
Home » 2020 APPROVALS
Category Archives: 2020 APPROVALS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Imdevimab
| (Heavy chain) QVQLVESGGG VVQPGRSLRL SCAASGFTFS NYAMYWVRQA PGKGLEWVAV ISYDGSNKYY ADSVKGRFTI SRDNSKNTLY LQMNSLRTED TAVYYCASGS DYGDYLLVYW GQGTLVTVSS ASTKGPSVFP LAPSSKSTSG GTAALGCLVK DYFPEPVTVS WNSGALTSGV HTFPAVLQSS GLYSLSSVVT VPSSSLGTQT YICNVNHKPS NTKVDKKVEP KSCDKTHTCP PCPAPELLGG PSVFLFPPKP KDTLMISRTP EVTCVVVDVS HEDPEVKFNW YVDGVEVHNA KTKPREEQYN STYRVVSVLT VLHQDWLNGK EYKCKVSNKA LPAPIEKTIS KAKGQPREPQ VYTLPPSRDE LTKNQVSLTC LVKGFYPSDI AVEWESNGQP ENNYKTTPPV LDSDGSFFLY SKLTVDKSRW QQGNVFSCSV MHEALHNHYT QKSLSLSPGK (Light chain) QSALTQPASV SGSPGQSITI SCTGTSSDVG GYNYVSWYQQ HPGKAPKLMI YDVSKRPSGV SNRFSGSKSG NTASLTISGL QSEDEADYYC NSLTSISTWV FGGGTKLTVL GQPKAAPSVT LFPPSSEELQ ANKATLVCLI SDFYPGAVTV AWKADSSPVK AGVETTTPSK QSNNKYAASS YLSLTPEQWK SHRSYSCQVT HEGSTVEKTV APTECS (Disulfide bridge: H22-H96, H147-H203, H223-L215, H229-H’229, H264-H324-H370-H428, H’22-H’96, H’147-H’203, H’223-L’215, H’264-H’324, H’370-H’428, L22-L90, L138-L197, L’22-L’90, L’138-L’197) |
イムデビマブ;
- Immunoglobulin G1, anti-(severe acute respiratory syndrome coronavirus 2 spike glycoprotein) (human monoclonal REGN10987 γ1-chain), disulfide with human monoclonal REGN10987 λ-chain, dimer
| Formula | C6396H9882N1694O2018S42 |
|---|---|
| CAS | 2415933-40-1 |
| Mol weight | 144141.7693 |
Monoclonal antibody
Treatment and prophylaxis of SARS-CoV-2 infection
ANTIVIRAL
SARS-CoV-2 spike glycoprotein
- REGN 10987
- RG 6412
Fact Sheet – US Food and Drug Administration
https://www.fda.gov › media › download
PDFBenefit of treatment with casirivimab and imdevimab has not been observed in patients hospitalized due to COVID-19. Monoclonal antibodies, such as casirivimab.
Casirivimab/imdevimab, sold under the brand name REGEN-COV,[1] is an experimental medicine developed by the American biotechnology company Regeneron Pharmaceuticals. It is an artificial “antibody cocktail” designed to produce resistance against the SARS-CoV-2 coronavirus responsible for the COVID-19 pandemic.[3][4] It consists of two monoclonal antibodies, casirivimab (REGN10933) and imdevimab (REGN10987) that must be mixed together.[1][5][6] The combination of two antibodies is intended to prevent mutational escape.[7]
Trials
In a clinical trial of people with COVID-19, casirivimab and imdevimab, administered together, were shown to reduce COVID-19-related hospitalization or emergency room visits in people at high risk for disease progression within 28 days after treatment when compared to placebo.[2] The safety and effectiveness of this investigational therapy for use in the treatment of COVID-19 continues to be evaluated.[2]
The data supporting the emergency use authorization (EUA) for casirivimab and imdevimab are based on a randomized, double-blind, placebo-controlled clinical trial in 799 non-hospitalized adults with mild to moderate COVID-19 symptoms.[2] Of these participants, 266 received a single intravenous infusion of 2,400 milligrams casirivimab and imdevimab (1,200 mg of each), 267 received 8,000 mg casirivimab and imdevimab (4,000 mg of each), and 266 received a placebo, within three days of obtaining a positive SARS-CoV-2 viral test.[2]
The prespecified primary endpoint for the trial was time-weighted average change in viral load from baseline.[2] Viral load reduction in participants treated with casirivimab and imdevimab was larger than in participants treated with placebo at day seven.[2] However, the most important evidence that casirivimab and imdevimab administered together may be effective came from the predefined secondary endpoint of medically attended visits related to COVID-19, particularly hospitalizations and emergency room visits within 28 days after treatment.[2] For participants at high risk for disease progression, hospitalizations and emergency room visits occurred in 3% of casirivimab and imdevimab-treated participants on average compared to 9% in placebo-treated participants.[2] The effects on viral load, reduction in hospitalizations and ER visits were similar in participants receiving either of the two casirivimab and imdevimab doses.[2]
As of September 2020, REGEN-COV is being evaluated as part of the RECOVERY Trial.[8]
On 12 April 2021, Roche and Regeneron announced that the Phase III clinical trial REGN-COV 2069 met both primary and secondary endpoints, reducing risk of infection by 81% for the non-infected patients, and reducing time-to-resolution of symptoms for symptomatic patients to one week vs. three weeks in the placebo group.[9]
Authorization
On 21 November 2020, the U.S. Food and Drug Administration (FDA) issued an emergency use authorization (EUA) for casirivimab and imdevimab to be administered together for the treatment of mild to moderate COVID-19 in people twelve years of age or older weighing at least 40 kilograms (88 lb) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19.[2][10][11] This includes those who are 65 years of age or older or who have certain chronic medical conditions.[2] Casirivimab and imdevimab must be administered together by intravenous (IV) infusion.[2]
Casirivimab and imdevimab are not authorized for people who are hospitalized due to COVID-19 or require oxygen therapy due to COVID-19.[2] A benefit of casirivimab and imdevimab treatment has not been shown in people hospitalized due to COVID-19.[2] Monoclonal antibodies, such as casirivimab and imdevimab, may be associated with worse clinical outcomes when administered to hospitalized people with COVID-19 requiring high flow oxygen or mechanical ventilation.[2]
The EUA was issued to Regeneron Pharmaceuticals Inc.[2][10][12]
On 1 February 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) started a rolling review of data on the REGN‑COV2 antibody combination (casirivimab/imdevimab), which is being co-developed by Regeneron Pharmaceuticals, Inc. and F. Hoffman-La Roche, Ltd (Roche) for the treatment and prevention of COVID‑19.[13][14] In February 2021, the CHMP concluded that the combination, also known as REGN-COV2, can be used for the treatment of confirmed COVID-19 in people who do not require supplemental oxygen and who are at high risk of progressing to severe COVID-19.[15]
The Central Drugs Standards Control Organisation (CDSCO) in India, on 5 May 2021, granted an Emergency Use Authorisation to Roche (Genentech)[16] and Regeneron[17] for use of the casirivimab/imdevimab cocktail in the country. The announcement came in light of the second wave of the COVID-19 pandemic in India. Roche India maintains partnership with Cipla, thereby permitting the latter to market the drug in the country.[18]
Deployment
Although Regeneron is headquartered in Tarrytown, New York (near New York City), REGEN-COV is manufactured at the company’s primary U.S. manufacturing facility in Rensselaer, New York (near the state capital at Albany).[19] In September 2020, to free up manufacturing capacity for REGEN-COV, Regeneron began to shift production of its existing products from Rensselaer to the Irish city of Limerick.[20]
Regeneron has a deal in place with Roche (Genentech)[21]to manufacture and market REGEN-COV outside the United States.[10][22]
On 2 October 2020, Regeneron Pharmaceuticals announced that US President Donald Trump had received “a single 8 gram dose of REGN-COV2” after testing positive for SARS-CoV-2.[23][24] The drug was provided by the company in response to a “compassionate use” (temporary authorization for use) request from the president’s physicians.[23]
References
- ^ Jump up to:a b c “REGEN-COV- casirivimab and imdevimab kit”. DailyMed. Retrieved 18 March 2021.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “Coronavirus (COVID-19) Update: FDA Authorizes Monoclonal Antibodies for Treatment of COVID-19”. U.S. Food and Drug Administration (FDA) (Press release). 21 November 2020. Retrieved 21 November 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Kelland K (14 September 2020). “Regeneron’s antibody drug added to UK Recovery trial of COVID treatments”. Reuters. Retrieved 14 September 2020.
- ^ “Regeneron’s COVID-19 Response Efforts”. Regeneron Pharmaceuticals. Retrieved 14 September 2020.
- ^ Morelle R (14 September 2020). “Antibody treatment to be given to Covid patients”. BBC News Online. Retrieved 14 September2020.
- ^ “Safety, Tolerability, and Efficacy of Anti-Spike (S) SARS-CoV-2 Monoclonal Antibodies for Hospitalized Adult Patients With COVID-19”. ClinicalTrials. 3 September 2020. Retrieved 14 September2020.
- ^ Baum A, Fulton BO, Wloga E, Copin R, Pascal KE, Russo V, et al. (August 2020). “Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies”. Science. 369 (6506): 1014–1018. Bibcode:2020Sci…369.1014B. doi:10.1126/science.abd0831. PMC 7299283. PMID 32540904.
- ^ “RECOVERY COVID-19 phase 3 trial to evaluate Regeneron’s REGN-COV2 investigational antibody cocktail in the UK”. Recovery Trial. Retrieved 14 September 2020.
- ^ “Phase III prevention trial showed subcutaneous administration of investigational antibody cocktail casirivimab and imdevimab reduced risk of symptomatic COVID-19 infections by 81%”. streetinsider.com. Archived from the original on 2021-04-12. Retrieved 2021-04-12.
- ^ Jump up to:a b c “Regeneron Reports Positive Interim Data with REGEN-COV Antibody Cocktail used as Passive Vaccine to Prevent COVID-19”(Press release). Regeneron Pharmaceuticals. 26 January 2021. Retrieved 19 March 2021 – via PR Newswire.
- ^ “Fact Sheet For Health Care Providers Emergency Use Authorization (EUA) Of Casirivimab And Imdevimab” (PDF). U.S. Food and Drug Administration (FDA).
- ^ “Casirivimab and Imdevimab”. Regeneron Pharmaceuticals. Retrieved 19 March 2021.
- ^ “EMA starts rolling review of REGN‑COV2 antibody combination (casirivimab / imdevimab)” (Press release). European Medicines Agency (EMA). 1 February 2021. Retrieved 1 February 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “EMA reviewing data on monoclonal antibody use for COVID-19” (Press release). European Medicines Agency (EMA). 4 February 2021. Retrieved 4 March 2021.
- ^ “EMA issues advice on use of REGN-COV2 antibody combination (casirivimab / imdevimab)” (Press release). European Medicines Agency (EMA). 26 February 2021. Retrieved 5 March 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^https://www.businesswire.com/news/home/20200818005847/en/Genentech-and-Regeneron-Collaborate-to-Significantly-Increase-Global-Supply-of-REGN-COV2-Investigational-Antibody-Combination-for-COVID-19
- ^ https://timesofindia.indiatimes.com/india/india-approves-roche/regeneron-antibody-cocktail-to-treat-covid-19/articleshow/82407551.cms
- ^ “Roche receives Emergency Use Authorisation in India for its investigational Antibody Cocktail (Casirivimab and Imdevimab) used in the treatment of Covid-19 | Cipla”. http://www.cipla.com. Retrieved 2021-05-06.
- ^ Williams, Stephen (3 October 2020). “Experimental drug given to President made locally”. The Daily Gazette.
- ^ Stanton, Dan (11 September 2020). “Manufacturing shift to Ireland frees up US capacity for Regeneron’s COVID antibodies”. BioProcess International.
- ^https://www.businesswire.com/news/home/20200818005847/en/Genentech-and-Regeneron-Collaborate-to-Significantly-Increase-Global-Supply-of-REGN-COV2-Investigational-Antibody-Combination-for-COVID-19
- ^ “Roche and Regeneron link up on a coronavirus antibody cocktail”. CNBC. 19 August 2020. Retrieved 14 September 2020.
- ^ Jump up to:a b Thomas K (2 October 2020). “President Trump Received Experimental Antibody Treatment”. The New York Times. ISSN 0362-4331. Retrieved 2 October 2020.
- ^ Hackett DW (3 October 2020). “8-Gram Dose of COVID-19 Antibody Cocktail Provided to President Trump”. http://www.precisionvaccinations.com. Archived from the original on 3 October 2020.
External links
- “Casirivimab”. Drug Information Portal. U.S. National Library of Medicine.
- “Imdevimab”. Drug Information Portal. U.S. National Library of Medicine.
- “Casirivimab and Imdevimab EUA Letter of Authorization” (PDF). U.S. Food and Drug Administration (FDA).
- “Frequently Asked Questions on the Emergency Use Authorization of Casirivimab + Imdevimab” (PDF). U.S. Food and Drug Administration (FDA).
| REGN10933 (blue) and REGN10987 (orange) bound to SARS-CoV-2 spike protein (pink). From PDB: 6VSB, 6XDG. | |
| Combination of | |
|---|---|
| Casirivimab | Monoclonal antibody against spike protein of SARS-CoV-2 |
| Imdevimab | Monoclonal antibody against spike protein of SARS-CoV-2 |
| Clinical data | |
| Trade names | REGEN-COV |
| Other names | REGN-COV2 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Casirivimab |
| Routes of administration | Intravenous |
| ATC code | None |
| Legal status | |
| Legal status | US: Unapproved (Emergency Use Authorization)[1][2] |
| Identifiers | |
| DrugBank | DB15691 |
| KEGG | D11938D11939 |
////////Imdevimab, ANTI VIRAL, PEPTIDE, CORONA VIRUS, COVID19, APPROVALS 2020, FDA 2020, イムデビマブ, REGN 10987, RG 6412,
NEW DRUG APPROVALS
one time
$10.00
Casirivimab
(Heavy chain)
QVQLVESGGG LVKPGGSLRL SCAASGFTFS DYYMSWIRQA PGKGLEWVSY ITYSGSTIYY
ADSVKGRFTI SRDNAKSSLY LQMNSLRAED TAVYYCARDR GTTMVPFDYW GQGTLVTVSS
ASTKGPSVFP LAPSSKSTSG GTAALGCLVK DYFPEPVTVS WNSGALTSGV HTFPAVLQSS
GLYSLSSVVT VPSSSLGTQT YICNVNHKPS NTKVDKKVEP KSCDKTHTCP PCPAPELLGG
PSVFLFPPKP KDTLMISRTP EVTCVVVDVS HEDPEVKFNW YVDGVEVHNA KTKPREEQYN
STYRVVSVLT VLHQDWLNGK EYKCKVSNKA LPAPIEKTIS KAKGQPREPQ VYTLPPSRDE
LTKNQVSLTC LVKGFYPSDI AVEWESNGQP ENNYKTTPPV LDSDGSFFLY SKLTVDKSRW
QQGNVFSCSV MHEALHNHYT QKSLSLSPGK
(Light chain)
DIQMTQSPSS LSASVGDRVT ITCQASQDIT NYLNWYQQKP GKAPKLLIYA ASNLETGVPS
RFSGSGSGTD FTFTISGLQP EDIATYYCQQ YDNLPLTFGG GTKVEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Disulfide bridge: H22-H96, H147-H203, H223-L214, H229-H’229, H232-H’232, H264-H324, H370-H428, H’22-H’96, H’147-H’203, H’223-L’214, H’264-H’324, H’370-H’428, L23-L88, L134-L194, L’23-L’88, L’134-L’194)
Casirivimab
カシリビマブ;
- Immunoglobulin G1, anti-(severe acute respiratory syndrome coronavirus 2 spike glycoprotein) (human monoclonal REGN10933 γ1-chain), disulfide with human monoclonal REGN10933 κ-chain, dimer
| Formula | C6454H9976N1704O2024S44 |
|---|---|
| CAS | 2415933-42-3 |
| Mol weight | 145233.3296 |
Monoclonal antibody
Treatment and prophylaxis of SARS-CoV-2 infection (COVID-19)
SARS-CoV-2 spike glycoprotein
- Protein Sequence
- Sequence Length: 1328, 450, 450, 214, 214
- REGN 10933
- RG 6413
https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-monoclonal-antibodies-treatment-covid-19 November 21, 2020
Today, the U.S. Food and Drug Administration issued an emergency use authorization (EUA) for casirivimab and imdevimab to be administered together for the treatment of mild to moderate COVID-19 in adults and pediatric patients (12 years of age or older weighing at least 40 kilograms [about 88 pounds]) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19. This includes those who are 65 years of age or older or who have certain chronic medical conditions.
In a clinical trial of patients with COVID-19, casirivimab and imdevimab, administered together, were shown to reduce COVID-19-related hospitalization or emergency room visits in patients at high risk for disease progression within 28 days after treatment when compared to placebo. The safety and effectiveness of this investigational therapy for use in the treatment of COVID-19 continues to be evaluated.
Casirivimab and imdevimab must be administered together by intravenous (IV) infusion.
Casirivimab and imdevimab are not authorized for patients who are hospitalized due to COVID-19 or require oxygen therapy due to COVID-19. A benefit of casirivimab and imdevimab treatment has not been shown in patients hospitalized due to COVID-19. Monoclonal antibodies, such as casirivimab and imdevimab, may be associated with worse clinical outcomes when administered to hospitalized patients with COVID-19 requiring high flow oxygen or mechanical ventilation.
“The FDA remains committed to advancing the nation’s public health during this unprecedented pandemic. Authorizing these monoclonal antibody therapies may help outpatients avoid hospitalization and alleviate the burden on our health care system,” said FDA Commissioner Stephen M. Hahn, M.D. “As part of our Coronavirus Treatment Acceleration Program, the FDA uses every possible pathway to make new treatments available to patients as quickly as possible while continuing to study the safety and effectiveness of these treatments.”
Monoclonal antibodies are laboratory-made proteins that mimic the immune system’s ability to fight off harmful pathogens such as viruses. Casirivimab and imdevimab are monoclonal antibodies that are specifically directed against the spike protein of SARS-CoV-2, designed to block the virus’ attachment and entry into human cells.
“The emergency authorization of these monoclonal antibodies administered together offers health care providers another tool in combating the pandemic,” said Patrizia Cavazzoni, M.D., acting director of the FDA’s Center for Drug Evaluation and Research. “We will continue to facilitate the development, evaluation and availability of COVID-19 therapies.”
The issuance of an EUA is different than an FDA approval. In determining whether to issue an EUA, the FDA evaluates the totality of available scientific evidence and carefully balances any known or potential risks with any known or potential benefits of the product for use during an emergency. Based on the FDA’s review of the totality of the scientific evidence available, the agency has determined that it is reasonable to believe that casirivimab and imdevimab administered together may be effective in treating patients with mild or moderate COVID-19. When used to treat COVID-19 for the authorized population, the known and potential benefits of these antibodies outweigh the known and potential risks. There are no adequate, approved and available alternative treatments to casirivimab and imdevimab administered together for the authorized population.
The data supporting this EUA for casirivimab and imdevimab are based on a randomized, double-blind, placebo-controlled clinical trial in 799 non-hospitalized adults with mild to moderate COVID-19 symptoms. Of these patients, 266 received a single intravenous infusion of 2,400 milligrams casirivimab and imdevimab (1,200 mg of each), 267 received 8,000 mg casirivimab and imdevimab (4,000 mg of each), and 266 received a placebo, within three days of obtaining a positive SARS-CoV-2 viral test.
The prespecified primary endpoint for the trial was time-weighted average change in viral load from baseline. Viral load reduction in patients treated with casirivimab and imdevimab was larger than in patients treated with placebo at day seven. However, the most important evidence that casirivimab and imdevimab administered together may be effective came from the predefined secondary endpoint of medically attended visits related to COVID-19, particularly hospitalizations and emergency room visits within 28 days after treatment. For patients at high risk for disease progression, hospitalizations and emergency room visits occurred in 3% of casirivimab and imdevimab-treated patients on average compared to 9% in placebo-treated patients. The effects on viral load, reduction in hospitalizations and ER visits were similar in patients receiving either of the two casirivimab and imdevimab doses.
Under the EUA, fact sheets that provide important information about using casirivimab and imdevimab administered together in treating COVID-19 as authorized must be made available to health care providers and to patients and caregivers. These fact sheets include dosing instructions, potential side effects and drug interactions. Possible side effects of casirivimab and imdevimab include: anaphylaxis and infusion-related reactions, fever, chills, hives, itching and flushing.
The EUA was issued to Regeneron Pharmaceuticals Inc.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
Related Information
- Casirivimab and Imdevimab EUA Letter of Authorization
- Frequently Asked Questions on the Emergency Use Authorization for Casirivimab and Imdevimab
- Emergency Use Authorization: Therapeutics
- Coronavirus Disease (COVID-19)
Casirivimab/imdevimab, sold under the brand name REGEN-COV,[1] is an experimental medicine developed by the American biotechnology company Regeneron Pharmaceuticals. It is an artificial “antibody cocktail” designed to produce resistance against the SARS-CoV-2 coronavirus responsible for the COVID-19 pandemic.[3][4] It consists of two monoclonal antibodies, casirivimab (REGN10933) and imdevimab (REGN10987) that must be mixed together.[1][5][6] The combination of two antibodies is intended to prevent mutational escape.[7]
Trials
In a clinical trial of people with COVID-19, casirivimab and imdevimab, administered together, were shown to reduce COVID-19-related hospitalization or emergency room visits in people at high risk for disease progression within 28 days after treatment when compared to placebo.[2] The safety and effectiveness of this investigational therapy for use in the treatment of COVID-19 continues to be evaluated.[2]
The data supporting the emergency use authorization (EUA) for casirivimab and imdevimab are based on a randomized, double-blind, placebo-controlled clinical trial in 799 non-hospitalized adults with mild to moderate COVID-19 symptoms.[2] Of these participants, 266 received a single intravenous infusion of 2,400 milligrams casirivimab and imdevimab (1,200 mg of each), 267 received 8,000 mg casirivimab and imdevimab (4,000 mg of each), and 266 received a placebo, within three days of obtaining a positive SARS-CoV-2 viral test.[2]
The prespecified primary endpoint for the trial was time-weighted average change in viral load from baseline.[2] Viral load reduction in participants treated with casirivimab and imdevimab was larger than in participants treated with placebo at day seven.[2] However, the most important evidence that casirivimab and imdevimab administered together may be effective came from the predefined secondary endpoint of medically attended visits related to COVID-19, particularly hospitalizations and emergency room visits within 28 days after treatment.[2] For participants at high risk for disease progression, hospitalizations and emergency room visits occurred in 3% of casirivimab and imdevimab-treated participants on average compared to 9% in placebo-treated participants.[2] The effects on viral load, reduction in hospitalizations and ER visits were similar in participants receiving either of the two casirivimab and imdevimab doses.[2]
As of September 2020, REGEN-COV is being evaluated as part of the RECOVERY Trial.[8]
On 12 April 2021, Roche and Regeneron announced that the Phase III clinical trial REGN-COV 2069 met both primary and secondary endpoints, reducing risk of infection by 81% for the non-infected patients, and reducing time-to-resolution of symptoms for symptomatic patients to one week vs. three weeks in the placebo group.[9]
Authorization
On 21 November 2020, the U.S. Food and Drug Administration (FDA) issued an emergency use authorization (EUA) for casirivimab and imdevimab to be administered together for the treatment of mild to moderate COVID-19 in people twelve years of age or older weighing at least 40 kilograms (88 lb) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19.[2][10][11] This includes those who are 65 years of age or older or who have certain chronic medical conditions.[2] Casirivimab and imdevimab must be administered together by intravenous (IV) infusion.[2]
Casirivimab and imdevimab are not authorized for people who are hospitalized due to COVID-19 or require oxygen therapy due to COVID-19.[2] A benefit of casirivimab and imdevimab treatment has not been shown in people hospitalized due to COVID-19.[2] Monoclonal antibodies, such as casirivimab and imdevimab, may be associated with worse clinical outcomes when administered to hospitalized people with COVID-19 requiring high flow oxygen or mechanical ventilation.[2]
The EUA was issued to Regeneron Pharmaceuticals Inc.[2][10][12]
On 1 February 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) started a rolling review of data on the REGN‑COV2 antibody combination (casirivimab/imdevimab), which is being co-developed by Regeneron Pharmaceuticals, Inc. and F. Hoffman-La Roche, Ltd (Roche) for the treatment and prevention of COVID‑19.[13][14] In February 2021, the CHMP concluded that the combination, also known as REGN-COV2, can be used for the treatment of confirmed COVID-19 in people who do not require supplemental oxygen and who are at high risk of progressing to severe COVID-19.[15]
The Central Drugs Standards Control Organisation (CDSCO) in India, on 5 May 2021, granted an Emergency Use Authorisation to Roche (Genentech)[16] and Regeneron[17] for use of the casirivimab/imdevimab cocktail in the country. The announcement came in light of the second wave of the COVID-19 pandemic in India. Roche India maintains partnership with Cipla, thereby permitting the latter to market the drug in the country.[18]
Deployment
Although Regeneron is headquartered in Tarrytown, New York (near New York City), REGEN-COV is manufactured at the company’s primary U.S. manufacturing facility in Rensselaer, New York (near the state capital at Albany).[19] In September 2020, to free up manufacturing capacity for REGEN-COV, Regeneron began to shift production of its existing products from Rensselaer to the Irish city of Limerick.[20]
Regeneron has a deal in place with Roche (Genentech)[21]to manufacture and market REGEN-COV outside the United States.[10][22]
On 2 October 2020, Regeneron Pharmaceuticals announced that US President Donald Trump had received “a single 8 gram dose of REGN-COV2” after testing positive for SARS-CoV-2.[23][24] The drug was provided by the company in response to a “compassionate use” (temporary authorization for use) request from the president’s physicians.[23]
References
- ^ Jump up to:a b c “REGEN-COV- casirivimab and imdevimab kit”. DailyMed. Retrieved 18 March 2021.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “Coronavirus (COVID-19) Update: FDA Authorizes Monoclonal Antibodies for Treatment of COVID-19”. U.S. Food and Drug Administration (FDA) (Press release). 21 November 2020. Retrieved 21 November 2020. This article incorporates text from this source, which is in the public domain.
- ^ Kelland K (14 September 2020). “Regeneron’s antibody drug added to UK Recovery trial of COVID treatments”. Reuters. Retrieved 14 September 2020.
- ^ “Regeneron’s COVID-19 Response Efforts”. Regeneron Pharmaceuticals. Retrieved 14 September 2020.
- ^ Morelle R (14 September 2020). “Antibody treatment to be given to Covid patients”. BBC News Online. Retrieved 14 September2020.
- ^ “Safety, Tolerability, and Efficacy of Anti-Spike (S) SARS-CoV-2 Monoclonal Antibodies for Hospitalized Adult Patients With COVID-19”. ClinicalTrials. 3 September 2020. Retrieved 14 September2020.
- ^ Baum A, Fulton BO, Wloga E, Copin R, Pascal KE, Russo V, et al. (August 2020). “Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies”. Science. 369 (6506): 1014–1018. Bibcode:2020Sci…369.1014B. doi:10.1126/science.abd0831. PMC 7299283. PMID 32540904.
- ^ “RECOVERY COVID-19 phase 3 trial to evaluate Regeneron’s REGN-COV2 investigational antibody cocktail in the UK”. Recovery Trial. Retrieved 14 September 2020.
- ^ “Phase III prevention trial showed subcutaneous administration of investigational antibody cocktail casirivimab and imdevimab reduced risk of symptomatic COVID-19 infections by 81%”. streetinsider.com. Archived from the original on 2021-04-12. Retrieved 2021-04-12.
- ^ Jump up to:a b c “Regeneron Reports Positive Interim Data with REGEN-COV Antibody Cocktail used as Passive Vaccine to Prevent COVID-19”(Press release). Regeneron Pharmaceuticals. 26 January 2021. Retrieved 19 March 2021 – via PR Newswire.
- ^ “Fact Sheet For Health Care Providers Emergency Use Authorization (EUA) Of Casirivimab And Imdevimab” (PDF). U.S. Food and Drug Administration (FDA).
- ^ “Casirivimab and Imdevimab”. Regeneron Pharmaceuticals. Retrieved 19 March 2021.
- ^ “EMA starts rolling review of REGN‑COV2 antibody combination (casirivimab / imdevimab)” (Press release). European Medicines Agency (EMA). 1 February 2021. Retrieved 1 February 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “EMA reviewing data on monoclonal antibody use for COVID-19” (Press release). European Medicines Agency (EMA). 4 February 2021. Retrieved 4 March 2021.
- ^ “EMA issues advice on use of REGN-COV2 antibody combination (casirivimab / imdevimab)” (Press release). European Medicines Agency (EMA). 26 February 2021. Retrieved 5 March 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^https://www.businesswire.com/news/home/20200818005847/en/Genentech-and-Regeneron-Collaborate-to-Significantly-Increase-Global-Supply-of-REGN-COV2-Investigational-Antibody-Combination-for-COVID-19
- ^ https://timesofindia.indiatimes.com/india/india-approves-roche/regeneron-antibody-cocktail-to-treat-covid-19/articleshow/82407551.cms
- ^ “Roche receives Emergency Use Authorisation in India for its investigational Antibody Cocktail (Casirivimab and Imdevimab) used in the treatment of Covid-19 | Cipla”. http://www.cipla.com. Retrieved 2021-05-06.
- ^ Williams, Stephen (3 October 2020). “Experimental drug given to President made locally”. The Daily Gazette.
- ^ Stanton, Dan (11 September 2020). “Manufacturing shift to Ireland frees up US capacity for Regeneron’s COVID antibodies”. BioProcess International.
- ^https://www.businesswire.com/news/home/20200818005847/en/Genentech-and-Regeneron-Collaborate-to-Significantly-Increase-Global-Supply-of-REGN-COV2-Investigational-Antibody-Combination-for-COVID-19
- ^ “Roche and Regeneron link up on a coronavirus antibody cocktail”. CNBC. 19 August 2020. Retrieved 14 September 2020.
- ^ Jump up to:a b Thomas K (2 October 2020). “President Trump Received Experimental Antibody Treatment”. The New York Times. ISSN 0362-4331. Retrieved 2 October 2020.
- ^ Hackett DW (3 October 2020). “8-Gram Dose of COVID-19 Antibody Cocktail Provided to President Trump”. http://www.precisionvaccinations.com. Archived from the original on 3 October 2020.
External links
- “Casirivimab”. Drug Information Portal. U.S. National Library of Medicine.
- “Imdevimab”. Drug Information Portal. U.S. National Library of Medicine.
- “Casirivimab and Imdevimab EUA Letter of Authorization” (PDF). U.S. Food and Drug Administration (FDA).
- “Frequently Asked Questions on the Emergency Use Authorization of Casirivimab + Imdevimab” (PDF). U.S. Food and Drug Administration (FDA).
| REGN10933 (blue) and REGN10987 (orange) bound to SARS-CoV-2 spike protein (pink). From PDB: 6VSB, 6XDG. | |
| Combination of | |
|---|---|
| Casirivimab | Monoclonal antibody against spike protein of SARS-CoV-2 |
| Imdevimab | Monoclonal antibody against spike protein of SARS-CoV-2 |
| Clinical data | |
| Trade names | REGEN-COV |
| Other names | REGN-COV2 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Casirivimab |
| Routes of administration | Intravenous |
| ATC code | None |
| Legal status | |
| Legal status | US: Unapproved (Emergency Use Authorization)[1][2] |
| Identifiers | |
| DrugBank | DB15691 |
| KEGG | D11938 |
//////////// Casirivimab, ANTI VIRAL, PEPTIDE, SARS-CoV-2, MONOCLONAL ANTIBODY, FDA 2020, 2020APPROVALS, CORONA VIRUS, COVID 19, カシリビマブ, REGN-COV2, REGN10933+REGN10987 combination therapy, REGN 10933, RG 6413
NEW DRUG APPROVALS
ONE TIME
$10.00
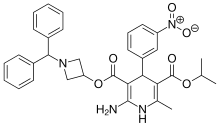
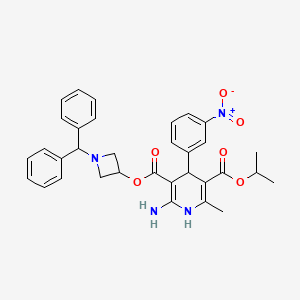
Azelnidipine
Azelnidipine
C33H34N4O6, 582.6 g/mol
CAS 123524-52-7
3-(1-Benzhydrylazetidin-3-yl) 5-isopropyl 2-amino-6-methyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate
CS-905, RS-9054
Approved India cdsco 2020
SYN REF https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4245158/
MP 95-98 °C AND NMR WO 2004058745 . EP 266922
Azelnidipine is a dihydropyridine calcium channel blocker. It is marketed by Daiichi-Sankyo pharmaceuticals, Inc. in Japan. It has a gradual onset of action and produces a long-lasting decrease in blood pressure, with only a small increase in heart rate, unlike some other calcium channel blockers. It is currently being studied for post-ischemic stroke management.
Azelnidipine (INN; marketed under the brand name CalBlock — カルブロック) is a dihydropyridine calcium channel blocker. Azelnidipine is L and T calcium channel blocker. It is sold in Japan by Daiichi-Sankyo pharmaceuticals, Inc. Unlike nicardipine, it has a gradual onset and has a long-lasting hypotensive effect, with little increase in heart rate. Drug Controller General Of India (DCGI) has approved the use of azelnipine in India. It is launched under the brand name Azusa (ajanta pharma ltd.)[1] In 2020.
Chemical Synthesis
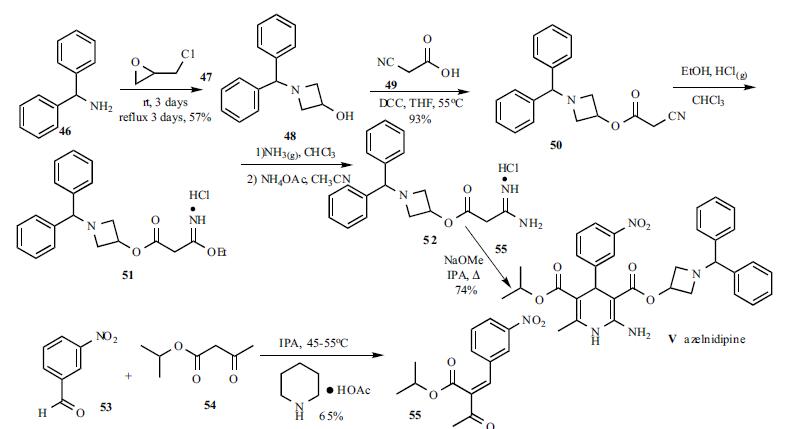
A solution of benzhydrylamine (46) and epichlorohydrin (47) was mixed without adding solvent to give azetidinol 48 in 57% yield. DCC coupling between cyanoacetic acid (49) and azetidinol 48 in hot THF gave ester 50 in 93% yield. Cyanoester 50 was treated with ethanol and HCl gas in chloroform to give imidate HCl salt 51, which was treated with ammonia gas in chloroform and ammonium acetate in acetonitrile to give the corresponding amidinoacetate 52. A modified Hantzsch reaction was employed to construct the 2-amino-1,4- dihydropyridine core structure. Compound 52 was condensed with 2-(3-nitrobenzylidene)acetic acid isopropyl ester (55) in the presence of NaOMe in refluxing isopropanol to give the cyclized product, azelnidipine (V) in 74% yield. Benzylideneacetoacetate 55 was obtained through the Knoevenagel reaction employing 3-nitrobenzaldehyde (53) and isopropyl acetoacetate (54) in isopropanol containing a catalytic amount of piperidinium acetate at 45-55oC in 65% yield.
PATENT
EP 266922
IN 201621044802
CN 106279109
CN 107188885
CN 105461691
CN 103509003
CN 103183663
CN 102382104
JP 2012020970 A
PAPER
Bioanalysis (2019), 11(4), 251-266.
PAPER
Asian Journal of Chemistry (2014), 26(15), 4675-4678.
PAPER
http://www.asianjournalofchemistry.co.in/User/ViewFreeArticle.aspx?ArticleID=26_16_30

Azelnidipine is designated chemically as 3-(1-benzhydrylazetidin-3-yl)-5-isopropyl-2-amino-6-methyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate. Its literature synthesis (Scheme-I) involves 3-nitrobenzaldehyde 5 with isopropyl acetoacetate 6. The product of (Z)-isopropyl 2-(3- nitrobenzylidene)-3-oxobutanoate (7a, b, c), on treatment with piperidine and acetic acid, coupling of (7) and 1-benzhydrylazetidin-3-yl 3-amino-3-iminopropanoate acetate (8) gave azelnidipine (1).
PAPER
International Research Journal of Pharmacy (2012), 3(8), 191-192.
Chemical & Pharmaceutical Bulletin (1995), 43(5), 797-817.
PATENT
https://patents.google.com/patent/WO2014139410A1/en
The invention belongs to the technical field of medicine and provides an important intermediate of dihydropyridine calcium antagonist adipine, 3-amino-3-iminopropionic acid-1-(diphenylhydrazinyl)-3-azetidine The synthesis process of ester acetate. Background technique
Azelnidipine is a new type of dihydropyridine calcium channel blocker developed by Sankyo and Ube Industries of Japan. It was approved for sale in Japan in late May 2003 under the trade name Calblock. Adipine has a selective blockade of calcium channels in arterial smooth muscle cells, it can dilate blood vessels, reduce peripheral vascular resistance and arterial pressure, and is widely used clinically for mild or moderate essential hypertension, renal disorders with hypertension And treatment of severe hypertension. Compared with nicardipine and nifedipine dihydropyridine calcium channel blockers, adipine is superior in selectivity, long-lasting and long-lasting, and has little effect on the heart.

阿折地平的结构式
A flat floor structure
At present, references to the preparation of agdipine include: European patents EP0266922; Chinese patent CN201010516967.7; Chinese Journal of Medicinal Chemistry, 2010, 20 (3): 192-194; Chinese Journal of Pharmaceutical Industry, 2008, 39 (3): 163-165; Chemical Industry and Engineering, 2009, 26 ( 1 ): 15-18; Qilu Pharmacy, 2005, 24 (6): 365-366. The preparation method of adipine in these literatures is based on the reaction of epichlorohydrin and diphenylamine with N-alkylation, cyclization, esterification, Pinner synthesis, neutralization, and oxime reaction. The intermediate 3-amino-3-iminopropionic acid-1-(diphenylfluorenyl)-3-azetidinyl acetate is prepared first, followed by 2-(3-nitrobenzylidene)acetyl Acepinedipine was obtained by the Hantzsch condensation of isopropyl acetate.
The control of the solvent and reaction conditions in the esterification, Pinner synthesis and neutralization three-step reaction in this route is critical. Using the preparation methods provided by these documents, we found that the operation was cumbersome and the yield and purity were not satisfactory.
In the esterification reaction, according to the method specifically reported in the above literature, the highest yield of the obtained product is only 85%, and the purity is poor, it is difficult to purify, and it is difficult to obtain a solid product.

副产物 (7 )和(8 )结构式 发明内容 We have found that 3-amino-3-iminopropionic acid-1- (3) is prepared by a three-step reaction from cyanoacetate-1-diphenylhydrazin-3-azetidinyl ester (3) according to the method specifically reported in the above literature. Diphenylhydrazino)-3-azetidinyl acetate (6), the reaction operation is cumbersome, and it is easy to produce by-products of hydrolysis of ester bonds and hydrolysis of imid bonds (7) and (8), three-step reaction. The total yield is only 20~30%, and the purification of the product is difficult, which seriously affects the quality of the final product and greatly increases the production cost.
Byproducts (7) and (8) structural formula Summary of the invention
It is an object of the present invention to provide a process for the preparation of the key intermediate of adipine, 3-amino-3-iminopropionic acid-1-(diphenylhydrazinyl)-3-azetidinyl acetate. The adipine intermediate of the present invention 3-amino-3-iminopropionic acid-1-(diphenylhydrazinyl)-3-azetidinyl acetate acetate has the following structural formula:

The preparation method of 3-amino-3-iminopropionic acid-1-(diphenylindenyl)-3-azetidinyl acetate of the present invention comprises the following steps: 1) Esterification: 1-diphenylhydrazin-3-azetidinol (2), cyanoacetic acid (1) and N,N-dicyclohexylcarbodiimide (DCC) in organic solvent at 0~ Reacting at 80 ° C, to obtain 7-diphenylindolyl-3-azetidinyl cyanoacetate (3);
2) Pinner reaction: Add intermediate (3), absolute ethanol to dichlorosilane, stir and cool
To -20~25 °C, dry hydrogen chloride gas is passed, and then the reaction solution is kept sealed at -20~25 °C to obtain 3-imino-3-ethoxypropionic acid-1-(diphenylfluorenyl) -3-azetidinyl ester hydrochloride (4);
3) Neutralization reaction: The intermediate (4) is dissolved in dichloromethane, and the base is added at -5 to 25 ° C to obtain 3-imino-3-ethoxypropionic acid-1-(diphenylhydrazine). Benzyl-3-azetidinyl ester (5);
4) Formation reaction: The intermediate (5) is dissolved in acetonitrile, ammonium acetate is added, and the temperature is raised to 40 to 60 ° C to obtain 3-amino-3-iminopropionic acid-1-(diphenylfluorenyl)-3. – azetidinium acetate compound (6). detailed description
Example
1. Preparation of cyanoacetic acid-1-diphenylhydrazine-3-azetidine (esterification)

Method 1: Add 1-diphenylhydrazin-3-azetidinol (2, 235 g, 0.983 mol) and cyanoacetic acid (1, 100 g, 1.18 mol) to 1.5 mL of dichloromethane, and stir until fully dissolved. Ν, Ν-dicyclohexylcarbodiimide (DCC, 243 g, 1.18 mol) was added at 0-10 ° C and allowed to react at room temperature for 3 h. After the completion of the reaction, the reaction mixture was cooled to 0 to 5 ° C, and filtered, filtered, washed with a small portion of dichloromethane. The organic solvent was evaporated to dryness under reduced pressure and dried to give 275 g of white solid.
Method 2: chloroform was used as the reaction solvent, and the operation was the same as above, and the reaction was carried out at 55 ° C for 5 hours, the HPLC purity was 98.7%, and the product yield was 95.3%.
Method 3: Ethyl acetate was used as the reaction solvent, and the operation was the same as above, and the reaction was carried out at 55 ° C for 2 h, the HPLC purity was 98.9%, and the product yield was 96.1%.

