







- USUS5856529 A
- USUS8785492 B2
- US5856529
- US8785492
- US9060995
- US9549913
- US9539234
- US9730910
- USRE46604
- US9855241
Home » Posts tagged 'FDA 2014'
Tag Archives: FDA 2014
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
DAPAGLIFLOZIN

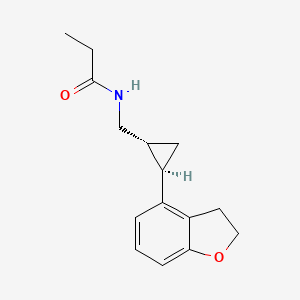
DAPAGLIFLOZIN, BMS-512148
ダパグリフロジン;
(2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol,
Cas 461432-26-8
| Molecular Formula: C21H25ClO6 |
| Molecular Weight: 408.87 |
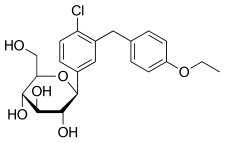
Dapagliflozin propandiol monohydrate; 960404-48-2
| Molecular Weight | 502.98 |
| Formula | C21H25ClO6•C3H8O2•H2O |
Bristol-Myers Squibb (Originator)
AstraZeneca
TYPE 2 DIABETES,SGLT-2 Inhibitors
launched 2012, as forxiga in EU, FDA 2014, JAPAN PMDA 2014
Dapagliflozin propanediol monohydrate was first approved by European Medicine Agency (EMA) on November 12, 2012, then approved by the U.S. Food and Drug Administration (FDA) on January 8, 2014, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on March 24, 2014. It was co-developed and co-marketed as Forxiga® by Bristol-Myers Squibb and AstraZeneca in EU.
Dapagliflozin propanediol monohydrate is a sodium-glucose co-transporter 2 (SGLT2) inhibitor indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
Forxiga® is available as tablet for oral use, containing 5 mg or 10 mg of free Dapagliflozin. The recommended starting dose is 5 mg once daily in the morning.

Dapagliflozin propanediol is a solvate containing 1:1:1 ratio of the dapagliflozin, (S)-(+)-1,2-propanediol, and water.
US——-In 2011, the product was not recommended for approval by the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee. In 2011, the FDA assigned a complete response letter to the application. A new application was resubmitted in 2013 by Bristol-Myers Squibb and AstraZeneca in the U.S
WILMINGTON, Del. & PRINCETON, N.J.--(BUSINESS WIRE)--December 12, 2013-- USFDA



Sales:$518.7 Million (Y2015); 
$235.8 Million (Y2014);
$33 Million (Y2013);ATC Code:A10BX09
Approved Countries or AreaUpdate Date:2015-07-29
- US
- EU
- JP
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-01-08 | Marketing approval | Farxiga | Type 2 diabetes | Tablet | 5 mg/10 mg | AstraZeneca |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2012-11-12 | Marketing approval | Forxiga | Type 2 diabetes | Tablet, Film coated | Eq. 5 mg/10 mg Dapagliflozin | Bristol-Myers Squibb, AstraZeneca |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-03-24 | Marketing approval | Forxiga | Type 2 diabetes | Tablet, Film coated | 5 mg/10 mg | Bristol-Myers Squibb, AstraZeneca, Ono |
MoreChemical Structure
AstraZeneca (NYSE:AZN) and Bristol-Myers Squibb Company (NYSE:BMY) today announced the U.S. Food and Drug Administration’s (FDA) Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) voted 13-1 that the benefits of dapagliflozin use outweigh identified risks and support marketing of dapagliflozin as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. The Advisory Committee also voted 10-4 that the data provided sufficient evidence that dapagliflozin, relative to comparators, has an acceptable cardiovascular risk profile.
The FDA is not bound by the Advisory Committee’s recommendation but takes its advice into consideration when reviewing the application for an investigational agent. The Prescription Drug User Fee Act (PDUFA) goal date for dapagliflozin is Jan. 11, 2014.

Dapagliflozin is being reviewed by the FDA for use as monotherapy, and in combination with other antidiabetic agents, as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. It is a selective and reversible inhibitor of sodium-glucose cotransporter 2 (SGLT2) that works independently of insulin to help remove excess glucose from the body. Dapagliflozin, an investigational compound in the U.S., was the first SGLT2 inhibitor to be approved anywhere in the world. Dapagliflozin is currently approved under the trade name [Forxiga](TM) for the treatment of adults with type 2 diabetes, along with diet and exercise, in 38 countries, including the European Union and Australia.
http://online.wsj.com/article/PR-CO-20131212-910828.html?dsk=y
Reference:1. WO03099836A1 / US6515117B2.
2. WO2010048358.
3. J. Med. Chem. 2008, 51, 1145–1149.
4. WO2004063209A2 / US7375213B2.
5. WO2008002824A1 / US7919598B2.Route 2
Reference:1. WO2010022313 / US8283454B2.Route 3
Reference:1. WO2013068850.Route 4
Reference:1. Org. Lett. 2012, 14, 1480-1483.
PAPER
https://www.future-science.com/doi/10.4155/fmc-2020-0154
Patent
https://patents.google.com/patent/WO2016178148A1/en
1. A process for the preparation of dapagliflozin in amorphous form, the process comprising:
(a) reducing a compound of formula II to a compound of formula ΠΙ in the presence of a Lewis acid;

(b) silylating a compound of formula IV with hexamethyldisilazane to form a compound of formula V;

(c) reacting the compound of formula III with the compound of formula V in the presence of a strong base followed by treatment with an acid in the presence of an alcohol to prepare a compound of formula VII, wherein R is an alkyl group selected from C1-5 alkyl;

(d) converting the compound of formula VII to dapagliflozin;
(e) acetylating dapagliflozin to give D-glucitol, l ,5-anhydro-l -C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]-, 2,3,4,6-tetraacetate, (IS)-, a compound of formula VIII;

(f) optionally, purifying the compound of formula VIII with a solvent selected from halogenated hydrocarbons, alcohols, ethers, or mixtures thereof; (g) hydrolyzing the compound of formula VIII obtained in step (f) to give dapagliflozin;
EXAMPLE 1: Preparation of 5-bromo-2-chlorobenzoyl chloride
To a suspension of 5-bromo-2-chiorobenzoic acid (lOg) in methylene di chloride (40niL), dimethylformamide (0.2g) and thionyl chloride were added and the reaction mixture was refluxed for about 2h. After completion of reaction, the solvent was distilled out. The mass obtained was degassed under vacuum followed by stripping with cyclohexane to give crude 5-bromo-2-chlorobenzoyl chloride (10.8g).
[0189] EXAMPLE 2: Preparation of 5-bromo-2-chloro-4′-ethoxybenzophenone (compound of Formula II)
5-bromo-2-chlorobenzoyl chloride (10.7g) was dissolved in methylene dichloride (40mL) and the reaction mixture was cooled to about -8°C to about -12°C under inert atmosphere. Aluminum chloride (5.65g) was added to the reaction mixture followed by addition of a solution of ethoxybenzene in methylene dichloride. The reaction mixture was stirred for about lh at about -8°C to -12°C and then quenched in dilute hydrochloric acid followed by extraction with methylene dichloride. The organic layer was washed with sodium bicarbonate solution and concentrated. The residue obtained was crystallized from methanol to give 5-bromo-2-chloro-4′-ethoxybenzophenone (8.5g). HPLC purity: 99.34%
[0190] EXAMPLE 3: Preparation of 5-bromo-2-chloro-4′-ethoxydiphenylmethane (compound of formula III)
To a mixture of 5-bromo-2-chloro-4′-ethoxybenzophenone (lOg) and methylene dichloride (50mL), cooled to about 0°C to about 5°C, triethylsilane (11.98g) and titanium chloride (22.3g) were added. The reaction mixture was stirred for about 3h at about 10°C to about 15°C. The reaction mixture was quenched into chilled water. The organic layer was separated, washed with water and sodium bicarbonate solution and concentrated under vacuum followed by stripping with toluene. The residue obtained was stirred with methanol, filtered and dried to give 5-bromo-2-chloro-4′-ethoxydiphenylmethane (9g). HPLC purity: 99.4%
[0191] EXAMPLE 4: Preparation of 2,3,4,6-tetra-0-(trimethylsilyl)-D-glucono-l,5- lactone (compound of Formula V)
To a mixture of D-glucono- 1,5 -lactone (lOg) and iodine (0.28g) in methylene dichloride (80mL), hexamethyldisilazane (36.1g) was added and the reaction mixture was refluxed. After completion of reaction, the reaction mixture was concentrated and degassed to give 2,3,4,6-tetra-0-(trimethylsilyl)-D-glucono-l,5-lactone as liquid (25g). HPLC purity: 95%
[0192] EXAMPLE 5: Preparation of D-glucopyranoside, methyl l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl] (compound of Formula VII wherein R is methyl)
To a mixture of 2,3,4,6-tetra-0-(trimethylsilyl)-D-glucono-l ,5-lactone (25g) and 5- bromo-2-chloro-4′-ethoxydiphenylmethane (8.7g) in tetrahydrofuran (174mL), cooled to about -75°C to about -88 °C under nitrogen atmosphere, n-butyl lithium in hexane (50mL) was slowly added. The reaction mixture was stirred at about the same temperature and then mixture of methanol and methanesulphonic acid was added to it. The reaction mixture was quenched into sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was separated, washed with saturated sodium chloride solution and concentrated under vacuum to obtain a residue. The residue was purified with a mixture of toluene and cyclohexane. Yield: 1 lg as thick mass with 80-85% HPLC purity.
reacting the compound of formula III with the compound of formula V in the presence of a strong base followed by treatment with an acid in the presence of an alcohol to prepare a compound of formula VII, wherein R is an alkyl group selected from C1-5 alkyl;
[0193] EXAMPLE 6: Preparation of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl) methyl] phenyl]
To a mixture of D-glucopyranoside, methyl l -C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl] in methylene di chloride (40mL) and acetonitrile (40mL), cooled to about -40°C to about -45°C, triethylsilane (8.74g) was added followed by addition of boron trifluoride etherate (10.67g) maintaining the temperature at about -40°C to about -45°C. The reaction mixture was quenched in sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was separated, concentrated and degassed under vacuum to give title compound (1 lg) as thick residue with 80-85% HPLC purity.
[0194] EXAMPLE 7: Preparation of D-Glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl]phenyl]-, 2,3,4,6-tetraacetate, (lS)-
To a cooled solution of D-glucitol, l,5-anhydro-l -C-[4-chloro-3-[(4-ethoxyphenyl) methyl] phenyl]- (l lg) in methylene dichloride (55mL) at about 0°C to about 5°C, diisopropylethylamme, dmiethylaminopyridine and acetic anhydride were added and the reaction mixture was stirred. After completion of reaction, the reaction mixture was quenched by adding water. The aqueous layer was separated and extracted with methylene dichloride. The organic layer was separated, washed with sodium bicarbonate solution and concentrated under vacuum to obtain residue which was stripped out with methanol. The residue was purified with methanol and charcoal, followed by diisopropyl ether and methanol crystallization. Yield: lOg; HPLC purity: 99.6%
acetylating dapagliflozin to give D-glucitol, l ,5-anhydro-l -C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]-, 2,3,4,6-tetraacetate, (IS)-, a compound of formula VIII;
[0195] EXAMPLE 8: Preparation of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl] (Dapagliflozin)
To a stirred solution of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]-, 2,3,4,6-tetraacetate, (IS)-, (lOg) in THF: methanol: water mixture (50mL: 50mL:30mL), sodium hydroxide was added and the reaction mixture was stirred. After completion of reaction, the solvents were distilled out under vacuum and the residue obtained was dissolved in methylene dichloride and washed with water and brine and dried over sodium sulfate. The reaction mixture was concentrated and degassed to give off- white to white solids of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]- (dapagliflozin) Yield: 7g (XRD matches with amorphous form) HPLC purity: 99.8%
[0197] EXAMPLE 10: Preparation of D-Glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl]phenyl]-, 2,3,4,6-tetraacetate, (IS)- from D-glucono-1,5- lactone (One-pot Synthesis)
To a mixture of D-glucono-l,5-lactone (lOg) in methylene dichloride (80mL), hexamethyldisilazane (36. lg) was added and the reaction mixture was refluxed. After completion of reaction, the reaction mixture was concentrated and degassed. The residue obtained was dissolved in tetrahydrofuran. 5-Bromo-2-chloro-4′-ethoxydiphenylmethane (8.7g) was added to the reaction mixture which was cooled to about -75°C to about-85°C under nitrogen atmosphere. n-Butyl lithium in hexane (50mL) was slowly added to the reaction mixture maintaining the temperature between -75°C to about -85°C. The reaction mixture was stirred at about the same temperature and then mixture of methanol and methanesulphonic acid was added to it. The reaction mixture was quenched into sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was separated, washed with saturated sodium chloride solution and concentrated under vacuum to obtain a residue. This residue was purified by a mixture of toluene and cyclohexane. To the product obtained, methylene dichloride and acetonitrile were added and the reaction mixture was cooled to about -40°C to about -45°C. Triethylsilane (8.74g) was added to the reaction mixture followed by addition of boron trifluoride etherate (10.67g) maintaining temperature at about -40°C to about -45°C. The reaction mixture was quenched in sodium bicarbonate solution. The aqueous layer was separated and extracted with ethyl acetate. The organic layer was separated, concentrated and degassed under vacuum. The thick residue obtained was dissolved in methylene dichloride and cooled to about 0°C to about 5°C. Diisopropylethylamine, dimethylaminopyridine and acetic anhydride were added to the reaction mixture which was stirred. After completion of reaction, the reaction mixture was quenched by adding water. The aqueous layer was separated and extracted with methylene dichloride. The organic layer was separated, washed with sodium bicarbonate solution and concentrated under vacuum to obtain residue which was stripped out with methanol The residue obtained was recrystallized with methanol and charcoal to give title compound (iOg) with 99.7% HPLC purity.
PAPER
Bioorganic & Medicinal Chemistry, 26(14), 3947-3952; 2018
https://www.sciencedirect.com/science/article/abs/pii/S0968089618309386?
Abstract
The cardiovascular complications were highly prevalent in type 2 diabetes mellitus (T2DM), even at the early stage of T2DM or the state of intensive glycemic control. Therefore, there is an urgent need for the intervention of cardiovascular complications in T2DM. Herein, the new hybrids of NO donor and SGLT2 inhibitor were design to achieve dual effects of anti-hyperglycemic and anti-thrombosis. As expected, the preferred hybrid 2 exhibited moderate SGLT2 inhibitory effects and anti-platelet aggregation activities, and its anti-platelet effect mediated by NO was also confirmed in the presence of NO scavenger. Moreover, compound 2 revealed significantly hypoglycemic effects and excretion of urinary glucose during an oral glucose tolerance test in mice. Potent and multifunctional hybrid, such as compound 2, is expected as a potential candidate for the intervention of cardiovascular complications in T2DM.
Graphical abstract

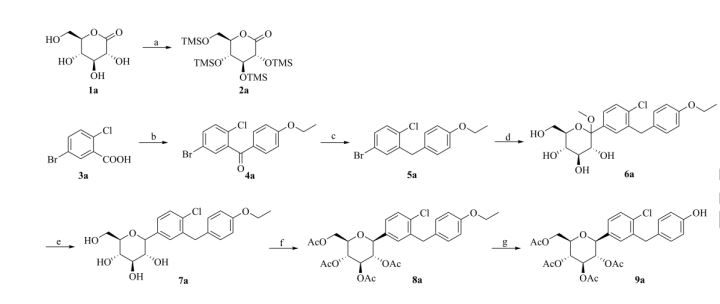
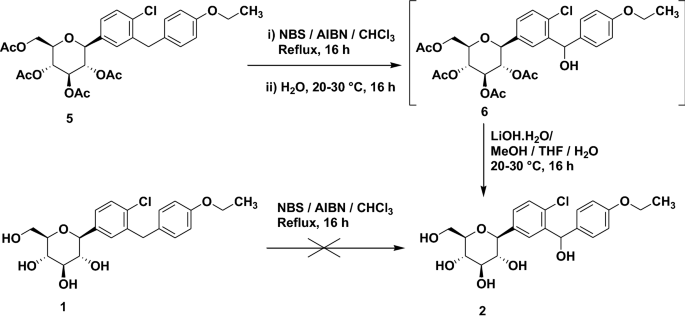
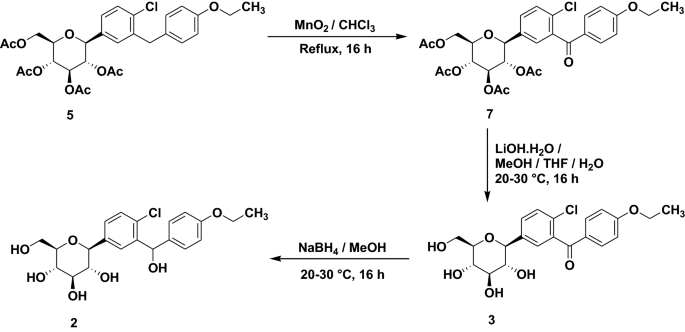
Scheme 1. Synthesis of target compounds 1-3. Reagents and conditions: (a) TMSCl, NMM, THF, 35 °C; (b) (COCl)2, CH2Cl2, DMF, then phenetole, AlCl3, 0 °C; (c) Et3SiH, BF3·OEt2, CH2Cl2, CH3CN, 25 °C; (d) n-BuLi, THF, toluene, -78 °C, then 2a followed by MeOH, CH3SO3H; (e) Et3SiH, BF3·OEt2, CH2Cl2, CH3CN, -10 °C; (f) Ac2O, pyridine, CH2Cl2, DMAP;
PATENT
Indian Pat. Appl., 2014MU03972,
PATENT
| Dapagliflozin, also known as SGLT2 inhibitor, chemical name is (2S,3R,4R,5S,6R)-2-[3-(4-ethoxybenzyl)-4-chlorophenyl]-6- Hydroxymethyltetrahydro-2H-pyran-3,4,5-triol, a sodium-glucose cotransporter 2 inhibitor, announced by the U.S. Food and Drug Administration (FDA) on January 8, 2014 , approved the use of dapagliflozin for the treatment of type 2 diabetes, the specific structural formula is as follows: |
| |
| Dapagliflozin works by inhibiting sodium-glucose transporter 2 (SGLT2), a protein in the kidney that allows glucose to be reabsorbed into the blood. This allows excess glucose to be excreted through the urine, thereby improving blood sugar control without increasing insulin secretion. |
| At present, there are two main methods for synthesizing dapagliflozin. One uses 5-bromo-2-chlorobenzoic acid as the starting material, which is chlorinated, Falk acylated, reduced, and then combined with 2,3,4 ,6-tetra-0-trimethylsilyl-D-glucopyranosic acid 1,5-lactone is condensed, methyl etherified, and demethoxylated to obtain dapagliflozin. The specific process route is as follows: |
| |
| The method has expensive starting materials and too many process steps, so it is not suitable for industrial production, and dangerous n-butyllithium needs to be used in the reaction process, so the requirements for experimental conditions are too high; |
| Another method is to use o-toluidine as the starting material, undergo bromination, diazotization, chlorination, and alkylation reactions, and then react with 2,3,4,6-tetra-0-trimethylsilyl -D-glucopyranosic acid 1,5-lactone is condensed, then methyl etherified and demethoxylated to obtain dapagliflozin. The specific process route is as follows: |
| |
| AIBN will be used in this reaction, which will produce highly toxic cyanide, which will seriously pollute the environment, and also requires the use of n-butyllithium, which requires high experimental conditions and is dangerous to operate, and is not suitable for large-scale production. |
| Example 1 |
| Weigh 16g (0.8mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodobenzene to it 2000mL of THF solution (total 2000mL: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsided, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene. |
| 433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 545.5mL of 33% hydrogen bromide in acetic acid solution (2.2mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 423.6 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose, and the yield was 93%. |
| Weigh 217.83g of 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 2500mL of toluene, then add 5.6g of europium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, and control the temperature at -20~-15°C. After the dropwise addition, the temperature is raised to 5°C for reaction for 2h, vacuum concentrated to an oily substance, and then added to it. 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, and 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , and dried to obtain 259.49 g of solid with a yield of 85%. |
| The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was finished, and then 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, dried the organic phase, concentrated in vacuo to an oil, to which was added 200 mL of 1:3 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 147.1 g of white rice-like crystals with a yield of 80.2% and a purity of 99.2% (determined by high performance liquid chromatography, external standard method). |
| IR(cm-1):1689,1612,1588,1523,1455,1253,1089,820。1H NMR(500MHz,CDCl 3 ):δ:7.36(d,J=8.2Hz,1H),7.32(d,J=1.9Hz,1H),7.23(dd,J=8.3,2.0Hz,1H),7.09(d,J=8.6Hz,2H),6.82(d,J=8.6Hz,2H),4.85(s,1H),4.41(s,3H),3.93~4.02(m,5H),3.70(dd,J=11.7,1.3Hz,1H),3.44(dd,J=11.7,5.6Hz,1H),3.26~3.28(m,1H)。LC-MS,m/z:[M+Na]+=431。 |
| Example 2 |
| Weigh 26.7g (1.33mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodine to it The THF solution of benzene (2000mL in total: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsides, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene . |
| 433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 272.75mL of 33% hydrogen bromide in acetic acid solution (1.1mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 381.5 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose, and the yield was 83.8%. |
| Weigh 217.83g of 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 5000mL of toluene, then add 5.76g of gadolinium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, and control the temperature at -20~-15°C. After the dropwise addition, the temperature is raised to 5°C for reaction for 2h, vacuum concentrated to an oily substance, and then added to it. 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, and 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , dried to obtain solid 281.2g, yield 92.1%. |
| The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was finished, and then 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, dried the organic phase, concentrated in vacuo to an oil, to which was added 200 mL of 1:4 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 150.9 g of white rice-like crystals with a yield of 88.2% and a purity of 99.5% (determined by high performance liquid chromatography, external standard method). |
| Example 3 |
| Weigh 37.4g (1.862mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodine to it The THF solution of benzene (2000mL in total: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsides, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene . |
| 433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 818.25mL of 33% hydrogen bromide in acetic acid solution (3.3mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 375.3 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose, and the yield was 82.4%. |
| Weigh 217.83g 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 7500mL toluene, then add 5.6g europium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, and control the temperature at -20~-15°C. After the dropwise addition, the temperature is raised to 5°C for reaction for 2h, vacuum concentrated to an oily substance, and then added to it. 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, and 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , and dried to obtain 273.25 g of solid with a yield of 89.5%. |
| The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was finished, and then 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, the organic phase was dried, concentrated in vacuo to an oil, to which was added 200 mL of 1:5 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 152.7 g of white rice-like crystals with a yield of 82.9% and a purity of 99.4% (determined by high performance liquid chromatography, external standard method). |
| Example 4 |
| Weigh 26.7g (1.33mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodine to it The THF solution of benzene (2000mL in total: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsides, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene . |
| 433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 545.5mL of 33% hydrogen bromide in acetic acid solution (2.2mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 425.1 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose in 94% yield. |
| Weigh 217.83g of 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 5000mL of toluene, then add 5.76g of gadolinium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, control the temperature at -20~-15°C, after the dropwise addition, raise the temperature to 5°C for 2 hours, concentrate in vacuo to an oily substance, then add to it 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, then 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , and dried to obtain 278.6 g of solid with a yield of 91.2%. |
| The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was completed, and 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, the organic phase was dried, concentrated in vacuo to an oil, to which was added 200 mL of 1:4 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 155.62 g of white rice-like crystals with a yield of 84.5% and a purity of 99.7% (determined by high performance liquid chromatography, external standard method). |
Patent
Sodium-glucose co-transporter-2 (SGLT2) inhibitors are a group of oral medicines used for treating diabetes that have been approved since 2013. SGLT2 inhibitors prevent the kidneys from re-absorbing glucose back into the blood by passing into the bladder. Glucose is re-absorbed back into the blood via the renal proximal tubules. SGLT2 is a protein predominantly expressed in the renal proximal tubules and is likely to be major transporter responsible for this uptake. Glucose-lowering effect of SGLT-2 inhibitors occurs via an insulin-independent mechanism mostly through glucosuria by increasing the urinary excretion of glucose.
It has been shown that the treatment with SGLT2 inhibitors in patients with type II diabetes lowers HbAlc, reduces body weight, lowers systemic blood pressure (BP) and induces a small increase in LDL-C and HDL-C levels.
SGLT2 inhibitors inhibit the reabsorption of sodium and glucose from the tubule and hence, more sodium is delivered in the macula densa causing arteriole dilation, reduced intraglomerular pressure and decreased hyperfiltration. SGLT2 inhibitors cause natriuresis and volume depletion, and an increase in circulating levels of renin, angiotensin and aldosterone. They also reduce albuminuria and slow GFR loss through mechanisms that appear independent of glycemia.
Dapagliflozin is a highly potent and reversible SGLT2 inhibitor, which increases the amount of glucose excreted in the urine and improves both fasting and post-prandial plasma glucose levels in patients with type 2 diabetes. Dapagliflozin has also been shown to tend to reduce liver fat content in some studies in a diabetic population.
Dapagliflozin is available on the market in the form of dapagliflozin propanediol monohydrate and is sold under trade name Forxiga or Farxiga in the form of fdm-coated tablets. Further it is available on the market as a combination product with metformin hydrochloride which is sold under trade name Xigduo IR or Xigduo XR in the form of film-coated tablets. In addition, it is available on the market as a combination product with saxagliptin hydrochloride which is sold under trade name Qtem in the form of film-coated tablets. Moreover, it is available on the market as a combination product with saxagliptin hydrochloride and metformin hydrochloride which is sold under trade name Qtemmet XR in the form of film-coated tablets.
Dapagliflozin as a monotherapy and in a combination with other active substances has demonstrated its efficacy in improving glycaemic control and reducing body weight and blood pressure in a broad spectrum of patients with type II diabetes, including those with high baseline HbAlc and the elderly. A sustained reduction in serum uric acid concentration was also observed. Dapagliflozin provides significant improvement in HbAlc, reduction in insulin dose and reduction in body weight in patients with type 1 diabetes as adjunct therapy to adjustable insulin.
Dapagliflozin can be in its free form or any stereoisomer or any pharmaceutically acceptable salt or co crystal complex or a hydrate or a solvate thereof and in any polymorphic forms and any mixtures thereof.
Dapagliflozin as a substance was first disclosed in US 6,515,117. The process for the preparation of dapagliflozin involves the reaction of 4-bromo- 1 -chloro-2-(4-ethoxybenyl)benzene with 2,3,4,6-tetra-O-trimethyl silyl -D-gluconolactone, the obtained compound 3 on demethoxylation yields diastereomeric mixture of Dapagliflozin. Hie diastereomeric mixture of dapagliflozin is further acetylated with acetic anhydride in the presence of pyridine and dimethylaminopyridine yields, then recrystallized from absolute ethanol to yield the desired tetra-acetyJated b-C-glucoside as a white solid. Compound tetra-acetylated b-C-glucoside is treated with lithium hydroxide hydrate which undergoes deprotection to yield the compound dapagliflozin.
Several other documents, patents and applications disclose the process for the preparation of dapagliflozin such as for example WOO 127128, WO03099836, W02004063209, W02006034489,
W02010022313, WO2012019496, W02013064909, W02013068850, W02013079501, WO2014094544, WO2014159151, WO2014206299, W02015040571, WO2015044849, WO2015063726, WO2015132803, WO2015155739, W02016098016, WO2016128995, WO2016178148, WO2017042683, WO2017063617, W02018029611, WO2018029264, WO2018142422.
Prior art documents already provided some compositions of SGLT2 inhibitor dapagliflozin.
W02008116179 discloses immediate release formulation in the form of a stock granulation or in the form of a capsule or a tablet which comprises dapagliflozin propylene glycol hydrate, one or more bulking agent, one or more binder and one or more disintegrant.
WO2011060256 describes the bilayer tablet comprising dapagliflozin having sustained release profde in one layer and metformin in another layer while WO2011060290 describes immediate release formulation of dapagliflozin and metformin.
WO2012163546 discloses the pharmaceutical composition comprising cyclodextrin and dapagliflozin.
Co-crystals of dapagliflozin with lactose are described in WO2014178040.
Solid dispersion compositions comprising amorphous dapagliflozin and at least one polymer are disclosed in W02015011113 and in WO2015128853.
CN103721261 discloses the combination of SGLT2 inhibitor with vitamins such as vitamin B.
Pharmaceutical composition preparation comprising dapagliflozin L-proline and metformin and/or DPP-IV inhibitor is disclosed in WO2018124497.
EP2252289A1 provides a combination of SGLT inhibitor with DPP4 inhibitor showing synergistic effect in increasing plasma active GLP-1 level in a patient over that provided by administration of the SGLT inhibitor or the DPP4 inhibitor alone.
EP2395983A1 relates to a pharmaceutical composition comprising a SGLT2 inhibitor, a DPP4 inhibitor and a third antidiabetic agent which is suitable in the treatment or prevention of one or more conditions selected from type 1 diabetes mellitus, type 2 diabetes mellitus, impaired glucose tolerance and hyperglycemia.
Example A: HPLC method
The purity of Dapagliflozine in general may be determined with the following HPLC method: column: XBridge C18, 150×4.6mm, 3.5; flow-rate: 0.9ml/min; column temperature: 50°C, wavelength: UV 225 nm; mobile phase: eluent A: 0.1% H3PO4, Eluent B: methanol; gradient:
Sample preparation: Accurately weigh about 40mg of sample and dissolve in 50 ml of solvent. Calculation: Use area per cent method. Do not integrate solvent peaks.
Example 1: Preparation of 5-bromo-2-chlorobenzoyl chloride
5-bromo-2-chlorobenzoic acid (450 g) was suspended in dichloromethane (2.25 L) and dimethylformamide (0.74 ml). At 15 – 30°C oxalyl chloride (180.3 ml) was slowly added. During addition gas evolution of HC1 and CO2 occurred. The reaction was performed at 20-30°C. The reaction was considered to be complete if 2-chloro-5-bromobenzoic acid was below 1% (area percent purity). The mixture was concentrated at elevated temperature until oily residue was obtained.
Example 2: Preparation of (5-bromo-2-chlorophenyl)(4-ethoxyphenyl)methanone
Dichloromethane (900 ml) was charged into reactor and then aluminum chloride (267.6 g) was added. The reaction mixture was cooled below 5°C and ethoxybenzene (256.1 ml) was slowly added. After complete addition, the mixture was gradually cooled below -5°C. In a separate reactor, 5-bromo-2-chlorobenzoyl chloride (485g) was dissolved in dichloromethane (900 ml). This solution was slowly added to the mixture of aluminum chloride and ethoxybenzene with such rate that temperature was kept below -5°C. After complete addition the mixture was stirred below -5°C until reaction was finished. The reaction was considered to be complete if methyl ester was below 1 % (the reaction mixture is sampled in methanol). After reaction was completed the reaction mixture was slowly added into cooled 1M HC1 solution and flushed with of dichloromethane (450 ml). The organic phase was separated and water phase was washed again with dichloromethane. Organic phases were combined and washed with water and NaHC03 solution. So obtained organic phase was concentrated to oily residue and dissolved in methanol ethyl acetate mixture in 10 to 1 ratio at reflux temperature. The clear solution was gradually cooled down to 35-45 °C and seeded with pure 5-bromo-2-chlorophenyl(4-ethoxyphenyl)methanone. The reaction mass was gradually cooled down to 0-10°C and stirred at that temperature up to 4 hours. The precipitate was isolated and washed with precooled methanol. The product was dried to a final LOD (Loss on drying) content of less than 1.0% with a yield of 564g (87% mass yield).
Example 3: Preparation of 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene
5-bromo-2-chlorophenyl(4-ethoxyphenyl)methanone (400g) was dissolved in 1.62L tetrahydrofuran. Into solution NaBTL (53.5g ) was added. After addition, the mixture was stirred at ambient temperature for 30-60 min followed by cooling of reaction mixture below -5°C. Aluminum chloride (314g) was added in portion and reaction mixture maintained below 5°C. After addition, the reaction mixture was gradually heated to reflux temperature and stirred until reaction was complete. Reaction mixture was cooled to ambient temperature and mixture of THF/water was slowly added into reaction mixture followed by addition of water and stirred at ambient temperature. Organic phase was collected and washed with saturated NaCl solution. Organic phase was concentrated to oily residue and dissolved in ethanol (800ml) at elevated temperature. Solution was cooled to 25-30°C and seeded with pure 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene. The reaction mass was gradually cooled to -2 to 10°C and stirred at that temperature. The product was isolated and washed with precooled ethanol and dried until final LOD (Loss on drying) content was less than 1.0%. Yield was 322 g (89%).
Example 4: Preparation of 3R,4S,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6- (hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol
4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene (97.5g ) and toluene (1.46L) was charge into reactor. Solution was heated to reflux temperature and approximately half of the solvent was distilled out. Tetrahydrofuran (195 mL) was charged into the solution and mixture was cooled below -70°C. Solution of 15% «-Buli in hexane (227.5 ml) was slowly added and temperature was kept below -70°C. After complete addition solution was stirred at temperature below -70°C to complete reaction. Solution of 2,3,4,6-tetra-O-trimethylsilyl-D-gluconolactone (182 g) in toluene (243 mL) was added into reaction mixture at temperature below -70°C. After complete addition, the mixture was stirred below -70°C, warmed to approximately -65°C and then mixture of 57.6 g methanesulfonic acid in 488 ml methanol was added. After addition, the mixture was gradually warmed to ambient temperature and stirred until reaction was complete. After reaction was finished reaction mixture was slowly added into saturated NaHCCL solution (630ml) and stirred. Into quenched mixture 975 ml of heptane and 585 ml methanol was added. The mixture was stirred for additional 15 min. Organic phase was washed with water/methanol mixture several times. Water phases were combined and distilled to remove organic solvents. Into the residual water phase, toluene was added to perform extraction. Organic phases were combined and washed with water. Organic phase was distillated at elevated temperature until oily residue was obtained.
Example 5: Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl) tetrahydro2H-pyran-3,4,5-triol – Dapagliflozin
Dichloromethane (656 mL) was charged into oily residue from step 4 and stirred at ambient temperature until clear solution was obtained. Triethysilane (122 mL) was added into the so obtained solution. Reaction mixture was cooled below -30°C and 94.2 mL of boron trifluoride etherate was slowly added at temperatures below -30°C. After complete addition, the mixture was stirred below -30°C for one hour and gradually warmed to -5 to 0°C until reaction was completed. After reaction was finished saturated NaHCCL solution (468 mL) was slowly added. Reaction mixture was distilled to remove organic solvents followed by addition ethyl acetate into the residue. Organic phase was collected and washed again with saturated NaHC03 and water. So obtained organic phase was distillated at elevated temperature until oily residue is obtained.
Example 6: Preparation of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate.
Oily residual from example 5 was dissolved in dichloromethane (602 mL) at ambient temperature followed by addition of DMAP (6.22g). Reaction mixture was cooled to 0 – 10°C and 144.3 mL of acetic anhydride was added at temperatures below 10°C. Reaction mixture was gradually warmed to ambient temperature and stirred until reaction was completed. Reaction mass was washed with water with saturated NaHC03. Organic phase was collected and concentrated to oily residue to which ethanol (1.68L) was charged and approximately 300 ml ethanol was removed by distillation. The clear solution was gradually cooled to 60-65 °C and seeded. The reaction mass was gradually cooled to 20-25 °C and product was isolated. The product was dried at 50°C in vacuum until LOD (Loss on drying) below 1.0% 123g of product was obtained with yield 71%. HPLC purity: 99.97 %.
Formula 9 Formula 2
(2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (740 g) as prepared according to process described in examples 1 to 6 was charged into solution of methanol (2.27L), water (0.74L) and NaOH (23 lg) at 35-45°C and stirred at 35-45°C until reaction was completed. After reaction was finished 1M HC1 (1.63L) was slowly added. Reaction mass was distilled to remove organic solvents and product was extracted by tert-butyl methyl ether. Combined organic phases were washed with water and concentrated at elevated temperature until oily residue was obtained. Content of impurity IMP A was below 0.02%.
Example 7a: Preparation of amorphous dapagliflozin.
Oily residue as prepared according to example 7 comprising approximately 262 g of dapagliflozin was dissolved in toluene (2.5L) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (5.3L) at temperature between 10 to 15°C and stirring rate with P/V at 4 W/m3. After complete addition, the suspension was cooled to 0°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Filtration rate was 14 · 104 m/s. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of impurity IMP A was below 0.02% and residual heptane and toluene were 1673 ppm and below 89 ppm.
(2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (30 g) of 4 different qualities obtained by the process known in the prior art was charged into solution of methanol (90 mL), water (30 mL) and NaOH (9.36 g) and stirred at 35-45°C until reaction was completed and sampled for HPLC analysis (Sample 1).
After reaction was finished 1M HC1 (66 mL) was slowly added. Reaction mass was distilled to remove organic solvents and product was extracted by tert-butyl methyl ether. To the combined organic phases 68ml of 1M NaOH was added and pH was set to 12.5 to 13.5. Phases were separated and organic phase is washed again with 68ml of water without pH correction. So obtained organic phase was sampled for HPLC analysis (Sample 2) and concentrated at elevated temperature until oily residue was obtained.
Oily residue comprising approximately 21 g of dapagliflozin was dissolved in toluene (210 mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (420 mL) at temperature between 10 to 15°C and stirring rate with P/V as defined in Table 1. After complete addition, the suspension was cooled to 0°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Filtration rate was as defined in Table 1. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Amorphous dapagliflozin with content of impurity IMP A as shown in Table 1 and residual heptane and toluene as shown in Table 1 was obtained for each cases.
Table 1 : Process parameters used in the preparation of four different starting materials (cases).
As it is evident from Table 2 the final amorphous dapagliflozin prepared by the extraction process according to the present invention contains less than 0.02% of impurity IMP A irrespective of the level of impurity IMP A present in the starting material.
Table 2: Content of impurity IMP A in the final amorphous dapagliflozin obtained with and without extraction.
Example 9
Oily residue, as obtained by the procedure described in example 8 case 1, containing approximately 2 g of dapagliflozin was dissolved in 1.5ml of isopropyl acetate and 6ml of tert-butyl methyl ether at temperature 50-55 °C. So prepared solution was charged into 25 mL of heptane at 0°C. After complete addition the suspension was stirred at -10 to 0°C. Suspension was isolated and washed with precooled heptane at temperatures between 25°C to 50 °C. 1.5 g of dapagliflozin was obtained with content of impurity IMP A was below 0.02%.
Example 10
Oily residue, as obtained by the procedure described in example 8 case 1, containing approximately 2 g of dapagliflozin was dissolved in 1.5ml of isopropyl acetate and 6ml of tert-butyl methyl ether at temperature 50-55 °C. So prepared solution was charged into 40 mL of heptane at 0°C. After complete addition the suspension was stirred at -10 to 0°C. Suspension was isolated and washed with precooled heptane at temperatures between 25°C to 50 °C. 1.5 g of dapagliflozin was obtained with content of impurity IMP A was below 0.02%.
Example 11
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 5°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to -10°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 1480 ppm and 732 ppm.
Example 12
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 20°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to 5°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled hcptanc Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 2873 ppm and 639 ppm.
Comparative Example 1
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at -5°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to -15°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 1940 ppm and 1557 ppm.
Comparative Example 2
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 25°C and stirring rate with P V at 16 W/m3. After complete addition, the suspension was cooled to 20°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 3663 ppm and 2047 ppm.
Comparative Example 3
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 30°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to 15°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 2425 ppm and 1812 ppm.
///////////////////////////////////////////
POST YOUR INTERMEDIATES LIST HERE FOR WORLDWIDE REACH

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
PATENT
https://patents.google.com/patent/WO2017206808A1/en
Daggliflozin (English name: Dapagliflozin) is a new Sodium glucose co-transporters 2 (SGLT-2) inhibitor developed by Bristol-Myers Squibb and AstraZeneca. Approved by the European Commission on November 14, 2012, and marketed in the United States on January 8, 2014, to improve glycemic control in adult patients with type 2 diabetes by combining diet and exercise; the trade name is Farxiga, currently offering 5 mg and 10 mg tablets. At the same time, a combination of dapagliflozin and metformin hydrochloride has also been marketed.The chemical name of dapagliflozin is (2S,3R,4R,5S,6R)-2-(3-(4-ethoxybenzyl)-4-chlorophenyl)-6-hydroxymethyltetrahydro-2H – pyran-3,4,5-triol, the chemical formula is C 21 H 25 ClO 6 , CAS No. 461432-26-8, the structural formula is shown as 2, clinically used as a pharmaceutical for dapagliflozin (S) -1,2-propanediol monohydrate, the structural formula is as shown in 1.

The synthesis of β-type C-aryl glycosidic bonds is a key point in the synthetic route during the preparation of dapagliflozin. At present, there are four synthetic methods for the synthesis of dapagliflozin reported in the literature and patents.Route 1: The synthetic route of dapagliflozin reported in patent WO03099836A1 is as follows:

The route uses 2-chloro-5-bromobenzoic acid (12) as raw material to react with phenethyl ether to form intermediate 11 and then triethylsilane to obtain intermediate 10; intermediate 10 and n-butyl The lithium is reacted at -78 ° C, and then subjected to a nucleophilic addition reaction with the intermediate 9, and then methoxylated to obtain the intermediate 8; the intermediate 8 is subjected to acylation reduction and deprotection to obtain the intermediate 2. The disadvantage of this method is that the β-type C-aryl glycosidic bond synthesis of the compound is carried out at a low temperature of -78 ° C, which is obviously difficult to meet the needs of industrial production; and, through nucleophilic addition, methoxylation, The five-step reaction of acetylation, reduction and hydrolysis can synthesize the β-type C-aryl glycosidic bond. The procedure is relatively long, and the purity of the intermediate 2 is only 94%.Route 2: The synthetic route of dapagliflozin reported in the literature OrgLett.2012, 14, 1480 is as follows:

The intermediate 14 of the route is reacted with di-n-butyl-n-hexylmagnesium for 48 hours at 0 ° C, and then reacted with zinc bromide to prepare an organozinc reagent by Br/Mg/Zn exchange reaction, and then with intermediate 4 Intermediate 3 was prepared by nucleophilic substitution reaction; finally, intermediate 2 was obtained by deprotection with sodium methoxide. The synthesis method is relatively novel, and the synthesis step is short. However, the research experiment is conducted only as a synthesis method, and the post treatment of the intermediate 3 is performed by column chromatography. The purity of the intermediate 2 produced was not reported. Moreover, the di-n-butyl-n-hexylmagnesium reagent used in the route is not a commonly used reagent, and is not commercially available in China. It can only be prepared by reacting dibutylmagnesium with n-hexyllithium reagent before the test, and the operation is cumbersome and difficult to mass. use.Route 3: The synthetic route of dapagliflozin reported in patent WO2013068850A2 is as follows:

The route uses 1,6-anhydroglucose (20) as a raw material, protects the 2,4-hydroxyl group by tert-butyldiphenylchlorosilane, and then protects the 3-position hydroxyl group with phenylmagnesium bromide. Intermediate 18. The intermediate 14 is subjected to an Br/Mg/Al exchange reaction to prepare an organoaluminum reagent 16, which is reacted with an intermediate 18 to form an intermediate 15, and finally, deprotected to obtain an intermediate 2. The synthesis method is very novel and is also used as a synthetic methodological study. The purification of the intermediates is carried out by column chromatography. The 1,6-anhydroglucose (20) used in the route is very expensive; and the multi-step reaction in the route uses a format reagent, a preparation format reagent or an organoaluminum reagent, which is cumbersome and cumbersome to perform, and is difficult to scale synthesis. The purity of the intermediate 2 produced was not reported.Route 4: The synthetic route of dapagliflozin reported in patent WO2013152476A1 is as follows:

The route uses 2-chloro-5-iodobenzoic acid (24) as raw material to form intermediate 22 by Friedel acylation and reduction reaction, and exchange with I-Mg at -5 ° C with isopropyl magnesium chloride lithium chloride. The intermediate 8 is obtained by nucleophilic addition and methoxylation with the intermediate 9, and then the intermediate 2 is obtained by reduction with triethylsilane, and the intermediate 2 is further purified by co-crystallizing with L-valine. Finally, The pure intermediate 2 was obtained by removing L-valine. This route is a modified route of Route 1, which replaces n-butyllithium with isopropylmagnesium chloride chloride to raise the reaction temperature of the reaction from -78 °C to -5 °C. However, the problem of a long step of synthesizing a β-type C-aryl glycosidic bond still exists. The obtained intermediate 2 is not optically pure, and needs to be purified by co-crystallizing with L-valine, and the work amount of post-treatment is increased, and finally the purity of the intermediate 2 is 99.3%.Among the four synthetic routes described above for dapagliflozin, route one and route four are commonly used synthetic methods for β-type C-aryl glycosidic bonds, and the route is long, and the optical purity of the obtained product is not high, and further purification is required. Post processing is cumbersome. Moreover, the reaction required at -78 °C in Route 1 requires high equipment and high energy consumption, which undoubtedly increases the cost. Although both Route 2 and Route 3 are new methods, most of the purification of intermediates used is column chromatography. Such a process is not suitable for scale production in factories; and some of the synthetic routes are used. Reagents are not commercially available or expensive, and there is no advantage in such route costs. Therefore, there is an urgent need to find a new method for the synthesis of dapagliflozin, and to enable industrial production, and the route has a cost advantage.Repeating the procedure reported in the literature in Equation 2, the yield of Intermediate 3 was only 46%. The organic zinc reagent is prepared by Br/Mg/Zn exchange reaction, and the exchange reaction yield is 78%; and the raw material is prepared by X/Li/Zn exchange reaction to prepare an organic zinc reagent, and the exchange reaction yield is 98.5%, which is also the two Different reaction pathways lead to the essential reason for the different yields of intermediate 3. Moreover, the price of commercially available 1.0 mol/L di-n-butyl magnesium n-heptane solution 500 mL is 1380 yuan, and the price of 1.6 mol/L n-hexyl lithium n-hexane solution 500 mL is 950 yuan, and 2.5 mol/L n-butyl lithium. The price of 500 mL of n-hexane solution is only 145 yuan. Therefore, the method for preparing dapagliflozin by preparing an organozinc reagent by X/Li/Zn and then synthesizing the β-type C-aryl glycosidic bond designed by the invention has the advantages of cost, ease of operation and industrialization. Very obvious advantage.In order to solve this problem, the original compound company uses a eutectic method in the production of dapagliflozin to make dapagliflozin together with a solvent or an amino acid compound, since the compound 2 sugar ring structure contains four hydroxyl groups and is easy to absorb moisture and deteriorate. The crystal is made into a relatively stable solid, easy to store, stable and controllable in quality, and easy to prepare. Among them, the marketed dapagliflozin forms a stable eutectic with (S)-1,2-propanediol and water (1). The original crystal form patent (CN101479287B, CN103145773B) reported that all 11 crystal forms are dapagliflozin solvate or dapagliflozin. Crystal. Among them, there are two preparation methods for the da forme (S)-1,2-propanediol monohydrate (1) having a crystal structure of type Ia:Method 1: The preparation method is as follows:

Compound 7 is deprotected with sodium hydroxide to obtain compound 2, then compound 2 is extracted with isopropyl acetate, (S)-1,2-propanediol ((S)-PG) is added, and seed crystal of compound 1 is added. Then, cyclohexane was added to crystallize and separated to obtain a eutectic of the compound (1) of the type Ia.Method 2: The preparation method is as follows:

Compound 8 is subjected to reduction of methoxy group by triethylsilane and boron trifluoride diethyl ether complex, and then the reaction solution is extracted with methyl tert-butyl ether (MTBE), and (S)-1,2-propanediol ( (S)-PG), a seed crystal of the compound 1 is added, and then cyclohexane is added to crystallize, and the mixture is separated and dried to obtain a eutectic of the compound (1) of the type Ia.The above two methods for preparing the eutectic are all used in the cyclohexane solvent, which is listed in the appendix of the 2015 edition of the Pharmacopoeia (four parts) as the second type of solvent that should be restricted, with a residual limit of 0.388%. The solvent residue of the final product obtained must reach the specified limit, and the post-treatment process is complicated, time-consuming and labor-intensive, and the production cost is correspondingly increased. The invention finds a suitable solvent on the basis of the synthetic route to prepare a medicinal crystal form, and has obvious advantages in both the method and the process operation steps.The synthetic route is as follows:

Comparative Example 1, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4- Preparation of chlorophenyl]glucosamine (Compound 3)Under nitrogen protection, 1.0 mol/L di-n-butylmagnesium-n-heptane solution (16 mL) was cooled to 0 ° C, and 1.6 mol/L n-hexane lithium n-hexane solution (10 mL) was slowly added dropwise. After the addition was completed, 0 ° C After stirring for 15 h, dry n-butyl ether (2.5 mL) was added to prepare a solution of di-n-butyl-n-hexylmagnesium lithium solution, which was calibrated with iodine and stored for use.Zinc bromide (2.7 g) and lithium bromide (1.04 g) were added with n-butyl ether (20 mL), heated to 50 ° C for 4 h, and cooled for use. 4-(2-Chloro-5-bromo-benzyl) phenyl ether (6.513 g) was added with toluene (8 mL) and n-butyl ether (5 mL) under nitrogen, cooled to 0 ° C, and 0.61 mol/L was added dropwise. n-Butyl-n-hexylmagnesium lithium solution (13.1 mL), after the addition is completed, the reaction was kept at 0 ° C for 48 h, and the above-mentioned alternate zinc bromide and lithium bromide n-butyl ether solution were added, and the reaction was kept at 0 ° C for 1 h, and added 2 , 3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (14.49 g) in toluene (25 mL), heated to 100 ° C to stir the reaction, after TLC detection reaction, add 1 mol / L diluted hydrochloric acid (60 mL), taken after stirring extraction, the organic phase was washed with water (40 mL), then washed with saturated brine (40 mL), dried over anhydrous Na 2 SO 4, concentrated under reduced pressure, column chromatography (petroleum ether / Ethyl acetate = 20:1) 10.38 g of Compound 3 as a pale yellow oil. Yield: 46%. Purity: 99.02%. The organozinc reagent prepared by the method has an iodine calibration yield of 78%.The calibration method of the concentration of the prepared organic zinc reagent: accurately weighed iodine (1 mmol), placed in a three-necked flask, replaced nitrogen, and added anhydrous 0.5 mol/L LiCl tetrahydrofuran solution (5 mL), stirred and dissolved, and cooled to 0 ° C. The prepared organozinc reagent was slowly added dropwise until the color of the brownish yellow solution disappeared.Example 2 (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)Zinc bromide (2.25 g) and lithium bromide (0.87 g) were added with n-butyl ether (30 mL), heated to 50 ° C for 2 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (10 mL) and n-butyl ether (10 mL) under nitrogen, cooled to -20 ° C, and slowly added dropwise 1.6 mol / L-n-hexyl lithium n-hexane solution (14mL), control the internal temperature does not exceed -10 ° C, after the completion of the addition, the temperature is incubated at -20 ° C for 0.5 h, adding the above-mentioned spare zinc bromide and lithium bromide n-butyl ether solution, The reaction was stirred at 20 ° C for 3 h. Add 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (11.59g) toluene (50mL) solution, heat to 120 ° C and stir the reaction for 4h, after TLC detection reaction, was added 1mol / L diluted hydrochloric acid (40 mL), water (20 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, concentrated with n-heptane (15mL) and methanol (60 mL) and recrystallized 10.8 g of Compound 3 as a white solid was obtained in a yield: 72.42%. Purity: 99.47%. Melting point: 99.5 to 101.6 °C. (The organic zinc reagent prepared by this method was iodine-calibrated in a yield of 98.5%.) ESI-MS (m/z): 767.30 [M+Na] + . 1 H-NMR (400 MHz, CDCl 3 ): δ 7.33 (1H, d), 7.14-7.17 (2H, m), 7.05 (2H, d), 6.79-6.81 (2H, dd), 5.39 (1H, t ), 5.21-5.31 (2H, m), 4.33 (1H, d), 4.17-4.20 (1H, dd), 3.94-4.11 (5H, m), 3.79-3.83 (1H, m), 1.39 (3H, t ), 1.20 (9H, s), 1.16 (9H, s), 1.11 (9H, s), 0.86 (9H, s).Example 3, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3) PrepareZinc bromide (3.38 g) and lithium bromide (1.3 g) were added with n-butyl ether (40 mL), heated to 50 ° C for 2 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (20 mL) and n-butyl ether (5 mL) under nitrogen, cooled to -50 ° C, and slowly added dropwise 2.5 mol / L-butyllithium hexane solution (8mL), control the internal temperature does not exceed -30 ° C, after the addition is completed, the reaction is kept at -50 ° C for 10 h, adding the above-mentioned alternate zinc bromide and lithium bromide n-butyl ether solution, The reaction was stirred at -20 ° C for 10 h. Add 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (34.77g) toluene (80mL) solution, heat to 100 ° C and stir the reaction for 24h, after TLC detection reaction, was added 1mol / L diluted hydrochloric acid (60 mL), water (50 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, concentrated with n-heptane (15mL) and methanol (60 mL) and recrystallized 10.854 g of Compound 3 as a white solid. Yield: 72.81%. Purity: 99.53%.Example 4, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)N-butyl ether (50 mL) was added to zinc iodide (3.19 g) and lithium iodide (1.34 g), and the mixture was heated to 50 ° C for 1.5 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (15 mL) and n-butyl ether (5 mL) under nitrogen, cooled to -60 ° C, and slowly added dropwise 1.6 mol / L-n-hexyl lithium n-hexane solution (13.8mL), control the internal temperature does not exceed -20 ° C, after the addition is completed, the reaction is kept at -60 ° C for 5 h, and the above-mentioned alternate zinc iodide and lithium iodide n-butyl ether solution is added. The reaction was stirred at 25 ° C for 1 h. Add 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (23.2g) toluene (50mL) solution, heat to 140 ° C reflux reaction for 0.5h, after TLC detection reaction was added 1mol / L diluted hydrochloric acid (50 mL), water (50 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous SO 4 Na 2, concentrated by weight of n-heptane (15mL) and methanol (60 mL) Crystallization gave 10.51 g of Compound 3 as a white solid, yield 70.5%. Purity: 99.41%.Example 5, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)To the zinc bromide (2.25 g) and lithium bromide (0.87 g), cyclopentyl methyl ether (30 mL) was added, and the mixture was heated to 50 ° C for 3 hours, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (10 mL) and cyclopentyl methyl ether (10 mL) under nitrogen, cooled to -5 ° C, and slowly added dropwise. Mol / L n-hexyl lithium n-hexane solution (12.5mL), control the internal temperature does not exceed 0 ° C, after the addition is completed, the reaction is kept at -5 ° C for 3 h, adding the above-mentioned spare zinc bromide and lithium bromide cyclopentyl methyl ether The solution was incubated at -5 ° C for 4 h, and a solution of 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (17.39 g) in toluene (40 mL) was added and heated to 80 ℃ reaction was stirred 6h, after completion of the reaction by TLC, was added 1mol / L diluted hydrochloric acid (50 mL), water (50 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous 2 SO 4 Na, and concentrated under reduced pressure, Recrystallization of n-heptane (15 mL) and methanol (60 mL) gave 8.15 g of Compound 3 as a white solid. Purity: 99.39%.Example 6, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)Zinc bromide (4.5 g) and lithium bromide (1.74 g) were added with n-butyl ether (60 mL), heated to 50 ° C for 3 h, and cooled for use. 4-(2-Chloro-5-bromo-benzyl) phenyl ether (6.513 g) was added with toluene (15 mL) and n-butyl ether (5 mL) under nitrogen, cooled to -30 ° C, and slowly added dropwise 2.5 mol / L-butyllithium n-hexane solution (8.4mL), control the internal temperature does not exceed -20 ° C, after the addition is completed, the reaction is kept at -30 ° C for 3 h, and the above-mentioned alternate zinc bromide and lithium bromide n-butyl ether solution is added. The reaction was incubated at -5 ° C for 4 h, and a solution of 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (14.49 g) in toluene (50 mL) was added and heated to 120 ° C for stirring. the reaction 4h, after completion of the reaction by TLC, was added 1mol / L diluted hydrochloric acid (50 mL), water (40 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, and concentrated under reduced pressure, n-heptyl Recrystallization of the alkane (15 mL) and methanol (60 mL) gave 10.38 g of Compound 3 as a white solid. Purity: 99.54%.Example 7, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)Methyl bromide (40 mL) was added to zinc bromide (2.25 g) and lithium bromide (0.87 g), and the mixture was heated to 50 ° C for 3 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (15 mL), methyl tert-butyl ether (15 mL), cooled to -40 ° C, and slowly added dropwise. 1.6mol/L n-hexyl lithium n-hexane solution (13.8mL), control the internal temperature does not exceed -30 ° C, after the addition is completed, the reaction is kept at -40 ° C for 4 h, and the above-mentioned alternate zinc bromide and lithium bromide are added. The butyl ether solution was incubated at 5 ° C for 7 h, and a solution of 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (17.39 g) in toluene (50 mL) was added and heated. to 90 deg.] C the reaction was stirred 8h, after completion of the reaction by TLC, was added 1mol / L diluted hydrochloric acid (40 mL), water (40 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, and concentrated under reduced pressure Recrystallization from n-heptane (15 mL) and methanol (60 mL) gave 9.41 g of Compound 3 as a white solid. Purity: 99.42%. Example 8. Preparation of dapagliflozin (S)-1,2-propanediol monohydrate eutectic (Compound 1)To the compound 3 (37.27 g), methanol (190 mL) was added, and sodium methoxide (10.8 g) was added thereto, and the mixture was heated under reflux for 3 hours. After the TLC reaction was completed, methanol was concentrated, and isopropyl acetate (100 mL) was added to the residue, and water was added. (60 mL), extracted with stirring and the organic phase washed with water (50 mL). (S)-1,2-propanediol (3.8g) and water (0.9g) were added to the organic phase, stirred until it was dissolved, and n-heptane (200 mL) was added, and the mixture was stirred for 2 hours under ice-cooling, suction filtration, filter cake Washing with n-heptane and drying at 30 ° C gave 23.89 g of Compound 1 as a white solid. Yield: 95%. Purity: 99.79%. Melting point: 69.1 to 75.6 °C. The product obtained was subjected to KF = 3.74% (theoretical value: 3.58%). ESI-MS (m/z): 431.22 [M+Na] + . 1 H-NMR (400 MHz, CD 3 OD): δ 7.33 – 7.37 (2H, m), 7.28-7.30 (1H, dd), 7.11 (2H, d), 6.80-6.83 (2H, dd), 4.1 ( 1H, d), 3.98-4.05 (4H, m), 3.88-3.91 (1H, dd), 3.74-3.82 (1H, m), 3.68-3.73 (1H, m), 3.37-3.49 (5H, m), 3.28-3.34 (1H, m), 1.37 (1H, t), 1.15 (3H, d).The crystal form of the obtained product was subjected to thermogravimetric analysis (TGA) by a Universal V4.7A TA instrument, and the TGA curve (Fig. 1) showed a weight loss of about 18.52% from about room temperature to about 240 ° C. The original form Ia crystal form The TGA plot shows a value of 18.7%.The crystal form of the obtained product was subjected to differential scanning calorimetry (DSC) by a Universal V4.7A TA instrument, and the DSC curve (Fig. 2) showed endotherm in the range of about 60 ° C to 85 ° C. The DSC plot shows a range of approximately 50 ° C to 78 ° C.
The crystal form of the obtained product was examined by a Bruker D8advance instrument for powder X-ray diffraction (PXRD), and the 2X value of the PXRD pattern (Fig. 3) (CuKα).

