

Note: Compound name must be entered under “Substance Identification” and then “Names and Synonyms” selected to view synonyms.
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Simple and effective method for two-step synthesis of 2-(1,3-dithian-2-ylidene)-acetonitrile

Simple and effective method for two-step synthesis of 2-(1,3-dithian-2-ylidene)-acetonitrile (75% overall yield) and molecular modeling calculation of the mechanism by B3LYP and the 6-311++G(2df,2p) basis set.
http://dx.doi.org/10.5935/0100-4042.20140308
Publicado online: dezembro 12, 2014
Método alternativo para a síntese e mecanismo de 2-(1,3-ditiano-2-ilideno)-acetonitrila
Marcelle S. Ferreira; José D. Figueroa-Villar*
Quim. Nova, Vol. 38, No. 2, 233-236, 2015
Artigo http://dx.doi.org/10.5935/0100-4042.20140308
*e-mail: jdfv2009@gmail.com
MÉTODO ALTERNATIVO PARA A SÍNTESE E MECANISMO DE 2-(1,3-DITIANO-2-ILIDENO)-ACETONITRILA
Marcelle S. Ferreira e José D. Figueroa-Villar* Departamento de Química, Instituto Militar de Engenharia, Praça General Tiburcio 80, 22290-270
Rio de Janeiro – RJ, Brasil
Recebido em 18/08/2014; aceito em 15/10/2014; publicado na web em 12/12/2014
ALTERNATIVE METHOD FOR SYNTHESIS AND MECHANISM OF 2-(1,3-DITHIAN-2-YLIDENE)-ACETONITRILE. We report an alternative method for the synthesis of 2-(1,3-dithian-2-ylidene)-acetonitrile using 3-(4-chlorophenyl)-3-oxopropanenitrile and carbon disulfide as starting materials. The methanolysis of the intermediate 3-(4-chlorophenyl)-2-(1,3-dithian-2-ylidene)-3- oxopropanenitrile occurs via three possible intermediates, leading to the formation of the product at a 75% overall yield. Molecular modeling simulation of the reaction pathway using B3LYP 6-311G++(2df,2p) justified the proposed reaction mechanism. Keywords: 2-(1,3-dithian-2-ylidene)-acetonitrile; reaction mechanism; methanolysis; molecular modeling.
3-(4-clorofenil)-2-(1,3-ditiano-2-ilideno)-3-oxopropanonitrila (3): Cristal amarelo. Rendimento: 95%, 2,80 g, pf 158-160 °C, lit.21 159-160 °C;
IV (KBr, cm-1): 2198 (CN), 1612 (C=O), 1585, 1560 (aromático), 678 cm -1 (C-S);
1H RMN (300 MHz, CDCl3) δ 2,38 (m, J 6,9, 2H, CH2); 3,01 (t, J 6,6, 2H, SCH2); 3,17 (t, J 7,2 , 2H, SCH2); 7,43 (d, J 8,5, 2H); 7,83 (d, J 8,5, 2H);
13C RMN (75 MHz, CDCl3) δ 23,9 (CH2), 30,4 (SCH2), 104,2 (CCO), 117,5 (CN), 128,9, 130,5, 135,6, 139,2 (aromático), 185,2 (C=CS), 185,4 (CO).
21…….Rudorf, W. D.; Augustin, M.; Phosphorus Sulfur Relat. Elem. 1981, 9, 329.
…………………………………….
Síntese da 2-(1,3-ditiano-2-ilideno)-acetonitrila (1) Em um balão de fundo redondo de 100 mL foram adicionados 0,400 g (1,4 mmol) de 3-(4-clorofenil)-2-(1,3-ditiano-2-ilideno)-3- -oxopropanonitrila (2) dissolvidos em 15 mL de THF seco, 0,140 g (20 mmol) de sódio e 15 mL de metanol seco sob atmosfera de nitrogênio. A mistura reacional foi mantida sob agitação à 25 °C por 48 h. Em seguida, a mistura reacional foi dissolvida em 30 mL de água destilada e extraída com acetato de etila (3 x 20 mL). A fase orgânica foi seca em sulfato de sódio anidro, filtrada e concentrada a vácuo para se obter o produto bruto, que foi purificado por cromatografia em coluna (silica gel e hexano:acetato de etila 7:3).
2-(1,3-ditiano-2-ilideno)-acetonitrila (1): Cristal branco. Rendimento: 75%, 165 mg, pf. 60-63 °C, lit1 60-62 °C;
1 H RMN (300 MHz, CDCl3) δ 2,23 (m, J 6,8, 2H, CH2); 3,01 (t, J 7,5, 2H, SCH2); 3,06 (t, J 6,9, 2H, SCH2), 5,39 (s, 1H, CH);
13C RMN (75 MHz, CDCl3) δ 22,9 (CH2), 28,7 (SCH2), 28,8 (SCH2), 90,4 (CHCN), 116,3 (CN), 163,8 (C=CS).
1………Yin, Y.; Zangh, Q.; Liu, Q.; Liu, Y.; Sun, S.; Synth. Commun. 2007, 37, 703.

