
Home » Phase3 drugs
Category Archives: Phase3 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
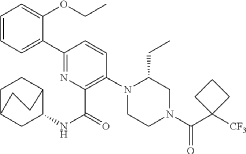
Atumelnant
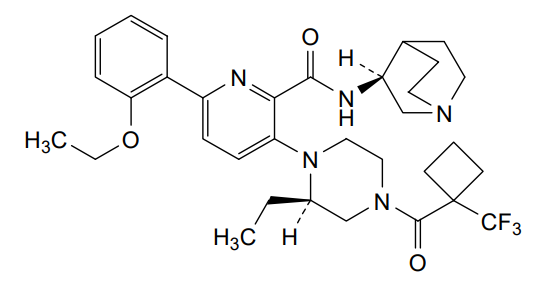
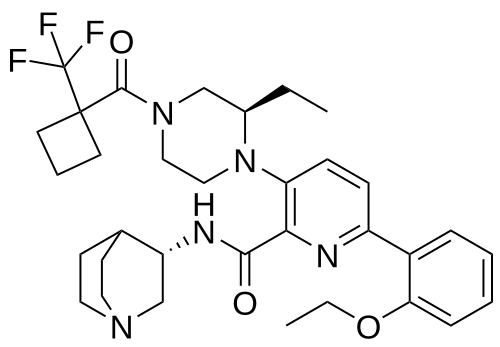
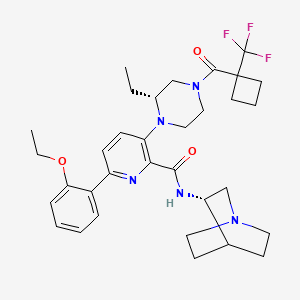
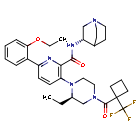
Atumelnant
CAS 2392970-97-5
MF C33H42F3N5O3 MW 613.7 g/mol
CRN04894, NR57FH6U1N
CRINETICS PHARMA, Orphan Drug Status, Congenital adrenal hyperplasia
N-[(3S)-1-azabicyclo[2.2.2]octan-3-yl]-6-(2-ethoxyphenyl)-3-[(2R)-2-ethyl-4-[1-(trifluoromethyl)cyclobutanecarbonyl]piperazin-1-yl]pyridine-2-carboxamide
N-[(3S)-1-azabicyclo[2.2.2]octan-3-yl]-6-(2-ethoxyphenyl)-3-{(2R)-2-ethyl-4-[1-(trifluoromethyl) cyclobutane-1-carbonyl]piperazin-1-yl}pyridine-2-carboxamide
Adrenocorticotropic hormone receptor antagonist
- OriginatorCrinetics Pharmaceuticals
- ClassAmides; Antineoplastics; Antisecretories; Benzene derivatives; Cyclobutanes; Ethers; Fluorocarbons; Ketones; Piperazines; Pyridines; Quinuclidines; Small molecules
- Mechanism of ActionMelanocortin type 2 receptor antagonists
- Orphan Drug StatusYes – Congenital adrenal hyperplasia
- Phase IICongenital adrenal hyperplasia; Cushing syndrome
- No development reportedEctopic ACTH syndrome
- 21 Aug 2025Atumelnant receives Orphan Drug status for Congenital adrenal hyperplasia in the US
- 07 Aug 2025Crinetics pharmaceuticals plans phase II/III clinical trial for Cushing’s disease in 1H 2026
- 08 May 2025Crinetics Pharmaceuticals plans the phase III CALM-CAH trial for Congenital adrenal hyperplasia (In adults) (PO), in the second half of 2025
Atumelnant (INNTooltip International Nonproprietary Name; developmental code name CRN04894) is an investigational new drug developed by Crinetics Pharmaceuticals for the treatment of adrenocorticotropic hormone (ACTH)-dependent endocrine disorders.[1] It is a selective antagonist of the melanocortin type 2 receptor (MC2R), also known as the ACTH receptor, which is primarily expressed in the adrenal glands.[1][2] The drug is orally active.[1] Atumelnant is being evaluated to treat conditions such as congenital adrenal hyperplasia (CAH) and ACTH-dependent Cushing’s syndrome caused for example by pituitary adenomas.[3]
Atumelnant is an orally bioavailable nonpeptide antagonist of the adrenocorticotropic hormone (ACTH) receptor (ACTHR; melanocortin receptor 2; MC2R), with potential steroid hormone production inhibitory activity. Upon oral administration, atumelnant competes with ACTH for receptor binding to MC2R in the adrenal cortex and inhibits ACTH signaling. This may inhibit the synthesis and secretion of steroid hormones. MC2R, a member of the melanocortin receptor subfamily of type 1 G protein-coupled receptors, plays a key role in adrenal steroidogenesis.
PAPER
Discovery of CRN04894: A Novel Potent Selective MC2R Antagonist
Publication Name: ACS Medicinal Chemistry Letters
Publication Date: 2024-03-19, PMCID: PMC11017392, PMID: 38628803
DOI: 10.1021/acsmedchemlett.3c00514
PATENTS
- Melanocortin subtype-2 receptor antagonists and uses thereofPublication Number: IL-279152-B2Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2024300920-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor antagonists and uses thereofPublication Number: IL-279152-B1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: JP-2024009837-APriority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: KR-102695210-B1Priority Date: 2018-06-05Grant Date: 2024-08-13
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2024109866-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: CN-112533904-BPriority Date: 2018-06-05Grant Date: 2024-10-29
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-10981894-B2Priority Date: 2018-06-05Grant Date: 2021-04-20
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2021002254-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2021238164-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-11566015-B2Priority Date: 2018-06-05Grant Date: 2023-01-31
- Melanocortin subtype-2 receptor (MC2R) antagonists and their usesPublication Number: JP-7359783-B2Priority Date: 2018-06-05Grant Date: 2023-10-11
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2020216415-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-10766877-B2Priority Date: 2018-06-05Grant Date: 2020-09-08
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: CN-112533904-APriority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: EP-3802500-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: KR-20210005995-APriority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists for the treatment of diseasePublication Number: CN-117043146-APriority Date: 2021-03-19
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-10562884-B2Priority Date: 2018-06-05Grant Date: 2020-02-18
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-10604507-B2Priority Date: 2018-06-05Grant Date: 2020-03-31
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2019367481-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2020010452-A1Priority Date: 2018-06-05
Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: US-2022313691-A1Priority Date: 2021-03-19 - Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: WO-2022197798-A1Priority Date: 2021-03-19
- Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: TW-202302108-APriority Date: 2021-03-19
- Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: AU-2022240609-A1Priority Date: 2021-03-19
- Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: EP-4308553-A1Priority Date: 2021-03-19
- Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of acth-dependent cushing’s syndromePublication Number: WO-2024211343-A1Priority Date: 2023-04-05
- Crystalline melanocortin subtype-2 receptor (mc2r) antagonistPublication Number: TW-202430167-APriority Date: 2022-12-16
- Crystalline melanocortin subtype-2 receptor (mc2r) antagonistPublication Number: US-2024208963-A1Priority Date: 2022-12-16
- Crystalline melanocortin subtype-2 receptor (mc2r) antagonistPublication Number: WO-2024130091-A1Priority Date: 2022-12-16
- Treatment of congenital adrenal hyperplasia and polycystic ovary syndromePublication Number: WO-2023163945-A1Priority Date: 2022-02-2
SYN
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US278278493&_cid=P22-MFXDN2-76849-1
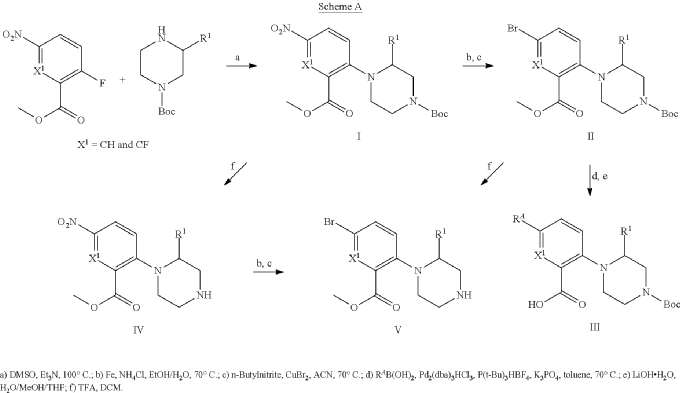
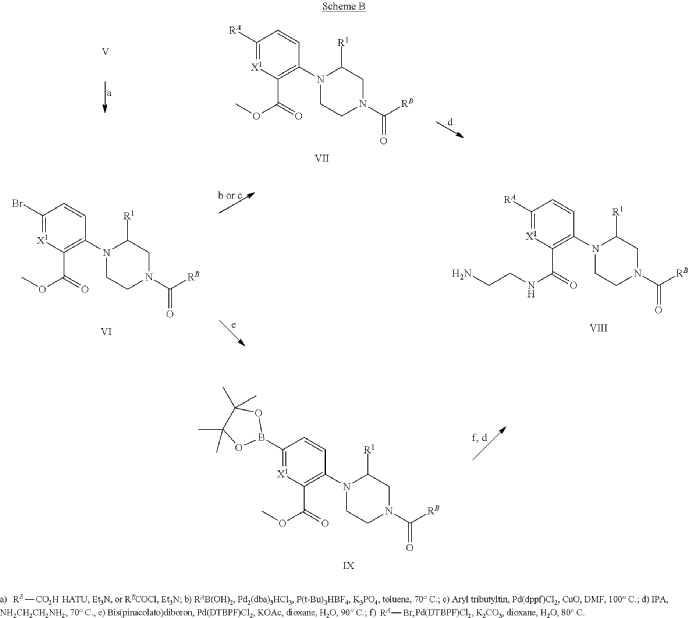
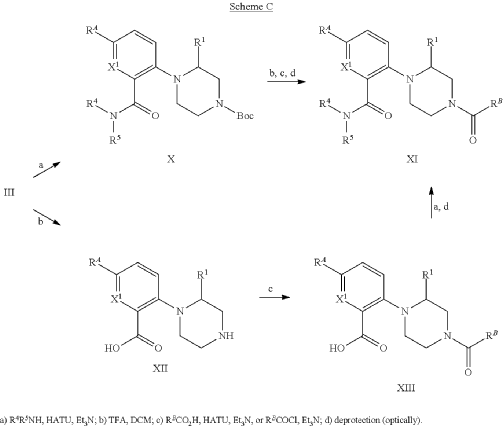
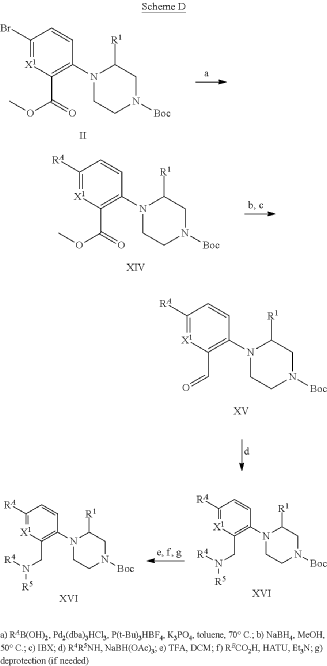
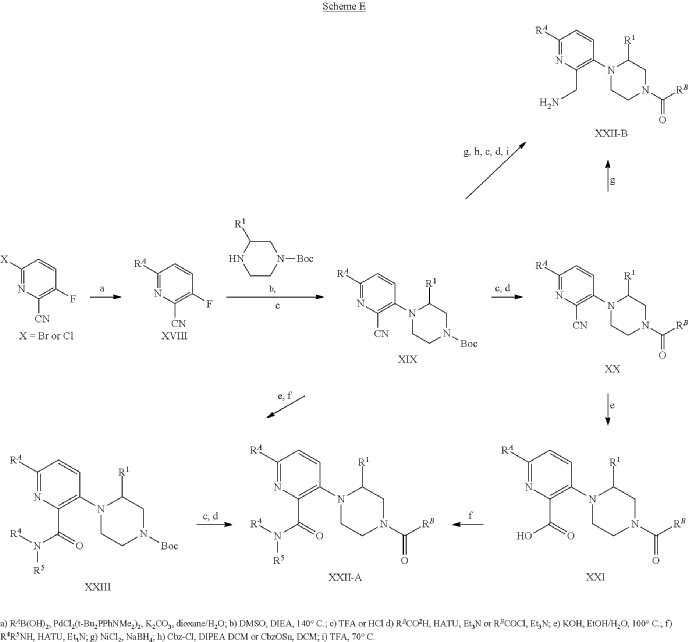
Example 31: N-[(3S)-1-azabicyclo[2.2.2]octan-3-yl]-6-(2-ethoxyphenyl)-3-[(2R)-2-ethyl-4-[1-(trifluoromethyl)cyclobutanecarbonyl]piperazin-1-yl]pyridine-2-carboxamide (Compound 1-410)
Step 31-1, Preparation of 6-(2-ethoxyphenyl)-3-[(2R)-2-ethyl-4-[1-(trifluoromethyl)cyclobutanecarbonyl]piperazin-1-yl]pyridine-2-carboxylic acid
Step 31-2, Preparation of N-[(3S)-1-azabicyclo[2.2.2]octan-3-yl]-6-(2-ethoxyphenyl)-3-[(2R)-2-ethyl-4-[1-(trifluoromethyl)cyclobutanecarbonyl]piperazin-1-yl]pyridine-2-carboxamide



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- “Crinetics Pharmaceuticals”. AdisInsight. 21 January 2025. Retrieved 25 February 2025.
- “Atumelnant (CRN04894)”. crinetics.com. 14 August 2020.
- Varlamov EV, Gheorghiu ML, Fleseriu M (December 2024). “Pharmacological management of pituitary adenomas – what is new on the horizon?”. Expert Opinion on Pharmacotherapy. 26 (2): 119–125. doi:10.1080/14656566.2024.2446625. PMID 39718553.
| Clinical data | |
|---|---|
| Other names | CRN04894 |
| Routes of administration | Oral[1] |
| Drug class | Melanocortin MC2 receptor antagonist[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2392970-97-5 |
| PubChem CID | 146361282 |
| IUPHAR/BPS | 13339 |
| ChemSpider | 129750231 |
| UNII | NR57FH6U1N |
| KEGG | D13102 |
| Chemical and physical data | |
| Formula | C33H42F3N5O3 |
| Molar mass | 613.726 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////Atumelnant, CRN04894, CRN 04894, NR57FH6U1N, CRINETICS PHARMA, Orphan Drug Status, Congenital adrenal hyperplasia, PHASE 3
Ebselen
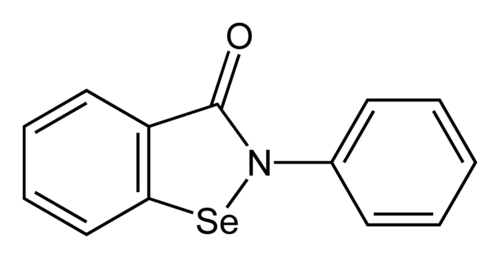
Ebselen
274.19 g/mol,
C13H9NOSe
2-phenyl-1,2-benzoselenazol-3-one
- CAS 60940-34-3
- 2-phenyl-1,2-benzoselenazol-3-one
- 2-Phenyl-1,2-benzisoselenazol-3(2H)-one
- Ebselene
- PZ 51, DR3305, and SPI-1005
- 40X2P7DPGH
Ebselen is a benzoselenazole that is 1,2-benzoselenazol-3-one carrying an additional phenyl substituent at position 2. Acts as a mimic of glutathione peroxidase. It has a role as a neuroprotective agent, an apoptosis inducer, an anti-inflammatory drug, an antioxidant, a hepatoprotective agent, a genotoxin, a radical scavenger, an enzyme mimic, an EC 1.3.1.8 [acyl-CoA dehydrogenase (NADP(+))] inhibitor, an EC 1.8.1.12 (trypanothione-disulfide reductase) inhibitor, an EC 1.13.11.33 (arachidonate 15-lipoxygenase) inhibitor, an EC 1.13.11.34 (arachidonate 5-lipoxygenase) inhibitor, an EC 2.5.1.7 (UDP-N-acetylglucosamine 1-carboxyvinyltransferase) inhibitor, an EC 2.7.10.1 (receptor protein-tyrosine kinase) inhibitor, an EC 3.5.4.1 (cytosine deaminase) inhibitor, an EC 5.1.3.2 (UDP-glucose 4-epimerase) inhibitor, a ferroptosis inhibitor, an antifungal agent, an EC 3.4.22.69 (SARS coronavirus main proteinase) inhibitor, an anticoronaviral agent, an antibacterial agent, an antineoplastic agent and an EC 3.1.3.25 (inositol–phosphate phosphatase) inhibitor.
Ebselen (also called PZ 51, DR3305, and SPI-1005), is a synthetic organoselenium molecule under preliminary investigation as a drug candidate.[1] It belongs to the class of compounds related to benzene and its derivatives.[1] It is being developed by the Seattle biotechnology company, Sound Pharmaceuticals, Inc.[1] It has also been reported to target tubulin, blocking its polymerization.[2]
Ebselen has been investigated for the treatment and basic science of Meniere’s Disease, Type 2 Diabetes Mellitus, and Type 1 Diabetes Mellitus.
Ebselen has been entered into clinical trials as a lead compound intended for the potential treatment of various diseases.[3] Its most advanced clinical trial is a Phase III study in people with Meniere’s disease, completed in July 2024.[4]
In vitro, ebselen is a mimic of glutathione peroxidase and reacts with peroxynitrite.[5] It is purported to have antioxidant and anti-inflammatory properties.[1][5]
Synthesis
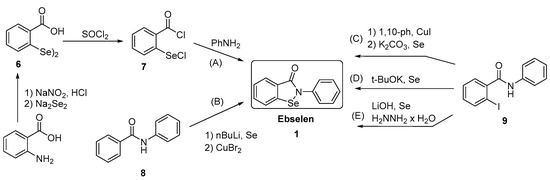
Generally, synthesis of the characteristic scaffold of ebselen, the benzoisoselenazolone ring system, can be achieved either through reaction of primary amines (RNH2) with 2-(chloroseleno)benzoyl chloride (Route I),[6] by ortho-lithiation of benzanilides followed by oxidative cyclization (Route II) mediated by cupric bromide (CuBr2),[7] or through the efficient Cu-catalyzed selenation / heterocyclization of o-halobenzamides, a methodology developed by Kumar et al.[8] (Route III).

SYN
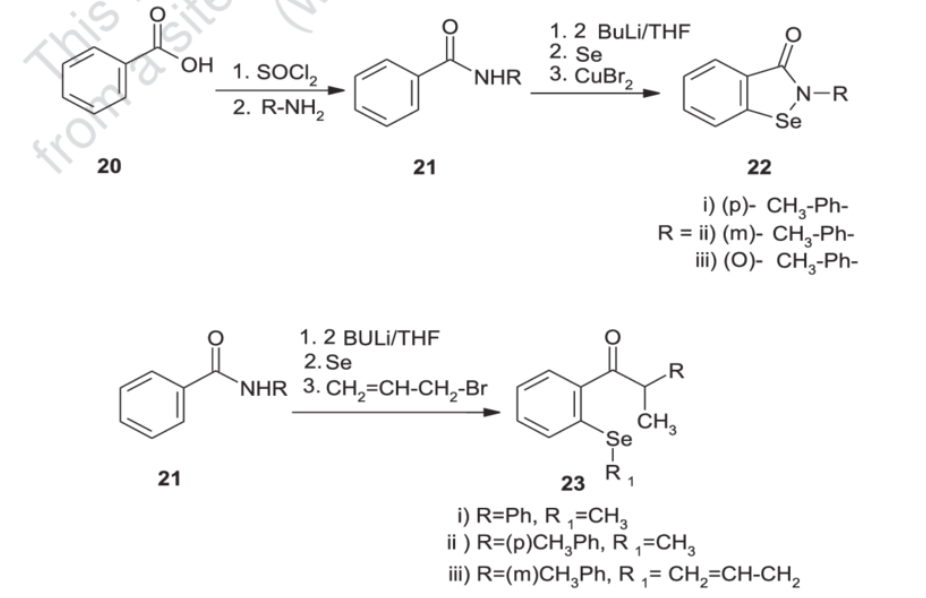
Synthesis of ebselen from benzoic acid by ortholithiation of benzanilide SOCl 2 =Thionyl chloride, R-NH 2 =Substituted aryl mine, BuLi/THF=n-butyllithium/ tetrahydrofuran, CuBr 2 =Cupper bromide, CH 2 =CH- CH 2 -Br = Allyl bromide.
SYN
New Chiral Ebselen Analogues with Antioxidant and Cytotoxic Potential
Molecules, March 2017, 22(3):492
SYN
https://pubs.acs.org/doi/10.1021/ol102027j
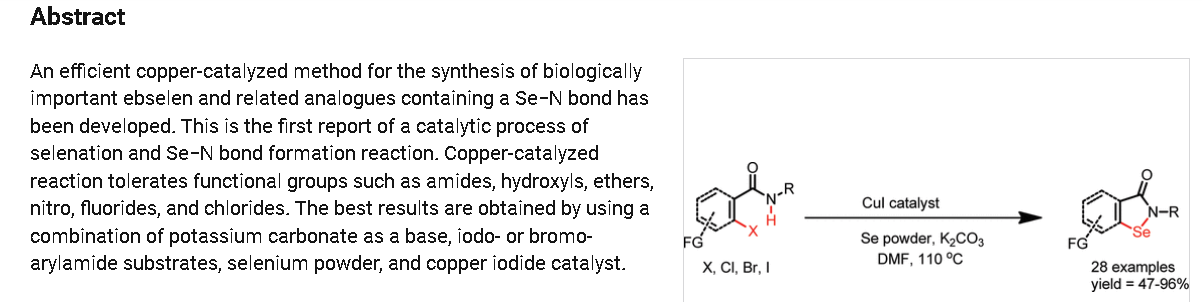

2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Iodo-N-phenylbenzamide (Typical
Procedure): Copper iodide (114 mg, 0.6 mmol) and 1,10-phenanthroline (108 mg, 0.6 mmol)
were added into DMF (3 mL) in a single neck flask. Resulted brownish solution was stirred for
15 min and then 2-iodo-N-phenylbenzamide1 (0.97 g, 3.0 mmol), selenium powder (0.29 g, 3.6
mmol), and potassium carbonate powder (0.65 g, 4.7 mmol) were added sequentially to same reaction flask. Brown colored reaction mixture was refluxed at 110oC using refluxing condenser
under nitrogen atmosphere. Progress of reaction was monitored by TLC. Reaction mixture was
refluxed for 8h. After this, reaction mixture poured over brine solution (60 mL) and stirred for 3
h. Product was precipitated as white solid which was collected by filtration over Buchner funnel,
product was washed with water (15 mL x 2), dried in air, dissolved in ethyl acetate, concentrated
over rotary evaporator, resulted brown solid which was purified by column chromatography
using hexane/ ethyl acetate (8:2) over silica gel. Yield 0.69 g (84%), mp 182-183 °C (180-181
°C).14,15 1H NMR (400 MHz, DMSO-d6) 8.09 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H),
7.71-7.62 (m, 3H), 7.51-7.43 (m, 3H), 7.28 (t, J = 8.0 Hz, 1H). 1H NMR (400 MHz, CDCl3)
8.12 (d, 7.6 Hz, 1H), 7.68-7.62 (m, 4H), 7.52-7.41 (m, 3H), 7.29 (m, 1H). IR (plate): 3057, 2921,
1598, 1443, 1346, 1263, 1028 cm-1; ESMS m/z: 276 (M+H+).
2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Iodo-N-phenylbenzamide at 74 mmol
scale: Reaction was carried out at 74 mmol scale using 2-iodo-N-phenylbenzamide (24.00 g,
74.3 mmol), selenium powder (7.04 g, 89.1 mmol), CuI (2.83 g, 14.9 mmol), 1,10
phenanthroline (2.69 g, 14.9 mmol), and anhydrous potassium carbonate powder (15.40 g, 111.4
mmol) in DMF (50 mL) and procedure and workup followed are similar to 3.6 mmol scale
reaction. Yield 16.28 g (80%), Figure S1.
2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Bromo-N-phenylbenzamide: Ebselen 1
was prepared from 2-bromo-N-phenylbenzamide2 (1.00 g, 3.6 mmol), selenium powder (0.34 g,
4.3 mmol), K2CO3 powder (0.74 g, 5.4 mmol), CuI (137 mg, 0.7 mmol), and 1,10-phenanthroline
(130 mg, 0.7 mmol) in DMF (3 mL). Reaction mixture was refluxed for 16 h at 110oC. Progress of reaction was monitored by TLC. After completion of reaction, mixture was poured into brine
solution (60 mL) and the resulted white precipitate was washed with water (20 mL x 2), and
dried in air. Purification by column chromatography on silica gel using CH2Cl2 provided white
crystalline solid (0.77 g, 78%).
2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Chloro-N-phenylbenzamide: Reaction
was carried out at 4 mmol scale using 2-chloro-N-phenylbenzamide3 (1.00 g, 4.3 mmol), CuI
(172 mg, 0.9 mmol), 1,10-phenanthroline (162 mg, 0.9 mmol), selenium powder (0.41 g, 5.2
mmol), K2CO3 (0.89 g, 6.4 mmol) in DMF (4 mL). Reaction mixture was refluxed for 24 h at
110oC. Workup procedure is similar as followed for bromo substrate. Yield 0.55 g (47%).
History
The first patent for 2-phenyl-1,2-benzoselenazol-3(2H)-one was filed in 1980 and granted in 1982.[9]
Research
Ebselen is in preliminary clinical development for the potential treatment of hearing loss and depression, among other medical indications.[3][10]
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Ebselen”. DrugBank. 29 January 2025. Retrieved 4 February 2025.
- Baksheeva VE, La Rocca R, Allegro D, Derviaux C, Pasquier E, Roche P, Morelli X, Devred F, Golovin AV, Tsvetkov PO (2025). “NanoDSF Screening for Anti-tubulin Agents Uncovers New Structure–Activity Insights”. Journal of Medicinal Chemistry. doi:10.1021/acs.jmedchem.5c01008.
- “Ebselen pipeline”. Sound Pharmaceuticals, Inc. 2025. Retrieved 4 February 2025.
- “SPI-1005 for the Treatment of Meniere’s Disease (STOPMD-3)”. ClinicalTrials.gov, US National Library of Medicine. 1 August 2024. Retrieved 4 February 2025.
- Schewe T (October 1995). “Molecular actions of ebselen – an antiinflammatory antioxidant”. General Pharmacology. 26 (6): 1153–69. doi:10.1016/0306-3623(95)00003-J. PMID 7590103.
- Kamigata N, Iizuka H, Izuoka A, Kobayashi M (July 1986). “Photochemical Reaction of 2-Aryl-1, 2-benzisoselenazol-3 (2 H)-ones”. Bulletin of the Chemical Society of Japan. 59 (7): 2179–83. doi:10.1246/bcsj.59.2179.
- Engman L, Hallberg A (1989-06-01). “Expedient synthesis of ebselen and related compounds”. The Journal of Organic Chemistry. 54 (12): 2964–2966. doi:10.1021/jo00273a035. ISSN 0022-3263.
- Balkrishna SJ, Bhakuni BS, Chopra D, Kumar S (December 2010). “Cu-catalyzed efficient synthetic methodology for ebselen and related Se-N heterocycles”. Organic Letters. 12 (23): 5394–7. doi:10.1021/ol102027j. PMID 21053969.
- DE3027073A1, Etschenberg, Eugen Dr; Renson, Marcel Prof Dipl-Chem Jupille & Winkelmann, Johannes Dr 5000 Köln, “2-phenyl-1,2-benzisoselenazol-3(2h)-on enthaltende pharmazeutische praeparate und ihre verwendung”, issued 1982-02-18
- “Ebselen search: list of clinical trials sponsored by Sound Pharmaceuticals”. ClinicalTrials.gov, US National Library of Medicine. 2025. Retrieved 4 February 2025.
External links
| Names | |
|---|---|
| Preferred IUPAC name2-Phenyl-1,2-benzoselenazol-3(2H)-one | |
| Identifiers | |
| CAS Number | 60940-34-3 |
| 3D model (JSmol) | Interactive imageInteractive image |
| ChEBI | CHEBI:77543 |
| ChEMBL | ChEMBL51085 |
| ChemSpider | 3082 |
| ECHA InfoCard | 100.132.190 |
| PubChem CID | 3194 |
| UNII | 40X2P7DPGH |
| CompTox Dashboard (EPA) | DTXSID7045150 |
| InChI | |
| SMILES | |
| Properties | |
| Chemical formula | C13H9NOSe |
| Molar mass | 274.17666 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
References
- Zhou Y, Zhang Y, Zhao D, Yu X, Shen X, Zhou Y, Wang S, Qiu Y, Chen Y, Zhu F: TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 2024 Jan 5;52(D1):D1465-D1477. doi: 10.1093/nar/gkad751. [Article]
////////Ebselen, Ebselene, PZ 51, DR 3305, SPI 1005, PHASE 3, 40X2P7DPGH, Meniere’s Disease, Type 2 Diabetes Mellitus, Type 1 Diabetes Mellitus
Sergliflozin Etabonate

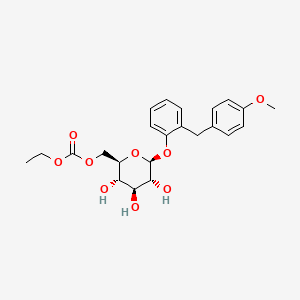
Sergliflozin Etabonate
408504-26-7 Cas no
Ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]oxan-2-yl]methyl carbonate
2-(4-methoxybenzyl)phenyl 6-O-ethoxycarbonyl-beta-D-glucopyranoside
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]tetrahydropyran-2-yl]methyl carbonate
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-{2-[(4-methoxyphenyl)methyl]phenoxy}oxan-2-yl]methyl carbonate
PHASE 2……….TYPE 3 DIABETES AND OBESITY
A SGLT-2 inhibitor potentially for the treatment of type 2 diabetes and obesity.
GW-869682; GW-869682X; KGT-1251
- etabonate de sergliflozine
- etabonato de sergliflozina
MW 448.4, C23H28O9
KISSEI INNOVATOR
GSK DEVELOPER
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.
Sergliflozin etabonate (INN/USAN,[1][2] codenamed GW869682X) is an investigational anti-diabetic drug being developed by GlaxoSmithKline. It did not undergo further development after phase II
Sergliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine.[3][4]
Chemistry
Etabonate refers to the ethyl carbonate group. The remaining structure, which is the active substance, is called sergliflozin.