There are characteristic peaks at 3.749°, 7.52°, 7.995°, 8.664°, 15.134°, 15.708°, 17.069°, 18.946°, 20.049°, which are completely consistent with the characteristic peaks of the PXRD pattern of the Ia crystal form in the original patent.In combination with the nuclear magnetic data and melting point of the prepared crystal form, the crystal form of the product (Compound 1) obtained by the present invention is consistent with the pharmaceutically acceptable crystalline form Ia reported in the original patent.
Patent Citations
Publication numberPriority datePublication dateAssigneeTitleCN101479287A *2006-06-282009-07-08布里斯托尔-迈尔斯斯奎布公司Crystalline solvates and complexes of (is) -1, 5-anhydro-l-c- (3- ( (phenyl) methyl) phenyl) -d-glucitol derivatives with amino acids as sglt2 inhibitors for the treatment of diabetesCN104496952A *2014-11-282015-04-08深圳翰宇药业股份有限公司Synthesis method of dapagliflozinCN105153137A *2015-09-172015-12-16上海应用技术学院Preparation method of empagliflozinFamily To Family CitationsCN104829572B *2014-02-102019-01-04江苏豪森药业集团有限公司Dapagliflozin novel crystal forms and preparation method thereofCN105399735A *2015-12-292016-03-16上海应用技术学院Empagliflozin intermediate, and preparation method and application thereof* Cited by examiner, † Cited by third party
Non-Patent Citations
TitleCHEN DEJIN ET AL., CHINA MASTER’S THESES FULL-TEXT DATABASE, ENGINEERING TECHNOLOGY I, vol. B016-731, no. 3, 15 March 2016 (2016-03-15) *LEMAIRE S. ET AL.: “Stereoselective C-glycosylation of furanosyl halides with arylzinc reagents”, PURE APPL. CHEM., vol. 86, no. 3, 4 March 2014 (2014-03-04), pages 329 – 333 *LEMAIRE S. ET AL.: “Stereoselective C-Glycosylation Reactions with Arylzinc Reagents”, ORGANIC LETTERS, vol. 14, no. 6, 2 March 2012 (2012-03-02), pages 1480 – 1483, XP055069093 ** Cited by examiner, † Cited by third partyCLIP
Chemical Synthesis
Dapagliflozin propanediol hydrate, an orally active sodium glucose cotransporter type 2 (SGLT-2) inhibitor, was developed by Bristol-Myers Squibb (BMS) and AstraZeneca for the once-daily treatment of type 2 diabetes. As opposed to competitor SGLT-2 inhibitors, dapagliflozin was not associated with renal toxicity or long-term deterioration of renal function in phase III clinical trials. The drug exhibits excellent SGLT2 potency with more than 1200 fold selectivity over the SGLT1 enzyme.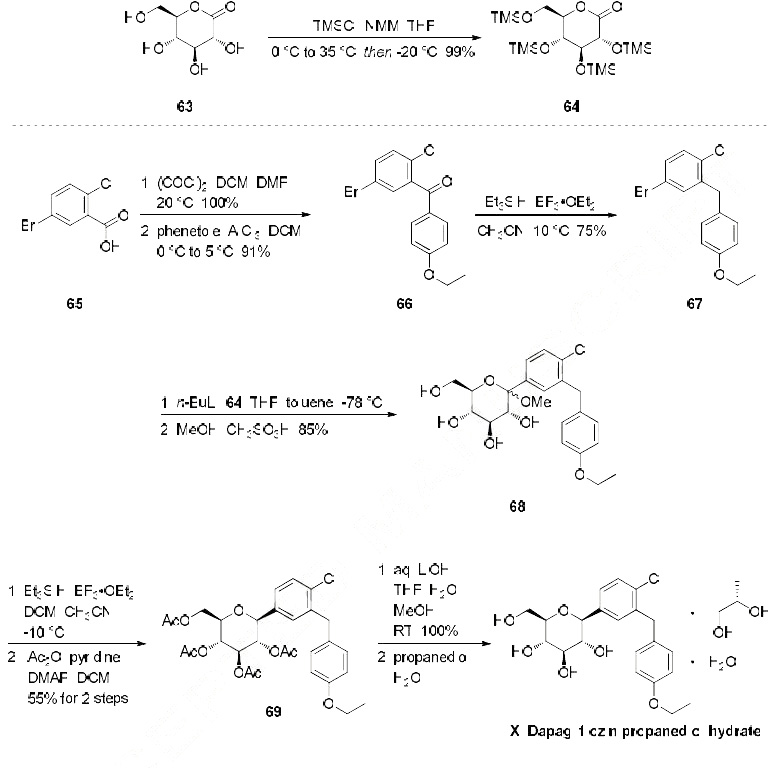
Dapagliflozin propanediol monohydrate
PAPER
https://link.springer.com/article/10.1007/s12039-020-1747-x

PATENTS
WO 2010138535
WO 2011060256
WO 2012041898
WO 2012163990
WO 2013068850
WO 2012163546
WO 2013068850
WO 2013079501
The IC50 for SGLT2 is less than one thousandth of the IC50 for SGLT1 (1.1 versus 1390 nmol/l), so that the drug does not interfere with the intestinal glucose absorption.[7

dapagliflozin being an inhibitor of sodiumdependent glucose transporters found in the intestine and kidney (SGLT2) and to a method for treating diabetes, especially type II diabetes, as well as hyperglycemia, hyperinsulinemia, obesity, hypertriglyceridemia, Syndrome X, diabetic
complications, atherosclerosis and related diseases, employing such C-aryl glucosides alone or in combination with one, two or more other type antidiabetic agent and/or one, two or more other type therapeutic agents such as hypolipidemic agents.
Approximately 100 million people worldwide suffer from type II diabetes (NIDDM – non-insulin-dependent diabetes mellitus), which is characterized by hyperglycemia due to excessive hepatic glucose production and peripheral insulin resistance, the root causes for which are as yet unknown. Hyperglycemia is considered to be the major risk factor for the development of diabetic complications, and is likely to contribute directly to the impairment of insulin secretion seen in advanced NIDDM. Normalization of plasma glucose in NIDDM patients would be predicted to improve insulin action, and to offset the development of diabetic complications. An inhibitor of the sodium-dependent glucose transporter SGLT2 in the kidney would be expected to aid in the normalization of plasma glucose levels, and perhaps body weight, by enhancing glucose excretion.
Dapagliflozin can be prepared using similar procedures as described in U.S. Pat. No. 6,515,117 or international published applications no. WO 03/099836 and WO 2008/116179
WO 03/099836 A1 refers to dapagliflozin having the structure according to formula 1 .

formula 1
WO 03/099836 A1 discloses a route of synthesis on pages 8-10, whereby one major step is the purification of a compound of formula 2

formula 2
The compound of formula 2 provides a means of purification for providing a compound of formula 1 since it crystallizes. Subsequently the crystalline form of the compound of formula 2 can be deprotected and converted to dapagliflozin. Using this process, dapagliflozin is obtained as an amorphous glassy off-white solid containing 0.1 1 mol% of EtOAc. Crystallization of a pharmaceutical drug is usually advantageous as it provides means for purification also suitable for industrial scale preparation. However, for providing an active pharmaceutical drug a very high purity is required. In particular, organic impurities such as EtOAc either need to be avoided or further purification steps are needed to provide the drug in a
pharmaceutically acceptable form, i.e. substantially free of organic solvents. Thus, there is the need in the art to obtain pure and crystalline dapagliflozinwhich is substantially free of organic solvents.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Crystalline forms (in comparision to the amorphous form) often show desired different physical and/or biological characteristics which may assist in the manufacture or formulation of the active compound, to the purity levels and uniformity required for regulatory approval. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps.
PATENT
WO 2008/ 1 16179 Al seems to disclose an immediate release formulation comprising dapagliflozin and propylene glycol hydrate. WO 2008/ 116195 A2 refers to the use of an SLGT2 inhibitor in the treatment of obesity
http://www.google.com/patents/US20120282336
http://www.tga.gov.au/pdf/auspar/auspar-dapagliflozin-propanediol-monohydrate-130114.pdf
Example 2 Dapagliflozin (S) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (S)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (S) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in published applications WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
Example 3 Dapagliflozin (R) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (R)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (R) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Surprisingly, amorphous dapagliflozin can be purified with the process of the present invention. For instance amorphous dapagliflozin having a purity of 99,0% can be converted to crystalline dapagliflozin hydrate having a purity of 100% (see examples of the present application). Moreover, said crystalline dapagliflozin hydrate does not contain any additional solvent which is desirable. Thus, the process of purifying dapagliflozin according to the present invention is superior compared with the process of WO 03/099836 A1 .
Additionally, the dapagliflozin hydrate obtained is crystalline which is advantageous with respect to the formulation of a pharmaceutical composition. The use of expensive diols such as (S)-propanediol for obtaining an immediate release pharmaceutical composition as disclosed in WO 2008/1 16179 A1 can be avoided
PAPER
In Vitro Characterization and Pharmacokinetics of Dapagliflozin …
dmd.aspetjournals.org/content/…/DMD29165_supplemental_data_.doc
Dapagliflozin (BMS-512148), (2S,3R,4R,5S,6R)-2-(3-(4-Ethoxybenzyl)-4-chlorophenyl)
-6-hydroxymethyl-tetrahydro-2H-pyran-3,4,5-triol. 1H NMR (500 MHz, CD3OD) δ 7.33
(d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H),
6.78 (d, J = 8.8, 2H), 4.07-3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6,
1H), 3.42-3.25 (m, 4H), 1.34 (t, J = 7.0, 3H). 13C NMR (125 MHz, CD3OD) δ 158.8,
140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9,
64.5, 63.1, 39.2, 15.2.
HRMS calculated for C21H25ClNaO6 (M+Na)+
For C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.
: 431.1237; found 431.1234. Anal. Calcd
SECOND SETJ. Med. Chem., 2008, 51 (5), pp 1145–1149DOI: 10.1021/jm701272q
1H NMR (500 MHz, CD3OD) δ 7.33 (d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H), 6.78 (d, J = 8.8, 2H), 4.07–3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6, 1H), 3.42–3.25 (m, 4H), 1.34 (t, J = 7.0, 3H);
13C NMR (125 MHz, CD3OD) δ 158.8, 140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9, 64.5, 63.1, 39.2, 15.2;
HRMS calcd for C21H25ClNaO6 (M + Na)+ 431.1237, found 431.1234. Anal. Calcd for C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.
HPLC
- HPLC measurements were performed with an Agilent 1100 series instrument equipped with a UV-vis detector set to 240 nm according to the following method:
Column: Ascentis Express RP-Amide 4.6 x 150 mm, 2.7 mm;
Column temperature: 25 °C
– Eluent A: 0.1 % formic acid in water
– Eluent B: 0.1 % formic acid in acetonitrile
– Injection volume: 3 mL
– Flow: 0.7 mL/min
– Gradient:Time [min][%] B0.02525.06526.07029.07029.52535.025……………………..
PATENT
http://www.google.com/patents/WO2013068850A2?cl=en
EXAMPLE 24 – Synthesis of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside 2,4-di-6>-TBDPS-dapagliflozin; (IVj”))

[0229] l-(5-Bromo-2-chlorobenzyl)-4-ethoxybenzene (1.5 g, 4.6 mmol) and magnesium powder (0.54 g, 22.2 mmol) were placed in a suitable reactor, followed by THF (12 mL) and 1,2- dibromoethane (0.16 mL). The mixture was heated to reflux. After the reaction had initiated, a solution of l-(5-bromo-2-chlorobenzyl)-4-ethoxybenzene (4.5 g, 13.8 mmol) in THF (28 mL) was added dropwise. The mixture was allowed to stir for another hour under reflux, and was then cooled to ambient temperature, and then titrated to determine the concentration. The above prepared 4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl magnesium bromide (31 mL, 10 mmol, 0.32 M in THF) and A1C13 (0.5 M in THF, 8.0 mL, 4.0 mmol) were mixed at ambient temperature to give a black solution, which was stirred at ambient temperature for 1 hour. To a solution of
I, 6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (0.64 g, 1.0 mmol) in PhOMe (3.0 mL) at ambient temperature was added phenylmagnesium bromide (0.38 mL, 1.0 mmol, 2.6 M solution in Et20). After stirring for about 5 min the solution was then added into the above prepared aluminum mixture via syringe, followed by additional PhOMe (1.0 mL) to rinse the flask. The mixture was concentrated under reduced pressure (50 torr) at 60 °C (external bath temperature) to remove low-boiling point ethereal solvents and then PhOMe (6mL) was added. The reaction mixture was heated at 130 °C (external bath temperature) for 8 hours at which time HPLC assay analysis indicated a 51% yield of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3- (4-ethoxybenzyl)phenyl)- -D-glucopyranoside. After cooling to ambient temperature, the reaction was treated with 10% aqueous NaOH (1 mL), THF (10 mL) and diatomaceous earth at ambient temperature, then the mixture was filtered and the filter cake was washed with THF. The combined filtrates were concentrated and the crude product was purified by silica gel column chromatography (eluting with 1:30 EtOAc/77-heptane) affording the product 2,4-di-6>- ieri-butyldiphenylsilyl- 1 – -(4-chloro-3 -(4-ethoxybenzyl)phenyl)- β-D-glucopyranoside (0.30 g, 34%) as a white powder.
1H NMR (400 MHz, CDC13) δ 7.56-7.54 (m, 2H), 7.43-7.31 (m, 13H), 7.29-7.22 (m, 6H), 7.07- 7.04 (m, 2H), 7.00 (d, J= 2.0 Hz, IH), 6.87 (dd, J= 8.4, 2.0 Hz, IH), 6.83-6.81 (m, 2H), 4.18 (d, J= 9.6 Hz, IH), 4.02 (q, J= 6.9 Hz, 2H), 3.96 (d, J= 10.8 Hz, 2H), 3.86 (ddd, J= 11.3, 7.7, 1.1 Hz, IH), 3.76 (ddd, J= 8.4, 8.4, 4.8 Hz, IH), 3.56 (ddd, J= 9.0, 6.4, 2.4 Hz, IH), 3.50 (dd, J=
I I.4, 5.4 Hz, IH), 3.44 (dd, J= 9.4, 8.6 Hz, IH), 3.38 (dd, J= 8.8, 8.8 Hz, IH), 1.70 (dd, J= 7.8, 5.4 Hz, IH, OH), 1.42 (t, J= 6.8 Hz, 3H), 1.21 (d, J= 5.2 Hz, IH, OH), 1.00 (s, 9H), 0.64 (s, 9H); 13C NMR (100 MHz, CDC13) δ 157.4 (C), 138.8 (C), 137.4 (C), 136.3 (CH x2), 136.1 (CH x2), 135.2 (CH x2), 135.0 (C), 134.9 (CH x2), 134.8 (C), 134.2 (C), 132.8 (C), 132.0 (C), 131.6 (CH), 131.1 (C), 129.9 (CH x2), 129.7 (CH), 129.6 (CH), 129.5 (CH), 129.4 (CH), 129.2 (CH), 127.58 (CH x2), 127.57 (CH x2), 127.54 (CH x2), 127.31 (CH), 127.28 (CH x2), 114.4 (CH x2), 82.2 (CH), 80.5 (CH), 79.3 (CH), 76.3 (CH), 72.7 (CH), 63.4 (CH2), 62.7 (CH2), 38.2 (CH2), 27.2 (CH3 x3), 26.6 (CH3 x3), 19.6 (C), 19.2 (C), 14.9 (CH3). EXAMPLE 25 -Synthesis of dapagliflozin ((25,3R,4R,55,6/?)-2-[4-chloro-3-(4- ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol; (Ij))

IVj’ U
[0230] A solution of the 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside (60 mg, 0.068 mmol) in THF (3.0 mL) and TBAF (3.0 mL, 3.0 mmol, 1.0 M in THF) was stirred at ambient temperature for 15 hours. CaC03 (0.62 g), Dowex^ 50WX8-400 ion exchange resin (1.86 g) and MeOH (5mL) were added to the product mixture and the suspension was stirred at ambient temperature for 1 hour and then the mixture was filtrated through a pad of diatomaceous earth. The filter cake was rinsed with MeOH and the combined filtrates was evaporated under vacuum and the resulting residue was purified by column chromatography (eluting with 1 : 10 MeOH/DCM) affording dapagliflozin (30 mg).
1H NMR (400 MHz, CD3OD) δ 7.37-7.34 (m, 2H), 7.29 (dd, J= 8.2, 2.2 Hz, 1H), 7.12-7.10 (m, 2H), 6.82-6.80 (m, 2H), 4.10 (d, J= 9.6 Hz, 2H), 4.04 (d, J= 9.2 Hz, 2H), 4.00 (q, J= 7.1 Hz, 2H), 3.91-3.87 (m, 1H), 3.73-3.67(m, 1H), 3.47-3.40 (m, 3H), 3.31-3.23 (m, 2H), 1.37 (t, J= 7.0 Hz, 3H);
13C NMR (100 MHz, CD3OD) δ 157.4 (C), 138.6 (C), 138.5 (C), 133.1 (C), 131.5 (C), 130.5 (CH), 129.4 (CH x2), 128.7 (CH), 126.8 (CH), 114.0 (CH x2), 80.5 (CH), 80.8 (CH), 78.3 (CH), 75.0 (CH), 70.4 (CH), 63.0 (CH2), 61.7 (CH2), 37.8 (CH2), 13.8 (CH3);
LCMS (ESI) m/z 426 (100, [M+NH4]+), 428 (36, [M+NH4+2]+), 447 (33, [M+K]+).
Example 1 – Synthesis of l,6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (II”)

III II”
[0206] To a suspension solution of l,6-anhydro- -D-glucopyranose (1.83 g, 11.3 mmol) and imidazole (3.07 g, 45.2 mmol) in THF (10 mL) at 0 °C was added dropwise a solution of TBDPSC1 (11.6 mL, 45.2 mmol) in THF (10 mL). After the l,6-anhydro-P-D-gJucopyranose was consumed, water (10 mL) was added and the mixture was extracted twice with EtOAc (20 mL each), washed with brine (10 mL), dried (Na2S04) and concentrated. Column
chromatography (eluting with 1 :20 EtOAc/rc-heptane) afforded 2,4-di-6>-ieri-butyldiphenylsilyl- l,6-anhydro- “D-glucopyranose (5.89 g, 81%).
1H NMR (400 MHz, CDC13) δ 7.82-7.70 (m, 8H), 7.49-7.36 (m, 12H), 5.17 (s, IH), 4.22 (d, J= 4.8 Hz, IH), 3.88-3.85 (m, IH), 3.583-3.579 (m, IH), 3.492-3.486 (m, IH), 3.47-3.45 (m, IH), 3.30 (dd, J= 7.4, 5.4 Hz, IH), 1.71 (d, J= 6.0 Hz, IH), 1.142 (s, 9H), 1.139 (s, 9H); 13C NMR (100 MHz, CDCI3) δ 135.89 (CH x2), 135.87 (CH x2), 135.85 (CH x2), 135.83 (CH x2), 133.8 (C), 133.5 (C), 133.3 (C), 133.2 (C), 129.94 (CH), 129.92 (CH), 129.90 (CH), 129.88 (CH), 127.84 (CH2 x2), 127.82 (CH2 x2), 127.77 (CH2 x4), 102.4 (CH), 76.9 (CH), 75.3 (CH), 73.9 (CH), 73.5 (CH), 65.4 (CH2), 27.0 (CH3 x6), 19.3 (C x2).
PATENT
WO 2016147197, DAPAGLIFLOZIN, NEW PATENT, HARMAN FINOCHEM LIMITED
LINK>>> (WO2016147197) A NOVEL PROCESS FOR PREPARING (2S,3R,4R,5S,6R)-2-[4-CHLORO-3-(4-ETHOXYBENZYL)PHENY 1] -6-(HY DROXY METHYL)TETRAHYDRO-2H-PY RAN-3,4,5-TRIOL AND ITS AMORPHOUS FORM
PATENT
PATENT
WO2016018024, CRYSTALLINE COMPOSITE COMPRISING DAPAGLIFLOZIN AND METHOD FOR PREPARING SAME
HANMI FINE CHEMICAL CO., LTD. [KR/KR]; 59, Gyeongje-ro, Siheung-si, Gyeonggi-do 429-848 (KR)
Dapagliflozin, sold under the brand name Farxiga among others, is a medication used to treat type 2 diabetes and, with certain restrictions, type 1 diabetes.[2] It is also used to treat adults with certain kinds of heart failure.[3][4][5]
Common side effects include hypoglycaemia (low blood sugar), urinary tract infections, genital infections, and volume depletion (reduced amount of water in the body).[6] Diabetic ketoacidosis is a common side effect in type 1 diabetic patients.[7] Serious but rare side effects include Fournier gangrene.[8] Dapagliflozin is a sodium-glucose co-transporter-2 (SGLT-2) inhibitor and works by removing sugar from the body with the urine.[9]
It was developed by Bristol-Myers Squibb in partnership with AstraZeneca. In 2018, it was the 227th most commonly prescribed medication in the United States, with more than 2 million prescriptions.[10][11]
Medical uses
Dapagliflozin is used along with diet and exercise to improve glycemic control in adults with type 2 diabetes and to reduce the risk of hospitalization for heart failure among adults with type 2 diabetes and known cardiovascular disease or other risk factors.[12][3] It appears more useful than empagliflozin.[13][verification needed]
In addition, dapagliflozin is indicated for the treatment of adults with heart failure with reduced ejection fraction to reduce the risk of cardiovascular death and hospitalization for heart failure.[3][4][5] It is also indicated to reduce the risk of kidney function decline, kidney failure, cardiovascular death and hospitalization for heart failure in adults with chronic kidney disease who are at risk of disease progression.[14]
In the European Union it is indicated in adults:
- for the treatment of insufficiently controlled type 2 diabetes mellitus as an adjunct to diet and exercise:
- as monotherapy when metformin is considered inappropriate due to intolerance;
- in addition to other medicinal products for the treatment of type 2 diabetes;
- for the treatment of insufficiently controlled type 1 diabetes mellitus as an adjunct to insulin in patients with BMI ≥ 27 kg/m2, when insulin alone does not provide adequate glycaemic control despite optimal insulin therapy; and
- for the treatment of heart failure with reduced ejection fraction.[5]
Adverse effects
Since dapagliflozin leads to heavy glycosuria (sometimes up to about 70 grams per day) it can lead to rapid weight loss and tiredness. The glucose acts as an osmotic diuretic (this effect is the cause of polyuria in diabetes) which can lead to dehydration. The increased amount of glucose in the urine can also worsen the infections already associated with diabetes, particularly urinary tract infections and thrush (candidiasis). Rarely, use of an SGLT2 drug, including dapagliflozin, is associated with necrotizing fasciitis of the perineum, also called Fournier gangrene.[15]
Dapagliflozin is also associated with hypotensive reactions. There are concerns it may increase the risk of diabetic ketoacidosis.[16]
Dapagliflozin can cause dehydration, serious urinary tract infections and genital yeast infections.[3] Elderly people, people with kidney problems, those with low blood pressure, and people on diuretics should be assessed for their volume status and kidney function.[3] People with signs and symptoms of metabolic acidosis or ketoacidosis (acid buildup in the blood) should also be assessed.[3] Dapagliflozin can cause serious cases of necrotizing fasciitis of the perineum (Fournier gangrene) in people with diabetes and low blood sugar when combined with insulin.[3]
To lessen the risk of developing ketoacidosis (a serious condition in which the body produces high levels of blood acids called ketones) after surgery, the FDA has approved changes to the prescribing information for SGLT2 inhibitor diabetes medicines to recommend they be stopped temporarily before scheduled surgery. Canagliflozin, dapagliflozin, and empagliflozin should each be stopped at least three days before, and ertugliflozin should be stopped at least four days before scheduled surgery.[17]
Symptoms of ketoacidosis include nausea, vomiting, abdominal pain, tiredness, and trouble breathing.[17]
Use is not recommended in patients with eGFR < 45ml/min/1.73m2, though data from 2021 shows the reduction in the kidney failure risks in people with chronic kidney disease using dapagliflozin.[18]
Mechanism of action
Dapagliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2) which are responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter mechanism causes blood glucose to be eliminated through the urine.[19] In clinical trials, dapagliflozin lowered HbA1c by 0.6 versus placebo percentage points when added to metformin.[20]
Regarding its protective effects in heart failure, this is attributed primarily to haemodynamic effects, where SGLT2 inhibitors potently reduce intravascular volume through osmotic diuresis and natriuresis. This consequently may lead to a reduction in preload and afterload, thereby alleviating cardiac workload and improving left ventricular function.[21]
Selectivity
The IC50 for SGLT2 is less than one thousandth of the IC50 for SGLT1 (1.1 versus 1390 nmol/L), so that the drug does not interfere with intestinal glucose absorption.[22]
Names
Dapagliflozin is the International nonproprietary name (INN),[23] and the United States Adopted Name (USAN).[24]
There is a fixed-dose combination product dapagliflozin/metformin extended-release, called Xigduo XR.[25][26][27]
In July 2016, the fixed-dose combination of saxagliptin and dapagliflozin was approved for medical use in the European Union and is sold under the brand name Qtern.[28] The combination drug was approved for medical use in the United States in February 2017, where it is sold under the brand name Qtern.[29][30]
In May 2019, the fixed-dose combination of dapagliflozin, saxagliptin, and metformin hydrochloride as extended-release tablets was approved in the United States to improve glycemic control in adults with type 2 diabetes when used in combination with diet and exercise. The FDA granted the approval of Qternmet XR to AstraZeneca.[31] The combination drug was approved for use in the European Union in November 2019, and is sold under the brand name Qtrilmet.[32]
History
In 2012, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) issued a positive opinion on the drug.[5]
Dapagliflozin was found effective in several studies in participants with type 2 and type 1 diabetes.[5] The main measure of effectiveness was the level of glycosylated haemoglobin (HbA1c), which gives an indication of how well blood glucose is controlled.[5]
In two studies involving 840 participants with type 2 diabetes, dapagliflozin when used alone decreased HbA1c levels by 0.66 percentage points more than placebo (a dummy treatment) after 24 weeks.[5] In four other studies involving 2,370 participants, adding dapagliflozin to other diabetes medicines decreased HbA1c levels by 0.54-0.68 percentage points more than adding placebo after 24 weeks.[5]
In a study involving 814 participants with type 2 diabetes, dapagliflozin used in combination with metformin was at least as effective as a sulphonylurea (another type of diabetes medicines) used with metformin.[5] Both combinations reduced HbA1c levels by 0.52 percentage points after 52 weeks.[5]
A long-term study, involving over 17,000 participants with type 2 diabetes, looked at the effects of dapagliflozin on cardiovascular (heart and circulation) disease.[5] The study indicated that dapagliflozin’s effects were in line with those of other diabetes medicines that also work by blocking SGLT2.[5]
In two studies involving 1,648 participants with type 1 diabetes whose blood sugar was not controlled well enough on insulin alone, adding dapagliflozin 5 mg decreased HbA1c levels after 24 hours by 0.37% and by 0.42% more than adding placebo.[5]
Dapagliflozin was approved for medical use in the European Union in November 2012.[5] It is marketed in a number of European countries.[33]
Dapagliflozin was approved for medical use in the United States in January 2014.[34][14]
In 2020, the U.S. Food and Drug Administration (FDA) expanded the indications for dapagliflozin to include treatment for adults with heart failure with reduced ejection fraction to reduce the risk of cardiovascular death and hospitalization for heart failure.[3] It is the first in this particular drug class, sodium-glucose co-transporter 2 (SGLT2) inhibitors, to be approved to treat adults with New York Heart Association’s functional class II-IV heart failure with reduced ejection fraction.[3]
Dapagliflozin was shown in a clinical trial to improve survival and reduce the need for hospitalization in adults with heart failure with reduced ejection fraction.[3] The safety and effectiveness of dapagliflozin were evaluated in a randomized, double-blind, placebo-controlled study of 4,744 participants.[3] The average age of participants was 66 years and more participants were male (77%) than female.[3] To determine the drug’s effectiveness, investigators examined the occurrence of cardiovascular death, hospitalization for heart failure, and urgent heart failure visits.[3] Participants were randomly assigned to receive a once-daily dose of either 10 milligrams of dapagliflozin or a placebo (inactive treatment).[3] After about 18 months, people who received dapagliflozin had fewer cardiovascular deaths, hospitalizations for heart failure, and urgent heart failure visits than those receiving the placebo.[3]
In July 2020, the FDA granted AstraZeneca a Fast Track Designation in the US for the development of dapagliflozin to reduce the risk of hospitalisation for heart failure or cardiovascular death in adults following a heart attack.[35]
In August 2020, it was reported that detailed results from the Phase III DAPA-CKD trial showed that AstraZeneca’s FARXIGA® (dapagliflozin) on top of standard of care reduced the composite measure of worsening of renal function or risk of cardiovascular (CV) or renal death by 39% compared to placebo (p<0.0001) in patients with chronic kidney disease (CKD) Stages 2-4 and elevated urinary albumin excretion. The results were consistent in patients both with and without type 2 diabetes (T2D)[36]
In April 2021, the FDA expanded the indications for dapagliflozin (Farxiga) to include reducing the risk of kidney function decline, kidney failure, cardiovascular death and hospitalization for heart failure in adults with chronic kidney disease who are at risk of disease progression.[14] The efficacy of dapagliflozin to improve kidney outcomes and reduce cardiovascular death in people with chronic kidney disease was evaluated in a multicenter, double-blind study of 4,304 participants.[14]
///////////////////////////////////////////
POST YOUR INTERMEDIATES LIST HERE FOR WORLDWIDE REACH
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
SYN
https://pharmacia.pensoft.net/article/70626/
Synthesis
Dapagliflozin is an approved drug by U.S. Food and Drug Administration (FDA). Dapagliflozin is a representative of SGLT-2 inhibitors, actively considered to cure diabetes type 2. Thus, methodology of dapagliflozin synthesis has rarely published (Ellsworth et al. 2002; Meng 2008). Scheme 1 have shown the general synthetic route for the synthesis of dapagliflozin. Gluconolactone 3 which was protected by trimethylsilyl TMS was treated with aryl lithium. Aryl lithium was obtained by reacting aryl bromide 2 (exchange of Li/Br) with n-BuLi. Methyl C-aryl glucoside 4 was produced by treatment of resulting mixture with methane sulfonic acid in the presence of methanol. Compound 4 was subjected to acetylation in the presence of Ac2O, resulted in the formation of 5 followed by reduction of 5 to 6 with the help of Et3SiH and BF3.OEt2. Finally, dapagliflozin 1 was produced via hydrolysis of 6 by LiOH (Deshpande et al. 2008; Meng 2008).
Ellsworth B, Washburn WN, Sher PM, Wu G, Meng W (2002) C-Aryl glucoside SGLT2 inhibitors and method, Google Patents. https://patents.google.com/patent/US6515117B2/en
Meng W, Ellsworth BA, Nirschl AA, McCann PJ, Patel M, Girotra RN, Wu G, Sher PM, Morrison EP, Biller SA, Zahler R, Deshpande PP, Pullockaran A, Hagan DL, Morgan N, Taylor JR, Obermeier MT, Humphreys WG, Khanna A, Discenza L, Robertson JG, Wang A, Han S, Wetterau JR, Janovitz EB, Flint OP, Whaley JM, Washburn WN (2008) “Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. ” Journal of Medicinal Chemistry 51(5): 1145–1149. https://doi.org/10.1021/jm701272q
Deshpande PP, Ellsworth BA, Singh J, Denzel TW, Lai C, Crispino G, Randazzo ME, Gougoutas JZ (2008) Methods of producing C-aryl glucoside SGLT2 inhibitors, Google Patents. https://patents.google.com/patent/US7375213B2/en

Jun et al. has reported a few improvements to the scheme 1. In the improved methodology scheme 2, trimethylsilyl chloride was added to gluconolactone 7 in the presence of N-methylmorpholine and tetrahydrofuran THF (Horton et al. 1981), followed by the formation of persilyated lactone 3. After completing the reaction of aryl bromide 2 with n-BuLi, added to persilyated lactone 3. Intermediate lactol 8 was produced by treating resulting reaction mixture with trifluoroacetic acid in aqueous form. Then ethyl C-aryl glycoside 9 yielded when subsequently compound 8 was subjected to methane-sulfonic acid in ethyl alcohol. Crude product 9 in the form of oil was secured after the screening of solvents. Jun et al. proposed that more than 98% pure 9 was collected as crystalline solvate after the crystallization of crude oil from n-propanol and n-heptane mixture (Yu et al. 2019). Moreover, Wang et al. proposed that a high extent of diastereoselectivity obtained after the reduction of tetra-O-unprotected methyl C-aryl glucoside by utilizing Et3SiH and BF3.Et2O (Wang et al. 2014). The nature of active pharmaceutical ingredient is amorphous foam which is isolated after the reduction of 9. Production of cocrystalline complex facilitate the isolation and purification of API (Deng et al. 2017). It is concluded that more than 99.7% pure dapagliflozin produced in overall 79% yield, after the crystallization of a mixture consists of n-heptane and ethyl acetate (Yu et al. 2019).
Zheng et al. designed the production methodology of dapagliflozin by introducing NO donor group at the last steps of general route of dapagliflozin synthesis (scheme 1). Novel hybrids achieved by the combination of dapagliflozin and NO donor, having excellent dual characteristics of anti-hyperglycemic and anti-thrombosis. The figure 2 represent the modifiable site (4-position) of dapagliflozin.
Horton D, Priebe W (1981) “Synthetic routes to higher-carbon sugars. Reaction of lactones with 2-lithio-, 3-dithiane. ” Carbohydrate Research 94(1): 27–41. https://doi.org/10.1016/S0008-6215(00)85593-7
Yu J, Cao Y, Yu H, Wang JJ (2019) “A Concise and Efficient Synthesis of Dapagliflozin. ” Organic Process Research & Development 23(7): 1458–1461. https://doi.org/10.1021/acs.oprd.9b00141
Wang X-j, Zhang L, Byrne D, Nummy L, Weber D, Krishnamurthy D, Yee N, Senanayake CH (2014) “Efficient synthesis of empagliflozin, an inhibitor of SGLT-2, utilizing an AlCl3-promoted silane reduction of a β-glycopyranoside. ” Organic Letters 16(16): 4090–4093. https://doi.org/10.1021/ol501755h
Deng J-H, Lu T-B, Sun CC, J-M Chen (2017) “Dapagliflozin-citric acid cocrystal showing better solid state properties than dapagliflozin. ” European Journal of Pharmaceutical Sciences 104: 255–261. https://doi.org/10.1016/j.ejps.2017.04.008

Scheme 3 has shown the formation strategy of new hybrids of nitric oxide with dapagliflozin. During the synthesis process, the compound 6 was treated with BBr3 that result in the formation of phenol 10, which was further subjected to condensation with bromoalkane and then undergo hydrolysis and produce 11 intermediates. Target compound was obtained by the reacting 11 with silver nitrate in acetonitrile (Li et al. 2018).
Li Z, Xu X, Deng L, Liao R, Liang R, Zhang B, Zhang LJB (2018) Design, synthesis and biological evaluation of nitric oxide releasing derivatives of dapagliflozin as potential anti-diabetic and anti-thrombotic agents. Bioorganic & Medicinal Chemistry 26(14): 3947–3952. https://doi.org/10.1016/j.bmc.2018.06.017

Lin et al. fabricated green route (scheme 4) for the production of dapagliflozin. 5-bromo-2-chlorobenzoic acid 12 and gluconolactone were utilized to initiate the synthesis. By taking BF3.Et2O in catalytic amount to produce 13, overall yield of 76% was obtained in one-pot way via considering the Friedel-Craft acylation and ketallization. There was no need to do work-up operations to separate the diaryl ketal 13 as it was easily crystallized from the mixture. Compound 14 was produce as a result of condensation between 13 and 3. Overall yield of 68% of compound 15 was produced by the deprotection of silyl group in ethyl alcohol media. In THF presence, single crystals of 15 was achieved and characterized by XRD-analysis. High yield of dapagliflozin was obtained after the reduction of 15 that was carried out by triethylsilane in the presence of boron trifluoride diethyl etherate in dichloromethane. Upon crystallization from the mixture having heptane and ethyl acetate, greater than 98% pure dapagliflozin was produced by green synthetic pathway (Hu et al. 2019).
Hu L, Zou P, Wei W, Yuan X-M, Qiu X-L, Gou S-H (2019) “Facile and green synthesis of dapagliflozin. ” Synthetic Communications 49(23): 3373–3379. https://doi.org/10.1080/00397911.2019.1666283
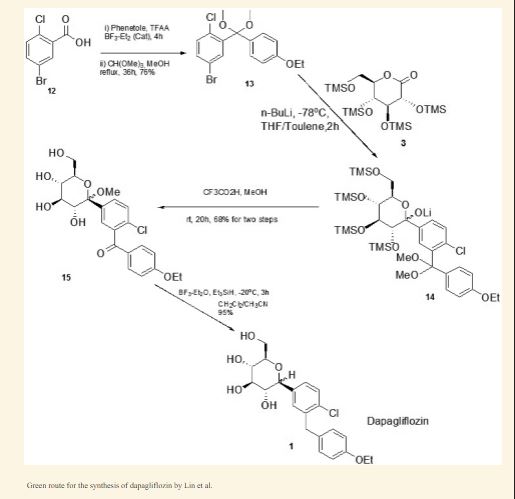
PAPER
A Concise and Efficient Synthesis of Dapagliflozin
Cite this: Org. Process Res. Dev.2019, 23, 7, 1458–1461
Publication Date:June 27, 2019
https://doi.org/10.1021/acs.oprd.9b00141
https://pubs.acs.org/doi/10.1021/acs.oprd.9b00141
file:///C:/Users/Inspiron/Downloads/op9b00141_si_001.pdf SUPP
Abstract

A concise and efficient synthesis of the SGLT-2 inhibitor dapagliflozin (1) has been developed. This route involves ethyl C-aryl glycoside 9 as the key intermediate, which is easily crystallized and purified as the crystalline n-propanol solvate with high purity (>98.5%). The tetra-O-unprotected compound 9 could be directly reduced to crude dapagliflozin with high diastereoselectivity. The final pure API product 1 was isolated and purified with high purity (>99.7%). The process has been implemented on a multikilogram scale.
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetra-hydro-2Hpyran-3,4,5-triol (1)
Compound 1 has a melting point of 88.3oC.
The MsOH solution should be used immediately to avoid sulfonate formation and we have no data on the presence of sulfonates in the API.
1HNMR (400 MHz, DMSO-d6) δ (ppm) 7.32-7.37 (m, 2H), 7.22-7.24 (m, 1H), 7.08-7.10 (m, 2H), 6.80-6.84 (m, 2H), 4.94-4.96 (m, 2H), 4.82-4.83 (d, J=5.6 Hz, 1H), 4.43-4.46 (m, 1H), 3.93-4.02 (m, 5H), 3.68-3.73 (m, 1H), 3.42-3.48 (m, 1H), 3.10-3.28 (m, 4H), 1.27-1.30 (m, 3H).
13C NMR (100 MHz, DMSOd6) δ(ppm) 157.38, 140.14, 138.27, 132.40, 131.69, 131.27, 130.03, 129.13, 127.81, 114.78, 81.67, 81.18, 78.80, 75.19, 70.80, 63.37, 61.86, 38.14, 15.15.
IR(KBr): 3415, 2979, 2918, 1617, 1512, 1475, 1391, 1242, 1103, 1045, 913, 825 cm-1.
MS(m/z):431.12[M + Na]+ .
1H NMR and 13C NMR spectra for Compound
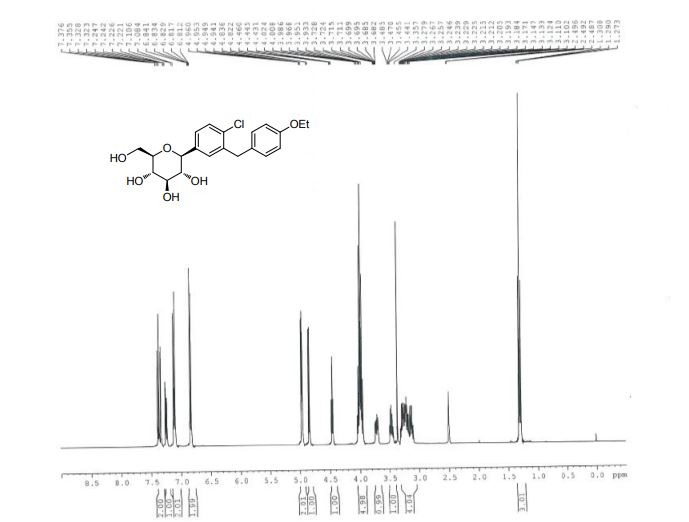
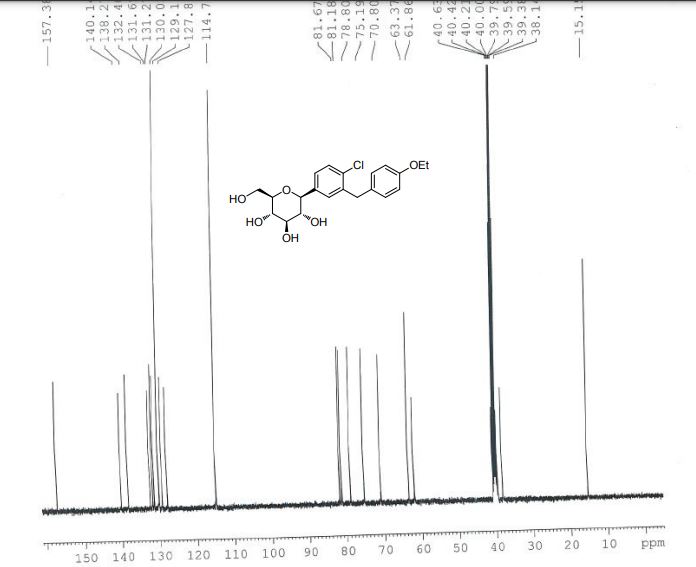
IR and Mass spectra for Compound
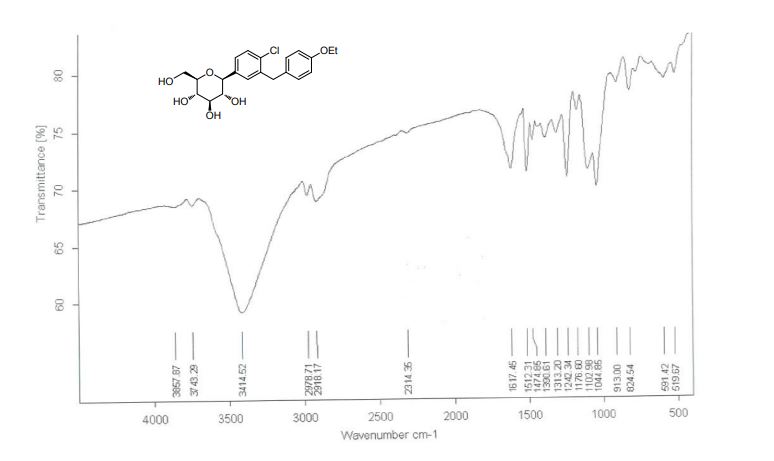
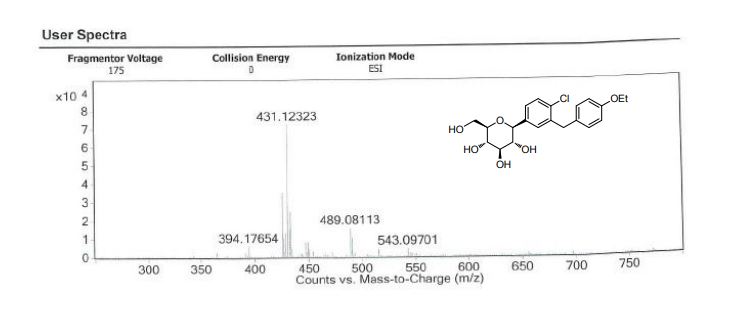
DSC spectra for Compound
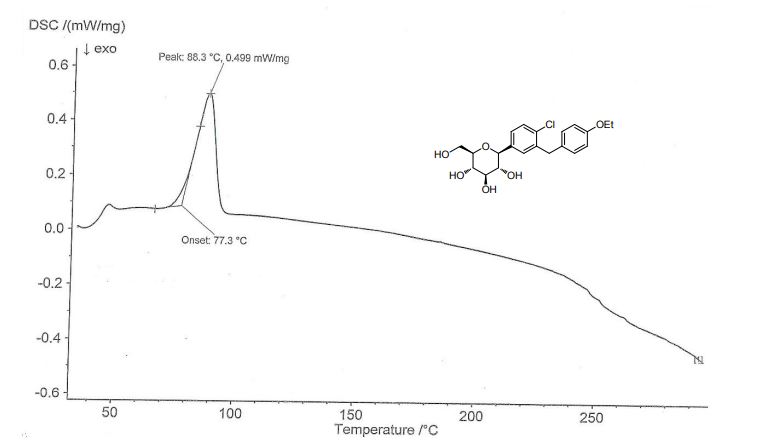
PAPER
https://pubs.acs.org/doi/10.1021/ol300220p
rg. Lett.2012, 14, 6, 1480–1483
Publication Date:March 2, 2012
https://doi.org/10.1021/ol300220p
Abstract

A general, transition-metal-free, highly stereoselective cross-coupling reaction between glycosyl bromides and various arylzinc reagents leading to β-arylated glycosides is reported. The stereoselectivity of the reaction is explained by invoking anchimeric assistance via a bicyclic intermediate. Stereochemical probes confirm the participation of the 2-pivaloyloxy group. Finally, this new method was applied to a short and efficient stereoselective synthesis of Dapagliflozin and Canagliflozin.
CROSS REF J. Med. Chem 2008, 51, 1145
H NMR (360 MHz, MeOD): δ 7.35‐7.28 (m, 3H); 7.09 (d, J=8.4Hz, 2H), 6.80 (d, J=8.8Hz, 2H), 4.09 (d, J=9.5Hz, 1H); 4.03‐3.96 (m, 4H); 3.89‐3.85 (m, 1H); 3.71‐3.66 (m, 1H); 3.45‐3.36 (m, 4H), 3.28‐3.26 (m, 2H); 1.36 (t, J=6.9Hz, 3H).
13CNMR (90 MHz, MeOD): δ 14.80, 38.31, 61.80, 63.38, 69.88, 74.67, 77.93, 79.35, 81.08, 114.48, 126.35, 128.20, 129.01, 129.72, 130.62, 131.14, 134.15, 137.04, 139.01, 157.31.
PAPER
https://pubs.acs.org/doi/10.1021/ja00199a028
Research
One study found that it had no benefit on heart disease risk or overall risk of death in people with diabetes.[37] Another study found that in heart failure with a reduced ejection fraction, dapagliflozin reduced the risk of worsening of heart failure or progression to death from cardiovascular causes, irrespective of diabetic status.[38]
References
- ^ Jump up to:a b “Dapagliflozin (Farxiga) Use During Pregnancy”. Drugs.com. 30 August 2018. Retrieved 5 May 2020.
- ^ Jump up to:a b “Farxiga- dapagliflozin tablet, film coated”. DailyMed. 3 February 2020. Retrieved 5 May 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA approves new treatment for a type of heart failure”. U.S. Food and Drug Administration (FDA) (Press release). 5 May 2020. Retrieved 5 May 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b National Institute for Health and Care Excellence (24 February 2021). “Dapagliflozin for treating chronic heart failure with reduced ejection fraction”. NICE Technology Appraisal Auidance [TA679]. NICE. Retrieved 9 May 2021.
- ^ Jump up to:a b c d e f g h i j k l m n “Forxiga EPAR”. European Medicines Agency (EMA). Retrieved 17 February 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Ptaszynska, Agata; Johnsson, Kristina M.; Parikh, Shamik J.; De Bruin, Tjerk W. A.; Apanovitch, Anne Marie; List, James F. (2014). “Safety Profile of Dapagliflozin for Type 2 Diabetes: Pooled Analysis of Clinical Studies for Overall Safety and Rare Events”. Drug Safety. 37 (10): 815–829. doi:10.1007/s40264-014-0213-4. PMID 25096959. S2CID 24064402.
- ^ Dandona, Paresh; Mathieu, Chantal; Phillip, Moshe; Hansen, Lars; Tschöpe, Diethelm; Thorén, Fredrik; Xu, John; Langkilde, Anna Maria; DEPICT-1 Investigators (2018). “Efficacy and Safety of Dapagliflozin in Patients with Inadequately Controlled Type 1 Diabetes: The DEPICT-1 52-Week Study”. Diabetes Care. 41(12): 2552–2559. doi:10.2337/dc18-1087. PMID 30352894. S2CID 53027785.
- ^ Hu, Yang; Bai, Ziyu; Tang, Yan; Liu, Rongji; Zhao, Bin; Gong, Jian; Mei, Dan (2020). “Fournier Gangrene Associated with Sodium-Glucose Cotransporter-2 Inhibitors: A Pharmacovigilance Study with Data from the U.S. FDA Adverse Event Reporting System”. Journal of Diabetes Research. 2020: 1–8. doi:10.1155/2020/3695101. PMC 7368210. PMID 32695827.
- ^ FARXIGA- dapagliflozin tablet, film coated. DailyMed. Retrieved 6 May 2021.
- ^ “The Top 300 of 2021”. ClinCalc. Retrieved 18 February 2021.
- ^ “Dapagliflozin – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ “FDA Approves Farxiga to Treat Type 2 Diabetes” (Press release). U.S. Food and Drug Administration (FDA). 8 January 2014. Archived from the original on 9 January 2014. Retrieved 15 November 2016. This article incorporates text from this source, which is in the public domain.
- ^ Zelniker TA, Wiviott SD, Raz I, et al. (January 2019). “SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials”. Lancet. 393(10166): 31–9. doi:10.1016/S0140-6736(18)32590-X. PMID 30424892. S2CID 53277899.
However, in patients with atherosclerotic cardiovascular disease, the effect of empagliflozin on cardiovascular death was more pro-nounced than that of canagliflozin or dapagliflozin
- ^ Jump up to:a b c d “FDA Approves Treatment for Chronic Kidney Disease”. U.S. Food and Drug Administration (FDA) (Press release). 30 April 2021. Retrieved 30 April 2021. This article incorporates text from this source, which is in the public domain.
- ^ “FDA warns about rare occurrences of a serious infection of the genital area with SGLT2 inhibitors for diabetes”. U.S. Food and Drug Administration (FDA). 9 February 2019. This article incorporates text from this source, which is in the public domain.
- ^ “SGLT2 inhibitors: Drug Safety Communication – FDA Warns Medicines May Result in a Serious Condition of Too Much Acid in the Blood”. U.S. Food and Drug Administration (FDA). 15 May 2015. Archived from the original on 27 October 2016. Retrieved 15 November 2016. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “FDA revises labels of SGLT2 inhibitors for diabetes to include warning”. U.S. Food and Drug Administration. 19 March 2020. Retrieved 6 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ McMurray, John J.V.; Wheeler, David C.; Stefánsson, Bergur V.; Jongs, Niels; Postmus, Douwe; Correa-Rotter, Ricardo; Chertow, Glenn M.; Greene, Tom; Held, Claes; Hou, Fan-Fan; Mann, Johannes F.E.; Rossing, Peter; Sjöström, C. David; Toto, Roberto D.; Langkilde, Anna Maria; Heerspink, Hiddo J.L.; DAPA-CKD Trial Committees Investigators (2021). “Effect of Dapagliflozin on Clinical Outcomes in Patients with Chronic Kidney Disease, with and Without Cardiovascular Disease” (PDF). Circulation. 143 (5): 438–448. doi:10.1161/CIRCULATIONAHA.120.051675. PMID 33186054. S2CID 226948086.
- ^ “Life Sciences – Clarivate”. Clarivate. Archived from the original on 5 November 2007.
- ^ “UEndocrine: Internet Endocrinology Community”. uendocrine.com. Archived from the original on 5 February 2013.
- ^ Lan NS, Fegan PG, Yeap BB, Dwivedi G (October 2019). “The effects of sodium-glucose cotransporter 2 inhibitors on left ventricular function: current evidence and future directions”. ESC Heart Fail. 6 (5): 927–935. doi:10.1002/ehf2.12505. PMC 6816235. PMID 31400090.
- ^ Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 59” (PDF). World Health Organization. 2008. p. 50. Retrieved 15 November 2016.
- ^ “Statement on a Nonproprietary Name Adopted by the USAN Council” (PDF). American Medical Association. Archived from the original (PDF) on 7 February 2012. Retrieved 15 November2016.
- ^ “US FDA Approves Once-Daily Xigduo XR Tablets for Adults with Type 2 Diabetes”. AstraZeneca. 30 October 2014.
- ^ “Drug Approval Package: Xigduo XR (dapagliflozin and metformin HCl) Extended-Release Tablets”. U.S. Food and Drug Administration (FDA). 7 April 2015. Retrieved 5 May 2020.
- ^ “Xigduo XR- dapagliflozin and metformin hydrochloride tablet, film coated, extended release”. DailyMed. 3 February 2020. Retrieved 5 May 2020.
- ^ “Qtern EPAR”. European Medicines Agency (EMA). Retrieved 7 May 2020.
- ^ “Drug Approval Package: Qtern (dapagliflozin and saxagliptin)”. U.S. Food and Drug Administration (FDA). 10 October 2018. Retrieved 8 May 2020.
- ^ “Qtern- dapagliflozin and saxagliptin tablet, film coated”. DailyMed. 24 January 2020. Retrieved 17 February 2020.
- ^ “Drug Approval Package: Qternmet XR”. U.S. Food and Drug Administration (FDA). 27 January 2020. Retrieved 17 February2020.
- ^ “Qtrilmet EPAR”. European Medicines Agency (EMA). Retrieved 30 March 2020.
- ^ “Forxiga”. Drugs.com. 4 May 2020. Retrieved 5 May 2020.
- ^ “Drug Approval Package: Farxiga (dapagliflozin) Tablets NDA #202293”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 5 May 2020.
- ^ “FARXIGA Granted Fast Track Designation in the US for Heart Failure Following Acute Myocardial Infarction Leveraging an Innovative Registry-Based Trial Design”. http://www.businesswire.com. 16 July 2020. Retrieved 20 July 2020.
- ^https://www.businesswire.com/news/home/20200830005009/en/FARXIGA-Demonstrated-Unprecedented-Reduction-Risk-Kidney-Failure
- ^ “Type 2 diabetes. Cardiovascular assessment of dapagliflozin: no advance”. Prescrire International. 29 (211): 23. January 2020. Retrieved 2 February 2020.
- ^ McMurray JJ, Solomon SD, Inzucchi SE, et al. (November 2019). “Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction”. New England Journal of Medicine. 381 (21): 1995–2008. doi:10.1056/NEJMoa1911303. PMID 31535829.
External links
- “Dapagliflozin”. Drug Information Portal. U.S. National Library of Medicine.
- “Dapagliflozin mixture with metformin hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
- “Dapagliflozin mixture with saxagliptin”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trials
- Clinical trial number NCT00528372 for “A Phase III Study of BMS-512148 (Dapagliflozin) in Patients With Type 2 Diabetes Who Are Not Well Controlled With Diet and Exercise” at ClinicalTrials.gov
- Clinical trial number NCT00643851 for “An Efficacy & Safety Study of BMS-512148 in Combination With Metformin Extended Release Tablets” at ClinicalTrials.gov
- Clinical trial number NCT00859898 for “Study of Dapagliflozin in Combination With Metformin XR to Initiate the Treatment of Type 2 Diabetes” at ClinicalTrials.gov
- Clinical trial number NCT00528879 for “A Phase III Study of BMS-512148 (Dapagliflozin) in Patients With Type 2 Diabetes Who Are Not Well Controlled on Metformin Alone” at ClinicalTrials.gov
- Clinical trial number NCT00660907 for “Efficacy and Safety of Dapagliflozin in Combination With Metformin in Type 2 Diabetes Patients” at ClinicalTrials.gov
- Clinical trial number NCT00680745 for “Efficacy and Safety of Dapagliflozin in Combination With Glimepiride (a Sulphonylurea) in Type 2 Diabetes Patients” at ClinicalTrials.gov
- Clinical trial number NCT01392677 for “Evaluation of Safety and Efficacy of Dapagliflozin in Subjects With Type 2 Diabetes Who Have Inadequate Glycaemic Control on Background Combination of Metformin and Sulfonylurea” at ClinicalTrials.gov
- Clinical trial number NCT00683878 for “Add-on to Thiazolidinedione (TZD) Failures” at ClinicalTrials.gov
- Clinical trial number NCT00984867 for “Dapagliflozin DPPIV Inhibitor add-on Study” at ClinicalTrials.gov
- Clinical trial number NCT00673231 for “Efficacy and Safety of Dapagliflozin, Added to Therapy of Patients With Type 2 Diabetes With Inadequate Glycemic Control on Insulin” at ClinicalTrials.gov
- Clinical trial number NCT02229396 for “Phase 3 28-Week Study With 24-Week and 52-week Extension Phases to Evaluate Efficacy and Safety of Exenatide Once Weekly and Dapagliflozin Versus Exenatide and Dapagliflozin Matching Placebo” at ClinicalTrials.gov
- Clinical trial number NCT02413398 for “A Study to Evaluate the Effect of Dapagliflozin on Blood Glucose Level and Renal Safety in Patients With Type 2 Diabetes (DERIVE)” at ClinicalTrials.gov
- Clinical trial number NCT01730534 for “Multicenter Trial to Evaluate the Effect of Dapagliflozin on the Incidence of Cardiovascular Events (DECLARE-TIMI58)” at ClinicalTrials.gov
- Clinical trial number NCT03036124 for “Study to Evaluate the Effect of Dapagliflozin on the Incidence of Worsening Heart Failure or Cardiovascular Death in Patients With Chronic Heart Failure (DAPA-HF)” at ClinicalTrials.gov
///////////DAPAGLIFLOZIN, ダパグリフロジン, BMS 512148, TYPE 2 DIABETES, SGLT-2 Inhibitors, EU 2012, forxiga, FDA 2014, JAPAN 2014, DIABETES
- Statement on a nonproprietory name adopted by the USAN council
- Efficacy and Safety of Dapagliflozin, Added to Therapy of Patients With Type 2 Diabetes With Inadequate Glycemic Control on Insulin, ClinicalTrials.gov, April 2009
- Trial Details for Trial MB102-020, Bristol-Myers Squibb, May 2009
- “FDA panel advises against approval of dapagliflozin”. 19 July 2011.
- Prous Science: Molecule of the Month November 2007
- UEndocrine: Internet Endocrinology Community
- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- more1) Pal, Manojit et al; Improved Process for the preparation of SGLT2 inhibitor dapagliflozin via glycosylation of 5-bromo-2-Chloro-4′-ethoxydiphenylmethane with Gluconolactone ;. Indian Pat Appl,. 2010CH03942 , 19 Oct 20122) Lemaire, Sebastien et al; Stereoselective C-Glycosylation Reactions with Arylzinc Reagents ;
- Organic Letters , 2012, 14 (6), 1480-1483;3) Zhuo, Biqin and Xing, Xijuan; Process for preparation of Dapagliflozin amino acid cocrystals ;
- Faming Zhuanli Shenqing , 102 167 715, 31 Aug 20114) Shao, Hua et al; Total synthesis of SGLT2 inhibitor Dapagliflozin ;
- Hecheng Huaxue , 18 (3), 389-392; 20105) Liou, Jason et al; Processes for the preparation of C-Aryl glycoside amino acid complexes as potential SGLT2 Inhibitors ;. PCT Int Appl,.
- WO20100223136) Seed, Brian et al; Preparation of Deuterated benzyl-benzene glycosides having an inhibitory Effect on sodium-dependent glucose co-transporter; . PCT Int Appl,.
- WO20100092437) Song, Yanli et al; Preparation of benzylbenzene glycoside Derivatives as antidiabetic Agents ;. PCT Int Appl,.
- WO20090265378) Meng, Wei et al; D iscovery of Dapagliflozin: A Potent, Selective Renal Sodium-Dependent Glucose cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes ;
- Journal of Medicinal chemistr y, 2008, 51 (5), 1145 -1149;9) Gougoutas, Jack Z. et al; Solvates Crystalline complexes of amino acid with (1S)-1 ,5-anhydro-LC (3 – ((phenyl) methyl) phenyl)-D-glucitol were prepared as for SGLT2 Inhibitors the treatment of Diabetes ;. PCT Int Appl,.
- WO200800282410) Deshpande, Prashant P. et al; Methods of producing C-Aryl glucoside SGLT2 Inhibitors ;..
- U.S. Pat Appl Publ,. 20,040,138,439
NEW DRUG APPROVALS
one time
$10.00
Fosnetupitant

Fosnetupitant
- Molecular FormulaC31H35F6N4O5P
- Average mass688.598 Da
[4-[5-[[2-[3,5-bis(trifluoromethyl)phenyl]-2-methylpropanoyl]-methylamino]-4-(2-methylphenyl)pyridin-2-yl]-1-methylpiperazin-1-ium-1-yl]methyl hydrogen phosphate(4-{5-[{2-[3,5-Bis(trifluoromethyl)phenyl]-2-methylpropanoyl}(methyl)amino]-4-(2-methylphenyl)-2-pyridinyl}-1-methylpiperazin-1-ium-1-yl)methyl hydrogen phosphate07-PNET10146
CAS 1703748-89-3
HCL 1643757-72-5
FDA 2014 AND EMA 2015Фоснетупитант [Russian] [INN]فوسنيتوبيتانت [Arabic] [INN]磷奈匹坦 [Chinese] [INN]
- 07-PNET
In April 2018, the U.S. Food and Drug Administration (FDA) and the Swiss company Helsinn approved the intravenous formulation of AKYNZEO® (NEPA, a fixed antiemetic combination of fosnetupitant, 235mg, and palonosetron, 0.25mg) as an alternative treatment option for patients experiencing chemotherapy-induced nausea and vomiting. Fosnetupitant is the pro-drug form of netupitant. Generally, 25% to 30% of patients with a diagnosis of cancer receive chemotherapy as a treatment modality and 70% to 80% of these patients undergoing chemotherapy treatment may experience nausea and vomiting as major side effects. Considered one of the most distressing side effects of chemotherapy, nausea and vomiting has an immense impact on the quality of life of patients receiving certain antineoplastic therapies.
In April 2018, the U.S. Food and Drug Administration (FDA) and the Swiss company Helsinn approved the intravenous formulation of AKYNZEO® (NEPA, a fixed antiemetic combination of fosnetupitant, 235mg, and palonosetron, 0.25mg) as an alternative treatment option for patients experiencing chemotherapy-induced nausea and vomiting 3. Fosnetupitant is the pro-drug form of netupitant Label.
Generally, 25% to 30% of patients with a diagnosis of cancer receive chemotherapy as a treatment modality and 70% to 80% of these patients undergoing chemotherapy treatment may experience nausea and vomiting as major side effects. Considered one of the most distressing side effects of chemotherapy, nausea and vomiting has an immense impact on the quality of life of patients receiving certain antineoplastic therapies 1.
Fosnetupitant: Fosnetupitant is a selective antagonist of human substance P/neurokinin 1 (NK-1) receptors. Upon intravenous administration, Fosnetupitant is converted by phosphatases to its active form. It competitively binds to and blocks the activity of NK-1 receptors in the central nervous system, by inhibiting binding of substance P (SP) to NK-1 receptors. This prevents delayed emesis, which is associated with SP secretion. AKYNZEO is a combination of palonosetron, a serotonin-3 receptor antagonist, and Fosnetupitant (capsules for oral use) or Fosnetupitant (injections for intravenous use). AKYNZEO for injection is indicated in combination with dexamethasone in adults for the prevention of acute and delayed nausea and vomiting associated with initial and repeat courses of highly emetogenic cancer chemotherapy.
EMA
The chemical name of fosnetupitant chloride hydrochloride is 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)- N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1- ium chloride hydrochloride is corresponding to the molecular formula C31H37Cl2N4O5P. It has a relative molecular mass of 761.53 g/mol and the following structure:
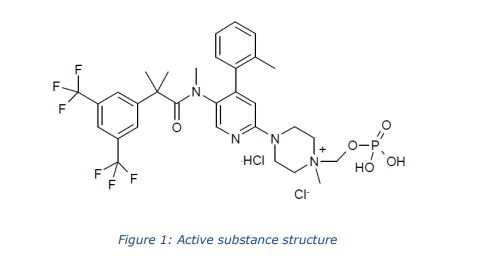
The chemical structure of fosnetupitant chloride hydrochloride was elucidated by a combination of 1H and 13C NMR spectroscopy, infrared spectroscopy, mass spectrometry and elemental analysis. The active substance is achiral. The solid state properties of the active substance were measured by gravimetric vapour sorption and x-ray powder diffraction (XRPD). It is a white to off-white to yellowish, crystalline, hygroscopic solid. Three polymorphic forms have been identified following extensive screening, requiring isolation from different solvent mixtures. Fosnetupitant chloride hydrochloride is always isolated as form I following the commercial manufacturing process. Since it is dissolved and lyophilised during finished product manufacture, particle size and polymorphic form are not considered critical quality attributes (CQAs) of the active substance and are not included in the specification.
RX
AKYNZEO (300 mg netupitant/0.5 mg palonosetron) capsules are an oral combination product of netupitant, a substance P/neurokinin 1 (NK-1) receptor antagonist, and palonosetron hydrochloride, a serotonin-3 (5-HT3) receptor antagonist. Both netupitant and palonosetron hydrochloride are anti-nausea and anti-emetic agents.
Netupitant is chemically described: 2-[3,5-bis(trifluoromethyl)phenyl]-N, 2 dimethyl-N-[4-(2methylphenyl)-6-(4-methylpiperazin-1-yl)pyridin-3-yl] propanamide. The empirical formula is C30H32F6N4O, with a molecular weight of 578.61. Netupitant exists as a single isomer and has the following structural formula:
 |
Palonosetron hydrochloride is chemically described: (3aS)-2-[(S)-1-Azabicyclo [2.2.2]oct-3-yl]2,3,3a,4,5,6-hexahydro-1-oxo-1H-benz[de]isoquinoline hydrochloride. The empirical formula is C19H24N2O.HCl, with a molecular weight of 332.87. Palonosetron hydrochloride exists as a single isomer and has the following structural formula:
 |
Netupitant is white to off-white crystalline powder. It is freely soluble in toluene and acetone, soluble in isopropanol and ethanol, and very slightly soluble in water.
Palonosetron hydrochloride is a white to off-white crystalline powder. It is freely soluble in water, soluble in propylene glycol, and slightly soluble in ethanol and 2-propanol.
Each AKYNZEO capsule is composed of one white-caramel hard gelatin capsule which contains three tablets each containing 100 mg netupitant and one gelatin capsule containing 0.5 mg palonosetron (equivalent to 0.56 mg palonosetron hydrochloride). The inactive ingredients are butylated hydroxyanisole (BHA), croscarmellose sodium, gelatin, glycerin, magnesium stearate, microcrystalline cellulose, mono-and di-glycerides of capryl/capric acid, polyglyceryl dioleate, povidone K-30, purified water, red iron oxide, silicon dioxide, sodium stearyl fumarate, sorbitol, sucrose fatty acid esters, titanium dioxide and yellow iron oxide. It may contain traces of medium-chain triglycerides, lecithin, and denatured ethanol.
AKYNZEO (235 mg fosnetupitant/0.25 mg palonosetron) for injection is a combination product of fosnetupitant, a prodrug of netupitant, which is a substance P/neurokinin 1 (NK-1) receptor antagonist, and palonosetron hydrochloride, a serotonin-3 (5-HT3) receptor antagonist.
Fosnetupitant chloride hydrochloride is chemically described as 2-(3,5-bistrifluoromethylphenyl)-N-methyl-N-[6-(4-methyl-4-O-methylene-phosphatepiperazinium-1-yl)4-o-tolyl-pyridin-3-yl]-isobutyramide chloride hydrochloride. The empirical formula is C31H36F6N4O5P•Cl•HCl, with a molecular weight of 761.53. Fosnetupitant chloride hydrochloride exists as a single isomer and has the following structural formula:
 |
Fosnetupitant chloride hydrochloride is white to off-white to yellowish solid or powder. Its solubility is pH dependent: at acidic pH (pH 2), its solubility is 1.4 mg/mL; at basic pH (pH 10), its solubility is 11.5 mg/mL.
Palonosetron hydrochloride is described above in this section.
AKYNZEO for injection is available for intravenous infusion, and is supplied as a sterile lyophilized powder in a single-dose vial. Each vial contains 235 mg of fosnetupitant (equivalent to 260 mg fosnetupitant chloride hydrochloride) and 0.25 mg of palonosetron (equivalent to 0.28 mg of palonosetron hydrochloride). The inactive ingredients are edetate disodium (6.4 mg), mannitol (760 mg), sodium hydroxide and/or hydrochloric acid (for pH adjustment).
PATENT
WO 2013082102
https://patents.google.com/patent/WO2013082102A1/un



