
Home » Phase2 drugs
Category Archives: Phase2 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Tebapivat
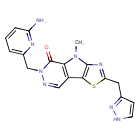
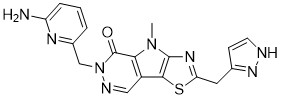
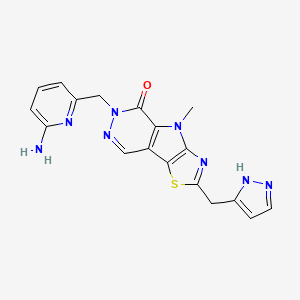
Tebapivat
CAS 2283422-04-6
WeightAverage: 392.44
Monoisotopic: 392.116778341
Chemical FormulaC18H16N8OS
10-[(6-aminopyridin-2-yl)methyl]-7-methyl-4-(1H-pyrazol-5-ylmethyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one
6-[(6-aminopyridin-2-yl)methyl]-4-methyl-2-[(1H-pyrazol-3-yl)methyl]-4,6-dihydro-5H-[1,3]thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5-one
6-[(6-aminopyridin-2-yl)methyl]-4-methyl-2-[(1H-pyrazol-3-yl)methyl]-4,6-dihydro-5H-[1,3]thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5-one
- AG946
- CS-0115951
- HY-135884
- ORG4KGP5ZS
- OriginatorAgios Pharmaceuticals
- ClassAntianaemics; Small molecules
- Mechanism of ActionPyruvate kinase stimulants
- Orphan Drug StatusYes – Myelodysplastic syndromes
- Phase IIAnaemia; Sickle cell anaemia
- 01 May 2025Phase-II clinical trials in Sickle cell anaemia in USA (PO) (NCT06924970)
- 01 May 2025Agios plans to initiate a phase II clinical trial for Sickle cell disease(PO) in mid-2025.
- 21 Feb 2025Agios Pharmaceuticals completes a phase I bioavailability trial (In volunteers) in USA (PO, capsule) (NCT06745271)
Tebapivat is under investigation in clinical trial NCT05490446 (A Study of Tebapivat (AG-946) in Participants With Anemia Due to Lower-risk Myelodysplastic Syndromes (LR-MDS)).
Tebapivat is an orally available activator of the red cell isoform of pyruvate kinase (PK-R; PKR), with potential to improve hemolytic anemia and related-symptoms in patients with pyruvate kinase deficiency (PKD). Upon oral administration, tebapivat binds to and activates PKR, thereby enhancing glycolytic pathway activity in red blood cells (RBCs), improving adenosine triphosphate (ATP) levels and reducing 2,3-diphosphoglycerate (2,3-DPG) levels. This may result in increased oxygen affinity, improved RBC deformability, decreased sickle RBC hemolysis, increased hemoglobin (Hb) levels and improved RBC membrane function. Mutations in PKR cause deficiency in PKR which prevents adequate RBC glycolysis, leading to a build-up of the upstream glycolytic intermediate 2,3-DPG and deficiency in the PKR product ATP.
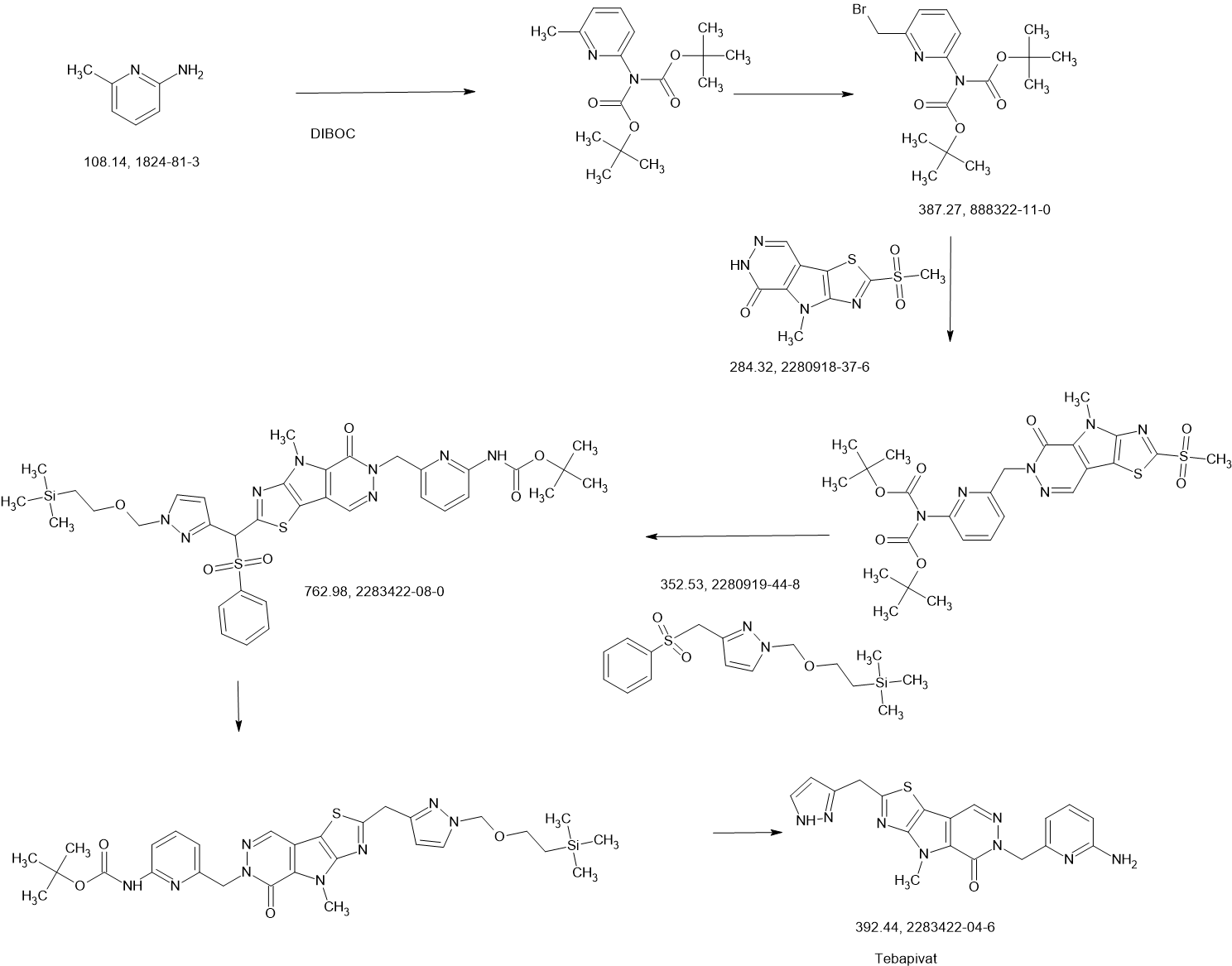
SCHEME
COUPLER
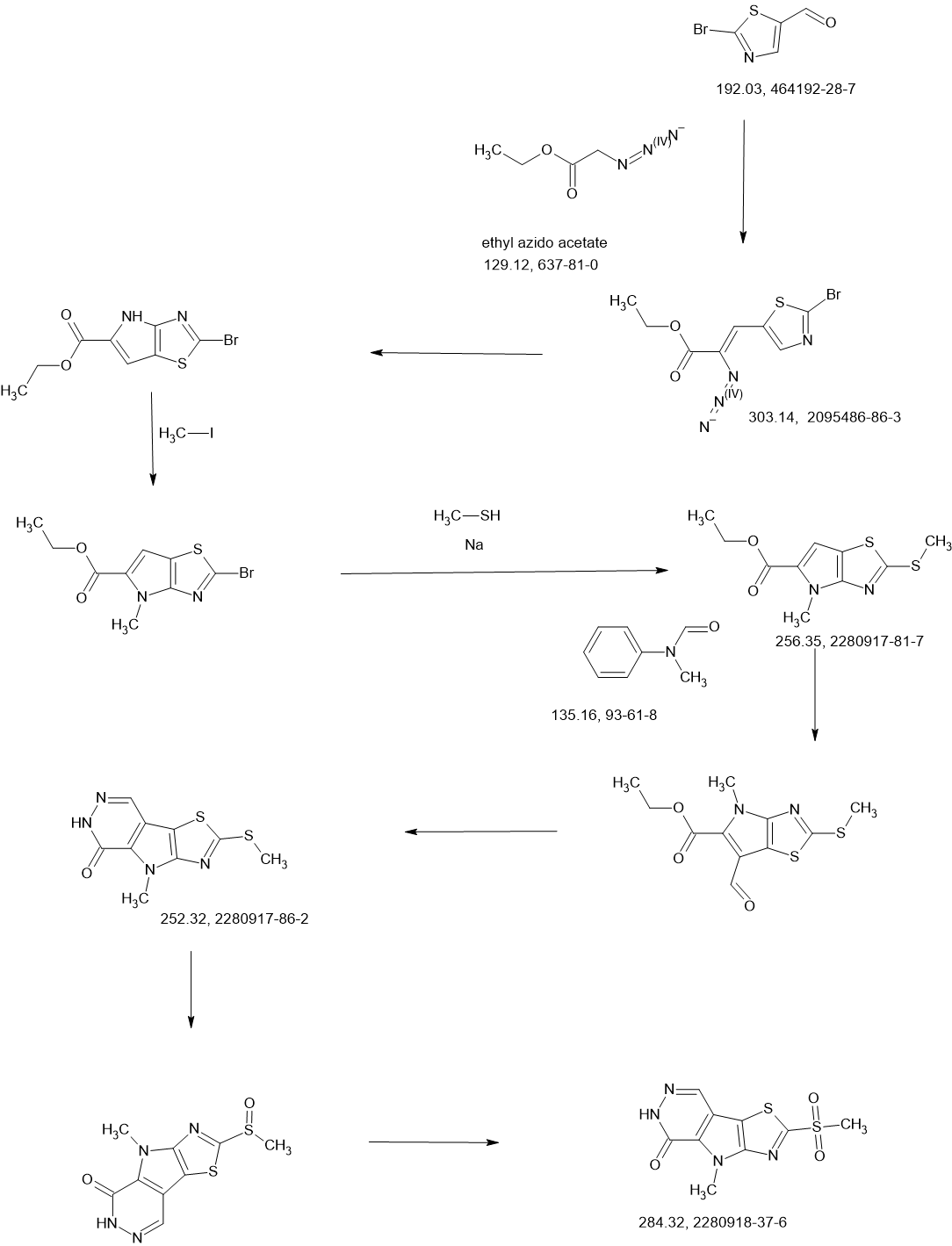
COUPLER
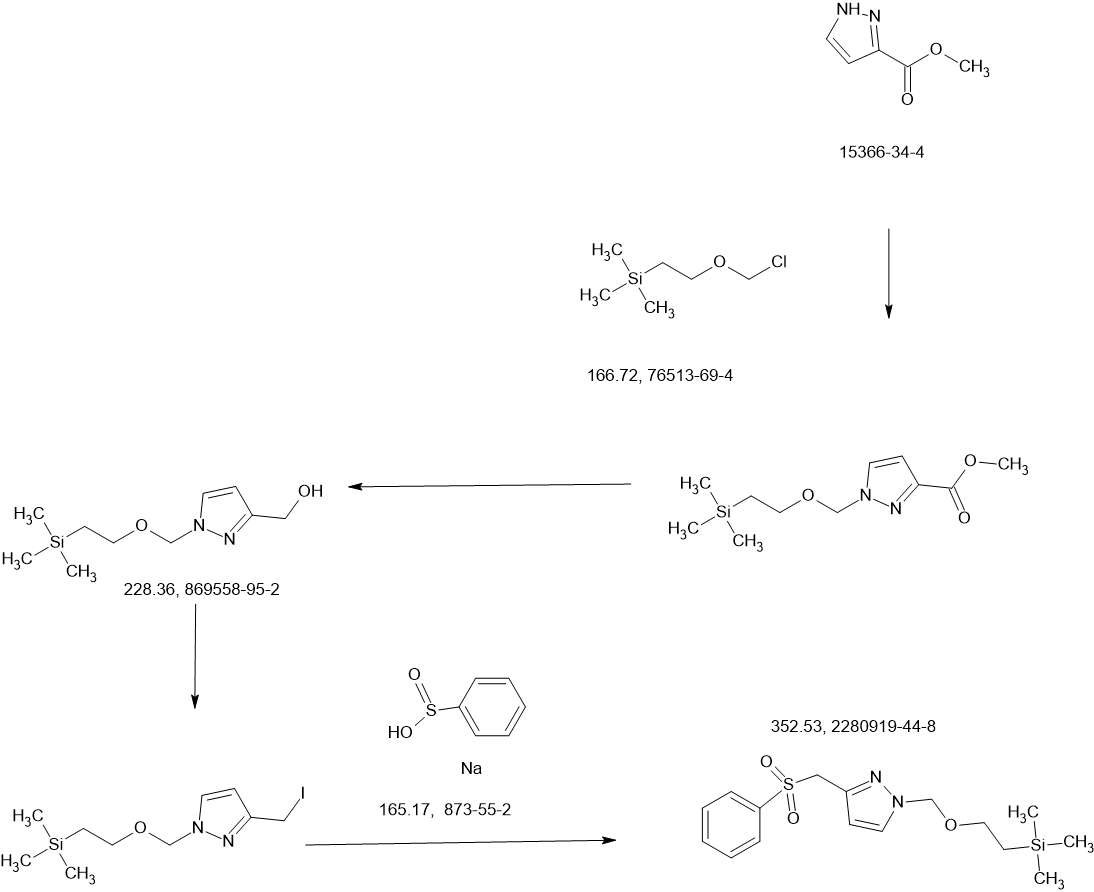
MAIN
PATENT
Agios Pharmaceuticals, Inc.
WO2019035864
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019035864&_cid=P22-MDGSEF-03229-1
Example 8A. Synthesis of 2-((1H-pyrazol-3-yl)methyl)-6-((6-aminopyridin-2-yl)methyl)- 4-methyl-4H-thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5(6H)-one and 6-((6- aminopyridin-2-yl)methyl)-4-methyl-2-(1H-pyrazole-3-carbonyl)-4H- thiazolo[5′,4′:4,5]pyrroIo[2,3-d]pyridazin-5(6H)-one
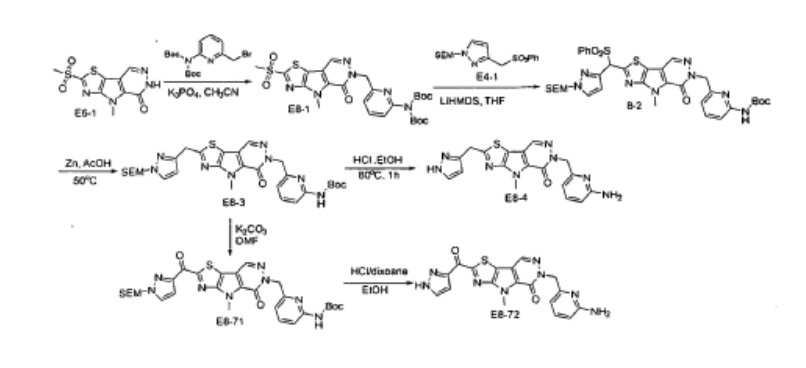


Step F. Synthesis of 6-((6-aminopyridin-2-yl)methyl)-4-methyl-2-(1H-pyrazole-3- carbonyl)-4H-thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5(6H)-one To a solution of tert- butyl (6-((4-methyl-5-oxo-2-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-3-carbonyl)- 4H-thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-6(5H)-yl)methyl)pyridin-2-yl)carbamate (20 mg, 0.03 mmol) in EtOH (1 mL) was added HCl (1 mL, 4 mol/L in dioxane). The mixture was stirred at 80 °C for lhr and cooled down. The precipitate was collected by filtration and neutralized with sat. NaHCO3, washed with water and dried to afford 5 mg of 6-((6- aminopyridin-2-yl)methyl)-4-methyl-2-(1H-pyrazole-3-carbonyl)-4H- thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5(6H)-one. LC-MS (ESI): m/z 407 (M+H)+. 1H NMR (400 MHz, DMSO-d6) δ: 8.75 (s, 1H), 7.96 (s, 1H), 7.50 (s, 1H), 7.31-7.22 (m, 1H), 6.31 (d, 1H), 6.14 (d, 1H), 5.91 (s, 2H), 5.23 (s, 2H), 4.38 (s, 3H).
PATENT
WO2023091414
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023091414&_cid=P22-MDGSRV-15431-1
PATENT
WO2019035863
WO2019035865
WO2019035864



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
/////////Tebapivat, 2283422-04-6, AG946, CS-0115951, HY-135884, AG 946, CS 0115951, HY 135884, ORG4KGP5ZS, AGIOS, Orphan Drug, PHASE 2,
NERIGLIATIN

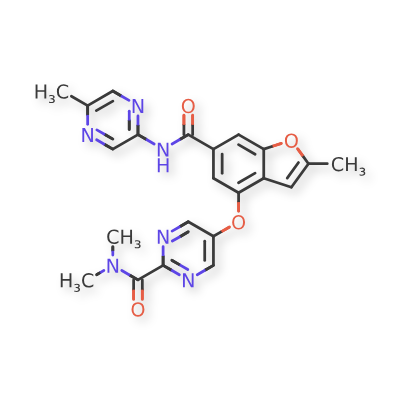
PF 04937319, NERIGLIATIN
N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide
MW 432.43, MF C22 H20 N6 O4
CAS 1245603-92-2
2-Pyrimidinecarboxamide, N,N-dimethyl-5-[[2-methyl-6-[[(5-methyl-2-pyrazinyl)amino]carbonyl]-4-benzofuranyl]oxy]-
N,N-Dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)carbamoyl)-benzofuran-4- yloxy)pyrimidine-2-carboxamide
- N,N-Dimethyl-5-({2-Methyl-6-[(5-Methylpyrazin-2-Yl)carbamoyl]-1-Benzofuran-4-Yl}oxy)pyrimidine-2-Carboxamide
- 2-Pyrimidinecarboxamide, N,N-dimethyl-5-[[2-methyl-6-[[(5-methyl-2-pyrazinyl)amino]carbonyl]-4-benzofuranyl]oxy]-
- 7E99B9ZM19
Pfizer Inc. clinical candidate currently in Phase 2 development.
SCHEME
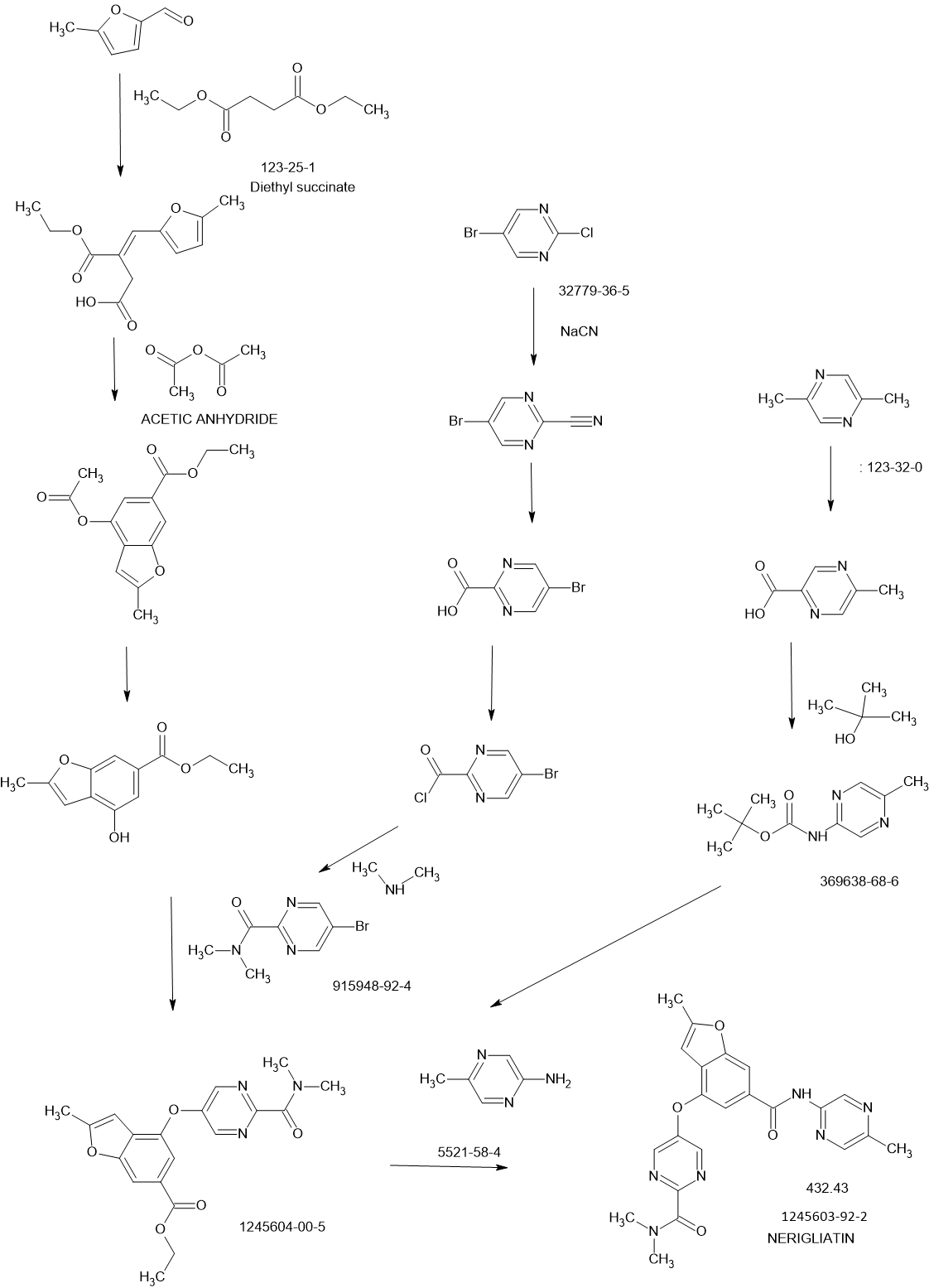
REF
MedChemComm (2011), 2(9), 828-839 81%
WO2010103437
CLINICAL TRIALS
A trial to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of single doses of PF-04937319 in subjects with type 2 diabetes mellitus (NCT01044537)
Multiple dose study of PF-04937319 in patients with type 2 diabetes (NCT01272804)
Phase 2 study to evaluate safety and efficacy of investigational drug – PF04937319 in patients with type 2 diabetes (NCT01475461)
SYNTHESIS

