
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
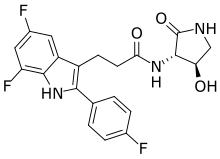
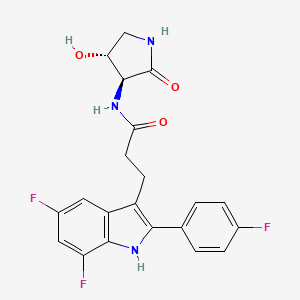
INAXAPLIN, 2446816-88-0
- 2446816-88-0
- S2SJ2RVZ6Y
- UNII-S2SJ2RVZ6Y
- C21H18F3N3O3
3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxopyrrolidin-3-yl]propanamide
Inaxaplin (VX-147) is a small-molecule apolipoprotein L1 inhibitor developed by Vertex Pharmaceuticals for APOL1-mediated kidney disease. In preliminary studies the drug has shown promise in treating people with kidney disease and multiple gain of function mutations on the APOL1 gene.[1][2]
FSGS is a disease of the podocyte (glomerular visceral epithelial cells) responsible for proteinuria and progressive decline in kidney function. NDKD is a disease characterized by hypertension and progressive decline in kidney function. Human genetics support a causal role for the G1 and G2 APOL1 variants in inducing kidney disease. Individuals with 2 APOL1 risk alleles are at increased risk of developing primary (idiopathic) FSGS, human immunodeficiency virus (HIV)-associated FSGS, and NDKD. Currently, FSGS and NDKD are managed with symptomatic treatment (including blood pressure control using blockers of the renin angiotensin system), and patients with FSGS and heavy proteinuria may be offered high dose steroids. Corticosteroids induce remission in a minority of patients and are associated with numerous side effects. These patients, in particular individuals of recent sub-Saharan African ancestry with 2 APOL1 risk alleles, experience rapid disease progression leading to end-stage renal disease (ESRD). Thus, there is an unmet medical need for treatment for FSGS and NDKD.
SCHEME
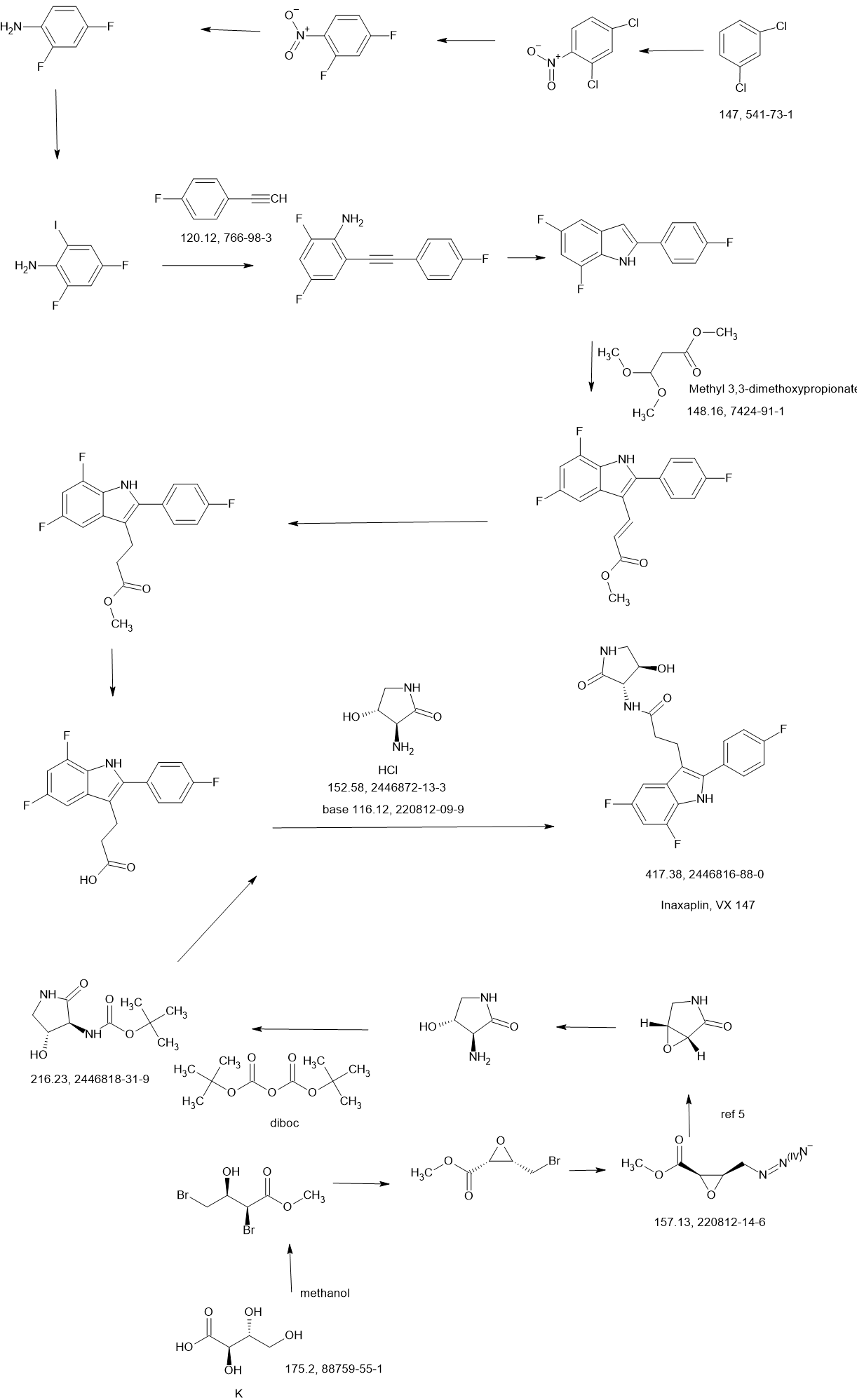
WO2021252863
US20210275496
WO2020131807
WO2021252849
PATENT
Compound 2