Method 4: Using hydrazine as the reaction solvent, the operation was the same as above, and the reaction was carried out at 55 ° C for 7 h, the HPLC purity was 98.5%, and the product yield was 94.7%. 2. Preparation of 3-imino-3-ethoxypropionic acid-1-(diphenylfluorenyl)-3-azetidinyl ester hydrochloride (Pinner reaction)
Intermediate 3 (270 g, 0.882 mol), absolute ethanol (61.8 mL, 1.06 mol) was added to 1.5 L of dry dichloromethane, cooled to -5 to 0 ° C in a water salt bath, and dried. HC1 gas for 2.5 h, after the completion of the aeration, the reaction solution was kept under stirring at 0 ° C for 6 h.
Allow to stand overnight at 0-4 °C. After completion of the reaction, the solvent was evaporated under reduced pressure to give an oily viscous intermediate 4 .
3. Preparation of 3-imino-3-ethoxypropionic acid-1-(diphenylfluorenyl)-3-azetidinyl ester

Method 1: Add 1.4 L of dichloromethane to Intermediate 4, cool to 0-5 ° C, add dry diethylamine (182 mL, 1.76 mol) to the solution, adjust pH 7-8, continue to stir after the dropwise addition. 2h. The mixture was suction filtered, and the filtrate was evaporated to dryness vacuo.
Method 2: Diamine is used for neutralization, and the operation is the same as above.
Method 3: Triethylamine is used for neutralization, and the operation is the same as above.
Method 4: Ethylenediamine is used for neutralization, and the operation is the same as above.
Method 5: Add 1.4 L of dichloromethane to Intermediate 4, cool to 0-5 ° C, add potassium carbonate (242.88 g, 1.76 mol) to the solution in portions, adjust pH 7-8, continue stirring for 2 h. . The mixture was suction filtered, and the filtrate was evaporated to dryness vacuo. Method 6: Neutralize with sodium carbonate, and operate as above.
Method 7: Neutralize with sodium hydroxide, and operate as above.

4. Preparation of 3-amino-3-iminopropionic acid-1-(diphenylindenyl)-3-azetidinyl acetate (formed into 脒)
To the intermediate 5, 1.2 L of acetonitrile was added, and after dissolution, ammonium acetate (68.0 g, 0.882 mol) was added, and the mixture was heated to 55 ° C for 6 h. After the reaction, it was naturally cooled, crystallization, suction filtration, acetonitrile washing cake, and dried to give 236 g of a white solid. The total yield of the three-step reaction was 69.9 73.1%.
PAPER
https://pubs.rsc.org/en/content/articlelanding/2015/cc/c4cc09337b#!divAbstract
Abstract
A protocol for the coupling of 3-iodoazetidines with Grignard reagents in the presence of an iron catalyst has been developed. A variety of aryl, heteroaryl, vinyl and alkyl Grignards were shown to participate in the coupling process to give the products in good to excellent yields. Furthermore, a short formal synthesis towards a pharmacologically active molecule was shown.

http://www.rsc.org/suppdata/cc/c4/c4cc09337b/c4cc09337b1.pdfPATENThttps://patents.google.com/patent/CN103509003A/zhAzelnidipine, whose chemical name is 3-(1-diphenylmethylazetidin-3-yl) 5-isopropyl 2-amino-1,4-dihydro-6-methyl 4-(3-nitrophenyl)-3,5-pyridinedicarboxylate, developed by Japan Sankyo Co., Ltd. and approved to be marketed in Japan in late May 2003. The existing synthesis method of azedipine is cumbersome, and the preparation of intermediate (VI) adopts column chromatography method, and the purification of product (I) also uses column chromatography method, which is not suitable for industrial production.
A method for preparing azeldipine, which is characterized in that it is prepared by the following steps.
[0006]

Description of the drawings:
Figure 1 is a flow chart of the synthesis process of azeldipine.
[0025] Example 12-Preparation of (3-nitrobenzylidene) isopropyl acetoacetate (III)
[0026] Add 2.1kg of 3-nitrobenzaldehyde and 5L of isopropanol to the reaction kettle, start stirring, add 3kg of isopropyl acetoacetate, and stir. Add 43ml of anhydrous piperidine and 12ml of glacial acetic acid, and continue to stir until the solid is completely dissolved. Heat the temperature to 45°C and keep the reaction for 6h, then lower the temperature, stir and crystallize for 16h. Filter and collect the resulting filter cake. Put the obtained filter cake and 16L ethanol (industrial) into the reaction kettle, start stirring, beating, filtering, and collecting the filter cake. Put the filter cake in the baking tray, put it in the oven, and dry at 70-80°C. Collect the product 2-(3-nitrobenzylidene) isopropyl acetoacetate (III), about 2.7 kg.
[0027] Example 21-Preparation of benzhydryl-3-hydroxyazetidine (Intermediate V)
[0028] 9.6L of methanol, 5.4kg of benzhydrylamine (IV) and 3.33kg of epichlorohydrin were added to the reaction kettle, stirred at room temperature for 48 hours, the reaction was completed, the temperature was raised to 68°C, and the reaction was refluxed for 72h. Cool to room temperature. Concentrate under reduced pressure to remove methanol, and collect the filter cake by filtration. The filter cake was put into the reaction kettle, 19.2L of ether and 13.75L of 3mol/L NaOH solution were added, stirred, and the water layer was released after standing still. The ether layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was collected. The ether was recovered under reduced pressure to dryness to obtain about 3.05 kg of 1-benzyl-3-hydroxyazetidine (Intermediate V).
[0029] Example 3 Preparation of cyanoacetic acid (1-diphenylmethylazetidin-3-yl) ester (Intermediate VI)
[0030] Put about 3.05g of intermediate (V), 27L of tetrahydrofuran and 1.7kg of cyanoacetic acid into the reactor, start stirring, turn on the chilled water of the reactor to cool down, and slowly add 3.1kgN, N’-dicyclohexyl to the reactor Diimine, control the temperature at IO0C -15°C, after the addition, close the chilled water in the reactor. Turn on the heating system, slowly increase the temperature to 55-60°C, and react for 10 hours. The material liquid was cooled to room temperature, filtered, and the filtrate was concentrated to dryness. Put 16.8L of ethyl acetate into the reaction kettle, stir to dissolve, then wash with water, dry with anhydrous sodium sulfate, filter, and collect the filtrate. Ethyl acetate was recovered under reduced pressure, petroleum ether was added to the solid residue, stirred, and filtered to obtain cyanoacetic acid (1-diphenylmethylazetidin-3-yl) ester (Intermediate VI), about 3.19 kg.
[0031] Example 4 Preparation of amidinoacetic acid (1-diphenylmethylazetidin-3-yl) ester acetate (VII)
[0032] Put 25L of dichloromethane, about 3.19kg of intermediate (VI), and 430g of ethanol into the reactor, start stirring, cool to below 0°C, and pass in hydrogen chloride gas until the temperature stabilizes below 0°C, at 0°C Let stand for 14 hours at °C. Concentrate under reduced pressure to remove most of the hydrogen chloride gas and recover the solvent dichloromethane. Add 25L of dichloromethane to the residue of the reaction kettle, stir, cool to below 0°C, and pass in ammonia until the temperature stabilizes below 0°C, and filter . The filtrate was poured into the reactor, concentrated under reduced pressure to recover the solvent to obtain a colorless liquid, added 22.8L of acetonitrile and 905g of amine acetate, heated to 55-60°C for 1.5 hours, stopped the reaction, filtered while hot, and recovered the filtrate under reduced pressure Solvent to dryness, add 3L of ether to the residue to crystallize, filter, and dry to obtain amidinoacetic acid (1-diphenylmethylazetidin-3-yl) ester acetate (Intermediate VII) about 3.2kg .
[0033] Example 5. Add about 3.2kg of Intermediate (VII), about 2.7kg of Intermediate (III), 21L of isopropanol and 585g of sodium methoxide to the reaction kettle, start stirring, heat to reflux and react for 4 hours, and cool to Below 10°C, filter, the filtrate is decompressed to recover the solvent to dryness, add 35L ethyl acetate to the residue to dissolve, wash with 6.5LX3 water, release the water layer, add anhydrous sodium sulfate to the ethyl acetate layer to dry, filter , Collect the filtrate, recover ethyl acetate under reduced pressure, add 4.2L of toluene to the residue,
3.4L of n-hexane was heated to dissolve, filtered, the filtrate was stirred to room temperature to crystallize, filtered and collected and dried, and the product was placed in an oven at 45-55°C to dry to obtain the crude azedipine (I), about 2.3kg.
[0034] Example 6, Refining
[0035] Put 8.8L ethyl acetate and 8.8L n-hexane into the reaction kettle, turn on the stirring, put about 2.3kg of the crude azeldipine into the reaction kettle, slowly heat up until the material is dissolved, add 180g of activated carbon and stir for 0.5h, while it is hot Filter, hydraulically filter the material to the crystallization dad, wash the filter cake with 5.5L ethyl acetate and 4.5L n-hexane solution, combine with the filtrate, cool to 0~5°C to crystallize, filter, collect the product, and place it in a hot air circulating oven After drying at 45-55°C, 2.2 g of azeldipine is obtained. The purity is 99.6% as measured by high performance liquid chromatography. The refined yield is 96.0%.
[0036] Example 7 Azedipin Refining
[0037] The mixed solvent was prepared according to the volume ratio of ethyl acetate and n-hexane of 2:1, 22L of the mixed solvent was put into the reactor, about 2.3kg of azedipine crude product was put into the reactor, and the temperature was slowly heated until the material was dissolved, Add 180g of activated carbon and stir for 0.5h, filter while hot, filter the material hydraulically into a crystallization kettle, wash the filter cake with a mixed solvent, combine the washing liquid with the filtrate, cool to 0~5°C for crystallization, filter, collect the product, and circulate the hot air Dry in an oven at a temperature of 45-55°C to obtain 2.2 g of azeldipine fine product, with a purity of 99.7% measured by high performance liquid chromatography.
[0038] Example 8 prepared a mixed solvent at a volume ratio of ethyl acetate and n-hexane of 1.5:1, put 22L of the mixed solvent into the reactor, put about 2.3kg of crude azeldipine into the reactor, and slowly heated to Dissolve the material, add 180g of activated carbon and stir for 0.5h, filter while it is hot, filter the material hydraulically into a crystallization kettle, wash the filter cake with a mixed solvent, combine the washing liquid and the filtrate, cool to 0~5°C to crystallize, filter, and collect the product. Dry in a hot air circulating oven at a temperature of 45-55°C to obtain
2.2g azeldipine is a fine product with a purity of 99.6% measured by high performance liquid chromatography.
PATENT
https://patents.google.com/patent/CN103183663B/zh
Azelnidipine (Azelnidipine) is a new type of dihydropyridine calcium channel blocker jointly developed by Sankyo Co., Ltd. and Ube Industries Co., Ltd., which inhibits the entry of calcium ions into excitable tissues and causes peripheral blood vessels And coronary artery vasodilation plays a role in lowering blood pressure. Clinically, it is widely used in patients with mild or moderate symptoms of primary hypertension, hypertension with renal dysfunction, and severe hypertension. Compared with similar antihypertensive drugs, azeldipine has a slow and long-lasting antihypertensive effect.
[0004] The chemical structure of azeldipine is similar to that of nifedipine:

[0006] The Chinese patent CN87107150.9 reported the compound earlier and gave a detailed introduction to its synthesis; afterwards, most of the synthesis of azeldipine adopts this route:

[0008] The reaction takes o-nitrobenzaldehyde and isopropyl acetoacetate as raw materials to prepare intermediate compound 5; takes benzhydrylamine and epichlorohydrin as raw materials to prepare compound 2, compound 2 and cyanoacetic acid act in DCC Compound 3 is prepared by the next reaction. Compound 3 is added with ethanol under the action of hydrogen chloride gas, ammonia gas ammonolysis, and acetate anion exchange to obtain compound 4. Compound 4 and compound 5 are under the action of sodium methoxide to obtain compound 1, namely azeldipine.
[0009] Wherein: Compound 3 can be purchased as an industrial product, or can be prepared according to the traditional method reported in the literature; Compound 5 is prepared according to the traditional method reported in the literature.
[0010] In the process of preparing amidine 4 in the traditional reaction route, hydrogen chloride gas and ammonia gas need to be passed in successively. Therefore, the reaction requires anhydrous reagents. According to literature reports, the reaction yield is about 70%. From the perspective of industrial synthesis, The application of anhydrous reagents will undoubtedly increase the cost, while the use of gas will increase the difficulty of operation and require the use of high-pressure equipment. At the same time, post-reaction processing is difficult and industrial production is difficult. Therefore, this step of the reaction requires further improvement.
With acetonitrile as a solvent, the crude product of reaction 2) was stirred until dissolved, ammonium acetate was added, and acetate anion exchange was performed to obtain the amidine compound 4;

[0018] The second step: use toluene as a solvent, compound 4 and compound 5 in the use of sodium amide to obtain compound 1, namely azedipine

[0020] The preferred technical solution of the present invention is characterized in that the temperature of reaction 1) is controlled below _5°C
Example 1: Preparation of azeldipine
[0030] Add 50 g of compound 3, 1500 mL of dichloromethane, and 16.64 mL of absolute ethanol to a 5L three-necked flask, and under mechanical stirring, pass HC1 gas below -5 °C to saturation, and after saturation, keep the reaction at -5 °C for 24 hours. Protect from light and nitrogen, slowly add the above reaction system to 1665ml of ammonia water with a concentration of 2.5-3.0% under the control of 0-5°C. After the addition, stir for 0.5h, stand for 0.5h, and separate the liquids. The dichloromethane layer was washed once with 2000 mL of saturated brine, left standing for 1.0 h, separated, and the dichloromethane layer was drained under reduced pressure to obtain a white solid. Without drying, it was directly added to 2000 mL of acetonitrile, and the temperature was slowly heated to dissolve. Add 11.7g of ammonium acetate, control the temperature at 55°C -60°C, and react for 2h under mechanical stirring. After cooling, the solid precipitated, filtered, and dried to obtain 57.55 g of amidine 4, the yield was 91.2%, the HPLC purity was 99.63%, and the melting point was 130-132.3°C.
[0031] 50g amidine 4, 43.5g compound 5, 1000mL toluene, and 7.7g sodium amide were added into a 1000mL three-necked flask, mechanically stirred, heated to reflux, and reacted for 4 hours. TLC detects that the reaction is complete and cools to room temperature to crystallize. Filter, put the solid directly into the mixed solution of toluene and n-hexane (1:1.2-1.5) without drying, heat up to reflux to clear, cool to 56°C naturally, add seed crystals, stop stirring, and cool to 25° C, filter. The solid was purified once more according to the above method, and dried under reduced pressure at 40°C for 48 hours to obtain 66.87g of α-crystal form of Azedipine, yield 88.2%, melting point: 121-123°C.
[0032] Example 2; Preparation of Azeldipine
[0033] Add 50g of compound 3, 1500mL of dichloromethane, 16·64mL of absolute ethanol into a 5L three-necked flask, and under mechanical stirring, pass HC1 gas below -5°C to saturation, and after saturation, -6°C to -8°C Incubate the reaction for 24h. Under the control of 0-5 °C, slowly add the above reaction system to ammonia water with a concentration of 2.5-3.0%, adjust the pH to 7.8-8.5, after adding, stir for 0.5h, stand for 0.5h, and separate. The dichloromethane layer was washed once with 2000 mL of saturated brine, left standing for 1.0 h, separated, and the dichloromethane layer was drained under reduced pressure to obtain a white solid. Without drying, it was directly added to 2000 mL of acetonitrile, and the temperature was slowly heated to dissolve. Add 11.7g of ammonium acetate, control the temperature at 55°C-60°C, and react for 2h under mechanical stirring. After cooling, the solid precipitated, filtered, and dried to obtain 59.0 lg of amidine 4 with a yield of 93.5%, an HPLC purity of 99.52%, and a melting point of 130.1-132.0°C.
[0034] 50g amidine 4, 43.5g compound 5, 1000mL toluene and 7.7g sodium amide were added to a 1000mL three-necked flask, mechanically stirred, heated to reflux, and reacted for 4 hours. TLC detects that the reaction is complete and cools to room temperature to crystallize. Filter, put the solid directly into the mixed solution of toluene and n-hexane (1:1.2-1.5) without drying, heat up to reflux to clear, cool to 56°C naturally, add seed crystals, stop stirring, and cool to 25° C, filter. The solid was refined once more according to the above method, and dried under reduced pressure at 40°C for 48 hours to obtain 68.31 g of α-crystal azedipine, yield 90.01%, melting point: 121 -123 °C.
[0035] Example 3: Preparation of Amidine 4
[0036] Add 50g of compound 3, 1500mL of dichloromethane, 16·64mL of absolute ethanol into a 5L three-necked flask, and under mechanical stirring, pass HC1 gas below -5°C to saturation, and after saturation, -7°C to -9°C Incubate the reaction for 24h. Under the control of 0-5 °C, slowly add the above reaction system to the ammonia water with a concentration of 2.5-3.0%, adjust the pH to 8.5-9.5, after adding, stir for 0.5h, stand for 0.5h, and separate. The dichloromethane layer was washed once with 2000 mL of saturated brine, left standing for 1.0 h, separated, and the dichloromethane layer was drained under reduced pressure to obtain a white solid. Without drying, it was directly added to 2000 mL of acetonitrile, and the temperature was slowly heated to dissolve. Add 11.7g of ammonium acetate, control the temperature at 55°C-60°C, and react for 2h under mechanical stirring. After cooling, the solid precipitated, filtered, and dried to obtain 59.5 g of amidine 4, HPLC purity 99.78%, melting point: 130.7-132·2°C.

Step 2: Using toluene as a solvent, compound 4 and compound 5 under the action of sodium amide to obtain compound 1, namely azeldipine

PATENThttps://patents.google.com/patent/CN102453023A/zh
detailed description
[0007] In the synthesis workshop, benzhydrylamine is used as a raw material to be synthesized by addition, cyclization, esterification, acidification, ammoniation, condensation and other reactions. The crude azeodipine is refined, dried, mixed and packaged in a clean area. Fold the ground. The specific response is as follows:
[0008] 1. Addition and cyclization reaction
[0009] Methanol, benzhydrylamine, and epichlorohydrin were added to the reaction kettle, stirred at room temperature for 24hr, the reaction was completed, the reaction was heated to reflux for 24hr, cooled, filtered to collect the precipitated solid, and then the mother liquor was concentrated to recover the raw materials, and the heating was continued to reflux 18 After hours, collect the product, add dichloromethane and H2O to the obtained solid, adjust the pH to 10-11 with 40% NaOH while stirring in an ice bath, stand still, separate the organic layer, dry with anhydrous magnesium sulfate, and recover the dichloromethane under reduced pressure To dryness, a colorless solid compound III (1-benzyl-3-hydroxyazetidine) is obtained. After improvement, the raw materials are fully reacted, and the reaction yield of this step is improved. The mass yield is 75%. % Mentioned 85%.
[0010]

[0011] 2. Esterification reaction
[0012] Add THF, compound (III), and cyanoacetic acid to the reaction kettle, stir evenly, add DCC in batches under ice bath stirring, control the temperature at 10°C~15°C, after the addition, remove the ice water bath, and slowly heat up React at 55°C~60°C for 18h. After the reaction is complete, cool, filter to remove insoluble materials, concentrate the filtrate to dryness, add ethyl acetate to the residue to dissolve, wash with water, dry with anhydrous magnesium sulfate, and recover ethyl acetate under reduced pressure. The residue was added with petroleum ether and stirred for crystallization, and the solid was collected by filtration to obtain compound IV (1-diphenylmethyl-3-azetidinyl cyanoacetate).
[0013]

[0014] 3. Acidification and amination reaction
[0015] Dichloromethane, ethanol and intermediate (IV) were added to the reaction kettle respectively, mixed and stirred, cooled to about _5 ° C in an ice salt bath, and dried hydrogen chloride gas was introduced until saturation (about 1.5 hours) after . Let stand overnight at about -5°C, recover the solvent under reduced pressure at room temperature, add dichloromethane to the residue and stir, cool to about _5°C in an ice-salt bath, pass in the dried ammonia gas until saturation (about 3 hours) , Filtration to remove the insoluble matter, and the filtrate was decompressed to recover solvent at room temperature. Acetonitrile and ammonium acetate were added to the residue respectively, and the temperature was raised to 55~60°C for 2 hours with stirring. After the reaction was completed, it was cooled and filtered. 3-Azacyclobutanylamidinoacetate acetate), the reaction in this step is controlled at about _5°C, and the transesterification
The side reaction is reduced, and the reaction yield is improved.
[0016]

[0017] 4. Condensation reaction
[0018] Add isopropanol, intermediate (III’), sodium methoxide and compound V to the reaction kettle, mix and stir, heat to reflux and react for 5 hours. After the reaction is complete, cool and filter, and the filtrate is decompressed to recover the solvent to dryness, leaving residue Add ethyl acetate to dissolve, wash with water, dry with anhydrous magnesium sulfate, recover ethyl acetate under reduced pressure to 1/4 of the total volume, add n-hexane, and stir at 50°C for 30 min. After cooling and crystallization, the solid was collected by filtration, and air-dried at 45°C to obtain the crude azedipine (I). After the crude product was dissolved in ethyl acetate-n-hexane mixed solvent, activated carbon was added for decolorization and impurity removal to achieve the purpose of purification.

[0020] The refined product is dissolved in dioxane, refluxed with n-hexane, cooled and crystallized, and dried to obtain a solid that is boiled in cyclohexane, cooled and filtered, and dried to obtain α-crystalline form Azedipine.
Patent
Publication numberPriority datePublication dateAssigneeTitleCN102453023A *2010-10-212012-05-16大丰市天生药业有限公司Process for producing azelnidipineCN103130700A *2013-03-142013-06-05沈阳中海药业有限公司Preparation method of azelnidipine intermediateCN103509003A *2012-06-272014-01-15威海威太医药技术开发有限公司Preparation method of azelnidipine
JP3491506B2 *1997-10-142004-01-26宇部興産株式会社Method for producing dihydropyridine derivativeCN101475521B *2008-11-132010-11-10青岛黄海制药有限责任公司Method for synthesizing acetate of 1-benzhydryl-3-azetidine amidino acetic ester
TitleLIU, JIAN-FENG ET AL.: “Improved Synthesis of Azelnidipine”, CHINESE JOURNAL OF MEDICINAL CHEMISTRY, vol. 20, no. 3, 30 June 2010 (2010-06-30), pages 192 – 194 *ZHANG, KAI ET AL.: “Synthesis of Azelnidipine”, CHINESE JOURNAL OF PHARMACEUTICALS, vol. 39, no. 3, 31 March 2008 (2008-03-31), pages 163 – 165, XP025959789, DOI: doi:10.1016/j.ejphar.2008.12.041 *
CN103130700B *2013-03-142015-04-29沈阳中海药业有限公司Preparation method of azelnidipine intermediateCN104860855B *2014-12-082017-06-16宁夏紫光天化蛋氨酸有限责任公司A kind of preparation method of the methylmercapto butyric acid ester of 2 hydroxyl of the D of high-purity, L 4CN105949102A *2016-06-202016-09-21许昌豪丰化学科技有限公司Production method of azelnidipine intermediatePublication numberPriority datePublication dateAssigneeTitleWO2014139410A1 *2013-03-142014-09-18Shenyang Zhonghai Pharmaceutical Co., Ltd.A kind of preparation method of azeldipine intermediateCN105461691A *2015-12-312016-04-06Weihai Disu Pharmaceutical Co., Ltd.A kind of preparation method of azeldipineCN106279109A *2016-08-182017-01-04Weihai Disu Pharmaceutical Co., Ltd.A kind of preparation method of azeldipineCN106543061A *2016-10-202017-03-29Weihai Disu Pharmaceutical Co., Ltd.Preparation method of N-diphenylmethylcyclobutane-3-alcohol
References
- ^ Oizumi K, Nishino H, Koike H, Sada T, Miyamoto M, Kimura T (September 1989). “Antihypertensive effects of CS-905, a novel dihydropyridine Ca++ channel blocker”. Jpn. J. Pharmacol. 51 (1): 57–64. doi:10.1254/jjp.51.57. PMID 2810942.
| Clinical data | |
|---|---|
| Trade names | CalBlock,AZUSA,Azovas |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | Oral |
| ATC code | none |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 123524-52-7 |
| PubChem CID | 65948 |
| ChemSpider | 59352 |
| UNII | PV23P19YUG |
| KEGG | D01145 |
| ChEMBL | ChEMBL1275868 |
| CompTox Dashboard (EPA) | DTXSID3020120 |
| ECHA InfoCard | 100.162.151 |
| Chemical and physical data | |
| Formula | C33H34N4O6 |
| Molar mass | 582.657 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILES[O-][N+](=O)c1cccc(c1)C5C(/C(=O)OC(C)C)=C(\NC(\N)=C5\C(=O)OC4CN(C(c2ccccc2)c3ccccc3)C4)C | |
| hideInChIInChI=1S/C33H34N4O6/c1-20(2)42-32(38)27-21(3)35-31(34)29(28(27)24-15-10-16-25(17-24)37(40)41)33(39)43-26-18-36(19-26)30(22-11-6-4-7-12-22)23-13-8-5-9-14-23/h4-17,20,26,28,30,35H,18-19,34H2,1-3H3 Key:ZKFQEACEUNWPMT-UHFFFAOYSA-N |
/////////Azelnidipine, CS-905, RS-9054, INDIA 2020, APPROVALS 2020
#Azelnidipine, #CS-905, #RS-9054, #INDIA 2020, #APPROVALS 2020
CC1=C(C(C(=C(N1)N)C(=O)OC2CN(C2)C(C3=CC=CC=C3)C4=CC=CC=C4)C5=CC(=CC=C5)[N+](=O)[O-])C(=O)OC(C)C
Fenfluramine Hydrochloride