Glucokinase is a key regulator of glucose homeostasis and small molecule activators of this enzyme represent a promising opportunity for the treatment of Type 2 diabetes. Several glucokinase activators have advanced to clinical studies and demonstrated promising efficacy; however, many of these early candidates also revealed hypoglycemia as a key risk. In an effort to mitigate this hypoglycemia risk while maintaining the promising efficacy of this mechanism, we have investigated a series of substituted 2-methylbenzofurans as “partial activators” of the glucokinase enzyme leading to the identification of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as an early development candidate.
Diabetes is a major public health concern because of its increasing prevalence and associated health risks. The disease is characterized by metabolic defects in the production and utilization of carbohydrates which result in the failure to maintain appropriate blood glucose levels. Two major forms of diabetes are recognized. Type I diabetes, or insulin-dependent diabetes mellitus (IDDM), is the result of an absolute deficiency of insulin. Type Il diabetes, or non-insulin dependent diabetes mellitus (NIDDM), often occurs with normal, or even elevated levels of insulin and appears to be the result of the inability of tissues and cells to respond appropriately to insulin. Aggressive control of NIDDM with medication is essential; otherwise it can progress into IDDM. As blood glucose increases, it is transported into pancreatic beta cells via a glucose transporter. Intracellular mammalian glucokinase (GK) senses the rise in glucose and activates cellular glycolysis, i.e. the conversion of glucose to glucose-6-phosphate, and subsequent insulin release. Glucokinase is found principally in pancreatic β-cells and liver parenchymal cells. Because transfer of glucose from the blood into muscle and fatty tissue is insulin dependent, diabetics lack the ability to utilize glucose adequately which leads to undesired accumulation of blood glucose (hyperglycemia). Chronic hyperglycemia leads to decreases in insulin secretion and contributes to increased insulin resistance. Glucokinase also acts as a sensor in hepatic parenchymal cells which induces glycogen synthesis, thus preventing the release of glucose into the blood. The GK processes are thus critical for the maintenance of whole body glucose homeostasis.
It is expected that an agent that activates cellular GK will facilitate glucose-dependent secretion from pancreatic beta cells, correct postprandial hyperglycemia, increase hepatic glucose utilization and potentially inhibit hepatic glucose release. Consequently, a GK activator may provide therapeutic treatment for NIDDM and associated complications, inter alia, hyperglycemia, dyslipidemia, insulin resistance syndrome, hyperinsulinemia, hypertension, and obesity. Several drugs in five major categories, each acting by different mechanisms, are available for treating hyperglycemia and subsequently, NIDDM (Moller, D. E., “New drug targets for Type 2 diabetes and the metabolic syndrome” Nature 414; 821 -827, (2001 )): (A) Insulin secretogogues, including sulphonyl-ureas (e.g., glipizide, glimepiride, glyburide) and meglitinides (e.g., nateglidine and repaglinide) enhance secretion of insulin by acting on the pancreatic beta-cells. While this therapy can decrease blood glucose level, it has limited efficacy and tolerability, causes weight gain and often induces hypoglycemia. (B) Biguanides (e.g., metformin) are thought to act primarily by decreasing hepatic glucose production. Biguanides often cause gastrointestinal disturbances and lactic acidosis, further limiting their use. (C) Inhibitors of alpha-glucosidase (e.g., acarbose) decrease intestinal glucose absorption. These agents often cause gastrointestinal disturbances. (D) Thiazolidinediones (e.g., pioglitazone, rosiglitazone) act on a specific receptor (peroxisome proliferator-activated receptor-gamma) in the liver, muscle and fat tissues. They regulate lipid metabolism subsequently enhancing the response of these tissues to the actions of insulin. Frequent use of these drugs may lead to weight gain and may induce edema and anemia. (E) Insulin is used in more severe cases, either alone or in combination with the above agents. Ideally, an effective new treatment for NIDDM would meet the following criteria: (a) it would not have significant side effects including induction of hypoglycemia; (b) it would not cause weight gain; (c) it would at least partially replace insulin by acting via mechanism(s) that are independent from the actions of insulin; (d) it would desirably be metabolically stable to allow less frequent usage; and (e) it would be usable in combination with tolerable amounts of any of the categories of drugs listed herein.
Substituted heteroaryls, particularly pyridones, have been implicated in mediating GK and may play a significant role in the treatment of NIDDM. For example, U.S. Patent publication No. 2006/0058353 and PCT publication No’s. WO2007/043638, WO2007/043638, and WO2007/117995 recite certain heterocyclic derivatives with utility for the treatment of diabetes. Although investigations are on-going, there still exists a need for a more effective and safe therapeutic treatment for diabetes, particularly NIDDM.
Designing glucokinase activators with reduced hypoglycemia risk: discovery of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as a clinical candidate for the treatment of type 2 diabetes mellitus
Jeffrey A. Pfefferkorn,*a et al
*Corresponding authors
aPfizer Worldwide Research & Development, Eastern Point Road, Groton
E-mail: jeffrey.a.pfefferkorn@pfizer.com
Tel: +860 686 3421
Med. Chem. Commun., 2011,2, 828-839
DOI: 10.1039/C1MD00116G
http://pubs.rsc.org/en/content/articlelanding/2011/md/c1md00116g/unauth#!divAbstract
http://www.rsc.org/suppdata/md/c1/c1md00116g/c1md00116g.pdf
Glucokinase is a key regulator of glucose homeostasis and small molecule activators of this enzyme represent a promising opportunity for the treatment of Type 2 diabetes. Several glucokinase activators have advanced to clinical studies and demonstrated promising efficacy; however, many of these early candidates also revealed hypoglycemia as a key risk. In an effort to mitigate this hypoglycemia risk while maintaining the promising efficacy of this mechanism, we have investigated a series of substituted 2-methylbenzofurans as “partial activators” of the glucokinase enzyme leading to the identification of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as an early development candidate.
N,N-Dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)carbamoyl)-benzofuran-4- yloxy)pyrimidine-2-carboxamide (28). To a solution of the 5-methyl-2-aminopyrazine (38.9 g, 356 mmol) in dimethoxyethane (315 mL) in a 3-neck flask equipped with overhead stirring and a condenser at 0 o C was added Me2AlCl (1 M solution in hexanes) (715 mL). The mixture was warmed to room temperature and stirred for 1.5 h. In a separate flask, 26 (52.6 g, 142.5 mmol) was dissolved in dimethoxyethane (210 mL). This mixture was then added to the amine mixture. A gum precipitated and upon scratching the flask it dissipated into a solid. The reaction was refluxed for 3.5 h. Aq. Rochelle’s salt (5 L) and 2-MeTHF (2 L) was added to the mixture and this was allowed to stir with overhead stirring for 14 h, after which time, a yellow solid precipitated. The solid was collected by filtration, washing with 2-MeTHF. The resulting solid was dried in a vacuum oven overnight to afford the desired material (50.0g) in 81% yield.
1 H NMR (400MHz, CDCl3) δ 9.54 (d, J = 1.56 Hz, 1H), 8.50 (s, 2H), 8.37 (s, 1H), 8.14 (d, J = 0.78 Hz, 1H), 7.88 – 7.92 (m, 1H), 7.52 (d, J = 1.37 Hz, 1H), 6.28 (t, J = 0.98 Hz, 1H), 3.14 (s, 3H), 2.98 (s, 3H), 2.55 (s, 3H), 2.49 (d, J = 1.17 Hz, 3H);
MS(ES+ ): m/z 433.4 (M+1), MS(ES- ): m/z 431.3 (M-1).
PAPER

http://pubs.rsc.org/en/content/articlelanding/2013/md/c2md20317k#!divAbstract
PAPER
Bioorganic & Medicinal Chemistry Letters (2013), 23(16), 4571-4578
http://www.sciencedirect.com/science/article/pii/S0960894X13007452

Figure 1.
Glucokinase activators 1 and 2.
PATENT
WO 2010103437
https://www.google.co.in/patents/WO2010103437A1?cl=en
Scheme I outlines the general procedures one could use to provide compounds of the present invention having Formula (I).


Preparations of Starting Materials and Key Intermediates
Preparation of Intermediate (E)-3-(ethoxycarbonyl)-4-(5-methylfuran-2-yl)but- 3-enoic acid (I- 1a):

(Ma) To a vigorously stirred solution of 5-methyl-2-furaldehyde (264 ml_, 2650 mmol) and diethyl succinate (840 ml_, 5050 mmol) in ethanol (1.820 L) at room temperature was added sodium ethoxide (0.93 L of a 21 weight % solution in ethanol) in one portion. The reaction mixture was then heated at reflux for 13 hours. After cooling to room temperature, the mixture was concentrated in vacuo (all batches were combined at this point). The resulting residue was partitioned between ethyl acetate (1 L) and hydrochloric acid (1 L of a 2M aqueous solution). After separation, the aqueous layer was extracted with ethyl acetate (2 x 1 L). The combined organic extracts were then extracted with sodium hydrogen carbonate (2 x 1 L of a saturated aqueous solution). These aqueous extracts were combined and adjusted to pH 2 with hydrochloric acid (2M aqueous solution) then extracted with ethyl acetate (2 x 1 L). These organic extracts were combined and concentrated in vacuo to give desired (E)-3-(ethoxycarbonyl)-4-(5-methylfuran-2-yl)but-3-enoic acid (J1 Ia: 34.34 g, 5%). The original organic extract was extracted with sodium hydroxide (2 L of a 2M aqueous solution). This aqueous extract was adjusted to pH 2 with hydrochloric acid (2M aqueous solution) then extracted with ethyl acetate (2 x 1 L). These organic extracts were combined and concentrated in vacuo to give additional desired materials (395.2 gram, 63%) as red liquid. 1H NMR (CDCI3, 300 MHz) δ ppm 7.48 (s, 1 H), 6.57 (d, 1 H), 6.09 (d, 1 H), 4.24 (q, 2H), 3.87 (s, 2H), 2.32 (s, 3H), 1.31 (t, 3H).
Preparation of Intermediate ethyl 4-acetoxy-2-methylbenzofuran-6- carboxylate (1-1 b):

(M b) To a vigorously stirred solution of (E)-3-(ethoxycarbonyl)-4-(5- methylfuran-2-yl)but-3-enoic acid (1-1 a: 326.6 g, 1 .371 mol) in acetic anhydride (1 .77 L, 18.72 mol) at room temperature was added sodium acetate (193 g, 2350 mmol) in one portion. The reaction mixture was then heated at reflux for 2.5 hours. After cooling to room temperature, the mixture was concentrated in vacuo (all batches were combined at this point). The resulting residue was suspended in dichloromethane (1 .5 L) and filtered, washing the solids with dichloromethane (3 x 500 ml_). The combined filtrate and washings were then washed with sodium hydrogencarbonate (2 x 1 L of a saturated aqueous solution) and brine (2 L), then concentrated in vacuo to give desired ethyl 4-acetoxy-2-methylbenzofuran-6-carboxylate (H b: 549.03 g, quantitative). 1H NMR (CDCI3, 300 MHz) δ ppm 8.00-7.99 (m, 1 H), 7.64 (d, 1 H), 6.32-6.32 (m, 1 H), 4.38 (q, 2H), 2.47 (d, 3H), 2.37 (s, 3H), 1 .39 (t, 3H).
Preparation of Intermediate ethyl 4-hydroxy-2-methylbenzofuran-6- carboxylate (1- 1 c):

(He) To a stirred solution of ethyl 4-acetoxy-2-methylbenzofuran-6- carboxylate (Hb: 549.03 g, 1 .37 mol) in ethanol (4.00 L) at room temperature was added potassium carbonate (266 g, 1 .92 mol) in one portion. The reaction mixture was then heated at 600C for 3 hours. Potassium carbonate (100 g, 0.720 mol) was then added in one portion and the reaction mixture was heated at 600C for a further 3 hours. After cooling to room temperature the mixture was diluted with dichloromethane (2 L) and the suspension filtered, washing the solids with dichloromethane (2 x 1 L) (all batches were combined at this point). The combined filtrate and washings were then washed with citric acid (2.5 L of a 1 M aqueous solution), then concentrated in vacuo and the resulting residue purified by dry flash chromatography (hexane then 2:1 hexane:ethyl acetate). All fractions containing the desired product were combined and concentrated in vacuo. The resulting residue, which solidified on standing, was slurried with cold toluene and filtered. The solids were then stirred with hot toluene and decolourising charcoal for 1 hour, followed by filtration of the hot mixture through a pad of celite. The filtrate was allowed to cool and the resulting precipitate isolated by filtration to give desired ethyl 4-hydroxy-2- methylbenzofuran-6-carboxylate (1-1 c: 360 g, 90%) as orange powder.
1H NMR (CDCI3, 300 MHz) δ ppm 7.73-7.73 (m, 1 H), 7.45 (d, 1 H), 6.51 -6.50 (m, 1 H), 5.85 (s, 1 H), 4.39 (q, 2H), 2.48 (d, 3H), 1.40 (t, 3H). LCMS (liquid chromatography mass spectrometry): m/z 221.06 (96.39 % purity).
Preparation of SM-25-bromo-N,N-dimethylpyrimidine-2-carboxamide (SM-
£1:

(SM-2) Oxalyl chloride (47.4g, 369mmol) was added to a suspension of 5-
Bromo-pyrimidine-2-carboxylic acid (5Og, 250mmol) in dichloromethane (821 ml) at room temperature followed by 1 -2 drop of dimethylformamide. The reaction mixture was stirred under nitrogen for 2 hours LCMS in methanol indicated the presence of the methyl ester and some acid. Dimethylformamide (0.2ml) was added to the reaction mixture. The acid dissolved after 30 minutess. LCMS showed corresponding methyl ester and no starting material peak was observed. The solvent was removed and dried in vacuo to afford the crude 5-Bromo-pyrimidine-2-carbonyl chloride (55g, 100%). The 5-Bromo-pyrimidine-2-carbonyl chloride (55g, 250mmol) was dissolved in tetrahydrofuran (828ml) and dimethyl-amine (2M solution in tetrahydrofuran) (373ml, 745mmol) was added portionwise at room temperature. The reaction was stirred at room temperature under nitrogen for 16 hours, after which time, LCMS indicated completion. The mixture was diluted with ethyl acetate (500ml) and washed with H2O (500ml). The water layer was further extracted with CH2CI2 (5x500ml), all organics combined, and dried over magnesium sulfate. The filtrate was concentrated in vacuo and then suspended in methyl-/-butylether (650ml). The solution was then heated to reflux. The hot solution was allowed to cool overnight to afford pink crystals. The crystals were filtered and washed with cold methyl-t-butylether (100ml) the solid was dried in a vacuum oven at 550C for 12 hourrs to afford the title compound 5-bromo-N,N-dimethylpyhmidine-2-carboxamide (SM-2: 44g, 77%) as a pink solid.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 2.94 (s, 3 H) 3.13 (s, 3 H) 8.85 (s, 2 H) m/z (M+1 ) = 232.
Preparation of Intermediate Ethyl 4-(2-(dimethylcarbamoyl)Dyrimidin-5- yloxy)-2-methylbenzofuran-6-carboxylate (l-2a):

A mixture of Cs2CO3 (62.1 g, 191 mmol), 5-bromo-N,N- dimethylpyrimidine-2-carboxamide (SM-2: 24g, 104mmol) and ethyl 4- hydroxy-2-methylbenzofuran-6-carboxylate (1-1 c: 2Og, 91 mmol); 1 ,10- phenanthroline (1.64g, 9.07mmol) and copper iodide (864mg, 4.54mmol) in dimethylformamide (200ml) was purged with N2 gas and then heated to 90°C using a mechanical stirrer. The heterogeneous reaction mixture was stirred at this temperature for 18 hours. HPLC indicated near completion. The reaction mixture was cooled to 350C and diluted with ethyl acetate (300ml). The mixture was filtered to remove any cesium carbonate. The filtrate was then partitioned between water (500ml) and ethyl acetate (500ml); however, no separation was observed. Concentrated HCL (20ml) was added to the mixture. When the aqueous phase was about pH1 , the phases separated. The organics were separated and the aqueous layer reextracted with ethyl acetate (2x500ml). All organics were combined and back extracted with water (200ml) and brine (500ml). The organics were separated and treated with activated charcoal (10g) and magnesium sulfate. The mixture was allowed to stir for 10 minutes and then filtered through a plug of celite to afford a crude yellow solution. The filter cake was washed with ethyl acetate (100 ml_). The organics were concentrated in vacuo to afford a crude solid this was dried under high vacuum for 4 days. The dry crude solid was triturated using methanol (80 ml_). The solids were dispersed into a fine light orange crystalline powder with a red liquor. The solids were isolated by filtration and rinsed with methanol (20 ml_). The solid was dried in the vacuum oven at 550C for 12 hours to afford ethyl 4-(2- (dimethylcarbamoyl)pyrimidin-5-yloxy)-2-methylbenzofuran-6-carboxylate (J1 2a) as a yellow solid (18.2g, 54%)
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.41 (t, J=7.12 Hz, 3 H) 2.50 (d, J=0.98 Hz, 3 H) 3.00 (s, 3 H) 3.17 (s, 3 H) 4.41 (d, J=7.22 Hz, 2 H) 6.29 (s, 1 H) 7.62 (d, J=1.17 Hz, 1 H) 8.06 (s, 1 H) 8.50 (s, 2 H). m/z (M+1 ) = 370.5
Preparation of Starting material 5-bromo-N-ethyl-N-methylpyrimidine-2- carboxamide (SM-3):

(SM-3) Oxalyl chloride (1 .45g, 1 1 .1 mmol) was added to a suspension of 5-
Bromo-pyrimidine-2-carboxylic acid (1 .5g, 7.4mmol) in dichloromethane (50ml) at room temperature followed by 1 -2 drop of dimethylformamide. The reaction mixture was stirred under nitrogen for 2 hours LCMS in methanol indicated the presence of the methyl ester and some acid. Dimethylformamide (0.2ml) was added to the reaction mixture and all of the acid dissolved after 30 minutes. LCMS showed corresponding methyl ester and no starting material peak was observed. The solvent was removed and dried in vacuo to afford the crude 5-Bromo-pyrimidine-2-carbonyl chloride (1 -6g). 5-Bromo-pyrinnidine-2-carbonyl chloride (1600mg, 7.225mnnol) was dissolved in dichloromethane (25ml) and triethylamine (4.03ml, 28.9mmol) was added followed by ethyl-methyl-amine (0.68 mL, 7.92 mmol). The reaction was stirred at room temperature under nitrogen for 16 ours, after which time, LCMS indicated completion. The mixture was diluted with dichloromethane (50ml) and washed with water (50ml) followed by 10% citric acid (50ml) and brine (50ml). The organic layer was separated and dried over MgSO4, the residue was filtered and the solvent was removed in vacuo to afford the title compound 5-bromo-N-ethyl-N-methylpyrimidine-2- carboxamide (SM-3): (1.4g, 79.4%) as a brown oil.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.08 – 1.31 (m, 3 H) 2.99 (d, J=79.05 Hz, 3 H) 3.19 (q, J=7.22 Hz, 1 H) 3.59 (q, J=7.22 Hz, 1 H) 8.84 (d, J=3.12 Hz, 2 H)
Example 2
Preparation of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2- yl)carbamoyl)-benzofuran-4-yloxy)Dyrimidine-2-carboxamide (2):

(2)
To a solution of the 5-methyl-2-aminopyrazine (38.9 g, 356 mmol) in dimethylether (315 ml_) in a 3-neck flask equipped with overhead stirring and a condensor at O0C was added Me2AICI (1 M solution in hexanes) (715 ml_). The mixture was warmed at room temperature and stirred for 1.5 hours. In a separate flask, ethyl 4-(2-(dimethylcarbamoyl)pyrimidin-5-yloxy)-2- methylbenzofuran-6-carboxylate (l-2a: 52.6g, 142.5mmol) was dissolved in dimethylether (210 ml_). This mixture was then added to the complexed amine. A gum precipitated upon scratching the flask and dissipated into a solid. The resultant reaction was refluxed for 3.5 hours HPLC indicated 93% complete. Five liters of Rochelles salt made up in water and 2 liters of 2- methyltetrahydrofuran was added to the mixture. The reaction mixture was then poured into the biphasic system. The mixture was allowed to stir with overhead stirring for 14 hours, after which time, a yellow solid precipitated. The solid was collected through filteration. The solid retained was washed with 2-methyltetrahydrofuran. The resultant solid was dried in vacuo oven overnight to afford the title compound N,N-dimethyl-5-(2-methyl-6-((5- methylpyrazin-2-yl)carbamoyl)benzofuran-4-yloxy)pyhmidine-2-carboxamide (2): (49.98g, 81 %)
1H NMR (400 MHz, CHLOROFORM-d) d ppm 2.49 (d, J=1 .17 Hz, 3H) 2.55 (s, 3H) 2.98 (s, 3 H) 3.14 (s, 3 H) 6.28 (t, J=0.98 Hz, 1 H) 7.52 (d, J=1 .37 Hz, 1 H) 7.88 – 7.92 (m, 1 H) 8.14 (d, J=0.78 Hz, 1 H) 8.37 (s, 1 H) 8.50 (s, 2 H) 9.54 (d, J=1 .56 Hz, 1 H).
m/z (M+1 ) = 433.4, m/z (M-1 )= 431 .5
REFERENCES
Beebe, D.A.; Ross, T.T.; Rolph, T.P.; Pfefferkorn, J.A.; Esler, W.P.
The glucokinase activator PF-04937319 improves glycemic control in combination with exercise without causing hypoglycemia in diabetic rats
74th Annu Meet Sci Sess Am Diabetes Assoc (ADA) (June 13-17, San Francisco) 2014, Abst 1113-P
Amin, N.B.; Aggarwal, N.; Pall, D.; Paragh, G.; Denney, W.S.; Le, V.; Riggs, M.; Calle, R.A.
Two dose-ranging studies with PF-04937319, a systemic partial activator of glucokinase, as add-on therapy to metformin in adults with type 2 diabetes
Diabetes Obes Metab 2015, 17(8): 751
Study to compare single dose of three modified release formulations of PF-04937319 with immediate release material-sparing-tablet (IR MST) formulation previously studied in adults with type 2 diabetes mellitus (NCT02206607)
OTHERS

///////////Pfizer , PF 04937319, glucokinase activators, Type 2 diabetes, NERIGLIATIN, 7E99B9ZM19
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

NEW DRUG APPROVALS
ONE TIME
$10.00
//////////
Alflutinib, Furmonertinib, Firmonertinib



FIRMOMERTINIB, Furmonertinib, Alflutinib
CAS 1869057-83-9
, AST 2818, UNII-A49A7A5YN4
N-[2-[[2-(Dimethylamino)ethyl]methylamino]-5-[[4-(1-methyl-1H-indol-3-yl)-2-pyrimidinyl]amino]-6-(2,2,2-trifluoroethoxy)-3-pyridinyl]-2-propenamide
N-[2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]-6-(2,2,2-trifluoroethoxy)pyridin-3-yl]prop-2-enamide
C28H31F3N8O2 568.6 g/mol
2-Propenamide, N-[2-[[2-(dimethylamino)ethyl]methylamino]-5-[[4-(1-methyl-1H-indol-3-yl)-2-pyrimidinyl]amino]-6-(2,2,2-trifluoroethoxy)-3-pyridinyl]-
Alflutinib is under investigation in clinical trial NCT03452592 (Efficacy and Safety of Alflutinib in Locally Advanced or Metastatic Non-small Cell Lung Cancer Patients With T790M).
Firmonertinib is an orally available selective inhibitor of the epidermal growth factor receptor (EGFR) mutant form T790M, with potential antineoplastic activity. Upon administration, firmonertinib specifically binds to and inhibits the tyrosine kinase activity of EGFR T790M, a secondarily acquired resistance mutation. This prevents EGFR T790M-mediated signaling and leads to cell death in EGFR T790M-expressing tumor cells. EGFR, a receptor tyrosine kinase that is mutated in many tumor cell types, plays a key role in tumor cell proliferation and tumor vascularization. Compared to some other EGFR inhibitors, alflutinib may have therapeutic benefits in tumors with T790M-mediated drug resistance.
FIRMONERTINIB is a small molecule drug with a maximum clinical trial phase of III (across all indications) and has 4 investigational indications.
SCHEME

CONTD……..