CAS 113998-04-2
- C6 H7 N S2
- Acetonitrile, 2-(1,3-dithian-2-ylidene)-
- 157.26
| Melting Point | 60-62 °C |
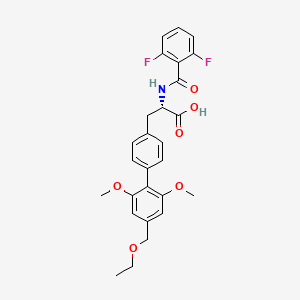
1H NMR predict
2-(1,3-dithian-2-ylidene)-acetonitrile

ACTUAL 1H NMR VALUES
1 H RMN (300 MHz, CDCl3)
δ 2,23 (m, J 6,8, 2H, CH2);
3,01 (t, J 7,5, 2H, SCH2);
3,06 (t, J 6,9, 2H, SCH2),
5,39 (s, 1H, CH);
……………………..
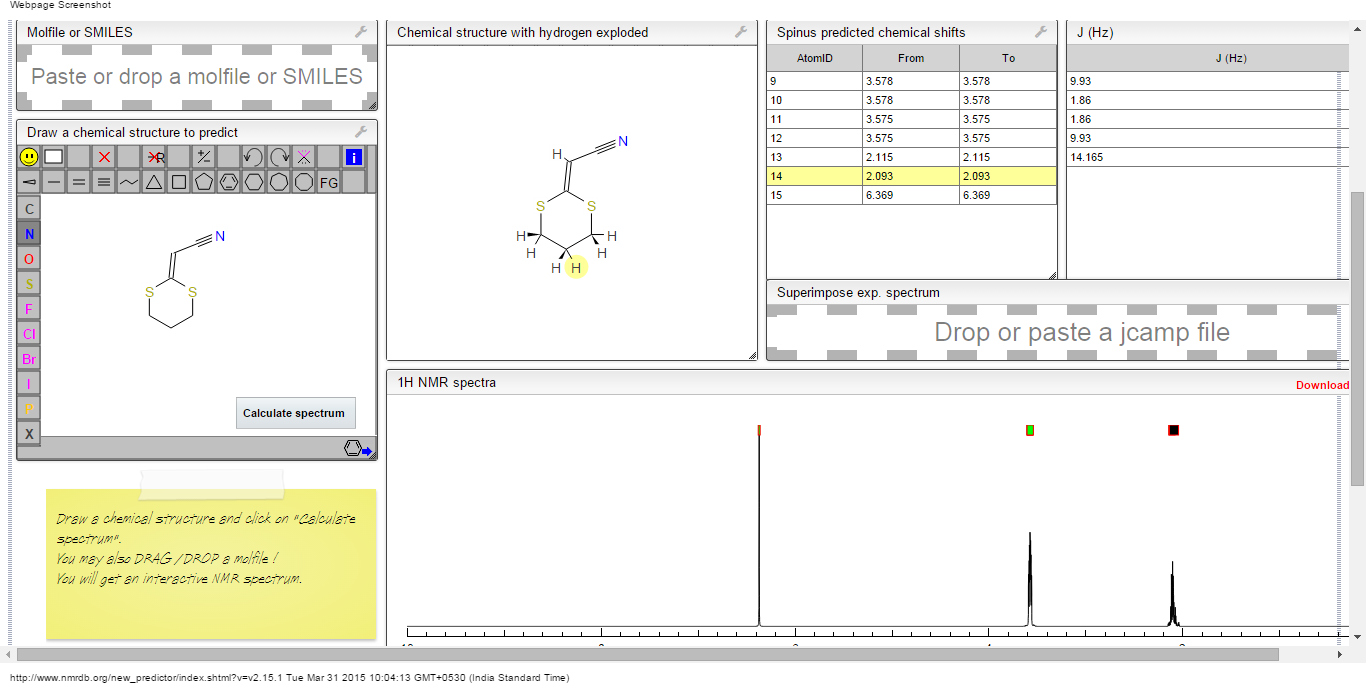
13C NMR PREDICT

ACTUAL 13C NMR VALUE
13C RMN (75 MHz, CDCl3)
δ 22,9 (CH2),
28,7 (SCH2),
28,8 (SCH2),
90,4 (CHCN),
116,3 (CN),
163,8 (C=CS)
COSY NMR PREDICT

SYNTHESIS
2-(1,3-ditiano-2-ilideno)-acetonitrila (1): Cristal branco. Rendimento: 75%, 165 mg, pf. 60-63 °C, lit1 60-62 °C;
1 H RMN (300 MHz, CDCl3) δ 2,23 (m, J 6,8, 2H, CH2); 3,01 (t, J 7,5, 2H, SCH2); 3,06 (t, J 6,9, 2H, SCH2), 5,39 (s, 1H, CH);
13C RMN (75 MHz, CDCl3) δ 22,9 (CH2), 28,7 (SCH2), 28,8 (SCH2), 90,4 (CHCN), 116,3 (CN), 163,8 (C=CS).
WILL BE UPDATED WATCH OUT…………………
Departamento de Química, Instituto Militar de Engenharia, Praça General Tiburcio

Instituto Militar de Engenharia, Rio de Janeiro. BELOW


Entrada do antigo Instituto de Química da UFRGS, um prédio histórico

Equipe – Os módulos foram fabricados na Unisanta sob a supervisão do professor Luiz Renato Lia, coordenador do Curso de Engenharia Química, …

Instituto de Florestas da Universidade Federal Rural do Rio de Janeiro

Praça General Tibúrcio

Praça General Tibúrcio com o Morro da Urca ao fundo
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