PATENT
US 20150011510
https://patents.google.com/patent/US20150011510A1/en
Step 1:
- [0160]
13.0 g (82.5 mMol) 6-Chloro-nicotinic acid in 65 ml THF were cooled to 0° C. and 206.3 ml (206.3 mMol) o-tolylmagnesium chloride solution (1M in THF) were added over 45 minutes. The solution obtained was further stirred 3 hours at 0° C. and overnight at room temperature. It was cooled to −60° C. and 103.8 ml (1.8 Mol) acetic acid were added, followed by 35 ml THF and 44.24 g (165 mMol) manganese(III) acetate dihydrate. After 30 minutes at −60° C. and one hour at room temperature, the reaction mixture was filtered and THF removed under reduced pressure. The residue was partitioned between water and dichloromethane and extracted. The crude product was filtered on silica gel (eluent: ethyl acetate/toluene/formic acid 20:75:5) then partitioned between 200 ml aqueous half-saturated sodium carbonate solution and 100 ml dichloromethane. The organic phase was washed with 50 ml aqueous half-saturated sodium carbonate solution. The combined aqueous phases were acidified with 25 ml aqueous HCI 25% and extracted with dichloromethane. The organic extracts were dried (Na2SO4) and concentrated under reduced pressure to yield 10.4 g (51%) of 6-chloro-4-o-tolyl-nicotinic acid as a yellow foam. MS (ISN): 246 (M−H, 100), 202 (M-CO2H, 85), 166 (36).
Step 2:
- [0161]
To a solution of 8.0 g (32.3 mMol) 6-chloro-4-o-tolyl-nicotinic acid in 48.0 ml THF were added 3.1 ml (42.0 mMol) thionylchloride and 143 .mu.l (1.8 mMol) DMF. After 2 hours at 50° C., the reaction mixture was cooled to room temperature and added to a solution of 72.5 ml aqueous ammonium hydroxide 25% and 96 ml water cooled to 0° C. After 30 minutes at 0° C., THF was removed under reduced pressure and the aqueous layer was extracted with ethyl acetate. Removal of the solvent yielded 7.8 g (98%) 6-chloro-4-o-tolyl-nicotinamide as a beige crystalline foam. MS (ISP): 247 (M+H+, 100).
Step 3:
- [0162]
1.0 g (4.05 mMol) 6-Chloro-4-o-tolyl-nicotinamide in 9.0 ml 1-methyl-piperazine was heated to 100° C. for 2 hours. The excess N-methyl-piperazine was removed under high vacuum and the residue was filtered on silica gel (eluent: dichloromethane) to yield 1.2 g (95%) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-nicotinamide as a light yellow crystalline foam. - [0163]
MS (ISP): 311 (M+H+, 100), 254 (62).
Step 4:
- [0164]
A solution of 0.2 g (0.6 mMol) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-nicotinamide in 1.0 ml methanol was added to a solution of 103 mg (2.6 mMol) sodium hydroxide in 1.47 ml (3.2 mMol) NaOCl (13%) and heated for 2 hours at 70° C. After removal of methanol, the aqueous layer was extracted with ethyl acetate. The combined organic extracts were dried (Na2SO4), concentrated under reduced pressure and the residue filtered on silica gel (eluent: dichloromethane/methanol 4:1) to yield 100 mg (70%) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-ylamine as a brown resin. MS (ISP): 283 (M+H+, 100), 226 (42).
Step 5:
- [0165]
2.15 mil (11.6 mMol) Sodium methoxide in methanol were added over 30 minutes to a suspension of 0.85 g (4.6 mMol) N-bromosuccinimide in 5.0 ml dichloromethane cooled to −5° C. The reaction mixture was stirred 16 hours at −5° C. Still at this temperature, a solution of 1.0 g (3.1 mMol) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-nicotinamide in 5.0 ml methanol was added over 20 minutes and stirred for 5 hours. 7.1 ml (7.1 mMol) Aqueous HCl 1N and 20 ml dichloromethane were added. The phases were separated and the organic phase was washed with deionized water. The aqueous phases were extracted with dichloromethane, brought to pH=8 with aqueous NaOH 1N and further extracted with dichloromethane. The latter organic extracts were combined, dried (Na2SO4) and concentrated to yield 1.08 g (quant.) [6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-carbamic acid methyl ester as a grey foam. - [0166]
MS (ISP): 341 (M+H+, 100), 284 (35).
Step 6:
- [0167]
A solution of 0.5 g (1.4 mMol) [6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-carbamic acid methyl ester in 3.0 ml dichloromethane was added over 10 minutes to a solution of 1.98 ml (6.9 mMol) Red-Al®. (70% in toluene) and 2.5 ml toluene (exothermic, cool with a water bath to avoid temperature to go >50° C.). The reaction mixture was stirred 2 hours at 50° C. in CH2Cl2, extracted with ethyl acetate and cooled to 0° C. 4 ml Aqueous NaOH 1N were carefully (exothermic) added over 15 minutes, followed by 20 ml ethyl acetate. The phases were separated and the aqueous phase was extracted with ethyl acetate. The combined organic extracts were washed with deionized water and brine, dried (Na2SO4) and concentrated under reduced pressure to yield 0.37 g (89%) methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine as an orange resin. MS (ISP): 297 (M+H+, 100).
Synthesis of 2-(3,5-bis-Trifluoromethyl-phenyl)-2-methyl-propionyl Chloride
- [0168]
- [0169]
15.0 g (50 mmol) 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionic acid were dissolved in 127.5 ml dichloromethane in the presence of 0.75 ml DMF. 8.76 ml (2 eq.) Oxalyl chloride were added and after 4.5 hours, the solution was rotary evaporated to dryness. 9 ml Toluene were added and the resulting solution was again rotary evaporated, then dried under high vacuum yielding 16.25 g (quant.) of 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride as a yellow oil of 86% purity according to HPLC analysis. NMR (250 MHz, CDCl3): 7.86 (br s, 1H); 7.77, (br s, 2H, 3Harom); 1.77 (s, 6H, 2 CH3).
Synthesis of 2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide (Netupitant)
- [0170]
- [0171]
A solution of 20 g (67.5 mmol) methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine and 17.5 ml (101 mmol) N-ethyldiisopropylamine in 200 ml dichloromethane was cooled in an ice bath and a solution of 24 g (75 mmol)2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride in 50 ml dichloromethane was added dropwise. The reaction mixture was warmed to 35-40° C. for 3 h, cooled to room temperature again and was stirred with 250 ml saturated sodium bicarbonate solution. The organic layer was separated and the aqueous phase was extracted with dichloromethane. The combined organic layers were dried (magnesium sulfate) and evaporated. The residue was purified by flash chromatography to give 31.6 g (81%) of 2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide as white crystals. - [0172]
M.P. 155-157° C.; MS m/e (%): 579 (M+H+, 100).
Synthesis of 5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-2-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridine 1-oxide
Step 1:
- [0174]
The solution of 6-chloropyridin-3-amine (115 g, 0.898 mol) and (Boc)2O (215.4 g, 0.988 mol) in 900 mL of dioxane was refluxed overnight. The resulting solution was poured into 1500 mL of water. The resulting solid was collected, washed with water and re-crystallized from EtOAc to afford 160 g tert-butyl (6-chloropyridin-3-yl)carbamate as a white solid (Yield: 78.2%).
Step 2:
- [0175]
To the solution of tert-butyl (6-chloropyridin-3-yl)carbamate (160 g, 0.7 mol) in 1 L of anhydrous THF was added n-BuLi (600 mL, 1.5 mol) at −78° C. under N2 atmosphere. After the addition was finished, the solution was stirred at −78° C. for 30 min, and the solution of I2 (177.68 g, 0.7 mol) in 800 mL of anhydrous THF was added. Then the solution was stirred at −78° C. for 4 hrs. TLC indicated the reaction was over. Water was added for quench, and EtOAc was added to extract twice. The combined organic phases were washed with brine, dried over Na2SO4, filtered and purified by flash chromatography to afford 80 g of tert-butyl (6-chloro-4-iodopyridin-3-yl)carbamate as a yellow solid (32.3%).
Step 3:
- [0176]
To the solution of tert-butyl (6-chloro-4-iodopyridin-3-yl)carbamate (61 g, 0.172 mol) in 300 mL of anhydrous THF was added 60% NaH (7.6 g, 0.189 mol) at 0° C. under N2 atmosphere. After the addition was finished, the solution was stirred for 30 min, and then the solution of MeI (26.92 g, 0.189 mol) in 100 mL of dry THF was added. Then the solution was stirred at 0° C. for 3 hrs. TLC indicated the reaction was over. Water was added for quench, and EtOAc was added to extract twice. The combined organic phases were washed with brine, dried over Na2SO4, filtered and concentrated to afford 63 g of crude tert-butyl (6-chloro-4-iodopyridin-3-yl)(methyl)carbamate used into the following de-protection without the further purification.
Step 4:
- [0177]
To the solution of tert-butyl (6-chloro-4-iodopyridin-3-yl)(methyl)carbamate (62.5 g, 0.172 mol) in 500 mL of anhydrous DCM was added 180 mL of TFA. Then the solution was stirred at room temperature for 4 hrs. Concentrated to remove the solvent, and purified by flash chromatography to afford 45.1 g 6-chloro-4-iodo-N-methylpyridin-3-amine as a yellow solid (Yield: 97.3%).
Step 5:
- [0178]
To the solution of 6-chloro-4-iodo-N-methylpyridin-3-amine (40.3 g, 0.15 mol) and 2-methylbenzene boric acid (24.5 g, 0.18 mol) in 600 mL of anhydrous toluene was added 400 mL of 2 N aq. Na2CO3 solution, Pd(OAc)2 (3.36 g, 15 mmol) and PPh3 (7.87 g, 0.03 mmol). The solution was stirred at 100° C. for 2 hrs. Cooled to room temperature, and diluted with water. EtOAc was added to extract twice. The combined organic phases were washed with water and brine consecutively, dried over Na2SO4, concentrated and purified by flash chromatography to afford 19 g 6-chloro-N-methyl-4-(o-tolyl)pyridin-3-amine as a white solid (Yield: 54.6%).
Step 6:
- [0179]
To the solution of 6-chloro-N-methyl-4-(o-tolyl)pyridin-3-amine (18.87 g, 81.3 mmol) and DMAP (29.8 g, 243.9 mmol) in 200 mL of anhydrous toluene was added the solution of 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride (28.5 g, 89.4 mmol) in toluene under N2 atmosphere. The solution was heated at 120° C. for 23 hrs. Cooled to room temperature, poured into 1 L of 5% aq. NaHCO3 solution, and extracted with EtOAc twice. The combined organic phases were washed by water and brine consecutively, dried over Na2SO4, filtered and purified by flash chromatography to afford 35 g 2-(3,5-bis(trifluoromethyl)phenyl)-N-(6-chloro-4-(o-tolyl)pyridin-3-yl)-N,2-dimethylpropanamide as a white solid (Yield: 83.9%).
Step 7:
- [0180]
To the solution of 2-(3,5-bis(trifluoromethyl)phenyl)-N-(6-chloro-4-(o-tolyl)pyridin-3-yl)-N,2-dimethylpropanamide (5.14 g, 10 mmol) in 60 mL of DCM was added m-CPBA (6.92 g, 40 mmol) at 0° C. under N2 atmosphere. Then the solution was stirred overnight at room temperature. 1 N aq. NaOH solution was added to wash twice for removing the excess m-CPBA and a side product. The organic phase was washed by brine, dried over Na2SO4, filtered and concentrated to afford 5.11 g of crude 5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-2-chloro-4-(o-tolyl)pyridine 1-oxide as a white solid (Yield: 96.4%).
Step 8:
- [0181]
To the solution of crude 5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-2-chloro-4-(o-tolyl)pyridine 1-oxide (5.1 g, 9.62 mmol) in 80 mL of n-BuOH was added N-methylpiperazine (7.41 g, 74.1 mmol) under N2 atmosphere. Then the solution was stirred at 80° C. overnight. Concentrated and purified by flash chromatography to afford 4.98 g 5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-2-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridine 1-oxide as a white solid (Yield: 87.2%). 1HNMR (CDCl3, 400 MHz) δ 8.15 (s, 1H), 7.93 (s, 1H), 7.78 (s, 2H), 7.38 (m, 2H), 7.28 (m, 1H), 7.17 (m, 1H), 7.07 (s, 1H), 5.50 (s, 3H), 2.72 (d, J=4.4 Hz, 4H), 2.57 (m, 3H), 2.40 (s, 3H), 2.23 (s, 3H), 1.45˜1.20 (m, 6H).
Synthesis of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-1-oxido-4-(o-tolyl)pyridin-2-yl)-1-methylpiperazine 1-oxide
- [0182]
- [0183]
To a solution of 5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-2-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridine 1-oxide (3 g, 5.05 mmol) and NaHCO3 (0.354 g, 12.66 mmol) in 60 mL of MeOH and 15 mL of H2O were added potassium monopersulfate triple salt (1.62 g, 26.25 mmol) at room temperature during 15 min. After stirring for 4 hrs at room temperature under N2 atmosphere, the reaction mixture was concentrated in vacuo and purified by flash chromatography (eluent: MeOH). The product was dissolved into DCM, the formed solid was filtered off, and the solution was concentrated under reduced pressure to afford 1.77 g 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-1-oxido-4-(o-tolyl)pyridin-2-yl)-1-methylpiperazine 1-oxide as a white solid (Yield: 57.4%). 1HNMR (CDCl3, 400 MHz) δ 8.06 (s, 1H), 7.78 (s, 1H), 7.60 (s, 2H), 7.37˜7.20 (m, 4H), 6.81 (s, 1H), 3.89 (s, 21H), 3.74 (m, 4H), 3.31 (m, 5H), 2.48 (s, 3H), 2.18 (s, 3H), 1.36 (s, 6H).
Synthesis of 1-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-4-methylpiperazine 1,4-dioxide
- [0184]
- [0185]
To the solution of 2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide (11.1 g, 19.2 mmol) in 75 ml of Methanol was added sodium bicarbonate (3.38 g, 40.3 mmol) dissolved in 20 ml of water. Then Oxone (14.75 g, 48.0 mmol) was added to the stirred solution at room temperature in 3-4 portions. The suspension was heated for 4 h at 50° C. After filtration of the salts (washed with 3×8 ml of methanol), the solvent has been evaporated under reduced pressure and substituted by DCM (30 ml). The organic phase was washed with water (5×30 ml), dried over Na2SO4, filtered, concentrated and purified by precipitation in toluene to afford 9.3 g 1-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-4-methylpiperazine 1,4-dioxide as a white solid (Yield: 80%). 1H-NMR (CDCl3, 400 MHz, at 333K) δ 8.27 (s, 2H), 7.75 (s, 1H), 7.63 (s, 2H), 7.26˜7.19 (m, 2H), 7.14 (t, 1H, J=7.4 Hz), 7.09 (d, 1H, J=7.4 Hz), 4.93 (t, 2H, J=11.6 Hz), 4.70 (t, 2H, J=11.6 Hz), 4.12 (d, 2H, J=10.7 Hz), 3.84 (s, 3H), 3.50 (d, 2H, J=10.3 Hz), 2.47 (s, 3H), 2.12 (s, 3H), 1.40 (s, 6H).
Synthesis (A) of di-tert-butyl (chloromethyl)phosphate
- [0186]
- [0187]
Di-tert-butyl phosphohite (40.36 mmole) was combined with potassium bicarbonate (24.22 mmole) in 35 ml of water. The solution was stirred in an ice bath and potassium permanganate (28.25 mmole) was added in three equal portions over one hour’s time. The reaction as then allowed to continue at room temperature for an additional half hour. - [0188]
Decolorizing carbon (600 mg) was then incorporated as the reaction was heated to 60° C. for 15 minutes. The reaction was then vacuum filtered to remove solid magnesium dioxide. The solid was washed several times with water. The filtrate was then combined with one gram of decolorizing carbon and heated at 60° C. for an additional twenty minutes. The solution was again filtered to yield a colorless solution, which was then evaporated under vacuum to afford crude Di-tert-butyl phosphate potassium salt. Di-tert-butyl phosphate potassium salt (5 g, 20.14 mmole) was dissolved in methanol (15 g): to this solution at 0° C. a slight excess of concentrated HCl is slowly added with efficient stirring at 0° C. The addition of acid causes the precipitation of potassium chloride. The solid is then filtered and washed with methanol. The compound in the mother liquor is then converted to the ammonium form by adding an equal molar amount of tetramethylammonium hydroxide (3.65 g, 20.14 mmole) while keeping the reaction cooled by a salt/ice bath with efficient stirring. The resulting clear solution is placed under reduced pressure to give the crude product. To the tetramethylammonium di-tert-butyl-phosphate dissolved in refluxing dimethoxyethane is then added 4.3 grams of chloroiodomethane (24.16 mmole) and stirred for 1-2 hours. The reaction is then filtered and the filtrate is placed under reduced pressure to concentrate the solution in DME. The chloromethyl di-tert-butyl phosphate 12-16% in DME is used in the synthesis of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1-ium without further purifications (60% yield): 1HNMR (CD3OD, 300 MHz) δ 1.51 (s, 12H), 5.63 (d, 2H, J=14.8). 31P-NMR (CD3OD, 300 MHz) δ −11.3 (s, 1P).
Synthesis (B) of di-tert-butyl (chloromethyl)phosphate
- [0189]
- [0190]
Di-tert-butyl phosphate potassium salt (5 g, 20.14 mmole) is dissolved in methanol (15 g): to this solution at 0° C. a slight excess of concentrated HCl is slowly added with efficient stirring at 0° C. The addition of acid causes the precipitation of potassium chloride. The solid is then filtered and washed with methanol. The compound in the mother liquor is then converted to the ammonium form by adding an equal molar amount of tetrabuthylammonium hydroxide 1 M in methanol (20.14 mmole) while keeping the reaction cooled at 0° C. with efficient stirring. The resulting clear solution is placed under reduced pressure to give the intermediate product. The tetrabuthylammonium di-tert-butyl-phosphate dissolved in acetone is then added dropwise to 53.3 grams of chloroiodomethane (302.1 mmole) and stirred at 40° C. for 1-2 hours. The solvent and excess of chloroiodomethane are distilled off, the reaction mass suspended in TBME and then filtered. The filtrate is washed by a saturated solution of sodium bicarbonate and water and then placed under reduced pressure to substitute the solvent by acetone, i.e., to remove the solvent after which it is replaced with acetone. The chloromethyl di-tert-butyl phosphate 7-20% in acetone is used in the next step without further purifications (70-80% yield): 1H-NMR (CD3OD, 300 MHz) δ 1.51 (s, 12H), 5.63 (d, 2H, J=14.8). 31P-NMR (CD3OD, 300 MHz) δ −11.3 (s, 1P).
Stability studies of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1-ium salts
- [0191]
In order to further improve the stability and solubility of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1-ium, a variety of its derivative salts were synthesized and tested. Their synthesis employed either a) neutralization of the dried diacid phosphate species and its corresponding base salts or b) a direct acid deprotection starting from the dried di(tert-butyl)-protected phosphate species. Neutralization was performed with L-histidine, magnesium salt, N-methyl-D-glucamine (dimeglumine), and L-lysine. Both procedures were tried in the synthesis of citric derivatives whereas with other acids the direct deprotection reaction was used. The figures below show the most relevant structures. - [0192]
When the parent acid species was not stored in dry condition it was found to undergo over 8% degradation in the first week and over 65% degradation in the first six months. When the dried parent acid species was held at 30° C. in air it underwent 0.05% degradation in the first 7 days and at total of 7.03% degradation in six months. When the dried parent acid species was held under argon at room temperature it underwent up to 0.13% degradation in the first 7 days but then was essentially stable for six months. Results for various derivative salts are shown in Table 1 below. - TABLE 1 Representative Degradation Results for Salts Purity A % Solvents Additives Yield % HPLC Comments MeOH L-Histidine, 2 eq. 26.6% 95.94% Degradation: +0.70% in 6 days (in air) +0.46% in 6 days (in argon) MeOH Mg(OH)2, 2 eq. 48.6% 94.11% Degradation: +0.81% in 6 days (in air) +0.29% in 6 days (in argon) MeOH + Citric acid, 2 eq. N.A. 94.40% From protected species. DCM, 1:1 MeOH 1. HCl dioxane, 4 eq. >90% 94.50% From protected species. 2. Ca(OH)2 MeOH H3PO4, 85%, 2 eq. >90% 98.81% From protected species and retains 0.39% of that species. MeOH HBr, 48%, 4 eq. 84.6% 96.11% From protected species. Product degrades rapidly, MeOH + CH3SO3H N.A. 61.54% From protected species. DCM, Product NOT stable: contains 1:4 32.45% decomposition species. MeOH NaH2PO4, 4 eq. N.A. n.d. Only 1.27 of parent species formed. Poor reaction. MeOH N-methyl-D- N.A. 96.88% Degradation: glucamine +0.87% in 6 days (in air) (Meglumine), 2 eq. +1.52% in 11 days (in argon) MeOH N-methyl-D- >99% 97.42% Degradation: glucamine +0.77% in 6 days (in air) (Meglumine), 1 eq. +0.83% in 7 days (in argon) MeOH+ 1. NaOH, 3 eq 96.5% 97.49% Degradation: DCM, 2. Citric acid, 1 eq. +0.09% in 2 days (in argon) 5:2 +0.59% in 89 days (in argon) MeOH+ 1. NaOH, 3 eq. 93.8% 97.46% Degradation: DCM, 2. Fumaric acid, 1 eq. +1.95% in 14 days (in air) 5:2 +1.80% in 12 days (in argon) MeOH L-lysine, 1 eq. >99% 97.62% Degradation: +0.69% in 14 days (in air) +0.48% in 12 days (in argon)
- [0194]
- [0195]
The solution of chloromethyl di-tert-butyl phosphate in DME (250 g from a 10% solution, 96.64 mmole) was evaporated under reduced pressure until the formation of pale yellow oil, dissolved then at 50° C. with 318 ml of Acetonitrile. 17.2 g (80.54 mmole) of 1,8-bis(dimethylamino)naphtalene and 46.6 g (80.54 mmole) of 2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide were added and the solution heated at 90° C. for at least 12 h. After the addition of 75 g of isopropylether, the precipitated crude product was cooled at room temperature, filtered and washed with acetonitrile, isopropylether/acetone, 3:1 and isopropylether, and dried under reduced pressure to afford 20-33 g of the 4-(5-{2-[3,5-bis(trifluoromethyl)phenyl]-N,2-dimethylpropanamido}-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-{[(tert-butoxy)phosphoryl]oxymethyl}piperazin-1-ium as white solid (Yield: 30-50%). 1H-NMR (CD3OD, 400 MHz) δ 7.98 (s, 1H), 7.86 (s, 1H), 7.76 (s, 2H), 7.33-7.10 (m, 4H), 6.80 (s, 1H), 5.03 (d, 2H, JPH=8.5 Hz), 4.52 (s, 2H), 4.13 (m, 2H), 3.83 (m, 2H), 3.69 (m, 2H), 3.52 (m. 2H), 3.23 (s, 3H), 2.53 (s, 3H), 2.18 (s, 3H), 1.46 (s, 18H), 1.39 (s, 6H). 31P-NMR (CD3OD, 161 MHz) δ −5.01 (s, 1P). To 20 g (23.89 mmole) of the 4-(5-{2-[3,5-bis(trifluoromethyl)phenyl]-N,2-dimethylpropanamido}-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-{[(tert-butoxy)phosphoryl]oxymethyl}piperazin-1-ium dissolved in 180 g of methanol and 400 g of dichloromethane was added HCl 4M in dioxane (18.8 g, 71.66 mmole) and the solution was heated for 3 h at reflux. After the addition of 200 g of dioxane, DCM and methanol were distilled under reduced pressure until precipitation of the product, which was filtered and washed with isopropylether (100 g), acetone (30 g) and pentane (2×60 g). The product was finally dried under reduced pressure at 55° C. to afford 15-17 g of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1-ium chloride hydrochloride as white solid (Yield: 88-93%). 1H-NMR (CD3OD, 400 MHz) δ 7.02 (s, 1H), 7.87 (s, 1H), 7.74 (s, 2H), 7.33-7.40 (m, 2H), 7.27 (m, 1H), 7.21 (s, 1H), 7.16 (d, 1H, J=8.2 Hz), 5.27 (d, 2H, JPH=7.9 Hz), 4.29 (m, 2H), 4.05 (m, 2H), 3.85 (m, 2H), 3.74 (m, 2H), 3.35 (s, 3H), 2.62 (s, 3H), 2.23 (s, 3H), 1.38 (s, 6H). 31P-NMR (CD3OD, 161 MHz) δ −2.81 (t, 1P, JPH=7.9 Hz).
Synthesis (B) of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-1-methyl-1-((phosphonooxy)methyl)piperazin-1-ium chloride hydrochloride
- [0196]
- [0197]
To the solution of chloromethyl di-tert-butyl phosphate in Acetone (22.1 g from a 10% solution, 85.58 mmole), 15.5 g (103.24 mmole) of sodium iodide and 33.0 g (57.00 mmole) of netupitant were added and the solution heated at 50° C. for at 6-16 h. The precipitated salts were filtered off, the acetone distilled under reduced pressure and the crude product dissolved in 43.0 g of methanol and 43.0 g 1,4-dioxane. 12.6 g of HCl 4M in dioxane (113.85 mmole) were added, and then methanol is distilled off at 40° C. under reduced pressure. The solution is cooled at 5° C. and stirred at 5° C. for at least 2 h at 5° C. The product was isolated by filtration, purified by additional slurry in acetone (238 g), and filtered and washed with acetone (47 g) and pentane (2×72 g). - [0198]
The product was finally dried under reduced pressure at 60° C. to afford 22-30 g of white-yellowish solid (Yield: 50-70%) - [0199]
1H-NMR (CD3OD, 400 MHz) δ 7.02 (s, 1H), 7.87 (s, 1H), 7.74 (s, 2H), 7.33-7.40 (m, 2H), 7.27 (m, 1H), 7.21 (s, 1H), 7.16 (d, 1H, J=8.2 Hz), 5.27 (d, 2H, JPH=7.9 Hz), 4.29 (m, 2H), 4.05 (m, 2H), 3.85 (m, 2H), 3.74 (m, 2H), 3.35 (s, 3H), 2.62 (s, 3H), 2.23 (s, 3H), 1.38 (s, 6H). 31P-NMR (CD3OD, 161 MHz) δ −2.81 (t, IP, JPH=7.9 Hz).
PATENT
US 8,426,450
PATENT
US 9,403,772
SYN
https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/chem.201901840
The synthesis of fosnetupitant (195) was developed by the Swiss company Helsinn (Scheme 34).[58] The synthesis started with the reaction of 6-chloronicotinic acid (196) with o-tolylmagnesium chloride followed by manganese(III) acetate to give acid derivative 197. This was converted to amide 198 after reaction with thionyl chloride and ammonium hydroxide. Next, reaction with N-methylpiperazine furnished intermediate 199, which was then transformed into carbamate 200 after reaction with NBS in methanol. Reduction with Red-Al followed by acylation with acyl chloride 202 afforded netupitant (203).
Finally, reaction with di-tert-butyl chloromethyl phosphate followed by the removal of the tert-butyl groups by treatment with HCl in dioxane afforded fosnetupitant (195).
L. Fadini, P. Manini, C. Pietra, C. Giuliano, E. Lovati, R. Cannella, S. Venturini, V. J. Stella, WO 082102 A1, 2013.


SYN
Fosnetupitant chloride HCl

PATENT
Fosnetupitant is a neurokynin-1 (“NK-1”) antagonist under development by Helsinn Healthcare SA, Lugano/Pazzallo Switzerland, for the treatment of chemotherapy induced nausea and vomiting. The compound is known chemically as 4-(5-(2-(3,5- bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-l-methyl- 1 -((phosphonooxy)methyl)piperazin- 1 -ium, and has the following chemical structure in its acidic/free base form:

[004] The chloride monohydrochloride salt, and a method for its preparation, is described in WO 2013/082102. The chemical structure for this salt is reported as follows:

[005] The molecule can be challenging to manufacture, particularly in a highly pure crystalline form in a commercially acceptable yield. Solvents used in the manufacture of the product pose special challenges. Prior art processes have removed these solvents via evaporative techniques, which can degrade the fosnetupitant due to the excessive heat.
EXAMPLES
[089] In all the examples reported, unless otherwise reported, the starting compound was Form I of the chloride hydrochloride salt of 4-(5-(2-(3,5-bis(trifluoromethyl)phenyl)- N,2-dimethylpropanamido)-4-(o-tolyl)pyridin-2-yl)-l-methyl-l – ((phosphonooxy)methyl)piperazin-l-ium, produced substantially according to the methods described in WO 2013/082102.
EXAMPLE 1 : CHARACTERIZATION OF FOSNETUPITANT
1 . Experimental Methods
1.1 Solubility
[090] The solubility of the starting compound was determined in 25 pharmaceutically acceptable solvents (class II and III) of differing polarity. The procedure was as follows:
[091] Approximately 20 mg of material was weighed out into each glass vial.
[092] 5 volume aliquots of each solvent were added separately with stirring (i.e. 1 volume = 20 μΐ; hence, 5 volume = 100 μΐ (5 x 20 μΐ)).
[093] The mixture was stirred at RT for 5- 10 minutes. Visual checks were then made for solubility.
[094] If no solubility was achieved then steps (ii) and (iii) were repeated until either the solubility was achieved or the 50 volume aliquots of that solvent were added.
[095] Solubility was then approximated.
[096] Solubility was finally checked at the elevated temperature (40°C).
1.2 Polymorph Screen (including slurry studies)
[097] Using the information from the solubility study, the compound was slurried in the solvents outlined in Table I and two more mixtures of water/ MeOH (10:90) and water/ Acetone (1 :20) respectively with temperature cycling between 40°C and RT (4 hour periods at each temperature) over 48 hours. After the slurries the resulting solids were isolated and analyzed by Raman and XRPD (where enough material was available) for any change in physical form.
[098] The compound was also dissolved in the listed solvents and two more mixtures of water/organic solvent to yield saturated solutions, and crystallization was induced by: crash cooling (at ca. -1 8°C); evaporation (at RT); and addition of an anti-solvent. Solid materials generated were then isolated and examined by Raman and XRPD (where enough material was available).
1.3 Scale-up of any new polymorphic forms
[099] Any new potential polymorphic forms of the Form I fosnetupitant were then scaled-up to ~500mg level for further characterizations by PLM, SEM, DSC, TGA, GVS (XRPD post GVS) and NMR. Further studies of conversion between each polymorphic form were also performed. From this information, an understanding of the polymorphic space was achieved.
Synthetic Reference
Fadini, Luca; Manini, Peter; Pietra, Claudio; Giuliano, Claudio; Lovati, Emanuela; Cannella, Roberta; Venturini, Alessio; Stella, Valentino. (Assignee: Helsinn Healthcare SA, Switz). Substituted 4 – phenyl – pyridines for the treatment of nk-1 receptor related diseases. WO2013082102 (2013).
//////////Fosnetupitant, 07-PNET, Фоснетупитант , فوسنيتوبيتانت , 磷奈匹坦 , FDA 2014, EMA 2015
Tasimelteon, タシメルテオン

Tasimelteon
N-([(1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl]methyl)propanamide,
609799-22-6 [RN]
8985
Hetlioz [Trade name]
N-{[(1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl]methyl}propanamide [ACD/IUPAC Name]
Propanamide, N-[[(1R,2R)-2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]methyl]- [ACD/Index Name]
SHS4PU80D9
609799-22-6 cas, BMS-214778; VEC-162, ATC:N05CH03
- Use:Treatment of sleep disorder; Melatonin receptor agonist
- (1R,2R)-N-[2-(2,3-dihydrobenzofuran-4-yl)cyclopropylmethyl]propanamide
- Formula:C15H19NO2, MW:245.3 g/mol
-
Hetlioz Vanda Pharmaceuticals, 2014
Approved fda 2014
EMA
Tasimelteon is a white to off-white crystalline powder, it is non hygroscopic, soluble in water across relevant pH values and freely soluble in alcohols, cyclohexane, and acetonitrile. Conducted in vivo studies demonstrate that tasimelteon is highly permeable substance. Photostability testing and testing on stress conditions demonstrated that the active substance degrades in light.
Tasimelteon exhibits stereoisomerism due to the presence of two chiral centres. Active substance is manufactured as a single, trans-1R,2R isomer. Enantiomeric purity is controlled routinely during manufacture of active substance intermediates by chiral HPLC/specific optical rotation and additionally controlled in the active substance. Stability data indicates tasimelteon is isomerically stable.
Polymorphism has been observed in polymorphic screening studies for tasimelteon and two forms have been identified. The thermodynamically more stable form has been chosen for development and the manufacturing process consistently yields active substance of single, desired polymorphic form. It was demonstrated that milling of the active substance does not affect polymorphic form. Polymorphism is additionally controlled in active substance release and shelf-life specifications using X-ray powder diffraction analysis.
Tasimelteon is synthesized in nine main steps using linear synthesis and using commercially available well-defined starting materials with acceptable specifications. Three intermediates are isolated for control of active substance quality including stereochemical control. The active substance is isolated by slow recrystallisation or precipitation of tasimelteon from an ethanol/water mixture which ensures the formation of desired polymorphic form. Up to two additional, optional recrystallisations may be performed for unmilled tasimelteon to ensure that milled tasimelteon active substance is of high purity. Seed crystals complying with active substance specifications can be used optionally. Active substance is jet milled (micronised) to reduce and control particle size, which is critical in finished product performance with regards to content uniformity and dissolution…….http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003870/WC500190309.pdf
launched in 2014 in the U.S. by Vanda Pharmaceuticals for the treatment of non-24-hour sleep-wake disorder in totally blind subjects. In 2015, the European Committee for Medicinal Products of the European Medicines Agency granted approval for the same indication. In 2010 and 2011, orphan drug designations were assigned for the treatment of non-24 hour sleep/wake disorder in blind individuals without light perception in the U.S. and the E.U., respectively.
Tasimelteon (trade name Hetlioz) is a drug approved by the U.S. Food and Drug Administration (FDA)[2] in January 2014 for the treatment of non-24-hour sleep–wake disorder (also called Non-24, N24 and N24HSWD).[3] In June 2014, the European Medicines Agency accepted an EU filing application for tasimelteon[4] and in July 2015, the drug was approved in Europe for the treatment of non-24-hour sleep-wake rhythm disorder in totally blind adults,[5] but not in the rarer case of non-24 in sighted people.
Tasimelteon is a selective agonist for the melatonin receptors MT1 and MT2, similar to other members of the melatonin receptor agonistclass of which ramelteon (2005) and agomelatine (2009) were the first approved.[6] As a treatment for N24HSWD, as with melatonin or other melatonin derivatives, the patient may experience improved sleep timing while taking the drug. Reversion to baseline sleep performance occurs within a month of discontinuation.[7]
![]()
Development
Tasimelteon (previously known as BMS-214,778) was developed for the treatment of insomnia and other sleep disorders. A phase II trial on circadian rhythm sleep disorders was concluded in March 2005.[8] A phase III insomnia trial was conducted in 2006.[9] A second phase III trial on insomnia, this time concerning primary insomnia, was completed in June 2008.[10] In 2010, the FDA granted orphan drug status to tasimelteon, then regarded as an investigational medication, for use in totally blind adults with N24HSWD.[11] (Through mechanisms such as easing the approval process and extending exclusivity periods, orphan drug status encourages development of drugs for rare conditions that otherwise might lack sufficient commercial incentive.)
On completion of Phase III trials, interpretations of the clinical trials by the research team concluded that the drug may have therapeutic potential for transient insomnia in circadian rhythm sleep disorders.[12] A year-long (2011–2012) study at Harvard tested the use of tasimelteon in blind subjects with non-24-hour sleep-wake disorder. The drug has not been tested in children nor in any non-blind people.
FDA approval
In May 2013 Vanda Pharmaceuticals submitted a New Drug Application to the Food and Drug Administration for tasimelteon for the treatment of non-24-hour sleep–wake disorder in totally blind people. It was approved by the FDA on January 31, 2014 under the brand name Hetlioz.[3] In the opinion of Public Citizen, an advocacy group, the FDA erroneously allowed it to be labelled without stating that it is only approved for use by totally blind people.[13] However, FDA updated its press release on Oct. 2, 2014 to clarify the approved use of Hetlioz, which includes both sighted and blind individuals. The update did not change the drug labeling (prescribing information).[14]
Toxicity
Experiments with rodents revealed fertility impairments, an increase in certain cancers, and serious adverse events during pregnancy at dosages in excess of what is considered the “human dose”.[15][16]
As expected, advisors to the US Food and Drug Administration have recommended approval of Vanda Pharmaceuticals’ tasimelteon, to be sold as Hetlioz, for the treatment of non-24-hour disorder in the totally blind.http://www.pharmatimes.com/Article/13-11-14/FDA_panel_backs_Vanda_body_clock_drug_for_blind.aspx
The master body clock controls the timing of many aspects of physiology, behavior and metabolism that show daily rhythms, including the sleep-wake cycles, body temperature, alertness and performance, metabolic rhythms and certain hormones which exhibit circadian variation. Outputs from the
suprachiasmatic nucleus (SCN) control many endocrine rhythms including those of melatonin secretion by the pineal gland as well as the control of Cortisol secretion via effects on the hypothalamus, the pituitary and the adrenal glands. This master body clock, located in the SCN, spontaneously generates rhythms of approximately 24.5 hours. These non-24-hour rhythms are synchronized each day to the 24-hour day-night cycle by light, the primary environmental time cue which is detected by specialized cells in the retina and transmitted to the SCN via the retino-hypothalamic tract. Inability to detect this light signal, as occurs in most totally blind individuals, leads to the inability of the master body clock to be reset daily and maintain entrainment to a 24-hour day.
Non-24-Hour Disorder, Non-24, also referred to as Non-24-Hour Sleep-Wake Disorder, (N24HSWD) or Non-24-Hour Disorder, is an orphan indication affecting approximately 65,000 to 95,000 people in the U.S. and 140,000 in Europe. Non- 24 occurs when individuals, primarily blind with no light perception, are unable to synchronize their endogenous circadian pacemaker to the 24-hour light/dark cycle. Without light as a synchronizer, and because the period of the internal clock is typically a little longer than 24 hours, individuals with Non-24 experience their circadian drive to initiate sleep drifting later and later each day. Individuals with Non-24 have abnormal night sleep patterns, accompanied by difficulty staying awake during the day. Non-24 leads to significant impairment, with chronic effects impacting the social and occupational functioning of these individuals.
In addition to problems sleeping at the desired time, individuals with Non-24 experience excessive daytime sleepiness that often results in daytime napping.
The severity of nighttime sleep complaints and/or daytime sleepiness complaints varies depending on where in the cycle the individual’s body clock is with respect to their social, work, or sleep schedule. The “free running” of the clock results in approximately a 1-4 month repeating cycle, the circadian cycle, where the circadian drive to initiate sleep continually shifts a little each day (about 15 minutes on average) until the cycle repeats itself. Initially, when the circadian cycle becomes desynchronous with the 24h day-night cycle, individuals with Non-24 have difficulty initiating sleep. As time progresses, the internal circadian rhythms of these individuals becomes 180 degrees out of synchrony with the 24h day-night cycle, which gradually makes sleeping at night virtually impossible, and leads to extreme sleepiness during daytime hours.
Eventually, the individual’s sleep-wake cycle becomes aligned with the night, and “free-running” individuals are able to sleep well during a conventional or socially acceptable time. However, the alignment between the internal circadian rhythm and the 24-hour day-night cycle is only temporary.
In addition to cyclical nighttime sleep and daytime sleepiness problems, this condition can cause deleterious daily shifts in body temperature and hormone secretion, may cause metabolic disruption and is sometimes associated with depressive symptoms and mood disorders.
It is estimated that 50-75% of totally blind people in the United States (approximately 65,000 to 95,000) have Non-24. This condition can also affect sighted people. However, cases are rarely reported in this population, and the true rate of Non-24 in the general population is not known.
The ultimate treatment goal for individuals with Non-24 is to entrain or synchronize their circadian rhythms into an appropriate phase relationship with the 24-hour day so that they will have increased sleepiness during the night and increased wakefulness during the daytime. Tasimelteon
Tasimelteon is a circadian regulator which binds specifically to two high affinity melatonin receptors, Mella (MT1R) and Mellb (MT2R). These receptors are found in high density in the suprachiasmatic nucleus of the brain (SCN), which is responsible for synchronizing our sleep/wake cycle. Tasimelteon has been shown to improve sleep parameters in prior clinical studies, which simulated a desynchronization of the circadian clock. Tasimelteon has so far been studied in hundreds of individuals and has shown a good tolerability profile.
Tasimelteon has the chemical name: tr ns-N-[[2-(2,3-dihydrobenzofuran- 4-yl)cycloprop-lyl] methyl] propanamide, has the structure of Formula I:

Formula I
and is disclosed in US 5856529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78°C (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG- 400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MTIR. It’s affinity (¾) for MTIR is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
Metabolites of tasimelteon include, for example, those described in “Preclinical Pharmacokinetics and Metabolism of BMS-214778, a Novel
Melatonin Receptor Agonist” by Vachharajani et al., J. Pharmaceutical Sci., 92(4):760-772, which is hereby incorporated herein by reference. The active metabolites of tasimelteon can also be used in the method of this invention, as can pharmaceutically acceptable salts of tasimelteon or of its active metabolites. For example, in addition to metabolites of Formula II and III, above, metabolites of tasimelteon also include the monohydroxylated analogs M13 of Formula IV, M12 of Formula V, and M14 of Formula VI.
Formula IV

Formula V
MO

Formula VI
Thus, it is apparent that this invention contemplates entrainment of patients suffering free running circadian rhythm to a 24 hour circadian rhythm by administration of a circadian rhythm regulator (i.e., circadian rhythm modifier) capable of phase advancing and/or entraining circadian rhythms, such as a melatonin agonist like tasimelteon or an active metabolite oftasimelteon or a pharmaceutically acceptable salt thereof. Other MT1R and MT2R agonists, i.e., melatonin agonists, can have similar effects on the master body clock. So, for example, this invention further contemplates the use of melatonin agonists such as but not limited to melatonin, N-[l-(2,3-dihydrobenzofuran-4- yl)pyrrolidin-3-yl]-N-ethylurea and structurally related compounds as disclosed in US 6,211,225, LY-156735 ((R)-N-(2-(6-chloro-5-methoxy-lH-indol- 3yl) propyl) acetamide) (disclosed in U.S. Patent No. 4,997,845), agomelatine (N- [2-(7-methoxy-l-naphthyl)ethyl]acetamide) (disclosed in U.S. Patent No.
5,225,442), ramelteon ((S)-N-[2-(l,6,7,8-tetrahydro-2H-indeno- [5,4-b] furan-8- yl)ethyl]propionamide), 2-phenylmelatonin, 8-M-PDOT, 2-iodomelatonin, and 6- chloromelatonin.
Additional melatonin agonists include, without limitation, those listed in U.S. Patent Application Publication No. 20050164987, which is incorporated herein by reference, specifically: TAK-375 (see Kato, K. et al. Int. J.
Neuropsychopharmacol. 2000, 3 (Suppl. 1): Abst P.03.130; see also abstracts P.03.125 and P.03.127), CGP 52608 (l-(3-allyl-4-oxothiazolidine-2-ylidene)-4- met- hylthiosemicarbazone) (See Missbach et al., J. Biol. Chem. 1996, 271, 13515-22), GR196429 (N-[2-[2,3,7,8-tetrahydro-lH-fur-o(2,3-g)indol-l- yl] ethyl] acetamide) (see Beresford et al., J. Pharmacol. Exp. Ther. 1998, 285, 1239-1245), S20242 (N-[2-(7-methoxy napth-l-yl) ethyl] propionamide) (see Depres-Brummer et al., Eur. J. Pharmacol. 1998, 347, 57-66), S-23478 (see Neuropharmacology July 2000), S24268 (see Naunyn Schmiedebergs Arch. June 2003), S25150 (see Naunyn Schmiedebergs Arch. June 2003), GW-290569, luzindole (2-benzyl-N-acetyltryptamine) (see U.S. Patent No. 5,093,352), GR135531 (5-methoxycarbonylamino-N-acetyltrypt- amine) (see U.S. Patent Application Publication No. 20010047016), Melatonin Research Compound A, Melatonin Agonist A (see IMSWorld R&D Focus August 2002), Melatonin
Analogue B (see Pharmaprojects August 1998), Melatonin Agonist C (see Chem. Pharm. Bull. (Tokyo) January 2002), Melatonin Agonist D (see J. Pineal Research November 2000), Melatonin Agonist E (see Chem. Pharm. Bull. (Tokyo) Febrary 2002), Melatonin Agonist F (see Reprod. Nutr. Dev. May 1999), Melatonin Agonist G (see J. Med. Chem. October 1993), Melatonin Agonist H (see Famaco March 2000), Melatonin Agonist I (see J. Med. Chem. March 2000), Melatonin Analog J (see Bioorg. Med. Chem. Lett. March 2003), Melatonin Analog K (see MedAd News September 2001), Melatonin Analog L, AH-001 (2-acetamido-8- methoxytetralin) (see U.S. Patent No. 5,151,446), GG-012 (4-methoxy-2- (methylene propylamide)indan) (see Drijfhout et al., Eur. J. Pharmacol. 1999, 382, 157-66), Enol-3-IPA, ML-23 (N-2,4-dinitrophenyl-5-methoxy-tryptamine ) (see U.S. Patent No. 4,880,826), SL-18.1616, IP-100-9 (US 5580878), Sleep Inducing Peptide A, AH-017 (see U.S. Patent No. 5,151,446), AH-002 (8-methoxy- 2-propionamido-tetralin) (see U.S. Patent No. 5,151,446), and IP-101.
Metabolites, prodrugs, stereoisomers, polymorphs, hydrates, solvates, and salts of the above compounds that are directly or indirectly active can, of course, also be used in the practice of this invention.
Melatonin agonists with a MT1R and MT2R binding profile similar to that of tasimelteon, which has 2 to 4 time greater specificity for MT2R, are preferred.
Tasimelteon can be synthesized by procedures known in the art. The preparation of a 4-vinyl-2,3-dihydrobenzofuran cyclopropyl intermediate can be carried out as described in US7754902, which is incorporated herein by reference as though fully set forth.
Pro-drugs, e.g., esters, and pharmaceutically acceptable salts can be prepared by exercise of routine skill in the art.
In patients suffering a Non-24, the melatonin and Cortisol circadian rhythms and the natural day/night cycle become desynchronized. For example, in patients suffering from a free-running circadian rhythm, melatonin and Cortisol acrophases occur more than 24 hours, e.g., >24.1 hours, prior to each previous day’s melatonin and Cortisol acrophase, respectively, resulting in desynchronization for days, weeks, or even months, depending upon the length of a patient’s circadian rhythm, before the melatonin, Cortisol, and day /night cycles are again temporarily synchronized.
Chronic misalignment of Cortisol has been associated with metabolic, cardiac, cognitive, neurologic, neoplastic, and hormonal disorders. Such disorders include, e.g., obesity, depression, neurological impairments.

SAR
Figure : Melatonin receptor agonists. The applied colors indicate the mutual properties with the general melatonin receptor agonists pharmacophore.

INTRODUCTION
Tasimelteon has the chemical name: trans-N-[[2-(2,3-dihydrobenzofuran-4-yl)cycloprop-1yl]methyl]propanamide, has the structure of Formula I:

and is disclosed in U.S. Pat. No. 5,856,529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78° C. (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG-400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MT1R. It’s affinity (Ki) for MT1R is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
SYNTHESIS
(1R-trans)-N-[[2 – (2,3-dihydro-4 benzofuranyl) cyclopropyl] methyl] propanamide PATENT: BRISTOL-MYERS SQUIBB PRIORITY DATE: 1996 HYPNOTIC


PREPARATION OF XV
XXIV D-camphorsulfonic acid IS REACTED WITH THIONYL CHLORIDE TO GIVE
…………XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
TREATED WITH
XXVI ammonium hydroxide
TO GIVE
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
TREATED WITH AMBERLYST15
….XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
TREATED WITH LAH, ie double bond is reduced to get
…..XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide

Intermediate
I 3-hydroxybenzoic acid methyl ester
II 3-bromo-1-propene
III 3 – (2-propenyloxy) benzoic acid methyl ester
IV 3-hydroxy-2-(2-propenyl) benzoic acid methyl ester
V 2,3-dihydro-4-hydroxy-2-benzofurancarboxylic acid methyl ester
VI benzofuran-4-carboxylic acid methyl ester
VII benzofuran-4-carboxylic acid
VIII 2,3-dihydro-4-benzofurancarboxylic acid
IX 2,3-dihydro-4-benzofuranmethanol
X 2,3-dihydro-4-benzofurancarboxaldehyde
XI Propanedioic acid
XII (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoic acid
XIII thionyl chloride
XIV (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoyl chloride
XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
XVI (3aS,6R,7aR)-1-[(E)-3-(2,3-dihydro-4-benzofuranyl)-1-oxo-2-propenyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVII (3aS,6R,7aR)-1-[[(1R,2R)-2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]carbonyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVIII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanol
XIX [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarboxaldehyde
XX hydroxylamine hydrochloride
XXI [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarbaldehyde oxime
XXII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanamine
XXIII propanoyl chloride
XXIV D-camphorsulfonic acid
XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
XXVI ammonium hydroxide
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
Bibliography
– Patents: Benzofuran and dihydrobenzofuran melatonergic agents: US5856529 (1999)
Priority: US19960032689P, 10 Dec. 1996 (Bristol-Myers Squibb Company, U.S.)
– Preparation III (quinazolines): US2004044015 (2004) Priority: EP20000402845, 13 Oct. 2000
– Preparation of VII (aminoalkylindols): Structure-Activity Relationships of Novel Cannabinoid Mimetics Eissenstat et al, J.. Med. Chem. 1995, 38, 3094-3105
– Preparation XXVIII: Towson et al. Organic Syntheses, Coll. Vol. 8, p.104 (1993) Vol. 69, p.158 (1990)
– Preparation XV: Weismiller et al. Organic Syntheses, Coll. Vol. 8, p.110 (1993) Vol. 69, p.154 (1990).
– G. Birznieks et al. Melatonin agonist VEC-162 Improves sleep onset and maintenance in a model of transient insomnia. Sleep 2007, 30, 0773 Abstract.
-. Rajaratnam SM et al, The melatonin agonist VEC-162 Phase time immediately advances the human circadian system, Sleep 2006, 29, 0159 Abstract.
-. AK Singh et al, Evolution of a manufacturing route for a highly potent drug candidate, 229th ACS Natl Meet, March 13-17, 2005, San Diego, Abstract MEDI 576.
– Vachharajani NN et al, Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist, J Pharm Sci. 2003 Apr; 92 (4) :760-72.
. – JW Scott et al, Catalytic Asymmetric Synthesis of a melotonin antagonist; synthesis and process optimization. 223rd ACS Natl Meet, April 7-11, Orlando, 2002, Abstract ORGN 186.
SYNTHESIS CONSTRUCTION AS IN PATENT
GENERAL SCHEMES
Reaction Scheme 1

The syntheses of the 4-aryl-propenoic acid derivatives, 2 and 3, are shown in Reaction Scheme 1. The starting aldehydes, 1 , can be prepared by methods well known to those skilled in the art. Condensation of malonic acid with the aldehydes, 1, in solvents such as pyridine with catalysts such as piperidine or pyrrolidine, gives the 4-aryl- propenoic acid, 2. Subsequent conversion of the acid to the acid chloride using reagents such as thionyl chloride, phosphoryl chloride, or the like, followed by reaction with N,0-dimethyl hydroxylamine gives the amide intermediate 3 in good yields. Alternatively, aldehyde 1 can be converted directly to amide 3 using reagents such as diethyl (N-methoxy- N-methyl-carbamoylmethyl)phosphonate with a strong base such as sodium hydride.
Reaction Scheme 2

The conversion of the amide intermediate 3 to the racemic, trans- cyclopropane carboxaldehyde intermediate, 4, is shown in Reaction Scheme 2. Intermediate 3 was allowed to react with cyclopropanating reagents such as trimethylsulfoxonium iodide and sodium hydride in solvents such as DMF, THF, or the like. Subsequent reduction using reagents such as LAH in solvents such as THF, ethyl ether, or the like, gives the racemic, trans-cyclopropane carboxaldehyde intermediates, 4.
Reaction Scheme 3

Racemic cyclopropane intermediate 5 (R = halogen) can be prepared from intermediate 2 as shown in Reaction Scheme 3. Intermediate 2 was converted to the corresponding allylic alcohol by treatment with reducing agents such as sodium borohydride plus iodine in solvents such as THF. Subsequent acylation using reagents such as acetic anhydride in pyridine or acetyl chloride gave the allylic acetate which was allowed to react with cyclopropanating reagents such as sodium chloro-difluoroacetate in diglyme to provide the racemic, trans- cyclopropane acetate intermediates, 5. Reaction Scheme 4

The conversion of the acid 2 to the chiral cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, is shown in Reaction Scheme 4. Intermediate 2 is condensed with (-)-2,10-camphorsultam under standard conditions, and then cyclopropanated in the presence of catalysts such as palladium acetate using diazomethane generated from reagents such as 1-methyl-3-nitro-1-nitrosoguanidine. Subsequent reduction using reagents such as LAH in solvents such as THF, followed by oxidation of the alcohol intermediates using reagents such as DMSO/oxalyl chloride, or PCC, gives the cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, in good yields. The enantiomer, (+)-(trans)-4, can also be obtained employing a similar procedure using (+)-2,10- camphorsultam in place of (-)-2,10-camphorsultam.
When it is desired to prepare compounds of Formula I wherein m = 2, the alcohol intermediate may be activated in the conventional manner such as with mesyl chloride and treated with sodium cyanide followed by reduction of the nitrile group with a reducing agent such as LAH to produce the amine intermediate 6.
Reaction Scheme 5


Reaction Scheme 5 shows the conversion of intermediates 4 and 5 to the amine intermediate, 7, and the subsequent conversion of 6. or 7 to compounds of Formula I. The carboxaldehyde intermediate, 4, is condensed with hydroxylamine and then reduced with reagents such as LAH to give the amine intermediate, 7. The acetate intermediate 5 is hydrolyzed with potassium hydroxide to the alcohol, converted to the mesylate with methane sulfonyl chloride and triethyl amine in CH2CI2and then converted to the azide by treatment with sodium azide in solvents such as DMF. Subsequent reduction of the azide group with a reducing agent such as LAH produced the amine intermediate 7. Further reaction of 6 or 7 with acylating reagents gives compounds of Formula I. Suitable acylating agents include carboxylic acid halides, anhydrides, acyl imidazoles, alkyl isocyanates, alkyl isothiocyanates, and carboxylic acids in the presence of condensing agents, such as carbonyl imidazole, carbodiimides, and the like. Reaction Scheme 6

Reaction Scheme 6 shows the alkylation of secondary amides of Formula I (R2 = H) to give tertiary amides of Formula I (R2 = alkyl). The secondary amide is reacted with a base such as sodium hydride, potassium tert-butoxide, or the like, and then reacted with an alkylating reagent such as alkyl halides, alkyl sulfonate esters, or the like to produce tertiary amides of Formula I.
Reaction Scheme 7

Reaction Scheme 7 shows the halogenation of compounds of Formula I. The carboxamides, i (Q1 = Q2 = H), are reacted with excess amounts of halogenating agents such as iodine, N-bromosuccinimide, or the like to give the dihalo-compounds of Formula I (Q1 = Q2 = halogen). Alternatively, a stoichiometric amount of these halogenating agents can be used to give the monohalo-compounds of Formula I (Q1 = H, Q2 = halogen; or Q1 = halogen, Q2 = H). In both cases, additives such as lead IV tetraacetate can be used to facilitate the reaction. Biological Activity of the Compounds
The compounds of the invention are melatonergic agents. They have been found to bind human melatonergic receptors expressed in a stable cell line with good affinity. Further, the compounds are agonists as determined by their ability, like melatonin, to block the forskolin- stimulated accumulation of cAMP in certain cells. Due to these properties, the compounds and compositions of the invention should be useful as sedatives, chronobiotic agents, anxiolytics, antipsychotics, analgesics, and the like. Specifically, these agents should find use in the treatment of stress, sleep disorders, seasonal depression, appetite regulation, shifts in circadian cycles, melancholia, benign prostatic hyperplasia and related conditions
EXPERIMENTAL PROCEDURES
SEE ORIGINAL PATENT FOR CORECTIONS
Preparation 1
Benzofuran-4-carboxaldehyde
Step 1 : N-Methoxy-N-methyl-benzofuran-4-carboxamide
A mixture of benzofuran-4-carboxylic acid [Eissenstat, et al.. J. Medicinal Chemistry, 38 (16) 3094-3105 (1995)] (2.8 g, 17.4 mmol) and thionyl chloride (25 mL) was heated to reflux for 2 h and then concentrated in vacuo. The solid residue was dissolved in ethyl acetate (50 mL) and a solution of N,O-dimethylhydroxylamine hydrochloride (2.8 g) in saturated NaHC03(60 mL) was added with stirring. After stirring for 1.5 h, the ethyl acetate layer was separated. The aqueous layer was extracted with ethyl acetate. The ethyl acetate extracts were combined, washed with saturated NaHCO3 and concentrated in vacuo to give an oil (3.2 g, 95.4%).
Step 2: Benzofuran-4-carboxaldehyde
A solution of N-methoxy-N-methyl-benzofuran-4-carboxamide (3.2 g, 16.6 mmol) in THF (100 mL) was cooled to -45°C and then LAH (0.7 g, 18.7 mmol) was added. The mixture was stirred for 15 min, allowed to warm to -5°C, and then recooled to -45°C. Saturated KHS04 (25 mL) was added with vigorous stirring, and the mixture was allowed to warm to room temperature. The precipitate was filtered and washed with acetone. The filtrate was concentrated in vacuo to give an oil (2.3 g, 94%). Preparation 2
2,3-Dihydrobenzofuran-4-carboxaldehyde
Step 1 : 2,3-Dihydrobenzofuran-4-carboxylic acid
Benzofuran-4-carboxylic acid (10.0 g, 61 .7 mmol) was hydrogenated (60 psi) in acetic acid (100 mL) over 10% Pd/C (2 g) for 12 hr. The mixture was filtered and the filtrate was diluted with water (500 mL) to give 2,3- dihydrobenzofuran-4-carboxylic acid as a white powder (8.4 g, 83%). A sample was recrystallized from isopropanol to give fine white needles (mp: 185.5-187.5°C).
Step 2: (2,3-Dihydrobenzofuran-4-yl)methanol
A solution of 2,3-dihydrobenzofuran-4-carboxylic acid (10 g, 61 mmol) in THF (100 mL) was stirred as LAH (4.64 g, 122 mmol) was slowly added. The mixture was heated to reflux for 30 min. The mixture was cooled and quenched cautiously with ethyl acetate and then with 1 N HCI (150 mL). The mixture was then made acidic with 12 N HCI until all the inorganic precipitate dissolved. The organic layer was separated, and the inorganic layer was extracted twice with ethyl acetate. The organic layers were combined, washed twice with brine, and then concentrated in vacuo. This oil was Kϋgelrohr distilled to a clear oil that crystallized upon cooling (8.53 g, 87.6%).
Step 3: 2.3-Dihydrobenzofuran-4-carboxaldehyde
DMSO (8.10 mL, 1 14 mmol) was added at -78°C to a stirred solution of oxalyl chloride in CH2CI2 (40 mL of a 2M solution). A solution of (2,3- dihydrobenzofuran-4-yl)methanol (8.53 g, 56.9 mmol) in CH2CI2 (35 mL) was added dropwise, and the solution stirred at -78°C for 30 min. Triethyl amine (33 mL, 228 mmol) was added cautiously to quench the reaction. The resulting suspension was stirred at room temperature for 30 min and diluted with CH2CI2 (100 mL). The organic layer was washed three times with water, and twice with brine, and then concentrated in vacuo to an oil (8.42 g, 100%) that was used without purification.
Preparation 16
(±)-(trans)-2-(2,3-Dihyd robenzofuran-4-yl)cyclopropane- carboxaldehyde
Step 1 : (±Htrans)-N-Methoxy-N-methyl-2-(2.3-dihydrobenzofuran-4- yhcyclopropanecarboxamide
Trimethylsulfoxonium iodide (9.9 g, 45 mmol) was added in small portions to a suspension of sodium hydride (1 .8 g, 45 mmol) in DMF (120 mL). After the foaming had subsided (10 min), a solution of (trans)- N-methoxy-N-methyl-3-(2,3-dihydrobenzofuran-4-yl)propenamide (3.5 g, 15 mmol) in DMF (60 mL) was added dropwise, with the temperature maintained between 35-40°C. The mixture was stirred for 3 h at room temperature. Saturated NH4CI (50 mL) was added dropwise and the mixture was extracted three times with ethyl acetate. The organic extracts were combined, washed with H2O and brine, dried over K2CO3, and concentrated in vacuo to give a white wax (3.7 g, 100%).
Step 2: (±)-(trans)- 2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde
A solution of (±)-(trans)-N-methoxy-N-methyl-2-(2,3-dihydrobenzofuran- 4-yl)cyclopropanecarboxamide (3.7 g, 15 mmol) in THF (10 mL) was added dropwise to a rapidly stirred suspension of LAH (683 mg, 18 mmol) in THF (50 mL) at -45°C, maintaining the temperature below -40°C throughout. The cooling bath was removed, the reaction was allowed to warm to 5°C, and then the reaction was immediately recooled to -45°C. Potassium hydrogen sulfate (3.4 g, 25.5 mmol) in H20 (50 mL) was cautiously added dropwise, the temperature maintained below – 30°C throughout. The cooling bath was removed and the suspension was stirred at room temperature for 30 min. The mixture was filtered through Celite and the filter cake was washed with ether. The combined filtrates were then washed with cold 1 N HCI, 1 N NaOH, and brine. The filtrates were dried over MgSO4, and concentrated in vacuo to give a clear oil (2.6 g, 99%).
Preparation 18
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde
Step 1 : (-Htrans)-N-[3-(2.3-Dihvdrobenzofuran-4-yl)-propenoyll-2.10- camphorsultam
To a solution of (-)-2,10-camphorsultam (8.15 g, 37.9 mmol) in 50 mL toluene at 0°C was added sodium hydride (1.67 g, 41.7 mmol). After stirring for 0.33 h at 0°C and 0.5 h at 20°C and recooling to 0°C, a solution of 3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl chloride
(37.9 mmol), prepared in situ from the corresponding acid and thionyl chloride (75 mL), in toluene (50 mL), was added dropwise. After stirring for 18 h at 20°C, the mixture was diluted with ethyl acetate and washed with water, 1 N HCI, and 1 N NaOH. The organic solution was dried and concentrated in vacuo to give 15.8 g of crude product. Recrystallization form ethanol-methanol (600 mL, 1 :1) gave the product (13.5 g, 92%, mp 199.5-200°C).
Step 2: (-)-N-[[(trans)-2-(2,3-Dihydrobenzofuran-4-yl)-cyclopropylj- carbonylj-2, 10-camphorsultam
1 -Methyl-3-nitro-1 -nitrosoguanidine (23.88g 163 mmol) was added in portions to a mixture of 10 N sodium hydroxide (60 mL) and ether (200 mL) at 0°C. The mixture was shaken vigorously for 0.25 h and the ether layer carefully decanted into a solution of (-)-N-[3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl]-2,10-camphorsultam (9.67 g, 25 mmol) and palladium acetate (35 mg) in methylene chloride (200 mL). After stirring for 18 h, acetic acid (5 mL) was added to the reaction and the mixture stirred for 0.5 h. The mixture was washed with 1 N HCI, 1 N NaOH and brine. The solution was dried, concentrated in vacuo and the residue crystallized twice from ethanol to give the product (6.67 g, 66.5%, mp 157-159°C).
Step 3: (-)-(trans)-2-(2,3-Dihydrobenzofuran-4-yl)cyclopropane- methanol
A solution of (-)-N-[(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclo-propanecarbonylj-2,10-camphorsultam (4.3 g, 10.7 mmol) in THF (50 mL) was added dropwise to a mixture of LAH (0.81 g, 21.4 mmol) in THF (50 mL) at -45°C. The mixture was stirred for 2 hr while it warmed to 10°C. The mixture was recooled to -40°C and hydrolyzed by the addition of saturated KHS0 (20 mL). The mixture was stirred at room temperature for 30 minutes and filtered. The precipitate was washed twice with acetone. The combined filtrate and acetone washes were concentrated in vacuo. The gummy residue was dissolved in ether, washed with 1 N NaOH and 1 N HCI, and then dried in vacuo to give the product (2.0 g, 98.4%).
Step 4: (-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde DMSO (1.6 g, 21 mmol) was added to oxalyl chloride in CH2CI2(7.4 mL of 2 M solution, 14.8 mmole) at -78°C. The (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)-cyclopropylmethanol (2.0 g, 10.5 mmol) in CH2CI2(15 mL) was added. The mixture was stirred for 20 min and then triethylamine (4.24 g, 42 mmol) was added. The mixture was warmed to room temperature and stirred for 30 min. The mixture was diluted with CH2CI2 and washed with water, 1 N HCI, and then 1 N NaOH. The organic layer was dried and concentrated iι> vacuo to give the aldehyde product (1.98 g, 100%).
Preparation 24
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-methanamine A mixture of (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde (1.98 g, 10.5 mmol), hydroxylamine hydrochloride (2.29 g, 33 mmol), and 30% NaOH (3.5 mL, 35 mmol), in 5:1
ethanol/water (50 mL) was heated on a steam bath for 2 h. The solution was concentrated in vacuo. and the residue mixed with water. The mixture was extracted with CH2CI2. The organic extracts were dried and concentrated in vacuo to give a solid which NMR analysis showed to be a mixture of the cis and trans oximes. This material was dissolved in THF (20 mL) and added to solution of alane in THF [prepared from LAH (1.14 g, 30 mmol) and H2S04 (1.47 g, 15 mmol) at 0°Cj. The reaction was stirred for 18 h, and quenched successively with water (1.15 mL), 15% NaOH (1.15 mL), and then water (3.45 mL). The mixture was filtered and the filtrate was concentrated in vacuo. The residue was mixed with ether and washed with water and then 1 N HCI. The acid washes were made basic and extracted with CH2CI . The extracts were dried and concentrated in vacuo to give the amine product (1.4 g, 70.5%). The amine was converted to the fumarate salt in ethanol (mp: 197-198°C).
Anal. Calc’d for C12H15NO • C4H404: C, 62.94; H, 6.27; N, 4.59.
Found: C, 62.87; H, 6.31 ; N, 4.52.
FINAL PRODUCT TASIMELTEON
Example 2
(-)-(trans)-N-[[2-(2,3-Dihydrobenzofuran-4-yl)cycloprop-1-yl]methyl]propanamide
This compound was prepared similar to the above procedure using propionyl chloride and (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)- cyclopropanemethanamine to give an oil that solidified upon standing to an off-white solid (61 %, mp: 71-72°C). IR (NaCI Film): 3298, 1645, 1548, 1459, 1235 cm“1.
Mo5 : -17.3°
Anal. Calc’d for C15H19N02: C, 73.44; H, 7.87; N, 5.71 . Found: C, 73.28; H, 7.68; N, 5.58
SYNTHESIS
Synthesis Path
SYN