REF
https://patentscope.wipo.int/search/en/detail.jsf?docId=US201062358&_cid=P22-MBFXFH-62339-1
Example 3: N-{2-{[2-(dimethylamino)ethyl](methyl)amino}-6-(2,2,2-trifluoroethoxyl)-5-{[4-(1-methyl-H-indol-3-yl)pyrimidin-2-yl]amino}pyridin-3-yl}acrylamide
Step 1: Synthesis of N2-methyl-N2-[2-(dimethylamino)ethyl]-6-(2,2,2-trifluoroethoxyl)-N5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl]-3-nitropyridin-2,5-diamine
| The compound was synthesized in the same manner as those in Step 1 of Example 1 with a yield of 86%. MS m/z: 545 [M+1]. |
Step 2: Synthesis of N2-methyl-N2-[2-(dimethylamino)ethyl]-6-(2,2,2-trifluoroethoxyl)-N5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl]pyridin-2,3,5-triamine
| The compound was synthesized in the same manner as those in Step 2 of Example 2 with a yield of 56%. MS m/z: 515 [M+1]. |
Step 3: Synthesis of N-{2-{[2-(dimethylamino)ethyl](methyl)amino}-6-(2,2,2-trifluoroethoxyl)-5-{[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl]amino}pyridin-3-yl}acrylamide
| The compound was synthesized in the same manner as those in Step 3 of Example 1 with a yield of 23%. MS m/z: 569 [M+1]. |
PATENT
CN110606842
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN280196686&_cid=P22-MBFXJY-67679-1
Patent application CN105315259A protects the compound of formula I and discloses its preparation method as follows:



| Example 1: Preparation of 6-chloro-3-nitro-2-(2,2,2-trifluoroethoxy)pyridine (XI-1) |
| Add toluene (24.0L) to the reactor, then add 2,6-dichloro-3-nitropyridine (3000g, 15.54mol), adjust the internal temperature between -20℃ and -10℃, and add sodium hydrogen (933g, 23.33mol) in batches. Add 2,2,2-trifluoroethanol (1586g, 16.00mol) toluene (6.0L) solution dropwise. React for 2h, and monitor the reaction end point by TLC and HPLC. After the reaction is completed, add 10% ammonium chloride solution (6.0L) dropwise. Let stand and separate. Wash the organic phase with water (6.0L) and concentrate under reduced pressure. Add ethyl acetate (0.3L), heat to 40-50℃, add n-heptane (2.7L) dropwise, cool to -15 to -5℃ after dripping, and continue crystallization for 3 hours, and filter with suction. Obtain 3017g of product solid, with a yield of 75.65%. |
| 1H NMR(500MHz,DMSO-d6)δ8.60(d,J=8.0Hz,1H),7.50(d,J=8.5Hz,lH),5.13(q,J=9.0Hz,2H); |
| 13C NMR(126MHz,DMSO-d6)δ153.20,151.09,139.34,132.67,123.38(q,J=277.2Hz),119.14,63.34(q,J=36Hz); |
| MS m/z:256.99[M+1]。 |
| Example 2: Preparation of 6-chloro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (X-1) |
| At room temperature, add acetonitrile (21.0L) and water (21.0L) to the reactor, start stirring, add 6-chloro-3-nitro-2-(2,2,2-trifluoroethoxy)pyridine (3017.0g, 11.76mol) obtained in Example 1, and add hydrosulfite (15.1Kg, 70.54mol). Control the temperature at 27-33°C to react for 2 hours. Add 36% concentrated hydrochloric acid (11.9Kg, 117.60mol) dropwise, and continue to react for 1.5 hours. Add solid sodium bicarbonate (12.8Kg, 12.96mol). Filter, separate the mother liquor, wash the organic phase with saturated brine (21.0L), and concentrate under reduced pressure to obtain an oily substance. Theoretically calculated for the next step reaction. |
| 1H NMR(500MHz,DMSO-d6)δ7.03(d,J=8.0Hz,1H),6.90(d,J=8.0Hz,1H),5.21(s,2H),4.93(q,J=9.0Hz,2H); |
| 13C NMR(126MHz,DMSO-d6)δ148.16,131.72,130.55,123.93(q,J=278.5Hz),121.02,118.42,61.72(q,J=34.0Hz); |
| MS m/z:227.01[M+1]。 |
| Example 3: Preparation of 6-chloro-3-(2,2,2-trifluoroacetamido)-2-(2,2,2-trifluoroethoxy)pyridine (IX-1) |
| At room temperature, dichloromethane (10.4 L) was added to the reaction kettle, stirring was started, 6-chloro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (2664 g, 11.76 mol) obtained in Example 2 was added, diisopropylethylamine (2279 g, 17.64 mol) was added, the temperature was controlled at -15 to -10°C, a dichloromethane (5.2 L) solution of trifluoroacetic anhydride (2963 g, 14.11 mol) was added dropwise, and stirring was continued for 20 minutes after the addition was completed. Water (13.0 L) was added dropwise, the layers were separated, the organic phase was concentrated under reduced pressure, and the next step reaction was theoretically calculated. |
| 1 H NMR(400MHz,DMSO-d6)δ11.23(s,7H),7.95(d,J8.0Hz,1H),7.34(d,J8.0Hz,1H),5.03(q,J8.9Hz,2H) |
| 13C NMR(101MHz,DMSO-d6)δ155.74(q,J=46.6Hz),155.60,145.37,140.24,124.01(q,J=278.8Hz),119.07,118.30,116.19(q,J=289.9Hz),62.99(q,J=35.4Hz); |
| MS m/z.322.99[M+1]。 |
| Example 4: Preparation of 6-chloro-5-nitro-3-(2,2,2-trifluoroacetamido)-2-(2,2,2-trifluoroethoxy)pyridine (VIII-1) |
| At room temperature, concentrated sulfuric acid (11.7 L) was added to the reaction kettle, stirring was started, 6-chloro-3-(2,2,2-trifluoroacetamido)-2-(2,2,2-trifluoroethoxy)pyridine (3.9 Kg, 11.76 mol) obtained in Example 3 was added, and potassium nitrate solid (1783.4 g, 17.64 mol) was added in batches. After the addition, stirring was continued for about 40 minutes. After monitoring the reaction, the temperature was lowered to control the internal temperature at 10-25°C, and dichloromethane (27.3 L) was added dropwise. Stirring was continued, stirring was continued for 45 minutes, and the layers were separated. The organic phase was taken and washed once with water (11.7 L). The organic phase was concentrated under reduced pressure and theoretically calculated for the next step reaction. |
| 1H NMR(500MHz,DMSO-d6)δ11.58(s,1H),8.78(s,1H),5.17(q,J=8.7Hz,2H); |
| 13C NMR(126MHz,DMSO-d6)δ155.89,155.43(q,J=37.8Hz),138.84,138.57,135.05,123.22(q,J=273.4Hz),118.47,115.51(q,J=278.5Hz),63.65(q,J=35.3Hz); |
| MS m/z:367.98[M+1]。 |
| Example 5: Preparation of 6-chloro-5-nitro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (VII-1) |
| At room temperature, methanol (13.0 L) was added to the reactor, 6-chloro-5-nitro-3-(2,2,2-trifluoroacetamido)-2-(2,2,2-trifluoroethoxy)pyridine (4322 g, 11.76 mol) obtained in Example 4 was added, p-toluenesulfonic acid monohydrate (3355 g, 17.64 mol) was added, the temperature was controlled at 60-65°C for 15 hours, and the methanol was removed under reduced pressure. Methyl tert-butyl ether (13.0 L) and water (6.5 L) were added, and the pH was adjusted to 7-8 with potassium carbonate. Layering was performed, the organic phase was washed once with water (8.6 L), separated, and concentrated under reduced pressure. n-heptane (21.5 L) was added, the temperature was controlled at 60-65°C and stirred for 1 hour, cooled to room temperature, filtered, and the filter cake was dried with air at 50°C for 18 hours to obtain 1475 g of the product. |
| The total yield of the five-step reaction from Example 1 to Example 5 is 34.9%. |
| 1H NMR(500 MHz,DMSO-d6)δ7.62(s,1H),5.92(s,2H),5.05(q,J=8.9Hz,2H). |
| 13C NMR(126MHz,DMSO-d6)δ149.30,139.53,132.84,123.46,123.44(q,J=278.5Hz),116.25,62.52(q,J=35.3Hz); |
| MS m/z:272.00[M+1]。 |
| Example 6: Preparation of 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (V-1) |
| Toluene (50 mL) was added to a 100 mL reaction bottle, and the compound of formula VII-1, 6-chloro-5-nitro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (5.0 g, 18.4 mmol), the compound of formula VI, 3-(2-chloropyrimidin-4-yl)-1-methyl-1H-indole (5.8 g, 23.8 mmol), p-toluenesulfonic acid monohydrate (1.8 g, 9.2 mmol) were added in sequence, and the reaction mixture was heated to 110-115°C and reacted for 24 hours. The temperature was lowered to 22°C, filtered by suction, and the filter cake was dried at 50°C for 20 hours to obtain the compound of formula V-1, 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (10.4 g, 74.7 HPLC area% purity). According to the HPLC purity conversion, the next step reaction was carried out. |
| 1H NMR(400MHz,DMSO-d6)δ9.43(s,1H),8.76(s,1H),8.46-8.45(d,J=5.4Hz,1H),8.39(s,1H),8.38-8.36(d,J=7.8Hz,1H),7.57-7.55(d,J=8.2Hz,1H),7.41-7.40(d,J=5.4Hz,1H),7.31-7.27(t,J=7.5Hz,1H),7.20-7.16(t,J=7.5Hz,1H),5.23-5.16(q,J=8.8Hz,2H),3.90(s,3H); |
| MS m/z:479.08[M+1]。 |
| Example 7: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (IV) |
| Add N,N-dimethylformamide (30 mL) to a 250 mL reaction bottle, add the compound of formula V-1 obtained in Example 6, 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (10.4 g, 16.22 mmol), stir, add potassium carbonate (4.48 g, 32.44 mol), N,N,N’-trimethylethylenediamine (2.48 g, 24.33 mol) in sequence, heat the reaction mixture to 77-82°C, keep warm for 1-1.5 hours. Add water (60 mL), and cool to room temperature after addition. Filter by suction, transfer the filter cake to a 50 L reactor, add acetonitrile (40 mL), and heat to reflux for 2 hours. The mixture was cooled to room temperature and filtered with suction. The filter cake was dried at 50°C for 18 hours to give a compound of formula IV, 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (6.7 g). The total yield of the two-step reaction with Example 6 was 66.8%. |
| 1H NMR(500MHz,DMSO-d6)δ8.62(s,1H),8.41(s,1H),8.26(s,2H),8.24(s,1H),7.48(d,J=8.2Hz,1H),7.21(t,J=7.6Hz,1H),7.16(d,J=5.3Hz,1H),7.05(t,J=7.3Hz,1H),5.04(q,J=8.9Hz,2H),3.84(s,3H),3.69(t,J=6.9Hz,2H),2.89(s,3H),2.55(t,J=6.9Hz,2H),2.17(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ162.15,160.55,156.99,154.98,148.42,137.53,132.83,132.68,125.50,123.58(q,J=279.7Hz),124.38,122.11,122.06,120.67,113.38,112.27,110.30,107.11,62.14(q,J=35.3Hz),56.10,49.51,45.34,45.33,39.35,32.98。 |
| MS m/z.:545.22[M+1]。 |
| Example 8: Preparation of 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine p-toluenesulfonate (V-1′) |
| Toluene (7.43 L) was added to a 20 L reactor, and compound VII-1 6-chloro-5-nitro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (743.0 g, 2.74 mol), compound VI 3-(2-chloropyrimidin-4-yl)-1-methyl-1H-indole (866.7 g, 3.56 mol), p-toluenesulfonic acid monohydrate (780.7 g, 4.10 mol) were added in sequence, stirred, and the reaction mixture was heated to 110-115°C and reacted for 36 hours. The temperature was controlled at 15-30°C, tetrahydrofuran (3.72 L) was added and stirred for 30 minutes. Filtered by suction, the filter cake was transferred to a 50 L reactor, tetrahydrofuran (4.46 L) was added, and heated to reflux for 3 hours. The temperature was lowered to 15-25°C, filtered, and the filter cake was dried at 50°C for 17 hours to obtain 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine p-toluenesulfonate (1719 g, 85.96 HPLC area% purity). The purity was calculated according to HPLC and used for the next step reaction. |
| Melting point: 216.0-218.3℃ |
| 1H NMR(500MHz,DMSO-d6)δ9.70(s,1H),9.21(s,1H),8.62(s,1H),8.40(d,J=6.2Hz,1H),8.24(d,J=7.8Hz,1H),7.59(d,J=8.3Hz,1H),7.50(d,J=6.5Hz,1H),7.49(d,J=8.3Hz,2H),7.32(t,J=7.6Hz,1H),7.18(t,J=7.5Hz,1H),7.12(d,J=7.9Hz,2H),5.17(q,J=8.8Hz,2H),3.91(s,3H),2.29(d,J=5.2Hz,3H); |
| 13C NMR(126MHz,DMSO-d6)δ166.66,157.35,155.72,147.40,140.87,139.90,139.72,138.59,135.83,130.09,129.99,129.98,129.97,127.39,127.38,127.37,127.15,125.22(q,J=278.5Hz),124.97,123.85,123.69,113.63,112.97,110.27,63.58(q,J=35.3Hz),35.57,22.81。 |
| Example 9: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (IV) |
| Add N,N-dimethylformamide (5.14L) to a 50L reactor, add 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine p-toluenesulfonate (1714.0g, 2.261mol) obtained in Example 8, stir, add potassium carbonate (624.7g, 4.52mol), N,N,N’-trimethylethylenediamine (346.2g, 3.39mol) in sequence, heat the reaction mixture to 77-82°C, keep warm for 1-1.5 hours. Add water (10.28L), and cool to room temperature after adding. Filter by suction, transfer the filter cake to a 50L reactor, add acetonitrile (6.86L), and heat to reflux for 2 hours. The temperature was lowered to 15-25°C, filtered with suction, and the filter cake was dried at 50°C for 18 hours to obtain the compound of formula IV, 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1142 g). The total yield of the two-step reaction with Example 8 was 76.54%. |
| 1H NMR(500MHz,DMSO-d6)δ8.62(s,1H),8.41(s,1H),8.26(s,2H),8.24(s,1H),7.48(d,J=8.2Hz,1H),7.21(t,J=7.6Hz,1H),7.16(d,J=5.3Hz,1H),7.05(t,J=7.3Hz,1H),5.04(q,J=8.9Hz,2H),3.84(s,3H),3.69(t,J=6.9Hz,2H),2.89(s,3H),2.55(t,J=6.9Hz,2H),2.17(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ162.15,160.55,156.99,154.98,148.42,137.53,132.83,132.68,125.50,123.58(q,J=279.7Hz),124.38,122.11,122.06,120.67,113.38,112.27,110.30,107.11,62.14(q,J=35.3Hz),56.10,49.51,45.34,45.33,39.35,32.98。 |
| MS m/z:545.22[M+1]。 |
| Example 10: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (IV) |
| Acetonitrile (10 mL) was added to a 50 L reactor, and 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine p-toluenesulfonate (1.0 g, 1.5 mmol) obtained in Example 8 was added, and stirred. Potassium carbonate (577 mg, 3 mmol) and N,N,N’-trimethylethylenediamine (320 mg, 2.25 mmol) were added in sequence. The reaction mixture was heated to 77-82°C and kept for 1-2 hours. Water (10 mL) was added and the temperature was cooled to room temperature after the addition. The product was filtered to give a compound of formula IV, 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (629 mg) with a purity of 95.94%. The total yield of the two-step reaction with Example 8 was 77%. |
| Example 11: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (III’) |
| Add the compound of formula IV 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.0 g, 7.34 mmol) to a 100 mL reaction bottle at room temperature, add tetrahydrofuran (27 mL) and water (13 mL), and stir for 10 to 20 minutes. Add hydrosulfite (9.6 g, 44.1 mmol) to the reactor in batches. After addition, continue stirring for 10 to 20 minutes. Control the temperature of the reactor to 30 to 35 ° C for reaction. The purity of the product compound of formula III’ was 64.68% after sampling the liquid phase after 2 hours of reaction. The reaction was continued until 17 hours after the reaction. 40 mL of water was added to the reaction solution, and the layers were separated by standing. The tetrahydrofuran phase was taken, and the aqueous phase was extracted twice with 100 mL of dichloromethane. The organic phases were combined, washed with saturated brine, separated by standing, and concentrated under reduced pressure to obtain 3.2 g of solid with a purity of 62.32%. |
| 1H NMR(500MHz,DMSO)δ10.67(s,1H),10.36(s,1H),8.82(s,1H),8.18(s,1H),8.01(s,1H),7.59(d,J=8.2Hz,1H),7.45(d,J=6.8Hz,1H),7.32(t,J=7.5Hz,1H),7.24(s,1H),4.97(q,J=8.7Hz,2H),3.93(s,3H),3.75(s,2H),3.41(s,2H),3.10(s,3H),2.78(s,6H); |
| MS m/z:515.24[M+1]。 |
| Example 12: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (III’) |
| In a 100mL single-mouth bottle, there is 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (2.0g, 3.67mmol), palladium carbon (200mg), ethanol (20mL), hydrogen balloon replacement twice, hydrogen gas, magnetic stirring, room temperature overnight (17 hours). After the reaction is completed, suction filtration, the filtrate is taken, and it is concentrated to dryness under reduced pressure to obtain 2.1g of product with a purity of 56.93%. |
| Example 13: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (III’) |
| At room temperature, add the compound of formula IV 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1317.0 g, 2.42 mol) to a 50 L reactor, add tetrahydrofuran (8.8 L) and water (4.3 L), and stir for 10 to 20 minutes. Add hydrosulfite (2970.0 g, 14.52 mol) to the reactor in batches. After adding, continue stirring for 10 to 20 minutes. Control the temperature of the reactor to 40-45 ° C and react for 2 hours. Add concentrated hydrochloric acid (5882.2 g, 58.08 mol) to the reactor. After the addition is complete, heat to 42 to 47 ° C and react for 15 hours. Add 30% sodium hydroxide (2323.2g, 58.08mol) aqueous solution dropwise, and then add solid sodium bicarbonate (1219.7g, 14.52mol) in batches to adjust the pH value to 6-8. After stirring for 20 minutes, filter with suction, let the filtrate stand and separate. The organic phase is concentrated under reduced pressure to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine, with a purity of 97.1%. Calculated based on the theoretical yield of 100%, it is directly used in the next step reaction. |
| 1H NMR(500MHz,DMSO)δ10.67(s,1H),10.36(s,1H),8.82(s,1H),8.18(s,1H),8.01(s,1H),7.59(d,J=8.2Hz,1H),7.45(d,J=6.8Hz,1H),7.32(t,J=7.5Hz,1H),7.24(s,1H),4.97(q,J=8.7Hz,2H),3.93(s,3H),3.75(s,2H),3.41(s,2H),3.10(s,3H),2.78(s,6H); |
| MS m/z:515.24[M+1]。 |
| Example 14: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (III-1) |
| To the 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine obtained in Example 13, THF (5.3 L) and ethanol (4.0 L) were added, the temperature was raised to 50-70°C, and concentrated hydrochloric acid (617.8 g, 6.1 mol) was added dropwise. After the addition was completed, the mixture was cooled to room temperature and stirred for 12 hours. Filtered by suction, the filter cake was dried by air at 50°C to obtain 1507.4 g of a crude product. Methanol (6.0 L) and ethanol (4.5 L) were added to a 20 L reaction bottle, and the above crude product was added, the temperature was raised to 55-60 ° C, hot slurry was added for 1-2 hours, the temperature was lowered to room temperature, and suction was filtered to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (1335.6 g), the liquid phase purity was 99.80%, and the total yield of the two-step reaction with Example 13 was 94.0%. Melting point: 236.6-240.8 ° C. |
| 1H NMR(500MHz,DMSO-d6)δ10.67(s,1H),10.36(s,1H),8.82(s,1H),8.18(s,1H),8.01(s,1H),7.59(d,J=8.2Hz,1H),7.45(d,J=6.8Hz,1H),7.32(t,J=7.5Hz,1H),7.24(s,1H),4.97(q,J=8.7Hz,1H),3.93(s,3H),3.75(s,2H),3.41(s,2H),3.10(s,3H),2.78(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ166.81,153.27,152.17,150.76,138.61,138.16,138.15,125.46,124.94,123.83(q.J=278.5Hz),123.42,123.41,122.60,122.59,120.52,111.34,111.17,106.29,62.14(q,J=35.3Hz),53.53,46.28,42.27,42.26,40.92,33.67。 |
| Example 15: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (III-1) |
| At room temperature, add the compound of formula IV 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1136.0 g, 2.09 mol) to a 50 L reactor, add acetonitrile (7.95 L) and water (7.95 L), and stir for 10 to 20 minutes. Add hydrosulfite (2563.9 g, 12.50 mol) to the reactor in batches. After adding, continue stirring for 10 to 20 minutes. Control the temperature of the reactor to 35 to 40 ° C and react for 3 hours. Add concentrated hydrochloric acid (2505.3 g, 25.08 mol) to the reactor. After the addition is complete, heat to 35 to 45 ° C and react for 18 hours. 30% sodium hydroxide (1003.2 g, 25.08 mol) aqueous solution was added dropwise to adjust the pH value to 6-8. Solid sodium bicarbonate (1053.5 g, 12.54 mol) was added to adjust the pH value to 7-8. After stirring for 40 minutes, the mixture was filtered, the filtrate was allowed to stand, the layers were separated, and the organic phase was concentrated under reduced pressure. The purity of the liquid phase was detected to be 97.60%. |
| Add ethanol (5.68 L) to the product of the previous step, raise the temperature to 50-70°C, and drop concentrated hydrochloric acid (522 g, 5.23 mol). After the dropwise addition is completed, cool to room temperature and stir for 15 hours. Filter by suction, and air dry the filter cake at 50°C to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (780 g), with a liquid phase purity of 98.74%. |
| Example 16: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride (II-1) |
| 2-[2-(Dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (1543.5 g, 2.63 mol) was added to a 50 L reactor, and dichloromethane (13.1 L) and triethylamine (532.2 g, 5.26 mol) were added. The mixture was stirred and cooled to -10 to -5 °C, and a solution of 3-chloropropionyl chloride (501.5 g, 3.95 mol) in dichloromethane (10.0 L) was added dropwise. After the addition is completed, keep warm and stir for 10 to 20 minutes, filter with suction, and the filter cake is formula II-12-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamide)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride wet product (2683.5g), which is calculated based on the theoretical yield of 100% and is directly used in the next reaction. |
| Melting point: 233.2-238.7℃ |
| 1H NMR(500MHz,DMSO-d6)δ10.18(s,1H),8.57(s,1H),8.42(s,1H),8.27(t,J=6.6Hz,2H),8.17(s,1H),7.51(d,J=8.1Hz,1H),7.26-7.22(m,1H),7.22-7.17(m,2H),4.99(q,J=9.1Hz,2H),3.91(d,J=6.3Hz,2H),3.89(s,3H),3.55(s,2H),3.13(s,2H),3.02(t,J=6.1Hz,2H),2.85(s,3H),2.64(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ168.41,161.88,160.22,157.34,148.05,146.73,137.62,133.25,130.86,125.43,124.09(q,J=279.2Hz),122.04,121.74,120.88,118.51,116.60,112.33,110.40,107.09,61.65(q,J=35.3Hz),54.90,40.96,40.95,40.60,38.71,32.96,32.95,32.94。 |
| Example 17: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I, crude product) |
| The wet product (2683.5 g) of Formula II 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamide)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride obtained in Example 16 was added to a 20L reactor, and acetonitrile (16.8L) and triethylamine (1329.3g, 13.15mol) were added, stirred, and heated to reflux for 4 hours. Cooled to room temperature, purified water (4.20L) was added, stirred at room temperature for 3-4 hours, and filtered. The filter cake was transferred to a 50L reactor, dichloromethane (17L) was added, and the pH value was adjusted to 7-8 with saturated sodium bicarbonate aqueous solution (17L). Liquid separation, the organic phase was transferred to a 20L reactor, activated carbon (84.3g) was added, refluxed for 1 hour, cooled to 20-30°C, and filtered. The filtrate was concentrated to dryness under reduced pressure to obtain the compound of formula I 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1390g), with a total yield of 92.9% and a purity of 99.21% for the two-step reaction with Example 16. |
| 1H NMR(500MHz,DMSO-d6)δ9.96(s,1H),8.71(s,1H),8.44(s,1H),8.29(d,J=5.3Hz,1H),8.26(d,J=7.7Hz,1H),8.13(s,1H),7.51(d,J=8.2Hz,1H),7.24(t,J=7.2Hz,1H),7.20(d,J=5.3Hz,1H),7.15(t,J=7.2Hz,1H),6.51(dd,J=17.0,10.2Hz,1H),6.28(dd,J=17.0,1.8Hz,1H),5.78(dd,J=10.2,1.8Hz,1H),5.00(q,J=9.1Hz,2H),3.89(s,3H),3.18(t,J=6.5Hz,2H),2.87(s,3H),2.48(t,J=6.5Hz,2H),2.22(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ163.40,161.84,160.26,157.35,148.07,147.15,137.60,133.23,131.61,130.07,126.67,125.41,124.03(q,J=278.5Hz),122.00,121.68,120.80,118.39,116.13,112.36,110.37,107.02,61.29(q,J=35.3Hz),56.57,52.44,45.60,45.59,38.54,32.93; |
| MS m/z:569.25[M+1]。 |
| Example 18: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (III’) |
| At room temperature, add the compound of formula IV 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (20.0 g, 36.73 mmol) to a 1L reaction bottle, add tetrahydrofuran (134 mL) and water (66 mL), and stir for 10 to 20 minutes. Add hydrosulfite (47.9 g, 220.38 mmol) to the reaction bottle in batches. After addition, continue stirring for 10 to 20 minutes. Control the internal temperature to 35-40°C and react for 3 hours. Add concentrated hydrochloric acid (89.3 g, 881.52 mmol) to the reaction bottle. After the addition is complete, heat to 42 to 47°C and react for 17 hours. 30% sodium hydroxide (35.26 g, 881.52 mmol) aqueous solution was added dropwise, and solid sodium bicarbonate (18.5 g, 220.38 mmol) was added in batches to adjust the pH value to 6-8. After stirring for 30 minutes, the mixture was filtered, and the filtrate was allowed to stand and separated. The organic phase was concentrated to dryness under reduced pressure to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (19.2 g) with a purity of 95.8% and a yield of 97.12%. |
| Example 19: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride (II-1) |
| Add 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5 g, 9.72 mmol) to a 250 mL reaction bottle, add dichloromethane (42 mL), stir, protect with argon, cool to -5 to 0°C, and add 3-chloropropionyl chloride (1.851 g) and dichloromethane (33 mL) dropwise. After the addition is complete, the mixture is stirred for 10-20 minutes at a temperature maintained at room temperature. After the reaction is complete, the mixture is concentrated under reduced pressure to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride (7.0 g) with a purity of 85.67%. Melting point: 233.5-238.9°C. |
| 1H NMR(500MHz,DMSO-d6)δ10.18(s,1H),8.57(s,1H),8.42(s,1H),8.27(t,J=6.6Hz,2H),8.17(s,1H),7.51(d,J=8.1Hz,1H),7.26-7.22(m,1H),7.22-7.17(m,2H),4.99(q,J=9.1Hz,2H),3.91(d,J=6.3Hz,2H),3.89(s,3H),3.55(s,2H),3.13(s,2H),3.02(t,J=6.1Hz,2H),2.85(s,3H),2.64(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ168.41,161.88,160.22,157.34,148.05,146.73,137.62,133.25,130.86,125.43,124.09(q,J=279.2Hz),122.04,121.74,120.88,118.51,116.60,112.33,110.40,107.09,61.65(q,J=35.3Hz),54.90,40.96,40.95,40.60,38.71,32.96,32.95,32.94。 |
| Example 20: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I, crude product) |
| The 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride obtained in Example 19 was added to a 250 mL reaction bottle, and acetonitrile (45 mL) and triethylamine (4.9 g) were added. The mixture was stirred magnetically and protected by argon. The temperature was raised to reflux in an oil bath. The reaction was allowed to react for 6 h. Water (23 mL) was added dropwise, and the mixture was naturally cooled to room temperature in an oil bath. The mixture was filtered with suction, and the filter cake was transferred to a 500 mL reaction bottle. Dichloromethane (100 mL) was added, and the pH value was adjusted to 7-8 with saturated aqueous sodium bicarbonate solution (100 mL). The liquids were separated and the organic phase was concentrated under reduced pressure. The solid was dried in an oven at 50°C to give 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamide)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.1 g) with a purity of 97.7%. The total yield of the two-step reaction with Example 19 was 74.17%. |
| Comparative Example 1: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I, crude product) |
| 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1 g, 1.94 mmol) was added to a 50 mL multi-necked flask, tetrahydrofuran (10 mL) was used as the solvent, argon was replaced three times, and stirring was maintained at 0-5°C under argon protection, and 3-chloropropionyl chloride (0.37 g, 2.92 mmol), the addition was completed in 15 minutes, and the mixture was stirred at 0-5°C for 1 hour. Sodium hydroxide (0.31 g, 7.77 mmol) and water (1 mL) were added to the reaction solution, and the temperature was raised to 65°C and stirred for 15 hours. Saturated ammonium chloride solution (10 mL) was added, and the liquids were separated. The organic phase was washed with saturated sodium bicarbonate solution (10 mL). The liquids were separated and the organic phase was concentrated to dryness to obtain 1.04 g of a yellow solid with a yield of 94.9% and a purity of 87.35%. |
| Comparative Example 2: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I, crude product) |
| Add 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0 g) to a 250 mL reaction bottle, add acetone (50 mL) and potassium carbonate (940 mg), stir, protect with argon, cool to -50°C, and add 3-chloropropionyl chloride (1.481 g) dropwise. After the addition is completed, the temperature is raised to -20°C and stirred for 30 minutes. A solution of sodium hydroxide (350 mg) and water (60 ml) is added dropwise over 10 minutes. The mixture is stirred at room temperature for 3 to 4 hours. The mixture is filtered and the filter cake is dried in an oven at 50°C to obtain a compound of formula II-1′, 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamide)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.28 g) with a purity of 64.18%. |
| 1H NMR(400MHz,DMSO-d 6 )δ10.32(s,1H),10.21(s,1H),8.54(s,1H),8.43(s,1H)8.29-8.28(d,J=5.1Hz,1H),8.28-8.26(d,J=6.2Hz,1H),8.19(s,1H),7.54-7.52(d,J=8.0Hz,1H),7.27-7.18(m,3H),5.77(s,2H),5.00(q,J=9.1Hz,1H),3.92(t,J=6.2Hz,1H),3.63(t,J=5.7Hz,2H),3.28(t,J=5.7Hz,2H),3.06-3.03(t,J=6.2Hz,2H),2.85(s,3H),2.74(s,6H). |
| MS m/z:605.23[M+1]。 |
| Add 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.28 g) to a 250 mL reaction bottle, add acetonitrile (45 ml) and triethylamine (3.606 g), stir magnetically, protect with argon, heat in an oil bath to reflux, and react for 6 h. Water (23 ml) was added dropwise, the temperature was naturally lowered in an oil bath and stirring was continued overnight (16 h), filtered with suction, and the solid was dried to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.3 g) with a purity of 95.13% and a two-step yield of 59.42%. |
| Example 21: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| The crude product (1390 g) of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine was transferred to a 50L reactor, acetone (25.0L) was added, argon was replaced 3 times, the temperature was raised to 45-50°C, all the solids were dissolved, and purified water (6.95L) was added dropwise. After the addition was completed, the mixture was cooled to 20-25°C and stirred for 2 hours. The mixture was filtered and the filter cake was vacuum dried at 50°C for 24 hours to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (895g). The reaction yield is 66.7% and the purity is 99.89%. |
| Example 22: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (5.0g), add ethyl acetate 100mL, heat to 70-75°C in an oil bath to dissolve all the solids, then cool naturally to 25°C in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.1g) with a purity of 99.73% and a yield of 62.0%. |
| Example 23: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add ethyl acetate 100mL, heat to 70-75°C in an oil bath, dissolve all the solids, continue to stir for 30min, and drop 150mL of n-heptane. After the drop is complete, cool to 25°C in an oil bath, filter by suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.0g), with a purity of 99.32% and a yield of 80%. |
| Example 24: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add acetonitrile 75mL, heat in an oil bath to 77-82°C, dissolve all the solids, and drop 25mL of water. After dripping, naturally cool to 25°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.3g), with a purity of 99.64% and a yield of 86%. |
| Example 25: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add acetonitrile 75mL, heat in an oil bath to 77-82°C, dissolve all the solids, and continue to stir for 30min. Cool naturally to 25°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.0g), with a purity of 99.45% and a yield of 80%. |
| Example 26: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0 g), add 20mL of tetrahydrofuran, heat in an oil bath to 45-50°C to dissolve all the solids, continue to stir and maintain the temperature for 30 minutes, and add 40mL of n-heptane dropwise. After the addition was completed, the mixture was naturally cooled to 25°C in an oil bath, filtered and dried to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.23 g) with a purity of 99.51% and a yield of 84.6%. |
| Example 27: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add 100mL of isopropanol, heat to 50°C in an oil bath, dissolve all the solids, and continue to stir for 30 minutes. Cool naturally to 22°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.25g), with a purity of 99.51% and a yield of 85%. |
| Example 28: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add 75mL of methanol, and heat to 55-60°C in an oil bath to dissolve all the solids. Cool naturally to 17°C in the oil bath, stir overnight, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.55g), with a purity of 99.63% and a yield of 71%. |
| Example 29: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0 g), add 50mL of dichloromethane, heat in an oil bath to 40°C to dissolve all the solids, continue to stir and maintain the temperature for 30 minutes, and add 100mL of n-heptane dropwise. The mixture was naturally cooled to 15°C in an oil bath, stirred overnight, filtered and dried to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.78 g) with a purity of 99.56% and a yield of 75.6%. |
| Example 30: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add 100mL of toluene, heat to 65°C in an oil bath, dissolve all the solids, and continue to stir for 30 minutes. Cool naturally to 20°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.27g), with a purity of 99.57% and a yield of 65.4%. |
| Example 31: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (5.0g), add DMF50mL, heat to 80°C in an oil bath, dissolve all the solids, continue to stir for 30min, and drop 25mL of water. Naturally cool to 20°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.84g), with a purity of 99.77% and a yield of 76.8%. |
| Example 32: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 25 mL single-necked bottle, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (1.0 g), add tetrahydrofuran (6 mL), protect with argon, heat in an oil bath to 40-45°C until all the solution is dissolved, continue to stir and keep warm for 30 min, cool naturally to 22°C in an oil bath, filter and obtain a solid. The solid was transferred to a crystallization dish and dried to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (622 mg) with a purity of 99.83% and a yield of 62.2%. |
| Example 33: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 50 mL single-mouth bottle, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (1.0 g), add acetone (15 mL), protect with argon, heat in an oil bath to 45-50° C. until all the solution is dissolved, and then continue to stir and keep warm for 30 min, cool naturally to 22° C. in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (537 mg) with a purity of 99.83% and a yield of 53.7%. |
| Example 34: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 50 mL single-mouth bottle, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (1.0 g), add tetrahydrofuran (8 mL), protect with argon, heat in an oil bath to 40-45° C. until all the solution is dissolved, and continue to stir and keep warm for 30 min. Add water (16 mL) dropwise, cool naturally to 21° C. in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (880 mg) with a purity of 99.68% and a yield of 88.0%. |
| Example 35: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 100 mL single-mouth bottle, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1.0 g), add ethanol (35 mL), protect with argon, heat in an oil bath to 75-80° C. until all the solution is dissolved, add water (10 mL) dropwise over 10 min, cool naturally to 20° C. in an oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (915 mg), with a yield of 91.5% and a purity of 99.49%. |
| Example 36: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 50 mL single-mouth bottle, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (1.0 g), add xylene (20 mL), protect with argon, heat in an oil bath to 80° C. until all the solution is dissolved, cool naturally to 20° C. in an oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (798 mg), with a yield of 79.8% and a purity of 99.48%. |
| Example 37: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (5.0g), add ethanol 125mL, heat in an oil bath to 75-80°C to dissolve all the solids, continue to stir and keep warm for 30min, then cool naturally to 25°C in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.7g) with a purity of 99.66% and a yield of 74%. |
| Example 38: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 100 mL single-mouth bottle, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1.0 g), add methanol (35 mL), protect with argon, heat in an oil bath to 80° C. until all the solution is dissolved, add water (10 mL) dropwise over 10 min, cool naturally to 20° C. in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (912 mg), with a yield of 91.2% and a purity of 99.53%. |
PATENT
CN110606842
WO2019238103
PAPER
https://www.nature.com/articles/s41401-020-0389-3
| NCT Number | Sponsor | Condition | Start Date | Phase |
|---|---|---|---|---|
| NCT02973763 | Allist Pharmaceuticals, Inc. | NSCLC | December 30, 2016 | Phase 1 |
| NCT03787992 | Allist Pharmaceuticals, Inc. | Locally Advanced or Metastatic EGFR Sensitising Mutation Positive Non-small Cell Lung Cancer | May 30, 2019 | Phase 3 |
| NCT03452592 | Allist Pharmaceuticals, Inc. | Advanced NSCLC Patients With T790M | April 30, 2018 | Phase 2 |
- [1]. Y. Shi, et al. P2.03-028 Third Generation EGFR Inhibitor AST2818 (Alflutinib) in NSCLC Patients with EGFR T790M Mutation: A phase1/2 Multi-Center Clinical Trial.[2]. Alexander I. Spira, et al. FURVENT: Phase 3 trial of furmonertinib vs chemotherapy as first-line treatment for advanced NSCLC with EGFR exon 20 insertion mutations (FURMO-004). Journal of Clinical Oncology. Volume 42, Number 16_suppl.
/////////FIRMOMERTINIB, Furmonertinib, Alflutinib, AST 2818, UNII-A49A7A5YN4, PHASE 2, CANCER
Ervogastat