Firategrast, T-0047

 Japan
Japan
Firategrast, 402567-16-2;
Firategrast, MS, Alpha4beta1 integrin
PHASE 2 GSK
Mitsubishi Tanabe Pharma INNOVATOR
Glaxo Group Limited, Mitsubishi Tanabe Pharma Corporation
SB 683699, SB-683699, UNII-OJY3SK9H5F
Firategrast; UNII-OJY3SK9H5F; SB-683699; Firategrast (USAN); 402567-16-2; SB683699; T-0047
Molecular Formula: C27H27F2NO6
Molecular Weight: 499.503186 g/mol
SYSTEMATIC NAME:
1,1′-Biphenyl)-4-propanoic acid, alpha-((2,6-difluorobenzoyl)amino)-4′-(ethoxymethyl)-2′,6′-dimethoxy-, (alphaS)-
N-(2,6-Difluorobenzoyl)-4-[4-(ethoxymethyl)-2,6-dimethoxyphenyl]-L-phenylalanine
N- (2 , 6-Difluorobenzoyl) -4- (2 , 6-dimethoxy-4- ethoxymethylphenyl) -L-phenylalanine .
2S)-2-((2,6-Difluorobenzoyl)amino)-3-(4′-(ethoxymethyl)-2′,6′-dimethoxybiphenyl-4- yl)propanoic acid
(2S)-2-{[(2,6- difluorophenyl)carbonyl]amino}-3-[4′-[(ethyloxy)methyl]-2′,6′-bis(methyloxy)-4- biphenylyl]propanoic acid
(2S)-2-[[2,6-bis(fluoranyl)phenyl]carbonylamino]-3-[4-[4-(ethoxymethyl)-2,6-dimethoxy-phenyl]phenyl]propanoic acid
Pharmacological half-life is 2.5 – 4.5 hours, compared to 11 days for natalizumab, a drug in the same class
Orally bioavailable small molecule α4-integrin antagonist
see
http://www.msdiscovery.org/node/1377#node-biblio-1338
http://multiple-sclerosis-research.blogspot.com/2012/01/research-oral-tysabri-analogue.html
SB683699 is an alpha4 integrin antagonist that had been studied in phase II trials at GlaxoSmithKline under a license from Mitsubishi Tanabe Pharma for the oral treatment of multiple sclerosis (MS) in Europe. GlaxoSmithKline and Tanabe Seiyaku (now Mitsubishi Tanabe Pharma) had been studying the drug candidate for the treatment of asthma, rheumatoid arthritis (RA) and Crohn’s disease
MECHANISMS/EFFECTS
HUMAN:
Similar mechanism of action to natalizumab (α4-integrin blocker), but its faster elimination could improve safety profile

Firategrast
SYNTHESIS
………………….
PATENT
Scheme 1

Scheme 2

In a further aspect the present invention provides for a process for the preparation of compound of formula (II) which comprises coupling the compound of formula (V)

Suitable coupling conditions for the compound of formula (V) and the compound of formula (VI) include those shown in Scheme 2. In a further aspect of the invention there is provided the compound of formula (V):

1H NMR characterisation data for the compound of formula (V) were generated on an isolated and purified batch. 1H-NMR spectra were recorded on a Bruker Avance 400 at 400MHz, using TMS as an internal reference.1H NMR (400 MHz, DMSO-D6) δ ppm 1.17 (t, J=7.09 Hz, 3 H) 2.96 (dd, J=13.82, 9.90 Hz, 1 H) 3.1 1 (dd, J=13.82, 5.26 Hz, 1 H) 4.12 (q, J=7.09 Hz, 2 H) 4.63 (ddd, J=9.78, 7.82, 5.38 Hz, 1 H) 7.15 (t, J=7.95 Hz, 2 H) 7.25 (d, J=8.31 Hz, 2 H) 7.47 – 7.55 (m, 3 H) 9.23 (d, J=7.83 Hz, 1 H).
The present invention provides a process for the preparation of the compound of formula

which process comprises the steps: a) hydrolysis of an ester of formula (I la):

Recrvstallisation of (2S)-2-{r(2,6-difluorophenyl)carbonyllamino)-3-r4′-r(ethyloxy)methyll- 2′,6′-bis(methyloxy)-4-biphenylyllpropanoic acid
(2S)-2-{[(2,6-difluorophenyl)carbonyl]amino}-3-[4′-[(ethyloxy)methyl]-2′,6′-bis(methyloxy)- 4-biphenylyl]propanoic acid (9.38Kg) was charged into a clean reactor, followed by ethyl acetate (46.9L). The solution was heated to 50°C and filtered into the pre-warmed (35°C) crystallizing vessel. A line-wash with ethyl acetate (9.4L) was carried out. The combined ethyl acetate solutions were heated to 50°C, stirred to ensure complete dissolution. Filtered heptane (9.4L) was added maintaining the temperature at 50°C then the solution cooled to 30°C and seeded with (2S)-2-{[(2,6-difluorophenyl)carbonyl]amino}-3-[4 – [(ethyloxy)methyl]-2′,6′-bis(methyloxy)-4-biphenylyl]propanoic acid (47g) slurried in 1 :9 ethyl acetate:heptane (0.47L). The slurry was aged for 2 hours at 30°C. Filtered heptane (75L) was added over 3 hours. The slurry was then cooled to 0°C over 1 hour. The mixture was aged at 0°C for 1 hour then the solid was filtered off, washed with isopropyl ether (29.6L and dried under vacuum at 50±3°C to give the product (8.55Kg, 91 %). Characterised by having an infrared absorption spectrum with significant absorption bands at about 754, 768, 800, 820, 849, 866, 1006, 1 100, 1 122, 1 157, 1 188, 1225, 1242, 1268, 1292, 1317, 1352, 1417, 1466, 1530, 1580, 1624, 1650, 1662, 171 1 , 1728, 2938, 3302cm“
…………………………………..
PATENT
Example 10: N- (2 , 6-Difluorobenzoyl) -4- (2 , 6-dimethoxy-4- ethoxymethylphenyl) -L-phenylalanine ethyl ester.
(1) The product obtained in Example l-(4) (2.1 g) was acylated with 2 , 6-difluorobenzoyl chloride in a similar manner as described in Example 1 -(5) to give N- (2, 6-difluorobenzoyl) – 4- (2 , 6-dimethoxy-4-hydroxymethylphenyl) -L-phenylalanine ethyl ester (2.75 g) . mp . 70-72 °C; IR (Nujol) 3400, 3263, 1735, 1654, 1624 cm“1; MS (APCI) m/z 500 (M+H) . (2) To a solution of the product obtained above (1.72 g) in DMSO (20 ml) were added Et3N (4.8 ml) and S03«pyridine (5.6 g) successively at room temperature. The whole mixture was stirred at room temperature for 25 minutes. The reaction mixture was poured into ice-water, and then the mixture was extracted with EtOAc. The organic layer was sequentially washed with 5% aqueous HCl, H20 and brine, dried (Na2S04) and then evaporated. The residue was purified by column chromatography (silica gel; eluent: n-hexane/EtOAc 5:1 to 1:1) to yield N-(2,6- difluorobenzoyl) -4- (2 , 6-dimethoxy-4-formylphenyl) -L- phenylalanine ethyl ester (1.54 g) . mp. 114-116°C; IR (Nujol)
3332, 1735, 1695, 1657, 1644, 1623 cm“1; MS (APCI) m/z 498 (M+H) .
(3) The product obtained above (716 mg) was converted into the title compound (428 mg) in a similar manner as described in Example 1- (7) . mp . 87-89°C; IR (Neat+CHC13) 3300, 1739, 1668 cm“ 1; MS (APCI) m/z 528 (M+H) .
Example 11: N- (2 , 6-Difluorobenzoyl) -4- (2 , 6-dimethoxy-4- ethoxymethylphenyl ) -L-phenylalanine methyl ester.
(1) The product obtained in Example 2- (4) (1.00 g) was acylated with 2 , 6-difluorobenzoyl chloride to give N-(2,6- difluorobenzoyl) -4- (2 , 6-dimethoxy-4-hydroxymethylphenyl) -L- phenylalanine methyl ester (873 mg) in a similar manner as described in Example l-(5). IR (Nujol) 3257, 1743, 1655, 1624 cm“ 1; MS (APCI +Q1MS) m/z 503 (M+NH4) , 486 (M+H) . (2) The product obtained above (860 mg) was converted into the title compound (220 mg) in a similar manner as described in Example 2- (6) and (7).
Example 12: N- (2 , 6-Difluorobenzoyl) -4- (2 , 6-dimethoxy-4- ethoxymethylphenyl) -L-phenylalanine .
The product obtained in Example 10 (200 mg) was hydrolyzed in a similar manner as described in Example 3 to give the title compound (160 mg) . The product obtained in Example 11 (220 mg) was also hydrolyzed in a similar manner as described in Example 3 to give the title compound (167 mg) . mp. 156-158°C; IR (Nujol) 1735, 1655 cm“1; MS (ESI) m/z 498 (M-H) .
…………………….
PATENT
https://www.google.com/patents/WO2003072536A1?cl=en
OUT LINE
phenylalanine derivative of the formula (I) :

