
Home » veterinary
Category Archives: veterinary
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Milpecitinib
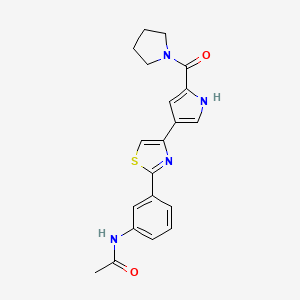
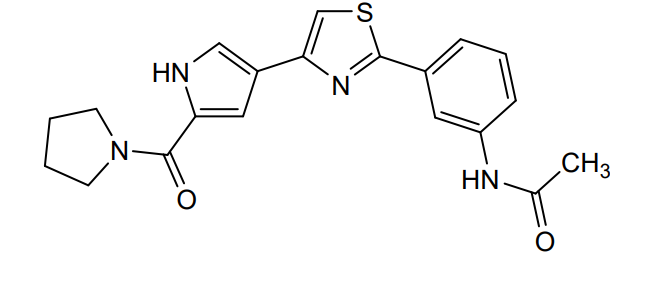
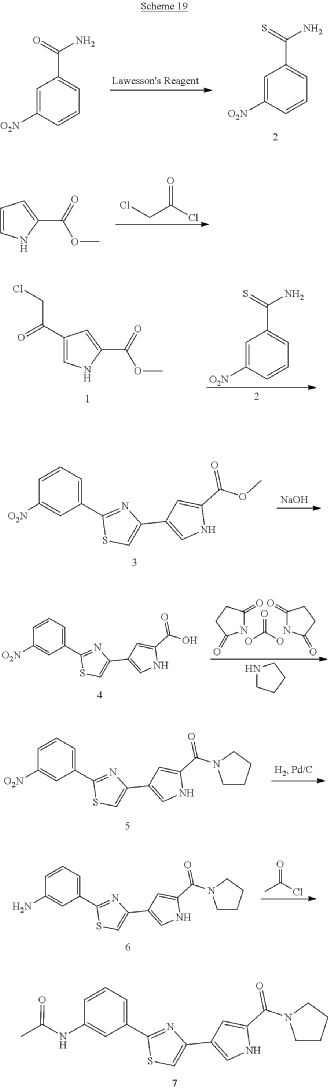
Milpecitinib
CAS 1415819-54-3
MF C20H20N4O2S MW380.5 g/mol
N-[3-[4-[5-(pyrrolidine-1-carbonyl)-1H-pyrrol-3-yl]-1,3-thiazol-2-yl]phenyl]acetamide
N-(3-{4-[5-(pyrrolidine-1-carbonyl)-1H-pyrrol-3-yl]-1,3-thiazol-2-yl}phenyl)acetamide
Janus tyrosine kinase inhibitor, anti-inflammatory, veterinary, PF-06263276, PF 06263276, Ph 1012, DNX 04013, CPh 1012, Ph-1012, CVXL 0074-02, 4Q8TT4B4GN
Milpecitinib is a small molecule drug. Milpecitinib has a monoisotopic molecular weight of 380.13 Da.
Milpecitinib (also known by its developmental codes PF-06263276, Ph-1012, and DNX-04013) is a potent, small-molecule Janus kinase (JAK) inhibitor used primarily in veterinary medicine and laboratory research. It functions as an ATP-competitive, broad-spectrum (pan-JAK) inhibitor that targets all four members of the JAK family: JAK1, JAK2, JAK3, and Tyrosine Kinase 2 (TYK2).
Primary Indication and Target
- Veterinary Use: Milpecitinib is designated for the control of pruritus (itching) associated with canine allergic dermatitis and the management of canine atopic dermatitis (CAD).
- Sponsorship: The United States Adopted Name (USAN) for this drug was officially adopted following sponsorship by Phibro Animal Health.
- Research Use: In laboratory settings, it is utilized to study complex inflammatory pathways, immune disorders, and certain cancers.
Mechanism of Action
Milpecitinib works by blocking the ATP-binding site of JAK enzymes. This inhibition halts the JAK-STAT signaling pathway, which plays a critical role in cellular responses to inflammatory cytokines. By blocking this cascade, the drug prevents the production and signaling of pro-inflammatory cytokines that cause severe itching and skin inflammation in dogs.
SYN
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
- Compositions and methods for modulating kinasesPublication Number: JP-6054379-B2Priority Date: 2011-06-07Grant Date: 2016-12-27
- Compositions and methods for modulating a kinasePublication Number: EP-2718290-B1Priority Date: 2011-06-07Grant Date: 2016-05-04
- Compositions and methods for modulating a kinasePublication Number: CA-2837268-CPriority Date: 2011-06-07Grant Date: 2020-05-12
- Compositions and Methods for Modulating a KinasePublication Number: US-2012316148-A1Priority Date: 2011-06-07
- Compositions and methods for modulating a kinasePublication Number: EP-2718290-A2Priority Date: 2011-06-07
- Compositions and methods for modulating a kinasePublication Number: WO-2012172438-A9Priority Date: 2011-06-07
- Compositions and methods for modulating a kinasePublication Number: US-8937065-B2Priority Date: 2011-06-07Grant Date: 2015-01-20
- Compositions and methods for modulating kinasesPublication Number: JP-2014520108-APriority Date: 2011-06-07
- Compositions and methods for modulating a kinasePublication Number: WO-2012172438-A2Priority Date: 2011-06-07
- Compositions and methods to modulate a kinasePublication Number: ES-2585244-T3Priority Date: 2011-06-07Grant Date: 2016-10-04
- Compound for modulating a kinase
- Publication Number: BR-112013031121-B1
- Priority Date: 2011-06-07
//////////milpecitinib, anax labs, Janus tyrosine kinase inhibitor, anti-inflammatory, veterinary, PF-06263276, PF 06263276, Ph 1012, DNX 04013, CPh 1012, Ph-1012, CVXL 0074-02, 4Q8TT4B4GN
Teprosulvose
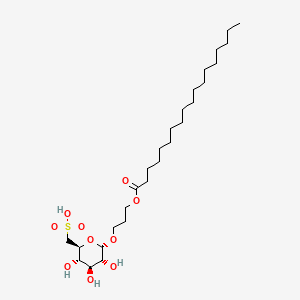
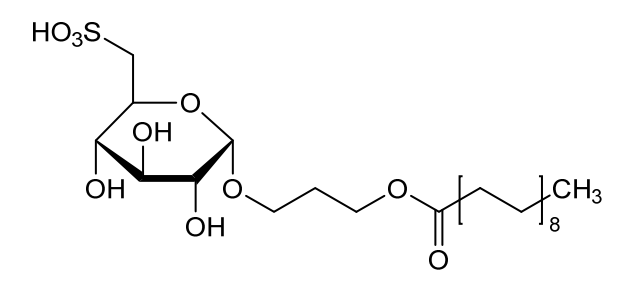
Teprosulvose
CAS 1983131-47-0
MF C27H52O10S MW568.761
| Sulfoquynovosylacylpropanediol [(2S,3S,4S,5R,6S)-3,4,5-trihydroxy-6-(3-octadecanoyloxypropoxy)oxan-2-yl]methanesulfonic acid |
3-(octadecanoyloxy)propyl 6-deoxy-6-sulfo-α-D-glucopyranoside
radiosensitizer (veterinary use), WV7377RGM8, SQAP
Teprosulvose (CAS 1983131-47-0) is a novel synthetic glycolipid, specifically a sulfoquinovosylacylpropanediol (SQAP). It is primarily developed for use in veterinary medicine as a radiosensitizer, intended to enhance the effectiveness of radiation therapy in treating malignant tumors.
1. Chemical Identity and Structure
- USAN/INN Name: Teprosulvose
- Systemic Name: 3-(octadecanoyloxy)propyl 6-deoxy-6-sulfo-$\alpha$-D-glucopyranoside
- Molecular Weight: 568.76 g/mol
- Structure: It consists of a glucose derivative (6-deoxy-6-sulfo-$\alpha$-D-glucopyranoside) linked via a propyl bridge to a long-chain fatty acid (stearic acid/octadecanoic acid).
Regulatory Data
Teprosulvose is currently in the investigational stage, primarily focused on veterinary oncology.
- USAN/INN Status: The name “Teprosulvose” was officially adopted by the USAN Council in 2024 (File LM-156).
- Classification: Radiosensitizer.
- Target Application: Adjuvant therapy for malignant tumors in animals (e.g., canine or feline cancers).
- Current Status: It has not yet received full FDA or EMA approval for human use. In the U.S., it is typically handled under Investigational New Animal Drug (INAD) protocols for clinical trials in veterinary patients.
Note: Because it is a specialized veterinary investigative agent, detailed safety data (LD50, pharmacokinetics) is generally found in specific FDA Freedom of Information (FOI) summaries or peer-reviewed veterinary oncology journals rather than standard human drug databases.
INN List 131 (WHO): Teprosulvose was officially included in the World Health Organization’s International Nonproprietary Names (INN) list in 2024. This confirms its unique status as a distinct drug substance.
USAN Council: The United States Adopted Names Council assigned the name in 2024, classifying it as a radiosensitizer.
FDA Status: It is currently under investigation (INAD) for canine oral melanoma and other solid tumors in veterinary medicine. Human clinical trial data is not yet widely available as the primary focus remains on the “Veterinary First” pathway.
Mechanism of Action: It is a potent inhibitor of DNA polymerase $\alpha$ and $\beta$. By inhibiting the repair of radiation-induced DNA damage, it effectively “locks in” the damage to tumor cells while sparing normal tissue due to differential uptake.
PAT
US Patent 10,206,942 (and related continuations): Covers the use of SQAP compounds in combination with radiation.
WO 2017/023812: International filing regarding the composition and therapeutic application of these glycolipids.
PAT
PAT
- Sulfonated sugar compounds, pharmaceutical compositions which contain the same, and methods of treating tumors with the samePublication Number: US-7973145-B2Priority Date: 2007-07-20Grant Date: 2011-07-05
- Sulfonated sugar compounds, pharmaceutical compositions which contain the same, and methods of treating tumors with the samePublication Number: US-2009209475-A1Priority Date: 2007-07-20
- Novel sulfonated sugar derivative, and use thereof for medicinal agentPublication Number: EP-2130834-A1Priority Date: 2007-07-20
- Sulfonated sugar compounds, pharmaceutical compositions which contain the same, and methods of treating tumors with the samePublication Number: US-2010298246-A1Priority Date: 2007-07-20
- Novel sulfonated sugar derivatives and their use as pharmaceuticalsPublication Number: JP-4435861-B2Priority Date: 2007-07-20Grant Date: 2010-03-24
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////teprosulvose, radiosensitizer (veterinary use), WV7377RGM8, SQAP
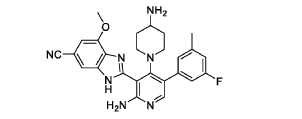
Branosotine
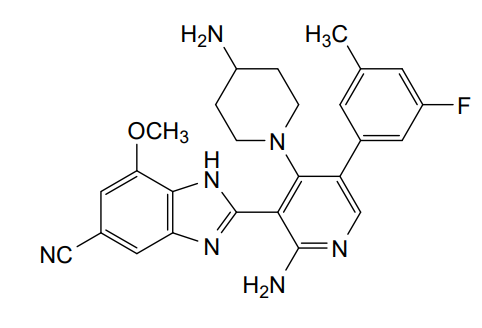
Branosotine
CAS 2412849-26-2
MF C26H26FN7O MW471.5 g/mol
2-[2-amino-4-(4-aminopiperidin-1-yl)-5-(3-fluoro-5-methylphenyl)-3-pyridinyl]-7-methoxy-3H-benzimidazole-5-carbonitrile
2-[2-amino-4-(4-aminopiperidin-1-yl)-5-(3-fluoro-5-
methylphenyl)pyridin-3-yl]-7-methoxy-1H-1,3-benzimidazole-5-
carbonitrile
somatostatin receptor agonist (veterinary use), 4L2VN6D3D8
Branosotine is a small molecule drug. The usage of the INN stem ‘-sotine’ in the name indicates that Branosotine is a non-peptidic somatostatin receptor agonist. Branosotine has a monoisotopic molecular weight of 471.22 Da.
SYN
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020061046&_cid=P21-MK9408-98104-1
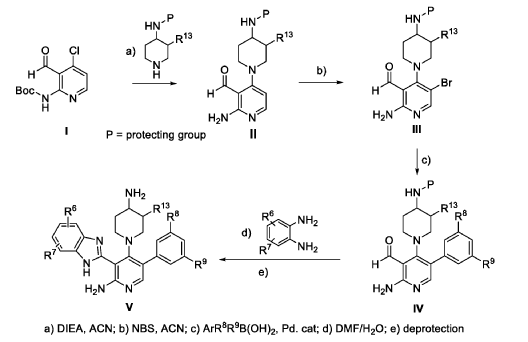
The following examples are provided for illustrative purposes only and not to limit the scope of the claims provided herein.
Example 1 : 2-[2-amino-4-(4-aminopiperidin-1-yl)-5-(3-fluoro-5-methylphenyl)pyridin- 3-yl]-4-methoxy-1H-1,3-benzodiazole-6-carbonitrile (1-1)



PAT
- Somatostatin modulators and uses thereofPublication Number: EP-4548974-A2Priority Date: 2018-09-18
- Somatostatin modulators and uses thereofPublication Number: EP-3853218-B1Priority Date: 2018-09-18Grant Date: 2025-02-19
- Somatostatin modulators and uses thereofPublication Number: TW-I852944-BPriority Date: 2018-09-18Grant Date: 2024-08-21
- Somatostatin modulator and its usePublication Number: JP-2022501342-APriority Date: 2018-09-18
- Somatostatin modulators and uses thereofPublication Number: US-11834462-B2Priority Date: 2018-09-18Grant Date: 2023-12-05
- Somatostatin modulators and uses thereofPublication Number: US-2022048924-A1Priority Date: 2018-09-18
- Somatostatin modulators and uses thereofPublication Number: US-2020283453-A1Priority Date: 2018-09-18
- Somatostatin modulators and their usesPublication Number: JP-7431813-B2Priority Date: 2018-09-18Grant Date: 2024-02-15
- Somatostatin modulators and uses thereofPublication Number: US-2023022513-A1Priority Date: 2018-09-18
- Somatostatin modulators for treating pituitary adenomasPublication Number: WO-2021076448-A1Priority Date: 2019-10-14
- Somatostatin modulators and uses thereofPublication Number: US-2020087318-A1Priority Date: 2018-09-18
- Somatostatin modulators and uses thereofPublication Number: US-10696689-B2Priority Date: 2018-09-18Grant Date: 2020-06-30
- Somatostatin modulators and uses thereofPublication Number: TW-202024095-APriority Date: 2018-09-18
- Somatostatin modulators and uses thereofPublication Number: US-11186590-B2Priority Date: 2018-09-18Grant Date: 2021-11-30

AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Branosotine, somatostatin receptor agonist (veterinary use), 4L2VN6D3D8
Velagliflozin
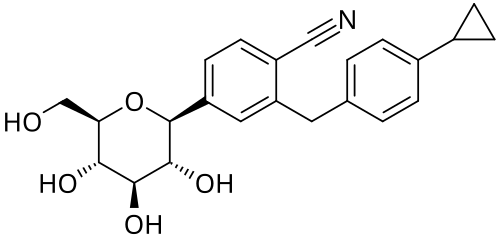
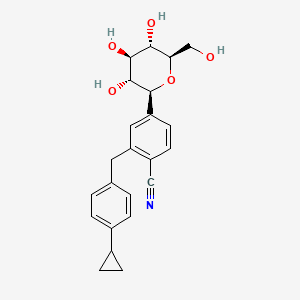
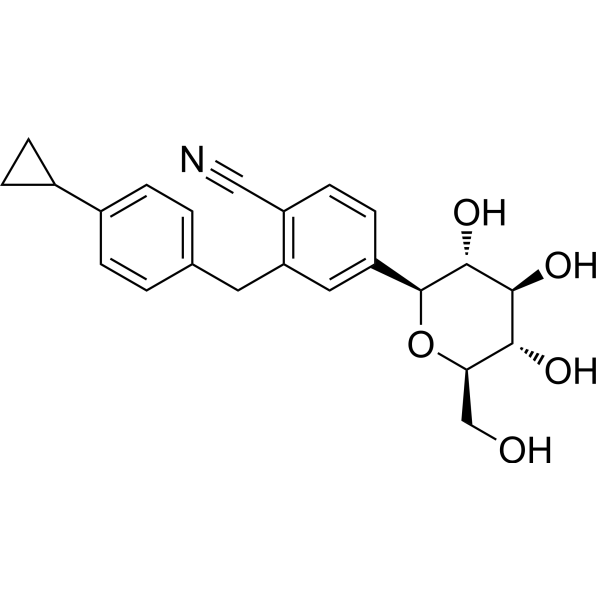
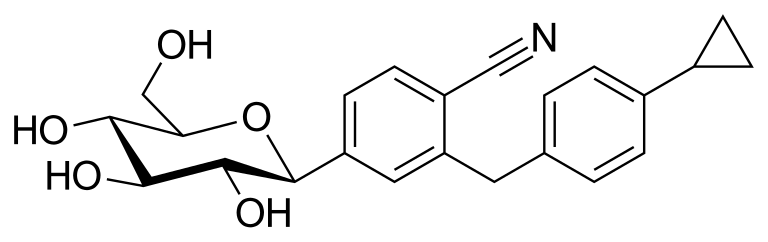
Velagliflozin
VETERINARY DRUG
- Cas 946525-65-1
- FV2YU8SL0P
- 2-((4-cyclopropylphenyl)methyl)-4-((2S,3R,4R,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl)benzonitrile
- 2-((4-Cyclopropylphenyl)methyl)-4-beta-D-glucopyranosylbenzonitrile
- 395.4 g/mol, C23H25NO5
2-[(4-cyclopropylphenyl)methyl]-4-[(2S,3R,4R,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]benzonitrile
- 2-((4-CYCLOPROPYLPHENYL)METHYL)-4-.BETA.-D-GLUCOPYRANOSYLBENZONITRILE
- BENZONITRILE, 2-((4-CYCLOPROPYLPHENYL)METHYL)-4-.BETA.-D-GLUCOPYRANOSYL-
Velagliflozin L-proline H2O
Velagliflozin, sold under the brand name Senvelgo, is an antidiabetic medication used for the treatment of cats.[2][4][5] Velagliflozin is a sodium-glucose cotransporter 2 (SGLT2) inhibitor.[6] It is taken by mouth.[2]
Velagliflozin is the active ingredient of the first oral liquid medication approved by the Food and Drug Administration for the treatment of diabetes in cats. This compound belongs to the known class of sodium-glucose cotransporter 2 inhibitors approved to treat diabetes in human.
- Application: NADA 141-568Drug: Senvelgo®Active Ingredient(s): VelagliflozinCompany: Boehringer lngelheim Animal Health USA Inc.Patent(s): 7776830 (Exp: 05/01/2027); 8557782 (Exp: 05/01/2027); 9145434 (Exp: 09/07/2033); 10617666 (Exp: 06/06/2035); 11896574 (Exp: 12/17/2034); 10220017 (Exp: 09/29/2036); 10709683 (Exp: 08/24/2036); 11225500 (Exp: 12/17/2038)
- [Indication for Use] To improve glycemic control in otherwise healthy cats with diabetes mellitus not previously treated with insulin.Application: NADA 141-568Active Ingredient(s): VelagliflozinCompany: Boehringer lngelheim Animal Health USA Inc.Freedom of Information: FOIA Summary 14320Approval Date: August 10, 2023
APPROVALS 2023, GDA 2023, EU 2023, EMA 2023, SENVELGO
Velagliflozin (brand name Senvelgo) is a veterinary medication approved for treating diabetes in cats, not humans.
Approved countries and years for velagliflozin:
- United States (US): Approved by the FDA in August 2023.
- European Union (EU): Received marketing authorization in November 2023.
- Switzerland: Approved in 2023.
- Great Britain: Approved in 2023.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US310904480&_cid=P11-METCZG-99171-1
SYN
US7776830
https://patentscope.wipo.int/search/en/detail.jsf?docId=US41880220&_cid=P11-METD0X-00376-1
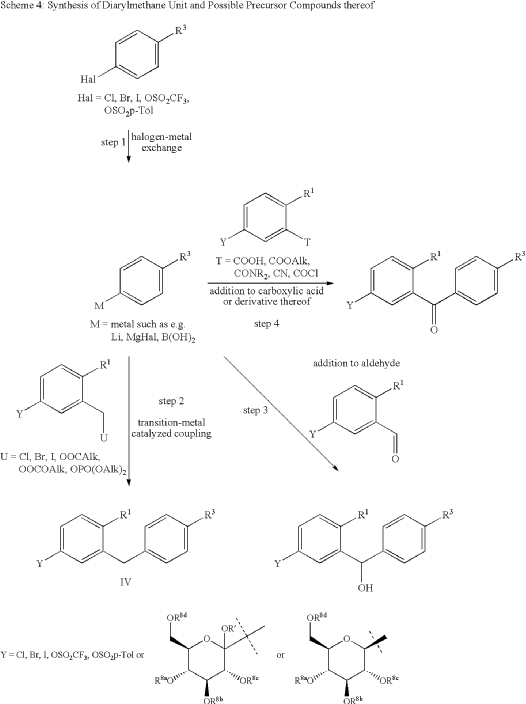
| The following compound is obtained analogously to Example XXIV: |
(1) 1-Cyano-2-(4-cyclopropyl-benzyl)-4-(β-D-glucopyranos-1-yl)-benzene
EXAMPLE 17
2-(4-Cyclopropyl-benzyl)-4-(β-D-glucopyranos-1-yl)-benzonitrile
| The compound is obtained according to example 6 using 4-cyclopropyl-phenylboronic acid as the coupling partner. |
SYN
WO2007128749
https://patents.google.com/patent/WO2007128749A1/en
The following compound is obtained analogously to Example XXIV:
(1 ) 1 -Cvano-2-(4-cvclopropyl-benzyl)-4-(3-D-glucopyranos-1 -vD-benzene

