
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Daclizumab
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from mouse) |
| Target | CD25 |
 DACLIZUMAB,
DACLIZUMAB,

Daclizumab is a humanized monoclonal antibody indicated in the United States for prophylaxis of acute organ rejection in patients receiving renal transplants.
It was marketed as Zenepax, but discontinued by Roche in 2009 due to diminishing market demand for that indication. Biogen Idec is currently conducting phase III trials for daclizumab in MS. A phase III trial started in March 2010 is being conducted to determine efficacy of preventing MS relapse.
Study dosing of daclizumab is 150 mg subcutaneously once every 4 weeks versus interferon beta-1a (Avonex) 30 mg intramuscularly given once weekly for 96 to 144 weeks.
Daclizumab (Zenapax®) (molecular wt = 144 kd.) is a humanized monoclonal antibody (IgG1) produced by recombinant DNA technology. It gained FDA approval in Dec 1997. It is known by several other names including HAT (Humanized Anti-Tac), SMART anti-Tac, anti-CD25, and humanized anti-IL2-receptor. It was developed and patented by Protein Design Laboratories (Mountain View, CA) and it is marketed by Hoffman LaRoche (Nutley, NJ ).
Daclizumab is a composite of human (90%) and murine (10%) antibody sequences. In the model below, the murine portions are shown in red and dark blue; the rest of the molecule (gray color) represents the human sequence
The study is aiming for enrollment of 1500 patients and is expected to be complete in January 2014.
more info
Daclizumab (trade name Zenapax) is a therapeutic humanized monoclonal antibody. It is used to prevent rejection in organ transplantation, especially in kidney transplants. The drug is also under investigation for the treatment of multiple sclerosis.
Daclizumab works by binding to CD25, the alpha subunit of the IL-2 receptor of T cells. The drug is marketed in the US, but not in Europe.
Uses
Prevention of organ transplants
Daclizumab is given in multiple doses, the first 1 hour before the transplant operation and 5 further doses given at two week intervals after the transplant. These saturate the receptors and prevent T cell activation and thus prevent formation of antibodiesagainst the transplant.
Like the similar drug basiliximab, daclizumab reduces the incidence and severity of acute rejection in kidney transplantation without increasing the incidence of opportunistic infections.
Daclizumab usage may also be indicated in place of a calcineurin-inhibitor (ciclosporin or tacrolimus) during the early phase after kidney transplantation, when the kidney is recovering and vulnerable to calcineurin-inhibitor toxicity. This has been shown to be beneficial in non-heart beating donor kidney transplantation.
In the United Kingdom, the National Institute for Health and Clinical Excellence (NICE) has recommended its use be considered for all kidney transplant recipients.[citation needed]
Multiple sclerosis
In 2006 it began a Phase II clinical trial that finished in 2007 as a possible multiple sclerosis (MS) treatment. Participants were nine patients with multiple sclerosis not controlled with interferon. Daclizumab was effective in reducing lesions and improving clinical scores.[1] As of June 2013, the drug is in Phase III trials for this indication.[2]
Autoimmune diseases
Daclizumab has also been used to slow the progression of autoimmune diseases, particularly that of birdshot chorioretinopathy.[3]
Common side effects with a frequency of at least 10% include sleeplessness, tremor, headache, arterial hypertension, dyspnoea, gastrointestinal side effects and oedema. In rare cases, the drug can cause severe anaphylaxis.[4]
Daclizumab must not be administered to lactating women.[4]
History
Daclizumab was developed by PDL Biopharma, building on research at the National Institutes of Health (NIH).[5] Since December 1997, it is marketed by Hoffmann-La Roche in the US.
In April 2008, Hoffmann-La Roche submitted an application to have its marketing authorisation withdrawn in the EU for commercial reasons. The drug faced diminishing market demand, according to the company. There were no safety concerns with its use. As of January 2009, its marketing authorisation has been withdrawn and the product discontinued completely.[6][7]
- Rose JW, Burns JB, Bjorklund J, Klein J, Watt HE, Carlson NG (2007). “Daclizumab phase II trial in relapsing and remitting multiple sclerosis: MRI and clinical results”.Neurology 69 (8): 785–789. doi:10.1212/01.wnl.0000267662.41734.1f.PMID 17709711.
- ClinicalTrials.gov NCT01462318 An Immunogenicity and Pharmacokinetics (PK) Study of DAC HYP Prefilled Syringe in Relapsing Remitting Multiple Sclerosis (RRMS) (OBSERVE)
- Sobrin L, Huang JJ, Christen W, Kafkala C, Choopong P, Foster CS (2008). “Daclizumab for treatment of birdshot chorioretinopathy”. Arch Ophthalmol. 126 (2): 186–191. doi:10.1001/archophthalmol.2007.49. PMID 18268208.
- “EPAR for Zenapax”. European Medicines Agency. 2007.
- Tsurushita, N.; Hinton, P. R.; Kumar, S. (2005). “Design of humanized antibodies: From anti-Tac to Zenapax”. Methods 36 (1): 69–83.doi:10.1016/j.ymeth.2005.01.007. PMID 15848076. edit
- British National Formulary, Edition 57
- EMEA: Withdrawal of the marketing authorisation in the European Union
Ipilimumab – Yervoy Battles Melanoma, but Can It Become a Blockbuster?
![]()
Ipilimumab
by Todd Campbell, The Motley Fool Sep 28th 2013 1:00PM
Updated Sep 28th 2013 1:02PM
In early 2011, the Food and Drug Administration approved Bristol-Myers Squibb‘s drug Yervoy as a treatment for skin cancer melanoma. The drug marked the first approved treatment proven to extend the life of a person diagnosed with the disease. It marked a big leap forward in medicine as an early leader in immunotherapy, or the unleashing of the body’s immune system on cancer.
read all at
http://www.dailyfinance.com/2013/09/28/yervoy-battles-melanoma-but-can-it-become-a-blockb/

Ipilimumab’s molecular target is CTLA-4 (Uniprot: P16410; canSAR ; PFAM: P16410), a negative regulator of T-cell activation. Ipilimumab augments T-cell activation and proliferation by binding to CTLA-4 and preventing its interaction with its ligands (CD80 and CD86). CTLA-4 is a membrane-bound, 223 amino acid long, T-cell protein. It contains an immunoglobulin V-type domain (PFAM:PF07686). The structure of CTLA-4 is determined (see e.g. PDBe:3osk)
Ipilimumab (i pi lim′ ue mab; also known as MDX-010 and MDX-101), marketed asYervoy, is a drug used for the treatment of melanoma, a type of skin cancer. It is a U.S. Food and Drug Administration (FDA) approved human monoclonal antibody developed byBristol-Myers Squibb, and works by activating the immune system by targeting CTLA-4.
Cytotoxic T lymphocytes (CTLs) can recognize and destroy cancer cells. However, there is also an inhibitory mechanism that interrupts this destruction. Ipilimumab turns off this inhibitory mechanism and allows CTLs to continue to destroy cancer cells.
In addition to melanoma, ipilimumab is undergoing clinical trials for the treatment of non-small cell lung carcinoma (NSCLC), small cell lung cancer (SCLC) and metastatic hormone-refractory prostate cancer.
Yervoy is a monoclonal antibody drug indicated for treating metastatic melanoma. The drug was developed by Bristol-Myers Squibb.
In March 2011, The US Food and Drug Administration (FDA) approved Yervoy to treat patients with newly diagnosed or previously-treated unresectable or metastatic melanoma. Yervoy is the first drug approved vor the treatment of metastatic melanoma in the US.
Bristol-Myers Squibb submitted a marketing authorisation application to the European Medicines Agency in May 2010. The drug received approval from the European Commission in July 2011.
Approval from Australia’s Therapeutic Goods Association was received in July 2011. The drug is currently being reviewed by Health Canada.
Metastatic melanoma
Melanoma responsible for majority of skin cancer deaths in the US. In metastatic melanoma the cancer spreads to other parts of the body from its starting point. It becomes difficult to treat the disease once it spreads beyond the skin to other parts of the body. The disease is also known as stage IV melanoma.
If the melanoma spreads to the lungs then the patient faces breathing problems. The patients with metastatic melanoma may feel symptoms of fatigue, loss of weight, and appetite and bowel problems.
The incidence of the disease has increased steadily in the US after 1970s. The American Cancer Society (ACS) estimated that more than 68,000 new cases of melanoma were registered in the US in 2009. The ACS estimated that the number of deaths occurred due to melanoma in 2010 was more than 8,700.
Yervoy mechanism
Yervoy treats metastatic melanoma by activating the immune system. The drug works by binding or inhibiting cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), a molecule that plays vital role in relating natural immune responses. The presence or absence of CTLA-4 can curb or increase the immune system’s T-cell response in fighting disease.
The drug also works by blocking a complex set of interactions in the immune system. It is designed to inhibit the activity of CTLA-4, thereby sustaining an active immune response in its attack on cancer cells.
Approvals and indications
Ipilimumab was approved by the FDA in March 2011 to treat patients with late-stage melanoma that has spread or cannot be removed by surgery. On February 1, 2012, Health Canada approved ipilimumab for “treatment of unresectable or metastatic melanoma in patients who have failed or do not tolerate other systemic therapy for advanced disease.” Additionally Ipilimumab was approved in the European Union (EU), for second line treatment of metastatic melanoma, November 2012