wherein X1 is a halogen atom, X2 is a halogen atom, Q is a group of the formula -CH2– or -(CH2)2– and Y is a lower alkyl group, or a pharmaceutically acceptable salt thereof, which has excellent inhibitory activity against α4 integrin-mediated cell adhesion.
Thus, the present invention relates to a process for preparing a compound of the formula (I) :

wherein the symbols are the same as defined above, or a pharmaceutically acceptable salt thereof, comprising : (1) coupling a compound of the formula (VI) :

wherein Z is a leaving group, R1NH is a protected amino group and C02R is a protected carboxyl group with a compound of the formula (V) :

wherein the symbols are the same as defined above, removing the protecting group from the protected amino group, and if necessary, converting the resulting compound into a salt, to yield a compound of the formula (IV) :

wherein the symbols are the same as defined above, or a salt thereof,
(2) condensing the compound (IV) or a salt thereof with a compound of the formula (III) :

wherein the symbols are the same as defined above, a salt or a reactive derivative thereof to yield a compound of the formula (II) :

Ethyl (ocS) – – [ [ (1, 1-dimethylethoxy) carbonyl] amino] -4- hydroxybenzene propionate and ethyl (otS) -α- [ [ (1, 1- dimethylethoxy) carbonyl] amino] -4-
(trifluoromethanesulfonyloxy) benzene propionate are described in J. Med. Chem. , 33: 1620 (1990) and JP-A-7- 157472, respectively. 4-Bromo-3, 5-dimethoxybenzyl alcohol is described in, for example, J. Med. Chem. , 20: 299 (1977), and can also be prepared according to the following process.