3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (2)
Synthesis of 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (2)
Alternative Procedure for Synthesis of Compound (2)
Step 1. Synthesis of Methyl (E)-3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]prop-2-enoate (C51)
Step 2. Synthesis of Methyl 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]propanoate (C52)
Step 3. Synthesis of 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]propanoic Acid (S12)
Step 4. Synthesis of 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (2)
Re-Crystallization of Compound 2
Form A of Compound 2
Hydrate Form A of Compound 2
| 200 mg of Compound 2 was charged with 10 mL of water. The slurry was cooled to 5° C. and allowed to stir. Hydrate A was observed after 3 days of stirring. |
PATENT
PATENT

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////
| Clinical data | |
|---|---|
| Other names | VX-147 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2446816-88-0 |
| PubChem CID | 147289591 |
| ChemSpider | 115009344 |
| UNII | S2SJ2RVZ6Y |
| ChEMBL | ChEMBL5083170 |
| Chemical and physical data | |
| Formula | C21H18F3N3O3 |
| Molar mass | 417.388 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Gbadegesin, Rasheed; Lane, Brandon (August 2023). “Inaxaplin for the treatment of APOL1-associated kidney disease”. Nature Reviews Nephrology. 19 (8): 479–480. doi:10.1038/s41581-023-00721-0. PMC 10461697. PMID 37106136.
- ^ Egbuna, Ogo; Zimmerman, Brandon; Manos, George; Fortier, Anne; Chirieac, Madalina C.; Dakin, Leslie A.; Friedman, David J.; Bramham, Kate; Campbell, Kirk; Knebelmann, Bertrand; Barisoni, Laura; Falk, Ronald J.; Gipson, Debbie S.; Lipkowitz, Michael S.; Ojo, Akinlolu; Bunnage, Mark E.; Pollak, Martin R.; Altshuler, David; Chertow, Glenn M. (16 March 2023). “Inaxaplin for Proteinuric Kidney Disease in Persons with Two APOL1 Variants”. New England Journal of Medicine. 388 (11): 969–979. doi:10.1056/NEJMoa2202396. PMID 36920755. S2CID 257534251.
………///////INAXAPLIN, VX-147, VX 147


{kind=link}
{kind=link}