Vitamin A
, Arctic National Wildlife Ref...")
Vitamin A is actually a group of compounds that include retinol and beta-carotene among others. The science behind it dates back to 1816 when scientists noticed nutrition deprived dogs developed corneal ulcers. It was later found to be a fat soluble vitamin (as opposed to water-soluble) meaning it is stored in the fat (along with vitamins D, E, and K).
Vitamin A is indeed important in maintaining good vision. It is also important for cell growth and differentiation, skin health, and the immune system. For women the recommended daily allowance (RDA) is 700 mcg and 900 for men. The widely considered upper safe limit is considered to be 3000 mcg.
Animal sources of vitamin A include cheddar cheese, eggs, butter, and liver. Plant source include carrots, broccoli, kale and spinach. Dandelion greens have a particularly high amount of vitamin A and are used in natural medicine.
Worldwide Vitamin A deficiency is a huge problem. Approximately…
View original post 1,116 more words
ONE LAKH VIEWS ON ALL BLOGS—DR ANTHONY CRASTO

DR ANTHONY MELVIN CRASTO Ph.D
WORLDDRUGTRACKER
ANNOUNCING ONE LAKH PLUS VIEWS ON ALL BLOGS- DR ANTHONY CRASTO
- Eurekamoments in Organic Chemistry
- NEW DRUG APPROVALS
- WORLD DRUG TRACKER
- Green Chemistry International
- drug regulatory affairs international
- ORGANIC SPECTROSCOPY INTERNATIONAL
- ORGANIC SYNTHESIS INTERNATIONAL
- ALL ABOUT DRUGS
- ORGANIC CHEMISTRY INTERNATIONAL
- Drug Scaleup and Manufacturing International
SEE ALSO
- Organic Chemistry by Dr Anthony
- Technology Transfer
- Stereochemistry
- Spectroscopy
- Polymorphism
- Reactions in Org Chem
DR ANTHONY MELVIN CRASTO, Worlddrugtracker, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his PhD from ICT ,1991, Mumbai, India, in Organic chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with GLENMARK- GENERICS LTD, Research centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Prior to joining Glenmark, he worked with major multinationals like Hoechst Marion Roussel, now sSanofi, Searle India ltd, now Rpg lifesciences, etc. he is now helping millions, has million hits on google on all organic chemistry websites. His New Drug Approvals, Green Chemistry International, Eurekamoments in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules and implementation them on commercial scale over a 25 year tenure, good knowledge of IPM, GMP, Regulatory aspects, he has several international drug patents published worldwide . He gas good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, polymorphism etc He suffered a paralytic stroke in dec 2007 and is bound to a wheelchair, this seems to have injected feul in him to help chemists around the world, he is more active than before and is pushing boundaries, he has one lakh connections on all networking sites, He makes himself available to all, contact him on +91 9323115463, amcrasto@gmail.com
Personal Links
- my sites on the net
- DR ANTHONY MELVIN CRASTO
- GOOGLE GROUP ORGANIC PROCESS DEVELOPMENT
- mixxt
- epernicus
- scipeople
- jimdo
- yolasite
- my cv
- slidestaxx
- wordpress blog
- ABOUT ME
- BRANDSITE
- SKILLPAGES
- Academia.edu
- RESEARCHGATE
- DIIGO
- SLIDESHATE
- WIX
- WIX BLOG
- ISSUU
- SCRIBD
- BIZ
- GOOGLE BLOG
- APNACIRCLE
- Eurekamoments in Organic Chemistry
- Organic Chemistry by Dr Anthony
- Green Chemistry International
- Technology Transfer
- Stereochemistry
- Spectroscopy
- Polymorphism
- Reactions in Org Chem
- DR ANTHONY MELVIN CRASTO Ph.D
- Pharmaceuticals
- Medicinal chemistry
- Organic chemistry literature
- Patent related site
- Green chemistry
- Reagents
- R & D
- Molecules
- Heterocyclic chem
- Sourcing
- NEW DRUG APPROVALS
- WORLD DRUG TRACKER
- Green Chemistry International
- drug regulatory affairs international
- ORGANIC SPECTROSCOPY INTERNATIONAL
- ORGANIC SYNTHESIS INTERNATIONAL
- ALL ABOUT DRUGS
- ORGANIC CHEMISTRY INTERNATIONAL
- GOOGLE PLUS
- Drug Scaleup and Manufacturing International


amcrasto@gmail.com
email me if u like my posts
VINCRISTINE……..Chemistry, Isolation

VINCRISTINE
(3aR,3a1R,4R,5S,5aR,10bR)-methyl 4-acetoxy-3a-ethyl-9-((5S,7S,9S)-5-ethyl-5-hydroxy-9-(methoxycarbonyl)-2,4,5,6,7,8,9,10-octahydro-1H-3,7-methano[1]azacycloundecino[5,4-b]indol-9-yl)-6-formyl-5-hydroxy-8-methoxy-3a,3a1,4,5,5a,6,11,12-octahydro-1H-indolizino[8,1-cd]carbazole-5-carboxylate
…………………………………………………………..
………………………………………………………………….


Vincristine (brand name, Oncovin), formally known as leurocristine, sometimes abbreviated “VCR”, is a vinca alkaloid from the Catharanthus roseus (Madagascar periwinkle), formerly Vinca rosea and hence its name. It is amitotic inhibitor, and is used in cancer chemotherapy. Vincristine is created by the coupling of indole alkaloids vindoline and catharanthine in the vinca plant.[1]
Mechanism
Tubulin is a structural protein that polymerizes to microtubules. The cell cytoskeleton and mitotic spindle, among other things, are made of microtubules. Vincristine binds to tubulin dimers, inhibiting assembly of microtubule structures. Disruption of the microtubules arrests mitosis in metaphase. Therefore, the vinca alkaloids affect all rapidly dividing cell types including cancer cells, but also those of intestinal epithelium and bone marrow.