Mass spectrum (ESI“): m/z = 413 [M+H] + Advantageously, the reduction of the anomeric carbon center of the appropriate intermediate obtained during the synthesis of this compound is conducted with the oxygen functionalities on the pyranose ring protected. Preferred protective groups are benzyl, p-methoxybenzyl, trimethylsilyl, triethylsilyl, terfbutyldimethylsilyl, triisopropylsilyl and allyl.
Example XXV

1-Cyano-2-(4-cyclopropyl-benzyl)-4-(tetra-O-acetyl-β-D-glucopyranos-1-yl)-benzene To a flask charged with a stir bar, 4-(2,3,4,6-tetra-O-acetyl-D-glucopyranos-1-yl)-2-(4- trifluoromethylsulfonyloxy-benzyl)-benzonitrile (4.4 g), degassed toluene (12 ml.) and degassed water (8 ml.) and kept under argon atmosphere is added cyclopropylboronic acid (0.20 g), potassium phosphate (5.0 g), tricyclohexylphosphine (0.19 g) and at last palladium(ll)acetate (76 mg). The mixture is stirred at 1 10 °C for 6 h meanwhile cyclopropylboronic acid is added after each hour (5x 0.20 g). After cooling to room temperature, the mixture is diluted with aqueous sodium hydrogen carbonate solution and extracted with ethyl acetate. The combined extracts are dried (sodium sulphate) and the solvent is removed under reduced pressure. The residue is chromatographed on silica gel (cyclohexane/ethyl acetate 20:1 -> 1 :1 ). Yield: 3.2 g (87% of theory ) Mass spectrum (ESI+): m/z = 581 [M+NH4] +
Example XXVI

4-(1 -Hvdroxy-cvclopropyD-phenylboronic acid A 3.0 M solution of ethylmagnesium bromide in diethylether (7.6 ml.) is added to a stirred solution of titanium(IV) isopropoxide (2.2 ml.) in diethylether (70 ml.) chilled to -78 °C. The resultant solution is stirred at -78 °C for 1.5 h, before 4-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa borolan-2-yl)-benzoic acid methyl ester (2.0 g) is added. The reaction mixture is warmed to ambient temperature and stirred for an additional 12 h. Then, 1 M aqueous hydrochloric acid is added and the resulting mixture is extracted with ethyl acetate. The combined organic extracts are dried (sodium sulphate) and the solvent is evaporated. The residue is dissolved in acetone (60 ml.) and 0.1 M aqueous NH4OAc solution (50 ml.) followed by NaIO4 (2.3 g) is added. The resulting reaction mixture is stirred at room temperature for 18 h. After removal of the acetone, the residue is extracted with ethyl acetate. The combined extracts are dried (sodium sulphate) and the solvent is evaporated. The residue is purified by chromatography on silicagel (cyclohexane/ethyl acetate). Yield: 0.45 g (33% of theory) Mass spectrum (ESI“): m/z = 223 [M+HCOO]“ Preparation of the end compounds:
Example 17: 2-(4-Cyclopropyl-benzyl)-4-(β-D-glucopyranos-1-yl)-benzonitrile

Mass spectrum (ESI+): m/z = 413 [M+NH4]+
The compound is obtained according to example 6 using 4-cyclopropyl-phenylboronic acid as the coupling partner.
Yield: 83% of theory
Alternatively this compound is obtained as described in Example XXIV(I ).
The compound of example 17 is also obtained by employing the following procedure:
A solution of 2-(4-cyclopropyl-benzyl)-4-(2,3,4,6-tetra-O-acetyl-D-glucopyranos-1 -yl)- benzonitrile (0.80 g) in methanol (5 ml.) and THF (5 ml.) is treated with aqueous potassium hydroxide solution (4 mol/l, 5 ml_). The reaction solution is stirred at ambient temperature for 1 h and then neutralized with 1 M hydrochloric acid. The organic solvents are evaporated and the residue is diluted with brine and extracted with ethyl acetate. The organic extracts are dried (sodium sulphate) and the solvent is removed. The residue is chromatographed on silica gel (dichloromethane/methanol 1 :0 -> 9:1 ). Yield: 0.54 g (96% of theory)
SYN
Synthesis 2024, 56, 906–943
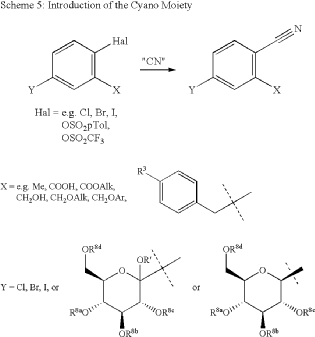
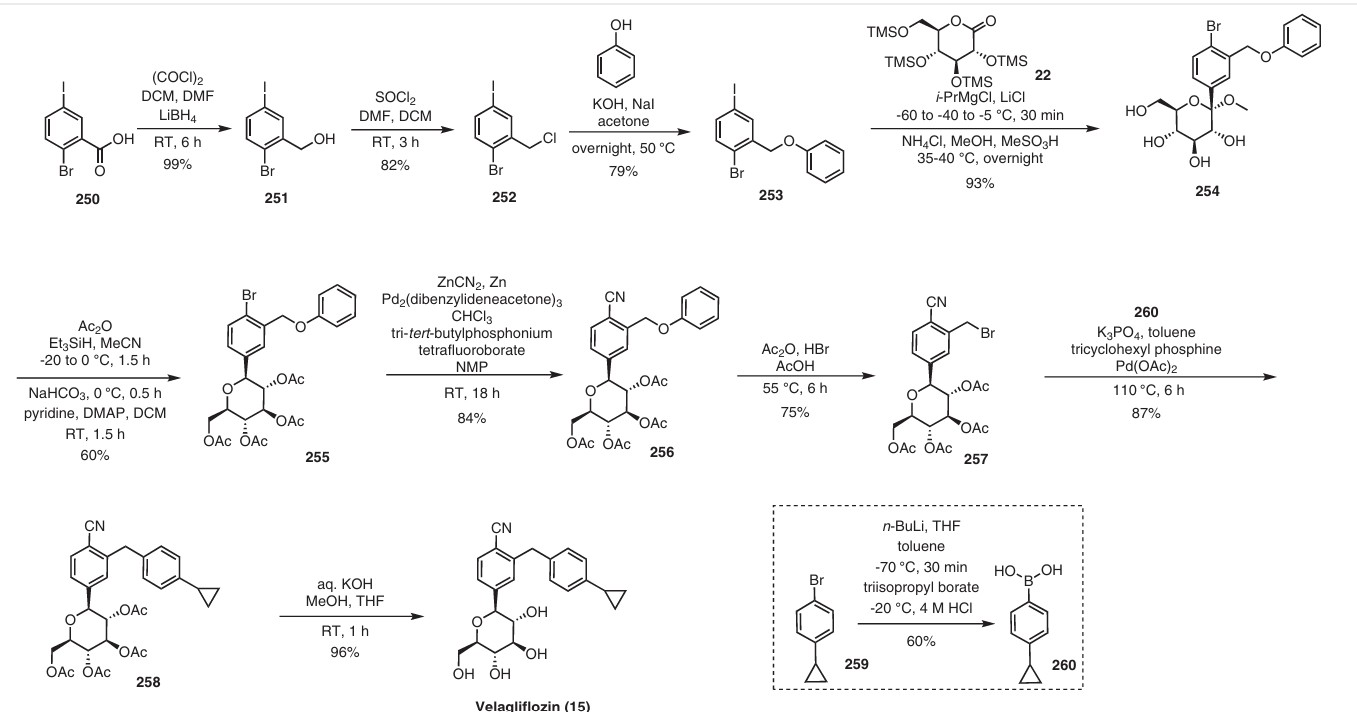
In 2007, Boehringer-Ingelheim Vetmedica GmbH pioneered the development of velagliflozin (15), subsequently submitting a patent application in the United States with the identification number US7776830B2.72a More recently, through clinical investigations, this compound has demonstrated its efficacy as an SGLT2 inhibitor, proving adept at curtailing glucose reabsorption, encouraging glucosuria,
and leading to reductions in both blood glucose and insulin levels.
The initial synthesis of velagliflozin (15) was also disclosed in the above patent,72a and in patent
WO2007128749A1.72b The synthesis, depicted in Scheme46, comprises of nine-steps starting with the readily available raw material 2-bromo-5-iodobenzoic acid (250), which undergoes reduction using LiBH4 to form the corresponding alcohol 251. Subsequently, chlorination is carried out using thionyl chloride, resulting in the formation of chloride 252. O-Alkylation of phenol with compound 252 is
then conducted in a basic medium, yielding intermediate 253.The C-glycosylation of 253 with 2,3,4,6-tetrakis-O(trimethylsilyl)-D-glucopyranone 22 in the presence of turbo Grignard reagent (isopropylmagnesium chloride and LiCl) and methanesulfonic acid in methanol gives compound
254 with an impressive 93% yield. The hydroxy group of in termediate 254 is protected using acetic anhydride, and themethoxy group is subsequently removed via Lewis acid (BF3·Et2O, Et3SiH) treatment, providing compound 255 in a yield of 60%. A metal-catalyzed cyano group installation is then performed on intermediate 255, leading to the formation of compound 256 in 84% yield. The subsequent steps involve benzylic bromination followed by coupling with cyclopropylphenyl boronic acid 260, resulting in the formation of intermediate 258. Finally, deacetylation of intermediate 258 using aqueous KOH produces the desired product
The overall yield obtained for velagliflozin (15) is calculated to be 11.3%, with this synthetic route providing a systematic and efficient approach. The highlight of the route is high-yielding chemical transformations. However, the drawback is the use of two palladium-mediated couplings
that increase the possibility of leaching of the toxic metal in scale-up batches. Additionally, the synthetic route requires a large number of chemical transformations and not best suited for commercial production.
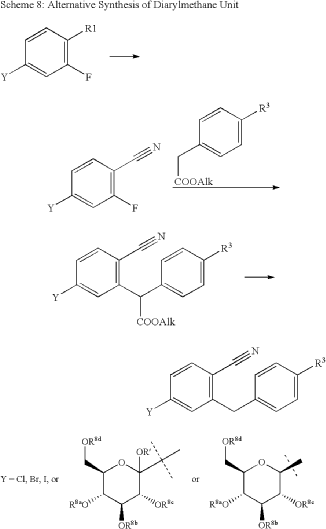
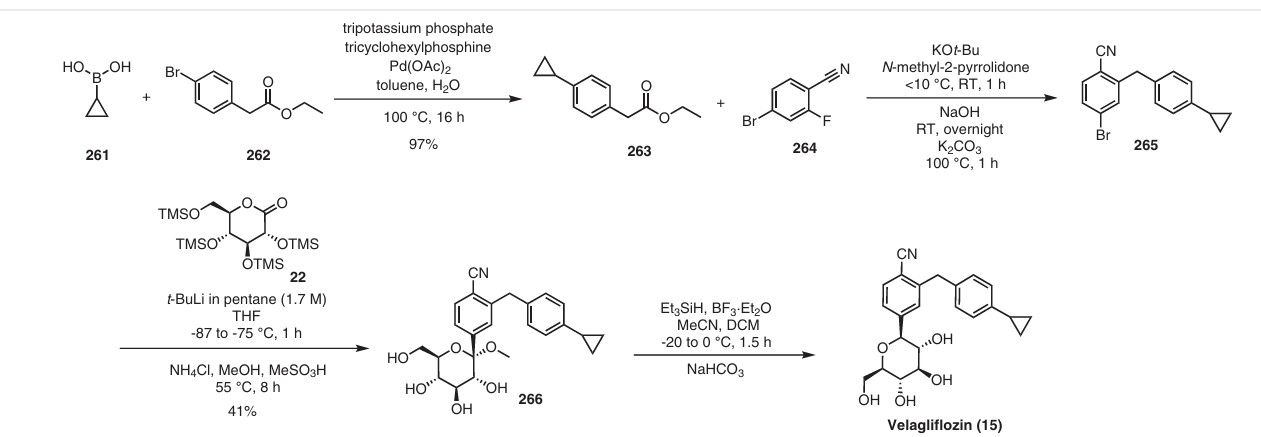
The same authors reported an alternative method (Scheme 47) for the synthesis of velagliflozin (15) in the product patent.72 The aglycone intermediate 265 is accessed in two steps starting from ethyl 2-(4-bromophenyl)acetate (262). O-Glycosylation takes place with the aglycone
4-bromo-2-(4-cyclopropylbenzyl)benzonitrile (265) using 2,3,4,6-tetrakis-O-(trimethylsilyl)-D-glucopyranone 22 in the presence of tert-butyllithium in pentane (1.7 M), resulting in the formation of compound 266. Reduction of compound 266 using boron trifluoride–diethyl etherate yields
the final API velagliflozin (15). This truncated synthetic route is well suited for scale-up due to the significantly low er number of transformations compared to the previous route. Unfortunately, the specific yields were not clearly in dicated for this process. This method presents an alternative approach to the synthesis of velagliflozin (15), providing a potential pathway for its preparation in 5 steps with
an overall yield of 40%.
(72) (a) Eckhardt, M.; Himmelsbach, F.; Eickelmann, P.; Sauer, A.;
Thomas, L. US7776830B2, 2010. (b) Eckhardt, M.; Himmelsbach,
F.; Eickelmann, P.; Sauer, A.; Thomas, L. WO2007128749A1,
2007.

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Medical uses
Velagliflozin is indicated to improve glycemic control in otherwise healthy cats with diabetes not previously treated with insulin.[2][4][6]
References
- “Notice: Multiple additions to the Prescription Drug List (PDL) [2024-10-18]”. Health Canada. 18 October 2024. Retrieved 25 October 2024.
- “Senvelgo- velagliflozin solution”. DailyMed. 8 November 2023. Retrieved 13 December 2023.
- “Senvelgo Product information”. Union Register of veterinary medicinal products. 22 November 2023. Retrieved 29 August 2024.
- “NADA 141-568 Senvelgo (velagliflozin oral solution) Cats”.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - Cook AK, Behrend E (January 2025). “SGLT2 inhibitor use in the management of feline diabetes mellitus”. Journal of Veterinary Pharmacology and Therapeutics. 48 Suppl 1 (Suppl 1): 19–30. doi:10.1111/jvp.13466. PMC 11736986. PMID 38954371.
- “Dear Veterinarian Letter regarding important safety conditions associated with the use of Senvelgo (velagliflozin oral solution) for improving glycemic control in certain cats with diabetes mellitus”. U.S. Food and Drug Administration. 4 December 2023. Retrieved 13 December 2023. This article incorporates text from this source, which is in the public domain.
| Clinical data | |
|---|---|
| Trade names | Senvelgo |
| License data | US DailyMed: Velagliflozin |
| Routes of administration | By mouth |
| ATCvet code | QA10BK90 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2]EU: Rx-only[3] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 946525-65-1 |
| PubChem CID | 24862817 |
| ChemSpider | 58827717 |
| UNII | FV2YU8SL0PEQE2P2T77I |
| Chemical and physical data | |
| Formula | C23H25NO5 |
| Molar mass | 395.455 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- SGLT2 inhibitors: a novel therapy for cognitive impairment via multifaceted effects on the nervous systemPublication Name: Translational NeurodegenerationPublication Date: 2024-08-09PMCID: PMC11312905PMID: 39123214DOI: 10.1186/s40035-024-00431-y
- Demographic, morphologic, hormonal and metabolic factors associated with the rate of improvement from equine hyperinsulinaemia-associated laminitisPublication Name: BMC Veterinary ResearchPublication Date: 2022-01-18PMCID: PMC8764787PMID: 35042535DOI: 10.1186/s12917-022-03149-z
- The efficacy and safety of velagliflozin over 16 weeks as a treatment for insulin dysregulation in poniesPublication Name: BMC Veterinary ResearchPublication Date: 2019-02-26PMCID: PMC6390376PMID: 30808423DOI: 10.1186/s12917-019-1811-2
- The sodium-glucose co-transporter 2 inhibitor velagliflozin reduces hyperinsulinemia and prevents laminitis in insulin-dysregulated poniesPublication Name: PLOS ONEPublication Date: 2018-09-13PMCID: PMC6136744PMID: 30212530DOI: 10.1371/journal.pone.0203655
- Effects of the sodium‐glucose cotransporter 2 (<scp>SGLT</scp>2) inhibitor velagliflozin, a new drug with therapeutic potential to treat diabetes in catsPublication Name: Journal of Veterinary Pharmacology and TherapeuticsPublication Date: 2017-11-15PMID: 29139146DOI: 10.1111/jvp.12467
/////////Velagliflozin, APPROVALS 2023, GDA 2023, EU 2023, EMA 2023, SENVELGO, DIABETES, SENVELGO,
CLOSANTEL

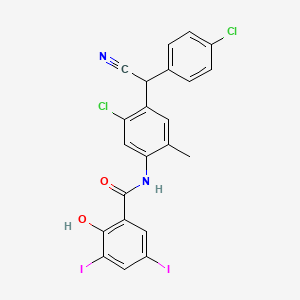
CLOSANTEL
Closantel
57808-65-8
N-(5-chloro-4-((4-chlorophenyl)(cyano)methyl)-2-methylphenyl)-2-hydroxy-3,5-diiodobenzamide
MW 663.1,

Closantel Sodium
CAS NO. 61438-64-0
| FORMULA | C22H13Cl2I2N2O2.Na |
|---|---|
| M. WT | 685.06 |
Closantel
CAS Registry Number: 57808-65-8
CAS Name:N-[5-Chloro-4-[(4-chlorophenyl)cyanomethyl]-2-methylphenyl]-2-hydroxy-3,5-diiodobenzamide
Manufacturers’ Codes: R-31520
Trademarks: Flukiver (Janssen); Seponver (Ethnor)
Molecular Formula: C22H14Cl2I2N2O2, Molecular Weight: 663.07
Percent Composition: C 39.85%, H 2.13%, Cl 10.69%, I 38.28%, N 4.22%, O 4.83%
Literature References: Salicylanilide derivative. Prepn: M. A. C. Janssen, V. K. Sipido, BE839481; eidem,US4005218 (1976, 1977 both to Janssen). Effectiveness against Taenia pisiformis in rabbits: R. A. F. Chevis et al.,Vet. Parasitol.7, 333 (1980); against Ancylostoma caninum: J. Guerrero et al.,J. Parasitol.68, 616 (1983); against Fasciola hepatica in sheep: B. E. Stromberg et al.,ibid.70, 446 (1984). Prolonged effect on Haemonchus contortus in sheep: C. A. Hall et al.,Res. Vet. Sci.31, 104 (1981). Acts by uncoupling oxidative phosphorylation: H. Van den Bossche et al.,Arch. Int. Physiol. Biochim.87, 851 (1979); H. J. Kane et al.,Mol. Biochem. Parasitol.1, 347 (1980).
Properties: Crystals from methanol, mp 217.8°.
Melting point: mp 217.8°
Therap-Cat-Vet: Anthelmintic.
N-{5-chloro-4-[(4-chlorophenyl)(cyano)methyl]-2-methylphenyl}-2-hydroxy-3,5-diiodobenzamide is an aromatic amide resulting from the formal condensation of the carboxy group of 3,5-diiodosalicylic acid with the amino group of aniline substituted at positions 2, 4, and 5 by methyl, (4-chlorophenyl)(cyano)methyl, and methyl groups respectively. It is a nitrile, a member of phenols, an organoiodine compound, a monocarboxylic acid amide, an aromatic amide and a member of monochlorobenzenes.
Closantel is a broad-spectrum antiparasitic agent used against
several species and developmental stages of trematodes, nematodes and
arthropods. The anti-trematode activity of closantel is mainly used
against liver fluke. The anti-nematode and anti-arthropod activity is
especially used against those species which feed on blood or plasma.
The drug is widely used in sheep and cattle and can be used
either parenterally (s.c. or i.m.) or orally for both prophylactic and
therapeutic purposes and is available as drench, bolus and injectable
formulations. Closantel has also been combined with mebendazole and
several other benzimidazoles in drench formulations for sheep and with
levamisole in a bolus for cattle (Marsboom et al., 1989).
Closantel has not been evaluated previously by the Joint FAO/WHO
Expert Committee on Food Additives.
PATENT
https://patents.google.com/patent/CN102180811B/en
Closantel sodium (Closantel Sodium) is a kind of very strong oxidative phosphorylation uncoupler, can suppress the mitochondrial phosphorylation process of polypide, nematode and insect etc. are contacted with blood circulation closely or sucking blood property worm all has and efficiently kills effect, be a kind of broad-spectrum de-worming medicine of efficient, low toxicity, it is huge on market a very large development potentiality.
And 4-chloro-phenyl–(the chloro-4-amino of 2–5-aminomethyl phenyl) cyano group methane is a kind of key intermediate for the synthesis of closantel sodium.But in prior art, the report of the synthetic method of relevant 4-chloro-phenyl–(the chloro-4-amino of 2–5-aminomethyl phenyl) cyano group methane is actually rare, is mainly the iron powder reducing synthetic method.As United States Patent (USP) (US4005218) relates to a kind of with the chloro-α of 4–[the chloro-4-of 2-(hydroxyl imido grpup)-5-methyl-2, the 5-phenylidene] benzyl cyanide (I) is raw material, with excessive iron powder, in ammonium chloride, water and toluene mixing solutions, heating reflux reaction, filter, clean filter cake with a large amount of solvents as tetrahydrofuran (THF) or 4-methyl-2 pentanone, filtrate boils off solvent, then adds the toluene recrystallization to obtain 4-chloro-phenyl–(the chloro-4-amino of 2–5-aminomethyl phenyl) cyano group methane (II).This reaction equation is:
But it is loaded down with trivial details that the shortcoming of the method is operating procedure, the supplementary material consumption is large, and, with producing a large amount of scrap iron powder after iron powder reducing, comparatively thickness, easily comprise product and impurity, and cost recovery is very high, and labour intensity is large, larger to the pollution effect of environment; And product yield and product quality lower.
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
PATENTS
CN103054846
WO2015048718
US2015164934
WO2016038035
CN105687172
WO2018013890
WO2018210449
WO2019222349
CN111150725
CN112294793
///////CLOSANTEL, veterinary
CC1=CC(=C(C=C1NC(=O)C2=C(C(=CC(=C2)I)I)O)Cl)C(C#N)C3=CC=C(C=C3)Cl

