
Home » japan 2019
Category Archives: japan 2019
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
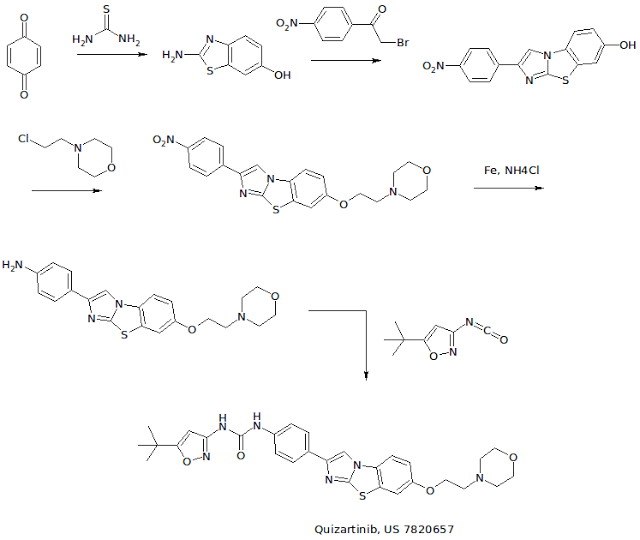
Quizartinib dihydrochloride, キザルチニブ塩酸塩
Quizartinib dihydrochloride
キザルチニブ塩酸塩
| Formula |
C29H32N6O4S. 2HCl
|
|---|---|
| CAS |
1132827-21-4
|
| Mol weight |
633.5891
|
JAPAN, PMDA APPROVED,. 2019/6/18, Vanflyta, To use as part of a treatment regimen for newly diagnosed acute myeloid leukemia that meets certain criteria
Drug Trials Snapshot
fda 2023, 7/20/2023
Quizartinib (AC220) is a small molecule receptor tyrosine kinase inhibitor, originated Ambit Biosciences, and acquired by Daiichi Sankyo, that is currently under development for the treatment of acute myeloid leukaemia. Its molecular target is FLT3, also known as CD135 which is a proto-oncogene.[1]
Flt3 mutations are among the most common mutations in acute myeloid leukaemia due to internal tandem duplication of Flt3. The presence of this mutation is a marker of adverse outcome.
Mechanism
Specifically, Quizartinib selectively inhibits class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), colony-stimulating factor 1 receptor (CSF1R/FMS), stem cell factor receptor (SCFR/KIT), and platelet derived growth factor receptors (PDGFRs).
Mutations cause constitutive action of Flt3 resulting in inhibition of ligand-independent leukemic cell proliferation and apoptosis.
Clinical trials
It reported good results in 2012 from a phase II clinical trial for refractory AML – particularly in patients who went on to have a stem cell transplant.[2]
As of 2017 it has completed 5 clinical trials and another 7 are active.[3]
SYN

References
- ^ Chao, Qi; Sprankle, Kelly G.; Grotzfeld, Robert M.; Lai, Andiliy G.; Carter, Todd A.; Velasco, Anne Marie; Gunawardane, Ruwanthi N.; Cramer, Merryl D.; Gardner, Michael F.; James, Joyce; Zarrinkar, Patrick P.; Patel, Hitesh K.; Bhagwat, Shripad S. (2009). “Identification of N-(5-tert-Butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea Dihydrochloride (AC220), a Uniquely Potent, Selective, and Efficacious FMS-Like Tyrosine Kinase-3 (FLT3) Inhibitor”. Journal of Medicinal Chemistry. 52 (23): 7808–7816. doi:10.1021/jm9007533.
- ^ Drug Tames Refractory AML. ASH Dec 2012
- ^ Quizartinib studies
 |
|
| Names | |
|---|---|
| IUPAC name
1-(5-(tert-Butyl)isoxazol-3-yl)-3-(4-(7-(2-morpholinoethoxy)benzo[d]imidazo[2,1-b]thiazol-2-yl)phenyl)urea
|
|
| Other names
AC220
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| KEGG | |
| UNII | |
|
CompTox Dashboard (EPA)
|
|
| Properties | |
| C29H32N6O4S | |
| Molar mass | 560.67 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
/////////////Quizartinib dihydrochloride, キザルチニブ塩酸塩, JAPAN 2019
OLD POST

N-(5-tert-butyl-isoxazol-3-yl)-N’-{ 4- [7-(2-morpholin-4-yl-ethoxy)imidazo [2, 1 -b] [ 1 ,3 ]benzothiazol-2-yl]phenyl } urea
| CAS | 950769-58-1 (free base) 1132827-21-4 (2HCl) |
| Formula | C29H32N6O4S |
| MW | 560.7 |
| Synonim | AC220, AC-010220 ASP-2689 |


Quizartinib
Ambit Biosciences
Ambit Biosciences (NASDAQ:AMBI) is a biotech company that focuses on treatments that inhibit kinases, which are drivers for diseases such as cancer. Three drugs are in development, with the lead one being quizartinib — a Phase 2B trial treatment for acute myeloid leukemia. However, AMBI’s collaboration agreement with Astellas Pharma is set to expire in September, and if it is not replaced, it could mean a delay in Phase 3 trials for quizartinib. Keep in mind that AMBI generated $23.8 million in collaboration revenues last year.
Quizartinib (AC220) is a small molecule receptor tyrosine kinase inhibitor that is currently under development by Ambit Biosciencesfor the treatment of acute myeloid leukaemia. Its molecular target is FLT3, also known as CD135 which is a proto-oncogene.[1]

AC-220 is an angiogenesis inhibitor that antagonizes several proteins involved in vascularization. It was engineered by Ambit Biosciences using KinomeScan technology to potently target FLT3, KIT, CSF1R/FMS, RET and PDGFR kinases. Ambit is developing oral AC-220 in phase III clinical studies for the treatment of relapsed/refractory acute myeloid leukemia (AML) patients with the FMS-like tyrosine kinase-3 (FLT3)-ITD mutation. Early clinical trials are also ongoing for the treatment of advanced solid tumors, for the treatment of refractory or relapsed myelodysplasia, in combination with induction and consolidation chemotherapy for previously-untreated de novo acute myeloid leukemia, and as a maintenance therapy of AML following hematopoietic stem cell transplantation (HSCT). In 2009, orphan drug designation was received both in the U.S. and in the EU for the treatment of AML. In 2009, Ambit Biosciences and Astellas Pharma have entered into a worldwide agreement to jointly develop and commercialize the drug candidate for the treatment of cancer and non-oncology indications. This agreement was terminated in 2013.
Flt3 mutations are among the most common mutations in acute myeloid leukaemia due to internal tandem duplication of Flt3. The presence of this mutation is a marker of adverse outcome.
| Quizartinib is a small molecule with potential anticancer activity. Quizartinib is a selective inhibitor of class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), stem cell factor receptor (SCFR / KIT), colony-stimulating factor 1 receptor (CSF1R/FMS) and platelet-derived growth factor receptors (PDGFRs .) Able to inhibition of ligand-independent cell proliferation and apoptosis. Mutations in FLT3 are the most frequent genetic alterations in acute myeloid leukemia (AML) and occur in approximately 30% of cases of AML. | |
| Quizartinib представляет собой малую молекулу с потенциальной противораковой активностью. Quizartinib является селективным ингибитором класса III рецепторов тирозин киназ, в том числе FMS-связанных тирозинкиназы 3 (FLT3/STK1), фактор стволовых клеток рецепторов (SCFR / KIT), колониестимулирующий фактор 1 рецепторов (CSF1R/FMS) и тромбоцитарный рецепторов фактора роста (PDGFRs). Способен к торможению лиганд-независимой клеточной пролиферации и апоптоза. Мутации в FLT3 являются наиболее частыми генетическими изменениями в остром миелобластном лейкозе (ОМЛ) и встречаются примерно в 30% случаев ОМЛ. |
Mechanism
Specifically, Quizartinib selectively inhibits class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), colony-stimulating factor 1 receptor (CSF1R/FMS), stem cell factor receptor (SCFR/KIT), and platelet derived growth factor receptors (PDGFRs).
Mutations cause constitutive action of Flt3 leading to resulting in inhibition of ligand-independent leukemic cell proliferation and apoptosis.

Clinical trials
It had good results in a phase II clinical trial for refractory AML – particularly in patients who went on to have a stem cell transplant.[2]

………………………..
WO 2007109120 COMPD B1
EXAMPLE 3: PREPARATION OF N-(5-TERT-BUTYL-ISOXAZOL-3-YL)-N’-{4-[7-(2- MORPHOLIN-4-YL-ETHOXY)IMIDAZO[2,1 -B3[1 ,3]BENZOTHIAZOL-2-YL]PHENYL}UREA [Compound B1]
[00426] A. The intermediate 2-amino-1,3-benzothiazol-6-ol was prepared according to a slightly modified literature procedure by Lau and Gompf. J. Org. Chem. 1970, 35, 4103-4108. To a stirred solution of thiourea (7.6 g, 0.10 mol) in a mixture of 200 ml_ ethanol and 9 ml_ concentrated hydrochloric acid was added a solution of 1 ,4-benzoquinone (21.6 g, 0.20 mol) in 400 mL of hot ethanol. The reaction was stirred for 24 hours at room temperature and then concentrated to dryness. The residue was triturated with hot acetonitrile and the resulting solid was filtered and dried.
[00427] The free base was obtained by dissolving the hydrochloride salt in water, neutralizing with sodium acetate, and collecting the solid by filtration. The product (2-amino-1 ,3-benzothiazol-6-ol) was obtained as a dark solid that was pure by LCMS (M+H = 167) and NMR. Yield: 13.0 g (78 %). NMR (DMSOd6) £7.6 (m, 2H ), 6.6 (d, 1H).
[00428] B. To prepare the intermediate 2-(4-nitrophenyl)imidazo[2,1- b][1 ,3]benzothiazoI-7-ol, 2-amino-1 ,3-benzothiazol-6-ol, (20.0 g, 0.12 mol) and 2-bromo-4′-nitroacetophenone (29.3 g, 0.12 mol) were dissolved in 600 mL ethanol and heated to reflux overnight. The solution was then cooled to 00C in an ice-water bath and the product was collected by vacuum filtration. After drying under vacuum with P2O5 , the intermediate (2-(4- nitrophenyl)imidazo[2,1-_D][1,3]benzothiazol-7-ol) was isolated as a yellow solid. Yield: 17.0 g (46 %) NMR (DMSO-CT6) δ 10 (s, 1 H), 8.9 (s, 1H), 8.3 (d, 2H), 8.1 (d, 2H), 7.8 (d, 1 H), 7.4 (s, 1 H), 6.9 (d, 1 H). [00429] C. To make the 7-(2-morpholin-4-yl-ethoxy)-2-(4-nttro- phenyl)imidazo[2,1-£>][1 ,3]benzothiazo!e intermediate: 2-(4- nitrophenyl)imidazo[2,1-jb][1 ,3]benzothiazol-7-ol, (3.00 g, 9.6 mmol) was suspended in 100 mL dry DMF. To this mixture was added potassium carbonate (4.15 g, 30 mmol, 3 eq), chloroethyl morpholine hydrochloride (4.65 g, 25 mmol, 2.5 eq) and optionally tetrabutyl ammonium iodide (7.39 g, 2 mmol). The suspension was then heated to 900C for 5 hours or until complete by LCMS. The mixture was cooled to room temperature, poured into 800 mL water, and allowed to stand for 1 hour. The resulting precipitate was collected by vacuum filtration and dried under vacuum. The intermediate, (7-(2- morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,1-jb][1 ,3]benzothiazole) was carried on without further purification. Yield: 3.87 g (95 %) NMR (DMSO-Cf6) δ 8.97 (s, 1 H), 8.30 (d, 2H), 8.0 (d, 2H), 7.9 (d, 1 H), 7.7 (s, 1 H), 7.2 (d, 1 H), 4.1 (t, 2H), 5.6 (m, 4H), 2.7 (t, 2H).
[00430] D. To make the intermediate 7-(2-morpholin-4-yl-ethoxy)-2-(4- amino-phenyl)!midazo[2, 1 -b][1 ,3]benzothiazole: To a suspension of 7-(2- morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,1-ib][1 ,3]benzothiazole (3.87g, 9.1 mmol) in 100 ml_ isopropyl alcohol/water (3:1 ) was added ammonium chloride (2.00 g, 36.4 mmol) and iron powder (5.04 g, 90.1 mmol). The suspension was heated to reflux overnight with vigorous stirring, completion of the reaction was confirmed by LCMS. The mixture was filtered through Celite, and the filtercake was washed with hot isopropyl alcohol (150 ml_). The filtrate was concentrated to approximately 1/3 of the original , volume, poured into saturated sodium bicarbonate, and extracted 3 times with dichloromethane. The combined organic phases were dried over MgSO4 and concentrated to give the product as an orange solid containing a small amount (4-6 %) of starting material. (Yield: 2.75 g 54 %). 80% ethanol/water may be used in the place of isopropyl alcohol /water — in which case the reaction is virtually complete after 3.5 hours and oniy traces of starting material are observed in the product obtained. NMR (DMSO-d6) δ 8.4 (s, 1 H), 7.8 (d, 1 H), 7.65 (d, 1 H), 7.5 (d, 2H), 7.1 (d, 1 H), 6.6 (d, 2H), 4.1 (t, 2H)1.3.6 (m, 4H), 2.7 (t, 2H).
[00431] E. A suspension of 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,1-b][1 ,3]benzothiazole (4.06 g, 10.3 mmol) and 5-tert- butylisoxazole-3-isocyanate (1.994 g, 12 mmol) in toluene was heated at 120 0C overnight. The reaction was quenched by pouring into a mixture of methylene chloride and water containing a little methanol and neutralized with saturated aqueous NaHCO3 solution. The aqueous phase was extracted twice with methylene chloride, the combined organic extracts were dried over MgSO4 and filtered. The filtrate was concentrated to about 20 ml volume and ethyl ether was added resulting in the formation of a solid. The precipitate was collected by filtration, washed with ethyl ether, and dried under vacuum to give the free base. Yield: 2.342 g (41 %) NMR (DMSO-Cf6) £9.6 (br, 1H), 8.9 (br, 1H), 8.61 (s, 1H), 7.86 (d, 1 H), 7.76 (d, 2H), 7.69 (d, 1 H), 7.51 (d, 2H), 7.18 (dd, 1H), 6.52 (s, 1H), 4.16 (t, 2H), 3.59 (t, 4H), 3.36 (overlapping, 4H), 2.72 (t, 2H), 1.30 (s, 9H). NMR (CDCI3) £9.3 (br, 1H), 7.84 (m, 4H), 7.59 (d, 2H), 7.49 (d, 1 H), 7.22 (d, 1 H), 7.03 (dd, 1 H)1 5.88 (s, 1 H), 4.16 (t, 2H), 3.76 (t, 4H), 2.84 (t, 2H), 2.61 (t, 4H), 1.37 (s, 9H).
[00432] F. For the preparation of the hydrochloride salt, N-(5-tert-butyl- isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2, 1 – b][1 ,3]benzothiazol-2-yl]phenyI}urea hydrochloride, the free base was dissolved in a mixture of 20 ml methylene chloride and 1 ml methanol. A solution of 1.0 M HCI in ethyl ether (1.1 eq.) was added dropwise, followed by addition of ethyl ether. The precipitate was collected by filtration or centrifugation and washed with ethyl ether to give the hydrochloride salt. Yield: 2.44 g (98 %) NMR (DMSO-d6) £11-0 (br, 1 H), 9.68 (s, 1H), 9.26 (s, 1H), 8.66 (s, 1 H), 7.93 (d, 1H), 7.78 (m, 3H), 7.53 (d, 2H), 7.26 (dd, 1H), 6.53 (S, 1 H), 4.50 (t, 2H), 3.97 (m, 2H), 3.81 (t, 2H), 3.6 (overlapping, 4H)13.23 (m, 2H)1 1.30 (s, 9H).
[00433] G. Alternatively, Compound B1 may be made by taking the intermediate from Example 4B and reacting it with chloroethyl morpholine hydrochloride under conditions described in Step C. [00434] H . Λ/-(5-tert-butyl-isoxazol-3-yl)-Λ/’-{4-[5-(2-morpholin-4-yl- ethoxy)imidazo[2,1-6][1 ,3]benzothiazol-2-yl]phenyl}urea hydrochloride, a compound having the general formula (I) where R1 is substituted on the 5 position of the tricyclic ring, was prepared in the manner described in Steps A- F but using the cyciization product 2-amino-benzothiazol-4-ol with 2-bromo-4′- nitroacetophenone in Step A. 1H NMR (DMSO-d6) δ 11.6 (br, 1 H)1 9.78 (br, 1H), 9.56 (br, 1 H), 8.64 (s, 1H)1 7.94 (d, 2H), 7.70 (s, 1H)1 7.56 (d, 2H), 7.45 (t, 1 H), 7.33 (d, 1H), 6.54 (s, 1 H), 4.79 (t, 2H), 3.87 (m, 6H), 3.60 (m, 2H), 3.34 (m, 2H)1 1.30 (s, 9H); LC-MS: ESI 561 (M+H)+. [Compound B11] [00435] I. N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[6-(2-morpholin-4-yl- ethoxy)imidazo[2,1-b][1 ,3]benzothiazol-2-yl]phenyl}urea hydrochloride [Compound B12] was also prepared by first preparing the benzothiazole starting material, 5 methoxy-benzothiazol-2yl~amine: [00436] To prepare the 5-methoxy-benzothiazol-2-ylamine starting material: To a suspension of (3-methoxy-phenyl)-thiourea (1.822g, 10 mmol) in CH2CI2 (20 ml_) at 0 0C was added dropwise a solution of bromine (1.76 g, 11 mmol) in 10 ml of trichloromethane over a period of thirty minutes. The reaction was stirred for 3 hours at room temperature then heated to 3 hours to reflux for one hour. The precipitate was filtered and washed with dichloromethane. The solid was suspended in saturated NaHCOsand extracted with CH2CI2. The extract was dried over MgSO4 and concentrated to give a white solid (1.716 g, 95%).
………………….
WO 2011056939
N-(5-ieri-butyl- isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof. N-(5-ieri-Butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- / ][!, 3]benzo

N- (5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-thiazol-2-yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, comprising any one, two, three, four, five, six, seven of the steps of:
(A) converting 2-amino-6-alkoxybenzothiazole (II), wherein R1 is a suitable phenolic hydroxyl protecting ;

(II) (III)
(B) reacting 2-amino-6-hydroxybenzothiazole (III) with compound (IV), wherein X is a leaving group, to yield 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol (V);

(C) reacting 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol (V) with compound (VI), wherein X2 is a leaving group, to yield 7-(2-morpholin-4-yl-ethoxy)-2-(4- nitrophenyl)imidazo[ -b]benzothiazole (VII);


(D) reducing 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo[2, 1- bjbenzothiazole (VII) to yield 7-(2-morpholin-4-yl-ethoxy)-2-(4- am

(E) converting 3-amino-5-£er£-butyl isoxazole (IX) to a 5-?er?-butylisoxazol-3- ylcarbamate derivative (X), wherein R2 is optionally substituted aryl, heteroaryl, alkyl, or cycloalkyl;

(IX) (X)
(F) reacting 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl)imidazo[2,l- bjbenzothiazole (VIII) with a 5-£er£-butylisoxazol-3-ylcarbamate derivative (X) to yield N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-

(G) converting N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2,l-&][l,3]benzo-thiazol-2-yl]phenyl}urea to an acid addition salt of N- (5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- b] [ 1 ,3]benzo-thiazol-2-yl]phenyl } urea.
[00128] In certain embodiments, provided herein are processes for the preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-thiazol-2-yl]phenyl}urea, or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, as depicted in Scheme 1, wherein R1, R2, X1, and X2 are defined herein elsewhere. In specific embodiments, provided herein are processes for the preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2,l-&][l,3]benzo-thiazol-2-yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, comprising any one, two, three, four, five, six, seven, of the Steps A, B, C, D, E, F, and G, as depicted in Scheme 1.
Scheme 1 :

A. Preparation of 2-amino-6-hydroxybenzothiazole
1. Example A-l[00252] To a 1-L 3-necked round bottom flask fitted with a condenser, heating mantle, and mechanical stirrer was charged aqueous hydrobromic acid (48%, 632 mL, 5.6 mol, 10 equiv). 2-Amino-6-methoxybenzothiazole (100 g, 0.55 mol, 1 equiv) was added to the above flask over 15 minutes. The reaction temperature was raised slowly to reflux (105-110 °C). A clear dark brown colored solution was observed at about 80 °C. The reflux was continued at 105-110 °C for about 4 hr. The progress of the reaction was monitored by HPLC. When 2-amino-6-methoxybenzothiazole was less than 2%, the reaction was substantially complete.
[00253] The reaction mass was cooled to 0-5 °C and at this point precipitation of a solid was observed. The mixture was maintained at 0-5 °C for 0.5 hr and filtered, and the cake was pressed to remove HBr. The wet cake was transferred to a 2-L round bottom flask fitted with a mechanical stirrer. Saturated aqueous sodium bicarbonate solution (-1500 mL) was added slowly at ambient temperature, whereupon considerable frothing was observed. The pH of the solution was found to be about 6.5 to 7. The mixture was stirred for 0.5 hr at ambient temperature and filtered. The filter cake was washed with water (500 mL), dried on the filter and then under vacuum at 30-35 °C for 10-12 hr to give the product 2-amino-6-hydroxybenzothiazole (82 g, 89% yield, HPLC purity = 99%). JH NMR (DMSO-if6, 500 MHz): δ 7.12 (d, 1H), 7.06 (S, 2H, NH2), 7.01 (d, 1H), 6.64 (dd, 1H); MS (m/z) = 167.1 [M+ + 1].
[00254] Table: Summary of Plant Batches

[00255] HPLC chromatographic conditions were as follows: The column used was XTerra RP8, 250 X 4.6 mm, 5μ or equivalent. Mobile Phase A was buffer, prepared by mixing 3.48 g of dipotassium hydrogen phosphate in 1.0 L of water, and adjusting the pH to 9.0 with phosphoric acid. Mobile Phase B was methanol. The flow rate was 1.0 mL/minute. Detection was set at UV 270 nm. The injection volume was 20 μΐ^, and the sample was diluted with a diluent (Mobile Phase A : Mobile Phase B = 70:30). Test solution was prepared by weighing accurately about 25 mg of sample and transferring it into a 100 mL volumetric flask, dissolving with 20-30 mL of diluent, making up the volume to the mark with diluent, and mixing. The HPLC was performed by separately injecting equal volumes of blank and test solution, and recording the chromatogram for all injections. The purity was calculated by area normalization method.
[00256] Table: HPLC Method

2. Example A-2
[00257] 2-Amino-6-methoxybenzothiazole was reacted with hot aqueous HBr at a temperature of >70 °C for about 3 hours and then the clear solution was cooled to ambient temperature overnight. The precipitated solids were collected, dissolved in hot water and the pH was adjusted to between 4.5-5.5. The resultant solids were collected, dried and re-crystallized from isopropanol. Second crop material was collected. The solids were vacuum dried to give 2-amino-6-hydroxybenzothiazole.
[00258] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a white solid, with 99.4% purity (HPLC area %). JH NMR (300 MHz, DMSO-if6) was collected, which conformed to structure.
3. Example A-3
[00259] A 22-L 3-neck round bottom flask was equipped with a mechanical agitator, thermocouple probe, a reflux condenser, and a heating mantle. The flask was charged with hydrobromic acid (14 L, 123.16 mol, 13.10 equiv). Heating was initiated and 2- amino-6-methoxybenzothiazole was added (1.7 kg, 9.4 mol, 1.00 equiv) over 10 minutes with stirring. The heating of the reaction mixture was continued to reflux, and maintained (>107 °C) for approximately 5 hours. The reaction mixture turned into a clear solution between 75 °C and 85 °C. The reaction progress was monitored by TLC until no starting material was observed (A -0.5 mL reaction mixture aliquot was diluted with -0.5 mL water as a clear solution, neutralized with sodium acetate to pH -5 and extracted with 1 mL dichloromethane. The organic layer was spotted: 5%
methanol/dichloromethane; Rf (product) = 0.35; Rf (starting material) = 0.40).
[00260] The reaction mixture was cooled to – 20 °C (overnight). White solids precipitated. The solids were filtered on a polypropylene filter and pressed to remove as much hydrobromic acid from the solids as possible to facilitate the subsequent pH adjustment step. The slightly wet crude product was dissolved in hot (50 °C) water (5 L). The clear solution was filtered to remove any insoluble material present, and the solids were washed with 50 °C water. The filtrate was cooled to 10 °C. The cooled filtrate was neutralized with sodium acetate (- 1.0 kg) to pH 4.5 to 5.5 with vigorous stirring. A thick white solid precipitated. The solids were collected by filtration, and washed with cool (-10 °C) water (2 x 1.0 L) and air dried.
[00261] The wet crude product was slurried in hot (50 °C) isopropanol (3 L) briefly and allowed to stand in a cool room (-5 °C) overnight. The solids were collected by filtration and washed with methyl ferf-butylether (2 x 500 mL). The solids were dried in a vacuum oven overnight (<30 mm Hg) at 30 °C (first crop). Yield: 1068 g (68%), white solid. HPLC: 99.4% (area). JH NMR (300 MHz, DMSO- ) conformed to structure.
[00262] The organic filtrate was collected in a total volume of 1.0 L, cooled to 10 °C. The off-white solids were precipitated and collected by filtration. The solids were dried in a vacuum oven overnight (<30 mm Hg) at 30 °C (second crop). Yield: 497 g (32%), off-white solid. HPLC: 99.8% (area).
[00263] The overall yield combining the first crop and the second crop was 1565 g, (99%).
B. Preparation of 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol

1. Example B-l[00264] A 3-L 3-neck round bottom flask fitted with a condenser, a heating mantle, and a mechanical stirrer was charged with H-butanol (1.5 L), followed by 2-amino-6- hydroxybenzothiazole (75 g, 0.45 mol, 1.0 equiv), 2-bromo-4′-nitroacetophenone (121 g, 0.50 mol, 1.1 equiv), and sodium bicarbonate (41.6 g, 0.50 mol, 1.0 equiv). The reaction temperature was gradually raised to reflux and maintained at reflux (110-115 °C) for 2-3 hr. During the temperature increase, the reaction mass turned into a clear solution and then immediately changed into an orange colored suspension at 65-75 °C. The progress of the reaction was monitored by HPLC analysis every 1 hr (reaction mass sample was submitted to QC). When the level of 2-amino-6-hydroxybenzothiazole was less than 2%, the reaction was substantially complete.
[00265] The reaction mass was slowly cooled to 50-60 °C and then further cooled to 0-5 °C and stirred for 15 min. The precipitated solids were collected by filtration, and dried on the filter. The wet cake was transferred in to a 1-L round bottom flask, and water (600 mL) was added. The suspension was stirred for 0.5 hr and filtered, and the solid was dried on the filter. The wet cake was again taken in to a 1-L round bottom flask and stirred with acetone (200 mL). The slurry was filtered and washed with acetone (2 X 100 mL), and the solid was dried on the filter, unloaded and further dried in a vacuum oven at ambient temperature to give the product 2-(4-nitrophenyl)imidazo[2,l- b]benzothiazol-7-ol (V) (120 g, 85.7% yield, HPLC purity = 98.7%). JH NMR (DMSO- d6, 500 MHz): δ 9.96 (s, 1H, OH), 8.93 (s, 1H), 8.27 (d, 2H), 8.06 (d, 2H), 7.78 (d, 1H), 7.38 (d, 1H), 6.97 (dd, 1H); MS (m/z) = 312 [M+ + 1].
[00266] Table: Summary of Plant Batches

* Input of 2-amino-6-hydroxybenzothiazole (III)
[00267] HPLC chromatographic conditions were as follows: The column used was XTerra RP8, 250 X 4.6 mm, 5μ or equivalent. Mobile Phase A was buffer, prepared by mixing 3.48 g of dipotassium hydrogen phosphate in 1.0 L of water, and adjusting the H to 9.0 with phosphoric acid. Mobile Phase B was methanol. The flow rate was 1.0 mL/minute. Detection was set at UV 235 nm. The injection volume was 10 μΐ^. The blank was prepared by transferring 200 μΐ. of DMSO and 200 μΐ. of 2M NaOH into a 10 mL volumetric flask, making up the volume to the mark with methanol, and mixing. The test solution was prepared by weighing accurately about 10 mg of sample and transferring it into a 50 mL volumetric flask, dissolving with 1 mL of DMSO and 1 mL of 2M NaOH, sonicating to dissolve, making up the volume to the mark with methanol, and mixing. The HPLC was performed by separately injecting equal volumes of blank and test solution, and recording the chromatogram for all injections. The purity was calculated by area normalization method.
[00268] Table: HPLC Method