Firstly, 4-bromo-3, 5-dihydroxybenzoic acid is methylated to give methyl 4-bromo-3, 5-dimethoxybenzoate, which is then reduced to yield 4-bromo-3, 5-dimethoxy benzyl alcohol. The methylation can be carried out by reacting with dimethyl sulfate in the presence of a base in a suitable solvent (e.g., ethyl acetate). The reduction can be carried out by reacting with an reducing agent (e.g., lithium alminium hydride, sodium borohydride and calcium borohydride) in a suitable solvent (e.g., tetrahydrofuran) .
EXAMPLES
The following Examples are provided to further illustrate the process of preparation according to the present invention. In the following examples, some compounds may be referred to by different compound name depending on the nomenclature, as illustrated below.
Ethyl (αS) -α-amino-4′ -ethoxymethyl-2′ , 6′ – dimethoxy (1, 1′ -biphenyl) -4-propionate
Another name: ethyl (2S) -2-amino-3- [4- (4-ethoxymethyl- 2, 6-dimethoxyphenyl) phenyl]propanoate
Ethyl (αS) – [ [1, 1-dimethylethoxy] carbonyl] amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1,1′ -biphenyl) -4-propionate
Another name 1: ethyl (2S) -2- [ (t-butoxycarbonyl) – amino] -3- [4- (4-ethoxymethyl-2, 6-dimethoxyphenyl) – phenyl]propanoate
Another name 2: Ethyl N- (t-butoxycarbonyl) -4- (4- ethoxymethyl-2, 6-dimethoxyphenyl) -L-phenylalanine
Ethyl (αS) – – [ (2, 6-difluorobenzoyl) amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1, 1′ -biphenyl) -4-propionate Another name 1: Ethyl (2S) -2- [ (2, 6- difluorobenzoyl) amino] -3- [4- (4-ethoxymethyl-2, 6- di ethoxyphenyl) phenyl] propanoate
Another name 2: Ethyl N- [2 , 6-difluorobenzoyl) -4- (4- ethoxymethyl-2, 6-dimethoxyphenyl) -L-phenylalanine
(ocS) – – [ (2, 6-Difluorobenzoyl) amino] -4′ -ethoxymethyl- 2′ , 6′ -dimethox (1,1′ -biphenyl) -4-propionic acid
Another name 1: (2S) -2- [ (2, 6-difluorobenzoyl) amino] -3- [4- (4-ethoxymethyl-2, 6-dimethoxyphenyl) phenyl]propanoic acid
Another name 2: N- [ 2 , 6-difluorobenzoyl) -4- (4- ethoxymethyl-2, 6-dimethoxyphenyl) -L-phenylalanine
EXAMPLE 1 (1) Under nitrogen atmosphere, pyridine (130.3 g) and trifluoromethanesulfonic anhydride (170.4 g) were added dropwise to a solution of ethyl (αS) -α- [ [ (1, 1- dimethylethoxy) carbonyl] amino] -4-hydroxybenzenepropionate
(170.0 g) in dichloromethane (1.7 L) at 10 ° C or below. After stirring for 1 hour at the same temperature, water
(850 ml) was added dropwise to the mixture and the mixture was stirred for 2 hours at the same temperature. The organic layer was washed with 10 % aqueous citric acid solution and aqueous saturated sodium hydrogen carbonate solution, and dried over magnesium sulfate. The solvent was removed in vacuo to yield ethyl (αS) -α- [ [ (1, 1- dimethylethoxy) carbonyl] amino] -4-
(trifluoromethanesulfonyloxy)benzenepropionate (242.5 g) as oil . MS (m/z) : 441 (M+) (2) Under nitrogen atmosphere, to a mixture of ethyl (αS)- – [ [ (1, 1-dimethylethoxy) carbonyl] amino] -4-
(trifluoromethanesulfonyloxy) benzenepropionate (66.2g), 4- ethoxymethyl-2, 6-dimethoxyphenylboric acid (54.0 g) , triphenylphosphine (9.83 g) and N-methylpyrrolidone (330 ml) were added palladium acetate (1.68 g) and diisopropylamine (24.9 g ), and the mixture was heated at 90 °C. After stirring for 1 hour at the same temperature, the mixture was cooled and toluene and water were added. The organic layers were washed with 10% aqueous citric acid solution and saturated aqueous NaCl solution and dried over magnesium sulfate. The solvent was removed in vacuo to yield ethyl (αS) -α- [[ (1, 1-dimethylethoxy) carbonyl] amino] – 4′ -ethoxymethyl-2′ , 6′ -dimethox (1,1′ -biphenyl) -4-propionate (90.1 g) as oil.
The product was dissolved in ethanol (330 ml) , and after addition of p-toluenesulfonic acid monohydrate (28.5 g) , the mixture was stirred for 2 hours at 75 °C. After cooling to room temperature, the mixture was filtrated over charcoal and the filtrate was concentrated under reduced pressure. The residue was dissolved in ethyl acetate with heating. After cooling, the crystalline precipitates were collected by filtration and dried to yield ethyl (αS)-α- amino-4′ -ethoxymethyl-2′ , 6′ -dimethoxy (1, 1′ -biphenyl) -4- propionate p-toluenesulfonate (63.4 g) .
MS (m/z) : 387 (M+-p-toluenesulfonic acid), M.p. 127-129°C
(3) To a mixture of ethyl (αS) -α-amino-4′ -ethoxymethyl- 2′ , 6′ -dimethox (1, 1′ -biphenyl) -4-propionate p- toluenesulfonate (29.0 g) , sodium hydrogen carbonate (15. 2 g) , water (290 ml) and ethyl acetate (290 ml) was added dropwise 2, 6-difluorobenzoyl chloride (9. 6 g) at 15 °C or below and the mixture was stirred for 30 minutes at the same temperature. The ethyl acetate layer was washed with saturated aqueous NaCl solution and dried over magnesium sulfate. The solvent was removed in vacuo. The residue was recrystallized from isopropanol-water to yield ethyl (αS) -oi- [ (2, 6-difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethox (1, 1′ -biphenyl) -4-propionate (26.4 g) . MS (m/z) : 527 (M+) , M.p. 87-89°C (4) To a solution of sodium hydroxide (2.9 g) in water- tetrahydrofuran (317 ml-159 ml) was added ethyl (oιS)-α- [ (2, 6-difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethoxy (1, 1′ -biphenyl) -4-propionate (31.7 g) at 15°C and the mixture was stirred for 4 hours at the same temperature. After neutralizing with IN HC1, the organic solvent was removed in vacuo. The aqueous layer was cooled, the crystalline precipitates were collected by filtration and recrystallized from ethanol-water to yield (αS) -a- [ (2, 6- difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethoxy (1, 1′ -biphenyl) -4-propionic acid (28.8 g) . MS (m/z): 499 (M+) , M.p. 154-155°C