NEW DRUG APPROVALS
ONE TIME HELP
$10.00
MONENSIN
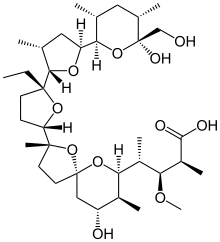
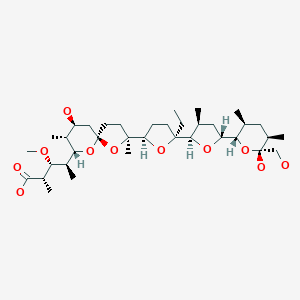
モネンシン;
MONENSIN
- Molecular FormulaC36H62O11
- Average mass670.871 Da
1,6-dioxaspiro[4.5]decane-7-butanoic acid, 2-[(2S,2’R,3’S,5R,5’R)-2-ethyloctahydro-3′-methyl-5′-[(2S,3S,5R,6R)-tetrahydro-6-hydroxy-6-(hydroxymethyl)-3,5-dimethyl-2H-pyran-2-yl][2,2′-bifuran]-5-yl]-9-hydroxy-β-methoxy-α,γ,2,8-tetramethyl-, (αS,βR,γS,2S,5R,7S,8R,9S)-
17090-79-8[RN]
241-154-0[EINECS]
(2S,3R,4S)-4-[(2S,5R,7S,8R,9S)-2-{(2S,2’R,3’S,5R,5’R)-2-Ethyl-5′-[(2S,3S,5R,6R)-6-hydroxy-6-(hydroxymethyl)-3,5-dimethyltetrahydro-2H-pyran-2-yl]-3′-methyloctahydro-2,2′-bifuran-5-yl}-9-hydroxy-2,8-di methyl-1,6-dioxaspiro[4.5]dec-7-yl]-3-methoxy-2-methylpentanoic acid
монензин[Russian]
مونانسين[Arabic]
莫能星[Chinese]
Antibiotic, Antifungal, Antiprotozoal

Synonym(s):
Monensin A sodium salt
Empirical Formula (Hill Notation):C36H61NaO11
CAS Number:22373-78-0
Molecular Weight:692.85
Beilstein:4122200
Title: Monensin
CAS Registry Number: 17090-79-8
CAS Name: 2-[5-Ethyltetrahydro-5-[tetrahydro-3-methyl-5-[tetrahydro-6-hydroxy-6-(hydroxymethyl)-3,5-dimethyl-2H-pyran-2-yl]-2-furyl]-2-furyl]-9-hydroxy-b-methoxy-a,g,2,8-tetramethyl-1,6-dioxaspiro[4.5]decane-7-butyric acid
Additional Names: monensic acid (obsolete)
Manufacturers’ Codes: A-3823A
Molecular Formula: C36H62O11, Molecular Weight: 670.87
Percent Composition: C 64.45%, H 9.32%, O 26.23%
Literature References: Polyether antibiotic. Major factor in antibiotic complex isolated from Streptomyces cinnamonensis. Discovery and isolation: Haney, Hoehn, Antimicrob. Agents Chemother.1967, 349. Production: Haney, Hoehn, US3501568 (1970 to Lilly). Structure: Agtarap et al.,J. Am. Chem. Soc.89, 5737 (1967). Crystal structure studies: Lutz et al.,Helv. Chim. Acta53, 1732 (1970); ibid.54, 1103 (1971). Fermentation studies: Stark et al.,Antimicrob. Agents Chemother.1967, 353. Chemistry: Agtarap, Chamberlin, ibid. 359. Stereocontrolled total synthesis: T. Fukuyama et al.,J. Am. Chem. Soc.101, 262 (1979); D. B. Collum et al.,ibid.102, 2117, 2118, 2120 (1980). 13C-NMR study: J. A. Robinson, D. L. Turner, Chem. Commun.1982, 148. Biosynthesis: Day et al.,Antimicrob. Agents Chemother.4, 410 (1973). Review: Stark, “Monensin, A New Biologically Active Compound Produced by a Fermentation Process”, in Fermentation Advances, Pap. Int. Ferment. Symp., 3rd, 1968, D. Perlman, Ed. (Academic Press, New York, 1969) pp 517-540.
Properties: Crystals, mp 103-105° (monohydrate). [a]D +47.7°. pKa 6.6 (in 66% DMF). Very stable under alkaline conditions. Slightly sol in water; more sol in hydrocarbons; very sol in other organic solvents. LD50 of monensin complex in mice, chicks (mg/kg): 43.8 ± 5.2, 284 ± 47 orally (Haney, Hoehn).
Melting point: mp 103-105° (monohydrate)
pKa: pKa 6.6 (in 66% DMF)
Optical Rotation: [a]D +47.7°
Toxicity data: LD50 of monensin complex in mice, chicks (mg/kg): 43.8 ± 5.2, 284 ± 47 orally (Haney, Hoehn)
Derivative Type: Sodium salt
Trademarks: Coban (Elanco); Romensin (Elanco); Rumensin (Elanco)
Molecular Formula: C36H61NaO11, Molecular Weight: 692.85
Percent Composition: C 62.41%, H 8.87%, Na 3.32%, O 25.40%
Properties: mp 267-269°. [a]D +57.3° (methanol). Slightly sol in water; more sol in hydrocarbons; very sol in other organic solvents.
Melting point: mp 267-269°
Optical Rotation: [a]D +57.3° (methanol)
Therap-Cat-Vet: Coccidiostat. Feed additive to improve feed efficiency in ruminants.
Monensin is a polyether antibiotic isolated from Streptomyces cinnamonensis.[1] It is widely used in ruminant animal feeds.[1][2]
The structure of monensin was first described by Agtarap et al. in 1967, and was the first polyether antibiotic to have its structure elucidated in this way. The first total synthesis of monensin was reported in 1979 by Kishi et al.[3]
SYN

SYN
Production / synthesis Monensin is produced in vivo by Streptomyces cinnamonensis as a natural defense against competing bacteria. Monensin presents a formidable challenge to synthetic chemists as it possesses 17 asymmetric centers on a backbone of only 26 carbon atoms. Although its total synthesis has been described (e.g., Kishi et al., 1979), the high complexity of monensin makes an extraction from the bacterium the most economical procedure for its production. The total synthesis has 56 steps and a yield of only 0.26%. The chemical precursors are 2-allyl-1,3-propanediol and 2- (furan-2-yl)acetonitrile. The method used for synthesizing monensin is based on the principle of “absolute asymmetric synthesis”. Molecules are constructed out of prefabricated building blocks in the correct conformation, aiming for higher yields of the desired enantiomer. New stereocenters are also introduced. Using this method, monensin is assembled in two parts, a larger right side and a smaller left one. The penultimate step is connecting the left and the right halves of monensin, which are independently generated, in an Aldol-condensation. The two halves’ keto end groups (C7/ C8) are linked by eliminating a water molecule. The C7 atom is favored over the C1 atom, because it is more reactive. For catalyzing this step, Yoshito Kishi’s group used iPr2NMgBr (Hauser base) and THF to coordinate it at a temperature of − 78°C. Thus, they were able to isolate the molecule in the right conformation at a ratio of 8:1. Due to the low temperature required for a high yield of the correct enantiomer, the reaction is very solw. One of the most difficult steps is the last one: the connection of the spiro center. This is due to a characteristic feature of spiro compounds; they open and close very easily. Therefore, the conditions for forming the right conformation must be optimal in the last step of synthesis. The biosynthesis in a cell culture of Streptomyces cinnamonensis involves a complex medium containing, among other components, glucose, soybean oil, and grit. Cultivation is carried out for a week at a temperature of 30°C and under constant aeration. Product isolation requires filtration, acidification to pH3, extraction with chloroform and purification with activated carbon. In this way, a few grams per liter of monensin are produced and isolated. For crystallization, azeotropic distillation is necessary. In vivo, polyether backbones are assembled by modular polyketide synthases and are modified by two key enzymes, epoxidase and epoxide hydrolase, to generate the product. Precursors of the polyketide pathway are acetate, butyrate and propionate.
SYN
The final-stage aldol addition in Yoshito Kishi‘s 1979 total synthesis of monensin. (1979). “Synthetic studies on polyether antibiotics. 6. Total synthesis of monensin. 3. Stereocontrolled total synthesis of monensin”. J. Am. Chem. Soc. 101 (1): 262–263. DOI:10.1021/ja00495a066.

SYN
A polyether antibiotic, Monensin was the first member of this class of molecules to be structurally characterized.1 The structural features of these polyethers comprise of a terminal carboxylic acid, multiple cyclic ether rings (ex. Tetrahydrofuran and tetrahydropyran), a large amount of stereocenters and (for many of these molecules) one or more spiroketal moieties.2 Monensin was introduced into the market in 1971 and is used to fight coccidial infections in poultry and as an additive in cattle feed.3 Of the 26 carbon atom’s in Monensin’s backbone, 17 are stereogenic and six of those are contiguous. Coupled with a spiroketal moiety, three hydrofuran rings and two hydropyran rings, the molecule was an attractive synthetic target.
1. Agtarap, A.; Chamberlain, J.W.; Pinkerton, M.; Stein-rauf, L. J. Am. Chem. Soc. 1967, 89, 5737 2. Polyether Antibiotics : Naturally Occurring Acid Ionophores. Westley J.W.; Marcel Dekker: New York (1982) Vol. 1-2. 3. Stark, W.M. In Fermentation Advances, Perlman, D., Ed., Academic Press: New York, 1969, 517
Retrosynthetic Analysis of Monensin
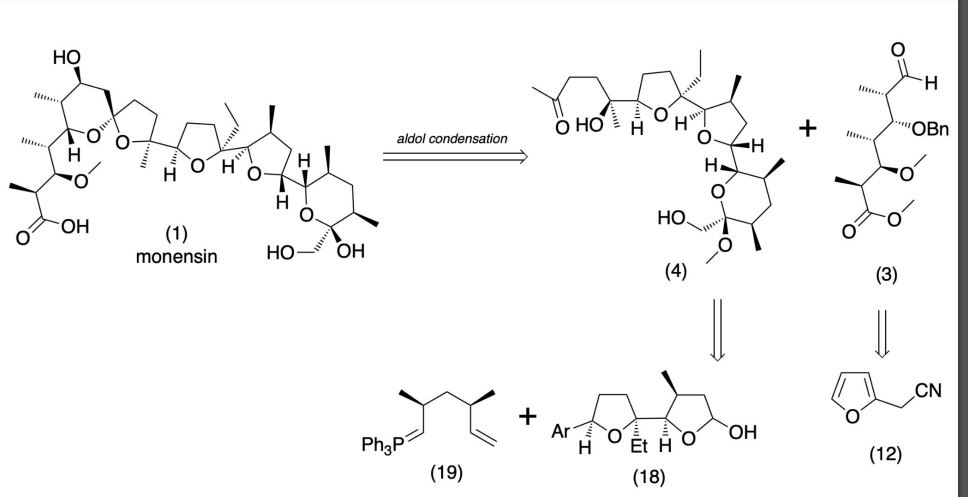
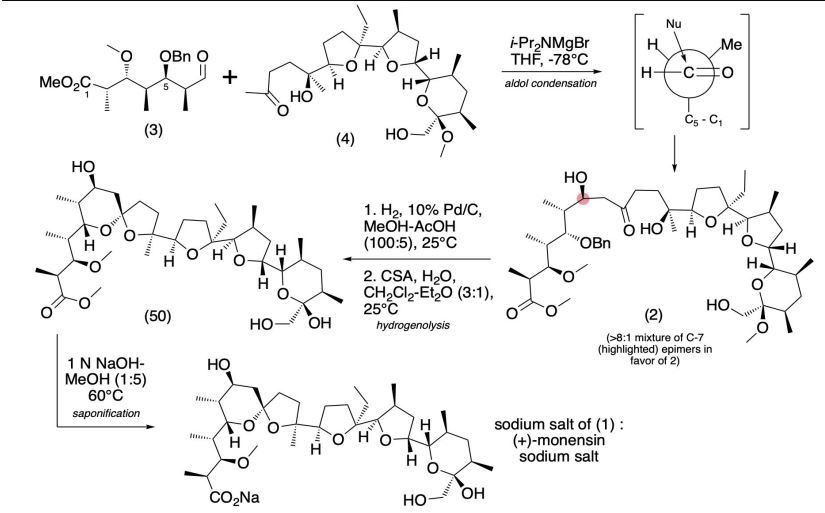
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Mechanism of action

The structure of the sodium (Na+) complex of monensin A.
Monensin A is an ionophore related to the crown ethers with a preference to form complexes with monovalent cations such as: Li+, Na+, K+, Rb+, Ag+, and Tl+.[4][5] Monensin A is able to transport these cations across lipid membranes of cells in an electroneutral (i.e. non-depolarizing) exchange, playing an important role as an Na+/H+ antiporter. Recent studies have shown that monensin may transport sodium ion through the membrane in both electrogenic and electroneutral manner.[6] This approach explains ionophoric ability and in consequence antibacterial properties of not only parental monensin, but also its derivatives that do not possess carboxylic groups. It blocks intracellular protein transport, and exhibits antibiotic, antimalarial, and other biological activities.[7] The antibacterial properties of monensin and its derivatives are a result of their ability to transport metal cations through cellular and subcellular membranes.[8]
Uses
Monensin is used extensively in the beef and dairy industries to prevent coccidiosis, increase the production of propionic acid and prevent bloat.[9] Furthermore, monensin, but also its derivatives monensin methyl ester (MME), and particularly monensin decyl ester (MDE) are widely used in ion-selective electrodes.[10][11][12]
In laboratory research, monensin is used extensively to block Golgi transport.[13][14][15]
Toxicity
Monensin has some degree of activity on mammalian cells and thus toxicity is common. This is especially pronounced in horses, where monensin has a median lethal dose 1/100th that of ruminants. Accidental poisoning of equines with monensin is a well-documented occurrence which has resulted in deaths.[16]
References
- ^ Jump up to:a b Daniel Łowicki and Adam Huczyński (2013). “Structure and Antimicrobial Properties of Monensin A and Its Derivatives: Summary of the Achievements”. BioMed Research International. 2013: 1–14. doi:10.1155/2013/742149. PMC 3586448. PMID 23509771.
- ^ Butaye, P.; Devriese, L. A.; Haesebrouck, F. (2003). “Antimicrobial Growth Promoters Used in Animal Feed: Effects of Less Well Known Antibiotics on Gram-Positive Bacteria”. Clinical Microbiology Reviews. 16 (2): 175–188. doi:10.1128/CMR.16.2.175-188.2003. PMC 153145. PMID 12692092.
- ^ Nicolaou, K. C.; E. J. Sorensen (1996). Classics in Total Synthesis. Weinheim, Germany: VCH. pp. 185–187. ISBN 3-527-29284-5.
- ^ Huczyński, A.; Ratajczak-Sitarz, M.; Katrusiak, A.; Brzezinski, B. (2007). “Molecular structure of the 1:1 inclusion complex of Monensin A lithium salt with acetonitrile”. J. Mol. Struct. 871 (1–3): 92–97. Bibcode:2007JMoSt.871…92H. doi:10.1016/j.molstruc.2006.07.046.
- ^ Pinkerton, M.; Steinrauf, L. K. (1970). “Molecular structure of monovalent metal cation complexes of monensin”. J. Mol. Biol. 49 (3): 533–546. doi:10.1016/0022-2836(70)90279-2. PMID 5453344.
- ^ Huczyński, Adam; Jan Janczak; Daniel Łowicki; Bogumil Brzezinski (2012). “Monensin A acid complexes as a model of electrogenic transport of sodium cation”. Biochim. Biophys. Acta. 1818 (9): 2108–2119. doi:10.1016/j.bbamem.2012.04.017. PMID 22564680.
- ^ Mollenhauer, H. H.; Morre, D. J.; Rowe, L. D. (1990). “Alteration of intracellular traffic by monensin; mechanism, specificity and relationship to toxicity”. Biochim. Biophys. Acta. 1031 (2): 225–246. doi:10.1016/0304-4157(90)90008-Z. PMC 7148783. PMID 2160275.
- ^ Huczyński, A.; Stefańska, J.; Przybylski, P.; Brzezinski, B.; Bartl, F. (2008). “Synthesis and antimicrobial properties of Monensin A esters”. Bioorg. Med. Chem. Lett. 18 (8): 2585–2589. doi:10.1016/j.bmcl.2008.03.038. PMID 18375122.
- ^ Matsuoka, T.; Novilla, M.N.; Thomson, T.D.; Donoho, A.L. (1996). “Review of monensin toxicosis in horses”. Journal of Equine Veterinary Science. 16: 8–15. doi:10.1016/S0737-0806(96)80059-1.
- ^ Tohda, Koji; Suzuki, Koji; Kosuge, Nobutaka; Nagashima, Hitoshi; Watanabe, Kazuhiko; Inoue, Hidenari; Shirai, Tsuneo (1990). “A sodium ion selective electrode based on a highly lipophilic monensin derivative and its application to the measurement of sodium ion concentrations in serum”. Analytical Sciences. 6 (2): 227–232. doi:10.2116/analsci.6.227.
- ^ Kim, N.; Park, K.; Park, I.; Cho, Y.; Bae, Y. (2005). “Application of a taste evaluation system to the monitoring of Kimchi fermentation”. Biosensors and Bioelectronics. 20 (11): 2283–2291. doi:10.1016/j.bios.2004.10.007. PMID 15797327.
- ^ Toko, K. (2000). “Taste Sensor”. Sensors and Actuators B: Chemical. 64 (1–3): 205–215. doi:10.1016/S0925-4005(99)00508-0.
- ^ Griffiths, G.; Quinn, P.; Warren, G. (March 1983). “Dissection of the Golgi complex. I. Monensin inhibits the transport of viral membrane proteins from medial to trans Golgi cisternae in baby hamster kidney cells infected with Semliki Forest virus”. The Journal of Cell Biology. 96 (3): 835–850. doi:10.1083/jcb.96.3.835. ISSN 0021-9525. PMC 2112386. PMID 6682112.
- ^ Kallen, K. J.; Quinn, P.; Allan, D. (1993-02-24). “Monensin inhibits synthesis of plasma membrane sphingomyelin by blocking transport of ceramide through the Golgi: evidence for two sites of sphingomyelin synthesis in BHK cells”. Biochimica et Biophysica Acta (BBA) – Lipids and Lipid Metabolism. 1166 (2–3): 305–308. doi:10.1016/0005-2760(93)90111-l. ISSN 0006-3002. PMID 8443249.
- ^ Zhang, G. F.; Driouich, A.; Staehelin, L. A. (December 1996). “Monensin-induced redistribution of enzymes and products from Golgi stacks to swollen vesicles in plant cells”. European Journal of Cell Biology. 71 (4): 332–340. ISSN 0171-9335. PMID 8980903.
- ^ “Tainted feed blamed for 4 horse deaths at Florida stable”. 2014-12-16.
| Names | |
|---|---|
| Preferred IUPAC name(2S,3R,4S)-4-[(2S,5R,7S,8R,9S)-2-{(2S,2′R,3′S,5R,5′R)-2-Ethyl-5′-[(2S,3S,5R,6R)-6-hydroxy-6-(hydroxymethyl)-3,5-dimethyloxan-2-yl]-3′-methyl[2,2′-bioxolan]-5-yl}-9-hydroxy-2,8-dimethyl-1,6-dioxaspiro[4.5]decan-7-yl]-3-methoxy-2-methylpentanoic acid | |
| Other namesMonensic acid | |
| Identifiers | |
| CAS Number | 17090-79-8 |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:27617 |
| ChEMBL | ChEMBL256105 |
| ChemSpider | 389937 |
| ECHA InfoCard | 100.037.398 |
| E number | E714 (antibiotics) |
| KEGG | D08228 |
| PubChemCID | 441145 |
| UNII | 906O0YJ6ZP |
| CompTox Dashboard (EPA) | DTXSID4048561 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C36H62O11 |
| Molar mass | 670.871 g/mol |
| Appearance | solid state, white crystals |
| Melting point | 104 °C (219 °F; 377 K) |
| Solubility in water | 3×10−6 g/dm3 (20 °C) |
| Solubility | ethanol, acetone, diethyl ether, benzene |
| Pharmacology | |
| ATCvet code | QA16QA06 (WHO) QP51AH03 (WHO) |
| Related compounds | |
| Related | antibiotics, ionophores |
| Related compounds | Monensin A methyl ester, |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). |
///////////MONENSIN, Elancoban, VETERINARY, Coccidiostat, A-3823A, A 3823A