Sergliflozin
[PDF] Design, Syntheses, and SAR Studies of Carbocyclic Analogues of …onlinelibrary.wiley.com974 × 740Search by imageDesign, Syntheses, and SAR Studies of Carbocyclic Analogues of Sergliflozin as Potent SodiumDependent Glucose Cotransporter 2 In
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.

sergliflozin and prodrugs of sergliflozin, in particular sergliflozin etabonate, including hydrates and solvates thereof, and crystalline forms thereof. Methods for its manufacture are described in the patent applications EP 1344780 and EP 1489089 for example.
The compounds are described in EP 1 329 456 A1 and a crystalline form ofSergliflozin etabonate is described in EP 1 489 089 A1.
PATENT
US6872706B2
https://patentscope.wipo.int/search/en/detail.jsf?docId=US40677423&_cid=P20-MF4ZUQ-42384-1
PATENT
WO2001068660A1
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2001068660&_cid=P20-MF4ZXC-45172-1
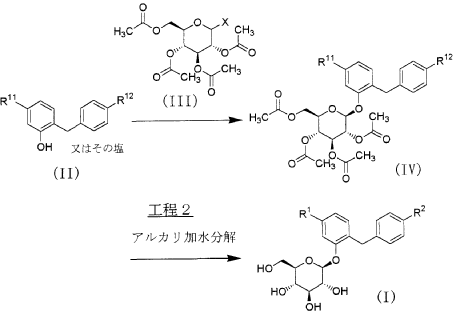
SYN
Heterocycles 2016, 92, 1599
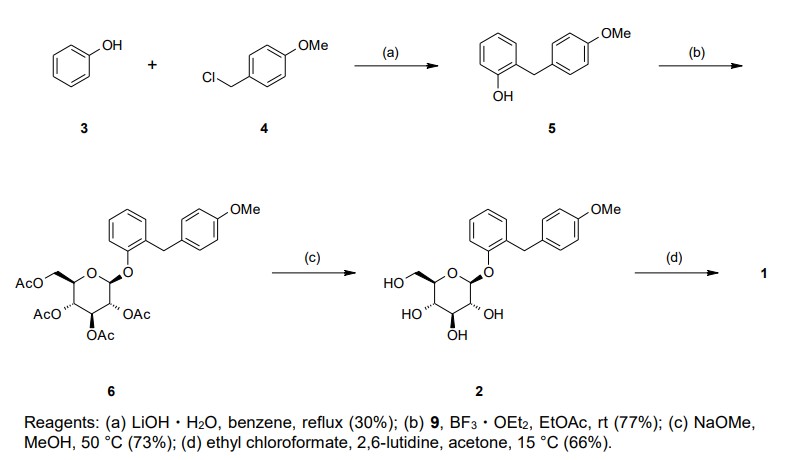
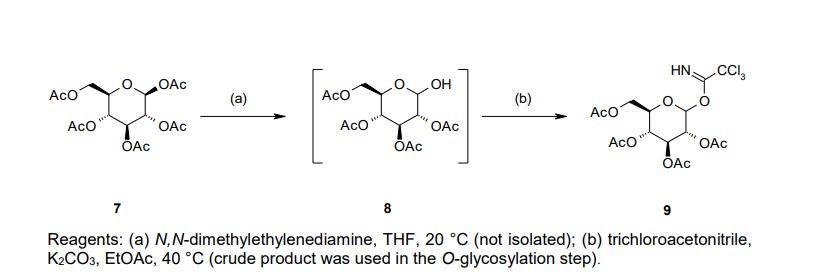
Our initial synthetic route of Serglifrozin etabonate (1) in early development consisted of six steps,
including synthesis of tetra-O-acetyl-D-glucopyranosyl trichloroacetimidate (9), as shown in Scheme 1
and Scheme 2 The first step is the coupling reaction of phenol (3) and 4-methoxybenzyl chloride (4) in the presence of
lithium hydroxide monohydrate (LiOH·H2O) to provide the aglycon 5 in a 30% yield following
chromatographic purification (Scheme 1). We prepared 9 separately by mono-deacetylation of
penta-O-acetyl-β-D-glucopyranose (7) with N,N-dimethylethylenediamine in THF followed by reaction of
the crude product of 8 with trichloroacetonitrile in the presence of potassium carbonate (K2CO3) in ethyl
acetate (EtOAc) (Scheme 2). Next, we carried out glycosylation of 5 with 9 in the presence of boron
trifluoride diethyl etherate (BF3·OEt2) in EtOAc to produce 6 in a 77% yield. The obtained 6 was
deacetylated with sodium methoxide (NaOMe) in MeOH to produce Serglifrozin (2) in a 73% yield, and
reaction of the isolated 2 with ethyl chloroformate in the presence of 2,6-lutidine in acetone provided 1 in
a 66% yield. The overall yield from 3 was 11%. While this route was capable of supplying small
amounts of 1, it suffered from several disadvantages.
The coupling reaction between 3 and 4 provided the aglycon 5 in low yield (30%); thus, chromatographic
purification was required to obtain highly pure 5. The trichloroimidation reaction of 8 is too hazardous
for large-scale manufacturing, because an excess amount of trichloroacetonitrile, a volatile and highly
toxic reagent, is required to obtain the trichloroacetimidate 9. Furthermore, 9 is too unstable to use
conveniently in large-scale manufacturing. Trichloroacetamide, a sublimation compound, is formed as a
by-product from the glycosylation of 5 with 9. Thus, the vacuum line and the vacuum pump of the
manufacturing equipment would be polluted by trichloroacetamide.
Because of these issues, this synthetic method is unsuitable for large-scale manufacturing. Therefore,
we investigated alternative processes for the preparation of 1, suitable for large-scale manufacturing. An improved synthetic method for 1 was achieved in a five-step procedure without purification of 6
(intermediate), as shown in Scheme 3.
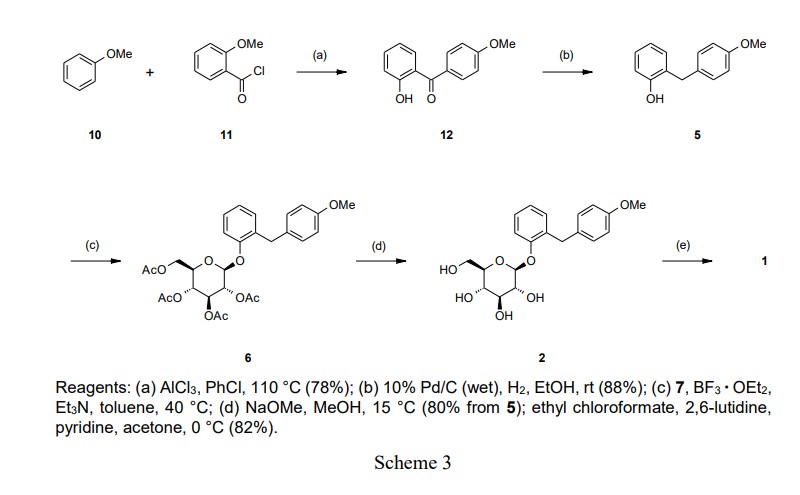
The Friedel-Crafts acylation of anisole (10) with 2-methoxybenzoyl chloride (11) in the presence of
aluminum chloride (AlCl3) at 110 °C provided benzophenone (12), which was selectively demethylated
on the methoxy group at the 2-position. The crude product of 12 was crystallized from MeOH to
provide highly pure 12 in a 78% yield. Hydrogenation of 12 in EtOH with 0.3–0.4 MPa H2 at room
temperature in the presence of 10% Pd/C provided 5. The crude product of 5 was crystallized from
toluene/n-heptane to provide highly pure 5 in an 88% yield.
The key step of the synthesis was the formation of the O-glycosylated product 6. In the initial synthesis,
it was necessary to isolate 6 to remove trichloroacetamide. Consequently, 2 was provided in a 56%
yield from 5. To obtain 6 efficiently without using the trichloroacetimidate (9), we evaluated several
conditions for the direct O-glycosylation of 5 with 7. The results are summarized in Table 1. The
O-glycosylation of 5 with 7 (200 mol%) in the presence of boron trifluoride diethyl etherate (BF3·OEt2;
100 mol%) in dichloromethane (DCM) at room temperature provided the crude product of 6 with a good
yield (80%) and β-selectivity (94/6), and then the deacetylation of the crude product of 6 in the presence
of sodium methoxide (NaOMe) in MeOH proceeded almost quantitatively to provide 2 in a 71% isolated
yield from 5 (run 1). Using this method, it was not necessary to isolate 6 because the excess amount of
7 was converted to glucose and removed to the aqueous layers in the deacetylation step. Use of DCM is
undesirable for large-scale manufacturing because quenching of O-glycosylation with water is highly
exothermic and washing of the DCM layer with water is a complicated procedure. Additionally, it is
strongly desirable to avoid using DCM in a manufacturing process due to environmental issues. For the reasons mentioned above, we attempted to use toluene as an alternative solvent. The O-glycosylation in
the presence of BF3·OEt2 (100 mol%) in toluene at 30 °C did not proceed completely, and the yield of 6
was lower than run 1 (run 2). We concluded that the lower solubility of 7 in toluene, compared with
DCM, caused the low yield. Because it was difficult to increase the amount of toluene from the
perspective of manufacturing efficiency, we tried to improve its solubility by optimizing the reagent
equivalent. Fortunately, we found that an excess amount of BF3·OEt2 enhanced the solubility of 7 in
toluene, and using 300 mol% of BF3·OEt2 in toluene provided 6 in a good yield (80%), similar to that
when using DCM (run 3). In contrast, reducing the amount of 7 provided 6 in an insufficient yield, and
2 was consequently provided in a lower yield (60%) (run 4). To achieve higher β-selectivity and an
increased yield, triethylamine (Et3N) was added to the O-glycosylation of 5 with 7 in the presence of
BF3·OEt2, according to the method of Lee et al.
9 Addition of Et3N (30 mol%) at 30 °C resulted in both
higher yield (89%) and higher β-selectivity (97/3) to provide 2 with a 79% isolated yield (run 5).
Increasing the amount of Et3N to 60 mol% at 30 °C resulted in a lower yield (85%) of 6 compared with
run 5, and the yield of 2 decreased (74%) (run 6). Increasing the reaction temperature to 40 °C in the
presence of 60 mol% of Et3N achieved the best results for both high yield (90%) and high β-selectivity
(99/1) to provide 2 in an 80% yield (run 7).
6-O-Ethoxycarbonyl-2-[(4-methoxyphenyl)methyl]phenyl-β-D-glucopyranoside (1). Ethyl
chloroformate (407 mg, 3.75 mol) was added drop-wise to the mixture of 2 (1.13 g, 3.0 mmol) and
2,6-lutidine (563 mg, 5.25 mmol) in acetone (4 mL) while maintaining the temperature between 12 and
18 °C. The reaction mixture was stirred at 15 °C for 23 h. Water (5 mL) was added drop-wise while
maintaining the temperature below 30 °C, and EtOAc (10 mL) was then added to the mixture. The
biphasic solution was transferred to a separating funnel for phase separation. The aqueous layer was
extracted with EtOAc (5 mL). The EtOAc layers were combined, washed successively with an aqueous
solution of 10% citric acid (5 mL × 2), an aqueous solution of 10% NaCl (5 mL), an aqueous solution of
5% NaHCO3 (5 mL × 2), and an aqueous solution of 10% NaCl (5 mL). They were then dried over
Na2SO4 and the filtrate was concentrated under reduced pressure. EtOH was added to the residue, and
the weight was adjusted to 7.2 g. The mixture was heated to 65 °C to dissolve solids. The solution was
cooled to 55 °C and seeded with 1. The solution was aged for 1 h at 50 °C, during which time the
product began to crystallize. After the slurry was cooled to 25 °C, n-heptane (11 mL) was added
drop-wise to the slurry at 25 °C followed by stirring for 1 h at 25 °C. The slurry was cooled to 3 °C and
then stirred for 2 h at 3 °C. The slurry was filtered, and the wet cake was washed with a mixed solvent
of EtOH (1.5 mL) and n-heptane (3 mL). The precipitate was dried in vacuo at 70 °C to give 888 mg
(66% yield) of 1 as a white solid. [α]
20
D -43.5 (c 1.0, DMSO). IR (KBr) cm-1
: 3495, 1744, 1514, 1488,
1454, 1467, 1411, 1372, 1340, 1266. 1H-NMR (CDCl3) δ: 1.27 (3H, t, J=7.0 Hz), 2.00 (1H, d, J=1.6
Hz), 3.46–3.54 (3H, m), 3.56–3.61 (2H, m), 3.72 (1H, d, J=2.1 Hz), 3.75 (3H, s), 3.82 (1H, d, J=15.5 Hz),
4.03 (1H, d, J=15.5 Hz), 4.11–4.22 (2H, m), 4.42 (2H, d, J=3.8 Hz), 4.69 (1H, d, J=7.4 Hz), 6.79–6.83
(2H, m), 6.97–7.02 (2H, m), 7.04–7.07 (2H, m), 7.15–7.22 (2H, m). 13C-NMR (CDCl3) δ: 14.2 (q), 36.1
(t), 55.4 (q), 64.4 (t), 66.4 (t), 69.6 (d), 73.4 (d), 73.8 (d), 75.7 (d), 100.8 (d), 114.1 (d×2), 114.4 (d), 122.7
(d), 128.0 (d), 129.2 (d×2), 130.0 (s), 131.1 (d), 133.4 (s), 155.2 (s), 155.4 (s), 157.8 (s). HRMS (ESI)
m/z: 466.2070 [M+NH4]
+
(Calcd for C23H32NO9: 466.2072)
6-O-Ethoxycarbonyl-2-[(4-methoxyphenyl)methyl]phenyl-β-D-glucopyranoside (1). Ethyl
chloroformate (21.6 g, 0.199 mol) was added drop-wise to the mixture of 2 (65.0 g, 0.173 mol),
2,6-lutidine (27.8 g, 0.259 mol) and pyridine (0.33 g, 4.2 mmol) in acetone (210 mL), maintaining the
temperature between -1 and 5 °C. The reaction mixture was stirred at 0 °C for 2 h. The reaction was
monitored by HPLC.15 Water (200 mL) was added drop-wise, maintaining the temperature below 30 °C,
and then EtOAc (220 mL) was added to the mixture. The biphasic solution was transferred to a
separating funnel for phase separation. The aqueous layer was extracted with EtOAc (140 mL). The
EtOAc layers were combined, washed successively with an aqueous solution of 10% citric acid (180 mL
× 2), an aqueous solution of 10% NaCl (66 g), an aqueous solution of 5% NaHCO3 (65 g × 2), and an aqueous solution of 10% NaCl (100 g), and then dried over Na2SO4 (65 g). After acetic acid (10 g,
0.167 mol) was added to the filtrate, the mixture was concentrated under reduced pressure. The residue
was dissolved in EtOH (660 mL) at 65 °C. The solution was concentrated under reduced pressure until
more than 330 mL distillate had been collected. EtOH was added to the residue, and the weight was
adjusted to 370 g. n-Heptane (120 mL) was added, and the resulting slurry was heated to 65 °C to
dissolve solids. The solution was cooled to 55 °C and seeded with 1. The solution was aged for 1 h at
50 °C, during which time the product began to crystallize. n-Heptane (480 mL) was added drop-wise to
the slurry, maintaining the temperature between 50 and 60 °C, and the slurry was stirred for 0.5 h at 55 °C.
The slurry was allowed to cool slowly over 2.5 h to 30 °C, then cooled to 3 °C, and then stirred for 1.5 h
at 3 °C. The slurry was filtered, and the wet cake was washed with a mixed solvent of EtOH (80 mL)
and n-heptane (180 mL). The precipitate was dried in vacuo at 70 °C to give 63.6 g (82% yield) of 1 as
a white solid.
REFERENCES (AND NOTES)
- W. N. Washburn, Expert Opin. Ther. Patents, 2009, 19, 1485.
- A. M. Pajor and E. M. Wright, J. Biol. Chem., 1992, 267, 3557.
- E. M. Wright, Am. J. Physiol. Renal Physiol., 2001, 280, F10.
- Y. Kanai, W. S. Lee, G. You, D. Brown, and M. A. Hediger, J. Clin. Invest., 1994, 93, 397.
- H. Fujikura, N. Fushimi, T. Nishimura, K. Tatani, and M. Isaji, PCT, WO 02/28872 (2002).
- H. Fujikura, N. Fushimi, T. Nishimura, K. Tatani, K. Katsuno, M. Hiratochi, Y. Tokutake, and M.
Isaji, PCT, WO 01/688660 (2001). - K. Katsuno, Y. Fujimori, Y. Takemura, M. Hiratochi, F. Itoh, Y. Komatsu, H. Fujikura, and M. Isaji,
J. Pharmacol. Exp. Ther., 2007, 320, 323. - M. Isaji, Curr. Opin. Investig. Drugs, 2007, 8, 285.
- S. Y. Lee, S. E. Rho, K. Y. Min, T. B. Kim, and H. K. Kim, J. Carbohydr. Chem., 2001, 20, 503.
- M. Yamaguchi, A. Horiguchi, A. Fukuda, and T. Minami, J. Chem. Soc., Perkin Trans. 1, 1990,
1079. - K. Ishihara, H. Kurihara, and H. Yamamoto, J. Org. Chem., 1993, 58, 3791.
- I. T. Akimova, A. V. Kaminsky, and V. I. Svistunova, Chem. Heterocycl. Compd., 2005, 41, 1374.
- B. N. Cook, S. Bhakta, T. Biegel, K. G. Bowman, J. I. Armstrong, S. Hemmerich, and C. R. Bertozzi,
J. Am. Chem. Soc., 2000, 122, 8612. - HPLC conditions: column, Inertsil ODS-3 (5 µm) 4.6 mm × 250 mm (GL Science Inc.); mobile
phase, isocratic elution with acetonitrile / 0.02 M KH2PO4, pH 3 = 6/4; flow rate, 1.0 mL/min;
column oven temperature, 40 °C; wave length, 225 nm; retention times, 5 = 16 min, α-anomer of 5 =18 min. - HPLC conditions: column, Inertsil ODS-3 (5 µm) 4.6 mm × 250 mm (GL Science Inc.); mobile
phase, gradient elution with 5 min 4/6 → 15 min 6/4 → 30 min 6/4 of acetonitrile/0.02 M KH2PO4,
pH 3; flow rate, 1.0 mL/min; column oven temperature, 40 °C; wavelength, 225 nm; retention times,
1 = 17 min, 2,6- and 4,6-bis-O-ethoxycarbonyl derivatives = 24 min, 3,6-bis-O-ethoxycarbonyl
derivative = 25 min.
SYN
Synthesis 2024, 56, 906–943
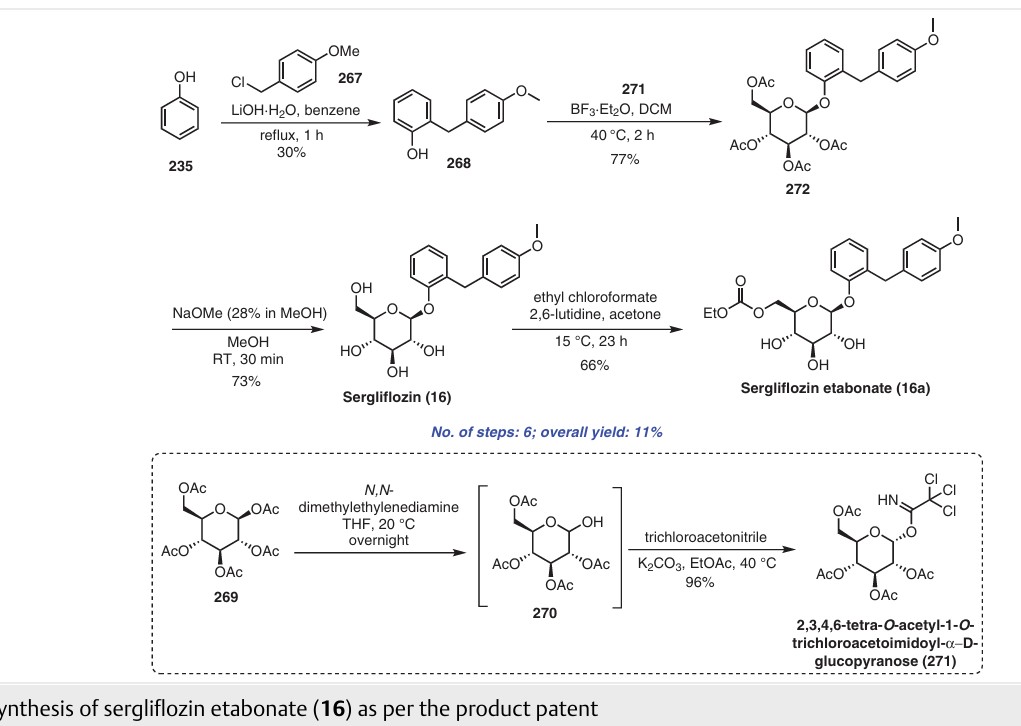
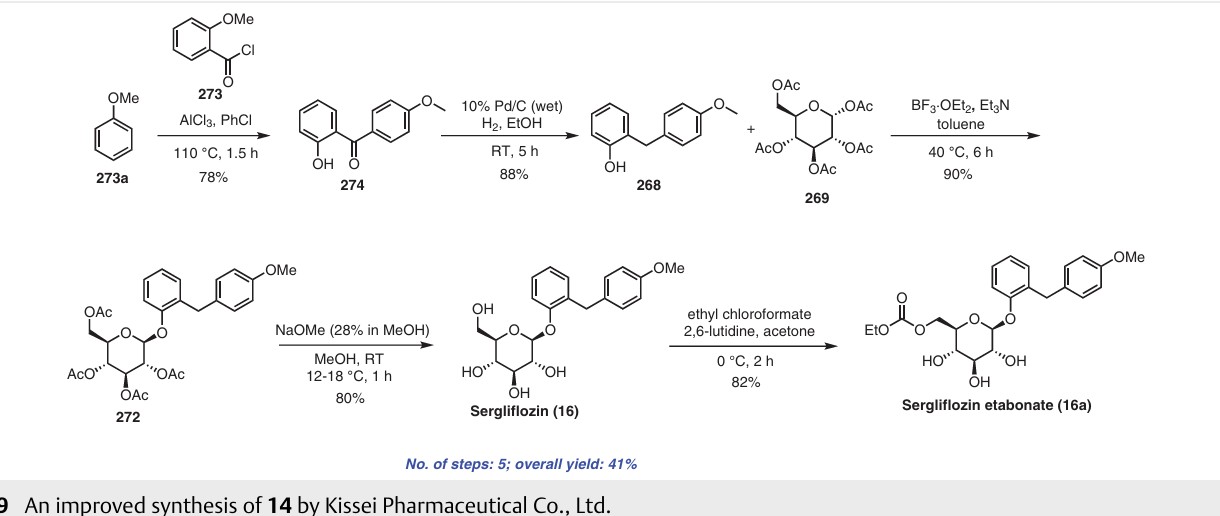
Sergliflozin etabonate (16), also known as GW869682X, was developed collaboratively by GlaxoSmithKline and Kissei Pharmaceutical (Japan). Unfortunately, it did not pass phase III trials. It belongs to the class of sodium–glucose linked transporter 2 (SGLT2) inhibitors and acts as a prodrug of sergliflozin, with the ethyl carbonate group referred to as etabonate. When compared to phlorizin, sergliflozin etabonate demonstrated significantly higher activity against SGLT2 than SGLT1. The initial synthetic route for the preparation of sergliflozin was described and patented by Kissei Pharmaceutical Co., Ltd. This particular route for Oaryl-glycoside-type derivatives was registered in the United States under patent application number US6872706B2.73 The first reported synthesis of sergliflozin etabonate
(16), which involves six steps, can be found in the patents US6872706B2 73a and WO2001068660A1 (Scheme 48).73b Compound 271 was prepared in a high yield of 96% follow ing a literature procedure. The selective monodeacetylationof penta-O-acetyl-b-D-glucopyranose, compound 269, was
achieved using N,N-dimethylethylenediamine in THF, resulting in the formation of compound 270. Subsequently, a reaction with trichloroacetonitrile and potassium carbonate led to the synthesis of intermediate 271 in excellent yield. To prepare the aglycone intermediate 268, phenol (235) was condensed with 4-methoxybenzyl chloride (267) using LiOH under reflux conditions. Further,O-glycosyla
tion of compound 268 with 271 was accomplished using boron trifluoride–diethyl etherate (BF3·OEt2), yielding intermediate 272. Removal of the acetyl groups from intermediate 272 was carried out using NaOMe in methanol to obtain sergliflozin (16a) in a yield of 73%. Finally, sergliflozin etabonate (16) was obtained by reacting compound 16a with ethyl chloroformate and 2,6-lutidine, resulting in a yield of
66%. The overall yield of sergliflozin etabonate (16a) was calculated to be 11%. It is important to note that the trichloroimidation reaction used in the synthesis of trichloroacetimidate 271 is considered hazardous and is not recommended for commercial use due to the highly toxic reagent, trichloroaceto
nitrile. Additionally, the process poses challenges in effectively removing the unwanted by-product, trichloroacetamide, formed during the preparation.A recently published approach presents an alternative synthesis of sergliflozin etabonate (16) that avoids the use of a trichloroacetimidate intermediate (Scheme 49).74a The five-step synthesis of compound 16a commenced from
readily available anisole (273a). An efficient Friedel–Crafts reaction was performed on anisole (273a) using the acid chloride 273 in the presence of aluminum chloride in chlorobenzene, leading to formation of benzophenone 274. Notably, demethylation of 274 was also observed under these
conditions. Next, ketone group reduction was achieved us ing 10% Pd/C and ethanol under 0.3–0.4 MPa of H2, providing compound 268 in 88% yield and high purity. Subsequently, O-glycosylation of 268 with penta-acetylated com pound 269 was carried out using BF3·Et2O and triethylamine, resulting in the formation of 272 in 90% yield with a high b-selectivity (99:1).74b Deacetylation of compound 272 was performed using NaOMe in methanol, affording sergliflozin (16a) in 80% yield. Further reaction with
ethyl chloroformate in the presence of 2,6-lutidine resulted in sergliflozin etabonate (16). The overall yield of compound 16 was calculated to be 41%. This novel synthetic route offers a promising alternative to the traditional method and demonstrates improved efficiency in the preparation of sergliflozin etabonate (16)
(73) (a) Fujikura, H.; Fushimi, N. US6872706B2, 2005. (b) Fujikura, H.; Fushimi, N.; Nishimura, T.; Tatani, K.; Katsuno, K.; Hiratochi, M.; Tokutake, Y.; Isaji, M. WO2001068660A1, 2001.
(74) (a) Kobayashi, M.; Isawa, H.; Sonehara, J.; Kubota, M. Heterocycles 2016, 92, 1599. (b) Lee, Y. S.; Rho, S. E.; Min, K. Y.; Kim, T. B.; Kim, H. K. J. Carbohydr. Chem. 2001, 20, 503.
| Patent | Submitted | Granted |
|---|---|---|
| Progression Inhibitor For Disease Attributed To Abnormal Accumulation Of Liver Fat [US2008045466] | 2008-02-21 | |
| NOVEL SUBSTITUTED TETRAHYDRONAPHTHALENES, PROCESS FOR THE PREPARATION THEREOF AND THE USE THEREOF AS MEDICAMENTS [US2010249097] | 2010-09-30 | |
| (CARBOXYLALKYLENEPHENYL)PHENYLOXAMIDES, METHOD FOR THE PRODUCTION THEREOF AND USE OF SAME AS A MEDICAMENT [US2010261645] | 2010-10-14 | |
| (CYCLOPROPYLPHENYL)PHENYLOXAMIDES, METHOD FOR THE PRODUCTION THEREOF, AND USE OF SAME AS A MEDICAMENT [US8148375] | 2010-10-14 | 2012-04-03 |
| Crystals of glucopyranosyloxybenzyl benzene derivative [US7371730] | 2005-06-02 | 2008-05-13 |
| CERTAIN CRYSTALLINE DIPHENYLAZETIDINONE HYDRATES, PHARMACEUTICAL COMPOSITIONS THEREOF AND METHODS FOR THEIR USE [US8003636] | 2009-08-13 | 2011-08-23 |
| NOVEL DIPHENYLAZETIDINONE SUBSTITUTED BY PIPERAZINE-1-SULFONIC ACID AND HAVING IMPROVED PHARMACOLOGICAL PROPERTIES [US2009264402] | 2009-10-22 | |
| Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, process for preparing them, medicaments comprising these compounds, and their use [US7759366] | 2009-08-27 | 2010-07-20 |
| Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same [US2005065098] | 2005-03-24 | |
| Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same [US6872706] | 2004-01-29 | 2005-03-29 |
| Patent | Submitted | Granted |
|---|---|---|
| PROGRESSION INHIBITOR FOR DISEASE ATTRIBUTED TO ABNORMAL ACCUMULATION OF LIVER FAT [US2009286751] | 2009-11-19 | |
| THERAPEUTIC USES OF SGLT2 INHIBITORS [US2011077212] | 2011-03-31 | |
| PHARMACEUTICAL COMPOSITION COMPRISING A SGLT2 INHIBITOR IN COMBINATION WITH A DPP-IV INHIBITOR [US2011098240] | 2011-04-28 | |
| Substituted imidazoline-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011112097] | 2011-05-12 | |
| Heterocycle-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising them and use thereof [US2011046105] | 2011-02-24 | |
| Arylchalcogenoarylalkyl-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011046185] | 2011-02-24 | |
| Arylchalcogenoarylalkyl-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011053947] | 2011-03-03 | |
| Novel aromatic fluoroglycoside derivatives, pharmaceuticals comprising said compounds and the use thereof [US2011059910] | 2011-03-10 | |
| Novel phenyl-substituted imidazolidines, process for preparation thereof, medicaments comprising said compounds and use thereof [US2011178134] | 2011-07-21 | |
| HETEROCYCLIC COMPOUNDS, PROCESSES FOR THEIR PREPARATION, MEDICAMENTS COMPRISING THESE COMPOUNDS, AND THE USE THEREOF [US2011183998] | 2011-07-28 |
| Systematic (IUPAC) name | |
|---|---|
| 2-(4-methoxybenzyl)phenyl 6-O-(ethoxycarbonyl)-β-D-glucopyranoside | |
| Clinical data | |
| Routes of administration | Oral |
| Identifiers | |
| CAS Number | 408504-26-7 |
| ATC code | None |
| PubChem | CID: 9824918 |
| IUPHAR/BPS | 4587 |
| ChemSpider | 21234810 |
| ChEMBL | CHEMBL450044 |
| Chemical data | |
| Formula | C23H28O9 |
| Molecular mass | 448.463 g/mol |
References
- World Health Organization (2008). “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 59” (PDF). WHO Drug Information. 22 (1): 66. Archived from the original (PDF) on February 19, 2009.
- “Statement on a nonproprietary name adopted by the USAN council: Sergliflozin etabonate” (PDF). American Medical Association. Retrieved 2008-08-10.
- Katsuno K, Fujimori Y, Takemura Y, et al. (January 2007). “Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level”. J Pharmacol Exp Ther. 320 (1): 323–30. doi:10.1124/jpet.106.110296. PMID 17050778. S2CID 8306408.
- “Prous Science: Molecule of the Month November 2007”. Archived from the original on 2007-11-05. Retrieved 2008-10-28.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////// etabonate, Sergliflozin etabonate, Sergliflozin, PHASE 3, GW869682X, GSK, KISSEI, GW-869682; GW-869682X; KGT-1251
CCOC(=O)OCC1C(C(C(C(O1)OC2=CC=CC=C2CC3=CC=C(C=C3)OC)O)O)O
CCOC(=O)OCC1C(C(C(C(O1)Oc2ccccc2Cc3ccc(cc3)OC)O)O)O
CEFILAVANCIN