Fenfluramine
- DEA No. 1670
- S 768
2020/12/18, FDA APPROVED, Fintepla
Fenfluramine hydrochloride
| Formula | C12H16F3N. HCl |
|---|---|
| CAS | 404-82-0458-24-2 (FREE) |
| Mol weight | 267.7183 |
Antiobesity
| Efficacy | Appetite suppressant |
|---|---|
| Disease | Dravet syndrome |
(+-)-Fenfluramine hydrochloride
Racemic fenfluramine hydrochloride
Fenfluramine hydrochloride [USAN]
1-(m-Trifluoromethylphenyl)-2-(ethylamino)propane hydrochloride
Research Code:ZX-008
MOA:Serotonin agonist
Indication:Dravet syndrome
Company:Zogenix (Originator)
Synonyms of Fenfluramine [INN:BAN]
- (+-)-Fenfluramine
- BRN 4783711
- dl-Fenfluramine
- DL-Fenfluramine
- EINECS 207-276-3
- Fenfluramina
- Fenfluramina [DCIT]
- Fenfluramine
- Fenfluraminum
- Fenfluraminum [INN-Latin]
- HSDB 3080
- Obedrex
- Pesos
- Ponderax PA
- Rotondin
- S 768
- UNII-2DS058H2CF
mp 160-161 °C, ethyl acetate US 3198834
nmr Salsbury, Jonathon S.; Magnetic Resonance in Chemistry 2005, V43(11), P910-917 C
IR BIORAD: Infrared spectral data from the Bio-Rad/Sadtler IR Data Collection was obtained from Bio-Rad Laboratories, Philadelphia, PA (US).FenfluramineCAS Registry Number: 458-24-2CAS Name:N-Ethyl-a-methyl-3-(trifluoromethyl)benzeneethanamineAdditional Names:N-ethyl-a-methyl-m-(trifluoromethyl)phenethylamine; 2-ethylamino-1-(3-trifluoromethylphenyl)propaneManufacturers’ Codes: S-768Molecular Formula: C12H16F3NMolecular Weight: 231.26Percent Composition: C 62.32%, H 6.97%, F 24.65%, N 6.06%Literature References: Prepn: L. G. Beregi et al.,FRM1658; eidem,US3198833 (1963, 1965 both to Sci. Union et Cie Soc. Franc. Recherche Méd.). Prepn of optical isomers: eidem,US3198834 (1965 to Sci. Union et Cie Soc. Franc. Recherche Med.). Pharmacology: Presse Med.71, 181 (1963). Pharmacology and toxicity of isomers and racemate: J. C. Le Douarec et al.,Arch. Int. Pharmacodyn. Ther.161, 206 (1966). Pharmacokinetics: S. Caccia et al.,Eur. J. Clin. Pharmacol.29, 221 (1985). Clinical trial of dextrofenfluramine in refractory obesity: N. Finer et al.,Curr. Ther. Res.38, 847 (1985). Comprehensive review: Pinder et al.,Drugs10, 241-323 (1975).Properties: bp12 108-112°. LD50 i.p. in mice: 144 mg/kg (US3198833).Boiling point: bp12 108-112°Toxicity data: LD50 i.p. in mice: 144 mg/kg
Derivative Type: HydrochlorideCAS Registry Number: 404-82-0Trademarks: Acino (IMA); Adipomin (Streuli); Obedrex (Beta); Pesos (Valeas); Ponderal (Servier); Ponderax (Selpharm); Ponderex (Robins); Pondimin (Robins); Rotondin (Casasco)Molecular Formula: C12H16F3N.HClMolecular Weight: 267.72Percent Composition: C 53.84%, H 6.40%, F 21.29%, N 5.23%, Cl 13.24%Properties: Crystals from ethanol + ether, mp 166°.Melting point: mp 166°
Derivative Type:d-FormCAS Registry Number: 3239-44-9Additional Names: Dexfenfluramine; dextrofenfluramineProperties: [a]D25 +9.5° (c = 8 in ethanol). LD50 orally in rats: 114.6 mg/kg (Le Douarec).Optical Rotation: [a]D25 +9.5° (c = 8 in ethanol)Toxicity data: LD50 orally in rats: 114.6 mg/kg (Le Douarec)
Derivative Type:d-Form hydrochlorideCAS Registry Number: 3239-45-0Trademarks: Adifax (Servier); Glypolix (Stroder); Isomeride (Ardix); Redux (Wyeth-Ayerst)Properties: Crystals from ethyl acetate, mp 160-161°.Melting point: mp 160-161°
Derivative Type:l-FormCAS Registry Number: 37577-24-5Properties: [a]D25 -9.6° (c = 8 in ethanol). LD50 orally in rats: 195 mg/kg (Le Douarec).Optical Rotation: [a]D25 -9.6° (c = 8 in ethanol)Toxicity data: LD50 orally in rats: 195 mg/kg (Le Douarec)
Derivative Type:l-Form hydrochlorideCAS Registry Number: 3616-78-2Properties: Crystals from ethyl acetate, mp 160-161°.Melting point: mp 160-161°
NOTE: This is a controlled substance: 21 CFR, 1308.14.Therap-Cat: Anorexic.Keywords: Anorexic.
A centrally active drug that apparently both blocks serotonin uptake and provokes transport-mediated serotonin release.
Fenfluramine Hydrochloride has been filed an IND application with the FDA in USA to initiate phase III trials by Brabant Pharma (acquired by Zogenix in 2014) for the treatment of dravets syndrome (also known as severe myoclonic epilepsy of infancy, SMEI), this compound has been granted orphan drug designation in Europe and U.S..
Fenfluramine Hydrochloride was launched in 1963 by Servier in France and in 1973 by Wyeth (now a wholly owned subsidiary of Pfizer) in US for the treatment of obesity. However, it was withdrawn from the market in 1997 due to heart disease.
Dravet syndrome is a pediatric encephalopathy that typically manifests within the first year of life following exposure to elevated temperatures. It is characterized by recurrent pharmacoresistant seizures, which increase in frequency and severity with disease progression. Concomitantly with these seizures, patients typically display delayed development and neurocognitive impairment.6,9,10,11 Fenfluramine is a serotonergic phenethylamine originally used as an appetite suppressant until concerns regarding cardiotoxicity in obese patients lead to its withdrawal from the market in 1997.6,12,13 Through its ability to modulate neurotransmission, fenfluramine has reemerged as an effective therapy against pharmacoresistant seizures, such as those involved in Dravet syndrome.3,5,8
Fenfluramine was granted initial FDA approval in 1973 prior to its withdrawal; it was granted a new FDA approval on June 25, 2020, for treatment of Dravet syndrome patients through the restricted FINTEPLA REMS program. It is currently sold under the name FINTEPLA® by Zogenix INC.16
Fenfluramine, sold under the brand name Fintepla, is a medication used for the treatment of seizures associated with Dravet syndrome in people age two and older.[2][3]
The most common adverse reactions include decreased appetite; drowsiness, sedation and lethargy; diarrhea; constipation; abnormal echocardiogram; fatigue or lack of energy; ataxia (lack of coordination), balance disorder, gait disturbance (trouble with walking); increased blood pressure; drooling, salivary hypersecretion (saliva overproduction); pyrexia (fever); upper respiratory tract infection; vomiting; decreased weight; risk of falls; and status epilepticus.[2]
Dravet syndrome is a pediatric encephalopathy that typically manifests within the first year of life following exposure to elevated temperatures. It is characterized by recurrent pharmacoresistant seizures, which increase in frequency and severity with disease progression. Concomitantly with these seizures, patients typically display delayed development and neurocognitive impairment.6,9,10,11 Fenfluramine is a serotonergic phenethylamine originally used as an appetite suppressant until concerns regarding cardiotoxicity in obese patients lead to its withdrawal from the market in 1997.6,12,13 Through its ability to modulate neurotransmission, fenfluramine has reemerged as an effective therapy against pharmacoresistant seizures, such as those involved in Dravet syndrome.3,5,8
Fenfluramine was granted initial FDA approval in 1973 prior to its withdrawal; it was granted a new FDA approval on June 25, 2020, for treatment of Dravet syndrome patients through the restricted FINTEPLA REMS program. It is currently sold under the name FINTEPLA® by Zogenix INC.16
Medical uses
Fenfluramine is indicated for the treatment of seizures associated with Dravet syndrome in people age two and older.[2][3]
Dravet syndrome is a life-threatening, rare and chronic form of epilepsy.[2] It is often characterized by severe and unrelenting seizures despite medical treatment.[2]
Adverse effects
The U.S. Food and Drug Administration (FDA) fenfluramine labeling includes a boxed warning stating the drug is associated with valvular heart disease (VHD) and pulmonary arterial hypertension (PAH).[2] Because of the risks of VHD and PAH, fenfluramine is available only through a restricted drug distribution program, under a risk evaluation and mitigation strategy (REMS).[2] The fenfluramine REMS requires health care professionals who prescribe fenfluramine and pharmacies that dispense fenfluramine to be specially certified in the fenfluramine REMS and that patients be enrolled in the REMS.[2] As part of the REMS requirements, prescribers and patients must adhere to the required cardiac monitoring with echocardiograms to receive fenfluramine.[2]
At higher therapeutic doses, headache, diarrhea, dizziness, dry mouth, erectile dysfunction, anxiety, insomnia, irritability, lethargy, and CNS stimulation have been reported with fenfluramine.[4]
There have been reports associating chronic fenfluramine treatment with emotional instability, cognitive deficits, depression, psychosis, exacerbation of pre-existing psychosis (schizophrenia), and sleep disturbances.[4][5] It has been suggested that some of these effects may be mediated by serotonergic neurotoxicity/depletion of serotonin with chronic administration and/or activation of serotonin 5-HT2A receptors.[5][6][7][8]
Heart valve disease
The distinctive valvular abnormality seen with fenfluramine is a thickening of the leaflet and chordae tendineae. One mechanism used to explain this phenomenon involves heart valve serotonin receptors, which are thought to help regulate growth. Since fenfluramine and its active metabolite norfenfluramine stimulate serotonin receptors, this may have led to the valvular abnormalities found in patients using fenfluramine. In particular norfenfluramine is a potent inhibitor of the re-uptake of 5-HT into nerve terminals.[9] Fenfluramine and its active metabolite norfenfluramine affect the 5-HT2B receptors, which are plentiful in human cardiac valves. The suggested mechanism by which fenfluramine causes damage is through over or inappropriate stimulation of these receptors leading to inappropriate valve cell division. Supporting this idea is the fact that this valve abnormality has also occurred in patients using other drugs that act on 5-HT2B receptors.[10][11]
According to a study of 5,743 former users conducted by a plaintiff’s expert cardiologist, damage to the heart valve continued long after stopping the medication.[12] Of the users tested, 20% of women, and 12% of men were affected. For all ex-users, there was a 7-fold increase of chances of needing surgery for faulty heart valves caused by the drug.[12]
Overdose
In overdose, fenfluramine can cause serotonin syndrome and rapidly result in death.[13][14]
Pharmacology
Pharmacodynamics
Fenfluramine acts primarily as a serotonin releasing agent.[15][16] It increases the level of serotonin, a neurotransmitter that regulates mood, appetite and other functions.[15][16] Fenfluramine causes the release of serotonin by disrupting vesicular storage of the neurotransmitter, and reversing serotonin transporter function.[17] The drug also acts as a norepinephrine releasing agent to a lesser extent, particularly via its active metabolite norfenfluramine.[15][16] At high concentrations, norfenfluramine, though not fenfluramine, also acts as a dopamine releasing agent, and so fenfluramine may do this at very high doses as well.[15][16] In addition to monoamine release, while fenfluramine binds only very weakly to the serotonin 5-HT2 receptors, norfenfluramine binds to and activates the serotonin 5-HT2B and 5-HT2C receptors with high affinity and the serotonin 5-HT2A receptor with moderate affinity.[18][19] The result of the increased serotonergic and noradrenergic neurotransmission is a feeling of fullness and reduced appetite.
The combination of fenfluramine with phentermine, a norepinephrine–dopamine releasing agent acting primarily on norepinephrine, results in a well-balanced serotonin–norepinephrine releasing agent with weaker effects of dopamine release.[15][16]
| Drug | NE | DA | 5-HT | Type | Ref |
|---|---|---|---|---|---|
| Fenfluramine | 739 | >10,000 | 79.3–108 | SRA | [20][15][16] |
| D-Fenfluramine | 302 | >10,000 | 51.7 | SNRA | [20][15] |
| L-Fenfluramine | >10,000 | >10,000 | 147 | SRA | [15][21] |
| Norfenfluramine | 168–170 | 1,900–1,925 | 104 | SNRA | [15][16] |
| Phentermine | 39.4 | 262 | 3,511 | NDRA | [20] |
Pharmacokinetics
The elimination half-life of fenfluramine has been reported as ranging from 13 to 30 hours.[4] The mean elimination half-lives of its enantiomers have been found to be 19 hours for dexfenfluramine and 25 hours for levfenfluramine.[13] Norfenfluramine, the major active metabolite of fenfluramine, has an elimination half-life that is about 1.5 to 2 times as long as that of fenfluramine, with mean values of 34 hours for dexnorfenfluramine and 50 hours for levnorfenfluramine.[13]
Chemistry
Fenfluramine is a substituted amphetamine and is also known as 3-trifluoromethyl-N-ethylamphetamine.[13] It is a racemic mixture of two enantiomers, dexfenfluramine and levofenfluramine.[13] Some analogues of fenfluramine include norfenfluramine, benfluorex, flucetorex, and fludorex.
History
Fenfluramine was developed in the early 1960s and was introduced in France in 1963.[13] Approximately 50 million Europeans were treated with fenfluramine for appetite suppression between 1963 and 1996.[13] Fenfluramine was approved in the United States in 1973.[13] The combination of fenfluramine and phentermine was proposed in 1984.[13] Approximately 5 million people in the United States were given fenfluramine or dexfenfluramine with or without phentermine between 1996 and 1998.[13]
In the early 1990s, French researchers reported an association of fenfluramine with primary pulmonary hypertension and dyspnea in a small sample of patients.[13] Fenfluramine was withdrawn from the U.S. market in 1997 after reports of heart valve disease[22][23] and continued findings of pulmonary hypertension, including a condition known as cardiac fibrosis.[24] It was subsequently withdrawn from other markets around the world. It was banned in India in 1998.[25]
Fenfluramine was an appetite suppressant which was used to treat obesity.[13] It was used both on its own and, in combination with phentermine, as part of the anti-obesity medication Fen-Phen.[13]
In June 2020, fenfluramine was approved for medical use in the United States with an indication to treat Dravet syndrome.[2][26]
The effectiveness of fenfluramine for the treatment of seizures associated with Dravet syndrome was demonstrated in two clinical studies in 202 subjects between ages two and eighteen.[2] The studies measured the change from baseline in the frequency of convulsive seizures.[2] In both studies, subjects treated with fenfluramine had significantly greater reductions in the frequency of convulsive seizures during the trials than subjects who received placebo (inactive treatment).[2] These reductions were seen within 3–4 weeks, and remained generally consistent over the 14- to 15-week treatment periods.[2]
The U.S. Food and Drug Administration (FDA) granted the application for fenfluramine priority review and orphan drug designations.[2][27][28] The FDA granted approval of Fintepla to Zogenix, Inc.[2]
On 15 October 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Fintepla, intended for the treatment of seizures associated with Dravet syndrome.[29] Fenfluramine was approved for medical use in the European Union in December 2020.[3]
Society and culture
Recreational use
Unlike various other amphetamine derivatives, fenfluramine is reported to be dysphoric, “unpleasantly lethargic“, and non-addictive at therapeutic doses.[30] However, it has been reported to be used recreationally at high doses ranging between 80 and 400 mg, which have been described as producing euphoria, amphetamine-like effects, sedation, and hallucinogenic effects, along with anxiety, nausea, diarrhea, and sometimes panic attacks, as well as depressive symptoms once the drug had worn off.[30][31][32] At very high doses (e.g., 240 mg, or between 200–600 mg), fenfluramine induces a psychedelic state resembling that produced by lysergic acid diethylamide (LSD).[32][33] Indirect (via induction of serotonin release) and/or direct activation of the 5-HT2A receptor would be expected to be responsible for the psychedelic effects of the drug at sufficient doses.
Research
Under the development code ZX008, the pharmaceutical company Zogenix is studying fenfluramine’s potential to treat seizures.[34] Clinical trials have studied the use of fenfluramine in patients with Dravet syndrome.[35] Results of a Phase III clinical trial showed a 64% reduction in seizures.[36]Route 1
Reference:1. J. Org. Chem.1979, 44, 3580-3583.Route 2
Reference:1. EP0810195A1.
2. Chem. Ind. Times 2002, 16, 33-34.Route 3
Reference:1. ACS Symp. Ser. 2009, 1003, 165-181.
ref
BE 609630
FR 1658 M 19630218
US 3198834
DE 1593595
US 3769319
NL 7215548
Ger. (East) (1974), DD 108971
EP 3170807
SYN
US20170174613
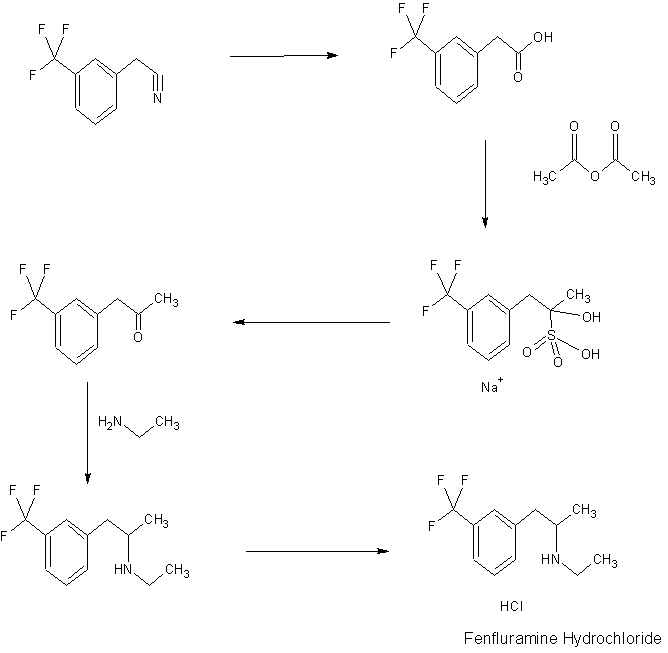
PATENT
US 20170174613
| Step 4.2: Crystallization of Fenfluramine Hydrochloride |
| Procedure: Charge Fenfluramine.HCl (crude) (1.00 wt, 1.0 eq.) and TBME (10.0 vol, 7.4 wt) to the vessel and commence stirring. Heat the suspension to reflux (50 to 58° C.). Charge ethanol (5.0 vol, 3.9 wt) maintaining the temperature at 50 to 58° C. Addition time 20 minutes. Stir at 50 to 58° C. for 5 to 10 minutes and check for dissolution. Stir the solution at 50 to 58° C. for 5 to 10 minutes, targeting 54 to 58° C. Clarify the reaction mixture through a 0.1 μm in-line filter at 54 to 58° C., followed by a line rinse with TBME (1 vol, 0.7 wt). Cool the solution to 48 to 50° C. Charge Fenfluramine.HCl, code FP0188 (0.01 wt). Check for crystallization. Allow the suspension to cool to 15 to 20° C., target 17° C. over 5 to 5.5 hours at an approximately constant rate. Stir the mixture at 15 to 20° C., target 17° C. for 2 to 3 hours. Filter the mixture and wash the filter-cake with clarified TBME (2×3.0 vol, 2×2.2 wt) at 5 to 15° C. Dry the solid at up to 40° C. until the TBME content is <0.5% w/w TBME and the ethanol content is <0.5% w/w EtOH by 1H-NMR analysis. 4 to 8 hours. Determine the w/w assay of the isolated Fenfluramine.HCl by 1H-NMR analysis. |
| Yields and Profiles: The yield for the stage 4 Demonstration batch is summarized in Table 1E below. Input: 750.0 g uncorr. Fenfluramine.HCl crude (1.00 eq, 1.00 wt uncorr.) for input calculation. FIG. 3 shows an exemplary HPLC chromatogram of a crystallized fenfluramine hydrochloride sample (210 nm UV absorbance). |
PATENT
US 20180208543
| Fenfluramine, i.e., 3-trifluoromethyl-N-ethylamphetamine, has the following chemical structure: |
| The marketing of fenfluramine as a pharmaceutical active ingredient in the United States began in 1973 and was used in a therapy in combination with phentermine to prevent and treat obesity. Anyway, in 1997 fenfluramine was withdrawn from the market in the United States and immediately thereafter in other countries, since its use was associated with the onset of cardiac fibrosis and pulmonary hypertension. As a consequence of this event, the pharmaceutical compounds containing this active ingredient were withdrawn from the market. However, fenfluramine, even after its exit from the market, has continued to attract scientific interest, as will become apparent from the discussion presented hereinafter. |
| In the literature, over the years, numerous syntheses or processes have been reported for preparing fenfluramine or its dextrorotatory enantiomer dexfenfluramine or an analog containing a highly electron-attractor group on the aromatic ring as in the fenfluramine molecule (see for example Pentafluorosulfanyl Serotonin Analogs: Synthesis, Characterization, and Biological Activity, John T. Welch and Dongsung Lim Chapter 8, pp 165-181 DOI: 10.1021/bk-2009-1003.ch008). Many of these synthesis paths are long and provide for multiple synthesis steps that can include reagents that are dangerous or scarcely environment-friendly and are therefore scarcely convenient for an industrial synthesis. Hereinafter, any reference to “fenfluramine” is understood to reference the racemic form, i.e., (RS)-N-ethyl-1-[3-(trifluoromethyephenyl]propan-2-amine. |
| To the best of the knowledge of the inventors, the first method for fenfluramine synthesis reported in the literature dates back to 1962 and is referenced in patent BE609630 and in similar patents U.S. Pat. No. 3,198,833 and FR1324220. All the synthesis methods reported in these patents provide for numerous synthesis steps. By way of example, one of the methods provides for the transformation into oxime of a ketone, 1-(3-trifluoromethyephenyl-propan-2-one, as shown here: |
| The oxime is then hydrogenated in the presence of Raney nickel catalyst so as to yield the corresponding primary amine, which is acetylated subsequently with ethanoic anhydride before being converted into fenfluramine by reduction with lithium aluminum hydride. |
| As can be seen, the final step of this chemical process provides for the use of lithium aluminum hydride and the persons skilled in the art will acknowledge that the use of this reagent should be avoided, if possible, on an industrial level, since it is extremely flammable and is the source of accidents. Furthermore, lithium is a potentially neurotoxic metal and therefore its use should be avoided where possible. Furthermore, the Raney nickel catalyst is used in the oxime reduction step and can contaminate the final active ingredient; the use of hydroxylamine also entails problems of toxicity for workers assigned to production. |
| A further disadvantage of this process is, as already mentioned earlier, the number of steps, not only because a large number of synthesis steps entails a reduction of the overall yield of active ingredient, but also because each synthesis step in principle can generate impurities and a larger number of steps can therefore entail a higher number of impurities in the final active ingredient. Many of these impurities, furthermore, due to their structural similarity to fenfluramine, are difficult to eliminate and remove from a fenfluramine preparation. One impurity for example that can be formed in the process described above and is difficult to eliminate is the following: |
| This impurity, which is a primary amine, shares physical-chemical properties that are similar to fenfluramine and therefore, like fenfluramine, it can form a hydrochloride salt by treatment with hydrochloric acid and thus contaminate the active ingredient fenfluramine hydrochloride. Furthermore, this impurity—as a free base—has a boiling point that is similar to that of fenfluramine (73° C. vs. 89° C. at 6 mmHg respectively), and therefore its elimination by distillation also can be problematic. |
| The process described above can in principle generate other impurities, which are listed in FIG. 1. |
| EP 0441160 claims a synthesis in 5 steps of dexfenfluramine, dextrorotatory enantiomer of fenfluramine. This synthesis can be adapted easily to produce fenfluramine instead of its dextrorotatory enantiomer simply by performing the first reduction step with a non-chiral reducing agent. In the first step, in fact: |
(MOL) (CDX) a ketone, 1-(3-trifluoromethyl)phenyl-propan-2-one, is first reduced to the corresponding alcohol in the presence of yeast, D-glucose, ethanol and water. Then the alcohol is converted into the tosylate in the second step:
(MOL) (CDX)
| This reaction occurs in the presence of triethylamine and tosyl chloride in methylene chloride as solvent. After purification, the tosylate is converted to fenfluramine by means of three successive steps: |
| In the first of these three steps, the tosylate is converted into an azide intermediate by reaction with sodium azide in dimethylformamide. The azide intermediate is then hydrogenated in the presence of a catalyst, palladium on carbon. Finally, the resulting primary amine is converted into fenfluramine by reaction with acetaldehyde and sodium borohydride. |
| Persons skilled in the art may see easily that this process is not desirable from an industrial standpoint due to reasons related to environmental risk, safety and costs. For example, the sodium azide used in the process is a notoriously explosive compound and its use at the industrial level is dangerous. Furthermore, palladium is an expensive material and its use in the process entails an increase in the production costs of fenfluramine. Furthermore, palladium can contaminate the finished active ingredient. |
| In another method for the synthesis of dexfenfluramine in 3-4 steps, reported by Goument et al. in Bulletin of the Chemical Society of France (1993), 130, p. 450-458, 3-bromobenzotrifluoride is subjected to a Grignard reaction with enantiopure 1,2-propylene-epoxide to yield 1-[3-(trifluoromethyl)phenyl]propan-2-ol as shown hereafter: |
| If this reaction is performed with racemic 1,2-propylene-epoxide, the synthesis can be adapted to the preparation of fenfluramine. |
| The alcohol thus obtained is first transformed into trifluoromethyl sulfonate by reaction with trifluoromethanesulfonic anhydride and then treated with ethylamine to yield fenfluramine, as shown in the diagram hereinafter: |
| In this article, the authors acknowledge that the main byproducts of the reaction are isomer alkenes having the following chemical structures: |
| The process proposed by Goument et al. is not interesting from the industrial standpoint for a series of reasons. First of all, it is known that the use of Grignard reagents, especially on an industrial scale, is problematic, because these compounds are often pyrophoric and corrosive. Furthermore, 1,2-propylene epoxide is a suspected carcinogenic compound. Finally, the formation of the three isomer alkenes as byproducts listed above is a disadvantage of the process. In the article, Goument presents methods for activation of the intermediate alcohol which are alternative to trifluoromethylsulfonate, for example by converting it to chloride (via thionyl chloride) or to mesylate (via mesyl chloride), but these process variations share the same disadvantages as the main process analyzed above. |
| In addition to the methods with multiple synthesis steps discussed so far in detail, the literature reports other methods or processes for producing fenfluramine or dexfenfluramine. In general, persons skilled in the art acknowledge that the syntheses in the literature for producing dexfenfluramine sometimes can be applied to the preparation of fenfluramine simply by replacing the initial materials and/or enantiopure reagents with the corresponding racemates while maintaining the reaction conditions. For example, patents that present long synthesis methods in multiple steps are the following: |
| DE1593595 and U.S. Pat. No. 3,769,319 |
| NL7215548 |
| EP810195 and EP882700 (dexfenfluramine) |
| EP0301925 (dexfenfluramine) |
| Other examples of preparation of fenfluramine, taken from non-patent literature, are the following: |
| Synthesis, November 1987, p. 1005-1007 |
| J. Org. Chem, 1991, 56, p. 6019 |
| Tetrahedron, 1994, 50(1), p. 171 |
| Bull. Soc. Chim. France, 1993, 130(4), p. 459-466 (dexfenfluramine) |
| Chirality, 2002, 14(4), p. 325-328 (dexfenfluramine) |
| Without analyzing in detail the individual methods described in these patents or articles, it can be stated in summary that all these methods are not attractive and interesting from the industrial standpoint because these are processes with many synthesis steps or because the initial materials described therein are not easily available and therefore have to be prepared separately, with a further expenditure of time and with further costs, or because they provide for the use of reagents that are dangerous/explosive/toxic or because they entail the use of catalysts based on heavy metals that can contaminate the final active ingredient. |
| One should consider that in the literature there are methods for the preparation of fenfluramine that did not provide for long syntheses and multiple steps but are shorter and consist of one or two steps. These processes, which therefore would be more interesting from the industrial standpoint, have other specific disadvantages, as will become apparent in detail hereinafter. For example, in the literature there is a first group of articles or patents that describe the reaction between 1-(3-trifluoromethyl)phenyl-propan-2-one and ethylamine in the presence of hydrogen gas and of a transition metal as catalyst: |
| In particular, in Huagong Shikan, 2002, 16(7), p. 33, the reaction is performed with hydrogen gas (2.9-3.38 atm), at 65-75° C., for 9 hours, in the presence of Raney nickel. Likewise, in patent DD108971 (1973), Raney nickel and hydrogen gas and methanol are used as solvent to perform this reaction. |
| In HU55343, instead, a similar reaction in one step is performed with hydrogen gas in the presence of another transition metal catalyst, such as palladium on carbon. |
| Although these three methods describe short single-step processes, they have the disadvantage of the use of hydrogen gas. As is known to persons skilled in the art, hydrogen gas is a dangerous gas due to the inherent danger of forming explosive mixtures with air and must be used by expert personnel in expensive facilities dedicated to its use and built with special precautions. Despite being used in purpose-built facilities, the use of hydrogen at the industrial level is inherently dangerous and to be avoided if possible. Another danger element that is shared by the processes described above is the fact that the reactions are performed under pressure. The third industrial disadvantage then arises from the use of heavy metal catalysts, which have a high cost and therefore increase the overall cost of the final active ingredient and may then contaminate the active ingredient fenfluramine even after filtration of the catalyst and purification of said active ingredient. |
| Analysis of the background art shows, however, that an attempt has been made to devise a process for the production or synthesis of fenfluramine that is short (one or two steps) and does not entail the use of hydrogen gas or of catalysts based on nickel or palladium or the like. In particular, for example, Synthesis 1987, 11, p. 1005, and then DECHEMA Monographien (1989), 112 (Org. Elektrochem.—Angew. Elektrothermie), 367-74, present a method for the synthesis of fenfluramine which starts from 1-(3-trifluoromethyl)phenyl-propan-2-one, which is made to react with ethylamine in great excess, in an electrochemical process, which uses a mercury cathode in a water/ethanol solution with pH 10-11. One obtains fenfluramine with 87% yield. This process has some drawbacks from an industrial standpoint: it is a process of the electrochemical type and therefore requires special equipment which is scarcely widespread, dedicated cells and reactors, and it is not possible to use the classic multipurpose reactors available in the pharmaceutical industry. Furthermore, the use of mercury at the industrial level poses severe environment safety problems, requiring constant health monitoring on workers who manage the equipment and systems for the management and destruction of wastewater that are particularly onerous; finally, mercury can be transferred from the cathode to the reaction environment and therefore to the active ingredient, and this obviously is to be considered very dangerous due to the accumulation of the metal in human beings; small traces of mercury are very toxic. |
| Another method for fenfluramine synthesis in a single step is the one presented in J. Org. Chem, 1979, 44(20), p. 3580. Here the reaction is described between an alkene derivative and ethylamine in the presence of sodium borohydride and mercury nitrate: |
| Again, this process is not interesting from an industrial standpoint since it has the same problems, if not even greater ones, related to the use of mercury (used here as a water-soluble salt) discussed previously. The complication introduced in this process with the use of mercury nitrate together with sodium borohydride highlights the level of innovation of the synthesis path found here. |
| In the past, therefore, it has not been possible to provide a process for synthesizing fenfluramine in a small number of steps by using modern reducing agents that are commonly and easily used. Indeed, while Gaodeng Xuexiao Huaxue Xuebao, 9(2), 1988, p. 134-139, describes and exemplifies the synthesis of 2-N-ethyl-1-phenyl propane by means of (1) the treatment of the precursor ketone with ethylamine followed by (2) sodium cyanoborohydride as reducing agent, Xuexiao Huaxue Xuebao provides no example for fenfluramine. Moreover, for the latter, Xuexiao Huaxue Xuebao indicates a melting point for the hydrochloride of 161° C., a data item that matches the value indicated in the literature initially (see BE609630); these facts prove thats fenfluramine synthesis with cyanoborohydride was not performed, otherwise one cannot explain why the author did not transcribe, in the document, the example of a product that at the time was very important. It should be noted in fact that 1-phenyl propan-2-one and 1-(3-trifluoromethyl)phenyl-propan-2-one can have different reactivities to reductive amination due to the presence of a highly electron-attractor-trifluoromethyl group, hence the need for an example to demonstrate its feasibility. The use of cyanoborohydride shares some disadvantages with other methods discussed in the preceding paragraphs. The excellent selectivity for reductive aminations of this reagent is highly appreciated, but its application can be less advantageous with respect to other reducing systems in the synthesis of fenfluramine, where the latter is intended for therapeutic application in human beings. The reasons for this are the possible contamination of the finished pharmaceutical active ingredient with cyanide ions, the toxicity of the reagent itself and finally the danger of its use. It is known to persons skilled in the art that sodium cyanoborohydride can release hydrocyanic acid if the pH of the reaction environment is acid enough and it is known that hydrocyanic acid is a powerful poison, since it competes with oxygen for hemoglobin coordination. As a consequence of this, particular care must be taken in its use and in the disposal of the production wastewater, which can be contaminated by cyanides. Not least, one must consider that the cost of sodium cyanoborohydride is considerable. |
| To conclude, it can be seen that more than 50 years after the publication of its first synthesis dated 1962, there are still numerous disadvantages or limitations in the synthesis paths developed in the past decades in the literature for the preparation of fenfluramine. |
| Moreover, recently there has been renewed pharmaceutical interest in the fenfluramine molecule, since the possibility of its therapeutic use in severe disorders of infancy has appeared in the medical literature. For example, mention can be made of Ceulemans et al., Epilepsia, 53(7), pages 1131 to 1139, 2012. |
| According to a certain part of medical literature, fenfluramine might therefore be interesting as a medication in a chronic therapy for the treatment of symptoms of epilepsy and other correlated severe disorders. |
| Based on recent medical developments, therefore, the need exists for a synthesis method that is better than the existing ones and can overcome in particular the disadvantages of the processes that are present in the literature. Particularly important, in view of use in chronic therapies for children such as epilepsy and other severe disorders, it would be fundamentally important to identify a path for synthesis of the active ingredient fenfluramine that does not entail the use of heavy metals and/or transition metals, which in a chronic therapy might accumulate in the body of the patients over the years, with severe consequences on health. |
| More generally, it is desirable to identify a synthesis path that uses reagents from which (or from the transformation products of which) it is then possible to easily purify fenfluramine. |
| It would be equally desirable to identify a synthesis path that comprises a small number of synthesis steps and uses reagents that are widely commercially available and easy to use. |
| At the same time, the new identified synthesis path should avoid if possible the formation of byproducts. |
DESCRIPTION OF THE FIGURES
| FIG. 1: impurities generated theoretically by means of the reagents used in the first fenfluramine synthesis according to BE609630. |
| FIG. 2: DSC of crude fenfluramine hydrochloride, obtained by reduction with sodium cyanoborohydride according to test 12 (table B) of the description that follows. |
| FIG. 3: DSC of fenfluramine hydrochloride recrystallized from 2-butanol as in example 2b (reduction with sodium borohydride). |
SUMMARY OF THE INVENTION
| The inventors of the present application have found surprisingly that the aim and objects indicated above are achieved by a new method for the synthesis of fenfluramine or of a pharmaceutically acceptable salt thereof, comprising the transformation of a ketone having the structure (I): |
(MOL) (CDX) wherein R 1 is CF 3, with ethylamine or with a salt thereof, and with a reducing agent chosen from the group consisting of alkaline cation or ammonium borohydride, alkaline cation or ammonium triacetoxyborohydride and alkaline cation or ammonium cyanoborohydride, in which the alkaline cation is always different from lithium cation and mixtures thereof, to yield fenfluramine, optionally followed by the transformation of the obtained fenfluramine into a pharmaceutically acceptable salt.
| Furthermore, the inventors of the present invention have also discovered a new preparation of fenfluramine, which can be obtained by means of the method described hereinafter, and new pharmaceutical compositions that contain it. |
Example 1
Synthesis of Fenfluramine
| A suspension of sodium hydroxide (34.62 g-0.866 mol, 3.5 eq) in 170 mL of methanol, under mechanical agitation, receives the addition, drop by drop, over the course of 30 minutes, of a solution of ethylamine hydrochloride (70.59 g-0.866 mol, 3.5 eq) in 165 mL of methanol, followed by 1-(3-trifluoromethyl)phenyl-propan-2-one (50 g-0.247 mol). The mixture is left under agitation at 20° C. for 4.5 hours, then cooling to 0° C. is performed and a solution of sodium borohydride (9.36 g-0.247 mol) in 19 mL of sodium hydroxide 1M in water is then added drop by drop, keeping the temperature below 10° C. The reaction is then left under agitation at 20° C. for another 2 hours. Once the reaction is complete, 270 mL of methanol are removed at a reduced pressure at 40° C. and then 200 mL of water are added and the mixture is extracted with heptane (200 mL). The aqueous phase is eliminated and the organic phase is washed with water (200 mL×3). The organic phase is concentrated at 50° C. at reduced pressure to yield free base fenfluramine as colorless oil. Yield: 72%; purity: 77%—as listed in test 3 of table A above. |
Example 2
Purification of Fenfluramine
| Purification of free base fenfluramine can be performed in two ways: |
| distillation of the free base |
| crystallization of the fenfluramine hydrochloride salt |
| Depending on the degree of purity that is desired, both purification processes are performed in sequence (distillation first and then crystallization), or only one of the two purification processes is performed. |
Example 2a
Distillation
| Free base fenfluramine (10 g), prepared as in Example 1, is distilled under reduced pressure with a distillation column of the Vigreux type: the distillation heads are eliminated, the fraction that is distilled at 89-90° C. at 6 mmHg, which is the active ingredient fenfluramine (8.5 g) with a high degree of purity, is collected. |
Example 2b
Conversion into Hydrochloride Salt and Crystallization
| Crude fenfluramine, prepared as in Example 1, or purified fenfluramine as in Example 2a, is dissolved in 125 mL of ethyl acetate, and cooling is performed to 0° Celsius under agitation. 272 mL of a solution of 1M HCl in ethyl acetate are added drop by drop at 0° C. The precipitate that forms is filtered and washed with ethyl acetate (125 mL×2) to yield approximately 55 g of solid fraction. The solid fraction is crystallized by 2-butanol (260 mL), keeping the solid for 22 hours at 3° C. under slow agitation before filtering it. Filtering is performed and washing is performed with cold 2-butanol. The solid fraction, fenfluramine hydrochloride, is dried in a vacuum stove, yielding 51.7 g of product. A DSC of the resulting product is shown in FIG. 3. |
PAPER
Journal of Organic Chemistry (1979), 44(20), 3580-3.J. Org. Chem. 1979, 44, 20, 3580–3583
Publication Date:September 1, 1979
https://doi.org/10.1021/jo01334a031https://pubs.acs.org/doi/abs/10.1021/jo01334a031
PATENT
https://patents.google.com/patent/US20170174613A1/en
- Fenfluramine is an amphetamine drug that was once widely prescribed as an appetite suppressant to treat obesity. Fenfluramine is devoid of the psychomotor stimulant and abuse potential of D-amphetamine and interacts with the 5-hydroxytryptamine (serotonin, 5-HT) receptors to release 5-HT from neurons. Fenfluramine has been investigated as having anticonvulsive activity in the treatment of Dravet Syndrome, or severe myoclonic epilepsy in infancy, a rare and malignant epileptic syndrome. This type of epilepsy has an early onset in previously healthy children.
- [0003]
Anorectic treatment with fenfluramine has been associated with the development of cardiac valvulopathy and pulmonary hypertension, including the condition cardiac fibrosis which led to the withdrawal of fenfluramine from world-wide markets. Interaction of fenfluramine’s major metabolite norfenfluramine with the 5-HT2B receptor is associated with heart valve hypertrophy. In the treatment of epilepsy, the known cardiovascular risks of fenfluramine are weighed against beneficial anticonvulsive activity.