2. Example B-2
[00269] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a reflux condenser, and a heating mantle. The flask was charged with 2-amino-6-hydroxybenzothiazole (1068 g, 6.43 mol, 1.0 equiv) and ethanol (200 proof, 32.0 L), and the suspension was stirred for 10 minutes. 2-Bromo-4- nitroacetophenone (1667 g, 6.49 mol, 1.01 equiv) was added in one portion. The reaction mixture was heated to reflux (78 °C). The reflux was maintained for approximately 25 hours, resulting in a yellow suspension. The reaction progress was monitored by TLC (20% methanol/ethyl acetate; Rf (product) = 0.85; Rf (starting material) = 0.30). TLC indicated -50% 2-amino-6-hydroxybenzothiazole after 24 hours of reflux. Tetrabutylammonium iodide (10 g) was added and reflux was maintained for an additional 12 hours. TLC indicated -50% starting material still present. Coupling was seen to occur at both the thiazole and the amine.
[00270] The reaction mixture was cooled to 0-5 °C. The solids were collected by filtration, and the yellow solid was washed with ethanol (200 proof, 2 X 1.0 L) and diethyl ether (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 40 °C. Yield: 930 g (46%), yellow solid. HPLC: 99.5% (area). JH NMR (300 MHz, DMSO-i¾) conformed to structure.
3. Example B-3
[00271] The reaction of Step B was carried out on multiple runs, varying solvents, temperature, and base. The results were summarized in the table below. The product (V) was isolated as yellow or green solids, with 1H NMR consistent with the structure and a purity of greater than about 98% by HPLC analysis.
[00272] Table: Reaction Condition Screening

TBAI = Tetrabutylammonium Iodide
C. Preparation of 7-(2-morpholin-4-yl-ethoxy)-2-(4- nitrophenyl)imidazo[2, 1 -bjbenzothiazole

1. Example C-l
[00273] To a 2000-L glass-lined (GL) reactor was charged DMF (298 kg), and the agitator was started. Under a nitrogen blanket, the reactor was charged with 2-(4- nitrophenyl)imidazo[2,l-&]benzothiazol-7-ol (36.8 kg, 118.2 mol, 1.0 equiv), 4-(2- chloroethyl)morpholine hydrochloride (57.2-66.0 kg, 307.3-354.6 mol, 2.6-3.0 equiv), tetrabutylammonium iodide (8.7 kg, 23.6 mol, 0.2 equiv) and potassium carbonate (49.0 kg, 354.6 mol, 3.0 equiv). The resulting yellow suspension was heated and stirred at 90 + 5 °C for at least 15 minutes, then the temperature was increased to 110 + 5 °C. The mixture was stirred for at least 1 hour and then sampled. The reaction was deemed complete if 2-(4-nitrophenyl) imidazo[2,l-&]benzothiazol-7-ol was <1% by HPLC. If the reaction was not complete, the heating was continued and the reaction sampled. If the reaction was incomplete after 6 hours, additional 4-(2-chloroethyl)morpholine hydrochloride may be charged. In general, additional charges of 4-(2- chloroethyl)morpholine hydrochloride had not been necessary at the given scale.
[00274] The reactor was cooled to 20 + 5 °C and charged with water (247 kg) and acetone (492 kg). The mixture was agitated at 20 + 5 °C for at least 1 hour. The product (VII) was isolated by filtration or centrifuge, and washed with water and acetone, and then dried in a vacuum oven at 45 °C to constant weight to give a yellow solid (46.2 kg, 92% yield, HPLC purity = 97.4% by area). JH NMR (300 MHz, DMSO- ) conformed to structure.
2. Example C-2
[00275] 2-(4-Nitrophenyl)imidazo[2, l-b]benzothiazol-7-ol, 4-(2-chloroethyl)- morpholine hydrochloride, potassium carbonate, and tetrabutylammonium iodide were added to N,N-dimethylformamide forming a yellow suspension that was heated at a temperature of >50 °C for over 3 hours. The reaction was cooled and the solids were collected, slurried into water, filtered, slurried into acetone, filtered and washed with acetone to give yellow solids that were dried under vacuum to give 7-(2-morpholin-4-yl- ethoxy)-2-(4-nitrophenyl)imidazo[2,l-b]benzothiazole.
[00276] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a yellow solid, with 99% purity (HPLC area %), and a water content of 0.20%. Infrared (IR) spectrum was collected, which conformed to structure.
3. Example C-3
[00277] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a drying tube, a reflux condenser, and a heating mantle. The flask was charged with 2-(4-nitrophenyl)imidazo [2,l-&]benzothiazol-7-ol (1.770 kg, 5.69 mol, 1.0 equiv), N,N-dimethylformamide (18.0 L), 4-(2-chloroethyl)morpholine hydrochloride (2.751 kg, 14.78 mol, 2.6 equiv), potassium carbonate (2.360 kg, 17.10 mol, 3.0 equiv), and tetrabutylammonium iodide (0.130 kg, 0.36 mol, 0.06 equiv) with stirring. The resulting yellow suspension was heated to about 90 °C to 95 °C, maintaining the temperature for approximately 5 hours. The reaction was monitored by TLC until no starting material was observed (20% methanol / ethyl acetate; Rf (product) = 0.15; Rf (starting material) = 0.85).
[00278] The reaction mixture was cooled to -10 °C, and the yellow solids were collected by filtration on a polypropylene filter pad. The solids were slurried in water (2 X 5 L) and filtered. The crude wet product was slurried in acetone (5 L), filtered, and the solids were rinsed with acetone (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 45 °C. Yield: 2.300 kg (95%), yellow solid. TLC: R/ = 0.15 (20% methanol / EtOAc). HPLC: 95.7% (area). JH NMR (300 MHz, DMSO-i¾) conformed to the structure.
[00279] Table: Yields from multiple batch runs

4. Example C-4
[00280] To a reactor were added 2-(4-nitrophenyl)imidazo [2,l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), tetrabutylammonium iodide (0.24 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) was added into the reactor. The DMF used had water content of no more than 0.05% w/w. The mixture was stirred for between 15 and 30 minutes to render a homogeneous suspension, which was heated to between 85 °C and 95 °C and stirred at between 85 °C and 95 °C for 15 to 30 minutes. The mixture was then heated to between 105 °C and 120 °C and stirred at between 105 °C and 120 °C (e.g. , 115 °C) until the reaction was complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction was not complete as indicated by HPLC analysis), additional 4-(2- chloroethyl)morpholine hydrochloride (0.03 kg) may be added and the reaction mixture stirred at between 105 °C and 120 °C (e.g. , 115 °C) until reaction completion.
[00281] The reaction mixture was cooled to between 20 °C and 30 °C (e.g. , over a period of 3 hours). To another reactor was added deionized water (7.6 L) and acetone (15 L). The mixture of water and acetone was then added into the reaction mixture while maintaining the temperature at between 20 °C and 30 °C. The mixture was then stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture was filtered, and the solid was washed with deionized water (e.g. , about 45x deionized water) until pH of washes was below 8. The solid was then washed with acetone (9.7 L). The solid was dried under vacuum at a temperature of less than 50 °C until the water content by Karl-Fischer was less than 0.30% w/w and TGA curve showed a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours). The desired product was obtained in about 89% yield having about 99% purity by HPLC.
5. Example C-5
[00282] To a reactor were added 2-(4-nitrophenyl)imidazo [2, l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) was added into the reactor. The DMF used had water content of no more than 0.05% w/w. The mixture was stirred for between 15 and 30 minutes to render a homogeneous suspension, which was heated to between 95 °C and 120 °C (e.g. , between 100 °C and 105 °C) and stirred at between 95 °C and 120 °C (e.g. , 105 °C) until the reaction was complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction was not complete as indicated by HPLC analysis), additional 4-(2-chloroethyl)morpholine hydrochloride (0.03 kg) and potassium carbonate (0.024 kg) may be added and the reaction mixture stirred at between 100 °C and 120 °C (e.g. , 105 °C) until reaction completion.
[00283] The reaction mixture was cooled to between 60 °C and 70 °C over a period of at least 60 minutes. Industrial water (6 L) was added to the reactor. The reaction mixture was cooled to between 20 °C and 30 °C. Acetone (6 L) was added to the reactor. The mixture was stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture was filtered, and the solid was washed with industrial water (e.g. , about 45 x industrial water) until pH of washes was below 8. The solid was then washed with acetone (9.7 L). The solid was dried under vacuum at a temperature of less than 50 °C, until the water content by Karl-Fischer was less than 0.30% w/w and TGA curve showed a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours).
6. Example C-6
[00284] To a reactor is added 2-(4-nitrophenyl)imidazo [2, l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) is added into the reactor. The DMF has a water content of no more than 0.05% w/w. The mixture is stirred for between 15 and 30 minutes to render a homogeneous suspension, which is heated to between 95 °C and 120 °C (e.g. , between 100 °C and 105 °C) and stirred at between 95 °C and 120 °C (e.g. , 105 °C) until the reaction is complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction is not complete as indicated by HPLC analysis), additional 4-(2-chloroethyl)morpholine hydrochloride (0.03 kg) and potassium carbonate (0.024 kg) may be added and the reaction mixture stirred at between 100 °C and 120 °C (e.g. , 105 °C) until reaction completion.
[00285] The reaction mixture is cooled to between 20 °C and 30 °C (e.g. , over a period of 3 hours). To another reactor is added deionized water (7.6 L) and acetone (15 L). The mixture of water and acetone is then added into the reaction mixture while maintaining the temperature at between 20 °C and 30 °C. The mixture is then stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture is filtered, and the solid is washed with deionized water (e.g. , about 45x deionized water) until pH of washes is below 8. The solid is then washed with acetone (9.7 L). The solid is dried under vacuum at a temperature of less than 50 °C until the water content by Karl-Fischer is less than 0.30% w/w and TGA curve shows a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours). D. Preparation of 7-(2-morpholin-4-yl-ethoxy)-2-(4- aminophenyl)imidazo [2, 1 -bjbenzothiazole

[00286] To a 200-L high pressure (HP) reactor was charged a slurry of 7-(2- morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2,l-&]benzothiazole (VII) (7.50 kg, 17.7 mol, 1.0 equiv) in methanol (30 kg). The container was rinsed with additional methanol (10 kg) and the rinse was charged to the reactor. The reactor was then charged with THF (67 kg) and methanol (19 kg). The contents were agitated and the reactor was flushed with nitrogen by alternating nitrogen and vacuum. Vacuum was applied to the reactor and Raney Ni catalyst (1.65 kg, 0.18 wt. equiv) was charged through a sample line. Water (1 kg) was charged through the sample line to rinse the line. The reactor was flushed with nitrogen by alternating nitrogen and vacuum. The reactor was then vented and heated to 50 °C. The reactor was closed and pressurized with hydrogen gas to 15 psi keeping the internal temperature below 55 °C. The reactor was vented and re- pressurized a total of 5 times, then pressurized to 150 psi with hydrogen gas. The contents were agitated at 50 °C for at least 4 hours. At this point a hydrogen uptake test was applied: The reactor was re-pressurized to 150 psi and checked after 1 hour. If a pressure drop of more than 5 psi was observed, the process was repeated. Once the pressure drop remained < 5 psi, the reactor was vented and sampled. The reaction was deemed complete when 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2,1- 6]benzothiazole (VII) was < 0.5% by HPLC.
[00287] The reactor was flushed with nitrogen as shown above. The 200-L HP reactor was connected to the 2000-L GL reactor passing through a bag filter and polish filter. The bag filter and polish filter were heated with steam. The 200-L HP reactor was pressurized (3 psi nitrogen) and its contents were filtered into the 2000-L reactor. The filtrates were hot. The 200-L reactor was vented and charged with THF (67 kg) and methanol (59 kg), the reactor agitated, and filtered into the 2000-L GL reactor.
[00288] A total of 6 reductions (46.2 kg processed) were carried out and the combined batches were concentrated by vacuum distillation (without exceeding an internal temperature of 40 °C) to a volume of -180 L. The reactor was cooled to 20 °C and charged with heptane (250 kg) and again vacuum distilled to a volume of -180 L. The reactor was charged with heptane (314 kg) and agitated at 20 °C for at least 1 hour, and then the product was isolated by centrifugation or collection on a Nutsche filter, washing with heptanes (2-5 kg per portion for centrifugation, followed by a 10-20 kg heptanes rinse of the reactor; or 94 kg for Nutsche filtration, rinsing the reactor first). The cake was blown dry, transferred to a vacuum oven and dried to constant weight maintaining a temperature < 50 °C to give the desired product (VIII) (34.45 kg, 80% yield, HPLC purity = 97.9%).
2. Example D-2
[00289] 7-(2-Morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo[2,l-b]benzothiazole was dissolved into methanol and THF and placed in a hydrogenator. Raney nickel was added and the vessel was pressurized with hydrogen and stirred for >24 hours. The reaction mixture was concentrated to a thick paste and diluted with methyl ferf-butyl ether. The resulting solids were filtered and washed with methyl ferf-butyl ether and dried under vacuum to give 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl) imidazo [2, 1 -bjbenzothiazole.
[00290] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a yellow solid, with 99% purity (HPLC area %). IR was collected, which conformed to structure.
3. Example D-3
[00291] Into a 5-gallon autoclave, 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl) imidazo[2,l-&]benzothiazole (580 g, 1.37 mol, 1.0 equiv), THF (7.5 L), methanol (7.5 L, AR) and -55 g of decanted Raney nickel catalyst were added. The reaction vessel was purged with nitrogen (3 X 50 psi) and hydrogen (2 X 50 psi), with stirring briefly under pressure and then venting. The autoclave was pressurized with hydrogen (150 psi). The mixture was stirred and the hydrogen pressure was maintained at 150 psi for over 24 hours with repressurization as necessary. The reaction progress was monitored by TLC (10% methanol / chloroform with 1 drop ammonium hydroxide; Rf (product) 0.20; Rf (SM) 0.80). The reaction was substantially complete when the TLC indicated no starting material present, typically after 24 hours of stirring at 150 psi. The hydrogenation was continued at 150 psi for a minimum of 4 hours or until completion if starting material was still present after the initial 4 hours.
[00292] The reaction mixture was filtered through a Buchner funnel equipped with a glass fiber filter topped with a paper filter. Unreacted starting material was removed together with the catalyst. The filtrate was concentrated to a total volume of 0.5 L, and the residue was triturated with methyl ferf-butyl ether (0.5 L). The resultant solids were collected by filtration, and washed with methyl ferf-butyl ether (0.3 L) (first crop).
[00293] The filtrate was concentrated to dryness and the residue was diluted with methyl ferf-butyl ether (2 L). The resultant solids were collected by filtration, washing with methyl ferf-butyl ether (0.5 L) (second crop).
[00294] The solids were dried in a vacuum oven (<10 mm Hg) at 25 °C. Yield: 510 g (95%), beige solid. TLC: R/ 0.2 (10% methanol / chloroform with one drop of ammonium hydroxide). HPLC: 99.0% (area). JH NMR (300 MHz, DMSO-i¾) conformed to the structure.
[00295] Table: Yields from multiple batch runs

4. Example D-4
[00296] The reaction of Step D was carried out in multiple runs under various conditions, such as, e.g. , varying catalyst loading, concentration of reactant, reaction temperature, and/or workup procedures. The results are summarized in the table below.

Description Run # l Run # 2 Run # 3 Run # 4 Run # 5Rxn Temp (°C) RT RT RT RT RT
Rxn Time (Hr) 24 hr 24 hr 24 hr 24 hr 24 hr
Filtered the Filtered the solution
Filtered the Filtered the Filtered the
solution through through celite. The solution through solution through solution through
celite, washed celite filter cake celite, celite, celite,
with THF, refluxed in THF concentrated, concentrated, concentrated,
concentrated, washed with hot solvent exchanged solvent exchanged solvent exchanged
Work Up solvent exchanged THF, concentrated, with heptane, with heptane, with heptane,
with heptane, solvent exchanged stirred the solids stirred the solids stirred the solids
stirred the solids with heptane, stirred and filtered and filtered and filtered
and filtered the solids and washed with washed with washed with
washed with filtered washed with heptane heptane heptane
heptane heptane
Produce (VIII) 1.9 g 3.88 g 1.11 g 2.6 g 4.4 g
Yield 88% 83.4% 56 94.6%
HPLC purity 95.6% 77.5% 91% 93.8%

5. Example D-5
[00297] To a pressure reactor under nitrogen atmosphere was added a slurry of Raney Nickel in water (0.22 kg) (e.g. about 0.14 kg dry catalyst in water) and the charging line was rinsed with deionized water (0.13 L). To another reactor (Reactor B) were added methanol (5.05 L) and 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2, 1- &]benzothiazole (1.0 kg), and the mixture was stirred for between 15 and 30 minutes to render a homogenous suspension. The suspension was transferred to the pressure reactor. Reactor B was washed with methanol (4.88 L) and the wash was transferred to the pressure reactor. Tetrahydrofuran (10.1 L) was added to the pressure reactor.
Hydrogen was charged to the pressure reactor to a pressure of between 2.0 bar and 3.0 bar. The reactor was heated to a temperature of between 45 °C and 55 °C. Hydrogen was then charged to the pressure reactor to a pressure of between 6.0 bar and 7.0 bar. The mixture was stirred at a temperature of between 45 °C and 55 °C (e.g. , 50 °C), while maintaining the hydrogen pressure between 6.0 bar and 7.0 bar until reaction completion (as determined by HPLC to monitor the consumption of starting material).
[00298] The mixture was filtered while maintaining the temperature at between 35 °C and 50 °C. The pressure reactor and the filter were washed with a mixture of THF (10.1 L) and methanol (9.93 L) preheated to a temperature of between 45 °C and 55 °C (e.g. , 50 °C). The combined filtrate was concentrated to a volume of between 2.4 L and 2.8 L under vacuum at a temperature of no more than 40 °C, and a precipitate was formed. Methanol (7.5 L) was added. The slurry was cooled to a temperature of between 5 °C and -5 °C (e.g. , over 3 hours) and stirred for at least 1 hour (e.g. , for 3 hours) while maintaining the temperature at between 5 °C and -5 °C. The suspension was filtered. The solid was washed with methanol (2 X 1.2 L). The solid was then dried under vacuum at a temperature of less than 50 °C until the methanol and THF contents were each less than 3000 ppm as analyzed by GC (e.g. , less than 1500 ppm). The desired product was obtained in about 88.5% yield having about 99% purity by HPLC.
E. Preparation of phenyl 5-£er£-butylisoxazol-3-ylcarbamate

[00299] The jacket temperature of a 200-L glass-lined (GL) reactor was set to 20 °C. To the reactor was charged 5-ieri-butylisoxazole-3-amine (15.0 kg, 107.0 mol, 1.0 equiv), then K2C03 (19.5 kg, 141.2 mol, 1.3 equiv) and anhydrous THF (62 kg).
Agitation was started and then phenyl chloroformate (17.6 kg, 112.4 mol, 1.05 equiv) was charged. The charging line was rinsed with additional anhydrous THF (5 kg). The reaction was agitated at 20 + 5 °C for at least 3 hours then sampled. The reaction was deemed complete if 5-£er£-butylisoxazole-3-amine was < 2% by HPLC. If the reaction was not complete after 6 hours, additional K2CO3 and phenyl chloroformate may be added to drive the reaction to completion.
[00300] Once complete, the reaction was filtered (Nutsche). The filter was rinsed with THF (80 kg). The filtrate was vacuum distilled without exceeding an internal temperature of 40 °C until -50 L remained. Water (188 kg) and ethanol (45 L) were charged, and the mixture was agitated for at least 3 hours with a jacket temperature of 20 °C. The resulting solid was isolated by centrifugation or collection on a Nutsche filter, rinsed with water (2-5 kg for each centrifugation portion; 30 kg for Nutsche filtration) and blow-dried. The solid was then dried to constant weight in a vacuum oven (45 °C) to give the desired product (19.4 kg, 92% yield, HPLC purity = 97.4%). On an 800 g scale, 1559 g of the desired product (98% yield) was obtained with a 99.9% HPLC purity. JH NMR (DMSO-i¾) δ 11.17 (s, 1H); 7.4 (m, 2H); 7.2 (m, 3H); 1.2 (s, 9H). LCMS (M+H)+ 261.
F. Preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, 1 -b] [ 1 ,3 ]benzothiazol-2-yl]phenyl } urea

1. Example F-l
[00301] The jacket of a 2000-L GL reactor was set to 20 °C and the reactor was charged with 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl)imidazo[2,l- &]benzothiazole (26.7 kg, 67.8 mol, 1.0 equiv), 3-amino-5-?-butylisoxazole phenyl carbamate (19.4 kg, 74.5 mol, 1.1 equiv), DMAP (0.5 kg, 4.4 mol, 0.06 equiv), and DCM (anhydrous, 260 kg). Agitation was started, triethylamine (1.0 kg, 10.2 mol, 0.15 equiv) was charged followed by additional DCM (5 kg) through the charging line. The reaction was heated to reflux (-40 °C) and agitated for at least 20 hours with complete dissolution observed followed by product crystallizing from solution after -30 minutes. The reaction was sampled and deemed complete when HPLC analysis showed a ratio of compound (VIII) to compound (I) < 1%.
[00302] The reactor was cooled to 0 °C and stirred for at least 2 hours. The content of the reactor were isolated by centrifuge. Each portion was rinsed with 2-3 kg of cold (0 °C) DCM and spun dry for at least 5 minutes with a 10 psi nitrogen purge. For the final portion, the reactor was rinsed with 10 kg of cold (0 °C) DCM and transferred to the centrifuge where it was spun dry for at least 5 minutes with a 10 psi nitrogen purge. The combined filter cakes were transferred to a vacuum tray dryer and dried to constant weight at 50 °C and at least >20 inches of Hg to give the desired product (I) (35.05 kg, 92% yield, HPLC purity = 98.8%). Phenol was the major impurity detected (0.99%); and three other impurities (<0.10%) were detected. JH NMR (300 MHz, DMSO- ) conformed to structure.
2. Example F-2
[00303] A variety of solvents were used in the reaction of Step F to optimize for better yields and purity profiles. The contents of the symmetrical urea impurity (XI) were compared and summarized in the table below:

http://www.google.com/patents/WO2011056939A1?cl=en SE THIS FOR DELETED CLIPS


4. Example F-4
[00305] To Reactor A under a nitrogen atmosphere was added 7-(2-morpholin-4-yl- ethoxy)-2-(4-aminophenyl)imidazo[2,l-&]benzothiazole (1 kg) and DMAP (0.02 kg). To Reactor B under a nitrogen atmosphere was added 3-amino-5-?-butylisoxazole phenyl carbamate (0.73 kg) and DCM (5.6 L). The DCM used had a water content of less than 0.05 % w/w. The mixture in Reactor B was stirred until dissolution. The solution was transferred into Reactor A (the solution can be filtered into Reactor A to remove any insoluble impurities in the carbamate starting material), and the mixture was stirred in Reactor A. Reactor B was washed with DCM (0.8 L) and the wash was transferred into Reactor A. Reactor A was washed with DCM (0.9 L). To Reactor A was added triethylamine (0.1 L) and the charging vessel and lines were rinsed with DCM (0.1 L) into Reactor A. The mixture was then heated to reflux and stirred at reflux until reaction completion (as determined by HPLC to monitor the consumption of starting material).
[00306] The reaction mixture was cooled (e.g. , over 2 to 3 hours) to a temperature of between -5 °C and 5 °C (e.g. , 0 °C). The mixture was then stirred for 2 to 3 hours at a temperature of between -5 °C and 5 °C (e.g. , 0 °C). The suspension was filtered. The solid was washed with cool DCM (2 X 1.5 L) (pre-cooled to a temperature of between -5 °C and 5 °C). The solid was dried under vacuum at a temperature of less than 45 °C until the DCM content was less than 1000 ppm (e.g., below 600 ppm) as analyzed by GC. The desired product was obtained having about 99% purity by HPLC.
G. Preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, l-b] [1 ,3]benzothiazol-2-yl]phenyl }urea dihydrochloride

1. Example G-l
[00307] The jacket of a 2000-L GL reactor was set to 20 °C and the reactor was charged with N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo [2, 1-&][1, 3]benzothiazol-2-yl]phenyl}urea (35.0 kg, 62.4 mol, 1.0 equiv) followed by methanol (553 kg). Agitation was started and the reaction mixture was heated to reflux (-65 °C). Concentrated aqueous HC1 (15.4 kg, 156.0 mol, 2.5 equiv) was charged rapidly (<5 minutes) and the charge line was rinsed into the reactor with methanol (12 kg). Addition of less than 2.0 equivalents of HC1 normally resulted in the formation of an insoluble solid. The reaction mixture was heated at reflux for at least 1 hour. Upon HC1 addition, the slurry dissolved and almost immediately the salt started to crystallize, leaving insufficient time for a polish filtration.
[00308] The reactor was cooled to 20 °C and the product was isolated by filtration (Nutsche) rinsing the reactor and then the cake with methanol (58 kg). The solid was then dried in a vacuum oven (50 °C) to constant weight to give the desired
dihydrochloride salt (35 kg, 89% yield, HPLC purity = 99.94%). JH NMR (300 MHz, DMSO-i¾) conformed to structure.
2. Example G-2
[00309] Concentrated HC1 was added to a suspension of N-(5-ieri-butyl-isoxazol-3- yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea in warm methanol forming a solution that slowly began to precipitate. The reaction mixture was refluxed for over 2 hours and then stirred overnight at ambient temperature. The dihydrochloride salt was collected and dried under vacuum.
3. Example G-3
[00310] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a nitrogen inlet, a drying tube, a reflux condenser, an addition funnel, and a heating mantle. The flask was charged with N-(5-ieri-butyl-isoxazol-3-yl)- N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2-yl]phenyl}urea (775 g, 1.38 mol, 1.0 equiv) and MeOH (40 L, AR). The resulting off-white suspension was heated to reflux (68 °C). A clear solution did not form. HC1 (37% aqueous) (228 mL, 3.46 mol, 2.5 equiv) was added over 5 minutes at 68 °C. The reaction mixture turned into a clear solution and then a new precipitate formed within approximately 3 minutes. Heating was continued at reflux for approximately 5 hours. The reaction mixture was allowed to cool to ambient temperature overnight. The off-white solids were collected by filtration on a polypropylene filter, washing with MeOH (2 X 1 L, AR). [00311] Two lots of material prepared in this manner were combined (740 g and 820 g). The combined solids were slurried in methanol (30 L) over 30 minutes at reflux and allowed to cool to the room temperature. The solids were collected by filtration on a polypropylene filter, rinsing with methanol (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 40 °C. Yield: 1598 g (84%), off-white solid. HPLC: 98.2% (area). MS: 561.2 (M+l)+. JH NMR (300 MHz, DMSO-i¾) conformed to the structure. Elemental Analysis (EA): Theory, 54.97 %C; 5.41 %H; 13.26 %N; 5.06 %S; 11.19 %C1; Actual, 54.45 %C; 5.46 %H; 13.09 %N; 4.99 %S; 10.91 %C1.
4. Example G-4
[00312] Into a 50-L 3-neck round bottom flask equipped with a mechanical stirrer, a heating mantle, a condenser and a nitrogen inlet, were charged N-(5-ieri-butyl-isoxazol- 3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea (1052.4 g, 1.877 mol, 1.0 equiv) and methanol (21 L). The reactor was heated and stirred. At an internal temperature of > 50 °C, cone. HC1 (398.63 mL, 4.693 mol, 2.5 equiv) was charged over 5 minutes through an addition funnel. During the addition, the mixture changed from a pale yellow suspension to a white suspension. The internal temperature was 55 °C at the conclusion of the addition. The mixture was heated to reflux for 1 hour, then heating was discontinued and the mixture was allowed to cool to room temperature. The mixture was filtered in two portions, and each filter cake was washed with methanol (2 X 1 L), transferred to trays and dried in a vacuum oven (45 °C) to constant weight. The dried trays were combined to produce 1141.9 g of the salt (96% yield, 99.1 % HPLC purity, 10.9% chloride by titration).
H. Analytical Data
1. N-(5-ieri-butyl-isoxazol-3-yl)-N’-{ 4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, l-&] [l ,3]benzothiazol-2-yl]phenyl}urea
dihydrochloride
[00314] A batch of about 30 grams of N-(5-ieri-butyl-isoxazol-3-yl)-N’- {4-[7-(2- morpholin-4-yl-ethoxy)imidazo[2, l-&] [l ,3]benzothiazol-2-yl]phenyl}urea
dihydrochloride was prepared using the methods described herein. This lot was
prepared in accordance with the requirements for production of clinical Active
Pharmaceutical Ingredients (APIs) under GMP conditions. The analytical data for this batch was obtained, and representative data were provided herein. [00315] Summary of analytical data for the dihydrochloride salt.