CEFILAVANCIN, TD-1792
CAS 722454-12-8
C87H96Cl3N16O28S2, 1984.28
F76229E21M
Vancomycin, 26-[[[3-[[(Z)-[1-(2-amino-5-chloro-4-thiazolyl)-2-[[(6R,7R)-2-carboxy-8-oxo-3-(pyridiniomethyl)-5-thia-1-azabicyclo[4.2.0]oct-2-en-7-yl]amino]-2-oxoethylidene]amino]oxy]propyl]amino]carbonyl]-26-decarboxy-
1-{[(6R,7R)-7-[(2Z)-2-(2-amino-5-chloro-1,3-thiazol-4-yl)-2-[(3-{[(1S,2R,18R,19R,22S,25R,28R,40S)-48-{[(2S,3R,4S,5S,6R)-3-{[(2S,4S,5S,6S)-4-amino-5-hydroxy-4,6-dimethyloxan-2-yl]oxy}-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy}-22-(carbamoylmethyl)-5,47-dichloro-2,18,32,35,37-pentahydroxy-19-[(2R)-4-methyl-2-(methylamino)pentanamido]-20,23,26,42,44-pentaoxo-7,13-dioxa-21,24,27,41,43-pentaazaoctacyclo[26.14.2.2^{3,6}.2^{14,17}.1^{8,12}.1^{29,33}.0^{10,25}.0^{34,39}]pentaconta-3,5,8,10,12(48),14,16,29(45),30,32,34(39),35,37,46,49-pentadecaen-40-yl]formamido}propoxy)imino]acetamido]-2-carboxylato-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl}pyridin-1-ium
Phase III Skin and soft tissue infections
- OriginatorGlaxoSmithKline; Theravance
- DeveloperR-Pharm; Theravance Biopharma
- ClassAcetamides; Antibacterials; Azabicyclo compounds; Beta-lactams; Cephalosporins; Peptide antibiotics; Pyridines; Thiazoles
- Mechanism of ActionCell wall inhibitors
BUILDING BLOCK
Vancomycin,

| Formula | C66H75Cl2N9O24 |
| Molar mass | 1449.27 g·mol−1 |
Cefilavancin (TD-1792) is an experimental antibiotic medication developed for the treatment of bacterial infections such as drug-resistant strains of Staphylococcus aureus. It is a prodrug which is also a codrug, injected intravenously and then cleaved inside the body to two active components, one of which is a modified form of vancomycin and the other a cephalosporin antibiotic. In clinical trials cefilavancin has shown similar efficacy with reduced side effects compared to vancomycin itself.[1][2][3][4][5][6][7][8]
- 31 Jan 2020Cefilavancin is still in phase III trials for Skin and soft tissue infection in Russia and Georgia (R-Pharm pipeline, January 2020)
- 17 Jun 2015Phase II development is ongoing the USA
- 02 Jun 2014Theravance Biopharma is formed as a spin-off of Theravance
SCHEME

SYN
WO2003031449
https://patentscope.wipo.int/search/en/WO2003031449
cheme A
REF
Li, Huijuan; ET AL, Medicine (Philadelphia, PA, United States) (2022), 101(34), e30120
References
- ^ Long DD, Aggen JB, Chinn J, Choi SK, Christensen BG, Fatheree PR, et al. (October 2008). “Exploring the positional attachment of glycopeptide/beta-lactam heterodimers”. The Journal of Antibiotics. 61 (10): 603–614. doi:10.1038/ja.2008.80. PMID 19168974.
- ^ Tyrrell KL, Citron DM, Warren YA, Goldstein EJ (April 2012). “In vitro activity of TD-1792, a multivalent glycopeptide-cephalosporin antibiotic, against 377 strains of anaerobic bacteria and 34 strains of Corynebacterium species”. Antimicrobial Agents and Chemotherapy. 56 (4): 2194–2197. doi:10.1128/AAC.06274-11. PMC 3318369. PMID 22290981.
- ^ Stryjewski ME, Potgieter PD, Li YP, Barriere SL, Churukian A, Kingsley J, et al. (November 2012). “TD-1792 versus vancomycin for treatment of complicated skin and skin structure infections”. Antimicrobial Agents and Chemotherapy. 56 (11): 5476–5483. doi:10.1128/aac.00712-12. PMC 3486540. PMID 22869571.
- ^ Douglas EJ, Laabei M (September 2023). “Staph wars: the antibiotic pipeline strikes back”. Microbiology. 169 (9). Reading, England. doi:10.1099/mic.0.001387. PMC 10569064. PMID 37656158.
- ^ Surur AS, Sun D (2021). “Macrocycle-Antibiotic Hybrids: A Path to Clinical Candidates”. Frontiers in Chemistry. 9: 659845. Bibcode:2021FrCh….9..317S. doi:10.3389/fchem.2021.659845. PMC 8120311. PMID 33996753.
- ^ Saxena D, Maitra R, Bormon R, Czekanska M, Meiers J, Titz A, et al. (December 2023). “Tackling the outer membrane: facilitating compound entry into Gram-negative bacterial pathogens”. npj Antimicrobials and Resistance. 1 (1): 17. doi:10.1038/s44259-023-00016-1. PMC 11721184. PMID 39843585.
- ^ Koh AJ, Thombare V, Hussein M, Rao GG, Li J, Velkov T (2023). “Bifunctional antibiotic hybrids: A review of clinical candidates”. Frontiers in Pharmacology. 14: 1158152. doi:10.3389/fphar.2023.1158152. PMC 10313405. PMID 37397488.
- ^ Homer JA, Johnson RM, Koelln RA, Moorhouse AD, Moses JE (2024). “Strategic re-engineering of antibiotics”. Nature Reviews Bioengineering. doi:10.1038/s44222-024-00250-w.
| Clinical data | |
|---|---|
| Other names | TD-1792 |
| Routes of administration | Intravenous |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 722454-12-8 |
| PubChem CID | 76960417 |
| DrugBank | DB05735 |
| ChemSpider | 34990483 |
| UNII | F76229E21M |
| ChEMBL | ChEMBL4297645 |
| Chemical and physical data | |
| Formula | C87H95Cl3N16O28S2 |
| Molar mass | 1983.27 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////////CEFILAVANCIN, TD-1792, TD 1792, F76229E21M, цефилаванцин, 头孢拉凡星, سيفيلافانسين , GlaxoSmithKline, Theravance, PHASE 3
Bezisterim, HE 3286; NE-3107




Bezisterim, HE 3286; NE-3107
CAS 1001100-69-1
(1R,3aS,3bR,4R,7S,9aR,9bS,11aS)-1-ethynyl-9a,11a-dimethyl-1H,2H,3H,3aH,3bH,4H,6H,7H,8H,9H,9aH,9bH,10H,11H,11aH-cyclopenta[a]phenanthrene-1,4,7-triol
- (3β,7β,17α)-Pregn-5-en-20-yne-3,7,17-triol
- 17α-Ethynyl-5-androstene-3β,7β,17β-triol
- 17α-Ethynyl-Δ5-androstene-3β,7β,17β-triol
- 17α-Ethynylandrost-5-ene-3β,7β,17β-triol
- 3β,7β,17β-Trihydroxy-17α-ethynylandrost-5-ene
- Bezisterim
- HE 3286
- NE 3107
- Triolex
(3S,7R,8R,9S,10R,13S,14S,17R)-17-ethynyl-10,13-dimethyl-1,2,3,4,7,8,9,11,12,14,15,16-dodecahydrocyclopenta[a]phenanthrene-3,7,17-triol
| Formula | C21H30O3 |
|---|---|
| Molar mass | 330.468 g·mol−1 |
Q27286562
(3beta,7beta,17alpha)-Pregn-5-en-20-yne-3,7,17-triol
17.ALPHA.-ETHYNYL-5-ANDROSTENE-3.BETA.,7.BETA.,17.BETA.-TRIOL
PREGN-5-EN-20-YNE-3,7,17-TRIOL, (3.BETA.,7.BETA.,17.ALPHA.)-
- (1R,3aS,3bR,4R,7S,9aR,9bS,11aS)-1-ethynyl-9a,11a-dimethyl-1H,2H,3H,3aH,3bH,4H,6H,7H,8H,9H,9aH,9bH,10H,11H,11aH-cyclopenta(a)phenanthrene-1,4,7-triol
- (1R,3aS,3bR,4R,7S,9aR,9bS,11aS)-1-ethynyl-9a,11a-dimethyl-1H,2H,3H,3aH,3bH,4H,6H,7H,8H,9H,9aH,9bH,10H,11H,11aH-cyclopenta[a]phenanthrene-1,4,7-triol
- 17-ethynyl-5-androstene-3, 7, 17-triol

Bezisterim (developmental code names NE3107, HE3286) is a synthetic analogue of androstenetriol that is believed to have anti-inflammatory and insulin-sensitizing effects in the brain.[1] The compound crosses the blood–brain barrier and does not activate any neurotransmitter receptors.[2] It has been tested as a treatment for Alzheimer’s disease,[3][4][5][6] Parkinson’s disease,[1] and traumatic brain injury.[7] The drug is under development for a variety of conditions and its highest developmental phase is phase 3 for Alzheimer’s disease.[1]
- Originator Hollis-Eden Pharmaceuticals
- Developer BioVie; Harbor Therapeutics; National Institutes of Health (USA); NeurMedix
- Class Anti-inflammatories; Antidementias; Antiepileptic drugs; Antifibrotics; Antiglaucomas; Antihyperglycaemics; Antimigraines; Antineoplastics; Antiparkinsonians; Antirheumatics; Hormones; Insulin sensitisers; Nootropics; Obesity therapies; Small molecules
- Mechanism of Action Adiponectin stimulants; Interleukin 23 inhibitors; Interleukin 6 inhibitors; Mitogen-activated protein kinase 1 inhibitors; Mitogen-activated protein kinase 3 inhibitors; NF-kappa B inhibitors; Tumour necrosis factor inhibitors
- Cystic fibrosis
- Phase III Alzheimer’s disease
- Phase II Parkinson’s disease; Traumatic brain injuries
- Preclinical Multiple myeloma; Prostate cancer
- No development reported Drug-induced dyskinesia
- Discontinued Amyotrophic lateral sclerosis; Cognition disorders; Cystic fibrosis; Epilepsy; Glaucoma; Huntington’s disease; Migraine; Myositis; Optic neuritis; Rheumatoid arthritis; Type 1 diabetes mellitus; Type 2 diabetes mellitus; Ulcerative colitis; Uveitis
28 Feb 2025BioVie plans the phase II ADdRESs-LC trial for Post-acute COVID-19 syndrome in USA (PO, Capsule), in February 2025 (NCT06847191)
- 18 Feb 2025Phase-II clinical trials in Parkinson’s disease (Early-stage disease, In the elderly) in USA (PO) (NCT06757010)
- 03 Jan 2025BioVie plans a phase II SUNRISE-PD trial for Parkinsons disease (Early stage disease) in February 2025 (PO) (NCT06757010)
SCHEME

US20100227841
https://patentscope.wipo.int/search/en/detail.jsf?docId=US43352763&_cid=P11-M9JSD6-84971-1
17α-Ethynylandrost-5-ene-3β,7β,17β-triol was prepared as follows
US20100222315 https://patentscope.wipo.int/search/en/detail.jsf?docId=US43344622&_cid=P11-M9JSIE-88638-1
WO2009149392
PATENT’
WO2009149392
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2009149392&_cid=P11-M9JSL7-90448-1

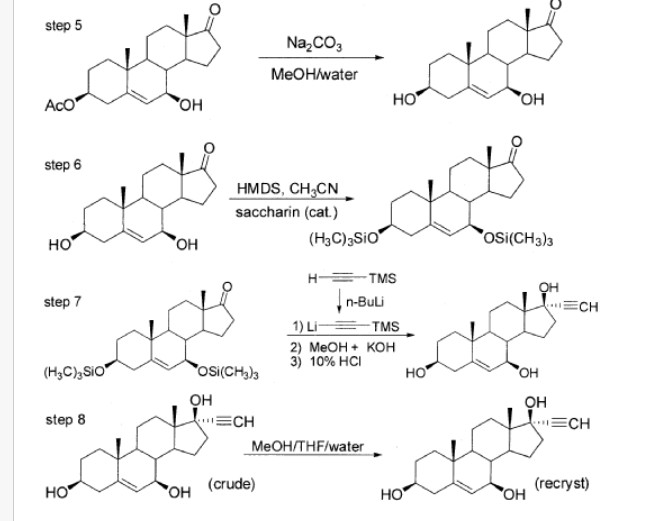
49] Example 7. Synthesis of 3β-acetoxy-androst-5-ene-17,17-ethylenedioxy: A 300L reactor was charged with 36 kg of triethylorthoformate, 20 kg of 3β-acetoxy-5-androsten-17-one, 12.6 kg of ethylene glycol and 400 g of p-toluenesulfonic acid. The mixture was heated to reflux under nitrogen until the reaction was complete (about 2-3 hours). The mixture was then cooled to 60 0C and 16 kg of anhydrous ethanol and 400 ml of pyridine were added. The resulting solution was transferred to a container and refrigerated overnight. The solids that formed were filtered and washed with 80 kg of 50% ethanol and dried at 40-50 0C to afford 18.5-21.0 kg (81.5-92.5%) of the title compound. [50] Example 8. Synthesis of 3β-acetoxy-androst-5-en-7-one-17,17-ethylenedioxy: A 500 L reactor was charged with 200 kg ethyl acetate and 25 kg of 3β-acetoxy-androst-5-en-17,17-ethylenedioxy. The mixture was stirred for 30 minutes whereupon 55 kg of 70% t-butyl peroxide and 9 kg of sodium bicarbonate were added. The reaction mixture was then cooled to 0 0C and 116 kg of 13% sodium perchlorate (aq.) was added over 10 hours so that a reaction temperature below 5 0C and pH between 7.5 and 8.5 were maintained. After the reaction was complete, the organic layer was separated and the aqueous phase was extracted with ethyl acetate (35 kg x 2). The combined organic phase was combined with a solution 33 kg of sodium sulfite in 167 kg of water, and the resulting mixture was stirred at 40 0C for 3 hours. The organic phase was washed with 50 kg of brine and concentrated to 55-60 kg whereupon 50 kg of methanol was added. After refrigeration overnight, a white solid was formed that was filtered and washed with 10 kg of methanol, and dried at 40-50 0C to yield 7.1-7.8 kg (27.4-30.1%) of the title compound.
[51] Example 9. Synthesis of 3β-acetoxy-androst-5-ene-17,17-ethylenedioxy-7β-ol. A 500 L reactor was charged with 48 kg of THF, 10 kg of 3β-acetoxy-androst-5-en-7-one-17,17-ethylenedioxy and a solution of 9.6 kg CeCI3-7H2O in 95 kg methanol. This mixture was cooled to 0 0C whereupon 2.0 kg of NaBH4 was added in batches over 3 hours in order to maintain the temperature below 5 0C. After stirring for 30 more minutes, 28 kg of acetone was added slowly in order to maintain the temperature below 5 0C, with stirring continued for another 30 minutes. To the mixture was added 240 kg water with stirring continued for 1 hour. The organic solvents were removed under vacuum and the residue was extracted with ethyl acetate (100 kg + 50 kg). The combined organic phase was washed with brine. Solvent was then removed to provide 8.6-8.9 kg (85.1-88.1 %) of the title compound. [52] Example 10. Synthesis of 3β-acetoxy-androst-5-en-17-one-7β-ol: A 500 L reactor was charged with 315 kg of acetone and 18 kg of 3β-acetoxy-androst-5-en-17,17-ethylenedioxy-7β-ol. The mixture was cooled to 5 0C and 2.34 kg of p-toluenesulfonic acid was added slowly to maintain the temperature below 10 0C. After stirring the mixture at 8-15 0C for 36-48 hours, 3.0 kg of sodium bicarbonate was added with stirring continued for 1 hour. Acetone was removed under vacuum, and to the residue was added 100 kg of water. The mixture was placed in a refrigerator overnight to give a white precipitate which was filtered to provide 33 kg (wet) of the title compound.
[53] Example 11. Synthesis of androst-5-en-17-one-3β,7β-diol: A 500 L reactor was charged 230 kg methanol, 33 kg (wet) 3β-acetoxy-7β-hydroxy-5-androsten-17-one, 108 kg water and 15 kg NaaCOβ. The mixture was heated to reflux for 3 hours. Methanol was removed under vacuum whereupon 250 kg of water was added to the residue. The mixture was put in refrigerator overnight to give a precipitate. The solids were collected by filtration, then washed with water and dried at 40-50 0C to yield 9.5-10.5 kg (67.9-75.0%) of the title compound as a white solid.
[54] Example 12. Purification of androst-5-en-17-one-3β,7β-diol: A 500 L reactor was charged with 20 kg crude 3β, 7β-dihydroxyandrost-5-en-17-one and 200 kg methanol and heated until all the solid dissolved. The solution was filtered while hot and after the filtrate cooled a white crystalline solid formed. The solids were collected by filtration, washed with small amount of methanol and dried at 40-50 0C. The solid was then refluxed in 50 kg of ethyl acetate for 20 minutes. After cooling the solid was filtered and dried at 40-50 0C under vacuum to provide 15.2 kg (76%) of purified title compound.
[55] Example 13. Synthesis of 3β,7β-bis-(trimethylsiloxy)-5-androsten-17-one: A mixture of 14.87 Kg of androst-5-en-17-one-3β,7β-diol, 23.8 Kg HMDS and 0.7 Kg saccharin catalyst in 100 L acetonitrile was heated to reflux for 8 hours with stirring under a nitrogen atmosphere. Liberated ammonia was purged under slight vacuum. The reaction volume was then reduced by distillation to collect 3OL of distillate (requires about 2 h). The reaction volume was further reduced to half of the original reaction volume by distillation under reduced pressure (700 mmHg), which requires about 2h of heating at 50 0C. The resulting uniform thick slurry is cooled to 5 0C (requires about 3 h), with additional acetonitrile added to maintain a minimum mixing volume, and held at that temperature for 1. The precipitated product was collected by filtration and dried at 45-50 0C under vacuum (29 mmHg) to a loss on drying (LOD) of not more than 1 % (requires 20 h) to provide 16 Kg (81 % yield) of the title compound (95% purity). [56] Example 14. Synthesis of 17α-ethynyl-5-androstene-3β,7β,17β-triol: To 11.02 Kg TMS-acetylene in 56.5 L tetrahydrofuran (THF) at -27 0C under a nitrogen atmosphere was added 8.51 L 10M n-BuLi. The n-butyl lithium was added very slowly to maintain a temperature at -7 to -27 0C (requires about 2 h) and the resulting reaction was stirred 10 min. at approximately 0°C to produce TMS-lithium-acetylide. To the TMS-lithium-acetylide solution was added a solution of 25.41 Kg of 3β,7β-bis-(trimethylsiloxy)-5-androsten-17-one in 95.3 L THF filtered through a 25 μm filter while allowing the reaction temperature to rise to 20-25 0C. After addition was completed, the reaction temperature was increased to 40-45 0C. To quench the reactor contents, 31.8 L of methanol was added over a period of about 1 h followed by 3.81 Kg KOH in 18.4 L of water giving a final reactor temperature of 50 0C. Liberated acetylene is purged under slight vacuum. The reactor contents were then concentrated by distillation at 80 0C for 1 h then under vacuum (175 mmHg) at about 70 0C (with an initial temperature of 25 0C to avoid bumping) to half of the original pot volume. The residue was cooled to about 10 0C and 35.0 Kg of deionized water was added, followed by 16.4 Kg 12N HCI while maintaining a pot temperature of about 10 0C and giving a final pH of 1. Additional 26.0 kg deionized water was added and the resulting mixture was stirred at about 5 0C for 1 h. The resulting slurry was filtered and washed with 75/25 mixture of methanol/water (16.9 L methanol, 5.6 L water). The collected solids were dried under vacuum (28 in Hg) at 45 0C for 12h for a loss on drying of no more than 0.5% to provide 9.6 Kg of the title compound (83% yield).
[57] Example 15. Recrystallization of 17α-ethynyl-5-androstene-3β,7β,17β-triol: Crude 9.6 Kg 17α-ethynyl-5-androstene-3β,7β,17β-triol prepared in
Example 14 was dissolved in refluxing 50/50 methanol/water (4.2 Kg methanol and 5.4 Kg water). To the solution was added 33.4 Kg methanol followed by 37.6 Kg of THF. The mixture was heated to reflux and stirring was continued until all solids have dissolved, whereupon 99.8 Kg of deionized water was added while maintaining a reactor temperature of 60-75 0C. The mixture was cooled to 0-5 0C over a period of 2 h and maintain at that temperature for 1 h while stirring was continued. The solids were recovered by filtration, washed with 9.6 Kg cold 50/50 methanol water and dried under vacuum (28 in Hg) at 50 0C for 8 h to provide 8.2 Kg of 17α-ethynyl-5-androstene-3β,7β,17β-triol. This first recrystallization is used to remove trace colored impurities from the initial product. A second recrystallization was conducted by heating the solid from the first recrystallization in ~10:1 methanohwater (145.8 Kg methanol and 18.2 Kg of water) to 80°C until all the solids have dissolved. The solution at 55-60 0C was filtered through a 25 μm filter to remove particulate impurities, whereupon 2.5 Kg of methanol at 55-60 0C (used to rinse the reactor) was added. Vacuum distillation at 125 mmHg at 70 0C was conducted until 0.9 to 1.2 times the volume of methanol that was added to the reactor was collected as distillate with water added as necessary to permit stirring (about 120-160 Kg water added). Final reaction volume was 200-225 L. The reactor mixture was cooled to 0-5 0C and maintained at that temperature for 1 h. The resulting slurry was filtered and the filter cake rinsed with 10 Kg deionized water and dried under vacuum (28 in Hg) at 50 0C for 12 h to a residual water content of less than 0.5%. This isolation procedure was used to reduce the THF content in the final product. The yield was 8.0 Kg of recrystallized title compound (83% yield).

[59] Example 16. Synthesis of 3β-acetoxy-androst-5-en-7-on-17-oxime: 3β-Acetoxy-androst-5-en-7,17-dione (45 g, 130 mmol) was dissolved in 800 ml_ methanol, 200 ml_ dichloromethane and 14.5g Et3N (144 mmol). To the solution at RT was added a solution of 10 g of hydroxylamine hydrochloride dissolved in 200 ml_ methanol. After stirring overnight, 200 ml_ of water was added followed by removal of volatile organics by evaporation under reduced pressure. To the resulting residue was added an additional 1 L of water to give a while solid that was filtered and washed well with water. Obtained was 45 g of crude title oxime in 95% purity by 1H-NMR, which was used in the next step without further purification.
[60] Example 17. Synthesis of 3β-acetoxy-androst-5-en-17-oxime-7β-ol: To a solution of 44 g of 3β-acetoxy-androst-5-en-7-on-17-oxime (100 mol%) in 800 ml_ methanol and 200 ml_ tetrahydrofuran was added 50 g of cerium chloride heptahydrate (110 mol%) in 20 ml_ of methanol. The resulting mixture was stirred until the solids were completely dissolved. To the solution cooled to about -5 0C was added 7 g sodium borohydride over 30 min. After stirring an additional 1.5 h at -5 0C, the reaction mixture was quenched with acetone (100 mL) and then allowed to warm to room temperature over a 30 min. period. The quenched reaction mixture was concentrated under vacuum to remove volatile organics. To the residue was added 800 mL of water followed by extraction with ethyl acetate (3 x 500 mL). The combined organic extracts were washed with brine, dried over Na2SO4, then concentrated to provide 42 g of the title compound as a white foam, which was used in the next step without further purification.. [61] Example 18. 3β-acetoxy-androst-5-en-17-one-7β-ol: To a solution of 42 g of 3β-acetoxy-androst-5-en-17-oxime-7β-ol (100 mol%) in 200 mL of ethanol was added 100 mL of water followed by 80 g (400 mol%) of sodium dithionite. The reaction was heated at 55 0C and stirred 16 h. After cooling, the reaction was concentrated under reduced pressure. The residue was diluted with 100 mL of water, and the resulting solid was collected by filtration and redissolved in 1 L dichloromethane. To the DCM solution was added 1 g activated carbon. After stirring overnight the mixture was filtered, and the resulting filtrate was washed with water, dried and concentrated to provide 25 g of crude product. Recrystallization from ethyl acetate gave 22g of the title compound. [62] Example 19. Estrogen receptor binding assay: A suitable example system is an estrogen receptor- kit manufactured by PanVera for ERβ, which contains recombinant estrogen receptor β ligand, FLUORMONE™ ES2 (ES2), a fluorescently labeled estrogen ligand, and appropriate buffer. The system was used in a fluorescence polarization competition assay in which a test article, such as a preparation of Compound 1 or a positive control displaces ES2 from its binding site. When bound to ERβ, ES2 tumbles slowly and has a high fluorescence polarization value. Unbound ES2 tumbles quickly and displays a low fluorescence polarization value. The change in polarization value in the presence of test compound then determines relative binding affinity of that test compound for ERβ as expressed by its IC50, which is the concentration of test compound that results in half-maximum shift in polarization. From IC50, K/ was calculated using the Cheng-Prusoff equation [Biochem. Pharmacol. 22: 3099-3108, (1973)]: K, = IC50Z(I + D/Kd) where D is the concentration of ES2 and Kd is the dissociation constant for binding of ES2 to ERβ (Kd = 4 ± 2 nM).
[63] The competition assay was conducted according to the manufacturer’s protocol (Lit. No. L0712, Rev. 10/03). Assay reagents used were bacculovirus expressed, full length human ERβ 4.5 pmol/μL in 50 mM Bis-Tris Propane (pH = 9), 400 mM KCI, 2 mM DTT, 1 mM EDTA, 10% glycerol, ES2 400 nM in methanol and E2 screening buffer consisting of 100 mM potassium phosphate (pH = 7.4), 100 μg/mL BGG, 0.02% NaN3. The ES2-ERβ complex was formed with 20 μL 20 nM ERβ (0.020 pmol/μL) and 20 μl_ 2 nM ES2 (0.002 pmol/μL). Positive control (estrogen) solution was prepared using 20 μL of a 1.0 mM stock solution in DMSO and 80 μL DMSO. In a first dilution, 50 μL of this solution is added to 50 μL of DMSO, which is followed by dilutions in 2-fold increments, to provide for a 14 point dilution curve. In a second dilution, to 4 μL of each DMSO solution from the first dilution is added 400 μL of ES2 screening buffer. To 20 μL of test compound, serially diluted in the manner described immediately above, in a 384 well black flat bottom microtiter plate, was added 20 μL of the ES2-ERβ complex (0.5% final DMSO concentration) followed by incubation in the dark at 20-30 0C for 1-4 h. Test compound was treated similarly except the starting concentration was 10 mM. Fluorescence polarization values are obtained using 485 nm excitation and 530 nm emission interference filters. Binding assay for ERa was conducted as for ERβ except bacculovirus expressed, full length human 2.8 pmol/μL ERa was used as reagent with the ERα-ES2 complex formed from 20 μL 30 nM (0.030 pmol/μL) and 20 μL 2 nM ES2 (0.002 pmol/μL). [64] Example 20. AR, GR and PR receptor binding assays. The AR competition assay was conducted according to the manufacturer’s protocol (Lit. No. L0844, Rev. 05/02) in the manner described for ERβ with the following exceptions. Reagents used were recombinant rat androgen receptor ligand binding domain tagged with His and GST [AR-LBD (His-GST)] 0.38 pmol/μL in buffer containing protein stabilizing agents and glycerol (pH = 7.5), 200 nM FLUORMONE™ AL Green, which is a fluorescently labeled androgen ligand, in 20 mM Tris, 90% methanol and AR screening buffer containing stabilizing agents and glycerol (pH = 7.5) with 2 μL of 1 mM DTT added per mL screening buffer (AR screening buffer 2 mM in added DTT) was used as the reagents. The AL Green-AR complex was formed with 20 μL 50 nM AR (0.050 pmol/μL) and 20 μL 2 nM AL Green (0.002 pmol/μL). K, was calculated using, for the dissociation constant for binding of the fluorophore to receptor, Kd = 20 ± 10 nM. [65] The PR competition assay was conducted according to the manufacturer’s protocol (Lit. No. L0503, Rev. 06/03) in the manner described for ERβ with the following exceptions. Reagents used were recombinant human progesterone receptor ligand binding domain tagged with GST [PR-LBD (GST)] 3.6 pmol/μL in 50 mM Tris (pH = 8.0), 500 mM KCI, 1 M urea, 5 mM DTT, 1 mM EDTA and 50% glycerol, 400 nM FLUORMONE™ PL Green, which is a fluorescently labeled progesterone ligand, in 20 mM Tris 90% methanol (pH = 6.8) and PR screening buffer containing protein stabilizing agents and glycerol (pH = 7.4) with 4 μL of 1 mM DTT added per mL screening buffer (PR screening buffer 4 mM in added DTT). The PL Green-PR complex was formed with 20 μL 80 nM PR (0.080 pmol/μL) and 20 μL 4 nM PL Green (0.004 pmol/μL). K, was calculated using, for the dissociation constant for binding of the fluorophore to receptor, Kd = 40 nM.
[66] The GR competition assay was conducted according to the manufacturer’s protocol (Lit. No. L0304, Rev. 12/01) in the manner described for ERβ with the following exceptions. Reagents used were recombinant full length human glucocorticoid receptor 0.240 pmol/μL in 10 mM phosphate buffer (pH = 7.4), 200 mM Na2MoO4, 0.1 mM EDTA, 5 mM DTT and 10% glycerol, 200 nM FLUORMONE™ GS1 , which is a fluorescently labeled glucocorticoid ligand, in 75% methanol, and GR screening buffer containing 100 mM potassium phosphate (pH = 7.4), 200 mM Na2MoO4, 1 mM EDTA, 20% DMSO with 5 μL of 1 mM DTT per mL screening buffer added (GR screening buffer 5 mM in added DTT), 1 mM GR stabilizing peptide, which is a co-activator related peptide [see Chang, CY. MoI. Cell Biol. 19: 8226-36 (1999)] in 10 mM phosphate buffer (pH = 7.4) and 1 M DTT in water were used as the reagents. To 2.5 mL of the GR screening buffer is added 2.5 mL GR stabilizing peptide solution and 125 μL of 1 M DTT to form the GR stabilizing peptide-glucocorticoid receptor complex. Order of addition to the microtiter plate was 20 μL test compound in 1 % DMSO, 10 μL of 16 nM GR (0.016 pmol/μL) and finally 10 μL of 4 nM GS1 , followed by incubation in the dark at 20-30 0C for 4 h (total experiment time should not exceed 7 h). K, was calculated using, for the dissociation constant for binding of the fluorophore to receptor, Kd = 0.3 ± 0.1 nM.
[67] Example 21. Impurity profiling of 17α-ethynyl-5-androstene-3β,7β,17β- triol (Compound 1) preparations.
[68] Process A: HPLC conditions for Impurity profiling of Compound 1 preparations form Process B are give in Table 1.
[69]
Table 1. HPLC Conditions for Impurity Profiling of Compound 1 Preparations form Process A
PATENT
Hollis-Eden Pharmaceuticals, Inc. WO2008039566
Zhejiang Xianju Junye Pharmaceutical Co., Ltd.; Jiangxi Junye Biopharmaceutical Co., Ltd.CN114478672
Harbor BioSciences, Inc.US20100227841
Harbor BioSciences, Inc. US20100222315 A1
Hollis-Eden Pharmaceuticals, Inc. US20100075937
Neurmedix Inc. US20080153792 A1
Hollis-Eden Pharmaceuticals, Inc.; Harbor Therapeutics, Inc. US20080146532 A1
Harbor Therapeutics, Inc.; Neurmedix, Inc. US20160045516 A1
Harbor Therapeutics, Inc. US8354396 B2
Hollis-Eden Pharmaceuticals, Inc. WO2009149392
| Clinical data | |
|---|---|
| Other names | NE3107; NE-3107; HE3286; HE-3286; 17α-Ethynyl-5-androstene-3β,7β,17β-triol; |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1001100-69-1 |
| PubChem CID | 16739648 |
| DrugBank | DB05212 |
| ChemSpider | 20571043 |
| UNII | PH8858757I |
| KEGG | D12932 |
| ChEMBL | ChEMBL4297284 |
| CompTox Dashboard (EPA) | DTXSID501267252 |
| Chemical and physical data | |
| Formula | C21H30O3 |
| Molar mass | 330.468 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Jump up to:a b c “Bezisterim”. AdisInsight. 5 September 2024. Retrieved 26 September 2024.
- ^ Reading, Chris L; Ahlem, Clarence N; Parameswaran, Narayanan (December 2021). “Rationale for an anti-inflammatory insulin sensitizer in a phase 3 Alzheimer’s disease trial”. Alzheimer’s & Dementia. 17 (S9). doi:10.1002/alz.057438.
- ^ Stoiljkovic, Milan; Horvath, Tamas L.; Hajós, Mihály (July 2021). “Therapy for Alzheimer’s disease: Missing targets and functional markers?”. Ageing Research Reviews. 68: 101318. doi:10.1016/j.arr.2021.101318. PMC 8131215. PMID 33711510.
- ^ Balzano, Tiziano; Esteban-García, Noelia; Blesa, Javier (2 January 2023). “Neuroinflammation, immune response and α-synuclein pathology: how animal models are helping us to connect dots”. Expert Opinion on Drug Discovery. 18 (1): 13–23. doi:10.1080/17460441.2023.2160440. PMID 36538833. S2CID 254959175.
- ^ Liu, Ping; Wang, Yunyun; Sun, Yan; Peng, Guoping (April 2022). “Neuroinflammation as a Potential Therapeutic Target in Alzheimer’s Disease”. Clinical Interventions in Aging. 17: 665–674. doi:10.2147/CIA.S357558. PMC 9064449. PMID 35520949.
- ^ Xi, Yilong; Chen, Yun; Jin, Yi; Han, Guochen; Song, Mingjie; Song, Tingting; Shi, Yang; Tao, Ling; Huang, Zewei; Zhou, Jianping; Ding, Yang; Zhang, Huaqing (May 2022). “Versatile nanomaterials for Alzheimer’s disease: Pathogenesis inspired disease-modifying therapy”. Journal of Controlled Release. 345: 38–61. doi:10.1016/j.jconrel.2022.02.034. PMID 35257810. S2CID 247285338.
- ^ “U.S. Clinical Trial: Neurological Associates of West Los Angeles Listed a New Clinical Trial to Study Insulin-sensitizing NE3107 in Improving Sleep and Fatigue in Subjects With Traumatic Brain Injury.” Contify Life Science News, 1 Aug. 2023, p. NA. Gale OneFile: Health and Medicine, link.gale.com/apps/doc/A759542006/HRCA?u=anon~bb46c85&sid=sitemap&xid=0c315c7e. Accessed 14 Dec. 2023.
/////Bezisterim, HE 3286, NE 3107, Triolex, NE3107, NE-3107, HE3286, HE-3286, PHASE 2
COBITOLIMOD
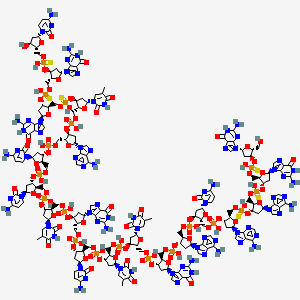