Tasimelteon (Hetlioz)Tasimelteon, which is marketed by Vanda Pharmaceuticals as Hetlioz and developed in partnership with Bristol-Myers Squibb,is a drug that was approved by the US FDA in January 2014 for the treatment of non-24-hour sleep–wake disorder (also called Non-24, N24 and N24HSWD).234 Tasimelteon is a melatonin MT1
and MT2 receptor agonist; because it exhibits a greater affinity to the MT2 receptor than MT1, is also known as Dual Melatonin
Receptor Agonist.234 Two randomized controlled trials (phases II
and III) demonstrated that tasimelteon improved sleep latency
and maintenance of sleep with a shift in circadian rhythms, and
therefore has the potential to treat patients with transient insomnia
associated with circadian rhythm sleep disorders.235 Preclinical
studies showed that the drug has similar phase-shifting properties
to melatonin, but with less vasoconstrictive effects.236 The most
likely scale preparation of the drug, much of which has been published
in the chemical literature, is described below in Scheme 44.
Activation of commercial bis-ethanol 250 with 2.5 equivalents
of the Vilsmeier salt 251 followed by treatment with base resulted
an intramolecular cyclization reaction with the proximal phenol
and concomitant elimination of the remaining imidate to deliver
the vinylated dihydrobenzofuran 252 in 76% yield.237 Interestingly,
this reaction could be performed on multi-kilogram scale, required
no chromatographic purification, and generated environmentallyfriendly
DMF and HCl as byproducts.237 Sharpless asymmetric
dihydroxylation of olefin 252 delivered diol 253 in 86% yield and
impressive enantioselectivity (>99% ee). This diol was then activated
with trimethylsilyl chloride and then treated with base to generate epoxide 254.238 Next, a modified Horner–Wadsworth–
Emmons reaction involving triethylphosphonoacetate (TEPA, 255)
was employed to convert epoxide 254 to cyclopropane 256.239
The reaction presumably proceeds through removal of the acidic
TEPA proton followed by nucleophilic attack at the terminal epoxide
carbon. The resulting alkoxide undergoes an intramolecular
phosphoryl transfer reaction resulting in an enolate, which then attacked the newly formed phosphonate ester in an SN2 fashion
resulting in the trans-cyclopropane ester, which was ultimately
saponified and re-acidified to furnish cyclopropane acid 256.239
Conversion of this acid to the corresponding primary amide preceded
carbonyl reduction with sodium borohydride. The resulting
amine was acylated with propionyl chloride to furnish tasimelteon
(XXXI) as the final product in 86% yield across the four-step
sequence.
PATENTS
| US2010261786 | 10-15-2010 | PREDICTION OF SLEEP PARAMETER AND RESPONSE TO SLEEP-INDUCING COMPOUND BASED ON PER3 VNTR GENOTYPE |
| US2009209638 | 8-21-2009 | TREATMENT FOR DEPRESSIVE DISORDERS |
| US6060506 | 5-10-2000 | Benzopyran derivatives as melatonergic agents |
| US5981571 | 11-10-1999 | Benzodioxa alkylene ethers as melatonergic agents |
| WO9825606 | 6-19-1998 | BENZODIOXOLE, BENZOFURAN, DIHYDROBENZOFURAN, AND BENZODIOXANE MELATONERGIC AGENTS |
| WO2007137244A1 * | May 22, 2007 | Nov 29, 2007 | Gunther Birznieks | Melatonin agonist treatment |
| US4880826 | Jun 25, 1987 | Nov 14, 1989 | Nava Zisapel | Melatonin antagonist |
| US4997845 | May 10, 1990 | Mar 5, 1991 | Eli Lilly And Company | β-alkylmelatonins as ovulation inhibitors |
| US5093352 | May 16, 1990 | Mar 3, 1992 | Whitby Research, Inc. | Antidepressant agents |
| US5151446 | Mar 28, 1991 | Sep 29, 1992 | Northwestern University | Substituted 2-amidotetralins as melatonin agonists and antagonists |
| US5225442 | Jan 3, 1992 | Jul 6, 1993 | Adir Et Compagnie | Compounds having a naphthalene structure |
| US5580878 | Jun 7, 1995 | Dec 3, 1996 | Interneuron Pharmaceuticals, Inc. | Substituted tryptamines phenalkylamines and related compounds |
| US5856529 | Dec 9, 1997 | Jan 5, 1999 | Bristol-Myers Squibb Company | Benzofuran and dihydrobenzofuran melatonergic agents |
| US6211225 | Jun 6, 2000 | Apr 3, 2001 | Bristol-Meyers Squibb | Heterocyclic aminopyrrolidine derivatives as melatonergic agents |
| US7754902 | May 18, 2006 | Jul 13, 2010 | Vanda Pharmaceuticals, Inc. | Ruthenium(II) catalysts for use in stereoselective cyclopropanations |
| US20010047016 | Apr 12, 2001 | Nov 29, 2001 | Gregory Oxenkrug | Method for treating depression |
| US20050164987 | Dec 22, 2004 | Jul 28, 2005 | Barberich Timothy J. | Melatonin combination therapy for improving sleep quality |
| US20090105333 | May 22, 2007 | Apr 23, 2009 | Gunther Birznieks | Melatonin agonist treatment |
extra info
Org. Synth.1990, 69, 154
(−)-D-2,10-CAMPHORSULTAM
[3H-3a,6-Methano-2,1-benzisothiazole, 4,5,6,7-tetrahydro-8,8-dimethyl-2,2-dioxide, (3aS)-]
|
Submitted by Michael C. Weismiller, James C. Towson, and Franklin A. Davis1.
Checked by David I. Magee and Robert K. Boeckman, Jr..
1. Procedure
(−)-2,10-Camphorsultam. A dry, 2-L, three-necked, round-bottomed flask is equipped with a 1.5-in egg-shaped Teflon stirring bar, a 250-mL addition funnel, and a 300-mL Soxhlet extraction apparatus equipped with a mineral oil bubbler connected to an inert-gas source. The flask is charged with 600 mL of dry tetrahydrofuran (THF) (Note 1) and6.2 g (0.16 mol) of lithium aluminum hydride (Note 2). Into the 50-mL Soxhlet extraction thimble is placed 35.0 g (0.16 mol) of (−)-(camphorsulfonyl)imine (Note 3) and the reaction mixture is stirred and heated at reflux. After all of the(camphorsulfonyl)imine has been siphoned into the reaction flask (3–4 hr), the mixture is allowed to cool to room temperature. The unreacted lithium aluminum hydride is cautiously hydrolyzed by dropwise addition of 200 mL of 1 Nhydrochloric acid via the addition funnel (Note 4). After the hydrolysis is complete the contents of the flask are transferred to a 1-L separatory funnel, the lower, silver-colored aqueous layer is separated, and the upper layer placed in a 1-L Erlenmeyer flask. The aqueous phase is returned to the separatory funnel and washed with methylene chloride (3 × 100 mL). After the reaction flask is rinsed with methylene chloride (50 mL), the organic washings are combined with the THF phase and dried over anhydrous magnesium sulfate for 10–15 min. Filtration through a 300-mL sintered-glass funnel of coarse porosity into a 1-L round-bottomed flask followed by removal of the solvent on arotary evaporator gives 33.5 g (95%) of the crude (−)-2,10-camphorsultam. The crude sultam is placed in a 250-mL Erlenmeyer flask and crystallized from approximately 60 mL of absolute ethanol. The product is collected on a 150-mL sintered-glass funnel of coarse porosity and dried in a vacuum desiccator to give 31.1 g (88%) of the pure sultam. A second crop of crystals can be gained by evaporating approximately half the filtrate; the residue is crystallized as above to give 1.4 g (4%). The combined yield of white crystalline solid, mp 183–184°C, [α]D −30.7° (CHCl3, c 2.3) is92% (Note 5) and (Note 6).
2. Notes
1. Tetrahydrofuran (Aldrich Chemical Company, Inc.) was distilled from sodium benzophenone.
2. Lithium aluminum hydride was purchased from Aldrich Chemical Company, Inc.
3. (−)-(Camphorsulfonyl)imine, [(7S)-(−)-10,10-dimethyl-5-thia-4-azatricyclo[5.2.1.03,7]dec-3-ene 5,5-dioxide] was prepared by the procedure of Towson, Weismiller, Lal, Sheppard, and Davis, Org. Synth., Coll. Vol. VIII, 1993, 104.
4. The addition must be very slow at first (1 drop/5 sec) until the vigorous reaction has subsided.
5. The NMR spectrum of (−)-2,10-camphorsultam is as follows: 1H NMR (CDCl3) δ: 0.94 (s, 3 H, CH3), 1.14 (s, 3 H, CH3), 1.33 (m, 1 H), 1.47 (m,, 1 H), 1.80–2.05 (5 H), 3.09 (d, 1 H, J = 14), 3.14 (d, 1 H, J = 14), 3.43 (m, 1 H), 4.05 (br s, 1 H, NH); 13C NMR (CDCl3) δ: 20.17 (q, CH3), 26.51 (t), 31.55 (t), 35.72 (t), 44.44 (d), 47.15 (s), 50.08 (t), 54.46 (s), 62.48 (d).
6. Checkers obtained material having the same mp (183–184°C) and [α]D − 31.8° (CHCl3, c 2.3).
3. Discussion
(−)-2,10-Camphorsultam was first prepared by the catalytic hydrogenation of (−)-(camphorsulfonyl)imine overRaney nickel.2 Lithium aluminum hydride reduction was used by Oppolzer and co-workers in their synthesis of the sultam.3,4 However, because of the low solubility of the sultam in tetrahydrofuran, a large amount of solvent was required.4 In the procedure described here the amount of solvent is significantly reduced by using a Soxhlet extractor to convey the imine slowly into the reducing medium.5
Oppolzer’s chiral auxiliary,6 (−)-2,10-camphorsultam, is useful in the asymmetric Diels–Alder reaction,3,4 and for the preparation of enantiomerically pure β-substituted carboxylic acids7 and diols,8 in the stereoselective synthesis of Δ2-isoxazolines,9 and in the preparation of N-fluoro-(−)-2,10-camphorsultam, an enantioselective fluorinating reagent.10
References and Notes
- Department of Chemistry, Drexel University, Philadelphia, PA 19104.
- Shriner, R. L.; Shotton, J. A.; Sutherland, H. J. Am. Chem. Soc.1938, 60, 2794.
- Oppolzer, W.; Chapuis, C.; Bernardinelli, G. Helv. Chim. Acta1984, 67, 1397.
- Vandewalle, M.; Van der Eycken, J.; Oppolzer, W.; Vullioud, C. Tetrahedron1986, 42, 4035.
- Davis, F. A.; Towson, J. C.; Weismiller, M. C.; Lal, G.; Carroll,, P. J. J. Am. Chem. Soc.1988, 110, 8477.
- Oppolzer, W. Tetrahedron1987, 43, 1969.
- Oppolzer, W.; Mills, R. J.; Pachinger, W.; Stevenson, T. Helv. Chim. Acta1986, 69, 1542; Oppolzer, W.; Schneider, P. Helv. Chim. Acta1986, 69, 1817; Oppolzer, W.; Mills, R. J.; Réglier, M. Tetrahedron Lett.1986, 27, 183; Oppolzer, W.; Poli. G.Tetrahedron Lett.1986, 27, 4717; Oppolzer, W.; Poli, G.; Starkemann, C.; Bernardinelli, G. Tetrahedron Lett.1988, 29, 3559.
- Oppolzer, W.; Barras, J-P. Helv. Chim. Acta1987, 70, 1666.
- Curran, D. P.; Kim, B. H.; Daugherty, J.; Heffner, T. A. Tetrahedron Lett.1988, 29, 3555.
- Differding, E.; Lang, R. W. Tetrahedron Lett.1988, 29, 6087.
Org. Synth.1990, 69, 158
(+)-(2R,8aS)-10-(CAMPHORYLSULFONYL)OXAZIRIDINE
[4H-4A,7-Methanooxazirino[3,2-i][2,1]benzisothiazole, tetrahydro-9,9-dimethyl-, 3,3-dioxide, [4aS-(4aα,7α,8aR*)]]
 |
Submitted by James C. Towson, Michael C. Weismiller, G. Sankar Lal, Aurelia C. Sheppard, Anil Kumar, and Franklin A. Davis1.
Checked by David I. Magee and Robert K. Boeckman, Jr..
1. Procedure
A. (+)-(1S)-10-Camphorsulfonamide. Into a 2-L, two-necked, round-bottomed flask, equipped with a 250-mL dropping funnel, a magnetic stirring bar, and a reflux condenser fitted with an outlet connected to a disposable pipettedipped in 2 mL of chloroform in a test tube for monitoring gas evolution, were placed 116 g (0.5 mol) ofcamphorsulfonic acid (Note 1) and 750 mL of reagent-grade chloroform. The suspension of camphorsulfonic acid was heated to reflux and 71.4 g (43.77 mL, 0.6 mol, 1.2 equiv) of freshly distilled thionyl chloride was added dropwise over a 1-hr period. Heating was continued until gas evolution (sulfur dioxide and hydrogen chloride) had ceased (approximately 9–10 hr). The resultant solution of camphorsulfonyl chloride in chloroform was converted tocamphorsulfonamide without further purification.
In a 5-L, two-necked, round-bottomed flask fitted with a 250-mL dropping funnel and a mechanical stirrer was placed a solution of 1.6 L of reagent-grade ammonium hydroxide solution and the flask was cooled to 0°C in an ice bath. The solution of the crude camphorsulfonyl chloride, prepared in the preceding section, was added dropwise to the ammonium hydroxide solution at 0–10°C over a period of 1 hr. The reaction mixture was warmed to room temperature, stirred for 4 hr, the organic layer separated, and the aqueous layer was extracted with methylene chloride (3 × 250 mL). The combined organic layers were washed with brine (250 mL) and dried over anhydrousmagnesium sulfate. Removal of the solvent on the rotary evaporator gave 104.0 g (90%) of the crudecamphorsulfonamide (Note 2) and (Note 3).
B. (−)-(Camphorsulfonyl)imine. A 1-L, round-bottomed flask is equipped with a 2-in. egg-shaped magnetic stirring bar, a Dean–Stark water separator, and a double-walled condenser containing a mineral oil bubbler connected to an inert gas source. Into the flask are placed 5 g of Amberlyst 15 ion-exchange resin (Note 4) and 41.5 g of the crude(+)-(1S)-camphorsulfonamide in 500 mL of toluene. The reaction mixture is heated at reflux for 4 hr. After the reaction flask is cooled, but while it is still warm (40–50°C), 200 mL of methylene chloride is slowly added to dissolve any(camphorsulfonyl)imine that crystallizes. The solution is filtered through a 150-mL sintered glass funnel of coarse porosity an the reaction flask and filter funnel are washed with an additional 75 mL of methylene chloride.
Isolation of the (−)-(camphorsulfonyl)imine is accomplished by removal of the toluene on the rotary evaporator. The resulting solid is recrystallized from absolute ethanol (750 mL) to give white crystals, 34.5–36.4 g (90–95%), mp225–228°C; [α]D −32.7° (CHCl3, c 1.9) (Note 5).
C. (+)-(2R, 8aS)-10-Camphorylsulfonyloxaziridine. A 5-L, three-necked, round-bottomed Morton flask is equipped with an efficient mechanical stirrer, a 125-mm Teflon stirring blade, a Safe Lab stirring bearing (Note 6), and a 500-mL addition funnel. Into the flask are placed the toluene solution of (−)-(camphorsulfonyl)imine (39.9 g, 0.187 mol)prepared in Step B and a room-temperature solution of 543 g (3.93 mol, 7 equiv based on oxone) of anhydrouspotassium carbonate dissolved in 750 mL of water. The reaction mixture is stirred vigorously and a solution of 345 g (0.56 mol, 6 equiv of KHSO5) of oxone dissolved in 1250 mL of water is added dropwise in three portions over 45 min(Note 7) and (Note 8). Completion of the oxidation is determined by TLC (Note 9) and the reaction mixture is filtered through a 150-mL sintered-glass funnel of coarse porosity to remove solids. The filtrate is transferred to a 3-L separatory funnel, the toluene phase is separated and the aqueous phase is washed with methylene chloride (3 × 100 mL). The filtered solids and any solids remaining in the Morton flask are washed with an additional 200 mL of methylene chloride. The organic extracts are combined and washed with 100 mL of saturated sodium sulfite, dried over anhydrousmagnesium sulfate for 15–20 min, filtered, and concentrated on the rotary evaporator. The resulting white solid is crystallized from approximately 500 mL of hot 2-propanol to afford, after drying under vacuum in a desiccator, 35.9 g(84%) of white needles, mp 165–167°C, [α]D +44.6° (CHCl3, c 2.2) (Note 10) and (Note 11).
(−)-(2S,8aR)-10-(camphorylsulfonyl)oxaziridine is prepared in a similar manner starting from (−)-10-camphorsulfonic acid; mp 166–167°C, [α]D +43.6° (CHCl3, c 2.2).
2. Notes
1. (1S)-(+)-10-Camphorsulfonic acid was purchased from Aldrich Chemical Company, Inc.
2. The crude sulfonamide is contaminated with 5–10% of the (camphorsulfonyl)imine, the yield of which increases on standing.
3. The 1H NMR spectrum of (+)-(1S)-10-camphorsulfonamide is as follows: (CDCl3) δ: 0.93 (s, 3 H, CH3), 1.07 (s, 3 H, CH3), 1.40–2.50 (m, 7 H), 3.14 and 3.53 (AB quartet, 2 H, CH2-SO2, J = 15.1), 5.54 (br s, 2 H, NH2).
4. Amberlyst 15 ion-exchange resin is a strongly acidic, macroreticular resin purchased from Aldrich Chemical Company, Inc.
5. The spectral properties of (−)-(camphorsulfonyl)imine are as follows: 1H NMR (CDCl3) δ: 1.03 (s, 3 H, CH3), 1.18 (s, 3 H, CH3), 1.45–2.18 (m, 6 H), 2.65 (m, 1 H), 3.10 and 3.28 (AB quartet, 2 H, CH2-SO2, J = 14.0); 13C NMR (CDCl3) δ: 19.01 (q, CH3), 19.45 (q, CH3), 26.64 (t), 28.44 (t), 35.92 (t), 44.64 (d), 48.00 (s), 49.46 (t), 64.52 (s), 195.52 (s); IR (CHCl3) cm−1: 3030, 2967, 1366. Checkers obtained material having identical melting point and [α]D−32.3° (CHCl3, c 1.8).
6. The SafeLab Teflon bearing can be purchased from Aldrich Chemical Company, Inc. A glass stirring bearing lubricated with silicone grease is unsatisfactory because the dissolved salts solidify in the shaft, causing freezing.
7. Efficient stirring is important and indicated by a milky white appearance of the solution.
8. Occasionally batches of oxone purchased from Aldrich Chemical Company, Inc., have exhibited reduced reactivity in this oxidation. Oxone exposed to moisture prior to use also gives reduced reactivity in this oxidation. If this occurs, oxone is added until oxidation is complete as determined by TLC (Note 9). Potassium carbonate is added as needed to maintain the pH at approximately 9.0. Oxone stored in the refrigerator under an inert atmosphere has shown no loss in reactivity for up to 6 months.
9. Oxidation is generally complete after addition of the oxone solution. The oxidation is monitored by TLC as follows. Remove approximately 0.5 mL of the toluene solution from the nonstirring solution, spot a 250-μm TLC silica gel plate, elute with methylene chloride, and develop with 10% molybdophosphoric acid in ethanol and heating(camphorsulfonyl)imine Rf = 0.28 and (camphorylsulfonyl)oxaziridine Rf = 0.62. If (camphorsulfonyl)imine is detected, stirring is continued at room temperature until the reaction is complete (see (Note 8)). If the reaction mixture takes on a brownish color after addition of oxone and has not gone to completion after 30 min, the reaction mixture is filtered through a 150-mL sintered-glass funnel of coarse porosity, and the solids are washed with 50 mL of methylene chloride. The aqueous/organic extracts are returned to the 5-L Morton flask and stirred vigorously and 52 g (0.08 mol, 1 equiv KHSO5) of oxone is added over 5 min and stirring continued until oxidation is complete (approximately 10–15 min).
10. The submitters employed a toluene solution of crude imine prepared in Part B and obtained somewhat higher yields (90–95%). However, the checkers obtained yields in this range on one half the scale using isolatedsulfonylimine.
11. The spectral properties of (+)-(camphorsulfonyl)oxaziridine are as follows: 1H NMR (CDCl3) δ: 1.03 (s, 3 H, CH3), 1.18 (s, 3 H, CH3), 1.45–2.18 (m, 6 H), 2.65 (d, 1 H), 3.10 and 3.28 (AB quartet, 2 H, CH2-SO2, J = 14.0); 13C NMR (CDCl3) δ: 19.45 (q, CH3), 20.42 (q, CH3), 26.55 (t), 28.39 (t), 33.64 (t), 45.78 (d), 48.16 (s), 48.32 (t), 54.07 (s), 98.76 (s). The checkers obtained material (mp 165–167°C) having [α]D +44.7° (CHCl3, c 2.2).
3. Discussion
Camphorsulfonamide, required for the preparation of the (camphorsulfonyl)imine, was previously prepared in two steps. The first step involved conversion of camphorsulfonic acid to the sulfonyl chloride with PCl5 or SOCl2. The isolated sulfonyl chloride was converted in a second step to the sulfonamide by reaction with ammonium hydroxide. This modified procedure is more efficient because it transforms camphorsulfonic acid directly to camphorsulfonamide, avoiding isolation of the camphorsulfonyl chloride.
(Camphorsulfonyl)imine has been reported as a by-product of reactions involving the camphorsulfonamide.2,3,4,5Reychler in 1898 isolated two isomeric camphorsulfonamides,2 one of which was shown to be the(camphorsulfonyl)imine by Armstrong and Lowry in 1902.3 Vandewalle, Van der Eycken, Oppolzer, and Vullioud described the preparation of (camphorsulfonyl)imine in 74% overall yield from 0.42 mol of the camphorsulfonyl chloride.6 The advantage of the procedure described here is that, by using ammonium hydroxide, the camphorsulfonyl chloride is converted to the sulfonamide in >95% yield.7 The sulfonamide is of sufficient purity that it can be used directly in the cyclization step, which, under acidic conditions, is quantitative in less than 4 hr. These modifications result in production of the (camphorsulfonyl)imine in 86% overall yield from the sulfonyl chloride.
In addition to the synthesis of enantiomerically pure (camphorylsulfonyl)oxaziridine7 and its derivatives,8 the(camphorsulfonyl)imine has been used in the preparation of (−)-2,10-camphorsultam (Oppolzers’ auxiliary),6,9 (+)-(3-oxocamphorysulfonyl) oxaziridine,10 and the N-fluoro-2,10-camphorsultam, an enantioselective fluorinating reagent.11
The N-sulfonyloxaziridines are an important class of selective, aprotic oxidizing reagents.121314 Enantiomerically pure N-sulfonyloxaziridines have been used in the asymmetric oxidation of sulfides to sulfoxides (30–91% ee),15selenides to selenoxides (8–9% ee).16 disulfides to thiosulfinates (2–13% ee),5 and in the asymmetric epoxidation of alkenes (19–65% ee).17,18 Oxidation of optically active sulfonimines (R*SO2N=CHAr) affords mixtures of N-sulfonyloxaziridine diastereoisomers requiring separation by crystallization and/or chromatography.3
(+)-(Camphorylsulfonyl)oxaziridine described here is prepared in four steps from inexpensive (1S)-(+)- or (1R)-(+)-10-camphorsulfonic acid in 77% overall yield.7 Separation of the oxaziridine diastereoisomers is not required because oxidation is sterically blocked from the exo face of the C-N double bond in the (camphorsulfonyl)imine. In general, (camphorsulfonyl)oxaziridine exhibits reduced reactivity compared to other N-sulfonyloxaziridines. For example, while sulfides are asymmetrically oxidized to sulfoxides (3–77% ee), this oxaziridine does not react with amines or alkenes.7 However, this oxaziridine is the reagent of choice for the hydroxylation of lithium and Grignard reagents to give alcohols and phenols because yields are good to excellent and side reactions are minimized.19 This reagent has also been used for the stereoselective oxidation of vinyllithiums to enolates.20
The most important synthetic application of the (camphorylsulfonyl)oxaziridines is the asymmetric oxidation of enolates to optically active α-hydroxy carbonyl compounds.14,21,22,23,24 Chiral, nonracemic α-hydroxy carbonylcompounds have been used extensively in asymmetric synthesis, for example, as chiral synthons, chiral auxiliaries, and chiral ligands. This structural array is also featured in many biologically active natural products. This oxidizing reagent gives uniformly high chemical yields regardless of the counterion, and stereoselectivities are good to excellent (50–95% ee).9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24 Since the configuration of the oxaziridine three-membered ring controls the stereochemistry, both α-hydroxy carbonyl optical isomers are readily available. Representative examples of the asymmetric oxidation of prochiral enolates by (+)-(2R,8aS)-camphorylsulfonyl)oxaziridine are given in Tables I and II.
This preparation is referenced from:
- Org. Syn. Coll. Vol. 8, 110
- Org. Syn. Coll. Vol. 9, 212
-
References and Notes
- Department of Chemistry, Drexel University, Philadelphia, PA 19104.
- Reychler, M. A. Bull. Soc. Chim. III1889, 19, 120.
- Armstrong, H. E.; Lowry, T. M. J. Chem. Soc., Trans.1902, 81, 1441.
- Dauphin, G.; Kergomard, A.; Scarset, A. Bull. Soc. Chim. Fr.1976, 862.
- Davis, F. A.; Jenkins, Jr., R. H.; Awad, S. B.; Stringer, O. D.; Watson, W. H.; Galloy, J. J. Am. Chem. Soc.1982, 104, 5412.
- Vandewalle, M.; Van der Eycken, J.; Oppolzer, W.; Vullioud, C. Tetrahedron, 1986, 42, 4035.
- Davis, F. A.; Towson, J. C.; Weismiller, M. C.; Lal, S.; Carroll, P. J. J. Am. Chem. Soc.1988, 110, 8477.
- Davis, F. A.; Weismiller, M. C.; Lal, G. S.; Chen, B. C.; Przeslawski, R. M. Tetrahedron Lett., 1989, 30, 1613.
- Oppolzer, W. Tetrahedron1987, 43, 1969.
- Glahsl, G.; Herrmann, R. J. Chem. Soc., Perkin Trans. I1988, 1753.
- Differding, E.; Lang, R. W. Tetrahedron Lett.1988, 29, 6087.
- For recent reviews on the chemistry of N-sulfonyloxaziridines, see: (a) Davis, F. A.; Jenkins, Jr., R. H. in “Asymmetric Synthesis,” Morrison, J. D., Ed.; Academic Press: Orlando, FL, 1984, Vol. 4, Chapter 4;
- Davis, F. A.; Haque, S. M. in “Advances in Oxygenated Processes,” Baumstark, A. L., Ed.; JAI Press: London, Vol. 2;
- Davis, F. A.; Sheppard, A. C. Tetrahedron1989, 45, 5703.
- Davis, F. A.; McCauley, Jr., J. P.; Chattopadhyay, S.; Harakal, M. E.; Towson, J. C.; Watson, W. H.; Tavanaiepour, I. J. Am. Chem. Soc.1987, 109, 3370.
- Davis, F. A.; Stringer, O. D.; McCauley, Jr., J. M. Tetrahedron1985, 41, 4747.
- Davis, F. A.; Chattopadhyay, S. Tetrahedron Lett.1986, 27, 5079.
- Davis, F. A.; Harakal, M. E.; Awad, S. B. J. Am. Chem. Soc.1983, 105, 3123.
- Davis, F. A.; Wei, J.; Sheppard, A. C.; Gubernick S. Tetrahedron Lett.1987, 28, 5115.
- Davis, F. A.; Lal, G. S.; Wei, J. Tetrahedron Lett.1988, 29, 4269.
- Davis, F. A.; Haque, M. S.; Ulatowski, T. G.; Towson, J. C. J. Org. Chem.1986, 51, 2402.
- Davis, F. A.; Haque, M. S. J. Org. Chem.1986, 51, 4083; Davis, F. A.; Haque, M. S.; Przeslawski, R. M. J. Org. Chem.1989, 54, 2021.
- Davis, F. A.; Ulatowski, T. G.; Haque, M. S. J. Org. Chem.1987, 52, 5288.
- Davis, F. A.; Sheppard, A. C., Lal, G. S. Tetrahedron Lett.1989, 30, 779.
- Davis, F. A.; Sheppard, A. C.; Chen, B. C.; Haque, M. S. J. Am. Chem. Soc.1990, 112, 6679.
a US 5 856 529 (Bristol-Myers Squibb; 5.1.1999; appl. 9.12.1997; USA-prior. 10.12.1996).
-
- b US 7 754 902 (Vanda Pharms.; 13.7.2010; appl. 18.5.2006).
-
treatment of circadian rhythm disorders:
- US 8 785 492 (Vanda Pharms.; 22.7.2014; appl. 25.1.2013; USA-prior. 26.1.2012).
-
synthesis cis-isomer:
- US 6 214 869 (Bristol-Myers Squibb; 10.4.2001; appl. 25.5.1999; USA-prior. 5.6.1998).
Patents
References
- Jump up^ “Tasimelteon Advisory Committee Meeting Briefing Materials”(PDF). Vanda Pharmaceuticals Inc. November 2013.
- Jump up^ “FDA transcript approval minutes” (PDF). FDA. November 14, 2013.
- ^ Jump up to:a b Food and Drug Administration (January 31, 2014). “FDA approves Hetlioz: first treatment for non-24 hour sleep-wake disorder”. FDA.
- Jump up^ “tasimelteon (Hetlioz) UKMi New Drugs Online Database”. Retrieved August 6, 2014.
- Jump up^ “HETLIOZ® Receives European Commission Approval for the Treatment of Non-24-Hour Sleep-Wake Disorder in the Totally Blind”. MarketWatch. PR Newswire. 7 July 2015. Retrieved 8 July 2015.
- Jump up^ Vachharajani, Nimish N.; Yeleswaram, Krishnaswamy; Boulton, David W. (April 2003). “Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist”. Journal of Pharmaceutical Sciences. 92 (4): 760–72. doi:10.1002/jps.10348. PMID 12661062.
- Jump up^ Sack, R. L.; Brandes, R. W.; Kendall, A. R.; Lewy, A. J. (2000). “Entrainment of Free-Running Circadian Rhythms by Melatonin in Blind People”. New England Journal of Medicine. 343 (15): 1070–7. doi:10.1056/NEJM200010123431503. PMID 11027741.
- Jump up^ “Safety and Efficacy of VEC-162 on Circadian Rhythm in Healthy Adult Volunteers”. ClinicalTrials.gov. |accessdate=May 15, 2014
- Jump up^ “VEC-162 Study in Healthy Adult Volunteers in a Model of Insomnia”. ClinicalTrials.gov. Retrieved May 15, 2014.
- Jump up^ “VEC-162 Study in Adult Patients With Primary Insomnia”. ClinicalTrials.gov. Retrieved May 15, 2014.
- Jump up^ Lynne Lamberg. “Improving Sleep and Alertness in the Blind (Part 5)”. Matilda Ziegler Magazine for the Blind. Retrieved May 15, 2014.
- Jump up^ Shantha MW Rajaratnam; Mihael H Polymeropoulos; Dennis M Fisher; Thomas Roth; Christin Scott; Gunther Birznieks; Elizabeth B Klerman (2009-02-07). “Melatonin agonist tasimelteon (VEC-162) for transient insomnia after sleep-time shift: two randomised controlled multicentre trials”. The Lancet. 373 (9662): 482–491. doi:10.1016/S0140-6736(08)61812-7. PMID 19054552. Retrieved 2010-02-23.
- Jump up^ Carome, Michael (1 July 2015). “Outrage of the Month: FDA Makes Major Blunder After Approving Drug for Rare Sleep Disorder”. Huffington Post. Retrieved 8 July 2015.
- Jump up^ Food and Drug Administration (January 31, 2014). “FDA NEWS RELEASE: FDA approves Hetlioz: first treatment for non-24 hour sleep–wake disorder in blind individuals”. FDA.
- Jump up^ “Side Effects Drug Center: Hetlioz Clinical Pharmacology”. RxList. February 10, 2014.
- Jump up^ “Side Effects Drug Center: Hetlioz Warnings and Precautions”. RxList. February 10, 2014.
In animal studies, administration of tasimelteon during pregnancy resulted in developmental toxicity (embryofetal mortality, neurobehavioral impairment, and decreased growth and development in offspring) at doses greater than those used clinically.
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Hetlioz |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | not determined in humans[1] |
| Protein binding | 89–90% |
| Metabolism | extensive hepatic, primarily CYP1A2 and CYP3A4-mediated |
| Elimination half-life | 0.9–1.7 h / 0.8–5.9 h (terminal) |
| Excretion | 80% in urine, 4% in feces |
| Identifiers | |
| CAS Number | |
| PubChemCID | |
| IUPHAR/BPS | |
| ChemSpider | |
| UNII | |
| ChEBI | |
| ECHA InfoCard | 100.114.889 |
| Chemical and physical data | |
| Formula | C15H19NO2 |
| Molar mass | 245.32 g/mol |
| 3D model (JSmol) | |

DR ANTHONY MELVIN CRASTO Ph.D
MOBILE-+91 9323115463
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
//////////////BMS-214778, VEC-162, Tasimelteon, Hetlioz, FDA 2014, 609799-22-6 , BMS-214778, VEC-162, ATC N05CH03, タシメルテオン , EU 2015, VANDA, BMS, orphan drug designations
CCC(=O)NCC1CC1C2=C3CCOC3=CC=C2
Chemical and physical properties
Tasimelteon has two stereogenic centers. Besides the medically used trans-1 R , 2 R isomer (in the picture above left), there are thus three further stereoisomers that do not arise in the synthesis.

Tasimelteon is a white to off-white crystalline non-hygroscopic substance, soluble in water at physiologically relevant pH levels and readily soluble in alcohols, cyclohexane and acetonitrile. The compound occurs in two crystal forms. It is an anhydrate melting at 74 ° C and a hemihydrate . [4] The hemihydrate is from about 35 ° C the water of hydration and converts thereby in the anhydrate form to. [4] The anhydrate crystallizes in a monoclinic lattice with the space group P 2 1 , and the hemihydrate crystallizes in a tetragonal lattice with the space group P 4 3 21 2. [4]
4 Kaihang Liu, Zhou Xinbo, Zhejing Xu, Bai Hongzhen, Jianrong Zhu Jianming Gu, Guping Tang, Liu Xingang, Hu Xiurong: anhydrate and hemihydrate of Tasimelteon: Synthesis, structure, and pharmacokinetic study in J. Pharm. Biomed. Anal. 151 (2018) 235-243, doi : 10.1016 / j.jpba.2017.12.035 .
Umeclidinium bromide, ウメクリジニウム臭化物

Umeclidinium bromide
GSK-573719A, ウメクリジニウム臭化物
- Molecular FormulaC29H34BrNO2
- Average mass508.490 Da
1-[2-(Benzyloxy)ethyl]-4-[hydroxy(diphenyl)methyl]-1-azoniabicyclo[2.2.2]octane bromide
1-Azoniabicyclo[2.2.2]octane, 4-(hydroxydiphenylmethyl)-1-[2-(phenylmethoxy)ethyl]-, bromide (1:1)
diphenyl-[1-(2-phenylmethoxyethyl)-1-azoniabicyclo[2.2.2]octan-4-yl]methanol;bromide
7AN603V4JV
869113-09-7 [RN]
9551
GSK573719A; UNII-7AN603V4JV
Umeclidinium bromide (trade name Incruse Ellipta) is a long-acting muscarinic antagonist approved for the maintenance treatment of chronic obstructive pulmonary disease (COPD).[1] It is also approved for this indication in combination with vilanterol (as umeclidinium bromide/vilanterol).[2][3]
In the 2014, the drug was also approved in the E.U. and in the U.S. for the maintenance treatment to relieve symptoms in adult patients with chronic obstructive pulmonary disease (COPD). It was launched in the U.K. in October 2014 and in the U.S. in January 2015. In Japan, the product candidate was approved in 2015 as monotherapy for the maintenance bronchodilator treatment to relieve symptoms in adult patients with chronic obstructive pulmonary disease (COPD) and launched on October in the same year.

Umeclidinium bromide (Ellipta)
Umeclidinium bromide is a long-acting muscarinic acetylcholine antagonist developed by GlaxoSmithKline and approved by the US FDA at the end of 2013 for use in combination with vilanterol, a b2 agonist, for the treatment of chronic obstructive pulmonary disease.269 Due to umeclidinium’s poor oral bioavailability, the drug is administrated by inhalation as dry powder.269
The most likely scale preparation of the drug is described in Scheme .270
Commercially available ethyl isonipecotate (278) was alkylated with 1-bromo-2-chloroethane in the presence of K2CO3 in acetone to give ethyl 1-(2-chloroethyl)piperidine-4-carboxylate (279). This material was then treated with lithium diisopropylamine (LDA) in THF to affect a transannular substitution reaction resulting in the cyclized quinuclidine 280 in 96% yield.270 Excess of phenyllithium was added to ester 280 in THF starting at low temperature then gradually warming to room temperature to give tertiary alcohol 281 in 61% yield. Amine 281 was finally alkylated with benzyl 2-bromoethyl ether (282) in MeCN/CHCl3 at elevated temperatures
to afford umeclidinium bromide (XXXV) in 69% yield.
269. Tal-Singer, R.; Cahn, A.; Mehta, R.; Preece, A.; Crater, G.; Kelleher, D.;Pouliquen, I. J. Eur. J. Pharmacol. 2013, 701, 40.
270. Laine, D. I.; McCleland, B.; Thomas, S.; Neipp, C.; Underwood, B.; Dufour, J.;Widdowson, K. L.; Palovich, M. R.; Blaney, F. E.; Foley, J. J.; Webb, E. F.;Luttmann, M. A.; Burman, M.; Belmonte, K.; Salmon, M. J. Med. Chem. 2009, 52, 2493.
FDA
https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/203975Orig1s000ChemR.pdf
1-[2-(benzyloxy)ethyl]-4-(hydroxydiphenylmethyl)-1-azoniabicyclo[2.2.2]octane bromide

PATENT
https://patents.google.com/patent/CN105461710A/en
umeclidinium bromide prepared patent US7439393, US RE44874, US 7488827, US 7498440, US7361787 and the like using phenyllithium prepared by reaction of intermediate 4 – [(diphenyl) hydroxymethyl] azabicyclo [2.2.2 ] octane.Specific methods: azabicyclo [2.2.2] octane-nucleophilic addition reaction with 4-carboxylate-fold amount of 2.02-2.5 phenyllithium occurs, the reaction temperature is controlled to -78 ° 0_15 ° C ο lithium Reagents expensive, difficult to store, use of harsh conditions, relatively high cost.
Example 1
Phenyl magnesium chloride: Under nitrogen atmosphere to 55g (2.3mol) of metallic magnesium sandpaper lit with 3 L of tetrahydrofuran was added dropwise 215g (1.91mol) chlorobenzene, micro-thermal reaction proceeds, controlled dropping, the reaction was kept boiling, dropwise for about 1.5 hours, after the dropping was heated slightly under reflux for 30min. Cool reserve.
[0008] Example 2
Phenyl magnesium bromide: The under argon 50.4g (2.lmol) sandpaper lit magnesium metal with 4.2 liters of anhydrous ethyl ether was added a solution of 300g (1.91mol) of bromobenzene, was added an iodine initiator, electrical hair fever reaction proceeds, controlled dropping, the reaction was kept boiling, about 1.5 hours dropwise was added dropwise to a gentle reflux heated 30min. Cool reserve.
[0009] Example 3
Preparation of crude product: azabicyclo [2.2.2] octane-4-carboxylate (135g, 0.736mo 1) was dissolved in 3L of tetrahydrofuran, under nitrogen, was cooled to -5~0 ° C, was added dropwise 300g preparation of benzyl bromide Grignard reagent. After incubation -5~0 ° C stirred for 1 hour (progress of the reaction was monitored by TLC sample). Adding 50ml of water quenching. Liquid separation, the aqueous phase was extracted twice with 500ml of tetrahydrofuran, and the combined organic phases were washed with water, dried and filtered. The solvent was partially removed under reduced pressure, the balance maintaining approximately 1L, the residue was stirred overnight at 20 ° C crystallization.Filtered, washed (petroleum ether 2 X 200 ml), the filter cake was dried at 40 ° C in vacuo to give a yellowish white crystals 121.2 g, yield 54.2%.
[0010] Example 4
Preparation of crude product: azabicyclo [2.2.2] octane-4-carboxylate (18.3g, 0.lOmo 1) was dissolved in 3L of tetrahydrofuran, under nitrogen, was cooled to 0~5 ° C, was added dropwise 0.25 mol phenyl magnesium chloride. After incubation 0~5 ° C stirred for 1 hour (progress of the reaction was monitored by TLC sample) o quenched with 10ml of water was added. Liquid separation, the aqueous phase was extracted twice with 100ml of tetrahydrofuran, and the combined organic phases were washed with water, dried and filtered. The solvent was partially removed under reduced pressure, the balance maintaining approximately 50mL, the residue was stirred overnight at 20 ° C crystallization.Filtered, washed (petroleum ether 2X20 ml), the filter cake was dried at 40 ° C in vacuo to give a yellowish white crystals 14.63 g, yield 48.1%.
[0011] Example 5
Preparation of crude product: azabicyclo [2.2.2] octane-4-carboxylate (18.38,0.1011101) ^ 31 was dissolved in tetrahydrofuran, under nitrogen, was cooled to 5~15 ° C, was added dropwise 0.30 mol of benzene bromide. After incubation 5~15 ° C stirred for 1 hour (progress of the reaction was monitored by TLC sample) o quenched with 10ml of water was added. Liquid separation, the aqueous phase was extracted twice with 100ml of tetrahydrofuran, and the combined organic phases were washed with water, dried and filtered. The solvent was partially removed under reduced pressure, the balance maintaining approximately 50mL, the residue was stirred overnight at 20 ° C crystallization.Filtered, washed (petroleum ether 2 X 20 ml), the filter cake was dried at 40 ° C in vacuo to yield 13.80 g of yellow-white crystals, yield 47.1%.
[0012] Example 6
Umeclidinium bromide purification: 100g crude product was dissolved in 320ml of water to 80 ° C a mixture of 640ml of acetone, add 5g active carbon, and filtered.The filtrate was cooled to 25 ° C, for 1 hour. Within 1 to 2 hours and cooled to 0~5 ° C for 3 hours. The filter cake with chilled 1: 2 acetone – washed twice with water (2x20ml). The filter cake was dried in vacuo at 60 ° C to give white crystalline solid (92 g, yield 92%). Purity (HPLC normalization method) 99.25%.
[0013] Example 7
Umeclidinium bromide purification: 100g crude product was dissolved in 180ml water at 50 ° C a mixture of 360ml of acetone, add 5g active carbon, and filtered.The filtrate was ~ 2 hours to 25 ° C, for 1 hour. Within 1 to 2 hours cooled to 0 ° C and left overnight protection. The filter cake with chilled 1: 2 acetone – washed twice with water (2x20ml). The filter cake was dried at 60 ° C in vacuo to give fine (98.3 g, yield 98.3%). Purity (HPLC normalization method) 97.75%.
PATENT
https://patents.google.com/patent/WO2014027045A1
International Patent Publication Number WO 2005/104745 (Glaxo Group Limited), filed 27th April 2005, discloses muscarinic acetylcholine receptor antagonists. In particular, WO 2005/104745 discloses 4- [hydroxy(diphenyl)methyl]-l-{2-[(phenylmethyl)oxy]ethyl}-l-azoniabicyclo[2.2.2]octane bromide, of formula (I), and a process for the preparation of this compound (Example 84):

4-[Hydroxy(diphenyl)methyl]-l-{2-[(phenylmethyl)oxy]ethyl}-l-azoniabicyclo[2.2.2]octane bromide may also be referred to as umeclidinium bromide.
International Patent Publication Number WO 2011/029896 (Glaxo Group Limited), filed 10th September 2010, discloses an alternative preparation for an early intermediate, ethyl-l-azabicyclo[2.2.2] octane-4-carboxylate, in the multi-step synthesis of umeclidinium bromide.
There exists a need for an alternative process for the preparation of umeclidinium bromide. In particular, a process that offers advantages over those previously disclosed in WO 2005/104745 and WO 2011/029896 is desired. Advantages may include, but are not limited to, improvements in safety, control (i.e of final product form and physical characteristics), yield, operability, handling, scalability, and efficiency.
Summary of the Invention
The present invention provides, in a first aspect, a process for the preparation of umeclidinium bromide, which comprises: a) reacting ((2-bromoethoxy)methyl)benzene, of formula (II)

in a dipolar aprotic solvent with a boiling point greater than about 90°C or an alcohol with a boiling point greater than about 80°C; and optionally
b) re-crystallising the product of step (a).
The present invention is further directed to intermediates used in the preparation of the compound of formula (III), and hence of umeclidinium bromide. The process disclosed herein provides a number of advantages over prior art processes of WO 2005/104745 and WO 2011/029896.

PATENT
EP 3248970
FORM A B AND AMORPHOUS
https://patents.google.com/patent/EP3248970A1/en
The invention relates to novel solid forms of umeclidinium bromide (I), chemically 1-[2-(benzyloxy)ethyl]-4-(hydroxydiphenylmethyl)-1-azabicyclo[2.2.2]octane bromide. In particular, to its novel crystalline forms, identified as form A and form B, as well as to an amorphous form, and to their characterization by means of analytic methods. The invention further relates to methods of their preparation and their use for the preparation of umeclidinium bromide in the API quality.

Umeclidinium bromide is indicated as an inhalation anticholinergic drug with an ultra-long-term effect in cooperating patients with the diagnosis of COPD (chronic obstructive pulmonary disease). COPD is defined as a preventable and treatable disease that is characterized by a persistent obstruction of air flow in the bronchi (bronchial obstruction), which usually progresses and is related to an intensified inflammatory response of the airways to harmful particles or gases. The main goal of the treatment of COPD is an improvement of the current control, i.e. elimination of symptoms, improvement of toleration of physical effort, improvement of the health condition and reduction of future risks, i.e. prevention and treatment of exacerbations, prevention of progression of the disease and mortality reduction
The structure of umeclidinium bromide, 1-[2-(benzyloxy)ethyl]-4-(hydroxydiphenylmethyl)-1-azabicyklo[2.2.2]octane bromide, is first mentioned in the general patent application WO2005009362 of 2003 .
Preparation of umeclidinium bromide is first disclosed in the patent EP 1 740 177B ( WO2005104745 ), where two methods (A and B) are mentioned, differing in the final processing and the product yield (method B included in Scheme 1). There, the last steps of the synthesis are described, the product being described by means of EI-MS, 1H NMR and elementary analysis. There is no information concerning the chemical purity or polymorphic form.

Another preparation method of umeclidinium bromide is disclosed in the patent application WO 2014027045 , where three forms are also described (identified as forms 1 to 3), prepared using a method that is different from the procedure disclosed in the patent EP 1 740 177B .
-
- Example 5
Preparation of the amorphous form of umeclidinium bromide
1-[2-(benzyloxy)ethyl]-4-(hydroxydiphenylmethyl)-1-azabicyclo[2.2.2]octane bromide (100 mg, 0.197 mmol, purity UPLC 98.89%) is dissolved at the temperature of 25°C in a water: tert-butanol mixture in the volume ratio of 6:4 (total 70 ml). The clear solution is freeze-dried (a bath with a mixture of dry ice and ethanol, -70°C) and lyophilized (vacuum: 1.8 Pa for 72 h). An amorphous form of umeclidinium bromide was obtained (100 mg). This amorphous form was confirmed with DSC and X-ray powder diffraction. The X-ray powder diffraction pattern is shown in Fig. 8 and the DSC record in Fig. 9.
PAPER
Synthetic Communications An International Journal for Rapid Communication of Synthetic Organic Chemistry , Volume 48, 2018 – Issue 9, Convenient new synthesis of umeclidinium bromide
Umeclidinium bromide, a drug used for chronic obstructive pulmonary disease, is synthesized through a new intermediate of phenyl(quinuclidin-4-yl)methanone. This novel method with simple operation flow and cheap reagents, makes it suitable for scale up. The overall four-step process provides umeclidinium bromide in 29% yield and the purity up to 99.83%. The X-ray crystal structure of the drug molecule was first reported.



External links
References
- ^ Jump up to:a b “Incruse Ellipta (umeclidinium inhalation powder) for Oral Inhalation Use. Full Prescribing Information” (PDF). GlaxoSmithKline, Research Triangle Park, NC 27709. Retrieved 22 February 2016.
- Jump up^ Feldman, GJ; Edin, A (2013). “The combination of umeclidinium bromide and vilanterol in the management of chronic obstructive pulmonary disease: Current evidence and future prospects”. Therapeutic advances in respiratory disease. 7 (6): 311–9. doi:10.1177/1753465813499789. PMID 24004659.
- Jump up^ “FDA Approves Umeclidinium and Vilanterol Combo for COPD”. Medscape. December 18, 2013.
|
|
| Clinical data | |
|---|---|
| Trade names | Incruse Ellipta |
| Synonyms | GSK573719A |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Inhalation (DPI) |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | ~89%[1] |
| Metabolism | Hepatic (CYP2D6) |
| Elimination half-life | 11 hours |
| Excretion | Feces (58%) and urine(22%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| KEGG | |
| ChEBI | |
| ECHA InfoCard | 100.166.375 |
| Chemical and physical data | |
| Formula | C29H34BrNO2 |
| Molar mass | 508.49 g/mol |
| 3D model (JSmol) | |
//////////////Umeclidinium bromide, Incruse Ellipta, ウメクリジニウム臭化物 , GSK573719A, UNII-7AN603V4JV, FDA 2014
C1C[N+]2(CCC1(CC2)C(C3=CC=CC=C3)(C4=CC=CC=C4)O)CCOCC5=CC=CC=C5.[Br-]
Synthesis
| FDA Orange Book Patents: 1 of 15 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 9750726 |
| Expiration | Nov 29, 2030 |
| Applicant | GLAXOSMITHKLINE |
| Drug Application |
|
from FDA Orange Book
| FDA Orange Book Patents: 2 of 15 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 6759398 |
| Expiration | Aug 3, 2021 |
| Applicant | GLAXOSMITHKLINE |
| Drug Application |
|
from FDA Orange Book
| FDA Orange Book Patents: 3 of 15 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 7439393 |
| Expiration | May 21, 2025 |
| Applicant | GLAXOSMITHKLINE |
| Drug Application |
|
from FDA Orange Book
| FDA Orange Book Patents: 4 of 15 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 7629335 |
| Expiration | Aug 3, 2021 |
| Applicant | GLAXOSMITHKLINE |
| Drug Application |
|
from FDA Orange Book
| FDA Orange Book Patents: 5 of 15 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 7776895 |
| Expiration | Sep 11, 2022 |
| Applicant | GLAXOSMITHKLINE |
| Drug Application |
|
from FDA Orange Book
| FDA Orange Book Patents: 6 of 15 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 8161968 |
| Expiration | Feb 5, 2028 |
| Applicant | GLAXOSMITHKLINE |
| Drug Application |
|
from FDA Orange Book
| FDA Orange Book Patents: 7 of 15 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 8201556 |
| Expiration | Feb 5, 2029 |
| Applicant | GLAXO GRP ENGLAND |
| Drug Application | N205382 (Prescription Drug: INCRUSE ELLIPTA . Ingredients: UMECLIDINIUM BROMIDE) |
from FDA Orange Book
| FDA Orange Book Patents: 8 of 15 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 6537983 |
| Expiration | Aug 3, 2021 |
| Applicant | GLAXOSMITHKLINE |
| Drug Application |
|
from FDA Orange Book
| FDA Orange Book Patents: 9 of 15 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 7498440 |
| Expiration | Apr 27, 2025 |
| Applicant | GLAXOSMITHKLINE |
| Drug Application |
|
from FDA Orange Book
| FDA Orange Book Patents: 10 of 15 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 7488827 |
| Expiration | Dec 18, 2027 |
| Applicant | GLAXOSMITHKLINE |
| Drug Application |
|
from FDA Orange Book
| FDA Orange Book Patents: 11 of 15 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 8183257 |
| Expiration | Jul 27, 2025 |
| Applicant | GLAXOSMITHKLINE |
| Drug Application |
|
from FDA Orange Book
| FDA Orange Book Patents: 12 of 15 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 6878698 |
| Expiration | Aug 3, 2021 |
| Applicant | GLAXOSMITHKLINE |
| Drug Application |
|
from FDA Orange Book
| FDA Orange Book Patents: 13 of 15 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 8511304 |
| Expiration | Jun 14, 2027 |
| Applicant | GLAXOSMITHKLINE |
| Drug Application |
|
from FDA Orange Book
| FDA Orange Book Patents: 14 of 15 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | RE44874 |
| Expiration | Mar 23, 2023 |
| Applicant | GLAXOSMITHKLINE |
| Drug Application |
|
from FDA Orange Book
| FDA Orange Book Patents: 15 of 15 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 8309572 |
| Expiration | Apr 27, 2025 |
| Applicant | GLAXOSMITHKLINE |
| Drug Application |
|
from FDA Orange Book
VORAPAXAR SULPHATE

VORAPAXAR
Thrombosis, Antiplatelet Therapy, PAR1 Antagonists , MERCK ..ORIGINATOR
Ethyl N-[(3R,3aS,4S,4aR,7R,8aR,9aR)-4-[(E)-2-[5-(3-fluorophenyl)-2-pyridyl]vinyl]-3-methyl-1-oxo-3a,4,4a,5,6,7,8,8a,9,9a-decahydro-3H-benzo[f]isobenzofuran-7-yl]carbamate
Carbamic acid, [(1R,3aR,4aR,6R,8aR,9S,9aS)-9-[(1E)-2-[5-(3-fluorophenyl)-2- pyridinyl]ethenyl]dodecahydro-1-methyl-3-oxonaphtho[2,3-c]furan-6-yl]-, ethyl ester
Carbamic acid, N-[(1R,3aR,4aR,6R,8aR,9S,9aS)-9-[(E)-2-[5-(3-fluorophenyl)-2-pyridinyl]ethenyl]dodecahydro-1-methyl-3-oxonaphtho[2,3-c]furan-6-yl]-, ethyl ester
Ethyl [(1R,3aR,4aR,6R,8aR,9S,9aS)-9-{(E)-2-[5-(3-fluorophenyl)-2-pyridinyl]vinyl}-1-methyl-3-oxododecahydronaphtho[2,3-c]furan-6-yl]carbamate
Ethyl ((1R,3aR,4aR,6R,8aR,9S,9aS)-9-((1E)-2-(5-(3-fluorophenyl)pyridin-2-yl)ethenyl)- 1-methyl-3-oxododecahydronaphtho(2,3-c)furan-6-yl)carbamate
Carbamic acid, ((1R,3aR,4aR,6R,8aR,9S,9aS)-9-((1E)-2-(5-(3-fluorophenyl)-2- pyridinyl)ethenyl)dodecahydro-1-methyl-3-oxonaphtho(2,3-c)furan-6-yl)-, ethyl ester
618385-01-6 CAS NO FREE FORM
CAS Number: 705260-08-8 SULPHATE
Has antiplatelet activity.
Also known as: SCH-530348, MK-5348
Molecular Formula: C29H33FN2O4
Molecular Weight: 492.581723
ZCE93644N2
- UNII-ZCE93644N2
- Zontivity
Registered – 2015 MERCK Thrombosis
Vorapaxar (formerly SCH 530348) is a thrombin receptor (protease-activated receptor, PAR-1) antagonist based on the natural product himbacine. Discovered by Schering-Plough and currently being developed by Merck & Co., it is an experimental pharmaceutical treatment for acute coronary syndrome chest pain caused by coronary artery disease.[1]
In January 2011, clinical trials being conducted by Merck were halted for patients with stroke and mild heart conditions.[2] In a randomized double-blinded trial comparing vorapaxar with placebo in addition to standard therapy in 12,944 patients who had acute coronary syndromes, there was no significant reduction in a composite end point of death from cardiovascular causes, myocardial infarction, stroke, recurrent ischemia with rehospitalization, or urgent coronary revascularization. However, there was increased risk of major bleeding.[3]
A trial published in February 2012, found no change in all cause mortality while decreasing the risk of cardiac death and increasing the risk of major bleeding.[4]
SCH-530348 is a protease-activated thrombin receptor (PAR-1) antagonist developed by Schering-Plough and waiting for approval in U.S. for the oral secondary prevention of cardiovascular events in patients with a history of heart attack and no history of stroke or transient ischemic attack. The drug candidate is being investigated to determine its potential to provide clinical benefit without the liability of increased bleeding; a tendency associated with drugs that block thromboxane or ADP pathways. In April 2006, SCH-530348 was granted fast track designation in the U.S. for the secondary prevention of cardiovascular morbidity and mortality outcomes in at-risk patients.
Vorapaxar was recommended for FDA approval on January 15, 2014.[5]
Vorapaxar is a protease-activated thrombin receptor (PAR-1) antagonist developed by Schering-Plough (now, Merck & Co.) and approved in the U.S. in 2014 for the reduction of thrombotic cardiovascular events in patients with a history of myocardial infarction or with peripheral arterial disease. However, in 2018 Aralez discontinued U.S. commercial operations. In 2015, the product was approved in the E.U. for the reduction of atherothrombotic events in adult patients with a history of myocardial infarction. In April 2006, vorapaxar was granted fast track designation in the U.S. for the secondary prevention of cardiovascular morbidity and mortality outcomes in at-risk patients. In 2016, Aralez Pharmaceuticals acquired the U.S. and Canadian rights to the product pursuant to an asset purchase agreement entered into between this company and Merck & Co.
Merck & Co (following its acquisition of Schering-Plough) has developed and launched vorapaxar (Zontivity; SCH-530348; MK-5348), an oral antagonist of the thrombin receptor (protease-activated receptor-1; PAR1); the product is marketed in the US by Aralez Pharmaceuticals
WO-03089428, published in October 2003, claims naphtho[2,3-c]furan-3-one derivatives as thrombin receptor antagonists. WO-03033501 and WO-0196330, published in April 2003 and December 2001, respectively, claim himbacine analogs as thrombin receptor antagonists. WO-9926943 published in June 1999 claims tricyclic compounds as thrombin receptor antagonists
 VORAPAXAR
VORAPAXAR17 JAN 2014
FDA advisory panel votes to approve Merck & Co’s vorapaxar REF 6
https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/204886Orig1s000ChemR.pdf
Zontivity (vorapaxar) tablets NDA 204886



VORAPAXAR SULPHATE

CAS Number: 705260-08-8 SULPHATE
Molecular Formula: C29H33FN2O4.H2O4S
Molecular Weight: 590.7
Chemical Name: Ethyl [(1R,3aR,4aR,6R,8aR,9S,9aS)-9-[(1E)-2-[5-(3-fluorophenyl)pyridin-2- yl]ethenyl]-1-methyl-3-oxododecahydronaphtho[2,3-c]furan-6-yl]carbamate sulfate
Synonyms: Carbamic acid, [(1R,3aR,4aR,6R,8aR,9S,9aS)-9-[(1E)-2-[5-(3-fluorophenyl)-2- pyridinyl]ethenyl]dodecahydro-1-methyl-3-oxonaphtho[2,3-c]furan-6-yl]-,ethyl ester,sulfate; SCH-530348
Vorapaxar Sulfate (SCH 530348) a thrombin receptor (PAR-1) antagonist for the prevention and treatment of atherothrombosis.
POLYMORPH
U.S.Pat. No. 7,304,078 discloses Vorapaxar base. U.S.Pat. No. 7,235,567 discloses Polymorph I and II of vorapaxar sulphate
CN 106478608 provides a crystalline polymorph A
EMA
Atherosclerosis and ischemic cardiovascular (CV) diseases like coronary artery disease (CAD) are progressive systemic disorders in which clinical events are precipitated by episodes of vascular thrombosis. Patients with an established history of atherothrombotic or athero-ischemic disease are at particular risk of future cardiac or cerebral events, and vascular death. Anti-thrombotic therapy options in patients with stable atherosclerosis are not well-established. Long-term therapies to effectively modulate the key components responsible for atherothrombosis in secondary prevention of ischemic CV disease are therefore required. Vorapaxar is a first – in – class selective antagonist of the protease-activated receptor 1 (PAR-1), the primary thrombin receptor on human platelets, which mediates the downstream effects of this critical coagulation factor in hemostasis and thrombosis. Thrombin-induced platelet activation has been implicated in a variety of cardiovascular disorders including thrombosis, atherosclerosis, and restenosis following percutaneous coronary intervention (PCI). As an antagonist of PAR-1, vorapaxar blocks thrombin-mediated platelet aggregation and thereby has the potential to reduce the risk of atherothrombotic complications of coronary disease. The applicant has investigated whether a new class of antiplatelet agents, PAR-1 antagonists, can further decrease the risk of cardiovascular events in a population of established atherothrombosis when added to standard of care, in secondary prevention of ischemic diseases. The following therapeutic indication has been submitted for vorapaxar: Vorapaxar is indicated for the reduction of atherothrombotic events in patients with a history of MI. Vorapaxar has been shown to reduce the rate of a combined endpoint of cardiovascular death, MI, stroke, and urgent coronary revascularization. Vorapaxar will be contraindicated in patients with a history of stroke or TIA. The indication sought in the current application is supported by the efficacy results of the TRA 2P-TIMI, which is considered the pivotal trial for this indication. During the procedure, the applicant requested the possibility of extending the indication initially sought for, to extend it to the population of PAD patients. This request was discussed at the CHMP and not accepted by the Committee.
Introduction The finished product is presented as immediate release film-coated tablets containing 2.5 mg of vorapaxar sulfate as active substance per tablet, corresponding to 2.08 mg vorapaxar. Other ingredients are: lactose monohydrate, microcrystalline cellulose (E460), croscarmellose sodium (E468), povidone (E1201) , magnesium stearate (E572), hypromellose (E464), titanium dioxide (E171), triacetin (glycerol triacetate) (E1518), iron oxide yellow (E172), as described in section 6.1 of the SmPC. The product is available in Aluminium–Aluminium blisters (Alu-Alu) as described in section 6.5 of the SmPC.
General information The chemical name of the active substance vorapaxar sulfate is ethyl[(1R,3aR,4aR,6R,8aR,9S,9aS)- -9-{(1E)-2-[5-(3-fluorophenyl)pyridin-2-yl]ethen-1-yl}-1-methyl-3-oxododecahydronaphtho[2,3-c] furan-6-yl]carbamate sulfate, corresponding to the molecular formula C29H33FN2O4 • H2SO4 and has a relative molecular mass 590.7. It has the following structure:
The structure of the active substance has been confirmed by mass spectrometry, infrared spectroscopy, 1H- and 13C-NMR spectroscopy and X-ray crystallography, all of which support the chemical structure elemental analysis. It appears as a white to off-white, slightly hygroscopic, crystalline powder. It is freely soluble in methanol and slightly soluble in ethanol and acetone but insoluble to practically insoluble in aqueous solutions at pH above 3.0. The highest solubility in aqueous solution can be achieved at pH 1.0 or in simulated gastric fluids at pH 1.4. The dissociation constant of vorapaxar sulfate was determined to be pKa = 4.7 and its partition coefficient LogP was determined to be 5.1. Vorapaxar sulfate contains seven chiral centers and a trans double bond. The seven chiral centres are defined by the manufacturing process of one of the intermediates in the vorapaxar synthesis and potential enantiomers are controlled by appropriate specifications. The cis-isomer of the double bond is controlled by a highly stereo-specific process reaction resulting in non-detectable levels of cis-isomer impurity. The cis-isomer impurity is controlled in one of the intermediates as an unspecified impurity. A single crystalline stable anhydrous form has been observed.
GENERAL INTRODUCTION
SIMILAR NATURAL PRODUCT
+ HIMBACINE


Himbacine is an alkaloid muscarinic receptor antagonist displaying more potent activity associated with M2 and M2 subtypes over M1 or M3. Observations show himbacine bound tightly to various chimeric receptors in COS-7 cells as well as possessed the ability to bind to cardiac muscarinic receptors allosterically. Recent studies have produced series of thrombin receptor (PAR1) antagonists derived from himbacine Himbacine is an inhibitor of mAChR M2 and mAChR M4.
Technical Information
| Physical State: | Solid |
| Derived from: | Australian pine Galbulimima baccata |
| Solubility: | Soluble in ethanol (50 mg/ml), methanol, and dichloromethane. Insoluble in water. |
| Storage: | Store at -20° C |
| Melting Point: | 132-134 °C |
| Boiling Point: | 469.65 °C at 760 mmHg |
| Density: | 1.08 g/cm3 |
| Refractive Index: | n20D 1.57 |
| Optical Activity: | α20/D +51.4º, c = 1.01 in chloroform |
| Application: | An alkaloid muscarinic receptor antagonist |
| CAS Number: | 6879-74-9 |
| Molecular Weight: | 345.5 |
| Molecular Formula: | C22H35NO2 |
General scheme:

PATENT
WO 2006076415
WO 2006076452
WO 2003089428
US 6063847
CN 107540564
WO 2008005344
CN 106749138
PATENT
Example 1:
[0027] The steel shed amide (300mg, 7. 93mmol) and 15 blood THF was added to 100 blood Ξ jar. The starting material II (2.OOg, 5. 89mmol) was dissolved in 15mL of THF dropwise via pressure-equalizing dropping funnel to the reaction system, the process temperature will produce a large number of bubbles -2 ~ 0 ° C, in the process, Lan mix of about 0.1 until no bubbles generate. THF solution containing 13 Blood Ship (0.75 Yap, 2. 95mmol) is transferred to a pressure-equalizing dropping funnel. It was slowly added dropwise to the reaction system. After the completion of dropwise continue to embrace mix ratio. After the treatment, at 0 ° C under 0.8 blood, Imol / L 1 fat slowly dropped into the embrace mixed reaction system, after adding the right amount of water, acetic acid extraction. The combined organic phase with Imol / L of 0H (17mLX3) washing the organic phase coating. Tu brine, dried over anhydrous sulfate steel, 25 ° C under reduced pressure to spin dry to give 1. 75g light yellow oil, yield 91%.
[0028] After the content was determined using the external standard method, first prepared by a qualified reference determine its content, W this as a standard substance, measuring the external standard method to get the content of 99%.
[0029] Zan NMR: (400MHz, CD3CN):… 5 46 of r, 1H), 4 70 (td, 1H), 4 03 based 2H), 3 69-3 57 (m, 2 Η).. , 3. 45-3. 32 (based, IH), 2. 77 (br, IH), 2. 61-2. 51 (m, IH), 2. 49-2. 39 (m, 1 field, 2 30 of r IH), 2 .12-1. 92 (m, IH), 1. 87 (dt, IH), 1. 81-1. 72 (m, IH), 1. 61-1. 50 ( …. m, IH), 1 48 (d, 3H), 1 23-1 09 (m, 7H), 1. 05-0 90 (m, 2H);
[0030] MS (ES +) m / z: 326. 24 [M + + field.
[Cited 00] Example 2:
[003 cited the steel shed amide (312mg, 8. 25mmol) and 16 blood THF was added to the lOOmL Ξ jar. The starting material II (2.OOg, 5. 89mmol) was dissolved in 15mL of THF dropwise via pressure-equalizing dropping funnel to the reaction system, the process temperature will produce a large number of bubbles -2 ~ -5 ° C, in the process and takes about 45min mix until no bubbles generate. The 13 ships of blood containing 60g, 2. 36mmol) in THF solution was transferred to a pressure-equalizing dropping funnel. It was slowly added dropwise to the reaction system. After the completion of dropwise continue to embrace mix ratio. After the treatment, at 0 ° C under 0.8 blood, Imol / L 1 fat slowly dropped into the embrace mixed reaction system, after adding the right amount of water, acetic acid extraction. The combined organic phase with llmol / L of 0H (17mLX3) washing the organic phase coating. Tu brine, dried over anhydrous sulfate steel, 25 ° C under reduced pressure to spin dry to give 1. 65g light yellow oil.
[0033] Determination of Reference Example 1 in an amount of 98.7%.
[0034] MS (ES +) m / z: 326. 24 [M + + field.
[003 cited Example 3:
[0036] 50 single jar of blood, condenser. Intermediate inb (l.〇〇g, 3. 07mmol) was dissolved in 10ml of dichloromethane burn during and after the blood was added to a 50-port flask, make dioxide of 32g, 3.68mmol), the reaction of reflux. After completion of the reaction by TLC, cooled to 20 ~ 25 ° C after suction filtration, the filter cake rinsed with methylene burning (the X3 3 blood), at 30 ° CW and the filtrate was concentrated to dryness. To the residue was added 5 blood acetic acid, at 20 ~ 25 ° C after mixing 0. embrace of suction, the resulting cake was vacuum dried at 30 ° C 10 ~ 12h. Give 0. 87g of white solid.
[0037] Electric NMR: (400MHz, CD3CN):. 9 74 oriented 1H), 5 40 of r, 1H), 4 77-4.66 (m, 1H), 4 09-3 98 (m, 2H…. ), 3. 49-3. 37 (m, IH), 2. 75-2. 64 (m, 2H), 2. 55-2. 48 (m, IH), 1. 95-1. 87 (m , 2H), 1. 89-1 .77 (m, 2H), 1. 61-1. 49 (m, IH), 1. 32-1. 13 (m, 9H), 1. 08-0. 82 (m, 2H);
[0038] MS (ES +) m / z: 324. 33 [M + + field.
PATENT
CN 106478608 crystal
https://patents.google.com/patent/CN106478608A/en
The present invention provides a crystalline polymorph A one kind of the compound of formula I:

In another embodiment, the present invention provides a method of preparing a crystalline polymorph of compound A I,

Which comprising, a) the compound II is dissolved in acetonitrile and stirred to form a mixture; b) heating the mixture to 50 ° C ~ 70 ° C; c) adding sulfuric acid to the heated mixture; d) evaluating the temperature was lowered to 0 ° C ~ 20 ° C, seeded and stirred to precipitate crystals.
Preparation [0042] A crystalline polymorph of the compound of Example 1 I

Compound II (1. 0g) was dissolved in 5. 0ml of acetonitrile, stirred and heated to 50 ° C ~ 70 ° C was added and this temperature was added 1.2ml 2N H2S04 / acetonitrile solution and then lowering the temperature of the system to 15 ° C ~ 20 ° C, the system was added to the appropriate amount of seed crystals and stirred for 2h, the precipitated solid was filtered and the cake washed twice with 2. 5ml of acetonitrile to give a white solid, the white solid was placed under 40 ° C desolventizing 2 hours and then dried at 80 ° C for vacuo to give a white solid 0. 83 g, 69. 3% yield, HPLC:. 99 94%. A powder X-ray diffraction spectrum shown in Figure 1, a DSC endothermic curve shown in Figure 2, which HPLC profile shown in Fig.
PATENT
https://patents.google.com/patent/CN106478608A/en
PATENT
WO 2009093972 synthesis
https://encrypted.google.com/patents/WO2009093972A1?cl=ko&hl=en&output=html_text
Clip
Vorapaxar sulfate (Zontivity)
Merck Sharp & Dohme successfully obtained approval in the EU in 2014 for vorapaxar sulfate, marketed as Zontivity. The drug is a first-in-class thrombin receptor (also referred to as a protease-activated or PAR-1) antagonist which, when used in conjunction with antiplatelet therapy, has been shown to reduce the chance of
myocardial infarction and stroke, particularly in patients with a history of cardiac events.277
Antagonism of PAR-1 allows for thrombin-mediated fibrin deposition while blocking thrombinmediated platelet activation.277 Although a variety of papers and patents describe the synthesis of vorapaxar sulfate (XXXVII),278–282 a combination of two patents describe the largest-scale synthesis reported in the literature, and this is depicted in Scheme 52.