EXAMPLE 2 (1) Under nitrogen atmosphere, a mixture of ethyl (oιS)-o:- [[ (1, 1-dimethylethoxy) carbonyl] amino] -4-bromobenzene propanoate (11.17 g) , 4-ethoxymethyl-2, 6- dimethoxyphenylboronic acid (10.80 g ), palladium acetate (0.34 g), triphenylphosphine (1.57 g) , anhydrous potassium carbonate (12.44 g) , iV-methylpyrrolidone (56 ml) and water (11 ml) was stirred for 50 minutes at 80 °C. After completion of the reaction, the mixture was cooled to room temperature and extracted with ethyl acetate and water. The organic layer was washed with 10% aqueous citric acid solution and saturated aqueous NaCl solution, dried over magnesium sulfate and filtrated. The filtrate was concentrated under reduced pressure to yield ethyl (αS)-α- [ [ (1, 1-dimethylethoxy) carbonyl] amino] -4′ -ethoxymethyl- 2′ , 6′ -dimethox (1, 1′ -biphenyl) -4-propionate (20.4 g) as oil. The product was dissolved in ethanol (100 ml) , and after addition of p-toluenesulfonic acid monohydrate (5.7 g) , the mixture was stirred for 1.5 hours at 75 °C. After cooling, the mixture was filtrated over charcoal and the filtrate was concentrated under reduced pressure. The residue was suspended in toluene with heating. After cooling, the crystalline precipitates were collected by filtration and dried to yield ethyl (αS) – -amino-4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1,1′ -biphenyl) -4-propionate p- toluenesulfonate (13.80 g) . (2) The compound obtained in the above step (1) was treated in the same manner as described in Example 1 (2) to (4) to yield (αS) -a- [ [2 , 6-difluorobenzoyl) amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1, 1′ -biphenyl) -4-propionic acid. The physicochemical data were the same as that obtained in Example 1.
EXAMPLE 3
To a solution of ethyl (αS) -α- [ (2, 6- difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethox (1, 1′ -biphenyl) -4-propionate (500 g ) in water (12.6 ml) and dioxane (50 ml) was added hydrochloric acid (12.4 g) and the mixture was stirred for 60 hours at 60 “C. The organic solvent was removed in vacuo and the aqueous layer was cooled. The crystalline precipitates were collected by filtration and recrystallized from ethanol- water to yield (αS) – – [ (2, 6-difluorobenzoyl) amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1,1′ -biphenyl) -4-propionic acid (426 mg) . The physicochemical data were the same as that obtained in Example 1.
REFERENCE EXAMPLE 1
(1) To a mixture of 4-bromo-3, 5-dimethoxybenzylalcohol (44.5 g) , triethylammonium benzyl chloride (2.05 g) and 20% aqueous sodium hydroxide solution (288 g) was added diethyl sulfate (41.7 g) under ice-cooling, and the mixture was stirred overnight at 25-30 °C. After stirring for 1 hour at 70 °C, the mixture was cooled and extracted with toluene. The toluene layer was washed with water and saturated aqueous NaCl solution and dried over magnesium sulfate. The solvent was removed in vacuo to yield 4-bromo-3, 5- dimethoxybenzyl ethyl ether (49.5 g) as colorless oil. MS (m/z): 276 (M++2) , 274 (M+)
(2) Under nitrogen atmosphere, to a solution of 4-bromo- 3, 5-dimethoxybenzyl ethyl ether (440.0 g) in tetrahydrofuran (4.0 L) was added dropwise n-butyl lithium (1.6 M n-hexane solution, 1.1 L) at -60°C. After stirring for 15 minutes at the same temperature, trimethyl borate (249.3 g) was added. The temperature of the mixture was gradually elevated, followed by stirring for 1 hour under ice-cooling. To the mixture was added dropwise 10% aqueous sulfuric acid solution (835 g ) . The mixture was extracted with ethyl acetate and the organic layer was washed with water and saturated aqueous NaCl solution. After drying over magnesium sulfate, the solvent was removed in vacuo. The residue was dissolved in isopropyl ether with heating and cooled. The crystalline precipitates were collected by filtration and dried to yield 4-ethyoxymethyl-2, 6- dimetoxyphenylboronic acid (312.9 g) . M.p. 59-61°C
REFERENCE EXAMPLE 2
(1) To a suspension of 4-bromo-3, 5-dihydroxybenzoic acid (95.0 kg) in ethyl acetate (950 L) were added anhydrous potassium carbonate (270.8 kg) and dimethyl sulfate (174.7 kg) . The mixture was heated at 50-80 ‘C for about 4 hours and partitioned by adding water. The organic layer was washed with water and saturated aqueous NaCl solution and concentrated under reduced pressure. The residue was suspended into methanol, stirred under heating and cooled. The crystalline precipitates were collected by filtration and dried to yield methyl 4-bromo-3, 5-dimethoxybenzoate (98.8 kg) as pale yellow crystals. MS (m/z): 277 (M++2) , 275 (M+) , M.p. 120-122°C
(2) To a solution of calcium chloride (46.5 kg) in ethanol (336 L) were added tetrahydrofuran (672 L) and methyl 4- bromo-3, 5-dimethoxybenzoate (96.0 kg) to obtain a suspension. To the suspension was added sodium borohydride
(31.7 kg) by portions at room temperature, and the mixture was stirred for about 9 hours at temperature of room temperature to 45 °C. The reaction mixture was added dropwise to aqueous HC1 solution and stirred for about 16 hours at room temperature. Organic solvent was removed in vacuo, and water (1440 L) was added to the residue and stirred for 1 hour at 50 °C. After cooling, the crystalline precipitates were collected by filtration and dried to yield 4-bromo-3, 5-dimethoxybenzyl alcohol (83.3 kg) as colorless crystals. MS (m/z): 249 (M++2), 247 (M+) , M.p. 100-102°C.
INDUSTRIAL APPLICABILITY The process for preparation of the present invention makes it possible to afford a compound of the formula (I) or a pharmaceutically acceptable salt thereof with high- purity, in a high yield and inexpensively, and, therefore, the process of the present invention is industrially very useful.
References
5. Firategrast
| WO2002018320A2 | 27 Ago 2001 | 7 Mar 2002 | Tanabe Seiyaku Co | INHIBITORS OF α4 MEDIATED CELL ADHESION |
| WO2003072536A1 | 27 Fev 2003 | 4 Set 2003 | Tanabe Seiyaku Co | A process for preparing a phenylalanine derivative and intermediates thereof |
| WO2003072537A2 | 6 Fev 2003 | 4 Set 2003 | Abbott Lab | Selective protein tyrosine phosphatatase inhibitors |
Mitsubishi Tanabe Pharma Corporation