Uses
Vincristine is delivered via intravenous infusion for use in various types of chemotherapy regimens. Its main uses are in non-Hodgkin’s lymphoma as part of the chemotherapy regimen CHOP, Hodgkin’s lymphoma as part of MOPP, COPP, BEACOPP, or the less popular Stanford V chemotherapy regimen, in acute lymphoblastic leukemia, and in treatment for nephroblastoma (Wilms tumor, a kidney tumor most common in young children). It is also used to induce remission in ALL with Dexamethasone and L-Asparaginase. Vincristine is occasionally used as an immunosuppressant, for example, in treating thrombotic thrombocytopenic purpura (TTP) or chronic idiopathic thrombocytopenic purpura (ITP). It is used in combination with prednisone to treat childhood leukemia.
The main side-effects of vincristine are peripheral neuropathy, hyponatremia, constipation, and hair loss.
Peripheral neuropathy can be severe, and hence a reason to avoid, reduce, or stop the use of vincristine. One of the first symptoms of peripheral neuropathy is foot drop: A person with a family history of foot drop and/or Charcot-Marie-Tooth disease (CMT) should avoid the taking of vincristine.[2]
Accidental injection of vinca alkaloids into the spinal canal (intrathecal administration) is highly dangerous, with a mortality rate approaching 100 percent. The medical literature documents cases of ascending paralysis due to massive encephalopathy and spinal nerve demyelination, accompanied by intractable pain, almost uniformly leading to death; a handful of survivors were left with devastating neurological damage with no hope of recovery. Rescue treatments consist of washout of the cerebrospinal fluid and administration of protective medications.[3] A significant series of inadvertent intrathecal vincristine administration occurred in China in 2007 when batches of cytarabine andmethotrexate (both often used intrathecally) manufactured by the company Shanghai Hualian were found to be contaminated with vincristine.[4]

Having been used as a folk remedy for centuries, studies in the 1950s revealed that C. roseus contained 70 alkaloids, many of which are biologically active. While initial studies for its use in diabetes mellitus were disappointing, the discovery that it caused myelosuppression (decreased activity of the bone marrow) led to its study in mice withleukemia, whose lifespan was prolonged by the use of a vinca preparation. Treatment of the ground plant with Skelly-B defatting agent and an acid benzene extract led to a fraction termed “fraction A”. This fraction was further treated withaluminium oxide, chromatography, trichloromethane, benz-dichloromethane, and separation by pH to yield vincristine.[5]
Vincristine was approved by the United States Food and Drug Administration (FDA) in July 1963 as Oncovin. The drug was initially discovered by a team led by Dr. J.G. Armstrong, then marketed by Eli Lilly and Company.
Like LSD, the microtubule toxin vincristine allegedly causes not-unpleasant visual hallucinations in humans. Other side-effects of vincristine include depression, agitation, and insomnia. Very small doses are needed for the effects of LSD or vincristine, for example, these drugs are active at concentrations of 4.3E-7 M-1 vincristine and 1.0E-8 M-1 LSD.
Many researchers have favored the drug-receptor theory to explain drug-induced hallucinations, usually at the 5-HT2A receptor. In the drug-receptor theory, signal amplification takes place when one molecule of drug binds to a receptor, which activates G-proteins, which affects more proteins, thus signaling cascades explain how a small amount of LSD can lead to widespread changes in the cell.
Van Woerkom suggests instead that LSD binds an element of the cytoskeleton, in a fashion similar to colchicine or vinblastine, which directly bind tubulin. The amount of LSD needed to produce hallucinations is so vanishly small, that it seems hard to believe that a submicromolar dosage of LSD could act on a substrate as vast as the cytoskeleton. However, some microtubule inhibitors such as vincristine are effective at very low dosages. The potency of vincristine may partly explain the success of this drug as a chemotherapeutic drug.
Three generic drug makers supply vincristine in the United States – APP, Mayne, and Sicor (Teva).
- ^ “Pharmacognosy of Vinca Alkaloids”.
- Graf, W. D.; Chance, P. F.; Lensch, M. W.; Eng, L. J.; Lipe, H. P.; Bird, T. D. (1996). “Severe Vincristine Neuropathy in Charcot-Marie-Tooth Disease Type 1A”. Cancer 77 (7): 1356–1362. doi:10.1002/(SICI)1097-0142(19960401)77:7<1356::AID-CNCR20>3.0.CO;2-#. PMID 8608515.
- Qweider, M.; Gilsbach, J. M.; Rohde, V. (2007). “Inadvertent Intrathecal Vincristine Administration: A Neurosurgical Emergency. Case Report”. Journal of Neurosurgery: Spine 6 (3): 280–283. doi:10.3171/spi.2007.6.3.280. PMID 17355029.
- Jake Hooker and Walt Bogdanich (January 31, 2008). “Tainted Drugs Tied to Maker of Abortion Pill”. New York Times.
- Johnson, I. S.; Armstrong, J. G.; Gorman, M.; Burnett, J. P. (1963). “The Vinca Alkaloids: A New Class of Oncolytic Agents” (pdf). Cancer Research 23 (8 Part 1): 1390–1427.PMID 14070392.
External links
- Vincristine chemotherapy
- Vincristine and vinblastine
- Description and Natural History of the Periwinkle
- The Boger Route to (-)-Vindoline
- U.S. National Library of Medicine: Drug Information Portal – Vincristine
-
Cytostatic Vinca alkaloids rosea L. Catharanthus roseus G.Don) are now well known anticancer and particularly useful. Given the small amount of vincristine in Catharanthus present, quite a number of ways of preparation have been proposed by chemists. Thus FR-A-2296418 describes the synthesis of vincristine by coupling Catha-ranthine and vindoline. Other laboratories have achieved the transformation of vinblastine vincristine oxidation under controlled conditions, very strict.
-
FR-A-2210393 and US-A-3899493 perform the oxidation by chromic acid at -30, -90 ° C in a mixture of acetic acid-acetone or chloroform-acetic acid at -55 ° C.
-
In U.S. 4,375,432, chromic compound is also used in acid medium at -65 ° C, -50 ° C in a medium based solvent THF. In addition, EP-A-37289 boasts an oxidation mixture ferrous salt, hydrogen peroxide, perchlorate in acetonitrile. ZA-A-82 08939 discloses a method with chromic acid and an ether-chloroform.
-
HU-A-23638 offers diterbutylchromate in pelargonic acid, and finally EP-A-117861 gets vinblastinel transformation vincristine oxidant potassium permanganate in acetic acid medium. It is clear that these dimeric alkaloids are a valuable material because of their low levels in vegetable raw materials, and therefore the processes of synthesis or semi-synthesis performance are of extreme interest.
-
Vincristine is used in cancer chemotherapy, particularly for the treatment of certain acute leukemias.
-
This alkaloid is obtained mainly by extraction from leaves of Catharanthus Ro-seus (U.S. Patent No. 3,205,220) where it is accompanied by other alkaloids bis-Indo-holic, especially vinblastine.Vinblastine (I, R = CH 3), however, is present at a concentration much higher than that of vincristine and is therefore a precursor of choice for the semisynthesis of the latter.
-
Several processes of vincristine from vinblastine were disclosed. We note in particular patents or patent applications include:
- a) Belgian Patent 739,337 (Gedeon Richter) which describes a method for the oxidation of vinblastine vincristine in a mixture chromic acid, acetic acid and acetone.
- b) Belgian Patent 823560 (Gedeon Richter) the oxidation is performed with oxygen in the presence of formic acid and of a catalyst based on platinum at room temperature.
- c) European Patent Application 18231 (Gedeon Richter): is carried out by oxidation with chromic acid or an alkali metal dichromate in the presence of acetic anhydride and, optionally, of ethanol and an organic solvent immis target with water.
- d) European Patent Application 37289 (Eli Lil-ly): the oxidation is effected by the perchlorate of iron (II) in the presence of hydrogen peroxide and acetonitrile.
-
In addition, the European patent application 37. 290 discloses a process for the oxidation of vinblastine base with Na 2 Cr 2 O 7 in the presence of sulfuric acid in tetrahydrofuran. This reaction led to -50 ° C, is achieved with a yield of 80-92% calculated for each estimation.
-
Observed yields or purity of the products obtained characterizing the processes described above are, however, significant disadvantages.
-
Frequently a secondary product formed is N-demethyl vinblastine need then reformulate for vincristine.
Thus Potier and Kutney obtained products with the C18’S-C2’R absolute configuration, which is critical for anti-tumor activity, by a coupling reaction of the N.sup.b -oxide of catharanthine, or its derivatives, with vindoline, in the presence of trifluoroacetic anhydride, followed by a reduction reaction. [See Potier et. al. J. Am. Chem. Soc. 98. 7017 (1976) and Kutney et. al. Helv. Chim. Acta, 59, 2858 (1976)].
The Potier and Kutney coupling process has disadvantages. The yields are not satisfactory except for the coupling of catharanthine N-oxide with vindoline and even there the preparative yield is low. While vindoline is the most abundant alkaloid of Vinca rosea and is thus readily available, the other possible components of the Potier-Kutney coupling process (catharanthine, allocatharanthine, voacangine,) are relatively inaccessible, costly, and they do not allow a wide range of structural variation of that component of the coupling process.
- …………………………………………………………………………………………………………………………………………………………………………………………………………..
-
EP 0117861 B1
-
clips
-
The process of the present invention produces a simple vincristine, in quantity and purity requiring little or no additional purification by recrystallization or chromatography.
-
[0009]The reagent used is oxidation permanganate ion dissolved in toluene or dichloromethane as solvent. An alternative consists in immobilizing the resin on a permanganate anion, for example a polymer such as polystyrene comprising ammonium groups. Solubilization can be achieved by the action of a complexing agent crown ether (“crown-ether”) of potassium permanganate.
-
[0010]The permanganate anion can also be solubilized by preparing an ammonium salt or quaternary phosphonium corresponding which is soluble in methylene chloride or toluene. For this purpose, it is preferable to use potassium permanganate benzyltriethylammonium.
-
[0011]Obtaining from vincristine vinblastine using a permanganate salt is unexpected since the potassium permanganate used in some acetone oxide derivatives of vinblastine at the portion of the molecule velbanamine (Kutney, Balsevich and Worth, Heterocycles, 11, 69, 1978). The N-methyl group of the vindoline part intact.
-
[0012]The formation of N-CHO indoline skeleton on a bis-indole group vinblastine using a permanganate salt has never been reported.
-
[0013]According to one embodiment of the method of the present invention, vinblastine, preferably in the form of sulphate, is treated in the presence of an organic acid such as acetic acid, with an excess of potassium permanganate dissolved in dichloromethane or toluene in the presence of “18-crown-6” or ether derivatives dibenzo-or di-cyclohexylcorrespondants. The reaction is conducted at a temperature between -40 ° C and -75 ° C and is preferably followed by thin layer chromatography. The reaction time generally ranges from 5 minutes to 3 hours.
-
[0014]Potassium permanganate is preferably dissolved in dichloromethane and the oxidation reaction is then carried out at -70 ° C.
-
[0015]The solubility of potassium permanganate is indeed substantially increased in the presence of a macrocyclic polyether as the “18-crown-6” ether (1, 4, 7, 10, 13, 16-hexaoxacy-clooctadécane) or derivative dibenzo – or corresponding dicyclohexyl-hexyl.
-
[0016]The reaction mixture is then treated simultaneously by a mild reducing and alkaline. For this purpose, use is preferably an aqueous solution of bisulfite, disulfite or sodium metabisulfite and ammonia.
-
[0017]The organic phase was separated and the aqueous phase is extracted several times with methylene chloride. The combined organic phases were concentrated in vacuo to give a residue containing 80-85% of base vincristine, a 90-95% yield.
-
[0018]Alternatively, you can proceed with the extraction of the reaction mixture after reduction without conducting a simultaneous alkalinization. The acidic aqueous solution was then extracted with dichloromethane. This route is a novel process for purification of vincristine formed in the reaction medium.
-
[0019]According to another embodiment of the present invention, vincristine is obtained by oxidation of vinblastine by reacting a quaternary ammonium permanganate. The ammonium cation is preferably benzyltriethylammonium group or benzyl trimethyl ammonium (see eg Angew. Chem., Intern. Ed. 13, 170, 1974). The reaction is carried out in 2 to 6 hours at -60 ° C in an inert solvent wherein the ammonium salt is soluble, and an acid, preferably an organic acid of low molecular weight. A mixture of dichloromethane and glacial acetic acid can be used. After treatment with a mild reducing agent in aqueous medium, the resulting acidic solution is extracted with dichloromethane, and the organic phase is made alkaline by washing with a basic aqueous solution and concentrated. Vincristine solvate is isolated with a yield higher than 90%.
-
[0020]The latest variant of the method of the invention is particularly advantageous in terms of economic and technical.
-
[0021]Purification or separation may be effected by crystallization and chromatography using techniques well known this from the crude product of the reaction. The product can also be lyophilized.
-
[0022]In most cases, vincristine thus obtained can be converted directly into an addition salt with an organic or inorganic acid, preferably pharmaceutically acceptable. This salt is preferably a sulfate that may arise in a more or less solvated or hydrated.
-
[0023]We can also prepare vincristine dissolved in a physiologically acceptable solvent and ready to be injected.
-
[0024]In particular, vincristine sulfate is obtained by addition of H 2 S0 4 to a solution of vincristine gross or recrystallized from ethanol, dissolved in a mixture of methylene chloride and anhydrous ethanol, partial removal in vacuo chloride methylene and crystallization.
-
[0025]Vincristine sulfate thus obtained has a purity sufficient for use as a medicament, particularly in the form of injectable solutions.