Ervogastat
CAS 2186700-33-2
Non-alcoholic Steatohepatitis (NASH) with Liver Fibrosis (FAST TRACK – U.S.)
- 2-[5-[(3-Ethoxy-2-pyridinyl)oxy]-3-pyridinyl]-N-[(3S)-tetrahydro-3-furanyl]-5-pyrimidinecarboxamide
- (S)-2-(5-((3-Ethoxypyridin-2-yl]oxy]pyridin-3-yl)-N-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide
- PF 06865571
- BSOIY5AKQW
407.4 g/mol, C21H21N5O4
2-[5-(3-ethoxypyridin-2-yl)oxypyridin-3-yl]-N-[(3S)-oxolan-3-yl]pyrimidine-5-carboxamide
- OriginatorPfizer
- ClassAmides; Ethers; Furans; Hepatoprotectants; Pyridines; Pyrimidines; Small molecules
- Mechanism of ActionDiacylglycerol O-acyltransferase inhibitors
Phase IINon-alcoholic fatty liver disease; Non-alcoholic steatohepatitis
- 08 Jan 2025Chemical structure information added.
- 21 Feb 2024Pfizer completes a phase II trial in Non-alcoholic steatohepatitis (Combination therapy) in Slovakia, Japan, Bulgaria, Canada, China, Hong Kong, India, Poland, Puerto Rico, South Korea, Taiwan (PO) (NCT04321031) (EudraCT2019-004775-39)
- 21 Feb 2024Pfizer completes a phase II trial in Non-alcoholic steatohepatitis (Monotherapy) in Slovakia, Japan, Bulgaria, Canada, China, Hong Kong, India, Poland, Puerto Rico, South Korea, Taiwan (PO) (NCT04321031) (EudraCT2019-004775-39)
Ervogastat is an experimental small-molecule drug and selective diacylglycerol O-acyltransferase 2 inhibitor developed by Pfizer for non-alcoholic steatohepatitis.[1] Its development was previously halted by the company but resumed in 2022.[2]
Scheme
SIDE CHAIN