COBITOLIMOD
| IUPAC Condensed | dGuo-sP-dGuo-sP-dAdo-sP-dAdo-P-dCyd-P-dAdo-P-dGuo-P-dThd-P-dThd-P-dCyd-P-dGuo-P-dThd-P-dCyd-P-dCyd-P-dAdo-P-dThd-sP-dGuo-sP-dGuo-sP-dCyd |
|---|---|
| Sequence | GGAACAGTTCGTCCATGGC |
| HELM | RNA1{[dR](G).[sp][dR](G).[sp][dR](A).[sp][dR](A).P[dR](C).P[dR](A).P[dR](G).P[dR](T).P[dR](T).P[dR](C).P[dR](G).P[dR](T).P[dR](C).P[dR](C).P[dR](A).P[dR](T).[sp][dR](G).[sp][dR](G).[sp][dR](C)}$$$$ |
| IUPAC | 2′-deoxy-P-thio-guanylyl-(3′->5′)-2′-deoxy-P-thio-guanylyl-(3′->5′)-2′-deoxy-P-thio-adenylyl-(3′->5′)-2′-deoxy-adenylyl-(3′->5′)-2′-deoxy-cytidylyl-(3′->5′)-2′-deoxy-adenylyl-(3′->5′)-2′-deoxy-guanylyl-(3′->5′)-thymidylyl-(3′->5′)-thymidylyl-(3′->5′)-2′-deoxy-cytidylyl-(3′->5′)-2′-deoxy-guanylyl-(3′->5′)-thymidylyl-(3′->5′)-2′-deoxy-cytidylyl-(3′->5′)-2′-deoxy-cytidylyl-(3′->5′)-2′-deoxy-adenylyl-(3′->5′)-P-thio-thymidylyl-(3′->5′)-2′-deoxy-P-thio-guanylyl-(3′->5′)-2′-deoxy-P-thio-guanylyl-(3′->5′)-2′-deoxy-cytidine |
[(2R,3S,5R)-3-[[(2R,3S,5R)-3-[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[(2R,3S,5R)-3-[[(2R,3S,5R)-3-[[(2R,3S,5R)-3-[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[(2R,3S,5R)-3-[[(2R,3S,5R)-3-[[(2R,3S,5R)-3-[[(2R,3S,5R)-3-[[(2R,3S,5R)-3-[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[(2R,3S,5R)-5-(4-amino-2-oxopyrimidin-1-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [(2R,3S,5R)-2-[[[(2R,3S,5R)-2-[[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-2-[[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-yl)-2-(hydroxymethyl)oxolan-3-yl]oxy-hydroxyphosphinothioyl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphinothioyl]oxymethyl]-5-(6-aminopurin-9-yl)oxolan-3-yl]oxy-hydroxyphosphinothioyl]oxymethyl]-5-(6-aminopurin-9-yl)oxolan-3-yl] hydrogen phosphate
DNA, d(G-sp-G-sp-A-sp-A-C-A-G-T-T-C-G-T-C-C-A-T-sp-G-sp-G-sp-C)
Molecular Formula, C185-H233-N73-O106-P18-S6
- Molecular Weight
- 5925.2087
MF C185H233N73O106P18S6
CAS 1226822-98-5
- WHO 10066,
- IDX 0150,
- DIMS 0150,
- Kappaproct
- DIMS 0150,
- IDX 0150,
- Treatment of Moderate to Severe Ulcerative Colitis
- DNA based oligonucleotide that activates toll-like receptor 9.
- UNII: 328101264R
- DNA, d(g-SP-g-SP-a-SP-a-c-a-g-t-t-c-g-t-c-c-a-t-SP-g-SP-g-SP-C)
Other Names
- DNA d(G-sp-G-sp-A-sp-A-C-A-G-T-T-C-G-T-C-C-A-T-sp-G-sp-G-sp-C)
- 1: PN: WO2007004977 SEQID: 1 claimed DNA
- 1: PN: WO2007050034 PAGE: 29 claimed DNA
- 1: PN: WO2013076262 SEQID: 1 claimed DNA
PATENT
WO/2022/112224COBITOLIMOD DOSAGE FOR SELF-ADMINISTRATION
Ulcerative colitis (UC) is a disease characterized by chronic inflammation of the rectal and colonic mucosa, affecting the innermost lining in the first stage. The disease is recurrent, with both active and inactive stages that differ in pathology, symptoms and treatment. The underlying cause of UC is not understood, nor is it known what triggers the disease to recur between its inactive and active forms (Irvine, EJ (2008) Inflamm Bowel Dis 14(4): 554-565). Symptoms of active UC include progressive loose stools with blood and increased frequency of bowel movements. Active mucosal inflammation is diagnosed by endoscopy.
The stools contain pus, mucous and blood and are often associated with abdominal cramping with urgency to evacuate (tenesmi). Diarrhoea may have an insidious onset or, more rarely, start quite suddenly. In severe cases the symptoms may include fever and general malaise. In severe stages, deep inflammation of the bowel wall may develop with abdominal tenderness, tachycardia, fever and risk of bowel perforation. Furthermore, patients with UC may suffer extra intestinal manifestations such as arthralgia and arthritis, erythema nodosum, pyoderma gangrenosum and inflammation in the eyes. In the case of remission or inactive UC, patients are usually free of bowel symptoms.
The extent of inflamed and damaged mucosa differs among patients with UC. UC that affects only the rectum is termed ulcerative proctitis. The condition is referred to as distal or left sided colitis when inflammatory changes are present in the left side of the colon up to the splenic flexure. In extensive UC the transverse colon is also affected, and pancolitis designates a disease involving the entire colon.
Active mucosal inflammation is diagnosed by endoscopy and is characterized by a loss of vascular patterning, oedema, petechia, spontaneous bleeding and fibrinous exudates. The endoscopic picture is that of continuous inflammation, starting in the rectum and extending proximally to a variable extent into the colon. Biopsies obtained at endoscopy and subjected to histological examination help to diagnose the condition. Infectious causes, including Clostridium difficile, camphylobacter, Salmonella and Shigella, may mimic UC and can be excluded by stool cultures.
The medical management of UC is divided into treatment of active disease and maintenance of remission.
The treatment of patients with active UC aims to reduce inflammation and promote colon healing and mucosal recovery. In milder cases the disease may be controlled with conventional drugs including sulphasalazine, 5 -aminosalicylic acid (5-ASA) (Sutherland, L., F. Martin, S. Greer, M. Robinson, N. Greenberger, F. Saibil, T Martin, J. Sparr, E. Prokipchuk and L. Borgn (1987) Gastroenterology 92: 1894-1898) and glucocorticosteroids (GCS) (Domenech, E., M. Manosa and E. Cabre (2014). Dig Dis 32( 4): 320-327).
GCS are generally used to treat disease flare-ups and are not recommended for maintenance of remission since there are significant side effects in long-term use, and the possible development of steroid dependent disease. Glucocorticoid drugs act non-selectively, so in the long run they may impair many healthy anabolic processes. As a result, maintenance treatment with systemic GCS is not advised (Prantera, C. and S.
Marconi (2013) Therap Adv Gastroenterol 6(2): 137-156).
For patients who become refractory to GCS and suffer from severe or moderately severe attacks of UC, the addition of immunomodulatory agents such as cyclosporine, 6-mercaptopurine and azathioprine may be used. However, immunomodulators are slow-
acting and the induction of remission in these patients is often temporary (Khan, KJ, MC Dubinsky, AC Ford, TA Ullman, NJ Talley and P. Moayyedi (2011) Am J Gastroenterol 106(4): 630-642).
Further treatment options for UC include biologic agents (Fausel, R. and A. Afzali (2015) Ther Clin Risk Manag 11: 63-73). The three TNF-α inhibitors currently approved for the treatment of moderate to severe UC are infliximab, adalimumab, and golimumab. All three carry potential risks associated with their use, and should be avoided in certain patients, eg those with uncontrolled infections, advanced heart failure, neurologic conditions and in patients with a history of malignancy, due to a potential risk of accelerating the growth of a tumor. Other potential adverse effects of TNF-α inhibitor therapy include neutropenia, hepatotoxicity, serum sickness, leukocytoclastic vasculitis, rash including psoriasiform rash, induction of autoimmunity, and injection or infusion site reactions, including anaphylaxis, convulsions, and hypotension.
All three TNF-α inhibitor agents and their related biosimilar/derivative counterparts may be used to induce and maintain clinical response and remission in patients with UC.
Combination therapy with azathioprine is also used for inducing remission.
However, more than 50% of patients receiving TNF-α inhibitor agents fail to respond to induction dosing, or lose response to the TNF-α inhibitor agents over time (Fausel, R. and A. Afzali (2015) Ther Clin Risk Manag 11 : 63-73).
Vedolizumab, an a4b7 integrin inhibitor, was recently approved for the treatment of UC. In the GEMINI 1 trial, vedolizumab was found to be more effective than placebo for inducing and maintaining clinical response, clinical remission, and mucosal healing (Feagan, BG, P. Rutgeerts, BE Sands, S. Hanauer, JF Colombel, WJ Sandbom, G. Van Assche, J. Axler, HJ Kim, S. Danese, I. Fox, C. Milch, S. Sankoh, T. Wyant, J. Xu, A. Parikh and GS Group (2013) “Vedolizumab as induction and maintenance therapy for ulcerative colitis.” N Engl J Med 369(8): 699-710.).
Ulcerative colitis patients, who are chronically active and refractory to known treatments pose a serious medical challenge and often the only remaining course of action is
colectomy. A total colectomy is a potentially curative option in severe UC, but is a life-changing operation that entails risks as complications, such as pouch failure, pouchitis, pelvic sepsis, infertility in women, and nocturnal faecal soiling, may follow. Therefore, surgery is usually reserved for patients with severe refractory disease, surgical or other emergencies, or patients with colorectal dysplasia or cancer.
An emerging third line treatment for UC is cobitolimod (Kappaproct/DIMS0150), a modified single strand deoxyribonucleic acid (DNA)-based synthetic oligonucleotide of 19 bases in length. Cobitolimod has the sequence 5′- G*G*A*ACAGTTCGTCCAT*G*G*C-3′ (SEQ ID NO:1), wherein the CG dinucleotide is unmethylated.
Cobitolimod functions as an immunomodulatory agent by targeting the Toll-like receptor 9 (TLR9) present in immune cells. These immune cells (ie, B-cells and plasmacytoid dendritic cell (pDCs) reside in high abundance in mucosal surfaces, such as colonic and nasal mucosa. The immune system is the key mediator of the changes of UC. The mucosa of the colon and rectum of patients with UC is chronically inflamed and contains active immune cells. Cobitolimod may be topically administered in the region of inflammation, which places the drug in close contact with a high number of intended target cells, ensuring that the drug will reach an area rich in TLR9 expressing cells.The activation of these cells by cobitolimod induces various cytokines,
The clinical efficacy of cobitolimod has been demonstrated in the “COLLECT” (CSUC-01/10 ) clinical trial, which involved the administration to patients of 30 mg doses of cobitolimod, at 4 week intervals and also in the “CONDUCT” (CSUC- 01/16 ) clinical trial, which involved testing different dosage regimes. The details of the “COLLECT” trial were published in Journal of Crohn’s and Colitis (Atreya et al. J Crohn’s Colitis, 2016 May 20) and are summarized in Reference Example 1. The details of the “CONDUCT” clinical trial were published in The Lancet Gastroenterology and Hepatology (Atreya et al 2020. Lancet Gastroenterol Hepatol. 2020 Dec;5(12): 1063-1075) and are summarized in Reference Example 2. Overall, data on cobitolimod support a positive benefit-risk
assessment for patients with chronic UC which is in an active phase (occasionally referred to herein as “chronic active UC”). Cobitolimod is safe and well tolerated and has been shown to be effective to induce clinical response and remission in patients with chronic UC which is in an active phase, as well as symptomatic and endoscopic remission in patients with treatment refractory, moderate to severe chronic UC which is in an active phase. Despite the clinical trial results obtained this far, there still remains a need for additional effective dosages of cobitolimod which exhibit both good efficacy and safety.
In the COLLECT study, which involved administration of a relatively low (30mg) dose of cobitolimod, topical administration of cobitolimod was performed using a spray catheter device, administered during an endoscopy. This is an invasive medical procedure which is necessarily carried out by a medical professional. Further, before the topical administration of the cobitolimod to the patients, the colon of each patient was cleaned to remove faecal matter. That was done to enable the cobitolimod to reach the intestinal epithelial cells within the colon and to enable the endoscopist to view the colonic mucosa. Thus, it is well known in the art that oligonucleotides such as cobitolimod bind to organic matter such as faeces.
As noted above, patients suffering from chronic ulcerative colitis, who are in an active disease state and refractory to known treatments pose a serious medical challenge and often the only remaining course of action is colectomy. For this reason, patients will tolerate medical intervention which requires both colonic cleaning to remove faecal matter and topical administration via spray catheter, despite the inconvenience and discomfort involved in such invasive procedures. However, it would be therapeutically desirable to provide a topical treatment for ulcerative colitis patients which does not require colonic cleaning to remove faecal matter and which, preferably, can be self-administered by the patient.
PATENTS
- WO2001074344
- WO2005080568
- WO2007004977
- WO2007004979
- WO2007050034
- EP2596806
- WO2018206722
- WO2018206713
- WO2018206711
- WO2020099585
- WO2021037764
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
InDex Pharmaceuticals enters phase III study of the drug candidate cobitolimod
InDex Pharmaceuticals enters agreement with Parexel Biotech for phase III clinical study of cobitolimod for ulcerative colitis
InDex Pharmaceuticals Holding AB (publ) announced that the company has entered an agreement for services with global clinical research organisation (CRO) Parexel Biotech for the phase III study CONCLUDE. The study will evaluate the efficacy and safety of the drug candidate cobitolimod for the treatment of moderate to severe left-sided ulcerative colitis.
“We are excited to advance cobitolimod into phase III, which is the final stage of development before applying for market approval. After the successful collaboration in our recent phase IIb study CONDUCT, we are very pleased to collaborate once again with Parexel Biotech as our clinical development partner”, says Peter Zerhouni, CEO of InDex Pharmaceuticals. “Parexel Biotech is a leading global CRO with considerable experience managing phase III studies in inflammatory bowel disease, which will ensure an efficient execution of the study.”
CONCLUDE is a randomised, double-blind, placebo-controlled, global phase III study to evaluate cobitolimod as a novel treatment for patients with moderate to severe left-sided ulcerative colitis. The induction study will include approximately 400 patients, and the primary endpoint will be clinical remission at week 6. Patients responding to cobitolimod in the induction study will be eligible to continue in a one-year maintenance study, where they will be treated with either cobitolimod or a placebo.
Apart from the dosing 250 mg x 2, which was the highest dose and the one that showed the best efficacy in the phase IIb study CONDUCT, the phase III study will also evaluate a higher dose, 500 mg x 2, in an adaptive study design. This higher dose has the potential to provide even better efficacy than what was observed in the phase IIb study.
“We are pleased to partner with InDex Pharmaceuticals on phase III clinical trial CONCLUDE to evaluate a potential new therapy for patients with moderate to severe ulcerative colitis,” said Jim Anthony, Senior Vice President and Global Head, Parexel Biotech. “Our collaboration with InDex Pharmaceuticals demonstrates our commitment to designing innovative solutions that draw from our global clinical experience and therapeutic expertise to fulfil unmet medical needs on behalf of patients worldwide.”
///////////COBITOLIMOD, WHO 10066, IDX 0150, DIMS 0150, Kappaproct
CC1=CN(C(=O)NC1=O)C2CC(C(O2)COP(=O)(O)OC3CC(OC3COP(=O)(O)OC4CC(OC4COP(=O)(O)OC5CC(OC5COP(=O)(O)OC6CC(OC6COP(=O)(O)OC7CC(OC7COP(=S)(O)OC8CC(OC8COP(=S)(O)OC9CC(OC9COP(=S)(O)OC1CC(OC1CO)N1C=NC2=C1N=C(NC2=O)N)N1C=NC2=C1N=C(NC2=O)N)N1C=NC2=C(N=CN=C21)N)N1C=NC2=C(N=CN=C21)N)N1C=CC(=NC1=O)N)N1C=NC2=C(N=CN=C21)N)N1C=NC2=C1N=C(NC2=O)N)N1C=C(C(=O)NC1=O)C)OP(=O)(O)OCC1C(CC(O1)N1C=CC(=NC1=O)N)OP(=O)(O)OCC1C(CC(O1)N1C=NC2=C1N=C(NC2=O)N)OP(=O)(O)OCC1C(CC(O1)N1C=C(C(=O)NC1=O)C)OP(=O)(O)OCC1C(CC(O1)N1C=CC(=NC1=O)N)OP(=O)(O)OCC1C(CC(O1)N1C=CC(=NC1=O)N)OP(=O)(O)OCC1C(CC(O1)N1C=NC2=C(N=CN=C21)N)OP(=O)(O)OCC1C(CC(O1)N1C=C(C(=O)NC1=O)C)OP(=S)(O)OCC1C(CC(O1)N1C=NC2=C1N=C(NC2=O)N)OP(=S)(O)OCC1C(CC(O1)N1C=NC2=C1N=C(NC2=O)N)OP(=S)(O)OCC1C(CC(O1)N1C=CC(=NC1=O)N)O
Smiles
CC1=CN([C@H]2C[C@H](OP(=O)(O)OC[C@H]3O[C@H](C[C@@H]3OP(=O)(O)OC[C@H]4O[C@H](C[C@@H]4OP(=O)(O)OC[C@H]5O[C@H](C[C@@H]5OP(=O)(O)OC[C@H]6O[C@H](C[C@@H]6OP(=O)(O)OC[C@H]7O[C@H](C[C@@H]7OP(=O)(O)OC[C@H]8O[C@H](C[C@@H]8OP(=O)(O)OC[C@H]9O[C@H](C[C@@H]9OP(=S)(O)OC[C@H]%10O[C@H](C[C@@H]%10OP(=S)(O)OC[C@H]%11O[C@H](C[C@@H]%11OP(=S)(O)OC[C@H]%12O[C@H](C[C@@H]%12O)N%13C=CC(=NC%13=O)N)n%14cnc%15C(=O)NC(=Nc%14%15)N)n%16cnc%17C(=O)NC(=Nc%16%17)N)N%18C=C(C)C(=O)NC%18=O)n%19cnc%20c(N)ncnc%19%20)N%21C=CC(=NC%21=O)N)N%22C=CC(=NC%22=O)N)N%23C=C(C)C(=O)NC%23=O)n%24cnc%25C(=O)NC(=Nc%24%25)N)N%26C=CC(=NC%26=O)N)[C@@H](COP(=O)(O)O[C@H]%27C[C@@H](O[C@@H]%27COP(=O)(O)O[C@H]%28C[C@@H](O[C@@H]%28COP(=O)(O)O[C@H]%29C[C@@H](O[C@@H]%29COP(=O)(O)O[C@H]%30C[C@@H](O[C@@H]%30COP(=O)(O)O[C@H]%31C[C@@H](O[C@@H]%31COP(=S)(O)O[C@H]%32C[C@@H](O[C@@H]%32COP(=S)(O)O[C@H]%33C[C@@H](O[C@@H]%33COP(=S)(O)O[C@H]%34C[C@@H](O[C@@H]%34CO)n%35cnc%36C(=O)NC(=Nc%35%36)N)n%37cnc%38C(=O)NC(=Nc%37%38)N)n%39cnc%40c(N)ncnc%39%40)n%41cnc%42c(N)ncnc%41%42)N%43C=CC(=NC%43=O)N)n%44cnc%45c(N)ncnc%44%45)n%46cnc%47C(=O)NC(=Nc%46%47)N)N%48C=C(C)C(=O)NC%48=O)O2)C(=O)NC1=O

NEW DRUG APPROVALS
TO PAY YEARLY SUBSCRIPTION OF THIS BLOG
$10.00
ABX 464

ABX-464
- Molecular FormulaC16H10ClF3N2O
- Averrage mass338.712 Da
SPL-4641258453-75-6[RN]26RU378B9V2-Quinolinamine, 8-chloro-N-[4-(trifluoromethoxy)phenyl]-8-Chloro-N-[4-(trifluoromethoxy)phenyl]-2-quinolinamine
EX-A3322, DB14828, SB18690, BS-14770
Abivax is developing ABX464 a lead from HIV-1 splicing inhibitors, which modulates biogenesis of viral RNA, and acts by targeting the Rev protein, for treating HIV infection, rheumatoid arthritis, ulcerative colitis and COVID-19 infection.
In August 2021, ABX464 was reported to be in phase 3 clinical development.
ABX464 is an oral, first-in-class, small molecule that has demonstrated safety and profound anti-inflammatory activity in preclinical trials and in Phase 2a and Phase 2b induction trials to treat ulcerative colitis (UC). Patients who completed the induction studies had the option to roll over into the respective open-label extension studies.
In May 2021, Abivax communicated the top-line results of its randomized, double-blind and placebo-controlled Phase 2b induction trial conducted in 15 European countries, the US and Canada in 254 patients. The primary endpoint (statistically significant reduction of Modified Mayo Score) was met with once-daily ABX464 (25mg, 50mg, 100mg) at week 8.
Further, all key secondary endpoints, including endoscopic improvement, clinical remission, clinical response and the reduction of fecal calprotectin showed significant difference in patients dosed with ABX464 compared to placebo. Importantly, ABX464 also showed rapid efficacy in patients who were previously exposed to biologics and/or JAK inhibitors treatment.
In addition to the top-line induction results, preliminary data from the first 51 patients treated with 50mg ABX464 in the Phase 2b open-label maintenance study showed increased and durable clinical remission and endoscopic improvement after 48 weeks of treatment.
Based on the positive results from the Phase 2a and Phase 2b studies, Abivax plans to advance ABX464 into a Phase 3 clinical program by the end of 2021.
- Originator Splicos
- Developer Abivax
- Class Anti-inflammatories; Antirheumatics; Antivirals; Small molecules
- Mechanism of Action MicroRNA stimulants; Rev gene product inhibitors; RNA cap-binding protein modulators
- Phase II/III COVID 2019 infections
- Phase II Crohn’s disease; Rheumatoid arthritis; Ulcerative colitis
- DiscontinuedHIV infections
- 24 Jun 2021 Discontinued – Phase-II for HIV infections (Adjunctive treatment, Treatment-experienced) in France (PO) (Abivax pipeline, June 2021)
- 24 Jun 2021 Discontinued – Phase-II for HIV infections (Treatment-experienced, Adjunctive treatment) in Belgium (PO) (Abivax pipeline, June 2021)
- 24 Jun 2021
- Discontinued – Phase-II for HIV infections (Treatment-experienced, Adjunctive treatment) in Spain (PO) (Abivax pipeline, June 2021)
Evotec and Abivax in small-molecule pact
by Michael McCoy
September 18, 2017 | A version of this story appeared in Volume 95, Issue 37

The contract research firm Evotec will work with Abivax, a French biotech company, to develop new treatments for viral diseases. Abivax has developed a library of more than 1,000 small molecules designed to inhibit mRNA biogenesis. At its facility in Toulouse, France, Evotec will optimize Abivax’s drug candidates and help develop new drugs for influenza, Dengue, and other viral infections. Abivax’s lead candidate, ABX464, is in Phase II clinical trials as an HIV/AIDS treatment.
PATENT
WO 2010143170
WO 2010143168
WO 2010143169
EP 2974729
WO 2016009065
WO 2017158201
PATENT
WO2016009065
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016009065

Buchwald-Hartwig coupling of 2,8-dichloroquinoline (I) with 4-(trifluoromethoxy)aniline (II) using Pd(OAc)2, Cs2CO3 and xantphos or Pd2dba3, K2CO3 and xphos in t-BuOH
PATENT
https://patents.google.com/patent/US10253020B2/en
US 20170226095
COMPD 90
- (90) 8-chloro-N-[4-(trifluoromethoxy)phenyl]quinolin-2-amine
Example 5: Compound (90) of the Table IAccording to route (A), a mixture of 2,8-dichloroquinoline (984 mg) and 4-(trifluoromethoxy)aniline (743 μL), Pd(OAc)2 (22 mg), XantPhos (58 mg) and Cs2CO3 (4.6 g) in 20 mL of t-BuOH gave compound (90) (1.1 g).1H NMR (300 MHz, CDCl3) δ 7.84 (d, J=9.1, 2H), 7.79 (d, J=8.9, 1H), 7.67 (dd, J=1.2, 7.6, 1H), 7.48 (dd, J=1.1, 8.0, 1H), 7.18 (s, 3H), 6.89 (s, 1H), 6.75 (d, J=8.9, 1H).13C NMR (75 MHz, CDCl3) δ 153.88, 144.30, 143.91, 139.00, 138.25, 131.13, 130.13, 126.55, 125.42, 123.45, 122.50, 122.17, 120.49, 119.10, 113.24.
| 90 | 1H NMR (300 MHz, CDCl3) δ 7.84 (d, J = 9.1, 2H), 7.79 (d, J = 8.9, 1H), 7.67 (dd, J = 1.2, |
| 7.6, 1H), 7.48 (dd, J = 1.1, 8.0, 1H), 7.18 (s, 3H), 6.89 (s, 1H), 6.75 (d, J = 8.9, | |
| 1H) | |
| 13C NMR (75 MHz, CDCl3) δ 153.88, 144.30, 143.91, 139.00, 138.25, 131.13, | |
| 130.13, 126.55, 125.42, 123.45, 122.50, 122.17, 120.49, 119.10, 113.24. | |
| MS (ESI) [M + H]+ = 339 |
PAPER
Tetrahedron Letters (2018), 59(23), 2277-2280.
https://www.sciencedirect.com/science/article/abs/pii/S0040403918305641
Abstract
A solvent-free Buchwald-Hartwig amination had been developed under high-speed ball-milling conditions, which afforded the desired products with moderate to high yields. The addition of sodium sulfate was found to be crucial for improving both the performance and the reproducibility. Comparative solvent-free stirring experiments implicated the importance of mechanical interaction for the transformation, and the inert gas was proved to be unnecessary for this amination.
Graphical abstract

PATENT
WO2015001518
COMPD 90
PATENT
WO-2021152131
Novel co-crystalline polymorphic forms and salts of ABX464 , useful for treating inflammatory diseases, cancer, and diseases caused by viruses eg HIV, severe acute respiratory syndrome caused by SARS-CoV or SARS-CoV-2 infection including strains responsible for COVID-19 and their mutants.
W02010/143169 application describes the preparation and use of compounds, and in particular quinoline derivatives including certain pharmaceutically acceptable salts useful in the treatment of HIV infection. Said application in particular discloses 8-Chloro-N-(4-(trifluoromethoxy)phenyl)quinolin-2-amine also named (8-chloro-quinoline-2-yl)-(4-trifluoromethoxy-phenyl) -amine which is currently under clinical development. The inventors have stated that ABX464 is naturally highly crystalliferous and thus is spontaneously present under a specific unique stable and crystalline form named “crystalline form I”.
W02017/158201 application deals with certain mineral acid or sulfonic acid salts of ABX464.
ABX464 has a poor solubility in aqueous solutions. The main drawback of said poor solubility is that the active ingredient cannot entirely reach their targets in the body if the drug remains undissolved in the gastrointestinal system.
PATENT
WO2021152129 ,
amorphous solid dispersion (eg tablet) comprising ABX464.
PATENT
WO2020127839
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020127839
use of quinoline derivatives (ie ABX464) for treating cancer and dysplasia.
///////////ABX464, ABX 464, phase 3 , SPL 464, EX A3322, DB14828, SB18690, BS 14770

NEWDRUG APPROVALS
ONE TIME
$10.00
OTESECONAZOLE

OTESECONAZOLE
VT 1161
オテセコナゾール;
(2R)-2-(2,4-difluorophenyl)-1,1-difluoro-3-(tetrazol-1-yl)-1-[5-[4-(2,2,2-trifluoroethoxy)phenyl]pyridin-2-yl]propan-2-ol
| C23H16F7N5O2 527.4 | |
| Synonyms | VT 1161 Oteseconazole CAS1340593-59-0 |
|---|
Other Names
- (αR)-α-(2,4-Difluorophenyl)-β,β-difluoro-α-(1H-tetrazol-1-ylmethyl)-5-[4-(2,2,2-trifluoroethoxy)phenyl]-2-pyridineethanol
- (2R)-2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-1,2,3,4-tetrazol-1-yl)- 1-{5-[4-(2,2,2-trifluoroethoxy)phenyl]pyridin-2-yl}propan-2-ol
UPDATE MAY 2022… FDA APPROVED 2022/4/26, Vivjoa
Oteseconazole, sold under the brand name Vivjoa, is a medication used for the treatment of vaginal yeast infections.[1]
It was approved for medical use in the United States in April 2022.[2][3] It was developed by Mycovia Pharmaceuticals.[3]
Names
Oteseconazole is the international nonproprietary name (INN).[4]
Oteseconazole is an azole antifungal used to prevent recurrent vulvovaginal candidiasis in females who are not of reproductive potential.
Oteseconazole, also known as VT-1161, is a tetrazole antifungal agent potentially for the treatment of candidal vaginal infection. VT-1161 Protects Immunosuppressed Mice from Rhizopus arrhizus var. arrhizus Infection. VT-1161 dosed once daily or once weekly exhibits potent efficacy in treatment of dermatophytosis in a guinea pig model.
Oteseconazole has been used in trials studying the treatment of Tinea Pedis, Onychomycosis, Candidiasis, Vulvovaginal, and Recurrent Vulvovaginal Candidiasis.
Mycovia Pharmaceuticals is developing oteseconazole, the lead from a program of metalloenzyme Cyp51 (lanosterol demethylase) inhibitors, developed using the company’s Metallophile technology, for treating fungal infections including onychomycosis and recurrent vulvovaginal candidiasis (RVVC). In July 2021, oteseconazole was reported to be in phase 3 clinical development. Licensee Jiangsu Hengrui Medicine is developing otesaconazole, as an oral capsule formulation, for treating fungal conditions, including RVVC, onychomycosis and invasive fungal infections, in Greater China and planned for a phase 3 trial in April 2021 for treating VVC.
- OriginatorViamet Pharmaceuticals
- DeveloperMycovia Pharmaceuticals; Viamet Pharmaceuticals
- ClassAntifungals; Foot disorder therapies; Pyridines; Small molecules; Tetrazoles
- Mechanism of Action14-alpha demethylase inhibitors
- PreregistrationVulvovaginal candidiasis
- Phase IIOnychomycosis
- No development reportedTinea pedis
- 01 Jun 2021Preregistration for Vulvovaginal candidiasis (In adolescents, In adults, In children, Recurrent) in USA (PO)
- 01 Jun 2021Mycovia intends to launch otesaconazole (Recurrent) for Vulvovaginal candidiasis in the US in early 2022
- 06 Jan 2021Interim efficacy and adverse events data from a phase III ultraVIOLET trial in Vulvovaginal candidiasis released by Mycovia Pharmaceuticals