- [0097]
Chemical Abstract Service (CAS) Registry Number (RN): 404-82-0 (HCl Salt), 458-24-2 (Parent Free Base) - [0098]
Chemical Name: N-ethyl-α-methyl-3-(trifluoromethyl)-benzeneethanamine hydrochloride (1:1). Other Names: Fenfluramine HCl, DL-Fenfluramine, (±)-Fenfluramine - [0099]
Structure of Hydrochloride Salt: - [0100]
Stereochemistry: Fenfluramine HCl has one chiral center and is being developed as the racemate and contains dexfenfluramine and levofenfluramine - [0101]
Molecular Formula of hydrochloride salt: C12H16F3N.HCl - [0102]
Molecular Mass/Weight: 267.72 g/mol
2. General Properties
- [0103]
Table 1 summarizes the chemical and physical properties of Fenfluramine HCl. - TABLE 1 General Properties of Fenfluramine HCl Drug Substance Property Result Appearance (color, White to off-white powder physical form) DSC (melting 170° C. (melt/sublimation) point)a TGA Onset 147° C. 0.03% at 150° C. 91% at 220° C. (evaporation) pKa (water) 10.15-10.38 Solubility (mg/mL) Resultant pH 25° C. 37° C. Solubility pH 6.69 (water) 54.13 71.22 (Aqueous) pH 1.73 buffer 25.34 53.68 pH 3.43 buffer 29.50 61.97 pH 6.41 buffer 37.42 95.60 0.9% NaCl (water) 22.98 — Solvent Solubility 25° C. (mg/mL) Solubility (Organic Ethanol 150 Solvents) Dichloromethane 30-35 Ethyl Acetate, 1-5 mg Tetrahydrofuran, Toluene, Acetonitrile UV Absorption Maxima: 210, 265 nm Solution pH (water) 6.69 Hygroscopicity @30% RH: ~0.05% (Dynamic Vapor @60% RH: ~0.07% Sorption (DVS) @90% RH: ~0.20%a) Polymorphism Fenfluramine HCl has been consistently isolated as a single crystalline Form 1 as determined by DSC and x-ray powder diffraction (XRPD) Solvation/Hydration Fenfluramine HCl is isolated as a nonhydrated, nonsolvated solid Solution Stability 8 weeks @ pH 6.7 phosphate buffer medium at 40° C. and 60° C. using concentrations of 0.5, 2.5 and 5.0 mg/ml. All conditions, no new impurities >0.1% by HPLC. Solid Stability 8 weeks @ 40° C., 60° C. and 80° C. 7 days at 150° C. All conditions, no new impurities >0.1% by HPLC.
3. Synthesis of Fenfluramine Drug Substance
- [0104]
Scheme 3.1 shows a 2-step route of synthesis used to manufacture initial clinical supplies of Fenfluramine HCl from ketone (2). The batch size is 4 kg performed in laboratory glassware (kilo lab). No chromatography is required and the process steps are amenable to scale-up. In process 1 there is one isolated intermediate Fenfluramine Free Base (1) starting from commercially supplied 1-(3-(trifluoromethyl)phenyl) acetone (Ketone 2). All steps are conducted under cGMPs starting from Ketone (2). - [0105]
Scheme 3.2 shows a 4-step route of synthesis to Fenfluramine HCl that can be used for commercial supply. Route 2 utilizes the same 2-step process used by Route 1 to convert Ketone (2) to Fenfluramine HCl with the exception that Ketone (2) is synthesized under cGMP conditions starting from 3-(Trifluoromethyl)-phenyl acetic acid (Acid 4). Bisulfate Complex (3) is an isolatable solid and can be purified before decomplexation to Ketone (2). In-situ intermediates which are oils are shown in brackets. Batch sizes of 10 Kg are performed. Commercial batch sizes of 20 kg are performed in fixed pilot plant equipment. Steps 1-2 of Scheme 3.2 to manufacture Ketone (2) have been demonstrated on a 100 g scale to provide high purity ketone (2) of >99.8% (GC & HPLC). Conversion of Ketone (2) to Fenfluramine using either Route 1 or 2 has provided similar purity profiles. - Starting materials are designated by enclosed boxes. Bracketed and non bracketed compounds respectively indicate proposed in-situ and isolated intermediates. NMI=N-Methyl Imidazole.
4.1. Narrative Description (Route 1)
- [0106]
Step 1: Reductive Amination (Preparation of Fenfluramine Free Base 1) - [0107]
A solution of ethylamine, water, methanol, and 1-(3-(trifluoromethyl)phenyl) acetone (Ketone 2) was treated with sodium triacetoxyborohydride and stirred for 16 h at 25° C. at which time HPLC analysis (IPC-1; In Process Control No. 1) showed the reaction to be complete and sodium hydroxide solution was added until pH>10. Toluene was added and the phases separated, and the aqueous phase (IPC-2) and organic phase (IPC-3) are checked for remaining Fenfluramine and Fenfluramine alcohol and the organic phase was reduced. Purified water was added and the pH adjusted to <2 using conc. HCl and the phases were separated. The aqueous phase was washed with toluene and the toluene phase (IPC-4) and the aqueous phase (IPC-5) was checked for Fenfluramine and Fenfluramine alcohol content. The aqueous phase containing product is pH adjusted to >10 using sodium hydroxide solution. The basic aqueous phase was extracted with MTBE until removal of Fenfluramine from the aqueous phase was observed by HPLC (<0.5 mg/ml) (IPC-6). The organic phase was dried over sodium sulfate and filtered. The filtrate was concentrated in vacuo to give the intermediate product Fenfluramine Free Base 1 as a pale yellow oil tested per specifications described herein which showed by NMR the material to contain 2.93% toluene giving an active yield of 88.3% with a purity of 98.23% by HPLC (0.67% Fenfluramine alcohol). - [0108]
Step 2: Salt Formation (Preparation of Fenfluramine HCl) - [0109]
To a flask was charged ethanol and acetyl chloride. The solution was stirred slowly overnight before ethyl acetate was added. The HCl in ethyl acetate solution formed was polish filtered into a clean carboy and retained for later use. To a vessel was added Fenfluramine free base 1 and MTBE. The Fenfluramine solution in MTBE was collected in two carboys before the vessel was cleaned and checked for particulate residue. The Fenfluramine solution was polish filtered into a vessel and cooled and HCl in ethyl acetate solution was added giving a final pH of 6-7. The batch was stirred for 1 h and filtered. The product was dried under vacuum at 40° C. The product (96.52% yield) was tested per IPC-7 had a purity of 99.75% by HPLC and GC headspace analysis showed MTBE (800 ppm) and EtOAc (150 ppm) to be present. The product was then tested per specifications shown herein.
4.2. Narrative Description (Route 2)
- [0110]
Step 1: Preparation of Ketone Bisulfite Adduct - [0111]
Procedure: Charge acetic anhydride, (2.8 vol, 3.0 wt, 5.0 eq.) to a vessel and commence stirring. Cool the solution to −5 to 5° C., targeting −4° C. Charge 1-methylimidazole, (0.2 vol, 0.21 wt, 0.5 eq.) to the mixture at −5 to 5° C. Caution: very exothermic. If necessary, adjust the temperature to 0 to 5° C. Charge ZX008 acid, (1.00 wt, 1.0 eq.) to the mixture at 0 to 5° C. Caution: exothermic. Stir the mixture at 0 to 5° C. until ≦2.1% area ZX008 acid by HPLC analysis, typically 7 to 9 hours. Charge 15% w/w sodium chloride solution (2.0 vol) to the mixture at 0 to 5° C., 60 to 90 minutes. Caution: very exothermic which will be slightly delayed. Warm the mixture to 18 to 23° C. over 45 to 60 minutes and continue stirring for a further 30 to 45 minutes at 18 to 23° C. Charge TBME, (5.0 vol, 3.7 wt) to the mixture and stir for 10 to 15 minutes at 18 to 23° C. Separate the aqueous layer and retain the organic layer. Back-extract the aqueous layer with TBME, (2×3.0 vol, 2×2.2 wt) at 18 to 23° C. retaining each organic layer. Adjust the pH of the combined organic layer to pH 6.5 to 9.0, targeting 7.0 by charging 20% w/w sodium hydroxide solution (5.3 to 8.3 vol) at 18 to 23° C. Caution: exothermic. Separate the aqueous layer and retain the organic layer. Wash the organic layer with 4% w/w sodium hydrogen carbonate solution (2×3.0 vol) at 18 to 23° C. Determine the residual ZX008 acid content in the organic layer by HPLC analysis, pass criterion ≦0.10% area ZX008 acid. Wash the organic layer with purified water, (2×3.0 vol) at 18 to 23° C. Concentrate the organic layer under reduced pressure to ca. 2 vol at 40 to 45° C., targeting 43° C. - [0112]
Determine the w/w assay of ZX008 ketone (WIP) in the mixture by 1H-NMR analysis for information only and calculate the contained yield of ZX008 ketone (WIP) in the mixture. Note: This step can be removed from the process since the process is robust and consistently delivers 80 to 90% th yield. The achieved yield was factored into the charges of the subsequent steps. - [0113]
Charge n-heptane, (4.0 vol, 2.7 wt) to the mixture at 40 to 45° C., targeting 43° C. Concentrate the mixture to ca. 2 vol at 40 to 45° C., targeting 43° C. Determine the TBME content in the mixture by 1H-NMR analysis, (pass criterion ≦5.0% w/w TBME vs. ZX008 ketone). Charge n-heptane, (2.4 vol, 1.6 wt) at 40 to 45° C., targeting 43° C., vessel A. To vessel B, charge sodium metabisulfite, (0.82 wt, 0.88 eq.) at 18 to 23° C. To vessel B, charge a solution of sodium hydrogen carbonate, (0.16 wt, 0.4 eq.) in purified water, code RM0120 (2.0 vol) at 18 to 23° C. followed by a line rinse with purified water, code RM0120 (0.4 vol) at 18 to 23° C. Caution: gas evolution. Heat the contents of vessel B to 40 to 45° C., targeting 43° C. Charge the contents from vessel A to vessel B followed by a line rinse with n-heptane, (0.8 vol, 0.5 wt) at 40 to 45° C., targeting 43° C. Stir the mixture for 1 to 1.5 hours at 40 to 45° C., targeting 43° C. Charge n-heptane, code RM0174 (3.2 vol, 2.2 wt) to the mixture with the temperature being allowed to cool to 18 to 45° C. at the end of the addition. Cool the mixture to 18 to 23° C. at approximately constant rate over 45 to 60 minutes. Stir the mixture at 18 to 23° C. for 1.5 to 2 hours. - [0114]
Sample the mixture to determine the residual ZX008 ketone content by 1H-NMR analysis, (pass criterion ≦10.0% mol, target 5.0% mol ZX008 ketone vs. ZX008 ketone bisulfite adduct). Filter the mixture and slurry wash the filter-cake with n-heptane, (2×2.0 vol, 2×1.4 wt) at 18 to 23° C. Dry the solid at up to 23° C. until the water content is <10.0% w/w water by KF analysis according to AKX reagent. At least 16 hours. Determine the w/w assay of the isolated ZX008 ketone bisulfite adduct by 1H-NMR analysis and calculate the contained yield of ZX008 ketone bisulfite adduct. - [0115]
Yields and Profiles: The yield for the stage 1 Demonstration batch is summarized Table below. Input: 1700.0 g uncorr., acid, 99.50% area (QC, HPLC), 2-isomer not detected, 4-isomer 0.02% area, RRT1.58 (previously not observed) 0.48% area as per the preparative method. The analytical data is summarized in Table 1A below. - TABLE 1A Table for isolated yields for step 1 Demonstration batch Corr. % area Reference Corr. Yield % w/w (HPLC, number Input Output (% th)** (1H-NMR)* QC) Comments Batch A1 1700.0 g 1500.1 g 89.1 45.0 —.— Crude ketone as TBME sol. Batch A2 1500.1 g 1716.1 77.8 76.0 98.15 Bisulfite adduct only 67.3 Overall product
- [0116]
Step 2: Preparation of Ketone - [0117]
Procedure: Charge toluene, (5.0 vol, 4.3 wt), and purified water, (5.0 vol) to the vessel and commence stirring. If necessary, adjust the temperature to 18 to 23° C. and charge ZX008 ketone bisulfite adduct, (1.00 wt corrected for % w/w assay) to the mixture at 18 to 23° C. Charge 20% w/w sodium hydroxide solution to the mixture at 18 to 23° C. adjusting the pH of the mixture to pH 8.0 to 12.0, targeting 9.0 (0.5 to 1.0 vol). - Separate the lower aqueous layer and retain the top organic layer. Wash the organic layer with purified water, (3.0 vol) at 18 to 23° C. Concentrate the organic layer under reduced pressure to ca. 2 vol at 45 to 50° C., targeting 48° C. Charge methanol, (5.0 vol, 4.0 wt) to the mixture at 45 to 50° C., targeting 48° C. Re-concentrate the mixture under reduced pressure to ca. 2 vol at 45 to 50° C., targeting 48° C. Repeat steps 7 and 8 once before continuing with step 9. Cool the mixture to 18 to 23° C. Clarify the mixture into a tared, suitably-sized drum followed by a methanol (1.0 vol, 0.8 wt) line rinse at 18 to 23° C. Determine the w/w assay of ZX008 ketone (WIP) in the mixture by 1H-NMR analysis and calculate the contained yield of ZX008 ketone (WIP) in the mixture. Determine the toluene content in the mixture by 1H-NMR analysis.
- [0118]
Yields and Profiles: The yield for the step 2 Demonstration batch is summarized in Table 1B below. Input: 1200.0 g corr. Ketone bisulfite adduct, 76.0% w/w assay (NMR, using DMB as internal standard in d6-DMSO), (1.00 eq, 1.00 wt corr. for w/w assay) for input calculation. - TABLE 1B Table for isolated yields for step 2 Demonstration batch % w/w % area Corr. Corr. Corr. Yield (1H- (HPLC, Input Output (% th) NMR)* QC) Comments 1200.0 g 858.15 g 108.3 25.5 99.31 Purified ketone
- [0119]
Step 3: Preparation of Fenfluramine HCl Crude - [0120]
Procedure: Charge the ZX008 ketone (corr. for assay, 1.00 wt, 1.00 eq. isolated as solution in MeOH in stage 2) to a vessel. Charge methanol, code RM0036 (5.0 vol, 4.0 wt) to the mixture at 18 to 23° C. Cool the solution to 0 to 5° C. Charge 70 wt % aqueous ethylamine solution (1.3 vol, 1.6 wt, 4.0 eq) to the mixture at 0 to 10° C., over 15 to 30 minutes, followed by a line rinse with methanol (1.0 vol, 0.8 wt). Warm the mixture to 15 to 20° C. and stir the mixture for a further 60 to 70 minutes at 15 to 20° C. Adjust the mixture to 15 to 18° C. if required, targeting 15° C. Charge sodium triacetoxyborohydride (2.4 wt, 2.25 eq.) to the mixture in approximately 10 portions, keeping the mixture at 15 to 20° C., targeting 17° C. Addition time 1.5 to 2 hours. Caution: Exothermic. Stir the mixture at 15 to 20° C. until complete by HPLC analysis, pass criterion ≦3.0% area ZX008 ketone, typically 2 to 3 hours. Adjust the pH of the mixture to pH>12 by charging 20% w/w aqueous sodium hydroxide solution (5.0 to 6.0 vol) to the mixture at 15 to 40° C. Addition time 10 to 30 minutes. Caution: Exothermic. If necessary, adjust the temperature to 18 to 23° C. Extract the mixture with toluene (3×3.0 vol, 3×2.6 wt) at 18 to 23° C., retaining and combining the top organic layer after each extraction. Wash the combined organic layer with purified water, (1.0 vol) at 18 to 23° C. Heat the mixture to 40 to 50° C., targeting 48° C. Concentrate the mixture under reduced pressure at constant volume maintaining ca. 5 vol by charging the organic layer at approximately the same rate as the distillation rate at 40 to 50° C., targeting 48° C. Cool the mixture to 18 to 23° C. Charge purified water (10.0 vol) to the mixture at 18 to 23° C. Adjust the pH of the mixture to 0.1<pH<1.5 at 18 to 23° C. by charging concentrated hydrochloric acid, 0.5 vol. Do not delay from this step until neutralization. - [0121]
Separate the layers at 18 to 23° C. retaining the bottom aqueous layer. Wash the aqueous layer with toluene, (3.0 vol, 2.6 wt) at 18 to 23° C. retaining the aqueous layer. Adjust the pH of the aqueous layer to pH>12 by charging 20% w/w sodium hydroxide solution at 18 to 23° C. 0.8 to 0.9 vol. Caution: Exothermic. Charge TBME, code RM0002 (2.0 vol, 1.5 wt) to the basic aqueous layer. Separate the layers at 18 to 23° C. retaining the top organic layer. Back-extract the aqueous layer with TBME (2×2.0 vol, 2×1.5 wt) at 18 to 23° C. retaining the organic layers. Wash the combined organic layer with purified water, (2×1.0 vol) at 18 to 23° C. Concentrate the combined organic layers under reduced pressure at 40 to 50° C., targeting 48° C. to ca. 3 vol. Determine the residual toluene content of the mixture by 1H-NMR analysis. Sample for determination of residual water content by KF analysis, AKX reagent. Charge TBME (8.7 vol, 6.4 wt) to the mixture at 40 to 50° C. Cool the solution to 0 to 5° C., targeting 2° C. Charge concentrated hydrochloric acid (0.54 vol, 0.46 wt) maintaining the temperature <15° C. Caution: Exothermic. Line rinse with TBME (1.0 vol, 0.7 wt). If necessary, adjust the temperature to 0 to 10° C. and stir the mixture at 0 to 10° C. for a further 2 to 3 hours. Filter the mixture and wash the filter-cake with TBME (2×4.4 vol, 2×3.3 wt) at 0 to 10° C. Dry the solid at up to 40° C. until the TBME content is <0.5% w/w TBME by 1H-NMR analysis. 4 to 8 hours. - [0122]
Yields and Profiles: The yield for the step 3 Demonstration batch is summarized in Table 1C below. Input: 856.8 g corr. Ketone, 44.2% w/w assay (NMR, using TCNB as internal standard in CDCl3), (1.00 eq, 1.00 wt corr. for w/w assay) for input calculation. FIG. 2 and Table 1D shows an exemplary HPLC chromatogram of a crude preparation of fenfluramine hydrochloride (210 nm UV absorbance). - TABLE 1C Table for isolated yields for step 3 Demonstration batch Corr. % area Reference Corr. Corr. Yield % w/w (HPLC, number Input Output (% th) (1H-NMR)* QC) Comments Batch A1 856.8 g 836.31 g 85.3 44.2 99.15 Fenfluramine free base (in situ intermediate) Batch A2 880.7 84.0 based 99.5 100.00 Fenfluramine•HCl on ketone crude (step 3 an bisulfite d 4.1) adduct (77.6 based on purified ketone)
- TABLE 1D Purity of crude fenfluramine hydrochloride by HPLC (see FIG. 2) Processed Channel Descr. DAD AU Ch 1 Sample 210, Bw 4 Peak Results USP USP USP Name RT RelRT Area Height Tailing Resolution Plate Count EP s/n % Area 1 NorFenfluramine 7.46 2 2-Fenfluramine 7.68 3 Fenfluramine 8.67 1.000 3789064 778178 1.7 70796 2549.8 99.15 4 4-Fenfluramine 8.95 5 11 34 1.308 6073 1449 1.2 23.5 215529 3 8 0.16 6 ZX008 acid 12.93 7 Fenfluramine alcohol 14.16 1.633 15266 2972 1.3 24.8 215040 8.7 0.40 8 ZX008 ketone 14.83 9 Fenfluramine acetamide 15.55 10 TOLUENE 15 75 11 15.92 1.836 4110 1122 2.7 0.11 12 16.60 1.915 6861 1630 1.5 451209 4.3 0.18 Sum 3821374 100.00
- [0123]
Step 4.2: Crystallization of Fenfluramine Hydrochloride - [0124]
Procedure: Charge Fenfluramine.HCl (crude) (1.00 wt, 1.0 eq.) and TBME (10.0 vol, 7.4 wt) to the vessel and commence stirring. Heat the suspension to reflux (50 to 58° C.). Charge ethanol (5.0 vol, 3.9 wt) maintaining the temperature at 50 to 58° C. Addition time 20 minutes. Stir at 50 to 58° C. for 5 to 10 minutes and check for dissolution. Stir the solution at 50 to 58° C. for 5 to 10 minutes, targeting 54 to 58° C. Clarify the reaction mixture through a 0.1 μm in-line filter at 54 to 58° C., followed by a line rinse with TBME (1 vol, 0.7 wt). Cool the solution to 48 to 50° C. Charge Fenfluramine HCl, code FP0188 (0.01 wt). Check for crystallization. Allow the suspension to cool to 15 to 20° C., target 17° C. over 5 to 5.5 hours at an approximately constant rate. Stir the mixture at 15 to 20° C., target 17° C. for 2 to 3 hours. Filter the mixture and wash the filter-cake with clarified TBME (2×3.0 vol, 2×2.2 wt) at 5 to 15° C. Dry the solid at up to 40° C. until the TBME content is <0.5% w/w TBME and the ethanol content is <0.5% w/w EtOH by 1H-NMR analysis. 4 to 8 hours. Determine the w/w assay of the isolated Fenfluramine.HCl by 1H-NMR analysis. - [0125]
Yields and Profiles: The yield for the stage 4 Demonstration batch is summarized in Table 1E below. Input: 750.0 g uncorr. Fenfluramine HCl crude (1.00 eq, 1.00 wt uncorr.) for input calculation. FIG. 3 shows an exemplary HPLC chromatogram of a crystallized fenfluramine hydrochloride sample (210 nm UV absorbance). - TABLE 1E Table for isolated yields for stage 4 Demonstration batch Uncorr. Uncorr. Uncorr. Yield HPLC (% area, Input Output (% th) QC) Comments 750.0 g 608.0 81.1 100.00* Fenfluramine•HCl
PATENT
https://patents.google.com/patent/EP3170807A1/en
- Fenfluramine, i.e., 3-trifluoromethyl-N-ethylamphetamine, has the following chemical structure:
- [0003]
The marketing of fenfluramine as a pharmaceutical active ingredient in the United States began in 1973 and was used in a therapy in combination with phentermine to prevent and treat obesity. However, in 1997 fenfluramine was withdrawn from the market in the United States and immediately thereafter in other countries, since its ingestion was associated with the onset of cardiac fibrosis and pulmonary hypertension. As a consequence of this event, the pharmaceutical compounds containing this active ingredient were withdrawn from the market. However, fenfluramine, even after its exit from the market, has continued to attract scientific interest, as will become apparent from the discussion presented hereinafter. - [0004]
In the literature, over the years, numerous syntheses or processes have been reported for preparing fenfluramine or its dextrorotatory enantiomer dexfenfluramine or an analog containing a highly electron-attractor group on the aromatic ring as in the fenfluramine molecule (see for example Pentafluorosulfanyl Serotonin Analogs: Synthesis, Characterization, and Biological Activity, John T. Welch and Dongsung Lim Chapter 8, pp 165-181 DOI: 10.1021/bk-2009-1003.ch008). Many of these synthesis paths are long and foresee multiple stages or synthesis steps that can include reagents that are dangerous or scarcely environment-friendly and are therefore scarcely convenient for an industrial synthesis. Hereinafter, any reference to “fenfluramine” is understood to referto the racemic form, i.e, (RS)-N-ethyl-1-[3-(trifluoromethyl)phenyl]propan-2-amine. - [0005]
To the best of the knowledge of the inventors, the first method for fenfluramine synthesis reported in the literature dates back to 1962 and is referenced in patent BE609630 and in analogous patents US3198833 and FR1324220 . All the synthesis methods reported in these patents provide for numerous synthesis steps. By way of example, one of the methods provides for the transformation into oxime of a ketone, 1-(3-trifluoromethyl)phenyl-propan-2-one, as shown here: - [0006]
The oxime is then hydrogenated in the presence of Raney nickel catalyst so as to yield the corresponding primary amine, which is acetylated subsequently with ethanoic anhydride before being converted into fenfluramine by reduction with lithium aluminum hydride. - [0007]
As can be seen, the final step of this chemical process provides for the use of lithium aluminum hydride and the persons skilled in the art will acknowledge that the use of this reagent should be avoided, if possible, on an industrial level, since it is extremely flammable and is the source of accidents. Furthermore, lithium is a potentially neurotoxic metal and therefore its use should be avoided where possible. Furthermore, the Raney nickel catalyst is used in the oxime reduction step and can contaminate the final active ingredient; the use of hydroxylamine also entails problems of toxicity for workers assigned to production. - [0008]
A further disadvantage of this process is, as already mentioned earlier, the number of steps, not only because a large number of synthesis steps entails a reduction of the overall yield of active ingredient, but also because each synthesis step in principle can generate impurities and a larger number of steps can therefore entail a higher number of impurities in the final active ingredient. Many of these impurities, furthermore, due to their structural similarity to fenfluramine, are difficult to eliminate and remove from a fenfluramine preparation. One impurity for example that can be formed in the process described above and is difficult to eliminate is the following: - [0009]
This impurity, which is a primary amine, shares physical-chemical properties that are similar to fenfluramine and therefore, like fenfluramine, it can form a hydrochloride salt by treatment with hydrochloric acid and thus contaminate the active ingredient fenfluramine hydrochloride. Furthermore, this impurity – as a free base – has a boiling point that is similar to that of fenfluramine (73°C vs. 89°C at 6 mmHg respectively), and therefore its elimination by distillation also can be problematic. - [0010]
The process described above can in principle generate other impurities, which are listed in Figure 1 . - [0011]
EP 0441160 claims a synthesis in 5 steps of dexfenfluramine, dextrorotatory enantiomer of fenfluramine. This synthesis can be adapted easily to produce fenfluramine instead of its dextrorotatory enantiomer simply by performing the first reduction step with a non-chiral reducing agent. In the first step, in fact:a ketone, 1-(3-trifluoromethyl)phenyl-propan-2-one, is first reduced to the corresponding alcohol in the presence of yeast, D-glucose, ethanol and water. Then the alcohol is converted into the tosylate in the second step: - [0012]
This reaction occurs in the presence of triethylamine and tosyl chloride in methylene chloride as solvent. After purification, the tosylate is converted to fenfluramine by means of three successive steps: - [0013]
In the first of these three steps, the tosylate is converted into an azide intermediate by reaction with sodium azide in dimethylformamide. The azide intermediate is then hydrogenated in the presence of a catalyst, palladium on carbon. Finally, the resulting primary amine is converted into fenfluramine by reaction with acetaldehyde and sodium borohydride. - [0014]
Persons skilled in the art may see easily that this process is not desirable from an industrial standpoint due to reasons related to environmental risk, safety and costs. For example, the sodium azide used in the process is a notoriously explosive compound and its use at the industrial level is dangerous. Furthermore, palladium is an expensive material and its use in the process entails an increase in the production costs of fenfluramine. Furthermore, palladium can contaminate the finished active ingredient. - [0015]
In another method for the synthesis of dexfenfluramine in 3-4 steps, reported by Goument et al. in Bulletin of the Chemical Society of France (1993), 130, p. 450-458, 3-bromobenzotrifluoride is subjected to a Grignard reaction with enantiopure 1,2-propylene-epoxide to yield 1-[3-(trifluoromethyl)phenyl]propan-2-ol as shown hereafter: - [0016]
If this reaction is performed with racemic 1,2-propylene-epoxide, the synthesis can be adapted to the preparation of fenfluramine. - [0017]
The alcohol thus obtained is first transformed into trifluoromethyl sulfonate by reaction with trifluoromethanesulfonic anhydride and then treated with ethylamine to yield fenfluramine, as shown in the diagram hereinafter: - [0018]
In this article, the authors acknowledge that the main byproducts of the reaction are isomer alkenes having the following chemical structures: - [0019]
The process proposed by Goument et al. is not interesting from the industrial standpoint for a series of reasons. First of all, it is known that the use of Grignard reagents, especially on an industrial scale, is problematic, because these compounds are often pyrophoric and corrosive. Furthermore, 1,2-propylene epoxide is a suspected carcinogenic compound. Finally, the formation of the three isomer alkenes as byproducts listed above is a disadvantage of the process. In the article, Goument presents methods for activation of the intermediate alcohol which are alternative to trifluoromethylsulfonate, for example by converting it to chloride (via thionyl chloride) or to mesylate (via mesyl chloride), but these process variations share the same disadvantages as the main process analyzed above. - [0020]
In addition to the methods with multiple synthesis steps discussed so far in detail, the literature reports other methods or processes for producing fenfluramine or dexfenfluramine. In general, persons skilled in the art acknowledge that the syntheses in the literature for producing dexfenfluramine sometimes can be applied to the preparation of fenfluramine simply by replacing the initial materials and/or enantiopure reagents with the corresponding racemates while maintaining the reaction conditions. For example, patents that present long synthesis methods in multiple steps are the following: - [0021]
Other examples of preparation of fenfluramine, taken from non-patent literature, are the following:- Synthesis, Nov.1987, p. 1005-1007
- J.Org.Chem, 1991, 56, p. 6019
- Tetrahedron, 1994, 50(1), p. 171
- Bull. Soc. Chim. France, 1993, 130(4), p. 459-466 (dexfenfluramine)
- Chirality, 2002, 14(4), p. 325-328 (dexfenfluramine)
- [0022]
Without analyzing in detail the individual methods described in these patents or articles, it can be stated in summary that all these methods are not attractive and interesting from the industrial standpoint because these are processes with many synthesis steps or because the initial materials described therein are not easily available and therefore have to be prepared separately, with a further expenditure of time and with further costs, or because they provide for the use of reagents that are dangerous/explosive/toxic or because they entail the use of catalysts based on heavy metals that can contaminate the final active ingredient. - [0023]
One should consider that in the literature there are methods for the preparation of fenfluramine that do not provide for long syntheses and multiple steps but are shorter and consist of one or two steps. These processes, which therefore would be more interesting from the industrial standpoint, have other specific disadvantages, as will become apparent in detail hereinafter. For example, in the literature there is a first group of articles or patents that describe the reaction between 1-(3-trifluoromethyl)phenyl-propan-2-one and ethylamine in the presence of hydrogen gas and of a transition metal as catalyst: - [0024]
In particular, in Huagong Shikan, 2002, 16(7), p. 33, the reaction is performed with hydrogen gas (2.9 – 3.38 atm), at 65-75°C, for 9 hours, in the presence of Raney nickel. Likewise, in patent DD108971 (1973), Raney nickel and hydrogen gas and methanol are used as solvent to perform this reaction. - [0025]
In HU55343 , instead, a similar reaction in one step is performed with hydrogen gas in the presence of another transition metal catalyst, such as palladium on carbon. - [0026]
Although these three methods describe short single-step processes, they have the disadvantage of the use of hydrogen gas. As is known to persons skilled in the art, hydrogen gas is a dangerous gas due to the inherent danger of forming explosive mixtures with air and must be used by expert personnel in expensive facilities dedicated to its use and built with special precautions. Despite being used in purpose-built facilities, the use of hydrogen at the industrial level is inherently dangerous and to be avoided if possible. Another danger element that is shared by the processes described above is the fact that the reactions are performed under pressure. The third industrial disadvantage then arises from the use of heavy metal catalysts, which have a high cost and therefore increase the overall cost of the final active ingredient and -on the other hand- may contaminate the active ingredient fenfluramine even after filtration of the catalyst and purification of said active ingredient. - [0027]
Analysis of the background art shows, however, that an attempt has been made to devise a process for the production or synthesis of fenfluramine that is short (one or two steps) and does not entail the use of hydrogen gas or of catalysts based on nickel or palladium or the like. In particular, for example, Synthesis 1987, 11, p. 1005, and then DECHEMA Monographien (1989), 112 (Org. Elektrochem.–Angew. Elektrothermie), 367-74, present a method for the synthesis of fenfluramine which starts from 1-(3-trifluoromethyl)phenyl-propan-2-one, which is made to react with ethylamine in great excess, in an electrochemical process, which uses a mercury cathode in a water/ethanol solution with pH 10-11. One obtains fenfluramine with 87% yield. This process has some drawbacks from an industrial standpoint: it is a process of the electrochemical type and therefore requires special equipment which is scarcely widespread, dedicated cells and reactors, and it is not possible to use the classic multipurpose reactors available in the pharmaceutical industry. Furthermore, the use of mercury at the industrial level poses severe environment safety problems, requiring constant health monitoring on workers who manage the equipment and systems for the management and destruction of wastewater that are particularly onerous; finally, mercury can be transferred from the cathode to the reaction environment and therefore to the active ingredient, and this obviously is to be considered very dangerous due to the accumulation of the metal in human beings; small traces of mercury are very toxic. - [0028]
Another method for fenfluramine synthesis in a single step is the one presented in J.Org.Chem, 1979, 44(20), p. 3580. Here the reaction is described between an alkene derivative and ethylamine in the presence of sodium borohydride and mercury nitrate: - [0029]
Again, this process is not interesting from an industrial standpoint since it has the same problems, if not even greater ones, related to the use of mercury (used here as a water-soluble salt) discussed previously. The complication introduced in this process with the use of mercury nitrate together with sodium borohydride highlights the level of innovation of the synthesis path found here. - [0030]
In past years, therefore, it has not been possible to provide a process for synthesizing fenfluramine in a small number of steps by using modern reducing agents that are commonly and easily used. Indeed, while Gaodeng Xuexiao Huaxue Xuebao, 9(2), 1988, p. 134-139, describes and exemplifies the synthesis of 2-N-ethyl-1-phenyl propane by means of (1) the treatment of the precursor ketone with ethylamine followed by (2) sodium cyanoborohydride as reducing agent, Xuexiao Huaxue Xuebao provides no example for fenfluramine. Moreover, for the latter, Xuexiao Huaxue Xuebao indicates a melting point for the hydrochloride of 161°C, a data item that matches the value indicated in the literature initially (see BE609630 ); these facts prove that fenfluramine synthesis with cyanoborohydride was not performed, otherwise one cannot explain why the author did not transcribe, in the document, the example of a product that at the time was very important. It should be noted in fact that 1-phenyl propan-2-one and 1-(3-trifluoromethyl)phenyl-propan-2-one can have different reactivities to reductive amination due to the presence of a highly electron-attractor -trifluoromethyl group, hence the need for an example to demonstrate its feasibility. The use of cyanoborohydride shares some disadvantages with other methods discussed in the preceding paragraphs. The excellent selectivity for reductive aminations of this reagent is highly appreciated, but its application can be less advantageous with respect to other reducing systems in the synthesis of fenfluramine, where the latter is intended for therapeutic application in human beings. The reasons for this are the possible contamination of the finished pharmaceutical active ingredient with cyanide ions, the toxicity of the reagent itself and finally the danger of its use. It is known to persons skilled in the art that sodium cyanoborohydride can release hydrocyanic acid if the pH of the reaction environment is acid enough and it is known that hydrocyanic acid is a powerful poison, since it competes with oxygen for hemoglobin coordination. As a consequence of this, particular care must be taken in its use and in the disposal of the production wastewater, which can be contaminated by cyanides. Not least, one must consider that the cost of sodium cyanoborohydride is considerable. - [0031]
To conclude, it can be seen that more than 50 years after the publication of its first synthesis dated 1962, there are still numerous disadvantages or limitations in the synthesis paths developed in the past decades in the literature for the preparation of fenfluramine. - [0032]
Moreover, recently there has been renewed pharmaceutical interest in the fenfluramine molecule, since the possibility of its therapeutic use in severe disorders of infancy has appeared in the medical literature. For example, mention can made of Ceulemans et al., Epilepsia, 53(7), pages 1131 to 1139, 2012. - [0033]
According to a certain part of medical literature, fenfluramine might therefore be interesting as a medication in a chronic therapy for the treatment of symptoms of epilepsy and other correlated severe disorders. - [0034]
Based on recent medical developments, therefore, the need exists for a synthesis method that is better than the existing ones and can overcome in particular the disadvantages of the processes that are present in the literature. Particularly important, in view of use in chronic therapies for children such as epilepsy and other severe disorders, it would be fundamentally important to identify a path for synthesis of the active ingredient fenfluramine or of isomers thereof and/or analogs thereof that does not entail the use of heavy metals and/or transition metals, which in a chronic therapy might accumulate in the body of the patients over the years, with severe consequences on health. - [0035]
More generally, it is desirable to identify a synthesis path that uses reagents from which (or from the transformation products of which) it is then possible to easily purify fenfluramine (or isomers and/or analogs thereof). - [0036]
It would be equally desirable to identify a synthesis path that comprises a small number of synthesis steps and uses reagents that are widely commercially available and easy to use. - [0037]
At the same time, the new identified synthesis path should avoid if possible the formation of byproducts.
EXAMPLES
- [0082]
The present invention is exemplified by, but not limited to, the following examples:
Example 1 – Synthesis of fenfluramine
- [0083]
A suspension of sodium hydroxide (34.62 g – 0.866 mol, 3.5 eq) in 170 mL of methanol, under mechanical agitation, receives the addition, drop by drop, over the course of 30 minutes, of a solution of ethylamine hydrochloride (70.59 g – 0.866 mol, 3.5 eq) in 165 mL of methanol, followed by 1-(3-trifluoromethyl)phenyl-propan-2-one (50 g – 0.247 mol). The mixture is left under agitation at 20°C for 4.5 hours, then cooling to 0°C is performed and a solution of sodium borohydride (9.36 g – 0.247 mol) in 19 mL of sodium hydroxide 1M in water is then added drop by drop, keeping the temperature below 10°C. The reaction is then left under agitation at 20°C for another 2 hours. Once the reaction is complete, 270 mL of methanol are removed at a reduced pressure at 40°C and then 200 mL of water are added and the mixture is extracted with heptane (200 mL). The aqueous phase is eliminated and the organic phase is washed with water (200 mL x 3). The organic phase is concentrated at 50°C at reduced pressure to yield free base fenfluramine as colorless oil. Yield: 72%; purity: 77% – as listed in test 3 of table A above.
Example 2 – Purification of fenfluramine
- [0084]
Purification of free base fenfluramine can be performed in two ways: - distillation of the free base
- crystallization of the fenfluramine hydrochloride salt
- [0085]
Depending on the degree of purity that is desired, both purification processes are performed in sequence (distillation first and then crystallization), or only one of the two purification processes is performed.
Example 2a – Distillation:
- [0086]
Free base fenfluramine (10 g), prepared as in Example 1, is distilled under reduced pressure with a distillation column of the Vigreux type: the distillation heads are eliminated, the fraction that is distilled at 89-90°C at 6 mmHg, which is the active ingredient fenfluramine (8.5 g) with a high degree of purity, is collected.
Example 2b – Conversion into hydrochloride salt and crystallization:
- [0087]
Crude fenfluramine, prepared as in Example 1, or purified fenfluramine as in Example 2a, is dissolved in 125 mL of ethyl acetate, and cooling is performed to 0°Celsius under agitation. 272 mL of a solution of 1M HCl in ethyl acetate are added drop by drop at 0°C. The precipitate that forms is filtered and washed with ethyl acetate (125 mL x 2) to yield approximately 55 g of solid fraction. The solid fraction is crystallized by 2-butanol (260 mL), keeping the solid for 22 hours at 3°C under slow agitation before filtering it. Filtering is performed and washing is performed with cold 2-butanol. The solid fraction, fenfluramine hydrochloride, is dried in a vacuum stove, yielding 51.7 g of product. A DSC of the resulting product is shown in Figure 3 .
CLIP
- Synthetic Method of Dexfenfluramine hydrochloride
- (CAS NO.: ), with its systematic name of (S)-N-Ethyl-alpha-methyl-m-(trifluoromethyl)phenethylamine hydrochloride, could be produced through many synthetic methods.Following is one of the synthesis routes:
 The action of d-camphoric acid on (rac)-fenfluramine (I) affords the camphorate of (+)-fenfluramine (II). After purification of this salt by crystallization, sodium hydroxide in methylene chloride is added, forming (+)-fenfluramine (III) after removal of camphoric acid. Finally, the action of hydrogen chloride in methyl cyclohexane on (+)-fenfluramine produces the corresponding salt: (+)-fenfluramine hydrochloride.
The action of d-camphoric acid on (rac)-fenfluramine (I) affords the camphorate of (+)-fenfluramine (II). After purification of this salt by crystallization, sodium hydroxide in methylene chloride is added, forming (+)-fenfluramine (III) after removal of camphoric acid. Finally, the action of hydrogen chloride in methyl cyclohexane on (+)-fenfluramine produces the corresponding salt: (+)-fenfluramine hydrochloride.
PAPER
https://www.designer-drug.com/pte/12.162.180.114/dcd/chemistry/fenfluramine.html
Fenfluramine 1 is the active ingredient of a obesity drug acting on the digestion of carbohydrates, the activity being restricted mainly to the S enantiomer [1, 2], which can be obtained by separation of the diastereoisomers [3] or by preferential crystallisation of derivates, which were identified of being conglomerates [4]. Only two syntheses of optical active fenfluramine have been described until now: one by stereoselective reduction of the imine derived from the ketone 2 and (R) or (S)-alpha-phenylethylamine [5], the other starting from (S)-alanine [6]. Two recent publications [7, 8] about the synthesis of (S)-fenfluramine via the intermediate alcohol (S)-3 (scheme 1) made us publish our previous results [9]. Through yeast reduction of the ketone 2, the authors obtain the alcohol (S)-3, the configuration of which they inverse in three steps. The alcohol (R)-3, via the intermediate tosylate (R)-4a and further the azide (S)-5 leads to the amine (S)-6 after reduction and finally to the (S)-fenfluramine (S)-1 after reductive amination in presence of acetaldehyde:
The (S)-fenfluramine is such obtained in 7 steps starting from the alcohol (S)-3 or in 4 steps from the alcohol (R)-3.
In this article we present a new way of preparing the two enantiomers of the alcohol 3, a new two step synthesis of (S)-fenfluramine starting from the alcohol (R)-3, a one step synthesis of (S)-fenfluramine starting from the azide (S)-5, which doesn’t pass over the intermediate primary amine (S)-6 and finally a much faster process (3 steps) of preparing (S)-fenfluramine starting from the alcohol (S)-3.
Results and discussion
Synthesis of 1-[3-(trifluoromethyl)phenyl]propan-2-ol (R)-3
The racemic alcohol is seldom mentioned. It’s one of the metabolites of fenfluramine in the human body, secreted in urine [10]. It can also be obtained by metabolic transformations of an oxime by different kinds of microorganisms [11]. It was used as an intermediate for the synthesis of a family of anorexics [12] and a family of antispasmodic and psychotherapeutic agents [13]. It was obtained by the reaction of methyloxirane 7 with the magnesium compound 8, with a yield of 50%. This same reaction was described earlier as being little regioselective [14], a fact we observed too [9].
The only synthesis of the optically active alcohol 3 is the reduction of the ketone 2 with yeast as described above. One abtains the S enantiomer, the R enantiomer is obtained by inversion.
On our part we used the condensation of the commercial [15] methyloxirane (R)-7, of which many syntheses are known [16], with the magnesium compound 8 and cuprous chloride [17, 18].
The yield is about 90% and the reaction very selective (purity GC: 93%). The optical purity of the methyloxirane 7 was determined by 1H-NMR in presence of the europium complex Eu(hfc)3 [19]. The optical purity of the alcohol (R)-3 was obtained by 1H-NMR and HPLC over silica of the Mosher derivate [20]. The comparison of these values show that the chiral centre is preserved. This procedure has the advantage of allowing us the preparation of the alcohol (S)-3 with the same reaction, because the methyloxirane (S)-7 is also commercially available and multiple syntheses are known [21].
Two step synthesis of the fenfluramine (S)-1 starting from the alcohol (R)-3
With the goal of obtaining the simplest procedure we have studied at first the transformation of the alcohol (R)-3 into fenfluramine (S)- 1 in two steps via the intermediate of the easily obtained sulfonates (R)-4: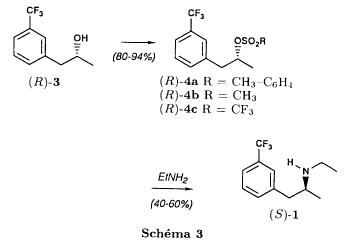
The substitution of the mesitylate (R)-4b and the tosylate (R)-4a with ethylamine was realised with medium yields always between 40 and 50% in spite of the large number of conditions tested: solvents (DMSO, DMF, ethanol, ethylamine), different dilutions (in proportions from 1 to 5) and temparatures from 50 to 160°C (with different times of contact). With the triflate (R)-4c the yield of the substitution is 60% but under non comparable conditions (-20°C in acetonitrile) because of its higher reactivity. In all cases the non aminated, and thus easily separated, byproducts are mainly the alkenes 9, 10Z and 10E (10E >> 10Z > 9).
The enantiomeric purity of the amine (S)-1 is analysed by HPLC chromatography through silica of the camphanylated derivate [22] and compared to the previously analysed alcohol (R)-3: we have thus shown that the optical centre is conserved during the nucleophilic substitution. One had indeed to fear that due to the participation of the aromatic ring as neighbour group there could be partial or complete racemisation with an phenonium ion as intermediate. With the results obtained, which match with the literature [23-26], one can suppose that the trifluoromethyl group in meta position is sufficiently deactivating the aromatic ring in order to prevent participation in the substitution. We probably have thus in our case a pure nucleophilic SN2 substitution in competition with an elimination reaction. We believe that this elimination reaction is due to the simultaneous nucleophilic and basic properties of the ethylamine.
Although the yields are medium, this method has the advantage of being relatively fast because it permits to prepare fenfluramine (S)-1 starting from the alcohol (R)-2 in two steps instead of four [7, 8]. As far as we know it was never mentioned in literature.
Synthesis of fenfluramine (S)-1 from 2-azido-1-[3-(trifluoromethyl)phenyl]propane (S)-5
The substitution of the mesylate (R)-4b by sodium azide (scheme 4), an only slightly basic nucleophile compared to ethylamine, forms no elimination side products. One obtains the optically pure azide (S)-5 with a yield of 95%.
The enantiomeric purity couldn’t be directly analysed on the azide (S)-5. Only for analytical purposes did we reduce it into the amine (S)- 6. Among the numerous methods for the reductions of azides to amines mentioned in the literature [27] we chose the catalytic hydrogenation with 5% Pd on calcium carbonate at standard temperature and pressure [27f]. The HPLC analysis through silica column of the champhanyl derivate [22] of the amine (S)-6 such obtained shows that the enantiomeric centre was totally inverted during the substitution when compared to the enantiomeric purity of the alcohol (R)-3.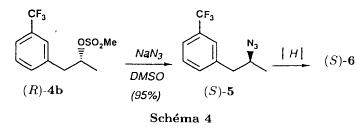
The reductive amination of the amine 6 in presence of acetaldehyde is known for a long time [28]. It was used recently in the works listed in the introduction [7, 8]. On our part, we propose another synthetic route for fenfluramine (S)-1 starting from the azide (S)-5 which does not go via the primary amine (S)-6 (Schema 5).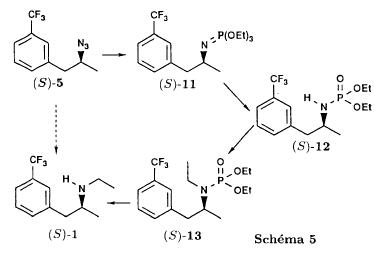
The reaction of Staudinger, reacting a stoichiometric quantity of triethylphosphite on the azide (S)-5 in THF at room temperature [29], gives quantitative yields of the phosphorimide 11 in 48 hours. It’s total conversion into the phosphoramide 13, by reacting with ethyl iodide [30] could not be realised [9]. We always obtained different mixtures of the phosphoramides 12 and 13 (referential compounds prepared from the amines 6 and 1). We also noted that the phosphorimide 11 can’t be isolated. When the solvent is evaporated, a partly transformation into the phosphoramide 12 takes place. This transformation is completed in less then 2h by simple heating to 100°C under argon after evaporation of the solvent. Because the phosphorimides are strongly basic compounds, we believe that an intramolecular arrangement of the phosphorimide, pictured in Schema 6, takes place.
Having the phosphoramide 12, we investigated the alkylation into the phosphoramide 13 in DMF at room temperature [31, 32]: one deprotonates with sodium hydride then alkylates with diethyl sulfate. After treatment with hydrogen bromide [33], one obtains fenfluramine 1 with a yield of 85% and a purity of 97% (GC).
With the goal of simplifying the reaction scheme by avoiding the isolation of the intermediates we have again studied the transformation 5 -> 11 -> 12 in DMF (Schema 7). First, we noted that the reaction of Staudinger can be directly realised in this solvent. Thereafter we pinned down the transformation of the phosphorimide 11 into the phosphoramide 12 by reaction with water [34]. One then proceeds as described above. The transformation is thus performed without isolation of a single intermediate with a yield of 83%.
HPLC analysis on silica column of the camphanyl derivate of the amine (S)-1 [22] shows that the optical centre is conserved during the whole transformation.
Synthesis via the intermediate 2-chloro-1-[3-(trifluoromethyl)phenyl]propane 14
The yeast reduction of the ketone 2 gives the alcohol (S)-3, of which the authors have inverted the configuration to get the pharmacological active S enantiomer of fenfluramine [7, 8]. Independent research, using the epoxidation method of Sharpless [9, 35] lead us too to the alcohol (S)- 3 which we tried to convert into fenfluramine (S)-1 using a different method. The reaction scheme we kept uses the chloride 14 and proceeds via two inversions of the optical centre (scheme 8). Not owning enough alcohol (S)-3 during the studies, we tested the principle starting with the alcohol (R)-3, produced earlier, and studied the transformation into the azide (R)-5 (scheme 8), the latter being able to lead to (R)-fenfluramine using different methods, like the one outlined above:
It is well known that the action of thionylchloride on an optically active alcohol gives the corresponding chlorine derivate, with inversion of the configuration in presence of bases and with retention of the configuration in the other case. We have performed the reaction with a catalytical amount of pyridine. One thus obtains the chloride (S)-14 with 91% yield and a purity of 91% (GC): it contains 9% of the elimination products 9, 10Z and 10E which are not separable by chromatography on silica.
The direct substitution of the chloride 14 with ethylamine with similar conditions to those used for the mesylate (R)-4b (EtNH2, DMSO, 110°C, 5h30 or EtNH2 (solvent and reactant), 140°C, 5h), gives mainly the elimination products. The yield of fenfluramine is below 10%.
By action of sodium azide in DMSO, on the other hand, one obtains the azide (R)-5 with a yield of 78%, the elimination products formed here or in the last step can be removed by chromatography on silica. HPLC analysis on silica of the camphanyl derivate of the amine (R)-6 [22] obtained by catalytic reduction of the azide (R)-5 has confirmed the double inversion without racemisation after comparison with the starting alcohol (R)-3. Then the fenfluramine (R)-1 is prepared without racemisation with a 83% yield starting from the azide (R)-5 like detailed above.
This procedure with two inversions allows to transform the alcohol 3 in the azide 5 with the same configuration in two steps with a global yield (non optimised) of 70% and without racemisation. It’s thus preferred over the recently published one [7, 8], which needs 5 steps for a lower global yield (55%) and in addition features an epimerisation of 10% [8]. It’s a promising way to fenfluramine (S)-1 starting from the alcohol (S)- 3.
References
- ^ “Fintepla- fenfluramine solution”. DailyMed. Retrieved 8 January 2021.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “FDA Approves New Therapy for Dravet Syndrome”. U.S. Food and Drug Administration (FDA) (Press release). 25 June 2020. Retrieved 25 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d “Fintepla EPAR”. European Medicines Agency. Retrieved 8 January2021.
- ^ Jump up to:a b c d Richard C. Dart (2004). Medical Toxicology. Lippincott Williams & Wilkins. pp. 874–. ISBN 978-0-7817-2845-4. Archived from the original on 2018-05-09.
- ^ Jump up to:a b James O’Donnell (Pharm. D.); Gopi Doctor Ahuja (2005). Drug Injury: Liability, Analysis, and Prevention. Lawyers & Judges Publishing Company. pp. 276–. ISBN 978-0-913875-27-8.
- ^ Integrating the Neurobiology of Schizophrenia. Academic Press. 27 February 2007. pp. 142–. ISBN 978-0-08-047508-0.
- ^ The Pharmacology of Corticotropin-releasing Factor (CRF). Effects on Sensorimotor Gating in the Rat. 2006. pp. 12–. ISBN 978-0-549-53661-1.
- ^ Christian P. Muller; Barry Jacobs (30 December 2009). Handbook of the Behavioral Neurobiology of Serotonin. Academic Press. pp. 630–. ISBN 978-0-08-087817-1.
- ^ Vickers SP, Dourish CT, Kennett GA (2001). “Evidence that hypophagia induced by d-fenfluramine and d-norfenfluramine in the rat is mediated by 5-HT2C receptors”. Neuropharmacology. 41 (2): 200–9. doi:10.1016/s0028-3908(01)00063-6. PMID 11489456. S2CID 23374227.
- ^ Roth, B. L. (2007). “Drugs and Valvular Heart Disease”. New England Journal of Medicine. 356 (1): 6–9. doi:10.1056/NEJMp068265. PMID 17202450.
- ^ Rothman, R. B.; Baumann, M. H. (2009). “Serotonergic Drugs and Valvular Heart Disease”. Expert Opinion on Drug Safety. 8 (3): 317–329. doi:10.1517/14740330902931524. PMC 2695569. PMID 19505264.
- ^ Jump up to:a b Dahl, C. F.; Allen, M. R.; Urie, P. M.; Hopkins, P. N. (2008). “Valvular Regurgitation and Surgery Associated with Fenfluramine Use: An Analysis of 5743 Individuals”. BMC Medicine. 6: 34. doi:10.1186/1741-7015-6-34. PMC 2585088. PMID 18990200.
- ^ Jump up to:a b c d e f g h i j k l m Donald G. Barceloux (3 February 2012). Medical Toxicology of Drug Abuse: Synthesized Chemicals and Psychoactive Plants. John Wiley & Sons. pp. 255–262. ISBN 978-1-118-10605-1. Archived from the original on 9 May 2018.
- ^ Mann SC, Caroff SN, Keck PE, Lazarus A (20 May 2008). Neuroleptic Malignant Syndrome and Related Conditions. American Psychiatric Pub. pp. 111–. ISBN 978-1-58562-744-8.
- ^ Jump up to:a b c d e f g h i Rothman RB, Clark RD, Partilla JS, Baumann MH (2003). “(+)-Fenfluramine and its major metabolite, (+)-norfenfluramine, are potent substrates for norepinephrine transporters”. J. Pharmacol. Exp. Ther. 305 (3): 1191–9. doi:10.1124/jpet.103.049684. PMID 12649307. S2CID 21164342.
- ^ Jump up to:a b c d e f g Setola V, Hufeisen SJ, Grande-Allen KJ, Vesely I, Glennon RA, Blough B, Rothman RB, Roth BL (2003). “3,4-methylenedioxymethamphetamine (MDMA, “Ecstasy”) induces fenfluramine-like proliferative actions on human cardiac valvular interstitial cells in vitro”. Mol. Pharmacol. 63 (6): 1223–9. doi:10.1124/mol.63.6.1223. PMID 12761331. S2CID 839426.
- ^ Nestler, E. J. (2001). Molecular Neuropharmacology: A Foundation for Clinical Neuroscience. McGraw-Hill.
- ^ Giuseppe Di Giovanni; Vincenzo Di Matteo; Ennio Esposito (2008). Serotonin-dopamine Interaction: Experimental Evidence and Therapeutic Relevance. Elsevier. pp. 393–. ISBN 978-0-444-53235-0.
- ^ Fitzgerald LW, Burn TC, Brown BS, Patterson JP, Corjay MH, Valentine PA, Sun JH, Link JR, Abbaszade I, Hollis JM, Largent BL, Hartig PR, Hollis GF, Meunier PC, Robichaud AJ, Robertson DW (2000). “Possible role of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with fenfluramine”. Mol. Pharmacol. 57 (1): 75–81. PMID 10617681.
- ^ Jump up to:a b c Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI, Partilla JS (January 2001). “Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin”. Synapse. 39 (1): 32–41. doi:10.1002/1098-2396(20010101)39:1<32::AID-SYN5>3.0.CO;2-3. PMID 11071707.
- ^ Rothman RB, Baumann MH (2002). “Therapeutic and adverse actions of serotonin transporter substrates”. Pharmacol. Ther. 95 (1): 73–88. doi:10.1016/s0163-7258(02)00234-6. PMID 12163129.
- ^ Connolly, H. M.; Crary, J. L.; McGoon, M. D.; Hensrud, D. D.; Edwards, B. S.; Edwards, W. D.; Schaff, H. V. (1997). “Valvular Heart Disease Associated with Fenfluramine-Phentermine”. New England Journal of Medicine. 337 (9): 581–588. doi:10.1056/NEJM199708283370901. PMID 9271479.
- ^ Weissman, N. J. (2001). “Appetite Suppressants and Valvular Heart Disease”. The American Journal of the Medical Sciences. 321 (4): 285–291. doi:10.1097/00000441-200104000-00008. PMID 11307869. S2CID 46466276.
- ^ FDA September 15, 1997. FDA Announces Withdrawal Fenfluramine and Dexfenfluramine (Fen-Phen) Archived 2013-07-19 at the Wayback Machine
- ^ “Drugs banned in India”. Central Drugs Standard Control Organization, Dte.GHS, Ministry of Health and Family Welfare, Government of India. Archived from the original on 2015-02-21. Retrieved 2013-09-17.
- ^ “FDA Approves FINTEPLA (fenfluramine) for the Treatment of Seizures Associated with Dravet Syndrome” (Press release). Zogenix Inc. 25 June 2020. Retrieved 25 June 2020 – via GlobeNewswire.
- ^ “Fenfluramine Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 25 June 2020.
- ^ “Fenfluramine Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 25 June 2020.
- ^ “Fintepla: Pending EC decision”. European Medicines Agency (EMA). 16 October 2020. Retrieved 16 October 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b John Calvin M. Brust (2004). Neurological Aspects of Substance Abuse. Butterworth-Heinemann. pp. 117–. ISBN 978-0-7506-7313-6.
- ^ United States. Congress. Senate. Select Committee on Small Business. Subcommittee on Monopoly and Anticompetitive Activities (1976). Competitive problems in the drug industry: hearings before Subcommittee on Monopoly and Anticompetitive Activities of the Select Committee on Small Business, United States Senate, Ninetieth Congress, first session. U.S. Government Printing Office. pp. 2–.
- ^ Jump up to:a b Drug Addiction II: Amphetamine, Psychotogen, and Marihuana Dependence. Springer Science & Business Media. 27 November 2013. pp. 258–. ISBN 978-3-642-66709-1.
- ^ Drug Addiction II: Amphetamine, Psychotogen, and Marihuana Dependence. Springer Science & Business Media. 27 November 2013. pp. 249–. ISBN 978-3-642-66709-1.
- ^ “Zogenix ends enrolment in second Phase lll trial of ZX008”. Drug Development Technology. Kable. 1 February 2018. Archived from the original on 2018-02-04. Retrieved 3 February 2018.
- ^ “A Two-Part Study to Investigate the Dose-Ranging Safety and Pharmacokinetics, Followed by the Efficacy and Safety of ZX008 (Fenfluramine Hydrochloride) Oral Solution as an Adjunctive Therapy in Children ≥2 Years Old and Young Adults With Dravet Syndrome”. ClinicalTrials.gov. Archived from the original on 29 September 2017. Retrieved 9 May 2018.
- ^ “Zogenix drug cuts seizure rate in patients with severe epilepsy”. statnews.com. 29 September 2017. Archived from the original on 30 April 2018. Retrieved 9 May 2018.
Further reading
- Gustafsson BI, Tømmerås K, Nordrum I, Loennechen JP, Brunsvik A, Solligård E, Fossmark R, Bakke I, Syversen U, Waldum H (March 2005). “Long-term serotonin administration induces heart valve disease in rats”. Circulation. 111 (12): 1517–22. doi:10.1161/01.CIR.0000159356.42064.48. PMID 15781732.
- Welch, J. T.; Lim, D. S. (2007). “The Synthesis and Biological Activity of Pentafluorosulfanyl Analogs of Fluoxetine, Fenfluramine, and Norfenfluramine”. Bioorganic & Medicinal Chemistry. 15 (21): 6659–6666. doi:10.1016/j.bmc.2007.08.012. PMID 17765553.
External links
- “Fenfluramine”. Drug Information Portal. U.S. National Library of Medicine.
- “Fenfluramine hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
- Inchem.org – Fenfluramine hydrochloride
| Clinical data | |
|---|---|
| Trade names | Fintepla |
| Other names | ZX008 |
| AHFS/Drugs.com | Professional Drug Facts |
| MedlinePlus | a620045 |
| License data | US DailyMed: Fenfluramine |
| Pregnancy category | AU: B2 |
| Routes of administration | By mouth |
| ATC code | A08AA02 (WHO) N03AX26 (WHO) |
| Legal status | |
| Legal status | US: Schedule IV [1][2]EU: Rx-only [3] |
| Pharmacokinetic data | |
| Elimination half-life | 13–30 hours[4] |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 458-24-2 |
| PubChem CID | 3337 |
| IUPHAR/BPS | 4613 |
| DrugBank | DB00574 |
| ChemSpider | 3220 |
| UNII | 2DS058H2CF |
| KEGG | D07945 C06996 |
| ChEBI | CHEBI:5000 |
| ChEMBL | ChEMBL87493 |
| CompTox Dashboard (EPA) | DTXSID4023044 |
| ECHA InfoCard | 100.006.616 |
| Chemical and physical data | |
| Formula | C12H16F3N |
| Molar mass | 231.262 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Chirality | Racemic mixture |
| SMILES[hide]FC(F)(C1=CC(CC(C)NCC)=CC=C1)F | |
| InChI[hide]InChI=1S/C12H16F3N/c1-3-16-9(2)7-10-5-4-6-11(8-10)12(13,14)15/h4-6,8-9,16H,3,7H2,1-2H3 Key:DBGIVFWFUFKIQN-UHFFFAOYSA-N |
CLIP
http://www.inchem.org/documents/pims/pharm/pim938.htm
////////////Fenfluramine, 塩酸フェンフルラミン , dravet, AHR-3002, ZX-008, Fintepla
CCNC(C)CC1=CC(=CC=C1)C(F)(F)F.Cl
PATENT
https://patents.google.com/patent/US20170174613A1/en
- [0093]
Many general references providing commonly known chemical synthetic schemes and conditions useful for synthesizing the disclosed compounds are available (see, e.g., Smith and March, March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Fifth Edition, Wiley-Interscience, 2001; or Vogel, A Textbook of Practical Organic Chemistry, Including Qualitative Organic Analysis, Fourth Edition, New York: Longman, 1978). - [0094]
Compounds as described herein can be purified by any purification protocol known in the art, including chromatography, such as HPLC, preparative thin layer chromatography, flash column chromatography and ion exchange chromatography. Any suitable stationary phase can be used, including normal and reversed phases as well as ionic resins. In certain embodiments, the disclosed compounds are purified via silica gel and/or alumina chromatography. See, e.g., Introduction to Modern Liquid Chromatography, 2nd Edition, ed. L. R. Snyder and J. J. Kirkland, John Wiley and Sons, 1979; and Thin Layer Chromatography, ed E. Stahl, Springer-Verlag, New York, 1969. - [0095]
During any of the processes for preparation of the subject compounds, it may be necessary and/or desirable to protect sensitive or reactive groups on any of the molecules concerned. This may be achieved by means of conventional protecting groups as described in standard works, such as J. F. W. McOmie, “Protective Groups in Organic Chemistry”, Plenum Press, London and New York 1973, in T. W. Greene and P. G. M. Wuts, “Protective Groups in Organic Synthesis”, Third edition, Wiley, New York 1999, in “The Peptides”; Volume 3 (editors: E. Gross and J. Meienhofer), Academic Press, London and New York 1981, in “Methoden der organischen Chemie”, Houben-Weyl, 4th edition, Vol. 15/1, Georg Thieme Verlag, Stuttgart 1974, in H.-D. Jakubke and H. Jescheit, “Aminosauren, Peptide, Protein”, Verlag Chemie, Weinheim, Deerfield Beach, and Basel 1982, and/or in Jochen Lehmann, “Chemie der Kohlenhydrate: Monosaccharide and Derivate”, Georg Thieme Verlag, Stuttgart 1974. The protecting groups may be removed at a convenient subsequent stage using methods known from the art. - [0096]
The subject compounds can be synthesized via a variety of different synthetic routes using commercially available starting materials and/or starting materials prepared by conventional synthetic methods. A variety of examples of synthetic routes that can be used to synthesize the compounds disclosed herein are described in the schemes below.
Example 11. Fenfluramine Nomenclature & Structure
- [0097]
Chemical Abstract Service (CAS) Registry Number (RN): 404-82-0 (HCl Salt), 458-24-2 (Parent Free Base) - [0098]
Chemical Name: N-ethyl-α-methyl-3-(trifluoromethyl)-benzeneethanamine hydrochloride (1:1). Other Names: Fenfluramine HCl, DL-Fenfluramine, (±)-Fenfluramine - [0099]
Structure of Hydrochloride Salt: - [0100]
Stereochemistry: Fenfluramine HCl has one chiral center and is being developed as the racemate and contains dexfenfluramine and levofenfluramine - [0101]
Molecular Formula of hydrochloride salt: C12H16F3N.HCl - [0102]
Molecular Mass/Weight: 267.72 g/mol
2. General Properties
- [0103]
Table 1 summarizes the chemical and physical properties of Fenfluramine HCl. - TABLE 1 General Properties of Fenfluramine HCl Drug Substance Property Result Appearance (color, White to off-white powder physical form) DSC (melting 170° C. (melt/sublimation) point)a TGA Onset 147° C. 0.03% at 150° C. 91% at 220° C. (evaporation) pKa (water) 10.15-10.38 Solubility (mg/mL) Resultant pH 25° C. 37° C. Solubility pH 6.69 (water) 54.13 71.22 (Aqueous) pH 1.73 buffer 25.34 53.68 pH 3.43 buffer 29.50 61.97 pH 6.41 buffer 37.42 95.60 0.9% NaCl (water) 22.98 — Solvent Solubility 25° C. (mg/mL) Solubility (Organic Ethanol 150 Solvents) Dichloromethane 30-35 Ethyl Acetate, 1-5 mg Tetrahydrofuran, Toluene, Acetonitrile UV Absorption Maxima: 210, 265 nm Solution pH (water) 6.69 Hygroscopicity @30% RH: ~0.05% (Dynamic Vapor @60% RH: ~0.07% Sorption (DVS) @90% RH: ~0.20%a) Polymorphism Fenfluramine HCl has been consistently isolated as a single crystalline Form 1 as determined by DSC and x-ray powder diffraction (XRPD) Solvation/Hydration Fenfluramine HCl is isolated as a nonhydrated, nonsolvated solid Solution Stability 8 weeks @ pH 6.7 phosphate buffer medium at 40° C. and 60° C. using concentrations of 0.5, 2.5 and 5.0 mg/ml. All conditions, no new impurities >0.1% by HPLC. Solid Stability 8 weeks @ 40° C., 60° C. and 80° C. 7 days at 150° C. All conditions, no new impurities >0.1% by HPLC.
3. Synthesis of Fenfluramine Drug Substance
- [0104]
Scheme 3.1 shows a 2-step route of synthesis used to manufacture initial clinical supplies of Fenfluramine HCl from ketone (2). The batch size is 4 kg performed in laboratory glassware (kilo lab). No chromatography is required and the process steps are amenable to scale-up. In process 1 there is one isolated intermediate Fenfluramine Free Base (1) starting from commercially supplied 1-(3-(trifluoromethyl)phenyl) acetone (Ketone 2). All steps are conducted under cGMPs starting from Ketone (2). - [0105]
Scheme 3.2 shows a 4-step route of synthesis to Fenfluramine HCl that can be used for commercial supply. Route 2 utilizes the same 2-step process used by Route 1 to convert Ketone (2) to Fenfluramine HCl with the exception that Ketone (2) is synthesized under cGMP conditions starting from 3-(Trifluoromethyl)-phenyl acetic acid (Acid 4). Bisulfate Complex (3) is an isolatable solid and can be purified before decomplexation to Ketone (2). In-situ intermediates which are oils are shown in brackets. Batch sizes of 10 Kg are performed. Commercial batch sizes of 20 kg are performed in fixed pilot plant equipment. Steps 1-2 of Scheme 3.2 to manufacture Ketone (2) have been demonstrated on a 100 g scale to provide high purity ketone (2) of >99.8% (GC & HPLC). Conversion of Ketone (2) to Fenfluramine using either Route 1 or 2 has provided similar purity profiles. - Starting materials are designated by enclosed boxes. Bracketed and non bracketed compounds respectively indicate proposed in-situ and isolated intermediates. NMI=N-Methyl Imidazole.
4.1. Narrative Description (Route 1)
- [0106]
Step 1: Reductive Amination (Preparation of Fenfluramine Free Base 1) - [0107]
A solution of ethylamine, water, methanol, and 1-(3-(trifluoromethyl)phenyl) acetone (Ketone 2) was treated with sodium triacetoxyborohydride and stirred for 16 h at 25° C. at which time HPLC analysis (IPC-1; In Process Control No. 1) showed the reaction to be complete and sodium hydroxide solution was added until pH>10. Toluene was added and the phases separated, and the aqueous phase (IPC-2) and organic phase (IPC-3) are checked for remaining Fenfluramine and Fenfluramine alcohol and the organic phase was reduced. Purified water was added and the pH adjusted to <2 using conc. HCl and the phases were separated. The aqueous phase was washed with toluene and the toluene phase (IPC-4) and the aqueous phase (IPC-5) was checked for Fenfluramine and Fenfluramine alcohol content. The aqueous phase containing product is pH adjusted to >10 using sodium hydroxide solution. The basic aqueous phase was extracted with MTBE until removal of Fenfluramine from the aqueous phase was observed by HPLC (<0.5 mg/ml) (IPC-6). The organic phase was dried over sodium sulfate and filtered. The filtrate was concentrated in vacuo to give the intermediate product Fenfluramine Free Base 1 as a pale yellow oil tested per specifications described herein which showed by NMR the material to contain 2.93% toluene giving an active yield of 88.3% with a purity of 98.23% by HPLC (0.67% Fenfluramine alcohol). - [0108]
Step 2: Salt Formation (Preparation of Fenfluramine HCl) - [0109]
To a flask was charged ethanol and acetyl chloride. The solution was stirred slowly overnight before ethyl acetate was added. The HCl in ethyl acetate solution formed was polish filtered into a clean carboy and retained for later use. To a vessel was added Fenfluramine free base 1 and MTBE. The Fenfluramine solution in MTBE was collected in two carboys before the vessel was cleaned and checked for particulate residue. The Fenfluramine solution was polish filtered into a vessel and cooled and HCl in ethyl acetate solution was added giving a final pH of 6-7. The batch was stirred for 1 h and filtered. The product was dried under vacuum at 40° C. The product (96.52% yield) was tested per IPC-7 had a purity of 99.75% by HPLC and GC headspace analysis showed MTBE (800 ppm) and EtOAc (150 ppm) to be present. The product was then tested per specifications shown herein.
4.2. Narrative Description (Route 2)
- [0110]
Step 1: Preparation of Ketone Bisulfite Adduct - [0111]
Procedure: Charge acetic anhydride, (2.8 vol, 3.0 wt, 5.0 eq.) to a vessel and commence stirring. Cool the solution to −5 to 5° C., targeting −4° C. Charge 1-methylimidazole, (0.2 vol, 0.21 wt, 0.5 eq.) to the mixture at −5 to 5° C. Caution: very exothermic. If necessary, adjust the temperature to 0 to 5° C. Charge ZX008 acid, (1.00 wt, 1.0 eq.) to the mixture at 0 to 5° C. Caution: exothermic. Stir the mixture at 0 to 5° C. until ≦2.1% area ZX008 acid by HPLC analysis, typically 7 to 9 hours. Charge 15% w/w sodium chloride solution (2.0 vol) to the mixture at 0 to 5° C., 60 to 90 minutes. Caution: very exothermic which will be slightly delayed. Warm the mixture to 18 to 23° C. over 45 to 60 minutes and continue stirring for a further 30 to 45 minutes at 18 to 23° C. Charge TBME, (5.0 vol, 3.7 wt) to the mixture and stir for 10 to 15 minutes at 18 to 23° C. Separate the aqueous layer and retain the organic layer. Back-extract the aqueous layer with TBME, (2×3.0 vol, 2×2.2 wt) at 18 to 23° C. retaining each organic layer. Adjust the pH of the combined organic layer to pH 6.5 to 9.0, targeting 7.0 by charging 20% w/w sodium hydroxide solution (5.3 to 8.3 vol) at 18 to 23° C. Caution: exothermic. Separate the aqueous layer and retain the organic layer. Wash the organic layer with 4% w/w sodium hydrogen carbonate solution (2×3.0 vol) at 18 to 23° C. Determine the residual ZX008 acid content in the organic layer by HPLC analysis, pass criterion ≦0.10% area ZX008 acid. Wash the organic layer with purified water, (2×3.0 vol) at 18 to 23° C. Concentrate the organic layer under reduced pressure to ca. 2 vol at 40 to 45° C., targeting 43° C. - [0112]
Determine the w/w assay of ZX008 ketone (WIP) in the mixture by 1H-NMR analysis for information only and calculate the contained yield of ZX008 ketone (WIP) in the mixture. Note: This step can be removed from the process since the process is robust and consistently delivers 80 to 90% th yield. The achieved yield was factored into the charges of the subsequent steps. - [0113]
Charge n-heptane, (4.0 vol, 2.7 wt) to the mixture at 40 to 45° C., targeting 43° C. Concentrate the mixture to ca. 2 vol at 40 to 45° C., targeting 43° C. Determine the TBME content in the mixture by 1H-NMR analysis, (pass criterion ≦5.0% w/w TBME vs. ZX008 ketone). Charge n-heptane, (2.4 vol, 1.6 wt) at 40 to 45° C., targeting 43° C., vessel A. To vessel B, charge sodium metabisulfite, (0.82 wt, 0.88 eq.) at 18 to 23° C. To vessel B, charge a solution of sodium hydrogen carbonate, (0.16 wt, 0.4 eq.) in purified water, code RM0120 (2.0 vol) at 18 to 23° C. followed by a line rinse with purified water, code RM0120 (0.4 vol) at 18 to 23° C. Caution: gas evolution. Heat the contents of vessel B to 40 to 45° C., targeting 43° C. Charge the contents from vessel A to vessel B followed by a line rinse with n-heptane, (0.8 vol, 0.5 wt) at 40 to 45° C., targeting 43° C. Stir the mixture for 1 to 1.5 hours at 40 to 45° C., targeting 43° C. Charge n-heptane, code RM0174 (3.2 vol, 2.2 wt) to the mixture with the temperature being allowed to cool to 18 to 45° C. at the end of the addition. Cool the mixture to 18 to 23° C. at approximately constant rate over 45 to 60 minutes. Stir the mixture at 18 to 23° C. for 1.5 to 2 hours. - [0114]
Sample the mixture to determine the residual ZX008 ketone content by 1H-NMR analysis, (pass criterion ≦10.0% mol, target 5.0% mol ZX008 ketone vs. ZX008 ketone bisulfite adduct). Filter the mixture and slurry wash the filter-cake with n-heptane, (2×2.0 vol, 2×1.4 wt) at 18 to 23° C. Dry the solid at up to 23° C. until the water content is <10.0% w/w water by KF analysis according to AKX reagent. At least 16 hours. Determine the w/w assay of the isolated ZX008 ketone bisulfite adduct by 1H-NMR analysis and calculate the contained yield of ZX008 ketone bisulfite adduct. - [0115]
Yields and Profiles: The yield for the stage 1 Demonstration batch is summarized Table below. Input: 1700.0 g uncorr., acid, 99.50% area (QC, HPLC), 2-isomer not detected, 4-isomer 0.02% area, RRT1.58 (previously not observed) 0.48% area as per the preparative method. The analytical data is summarized in Table 1A below. - TABLE 1A Table for isolated yields for step 1 Demonstration batch Corr. % area Reference Corr. Yield % w/w (HPLC, number Input Output (% th)** (1H-NMR)* QC) Comments Batch A1 1700.0 g 1500.1 g 89.1 45.0 —.— Crude ketone as TBME sol. Batch A2 1500.1 g 1716.1 77.8 76.0 98.15 Bisulfite adduct only 67.3 Overall product
- [0116]
Step 2: Preparation of Ketone - [0117]
Procedure: Charge toluene, (5.0 vol, 4.3 wt), and purified water, (5.0 vol) to the vessel and commence stirring. If necessary, adjust the temperature to 18 to 23° C. and charge ZX008 ketone bisulfite adduct, (1.00 wt corrected for % w/w assay) to the mixture at 18 to 23° C. Charge 20% w/w sodium hydroxide solution to the mixture at 18 to 23° C. adjusting the pH of the mixture to pH 8.0 to 12.0, targeting 9.0 (0.5 to 1.0 vol). - Separate the lower aqueous layer and retain the top organic layer. Wash the organic layer with purified water, (3.0 vol) at 18 to 23° C. Concentrate the organic layer under reduced pressure to ca. 2 vol at 45 to 50° C., targeting 48° C. Charge methanol, (5.0 vol, 4.0 wt) to the mixture at 45 to 50° C., targeting 48° C. Re-concentrate the mixture under reduced pressure to ca. 2 vol at 45 to 50° C., targeting 48° C. Repeat steps 7 and 8 once before continuing with step 9. Cool the mixture to 18 to 23° C. Clarify the mixture into a tared, suitably-sized drum followed by a methanol (1.0 vol, 0.8 wt) line rinse at 18 to 23° C. Determine the w/w assay of ZX008 ketone (WIP) in the mixture by 1H-NMR analysis and calculate the contained yield of ZX008 ketone (WIP) in the mixture. Determine the toluene content in the mixture by 1H-NMR analysis.
- [0118]
Yields and Profiles: The yield for the step 2 Demonstration batch is summarized in Table 1B below. Input: 1200.0 g corr. Ketone bisulfite adduct, 76.0% w/w assay (NMR, using DMB as internal standard in d6-DMSO), (1.00 eq, 1.00 wt corr. for w/w assay) for input calculation. - TABLE 1B Table for isolated yields for step 2 Demonstration batch % w/w % area Corr. Corr. Corr. Yield (1H- (HPLC, Input Output (% th) NMR)* QC) Comments 1200.0 g 858.15 g 108.3 25.5 99.31 Purified ketone
- [0119]
Step 3: Preparation of Fenfluramine HCl Crude - [0120]
Procedure: Charge the ZX008 ketone (corr. for assay, 1.00 wt, 1.00 eq. isolated as solution in MeOH in stage 2) to a vessel. Charge methanol, code RM0036 (5.0 vol, 4.0 wt) to the mixture at 18 to 23° C. Cool the solution to 0 to 5° C. Charge 70 wt % aqueous ethylamine solution (1.3 vol, 1.6 wt, 4.0 eq) to the mixture at 0 to 10° C., over 15 to 30 minutes, followed by a line rinse with methanol (1.0 vol, 0.8 wt). Warm the mixture to 15 to 20° C. and stir the mixture for a further 60 to 70 minutes at 15 to 20° C. Adjust the mixture to 15 to 18° C. if required, targeting 15° C. Charge sodium triacetoxyborohydride (2.4 wt, 2.25 eq.) to the mixture in approximately 10 portions, keeping the mixture at 15 to 20° C., targeting 17° C. Addition time 1.5 to 2 hours. Caution: Exothermic. Stir the mixture at 15 to 20° C. until complete by HPLC analysis, pass criterion ≦3.0% area ZX008 ketone, typically 2 to 3 hours. Adjust the pH of the mixture to pH>12 by charging 20% w/w aqueous sodium hydroxide solution (5.0 to 6.0 vol) to the mixture at 15 to 40° C. Addition time 10 to 30 minutes. Caution: Exothermic. If necessary, adjust the temperature to 18 to 23° C. Extract the mixture with toluene (3×3.0 vol, 3×2.6 wt) at 18 to 23° C., retaining and combining the top organic layer after each extraction. Wash the combined organic layer with purified water, (1.0 vol) at 18 to 23° C. Heat the mixture to 40 to 50° C., targeting 48° C. Concentrate the mixture under reduced pressure at constant volume maintaining ca. 5 vol by charging the organic layer at approximately the same rate as the distillation rate at 40 to 50° C., targeting 48° C. Cool the mixture to 18 to 23° C. Charge purified water (10.0 vol) to the mixture at 18 to 23° C. Adjust the pH of the mixture to 0.1<pH<1.5 at 18 to 23° C. by charging concentrated hydrochloric acid, 0.5 vol. Do not delay from this step until neutralization. - [0121]
Separate the layers at 18 to 23° C. retaining the bottom aqueous layer. Wash the aqueous layer with toluene, (3.0 vol, 2.6 wt) at 18 to 23° C. retaining the aqueous layer. Adjust the pH of the aqueous layer to pH>12 by charging 20% w/w sodium hydroxide solution at 18 to 23° C. 0.8 to 0.9 vol. Caution: Exothermic. Charge TBME, code RM0002 (2.0 vol, 1.5 wt) to the basic aqueous layer. Separate the layers at 18 to 23° C. retaining the top organic layer. Back-extract the aqueous layer with TBME (2×2.0 vol, 2×1.5 wt) at 18 to 23° C. retaining the organic layers. Wash the combined organic layer with purified water, (2×1.0 vol) at 18 to 23° C. Concentrate the combined organic layers under reduced pressure at 40 to 50° C., targeting 48° C. to ca. 3 vol. Determine the residual toluene content of the mixture by 1H-NMR analysis. Sample for determination of residual water content by KF analysis, AKX reagent. Charge TBME (8.7 vol, 6.4 wt) to the mixture at 40 to 50° C. Cool the solution to 0 to 5° C., targeting 2° C. Charge concentrated hydrochloric acid (0.54 vol, 0.46 wt) maintaining the temperature <15° C. Caution: Exothermic. Line rinse with TBME (1.0 vol, 0.7 wt). If necessary, adjust the temperature to 0 to 10° C. and stir the mixture at 0 to 10° C. for a further 2 to 3 hours. Filter the mixture and wash the filter-cake with TBME (2×4.4 vol, 2×3.3 wt) at 0 to 10° C. Dry the solid at up to 40° C. until the TBME content is <0.5% w/w TBME by 1H-NMR analysis. 4 to 8 hours. - [0122]
Yields and Profiles: The yield for the step 3 Demonstration batch is summarized in Table 1C below. Input: 856.8 g corr. Ketone, 44.2% w/w assay (NMR, using TCNB as internal standard in CDCl3), (1.00 eq, 1.00 wt corr. for w/w assay) for input calculation. FIG. 2 and Table 1D shows an exemplary HPLC chromatogram of a crude preparation of fenfluramine hydrochloride (210 nm UV absorbance). - TABLE 1C Table for isolated yields for step 3 Demonstration batch Corr. % area Reference Corr. Corr. Yield % w/w (HPLC, number Input Output (% th) (1H-NMR)* QC) Comments Batch A1 856.8 g 836.31 g 85.3 44.2 99.15 Fenfluramine free base (in situ intermediate) Batch A2 880.7 84.0 based 99.5 100.00 Fenfluramine•HCl on ketone crude (step 3 an bisulfite d 4.1) adduct (77.6 based on purified ketone)
- TABLE 1D Purity of crude fenfluramine hydrochloride by HPLC (see FIG. 2) Processed Channel Descr. DAD AU Ch 1 Sample 210, Bw 4 Peak Results USP USP USP Name RT RelRT Area Height Tailing Resolution Plate Count EP s/n % Area 1 NorFenfluramine 7.46 2 2-Fenfluramine 7.68 3 Fenfluramine 8.67 1.000 3789064 778178 1.7 70796 2549.8 99.15 4 4-Fenfluramine 8.95 5 11 34 1.308 6073 1449 1.2 23.5 215529 3 8 0.16 6 ZX008 acid 12.93 7 Fenfluramine alcohol 14.16 1.633 15266 2972 1.3 24.8 215040 8.7 0.40 8 ZX008 ketone 14.83 9 Fenfluramine acetamide 15.55 10 TOLUENE 15 75 11 15.92 1.836 4110 1122 2.7 0.11 12 16.60 1.915 6861 1630 1.5 451209 4.3 0.18 Sum 3821374 100.00
- [0123]
Step 4.2: Crystallization of Fenfluramine Hydrochloride - [0124]
Procedure: Charge Fenfluramine.HCl (crude) (1.00 wt, 1.0 eq.) and TBME (10.0 vol, 7.4 wt) to the vessel and commence stirring. Heat the suspension to reflux (50 to 58° C.). Charge ethanol (5.0 vol, 3.9 wt) maintaining the temperature at 50 to 58° C. Addition time 20 minutes. Stir at 50 to 58° C. for 5 to 10 minutes and check for dissolution. Stir the solution at 50 to 58° C. for 5 to 10 minutes, targeting 54 to 58° C. Clarify the reaction mixture through a 0.1 μm in-line filter at 54 to 58° C., followed by a line rinse with TBME (1 vol, 0.7 wt). Cool the solution to 48 to 50° C. Charge Fenfluramine HCl, code FP0188 (0.01 wt). Check for crystallization. Allow the suspension to cool to 15 to 20° C., target 17° C. over 5 to 5.5 hours at an approximately constant rate. Stir the mixture at 15 to 20° C., target 17° C. for 2 to 3 hours. Filter the mixture and wash the filter-cake with clarified TBME (2×3.0 vol, 2×2.2 wt) at 5 to 15° C. Dry the solid at up to 40° C. until the TBME content is <0.5% w/w TBME and the ethanol content is <0.5% w/w EtOH by 1H-NMR analysis. 4 to 8 hours. Determine the w/w assay of the isolated Fenfluramine.HCl by 1H-NMR analysis. - [0125]
Yields and Profiles: The yield for the stage 4 Demonstration batch is summarized in Table 1E below. Input: 750.0 g uncorr. Fenfluramine HCl crude (1.00 eq, 1.00 wt uncorr.) for input calculation. FIG. 3 shows an exemplary HPLC chromatogram of a crystallized fenfluramine hydrochloride sample (210 nm UV absorbance). - TABLE 1E Table for isolated yields for stage 4 Demonstration batch Uncorr. Uncorr. Uncorr. Yield HPLC (% area, Input Output (% th) QC) Comments 750.0 g 608.0 81.1 100.00* Fenfluramine•HCl
5. In-Process Controls
- [0126]
Table 2 summarizes the in-process controls (IPCs) by IPC number as cited in the narrative procedures above used for Process 1. - TABLE 2 In-Process Controls Performed during Process 1 Critical IPC Synthesis Process No. Step Sample Description Method Acceptance Criteria 1 1 Reaction Reaction HPLC NMT 3.0% Ketone (1) Mixture Completion 2 1 Extraction Purity HPLC Report percent Aqueous Fenfluramine Free Base and Phase Fenfluramine Alcohol 3 1 Extraction Purity HPLC Report percent Organic Fenfluramine Free Base and Phase Fenfluramine Alcohol 4 1 Extraction Purity HPLC Report percent Organic Fenfluramine Free Base and Phase Fenfluramine Alcohol 5 1 Extraction Purity HPLC NLT 98.0% Fenfluramine Aqueous HCl Phase LT 1.0% Fenfluramine Alcohol 6 1 Extraction Purity HPLC Report percent result of Aqueous Fenfluramine HCl Phase Fenfluramine Alcohol 7 2 Reaction Purity 1H-NMR Residual Solvents by 1H- Mixture NMR Ethanol NMT 0.50% w/w Ethyl Acetate NMT 0.50% w/w Methanol NMT 0.50% w/w Toluene NMT 0.50% w/w MTBE NMT 0.50% w/w
6. Starting Materials
- [0127]
This section provides information and specification controls for the starting materials used to produce clinical supplies of fenfluramine per the routes shown herein. - TABLE 3 Starting Materials via the Route 1 Chemical Name Code [CAS. No.] Name Structure Source Step 1-(3- (Trifluoromethyl)- phenylacetone [21906-39-8] Ketone (1)Fluorochem 1 Ethyl Amine Ethyl EtNH2 Alfa Aesar 1 (70% in water) Amine [75-04-7]
- TABLE 4 Starting Materials via Route 2 Chemical Name Code [CAS. No.] Name Structure Source Step 3-(Trifluoromethyl)- phenylacetic acid [351-35-9] Acid (1a)To be determined 1 Acetic Anhydride [108-24-7] Acetic AnhydrideVarious 1 Ethyl Amine Ethyl EtNH2 Various 3 (70% in water) Amine [75-04-7]
- [0128]
Table 5 provides a list of the intermediates for the Route 2 synthesis. Both routes share the same intermediate Fenfluramine Free Base (1). Fenfluramine Free Base (1) was treated as an isolated intermediate in the Route 1 process however the Route 2 process uses fixed equipment where both Ketone (2) and Fenfluramine Free Base 1, both non-isolatable oils, are telescoped as a solution and controlled as in-situ intermediates. The Bisulfate Complex (3) is isolated as a solid thus is amenable to treatment as an isolated intermediate and released as such. Crude Fenfluramine HCl can be isolated as an intermediate before recrystallization. - [0129]
A Specification and Testing Strategy for Intermediates is used. Additional tests and acceptance criteria are be added based upon review of data from the primary stability batches and process validation critical parameter studies. Analytical reference standards are used in full characterization of each intermediate. HPLC methods to determine assay and impurities are the same as the drug substance release method and are validated for Accuracy, Precision: Repeatability, Intermediate Precision, Selectivity/Specificity, Detection limit, Quantitation limit, Linearity, Range, and Robustness. - TABLE 5 In-Situ and Isolated Intermediates Chemical Name [CAS No] Code Name Step No. Control Structure Bisulfate Complex of Ketone 1 Bisulfate Complex (3) Step 1 Isolated (Solid)1-(3- (Trifluoromethyl)- phenylacetone [21906-39-8] Ketone (2) Step 2 In-Situ (oil)Fenfluramine Free Base [458-24-2] Fenfluramine Free Base (1) Step 3 In-Situ (oil)Fenfluramine HCl [404-82-0] Crude Fenfluramine HCl Step 4 Isolated (Solid)
7. Characterization
- [0130]
Physiochemical Characteristics of Drug Substance. - [0131]
Fenfluramine HCl is developed as a single polymorph Form 1. A polymorphism and pre-formulation study has been conducted. Under a wide range of solvents and conditions crystalline material is produced of the same polymorph Form 1 based on a well-defined XRPD pattern and a consistent reproducible endotherm by DSC analysis. A summary of the chemophysical properties of Fenfluramine HCl from this study is provided below. Tabulated data includes example diffractograms, DSCs, and micrographs. - [0132]
The input Fenfluramine HCl (from precipitative isolation) was characterized to provide reference data and also to determine if the salt was of the same form as that identified from previous salt formations. The XRPD pattern of the salt reveals a crystalline solid that visually matches the reflection patterns obtained from formal crystallization of Fenfluramine HCl and has been arbitrarily termed Form 1. Comparison of the μATR-FTIR data for the salt from various batches gave profiles that had a 99.95% match. - [0133]
Thermal data analysis matched previous data obtained with only one major endotherm on the DSC thermograph peaking at 172.3° C. that matches the beginning of potential decomposition shown in a TGA thermograph. This also matches the reported melting point quoted for the reference standard. - [0134]
Isolation of the amorphous form has been shown to be difficult, with attempts using three common methods (rapid solvent evaporation, anti-solvent precipitation and lyophilization) all yielding highly crystalline solids that very closely share the same XRPD pattern of the input Form 1. - [0135]
Stability analysis of the salt after one week at 40° C./0% RH, three weeks at 40° C./75% RH, and under photostability conditions revealed that the input Form 1 has been maintained with no new impurities observed at 0.1% threshold. - [0136]
Results from DSC heat cycling analysis of Fenfluramine HCl are comparable to results generated when the material was held at 170° C. No crystallization event was noted and the amorphous was not generated but rather Form 1 was returned. - [0137]
Holding Fenfluramine HCl at approximately 170° C. for several hours causes a melt and evaporation event to take place with recombination and cooling to provide a white solid. Analysis of the white solid by XRPD, DSC and 1H NMR indicates no change in chemical or physical form, purity, or dissociation. - [0138]
Forced degradation studies carried out have proven Fenfluramine HCl to be stable under a range of conditions. Thermal modulation of Fenfluramine HCl repeatedly yielded the input Form 1.
8. Impurities
- [0139]
Impurities in a drug substance can be organic impurities (process impurities or drug substance-related degradants), inorganic impurities (salt residues or metals) and residual solvents; some of these impurities must be evaluated as to whether or not they are genotoxic agents. These impurities are taken into consideration and controlled in Fenfluramine HCl preparation by using either compendia or validated analytical methods per the specifications or by separate “for information only” testing. The following sections address the actual and potential impurities in Fenfluramine HCl. - [0140]
Actual Impurities and the Qualification of Synthesis Batch - [0141]
No impurities reported in cGMP drug substance batches intended for use in humans have exceeded the ICHQ3A qualification thresholds of 0.15% (Table 8). All impurities >0.1% are identified and handled as described in ICH Q3A unless they are genotoxic impurities. - [0142]
Process Impurities - [0143]
Table 6 lists the known potential impurities arising from the route of synthesis. All of these impurities are controlled to below ICHQ3A qualification threshold of 0.15% by either process changes and/or control of starting material input purities. - TABLE 6 Fenfluramine HCl Known Potential Process Impurities (Route 1) Observed Observed in in Development cGMP Name PLC Batches Batches [Cas. No.] Source (RRT) ≧0.10%1) ≧0.10%1) Ketone (2) Starting RRT No No [351-35-9] Material or 0.89 Intermediate Fenfluramine By-product RRT Yes No Alcohol 1.60 [621-45-4] Norfenfluramine By-product RRT Yes Yes [1886-26-6] 1.67 2-Fenfluramine Starting RRT No No [172953-70-7] Material 0.89 (isomer) 4-Fenfluramine Starting RRT Yes Yes [1683-15-4] Material 1.02 (isomer) N-(3- By-product RRT Yes Yes (trifluoromethyl)- 0.53-0.57 benzyl)ethanamine [90754-95-3] 1)ICH Q3A Identification threshold. The Reporting threshold (LOQ) for the HPLC method is 0.05%.
- [0144]
Degradation Impurities - [0145]
No change in impurity profile is observed upon long-term storage based on forced degradation studies under the ICH Q1A(R2) conditions of heat (solid, solution), acid, base, oxidizing, and ICH Q1B photostability conditions (solid, solution). Fenfluramine HCL is stable for 7 days as a solid at 150° C. (99.90 parent area %), as a solution in water-acetonitrile at 70° C. (99.73 parent area %), as a solution in acid, base, or photosensitizing conditions at ambient. Only oxidizing conditions (peroxide conditions) produced degradation of Fenfluramine HCl to 94.42% after 1 day producing several new related substances at −1% each consistent by LC-MS with +16 oxidation by-products - [0146]
Organic Volatiles/Residual Solvents - [0147]
Table 11 in the Batch Analysis section summarizes the solvents used in the process and the resulting amounts found in drug substance. All solvents used in the GMP steps are controlled at ICH Q3A limits using a suitably qualified Head-Space (HS) GC method. - [0148]
Inorganic Impurities - [0149]
Heavy Metals conform to either USP <231> or ICP method USP <233> as well as ICH Q3D. - [0150]
Genotoxic Impurities - [0151]
The ICH guidelines Q3A and Q3B are not sufficient to provide guidance on impurities that are DNA-reactive. The European Medicines Agency (EMA) guideline (2006) “Guideline on the Limits of Genotoxic Impurities” (EMA 2006) and the ICH Guideline M7 (2014) “Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk” (ICH Guideline M7) are taken into consideration in controlling for potential genotoxic impurities. The diazonium route to prepare ketone (2) described in FIG. 5 has a disadvantage due to the potential formation of genotoxic intermediates shown as boxed compounds (e.g., N-hydroxyaryl, N-nitrosamine and Nitro compound). Muller et al. (Regulatory Toxicology and Pharmacology 44 (2006) 198-211) list potential functional alert groups that can be genotoxic. Safety guidances and regulations indicate that analysis of a process and identification of potential genotoxic agents, and control of such impurities at sub 10 parts per million levels is critical for safety. Often removal of such impurities and/or demonstrating their absence is costly and time consuming and sometimes difficult to achieve technically. For these reasons, selecting synthetic routes that circumvent the potential for such toxic intermediates is important. Because of the potential problems with the diazo route discussed above, as well as potential safety issues using diazo (shock-sensitive) intermediates, as well as the lower purity profiles with this route, this route is less preferred than the preferred route to ketone (2) starting from Nitrile (5). This route produces no potential genotoxic agents and leads to high purity Ketone (2) after isolation by distillation or via the bisulfite salt adduct—hydrolysis sequence. - [0152]
Additionally, attempts to remove isomer by-products present in commercial supplies of Aniline were unsuccessful whereas crystallization the Acid (4) resulting from hydrolysis of the nitrile (5) provides crystalline Acid (4) which can be purified to remove isomers early in synthesis. Removing impurities and/or isomers early in a synthesis is preferred if it is known such impurities track to final product, as the need to crystallize a final product at the end of a synthesis is more costly in losses and impacts cost of goods more greatly than removing early in synthesis before raw materials are invested along the process. - TABLE 7 Potential Impurities in Fenfluramine Synthesis Synthesis Route No. Compound Route 1 Route 2 CAS. No. 1No Starting Material [351-35-9] 2Starting Material Intermediate [21906-39-8] 4No Intermediate Not Available 5Potential Impurity Potential Impurity [621-45-4] 6Potential Impurity Potential Impurity [1886-26-6] 7Potential Impurity Potential Impurity [172953-70-7] 8Potential Impurity Potential Impurity [1683-15-4] 9Potential Impurity Potential Impurity [90754-95-3]
- TABLE 8 Batch Analyses of Fenfluramine HCl Drug Substance Test Batch 1 Batch 2 Batch 3 Batch 4 Appearance* White solid White solid White solid White solid Identification: FTIR* a) a Conforms Conforms Identification: 1H-NMR Conforms Conforms Conforms Conforms Identification: 13C-NMR Conforms Conforms Conforms Conforms Identification: MS Conforms Conforms Conforms Conforms Purity (HPLC area %) 99.57 99.77 b) b Assay (w/w %)* 99.49 100.37 100.79 100.13 Anhydrous Basis (HPLC) Impurities 2-Fenfluramine ND ND ND ND (HPLC 4-Fenfluramine) 0.16 0.15 0.11 0.12 area %) Fenfluramine Alcohol ND ND ND ND 1-((3-trifluoromethyl)phenyl)acetone ND ND ND ND Acetamide 0.27 ND ND ND N-(3-(trifluoromethyl)- ND 0.08 0.07 0.13 benzyl)ethanamine (RRT 0.53-0.57) Total 0.43 0.23 0.19 0.25 Residual Solvents Methanol ND ND ND ND (GC): ppm Ethanol ND ND ND ND MTBE 597 844 472 800 Ethyl Acetate 115 164 79 150 Toluene 4 7 ND ND Residue on Ignition (w/w %) 0.01 0.02 0.04 ND Heavy Metals (as Pb) <10 ppm <10 ppm b b Heavy Metals ICP (ppm) As a a <0.1 <0.1 Cd a a 0.1 0.1 Hg a a <0.1 <0.1 Pb a a 0.2 <0.4 Co a a <0.1 0.1 Mo a a <0.1 <0.1 Se a a <0.1 <0.1 V a a <0.1 <0.1 Water Determination* 0.21 0.08 0.02 0.03 (Karl Fischer) Chloride content by titration 13.19 13.09 12.92 12.93 XRPD* Form 1 Form 1 Form 1 Form 1 Differential Scanning Onset 169.42° C. 169.23° C. 169.85° C. 168.70° C. Calorimetry (DSC)* Peak 172.82° C. 171.55° C. 172.22° C. 171.97° C. Particle Size % Volume mean (D) a 11 11 19 Malvern (μm) D10 a 1 1 1 D50 a 5 7 9 D90 a 17 26 32 Microbial Total aerobic a a LT 100 CFU/g LT 100 CFU/g Limits Tests* microbial Count USP <61> Total yeast and a a LT 100 CFU/g LT 100 CFU/g molds count USP <62> Absence of a a Absent Absent Pathogens a)These tests were added to the specifications recently thus only recent lots have been tested using this test. b)These tests have been dropped from the specifications thus only historical lots have been tested using this test.
Example 3Method for Hydrolysis of Nitrile (5) to Acid
- [0153]
- TABLE 9 Step Operation 1. Charge 3-(trifluoromethyl)phenyl acetonitrile (1.0 eq., 1.00 wt) and purified water (5.0 vol) to a vessel and commence stirring. 2. Dissolve sodium hydroxide (1.1 wt, 5.0 eq.) in purified water (4.0 vol) at up to 40° C. in a suitable make-up vessel. Caution very exothermic. 3. Charge the aqueous sodium hydroxide solution to the mixture from step 1 at up to 40° C. followed by a line rinse with purified water, code RM0120 (1.0 vol) at up to 40° C. 4. Heat the mixture to 75 to 85° C., target 80° C. over 1 to 2 hours. 5. Heat the mixture at 80° C. until ≦0.1% area nitrile by HPLC analysis, expected 4 to 6 hours. 6. Cool the mixture to 18 to 23° C. 7. Adjust the pH of the mixture to pH ≦2 by charging 6M HCl (expected 7.0 vol) to the mixture at 18 to 23° C. Caution exothermic. 8. Stir the mixture for 15 to 30 minutes at 18 to 23° C. 9. Filter and wash the filter-cake with purified water (2 × 5.0 vol) at 18 to 23° C. 10. Slurry wash the filter-cake with n-heptane, code RM (2 × vol) at 0 to 5° C. 11. Dry the isolated solid at up to 45° C. until the water content is ≦.0.2% w/w by KF analysis according to MET/AN/0163, AKX-reagent. 12. Crystallization of crude stage 1 acid (1.00 wt for input calculation) 13. Charge the crude stage 1 acid (1.00 wt), ethyl acetate (0.75 vol) and n-heptane (10.5 vol) to a vessel and commence stirring. 14. Heat the mixture to 50° C. to achieve dissolution. 15. Cool the mixture to 5° C. and age at 5° C. for at least 30 mins. 16. Filter and wash the filter-cake with n-heptane (2 × 5.0 vol). 17. Dry the isolated solid at up to 45° C. until the residual solvent content by 1H-NMR analysis is ≦.0% w/w EtOAc and ≦.0% w/w n-heptane. Expected yield: 60 to 90% th uncorr. 68 to 103% w/w Expected purity: 93.00 to 99% area by HPLC
Example 4Evaluation of Minor Components Formed During Dakin-West Reaction in Preparation of Ketone (2)
- [0154]
The impurities formed during the Dakin-West chemistry and their subsequent removal using the distillation or via isolation of the product ketone as the bisulfite salt are described. The two major impurities found are shown below. - [0155]
Table 10 shows a table of analytical data for crude Ketone (2) isolated from Dakin-West reaction before and after bisulfite purification. In entry 1 crude Ketone isolated directly from the Dakin-West step (pre-bisulfite treatment) is 61.66% purity (e.g. about 62%) and contains 1.98% (e.g., about 2%) and 4.64% (e.g., about 5%) respectively of impurities having RRTs 1.20 and 1.34, which are proposed to be the acetate and dimer impurities (e.g., depicted above), respectively. In entry 2 which is post bisulfite treatment these are other impurities are removed leading to an overall purity of 95.55% (e.g., about 96%). Other entries shown in Table 10 provide other examples of this impurity enhancement by bisulfite treatment of crude Dakin-West ketone. The last two entries use pure Fluorchem ketone as input to the salt formation step and re-isolation of ketone thus illustrating that the salt formation and re-isolation does not produce any impurities itself. Additionally use of bicarbonate extraction procedure during reaction workup provides an improvement in purity of the resulting composition as it serves to remove any unreacted acid. Crude Ketone (2) made by the Diazo route showed similar improvements in purity when treated with bisulfite and isolated. - TABLE 10 Analytical purity data for crude Ketone (2) isolated from Dakin-West reaction before and after bisulfite purification. RRT is relative retention time (min) in chromatogram. RRT 0.93 1.00 1.009 1.06 Entry 0.85 Aniline 0.95 0.99 Ketone 1.004 Nitrile 1.02 Acid 1.10 1.15 1.34 1.38 1 1.38 1.76 0.04 0.49 61.66 nd 0.29 0.29 0.26 1.98 0.66 4.64 0.14 2 0.82 nd nd nd 95.53 0.31 0.14 nd 0.23 0.01 0.10 0.43 0.27 3 nd nd nd nd 77.82 nd nd nd nd 3.12 0.01 7.76 6.16 4 nd nd nd nd 98.82 nd 0.63 nd nd nd 0.02 0.30 0.22 5 0.08 nd nd 0.05 72.02 nd 0.02 nd nd 7.11 0.04 3.58 10.33 6 nd nd nd nd 99.49 nd 0.02 nd nd nd 0.02 0.11 0.24 7 0.15 0.23 nd nd 98.35 nd nd nd nd nd nd nd 0.24 8 nd nd nd nd 99.84 nd nd nd nd nd nd nd nd Entry 1 (Crude ketone from Route 1); Entry 2 (Ketone Route 1 post bisulfite release); Entry 3 (Crude ketone using crude acid); Entry 4 (Ketone using crude acid Post bisulfite); Entry 5 (Crude ketone using cryst. acid); Entry 6 (Crude ketone using cryst. acid post bisulfite); Entry 7 (Crude ketone using cryst. acid); Entry 8 (Fluorochem ketone); Entry 9 (Fluorochem ketone post bisulfite).
Example 5
- [0156]
- Additional Method for Preparation of 1-(3-trifluoromethyl)phenyl-propan-2-one
- [0157]
35 mL of water and 45 g of 37% (w/w) aqueous hydrochloric acid are put in a flask equipped with stirrer and dropping funnel. 24.25 Grams (0.151 moles) of m-trifluoromethylaniline are added after having cooled to 10 degree C. with an ice bath and then, at 5 degree C., an aqueous solution containing 12.43 g (0.180 moles) of sodium nitrite in 150 ml of water is slowly added. The reaction mixture is stirred for 30 minutes and then is poured during 30 minutes into a mixture made by 90 ml of water, 1.35 g (0.014 moles) of cuprous chloride, 2.30 g (0.013 moles) of cupric chloride dihydrate, 50 ml of acetone, 40.8 g (0.300 moles) of sodium acetate trihydrate and 23 g (0.230 moles) of isopropenyl acetate while keeping the reaction temperature at 30 degree C. After further 30 minutes of stirring, the reaction mixture is brought to 20 degree C., 50 ml of methylene chloride are added and the two layers are separated. - The aqueous layer is discarded while the organic layer is concentrated under vacuum until an oil is obtained which is treated with 35 g of sodium metabisulfite, 70 ml of water and 150 ml of heptane under stirring at room temperature for 12 hours. The suspension is filtered, the bisulfite complex is washed on the filter with 50 ml of heptane and then suspended in a two-phase mixture made by 100 ml of methylene chloride and 150 ml of a 10% (w/v) aqueous solution of sodium hydroxide. The layers are separated after one hour of stirring at room temperature, the aqueous phase is discarded while the organic layer is washed with water and evaporated under vacuum to give pure ketone.
Centhaquine
Centhaquine
PMZ-2010
CAS 57961-90-7
2-[2-[4-(3-methylphenyl)piperazin-1-yl]ethyl]quinoline
INDIA 2020, 14.05.2020, Centhaquine citrate bulk and Centhaquine citrate injection 1.0mg/vial, Add on resuscitative agent for hypovolemic shock
- OriginatorMidwestern University; Pharmazz
- DeveloperPharmazz
- ClassAnalgesics; Antihaemorrhagics; Antihypertensives; Cardiovascular therapies; Piperazines; Quinolines; Small molecules
- Mechanism of ActionAlpha 1 adrenergic receptor antagonists; Alpha 2 adrenergic receptor agonists
- RegisteredHaemorrhagic shock
- Phase IHeart arrest; Postoperative pain
- 20 Jul 2020Pharmazz plans to launch centhaquin for Haemorrhagic shock (Adjuvant therapy) in India by the middle of September 2020
- 20 Jul 2020Efficacy data from a phase III trial in Haemorrhagic shock released by Pharmazz
- 02 Jun 2020Centhaquine is still in phase I trials for Postoperative pain in USA (Pharmazz pipeline, June 2020)
SYNCenthaquin is a compound that produces hypotension and bradycardia in higher doses and resuscitation in lower doses. It is water insoluble, and is unsuitable for intravenous use. We prepared the citrate salt of centhaquin and evaluated its cardiovascular efficacy vs. centhaquin. Centhaquin citrate was prepared and characterized; its purity was determined by HPLC. Mean arterial pressure (MAP), heart rate (HR), pulse pressure (PP), cardiac output (CO), stroke volume (SV) and stroke work (SW) following intravenous administration of centhaquin and the citrate (0.05, 0.15 and 0.45 mg.kg(-1)) were determined in anaesthetized male Sprague-Dawley rats. Centhaquin citrate was 99.8% pure and water soluble. Centhaquin (0.05, 0.15 and 0.45 mg.kg(-1)) produced a maximal decrease in MAP of 15.6, 25.2 and 28.1%, respectively; while centhaquin citrate produced a greater (p<0.001) decrease of 35.7, 47.1 and 54.3%, respectively. The decrease in PP and HR produced by the citrate was greater than centhaquin (p<0.001). At 0.45 mg.kg(-1) centhaquin produced a maximal decrease of 20.9% (p<0.01) in CO, while centhaquin citrate produced a decrease of 42.1% (p<0.001). Reduction in SV (p<0.01) and SW (p<0.001) produced by centhaquin citrate were greater than centhaquin. Centhaquin citrate has greater cardiovascular activity compared to centhaquin.https://www.semanticscholar.org/paper/Synthesis-and-characterization-of-centhaquin-and-a-Reniguntala-Lavhale/6ca3975b114b0f23753e7a47710eff2467bc2dae