MAIN

https://doi.org/10.1021/acs.jmedchem.2c01200

SYN
WO2023026180
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023026180&_cid=P11-MB5XF4-40032-1
Preparation of Intermediates and Examples
Preparation of Intermediate 1 and Example 1 (Forms 1 and 2) were described in WO2018/033832 and are reproduced below.
Intermediate 1 : 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylic acid
Step 1 : 3-Ethoxypyridine
Cesium carbonate (12 mol, 1.5 equiv) and ethyl iodide (9.7 mol, 1.2 equiv) were added to a solution of 3-hydroxypyrdine (8.10 mol, 1.0 equiv) in acetone (12 L) at 15 °C. The reaction mixture was stirred at room temperature for 24 hours. The reaction mixture was filtered and the organic layer was concentrated to give crude product. Ethyl acetate (20 L) was added and washed with water (3×5 L). The organic layer was dried over sodium sulfate, filtered and concentrated to give 3-ethoxypyridine (620 g, 62%) as an oil. 1H NMR (400 MHz, CDCh) 5 1.44 (t, 3H), 4.07 (q, 2H), 7.15-7.23 (m, 2H), 8.20 (dd, 1 H), 8.30 (d, 1 H).
Step 2: 3-Ethoxypyridine-1 -oxide
m-Chloroperoxybenzoic acid (6.5 mol, 1.3 equiv) was added to a solution of 3-ethoxypyridine (5.0 mol, 1.0 equiv) in dichloromethane (12 L) at 10 °C. The reaction mixture was stirred at room temperature for 24 hours. Sodium thiosulfate (4 kg, in 5 L of water) was added. The reaction mixture was stirred at 15 °C for 2 hours. Another portion of sodium thiosulfate (1.5 kg, in 5 L of water) was added. The reaction mixture was stirred at 15 °C for 1 hour. The mixture was extracted with dichloromethane (16×10 L). The combined organic layers were concentrated to give crude product. The crude product was purified by silica gel column chromatography (dichloromethane:methanol; 100:1-10:1) to give the title compound (680 g, 97%) as brown oil. This was further purified by trituration with petroleum ether (4 L) at room temperature for 24 hours to give 3-ethoxypyridine-1 -oxide (580 g, 83%) as yellow solid. 1H NMR (400 MHz, CDCh) 5 1.41 (t, 3H), 4.02 (q, 2H), 6.84 (dd, 1 H), 7.12 (dd, 1 H), 7.85 (d, 1 H), 7.91-7.95 (m, 1 H).
Step 3: 2-((5-Bromopyridin-3-yl)oxy)-3-ethoxypyridine
This reaction was carried out in five parallel batches.
Diisopropylethylamine (2.69 mol, 3.7 equiv) and bromotripyrrolidinophosphonium hexafluorophosphate (0.93 mol, 1.3 equiv) were added to a stirred solution of 3-ethoxypyridine-1-oxide (0.72 mol, 1.0 equiv) and 3-bromo-5-hydroxypyridine (0.72 mol, 1.0 equiv) in tetrahydrofuran (2500 mL) at room temperature. The reaction mixture was stirred at room temperature for 2 days then the separate batches were combined to a single batch. The resulting suspension was concentrated to dryness and dissolved in dichloromethane (25 L). The organic layer was washed with 1 N sodium hydroxide (15 L), water (3×20 L), and brine (20 L). The organic layer was dried over sodium sulfate, filtered and concentrated to give an oil. The crude oil was purified by silica gel column chromatography (petroleum ether : ethyl acetate; 10:1-1 :1) to give crude product as brown solid. This solid was triturated with methyl tert-butyl ether: petroleum ether (1 :10; 11 L) to afford 2-((5-bromopyridin-3-yl)oxy)-3-ethoxypyridine (730 g, 69%) as off yellow solid. 1H NMR (400 MHz, CDCh) 5 1.49 (t, 3H), 4.16 (q, 2H), 7.04 (dd, 1 H), 7.25 (dd, 1 H), 7.68-7.73 (m, 2H), 8.44 (d, 1 H), 8.49 (d, 1 H). MS (ES+) 297.1 (M+H).
Step 4: Ethyl 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylate
A solution of 2-((5-bromopyridin-3-yl)oxy)-3-ethoxypyridine (300 mmol, 1.0 equiv) in tetrahydrofuran (1.3 L) was degassed with nitrogen for 30 minutes. Turbo Grignard
(390 mmol, 1.3 equiv, 1.3 M in tetrahydrofuran) was added at room temperature at a rate to maintain the internal temperature below 30 °C. The reaction mixture was allowed to cool to room temperature and stirred for 3 hours. The reaction was cooled to 10 °C and zinc chloride (390 mmol, 1.3 equiv, 1.9 M in 2-methyltetrahydrofuran) was added at a rate to maintain the temperature below 15 °C. The resulting suspension was warmed to room temperature until all the precipitate was dissolved and then cooled back to 10 °C. Ethyl 2-chloropyrimidine-5-carboxylate (360 mmol, 1.2 equiv) and dichloro[bis(2-(diphenylphosphino)phenyl)ether]palladium(ll) (6.00 mmol, 0.02 equiv) were added as solids. The resulting suspension was degassed with nitrogen for 30 minutes then heated to 50 °C for 16 hours. The reaction was worked up under aqueous conditions then treated sequentially with ethylenediaminetetraacetic acid disodium salt, thiosilica, and charcoal to remove metal impurities. The crude compound was recrystallized from methanol (450 mL) to yield ethyl 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylate (77 g, 70%) as a pale, yellow solid. 1H NMR (400 MHz, CDCI3) 5 1.44 (t, 3H), 1.50 (t, 3H), 4.19 (q, 2H), 4.46 (q, 2H), 7.00-7.04 (m, 1 H), 7.25 (s, 1 H), 7.71 (d, 1 H), 8.59 (s, 1 H), 8.66 (d, 1 H), 9.32 (s, 2H), 9.55 (s, 1 H).
Step 5: 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylic acid
Sodium hydroxide (307 mmol, 1.5 equiv, 4M aqueous) and methanol (50 mL) were added to a suspension of 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylate (205 mmol, 1.0 equiv) in tetrahydrofuran (300 mL). The resulting solution was stirred at room temperature for 3 hours. The reaction mixture was diluted with water (400 mL) and extracted with 2:1 diethyl ether: heptanes (2x 300 mL). The aqueous layer was acidified to pH of 4 with 4M hydrochloric acid. The resulting suspension was stirred at room temperature for 1 hour. The solid was filtered, washed with water, and dried to yield 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylic acid (69 g, 100%) as a pale, yellow solid. 1H NMR (400 MHz, DMSO-de) 51.37 (t, 3H), 4.18 (q, 2H), 7.19 (dd, 1 H), 7.58 (dd, 1 H), 7.70 (dd, 1 H), 8.35-8.40 (m, 1 H), 8.66 (d, 1 H), 9.33 (s, 2H), 9.41 (d, 1 H), 13.9 (br. s, 1 H).
Example 1 : (S)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-/V-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide
Preparation of Form 1 of (S)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-/\/- (tetrahydrofuran-3-yl)pyrimidine-5-carboxamide
Oxalyl chloride (13.8 mL, 160 mmol, 1.2 equiv) and dimethylformamide (0.510 mL, 6.65 mmol, 0.05 equiv) were added to a suspension of 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylic acid (45.0 g, 133 mmol, 1.0 equiv) in dichloromethane (500 mL). The suspension was stirred for 2 hours when a solution was achieved. The reaction mixture was concentrated to yield crude acid chloride as a red solid. A solution of (S)-tetrahydrofuran-3-amine (12.2 g, 140 mmol, 1.05 equiv) and diisopropylethylamine (51.0 mL, 293 mmol, 2.2 equiv) in tetrahydrofuran (100 mL) was added dropwise to a solution of the crude acid chloride in dichloromethane (200 mL) at 0 °C. The reaction was allowed to warm to room temperature and stirred for 16 hours. Water (1.0 L) and ethyl acetate (600 mL) were added and the organic layer was separated, washed with saturated sodium bicarbonate, dried over magnesium sulfate, and filtered. The filtrate was treated with activated charcoal (20 g) was stirred at 65 °C for 20 minutes. The suspension was filtered warm and filtrate was concentrated to a pale, yellow solid which was recrystallized from methanol in ethyl acetate (1 :4, 1 L) to yield (S)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-/\/-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide (43.5 g, 81%) as a colorless solid. The title compound was combined with previous batches (108.7 g, 266.8 mmol) prepared in the same manner and slurried with ethyl acetate (1 .0 L) at 80 °C for 4 hours. The suspension was allowed to cool to room temperature and stirred for 4 days. The solid was filtered, washed with ethyl acetate (3×200 mL) and dried under high vacuum at 50 °C for 24 hours to yield (S)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-/\/-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide (100.5 g, 92%) as a colorless solid. 1H NMR (300 MHz, DMSO-de) 5 1.38 (t, 3H), 1.89-1.98 (m, 1 H), 2.15-2.26 (m, 1 H), 3.65 (dd, 1 H), 3.70-3.78 (m, 1 H), 3.85-3.92 (m, 2H), 4.18 (q, 2H), 4.46-4.55 (m, 1 H), 7.18 (dd, 1 H), 7.58 (dd, 1 H), 7.69 (dd, 1 H), 8.37 (dd, 1 H), 8.64 (d, 1 H), 8.95 (d, 1 H), 9.28 (s, 2H), 9.39 (d, 1 H). MS (ES+) 408.4 (M+H). Melting point 177.5 °C. Elemental analysis for C21H21N5O4: calculated C, 61.91 ; H, 5.20; N, 17.19; found C, 61.86; H, 5.18; N, 17.30.
PATENT
WO2020234726
WO2020044266
WO2018033832
WO2021171164
WO2016036636 EG 1
References
- ^ Futatsugi, Kentaro; Cabral, Shawn; Kung, Daniel W.; Huard, Kim; Lee, Esther; Boehm, Markus; Bauman, Jonathan; Clark, Ronald W.; Coffey, Steven B.; Crowley, Collin; Dechert-Schmitt, Anne-Marie; Dowling, Matthew S.; Dullea, Robert; Gosset, James R.; Kalgutkar, Amit S.; Kou, Kou; Li, Qifang; Lian, Yajing; Loria, Paula M.; Londregan, Allyn T.; Niosi, Mark; Orozco, Christine; Pettersen, John C.; Pfefferkorn, Jeffrey A.; Polivkova, Jana; Ross, Trenton T.; Sharma, Raman; Stock, Ingrid A.; Tesz, Gregory; Wisniewska, Hanna; Goodwin, Bryan; Price, David A. (24 November 2022). “Discovery of Ervogastat (PF-06865571): A Potent and Selective Inhibitor of Diacylglycerol Acyltransferase 2 for the Treatment of Non-alcoholic Steatohepatitis”. Journal of Medicinal Chemistry. 65 (22): 15000–15013. doi:10.1021/acs.jmedchem.2c01200. PMID 36322383. S2CID 253257260.
- ^ “With the right partner, Pfizer gains fast-track tag for previously shelved NASH drug”. Retrieved 20 November 2023.
| Clinical data | |
|---|---|
| Other names | PF-06865571 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2186700-33-2 |
| PubChem CID | 134262752 |
| ChemSpider | 114929473 |
| UNII | BSOIY5AKQW |
| ChEMBL | ChEMBL4760665 |
| Chemical and physical data | |
| Formula | C21H21N5O4 |
| Molar mass | 407.430 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////////Ervogastat, PF 06865571, fast track, BSOIY5AKQW, PFIZER, PHASE 2,
Eclitasertib
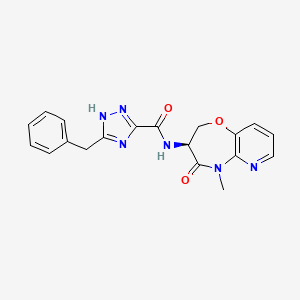
Eclitasertib
CAS 2125450-76-0
5-benzyl-N-[(3S)-5-methyl-4-oxo-2,3-dihydropyrido[3,2-b][1,4]oxazepin-3-yl]-1H-1,2,4-triazole-3-carboxamide
- DNL-758
- SAR-443122
- Eclitasertib (DNL-758) is a potent receptor-interacting protein kinase 1 (RIPK1) inhibitor with an IC50 of 0.0375 µΜ.
- UNII-975AT1P9J6
| Molecular Weight | 378.38 |
|---|---|
| Formula | C19H18N6O3 |
- OriginatorHarvard University
- DeveloperDenali Therapeutics Inc; Sanofi
- Class2 ring heterocyclic compounds; Amides; Anti-inflammatories; Antipsoriatics; Antirheumatics; Oxazepines; Pyridines; Skin disorder therapies; Small molecules; Triazoles
- Mechanism of ActionRIPK1 protein inhibitors
- Phase IIUlcerative colitis
- DiscontinuedCutaneous lupus erythematosus; Psoriasis; Rheumatoid arthritis; SARS-CoV-2 acute respiratory disease
- 12 Mar 2024Discontinued – Phase-I for Psoriasis (In volunteers) in USA (unspecified route) (Denali pipeline, February 2024)
- 12 Mar 2024Discontinued – Phase-I for Rheumatoid arthritis (In volunteers) in USA (unspecified route) (Denali pipeline, February 2024)
- 27 Feb 2024Efficacy and adverse events data from phase II trial in Cutaneous lupus erythematosus released by Sanofi
SAR443122, was investigated in several clinical trials to evaluate its safety and efficacy. NCT04469621 was studied in severe COVID-19 patients, while NCT05588843 is currently recruiting participants with ulcerative colitis. Additionally, NCT04781816, which was completed with results, focused on patients with cutaneous lupus erythematosus.
Eclitasertib is an orally bioavailable, small-molecule inhibitor of receptor-interacting serine/threonine-protein kinase 1 (RIPK1; receptor-interacting protein 1; RIP1), with potential anti-inflammatory and immunomodulatory activities. Upon oral administration, eclitasertib disrupts RIPK1-mediated signaling, and may attenuate inflammation and the resulting tissue damage. RIPK1, a signaling protein in the tumor necrosis factor (TNF) receptor pathway, plays a key role in inflammation and cell death in response to tissue damage and pathogen recognition.
SCHEME
SIDE CHAIN
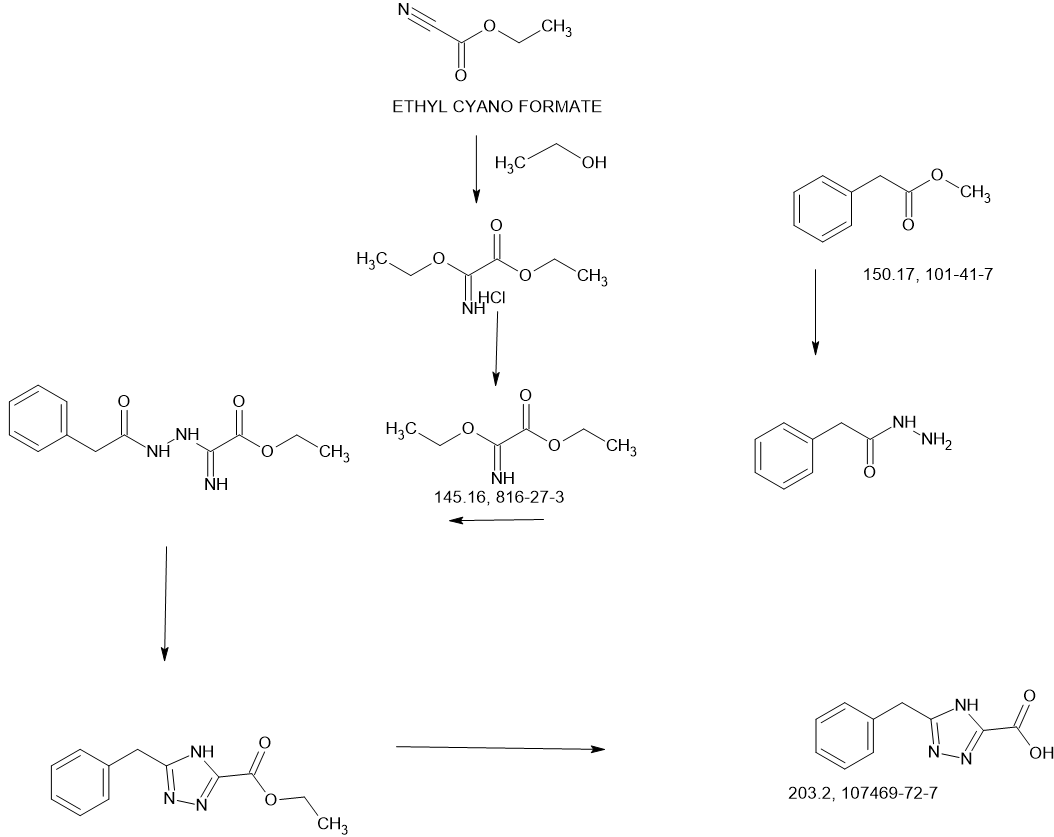
MAIN
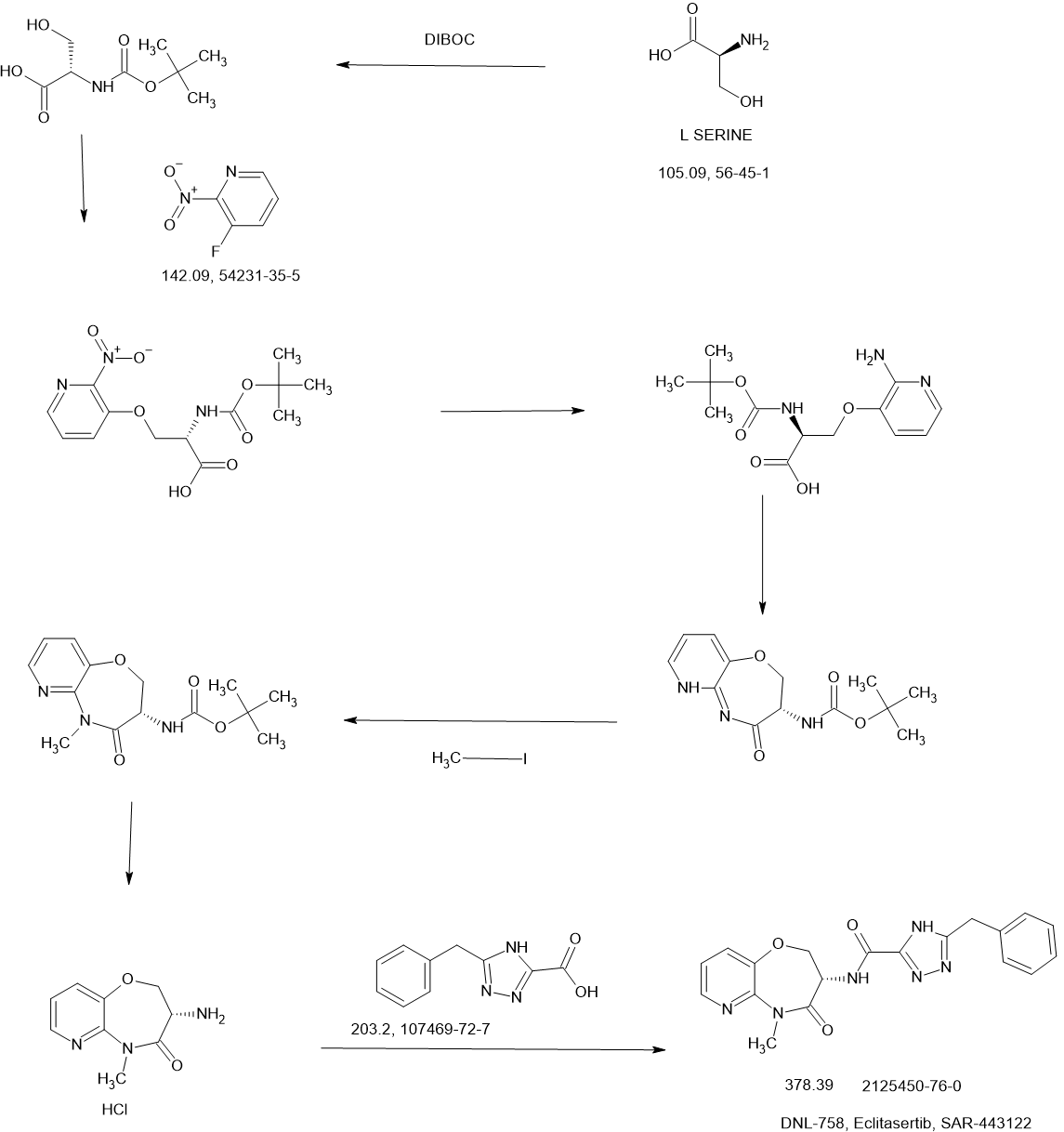
REF
WO2017136727
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017136727&_cid=P22-MAYSGO-11421-1
Example 42: (S)-5-benzyl-N-(5-methyl-4-oxo-2,3,4,5-tetrahydropyrido[3,2-b][1,4]oxazepin-3-yl)- 4H-1,2,4-triazole-3-carboxamide
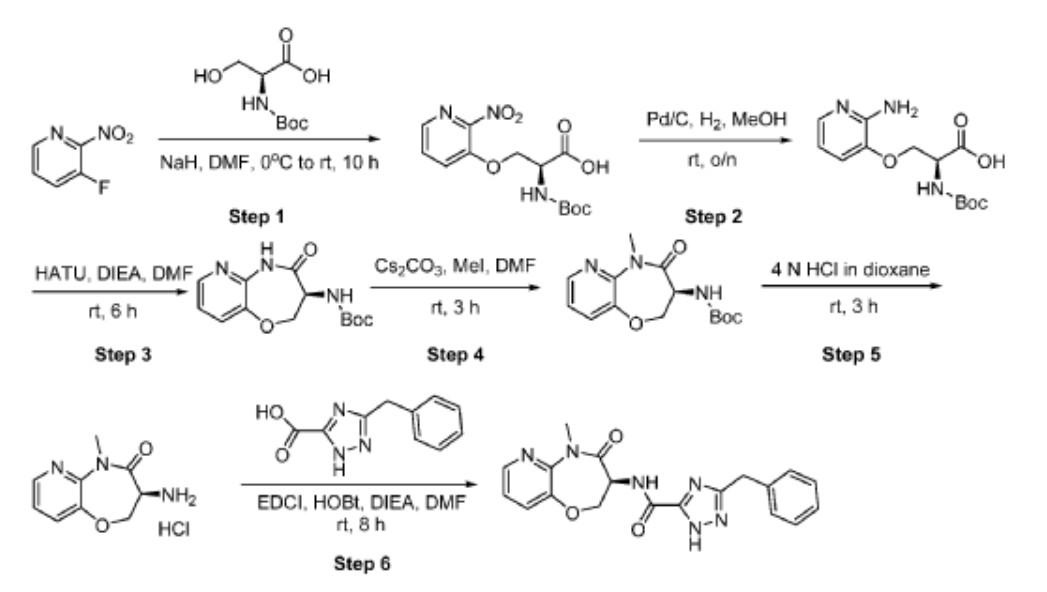
Step 1: Preparation oƒ (2S)-2-(((tert-butoxy)carbonyl)amino)-3-((2-nitropyridin-3-yl)oxy)propanoic acid
[0535] Sodium hydride (60%, 2 g, 50 mmol) was added into a stirring solution of (2S)-2-(tert-butoxycarbonylamino)-3-hydroxypropanoic acid (5 g, 25.0 mmol) in N,N-dimethylformamide (100 mL). The resulting mixture was stirred at 0 °C for 2 hours. 3-Fluoro-2-nitropyridine (3.6 g, 25.3 mmol) was added and the reaction mixture was stirred at room temperature for an additional 8 hours before quenching with hydrochloric acid (3 N, 5 mL). After adjusting the pH to 3-4 with hydrochloric acid (3 N, 20 mL), the resulting mixture was extracted with ethyl acetate (3 x 100 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by reversed phase chromatography with a RP-C18 column (acetonitrile/water, 1/2) to afford the title compound (3.2 g, 39%) as a light yellow oil. LC-MS (Method C): m/z = 272.1 [M+H-(t-BuO)]+, 1.269 min.
Step 2: Preparation oƒ (2S)-3-((2-aminopyridin-3-yl)oxy)-2-(((tert-butoxy)carbonyl)amino)propanoic acid
[0536] (2S)-2-(((tert-butoxy)carbonyl)amino)-3-((2-nitropyridin-3-yl)oxy)propanoic acid (0.45 g, 1.4 mmol) in methanol (20 mL) was aged overnight at room temperature in the presence of palladium on carbon (10%, 0.5 g) under hydrogen atmosphere (2-3 atm). The reaction mixture was filtered through Celite and the filtrate was concentrated under reduced pressure to afford the title compound (0.32 g, 78%) as a yellow oil. LC-MS (Method C): m/z = 298.1 [M+H]+, 0.982 min.
Step 3: Preparation oƒ tert-butyl N-((3S)-4-oxo-2H,3H,4H,5H-pyrido[3,2-b][1,4]oxazepin-3-yl)carbamate
[0537] N,N,N’,N’-tetramethyl-O-(7-azabenzotriazol-1-yl)uronium hexafluorophospate (0.73 g, 1.92 mmol) and N,N-diisopropylethylamine (0.25 g, 1.93 mmol) were added to a stirring solution of (2S)-3-((2-aminopyridin-3-yl)oxy)-2-(((tert-butoxy)carbonyl)amino)propanoic acid (0.45 g, 1.51 mmol) in N,N-dimethylformamide (5 mL). After stirring for 6 hours at room temperature, the reaction mixture was quenched by the addition of water (20 mL), and extracted with ethyl acetate (3 x 100 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by column chromatography (methanol/dichloromethane, 1/10) to afford the title compound (0.11 g, 26%) as a white solid. LC-MS (Method C): m/z = 280.1 [M+H]+, 1.248 min.
tep 4: Preparation oƒ tert-butylN-((3S)-5-methyl-4-oxo-2H,3H,4H,5H-pyrido[3,2-b][1,4]oxazepin-3-yl)carbamate
[0538] Iodomethane (50 mg, 0.35 mmol) was added dropwise to a stirring solution of tert-butyl N-((3S)-4-oxo-2H,3H,4H,5H-pyrido[3,2-b][1,4]oxazepin-3-yl)carbamate (100 mg, 0.36 mmol) and cesium carbonate (120 mg, 0.36 mmol) in N,N-dimethylformamide (5 mL). After stirring for 3 hours at room temperature, the reaction mixture was diluted with water (20 mL), and extracted with ethyl acetate (3 x 100 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (methanol/dichloromethane, 1/10) to afford the title compound (90 mg, 86%) as a white solid. LC-MS (Method C): m/z = 294.1 [M+H]+, 1.333 min.
Step 5: Preparation oƒ (3S)-3-amino-5-methyl-2H,3H,4H,5H-pyrido-[3,2-b][1,4]oxazepin-4-one hydrochloride
[0539] tert-butyl N-((3S)-5-methyl-4-oxo-2H,3H,4H,5H-pyrido[3,2-b][1,4]oxazepin-3-yl)carbamate (90 mg, 0.31 mmol) was added to a solution of hydrogen chloride in dioxane (4 M, 10 mL). The reaction mixture was stirred for 3 hours at room temperature and concentrated under reduced pressure to afford the title compound (65 mg, 93%) as a white solid, which was used directly in the next step without further purification. LC-MS (Method C): m/z = 194.1 [M+H]+, 0.847 min.
Step 6: Preparation oƒ (S)-5-benzyl-N-(5-methyl-4-oxo-2,3,4,5-tetrahydropyrido[3,2-b][1,4]oxazepin-3-yl)-4H-1,2,4-triazole-3-carboxamide
[0540] A solution of (3S)-3-amino-5-methyl-2H,3H,4H,5H-pyrido-[3,2-b][1,4]oxazepin-4-one hydrochloride (55 mg, 0.24 mmol) in N,N-dimethylformamide (1 mL) was added to a stirring solution of 5-benzyl-2H-1,2,4-triazole-3-carboxylic acid (80 mg, 0.40 mmol), 1-hydroxy-benzotrizole (70 mg, 0.53 mmol), N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (100 mg, 0.52 mmol) and N,N-
diisopropylethylamine (160 mg, 1.21 mmol) in N,N-dimethylformamide (2 mL). After stirring for 8 hours at room temperature, the reaction mixture was quenched by the addition of water (20 mL), and extracted with ethyl acetate (3 x 50 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The resulting residue was purified by Prep-HPLC with the following conditions: Column, XBridge Shield RP18 OBD Column, 5 μm, 19 x 150 mm; mobile phase, water (0.1% formic acid) and ACN (30.0% ACN to 60.0% over 7 min); Detector, UV 254 & 220 nm to afford the title compound. 1H NMR (300 MHz, DMSO-d6) δ 14.45 (s, 1H), 8.67 (d, J= 7.2 Hz, 1H), 8.37 (dd, J= 4.8, 1.8 Hz, 1H), 7.71 (dd, J= 7.8, 1.5 Hz, 1H), 7.37-7.21 (m, 6H), 4.92-4.82 (m, 1H), 4.73 (dd, J= 11.4, 9.6 Hz, 1H), 4.53 (dd, J= 9.6, 7.5 Hz, 1H), 4.14 (s, 2H), 3.37 (s, 3H). LC-MS (Method D): m/z = 379.1 [M+H]+, 1.611 min.
PATENT
WO2023182512
WO2023137035
WO2022208262
WO2021211919
WO2021209740
WO2021205298
WO2021205296
, WO2017136727
PAPER
European Journal of Medicinal Chemistry (2021), 220, 113484
Structure-based bioisosterism design of thio-benzoxazepinones as novel necroptosis inhibitors
Publication Name: European Journal of Medicinal Chemistry
Publication Date: 2021-08-05
PMID: 33930803
DOI: 10.1016/j.ejmech.2021.113484
PATENT
WO2021203011
- [1]. Anthony A. ESTRADA, et al. Compounds, compositions and methods. WO2017136727A2.[2]. Darwish I, et al., Rip1k inhibitors. WO2021203011
//////////Eclitasertib, DNL-758, SAR-443122, DNL 758, SAR 443122, UNII-975AT1P9J6, Phase 2, Ulcerative colitis
Davelizomib



Davelizomib
| Molecular Weight | 481.25 |
|---|---|
| Formula | C21H26BF2N3O7 |
| CAS No. | 2409841-51-4 |
{(4S)-2-[(1R)-1-{2-[(2S)-1-(2,4-difluorophenyl)azetidine-2- carboxamido]acetamido}-3-methylbutyl]-5-oxo-1,3,2- dioxaborolan-4-yl}acetic acid proteasome inhibitor, antineoplastic
2-[(4S)-2-[(1R)-1-[[2-[[(2S)-1-(2,4-difluorophenyl)azetidine-2-carbonyl]amino]acetyl]amino]-3-methylbutyl]-5-oxo-1,3,2-dioxaborolan-4-yl]acetic acid
- 1,3,2-Dioxaborolane-4-acetic acid, 2-[(1R)-1-[[2-[[[(2S)-1-(2,4-difluorophenyl)-2-azetidinyl]carbonyl]amino]acetyl]amino]-3-methylbutyl]-5-oxo-, (4S)-
- 2-[(4S)-2-[(1R)-1-[[2-[[(2S)-1-(2,4-difluorophenyl)azetidine-2-carbonyl]amino]acetyl]amino]-3-methylbutyl]-5-oxo-1,3,2-dioxaborolan-4-yl]acetic acid
T3LN9U6BRF
Davelizomib is proteasome inhibitor with antineoplastic effect.
DAVELIZOMIB is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
Multiple myeloma (MM) is a malignant proliferative disease of plasma cells, characterized by abnormal proliferation of clonal plasma cells in the bone marrow, destruction of hematopoietic function, stimulation of osteolytic lesions in the bones, detection of monoclonal immunoglobulins or their fragments (M protein) in serum and/or urine, and clinical manifestations of bone pain, anemia, hypercalcemia, renal impairment, infection, and bleeding. Bortezomib is a reversible proteasome inhibitor that achieves the purpose of treating multiple myeloma by promoting apoptosis of myeloma cells. However, in the long-term treatment process, some multiple myeloma patients have developed resistance to bortezomib. Therefore, there is still a need for new, safe, and highly stable drugs for the treatment of multiple myeloma.
SCHEME

PATENT
Borate of azetidine derivative
Publication Number: JP-2021531302-A
Priority Date: 2018-08-02
WO2020025037

