
Home » GLIPTIN
Category Archives: GLIPTIN
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Gemigliptin tartrate sesquihydrate, ゲミグリプチン
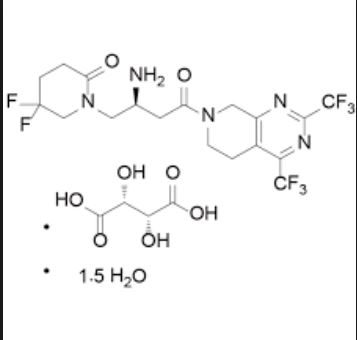

Gemigliptin tartrate sesquihydrate;
ゲミグリプチン
| Molecular Weight | 666.47 |
| Formula | C18H19F8N5O2 • C4H6O6 • 1.5H2O |
911637-19-9 (Gemigliptin);
1375415-82-9 (Gemigliptin L-tartrate Sesquihydrate)
Gemiglo®; LC-150444; Zemiglo
Gemigliptin L-tartrate sesquihydrate was approved by Korean food and Drug Administration on June 27, 2012. It was developed and marketed as Zemiglo® by LG Life Sciences in KR.
Gemigliptin L-tartrate sesquihydrate is a dipeptidyl peptidase-4 inhibitor indicated for the treatment of type 2 diabetes mellitus.
Zemiglo® is available as tablet for oral use, containing 50 mg of free Gemigliptin. The recommended dose is 50 mg once daily taken regardless of meals.
- Originator LG Life Sciences
- Developer LG Chem; Sanofi
- Class Antihyperglycaemics; Piperidines; Pyrimidines; Small molecules
- Mechanism of Action CD26 antigen inhibitors
- Marketed Type 2 diabetes mellitus
- Phase II/III Acute kidney injury
- 24 Jun 2018 Biomarkers information updated
- 05 Apr 2018 LG Chem initiates enrolment in a phase I trial for Type-2 diabetes mellitus (Combination therapy) in South Korea (NCT03565458)
- 08 Feb 2018 LG Life Sciences completes the phase III ZEUS II trial in Type-2 diabetes mellitus (Adjunctive treatment) in South Korea (PO) (NCT02831361)
- Research Code:LC-15-0444
- Trade Name:Zemiglo®
- MOA:Dipeptidyl peptidase-4 (DPP-4) inhibitor
- Indication:Type 2 diabetes
- Status:Approved 2012-06-27 korea
- Company:LG Life Sciences (Originator)
- Sales:ATC Code:A10BH06
Mechanism of Action
● Gemigliptin is a selective DPP-4 inhibitor[4-7].
● DPP-4 inhibition IC50=16 nM.
● Selectivity compared against DPP8/9 >3000 fold.
● Gemigliptin bound to DPP-4 enzyme with a Ki=15.2 nM.
In Vivo Efficacy
Minimum effective dose of gemigliptin in animal models:
● DPP-4 activity inhibition: Mice:0.3 mg/kg; rats: 3 mg/kg; dog: 0.3 mg/kg; monkeys: 1 mg/kg.
● Active GLP-1 concentration increase:Rats:1 mg/kg; dogs: 1 mg/kg.
● Plasma glucagon concentration decrease: Dogs: 1 mg/kg in.
● Blood glucose reduce: DIO mice:0.3 mg/kg; beagle dog: 1 mg/kg.
● HbA1c reduce: DIO mice: 3 mg/kg/day for 4 weeks.
Absorption
● The oral bioavailability of gemigliptin in the rats, dogs and monkeys are species-dependent with the values of 94 %, 73 %, and 26 %, respectively.
● Gemigliptin is rapidly absorbed after single oral dose administration, with Tmax occurring 0.3 to 0.5 hr postdose in rats and dogs.
● Half-life of gemigliptin is moderate in rats, dogs, monkeys (3.6 – 5.8 hrs) and long in humans (30.8 hrs), clearance of gemigliptin ranged from 0.6 L/hr/kg (32 % of liver blood flow) in dogs to 4.1 L/hr/kg (123 % of liver blood flow).
● Volume of distribution of gemigliptin is greater than body water volume, occurring 3.7 to 14 L/kg, which suggested extensive extravascular distribution.
Distribution
● [14C] Gemigliptin and metabolites were not extensively bound to plasma proteins.
● Following oral administration of gemigliptin, [14C] gemigliptin-derived radioactivity was widely distributed to most tissues and organs in rats.
● In most tissues and organs, the total radioactivity decreased with time and was almost entirely eliminated 24 hrs after administration.
● The highest concentrations were observed in the tissues of the alimentary (the small intestine and large intestine) and the excretory or metabolic (the kidney, pancreas and liver) systems.
● Accumulation of the drug was not observed in most of the tissues or organs tested but the time course of radioactivity in testis showed some possibility of drug accumulation.
Metabolism
● Following oral administration of 10 mg/kg [14C] gemigliptin to male rats: The major circulating metabolites were LC15-0516 (dehydrated), LC15-0635 and LC15-0636 (hydroxylated), but the majority of the radioactivity in plasma was associated with the parent compound. The metabolite profile in urine was similar to that in plasma, but the profile in bile was somewhat different from that in urine or plasma. The major metabolic pathway was hydroxylation.
● Following oral administration of 50 mg [14C] gemigliptin to healthy male subjects: Gemigliptin was the most abundant component accounting for 67.2%-100% of plasma radioactivity. Unchanged gemigliptin accounted for 44.8%-67.2% of urinary radioactivity and 27.7%-51.8% of fecal radioactivity. LC15-0636 was the most abundant metabolite in plasma, accounting for 9.1%–17.5 % of plasma radioactivity. LC15-0516 and LC15-0635 were not detected in plasma samples. LC15-0636 was the only human metabolite with systemic exposure more than 10% of total drug-related exposure.
● CYP3A4 was identified as the dominant CYP isozyme converting gemigliptin to LC15-0636 in recombinant CYP/FMO enzymes.