Mitsubishi Tanabe Pharma Corporation
Pharmacological research building

 |
||
| ■Mitsubishi Tanabe Pharma Corporation Pharmacological research building |
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
Masitinib

Masitinib
Masitinib; 790299-79-5; Masivet; AB1010; AB-1010;
CLASS:Immunomodulator
TARGET:KIT (a stem cell factor, also called c-KIT) receptor as well as select other tyrosine kinases
STATUS FOR MS:Phase III
COMMERCIAL:Under development by AB Science..Ab Science
4-((4-Methylpiperazin-1-yl)methyl)-N-(4-methyl-3-((4-(pyridin-3-yl)-1,3-thiazol-2-yl)amino)phenyl)benzamide
AB 1010
UNII-M59NC4E26P
4-((4-Methylpiperazin-1-yl)methyl)-N-(4-methyl-3-((4-(pyridin-3-yl)-1,3-thiazol-2-yl)amino)phenyl)benzamide
Regulatory and Commercial Status
STATUS FOR MS:Phase III
HIGHEST STATUS ACHIEVED (FOR ANY CONDITION):
Marketing Authorization Application for the treatment of pancreatic cancer has been filed with the European Medicines Agency (16 October 2012)
Marketing Authorization Application for the conditional approval in the treatment of pancreatic cancer has been accepted by the European Medicines Agency (30 October 2012)
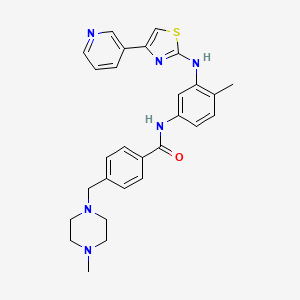
Masitinib is a tyrosine-kinase inhibitor used in the treatment of mast cell tumors in animals, specifically dogs.[1][2] Since its introduction in November 2008 it has been distributed under the commercial name Masivet. It has been available in Europe since the second part of 2009. In the USA it is distributed under the name Kinavet and has been available for veterinaries since 2011.
Masitinib is being studied for several human conditions including cancers. It is used in Europe to fight orphan diseases.[3]
Mechanism of action
Masitinib inhibits the receptor tyrosine kinase c-Kit which is displayed by various types of tumour.[2] It also inhibits the platelet derived growth factor receptor (PDGFR) and fibroblast growth factor receptor (FGFR).

http://www.google.com/patents/WO2012136732A1?cl=en
In a preferred embodiment of the above-depicted treatment, the active ingredient masitinib is administered in the form of masitinib mesilate; which is the orally bioavailable mesylate salt of masitinib – CAS 1048007-93-7 (MsOH); C28H30N6OS.CH3SO3H; MW 594.76:

http://www.google.com/patents/WO2004014903A1?cl=en

003 : 4-(4-Methyl-piperazin-l-ylmethyl)-N-[3-(4-pyridin-3-yl-thiazol-2-ylamino)- phenyl] -benzamide
4-(4-Methyl-piperazin-l-yl)-N-[4-methyl-3-(4-pyridin-3-yl-thiazol-2-ylmethyl)- phenyl] -benzamide

beige brown powder mp : 128-130°C
1H RMN (DMSO-d6) δ = 2.15 (s, 3H) ; 2.18 (s, 3H) ; 2.35-2.41 (m, 4H) ; 3.18-3.3.24 (m, 4H) ; 6.94 (d, J = 8.9 Hz, 2H) ; 7.09 (d, J = 8.4 Hz, IH) ; 7.28-7.38 (m, 3H) ; 7.81 (d, J = 8.9 Hz, 2H) ; 8.20-8.25 (m, IH) ; 8.40 (dd, J = 1.6 Hz, J = 4.7 , IH) ; 8.48 (d, J = 1.9 Hz, IH) ; 9.07 (d, J = 1.5 Hz, IH) ; 9.35 (s, IH) ; 9.84 (s, IH)

……………
http://www.google.com/patents/WO2008098949A2?cl=en
EXAMPLE 4 N- [4-Methyl-3 -(4-pyridin-3 -yl-thiazol-2-ylamino)-phenyl] -benzamide derivatives
Method A In a reactor and under low nitrogen pressure, add 4-Methyl-N3-(4-pyridin-3-yl-thiazol- 2-yl)-benzene-l,3-diamine (95 g, 336.45 mmol), dichloromethane (2 L). To this suspension cooled to temperature of 5°C was added dropwise 2M/n-hexane solution of trimethylaluminium (588 mL). The reaction mixture was brought progressively to 15°C, and maintained for 2 h under stirring. 4-(4-Methyl-piperazin-l-ylmethyl)-benzoic acid methyl ester (100 g, 402.71 mmol) in dichloromethane (200 mL) was added for 10 minutes. After 1 h stirring at room temperature, the reaction mixture was heated to reflux for 20 h and cooled to room temperature. This solution was transferred dropwise via a cannula to a reactor containing 2N NaOH (2.1 L) cooled to 5°C. After stirring for 3 h at room temperature, the precipitate was filtered through Celite. The solution was extracted with dichloromethane and the organic layer was washed with water and saturated sodium chloride solution, dried over MgSO4 and concentrated under vacuum. The brown solid obtained was recrystallized from /-Pr2O to give 130.7 g (78%) of a beige powder.
Method B Preparation of the acid chloride
To a mixture of 4-(4-Methyl-piperazin-l-ylmethyl)-benzoic acid dihydrochloride (1.0 eq), dichloromethane (7 vol) and triethylamine (2.15 eq), thionyl chloride (1.2 eq) was added at 18-28°C . The reaction mixture was stirred at 28-32°C for 1 hour. Coupling of acid chloride with amino thiazole To a chilled (0-50C) suspension of 4-Methyl-N3-(4-pyridin-3-yl-thiazol-2-yl)-benzene- 1,3-diamine (0.8 eq) and thiethylamine (2.2 eq) in dichloromethane (3 vol), the acid chloride solution (prepared above) was maintaining the temperature below 5°C. The reaction mixture was warmed to 25-300C and stirred at the same temperature for 1O h. Methanol (2 vol) and water (5 vol) were added to the reaction mixture and stirred. After separating the layers, methanol (2 vol), dihloromethane (5 vol) and sodium hydroxide solution (aqueous, 10%, till pH was 9.5-10.0) were added to the aqueous layer and stirred for 10 minutes. The layers were separated. The organic layer was a washed with water and saturated sodium chloride solution. The organic layer was concentrated and ethanol (2 vol) was added and stirred. The mixture was concentrated. Ethanol was added to the residue and stirred. The product was filtered and dried at 50-550C in a vaccum tray drier. Yield = 65-75%.
Method C
To a solution of 4-methyl-N3-(4-pyridin-3-yl-thiazol-2-yl)-benzene-l,3-diamine (1.0 eq) in DMF (20 vol) were added successively triethylamine (5 eq), 2-chloro-l- methylpyridinium iodide (2 eq) and 4-(4-methyl-piperazin-l-ylmethyl)-benzoic acid (2 eq). The reaction mixture was stirred for 7 h at room temperature. Then, the mixture was diluted in diethyl ether and washed with water and saturated aqueous NaHCO3, dried over Na2SO4 and concentrated. The crude product was purified by column chromatography using an elution of 100% EtOAc to give a yellow solid.
Yield = 51%.
1H NMR (CDCl3) : δ = 9.09 (IH, s, NH); 8.52 (IH, br s); 8.27 (IH, s); 8.13 (IH, s);
8.03 (IH, s); 7.85 (2H, d, J= 8.3Hz); 7.45 (2H, m); 7.21-7.38 (4H, m); 6.89 (IH, s);
3.56 (2H, s); 2.50 (8H, br s); 2.31 (6H, br s).
MS (CI) m/z = 499 (M+H)+.
An additional aspect of the present invention relates to a particular polymorph of the methanesulfonic acid salt of N-[4-Methyl-3-(4-pyridin-3-yl-thiazol-2-ylamino)-phenyl]- benzamide of formula (IX).