Step 1: Synthesis of compound 4-3
[0252]N, N-diisopropylethylamine (22.02 g) was added to a solution of acetonitrile (200 mL) containing compound 4-1 (10 g) and compound 4-2 (20.13 g) at room temperature. The reaction mixture was stirred at 100 ° C for 16 hours, then cooled to room temperature and then added to ethyl acetate. The organic layer was washed with water and saturated brine, respectively, and then the organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated to remove the solvent, and the residue was purified by silica gel column chromatography (mobile phase: petroleum ether: ethyl acetate = 10: 1) to obtain compound 4-3. Compound 4-3: MS (ESI) m/z: 227.9 [M+1].
[0253]Step 2: Synthesis of compound 4-4
[0254]
LiOH·H 2 O (6.65 g) was added to a mixed solution of compound 4-3 (7.2 g) in methanol (20 mL), tetrahydrofuran (20 mL) and water (10 mL) at 0°C. The reaction mixture was stirred at room temperature for 1 hour, then concentrated under reduced pressure, diluted with water and ethyl acetate, and separated. The aqueous layer was adjusted to pH=6 with 1 mol/L hydrochloric acid, and then extracted with ethyl acetate. The organic phases were combined and washed with saturated brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated to remove the solvent to obtain compound 4-4, which was directly used in the next step. Compound 4-4: MS (ESI) m/z: 213.9 [M+1].
[0255]Step 3: Synthesis of compound 4-5
[0256]Glycine methyl ester hydrochloride (1.06 g), TBTU (2.71 g) and N,N-diisopropylethylamine (3.64 g) were added to a solution of compound 4-4 (1.5 g) in dichloromethane (50 mL) at -10°C. The reaction mixture was stirred at -10°C to 0°C for 3 hours, then diluted with water (40 mL) and extracted with dichloromethane. The organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated to remove the solvent, and the residue was purified by silica gel column chromatography (mobile phase: petroleum ether: ethyl acetate = 5:1) to obtain compound 4-5. Compound 4-5: MS (ESI) m/z: 284.9 [M+1].
[0257]Step 4: Synthesis of Compound 4-6
[0258]To a mixed solution of compound 4-5 (0.5 g) in tetrahydrofuran (2 mL), methanol (2 mL) and water (1 mL) was added LiOH·H
2 O (369.03 mg) at 0°C. The reaction mixture was stirred at 0°C to 20°C for 2 hours, then concentrated, diluted with water (3 mL), and separated. The aqueous layer was adjusted to pH=6 with 1 mol/L hydrochloric acid and extracted with ethyl acetate. The organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated to remove the solvent to obtain compound 4-6, which was directly used in the next step. Compound 4-6: MS (ESI) m/z: 270.9 [M+1].
[0259]Step 5: Synthesis of Compound 4-8
[0260]N,N-diisopropylethylamine (273.56 mg) was added to a solution of compound 4-6 (0.26 g), compound 2-6 (437.84 mg) and TBTU (370.71 mg) in dichloromethane (10 mL) at -10 ° C. The reaction mixture was slowly warmed to room temperature and continued to stir for 2 hours, then the reaction mixture was added to water (10 mL) for dilution and extracted with dichloromethane. The organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated to remove the solvent, and the residue was purified by silica gel column chromatography (mobile phase: petroleum ether: ethyl acetate = 1: 1) to obtain compound 4-8. Compound 4-8: MS (ESI) m/z: 518.2 [M+1].
[0261]Step 6: Synthesis of Compound 4-9
[0262]Isobutylboric acid (234.45 mg) and 1 mol/L HCl (1.31 mL) were added to a mixed solution of methanol (4 mL) and n-hexane (6 mL) of compound 4-8 (0.17 g) at 0°C. The reaction mixture was slowly warmed to room temperature and stirred for 12 hours, then concentrated under reduced pressure to remove the solvent to obtain a residue. The residue was purified by preparative HPLC and separated by SFC to obtain compound 4-9. Compound 4-9:
1 H NMR (400MHz, METHANOL-d4) δ6.83(br s,2H),6.61(br s,1H),4.49(br s,1H),4.10(br s,3H),3.84(br s,1H),2.75(br s,1H),2.59(br s,1H),2.48(br s,1H),1.62(br s,1H),1.30(br s,2H),0.92(br s,6H). MS(ESI)m/z:366.1[M-17].
[0263]Preparative HPLC separation method of compound 4-9:
[0264]Column: Xtimate C18 150×25mm, 5μm;
[0265]Mobile phase: water (0.225% FA)-MeOH;
[0266]Elution gradient: 61%-85%;
[0268]Preparation of compound 4-9 SFC separation method:
[0269]Chromatographic column: C2 250mm×30mm, 10μm;
[0270]Mobile phase: A: carbon dioxide, B: methanol;
[0271]Elution gradient B%: 30%-30%;
[0273]The elution order of compound 4-9 is the second peak appearing in high performance chiral liquid column chromatography.
[0274]Step 7: Synthesis of Compound I-1
[0275]Method 1: Add L-malic acid (332 mg) to isopropyl acetate (2.5 mL), heat to 70°C and stir, and after 10 minutes, add compound 4-9 (1.0 g) dissolved in 2.5 mL isopropyl acetate solution. Then stop heating, cool to 25°C and continue stirring at this temperature for 5 days. Filter, collect the filter cake, and vacuum dry to obtain compound I-1, which is Form I crystal of compound I-1.
[0276]Method 2: Add compound I-1 (68.9 g) to a reaction flask, then add 440 mL of isopropyl acetate, and stir the mixture at room temperature for 24 h under nitrogen protection. Filter and dry to obtain Form I crystals of compound I-1 (64.4 g). The X-ray powder diffraction pattern of the obtained crystals using Cu Kα rays is shown in Figure 1.
[0277]
化合物I-1: 1H NMR(400MHz,DMSO-d 6)δ12.30(br s,1H),10.65(br s,1H),8.57(br t,J=5.77Hz,1H),7.11(ddd,J=2.64,9.16,12.30Hz,1H),6.91(br t,J=8.16Hz,1H),6.53(dt,J=5.65,9.60Hz,1H),4.44(br t,J=7.91Hz,1H),4.37(dd,J=3.89,7.65Hz,1H),4.10(br s,2H),3.91-4.01(m,1H),3.76(q,J=7.36Hz,1H),2.61(br d,J=10.79Hz,2H),2.19-2.44(m,3H),1.61(td,J=6.71,13.68Hz,1H),1.20-1.36(m,2H),0.86(t,J=6.02Hz,6H)。
///////Davelizomib, T3LN9U6BRF, PHASE 2
Crisugabalin



Crisugabalin
Cas 2209104-84-5
2-[(1S,2S,3R,6S,8S)-2-(aminomethyl)-2-tricyclo[4.2.1.03,8]nonanyl]acetic acid
WeightAverage: 209.289
Monoisotopic: 209.141578856
Chemical FormulaC12H19NO2
- HSK 16149
- HSK-16149
- HSK16149
- Q3MK7E8686
- PHASE 2
Tricyclo[4.2.1.03,8]nonane-2-acetic acid, 2-(aminomethyl)-, (1S,2S,3R,6S,8S)-
(1S,2S,3R,6S,8S)-2-(Aminomethyl)tricyclo[4.2.1.03,8]nonane-2-acetic acid
- (1S,2S,3R,6S,8S)-2-(Aminomethyl)tricyclo[4.2.1.03,8]nonane-2-acetic acid
- 2-[(1S,2S,3R,6S,8S)-2-(aminomethyl)-2-tricyclo[4.2.1.03,8]nonanyl]acetic acid
- Tricyclo[4.2.1.03,8]nonane-2-acetic acid, 2-(aminomethyl)-, (1S,2S,3R,6S,8S)-
Crisugabalin (HSK16149) is a selective GABA analog in development for the treatment of chronic pain. It has a wider therapeutic index than pregabalin, which has a similar mechanism of action. In China, it was approved in May 2024 for the treatment of diabetic peripheral neuropathic pain[1] and approved in July 2024 for the treatment of postherpetic neuralgia.[2] In the United States, it is in Phase III trials as of 2023.[3][4] The drug can be administered with or without food.[5]
Crisugabalin is under investigation in clinical trial NCT06490484 (Efficacy and Safety of HSK16149 Capsule in Chinese Patients With Diabetic Peripheral Neuropathic Pain Who Had an Inadequate Response to Pregabalin).
SCHEME

PATENTS
WO2020029762
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020029762&_cid=P10-MAI1TM-34428-1


[0414]2-((1S,2S,3R,6S,8S)-2-(aminomethyl)tricyclo[4.2.1.0
3,8 ]nonanyl-2-yl)acetate benzenesulfonate (1:1) (Compound 1)
[0415]
2-((1S,2S,3R,6S,8S)-2-(aminomethyl)tricyclo[4.2.1.0 3,8]nonan-2-yl)acetic acid compound with benzenesulfonic acid(1:1)
[0416]
[0417]
[0418]Step 1: 3-(Cyclohexyl-3-en-1-yl)propanoic acid (1B)
[0419]
3-(cyclohex-3-en-1-yl)propanoic acid
[0420]
[0421]Anhydrous formic acid (18.82kg, 409.09mol) was pumped into a 100-liter reactor. The temperature was lowered to 10°C. Triethylamine (16.53kg, 163.64mol) was added dropwise to the reaction solution. After addition, it was stirred for 20 minutes. When the internal temperature was 10°C, cycloisopropyl malonate (7.86kg, 54.55mol) was added to the reactor. Then 3-cyclohexene-1-carboxaldehyde (6.00kg, 54.55mol) was added dropwise to the reaction solution at an internal temperature of 40°C. After addition, the temperature was raised to 140-150°C and the reaction was continued until no gas was released. The pH of the reaction solution was adjusted to 1-2 with 6N hydrochloric acid (24.0L). The aqueous phase was extracted with dichloromethane (12L×2), and the organic phases were combined and washed with saturated brine (10L×2). The organic phase was dried over anhydrous sodium sulfate (2.0 kg) for 1 hour, filtered, and the filtrate was concentrated and evaporated to dryness to obtain a yellow oil 1B (8.80 kg).
[0422]
1H NMR(400MHz,CDCl 3)δ10.23(s,1H),5.73–5.55(m,2H),2.46–2.30(m,2H),2.09–1.96(m,2H),1.81–1.53(m,6H),1.35–1.17(m,1H)。
[0423]
[0424]Step 2: 3-(Cyclohexyl-3-en-1-yl)-1-(pyrrolidin-1-yl)propyl-1-one (1C)
[0425]
3-(cyclohex-3-en-1-yl)-1-(pyrrolidin-1-yl)propan-1-one
[0426]
[0427]Dissolve 1B (11.20kg, 72.727mol) in dichloromethane (60.0L) and pump into a 100L reactor. Add DMF (3.0mL) and drop oxalyl chloride (9.046kg, 71.272mol) into the reaction solution. After addition, stir at room temperature for 2.0 hours. Add tetrahydropyrrole (5.689kg, 79.999mol) and triethylamine (8.814kg, 87.272mol) dropwise into the reactor. Control the internal temperature below 10℃, after addition, stir at room temperature overnight. Cool the reaction solution to 10℃. Add 3N hydrochloric acid (20.0L) dropwise to adjust the pH of the reaction solution to between 1-2. Let stand, separate the liquids, and extract the aqueous phase with dichloromethane (10.0L×1). The organic phases were combined and washed with 5% sodium hydroxide solution (10.0 L x 1) and saturated ammonium chloride solution (20.0 L x 1) in sequence. The organic phase was dried over anhydrous sodium sulfate (2.0 kg) for 30 minutes, filtered, and the filtrate was concentrated to obtain brown liquid 1C (15.00 kg, yield 99.6%).
[0428]
1H NMR(400MHz,CDCl 3)δ5.73–5.56(m,2H),3.43(dd,4H),2.37–2.22(m,2H),2.16–2.01(m,4H),1.90(dt,4H),1.81–1.51(m,6H),1.30–1.15(m,2H)。
[0429]
[0430]Step 3: Tricyclo[4.2.1.0
3,8 ]nonanyl-2-one (1R,3S,6R,8R and 1S,3R,6S,8S racemate) (1D)
[0431]
tricyclo[4.2.1.0 3,8]nonan-2-one(1R,3S,6R,8R and 1S,3R,6S,8S racemate)
[0432]
[0433]Dissolve 1C (5.64kg, 27.22mol) in dichloromethane (40.0L) and pump it into a 100L reactor. Cool to -10°C and add 2,4,6-trimethylpyridine (4.94kg, 40.83mol). Add a dichloromethane solution (16.0L) of trifluoromethanesulfonic anhydride (11.50kg, 40.83mol) dropwise to the reaction solution until complete. Heat and reflux for 12 hours. After the reaction is complete as detected by the central control, add an aqueous solution (23.0L) of sodium hydroxide (3.10kg, 77.5mol) dropwise to the reaction solution and adjust the pH of the reaction solution to between 10-11. Continue to reflux for 5-6 hours. Stand and separate the liquids, extract the aqueous phase with dichloromethane (5.0L×1), and combine the organic phases. Pump the organic phase into the reactor and cool to 10°C. 2.0N hydrochloric acid solution (20.0L) was added dropwise to adjust the pH of the reaction solution to between 1 and 2. The solution was separated by standing, and the organic phase was washed with saturated brine (20L×1), concentrated, and the residue was dissolved with acetone (20.0L), then pumped into a 100L reactor and stirred, and a solution of concentrated sulfuric acid (4.0L) and water (20.0L) was added dropwise, and refluxed for 2 hours after addition. The temperature was lowered to 15°C, saturated brine (20.0L) was added to the reaction solution, and extracted with n-hexane (15.0L×2). The organic phases were combined, washed with saturated brine (20.0L×1), and the organic phase was dried over anhydrous sodium sulfate overnight. After filtration, the filtrate was concentrated under reduced pressure to obtain a yellow solid crude product 1D (3.00kg, yield 81%) with a purity of 50%.
[0434]1D Further purification steps:
[0435]Method 1: Anhydrous sodium bisulfite (5.735 kg, 55.147 mol) was dissolved in 66 L of purified water and added to a 100 L reactor. A solution of crude 1D (3.00 kg, 22.059 mol) in ethanol (3.0 L) was added under stirring at room temperature. The mixture was stirred overnight at room temperature and extracted with ethyl acetate (20 L × 2). The aqueous phase was added to the reactor, stirred and cooled to 10°C. A solution of sodium hydroxide (2.250 kg, 56.250 mol) in water (10 L) was added dropwise. The pH was adjusted to 10-12. The mixture was stirred at room temperature for 2 hours. The mixture was extracted with n-hexane (20 L × 2). The organic phases were combined and washed with purified water (20 L × 1). The organic phases were dried with anhydrous sodium sulfate (2 kg) for 1 hour, filtered, and the filtrate was evaporated to dryness to obtain 1D as a colorless crystalline solid (2.7 kg, yield 90%) with a purity of 98.3%.
[0436]Method 2: Sodium bisulfite (1529g, 14.706mol) was dissolved in 22L water, and a solution of 1D crude product (1000g, 7.353mol) in anhydrous ethanol (1000mL) was added dropwise under stirring, and stirred overnight at room temperature (24 hours). The reaction solution was extracted with dichloromethane (5L×2) to remove impurities, and sulfuric acid solution (prepared with 6.4L concentrated sulfuric acid and 6kg crushed ice) was added dropwise to the aqueous phase, and stirred at room temperature for 5 hours. The reaction solution was extracted with n-hexane (extracted 3-4 times, 4L each time), the organic phases were combined and washed with saturated sodium chloride aqueous solution (5L×2), the organic phases were dried with 1kg anhydrous sodium sulfate for 2 hours, filtered, and the filtrate was evaporated to dryness to obtain 1D, a white solid (900g, yield: 90%), and the purity was determined to be 98.1%.
[0437]
1H NMR(400MHz,CDCl 3)δ3.39(m,1H),3.19(m,1H),2.77(m,1H),2.38(m,1H),2.05(m,1H),1.93(d,1H),1.77(m,1H),1.45(m,4H),1.20(m,1H)。
[0438]
[0439]Step 4: tert-Butyl 2-(tricyclo[4.2.1.0
3,8 ]nonanyl-2-ylidene) acetate (1R,3S,6R,8R and 1S,3R,6S,8S racemate) (1E)
[0440]
tert-butyl 2-tricyclo[4.2.1.0 3,8]nonan-2-ylidene)acetate(1R,3S,6R,8R and 1S,3R,6S,8S racemate)
[0441]
[0442]Potassium tert-butoxide (742.0g, 6.62mol) and tetrahydrofuran (6.20L) were added to a 20L reactor. The temperature was lowered to 5°C, and tert-butyl dimethoxyphosphonoacetate (1480g, 6.62mol, 1.1eq) was added dropwise to the reaction solution. The reaction temperature was controlled at 10°C-15°C, and stirring was continued for 1 hour. Then, a solution of 1D (820.0g, 6.02mol, 1.0eq) in tetrahydrofuran (2.0L) was added dropwise to the reaction solution. The addition was completed within 1 hour, and the temperature was naturally raised to room temperature for reaction for 2 hours. Saturated ammonium chloride solution (2.0L) and purified water (2.0L) were added to the reactor in sequence. After stirring for 20 minutes, the mixture was allowed to stand for stratification, and the aqueous phase was extracted with methyl tert-butyl ether (1.5L×2). The organic phases were combined, washed with saturated brine (2L×2), and dried over anhydrous sodium sulfate. Filtration and concentration afforded 1E as a yellow liquid (1.50 kg).
[0443]
[0444]Step 5: tert-Butyl 2-(2-(nitromethyl)tricyclo[4.2.1.0
3,8 ]nonanyl-2-yl)acetate (1R,2R,3S,6R,8R and 1S,2S,3R,6S,8S racemate) (1F)
[0445]
tert-butyl 2-(2-(nitromethyl)tricyclo[4.2.1.03,8]nonan-2-yl)acetate(1R,2R,3S,6R,8R and 1S,2S,3R,6S,8S racemate)
[0446]
[0447]1E (1.40 kg, 5.97 mol, 1.0 eq), nitromethane (1.82 kg, 29.85 mol, 5.0 eq) and dimethyl sulfoxide (9.8 L) were added to a 20 L reactor in sequence. Stir and add cesium carbonate (2.34 kg, 7.16 mol, 1.2 eq) to the reaction solution. After the addition, heat to 80°C-85°C, continue to keep the reaction for 5 hours, then cool to room temperature, add purified water (20.0 L) to the reactor, and extract the aqueous phase with methyl tert-butyl ether (8.0 L × 3). Combine the organic phases, wash with saturated brine (8.0 L × 2), and dry over anhydrous sodium sulfate. Filter and concentrate to obtain a brown liquid 1F (1.62 kg, yield: 92%).
[0448]
[0449]Step 6: tert-Butyl 2-((1S,2S,3R,6S,8S)-2-(aminomethyl)tricyclo[4.2.1.0
3,8 ]nonanyl-2-yl)acetate (S)-2-acetoxy-2-phenylacetic acid (1H)
[0450]
tert-butyl2-((1S,2S,3R,6S,8S)-2-(aminomethyl)tricyclo[4.2.1.03,8]nonan-2-yl)acetate(S)-2-acetoxy-2-phenylacetate
[0451]
[0452]Add 1F (730.0 g, 2.47 mol) and methanol (7.3 L) to a 50 L reactor. Stir, add nickel chloride hexahydrate (118 g, 0.49 mol, 0.2 eq) to the reaction, cool the reaction solution to 5 ° C, add sodium borohydride (374 g, 9.88 mol, 4.0 eq) in batches, keep the reaction system temperature at 20 ° C-30 ° C, and add it in about 3 hours. After the addition, continue to stir and react for 2 hours. Add ice water (16.4 L) to the reactor, and filter the aqueous phase with diatomaceous earth. Extract the filtrate with dichloromethane (3.0 L × 2) and combine the organic phases, wash with saturated brine (4 L × 1), and dry over anhydrous sodium sulfate. Filter, add (S)-(+)-O-acetyl-L-mandelic acid (384 g, 1.97 mol, 0.8 eq) to the filtrate, and stir for 20 minutes after the addition. The organic phase was concentrated by distillation until no solvent was evaporated, and then stirred with isopropanol (5.9 L) for 2 hours, cooled to 5°C and stirred for 1 hour. Filtered, the filter cake was washed with isopropanol (0.4 L × 1), and dried to obtain a white solid product 1H crude product (422 g, yield: 34.96%). The solid was taken and the ee value was determined to be 48.35% after derivatization.
[0453]First crystallization: Add crude product 1H (420.0 g, 0.92 mol), isopropanol (4.20 L) and water (0.21 L) to the reactor in sequence. Raise the temperature to 82 °C to completely dissolve the solid and keep warm for 0.5 hours. Cool down to 20 °C for crystallization for about 6 hours. When the internal temperature reaches 20 °C, filter and wash the filter cake with isopropanol (0.40 L × 1). Combine the solids and dry them at 60-65 °C for 4 hours to constant weight. Obtain the first crystal of 1H (260 g, yield: 62%). After taking the solid for derivatization, the ee value is 81.25%.
[0454]Second crystallization: Add the first crystal of 1H (177g, 0.39mol), isopropanol (2.53L) and water (0.126L) to the reactor in sequence. Raise the temperature to 82℃ to completely dissolve the solid and keep warm for 0.5 hours. Cool down to 20℃ for crystallization for about 4.5 hours. When the internal temperature reaches 30℃, filter and wash the filter cake with isopropanol (0.10L×1). Combine the solids and dry them at 60-65℃ for 4 hours to constant weight. Obtain pure white solid 1H (128g, yield: 72%). After taking the solid derivative, the ee value is determined to be 99.73%.
[0455]
[0456]Step 7: 2-((1S,2S,3R,6S,8S)-2-(aminomethyl)tricyclo[4.2.1.0
3,8 ]nonanyl-2-yl)acetic acid benzenesulfonic acid compound (1:1) (Compound 1)
[0457]
2-((1S,2S,3R,6S,8S)-2-(aminomethyl)tricyclo[4.2.1.0 3,8]nonan-2-yl)acetic acid compound with benzenesulfonic acid(1:1)
[0458]
[0459]Add 1H pure product (100.0g, 0.218mol) and purified water (0.8L) to the reactor in sequence and cool to 0-10℃. When the internal temperature reaches 0-10℃, add 1mol/L NaOH (218mL) aqueous solution to the reactor and adjust the pH of the reaction solution to 9-10. Let stand for stratification and extract the aqueous phase with dichloromethane (0.30L×2). Combine the organic phases and wash with 1mol/L NaOH (0.10L×1) solution and saturated brine (0.15L×1) in sequence. Add activated carbon (5.0g) to the organic phase for decolorization and dry with anhydrous sodium sulfate. Filter, concentrate the filtrate, and dissolve the residue in the concentration kettle with acetonitrile (280mL). Prepare a solution of benzenesulfonic acid monohydrate (77.0g, 0.437mol) with purified water (280mL) and add it dropwise to the above acetonitrile solution until complete. The temperature was raised to 80-85°C and kept for 4-6 hours. The reaction solution was cooled to 10-20°C for crystallization for about 4-6 hours. When the internal temperature reached 10-20°C, the solution was filtered and the filter cake was washed with water (30 mL × 1) and acetonitrile (50 mL × 1) in turn. After drying, compound 1 was obtained as a white solid (72 g, yield: 90%).
[0460]
1H NMR(400MHz,MeOD)δ7.83(m,2H),7.42(m,3H),3.31(dt,4H),2.86(m,1H),2.55(d,2H),2.48(ddd,1H),2.32(dd,1H),2.15(m,1H),2.04(m,1H),1.77(m,1H),1.62(m,4H),1.45(m,1H),1.28(dt,1H)。
[0461]
LCMS m/z=210.1[M+1]。
References
^ “Monthly Report: New Drug Approval in China, May 2024”.
- ^ “海思科苯磺酸克利加巴林胶囊获批新适应症”. PhIRDA. 19 July 2024. Retrieved 26 April 2025.
- ^ Gou, Xiaoli; Yu, Xiaojuan; Bai, Dongdong; Tan, Bowei; Cao, Pingfeng; Qian, Meilin; Zheng, Xiaoxiao; Chen, Lei; Shi, Zongjun; Li, Yao; Ye, Fei; Liang, Yong; Ni, Jia (March 2021). “Pharmacology and Mechanism of Action of HSK16149, a Selective Ligand of α2δ Subunit of Voltage-Gated Calcium Channel with Analgesic Activity in Animal Models of Chronic Pain”. The Journal of Pharmacology and Experimental Therapeutics. 376 (3): 330–337. doi:10.1124/jpet.120.000315. ISSN 1521-0103. PMID 33293377.
- ^ Guo, Xiaohui; Zhang, Tingting; Yuan, Geheng; Yukun, LI; Hua Ma, Jian; Hong-Mei, LI (2023). “224-OR: The Efficacy and Safety of HSK 16149 in Chinese with Diabetic Peripheral Neuropathic Pain—A Randomized, Double-Blinded, Placebo and Pregabalin-Controlled Phase II/III Study”. Diabetes. 72. doi:10.2337/db23-224-OR.
- ^ Wu, Qingqing; Zhu, Huijuan; Song, Rong; Zhang, Mengqi; Li, Fangqiong; Zeng, Weifang; Wang, Wei; Jia, Jingying; Yu, Chen; Liu, Yanmei (June 2023). “Effect of a high-fat and high-calorie food on the pharmacokinetics of a novel, potent GABA analog HSK16149 in healthy subjects”. Pharmacology Research & Perspectives. 11 (3): e01102. doi:10.1002/prp2.1102. PMC 10199234. PMID 37208866.
| Legal status | |
|---|---|
| Legal status | Investigational |
| Identifiers | |
| CAS Number | 2209104-84-5 |
| UNII | Q3MK7E8686 |
| Chemical and physical data | |
| Formula | C12H19NO2 |
| Molar mass | 209.289 g·mol−1 |
//////////Crisugabalin, HSK 16149, HSK-16149, HSK16149, Q3MK7E8686, PHASE 2
Clesacostat



Clesacostat
PF 05221304, 752DF9PPPI
CAS 1370448-25-1
WeightAverage: 502.571
Monoisotopic: 502.221620082
Chemical FormulaC28H30N4O5
4-[6-methoxy-4-(7-oxo-1-propan-2-ylspiro[4,6-dihydroindazole-5,4′-piperidine]-1′-carbonyl)pyridin-2-yl]benzoic acid
- Originator Pfizer
- ClassBenzoic acids; Carboxylic acids; Ethers; Hepatoprotectants; Indazoles; Piperidines; Pyridines; Small molecules; Spiro compounds
- Mechanism of ActionAcetyl-CoA carboxylase inhibitors
- Phase IINon-alcoholic fatty liver disease; Non-alcoholic steatohepatitis
- 21 Feb 2024Pfizer completes a phase II trial in Non-alcoholic steatohepatitis (Combination therapy) in Slovakia, Japan, Bulgaria, Canada, China, Hong Kong, India, Poland, Puerto Rico, South Korea, Taiwan (PO) (NCT04321031) (EudraCT2019-004775-39)
- 26 May 2022Clesacostat – Pfizer receives Fast Track designation for Non-alcoholic steatohepatitis [PO] (Combination therapy) in USA
- 28 Apr 2022Pfizer completes a phase II trial for Non-alcoholic fatty liver disease (Combination therapy) in USA and Canada (PO) (NCT04399538)
Clesacostat is under investigation in clinical trial NCT04321031 (Metabolic Interventions to Resolve Non-alcoholic Steatohepatitis (NASH) With Fibrosis (MIRNA)).
CLESACOSTAT is a small molecule drug with a maximum clinical trial phase of II (across all indications) and has 4 investigational indications.
SCHEME
SIDECHAIN