Gemigliptin (rINN), previously identified as LC15-0444, is an oral anti-hyperglycemic agent (anti-diabetic drug) of the new dipeptidyl peptidase-4 (DPP-4) inhibitor class of drugs.[1] It is well known thatglucose lowering effects of DPP-4 inhibitors are mainly mediated by GLP-1 and gastric inhibitory polypeptide (GIP) incretin hormones which are inactivated by DPP-4.
Gemigliptin was initially developed solely by LG Life Sciences. In 2010, Double-Crane Pharmaceutical Co. (DCPC) joined with LGLS to co-develop the final compound and collaborate on the marketing of the drug in China. LGLS also announced in November 2010 that NOBEL Ilac has been granted rights to develop and commercialize gemigliptin in Turkey.
A New Drug Application (NDA) for gemigliptin in the treatment of type 2 diabetes was submitted to the Korea Food & Drug Administration (KFDA) in July 2011. Then on June 27, 2012, the KFDA has approved the manufacture and distribution of LG Life Sciences’ diabetes treatment, Zemiglo, the main substance of which is gemigliptin. LG Life Sciences signed a licensing agreement with multinational pharmaceutical companies such as Sanofi (Paris, France) and Stendhal (Mexico City, Mexico) for 104 countries. Currently, gemigliptin has been approved in 11 countries such as India, Columbia, Costa Rica, Panama, and Ecuador, and several clinical studies are in progress in Russia, Mexico, and Thailand.
History
The NDA for gemigliptin was submitted to KFDA in July, 2011 and it was approved on June 27, 2012. By the end of 2012, gemigliptin will be marketed in Korea as Zemiglo which is the fifth new DPP-4 inhibitor diabetes treatment in the world. Sanofi-Synthelabo India Private Limited announced the launch of drug for type 2 diabetes patients in India: Zemiglo (gemigliptin) on Juty 19, 2016. Zemiglo is a once daily, oral tablet. As per the International Diabetes Federation Diabetes Atlas 2015, India is home to the second largest number of adults living with diabetes worldwide, after China, with 69.1 million patients and expected to rise to 1401 million in 2040. India is the largest contributor to South East Asia regional mortality, with 1 million deaths attributable to diabetes. These statistics reveal how diabetes is fast gaining the status of a potential epidemic in India and establishes the need for treatment compliance and effective control through diet, exercise and drugs for long-term positive effects in disease management.
Mechanism of action
DPP-4 is a serine protease located on the cell surfaces throughout the body. In plasma, DPP-4 enzyme rapidly inactivates incretins including GLP-1 and GIP which are produced in the intestine depending on the blood glucose level and contribute to the physiological regulation of glucose homeostatis. Active GLP-1 and GIP increase the production and release of insulin by pancreatinc beta cells. GLP-1 also reduces the secretion of glucacon by pancreatic alpha cells, thereby resulting in a decreased hepatic glucose production. However these incretins are rapidly cleaved by DPP-4 and their effects last only for a few minutes. DPP-4 inhibitors block the cleavage of the gliptins and thus lead to an increasee insulin level and a reduced glucagon level in a glucose-dependent way. This results in a decrease of fasting and postprandial glycemia, as well as HbA1c levels.[2]
Preclinical studies
Gemigliptin is a competitive, reversible DPP-4 inhibitor (Ki = 7.25 ± 0.67 nM) with excellent selectivity over other critical human proteases such as DPP-2, DPP-8, DPP-9, elastase, trypsin, urokinase and cathepsin G. The kinetics of DPP-4 inhibition by gemigliptin was characterized by a fast association and a slow dissociation rate compared to sitagliptin (fast on and fast off rate) or vildagliptin (slow on and slow off rate). Gemigliptin was rapidly absorbed after single oral dosing and the compound was eliminated with a half-life of 3.6 h, 5.2 h, and 5.4 h in the rat, dog, and monkey, respectively.
The bioavailability of gemigliptin in the rat, dog, and monkey was species-dependent with the values of 94%, 73%, and 26%, respectively. Following the oral administration of gemigliptin in the rat, dog and monkey, about 80% inhibition of plasma DPP-4 activity were observed at the plasma levels of 18 nM, 14 nM and 4 nM, respectively.
In a diet-induced obesity model, gemigliptin reduced glucose excursion during OGTT in a dose dependent manner with the minimum effective dose of 0.3 mg/kg and enhanced glucose-stimulated plasma GLP-1 increase in a dose dependent manner reaching the maximum effect at the dose of 1 mg/kg.
Following 4 week oral repeat dosing in the DIO mice, gemigliptin reduced significantly HbA1c with the minimum effective dose of 3 mg/kg. In the beagle dog, gemigliptin significantly enhanced active GLP-1, decreased glucagon, and reduced glucose excursion during OGTT following a single dosing.
Studies on animals suggest its positive effect on hepatic and renal fibrosis .[3][4] Data on human patients are still inconclusive .[5]
Clinical studies
Monotherapy
The efficacy and safety of gemigliptin monotherapy were evaluated in two double-blind placebo controlled studies and one double-blind active-controlled study. A phase II study (study identifier: LG-DPCL002) of gemigliptin was conducted in a randomized, double-blind, placebo-controlled, parallel group design with three doses of 50, 100, and 200 mg qd for the purpose of finding a dose responsiveness and an optimal dose in patients with T2DM. The mean changes of HbA1c at week 12 from the baseline were –0.98%, –0.74%, –0.78% (when adjusted with placebo data, –0.92%, –0.68%, and –0.72%) at 50, 100, and 200 mg, respectively. Among the effective doses obtained from the phase II study in patients with T2DM, the 50 mg dose showed a similar efficacy as the 100 and 200 mg doses, within the maximum safety margin. Similar findings were reported from two phase III studies. Patients were randomized to receive gemigliptin, either a 50 mg qd (n=90) or a placebo (n=92) for 24 weeks (study identifier: LG-DPCL005; ClinicalTrials.gov registration number: NCT01601990). The placebo-subtracted changes from baseline in HbA1c were reported to be −0.71% (95% confidence interval [CI], −1.04 to −0.37) with gemigliptin 50 mg. In addition, a 28-week open-label extension study was designed to evaluate the long-term safety and efficacy of gemigliptin. Among 165 patients who consented to participate in the extension period of study LG-DPCL005, 158 patients (96%) completed their treatments for 52 weeks. All patients were switched to or continued their treatments only with gemigliptin 50 mg qd during the extension period. A further decrease in HbA1c was observed in the continued treatment with gemigliptin 50 mg in this extension period, and the mean change from baseline at 52 weeks (–0.87%) was still clinically and statistically significant (full analysis set analysis, P<0.0001). In another double-blind, active-controlled, phase III trial (study identifier: LG-DPCL011), eligible patients with HbA1c greater than 7.5% were randomized to receive gemigliptin 50 mg qd with metformin slow release (SR) qd (n=141), gemigliptin 50 mg qd (n=142), or metformin SR qd (n=150) for 24 weeks. After 24 weeks, the reduction from the baseline in HbA1c was –1.24% for gemigliptin monotherapy.
Initial combination therapy with metformin
In this randomized, double-blind, active-controlled, phase III trial (study identifier: LG-DPCL011, INICOM study; ClinicalTrials.gov registration number: NCT01787396), eligible patients with an HbA1c greater than 7.5% were randomized to gemigliptin 50 mg qd+metformin SR qd (n=141), gemigliptin 50 mg qd (n=142), or metformin SR qd (n=150). From weeks 2 to 6, metformin SR was uptitrated incrementally from 500 to 2,000 mg/day maximum in the gemigliptin/metformin and metformin groups. The mean daily doses of metformin at week 24 were 1,699 and 1,868 mg for the gemigliptin/metformin group and the metformin group, respectively. Mean change in HbA1c from baseline was –2.06% for gemigliptin/metformin group versus –1.24% for the gemigliptin group and –1.47% for the metformin group, respectively (P<0.0001 for all comparisons of combination therapy vs. monotherapy). The differences in proportions achieving an HbA1c <7% or <6.5% were also statistically significant (P<0.0001) between the combination therapy and the respective monotherapy groups.
Add-on to metformin
A 24-week, multinational, randomized, double-blind, active-controlled study (study identifier: LG-DPCL006; ClinicalTrials.gov registration number: NCT01602003) was designed to assess the efficacy and safety of gemigliptin 50 mg compared to the active control (sitagliptin) added to ongoing metformin therapy in patients with T2DM inadequately controlled with metformin alone (HbA1c, 7% to 11%). After 24 weeks, the reduction from baseline for HbA1c was 0.81% for gemigliptin 25 mg twice a day (bid) and 0.77% for gemigliptin 50 mg qd, and the differences in the least square mean changes from baseline between groups (each group of gemigliptin-sitagliptin group) were −0.011% in gemigliptin 25 mg bid and 0.004% in gemigliptin 50 mg qd. The proportion of patients achieving an HbA1c <7% at week 24 (gemigliptin 25 mg bid group, 50%; gemigliptin 50 mg qd group, 54.07%) was comparable to the results with sitagliptin 100 mg qd (48.87%). The efficacy of lowering HbA1c in the gemigliptin group was generally consistent across the subgroups based on age (<65 or ≥65 years), gender, duration of T2DM (5, >5 to 10, or >10 years), and baseline body mass index (BMI, <25 or ≥25 kg/m2). In addition, gemigliptin groups led to a significantly greater inhibition of plasma DPP-4 compared to sitagliptin. This study was extended by 28 weeks in order to evaluate the long-term efficacy and safety of gemigliptin. All treatment groups showed clinically and statistically (P<0.0001) significant improvement in glycemic control from baseline after 52 weeks. The reduction from the baseline in HbA1c was –1.06 (95% CI, –1.28 to –0.85) in the patients who continued to receive gemigliptin 50 mg qd.
Add-on to metformin and glimepiride
In this multicenter, randomized, double-blind, phase III study (study identifier: LG-DPCL010, TROICA study; ClinicalTrials.gov registration number: NCT01990469), eligible patients with inadequate glycemic control (7%≤HbA1c≤11%) were randomized to gemigliptin 50 mg qd (n=109) or placebo (n= 110). The baseline demographics were similar between groups (age, 60.9 years; BMI, 24.9 kg/m2; duration of T2DM, 12.9 years), with mean±standard deviation (SD) baseline HbA1c of 8.12%± 0.82% in the gemigliptin group and 8.15%±0.89% in the placebo group. At week 24, the adjusted mean±standard error change for HbA1c with gemigliptin was –0.88%±0.17% (change with placebo –0.01%±0.18%; difference –0.87%±0.12%; 95% CI, –1.09 to –0.64; P<0.0001).
Add-on therapy in patients with renal impairment
RI in T2DM limits the usable medications for lowering glucose level and requires frequent monitoring of renal function. Gemigliptin has balanced elimination between urinary/fecal excretion and hepatic metabolism; therefore, it does not require dose adjustment in patient with moderate to severe RI. This study evaluated the efficacy and safety of gemigliptin in T2DM patients with moderate to severe RI. This randomized, double-blind, parallel group, phase IIIb study (study identifier: LG-DPCL015, GUARD study; ClinicalTrials.gov registration number: NCT01968044) was composed of a 12-week, placebo controlled period, followed by a 40-week, double-blind active controlled extension period (placebo switched to linagliptin). A total of 132 patients with moderate or severe RI were randomized to receive gemigliptin (n=66) or placebo (n=66). Insulin was used as predominant background therapy (63.1%). At week 12, the placebo-adjusted mean change in HbA1c from the baseline was –1.20% (95% CI, –1.53 to –0.87; P<0.0001). A similar profile was also observed in other glycemic control parameters (fasting plasma glucose, glycated albumin, and fructosamine).
Effects on glycemic variability
Glycemic variability and chronic sustained hyperglycemia are the main components of dysglycemia in diabetes. The previous studies suggested that different pharmacodynamic profiles between DPP-4 inhibitors have been associated with the different effects on glycemic variability. In this study, a multicenter, randomized, active-controlled, parallel group, open-label, exploratory study was designed to evaluate the efficacy on glycemic variability and safety of initial combination therapy of gemigliptin 50 mg qd versus sitagliptin 100 mg qd, or glimepiride 2 mg qd with metformin in patients with T2DM (study identifier: LG-DPCL012, STABLE study; ClinicalTrials.gov registration number: NCT01890629). The mean amplitude of glycemic excursions (MAGE) and SD of glucose were used for assessing glucose fluctuations from the baseline after 12 weeks of treatment. At 12 weeks, MAGE was significantly lower in the DPP-4 inhibitor groups (gemigliptin and sitagliptin) than in the glimepiride group (–43.1, –38.3, and –21.7 mg/dL, respectively). Furthermore, the SD of mean glucose was significantly lower in patients with gemigliptin when compared with sitagliptin (P=0.023) and glimepiride (P=0.0058).
Ongoing studies
Several clinical studies in LG Life Sciences are actively underway to assess the efficacy and safety as an add-on combination therapy with insulin (with or without metformin) (ClinicalTrials.gov registration number: NCT02831361), to evaluate the efficacy and safety of gemigliptin-rosuvastatin fixed-dose combination in patients with T2DM and dyslipidemia in phase III clinical trials (ClinicalTrials.gov registration number: NCT02126358), and to evaluate the efficacy and safety of gemigliptin compared with vildagliptin in Russian patients with T2DM (ClinicalTrials.gov registration number: NCT02343926).
Key Characteristics
·Gemigliptin is a reversible, potent, selective, competitive, and long-acting inhibitor of DPP-4.
·Gemigliptin is orally administered 50 mg once daily either as monotherapy or in combination with other drugs. It can be taken with or without food.
·No dose adjustment is recommended for patients with renal or hepatic impairment.
·Gemigliptin shows a low propensity of drug interactions with metformin, pioglitazone, glimepiride, CYP3A4 inhibitors, rosuvastatin, or irbesartan, and dose adjustment of gemigliptin is not required for the patients who are concomitantly receiving these drugs.
·Gemigliptin decreases the mean level of HbA1c from baseline by 1.24% in monotherapy and 0.8% in add-on therapy with metformin. For gemigliptin as an initial combination with metformin, the mean reduction from baseline in HbA1c was 2.8%. In head-to-head comparisons, the mean reduction from baseline in HbA1c was 0.8% for gemigliptin with metformin and 0.8% for sitagliptin with metformin, hence the efficacy of gemigliptin is found to be comparable to sitagliptin.
·Gemigliptin was shown to be more effective in reduction of glycemic variability than glimepiride and sitagliptin with metformin as an initial combination therapy for drug naïve patients with T2DM.
·Gemigliptin is generally well tolerated in controlled clinical studies as monotherapy and as part of combination therapy. The incidences of AEs are generally similar to those of placebo and active control groups.
PATENT
CN103189375A
https://patents.google.com/patent/CN103189375A/en
The present invention relates to the following formula 1- {(2S) -2- amino-I shown 4- [2,4-bis (trifluoromethyl) _5,8_ dihydro-pyrido [3,4-d ] pyrimidin -7 (6H) – yl] -4-oxo – 1.5 hydrate butyl} -5,5-difluoropiperidin-2-one (hereinafter, referred to as “compound I”) and its tartrate Preparation.
Preparation of Hydrate (Form I) of Example 1 I tartrate salt of the compound of embodiment [0072]
[0073]