PATENT
https://patents.google.com/patent/WO2014035446A1/en
Shock due to severe hemorrhage accounts for a large proportion of posttraumatic deaths, particularly during early stages of injury (Wu, Dai et al. 2009). A majority of deaths due to hemorrhage occur within the first six hours after trauma (Shackford, Mackersie et al. 1993), but many of these deaths can be prevented (Acosta, Yang et al. 1998).
[0003] Shock is accompanied by circulatory failure which is the primary cause of mortality and morbidity. Presently, the recommended fluid therapy uses large volumes of Lactated Ringer’s solution (LR), which is effective in restoring hemodynamic parameters, but presents logistic and physiologic limitations (Vincenzi, Cepeda et al. 2009). For example, resuscitation using a large volume of crystalloids, like LR, has been associated with secondary abdominal compartment syndrome, pulmonary edema, cardiac dysfunction, and paralytic ileus (Balogh, McKinley et al. 2003). Therefore, a need exists in the art for a resuscitation agent that improves survival time, and can be used with a small volume of resuscitation fluid, for resuscitation in hypovolemic shock.
[0004] Centhaquin (2-[2-(4-(3-methyphenyl)-l-piperazinyl) ethyl-quinoline) is a centrally acting antihypertensive drug. The structure of centhaquin was determined (Bajpai et al., 2000) and the conformation of centhaquin was confirmed by X-ray diffraction (Carpy and Saxena, 1991).