MAIN

PATENT
WO2021171164 89%
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021171164&_cid=P20-MAF4R3-69728-1
4-(4-(1-lsopropyl-7-oxo-1 ,4,6,7-tetrahydrospiro[indazole-5,4′-piperidine]-1′-carbonyl)-6-methoxypyridin-2-yl)benzoic acid,
A preparation of (S)- 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-N-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide is presented in Example 1 of US 2018-0051012A1 , hereby incorporated herein by reference in its entireties for all purposes. A preparation of 4-(4-(1-lsopropyl-7-oxo-1 ,4,6,7-tetrahydrospiro[indazole-5,4′-piperidine]-1 ‘-carbonyl)-6-methoxypyridin-2-yl)benzoic acid is in Example 9 of US 8,859,577, hereby incorporated herein by reference in its entireties for all purposes. Preparation of [(1 R,5S,6R)-3-{2-[(2S)-2-methylazetidin-1-yl]-6-(trifluoromethyl)pyrimidin-4-yl}-3-azabicyclo[3.1 0]hex-6-yl]acetic acid (including a crystalline free acid form thereof) is described in Example 4 of U.S. Patent No. 9,809,579. Preparation of GLP-1 R agonists are described in U.S. Patent No.10,208,019.
Step 6: (S)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-N-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide (Example 1 (DGAT2i Compound))
Oxalyl chloride (13.8 ml_, 160 mmol, 1.2 equiv) and dimethylformamide (0.510 ml_, 6.65 mmol, 0.05 equiv) were added to a suspension of 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylic acid (45.0 g, 133 mmol, 1.0 equiv) in dichloromethane (500 ml_). The suspension was stirred for 2 hours when a solution was achieved. The reaction mixture was concentrated to yield crude acid chloride as a red solid. A solution of (S)-tetrahydrofuran-3-amine (12.2 g, 140 mmol, 1.05 equiv) and diisopropylethylamine (51.0 ml_, 293 mmol, 2.2 equiv) in tetrahydrofuran (100 ml_) was added dropwise to a solution of the crude acid chloride in dichloromethane (200 ml_) at 0 °C. The reaction was allowed to warm to room temperature and stirred for 16 hours. Water (1 .0 L) and ethyl acetate (600 ml_) were added and the organic layer was separated, washed with saturated sodium bicarbonate, dried over magnesium sulfate, and filtered. The filtrate was treated with activated charcoal (20 g) was stirred at 65 °C for 20 minutes. The suspension was filtered warm and filtrate was concentrated to a pale, yellow solid which was recrystallized from methanol in ethyl acetate (1 :4, 1 L) to yield (S)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-A/-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide (43.5 g, 81%) as a colorless solid. The title compound was combined with previous batches (108.7 g, 266.8 mmol) prepared in the same manner and slurried with ethyl acetate (1.0 L) at 80 °C for 4 hours. The suspension was allowed to cool to room temperature and stirred for 4 days. The solid was filtered, washed with ethyl acetate (3×200 ml_) and dried under high vacuum at 50 °C for 24 hours to yield (S)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-A/-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide (100.5 g, 92%) as a colorless solid. 1H NMR (300 MHz, DMSO-d6) 6 1.38 (t, 3H), 1.89-1.98 (m, 1H), 2.15-2.26 (m, 1H), 3.65 (dd, 1H), 3.70-3.78 (m, 1H), 3.85-3.92 (m, 2H), 4.18 (q, 2H), 4.46-4.55 (m, 1H), 7.18 (dd, 1H), 7.58 (dd, 1H), 7.69 (dd, 1H), 8.37 (dd,
1 H), 8.64 (d, 1 H), 8.95 (d, 1 H), 9.28 (s, 2H), 9.39 (d, 1 H). MS (ES+) 408.4 (M+H). Melting point 177.5 °C. Elemental analysis for C21H21N5O4: calculated C, 61.91 ; H, 5.20; N, 17.19; found C, 61.86; H, 5.18; N, 17.30.
PATENT
WO2021171163 65%
WO2020234726 65%
Journal of Medicinal Chemistry (2020), 63(19), 10879-10896
WO2020044266 89%
WO2019102311 89%
//////////Clesacostat, PF 05221304, PHASE 2, 752DF9PPPI
Bezisterim, HE 3286; NE-3107




Bezisterim, HE 3286; NE-3107
CAS 1001100-69-1
(1R,3aS,3bR,4R,7S,9aR,9bS,11aS)-1-ethynyl-9a,11a-dimethyl-1H,2H,3H,3aH,3bH,4H,6H,7H,8H,9H,9aH,9bH,10H,11H,11aH-cyclopenta[a]phenanthrene-1,4,7-triol
- (3β,7β,17α)-Pregn-5-en-20-yne-3,7,17-triol
- 17α-Ethynyl-5-androstene-3β,7β,17β-triol
- 17α-Ethynyl-Δ5-androstene-3β,7β,17β-triol
- 17α-Ethynylandrost-5-ene-3β,7β,17β-triol
- 3β,7β,17β-Trihydroxy-17α-ethynylandrost-5-ene
- Bezisterim
- HE 3286
- NE 3107
- Triolex
(3S,7R,8R,9S,10R,13S,14S,17R)-17-ethynyl-10,13-dimethyl-1,2,3,4,7,8,9,11,12,14,15,16-dodecahydrocyclopenta[a]phenanthrene-3,7,17-triol
| Formula | C21H30O3 |
|---|---|
| Molar mass | 330.468 g·mol−1 |
Q27286562
(3beta,7beta,17alpha)-Pregn-5-en-20-yne-3,7,17-triol
17.ALPHA.-ETHYNYL-5-ANDROSTENE-3.BETA.,7.BETA.,17.BETA.-TRIOL
PREGN-5-EN-20-YNE-3,7,17-TRIOL, (3.BETA.,7.BETA.,17.ALPHA.)-
- (1R,3aS,3bR,4R,7S,9aR,9bS,11aS)-1-ethynyl-9a,11a-dimethyl-1H,2H,3H,3aH,3bH,4H,6H,7H,8H,9H,9aH,9bH,10H,11H,11aH-cyclopenta(a)phenanthrene-1,4,7-triol
- (1R,3aS,3bR,4R,7S,9aR,9bS,11aS)-1-ethynyl-9a,11a-dimethyl-1H,2H,3H,3aH,3bH,4H,6H,7H,8H,9H,9aH,9bH,10H,11H,11aH-cyclopenta[a]phenanthrene-1,4,7-triol
- 17-ethynyl-5-androstene-3, 7, 17-triol

Bezisterim (developmental code names NE3107, HE3286) is a synthetic analogue of androstenetriol that is believed to have anti-inflammatory and insulin-sensitizing effects in the brain.[1] The compound crosses the blood–brain barrier and does not activate any neurotransmitter receptors.[2] It has been tested as a treatment for Alzheimer’s disease,[3][4][5][6] Parkinson’s disease,[1] and traumatic brain injury.[7] The drug is under development for a variety of conditions and its highest developmental phase is phase 3 for Alzheimer’s disease.[1]
- Originator Hollis-Eden Pharmaceuticals
- Developer BioVie; Harbor Therapeutics; National Institutes of Health (USA); NeurMedix
- Class Anti-inflammatories; Antidementias; Antiepileptic drugs; Antifibrotics; Antiglaucomas; Antihyperglycaemics; Antimigraines; Antineoplastics; Antiparkinsonians; Antirheumatics; Hormones; Insulin sensitisers; Nootropics; Obesity therapies; Small molecules
- Mechanism of Action Adiponectin stimulants; Interleukin 23 inhibitors; Interleukin 6 inhibitors; Mitogen-activated protein kinase 1 inhibitors; Mitogen-activated protein kinase 3 inhibitors; NF-kappa B inhibitors; Tumour necrosis factor inhibitors
- Cystic fibrosis
- Phase III Alzheimer’s disease
- Phase II Parkinson’s disease; Traumatic brain injuries
- Preclinical Multiple myeloma; Prostate cancer
- No development reported Drug-induced dyskinesia
- Discontinued Amyotrophic lateral sclerosis; Cognition disorders; Cystic fibrosis; Epilepsy; Glaucoma; Huntington’s disease; Migraine; Myositis; Optic neuritis; Rheumatoid arthritis; Type 1 diabetes mellitus; Type 2 diabetes mellitus; Ulcerative colitis; Uveitis
28 Feb 2025BioVie plans the phase II ADdRESs-LC trial for Post-acute COVID-19 syndrome in USA (PO, Capsule), in February 2025 (NCT06847191)
- 18 Feb 2025Phase-II clinical trials in Parkinson’s disease (Early-stage disease, In the elderly) in USA (PO) (NCT06757010)
- 03 Jan 2025BioVie plans a phase II SUNRISE-PD trial for Parkinsons disease (Early stage disease) in February 2025 (PO) (NCT06757010)
SCHEME

US20100227841
https://patentscope.wipo.int/search/en/detail.jsf?docId=US43352763&_cid=P11-M9JSD6-84971-1
17α-Ethynylandrost-5-ene-3β,7β,17β-triol was prepared as follows
US20100222315 https://patentscope.wipo.int/search/en/detail.jsf?docId=US43344622&_cid=P11-M9JSIE-88638-1
WO2009149392
PATENT’
WO2009149392
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2009149392&_cid=P11-M9JSL7-90448-1

49] Example 7. Synthesis of 3β-acetoxy-androst-5-ene-17,17-ethylenedioxy: A 300L reactor was charged with 36 kg of triethylorthoformate, 20 kg of 3β-acetoxy-5-androsten-17-one, 12.6 kg of ethylene glycol and 400 g of p-toluenesulfonic acid. The mixture was heated to reflux under nitrogen until the reaction was complete (about 2-3 hours). The mixture was then cooled to 60 0C and 16 kg of anhydrous ethanol and 400 ml of pyridine were added. The resulting solution was transferred to a container and refrigerated overnight. The solids that formed were filtered and washed with 80 kg of 50% ethanol and dried at 40-50 0C to afford 18.5-21.0 kg (81.5-92.5%) of the title compound. [50] Example 8. Synthesis of 3β-acetoxy-androst-5-en-7-one-17,17-ethylenedioxy: A 500 L reactor was charged with 200 kg ethyl acetate and 25 kg of 3β-acetoxy-androst-5-en-17,17-ethylenedioxy. The mixture was stirred for 30 minutes whereupon 55 kg of 70% t-butyl peroxide and 9 kg of sodium bicarbonate were added. The reaction mixture was then cooled to 0 0C and 116 kg of 13% sodium perchlorate (aq.) was added over 10 hours so that a reaction temperature below 5 0C and pH between 7.5 and 8.5 were maintained. After the reaction was complete, the organic layer was separated and the aqueous phase was extracted with ethyl acetate (35 kg x 2). The combined organic phase was combined with a solution 33 kg of sodium sulfite in 167 kg of water, and the resulting mixture was stirred at 40 0C for 3 hours. The organic phase was washed with 50 kg of brine and concentrated to 55-60 kg whereupon 50 kg of methanol was added. After refrigeration overnight, a white solid was formed that was filtered and washed with 10 kg of methanol, and dried at 40-50 0C to yield 7.1-7.8 kg (27.4-30.1%) of the title compound.
[51] Example 9. Synthesis of 3β-acetoxy-androst-5-ene-17,17-ethylenedioxy-7β-ol. A 500 L reactor was charged with 48 kg of THF, 10 kg of 3β-acetoxy-androst-5-en-7-one-17,17-ethylenedioxy and a solution of 9.6 kg CeCI3-7H2O in 95 kg methanol. This mixture was cooled to 0 0C whereupon 2.0 kg of NaBH4 was added in batches over 3 hours in order to maintain the temperature below 5 0C. After stirring for 30 more minutes, 28 kg of acetone was added slowly in order to maintain the temperature below 5 0C, with stirring continued for another 30 minutes. To the mixture was added 240 kg water with stirring continued for 1 hour. The organic solvents were removed under vacuum and the residue was extracted with ethyl acetate (100 kg + 50 kg). The combined organic phase was washed with brine. Solvent was then removed to provide 8.6-8.9 kg (85.1-88.1 %) of the title compound. [52] Example 10. Synthesis of 3β-acetoxy-androst-5-en-17-one-7β-ol: A 500 L reactor was charged with 315 kg of acetone and 18 kg of 3β-acetoxy-androst-5-en-17,17-ethylenedioxy-7β-ol. The mixture was cooled to 5 0C and 2.34 kg of p-toluenesulfonic acid was added slowly to maintain the temperature below 10 0C. After stirring the mixture at 8-15 0C for 36-48 hours, 3.0 kg of sodium bicarbonate was added with stirring continued for 1 hour. Acetone was removed under vacuum, and to the residue was added 100 kg of water. The mixture was placed in a refrigerator overnight to give a white precipitate which was filtered to provide 33 kg (wet) of the title compound.
[53] Example 11. Synthesis of androst-5-en-17-one-3β,7β-diol: A 500 L reactor was charged 230 kg methanol, 33 kg (wet) 3β-acetoxy-7β-hydroxy-5-androsten-17-one, 108 kg water and 15 kg NaaCOβ. The mixture was heated to reflux for 3 hours. Methanol was removed under vacuum whereupon 250 kg of water was added to the residue. The mixture was put in refrigerator overnight to give a precipitate. The solids were collected by filtration, then washed with water and dried at 40-50 0C to yield 9.5-10.5 kg (67.9-75.0%) of the title compound as a white solid.
[54] Example 12. Purification of androst-5-en-17-one-3β,7β-diol: A 500 L reactor was charged with 20 kg crude 3β, 7β-dihydroxyandrost-5-en-17-one and 200 kg methanol and heated until all the solid dissolved. The solution was filtered while hot and after the filtrate cooled a white crystalline solid formed. The solids were collected by filtration, washed with small amount of methanol and dried at 40-50 0C. The solid was then refluxed in 50 kg of ethyl acetate for 20 minutes. After cooling the solid was filtered and dried at 40-50 0C under vacuum to provide 15.2 kg (76%) of purified title compound.
[55] Example 13. Synthesis of 3β,7β-bis-(trimethylsiloxy)-5-androsten-17-one: A mixture of 14.87 Kg of androst-5-en-17-one-3β,7β-diol, 23.8 Kg HMDS and 0.7 Kg saccharin catalyst in 100 L acetonitrile was heated to reflux for 8 hours with stirring under a nitrogen atmosphere. Liberated ammonia was purged under slight vacuum. The reaction volume was then reduced by distillation to collect 3OL of distillate (requires about 2 h). The reaction volume was further reduced to half of the original reaction volume by distillation under reduced pressure (700 mmHg), which requires about 2h of heating at 50 0C. The resulting uniform thick slurry is cooled to 5 0C (requires about 3 h), with additional acetonitrile added to maintain a minimum mixing volume, and held at that temperature for 1. The precipitated product was collected by filtration and dried at 45-50 0C under vacuum (29 mmHg) to a loss on drying (LOD) of not more than 1 % (requires 20 h) to provide 16 Kg (81 % yield) of the title compound (95% purity). [56] Example 14. Synthesis of 17α-ethynyl-5-androstene-3β,7β,17β-triol: To 11.02 Kg TMS-acetylene in 56.5 L tetrahydrofuran (THF) at -27 0C under a nitrogen atmosphere was added 8.51 L 10M n-BuLi. The n-butyl lithium was added very slowly to maintain a temperature at -7 to -27 0C (requires about 2 h) and the resulting reaction was stirred 10 min. at approximately 0°C to produce TMS-lithium-acetylide. To the TMS-lithium-acetylide solution was added a solution of 25.41 Kg of 3β,7β-bis-(trimethylsiloxy)-5-androsten-17-one in 95.3 L THF filtered through a 25 μm filter while allowing the reaction temperature to rise to 20-25 0C. After addition was completed, the reaction temperature was increased to 40-45 0C. To quench the reactor contents, 31.8 L of methanol was added over a period of about 1 h followed by 3.81 Kg KOH in 18.4 L of water giving a final reactor temperature of 50 0C. Liberated acetylene is purged under slight vacuum. The reactor contents were then concentrated by distillation at 80 0C for 1 h then under vacuum (175 mmHg) at about 70 0C (with an initial temperature of 25 0C to avoid bumping) to half of the original pot volume. The residue was cooled to about 10 0C and 35.0 Kg of deionized water was added, followed by 16.4 Kg 12N HCI while maintaining a pot temperature of about 10 0C and giving a final pH of 1. Additional 26.0 kg deionized water was added and the resulting mixture was stirred at about 5 0C for 1 h. The resulting slurry was filtered and washed with 75/25 mixture of methanol/water (16.9 L methanol, 5.6 L water). The collected solids were dried under vacuum (28 in Hg) at 45 0C for 12h for a loss on drying of no more than 0.5% to provide 9.6 Kg of the title compound (83% yield).
[57] Example 15. Recrystallization of 17α-ethynyl-5-androstene-3β,7β,17β-triol: Crude 9.6 Kg 17α-ethynyl-5-androstene-3β,7β,17β-triol prepared in
Example 14 was dissolved in refluxing 50/50 methanol/water (4.2 Kg methanol and 5.4 Kg water). To the solution was added 33.4 Kg methanol followed by 37.6 Kg of THF. The mixture was heated to reflux and stirring was continued until all solids have dissolved, whereupon 99.8 Kg of deionized water was added while maintaining a reactor temperature of 60-75 0C. The mixture was cooled to 0-5 0C over a period of 2 h and maintain at that temperature for 1 h while stirring was continued. The solids were recovered by filtration, washed with 9.6 Kg cold 50/50 methanol water and dried under vacuum (28 in Hg) at 50 0C for 8 h to provide 8.2 Kg of 17α-ethynyl-5-androstene-3β,7β,17β-triol. This first recrystallization is used to remove trace colored impurities from the initial product. A second recrystallization was conducted by heating the solid from the first recrystallization in ~10:1 methanohwater (145.8 Kg methanol and 18.2 Kg of water) to 80°C until all the solids have dissolved. The solution at 55-60 0C was filtered through a 25 μm filter to remove particulate impurities, whereupon 2.5 Kg of methanol at 55-60 0C (used to rinse the reactor) was added. Vacuum distillation at 125 mmHg at 70 0C was conducted until 0.9 to 1.2 times the volume of methanol that was added to the reactor was collected as distillate with water added as necessary to permit stirring (about 120-160 Kg water added). Final reaction volume was 200-225 L. The reactor mixture was cooled to 0-5 0C and maintained at that temperature for 1 h. The resulting slurry was filtered and the filter cake rinsed with 10 Kg deionized water and dried under vacuum (28 in Hg) at 50 0C for 12 h to a residual water content of less than 0.5%. This isolation procedure was used to reduce the THF content in the final product. The yield was 8.0 Kg of recrystallized title compound (83% yield).