NEW DRUG APPROVALS
ONE TIME TO SUSTAIN THIS BLOG SUBSCRIPTION AND KEEP GOING
$10.00
DETOMIDINE

DETOMIDINE1H-Imidazole, 4-[(2,3-dimethylphenyl)methyl]-
4-(2,3-Dimethylbenzyl)-1H-imidazole
507876631-46-4[RN]7N8K34P2XH
- Molecular FormulaC12H14N2
- Average mass186.253 Da
UNII-7N8K34P2XHдетомидинديتوميدين地托咪定
Formal Name5-[(2,3-dimethylphenyl)methyl]-1H-imidazole, monohydrochlorideCAS Number90038-01-0Synonyms
- Domosedan
- MPV 253AII
Molecular FormulaC12H14N2 • HClFormula Weight222.7DetomidineCAS Registry Number: 76631-46-4CAS Name: 4-[(2,3-Dimethylphenyl)methyl]-1H-imidazoleAdditional Names: 4-(2¢,3¢-dimethylbenzyl)imidazoleMolecular Formula: C12H14N2Molecular Weight: 186.25Percent Composition: C 77.38%, H 7.58%, N 15.04%Literature References: a2-Adrenoceptor agonist with sedative and analgesic activity. Prepn: A. J. Karjalayne, K. O. A. Kurkela, EP24829; eidem,US4443466 (1981, 1984 both to Farmos). Physical studies: E. Laine et al.,Acta Pharm. Suec.20, 451 (1983). Crystal structure: L. H. J. Lajunen et al.,ibid.21, 163 (1984). Pharmacology: R. Virtanen, L. Nyman, Eur. J. Pharmacol.108, 163 (1985); R. Virtanen, E. MacDonald, ibid.115, 277 (1985). Mechanism of action: eidem,J. Vet. Pharmacol. Ther.8, 30 (1985).Properties: Crystals from acetone, mp 114-116°. LD50 i.v. in mice: 35 mg/kg (Karjalayne, Kurkela).Melting point: mp 114-116°Toxicity data: LD50 i.v. in mice: 35 mg/kg (Karjalayne, Kurkela) Derivative Type: HydrochlorideTrademarks: Domosedan (Farmos)Molecular Formula: C12H14N2.HClMolecular Weight: 222.71Percent Composition: C 64.72%, H 6.79%, N 12.58%, Cl 15.92%Properties: Crystals, mp 160°. Converts reversibly to monohydrate at room temp, 80% humidity.Melting point: mp 160° Therap-Cat-Vet: Sedative.
Detomidine is an imidazole derivative and α2-adrenergic agonist,used as a large animal sedative, primarily used in horses. It is usually available as the salt detomidine hydrochloride. It is a prescription medication available to veterinarians sold under the trade name Dormosedan.
Currently, detomidine is only licensed for use in horses in the US but it is also licensed for use in cattle in Europe and Australasia.[1]
Properties
Detomidine is a sedative with analgesic properties.[2] α2-adrenergic agonists produce dose-dependent sedative and analgesic effects, mediated by activation of α2 catecholamine receptors, thus inducing a negative feedback response, reducing production of excitatory neurotransmitters. Due to inhibition of the sympathetic nervous system, detomidine also has cardiac and respiratory effects and an antidiuretic action.[3]
Effects
UsesA profound lethargy and characteristic lowering of the head with reduced sensitivity to environmental stimuli (sound, pain, etc.) are seen with detomidine. A short period of reduced coordination is characteristically followed by immobility and a firm stance with front legs spread. Following administration there is an initial increase in blood pressure, followed by bradycardia and second degree atrioventricular block (this is not pathologic in horses). The horse commonly sweats to excess, especially on the flanks and neck. Other side effects reported include pilo erection (hair standing erect), ataxia, salivation, slight muscle tremors, and (rarely) penile prolapse.
Sedation and anaesthetic premedication in horses and other large animals, commonly combined with butorphanol for increased analgesia and depth of sedation. In conjunction with ketamine it may also be used for intravenous anaesthesia of short duration.
The drug is normally administered by the intravenous route, and is fastest and most efficient when given intravenously . However, in recalcitrant animals, detomidine may be administered by the intramuscular or sublingual routes. The dose range advised by the manufacturers is 20–40 µg/kg intravenous for moderate sedation, but this dose may need to be higher if given intramuscularly.
When given intravenously, detomidine usually takes effect in 2–5 minutes, and recovery is full within 30–60 minutes. However, this is highly dependent upon the dosage, environment, and the individual animal; some horses are highly resistant to sedation.
Detomidine is a poor premedication when using ketamine as an anesthetic in horses.As detomidine is an arrhythmogenic agent, extreme care should be exercised in horses with cardiac disease, and in the concurrent administration of other arrhythmogenics. The concurrent use of potentiated sulfonamide antibiotics is considered particularly dangerous.
Anesthetic recoveries in horses that have received ketamine following a detomidine premedication are often violent with the horse having multiple failures to stand resulting in trauma to itself. Xylazine is a superior premedication with ketamine resulting in safer recoveries.
PATENT
EP-03782989
Novel crystalline forms of detomidine hydrochloride monohydrate, processes for their preparation and compositions comprising them are claimed. Also claimed is their use as alpha2-adrenoreceptor agonists.
Detomidine hydrochloride (1H imidazole,4-[(2,3-dimethylphenyl)methyl]-hydrochloride (CAS Number: 90038-01-0) is a synthetic alpha 2-adrenoreceptor agonist with sedative and analgesic properties widely used for sedation of large animals like horses and cattle. This substance displays various other pharmacologic effects related to the cardiovascular and respiratory system as well as on muscles. Detomidine hydrochloride is available as a parenteral solution with 10 mg/ml as active ingredient which is indicated for use as a sedative and analgesic to facilitate minor surgical and diagnostic procedures in mature horses and yearlings (e.g. DORMOSEDAN®). Furthermore, detomidine hydrochloride is supplied as an oromucosal (i.e. sublingual) gel (e.g. DORMOSEDAN GEL®) with 7.6 mg/ml as active ingredient which is indicated for sedation and restraint in horses.
Further details regarding the clinical pharmacology and side effects as well as contraindications for this drug substance (i.e. active pharmaceutical ingredient) can be found in: Veterinary Psychopharmacology; Sharon L. et al., 2nd edition (2019), Wiley & Sons (pages 161 – 162). According to these authors detomidine has not been used in humans to date.
Detomidini hydrochloridum ad usum veterinarium is included in the EUROPEAN PHARMACOPOEIA (Ph. Eur. 9.0) but currently not included in the United States Pharmacopoeia (USP). It has to be noted that in the absence of a statement regarding a specific hydrate form, like a degree of hydration or mono-, di-, etc., in the title of the monograph – as is the case for detomidine hydrochloride – the anhydrous form is indicated for this substance.
According to a prior version of the respective monograph, namely Ph. Eur. 8.0, the substance exists as a white or almost white, hygroscopic, crystalline powder. The substance is soluble in water, freely soluble in ethanol (96 %), very slightly soluble in methylene chloride and practically insoluble in acetone. The molecular weight (M r) amounts to 222.7. The melting point (mp) is specified at about 160 °C. In the current monograph (Ph. Eur. 9.0) the content of detomidine hydrochloride is specified at 99.0 % to 101.0 percent (anhydrous substance).
[0003] In the current monograph (Ph. Eur. 9.0) the content of detomidine hydrochloride is specified at 99.0 % to 101.0 % (anhydrous substance).
The current monograph includes the three following known impurities:
Impurity A: (RS)-(2,3-dimethylphenyl) (1H-imidazol-4-yl)-methanol
Impurity B: (RS)-(1-benzyl-1H-imidazol-5-yl)(2,3-dimethylphenyl)-methanol
Impurity C: 4-[(2,3-dimethylcylohexyl)methyl]-1H-imidazole
The related substances are specified at ≤ 0.20 % for any unspecified impurities and ≤ 0.5 % for total impurities with a reporting threshold of 0.10 %.
The water content of detomidine hydrochloride as determined by Karl Fischer (KF) titration is limited to ≤ 2.0 % for release as well as shelf-life testing. As detomidine hydrochloride is hygroscopic, the compound has to be stored in airtight containers.
[0004] A synthesis method for detomidine was disclosed in US 4,584,383.
Specific details on the last two steps of a synthesis method for detomidine hydrochloride (including a reaction scheme) were published in Drugs Future 10, 17 (1985).
[0005] Detomidine hydrochloride is known to exist in two crystalline forms, namely the anhydrous form, as described above, and the monohydrate form B (M r: 240.7, CAS Number: 90038-00-9) which can easily interconvert, depending on ambient temperature and air humidity ( Veldre, K. et al., Eur. Journ. Pharm. 44, 273-280 (2011)). At 80 % air humidity and room temperature the monohydrate is reversibly formed. The theoretical water content of detomidine hydrochloride monohydrate amounts to 7.48 %.
[0006] To date, all commercially available (i.e. veterinary) drug products (i.e. parenteral solutions and oromucosal gels) only contain the anhydrous form. In general, hygroscopic substances like detomidine hydrochloride tend to absorb moisture so that they have to be protected from a humid environment during production and storage of the drug substance and corresponding drug product to avoid an inacceptable uptake of water. It has to be noted that such uptake during storage will reduce the content of the drug substance so that this would have to be taken into consideration during production of the corresponding drug product, like pharmaceutical preparation.
[0007] The problem to be solved is to provide a pure and stable active pharmaceutical ingredient (API), namely detomidine hydrochloride monohydrate, that can advantageously be used for the production of pharmaceutical compositions comprising the active pharmaceutical ingredient detomidine hydrochloride.
Example 1
Preparation of detomidine hydrochloride monohydrate (DHM)
[0053] Detomidine hydrochloride was synthesized starting from 1-benzyl-imidazole-4-carboxyaldehyde and 2,3-dimethylphenlymagnesiumbromide according to the two-step synthesis described in Drugs Future 10, 17 (1985).
[0054] For the second step of this synthesis (RS)-(3-Benzyl-3 H-imidazol-4-yl)-(2,3-dimethyl-phenyl)-methanol (HCl) was suspended in a mixture of water and hydrochloric acid. The catalyst (i. e. palladium on activated carbon) suspended in demineralized water was added. Hydrogenation (i.e. removal of the benzyl group and reduction of the hydroxyl group with hydrogen (H 2/Pd-C in HCl)) was performed at elevated temperature (50 – 80 °C) and the obtained suspension was filtered after the hydrogenation was finished. Subsequently ethyl acetate and a solution of ammonium hydroxide were added under continuous stirring. After discontinuation of stirring, phase separation occured after which the aqueous phase was repeatedly extracted with ethyl acetate. The combined organic phases were washed with demineralized water and filtered.
[0055] After addition of 5 – 6 N hydrogen chloride in 2-propanol and cooling precipitation of detomidine hydrochloride occured. After filtration the filtercake (i.e. raw product) was washed with ethyl acetate and dried.
[0056] A fraction of the resulting raw product (i.e. 5 g batch RSO E-190604 RP) was recrystallized from 5 g demineralized water by heating (until complete dissolution was obtained) and subsequent cooling on an ice bath. The resulting crystals were separated by filtration and the resulting filter cake washed with 2-propanol. Subsequently, the washed product was dried under vacuum (10 mbar) at 23 °C. The obtained yield for the white crystalline substance amounted to 66.0 % of the theory.
[0057] The resulting drug substance showed a water content (KF) of 7.49 %. The corresponding DSC curve was in line with the expectation (see for example Figure 1) and showed the two typical peaks routinely observed for DHM. Other than 2-propanol used for final washing none of the other solvents employed during the overall synthesis of this compound were found above the respective LOQ by GC-FID.
Example 2
Impurities after preparation of detomidine hydrochloride monohydrate (DHM)
[0058] A larger batch of detomidine hydrochloride (i.e. 50 g NK E-190709-I A K1) was synthesized in line with Example 1. However, the final crystals obtained after recrystallization from 50 ml demineralized water were washed with 25 ml demineralized water instead of 2-propanol. Drying was performed at 21 °C and 10 mbar until constant weight. The obtained yield for the white crystalline substance amounted to 87.2 % of the theory which was markedly higher than the yield obtained in Example 1. The water content of this substance was determined at 7.54 % (KF) and the corresponding DSC curve showed two peaks with an onset at 95.7 °C and 159.3 °C.
[0059] As shown below, recrystallization of the initial raw product from water (incl. washing) resulted in significant removal/reduction of impurities eluting before the detomidine peak (i.e. more polar compounds, e.g. Impurity A) as well as impurities eluting behind the detomidine peak (i.e. less polar compounds, e.g. Impurity C).
| Sample | Relevant compounds as detected by HPLC [area%]* | ||||
| Impurity A | Impurity RRT 0.84 | Detomidine | Impurity RRT 1.75 | Impurity C | |
| Raw product | 0.11 | 0.33 | 99.40 | 0.04 | 0.04 |
| Final crystallizate (K1) | 0.06 | 0.06 | 99.80 | 0.01 | 0.02 |
| *Table includes all compounds found at or above 0.04 area% in the initial raw product in the order in which they eluted from the HPLC column |
[0060] The final substance showed a very high HPLC purity of 99.80 area% (Ph. Eur. test method) and only a limited number of unknown impurities in addition to those
PATENT
https://patents.google.com/patent/WO2006108910A1/en
Example 1. Preparation of 4-[(2,3-dimethylbenzyl)]imidazole hydrochloride
(detomidine HCl)
l-Benzyl-5-(2,3-dimethylphenylhydroxymethyl)imidazole (20 kg), water (225 1), 30 % HCl (20 1), ethanol (5 1) and palladium on charcoal 10 % (4.4 kg) are charged. The mixture is stirred under 2.2 bar overpressure of hydrogen at 75 ± 5 °C for 24 hours. The reaction mixture is filtered at 45 ± 3 0C and the filter cake is washed with water (30 1). 170 1 of water is distilled off under reduced pressure and 30 % HCl (8 1) is added. The solution is cooled to 3 ± 3 0C during 2 h. The solution is seeded with crystals of detomidine HCl at 40 ± 5 °C, 30 ± 5 0C, 20 ± 5 °C and at 10 ± 5 0C, until the crystallization starts. The mixture is agitated for two hours. The crystalline compound is collected by centrifugation and washed with toluene. The crude product and water (250 1) are charged. The solution is heated to about 50 °C and stirred for 1 hour. The solution is cooled to 10 °C during 1.5 hour. The solution is filtered and 180 1 of water is distilled off under vacuum. 30 % HCl (20 1) is added and the solution is warmed to 60 0C, and then cooled to 3 ± 3 °C during 2 hours. The solution is seeded as above until the crystallization starts and agitated for two hours. The crystalline compound is collected by centrifogation and washed with toluene. The product is dried under vacuum at 39 ± 5 °C for 20 hours, at 61 ± 5 °C for 6 hours and at 85 ± 5 °C for 16 hours. The yield is 10.5 kg (78 %).
PATENT
https://patents.google.com/patent/US20080287685A1/en
- Detomidine which is 4-[(2,3-dimethylbenzyl)]imidazole of formula I
- is a well known pharmaceutical agent currently used as its hydrochloride salt in animal sedation.
- [0003]The synthesis of detomidine is described in U.S. Pat. Nos. 4,443,466 and 4,584,383. The preparation of detomidine hydrochloride salt is described in U.S. Pat. No. 4,584,383, wherein detomidine obtained from the hydrogenation step is first recovered from alkaline solution as a free base after which the crystalline product is converted into its hydrochloride salt by treatment with HCl-isopropanol in ethyl acetate.
- [0020]1-Benzyl-5-(2,3-dimethylphenylhydroxymethyl)imidazole (20 kg), water (225 l), 30% HCl (20 l), ethanol (5 l) and palladium on charcoal 10% (4.4 kg) are charged. The mixture is stirred under 2.2 bar overpressure of hydrogen at 75±5° C. for 24 hours. The reaction mixture is filtered at 45±3° C. and the filter cake is washed with water (30 l). 170 l of water is distilled off under reduced pressure and 30% HCl (8 l) is added. The solution is cooled to 3±3° C. during 2 h. The solution is seeded with crystals of detomidine HCl at 40±5° C., 30±5° C., 20±5° C. and at 10±5° C., until the crystallization starts. The mixture is agitated for two hours. The crystalline compound is collected by centrifugation and washed with toluene. The crude product and water (250 l) are charged. The solution is heated to about 50° C. and stirred for 1 hour. The solution is cooled to 10° C. during 1.5 hour. The solution is filtered and 180 l of water is distilled off under vacuum. 30% HCl (20 l) is added and the solution is warmed to 60° C., and then cooled to 3±3° C. during 2 hours. The solution is seeded as above until the crystallization starts and agitated for two hours. The crystalline compound is collected by centrifugation and washed with toluene. The product is dried under vacuum at 39±5° C. for 20 hours, at 61±5° C. for 6 hours and at 85±5° C. for 16 hours. The yield is 10.5 kg (78%).
PATENT
https://patents.google.com/patent/WO2020016827A1/en
Detomidine
Detomidine, 4-[(2,3-dimethylphenyl)methyl]-lH-Imidazole, is an a-2-andregenic agonist available under the brand name Equimidine® and Dormosedan® for use as a veterinary sedative. Detomidine is not currently approved for human use.
Detomidine and related compounds, including its 3,4 dimethyl isomer, iso-detomidine (4-(3,4- Dimethylbenzyl)-lH-imidazole) were first described in US4,443,466. Both the‘466 patent and the later US4, 584,383 describe the synthetic method of manufacturing detomidine as being based on coupling of an imidazole moiety with l-Bromo-2, 3-dimethyl benzene using a Grignard reaction. RU2448095 describes an alternative route of synthesis of the molecule based on coupling in presence of a Titanium catalyst. According to both the‘383 and‘095 patents, detomidine is purified by crystallization of its hydrochloride salt from water. The chemical structures of detomidine HC1 and iso-detomidine are shown below:

Detomidine HC1 Iso-detomidine
Two solid state forms of detomidine HC1 are known, the anhydrous and monohydrate forms.
Synthesis of the anhydrous form by crystallization of the monohydrate and further decomposition at elevated temperatures is described in US7,728,l47. Synthesis of the anhydrous form via decomposition of the monohydrate in reduced pressure is described in Laine et al (1983). According to Veldre et al (2011), the anhydrous and monohydrate forms of detomidine HC1 can easily interconvert depending on temperature and humidity.
The European Pharmacopeia 9.0 monograph (January 2014) describes detomidine HC1 for veterinary use. The monograph lists the established HPLC method for identification of detomidine and its impurities as using a Symmetry C8, 5 pm, 4.6 x 150 mm column, with a mobile phase of Ammonium phosphate buffer pH 7.9 – 65% and Acetonitrile – 35% at a flow rate of 1.0 mL/min and UV detection at 220 nm. That procedure is listed as recording three distinct impurities of detomidine:
Impurity A: (RS)-(2, 3 -dimethylphenyl)(l/f-imidazol-4-yl)m ethanol
– l/f-imidazol-5-yl)(2,3-dimethylphenyl)m ethanol

Impurity C: 4-| (2.3 -dimcthy ley clohcxyl)m ethyl |- 1 /7-im ida/olc

PCT/US18/012579 discloses topical formulations of detomidine and their uses in treating pain.
Purified detomidine for use in human pharmaceutical formulations is not known in the art.
EXAMPLE 5: Purification of organic impurities from detomidine HC1 monohvdrate
Two potential procedures for purification of organic impurities from sourced monohydrate were compared. The first attempted procedure was by direct re-crystallization of detomidine HC1 from 2.88 volumes of water, while the second included carbon treatment and precipitation of detomidine free base followed by the free base being reacted with HC1 and crystallized as monohydrate. Both procedures used the same non-GMP, off white anhydrous detomidine HC1 starting material which had previously been shown in Table 7 to contain 0.21% of iso-detomidine and 0.07% of Impurity A. All the re-crystallized materials were found to have practically the same purity level. The direct re-crystallization procedure was found to provide a product with a high yield and purity and at the same time provides a practical and scalable crystallization process which could be controlled by process parameters such as seeding and cooling rate.
Example 5 a: Direct recrvstallization
Anhydrous detomidine HC1 (4.5g) was introduced to a round-bottom flask with a magnetic stirrer and thermometer. Deionized water (l3ml) was then added and the mixture stirred and heated in a water bath. At 39°C, the complete dissolution of solids was observed, providing a clear yellow solution with a pH = 4.
The batch was gradually cooled by stirring. At 3 l°C, intensive crystallization was observed. The resulting slurry was cooled in an ice-water bath for 20 min and filtered. Flask and cake were then washed with 2 ml of cold deionized water and 3.97g of a white to cream colored solid was collected. 2.03g of the material was dried in a vacuum desiccator at ambient temperature and 20 mbar to a constant weight over 23 hrs producing a dry monohydrate – l .96g off-white crystalline solid (sample 1).
An additional l .9 lg of the material was dried in a vacuum oven at 90°C under house vacuum to a constant weight over about 24.5 hrs producing a dry anhydrate , l .68g off-white solid (sample 2)
The two samples were subjected to physical characterization and purity analysis by HPLC. The XRPD spectra and DSC and TGA thermograms of sample 1 are presented in Figures 8 -10 and of sample 2 are presented in Figures 11-13, respectively.
As shown in Table 11, direct re-crystallization resulted in the effective purification from all organic impurities, but was not effective for color. The content of iso-detomidine and of Impurity A was reduced to a level below the QL, but the off white color remained after re-crystallization.
Table 11 : properties following direct recrystallization (sample 1)