Structure of centhaquin (2-[2-(4-(3-methyphenyl)- 1 -piperazinyl) ethyl] -quinoline) (as free base)
[0005] Centhaquin is an active cardiovascular agent that produces a positive inotropic effect and increases ventricular contractions of isolated perfused rabbit heart (Bhatnagar, Pande et al. 1985). Centhaquin does not affect spontaneous contractions of the guinea pig right auricle, but significantly potentiates positive inotropic effect of norepinephrine (NE) (Srimal, Mason et al. 1990). The direct or indirect positive inotropic effect of centhaquin can lead to an increase in cardiac output (CO). Centhaquin produces a decrease in mean arterial pressure (MAP) and heart rate (HR) in anesthetized rats and conscious freely moving cats and rats (Srimal, Gulati et al. 1990) due to its central sympatholytic activity (Murti, Bhandari et al. 1989; Srimal, Gulati et al. 1990; Gulati, Hussain et al. 1991). When administered locally into a dog femoral artery, centhaquin (10 and 20 μg) increased blood flow, which was similar to that observed with acetylcholine and papaverine. However, the vasodilator effect of centhaquin could not be blocked by atropine or dibenamine (Srimal, Mason et al. 1990). The direct vasodilator or central sympatholytic effect of centhaquin is likely to decrease systemic vascular resistance (SVR).
[0006] It was found that centhaquin enhances the resuscitative effect of hypertonic saline (HS) (Gulati, Lavhale et al. 2012). Centhaquin significantly decreased blood lactate and increases MAP, stroke volume, and CO compared to hypertonic saline alone. It is theorized, but not relied upon, that the cardiovascular actions of hypertonic saline and centhaquin are mediated through the ventrolateral medulla in the brain (Gulati, Hussain et al. 1991 ; Cavun and Millington 2001) and centhaquin may be augmenting the effect of hypertonic saline.
[0007] A large volume of LR (i.e., about three times the volume of blood loss) is the most commonly used resuscitation fluid therapy (Chappell, Jacob et al. 2008), in part because LR does not exhibit the centrally mediated cardiovascular effects of hypertonic saline. Large volume resuscitation has been used by emergency medical personnel and surgeons to reverse hemorrhagic shock and to restore end-organ perfusion and tissue oxygenation. However, there has been a vigorous debate with respect to the optimal methods of resuscitation (Santry

ased on the molecular weight of centhaquin (free base) (MW-332) and centhaquin citrate (MW-523), for identical doses of centhaquin (as free base) and centhaquin citrate, centhaquin citrate provides only 63.5% of centhaquin free base compared to the dose of centhaquin free base, e.g., a 0.05 mg dose of centhaquin citrate contains a 0.0318 mg of centhaquin (as free base). Similarly, a dose of centhaquin citrate dihydrate (MW-559) provides 59.4% centhaquin (free base) of the same dose as centhaquin (as free base), i.e., a 0.0005 mg dose of centhaquin citrate dihydrate contains 0.030 mg of centhaquin (as free base). Surprisingly, and as demonstrated below, at the same mg/kg dose centhaquin citrate and centhaquin citrate dihydrate provides greater cardiovascular effects than centhaquin free base.

Synthesis of Centhaquin

[0061] The synthesis of centhaquin was reported by Murthi and coworkers (Murthi et al U.S. Patent No. 3,983,121 ; Murti, Bhandari et al. 1989). In one procedure, reactants 1 and 2 were stirred at reflux for 15 hours. The resulting product was purified by evaporating the solvents to obtain an oil, which was heated in vacuo (100°C, 1 mm Hg). The remaining residue was recrystallized from ether-petroleum ether to obtain the final centhaquin product 3. The melting point reported for centhaquin was 76-77°C. In a subsequent publication (Murti, Bhandari et al. 1989), the reaction mixture was concentrated following 24 hours of reflux, diluted with water, and basified with aqueous NaOH. The basic mixture was extracted with ethyl acetate, and the ethyl acetate extracts were dried over anhydrous sodium sulfate and evaporated in vacuo to give centhaquin which was crystallized from hexane. The melting point of centhaquin (free base) obtained in this procedure was 82°C. The product obtained using either purification method is light tan in color, which is indicative of small amounts of impurities that were not completely removed using previously reported purification methods.
[0062] In accordance with the present invention, an improved purification method was found. According to the improved method, reactants 1 and 2 were stirred at reflux for 24 hours. The solvents were evaporated in vacuo and the resulting mixture was diluted with water and basified (10% NaOH). The basic mixture was extracted with ethyl acetate and the combined ethyl acetate extracts are dried over anhydrous sodium sulfate and evaporated in vacuo to obtain a residue, which was further purified with column chromatography (Si02, ethyl acetate). The resulting product can be decolorized using activated charcoal or directly crystallized from hot hexane to yield pure centhaquin. The resulting product is an off-white crystalline solid having a melting point of 94-95°C (free base). The product was
characterized using proton NMR, mass spectral, and elemental analysis and indicated high purity and superior quality.
[0063] Synthesis and characterization of centhaquin (free base): A mixture of 2- vinylquinoline (1) (5.0 g, 32.2 mmol, 98.5%) and 1 -(3-methylphenyl)piperazine (2) (5.68 g, 32.2 mmol, 99.0%) in absolute ethyl alcohol (150 ml) and glacial acetic acid (3.5 ml) was stirred at reflux for 24 hours in a round bottom flask. The reaction mixture was concentrated in vacuo, diluted with water (150 ml) and treated with 10% aqueous NaOH (150 ml). The residue was extracted with ethyl acetate (4 x 125 ml), dried with anhydrous Na2S04, and concentrated under reduced pressure to yield a crude product which was purified by column chromatography using silica gel (100-200 mesh) with ethyl acetate as an eluent. The resulting compound was recrystallized from hot hexane and filtered, to yield centhaquin as an off- white crystalline solid (7.75 g, 23.4 mmol, 73% yield); mp. 94-95°C; i? 0.30 (100% ethyl acetate); 1H NMR (300 MHz, CDC13): δ 8.07 (t, J= 7.5 Hz, 2 H), 7.78 (d, J= 7.8 Hz, 1 H), 7.70 (t, J= 7.8 Hz, 1 H), 7.50 (t, J= 7.5 Hz, 1 H), 7.36 (d, J= 8.4 Hz, 1 H), 7.16 (t, J = 7.5 Hz, 1 H), 6.77 – 6.74 (m, 2 H), 6.69 (d, J= 7.2 Hz, 1 H), 3.26- 3.21 (m, 6 H), 2.97 – 2.92 (m, 2 H), 2.76 – 2.73 (m, 4 H), 2.32 (s, 3 H); HRMS (ESI) m/z 332.2121 [M+l]+ (calcd for C22H26N3 332.2122); Anal. (C22H25N3) C, H, N.
[0064] Preparation of centhaquin citrate: Centhaquin (free base) (5.62 g, 16.98 mmol) was treated with citric acid (3.26 g, 16.98 mmol) in a suitable solvent and converted to the citrate salt obtained as an off-white solid (7.96 g, 15.2 mmol, 90%); m.p. 94-96°C ; Anal.
10065] Figs. 1(a) and 1(b) are high resolution mass spectral analyses of centhaquin free base (Fig 1(a)) and centhaquin citrate (Fig. 1(b)). Compound samples were analyzed following ionization using electrospray ionization (ESI).
[0066J For centhaquin free base in Fig 1(a), a base peak [M+l]+ was observed at m z 332.2141 (theory: 332.2121) consistent with the elemental composition of protonated centhaquin (C22H26N3).
[0067] For centhaquin citrate in Fig 1(b), the mass spectrum was identical to the mass spectrum obtained for the free base. An [M+l]+base peak was observed at m z 332.2141 (theory: 332.2121), which corresponds to the elemental composition of protonated centhaquin (C22H26N3). This result is typical of salts of organic bases to yield the [M+l]+ of the free base as observed here with centhaquin citrate.
[0068] Mass spectrometry is one of the most sensitive analytical methods, and examination of the mass spectra of Fig. 1 indicate that the samples are devoid of any extraneous peaks and are of homogeneous purity (>99.5).
PATENT
https://patents.google.com/patent/WO2014035446A1/en
////////////Centhaquine, PMZ-2010, PMZ 2010, INDIA 2020, 2020 APPROVALS
Vonicog alfa
>>von Willebrand factor<<<
MIPARFAGVLLALALILPGTLCAEGTRGRSSTARCSLFGSDFVNTFDGSMYSFAGYCSYL
AGGCQKRSFSIIGDFQNGKRVSLSVYLGEFFDIHLFVNGTVTQGDQRVSMPYASKGLYLE
TEAGYYKLSGEAYGFVARIDGSGNFQVLLSDRYFNKTCGLCGNFNIFAEDDFMTQEGTLT
SDPYDFANSWALSSGEQWCERASPPSSSCNISSGEMQKGLWEQCQLLKSTSVFARCHPLV
DPEPFVALCEKTLCECAGGLECACPALLEYARTCAQEGMVLYGWTDHSACSPVCPAGMEY
RQCVSPCARTCQSLHINEMCQERCVDGCSCPEGQLLDEGLCVESTECPCVHSGKRYPPGT
SLSRDCNTCICRNSQWICSNEECPGECLVTGQSHFKSFDNRYFTFSGICQYLLARDCQDH
SFSIVIETVQCADDRDAVCTRSVTVRLPGLHNSLVKLKHGAGVAMDGQDIQLPLLKGDLR
IQHTVTASVRLSYGEDLQMDWDGRGRLLVKLSPVYAGKTCGLCGNYNGNQGDDFLTPSGL
AEPRVEDFGNAWKLHGDCQDLQKQHSDPCALNPRMTRFSEEACAVLTSPTFEACHRAVSP
LPYLRNCRYDVCSCSDGRECLCGALASYAAACAGRGVRVAWREPGRCELNCPKGQVYLQC
GTPCNLTCRSLSYPDEECNEACLEGCFCPPGLYMDERGDCVPKAQCPCYYDGEIFQPEDI
FSDHHTMCYCEDGFMHCTMSGVPGSLLPDAVLSSPLSHRSKRSLSCRPPMVKLVCPADNL
RAEGLECTKTCQNYDLECMSMGCVSGCLCPPGMVRHENRCVALERCPCFHQGKEYAPGET
VKIGCNTCVCRDRKWNCTDHVCDATCSTIGMAHYLTFDGLKYLFPGECQYVLVQDYCGSN
PGTFRILVGNKGCSHPSVKCKKRVTILVEGGEIELFDGEVNVKRPMKDETHFEVVESGRY
IILLLGKALSVVWDRHLSISVVLKQTYQEKVCGLCGNFDGIQNNDLTSSNLQVEEDPVDF
GNSWKVSSQCADTRKVPLDSSPATCHNNIMKQTMVDSSCRILTSDVFQDCNKLVDPEPYL
DVCIYDTCSCESIGDCACFCDTIAAYAHVCAQHGKVVTWRTATLCPQSCEERNLRENGYE
CEWRYNSCAPACQVTCQHPEPLACPVQCVEGCHAHCPPGKILDELLQTCVDPEDCPVCEV
AGRRFASGKKVTLNPSDPEHCQICHCDVVNLTCEACQEPGGLVVPPTDAPVSPTTLYVED
ISEPPLHDFYCSRLLDLVFLLDGSSRLSEAEFEVLKAFVVDMMERLRISQKWVRVAVVEY
HDGSHAYIGLKDRKRPSELRRIASQVKYAGSQVASTSEVLKYTLFQIFSKIDRPEASRIA
LLLMASQEPQRMSRNFVRYVQGLKKKKVIVIPVGIGPHANLKQIRLIEKQAPENKAFVLS
SVDELEQQRDEIVSYLCDLAPEAPPPTLPPHMAQVTVGPGLLGVSTLGPKRNSMVLDVAF
VLEGSDKIGEADFNRSKEFMEEVIQRMDVGQDSIHVTVLQYSYMVTVEYPFSEAQSKGDI
LQRVREIRYQGGNRTNTGLALRYLSDHSFLVSQGDREQAPNLVYMVTGNPASDEIKRLPG
DIQVVPIGVGPNANVQELERIGWPNAPILIQDFETLPREAPDLVLQRCCSGEGLQIPTLS
PAPDCSQPLDVILLLDGSSSFPASYFDEMKSFAKAFISKANIGPRLTQVSVLQYGSITTI
DVPWNVVPEKAHLLSLVDVMQREGGPSQIGDALGFAVRYLTSEMHGARPGASKAVVILVT
DVSVDSVDAAADAARSNRVTVFPIGIGDRYDAAQLRILAGPAGDSNVVKLQRIEDLPTMV
TLGNSFLHKLCSGFVRICMDEDGNEKRPGDVWTLPDQCHTVTCQPDGQTLLKSHRVNCDR
GLRPSCPNSQSPVKVEETCGCRWTCPCVCTGSSTRHIVTFDGQNFKLTGSCSYVLFQNKE
QDLEVILHNGACSPGARQGCMKSIEVKHSALSVELHSDMEVTVNGRLVSVPYVGGNMEVN
VYGAIMHEVRFNHLGHIFTFTPQNNEFQLQLSPKTFASKTYGLCGICDENGANDFMLRDG
TVTTDWKTLVQEWTVQRPGQTCQPILEEQCLVPDSSHCQVLLLPLFAECHKVLAPATFYA
ICQQDSCHQEQVCEVIASYAHLCRTNGVCVDWRTPDFCAMSCPPSLVYNHCEHGCPRHCD
GNVSSCGDHPSEGCFCPPDKVMLEGSCVPEEACTQCIGEDGVQHQFLEAWVPDHQPCQIC
TCLSGRKVNCTTQPCPTAKAPTCGLCEVARLRQNADQCCPEYECVCDPVSCDLPPVPHCE
RGLQPTLTNPGECRPNFTCACRKEECKRVSPPSCPPHRLPTLRKTQCCDEYECACNCVNS
TVSCPLGYLASTATNDCGCTTTTCLPDKVCVHRSTIYPVGQFWEEGCDVCTCTDMEDAVM
GLRVAQCSQKPCEDSCRSGFTYVLHEGECCGRCLPSACEVVTGSPRGDSQSSWKSVGSQW
ASPENPCLINECVRVKEEVFIQQRNVSCPQLEVPVCPSGFQLSCKTSACCPSCRCERMEA
CMLNGTVIGPGKTVMIDVCTTCRCMVQVGVISGFKLECRKTTCNPCPLGYKEENNTGECC
GRCLPTACTIQLRGGQIMTLKRDETLQDGCDTHFCKVNERGEYFWEKRVTGCPPFDEHKC
LAEGGKIMKIPGTCCDTCEEPECNDITARLQYVKVGSCKSEVEVDIHYCQGKCASKAMYS
IDINDVQDQCSCCSPTRTEPMQVALHCTNGSVVYHEVLNAMECKCSPRKCSK
Vonicog alfa
ボニコグアルファ (遺伝子組換え) ;
フォン・ヴィレブランド因子;
| Formula | C9712H15373N2737O3032S210 |
|---|---|
| CAS | 109319-16-6 |
| Mol weight | 225723.1487 |
JAPAN 2020, APPROVED 2020/3/25, VONVENDI
Anticoagulant, Hemostatic, Replenisher (von Willebrand factor)
Active Substance
General information Recombinant von Willebrand Factor (rVWF) is co-expressed with recombinant Factor VIII (rFVIII) in Chinese hamster ovary (CHO) cells as part of the ADVATE (Centrally authorised product) manufacturing process. The rVWF protein is separated from the FVIII and further purified.
Structural formula
Vonicog alfa is expressed as a 2813 amino acid pro-VWF molecule. The pro-VWF is composed of A, B, C and D repeats, which contain various functional domains that have been identified. The mature VWF monomer is a 2050 amino acid protein. Every monomer contains a number of specific domains with a specific function. Elements of note are: • The D’/D3 domain, which binds to Factor VIII • The A1 domain, which binds to: Platelet gp1b-receptor, Heparin, Collagen • The A3 domain, which binds to collagen • The C1 domain, in which the RGD domain binds to platelet integrin αIIbβ3 when this is activated • The “cysteine knot” domain Monomers of pro-VWF are subsequently N-glycosylated, arranged into dimers via a C-terminal disulfide bond in the endoplasmic reticulum and into multimers by crosslinking of N-terminal cysteine residues via disulfide bonds.
Figure 1. Structure of Von Willebrand Factor Monomer/Dimer

After reduction of disulfide bonds in electrophoretic analysis, rVWF appears as a single predominant band having an apparent molecular weight of approximately 260 kDa. In low resolution agarose gel electrophoresis, rVWF shows a characteristic ladder of bands also known as multimers. In this analysis, rVWF contains as many distinct bands as VWF detectable in normal human plasma or VWF isolated from human plasma but in addition, has a zone with unresolved bands in the ultra-high molecular weight range. Highresolution electrophoresis shows a single band for all multimer levels without any satellite bands, as rVWF has never been exposed to ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) cleavage.

Vonicog alfa, sold under the brand names Vonvendi and Veyvondi, is a medicine used to control bleeding in adults with von Willebrand disease (an inherited bleeding disorder).[5][4][6] It is a recombinant von Willebrand factor.[5][4]
The most common adverse reactions are generalized itching, vomiting, nausea, dizziness, and vertigo.[5]
Vonicog alfa should not be used in the treatment of Hemophilia A.[4]
In the UK it is available only via a named patient access program.[7]
Vonicog alfa was approved for medical use in the United States in December 2015, in the European Union in August 2018, and in Australia in April 2020.[3][5][4][8] It was granted orphan drug designations in both the United States and the European Union.[4][1]
Medical uses
Vonicog alfa is indicated in adults with von Willebrand Disease (VWD), when desmopressin (DDAVP) treatment alone is ineffective or not indicated for the
Adverse effects
The following side effects may occur during treatment with vonicog alfa: hypersensitivity (allergic) reactions, thromboembolic events (problems due to the formation of blood clots in the blood vessels), development of inhibitors (antibodies) against von Willebrand factor, causing the medicine to stop working and resulting in a loss of bleeding control.[4] The most common side effects with vonicog alfa (which may affect up to 1 in 10 patients) are dizziness, vertigo (a spinning sensation), dysgeusia (taste disturbances), tremor, rapid heartbeat, deep venous thrombosis (blood clot in a deep vein, usually in the leg), hypertension (high blood pressure), hot flush, vomiting, nausea (feeling sick), pruritus (itching), chest discomfort, sensations like numbness, tingling, pins and needles at the site of infusion, and an abnormal reading on the electrocardiogram (ECG).[4]
References
- ^ Jump up to:a b c “Veyvondi Australian prescription medicine decision summary”. Therapeutic Goods Administration (TGA). 29 April 2020. Retrieved 16 August 2020.
- ^ “Vonvendi 650 IU powder and solvent for solution for injection – Summary of Product Characteristics (SmPC)”. (emc). 7 May 2020. Retrieved 16 August 2020.
- ^ Jump up to:a b “Vonvendi”. U.S. Food and Drug Administration (FDA). 9 May 2018. Archived from the original on 23 April 2019. Retrieved 15 April 2020.
- ^ Jump up to:a b c d e f g h i j “Veyvondi EPAR”. European Medicines Agency (EMA). 20 September 2018. Retrieved 27 March 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c d “Vonvendi (von willebrand factor- recombinant kit”. DailyMed. 13 February 2019. Retrieved 27 March 2020.
- ^ “Veyvondi-epar product information” (PDF). European Medicines Agency.
- ^ “Vonicog alfa”. Specialist Pharmacy Service. 15 January 2020. Retrieved 27 March 2020.
- ^ “Vonvendi”. U.S. Food and Drug Administration (FDA). 13 April 2018. STN: 125577. Retrieved 27 March 2020.
Further reading
- AusPAR: Vonicog alfa. Therapeutic Goods Administration (TGA) (Report). October 2020.
External links
- “Vonicog alfa”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Vonvendi, Veyvondi |
| Other names | BAX-111 |
| AHFS/Drugs.com | Monograph |
| License data | EU EMA: by INNUS DailyMed: Vonvendi |
| Pregnancy category | AU: B2[1] |
| Routes of administration | Intravenous |
| Drug class | Hemostatic |
| ATC code | B02BD10 (WHO) B02BD06 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only) [1]UK: POM (Prescription only) [2]US: ℞-only [3]EU: Rx-only [4]In general: ℞ (Prescription only) |
| Identifiers | |
| CAS Number | 109319-16-6 |
| DrugBank | DB12872 |
| UNII | 5PKM8P0G5I |
| KEGG | D08681 |
| Chemical and physical data | |
| Formula | C9712H15373N2737O3032S210 |
| Molar mass | 225725.54 g·mol−1 |
General References
- Singal M, Kouides PA: Recombinant von Willebrand factor: a first-of-its-kind product for von Willebrand disease. Drugs Today (Barc). 2016 Dec;52(12):653-664. doi: 10.1358/dot.2016.52.12.2570978. [PubMed:28276537]
- Brown R: Recombinant von Willebrand factor for severe gastrointestinal bleeding unresponsive to other treatments in a patient with type 2A von Willebrand disease: a case report. Blood Coagul Fibrinolysis. 2017 Oct;28(7):570-575. doi: 10.1097/MBC.0000000000000632. [PubMed:28379876]
- Gill JC, Castaman G, Windyga J, Kouides P, Ragni M, Leebeek FW, Obermann-Slupetzky O, Chapman M, Fritsch S, Pavlova BG, Presch I, Ewenstein B: Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood. 2015 Oct 22;126(17):2038-46. doi: 10.1182/blood-2015-02-629873. Epub 2015 Aug 3. [PubMed:26239086]
- Lenting PJ, Christophe OD, Denis CV: von Willebrand factor biosynthesis, secretion, and clearance: connecting the far ends. Blood. 2015 Mar 26;125(13):2019-28. doi: 10.1182/blood-2014-06-528406. Epub 2015 Feb 23. [PubMed:25712991]
- Chung MC, Popova TG, Jorgensen SC, Dong L, Chandhoke V, Bailey CL, Popov SG: Degradation of circulating von Willebrand factor and its regulator ADAMTS13 implicates secreted Bacillus anthracis metalloproteases in anthrax consumptive coagulopathy. J Biol Chem. 2008 Apr 11;283(15):9531-42. doi: 10.1074/jbc.M705871200. Epub 2008 Feb 8. [PubMed:18263586]
- Boston Children’s Hospital [Link]
- EMA [Link]
- FDA application [Link]
- National Institute for Health Research [Link]
- Hemophilia [Link]
////////Vonicog alfa, JAPAN 2020, APPROVALS 2020,, VONVENDI, BAX 111,
DOFETILIDE

Dofetilide
115256-11-6[RN]
6756
b-((p-Methanesulfonamidophenethyl)methylamino)methanesulfono-p-phenetidide
Methanesulfonamide, N-[4-[2-[methyl[2-[4-[(methylsulfonyl)amino]phenoxy]ethyl]amino]ethyl]phenyl]-
MFCD00869707 [MDL number]
- Molecular FormulaC19H27N3O5S2
- Average mass441.565 Da
- UK68798UNII:R4Z9X1N2NDUNII-R4Z9X1N2NDXelideβ-((p-Methanesulfonamidophenethyl)methylamino)methanesulfono-p-phenetidideдофетилидدوفيتيليد多非利特
N-[4-[2-[methyl[2-[4-[(methylsulfonyl)amino]phenoxy]ethyl]amino]ethyl]phenyl]-methanesulfonamideNCGC00164549-01PB0478000SMR000466333Tikosyn (TN)Research Code:UK-68798Trade Name:Tikosyn®MOA:Atrial potassium channel blockerIndication:Atrial flutter; Atrial fibrillationStatus:ApprovedCompany:Pfizer (Originator)Sales:ATC Code:C01BD04
INDIA 31/7/2020 APPROVED CDSCO
Dofetilide was first approved by the U.S. Food and Drug Administration (FDA) on Oct 1, 1999, then approved by European Medicine Agency (EMA) on Nov 29, 1999. It was developed and marketed as Tikosyn® by Pfizer.
Dofetilide is a selective blocker of delayed rectifier outward potassium current (IKr). It is indicated for the maintenance of normal sinus rhythm (delay in time to recurrence of atrial fibrillation/atrial flutter [AF/AFl]) in patients with atrial fibrillation/atrial flutter of greater than one week duration who have been converted to normal sinus rhythm.
Tikosyn® is available capsule for oral use, containing 0.125, 0.25 or 0.5 mg of free Dofetilide. The recommended dose is 500 µg orally twice daily.
Dofetilide is a class III antiarrhythmic agent.[1] It is marketed under the trade name Tikosyn by Pfizer, and is available in the United States in capsules containing 125, 250, and 500 µg of dofetilide. It is not available in Europe[2] or Australia.[3] In the United States it is only available by mail order or through specially trained local pharmacies.[4]
Medical uses
Dofetilide is used for the maintenance of sinus rhythm in individuals prone to the occurrence of atrial fibrillation and flutter arrhythmias, and for chemical cardioversion to sinus rhythm from atrial fibrillation and flutter.[5][6]
Based on the results of the Danish Investigations of Arrhythmias and Mortality on Dofetilide (“DIAMOND”) study,[7] dofetilide does not affect mortality in the treatment of patients post-myocardial infarction with left ventricular dysfunction, however it was shown to decrease all-cause readmissions as well as CHF-related readmissions.[8] Because of the results of the DIAMOND study, some physicians use dofetilide in the suppression of atrial fibrillation in individuals with LV dysfunction, however use appears limited: After initially receiving marketing approval in Europe in 1999, Pfizer voluntarily withdrew this approval in 2004 for commercial reasons[2] and it is not registered in other first world countries.
It has clinical advantages over other class III antiarrhythmics in chemical cardioversion of atrial fibrillation, and maintenance of sinus rhythm, and does not have the pulmonary or hepatotoxicity of amiodarone, however atrial fibrillation is not generally considered life-threatening, and dofetilide causes an increased rate of potentially life-threatening arrhythmias in comparison to other therapies.[9]
Contraindications
Prior to administration of the first dose, the corrected QT (QTc) must be determined. If the QTc is greater than 440 msec (or 500 msec in patients with ventricular conduction abnormalities), dofetilide is contraindicated. If heart rate is less than 60 bpm, the uncorrected QT interval should be used. After each subsequent dose of dofetilide, QTc should be determined and dosing should be adjusted. If at any time after the second dose of dofetilide the QTc is greater than 500 msec (550 msec in patients with ventricular conduction abnormalities), dofetilide should be discontinued. [4]
Adverse effects
Torsades de pointes is the most serious side effect of dofetilide therapy. The incidence of torsades de pointes is 0.3-10.5% and is dose-related, with increased incidence associated with higher doses. The majority of episodes of torsades de pointes have occurred within the first three days of initial dosing. Patients should be hospitalized and monitored for the first three days after starting dofetilide.[10]
The risk of inducing torsades de pointes can be decreased by taking precautions when initiating therapy, such as hospitalizing individuals for a minimum of three days for serial creatinine measurement, continuous telemetry monitoring and availability of cardiac resuscitation.
Pharmacology
Mechanism of action
Dofetilide works by selectively blocking the rapid component of the delayed rectifier outward potassium current (IKr).[11]
This causes the refractory period of atrial tissue to increase, hence its effectiveness in the treatment of atrial fibrillation and atrial flutter.
Dofetilide does not affect dV/dTmax (the slope of the upstroke of phase 0 depolarization), conduction velocity, or the resting membrane potential.