………………………
EXAMPLE 1. SYNTHESIS OF N-(5-TERT-BUTYL-ISOXAZOL-3-YU- N>-{4-f7-(2-MORPHOLIN-4- YL-ETHOXY)IMID AZO[2,1- BlH,31BENZOTHIAZOL-2-YL|PHENYLiUREA (“COMPOUND Bl”)
[00357] A. The intermediate 2-amino-l,3-benzothiazol-6-ol was prepared according to a slightly modified literature procedure by Lau and Gompf: J. Org. Chem. 1970, 35, 4103- 4108. To a stirred solution of thiourea (7.6 g, 0.10 mol) in a mixture of 200 mL ethanol and 9 mL concentrated hydrochloric acid was added a solution of 1 ,4-benzoquinone (21.6 g, 0.20 mol) in 400 mL of hot ethanol. The reaction was stirred for 24 hours at room temperature and then concentrated to dryness. The residue was triturated with hot acetonitrile and the resulting solid was filtered and dried.
[00358] The free base was obtained by dissolving the hydrochloride salt in water, neutralizing with sodium acetate, and collecting the solid by filtration. The product (2- amino-l,3-benzothiazol-6-ol) was obtained as a dark solid that was pure by LCMS (M+H = 167) and NMR. Yield: 13.0 g (78 %). NMR (DMSO-^) Sl.6 (m, 2H), 6.6 (d, IH). [00359] B. To prepare the 2-(4-nitrophenyl)imidazo[2,l-b][l,3]benzothiazol-7-ol intermediate, 2-amino-l,3-benzothiazol-6-ol (20.0 g, 0.12 mol) and 2-bromo-4′- nitroacetophenone (29.3 g, 0.12 mol) were dissolved in 600 mL ethanol and heated to reflux overnight. The solution was then cooled to O0C in an ice-water bath and the product was collected by vacuum filtration. After drying under vacuum with P2O5 , the intermediate (2- (4-nitrophenyl)imidazo[2,l-£][l,3]benzothiazol-7-ol) was isolated as a yellow solid. Yield: 17.0 g (46 %) NMR (DMSO-(I6) δ 10 (s, IH), 8.9 (s, IH), 8.3 (d, 2H), 8.1 (d, 2H), 7.8 (d, IH), 7.4 (s, IH), 6.9 (d, IH).
[00360] C. To make the 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,l-
6][l,3]benzothiazole intermediate: 2-(4-nitrophenyl)imidazo[2,l-6][l,3]benzothiazol-7-ol,
NYI-4144519vl 84 (3.00 g, 9.6 mmol) was suspended in 100 mL dry DMF. To this mixture was added potassium carbonate (4.15 g, 30 mmol, 3 eq), chloroethyl morpholine hydrochloride (4.65 g, 25 mmol, 2.5 eq) and optionally tetrabutyl ammonium iodide (7.39 g, 2 mmol). The suspension was then heated to 900C for 5 hours or until complete by LCMS. The mixture was cooled to room temperature, poured into 800 mL water, and allowed to stand for 1 hour. The resulting precipitate was collected by vacuum filtration and dried under vacuum. The intermediate, (7-(2-morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2, 1 – b][\, 3]benzothiazole) was carried on without further purification. Yield: 3.87 g (95 %) NMR (DMSO-d6) δ 8.97 (s, IH), 8.30 (d, 2H), 8.0 (d, 2H), 7.9 (d, IH), 7.7 (s, IH), 7.2 (d, IH), 4.1 (t, 2H), 5.6 (m, 4H), 2.7 (t, 2H).
[00361] D. To make the intermediate 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,l-b][l,3]benzothiazole: To a suspension of 7-(2-morpholin-4-yl-ethoxy)- 2-(4-nitro-phenyl)imidazo[2,l -b][\ , 3]benzothiazole (3.87g, 9.1 mmol) in 100 mL isopropyl alcohol/water (3:1) was added ammonium chloride (2.00 g, 36.4 mmol) and iron powder (5.04 g, 90.1 mmol). The suspension was heated to reflux overnight with vigorous stirring, completion of the reaction was confirmed by LCMS. The mixture was filtered through Celite, and the filtercake was washed with hot isopropyl alcohol (150 mL). The filtrate was concentrated to approximately 1/3 of the original volume, poured into saturated sodium bicarbonate, and extracted 3 times with dichloromethane. The combined organic phases were dried over MgSO4 and concentrated to give the product as an orange solid containing a small amount (4-6 %) of starting material. (Yield: 2.75 g 54 %). 80% ethanol/water may be used in the place of isopropyl alcohol /water – in which case the reaction is virtually complete after 3.5 hours and only traces of starting material are observed in the product obtained. NMR (DMSO-Λfc) δ 8.4 (s, IH), 7.8 (d, IH), 7.65 (d, IH), 7.5 (d, 2H), 7.1 (d, IH), 6.6 (d, 2H), 4.1 (t, 2H), 3.6 (m, 4H), 2.7 (t, 2H).
[00362] E. A suspension of 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,l-b][l,3]benzothiazole (4.06 g, 10.3 mmol) and 5-tert-butylisoxazole-3- isocyanate (1.994 g, 12 mmol) in toluene was heated at 120 0C overnight. The reaction was quenched by pouring into a mixture of methylene chloride and water containing a little methanol and neutralized with saturated aqueous NaHCO3 solution. The aqueous phase was extracted twice with methylene chloride, the combined organic extracts were dried over
NYI-4144519vl 85 MgSO4 and filtered. The filtrate was concentrated to about 20 ml volume and ethyl ether was added resulting in the formation of a solid. The precipitate was collected by filtration, washed with ethyl ether, and dried under vacuum to give the free base of Compound B 1. Yield: 2.342 g (41 %) NMR (DMSO-J6) £9.6 (br, IH), 8.9 (br, IH), 8.61 (s, IH), 7.86 (d, IH), 7.76 (d, 2H), 7.69 (d, IH), 7.51 (d, 2H), 7.18 (dd, IH), 6.52 (s, IH), 4.16 (t, 2H), 3.59 (t, 4H), 3.36 (overlapping, 4H), 2.72 (t, 2H), 1.30 (s, 9H). NMR (CDCl3) £9.3 (br, IH), 7.84 (m, 4H), 7.59 (d, 2H), 7.49 (d, IH), 7.22 (d, IH), 7.03 (dd, IH), 5.88 (s, IH), 4.16 (t, 2H), 3.76 (t, 4H), 2.84 (t, 2H), 2.61 (t, 4H), 1.37 (s, 9H).
6.2 EXAMPLE 2. ALTERNATIVE SYNTHESIS QF N-(5-TERT-BUTYL- ISOXAZQL-3- YL)-N -{4-[7-q-MORPHOLIN-4- YL- ETHOXYUMID AZOf2,l-BUl,31BENZOTHIAZOL-2- YLIPHENYLIUREA (“COMPOUND Bl”)
[00363] A. To a suspension of the intermediate 2-(4-Nitrophenyl)imidazo[2,l- b][l,3]benzothiazol-7-ol from Example IB (2.24 g, 7.2 mmol) in ethanol (40 mL) was added SnCl2 1H2O (7.9Og, 35 mmol) and heated to reflux. Concentrated HCl was added to the reaction mixture and the precipitate formed gradually. The reaction mixture was heated to reflux for 20 hours and then allowed to cool to room temperature. The solution was poured into ice and neutralized with 10% NaOH and adjusted to approximately pH 6. The organic phase was extracted three times with ethyl acetate (80 mL x 3). Extracts were dried over MgSθ4 and concentrated to give a yellow solid. (1.621 g, 80%). The solid was recrystallized from methanol to give a pure product (1.355 g, 67%).
[00364] B. To a suspension of the intermediate from Step 2A (0.563 g, 2 mmol) in toluene (30 mL) was added 5-tert-butylisoxazole-3-isocyanate (0.332g, 2 mmol) and heated to reflux overnight. LC-MS analysis showed presence of the intermediate but no trace of 5- tert-butylisoxazole-3-isocyanate and an additional 0.166 g of the isocyanate was added. The reaction was again heated to reflux overnight. Completion of reaction was verified by LC- MS. The solvent was removed and the resulting mixture was dissolved in methanol which was removed to give the second intermediate as a solid.
[00365] The mixture was dissolved in CH2Cl2 (150 mL) and washed with saturated
NaHCO3. The organic layer was dried over MgSO4, concentrated, and purified by silica gel chromatography three times, first using a methanol/CH2Cl2 gradient, the second time using a
NYI-4144519vl 86 hexane/ethyl acetate gradient followed by a methanol/ethyl acetate gradient, and a third time using a methanol/CH2Cl2 gradient.
[00366] C. To a suspension of the intermediate from Step 2B (0.1 10 g, 0.25 mmol) in
THF (5mL) was added Ph3P (0.079g, 0.3 mmol), diisopropylazodicarboxylate (0.06 Ig, 0.3 mmol) and 4-morpholinoethanol (0.039 g, 0.3 mmol). The reaction mixture was stirred at room temperature overnight. Completion of the reaction was verified by LC-MS. The solvent was removed and the final product was purified using silica gel chromatography, with methanol in CH2Cl2 (0.030g, 21%).
6.3 EXAMPLE 3. BULK SYNTHESIS OF N-(5-TERT-BUTYL- ISOXAZOL-3-YL)-N’-f4-[7-(2-MORPHOLIN-4-YL- ETHOXY^IMID AZO[2α-BUlJlBENZOTHIAZOL-2- YLlPHENYLiUREA (“COMPOUND Bl”)
[00367] A multi-step reaction scheme that was used to prepare bulk quantities of
Compound Bl is depicted in FIG. 66a and FIG. 66b, and is described further below. [00368] Step 1 : Preparation of 2- Amino-6-hydroxybenzothiazole (Intermediate 1). 2-
Amino-6-methoxybenzothiazole is reacted with hot aqueous HBr for about 3 hrs and then the clear solution is cooled to ambient temperature overnight. The precipitated solids are collected, dissolved in hot water and the pH is adjusted to between 4.5-5.5. The resultant solids are collected, dried and recrystallized from Isopropanol. Second crop material is collected. The solids are vacuum dried to give Intermediate 1.
[00369] Step 2: Preparation of 2-(4-Nitrophenyl) imidazo [2J-b]benzothiazol-7-ol
(Intermediate 2). 2-Amino-6-hydroxybenzothiazole, 2-Bromo-4-nitroacetophenone and absolute Ethanol are added together and heated to reflux for approximately 24 hours. Tetrabutylammonium iodide is added and the reaction is refluxed an additional 12 hours. The resulting yellow suspension is cooled and the solids collected and washed with Ethanol and Diethyl ether. The solids are dried under vacuum to give Intermediate 2. [00370] Step 3: Preparation of 7-(2-Morpholin-4-yl-ethoxy)-2-(4-nitrophenyl) imidazo
[2,1-b] benzothiazole (Intermediate 3). Intermediate 2, 4-(2-Chloroethyl)morpholine hydrochloride, Potassium carbonate and Tetrabutylammonium iodide are added to N,N- Dimethylformamide forming a yellow suspension that is heated for over 3 hours. The reaction is cooled and the solids are collected, slurried into water, filtered, slurried into
NYl-4 l4451′)v l 87 acetone, filtered and washed with Acetone to give yellow solids that are dried under vacuum to give Intermediate 3.
[O0371] Step 4: Preparation of 7-(2-Moφholin-4-yl-ethoxy)-2-(4-aminophenyl) imidazo [2,1 -b] benzothiazole (Intermediate 4). Intermediate 3 is dissolved into Methanol and THF and placed in a Hydrogenator. Raney Nickel is added and the vessel is pressurized with Hydrogen and stirred for >24 hrs. The reaction mixture is concentrated to a thick paste and diluted with Methyl tert-butyl ether. The resulting solids are filtered and washed with Methyl tert-butyl ether and dried under vacuum to give Intermediate 4. [O0372] Step 5: Preparation of {[5-(tert-Butyl) isoxazol-3-vnatnino}-N-{4-r7-(2- morpholin-4-yl-ethoxy)(4-hvdroimidazolo[2J-blbenzothiazol-2-yl)]phenyl|carboxamide (Compound Bl). 3 -Amino- 5 -tert-butyl isoxazole in Methylene chloride is added to a vessel containing toluene which is cooled to approx 0 0C. Triphosgene is then added and the reaction mixture is cooled to below -15 0C. Triethylamine is added, followed by Intermediate 4. The mixture is heated to distill off the Methylene chloride and then heated to over 60 0C for over 12 hours and cooled to 50-60 °C. The resulting solids are filtered, washed with Heptane, slurried with 4% sodium hydroxide solution, and filtered. The solids are then washed with Methyl tert-butyl ether followed by Acetone and dried under vacuum to give Compound Bl.
6.4 EXAMPLE 4. EXAMPLES OF PREPARATION OF COMPOUND Bl HCL SALT
[00373] Example A: For the preparation of a hydrochloride salt of Compound Bl5 N-
(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- b][l,3]benzothiazol-2-yl]phenyl}urea hydrochloride, the free base was dissolved in a mixture of 20 ml methylene chloride and 1 ml methanol. A solution of 1.0 M HCl in ethyl ether (1.1 eq.) was added dropwise, followed by addition of ethyl ether. The precipitate was collected by filtration or centrirugation and washed with ethyl ether to give a hydrochloride salt of Compound Bl. Yield: 2.44 g (98 %) NMR (DMSO-^) S X 1.0 (br, IH), 9.68 (s, IH), 9.26 (s, IH), 8.66 (s, IH), 7.93 (d, IH), 7.78 (m, 3H), 7.53 (d, 2H), 7.26 (dd, IH), 6.53 (s, IH), 4.50 (t, 2H), 3.97 (m, 2H), 3.81 (t, 2H), 3.6 (overlapping, 4H), 3.23 (m, 2H), 1.30 (s, 9H). [00374] Example B: Concentrated HCl is added to a suspension of Compound Bl in warm methanol forming a solution that slowly begins to precipitate. The reaction mixture is
NYI-4144519vl 88 refluxed for over 2 hrs and then stirred overnight at ambient temperature. The HCl salt is collected and dried under vacuum.
[00375] Example C: Materials: {[5-(tert-Butyl) isoxazol-3-yl]amino}-N-{4-[7-(2- morpholin-4-yl-ethoxy)(4-hydroimidazolo[2,l-6]benzothiazol-2-yl)] phenyl }carboxamide (775 g, 1.38 mol, 1.0 eq); HCl 37% aqueous (288 mL, 3.46 mol, 2.5 eq); Methanol (MeOH, AR) (40L). Procedure: (Step 1) Equipped a 5OL 3-neck round bottom flask with a mechanical agitator, thermocouple probe, Nitrogen inlet, drying tube, reflux condenser, addition funnel and in a heating mantle. (Step 2) Charged the flask with {[5-(tert-Butyl) isoxazol-3-yl] amino}-N-{4-[7-(2-morpholin-4-yl-ethoxy)(4-hydroimidazolo[2,l- b]benzothiazol-2-yl)] phenyl jcarboxamide (775g) and MeOH, AR (40L). Heat the resulting off-white suspension to reflux (680C). A clear solution did not form. (Step 3) Added HCl (37% aqueous) (228 mL) over 5 minutes at 68°C. The reaction mixture turned into a clear solution and then a new precipitate formed within approximately 3 minutes. Continued heating at reflux for approximately 5 hours. Allowed the reaction mixture to cool to ambient temperature overnight. (Step 4) Collected the off-white solids by filtration onto a polypropylene filter, washing the solids with MeOH, AR (2 x 1 L). (Step 5) Combined two lots of material prepared in this manner (74Og and 82Og). Slurried the combined solids in Methanol (30 L) over 30 minutes at reflux and cool to the room temperature. (Step 6) Collected the solids by filtration onto a polypropylene filter, rinsing with Methanol (2 x 1.5L). (Step 7) Dried the solids in a vacuum oven (<10mniHg) at 400C. Yield: 1598 g (84%), off-white solid; HPLC: 98.2% (area); MS: 561.2 (M+l); IH NMR: conforms (300 MHz, DMSO-d6); Elemental Analysis (EA): Theory = 54.97 %C; 5.41 %H; 13.26 %N; 5.06 %S; 11.19 %C1; Actual = 54.45 %C; 5.46 %H; 13.09 %N; 4.99 %S; 10.91 %C1.
NYl-4I44519v! 89 [00376] Examples of Compound Bl HCl salt synthesis

[00377] Example D: In a 50-L 3-neck round bottom flask equipped with a mechanical stirrer, heating mantle, condenser and nitrogen inlet was charged Compound Bl (1052.4 g, 1.877 mol, 1.00 equiv.) and methanol (21 L). The reactor was heated and stirred. At an internal temperature > 50 0C, cone. HCl (398.63 mL, 4.693 mol, 2.5 equiv.) was charged over 5 minutes through an addition funnel. With the addition, the reaction changed from a pale yellow suspension to a white suspension. The internal temperature was 55 0C at the conclusion of the addition. The reaction was heated to reflux for 1 hour, then heating discontinued and the reaction allowed to cool to room temperature. The reaction was filtered in two portions, each filter cake washed with methanol (2 x 1 L), transferred to trays and dried in a vacuum oven (45 0C) to constant weight. The dried trays were combined to produce 1141.9 g, 96% yield, 99.1 % HPLC purity, 10.9% chloride by titration.
Solid Forms Comprising the HCl Salt of Compound Bl 6.6.2.1 Preparation of Solid Forms

6.6.2.2 Cold Precipitation Experiments

NYl-4144519vl 102 6.6.2.3 Slurry Experiments
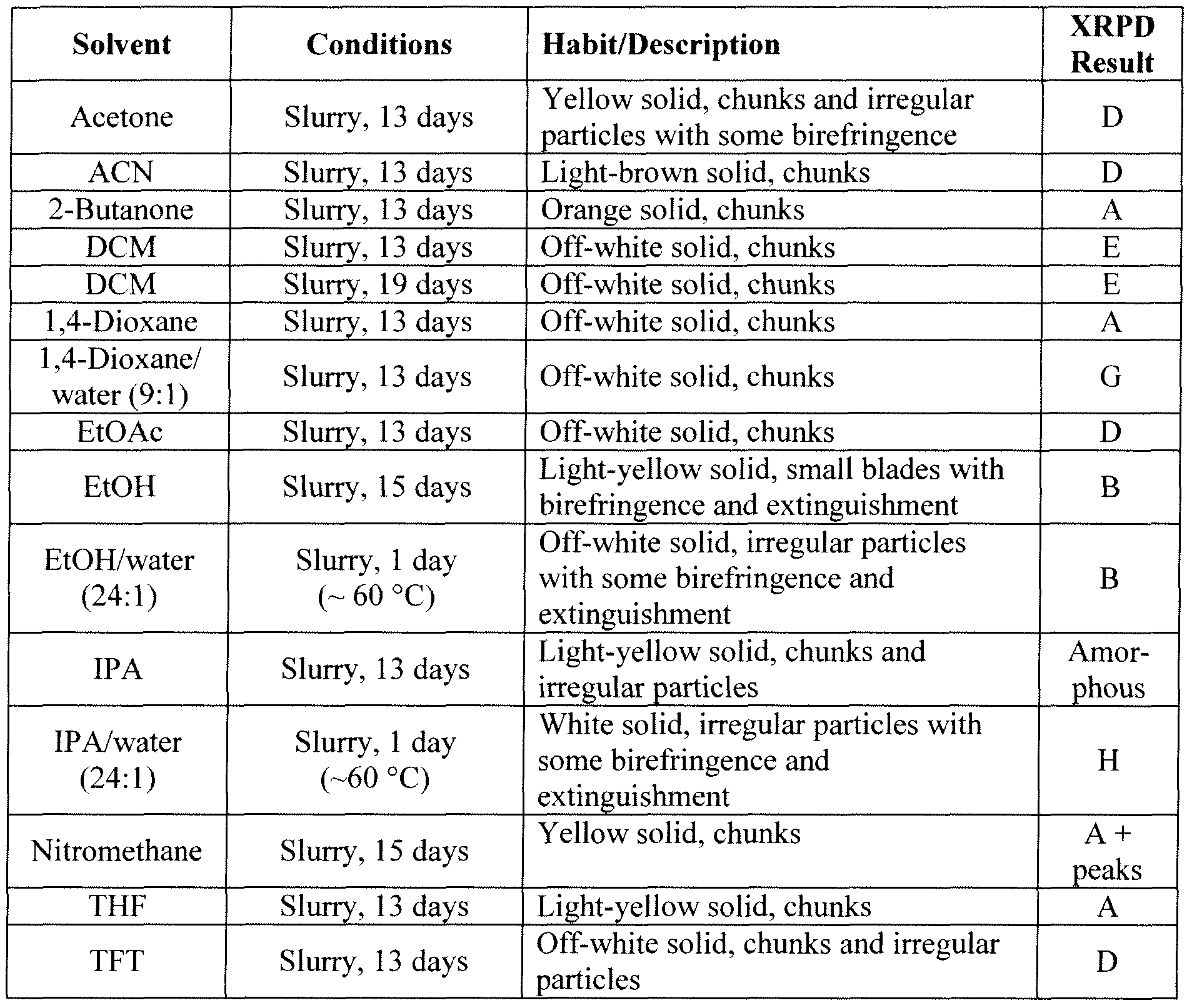
NYI-41445 l9vl 103 6.6.2.4 Additional Preparation of Solid Forms Comprising the HCI Salt of Compound Bl

NYl-4144519v l 104

NYM 144519vl 105

N Y l -4 1 4 4 5 1 9 v l 1 0 6

NYI-4I44519vi 107

N V I 4 1 4 4 5 1 9 1 0 8

“Abbreviations in Table: CC = crash cool, CP = crash precipitation, EtOAc = ethyl acetate, FE = fast evaporation, VD = vapor diffusion, IPA = isopropanol, MEK = methyl ethyl ketone (2-butanone), RE = rotary evaporation, RT = room (ambient) temperature, SC = slow cool, SE = slow evaporation, THF = tetrahydrofuran, TFE = 2,2,2=trifluoroethanol.
6.6.2.5 Scale-up Experiments of Involving Crystal Forms Comprising the HCl Salt of Compound Bl

NYI-4144519v l 109

Abbreviations in Table: CC = crash cool, CP = crash precipitation, EtOAc = ethyl acetate, FE = fast evaporation, IPA = isopropanol, MEK = methyl ethyl ketone (2-butanone), RE = rotary evaporation, RT = room (ambient) temperature, SC = slow cool, SE = slow evaporation, THF = tetrahydrofuran, TFE = 2,2,2=trifluoroethanol.
……………………
Identification of N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea dihydrochloride (AC220), a uniquely potent, selective, and efficacious FMS-like tyrosine kinase-3 (FLT3) inhibitor
J Med Chem 2009, 52(23): 7808
http://pubs.acs.org/doi/full/10.1021/jm9007533

General Procedure E for Preparation of Hydrochloride Salt
References
- Chao, Qi; Sprankle, Kelly G.; Grotzfeld, Robert M.; Lai, Andiliy G.; Carter, Todd A.; Velasco, Anne Marie; Gunawardane, Ruwanthi N.; Cramer, Merryl D.; Gardner, Michael F.; James, Joyce; Zarrinkar, Patrick P.; Patel, Hitesh K.; Bhagwat, Shripad S. (2009). “Identification of N-(5-tert-Butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea Dihydrochloride (AC220), a Uniquely Potent, Selective, and Efficacious FMS-Like Tyrosine Kinase-3 (FLT3) Inhibitor”. Journal of Medicinal Chemistry 52 (23): 7808–7816.
- Drug Tames Refractory AML. ASH Dec 2012
- NMR……….http://file.selleckchem.com/downloads/nmr/S152601-AC-220-HNMR-Selleck.pdf
- HPLC………http://file.selleckchem.com/downloads/hplc/S152601-AC-220-HPLC-Selleck.pdf


Beperminogene perplasmid, ベペルミノゲンペルプラスミド
Beperminogene perplasmid
ベペルミノゲンペルプラスミド
HGF plasmid
- DNA (human hepatocyte growth factor plasmid pVAX1 cDNA)
- DNA (plasmid pVAX1HGF/MGBI)
- AMG-0001
DS-992
Nucleic Acid Sequence
Sequence Length: 51811342 a 1223 c 1314 g 1302 t
APPROVED, japan 2019, Collategene, 2019/3/29
Antiparkinsonian, Angiogenesis inducing agent
CAS: 627861-07-8
- Originator AnGes MG
- Developer AnGes MG; Osaka University Hospital
- Class Antiparkinsonians; Gene therapies; Ischaemic heart disorder therapies; Vascular disorder therapies
- Mechanism of Action Angiogenesis inducing agents; Gene transference; Hepatocyte growth factor expression stimulants
- Available For Licensing Yes – Ischaemic heart disorders; Lymphoedema; Parkinson’s disease
- Registered Peripheral arterial disorders
- Phase I/II Lymphoedema
- No development reported Arteriosclerosis obliterans; Ischaemic heart disorders; Parkinson’s disease; Thromboangiitis obliterans
- 26 Mar 2019 Registered for Peripheral arterial disorders in Japan (IM)
- 21 Feb 2019 The Pharmaceutical Affairs and Food Sanitation Council recommends conditional and time-limited approval of beperminogene perplasmid for the improvement of ulcers associated with chronic peripheral arterial disease
- 21 Feb 2019 AnGes plans a clinical study to assess the efficacy of beperminogene perplasmid in improvement of pain at rest in chronic peripheral arterial disorders
- In 2010, the product received fast track designation in the U.S. for the treatment of critical limb ischemia
HGF Plasmid (Beperminogene Perplasmid)Critical Limb Ischemia (Arteriosclerosis Obliterans & Buerger’s Disease) AMG0001 Injection, JAPAN AND US ALLIANCE Mitsubishi Tanabe Pharma
PATENT
WO 2017126488
US 20170283446
Expert Review of Cardiovascular Therapy (2014), 12(10), 1145-1156.
////////////Beperminogene perplasmid, japan 2019, ベペルミノゲンペルプラスミド , AnGes MG, Osaka University Hospital, Critical Limb Ischemia, Arteriosclerosis Obliterans, Buerger’s Disease, AMG0001, AMG-0001, DS-992 , HGF plasmid , fast track designation
Ferric carboxymaltose , カルボキシマルトース第二鉄