Retrosynthetically, the drug can be divided into olefination partners 306 and 305.283,284 Lactone 305
is further derived from synthons 300 and 299, which are readily prepared from commercially available starting materials. Dienyl acid 300 was constructed in two steps starting from commercial vinyl bromide 307, which first undergoes a Heck reaction with methacrylate (308) followed by saponification of the ester to afford the desired acid 300 in 71% over two steps (Scheme 53).
The synthesis of alcohol 299 begins with tetrahydropyranyl (THP) protection of enantioenriched alcohol 295 to afford butyne 297 (Scheme 52). Lithiation of this system followed by trapping with (benzyloxy)chloroformate and Dowex work-up to remove the protective functionality provided acetyl ester 298. Hydrogenation of the alkyne with Lindlar’s catalyst delivered cis-allylic alcohol 299 in 93% yield. Acid 300 was then esterified with alcohol 299 by way of a 1,3-dicyclohexylcarbodiimide (DCC) coupling and, upon heating in refluxing xylenes, an intramolecular Diels–
Alder reaction occurred. Subsequent subjection to DBU secured the tricyclic system 301 in 38% over three steps as a single enantiomer.
Diastereoselective hydrogenation reduced the olefin with concomitant benzyl removal to give key fragment 302. Next, acidic revelation of the ketone followed by reductive amination with ammonium formate delivered primary amines 303a/303b as a mixture of diastereomers. These amines were then converted to the corresponding carbamates, and resolution by means of recrystallization yielded 50% of 304 as the desired diastereomer. Acid 304
was treated with oxalyl chloride and the resulting acid chloride was reduced to aldehyde 305 in 66% overall yield. Finally, deprotonation of phosphonate ester 306 (whose synthesis is described in Scheme 54) followed by careful addition of 305 and acidic quench delivered vorapaxar sulfate (XXXVII) in excellent yield over the
two-step protocol.
The preparation of vorapaxar phosponate ester 306 (Scheme 54)commenced from commercial sources of 5-(3-fluorophenyl)-2-methylpyridine (310). Removal of the methyl proton with LDA followed by quench with diethyl chlorophosphonate resulted in phosponate ester 306.

277. Frampton, J. E. Drugs 2015, 75, 797.
278. Chackalamannil, S.; Wang, Y.; Greenlee, W. J.; Hu, Z.; Xia, Y.; Ahn, H.; Boykow,G.; Hsieh, Y.; Palamanda, J.; Agans-Fantuzzi, J.; Kurowski, S.; Graziano, M.;Chintala, M. J. Med. Chem. 2008, 51, 3061.
279. Sudhakar, A.; Kwok, D.; Wu, G. G.; Green, M. D. WO Patent 2006076452A2,2006.
280. Wu, G. G.; Sudhakar, A.; Wang, T.; Ji, X.; Chen, F. X.; Poirier, M.; Huang, M.;Sabesan, V.; Kwok, D.; Cui, J.; Yang, X.; Thiruvengadam, T.; Liao, J.; Zavialov, I.;Nguyen, H. N.; Lim, N. K. WO Patent 2006076415A2, 2006.
281. Yong, K. H.; Zavialov, I. A.; Yin, J.; Fu, X.; Thiruvengadam, T. K. US Patent20080004449A1, 2008.
282. Chackalamannil, S.; Clasby, M.; Greenlee, W. J.; Wang, Y.; Xia, Y.; Veltri, E.;Chelliah, M. WO Patent 03089428A1, 2003.
283. Thiruven-Gadam, T. K.; Wang, T.; Liao, J.; Chiu, J. S.; Tsai, D. J. S.; Lee, H.; Wu,W.; Xiaoyong, F. WO Patent 2006076564A1, 2006.
284. Chackalamannil, S.; Asberon, T.;Xia, Y.; Doller, D.; Clasby, M. C.; Czarniecki,M. F. US Patent 6,063,847, 2000.
PRODUCT PATENT
SYNTHESIS
Inventor Samuel ChackalamannilMartin C. ClasbyWilliam J. GreenleeYuguang WangYan XiaEnrico P. VeltriMariappan ChelliahWenxue Wu
Original Assignee Schering Corporation
Priority date 2002-04-16
THE EXACT BELOW COMPD IS 14
Example 2
Step 1 :

Phosphonate 7, described in US 6,063,847, (3.27 g, 8.1 mmol) was dissolved in THF (12 ml) and C(O)Oled to 0 °C, followed by addition of 2.5 M n- BuLi (3.2 ml, 8.1 mmol). The reaction mixture was stirred at 0 °C for 10 min and warmed up to rt. A solution of aldehyde 6, described in US 6,063,847, in THF (12 ml) was added to the reaction mixture. The reaction mixture was stirred for 30 min. Standard aqueous work-up, followed by column chromatography (30-50% EtOAc in hexane) afforded product 8. 1HNMR (CDCI3): δ 0.92-1.38 (m, 31 H), 1.41 (d, J= 6 Hz, 3H), 1.40-1.55 (m, 2H), 1.70-1.80 (m, 2H), 1.81-1.90 (m, 2H), 2.36 (m, 2H), 2.69 (m, 1 H), 3.89 (m, 4H), 4.75 (m, 1 H), 6.28-6.41 (m, 2H), 7.05-7.15 (m, 2H), 8.19 (br s, 1 H). Step 2:

Compound 8 (2.64 g, 4.8 mmol) was dissolved in THF (48 ml). The reaction mixture was C(O)Oled to 0 °C followed by addition of 1 M TBAF (4.8 ml). The reaction mixture was stirred for 5 min followed by standard aqueous work-up. Column chromatography (50% EtOAc/hexane) afforded product 9 (1.9 g, 100%). 1HNMR (CDCI3): δ 1.15-1.55 (m, 6H), 1.41 (d, J= 6 Hz, 3H), 1.70-1.82 (m, 3H), 1.85-1.90 (m, 1 H), 2.36 (m, 2H), 2.69 (m, 1 H), 3.91 (m, 4H), 4.75 (m, 1 H), 6.18- 6.45 (m, 2H), 7.19 (br s, 2H), 8.19 (br s, 1 H). Step 3:

To a solution of compound 9 (250 mg, 0.65 mmol) in pyridine (5 ml) C(O)Oled to 0 °C was added Tf2O (295 μL, 2.1 mmol). The reaction mixture was stirred overnight at rt. Standard aqueous work-up followed by column chromatography afforded product 10 (270 mg, 80%). 1HNMR (CDCI3): δ 1.15-1.55 (m, 6H), 1.41 (d, J= 6 Hz, 3H), 1.70-1.82 (m, 3H), 1.85-1.90 (m, 1 H), 2.36 (m, 2H), 2.69 (m, 1 H), 3.91 (m, 4H), 4.75 (m, 1 H), 6.42-6.68 (m, 2H), 7.25 (m, 1 H), 7.55 (m, 1 H), 8.49 (d, J= 2.8 Hz, 1 H).

Compound 10 (560 mg, 1.1 mmol), 3-fluorophenyl boronic acid (180 mg, 1.3 mmol) and K2CO3 (500 mg, 3.6 mmol) were mixed with toluene (4.4 ml), H2O (1.5 ml) and EtOH (0.7 ml) in a sealed tube. Under an atmosphere of N2, Pd(Ph3P)4 (110 mg, 0.13 mmol) was added. The reaction mixture was heated at 100 °C for 2 h under N2. The reaction mixture was C(O)Oled down to rt, poured to EtOAc (30 ml) and washed with water (2X20 ml). The EtOAc solution was dried with NaHCO3 and concentrated at reduced pressure to give a residue. Preparative TLC separation of the residue (50% EtOAc in hexane) afforded product 11 (445 mg, 89%). 1HNMR (CDCI3): δ 1.15-1.59 (m, 6H), 1.43 (d, J= 6 Hz, 3H), 1.70-1.79 (m, 2H), 1.82 (m, 1H), 1.91 (m, 2H), 2.41 (m, 2H), 2.69 (m, 1 H), 3.91 (m, 4H), 4.75 (m, 1 H), 6.52-6.68 (m, 2H), 7.15 (m, 1 H), 7.22 (m, 2H), 7.35 (m, 1 H), 7.44 (m, 1 H), 7.81 (m, 1 H), 8.77 (d, J= 1.2 Hz, 1 H). Step 5:

Compound 11 (445 mg, 0.96 mmol) was dissolved in a mixture of acetone (10 ml) and 1 N HCI (10 ml). The reaction mixture was heated at 50 °C for 1 h.
Standard aqueous work-up followed by preparative TLC separation (50% EtOAc in hexane) afforded product 12 (356 mg, 89%). 1HNMR (CDCI3): δ 1.21-1.45 (m, 2H), 1.47 (d, J= 5.6 Hz, 3H), 1.58-1.65 (m, 2H), 2.15 (m, 1 H), 2.18-2.28 (m, 2H), 2.35- 2.51 (m, 5H), 2.71 (m, 1 H), 4.79 (m, 1 H), 6.52-6.68 (m, 2H), 7.15 (m, 1 H), 7.22 (m, 2H), 7.35 (m, 1 H), 7.44 (m, 1 H), 7.81 (m, 1 H), 8.77 (d, J= 1.2 Hz, 1 H). Step 6:

Compound 12 (500 mg, 4.2 mmol) was dissolved in EtOH (40 ml) and CH2CI2 (15 ml) NH3 (g) was bubbled into the solution for 5 min. The reaction mixture was C(O)Oled to 0 °C followed by addition of Ti(O/‘Pr)4 (1.89 ml, 6.3 mmol). After stirring at 0 °C for 1 h, 1 M TiCI (6.3 ml, 6.3 mmol) was added. The reaction mixture was stirred at rt for 45 min and concentrated to dryness under reduced pressure. The residue was dissolved in CH3OH (10 ml) and NaBH3CN (510 mg, 8 mmol) was added. The reaction mixture was stirred overnight at rt. The reaction mixture was poured to 1 N NaOH (100 ml) and extracted with EtOAc (3x 100 ml). The organic layer was combined and dried with NaHC03. Removal of solvent and separation by PTLC (5% 2 M NH3 in CH3OH/ CH2CI2) afforded β-13 (spot 1 , 30 mg, 6%) and α-13 (spot 2, 98 mg, 20%). β-13: 1HNMR (CDCI3): δ 1.50-1.38 (m, 5H), 1.42 (d, J= 6 Hz, 3H), 1.51-1.75 (m, 5H), 1.84 (m, 2H), 2.38 (m, 1 H), 2.45 (m, 1 H), 3.38 (br s, 1 H), 4.78 (m, 1 H), 6.59 (m, 2H), 7.15 (m, 1 H), 7.26 (m, 2H), 7.36 (m, 1 H), 7.42 (m, 1 H), 7.82 (m, 1 H), 8.77 (d, J= 2 Hz, 1 H). α-13:1HNMR (CDCI3): δ 0.95 (m, 2H), 1.02-1.35 (m, 6H), 1.41 (d, J= 6 Hz, 3H), 1.82-1.95 (m, 4H), 2.37 (m; 2H), 2.69 (m, 2H), 4.71 (m, 1 H), 6.71 (m, 2H), 7.11 (m, 1 H), 7.25 (m, 2H), 7.38 (m, 1 H), 7.42 (m, 1 H), 7.80 (m, 1 H), 8.76 (d, J= 1.6 Hz, 1 H). Step 7:
Compound α-13 (300 mg, 0.71 mmol) was dissolved in CH2CI2 (10 ml) followed by addition of Et3N (0.9 ml). The reaction mixture was C(O)Oled to 0 °C and ethyl chloroformate (0.5 ml) was added. The reaction mixture was stirred at rt for 1 h. The reaction mixture was directly separated by preparative TLC (EtOAc/ hexane, 1 :1) to give the title compound (14) VORAPAXAR (300 mg, 86%). MS m/z 493 (M+1).
HRMS Calcd for C29H34N2O4F (M+1 ): 493.2503, found 493.2509.
PATENT
SYNTHESIS 1
http://www.google.com/patents/WO2006076564A1
VORAPAXAR= COMPD A
Example 6 – Preparation of Compound A

To a three-neck flask equipped with an agitator, thermometer and nitrogen inertion was added 7A (13.0 g), THF (30 mL). The mixture was cooled to below -200C after which lithium diisopropylamide (2M, 20 mL) was slowly added. The reaction mixture was agitated for an additional hour (Solution A). To another flask was added 6 (10.0 g) and THF (75 mL) . The mixture was stirred for about 30 minutes and then slowly transferred into the solution A while maintaining the temperature below 200C. The mixture was stirred at below -200C for an additional hour before quenching the reaction by adding 20 mL of water. The reaction mixture was warmed to 00C and the pH was adjusted to about 7 by addition of 25% HaSO4 (11 mL). The mixture was further warmed to 200C and then diluted with 100 mL of ethyl acetate and 70 mL of water. The two phases that had formed were separated and the aqueous layer was extracted with 50 mL of ethyl acetate. The solvents THF and ethyl acetate were then replaced with ethanol, and the Compound A was precipitated out as a crystalline solid from ethanol with seeding at 35 to 4O0C. After cooling to O0C, the suspension was stirred for an additional hour and then the product was filtered and washed with cold ethanol. The product was dried at 50 – 600C under vacuum to provide an off-white solid. VORAPAXAR
Yield: 12.7 g, (90%). m.p. 104.90C (DSC onset point).
1H NMR (CDCl3) δ 8.88 (d, J = 2.4 Hz, IH), 8.10 (dd, J = 8.2, 2.4 Hz, IH), 7.64 (IH), 7.61 (d, J = 8.8 Hz, IH), 7.55 (m, J = 8.2, 6.2 Hz, IH), 7.51 (d, J = 8.0 Hz, IH), 7.25 (dt, J = 9.0, 2.3 Hz, IH), 7.08 (d, J = 8.0 Hz, IH), 6.68 (dd, J = 15.4, 9.4 Hz, IH), 6.58 (d, J = 9.6 Hz, IH), 4.85 (dd, J = 14.2, 7.2 Hz, IH), 3.95 (dd, J = 14.2, 7.1 Hz, 2H), 3.29 (m, IH), 2.66 (m, J = 12.0, 6.4 Hz, IH), 2.33 (m, 2H), 1.76 (m, 4H), 1.30 (d, J = 5.6 Hz, 3H), 1.19 (m, 4H), 1.14 (t, J = 7.2 Hz, 3H), 0.98 (m, IH), 0.84 (m, IH). MS (EI) m/z: calcd. 492, found 492.
BISULPHATE SALT
Example 7 – Preparation of an Acid Salt (bisulfate) of Compound A:
Compound IA (5 g) was dissolved in about 25 mL of acetonitrile.
The solution was agitated for about 10 minutes and then heated to about 50 0C. About 6 mL of 2M sulfuric acid in acetonitrile was added into the heated reaction mixture. The solid salt of Compound A precipitated out during the addition of sulfuric acid in acetonitrile. After addition of sulfuric acid solution, the reaction mixture was agitated for 1 hour before cooling to room temperature. The precipitated solid was filtered and washed with about 30 mL of acetonitrile. The wet solid was dried under vacuum at room temperature for 1 hour and at 80 0C for about 12 hours to provide about 5 g white solid (yield 85%). m.p. 217.0 0C. 1H NMR (DMSO) 9.04 (s, IH), 8.60 (d, J = 8.1 Hz, IH), 8.10 (d, J = 8.2 Hz, IH), 7.76 (d, J = 10.4, IH), 7.71 (d, J = 7.8 Hz, IH), 7.60 (dd, J = 8.4, 1.8 Hz, IH), 7.34 (dd, 8.4, 1.8 Hz, IH), 7.08 (d, J = 8.0 Hz, IH), 7.02 (m, IH), 6.69 (d, J = 15.8 Hz, IH), 4.82 (m, IH), 3.94 (dd, J = 14.0, 7.0 Hz, 2H), 3.35 (brs, IH), 2.68 (m, IH), 2.38 (m, 2H), 1.80-1.70 (m, 4H), 1.27 (d, J = 5.8 Hz, 3H), 1.21 (m, 2H), 1.13 (t, J = 7.0 Hz, 3H), 0.95 (m, IH, 0.85 (m, IH). MS (EI) m/z calcd. 590, found 492.
INTERMEDIATE 6
Example 5- Preparation of Compound 6

To a three-neck flask equipped with an agitator, thermometer and nitrogen inert were added the crude product solution of Compound 5 (containing about 31 g. of Compound 5 in 300 mL solution) and anhydrous DMF (0.05 mL). After the mixture was agitated for 5 minutes, oxalyl chloride (12.2 mL) was added slowly while maintaining the batch temperature between 15 and 25°C. The reaction mixture was agitated for about an hour after the addition and checked by NMR for completion of reaction. After the reaction was judged complete, the mixture was concentrated under vacuum to 135 mL while maintaining the temperature of the reaction mixture below 300C. The excess oxalyl chloride was removed completely by two cycles of vacuum concentration at below 500C with replenishment of toluene (315 mL) each time, resulting in a final volume of 68 mL. The reaction mixture was then cooled to 15 to 25°C, after which THF (160 mL) and 2,6-lutidine (22 mL) were added. The mixture was agitated for 16 hours at 20 to 25°C under 100 psi hydrogen in the presence of dry 5% Pd/C (9.0 g). After the reaction was judged complete, the reaction mixture was filtered through celite to remove catalyst. More THF was added to rinse the hydrogenator and catalyst, and the reaction mixture was again filtered through celite. Combined filtrates were concentrated under vacuum at below 25°C to 315 mL. MTBE (158 mL) and 10% aqueous solution of phosphoric acid (158 mL) were added for a thorough extraction at 100C to remove 2,6- lutidine. Then phosphoric acid was removed by extracting the organic layer with very dilute aqueous sodium bicarbonate solution (about 2%), which was followed by a washing with dilute brine. The organic solution was concentrated atmospherically to a volume of 90 mL for solvent replacement. IPA (315 mL) was added to the concentrated crude product solution. The remaining residual solvent was purged to <_ 0.5% of THF (by GC) by repeated concentration under vacuum to 68 mL, with replenishment of IPA (315 mL) before each concentration. The concentrated (68 mL) IPA solution was heated to 50°C, to initiate crystallization. To this mixture n-heptane (68 mL) was added very slowly while maintaining the batch temperature at 50°C. The crystallizing mixture was cooled very slowly over 2.5 hours to 25°C. Additional n- heptane (34 mL) was added very slowly into the suspension mixture at 250C. The mixture was further cooled to 200C, and aged at that temperature for about 20 hours. The solid was filtered and washed with a solvent mixture of 25% IPA in n-heptane, and then dried to provide
19.5 g of a beige colored solid of Compound 6. (Yield: 66%) m.p. 169.30C. IH NMR (CD3CN) δ 9.74 (d, J = 3.03 Hz, IH), 5.42 (br, IH), 4.69 (m, IH), 4.03 (q, J = 7.02 Hz, 2H), 3.43 (qt, J = 3.80, 7.84 Hz, IH), 2.67 (m, 2H), 2.50 (dt, J = 3.00, 8.52 Hz, IH), 1.93 (d, J = 12.0 Hz, 2H), 1.82 (dt, J = 3.28, 9.75 Hz, 2H), 1.54 (qd, J = 3.00, 10.5 Hz, IH), 1.27 (d, J = 5.97 Hz, 3H), 1.20 (m, 6H), 1.03 – 0.92 (m, 2H). MS (ESI) m/z (M++1): calcd. 324, found 324.
INTERMEDIATE 7A
Example 4 – Preparation of Compound 7A
+ 1-Pr2NLi + (EtO)2POCI – + LiCI
![]()
8

7A
To a 10 L three-necked round bottomed flask equipped with an agitator, thermometer and a nitrogen inlet tube, was added 20Og of
Compound 8 (1.07 mol, from Synergetica, Philadelphia, Pennsylvania). THF (1000 mL) was added to dissolve Compound 8. After the solution was cooled to -80 0C to -50 0C, 2.0 M LDA in hexane/THF(1175 mL, 2.2 eq) was added while maintaining the batch temperature below -50 0C. After about 15 minutes of agitation at -800C to -50 0C, diethyl chlorophosphate (185 mL, 1.2 eq) was added while maintaining the batch temperature below -50 0C. The mixture was agitated at a temperature from -800C to – 50 0C for about 15 minutes and diluted with n-heptane (1000 mL). This mixture was warmed up to about -35 0C and quenched with aqueous ammonium chloride (400 g in 1400 mL water) at a temperature below -10 0C. This mixture was agitated at -150C to -10 0C for about 15 minutes followed by agitation at 150C to 25 0C for about 15 minutes. The aqueous layer was split and extracted with toluene (400 mL). The combined organic layers were extracted with 2N hydrochloric acid (700 mL) twice. The product-containing hydrochloric acid layers were combined and added slowly to a mixture of toluene (1200 mL) and aqueous potassium carbonate (300 g in 800 mL water) at a temperature below 30 0C. The aqueous layer was extracted with toluene (1200 mL). The organic layers were combined and concentrated under vacuum to about 600 ml and filtered to remove inorganic salts. To the filtrate was added n-heptane (1000 ml) at about 55 0C. The mixture was cooled slowly to 40 0C, seeded, and cooled further slowly to -10 0C. The resulting slurry was aged at about -10 0C for 1 h, filtered, washed with n- heptane, and dried under vacuum to give a light brown solid (294 g, 85% yield), m.p. 52 0C (DSC onset point).1H NMR (CDCl3) δ 8.73 (d, J = 1.5 Hz, IH), 7.85 (dd, Ji = 8.0 Hz, J2 = 1.5 Hz, IH), 7.49 (dd, Ji = 8.0 Hz, J2 = 1.3 Hz, IH), 7.42 (m, IH), 7.32 (d, J = 7.8 Hz, IH), 7.24 (m, IH), 7.08 (dt, Ji = 8.3 Hz, J2 = 2.3 Hz, IH), 4.09 (m, 4H), 3.48 (d, J = 22.0 Hz, 2H), 1.27 (t, J = 7.0 Hz, 6H). MS (ESI) for M+H calcd. 324, found 324.
Example 3 – Preparation of Compound 5:

4 5
To a three-necked round bottomed flask equipped with an agitator, thermometer and a nitrogen inlet tube was added a solution of Compound 4 in aqueous ethanol (100 g active in 2870 ml). The solution was concentrated to about 700 ml under reduced pressure at 350C to 40°C to remove ethyl alcohol. The resultant homogeneous mixture was cooled to 200C to 300C and its pH was adjusted to range from 12 to 13 with 250 ml of 25% sodium hydroxide solution while maintaining the temperature at 20-300C. Then 82 ml of ethyl chloroformate was slowly added to the batch over a period of 1 hour while maintaining the batch temperature from 200C to 300C and aged for an additional 30 minutes. After the reaction was judged complete, the batch was acidified to pH 7 to 8 with 10 ml of concentrated hydrochloric acid (37%) and 750 ml of ethyl acetate. The pH of the reaction mixture was further adjusted to pH 2 to 3 with 35% aqueous hydrochloric acid solution. The organic layer was separated and the aqueous layer was extracted again with 750 ml of ethyl acetate. The combined organic layers were washed twice with water (200 ml) . Compound 5 was isolated from the organic layer by crystallization from ethyl acetate and heptane mixture (1: 1 mixture, 1500 ml) at about 700C to 80 0C. The solid was filtered at 500C to 60 °C, washed with heptane and then dried to provide an off-white solid (yield 50%). m.p. 197.7°C. 1HNMR (CD3CN) δ 5.31 (brs, IH), 4.67 (dt, J = 16.1, 5.9 Hz, IH), 4.03 (q, J = 7.1 Hz, 2H), 3.41 (m, IH), 2.55 – 2.70 (m, 2H), 1.87 – 1.92 (m, IH), 1.32 – 1.42 (m, IH), 1.30 (d, J = 5.92 Hz, 3H), 1.30 – 1.25 (m, 6H), 0.98 (qt, J = 15.7, 3.18 Hz, 2H). MS (ESI) M+l m/z calculated 340, found 340.
Example 2 – Preparation of Compound 4;

3 4
7.4 kg of ammonium formate was dissolved in 9L of water at 15- 250C, and then cooled to 0-100C. 8.9 kg of Compound 3 was charged at 0-150C followed by an addition of 89L of 2B ethyl alcohol. The batch was cooled to 0-50C 0.9 kg of 10% Palladium on carbon (50% wet) and 9 L of water were charged. The batch was then warmed to 18-280C and agitated for 5 hours, while maintaining the temperature between 18-28 0C. After the reaction was judged complete, 7 IL of water was charged. The batch was filtered and the wet catalyst cake was then washed with 8OL of water. The pH of the filtrate was adjusted to 1-2 with 4N aqueous hydrochloric acid solution. The solution was used in the next process step without further isolation. The yield is typically quantiative. m.p. 216.40C. IH NMR (D2O+1 drop HCl) δ 3.15 (m, IH), 2.76 (m, IH), 2.62 (m, IH), 2.48 (dd,J-5.75Hz, IH), 1.94 (m, 2H), 1.78 (m, 2H), 1.38 (m, 2H), 1.20 (m, 6H), 1.18 (m, IH), 0.98 (q,J=2.99Hz, IH).
Example 1 – Preparation of Compound 3

2B 3
To a reactor equipped with an agitator, thermometer and nitrogen, were added about 10.5 kg of 2B, 68 L of acetone and 68 L of IN aqueous hydrochloric acid solution. The mixture was heated to a temperature between 50 and 600C and agitated for about 1 hour before cooling to room temperature. After the reaction was judged complete, the solution was concentrated under reduced pressure to about 42 L and then cooled to a temperature between 0 and 50C. The cooled mixture was agitated for an additional hour. The product 3 was filtered, washed with cooled water and dried to provide an off-white solid (6.9 kg, yield 76%). m.p. 2510C. Η NMR (DMSO) δ 12.8 (s, IH), 4.72 (m, J = 5.90 Hz, IH), 2.58 (m, 2H), 2.40 (m, J = 6.03 Hz, 2H), 2.21 (dd, J = 19.0, 12.8 Hz, 3H), 2.05 (m, IH), 1.87 (q, J = 8.92 Hz, IH), 1.75 (m, IH), 1.55 (m, IH), 1.35 (q, J = 12.6 Hz, IH), 1.27 (d, J = 5.88 Hz, 3H). MS (ESI) M+l m/z calcd. 267, found 267.
NOTE
Compound 7A may be prepared from Compound 8 by treating Compound 8 with diethylchlorophosphate:

Compound 8 may be obtained by the process described by Kyoku, Kagehira et al in “Preparation of (haloaryl)pyridines,” (API Corporation, Japan). Jpn. Kokai Tokkyo Koho (2004). 13pp. CODEN: JKXXAF JP
2004182713 A2 20040702. Compound 8 is subsequently reacted with a phosphate ester, such as a dialkyl halophosphate, to yield Compound 7A. Diethylchlorophosphate is preferred. The reaction is preferably conducted in the presence of a base, such as a dialkylithium amide, for example diisopropyl lithium amide.
Paper
J Med Chem 2008, 51(11): 3061
http://pubs.acs.org/doi/abs/10.1021/jm800180e
The discovery of an exceptionally potent series of thrombin receptor (PAR-1) antagonists based on the natural product himbacine is described. Optimization of this series has led to the discovery of 4 (SCH 530348), a potent, oral antiplatelet agent that is currently undergoing Phase-III clinical trials for acute coronary syndrome (unstable angina/non-ST segment elevation myocardial infarction) and secondary prevention of cardiovascular events in high-risk patients.
Ethyl [(3aR,4aR,8aR,9aS)-9(S)-[(E)-2-[5-(3-fluorophenyl)-2-
pyridinyl]ethenyl]dodecahydro-1(R)-methyl-3-oxonaphtho[2,3-c]furan-6(R)-yl]carbamate (4).
4 (300 mg, 86%). MS m/z 493 (M+1).
HRMS Calcd for C29H34N2O4F
(M+1): 493.2503, found 493.2509; mp125 °C;
[]D20 6.6 (c 0.5, MeOH).
1HNMR (CDCl3):
http://pubs.acs.org/doi/suppl/10.1021/jm800180e/suppl_file/jm800180e-file002.pdf
0.88-1.18 (m, 5 H), 1.22-1.30 (m, 3 H), 1.43 (d, J = 5.85 Hz, 3 H), 1.88-2.10 (m, 4 H), 2.33-2.42 (m, 2 H),
2.75-2.67 (m, 1 H), 3.52-3.60 (m, 1 H), 4.06-4.14 (m, 2 H), 4.54-4.80 (m, 1 H), 4.71-4.77 (m, 1 H),
6.55-6.63 (m, 2 H), 7.07-7.12 (m, 1 H), 7.26-7.29 (m, 2 H), 7.34 (d, J = 8.05 Hz, 1 H), 7.41-7.46 (m, 1 H), 7.80-7.82 (m, 1 H), 8.76-8.71 (m, 1 H).
PATENT
IN 201621010411
An improved process for preparation of Vorapaxar intermediates and a novel polymorphic form of Vorapaxar
ALEMBIC PHARMACEUTICALS LIMITED
Vorapaxar Sulfate is indicated for the reduction of thrombotic cardiovascular events in patients with a history of myocardial infarction (MI) or with peripheral arterial disease (PAD). ZONTIVITY has been shown to reduce the rate of a combined endpoint of cardiovascular death, MI, stroke, and urgent coronary revascularization (UCR).
According to present invention Vorapaxar sulfate is synthesized from compound of formula 1.
wherein R1 and R2 are each independently selected from the group consisting of H, alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, aryl, alkylaryl, arylalkyl, and heteroaryl groups. Process for the preparation of compound of formula 1 is disclosed in U.S Pat. No. 7,605,275. It disclosed preparation of compound of formula 1 via cyclization of compound 2 in presence of solvent selected from xylene, N-methylpyrrolidinone, Dimethylsulfoxide, diphenyl ether, dimethylacetamide. This cyclization step takes approximately 6-8 hrs.
There is need to develop a process which takes less time for cyclization step to prepare compound of formula 1. Therefore, our scientist works tenaciously to develop process which takes approximately 1-2 hrs for cyclization of compound 1.
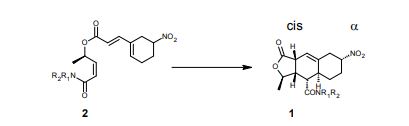
5 According to present invention Vorapaxar sulfate is synthesized from intermediate compound of formula-II.
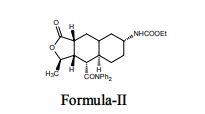
Formula-II Compound of formula-II is critical intermediate in the preparation of Vorapaxar Sulfate.
10 Patent WO2006076415 discloses the process of preparation of above Formula-II in example 7, in which purification/crystallisation step involves treating the reaction mixture having compound of Formula-II with an ethanol/water mixture followed by azeotropic distillation of the mixture. This process yielded formula-II with low yields and with low purities. WO2009055416 (page 9, second paragraph) discloses that use of various solvent systems for
15 formula-II purification such as Methyl-tert-Butyl Ether (MTBE) and various solvent/antisolvent systems, for example, ethylacetate/heptane and toluene/heptane and by using these solvent systems, compound of formula-II are obtained as oil. These oils did not yield a reduced impurity profile in synthesis of the compound of Formula II, nor provide an improvement in the quality of the product compound of Formula II.
20 The inventors surprisingly found that using the process according to the invention provides formula-II with improved yield and high purity. Further, present invention provides a process for the preparation of novel crystalline form of Vorapaxar base. The present invention also relates to novel impurity and process for its preparation.
U.S.Pat. No. 7,304,078 discloses Vorapaxar base. U.S.Pat. No. 7,235,567 discloses Polymorph I and II of vorapaxar sulphate
Example 1- Preparation of compound 1a:
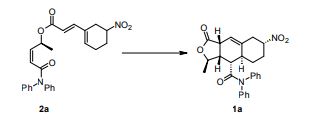
Process A: 5.0 g of compound 2a was suspended in 10.0 ml silicone oil at room temperature. The reaction mixture was then heated to 125°C and stirred for 30 min. Then reaction mass was further heated up to 150°C and stirred for 30 min. After completion of reaction, the reaction mass was cooled to 50-60°C and 25 ml of cyclohexane was added to the reaction mass. The reaction mass was cooled slowly up to room temperature and stirred for 30 min.
15 The precipitated product was filtered off and washed with 5.0 ml Cyclohexane. Wet solid was suspended in mixture of 45.0 ml isopropyl alcohol and 20.0 ml denatured ethanol at 40-45°C and further epimerized with 0.17 ml DBU. The crystallized solid was filtered off with suction, washed with mixture of 1.5 ml Isopropyl alcohol and 0.67 ml denatured ethanol and dried.
20 Process B: 5.0 g of compound 2a was suspended in 10.0 ml paraffin oil at room temperature. The reaction mixture was then heated to 125°C and stirred for 30 min. Then reaction mass was further heated up to 150°C and stirred for 30 min. After completion of reaction, the reaction mass was cooled to 50-60°C and 25 ml of cyclohexane was added to the reaction mass. The reaction mass was cooled slowly up to room temperature and stirred for 30 min.
25 The precipitated product was filtered off and washed with 5.0 ml Cyclohexane. Wet solid was suspended in mixture of 45.0 ml isopropyl alcohol and 20.0 ml denatured ethanol at 40-45°C and further epimerized with 0.17 ml DBU. The crystallized solid was filtered off with suction, washed with mixture of 1.5 ml Isopropyl alcohol and 0.67 ml denatured ethanol and dried. Yield: 4.3 g
Process C: 5.0 g of compound 2a was charged in reaction vessel at room temperature. The solid was then heated to 125°C and stirred for 30 min. Then reaction mass was further heated up to 150°C and stirred for 30 min. After completion of reaction, the reaction mass was cooled to 50-60°C and was added mixture of 45.0 ml isopropyl alcohol and 20.0 ml
5 denatured ethanol at 50-60°C. This was cooled to 40-45°C and further epimerized with 0.17 ml DBU. The crystallized solid was filtered off with suction, washed with mixture of 1.5 ml Isopropyl alcohol and 0.67 ml denatured ethanol and dried. Yield: 4.5 g Example 2: Preparation of Intermediate (Formula-II) of vorapaxar
10 Example 2(a): 50.0g of 1,3,3a,4,4a,5,6,7,8,9a-Decahydro-3-methyl-7-nitro-1-oxo-N,Ndiphenylnaphtho[2,3-c]furan-4-carboxamide compound was suspended in 300.0 ml THF, 15 g 10% Pd/C (50% wet) and 200 ml Process water at room temperature. The reaction mixture was heated to 45°C and drop wise formic acid (35 ml) was added and then stirred for 15 hrs. After completion of reaction, the reaction mass was cooled to 25-30°C and 100 ml THF was
15
added and pH was made acidic with 2M sulfuric acid solution. The reaction mass was filtered and washed with 150 ml THF, 150 ml water. Organic and aqueous layer were separated and aqueous layer was extracted with THF. Organic layers were combined and washed with water. The organic layer was cooled up to 5-10°C, 20 ml of TEA and 13 ml of Ethyl chloro formate were added. The reaction mass was stirred for 30 min. After completion of reaction,
20
reaction mass was washed with 2M sulfuric acid solution and distilled out reaction mass completely under vacuum. Acetonitrile (50 ml) was added to residue and heated up to 40- 45°C. Cooled the reaction mass up to 25-30°C and filtered the solid. Purity: 94-96% Example 2(b): Crystallization with Acetonitrile Acetonitrile (50 ml) was added to above obtained solid and heated to 40-45°C. Cooled the
25 reaction mass slowly up to 25-30°C and then up to 5-10°C. The reaction mass was stirred and the solid was filtered. XRD: Fig-1 Purity: 98-99% Example 2(c): Crystallization with Ethyl acetate To the solid obtained in example-1(a) Ethyl acetate (30 ml) was added. The reaction mass was heated up to 70-75°C and stirred for 10-15 min. The reaction mass was cooled slowly up 30 to 25-30°C and then up to 5-10°C. The reaction mass was stirred for 30 min. The solid was filtered and washed with Ethyl acetate. XRD: Fig-2 Purity: 98-99%
Example 3: Preparation of Amorphous Form of Vorapaxar base Vorapaxar base (10.0 g) was dissolved in 500 ml of 40% Ethyl acetate in Cyclohexane. The solvent was then completely removed under vacuum at 45-50o C to give a solid. Yield: 9.8 g
Example 3 (a): Preparation of crystalline vorapaxar base 5 (2-{[Ethyl (ethylperoxy)phosphory]methyl}-5-(3-fluorophenyl)pyridine) (10 g) was dissolved in THF (30ml) at 25±5°C under Nitrogen. Cool the reaction mass up to -30 to – 50°C. Add drop wise LDA (2.0 M solution in THF). After 1 hr add drop wise (N- [(1R,3aR,4aR,6R,8aR,9S,9aS)-9-formyl dodecahydro-1-methyl-3-oxonaphtho[2,3-c]furan-6- yl]-ethyl ester Carbamic acid) solution (10 g dissolved in 70 ml THF). After completion of 10 reaction mass quench the reaction mass to sulphuric acid solution. Separate the layers and distilled out organic layer under vacuum get foamy residue. (purity 82%) Add MIBK (10 ml) in above residue and stir it at 40-50°C till clear solution. Add drop wise n-Heptane (10 ml) and stir the reaction mass for 30 min. Gradually cool the reaction mass up to 25-30°C. Stir the reaction mass for 24 hrs. Filter the solid and washed it with n-Heptane (5.0 ml). Dry the 15 solid. Yield: 7.0 g. XRD: Fig-3 purity 96%
Example 3(b): Preparation of crystalline vorapaxar base Vorapaxar advance intermediate (2-{[Ethyl (ethylperoxy)phosphory]methyl}-5-(3- fluorophenyl)pyridine) (10 g) was dissolved in THF (30ml) at 25±5°C under Nitrogen. Cool the reaction mass up to -30 to -50°C. Add drop wise LDA (2.0 M solution in THF). After a 1
20 hr add drop wise VORA-Aldehyde (N-[(1R,3aR,4aR,6R,8aR,9S,9aS)-9-formyl dodecahydro1-methyl-3-oxonaphtho[2,3-c]furan-6-yl]-ethyl ester Carbamic acid) solution (10 g dissolved in 70 ml THF). After completion of reaction mass quench the reaction mass to sulphuric acid solution. Separate the layers and distilled out organic layer under vacuum get foamy residue (purity 82%). Add MTBE (10 ml) in above residue and stir it at 40-50°C till clear solution.
25 Add drop wise n-Heptane (30 ml) and stir the reaction mass for 30 min. Gradually cool the reaction mass up to 25-30°C. Stir the reaction mass for 24 hrs. Filter the solid and washed it with n-Heptane (5.0 ml). Dry the solid. Yield: 8.5.0 g. XRD: Fig-4 purity 97%
References
- Samuel Chackalamannil; Wang, Yuguang; Greenlee, William J.; Hu, Zhiyong; Xia, Yan; Ahn, Ho-Sam; Boykow, George; Hsieh, Yunsheng et al. (2008). “Discovery of a Novel, Orally Active Himbacine-Based Thrombin Receptor Antagonist (SCH 530348) with Potent Antiplatelet Activity”. Journal of Medicinal Chemistry 51 (11): 3061–4.doi:10.1021/jm800180e. PMID 18447380.
- Merck Blood Thinner Studies Halted in Select Patients, Bloomberg News, January 13, 2011
- Tricoci et al. (2012). “Thrombin-Receptor Antagonist Vorapaxar in Acute Coronary Syndromes”. New England Journal of Medicine 366 (1): 20–33.doi:10.1056/NEJMoa1109719. PMID 22077816.
- Morrow, DA; Braunwald, E; Bonaca, MP; Ameriso, SF; Dalby, AJ; Fish, MP; Fox, KA; Lipka, LJ; Liu, X; Nicolau, JC; Ophuis, AJ; Paolasso, E; Scirica, BM; Spinar, J; Theroux, P; Wiviott, SD; Strony, J; Murphy, SA; TRA 2P–TIMI 50 Steering Committee and, Investigators (Apr 12, 2012). “Vorapaxar in the secondary prevention of atherothrombotic events.”. The New England Journal of Medicine 366 (15): 1404–13. doi:10.1056/NEJMoa1200933.PMID 22443427.
- “Merck Statement on FDA Advisory Committee for Vorapaxar, Merck’s Investigational Antiplatelet Medicine”. Merck. Retrieved 16 January 2014.
- http://www.forbes.com/sites/larryhusten/2014/01/15/fda-advisory-panel-votes-in-favor-of-approval-for-mercks-vorapaxar/
- SCH-530348 (Vorapaxar) is an investigational candidate for the prevention of arterial thrombosis in patients with acute coronary syndrome and peripheral arterial disease. “Convergent Synthesis of Both Enantiomers of 4-Hydroxypent-2-ynoic Acid Diphenylamide for a Thrombin Receptor Antagonist Sch530348 and Himbacine Analogues.” Alex Zaks et al.: Adv. Synth. Catal. 2009, 351: 2351-2357 Full text;
- Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplatelet activity
J Med Chem 2008, 51(11): 3061
- Stu Borman (2005). “Hopes Ride on Drug Candidates: Researchers reveal potential new medicines for thrombosis, anxiety, diabetes, and cancer”. Chemical & Engineering News 83 (16): 40–44.
PATENTS
- WO 2003089428
- WO 2006076452
- US 6063847
- WO 2006076565
- WO 2008005344
- WO2010/141525
- WO2008/5353
- US2008/26050
- WO2006/76564 mp, nmr
|
3-21-2012
|
EXO-SELECTIVE SYNTHESIS OF HIMBACINE ANALOGS
|
|
|
10-14-2011
|
EXO- AND DIASTEREO- SELECTIVE SYNTHESIS OF HIMBACINE ANALOGS
|
|
|
8-3-2011
|
Exo- and diastereo-selective syntheses of himbacine analogs
|
|
|
3-18-2011
|
COMBINATION THERAPIES COMPRISING PAR1 ANTAGONISTS WITH NAR AGONISTS
|
|
|
8-11-2010
|
Exo-selective synthesis of himbacine analogs
|
|
|
6-4-2010
|
SYNTHESIS Of DIETHYLPHOSPHONATE
|
|
|
5-12-2010
|
THROMBIN RECEPTOR ANTAGONISTS
|
|
|
3-31-2010
|
Synthesis of diethyl{[5-(3-fluorophenyl)-pyridine-2yl]methyl}phosphonate
|
|
|
12-4-2009
|
Local Delivery of PAR-1 Antagonists to Treat Vascular Complications
|
|
|
12-2-2009
|
SYNTHESIS OF HIMBACINE ANALOGS
|
|
10-21-2009
|
Exo- and diastereo- selective syntheses of himbacine analogs
|
|
|
6-31-2009
|
Synthesis of 3-(5-nitrocyclohex-1-enyl) acrylic acid and esters thereof
|
|
|
6-3-2009
|
Synthesis of himbacine analogs
|
|
|
1-23-2009
|
METHODS AND COMPOSITIONS FOR TREATING CARDIAC DYSFUNCTIONS
|
|
|
9-26-2008
|
REDUCTION OF ADVERSE EVENTS AFTER PERCUTANEOUS INTERVENTION BY USE OF A THROMBIN RECEPTOR ANTAGONIST
|
|
|
2-8-2008
|
IMMEDIATE-RELEASE TABLET FORMULATIONS OF A THROMBIN RECEPTOR ANTAGONIST
|
|
|
1-32-2008
|
SOLID DOSE FORMULATIONS OF A THROMBIN RECEPTOR ANTAGONIST
|
|
|
12-5-2007
|
Thrombin receptor antagonists
|
|
|
11-23-2007
|
THROMBIN RECEPTOR ANTAGONISTS
|
|
|
8-31-2007
|
THROMBIN RECEPTOR ANTAGONISTS AS PROPHYLAXIS TO COMPLICATIONS FROM CARDIOPULMONARY SURGERY
|
|
8-31-2007
|
CRYSTALLINE POLYMORPH OF A BISULFATE SALT OF A THROMBIN RECEPTOR ANTAGONIST
|
|
|
6-27-2007
|
Crystalline polymorph of a bisulfate salt of a thrombin receptor antagonist
|
|
|
8-4-2006
|
Preparation of chiral propargylic alcohol and ester intermediates of himbacine analogs
|
|
|
9-31-2004
|
Methods of use of thrombin receptor antagonists
|
| US6063847 * | Nov 23, 1998 | May 16, 2000 | Schering Corporation | Thrombin receptor antagonists |
| US6326380 * | Apr 7, 2000 | Dec 4, 2001 | Schering Corporation | Thrombin receptor antagonists |
| US20030216437 * | Apr 14, 2003 | Nov 20, 2003 | Schering Corporation | Thrombin receptor antagonists |
| US20040176418 * | Jan 9, 2004 | Sep 9, 2004 | Schering Corporation | Crystalline polymorph of a bisulfate salt of a thrombin receptor antagonist |
| WO2011128420A1 | Apr 14, 2011 | Oct 20, 2011 | Sanofi | Pyridyl-vinyl pyrazoloquinolines as par1 inhibitors |
//////////////fast track designation , VORAPAXAR, FDA 2014, EU 2016, Zontivity, NDA 204886, MERCK, VORAPAXAR SULPHATE
CCOC(=O)NC1CCC2C(C1)CC3C(C2C=CC4=NC=C(C=C4)C5=CC(=CC=C5)F)C(OC3=O)C
Naloxegol
Naloxegol
Movantik; NKTR-118; NKTR118; UNII-44T7335BKE; NKTR 118
854601-70-0 cas
1354744-91-4 (Naloxegol Oxalate)
(4R,4aS,7S,7aR,12bS)-7-[2-[2-[2-[2-[2-[2-(2-methoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]-3-prop-2-enyl-1,2,4,5,6,7,7a,13-octahydro-4,12-methanobenzofuro[3,2-e]isoquinoline-4a,9-diol
| MF | C34H53NO11 |
|---|---|
| MW | 651.78472 g/mol |
Morphinan-3,14-diol, 4,5-epoxy-6-(3,6,9,12,15,18,21-heptaoxadocos-1-yloxy)-17-(2-propen-1-yl)-, (5α,6α)-, ethanedioate (1:1) (salt)
Naloxegol oxalate [USAN]
UNII-65I14TNM33
AZ-13337019 oxalate
Naloxegol (oxalate)
NKTR-118 oxalate;AZ-13337019 oxalate
UNII:65I14TNM33
Naloxegol oxalate (MovantikTM, Moventig)
Naloxegol (INN; PEGylated naloxol;[1] trade names Movantik and Moventig) is a peripherally–selective opioid antagonistdeveloped by AstraZeneca, licensed from Nektar Therapeutics, for the treatment of opioid-induced constipation.[2] It was approved in 2014 in adult patients with chronic, non-cancer pain.[3] Doses of 25 mg were found safe and well tolerated for 52 weeks.[4] When given concomitantly with opioid analgesics, naloxegol reduced constipation-related side effects, while maintaining comparable levels of analgesia.[5]

Naloxegol Oxalate was approved by the U.S. Food and Drug Administration (FDA) on Sept 16, 2014, then approved by European Medicine Agency (EMA) on Dec 8, 2014. It was developed and marketed as Movantik®(in the US)/Moventig®(in EU) by AstraZeneca.
Naloxegol oxalate is an antagonist of opioid binding at the mu-opioid receptor. It is indicated for the treatment of opioid-induced constipation (OIC) in adult patients with chronic non-cancer pain.
Movantik® is available as tablets for oral use, containing 12.5 mg or 25 mg of free Naloxegol. The recommended dose is 25 mg once daily (reduce to 12.5 mg if not tolerated).
Chemically, naloxegol is a pegylated (polyethylene glycol-modified) derivative of α-naloxol. Specifically, the 5-α-hydroxyl group of α-naloxol is connected via an ether linkage to the free hydroxyl group of a monomethoxy-terminated n=7 oligomer of PEG, shown extending at the lower left of the molecule image at right. The “n=7” defines the number of two-carbon ethylenes, and so the chain length, of the attached PEG chain, and the “monomethoxy” indicates that the terminal hydroxyl group of the PEG is “capped” with amethyl group.[6] The pegylation of the 5-α-hydroxyl side chain of naloxol prevents the drug from crossing the blood-brain barrier(BBB).[5] As such, it can be considered the antithesis of the peripherally-acting opiate loperamide which is utilized as an opiate-targeting anti-diarrheal agent that does not cause traditional opiate side-effects due to its inability to accumulate in the central nervous system in normal subjects.
Naloxegol was previously a Schedule II drug in the United States because of its chemical similarity to opium alkaloids, but was recently reclassified as a prescription drug after the FDA concluded that the impermeability of the blood-brain barrier to this compound made it non-habit-forming, and so without the potential for abuse — specifically, naloxegol was officially decontrolled on 23. January 2015. [7]

As an opiate antagonist, it is not expected to be capable of inducing the euphoria and anxiolytic effects which are generally cited as the desirable effects of commonly abused opiates (all of which are opiate agonists) if it were to cross the BBB; it would in fact reverse the effects of opiate drugs of abuse if it entered the central nervous system.
Naloxegol is an oral polyethylene glycol (PEG) derivative of naloxone, a peripherally acting µ-opioid receptor antagonist (PAMORA) with limited potential for interfering with centrally mediated opioid analgesia. The incorporation of a polyethylene glycol moiety aims at inhibiting naloxone’s capacity to cross the blood-brain barrier, while preserving the affinity for the µ-opioid receptor [1].

Opioid-induced bowel dysfunction (OIBD) represents a broad spectrum of symptoms that result from the actions of opioids on the CNS as well as the gastrointestinal tract. The majority of gastrointestinal effects seem to be mediated by the high number of µ-receptors that are expressed in the enteric nervous system. Naloxegol was more effective than placebo in increasing the number of spontaneous bowel movements in patients with opioid-induced constipation, including those with an inadequate response to laxatives.
Recognition of Naloxegol as a useful option in the treatment of opioid-induced constipation resulted in its approval by US-FDA for adult patients with chronic, non-cancer pain in 2014.
Naloxegol oxalate (XXI) is a peripherally acting l-opioid receptor antagonist that was approved in the USA and EU for the treatment of opioid-induced constipation in adults with chronic non-cancer pain. The drug, a pegylated version of naloxone, has significantly reduced central nervous system (CNS) penetration and works by inhibiting the binding of opioids in the gastrointestinal tract.152–154 Naloxegol oxalate was developed by Nektar and licensed to AstraZeneca. Although we were unable to find a single report in the primary or patent literature that describes the exact experimental procedures to prepare naloxegol oxalate, there havebeen reports on the preparation of closely related analogs155 with specific reports on improving the selectivity of the reduction step156 and the salt formation of the final drug substance.157 Taken together, the likely synthesis of naloxegol oxalate (XXI) is
described in Scheme 28. Naloxone (180) was treated with methoxyethyl chloride in the presence of Hunig’s base to give the protected ketone 181. Reduction of the ketone with potassium trisec-butylborohydride exclusively provided the a-alcohol 182 in 85% yield. Alternatively, sodium trialkylborohydrides could also be used to provide similar a-selective reduction in high yield.
Deprotonation of the alcohol with sodium hydride followed by alkylation with CH3(OCH2CH2)7Br (183) provided the pegylated intermediate 184 in 88% yield. Acidic removal of the methoxyethyl ether protecting group followed by treatment with oxalic acid and crystallization provided naloxegol oxalate (XXI) in good yield.
152. Corsetti, M.; Tack, J. Expert Opin. Pharmacol. 2015, 16, 399.
153. Garnock-Jones, K. P. Drugs 2015, 75, 419.
154. Leonard, J.; Baker, D. E. Ann. Pharmacother. 2015, 49, 360.
155. Bentley, M. D.; Viegas, T. X.; Goodin, R. R.; Cheng, L.; Zhao, X. US Patent
2005136031A1, 2005.
156. Cheng, L.; Bentley, M. D. WO Patent 2007124114A2, 2007.
157. Aaslund, B. L.; Aurell, C.-J.; Bohlin, M. H.; Sebhatu, T.; Ymen, B. I.; Healy, E. T.;
Jensen, D. R.; Jonaitis, D. T.; Parent, S. WO Patent 2012044243A1, 2012.
158. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm4183
Naloxegol Synthesis
CREDIT
https://ayurajan.blogspot.in/2016/08/naloxegol.html
US20050136031A1: The patent reports detailed synthetic procedures to manufacture gram quantities of Naloxegol. The synthesis starts with Naloxone which was treated with methoxyethyl chloride in the presence of Hunig’s base to give the protected ketone. Reduction of the ketone with potassium tri-sec-butylborohydride exclusively provided the α-alcohol in 85% yield. Deprotonation of the alcohol with sodium hydride followed by alkylation with CH3(OCH2CH2)7Br provided the pegylated Naloxone in 88% yield.
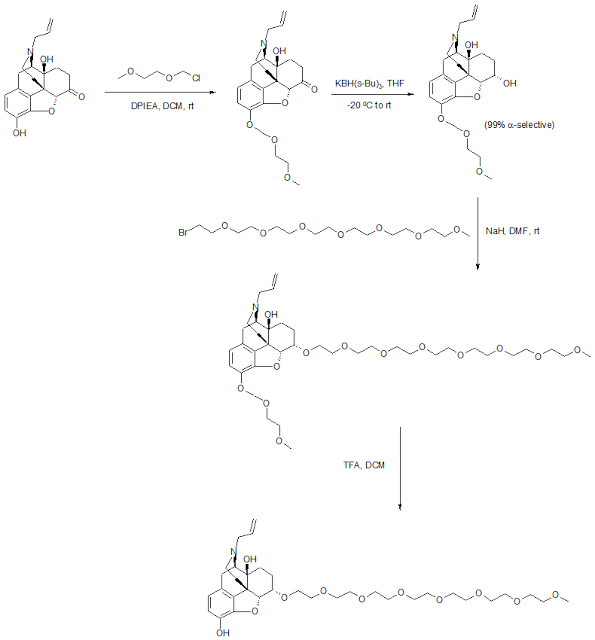
Identifications:
| 1H NMR (Estimated) for Naloxegol |


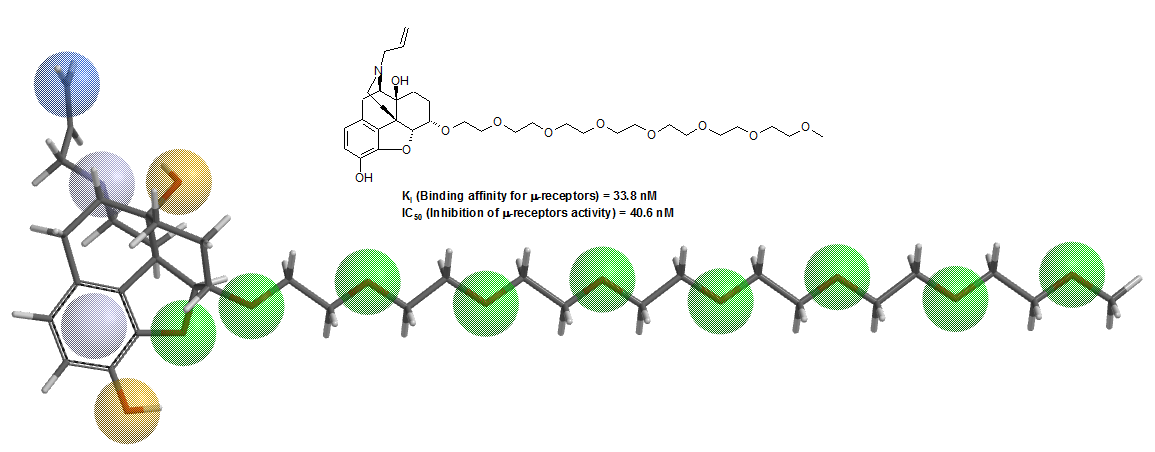

PATENT

PATENT
http://www.google.co.in/patents/WO2005058367A2?cl=en
EXAMPLE 4 SYNTHESIS OF PEG 3-NALθxoL [0211] The structure of the naloxol, an exemplary small molecule drug, is shown below.

Naloxol [0212] This molecule was prepared (having a protected hydroxyl group) as part of a larger synthetic scheme as described in Example 5.
EXAMPLE 5
[0213] α,β-PEGι-naloxol was prepared. The overview of the synthesis is provided below.