1 – below the QL
2 – system peak
Example 5b(i): Carbon treatment and detomidine free base isolation
Anhydrous detomidine HC1 (70.3g) and deionized water (220ml) were introduced to a 0.5 liter jacketed glass reactor equipped with a mechanical stirrer, thermocoupler and a circulating oil bath for heating and cooling.
The mixture was heated while stirring. At 40°C, complete dissolution was observed. Active carbon (CXV type, 5.2g) was added to the clear yellow solution and the batch stirred at 45°C for 50 minutes. Following this, the batch was filtered on through paper filter on Buchner funnel, reactor and filter washed with deionized water (20ml).
The slightly yellowish clear filtrate was reintroduced to the 0.5 liter reactor, stirred and 40% NaOH solution was added at 40°C. After 10ml NaOH solution was added, a pH of 7 was reached and precipitation began. An additional 13ml of NaOH was added over 1 hour at 42 – 52°C and intensive stirring (400 – 450 rpm) performed. The mixture at the end of the addition of NaOH had a pH of 13.
The batch was stirred at 33 – 35°C overnight then cooled to l6°C over 4 hours and stirred at this temperature for an additional hour. The resultant solid was filtered on Buchner filter, reactor and cake washed with two portions of deionized water (2><200ml). The wet solid (86g) was dried in a vacuum oven at 45°C to constant weight to produce a dry product (53.2g, Yield 90.7%) – white powder, m.p.=l 18.6 – 119.2
The dry detomidine base was analyzed for purity by HPLC, the results presented in Table 12. Table 12: Properties of detomidine base (intermediate in sample 2)

1 – system peak
Example 5b(nT Monohvdrate crystallization from detomidine base
The dry detomidine free base (53.0g) from Example 5b(i) was introduced together with 37% HC1 (29.7g) and deionized water (159g) into a 0.5 liter jacketed glass reactor equipped with a mechanical stirrer, a thermocoupler and a circulating oil bath for heating and cooling. The batch was stirred and heated to 45°C, at 37°C complete dissolution of solid was observed. The clear solution had a pH of 1. The solution was cooled gradually to 37°C and seeded with detomidine HC1 monohydrate and cooled gradually to 3°C over 4 hours, and then the batch was stirred for 45 minutes at this temperature. The solid was filtered on Buchner filter, reactor and cake washed with cold deionized water (80ml). The wet solid (61.9g) was dried in vacuum oven for 16 hours at 45°C to produce a dry product (57.8g, Yield 84.3%) – white crystalline powder (sample 2)
The dry detomidine HC1 monohydrate was analyzed for water by CKF (¾0 = 7.46%) and for purity by HPLC with the results presented in Table 13. Microscopic observation for particle morphology (regular prisms) was performed and the microscopic photograph is shown in Figure
14.
Table 13 : Properties of detomidine HC1 (sample 2)

1 – system peak
Example 5c: Re-crvstallization of detomidine HC1 to monohvdrate. bench scale experiment Anhydrous detomidine HC1 (754.6g) 37% HC1 (116. Og) and deionized water (2008g) were introduced to a 3 liter glass jacketed reactor equipped with a mechanical stirrer, two baffles, a thermocoupler and a circulating oil bath for heating and cooling. The batch was stirred and heated to 52°C, at 47°C complete dissolution was observed and the clear solution was found to have a pH of 0-0.5.
The solution was cooled gradually and at 45°C seeded with detomidine HC1 monohydrate (0.5g). Crystallization initiation was observed at 43°C and the batch was then cooled to 1.5°C during 5 hours and stirred for 12 hours at this temperature. The solid was filtered on Buchner filter and conditioned on the filter with vacuum for 40 minutes. The wet product (817g) was dried in vacuum oven to constant weight. For the first 13 hours, the material was dried at 30°C and 35-27 mbar, then for an additional 7 hours at 40°C and 30-18 mbar to produce a dry product (771.2g, Yield 94.6%) – white crystalline powder (Batch“90” in Tables 8-9; sample 3)
Dry detomidine HC1 monohydrate was analyzed for water by CKF (FhO = 7.37%) and for purity by HPLC, the results presented in Table 14. The physical characterization results are shown in Table 10 above.
The material was subjected to physical characterization and microscopic observation for particle morphology (regular prisms) microscopic photograph presented in Figure 7.
Table 14: Properties of detomidine HC1 (sample 3)
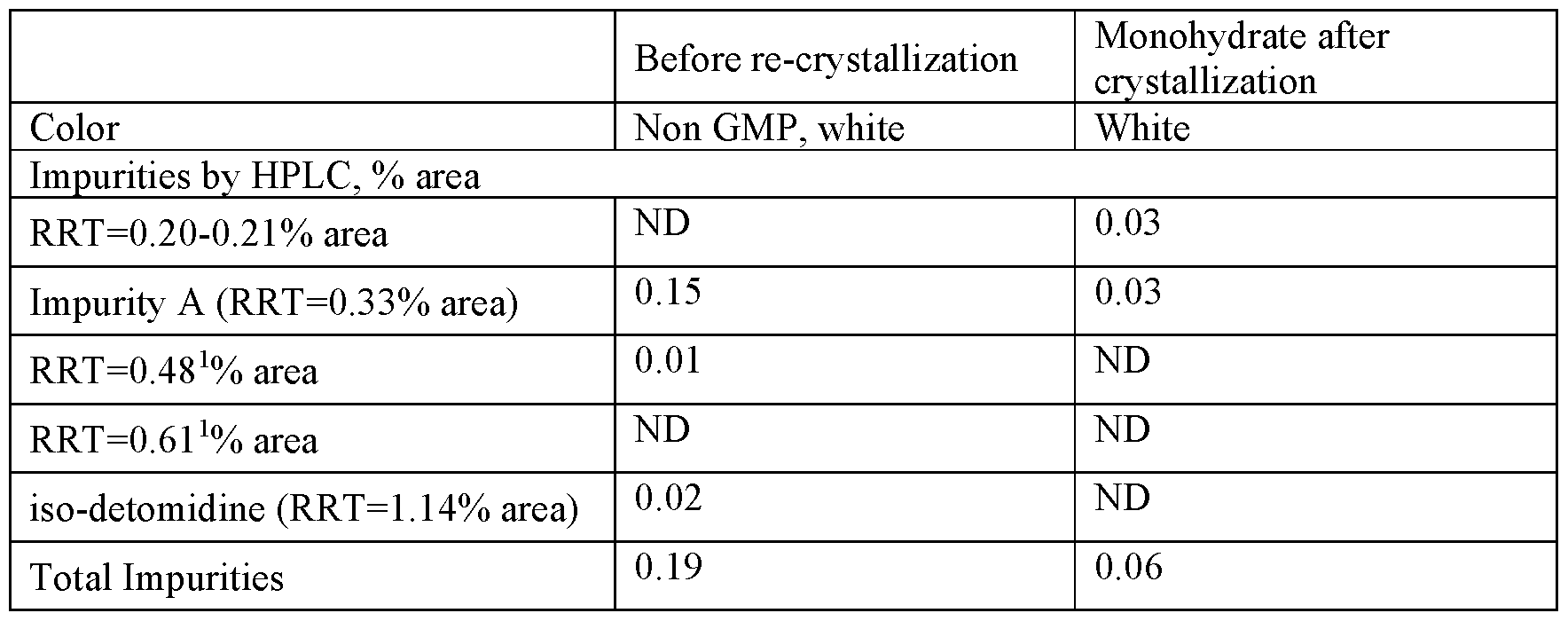
1 – system peak
EXAMPLE 6: Synthesis of iso -detomidine
Scheme 1 outlines a process for the synthesis of iso-detomidine was developed to produce a solid iso-detomidine HC1 in high yield and substantially free of impurities.

Scheme 1 : Route of synthesis of iso-detomidine
Example 6a: Sandmever Reaction
3,4 dimethyl aniline (150g, 1.24M) was mixed with acetonitrile (0.6 liter) in a 5 liter flask, chilled to lO°C and water (1.2 liter) added dropwise over 5 minutes. The mixture was cooled to 5°C with ice-ethanol bath and concentrated H2SO4 (98% wt, 363g 3.71M) was added dropwise over 30 min at 5-l0°C. Sodium nitrite (NaNC ) aqueous solution (89.7g in 300 ml water, 1.30M) was then added dropwise over 30 min at 0-5°C to give a brown solution. The resulting solution of diazonium salt was stirred at 0-5°C for an additional 30 min.
In another 5 liter flask KI (225g, 1.36M) was dissolved in water (0.8 liter) during stirring and cooled. The diazonium salt solution was added dropwise to the KI solution at 7-l3°C during 35 min, the batch stirred at 7-l3°C for 1.25 hr to give a black solution. MTBE (2.0 liter) was then added to the reaction mixture and Na2SC>4 (23.4g) was introduced in small portions during 5 min.
The mixture was settled and the organic phase separated and washed with two portions of brine (2 500ml). The organic solution was concentrated under vacuum to a volume of about 250ml.
The product was purified by vacuum distillation at ca. 40Pa, BP = 52 – 60°C to give 246g of intermediate 1 as a brown oil with a product yield of 86%.
Example 6b: TRT protection reaction
lH-Imidazole-4-carbaldehyde (45.2g, 0.47M) and acetonitrile (0.8 liter) are introduced into a 2 liter flack and cooled to 8°C, then TRT-C1 (131. Og, 0.47M) was added at 8°C and TEA (57. lg, 0.56M) was added dropwise during 20 min. The reaction mixture was stirred at 8 to l8°C for 2 hrs.
The reaction mixture was poured into a stirring mixture of water (0.72 liter) and MTBE (0.72 liter) and stirred for 10 minutes. The resulting solid was isolated by filtration on Buchner funnel and dissolved with THF (3 liter). The solution was dried over Na2SC>4 and concentrated to remove most of the solvent.
MTBE (400 ml) and PE (200ml) was added to the residue, the mixture stirred at 8°C for 16 hrs. The precipitated solid was isolated by filtration on Buchner filter and dried in air for 16 hrs at room temperature. Then the filter cake is dried by azeotropic drying with 2-Me-THF (2×500 ml) to give l29g of intermediate 2 as white solid with a yield of 66.5%.
Example 6c: Grignard reaction
A 2M solution of i-PrMgCl in THF (0.275 liter, 0.55M) and THF (1.0 liter) was introduced to a 2 liter flask at l2°C. Intermediate 1 (121.8g, 0.525M) was added dropwise during 20 min. The mixture was stirred at l2-l5°C for 3 hrs.
Intermediate 2 (84.6g, 0.25M) was added in small portions without cooling during 30 min, with a temperature rise to 25°C, to give a light brown solution. The solution was stirred for 2.5hrs at l5°C and added to aqueous solution of NH4CI (117g in 0.7 liter water) during 10 min at 5°C. PE (1.6 liter) was added during 5 min and the mixture stirred for extra 25 min.
Precipitated solid filtered on Buchner funnel and then re-slurred with mixture of MTBE (400 ml), water (600 ml) and PE (200 ml). Then the solid was filtered on Buchner funnel and re-slurred with MeOH (700 ml) at 60°C for 10 min, cooled to 20°C with cold water bath and filtered again on Buchner funnel. The solid product was dried in an air oven at 45 °C for 2 hrs to give 112 g of intermediate 3 as a white solid with a yield of 89.9%.
Example 6d: Reductive dehvdroxylation and de-protection
Intermediate 3 (l07g, 0.240M) and DCM (1.10 liter) were introduced to a 2 liter flask at 1 l°C, TFA (214 ml) was added dropwise over 5 mins with a temperature rise to l4°C.
The mixture was stirred for about 5 mins and EhSiH (94.4g, 0.794M) added dropwise during 5 mins. After stirring at 25-30°C for 16 hrs the mixture was concentrated by rotary evaporation at 40°C to a residue.
The residue of evaporation was dissolved in DCM (600 ml) and washed with 1.5M aq. HC1 (0.241iter). Organic phase was separated and washed with aq. NaOH (11.5g in 200ml water), pH of aqueous phase 13. Two phases were separated and the organic phase washed with brine (200 ml) dried over Na2S04 and filtered. The resulting solution was concentrated by rotary evaporation.
The evaporation residue was dissolved in mixture of EtOAc (500 ml) and EtOH (30 ml) and then 4M HC1 solution in dioxane (40 ml) was added dropwise in 5 minutes, pH = 1 – 2 adjusted and a white solid precipitated out.
The solid product was filtered on Buchner funnel, the cake dried in air for 16 hrs to give 36g of white solid.
The solid product was re-crystallized from iPrOH / Acetone. The dry cake (36g) and iPrOH were introduced into a 1 liter flask and heated to dissolution. Acetone (360 ml) was added to the resulting colorless solution at reflux during 10 mins. The mixture was cooled to 8°C and stirred at this temperature for additional 4.5 hrs. The solid product was filtered on Buchner funnel and dried in air for 36 hrs. 29.2g of iso-detomidine as a white solid was obtained with a yield of 54.4%. The 1H-NMR spectra of iso-detomidine is shown in Figure 15. EXAMPLE 7 : Re-crvstallization of detomidine HC1 spiked with 2% iso-detomidine
Detomidine HC1 monohydrate (26. Og), iso-detomidine HC1 (0.52g) and deionized water (68.7g) were introduced to a 100 ml glass jacketed reactor equipped with a mechanical stirrer, a thermocouple and a circulating oil bath for heating and cooling. The batch was stirred and heated to 51°C, at 47°C complete dissolution was observed.
The solution was cooled gradually and at 42°C seeded with detomidine HC1 monohydrate. Crystallization initiation was observed at 39°C and then the batch was cooled to 3°C for 5 hours, filtered on Buchner filter and conditioned on the filter with vacuum. The wet product (20.7 g) was dried in vacuum oven to constant weight to produce a dry product (20.13g, Yield 75.9%) – white crystalline powder
Dry detomidine HC1 monohydrate was analyzed for PSD and morphology, the results are presented in Table 8 (Sample. No. 91). The purity of re-crystallized material was analyzed using the optimized HPLC process disclosed herein, and the results are presented in Table 15.
Table 15 : Properties of detomidine HC1 following recrystallization from iso-detomidine spiked material

a area %
b Spiked amount, calculated
References
- ^ Clarke, Kathy W.; Hall, Leslie W.; Trim, Cynthia M., eds. (2014). “Principles of sedation, anticholinergic agents, and principles of premedication”. Veterinary Anaesthesia. pp. 79–100. doi:10.1016/B978-0-7020-2793-2.00004-9. ISBN 978-0-7020-2793-2.
- ^ England GC, Clarke KW (November 1996). “Alpha 2 adrenoceptor agonists in the horse–a review”. The British Veterinary Journal. 152 (6): 641–57. doi:10.1016/S0007-1935(96)80118-7. PMID 8979422.
- ^ Fornai F, Blandizzi C, del Tacca M (1990). “Central alpha-2 adrenoceptors regulate central and peripheral functions”. Pharmacological Research. 22 (5): 541–54. doi:10.1016/S1043-6618(05)80046-5. PMID 2177556.
External links
- “Medication Protocols for Horses”. The Ontario Association of Equine Practitioners. 2005. Archived from the original on 2007-04-17.
- Compendium of data sheets for animal medicines. National Office of Animal Health. 2005. ISBN 978-0-9548037-0-4.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| ATCvet code | QN05CM90 (WHO) |
| Legal status | |
| Legal status | Veterinary use only |
| Pharmacokinetic data | |
| Elimination half-life | 30 min |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 76631-46-4 |
| PubChem CID | 56032 |
| ChemSpider | 50586 |
| UNII | 7N8K34P2XH |
| KEGG | D07795 |
| ChEMBL | ChEMBL2110829 |
| CompTox Dashboard (EPA) | DTXSID00227457 |
| Chemical and physical data | |
| Formula | C12H14N2 |
| Molar mass | 186.258 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESCc2cccc(Cc1cnc[nH]1)c2C | |
| hideInChIInChI=1S/C12H14N2/c1-9-4-3-5-11(10(9)2)6-12-7-13-8-14-12/h3-5,7-8H,6H2,1-2H3,(H,13,14) Key:RHDJRPPFURBGLQ-UHFFFAOYSA-N |
////////////// DETOMIDINE, UNII-7N8K34P2XH , детомидин ,ديتوميدين, 地托咪定 , Domosedan, Farmos, SEDATIVE
#DETOMIDINE, #UNII-7N8K34P2XH , #детомидин ,#ديتوميدين, #地托咪定 , #Domosedan, #Farmos, #SEDATIVE
https://patents.google.com/patent/WO2020016827A1/en
EXAMPLES
EXAMPLE 1 : Elemental analysis of impurities found in commercially available anhydrous detomidine HC1
Example la: Anhydrous detomidine HC1 was sourced from two commercial API suppliers. Properties of the commercial batches, GMP1, GMP2 and GMP3, are presented below.
Elemental impurity analysis was performed by inductively coupled plasma mass spectrometry (ICP-MS) on four different batches of sourced anhydrate. The results of the analysis are found in Table 1.
Table 1 : Elemental impurities in anhydrous detomidine HC1
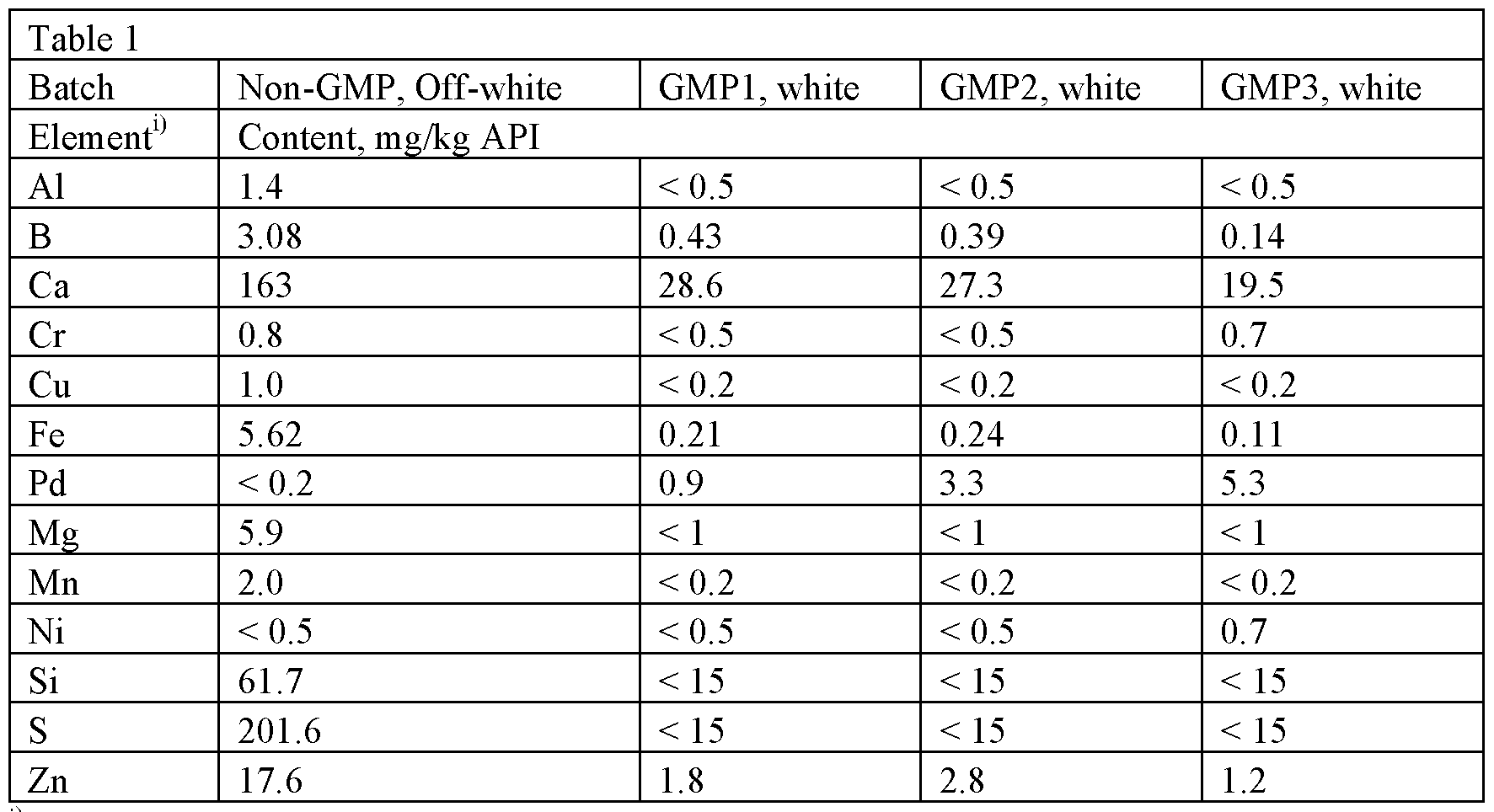
11 Elements having levels L.T. 0.5 mg/kg (Ti, As, Hg, Pb, Mo, Pt, etc) are not presented in the table
The screening of elemental impurities shows that the GMP products contained significant levels of Pd (0.9 – 5.3 mg/kg). Pd is understood to be a catalyst used in the synthesis of detomidine (e.g., in reduction/hydrogenation methods).
Example lb: Characterization of commercially sourced material
Samples of the anhydrous detomidine products described in Table 1 were analyzed for water content and characterized by microscope, XRPD and thermal analyses. The results are summarized in Table 2.
Table 2: Characterization of commercial anhydrous detomidine HC1

a Anhydrous + mono hydrate The values presented in T able 2 demonstrate that the commercial samples of detomidine HC1 labeled as anhydrous contain some amount of monohydrate and this amount varied depending on storage conditions and packaging.
EXAMPLE 2: Stability assessment of anhvdrate and monohvdrate forms of detomidine base and detomidine HC1
Pure forms of crystalline free base, and HC1 salt (both monohydrate and anhydrate) were prepared from commercially sourced anhydrous detomidine HC1 as outlined in Table 3, and characterized using XRPD and thermal analysis. The solids were crystallized from aqueous solutions and then dried under different conditions. The crystallization and drying conditions are summarized in Table 3.
Table 3: Preparation of detomidine HC1 crystalline forms

