOOMHz): 8 (ppm) I. 85 (d, J = 6. 8Hz, 3H), I 88-1 96 (m, 4H), 2 48 (s… , 3H), 2. 76-2. 82 (m, 2H), 2. 98-3. 05 (m, 2H), 6. 75 (q, J = 6. 4Hz, 1H), 7. 16 (t , J = 8. 8Hz, 1H), 7. 31 (d, J = 2. OHz, 1H), 7. 36-7. 43 (m, 3H), 7. 88 (s, 1H).
Mass spectrum m / z:. 515 99 [M + H, 35Clj35Cl], 517 90 [M + H, 35Cl, 37Cl]..
SEE
Bioorganic & Medicinal Chemistry Letters (2014), 24(16), 3673-3682.
School of Pharmaceutical Sciences, Southern Medical University,



 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


 amcrasto@gmail.com
amcrasto@gmail.com





 Stockholm, Sweden
Stockholm, Sweden Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …
Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …
