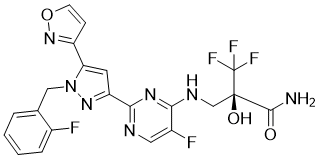

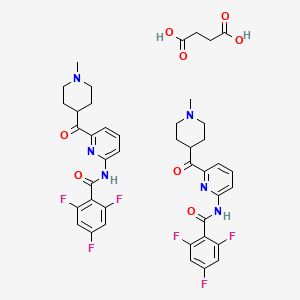
OLINCIGUAT
cas 1628732-62-6
Chemical Formula: C21H16F5N7O3
UNII-PD5F4ZXD21
Molecular Weight: 509.4
Olinciguat is a guanylate cyclase activator drug candidate.
(2R)-3,3,3-trifluoro-2-{[(5-fluoro-2-{1-[(2-fluorophenyl)methyl]- 5-(1,2-oxazol-3-yl)-1H-pyrazol- 3-yl}pyrimidin-4-yl)amino]methyl}-2-hydroxypropanamide
- Originator Ironwood Pharmaceuticals
- Class Antifibrotics; Cardiovascular therapies
- Mechanism of Action Soluble guanylyl cyclase agonists
- Orphan Drug StatusNo
- New Molecular EntityYes
Highest Development Phases
- Phase II Gastrointestinal disorders; Sickle cell anaemia
- Phase I Cardiovascular disorders; Fibrosis
Most Recent Events
- 03 Jan 2018 Pharmacodynamics data from a preclinical trial in Cardiovascular disorders presented at the 59th Annual Meeting and Exposition of the American Society of Hematology (ASH-2017)
- 21 Dec 2017 Phase-II clinical trials in Sickle cell anaemia in USA (PO)
- 09 Dec 2017 Adverse events, pharmacokinetic and pharmacodynamics data from a phase Ib trial in healthy volunteers presented at the 59th Annual Meeting and Exposition of the American Society of Hematology

IW-1701
Currently in Phase II Clinical Development
Area of focus:
Achalasia and Sickle Cell Disease
Dysregulation of the nitric oxide-soluble guanylate cyclase-cyclical guanosine monophosphate (NO-sGC-cGMP) signaling pathway is believed to be linked to multiple vascular and fibrotic diseases, such as achalasia and sickle cell disease.
Our candidate:
IW-1701 is an investigational soluble guanylate cyclase (sGC) stimulator from Ironwood’s diverse library of sGC stimulators, which are being investigated for their potential effects on vascular and fibrotic diseases. The compound has been shown in nonclinical studies to modulate the NO-sGC-cGMP signaling pathway and is currently being evaluated in a Phase II study in achalasia. IW-1701 is wholly-owned by Ironwood Pharmaceuticals.
 IW-1701 is being evaluated to determine its potential effects on vascular and fibrotic diseases.
IW-1701 is being evaluated to determine its potential effects on vascular and fibrotic diseases.
sGC is the primary receptor for NO in vivo. sGC can be activated via both NO-dependent and NO-independent mechanisms. In response to this activation, sGC converts Guanosine-5′-triphosphate (GTP) into the secondary messenger cGMP. The increased level of cGMP, in turn, modulates the activity of downstream effectors including protein kinases, phosphodiesterases (PDEs) and ion channels.
In the body, NO is synthesized from arginine and oxygen by various nitric oxide synthase (NOS) enzymes and by sequential reduction of inorganic nitrate. Three distinct isoforms of NOS have been identified: inducible NOS (iNOS or NOS II) found in activated macrophage cells; constitutive neuronal NOS (nNOS or NOS I), involved in neurotransmission and long term potentiation; and constitutive endothelial NOS (eNOS or NOS III) which regulates smooth muscle relaxation and blood pressure. Experimental and clinical evidence indicates that reduced concentrations orbioavailability of NO and/or diminished responsiveness to endogenously produced NO contributes to the development of disease.
NO-independent, heme -dependent sGC stimulators, have shown several important differentiating characteristics, when compared to sGC activators, including crucial dependency on the presence of the reduced prosthetic heme moiety for their activity, strong synergistic enzyme activation when combined with NO and stimulation of the synthesis of cGMP by direct stimulation of sGC, independent of NO. The benzylindazole compound YC-1 was the first sGC stimulator to be identified. Additional sGC stimulators with improved potency and specificity for sGC have since been developed.
Compounds that stimulate sGC in an NO-independent manner offer considerable advantages over other current alternative therapies that target the aberrant NO pathway. There is a need to develop novel, well-characterized stimulators of sGC. Compound I is an sGC stimulator that has demonstrated efficacy for the treatment of a number of NO related disorders in preclinical models. Compound I was previously described in WO2014144100, Example 1, as a light orange solid. Compound I may be present in various crystalline forms and may also form several pharmaceutically acceptable salts.
Compounds which enhance eNOS transcription: for example those described in WO
02/064146, WO 02/064545, WO 02/064546 and WO 02/064565, and corresponding patent documents such as US2003/0008915, US2003/0022935, US2003/0022939 and US2003/0055093. Other eNOS transcriptional enhancers including those described in US20050101599 (e.g. 2,2-difluorobenzo[l,3]dioxol-5-carboxylic acid indan-2-ylamide, and 4-fluoro-N-(indan-2-yl)-benzamide), and Sanofi-Aventis compounds AVE3085 and AVE9488 (CA Registry NO. 916514-70-0; Schafer et al., Journal of Thrombosis and Homeostasis 2005; Volume 3, Supplement 1 : abstract number P 1487);
NO independent heme-independent sGC activators, including, but not limited to: -2667 (see patent publication DE19943635)

HMR-1766 (ataciguat sodi

S 3448 (2-(4-chloro-phenylsulfonylamino)-4,5-dimethoxy-N-(4-(thiomoφholine-4-sulfonyl)-phenyl)-benzamide (see patent publi

HMR-1069 (Sanofi-Aventis).
(7) Heme-dependent sGC stimulators including, but not limited to:
YC-1 (see patent publications EP667345 and DE19744026)

Riociguat (BAY 63-2521, Adempas, commercial product, described in DE19834044)

Neliciguat (BAY 60-4552, described in WO 2003095451)

Vericiguat (BAY 1021189, clinical backup to Riociguat),
BAY 41-2272 (described in DE19834047 and DE19942809)

BAY 41-8543 (described in DE I 9834044)

Etriciguat (described in WO 2003086407)

CFM-1571 (see patent publicatio
A-344905, its acrylamide analo analogue A-778935.

A-344905;

Compounds disclosed in one of publications: US20090209556, US8455638, US20110118282 (WO2009032249), US20100292192, US20110201621, US7947664, US8053455 (WO2009094242), US20100216764, US8507512, (WO2010099054) US20110218202 (WO2010065275),
US20130012511 (WO2011119518), US20130072492 (WO2011149921), US20130210798
(WO2012058132) and other compounds disclosed in Tetrahedron Letters (2003), 44(48): 8661-8663.
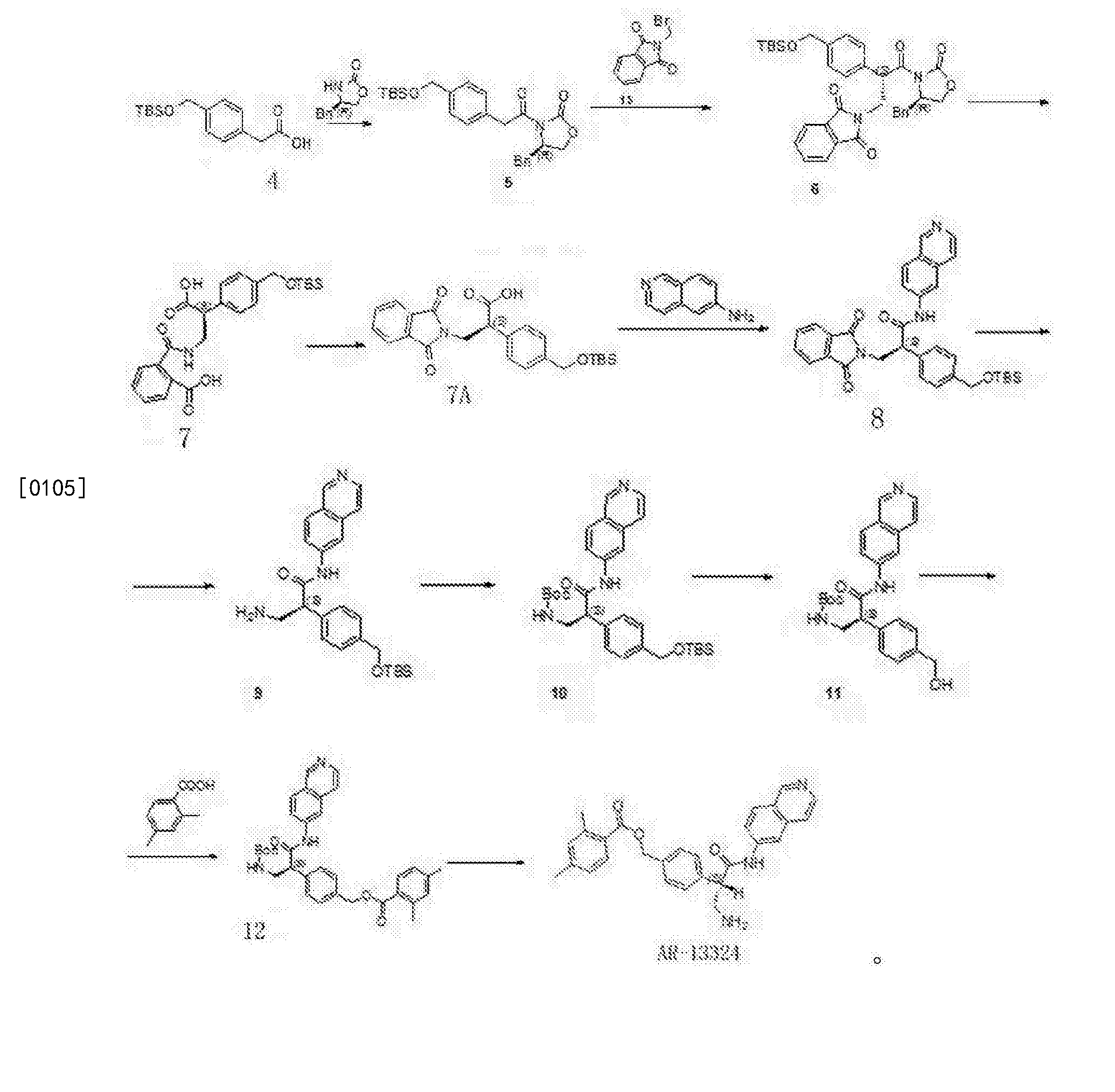
Pictorial synthesis
FROM PATENTS
CONSTRUCT YOUR OWN




SIDE CHAIN SHOWN ABOVE

FINAL STEP SHOWN ABOVE OLINCIGUAT










PATENT
WO2014144100, Example 1
| Inventors |
Takashi Nakai, Joel Moore, Nicholas Robert Perl, Rajesh R. Iyengar, Ara Mermerian, G-Yoon Jamie Im, Thomas Wai-Ho Lee, Colleen Hudson, Glen Robert RENNIE, James Jia, Paul Allen RENHOWE, Timothy Claude Barden, Xiang Y Yu, James Edward SHEPPECK, Karthik Iyer, Joon Jung, Less « |
| Applicant |
Takashi Nakai, Joel Moore, Nicholas Robert Perl, Iyengar Rajesh R, Ara Mermerian, G-Yoon Jamie Im, Thomas Wai-Ho Lee, Colleen Hudson, Rennie Glen Robert, James Jia, Renhowe Paul Allen, Timothy Claude Barden, Xiang Y Yu, Sheppeck James Edward, Karthik Iyer, Joon Jung, Less « |
PATENT
WO 2016044447
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016044447
| Inventors |
Timothy Claude Barden, James Edward SHEPPECK, Glen Robert RENNIE, Paul Allan Renhowe, Nicholas Perl, Takashi Nakai, Ara Mermerian, Thomas Wai-Ho Lee, Joon Jung, James Jia, Karthik Iyer, Rajesh R. Iyengar, G-Yoon Jamie Im |
| Applicant |
Ironwood Pharmaceuticals, Inc. |
Compound 195

lntermediate-36 Compound 195
[00463] lntermediate-36 (35 mg, 0.09 mmol),
(R)-2-(aminomethyl)-3,3,3-trifluoro-2-hydroxypropanamide (60 mg, 0.35 mmol) and
N-ethyl-N-isopropylpropan-2-amine (0.10 mL, 0.56 mmol) were mixed in dimethylsulfoxide (1.5 mL) and heated at 95°C for 8 hr. The solution was cooled to room temperature, diluted with water (2 mL) and the pH taken to 2-3 with 1 N (aq) HC1. The solution was mixed with ethyl acetate (50 mL) and the organic phase was washed with water (2 x 5 mL), brine, then dried over Na2S04, filtered and concentrated by rotary evaporation. The residue was subjected to preparative reverse phase HPLC
. . . t . + + . using a giauiciu ui water acetonitri e . tni uoroacetic aci as e uant to give me iouu i s a wnite solid (11 mg, 23% yield). ¾-NMR (400 MHz, CD3OD) δ 8.83 (br s, 1H), 8.27 (br s, 1H), 7.49 (br s,
1H), 6.9-7.0 (m, 2H), 6.5-6.6 (m, 2H), 5.86 (s, 2H), 4.35 (d, 1H), 4.16 (d, 1H) ppm. Note: exchangable protons all appeared under the residual HOD peak at 4.91 ppm.
PATENT
WO-2018009609
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018009609&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
Novel crystalline solid forms of olinciguat (presumed to be IW-1701), an SGC stimulator and their salts, such as hydrochloride acid (designated as Forms A, B, D, E, F, H and G), processes for their preparation and compositions comprising them are claimed. Also claimed are processes for preparing the crystalline forms. Further claimed are their use for treating cancer, sickle cell disease, osteoporosis, dyspepsia, Duchenne muscular dystrophy, amyotrophic lateral sclerosis and spinal muscle atrophy
In one aspect, the invention relates to crystalline solid forms of Compound I, depicted below:

Compound I
[0009] For purposes of this disclosure, “Compound I,” unless otherwise specifically indicated, refers to the free base or to the hydrochloric acid salt of the structure denoted above. Compound I, as its crystalline free base, is highly polymorphic and known to have seven crystalline forms (Forms A, B, D, E, F, G and H) as well as multiple solvates. Compound I was previously described in
WO2014144100, Example 1, as a light orange solid.
[0010] In one embodiment, the crystalline solid forms of Compound I here disclosed are polymorphs of the free base. In another embodiment, a crystalline solid form of Compound I is the hydrochloric acid salt. In one embodiment, the polymorphs of Compound I are crystalline free base forms. In another embodiment, they are solvates.
[001 1] In another aspect, also provided herein are methods for the preparation of the above described crystalline free forms and salts of Compound I.
[0012J In another aspect, the invention relates to pharmaceutical compositions comprising one or more of the polymorphs of Compound I herein disclosed, or the hydrochloric acid salt of Compound I, and at least one pharmaceutically acceptable excipient or carrier. In another embodiment, the invention relates to pharmaceutical dosage forms comprising said pharmaceutical compositions.
[0013] In another embodiment, the invention relates to a method of treating a disease, health condition or disorder in a subject in need thereof, comprising administering, alone or in combination therapy, a therapeutically effective amount of a polymorph of Compound I herein disclosed, or a mixture of polymorphs thereof, or its hydrochloric acid salt , to the subject; wherein the disease or disorder is one that may benefit from sGC stimulation or from an increase in the concentration of NO and/or cGMP.
EXAMPLES
Example 1: Preparation of crude Compound I
i): Coupling of Compound (1′) and 7V,0-Dimethylhydroxylamine to provide N-methoxy-N-methylisoxazole-3-carboxamide (2′)
[00238] Isooxazole-3-carboxylic acid ((l’)> 241.6 g, 2137 mmoles, 1.0 equiv.), toluene (1450 mL) and DMF (7.8 g, 107 mmoles, 0.05 equiv.) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The resulting slurry was heated to 45-50 °C. Oxalyl chloride (325 g, 2559 mmoles, 1.2 equiv.) was then charged via an addition funnel over the course of 2 h while maintaining the reaction temperature between 45 to 50 °C and a vigorous gas evolution was observed. A brown mixture was obtained after addition. The brown mixture was heated to 87 to 92 °C over 1 h and stirred at 87 to 92 °C for 1 h. The reaction was completed as shown by HPLC. During heating, the brown mixture turned into a dark solution. The reaction was monitored by quenching a portion of the reaction mixture into piperidine and monitoring the piperidine amide by HPLC. The dark mixture was cooled to 20-25 °C and then filtered through a sintered glass funnel to remove any insolubles. The dark filtrate was concentrated under reduced pressure to a volume of 400 mL dark oil.
[00239] Potassium carbonate (413 g, 2988 mmoles, 1.4 equiv.) and water (1000 mL) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The reaction solution was cooled to -10 to -5 °C. N,0-dimethylhydroxyamine hydrochloride (229 g, 2348 mmoles, 1.1 equiv.) was charged to a suitable reaction vessel and dissolved in water (1000 mL). The N,0-dimethylhydroxyamine solution and dichloromethane (2500 mL) were then charged to the potassium carbonate solution.
[00240] The above dark oil (400 mL) was then charged slowly via an addition funnel while maintaining the reaction temperature -10 to 0 °C. The addition was slightly exothermic and a brown mixture was obtained after addition. The mixture was stirred at 0 to 5 °C over 20 min. and then warmed to 20 to 25 °C. The bottom organic layer was collected and the top aq. layer was extracted with dichloromethane (400 mL). The combined organic layers were washed with 15% sodium chloride solution (1200 mL). The organic layer was dried over magnesium sulfate and then filtered. The filtrate was concentrated under reduced pressure to give intermediate (2′) as a dark oil (261.9 g, 97 wt%, 76% yield, 3 wt% toluene by Ή-ΝΜΡν, 0.04 wt % water content by KF). Ή-ΝΜΡν (500 MHz, CDC13) δ ppm 8.48 (s, 1 H); 6.71(s, 1 H); 3.78 (s, 3 H); 3.38 (s, 3 H).
ii): alkylation of Compound (2′) and ethyl propiolate to provide (E)-ethyl 4-(isoxazol-3-yl)-2-(methox methyl)amino)-4-oxobut-2-enoate (3′)
(2′) (3′)
[00241] Intermediate (2′) (72.2 g, 96 wt%, 444 mmoles, 1.0 equiv.), ethyl propiolate (65.7 g, 670 mmoles, 1.5 equiv.) and anhydrous THF (650 mL) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The solution was cooled to -65 to -55 °C. Sodium bis(trimethylsilyl)amide in THF (1 M, 650 mL, 650 mmoles, 1.46 equiv.) was then charged slowly via an addition funnel while maintaining the reaction temperature -65 to -55 °C. The mixture was stirred below -55 °C over 10 min. after addition was complete. Then 1 N HC1 (650 mL, 650 mmoles, 1.46 equiv.) was charged to quench the reaction while maintaining the reaction temperature below -20 °C followed immediately with the addition of ethyl acetate (1500 mL) and water (650 mL). The top ethyl acetate layer was collected and the bottom aqueous layer was extracted with ethyl acetate (800 mL). The combined organic layers were washed with 10% citric acid (1000 mL) and saturated sodium chloride solution (650 mL). The organic layer was concentrated under reduced pressure to give a dark oil.
[00242] The dark oil was dissolved in a solution of dichloromethane/ethyl acetate/heptane
(150mL/100mL/100mL). The solution was loaded on a silica pad (410 g) and the silica pad was eluted with ethyl acetate/heptane (1/1 v/v). The filtrate (~ 3000 mL) was collected and then concentrated under reduced pressure to a volume of 150 mL to give a slurry upon standing. Heptane (200 mL) was then added to the slurry and the slurry was concentrated under reduced pressure to a volume of 150 mL. The resulting slurry was filtered, and the filter cake was washed with heptane (150 mL). The filter cake was then air dried overnight to furnish intermediate (3′) as a brown solid (63.4 g, 56% yield, >99% pure by HPLC). i-NMR (500 MHz, CDC13) δ ppm 8.42 (d, J=1.53 Hz, 1 H); 6.76 (d, J=1.53 Hz, 1 H); 6.18 (s, 1 H); 4.47 (q, J=7.07 Hz, 2H); 3.75 (s, 3 H); 3.21 (s, 3 H); 1.41 (t, J=7.17 Hz, 3 H). iii): cyclization of Compound 3′ and 2-fluorobenzylhydrazine to provide ethyl l-(2-fluorobenz l)-5-(isoxazol-3-yl)-lH-pyrazole-3-carboxylate (4′)
[00243] Intermediate (3′) (72.9 g, 287 mmoles, 1.0 equiv.) and absolute ethanol (730 mL) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The mixture was cooled to 0 to 5 °C. 2-Fluorobenzylhydrazine (48.2 g, 344 mmoles, 1.2 equiv.) was then charged to the mixture. The mixture was stirred at 0 to 10 °C over 1 h and then warmed to 20 to 25 °C and stirred at 20 to 25 °C over 16 h. The reaction was completed by HPLC. Concentrated HCl (33.9 g, 37 wt%, 344 mmoles, 1.2 equiv.) was charged to the reaction mixture over 1 min and the batch temperature exothermed from 20 °C to 38 °C. A slurry was obtained. The mixture was cooled to 0 to 10 °C over 1 h and stirred at 0-10 °C for 1 h. The resulting slurry was filtered, and the filter cake was washed with ethanol (200 mL). The filter cake was dried under vacuum at 30 to 40 °C over 16 h to furnish intermediate (4′) as an off-white solid (81.3 g, 90% yield, >99% pure by HPLC). ¾-NMR (500 MHz, CDC13) δ ppm 8.47 (d, J=1.68 Hz, 1 H); 7.15 – 7.26 (m, 2 H); 6.94 – 7.08 (m, 2H); 6.77 – 6.87 (m, 1 H); 6.55 (d, J=1.68 Hz, 1 H); 5.95 (s, 2 H); 4.43 (q, J=7.02 Hz, 2 H); 1.41 (t, J=7.17 Hz, 3 H).
iv): amination of Compound (4′) to provide l-(2-fluorobenzyl)-5-(isoxazol-3-yl)-lH-pyrazole-3-carboximidamide hydrochloride (5’B)
[00244] Anhydrous ammonium chloride (267 g, 4991 mmoles, 5.0 equiv.) and toluene (5400 mL) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. Trimethylaluminum in toluene (2 M, 2400 mL, 4800 mmoles, 4.8 equiv.) was charged
slowly via an addition funnel while maintaining the reaction temperature at 20 to 40 °C (Note:
Methane gas evolution was observed during addition). Then the mixture was heated to 75 to 80 °C over 30 min. and a clear white solution was obtained. Intermediate (4′) (315 g, 999 mmoles, 1.0 equiv.) was charged to reaction mixture in four equal portions over 1 h at 75 to 90 °C. The reaction was stirred at 80 to 90 °C over 30 min. and then heated to 100 to 110 °C and stirred at 100 to 110 °C over 3 h. The reaction was completed by HPLC. The reaction mixture was cooled to 10 to 20 °C and methanol (461 g, 14.4 moles, 14.4 equiv.) was charged slowly via an addition funnel while
maintaining the reaction temperature 10-40 °C. Note the quenching was very exothermic and a lot gas evolution was observed. A thick slurry was obtained. A 3N HQ (6400 mL, 3 N, 19.2 moles, 19.2 equiv.) was then charged slowly via an addition funnel while maintaining the reaction temperature at 20 to 45 °C. The mixture was heated to 80 to 85 °C and stirred at 80 to 85 °C over 10 min. to obtain a clear biphasic mixture. The mixture was cooled to 0 to 5 °C over 3 h and stirred at 0 to 5 °C over 1 h. The resulting slurry was filtered, and the filter cake was washed with water (3000 mL). The filter cake was dried under vacuum at 40 to 50 °C over 24 h to furnish intermediate (5’B) as an off-white solid (292 g, 91% yield, >99% pure by HPLC). ¾-ΝΜΡν (500 MHz, DMSO- 6) δ ppm 9.52 (s, 2 H); 9.33 (s, 2 H); 9.18 (d, J=1.53 Hz, 1 H); 7.88 (s, 1 H); 7.29 – 7.38 (m, 1 H); 7.19 – 7.25 (m, 1 H); 7.10 – 7.16 (m, 1 H); 7.03 (d, J=1.53 Hz, 1 H); 6.92 – 6.98 (m, 1 H); 5.91 (s, 2 H). M.P. 180-185 °C.
v): cyclization of Compound (5’B) and diethyl fluoromalonate to provide 5-fluoro-2-(l-(2-fluorobenz l)-5-(isoxazol-3-yl)-lH-pyrazol-3-yl)pyrimidine-4,6-diol (6′)
(5’B) (6·)
[00245] Intermediate (5’B) (224.6 g, 698 mmoles, 1.0 equiv.), methanol (2250 mL) and diethyl fluoromalonate (187 g, 1050 mmoles, 1.5 equiv.) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. Then sodium methoxide in methanol solution (567 g, 30 wt %, 3149 mmoles, 4.5 equiv.) was charged via an addition funnel while maintaining the reaction temperature 20 to 35 °C. The mixture was stirred at 20 to 35 °C over 30 min. and a light suspension was obtained. The reaction was completed by HPLC. A solution of 1.5 N HQ (2300 mL, 3450 mmoles, 4.9 equiv.) was charged via an addition funnel over 1 h while maintaining the reaction temperature 20 to 30 °C. A white suspension was obtained. The pH of the reaction mixture was to be ~1 by pH paper. The slurry was stirred at 20 to 30 °C over 30 min. The resulting slurry was filtered, and the filter cake was washed with a pre-mixed solution of methanol and water (500 mL/500 mL), and then with water (1000 mL). The filter cake was dried under vacuum at 50 to 60 °C over 16 h to furnish intermediate (6′) as an off-white solid (264 g, 97% yield, >99% pure by HPLC). ¾-NMR (500 MHz,
DMSO- s) δ ppm 12.82 (br. s., 1 H); 12.31 (br. s., 1 H); 9.14 (d, J=1.53 Hz, 1 H); 7.55 (s, 1 H); 7.31 -7.37 (m, 1 H); 7.18 – 7.25 (m, 1 H); 7.10 – 7.15 (m, 2 H); 6.97 – 7.02 (t, J=7.55 Hz, 1 H); 5.88 (s, 2 H).
vi): chlorination of Compound (6′) to provide 3-(3-(4,6-dichloro-5-fluoropyrimidin-2-yl)-l-(2-fluorobenz l)-lH-pyrazol-5-yl)isoxazole (7′)
(6«) (7«)
[00246] Intermediate (6′) (264 g, 71 1 mmoles, 1.0 equiv.), acetonitrile (4000 mL) and N,N-dimethylaniline (138 g, 1 137 mmoles, 1.6 equiv.) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The slurry mixture was heated to 70-80 °C. Then phosphorous oxychloride (655 g, 4270 mmoles, 6.0 equiv.) was charged via an addition funnel over 1 h while maintaining the reaction temperature 70 to 80 °C. The mixture was stirred at 75 to 80 °C over 22 h and a brown solution was obtained. The reaction was completed by HPLC. Then the mixture was cooled to between 0 and 5 °C and cotton like solids precipitated out at 25 °C. Water (3000 mL) was charged slowly via an addition funnel while maintaining the reaction temperature at 0 to 10 °C. The slurry was stirred at 0 to 10 °C over 30 min. The resulting slurry was filtered, and the filter cake was washed with a pre-mixed solution of acetonitrile and water (500 mL/500 mL). The filter cake was dried under vacuum at 35 to 45 °C over 16 h to furnish intermediate (7′) as an off-white solid (283 g, 98% yield, >99% pure by HPLC). ‘H-NMR (500 MHz, CDC13) δ ppm 8.48 (d, J=1.68 Hz, 1 H); 7.44 (s, 1 H); 7.19 – 7.25 (m, 1 H); 6.96 – 7.08 (m, 2 H); 6.81 – 6.88 (m, 1 H); 6.60 (d, J=1.68 Hz, 1 H); 6.03 (s, 2 H).
vii): substitution of Compound (7′) with meth oxide to provide 3-(3-(4-chloro-5-fluoro-6-m
(7′) (8′)
[00247] Methanol (3400 mL) and sodium methoxide in methanol (154 mL, 5.4 M, 832 mmoles,
1.2 equiv.) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The reaction mixture was heated to 23 to 27 °C. Intermediate (7′) (283 g, 693 mmoles, 1.0 equiv.) was charged to the mixture in small portions (5-10 g each portion) over 40 min while maintaining the reaction temperature 23 to 27 °C. The slurry was stirred at 23 to 27 °C over 30 min. The reaction was completed by HPLC. The resulting slurry was filtered, and the filter cake was washed with methanol (850 mL) and then water (850 mL). The filter cake was dried under vacuum at 35 to 45 °C over 16 h to furnish intermediate (8′) as an off-white solid (277 g, 99% yield, 97% pure by HPLC). i-NMR (500 MHz, CDCl3) 5 ppm 8.47 (d, J=1.83 Hz, 1 H); 7.38 (s, 1 H); 7.18 – 7.25 (m, 1 H); 7.01 – 7.08 (m, 1 H); 6.94 – 7.00 (m, 1 H); 6.81 – 6.88 (m, 1 H); 6.60 (d, J=1.68 Hz, 1 H); 6.00 (s, 2 H); 4.21 (s, 3 H).
viii): hydrogenation of Compound (8′) to provide 3-(3-(5-fluoro-4-methoxypyrimidin-2-yl)-l-(2-fluorobenz l)-lH-pyrazol-5-yl)isoxazole (9′)
[00248] Intermediate (8′) (226 g, 560 mmoles, 1.0 equiv.), palladium (10% on activated carbon, nominally 50% water wet, 22.6 g, 0.01 moles, 0.018 equiv), tetrahydrofuran (3400 mL) and triethylamine (91 g, 897 mmoles, 1.6 equiv.) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. Nitrogen was bubbled into the reaction mixture via teflon tubing over 10 min. at 20 to 30 °C. Then the mixture was heated to 40 to 50 °C and hydrogen gas was bubbled into the reaction mixture via teflon tubing over 6 h while maintaining the reaction temperature 40 to 50 °C. The reaction was completed by HPLC. Nitrogen was then bubbled into the reaction mixture via teflon tubing over 10 min. at 40 to 50 °C The reaction mixture was hot filtered through Hypo Supercel™ and the filter cake was washed with tetrahydrofuran (2000 mL). The filtrate was concentrated under reduced pressure to a volume of -1300 mL to give a slurry. Tetrahydrofuran was then solvent exchanged to methanol under reduced pressure via continuously feeding methanol (3000 mL). The final volume after solvent exchange was 1300 mL. The resulting slurry was filtered, and the filter cake was washed with methanol (500 mL). The filter cake was dried under vacuum at 20 to 25 °C over 16 h to furnish intermediate (9′) as a white solid (192 g, 93% yield, 98% pure by HPLC). ¾-NMR (500 MHz, CDC13) δ ppm 8.47 (d, J=1.68 Hz, 1 H); 8.41 (d, J=2.59 Hz, 1 H); 7.36 (s, 1 H); 7.17 – 7.24 (m, 1 H); 6.95 – 7.07 (m, 2 H); 6.83 – 6.90 (m, 1 H); 6.60 (d, J=1.68 Hz, 1 H); 5.99 (s, 2 H); 4.19 (s, 3 H).
ix: demethylation of Compound (9′) to provide 5-fluoro-2-(l-(2-fluorobenzyl)-5-(isoxazol-3-yl)-lH-pyrazol-3-yl)pyrimidin-4-ol (10′)
[00249] Intermediate (9′) (230 g, 623 mmoles, 1.0 equiv.), Me OH (3450 mL) and cone. HC1
(307 g, 37 wt%, 3117 mmoles, 5.0 equiv.) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The mixture was heated to 60 to 65 °C and a solution was obtained. The mixture was then stirred at 60 to 65 °C over 17 h and a slurry was obtained. The reaction was completed by HPLC. The slurry was cooled to 20 to 25 °C over 2 h and stirred at 20 to 25 °C over 30 min. The resulting slurry was filtered, and the filter cake was washed with methanol (1000 mL). The filter cake was dried under vacuum at 35 to 45 °C over 16 h to furnish intermediate (10′) as a white solid (214 g, 97% yield, >99% pure by HPLC). ¾-NMR (500 MHz, DMSO-t/6) δ ppm 12.90 – 13.61 (br. s., 1 H); 9.11 (d, J=1.68 Hz, 1 H); 8.16 (s, 1 H); 7.64 (s, 1 H); 7.29 – 7.42 (m, 1 H); 7.17 – 7.28 (m, 2 H); 7.08 – 7.15 (m, 1 H); 6.97 (s, 1 H); 5.91 (s, 3 H).
x): chlorination of Compound (10′) to provide 3-(3-(4-chloro-5-fluoropyrimidin-2-yl)-l-(2-fluorobenzyl)-lH-pyrazol-5-yi)isoxazole (Formula IV)
Formula IV
[00250] Intermediate (10′) (214 g, 602 mmoles, 1.0 equiv.), acetonitrile (3000 mL) and NN-dimethylaniline (109 g, 899 mmoles, 1.5 equiv.) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The slurry mixture was heated to 70 to 80 °C. Then phosphorous oxychloride (276 g, 1802 mmoles, 3.0 equiv.) was charged via an addition funnel over 30 min. while maintaining the reaction temperature 70-80 °C. The mixture was stirred at 75 to 80 °C over 2 h and a green solution was obtained. The reaction was completed by HPLC. Then the mixture was cooled to 0 to 5 °C. Water (1500 mL) was charged slowly via an addition funnel while maintaining the reaction temperature at 0 to 10 °C. The slurry was stirred at 0 to 10 °C over 30 min. The resulting slurry was filtered, and the filter cake was washed with a pre-mixed solution of
acetonitrile and water (500 mL/500 mL) and water (500 mL). The filter cake was dried under vacuum at 30 to 40 °C over 16 h to furnish intermediate of Formula IV as an off-white to pink solid (214 g, 95% yield, >99% pure by HPLC). 1H NMR (500 MHz, CDC13) 5 ppm 8.65 (s, 1 H); 8.48 (d, J=1.68 Hz, 1 H); 7.44 (s, 1 H); 7.21 – 7.25 (m, 1 H); 6.97 – 7.06 (m, 2 H); 6.83 – 6.87 (m, 1 H); 6.61 (d, J=1.68 Hz, 1 H); 6.03 (s, 2 H).
a): Cyanation of intermediate (15) to provide 2-(bromomethyl)-3,3,3-trifluoro-2-((trimethylsilyl)oxy)propanenitrile (16)
(15) (16)
[00251 ] Trimethylsilanecarbonitrile ( 153 g, 1.54 moles, 0.97 equiv) and triethylamine (4.44 mL,
3.22 g, 0.032 mole, 0.02 equiv) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The mixture was cooled to 5 °C. 3-Bromo-l, l, l-trifluoropropan-2-one ((15), 304 g, 1.59 moles, 1.0 equiv) was charged via an addition funnel over 35 min, while maintaining the reaction temperature between 10 to 20 °C. The mixture was stirred at 20 to 30 °C over 3 h after the addition to furnish intermediate (16) as a dense oil which was used directly in the next step. 1H-NMR (500 MHz, CDC13) δ ppm 3.68 (d, J=1 1.14 Hz, 1 H); 3.57 (d, J=11.14 Hz, 1 H), 0.34 – 0.37 (m, 9 H).
b): Conversion of nitrile Compound (16) to amide to provide 2-(bromomethyl)-3,3,3-trifluoro-2-hydroxypropanamide (17)
2
(16) (17)
[00252] Concentrated sulfuric acid (339 mL, 6.37 moles, 4.0 equiv) was stirred in a suitable reaction vessel equipped with a mechanical stirrer, digital thermometer and an addition funnel. The sulfuric acid was heated to 45 °C. The above intermediate (16) was added via an addition funnel over 50 min, while keeping the temperature below 75 °C. The reaction mixture was stirred at 75 °C for 2 h and then allowed to cool to room temperature. ¾-NMR indicated reaction complete. The reaction mixture was cooled to -15 °C and diluted with ethyl acetate (1824 mL) via an addition funnel over 45 min (very exothermic), while keeping the temperature between -15 to 5 °C. Water ( 1520 mL) was added slowly via an addition funnel for 1 h 20 min. (very exothermic) between -10 to 0 °C. The layers were separated and the organic layer was washed with 15% aqueous sodium chloride solution ( 1520
mL), 25% aqueous sodium carbonate solution (911 mL) followed by 15% aqueous sodium chloride solution (911 mL). The organic layer was filtered and concentrated under reduced pressure to get 348 g of intermediate (17) as light yellow oil. This oil was dissolved in methanol (1200 mL) and concentrated to furnish 380 g of intermediate (17). (296 g adjusted weight, 79% yield). i-NMR (500 MHz, CDC13) 5 6.61 – 6.94 (m, 1 H); 5.92 – 6.26 (m, 1 H); 3.93 – 4.00 (m, 1 H); 3.68 (d, J=l 1.14 Hz, 1 H).
c): N-Alkylation of compound (17) to provide of 2-(aminomethyl)-3,3,3-trifluoro-2-hydroxypropanamide (14)
(17) (14)
[00253] A 7 N solution of ammonia in methanol (600 mL, 4.28 moles, 10 equiv) was charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The solution was cooled to 0 to 5 °C. Then the intermediate (17) (102 g, 0.432 moles, 1 equiv) was added via an addition funnel over 30 min at 0 to 5 °C. The reaction mixture was warmed to 20 to 25 °C over 1 h and held for 72 h. The reaction was completed by HPLC. The reaction mixture was cooled to 0 to 5 °C and sodium methoxide (78 mL, 5.4 M, 0.421 moles, 0.97 equiv) was added over 2 min. The reaction mixture was then concentrated under reduced pressure to a volume of 300 mL. 2 L of ethyl acetate was added and concentration was continued under reduced pressure to a volume to 700 mL to get a slurry. 700 mL of ethyl acetate was added to the slurry to make the final volume to 1400 mL. 102 mL of water was added and stirred for 2 min to get a biphasic solution. The layers were separated. The ethyl acetate layer was concentrated under reduced pressure to a volume of 600 mL. Then the ethyl acetate layer was heated to > 60 °C and heptane (600 mL) was added slowly between 55 to 60 °C. The mixture was cooled to 15 to 20 °C to give a slurry. The slurry was stirred at 15 to 20 °C for 2 h and filtered. The solids were dried under vacuum at 25 °C for 16 h to furnish amine (14) as white solid (48 g, 64% yield). ‘H-NMR (500 MHz, MeOH-d4) δ ppm 2.94 (d, J= 13.73 Hz, 1H); 3.24 (d, J= 13.58 Hz, 1H).
d): chiral resolution of amine (14) as the 1:1 salt of (R)-2,2-dimethyl-5- (trifluoromethyl)oxazolidine-5-carboxamide (R)-2-hydroxysuccinate (18A) and (D)-malic acid.
(14) (ISA)
[00254] Amine (14) (105 g, 0.608 moles, 1.0 equiv.), (D)-Malic acid (82 g, 0.608 moles, 1.0 equiv.) and acetone (1571 mL) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The reaction mixture was stirred at 20 to 25 °C for 16 h. The resulting slurry was filtered, and the wet cake was washed with acetone (300 mL). The wet cake was charged back to the reaction vessel, and acetone (625 mL) was charged. The slurry was heated to 53 °C and held for 6 h. The slurry was cooled to 20 to 25 °C and held at this temperature for 16 h. The slurry was filtered, and the wet cake was washed with acetone (200 mL). The wet cake was dried under vacuum at 40 °C for 4 h to furnish 82.4 g of the 1 : 1 salt of (18A) and (D)-malic acid as a white solid (82.4 g, 39% yield, 97% ee). i-NMR (500 MHz, D20) δ ppm 4.33 (br, s, 1H); 3.61 (br, d, J= 13.58 Hz, 1H); 3.40 – 3.47 (m, 1H); 2.76 (br, d, J= 15.87 Hz, 1H); 2.53 – 2.63 (m, 1H); 2.16 (br, s, 4H).
e): Coupling of the 1:1 (D)-malic acid salt of intermediate (18A) and Formula IV to provide (R)-3,3,3-trifluoro-2-(((5-fluoro-2-(l-(2-fluorobenzyl)-5-(isoxazol-3-yl)-lH-pyrazol-3-yl)pyrimidin-4-yl)amino)methyl)-2-hydroxypropanamide (Compound I)
Formula IV Compound I
[00255] The 1: 1 salt of intermediate (18A) and (D)-malic acid (74.1 g, 0.214 moles, 2.5 equiv) and water (44.8 mL) were charged to a suitable reaction vessel equipped with a mechanical stirrer and a digital thermometer. The reaction mixture was heated to 70 °C and stirred for 20 min. Acetone generated during the reaction was removed by blowing with nitrogen. The reaction mixture was cooled to 30 to 40 °C and Formula IV (32 g, 0.086 moles, 1.0 equiv), DMSO (448 mL) and Hunig’s base (44.7 mL, 0.257 moles, 3.0 equiv) were charged. The reaction mixture was heated to 90 °C and stirred at 90 °C over 17 h. The reaction was complete by HPLC. Then the mixture was cooled to 60 °C. Another portion of Hunig’s base (104 mL, 0.599 moles, 7.0 equiv) was charged followed by water (224 mL) at 55 to 62 °C. The reaction mixture was stirred for 15 min at 55 to 60 °C to form the seed bed. Water (320 mL) was added via addition funnel at 55 to 62 °C over the course of 30 min, and the resultant slurry was stirred for 1 h at 55 to 60 °C. The resulting slurry was filtered, and the filter cake was washed with a pre-mixed solution of methanol and water (320 mL/320 mL) followed by water (640 mL). The filter cake was then dried under vacuum at 40 °C over 16 h to furnish Compound I as an off-white solid (40 g, 92% yield, 99% pure by HPLC, 98% ee). ¾-NMR (500 MHz, DMSO-t/6) δ ppm 9.10 (s, 1 H); 8.33 (d, J=2.90 Hz, 1 H); 7.93 (s, br, 1 H); 7.90 (s, 1 H); 7.78 (s, br, 1 H); 7.69 (s, br, 1 H); 7.52 (s, 1 H); 7.33 (q, J=7.02 Hz, 1 H); 7.17 – 7.25 (m, 1 H); 7.17 – 7.25
(m, 1 H); 7.10 (t, J=7.48 Hz, l H); 6.98 (t, J=7.55 Hz, 1 H); 5.90 (s, 2 H); 3.92-4.05 (m, 2 H).
////////////OLINCIGUAT, IW-1701, phase 2, ironwood
NC(=O)[C@](O)(CNc1nc(ncc1F)c2cc(c3ccon3)n(Cc4ccccc4F)n2)C(F)(F)F

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....



























































































 Open Access
Open Access