Madagascar Periwinkle: Public Domain Illustration by Sydenham Edwards
The Madagascar periwinkle, an attractive flowering plant, contains the powerful anti-cancer chemicals vinblastine and vincristine. Velvet beans, which are named from the covering of soft hairs on the young plant, contain L-dopa, a very helpful chemical in the treatment of Parkinson’s disease. The Madagascar periwinkle and the velvet bean are just two of the large number of plants that have been found to contain medicinal chemicals. There are almost certainly many more plants that have undiscovered health benefits.
The Madagascar Periwinkle
The Madagascar periwinkle is native to Madagascar and India, but is now grown in many countries as a garden plant. It has also escaped from gardens and grows as a weed. The red, purple, pink or white flowers often have a center which is a different color from the rest of the flower. Madagascar periwinkles may grow up to one meter tall and have glossy green leaves.
The sap of the Madagascar periwinkle, which has a milky appearance and is poisonous, contains vinblastine, vincristine and many other alkaloids. Researchers are discovering that many of these alkaloids are biologically active inside the human body.
Vinblastine and Vincristine
Vinblastine and vincristine have very similar chemical structures, but their effects on the body are not the same. Vinblastine is used to treat specific types of cancer, such as Hodgkin’s disease, breast cancer, testicular cancer and non-small cell lung cancer. Vincristine is used in the treatment of acute lymphoblastic leukemia (ALL) and has provided a great breakthrough in successful treatment of this disease in children. When vincristine is added to the treatment regimen for children suffering from ALL, the survival rate reaches eighty percent. Vincristine is not so impressive in the treatment of ALL in adults.
Cells contain a supporting network of protein tubules, which are known as microtubules. Microtubules also play a vital role in the process of cell division. Before a cell divides, each chromosome in the cell is replicated. The replicated chromosomes are separated from their partners and pulled to opposite ends of the cell by microtubules during a process called mitosis. The cell then divides down the middle.
Vinblastine and vincristine stop microtubule formation during mitosis and therefore prevent cells from reproducing. This effect is strongest in cells that have a high rate of division, such as cancer cells. However, vinblastine and vincristine also affect cells lining the intestine, the cells in the bone marrow that produce blood cells, and the cells in the hair follicles, since these too have a high rate of cell division.
Possible vinblastine or vincristine side effects include constipation, hair loss, a low platelet count, which can cause increased bleeding, a low white blood cell count, which can lead to increased infections, or a low red blood cell count, resulting in anemia. There may occasionally be nerve damage, possibly due to the effect of the medicines on the microctubules in the nerve cells. Vincristine is more likely to cause nerve damage than vinblastine.