[0074] The compound 2 was dissolved in about 1.87Kg 9L of ethanol. Was added 0.94Kg at O~10 ° C SOCl2, and then stirred while maintaining a low temperature.After concentration under reduced pressure, the concentrate was dissolved in MTBE 11.2L (MTBE), and the resulting mixture was adjusted to pH7~8 with ION NaOH solution. After the layers were separated, and the aqueous layer was extracted with MTBE about 3.7L and 3.7L extracted twice with MTBE, and then concentrated under reduced pressure. The resulting brown turbid solution was dissolved in 12L of ethanol, to which was added 0.47kg of dissolved water of about 1.5L of L- tartaric acid, and then stirred for I hour. The resulting crystalline slurry was filtered, washed with water and ethanol: washing (18), and then dried to obtain the title compound 1.13kg (yield 97.5%) of.
[0075] 1H NMR (500MHz, CD3OD) δ 2.38 (m, 2H), 2.59 (m, 2H), 2.82 –
2.99 (m, 2Η), 3.11 (bt, 1H), 3.21 (bt, 1Η), 3.50 – 3.55 (m, 1Η), 3.72 – 3.91 (m, 5Η), 3.98 (t, J = 5.2Hz, 1Η) , 4.38 (s, 2Η), 4.97 -. 5.00 (m, 2Η) [0076] Example 2 compound I tartrate embodiment 1.5 hydrate (Form I) was recrystallized from water
[0077] obtained from Example 1 50g of compound I was added 250~500ml tartrate dissolved in water and water, while with ION NaOH solution was adjusted to pH6~7. Was dissolved in 23.5ml of water was added 11.7g of L- tartaric acid, and shown in Table I below, with changes in temperature, stirring speed and stirring time to obtain crystals. Then, the crystals were filtered and dried to obtain Form I.Stirring rate of change in 50~400rpm range, the temperature change in the range of 5~32 ° C. The volume of water for the recrystallization, stirring rate, temperature and mixing time shown in Table I below.
[0078] TABLE I
[0079]

PAPER
CN101151265A
https://patents.google.com/patent/CN103189375A/en
PATENT
WO2012060590
https://patents.google.com/patent/WO2012060590A2/en
Example 1
Preparation of 1.5 hydrate of tartrate salt of Compound 1 (crystal form I)

1.87 kg of the compound 2 was dissolved in about 9 L of ethanol. 0.94 kg of SOCl2was added at 0~10℃ and then stirred while maintaining low temperature. After concentrating under reduced pressure, the concentrate was dissolved in 11.2 L of MTBE (methyl t-butyl ether), and the resulting mixture was adjusted with 10 N NaOH solution to pH 7~8. After separating the layers, the aqueous layer was extracted with about 3.7 L of MTBE and twice with 3.7 L of MTBE, and then concentrated under reduced pressure. The resulting brown turbid solution was dissolved in 12 L of ethanol, 0.47 kg of L-tartaric acid dissolved in about 1.5 L of water was added thereto, and then stirred for 1 hour. The resulting crystalline slurry was filtered, washed with water and ethanol (1:8), and then dried to obtain 1.13 kg (yield 97.5%) of the title compound.
1H NMR (500 MHz, CD3OD) δ 2.38 (m, 2H), 2.59 (m, 2H), 2.82 – 2.99 (m, 2H), 3.11 (bt, 1H), 3.21 (bt, 1H), 3.50 – 3.55 (m, 1H), 3.72 – 3.91 (m, 5H), 3.98 (t, J=5.2 Hz, 1H), 4.38 (s, 2H), 4.97 – 5.00 (m, 2H).
Example 2
Recrystallization of 1.5 hydrate of tartrate salt of Compound 1 (crystal form I) from water
50 g of tartrate salt of Compound 1 obtained from Example 1 was added to 250~500 ml of water, and dissolved in water while adjusting the solution with 10 N NaOH to pH 6~7. 11.7 g of L-tartaric acid dissolved in 23.5 ml of water was added, and crystals were obtained with varying the temperature, stirring rate and stirring time as shown in the following Table 1. Then, the crystals were filtered and dried to obtain the crystal form I. The stirring rate was varied in the range of 50~400 rpm, and the temperature was varied in the range of 5~32℃. The volume of water used for recrystallization, the stirring rate, temperature and stirring time are represented in the following Table 1.
Table 1

Conditions for HPLC analysis
Column: Atlantis dC18 (4.6 mm I.D x 250 mm L, Particle Size 5㎛, Waters)
Column Temperature: 10℃
Mobile phase:
Mobile phase A: MeCN/TFA = 100/0.1 (v/v)
Mobile phase B: H2O/TFA = 100/0.1 (v/v)
Gradient condition:

Flow rate: 0.7 ml/min.
Detection: 256 nm, UV
Injection volume: 10㎕
Total analysis time: 55 min.
The results of the stability for the crystal form I and the crystal form II are shown in the following Table 4.
Table 4
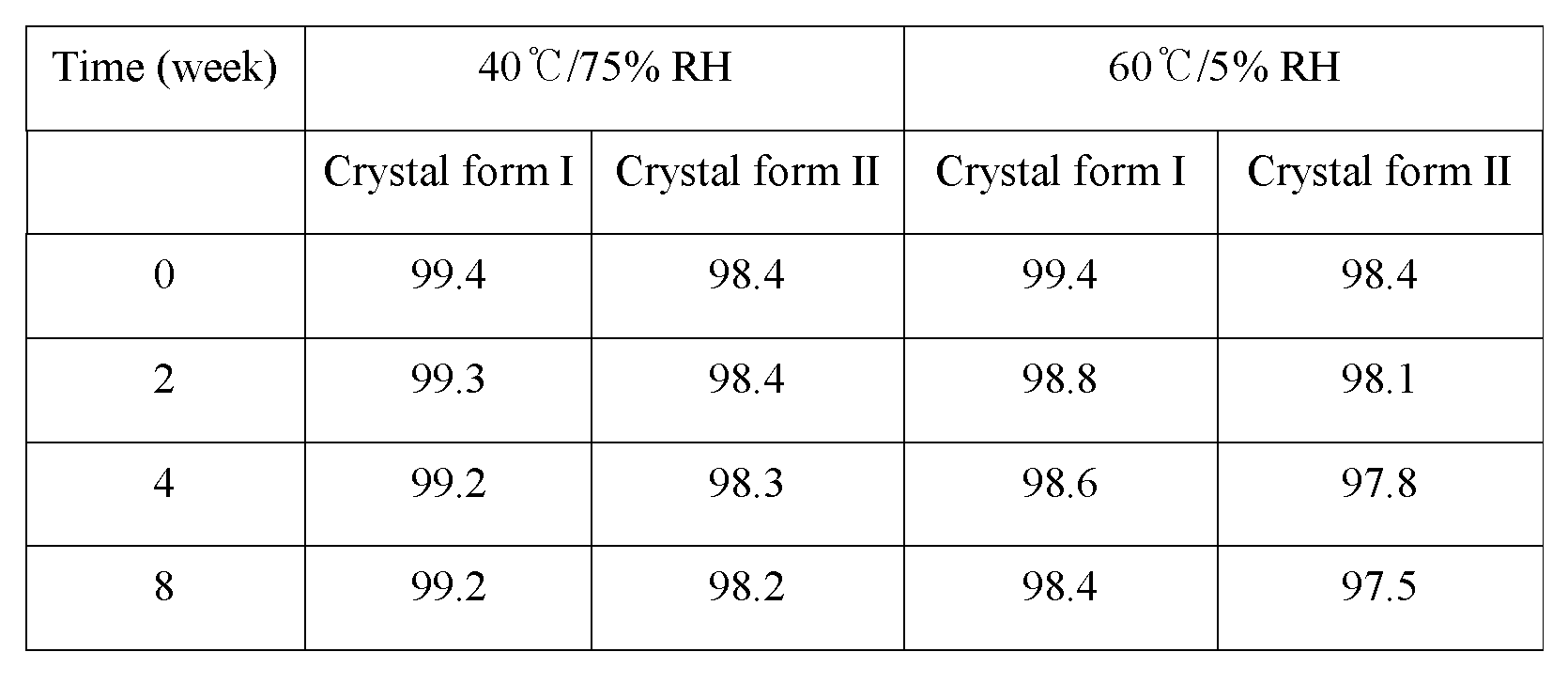
As shown in Table 4, it could be confirmed that upon keeping the crystal form I and the crystal form II at 40±2℃, 75±5% RH or 60±2℃, 5±5% RH they exhibit a superior stability up to 8 weeks. However, according to the result of XRD analysis the crystal form I did not show any change up to 8 weeks, but the crystal form II was converted into the crystal form I at 8 week under the condition of 40℃/75% RH (see Figure 16).
PAPER
https://link.springer.com/article/10.1007%2Fs12272-013-0171-x
Archives of Pharmacal Research
Gemigliptin, a potent, selective and long-acting DPP 4 inhibitor was developed by LG Life Sciences and approved for use in patients with type 2 diabetes mellitus by the Korean Food and Drug Administration in June 2012 under the trade name Zemiglo®. Clinical pharmacokinetic and pharmacodynamic data suggest the efficacy and once daily dosing of gemigliptin. In clinical phase III studies, gemigliptin was efficacious as either monotherapy or combination therapy (add-on to metformin) and well tolerated in patients with type 2 diabetes. Further development of combination therapy is on-going.
PAPER
https://pubs.acs.org/doi/pdfplus/10.1021/jm400658e
Dipeptidyl Peptidase IV and Its Inhibitors: Therapeutics for Type 2 Diabetes and What Else?
Lucienne Juillerat-Jeanneret* University Institute of Pathology, CHUV-UNIL, Bugnon 25, CH-1011 Lausanne, Switzerland
PAPER
J. Med. Chem. 1999, 42, 3557-3571

The discovery of a series of non-peptide factor Xa (FXa) inhibitors incorporating 3-(S)-amino-2-pyrrolidinone as a central template is described. After identifying compound 4, improvements in in vitro potency involved modifications of the liphophilic group and optimizing the angle of presentation of the amidine group to the S1 pocket of FXa. These studies ultimately led to compound RPR120844, a potent inhibitor of FXa (Ki = 7 nM) which shows selectivity for FXa over trypsin, thrombin, and several fibrinolytic serine proteinases. RPR120844 is an effective anticoagulant in both the rat model of FeCl2-induced carotid artery thrombosis and the rabbit model of jugular vein thrombus formation.
PATENT
http://www.google.co.in/patents/WO2006104356A1?cl=enWO 2006104356
EXAMPLE 83: Synthesis of l-(f2SV2-amino-4-r2.4-bisftrifluoromethylV5.8-dihvdropyridor3.4-d]pyrimidin-7f6H‘)
-yl1-4-oxobutyll-5.5-difluoropiperidin-2-one [1960]

[1961] 21 mg of the title compound was obtained in a yield of 56% at the same manner as in EXAMPLE 1, except that 42 mg (0.071 mmol) of t-butyl
{(lS)-3-[2,4-bis(trifluoromethyl)-5,8-dihydropyrido[3,4-d]pyrimidin-7(6H)-yl]-l-[(5,5
-difluoro-2-oxpiperidin-l-yl)methyl]-3-oxpropyl}carbamate obtained in
PREPARATION 143 was used. [1962] 1K NMR (CD3OD) δ 5.05-4.92 (2H, m), 3.98-3.91 (2H, m), 3.85-3.79 (2H, m),
3.70-3.59 (2H, m), 3.54-3.48 (IH, m), 3.36-3.33 (2H, m), 3.24 (IH, bra), 3.14 (IH, bra), 2.83-2.76 (IH, m), 2.72-2.53 (3H, m), 2.43-2.34 (2H, m) [1963] Mass (m/e) 490 (M+l)
[1964]
[1965] PREPARATION 144: Synthesis of t-butyl
(riSV3-r2.4-bisrtrifluoromethylV5.8-dihvdropyridor3.4-d]pyrimidin-7r6HVyl]-l-(rr2 S)-2-methyl-5-oxomorpholin-4-yl1methyl 1 -3-oxpropyl 1 carbamate
[1966] 14 mg of the title compound was obtained in a yield of 17% at the same manner as in PREPARATION 45, except that 43.7 mg (0.138 mmol) of (3S)-3-[(t-butoxycarbonyl)amino]-4-[2(S)-2-methyl-5-oxomoφholin-4-yl]-butanoic acid obtained in PREPARATION 55 and 42.5 mg (0.138 mmol) of 2,4-bis(trifluoromethyl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidine hydrochloric acid salt (product of PREPARATION 127) were used.
[1967] 1K NMR (CDCl3) δ 5.85-5.83 (IH, m), 5.09-4.92 (IH, m), 4.95-4.78 (IH, m),
4.23-4.08 (3H, m), 4.04-3.76 (3H, m), 3.73-3.66 (IH, m), 3.46-3.38 (IH, m), 3.36-3.21 (2H, m), 3.18-3.10 (2H, m), 2.96-2.81 (IH, m), 2.61-2.50 (IH, m), 1.43-1.41 (9H, m), 1.28-1.24 (3H, m)
[1968] Mass (m/e) 470 (M+l-Boc)
PATENT
WO 2012030106
https://www.google.com/patents/WO2012030106A2?cl=en
Reaction Scheme 1