The properties of the solids crystallized according to Table 3 are described in Table 4.
Table 4: Properties of Detomidine HC1 crystalline forms


These results demonstrate that crystallization from 2.8 – 2.9 volumes of water is effective for isolation and purification of the detomidine HC1 monohydrate drug substance. Drying of the monohydrate under mild conditions (20-40 mbar and temperatures from at least ambient to about 45 °C) provided pure monohydrate without traces of the anhydrous form.
The same monohydrate dried at elevated temperature (30-40 mbar 90°C) converted completely into the anhydrous form. The vacuum dried, hermetically closed anhydrate did not absorb water from the atmosphere and did not convert into the monohydrate. After exposure to atmospheric air, however, the anhydrate absorbed water and converted to a mixture of anhydrate and
monohydrate.
Melting points (m.p.) of the intermediate detomidine free base and hydrochloride of Sample 5 measured in open capillary corresponded with the published literature and the DSC data and are presented in Table 5. In order to evaluate effect of humidity on different forms of detomidine, a hydration study was performed. Samples of detomidine free base and hydrochloride salt were subjected to DVS analysis. These observations are in accordance with the DVS results shown in Figures 5 and 6, for detomidine free base and detomidine HC1, respectively.
Table 5: Composition and properties of known solid forms of detomidine



a -literature data
The free base was found to be crystalline and insoluble in water but it reacted readily with aqueous HC1 giving soluble detomidine hydrochloride.
Crystallization from water provided effective purification of the detomidine HC1 and formation of large regular crystals. Anhydrous detomidine hydrochloride appeared as small irregular particles whereas the possibility to control particle size distribution by crystallization parameters existed for the monohydrate.
The detomidine free base was found to be non-hygroscopic, but also able to absorb more than 1% of water at relative humidity (RH) >50%. An increase of humidity from RH 70% to RH >90% did not lead to absorption of additional water to monohydrate. During the dehydration cycle, the monohydrate began to lose water at RH -10% and converted into the anhydrate at RH =0%. Anhydrate did not absorb water at RH <30% and transformed completely to into the monohydrate at RH between 30% and 50%.
Four cycles of hydration-dehydration demonstrated good reproducibility of anhydrate- monohydrate interconversion.
An anhydrous detomidine HC1 of Sample 2 was shown to absorb water to a level of cKF 7.7% which corresponds well to the theoretical amount of water in the monohydrate form (Table 5).
The hydration profile of detomidine hydrochloride showed that the monohydrate is stable in a wide range of humidity between 10% and >90% RH. At the same time, the anhydrous form is not stable in atmospheric air and absorbs water at RH = 30 – 50%.
This data demonstrates that the anhydrous form is challenging in the aspects of water content and solid form stability and that detomidine HC1 monohydrate is more suitable for pharmaceutical development.
Example 3 : Impurity analysis of commercially sourced detomidine HC1
Using the established Pharmacopeia HPLC protocol (Symmetry C8, 5 pm, 4.6 x 150 mm column, with a mobile phase of 65% Ammonium phosphate buffer pH 7.9 and 35% Acetonitrile at a flow rate of 1.0 mL/min and UV detection at 220 nm), sourced samples of detomidine HC1 were assayed for impurities. As shown in Figure 1, a previously unreported peak was identified, which partially overlapped with that of detomidine. By LC-MS/MS analysis, this impurity was shown to have the same molecular weight as detomidine.
The established Pharmacopeia HPLC protocol did not separate the detomidine from the impurity. Therefore, for further identification of the elusive impurity, new HPLC protocols for assaying detomidine HC1 were developed. One protocol (“HPLC Protocol A”) comprised using a SunFire C8 column, IOqA, 3.5 pm, 4.6 x l50mm column with an initial mobile phase of 70% Ammonium Phosphate buffer solution, pH 7.9 and 30% Acetonitrile, at a flow rate of 1.0 mL/min and UV detection at 220 nm. To remove late eluting peaks, the flush gradient shown in Table 6 was applied after each run. This HPLC protocol allowed for a resolution factor of 3.9 between detomidine and the unidentified impurity. The quantitation level (QL) for impurities and degradation products is 0.025%. The detection level (DL) for impurities and degradation products is 0.01%.
Table 6: Flush gradient for HPLC protocol

Given its molecular weight, it was hypothesized that the impurity was iso-detomidine.
A solution of 100 pg/ml detomidine HC1 and about 1 pg/mL (about 1% of the working concentration) of detomidine impurity A and iso-detomidine were prepared and assayed using the new HPLC protocol (HPLC Protocol A), disclosed hereinabove. Figure 2 is a chromatogram showing that the previously unreported peak is confirmed as being iso-detomidine.
The analysis of commercially sourced detomidine HC1 revealed a significant additional impurity. Table 7 provides levels of the various detomidine impurities in different commercial batches. In all batches, total impurities were observed at levels of > 0.1% area.
Table 7: Impurity levels (% area) in commercial batches of detomidine.


provided by commercial supplier after undergoing the reciystallization process of Example 5, provided by inventors.
Further analysis of the peak at RRT=0.38 showed that it actually consisted of 2 separate, overlapping peaks. As shown in Figure 3, LC-MS/MS analysis confirmed one of these peaks as iso-impurity A. Further analysis, as shown in Figure 4, identified the second peak as (2,3- dimcthylphcnylX 1 //-imidazol-4-yl) methanone.
EXAMPLE 4: Optimization of the crystallization method of detomidine HC1 monohvdrate from commercial batches of anhydrous detomidine HC1
Crystallization experiments on 25, 65, and 770 gram scale were performed in 100 ml, 500 ml and 3 liter jacketed glass reactors, respectively, equipped with CBT (curved blade turbine) mechanical stirrers, circulating oil bath, thermocouples, and condensers. Stirrer speed in all experiments was between 300 – 600 rpm. Variable process parameters were: amounts of HC1, solvent ratio, cooling time/rate, seeding and cake wash. The parameters and the variation ranges were chosen according to production conditions. The crystallization parameters are summarized in Table 8.
Table 8: Crystallization parameters

a Seeding with detomidine HC1 monohydrate
b Time 24 hrs
c Seeding with anhydrous detomidine HC1
d 5.5 hrs cooling and overnight stirring at 1-3° C
e Spiked with 2% iso -detomidine
The drying parameters and solid properties of batches shown in Table 8 are described in Table 9. Table 9: Drying parameters and solid properties of detomidine monohydrate crystals

microscopic observation: Rods – aspect ratio > 2; prisms – aspect ratio < 2
u)M = mono hydrate
The data presented in Tables 8 and 9 demonstrate that crystallization from water and drying under technical vacuum gives pure detomidine HC1 monohydrate without traces of the detomidine HC1 anhydrous form. Variations of HC1 excess from 0 to 0.5 mole/mole base, cooling time from 1.5 to 24 hours and drying time from 15 to 33 hours appear to have no effect on the obtained properties of the solid form. All crystallization products appeared as pure detomidine HC1 monohydrate.
The crystallization initiation method also had no effect on crystalline form. The batches seeded with anhydrous material gave the same monohydrate as batches seeded with monohydrate and batches which crystallized spontaneously.
Contact with water for 24 hrs completely converted the anhydrous form into the monohydrate, even without complete dissolution (re-slurry).
Crystallization of the monohydrate from water gave large clear crystalline particles with a mean crystal size 0.3 – 0.7 mm, with some crystals larger than 2 mm in size. The shape of the crystals was rod-like or prism-like, if the aspect ratio of the crystals was < 2 the crystals were reported in Table 8 as prisms. A ratio of HC1 to base within the range 1.0 – 1.5 mole : mole and water to solid ratio within the range 2.1 – 2.8 V/wt were found to have no significant effect on the particle size distribution (PSD). However, a ratio of HC1 to base of about 1.5 were found to increase yields of highly pure detomidine HC1 monohydrate from under 90% (60.8%-86.4%) to over 90% (9l .4%-95.9%). Seeding also appeared to have no significant effect on PSD.
The cooling rate was found to have a weak effect on PSD. There was no effect observed for cooling over a time range between 1.5 and 5.5 hrs (mean cooling rate 0.10-0.3 l°C/min).
Slurry -to-slurry recrystallization of anhydrous material resulted in a strong reduction in particle size with the d(0.5) decreasing from 300-500m to 87m. These crystals were found irregular with no signs of prism-like or rod-like habit. In contrast, the re-slurry procedure applied to a mixture of anhydrate and monohydrate (15:85) gave a mixture of rod and prism-like crystals with d(0.5)=4l5p.
Batch size was found to have no significant effect on crystal size and shape. After scaling up from a 26g batch in 100 ml reactor to 770g in a 3 liter reactor, the PSD was very similar to that of small scale batches.
Prolonged cooling resulted in a “rounded” form of crystals. This effect was observed in two experiments, as seen in the microscopic photograph in Figure 7. In the first experiment the crystallizing suspension was cooled for 8 hrs, and in the second one it was stirred at low temperature for 12 hrs (batches 83 and 90 in Tables 8 and 9).
Under the conditions described, cooling had a strong effect on the process yield. Two re-slurry experiments were performed at the same water volume ratio as most of experiments (2.80 V/wt) but these two batches were not cooled and filtered at 24°C. In these experiments the yield dropped from 86% to 60-65% (batches 84, 85 in Tables 8 and 9).
Acceptable yields were obtained in cooled batches within the solvent volume ratio range 2.1 – 2.8 V/wt with the cooling temperature between about l.5°C – 4°C
An increase of HC1 to base molar ratio from 1 to 1.5 was found to raise the yield from 86% to 95%. Cake wash reduced the yield by 2 – 3%. Re-crystallization in presence of 2% iso- detomidine reduced the yield from 84 – 85% to 76%. The purity of the samples prepared according to methods disclosed in Tables 8 and 9, determined using the optimized HPLC method, are presented in Table 10. Table 10

E
Fipronil, 芬普尼 , フィプロニル

Fipronil
- Molecular Formula C12H4Cl2F6N4OS
- Average mass 437.148 Da
APPROVED CDSCO INDIA 25.06.2018
Fipronil 50mg/134mg/268mg/402 mg spot on solution for cats and dogs , For treatment of flea and tick infestation in cats and dogs (for veterinary use only)
Fipronil is a broad-spectrum insecticide that belongs to the phenylpyrazole chemical family. Fipronil disrupts the insect central nervous system by blocking GABA-gated chloride channels and glutamate-gated chloride (GluCl) channels. This causes hyperexcitation of contaminated insects’ nerves and muscles. Fipronil’s specificity towards insects is believed to be due to its greater affinity to the GABA receptor in insects relative to mammals and its effect on GluCl channels, which do not exist in mammals.[1]
Because of its effectiveness on a large number of pests, fipronil is used as the active ingredient in flea control products for pets and home roach traps as well as field pest control for corn, golf courses, and commercial turf. Its widespread use makes its specific effects the subject of considerable attention. This includes ongoing observations on possible off-target harm to humans or ecosystems as well as the monitoring of resistance development.[2]
Use
Fipronil is or has been used in:
- Under the trade name Regent, it is used against major lepidopteran (moth, butterfly, etc.) and orthopteran (grasshopper, locust, etc.) pests on a wide range of field and horticultural crops and against coleopteran (beetle) larvae in soils. In 1999, 400,000 hectares were treated with Regent. It became the leading imported product in the area of rice insecticides, the second-biggest crop protection market after cotton in China.[3]
- Under the trade names Goliath and Nexa, it is employed for cockroach and ant control, including in the US. It is also used against pests of field corn, golf courses, and commercial lawn care under the trade name Chipco Choice.[3]
- It has been used under the trade name Adonis for locust control in Madagascar and Kazakhstan.[3]
- Marketed under the names Termidor, Ultrathor, and Taurus in Africa and Australia, fipronil effectively controls termite pests, and was shown to be effective in field trials in these countries.[3]
- Termidor has been approved for use against the Rasberry crazy ant in the Houston, Texas, area, under a special “crisis exemption” from the Texas Department of Agriculture and the Environmental Protection Agency. The chemical is only approved for use in Texascounties experiencing “confirmed infestations” of the newly discovered ant species.[4] Use of Termidor is restricted to certified pest control operators in the following states: Alaska, Connecticut, Nebraska, South Carolina, Massachusetts, Indiana, New York, and Washington.[citation needed]
- In Australia, it is marketed under numerous trade names, including Combat Ant-Rid, Radiate and Termidor, and as generic fipronil
- In the UK, provisional approval for five years has been granted for fipronil use as a public hygiene insecticide.[3]
- Fipronil is the main active ingredient of Frontline TopSpot, Fiproguard, Flevox, and PetArmor (used along with S-methoprene in the ‘Plus’ versions of these products); these treatments are used in fighting tick and flea infestations in dogs and cats.
- In New Zealand, fipronil was used in trials to control wasps (Vespula spp.), which are a threat to indigenous biodiversity.[5] It is now being used by the Department of Conservation to attempt local eradication of wasps,[6].[7][8]
Effects
Toxicity
Fipronil is classed as a WHO Class II moderately hazardous pesticide, and has a rat acute oral LD50 of 97 mg/kg.
It has moderate acute toxicity by the oral and inhalation routes in rats. Dermal absorption in rats is less than 1% after 24 h and toxicity is considered to be low. It has been found to be very toxic to rabbits.
The photodegradate MB46513 or desulfinylfipronil, appears to have a higher acute toxicity to mammals than fipronil itself by a factor of about 10.[9]
Symptoms of acute toxicity via ingestion includes sweating, nausea, vomiting, headache, abdominal pain, dizziness, agitation, weakness, and tonic-clonic seizures. Clinical signs of exposure to fipronil are generally reversible and resolve spontaneously. As of 2011, no data were available regarding the chronic effects of fipronil on humans. The U.S. EPA has classified fipronil as a group C (possible human) carcinogen based on an increase in thyroid follicular cell tumors in both sexes of the rat. However, as of 2011, no human data is available regarding the carcinogenic effects of fipronil.[10]
Two Frontline TopSpot products were determined by the New York State Department of Environmental Conservation to pose no significant exposure risks to workers applying the product. However, concerns were raised about human exposure to Frontline spray treatment in 1996, leading to a denial of registration for the spray product. Commercial pet groomers and veterinarians were considered to be at risk from chronic exposure via inhalation and dermal absorption during the application of the spray, assuming they may have to treat up to 20 large dogs per day.[3] Fipronil is not volatile, so the likelihood of humans being exposed to this compound in the air is low.[10]
In contrast to neonicotinoids such as acetamiprid, clothianidin, imidacloprid, and thiamethoxam, which are absorbed through the skin to some extent, fipronil is not absorbed substantially through the skin.[11]
Detection in body fluids
Fipronil may be quantitated in plasma by gas chromatography-mass spectrometry or liquid chromatography-mass spectrometry to confirm a diagnosis of poisoning in hospitalised patients or to provide evidence in a medicolegal death investigation.[12]
Ecological toxicity
Fipronil is highly toxic for crustaceans, insects and zooplankton,[13] as well as bees, termites, rabbits, the fringe-toed lizard, and certain groups of gallinaceous birds. It appears to reduce the longevity and fecundity of female braconid parasitoids. It is also highly toxic to many fish, though its toxicity varies with species. Conversely, the substance is relatively innocuous to passerines, wildfowl, and earthworms.
Its half-life in soil is four months to one year, but much less on soil surface because it is more sensitive to light (photolysis) than water (hydrolysis).[14]
Few studies of effects on wildlife have been conducted, but studies of the nontarget impact from emergency applications of fipronil as barrier sprays for locust control in Madagascar showed adverse impacts of fipronil on termites, which appear to be very severe and long-lived. Also, adverse effects were indicated in the short term on several other invertebrate groups, one species of lizard (Trachylepis elegans), and several species of birds (including the Madagascar bee-eater).
Nontarget effects on some insects (predatory and detritivorous beetles, some parasitic wasps and bees) were also found in field trials of fipronil for desert locust control in Mauritania, and very low doses (0.6-2.0 g a.i./ha) used against grasshoppers in Niger caused impacts on nontarget insects comparable to those found with other insecticides used in grasshopper control. The implications of this for other wildlife and ecology of the habitat remain unknown, but appear unlikely to be severe.[3] Unfortunately, this lack of severity was not observed in bee species in South America. Fipronil is also used in Brazil and studies on the stingless bee Scaptotrigona postica have shown adverse reactions to the pesticide, including seizures, paralysis, and death with a lethal dose of .54 ng a.i./bee and a lethal concentration of .24 ng a.i./μl diet. These values are highly toxic in Scaptotrigona postica and bees in general.[15] Toxic baiting with fipronil has been shown to be effective in locally eliminating German wasps. All colonies within foraging range were completely eliminated within one week.[16][17][5]
In May 2003, the French Directorate-General of Food at the Ministry of Agriculture determined that a case of mass bee mortality observed in southern France was related to acute fipronil toxicity. Toxicity was linked to defective seed treatment, which generated dust. In February 2003, the ministry decided to temporarily suspend the sale of BASF crop protection products containing fipronil in France.[18] The seed treatment involved has since been banned.[citation needed] Fipronil was used in a broad spraying to control locusts in Madagascar in a program that began in 1997.[19]
Notable results from wildlife studies include:
- Fipronil is highly toxic to fish and aquatic invertebrates. Its tendency to bind to sediments and its low water solubility may reduce the potential hazard to aquatic wildlife.[20]
- Fipronil is toxic to bees and should not be applied to vegetation when bees are foraging.[20]
- Based on ecological effects, fipronil is highly toxic to upland game birds on an acute oral basis and very highly toxic on a subacute dietary basis, but is practically nontoxic to waterfowl on both acute and subacute bases.[21]
- Chronic (avian reproduction) studies show no effects at the highest levels tested in mallards (NOEC) = 1000 ppm) or quail (NOEC = 10 ppm). The metabolite MB 46136 is more toxic to the parent than avian species tested (very highly toxic to upland game birds and moderately toxic to waterfowl on an acute oral basis).[21]
- Fipronil is very highly toxic to bluegill sunfish and highly toxic to rainbow trout on an acute basis.[21]
- An early-lifestage toxicity study in rainbow trout found that fipronil affects larval growth with a NOEC of 0.0066 ppm and an LOEC of 0.015 ppm. The metabolite MB 46136 is more toxic than the parent to freshwater fish (6.3 times more toxic to rainbow trout and 3.3 times more toxic to bluegill sunfish). Based on an acute daphnia study using fipronil and three supplemental studies using its metabolites, fipronil is characterized as highly toxic to aquatic invertebrates.[21]
- An invertebrate lifecycle daphnia study showed that fipronil affects length in daphnids at concentrations greater than 9.8 ppb.[21]
- A lifecycle study in mysids shows fipronil affects reproduction, survival, and growth of mysids at concentrations less than 5 ppt.[21]
- Acute studies of estuarine animals using oysters, mysids, and sheepshead minnows show that fipronil is highly acutely toxic to oysters and sheepshead minnows, and very highly toxic to mysids. Metabolites MB 46136 and MB 45950 are more toxic than the parent to freshwater invertebrates (MB 46136 is 6.6 times more toxic and MB 45950 is 1.9 times more toxic to freshwater invertebrates).[21]
Colony collapse disorder
Fipronil is one of the main chemical causes blamed for the spread of colony collapse disorder among bees. It has been found by the Minutes-Association for Technical Coordination Fund in France that even at very low nonlethal doses for bees, the pesticide still impairs their ability to locate their hive, resulting in large numbers of forager bees lost with every pollen-finding expedition.[22] A synergistic toxic effect of fipronil with the fungal pathogen Nosema ceranae was recently reported[23]. The functional basis for this toxic effect is now understood: the synergy between fipronil and the pathogenic fungus induces changes in male physiology leading to infertility[24] A 2013 report by the European Food Safety Authorityidentified fipronil as “a high acute risk to honeybees when used as a seed treatment for maize and on July 16, 2013 the EU voted to ban the use of fipronil on corn and sunflowers within the EU. The ban took effect at the end of 2013.”[25][26]
Pharmacodynamics
Fipronil acts by binding to allosteric sites of GABAA receptors and GluCl receptors (of insects) as an antagonist (a form of noncompetitive inhibition). This prevents the opening of chloride ion channels normally encouraged by GABA, reducing the chloride ions’ ability to lower a neuron’s membrane potential. This results in an overabundance of neurons reaching action potential and likewise CNS toxicity via overstimulation.[27][28][29][30]
- Acute oral LD50 (rat) 97 mg/kg
- Acute dermal LD50 (rat) >2000 mg/kg
In animals and humans, fipronil poisoning is characterized by vomiting, agitation, and seizures, and can usually be managed through supportive care and early treatment of seizures, generally with benzodiazepine use.[31][32]
History
Fipronil was discovered and developed by Rhône-Poulenc between 1985 and 1987, and placed on the market in 1993 under the B2 U.S. Patent 5,232,940 B2. Between 1987 and 1996, fipronil was evaluated on more than 250 insect pests on 60 crops worldwide, and crop protection accounted for about 39% of total fipronil production in 1997. Since 2003, BASF holds the patent rights for producing and selling fipronil-based products in many countries.
2017 Fipronil eggs contamination
The 2017 Fipronil eggs contamination is an incident in Europe and South Korea involving the spread of insecticide contaminated eggs and egg products. Chicken eggs were found to contain Fipronil and distributed to 15 European Union countries, Switzerland, and Hong Kong.[33][34] Approximately 700,000 eggs are thought to have reached shelves in the UK alone.[35] Eggs at 44 farms in Taiwan were also found with excessive Fipronil levels.[36]
SYN