Ferric carboxymaltose
カルボキシマルトース第二鉄
CAS: 9007-72-1
(2S,3S,4S,5R)-4-[(2R,3R,4R,5S,6R)-5-[(2R,3R,4R,5S,6R)-3,4-dihydroxy-6-(hydroxymethyl)-5-[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoxan-2-yl]oxy-3,4-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-2,3,5,6-tetrahydroxyhexanoate;iron(3+);oxygen(2-);hydroxide;hydrate
Iron dextri-maltose
Iron(3+) hydroxide oxide poly-(1–4)-alpha-D-glucopyranosyl-(1–4)-D-gluconate hydrate
Polynuclear iron (III)-hydroxide 4(R)-(poly-(1–4)-O-alpha-D-glucopyranosyl)-oxy-2(R),3(S),5(R),6-tetrahydroxy-hexanoate
Poly[D-glucopyranosyl(1–4)]-D-gluconic acid complex of hydrated iron(III) oxide
japan pmda approved, 2019/3/26, Ferinject
Treatment of patients with iron deficiency anemia
Hematinic, Supplement (iron)
LAUNCHED, 2007, Vifor Pharma, Anemia, iron deficiency
1 Injectafer
2. Ferinject
3. Iron Dextri-maltose
4. Unii-6897gxd6oe
5. Vit 45
6. Vit-45
7. Ferric Carboxymaltose [usan:inn:ban]
8. Iron(3+) Hydroxide Oxide Poly-(1?4)-alpha-d-glucopyranosyl-(1?4)-d-gluconate Hydrate
9. 889138-31-2
10. 9007-72-1
11 Z-213
In 2013, Vifor Pharma and Zeria Pharmaceutical signed an exclusive licensing agreement for the product’s development and commercialization in Japan for the treatment of iron deficiency anemia.
Ferric carboxymaltose is an intravenously-administered iron complex which was first launched in Germany following E.U. approval in 2007 for the treatment of iron deficiency anemia (IDA)
PATENT
WO 2011055374
US 20120214986
IN 2011MU03463
IN 2013CH03474
WO 2016181195
IN 2015CH02360
CN 106236707
CN 106977621
EP 3339329
PATENT
Iron deficiency anaemia (IDA) is a common haematological complication with potentially serious clinical consequences that may require intravenous iron therapy.
Ferric carboxymaltose (FCM) is a stable, non-dextran iron formulation administered intravenously in large single doses to treat IDA. It is an iron complex that consists of a ferric hydroxide core stabilized by a carbohydrate shell. It is commercially available in the market under the trade name Ferinject®
Ferric carboxymaltose has been designed to provide high iron utilisation and to have a better benefit to risk profile than iron dextran and iron sucrose therapy. In the case of iron dextran, a key risk is the reaction with anti-dextran antibodies leading to the well known dextran induced anaphylactic reactions. In the case of iron sucrose, the negative characteristics include high pH, high osmolarity, low dosage limits and the long duration of administration.
Ferric carboxymaltose allows for controlled delivery of iron within the cells of the reticuloendothelial system and subsequent delivery to the iron-binding proteins ferritin and transferrin, with minimal risk of release of large amounts of ionic iron in the serum.
U.S. Pat. No. 3,076,798 discloses a process for the preparation of iron(III)-polymaltose complex compounds. The iron(III)-polymaltose complex compound
preferably has a molecular weight in the range from 20,000 to 500,000 daltons, preferably from 30,000 to 80,000 daltons.
U.S. Patent No. 7,612,109 discloses water-soluble iron carbohydrate complexes (ferric carboxymaltose complexes) obtainable from an aqueous solution of an iron (III) salt, preferably iron (III) chloride, and an aqueous solution of the oxidation product of one or more maltodextrins using an aqueous hypochlorite solution.
PCT application No.WO2011/055374, discloses a process for the preparation of iron (III) carboxymaltose complex using ferric hydroxide.
In Netherlands article, starch 41 (1989) Nr .8, S. 303-309 transition metal ions enhance the selectivity of oxidations by H2O2 to produce polysaccharides to polydicarbonates by glycol cleavage of the C2-C3 vicinal diol moiety.
Even though many prior art processes reported methods for the preparation of Iron(III) carboxymaltose, each process has some limitations with respect to yield, purity and scale-up etc.
EXAMPLES
Example- 1: Preparation of trivalent iron carboxymaltose
Step (i)
20grams of anhydrous iron(III)chloride was dissolved in 50ml of purified water at room temperature for 10 minutes stirring. To this 2gm of maltodextrin (13-17 dextrose equivalents) was added and stirred for 10 minutes at room temperature. The obtained brownish-yellow clear solution was cooled to 0-5°C and the pH of the reaction mixture was adjusted to 7.0 by adding 20% aqueous sodium hydroxide solution. A brown colour precipitate obtained was maintained for 1 hour at 0-5°C and collected through filtration (Wet cake wt. ~ 65. Og). The cake was suck dried and used for next step.
Step (ii)
20grams of maltodextrin having a dextrose equivalents of 13-17 were dissolved in 50ml of purified water and the solution was metered in the course of 20 minutes to a stirred mixture of 2.66gm of Starks catalyst (methyl trioctyl ammonium hydrogen sulfate prepared in-situ from 2gm of Aliquat 336 and 0.66gm of NaHSO4.H2O), 0.8gm of sodium tungstate dihydrate and 0.37gm of TEMPO at RT. 31.12gm of hydrogen peroxide solution (50-55% w/v) was then added drop wise over a period of 40 minutes at 25-30°C and raised the temperature to90-95°C and stirred for 3 hours. After cooling to room temperature, a second portion of 15.5gm of H2O2 solution was metered in the course of 15 minutes at 25-30°C and the resulting solution was again refluxed at 90-95°C for 1 hour. After cooling to 35-40°C, wet cake of step (i) (ferric hydroxide maltodextrin complex) was added, with stirring. 14.0ml of 20% aqueous sodium hydroxide solution was added to adjust the reaction mass pH to 10- 10.5 and the slurry was heated to 50°C, stirred for 30 minutes. Then the reaction mixture was acidified to pH 5.5 by adding hydrochloric acid solution and the mixture was maintained at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 14 hours. Then the reaction mixture was cooled to room temperature and filtered through a celite pad. Thereafter, the iron(III)complex was isolated by precipitation by adding ethanol (237. Og) drop wise at room temperature. The obtained brown amorphous solid was dried under vacuum at 50°C for 2-3 hours. Molecular weight = 202 kDa. Iron content = 23.38% w/w
Example-2:
20grams of maltodextrin (13-17 dextrose equivalents) were dissolved in 50ml of purified water and the solution was added to a stirred mixture of 2.66gm of Starks catalyst and 0.2gm of Na2WO4.2H2O at room temperature in the course of 20 minutes. 24grams of H2O2 solution was metered in the course of 45 minutes at 25-30°C and raised the temperature to 90-95°C and stirred for 2 hours and cooled to room temperature.
The solution was added to another portion of a stirred mixture of 1.33gm of Starks catalyst and 0.2gm of Na2WO4.2H2O at room temperature. Thereafter, 12gm of
H2O2solution was added drop wise over a period of 20 minutes at 25-30°C and the resulting reaction mixture was again refluxed at 90-95°C for 2 hours. After cooling to 25-30°C, wet cake of step (i) from example- 1 was added and stirred for 10 minutes. 14ml of 20% NaOH solution was added to adjust the reaction mass pH to 10- 10.5 and the slurry was heated to 50°C, stirred for 30 minutes. Then the mixture was acidified to pH 5.5 by adding hydrochloric acid solution and the solution was maintained at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 13 hours. Then the reaction solution was cooled to room temperature, adjusted pH to 5.5 to 6.0 with 20% NaOH solution and filtered through a celite pad. Then the iron(III)complex was isolated by precipitating with ethanol (331.0g) addition drop wise at room temperature. The obtained brown amorphous solid was dried in vacuum at 50°C for 2-3 hours. Molecular weight = 200 kDa. Iron content = 25.57 % w/w
Example-3:
20grams of maltodextrin (13-17 dextrose equivalents) were dissolved in 100ml of purified water and the solution was added to a stirred mixture of 2.66gm of Starks catalyst, 0.8gm of Na2WO4.2H2O and 0.37gm of TEMPO at room temperature over a period of 15 minutes. 30grams of H2O2solution was added drop wise in the course of 1 hour at 25-30°C and raised the temperature to 90-95°C, stirred for 3 hours and cooled to room temperature.
At 25-30°C, wet cake of step (i) from example- 1 was added and stirred for 10 minutes. A pH of 10-10.5 was established by adding 12ml of 20% NaOH solution and the slurry was heated to 50°C, stirred at this temperature for 30 minutes. Then the reaction mixture was acidified to pH 5.5 with hydrochloric acid addition and the mixture was maintained at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 14 hours. The reaction mixture was allowed to cool to room temperature, adjusted pH to 6.0 to 6.5 with 20% NaOH solution and filtered through a celite pad. Then the iron(III)complex was isolated by precipitating with ethanol (343.0g) addition drop wise at room temperature. The obtained brown
amorphous solid was dried in vacuum at 50°C for 2-3 hours. Molecular weight = 260 kDa. Iron content = 23.67 % w/w
Example-4:
20grams of maltodextrin (13-17 dextrose equivalents) were dissolved in 50ml of purified water and the solution was added to a stirred mixture of 2.66gm of Starks catalyst, 0.8gm of Na2WO4.2H2O and 0.37g of TEMPO at room temperature over a period of 15 minutes. 30grams of H2O2 solution was added drop wise over a period of 1 hour at 55-60°C and the temperature was raised to 90-95 °C, stirred for 3 hours and cooled to room temperature. After cooling to 25-30°C, wet cake of step (i) from example- 1 was added and stirred for 10 minutes. A pH of 10-10.5 was established by adding 12ml of 20% NaOH solution and the slurry was heated to 50°C, stirred at this temperature for 30 minutes. Then the reaction mixture was acidified to pH 5.5 with hydrochloric acid addition and was maintained at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 12 hours. The reaction mixture was allowed to cool to room temperature, adjusted pH to 6.0 to 6.5 with 20% NaOH solution and filtered through a celite pad. Then the iron(III)complex was isolated by precipitating with ethanol (343.0g) addition drop wise at room temperature. The obtained brown amorphous solid was dried under vacuum at 50°C for 2-3 hours. Molecular weight = 261 kDa. Iron content = 22.85 % w/w
Example-5:
Step (i)
16grams of anhydrous iron(III)chloride was dissolved in 50ml of purified water at room temperature for 10 min stirring. The obtained brownish-yellow clear solution was cooled to 0-5°C and the pH was adjusted to 7.0 first by adding aqueous sodium carbonate solution (21gm of Na2CO3dissolved in 102 ml of purified water) and then by adding 20% NaOH solution. A brown colour precipitate obtained was maintained for 1 hour at 0-5°C and collected through filtration (Wet wt. ~54.0g). The cake was suck dried and used for next step.
Step (ii)
20grams of maltodextrin (13-17 dextrose equivalents) were dissolved in 50ml of purified water and the solution was added to a stirred mixture of 2.66gm of Starks catalyst, 0.8gm of Na2WO4.2H2O and 0.37gm of TEMPO at room temperature over a period of 15 minutes. 30gm of H2O2solution was added drop wise over a period of 1 hour at 25-30°C and the temperature was raised to 90-95°C, stirred for 3 hours and cooled to room temperature.
At 25-30°C, wet cake of step (i) added and stirred for 10 minutes. 20% NaOH solution was added drop wise to adjust the reaction mass pH tolO-10.5 and the slurry was heated to 50°C, stirred for 30 minutes. Then the solution was acidified to pH 5.5 with hydrochloric acid addition and the solution was kept at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 12 hours. The reaction mixture was allowed to cool to room temperature, adjusted pH to 6.0 to 6.5 with 20% NaOH solution and filtered through a celite pad. Then the iron(III)complex was isolated by precipitating with ethanol (315.0g) addition drop wise at room temperature. The obtained brown amorphous solid was dried under vacuum at 50°C for 2-3 hours. Molecular weight = 236 kDa. Iron content = 22.35 % w/w
Example-6:
Step (i)
20grams of anhydrous ferric chloride was dissolved in 50ml of purified water at room temperature for 10 min stirring. The obtained brownish-yellow clear solution was cooled to 0-5°C and the pH was adjusted to 7.0 by adding 20% NaOH solution. A brown colour precipitate obtained was stirred for 1 hour at 0-5°C and collected through filtration. The cake was suck dried and used for next step.
Step (ii)
20grams of maltodextrin (13-17 dextrose equivalents) were dissolved in 50ml of purified water and the solution was added to a stirred mixture of 2.66gm of Starks catalyst, 0.8gm of Na2WO4.2H2O and 0.37g of TEMPO at room temperature over a
period of 15 minutes. 36gm of H2O2 solution was metered in the course of 1 hour at 25-30°C and the resulting solution was heated to 90-95°C, stirred for 3 hours and cooled to room temperature.
After cooling to 25-30°C, wet cake of step (i) was added and stirred for 10 min. 12ml of 20% NaOH solution was added drop wise to adjust the reaction mass pH to 10-10.5 and the slurry was heated to 50°C, kept at this temperature for 30 minutes. Then the solution was acidified to pH 5.5 with hydrochloric acid addition and the solution was maintained at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 12 hours. The reaction mixture was allowed to cool to room temperature, adjusted pH to 6.0 to 6.5 with 20% NaOH solution and filtered through a celite pad. Then the iron(III)complex was isolated by precipitating with ethanol (315.0g) addition at room temperature. The obtained brown amorphous solid was dried under vacuum at 50°C for 2-3 hours. Molecular weight = 365 kDa. Iron content = 23.93 % w/w
Example-7:
20grams of maltodextrin (13-17 dextrose equivalents) were dissolved in 50ml of purified water and the solution was added to a stirred mixture of 2.66gm of Starks catalyst, 0.2gm of Na2WO4.2H2O and 0.37gm of TEMPO at room temperature over a period of 15 minutes. 30gm of H2O2 solution was added drop wise in the course of 1 hour at 25-30°C and the temperature was raised to 90-95°C, stirred for 3 hours and cooled to room temperature.
At 25-30°C, wet cake of step (i) from example-6 was added and stirred for 10 minutes. A pH of 10-10.5 was established by adding 12.0ml of 20% NaOH solution and the slurry was heated to 50°C, stirred at this temperature for 30 minutes. Then the solution was acidified to pH 5.5 with hydrochloric acid addition and the solution was kept at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 12 hours. The reaction mixture was allowed to cool to room temperature; pH was adjusted to 6.0 to 6.5 with 20% NaOH solution and filtered through a celite pad. Then the iron(III)complex was isolated by precipitating with ethanol (276.0g) addition drop wise at room temperature. The obtained brown amorphous solid was dried in vacuum at 50°C for 2-3 hours. Molecular weight = 366 kDa. Iron content = 21.2 % w/w
Example-8:
20grams of maltodextrin (13-17 dextrose equivalents) were dissolved in 50ml of purified water and the solution was added to a stirred mixture of 2.66gm of Starks catalyst and 0.8gm of Na2WO4.2H2O at room temperature over a period of 15 minutes. 30grams of H2O2 solution was metered in the course of 1 hour at 25-30°C and the temperature was raised to 90-95°C, stirred for 3 hours and cooled to room temperature.
At 25-30°C, wet cake of step (i) from example-6 was added and stirred for 10 minutes. A pH of 10-10.5 was established by adding 12ml of 20% NaOH solution and the slurry was heated to 50°C, stirred at this temperature for 30 minutes. Then the solution was acidified to pH 5.5 with hydrochloric acid addition and the solution was maintained at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 12 hours. The reaction mixture was allowed to cool to 25-30°C, adjusted pH to 6.0 to 6.5 with 20% NaOH solution and filtered through a celite pad. Then the iron(III)complex was isolated by precipitating with ethanol (315.0g) addition drop wise at room temperature. The obtained brown amorphous solid was dried in vacuum at 50°C for 2-3 hours. Molecular weight = 340 kDa. Iron content = 23.28 % w/w
Example-9:
20grams of maltodextrin (13-17 dextrose equivalents) were dissolved in 50ml of purified water and the solution was added to a stirred mixture of 2.66gm of Starks catalyst, 0.8gm of Na2WO4.2H2O and 0.37gm of TEMPO at room temperature over a period of 15 minutes. 30grams of H2O2 solution was added drop wise over a period of 1 hour at 25-30°C and the resulting solution was heated to 90-95°C, stirred for 3 hours and cooled to room temperature.
At 25-30°C, wet cake of step (i) from example- 1 was added and stirred for 10 minutes. A pH of 10-10.5 was established by adding 12ml of 20% NaOH solution and the slurry was heated to 50°C, stirred at this temperature for 30 minutes. Then the solution was acidified to pH 5.5 with hydrochloric acid addition and the solution was maintained at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 12 hours. The reaction mixture was allowed to cool to room temperature, adjusted pH to 6.0 to 6.5 with 20% NaOH solution and filtered through a celite pad. Then the iron(III)complex was isolated by precipitating with ethanol (304.0g) addition drop wise at room temperature. The obtained brown amorphous solid was dried under vacuum at 50°C for 2-3 hours. Molecular weight = 352 kDa. Iron content = 23.0 % w/w
Example-10:
20grams of maltodextrin (13-17 dextrose equivalents) were dissolved in 50ml of purified water and the solution was added to a stirred mixture of 2.66gm of Starks catalyst, 0.8gm of Na2WO4.2H2O and 0.37gm of TEMPO at room temperature over a period of 15 minutes. 30grams of H2O2 solution was added drop wise in the course of 60 minutes at 25-30°C and the temperature was raised to 90-95°C, stirred for 3 hours and cooled to room temperature.
At 25-30°C, wet cake of step (i) from example- 1 was added and stirred for 10 minutes. 12ml of 20% NaOH solution was added drop wise to adjust the reaction mixture pH to 10-10.5 and the temperature of the slurry was raised to 50°C, stirred at this temperature for 30 minutes. Then the reaction mixture was acidified to pH 5.5 with hydrochloric acid addition and was maintained at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 12 hours. The reaction mixture was allowed to cool to room temperature, adjusted pH to 6.0 to 6.5 with 20% NaOH solution and filtered through a celite pad. Then the iron(III)complex was
isolated by precipitating with ethanol (276.0g) addition drop wise at room temperature. The obtained brown amorphous solid was dried in vacuum at 50°C for 2-3 hours. Molecular weight = 348 kDa. Iron content = 24.6 % w/w
/////////Ferric carboxymaltose , カルボキシマルトース第二鉄 ,Injectafer, Ferinject, Iron dextri-maltose, Unii-6897gxd6oe, Vit 45, Vit-45, japan 2019, Z-213
C(C1C(C(C(C(O1)OC2C(OC(C(C2O)O)OC3C(OC(C(C3O)O)OC(C(CO)O)C(C(C(=O)[O-])O)O)CO)CO)O)O)O)O.O.[OH-].[O-2].[Fe+3]
Peficitinib hydrobromide, ペフィシチニブ臭化水素酸塩


Peficitinib hydrobromide
ペフィシチニブ臭化水素酸塩
ASP015K,
Rheumatoid Arthritis
1H-Pyrrolo(2,3-b)pyridine-5-carboxamide, 4-((5-hydroxytricyclo(3.3.1.13,7)dec-2-yl)amino)-, hydrobromide (1:1), stereoisomer
4-{[(1R,2s,3S,5r)-5-Hydroxyadamantan-2-yl]amino}-1H-pyrrolo[2,3-b]pyridine-5-carboxamide hydrobromide (1:1)
1H-Pyrrolo[2,3-b]pyridine-5-carboxamide, 4-[[(1R,3S)-5-hydroxytricyclo[3.3.1.13,7]dec-2-yl]amino]-, hydrobromide (1:1)
U55XHZ5X6P
| Formula |
C18H22N4O2. HBr
|
|---|---|
| CAS |
1353219-05-2 HBR
944118-01-8 BASE
|
| Mol weight |
407.3048
|
PMDA, 2019/3/26 JAPAN APPROVED, Smyraf

Peficitinib hydrobromide is used in the treatment of Psoriasis and Rheumatoid Arthritis
Peficitinib (formerly known as ASP015K) is a pyrrolo[2,3-b]pyridine derivative orally administered once-daily JAK inhibitor in development for the treatment of Rheumatoid Arthritis. In preclinical studied Peficitinib inhibited JAK1 and JAK3 with IC50 of 3.9 and 0.7 nM, respectively. Peficitinib also inhibited IL-2-dependent T cell proliferation in vitro and STAT5 phosphorylation in vitro and ex vivo. Furthermore, Peficitinib dose-dependently suppressed bone destruction and paw swelling in an adjuvant-induced arthritis model in rats via prophylactic or therapeutic oral dosing regimens.In clinical trials, Peficitinib treatment prescribed at 50, 100 and 150 mg amounts each showed statistically significantly higher ACR20 response rates compared to the placebo and response rates increased up to the 150 mg dosage. Adverse events included neutropenia, headache, and abdominal pain. The treatment-emergent adverse events occurring more frequently in the Peficitinib group compared with the placebo group included diarrhea, nasopharyngitis, and increased serum creatine phosphokinase activity. No cases of serious infections were reported. Herpes zoster occurred in four patients (two each in the peficitinib 25 and 100 mg cohorts). The authors concluded that treatment with peficitinib as monotherapy for 12 weeks in Japanese patients with moderate to severe RA is efficacious and showed an acceptable safety profile.
SYN

CLIP
Bioorganic & Medicinal Chemistry
Discovery and structural characterization of peficitinib (ASP015K) as a novel and potent JAK inhibitor
Abstract
Janus kinases (JAKs) are considered promising targets for the treatment of autoimmune diseases including rheumatoid arthritis (RA) due to their important role in multiple cytokine receptor signaling pathways. Recently, several JAK inhibitors have been developed for the treatment of RA. Here, we describe the identification of the novel orally bioavailable JAK inhibitor 18, peficitinib (also known as ASP015K), which showed moderate selectivity for JAK3 over JAK1, JAK2, and TYK2 in enzyme assays. Chemical modification at the C4-position of lead compound 5 led to a large increase in JAK inhibitory activity and metabolic stability in liver microsomes. Furthermore, we determined the crystal structures of JAK1, JAK2, JAK3, and TYK2 in a complex with peficitinib, and revealed that the 1H-pyrrolo[2,3–b]pyridine-5-carboxamide scaffold of peficitinib forms triple hydrogen bonds with the hinge region. Interestingly, the binding modes of peficitinib in the ATP-binding pockets differed among JAK1, JAK2, JAK3, and TYK2. WaterMap analysis of the crystal structures suggests that unfavorable water molecules are the likely reason for the difference in orientation of the 1H-pyrrolo[2,3-b]pyridine-5-carboxamide scaffold to the hinge region among JAKs.