PREPARATION 1: Synthesis of diethyl 2,2-difluoropentanedioate

To a solution of ethyl bromodifluoroacetate (33.2 g) in tetrahydrofuran (94.0 g) was added ethyl acrylate (8.2 g) and copper powder (10.9 g). After heating to 50℃, TMEDA (9.5 g) was added dropwise and the reaction mixture was then stirred for 3 hours at the same temperature. Upon disappearance of ethyl acrylate as the starting material, to the reaction solution was added methyl t-butyl ether (MTBE, 73.7 g) followed by addition of 10% aqueous ammonium chloride solution (49.8 g) dropwise, and the mixture was then stirred for 30 minutes. The remaining copper residue was removed by filtration through a celite, and methyl t-butyl ether (MTBE, 66.3 g) was added to separate the layers. The separated organic layer was washed successively with 10% aqueous NH4Cl solution (66.3 g) and 3 N aqueous hydrochloric acid solution (99.6 g) in order and then distilled under reduced pressure to obtain 55.0 g of the desired title compound.
1H NMR (400 MHz, CDCl3) δ 1.26 (t, J=7.2 Hz, 3H), 1.37 (t, J=7.2 Hz, 3H), 2.37-2.49 (m, 2H), 2.55 (t, J=7.2 Hz, 2H), 4.16 (q, J=7.2 Hz, 2H), 4.29 (q, J=7.2 Hz, 2H).
PREPARATION 2: Synthesis of ethyl 4,4-difluoro-5-hydroxypentanoate

14.8 g of the compound obtained from the above Preparation 1 was diluted with ethanol (20.4 g) and tetrahydrofuran (69.1 g) and then cooled to 0℃. To this solution was slowly added sodium borohydride (NaBH4, 3.5 g) stepwise while keeping the internal temperature below 30℃. After confirming completion of the reaction by 1H NMR, the reaction solution was cooled to the temperature of 10℃ and 10% aqueous ammonium chloride solution (77.7 g) was slowly added. The remaining boron compound was filtered through celite, and the filtrate was distilled under reduced pressure to remove tetrahydrofuran. Then, ethyl acetate (105.2 g) was added to separate the layers, and the organic layer was distilled under reduced pressure to obtain 10.8 g of the title compound.
1H NMR (400 MHz, CDCl3) δ 1.23 (t, J=7.2 Hz, 3H), 2.15-2.29 (m, 2H), 2.49 (t, J=7.2 Hz, 2H), 3.69 (t, J=12.0 Hz, 2H), 4.12 (q, J=4.0 Hz, 2H).
EXAMPLE 1: Synthesis of ethyl 4,4-difluoro-5-{[(trifluoromethyl)sulfonyl]oxy}- pentanoate

To the solution of 10.8 g of the compound, as obtained from the above Preparation 2, dissolved in dichloromethane (100.2 g) was added pyridine (7.0 g), and then the mixture was cooled to -5.0℃. After completion of cooling, trifluoromethane sulfonic acid anhydride (20.1 g) was slowly added dropwise while keeping the reaction temperature below 6.3℃. After stirring the reaction solution for 30 minutes, 1.5 N hydrochloric acid solution was added dropwise at 0℃ to separate the layers. The aqueous layer as separated was back-extracted twice with dichloromethane (33.4 g), and the extracts were combined with the organic layer separated from the above and then distilled under reduced pressure to obtain 19.7 g of the title compound as a yellow oil.
1H NMR (500 MHz, CDCl3) δ 1.27 (t, J=7.2 Hz, 3H), 2.29-2.39 (m, 2H), 2.59 (t, J=7.6 Hz, 2H), 4.18 (q, J=7.2 Hz, 2H), 4.55 (t, J=11.6 Hz, 2H).
EXAMPLE 2-1: Synthesis of ethyl 4,4-difluoro-5-{[(nonafluorobutyl)sulfonyl]- oxy}pentanoate

To the solution of 100.0 g of the compound, as obtained from the above Preparation 2, dissolved in dichloromethane (300.0 ml) was added pyridine (65.7 g), and the mixture was then cooled to -10.0℃. After completion of cooling, nonafluorobutanesulfonic anhydride (477.4 g) was slowly added dropwise. After stirring the reaction solution for 3 hours, 1.0 N hydrochloric acid solution (300.0 ml) was added dropwise to separate the layers. The aqueous layer as separated was back extracted once with dichloromethane (500.0 ml), and the extracts were combined with the organic layer separated from the above and then distilled under reduced pressure to obtain 177.5 g of the title compound.
1H NMR (500 MHz, CDCl3) δ 1.26 (t, 3H, J=7.3 Hz), 2.30-2.36 (m, 2H), 2.58 (t, 2H, J=7.4 Hz), 4.16 (q, 2H, J=7.3 Hz), 4.57 (t, 2H, J=11 Hz).
EXAMPLE 2-2: Synthesis of ethyl 4,4-difluoro-5-{[(nonafluorobutyl)sulfonyl]- oxy}pentanoate
To the solution of 500.0 g of the compound, as obtained from the above Preparation 2, dissolved in dichloromethane (1000.0 ml) was added triethylamine (389.0 g), and the mixture was then cooled to 0℃. After completion of cooling, perfluorobutanesulfonyl chloride (948.80 g) was slowly added dropwise. The reaction solution was stirred for 3 hours at room temperature, distilled under reduced pressure, dissolved in methyl t-butyl ether (MTBE, 3000.0 ml) and then washed three times with water. The organic layer thus obtained was dehydrated with magnesium sulfate, filtered through a celite and then distilled under reduced pressure to obtain 960.0 g of the title compound.
EXAMPLE 3: Synthesis of methyl (2S)-2-[(tert-butoxycarbonyl)amino]-4-oxo- pentanoate

To 25.0 g of the starting material, (3S)-3-[(t-butoxycarbonyl)amino]-4-oxo- pentanoic acid, was added t-butanol (96.9 g) followed by the addition of Boc2O (25.4 g) and dimethylaminopyridine (DMAP, 62.0 g, 0.5 mol%) at room temperature, and the reaction mixture was then stirred for 23 hours at 40℃. Upon completion of the reaction, ethylene dichloride (62.3 g) in t-butanol was added, and the mixture was then distilled under reduced pressure to obtain 30.7 g of the title compound.
1H NMR (400 MHz, CDCl3) δ 1.45 (s, 9H), 1.47 (s, 9H), 2.71 (dd, J=4.8, 16.4 Hz, 1H), 2.88 (dd, J=4.4, 16.4 Hz, 1H), 3.75 (s, 3H), 4.53 (m, 1H), 5.44 (br d, J=8.0 Hz, 1H).
EXAMPLE 4: Synthesis of tert-butyl (3S)-3-[(tert-butoxycarbonyl)amino]-4-hydroxy- butanoate

30.7 g of the compound obtained from the above Example 3 was dissolved in ethanol (112.3 g) and, after lowering the internal temperature to 10.5℃ sodium borohydride (NaBH4, 5.7 g) was slowly added dropwise. This reaction solution was stirred while maintaining the temperature below 22℃. After confirming completion of the reaction by 1H NMR and TLC, to the reaction solution was slowly added 3.0 N hydrochloric acid solution (30.7 g) dropwise at the internal temperature of 10℃ followed by addition of diluted 0.2% hydrochloric acid solution (100.0 g). The reaction solution was adjusted to pH 3~4 with addition of 9.0% aqueous hydrochloric acid solution, and then back-extracted twice with ethyl acetate (100.0 g) and toluene (44.0 g). The organic layer thus obtained was distilled under reduced pressure to obtain 25.1 g of the title compound.
1H NMR (500 MHz, CDCl3) δ 1.44 (s, 9H), 1.45 (s, 9H), 2.48-2.57 (m, 2H), 3.69 (d, J=4.9 Hz, 1H), 3.97 (m, 1H), 5.22 (bs, 1H).
EXAMPLE 5: tert-butyl (3S)-[(tert-butoxycarbonyl)amino]-4-[(methylsulfonyl)oxy]- butanoate

To 25.1 g of the compound obtained from the above Example 4 was added dichloromethane (133.0 g) and triethylamine (148.0 g), and the mixture was then cooled to 0℃. To this reaction solution was slowly added methanesulfonyl chloride (11.8 g) diluted with dichloromethane (39.9 g) dropwise for 50 minutes while maintaining the internal temperature below 12℃. After completion of the reaction, the reaction solution was washed with 0.5 N aqueous hydrochloric acid solution (120.0 g) and water (100.4 g), and then distilled under reduced pressure to obtain 31.5 g of the title compound.
1H NMR (500 MHz, CDCl3) δ 1.44 (s, 9H), 1.46 (s, 9H), 2.62 (d, J=6.0 Hz, 2H), 3.04 (s, 3H), 4.21 (m, 1H), 4.30 (d, J=5.2 Hz, 2H), 5.16 (br d, J=7.2 Hz, 1H).
EXAMPLE 6: Synthesis of tert-butyl (3S)-4-azido-3-[(tert-butoxycarbonyl)amino]- butanoate

Sodium azide (NaN3, 11.6 g) was diluted with dimethylacetamide (DMAc, 260.0 g). After elevating the internal temperature to 80℃, a solution of 31.5 g of the compound, as obtained from the above Example 5, diluted with dimethylacetamide (DMAc, 45.0 g) was added thereto. The reaction proceeded at 80℃ for 2 hours. To the reaction solution were added toluene (251.0 g) and water (320.0 g) to separate the layers. The organic layer thus obtained was distilled under reduced pressure to obtain 24.0 g of the title compound.
1H NMR (500 MHz, CDCl3) δ 1.47 (s, 9H), 1.49 (s, 9H), 2.49 (d, J=6.0 Hz, 2H), 3.44-3.55 (m, 2H), 4.09 (br s, 1H), 5.14 (br s, 1H).
EXAMPLE 7: Synthesis of tert-butyl (3S)-4-amino-3-[(tert-butoxycarbonyl)amino]- butanoate