,,,,,,,,,,,,,


Total synthesis of (+)-vincristine (2). TFA, trifluoroacetic acid or trifluoroacetyl; DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene.
Stereocontrolled total synthesis of (+)-vincristine

………………….
see docstoc presentation
click below
Vincristine
var docstoc_docid=”51697405″;var docstoc_title=”Vincristine”;var docstoc_urltitle=”Vincristine”;
…………………….
isolation
Kumar A, Patil D, Rajamohanan PR, Ahmad A (2013)
Isolation, Purification and Characterization of Vinblastine and Vincristine from Endophytic Fungus Fusarium oxysporumIsolated from Catharanthus roseus. PLoS ONE 8(9): e71805. doi:10.1371/journal.pone.0071805
http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0071805
Isolation, purification and characterization of vinblastine and vincristine from the endophytic fungus Fusarium oxysporum
A two stage fermentation procedure was employed for the isolation of vinblastine and vincristine by Fusarium oxysporum. In the first stage, 500 ml Erlenmeyer flasks containing 100 ml medium (MGYP, (0.3%) malt extract, (1.0%) glucose, (0.3%) yeast extract and (0.5%) peptone) were inoculated with 7 days old culture and incubated at 28°C on a rotary shaker (240 rpm) for 4–5 days, which was used as seed culture (I stage). Later, 10 ml seed culture was transferred to 500 ml Erlenmeyer flask containing 100 ml production medium called as vinca medium-1 (Glucose: 3%, Succinic acid: 1%, Sodium benzoate: 100 mg, Peptone: 1%, Magnesium sulphate: 3.6 mg, Biotin: 1 mg, Thiamine: 1 mg, Pyridoxal: 1 mg, Calcium pentothenate: 1 mg, Phosphate buffer: 1 ml (pH 6.8), L-Tryptophan: 0.1%, Geranium oil: 0.05%.) which were incubated at 28°C for 20 days as shake culture (II stage), after which it was harvested and used for further study. Culture filtrates and mycelia were separated with the help of muslin cloth and then lyophilized. Lyophilized culture filtrate was extracted using ethyl acetate as a solvent system. The organic layer was separated from the aqueous layer using separating funnel. The extraction was repeated thrice and the solvent was dried using anhydrous sodium sulphate and concentrated under vacuum using rotavapour at 40°C in order to get crude extract. A small amount of crude extract was dissolved in ethyl acetate and subjected to thin layer chromatography (TLC) on silica gel-G (0.5 mm thickness) using chloroform:methanol (8:2) as a solvent system. The TLC plates were sprayed with ceric ammonium sulphate reagent. Vinca alkaloids spots produced brilliant violet color as well as purple color with above spraying reagent. Purification of fungal vinblastine and vincristine were done by silica gel column chromatography. The crude extract was loaded on silica gel column (60–120 mesh size, 40 cm×2 cm length width) pre-equilibrated with chloroform and eluted with a gradient of chloroform:methanol (100% chloroform, 9:1, 8:2, 7:3, 1:1 and 3:7 and 100% methanol). Fractions containing compounds with Rf values similar to that of the standard vinblastine and vincristine were pooled and subjected to preparative TLC on a 0.5 mm thick (20 cm×20 cm) silica plate and developed in chloroform:methanol (8:2) solvent system. The putative bands of fungal vinblastine and vincristine were scraped and eluted out with methanol. Purity of the isolated compounds was checked on TLC in the solvent systems such as (a) chloroform:methanol (8:2) (b) chloroform:methanol (9:1) and (c) ethyl acetate: acetonitrile (8:2).
http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0071805
see also
http://www.ncbi.nlm.nih.gov/pubmed/20209002
ALSO
large-scale isolation of native catharantine, vindoline and 3′,4′-anhydrovinblastine whereby the isolation of vincristine, vinblastine, leurosine and the corresponding desacetoxy, desacetyl and N-desmethyl derivatives in a manner known per se can also be accomplished.
For the isolation of the two monoindole alkaloids: vindoline and catharantine from the dried plant Vinca rosea L. Svoboda [J. Am. Pharm. Assoc. 48, (11), 659 (1959)] described a method, which can be accomplished only with a very modest yield. From 1 kg. of the dried plant–subjecting the whole plant to a suitable treatment–approximately 0.6 g. of vindoline and 0.05 g. of catharantine were obtained.
3′,4′-ANHYDROVINBLASTINE UNTIL NOW HAS NEITHER BEEN ISOLATED FROM THE PLANT Vinca rosea L. nor identified in it.
For the preparation of the diindole alkaloid components starting from the leaves of Vinca rosea L. there are more methods known in the art (U.S. Pat. nos. 3,097,137; 3,205,220; 3,225,030 and Hungarian Pat. Nos. 153,200; 154,715; 160,967 and 164,958 as well as Austrian Pat. Nos. 313,435, 313,485, Australian pat. No. 458,629 and Swiss Pat. No. 572,488 and British Pat Nos. 1,412,932, 1,382,460 corresponding to the preceding two patents). According to these known processes from 1 kg. of the dried leaves of Vinca rosea L. about 0.1 to 0.2 g. of leurosine can be obtained and vinblastine, vincristine and optionally the corresponding N-desmethyl, desacetyl and desacetoxy derivatives are also simultaneously isolated.
Further on it is well known that the synthetic catharantine and vindoline may be coupled by the Polonovszky reaction to give 3′,4′-anhydrovinblastine which can thereafter be epoxidized to leurosine [Potier et al. Tetrahedron Letters 3945 (1976); DT-OS 25 58,124; Helv. Chim. Acta 59, 2858 (1976); Heterocycles 4, 997 (1976), Belgian patent specification No. 842,200 equivalent to U.S. patent application Ser. No. 582,372]. Leurosine itself has a valuable tumour growth inhibiting activity and the N-desmethyl-N-formyl derivative thereof is the most promising substance against leukemia (Hungarian Pat. No. 165,986 equivalent to U.S. patent application Ser. No. 422,100, and Austrian Pat. No. 332,566 which has issued as British Pat. No. 1,412,932).
Simeprevir has been approved in Japan for the treatment of genotype 1 chronic hepatitis C infection

simeprevir
| CAS number | 923604-59-5 | ||
| Formula | C38H47N5O7S2 | ||
| Weight | 749.93908 |
Stockholm, Sweden — Medivir AB (OMX: MVIR) today reports that Janssen Pharmaceutical R&D Ireland (Janssen) has been informed by the Japanese Ministry of Health, Labour and Welfare (MHLW) that simeprevir has been approved for the treatment of genotype 1 chronic hepatitis C virus (HCV) infection.
read all at
http://www.pharmalive.com/japan-approves-simeprevir
Hepatitis C virus (HCV) infections affect approximately 3 percent of the worldwide population and often lead to cirrhosis and hepatocellular carcinoma. The standard therapy of pegylated- interferon and ribavirin induces serious side effects and provides viral eradication in less than 50% of patients. Combination therapy of HCV including ribavirin and interferonare currently is the approved therapy for HCV. Unfortunately, such combination therapy also produces side effects and is often poorly tolerated, resulting in major clinical challenges in a significant proportion of patients. Numerous direct acting agents (DAAs) have been or are being developed for treatment of HCV, such as telaprevir and boceprevir (both received MA approved in 2011 for use with interferon and ribavirin based therapy), however direct acting agents are linked to increased toxicity of treatment, the emergence of resistance, and to date do not provide a standard of care which is interferon free. The combination of direct acting agents can also result in drug-drug interactions. To date, no HCV therapy has been approved which is interferon free. There is therefore a need for new combination therapies which have reduced side effects, and interferon free, have a reduced emergence of resistance, reduced treatment periods and/or and enhanced cure rates.
Simeprevir (formerly TMC435) is an experimental drug candidate for the treatment of hepatitis C. It is being developed byMedivir and Johnson & Johnson‘s pharmaceutical division Janssen Pharmaceutica and is currently in Phase III clinical trials.[1]
Simeprevir is a hepatitis C virus protease inhibitor.[2]
Simeprevir is being tested in combination regimens with pegylated interferon alfa-2a and ribavirin,[3] and in interferon-free regimens with other direct-acting antiviral agents including daclatasvir[4] and sofosbuvir [5]
Food and Drug Administration (FDA) has granted Priority Review to the New Drug Application (NDA) for simeprevir (TMC435). Simeprevir is an investigational NS3/4A protease inhibitor taken orally (150 mg capsule) once a day along with pegylated interferon and ribavirin for genotype 1 chronic hepatitis C virus (HCV) infection in adult patients with compensated liver disease (meaning the liver is heavily scarred but still functional).
“Hepatitis C is a complex disease and Janssen is committed to working with the HCV community, caregivers, and health care systems to address this global epidemic,” said Gaston Picchio, Hepatitis Disease Area Leader, Janssen Research & Development. “We are pleased that the FDA has granted simeprevir Priority Review, as it is a significant step forward in making this therapy available to physicians and their hepatitis C patients.”
The FDA grants Priority Review to medicines that may offer major advances in care or provide a treatment option where no adequate therapy exists. Under the Prescription Drug User Fee Act, FDA review will begin approximately 60 days after receipt of the application and will aim to be completed within six months from when the review period begins.
The regulatory submission for simeprevir is supported in part by data from three pivotal Phase 3 studies: QUEST-1 and QUEST-2 in treatment-naïve patients and PROMISE in patients who have relapsed after prior interferon-based treatment. Janssen also recently submitted simeprevir for marketing authorization to regulatory authorities in Japan and Europe.