(3)

(4)
5.A. Synthesis of 3-MEM-naloxone
[0214] Diisopropylethylamine (390 mg, 3.0 mmole) was added to a solution of naloxone ■ HCl • 2H2O (200 mg, 0.50 mmole) in CH2C12 (10 mL) with stining. Methoxyethyl chloride (“MEMCl,” 250 mg, 2.0 mmole) was then added dropwise to the above solution. The solution was stined at room temperature under N2 overnight.
[0215] The crude product was analyzed by HPLC, which indicated that 3-
MEM-O-naloxone (1) was formed in 97% yield. Solvents were removed by rotary evaporation to yield a sticky oil.
5.B. Synthesis of α and β epimer mixture of 3-MEM-naloxoI (2)
[0216] 3 mL of 0.2 N NaOH was added to a solution of 3-MEM-naloxone
(1) (obtained from 5.A. above, and used without further purification) in 5mL of ethanol. To this was added a solution of NaBHLt (76 mg, 2.0 mmole) in water (1 mL) dropwise. The resulting solution was stined at room temperature for 5 hours. The ethanol was removed by rotary evaporation followed by addition of a solution of 0.1 N HCl solution to destroy excess NaBKj and adjust the pH to a value of 1. The solution was washed with CHC13 to remove excess methoxyethyl chloride and its derivatives (3 x 50 mL), followed by addition of K2OO3 to raise the pH of the solution to 8.0. The product was then extracted with CHC13 (3 x 50 mL) and dried over Na2SO4. The solvent was removed by evaporation to yield a colorless sticky solid (192 mg, 0.46 mmole, 92% isolated yield based on naloxone • HCl • 2H2O).
[0217] HPLC indicated that the product was an α and β epimer mixture of
3-MEM-naloxol (2).
5.C. Synthesis of α and β epimer mixture of 6-CH3-OCH2CH2-O-3-MEM- naloxol (3a).
[0218] NaH (60% in mineral oil, 55 mg, 1.38 mmole) was added into a solution of 6-hydroxyl-3-MEM-naloxol (2) (192 mg, 0.46 mmole) in dimethylformamide (“DMF,” 6 mL). The mixture was stined at room temperature under N2 for 15 minutes, followed by addition of 2-bromoethyl methyl ether (320 mg, 2.30 mmole) in DMF (1 mL). The solution was then stirred at room temperature under N2 for 3 hours.
[0219] HPLC analysis revealed formation of a mixture of α- and β-6-CH3-OCH2CH2-0-3-MEM-naloxol (3) in about 88% yield. DMF was removed by a rotary evaporation to yield a sticky white solid. The product was used for subsequent transformation without further purification.
5.D. Synthesis of α and β epimer mixture of 6-CH3-OCH2CH2-naloxoI (4)
[0220] Crude α- and β-6-CH3-OCH2CH2-O-3-MEM-naloxol (3) was dissolved in 5 mL of CH2C12 to form a cloudy solution, to which was added 5 mL of trifluoroacetic acid (“TFA”). The resultant solution was stined at room temperature for 4 hours. The reaction was determined to be complete based upon HPLC assay. CH2C12 was removed by a rotary evaporator, followed by addition of 10 mL of water. To this solution was added sufficient K2OO3 to destroy excess TFA and to adjust the pH to 8. The solution was then extracted with CHC13 (3 x 50 mL), and the extracts were combined and further extracted with 0.1 N HCl solution (3 x 50 mL). The pH of the recovered water phase was adjusted to a pH of 8 by addition of K2CO3>followed by further extraction with CHC13 (3 x 50 mL). The combined organic layer was then dried with Na2SO4. The solvents were removed to yield a colorless sticky solid.
[0221] The solid was purified by passage two times through a silica gel column (2 cm x 30 cm) using CHCl3/CH3OH (30:1) as the eluent to yield a sticky solid. The purified product was determined by 1H NMR to be a mixture of α- and β epimers of 6-CH3-OCH2CH2-naloxol (4) containing ca. 30% α epimer and ca. 70% β epimer [100 mg, 0.26 mmole, 56% isolated yield based on 6-hydroxyl-3-MEM- naloxol (2)].
[0222] 1H NMR (δ, ppm, CDC13): 6.50-6.73 (2 H, multiplet, aromatic proton of naloxol), 5.78 (1 H, multiplet, olefinic proton of naloxone), 5.17 (2 H, multiplet, olefinic protons of naloxol), 4.73 (1 H, doublet, C5 proton of α naloxol), 4.57 (1 H, doublet, C5 proton of β naloxol), 3.91 (1H, multiplet, C6 proton of naloxol), 3.51-3.75 (4 H, multiplet, PEG), 3.39 (3 H, singlet, methoxy protons of PEG, α epimer), 3.36 (3 H, singlet, methoxy protons of PEG, β epimer), 3.23 (1 H, multiplet, C6 proton of β naloxol), 1.46-3.22 (14 H, multiplet, protons of naloxol).
SYN 1

PATENT
https://www.google.com/patents/WO2012044243A1?cl=en
Naloxol-polyethylene glycol conjugates are provided herein in solid phosphate and oxalate salt forms. Methods of preparing the salt forms, and pharmaceutical compositions comprising the salt forms are also provided herein. ACKGROUND
Effective pain management therapy often calls for an opioid analgesic. In addition to the desired analgesic effect, however, certain undesirable side effects, such as bowel dysfunction, nausea, constipation, among others, can accompany the use of an opioid analgesic. Such side effects may be due to opioid receptors being present outside of the central nervous system, principally in the gastrointestinal tract. Clinical and preclinical studies support the use of mPEG7-0-naloxol, a conjugate of the opioid antagonist naloxol and polyethylene glycol, to counteract undesirable side effects associated with use of opioid analgesics. When administered orally to a patient mPEG7-0-naloxol largely does not cross the blood brain barrier into the central nervous system, and has minimal impact on opioid- induced analgesia. See, e.g., WO 2005/058367; WO 2008/057579; Webster et al., “NKTR-118 Significantly Reverses Opioid-Induced Constipation,” Poster 39, 20th AAPM Annual Clinical Meeting (Phoenix, AZ), October 10, 2009.
To move a drug candidate such as mPEG7-O-naloxol to a viable pharmaceutical product, it is important to understand whether the drug candidate has polymorphic forms, as well as the relative stability and interconversions of these forms under conditions likely to be encountered upon large-scale production, transportation, storage and pre-usage preparation. Solid forms of a drug substance are often desired for their convenience in formulating a drug product. No solid form of mPEG7-O-naloxol drug substance has been made available to date, which is currently manufactured and isolated as an oil in a free base form. Exactly how to accomplish this is often not obvious. For example the number of pharmaceutical products that are oxalate salts is limited. The free base form of mPEG7-0-naloxol has not been observed to form a crystalline phase even when cooled to -60 °C and has been observed to exist as a glass with a transition temperature of
approximately -45 °C. Furthermore, mPEG7-0-naloxol in its free base form can undergo oxidative degradation upon exposure to air. Care can be taken in handling the free base, for example, storing it under inert gas, to avoid its degradation. However, a solid form of mPEG7-0-naloxol, preferably one that is stable when kept exposed to air, is desired
a naloxol-polyethlyene glycol conjugate oxalate salt, the salt comprising ionic species of mPEG7-0-naloxol and oxalic acid. The formulas of mPEG7-0-naloxol and oxalic acid are as follows:

In certain embodiments, the methods provided comprise dissolving mPEG7-0- naloxol free base in ethanol; adding methyl t-butyl ether to the dissolved mPEG?–
O-naloxol solution; adding oxalic acid in methyl t-butyl ether to the dissolved mPEG7-0-naloxol over a period of at least 2 hours to produce a slurry; and filtering the slurry to yield the naloxol-polyethlyene glycol conjugate oxalate salt in solid form.
In certain embodiments, the methods provided comprise dissolving mPEG7-0- naloxol free base in acetonitrile; adding water to the dissolved mPEG7-0-naloxol solution; adding oxalic acid in ethyl acetate to the dissolved mPEG7-0-naloxol over a period of at least 2 hours to produce a slurry; and filtering the slurry to yield the naloxol-polyethlyene glycol conjugate oxalate salt in solid form.
In some embodiments, the solid salt form of mPEG7-0-naloxol is a crystalline form.
In certain embodiments a solid crystalline salt provided herein is substantially pure, having a purity of at least about 80%, at least about 85%, at least about 90%, at least about 92%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, or at least about 99%.
In certain embodiments, the solid salt form of mPEG7-0-naloxol is a phosphate salt.
In other embodiments, the solid mPEG7-0-naloxol salt form is an oxalate salt. For instance, in some embodiments of solid oxalate salt forms provided herein, the solid mPEG7-0-naloxol oxalate salt form is in Form A, as described herein. As another example, in some embodiments of solid oxalate salt forms provided herein, the solid mPEG7-0-naloxol oxalate salt form is in Form B, as described herein. In yet other embodiments, an oxalate salt of mPEG7-0-naloxol in solid form prepared according to the methods described herein is provided.
In yet other embodiments, an dihydrogenphosphate salt of mPEG7-0-naloxol in solid form prepared according to the methods described herein is provided.
In certain embodiments of a solid mPEG7-0-naloxol oxalate salt Form B provided herein, the salt form exhibits a single endothermic peak on differential scanning calorimetry between room temperature and about 150 °C. The single endothermic peak can occur, for instance, between about 91 °C to about 94 °C. For example, in some embodiments the endothermic peak is at about 92 °C, about 92.5 °C, or about93 °C.
PATENT
http://www.google.co.in/patents/WO2005058367A2?cl=en
PATENT
PATENT
https://www.google.com/patents/CN102174049A?cl=en
References and notes
- Roland Seifert; Thomas Wieland; Raimund Mannhold; Hugo Kubinyi; Gerd Folkers (17 July 2006). G Protein-Coupled Receptors as Drug Targets: Analysis of Activation and Constitutive Activity. John Wiley & Sons. p. 227. ISBN 978-3-527-60695-5. Retrieved 14 May 2012.
- “Nektar | R&D Pipeline | Products in Development | CNS/Pain | Oral Naloxegol (NKTR-118) and Oral NKTR-119”. Retrieved2012-05-14.
- “FDA approves MOVANTIK™ (naloxegol) Tablets C-II for the treatment of opioid-induced constipation in adult patients with chronic non-cancer pain”. 16 September 2014.
- “Randomised clinical trial: the long-term safety and tolerability of naloxegol in patients with pain and opioid-induced constipation.”. Aliment Pharmacol Ther. 40: 771–9. Oct 2014.doi:10.1111/apt.12899. PMID 25112584.
- ^ Jump up to:a b Garnock-Jones KP (2015). “Naloxegol: a review of its use in patients with opioid-induced constipation”. Drugs. 75 (4): 419–425. doi:10.1007/s40265-015-0357-2.
- Technically, the molecule that is attached via the ether link is O-methyl-heptaethylene glycol [that is, methoxyheptaethylene glycol, CH3OCH2CH2O(CH2CH2O)5CH2CH2OH], molecular weight 340.4, CAS number 4437-01-8. See Pubchem Staff (2016). “Compound Summary for CID 526555, Pubchem Compound 4437-01”. PubChem Compound Database. Bethesda, MD, USA: NCBI, U.S. NLM. Retrieved 28 January2016.
- ^http://www.deadiversion.usdoj.gov/fed_regs/rules/2015/fr0123_3.htm
1. WO2012044243A / US12015038524A1.
2. WO2005058367A2 / US7786133B2.
3. US20060182692A1 / US8067431B2.
4. CN101033228A.
5. Fudan Univ. J. Med. Sci. 2007, 34, 888-890.
| WO2008057579A2 * | Nov 7, 2007 | May 15, 2008 | Nektar Therapeutics Al, Corporation | Dosage forms and co-administration of an opioid agonist and an opioid antagonist |
| WO2009137086A1 * | May 7, 2009 | Nov 12, 2009 | Nektar Therapeutics | Oral administration of peripherally-acting opioid antagonists |
| US20050136031 * | Dec 16, 2004 | Jun 23, 2005 | Bentley Michael D. | Chemically modified small molecules |
Patents
7056500 United States
7662365 United States
8067431 United States
8617530 United States
9012469 United States
FDA Orange Book Patents
| FDA Orange Book Patents: 1 of 6 | |
|---|---|
| Patent | 7056500 |
| Expiration | Jun 29, 2024 |
| Applicant | ASTRAZENECA PHARMS |
| Drug Application | N204760 (Prescription Drug: MOVANTIK. Ingredients: NALOXEGOL OXALATE) |
| FDA Orange Book Patents: 2 of 6 | |
|---|---|
| Patent | 7662365 |
| Expiration | Oct 18, 2022 |
| Applicant | ASTRAZENECA PHARMS |
| Drug Application | N204760 (Prescription Drug: MOVANTIK. Ingredients: NALOXEGOL OXALATE) |
| FDA Orange Book Patents: 3 of 6 | |
|---|---|
| Patent | 8617530 |
| Expiration | Oct 18, 2022 |
| Applicant | ASTRAZENECA PHARMS |
| Drug Application | N204760 (Prescription Drug: MOVANTIK. Ingredients: NALOXEGOL OXALATE) |
| FDA Orange Book Patents: 4 of 6 | |
|---|---|
| Patent | 9012469 |
| Expiration | Apr 2, 2032 |
| Applicant | ASTRAZENECA PHARMS |
| Drug Application | N204760 (Prescription Drug: MOVANTIK. Ingredients: NALOXEGOL OXALATE) |
| FDA Orange Book Patents: 5 of 6 | |
|---|---|
| Patent | 7786133 |
| Expiration | Dec 19, 2027 |
| Applicant | ASTRAZENECA PHARMS |
| Drug Application | N204760 (Prescription Drug: MOVANTIK. Ingredients: NALOXEGOL OXALATE) |
| FDA Orange Book Patents: 6 of 6 | |
|---|---|
| Patent | 8067431 |
| Expiration | Dec 16, 2024 |
| Applicant | ASTRAZENECA PHARMS |
| Drug Application | N204760 (Prescription Drug: MOVANTIK. Ingredients: NALOXEGOL OXALATE) |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(5α,6α)-4,5-epoxy-6-(3,6,9,12,15,18,21-heptaoxadocos-1-yloxy)-17-(2-propen-1-yl)morphinan-3,14-diol
|
|
| Clinical data | |
| Trade names | Movantik, Moventig |
| AHFS/Drugs.com | movantik |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | ~4.2% |
| Metabolism | Hepatic (CYP3A) |
| Biological half-life | 6–11 h |
| Excretion | Feces (68%), urine (16%) |
| Identifiers | |
| CAS Number | 854601-70-0 |
| ATC code | A06AH03 (WHO) |
| PubChem | CID 56959087 |
| ChemSpider | 28651656 |
| ChEBI | CHEBI:82975 |
| Synonyms | NKTR-118 |
| Chemical data | |
| Formula | C34H53NO11 |
| Molar mass | 651.785 g/mol |
//////////////Naloxegol, Movantik, NKTR-118, NKTR118, UNII-44T7335BKE, NKTR 118, 854601-70-0, EU 2014, FDA 2014
COCCOCCOCCOCCOCCOCCOCCO[C@H]1CC[C@]2([C@H]3Cc4ccc(c5c4[C@]2([C@H]1O5)CCN3CC=C)O)O
Naloxegol oxalate (MovantikTM, Moventig)
Naloxegol oxalate (XXI) is a peripherally acting l-opioid receptor antagonist that was approved in the USA and EU for the treatment of opioid-induced constipation in adults with chronic non-cancer pain. The drug, a pegylated version of naloxone, has significantly reduced central nervous system (CNS) penetration and works by inhibiting the binding of opioids in the gastrointestinal tract.152–154 Naloxegol oxalate was developed by Nektar and licensed to AstraZeneca. Although we were unable to find a single report in the primary or patent literature that describes the exact experimental procedures to prepare naloxegol oxalate, there have been reports on the preparation of closely related analogs155 with specific reports on improving the selectivity of the reduction step156 and the salt formation of the final drug substance.157 Taken together, the likely synthesis of naloxegol oxalate (XXI) is described in Scheme 28. Naloxone (180) was treated with methoxyethyl chloride in the presence of Hunig’s base to give the protected ketone 181. Reduction of the ketone with potassium trisec-butylborohydride exclusively provided the a-alcohol 182 in 85% yield. Alternatively, sodium trialkylborohydrides could also be used to provide similar a-selective reduction in high yield.
Deprotonation of the alcohol with sodium hydride followed by alkylation with CH3(OCH2CH2)7Br (183) provided the pegylated intermediate 184 in 88% yield. Acidic removal of the methoxyethyl ether protecting group followed by treatment with oxalic acid and crystallization provided naloxegol oxalate (XXI) in good yield.

152. Corsetti, M.; Tack, J. Expert Opin. Pharmacol. 2015, 16, 399.
153. Garnock-Jones, K. P. Drugs 2015, 75, 419.
154. Leonard, J.; Baker, D. E. Ann. Pharmacother. 2015, 49, 360.
155. Bentley, M. D.; Viegas, T. X.; Goodin, R. R.; Cheng, L.; Zhao, X. US Patent
2005136031A1, 2005.
156. Cheng, L.; Bentley, M. D. WO Patent 2007124114A2, 2007.
157. Aaslund, B. L.; Aurell, C.-J.; Bohlin, M. H.; Sebhatu, T.; Ymen, B. I.; Healy, E. T.;
Jensen, D. R.; Jonaitis, D. T.; Parent, S. WO Patent 2012044243A1, 2012.
Belinostat (PXD101), a novel HDAC inhibitor

Belinostat (PXD101)
FAST TRACK FDA , ORPHAN STATUS
PXD101;PX105684;PXD-101;PXD 101;PX-105684
UNII:F4H96P17NZ
N-Hydroxy-3-(3-phenylsulphamoylphenyl)acrylamide
N-HYDROXY-3-[3-[(PHENYLAMINO)SULFONYL]PHENYL]-2-PROPENAMIDE
NSC726630

July 3, 2014 — The U.S. Food and Drug Administration today approved Beleodaq (belinostat) for the treatment of patients with peripheral T-cell lymphoma (PTCL), a rare and fast-growing type of non-Hodgkin lymphoma (NHL). The action was taken under the agency’s accelerated approval program.
Belinostat (PXD101) is a novel HDAC inhibitor with IC50 of 27 nM, with activity demonstrated in cisplatin-resistant tumors.
CLINICAL TRIALS…http://clinicaltrials.gov/search/intervention=Belinostat+OR+PXD101
MP 172–174 °C, (lit.(@) 172 °C). 1H NMR (400 MHz, DMSO-d6) δ = 10.75–10.42 (m, 2H), 9.15 (s, 1H), 7.92 (s, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.56 (d, J = 7.8 Hz, 1H),7.47 (d, J = 15.8 Hz, 1H), 7.24 (m, 2H), 7.10–7.01 (m, 3H), 6.51 (d, J = 15.8 Hz, 1H). MS (ESI): m/z = 318.6 [M+H] +.
@ Finn, P. W.; Bandara, M.; Butcher, C.; Finn, A.; Hollinshead, R.; Khan, N.; Law, N.; Murthy, S.; Romero,R.; Watkins, C.; Andrianov, V.; Bokaldere, R. M.; Dikovska, K.; Gailite, V.; Loza, E.; Piskunova, I.;Starchenkov, I.; Vorona, M.; Kalvinsh, I. Helv. Chim. Acta 2005, 88, 1630, DOI: 10.1002/hlca.200590129
Beleodaq and Folotyn are marketed by Spectrum Pharmaceuticals, Inc., based in Henderson, Nevada. Istodax is marketed by Celgene Corporation based in Summit, New Jersey.
Belinostat was granted orphan drug status for the treatment of Peripheral T-cell lymphoma (PTCL) in the US in September 2009 and the EU in October 2012. In July 2015, an orphan drug designation has also been granted for malignant thymoma in the EU.
Belinostat received its first global approval in the US-FDA on 3 July 2014 for the intravenous (IV) treatment of relapsed or refractory PTCL in adults.
Belinostat was approved by the U.S. Food and Drug Administration (FDA) on July 3, 2014. It was originally developed by CuraGen Pharma,then developed by Spectrum Pharmaceuticals cooperating with Onxeo, then marketed as Beleodaq® by Spectrum.
Beleodaq is a pan-histone deacetylase (HDAC) inhibitor selectively causing the accumulation of acetylated histones and other proteinsin tumor cells. It is indicated for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL).
Beleodaq® is available as lyophilized powder for intravenous infusion, containing 500 mg of free Belinostat. The recommended dose is 1,000 mg/m2 once daily on days 1-5 of a 21-day cycle.

Index:
| MW 318.07 | |
| MF | C15H14N2O4S |
414864-00-9 cas no
866323-14-0
(2E)-N-hydroxy-3-[3-(phenylsulfamoyl)phenyl]acrylamide
A novel HDAC inhibitor
PTCL comprises a diverse group of rare diseases in which lymph nodes become cancerous. In 2014, the National Cancer Institute estimates that 70,800 Americans will be diagnosed with NHL and 18,990 will die. PTCL represents about 10 to 15 percent of NHLs in North America.Belinostat inhibits the growth of tumor cells (A2780, HCT116, HT29, WIL, CALU-3, MCF7, PC3 and HS852) with IC50 from 0.2-0.66 μM. PD101 shows low activity in A2780/cp70 and 2780AD cells. Belinostat inhibits bladder cancer cell growth, especially in 5637 cells, which shows accumulation of G0-G1 phase, decrease in S phase, and increase in G2-M phase. Belinostat also shows enhanced tubulin acetylation in ovarian cancer cell lines. A recent study shows that Belinostat activates protein kinase A in a TGF-β signaling-dependent mechanism and decreases survivin mRNA.


Beleodaq works by stopping enzymes that contribute to T-cells, a type of immune cell, becoming cancerous. It is intended for patients whose disease returned after treatment (relapsed) or did not respond to previous treatment (refractory).
“This is the third drug that has been approved since 2009 for the treatment of peripheral T-cell lymphoma,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval expands the number of treatment options available to patients with serious and life-threatening diseases.”
The FDA granted accelerated approval to Folotyn (pralatrexate) in 2009 for use in patients with relapsed or refractory PTCL and Istodax (romidepsin) in 2011 for the treatment of PTCL in patients who received at least one prior therapy.
The safety and effectiveness of Beleodaq was evaluated in a clinical study involving 129 participants with relapsed or refractory PTCL. All participants were treated with Beleodaq until their disease progressed or side effects became unacceptable. Results showed 25.8 percent of participants had their cancer disappear (complete response) or shrink (partial response) after treatment.
The most common side effects seen in Beleodaq-treated participants were nausea, fatigue, fever (pyrexia), low red blood cells (anemia), and vomiting.
The FDA’s accelerated approval program allows for approval of a drug based on surrogate or intermediate endpoints reasonably likely to predict clinical benefit for patients with serious conditions with unmet medical needs. Drugs receiving accelerated approval are subject to confirmatory trials verifying clinical benefit. Beleodaq also received orphan product designation by the FDA because it is intended to treat a rare disease or condition.
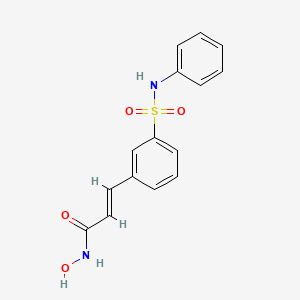
Belinostat (trade name Beleodaq, previously known as PXD101) is a histone deacetylase inhibitor drug developed by TopoTargetfor the treatment of hematological malignancies and solid tumors.[2]
It was approved in July 2014 by the US FDA to treat peripheral T-cell lymphoma.[3]
In 2007 preliminary results were released from the Phase II clinical trial of intravenous belinostat in combination with carboplatin andpaclitaxel for relapsed ovarian cancer.[4] Final results in late 2009 of a phase II trial for T-cell lymphoma were encouraging.[5]Belinostat has been granted orphan drug and fast track designation by the FDA,[6] and was approved in the US for the use againstperipheral T-cell lymphoma on 3 July 2014.[3] It is not approved in Europe as of August 2014.[7]
The approved pharmaceutical formulation is given intravenously.[8]:180 Belinostat is primarily metabolized by UGT1A1; the initial dose should be reduced if the recipient is known to be homozygous for the UGT1A1*28 allele.[8]:179 and 181
NCI: A novel hydroxamic acid-type histone deacetylase (HDAC) inhibitor with antineoplastic activity. Belinostat targets HDAC enzymes, thereby inhibiting tumor cell proliferation, inducing apoptosis, promoting cellular differentiation, and inhibiting angiogenesis. This agent may sensitize drug-resistant tumor cells to other antineoplastic agents, possibly through a mechanism involving the down-regulation of thymidylate synthase


The study of inhibitors of histone deacetylases indicates that these enzymes play an important role in cell proliferation and differentiation. The inhibitor Trichostatin A (TSA) (Yoshida et al., 1990a) causes cell cycle arrest at both G1 and G2 phases (Yoshida and Beppu, 1988), reverts the transformed phenotype of different cell lines, and induces differentiation of Friend leukaemia cells and others (Yoshida et al., 1990b). TSA (and SAHA) have been reported to inhibit cell growth, induce terminal differentiation, and prevent the formation of tumours in mice (Finnin et al., 1999).
Trichostatin A (TSA)

Suberoylanilide Hydroxamic Acid (SAHA)

Cell cycle arrest by TSA correlates with an increased expression of gelsolin (Hoshikawa et al., 1994), an actin regulatory protein that is down regulated in malignant breast cancer (Mielnicki et al., 1999). Similar effects on cell cycle and differentiation have been observed with a number of deacetylase inhibitors (Kim et al., 1999). Trichostatin A has also been reported to be useful in the treatment of fibrosis, e.g., liver fibrosis and liver cirrhosis. See, e.g., Geerts et al., 1998.
Recently, certain compounds that induce differentiation have been reported to inhibit histone deacetylases. Several experimental antitumour compounds, such as trichostatin A (TSA), trapoxin, suberoylanilide hydroxamic acid (SAHA), and phenylbutyrate have been reported to act, at least in part, by inhibiting histone deacetylase (see, e.g., Yoshida et al., 1990; Richon et al., 1998; Kijima et al., 1993). Additionally, diallyl sulfide and related molecules (see, e.g., Lea et al., 1999), oxamflatin (see, e.g., Kim et al., 1999), MS-27-275, a synthetic benzamide derivative (see, e.g., Saito et al., 1999; Suzuki et al., 1999; note that MS-27-275 was later re-named as MS-275), butyrate derivatives (see, e.g., Lea and Tulsyan, 1995), FR901228 (see, e.g., Nokajima et al., 1998), depudecin (see, e.g., Kwon et al., 1998), and m-carboxycinnamic acid bishydroxamide (see, e.g., Richon et al., 1998) have been reported to inhibit histone deacetylases. In vitro, some of these compounds are reported to inhibit the growth of fibroblast cells by causing cell cycle arrest in the G1 and G2 phases, and can lead to the terminal differentiation and loss of transforming potential of a variety of transformed cell lines (see, e.g., Richon et al, 1996; Kim et al., 1999; Yoshida et al., 1995; Yoshida & Beppu, 1988). In vivo, phenybutyrate is reported to be effective in the treatment of acute promyelocytic leukemia in conjunction with retinoic acid (see, e.g., Warrell et al., 1998). SAHA is reported to be effective in preventing the formation of mammary tumours in rats, and lung tumours in mice (see, e.g., Desai et al., 1999).
The clear involvement of HDACs in the control of cell proliferation and differentiation suggest that aberrant HDAC activity may play a role in cancer. The most direct demonstration that deacetylases contribute to cancer development comes from the analysis of different acute promyelocytic leukaemias (APL). In most APL patients, a translocation of chromosomes 15 and 17 (t(15;17)) results in the expression of a fusion protein containing the N-terminal portion of PML gene product linked to most of RARσ (retinoic acid receptor). In some cases, a different translocation (t(11 ;17)) causes the fusion between the zinc finger protein PLZF and RARα. In the absence of ligand, the wild type RARα represses target genes by tethering HDAC repressor complexes to the promoter DNA. During normal hematopoiesis, retinoic acid (RA) binds RARα and displaces the repressor complex, allowing expression of genes implicated in myeloid differentiation. The RARα fusion proteins occurring in APL patients are no longer responsive to physiological levels of RA and they interfere with the expression of the RA- inducible genes that promote myeloid differentiation. This results in a clonal expansion of promyelocytic cells and development of leukaemia. In vitro experiments have shown that TSA is capable of restoring RA-responsiveness to the fusion RARα proteins and of allowing myeloid differentiation. These results establish a link between HDACs and oncogenesis and suggest that HDACs are potential targets for pharmaceutical intervention in APL patients. (See, for example, Kitamura et al., 2000; David et al., 1998; Lin et al., 1998).
BELINOSTAT
Furthermore, different lines of evidence suggest that HDACs may be important therapeutic targets in other types of cancer. Cell lines derived from many different cancers (prostate, coloreetal, breast, neuronal, hepatic) are induced to differentiate by HDAC inhibitors (Yoshida and Horinouchi, 1999). A number of HDAC inhibitors have been studied in animal models of cancer. They reduce tumour growth and prolong the lifespan of mice bearing different types of transplanted tumours, including melanoma, leukaemia, colon, lung and gastric carcinomas, etc. (Ueda et al., 1994; Kim et al., 1999).
Psoriasis is a common chronic disfiguring skin disease which is characterised by well-demarcated, red, hardened scaly plaques: these may be limited or widespread. The prevalence rate of psoriasis is approximately 2%, i.e., 12.5 million sufferers in the triad countries (US/Europe/Japan). While the disease is rarely fatal, it clearly has serious detrimental effects upon the quality of life of the patient: this is further compounded by the lack of effective therapies. Present treatments are either ineffective, cosmetically unacceptable, or possess undesired side effects. There is therefore a large unmet clinical need for effective and safe drugs for this condition. Psoriasis is a disease of complex etiology. Whilst there is clearly a genetic component, with a number of gene loci being involved, there are also undefined environmental triggers. Whatever the ultimate cause of psoriasis, at the cellular level, it is characterised by local T-cell mediated inflammation, by keratinocyte hyperproliferation, and by localised angiogenesis. These are all processes in which histone deacetylases have been implicated (see, e.g., Saunders et al., 1999; Bernhard et al, 1999; Takahashi et al, 1996; Kim et al , 2001 ). Therefore HDAC inhibitors may be of use in therapy for psoriasis. Candidate drugs may be screened, for example, using proliferation assays with T-cells and/or keratinocytes.
CLIP
PXD101/Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.


PATENT
GENERAL SYNTHESIS
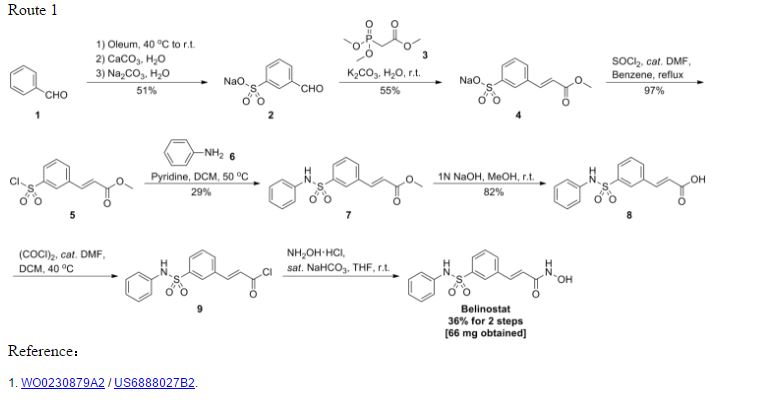
IGNORE 10

ENTRY 45 IS BELINOSTAT
Scheme 1

By using amines instead of aniline, the corresponding products may be obtained. The use of aniline, 4-methoxyaniline, 4-methylaniline, 4-bromoaniline, 4-chloroaniline, 4-benzylamine, and 4-phenethyamine, among others, is described in the Examples below.
In another method, a suitable amino acid (e.g., ω-amino acid) having a protected carboxylic acid (e.g., as an ester) and an unprotected amino group is reacted with a sulfonyl chloride compound (e.g., RSO2CI) to give the corresponding sulfonamide having a protected carboxylic acid. The protected carboxylic acid is then deprotected using base to give the free carboxylic acid, which is then reacted with, for example, hydroxylamine 2-chlorotrityl resin followed by acid (e.g., trifluoroacetic acid), to give the desired carbamic acid.
One example of this approach is illustrated below, in Scheme 2, wherein the reaction conditions are as follows: (i) RSO2CI, pyridine, DCM, room temperature, 12 hours; (ii) 1 M LiOH or 1 M NaOH, dioxane, room temperature, 3-48 hours; (iii) hydroxylamine 2-chlorotrityl resin, HOAt, HATU, DIPEA, DCM, room temperature, 16 hours; and (iv) TFA/DCM (5:95, v/v), room temperature, 1.5 hours.
Scheme 2

Additional methods for the synthesis of compounds of the present invention are illustrated below and are exemplified in the examples below.
Scheme 3A

Scheme 3B

Scheme 4


Scheme 8

Scheme 9

PATENT
SYNTHESIS
Example 1
3-Formylbenzenesulfonic acid, sodium salt (1)

Oleum (5 ml) was placed in a reaction vessel and benzaldehyde (2.00 g, 18.84 mmol) was slowly added not exceeding the temperature of the reaction mixture more than 30°C. The obtained solution was stirred at 40°C for ten hours and at ambient temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate. The aqueous phase was treated with CaC03 until the evolution of C02 ceased (pH~6-7), then the precipitated CaSO4was filtered off and washed with water. The filtrate was treated with Na2CO3 until the pH of the reaction medium increased to pH 8, obtained CaCO3 was filtered off and water solution was evaporated in vacuum. The residue was washed with methanol, the washings were evaporated and the residue was dried in desiccator over P2Oβ affording the title compound (2.00 g, 51%). 1H NMR (D20), δ: 7.56-8.40 (4H, m); 10.04 ppm (1 H, s).
Example 2 3-(3-Sulfophenyl)acrylic acid methyl ester, sodium salt (2)

Sodium salt of 3-formylbenzenesulfonic acid (1) (1.00 g, 4.80 mmol), potassium carbonate (1.32 g, 9.56 mmol), trimethyl phosphonoacetate (1.05 g, 5.77 mmol) and water (2 ml) were stirred at ambient temperature for 30 min., precipitated solid was filtered and washed with methanol. The filtrate was evaporated and the title compound (2) was obtained as a white solid (0.70 g, 55%). 1H NMR (DMSO- dβl HMDSO), δ: 3.68 (3H, s); 6.51 (1 H, d, J=16.0 Hz); 7.30-7.88 (5H, m).
Example 3 3-(3-Chlorosulfonylphenyl)acrylic acid methyl ester (3)

To the sodium salt of 3-(3-sulfophenyl)acrylic acid methyl ester (2) (0.670 g, 2.53 mmol) benzene (2 ml), thionyl chloride (1.508 g, 0.9 ml, 12.67 mmol) and 3 drops of dimethylformamide were added and the resultant suspension was stirred at reflux for one hour. The reaction mixture was evaporated, the residue was dissolved in benzene (3 ml), filtered and the filtrate was evaporated to give the title compound (0.6’40 g, 97%).
Example 4 3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a)

A solution of 3-(3-chlorosulfonylphenyl)acrylic acid methyl ester (3) (0.640 g, 2.45 mmol) in dichloromethane (2 ml) was added to a mixture of aniline (0.465 g, 4.99 mmol) and pyridine (1 ml), and the resultant solution was stirred at 50°C for one hour. The reaction mixture was evaporated and the residue was partitioned between ethyl acetate and 10% HCI. The organic layer was washed successively with water, saturated NaCl, and dried (Na2S0 ). The solvent was removed and the residue was chromatographed on silica gel with chloroform-ethyl acetate (7:1 , v/v) as eluent. The obtained product was washed with diethyl ether to give the title compound (0.226 g, 29%). 1H NMR (CDCI3, HMDSO), δ: 3.72 (3H, s); 6.34 (1H, d, J=16.0 Hz); 6.68 (1 H, br s); 6.92-7.89 (10H, m).
Example 5 3-(3-Phenylsulfamoylphenyl)acrylic acid (5a)

3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a) (0.220 g, 0.69 mmol) was dissolved in methanol (3 ml), 1N NaOH (2.08 ml, 2.08 mmol) was added and the resultant solution was stirred at ambient temperature overnight. The reaction mixture was partitioned between ethyl acetate and water. The aqueous layer was acidified with 10% HCI and stirred for 30 min. The precipitated solid was filtered, washed with water and dried in desiccator over P2Os to give the title compound as a white solid (0.173 g, 82%). Example 6 3-(3-Phenylsulfamoylphenyl)acryloyl chloride (6a)

To a suspension of 3-(3-phenylsulfamoylphenyl)acrylic acid (5a) (0.173 g, 0.57 mmol) in dichloromethane (2.3 ml) oxalyl chloride (0.17 ml, 1.95 mmol) and one drop of dimethylformamide were added. The reaction mixture was stirred at 40°C for one hour and concentrated under reduced pressure to give crude title compound (0.185 g).
Example 7
N-Hydroxy-3-(3-phenylsulfamoylphenyl)acrylamide (7a) (PX105684) BELINOSTAT

To a suspension of hydroxylamine hydrochloride (0.200 g, 2.87 mmol) in tetrahydrofuran (3.5 ml) a saturated NaHCOβ solution (2.5 ml) was added and the resultant mixture was stirred at ambient temperature for 10 min. To the reaction mixture a 3-(3-phenylsulfamoylphenyl)acryloyl chloride (6a) (0.185 g) solution in tetrahydrofuran (2.3 ml) was added and stirred at ambient temperature for one hour. The reaction mixture was partitioned between ethyl acetate and 2N HCI. The organic layer was washed successively with water and saturated NaCl, the solvent was removed and the residue was washed with acetonitrile and diethyl ether.
The title compound was obtained as a white solid (0.066 g, 36%), m.p. 172°C. BELINOSTAT

1H NMR (DMSO-d6, HMDSO), δ: 6.49 (1 H, d, J=16.0 Hz); 7.18-8.05 (10H, m); 9.16 (1 H, br s); 10.34 (1 H, s); 10.85 ppm (1 H, br s).
HPLC analysis on Symmetry C18column: impurities 4% (column size 3.9×150 mm; mobile phase acetonitrile – 0.1 M phosphate buffer (pH 2.5), 40:60; sample concentration 1 mg/ml; flow rate 0.8 ml/ min; detector UV 220 nm).
Anal. Calcd for C15Hι4N204S, %: C 56.59, H 4.43, N 8.80. Found, %: C 56.28, H 4.44, N 8.56.
PATENT
https://www.google.com/patents/CN102786448A?cl=en
Example: belinostat (compound of formula I) Preparation of

Methods of operation:
The compound of formula II (4. Og) added to the reactor, was added methanol 30ml, and stirred to dissolve, was added IM aqueous sodium hydroxide solution (38mL), stirred at room temperature overnight, the reaction was completed, ethyl acetate was added (IOmL) ^ K (20mL), stirred for 5 minutes, phase separation, the ethyl acetate phase was discarded, the aqueous phase was acidified with 10% hydrochloric acid to pH2, stirred at room temperature for 30 minutes, filtered, washed with water, and dried to give hydrolyzate 3. lg, yield rate of 81.6%.
The hydrolyzate (3. Og) added to the reactor, was added methylene chloride (53. 2g), dissolved with stirring, was added oxalyl chloride (2.8mL, 0.0032mol) at room temperature was added I drop DMF, reflux I hours, concentrated and the residue was dissolved in THF (30mL) alternate, the other to take a reaction flask was added hydroxylamine hydrochloride (3. 5g, 0.05mol), THF (50mL), saturated aqueous sodium bicarbonate (40mL), the mixture at room temperature under stirring for 10 minutes, then was added to spare, stirred at room temperature for I hour, the reaction was complete, at – at room temperature was added ethyl acetate (50mL), 2M hydrochloric acid (50mL), stirred for 5 minutes the phases were separated, the aqueous phase was discarded, the organic layer was washed with water, saturated brine, dried, filtered and concentrated to give crude product belinostat, recrystallized from ethyl acetate, 50 ° C and dried for 8 hours to give white crystals 2. 6g, yield 83.8%. . 1H-NMR (DMS0-d6, 400MHz) δ: 6 50 (1H, d, J = 16. OHz); 7 07 (d, J = 7. 8Hz, 2H); 7 16 (t.. , J = 7. 3Hz, 1H);. 7 25 (m, 2H);. 7 45 (t, J = 7. 8Hz, 1H);. 7 60 (d, J = 15. 9Hz, 1H); 7 . 62 (d, J = 7. 7Hz, 1H);. 7 75 (d, J = 7. 8Hz, 1H);. 7 88 (br s. ‘1H);. 9 17 (br s’ 1H); 10. 35 (s, 1H);. 10 82ppm (br s, 1H). ·
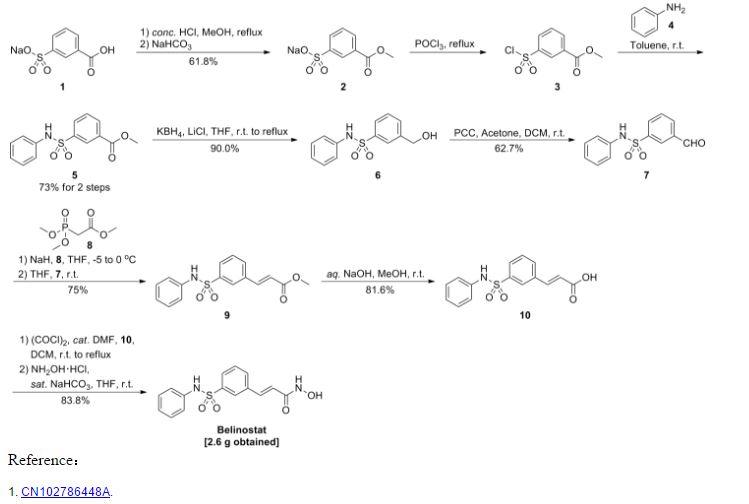
Step a): Preparation of Compound III

The carboxy benzene sulfonate (224g, Imol), anhydrous methanol (2300g), concentrated hydrochloric acid (188. 6g) refluxing
3-5 hours, filtered and the filtrate was added anhydrous sodium bicarbonate powder (200g), stirred for I hour, filtered, the filter residue was discarded, the filtrate was concentrated. The concentrate was added methanol (2000g), stirred at room temperature for 30 minutes, filtered and the filtrate was concentrated to dryness, 80 ° C and dried for 4 hours to give a white solid compound III147g, yield 61.8%.
Step b): Preparation of Compound IV

Compound III (50g, 0. 21mol), phosphorus oxychloride (250mL) was refluxed for 2_6 hours, completion of the reaction, cooled to
0-5 ° C, was slowly added to ice water, stirred for 2 hours and filtered to give a brown solid compound IV40 g, due to the instability of Compound IV, directly into the next reaction without drying.
Preparation of Compound V: [0040] Step c)

The aniline (5. 58g, 0. 06mol) and 30mL of toluene added to the reactor, stirred to dissolve, in step b) the resulting compound IV (7. 05g, O. 03mol) was dissolved in 60 ml of toluene, at room temperature dropwise added to the reactor, the addition was completed, stirring at room temperature for 1-2 hours, the reaction was completed, the filtered solid washed with water, and then recrystallized from toluene, 50 ° C and dried for 4 hours to obtain a white crystalline compound V6. Og, yield 73%. mp:.. 144 4-145 2. . .
1H- bandit R (CDCl3, 400MHz) δ:…. 3 92 (s, 3H); 6 80 (. Br s, 1H); 7 06-7 09 (m, 2H); 7 11. . -7 15 (m, 1H);.. 7 22-7 26 (m, 2H);. 7 51 (t, J = 7. 8Hz, 1H);.. 7 90-7 93 (dt, J = . 1.2,7 8Hz, 1H); 8 18-8 21 (dt, J = I. 4, 7. 8Hz, 1H);… 8 48 (t, J = L 6Hz, 1H).
IR v ™ r: 3243,3198,3081,2953,1705,1438,1345,766,702,681cm-1.
Step d): Preparation of Compound VI

The anhydrous lithium chloride 2. 32g, potassium borohydride 2. 96g, THF50mL added to the reactor, stirring evenly, Compound V (8g, 0. 027mol) was dissolved in 7mL of tetrahydrofuran, was slowly dropped into the reactor was heated under reflux for 5 hours, the reaction was completed, the force mouth 40mL water and ethyl acetate 40mL, stirred for half an hour, allowed to stand for separation, the organic layer was washed with 40mL water, concentrated under reduced pressure to give the crude product, the crude product was recrystallized from toluene, solid 50 V dried for 4 hours to give a white crystalline compound VI6. 82g, yield 90. O%. mp:.. 98 2-98 6. . .
1H-NMR (DMS0-d6, 400ΜΗζ) δ:….. 4 53 (s, 2H); 5 39 (s, 1H); 6 99-7 03 (m, 1H); 7 08- 7. ll (m, 2H);.. 7 19-7 24 (m, 2H);.. 7 45-7 52 (m, 2H);.. 7 61-7 63 (dt, J = I. 8 , 7 4Hz, 1H);.. 7 79 (br s, 1H);. 10. 26 (s, 1H).
IRv =: 3453,3130,2964,1488,1151,1031, 757,688cm_10
Step e): Preparation of Compound VII

After Compound VI (7.5g, 0.028mol) dissolved in acetone was added 7ml, dichloromethane was added 60mL, supported on silica gel was added PCC at room temperature 20g, stirred at room temperature for 12-24 hours, the reaction was complete, filtered and the filtrate was purified The layers were separated and the aqueous layer was discarded after the organic phase is washed 30mL5% aqueous sodium bicarbonate, evaporated to dryness under reduced pressure to give the crude product, the crude product was recrystallized from toluene, 50 ° C and dried for 8 hours to give white crystalline compound VII4. 7g, yield 62.7%. mp:.. 128 1-128 5 ° C.
1H- bandit R (CDCl3,400MHz) δ:…. 7 08-7 15 (m, 4Η); 7 · 23-7 27 (m, 2H); 7 · 60-7 64 (t, J = 7 7Hz, 1Η);.. 8 00 (d, J = 7. 6Hz, 1Η);. 8 04 (d, J = 7. 6Hz, 1Η);. 8 30 (br s’ 1Η).; 10. 00 (S, 1Η).
IR ν ™ Γ: 3213,3059,2964,2829,1687,1480,1348,1159,1082,758,679cm_10
Preparation of compounds of formula II: [0055] Step f)

phosphoryl trimethylorthoacetate (2. 93g, 0. 0161mol) added to the reaction vessel, THF30mL, stirring to dissolve, cooled to -5-0 ° C, was added sodium hydride (O. 8g, content 80%) , the addition was completed, stirring for 10-20 minutes, was added dropwise the compound VII (4g, O. 0156mol) and THF (20mL) solution, stirred for 1_4 hours at room temperature, the reaction was complete, 10% aqueous ammonium chloride solution was added dropwise 50mL, and then After addition of 50mL of ethyl acetate, stirred 30min rested stratification, the aqueous layer was discarded, the organic phase was concentrated under reduced pressure to give the crude product, the crude product was recrystallized from methanol 60mL, 50 ° C and dried for 8 hours to give white crystalline compound 113. 6g, yield 75%. mp:.. 152 0-152 5 ° C.
1H-Nmr (Cdci3JOOmHz) δ:…. 3 81 (s, 3H); 6 40 (d, J = 16. 0Hz, 1H); 6 79 (. Br s, 1H); 7 08 ( d, J = 7. 8Hz, 2H);. 7 14 (t, J = 7. 3Hz, 1H);. 7 24 (m, 2H);. 7 46 (t, J = 7. 8Hz, 1H); 7. 61 (d, J = 16. ΟΗζ, ΙΗ);. 7 64 (d, J = 7. 6Hz, 1H);. 7 75 (d, J = 7. 8Hz, 1H);. 7 89 (br . s, 1H).
IR v ^ :: 3172,3081,2954,2849,1698,1475,1345,1157,773,714,677cm-1.
PATENT
SYNTHESIS
US20100286279

CLIP
SYNTHESIS AND SPECTRAL DATA
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
(E)-N-Hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (28, belinostat, PXD101).
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
The methyl ester (27) (8.0 g) was prepared according to reported synthetic route,
(Watkins, C. J.; Romero-Martin, M.-R.; Moore, K. G.; Ritchie, J.; Finn, P. W.; Kalvinsh, I.;
Loza, E.; Dikvoska, K.; Gailite, V.; Vorona, M.; Piskunova, I.; Starchenkov, I.; Harris, C. J.;
Duffy, J. E. S. Carbamic acid compounds comprising a sulfonamide linkage as HDAC
inhibitors. PCT Int. Appl. WO200230879A2, April 18, 2002.)
but using procedure D (Experimental Section) or method described for 26 to convert the methyl ester to crude
hydroxamic acid which was further purified by chromatography (silica, MeOH/DCM = 1:10) to
afford 28 (PXD101) as off-white or pale yellow powder (2.5 g, 31%).
LC–MS m/z 319.0 ([M +H]+).
1H NMR (DMSO-d6) 12–9 (very broad, 2H), 7.90 (s, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.70 (d, J
= 7.8 Hz, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 2H), 7.08 (d,J = 7.8 Hz, 2H), 7.01 (t, J = 7.3 Hz, 1H), 6.50 (d, J = 15.8 Hz, 1H);
13C NMR (DMSO-d6) 162.1, 140.6, 138.0, 136.5, 135.9, 131.8, 130.0, 129.2, 127.1, 124.8, 124.1, 121.3, 120.4.
Anal.
(C15H14N2O4S) C, H, N
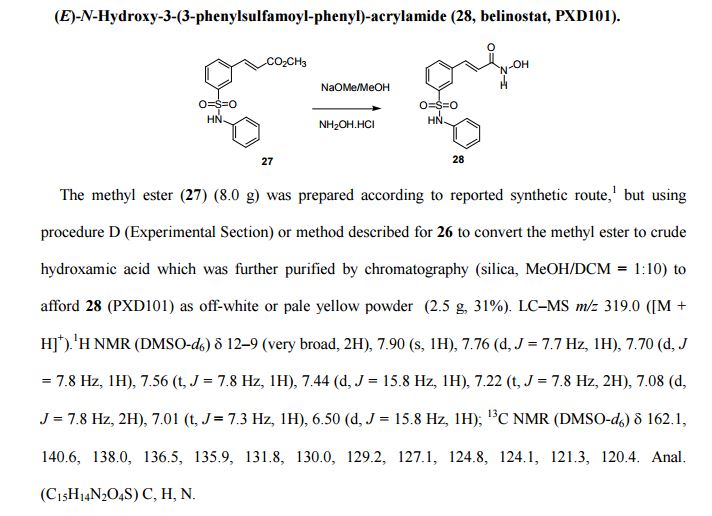
PATENT
SYNTHESIS
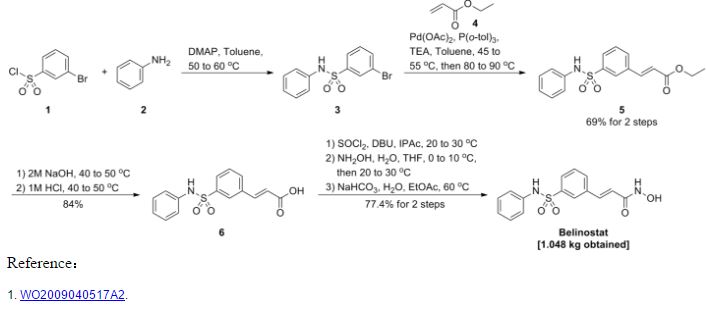
PXDIOI / Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.
Scheme 1
Not isolated

ed on (A)
on (D)

d on (H)

There is a need for alternative methods for the synthesis of PXD101 and related compounds for example, methods which are simpler and/or employ fewer steps and/or permit higher yields and/or higher purity product.
Scheme 5

DMAP, toluene



Synthesis 1 3-Bromo-N-phenyl-benzenesulfonamide (3)

To a 30 gallon (-136 L) reactor was charged aniline (2) (4.01 kg; 93.13 g/mol; 43 mol), toluene (25 L), and 4-(dimethylamino)pyridine (DMAP) (12 g), and the mixture was heated to 50-600C. 3-Bromobenzenesulfonyl chloride (1) (5 kg; 255.52 g/mol; 19.6 mol) was charged into the reactor over 30 minutes at 50-600C and progress of the reaction was monitored by HPLC. After 19 hours, toluene (5 L) was added due to losses overnight through the vent line and the reaction was deemed to be complete with no compound (1) being detected by HPLC. The reaction mixture was diluted with toluene (10 L) and then quenched with 2 M aqueous hydrochloric acid (20 L). The organic and aqueous layers were separated, the aqueous layer was discarded, and the organic layer was washed with water (20 L), and then 5% (w/w) sodium bicarbonate solution (20 L), while maintaining the batch temperature at 45-55°C. The batch was then used in the next synthesis.
Synthesis 2 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrylic acid ethyl ester (5)

To the batch containing 3-bromo-N-phenyl-benzenesulfonamide (3) (the treated organic layer obtained in the previous synthesis) was added triethylamine (2.97 kg; 101.19 g/mol; 29.4 mol), tri(o-tolyl)phosphine (119 g; 304.37 g/mol; 0.4 mol), and palladium (II) acetate (44 g; 224.51 g/mol; 0.2 mol), and the resulting mixture was degassed four times with a vacuum/nitrogen purge at 45-55°C. Catalytic palladium (0) was formed in situ. The batch was then heated to 80-900C and ethyl acrylate (4) (2.16 kg; 100.12 g/mol; 21.6 mol) was slowly added over 2.75 hours. The batch was sampled after a further 2 hours and was deemed to be complete with no compound (3) being detected by HPLC. The batch was cooled to 45-55°C and for convenience was left at this temperature overnight.
The batch was then reduced in volume under vacuum to 20-25 L, at a batch temperature of 45-55°C, and ethyl acetate (20 L) was added. The batch was filtered and the residue washed with ethyl acetate (3.5 L). The residue was discarded and the filtrates were sent to a 100 gallon (-454 L) reactor, which had been pre-heated to 600C. The 30 gallon (-136 L) reactor was then cleaned to remove any residual Pd, while the batch in the 100 gallon (-454 L) reactor was washed with 2 M aqueous hydrochloric acid and water at 45-55°C. Once the washes were complete and the 30 gallon (-136 L) reactor was clean, the batch was transferred from the 100 gallon (-454 L) reactor back to the 30 gallon (-136 L) reactor and the solvent was swapped under vacuum from ethyl acetate/toluene to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it. The batch was then cooled to 0-100C and held at this temperature over the weekend in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). A sample of the wet-cake was taken for Pd analysis. The Pd content of the crude product (5) was determined to be 12.9 ppm.
The wet-cake was then charged back into the 30 gallon (-136 L) reactor along with ethyl acetate (50 L) and heated to 40-500C in order to obtain a solution. A sparkler filter loaded with 12 impregnated Darco G60® carbon pads was then connected to the reactor and the solution was pumped around in a loop through the sparkler filter. After 1 hour, a sample was taken and evaporated to dryness and analysed for Pd content. The amount of Pd was found to be 1.4 ppm. A second sample was taken after 2 hours and evaporated to dryness and analysed for Pd content. The amount of Pd had been reduced to 0.6 ppm. The batch was blown back into the reactor and held at 40-500C overnight before the solvent was swapped under vacuum from ethyl acetate to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it and the batch was cooled to 0-100C and held at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum for 25 hours. A first lot of the title compound (5) was obtained as an off-white solid (4.48 kg, 69% overall yield from 3-bromobenzenesulfonyl chloride (1)) with a Pd content of 0.4 ppm and a purity of 99.22% (AUC) by HPLC.
Synthesis 3 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrvlic acid (6)

To the 30 gallon (-136 L) reactor was charged the (E)-3-(3-phenylsulfamoyl-phenyl)- acrylic acid ethyl ester (5) (4.48 kg; 331.39 g/mol; 13.5 mol) along with 2 M aqueous sodium hydroxide (17.76 L; -35 mol). The mixture was heated to 40-50°C and held at this temperature for 2 hours before sampling, at which point the reaction was deemed to be complete with no compound (5) being detected by HPLC. The batch was adjusted to pH 2.2 using 1 M aqueous hydrochloric acid while maintaining the batch temperature between 40-500C. The product had precipitated and the batch was cooled to 20-300C and held at this temperature for 1 hour before filtering and washing the cake with water (8.9 L). The filtrate was discarded. The batch was allowed to condition on the filter overnight before being charged back into the reactor and slurried in water (44.4 L) at 40-500C for 2 hours. The batch was cooled to 15-20°C, held for 1 hour, and then filtered and the residue washed with water (8.9 L). The filtrate was discarded. The crude title compound (6) was transferred to an oven for drying at 45-55°C under vacuum with a slight nitrogen bleed for 5 days (this was done for convenience) to give a white solid (3.93 kg, 97% yield). The moisture content of the crude material was measured using Karl Fischer (KF) titration and found to be <0.1% (w/w). To the 30 gallon (-136 L) reactor was charged the crude compound (6) along with acetonitrile (47.2 L). The batch was heated to reflux (about 80°C) and held at reflux for 2 hours before cooling to 0-10°C and holding at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with cold acetonitrile (7.9 L). The filtrate was discarded and the residue was dried under vacuum at 45-55°C for 21.5 hours. The title compound (6) was obtained as a fluffy white solid (3.37 kg, 84% yield with respect to compound (5)) with a purity of 99.89% (AUC) by HPLC.
Synthesis 4 (E)-N-Hvdroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (PXD101) BELINOSTAT

To the 30 gallon (-136 L) reactor was charged (E)-3-(3-phenylsulfamoyl-phenyl)-acrylic acid (6) (3.37 kg; 303.34 g/mol; 11.1 mol) and a pre-mixed solution of 1 ,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in isopropyl acetate (IPAc) (27 g in 30 L; 152.24 g/mol; 0.18 mol). The slurry was stirred and thionyl chloride (SOCI2) (960 mL; density ~1.631 g/mL; 118.97 g/mol; -13 mol) was added to the reaction mixture and the batch was stirred at 20-300C overnight. After 18.5 hours, the batch was sampled and deemed to be complete with no compound (6) being detected by HPLC. The resulting solution was transferred to a 100 L Schott reactor for temporary storage while the
30 gallon (-136 L) reactor was rinsed with isopropyl acetate (IPAc) and water. Deionized water (28.9 L) was then added to the 30 gallon (-136 L) reactor followed by 50% (w/w) hydroxylamine (6.57 L; -1.078 g/mL; 33.03 g/mol; -214 mol) and another charge of deionized water (1.66 L) to rinse the lines free of hydroxylamine to make a 10% (w/w) hydroxylamine solution. Tetrahydrofuran (THF) (6.64 L) was then charged to the
30 gallon (-136 L) reactor and the mixture was stirred and cooled to 0-100C. The acid chloride solution (from the 100 L Schott reactor) was then slowly charged into the hydroxylamine solution over 1 hour maintaining a batch temperature of 0-10°C during the addition. The batch was then allowed to warm to 20-300C. The aqueous layer was separated and discarded. The organic layer was then reduced in volume under vacuum while maintaining a batch temperature of less than 300C. The intention was to distill out 10-13 L of solvent, but this level was overshot. A larger volume of isopropyl acetate (IPAc) (16.6 L) was added and about 6 L of solvent was distilled out. The batch had precipitated and heptanes (24.9 L) were added and the batch was held at 20-30°C overnight. The batch was filtered and the residue was washed with heptanes (6.64 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum with a slight nitrogen bleed over the weekend. The title compound (PXD101) was obtained as a light orange solid (3.11 kg, 89% yield with respect to compound (6)) with a purity of 99.25% (AUC) by HPLC.
The title compound (PXD101) (1.2 kg, 3.77 mol) was dissolved in 8 volumes of 1:1 (EtOH/water) at 600C. Sodium bicarbonate (15.8 g, 5 mol%) was added to the solution. Water (HPLC grade) was then added at a rate of 65 mL/min while keeping the internal temperature >57°C. After water (6.6 L) had been added, crystals started to form and the water addition was stopped. The reaction mixture was then cooled at a rate of 10°C/90 min to a temperature of 0-10cC and then stirred at ambient temperature overnight. The crystals were then filtered and collected. The filter cake was washed by slurrying in water (2 x 1.2 L) and then dried in an oven at 45°C for 60 hours with a slight nitrogen bleed. 1.048 kg (87% recovery) of a light orange solid was recovered. Microscopy and XRPD data showed a conglomerate of irregularly shaped birefringant crystalline particles. The compound was found to contain 0.02% water.
As discussed above: the yield of compound (5) with respect to compound (1) was 69%. the yield of compound (6) with respect to compound (5) was 84%. the yield of PXD101 with respect to compound (6) was 89%.
PAPER
Synthetic Commun. 2010, 40, 2520-2524.
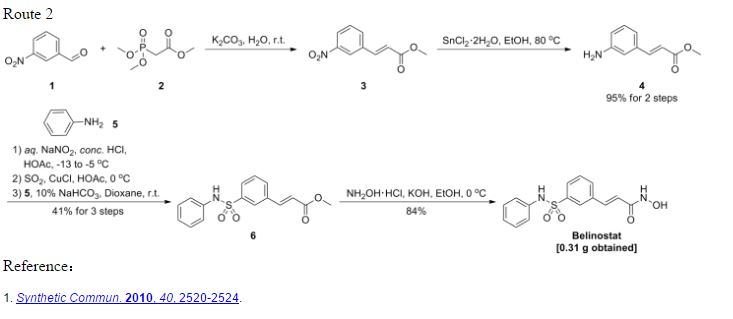
PATENT
FORMULATION
Formulation Studies
These studies demonstrate a substantial enhancement of HDACi solubility (on the order of a 500-fold increase for PXD-101) using one or more of: cyclodextrin, arginine, and meglumine. The resulting compositions are stable and can be diluted to the desired target concentration without the risk of precipitation. Furthermore, the compositions have a pH that, while higher than ideal, is acceptable for use.

UV Absorbance
The ultraviolet (UV absorbance E\ value for PXD-101 was determined by plotting a calibration curve of PXD-101 concentration in 50:50 methanol/water at the λmax for the material, 269 nm. Using this method, the E1i value was determined as 715.7.
Methanol/water was selected as the subsequent diluting medium for solubility studies rather than neat methanol (or other organic solvent) to reduce the risk of precipitation of the cyclodextrin.
Solubility in Demineralised Water
The solubility of PXD-101 was determined to be 0.14 mg/mL for demineralised water. Solubility Enhancement with Cvclodextrins
Saturated samples of PXD-101 were prepared in aqueous solutions of two natural cyclodextrins (α-CD and γ-CD) and hydroxypropyl derivatives of the α, β and Y cyclodextrins (HP-α-CD, HP-β-CD and HP-γ-CD). All experiments were completed with cyclodextrin concentrations of 250 mg/mL, except for α-CD, where the solubility of the cyclodextrin was not sufficient to achieve this concentration. The data are summarised in the following table. HP-β-CD offers the best solubility enhancement for PXD-101.

Phase Solubility Determination of HP-β-CD
The phase solubility diagram for HP-β-CD was prepared for concentrations of cyclodextrin between 50 and 500 mg/mL (5-50% w/v). The calculated saturated solubilities of the complexed HDACi were plotted against the concentration of cyclodextrin. See Figure 1.

CLIP
SPECTRUM
Tiny Biotech With Three Cancer Drugs Is More Alluring Takeover Bet Now
Forbes
The drug is one of Spectrum’s two drugs undergoing phase 3 clinical trials. Allergan paid Spectrum $41.5 million and will make additional payments of up to $304 million based on achieving certain milestones. So far, Raj Shrotriya, Spectrum’s chairman, …
CLIP
Copenhagen, December 10, 2013
Topotarget announces the submission of a New Drug Application (NDA) for belinostat for the treatment of relapsed or refractory (R/R) peripheral T-cell lymphoma (PTCL) to the US Food and Drug Administration (FDA). The NDA has been filed for Accelerated Approval with a request for Priority Review. Response from the FDA regarding acceptance to file is expected within 60 days from the FDA receipt date.
read all this here
PAPER
The Development of an Effective Synthetic Route of Belinostat
Key Laboratory of Structure-Based Drug Design & Discovery, Ministry of Education, Shenyang Pharmaceutical University, Shenyang 110016, China
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.6b00170
Publication Date (Web): July 12, 2016
Copyright © 2016 American Chemical Society
*E-mail: guoliang222@gmail.com.

A practical synthetic route of belinostat is reported. Belinostat was obtained via a five-step process starting from benzaldehyde and including addition reaction with sodium bisulfite, sulfochlorination with chlorosulfonic acid, sulfonamidation with aniline, Knoevenagel condensation, and the final amidation with hydroxylamine. Key to the strategy is the preparation of 3-formylbenzenesulfonyl chloride using an economical and practical protocol. The main advantages of the route include inexpensive starting materials and acceptable overall yield. The scale-up experiment was carried out to provide 169 g of belinostat with 99.6% purity in 33% total yield.
(E)-N-Hydroxy-3-((phenylamino)sulfonyl)phenyl)acrylamide (Belinostat, 1)
1
mp 172–174 °C, (lit.(@) 172 °C). 1H NMR (400 MHz, DMSO-d6) δ = 10.75–10.42 (m, 2H), 9.15 (s, 1H), 7.92 (s, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.56 (d, J = 7.8 Hz, 1H),7.47 (d, J = 15.8 Hz, 1H), 7.24 (m, 2H), 7.10–7.01 (m, 3H), 6.51 (d, J = 15.8 Hz, 1H). MS (ESI): m/z = 318.6 [M+H] +.
@ Finn, P. W.; Bandara, M.; Butcher, C.; Finn, A.; Hollinshead, R.; Khan, N.; Law, N.; Murthy, S.; Romero,R.; Watkins, C.; Andrianov, V.; Bokaldere, R. M.; Dikovska, K.; Gailite, V.; Loza, E.; Piskunova, I.;Starchenkov, I.; Vorona, M.; Kalvinsh, I. Helv. Chim. Acta 2005, 88, 1630, DOI: 10.1002/hlca.200590129
Clip
Belinostat (Beleodaq),
Belinostat is a drug which was developed by Spectrum Pharmaceuticals and is currently marketed by Onxeo as Beleodaq. The
drug, which received fast track designation by the United States Food and Drug Administration (US FDA) and was approved for
the treatment of hematological malignancies and solid tumors associated with peripheral T-cell lymphoma (PTCL) in 2014,58 is a histone deacetylase (HDAC) inhibitor and is the third such treatment to receive accelerated approval for PTCL, the others being
vorinostat (Zolinza) and pralatrexate (Folotyn).58 Although belinostat was not yet approved in Europe as of August 2014,58 the
compound exhibits a safety profile considered to be acceptable for HDAC inhibitors–less than 25% of patients reported adverse
effects and these most frequently were nausea, fatigue, pyrexia,anemia, and emesis.58 While several different synthetic approaches
have been reported for the preparation of belinostat and related HDAC inhibitors,59–62 the most likely process-scale approach has
been described in a patent application filed by Reisch and co-workers at Topotarget UK, which exemplifies the synthesis described in
Scheme 8 on kilogram scale.63
Commercially available 3-bromobenzenesulfonyl chloride (41) was reacted with aniline in the presence of aqueous sodium carbonate
to deliver sulfonamide 42 in 94% yield. Next, this aryl bromide was subjected to a Heck reaction involving ethyl acrylate to
give rise to cinnamate ester 43, which was immediately saponified under basic conditions and acidic workup to furnish the corresponding acid 44. This acid was activated as the corresponding acid chloride prior to subjection to hydroxylamine under basic conditions to form the hydroxamic acid, which was then recrystallized from an 8:1 ethanol/water mixture in the presence of a catalytic
amount of sodium bicarbonate to furnish crystalline belinostat (VI) in 87% overall yield from acid 44.61
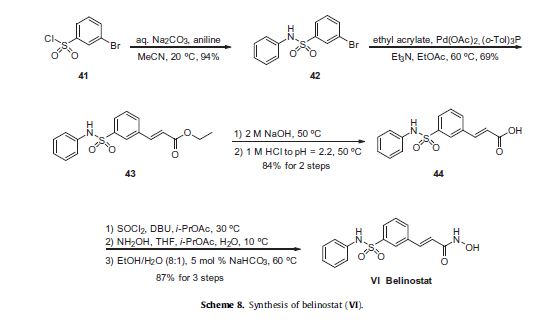
Lee, H. Z.; Kwitkowski, V. E.; Del Valle, P. L.; Ricci, M. S.; Saber, H.;Habtemariam, B. A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X. H.;Brown, J.; Mehrotra, N.; Dorff, S.; Charlab, R.; Kane, R. C.; Kaminskas, E.;Justice, R.; Farrell, A. T.; Pazdur, R. Clin. Cancer Res. 2015, 21, 2666.
59. Qian, J.; Zhang, G.; Qin, H.; Zhu, Y.; Xiao, Y. CN Patent 102786448A, 2012.
60. Wang, H.; Yu, N.; Chen, D.; Lee, K. C.; Lye, P. L.; Chang, J. W.; Deng, W.; Ng, M.C.; Lu, T.; Khoo, M. L.; Poulsen, A.; ngthongpitag, K.; Wu, X.; Hu, C.; Goh, K.C.; Wang, X.; Fang, L.; Goh, K. L.; Khng, H. H.; Goh, S. K.; Yeo, P.; Liu, X.; Bonday, Z.; Wood, J. M.; Dymock, B. W.; Kantharaj, E.; Sun, E. T. J. Med. Chem.2011, 54, 4694.
61. Yang, L.; Xue, X.; Zhang, Y. Synth. Comm. 2010, 40, 2520.
CLIP
Let’s Research !!!!!
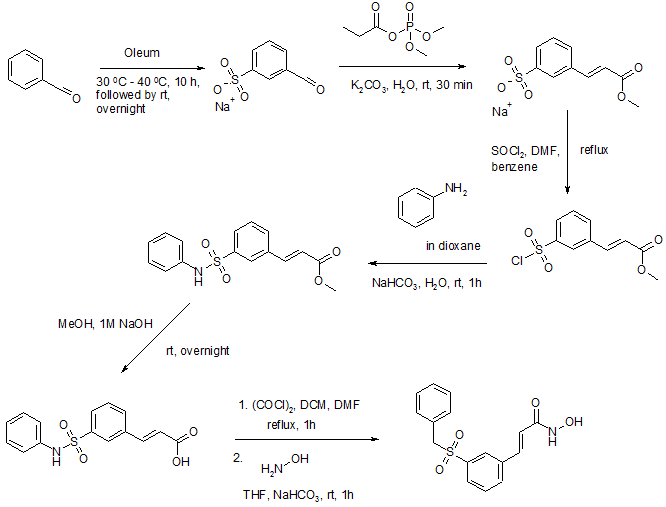
Helv Chim Acta 2005, 88(7), 1630-1657: It is first reported synthesis for Belinostat and many other derivatives. The procedure uses oleum, thionyl chloride (SOCl2) as well as oxalyl chloride (COCl)2, no wonder better procedures were derived from it. ABOVE
Synth Comm 2010, 40(17), 2520–2524: The synthesis avoids the use of the extremely corrosive oleum and thionyl chloride (SOCl2) and therefore is possibly better for scaled-up production. Second, synthetic steps do not involve tedious separations and give a better overall yield. BELOW Identifications:
Identifications:
Synth Comm 2010, 40(17), 2520–2524: The synthesis avoids the use of the extremely corrosive oleum and thionyl chloride (SOCl2) and therefore is possibly better for scaled-up production. Second, synthetic steps do not involve tedious separations and give a better overall yield. BELOW
Identifications:| 1H NMR (Estimated) for Belinostat |
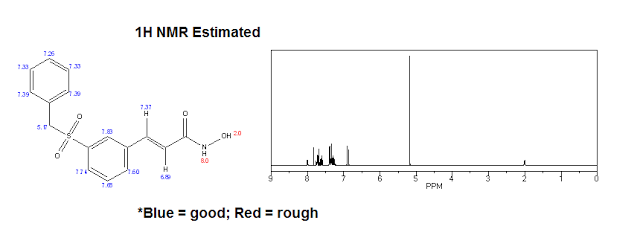
Experimental: 1H NMR (300 MHz, DMSO-d6): δ 6.52 (d, J=15.9 Hz, 1H), 6.81–7.12 (m, 6H), 7.33 (d, J=15.9 Hz, 1H), 7.47–7.67 (m, 3 H), 7.87 (s, 1H), 9.00–11.20 (br, 3H).