To 21.0 g of the compound obtained from the above Example 6 was added tetrahydrofuran (93.3 g) followed by the addition of triphenylphosphine (PPh3, 21.0 g) at 40℃, the mixture was stirred for 2 hours at the same temperature, and water (3.8 g) was then added thereto. The reaction solution was distilled under reduced pressure, and the resulting triphenylphosphine oxide solid was diluted with toluene (26.0 g) and n-hexane (41.0 g), and then filtered off. The filtrate was adjusted to pH 2~3 with 1.0 N aqueous hydrochloric acid solution (110.0 g) and then subjected to separation of the layers. To remove any residual triphenylphosphine oxide solid, the aqueous layer obtained above was washed with dichloromethane (100.0 g) and then adjusted to pH 8~9 with 28% aqueous ammonia solution (7.6 g). The aqueous solution thus obtained was extracted with dichloromethane (100.0 g) and distilled under reduced pressure to obtain 8.5 g of the title compound as a white solid.
1H NMR (500 MHz, CDCl3) δ 1.44 (s, 9H), 1.45 (s, 9H), 2.45 (d, J=6.1 Hz, 2H), 2.77 (d, J=5.5 Hz, 2H), 3.87 (br s, 1H), 5.22 (br s, 1H).
EXAMPLE 8: Synthesis of N,N-dibenzyl-L-N(Boc)-aspartamide 4-tert-butyl ester

N-Boc-L-aspartic acid 4-t-butyl ester (29.0 g, 0.10 mol) was added to THF (200 ml). After cooling to temperature below -5℃, to the reaction solution was added isobutylchloroformate (13.0 ml, 0.10 mol) followed by addition of N-methyl morpholine (12.0 ml, 0.10 mol) dropwise, and the reaction mixture was stirred for over 30 minutes. To the reaction mixture was added dropwise dibenzylamine (21.1 ml, 0.11 mol), and the mixture was then stirred for over 3 hours and monitored for the reaction progress by TLC (EtOAc: Hexane=1:4). Upon completion of the reaction, the reaction solution was stirred with addition of ethyl acetate (300.0 mL) and 1 N hydrochloric acid to separate the layers, and distilled under reduced pressure to precipitate a solid. The solid was filtered and washed with ethyl acetate (100 ml), and then the washings were concentrated by distillation again under reduced pressure. The residue was then subjected to silica gel column to obtain the purified desired product (41.7 g, 0.89 mol).
1H NMR (400 MHz, CDCl3) δ: 7.32 (m, 5H), 7.20 (m, 5H), 5.39 (d, J=7.2 Hz, 1H), 5.30 (m, 1H), 4.87-4.77 (m, 2H), 4.48-4.39 (m, 2H), 2.72 (dd, J=15.8 Hz, J=8.0 Hz, 1H), 2.56 (dd, J=15.8 Hz, J=6.4 Hz, 1H), 1.43 (s, 9H), 1.37 (s, 9H).
Mass (ESI, m/z): 491 (M+Na), 469 (M+H), 413 (M-55).
EXAMPLE 9: Synthesis of N, N-diallyl-L-N(Boc)-aspartamide 4-tert-butyl ester

L-N(Boc)-aspartic acid 4-t-butyl ester (5.00 g, 17.3 mol) was added to THF (50 ml). After cooling to temperature below -5℃, to the reaction solution was added isobutylchloroformate (2.26 ml, 17.3 mol) followed by addition of N-methyl morpholine (1.90 ml, 17.3 mol) dropwise, and the reaction mixture was stirred for over 30 minutes. To the reaction mixture was added dropwise diallylamine (2.35 ml, 19.0 mol), and the mixture was then stirred for over 3 hours and monitored for the reaction progress by TLC (EtOAc: Hexane=1:4). Upon completion of the reaction, the reaction solution was stirred with addition of ethyl acetate (60 ml) and 1 N hydrochloric acid and, after separating the layers, concentrated by distillation under reduced pressure. The residue was then subjected to silica gel column to obtain the purified desired product (6.0 g, 16.3 mol).
1H NMR (400 MHz, CDCl3) δ: 5.78 (m, 2H), 5.30 (m, 1H), 5.23-5.11 (m, 1H), 5.30 (m, 1H), 4.93 (m, 1H), 4.11-3.84 (m, 4H), 2.68 (dd, J=15.8 Hz, J=8.0 Hz, 1H), 2.51 (dd, J=15.8 Hz, J=8.0 Hz, 1H), 1.44 (s, 9H), 1.42 (s, 9H).
Mass (ESI, m/z): 391 (M+Na), 369 (M+H), 313 (M-55).
EXAMPLE 10: Synthesis of N,N-dibenzyl-4-amino-3(S)-N(Boc)-aminobutanoic acid 4-tert-butyl ester

10.0 g of the compound obtained from the above Example 8, Ru3(CO)12 (136 mg, 1mol%), and diphenylsilane (19.7 ml, 106.7 mmol) were added to tetrahydrofuran (50 ml), and the reaction solution was stirred under reflux for over 40 hours. The reaction solution was extracted with ethyl acetate (200 ml) and concentrated by distillation under reduced pressure. The residue was then subjected to silica gel column to obtain the purified desired product (4.7 g, 10.5 mmol).
1H NMR (400 MHz, CDCl3) δ: 7.31-7.20 (m, 10H), 5.12 (bs, 1H), 3.90 (bs, 1H), 3.63 (d, J=12.0 Hz, 2H), 3.48 (d, J=12.0 Hz, 2H), 3.24 (m, 1H), 3.16 (bs, 1H), 2.42 (m, 2H), 1.81 (m, 1H), 1.59 (m, 9H), 1.46 (s, 9H), 1.06 (s, 9H).
Mass (ESI, m/z): 455 (M+H), 441 (M-13).
EXAMPLE 11: Synthesis of tert-butyl (3S)-4-amino-3-[(tert-butoxycarbonyl)amino]- 4-oxobutanoate

360.0 g of the starting material, N-Boc-Asp(O-t-Bu)OH, together with Boc2O (353.0 g) and ammonium bicarbonate (NH4HCO3, 123.9 g) was added to dimethylformamide (1174.6 g), and pyridine (61.0 g) was added dropwise thereto at room temperature, and the reaction mixture was then stirred for about 3 hours. Upon completion of the reaction, water (1440 ml) and toluene (1800 ml) were added to the reaction solution and stirred for 30 minutes to separate the layers. The organic layer thus obtained was distilled under reduced pressure to remove t-butanol and toluene to obtain the title compound, which was directly used in the next reaction.
EXAMPLE 12: Synthesis of (S)-tert-butyl 3-(tert-butoxycarbonylamino)-3-cyanopropanoate

To the compound obtained from Example 11 was added dimethylformamide (1019.5 g) followed by addition of cyanuric chloride (112.0 g) dropwise for 1.5 hours at temperature below 25℃. The reaction solution was stirred for one hour at room temperature, and then 0.1 N aqueous sodium hydroxide solution (1850.0 g) and toluene (1860 ml) were added thereto to separate the layers. The organic layer thus obtained was washed once again with water (700 ml) and then distilled under reduced pressure to obtain 318.3 g of the title compound.
1H NMR (500 MHz, CDCl3) δ: 1.44 (s, 9H), 1.45 (s, 9H), 2.45 (d, J=6.1 Hz, 2H), 2.77 (d, J=5.5 Hz, 2H), 3.87 (br s, 1H), 5.22 (br s, 1H).
EXAMPLE 13: Synthesis of tert-butyl (3S)-4-amino-3-[(tert-butoxycarbonyl)amino]- butanoate

To 212.1 g of the compound obtained from the above Example 12 was added acetic acid (4000 ml) followed by addition of 20 wt% Pd(OH)2 (1.1 g) at 40℃. The mixture was stirred for 8 hours while keeping the internal temperature below 45℃ and 3 atmospheric pressure of hydrogen. Upon completion of the reaction, the reaction solution was distilled under reduced pressure to remove acetic acid, diluted with toluene (640 L) and then filtered through a celite. To the filtrate was added 0.25 N aqueous hydrochloric acid solution (1060 ml) to separate the layers. The aqueous layer thus obtained was basified with aqueous ammonia solution (543.1 g) and then extracted with methyl t-butyl ether (MTBE, 1000 ml). The organic layer thus obtained was distilled under reduced pressure to obtain 185.0 g of the title compound.
EXAMPLE 14: Synthesis of 3-t-butoxycarbonylamino-4-(5,5-difluoro-2-oxo- piperidin-1-yl)-butyric acid t-butyl ester