- “Medivir Announces That Simeprevir (TMC435) Data Will Be Presented at the Upcoming AASLD Meeting”. Yahoo News. October 1, 2012. Retrieved November 6, 2012.
- Lin, TI; Lenz, O; Fanning, G; Verbinnen, T; Delouvroy, F; Scholliers, A; Vermeiren, K; Rosenquist, A et al. (2009). “In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor”. Antimicrobial agents and chemotherapy 53 (4): 1377–85. doi:10.1128/AAC.01058-08. PMC 2663092. PMID 19171797.
|displayauthors=suggested (help) - “Phase 3 Studies Show Simeprevir plus Interferon/Ribavirin Cures Most Patients in 24 Weeks”. hivandhepatitis.com. December 27, 2012.
- Medivir announces TMC435 in an expanded clinical collaboration. Medivir. 18 April 2012.
- Results from a phase IIa study evaluating Simeprevir and Sofosbuvir in prior null responder Hepatitis C patients have been presented at CROI. 6 March 2013.
IUPAC standard name
(1R, 4R, 6S, 15R, 17R)-N-(cyclopropanesulfonyl) -17 – ({7-methoxy-8-methyl-2-[4 – (propan-2-yl) -1,3-thiazol-2 -yl] quinolin-4-yl} oxy)-13-methyl-2 ,14-dioxo-3 ,13-diazatricyclo [13.3.0.0 4 , 6 ] octadec-7-ene-4-carboxamide
IUPAC traditional name
(1R, 4R, 6S, 15R, 17R)-N-(cyclopropanesulfonyl) -17 – {[2 – (4-isopropyl-1 ,3-thiazol-2-yl)-7-methoxy-8-methylquinolin-4- yl] oxy}-13-methyl-2 ,14-dioxo-3 ,13-diazatricyclo [13.3.0.0 4 , 6 ] octadec-7-ene-4-carboxamide
Aliases
TMC435
TMC435350

,,,,,,,,,,,,,
NS3/4A protease inhibitors
Ciluprevir (BILN 2061) Boehringer Ingelheim
Boceprevir (SCH503034) Merck
Telaprevir (VX-950) Vertex
Danoprevir (RG7227) Roche
simeprevir /TMC435 Tibotec / Medivir
Vaniprevir (MK-7009) Merck
Bl 201335 Boehringer Ingelheim
BMS-650032 Bristol-Myers Squibb
GS-9256 Gilead
ABT-450 Abbott / Enanta
Narlaprevir (SCH900518) Merck
PHX1766 Phenomix
ACH-1625 Achillion
IDX320 Idenix
MK-5172 Merck
VX-985 Vertex Drug name Company
GS-9451 Gilead
Telaprevir
Accordin to http://en.wikipedia.Org/wiki/File:Telaprevir.svg, Teaprevir has the structure

Systematic lUPAC Name: (1 S,3aR,6aS)-2-[(2S)-2-[[(2S)-2-Cyclohexyl-2-(pyrazine-2- carbonylamino)acetyl]amino]-3,3-dimethylbutanoyl]-/\/-[(3S)-1-(cyclopropylamino)-1 ,2- dioxohexan-3-yl]-3,3a,4,5,6,6a-hexahydro-1/-/-cyclopenta[c]pyrrole-1-carboxamide
Telaprevir may be administered in a unit dose of, for example between about 250 and about l OOOmg, such as about 750mg/kg. Typically once, twice, three or four times daily, such as three times daily for the duration of the pre-treatment period and/or combination treatment period.
Boceprevir
Accordin to http://en.wikipedia.0rg/wiki/File:B0ceprevir.svg, Boceprevir has the structure:

Systematic lUPAC Name: (1 R,2S,5S)-N-[(2≡)-4-amino-1-cyclobutyl-3,4-dioxobutan-2-yl)]- 3-{(2S)-2-[(tert-butylcarbamoyl)amino]-3,3-dimethylbutanoyl}- 6,6-dimethyl-3- azabicyclo[3.1.0]hexane-2-carboxamide
Boceprevir may be administered in a unit dose of, for example between about 250 and about 1000mg, such as about 800mg/kg. Typically once, twice, three or four times daily, such as three times daily for the duration of the pre-treatment period and/or combination treatment period.
Compound 1: miR-122 inhibitor
As reported in Young et al., JACS 2010, 132, 7976-7981) (hereby incorporated by reference), it is possible to assay for small molecule inhibitors of miR122 and small molecule are known, such as those illustrated below:



» {7.02 ± 1.40) If (4. S3 * 0.45)

The numerical values refer to luciferase expression due to miR-122 deprepression, and values greater than 1 indicate miR-122 inhibition.
EMA Accepts AstraZeneca’s Naloxegol Application

naloxegol
http://www.ama-assn.org/resources/doc/usan/naloxegol.pdf
Morphinan-3,14-diol, 4,5-epoxy-6-(3,6,9,12,15,18,21-heptaoxadocos-1-yloxy)-17-(2-
propen-1-yl)-, (5α,6α)-
4,5α-epoxy-6α-[(3,6,9,12,15,18,21-heptaoxadocosan-1-yl)oxy]-17-(prop-2-en-1-
yl)morphinan-3,14-diol
MOLECULAR FORMULA C34H53NO11
MOLECULAR WEIGHT 651.8
SPONSOR AstraZeneca
CODE DESIGNATION NKTR-118
CAS REGISTRY NUMBER 854601-70-0
WHO NUMBER 9434
Marketing Authorisation Application for naloxegol accepted by European Medicines Agency
Friday, 27 September 2013
AstraZeneca today announced that the European Medicines Agency (EMA) has accepted the Marketing Authorisation Application (MAA) for naloxegol, an investigational peripherally-acting mu-opioid receptor antagonist, which has been specifically designed for the treatment of opioid-induced constipation (OIC) for adult patients 18 years and older, including patients with inadequate response to laxatives.
read more
http://www.pharmalive.com/ema-accepts-astrazeneca-s-naloxegol-application
Naloxegol (INN; NKTR-118), or PEGylated naloxol,[1] is a peripherally–selective opioid antagonist under development byAstraZeneca, licensed from Nektar, for the treatment of opioid-induced constipation.[2]
- ^ Roland Seifert; Thomas Wieland; Raimund Mannhold; Hugo Kubinyi, Gerd Folkers (17 July 2006). G Protein-Coupled Receptors as Drug Targets: Analysis of Activation and Constitutive Activity. John Wiley & Sons. p. 227. ISBN 978-3-527-60695-5. Retrieved 14 May 2012.
- ^ “Nektar | R&D Pipeline | Products in Development | CNS/Pain | Oral Naloxegol (NKTR-118) and Oral NKTR-119”. Retrieved 2012-05-14.
Naloxegol (NKTR-118) is an investigational drug candidate in Phase 3 studies being developed as a once-daily oral tablet for the treatment of opioid-induced constipation. Naloxegol (NKTR-118) was designed using Nektar’s proprietary small molecule polymer conjugate technology. Results of the Phase 2 study of naloxegol (NKTR-118) were presented in October 2009 at the American College of Gastroenterology Annual Clinical Meeting and the American Academy of Pain Management. NKTR-119 is an early stage drug development program that is intended to combine oral naloxegol (NKTR-118) with selected opioids, with the goal of treating pain without the side effect of constipation traditionally associated with opioid therapy.
Nektar and AstraZeneca have a global agreement for both naloxegol (NKTR-118) and NKTR-119. Under the agreement, AstraZeneca has responsibility for the development, global manufacturing and marketing of both naloxegol (NKTR-118) and NKTR-119. For naloxegol (NKTR-118), Nektar is eligible to receive up to $235 million in aggregate payments upon the achievement of certain regulatory milestones, as well as additional tiered sales milestone payments of up to $375 million if the product achieves considerable levels of commercial success. Nektar will also be eligible to receive significant double-digit royalty payments on net sales of naloxegol (NKTR-118) worldwide. For NKTR-119, Nektar would receive development milestone payments as well as tiered sales milestone payments. Nektar will also receive significant double-digit royalty payments on NKTR-119 net sales worldwide.
oxalate derivative
http://www.ama-assn.org/resources/doc/usan/naloxegol-oxalate.pdf
Morphinan-3,14-diol, 4,5-epoxy-6-(3,6,9,12,15,18,21-heptaoxadocos-1-yloxy)-
17-(2-propen-1-yl)-, (5α,6α)-, ethanedioate (1:1)
4,5α-epoxy-6α-[(3,6,7,12,15,18,21-heptaoxadocosyl)oxy]-17-(prop-2-
enyl)morphinan-3,14-diol hydrogen ethanedioate
MOLECULAR FORMULA C34H53NO11 . C2H2O4
MOLECULAR WEIGHT 741.8
TRADEMARK None as yet
SPONSOR AstraZeneca
CODE DESIGNATIONS NKTR-118 oxalate, AZ13337019 oxalate
CAS REGISTRY NUMBER 1354744-91-4
Janssen signs licensing agreement with PATH for development of HIV-1 drug

rilpivirine.
Janssen R&D Ireland has signed a licensing agreement with PATH for the early development of a long-acting depot formulation of the human immunodeficiency virus type 1 (HIV-1) drug rilpivirine.