PATENT
WO 2011162300
[Chemical formula 1]
(Production Method of Hydrobromide Salt Form B 45)
(In the Case of Addition of Seed Crystals )
After nitrogen substitution, the reaction vessel was charged with 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy- 2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide (145.0 kg), water (377 L), EtOH (1508 L), 48% hydrobromic acid (74.9 kg) It charged sequentially at room temperature and started stirring. 48% hydrobromic acid was added, taking care that the pH was in the range of 1.5 to 1.9. The reaction mixture was heated and stirred until the internal temperature reached 70 ° C. or higher. After confirming that the solution was completely dissolved, the solution was stirred for 5 minutes or more, and the solution was subjected to clear filtration at an internal temperature of 70 ° C. or higher, and the pot and line were washed with warm EtOH (290 L). At an internal temperature of about 50 ° C., 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide odor Hydrochloric acid salt seed crystals (B45, 145 g) were added, and the mixture was ripened and stirred overnight at an internal temperature of 40 to 50 ° C. Subsequently, the mixture was cooled to an internal temperature of 20 to 30 ° C. over 1 hour or more, and the mixture was aged and stirred at the same temperature for 1 hour or more. At an internal temperature of 20 to 30 ° C., EtOAc (4350 L) was added dropwise over 1 hour, and the mixture was aged and stirred overnight at the same temperature. The precipitated crystals were filtered. The wet crystals were washed with a solution of EtOH / EtOAc (145 L / 290 L). The wet crystals are dried under reduced pressure at an external temperature of 40 ° C. overnight under reduced pressure to give 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3 -b] Pyridine-5-carboxamide hydrobromide crystal (B45, 161 kg) was obtained.
(In the case of no addition of seed crystals) After
sufficiently drying the reaction vessel and replacing with nitrogen, water (585 L) is charged and subsequently 4- {[ (1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide (225 kg), EtOH (2250 L) was charged Stirring was started. The internal temperature was adjusted to 25 ° C., and 48% hydrobromic acid (127.8 kg) was charged at the same temperature, and the vessel and kettle wall were washed with EtOH (90 L). After the completion of the charging, it was confirmed that the reaction solution had been dissolved, the pH was measured, and the pH was confirmed to be in the range of 1.5 to 1.9. When the pH was out of the range, the pH was adjusted to a predetermined pH using 48% hydrobromic acid (48% hydrobromic acid: about 11.6 kg). The temperature was raised until the internal temperature reached 70 ° C., and after confirmation of dissolution, the mixture was stirred for 5 minutes or more. The solution was subjected to clear filtration while maintaining the internal temperature at 60 ° C. or higher, and washed through a filter from a dissolution vessel with warm EtOH (450 L) preheated to 50 ° C. or higher. The clarified filtrate was gradually cooled to an internal temperature of 45 ° C., and filtered EtOAc (6750 L) was added dropwise over 6 hours at an internal temperature of 45 ° C. After the dropping was completed, the mixture was stirred at an internal temperature of 45 ° C. for 10 hours or more. Subsequently, it was cooled to an internal temperature of 25 ° C. using a follow-up temperature control cooler, and stirred at an internal temperature of 25 ° C. for 3 hours. The predetermined supernatant concentration and the crystal form of the precipitated crystals were confirmed and filtered. A mixed solvent of EtOH / EtOAc (225 L / 450 L) was prepared and cake washed using this mixed solvent. The obtained wet crystals are dried under reduced pressure at an external temperature of 40 ° C. for 10 hours or more, and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [ 2,3-b] pyridine-5-carboxamido hydrobromide crystal (B45, 250 kg) was obtained.
Elemental analysis: theoretical value: C 53.08%, H 5.69% , N 13.76%, O 7.86% , Br 19.62%;
Found:. C 53.02%, H 5.74 %, N 13.73%, Br 19.42%
molecular composition: C 18 H 22 N 4 O 2 . HBr
MS: 327.0 (M From the result of + H) +
elemental analysis, 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5 The carboxamido hydrobromide was a monohydrobromide.
(hydrobromide A87 crystal)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] Pyridine-5-carboxamide (6.0 g) was charged in EtOH / water (57.6 mL / 14.4 mL). At 50-60 ° C., 48% hydrobromic acid was added, stirred for 15 minutes more, and washed with EtOH (18 mL). At 45 ° C.-55 ° C. EtOAc (180 mL) was added dropwise over 30 minutes. Crystals were precipitated upon stirring at 15 ° C to 25 ° C. The crystals were collected by filtration and washed with a mixed solvent of EtOH / EtOAc (6 mL / 12 mL). The crystals are dried under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide odor Seed crystals of hydrofluoride (Form A87, 6.11 g) were obtained.
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide (3.0 g), EtOH (24 mL), water (6 mL), and 48% hydrobromic acid (1.55 g) were charged sequentially at room temperature. After charging, the mixture was heated to an internal temperature of 60 ° C. or higher and stirred. After confirming that the solution was completely in solution, the solution was subjected to clear filtration at an internal temperature of 60 ° C. or higher, and washed with warm EtOH (9 mL). EtOH (21 mL) is added dropwise at an internal temperature of 70 ° C. or higher, and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H at an internal temperature of 70 ° C. Seed crystals (A87, 30 mg) of pyrrolo [2,3-b] pyridine-5-carboxamide hydrobromide were added, and the mixture was ripened and stirred overnight at an internal temperature of 65 to 70 ° C. Subsequently, it was cooled to an internal temperature of 20 to 30 ° C., and ripening stirring was carried out at the same temperature overnight. At an internal temperature of 20 to 30 ° C., EtOAc (90 mL) was added dropwise over 1 hour, and the mixture was aged and stirred at the same temperature for 1 hour or more. The precipitated crystals were collected by filtration. The wet crystals were washed with a solution of EtOH / EtOAc (3 mL / 12 mL). The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide crystal (Form A87, 3.09 g) was obtained.
(hydrobromide A61 crystal)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] Pyridine-5-carboxamide (5.0 g), EtOH (48 mL), water (12 mL), and 48% hydrobromic acid (2.58 g) were charged sequentially at room temperature. After charging, the mixture was heated to an internal temperature of 70 ° C. and stirred. After confirming complete dissolution, the solution was clarified by filtration at an internal temperature of 70 ° C., and washed with warm EtOH (15 mL). The internal temperature was cooled to 50 to 60 ° C., and EtOAc (150 mL) was added dropwise over 1 hour at the same temperature. After the addition was completed, the solution was gradually cooled to 20 to 30 ° C., and the mixture was aged and stirred at the same temperature for 1 hour or more. The precipitated crystals were collected by filtration. The wet crystals were washed with a solution of EtOH / EtOAc (5 mL / 10 mL). The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide crystal (Form A61, 5.19 g) was obtained.
(hydrobromide A36 type crystal)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] To a suspension of pyridine-5-carboxamide (500 mg) in EtOAc, 48% hydrobromic acid (258 μL) was added, and the mixture was stirred with heating under reflux for 1 hour, and further allowed to cool to room temperature. The precipitated crystals were collected by filtration and washed with EtOAc. The resulting crystals are dried at 60 ° C. under reduced pressure to give 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-]. b] Pyridine-5-carboxamide monohydrobromide crystal (Form A36, 625 mg) was obtained.
(B11-type crystal of hydrobromide monohydrate)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2 , 3-b] Pyridine-5-carboxamide (5.0 g), EtOH (48 mL), water (12 mL), 48% hydrobromic acid (2.58 g) were sequentially charged at room temperature. After charging, the mixture was heated to an internal temperature of 70 ° C. or higher and stirred. After confirming that the solution had completely dissolved, the solution was subjected to clear filtration at an internal temperature of 70 ° C. or higher, and washed with warm EtOH (15 mL). 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide odor at an internal temperature of about 35 ° C. Hydrochloric acid salt seed crystals (A87, 49.0 mg) were added, and the mixture was aged with an internal temperature of 30 to 40 ° C. for 4 hours. Subsequently, the mixture was cooled to an internal temperature of 20 to 30 ° C., and aged and stirred overnight at the same temperature. At an internal temperature of 20-25 ° C., EtOAc (150 mL) was added dropwise over 1 hour, and the mixture was aged and stirred at the same temperature for 30 minutes or longer. The precipitated crystals were collected by filtration. The wet crystals were washed with a solution of EtOH / EtOAc (5 mL / 10 mL). The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide monohydrate crystal (Form B11, 5.24 g) was obtained.
(B21-type crystal of hydrobromide dihydrate)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2 , 3-b] Pyridine-5-carboxamide (5.0 g), EtOH (18 mL), water (12 mL), 48% hydrobromic acid (2.58 g) were sequentially charged at room temperature. After charging, the mixture was heated to an internal temperature of 60 ° C. or higher and stirred. After confirming that the solution was completely dissolved, the solution was subjected to clear filtration at an internal temperature of 60 ° C. or higher, and washed with warm EtOH (10 mL). The mixture was cooled to an internal temperature of about 45 to 50 ° C. and aged for 2 hours while stirring. Subsequently, the reaction solution is cooled to an internal temperature of 20 to 30 ° C., and aged at the same temperature and stirred overnight. At an internal temperature of 20 to 30 ° C., EtOAc (160 mL) was added dropwise over 1 hour, and the mixture was aged and stirred for 1 hour or more at the same temperature. The precipitated crystals were filtered. The wet crystals were washed with a solution of EtOH / EtOAc (3 mL / 12 mL). The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide dihydrate crystals (Form B21, 6.05 g) were obtained.
(Tautomerism of each crystal)
(Crystal form conversion; hydrobromide B21 → A61)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H- Pyrrolo [2,3-b] pyridine-5-carboxamide hydrobromide dihydrate (Form B21, 300 mg) and EtOH (3 mL) were sequentially charged at room temperature and suspended overnight. After suspension, the crystals were collected by filtration at room temperature and the wet crystals were washed with EtOH. The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide crystal (Form A61, 258 mg) was obtained.
(Crystal form conversion; hydrobromide B11 → B21)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H- Pyrrolo [2,3-b] pyridine-5-carboxamide hydrobromide monohydrate (form B11, 2.0 g), EtOH (7 mL), water (3 mL) were sequentially charged at room temperature and suspended overnight. It became cloudy. After suspension, the crystals were collected by filtration at room temperature and the wet crystals were washed with 70% aqueous EtOH. The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide dihydrate crystals (Form B21, 1.54 g) were obtained.
(Crystal form conversion; hydrobromide A61 form → B21 form)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H- Pyrrolo [2,3-b] pyridine-5-carboxamide hydrobromide (form A61, 1.0 g), EtOH (3.5 mL) and water (1.5 mL) were sequentially charged at room temperature and suspended overnight. After suspension, the crystals were filtered at room temperature and the wet crystals were washed with 70% aqueous EtOH. The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide dihydrate crystals (Form B21, 827 mg) were obtained.
( Example of Preparation of Monohydrate Crystalline Compound (I) Free Form)
4-Chloro-1H-pyrrolo [2,3-b] pyridine-5-carboxamide (44.5 g) under nitrogen atmosphere 1s, 3R, 4s, 5S) -4-aminoadamantan-1-ol (57.0 g) and tributylamine (162.6 mL) were charged in NMP (222.5 mL), and heated and stirred at a bath temperature of 200 ° C. for 2.5 hours. The reaction solution was allowed to cool, and then the reaction solution was added dropwise while stirring in water / Et 2 O (6 L / 0.5 L), followed by stirring for 30 minutes. The obtained solid was collected by filtration, washed twice with water (400 mL), washed twice with Et 2 O (300 mL), and dried. The resulting solid was warmed to dissolve in MeOH (1.8 L) and filtered hot. The resulting mother liquor was concentrated under reduced pressure and MeOH (1.8 L) was added to the residue and heated to dissolve. The resulting solution was allowed to cool and stir, and then stirred at room temperature and aged overnight. The precipitated solid was collected by filtration, washed with EtOH and dried under reduced pressure. The resulting solid was suspended in EtOH (250 mL) and stirred at room temperature for 1 h. The solid was collected by filtration, washed with EtOH and dried under reduced pressure. The obtained solid was suspended in water (900 mL) and stirred at a bath temperature of 70 ° C. for 2 hours. The solid was collected by filtration, washed with water and dried under reduced pressure. Furthermore, the solid was suspended in water (900 mL) and stirred at a bath temperature of 70 ° C. for 2 hours. The solid is collected by filtration, washed with water and then dried under reduced pressure to give 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2 , 3-b] Pyridine-5-carboxamide monohydrate crystal (Form A01, 44 g) was obtained.
REFERENCES
1: D’Amico F, Fiorino G, Furfaro F, Allocca M, Danese S. Janus kinase inhibitors for the treatment of inflammatory bowel diseases: developments from phase I and phase II clinical trials. Expert Opin Investig Drugs. 2018 Jul;27(7):595-599. doi: 10.1080/13543784.2018.1492547. Epub 2018 Jul 6. Review. PubMed PMID: 29938545.
2: Sands BE, Sandborn WJ, Feagan BG, Lichtenstein GR, Zhang H, Strauss R, Szapary P, Johanns J, Panes J, Vermeire S, O’Brien CD, Yang Z, Bertelsen K, Marano C; Peficitinib-UC Study Group. Peficitinib, an Oral Janus Kinase Inhibitor, in Moderate-to-Severe Ulcerative Colitis: Results From a Randomized, Phase 2 Study. J Crohns Colitis. 2018 Jun 15. doi: 10.1093/ecco-jcc/jjy085. [Epub ahead of print] PubMed PMID: 29917064.
3: Veale DJ, McGonagle D, McInnes IB, Krueger JG, Ritchlin CT, Elewaut D, Kanik KS, Hendrikx T, Berstein G, Hodge J, Telliez JB. The rationale for Janus kinase inhibitors for the treatment of spondyloarthritis. Rheumatology (Oxford). 2018 Apr 3. doi: 10.1093/rheumatology/key070. [Epub ahead of print] PubMed PMID: 29618084.
4: Cline A, Cardwell LA, Feldman SR. Advances in treating psoriasis in the elderly with small molecule inhibitors. Expert Opin Pharmacother. 2017 Dec;18(18):1965-1973. doi: 10.1080/14656566.2017.1409205. Epub 2017 Nov 27. Review. PubMed PMID: 29171774.
5: Baker KF, Isaacs JD. Novel therapies for immune-mediated inflammatory diseases: What can we learn from their use in rheumatoid arthritis, spondyloarthritis, systemic lupus erythematosus, psoriasis, Crohn’s disease and ulcerative colitis? Ann Rheum Dis. 2018 Feb;77(2):175-187. doi: 10.1136/annrheumdis-2017-211555. Epub 2017 Aug 1. Review. PubMed PMID: 28765121.
6: Zhu T, Howieson C, Wojtkowski T, Garg JP, Han D, Fisniku O, Keirns J. The Effect of Verapamil, a P-Glycoprotein Inhibitor, on the Pharmacokinetics of Peficitinib, an Orally Administered, Once-Daily JAK Inhibitor. Clin Pharmacol Drug Dev. 2017 Nov;6(6):548-555. doi: 10.1002/cpdd.344. Epub 2017 Mar 16. PubMed PMID: 28301084.
7: Genovese MC, Greenwald M, Codding C, Zubrzycka-Sienkiewicz A, Kivitz AJ, Wang A, Shay K, Wang X, Garg JP, Cardiel MH. Peficitinib, a JAK Inhibitor, in Combination With Limited Conventional Synthetic Disease-Modifying Antirheumatic Drugs in the Treatment of Moderate-to-Severe Rheumatoid Arthritis. Arthritis Rheumatol. 2017 May;69(5):932-942. doi: 10.1002/art.40054. PubMed PMID: 28118538.
8: Ito M, Yamazaki S, Yamagami K, Kuno M, Morita Y, Okuma K, Nakamura K, Chida N, Inami M, Inoue T, Shirakami S, Higashi Y. A novel JAK inhibitor, peficitinib, demonstrates potent efficacy in a rat adjuvant-induced arthritis model. J Pharmacol Sci. 2017 Jan;133(1):25-33. doi: 10.1016/j.jphs.2016.12.001. Epub 2016 Dec 23. PubMed PMID: 28117214.
9: Zhu T, Parker B, Wojtkowski T, Nishimura T, Garg JP, Han D, Fisniku O, Keirns J. Drug Interactions Between Peficitinib, an Orally Administered, Once-Daily Janus Kinase Inhibitor, and Rosuvastatin in Healthy Subjects. Clin Pharmacokinet. 2017 Jul;56(7):747-757. doi: 10.1007/s40262-016-0474-4. PubMed PMID: 27878567.
10: Semerano L, Decker P, Clavel G, Boissier MC. Developments with investigational Janus kinase inhibitors for rheumatoid arthritis. Expert Opin Investig Drugs. 2016 Dec;25(12):1355-1359. Epub 2016 Oct 31. PubMed PMID: 27748152.
11: Kivitz AJ, Gutierrez-Ureña SR, Poiley J, Genovese MC, Kristy R, Shay K, Wang X, Garg JP, Zubrzycka-Sienkiewicz A. Peficitinib, a JAK Inhibitor, in the Treatment of Moderate-to-Severe Rheumatoid Arthritis in Patients With an Inadequate Response to Methotrexate. Arthritis Rheumatol. 2017 Apr;69(4):709-719. doi: 10.1002/art.39955. PubMed PMID: 27748083.
12: Lam S. JAK inhibitors: A broadening approach in rheumatoid arthritis. Drugs Today (Barc). 2016 Aug;52(8):467-469. PubMed PMID: 27722215.
13: Roskoski R Jr. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol Res. 2016 Sep;111:784-803. doi: 10.1016/j.phrs.2016.07.038. Epub 2016 Jul 26. Review. PubMed PMID: 27473820.
14: Iwata S, Tanaka Y. Progress in understanding the safety and efficacy of Janus kinase inhibitors for treatment of rheumatoid arthritis. Expert Rev Clin Immunol. 2016 Oct;12(10):1047-57. doi: 10.1080/1744666X.2016.1189826. Epub 2016 Jun 6. Review. PubMed PMID: 27253519.
15: Cao YJ, Sawamoto T, Valluri U, Cho K, Lewand M, Swan S, Lasseter K, Matson M, Holman J Jr, Keirns J, Zhu T. Pharmacokinetics, Pharmacodynamics, and Safety of ASP015K (Peficitinib), a New Janus Kinase Inhibitor, in Healthy Subjects. Clin Pharmacol Drug Dev. 2016 Nov;5(6):435-449. doi: 10.1002/cpdd.273. Epub 2016 Jun 30. PubMed PMID: 27162173.
16: Nielsen OH, Seidelin JB, Ainsworth M, Coskun M. Will novel oral formulations change the management of inflammatory bowel disease? Expert Opin Investig Drugs. 2016 Jun;25(6):709-18. doi: 10.1517/13543784.2016.1165204. Epub 2016 Mar 28. Review. PubMed PMID: 26967267.
17: Yiu ZZ, Warren RB. Novel Oral Therapies for Psoriasis and Psoriatic Arthritis. Am J Clin Dermatol. 2016 Jun;17(3):191-200. doi: 10.1007/s40257-016-0179-3. Review. PubMed PMID: 26923915.
18: Takeuchi T, Tanaka Y, Iwasaki M, Ishikura H, Saeki S, Kaneko Y. Efficacy and safety of the oral Janus kinase inhibitor peficitinib (ASP015K) monotherapy in patients with moderate to severe rheumatoid arthritis in Japan: a 12-week, randomised, double-blind, placebo-controlled phase IIb study. Ann Rheum Dis. 2016 Jun;75(6):1057-64. doi: 10.1136/annrheumdis-2015-208279. Epub 2015 Dec 15. PubMed PMID: 26672064; PubMed Central PMCID: PMC4893099.
19: Oda K, Cao YJ, Sawamoto T, Nakada N, Fisniku O, Nagasaka Y, Sohda KY. Human mass balance, metabolite profile and identification of metabolic enzymes of [¹⁴C]ASP015K, a novel oral janus kinase inhibitor. Xenobiotica. 2015;45(10):887-902. doi: 10.3109/00498254.2015.1026864. Epub 2015 May 19. PubMed PMID: 25986538.
20: Nakada N, Oda K. Identification and characterization of metabolites of ASP015K, a novel oral Janus kinase inhibitor, in rats, chimeric mice with humanized liver, and humans. Xenobiotica. 2015;45(9):757-65. doi: 10.3109/00498254.2015.1019594. Epub 2015 Jun 12. PubMed PMID: 25869242.
/////////////////Peficitinib hydrobromide, Smyraf, JAPAN 2019, ペフィシチニブ臭化水素酸塩 , ASP015K, Rheumatoid Arthritis
O=C(C1=CN=C(NC=C2)C2=C1N[C@@H]3[C@]4([H])C[C@@]5([H])C[C@](C4)(O)C[C@]3([H])C5)N.[H]Br
Romosozumab, ロモソズマブ (遺伝子組換え)
Romosozumab
ロモソズマブ (遺伝子組換え)
AMG 785
Immunoglobulin G2, anti-(human sclerostin) (human-mouse monoclonal 785A070802 heavy chain), disulfide with human-mouse monoclonal 785A070802 κ-chain, dimer
- Immunoglobulin G2, anti-(human sclerostin) (humanized monoclonal 785A070802 heavy chain), disulfide with humanized monoclonal 785A070802 κ-chain, dimer
| Formula |
C6452H9926N1714O2040S54
|
|---|---|
| CAS |
909395-70-6
|
| Mol weight |
145875.6186
|
Monoclonal antibody
Treatment of osteoporosis
Osteoporosis agent, Sclerostin activity inhibitor
JAPAN APPROVED 2019/1/8, Evenity
Romosozumab (AMG 785) is a humanized monoclonal antibody that targets sclerostin for the treatment of osteoporosis.[1]
Romosozumab was originally discovered by Chiroscience,[2] which was acquired by Celltech (now owned by UCB).[3] Celltech entered in a partnership with Amgen in 2002 for the product’s development.[4]
In 2016 results from 12 months of a clinical study were reported.[5]
Some results from the FRAME[6] and ARCH clinical studies were reported on in 2017.[7]
Japan’s Ministry of Health, Labor and Welfare has granted a marketing authorization for romosozumab (EVENITY) for the treatment of osteoporosis in patients at high risk of fracture. Developed by Amgen and UCB, romosozumab is a humanized IgG2 monoclonal antibody that targets sclerostin. The approval in Japan is based on results from the Phase 3 FRAME and BRIDGE studies, which included 7,180 postmenopausal women with osteoporosis and 245 men with osteoporosis, respectively.
A biologics license application (BLA) for romosozumab as a treatment of osteoporosis in postmenopausal women at high risk for fracture was submitted to the U.S. Food and Drug Administration (FDA) in July 2016, but additional safety and efficacy data was requested in the FDA’s complete response letter, as announced by Amgen and UCB in July 2017. In July 2018, Amgen and UCB announced that the BLA had been resubmitted. In addition to data from early-stage clinical studies, the original BLA included data from the Phase 3 FRAME study. The resubmitted BLA includes results from the more recent Phase 3 ARCH study, an alendronate-active comparator trial including 4,093 postmenopausal women with osteoporosis who experienced a fracture, and the Phase 3 BRIDGE study. The FDA’s Bone, Reproductive and Urologic Drugs Advisory Committee is scheduled to review data supporting the BLA for romosozumab at a meeting on January 16, 2019.
The European Medicines Agency is also currently reviewing a marketing application for romosozumab.
|
Commercial production of cell culture-derived products (for example, protein-based products, such as monoclonal antibodies (mAbs)), requires optimization of cell culture parameters in order for the cells to produce enough product to meet clinical and commercial demands. However, when cell culture parameters are optimized for improving productivity of a protein product, it is also necessary to maintain desired quality specifications of the product such as glycosylation profile, aggregate levels, charge heterogeneity, and amino acid sequence integrity (Li, et al., 2010 , mAbs., 2(5):466-477).
|
References
- ^ “Statement On A Nonproprietary Name Adopted By The USAN Council: Romosozumab” (PDF). American Medical Association.
- ^ Quested, Tony (June 7, 2015). “Cream of life science entrepreneurs’ first venture was selling doughnuts”. Business Week. Cambridge, England: Q Communications. Retrieved December 24, 2018.
- ^ Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003 Dec 1;22(23):6267-76.
- ^ Celltech group Annual Report and Accounts 2002
- ^ Cosman; et al. (2016). “Romosozumab Treatment in Postmenopausal Women with Osteoporosis”. The New England Journal of Medicine. 375: 1532–1543. doi:10.1056/NEJMoa1607948. PMID 27641143.
- ^ Efficacy and Safety of Romosozumab Treatment in Postmenopausal Women With Osteoporosis (FRAME)
- ^ Bone Loss Drug Effective, But is it Safe? Oct 2017
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from mouse) |
| Target | Sclerostin |
| Clinical data | |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| ChemSpider |
|
| KEGG | |
| Chemical and physical data | |
| Formula | C6452H9926N1714O2040S54 |
| Molar mass | 145.9 kg/mol |
///////////Romosozumab, ロモソズマブ (遺伝子組換え) , JAPAN 2019, Monoclonal antibody, Osteoporosis, AMG 785
|
Monoclonals for bone, musculoskeletal, circulatory, and neurologic systems
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Bone (“-os-“, “-s(o)-“) |
|
||||||||
| Musculoskeletal (“-mul-“) |
|
||||||||
| Circulatory (“-c(i[r])-“) |
|
||||||||
| Neurologic (“-ne(u)(r)-“) |
|
||||||||
| Angiogenesis inhibitor (“-anibi-“) |
|
||||||||
| Growth factor (“-gr(o)-“) |
|
||||||||
|
|||||||||
Esaxerenone エサキセレノン , эсаксеренон , إيساكسيرينون , 艾沙利 酮 ,
Esaxerenone
エサキセレノン , эсаксеренон , إيساكسيرينون , 艾沙利 酮 ,
CS-3150, XL-550
| Formula |
C22H21F3N2O4S
|
|---|---|
| CAS |
1632006-28-0
|
| Mol weight |
466.4734
|
Pmda approved japan, 2019/1/8, Minebro
Antihypertensive, Aldosterone antagonist
- Originator X-Ceptor Therapeutics
- Developer Daiichi Sankyo Company
- Class Antihyperglycaemics; Antihypertensives; Pyrroles; Small molecules; Sulfones
- Mechanism of Action Mineralocorticoid receptor antagonists
- Registered Hypertension
- Phase III Diabetic nephropathies
- No development reported Cardiovascular disorders; Heart failure
- 09 Jan 2019 Registered for Hypertension in Japan (PO) – First global approval
- 27 Nov 2018 Daiichi Sankyo completes a phase III trial in Diabetic nephropathies in Japan (PO) (JapicCTI-173696)
- 08 Jun 2018 Efficacy and adverse events data from the phase III ESAX-HTN trial in Essential hypertension presented 28th European Meeting on Hypertension and Cardiovascular Protection (ESH-2018)
CS 3150, angiotensin II receptor antagonist, for the treatment or prevention of such hypertension and heart disease similar to olmesartan , losartan, candesartan , valsartan, irbesartan, telmisartan, eprosartan,
Cas name 1H-Pyrrole-3-carboxamide, 1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-, (5S)-
CAS 1632006-28-0 for S conf
MF C22 H21 F3 N2 O4 S
MW 466.47
(S)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
CAS 1632006-28-0 for S configuration
1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide
(S) -1- (2- hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide
(+/-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide, CAS 880780-76-7
(+)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide..1072195-82-4
(-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide..1072195-83-5
WO2008 / 126831 (US Publication US2010-0093826)http://www.google.co.in/patents/EP2133330A1?cl=en
WO 2006012642..compound A;..http://www.google.com/patents/WO2006012642A2?cl=en
WO2006 / 012642 (US Publication US2008-0234270)
WO 2015030010…http://www.google.com/patents/WO2015030010A1?cl=en
How to synthesis Esaxerenone 1632006-28-0 – YouTube

Esaxerenone 1632006-28-0, FDA approved new drug will be a big potential drug. Original Route of Synthesis …

Esaxerenone (INN) (developmental code names CS-3150, XL-550) is a nonsteroidal antimineralocorticoid which was discovered by Exelixis and is now under development by Daiichi Sankyo Company for the treatment of hypertension, essential hypertension, hyperaldosteronism, and diabetic nephropathies.[1][2][3] It acts as a highly selective silent antagonist of the mineralocorticoid receptor(MR), the receptor for aldosterone, with greater than 1,000-fold selectivity for this receptor over other steroid hormone receptors, and 4-fold and 76-fold higher affinity for the MR relative to the existing antimineralocorticoids spironolactone and eplerenone.[1][2][3] As of 2017, esaxerenone is in phase III clinical trials for hypertension, essential hypertension, and hyperaldosteronism and is in phase IIclinical trials for diabetic nephropathies.[1]
- Mechanism of Action Mineralocorticoid receptor antagonists

JAPAN PHASE 2……….Phase 2 Study to Evaluate Efficacy and Safety of CS-3150 in Patients with Essential Hypertension
http://www.clinicaltrials.jp/user/showCteDetailE.jsp?japicId=JapicCTI-121921
Phase II Diabetic nephropathies; Hypertension
- 01 Jan 2015 Daiichi Sankyo initiates a phase IIb trial for Diabetic nephropathies in Japan (NCT02345057)
- 01 Jan 2015 Daiichi Sankyo initiates a phase IIb trial for Hypertension in Japan (NCT02345044)
- 01 May 2013 Phase-II clinical trials in Diabetic nephropathies in Japan (PO)
-
Currently, angiotensin II receptor antagonists and calcium antagonists are widely used as a medicament for the treatment or prevention of such hypertension or heart disease.Mineralocorticoid receptor (MR) (aldosterone receptor) has been known to play an important role in the control of body electrolyte balance and blood pressure, spironolactone having a steroid structure, MR antagonists such as eplerenone, are known to be useful in the treatment of hypertension-heart failure.Renin – angiotensin II receptor antagonists are inhibitors of angiotensin system is particularly effective in renin-dependent hypertension, and show a protective effect against cardiovascular and renal failure. Also, the calcium antagonists, and by the function of the calcium channel antagonizes (inhibits), since it has a natriuretic action in addition to the vasodilating action, is effective for hypertension fluid retention properties (renin-independent) .Therefore, the MR antagonist, when combined angiotensin II receptor antagonists or calcium antagonists, it is possible to suppress the genesis of multiple hypertension simultaneously, therapeutic or prophylactic effect of the stable and sufficient hypertension irrespective of the etiology is expected to exhibit.Also, diuretics are widely used as a medicament for the treatment or prevention of such hypertension or heart disease. Diuretic agent is effective in the treatment of hypertension from its diuretic effect. Therefore, if used in combination MR antagonists and diuretics, the diuretic effect of diuretics, it is possible to suppress the genesis of multiple blood pressure at the same time, shows a therapeutic or prophylactic effect of the stable and sufficient hypertension irrespective of the etiology it is expected.1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide (hereinafter, compound ( I)) is, it is disclosed in Patent Documents 1 and 2, hypertension, for the treatment of such diabetic nephropathy are known to be useful.
CS-3150 (XL550) is a small-molecule antagonist of the mineralocorticoid receptor (MR), a nuclear hormone receptor implicated in a variety of cardiovascular and metabolic diseases. MR antagonists can be used to treat hypertension and congestive heart failure due to their vascular protective effects. Recent studies have also shown beneficial effects of adding MR antagonists to the treatment regimen for Type II diabetic patients with nephropathy. CS-3150 is a non-steroidal, selective MR antagonist that has the potential for the treatment of hypertension, congestive heart failure, or end organ protection due to vascular damage.
Useful as a mineralocorticoid receptor (MR) antagonist, for treating hypertension, cardiac failure and diabetic nephropathy. It is likely to be CS-3150, a non-steroidal MR antagonist, being developed by Daiichi Sankyo (formerly Sankyo), under license from Exelixis, for treating hypertension and diabetic nephropathy (phase 2 clinical, as of March 2015). In January 2015, a phase II trial for type 2 diabetes mellitus and microalbuminuria was planned to be initiated later that month (NCT02345057).
Exelixis discovered CS-3150 and out-licensed the compound to Daiichi-Sankyo. Two phase 2a clinical trials, one in hypertensive patients and the other in type 2 diabetes with albuminuria, are currently being conducted in Japan by Daiichi-Sankyo.


Mineralocorticoid receptor (MR) (aldosterone receptor) has been known to play an important role in the control of body electrolyte balance and blood pressure, spironolactone having a steroid structure, MR antagonists such as eplerenone, are known to be useful in the treatment of hypertension-heart failure.
CS-3150 (XL550) is a small-molecule antagonist of the mineralocorticoid receptor (MR), a nuclear hormone receptor implicated in a variety of cardiovascular and metabolic diseases. MR antagonists can be used to treat hypertension and congestive heart failure due to their vascular protective effects. Recent studies have also shown beneficial effects of adding MR antagonists to the treatment regimen for Type II diabetic patients with nephropathy. CS-3150 is a non-steroidal, selective MR antagonist that has the potential for the treatment of hypertension, congestive heart failure, or end organ protection due to vascular damage.
Exelixis discovered CS-3150 and out-licensed the compound to Daiichi-Sankyo. Two phase 2a clinical trials, one in hypertensive patients and the other in type 2 diabetes with albuminuria, are currently being conducted in Japan by Daiichi-Sankyo.
Daiichi Sankyo (formerly Sankyo), under license from Exelixis, is developing CS-3150 (XL-550), a non-steroidal mineralocorticoid receptor (MR) antagonist, for the potential oral treatment of hypertension and diabetic nephropathy, microalbuminuria , By October 2012, phase II development had begun ; in May 2014, the drug was listed as being in phase IIb development . In January 2015, a phase II trial for type 2 diabetes mellitus and microalbuminuria was planned to be initiated later that month. At that time, the trial was expected to complete in March 2017 .
Exelixis, following its acquisition of X-Ceptor Therapeutics in October 2004 , was investigating the agent for the potential treatment of metabolic disorders and cardiovascular diseases, such as hypertension and congestive heart failure . In September 2004, Exelixis expected to file an IND in 2006. However, it appears that the company had fully outlicensed the agent to Sankyo since March 2006 .
| Description | Small molecule antagonist of the mineralocorticoid receptor (MR) |
| Molecular Target | Mineralocorticoid receptor |
| Mechanism of Action | Mineralocorticoid receptor antagonist |
| Therapeutic Modality | Small molecule |
In January 2015, a multi-center, placebo-controlled, randomized, 5-parallel group, double-blind, phase II trial (JapicCTI-152774; NCT02345057; CS3150-B-J204) was planned to be initiated later that month in Japan, in patients with type 2 diabetes mellitus and microalbuminuria, to assess the efficacy and safety of different doses of CS-3150 compared to placebo. At that time, the trial was expected to complete in March 2017; later that month, the trial was initiated in the Japan
By October 2012, phase II development had begun in patients with essential hypertension
By January 2011, phase I trials had commenced in Japan
Several patents WO-2014168103,
WO-2015012205 and WO-2015030010
XL-550, claimed in WO-2006012642,
PATENT
http://www.google.co.in/patents/EP2133330A1?cl=en
(Example 3)(+/-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
-
After methyl 4-methyl-5-[2-(trifluoromethyl) phenyl]-1H-pyrrole-3-carboxylate was obtained by the method described in Example 16 of WO 2006/012642 , the following reaction was performed using this compound as a raw material.
-
Methyl 4-methyl-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylate (1.4 g, 4.9 mmol) was dissolved in methanol (12 mL), and a 5 M aqueous sodium hydroxide solution (10 mL) was added thereto, and the resulting mixture was heated under reflux for 3 hours. After the mixture was cooled to room temperature, formic acid (5 mL) was added thereto to stop the reaction. After the mixture was concentrated under reduced pressure, water (10 mL) was added thereto to suspend the resulting residue. The precipitated solid was collected by filtration and washed 3 times with water. The obtained solid was dried under reduced pressure, whereby 4-methyl-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylic acid (1.1 g, 83%) was obtained as a solid. The thus obtained solid was suspended in dichloromethane (10 mL), oxalyl chloride (0.86 mL, 10 mmol) was added thereto, and the resulting mixture was stirred at room temperature for 2 hours. After the mixture was concentrated under reduced pressure, the residue was dissolved in tetrahydrofuran (10 mL), and 4-(methylsulfonyl)aniline hydrochloride (1.0 g, 4.9 mmol) and N,N-diisopropylethylamine (2.8 mL, 16 mmol) were sequentially added to the solution, and the resulting mixture was heated under reflux for 18 hours. After the mixture was cooled to room temperature, the solvent was distilled off under reduced pressure, and acetonitrile (10 mL) and 3 M hydrochloric acid (100 mL) were added to the residue. A precipitated solid was triturated, collected by filtration and washed with water, and then, dried under reduced pressure, whereby 4-methyl-N-[4-(methylsulfonyl) phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide (1.4 g, 89%) was obtained as a solid.
1H-NMR (400 MHz, DMSO-d6) δ11.34 (1H, brs,), 9.89 (1H, s), 7.97 (2H, d, J = 6.6 Hz), 7.87-7.81 (3H, m), 7.73 (1H, t, J = 7.4 Hz), 7.65-7.61 (2H, m), 7.44 (1H, d, J = 7.8 Hz), 3.15 (3H, s), 2.01 (3H, s). -
Sodium hydride (0.12 g, 3 mmol, 60% dispersion in mineral oil) was dissolved in N,N-dimethylformamide (1.5 mL), and 4-methyl -N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide (0.47 g, 1.1 mmol) was added thereto, and then, the resulting mixture was stirred at room temperature for 30 minutes. Then, 1,3,2-dioxathiolane-2,2-dioxide (0.14 g, 1.2 mmol) was added thereto, and the resulting mixture was stirred at room temperature. After 1 hour, sodium hydride (40 mg, 1.0 mmol, oily, 60%) was added thereto again, and the resulting mixture was stirred for 30 minutes. Then, 1,3,2-dioxathiolane-2,2-dioxide (12 mg, 0.11 mmol) was added thereto, and the resulting mixture was stirred at room temperature for 1 hour. After the mixture was concentrated under reduced pressure, methanol (5 mL) was added to the residue and insoluble substances were removed by filtration, and the filtrate was concentrated again. To the residue, tetrahydrofuran (2 mL) and 6 M hydrochloric acid (2 mL) were added, and the resulting mixture was stirred at 60°C for 16 hours. The reaction was cooled to room temperature, and then dissolved in ethyl acetate, and washed with water and saturated saline. The organic layer was dried over anhydrous sodium sulfate and filtered. Then, the filtrate was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (ethyl acetate), whereby the objective compound (0.25 g, 48%) was obtained.
1H-NMR (400 MHz, CDCl3) δ: 7.89-7.79 (m, 6H), 7.66-7.58 (m, 2H), 7.49 (s, 1H), 7.36 (d, 1H, J = 7.4Hz), 3.81-3.63 (m, 4H), 3.05 (s, 3H), 2.08 (s, 3H).
HR-MS (ESI) calcd for C22H22F3N2O4S [M+H]+, required m/z: 467.1252, found: 467.1246.
Anal. calcd for C22H21F3N2O4S: C, 56.65; H, 4.54; N, 6.01; F, 12.22; S, 6.87. found: C, 56.39; H, 4.58; N, 5.99; F, 12.72; S, 6.92.
(Example 4)
Optical Resolution of Compound of Example 3
-
Resolution was performed 4 times in the same manner as in Example 2, whereby 74 mg of Isomer C was obtained as a solid from a fraction containing Isomer C (tR = 10 min), and 71 mg of Isomer D was obtained as a solid from a fraction containing Isomer D (tR = 11 min).
-
Isomer C: (+)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
[α]D 21: +7.1° (c = 1.0, EtOH) .
1H-NMR (400 MHz, CDCl3) δ: 7.91 (s, 1H), 7.87-7.79 (m, 5H), 7.67-7.58 (m, 2H), 7.51 (s, 1H), 7.35 (d, 1H, J = 7.0 Hz), 3.78-3.65 (m, 4H), 3.05 (s, 3H), 2.07 (s, 3H).
HR-MS (ESI) calcd for C22H22F3N2O4S [M+H]+, required m/z: 467.1252, found: 467.1260.
Retention time: 4.0 min. -
Isomer D: (-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
[α]D 21: -7.2° (c = 1.1, EtOH) .
1H-NMR (400 MHz, CDCl3) δ: 7.88-7.79 (m, 6H), 7.67-7.58 (m, 2H), 7.50 (s, 1H), 7.36 (d, 1H, J = 7.5 Hz), 3.79-3.65 (m, 4H), 3.05 (s, 3H), 2.08 (s, 3H).
HR-MS (ESI) calcd for C22H22F3N2O4S [M+H]+, required m/z: 467.1252, found: 467.1257.
Retention time: 4.5 min.
……………………………………………….
WO 2014168103
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014168103
(7-1) (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid quinine salt
obtained by the method of Example 6 the (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid 50.00 g (160 mmol), N, N- dimethylacetamide (25 mL), ethyl acetate (85 mL) was added and dissolved at room temperature (solution 1).
Quinine 31.05 g (96 mmol) in N, N- dimethylacetamide (25 mL), ethyl acetate (350 mL), was heated in water (15 mL) 65 ~ 70 ℃ was added, was added dropwise a solution 1. After about 1 hour stirring the mixture at 65 ~ 70 ℃, and slowly cooled to 0 ~ 5 ℃ (cooling rate standard: about 0.3 ℃ / min), and stirred at that temperature for about 0.5 hours. The crystals were filtered, 5 ℃ using ethyl acetate (100 mL) which was cooled to below are washed crystals, the resulting wet product crystals was obtained and dried under reduced pressure to give the title compound 43.66 g at 40 ℃ (Yield 42.9%). Furthermore, the diastereomeric excess of the obtained salt was 98.3% de. 1 H NMR (400 MHz, DMSO-D 6 ) delta: 1.30 ~ 2.20 (10H, M), 2.41 ~ 2.49 (2H, M), 2.85 ~ 3.49 (6H, M), 3.65 ~ 3.66 (1H, M), 3.88 (3H, s), 4.82 (1H, broad singlet), 4.92 ~ 5.00 (2H, m), 5.23 ~ 5.25 (1H, m), 5.60 (1H, br), 5.80 ~ 6.00 (1H, m), 7.36 ~ 7.92 (9H, M), 8.67 (1H, D, J = 4.6 Hz) (7-2) (S)-1-(2-hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3 diastereomeric excess of the carboxylic acid quinine salt HPLC measurements (% de) that the title compound of about 10 mg was collected, and the 10 mL was diluted with 50v / v% aqueous acetonitrile me was used as a sample solution.
mobile phase A: 0.02mol / L phosphorus vinegar buffer solution (pH 3)
mobile phase B: acetonitrile
solution sending of mobile phase: mobile phase A and I indicates the mixing ratio of mobile phase B in Table 1 below.
flow rate: about 0.8 mL / min
column temperature: 30 ℃ constant temperature in the vicinity of
measuring time: about 20 min
Injection volume: 5 μL
diastereomeric excess (% de), the title compound (retention time about 12 min), was calculated by the following equation using a peak area ratio of R-isomer (retention time of about 13 min).
% De = {[(the title compound (S body) peak area ratio) – (R body peak area ratio)] ÷ [(the title compound (S body) peak area ratio) + (R body peak area ratio)]} × 100
(8-1) (S)-1-(2-hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole -3 – carboxylic acid
obtained by the method of Example 7 (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid (8α, 9R) -6′- methoxycinnamate Conan-9-ol 40.00 g (63 mmol) in ethyl acetate (400 mL), was added 2N aqueous hydrochloric acid (100 mL) was stirred at room temperature and separated . The resulting organic layer was concentrated under reduced pressure (120 mL), and added ethyl acetate (200 mL), and further concentrated under reduced pressure to obtain a solution containing the title compound (120 mL).
ethyl acetate (240 mL), was mixed tetrahydrofuran (80 mL) and oxalyl chloride 20.72 g (163 mmol), and cooled to 10 ~ 15 ℃ was. Then the resulting solution was added while keeping the 10 ~ 15 ℃ Example (8-1) and stirred for about 1 hour by heating to 15 ~ 20 ℃. After stirring, acetonitrile (120 mL) and pyridine 2.46 g (31 mmol) was added and the reaction mixture was concentrated under reduced pressure (120 mL), acetonitrile (200 mL) was added and further concentrated under reduced pressure (120 mL).
obtained by the method of Example 7 (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole 3-carboxylic acid (8α, 9R) -6′- methoxycinnamate Conan-9-ol 10.00 g (16 mmol) in t- butyl methyl ether (90 mL), water (10 mL) 36w / w% aqueous hydrochloric acid ( 5 mL) was added and stirring at room temperature and separated. The resulting organic layer was concentrated under reduced pressure (30 mL), was added ethyl acetate (50 mL), and further concentrated under reduced pressure to obtain a solution containing the title compound (30 mL).
ethyl acetate (50 mL), was mixed with tetrahydrofuran (20 mL) and oxalyl chloride 5.18 g (41 mmol), and cooled to 0 ~ 5 ℃ was.Then the resulting solution was added in Examples while maintaining the 0 ~ 5 ℃ (12-1), and the mixture was stirred for 6 hours at 0 ~ 10 ℃. After stirring, acetonitrile (30 mL) and pyridine 0.62 g (8 mmol) was added and the reaction mixture was concentrated under reduced pressure (30 mL), acetonitrile (50 mL) was added, and further concentrated under reduced pressure (30 mL).
Patent literature
Patent Document 2: International Publication WO2008 / 056907 (US Publication US2010-0093826)
Patent Document 3: Pat. No. 2,082,519 JP (US Patent No. 5,616,599 JP)
Patent Document 4: Pat. No. 1,401,088 JP (US Pat. No. 4,572,909)
Patent Document 5: US Pat. No. 3,025,292
Angiotensin II receptor 桔抗 agent
is described in Japanese or the like, its chemical name is (5-methyl-2-oxo-1,3-dioxolen-4-yl ) methyl 4- (1-hydroxy-1-methylethyl) -2-propyl-1 – in [2 ‘(1H- tetrazol-5-yl) biphenyl-4-ylmethyl] imidazole-5-carboxylate, Yes, olmesartan medoxomil of the present application includes its pharmacologically acceptable salt.
 OLMESARTAN
OLMESARTAN
References
- ^ Jump up to:a b c http://adisinsight.springer.com/drugs/800021527
- ^ Jump up to:a b Yang J, Young MJ (2016). “Mineralocorticoid receptor antagonists-pharmacodynamics and pharmacokinetic differences”. Curr Opin Pharmacol. 27: 78–85. doi:10.1016/j.coph.2016.02.005. PMID 26939027.
- ^ Jump up to:a b Kolkhof P, Nowack C, Eitner F (2015). “Nonsteroidal antagonists of the mineralocorticoid receptor”. Curr. Opin. Nephrol. Hypertens. 24 (5): 417–24. doi:10.1097/MNH.0000000000000147. PMID 26083526.
External links
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| Drug class | Antimineralocorticoid |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C22H21F3N2O4S |
| Molar mass | 466.475 g/mol |
| 3D model (JSmol) | |
///////////JAPAN 2019, Esaxerenone, Minebro, エサキセレノン ,Phase III, Diabetic nephropathies, HYPERTENSION. PHASE 3, N62TGJ04A1, UNII:N62TGJ04A1, эсаксеренон , إيساكسيرينون , 艾沙利 酮 , CS-3150, XL-550, CS 3150, XL 550
Relugolix レルゴリクス