Triethylamine (13.2 g) was added to 16.0 g of the compound obtained from the above Example 1 or 2-1 or 2-2, and 14.1 g of the compound obtained from the above Example 7 or 13, and the mixture was then stirred for 21 hours at 40℃. Then, dichloromethane (154.8 g) and acetic acid (18.3 g) were added, and the mixture was stirred for 5 hours at room temperature. To the resulting reaction solution was added 0.5 N aqueous hydrochloric acid solution (116.8 g) and then, the mixture was stirred for 30 minutes to separate the layers. The organic layer thus obtained was distilled under reduced pressure to obtain 23.6 g of the title compound.
1H NMR (500 MHz, CDCl3) δ: 1.42 (s, 9H), 1.46 (s, 9H), 2.27 (m, 2H), 2.40-2.64 (m, 4H), 3.20 (dd, J=4.3, 13.5 Hz, 1H), 3.56-3.70 (m, 2H), 3.76-3.91 (m, 2H), 4.16 (m, 1H), 5.20 (d, J=8.6 Hz, 1H).
EXAMPLE 15: Synthesis of 3-t-butoxycarbonylamino-4-(5,5-difluoro-2-oxo- piperidin-1-yl)-butyric acid

23.6 g of the compound obtained from the above Example 14 was added to dichloromethane (20.0 g) followed by addition of H3PO4 (30.0 g), and the mixture was stirred for 16 hours at room temperature. After confirming the detachment of all of t-butyl group and t-butyloxycarbonyl group, the reaction solution was adjusted to pH 7.0~8.0 with 10 N aqueous hydrogen peroxide, and Boc2O (16.0 g) was added thereto. After completion of the addition, 10 N aqueous hydrogen peroxide was used to maintain the pH of the reaction solution at 8.0~9.0. After stirring for 3 hours, the resulting sodium phosphate was filtered off, and the filtrate was then adjusted to pH 2.0~3.0 with 3.0 N aqueous hydrochloric acid solution. The resulting solid was filtered and dried under nitrogen to obtain 14.5 g of the title compound.
1H NMR (500 MHz, CDCl3) δ: 1.32 (s, 9H), 2.20-2.43 (m, 6H), 3.26-3.31 (m, 2H), 3.61 (m, 1H), 3.81 (m, 1H), 4.02 (m, 1H), 6.73 (d, J=8.6 Hz, 1H), 12.16 (s, 1H).
For the title compound resulting from the above, its enantiomeric isomers―i.e. S-form and R-form―were measured by HPLC (high-performance liquid chromatography), and an excess of the enantiomeric isomers (S vs. R form) (enantiomeric excess; ee) was then calculated as being ee > 99%. On the other hand, in case of the Comparative Example prepared according to the prior method based on WO 06/104356, as described below, the excess (ee) of enantiomeric isomers (S vs. R form) was 80%. From this, it can be identified that the compound of formula (2) having an optically high purity could be obtained according to the method of the present invention.
COMPARATIVE EXAMPLE 1: Synthesis of 3-t-butoxycarbonylamino-4-(5,5- difluoro-2-oxo-piperidin-1-yl)-butyric acid t-butyl ester
COMPARATIVE EXAMPLE 1-1: Synthesis of methyl 5-amino-4,4-difluoro- pentanoate HCl

To 10.0 g of the compound obtained from Example 1 was added 40 ml of anhydrous ammonia solution (7 M solution in methanol), and the mixture was stirred for 3 hours. The reaction solution was distilled and 30 ml of hydrochloric acid solution saturated with methanol was added dropwise thereto. The reaction mixture was stirred at room temperature and then distilled to obtain 7.2 g of the title compound as a white solid.
1H NMR (500 MHz, CD3OD) δ: 2.35 (m, 2H), 2.59 (t, J=7.6 Hz, 2H), 3.49 (t, J=15.3 Hz, 2H), 3.68 (s, 3H).
COMPARATIVE EXAMPLE 1-2: Synthesis of 3-t-butoxycarbonylamino-4-(5,5- difluoro-2-oxo-piperidin-1-yl)-butyric acid t-butyl ester
To the solution of the compound (1.93 g), as obtained from the above Example 4, dissolved in dichloromethane (20.0 g) and H2O (4.0 g) were added NaBr (0.8 g) and TEMPO (11 mg, 1 mol%). To this reaction solution was slowly added a solution of 5% NaOCl (11.5 g) and NaHCO3 (1.7 g) dissolved in H2O (12.0 g) dropwise for about 2 hours while maintaining the temperature below 5℃. Upon completion of dropwise addition, the reaction solution was stirred for 30 minutes to separate the layers. To the organic layer thus obtained was added the compound (1.6 g) obtained from the above Comparative Example 1-1. After stirring for 15 minutes at room temperature, NaBH(OAc)3 (2.23 g) was added to the reaction solution. After stirring for about 19 hours, 10% aqueous NaHCO3 solution (20.0 g) and 0.5 N aqueous hydrochloric acid solution (20.0 g) were added dropwise to the reaction solution to separate the layers. The organic layer thus obtained was dehydrated under anhydrous MgSO4 to obtain 2.0 g (yield 73%) of the same title compound as Example 14, as a yellow solid. For the title compound resulting from the above, its enantiomeric isomers―i.e., S-form and R-form―were measured by HPLC (high-performance liquid chromatography), and an excess (ee) of the enantiomeric isomers (S vs. R form) was then calculated as being ee = 80%.
PAPER
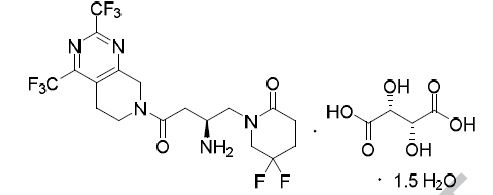
Gemigliptin is a prolyl-specific dipeptidyl aminopeptidase IV (DPP IV, DPP-4, CD26) inhibitor
approved for the treatment of type 2 diabetes mellitus by the Korean Food and Drug Administration in
2012. Gemigliptin was discovered and developed by LG Life Sciences81 and is now the sixth DPP-4
inhibitor approved for the treatment of type 2 diabetes.82 At the time this review was prepared, there
were no publications describing the discovery strategy and preclinical data that led to the advancement
of gemigliptin to the clinic. Additionally, the synthesis of the drug has only been described in the patent
literature.83-85
The molecule was prepared via a convergent route and the synthesis of the dihydropyridopyrimidine
fragment is described in Scheme 11.85 Commercial N-Boc-3-piperidone (71) was treated with lithium
hexamethyldisilazane (LHMDS) followed by ethyl trifluoroacetate to effect a Claisen condensation,
producing diketone 72 in 81% yield. Cyclization of 72 with 2,2,2-trifluoroacetamide (73) gave bistrifluoromethyl
dihydropyridopyrimidine 74 in 23% yield. Removal of the Boc protecting group
efficiently provided amine 75 in 96% yield.
SCHEME 11

The synthesis of the carbon skeleton of the difluoropyridone fragment 80 is described in Scheme
12.84 1,4-Addition of ethyl bromodifluoroacetate (76) to ethyl acrylate (77) in the presence of copper powder and tetramethylethylenediamine (TMEDA) gave diester 78, which was selectively reduced with
sodium borohydride (NaBH4) to give alcohol 79 in 90% overall yield for the two-step procedure.
Alcohol 79 was then treated with perfluorobutanesulfonyl chloride and triethylamine to give activated
alcohol 80 in 75% yield.
87 in 51% yield. Removal of the Boc group with thionyl chloride in ethanol followed by neutralization
with aqueous sodium hydroxide and salt formation with L-tartaric acid provided gemigliptin L-tartrate
hydrate (X) in 97.5% yield.83

The completion of the synthesis of gemigliptin is described in Scheme 13.83, 84 Boc-L-aspartic acid
4-tert-butyl ester (81) was treated with ammonium bicarbonate and pyridine in the presence of di-tertbutyl
dicarbonate to give formamide 82. Dehydration of 82 to give nitrile 83 was accomplished through
reaction with cyanuric chloride in 95% overall yield for the two-step sequence. Hydrogenation of 83 in
the presence of Pearlman’s catalyst provided butyl amine 84. Alkylation of 84 with activated alcohol 80
in triethylamine followed by cyclization in acetic acid afforded difluoropyridone 85. Acidic hydrolysis
of the ester proceeded with concomitant removal of the Boc protecting group, and was followed by
reprotection of the amine with di-tert-butyl dicarbonate to give acid 86 in 84% overall yield for the
three-step procedure in >97% ee. Coupling of 86 with fragment 75 in the presence of
hydroxybenzotriazole (HOBT) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) gave amide