Rilpivirine, a non-nucleoside reverse transcriptase inhibitor (NNRTI), is being developed as potential pre-exposure prophylaxis against HIV infection
read all at
Rilpivirine (TMC278, trade name Edurant) is a pharmaceutical drug, developed by Tibotec, for the treatment of HIVinfection.[1][2] It is a second-generation non-nucleoside reverse transcriptase inhibitor (NNRTI) with higher potency, longer half-lifeand reduced side-effect profile compared with older NNRTIs, such as efavirenz.[3][4]
Rilpivirine entered phase III clinical trials in April 2008,[5][6] and was approved for use in the United States in May 2011.[7] A fixed-dose drug combining rilpivirine with emtricitabine and tenofovir, was approved by the U.S. Food and Drug Administration in August 2011 under the brand name Complera.[8]

Like etravirine, a second-generation NNRTI approved in 2008, rilpivirine is a diarylpyrimidine (DAPY). Rilpivirine in combination with emtricitabine and tenofovir has been shown to have higher rates of virologic failure than Atripla in patients with baseline HIV viral loads greater than 100,000 copies.
- Rilpivirine bound to proteins in the PDB

- “TMC278 – A new NNRTI”. Tibotec. Retrieved 2010-03-07.
- Stellbrink HJ (2007). “Antiviral drugs in the treatment of AIDS: what is in the pipeline ?”. Eur. J. Med. Res. 12 (9): 483–95.PMID 17933730.
- Goebel F, Yakovlev A, Pozniak AL, Vinogradova E, Boogaerts G, Hoetelmans R, de Béthune MP, Peeters M, Woodfall B (2006).“Short-term antiviral activity of TMC278–a novel NNRTI–in treatment-naive HIV-1-infected subjects”. AIDS 20 (13): 1721–6.doi:10.1097/01.aids.0000242818.65215.bd. PMID 16931936.
- Pozniak A, Morales-Ramirez J, Mohap L et al. 48-Week Primary Analysis of Trial TMC278-C204: TMC278 Demonstrates Potent and Sustained Efficacy in ART-naïve Patients. Oral abstract 144LB.
- ClinicalTrials.gov A Clinical Trial in Treatment naïve HIV-1 Patients Comparing TMC278 to Efavirenz in Combination With Tenofovir + Emtricitabine
- ClinicalTrials.gov A Clinical Trial in Treatment naïve HIV-Subjects Patients Comparing TMC278 to Efavirenz in Combination With 2 Nucleoside/Nucleotide Reverse Transcriptase Inhibitors
- “FDA approves new HIV treatment”. FDA. Retrieved 2011-05-20.
- “Approval of Complera: emtricitabine/rilpivirine/tenofovir DF fixed dose combination”. FDA. August 10, 2011.
-
Rilpivirine hydrochloride, 4-[[4-[[4-(2-Cyanoethenyl)-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile monohydrochloride, is a non-nucleoside reverse transcriptase inhibitor (NNRTI) of human immunodeficiency virus type 1 (HIV-1) and indicated for the treatment of HIV-1 infection in treatment-naïve adult patients in combination with other antiretroviral agents. The product received marketing approval in the US (brand name Edurant) and is represented by the following general formula (I):

-
[0003]EP1419152 B1 claims amongst others Rilpivirine base and Rilpivirinehydrochloride per se as well as pharmaceutical compositions comprising the same. However, only concrete examples for preparingRilpivirine base are given in said patent but no concrete examples describing the production of the hydrochloride salt are provided.
-
[0004]EP1632232 B1 claims amongst others a solid pharmaceutical composition comprising crystalline forms A, B, C or D of Rilpivirinehydrochloride. In addition said patent claims a process for the production of Rilpivirine hydrochloride by reacting Rilpivirine base with hydrochloric acid in the presence of a suitable acid, such as acetic acid.
-
[0005]Polymorphism is a phenomenon relating to the occurrence of different crystal forms for one molecule. There may be several different crystalline forms for the same molecule with distinct crystal structures and varying in physical properties like melting point, XRPD pattern and FTIR spectrum. These polymorphs are thus distinct solid forms which share the molecular formula of the compound from which the crystals are made up, however they may have distinct advantageous physical properties such as e.g. chemical stability, physical stability, hygroscopicity, solubility, dissolution rate, bioavailability, etc.
-
[0006]The bioavailability of a compound intended to be administered orally, is dependent on the compounds solubility in aqueous systems as well as the compounds permeability as mentioned in EP1632232 B1 . Hydrates are known to be less soluble in aqueous systems than anhydrous forms of the same compound. Hence anhydrous forms of Rilpivirinehydrochloride are preferred over hydrated forms. Rilpivirinehydrochloride form D of EP1632232 B1 is a hydrate and thus no suitable candidate for the preparation of an orally administered medicament, whereas form E of the present invention is an anhydrate.
-
[0007]The novel polymorph E of Rilpivirine hydrochloride of the present invention shows high solubility in aqueous systems e.g. a higher solubility than forms A and C of EP1632232 B1 and is thus especially suitable for the preparation of an orally administered medicament.
-
[0008]In addition the crystalline forms A and C of EP1632232 B1 are difficult to make in a reliable manner as these forms are obtained from the same solvent system. As the polymorphs A and C of Rilpivirinehydrochloride are obtainable from the same solvent system acetic acid/water, the production processes are especially critical and sensitive because the single crystalline forms are only obtainable in pure form in a quite narrow range of temperature as described in the concrete examples A.a) and A.c) of EP1632232 B1 . In contrast form E of the present invention is reliably obtained by crystallization from ethanol as form E is the only polymorph of Rilpivirine hydrochloride obtained from this solvent system.
-
[0009]According to example A.b) of EP1632232 B1 form B is obtained by recrystallizing Rilpivirine hydrochloride from propanone using an initial Rilpivirine hydrochloride concentration of 0.3 g/L. However, this concentration is not suitable for up-scaling as larger amounts of Rilpivirine hydrochloride would ask for tremendous solvent volumina and hence the usage of tremendously large reaction vessels. In contrast form E of the present invention is also obtained by applying higher initial Rilpivirine hydrochloride concentrations such as e.g. ≥10 g/L and is thus suitable for large scale production.
-
[0010]Hence, aim of the present invention is to circumvent the drawbacks of the known forms A, B, C and D ofEP1632232 B1 by providing an anhydrous polymorph of Rilpivirine hydrochloride, which is obtainable in an easy and reliable manner also in large scale. In addition the novel polymorph shows high solubility in aqueous systems making it especially suitable for the preparation of an orally administered medicament.