Dofetilide synthesis
There is a dose-dependent increase in the QT interval and the corrected QT interval (QTc). Because of this, many practitioners will initiate dofetilide therapy only on individuals under telemetry monitoring or if serial EKG measurements of QT and QTc can be performed.
Pharmacokinetics
Peak plasma concentrations are seen two to three hours after oral dosing when fasting. Dofetilide is well absorbed in its oral form, with a bioavailability of >90%. Intravenous administration of dofetilide is not available in the United States. [12]
The elimination half-life of dofetilide is roughly 10 hours; however, this varies based on many physiologic factors (most significantly creatinine clearance), and ranges from 4.8 to 13.5 hours. Due to the significant level of renal elimination (80% unchanged, 20% metabolites), the dose of dofetilide must be adjusted to prevent toxicity due to impaired renal function.[13]
Dofetilide is metabolized predominantly by CYP3A4 enzymes predominantly in the liver and GI tract. This means that it is likely to interact with drugs that inhibit CYP3A4, such as erythromycin, clarithromycin, or ketoconazole, resulting in higher and potentially toxic levels of dofetilide. [14]
Metabolism
A steady-state plasma level of dofetilide is achieved in 2–3 days.
80% of dofetilide is excreted by the kidneys, so the dose of dofetilide should be adjusted in individuals with chronic kidney disease, based on creatinine clearance.
In the kidneys, dofetilide is eliminated via cation exchange (secretion). Agents that interfere with the renal cation exchange system, such as verapamil, cimetidine, hydrochlorothiazide, itraconazole, ketoconazole, prochlorperazine, and trimethoprim should not be administered to individuals taking dofetilide.
About 20 percent of dofetilide is metabolized in the liver via the CYP3A4 isoenzyme of the cytochrome P450 enzyme system. Drugs that interfere with the activity of the CYP3A4 isoenzyme can increase serum dofetilide levels. If the renal cation exchange system is interfered with (as with the medications listed above), a larger percentage of dofetilide is cleared via the CYP3A4 isoenzyme system.
History
After its initial US FDA approval, due to the pro-arrhythmic potential it was only made available to hospitals and prescribers that had received education and undergone specific training in the risks of treatment with dofetilide; however, this restriction was subsequently removed in 2016. [15
SYN


REF
Reference:1. US5079248A / US4959366A.
2. J. Med. Chem. 1990, 33, 1151-1155.
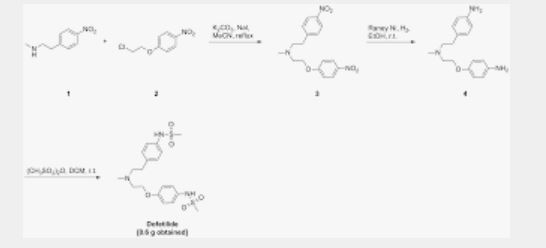
SYN
SYN
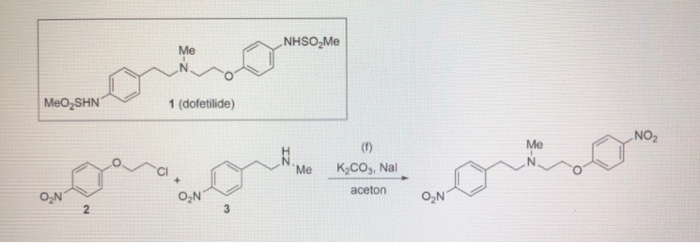
SYN
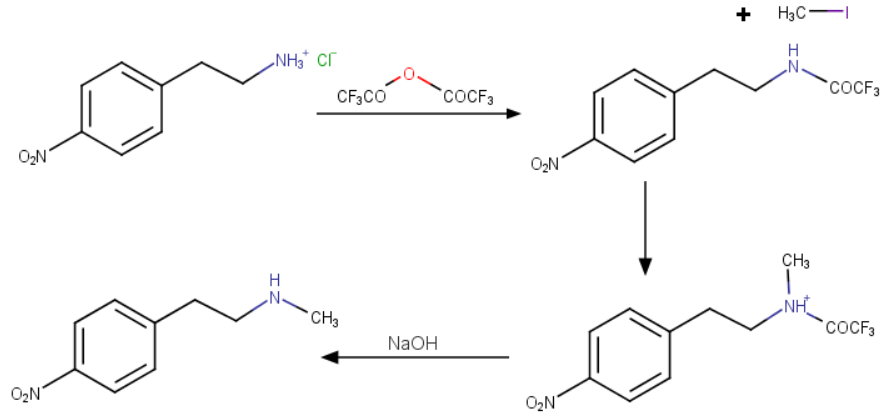
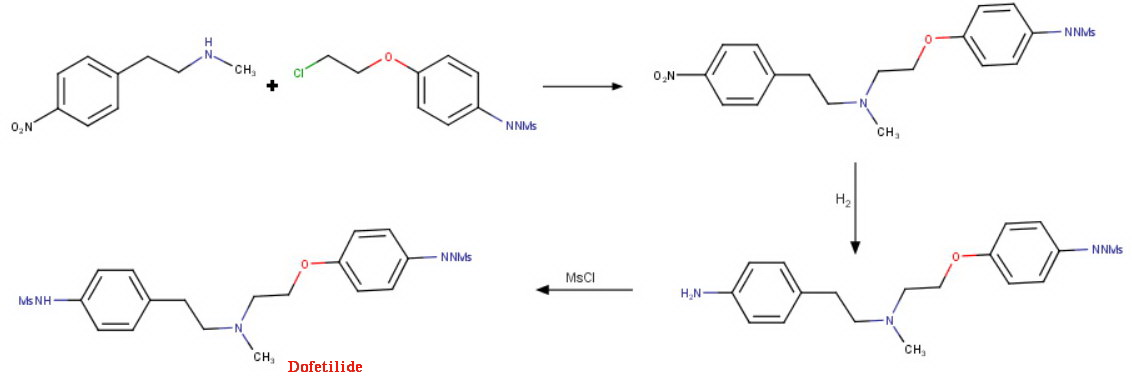
SYN
EP 0245997; JP 1987267250; US 4959366; US 5079248
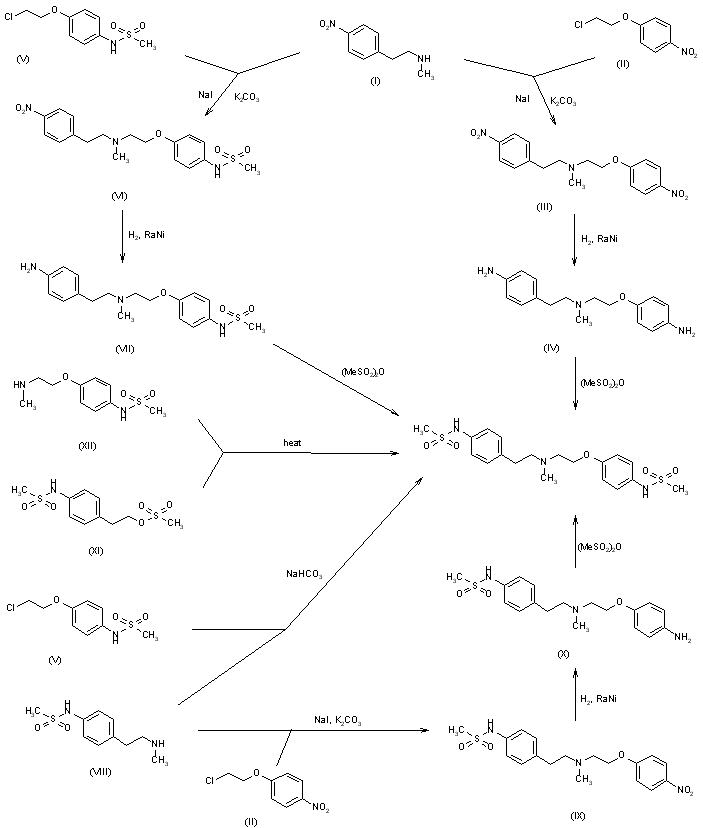
This compound can be prepared by several related ways: 1) The condensation of N-methyl-2-(4-nitrophenyl)ethylamine (I) with 4-(2-chloroethoxy)nitrobenzene (II) by means of NaI and K2CO3 in refluxing acetonitrile gives 1-(4-nitrophenoxy)-5-(4-nitrophenyl)-3-methyl-3-azapentane (III), which is reduced with H2 over Pd/C in ethanol, yielding the corresponding diamino derivative (IV). Finally, this compound is acylated with methanesulfonyl anhydride in dichloromethane. 2) The condensation of (I) with N-[4-(2-chloroethoxy)phenyl]methanesulfonamide (V) with NaI and K2CO3 as before gives 1-[4-(methanesulfonamide)phenoxy]-3-methyl-5-(4-nitrophenyl)-3-azapentane (VI), which is reduced with H2 over Pd/C as before, yielding the corresponding amino derivative (VII). Finally, this compound is acylated with methanesulfonyl anhydride as usual. 3) The condensation of (II) with N-[4-[2-(methylamino)ethyl]phenyl]methanesulfonamide (VIII) with NaI and K2CO3 as usual gives 1-[4-(methanesulfonamido)phenyl]-3-methyl-5-(4-nitrophenoxy)-3-azapentane (IX), which is reduced with H2 and RaNi to the corresponding amino derivative (X). Finally, this compound is acylated with methanesulfonyl chloride and pyridine. 4) By condensation of N-[4-[2-(methanesulfonyloxy)ethyl]phenyl]methanesulfonamide (XI) with N-[4-[2-(methylamino)ethoxy]phenyl]methanesulfonamide (XII) in refluxing ethanol. 5) By condensation of (V) with (VIII) by means of NaHCO3.
References
- ^ Lenz TL; Hilleman DE (July 2000). “Dofetilide, a new class III antiarrhythmic agent”. Pharmacotherapy. 20 (7): 776–86. doi:10.1592/phco.20.9.776.35208. PMID 10907968.
- ^ Jump up to:a b Wathion, Noel (2004-04-13). “Public Statement on Tikosyn (dofetilide): Voluntary Withdrawal of the Marketing Authorisation in the European Union” (PDF). European Agency for the Evaluation of Medicinal Products.
- ^ Australian Medicines Handbook 2014
- ^ Jump up to:a b TIKOSYN® (dofetilide). Pfizer. <http://www.tikosyn.com/>.
- ^ Banchs JE; Wolbrette DL; Samii SM; et al. (November 2008). “Efficacy and safety of dofetilide in patients with atrial fibrillation and atrial flutter”. J Interv Card Electrophysiol. 23(2): 111–5. doi:10.1007/s10840-008-9290-6. PMID 18688699. S2CID 25162347.
- ^ Lenz TL; Hilleman DE (November 2000). “Dofetilide: A new antiarrhythmic agent approved for conversion and/or maintenance of atrial fibrillation/atrial flutter”. Drugs Today. 36 (11): 759–71. doi:10.1358/dot.2000.36.11.601530. PMID 12845335.
- ^ Torp-Pedersen C, Møller M, Bloch-Thomsen PE, et al. (September 1999). “Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group”. The New England Journal of Medicine. 341 (12): 857–65. doi:10.1056/NEJM199909163411201. PMID 10486417.
- ^ Torp-Pedersen C; ller M; Mø Bloch-Thomsen PE; et al. (September 1999). “Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group”. N. Engl. J. Med. 341 (12): 857–65. doi:10.1056/NEJM199909163411201. PMID 10486417.
- ^ Micromedex Drugdex drug evaluations micromedex.com
- ^ Torp-Pedersen C, Møller M, Bloch-Thomsen PE, et al. Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group. N Engl J Med 1999; 341:857.
- ^ Roukoz H; Saliba W (January 2007). “Dofetilide: a new class III antiarrhythmic agent”. Expert Rev Cardiovasc Ther. 5 (1): 9–19. doi:10.1586/14779072.5.1.9. PMID 17187453. S2CID 11255636.
- ^ 1Rasmussen HS, Allen MJ, Blackburn KJ, et al. Dofetilide, a novel class III antiarrhythmic agent. J Cardiovasc Pharmacol 1992; 20 Suppl 2:S96.
- ^ “Dofetilide.” Lexicomp. Wulters Kluwer Health, n.d. Web. <online.lexi.com>.
- ^ Walker DK, Alabaster CT, Congrave GS, et al. Significance of metabolism in the disposition and action of the antidysrhythmic drug, dofetilide. In vitro studies and correlation with in vivo data. Drug Metab Dispos 1996; 24:447.
- ^ “Information for Tikosyn (dofetilide)”. US Food and Drug Administration. 2016-03-09.
DofetilideCAS Registry Number: 115256-11-6CAS Name:N-[4-[2-[Methyl[2-[4-[(methylsulfonyl)amino]phenoxy]ethyl]amino]ethyl]phenyl]methanesulfonamideAdditional Names: 1-(4-methanesulfonamidophenoxy)-2-[N-(4-methanesulfonamidophenethyl)-N-methylamino]ethaneManufacturers’ Codes: UK-68798Trademarks: Tikosyn (Pfizer)Molecular Formula: C19H27N3O5S2Molecular Weight: 441.56Percent Composition: C 51.68%, H 6.16%, N 9.52%, O 18.12%, S 14.52%Literature References: Potassium channel blocker. Prepn: J. E. Arrowsmith et al.,EP245997; P. E. Cross et al.,US4959366 (1987, 1990 both to Pfizer); idemet al.,J. Med. Chem.33, 1151 (1990). HPLC determn in urine: D. K. Walker et al.,J. Chromatogr.568, 475 (1991). Mechanism of action study: D. Carmeliet, J. Pharmacol. Exp. Ther.262, 809 (1992). Review of pharmacology and pharmacokinetics: H. S. Rasmussen et al.,ibid.20, Suppl. 2, S96-S105 (1992). Clinical trial in atrial fibrillation and flutter: B. L. Norgaard et al.,Am. Heart J.137, 1062 (1999); in congestive heart failure: C. Torp-Pedersen et al.,N. Engl. J. Med.341, 857 (1999).Properties: Crystals from ethyl acetate/methanol (10:1), mp 147-149° (Cross); from hexane/ethyl acetate, mp 151-152° (Arrowsmith). Also reported as white crystalline solid, mp 161° (Rasmussen). pKa 7.0, 9.0, 9.6. Distribution coefficient (pH 7.4): 0.96. Sol in 0.1M NaOH, acetone, 0.1M HCl; very slightly sol in water, propan-2-ol.Melting point: mp 147-149° (Cross); mp 151-152° (Arrowsmith); mp 161° (Rasmussen)pKa: pKa 7.0, 9.0, 9.6Therap-Cat: Antiarrhythmic (class III).Keywords: Antiarrhythmic; Potassium Channel Blocker.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a601235 |
| ATC code | C01BD04 (WHO) |
| Pharmacokinetic data | |
| Bioavailability | 96% (oral) |
| Protein binding | 60% -70% |
| Elimination half-life | 10 hours |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 115256-11-6 |
| PubChem CID | 71329 |
| IUPHAR/BPS | 2604 |
| DrugBank | DB00204 |
| ChemSpider | 64435 |
| UNII | R4Z9X1N2ND |
| KEGG | D00647 |
| ChEBI | CHEBI:4681 |
| ChEMBL | ChEMBL473 |
| CompTox Dashboard (EPA) | DTXSID5046433 |
| ECHA InfoCard | 100.166.441 |
| Chemical and physical data | |
| Formula | C19H27N3O5S2 |
| Molar mass | 441.56 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]O=S(=O)(Nc1ccc(cc1)CCN(CCOc2ccc(cc2)NS(=O)(=O)C)C)C | |
| InChI[hide]InChI=1S/C19H27N3O5S2/c1-22(13-12-16-4-6-17(7-5-16)20-28(2,23)24)14-15-27-19-10-8-18(9-11-19)21-29(3,25)26/h4-11,20-21H,12-15H2,1-3H3 Key:IXTMWRCNAAVVAI-UHFFFAOYSA-N |
////////////DOFETILIDE, 2020 APPROVALS, INDIA 2020, UK 68798, UNII:R4Z9X1N2ND, дофетилид , دوفيتيليد ,多非利特 , TIKOSYN
Lurbinectedin

Lurbinectedin
(1’R,6R,6aR,7R,13S,14S,16R)-5-(Acetyloxy)-2′,3′,4′,6,6a,7,9′-decahydro-8,14-dihydroxy-6′,9-dimethoxy-4,10,23-trimethyl-spiro(6,16-(epithiopropaneoxymethano)-7.13-imino-12H-1,3-dioxolo[7,8]soquino[3,2-b][3]benzazocine-20,1′-[1H]pyrido[3,4-b]indol]-19-one
| Molecular Weight | 784.87 |
| Formula | C41H44N4O10S |
| CAS No. | 497871-47-3 (Lurbinectedin); |
| Chemical Name | Spiro[6,16-(epithiopropanoxymethano)-7,13-imino-12H-1,3-dioxolo[7,8]isoquino[3,2-b][3]benzazocine-20,1′-[1H]pyrido[3,4-b]indol]-19-one, 5-(acetyloxy)-2′,3′,4′,6,6a,7,9′,13,14,16-decahydro-8,14-dihydroxy-6′,9-dimethoxy-4,10,23-trimethyl-, (1’R,6R,6aR,7R,13S,14S,16R)- (9CI) |
fda approved , 6/15/2020 , ZEPZELCA, Pharma Mar S.A.
To treat metastatic small cell lung cancer
Drug Trials Snapshot
Research Code:PM-01183; PM-1183
MOA:RNA polymerase inhibitor
Indication:Ovarian cancer; Breast cancer; Non small cell lung cancer (NSCLC)лурбинектединلوربينيكتيدين芦比替定(1R,1’R,2’R,3’R,11’S,12’S,14’R)-5′,12′-Dihydroxy-6,6′-dimethoxy-7′,21′,30′-trimethyl-27′-oxo-2,3,4,9-tetrahydrospiro[β-carboline-1,26′-[17,19,28]trioxa[24]thia[13,30]diazaheptacyclo[12.9.6.13,11. 02,13.04,9.015,23.016,20]triaconta[4,6,8,15,20,22]hexaen]-22′-yl acetate [ACD/IUPAC Name]2CN60TN6ZS497871-47-3[RN]9397
Lurbinectedin is in phase III clinical development for the treatment of platinum refractory/resistant ovarian cancer.
Phase II clinical trials are also ongoing for several oncology indications: non-small cell lung cancer, breast cancer, small cell lung cancer, head and neck carcinoma, neuroendocrine tumors, biliary tract carcinoma, endometrial carcinoma, germ cell tumors and Ewing’s family of tumors.
Lurbinectedin, sold under the brand name Zepzelca, is a medication for the treatment of adults with metastatic small cell lung cancer (SCLC) with disease progression on or after platinum-based chemotherapy.[1][2][3]
The most common side effects include leukopenia, lymphopenia, fatigue, anemia, neutropenia, increased creatinine, increased alanine aminotransferase, increased glucose, thrombocytopenia, nausea, decreased appetite, musculoskeletal pain, decreased albumin, constipation, dyspnea, decreased sodium, increased aspartate aminotransferase, vomiting, cough, decreased magnesium and diarrhea.[1][2][3]
Lurbinectedin is a synthetic tetrahydropyrrolo [4, 3, 2-de]quinolin-8(1H)-one alkaloid analogue with potential antineoplastic activity.[4] Lurbinectedin covalently binds to residues lying in the minor groove of DNA, which may result in delayed progression through S phase, cell cycle arrest in the G2/M phase and cell death.[4]
Lurbinectedin was approved for medical use in the United States in June 2020.[5][1][2][3][6]
Structure
Lurbinectedin is structurally similar to trabectedin, although the tetrahydroisoquinoline present in trabectedin is replaced with a tetrahydro β-carboline which enables lurbinectedin to exhibit increased antitumor activity compared with trabectedin.[7]
Biosynthesis
Lurbinectedin a marine agent isolated from the sea squirt species Ecteinascidia turbinata. Synthetic production is necessary because very small amounts can be obtained from sea organisms. For example, one ton (1000 kg) of sea squirts are required to produce one gram of trabectedin, which is analogue of lurbinectedin. Complex synthesis of lurbinectedin starts from small, common starting materials that require twenty-six individual steps to produce the drug with overall yield of 1.6%.[8][9]
Mechanism of action
According to PharmaMar,[10] lurbinectedin inhibits the active transcription of the encoding genes. This has two consequences. On one hand, it promotes tumor cell death, and on the other it normalizes tumor microenvironment. Active transcription is the process by which there are specific signal where information contained in the DNA sequence is transferred to an RNA molecule. This activity depends on the activity of an enzyme called RNA polymerase II. Lurbinectedin inhibits transcription through a very precise mechanism. Firstly, lurbinectedin binds to specific DNA sequences. It is at these precise spots that slides down the DNA to produce RNA polymerase II that is blocked and degraded by lurbinectedin. Lurbinectedin also has important role in tumor microenvironment. The tumor cells act upon macrophages to avoid them from behaving like an activator of the immune system. Literally, macrophages work in any tumor’s favor. Macrophages can contribute to tumor growth and progression by promoting tumor cell proliferation and invasion, fostering tumor angiogenesis and suppressing antitumor immune cells.[11][12] Attracted to oxygen-starved (hypoxic) and necrotic tumor cells they promote chronic inflammation. So, not only that macrophages inhibit immune system avoiding the destruction of tumor cells, but they also create tumor tissue that allows tumor growth. However, macrophages associated with tumors are cells that are addicted to the transcription process. Lurbinectedin acts specifically on the macrophages associated with tumors in two ways: firstly, by inhibiting the transcription of macrophages that leads to cell death and secondly, inhibiting the production of tumor growth factors. In this way, lurbinectedin normalizes the tumor microenvironment.
History
Lurbinectedin was approved for medical use in the United States in June 2020.[5][1][2][3][6]
Efficacy was demonstrated in the PM1183-B-005-14 trial (Study B-005; NCT02454972), a multicenter open-label, multi-cohort study enrolling 105 participants with metastatic SCLC who had disease progression on or after platinum-based chemotherapy.[3][6] Participants received lurbinectedin 3.2 mg/m2 by intravenous infusion every 21 days until disease progression or unacceptable toxicity.[3] The trial was conducted at 26 sites in the United States, Great Britain, Belgium, France, Italy, Spain and Czech Republic.[6]
The U.S. Food and Drug Administration (FDA) granted the application for lurbinectedin priority review and orphan drug designations and granted the approval of Zepzelca to Pharma Mar S.A.[3][13]
Research
Clinical Trials
Lurbinectedin can be used as monotherapy in the treatment of SCLC. Lurbinectedin monotherapy demonstrated the following clinical results in relapsed extensive stage SCLC:
- For sensitive disease (chemotherapy-free interval of ≥ 90 days) overall response rate (ORR) was 46.6% with 79.3% disease control rate and median overall survival (OS) being increased to 15.2 months.[14]
- For resistant disease (chemotherapy-free interval of < 90 days) overall response rate (ORR) was 21.3% with 46.8% disease control rate and 5.1 months median overall survival (OS).[14]
Lurbinectedin is also being investigated in combination with doxorubicin as second-line therapy in a randomized Phase III trial.[medical citation needed] While overall survival in this trial is not yet known, response rates at second line were
- 91.7% in sensitive disease with median progression-free survival of 5.8 months, and
- 33.3% in resistant disease with median progression-free of 3.5 months.[15]
Lurbinectedin is available in the U.S. under Expanded Access Program (EAP).[15][16]
SYN
SYN
WO2011/147828
Ecteinascidins is a group of naturally occurring marine compounds and analogs thereof, which are well identified and structurally characterized, and are disclosed to have antibacterial and cytotoxic properties. See for example, European Patent 309.477; WO 03/66638; WO 03/08423; WO 01 /771 15; WO 03/014127; R. Sakai et al., 1992, Proc. Natl. Acad. Sci. USA 89, pages 1 1456- 1 1460; R. Menchaca et al., 2003, J. Org. Chem. 68(23), pages 8859-8866; and I. Manzanares et al., 2001 , Curr. Med. Chem. Anti-Cancer Agents, 1 , pages 257-276; and references therein. Examples of ecteinascidins are provided by ET-743, ET-729, ET-745, ET-759A, ET-759B, ET-759C, ET-770, ET-815, ET-731 , ET-745B, ET-722, ET-736, ET-738, ET-808, ET-752, ET-594, ET-552, ET-637, ET-652, ET-583, ET-597, ET-596, ET-639, ET-641 , and derivatives thereof, such as acetylated forms, formylated forms, methylated forms, and oxide forms.
The structural characterizations of such ecteinascidins are not given again explicitly herein because from the detailed description provided in such references and citations any person of ordinary skill in this technology is capable of obtaining such information directly from the sources cited here and related sources.
At least one of the ecteinascidin compounds, ecteinascidin 743 (ET-743), has been extensively studied, and it will be referred to
specifically herein to illustrate features of this invention. ET-743 is being employed as an anticancer medicament, under the international nonproprietary name (INN) trabectedin, for the treatment of patients with advanced and metastatic soft tissue sarcoma (STS), after failure of anthracyclines and ifosfamide, or who are unsuited to receive such agents, and for the treatment of relapsed platinum- sensitive ovarian cancer in combination with pegylated liposomal doxorubicin.
ET-743 has a complex tris(tetrahydroisoquinoline) structure of formula
It was originally prepared by isolation from extracts of the marine tunicate Ecteinascidia turbinata. The yield was low, and alternative preparative processes had been sought.
The first synthetic process for producing ecteinascidin compounds was described in US Patent 5,721 ,362. This process employed sesamol as starting material and yielded ET-743 after a long and complicated sequence of 38 examples each describing one or more steps in the synthetic sequence.
An improvement in the preparation of one intermediate used in such process was disclosed in US Patent 6,815,544. Even with this improvement, the total synthesis was not suitable for manufacturing ET-743 at an industrial scale.
A hemisynthetic process for producing ecteinascidin compounds was described in EP 1.185.536. This process employs cyanosafracin B as starting material to provide ET-743. Cyanosafracin B is a pentacyclic antibiotic obtained by fermentation from the bacteria Pseudomonas fluorescens.
Cyanosafracin B
An improvement in such hemisynthetic process was disclosed in
EP 1.287.004.
To date four additional synthetic process (2 total and 2 formal synthesis) have been disclosed in patent applications JP 2003221395, WO 2007/045686, and WO 2007/087220 and in J. Org. Chem. 2008, 73, pages 9594-9600.
WO 2007/045686 also relates to the synthesis of Ecteinascidins-583 and 597 using intermediate compounds of formula:
Total synthesis strategies for the synthesis of the pentacyclic core -743 are overviewed in Figure I.
X = OH or CI
R = Protecting Group
WO2007087220 JOC 2008, 73, 9594-9600
EXAMPLE 3: SYNTHESIS OF COMPOUND 17.
Scheme X above provides an example of the synthesis of compound 17 from intermediate 10.
Compounds 16 and 17 are obtainable from intermediate 15 using the same procedures than those previously described in WO03/014127.
SYN

Reference:
1. WO2003014127A1.
https://patents.google.com/patent/WO2003014127A1/en
The ecteinascidins are exceedingly potent antitumour agents isolated from the marine tunicate Ecteinascidia turbinata. Several ecteinascidins have been reported previously in the patent and scientific literature. See, for example:
U.S. Patent No 5.256.663, which describes pharmaceutical compositions comprising matter extracted from the tropical marine invertebrate, Ecteinascidia turbinata, and designated therein as ecteinascidins, and the use of such compositions as antibacterial, antiviral, and/ or antitumour agents in mammals.
U.S. Patent No 5.089.273, which describes novel compositions of matter extracted from the tropical marine invertebrate, Ecteinascidia turbinata, and designated therein as ecteinascidins 729, 743, 745, 759A, 759B and 770. These compounds are useful as antibacterial and/or antitumour agents in mammals.
U.S. Patent No 5.149.804 which describes Ecteinascidins 722 and 736 (Et’s 722 and 736) isolated from the Caribbean tunicate Ecteinascidia turbinata and their structures. Et’s 722 and 736 protect mice in vivo at very low concentrations against P388 lymphoma, B 16 melanoma, and Lewis lung carcinoma.
U.S. Patent No 5.478.932, which describes ecteinascidins isolated from the Caribbean tunicate Ecteinascidia turbinata, which provide in vivo protection against P388 lymphoma, B 16 melanoma, M5076 ovarian sarcoma, Lewis lung carcinoma, and the LX- 1 human lung and MX- 1 human mammary carcinoma xenografts.
U.S. Patent No 5.654.426, which describes several ecteinascidins isolated from the Caribbean tunicate Ecteinascidia turbinata, which provide in vivo protection against P388 lymphoma, B 16 melanoma, M5076 ovarian sarcoma, Lewis lung carcinoma, and the LX-1 human lung and MX- 1 human mammary carcinoma xenografts.
U.S. Patent No 5.721.362 which describes a synthetic process for the formation of ecteinascidin compounds and related structures.
U.S. Patent No 6.124.292 which describes a series of new ecteinascidin- like compounds.
WO 0177115, WO 0187894 and WO 0187895, which describe new synthetic compounds of the ecteinascidin series, their synthesis and biological properties.
See also: Corey, E.J., J. Am. Chem. Soc, 1996, 118 pp. 9202-9203; Rinehart, et al., Journal of Natural Products, 1990, “Bioactive Compounds from Aquatic and Terrestrial Sources”, vol. 53, pp. 771- 792; Rinehart et al., Pure and Appl. Chem., 1990, “Biologically active natural products”, vol 62, pp. 1277- 1280; Rinehart, et al., J. Org. Chem., 1990, “Ecteinascidins 729, 743, 745, 759A, 759B, and 770: potent Antitumour Agents from the Caribbean Tunicate Ecteinascidia tuminata”, vol. 55, pp. 4512-4515; Wright et al., J. Org. Chem., 1990, “Antitumour Tetrahydroisoquinoline Alkaloids from the Colonial ascidian Ecteinascidia turbinata”, vol. 55, pp. 4508-4512; Sakai et al., Proc. Natl. Acad. Sci. USA 1992, “Additional anitumor ecteinascidins from a Caribbean tunicate: Crystal structures and activities in vivo”, vol. 89, 1 1456- 1 1460; Science 1994, “Chemical Prospectors Scour the Seas for Promising Drugs”, vol. 266, pp.1324; Koenig, K.E., “Asymmetric Synthesis”, ed. Morrison, Academic Press, Inc., Orlando, FL, vol. 5, 1985, p. 71; Barton, et al., J. Chem Soc. Perkin Trans., 1 , 1982, “Synthesis and Properties of a Series of Sterically Hindered Guanidine bases”, pp. 2085; Fukuyama et al., J. Am. Chem. Soc, 1982, “Stereocontrolled Total Synthesis of (+)-Saframycin B”, vol. 104, pp. 4957; Fukuyama et al., J. Am. Chem. Soc, 1990, “Total Synthesis of (+) – Saframycin A”, vol. 112, p. 3712; Saito, et al., J. Org. Chem., 1989, “Synthesis of Saframycins. Preparation of a Key tricyclic Lactam Intermediate to Saframycin A”, vol. 54, 5391; Still, et al., J Org. Chem., 1978, “Rapid Chromatographic Technique for Preparative Separations with Moderate Resolution”, vol. 43, p. 2923; Kofron, W.G.; Baclawski, L.M., J. Org. Chem., 1976, vol. 41, 1879; Guan et al., J. Biomolec Struc & Dynam., vol. 10, pp. 793-817 (1993); Shamma et al., “Carbon- 13 NMR Shift Assignments of Amines and Alkaloids”, p. 206 (1979); Lown et al., Biochemistry, 21, 419-428 (1982); Zmijewski et al., Chem. Biol. Interactions, 52, 361-375 (1985); Ito, CRC Crit. Rev. Anal. Chem., 17, 65- 143 (1986); Rinehart et al., “Topics in Pharmaceutical Sciences 1989”, pp. 613-626, D. D. Breimer, D. J. A. Cromwelin, K. K. Midha, Eds., Amsterdam Medical Press B. V., Noordwijk, The Netherlands (1989); Rinehart et al., “Biological Mass Spectrometry”, 233-258 eds. Burlingame et al., Elsevier Amsterdam (1990); Guan et al., Jour. Biomolec. Struct. & Dynam., vol. 10 pp. 793-817 (1993); Nakagawa et al., J. Amer. Chem. Soc, 11 1 : 2721-2722 (1989);; Lichter et al., “Food and Drugs from the Sea Proceedings” (1972), Marine Technology Society, Washington, D.C. 1973, 117- 127; Sakai et al., J. Amer. Chem. Soc, 1996, 1 18, 9017; Garcϊa-Rocha et al., Brit. J. Cancer, 1996, 73: 875-883; and pommier et al., Biochemistry, 1996, 35: 13303- 13309;
In 2000, a hemisynthetic process for the formation of ecteinascidin compounds and related structures such as phthalascidin starting from natural bis(tetrahydroisoquinoline) alkaloids such as the saframycin and safracin antibiotics available from different culture broths was reported; See Manzanares et al., Org. Lett., 2000, “Synthesis of Ecteinascidin ET-743 and Phthalascidin Pt-650 from Cyanosafracin B”, Vol. 2, No 16, pp. 2545-2548; and International Patent Application WO 00 69862.
Ecteinascidin 736 was first discovered by Rinehart and features a tetrahydro-β-carboline unit in place of the tetrahydroisoquinoline unit more usually found in the ecteinascidin compounds isolated from natural sources; See for example Sakai et al., Proc. Natl. Acad. Sci. USA 1992, “Additional antitumor ecteinascidins from a Caribbean tunicate: Crystal structures and activities in vivo”, vol. 89, 11456-11460.
Et-736
WO 9209607 claims ecteinascidin 736, as well as ecteinascidin 722 with hydrogen in place of methyl on the nitrogen common to rings C and D of ecteinascidin 736 and O-methylecteinascidin 736 with methoxy in place of hydroxy on ring C of ecteinascidin 736.
Despite the positive results obtained in clinical applications in chemotherapy, the search in the field of ecteinascidin compounds is still open to the identification of new compounds with optimal features of cytotoxicity and selectivity toward the tumour and with a reduced systemic toxicity and improved pharmacokinetic properties.
PATENT
WO2001087894A1.
PATENT
US 20130066067
https://patents.google.com/patent/US20130066067A1/en
- Ecteinascidins is a group of naturally occurring marine compounds and analogs thereof, which are well identified and structurally characterized, and are disclosed to have antibacterial and cytotoxic properties. See for example, European Patent 309.477; WO 03/66638; WO 03/08423; WO 01/77115; WO 03/014127; R. Sakai et al., 1992, Proc. Natl. Acad. Sci. USA 89, pages 11456-11460; R. Menchaca et al., 2003, J. Org. Chem. 68(23), pages 8859-8866; and I. Manzanares et al., 2001, Curr. Med. Chem. Anti–Cancer Agents, 1, pages 257-276; and references therein. Examples of ecteinascidins are provided by ET-743, ET-729, ET-745, ET-759A, ET-759B, ET-759C, ET-770, ET-815, ET-731, ET-745B, ET-722, ET-736, ET-738, ET-808, ET-752, ET-594, ET-552, ET-637, ET-652, ET-583, ET-597, ET-596, ET-639, ET-641, and derivatives thereof, such as acetylated forms, formylated forms, methylated forms, and oxide forms.
- [0003]
The structural characterizations of such ecteinascidins are not given again explicitly herein because from the detailed description provided in such references and citations any person of ordinary skill in this technology is capable of obtaining such information directly from the sources cited here and related sources. - [0004]
At least one of the ecteinascidin compounds, ecteinascidin 743 (ET-743), has been extensively studied, and it will be referred to specifically herein to illustrate features of this invention. ET-743 is being employed as an anticancer medicament, under the international nonproprietary name (INN) trabectedin, for the treatment of patients with advanced and metastatic soft tissue sarcoma (STS), after failure of anthracyclines and ifosfamide, or who are unsuited to receive such agents, and for the treatment of relapsed platinum-sensitive ovarian cancer in combination with pegylated liposomal doxorubicin. - [0005]
ET-743 has a complex tris(tetrahydroisoquinoline) structure of formula - [0006]
It was originally prepared by isolation from extracts of the marine tunicate Ecteinascidia turbinata. The yield was low, and alternative preparative processes had been sought. - [0007]
The first synthetic process for producing ecteinascidin compounds was described in U.S. Pat. No. 5,721,362. This process employed sesamol as starting material and yielded ET-743 after a long and complicated sequence of 38 examples each describing one or more steps in the synthetic sequence. - [0008]
An improvement in the preparation of one intermediate used in such process was disclosed in U.S. Pat. No. 6,815,544. Even with this improvement, the total synthesis was not suitable for manufacturing ET-743 at an industrial scale. - [0009]
A hemisynthetic process for producing ecteinascidin compounds was described in EP 1.185.536. This process employs cyanosafracin B as starting material to provide ET-743. Cyanosafracin B is a pentacyclic antibiotic obtained by fermentation from the bacteria Pseudomonas fluorescens. - [0010]
An improvement in such hemisynthetic process was disclosed in EP 1.287.004. - [0011]
To date four additional synthetic process (2 total and 2 formal synthesis) have been disclosed in patent applications JP 2003221395, WO 2007/045686, and WO 2007/087220 and in J. Org. Chem. 2008, 73, pages 9594-9600. - [0012]
WO 2007/045686 also relates to the synthesis of Ecteinascidins-583 and 597 using intermediate compounds of formula: - [0013]
Total synthesis strategies for the synthesis of the pentacyclic core of ET-743 are overviewed in FIG. 1.
PAPER
Angewandte Chemie, International Edition (2019), 58(12), 3972-3975.
https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.201900035
An efficient and scalable approach is described for the total synthesis of the marine natural product Et‐743 and its derivative lubinectedin, which are valuable antitumor compounds. The method delivers 1.6 % overall yield in 26 total steps from Cbz‐protected (S)‐tyrosine. It features the use of a common advanced intermediate to create the right and left parts of these compounds, and a light‐mediated remote C−H bond activation to assemble a benzo[1,3]dioxole‐containing intermediate.

Synthesis of lactone SI-5. A mixture of 19 (98.0 mg, 0.16 mmol, 1.0 equiv), 2-(5-methoxy-1H-indol-3-yl) ethanamine hydrochloride salt (357.8 mg, 1.58 mmol, 10.0 equiv) and NaOAc (144 mg, 1.74 mmol, 11.0 equiv) in anhydrous EtOH (5.0 mL) was stirred at 60 oC for 5 h. The cooled mixture was extracted with ethyl acetate, and the organic layer was dried over sodium sulfate and concentrated. The residue was purified by flash column chromatography (eluting with DCM/MeOH = 20:1) to afford compound SI-5 (109 mg, 87%). [α]𝐷 20 = -27.7 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.61 (s, 1H), 7.13 (d, J = 8.8 Hz, 1H), 6.82 (d, J = 2.2 Hz, 1H), 6.75 (dd, J = 8.8, 2.4 Hz, 1H), 6.66 (s, 1H), 6.22 (d, J = 1.0 Hz, 1H), 6.02 (d, J = 1.0 Hz, 1H), 5.78 (s, 1H), 5.08 (d, J = 11.7 Hz, 1H), 4.55 (s, 1H), 4.32 (s, 1H), 4.27 (d, J = 3.8 Hz, 1H), 4.23–4.15 (m, 2H), 3.81 (s, 3H), 3.79 (s, 3H), 3.47–3.39 (m, 2H), 3.20–3.10 (m, 1H), 3.06 (d, J = 18.1 Hz, 1H), 2.93 (dd, J = 18.2, 9.1 Hz, 1H), 2.86–2.76 (m, 1H), 2.62 (dt, J = 14.9, 4.8 Hz, 1H), 2.56–2.47 (m, 2H), 2.37 (s, 3H), 2.30–2.27 (m, 1H), 2.26 (s, 3H), 2.22 (s, 3H), 2.06 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 171.6, 168.8, 154.0, 148.2, 145.8, 143.1, 141.3, 140.5, 131.4, 130.8, 130.7, 129.4, 127.3, 120.9, 120.8, 118.4, 118.4, 113.9, 113.8, 112.2, 111.8, 110.2, 102.2, 100.5, 62.6, 61.4, 60.7, 60.5, 59.6, 59.6, 55.9, 54.9, 54.8, 42.1, 41.6, 39.9, 39.5, 29.5, 24.0, 20.8, 16.0, 9.9; HRMS (ESI) m/z calcd. for C42H43N5O9S [M + H]+ 794.2860, found 794.2858