Synthesis Reference
Hoekstra, WJ., et al. (2020). Antifungal compound process (U.S. Patent No. US 10,745,378 B2). U.S. Patent and Trademark Office. https://patentimages.storage.googleapis.com/f4/62/19/5ba525b1caad0e/US10745378.pdf
PATENT
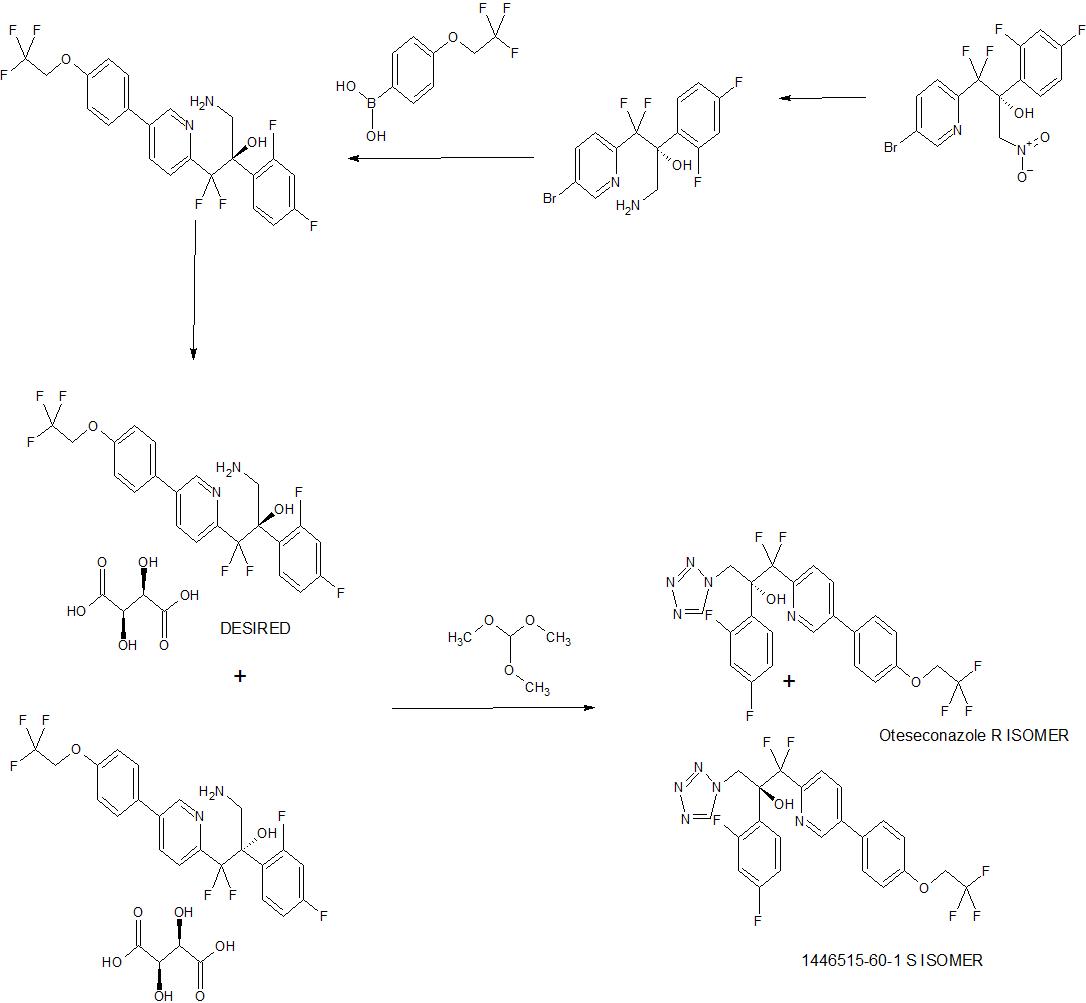
WO 2017049080
WO 2016149486
US 20150024938
WO 2015143172
WO 2015143184
WO 2015143180
WO 2015143142
WO 2013110002
WO 2013109998
WO 2011133875
PATENT
WO 2017049080,
Syn
J. Med. Chem. 2024, 67, 4376−4418
Oteseconazole was approved by the USFDA in April 2022 for the treatment of recurrent vulvovaginal candidiasis in women with a history of vulvovaginal candidiasis and who are not of reproductive
potential. Additional studies for other invasive and opportunistic infections and for onychomycosis are underway.40, The design and discovery of oteseconazole is published by a group from Viamet Pharmaceuticals, now part of Mycovia Pharmaceuticals. It details the racemic synthesis of the drug on
<1 g scale in which the metal-binding tetrazole is installed by treatment of ester 5.2 (Scheme 10) with diazomethane and tetrazole.42
A more scale-friendly asymmetric route that avoided the use of diazomethane was subsequently disclosed in patents and is detailed in Scheme 10 and Scheme 11.43
First, a mixture of ethyl bromodifluoroacetate, stoichiometric copper
powder, and 2,5-dibromopyridine (5.1) in DMSO provided ester 5.2 as an oil that was purified via distillation (Scheme10). Conversion to the aryl ketone 5.5 was achieved via direct addition of lithiated 5.3 or via a two-step process by first conversion to morpholine amide 5.4 followed by addition of
the Grignard generated from aryl bromide 5.3. The resulting ketone 5.5 was a liquid that was carried into the next step without purification.
The key step in the synthesis of 5 is an asymmetric Henry reaction using cinchona alkaloid catalyst 5.6. Addition of nitromethane to ketone 5.5 furnished alcohol 5.7 in 75% yield and ∼90:10 ratio of enantiomers. Next, reduction of the nitro group to the primary amine was accomplished using Pt
catalyzed hydrogenation. The chiral purity of the resulting amine was upgraded by classical resolution using di-p-toluoyl L-tartaric acid to provide 5.8·L-DTTA in 33% yield and >99% chiral purity.Conversion of amino alcohol 5.8 to oteseconazole (5) required two steps: cross coupling to introduce the aryltrifluoroethyl ether fragment and tetrazole formation. These steps were performed in either sequence in the patent. The route shown in Scheme 11 represents the largest scale demonstrated (>100 g input of 5.8). While the use of azide containing reagents presents significant safety risks, no information was provided on safe operation of the tetrazole forming step in the laboratory or on plant scale. Some of the
procedures for tetrazole formation described in the patent would likely require modification for safe scale-up.
To complete the synthesis of oteseconazole, resolved amino alcohol 5.8 first underwent a salt break followed by Suzuki coupling using boronic acid 5.9 to provide biaryl product 5.10 as the L-tartrate salt (Scheme 11). Conversion of 5.10 to 5 was accomplished using TMSN3 in acetic acid with sodium acetate and trimethoxy orthoformate. Treatment of the resulting solution with a Pd scavenger preceded crystallization of the product from EtOH and water after pH adjustment with potassium carbonate. The product was isolated in 85% yield as a hydrated form. Another patent described conversion of the oteseconazolehydrate totheanhydrous form byrecrystallizationfrom EtOHandn-heptanetofurnish5 in90%yield.45
(40) Hoy, S. M. Oteseconazole: First approval. Drugs 2022, 82,1017−1023.
(41) Sobel, J. D.; Nyirjesy, P. Oteseconazole: an advance in
treatment of recurrent vulvovaginal candidiasis. Future Microbiol 2021,
16, 1453−1461.
(42) Hoekstra, W. J.; Garvey, E. P.; Moore, W. R.; Rafferty, S. W.;
Yates, C. M.; Schotzinger, R. J. Design and optimization of highly
selective fungal CYP51 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24,
3455−3458.
(43) Wirth, D. D.; Yates, C. M.; Hoekstra, W. J.; Bindl, M. F.;
Hartmann, E. Process for enantioselective preparation of tetrazolyl
pyridinyl diaryl propanols as antifungal drugs and their precursors.
WO 2017049080, 2017.
(44) González-Bobes, F.; Kopp, N.; Li, L.; Deerberg, J.; Sharma, P.;
Leung, S.; Davies, M.; Bush, J.; Hamm, J.; Hrytsak, M. Scale-up of
Azide Chemistry: A Case Study. Org. Process Res. Dev. 2012, 16,
2051−2057.
(45) Hoekstra, W. J.; Wirth, D. D.; Ehiwe, T.; Bonnaud, T.
Antifungal compounds and processes for making. WO 2016149486,
2016.


.
PATENT
WO-2021143811
Novel crystalline polymorphic form of VT-1161 (also known as oteseconazole) phosphate disodium salt, useful as a prodrug of oteseconazole, for treating systemic fungal infection (eg Candida albicans infection) or onychomycosis.The function of metalloenzymes is highly dependent on the presence of metal ions in the active site of the enzyme. It is recognized that reagents that bind to and inactivate metal ions at the active site greatly reduce the activity of the enzyme. Nature uses this same strategy to reduce the activity of certain metalloenzymes during periods when enzyme activity is not needed. For example, the protein TIMP (tissue inhibitor of metalloproteinases) binds to zinc ions in the active sites of various matrix metalloproteinases, thereby inhibiting enzyme activity. The pharmaceutical industry has used the same strategy in the design of therapeutic agents. For example, the azole antifungal agents fluconazole and voriconazole contain 1-(1,2,4-triazole) group, which exists in the active site of the target enzyme lanosterol demethylase The heme iron binds, thereby inactivating the enzyme. Another example includes zinc-bound hydroxamic acid groups, which have been introduced into most of the published inhibitors of matrix metalloproteinases and histone deacetylases. Another example is the zinc-binding carboxylic acid group, which has been introduced into most of the published angiotensin converting enzyme inhibitors.
VT-1161, the compound 2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2, 2,2-Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol, is an antifungal drug developed by VIAMET, currently in the clinical research stage, its structure is as follows Shown:
This compound mainly acts on the CYP51 target of fungal cells. Compared with the previous triazole antifungal drugs, it has the advantages of wider antibacterial spectrum, low toxicity, high safety and good selectivity. However, this compound is not suitable for Liquid preparations (including or excluding the parenteral delivery carrier) are used to treat patients in need thereof.
2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2,2-trifluoro Ethoxy)phenyl)pyridin-2-yl)propan-2-yl dihydrogen phosphate is a prodrug of VT-1161.
On the other hand, nearly half of the drug molecules are in the form of salts, and salt formation can improve certain undesirable physicochemical or biological properties of the drug. Relative to 2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2,2- Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-yl dihydrogen phosphate, it is of great significance to develop salts with more excellent properties in terms of physical and chemical properties or pharmaceutical properties.To this end, the present disclosure provides a new pharmaceutically acceptable salt form of a metalloenzyme inhibitor.Example 1:[0161](R)-2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2, 2-Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-yl phosphate disodium salt (Compound 1)[0162]
[0163](R)-2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2 ,2-Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-yl phosphate (compound 1a, prepared according to the method of patent WO2013110002, 0.28g, 0.46mmol, 1.0eq) and ethanol (5mL ) Add to the reaction flask and stir evenly. A solution of NaOH (36.90 mg, 2.0 eq) dissolved in water (1 mL) was added dropwise into the above reaction flask, stirring was continued for 2 h, and concentrated to obtain compound 1, 300 mg of white solid.[0164]After X-ray powder diffraction detection, the XRPD spectrum has no sharp diffraction peaks, as shown in FIG. 10.[0165]Ms:608.10[M-2Na+3H] + .[0166]Ion chromatography detected that the sodium ion content was 6.23%.[0167]Example 2: (R)-((2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4 -(2,2,2-Trifluoroethoxy)phenyl)pyridin-2-yl)prop-2-yl)oxy)methyl phosphate disodium salt (compound 2)
[0169]Under ice-cooling, NaH (58mg, 0.87mmol) was added to the reaction flask, 1.5mL of N,N-dimethylformamide and 0.6mL of tetrahydrofuran were added, followed by iodine (38mg, 0.15mmol), and then Compound 2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2,2-tri Fluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (2b, prepared according to the method of patent WO2013110002, 158mg, 0.3mmol) tetrahydrofuran (1ml) solution was added to the reaction solution, stirred and reacted for 1-4h , And then add compound 2a (519mg, 2.01mmol) in tetrahydrofuran (1ml) solvent to the reaction, stir until the reaction is complete, 10% aqueous ammonium chloride solution to quench the reaction, extract, concentrate and drain, the crude product 2c is directly used for the next One-step reaction, Ms: 750.0[M+H] + .[0170]
[0171]Under ice-bath cooling, add trifluoroacetic acid (0.5mL) to the crude product 2c (300mg) in dichloromethane (2mL) solution, stir until the reaction is complete, and after concentration, the target compound 2d, 82mg, Ms was separated by high performance liquid phase separation. :638.0[M+H] + .[0172]
Add compound 2d (0.29g, 0.46mmol, 1.0eq) and ethanol (5mL) obtained in the previous step into the reaction flask, stir, and add NaOH (36.90mg, 2.0eq) water (1ml) solution dropwise to the aforementioned reaction solution , Stirred for 2-5 h, and concentrated to obtain 2,313 mg of the target compound.
Ms:638.10[M-2Na+3H] + .
PATENT
WO2011133875
https://patents.google.com/patent/WO2011133875A2/en
Product pat, WO2011133875 , protection in the EU states and the US April 2031.
PATENT
WO2015143184 ,
https://patents.google.com/patent/WO2015143184A1/en
Mycovia, claiming a process for preparing antifungal compounds, particularly oteseconazole.EXAMPLE 11

2-(2,4-Difluorophenyl)-l,l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4-(2,2,2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (11)Compound 11 was prepared using the conditions employed for 1: 0.33 g as a solid. The precursor l-bromo-4-(2,2,2-trifluoroethoxy)benzene was prepared as described below in one step.1H NMR (500 MHz, CDC13): δ 8.76 (s, 1 H), 8.70 (s, 1 H), 7.95 (d, / = 8.0 Hz, 1 H), 7.70 (s, 1 H), 7.64 (d, / = 8.5 Hz, 1 H), 7.54 (d, / = 8.5 Hz, 2 H), 7.42- 7.37 (m, 1 H), 7.08 (d, / = 8.5 Hz, 2 H), 6.79- 6.75 (m, 1 H), 6.69- 6.66 (m, 1 H), 5.58 (d, / = 14.0 Hz, 1 H), 5.14 (d, / = 14.0 Hz, 1 H), 4.44 – 4.39 (m, 2 H). HPLC: 99.1%. MS (ESI): m/z 528 [M++l].Chiral preparative HPLC Specifications for (+)-ll:Column: Chiralpak IA, 250 x 4.6mm, 5uMobile Phase: A) w-Hexane, B) IPAIsocratic: A: B (65:35)Flow Rte: l.OO mL/minOptical rotation [a]D: + 24° (C = 0.1 % in MeOH). 1 -Bromo-4-( 2,2,2-trifluoroethoxy )benzeneTo a stirred solution of trifluoroethyl tosylate (1.5 g, 5.8 mmol) in DMF (20 mL) was added K2CO3 (4 g, 29.4 mmol) followed by addition of p-bromo phenol (1.1 g, 6.46 mmol) at RT under inert atmosphere. The reaction mixture was stirred at 120 °C for 6 h. The volatiles were evaporated under reduced pressure; the residue was diluted with water (5 mL) and extracted with ethyl acetate (3 x 30 mL). The organic layer was washed with water, brine and dried over anhydrous Na2S04, filtered and concentrated in vacuo. The crude compound was purified by silica gel column chromatography eluting with 5% EtOAc/hexane to afford the desired product (0.8 g, 3.13 mmol, 53.3%) as semi solid. 1H NMR (200 MHz, CDC13): δ 7.44 – 7.38 (m, 2 H), 6.86-6.80 (m, 2 H), 4.38- 4.25 (m, 2 H).ExamplesThe present invention will now be demonstrated using specific examples that are not to be construed as limiting.General Experimental ProceduresDefinitions of variables in the structures in schemes herein are commensurate with those of corresponding positions in the formulae delineated herein.Synthesis of 1 or la

A process to prepare enantiopure compound 1 or la is disclosed. Syntheses of lor la may be accomplished using the example syntheses that are shown below (Schemes 1-4). The preparation of precursor ketone 3-Br is performed starting with reaction of 2,5-dibromo- pyridine with ethyl 2-bromo-difluoroacetate to produce ester 2-Br. This ester can be reacted with morpholine to furnish morpholine amide 2b-Br, followed by arylation to provide ketone 3-Br. Alternatively, ketone 3-Br can be afforded directly from ester 2-Br as shown in Scheme 1. Scheme 1. Synthesis of ketone 3-Br r

Ketone 3 may be prepared in an analogous fashion as described in Scheme 1 starting from corresponding substituted 2-bromo-pyridines, which can be prepared according to synthetic transformations known in the art and contained in the references cited herein (Scheme 2).Scheme 2. Synthesis of ketone 3

R-i = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, – 0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, – 0(S02)-aryl, or -0(S02)-substituted aryl.Alternatively, compound 1 can be prepared according to Scheme 3 utilizing diols 2-6b (or 2- 6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof). Olefins 2-5a and 2-5 can be prepared by reacting ketones 3 and 1-4 under Wittig olefination conditions (e.g., Ph3PCH3Br and BuLi). Also, as indicated in Scheme 5, any of pyridine compounds, 3, 2-5a, 2-6b, 2-7b, 4*, 4b, or 6 can be converted to the corresponding 4-CF3CH2O-PI1 analogs (e.g., 1-4, 2-5, 2-6a, 2-7a, 5*, 1-6*, or 1 or the corresponding enantiomers, or mixtures thereof) by cross-coupling with 4,4,5, 5-tetramethyl-2- (4-(2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (or the corresponding alkyl boronates or boronic acid or the like), in a suitable solvent system (e.g., an organic-aqueous solvent mixture), in the presence of a transition metal catalyst (e.g., (dppf)PdCl2), and in the presence of a base (e.g., KHCO3, K2C03, Cs2C03, or Na2C03, or the like). Olefins 2-5a and 2-5 can be transformed to the corresponding chiral diols, 2-6b (or 2-6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof), through exposure to Sharpless asymmetric dihydroxylation conditions: 1) commercially available AD- mix alpha or AD-mix beta with or without additional osmium oxidant and methanesulfonamide, 2) combination of a catalytic osmium oxidant (e.g., Os04 or K20sC>2(OH)4), a stoichiometric iron oxidant (e.g., K3Fe(CN)6), a base (e.g., KHCO3, K2CO3, Cs2C03, or Na2C03, or the like), and a chiral ligand (e.g., (DHQ)2PHAL, (DHQD)2PHAL, (DHQD)2AQN, (DHQ)2AQN, (DHQD)2PYR, or (DHQ)2PYR; preferably (DHQ)2PHAL, (DHQD)2PHAL, (DHQD)2AQN, and (DHQD)2PYR), or 3) option 2) with methanesulfonamide. The primary alcohol of the resultant chiral diols, 2-6b (or 2-6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof), can then be activated to afford compounds 2-7b (or 2-7d, the enantiomer of 2-7b, or mixtures thereof) or 2-7a (or 2-7c, the enantiomer of 2-7a, or mixtures thereof). For example, the mesylates can be prepared by exposing chiral diols, 2-6b (or 2-6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof), to methanesulfonyl chloride and a base. Epoxide formation can be affected by the base-mediated (e.g., KHCO3, K2CO3, CS2CO3, or Na2CC>3, or the like) ring closure of compounds 2-7b (or 2- 7d, the enantiomer of 2-7b, or mixtures thereof) or 2-7a (or 2-7c, the enantiomer of 2-7a, or mixtures thereof) to provide epoxides 4* (or 4c*, the enantiomer of 4*, or mixtures thereof) and 5* (or 5-b*, the enantiomer of 5*, or mixtures thereof). The epoxides can then be converted into amino-alcohols 4b (or 4c, the enantiomer of 4b, or mixtures thereof) and 1-6* (or 1-7*, the enantiomer of 1-6*, or mixtures thereof) through ammonia-mediated epoxide opening using ammonia in a suitable solvent (e.g., MeOH, EtOH, or water). Subsequent treatment with TMS-azide in the presence of trimethylorthoformate and sodium acetate in acetic acid would yield compounds 6 (or 6a, the enantiomer of 6, or mixtures thereof) or 1 (or la, the enantiomer of 1, or mixtures thereof) (US 4,426,531).Scheme 3. Synthesis of 1 via Asymmetric Dihydroxylation Method


Y is -OS02-alkyl, -OS02-substituted alkyl, -OS02-aryl, -OS02- substituted aryl, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, – 0(C=0)-aryl, -0(C=0)-substituted aryl, or halogen

R-i = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, -0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, -0(S02)-aryl, or -0(S02)-substituted aryl.Compound 1 (or la, the enantiomer of 1, or mixtures thereof) prepared by any of the methods presented herein can be converted to a sulfonic salt of formula IX (or IXa, the enantiomer of IX, or mixtures thereof), as shown in Scheme 4. This can be accomplished by a) combining compound 1 (or la, the enantiomer of 1, or mixtures thereof), a crystallization solvent or crystallization solvent mixture (e.g., EtOAc, i‘PrOAc, EtOH, MeOH, or acetonitrile, or oZ-S-OHcombinations thereof), and a sulfonic acid o (e.g., Z = Ph, p-tolyl, Me, or Et), b) diluting the mixture with an appropriate crystallization co-solvent or crystallization co-solvent mixture (e.g., pentane, methyl i-butylether, hexane, heptane, or toluene, or combinations thereof), and c) filtering the mixture to obtain a sulfonic acid salt of formula IX (or IXa, the enantiomer of IX, or mixtures thereof). cheme 4. Synthesis of a Sulfonic Acid Salt of Compound 1 or la

The following describes the HPLC method used in assessing HPLC purity of the examples and intermediates presented below:Column: Waters XBridge Shield RP18, 4.6 x 150 mm, 3.5 μιηMobile Phase: A = 0.05% TFA/H20, B = 0.05% TFA/ACNAutosampler flush: 1 : 1 ACN/H20Diluent: 1:1 ACN/H20Flow Rate: 1.0 ml/minTemperature: 45 °CDetector: UV 275 nmPump Parameters:

EXAMPLE 1Preparation of ethyl 2-(5-bromopyridin-2-yl)-2,2-difluoroacetate (2-Br)

2-Br Dialkylated impurity In a clean multi-neck round bottom flask, copper powder (274.7 g, 2.05 eq) was suspended in dimethyl sulfoxide (3.5 L, 7 vol) at 20 – 35 °C. Ethyl bromodifluoroacetate (449 g, 1.05 eq) was slowly added to the reaction mixture at 20 – 25 °C and stirred for 1 – 2 h. 2, 5- dibromopyridine (500 g, 1 eq) was added to the reaction mixture and the temperature was increased to 35 – 40 °C. The reaction mixture was maintained at this temperature for 18 – 24 h and the reaction progress was monitored by GC.After the completion of the reaction, ethyl acetate (7 L, 14 vol) was added to the reaction mixture and stirring was continued for 60 – 90 min at 20 – 35 °C. The reaction mixture was filtered through a Celite bed (100 g; 0.2 times w/w Celite and 1L; 2 vol ethyl acetate). The reactor was washed with ethyl acetate (6 L, 12 vol) and the washings were filtered through a Celite bed. The Celite bed was finally washed with ethyl acetate (1 L, 2 vol) and all the filtered mother liquors were combined. The pooled ethyl acetate solution was cooled to 8 – 10 °C, washed with the buffer solution (5 L, 10 vol) below 15 °C (Note: The addition of buffer solution was exothermic in nature. Controlled addition of buffer was required to maintain the reaction mixture temperature below 15 °C). The ethyl acetate layer was washed again with the buffer solution until (7.5 L; 3 x 5 vol) the aqueous layer remained colorless. The organic layer was washed with a 1: 1 solution of 10 % w/w aqueous sodium chloride and the buffer solution (2.5 L; 5 vol). The organic layer was then transferred into a dry reactor and the ethyl acetate was distilled under reduced pressure to get crude 2-Br.The crude 2-Br was purified by high vacuum fractional distillation and the distilled fractions having 2-Br purity greater than 93 % (with the dialkylated not more than 2 % and starting material less than 0.5 %) were pooled together to afford 2-Br.Yield after distillation: 47.7 % with > 93 % purity by GC (pale yellow liquid). Another 10 % yield was obtained by re-distillation of impure fractions resulting in overall yield of ~ 55 – 60 %.*H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz): 8.85 (1H, d, 1.6 Hz), 8.34 (1H, dd, J = 2.0 Hz, 6.8 Hz), 7.83 (1H, d, J = 6.8 Hz), 4.33 (2H, q, J = 6.0 Hz), 1.22 (3H, t, J = 6.0 Hz). 13C NMR: 162.22 (i, -C=0), 150.40 (Ar-C-), 149.35 (t, Ar-C), 140.52 (Ar-C), 123.01 (Ar-C), 122.07 (Ar-C), 111.80 (t, -CF2), 63.23 (-OCH2-), 13.45 (-CH2CH3).EXAMPLE 2
Preparation of2-( 5-bromopyridin-2-yl )-l -(2,4-difluorophenyl )-2, 2-difluoroethanone ( 3-Br ) A. One-step Method

l-Bromo-2,4-difluorobenzene (268.7 g; 1.3 eq) was dissolved in methyl tert butyl ether (MTBE, 3.78 L, 12.6 vol) at 20 – 35 °C and the reaction mixture was cooled to -70 to -65 °C using acetone/dry ice bath. n-Butyl lithium (689 rriL, 1.3 eq; 2.5 M) was then added to the reaction mixture maintaining the reaction temperature below -65 °C (Note: Controlled addition of the n-Butyl Lithium to the reaction mixture was needed to maintain the reaction mixture temperature below – 65 °C). After maintaining the reaction mixture at this temperature for 30 – 45 min, 2-Br (300 g, 1 eq) dissolved in MTBE (900 rriL, 3 vol) was added to the reaction mixture below – 65 °C. The reaction mixture was continued to stir at this temperature for 60 – 90 min and the reaction progress was monitored by GC.The reaction was quenched by slow addition of 20 % w/w ammonium chloride solution (750 mL, 2.5 vol) below -65 °C. The reaction mixture was gradually warmed to 20 – 35 °C and an additional amount of 20 % w/w ammonium chloride solution (750 mL, 2.5 vol) was added. The aqueous layer was separated, the organic layer was washed with a 10 % w/w sodium bicarbonate solution (600 mL, 2 vol) followed by a 5 % sodium chloride wash (600 mL, 2 vol). The organic layer was dried over sodium sulfate (60 g; 0.2 times w/w), filtered and the sodium sulfate was washed with MTBE (300 mL, 1 vol). The organic layer along with washings was distilled below 45 °C under reduced pressure until no more solvent was collected in the receiver. The distillation temperature was increased to 55 – 60 °C, maintained under vacuum for 3 – 4 h and cooled to 20 – 35 °C to afford 275 g (73.6 % yield, 72.71 % purity by HPLC) of 3-Br as a pale yellow liquid.*H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):8.63 (1H, d, 1.6 Hz, Ar-H), 8.07 – 8.01 (2H, m, 2 x Ar-H), 7.72 (1H, d, J = 6.8 Hz, Ar-H), 7.07 – 6.82 (1H, m, Ar-H), 6.81 – 6.80 (1H, m, Ar-H). 13C NMR: 185.60 (t, -C=0), 166.42 (dd, Ar-C-), 162.24 (dd, Ar-C),150.80 (Ar-C), 150.35 (Ar-C), 140.02 (Ar-C), 133.82 (Ar-C), 123.06 (Ar-C), 1122.33 (Ar-C), 118.44 (Ar-C), 114.07 (-CF2-), 122.07 (Ar-C), 105.09 (Ar-C).
B. Two-step Method via 2b-Br

2-Br (147.0 g) was dissolved in n-heptane (1.21 L) and transferred to a 5-L reactor equipped with overhead stirrer, thermocouple, condenser and addition funnel. Morpholine (202 ml) was added. The solution was heated to 60 °C and stirred overnight. The reaction was complete by HPLC analysis (0.2% 2-Br; 94.7% 2b-Br). The reaction was cooled to room temperature and 1.21 L of MTBE was added. The solution was cooled to ~4 °C and quenched by slow addition of 30% citric acid (563 ml) to maintain the internal temperature <15 °C. After stirring for one hour the layers were allowed to settle and were separated (Aq. pH=5). The organic layer was washed with 30% citric acid (322 ml) and 9% NaHC03 (322 ml, aq. pH 7+ after separation). The organic layer was concentrated on the rotary evaporator (Note 1) to 454 g (some precipitation started immediately and increased during concentration). After stirring at room temperature the suspension was filtered and the product cake was washed with n-heptane (200 ml). The solid was dried in a vacuum oven at room temperature to provide 129.2 g (77%) dense powder. The purity was 96.5% by HPLC analysis.To a 1-L flask equipped with overhead stirring, thermocouple, condenser and addition funnel was added magnesium turnings (14.65 g), THF (580 ml) and l-bromo-2,4-difluorobenzene (30.2 g, 0.39 equiv). The mixture was stirred until the reaction initiated and self-heating brought the reaction temperature to 44 °C. The temperature was controlled with a cooling bath as the remaining l-bromo-2,4-difluorobenzene (86.1 g, 1.11 equiv) was added over about 30 min. at an internal temperature of 35-40 °C. The reaction was stirred for 2 hours while gradually cooling to room temperature. The dark yellow solution was further cooled to 12 °C.During the Grignard formation, a jacketed 2-L flask equipped with overhead stirring, thermocouple, and addition funnel was charged with morpholine amide 2b-Br (129.0 g) and THF (645 ml). The mixture was stirred at room temperature until the solid dissolved, and then the solution was cooled to -8.7 °C. The Grignard solution was added via addition funnel over about 30 min. at a temperature of -5 to 0 °C. The reaction was stirred at 0 °C for 1 hour and endpointed by HPLC analysis. The reaction mixture was cooled to -5 °C and quenched by slow addition of 2N HC1 over 1 hour at <10 °C. The mixture was stirred for 0.5 h then the layers were allowed to settle and were separated. The aqueous layer was extracted with MTBE (280 ml). The combined organic layers were washed with 9% NaHCC>3 (263 g) and 20% NaCl (258 ml). The organic layer was concentrated on the rotary evaporator with THF rinses to transfer all the solution to the distillation flask. Additional THF (100 ml) and toluene (3 x 100 ml) were added and distilled to remove residual water from the product. After drying under vacuum, the residue was 159.8 g of a dark brown waxy solid (>theory). The purity was approximately 93% by HPLC analysis.EXAMPLE 3Preparation of 3-amino-l-(5-bromopyridin-2-yl)-2-(2,4-difluorophenyl)-l,l-difluoropropan- -ol (±ib-Br)