81. Kim, S.-H.; Lee, S.-H.; Yim, H.-J. Arch. Pharmacal Res. 2013, 36, 1185.
82. Juillerat-Jeanneret, L. J. Med. Chem. 2014, 57, 2197.
83. Park, K. S.; Yun, J. M.; Kim, B. C.; Kim, K. Y.; Lee, J. H. WO Patent 2012060590A2, 2012.
84. Kim, B. C.; Kim, K. Y.; Lee, H. B.; An, J. E.; Lee, K. W. WO Patent 2012030106A2, 2012.
85. Lee, C.-S.; Koh, J. S.; Koo, K. D.; Kim, G. T.; Kim, K.-H.; Hong, S. Y.; Kim, S.; Kim, M.-J.;
Yim, H. J.; Lim, D.; Kim, H. J.; Han, H. O.; Bu, S. C.; Kwon, O. H.; Kim, S. H.; Hur, G.-C.;
Kim, J. Y.; Yeom, Z.-H.; Yeo, D.-J. WO Patent 2006104356A1, 2006.
PAPER
Gemigliptin, (LC15-0444, LG Life Sciences)
Gemigliptin is a sitagliptin analogue discovered by LG Life sciences Ltd, Korea via the derivatization of the compounds. It is potent and long acting DPP-IV inhibitor with high selectivity profile (3000-fold) against isoenzymes. The binding mode of gemigliptin is not reported, but expected as sitagliptin due to structural similarity. It inhibited more than 80% of DPP-IV activity and exhibited the bioavailability of 94% in rats. It also showed the lowering of
blood glucose and elevating of GLP-1 levels in dose-dependent manner in the diet-induced obese mice. Gemigliptin displayed a noteworthy lowering in HbA1c level (0.77%) at a dose of 3.0 mg/kg.[57] It is approved by Korean FDA in June 2012 for the treatment of T2DM.[58]
Synthesis of gemigliptin involved the preparation of two key intermediates dihydropyrido[3,4-d]pyrimidine moiety 88 and β-amino acid moiety 92. Compound 86 was prepared by generating enolate from compound 85 using LHMDS and adding trifluoroacetate.
Compound 86 gave the 87 in reflux condition which after Boc deprotection afforded key amine intermediate 88. The β-amino derivative 91 was synthesized by cyclization reaction between 89 and 90, which on benzyl deprotection using Pd/C gave desired β-amino intermediate 92.
Coupling of this intermediate with 88 using EDC/HOBt followed by Boc deprotection offered
gemigliptin 94 via 93 (Scheme 13).[59,60]

[57] S. J. Yang, K. W. Min, S. K. Gupta, J. Y. Park, V. K. Shivane, S. U. Pitale, P. K.
Agarwal, A. Sosale, P. Gandhi, M. Dharmalingam, V. Mohan, U. Mahesh, D. M. Kim, Y.
S. Kim, J. A. Kim, P. K. Kim, and S. H. Baik, Diabetes, Obes. Metab., 2013, 15, 410–
416.
[58] S. H. Kim, S. H. Lee, and H. J. Yim, Arch. Pharm. Res., 2013, 36 (10), 1185-1188.
[59] C. S. Lee, J. S. Koh, K. D. Koo, G. T. Kim, K. H. Kim, S. Y. Hong, S. Kim, M. J. Kim,
H. J. Yim, D. Lim, H. J. Kim, H. O. Han, S. C. Bu, O. H. Kwon, S. H. Kim, G. C. Hur, J.
Y. Kim, Z. H. Yeom, D. J. Yeo, WO 2006/104356 A1, 2006.
[60] K. S. Park, J. M. Yun, B. C. Kim, Y. U. Kim, J. H. Lee, WO 2012/060590, 2012.
| WO2006104356A1 | Mar 30, 2006 | Oct 5, 2006 | Seong Cheol Bu | Dipeptidyl peptidase-iv inhibiting compounds, methods of preparing the same, and pharmaceutical compositions containing the same as an active agent |
| EP0279435A2 * | Feb 18, 1988 | Aug 24, 1988 | BASF Aktiengesellschaft | Process for the reduction of mono- and dicarboxylic acids |
| US5556982 * | Jul 12, 1993 | Sep 17, 1996 | Neorx Corporation | Metal radionuclide labeled proteins for diagnosis and therapy |
| US20080039517 * | Aug 7, 2007 | Feb 14, 2008 | Washburn David G | Pyrrolidinone anilines as progesterone receptor modulators |
Patent
Reference:
[1]. J. Med. Chem., Ahead of Print.
[2]. Clinical therapeutics 2008, 30, 1817-1830.
[3]. Int. J. Res. Dev. Pharm. Life Sci. 2013, 2, 602-610, 609 pp.
[4]. Xenobiotica; the fate of foreign compounds in biological systems 2014, 44, 627-634.
[5]. Poster presented at the annual meeting of American Diabetes Association, 2008, San Francisco: CA.
[6]. Arch. Pharmacal Res. 2013, 36, 1185-1188.
.svg) |
|
| Clinical data | |
|---|---|
| Synonyms | LC15-0444 |
| Routes of administration |
Oral |
| ATC code | |
| Pharmacokinetic data | |
| Bioavailability | 94% (rat), 73% (dog), 26% (monkey) |
| Elimination half-life | 3.6 h (rat), 5.2 h (dog), 5.4 h (monkey) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C18H19F8N5O2 |
| Molar mass | 489.36 g/mol |
| 3D model (JSmol) | |
Footnotes
- ^ Lim KS, Kim JR, Choi YJ, Shin KH, Kim KP, Hong JH, Cho JY, Shin HS, Yu KS, Shin SG, Kwon OH, Hwang DM, Kim JA, Jang IJ (October 2008). “Pharmacokinetics, pharmacodynamics, and tolerability of the dipeptidyl peptidase IV inhibitor LC15-0444 in healthy Korean men: a dose-block-randomized, double-blind, placebo-controlled, ascending single-dose, Phase I study”. Clin Ther. 30 (10): 1817–30. doi:10.1016/j.clinthera.2008.10.013. PMID 19014837.
- ^ Ábel T (2011). “A New Therapy of Type 2 Diabetes: DPP-4 Inhibitors”. In Rigobelo EC. Hypoglycemia – Causes and Occurrences. Croatia: InTech. pp. 3–52. doi:10.5772/23604. ISBN 978-953-307-657-7.
- ^ Kaji K (Mar 2014). “Dipeptidyl peptidase-4 inhibitor attenuates hepatic fibrosis via suppression of activated hepatic stellate cell in rats”. J Gastroenterol.. 49 (3): 481–91. doi:10.1007/s00535-013-0783-4. PMID 23475323.
- ^ Min HS (Jun 2014). “Dipeptidyl peptidase IV inhibitor protects against renal interstitial fibrosis in a mouse model of ureteral obstruction”. Lab. Invest. 94 (5): 598–607. doi:10.1038/labinvest.2014.50. PMID 24687121.
- ^ Gouni-Berthold I (2014). “The role of oral antidiabetic agents and incretin mimetics in type 2 diabetic patients with non-alcoholic Fatty liver disease”. Curr Pharm Des. 20 (5): 3705–15. PMID 24040873.
Further reading
- Kim SE, Yi S, Shin KH, Kim TE, Kim MJ, Kim YH, Yoon SH, Cho JY, Shin SG, Jang IJ, Yu KS (January 2012). “Evaluation of the pharmacokinetic interaction between the dipeptidyl peptidase IV inhibitor LC15-0444and pioglitazone in healthy volunteers”. Int J Clin Pharmacol Ther. 50 (1): 17–23. doi:10.5414/cp201568. PMID 22192641.
- Rhee EJ, Lee WY, Yoon KH, Yoo SJ, Lee IK, Baik SH, Kim YK, Lee MK, Park KS, Park JY, Cha BS, Lee HW, Min KW, Bae HY, Kim MJ, Kim JA, Kim DK, Kim SW (December 2010). “A multicenter, randomized, placebo-controlled, double-blind phase II trial evaluating the optimal dose, efficacy and safety of LC 15-0444 in patients with type 2 diabetes”. Diabetes Obes Metab. 12 (12): 1113–1119. doi:10.1111/j.1463-1326.2010.01303.x. PMID 20977584.
- Lim KS, Cho JY, Kim BH, Kim JR, Kim HS, Kim DK, Kim SH, Yim HJ, Lee SH, Shin SG, Jang IJ, Yu KS (December 2009). “Pharmacokinetics and pharmacodynamics of LC15-0444, a novel dipeptidyl peptidase IV inhibitor, after multiple dosing in healthy volunteers”. Br J Clin Pharmacol. 68 (6): 883–890. doi:10.1111/j.1365-2125.2009.03376.x. PMC 2810799. PMID 20002082.
External links
////////////////gemigliptin, LC15-0444, KOREA 2012, Gemigliptin tartrate sesquihydrate, GLIPTIN, ゲミグリプチン , LG Chem, Sanofi
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SON