Relugolix (TAK-385), RVT 601
レルゴリクス
UPDATE FDA APPROVED, 12/18/2020, Orgovyx
To treat advanced prostate cancer
Press Release
| Formula |
C29H27F2N7O5S
|
|---|---|
| CAS |
737789-87-6
|
| Mol weight
UNII |
623.6304
UNII-P76B05O5V6
|
2019/1/8 PMDA JAPAN APPROVED, Relumina

- Originator Takeda
- Developer Myovant Sciences; Takeda; Takeda Oncology
- Class Analgesics; Antineoplastics; Ketones; Pyrimidines; Small molecules
- Mechanism of Action LHRH receptor antagonists
- Preregistration Uterine leiomyoma
- Phase III Pain; Prostate cancer
- No development reported Solid tumours
- 08 Nov 2018 Myovant announces intention to submit NDA for Uterine leiomyoma in Q3 of 2019
- 08 Nov 2018 Myovant Sciences completes enrollment in the phase III LIBERTY 1 trial for Uterine leiomyoma (Combination therapy) in USA (PO)(NCT03049735)
- 25 Oct 2018 Myovant Sciences completes enrolment in its phase III HERO trial for Prostate cancer (Late-stage disease) in Denmark, Australia, Austria, Belgium, Canada, United Kingdom, USA, Japan, Taiwan, Sweden, Spain, Slovakia, New Zealand, Netherlands, South Korea, Germany, France and Finland (PO) (NCT03085095)

Relugolix has been used in trials studying the treatment of Endometriosis, Prostate Cancer, Uterine Fibroids, and Androgen Deprivation Treatment-naïve Nonmetastatic Prostate Cancer.
Relugolix (developmental code names RVT-601, TAK-385) is a gonadotropin-releasing hormone antagonist (GnRH antagonist) medication which is under development by Myovant Sciences and Takeda for the treatment of endometriosis, uterine fibroids, and prostate cancer.[1][2][3][4][5][6][7] Unlike most other GnRH modulators, but similarly to elagolix, relugolix is a non-peptide and small-molecule compound and is orally active.[6][7] As of July 2018, it is in the pre-registration phase of development for uterine fibroids and is in phase III clinical trials for endometriosis and prostate cancer.[1]
Pharmacology
Pharmacodynamics
Relugolix is a selective antagonist of the gonadotropin-releasing hormone receptor (GnRHR) (IC50 = 0.12 nM).[6][7][8]
A single oral administration of relugolix at a dose of 3 mg/kg has been found to suppress luteinizing hormone (LH) levels for more than 24 hours in castrated cynomolgus monkeys, indicating a long duration of action.[6] The drug (80–160 mg/day) has been found to reduce testosterone levels to sustained castrate levels in men with once-daily administration.[8] Lower dosages (10–40 mg/day) are being studied in the treatment of endometriosis and uterine fibroids to achieve partial sex hormone suppression.[4] The reasoning behind partial suppression for these conditions is to reduce the incidence and severity of menopausal symptoms such as hot flushes and to avoid bone mineral density changes caused by estrogen deficiency that can eventually lead to osteoporosis.[4][9]
History
Relugolix was first described in 2004.[10][6] It superseded sufugolix, which was developed by the same group.[6]
Society and culture
Generic names
Relugolix is the generic name of the drug and its INN and USAN.[11] It is also known by its developmental code names RVT-601 and TAK-385.[1][11]
SYN
Journal of Medicinal Chemistry, 54(14), 4998-5012; 2011

PATENT
http://www.google.co.in/patents/EP1591446A1?cl=en
(Production Method 1)

-
- (Production method 2)

-
-
- Example 83
-
http://www.google.co.in/patents/EP1591446A1?cl=en
- Production of N-(4-(1-(2,6-difluorobenzyl)-5-((dimethylamino)methyl)-3-(6-methoxy-3-pyridazinyl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl)phenyl)-N’-methoxyurea
-

-
The similar reaction as described in Example 4 by using the compound (100 mg, 0.164 mmol) obtained in Reference Example 54 and methyl iodide (0.010 ml, 0.164 mmol) gave the title compound (17.3 mg, 17 %) as colorless crystals.
1 H-NMR(CDCl3) δ: 2.15 (6H, s), 3.6-3.8 (2H, m), 3.82 (3H, s), 4.18 (3H, s), 5.35 (2H, s), 6.92 (2H, t, J = 8.2 Hz), 7.12 (1H, d, J = 8.8 Hz), 7.2-7.65 (7H, m), 7.69 (1H, s).

PAPER
Discovery of 1-{4-[1-(2,6-difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor
J Med Chem 2011, 54(14): 4998. http://pubs.acs.org/doi/full/10.1021/jm200216q
1-{4-[1-(2,6-Difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (16b)
 tak 385
tak 385
http://pubs.acs.org/doi/suppl/10.1021/jm200216q/suppl_file/jm200216q_si_001.pdf
PATENT
Method for the production of TAK-385 or its salt and crystals starting from 6-(4-aminophenyl)-1-(2,6-difluorobenzyl)-5-dimethylaminomethyl-3-(6-methoxypyridazin-3-yl) thieno[2,3-d] pyrimidine-2,4 (1H,3H)-dione or its salt. Takeda Pharmaceutical is developing relugolix (TAK-385), an oral LHRH receptor antagonist analog of sufugolix, for the treatment of endometriosis and uterine fibroids. As of April 2014, the drug is in Phase 2 trails. See WO2010026993 claiming method for improving the oral absorption and stability of tetrahydro-thieno[2,3-d]pyrimidin-6-yl]-phenyl)-N’-methoxy urea derivatives.
PATENT
https://patents.google.com/patent/WO2015062391A1/en
Endometriosis is a common estrogen-dependent gynecological diseases, often occurs in women during their childbearing years, and its mechanism is unclear. Complex and difficult to diagnose the cause of the symptoms of endometriosis is unknown, serious block to the discovery of effective therapies. Currently, endometriosis primarily by laparoscopy diagnosis, and treatment by surgery, or pill, or progesterone receptor agonists of GnRH reduce estrogen levels to control.
Currently the high incidence of endometriosis, Datamonitor 2009 year data show that only two countries, India and China, the number of female patients suffering from endometriosis had more than 68 million (31,288,000 India, China 3753.5 million) passengers, while the national prevalence of the number seven major markets have more than 17 million. Datamonitor expects 2009 to 2018, endometriosis market from 2009 to $ 764 million (US $ 596 billion and the EU $ 117 million, Japan US $ 051 million) in 2018 increased to US $ 1.156 billion (US 8.44 billion dollars, 206 million US dollars the European Union, Japan $ 106 million), while the Chinese market will have more room for growth.
Gonadotropin-releasing hormone (Gonadoliberin; gonadotropin releasing hormone; GnRH), also known as luteinizing hormone releasing hormone (LHRH), is synthesized by neuroendocrine cells of the hypothalamus hormones decapeptide (pGlu-His-Trp-Ser-Tyr-Gly- Leu-Arg-Pro-Gly-NH2), a central regulator of reproductive endocrine system. Which conveys the circulatory system through hypothalamus-pituitary portal to the pituitary, bind to the cells of the anterior pituitary GnRH receptor, such as gonadotropin luteinizing hormone (Luteinizing Hormone, LH) and FSH (Follicle-Stimulating Hormone, FSH ) secretion and release, regulation of normal development and corpus luteum of the ovary, hypothalamic – pituitary – gonadal axis plays an important role. GnRH receptors capable of activating the G protein coupled calcium phosphatidylinositol second messenger system exert their regulatory role, and LH is adjusted to produce steroids, FSH regulating development of the male and female follicle spermatogenesis.
LH and FSH are released into the circulation, and combined with the ovaries or testes specific cell receptors, stimulating the production of steroids. The presence of sex steroids, diseases such as endometriosis, uterine fibroids, prostate cancer and exacerbations, to be given long-acting GnRH receptor agonists and antagonists for treatment control peptides.
Peptide GnRH receptor antagonists include linear peptides (US 5,171,835) GnRH-derived, cyclic hexapeptide derivatives (US 2002/0065309), a bicyclic peptide derivative (Journal of Medicinal Chemistry, 1993; 36: 3265-73), etc. ; and GnRH receptor peptide agonists include leuprolide (leuprorelin, pGlu-His-Trp-Ser-Tyr-d-Leu-Leu-Arg-Pro-NHEt). However, there are many problems including oral absorbability, dosage form, dose volume, drug stability, sustained action, and metabolic stability of the peptide-type compound to be resolved. But the main reason small molecule GnRH receptor antagonists of peptide-based therapy is superior to the existing method is that small molecule GnRH receptor antagonist may be orally administered directly, convenient. Studies have shown that small molecule antagonists of endometriosis, precocious puberty, prostate cancer and other hormone-dependent diseases having a significant effect.
GnRH receptor agonist mediated indirect mechanisms of tumor suppression by long-term effects on the hypothalamic – pituitary – gonadal axis, leading to pituitary gonadotropins (FSH, LH) is reduced, thereby reducing the secretion of sex hormones and indirectly inhibit growth of tumor cells. And a GnRH receptor antagonist directly to inhibit the release of the pituitary gonadotropins, thereby inhibiting tumor cell growth.
Given the limitations of peptide GnRH receptor antagonists, non-peptide GnRH receptor antagonists have been proposed and into the development, clinical trials and launch phase, such as Elagolix (NBI-56418, or also known as ABT-620) is a Abbott and Neurocrine Biosciences Inc company co-developed small molecule GnRH receptor antagonist, is currently in phase III clinical stage, mainly used in the treatment of endometriosis (III phase) and uterine fibroids (II period). June 2012, data released results of a Phase II clinical endometrial endometriosis Houston, the 94th annual meeting of the Endocrine Society: 131 accepts elagolix (150 or 250mg qd), leuprorelin depot (3.75mg sc in, once a month, female patients with endometriosis endometrium 12 weeks) or placebo treatment, elagolix treatment groups in patients with serum hormone estrogen compared to leuprorelin therapy group and the placebo group was significantly reduced. At the same time, elagolix safety and tolerability have been well verified.
Relugolix also known as TAK-385, is a GnRH by the Japanese Takada Pharmaceutical company developed an oral small molecule receptor antagonist, for the treatment of endometriosis, uterine fibroids and prostate. 2011 entered endometriosis and uterine fibroids clinical phase II study, carried out a clinical study of prostate cancer in the same year.
It disclosed a series of current small molecule GnRH receptor antagonists including patent WO2006096785, WO2010026993, WO2011076687, WO2012175514 like.
Despite the large number of interesting studies have been conducted in this field, there remains a need to continue research and development of more effective small molecule GnRH receptor antagonists, the present invention provides a novel GnRH receptor antagonist structure, and found to have such a structure compounds having good activity, reproductive endocrine system effective to treat the disease.
PATENT
US 20120071486, https://patentscope.wipo.int/search/en/detail.jsf?docId=US73518712&redirectedID=true
Example 83
Production of N-(4-(1-(2,6-difluorobenzyl)-5-((dimethylamino)methyl)-3-(6-methoxy-3-pyridazinyl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl)phenyl)-N′-methoxyurea
References
Discovery of TAK-385, a thieno[2,3-d]pyrimidine-2,4-dione derivative, as a potent and orally bioavailable nonpeptide antagonist of gonadotropin releasing hormone (GnRH) receptor
238th ACS Natl Meet (August 16-20, Washington) 2009, Abst MEDI 386
Discovery of 1-{4-[1-(2,6-difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor
J Med Chem 2011, 54(14): 4998. http://pubs.acs.org/doi/full/10.1021/jm200216q
References
- ^ Jump up to:a b c http://adisinsight.springer.com/drugs/800028257
- ^ Goenka L, George M, Sen M (June 2017). “A peek into the drug development scenario of endometriosis – A systematic review”. Biomed. Pharmacother. 90: 575–585. doi:10.1016/j.biopha.2017.03.092. PMID 28407578.
- ^ Dellis A, Papatsoris A (October 2017). “Therapeutic outcomes of the LHRH antagonists”. Expert Rev Pharmacoecon Outcomes Res. 17 (5): 481–488. doi:10.1080/14737167.2017.1375855. PMID 28870102.
- ^ Jump up to:a b c Streuli I, de Ziegler D, Borghese B, Santulli P, Batteux F, Chapron C (March 2012). “New treatment strategies and emerging drugs in endometriosis”. Expert Opin Emerg Drugs. doi:10.1517/14728214.2012.668885. PMID 22439891.
- ^ Elancheran, R.; Maruthanila, V. L.; Ramanathan, M.; Kabilan, S.; Devi, R.; Kunnumakara, A.; Kotoky, Jibon (2015). “Recent discoveries and developments of androgen receptor based therapy for prostate cancer”. Med. Chem. Commun. 6 (5): 746–768. doi:10.1039/C4MD00416G. ISSN 2040-2503.
- ^ Jump up to:a b c d e f Miwa K, Hitaka T, Imada T, Sasaki S, Yoshimatsu M, Kusaka M, Tanaka A, Nakata D, Furuya S, Endo S, Hamamura K, Kitazaki T (July 2011). “Discovery of 1-{4-[1-(2,6-difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor”. J. Med. Chem. 54 (14): 4998–5012. doi:10.1021/jm200216q. PMID 21657270.
- ^ Jump up to:a b c Nakata D, Masaki T, Tanaka A, Yoshimatsu M, Akinaga Y, Asada M, Sasada R, Takeyama M, Miwa K, Watanabe T, Kusaka M (January 2014). “Suppression of the hypothalamic-pituitary-gonadal axis by TAK-385 (relugolix), a novel, investigational, orally active, small molecule gonadotropin-releasing hormone (GnRH) antagonist: studies in human GnRH receptor knock-in mice”. Eur. J. Pharmacol. 723: 167–74. doi:10.1016/j.ejphar.2013.12.001. PMID 24333551.
- ^ Jump up to:a b MacLean D, Shi H, Suri A, Faessel H, and Saad F (2013). “Safety and Testosterone-Lowering Effects of the Investigational, Oral, GnRH Antagonist, TAK-385 in Healthy Male Volunteers: Results of a Phase 1 Inpatient/Outpatient Study”. doi:10.1210/endo-meetings.2013.CN.1.SAT-318.
- ^ Struthers RS, Nicholls AJ, Grundy J, Chen T, Jimenez R, Yen SS, Bozigian HP (February 2009). “Suppression of gonadotropins and estradiol in premenopausal women by oral administration of the nonpeptide gonadotropin-releasing hormone antagonist elagolix”. J. Clin. Endocrinol. Metab. 94 (2): 545–51. doi:10.1210/jc.2008-1695. PMC 2646513. PMID 19033369.
- ^ https://patents.google.com/patent/US7300935/
- ^ Jump up to:a b https://chem.nlm.nih.gov/chemidplus/rn/737789-87-6
 |
|
 |
|
| Clinical data | |
|---|---|
| Synonyms | RVT-601; TAK-385 |
| Routes of administration |
By mouth |
| Drug class | GnRH antagonist |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C29H27F2N7O5S |
| Molar mass | 623.630 g/mol |
| 3D model (JSmol) | |
External links
| PHASE | STATUS | PURPOSE | CONDITIONS | COUNT |
|---|---|---|---|---|
| 1 | Completed | Basic Science | Healthy Volunteers | 1 |
| 1 | Completed | Treatment | Androgen Deprivation Treatment-naïve Nonmetastatic Prostate Cancer / Hormone Treatment-naïve Participants With Prostate Cancer | 1 |
| 1 | Completed | Treatment | Endometriosis / Prostate Cancer | 1 |
| 1 | Completed | Treatment | Japanese Premenopausal Healthy Adult Women | 1 |
| 2 | Completed | Treatment | Endometriosis | 2 |
| 2 | Completed | Treatment | Prostate Cancer | 2 |
| 2 | Completed | Treatment | Uterine Leiomyomas | 1 |
| 3 | Active Not Recruiting | Treatment | Heavy Menstrual Bleeding / Uterine Leiomyomas | 2 |
| 3 | Active Not Recruiting | Treatment | Uterine Leiomyomas | 1 |
| 3 | Completed | Treatment | Uterine Leiomyomas | 1 |
| PHASE | STATUS | PURPOSE | CONDITIONS | COUNT |
|---|---|---|---|---|
| 3 | Enrolling by Invitation | Treatment | Endometriosis / Endometriosis related pain | 1 |
| 3 | Enrolling by Invitation | Treatment | Heavy Menstrual Bleeding / Uterine Leiomyomas | 1 |
| 3 | Enrolling by Invitation | Treatment | Uterine Leiomyomas | 1 |
| 3 | Recruiting | Treatment | Endometriosis related pain | 2 |
| 3 | Recruiting | Treatment | Prostate Cancer | 1 |
///////////Relugolix, TAK-385, JAPAN 2019, Relumina, レルゴリクス , PHASE 3
CONC(=O)NC1=CC=C(C=C1)C1=C(CN(C)C)C2=C(S1)N(CC1=C(F)C=CC=C1F)C(=O)N(C2=O)C1=CC=C(OC)N=N1
Metyrosine, Metirosine メチロシン метирозин , ميتيروسين , 甲酪氨酸 ,

Metyrosine, Metirosine
メチロシン метирозин , ميتيروسين , 甲酪氨酸 ,
Metyrosine (USP);
Metirosine (JAN/INN);
Demser (TN)
CAS 672-87-7
- Molecular FormulaC10H13NO3
- Average mass195.215 Da
APPROVED P[MDA JAPAN 2019/1/8
- (-)-alpha-Methyl-L-tyrosine
- (–)-α-methyl-L-tyrosine
- (S)-alpha-Methyltyrosine
- Methyltyrosine
Synthesis ReferenceUS2868818
An inhibitor of the enzyme tyrosine 3-monooxygenase, and consequently of the synthesis of catecholamines. It is used to control the symptoms of excessive sympathetic stimulation in patients with pheochromocytoma. (Martindale, The Extra Pharmacopoeia, 30th ed)
Metirosine (INN and BAN; α-Methyltyrosine, Metyrosine USAN, AMPT) is an antihypertensive drug. It inhibits the enzyme tyrosine hydroxylase and, therefore, catecholamine synthesis, which, as a consequence, depletes the levels of the catecholamines dopamine, adrenaline and noradrenaline in the body.
For use in the treatment of patients with pheochromocytoma, for preoperative preparation of patients for surgery, management of patients when surgery is contraindicated, and chronic treatment of patients with malignant pheochromocytoma.