SEE COMPILATION ON SIMILAR COMPOUNDS AT …………..http://drugsynthesisint.blogspot.in/p/nostat-series.html
HPLC
ANALYTICAL HPLC TEST METHOD
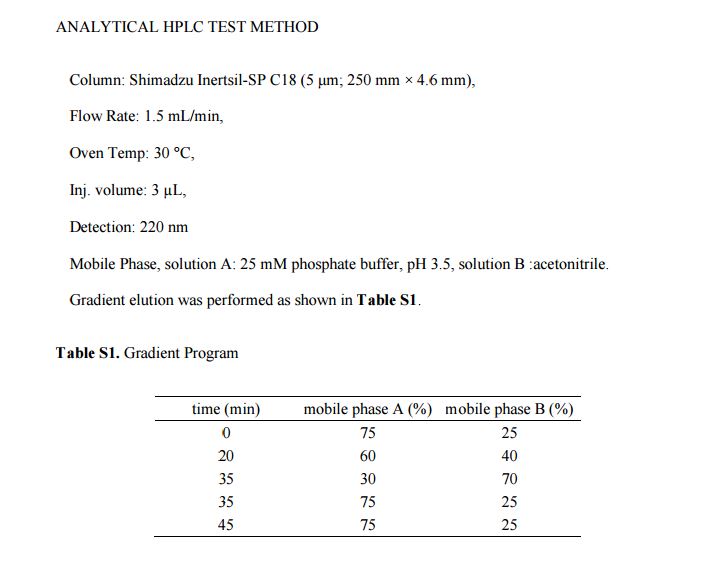
HPLC spectrum of Belinostat.
PATENT
http://www.google.si/patents/CN102531972A?cl=en
Belinostat synthesis process related to the first report of the literature of W002 / 30879 A2, including preparation for Belinostat described as follows:


Example 3:
3- (3-sulfonate-yl) phenyl – acrylate preparation:
First, 3-bromophenyl sulfonate 37. Ig (257. 90g / mol, 0. 1439mol) was dissolved with stirring in 260mL toluene IL reactor was then added triethylamine 36. 5g (101. 19g / mol, 0. 3604mol), tri (o-methylphenyl) phosphine 0. 875g (304. 37g / mol, 0. 002874mol), palladium acetate 0. 324g (224. 51g, 0. 001441mol), the reaction mixture was heated to 45- 55 ° C with nitrogen pumping ventilation four, this time in the reaction system to generate the catalytically active 1 ^ (0). The temperature of the reaction system was raised to 80-90 ° C, within 2. 75h dropwise methacrylate 13. 6g (86. 04g / mol, 0. 1586mol), the reaction was continued after the cell by HPLC 3- bromophenyl sulfonyl chloride was completion of the reaction. The temperature of the reaction system was reduced to 45-55 ° C.
[0021] In at 45-55 ° C, the reaction mixture was concentrated under reduced pressure, ethyl acetate and n-heptane and recrystallized to give the product 29. 4g, 83% yield.
[0022] The spectral data:
1HNMR (DMS0-d6, HMDS0), δ (ppm): 3. 65 (3H, S, H-1); 6. 47 (1H, d, J = 16 0 Hz, H-2.); 7. 30 -8 00 (5H, m, H-3, H_4, H_5, H_6, H_7) m / e:. 264. 23

References
-
- “Beleodaq (belinostat) For Injection, For Intravenous Administration. Full Prescribing Information” (PDF). Spectrum Pharmaceuticals, Inc. Irvine, CA 92618. Retrieved 21 November2015.
- Plumb JA; Finn PW; Williams RJ; et al. (2003). “Pharmacodynamic Response and Inhibition of Growth of Human Tumor Xenografts by the Novel Histone Deacetylase Inhibitor PXD101”. Molecular Cancer Therapeutics 2 (8): 721–728.PMID 12939461.
- “FDA approves Beleodaq to treat rare, aggressive form of non-Hodgkin lymphoma”. FDA. 3 July 2014.
- “CuraGen Corporation (CRGN) and TopoTarget A/S Announce Presentation of Belinostat Clinical Trial Results at AACR-NCI-EORTC International Conference”. October 2007.
- Final Results of a Phase II Trial of Belinostat (PXD101) in Patients with Recurrent or Refractory Peripheral or Cutaneous T-Cell Lymphoma, December 2009
- “Spectrum adds to cancer pipeline with $350M deal.”. February 2010.
- H. Spreitzer (4 August 2014). “Neue Wirkstoffe – Belinostat”.Österreichische Apothekerzeitung (in German) (16/2014): 27.
- Lexicomp, (corporate author) (2016). Bragalone, DL, ed.Drug Information Handbook for Oncology (14th ed.). Wolters Kluwer. ISBN 9781591953517.
- Helvetica Chimica Acta, 2005 , vol. 88, 7 PG. 1630 – 1657, MP 172
- WO2009/40517 A2, ….
- WO2006/120456 A1, …..
- Synthetic Communications, 2010 , vol. 40, 17 PG. 2520 – 2524, MP 172
- Journal of Medicinal Chemistry, 2011 , vol. 54, 13 PG. 4694 – 4720, NMR IN SUP INFO
Drug@FDA, NDA206256 Pharmacology Review(s).
Biochem. J. 2008, 409, 581-589.
J. Transl. Med. 2007, 5, 1-12.
Mol. Cancer Ther. 2006, 5, 2086-2095.
Int. J. Cancer 2008, 122, 1400-1410.
Synthetic Commun. 2010, 40, 2520-2524.
| CN101868446A * | Sep 23, 2008 | Oct 20, 2010 | 托波塔吉特英国有限公司 | Methods of synthesis of certain hydroxamic acid compounds |
| CN102531972A * | Dec 31, 2010 | Jul 4, 2012 | 北京万全阳光医药科技有限公司 | Preparation method of intermediate of antitumor medicament Belinostat |
| EP2093292A2 * | Mar 26, 2001 | Aug 26, 2009 | Methylgene, Inc. | Inhibition of specific histone deacetylase isoforms |
| GB2378179A * | Title not available | |||
| WO2002030879A2 * | Sep 27, 2001 | Apr 18, 2002 | Prolifix Limited | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2008068170A1 * | Nov 27, 2007 | Jun 12, 2008 | William Paul Jackson | Hdac inhibitors |
| WO2009146871A1 * | Jun 1, 2009 | Dec 10, 2009 | William Paul Jackson | 5-lipoxygenase inhibitors |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN104478769A * | Dec 22, 2014 | Apr 1, 2015 | 深圳万乐药业有限公司 | Belinostatsynthesis method suitable for industrial production |
| CN104478769B * | Dec 22, 2014 | Jan 6, 2016 | 深圳万乐药业有限公司 | 一种适合工业化生产的贝利司他合成方法 |
| CN104610100A * | Jan 9, 2015 | May 13, 2015 | 华东理工大学 | Nitrogen-chlorine type chlorination agent |
| US2008274120 | 11-7-2008 | Histone Deacetylase (Hdac) Inhibitors (Pxd101) for the Treatment of Cancer Alone or in Combination With Chemotherapeutic Agent |
| US2008227845 | 9-19-2008 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US2008213399 | 9-5-2008 | Combination Therapies Using Hdac Inhibitors |
| US2008194690 | 8-15-2008 | Pharmaceutical Formulations Of Hdac Inhibitors |
| US7407988 | 8-6-2008 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US7402603 | 7-23-2008 | Cyclooxygenase-2 inhibitor/histone deacetylase inhibitor combination |
| US7183298 | 2-28-2007 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US2005107445 | 5-20-2005 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US6888027 | 5-4-2005 | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising asulfonamide linkage as hdac inhibitors |
| US7973181 | 7-6-2011 | HYDROXAMIC ACID DERIVATIVES AS INHIBITORS OF HDAC ENZYMATIC ACTIVITY |
| US7928081 | 4-20-2011 | Combined Use of Prame Inhibitors and Hdac Inhibitors |
| US2011077305 | 3-32-2011 | 5-LIPOXYGENASE INHIBITORS |
| US2011003777 | 1-7-2011 | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
| US2010286279 | 11-12-2010 | Methods of Synthesis of Certain Hydroxamic Acid Compounds |
| US2010190694 | 7-30-2010 | Methods for identifying patients who will respond well to cancer treatment |
| US2010010010 | 1-15-2010 | HDAC INHIBITORS |
| US2009312311 | 12-18-2009 | COMBINATION OF ORGANIC COMPOUNDS |
| US2009192211 | 7-31-2009 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US7557140 | 7-8-2009 | CARBAMIC ACID COMPOUNDS COMPRISING A SULFONAMIDE LINKAGE AS HDAC INHIBITORS |
| WO1998038859A1 * | Mar 4, 1998 | Sep 11, 1998 | Thomas E Barta | Sulfonyl divalent aryl or heteroaryl hydroxamic acid compounds |
| WO1999024399A1 * | Nov 12, 1998 | May 20, 1999 | Darwin Discovery Ltd | Hydroxamic and carboxylic acid derivatives having mmp and tnf inhibitory activity |
| WO2000056704A1 * | Mar 22, 2000 | Sep 28, 2000 | Duncan Batty | Hydroxamic and carboxylic acid derivatives |
| WO2000069819A1 * | May 12, 2000 | Nov 23, 2000 | Thomas E Barta | Hydroxamic acid derivatives as matrix metalloprotease inhibitors |
| WO2001038322A1 * | Nov 22, 2000 | May 31, 2001 | Methylgene Inc | Inhibitors of histone deacetylase |
| EP0570594A1 * | Dec 7, 1992 | Nov 24, 1993 | SHIONOGI & CO., LTD. | Hydroxamic acid derivative based on aromatic sulfonamide |
| EP0931788A2 * | Dec 16, 1998 | Jul 28, 1999 | Pfizer Inc. | Metalloprotease inhibitors |
| GB2312674A * | Title not available |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2005063806A1 | Dec 30, 2003 | Jul 14, 2005 | Council Scient Ind Res | Arginine hydrochloride enhances chaperone-like activity of alpha crystallin |
| US4642316 | May 20, 1985 | Feb 10, 1987 | Warner-Lambert Company | Parenteral phenytoin preparations |
| WO2008090585A2 * | Jan 25, 2008 | Jul 31, 2008 | Univ Roma | Soluble forms of inclusion complexes of histone deacetylase inhibitors and cyclodextrins, their preparation processes and uses in the pharmaceutical field |
| WO2009109861A1 * | Mar 6, 2009 | Sep 11, 2009 | Topotarget A/S | Methods of treatment employing prolonged continuous infusion of belinostat |
| WO2010048332A2 * | Oct 21, 2009 | Apr 29, 2010 | Acucela, Inc. | Compounds for treating ophthalmic diseases and disorders |
| WO2011064663A1 | Nov 24, 2010 | Jun 3, 2011 | Festuccia, Claudio | Combination treatment employing belinostat and bicalutamide |
| US20110003777 * | Mar 6, 2009 | Jan 6, 2011 | Topotarget A/S | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
| CN102786448A * | 9 avg 2012 | 21 nov 2012 | 深圳万乐药业有限公司 | Method of synthesizing belinostat |
| CN102786448B | 9 avg 2012 | 12 mar 2014 | 深圳万乐药业有限公司 | Method of synthesizing belinostat |
CLIP
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(2E)-N-Hydroxy-3-[3-(phenylsulfamoyl)phenyl]prop-2-enamide
|
|
| Clinical data | |
| Trade names | Beleodaq |
| AHFS/Drugs.com | beleodaq |
| Pregnancy category |
|
| Routes of administration |
Intravenous (IV) |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 100% (IV) |
| Protein binding | 92.9–95.8%[1] |
| Metabolism | UGT1A1 |
| Excretion | Urine |
| Identifiers | |
| CAS Number | 866323-14-0 |
| ATC code | L01XX49 (WHO) |
| PubChem | CID 6918638 |
| ChemSpider | 5293831 |
| UNII | F4H96P17NZ |
| ChEBI | CHEBI:61076 |
| ChEMBL | CHEMBL408513 |
| Synonyms | PXD101 |
| Chemical data | |
| Formula | C15H14N2O4S |
| Molar mass | 318.348 g/mol |
////////////Belinostat, PXD101, novel HDAC inhibitor, Beleodaq, Folotyn, Spectrum Pharmaceuticals, Inc., Henderson, Nevada, Istodax, Celgene Corporation, Summit, New Jersey, CuraGen Pharma, FDA 2014
O=S(=O)(Nc1ccccc1)c2cc(\C=C\C(=O)NO)ccc2
SEE COMPILATION ON SIMILAR COMPOUNDS AT …………..http://drugsynthesisint.blogspot.in/p/nostat-series.html
Eliglustat tartrate (Cerdelga) エリグルスタット酒石酸塩 依利格鲁司特 エリグルスタット,サーデルガ

Eliglustat tartrate (Cerdelga) エリグルスタット酒石酸塩
依利格鲁司特
エリグルスタット,サーデルガ
FOR TREATMENT OF GAUCHERS DISEASE

ELIGLUSTAT; Cerdelga; Genz 99067; Genz-99067; UNII-DR40J4WA67; GENZ-112638;
CAS 491833-29-5 FREE FORM
| Molecular Formula: | C23H36N2O4 |
|---|---|
| Molecular Weight: | 404.54294 g/mol |
N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide
N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide;(2R,3R)-2,3-dihydroxybutanedioic acid
Mechanism of Action: glucosylceramide synthase inhibitor
Indication: Type I Gaucher Disease
Date of Approval: August 19, 2014 (US)
US patent number:US6916802 , US7196205 , US7615573
Patent Expiration Date: Apr 29, 2022 (US6916802, US7196205, US7615573)
Exclusivity Expiration Date:Aug 19, 2019(NCE), Aug 19, 2021 (ODE)
Originator:University of Michigan
Developer: Genzyme, a unit of Sanofi
Eliglustat, marketed by Genzyme as CERDELGA, is a glucosylceramide synthase inhibitor indicated for the long-term treatment of type 1 Gaucher disease. Patients selected for treatment with Eliglustat undergo an FDA approved genotype test to establish if they are CYP2D6 EM (extensive metabolizers), IM (intermediate metabolizers), or PM (poor metabolizers), as the results of this test dictate the dosage of Eliglustat recommended. Eliglustat was approved for use by the FDA in August 2014.
Eliglustat (INN, USAN;[1] trade name Cerdelga) is a treatment for Gaucher’s disease developed by Genzyme Corp that was approved by the FDA August 2014.[2] Commonly used as the tartrate salt, the compound is believed to work by inhibition ofglucosylceramide synthase.[3][4] According to an article in Journal of the American Medical Association the oral substrate reduction therapy resulted in “significant improvements in spleen volume, hemoglobin level, liver volume, and platelet count” in untreated adults with Gaucher disease Type 1.[5]
Cerdelga, capsule, 84 mg/1, oralGenzyme Corporation, 2014-09-03, ![]()

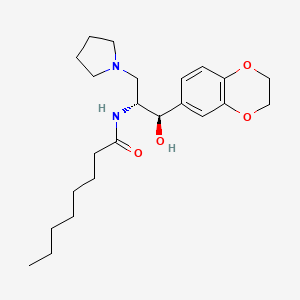
Eliglustat tartrate
- Molecular FormulaC50H78N4O14
- Average mass959.173 Da
- UNII-N0493335P3
-
Butanedioic acid, 2,3-dihydroxy-, (2R,3R)-, compd. with N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(1-pyrrolidinylmethyl)ethyl]octanamide (1:2)
-
eliglustat hemitartrate
-
eliglustat L-tartrate
CAS 928659-70-5
![]()


CERDELGA (eliglustat) capsules contain eliglustat tartrate, which is a small molecule inhibitor of glucosylceramide synthase that resembles the ceramide substrate for the enzyme, with the chemical name N-((1R,2R)-1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-1- hydroxy-3-(pyrrolidin-1-yl)propan-2-yl)octanamide (2R,3R)-2,3-dihydroxysuccinate. Its molecular weight is 479.59, and the empirical formula is C23H36N2O4+½(C4H6O6) with the following chemical structure:
|
Each capsule of CERDELGA for oral use contains 84 mg of eliglustat, equivalent to 100 mg of eliglustat tartrate (hemitartrate salt). The inactive ingredients are microcrystalline cellulose, lactose monohydrate, hypromellose and glyceryl behenate, gelatin, candurin silver fine, yellow iron oxide, and FD&C blue 2.
Cost
In 2014, the annual cost of Cerdelga hard gelatin capsules taken orally twice a day was $310,250. Genzyme’s flagship Imiglucerase(brand name Cerezyme) cost about $300,000 for the infusions if taken twice a month.[6] Manufacturing costs for Cerdelga are slightly lower than for Cerezyme. Genzyme’s maintains higher prices for orphan drugs—most often paid for by insurers— in order to remain financially sustainable.[6]
Chemically Eliglustat is named N-[(1 R,2R)-2-(2,3-dihydro-1 ,4-benzodioxin-6-yl)-2-hydroxy-1 -(1 -pyrrolidinylmethyl)ethyl]-Octanamide(2R!3R)-2,3-dihydroxybutanedioate and the hemitartarate salt of eliglustat has the structural formula as shown in Formula I.

Formula I
Eliglustat hemitartrate (Genz-1 12638), currently under development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy. Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase, and is currently under development by Genzyme.
U.S. patent No. 7,196,205 discloses a process for the preparation of Eliglustat or a pharmaceutically acceptable salt thereof.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 discloses process for preparation of Eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of Eliglustat, (ii) a hemitartrate salt of Eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
It has been disclosed earlier that the amorphous forms in a number of drugs exhibit different dissolution characteristics and in some cases different bioavailablity patterns compared to crystalline forms [Konne T., Chem pharm Bull., 38, 2003(1990)]. For some therapeutic indications one bioavailabihty pattern may be favoured over another. An amorphous form of Cefuroxime axetil is a good example for exhibiting higher bioavailability than the crystalline form.
CLIP
Eliglustat tartrate, developed and marketed by Genzyme Corporation (a subsidiary of Sanofi), was approved by the US FDA in August 2014 for the treatment of nonneuropathic (type 1) Gaucher disease (GD1) in both treatment-naïve and treatment-experienced adult patients.98
It is the first oral treatment to be approved for first-line use in patients with Gaucher disease type 1, which is a rare lysosomal storage disease characterized by accumulation of lipid glucosylceramide (GL-1) due to insufficient production of the enzyme glucosylceramidase.99,100
Clinical complications include hepatosplenomegaly, anemia, thrombocytopenia, and bone involvement.101 Eliglustat is a specific inhibitor of glucosylceramide synthase with an IC50 of 10 ng/mL and acts as substrate reduction therapy for GD1;102 it has demonstrated non-inferiority to enzyme replacement therapy, which is the current standard of care, in Phase III trials.99
While the process-scale route has not yet been disclosed,103 the largest scale route to eliglustat tartrate reported to date is described in Scheme 15.104
Condensation of commercially available S-(+)-2-phenyl glycinol (87) with phenyl bromoacetate (88) in acetonitrile in the presence of N,N-diisopropylethylamine (DIPEA) provided morpholin-2-one 89 upon treatment with HCl.Neutralization with NaHCO3 followed by coupling with aldehyde 90 in refluxing EtOAc/toluene yielded oxazine adduct 91, which was isolated as a precipitate from methyl-tert-butyl ether (MTBE).
The stereochemistry of the three new stereocenters in 91 can be rationalized through the cycloaddition of an ylide intermediate in the sterically-preferred S-configuration (generated by the reaction of the morpholinone 89 with aldehyde 90) with a second equivalent of the aldehyde. With the morpholinone in a chair conformation in which the phenyl group is equatorial, endo axial approach of the dipolarophile to the less-hindered face of the ylide and subsequent ring flip of the morpholinone ring to a boat conformation positions all exocyclic aryl substituents in a pseudoequatorial configuration. 105
Opening of oxazine 91 with pyrrolidine in refluxing THF followed by addition of HCl in refluxing MeOH gave amide 92, which was reduced to amine 93 using LiAlH4 in refluxing THF.
Subsequent hydrogenation with Pd(OH)2 in EtOH cleaved the phenylethanol group to give the free amine, which was converted to dioxalate salt 94 by treatment with oxalic acid in methyl isobutylketone (MIBK). Subjection of aminoethanol 94 to aqueous sodium hydroxide followed by coupling with palmitic acid Nhydroxysuccinimide (NHS)-ester (95) gave eliglustat as the corresponding freebase (96) in 9.5% overall yield from 87.
Salt formation with L-tartaric acid (0.5 equiv) then provided eliglustat tartrate (XII).106
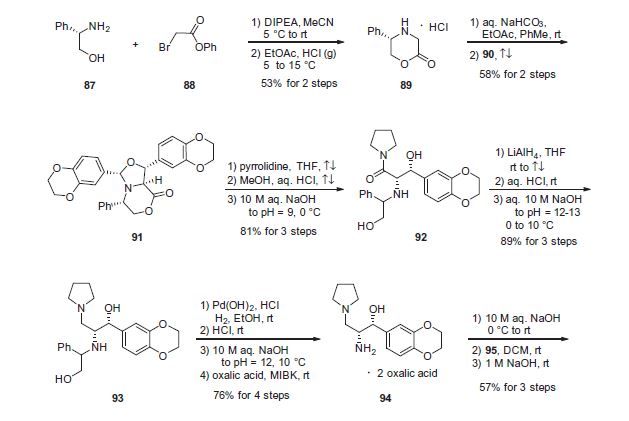
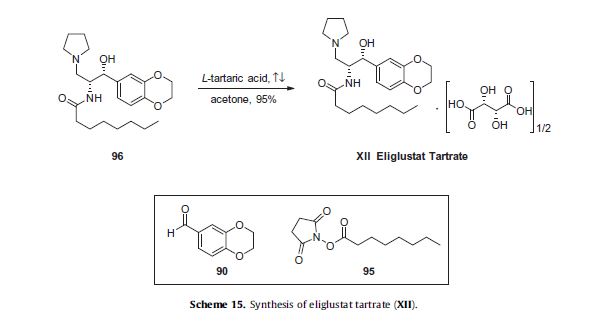
98. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm410585.htm.
99. Poole, R. M. Drugs 2014, 74, 1829.
100. Kaplan, P. Res. Rep. Endocr. Disord. 2014, 4, 1.
101. Pastores, G. M.; Hughes, D. Clin. Invest. 2014, 4, 45.
102. Shayman, J. A. Drugs Future 2010, 35, 613.
103. Javed, I.; Dahanukar, V. H.; Oruganti, S.; Kandagatla, B. WO Patent2,015,059,679, 2015.
104. Hirth, B.; Siegel, C. WO Patent 2,003,008,399, 2003.
105. Anslow, A. S.; Harwood, L. M.; Phillips, H.; Watkin, D.; Wong, L. F. Tetrahedron:Asymmetry 1991, 2, 1343.
106. Liu, H.; Willis, C.; Bhardwaj, R.; Copeland, D.; Harianawala, A.; Skell, J.;Marshall, J.; Kochling, J.; Palace, G.; Peterschmitt, J.; Siegel, C.; Cheng, S. WO Patent 2,011,066,352, 2011.
CLIP
TAKEN FROM
http://www.xinbiaopin.com/a/zuixindongtai/huaxuepinshuju/2015/0310/2383.html

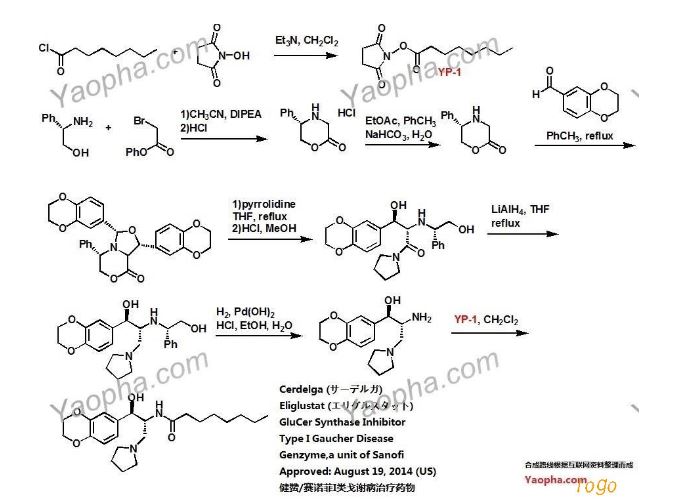
Nmr predict
![N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide NMR spectra analysis, Chemical CAS NO. 491833-29-5 NMR spectral analysis, N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-09-06/001/571/702/491833-29-5-1h.png)
13 C NMR
![N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide NMR spectra analysis, Chemical CAS NO. 491833-29-5 NMR spectral analysis, N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide C-NMR spectrum](https://i1.wp.com/pic11.molbase.net/nmr/nmr_image/2014-09-06/001/571/702/491833-29-5-13c.png)
CAS NO. 491833-29-5, N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide
C-NMR spectral analysis
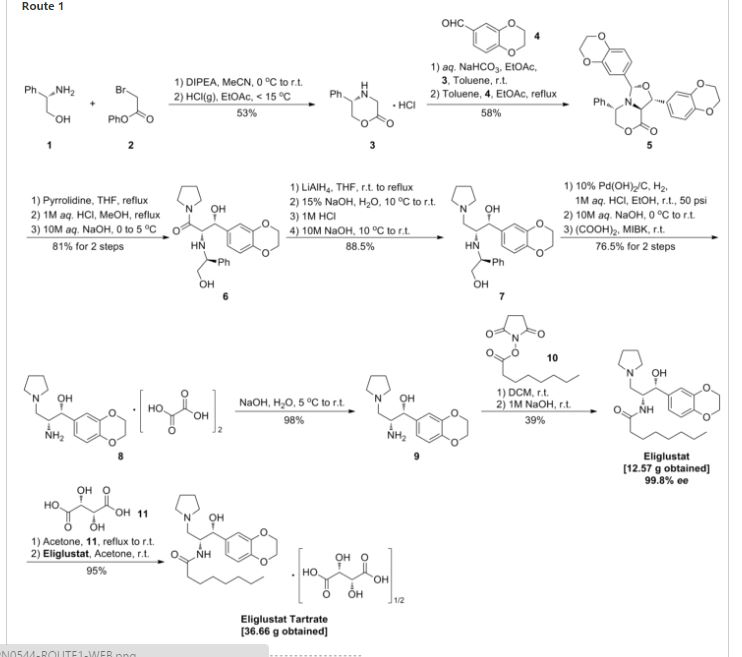
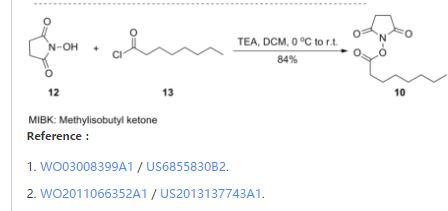
PATENT
http://www.google.com/patents/WO2013059119A1?cl=en

http://www.google.com/patents/US7196205
Compound 7
(1R,2R)-Nonanoic acid[2-(2′,3′-dihydro-benzo[1,4]dioxin-6′-yl)-2-hydroxy-1-pyrrolidin-1-ylmethyl-ethyl]-amide

This compound was prepared by the method described for Compound 6 using Nonanoic acid N-hydroxysuccinimide ester. Analytical HPLC showed this material to be 98.4% pure. mp 74–75° C.
1H NMR (CDCl3) δ 6.86–6.76 (m, 3H), 5.83 (d, J=7.3 Hz, 1H), 4.90 (d, J=3.3 Hz, 1H), 4.24 (s, 4H), 4.24–4.18 (m, 1H), 2.85–2.75 (m, 2H), 2.69–2.62 (m, 4H), 2.10 (t, J=7.3 Hz, 2H), 1.55–1.45 (m, 2H), 1.70–1.85 (m, 4H), 1.30–1.15 (m, 10H), 0.87 (t, J=6.9 Hz, 3H) ppm.
Intermediate 4(1R,2R)-2-Amino-1-(2′,3′-dihydro-benzo[1,4]dioxin-6′-yl)-3-pyrrolidin-1-yl-propan-1-ol

Intermediate 3 (5.3 g, 13.3 mmol) was dissolved in methanol (60 mL). Water (6 mL) and trifluoroacetic acid (2.05 m/L, 26.6 mmol, 2 equivalents) were added. After being placed under nitrogen, 20% Palladium hydroxide on carbon (Pearlman’s catalysis, Lancaster or Aldrich, 5.3 g) was added. The mixture was placed in a Parr Pressure Reactor Apparatus with glass insert. The apparatus was placed under nitrogen and then under hydrogen pressure 110–120 psi. The mixture was stirred for 2–3 days at room temperature under hydrogen pressure 100–120 psi. The reaction was placed under nitrogen and filtered through a pad of celite. The celite pad was washed with methanol (100 mL) and water (100 mL). The methanol was removed by rotoevaporation. The aqueous layer was washed with ethyl acetate three times (100, 50, 50 mL). A 10 M NaOH solution (10 mL) was added to the aqueous layer (pH=12–14). The product was extracted from the aqueous layer three times with methylene chloride (100, 100, 50 mL). The combined organic layers were dried with Na2SO4, filtered and rotoevaporated to a colorless oil. The foamy oil was vacuum dried for 2 h. Intermediate 4 was obtained in 90% yield (3.34 g).
Intermediate 3(1R,2R,1″S)-1-(2′,3′-Dihydro-benzo[1,4]dioxin-6′-yl)-2-(2″-hydroxy -1′-phenyl-ethylamino)-3-pyrrolidin-1-yl-propan-1-ol

To a 3-neck flask equipped with a dropping funnel and condenser was added LiAlH4 (Aldrich, 1.2 g, 31.7 mmol, 2.5 equivalents) and anhydrous THF (20 mL) under nitrogen. A solution of Intermediate 2 (5.23 g, 12.68 mmol) in anhydrous THF (75 mL) was added dropwise to the reaction over 15–30 minutes. The reaction was refluxed under nitrogen for 9 hours. The reaction was cooled in an ice bath and a 1M NaOH solution was carefully added dropwise. After stirring at room temperature for 15 minutes, water (50 mL) and ethyl acetate (75 mL) was added. The layers were separated and the aqueous layer was extracted twice with ethyl acetate (75 mL). The combined organic layers were washed with saturated sodium chloride solution (25 mL). After drying with Na2SO4 the solution was filtered and rotoevaporated to yield a colorless to yellow foamy oil. Intermediate 3 was obtained in 99% yield (5.3 g).
PATENT
EXAMPLES
Example 1 : Preparation of amorphous form of eliglustat hemitartarate.
500mg of eliglustat hemitartarate was dissolved in 14 mL of dichloromethane at 26°C and stirred for 15 min. The solution is filtered to remove the undissolved particles and the filtrate is distilled under reduced pressure at 45°C. After distillation the solid was dried under vacuum at 45°C.
PATENT
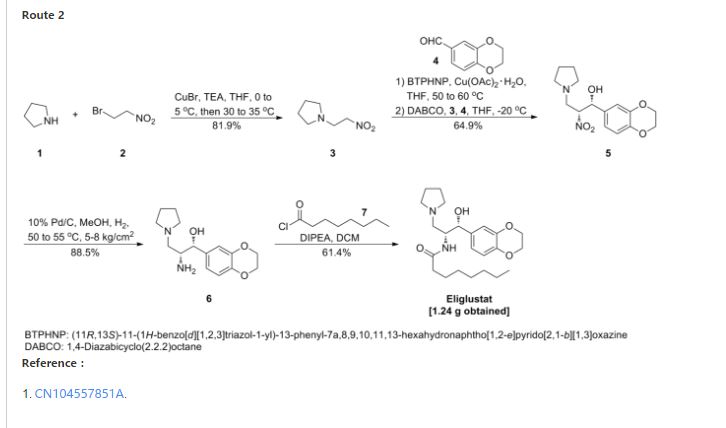
PAPER
Journal of Medicinal Chemistry (2012), 55(9), 4322-4335
OLD CLIPS
Genzyme Announces Positive New Data from Two Phase 3 Studies for Oral Eliglustat Tartrate for Gaucher Disease
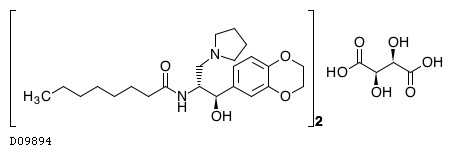
Eliglustat tartrate (USAN)
CAS:928659-70-5
February 15, 2013
Genzyme , a Sanofi company (EURONEXT: SAN and NYSE: SNY), today announced positive new data from the Phase 3 ENGAGE and ENCORE studies of eliglustat tartrate, its investigational oral therapy for Gaucher disease type 1. The results from the ENGAGE study were presented today at the 9th Annual Lysosomal Disease Network WORLD Symposium in Orlando, Fla. In conjunction with this meeting, Genzyme also released topline data from its second Phase 3 study, ENCORE. Both studies met their primary efficacy endpoints and together will form the basis of Genzyme’s registration package for eliglustat tartrateThe data presented at this year’s WORLD symposium reinforce our confidence that eliglustat tartrate may become an important oral option for patients with Gaucher disease”The company is developing eliglustat tartrate, a capsule taken orally, to provide a convenient treatment alternative for patients with Gaucher disease type 1 and to provide a broader range of treatment options for patients and physicians. Genzyme’s clinical development program for eliglustat tartrate represents the largest clinical program ever focused on Gaucher disease type 1 with approximately 400 patients treated in 30 countries.“The data presented at this year’s WORLD symposium reinforce our confidence that eliglustat tartrate may become an important oral option for patients with Gaucher disease,” said Genzyme’s Head of Rare Diseases, Rogerio Vivaldi MD. “We are excited about this therapy’s potential and are making excellent progress in our robust development plan for bringing eliglustat tartrate to the market.”ENGAGE Study Results:In ENGAGE, a Phase 3 trial to evaluate the safety and efficacy of eliglustat tartrate in 40 treatment-naïve patients with Gaucher disease type 1, improvements were observed across all primary and secondary efficacy endpoints over the 9-month study period. Results were reported today at the WORLD Symposium by Pramod Mistry, MD, PhD, FRCP, Professor of Pediatrics & Internal Medicine at Yale University School of Medicine, and an investigator in the trial.The randomized, double-blind, placebo-controlled study had a primary efficacy endpoint of improvement in spleen size in patients treated with eliglustat tartrate. Patients were stratified at baseline by spleen volume. In the study, a statistically significant improvement in spleen size was observed at nine months in patients treated with eliglustat tartrate compared with placebo. Spleen volume in patients treated with eliglustat tartrate decreased from baseline by a mean of 28 percent compared with a mean increase of two percent in placebo patients, for an absolute difference of 30 percent (p<0.0001).
Eliglustat tartate (Genz-112638)
What is Eliglustat?
- Eliglustat is a new investigational phase 3 compound from Genzyme Corporation that is being studied for type 1 Gaucher Disease.
- Eliglustat works as a substrate reduction therapy by reducing glucocerebroside. formation.
- This product is an oral agent (i.e. a pill) that is taken once or twice a day in contrast to an IV infusion for enzyme replacement therapy. Enzyme replacement therapy focuses on replenishing the enzyme that is deficient in Gaucher Disease and breaks down glucocerebroside that accumulates.
- The clinical trials for eliglustat tartate are sponsored by Genzyme Corporation.


Eliglustat tartrate (Genz-1 12638) is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of gaucher disease and other lysosomal storage disorders, which is currently under development.
Eliglustat is chemically known as 1 R, 2R-Octanoic acid [2-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1 -ylmethyl]-ethyl]-amide, having a structural formula I depicted here under.
Formula I
Eliglustat hemitartrate (Genz-1 12638) development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy.
Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase.
U.S. patent No. 7,196,205 (herein described as US’205) discloses a process for the preparation of eliglustat or a pharmaceutically acceptable salt thereof. In this patent, eliglustat was synthesized via a seven-step process involving steps in that sequence:
(i) coupling S-(+)-2-phenyl glycinol with phenyl bromoacetate followed by column chromatography for purification of the resulting intermediate,
(ii) reacting the resulting (5S)-5-phenylmorpholin-2-one with 1 , 4-benzodioxan-6-carboxaldehyde to obtain a lactone,
(iii) opening the lactone of the oxazolo-oxazinone cyclo adduct via reaction with pyrrolidine,
(iv) hydrolyzing the oxazolidine ring, (v) reducing the amide to amine to obtain sphingosine like compound, (vi) reacting the resulting amine with octanoic acid and N-hydroxysuccinimide to obtain crude eliglustat, (vii) purifying the crude eliglustat by repeated isolation for four times from a mixture of ethyl acetate and n-heptane.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 disclose processes for preparation of eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of eliglustat, (ii) a hemitartrate salt of eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
WO 2015059679
| Process for the preparation of eliglustat free base – comprising the reaction of S-(+)-phenyl glycinol with phenyl-alpha-bromoacetate to obtain 5-phenylmorpholin-2-one, which is further converted to eliglustat. | |
| Dr Reddy’s Laboratories Ltd | |
| New crystalline eliglustat free base Form R1 and a process for its preparation are claimed. Also claimed is a process for the preparation of eliglustat free base which comprises the reaction of S-(+)-phenyl glycinol with phenyl-alpha-bromoacetate to obtain 5-phenylmorpholin-2-one, which is further converted to eliglustat.Further eliglustat oxalate, its crystalline form, and a process for the preparation of crystalline eliglustat oxalate, are claimed. | |
Eliglustat tartrate (Genz-1 12638) is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of gaucher disease and other lysosomal storage disorders, which is currently under development.
Eliglustat is chemically known as 1 R, 2R-Octanoic acid [2-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1 -ylmethyl]-ethyl]-amide, having a structural formula I depicted here under.

Formula I
Eliglustat hemitartrate (Genz-1 12638) development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy.
Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase.
U.S. patent No. 7,196,205 (herein described as US’205) discloses a process for the preparation of eliglustat or a pharmaceutically acceptable salt thereof. In this patent, eliglustat was synthesized via a seven-step process involving steps in that sequence:
(i) coupling S-(+)-2-phenyl glycinol with phenyl bromoacetate followed by column chromatography for purification of the resulting intermediate,
(ii) reacting the resulting (5S)-5-phenylmorpholin-2-one with 1 , 4-benzodioxan-6-carboxaldehyde to obtain a lactone,
(iii) opening the lactone of the oxazolo-oxazinone cyclo adduct via reaction with pyrrolidine,
(iv) hydrolyzing the oxazolidine ring, (v) reducing the amide to amine to obtain sphingosine like compound, (vi) reacting the resulting amine with octanoic acid and N-hydroxysuccinimide to obtain crude eliglustat, (vii) purifying the crude eliglustat by repeated isolation for four times from a mixture of ethyl acetate and n-heptane.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 disclose processes for preparation of eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of eliglustat, (ii) a hemitartrate salt of eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
Example 1 : Preparation of 5-phenyl morpholine-2-one hydrochloride
To a (S) + phenyl glycinol (100g) add N, N-diisopropylethylamine (314ml) and acetonitrile (2000ml) under nitrogen atmosphere at room temperature. It was cooled to 10- 15° C. Phenyl bromoacetate (172.4g) dissolved in acetonitrile (500ml) was added to the above solution at 15° C over a period of 30 min. The reaction mixture is allowed to room temperature and stirred for 16-20h. Progress of the reaction was monitored by thin layer chromatography. After completion of the reaction, the reaction mixture was concentrated under reduced pressure at a water bath
temperature less than 25° C to get a residue. The residue was dissolved in ethyl acetate (1000ml) and stirred for 1 h at 15-20°C to obtain a white solid. The solid material obtained was filtered and washed with ethyl acetate (200ml). The filtrate was dried over anhydrous sodium sulphate (20g) and concentrated under reduced pressure at a water bath temperature less than 25° C to give crude compound (1000g) as brown syrup. The Crude brown syrup is converted to HCI salt by using HCI in ethyl acetate to afford 5-phenyl morpholine-2-one hydrochloride (44g) as a white solid. Yield: 50%, Mass: m/z = 177.6; HPLC (% Area Method): 90.5%
Example 2: Preparation of (1 R,3S,5S,8aS)-1 ,3-Bis-(2′,3′-dihydro-benzo[1 ,4] dioxin-6′-yl)-5-phenyl-tetrahydro-oxazolo[4,3-c][1 ,4]oxazin-8-one.
5-phenyl morpholine-2-one hydrochloride (100g) obtained from above stage 1 is dissolved in toluene (2500ml) under nitrogen atmosphere at 25-30°C. 1 ,4-benzodioxane-6-carboxaldehyde (185.3g) and sodium sulphate (400g) was added to the above solution and the reaction mixture was heated at 100-105°C for 72h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was concentrated under reduced pressure at a water bath temperature less than 25° C to get a residue. The residue was cooled to 10°C, ethyl acetate (2700ml) and 50% sodium bisulphate solution (1351 ml) was added to the residue and stirred for 1 h at 10°C to obtain a white solid. The obtained white solid was filtered and washed with ethyl acetate. The separated ethyl acetate layer was washed with water (1000ml), brine (1000ml) and dried over anhydrous sodium sulphate. The organic layer was concentrated under reduced pressure at a water bath temperature of 45-50°C to get a crude material. The obtained crude material is triturated with diethyl ether (1500ml) to get a solid material which is filtered and dried under vacuum at room temperature for 2-3h to afford (1 R,3S,5S,8aS)-1 ,3-Bis-(2′,3′-dihydro-benzo[1 ,4]dioxin-6′-yl)-5-phenyl-tetrahydro-oxazolo[4,3-c][1 ,4]oxazin-8-one (148g) as a yellow solid. Yield: 54%, Mass: m/z = 487.7; HPLC (% Area Method): 95.4 %
Example 3: Preparation of (2S,3R,1 “S)-3-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)-3-hydroxy-2-(2″-hydroxy-1 ”^henyl-ethy^
(1 R,3S,5S,8aS)-1 !3-Bis-(2′!3′-dihydro-benzo[1 ,4]dioxin-6′-yl)-5-phenyl-tetrahydro-oxazolo[4,3-c][1 ,4]oxazin-8-one (70g) obtained from above stage 2 was dissolved in chloroform (1400ml) at room temperature. It was cooled to 0-5°C and pyrrolidone (59.5ml) was added at 0-5°C over a period of 30 minutes. The reaction mixture was allowed to room temperature and stirred for 16-18h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was concentrated under reduced pressure at a water bath temperature of 40-45°C to obtain a crude. The obtained crude was dissolved in methanol (1190ml) and 1 N HCI (1 190ml) at 10-15° C, stirred for 10 minutes and heated at 80-85°C for 7h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, methanol was concentrated under reduced pressure at a water bath temperature of 50-55°C.The aqueous layer was extracted with ethyl acetate and the organic layer was washed with 1 N HCI (50ml). The aqueous layer was basified with saturated sodium bicarbonate solution up to pH 8-9 and extracted with ethyl acetate (3x70ml). The combined organic layers was washed with brine (100ml), dried over anhydrous sodium sulphate and concentrated under reduced pressure at a water bath temperature of 50-55°C to afford (2S,3R,1″S)-3-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)-3-hydroxy-2-(2″-hydroxy-1 “-phenyl-ethylamino)-1 -pyrrolidin-1 -yl-propan-1 -one (53g) as a yellow foamy solid. Yield: 90%, Mass: m/z = 412.7, HPLC (% Area Method): 85.1 %
Example 4: Preparation of (1 R,2R,1 “S)-1-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)2-hydroxy-2-(2”-hydroxy-1 ‘-phenyl-ethylamino)-3-pyrrolidin-1-yl-propan-1-ol.
(2S,3R,1 “S)-3-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6’-yl)-3-hydroxy-2-(2”-hydroxy-1 “-phenyl-ethylamino)-1 -pyrrolidin-1 -yl-propan-1 -one (2.5g) obtained from above stage 3 dissolved in Tetrahydrofuran (106ml) was added to a solution of Lithium aluminium hydride (12.2g) in tetrahydrofuran (795ml) at 0°C and the reaction mixture was heated at 60-65°C for 10h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was cooled to 5- 10°C and quenched in saturated sodium sulphate solution (100ml) at 5-10°C. Ethyl acetate was added to the reaction mass and stirred for 30-45 min. The obtained solid is filtered through celite bed and washed with ethyl acetate. Filtrate was dried over anhydrous sodium sulphate and concentrated under reduced pressure at a water bath temperature of 50°C to afford (1 R,2R, 1″S)-1 -(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)2-hydroxy-2-(2″-hydroxy-1 ‘-phenyl-ethylamino)-3-pyrrolidin-1 -yl-propan-1 -ol (43.51 g) as a yellow gummy liquid. The crude is used for the next step without further purification. Yield: 85%, Mass: m/z = 398.7, HPLC (% Area Method): 77 %
Example 5: Preparation of (1 R, 2R)-2-Amino-1-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol.
(1 R,2R,1 “S)-1 -(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6’-yl)2-hydroxy-2-(2”-hydroxy-1 ‘-phenyl-ethylamino)-3-pyrrolidin-1 -yl-propan-1 -ol (40g) obtained from above stage 4 was dissolved in methanol (400ml) at room temperature in a 2L hydrogenation flask. Trifluoroacetic acid (15.5ml) and 20% Pd (OH) 2(40g) was added to the above solution under nitrogen atmosphere. The reaction mixture was hydrogenated under H2, 10Opsi for 16-18h at room temperature. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was filtered through celite bed and washed with methanol (44ml) and water (44ml). Methanol was concentrated under reduced pressure at a water bath temperature of 50-55°C and the aqueous layer was washed with ethyl acetate. The aqueous layer was basified with 10M NaOH till the PH reaches 12-14 and then extracted with dichloromethane (2x125ml). The organic layer was dried over anhydrous sodium sulphate (3gm) and concentrated under reduced pressure at a water bath temperature of 45°C to obtain a gummy liquid. The gummy liquid was triturated with methyl tertiary butyl ether for 1 h to get a white solid, which is filtered and dried under vacuum at room temperature to afford (1 R, 2R)-2-Amino-1 -(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol (23g) as a white solid. Yield: 82.3%, Mass (m/zj: 278.8, HPLC (% Area Method): 99.5%, Chiral HPLC (% Area Method): 97.9%
Example 6: Preparation of Eliglustat {(1 R, 2R)-Octanoic acid[2-(2′,3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1-ylmethyl-ethyl]-amide}.
(1 R, 2R)-2-Amino-1 -(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol (15g) obtained from above stage 5 was dissolved in dry dichloromethane (150ml) at room temperature under nitrogen atmosphere and cooled to 10-15° C. Octanoic acid N-hydroxy succinimide ester (13.0 g)was added to the above reaction mass at 10-15° C and stirred for 15 min. The reaction mixture was stirred at room temperature for 16h-18h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was cooled to 15°C and diluted with 2M NaOH solution (100 ml_) and stirred for 20 min at 20 °C. The organic layer was separated and washed with 2M sodium hydroxide (3x90ml).The organic layer was dried over anhydrous sodium sulphate (30g) and concentrated under reduced pressure at a water bath temperature of 45°C to give the crude compound (20g).The crude is again dissolved in methyl tertiary butyl ether (25 ml_) and precipitated with Hexane (60ml). It is stirred for 10 min, filtered and dried under vacuum to afford Eliglustat as a white solid (16g). Yield: 74%, Mass (m/zj: 404.7 HPLC (% Area Method): 97.5 %, ELSD (% Area Method): 99.78%, Chiral HPLC (% Area Method): 99.78 %.
Example 7: Preparation of Eliglustat oxalate.
Eliglustat (5g) obtained from above stage 6 is dissolved in Ethyl acetate (5ml) at room temperature under nitrogen atmosphere. Oxalic acid (2.22g) dissolved in ethyl acetate (5ml) was added to the above solution at room temperature and stirred for 14h. White solid observed in the reaction mixture was filtered and dried under vacuum at room temperature for 1 h to afford Eliglustat oxalate as a white solid (4g). Yield: 65.46%, Mass (m/zj: 404.8 [M+H] +> HPLC (% Area Method): 95.52 %, Chiral HPLC (% Area Method): 99.86 %
References
- Eligustat (PDF), AMA By subscription only
- FDA approves new drug to treat a form of Gaucher disease, U.S. Food and Drug Administration, 19 August 2015, retrieved 18 July 2015
- Lee, L.; Abe, A.; Shayman, J. A. (21 May 1999). “Improved Inhibitors of Glucosylceramide Synthase”. Journal of Biological Chemistry 274(21): 14662–14669. doi:10.1074/jbc.274.21.14662.
- Shayman, JA (1 August 2010). “Eliglustat Tartrate: Glucosylceramide Synthase Inhibitor Treatment of Type 1 Gaucher Disease.”. Drugs of the future 35 (8): 613–620. PMID 22563139.
- Pramod K. Mistry, Elena Lukina, Hadhami Ben Turkia, Dominick Amato, Hagit Baris, Majed Dasouki, Marwan Ghosn, Atul Mehta, Seymour Packman, Gregory Pastores, Milan Petakov, Sarit Assouline, Manisha Balwani, Sumita Danda, Evgueniy Hadjiev, Andres Ortega, Suma Shankar, Maria Helena Solano, Leorah Ross, Jennifer Angell, M. Judith Peterschmitt (17 February 2015), “Effect of Oral Eliglustat on Splenomegaly in Patients With Gaucher Disease Type 1: The ENGAGE Randomized Clinical Trial”, Journal of the American Medical Association 313 (7): 695–706, doi:10.1001/jama.2015.459
- Robert Weisman (2 September 2014), New Genzyme pill will cost patients $310,250 a year, The Boston Globe, retrieved 18 July 2015
FDA Orange Book Patents
| FDA Orange Book Patents: 1 of 3 | |
|---|---|
| Patent | 6916802 |
| Expiration | Apr 29, 2022 |
| Applicant | GENZYME CORP |
| Drug Application | N205494 (Prescription Drug: CERDELGA. Ingredients: ELIGLUSTAT TARTRATE) |
from FDA Orange Book
| FDA Orange Book Patents: 2 of 3 | |
|---|---|
| Patent | 7196205 |
| Expiration | Apr 29, 2022 |
| Applicant | GENZYME CORP |
| Drug Application | N205494 (Prescription Drug: CERDELGA. Ingredients: ELIGLUSTAT TARTRATE) |
from FDA Orange Book
| Patent ID | Date | Patent Title |
|---|---|---|
| US8003617 | 2011-08-23 | Methods of Treating Diabetes Mellitus |
| US2010298317 | 2010-11-25 | METHOD OF TREATING POLYCYSTIC KIDNEY DISEASES WITH CERAMIDE DERIVATIVES |
| US7763738 | 2010-07-27 | SYNTHESIS OF UDP-GLUCOSE: N-ACYLSPHINGOSINE GLUCOSYLTRANSFERASE INHIBITORS |
| US7615573 | 2009-11-10 | Synthesis of UDP-glucose: N-acylsphingosine glucosyltransferase inhibitors |
| US2009105125 | 2009-04-23 | Methods of Treating Fatty Liver Disease |
| US7265228 | 2007-09-04 | Synthesis of UDP-glucose: N-acylsphingosine glucosyltransferase inhibitors |
| US7196205 | 2007-03-27 | Synthesis of UDP-glucose: N-acylsphingosine glucosyltransferase inhibitors |
| US6855830 | 2005-02-15 | Synthesis of UDP-glucose: N-acylsphingosine glucosyltransferase inhibitors |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2016068519 | 2016-03-10 | INHIBITORS OF THE ENZYME UDP-GLUCOSE: N-ACYL-SPHINGOSINE GLUCOSYLTRANSFERASE |
| US2015148534 | 2015-05-28 | SYNTHESIS OF UDP-GLUCOSE: N-ACYLSPHINGOSINE GLUCOSYL TRANSFERASE INHIBITORS |
| US2015051261 | 2015-02-19 | Methods of Treating Fatty Liver Disease |
| US8779163 | 2014-07-15 | Synthesis of UDP-Glucose: N-acylsphingosine glucosyl transferase inhibitors |
| US2013137743 | 2013-05-30 | AMORPHOUS AND A CRYSTALLINE FORM OF GENZ 112638 HEMITARTRATE AS INHIBITOR OF GLUCOSYLCERAMIDE SYNTHASE |
| US2013095089 | 2013-04-18 | GLUCOSYLCERAMIDE SYNTHASE INHIBITORS AND THERAPEUTIC METHODS USING THE SAME |
| US2012322786 | 2012-12-20 | 2-ACYLAMINOPROPOANOL-TYPE GLUCOSYLCERAMIDE SYNTHASE INHIBITORS |
| US8138353 | 2012-03-20 | SYNTHESIS OF UDP-GLUCOSE: N-ACYLSPHINGOSINE GLUCOSYLTRANSFERASE INHIBITORS |
| US2012022126 | 2012-01-26 | Method Of Treating Diabetes Mellitus |
| US8003617 | 2011-08-23 | Methods of Treating Diabetes Mellitus |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[(1R,2R)-1-(2,3-Dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-(1-pyrrolidinyl)-2-propanyl]octanamide
|
|
| Clinical data | |
| Trade names | Cerdelga |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | 491833-29-5 |
| ATC code | A16AX10 (WHO) |
| PubChem | CID 23652731 |
| ChemSpider | 28475348 |
| ChEBI | CHEBI:82752 |
| Chemical data | |
| Formula | C23H36N2O4 |
| Molar mass | 404.543 g/mol |
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US6916802 | No | 2002-04-29 | 2022-04-29 | |
| US7196205 | No | 2002-04-29 | 2022-04-29 | |
| US7615573 | No | 2002-04-29 | 2022-04-29 |
///////////491833-29-5, 928659-70-5, eliglustat hemitartrate, eliglustat L-tartrate, ELIGLUSTAT, Cerdelga, Genz 99067, Genz-99067, UNII-DR40J4WA67, GENZ-112638, エリグルスタット酒石酸塩 , FDA 2014, GAUCHERS DISEASE, 依利格鲁司特, エリグルスタット,サーデルガ
CCCCCCCC(=O)N[C@H](CN1CCCC1)[C@@H](C2=CC3=C(C=C2)OCCO3)O
Ombitasvir

Ombitasvir; ABT-267; ABT 267; UNII-2302768XJ8; 1258226-87-7;
| C50H67N7O8 | |
| Molecular Weight: | 894.10908 g/mol |
|---|
Anti-Viral Compounds [US2010317568]
Dimethyl (2S,2′S)-1,1′-((2S,2′S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-Butylphenyl)pyrrolidine-2,5-diyl)bis(4,1-phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-diyl)dicarbamate
methyl N-[(2S)-1-[(2S)-2-[[4-[(2S,5S)-1-(4-tert-butylphenyl)-5-[4-[[(2S)-1-[(2S)-2-(methoxycarbonylamino)-3-methylbutanoyl]pyrrolidine-2-carbonyl]amino]phenyl]pyrrolidin-2-yl]phenyl]carbamoyl]pyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl]carbamate
1258226-87-7 [RN]
2:9 hydrate cas= 1456607-70-7…… is the drug substance
ABT-267
Abbvie Inc. innovator
ombitasvir is Dimethyl ([(2S,5S)-1-(4-tert-butylphenyl) pyrrolidine-2,5diyl]bis{benzene-4,1-diylcarbamoyl(2S)pyrrolidine-2,1-diyl[(2S)-3-methyl-1-oxobutane-1,2diyl]})biscarbamate hydrate. The molecular formula is C50H67N7O8•4.5H2O (hydrate) and the molecular weight for the drug substance is 975.20 (hydrate).