Lurbinectedin: To a solution of SI-5 (80 mg, 0.1 mmol, 1.0 equiv) in acetonitrile and water (3:2, v/v, 10 mL) was added silver nitrate (514 mg, 3 mmol, 30.0 equiv). The suspension was stirred at 25 oC for 24 h before a mixture of saturated brine (5.0 mL) and saturated sodium hydrogen carbonate (5 mL) were added. The resultant mixture was stirred at 25 oC for 15 min before it was filtered through celite and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were dried over sodium sulfate and concentrated, and the residue was purified by flash column chromatography (eluting with DCM/MeOH = 20:1) to afford Lurbinectedin (71 mg, 89%). [α]𝐷 20 = -45.0 (c = 1.0, CHCl3) 1H NMR (400 MHz, CDCl3) δ 7.61 (s, 1H), 7.13 (d, J = 8.8 Hz, 1H), 6.82 (d, J = 2.2 Hz, 1H), 6.74 (dd, J = 8.8, 2.4 Hz, 1H), 6.67 (s, 1H), 6.19 (d, J = 1.1 Hz, 1H), 5.99 (d, J = 1.1 Hz, 1H), 5.77 (br s, 1H), 5.20 (d, J = 11.3 Hz, 1H), 4.82 (s, 1H), 4.53–4.40 (m, 2H), 4.18–4.08 (m, 2H), 3.81 (s, 3H), 3.79 (s, 3H), 3.49 (d, J = 4.2 Hz, 1H), 3.24–3.13 (m, 2H), 3.01 (d, J = 17.9 Hz, 1H), 2.88–2.79 (m, 2H), 2.63 (dt, J = 15.0, 4.9 Hz, 1H), 2.56–2.47 (m, 2H), 2.37 (s, 3H), 2.32–2.27 (m, 1H), 2.26 (s, 3H), 2.19 (s, 3H), 2.05 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 171.4, 168.8, 153.8, 147.9, 145.5, 142.9, 141.1, 140.7, 131.8, 131.3, 130.7, 129.1, 127.3, 121.4, 121.0, 118.2, 115.6, 112.9, 111.9, 111.7, 110.0, 101.8, 100.4, 82.0, 62.4, 61.9, 60.4, 57.8, 57.5, 56.0, 55.8, 55.0, 42.2, 41.3, 39.8, 39.3, 29.3, 23.6, 20.6, 15.9, 9.7; HRMS (ESI) m/z calcd. for C41H44N4O10S [M – OH]+ 767.2745, found 767.2742.
References
- ^ Jump up to:a b c d e “Zepzelca- lurbinectedin injection, powder, lyophilized, for solution”. DailyMed. 15 June 2020. Retrieved 24 September 2020.
- ^ Jump up to:a b c d “Jazz Pharmaceuticals Announces U.S. FDA Accelerated Approval of Zepzelca (lurbinectedin) for the Treatment of Metastatic Small Cell Lung Cancer” (Press release). Jazz Pharmaceuticals. 15 June 2020. Retrieved 15 June 2020 – via PR Newswire.
- ^ Jump up to:a b c d e f g “FDA grants accelerated approval to lurbinectedin for metastatic small”. U.S. Food and Drug Administration (FDA). 15 June 2020. Retrieved 16 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “Lurbinectedin”. National Cancer Institute. Retrieved 15 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “Zepzelca: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 15 June 2020.
- ^ Jump up to:a b c d “Drug Trials Snapshots: Zepzelca”. U.S. Food and Drug Administration (FDA). 15 June 2020. Retrieved 28 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ Takahashi, Ryoko; Mabuchi, Seiji; Kawano, Mahiru; Sasano, Tomoyuki; Matsumoto, Yuri; Kuroda, Hiromasa; Kozasa, Katsumi; Hashimoto, Kae; Sawada, Kenjiro; Kimura, Tadashi (17 March 2016). “Preclinical Investigations of PM01183 (Lurbinectedin) as a Single Agent or in Combination with Other Anticancer Agents for Clear Cell Carcinoma of the Ovary”. PLOS ONE. 11 (3): e0151050. Bibcode:2016PLoSO..1151050T. doi:10.1371/journal.pone.0151050. PMC 4795692. PMID 26986199.
- ^ Total synthesis of marine antitumor agents trabectedin and lurbinectedin | https://www.sciencedaily.com/releases/2019/02/190219111659.htm
- ^ A Scalable Total Synthesis of the Antitumor Agents Et‐743 and Lurbinectedin | https://onlinelibrary.wiley.com/doi/full/10.1002/anie.201900035
- ^ PharmaMar presentation of Lurbinectedin’s Mechanism of Action Lurbinectedin Mechanisim of Action | https://www.youtube.com/watch?v=8daELhxAXcQ
- ^ Qian BZ, Pollard JW (April 2010). “Macrophage diversity enhances tumor progression and metastasis”. Cell. 141 (1): 39–51. doi:10.1016/j.cell.2010.03.014. PMC 4994190. PMID 20371344.
- ^ Engblom C, Pfirschke C, Pittet MJ (July 2016). “The role of myeloid cells in cancer therapies”. Nature Reviews. Cancer. 16 (7): 447–62. doi:10.1038/nrc.2016.54. PMID 27339708. S2CID 21924175.
- ^ “Lurbinectedin Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 1 August 2018. Retrieved 16 June 2020.
- ^ Jump up to:a b Paz-Ares, Luis G.; Trigo Perez, Jose Manuel; Besse, Benjamin; Moreno, Victor; Lopez, Rafael; Sala, Maria Angeles; Ponce Aix, Santiago; Fernandez, Cristian Marcelo; Siguero, Mariano; Kahatt, Carmen Maria; Zeaiter, Ali Hassan; Zaman, Khalil; Boni, Valentina; Arrondeau, Jennifer; Martinez Aguillo, Maite; Delord, Jean-Pierre; Awada, Ahmad; Kristeleit, Rebecca Sophie; Olmedo Garcia, Maria Eugenia; Subbiah, Vivek (20 May 2019). “Efficacy and safety profile of lurbinectedin in second-line SCLC patients: Results from a phase II single-agent trial”. Journal of Clinical Oncology. 37 (15_suppl): 8506. doi:10.1200/JCO.2019.37.15_suppl.8506.
- ^ Jump up to:a b Calvo, E.; Moreno, V.; Flynn, M.; Holgado, E.; Olmedo, M.E.; Lopez Criado, M.P.; Kahatt, C.; Lopez-Vilariño, J.A.; Siguero, M.; Fernandez-Teruel, C.; Cullell-Young, M.; Soto Matos-Pita, A.; Forster, M. (October 2017). “Antitumor activity of lurbinectedin (PM01183) and doxorubicin in relapsed small-cell lung cancer: results from a phase I study”. Annals of Oncology. 28 (10): 2559–2566. doi:10.1093/annonc/mdx357. PMC 5834091. PMID 28961837. Lay summary.
- ^ Farago, Anna F; Drapkin, Benjamin J; Lopez-Vilarino de Ramos, Jose Antonio; Galmarini, Carlos M; Núñez, Rafael; Kahatt, Carmen; Paz-Ares, Luis (January 2019). “ATLANTIS: a Phase III study of lurbinectedin/doxorubicin versus topotecan or cyclophosphamide/doxorubicin/vincristine in patients with small-cell lung cancer who have failed one prior platinum-containing line”. Future Oncology. 15 (3): 231–239. doi:10.2217/fon-2018-0597. PMC 6331752. PMID 30362375.
External links
- “Lurbinectedin”. Drug Information Portal. U.S. National Library of Medicine.
- “Lurbinectedin”. NCI Dictionary of Cancer Terms. National Cancer Institute.
- Clinical trial number NCT02454972 for “Clinical Trial of Lurbinectedin (PM01183) in Selected Advanced Solid Tumors” at ClinicalTrials.gov
FDA grants accelerated approval to lurbinectedin for metastatic small cell lung cancer
On June 15, 2020, the Food and Drug Administration granted accelerated approval to lurbinectedin(ZEPZELCA, Pharma Mar S.A.) for adult patients with metastatic small cell lung cancer (SCLC) with disease progression on or after platinum-based chemotherapy.
Efficacy was demonstrated in the PM1183-B-005-14 trial (Study B-005; NCT02454972), a multicenter open-label, multi-cohort study enrolling 105 patients with metastatic SCLC who had disease progression on or after platinum-based chemotherapy. Patients received lurbinectedin 3.2 mg/m2 by intravenous infusion every 21 days until disease progression or unacceptable toxicity.
The main efficacy outcome measures were confirmed overall response rate (ORR) determined by investigator assessment using RECIST 1.1 and response duration. Among the 105 patients, the ORR was 35% (95% CI: 26%, 45%), with a median response duration of 5.3 months (95% CI: 4.1, 6.4). The ORR as per independent review committee was 30% (95% CI: 22%, 40%) with a median response duration of 5.1 months (95% CI: 4.9, 6.4).
The most common adverse reactions (≥20%), including laboratory abnormalities, were myelosuppression, fatigue, increased creatinine, increased alanine aminotransferase, increased glucose, nausea, decreased appetite, musculoskeletal pain, decreased albumin, constipation, dyspnea, decreased sodium, increased aspartate aminotransferase, vomiting, cough, decreased magnesium and diarrhea.
The recommended lurbinectedin dose is 3.2 mg/m2 every 21 days.
View full prescribing information for ZEPZELCA.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this application, a modified Project Orbis was undertaken because of the timing of submission to other regulatory agencies. FDA is collaborating with the Australian Therapeutic Goods Administration (TGA). FDA approved this application 2 months ahead of the goal date. The review is ongoing for the Australian TGA.
FDA granted lurbinectedin orphan drug designation for the treatment of SCLC and priority review to this application. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
REFERENCES
1: Calvo E, Moreno V, Flynn M, Holgado E, Olmedo ME, Lopez Criado MP, Kahatt C, Lopez-Vilariño JA, Siguero M, Fernandez-Teruel C, Cullell-Young M, Soto Matos-Pita A, Forster M. Antitumor activity of lurbinectedin (PM01183) and doxorubicin in relapsed small-cell lung cancer: results from a phase I study. Ann Oncol. 2017 Oct 1;28(10):2559-2566. doi: 10.1093/annonc/mdx357. PubMed PMID: 28961837.
2: Erba E, Romano M, Gobbi M, Zucchetti M, Ferrari M, Matteo C, Panini N, Colmegna B, Caratti G, Porcu L, Fruscio R, Perlangeli MV, Mezzanzanica D, Lorusso D, Raspagliesi F, D’Incalci M. Ascites interferes with the activity of lurbinectedin and trabectedin: Potential role of their binding to alpha 1-acid glycoprotein. Biochem Pharmacol. 2017 Nov 15;144:52-62. doi: 10.1016/j.bcp.2017.08.001. Epub 2017 Aug 4. PubMed PMID: 28782526.
3: Belgiovine C, Bello E, Liguori M, Craparotta I, Mannarino L, Paracchini L, Beltrame L, Marchini S, Galmarini CM, Mantovani A, Frapolli R, Allavena P, D’Incalci M. Lurbinectedin reduces tumour-associated macrophages and the inflammatory tumour microenvironment in preclinical models. Br J Cancer. 2017 Aug 22;117(5):628-638. doi: 10.1038/bjc.2017.205. Epub 2017 Jul 6. PubMed PMID: 28683469; PubMed Central PMCID: PMC5572168.
4: Jimeno A, Sharma MR, Szyldergemajn S, Gore L, Geary D, Diamond JR, Fernandez Teruel C, Soto Matos-Pita A, Iglesias JL, Cullell-Young M, Ratain MJ. Phase I study of lurbinectedin, a synthetic tetrahydroisoquinoline that inhibits activated transcription, induces DNA single- and double-strand breaks, on a weekly × 2 every-3-week schedule. Invest New Drugs. 2017 Aug;35(4):471-477. doi: 10.1007/s10637-017-0427-2. Epub 2017 Jan 20. PubMed PMID: 28105566.
5: Paz-Ares L, Forster M, Boni V, Szyldergemajn S, Corral J, Turnbull S, Cubillo A, Teruel CF, Calderero IL, Siguero M, Bohan P, Calvo E. Phase I clinical and pharmacokinetic study of PM01183 (a tetrahydroisoquinoline, Lurbinectedin) in combination with gemcitabine in patients with advanced solid tumors. Invest New Drugs. 2017 Apr;35(2):198-206. doi: 10.1007/s10637-016-0410-3. Epub 2016 Nov 21. PubMed PMID: 27873130.
6: Harlow ML, Maloney N, Roland J, Guillen Navarro MJ, Easton MK, Kitchen-Goosen SM, Boguslawski EA, Madaj ZB, Johnson BK, Bowman MJ, D’Incalci M, Winn ME, Turner L, Hostetter G, Galmarini CM, Aviles PM, Grohar PJ. Lurbinectedin Inactivates the Ewing Sarcoma Oncoprotein EWS-FLI1 by Redistributing It within the Nucleus. Cancer Res. 2016 Nov 15;76(22):6657-6668. doi: 10.1158/0008-5472.CAN-16-0568. Epub 2016 Oct 3. PubMed PMID: 27697767; PubMed Central PMCID: PMC5567825.
7: Céspedes MV, Guillén MJ, López-Casas PP, Sarno F, Gallardo A, Álamo P, Cuevas C, Hidalgo M, Galmarini CM, Allavena P, Avilés P, Mangues R. Lurbinectedin induces depletion of tumor-associated macrophages, an essential component of its in vivo synergism with gemcitabine, in pancreatic adenocarcinoma mouse models. Dis Model Mech. 2016 Dec 1;9(12):1461-1471. Epub 2016 Oct 20. PubMed PMID: 27780828; PubMed Central PMCID: PMC5200894.
8: Santamaría Nuñez G, Robles CM, Giraudon C, Martínez-Leal JF, Compe E, Coin F, Aviles P, Galmarini CM, Egly JM. Lurbinectedin Specifically Triggers the Degradation of Phosphorylated RNA Polymerase II and the Formation of DNA Breaks in Cancer Cells. Mol Cancer Ther. 2016 Oct;15(10):2399-2412. Epub 2016 Sep 14. PubMed PMID: 27630271.
9: Metaxas Y, Cathomas R, Mark M, von Moos R. Combination of cisplatin and lurbinectedin as palliative chemotherapy in progressive malignant pleural mesothelioma: Report of two cases. Lung Cancer. 2016 Dec;102:136-138. doi: 10.1016/j.lungcan.2016.07.012. Epub 2016 Jul 14. PubMed PMID: 27440191.
10: Lima M, Bouzid H, Soares DG, Selle F, Morel C, Galmarini CM, Henriques JA, Larsen AK, Escargueil AE. Dual inhibition of ATR and ATM potentiates the activity of trabectedin and lurbinectedin by perturbing the DNA damage response and homologous recombination repair. Oncotarget. 2016 May 3;7(18):25885-901. doi: 10.18632/oncotarget.8292. PubMed PMID: 27029031; PubMed Central PMCID: PMC5041952.
11: Takahashi R, Mabuchi S, Kawano M, Sasano T, Matsumoto Y, Kuroda H, Kozasa K, Hashimoto K, Sawada K, Kimura T. Preclinical Investigations of PM01183 (Lurbinectedin) as a Single Agent or in Combination with Other Anticancer Agents for Clear Cell Carcinoma of the Ovary. PLoS One. 2016 Mar 17;11(3):e0151050. doi: 10.1371/journal.pone.0151050. eCollection 2016. PubMed PMID: 26986199; PubMed Central PMCID: PMC4795692.
12: Pernice T, Bishop AG, Guillen MJ, Cuevas C, Aviles P. Development of a liquid chromatography/tandem mass spectrometry assay for the quantification of PM01183 (lurbinectedin), a novel antineoplastic agent, in mouse, rat, dog, Cynomolgus monkey and mini-pig plasma. J Pharm Biomed Anal. 2016 May 10;123:37-41. doi: 10.1016/j.jpba.2016.01.043. Epub 2016 Jan 21. PubMed PMID: 26871278.
13: Elez ME, Tabernero J, Geary D, Macarulla T, Kang SP, Kahatt C, Pita AS, Teruel CF, Siguero M, Cullell-Young M, Szyldergemajn S, Ratain MJ. First-in-human phase I study of Lurbinectedin (PM01183) in patients with advanced solid tumors. Clin Cancer Res. 2014 Apr 15;20(8):2205-14. doi: 10.1158/1078-0432.CCR-13-1880. Epub 2014 Feb 21. PubMed PMID: 24563480.
14: Romano M, Frapolli R, Zangarini M, Bello E, Porcu L, Galmarini CM, García-Fernández LF, Cuevas C, Allavena P, Erba E, D’Incalci M. Comparison of in vitro and in vivo biological effects of trabectedin, lurbinectedin (PM01183) and Zalypsis® (PM00104). Int J Cancer. 2013 Nov;133(9):2024-33. doi: 10.1002/ijc.28213. Epub 2013 May 25. PubMed PMID: 23588839.
15: Vidal A, Muñoz C, Guillén MJ, Moretó J, Puertas S, Martínez-Iniesta M, Figueras A, Padullés L, García-Rodriguez FJ, Berdiel-Acer M, Pujana MA, Salazar R, Gil-Martin M, Martí L, Ponce J, Molleví DG, Capella G, Condom E, Viñals F, Huertas D, Cuevas C, Esteller M, Avilés P, Villanueva A. Lurbinectedin (PM01183), a new DNA minor groove binder, inhibits growth of orthotopic primary graft of cisplatin-resistant epithelial ovarian cancer. Clin Cancer Res. 2012 Oct 1;18(19):5399-411. doi: 10.1158/1078-0432.CCR-12-1513. Epub 2012 Aug 15. PubMed PMID: 22896654.
| Clinical data | |
|---|---|
| Pronunciation | LOOR-bih-NEK-teh-din |
| Trade names | Zepzelca |
| Other names | PM-01183 |
| AHFS/Drugs.com | Professional Drug Facts |
| MedlinePlus | a620049 |
| License data | US DailyMed: Lurbinectedin |
| Pregnancy category | US: N (Not classified yet) |
| Routes of administration | Intravenous |
| Drug class | Antineoplastic agent |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 497871-47-3 |
| PubChem CID | 57327016 |
| DrugBank | 12674 |
| ChemSpider | 32701856 |
| UNII | 2CN60TN6ZS |
| KEGG | D11644 |
| ChEMBL | ChEMBL4297516 |
| CompTox Dashboard (EPA) | DTXSID30198065 |
| Chemical and physical data | |
| Formula | C41H44N4O10S |
| Molar mass | 784.88 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]CC1=CC2=C([C@@H]3[C@@H]4[C@H]5C6=C(C(=C7C(=C6[C@@H](N4[C@H]([C@H](C2)N3C)O)COC(=O)[C@@]8(CS5)C9=C(CCN8)C2=C(N9)C=CC(=C2)OC)OCO7)C)OC(=O)C)C(=C1OC)O | |
| InChI[hide]InChI=1S/C41H44N4O10S/c1-17-11-20-12-25-39(48)45-26-14-52-40(49)41(38-22(9-10-42-41)23-13-21(50-5)7-8-24(23)43-38)15-56-37(31(45)30(44(25)4)27(20)32(47)33(17)51-6)29-28(26)36-35(53-16-54-36)18(2)34(29)55-19(3)46/h7-8,11,13,25-26,30-31,37,39,42-43,47-48H,9-10,12,14-16H2,1-6H3/t25-,26-,30+,31+,37+,39-,41+/m0/s1Key:YDDMIZRDDREKEP-HWTBNCOESA-N |
//////////lurbinectedin, FDA 2020, 2020 APPROVALS, ORPHAN, priority review , ZEPZELCA, Pharma Mar, PM-1183, PM 1183, PM 01183, лурбинектедин , لوربينيكتيدين , 芦比替定
Cc1cc2c(c(c1OC)O)[C@@H]3[C@@H]4[C@H]5c6c(c7c(c(c6OC(=O)C)C)OCO7)[C@@H](N4[C@H]([C@H](C2)N3C)O)COC(=O)[C@@]8(CS5)c9c(c1cc(ccc1[nH]9)OC)CCN8
Naxitamab

(Heavy chain)
QVQLVESGPG VVQPGRSLRI SCAVSGFSVT NYGVHWVRQP PGKGLEWLGV IWAGGITNYN
SAFMSRLTIS KDNSKNTVYL QMNSLRAEDT AMYYCASRGG HYGYALDYWG QGTLVTVSSA
STKGPSVFPL APSSKSTSGG TAALGCLVKD YFPEPVTVSW NSGALTSGVH TFPAVLQSSG
LYSLSSVVTV PSSSLGTQTY ICNVNHKPSN TKVDKRVEPK SCDKTHTCPP CPAPELLGGP
SVFLFPPKPK DTLMISRTPE VTCVVVDVSH EDPEVKFNWY VDGVEVHNAK TKPREEQYNS
TYRVVSVLTV LHQDWLNGKE YKCKVSNKAL PAPIEKTISK AKGQPREPQV YTLPPSRDEL
TKNQVSLTCL VKGFYPSDIA VEWESNGQPE NNYKTTPPVL DSDGSFFLYS KLTVDKSRWQ
QGNVFSCSVM HEALHNHYTQ KSLSLSPGK
(Light chain)
EIVMTQTPAT LSVSAGERVT ITCKASQSVS NDVTWYQQKP GQAPRLLIYS ASNRYSGVPA
RFSGSGYGTE FTFTISSVQS EDFAVYFCQQ DYSSFGQGTK LEIKRTVAAP SVFIFPPSDE
QLKSGTASVV CLLNNFYPRE AKVQWKVDNA LQSGNSQESV TEQDSKDSTY SLSSTLTLSK
ADYEKHKVYA CEVTHQGLSS PVTKSFNRGE C
(Disulfide bridge: H22-H95, H146-H202, H222-L211, H228-H’228, H231-H’231, H263-H323, H369-H427, H’22-H’95, H’146-H’202, H’222-L’211, H’263-H’323, H’369-H’427, L23-L88, L131-L191, L’23-L’88, L’131-L’191)
Naxitamab
ナキシタマブ;
Antineoplastic, Anti-GD2 antibody
| Formula | C6414H9910N1718O1996S44 |
|---|---|
| CAS | 1879925-92-4 |
| Mol weight | 144434.4882 |
FDA APPROVED 2020/11/25, Danyelza
FDA grants accelerated approval to naxitamab for high-risk neuroblastoma in bone or bone marrow
On November 25, 2020, the Food and Drug Administration granted accelerated approval to naxitamab (DANYELZA, Y-mAbs Therapeutics, Inc.) in combination with granulocyte-macrophage colony-stimulating factor (GM-CSF) for pediatric patients one year of age and older and adult patients with relapsed or refractory high-risk neuroblastoma in the bone or bone marrow demonstrating a partial response, minor response, or stable disease to prior therapy.
Efficacy was evaluated in patients with relapsed or refractory neuroblastoma in the bone or bone marrow enrolled in two single-arm, open-label trials: Study 201 (NCT 03363373) and Study 12-230 (NCT 01757626). Patients with progressive disease following their most recent therapy were excluded. Patients received 3 mg/kg naxitamab administered as an intravenous infusion on days 1, 3, and 5 of each 4-week cycle in combination with GM-CSF subcutaneously at 250 µg/m2/day on days -4 to 0 and at 500 µg/m2/day on days 1 to 5. At the investigator’s discretion, patients were permitted to receive pre-planned radiation to the primary disease site in Study 201 and radiation therapy to non-target bony lesions or soft tissue disease in Study 12-230.
The main efficacy outcome measures were confirmed overall response rate (ORR) per the revised International Neuroblastoma Response Criteria (INRC) and duration of response (DOR). Among 22 patients treated in the multicenter Study 201, the ORR was 45% (95% CI: 24%, 68%) and 30% of responders had a DOR greater or equal to 6 months. Among 38 patients treated in the single-center Study 12-230, the ORR was 34% (95% CI: 20%, 51%) with 23% of patients having a DOR greater or equal to 6 months. For both trials, responses were observed in either the bone, bone marrow or both.
The prescribing information contains a Boxed Warning stating that naxitamab can cause serious infusion-related reactions and neurotoxicity, including severe neuropathic pain, transverse myelitis and reversible posterior leukoencephalopathy syndrome (RPLS). To mitigate these risks, patients should receive premedication prior to each naxitamab infusion and be closely monitored during and for at least two hours following completion of each infusion.
The most common adverse reactions (incidence ≥25% in either trial) in patients receiving naxitamab were infusion-related reactions, pain, tachycardia, vomiting, cough, nausea, diarrhea, decreased appetite, hypertension, fatigue, erythema multiforme, peripheral neuropathy, urticaria, pyrexia, headache, injection site reaction, edema, anxiety, localized edema, and irritability. The most common Grade 3 or 4 laboratory abnormalities (≥5% in either trial) were decreased lymphocytes, decreased neutrophils, decreased hemoglobin, decreased platelet count, decreased potassium, increased alanine aminotransferase, decreased glucose, decreased calcium, decreased albumin, decreased sodium and decreased phosphate.
The recommended naxitamab dose is 3 mg/kg/day (up to 150 mg/day) on days 1, 3, and 5 of each treatment cycle, administered after dilution as an intravenous infusion in combination with GM-CSF, subcutaneously at 250 µg/m2/day on days -4 to 0 and at 500 µg/m2/day on days 1 to 5. Treatment cycles are repeated every 4 to 8 weeks.
View full prescribing information for DANYELZA. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761171lbl.pdf
This review used the Real-Time Oncology Review (RTOR) pilot program and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted accelerated approval based on overall response rate and duration of response. Continued approval may be contingent upon verification and description of clinical benefit in confirmatory trials.
This application was granted priority review, breakthrough therapy, and orphan drug designation. A priority review voucher was issued for this rare pediatric disease product application. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
////////////Naxitamab, priority review, breakthrough therapy, orphan drug, FDA 2020, 2020 APPROVALS, Danyelza, MONOCLONAL ANTIBODY, PEPTIDE, ナキシタマブ,
Ansuvimab-zykl

Ansuvimab-zykl
FDA APPROVED, 12/21/2020, EBANGA
To treat ebola
https://www.fda.gov/drugs/drug-safety-and-availability/fda-approves-treatment-ebola-virus
The U.S. Food and Drug Administration approved Ebanga (Ansuvimab-zykl), a human monoclonal antibody, for the treatment for Zaire ebolavirus (Ebolavirus) infection in adults and children. Ebanga blocks binding of the virus to the cell receptor, preventing its entry into the cell.
Zaire ebolavirus is one of four Ebolavirus species that can cause a potentially fatal human disease. It is transmitted through blood, body fluids, and tissues of infected people or wild animals, and through surfaces and materials, such as bedding and clothing, contaminated with these fluids. Individuals who care for people with the disease, including health care workers who do not use correct infection control precautions, are at the highest risk for infection.
During an Ebola outbreak in the Democratic Republic of the Congo (DRC) in 2018-2019, Ebanga was evaluated in a clinical trial (the PALM trial). The PALM trial was led by the U.S. National Institutes of Health and the DRC’s Institut National de Recherche Biomédicale with contributions from several other international organizations and agencies.
In the PALM trial, the safety and efficacy of Ebanga was evaluated in a multi-center, open-label, randomized controlled trial. 174 participants (120 adults and 54 pediatric patients) with confirmed Ebolavirus infection received Ebanga intravenously as a single 50 mg/kg infusion and 168 participants (135 adults and 33 pediatric patients) received an investigational control. The primary efficacy endpoint was 28-day mortality. The primary analysis population was all patients who were randomized and concurrently eligible to receive either Ebanga or the investigational control during the same time period of the trial. Of the 174 patients who received Ebanga, 35.1% died after 28 days, compared to 49.4% of the 168 patients who received a control.
The most common symptoms experienced while receiving Ebanga include: fever, tachycardia (fast heart rate), diarrhea, vomiting, hypotension (low blood pressure), tachypnea (fast breathing) and chills; however, these are also common symptoms of Ebolavirus infection. Hypersensitivity, including infusion-related events, can occur in patients taking Ebanga, and treatment should be discontinued in the event of a hypersensitivity reaction.
Patients who receive Ebanga should avoid the concurrent administration of a live virus vaccine against Ebolavirus. There is the potential for Ebanga to inhibit replication of a live vaccine virus and possibly reduce the efficacy of this vaccine.
Ebanga was granted an Orphan Drug designation, which provides incentives to assist and encourage drug development for rare diseases. Additionally, the agency granted Ebanga a Breakthrough Therapy designation.
FDA granted the approval to Ridgeback Biotherapeutics, LP.
Ansuvimab, sold under the brand name Ebanga, is a monoclonal antibody medication for the treatment of Zaire ebolavirus (Ebolavirus) infection.[1][2]
The most common symptoms include fever, tachycardia (fast heart rate), diarrhea, vomiting, hypotension (low blood pressure), tachypnea (fast breathing) and chills; however, these are also common symptoms of Ebolavirus infection.[1]
Ansuvimab was approved for medical use in the United States in December 2020.[1][2]
Chemistry
The drug is composed of a single monoclonal antibody (mAb) and was initially isolated from immortalized B-cells that were obtained from a survivor of the 1995 outbreak of Ebola virus disease in Kikwit, Democratic Republic of Congo.[3] In work supported by the United States National Institutes of Health and the Defense Advanced Projects Agency, the heavy and light chain sequences of ansuvimab mAb was cloned into CHO cell lines and initial production runs were produced by Cook Phamica d.b.a. Catalent under contract of Medimmune.[4][5]
Mechanism of action
Neutralization
Ansuvimab is a monoclonal antibody therapy that is infused intravenously into patients with Ebola virus disease. Ansuvimab is a neutralizing antibody,[3] meaning it binds to a protein on the surface of Ebola virus that is required to infect cells. Specifically, ansuvimab neutralizes infection by binding to a region of the Ebola virus envelope glycoprotein that, in the absence of ansuvimab, would interact with virus’s cell receptor protein, Niemann-Pick C1 (NPC1).[6][7][8] This “competition” by ansuvimab prevents Ebola virus from binding to NPC1 and “neutralizes” the virus’s ability to infect the targeted cell.[6]
Effector function
Antibodies have antigen-binding fragment (Fab) regions and constant fragment (Fc) regions. The Neutralization of virus infection occurs when the Fab regions of antibodies binds to virus antigen(s) in a manner that blocks infection. Antibodies are also able to “kill” virus particles directly and/or kill infected cells using antibody-mediated “effector functions” such as opsonization, complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity and antibody-dependent phagocytosis. These effector functions are contained in the Fc region of antibodies, but is also dependent on binding of the Fab region to antigen. Effector functions also require the use of complement proteins in serum or Fc-receptor on cell membranes. Ansuvimab has been found to be capable of killing cells by antibody-dependent cell-mediated cytotoxicity.[3] Other functional killing tests have not been performed.
History
Ansuvimab is a monoclonal antibody that is being evaluated as a treatment for Ebola virus disease.[9] Its discovery was led by the laboratory of Nancy Sullivan at the United States National Institute of Health Vaccine Research Center and J. J. Muyembe-Tamfum from the Institut National pour la Recherche Biomedicale (INRB) in the Democratic Republic of Congo, working in collaboration with the Institute of Biomedical Research and the United States Army Medical Research Institute of Infectious Diseases.[3][10] Ansuvimab was isolated from the blood of a survivor of the 1995 outbreak of Ebola virus disease in Kikwit, Democratic Republic of Congo roughly ten years later.[3]
In 2018, a Phase 1 clinical trial of ansuvimab was conducted by Martin Gaudinski within the Vaccine Research Center Clinical Trials Program that is led by Julie E. Ledgerwood.[5][4][11] Ansuvimab is also being evaluated during the 2018 North Kivu Ebola outbreak.[12]
Ansuvimab has also shown success with lowering the mortality rate from ~70% to about 34%. In August 2019, Congolese health authorities, the World Health Organization, and the U.S. National Institutes of Health promoted the use of ansuvimab, alongside REGN-EB3, a similar Regeneron-produced monoclonal antibody treatment, over other treatments yielding higher mortality rates, after ending clinical trials during the outbreak.[13][14]
Discovery
A 2016 paper describes the efforts of how ansuvimab was originally developed as part of research efforts lead by Dr. Nancy Sullivan at the United States National Institute of Health Vaccine Research Center and Dr. J. J. Muyembe-Tamfum from the Institut National de Recherche Biomedicale (INRB) in the Democratic Republic of Congo.[3][10] This collaborative effort also involved researchers from Institute of Biomedical Research and the United States Army Medical Research Institute of Infectious Diseases.[3][10] A survivor from the 1995 outbreak of Ebola virus disease in Kikwit, Democratic Republic of Congo donated blood to the project that began roughly ten years after they had recovered.[3] Memory B cells isolated from the survivor’s blood were immortalized, cultured and screened for their ability to produce monoclonal antibodies that reacted with the glycoprotein of Ebola virus. Ansuvimab was identified from one of these cultures and the antibody heavy and light chain gene sequences were sequenced from the cells.[3] These sequences were then cloned into recombinant DNA plasmids and purified antibody protein for initial studies was produced in cells derived from HEK 293 cells.[3]
Ansuvimab and mAb100 combination
In an experiment described in the 2016 paper, rhesus macaques were infected with Ebola virus and treated with a combination of ansuvimab and another antibody isolated from the same subject, mAb100. Three doses of the combination were given once a day starting 1 day after the animals were infected. The control animal died and the treated animals all survived.[3]
Ansuvimab monotherapy
In a second experiment described in the 2016 paper, rhesus macaques were infected with Ebola virus and only treated with ansuvimab. Three doses of ansuvimab were given once a day starting 1 day or 5 days after the animals were infected. The control animals died and the treated animals all survived.[3] Unpublished data referred to in a publication of the 2018 Phase I clinical trial results of ansuvimab, reported that a single infusion of ansuvimab provided full protection of rhesus macaques and was the basis of the dosing used for human studies.[5][4]
Development
Ansuvimab was developed by the Vaccine Research Center with support of the United States National Institutes of Health and the Defense Advanced Projects Agency. The heavy and light chain sequences of ansuvimab mAb were cloned into CHO cell lines to enable large-scale production of antibody product for use in humans.[4][5]
Human safety testing
In early 2018,[9] a Phase 1 clinical trial of ansuvimab’s safety, tolerability and pharmacokinetics was conducted by Dr. Martin Gaudinski within the Vaccine Research Center Clinical Trials Program that is led by Dr. Julie E. Ledgerwood.[5][4][11] The study was performed in the United States at the NIH Clinical Center and tested single dose infusions of ansuvimab infused over 30 minutes. The study showed that ansuvimab was safe, had minimal side effects and had a half-life of 24 days.[5][4]
Ridgeback Biotherapeutics
A license for ansuvimab was obtained by Ridgeback Biotherapeutics in 2018, from the National Institutes of Health–National Institute of Allergy and Infectious Diseases.[15] Ansuvimab was given orphan drug status in May 2019 and March 2020.[16][17][18]
Experimental use in the Democratic Republic of Congo
During the 2018 Équateur province Ebola outbreak, ansuvimab was requested by the Democratic Republic of Congo (DRC) Ministry of Public Health. Ansuvimab was approved for compassionate use by the World Health Organization MEURI ethical protocol and at DRC ethics board. Ansuvimab was sent along with other therapeutic agents to the outbreak sites.[19][20][11] However, the outbreak came to a conclusion before any therapeutic agents were given to patients.[11]
Approximately one month following the conclusion of the Équateur province outbreak, a distinct outbreak was noted in Kivu in the DRC (2018–20 Kivu Ebola outbreak). Once again, ansuvimab received approval for compassionate use by WHO MEURI and DRC ethic boards and has been given to many patients under these protocols.[11] In November 2018, the Pamoja Tulinde Maisha (PALM [together save lives]) open-label randomized clinical control trial was begun at multiple treatment units testing ansuvimab, REGN-EB3 and remdesivir to ZMapp. Despite the difficulty of running a clinical trial in a conflict zone, investigators have enrolled 681 patients towards their goal of 725. An interim analysis by the Data Safety and Monitoring Board (DSMB) of the first 499 patient found that ansuvimab and REGN-EB3 were superior to the comparator ZMapp. Overall mortality of patients in the ZMapp and remdesivir groups were 49% and 53% compared to 34% and 29% for ansuvimab and REGN-EB3. When looking at patients who arrived early after disease symptoms appeared, survival was 89% for ansuvimab and 94% for REGN-EB3. While the study was not powered to determine whether there is any difference between REGN-EB3 and ansuvimab, the survival difference between those two therapies and ZMapp was significant. This led to the DSMB halting the study and PALM investigators dropping the remdesivir and ZMapp arms from the clinical trial. All patients in the outbreak who elect to participate in the trial will now be given either ansuvimab or REGN-EB3.[21][22][13][12]
In October 2020, the U.S. Food and Drug Administration (FDA) approved atoltivimab/maftivimab/odesivimab (Inmazeb, formerly REGN-EB3) with an indication for the treatment of infection caused by Zaire ebolavirus.[23]
FDA approves ansuvimab-zykl for Ebola virus infection
DECEMBER 21, 2020 BY JANICE REICHERThttps://www.antibodysociety.org/antibody-therapeutic/fda-approves-ansuvimab-zykl-for-ebola-virus-infection/embed/#?secret=zWW0Sr0BdW
On December 21, 2020, the US Food and Drug Administration approved Ebanga (ansuvimab-zykl) for the treatment for Zaire ebolavirus (Ebolavirus) infection in adults and children. Ebanga had been granted US Orphan Drug designation and Breakthrough Therapy designations. Ansuvimab is a human IgG1 monoclonal antibody that binds and neutralizes the virus.
The safety and efficacy of Ebanga were evaluated in the multi-center, open-label, randomized controlled PALM trial. In this study, 174 participants (120 adults and 54 pediatric patients) with confirmed Ebolavirus infection received Ebanga intravenously as a single 50 mg/kg infusion and 168 participants (135 adults and 33 pediatric patients) received an investigational control. The primary efficacy endpoint was 28-day mortality. Of the 174 patients who received Ebanga, 35.1% died after 28 days, compared to 49.4% of the 168 patients who received a control.
Ebanga is the 12th antibody therapeutic to be granted a first approval in the US or EU during 2020.
The Antibody Society maintains a comprehensive table of approved monoclonal antibody therapeutics and those in regulatory review in the EU or US. The table, which is located in the Web Resources section of the Society’s website, can be downloaded in Excel format.
References
- ^ Jump up to:a b c d “FDA Approves Treatment for Ebola Virus”. U.S. Food and Drug Administration. 21 December 2020. Retrieved 23 December 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “Ridgeback Biotherapeutics LP Announces the Approval of Ebanga for Ebola” (Press release). Ridgeback Biotherapeutics LP. 22 December 2020. Retrieved 23 December 2020– via Business Wire.
- ^ Jump up to:a b c d e f g h i j k l Corti D, Misasi J, Mulangu S, Stanley DA, Kanekiyo M, Wollen S, et al. (March 2016). “Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody”. Science. 351 (6279): 1339–42. Bibcode:2016Sci…351.1339C. doi:10.1126/science.aad5224. PMID 26917593.
- ^ Jump up to:a b c d e f Clinical trial number NCT03478891 for “Safety and Pharmacokinetics of a Human Monoclonal Antibody, VRC-EBOMAB092-00-AB (MAb114), Administered Intravenously to Healthy Adults” at ClinicalTrials.gov
- ^ Jump up to:a b c d e f Gaudinski MR, Coates EE, Novik L, Widge A, Houser KV, Burch E, et al. (March 2019). “Safety, tolerability, pharmacokinetics, and immunogenicity of the therapeutic monoclonal antibody ansuvimab targeting Ebola virus glycoprotein (VRC 608): an open-label phase 1 study”. Lancet. 393 (10174): 889–898. doi:10.1016/S0140-6736(19)30036-4. PMC 6436835. PMID 30686586.
- ^ Jump up to:a b Misasi J, Gilman MS, Kanekiyo M, Gui M, Cagigi A, Mulangu S, et al. (March 2016). “Structural and molecular basis for Ebola virus neutralization by protective human antibodies”. Science. 351 (6279): 1343–6. Bibcode:2016Sci…351.1343M. doi:10.1126/science.aad6117. PMC 5241105. PMID 26917592.
- ^ Côté M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, et al. (August 2011). “Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection”. Nature. 477 (7364): 344–8. Bibcode:2011Natur.477..344C. doi:10.1038/nature10380. PMC 3230319. PMID 21866101.
- ^ Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, et al. (August 2011). “Ebola virus entry requires the cholesterol transporter Niemann-Pick C1”. Nature. 477 (7364): 340–3. Bibcode:2011Natur.477..340C. doi:10.1038/nature10348. PMC 3175325. PMID 21866103.
- ^ Jump up to:a b “NIH begins testing Ebola treatment in early-stage trial”. National Institutes of Health (NIH). 2018-05-23. Retrieved 2018-10-15.
- ^ Jump up to:a b c Hayden EC (2016-02-26). “Ebola survivor’s blood holds promise of new treatment”. Nature. doi:10.1038/nature.2016.19440. ISSN 1476-4687.
- ^ Jump up to:a b c d e “NIH VideoCast – CC Grand Rounds: Response to an Outbreak: Ebola Virus Monoclonal Antibody (mAb114) Rapid Clinical Development”. videocast.nih.gov. Retrieved 2019-08-09.
- ^ Jump up to:a b Kingsley-Hall A. “Congo’s experimental mAb114 Ebola treatment appears successful: authorities | Central Africa”. http://www.theafricareport.com. Retrieved 2018-10-15.
- ^ Jump up to:a b McNeil DG (12 August 2019). “A Cure for Ebola? Two New Treatments Prove Highly Effective in Congo”. The New York Times. Retrieved 13 August 2019.
- ^ Molteni M (12 August 2019). “Ebola is Now Curable. Here’s How The New Treatments Work”. Wired. Retrieved 13 August 2019.
- ^ “Ridgeback Biotherapeutics LP announces licensing of mAb114, an experimental Ebola treatment, from the National Institute of Allergy and Infectious Diseases” (Press release). Ridgeback Biotherapeutics LP. Retrieved 2019-08-17 – via PR Newswire.
- ^ “Ansuvimab Orphan Drug Designations and Approvals”. accessdata.fda.gov. 8 May 2019. Retrieved 24 December 2020.
- ^ “Ansuvimab Orphan Drug Designations and Approvals”. accessdata.fda.gov. 30 March 2020. Retrieved 24 December 2020.
- ^ “Ridgeback Biotherapeutics LP Announces Orphan Drug Designation for mAb114”(Press release). Ridgeback Biotherapeutics LP. Retrieved 2019-08-17 – via PR Newswire.
- ^ Check Hayden, Erika (May 2018). “Experimental drugs poised for use in Ebola outbreak”. Nature. 557 (7706): 475–476. Bibcode:2018Natur.557..475C. doi:10.1038/d41586-018-05205-x. ISSN 0028-0836. PMID 29789732.
- ^ WHO: Consultation on Monitored Emergency Use of Unregistered and Investigational Interventions for Ebola virus Disease. https://www.who.int/emergencies/ebola/MEURI-Ebola.pdf
- ^ Mole B (2019-08-13). “Two Ebola drugs boost survival rates, according to early trial data”. Ars Technica. Retrieved 2019-08-17.
- ^ “Independent monitoring board recommends early termination of Ebola therapeutics trial in DRC because of favorable results with two of four candidates”. National Institutes of Health (NIH). 2019-08-12. Retrieved 2019-08-17.
- ^ “FDA Approves First Treatment for Ebola Virus”. U.S. Food and Drug Administration(FDA) (Press release). 14 October 2020. Retrieved 14 October 2020. This article incorporates text from this source, which is in the public domain.
External links
- “Ansuvimab”. Drug Information Portal. U.S. National Library of Medicine.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Zaire ebolavirus |
| Clinical data | |
| Trade names | Ebanga |
| Other names | Ansuvimab-zykl, mAb114 |
| License data | US DailyMed: Ansuvimab |
| Routes of administration | Intravenous |
| Drug class | Monoclonal antibody |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| CAS Number | 2375952-29-5 |
| DrugBank | DB16385 |
| UNII | TG8IQ19NG2 |
| KEGG | D11875 |
| Chemical and physical data | |
| Formula | C6368H9924N1724O1994S44 |
| Molar mass | 143950.15 g·mol−1 |
//////////Ansuvimab-zykl , EBANGA, FDA 2020, 2020 APPROVALS, MONOCLONAL ANTIBODY, Orphan Drug designation, , Breakthrough Therapy designation , Ridgeback Biotherapeutics,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
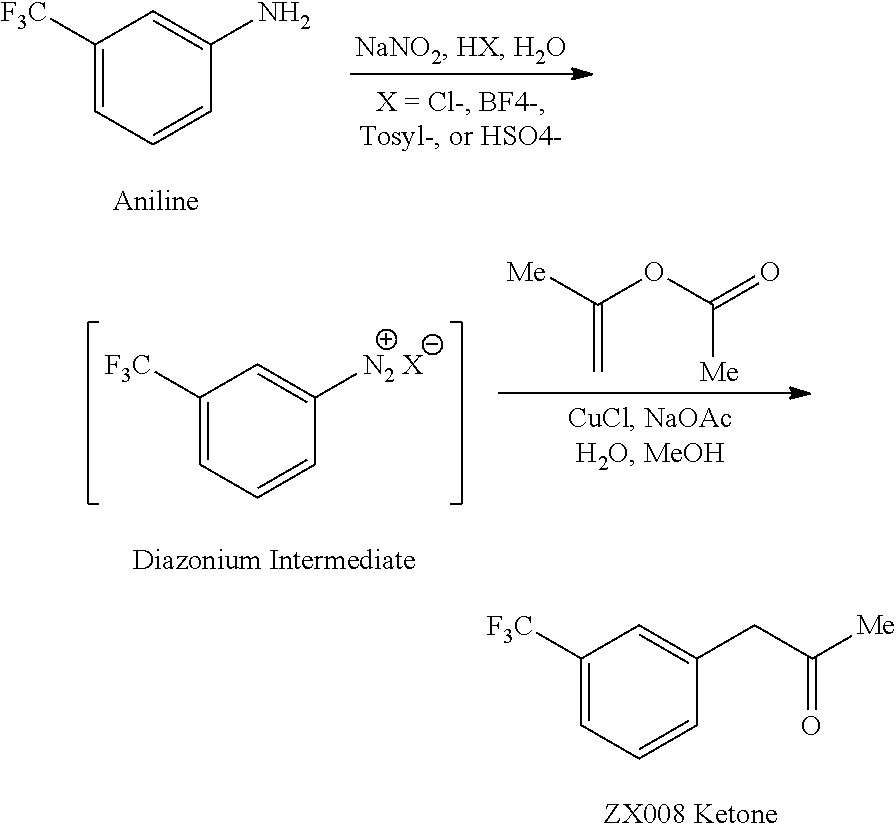{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}