SYN 2

SYN 3

SYN 4

PATENT
http://www.allindianpatents.com/patents/271132-a-process-for-the-synthesis-of-fipronil
5-Amino-l-(2,6-dichloro-4-trifluoromethylphenyl)-3-cyano-4-trifluoromethyl
sulfinyl pyrazole or 5-Amino-[2,6-dichloro-4-(trif]uoromethyl)phenyl]-4-[-(1 (R,S)-trifluoromethyl)sulfinyl]-1H-pyrazole-3-carbonitrile also known as Fipronil is a novel pesticide characterized by high efficiency, low toxicity and especially low residue.
There are various routes to synthesize Fipronil by oxidation of thiopyrazole with various other oxidizing agents in suitable solvents. Oxidation of sulfides is a very useful route for the preparation of sulfoxides. Literature is replete with the conversion of sulfides to sulfoxides and/or sulfones. However, most of the existing methods use expensive, toxic or rare oxidizing reagents, which are difficult to prepare, are very expensive and cannot be used on commercial scale. Many of these processes suffer from poor selectivity.
WO01/30760 describes oxidation of 5-amino-l-(2,6-dichloro-4-trifluoromethylphenyl)-3-cyano-4-trifluoromethylthio-pyrazole with trifluoro-acetic acid and hydrogen peroxide in the presence of boric acid. The quantity
of trifluoroacetic acid used is 14.5 molar equivalents. The patent also
discloses the preparation of 5-amino-1-(2,6-dichloro-4-trifluoromethyl
phenyl)-3-cyano-4-trifluoromethylthio-pyrazole from 5-amino-1-(2,6-
dichloro-4-trifluoromethyl phenyl)-3-cyano pyrazole-4-yl disulphide.
European Patent publication No.295117 describes the preparation of 5-amino-l-(2,6-dichloro-4-trifluoromethylphenyl)-3-cyano-4-trifluoromethylsulphinyl pyrazole starting from 2,6-Dichloro-4-trifluoromethylaniline to give an intermediate 5-amino-l-(2,6-dichloro-4-trifluoromethylphenyl)-3-cyano-4-trifluoromethylthiopyrazole which is oxidized with meta-chloroperbenzoic acid in chloroform to give desired product.
Oxidizing agents such as perbenzoic acids do not provide effective and regioselective oxidation of electron deficient sulfides such as trifluoromethylsulphides which are less readily oxidized than other sulfides. Trifluoroacetic acid and trichloroacetic acid are found to be very efficient and regioselective oxidation medium for oxidation of 5-amino-l-(2,6-dichloro-4-trifluoromethylphenyl)-3-cyano-4-trifluoromethylthio-pyrazole in presence of hydrogen peroxide. Trichloroacetic acid can not be used alone due to higher melting point. Trifluoroacetic acid on the other hand is very regioselective with respect to conversion and low by-products formation. However, it is expensive, water miscible, corrosive to metal as well as glass, comparatively lower boiling and it’s recovery (in anhydrous form) is complex in nature.
W000/35851/2000 talks about synthesis of 2,6-Dichloro-4-trifluoromethylaniline starting from 3,4,5-trichloro-benzotrifluoride in the presence of alkaline fluorides like lithium fluoride and ammonia in the
presence of N-methylpyrrolidone at 250°C to give 97% conversion and 87% selectivity. The main drawback of the above process is the synthesis of 3,4,5-trichlorobenzotrifluoride in high yield and purity. Chlorination of p-chlorobenzotrifluoride gives a mixture of 3,4,5-trichlorobenzotrifluoride in 72% GLC conversions, 3,4-dichloro and tetrachlorobenzotrifluoride. The process to get pure 3,4,5-isomer from this mixture by fractionation followed by crystallization is very tedious. Moreover in-spite of using very pure intermediates, substantial amount of an undesired isomer (3-amino-4,5-dichlorobenzotrifluoride) is also obtained.
Another approach to generate 3,4,5-trichlorobenzotrifluoride with high yield and purity is to perform denitrochlorination of 4-chloro-3,5-dinitrobenzotrifluoride in the presence of a catalyst as described in GB Patent 2154581A. Even though the process produces 3,4,5-trichlorobenzotrifluoide in high yield and purity, the reaction conditions are too drastic to be employed for an industrial process.
The known commercial processes for the manufacture of Fipronil uses corrosive and expensive chemical such as trifluoroaceticacid, hydrogen peroxide and m-chloroperbenzoicacid Trifluoroacetic acid is expensive and generally not used in large quantities, as well as of m-chloroperbenzoic acid is difficult to handle at commercial scale due to its un-stability and detonating effect. Also the raw material used such as 2,6-Dichloro-4-trifluoromethylaniline are not easily available or made. The overall process for the Fipronil as disclosed above is found to be unsatisfactory in one respect or the other.
Thus, there is felt a need for preparing Fipronil from easily available raw materials in a simple and economical manner at an industrial level, with high yields and purity.
PATENT
https://patents.google.com/patent/US20130030190A1/en
-
5-Amino-1-(2,6-dichloro-4-trifluoromethylphenyl)-3-cyano-4-trifluoromethyl sulfinyl pyrazole or 5-Amino-[2,6-dichloro-4-(trifluoromethyl)phenyl]-4-[-(1(R,S)-trifluoromethyl)sulfinyl]-1H-pyrazole-3-carbonitrile also known as Fipronil is a novel pesticide characterized by high efficiency, low toxicity and especially low residue.
- [0005]
There are various routes to synthesize Fipronil by oxidation of thiopyrazole with various other oxidizing agents in suitable solvents. Oxidation of sulfides is a very useful route for the preparation of sulfoxides. Literature is replete with the conversion of sulfides to sulfoxides and/or sulfones. However, most of the existing methods use expensive, toxic or rare oxidizing reagents, which are difficult to prepare, are very expensive and cannot be used on commercial scale. Many of these processes suffer from poor selectivity.
- [0006]
WO01/30760 describes oxidation of 5-amino-1-(2,6-dichloro-4-trifluoromethylphenyl)-3-cyano-4-trifluoromethylthio-pyrazole with trifluoro-acetic acid and hydrogen peroxide in the presence of boric acid. The quantity of trifluoroacetic acid used is 14.5 molar equivalents. The patent also discloses the preparation of 5-amino-1-(2,6-dichloro-4-trifluoromethyl phenyl)-3-cyano-4-trifluoromethylthio-pyrazole from 5-amino-1-(2,6-dichloro-4-trifluoromethyl phenyl)-3-cyano pyrazole-4-yl disulphide.
- [0007]
European Patent publication No. 295117 describes the preparation of 5-amino-1-(2,6-dichloro-4-trifluoromethylphenyl)-3-cyano-4-trifluoromethylsulphinyl pyrazole starting from 2,6-Dichloro-4-trifluoromethylaniline to give an intermediate 5-amino-1-(2,6-dichloro-4-trifluoromethylphenyl)-3-cyano-4-trifluoromethylthiopyrazole which is oxidized with meta-chloroperbenzoic acid in chloroform to give desired product.
- [0008]
Oxidizing agents such as perbenzoic acids do not provide effective and regioselective oxidation of electron deficient sulfides such as trifluoromethylsulphides which are less readily oxidized than other sulfides. Trifluoroacetic acid and trichloroacetic acid are found to be very efficient and regioselective oxidation medium for oxidation of 5-amino-1-(2,6-dichloro-4-trifluoromethylphenyl)-3-cyano-4-trifluoromethylthio-pyrazole in presence of hydrogen peroxide. Trichloroacetic acid can not be used alone due to higher melting point. Trifluoroacetic acid on the other hand is very regioselective with respect to conversion and low by-products formation. However, it is expensive, water miscible, corrosive to metal as well as glass, comparatively lower boiling and it’s recovery (in anhydrous form) is complex in nature.
- [0009]
WO00/35851/2000 talks about synthesis of 2,6-Dichloro-4-trifluoromethylaniline starting from 3,4,5-trichloro-benzotrifluoride in the presence of alkaline fluorides like lithium fluoride and ammonia in the presence of N-methylpyrrolidone at 250° C. to give 97% conversion and 87% selectivity. The main drawback of the above process is the synthesis of 3,4,5-trichlorobenzotrifluoride in high yield and purity. Chlorination of p-chlorobenzotrifluoride gives a mixture of 3,4,5-trichlorobenzotrifluoride in 72% GLC conversions, 3,4-dichloro and tetrachlorobenzotrifluoride. The process to get pure 3,4,5-isomer from this mixture by fractionation followed by crystallization is very tedious. Moreover in-spite of using very pure intermediates, substantial amount of an undesired isomer (3-amino-4,5-dichlorobenzotrifluoride) is also obtained.
- [0010]
Another approach to generate 3,4,5-trichlorobenzotrifluoride with high yield and purity is to perform denitrochlorination of 4-chloro-3,5-dinitrobenzotrifluoride in the presence of a catalyst as described in GB Patent 2154581A. Even though the process produces 3,4,5-trichlorobenzotrifluoide in high yield and purity, the reaction conditions are too drastic to be employed for an industrial process.
- [0011]
The known commercial processes for the manufacture of Fipronil uses corrosive and expensive chemical such as trifluoroaceticacid, hydrogen peroxide and m-chloroperbenzoicacid Trifluoroacetic acid is expensive and generally not used in large quantities, as well as of m-chloroperbenzoic acid is difficult to handle at commercial scale due to its un-stability and detonating effect. Also the raw material used such as 2,6-Dichloro-4-trifluoromethylaniline are not easily available or made. The overall process for the Fipronil as disclosed above is found to be unsatisfactory in one respect or the other.
- [0012]
Thus, there is felt a need for preparing Fipronil from easily available raw materials in a simple and economical manner at an industrial level, with high yields and purity.
-
- Example 18
- [0081]
A mixture of 700 g of dichloroacetic acid and trichloroacetic acid was taken along with 300 g of chlorobenzene, 2 g of boric acid and 280 g of 5-amino-1-(2,6-dichloro-4-trifluoromethylphenyl)-3-cyano-4-trifluoromethyl thiopyrazole, the content were cooled to 15-20° C. Aqueous H202 (44.2 g, 50%) was added and mass was stirred for 20 hrs. The mass was then processed and Fipronil was isolated by filtration. After work up as above, 269 g of Fipronil of purity 94% was obtained. The filtered Fipronil was then purified using chlorobenzene (5 ml/g) followed by mixture (1 ml/g, 80:20 v/v) of ethylacetate and chlorobenzene to get 232 g of Fipronil of greater than 97% purity.
- [0081]
Example 19 Purification of Fipronil
- [0082]
The fipronil prepared in example 18 of purity 97% was treated with a mixture (232 ml) of ethylacetate & chlorobenzene (80:20 v/v). This reaction mixture was heated to 85-90° C. & maintained for 1 hr. It was further cooled up to 30° C. in stages & filtered. Fipronil thus obtained had a purity of 98%. This cycle was repeated to obtain fipronil of above 98% purity.
- [0083]
The useful constituents from various streams of crystallization, leaching as above were reused and recycled, fipronil was isolated in 80-85% yield with purity of above 98%.
PATENT
CN 101250158 [2008 to Hunan Res Inst of of chemical Ind.]
WO2005/44806 A1, ; Page/Page column 7-8; 12 ;
WO2009/77853 A1, ; Page/Page column 28-29 ;
US 5,618,945 [1995, to Rhone-Poulenc]
CN 102060774
IN 178903 [1997, to Rallis India Ltd.]
WO 2009/077,853 [2009 to Vetoquinol SA ]
BG 109983 [2008 to BASF Agro B V]
US 8,507,693 [2013, to Gharda]
US 5,618,945 [1995, to Rhone-Poulenc]
WO 2007/122,440 [ 2007 to Gharda Chemicals Ltd.]
FR 2,925,493 [2009 to to Vetoquinol SA ]
CN 1176078 [ 2002 to Jiangsu Prov Inst of Pesticide]
EP 0,374,061 [ 1990 to Rhone Poulenc Agrochimie]
US 5,232,940 [1993, to May and Baker]
PAPER
Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), , # 24 p. 3371 – 3376
Synthesis 2008, 11, 1682-1684
Synthesis 2007, 22, 3507-3511
Tetrahedron Letters, , 2007, 48(48), 8518-8520
Tetrahedron Letters 2008 ,49. 3463-3465
References
- Jump up^ Raymond-Delpech, Valérie; Matsuda, Kazuhiko; Sattelle, Benedict M.; Rauh, James J.; Sattelle, David B. (2005-09-20). “Ion channels: molecular targets of neuroactive insecticides”. Invertebrate Neuroscience. 5 (3-4): 119–133. doi:10.1007/s10158-005-0004-9. ISSN 1354-2516.
- Jump up^ Maddison, Jill E.; Page, Stephen W. (2008). Small Animal Clinical Pharmacology(Second ed.). Elsevier Health Sciences. p. 229. ISBN 9780702028588.
- ^ Jump up to:a b c d e f g “Fipronil”. Pesticides News. 48: 20. 2000.
- Jump up^ “Rasberry Crazy Ant”. Texas A&M. 2008-04-12. Archived from the original on 2007-07-14. Retrieved 2012-08-06.
- ^ Jump up to:a b “Revive Rotoiti Autumn 2011”. Department of Conservation. 2011. Retrieved 11 April2012.
- Jump up^ “War on wasps in Abel Tasman”. 10 February 2016.
- Jump up^ “Vespex: Making wide-area wasp control a reality – WWF’s Conservation Innovation Awards”. wwf-nz.crowdicity.com.
- Jump up^ (DOC), corporatename = New Zealand Department of Conservation. “Wasp control using Vespex”. http://www.doc.govt.nz.
- Jump up^ “Fipronil insecticide: Novel photochemical desulfinylation with retention of neurotoxicity”. Proceedings of the National Academy of Sciences. 93: 12764–12767. doi:10.1073/pnas.93.23.12764. PMC 23994
 .
. - ^ Jump up to:a b “Fipronil Technical Fact Sheet, National Pesticide Information Center”. Retrieved 2015-12-07.
- Jump up^ “Cockroach Control”. Retrieved August 10, 2016.
- Jump up^ R. Baselt, Disposition of Toxic Drugs and Chemicals in Man, 11th edition, Biomedical Publications, Seal Beach, CA, 2017, pp. 894-895. ISBN 978-0-692-77499-1
- Jump up^ “Ecotoxicity for Fipronil”. Retrieved 2010-05-03.
- Jump up^ Amrith S. Gunasekara & Tresca Troung (March 5, 2007). “Environmental Fate of Fipronil” (PDF). Retrieved April 16, 2016.
- Jump up^ Jacob, CRO; Hellen Maria Soares; Stephen Malfitano Carvalho; Roberta Cornélio Ferreira Nocelli; Osmar Malspina (2013). “Acute Toxicity of Fipronil to the Stingless Bee Scaptotrigona postica Latreille”. Bulletin of Environmental Contamination and Toxicology. 90 (1): 69–72. doi:10.1007/s00128-012-0892-4. Retrieved 23 September 2015.
- Jump up^ Paula Sackmann, Mauricio Rabinovich and Juan Carlos Corley J. (2001). “Successful Removal of German Yellowjackets (Hymenoptera: Vespidae) by Toxic Baiting” (PDF). pp. 811–816.
- Jump up^ “Short and long-term control of Vespula pensylvanica in Hawaii by fipronil baiting”. Pest Management Science. 68: 1026–1033. doi:10.1002/ps.3262.
- Jump up^ Elise Kissling; BASF SE (2003). “BASF statement regarding temporary suspension of sales of crop protection products containing fipronil in France”.
- Jump up^ June 2000 BBC News story “Anti-locust drive ‘created havoc'”.
- ^ Jump up to:a b “Fipronil” (PDF). National Pesticides Communication Network. p. 3. Retrieved 19 June 2012.
- ^ Jump up to:a b c d e f g United States Environmental Protection Agency Office of Prevention, Pesticides and Toxic Substances (1996). “Fipronil. May 1996. New Pesticide Fact Sheet. US EPA Office of Prevention, Pesticides and Toxic Substances”.
- Jump up^ “Colony Collapse Disorder linked to Fipronil”. Retrieved 2010-06-17.
- Jump up^ Aufavure J., Biron D. G., Vidau C., Fontbonne R., Roudel M., Diogon M., Viguès B., Belzunces L. P., Delbac F., Blot N. (2012) Parasite – insecticide interactions: a case study of Nosema ceranae and fipronil synergy on honeybee. Scientific Reports 2:326 – DOI: 10.1038/srep00326
- Jump up^ Kairo G, Biron D.G, Ben A.F, Bonnet M, Tchamitchian S, Cousin M, … & Brunet J.L (2017) Nosema ceranae, Fipronil and their combination compromise honey bee reproduction via changes in male physiology]. Scientific reports, 7(1), 8556.
- Jump up^ “EFSA assesses risks to bees from fipronil”. 27 May 2013. Retrieved 29 May 2013.
- Jump up^ Carrington, Damian (16 July 2013). “EU to ban fipronil to protect honeybees”. The Guardian. London.
- Jump up^ Cole, L. M.; Nicholson, R. A.; Casida, J. E. (1993). “Action of Phenylpyrazole Insecticides at the GABA-Gated Chloride Channel”. Pestic. Biochem. Physiol. 46: 47–54. doi:10.1006/pest.1993.1035.
- Jump up^ Ratra, G. S.; Casida, J. E. (2001). “GABA receptor subunit composition relative to insecticide potency and selectivity”. Toxicol. Lett. 122: 215–222. doi:10.1016/s0378-4274(01)00366-6. PMID 11489356.
- Jump up^ WHO. Pesticide Residues in Food – 1997: Fipronil; International Programme on Chemical Safety, World Health Organization: Lyon, 1997.
- Jump up^ Olsen RW, DeLorey TM (1999). “Chapter 16: GABA and Glycine”. In Siegel GJ, Agranoff BW, Fisher SK, Albers RW, Uhler MD. Basic neurochemistry: molecular, cellular, and medical aspects (Sixth ed.). Philadelphia: Lippincott-Raven. ISBN 0-397-51820-X.
- Jump up^ Ramesh C. Gupta (2007). Veterinary Toxicology. pp. 502–503. ISBN 978-0-12-370467-2.
- Jump up^ Mohamed F, Senarathna L, Percy A, Abeyewardene M, Eaglesham G, Cheng R, Azher S, Hittarage A, Dissanayake W, Sheriff MH, Davies W, Buckley NA, Eddleston M., Acute human self-poisoning with the N-phenylpyrazole insecticide fipronil–a GABAA-gated chloride channel blocker, J Toxicol Clin Toxicol. 2004;42(7):955-63.
- Jump up^ “Eggs containing fipronil found in 15 EU countries and Hong Kong”. BBC News. 2017-08-11. Retrieved 2017-08-11.
- Jump up^ News, ABC. “EU: 17 nations get tainted eggs, products in growing scandal”. ABC News. Archived from the original on 2017-08-11. Retrieved 2017-08-11.
- Jump up^ Boffey, Daniel (11 August 2017). “Egg contamination scandal widens as 15 EU states, Switzerland and Hong Kong affected”. The Guardian. Retrieved 11 August 2017.
- Jump up^ “Eggs at 44 farms in Taiwan found with excessive insecticide levels”. Taiwan News. 26 August 2017. Retrieved 26 August 2017.
External links
- Fipronil Fact Sheet – National Pesticide Information Center
- Fipronil Toxicity & Regulatory Info – PANNA PesticideInfo database
- Fipronil in the Pesticide Properties DataBase (PPDB)
 |
|
 |
|
| Names | |
|---|---|
| IUPAC name
(RS)-5-Amino-1-[2,6-dichloro-4-(trifluoromethyl)phenyl]-4-(trifluoromethylsulfinyl)pyrazole-3-carbonitrile
|
|
| Other names
Fipronil
Fluocyanobenpyrazole Termidor |
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| ECHA InfoCard | 100.102.312 |
| KEGG | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C12H4Cl2F6N4OS | |
| Molar mass | 437.14 g·mol−1 |
| Density | 1.477-1.626 g/cm3 |
| Melting point | 200.5 °C (392.9 °F; 473.6 K) |
| Pharmacology | |
| QP53AX15 (WHO) | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
/////////////Fipronil, INDIA 2018, フィプロニル , HSDB 7051, RM 1601, veterinary, ind 2018
C1=C(C=C(C(=C1Cl)N2C(=C(C(=N2)C#N)S(=O)C(F)(F)F)N)Cl)C(F)(F)F
“ DRUG APPROVALS INTERNATIONAL” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
Veterinary- Atipamezole
Atipamezole

4-(2-Ethyl-1,3-dihydroinden-2-yl)-3H-imidazole, Atipamezole, cas 104054-27-5
hydrochloride cas no 104075-48-1
- MPV 1248 (IS: FarmosGroupLt)
- UNII-03N9U5JAF6 (IS)
- UNII-2W4279571X (IS)
Atipamezole is a synthetic alpha2-adrenergic antagonist, indicated for the reversal of the sedative and analgesic effects of dexmedetomidine and medetomidine in dogs. It has also been researched in humans as a potential anti-Parkinsonian drug.Atipamezole is more potent than yohimbine; it is very selective for alpha2-adrenergic vs alpha1sites, but not selelctive for alpha2 – subtypes.
Atipamezole (brand name Antisedan, Pfizer) is a synthetic alpha2–adrenergic antagonist, indicated for the reversal of the sedative and analgesic effects of dexmedetomidine andmedetomidine in dogs.[1] It has also been researched in humans as a potential anti-Parkinsonian drug.[2]
- Pfizer Animal Health ANTISEDAN Product Overview
- Pertovaara A, Haapalinna A, Sirviö J, Virtanen R (2005). “Pharmacological properties, central nervous system effects, and potential therapeutic applications of atipamezole, a selective alpha2-adrenoceptor antagonist”. CNS Drug Reviews 11 (3): 273–88.doi:10.1111/j.1527-3458.2005.tb00047.x. PMID 16389294.