Ombitasvir is an antiviral drug for the treatment of hepatitis C virus (HCV) infection. In the United States, it is approved by theFood and Drug Administration for use in combination with paritaprevir, ritonavir and dasabuvir in the product Viekira Pak for the treatment of HCV genotype 1,[1][2] and with paritaprevir and ritonavir in the product Technivie for the treatment of HCV genotype 4.[3][4]
Ombitasvir is in phase II clinical development at AbbVie for the treatment of chronic hepatitis C infection in combination with ABT-450/ritonavir and, in combination with peginterferon alpha-2a/ribavirin (pegIFN/RBV) in treatment naïve Hepatitis C virus (HCV) genotype 1 infected patients.
Ombitasvir is part of a fixed-dose formulation with ABT-450/ritonavir that is approved in the U.S. and the E.U.
Ombitasvir acts by inhibiting the HCV protein NS5A.[5]
In 2013, breakthrough therapy designation was assigned in the U.S. for the treatment of genotype 1 hepatitis C in combination with ABT-450, ritonavir and ABT-333, with and without ribavirin.
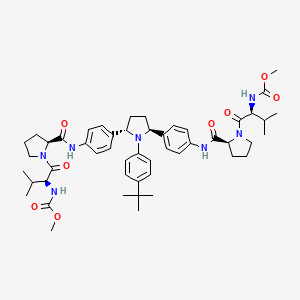


DeGoey, DA, Discovery of ABT-267, a Pan-genotypic Inhibitor of HCV NS5A, J. Med. Chem., 2014, 57 (5), pp 2047-2057
http://pubs.acs.org/doi/full/10.1021/jm401398x
http://pubs.acs.org/doi/suppl/10.1021/jm401398x/suppl_file/jm401398x_si_001.pdf

We describe here N-phenylpyrrolidine-based inhibitors of HCV NS5A with excellent potency, metabolic stability, and pharmacokinetics. Compounds with 2S,5S stereochemistry at the pyrrolidine ring provided improved genotype 1 (GT1) potency compared to the 2R,5Ranalogues. Furthermore, the attachment of substituents at the 4-position of the central N-phenyl group resulted in compounds with improved potency. Substitution with tert-butyl, as in compound 38 (ABT-267), provided compounds with low-picomolar EC50 values and superior pharmacokinetics. It was discovered that compound 38 was a pan-genotypic HCV inhibitor, with an EC50 range of 1.7–19.3 pM against GT1a, -1b, -2a, -2b, -3a, -4a, and -5a and 366 pM against GT6a. Compound 38 decreased HCV RNA up to 3.10 log10 IU/mL during 3-day monotherapy in treatment-naive HCV GT1-infected subjects and is currently in phase 3 clinical trials in combination with an NS3 protease inhibitor with ritonavir (r) (ABT-450/r) and an NS5B non-nucleoside polymerase inhibitor (ABT-333), with and without ribavirin.
Dimethyl (2S,2′S)-1,1′-((2S,2′S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-Butylphenyl)pyrrolidine-2,5-diyl)bis(4,1-phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-diyl)dicarbamate (38)…desired
and
Dimethyl (2S,2′S)-1,1′-((2S,2′S)-2,2′-(4,4′-((2R,5R)-1-(4-tert-Butylphenyl)pyrrolidine-2,5-diyl)bis(4,1-phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-diyl)dicarbamate (39)…….undesired
…………….. The resulting mixture was stirred at room temperature for 16 h. The mixture was partitioned between ethyl acetate and water, and the organic layer was washed with saturated aqueous NaHCO3, brine (2×) and dried with Na2SO4. The drying agent was filtered off and the solution was concentrated in vacuo to give a crude product that was purified by column chromatography on silica gel, eluting with a solvent gradient of 2–8% methanol in dichloromethane to give a 1:1 mixture of trans-pyrrolidine isomers (290 mg, 96%). The mixture was separated on a Chiralpak AD-H column, eluting with a mixture of 1 part (2:1 isopropanol/ethanol) and 2 parts hexanes (0.1% TFA).
Compound 38 was the first of two stereoisomers to elute (101 mg, 99% ee by chiral HPLC). 1H NMR (400 MHz, DMSO-d6) δ 0.88 (d, J = 6.61 Hz, 6H), 0.93 (d, J = 6.72 Hz, 6H), 1.11 (s, 9H), 1.63 (d, J = 5.42 Hz, 2H), 1.80–2.04 (m, 8H), 2.09–2.19 (m, 2H), 2.44–2.47 (m, 2H), 3.52 (s, 6H), 3.59–3.66 (m, 2H), 3.77–3.84 (m, 2H), 4.02 (t, J = 8.40 Hz, 2H), 4.42 (dd, J = 7.86, 4.83 Hz, 2H), 5.14 (d, J = 6.18 Hz, 2H), 6.17 (d, J = 8.67 Hz, 2H), 6.94 (d, J = 8.78 Hz, 2H), 7.13 (d, J = 8.46 Hz, 4H), 7.31 (d, J= 8.35 Hz, 2H), 7.50 (d, J = 8.35 Hz, 4H), 9.98 (s, 2H).
MS (ESI) m/z 894.9 (M + H)+.
Compound39 was the second of two stereoisomers to elute. 1H NMR (400 MHz, DMSO-d6) δ 0.87 (d, J = 6.51 Hz, 6H), 0.92 (d, J = 6.72 Hz, 6H), 1.11 (s, 9H), 1.63 (d, J = 5.53 Hz, 2H), 1.82–2.04 (m, 8H), 2.09–2.18 (m, 2H), 2.41–2.47 (m, 2H), 3.52 (s, 6H), 3.58–3.67 (m, 2H), 3.75–3.84 (m, 2H), 4.02 (t, J = 7.26 Hz, 2H), 4.43 (dd, J = 7.92, 4.88 Hz, 2H), 5.14 (d, J = 6.18 Hz, 2H), 6.17 (d, J = 8.78 Hz, 2H), 6.94 (d, J = 8.67 Hz, 2H), 7.12 (d, J = 8.46 Hz, 4H), 7.31 (d, J = 8.35 Hz, 2H), 7.49 (d, J = 8.46 Hz, 4H), 9.98 (s, 2H). MS (ESI) m/z 895.0 (M + H)+.
………..
PATENT
WO 2011156578
dimethyl (2S,2,S)-l,l ‘-((2S,2’S)-2,2′-(4,4’-((2S,5S)-l-(4-fert-butylphenyl)pyrrolidine- 2,5-diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3- methyl- l-oxobutane-2,l-diyl)dicarbamate

hereinafter Compound IA),..http://www.google.com/patents/WO2011156578A1?cl=en
……………………………..
PATENT
US 20100317568
https://www.google.co.in/patents/US20100317568
Example 34
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate and
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-ter/’-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate

Example 34A l-(4-fer?-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine The product from Example 1C (3.67 g, 7.51 mmol) and 4-tert-butylaniline (11.86 ml, 75 mmol) in DMF (40 ml) was stirred under nitrogen at 50 °C for 4 h. The resulting mixture was diluted into ethyl acetate, treated with IM HCl, stirred for 10 minutes and filtered to remove solids. The filtrate organic layer was washed twice with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (5% to 30%) to give a solid. The solid was triturated in a minimal volume of 1 :9 ethyl acetate/hexane to give a light yellow solid as a mixture of trans and cis isomers (1.21 g, 36%).
Example 34B 4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline and 4,4′-((2R,5R)-1-(4-fert- butylphenyl)pyrrolidine-2,5-diyl)dianiline To a solution of the product from Example 34A (1.1 g, 2.47 mmol) in ethanol (20 ml) and
THF (20 ml) was added PtC>2 (0.22 g, 0.97 mmol) in a 50 ml pressure bottle and stirred under 30 psi hydrogen at room temperature for 1 h. The mixture was filtered through a nylon membrane and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (20% to 60%). The title compound eluted as the first of 2 stereoisomers (trans isomer, 0.51 g, 54%).
Example 34C
(2S,2’S)-tert-Butyl 2,2′-(4,4′-((2S,5S)-1-(4-fer/’-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine- 1 -carboxylate and (2S,2’S)-tert-Butyl 2,2′- (4,4′-((2R,5R)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine-1-carboxylate To a mixture of the product from Example 34B (250 mg, 0.648 mmol), (S)-1-(tert- butoxycarbonyl)pyrrolidine-2-carboxylic acid (307 mg, 1.427 mmol) and HATU (542 mg, 1.427 mmol) in DMSO (10 ml) was added Hunig’s base (0.453 ml, 2.59 mmol). The reaction mixture was stirred at room temperature for 1 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (10% to 50%) to give the title compound (500 mg, 99%).
Example 34D
(2S,2’S)-N,N’-(4,4′-((2S,5S)-1-(4-ter/’-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))dipyrrolidine-2-carboxamide and (2S,2’S)-N,N’-(4,4′-((2R,5R)-1-(4-tert- butylphenyl)pyrrolidine-2,5-diyl)bis(4,l-phenylene))dipyrrolidine-2-carboxamide To the product from Example 34C (498 mg, 0.638 mmol) in dichloromethane (4 ml) was added TFA (6 ml). The reaction mixture was stirred at room temperature for 1 h and concentrated in vacuo. The residue was partitioned between 3: 1 CHCl3dsopropyl alcohol and saturated aq. NaHCO3. The aqueous layer was extracted by 3: 1 CHCl3:isopropyl alcohol again. The combined organic layers were dried over
filtered and concentrated to give the title compound (345 mg, 93%).
Example 34E Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate and
Dimethyl (2S,2’S)-1, r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate
The product from Example 34D (29.0 mg, 0.050 mmol), (S)-2-(methoxycarbonylamino)-3- methylbutanoic acid (19.27 mg, 0.110 mmol), EDAC (21.09 mg, 0.110 mmol), HOBT (16.85 mg,
0.110 mmol) and N-methylmorpholine (0.027 ml, 0.250 mmol) were combined in DMF (2 ml). The reaction mixture was stirred at room temperature for 3 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine twice, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (50% to 80%) to give a solid. The solid was triturated with ethyl acetate/hexane to give the title compound (13 mg, 29%). 1H NMR (400 MHz, DMSO-D6) δ ppm 0.85 – 0.95 (m, 12 H) 1.11 (s, 9 H) 1.59 – 1.65 (m, 2 H) 1.79 – 2.04 (m, 8 H) 2.10 – 2.18 (m, 2 H) 2.41-2.46 (m, 2H) 3.52 (s, 6 H)
3.57 – 3.67 (m, 2 H) 3.76 – 3.86 (m, 2 H) 4.00 (t, J=7.56 Hz, 2 H) 4.39 – 4.46 (m, 2 H) 5.15 (d, J=7.00
Hz, 2 H) 6.17 (d, J=7.70 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=7.37 Hz, 4 H) 7.30 (d, J=8.20
Hz, 2 H) 7.50 (d, J=8.24 Hz, 4 H) 9.98 (s, 2 H); (ESI+) m/z 895 (M+H)+. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 35
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate
 ………………desired
………………desiredThe product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the first of the 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 – 2.04 (m, 8 H) 2.09 – 2.19 (m, 2 H) 2.44 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 – 3.66 (m, 2 H) 3.77 – 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV Ib- Conl replicon assays in the presence of 5% FBS.
Example 36 Dimethyl (2S,2’S)-1, r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate
 …….undesired
…….undesiredThe product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the second of 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.87
(d, J=6.51 Hz, 6 H) 0.92 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.53 Hz, 2 H) 1.82 – 2.04 (m, 8
H) 2.09-2.18 (m, 2 H) 2.41 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.58 – 3.67 (m, 2 H) 3.75 – 3.84 (m, 2 H) 4.02
(t, J=7.26 Hz, 2 H) 4.43 (dd, J=7.92, 4.88 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.78 Hz, 2 H) 6.94 (d, J=8.67 Hz, 2 H) 7.12 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.49 (d, J=8.46 Hz, 4 H)
9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 37 Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate
 ……………desired
……………desiredExample 37A (S)-2,5-dioxopyrrolidin-1-yl 2-(methoxycarbonylamino)-3-methylbutanoate To a mixture of (S)-2-(methoxycarbonylamino)-3-methylbutanoic acid (19.66 g, 112 mmol) and N-hydroxysuccinimide (13.29g, 116 mmol) was added ethyl acetate (250 ml), and the mixture was cooled to 0-5 °C. Diisopropylcarbodiimide (13.88 g, 110 mmol) was added and the reaction mixture was stirred at 0-5 °C for about 1 hour. The reaction mixture was warmed to room temperature. The solids (diisopropylurea by-product) were filtered and rinsed with ethyl acetate. The filtrate was concentrated in vacuo to an oil. Isopropyl alcohol (200 ml) was added to the oil and the mixture was heated to about 50 °C to obtain a homogeneous solution. Upon cooling, crystalline solids formed. The solids were filtered and washed with isopropyl alcohol (3 x 20 ml) and dried to give the title compound as a white solid (23.2 g, 77% yield).
Example 37B
(S)- 1 -((S)-2-(methoxycarbonylamino)-3-methylbutanoyl)pyrrolidine-2-carboxylic acid To a mixture of L-proline (4.44g, 38.6 mmol), water (20 ml), acetonitrile (20 ml) and DIEA (9.5 g, 73.5 mmol) was added a solution of the product from Example 37A (1Og, 36.7 mmol) in acetonitrile (20 inL) over 10 minutes. The reaction mixture was stirred overnight at room temperature. The solution was concentrated under vacuum to remove the acetonitrile. To the resulting clear water solution was added 6N HCl (9 ml) until pH ~ 2 .The solution was transferred to a separatory funnel and 25% NaCl (10 ml) was added and the mixture was extracted with ethyl acetate (75 ml), and then again with ethyl acetate (6 x 20 ml), and the combined extracts were washed with 25% NaCl (2 x 10ml). The solvent was evaporated to give a thick oil. Heptane was added and the solvent was evaporated to give a foam, which was dried under high vacuum. Diethyl ether was added and the solvent was evaporated to give a foam, which was dried under high vacuum to give the title compound (10.67g) as a white solid.
The compound of Example 37B can also be prepreared according to the following procedure: To a flask was charged L- valine (35 g, 299 mmol), IN sodium hydroxide solution (526 ml,
526 mmol) and sodium carbonate (17.42 g, 164 mmol). The mixture was stirred for 15 min to dissolve solids and then cooled to 15 °C. Methyl chloroformate (29.6 g, 314 mmol) was added slowly to the reaction mixture. The mixture was then stirred at rt for 30 min. The mixture was cooled to 15 °C and pH adjusted to -5.0 with concentrated HCl solution. 100 inL of 2-methytetrahydrofuran (2- MeTHF) was added and the adjustment of pH continued until the pH reached ~ 2.0. 150 mL of 2- MeTHF was added and the mixture was stirred for 15 min. Layers were separated and the aqueous layer extracted with 100 mL of 2-MeTHF. The combined organic layer was dried over anhyd Na2SC^ and filtered, and Na2SC^ cake was washed with 50 mL of 2-MeTHF. The product solution was concentrated to ~ 100 mL, chased with 120 mL of IPAc twice. 250 mL of heptanes was charged slowly and then the volume of the mixture was concentrated to 300 mL. The mixture was heated to 45 °C and 160 mL of heptanes charged. The mixture was cooled to rt in 2h, stirred for 30 min, filtered and washed with 2-MeTHF/heptanes mixture (1:7, 80 inL). The wetcake was dried at 55 °C for 24 h to give 47.1 g of Moc-L- VaI-OH product as a white solid (90%).
Moc-L- VaI-OH (15O g, 856 mmol), HOBt hydrate (138 g, 899 mmol) and DMF (1500 ml) were charged to a flask. The mixture was stirred for 15 min to give a clear solution. EDC hydrochloride (172 g, 899 mmol) was charged and mixed for 20 min. The mixture was cooled to 13
°C and (L)-proline benzyl ester hydrochloride (207 g, 856 mmol) charged. Triethylamine (109 g,
1079 mmol) was then charged in 30 min. The resulting suspension was mixed at rt for 1.5 h. The reaction mixture was cooled to 15 °C and 1500 mL of 6.7% NaHCO3 charged in 1.5 h, followed by the addition of 1200 mL of water over 60 min. The mixture was stirred at rt for 30 min, filtered and washed with water/DMF mixture (1 :2, 250 mL) and then with water (1500 mL). The wetcake was dried at 55 °C for 24 h to give 282 g of product as a white solid (90%).
The resulting solids (40 g) and 5% Pd/ Alumina were charged to a Parr reactor followed by THF (160 mL). The reactor was sealed and purged with nitrogen (6 x 20 psig) followed by a hydrogen purge (6 x 30 psig). The reactor was pressurized to 30 psig with hydrogen and agitated at room temperature for approximately 15 hours. The resulting slurry was filtered through a GF/F filter and concentrated to approximately 135 g solution. Heptane was added (120 mL), and the solution was stirred until solids formed. After an addition 2 – 3 hours additional heptane was added drop-wise (240 mL), the slurry was stirred for approximately 1 hour, then filtered. The solids were dried to afford the title compound.
Example 37C
(lR,4R)-1,4-bis(4-nitrophenyl)butane-1,4-diyl dimethanesulfonate
The product from Example 32 (5.01 g, 13.39 mmol) was combined with 2- methyltetrahydrofuran (70 mL) and cooled to -5 °C, and N,N-diisopropylethylamine (6.81 g, 52.7 mmol) was added over 30 seconds. Separately, a solution of methanesulfonic anhydride (6.01 g, 34.5 mmol) in 2-methyltetrahydrofuran (30 mL) was prepared and added to the diol slurry over 3 min., maintaining the internal temperature between -15 °C and -25 °C. After mixing for 5 min at -15 °C, the cooling bath was removed and the reaction was allowed to warm slowly to 23 °C and mixed for 30 minutes. After reaction completion, the crude slurry was carried immediately into the next step.
Example 37D
(2S,5S)-1-(4-tert-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine
To the crude product solution from Example 37C (7.35 g, 13.39 mmol) was added 4-tert- butylaniline (13.4 g, 90 mmol) at 23 °C over 1 minute. The reaction was heated to 65 °C for 2 h. After completion, the reaction mixture was cooled to 23 °C and diluted with 2-methyltetrahydrofuran (100 mL) and 1 M HCl (150 mL). After partitioning the phases, the organic phase was treated with 1 M HCl (140 mL), 2-methyltetrahydrofuran (50 mL), and 25 wt% aq. NaCl (100 mL), and the phases were partitioned. The organic phase was washed with 25 wt% aq. NaCl (50 mL), dried over MgSO/t, filtered, and concentrated in vacuo to approximately 20 mL. Heptane (30 mL) and additional 2- methyltetrahydrofuran were added in order to induce crystallization. The slurry was concentrated further, and additional heptane (40 mL) was slowly added and the slurry was filtered, washing with 2- methyltetrahydrofuran:heptane (1:4, 20 mL). The solids were suspended in MeOH (46 mL) for 3 h, filtered, and the wet solid was washed with additional MeOH (18 mL). The solid was dried at 45 °C in a vacuum oven for 16 h to provide the title compound (3.08 g, 51% 2-step yield).
Example 37E
4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline
To a 160 ml Parr stirrer hydrogenation vessel was added the product from Example 37D (2 g, 4.49 mmol), followed by 60 ml of THF, and Raney Nickel Grace 2800 (1 g, 50 wt% (dry basis)) under a stream of nitrogen. The reactor was assembled and purged with nitrogen (8 x 20 psig) followed by purging with hydrogen (8 x 30 psig). The reactor was then pressurized to 30 psig with hydrogen and agitation (700 rpm) began and continued for a total of 16 h at room temperature. The slurry was filtered by vacuum filtration using a GF/F Whatman glass fiber filter. Evaporation of the filtrate to afford a slurry followed by the addition heptane and filtration gave the crude title compound, which was dried and used directly in the next step.
Example 37F dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4, l- phenylene)bis(azanediyl)bis(oxomethylene))bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diy 1) die arb amate To a solution of the product from Example 37E (1.64 g, 4.25 mmol) in DMF (20 ml), the product from Example 37B (2.89 g, 10.63 mmol), and HATU (4.04 g, 10.63 mmol) in DMF (15OmL) was added triethylamine (1.07 g, 10.63 mmol), and the solution was stirred at room temperature for 90 min. To the reaction mixture was poured 20 mL of water, and the white precipitate obtained was filtered, and the solid was washed with water (3×5 mL). The solid was blow dried for Ih. The crude material was loaded on a silica gel column and eluted with a gradient starting with ethyl acetate/ heptane (3/7), and ending with pure ethyl acetate. The desired fractions were combined and solvent distilled off to give a very light yellow solid, which was dried at 45 °C in a vacuum oven with nitrogen purge for 15 h to give the title compound (2.3 g, 61% yield). 1H NMR (400 MHz, DMSO- D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 – 2.04 (m, 8 H) 2.09 – 2.19 (m, 2 H) 2.44 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 – 3.66 (m, 2 H) 3.77 – 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H).
Alternately, the product from example 37E (11.7 g, 85 wt%, 25.8 mmol) and the product from example 37B (15.45 g, 56.7 mmol) are suspended in EtOAc (117 mL), diisopropylethylamine (18.67 g, 144 mmol) is added and the solution is cooled to 0 °C. In a separate flask, 1-propanephosphonic acid cyclic anhydride (T3P®) (46.0 g, 50 wt% in EtOAc, 72.2 mmol) was dissolved in EtOAc (58.5 mL), and charged to an addition funnel. The T3P solution is added to the reaction mixture drop-wise over 3-4 h and stirred until the reaction is complete. The reaction is warmed to room temperature,and washed with IM HCl/7.5 wt% NaCl (100 mL), then washed with 5% NaHCO3 (100 mL), then washed with 5% NaCl solution (100 mL). The solution was concentrated to approximately 60 mL, EtOH (300 mL) was added, and the solution was concentrated to 84 g solution.
A portion of the EtOH solution of product (29 g) was heated to 40 °C, and added 134 g 40 w% EtOH in H2O. A slurry of seeds in 58 wt/wt% EtOH/H2O was added, allowed to stir at 40 °C for several hours, then cooled to 0 °C. The slurry is then filtered, and washed with 58wt/wt% EtOH/H2O. The product is dried at 40 – 60 °C under vacuum, and then rehydrated by placing a tray of water in the vacuum oven to give the title compound. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
……………..
PATENT
http://www.google.com/patents/EP2337781A2?cl=en
Example 34
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate and
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-ter/’-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate

Example 34A l-(4-fer?-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine The product from Example 1C (3.67 g, 7.51 mmol) and 4-tert-butylaniline (11.86 ml, 75 mmol) in DMF (40 ml) was stirred under nitrogen at 50 °C for 4 h. The resulting mixture was diluted into ethyl acetate, treated with IM HCl, stirred for 10 minutes and filtered to remove solids. The filtrate organic layer was washed twice with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (5% to 30%) to give a solid. The solid was triturated in a minimal volume of 1 :9 ethyl acetate/hexane to give a light yellow solid as a mixture of trans and cis isomers (1.21 g, 36%).
Example 34B 4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline and 4,4′-((2R,5R)-1-(4-fert- butylphenyl)pyrrolidine-2,5-diyl)dianiline To a solution of the product from Example 34A (1.1 g, 2.47 mmol) in ethanol (20 ml) and
THF (20 ml) was added PtC>2 (0.22 g, 0.97 mmol) in a 50 ml pressure bottle and stirred under 30 psi hydrogen at room temperature for 1 h. The mixture was filtered through a nylon membrane and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (20% to 60%). The title compound eluted as the first of 2 stereoisomers (trans isomer, 0.51 g, 54%).
Example 34C
(2S,2’S)-tert-Butyl 2,2′-(4,4′-((2S,5S)-1-(4-fer/’-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine- 1 -carboxylate and (2S,2’S)-tert-Butyl 2,2′- (4,4′-((2R,5R)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine-1-carboxylate To a mixture of the product from Example 34B (250 mg, 0.648 mmol), (S)-1-(tert- butoxycarbonyl)pyrrolidine-2-carboxylic acid (307 mg, 1.427 mmol) and HATU (542 mg, 1.427 mmol) in DMSO (10 ml) was added Hunig’s base (0.453 ml, 2.59 mmol). The reaction mixture was stirred at room temperature for 1 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (10% to 50%) to give the title compound (500 mg, 99%).
Example 34D
(2S,2’S)-N,N’-(4,4′-((2S,5S)-1-(4-ter/’-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))dipyrrolidine-2-carboxamide and (2S,2’S)-N,N’-(4,4′-((2R,5R)-1-(4-tert- butylphenyl)pyrrolidine-2,5-diyl)bis(4,l-phenylene))dipyrrolidine-2-carboxamide To the product from Example 34C (498 mg, 0.638 mmol) in dichloromethane (4 ml) was added TFA (6 ml). The reaction mixture was stirred at room temperature for 1 h and concentrated in vacuo. The residue was partitioned between 3: 1 CHCl3dsopropyl alcohol and saturated aq. NaHCO3. The aqueous layer was extracted by 3: 1 CHCl3:isopropyl alcohol again. The combined organic layers were dried over
filtered and concentrated to give the title compound (345 mg, 93%).
Example 34E Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate and
Dimethyl (2S,2’S)-1, r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate
The product from Example 34D (29.0 mg, 0.050 mmol), (S)-2-(methoxycarbonylamino)-3- methylbutanoic acid (19.27 mg, 0.110 mmol), EDAC (21.09 mg, 0.110 mmol), HOBT (16.85 mg,
0.110 mmol) and N-methylmorpholine (0.027 ml, 0.250 mmol) were combined in DMF (2 ml). The reaction mixture was stirred at room temperature for 3 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine twice, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (50% to 80%) to give a solid. The solid was triturated with ethyl acetate/hexane to give the title compound (13 mg, 29%). 1H NMR (400 MHz, DMSO-D6) δ ppm 0.85 – 0.95 (m, 12 H) 1.11 (s, 9 H) 1.59 – 1.65 (m, 2 H) 1.79 – 2.04 (m, 8 H) 2.10 – 2.18 (m, 2 H) 2.41-2.46 (m, 2H) 3.52 (s, 6 H)
3.57 – 3.67 (m, 2 H) 3.76 – 3.86 (m, 2 H) 4.00 (t, J=7.56 Hz, 2 H) 4.39 – 4.46 (m, 2 H) 5.15 (d, J=7.00
Hz, 2 H) 6.17 (d, J=7.70 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=7.37 Hz, 4 H) 7.30 (d, J=8.20
Hz, 2 H) 7.50 (d, J=8.24 Hz, 4 H) 9.98 (s, 2 H); (ESI+) m/z 895 (M+H)+. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 35
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate
 ………….desired
………….desiredThe product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the first of the 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 – 2.04 (m, 8 H) 2.09 – 2.19 (m, 2 H) 2.44 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 – 3.66 (m, 2 H) 3.77 – 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV Ib- Conl replicon assays in the presence of 5% FBS.
Example 36 Dimethyl (2S,2’S)-1, r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate
 ……….undesired
……….undesiredThe product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the second of 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.87
(d, J=6.51 Hz, 6 H) 0.92 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.53 Hz, 2 H) 1.82 – 2.04 (m, 8
H) 2.09-2.18 (m, 2 H) 2.41 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.58 – 3.67 (m, 2 H) 3.75 – 3.84 (m, 2 H) 4.02
(t, J=7.26 Hz, 2 H) 4.43 (dd, J=7.92, 4.88 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.78 Hz, 2 H) 6.94 (d, J=8.67 Hz, 2 H) 7.12 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.49 (d, J=8.46 Hz, 4 H)
9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 37 Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate
 ………………desired
………………desiredExample 37A (S)-2,5-dioxopyrrolidin-1-yl 2-(methoxycarbonylamino)-3-methylbutanoate To a mixture of (S)-2-(methoxycarbonylamino)-3-methylbutanoic acid (19.66 g, 112 mmol) and N-hydroxysuccinimide (13.29g, 116 mmol) was added ethyl acetate (250 ml), and the mixture was cooled to 0-5 °C. Diisopropylcarbodiimide (13.88 g, 110 mmol) was added and the reaction mixture was stirred at 0-5 °C for about 1 hour. The reaction mixture was warmed to room temperature. The solids (diisopropylurea by-product) were filtered and rinsed with ethyl acetate. The filtrate was concentrated in vacuo to an oil. Isopropyl alcohol (200 ml) was added to the oil and the mixture was heated to about 50 °C to obtain a homogeneous solution. Upon cooling, crystalline solids formed. The solids were filtered and washed with isopropyl alcohol (3 x 20 ml) and dried to give the title compound as a white solid (23.2 g, 77% yield).
Example 37B
(S)- 1 -((S)-2-(methoxycarbonylamino)-3-methylbutanoyl)pyrrolidine-2-carboxylic acid To a mixture of L-proline (4.44g, 38.6 mmol), water (20 ml), acetonitrile (20 ml) and DIEA (9.5 g, 73.5 mmol) was added a solution of the product from Example 37A (1Og, 36.7 mmol) in acetonitrile (20 inL) over 10 minutes. The reaction mixture was stirred overnight at room temperature. The solution was concentrated under vacuum to remove the acetonitrile. To the resulting clear water solution was added 6N HCl (9 ml) until pH ~ 2 .The solution was transferred to a separatory funnel and 25% NaCl (10 ml) was added and the mixture was extracted with ethyl acetate (75 ml), and then again with ethyl acetate (6 x 20 ml), and the combined extracts were washed with 25% NaCl (2 x 10ml). The solvent was evaporated to give a thick oil. Heptane was added and the solvent was evaporated to give a foam, which was dried under high vacuum. Diethyl ether was added and the solvent was evaporated to give a foam, which was dried under high vacuum to give the title compound (10.67g) as a white solid.
The compound of Example 37B can also be prepreared according to the following procedure: To a flask was charged L- valine (35 g, 299 mmol), IN sodium hydroxide solution (526 ml,
526 mmol) and sodium carbonate (17.42 g, 164 mmol). The mixture was stirred for 15 min to dissolve solids and then cooled to 15 °C. Methyl chloroformate (29.6 g, 314 mmol) was added slowly to the reaction mixture. The mixture was then stirred at rt for 30 min. The mixture was cooled to 15 °C and pH adjusted to -5.0 with concentrated HCl solution. 100 inL of 2-methytetrahydrofuran (2- MeTHF) was added and the adjustment of pH continued until the pH reached ~ 2.0. 150 mL of 2- MeTHF was added and the mixture was stirred for 15 min. Layers were separated and the aqueous layer extracted with 100 mL of 2-MeTHF. The combined organic layer was dried over anhyd Na2SC^ and filtered, and Na2SC^ cake was washed with 50 mL of 2-MeTHF. The product solution was concentrated to ~ 100 mL, chased with 120 mL of IPAc twice. 250 mL of heptanes was charged slowly and then the volume of the mixture was concentrated to 300 mL. The mixture was heated to 45 °C and 160 mL of heptanes charged. The mixture was cooled to rt in 2h, stirred for 30 min, filtered and washed with 2-MeTHF/heptanes mixture (1:7, 80 inL). The wetcake was dried at 55 °C for 24 h to give 47.1 g of Moc-L- VaI-OH product as a white solid (90%).
Moc-L- VaI-OH (15O g, 856 mmol), HOBt hydrate (138 g, 899 mmol) and DMF (1500 ml) were charged to a flask. The mixture was stirred for 15 min to give a clear solution. EDC hydrochloride (172 g, 899 mmol) was charged and mixed for 20 min. The mixture was cooled to 13
°C and (L)-proline benzyl ester hydrochloride (207 g, 856 mmol) charged. Triethylamine (109 g,
1079 mmol) was then charged in 30 min. The resulting suspension was mixed at rt for 1.5 h. The reaction mixture was cooled to 15 °C and 1500 mL of 6.7% NaHCO3 charged in 1.5 h, followed by the addition of 1200 mL of water over 60 min. The mixture was stirred at rt for 30 min, filtered and washed with water/DMF mixture (1 :2, 250 mL) and then with water (1500 mL). The wetcake was dried at 55 °C for 24 h to give 282 g of product as a white solid (90%).
The resulting solids (40 g) and 5% Pd/ Alumina were charged to a Parr reactor followed by THF (160 mL). The reactor was sealed and purged with nitrogen (6 x 20 psig) followed by a hydrogen purge (6 x 30 psig). The reactor was pressurized to 30 psig with hydrogen and agitated at room temperature for approximately 15 hours. The resulting slurry was filtered through a GF/F filter and concentrated to approximately 135 g solution. Heptane was added (120 mL), and the solution was stirred until solids formed. After an addition 2 – 3 hours additional heptane was added drop-wise (240 mL), the slurry was stirred for approximately 1 hour, then filtered. The solids were dried to afford the title compound.
Example 37C
(lR,4R)-1,4-bis(4-nitrophenyl)butane-1,4-diyl dimethanesulfonate
The product from Example 32 (5.01 g, 13.39 mmol) was combined with 2- methyltetrahydrofuran (70 mL) and cooled to -5 °C, and N,N-diisopropylethylamine (6.81 g, 52.7 mmol) was added over 30 seconds. Separately, a solution of methanesulfonic anhydride (6.01 g, 34.5 mmol) in 2-methyltetrahydrofuran (30 mL) was prepared and added to the diol slurry over 3 min., maintaining the internal temperature between -15 °C and -25 °C. After mixing for 5 min at -15 °C, the cooling bath was removed and the reaction was allowed to warm slowly to 23 °C and mixed for 30 minutes. After reaction completion, the crude slurry was carried immediately into the next step.
Example 37D
(2S,5S)-1-(4-tert-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine
To the crude product solution from Example 37C (7.35 g, 13.39 mmol) was added 4-tert- butylaniline (13.4 g, 90 mmol) at 23 °C over 1 minute. The reaction was heated to 65 °C for 2 h. After completion, the reaction mixture was cooled to 23 °C and diluted with 2-methyltetrahydrofuran (100 mL) and 1 M HCl (150 mL). After partitioning the phases, the organic phase was treated with 1 M HCl (140 mL), 2-methyltetrahydrofuran (50 mL), and 25 wt% aq. NaCl (100 mL), and the phases were partitioned. The organic phase was washed with 25 wt% aq. NaCl (50 mL), dried over MgSO/t, filtered, and concentrated in vacuo to approximately 20 mL. Heptane (30 mL) and additional 2- methyltetrahydrofuran were added in order to induce crystallization. The slurry was concentrated further, and additional heptane (40 mL) was slowly added and the slurry was filtered, washing with 2- methyltetrahydrofuran:heptane (1:4, 20 mL). The solids were suspended in MeOH (46 mL) for 3 h, filtered, and the wet solid was washed with additional MeOH (18 mL). The solid was dried at 45 °C in a vacuum oven for 16 h to provide the title compound (3.08 g, 51% 2-step yield).
Example 37E
4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline
To a 160 ml Parr stirrer hydrogenation vessel was added the product from Example 37D (2 g, 4.49 mmol), followed by 60 ml of THF, and Raney Nickel Grace 2800 (1 g, 50 wt% (dry basis)) under a stream of nitrogen. The reactor was assembled and purged with nitrogen (8 x 20 psig) followed by purging with hydrogen (8 x 30 psig). The reactor was then pressurized to 30 psig with hydrogen and agitation (700 rpm) began and continued for a total of 16 h at room temperature. The slurry was filtered by vacuum filtration using a GF/F Whatman glass fiber filter. Evaporation of the filtrate to afford a slurry followed by the addition heptane and filtration gave the crude title compound, which was dried and used directly in the next step.
Example 37F dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4, l- phenylene)bis(azanediyl)bis(oxomethylene))bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diy 1) die arb amate To a solution of the product from Example 37E (1.64 g, 4.25 mmol) in DMF (20 ml), the product from Example 37B (2.89 g, 10.63 mmol), and HATU (4.04 g, 10.63 mmol) in DMF (15OmL) was added triethylamine (1.07 g, 10.63 mmol), and the solution was stirred at room temperature for 90 min. To the reaction mixture was poured 20 mL of water, and the white precipitate obtained was filtered, and the solid was washed with water (3×5 mL). The solid was blow dried for Ih. The crude material was loaded on a silica gel column and eluted with a gradient starting with ethyl acetate/ heptane (3/7), and ending with pure ethyl acetate. The desired fractions were combined and solvent distilled off to give a very light yellow solid, which was dried at 45 °C in a vacuum oven with nitrogen purge for 15 h to give the title compound (2.3 g, 61% yield). 1H NMR (400 MHz, DMSO- D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 – 2.04 (m, 8 H) 2.09 – 2.19 (m, 2 H) 2.44 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 – 3.66 (m, 2 H) 3.77 – 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H).
Alternately, the product from example 37E (11.7 g, 85 wt%, 25.8 mmol) and the product from example 37B (15.45 g, 56.7 mmol) are suspended in EtOAc (117 mL), diisopropylethylamine (18.67 g, 144 mmol) is added and the solution is cooled to 0 °C. In a separate flask, 1-propanephosphonic acid cyclic anhydride (T3P®) (46.0 g, 50 wt% in EtOAc, 72.2 mmol) was dissolved in EtOAc (58.5 mL), and charged to an addition funnel. The T3P solution is added to the reaction mixture drop-wise over 3-4 h and stirred until the reaction is complete. The reaction is warmed to room temperature,and washed with IM HCl/7.5 wt% NaCl (100 mL), then washed with 5% NaHCO3 (100 mL), then washed with 5% NaCl solution (100 mL). The solution was concentrated to approximately 60 mL, EtOH (300 mL) was added, and the solution was concentrated to 84 g solution.
A portion of the EtOH solution of product (29 g) was heated to 40 °C, and added 134 g 40 w% EtOH in H2O. A slurry of seeds in 58 wt/wt% EtOH/H2O was added, allowed to stir at 40 °C for several hours, then cooled to 0 °C. The slurry is then filtered, and washed with 58wt/wt% EtOH/H2O. The product is dried at 40 – 60 °C under vacuum, and then rehydrated by placing a tray of water in the vacuum oven to give the title compound. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Intermediates
Example 32
( 1 R,4R)- 1 ,4-bis(4-mtrophenyl)butane- 1 ,4-diol
To (S)-(-)-α,α-diphenyl-2-pyrrohdinemethanol (2 71 g, 10 70 mmol) was added THF (80 mL) at 23 °C The very thin suspension was treated with t11methyl borate (1 44 g, 13 86 mmol) over 30 seconds, and the resulting solution was mixed at 23 °C for 1 h The solution was cooled to 16-19 °C, and N,N-diethylanilme borane (21 45 g, 132 mmol) was added dropwise via syringe over 3-5 mm (caution vigorous H2 evolution), while the internal temperature was maintained at 16-19 °C After 15 mm, the H2 evolution had ceased To a separate vessel was added the product from Example IA (22 04 g, 95 wt%, 63 8 mmol), followed by THF (80 mL), to form an orange slurry After cooling the slurry to 11 °C, the borane solution was transferred via cannula into the dione slurry over 3-5 min During this period, the internal temperature of the slurry rose to 16 °C After the addition was complete, the reaction was maintained at 20-27 °C for an additional 2 5 h After reaction completion, the mixture was cooled to 5 °C and methanol (16 7 g, 521 mmol) was added dropwise over 5-10 mm, maintaining an internal temperature <20 °C (note vigorous H2 evolution) After the exotherm had ceased (ca 10 mm), the temperature was adjusted to 23 °C, and the reaction was mixed until complete dissolution of the solids had occurred Ethyl acetate (300 mL) and 1 M HCl (120 mL) were added, and the phases were partitioned The organic phase was then washed successively with 1 M HCl (2 x 120 mL), H2O (65 mL), and 10% aq NaCl (65 mL) The orgamcs were dried over MgSO4, filtered, and concentrated in vacuo Crystallization of the product occurred during the concentration The slurry was warmed to 50 °C, and heptane (250 inL) was added over 15 min. The slurry was then allowed to mix at 23 °C for 30 min and filtered. The wet cake was washed with 3: 1 heptane:ethyl acetate (75 mL), and the orange, crystalline solids were dried at 45 °C for 24 h to provide the title compound (15.35 g, 99.3% ee, 61% yield), which was contaminated with 11% of the meso isomer (vs. dl isomer).
References
- “VIEKIRA PAK™ (ombitasvir, paritaprevir and ritonavir tablets; dasabuvir tablets), for Oral Use. Full Prescribing Information”(PDF). AbbVie Inc., North Chicago, IL 60064. Retrieved 30 July 2015.
- “FDA approves Viekira Pak to treat hepatitis C”. Food and Drug Administration. December 19, 2014.
- “TECHNIVIE™ (ombitasvir, paritaprevir and ritonavir) Tablets, for Oral Use. Full Prescribing Information” (PDF). AbbVie Inc., North Chicago, IL 60064. Retrieved 28 July 2015.
- “FDA approves Technivie for treatment of chronic hepatitis C genotype 4”. Food and Drug Administration. July 24, 2015.
- Jordan J. Feld, Kris V. Kowdley, Eoin Coakley, Samuel Sigal, David R. Nelson, Darrell Crawford, Ola Weiland, Humberto Aguilar, Junyuan Xiong, Tami Pilot-Matias, Barbara DaSilva-Tillmann, Lois Larsen, Thomas Podsadecki, and Barry Bernstein (2014). “Treatment of HCV with ABT-450/r–Ombitasvir and Dasabuvir with Ribavirin”. N Engl J Med 370: 1594–1603.doi:10.1056/NEJMoa1315722.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
Dimethyl ({(2S,5S)-1-[4-(2-methyl-2-propanyl)phenyl]-2,5-pyrrolidinediyl}bis{4,1-phenylenecarbamoyl(2S)-2,1-pyrrolidinediyl[(2S)-3-methyl-1-oxo-1,2-butanediyl]})biscarbamate
|
|
| Clinical data | |
| Trade names | Viekira Pak (with ombitasvir, paritaprevir, ritonavir and dasabuvir), Technivie (with ombitasvir, paritaprevir, and ritonavir) |
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | not determined |
| Protein binding | ~99.9% |
| Metabolism | amide hydrolysis followed by oxidation |
| Onset of action | ~4 to 5 hours |
| Biological half-life | 21 to 25 hours |
| Excretion | mostly with feces (90.2%) |
| Identifiers | |
| CAS Registry Number | 1258226-87-7 |
| PubChem | CID: 54767916 |
| ChemSpider | 31136214 |
| ChEBI | CHEBI:85183 |
| Synonyms | ABT-267 |
| Chemical data | |
| Formula | C50H67N7O8 |
| Molecular mass | 894.11 g/mol |
rx list
VIEKIRA PAK is ombitasvir, paritaprevir, ritonavir fixed dose combination tablets copackaged with dasabuvir tablets.
Ombitasvir, paritaprevir, ritonavir fixed dose combination tablet includes ahepatitis C virus NS5A inhibitor (ombitasvir), a hepatitis C virus NS3/4Aprotease inhibitor (paritaprevir), and a CYP3A inhibitor (ritonavir) that inhibits CYP3A mediated metabolism of paritaprevir, thereby providing increased plasma concentration of paritaprevir. Dasabuvir is a hepatitis C virus nonnucleoside NS5B palm polymerase inhibitor, which is supplied as separate tablets in the copackage. Both tablets are for oral administration.
Ombitasvir
The chemical name of ombitasvir is Dimethyl ([(2S,5S)-1-(4-tert-butylphenyl) pyrrolidine-2,5diyl]bis{benzene-4,1-diylcarbamoyl(2S)pyrrolidine-2,1-diyl[(2S)-3-methyl-1-oxobutane-1,2diyl]})biscarbamate hydrate. The molecular formula is C50H67N7O8•4.5H2O (hydrate) and the molecular weight for the drug substance is 975.20 (hydrate). The drug substance is white to light yellow to light pink powder, and is practically insoluble in aqueous buffers but is soluble in ethanol. Ombitasvir has the following molecular structure:
![]()
Paritaprevir
The chemical name of paritaprevir is (2R,6S,12Z,13aS,14aR,16aS)-N-(cyclopropylsulfonyl)-6{[(5-methylpyrazin-2-yl)carbonyl]amino}-5,16-dioxo-2-(phenanthridin-6-yloxy)1,2,3,6,7,8,9,10,11,13a,14,15,16,16a-tetradecahydrocyclopropa[e]pyrrolo[1,2-a][1,4] diazacyclopentadecine-14a(5H)-carboxamide dihydrate. The molecular formula is C40H43N7O7S•2H2O (dihydrate) and the molecular weight for the drug substance is 801.91 (dihydrate). The drug substance is white to off-white powder with very low water solubility. Paritaprevir has the following molecular structure:
 |
Ritonavir
The chemical name of ritonavir is [5S-(5R*,8R*,10R*,11R*)]10-Hydroxy-2-methyl-5-(1methyethyl)-1-[2-(1-methylethyl)-4-thiazolyl]-3,6-dioxo-8,11-bis(phenylmethyl)-2,4,7,12tetraazatridecan-13-oic acid,5-thiazolylmethyl ester. The molecular formula is C37H48N6O5S2 and the molecular weight for the drug substance is 720.95. The drug substance is white to off white to light tan powder practically insoluble in water and freely soluble in methanol and ethanol. Ritonavir has the following molecular structure:
![]()
Ombitasvir, Paritaprevir, Ritonavir Fixed-Dose Combination Tablets
Ombitasvir, paritaprevir, and ritonavir film-coated tablets are co-formulated immediate release tablets. The tablet contains copovidone, K value 28,vitamin E polyethylene glycol succinate, propylene glycol monolaurate Type I, sorbitan monolaurate, colloidal silicon dioxide/colloidal anhydrous silica, sodium stearyl fumarate, polyvinyl alcohol, polyethylene glycol 3350/macrogol 3350, talc, titanium dioxide, and iron oxide red. The strength for the tablet is 12.5 mg ombitasvir, 75 mg paritaprevir, 50 mg ritonavir.
Dasabuvir
The chemical name of dasabuvir is Sodium 3-(3-tert-butyl-4-methoxy-5-{6[(methylsulfonyl)amino]naphthalene-2-yl}phenyl)-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-ide hydrate (1:1:1). The molecular formula is C26H26N3O5S•Na•H2O (salt, hydrate) and the molecular weight of the drug substance is 533.57 (salt, hydrate). The drug substance is white to pale yellow to pink powder, slightly soluble in water and very slightly soluble in methanol and isopropyl alcohol. Dasabuvir has the following molecular structure:
 |
Dasabuvir is formulated as a 250 mg film-coated, immediate release tablet containing microcrystalline cellulose (D50-100 um), microcrystalline cellulose (D50-50 um), lactose monohydrate, copovidone, croscarmellose sodium, colloidal silicon dioxide/anhydrous colloidal silica, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol 3350/macrogol 3350, talc, and iron oxide yellow, iron oxide red and iron oxide black. Each tablet contains 270.3 mg dasabuvir sodium monohydrate equivalent to 250 mg dasabuvir.
//////////fda 2014, Ombitasvir, orphan drug, Abbvie Inc.
Eliglustat

ELIGLUSTAT TARTRATE
THERAPEUTIC CLAIM Treatment of lysosomal storage disorders
CHEMICAL NAMES
1. Octanamide, N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(1-
pyrrolidinylmethyl)ethyl]-, (2R,3R)-2,3-dihydroxybutanedioate (2:1)
2. bis{N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(pyrrolidin-1-
ylmethyl)ethyl]octanamide} (2R,3R)-2,3-dihydroxybutanedioate
MOLECULAR FORMULA C23H36N2O4 . ½ C4H6O6
MOLECULAR WEIGHT 479.6
MANUFACTURER Genzyme Corp.
CODE DESIGNATION Genz-112638
CAS REGISTRY NUMBER 928659-70-5
Eliglustat (INN, USAN;[1] trade name Cerdelga) is a treatment for Gaucher’s disease developed by Genzyme Corp that was approved by the FDA August 2014.[2] Commonly used as the tartrate salt, the compound is believed to work by inhibition ofglucosylceramide synthase.[3][4]
In March 2015, eliglustat tartrate was approved in Japan for the treatment of Gaucher disease. Eliglustat tartrate was described specifically within the US FDA’s Orange Booked listed US6916802, which is set to expire in April 2022.
In May 2015, the Orange Book also listed that eliglustat tartrate had Orphan Drug Exclusivity and New Chemical Entity exclusivity until 2019 and 2021, respectively.
it having been developed and launched as eliglustat tartrate by Genzyme (a wholly owned subsidiary of Sanofi), under license from the University of Michigan.
Eliglustat tartrate is known to act as inhibitors of glucosylceramide synthase and glycolipid, useful for the treatment of Gaucher’s disease type I and lysosome storage disease.
Genzyme Announces Positive New Data from Two Phase 3 Studies for Oral Eliglustat Tartrate for Gaucher Disease
Eliglustat tartrate (USAN)
CAS:928659-70-5
February 15, 2013
Genzyme , a Sanofi company (EURONEXT: SAN and NYSE: SNY), today announced positive new data from the Phase 3 ENGAGE and ENCORE studies of eliglustat tartrate, its investigational oral therapy for Gaucher disease type 1. The results from the ENGAGE study were presented today at the 9th Annual Lysosomal Disease Network WORLD Symposium in Orlando, Fla. In conjunction with this meeting, Genzyme also released topline data from its second Phase 3 study, ENCORE. Both studies met their primary efficacy endpoints and together will form the basis of Genzyme’s registration package for eliglustat tartrateThe data presented at this year’s WORLD symposium reinforce our confidence that eliglustat tartrate may become an important oral option for patients with Gaucher disease”The company is developing eliglustat tartrate, a capsule taken orally, to provide a convenient treatment alternative for patients with Gaucher disease type 1 and to provide a broader range of treatment options for patients and physicians. Genzyme’s clinical development program for eliglustat tartrate represents the largest clinical program ever focused on Gaucher disease type 1 with approximately 400 patients treated in 30 countries.“The data presented at this year’s WORLD symposium reinforce our confidence that eliglustat tartrate may become an important oral option for patients with Gaucher disease,” said Genzyme’s Head of Rare Diseases, Rogerio Vivaldi MD. “We are excited about this therapy’s potential and are making excellent progress in our robust development plan for bringing eliglustat tartrate to the market.”ENGAGE Study Results:In ENGAGE, a Phase 3 trial to evaluate the safety and efficacy of eliglustat tartrate in 40 treatment-naïve patients with Gaucher disease type 1, improvements were observed across all primary and secondary efficacy endpoints over the 9-month study period. Results were reported today at the WORLD Symposium by Pramod Mistry, MD, PhD, FRCP, Professor of Pediatrics & Internal Medicine at Yale University School of Medicine, and an investigator in the trial.The randomized, double-blind, placebo-controlled study had a primary efficacy endpoint of improvement in spleen size in patients treated with eliglustat tartrate. Patients were stratified at baseline by spleen volume. In the study, a statistically significant improvement in spleen size was observed at nine months in patients treated with eliglustat tartrate compared with placebo. Spleen volume in patients treated with eliglustat tartrate decreased from baseline by a mean of 28 percent compared with a mean increase of two percent in placebo patients, for an absolute difference of 30 percent (p<0.0001).
Eliglustat tartate (Genz-112638)
What is Eliglustat?
- Eliglustat is a new investigational phase 3 compound from Genzyme Corporation that is being studied for type 1 Gaucher Disease.
- Eliglustat works as a substrate reduction therapy by reducing glucocerebroside. formation.
- This product is an oral agent (i.e. a pill) that is taken once or twice a day in contrast to an IV infusion for enzyme replacement therapy. Enzyme replacement therapy focuses on replenishing the enzyme that is deficient in Gaucher Disease and breaks down glucocerebroside that accumulates.
- The clinical trials for eliglustat tartate are sponsored by Genzyme Corporation.


Eliglustat tartrate (Genz-1 12638) is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of gaucher disease and other lysosomal storage disorders, which is currently under development.
Eliglustat is chemically known as 1 R, 2R-Octanoic acid [2-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1 -ylmethyl]-ethyl]-amide, having a structural formula I depicted here under.
Formula I
Eliglustat hemitartrate (Genz-1 12638) development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy.
Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase.
U.S. patent No. 7,196,205 (herein described as US’205) discloses a process for the preparation of eliglustat or a pharmaceutically acceptable salt thereof. In this patent, eliglustat was synthesized via a seven-step process involving steps in that sequence:
(i) coupling S-(+)-2-phenyl glycinol with phenyl bromoacetate followed by column chromatography for purification of the resulting intermediate,
(ii) reacting the resulting (5S)-5-phenylmorpholin-2-one with 1 , 4-benzodioxan-6-carboxaldehyde to obtain a lactone,
(iii) opening the lactone of the oxazolo-oxazinone cyclo adduct via reaction with pyrrolidine,
(iv) hydrolyzing the oxazolidine ring, (v) reducing the amide to amine to obtain sphingosine like compound, (vi) reacting the resulting amine with octanoic acid and N-hydroxysuccinimide to obtain crude eliglustat, (vii) purifying the crude eliglustat by repeated isolation for four times from a mixture of ethyl acetate and n-heptane.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 disclose processes for preparation of eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of eliglustat, (ii) a hemitartrate salt of eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
WO 2015059679
| Process for the preparation of eliglustat free base – comprising the reaction of S-(+)-phenyl glycinol with phenyl-alpha-bromoacetate to obtain 5-phenylmorpholin-2-one, which is further converted to eliglustat. | |
| Dr Reddy’s Laboratories Ltd | |
| New crystalline eliglustat free base Form R1 and a process for its preparation are claimed. Also claimed is a process for the preparation of eliglustat free base which comprises the reaction of S-(+)-phenyl glycinol with phenyl-alpha-bromoacetate to obtain 5-phenylmorpholin-2-one, which is further converted to eliglustat.Further eliglustat oxalate, its crystalline form, and a process for the preparation of crystalline eliglustat oxalate, are claimed. | |
Eliglustat tartrate (Genz-1 12638) is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of gaucher disease and other lysosomal storage disorders, which is currently under development.
Eliglustat is chemically known as 1 R, 2R-Octanoic acid [2-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1 -ylmethyl]-ethyl]-amide, having a structural formula I depicted here under.
Formula I
Eliglustat hemitartrate (Genz-1 12638) development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy.
Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase.
U.S. patent No. 7,196,205 (herein described as US’205) discloses a process for the preparation of eliglustat or a pharmaceutically acceptable salt thereof. In this patent, eliglustat was synthesized via a seven-step process involving steps in that sequence:
(i) coupling S-(+)-2-phenyl glycinol with phenyl bromoacetate followed by column chromatography for purification of the resulting intermediate,
(ii) reacting the resulting (5S)-5-phenylmorpholin-2-one with 1 , 4-benzodioxan-6-carboxaldehyde to obtain a lactone,
(iii) opening the lactone of the oxazolo-oxazinone cyclo adduct via reaction with pyrrolidine,
(iv) hydrolyzing the oxazolidine ring, (v) reducing the amide to amine to obtain sphingosine like compound, (vi) reacting the resulting amine with octanoic acid and N-hydroxysuccinimide to obtain crude eliglustat, (vii) purifying the crude eliglustat by repeated isolation for four times from a mixture of ethyl acetate and n-heptane.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 disclose processes for preparation of eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of eliglustat, (ii) a hemitartrate salt of eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
Example 1 : Preparation of 5-phenyl morpholine-2-one hydrochloride
To a (S) + phenyl glycinol (100g) add N, N-diisopropylethylamine (314ml) and acetonitrile (2000ml) under nitrogen atmosphere at room temperature. It was cooled to 10- 15° C. Phenyl bromoacetate (172.4g) dissolved in acetonitrile (500ml) was added to the above solution at 15° C over a period of 30 min. The reaction mixture is allowed to room temperature and stirred for 16-20h. Progress of the reaction was monitored by thin layer chromatography. After completion of the reaction, the reaction mixture was concentrated under reduced pressure at a water bath
temperature less than 25° C to get a residue. The residue was dissolved in ethyl acetate (1000ml) and stirred for 1 h at 15-20°C to obtain a white solid. The solid material obtained was filtered and washed with ethyl acetate (200ml). The filtrate was dried over anhydrous sodium sulphate (20g) and concentrated under reduced pressure at a water bath temperature less than 25° C to give crude compound (1000g) as brown syrup. The Crude brown syrup is converted to HCI salt by using HCI in ethyl acetate to afford 5-phenyl morpholine-2-one hydrochloride (44g) as a white solid. Yield: 50%, Mass: m/z = 177.6; HPLC (% Area Method): 90.5%
Example 2: Preparation of (1 R,3S,5S,8aS)-1 ,3-Bis-(2′,3′-dihydro-benzo[1 ,4] dioxin-6′-yl)-5-phenyl-tetrahydro-oxazolo[4,3-c][1 ,4]oxazin-8-one.
5-phenyl morpholine-2-one hydrochloride (100g) obtained from above stage 1 is dissolved in toluene (2500ml) under nitrogen atmosphere at 25-30°C. 1 ,4-benzodioxane-6-carboxaldehyde (185.3g) and sodium sulphate (400g) was added to the above solution and the reaction mixture was heated at 100-105°C for 72h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was concentrated under reduced pressure at a water bath temperature less than 25° C to get a residue. The residue was cooled to 10°C, ethyl acetate (2700ml) and 50% sodium bisulphate solution (1351 ml) was added to the residue and stirred for 1 h at 10°C to obtain a white solid. The obtained white solid was filtered and washed with ethyl acetate. The separated ethyl acetate layer was washed with water (1000ml), brine (1000ml) and dried over anhydrous sodium sulphate. The organic layer was concentrated under reduced pressure at a water bath temperature of 45-50°C to get a crude material. The obtained crude material is triturated with diethyl ether (1500ml) to get a solid material which is filtered and dried under vacuum at room temperature for 2-3h to afford (1 R,3S,5S,8aS)-1 ,3-Bis-(2′,3′-dihydro-benzo[1 ,4]dioxin-6′-yl)-5-phenyl-tetrahydro-oxazolo[4,3-c][1 ,4]oxazin-8-one (148g) as a yellow solid. Yield: 54%, Mass: m/z = 487.7; HPLC (% Area Method): 95.4 %
Example 3: Preparation of (2S,3R,1 “S)-3-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)-3-hydroxy-2-(2″-hydroxy-1 ”^henyl-ethy^
(1 R,3S,5S,8aS)-1 !3-Bis-(2′!3′-dihydro-benzo[1 ,4]dioxin-6′-yl)-5-phenyl-tetrahydro-oxazolo[4,3-c][1 ,4]oxazin-8-one (70g) obtained from above stage 2 was dissolved in chloroform (1400ml) at room temperature. It was cooled to 0-5°C and pyrrolidone (59.5ml) was added at 0-5°C over a period of 30 minutes. The reaction mixture was allowed to room temperature and stirred for 16-18h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was concentrated under reduced pressure at a water bath temperature of 40-45°C to obtain a crude. The obtained crude was dissolved in methanol (1190ml) and 1 N HCI (1 190ml) at 10-15° C, stirred for 10 minutes and heated at 80-85°C for 7h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, methanol was concentrated under reduced pressure at a water bath temperature of 50-55°C.The aqueous layer was extracted with ethyl acetate and the organic layer was washed with 1 N HCI (50ml). The aqueous layer was basified with saturated sodium bicarbonate solution up to pH 8-9 and extracted with ethyl acetate (3x70ml). The combined organic layers was washed with brine (100ml), dried over anhydrous sodium sulphate and concentrated under reduced pressure at a water bath temperature of 50-55°C to afford (2S,3R,1″S)-3-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)-3-hydroxy-2-(2″-hydroxy-1 “-phenyl-ethylamino)-1 -pyrrolidin-1 -yl-propan-1 -one (53g) as a yellow foamy solid. Yield: 90%, Mass: m/z = 412.7, HPLC (% Area Method): 85.1 %
Example 4: Preparation of (1 R,2R,1 “S)-1-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)2-hydroxy-2-(2”-hydroxy-1 ‘-phenyl-ethylamino)-3-pyrrolidin-1-yl-propan-1-ol.
(2S,3R,1 “S)-3-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6’-yl)-3-hydroxy-2-(2”-hydroxy-1 “-phenyl-ethylamino)-1 -pyrrolidin-1 -yl-propan-1 -one (2.5g) obtained from above stage 3 dissolved in Tetrahydrofuran (106ml) was added to a solution of Lithium aluminium hydride (12.2g) in tetrahydrofuran (795ml) at 0°C and the reaction mixture was heated at 60-65°C for 10h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was cooled to 5- 10°C and quenched in saturated sodium sulphate solution (100ml) at 5-10°C. Ethyl acetate was added to the reaction mass and stirred for 30-45 min. The obtained solid is filtered through celite bed and washed with ethyl acetate. Filtrate was dried over anhydrous sodium sulphate and concentrated under reduced pressure at a water bath temperature of 50°C to afford (1 R,2R, 1″S)-1 -(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)2-hydroxy-2-(2″-hydroxy-1 ‘-phenyl-ethylamino)-3-pyrrolidin-1 -yl-propan-1 -ol (43.51 g) as a yellow gummy liquid. The crude is used for the next step without further purification. Yield: 85%, Mass: m/z = 398.7, HPLC (% Area Method): 77 %
Example 5: Preparation of (1 R, 2R)-2-Amino-1-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol.
(1 R,2R,1 “S)-1 -(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6’-yl)2-hydroxy-2-(2”-hydroxy-1 ‘-phenyl-ethylamino)-3-pyrrolidin-1 -yl-propan-1 -ol (40g) obtained from above stage 4 was dissolved in methanol (400ml) at room temperature in a 2L hydrogenation flask. Trifluoroacetic acid (15.5ml) and 20% Pd (OH) 2 (40g) was added to the above solution under nitrogen atmosphere. The reaction mixture was hydrogenated under H2, 10Opsi for 16-18h at room temperature. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was filtered through celite bed and washed with methanol (44ml) and water (44ml). Methanol was concentrated under reduced pressure at a water bath temperature of 50-55°C and the aqueous layer was washed with ethyl acetate. The aqueous layer was basified with 10M NaOH till the PH reaches 12-14 and then extracted with dichloromethane (2x125ml). The organic layer was dried over anhydrous sodium sulphate (3gm) and concentrated under reduced pressure at a water bath temperature of 45°C to obtain a gummy liquid. The gummy liquid was triturated with methyl tertiary butyl ether for 1 h to get a white solid, which is filtered and dried under vacuum at room temperature to afford (1 R, 2R)-2-Amino-1 -(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol (23g) as a white solid. Yield: 82.3%, Mass (m/zj: 278.8, HPLC (% Area Method): 99.5%, Chiral HPLC (% Area Method): 97.9%
Example 6: Preparation of Eliglustat {(1 R, 2R)-Octanoic acid[2-(2′,3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1-ylmethyl-ethyl]-amide}.
(1 R, 2R)-2-Amino-1 -(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol (15g) obtained from above stage 5 was dissolved in dry dichloromethane (150ml) at room temperature under nitrogen atmosphere and cooled to 10-15° C. Octanoic acid N-hydroxy succinimide ester (13.0 g)was added to the above reaction mass at 10-15° C and stirred for 15 min. The reaction mixture was stirred at room temperature for 16h-18h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was cooled to 15°C and diluted with 2M NaOH solution (100 ml_) and stirred for 20 min at 20 °C. The organic layer was separated and washed with 2M sodium hydroxide (3x90ml).The organic layer was dried over anhydrous sodium sulphate (30g) and concentrated under reduced pressure at a water bath temperature of 45°C to give the crude compound (20g).The crude is again dissolved in methyl tertiary butyl ether (25 ml_) and precipitated with Hexane (60ml). It is stirred for 10 min, filtered and dried under vacuum to afford Eliglustat as a white solid (16g). Yield: 74%, Mass (m/zj: 404.7 HPLC (% Area Method): 97.5 %, ELSD (% Area Method): 99.78%, Chiral HPLC (% Area Method): 99.78 %.
Example 7: Preparation of Eliglustat oxalate.
Eliglustat (5g) obtained from above stage 6 is dissolved in Ethyl acetate (5ml) at room temperature under nitrogen atmosphere. Oxalic acid (2.22g) dissolved in ethyl acetate (5ml) was added to the above solution at room temperature and stirred for 14h. White solid observed in the reaction mixture was filtered and dried under vacuum at room temperature for 1 h to afford Eliglustat oxalate as a white solid (4g). Yield: 65.46%, Mass (m/zj: 404.8 [M+H] +> HPLC (% Area Method): 95.52 %, Chiral HPLC (% Area Method): 99.86 %
……………………………..
Nmr predict
13 C NMR
![N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide NMR spectra analysis, Chemical CAS NO. 491833-29-5 NMR spectral analysis, N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-09-06/001/571/702/491833-29-5-13c.png)
CAS NO. 491833-29-5, N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide
C-NMR spectral analysis
………………..
http://www.google.com/patents/WO2013059119A1?cl=en
http://www.google.com/patents/US7196205
Compound 7
(1R,2R)-Nonanoic acid[2-(2′,3′-dihydro-benzo[1,4]dioxin-6′-yl)-2-hydroxy-1-pyrrolidin-1-ylmethyl-ethyl]-amide
This compound was prepared by the method described for Compound 6 using Nonanoic acid N-hydroxysuccinimide ester. Analytical HPLC showed this material to be 98.4% pure. mp 74–75° C.
1H NMR (CDCl3) δ 6.86–6.76 (m, 3H), 5.83 (d, J=7.3 Hz, 1H), 4.90 (d, J=3.3 Hz, 1H), 4.24 (s, 4H), 4.24–4.18 (m, 1H), 2.85–2.75 (m, 2H), 2.69–2.62 (m, 4H), 2.10 (t, J=7.3 Hz, 2H), 1.55–1.45 (m, 2H), 1.70–1.85 (m, 4H), 1.30–1.15 (m, 10H), 0.87 (t, J=6.9 Hz, 3H) ppm.
Intermediate 4(1R,2R)-2-Amino-1-(2′,3′-dihydro-benzo[1,4]dioxin-6′-yl)-3-pyrrolidin-1-yl-propan-1-ol
Intermediate 3 (5.3 g, 13.3 mmol) was dissolved in methanol (60 mL). Water (6 mL) and trifluoroacetic acid (2.05 m/L, 26.6 mmol, 2 equivalents) were added. After being placed under nitrogen, 20% Palladium hydroxide on carbon (Pearlman’s catalysis, Lancaster or Aldrich, 5.3 g) was added. The mixture was placed in a Parr Pressure Reactor Apparatus with glass insert. The apparatus was placed under nitrogen and then under hydrogen pressure 110–120 psi. The mixture was stirred for 2–3 days at room temperature under hydrogen pressure 100–120 psi. The reaction was placed under nitrogen and filtered through a pad of celite. The celite pad was washed with methanol (100 mL) and water (100 mL). The methanol was removed by rotoevaporation. The aqueous layer was washed with ethyl acetate three times (100, 50, 50 mL). A 10 M NaOH solution (10 mL) was added to the aqueous layer (pH=12–14). The product was extracted from the aqueous layer three times with methylene chloride (100, 100, 50 mL). The combined organic layers were dried with Na2SO4, filtered and rotoevaporated to a colorless oil. The foamy oil was vacuum dried for 2 h. Intermediate 4 was obtained in 90% yield (3.34 g).
Intermediate 3(1R,2R,1″S)-1-(2′,3′-Dihydro-benzo[1,4]dioxin-6′-yl)-2-(2″-hydroxy -1′-phenyl-ethylamino)-3-pyrrolidin-1-yl-propan-1-ol
To a 3-neck flask equipped with a dropping funnel and condenser was added LiAlH4 (Aldrich, 1.2 g, 31.7 mmol, 2.5 equivalents) and anhydrous THF (20 mL) under nitrogen. A solution of Intermediate 2 (5.23 g, 12.68 mmol) in anhydrous THF (75 mL) was added dropwise to the reaction over 15–30 minutes. The reaction was refluxed under nitrogen for 9 hours. The reaction was cooled in an ice bath and a 1M NaOH solution was carefully added dropwise. After stirring at room temperature for 15 minutes, water (50 mL) and ethyl acetate (75 mL) was added. The layers were separated and the aqueous layer was extracted twice with ethyl acetate (75 mL). The combined organic layers were washed with saturated sodium chloride solution (25 mL). After drying with Na2SO4 the solution was filtered and rotoevaporated to yield a colorless to yellow foamy oil. Intermediate 3 was obtained in 99% yield (5.3 g).

|
|
| Systematic (IUPAC) name | |
|---|---|
| N-[(1R,2R)-1-(2,3-Dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-(1-pyrrolidinyl)-2-propanyl]octanamide | |
| Clinical data | |
| Trade names | Cerdelga |
|
|
| Identifiers | |
| 491833-29-5 | |
| A16AX10 | |
| PubChem | CID 23652731 |
| ChemSpider | 28475348 |
| ChEBI | CHEBI:82752 |
| Chemical data | |
| Formula | C23H36N2O4 |
| 404.543 g/mol | |
- https://download.ama-assn.org/resources/doc/usan/x-pub/eligustat.pdf
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm410585.htm
- Lee, L.; Abe, A.; Shayman, J. A. (21 May 1999). “Improved Inhibitors of Glucosylceramide Synthase”. Journal of Biological Chemistry 274(21): 14662–14669. doi:10.1074/jbc.274.21.14662.
- Shayman, JA (Aug 1, 2010). “ELIGLUSTAT TARTRATE: Glucosylceramide Synthase Inhibitor Treatment of Type 1 Gaucher Disease.”.Drugs of the future 35 (8): 613–620. PMID 22563139.
| WO2008150486A2 * | May 30, 2008 | Dec 11, 2008 | Genzyme Corp | 2-acylaminopropoanol-type glucosylceramide synthase inhibitors |
| WO2009045503A1 * | Oct 3, 2008 | Apr 9, 2009 | Genzyme Corp | Method of treating polycystic kidney diseases with ceramide derivatives |
| WO2010014554A1 * | Jul 27, 2009 | Feb 4, 2010 | Genzyme Corporation | Glucosylceramide synthase inhibition for the treatment of collapsing glomerulopathy and other glomerular disease |
| WO2010039256A1 * | Oct 2, 2009 | Apr 8, 2010 | Genzyme Corporation | 2-acylaminopropoanol-type glucosylceramide synthase inhibitors |
………………..
SWEDEN


Alfred Nobel had the unpleasant surprise of reading his own obituary, titled The merchant of death is dead, in a French newspaper.



Nyköping (Sweden)-houses.
Fjallbacka, a colorful fishing Village along the west coast of Sweden
Knights Island, Stockholm, Sweden
 Stockholm, Sweden
Stockholm, Sweden
Sweden Stockholm
Europe Örby Änger – Sweden
 Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …
Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
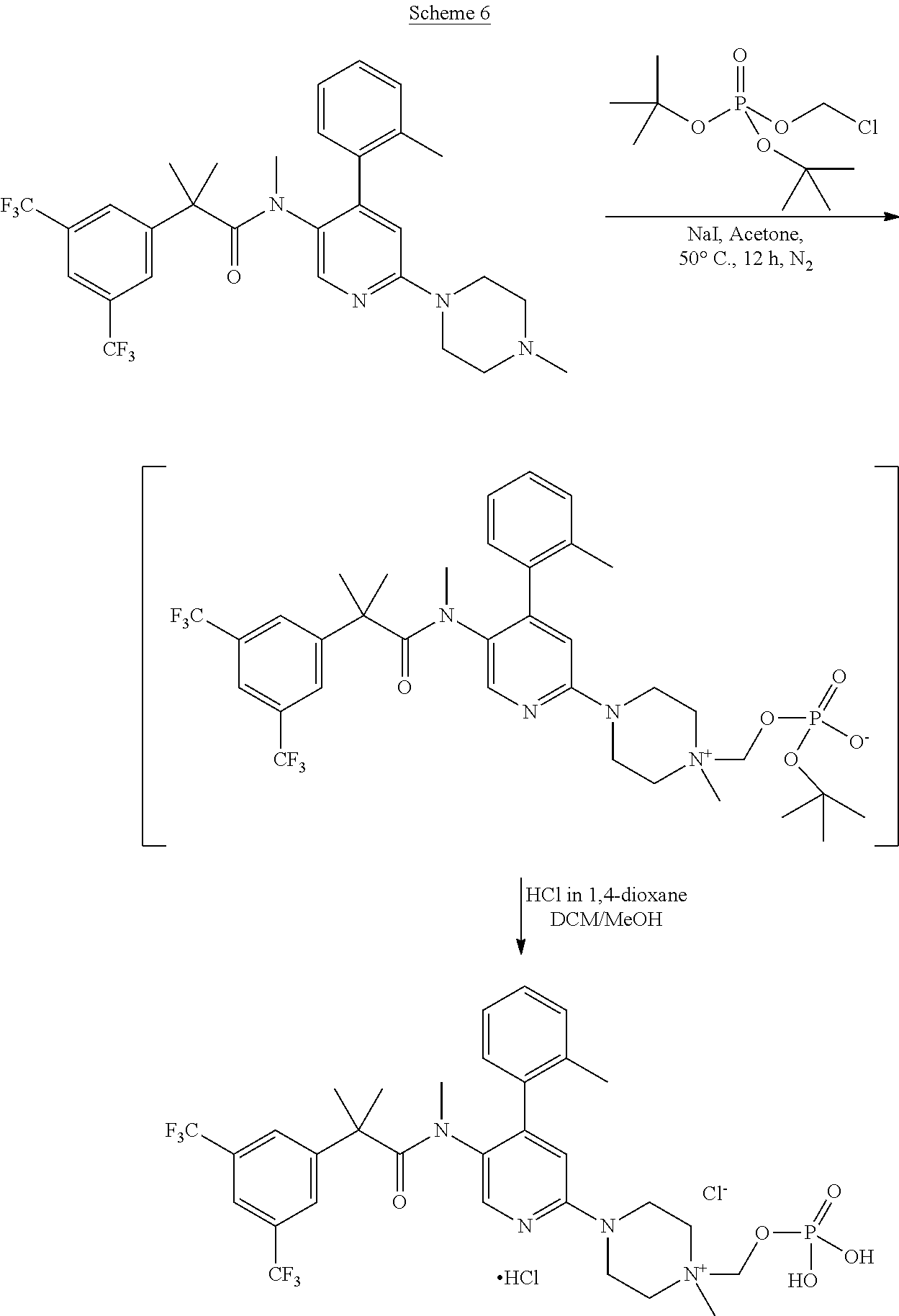{kind=link}
{kind=link}