Pharmacodynamics
In patients with pheochromocytoma, who produce excessive amounts of norepinephrine and epinephrine, administration of one to four grams of metyrosine per day has reduced catecholamine biosynthesis from about 35 to 80 percent as measured by the total excretion of catecholamines and their metabolites (metanephrine and vanillylmandelic acid). The maximum biochemical effect usually occurs within two to three days, and the urinary concentration of catecholamines and their metabolites usually returns to pretreatment levels within three to four days after metyrosine is discontinued. Most patients with pheochromocytoma treated with metyrosine experience decreased frequency and severity of hypertensive attacks with their associated headache, nausea, sweating, and tachycardia. In patients who respond, blood pressure decreases progressively during the first two days of therapy with metyrosine; after withdrawal, blood pressure usually increases gradually to pretreatment values within two to three days.
Mechanism of action
Metyrosine inhibits tyrosine hydroxylase, which catalyzes the first transformation in catecholamine biosynthesis, i.e., the conversion of tyrosine to dihydroxyphenylalanine (DOPA). Because the first step is also the rate-limiting step, blockade of tyrosine hydroxylase activity results in decreased endogenous levels of catecholamines and their synthesis. This consequently, depletes the levels of the catecholamines dopamine, adrenaline and noradrenaline in the body,usually measured as decreased urinary excretion of catecholamines and their metabolites. One main end result of the catecholamine depletion is a decrease in blood presure
Clinical use
Metirosine has been used in the treatment of pheochromocytoma.[1] It is contra-indicated for the treatment of essential hypertension.
However it is now rarely used in medicine, its primary use being in scientific research to investigate the effects of catecholamine depletion on behaviour.[2] Info on how catecholamine depletion from this medicine affects behavior needed in this srticle.
SYN

PAPER
Saito, Hiroyuki; Agricultural and Biological Chemistry 1988, 52(9), PG 2349-50
PATENT
https://patents.google.com/patent/WO2011053835A1
Metyrosine, which has the structure of Formula:

is useful in reducing elevated levels of catecholamines associated with pheochromocytoma, and preventing hypertension. Metyrosine, as shown, is a chiral compound. The synthesis of metyrosine in pure or substantially pure enantiomeric form requires a process that involves using substantially diastereomerically and/or enantiomerically pure intermediates. The Applicant has discovered, surprisingly, certain compounds that are substantially
diastereomerically or enantiomerically pure and processes to prepare them, and which compounds may be converted to metyrosine.
Scheme 1


Lower diastereomeric purity Higher diastereomeric purity

Example 1. (R)-Phenylglycinamide«HCl

[0091] To a 500 mL flask were charged (i?)-phenylglycinamide (20.0 g, 133 mmol, 1 eq. Amplachem ref: Aa-33365) and MeOH (160 mL). 4 M HCl/dioxane (50 mL, 200 mmol, 1.5 eq.) was then added dropwise resulting in the formation of a white precipitate. The mixture was stirred for 30 min, was filtered and was washed with MeOH (20 mL) and diethyl ether (20 mL). Drying in vacuo provided (i?)-phenylglycinamide.HCl (21.9 g, 89%) as a white solid. Ή NMR (D20, 400 MHz) 4.97 (s, 1 H); 7.36-7.41 (m, 5 H).
[0092] Example 2. 2-[l-(S)-Cyano-2-(4-methoxyphenyl)-l-methylethylamino]-2-
(R)-phenylacetamide 2

[0093] Method A. To a 500 mL flask were charged (j )-phenylglycinamide’HCl
(15.0 g, 80.6 mmol, 1 eq.), MeOH (104 mL), H20 (17 mL) and -methoxyphenylacetone (12.4 mL, 80.6 mmol, 1 eq, Aldrich, ref: 19917-6). To this mixture was added a solution of NaCN (3.95 g, 80.6 mmol, 1 eq.) in H20 (10 mL). The resulting solution was stirred for 4 days at room temperature while a white precipitate formed. The precipitate was filtered and washed with H20/MeOH (7:3) to provide 2-[l-(5)-cyano-2-(4-methoxyphenyl)-l- methylethylamino]-2-(i?)-phenylacetamide 2 (11 ,0 g) as a white solid. The filtrate was stirred for 3 d more at room temperature and the solid formed was filtered to provide 2 (3.30 g). The filtrate was stirred for 1 d more to provide 2 (1.70 g). The filtered solids were combined and dried in vacuo to provide 2 (16.0 g, 61 %, dr 98/2) as a white solid.
[0094] Method B. In a sealed tube were charged (i?)-phenylglycinamide.HCl (1.2 g,
6.45 mmol, 1 eq.), MeOH (4 mL), H20 (7 mL) and 7-methoxyphenylacetone (991 μL, 6.45 mmol, 1 eq.). A solution of NaCN (316 mg, 6.45 mmol, 1 eq.) in H20 (1 mL) was added. The mixture was stirred for 20 hours at 40°C resulting in the formation of a white precipitate. The precipitate was filtered, was washed with H20/MeOH (7:3 v/v, 2 x 2 mL) and was dried in vacuo to provide 2 (1.59 g, 76%, dr 98/2) as a white solid. Ή NMR (CDC13, 400 MHz) 1.14 (s, 3 H); 2.90 (d, J= 13.6 Hz, 1 H); 2.99 (d, J= 13.6 Hz, 1 H); 3.20 (bs, 1 H); 3.80 (s, 3 H); 4.51 (s, 1 H); 5.45 (bs, 1 H); 5.75 (bs, 1 H); 6.90 (d, J= 8.6 Hz, 2 H); 7.27 (d, J= 8.6 Hz, 2 H); 7.30-7.50 (m, 5 H). dr determination: Ή NMR comparing integration of peaks of 2 at 2.90/2.99 (1.98 H, formally 2 H) with those of its diastereoisomer or diastereomer
(prepared from racemic phenylglycinamide) at 2.82/2.85 (0.04 H, formally 2 H). Ή NMR diastereoisomer of 2 (CDC13, 400 MHz) 1.49 (s, 3 H); 2.82 (d, J = 13.8 Hz, 1 H); 2.85 (d, J = 13.8 Hz, 1 H); 3.78 (s, 3 H); 4.52 (s, 1 H); 5.55 (bs, 1 H); 6.60 (bs, 1 H); 6.84 (d, J = 8.6 Hz, 2 H); 7.17 (d, J = 8.6 Hz, 2 H); 7.30-7.40 (m, 5 H).
[0095] Example 3. 2-[(R)-(Carbamoylphenylmethyl)-amino]-3-(4- methoxyphenyl)-2-(S)-methylpropionamide 3

[0096] To a 500 mL flask were added nitrile 2 (10.0 g, 30.95 mmol) and CH2C12(130 mL). The solution was cooled to -10°C and H2S04 (10 mL) was added dropwise over 15 min. The mixture was stirred for 2 h at 0°C. Ice (200 g) was added and the mixture was stirred for 1 h. The mixture was basified with 32% aq NH3 to pH 8-9, EtOAc (400 mL) was added and the phases were separated. The aqueous phase was extracted with EtOAc (2 x 250 mL). The combined organic phases were dried over MgS04 and were concentrated to provide 3 (1 1.1 g (9.99 g theoretical, the sample contains 10% w/w of EtOAc by NMR) 94%, 92.9% chemical purity, 97% de by HPLC/MS) as a white solid. HPLC/MS tR – 3.04 min; m/z = 342.1 (M+l) de determination: HPLC/MS comparing integration of peaks at tR = 3.04 min (98.5 area%) and tR = 2.92 min (1.5 area% the other diastereoisomer of 3) HPLC/MS diastereoisomer of 3 tR = 2.92 min; m/z = 342.1 (M+l)
[0097] Example 4. Purification of 3 by Crystallization. Amide 3 (8.10 g, 23.7 mmol) and methyl isobutyl ketone (124 mL) were heated to reflux temperature until the solid was dissolved and the solution was cooled to room temperature. The solid formed was filtered, was washed with methyl isobutyl ketone (2 x 20 mL) and was dried in vacuo to provide 3 (5.45 g, 67%, >99.5% purity by HPLC/MS) as a white solid. 1H NMR (DMSO, 200 MHz) 1.00 (3H, s); 2.35 (bs, 1 H); 2.75 (d, J = 13.3 Hz, 1 H) 2.85 (d, J = 13.3 Hz, 1 H); 3.61 (s, 3 H); 4.17 (s, 1 H); 6.60 (d, J= 8.4 Hz, 2 H); 691 (d, J= 8.4 Hz, 2 H); 6.93 (bs, 1 H), 7.01 bs (1 H); 7.20-7.50 (m, 6 H); 7.6 (bs, 1 H). HPLC/MS m z = 342.1 (M+l ) Example 5. 2-(S)-Amino-3-(4-methoxyphenyl)-2-methyIpropionamide 4

[0099] Method A, Hydro genolysis: Amine 3 (6.00 g, 17.6 mmol, 1 eq) was dissolved in MeOH (60 mL) and 10% Pd/C (2.15 g, 56% moisture content, 16 wt%) was added. The mixture was stirred under H2 (3 bar) at 50°C for 16 h. The mixture was filtered through Celite, the filter pad was washed with MeOH (20 mL) and the filtrates were concentrated to provide 5.50 g of a 1 : 1 mixture of 4 (3.37 g, 91%) and phenylacetamide as a white solid.
[0100] Method B, Transfer hydrogenolysis: Amine 3 (500 mg, 1 ,47 mmol, 1 eq) was dissolved in i-PrOH (5 mL) under Argon. 10% Pd/C (200 mg, 56% moisture content, 18 wt%) and ammonium formate (601 mg, 9,56 mmol, 6.5 eq.) were added. The mixture was stirred at reflux temperature for 1 h. The mixture was filtered through Celite, the filter pad was washed with EtOH (10 mL) and the filtrates were concentrated to provide 480 mg of a 1 : 1 mixture of desired compound (293 mg, 96%) and phenylacetamide as a white solid. Ή NMR (DMSO, 400 MHz) 1.14 (s, 3 H); 2.10 (bs, 2 H); 2.50 (d, J= 13.1 Hz, 1 H); 2.95 (d, J = 13.1 Hz, 1 H); 3.69 (s, 3 H); 6.80 (d, J = 8.5 Hz, 2 H); 6.81 (bs, 1 H); 6.90 (bs, 1 H); 7.09 (d, J= 8.5 Hz, 2 H). (Phenylacetamide 3.34 (s, 2 H); 7.15-7.28 (m, 6 H); 7.42 (bs, 1 H).). HPLC/MS 4 tR = 1.96 min. MS (ESI (+)) m/z = 164.2 (M-CONH2). (Phenylacetamide tR = 1.76 min. MS (ESI (+)) m/z = 136.2 (M+l)).
[0101] Example 6. Metyrosine 1

[0102] To a 100 mL flask with a reflux condenser was charged amide 4 (5.50 g of a mixture containing 4 (3.37 g, 16.1 mmol) and phenylacetamide (2.13 g)) and 48% HBr (30 mL). The solution was heated for 5 h at 120 °C and was cooled to room temperature. H20 (60 mL) was added and the solution was washed with EtOAc (3 35 mL). The aqueous phase was concentrated in vacuo to provide a beige paste. The paste was dissolved in H20 (15 mL) and the resulting mixture was heated to 65 °C. Activated carbon (300 mg, Type NORIT SX) was added and the mixture was stirred for 15 min, was filtered and the filter pad was washed with water (2 4 mL). The combined filtrates were heated to 55 °C and the pH was adjusted to 5-6 using 32% aq. NH3. The mixture was cooled to 0 °C, was stirred for 15 min and was filtered. The collected solids were washed with cold water (2 x 5 mL) and were dried in vacuo to provide (-)-Q!-methyl-L-tyrosine (or metyrosine) 1 (2.65 g, 84%) as a white solid. HPLC (Zorbax C18, NaH2P04 10 mM pH = 3 / MeCN (100:0) 10 min, (100:0) to (0: 100) 15 min, 0: 100 5 min) tR = 10.1 min. Chiral HPLC (Nucleosil Chiral-1, CuS04 10 mM / MeCN 10:1) tR = 16.9 min. m.p. – 320-321°C. [a]5 6 = +201° (c – 0.5 Copper complex solution)(lit.2 + 185-190°) Copper complex solution preparation: Solution A
(anhydrous NaOAc dissolved in H20 (150 mL) in 250 mL volumetric flask, glacial acetic acid (50 mL) added and diluted to volume with H20) mixed with Solution B (cupric sulfate (62.5 g) diluted to volume with H20 in a 200 mL volumetric flask) in a 1 L volumetric flask and was diluted to volume with H20. Metyrosine solution (5 mg/mL) was prepared in this solution.
[0103] To obtain an NMR spectrum (taking into account the low solubility of the product), a small sample (10 mg) was transformed into its HC1 salt. The sample was dissolved in 2 M HC1 and the solution was evaporated to dryness. Ή NMR (D20, 400 MHz) 1.49 (s, 3 H); 2.90 (d, J= 14.5 Hz, 1 H); 3.16 (d, J = 14.5 Hz, 1 H); 6.75 (d, J= 8.2 Hz, 2 H), 7.01 ((d, J= 8.2 Hz, 2 H). 13C NMR (D20, 100.6MHz) 21.6; 41.6; 61.0; 116.0, 125.0; 131.7; 155.5; 173.8. [0104] Preparation of Metyrosine using (S)-phenylethylamine
Scheme 4

(S)~Phenylethy la mine. HCl


[0105] Example 7. (S)-Phenylethylamine hydrochloride.

[0106] To a 500 mL flask were added (5)-phenylethylamine (BASF, ref: UN2735,
40.0 g, 333 mmol, 1 eq.) and MeOH (160 mL). The solution was cooled to 0 °C and 37% HCl (40 mL, 480 mmol, 1.44 eq.) were added dropwise. Concentration of the reaction mixture gave a white solid. Diethyl ether (300 mL) was added and the suspension was stirred for 15 min. The solid was filtered and was washed with diethyl ether (2 x 60 mL) to provide (^-phenylethylamine hydrochloride (39.1 g, 75%) as a white solid. Ή NMR (D20, 400 MHz) 1.52 (d, J= 7.2 Hz, 3 H); 4.42 (q, J= 7.2 Hz, 1 H); 7.35-7.40 (m, 5 H).
Example 8. 3-(4-Methoxyphenyl)-2-methyl-2-(l-(S)-phenylethylamino)-

To a 500 mL flask were added (^-phenylethylamine.HCl (25.0 g, 159.2 mmol, 1 eq.), MeOH (125 mL), NaCN (7.80 g, 159.2 mmol, 1 eq.) and 4-methoxyphenylacetone (Aldrich, ref: 19917-6. 24.5 mL, 159.2 mmol, 1 eq.). The mixture was stirred for 14 h at room
temperature. The mixture was filtered, the filter cake was washed with MeOH (30 mL) and the filtrates were concentrated to an oil which was dissolved in CH2C12(370 mL) and washed with water (250 mL). The organic phase was dried (MgS04) and concentrated in vacuo to provide 3-(4-methoxyphenyl)-2-methyl-2-(l-(5 -phenylethylamino)-propionitrile (47.2 g, 100% as a 6/4 mixture of diastereoisomers (S,S)/(R,S)), containing 5% of 4- methoxyphenylacetone) as a yellow oil. Ή NMR (CDC13, 400 MHz) 1 ,05 (s, 0.62 x 3 H); 1.27 (d, J- 6.4 Hz, 0.6 x 3 H); 1.40-1.44 (m, 0.4 x 6 H); 2.47 (d, J= 13.6 Hz, 0.4 x 1 H); 2.74 (d, J= 13.6 Hz, 0.4 x 1 H); 2.84 (d, J= 14 Hz, 0.6 x 1 H) 2.94 (d, J= 14 Hz, 0.6 x 1 H); 3.78 (s, 0.4 x 3 H); 3.82 (s, 0.6 x 3 H); 4.02 (q, J= 6.4 Hz, 0.6 x 1 H); 4.16 (q, J= 6, 4 Hz, 0.4 x 1 H); 6.80-7.40 (m, 9H). [0108] Example 9. 3-(4-Methoxyphenyl)-2-methyl-2-(l-(S)-phenylethylam o)- propionamidc 6.

[0109] Method A. To a 1 L flask with mechanical stirring under argon was added 3-
(4-methoxyphenyl)-2-methyl-2-(l-(5)-phenylethylamino)-propionitrile 5 (40.0 g, 136.1 mmol) dissolved in CH2C12 (400 mL). The solution was cooled to -5 °C (using an ice salt bath) and cone. H2S04 (40 mL) was added dropwise maintaining the temperature between – 5°C and 5°C. The mixture was warmed to RT over 2 h and was stirred for 16 h. Ice (400 g) was added and the mixture was stirred for 40 min. The two phases were separated and the aqueous phase was neutralized to pH 8-9 with 32% aq. NH3. The aqueous phase was extracted with EtOAc (3 x 350 mL). The combined organic layers were dried (MgS04) and were concentrated in vacuo to provide 6 (14.2 g, 34%, 98% chemical purity by HPLC/MS) as a 6/4 mixture of diastereoisomers (S,S)/(R, S)) as a yellow oil.
[01 10] Method B. In a 250 mL flask was dissolved 3-(4-methoxyphenyl)-2-methyl-
2-(l-(«S -phenylethylamino)-propionitrile 5 (5.0 g, 17.0 mmol) in CH2C12 (50 mL). The solution was cooled to 0°C and cone. H2S04 (2.5 mL) was added dropwise. The mixture was stirred at 40 °C for 28 h, was cooled to RT and ice (50 g) was added. The mixture was stirred for 1 h and the phases separated. The aqueous phase was basified to pH 8-9 using 32% aq. NH3 and was extracted with EtOAc (3 x 50 mL). The combined organic layers were dried (MgS04) and concentrated in vacuo to provide 6 (3.32 g, 62%, 97% purity by HPLC/MS) as a 6/4 mixture of diastereoisomers (S,S)/(R,S)) as a yellow oil. Ή NMR (CDC13, 400 MHz) 1.13 (s, 0.4 x 3 H); 1.15 (s, 03 x 3 H) 1.24 (d, J= 6.6 Hz, 0.4 x 3 H); 1.30 (d, J = 6.6 Hz 0.6 x 3 H); 2.75 (d, J= 13.4 Hz, 0.6 x 1 H); 2.78 (d, J= 13.6 Hz, 0.4 x 1 H); 2.84 (d, J= 13.4 Hz, 0.6 x 1 H) 3.32 (d, J = 13.6 Hz, 0.4 x 1 H); 3.78 (s, 0.6 x 3 H); 3.80 (s, 0.4 x 3 H); 3.85 (q, J = 6.6 Hz, 0.6 x 1 H); 4.16 (q, J = 6.6 Hz, 0.4 x 1 H); 6.80-7.40 (m, 9 H). HPLC/MS tR = 4.21 min [(S,S)-6 MS (ESI (+)) m/z 313.2 (M+l )] and 4.34 min [(R,S)-6 MS (ESI (+)) m/z 313.2 (M+l)].
[01 1 1 ] Example 10. 3-(4-Methoxyphenyl)-2-(5)-methyl-2-(l-(S)- phenylethylamino)-propionamide hydrochloride.

[01 12] In a 500 mL flask was dissolved amide 6 (14.2 g, 45.5 mmol, 1 eq.) in z‘-PrOH
(140 mL). Cone. HCl (5.7 mL, 68.3 mmol, 1.5 eq.) was added dropwise and the mixture was stirred for 20 min. The solvent was evaporated in vacuo and methyl isobutyl ketone (200 mL) was added. The mixture was heated to reflux temperature, was cooled to room temperature and was stirred for 72 h. The solids were collected by filtration, washed with methyl isobutyl ketone (20 mL) and dried in vacuo to provide 6«HC1 (13.6 g, 86%, diasteremeric ratio (dr) 63/37( i.e., 63% diastereomeric purity of the S,S diastereomer)) as a white solid.
[01 13] Example 11. Purification (Enhancing Diastereomeric Purity) of 6.HC1 by
Crystallization. In a 250 mL flask were placed amide hydrochloride 6·ΗΟ (13.6 g, dr 37/63) and /-BuOH (136 mL). The mixture was heated to reflux temperature and z‘-BuOH (95 mL) was distilled. The mixture was cooled to room temperature and was stirred overnight. The solids were collected by filtration and were washed with z‘-BuOH to provide 6»HC1 (1 1.6 g, 85%, dr 73/27 as a white solid.
[01 14] This solid was dissolved in /-BuOH (139 mL) and was heated to reflux temperature. z‘-BuOH (70 mL) was distilled and the mixture was cooled to room temperature and was stirred for 3 h. Filtration provided 6»HC1 (7.5 g, 65%, dr 88/12) as a white solid. [01 15] This solid was dissolved in /-BuOH (130 mL) and was heated to reflux temperature. z‘-BuOH (65 mL) was distilled and the mixture was cooled to room temperature and was stirred for 3 h. Filtration provided 6·ΗΟ (6.0 g, 80%, dr 99/1) as a white solid.
[01 16] This solid was dissolved in z‘-BuOH (105 mL) and was heated to reflux temperature. z‘-BuOH (53 mL) was distilled and the mixture was cooled to room temperature and was stirred for 16 h. Filtration provided 6·ΗΟ (5.4 g, 90%, dr >99/l, 100% purity by HPLC/MS, 40% overall yield (67% theoretical yield)) as a white solid. Ή NMR (DMSO, 400 MHz) 1.06 (s, 3 H); 1.57 (d, J= 6.4 Hz, 3 H); 2.84 (d, J= 13.2 Hz, 1 H); 3.27 (d, J = 13.2 Hz, 1 H); 3. 69 (s, 3 H) 4.40 (bs, 1 H); 6.83 (d, J= 8.4 Hz, 2 H); 6.98 (d, J= 8.4 Hz, 2 H); 7.37-45 (m, 2 H); 7.50-7,65 (m, 2 H); 7.80 (bs, 1 H); 9.40 (bs, 2 H). HPLC/MS tR = 4.21 min [(S,S)- 6»HC1. MS (ESI (+)) m z 313.2 (M+l)].
Example 12. 2-(S)-Amino-3-(4-methoxyphenyl)-2-methyl-propionamide

[01 18] Amine 6-HC1 (5.40 g, 15.5 mmol, 1 eq) was dissolved in MeOH (60 mL) and
10%Pd/C (2.0 g, 56% moisture content, 16% w/w) was added. The mixture was stirred under H2 (3 bar) at 50 °C for 80 min. The mixture was filtered through celite and the filter pad was washed with MeOH (20 mL). The filtrates were concentrated in vacuo to provide 2-(S)- amino-3-(4-methoxyphenyl)-2-methyl-propionamide hydrochloride (3.80 g, 100%, 99.5% purity by HPLC/MS) as a yellow solid. Ή NMR (DMSO, 400 MHz) 1.46 (s, 3 H); 3.02 (d, J = 14 Hz, 1 H); 3.10 (d, J- 14 Hz, 1 H), 3.72 (s, 3 H); 6.87 (d, J= 8.8 Hz, 2 H); 7.15 (d, J = 8.8 Hz, 2 H); 7.64 (s, 1 H); 7.94 (s, 1 H); 8.08 (bs, 2 H). HPLC/MS tR = 1.95 min. MS (ESI (+)) m z = 164.2 (M – CONH2). [01 19] Example 13. Metyrosine 1

[0120] To a 100 mL flask with a reflux condenser were added 2-(iS -amino-3-(4- methox phenyl)-2-methyl-propionamide hydrochloride (3.80 g, 15.6 mmol) and 48% HBr (20 mL). The solution was heated for 4 h at 120 °C, was cooled to room temperature and was concentrated in vacuo to give a beige paste. The paste was dissolved in water (15 ml) and the solution was again concentrated under vacuum. The paste was dissolved in H20 (15 mL), the solution was heated to 65 °C and 300 mg of activated carbon were added. The mixture was stirred for 15 min, was filtered and the filter pad was washed with water (2 x 4 mL). The solution was heated to 55 °C and the pH was adjusted to 5-6 using 32% aq. NH3. The mixture was cooled to 0 °C and was stirred for 15 min. Filtration, washing with cold water (2 x 5 mL) and drying in vacuo provided Metyrosine 1 (2.55 g, 83% yield, 99.6% HPLC purity, >99,5% ee) as a white solid. HPLC ((Zorbax C18, NaH2P04 10 mM pH = 3/MeCN (100:0) 10 min, (100:0) to (0: 100) 15 min, 0: 100 5 min), tR = 10.1 min. Chiral HPLC (Nucleosil Chiral-1, CuS04 10 mM/MeCN 10: 1), tR = 16.9 min. m.p. = 321-322 °C. [α]546 = +187° (c = 0.5, Copper complex solution) Copper complex solution preparation: Solution A (anhydrous NaOAc dissolved in H20 (150 mL) in 250 mL volumetric flask, glacial acetic acid (50 mL) added and diluted to volume with H20) mixed with Solution B (cupric sulfate (62.5 g) diluted to volume with H20 in a 200 mL volumetric flask) in a 1 L volumetric flask and diluted to volume with H20. Sample prepared 5 mg/mL in this solution.
[0121] In order to obtain an NMR spectrum and taking into account the low solubility of the product, a small sample (10 mg) was transformed into its HC1 salt. The sample was dissolved in 2 M HC1 and the solution was evaporated to dryness. Ή NMR (D20, 400 MHz) 1.49 (s, 3 H); 2.90 (d, J= 14.5 Hz, 1 H); 3.16 (d, J= 14.5 Hz, 1 H); 6.75 (d, J- 8.2 Hz, 2 H), 7.01 (d, J= 8.2 Hz, 2 H). [0122] Preparation of Metyrosine using L-alanine tert-butyl ester:
Scheme 5

[0123] Example 14. Synthesis of Aldimine. 4-Chlorobenzaldehyde (3.87 g, 27.5 mmol) was dissolved in methanol (50 mL) and treated with triethylamine (3.87 g, 38.3 mmol, 1.39 equiv). The mixture was stirred for 7 min at ambient temperature followed by addition of L-alanine tert-butyl ester hydrochloride (5.00 g, 27.5 mmol). Magnesium sulfate (6.63 g, 55.1 mmol, 2 equiv) was added to this solution and the slurry was stirred for 17 h at ambient temperature. The solid was filtered and washed with methanol (6 mL). The filtrate was evaporated to dryness to result in an oily solid. This solid was dissolved in a biphasic MTBE/water (70 mL/20 mL) mixture. The organic phase was separated and washed with water (20 mL). The organic phase was dried over MgS04, the solid was filtered, and the filtrate was evaporated to dryness to afford aldimine 7 [7.09 g; 96.2%] as a clear oil, which became a solid when stored in a refrigerator. 1H NMR (500 MHz, CDC13): δ 8.25 (br. s, 1H, ArCH), 7.71 (d, J= 8.5 Hz, 2H, Ar), 7.38 (d, J= 8.5 Hz, 2H, Ar), 4.04 (dq, J, = 0.6 Hz, J2 = 6.8 Hz, 1H, CH), 1.48 (d, J= 6.8 Hz, 3H, CH3), 1.47 (s, 9H, 3 CH3).
[0124] Example 15. Synthesis of ferf-Butyl 2-Amino-3-(4-methoxyphenyl)-2- methylpropanoate. Aldimine (2.00 g, 7.47 mmol) and O-allyl-N-benzylcinchonidinium bromide (0.38 g, 0.75 mmol, 0.10 equiv) were mixed with toluene (20 mL) at ambient temperature. The mixture was stirred for 30 min and then was cooled to 0°C. Powdered KOH (2.10 g, 37.35 mmol, 5 equiv) was added at once to convert the thin slurry into a yellow solution. The mixture was stirred for 5 min and 4-methoxybenzyl bromide (7.51 g, 37.35 mmol, 5 equiv) was added at 0 to 1°C. The solution was allowed to warm and was stirred at ambient temperature for 16 h. The reaction mixture was sequentially washed with water (20 mL) and brine (20 mL), separated, and treated with a 5-6 N HC1 solution in IPA (7 mL) for 1 h at ambient temperature. The reaction mixture was washed with water (20 mL). The aqueous phase was separated and treated with toluene (20 mL). The aqueous phase was separated, treated with a 2 N NaOH solution until basic, and the product was extracted with toluene (20 mL). The toluene phase was washed with brine (20 mL), separated, and dried over Na2S04. The solid was filtered and the solvent was stripped to dryness to afford tert- butyl 2-amino-3-(4-methoxyphenyl)-2-methylpropanoate; 1.70 g; 85.8% as a clear oil. Ή NMR (500 MHz, CDC13): δ 7.13 (d, J= 8.7 Hz, 2H, Ar), 6.81 (d, J= 8.7 Hz, 2H, Ar), 3.78 (s, 3H, CH3), 3.05 (d, J= 13.3 Hz, 1H, CH2), 2.71 (d, J- 13.3 Hz, 1H, CH2), 1.62 (br. s, 2H, NH2), 1.45 (s, 9H, 3xCH3), 1.32 (s, 3H, CH3). Ή NMR analysis, carried out in the presence of 1.2 equiv of BINOL, resulted in 47.6% ee. Optical rotation (Q?5D, chloroform, c = 1.38): – 9.06°.
[0125] Example 16. Synthesis of 2-Amino-3-(4-methoxyphenyl)-2- methylpropanoic Acid Hydrochloride. Intermediate
2-amino-3-(4- methoxyphenyl)-2-methylpropanoate (0.60 g, 2.26 mmol) was mixed with toluene (6 mL) and a 5-6 N HC1 solution in IPA (2 mL). A clear yellow solution was heated to reflux and kept at that temperature for 7 h. The resulting slurry was cooled to ambient temperature and filtered. The solid was washed with toluene (3 mL) on a filter and air-dried to afford 2- amino-3-(4-methoxyphenyl)-2-methylpropanoic acid hydrochloride [0.37 g; 67%] as a white solid [HPLC 71.8% (AUC; ¾= 3.71 ]. Ή NMR (500 MHz, DMSO-i¾): <5 13.96 (br. s, 1H, COOH), 8.44 (br. s, 3H, NH3), 7.16 (d, J= 8.7 Hz, 2H, Ar), 6.90 (d, J= 8.7 Hz, 2H, Ar), 3.74 (s, 3H, CH3), 3.08 (s, 2H, CH2), 1.48 (s, 3H, CH3). Optical rotation (Λ, DMSO, c = 1.10) +7.27°. [0126] Example 17. Synthesis of Metyrosine 1. tert-Butyl 2-amino-3-(4- methoxyphenyl)-2-methylpropanoate (0.30 g, 1.13 mmol) was dissolved in CH2C12(3 mL) and BBr3 (0.85 g, 3.39 mmol, 3 equiv) was added at room temperature. The reaction mixture was stirred for 1.5 h and treated with a NaHC03 solution to a basic pH. The aqueous phase was isolated. Solid started to precipitate in the aqueous phase in 30 min. Solid was filtered in 16 h and was washed on a filter with CH2C12(2 mL) and water (2 mL). The solid was air- dried to afford metyrosine [0.10 g; 45.4%] as a white solid [HPLC 80.7% (Metyrosine; AUC; fR= 2.32 & 2.57]. Ή NMR (500 MHz, TFA-d): δ 8.60 (d, J= 8.7 Hz, 2H, Ar), 8.39 (d, J = 8.7 Hz, 2H, Ar), 4.91 (d, J= 15.0 Hz, 1H, CH2), 4.67 (d, J= 15.0 Hz, 1H, CH2), 3.30 (s, 3H, CH3). Optical rotation (ο?0ο, c = 1.080, 1 = 10 mm, NaOAc/CuS04/H20/AcOH) +148.1.
[0127] Larger scale (e.g. >100 g) Synthesis Metyrosine using (R)- phenylglycinamide (Examples 18-24). Scheme 6 provides the general synthetic outline.
Scheme 6