4-Br (200g, 1 eq) was added into methanolic ammonia (8.0 L; 40 vol; ammonia content: 15 – 20 % w/v) in an autoclave at 10 – 20 °C. The reaction mixture was gradually heated to 60 – 65 °C and at 3 – 4 kg/cm2 under sealed conditions for 10 – 12 h. The reaction progress was monitored by GC. After completion of the reaction, the reaction mixture was cooled to 20 – 30 °C and released the pressure gradually. The solvent was distilled under reduced pressure below 50 °C and the crude obtained was azeotroped with methanol (2 x 600 mL, 6 vol) followed by with isopropanol (600 mL, 2 vol) to afford 203 g (96.98 % yield, purity by HPLC: 94.04 %) of +4b-Br. EXAMPLE 4Preparation of3-amino-l-(5-bromopyridin-2-yl)-2-(2,4-difluorophenyl)-l,l-difluoropropan- -ol (4b-Br or 2c-Br)

Amino alcohol ±4b-Br (150 g, 1 eq) was dissolved in an isopropanol /acetonitrile mixture (1.5L, 8:2 ratio, 10 vol) and Di-p-toluoyl-L-tartaric acid (L-DPTTA) (84.05 g, 0.55 eq) was added into the reactor at 20 – 30 °C. The reaction mixture was heated to 45 – 50 °C for 1 – 1.5 h (Note: The reaction mixture becomes clear and then became heterogeneous). The reaction mixture was gradually cooled to 20 – 30 °C and stirred for 16 – 18 h. The progress of the resolution was monitored by chiral HPLC analysis.After the completion of the resolution, the reaction mixture was gradually cooled to 20 – 35 °C. The reaction mixture was filtered and the filtered solid was washed with a mixture of acetonitrile and isopropanol (8:2 mixture, 300 mL, 2 vol) and dried to afford 75 g of the L- DPTTA salt (95.37 % ee). The L-DPTTA salt obtained was chirally enriched by suspending the salt in isopropanol /acetonitrile (8:2 mixture; 750 mL, 5 vol) at 45 – 50 °C for 24 – 48 h. The chiral enhancement was monitored by chiral HPLC; the solution was gradually cooled to 20 – 25 °C, filtered and washed with an isoporpanol /acetonitrile mixture (8:2 mixture; 1 vol). The purification process was repeated and after filtration, the salt resulted in chiral purity greater than 96 % ee. The filtered compound was dried under reduced pressure at 35 – 40 °C to afford 62 g of the enantio-enriched L-DPPTA salt with 97.12% ee as an off-white solid. The enantio-enriched L-DPTTA salt (50 g, 1 eq) was dissolved in methanol (150 mL, 3 vol) at 20 – 30 °C and a potassium carbonate solution (18.05 g K2CO3 in 150 mL water) was slowly added at 20 – 30 °C under stirring. The reaction mixture was maintained at this temperature for 2 – 3 h (pH of the solution at was maintained at 9). Water (600 mL, 12 vol) was added into the reaction mixture through an additional funnel and the reaction mixture was stirred for 2 – 3 h at 20 – 30 °C. The solids were filtered; washed with water (150 mL, 3 vol) and dried under vacuum at 40 – 45 °C to afford 26.5 g of amino alcohol 4b-Br or 4c-Br with 99.54 % chemical purity, 99.28 % ee as an off-white solid. (Water content of the chiral amino alcohol is below 0.10 % w/w).1H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):8.68 (1H, d, J = 2.0 Hz, Ar- H), 8.16 (1H, dd, J = 8.0 Hz, 2.0 Hz, Ar-H), 7.49 – 7.43 (1H, m, Ar-H), 7.40 (1H, d, J = 8 Hz, Ar-H), 7.16 – 7.11 (1H, m, Ar-H), 7.11 – 6.99 (1H, m, Ar-H), 3.39 – 3.36 (1H, m, -OCHAHB– ), 3.25 – 3.22 (1H, m, -OCHAHB-).13C NMR: 163.87 -158.52 (dd, 2 x Ar-C-), 150.88 (Ar-C), 149.16 (Ar-C), 139.21 (Ar-C), 132.39 (Ar-C), 124.49 (Ar-C), 122.17 (Ar-C), 121.87 (d, Ar- C), 119.91 (t, -CF2-), 110.68 (Ar-C), 103.97 (i, Ar-C), 77.41 (i,-C-OH), 44.17 (-CH2-NH2).EXAMPLE 5
Preparation of l-(5-bromopyridin-2-yl)-2-(2,4-difluorophenyl)-l,l-difluoro-3-(lH-tetrazol-l- yl)propan-2-ol (l-6*-Br or l-7*-Br)

4b-Br or 4c-Br (20.0 g, 1 eq.) was added to acetic acid (50 mL, 2.5 vol) at 25 – 35 °C followed by the addition of anhydrous sodium acetate (4.32 g, 1 eq), trimethyl orthoformate (15.08 g, 2.7 eq). The reaction mixture was stirred for 15 – 20 min at this temperature and trimethylsilyl azide (12.74 g, 2.1 eq) was added to the reaction mixture (Chilled water was circulated through the condenser to minimize the loss of trimethylsilyl azide from the reaction mixture by evaporation). The reaction mixture was then heated to 70 – 75 °C and maintained at this temperature for 2 -3 h. The reaction progress was monitored by HPLC. Once the reaction was complete, the reaction mixture was cooled to 25 – 35 °C and water (200 mL, 10 vol) was added. The reaction mixture was extracted with ethyl acetate (400 mL, 20 vol) and the aqueous layer was back extracted with ethyl acetate (100 mL, 5 vol). The combined organic layers were washed with 10 % potassium carbonate solution (3 x 200 mL; 3 x 10 vol) followed by a 10 % NaCl wash (1 x 200 mL, 10 vol). The organic layer was distilled under reduced pressure below 45 °C. The crude obtained was azeotroped with heptanes (3 x 200 mL) to get 21.5g (94 % yield, 99.26 5 purity) of tetrazole 1-6* or 1-7* compound as pale brown solid (low melting solid).1H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz NMR instrument): 9.13 (1H, Ar-H), 8.74 (1H, Ar-H), 8.22 – 8.20 (1H, m, Ar-H), 7.44 (1H, d, J = 7.2 Hz, Ar-H), 7.29 (1H„Ar-H), 7.23 – 7.17 (1H, m, Ar-H), 6.92 – 6.88 (1H, Ar-H), 5.61 (1H, d, J = 1 1.2 Hz, – OCHAHB-), 5.08 (1H, d, J = 5.6 Hz, -OCHAHB-).13C NMR: 163.67 -161.59 (dd, Ar-C-), 160.60 – 158.50 (dd, Ar-C-), 149.65 (Ar-C), 144.99 (Ar-C), 139.75 (Ar-C), 131.65 (Ar-C), 124.26 (Ar-C), 122.32 (d, Ar-C), 119.16 (t, -CF2-), 118.70 (d, Ar-C), 1 11.05 (d, Ar-C) 104.29 (t, Ar-C), 76.79 (i,-C-OH), 59.72 (Ar-C), 50.23 (-OCH2N-). EXAMPLE 6Preparation of 2-(2,4-difluorophenyl)-l , 1 -difluoro-3-( 1 H-tetrazol-1 -yl)-l -(5-(4-(2,2,2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (1 or la)A. Preparation of 1 or la via l-6*-Br or l-7*-Br

Synthesis of 4,4,5, 5-tetramethyl-2-(4-(2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane Potassium carbonate (59.7 g, 2.2 eq.) was added to a slurry of DMF (190 mL, 3.8 Vol.), 4- Bromo phenol (37.4g, 1.1 eq.) and 2,2,2-trifluroethyl tosylate (50.0 g, 1.0 eq.) at 20 – 35 °C under an inert atmosphere. The reaction mixture was heated to 115 – 120 °C and maintained at this temperature for 15 – 18 h. The reaction progress was monitored by GC. The reaction mixture was then cooled to 20 – 35 °C, toluene (200 mL, 4.0 vol.) and water (365 mL, 7. 3 vol.) were added at the same temperature, stirred for 10 – 15 minutes and separated the layers. The aqueous layer was extracted with toluene (200 mL, 4.0 vol.). The organic layers were combined and washed with a 2M sodium hydroxide solution (175 mL, 3.5 vol.) followed by a 20 % sodium chloride solution (175 mL, 3.5 vol.). The organic layer was then dried over anhydrous sodium sulfate and filtered. The toluene layer was transferred into clean reactor, spurged with argon gas for not less than 1 h. Bis(Pinacolato) diborane (47 g, 1.1 eq.), potassium acetate (49.6 g, 3.0 eq.) and 1,4-dioxane (430 mL, 10 vol.) were added at 20 -35 °C, and spurged the reaction mixture with argon gas for at least 1 h. Pd(dppf)Cl2 (6.88 g, 0.05eq) was added to the reaction mixture and continued the argon spurging for 10 – 15 minutes. The reaction mixture temperature was increased to 70 – 75 °C, maintained the temperature under argon atmosphere for 15 – 35 h and monitored the reaction progress by GC. The reaction mixture was cooled to 20 – 35 °C, filtered the reaction mixture through a Celite pad, and washed with ethyl acetate (86 mL, 2 vol.). The filtrate was washed with water (430 mL, 10 vol.). The aqueous layer was extracted with ethyl acetate (258 mL, 6 vol.) and washed the combined organic layers with a 10 % sodium chloride solution (215 mL, 5 vol.). The organic layer was dried over anhydrous sodium sulfate (43g, 1 time w/w), filtered and concentrated under reduced pressure below 45 °C to afford crude 4,4,5, 5-tetramethyl-2-(4-(2,2,2- trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (65 g; 71 % yield with the purity of 85.18 % by GC). The crude 4,4,5,5-tetramethyl-2-(4-(2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (65 g, 1 eq.) was dissolved in 10 % ethyl acetate – n-Heptane (455 mL, 7 vol.) and stirred for 30 – 50 minutes at 20 – 35 °C. The solution was filtered through a Celite bed and washed with 10 % ethyl acetate in n-Heptane (195 mL, 3 vol.). The filtrate and washings were pooled together, concentrated under vacuum below 45 °C to afford 4,4,5, 5-tetramethyl-2-(4-(2,2,2- trifluoroethoxy)phenyl)-l,3,2-dioxaborolane as a thick syrup (45.5 g; 70 % recovery). This was then dissolved in 3 % ethyl acetate-n-heptane (4 vol.) and adsorbed on 100 – 200 M silica gel (2 times), eluted through silica (4 times) using 3 % ethyl acetate – n- heptane. The product rich fractions were pooled together and concentrated under vacuum. The column purified fractions (> 85 % pure) were transferred into a round bottom flask equipped with a distillation set-up. The compound was distilled under high vacuum below 180 °C and collected into multiple fractions. The purity of fractions was analyzed by GC (should be > 98 % with single max impurity < 1.0 %). The less pure fractions (> 85 % and < 98 % pure fraction) were pooled together and the distillation was repeated to get 19g (32% yield) of 4,4,5, 5-tetramethyl-2-(4- (2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane as a pale yellow liquid.*H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):7.64 (2H, d, 6.8 Hz), 7.06 (2H, d, J = 6.4 Hz), 4.79 (2H, q, J = 6.8 Hz), 1.28 (12H, s).13C NMR: 159.46 (Ar-C-O-), 136.24 (2 x Ar-C-), 127.77 – 120.9 (q, -CF3), 122.0 (Ar-C-B), 114.22 (2 x Ar-C-), 64.75 (q, J = 27.5 Hz).Synthesis of 2-(2.4-difluorophenyl)-l.l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4-(2.2.2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (1 or la)l-6*-Br or l-7*-Br (14 g, 0.03 mol, 1 eq) was added to tetrahydrofuran (168 mL, 12 vol) at 25 – 35 °C and the resulting solution was heated to 40 – 45 °C. The reaction mixture was maintained at this temperature for 20 – 30 min under argon bubbling. Sodium carbonate (8.59 g, 0.08 mol, 2.5 eq) and water (21 mL, 1.5 vol) were added into the reaction mixture and the bubbling of argon was continued for another 20 – 30 min. 4,4,5, 5-tetramethyl-2-(4-(2,2,2- trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (10.76 g, 1.1 eq) dissolved in tetrahydrofuran (42 mL, 3 vol) was added into the reaction mixture and argon bubbling was continued for 20 – 30 min. Pd(dppf)Cl2 (2.65 g, 0.1 eq) was added to the reaction mixture under argon bubbling and stirred for 20 – 30 min (Reaction mixture turned into dark red color). The reaction mixture was heated to 65 – 70 °C and maintained at this temperature for 3 – 4 h. The reaction progress was monitored by HPLC. The reaction mixture was cooled to 40 – 45 °C and the solvent was distilled under reduced pressure. Toluene (350 mL, 25 vol.) was added to the reaction mixture and stirred for 10 – 15 min followed by the addition of water (140 mL, 10 vol). The reaction mixture was filtered through Hyflo (42 g, 3 times), the layers were separated and the organic layer was washed with water (70 mL, 5 vol) and a 20 % w/w sodium chloride solution (140 mL, 10 vol). The organic layer was treated with charcoal (5.6 g, 0.4 times, neutral chalrcoal), filtered through Hyflo. (lS)-lO-Camphor sulfonic acid (7.2 g, 1 eq.) was added to the toluene layer and the resulting mixture was heated to 70 – 75 °C for 2 – 3 h. The reaction mixture was gradually cooled to 25 – 35 °C and stirred for 1 – 2 h. The solids were filtered, washed with toluene (2 x 5 vol.) and then dried under vacuum below 45 °C to afford 18.0 g of an off white solid. The solids (13.5 g, 1 eq.) were suspended in toluene (135 mL, 10 vol) and neutralized by adding 1M NaOH solution (1.48 vol, 1.1 eq) at 25 – 35 °C and stirred for 20 – 30 min. Water (67.5 mL, 5 vol) was added to the reaction mixture and stirred for 10 – 15 min, and then the layers were separated. The organic layer was washed with water (67.5 mL, 5 vol) to remove the traces of CSA. The toluene was removed under reduced pressure below 45 °C to afford crude 1 or la. Traces of toluene were removed by azeotroping with ethanol (3 x 10 vol), after which light brown solid of crude 1 or la (7.5 g, 80% yield) was obtained.The crude 1 or la (5 g) was dissolved in ethanol (90 mL, 18 vol.) at 20 – 35 °C, and heated to 40 – 45 °C. Water (14 vol) was added to the solution at 40 – 45 °C, the solution was maintained at this temperature for 30 – 45 min and then gradually cooled to 20 – 35 °C. The resulting suspension was continued to stir for 16 – 18 h at 20 – 35 °C, an additional amount of water (4 vol.) was added and the stirring continued for 3 – 4 h. The solids were filtered to afford 4.0 g (80% recovery) of 1 or la (HPLC purity >98%) as an off-white solid.1H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):9.15 (1H, s, Ar-H), 8.93 (1H, d, J = 0.8 Hz, Ar-H), .8.22 – 8.20 (1H, m, Ar-H), 7.80 (2H, d, J = 6.8 Hz, Ar-H), 7.52 (1H, d, J = 6.8 Hz, Ar-H), 7.29 (1H, d,J = 3.2Hz, Ar-H), 7.27 – 7.21 (1H, m, Ar-H), 7.23 – 7.21 (2H, d, J = 6.8 Hz, Ar-H), 7.19 (1H, d, J = 6.8 Hz, Ar-H), 6.93 – 6.89 (1H, m, Ar-H), 5.68 (1H, / = 12 Hz, -CHAHB), 5.12 (2H, d, J = 11.6 Hz, -CHAHB), 4.85 (2H, q, J = 1.6 Hz).13C NMR: 163.93 – 158.33 (m, 2 x Ar-C), 157.56 (Ar-C), 149.32 (i, Ar-C), 146.40 (Ar-C), 145.02 (Ar-C), 136.20 (Ar-C), 134.26 (2 x Ar-C), 131.88 – 131.74 (m, AR-C), 129.72 (Ar-C), 128.47 (2 x Ar-C), 123.97 (q, -CF2-), 122.41 (Ar-C), 119.30 (-CF3), 118.99 (Ar-C), 115.65 (2 x Ar-C), 110.99 (d, Ar-C), 104.22 (i, Ar-C), 77.41 – 76.80 (m, Ar-C), 64.72 (q, -OCH2-CF3), 50.54 (-CH2-N-).B. Preparation of 1 or la via 4b-Br or 4c-Br


Synthesis of 3-amino-2-(2.4-difluorophenyl)-l.l-difluoro-l-(5-(4-(2.2.2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (8a or 8b)Potassium carbonate (30.4 g) and water (53.3 g) were charged to a 1-L flask equipped with overhead stirring, thermocouple, and nitrogen/vacuum inlet valve, and stirred until dissolved. The boronic acid (19.37 g), a solution of 4b-Br or 4c-Br in 2-butanol (103.5 g, 27.8 g theoretical 4b-Br or 4c-Br)) and 2-BuOH (147.1 g) were added and stirred to form a clear mixture. The flask was evacuated and refilled with nitrogen 3 times. Pd(d f)2Cl2 (0.30 g) was added and stirred to form a light orange solution. The flask was evacuated and refilled with nitrogen 4 times. The mixture was heated to 85 °C and stirred overnight and endpointed by HPLC analysis. The reaction mixture was cooled to 60 °C and the layers were allowed to settle. The aqueous layer was separated. The organic layer was washed with 5% NaCl solution (5 x 100 ml) at 30-40 °C. The organic layer was filtered and transferred to a clean flask with rinses of 2-BuOH. The combined solution was 309.7 g, water content 13.6 wt% by KF analysis. The solution was diluted with 2-BuOH (189 g) and water (10 g). Theoretically the solution contained 34.8 g product, 522 ml (15 volumes) of 2-BuOH, and 52.2 ml (1.5 volumes) of water. L-Tartaric acid (13.25 g) was added and the mixture was heated to a target temperature of 70-75 °C. During the heat-up, a thick suspension formed. After about 15 minutes at 70-72 °C the suspension became fluid and easily stirred. The suspension was cooled at a rate of 10 °C/hour to 25 °C then stirred at 25 °C for about 10 hours. The product was collected on a vacuum filter and washed with 10:1 (v/v) 2-BuOH/water (50 ml) and 2- butanol (40 ml). The salt was dried in a vacuum oven at 60 °C with a nitrogen purge for 2 days. The yield was 40.08 g of 8a or 8b as a fluffy, grayish-white solid. The water content was 0.13 wt% by KF analysis. The yield was 87.3% with an HPLC purity of 99.48%. Synthesis of 2-(2,4-difluorophenyl)-l,l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4-(2,2,2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (1 or la)To a 350 ml pressure bottle were charged acetic acid (73 ml), 8a or 8b (34.8 g), sodium acetate (4.58 g) and trimethylorthoformate (16.0 g). The mixture was stirred for 18 min. at room temperature until a uniform suspension was obtained. Azidotrimethylsilane (8.88 g) was added and the bottle was sealed. The bottle was immersed in an oil bath and magnetically stirred. The oil bath was at 52 °C initially, and was warmed to 62-64 °C over about ½ hour. The suspension was stirred at 62-64 °C overnight. After 20.5 hours the suspension was cooled to room temperature and sampled. The reaction was complete by HPLC analysis. The reaction was combined with three other reactions that used the same raw material lots and general procedure (total of 3.0 g additional starting material). The combined reactions were diluted with ethyl acetate (370 ml) and water (368 ml) and stirred for about ½ hour at room temperature. The layers were settled and separated. The organic layer was washed with 10% K2C03 solution (370 ml/ 397 g) and 20% NaCl solution (370 ml/ 424 g). The organic layer (319 g) was concentrated, diluted with ethanol (202 g) and filtered, rinsed with ethanol (83 g). The combined filtrate was concentrated to 74 g of amber solution.The crude 1 or la solution in ethanol (74 g solution, containing theoretically 31.9 g 1 or la) was transferred to a 2-L flask equipped with overhead stirring, thermocouple, and addition funnel. Ethanol (335 g) was added including that used to complete the transfer of the 1 or la solution. The solution was heated to nominally 50 °C and water (392 g) was added over 12 minutes. The resulting hazy solution was seeded with 1 or la crystals and stirred at 50 °C. After about ½ hour the mixture was allowed to cool to 40 °C over about ½ hour during which time crystallization started. Some darker colored chunky solid separated out from the main suspension. The pH of the crystallizing mixture was adjusted from 4.5 to 6 using 41% KOH (1.7 g). After about 1 hour a good suspension had formed. Additional water (191 g) was added slowly over ½ hour. The suspension was heated to 50 °C and cooled at 5 °C/min to room temperature. After stirring overnight the suspension was cooled in a water bath to 16 °C and filtered after 1 hour. The wet cake was washed with 55:45 (v/v) water/ethanol (2 x 50 ml) and air-dried on the vacuum filter funnel overnight. Further drying at 40 °C in a vacuum oven with a nitrogen bleed resulted in no additional weight loss. The yield was 30.2 g of off-white fine powder plus some darker granular material. By in-process HPLC analysis there was no difference in the chemical purity of the darker and lighter materials. The purity was 99.4%. The water content was 2.16 wt% by KF analysis. The residual ethanol was 1.7 wt% estimated by ‘Ft NMR analysis. The corrected yield was 29.0 g, 91.0% overall yield for tetrazole formation and crystallization. The melting point was 65 °C by DSC analysis.
FDA Approves Mycovia Pharmaceuticals’ VIVJOA™ (oteseconazole), the First and Only FDA-Approved Medication for Recurrent Vulvovaginal Candidiasis (Chronic Yeast Infection)
– Approval of VIVJOA™ marks a significant therapeutic advancement for reducing the incidence of RVVC, a condition with substantial unmet need, in permanently infertile and postmenopausal women
– VIVJOA™ is the first FDA approval in Mycovia’s pipeline of novel treatments for fungal infections
– U.S. commercial launch of VIVJOA™ expected in Q2
April 28, 2022 07:55 AM Eastern Daylight Time
DURHAM, N.C.–(BUSINESS WIRE)–The U.S. Food and Drug Administration (FDA) approved VIVJOA™ (oteseconazole capsules), an azole antifungal indicated to reduce the incidence of recurrent vulvovaginal candidiasis (RVVC) in females with a history of RVVC who are NOT of reproductive potential. VIVJOA is the first and only FDA-approved medication for this condition and provides sustained efficacy demonstrated by significant long-term reduction of RVVC recurrence through 50 weeks versus comparators. VIVJOA is the first FDA-approved product for Mycovia Pharmaceuticals, Inc. (Mycovia), an emerging biopharmaceutical company dedicated to recognizing and empowering those living with unmet medical needs by developing novel therapies.
“We believe the market need for VIVJOA is strong, and we are eager to execute our commercial plans”Tweet this
RVVC, also known as chronic yeast infection, is defined by the Centers for Disease Control and Prevention (CDC) as three or more symptomatic acute episodes of yeast infection in 12 months. RVVC is a distinct condition from vulvovaginal candidiasis (VVC), and until now, there have been no FDA-approved medications specifically indicated for it. Nearly 75% of all adult women will have at least one yeast infection in their lifetime, with approximately half experiencing a recurrence. Of those women, up to 9% develop RVVC.
“After nearly two decades of living with chronic yeast infection and feeling like there was no hope from the itchiness, irritation and constant dread of when the next yeast infection would return, I was overjoyed to even be a part of this clinical trial,” said Leslie Ivey, RVVC patient and clinical trial participant. “It is gratifying to see RVVC finally get the attention it deserves.”
Symptoms of RVVC include vaginal itching, burning, irritation and inflammation. Some women may experience abnormal vaginal discharge and painful sexual intercourse or urination, causing variable but often severe discomfort and pain.
VIVJOA’s FDA approval is based upon the positive results from three Phase 3 trials of oteseconazole – two global, pivotal VIOLET studies and one U.S.-focused ultraVIOLET study, including 875 patients at 232 sites across 11 countries. In the two global VIOLET studies, 93.3% and 96.1% of women with RVVC who received VIVJOA did not have a recurrence for the 48-week maintenance period compared to 57.2% and 60.6% of patients who received placebo (p <0.001). In the ultraVIOLET study, 89.7% of women with RVVC who received VIVJOA cleared their initial yeast infection and did not have a recurrence for the 50-week maintenance period compared to 57.1% of those who received fluconazole followed by placebo (p <0.001). The most common side effects reported in Phase 3 clinical studies were headache (7.4%) and nausea (3.6%). VIVJOA is contraindicated in those with a hypersensitivity to oteseconazole, and based on data from rat studies, also in females who are of reproductive potential, pregnant, or lactating. Please see additional Important Safety Information below.
Patrick Jordan, CEO of Mycovia Pharmaceuticals and Partner at NovaQuest Capital Management, stated, “We celebrate this important milestone for Mycovia, as VIVJOA is the first antifungal in our pipeline to obtain FDA approval and achieves our goal to fulfill a previously unmet medical need among women suffering from RVVC. We are honored to lead this advancement in women’s health.”
“We believe the market need for VIVJOA is strong, and we are eager to execute our commercial plans,” Jordan continued. “As we enter a new chapter of our history as a commercial biopharmaceutical company, we will continue driving our mission forward to develop novel therapies for overlooked conditions.”
Oteseconazole is designed to inhibit fungal CYP51, which is required for fungal cell wall integrity, and this selective interaction is also toxic to fungi, resulting in the inhibition of fungal growth. Due to its chemical structure, oteseconazole has a lower affinity for human CYP enzymes as compared to fungal CYP enzymes. The FDA granted oteseconazole Qualified Infectious Disease Product and Fast Track designations.
“A medicine with VIVJOA’s sustained efficacy combined with the clinical safety profile has been long needed, as until now, physicians and their patients have had no FDA-approved medications for RVVC,” stated Stephen Brand, Ph.D., Chief Development Officer of Mycovia. “We are excited to be the first to offer a medication designed specifically for RVVC, a challenging and chronic condition that is expected to increase in prevalence over the next decade.”
Mycovia is planning its commercial launch of VIVJOA™ in the second quarter of 2022.
About Recurrent Vulvovaginal Candidiasis
RVVC is a debilitating, chronic infectious condition that affects 138 million women worldwide each year. RVVC, also known as chronic yeast infection, is a distinct condition from vulvovaginal candidiasis (VVC) and defined as three or more symptomatic acute episodes of yeast infection in 12 months. Primary symptoms include vaginal itching, burning, irritation and inflammation. Some women may experience abnormal vaginal discharge and painful sexual intercourse or urination, causing variable but often severe discomfort and pain.
About VIVJOA™
VIVJOA™ (oteseconazole) is an azole antifungal indicated to reduce the incidence of recurrent vulvovaginal candidiasis (RVVC) in females with a history of RVVC who are NOT of reproductive potential. VIVJOA is the first and only FDA-approved medication that provides sustained efficacy demonstrated by significant long-term reduction of RVVC recurrence through 50 weeks versus comparators. Oteseconazole is designed to inhibit fungal CYP51, which is required for fungal cell wall integrity, and this selective interaction is also toxic to fungi, resulting in the inhibition of fungal growth. Due to its chemical structure, oteseconazole has a lower affinity for human CYP enzymes as compared to fungal CYP enzymes. The FDA approved VIVJOA based upon the positive results from three Phase 3 clinical trials of oteseconazole – two global, pivotal VIOLET studies and one U.S.-focused ultraVIOLET study, including 875 patients at 232 sites across 11 countries.
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215888s000lbl.pdf
- ^ “Vivjoa: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 27 April 2022.
- ^ Jump up to:a b “FDA Approves Mycovia Pharmaceuticals’ VIVJOA (oteseconazole), the First and Only FDA-Approved Medication for Recurrent Vulvovaginal Candidiasis (Chronic Yeast Infection)” (Press release). Mycovia Pharmaceuticals. 28 April 2022. Retrieved 28 April 2022 – via Business Wire.
- ^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 76”. WHO Drug Information. 30 (3). hdl:10665/331020.
Further reading
- Sobel JD, Nyirjesy P (December 2021). “Oteseconazole: an advance in treatment of recurrent vulvovaginal candidiasis”. Future Microbiology. 16: 1453–1461. doi:10.2217/fmb-2021-0173. PMID 34783586.
External links
- “Oteseconazole”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03562156 for “A Study of Oral Oteseconazole for the Treatment of Patients With Recurrent Vaginal Candidiasis (Yeast Infection) (VIOLET)” at ClinicalTrials.gov
- Clinical trial number NCT03561701 for “A Study of Oral Oteseconazole (VT-1161) for the Treatment of Patients With Recurrent Vaginal Candidiasis (Yeast Infection) (VIOLET)” at ClinicalTrials.gov
- Clinical trial number NCT03840616 for “Study of Oral Oteseconazole (VT-1161) for Acute Yeast Infections in Patients With Recurrent Yeast Infections (ultraVIOLET)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Vivjoa |
| Other names | VT-1161 |
| License data | US DailyMed: Oteseconazole |
| Routes of administration | By mouth |
| Drug class | Antifungal |
| ATC code | J02AC06 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1340593-59-0 |
| PubChem CID | 77050711 |
| DrugBank | DB13055 |
| ChemSpider | 52083215 |
| UNII | VHH774W97N |
| KEGG | D11785 |
| ChEBI | CHEBI:188153 |
| ChEMBL | ChEMBL3311228 |
| ECHA InfoCard | 100.277.989 |
| Chemical and physical data | |
| Formula | C23H16F7N5O2 |
| Molar mass | 527.403 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////////OTESECONAZOLE, vt 1161, fungal infection, Candida albicans infection, onychomycosis, PHASE 3,
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
C1=CC(=CC=C1C2=CN=C(C=C2)C(C(CN3C=NN=N3)(C4=C(C=C(C=C4)F)F)O)(F)F)OCC(F)(F)F
NEW DRUG APPROVALS
ONE TIME
$10.00
Nangibotide