[59] Example 16. Synthesis of 3β-acetoxy-androst-5-en-7-on-17-oxime: 3β-Acetoxy-androst-5-en-7,17-dione (45 g, 130 mmol) was dissolved in 800 ml_ methanol, 200 ml_ dichloromethane and 14.5g Et3N (144 mmol). To the solution at RT was added a solution of 10 g of hydroxylamine hydrochloride dissolved in 200 ml_ methanol. After stirring overnight, 200 ml_ of water was added followed by removal of volatile organics by evaporation under reduced pressure. To the resulting residue was added an additional 1 L of water to give a while solid that was filtered and washed well with water. Obtained was 45 g of crude title oxime in 95% purity by 1H-NMR, which was used in the next step without further purification.
[60] Example 17. Synthesis of 3β-acetoxy-androst-5-en-17-oxime-7β-ol: To a solution of 44 g of 3β-acetoxy-androst-5-en-7-on-17-oxime (100 mol%) in 800 ml_ methanol and 200 ml_ tetrahydrofuran was added 50 g of cerium chloride heptahydrate (110 mol%) in 20 ml_ of methanol. The resulting mixture was stirred until the solids were completely dissolved. To the solution cooled to about -5 0C was added 7 g sodium borohydride over 30 min. After stirring an additional 1.5 h at -5 0C, the reaction mixture was quenched with acetone (100 mL) and then allowed to warm to room temperature over a 30 min. period. The quenched reaction mixture was concentrated under vacuum to remove volatile organics. To the residue was added 800 mL of water followed by extraction with ethyl acetate (3 x 500 mL). The combined organic extracts were washed with brine, dried over Na2SO4, then concentrated to provide 42 g of the title compound as a white foam, which was used in the next step without further purification.. [61] Example 18. 3β-acetoxy-androst-5-en-17-one-7β-ol: To a solution of 42 g of 3β-acetoxy-androst-5-en-17-oxime-7β-ol (100 mol%) in 200 mL of ethanol was added 100 mL of water followed by 80 g (400 mol%) of sodium dithionite. The reaction was heated at 55 0C and stirred 16 h. After cooling, the reaction was concentrated under reduced pressure. The residue was diluted with 100 mL of water, and the resulting solid was collected by filtration and redissolved in 1 L dichloromethane. To the DCM solution was added 1 g activated carbon. After stirring overnight the mixture was filtered, and the resulting filtrate was washed with water, dried and concentrated to provide 25 g of crude product. Recrystallization from ethyl acetate gave 22g of the title compound. [62] Example 19. Estrogen receptor binding assay: A suitable example system is an estrogen receptor- kit manufactured by PanVera for ERβ, which contains recombinant estrogen receptor β ligand, FLUORMONE™ ES2 (ES2), a fluorescently labeled estrogen ligand, and appropriate buffer. The system was used in a fluorescence polarization competition assay in which a test article, such as a preparation of Compound 1 or a positive control displaces ES2 from its binding site. When bound to ERβ, ES2 tumbles slowly and has a high fluorescence polarization value. Unbound ES2 tumbles quickly and displays a low fluorescence polarization value. The change in polarization value in the presence of test compound then determines relative binding affinity of that test compound for ERβ as expressed by its IC50, which is the concentration of test compound that results in half-maximum shift in polarization. From IC50, K/ was calculated using the Cheng-Prusoff equation [Biochem. Pharmacol. 22: 3099-3108, (1973)]: K, = IC50Z(I + D/Kd) where D is the concentration of ES2 and Kd is the dissociation constant for binding of ES2 to ERβ (Kd = 4 ± 2 nM).
[63] The competition assay was conducted according to the manufacturer’s protocol (Lit. No. L0712, Rev. 10/03). Assay reagents used were bacculovirus expressed, full length human ERβ 4.5 pmol/μL in 50 mM Bis-Tris Propane (pH = 9), 400 mM KCI, 2 mM DTT, 1 mM EDTA, 10% glycerol, ES2 400 nM in methanol and E2 screening buffer consisting of 100 mM potassium phosphate (pH = 7.4), 100 μg/mL BGG, 0.02% NaN3. The ES2-ERβ complex was formed with 20 μL 20 nM ERβ (0.020 pmol/μL) and 20 μl_ 2 nM ES2 (0.002 pmol/μL). Positive control (estrogen) solution was prepared using 20 μL of a 1.0 mM stock solution in DMSO and 80 μL DMSO. In a first dilution, 50 μL of this solution is added to 50 μL of DMSO, which is followed by dilutions in 2-fold increments, to provide for a 14 point dilution curve. In a second dilution, to 4 μL of each DMSO solution from the first dilution is added 400 μL of ES2 screening buffer. To 20 μL of test compound, serially diluted in the manner described immediately above, in a 384 well black flat bottom microtiter plate, was added 20 μL of the ES2-ERβ complex (0.5% final DMSO concentration) followed by incubation in the dark at 20-30 0C for 1-4 h. Test compound was treated similarly except the starting concentration was 10 mM. Fluorescence polarization values are obtained using 485 nm excitation and 530 nm emission interference filters. Binding assay for ERa was conducted as for ERβ except bacculovirus expressed, full length human 2.8 pmol/μL ERa was used as reagent with the ERα-ES2 complex formed from 20 μL 30 nM (0.030 pmol/μL) and 20 μL 2 nM ES2 (0.002 pmol/μL). [64] Example 20. AR, GR and PR receptor binding assays. The AR competition assay was conducted according to the manufacturer’s protocol (Lit. No. L0844, Rev. 05/02) in the manner described for ERβ with the following exceptions. Reagents used were recombinant rat androgen receptor ligand binding domain tagged with His and GST [AR-LBD (His-GST)] 0.38 pmol/μL in buffer containing protein stabilizing agents and glycerol (pH = 7.5), 200 nM FLUORMONE™ AL Green, which is a fluorescently labeled androgen ligand, in 20 mM Tris, 90% methanol and AR screening buffer containing stabilizing agents and glycerol (pH = 7.5) with 2 μL of 1 mM DTT added per mL screening buffer (AR screening buffer 2 mM in added DTT) was used as the reagents. The AL Green-AR complex was formed with 20 μL 50 nM AR (0.050 pmol/μL) and 20 μL 2 nM AL Green (0.002 pmol/μL). K, was calculated using, for the dissociation constant for binding of the fluorophore to receptor, Kd = 20 ± 10 nM. [65] The PR competition assay was conducted according to the manufacturer’s protocol (Lit. No. L0503, Rev. 06/03) in the manner described for ERβ with the following exceptions. Reagents used were recombinant human progesterone receptor ligand binding domain tagged with GST [PR-LBD (GST)] 3.6 pmol/μL in 50 mM Tris (pH = 8.0), 500 mM KCI, 1 M urea, 5 mM DTT, 1 mM EDTA and 50% glycerol, 400 nM FLUORMONE™ PL Green, which is a fluorescently labeled progesterone ligand, in 20 mM Tris 90% methanol (pH = 6.8) and PR screening buffer containing protein stabilizing agents and glycerol (pH = 7.4) with 4 μL of 1 mM DTT added per mL screening buffer (PR screening buffer 4 mM in added DTT). The PL Green-PR complex was formed with 20 μL 80 nM PR (0.080 pmol/μL) and 20 μL 4 nM PL Green (0.004 pmol/μL). K, was calculated using, for the dissociation constant for binding of the fluorophore to receptor, Kd = 40 nM.
[66] The GR competition assay was conducted according to the manufacturer’s protocol (Lit. No. L0304, Rev. 12/01) in the manner described for ERβ with the following exceptions. Reagents used were recombinant full length human glucocorticoid receptor 0.240 pmol/μL in 10 mM phosphate buffer (pH = 7.4), 200 mM Na2MoO4, 0.1 mM EDTA, 5 mM DTT and 10% glycerol, 200 nM FLUORMONE™ GS1 , which is a fluorescently labeled glucocorticoid ligand, in 75% methanol, and GR screening buffer containing 100 mM potassium phosphate (pH = 7.4), 200 mM Na2MoO4, 1 mM EDTA, 20% DMSO with 5 μL of 1 mM DTT per mL screening buffer added (GR screening buffer 5 mM in added DTT), 1 mM GR stabilizing peptide, which is a co-activator related peptide [see Chang, CY. MoI. Cell Biol. 19: 8226-36 (1999)] in 10 mM phosphate buffer (pH = 7.4) and 1 M DTT in water were used as the reagents. To 2.5 mL of the GR screening buffer is added 2.5 mL GR stabilizing peptide solution and 125 μL of 1 M DTT to form the GR stabilizing peptide-glucocorticoid receptor complex. Order of addition to the microtiter plate was 20 μL test compound in 1 % DMSO, 10 μL of 16 nM GR (0.016 pmol/μL) and finally 10 μL of 4 nM GS1 , followed by incubation in the dark at 20-30 0C for 4 h (total experiment time should not exceed 7 h). K, was calculated using, for the dissociation constant for binding of the fluorophore to receptor, Kd = 0.3 ± 0.1 nM.
[67] Example 21. Impurity profiling of 17α-ethynyl-5-androstene-3β,7β,17β- triol (Compound 1) preparations.
[68] Process A: HPLC conditions for Impurity profiling of Compound 1 preparations form Process B are give in Table 1.
[69]
Table 1. HPLC Conditions for Impurity Profiling of Compound 1 Preparations form Process A
PATENT
Hollis-Eden Pharmaceuticals, Inc. WO2008039566
Zhejiang Xianju Junye Pharmaceutical Co., Ltd.; Jiangxi Junye Biopharmaceutical Co., Ltd.CN114478672
Harbor BioSciences, Inc.US20100227841
Harbor BioSciences, Inc. US20100222315 A1
Hollis-Eden Pharmaceuticals, Inc. US20100075937
Neurmedix Inc. US20080153792 A1
Hollis-Eden Pharmaceuticals, Inc.; Harbor Therapeutics, Inc. US20080146532 A1
Harbor Therapeutics, Inc.; Neurmedix, Inc. US20160045516 A1
Harbor Therapeutics, Inc. US8354396 B2
Hollis-Eden Pharmaceuticals, Inc. WO2009149392
| Clinical data | |
|---|---|
| Other names | NE3107; NE-3107; HE3286; HE-3286; 17α-Ethynyl-5-androstene-3β,7β,17β-triol; |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1001100-69-1 |
| PubChem CID | 16739648 |
| DrugBank | DB05212 |
| ChemSpider | 20571043 |
| UNII | PH8858757I |
| KEGG | D12932 |
| ChEMBL | ChEMBL4297284 |
| CompTox Dashboard (EPA) | DTXSID501267252 |
| Chemical and physical data | |
| Formula | C21H30O3 |
| Molar mass | 330.468 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Jump up to:a b c “Bezisterim”. AdisInsight. 5 September 2024. Retrieved 26 September 2024.
- ^ Reading, Chris L; Ahlem, Clarence N; Parameswaran, Narayanan (December 2021). “Rationale for an anti-inflammatory insulin sensitizer in a phase 3 Alzheimer’s disease trial”. Alzheimer’s & Dementia. 17 (S9). doi:10.1002/alz.057438.
- ^ Stoiljkovic, Milan; Horvath, Tamas L.; Hajós, Mihály (July 2021). “Therapy for Alzheimer’s disease: Missing targets and functional markers?”. Ageing Research Reviews. 68: 101318. doi:10.1016/j.arr.2021.101318. PMC 8131215. PMID 33711510.
- ^ Balzano, Tiziano; Esteban-García, Noelia; Blesa, Javier (2 January 2023). “Neuroinflammation, immune response and α-synuclein pathology: how animal models are helping us to connect dots”. Expert Opinion on Drug Discovery. 18 (1): 13–23. doi:10.1080/17460441.2023.2160440. PMID 36538833. S2CID 254959175.
- ^ Liu, Ping; Wang, Yunyun; Sun, Yan; Peng, Guoping (April 2022). “Neuroinflammation as a Potential Therapeutic Target in Alzheimer’s Disease”. Clinical Interventions in Aging. 17: 665–674. doi:10.2147/CIA.S357558. PMC 9064449. PMID 35520949.
- ^ Xi, Yilong; Chen, Yun; Jin, Yi; Han, Guochen; Song, Mingjie; Song, Tingting; Shi, Yang; Tao, Ling; Huang, Zewei; Zhou, Jianping; Ding, Yang; Zhang, Huaqing (May 2022). “Versatile nanomaterials for Alzheimer’s disease: Pathogenesis inspired disease-modifying therapy”. Journal of Controlled Release. 345: 38–61. doi:10.1016/j.jconrel.2022.02.034. PMID 35257810. S2CID 247285338.
- ^ “U.S. Clinical Trial: Neurological Associates of West Los Angeles Listed a New Clinical Trial to Study Insulin-sensitizing NE3107 in Improving Sleep and Fatigue in Subjects With Traumatic Brain Injury.” Contify Life Science News, 1 Aug. 2023, p. NA. Gale OneFile: Health and Medicine, link.gale.com/apps/doc/A759542006/HRCA?u=anon~bb46c85&sid=sitemap&xid=0c315c7e. Accessed 14 Dec. 2023.
/////Bezisterim, HE 3286, NE 3107, Triolex, NE3107, NE-3107, HE3286, HE-3286, PHASE 2
Bexicaserin



Bexicaserin
CAS 2035818-24-5
| Formula | C15H19F2N3O |
|---|---|
| Molar mass | 295.334 g·mol−1 |
(3R)-N-(2,2-difluoroethyl)-3-methyl-1,10-diazatricyclo[6.4.1.04,13]trideca-4,6,8(13)-triene-5-carboxamide
- (3R)-N-(2,2-difluoroethyl)-3-methyl-1,10-diazatricyclo[6.4.1.04,13]trideca-4,6,8(13)-triene-5-carboxamide
- (7R)-N-(2,2-difluoroethyl)-7-methyl-1,2,3,4,6,7-hexahydro[1,4]diazepino[6,7,1-hi]indole-8-carboxamide
- Pyrrolo[3,2,1-jk][1,4]benzodiazepine-8-carboxamide, N-(2,2-difluoroethyl)-1,2,3,4,6,7-hexahydro-7-methyl-, (7R)-
Bexicaserin is under investigation in clinical trial NCT05626634 (Open-label, Long-term Safety Study of LP352 in Subjects With Developmental and Epileptic Encephalopathy).
PATENT
Arena Pharmaceuticals, Inc.WO2023172685
Arena Pharmaceuticals, Inc., WO2016176177
https://patents.google.com/patent/WO2016176177A1/en
Example 1: Syntheses of Compounds of Table A Example 1.1: Preparation of N-(2,2-difluoroethyl)-7-methyl-l,2,3,4,6,7-hexahydro- [l,4]diazepino[6,7,l-hi]indole-8-carboxamide (Compound 1)
Step A: Preparation of methyl 3-formyl-lH-indole-4-carboxylate
2M solution of oxalyl dichloride in dichloromethane (DCM) (1.712 ml, 3.425 mmol) was added to DCM (15 mL) cooled down in an ice-water bath. N,N-dimethylformamide (0.250 g, 3.425 mmol) was added dropwise under nitrogen. The reaction mixture was stirred at 0 °C for 30 min. Then methyl lH-indole-4-carboxylate (0.5 g, 2.854 mmol) in DCM (10 mL) was added. The reaction mixture was warmed to room temperature and stirred for 1 h. The solvent was removed. THF (15 mL) and 20% aqueous ammonium acetate were added. The reaction mixture was stirred under reflux (-70 °C) for 30 min. The reaction mixture was then extracted with ethyl acetate. The combined organics (organic phases) were concentrated; the residue was purified by silica gel column chromatography with 90% ethyl acetate/hexanes to give the title compound (551 mg, 95.0 %) as white solid. LCMS m/z = 204.2 [M+H]+; Ή NMR (400 MHz, CDC13) δ ppm 4.00 (s, 3H), 7.34 (t, / = 7.8 Hz, 1H), 7.63 (dd, / = 8.0 and 1.0 Hz, 1H), 7.87 (dd, / = 7.5 and 1.0 Hz, 1H), 8.10 (d, / = 3.2 Hz, 1H), 9.08 (br s, 1H), 10.53 (s, 1H).
Step B: Preparation of methyl 3-methyl-lH-indole-4-carboxylate
To a stirred solution of methyl 3-formyl-lH-indole-4-carboxylate (551 mg, 2.712 mmol) in DMF (8 mL) was added 4-methylbenzenesulfonohydrazide (0.657 g, 3.525 mmol) followed by p- toluenesulfonic acid monohydrate (77.37 mg, 0.407 mmol) and tetramethylene sulfone (sulfolane, 8 mL). The reaction mixture was stirred at 100 °C for 1 h, cooled to room temperature. Sodium cyanoborohydride (0.682 g, 10.85 mmol) was added portionwise. Then the mixture was stirred at 100 °C for 2 h. The reaction mixture was cooled down, diluted with water, and extracted with 50% ethyl acetate in hexanes. The organics were concentrated; the residue was purified by silica gel column chromatography with 20% ethyl acetate hexanes to give the title compound (355 mg, 69.2 %) as off- white solid. LCMS m/z = 190.4 [M+H]+; Ή NMR (400 MHz, CDC13) δ ppm 2.41 (d, / = 1.0 Hz, 3H), 3.96 (s, 3H), 7.07-7.10 (m, 1H), 7.18 (t, / = 7.8 Hz, 1H), 7.50 (dd, / = 8.0 and 1.0 Hz, 1H), 7.64 (dd, J = 7.5 and 1.0 Hz, 1H), 8.12 (bs, 1H).
Step C: Preparation of methyl 3-methylindoline-4-carboxylate
To a solution of methyl 3-methyl-lH-indole-4-carboxylate (1.253 g, 6.622 mmol) in TFA (trifluoroacetic acid) (4.06 mL) in an ice-water bath was added triethylsilane (4.231 ml, 26.49 mmol) drop wise under N2. The reaction mixture was warmed to room temperature and stirred overnight. The mixture was concentrated and added water. After adjusting pH to 8 with saturated aqueous NaHC03 solution, the mixture was extracted with ethyl acetate. The combined organics were concentrated. The residue was purified by silica gel column chromatography with 25% ethyl acetate/hexanes (column prewashed with 0.1% Et3N/hexanes) to give the title compound (1.013 g, 80.0 %) as orange-red oil. LCMS m/z = 192.2 [M+H]+; Ή NMR (400 MHz, CDC13) δ ppm 1.25 (d, J = 6.9 Hz, 3H), 3.25 (dd, J = 8.6 and 1.7 Hz, 1H), 3.68 (t, / = 8.5 Hz, 1H), 3.83-3.92 (m, 1H), 3.90 (s, 3H), 6.78 (dd, / = 7.8 and 1.0 Hz, 1H), 7.07 (t, / = 7.8 Hz, 1H), 7.34 (dd, / = 7.8 and 1.0 Hz, 1H).
Step D: Preparation of 2-tert-butyl 8-methyl 7-methyl-3,4,6,7-tetrahydro- [l,4]diazepino[6,7,l-hi]indole-2,8(lH)-dicarboxylate
A mixture of methyl 3-methylindoline-4-carboxylate (1.013 g, 5.297 mmol) and 2- bromoethanamine hydrobromide (1.302 g, 6.357 mmol) was heated at 115 °C overnight. The residue was dissolved in methanol and purified by preparative HPLC (5-60% CH3CN/H20 with 0.1% TFA over 30 min). The combined fractions were then concentrated to give methyl l-(2-aminoethyl)-3- methylindoline-4-carboxylate. LCMS m/z = 235.4 [M+H]+; Ή NMR (400 MHz, CDC13) δ ppm 1.24 (d, / = 7.0 Hz, 3H), 2.92-3.00 (m, 3H), 3.25 (dd, / = 8.5 and 1.7 Hz, 1H), 3.30-3.40 (m, 2H), 3.80-3.90 (m, 1H), 3.88 (s, 3H), 6.64 (d, / = 7.7 Hz, 1H), 7.11 (t, / = 7.8 Hz, 1H), 7.27 (dd, / = 7.9 and 0.8 Hz, 1H).
Methyl l-(2-aminoethyl)-3-methylindoline-4-carboxylate obtained above was dissolved in methanol (10 mL), 37% formaldehyde in water (1.183 ml, 15.89 mmol) was added, followed by TFA (1.217 ml, 15.89 mmol). The reaction mixture was heated at 80 °C for lh and concentrated. The residue was dissolved in THF (8 mL), and added saturated aqueous NaHC03 (8 mL) solution and di-tert-butyl dicarbonate (0.776 ml, 5.297 mmol). The reaction mixture was stirred at room temperature overnight, diluted with water, and extracted with ethyl acetate. The combined organics were concentrated. The residue was purified by silica gel column chromatography with 25% ethyl acetate/hexanes to give the title compound (1.212 g, 66.0 %) as colorless oil. LCMS m/z = 347.2 [M+H]+; Ή NMR (400 MHz, CDC13) δ ppm rotamers 1.20 (d, / = 6.9 Hz, 3H), 1.35-1.45 (br, 9H), 2.80-2.95 (m, 1H), 3.08-3.18 (m,lH), 3.24-3.35 (m, 2H), 3.35-3.45 (m, 1H), 3.85-3.95 (m, 1H), 3.86 (s, 3H), 3.97-4.08 (m, 2H), 4.62-4.88 (m, 1H), 6.91-7.06 (m, 1H), 7.36 (d, J = 8.0 Hz, 1H).
Step E: Preparation of 2-(tert-butoxycarbonyl)-7-methyl-l,2,3,4,6,7-hexahydro- [l,4]diazepino[6,7,l-hi]indole-8-carboxylic acid
To a solution of 2-tert-butyl 8-methyl 7-methyl-3,4,6,7-tetrahydro-[l,4]diazepino[6,7,l- hi]indole-2,8(lH)-dicarboxylate (1.212 g, 3.499 mmol) in dioxane (10 mL) was added a 1M solution of lithium hydroxide in water (13.99 ml, 13.99 mmol). The reaction mixture was stirred at 80 °C for 2 h. Organic solvent was evaporated. The residue was diluted with water, adjusted pH to 3-4 with aqueous 5% citric acid. The off-white precipitate was collected and dried to give the title compound (1.116 g, 96.0 %) as off-white solid. LCMS m/z = 333.4 [M+H]+.
Step F: Preparation of N-(2,2-difluoroethyl)-7-methyl-l,2,3,4,6,7-hexahydro- [l,4]diazepino[6,7,l-hi]indole-8-carboxamide
To the solution of 2-(tert-butoxycarbonyl)-7-methyl- 1,2, 3,4,6, 7-hexahydro- [l,4]diazepino[6,7,l-hi]indole-8-carboxylic acid (25 mg, 75.21 μιηοΐ), HATU (42.87 mg, 0.113 mmol) and triethylamine (20.97 μί, 0.150 mmol) in DMF (2 mL) was added 2,2-difluoroethanamine (9.146 mg, 0.113 mmol). The reaction was stirred at room temperature overnight. The mixture was purified by semi preparative HPLC (15-85% CH3CN/H20 with 0.1% TFA over 30 min). The combined fractions were lyophilized to give tert-butyl 8-((2,2-difluoroethyl)carbamoyl)-7-methyl-3,4,6,7-tetrahydro- [l,4]diazepino[6,7,l-hi]indole-2(lH)-carboxylate, which was dissolved in dioxane (0.5 mL). A solution of 4M HC1 in dioxane (0.5 mL) was added. The reaction mixture was stirred at room temperature for 4 h and concentrated. The residue was purified by semi preparative HPLC (5-60% CH3CN/H20 with 0.1% TFA over 30 min). The combined fractions were lyophilized to give the title compound as TFA salt (17 mg, 55.2 %). LCMS m z = 296.2 [M+H]+; Ή NMR (400 MHz, CD3OD) δ ppm 1.18 (d, / = 6.9 Hz, 3H), 3.10-3.20 (m, 1H), 3.26-3.40 (m, 2H), 3.40-3.64 (m, 3H), 3.65-3.85 (m, 3H), 4.21 (d, / = 14.9 Hz, 1H), 4.40 (d, J = 14.9 Hz, 1H), 6.00 (tt, J = 56.0 and 3.9 Hz, 1H), 6.99 (d, J = 7.8 Hz, 1H), 7.12 (d, / = 7.9 Hz, 1H).
Example 1.2: Preparation of (S)- N-(2,2-difluoroethyl)-7-methyl-l,2,3,4,6,7-hexahydro- [l,4]diazepino[6,7,l-hi]indole-8-carboxamide (Compound 2) and (R)- N-(2,2-difluoroethyl)-7- methyl-l,2,3,4,6,7-hexahydro-[l,4]diazepino[6,7,l-hi]indole-8-carboxamide (Compound 3)
Enantiomers of N-(2,2-difluoroethyl)-7-methyl- 1,2,3, 4,6, 7-hexahydro-[l, 4]diazepino[6,7,l-hi]indole-8- carboxamide were obtained by chiral HPLC separation using following conditions. Column: Chiralpak IC column 250 x 20 mm (L x I.D.)
Flow: 12 mL/min
Eluent: 12 % ethanol/8 % mTBE/80 % hexanes with 0.1 % Et3N
Detector: UV 254 nm
Retention time: 1st eluting enantiomer 22.0 min, 2nd eluting enantiomer 23.5 min
After separation, both enantiomers were further purified by semi preparative HPLC (5-60%
CH3CN/H20 with 0.1% TFA (trifluoroacetic acid) over 30 min). The combined fractions were lyophilized to give the title compounds as the TFA salt.
SCHEME

Bexicaserin (INNTooltip International Nonproprietary Name; developmental code names LP352 and AN352) is a selective serotonin 5-HT2C receptor agonist which is under development for the treatment of seizures in developmental disabilities such as Dravet syndrome and Lennox-Gastaut syndrome.[1][3][2] It is taken by mouth.[2][1]
The drug is highly selective for the serotonin 5-HT2C receptor, with negligible affinity for the serotonin 5-HT2A and 5-HT2B receptors.[2] Because it does not activate the serotonin 5-HT2B receptor, bexicaserin is not expected to pose a risk of cardiac valvulopathy, unlike the existing agent fenfluramine.[2]
As of October 2024, bexicaserin is in phase 3 clinical trials for treatment of developmental disabilities.[1][3] It is being developed by Longboard Pharmaceuticals.[1][3]
The activation of 5HT2c receptors has been shown to reduce epileptic seizure activity by inhibiting CaV3 calcium channels which mediate the T-type calcium current.[4] CaV3 calcium channels facilitate high frequency burst firing in princible neurons of the subiculum. This firing pattern is upregulated following status epilepticus, with these hyperactive neurons often serving as the initiation point for seizures.[5][6][7]
References
- ^ Jump up to:a b c d e f “Bexicaserin – Longboard Pharmaceuticals”. AdisInsight. 16 October 2024. Retrieved 29 October 2024.
- ^ Jump up to:a b c d e f Dell’isola GB, Verrotti A, Sciaccaluga M, Roberti R, Parnetti L, Russo E, et al. (June 2024). “Evaluating bexicaserin for the treatment of developmental epileptic encephalopathies”. Expert Opinion on Pharmacotherapy. 25 (9): 1121–1130. doi:10.1080/14656566.2024.2373350. PMID 38916481.
- ^ Jump up to:a b c “Delving into the Latest Updates on Bexicaserin with Synapse”. Synapse. 28 October 2024. Retrieved 29 October 2024.
- ^ Petersen AV, Jensen CS, Crépel V, Falkerslev M, Perrier JF (2017). “Serotonin Regulates the Firing of Principal Cells of the Subiculum by Inhibiting a T-type Ca2+ Current”. Frontiers in Cellular Neuroscience. 11: 60. doi:10.3389/fncel.2017.00060. PMC 5339341. PMID 28326015.
- ^ Menendez de la Prida L, Gal B (June 2004). “Synaptic contributions to focal and widespread spatiotemporal dynamics in the isolated rat subiculum in vitro”. The Journal of Neuroscience. 24 (24): 5525–36. doi:10.1523/JNEUROSCI.0309-04.2004. PMC 6729319. PMID 15201325.
- ^ Su H, Sochivko D, Becker A, Chen J, Jiang Y, Yaari Y, et al. (May 2002). “Upregulation of a T-type Ca2+ channel causes a long-lasting modification of neuronal firing mode after status epilepticus”. The Journal of Neuroscience. 22 (9): 3645–55. doi:10.1523/JNEUROSCI.22-09-03645.2002. PMC 6758371. PMID 11978840.
- ^ Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002 Nov 15;298(5597):1418-21. doi: 10.1126/science.1076510. PMID 12434059.
| Clinical data | |
|---|---|
| Other names | LP352; LP-352; AN352; AN-352 |
| Routes of administration | Oral[1] |
| Drug class | Serotonin 5-HT2C receptor agonist[1][2] |
| Pharmacokinetic data | |
| Elimination half-life | 5–7 hours[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2035818-24-5 |
| PubChem CID | 122662787 |
| DrugBank | DB18885 |
| ChemSpider | 129309383 |
| UNII | R8XR1D6SCB |
| KEGG | D13035 |
| ChEMBL | ChEMBL5314507 |
| Chemical and physical data | |
| Formula | C15H19F2N3O |
| Molar mass | 295.334 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////Bexicaserin, PHASE 2, LP 352, LP-352, AN 352, AN-352

{kind=link}
{kind=link}
{kind=link}
{kind=link}