l) NaCN, MeOH/H20, 24 h, 44 °C; 2) H2S04 cone, DCM, 15 °C to RT, 1.25 h: 3) a. 3 atm H2 6 wt% 10% Pd/C, MeOH, 55 °C, 16 h, b. HBr aq; 4) HBr 48%, 105 °C, 17 h; 5) NaOH aq; 6) 5 wt% 10% Pd/C, HC02H/H20 MeOH, 55 °C, 6 h
[0128] Example 18. 2-[l-(S)-Cyano-2-(4-methoxyphenyl)-l-methylethylamino]-
2-(R)-phenylacetamide 2. In a 5 L reactor equipped with anchor stirrer were charged (i?)-phenylglycinamide»HCl (330 g, 1.77 mol, 1 eq.), MeOH (1.1 L), H20 (1.9 L) and
7-methoxyphenylacetone (290 g, 1.77 mol, 1 eq.). A solution of NaCN (86.7 g, 1.77 mol, 1 eq.) in H20 (300 mL) was added over 15 min at room temperature. The mixture was stirred for 24 hours at 44 °C resulting in the formation of a yellow precipitate. The mixture was cooled to room temperature. The precipitate was filtered, was washed with H20/MeOH (7:3 v/v, 2 x 750 mL) and -PrOH (2 x 500 mL). The solid was dried in vacuo (3 days) at 35°C to provide 2-[l-(5)-Cyano-2-(4-methoxyphenyl)-l-methylethylamino]-2-( ?)-phenylacetamide 2 (460 g, 80%, dr 97/3) as a yellow solid. Ή NMR (400 MHz, CDC13) 1.14 (s, 3 H), 2.90 (d, J = 13.6 Hz, 1 H), 2.99 (d, J = 13.6 Hz, 1 H), 3.20 (bs, 1 H), 3.80 (s, 3 H), 4.51 (s, 1 H), 5.45 (bs, 1 H), 5.75 (bs, 1 H), 6.90 (d, J= 8.6 Hz, 2 H), 7.27 (d, J= 8.6 Hz, 2 H), 7.30-7.50 (m, 5 H). Ή NMR (R,R and S^-diastereoisomers of 2 (400 MHz, CDC13) 1.49 (s, 3 H), 2.82 (d, J = 13.8 Hz, 1 H), 2.85 (d, J= 13.8 Hz, 1 H), 3.78 (s, 3 H), 4.52 (s, 1 H), 5.55 (bs, 1 H), 6.60 (bs, 1 H), 6.84 (d, J = 8.6 Hz, 2 H), 7.17 (d, J= 8.6 Hz, 2 H), 7.30-7.40 (m, 5 H). dr determination: Ή NMR comparing integration of peaks of 2 at 2.90/2.99 (1.00 H, formally 2 H) with those of its (^.ii/S’^-diastereoisomeric pair (prepared from nearly rac- phenylglycinamide) at 2.82/2.85 (0.03 H, formally 2 H).
[0129] Example 19. 2-[(R)-(Carbamoylphenylmethyl)-amino]-3-(4- methoxyphenyl)-2-(S)-methylpropionamide 3. Into a 10 L reactor equipped with anchor stirrer was charged CH2C12 (1.64 L). The solvent was cooled to 15°C, then 95% H2S04 (492 mL) and 2-[l-(5)-cyano-2-(4-methoxyphenyl)-l-methylethylamino]-2-( ?)-phenylacetamide 2 (410 g, 1.27 mol) were added alternately in 9 portions over approximately 45 min
(specifically: 164 mL of H2S04 then 82 g of 2; subsequently, at approximately 5 min intervals, 8 x [41 mL of H2S04 then immediately 41 g of 2]). On addition of each portion, the suspension of 2 in the dense oily phase slowly dissolved (1-2 min) to provide a biphasic mixture. The resulting biphasic mixture (a red-brown dense oil with a pale yellow
supernatant CH2C12 layer) was stirred for 0.5 h at 25°C. Ice-cold water (4.1 L) was added over 30 min, very slowly initially (200 mL dropwise over 15 min) due to a violent exotherm, and the biphasic mixture was stirred for 0.5 h. The phases were separated and the organic phase discarded. The combined aqueous phases were washed with CH2C12 (450 mL), and residual CH2C12 was stripped from the aqueous phase by distillation under vacuum at 55°C (20-30 mBar). The aqueous solution was then cooled to 20°C and was basified with 32% aq NH3 (1 150 mL) to pH 8-9 at such a rate that the temperature was kept below 28°C
(approximately 120 min). The suspension was stirred for 30 min to ascertain a stable pH. The white solid which formed was separated by filtration, washed with H20 (2 x 2050 mL), and was thoroughly drained of water (but was not dried) to provide 2-[(R)- (carbamoylphenylmethyl)-amino]-3-(4-methoxyphenyl)-2-(5)-methylpropionarnide 3 (1087 g (391 g theoretical, the sample contains 64% w/w of H20), yield 90%, 97% HPLC purity, 96% de) as a wet white solid. HPLC (Luna C18, H20 / MeCN 95:5 to 0: 100 30 min, 254 nm, sample 2 mg/mL in MeOH). tR (3) = 15.3 min, 96% de. (2 degrades under these conditions: 3 peaks are detected at 17.7, 18.9 and 19.9 min). HPLC (#R/S,S)-diastereoisomer of 3, tR = 1 5.0 min. de determination: HPLC comparing integration of peaks at tR = 15.3 min (97.3 area% 3) and tR = 15.0 min (1 .6 area% ( ?, ?)-diastereoisomer of 3 (reference (R,R/S,S) prepared from nearly rac-phenylglycinamide).
[0130] Example 20. Purification of 2-[(R)-(carbamoylphenylmethyl)-amino]-3-
(4-methoxyphenyl)-2-(S)-methylpropionamide 3. 2-[( ?)-(Carbamoylphenylmethyl)- amino]-3-(4-methoxyphenyl)-2-(5)-methylpropionamide 3 (321 g, 941 mmol) and methyl isobutyl ketone (4173 mL) were heated to 72°C until the solid was dissolved and the biphasic mixture (the minor lower aqueous layer is only visible on stopping stirring) was allowed to cool to room temperature with constant stirring. Stirring was maintained for 2 h. The solid formed was filtered at room temperature, was washed with methyl isobutyl ketone (2 x 320 mL) and was dried in vacuo to provide 2-[( ?)-(carbamoylphenylmethyl)-amino]-3-(4- methoxyphenyl)-2-(5 methylpropionamide 3 (268 g, 84%, >99.5% purity by HPLC) as a white solid. Ή NMR (400 MHz, DMSO-d6) 1.04 (s, 3H), 2.35 (bs, 1 H), 2.79 (d, J- 12.8 Hz, 1 H), 2.95 (d, J = 12.8 Hz, 1 H), 3.65 (s, 3 H), 4.22 (s, 1 H), 6.65 (d, J – 8.4 Hz, 2 H), 6.96 (d, J = 8.4 Hz, 2 H), 7.02 (bs, 1 H), 7.05 (bs, 1 H), 7.33-7.30 (m, 3 H), 7.48 (d, J= 7.2 Hz, 2 H), 7.52 (bs, 1 H), 7.64 (bs, 1 H). HPLC (Luna CI 8, H20 / MeCN 95:5 to 0: 100 30 min, 254 nm, sample 2 mg/mL in MeOH) tR = 15.3 min, >99.5% purity. Mp: 106-108°C.
[0131 ] Example 21. Hydrogenolysis to provide 2-(S)-Amino-3-(4- methoxyphenyl)-2-methyl-propionamide hydrogen bromide salt 4»HBr. To a 1 L hydrogenation reactor were added 2-[( ?)-(carbamoylphenylmethyl)-amino]-3-(4- methoxyphenyl)-2-(5)-methylpropionarnide 3 (183.0 g, 537 mmol, 1 eq), MeOH (549 mL) and 10% Pd/C (19.4 g, 5 wt%). The mixture was stirred under H2 (3 bar) at 51 °C for 8 h. Further 10% Pd/C (3,88 g, 1 wt%) was added and the mixture was stirred for a further 8 h at 53°C. The mixture was cooled to room temperature, was filtered through Celite and the filter pad was washed with MeOH (2 x 50 mL). The combined filtrates were concentrated at 30°C under reduced pressure (rotary evaporator) to a dense white “stirrable” paste (250 mL) containing 2-(,S)-amino-3-(4-methoxyphenyl)-2-methyl-propionamide 4 and 5.
[0132] H20 (75 mL) and 48% HBr (75 mL, 667 mmol, 1 ,25 eq.) were then added resulting in a white suspension. Residual MeOH was stripped from the mixture (45 mL distilled) by distillation at 100°C (bath temperature) at reduced pressure (20-30 mBar). The resulting aqueous solution was cooled to room temperature and filtered; the solid was washed with H20 (50 mL). The white solid was discarded (containing phenylacetamide 5 and 4% 4-HBr by NMR) and the resulting solution of 4»HBr (approximately 400 mL, containing 23% 5 with respect to 4 by NMR) was used directly. 1H NMR (400 MHz, DMSO-6d) 4 1.14 (s, 3 H), 2.10 (bs, 2 H), 2.50 (d, J= 13.1 Hz, 1 H), 2.95 (d, J= 13.1 Hz, 1 H), 3.69 (s, 3 H), 6.80 (d, J= 8.5 Hz, 2 H), 6.81 (bs, 1 H), 6.90 (bs, 1 H), 7.09 (d, J- 8.5 Hz, 2 H).
Phenylacetamide 5 3.34 (s, 2 H), 7.15-7.28 (m, 6 H), 7.42 (bs, 1 H). HPLC (Kromasil C8, H2O/0.1 % TFA / MeCN/0.07% TFA 95:5 to 0: 100 30 min, 254 nm, 1 mg/mL in MeOH) phenylacetamide 5 tR = 1 1.21 min. 4 tR = 9.10 min. (3 tR = 1 1 ,95 min.).
[0133] Example 22. (-)-a-Methyl-L-tyrosine, Metyrosine 1. To a 2 L flask equipped with an anchor stirrer was charged the 4»HBr solution (519 mmol obtained from hydrogenolysis) and 48% HBr (648 mL) was added. The solution was heated for 17 h at 105°C and was cooled to room temperature. The solution was washed with CH2C12 (8 x 80 mL, to remove traces of phenylacetic acid) and the aqueous phase was stripped of residual CH2C12 by distillation at 65°C at reduced pressure (20-30 mBar). Activated carbon (10.5 g) was added and the mixture was stirred for 30 min at 60°C, was filtered at 60°C and the filter pad was washed with water (2 x 35 mL) at RT. The combined filtrates were cooled to room temperature and were basified with 12.5 M NaOH (430 mL) to pH 6-7 at such a rate as to maintain the temperature below 30°C (over approximately 2 h). The white solid formed was separated by filtration, was washed with H20 (2 x 315 mL) and was dried in vacuo to provide (-)-a-methyl-L-tyrosine, metyrosine 1 (87 g, 86%, >99.9% HPLC purity, no impurities detected, >99.9% ee the other enantiomer is not detected) as a white solid. HPLC (Zorbax CI 8, NaH2P04 10 mM pH = 3 / MeCN (100:0) 10 min, (100:0) to (0:100) 15 min, 0: 100 5 min, , 225 nm, sample 1 mg/mL in 0.1 M HC1) tR (1) = 7.6 min, tR (4) = 13,95 min. Chiral HPLC (Nucleosil Chiral-1 , CuS04 10 mM / MeCN 9: 1 , 254 nm, sample 1 mg/mL in eluant) tR = 14.4 min. tR enantiomer = 8.4 min. Mp: 309-313°C.
[0134] In order to obtain an NMR spectrum (taking into account the low solubility of the product), a small sample (10 mg) was transformed into its HC1 salt. The sample was dissolved in 2 M HC1 and the solution was evaporated to dryness. Ή NMR (400 MHz, D20) 1.49 (s, 3 H), 2.90 (d, J = 14.5 Hz, 1 H), 3.16 (d, J = 14.5 Hz, 1 H), 6.75 (d, J = 8.2 Hz, 2 H), 7.01 (d, J = 8.2 Hz, 2 H).
[0135] Example 23. Transfer hydrogenolysis to provide 2-(S)-Amino-3-(4- methoxyphenyl)-2-methylpropionamide formic acid salt 4-HCOOH. In a 5 L reactor equipped with anchor stirrer and oil bubbler, 2-[(i?)-(carbamoylphenylmethyl)-amino]-3-(4- methoxyphenyl)-2-(5)-methylpropionamide 3 (261.8 g, 766.8 mmol) was dissolved in MeOH (1570 mL). A first batch of 10% Pd/C (22.2 g, 4% w/w) was added and the mixture was heated to 56 °C. HCOOH (217 mL, 5.75 mol) was dissolved in H20 (393 mL) and 480 mL of the resulting solution were added to the mixture dropwise over 4.5 h, and the temperature was maintained between 54 and 60°C. When gas development ceased (as determined from the oil bubbler, approximately 30 min after complete addition of HC02H aq), the mixture was cooled to room temperature and a second batch of 10% Pd/C (5.6 g, 1 % w/w) was added. The mixture was heated again to 55°C and the remaining HCOOH solution (130 mL) was added dropwise over 45 min. Stirring was maintained for a further 30 min. The mixture was cooled to room temperature, was filtered over a pad of Celite and the filter pad was washed with MeOH (2 x 100 mL). The combined filtrates were concentrated under reduced pressure (rotary evaporator) to a white “stirrable” paste (approximate volume 300 mL) containing 4’HCOOH and phenylacetamide 5.
[0136] H20 (100 mL) was added and residual MeOH was stripped by distillation at reduced pressure (20-30 mBar) at 70 °C. The resulting solution (approximately 360 mL) was filtered and was washed with 100 mL of H20. The white solid (5 containing 1 % of
4«HCOOH by NMR) was discarded and the resulting solution of 4»HCOOH (containing 13% of 5 with respect to 4 by NMR) was used directly. Ή NMR (400 MHz, DMSO-d6) 1.43 (s, 3 H), 2.93 (d, J = 13.6 Hz, 1 H), 3.08 (d, J = 13.6 Hz, 1 H), 3.73 (s, 3 H), 6.88 (d, J = 8.8 Hz, 2 H), 7.17 (d, J = 8.8 Hz, 2 H), 7.56 (s, 1 H), 7.83 (s, 1 H). Phenylacetamide 5 (200 MHz, DMSO-d6) 3.36 (s, 2 H), 6.87 (bs, 1 H), 7.20-7.33 (m, 5 H), 7.45 (bs, 1 H). HPLC (Kromasil C8, H2O/0.1% TFA / MeCN/0.07% TFA 95:5 to 0:100 30 min, 254 nm, 1 mg/mL in MeOH) Phenylacetamide 5 tR = 1 1.38 min. 4.HC02H tR = 9.11 min. (3 tR = 11,95 min).
[0137] Example 24. (-)-a-Methyl-L-tyrosine, Metyrosine 1. Into a 2 L reactor equipped with anchor stirrer, a solution of 4»HCOOH (760 mmol from transfer
hydrogenolysis) and H20 (200 mL) was mixed with 48% aqueous HBr (948 mL, 8.43 mol). The resulting solution was heated to 105°C for 17 h. The mixture was cooled to room temperature and was washed with CH2C12 (6 x 125 mL, to remove traces of phenylacetic acid), the aqueous phase was stripped of residual CH2C12 at 60 °C at reduced pressure (20-30 mBar). The solution was mixed with activated carbon (15.6 g, 10% w/w) and was heated to 60°C for 30 min. The mixture was filtered at 60°C and the residue was rinsed with H20 (2 x 60 mL). The combined filtrates were cooled to 15°C and were basified with 12.5 M NaOH (730 mL) to pH 6-7 at such a rate as to maintain the temperature below 30°C (over approximately 100 min). The white solid formed was separated by filtration, was washed with H20 (2 x 460 mL) and IPA (490 mL and 245 mL) and was dried to provide (-)-ot- methyl-L-tyrosine, Metyrosine 1 (121.1 g, 82%, >99.9% HPLC no impurities >0.1% detected; >99.9% ee., the other enantiomer is not detected) as a white solid. HPLC (Zorbax CI 8, NaH2P04 10 mM pH = 3 / MeCN (100:0) 10 min, (100:0) to (0:100) 15 min, 0: 100 5 min, 225 nm, sample 1 mg/mL in 0.1 M HC1) tR (1) = 7.6 min; t (4) = 13,95 min. Chiral HPLC (Nucleosil Chiral-1, CuS04 10 mM / MeCN 9:1, 254 nm, sample 1 mg/mL in eluant) tR – 14.3 min. tR enantiomer = 8.4 min. Mp: 308-313 °C.
Patent
IN 2010/CHE/1204, IN 1204/CHE/2010,








EXAMPLE-1:
Step- (a): Preparation of (S)- [1-(3,5-Dichloro-phenylcarbamoyl)-ethyl]-carbamic acid tert-butyl ester(32).
Dissolved N-BOC-L-AIanine (200 gm, 1.057 mol) in methylene chloride (800 ml) under stirring. Cooled the resulting reaction mixture to -15 to -10°C, added N-methyl morpholine (128.3 gm, 1.268 mol) then further cooled to -35°C, added ethylchloroformate (131.9 gm, 1.215 mol) followed by 3,5-dichloroaniline (171.2 gm, 1.057 mol). Stirred the above reaction mixture at 0 to 5°C for a period of 16-18 hours. Quenched the reaction mixture with water (500 ml), stirred for 10-15 minutes and separated the organic layer. The organic Iayerwas washed with water (2×200 ml), dried with anhydrous sodium sulfate and filtered. The organic layer was distilled off under reduced pressure, added hexane (400 ml) and stirred for 30 minutes at 0-5°C. Filtered the resulting solid and washed with hexane (100ml). Dried until constant weight is reached. Dry weight of obtained (S)- [1-(3,5-Dichloro-phenylcarbamoyl)-ethyl]-carbamic acid tert-butyl ester is 300.0 gm.
Yield: 85.1%;
Melting point of the resulting compounds ranges from 138.1-140.5°C;
IR spectra (cm’1): 3321, 2981, 1671, 1589, 1539,1446, 1317, 1255, 1165, 1117, 1072, 858, 802;
32
1H NMR (400 MHz, CDCI3): 59.27(br.s, 1H), 7.37(s, 2H), 6.96(s, 1H), 5.42 (br.d, J=5.16HZ, 1H), 4.39(br.s, 1H),1.47 (s, 9H), 1.41(d, J=7.00HZ, 3H).
Mass (m/z): 334.2 [M+H]+.
Step- (b): Preparation of (S)- 2-Amino-N-(3,5-dichloro-phenyl)-propionamide (33).
Dissolved (S)- [1-(3,5-Dichloro-phenylcarbamoyl)-ethyl]-carbamic acid tert-butyl ester (200.0 gm, 0.599 mol) obtained from step-(a) in methanol (300 ml). To the resulting mixture Cone, hydrochloric acid (500 ml) and water (320 ml) are added at 20-25°C. The resulting reaction mixture was stirred for 16-18 hours at 20-25°C. Added water (200 ml) and toluene (400 ml), cooled to 5-100C1 then basified with 50% sodium hydroxide solution. Stirred for 15 minutes and separated the layers. Aqueous layer was washed with toluene (3x200ml). Combined organic layers and washed with water, dried with anhydrous sodium sulfate and filtered. The organic layer was distilled off under reduced pressure to get the title compound as brown colored syrup. Weight: 135.0 gm.
Yield: 96.4%;
IR spectra (cm-1): 3281, 2969, 2930, 1681, 1585, 1515, 1445, 1409, 1372, 1258, 1185, 1112, 925, 843, 798, 669;\
1H NMR (400 MHz, CDCI3) : 59.67(br.s, 1H), 7.50(s, 2H), 7.00(s, 1H), 3.51 (q,J=7.03HZ, 1H), 1.65(br.s, 2H),1.34(d, J=7.04HZ, 3H).
Mass (m/z): 234.09 [M+H]+.
Step- (c): Preparation of (S)- 3-(3,5-Dichloro-phenyl)-2-isopropyl-5-methyl-imidazolidin-4-one(34).

Dissolved (S)- 2-Amino-N-(3,5-dichloro-phenyl)-propionamide (135 gm, 0.579 mol) obtained in step-(b) in toluene (925 ml) under stirring. Cooled the resulting reaction mixture to 20°C, added isobutyraldehyde (83.5 gm, 1.157mol) over a period of 50 to 60 minutes. Stirred the above reaction mass at 50 to 55°C for a period of 18-19 hours. The organic layer was distilled off under reduced pressure, added hexane (270 ml) and stirred for 1.45 hours at 0-5°C. Filtered the resulting solid and washed with hexane (50ml). Dried until constant weight is reached. Dry weight of obtained (S)- 3-(3,5-Dichloro-phenyl)-2-isopropyl-5-methyl-imidazolidin-4-one is 138.0 gm.
Yield: 82.98%;
Melting point ranges from 128.9-131.8°C;
IR spectra (cm”1): 3305, 3051, 2962, 2877, 1689, 1587, 1450, 1383, 1278, 1223, 1055, 846, 798, 743;
1H NMR (400 MHz, CDCI3) : 57.40 (s, 2H), 7.15 (s, 1H), 5.00 (s, 1H), 3.68 (q, J=6.83HZ, 1H), 1.98 (br.s, 1H), 1.35 (d, J=6.84HZ, 3H), 0.96(d, J=6.87HZ, 3H), 0.76 (d, J=6.70HZ, 3H).
Mass (m/z): 288.1 [M+H]+.
Step- (d): Preparation of (2R, 5S)-2-isopropyl-3-(3,5-Dichloro-phenyl)-5-methyl-1 -(2,2,2-trifluoroacetyl)-imidazolidin-4-one (35).
Dissolved (S)-3-(3,5-Dichloro-phenyl)-2-isopropyl-5-methyl-imidazolidin-4-one (135 gm, 0.470 mol) obtained in step-(c) in methylene chloride (1080 ml) under stirring. Cooled the resulting reaction mixture to 0 to 5°C, added triethyl amine (66.48 gm, 0.658 mol) followed by trifluoroacetic anhydride (132.3 gm, 0.658 mol) over a period of 30 minutes. Stirred the above reaction mass at 0 to 5°C for a period of 2-3 hours. Quenched the reaction mixture with water (405 ml), stirred for 20-25 minutes and separated the organic layer. The organic layer was washed with water (2×270 ml), dried with anhydrous sodium sulfate and filtered. The organic layer was distilled off under reduced pressure, to get crude product as semi solid. The obtained semi solid mass was recrystalised using isopropyl alcohol to get pure product. Dried until constant weight is reached. Dry weight of obtained (2R, 5S)-2-isopropyl-3-(3,5-Dichloro-phenyl)-5-methyl-1-(2,2,2-trifluoroacetyl)-imidazolidin-4-one is 153.0 gm.
Yield: 84.93%;













Step- (c): Preparation of (S)- 3-(3,5-Dichloro-phenyl)-2-isopropyl-5-methyl-imidazolidin-4-one (34).
Dissolved (S)- 2-Amino-N-(3,5-dichloro-phenyl)-propionamide (104 gm, 0.446 mol) obtained in step-(b) in toluene (715 ml) under stirring. Cooled the resulting reaction mixture to 20°C, added isobutyraldehyde (64.34 gm, 0.892mol) over a period of 50 to 60 minutes. Stirred the above reaction mass at 50 to 55°C for a period of 18-19 hours. The organic layer was distilled off under reduced pressure, added hexane (200 ml) and stirred for 3 hours at 0-5°C. Filtered the resulting solid and washed with hexane (25ml). Dried until constant weight is reached. Dry weight of obtained (S)- 3-(3,5-Dichloro-phenyl)-2-isopropyl-5-methyl-imidazolidin-4-one is 101.0 gm. Yield: 78.90%;
Step- (d): Preparation of (2R, 5S)-2-isopropyl-3-(3,5-Dichloro-phenyl)-5-methyl-1-(2,2,2-trifluoroacetyl)-imidazolidin-4-one (35).
Dissolved (S)- 3-(3,5-Dichloro-phenyl)-2-isopropyl-5-methyl-imidazolidin-4-one (100 gm, 0.348 mol) obtained in step-(c) in methylene chloride (800 ml) under stirring. Cooled the resulting reaction mixture to 0 to 5°C, added triethyl amine (42.28 gm, 0.417 mol) followed by trifluoroacetic anhydride (87.73 gm, 0.417 mol) over a period of 30 minutes. Stirred the above reaction mass at 0 to 5°C for a period of 2-3 hours. Quenched the reaction mixture with water (300 ml), stirred for 20-25 minutes and separated the organic layer. The organic layer was washed with water (2×200 ml), dried with anhydrous sodium sulfate and filtered. The organic layer was distilled off under reduced pressure, to get crude product as semi solid. The obtained semi solid mass was recrystalised using isopropyl alcohol to get pure product. Dried until constant

reached. Dry weight of obtained (2S, 5S)-2-isopropyl-3-(3,5-Dichloro-phenyl)-5-methyl-1-(2,2,2-trifluoroacetyl)-5-(4-methoxy benzyl)-imidazolidin-4-one is 6.6 gm.
Yield: 61.10%;
Melting point ranges from 114.2-116.4°C;
IR spectra (cm’1): 2977, 1731, 1692, 1609, 1585, 1572, 1513, 1426, 1348, 1250, 1197, 1048, 890, 748;
1H NMR (400 MHz, CDCI3) : 57.29 (s,1H), 6.84(d, J=8.5HZ, 2H), 6.80(d, J=1.5HZ, 2H), 6.73(d, J=8.1HZ, 2H), 5.07(s, 1H), 3.99(q, J=6.9Hz, 2H), 3.66(d, J=13.8HZ, 1H), 2.99(d, J=13.8HZ, 1H), 2.05(t, J=6.94HZ, 1H), 1.95(s, 3H), 1.41 (t, J=6.94HZ, 3H), 0.91 (d, J=6.61HZ, 3H), 0.52(d, J=7.30HZ, 3H).
Mass (m/z): 517.3 [M+H]+.
Step- (f): Preparation of (S)-2-amino-N-(3,5-Dichloro-phenyl)-2-methyl-3-(4-ethoxy phenyi)-propionamide (45).
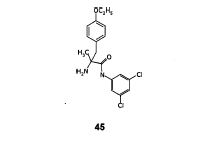
To the suspension of potassium hydroxide (1.12 gm, 0.0169 mol) in isopropyl alcohol (25 ml) , (2S, 5S)-2-isopropyl-3-(3,5-Dichloro-phenyl)-5-methyl-1 -(2,2,2-trifluoroacetyl)- 5-(4-ethoxy benzyl)-imidazolidin-4-one (5.0 gm, 0.009 mol) obtained in step-(e) was added at 25 to 30°C under stirring. The resulting reaction mixture was stirred at 40 to Cl 45 45°C for 3-4 hours. Cooled to 10 to 15°C,added 3M sulfuric acid (15 ml) over a period of 30 minutes. The resulting reaction mixture was heated to 70 to 75°C, stirred for 2 to 3 hours at same temperature. Distilled solvent completely under reduced pressure, added water (25 ml) and ethyl acetate (50 ml). Stirred for 15 minutes and basified with 20% sodium hydroxide solution. Stirred for 15 minutes at 25 to 30°C, separated the organic layer. Aqueous layer washed with ethyl acetate (50 ml). Combined the organic layers and washed with saturate sodium chloride solution (25 ml), dried over anhydrous sodium sulfate, filtered. Removed solvent completely under reduced pressure to get the title compound as brown colored syrup. Weight of (S)-2-amino-N-(3,5-Dichloro-phenyl)- 2-methyl-3-(4-ethoxy phenyl)-propionamide is 2.8 gm. Yield: 79.0%; IR spectra (cm”1 ): 2981, 1732, 1689, 1682, 1575, 1513, 1446, 1302, 1244, 1179, 1116, 1048, 843; 1H NMR (400 MHz, CDCI3) : 59.79(br.s, 1H), 7.52(s, 2H), 7.05(d, J=8.68HZ, 3H), 6.79(d, J=8.53HZ, 2H), 3.94(q, J=6.98, 2H), 3.38(d, J=13.25HZ, 1H), 2.57(d, J=13.56HZ, 1H), 2.03(s, 1H), 1.58(br.s, 2H), 1.43(s, 3H),1.39(t, J=5.42HZ, 3H), Mass (m/z): 368.2 [M+H]+ .
Step- (g): Preparation of (2S)-2-amino-3-(4-hydroxy phenyl)-2-methyl propanoic acid (Metyrosine). Dissolved the (S)-2-amino-N-(3,5-Dichloro-phenyl)-2-methyl-3-(4-ethoxy phenyl)- propionamide (2.8 gm, 0.007 mol) obtained in step-(f) in aqueous HBr (50 ml) under stirring. The resulting reaction mixture was heated to 120-125°C and stirred for 24 hours. Cooled to 50°C, added water (100 ml) stirred for 15 minutes then further cooled 54 to 10-15°C , pH adjusted to 5-6 with ammonium hydroxide solution. Stirred for 30 minutes at 10-15°C, filtered and cake washed with water (2×5 ml). Dried until constant weight is reached. Dry weight of obtained crude (2S)-2-amino-3- (4-hydroxy phenyl)-2-methyl propanoic acid (Metyrosine) of formula-1 is 1.8 gm.
Step- (h): Purification of (2S)-2-amino-3-(4-hydroxy phenyl)-2-methyl propanoic acid (Metyrosine). The crude (2S)-2-amino-3-(4-hydroxy phenyl)-2-methyl propanoic acid (Metyrosine) obtained in step ( g) 1.8 gm) was dissolved in water (180 ml) by heating the reaction mixture to 90°C. Darco (Charcoal) was added and stirred for 10-15 minutes at same temperature. The resulting reaction mixture was filtered through celite bed. The filtered reaction mass concentrated up to half volume reached under reduced pressure. Cooled to IO0C and stirred for a period of 30 minutes at same temperature. Filtered the resulting solid, washed with water. Dried until constant weight is reached. Dry weight of obtained pure (2S)-2-amino-3- (4-hydroxy phenyl)-2-methyl propanoic acid (Metyrosine) of formula-1 was 0.8 gm. The product is matching in all respects with compounds of Metyrosine obtained from EXAMPLE-1 (Step-h). Purity: 99.98%. Chiral purity by HPLC: 100.0%.
IR FROM NET


13 C NMR
References
- ^ Green KN, Larsson SK, Beevers DG, Bevan PG, Hayes B (August 1982). “Alpha-methyltyrosine in the management of phaeochromocytoma”. Thorax. 37 (8): 632–3. doi:10.1136/thx.37.8.632. PMC 459390. PMID 7179194.
- ^ O’Leary OF, Bechtholt AJ, Crowley JJ, Hill TE, Page ME, Lucki I. Depletion of serotonin and catecholamines block the acute behavioral response to different classes of antidepressant drugs in the mouse tail suspension test. Psychopharmacology. 2007 Jun;192(3):357-71. PMID 17318507
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Demser |
| AHFS/Drugs.com | Consumer Drug Information |
| ATC code | |
| Pharmacokinetic data | |
| Elimination half-life | 3.4–3.7 hours |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.010.546 |
| Chemical and physical data | |
| Formula | C10H13NO3 |
| Molar mass | 195.215 g/mol |
| 3D model (JSmol) | |
////////////Metyrosine, Metirosine, Demser, メチロシン , JAPAN 2019, α-Methyl-p-tyrosine, метирозин , ميتيروسين , 甲酪氨酸 ,

{kind=link}