Nangibotide
LQEEDAGEYGCM-amide
CAS 2014384-91-7
- Molecular FormulaC54H82N14O22S2
- Average mass1343.439 Da
- 2014384‐91‐7
- L-Leucyl-L-glutaminyl-L-α-glutamyl-L-α-glutamyl-L-α-aspartyl-L-alanylglycyl-L-α-glutamyl-L-tyrosylglycyl-L-cysteinyl-L-methioninamide
- LR 12 peptide
- LQEEDAGEYG CM
L-Leucyl-L-glutaminyl-L-glutaminyl-L-α-glutamyl-L-α-aspartyl-L-alanylglycyl-L-α-glutamyl-L-tyrosylglycyl-L-cysteinyl-L-methionine
L-Methionine, L-leucyl-L-glutaminyl-L-glutaminyl-L-α-glutamyl-L-α-aspartyl-L-alanylglycyl-L-α-glutamyl-L-tyrosylglycyl-L-cysteinyl-нангиботидمانغيبوتيد南吉博肽
| Sequence (one letter code) | LQEEDAGEYGCM-amide |
|---|---|
| Sequence (three letter code) | H-Leu-Gln-Glu-Glu-Asp-Ala-Gly-Glu-Tyr-Gly-Cys-Met-NH2 |
- OriginatorInotrem
- ClassAnti-infectives; Anti-inflammatories; Anti-ischaemics; Antivirals; Peptides
- Mechanism of ActionTREML1 protein inhibitors
- Phase II/IIICOVID 2019 infections
- Phase IISeptic shock
- Phase IMyocardial infarction
- 12 Jul 2021Inotrem has patents pending for nangibotide use in severe forms of COVID-19
- 12 Jul 2021Inotrem receives funding from French government by Bpifrance for nangibotide development in COVID-2019 infections
- 12 Jul 2021Inotrem receives authorization from both the French and Belgian authorities to proceed with clinical development of nangibotide up to registration in COVID-2019 infections
Nangibotide, also referred as LR12, is an antagonist of triggering receptor expressed on myeloid cells (TREM)-1, and was derived from residues 94 to 105 of TREM-like transcript-1 (TLT-1).
TREM-1 plays a crucial role in the onset of sepsis by amplifying the host immune response. TLT-1– and TLT-1–derived peptides therefore exhibit anti-inflammatory properties by dampening TREM-1 signalling. LR12 blocks TREM-1 by binding to the TREM-1 ligand and provides protective effects during sepsis such as inhibiting hyper-responsiveness, organ damage, and death, without causing deleterious effects. The protective effects of modulating TREM-1 signalling are also evident in other models of inflammation such as: pancreatitis; haemorrhagic shock; inflammatory bowel diseases and inflammatory arthritis
Inotrem is developing the peptide nangibotide, a triggering receptor expressed on myeloid cells 1 inhibitor, for treating sepsis and septic shock. In July 2021, this drug was reported to be in phase 3 clinical development.
Nangibotide is an inhibitor of TREM-1, a receptor found on certain white blood cells. Activation of TREM-1 stimulates inflammation. Nangibotide is therefore being investigated as a treatment for the overwhelming inflammation typically seen in severe sepsis.
Mode of action
TREM-1 is a receptor found on neutrophils, macrophages and monocytes, key elements of the immune system. Activation of TREM-1 results in expression of NF-κB, which promotes systemic inflammation. Nangibotide inhibits TREM-1, thereby preventing the inflammatory activation. Absence of TREM-1 results in vastly reduced inflammation without impairing the ability to fight infection.[2]
Animal models
LR17, a mouse equivalent of nangibotide, improves survival in mouse models of severe sepsis.[3] In a pig model of sepsis, LR12 – another animal equivalent of nangibotide – resulted in significantly improved haemodynamics and less organ failure.[4] In monkeys, LR12 also reduced the inflammatory and hypotensive effects of sepsis.[5]
Human studies
Nangibotide has demonstrated safety in Phase 1 (healthy volunteers)[6] and Phase 2 (sick patients with septic shock)[7] studies. The ASTONISH trial will examine clinical efficacy in 450 patients with septic shock.[8]
Inotrem Receives Approval to Expand Nangibotide Clinical Trial in Critically Ill COVID-19 Patients and Receives Additional Public Funding of €45 Million
- Inotrem’s phase 2/3 clinical trial “ESSENTIAL” will enroll up to 730 patients in Europe to demonstrate the safety and efficacy of nangibotide to treat critically ill COVID-19 patients with respiratory failure.
- Recent preclinical studies have strengthened the body of evidence for targeting the TREM-1 pathway which is activated in a subset of patients suffering from severe COVID-19.
July 12, 2021 03:00 AM Eastern Daylight Time
PARIS–(BUSINESS WIRE)–Inotrem S.A., a biotechnology company specializing in the development of immunotherapies targeting the TREM-1 pathway, announces that it has obtained authorization to pursue the clinical development of nangibotide up to registration in COVID-19 patients from both the French and Belgian competent authorities.
As part of this program, Inotrem receives additional 45 million euros in public funding under the “Capacity Building” Call for Expression of Interest, operated on behalf of the French government by Bpifrance, the French national investment bank, as part of the Programme d’investissements d’avenir (PIA) and the France Recovery Plan, bringing French state support for the project to a total of 52,5 million euros. This public funding will support Inotrem’s clinical program including the phase 2/3 study “ESSENTIAL” which aims to demonstrate the efficacy and safety of nangibotide in treating patients in respiratory distress with severe forms of COVID-19.
The primary endpoint is evaluation of the impact of nangibotide on the progression of disease in patients receiving ventilatory support due to COVID-19 as well as on the severity of the respiratory failure, duration of mechanical ventilation, length of stay in intensive care and mortality. In “ESSENTIAL”, a Phase 2/3 clinical program, up to 730 patients will be enrolled initially in France and Belgium and, possibly in other European countries. Pre-defined interim analyses will be conducted by an independent Data Monitoring Board to test futility and to allow for the study design to be adapted as necessary. “ESSNTIAL” is the continuation of a 60 patients phase 2a evaluating the safety and efficacy of nangibotide in patients suffering from severe COVID-19. In July 2020, the CoviTREM-1 consortium, which includes the Nancy and Limoges university hospitals and Inotrem, obtained public funding of 7,5 million euros under the “PSPC-COVID” call for projects, operated on behalf of the French government by Bpifrance
New pre-clinical studies with nangibotide have demonstrated that the administration of nangibotide in murine models infected with SARS-CoV-2 was associated with a decrease in inflammatory mediators and an improvement of clinical signs, in particular respiratory function, and survival. Inotrem also confirmed in 3 different and independent cohorts that sTREM-1, a marker of the activation of the TREM-1 biological pathway, is associated with both severity and mortality in critically ill COVID-19 patients.
Leveraging the results of these preclinical studies and the implications for the role of the TREM-1 pathway in COVID-19, Inotrem has filed additional patents to cover nangibotide use in severe forms of COVID-19 as well as the use of sTREM-1 as a biomarker and companion diagnostic. This significantly strengthens Inotrem’s already broad patent estate.
Jean-Jacques Garaud, Executive Vice-President, Head of Scientific and Medical Affairs and Inotrem’s co-founder said :“We are eager to pursue the development of nangibotide in these severe forms of COVID-19. Nangibotide is a TREM-1 inhibitor which has already demonstrated a trend towards efficacy in septic shock patients and has the potential to modulate the dysregulated immune response in critically ill COVID-19 patients. With this large clinical study, we can demonstrate efficacy for nangibotide in a further indication with the goals of reducing the duration of hospitalization and mortality.”
Sven Zimmerman, CEO of Inotrem, also declared: “The size of the financial support awarded to us as part of the French government’s initiative against COVID-19 is a testimony to the relevance of targeting the TREM-1 pathway with nangibotide in these severely ill patients. We are delighted by the confidence placed in our technology and our team. Everyone at Inotrem is fully committed to deliver on this ambitious program alongside nangibotide’s ongoing Phase 2b trial in septic shock patients.”
About Inotrem
Inotrem S.A. is a biotechnology company specialized in immunotherapy for acute and chronic inflammatory syndromes. The company has developed a new concept of immunomodulation that targets the TREM-1 pathway to control unbalanced inflammatory responses. Through its proprietary technology platform, Inotrem has developed the first-in-class TREM-1 inhibitor, LR12 (nangibotide), with potential applications in a number of therapeutic indications such as septic shock and myocardial infarction. In parallel, Inotrem has also launched another program to develop a new therapeutic modality targeting chronic inflammatory diseases. The company was founded in 2013 by Dr. Jean-Jacques Garaud, a former head of research and early development at the Roche Group, Prof. Sébastien Gibot and Dr. Marc Derive. Inotrem is supported by leading European and North American investors.
About TREM-1 pathway
TREM-1 pathway is an amplification loop of the immune response that triggers an exuberant and hyperactivated immune state which is known to play a crucial role in the pathophysiology of septic shock and acute myocardial infarction.
About Nangibotide
Nangibotide is the formulation of the active ingredient LR12, which is a 12 amino-acid peptide prepared by chemical synthesis. LR12 is a specific TREM-1 inhibitor, acting as a decoy receptor and interfering in the binding of TREM-1 and its ligand. In preclinical septic shock models, nangibotide was able to restore appropriate inflammatory response, vascular function, and improved animals’ survival post septic shock.
About ESSENTIAL study:
The Efficacy and Safety Study Exploring Nangibotide Treatment in COVID-19 pAtients with ventiLatory support, is a randomized, double-blind, placebo-controlled confirmatory study with adaptive features that will be performed in Europe. This is a pivotal study and it is expected that based on its results, nangibotide could be registered in this indication. The first part of the study (i.e.: 60 patients) has been already finalized and assessed by an independent data monitoring committee with excellent safety results. The study will recruit up to 730 patients in up to 40 sites. Several interim and futility analyses are foreseen as part of the adaptive design of the study.
About Bpifrance
Bpifrance is the French national investment bank: it finances businesses – at every stage of their development – through loans, guarantees, equity investments and export insurances. Bpifrance also provides extra-financial services (training, consultancy.). to help entrepreneurs meet their challenges (innovation, export…).
PATENT
WO-2021144388
Process for preparing nangibotide by solid phase synthesis, useful for treating acute inflammatory disorders such as septic shock. Also claims novel peptide fragments, useful in the synthesis of nangibotide.
Example 1
Preparation of nangibotide by full SPPS (Reference)
Step 1 : Loading of the first amino acid onto the Rink Amide Resin
2 g of MBHA resin (1.0-1.3 mmol/g) was swelled using 16 mL of DMF for 30 min. 2 eq Fmoc-Met-OH (2.4 mmol, 2.67 g), 2 eq DIC (2.4 mmol, 1.136 mL) and 2 eq OxymaPure (2.4 mmol, 1.023 g) were dissolved in 8 mL of DMF at 0.3 M cone, and added to the resin after 5 min. All the coupling steps were conducted in this way unless described differently. The loading step was carried out for 1.5 hour. After the loading, the resin was filtered and washed 3 times with 12 mL of DMF. The Fmoc deprotection step was carried out by addition of 12 mL of 20% piperidine solution in DMF for two 10 min cycles. This step was performed analogously for all the amino acid residues. The loading, calculated by UV absorption for the peptidyl resin, was 0.8 mmol/g.
Step 2: peptide elongation
For the coupling of all the amino acids involved in the synthesis of nangibotide, 3 eq of each amino acid were activated by 3 eq of DIC and OxymaPure dissolved in DMF at 0.3 M cone. At the end of the peptide elongation, a final Fmoc deprotection, as already described, was performed before moving to the cleavage step.
Step 3: Cleavage and precipitation of crude nangibotide
The cleavage of nangibotide off the resin was carried out using a solution of 16 mL of TFA/DODT/TIPS/water in 90/4/3/3 ratio cooled at 0°C. The peptidyl resin was added portionwise in 30 min keeping the internal temperature under 25°C. The cleavage was run for 3.5 hours, then the resin was filtered and washed by 10 mL of TFA for 10 min.
DIPE was used for the precipitation of the peptide, adding 12 volumes (300 mL) dropwise to the peptide TFA solution, keeping the temperature under 20°C. The suspension with nangibotide was filtered on a gooch funnel, the peptide washed again with 100 mL of DIPE and then dried under vacuum overnight. Molar yield 40%. Purity 61%.
Example 2
Preparation of nangibotide by three-fragment condensation
In the approach using three fragments, only the cysteine residue was coupled to the methionine on rink amide resin to prepare fragment 11-12, whereas protected peptide fragments 1-7 and 8-10 were synthesized using 2-CTC resin.
Step 1: Synthesis of fragment 11-12
2 g of MBHA resin (1.0-1.3 mmol/g) was swelled using 16 mL of DMF for 30 min 2 eq of Fmoc-Met-OH (2.4 mmol, 2.67 g), 2 eq DIC (2.4 mmol, 1.136 mL) and 2 eq OxymaPure (2.4 mmol, 1.023 g) were dissolved in 8 mL of DMF at 0.3 M cone, and added to the resin. The loading step was carried out for 1 and half hour. After the loading, the resin was filtered and washed 3 times with 12 mL of DMF. The Fmoc deprotection step was carried out by
addition of 12 mL of a 20% piperidine solution in DMF for two 10 min cycles. Same procedure was repeated for the coupling of Fmoc-Cys(Trt)-OH to obtain resin-attached Fmoc-deprotected fragment 11-12. The loading, calculated by UV absorption for the peptidyl resin relative to the first amino acid inserted, was 0.8 mmol/g.
Step 2: Synthesis of fragments 1-7 and 8-10
For the synthesis of both fragments the loading of 2-chloro trityl chloride resin was performed on 5 g (1.6 mmol/g) using 0.8 eq Fmoc-Gly-OH (6.40 mmol, 1.90 g) dissolved in 30 mL of DCM and addition of 3 eq DIPEA (24 mmol, 4.19 mL). The loading step was carried out for 1 hour, then the resin was washed by 30 mL DCM for three times and eventual Cl-groups were capped by two different capping solutions: first by 30 mL of methanol/DIPEA/DCM (1:2:7) and then by 30 mL AC2O/DIPEA/DCM in the same ratio. After the treatment with these solutions for 15 min and subsequent washing with DCM, the resin was washed three times with DMF, before deprotection of Fmoc and evaluation of the resin loading. Generally, this protocol gave a resin loaded with 1.1 mmol/g Fmoc-Gly-OH. The Fmoc deprotection and coupling step protocols were equally performed with all the amino acids in the respective sequences: Fmoc-Tyr(tBu)-OH and Fmoc-Glu(tBu)-OH for fragment 8-10, and Fmoc-Ala-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Glu(OtBu)-OH twice, Fmoc-Gln(Trt)-OH and Fmoc-Leu-OH for fragment 1-7.
For each coupling, 3 eq amino acid were activated by 3 eq DIC and 3 eq OxymaPure dissolved in DMF at 0.3 M cone.
Fragment Fmoc-Glu(tBu)-Tyr(tBu)-Gly-OH (8-10) was obtained by cleavage off the resin using 6 volumes (30 mL) of a TFA 1.5 % solution in DCM, 5 times for 2 min. The final TFA solution was neutralized by 1.2 eq pyridine (15.89 mmol, 1.3 mL) diluted in 30 mL methanol. The final solution was concentrated to 50 mL under vacuum then washed by water and brine. The organic layer was dried by anhydrous sodium sulphate, filtered and further concentrated before crystallization of the tripeptide with 5 volumes of petroleum ether at 0°C. The peptide was filtered, washed by petroleum ether and dried overnight in a vacuum oven at 37°C. Molar yield 65%. Purity 90%.
Fragment Fmoc-Leu-Gln(Trt)-Glu(OtBu)-Glu(OtBu)-Asp(OtBu)-Ala-Gly-OH (1-7) was obtained by cleavage off the resin using 6 volumes (30 mL) of a TFA 1.5 % solution in DCM, 5 times for 2 min. The final TFA solution was neutralized by 1.2 eq pyridine (15.89 mmol, 1.3 mL) diluted in 30 mL methanol. The DCM was evaporated and replaced by methanol, adding and evaporating 30 mL methanol a couple of times till one third of the volume. The peptide fragment was precipitated by adding 5 volumes (150 mL) water to the methanol solution at 0°C and filtered after stirring for 30 min. The full protected heptapeptide was washed by water and dried overnight in a vacuum oven at 37°C. Molar yield 85%. Purity 89%.
Step 3: Synthesis of fragment 8-12 (Fragment condensation 1)
The fragment condensation between Fmoc-Glu(tBu)-Tyr(tBu)-Gly-OH (8-10) and H-Cys(Trt)-Met-MBHA resin (11-12) was carried out activating 2 eq (1.6 mmol, 1.12 g) of fragment 8-10 dissolved in 6 mL of DMF at 40°C by using 2 eq OxymaPure (1.6 mmol, 0.22 g) and 2 eq DIC (1.6 mmol, 0.25 mL) for 10 min. The activated ester of tripeptide 8-10 was added to the resin-attached fragment 11-12 and stirred for 3 hours at 40°C. After filtration, the resin was washed three times by 15 mL DMF and then capped by 12 mL of AC2O 10% in DMF for 15 min. The resin was washed three timed by 12 mL DMF before deprotection of Fmoc to finally obtain resin-attached Fmoc-protected fragment 8-12. Molar yield 91%. Purity 89%.
Step 4: Synthesis of nanaibotide (Fragment condensation 2)
The fragment condensation between fragment 1-7 and H-Glu(OtBu)-Tyr(tBu)-Gly-Cys(Trt)-Met-MBHA resin (8-12) was carried out activating 1.5 eq (2.25 mmol, 2.64 g) of fragment 1-7 dissolved in 25 mL DMF at 40°C by using 2 eq OxymaPure (2.25 mmol, 0.32 g) and 2 eq DIC (2.25 mmol, 0.35 mL) for 15 min. The activated ester of fragment 1-7 was added to the resin-attached fragment 8-12 and stirred for 3.5 hours at 40°C. After filtration, the resin was washed three times by 12 mL DMF before deprotection of Fmoc with the standard procedure described above. After Fmoc deprotection, the resin was washed again by DMF and DCM and then dried at vacuum pump.
Step 5: Cleavage and precipitation of crude nanaibotide
The cleavage of nangibotide off the resin was carried out using a solution of 16 mL of TFA/DODT/TIPS/water in 90/4/3/3 ratio cooled at 0°C. The peptidyl resin was added portionwise in 30 min keeping the internal temperature under 25°C. The cleavage was run for 3.5 hours, then the resin filtered and washed by 10 mL of TFA for 10 min.
DIPE was used to precipitate the peptide, adding 12 volumes (300 mL) dropwise to the peptide TFA solution, keeping the temperature under 20°C. The suspension with nangibotide was filtered on a gooch funnel, the peptide washed again with 100 mL of DIPE and then dried at vacuum pump overnight. Molar yield 61%. Purity 73%.
Example 3
Preparation of nangibotide by two-fragment condensation
In the approach using two fragments, the SPPS elongation onto MBHA resin, as described in Example 2, step 1, was continued until Glu8 was attached to provide fragment 8-12, then fragment 1-7, synthesized on 2-CTC resin as described in example 2, step 2, was coupled to the resin-attached fragment 8-12 as described in example 2, step 4.
Step 1: Synthesis of fragment 8-12
2 g of MBHA resin (1.0-1.3 mmol/g) was swelled using 16 mL of DMF for 30 min 2 eq of Fmoc-Met-OH (2.4 mmol, 2.67 g), 2 eq DIC (2.4 mmol, 1.136 mL) and 2 eq OxymaPure (2.4 mmol, 1.023 g) were dissolved in 8 mL of DMF at 0.3 M cone, and added to the resin. The loading step was carried out for 1 and half hour. After the loading, the resin was filtered and washed 3 times with 12 mL of DMF. The Fmoc deprotection step was carried out by addition of 12 mL of a 20% piperidine solution in DMF for two 10 min cycles. Same procedure was repeated for the coupling of Fmoc-Cys(Trt)-OH; Fmoc-Glu(OtBu)-OH; Fmoc-Tyr(tBu)-OH; Fmoc-Gly-OH to obtain fragment 8-12. The loading, calculated by UV absorption for the peptidyl resin relative to the first amino acid inserted, was 0.8 mmol/g. Molar yield 88%. Purity 83%.
Step 2: Synthesis of nanaibotide (Fragment condensation 2)
The final fragment condensation was performed as described in example 2, step 4.
Step 3: Cleavage and precipitation of crude nanaibotide
The cleavage of nangibotide off the resin was carried out as described in example 2, step 5. Molar yield 60%. Purity 70%.
PAPER
Methods in enzymology (2000), 312, 293-304
Journal of the American College of Cardiology (2016), 68(25), 2776-2793
PATENT
https://patents.google.com/patent/WO2011124685A1/en
Product pat, WO2011124685 ,protection in the EU states and the US April 2031
References
- ^ Cuvier V, Lorch U, Witte S, Olivier A, Gibot S, Delor I, Garaud JJ, Derive M, Salcedo-Magguilli M (2018). “A first-in-man safety and pharmacokinetics study of nangibotide, a new modulator of innate immune response through TREM-1 receptor inhibition”. Br J Clin Pharmacol. 84 (10): 2270–2279. doi:10.1111/bcp.13668. PMC 6138490. PMID 29885068.
- ^ Weber B, Schuster S, Zysset D, Rihs S, Dickgreber N, Schürch C, Riether C, Siegrist M, Schneider C, Pawelski H, Gurzeler U, Ziltener P, Genitsch V, Tacchini-Cottier F, Ochsenbein A, Hofstetter W, Kopf M, Kaufmann T, Oxenius A, Reith W, Saurer L, Mueller C (2014). “TREM-1 deficiency can attenuate disease severity without affecting pathogen clearance”. PLOS Pathog. 10 (1): e1003900. doi:10.1371/journal.ppat.1003900. PMC 3894224. PMID 24453980.
- ^ Derive M, Bouazza Y, Sennoun N, Marchionni S, Quigley L, Washington V, Massin F, Max JP, Ford J, Alauzet C, Levy B, McVicar DW, Gibot S (1 June 2012). “Soluble TREM-like transcript-1 regulates leukocyte activation and controls microbial sepsis”. Journal of Immunology. 188 (11): 5585–5592. doi:10.4049/jimmunol.1102674. PMC 6382278. PMID 22551551.
- ^ Derive M, Boufenzer A, Bouazza Y, Groubatch F, Alauzet C, Barraud D, Lozniewski A, Leroy P, Tran N, Gibot S (Feb 2013). “Effects of a TREM-like transcript 1-derived peptide during hypodynamic septic shock in pigs”. Shock. 39 (2): 176–182. doi:10.1097/SHK.0b013e31827bcdfb. PMID 23324887. S2CID 23583753.
- ^ Derive M, Boufenzer A, Gibot S (April 2014). “Attenuation of responses to endotoxin by the triggering receptor expressed on myeloid cells-1 inhibitor LR12 in nonhuman primate”. Anaesthesiology. 120 (4): 935–942. doi:10.1097/ALN.0000000000000078. PMID 24270127. S2CID 10347527.
- ^ Cuvier V, Lorch U, Witte S, Olivier A, Gibot S, Delor I, Garaud JJ, Derive M, Salcedo-Magguilli M (2018). “A first-in-man safety and pharmacokinetics study of nangibotide, a new modulator of innate immune response through TREM-1 receptor inhibition”. Br J Clin Pharmacol. 84 (10): 2270–2279. doi:10.1111/bcp.13668. PMC 6138490. PMID 29885068.
- ^ François B, Wittebole X, Ferrer R, Mira JP, Dugernier T, Gibot S, Derive M, Olivier A, Cuvier V, Witte S, Pickkers P, Vandenhende F, Garaud JJ, Sánchez M, Salcedo-Magguilli M, Laterre PF (July 2020). “Nangibotide in patients with septic shock: a Phase 2a randomized controlled clinical trial”. Intensive Care Medicine. 46 (7): 1425–1437. doi:10.1007/s00134-020-06109-z. PMID 32468087. S2CID 218912723.
- ^ “Efficacy, Safety and Tolerability of Nangibotide in Patients With Septic Shock (ASTONISH)”. ClinicalTrials.gov. US National Library of Medicine. Retrieved 13 July 2020.
Derive et al (2013) Effects of a TREM-Like Transcript 1–Derived Peptide During Hypodynamic Septic Shock in Pigs. Shock39(2) 176 PMID: 23324887
Derive et al (2014) Attenuation of Responses to Endotoxin by the Triggering Receptor Expressed on Myeloid Cells-1 Inhibitor LR12 in Nonhuman Primate. Anesthesiology120(4) 935 PMID: 24270127
Derive et al (2012) Soluble Trem-like Transcript-1 Regulates Leukocyte Activation and Controls Microbial Sepsis. J. Immunol.188(11) 5585 PMID: 22551551
| Clinical data | |
|---|---|
| Routes of administration | Intravenous; intraperitoneal |
| Physiological data | |
| Receptors | TREM-1 |
| Metabolism | Enzymatic in bloodstream |
| Pharmacokinetic data | |
| Metabolism | Enzymatic in bloodstream |
| Elimination half-life | 3 minutes |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2014384‐91‐7 |
| ChemSpider | 64835227 |
| UNII | 59HD7BLX9H |
| ChEMBL | ChEMBL4297793 |
| Chemical and physical data | |
| Formula | C54H82N14O22S2 |
| Molar mass | 1343.439 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
//////////////Nangibotide, phase 3, нангиботид , مانغيبوتيد , 南吉博肽 , INOTREM, SEPTIC SHOCK, PEPTIDE
NEW DRUG APPROVALS
one time
$10.00
Rilzabrutinib
![(R)-2-(3-(4-Amino-3-(2-fluoro-4-phenoxyphenyl)-1H-pyrazolo[3,4-d]-pyrimidin-1-yl)piperidine-1-carbonyl)-4-methyl-4-(4-(oxetan-3-yl)piperazin-1-yl)pent-2-enenitrile.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=118325989&t=l)