Drugs for Chronic Thromboembolic Pulmonary Hypertension (CTEPH)
<div style=”margin-bottom:5px”> <strong> <a href=”https://www.slideshare.net/CTEPH/drugs-for-cteph-studi-farmacologici” title=”Drugs for CTEPH – studi farmacologici” target=”_blank”>Drugs for CTEPH – studi farmacologici</a> </strong> from <strong><a href=”http://www.slideshare.net/CTEPH” target=”_blank”>CTEPH</a></strong> </div>
Cobicistat – European Commission Approves Gilead Sciences’ TybostTM, a New Boosting Agent for HIV Therapy

cobicistat
1004316-88-4
| C 40 H 53 N 7 O 5 S 2 |
(1,3-thiazol-5-yl) methyl (5S, 8R, 11R) -8,11-dibenzyl-2-methyl-5-[2 – (morpholin-4-yl) ethyl] -1 – [2 – (propan-2-yl) -1,3-thiazol-4-yl] -3,6-dioxo-2 ,4,7,12-tetraazatridecan-13-oate
cytochrome P450 3A4 (CYP3A4) inhibitor
Tybost Facilitates Once-Daily Dosing of the Protease Inhibitors Atazanavir and Darunavir –
FOSTER CITY, Calif.–(BUSINESS WIRE)–Sep. 25, 2013– Gilead Sciences, Inc. (Nasdaq: GILD) today announced that the European Commission has granted marketing authorization for once-daily TybostTM (cobicistat 150 mg tablets), a pharmacokinetic enhancer that boosts blood levels of certain HIV medicines. Tybost is indicated as a boosting agent for the HIV protease inhibitors atazanavir 300 mg once daily and darunavir 800 mg once daily as part of antiretroviral combination therapy in adults with HIV-1 infection. Today’s approval allows for the marketing of Tybost in all 28 countries of the European Union (EU).
read all at
http://www.pharmalive.com/eu-oks-gileads-hiv-therapy-tybost

Cobicistat (formerly GS-9350) is a licensed drug for use in the treatment of infection with the human immunodeficiency virus (HIV).
Like ritonavir (Norvir), cobicistat is of interest not for its anti-HIV properties, but rather its ability to inhibit liver enzymes that metabolize other medications used to treat HIV, notablyelvitegravir, an HIV integrase inhibitor currently under investigation itself. By combining cobicistat with elvitegravir, higher concentrations of elvitgravir are achieved in the body with lower dosing, theoretically enhancing elvitgravir’s viral suppression while diminishing its adverse side-effects. In contrast with ritonavir, the only currently approved booster, cobicistat has no anti-HIV activity of its own.[1]
Cobicistat is a component of the four-drug, fixed-dose combination HIV treatmentelvitegravir/cobicistat/emtricitabine/tenofovir (known as the “Quad Pill” or Stribild).[1][2] The Quad Pill/Stribild was approved by the FDA in August 2012 for use in the United States and is owned by Gilead Sciences.
Cobicistat is a potent inhibitor of cytochrome P450 3A enzymes, including the importantCYP3A4 subtype. It also inhibits intestinal transport proteins, increasing the overall absorption of several HIV medications, including atazanavir, darunavir and tenofovir alafenamide fumarate.[3]
- Highleyman, L. Elvitegravir “Quad” Single-tablet Regimen Shows Continued HIV Suppression at 48 Weeks. HIV and Hepatitis.com
- R Elion, J Gathe, B Rashbaum, and others. The Single-Tablet Regimen of Elvitegravir/Cobicistat/Emtricitabine/Tenofovir Disoproxil Fumarate (EVG/COBI/FTC/TDF; Quad) Maintains a High Rate of Virologic Suppression, and Cobicistat (COBI) is an Effective Pharmacoenhancer Through 48 Weeks. 50th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC 2010). Boston, September 12–15, 2010.
- Lepist, E. -I.; Phan, T. K.; Roy, A.; Tong, L.; MacLennan, K.; Murray, B.; Ray, A. S. (2012). “Cobicistat Boosts the Intestinal Absorption of Transport Substrates, Including HIV Protease Inhibitors and GS-7340, in Vitro”. Antimicrobial Agents and Chemotherapy 56 (10): 5409–5413. doi:10.1128/AAC.01089-12. PMC 3457391. PMID 22850510.


Quad ® laboratoryGilead Sciences , which funded the two clinical trials that have been published, containing a mixture of three active ingredients:tenofovir ,emtricitabine and a new active antiretroviral elvitegravir , also a CYP3A4 inhibitor called cobicistat.
The chemical structures of some of these HCV inhibitors as reported by numerous sources are provided below:

Telaprevir

BI-201335

TMC-435 (TMC-435350)


BMS-650032 (Asunaprevir)

danoprevir

MK-5172

ANA-598 (Setrobuvir)

GS-333126 (GS-9190 or tegobuvir)

GS-9451

Mericitabine (R-4048 or RG7128 or R7128)

IDX-184

filibuvir (PF-00868554)

PSI-7977 (GS-7977)

BMS-790052 (daclatasvir)


BIT-225


[0153] BMS-791 As used herein, BMS-
791325 may also be
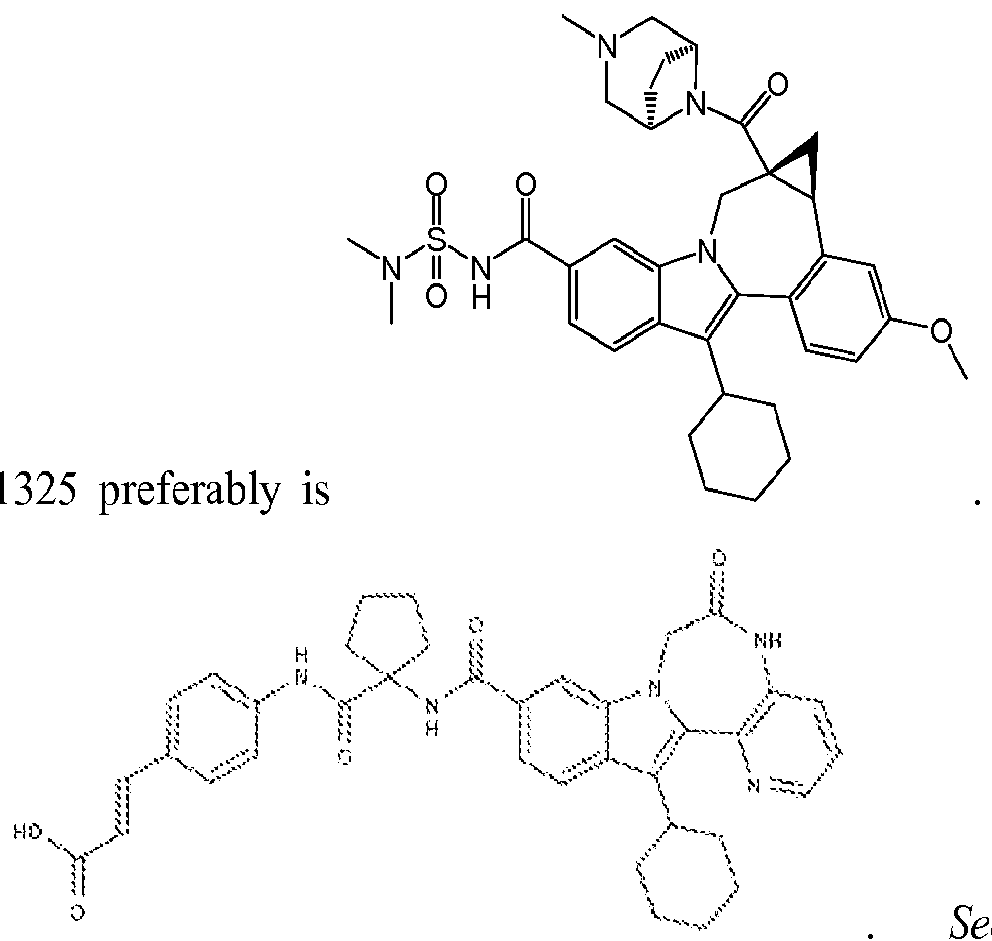
See also publications at http://wwwl .easl.eu/easl201 l/program/’Posters/Abstract680.htm; and http://clinicaltrials.gov/show/NCT00664625. For GS-5885, see publications at http://www.natap.org/201 l/EASL/EASL_68.htm; http://wwwl .easl.eu/easl2011/program/Posters/Abstractl 097.htm; and http://clinicaltrials.gov/ct2/show/NCT01353248.
| C 40 H 53 N 7 O 5 S 2 |

{kind=link}
{kind=link}
{kind=link}