- ANTISEDAN product information page (Pfizer Animal Health)
- Drugs Future, vol. 15, no. 5, 1990, “Atipamezole“, pages 448-452, see page 449
Synonyms
1H-imidazole, 4-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-
1H-imidazole, 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-
5-(2-Ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole
Atipamezole
4-(2-Ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole
4-(2-Ethyl-2-indanyl)imidazole
4-(2-Ethyl-indan-2-yl)-1H-imidazole(Atipamezole)
4-(2-ethylindan-2-yl)imidazole
Antisedan
Antisedan
Atipamezol
Atipamezolum
Atipamezole Hydrochloride CAS 104075-48-1
………………………………
Atipamezole is a selective alpha2 – adrenoceptor antagonist which is currently marketed under the trademark Antisedan® for the reversal of sedative- analgesic veterinary drugs. Atipamezole has been disclosed e.g. in the European Patent EP 183492 as useful for the reversal of detomidine. European Patent EP 0589957 discloses the use of atipamezole for the treatment of male sexual impotence. In US 4698692 the use of atipamezole for the attenuation of ethyl alcohol intoxication is disclosed.
US Patent No. US6543389 discloses insecticidal pet collars for dogs comprising amitraz and atipamezole. Atipamezole in the collar provides amelioration of amitraz toxicosis in combination with the amitraz in case the dogs ingests the collar. The pet collar comprises 0.01 to 1%, preferably 0.1 to 1 %, by weight of atipamezole. Safe, effective ways to eliminate ectoparasites are desired for the companion animal’s well-being, for the well-being and comfort of its human associate and for the prevention of losses in livestock
A substantial amount of work has been devoted to identifying the neurotransmitters involved in the facilitation and inhibition of male sexual behaviour (see e.g. Bitran and Hull 1987, Neuroscience and Behavioral reviews 11 , 365-389). Noradrenergic neuro-transmission seems to have an important role.
Atipamezole is a selective and potent a2*-adrenoceptor antagonist which is currently marketed for the reversal of sedative-analgesic veterinary drugs. Atipamezole has been disclosed e.g. in the European Patent EP 183492 as useful for the reversal of detomidine.
We have now found that this compound is also very effective in increasing male sexual capacity in a monkey model. These findings suggest that atipamezole would be an effective therapy in male impotence in humans as well.
Another a2-adrenoreceptor antagonist, yohimbine, is currently used for the treatment of male impotence. Yohimbine increases noradrenergic neurotransmission and has been reported to facilitate the sexual capacity of male animals, although the results of different studies are conflicting.
Atipamezole is, however clearly advantageous over yohimbine for this use because of its excellent selectivity. The a2/a-|selectivity ratio of atipamezole is
200-300 times higher than that of yohimbine.
- EP 0310745 B (FARMOS OY) 1989.04.12. disclosed preparation of 5-(2-ethyl-2,3-dihydro-1 H-inden-2-yl)-1 H-imidazole salt by two synthetic routes.
-
First synthetic route as starting material was used 2-acetyl-1-indanone, which was alkylated with ethylbromide in acetone in the presence of sodium carbonate to 2-acetyl-2-ethyl-1-indanone. The acetyl group was brominated with bromine in methanol and to imidazole by heating in formamide. Then the intermediate was hydrogenated in 2N hydrochloric acid in the presence of 10% palladium on carbon.
-
Second synthetic route disclosed in the same patent is following, as starting material was used 2,3-dihydro-1H-indene-2-carboxylic acid methyl ester, which was prepared by methylation of 2,3-dihydro-1H-indene-2-carboxylic acid in the presence of sulphuric acid. The 2,3-dihydro-1H-indene-2-carboxylic acid methyl ester was reacted with N-isopropylcyclohexylamide and ethylbromide yielding 2,3-dihydro-2-ethyl-1H-indene-2-carboxylic acid, then thionyl chloride was added and 2,3-dihydro-2-ethyl-1H-indene-2-carboxylic acid chloride was obtained. In the next step ethoxymagnesiummalonic acid ethyl ester in dry ether was added to 2,3-dihydro-2-ethyl-1H-indene-2-carboxylic acid chloride and reaction mixture was treated with sulphuric acid, and 1-(2,3-dihydro-2-ethyl-1H-inden-2-yl)ethanone was obtained, then the intermediate was stirred in methylene chloride and bromine was added by giving a new intermediate 2-bromo-1-(2,3-dihydro-2-methyl-1H-inden-2-yl)ethanone, to which was thereafter added formamide and hydrochloric acid yielding crude product of 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole. The last step involved hydrogenation of the crude product of 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1 H-imidazole with 10% palladium on carbon.
-
EP 0247764 B (ORION-YHTYMÄ OY) 1987.02.12. disclosed the following process for preparation of 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole hydrochloride. The process starts by reaction of alpha, alpha-dibromo-o-xylene with 4-penten-2-one to obtain 1-(2,3-dihydro-2-vinyl-1H-inden-2-yl)ethanone. The obtained intermediate was brominated, e.g. with bromine, methylene chloride was used as solvent and 2-bromo-1-(2,3-dihydro-2-vinyl-1H-inden-2-yl)-ethanone was obtained, which is thereafter reacted with formamide in excess formamide to give a 4(5)-(2,3-dihydro-2-vinyl-1H-inden-2-ylimidazole hydrochloride. As the last step the vinyl group was catalytically hydrogenated to an ethyl group so as to form a product 4(5)-(2,3-dihydro-2-ethyl-1 H-inden-2-yl) imidazole.
-
Another synthetic route for obtaining 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole is disclosed in WAI, Wonf, et al. A Concise Synthesis of Atipamezole. Synthesis. 1995, no.2, p.139-140. The cyclization of alpha, alpha’-dibromo-o-xylene with acetylacetone by means of NaOH and tetrabutylammonium bromide in toluene/water at 80°C under phase-transfer conditions gives the unstable diacetyl derivative, which presumably undergoes cleavage to afford 2-acetylindane. The alkylation of 2-acetylindane with ethyl iodide and potassium tert-butoxide yields 2-acetyl-2-ethylindan, which is brominated with Br2 to give 2-bromoacetyl-2-ethylindan. Finally, this compound is cyclised with formamide at 160°C (some 2-ethyl-2-(4-oxazolyl)indane is also formed but easily eliminated); the cyclization can also be carried out with formamidine in liquid ammonia. Although the substitution of formamide by formamidine acetate eliminates the oxazole formation, it does not increase the yield of Atipamezole (<30%) WAI, Wonf, et al. A Concise Synthesis of Atipamezole. Synthesis. 1995, no.2, p.139-140 in the final step.
The preparation of atipamezole hydrochloride salt is described in U.S. Patent 4,689,339, wherein atipamezole obtained from the hydrogenation step is first recovered from alkaline solution as free base. After the evaporation of methylene chloride solvent to dryness the isolated crystalline product is converted into its hydrochloride salt by treatment with dry hydrogen chloride in ethyl acetate
Other compounds having alpha-2 adrenoceptor antagonist properties which may be useful in accordance with the present invention include idazoxan related compounds [Reckitt & Colman] Doxey, et al., Br. J. Parmacol., Vol. 78, p.489-505 (1983); imiloxan [Syntex] Michel, et al., Br. J. Pharmacol., Vol. 74, p.255-256 (1981); WY 26703 and related compounds [Wyeth] Latimer, et al., Naunvn Schmiedeberg’s Arch. Pharmacol., Vol. 327, p. 312-318 (1984); CH-38083 [Chinoin] Vizi, et a., J. Pharmacol. Exp. Ther., Vol. 238, p. 701-706 (1986); GR 50360A and related compounds [Glaxo] Halliday, et al., Br. J. Pharmacol., Vol. 95, p. 715 (1988); DG 5128 and related compounds of Daiichi Seiyaku Co., Ltd., Tokyo, Japan; and Yohimbine [Sigma].
………………………………….

-
- 1. an essential process for obtaining 5-(2-ethyl-2,3-1H-inden-2-yl)-1H-imidazole, without bromination in any step of process, thus preventing the possibility of brominated by-products;
- 2. This process has given superior yields, compared to patents cited above;
- 3. This process is amenable to large scale production which does not require specialized equipment.
-
The condensing of commercially available 1-trityl-1H-imidazole-4-carboxaldehyde (I) with phtalide to form 2-(1-trityl-1H-imidazole-4-yl)indan-1,3-dione (II) is performed under the conditions that are similar to those used for synthesis of 4-(indane-1,3-dionyl) pyridine J. Org. Chem. 1971, vol.36, p.1563. surprisingly, the bulky 1-trityl-1H-imidazole-4-carboxaldehyde (I) reacted as expected and produced 2-(1-trityl-1H-imidazole-4-yl)indan-1,3-dione (II) in over 67% yield. Both ethyl acetate and dioxane can be used as reaction media.
-
The alkylation of (II) by ethyl iodide is performed in boiling acetone with potassium carbonate as basic agent. 2-Ethyl-2-(1-trityl-1H-imidazole-4-yl)indan-1,3-dione (III) is formed in over 67% yield and easily isolated from the acetone solution by concentrating it and diluting with water. A high purity (III) is obtained after crystallization from methanol or ethanol.
-
Removing the trityl group of 2-ethyl-2-(1-trityl-1H-imidazole-4-yl)indan-1,3-dione by acid hydrolysis to yield the deprotected 2-ethyl-2-(1H-imidazol-2-yl)indan-1,3-dione.
-
The reduction of (IV) to 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole hydrochloride (V) is performed in hydrogenation apparatus with Pd/C catalyst under hydrogen pressure in HCI solution. The reaction proceeds under variable pressure and temperature conditions, but a pressure of about 3 bar and the temperature of about 80-85°C is preferable. After removing the catalyst the product crystallizes on chilling in over 77% yield. It can be purified by additional crystallization.
-
EP 0310745 B (FARMOS OY) 1989.04.12. disclosed preparation of 5-(2-ethyl-2,3-dihydro-1 H-inden-2-yl)-1 H-imidazole salt by two synthetic routes.
-
First synthetic route as starting material was used 2-acetyl-1-indanone, which was alkylated with ethylbromide in acetone in the presence of sodium carbonate to 2-acetyl-2-ethyl-1-indanone. The acetyl group was brominated with bromine in methanol and to imidazole by heating in formamide. Then the intermediate was hydrogenated in 2N hydrochloric acid in the presence of 10% palladium on carbon.
-
Second synthetic route disclosed in the same patent is following, as starting material was used 2,3-dihydro-1H-indene-2-carboxylic acid methyl ester, which was prepared by methylation of 2,3-dihydro-1H-indene-2-carboxylic acid in the presence of sulphuric acid. The 2,3-dihydro-1H-indene-2-carboxylic acid methyl ester was reacted with N-isopropylcyclohexylamide and ethylbromide yielding 2,3-dihydro-2-ethyl-1H-indene-2-carboxylic acid, then thionyl chloride was added and 2,3-dihydro-2-ethyl-1H-indene-2-carboxylic acid chloride was obtained. In the next step ethoxymagnesiummalonic acid ethyl ester in dry ether was added to 2,3-dihydro-2-ethyl-1H-indene-2-carboxylic acid chloride and reaction mixture was treated with sulphuric acid, and 1-(2,3-dihydro-2-ethyl-1H-inden-2-yl)ethanone was obtained, then the intermediate was stirred in methylene chloride and bromine was added by giving a new intermediate 2-bromo-1-(2,3-dihydro-2-methyl-1H-inden-2-yl)ethanone, to which was thereafter added formamide and hydrochloric acid yielding crude product of 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole. The last step involved hydrogenation of the crude product of 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1 H-imidazole with 10% palladium on carbon.
-
EP 0247764 B (ORION-YHTYMÄ OY) 1987.02.12. disclosed the following process for preparation of 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole hydrochloride. The process starts by reaction of alpha, alpha-dibromo-o-xylene with 4-penten-2-one to obtain 1-(2,3-dihydro-2-vinyl-1H-inden-2-yl)ethanone. The obtained intermediate was brominated, e.g. with bromine, methylene chloride was used as solvent and 2-bromo-1-(2,3-dihydro-2-vinyl-1H-inden-2-yl)-ethanone was obtained, which is thereafter reacted with formamide in excess formamide to give a 4(5)-(2,3-dihydro-2-vinyl-1H-inden-2-ylimidazole hydrochloride. As the last step the vinyl group was catalytically hydrogenated to an ethyl group so as to form a product 4(5)-(2,3-dihydro-2-ethyl-1 H-inden-2-yl) imidazole.
-
Another synthetic route for obtaining 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole is disclosed in WAI, Wonf, et al. A Concise Synthesis of Atipamezole. Synthesis. 1995, no.2, p.139-140. The cyclization of alpha, alpha’-dibromo-o-xylene with acetylacetone by means of NaOH and tetrabutylammonium bromide in toluene/water at 80°C under phase-transfer conditions gives the unstable diacetyl derivative, which presumably undergoes cleavage to afford 2-acetylindane. The alkylation of 2-acetylindane with ethyl iodide and potassium tert-butoxide yields 2-acetyl-2-ethylindan, which is brominated with Br2 to give 2-bromoacetyl-2-ethylindan. Finally, this compound is cyclised with formamide at 160°C (some 2-ethyl-2-(4-oxazolyl)indane is also formed but easily eliminated); the cyclization can also be carried out with formamidine in liquid ammonia. Although the substitution of formamide by formamidine acetate eliminates the oxazole formation, it does not increase the yield of Atipamezole (<30%) WAI, Wonf, et al. A Concise Synthesis of Atipamezole. Synthesis. 1995, no.2, p.139-140 in the final step.
…………………………….
US Patent 8,431,717
Atipamezole [5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole, 1] is a veterinary drug that has been investigated for treating Parkinson’s disease in humans. V. Lusis and co-inventors summarize several ways to synthesize 1. Some routes give a low yield of 1 and produce large quantities of an oxazole byproduct. Other processes involve a sluggish bromination reaction that leads to many byproducts.
The inventors’ process is intended to overcome these problems. In particular, it does not use the bromination reaction and thus avoids forming brominated byproducts. The process, outlined in the figure, begins with the reaction of imidazole 2 with i-PrMgCl to form iodo Grignard reagent 3, which is treated with DMF to give 4. This intermediate is not isolated but is treated with aq NH4Cl to give aldehyde 5, isolated in 73.2% yield. The aldehyde is condensed with phthalide (6) in the presence of NaOMe to produce imidazolylindane 7, recovered in crude form in 67.2% yield.

In the next stage, compound 7 is alkylated with EtI in the presence of K2CO3. Product 8 is isolated in 50.9% yield after being recrystallized from EtOH. Product1 can be produced directly from 8 by making its HCl salt and hydrogenating the salt over Pd/C. Crude atipamezole is isolated as its HCl salt in 26.6% yield.
Alternatively, acid hydrolysis of 8 removes the trityl group to form dione 9, recovered as a white crystalline solid in 76.2% yield. The HCl salt of 9 is then hydrogenated to 1·HCl.
The patent’s claims cover the process to make 1 and new compounds 7 and 8. The overall yield of compound 1 is poor, partly because of the low yield from the hydrogenation step. The inventors claim, however, that the yield is higher than from earlier methods. They point out that the process is amenable to large-scale production without the use of specialized equipment. (JSC Grindeks [Riga, Latvia]. US Patent 8,431,717, April 30, 2013; Keith Turner), View the full-text here.
………………………………
nmr

Atipamezole Hydrochloride CAS 104075-48-1 HNMR
……………………………………………………….
Veterinary-Dirlotapide, drug used to treat obesity in dogs

1-Methyl-N-[(1S)-2-(methyl-(phenylmethyl)amino)-2-oxo-1-phenylethyl]-5-[[oxo-[2-[4-(trifluoromethyl)phenyl]phenyl]methyl]amino]-2-indolecarboxamide
Chemical Formula
C40-H33-F3-N4-O3
Molecular Weight
674
Therapeutic Category, dog
Antiobesity agent
Chemical Names
N-{(1S)-2-[Benzyl(methyl)amino]-2-oxo-1-phenylethyl}-1-methyl-5-[4′-(trifluoromethyl)biphenyl-2-carboxamido]-1H-indol-2-carboxamide (WHO)
1H-Indole-2-carboxamide, 1-methyl-N-[(1S)-2-[methyl(phenylmethyl)amino]-2-oxo-1-phenylethyl]-5-[[[4′-(trifluoromethyl)[1,1′-biphenyl]-2-yl]carbonyl]amino]- (USAN)
1-Methyl-5-[(4′-trifluormethylbiphenyl-2-carbonyl)amino]-1H-indol-2-carbonsäure-[(S)-(benzylmethylcarbamoyl)phenylmethyl]amid (IUPAC)
5-[4′-(Trifluoromethylbiphenyl-2-carbonyl)amino]-1H-indole-2-carboxylic acid benzylmethyl carbamoylamide
| Identifiers | |
|---|---|
| CAS number | 481658-94-0 |
| ATCvet code | QA08AB91 |
Dirlotapide is a drug used to treat obesity in dogs. It is manufactured by Pfizer and marketed as Slentrol.
It works as a selective microsomal triglyceride transfer protein (MTTP) inhibitor. This blocks the assembly and release of lipoproteins into the bloodstream, thereby reducing fat absorption. It also elicits a satiety signal from lipid-filled cells lining the intestine.
It is supplied as an oral solution. It is not intended for use in humans, cats, or parrots.
On January 5 2007, the U.S. Food and Drug Administration (FDA) approved Slentrol, the first time the FDA has approved a drug for obese dogs.[1]
Dirlotapide is used to manage obesity in dogs and helps by reducing appetite. It should be used as part of an overall weight control program that also includes proper diet and exercise, under the supervision of a veterinarian. Side effects may include vomiting, diarrhea, lethargy, drooling, or uncoordination. Allergic reaction to the medication may include, facial swelling, hives, scratching, sudden onset of diarrhea, vomiting, shock, seizures, pale gums, cold limbs, or coma. Contact your veterinarian if you observe any of these signs. The dose of dirlotapide will need to be recalculated each month, based on your dog’s weight.
Canine patient information sheet http://www.drsfostersmith.com/Rx_Info_Sheets/rx_dirlotapide.pdf

- “FDA approves 1st drug for obese dogs”. Yahoo. Archived from the original on January 8, 2007. Retrieved 2007-01-06.
Generic Names
- Dirlotapide (OS: USAN)
- CP-742033 (IS)
Brand Names
- Slentrol (veterinary use)
Pfizer, Poland; Pfizer Animal Health, Belgium; Pfizer Animal Health, Switzerland; Pfizer Animal Health, United Kingdom; Pfizer Animal Health, United States; Pfizer GmbH Tiergesundheit, Germany; Pfizer Limited, Austria; Pfizer Santé Animale, France


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}