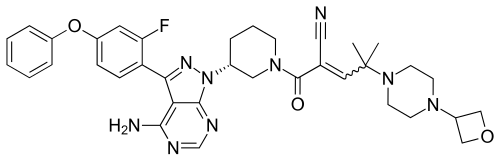
PRN 1008, Rilzabrutinib
CAS 1575591-66-0
| リルザブルチニブ; |
C36H40FN9O3,
| MW 665.7597 |
2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]piperidine-1-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin-1-yl]pent-2-enenitrile
Anti-inflammatory disease, Autoimmune disease treatment
Fda 2025, approvals 2025 8/29/2025, Wayrilz, To treat persistent or chronic immune thrombocytopenia that has not sufficiently responded to immunoglobulins, anti-D therapy, or corticosteroids
- OriginatorPrincipia Biopharma
- Class2 ring heterocyclic compounds; Amines; Anti-inflammatories; Fluorobenzenes; Nitriles; Phenyl ethers; Piperazines; Piperidines; Pyrazoles; Pyrimidines; Skin disorder therapies; Small molecules
- Mechanism of ActionAgammaglobulinaemia tyrosine kinase inhibitors
- Orphan Drug StatusYes – Idiopathic thrombocytopenic purpura; Pemphigus vulgaris
- Phase IIIIdiopathic thrombocytopenic purpura; Pemphigus vulgaris
- Phase IIAutoimmune disorders
- 02 Jun 2021Efficacy data from a phase IIa trial in Ankylosing spondylitis presented at the 22nd Annual Congress of the European League Against Rheumatism (EULAR-2021)
- 07 Apr 2021Sanofi initiates enrollment in a phase I pharmacokinetics trial in healthy volunteers in Australia (PO, Tablet, Capsule) (NCT04748926)
- 31 Mar 2021Sanofi announces intention to seek regulatory approval for Idiopathic thrombocytopenic purpura in 2023 (Sanofi pipeline, May 2021)
Rilzabrutinib, sold under the brand name Wayrilz, is an anti-cancer medication used for the treatment of immune thrombocytopenia.[1] Rilzabrutinib is a tyrosine kinase inhibitor.[1] It is taken by mouth.[1]
Rilzabrutinib may increase the risk of serious infections (including bacterial, viral, or fungal).[2] The most common side effects include diarrhea, nausea, headache, abdominal pain, and COVID-19.[2]
Rilzabrutinib was approved for medical use in the United States in August 2025.[2]
CLIP
Sanofi to acquire BTK inhibitor firm Principia for $3.7 billion
Principia is testing its small-molecule compounds in multiple sclerosis and immune system diseases
Sanofi will pay $3.7 billion to acquire Principia Biopharma, a San Francisco-based biotech firm developing small molecules that inhibit Bruton tyrosine kinase (BTK). The price represents about a 75% premium over Principia’s stock market value in early July, before reports surfaced that Sanofi was interested in buying the firm.
BTK is a protein important for both normal B cell development and the proliferation of lymphomas, which are B cell cancers. AbbVie, AstraZeneca, and BeiGene all market BTK inhibitors for treating specific kinds of lymphomas. Sales of AbbVie’s inhibitor, Imbruvica, approached $4.7 billion in 2019.
Other drug firms have been eager to get in on the action as well. In January, Merck & Co. spent $2.7 billion to acquire ArQule, whose experimental noncovalent BTK inhibitor is designed to overcome resistance that some cancers develop after treatment with current covalent BTK inhibitors. Eli Lilly and Company’s $8 billion acquisition of Loxo Oncology in 2019 also included a noncovalent BTK inhibitor.
BTK is also linked to inflammation, and Principia focuses on developing BTK inhibitors for immune system diseases and multiple sclerosis. Its compound rilzabrutinib is currently in clinical trials for pemphigus and immune thrombocytopenia. In 2017, Sanofi struck a deal to develop Principia’s brain-penetrant BTK inhibitor, SAR442168, for multiple sclerosis.
Sanofi announced in April of this year that the inhibitor reduced formation of new lesions—the scarred nervous tissue that gives multiple sclerosis its name—by 85% in a Phase II clinical trial. A Phase III trial of the compound began in June.
Upon announcing its deal to acquire Principia, Sanofi said that both rilzabrutinib and SAR442168 have the potential to become a “pipeline in a product,” indicating they can be used for many immune-related and neurological diseases, respectively.
The anti-inflammatory effects of BTK inhibitors have raised interest in the drugs as treatments for people hospitalized with COVID-19. Notably, the US National Cancer Institute conducted a small study suggesting acalabrutinib may help reduce the respiratory distress and inflammation in people with COVID-19. Based on that preliminary study, AstraZeneca—which markets acalabrutinib as Calquence—is conducting a 60-person randomized trial of the drug for COVID-19.
Sanofi has not indicated interest in investigating Principia’s BTK inhibitors as COVID-19 treatments.Chemical & Engineering NewsISSN 0009-2347
PATENT
WO 2021127231https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021127231&tab=PCTDESCRIPTION&_cid=P20-KRA0I9-18818-1
SOLID FORMS OF 2-[3-[4-AMTNO-3-(2-FT,TTORO-4-PHENOXY- PHEN¥L)PYRAZOLO[3,4 D]PYRIMIDIN l~YL]PIPERIDINE~l~CARBON¥L] 4~
METHYL-4-[4-(OXETAN-3-YL)PIPERAZIN-l-YLjPENT-2-ENENITRILE
[11 This application claims the benefit of priority to U.S. Provisional Application
No 62/951,958, filed December 20, 2019, and U.S Provisional Application No. 63/122,309, filed December 7, 2020, the contents of each of which are incorporated by reference herein in their entirety.
[2] Disclosed herein are solid forms of 2-[3-[4~amino-3~(2~fluoro-4-phenoxy-plienyl)pyrazolo[3,4-d]pyrimidin-l-yl]piperidine-l Carbonyl]~4-nietliyl-4~[4-(oxetaii~3-yl)piperazin-!~yi]pent-2~enenitriie (Compound (I)), methods of using the same, and processes for making Compound (I), including its solid forms. The solid forms of Compound (I) may be inhibitors of Bruton’s tyrosine kinase (BTK) comprising low residual solvent content.
[3| The enzyme BTK is a member of the Tec family non-receptor tyrosine kinases.
BTK is expressed in most hematopoietic cells, including B cells, mast cells, and macrophages BTK plays a role in the development and activation of B cells. BTK activity has been implicated in the pathogenesis of several disorders and conditions, such as B cell-related hematological cancers (e.g., non-Hodgkin lymphoma and B cell chronic lymphocytic leukemia) and autoimmune diseases (e.g., rheumatoid arthritis, Sjogren’s syndrome, pemphigus, IBD, lupus, and asthma).
[4] Compound (I), pharmaceutically acceptable salts thereof, and solid forms of any of the foregoing may inhibit BTK and be useful in the treatment of disorders and conditions mediated by BTK activity. Compound (I) is disclosed in Example 31 of WO 2014/039899 and has the following structure:
where *C is a stereochemical center. An alternative procedure for producing Compound (!) is described in Example 1 of WO 2015/127310.
[5] Compound (I) obtained by the procedures described in WO 2014/039899 and WO 2015/127310 comprises residual solvent levels well above the limits described in the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (“ICH”) guidelines. In general, manufacturing processes producing residual solvent levels near or above the ICH limits are not desirable for preparing active pharmaceutical ingredients (APIs).
Example 1: Spray Drying Process A
[311] A solution of Compound (I) in dichloromethane (prepared according to Example 31 on pages 86-87 of WO 2014/039899) was washed with pH 3 phosphate buffer to remove basic impurities that are more soluble than Compound (I) in the aqueous layer. The dichloromethane solution was then washed with pH 7 buffer and solvent exchanged into isopropyl acetate. The isopropyl acetate solution was then washed with pH 3 phosphate buffer, bringing Compound (I) into the aqueous layer and removing non-basic impurities. The pH of the aqueous layer was adjusted to pH 9 with 10% sodium hydroxide, and the aqueous layer was extracted with isopropyl acetate. Upon concentration under vacuum, Compound (I) was precipitated from heptane at 0 °C, filtered and dried to give a white amorphous solid as a mixture of the (E) and (Z) isomers, as wet Compound (I). Wet Compound (I) was dissolved in methanol and spray dried at dryer inlet temperature of 125 °C to 155 °C and dryer outlet temperature of 48 to 58 °C to obtain the stable amorphous Compound (I) free base with levels of isopropyl acetate and heptane below 0.5% and 0.05%, respectively.
Example 2: Spray Drying Process B
intermediate A
Compound (!)
[241] A jacketed reactor with overhead stirrer, condenser, nitrogen line, temperature probe, and recirculating fluid chiller/heater was charged with Intermediate A (20.2 kg) and Intermediate B (13.6 kg, 1.5 equiv). DCM (361.3 kg, 14.5 vol) was charged to the reactor. The mixture was agitated, and the batch cooled to 0 °C to 5 °C. The reactor was charged with pyrrolidine (18.3 kg, 6 equiv) and then charged with TMSC1 (18.6 kg, 4 eq). Stirring was continued at 0 °C to 5 °C for 0.5 to 1 hour
[242] At 0 °C to 5 °C, acetic acid (2.0 equiv) was charged to the reactor followed by water (5 equiv). Stirring was continued at 0 °C to 5 °C for 1 to 1.5 hours. Water (10 equiv) was charged to the reactor, and the solution was adjusted to 20 °C to 25 °C. The internal temperature was adjusted to 20 °C to 25 °C and the biphasic mixture was stirred for 15 to 20 mins. Stirring was stopped and phases allowed to separate for at least 0.5 h. The lower aqueous layer was removed.
[243] Water (7 vol) was charged to the reactor. The pH was adjusted to 2.8-3.3 with a 10 wt. % solution of citric acid. Stirring was continued at 0 to 5 °C for 1 to 1.5 hours. Stirring was stopped and phases allowed to separate for at least 0.5 h. The lower aqueous layer was removed.
[244] A jacketed reactor with overhead stirrer, condenser, nitrogen line, temperature probe, and recirculating fluid chiller/heater was charged with an approximately 9% solution of NaHCCri (1 vol) and the organic layer. The internal temperature was adjusted to 20 °C to 25 °C, and the biphasic mixture was stirred for 15 to 20 mins. Stirring was stopped and phases allowed to separate for at least 0.5 h. The lower aqueous layer was removed. The aqueous layer was measured to have a pH greater than 7.
[245] A jacketed reactor with overhead stirrer, condenser, nitrogen line, temperature probe and recirculating fluid chiller/heater was charged with the organic layer. The organic phase ¾s distilled under vacuum at less than 25 °C to 4 total volumes. IP AC (15 vol) was charged to the reactor. The organic phase was distilled under vacuum at less than 25 °C to 10 total volumes. Water (15 vol) followed by pH 2.3 phosphate buffer were charged to the reactor at an internal temperature of 20 °C to 25 °C. The pH adjusted to 3 Stirring was stopped and phases allowed to separate for at least 0.5 h. The organic phase was removed.
[246] The following steps were repeated twice: IP AC (5 vol) was charged to the reactor containing the aqueous layer. Stirring was continued for 0.25 to 0.5 hours. Stirring was stopped and phases allowed to separate for at least 0.5 h. The organic phase was removed. [247] IP AC (15 vol) was charged to the reactor containing the aqueous layer. A pH 10 phosphate buffer was charged to the reactor and the pH adjusted to 10 with 14% NaOH solution. Stirring was continued for 1.5 to 2 hours. Stirring was stopped and phases allowed to separate for at. least 0.5 h. The aqueous layer was discarded. The organic layer was dried over brine.
[248] The organic solution was distilled under vacuum at less than 25 °C to 5 total volumes.
[249] A jacketed reactor with overhead stirrer, condenser, nitrogen line, temperature probe and recirculating fluid chiller/heater was charged with n-heptane (20 vol). The internal temperature was adjusted to 0 to 5 °C, and the IP AC solution was added.
[250] The suspension was filtered. The filter cake was washed with n-heptane and the tray was dried at 35 °C. Compound (I) (24.6 kg) was isolated in 86% yield.
[251] Compound (1) was dissolved in methanol (6 kg) and spray dried to remove residual IP AC and n-heptane.
Example 3: Precipitation Process A
[252] A solution of Compound (I) in dichloromethane (prepared according to Example 31 on pages 86-87 of WO 2014/039899) was quenched with acetic acid and water, followed by washing with pH 3 aqueous solution to remove basic impurities that are more soluble than Compound (1) in the aqueous layer. Washing was repeated as needed to reduce impurities. Methanesulfonic acid was added to the dichloromethane solution, and the dichloromethane solution was concentrated by distillation under reduced pressure, followed by addition of 1% NaCi aqueous solution and isopropyl acetate before adjustment of pH to approximately 3 with potassium hydroxide. The isopropyl acetate layer was removed and discarded. The aqueous layer containing Compound (I) was washed with isopropyl acetate to remove hydrophobic impurities. Washing was repeated as needed to reduce related substance impurities. Residual isopropyl acetate was removed by distillation under reduced pressure. The aqueous solution containing Compound (I) was cooled to 0 to 5°C before adjusting the pH to approximately 9 with potassium hydroxide. The free base of Compound (I) was allowed to precipitate and maturate at 20 °C for 20 hours. The mixture temperature was then adjusted to 20 °C to 25 °C, and the hydrate impurity was verified to be less than 0.3% (< 0.3%). The cake of the free base of Compound (I) was filtered and washed as needed to reduce conductivity. The cake was then allowed to dry on the filter under vacuum and nitrogen swept to reduce water content by Karl-Fischer (KF < 50%) before transferring to the oven for drying. The wet cake of the free base of Compound (1) was dried under vacuum at 25 °C until water content by Karl -Fischer was less than 1.5% (KF < 1.5%), and then dehmiped by milling to yield a uniform white amorphous solid as a mixture of the (E) and (Z) isomers, with no detectible levels of isopropyl acetate or heptane.
Example 4: Precipitation Process 3B
[253] A solution of Compound (I) in dichloromethane (prepared according to Example 31 on pages 86-87 of WO 2014/039899) was quenched with acetic acid and water, followed by washing with pH 3 aqueous solution to remove basic impurities that are more soluble than Compound (I) in the aqueous layer. The washing was repeated as needed to reduce residual solvents and impurities. The dichloromethane solution was then washed with saturated sodium bicarbonate (pH > 7). Dichloromethane was removed by distillation under reduced pressure, followed by addition of water and isopropyl acetate. The pH of the aqueous layer was adjusted to pH to 2.8 – 3.3 with 2 M aqueous sulfuric acid (H2SQ4) at 0 – 5 °C, and the mixture rvas stirred and settled. After phase separation removal of the organic layer, the aqueous layer was washed with isopropyl acetate three times and the residual isopropyl acetate in aqueous layer was distilled out under vacuum at a temperature below 25 °C and the solution was basitied with 5% aqueous KOFI to pH 9 – 10 to a slurry . The resulting suspension was stirred and warmed up to 20 °C to 25 °C and aged for 20 h. The product was filtered and washed with water and dried to give white solid in 86% yield.
Example 5: Precipitation Process C
[254] A solution of Compound (I) in dichloromethane (prepared according to Example 31 on pages 86-87 of WO 2014/039899) was quenched with acetic acid and water, followed by washing to remove basic impurities that are more soluble than Compound (I) in the aqueous layer. Washing was repeated as needed to reduce impurities. Methanesulfonic acid was added to the d chloromethane solution, and the dichloromethane solution was concentrated under reduced pressure to obtain a thin oil. The concentrated oil was cooled to approximately 5°C before washing with an aqueous solution of sodium chloride. The organic phase was discarded. Washing of the aqueous layer was repeated as needed with dichloromethane to remove low level impurities. The pH of the aqueous solution was adjusted to approximately 3 with an aqueous solution of potassium hydroxide. Residual dichloromethane was removed
under reduced pressure. The level of residual acetic acid was determined by, for example, titration. The aqueous solution containing Compound (I) was cooled to a temperature between 0°C and 5°C. Acetic acid was present at 0 wt % to 8 wt. %. Acetic acid level was 0 wt % if the aqueous acid solution was washed with aqueous sodium bicarbonate or another aqueous inorganic base. Optionally, additional acetic acid was added to achieve a 0 wt.% to 8 wt. % acetic acid level. An aqueous solution of potassium hydroxide was constantly charged to the aqueous solution to obtain a pH to approximately 9.5. The free base of Compound (I) was allowed to precipitate and maturate at approximately 20 °C for least 3 hours. The cake (wet solid) of the free base of Compound (I) was filtered and washed with water. The wet cake was then dried under reduced vacuum with slight heat. Alternatively, instead of washing the wet cake with water, the wet cake was reslurried with water at approximately 15 °C for at least 1 hour before filtering. The free base of Compound (I) in the fomi of a wet cake was dried under vacuum with slight heat at 25°C.
[255] FIGs. 12-15 are example SEM images showing the variable morphologies of particles of Compound (I) during the filtration step to isolate Compound (I) based on the amount acetic acid added during the initial step in the precipitation of Compound (Ϊ) (FIG. 12: at 0 wt. % acetic acid; FIG 13: at 3 wt. % acetic acid; FIG. 14: at 5 wt. % acetic acid; FIG 15: at 8 wt. % acetic acid). Filtration speed depended on the morphology and was the fastest for 0 wt. % acetic acid. At 1 wt. % acetic acid, the filtration speed diminished considerably, improving at 2 wt. % to 3 wt. % acetic acid. Morphologies with more open holes (such as, e.g., more porous particles) resulted in improved filtration speeds, whereas more compact particles resulted in decreased filtration speed.
Example 6: Conversion of a Crystalline Form of Compound (Ϊ) to an Amorphous Form
[256] 9.8 grams of a crystalline form of Compound (I) were dissolved in approximately 20 mL of dichloromethane and approximately 120 ml. of brine solution. Then, approximately 1 equivalent of methanesulfonic acid was added. The pH w¾s approximately 2. The layers were separated. The aqueous layer was concentrated at a temperature between 0°C and 5°C to remove residual dichloromethane before slowly adding aqueous KOI I solution (approximately 5%) to adjust the pH to a value between 9 and 10. During aqueous KOH addition, an amorphous form of Compound (I) precipitated out. The slurry was slowly warmed to room temperature and then was stirred for approximately 24 hours before filtering and rinsing the wet cake with water. The wet cake was dried under vacuum with slight heat at approximately 30°C to provide 7 grams of a white to an off-white solid (87% yield and 98 4% purity). XRPD showed that the product was an amorphous solid form of Compound (I).
Example 7: Micronization of Compound (I) Particles Obtained by Precipitation Processes
[257] A fluid jet mill equipment was used during lab scale jet milling trials. The fluid jet mill equipment includes a flat cylindrical chamber with 1.5” diameter, fitted with four symmetric jet nozzles winch are tangentially positioned in the inner wall. Prior to feeding material to the fluid jet mill in each trial, the material was sieved in a 355 iim screen to remove any agglomerates and avoid blocking of the nozzles during the feed of material to the micronization chamber. The material to be processed was drawn into the grinding chamber through a vacuum created by the venturi (P vent ~ 0 5 – 1 0 bar above P grind). The feed flow rate of solids (F_feed) was controlled by a manual valve and an infinite screw volumetric feeder. Compressed nitrogen was used to inject the feed material; compressed nitrogen was also used for the jet nozzles in the walls of the milling chamber. Compressed fluid issuing from the nozzles expands from P grind and imparts very’ high rotational speeds in the chamber. Accordingly, material is accelerated by rotating and expanding gases and subjected to centrifugal forces. Particles move outward and are impacted by high velocity jets, directing the particles radially inward at very high speeds. Rapidly moving particles impact the slower moving path of particles circulating near the periphery of the chamber. Attrition takes place due to the violent impacts of particles against each other. Particles with reduced size resulting from this sequence of impacts are entrained in the circulating stream of gas and swept against the action of centrifugal force toward the outlet at the center. Larger particles in the gas stream are subjected to a centrifugal force and returned to the grinding zone. Fine particles are carried by the exhaust gas to the outlet and pass from the grinding chamber into a collector.
[258] The feeder has continuous feed rate control; however, to more precisely control the feed rate, the full scale of feed rates was arbitrary divided in 10 positions. To calibrate F feed, the feeder was disconnected from milling chamber and 10 g of Compound (I) powder was fed through the feeder operating at various feed rate positions. The mass of powder flowing through the feeder over 6 minutes was marked. The resulting feed rate was directly proportional to feeder position. After processing each of the four trials, the jet mill was stopped, micronized product removed from the container, and the milling chamber checked for any powder accumulation.
Variables/Parameters
F_feed Feed flow rate of solids [kg/h]
P grind Grinding pressure inside the
drying chamber [bar]
P vent Feed pressure in the venturi [bar]
Example 8: Residual Solvent Levels
[251] Retention of process solvents (/.<?., res dual solvents) depends on van der Waal s’ forces that are unique to and an inherent property of each molecule. Additionally, solvent retention depends how the API solid is formed, isolated, washed, and dried (i.e., during the manufacturing process). Because residual solvents may pose safety risks, pharmaceutical processes should be designed to minimize residual solvent levels (e.g , to result in residual solvent levels below the limits established in the ICH guidelines).
[252] Residual solvent analysis was performed using gas chromatography-mass spectrometry. The residual solvent levels in solid forms of Compound (I) prepared by spray drying processes described herein and precipitation processes described herein are provided in Table 2. The residual solvent levels in crude Compound (I) listed in Table 2 are comparable to the residual solvent levels in crude Compound (I) prepared according to the procedures detailed in Example 31 of WO 2014/039899 and Example 1 of WO 2015/127310.
Table 2: Residual solvent levels in solid forms of Compound (I)
PATENT
WO 2015127310
https://patents.google.com/patent/WO2015127310A1/enExample 1Synthesis of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4-d]pyrimidin-l- yl]-piperidine-l-carbonyl]-4-m iperazin-l-yl]pent-2-enenitrile

Step 1To a solution of 3-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4- d]pyrimidin-l -yl]-l-piperidyl]-3-oxo-propanenitrile (15 g, 3.12mmol), 2-methyl-2-[4- (oxetan-3-yl)piperazin-l-yl]propanal (794.25mg, 3.74mmol) in DCM (40mL), pyrrolidine (1.54mL,18.71mmol) at 0-5 °C was added, which is followed by TMS-Cl (1.58mL,12.47mmol). The reaction mixture was stirred at 0-5 °C for 3 h and was quenched with 1 M potassium phosphate buffer (pH 3). Layers were separated and the organic layer was washed once more with 1 M potassium phosphate buffer (pH 3). The organic layer was extracted withl M potassium Phosphate buffer at pH 1.5. Layers were separated. The aqueous phase contained the desired product while the impurities stayed in the organic phase. The aqueous phase was neutralized with 1 M potassium phosphate (pH 7) and was extracted with isopropylacetate (10 volumes). Upon concentration 2-[(3R)-3-[4-amino-3-(2-fluoro-4- phenoxy-phenyl)pyrazolo[3,4-d]pyrimidin-l-yl]piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin-l-yl]pent-2-enenitrile was obtained as a foam having >99% HPLC purity. MS (pos. ion) m/z: 666 (M+l ).The foam containing high levels of residual solvent was dissolved in 2 M HC1 and the resulting solution was placed under vacuum to remove residual organic solvents. pH of the solution was then adjusted to ~ 7 and the resulting paste was filtered and dried in vacuum without heat. This resulted in isolation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy- phenyl)pyrazolo[3,4-d]pyrimidin-l-yl]piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3- yl)piperazin- l-yl]pent-2-enenitrile containing residual water up to 10%. Drying under vacuum without heat reduces the water level but lead to generation of impurities.Step 1AAlternatively, the isopropylacetate solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4- phenoxy-phenyl)pyrazolo[3,4-d]pyrimidin- 1 -yl]piperidine- 1 -carbonyl]-4-methyl-4-[4- (oxetan-3-yl)piperazin-l -yl]pent-2-enenitrile can be concentrated to 4 vol and added to heptane (20 volume) at 0 °C. The resulting suspension was stirred at 0 °C overnight and the product was filtered, washed twice with heptane and dried at 45 °C for 2 days under vacuum to give 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4-d]pyrimidin-l – yl]piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin-l-yl]pent-2-enenitrile in 85 – 90 % yield as a free flowing solid. However, the solids obtained by this method contained high residual solvents (3.9 wt% isopropylacetate and 1.7 wt% heptane). In addition, the free base form was not very stable as degradation products were observed during the drying process at less than 45 °C.Salt formationExample 2Preparation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)-pyrazolo[3,4-d]pyrimidin- l-yl]-piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)-piperazin-l-yl]pent-2-enenitrile hemisulfate and sulfate saltHemisulfate: To the solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)-pyrazolo[3,4- d]pyrimidin-l-yl]-piperidine -carbonyl]-4-methyl-4-[4-(oxetan-3-yl)-piperazin-l-yl]pent-2- enenitrile (4.2 g) in EtOAc (60 mL, 15 vol) was added sulfuric acid (0.31 g, 0.17 mL, 0.5 eq) in EtOAc (20 mL, 5 vol) at ambient temperature. The suspension was stirred at ambient temperature for ~ 2 hr and then 40 °C for 4 hr and then at ambient temperature for at least 1 hr. After filtration and drying at ambient temperature under vacuum, 1.5 g of white powder was obtained. Solubility of the hemi-sulfate at ambient temperature was > 100 mg/mL in water.Sulfate saltTo the solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)-pyrazolo[3,4- d]pyrimidin-l-yl]-piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)-piperazin-l-yl]pent-2- enenitrile (810 mg) in EtOAc (8 mL, 10 vol) was added sulfuric acid (0.06 mL, 1.0 equiv.) in EtOAc (2.5 mL, 5 vol) at ambient temperature. The resulting suspension was stirred at 40 °C for 2 hr and then cooled to ambient temperature for at least 1 hr. After filtration, solids were dried by suction under Argon for 1 h to give a white powder (0.68 g) in 69% yield.
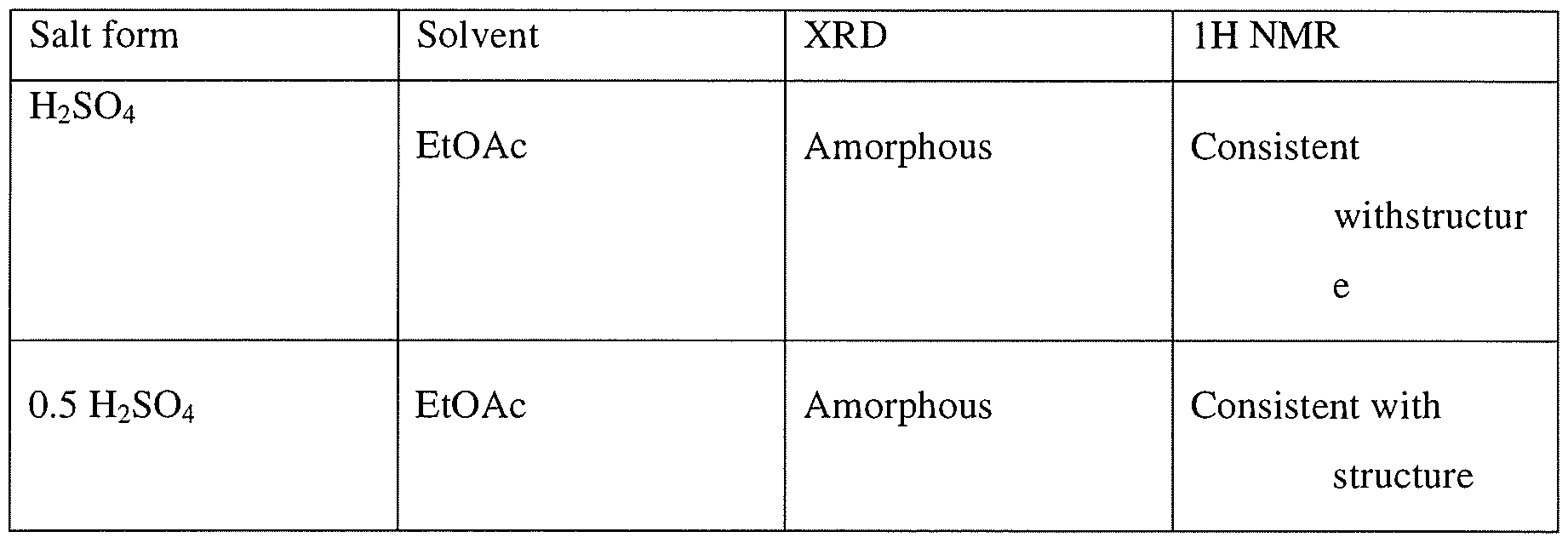
Example 3Preparation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)-pyrazolo[3,4- d]pyrimidin- 1 -yl]-piperidine- 1 -carbonyl] -4-methyl-4-[4-(oxetan-3-yl)-piperazin- 1 -yl]pent-2- enenitrile hydrochlorideTo a solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4- d]pyrimidin- 1 -yl]piperidine- 1 -carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin- 1 -yl]pent-2- enenitrile (100 mg, 0.15 mmol) in CH2CI2 (1ml) at ambient temperature was added 2 equivalent of HC1 (0.3 mmol, 0.15 ml of 2M HC1 in 1 : 1 dioaxane:CH2Cl2). The resulting homogeneous solution was stirred at ambient temperature for 1 h and was added dropwise to 15 volumes of ethylacetate (as compared to CH2C12) resulting in formation of a white solid. The mixtures was aged at ambient temperature for lh and placed at 2-8 C for 19 h. Upon filtration and washing of the filter cake with ethylacetate and drying a white solid was obtained. Analysis by XRPD indicated formation of an amorphous solid. Both Ή-NMR and IC analysis indicated formation of the salt. IC indicated formation mono-HCl salt.

Example 4General procedure for preparation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy- phenyl)pyrazolo[3,4-d]pyrimidin-l-yl]-piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)- piperazin-l-yl]pent-2-enenitrile mono- and di-mesylate saltsTo a solution of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4- d]pyrimidin-l-yl]piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin-l-yl]pent-2- enenitrile (100 mg, 0.15 mmol) in CH2C12 (1 ml) at ambient temperature was added either 1 equivalent of methanesulfonic acid (0.15 mmol, 0.2 ml of 74 mg/ml solution in CH2C12) or 2 equivalent of methanesulfonic acid (0.3 mmol, 0.4 ml of 74 mg/ml solution in CH2C12). The resulting homogeneous solution was stirred at ambient temperature for 1 h and was added dropwise to 10 volumes of antisolvents (ethylacetate, methyl tert-butylether (MTBE), or cyclohexane) (10 ml as compared to CH2C12) resulting in formation of a white solid. The mixture was aged at ambient temperature for lh and placed at 2-8 °C for 19 h. Upon filtration and washing of the filter cake with the antisolvent and drying, a white solid was obtained. Analysis by XRPD indicated formation of an amorphous solid. Both Ή-NMR and IC analysis indicated formation of the salt as well as counterion ratio.Alternatively 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4-d]- pyrimidin- 1 -yl]piperidine- 1 -carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin- 1 -yl]pent-2- enenitrile can be dissolved in 4 volumes of isopropylacetate and added to 2 equivalent of methanesulfonic acid in 6 volumes of isopropylacetate at 0 °C to generate the dimesylate salt.

1. Theoretical mesylate content, monomesylate=12.6% and dimesylate=22.4%, NO- not determinedExample 5 General procedure for the preparation of carboxylate salt Approximately 20 mg of the compound (I) was dissolved in minimum amount of the allocated solvent system. These were then mixed with the appropriate number of equivalents of counterion dissolved or slurried in the allocated solvent.If compound (I) was insoluble in the selected solvent, slurry of the sample was used after adding 300 μί.If the acid was insoluble in the selected solvent, slurry of the acid was used after adding 300 xL.If the acid was a liquid, the acid was added to the dissolved/slurried compound (I) from a stock solution in the allocated solvent.The suspensions/ precipitates resulting from the mixtures of compound (I) were temperature cycled between ambient (ca. 22°C) and 40°C in 4 hour cycles for ca. 48 hrs (the cooling/heating rate after each 4 hour period was ca. 1 °C/min). The mixtures were visually checked and any solids present were isolated and allowed to dry at ambient conditions prior to analysis. Where no solid was present, samples were allowed to evaporate at ambient. Samples which produced amorphous material, after the treatment outlined above, were re- dissolved and precipitated using anti-solvent (ter/-butylmethylether) addition methods at ambient conditions (ca. 22°C). i.e. the selected anti-solvent was added to each solution, until no further precipitation could be observed visually or until no more anti-solvent could be added. The solvents used in this preparation were acetonitrile, acetone, isopropyl acetate, THF and MTBE. The acid used were oxalic acid, L-aspartic acid, maleic acid, malonic acid, L-tartaric acid, and fumaric acid.Example 6General procedure for preparation of 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy- phenyl)pyrazolo[3,4-d]pyrimidin-l-yl]-piperidine-l-carbonyl]-4-methyl-4-[4-(oxetan-3-yl)- piperazin-l-yl]pent-2-enenitrile hemicitrate saltTo a solution 2-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)pyrazolo[3,4-d]- pyrimidin- 1 -yl]piperidine- 1 -carbonyl]-4-methyl-4-[4-(oxetan-3-yl)piperazin- 1 -yl]pent-2- enenitrile (5 g, 7.5 mmol) in ethanol (50 ml) was added citric acid (720.5 mg, 3.76 mmol) dissolved in 2 ml of water. Mixture was stirred at ambient temperature for 15 min, additional 0.5 ml of water was added and the mixture was stirred for 1 h, concentrated in vacuo to a gum. Ethanol was added and the mixture was concentrated. This process was repeated twice more and then CH2CI2 was added to the mixture. Upon concentration a white solid was obtained which was tumble dried under reduced pressure at 40 C for 4 h, then in a vacuum oven for 19h to give 5.4 g of a solid. Analysis by XRD indicated formation of an amorphous solid
PATENT
WO2014039899, Example 31
Rilzabrutinib (PRN1008) is an oral, reversible covalent inhibitor of Bruton’s tyrosine kinase (BTK) [1].
https://patents.google.com/patent/WO2014039899A1/enExample 31Synthesis of (R)-2-(3-(4-amino-3-(2-fluoro-4-phenoxyphenyl)- 1 H-pyrazolo[3,4-d]pyrimidin- 1 -yl)piperidine- 1 -carbonyl)-4-methyl-4-(4-(oxetan-3-yl)piperazin- 1 -yl)pent-2-enenitrile

Step 1A solution of 2-bromo-2-methyl-propanal (696.6 mg, 4.61 mmol) in DCM (10 mL) was cooled with an ice bath and l -(oxetan-3-yl)piperazine (328 mg, 2.31 mmol), diluted with 5-10 mL of DCM, was slowly added via addition funnel over a 15 min period. Next, Hunig’s base (0.4 mL, 2.31 mmol) was added and then the cooling bath was removed. The reaction mixture was stirred at room temperature overnight and the DCM layer was washed three times with 0.5N HC1. The combined aqueous layer was neutralized with NaOH to pH 10-11 and extracted with DCM. The combined organic layer was washed with brine and dried over Na?S04. Filtration and removal of solvent afforded 2-methyl-2-[4-(oxetan-3-yl)piperazin-l- yl]propanal as a light yellow liquid, which was used directly in the next step without further purification.Step 2To a cooled (0 °C) solution of 3-[(3R)-3-[4-amino-3-(2-fluoro-4-phenoxy-phenyl)- pyrazolo[3,4-d]pyrimidin-l-yl]-l-piperidyl]-3-oxo-propanenitrile (80 mg, 0.17 mmol), was added 2-methyl-2-[4-(oxetan-3-yl)piperazin-l-yl]propanal (-108 mg, 0.51 mmol) in DCM (10 mL) followed by pyrrolidine (0.08 mL, 1.02 mmol) and TMS-C1 (0.09 raL, 0.68 mmol.) The ice bath was removed, and the reaction stirred 1 hour. Most of the solvent was removed and the residues were purified by chromatography, using 95:5 CH2Cl2:MeOH to obtain 79 mg of (R)-2-(3-(4-amino-3-(2-fluoro-4-phenoxyphenyl)-lH-pyrazolo[3,4-d]-pyrimidin-l- yl)piperidine- 1 -carbonyl)-4-methyl-4-(4-(oxetan-3-yl)piperazin- 1 -yl)pent-2-enenitrile as a white solid. MS (pos. ion) m/z: 666 (M+l).
PAPER
https://www.sciencedirect.com/science/article/abs/pii/S0223523421001781?dgcid=rss_sd_all
Therapy based on Bruton’s tyrosine kinase (BTK) inhibitors one of the major treatment options currently recommended for lymphoma patients. The first generation of BTK inhibitor, Ibrutinib, achieved remarkable progress in the treatment of B-cell malignancies, but still has problems with drug-resistance or off-target induced serious side effects. Therefore, numerous new BTK inhibitors were developed to address this unmet medical need. In parallel, the effect of BTK inhibitors against immune-related diseases has been evaluated in clinical trials. This review summarizes recent progress in the research and development of BTK inhibitors, with a focus on structural characteristics and structure-activity relationships. The structure-refinement process of representative pharmacophores as well as their effects on binding affinity, biological activity and pharmacokinetics profiles were analyzed. The advantages and disadvantages of reversible/irreversible BTK inhibitors and their potential implications were discussed to provide a reference for the rational design and development of novel potent BTK inhibitors.

Research
Rilzabrutinib is an oral, reversible covalent inhibitor of Bruton’s tyrosine kinase, that may increase platelet counts in people with immune thrombocytopenia by means of dual mechanisms of action: decreased macrophage (Fcγ receptor)–mediated platelet destruction and reduced production of pathogenic autoantibodies.[5]
References
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/219685s000lbl.pdf
- “FDA Approves Drug to Treat Adults with Persistent or Chronic Immune Thrombocytopenia”. U.S. Food and Drug Administration. 2 September 2025. Retrieved 5 September 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - “Press Release: Sanofi’s Wayrilz approved in US as first BTK inhibitor for immune thrombocytopenia” (Press release). Sanofi. 29 August 2025. Retrieved 5 September 2025 – via GlobeNewswire.
- World Health Organization (2020). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 83”. WHO Drug Information. 34 (1). hdl:10665/339768.
- Kuter DJ, Efraim M, Mayer J, Trněný M, McDonald V, Bird R, et al. (April 2022). “Rilzabrutinib, an Oral BTK Inhibitor, in Immune Thrombocytopenia”. The New England Journal of Medicine. 386 (15): 1421–1431. doi:10.1056/NEJMoa2110297. PMID 35417637.
External links
- “Rilzabrutinib ( Code – C174769 )”. EVS Explore.
- Clinical trial number NCT04562766 for “Study to Evaluate Rilzabrutinib in Adults and Adolescents With Persistent or Chronic Immune Thrombocytopenia (ITP) (LUNA 3)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Wayrilz |
| Other names | PRN-1008 |
| AHFS/Drugs.com | Wayrilz |
| License data | US DailyMed: Rilzabrutinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1575591-66-0 |
| PubChem CID | 73388818 |
| DrugBank | DB17709 |
| ChemSpider | 58893525 |
| UNII | NWN58M4F5T |
| KEGG | D11873 |
| ChEMBL | ChEMBL3702854 |
| Chemical and physical data | |
| Formula | C36H40FN9O3 |
| Molar mass | 665.774 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////////////PRN-1008, PRN 1008, Rilzabrutinib, リルザブルチニブ, Fda 2025, approvals 2025 8/29/2025, Wayrilz,
N#CC(=CC(N(C1COC1)C)(C)C)C(=O)N1CCCC1Cn1nc(c2c1ncnc2N)c1ccc(cc1F)Oc1ccccc1
PAT
PAT
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}