(VI)
Hereinafter is described the polymorph form of (IX) which has the most advantageous properties concerning processability, storage and formulation. For example, this form remains, dry at 80% relative humidity and thermodynamically stable at temperatures below 2000C.
The polymorph of this form is characterized by an X-ray diffraction pattern illustrated in FIG.I, comprising characteristic peaks approximately 7.269, 9.120, 11.038, 13.704, 14.481, 15.483, 15.870, 16.718, 17.087, 17.473, 18.224, 19.248, 19.441, 19.940, 20.441, 21.469, 21.750, 22.111, 23.319, 23.763, 24.120, 24.681, 25.754, 26.777, 28.975, 29.609, 30.073 degrees θ, and is also characterized by differential scanning calorimetry (DSC) illustrated in FIG.II, which exhibit a single maximum value at approximately 237.49 ± 0.3 0C. X-ray diffraction pattern is measured using a Bruker AXS (D8 advance). Differential scanning calorimetry (DSC) is measured using a Perking Elmer Precisely (Diamond DSC).
This polymorph form can be obtained by treatement of 4-(4-Methyl-piperazin-l- ylmethyl)-N-[4-methyl-3-(4-pyridin-3-yl-thiazol-2-ylamino)-phenyl]-benzamide with 1.0 to 1.2 equivalent of methanesulfonic acid, at a suitable temperature, preferably between 20-800C.
The reaction is performed in a suitable solvent especially polar solvent such as methanol or ethanol, or ketone such as acetone, or ether such as diethylether or dioxane, or a mixture therof. This invention is explained in example given below which is provided by way of illustration only and therefore should not be construed to limit the scope of the invention. Preparation of the above-mentioned polymorph form of 4-(4-Methyl-piperazin-l- ylmethyl)-N- [4-methyl-3 -(4-pyridin-3 -yl-thiazol-2-ylamino)-phenyl] -benzamide methanesulfonate .
4-(4-Methyl-piperazin- 1 -ylmethyl)-N- [4-methyl-3 -(4-pyridin-3 -yl-thiazol-2-ylamino) phenyl] -benzamide (1.0 eq) was dissolved in ethanol (4.5 vol) at 65-700C. Methanesulfonic acid (1.0 eq) was added slowly at the same temperature. The mixture was cooled to 25-300C and maintained for 6 h. The product was filtered and dried in a vacuum tray drier at 55-600C. Yield = 85-90%. Starting melting point Smp = 236°C.
References
- Hahn, K.A.; Oglivie, G.; Rusk, T.; Devauchelle, P.; Leblanc, A.; Legendre, A.; Powers, B.; Leventhal, P.S.; Kinet, J.-P.; Palmerini, F.; Dubreuil, P.; Moussy, A.; Hermine, O. (2008). “Masitinib is Safe and Effective for the Treatment of Canine Mast Cell Tumors”. Journal of Veterinary Internal Medicine 22 (6): 1301–1309. doi:10.1111/j.1939-1676.2008.0190.x. ISSN 0891-6640.
- Information about Masivet at the European pharmacy agency website
- Orphan designation for Masitinib at the European pharmacy agency website
| WO2004014903A1 | Jul 31, 2003 | Feb 19, 2004 | Ab Science | 2-(3-aminoaryl)amino-4-aryl-thiazoles and their use as c-kit inhibitors |
| WO2008098949A2 | Feb 13, 2008 | Aug 21, 2008 | Ab Science | Process for the synthesis of 2-aminothiazole compounds as kinase inhibitors |
| EP1525200B1 | Jul 31, 2003 | Oct 10, 2007 | AB Science | 2-(3-aminoaryl)amino-4-aryl-thiazoles and their use as c-kit inhibitors |
| US7423055 | Aug 1, 2003 | Sep 9, 2008 | Ab Science | 2-(3-Aminoaryl)amino-4-aryl-thiazoles for the treatment of diseases |
| US20080207572 * | Jul 13, 2006 | Aug 28, 2008 | Ab Science | Use of Dual C-Kit/Fgfr3 Inhibitors for Treating Multiple Myeloma |
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 4-[(4-Methylpiperazin-1-yl)methyl]-N-(4-methyl-3-{[4-(pyridin-3-yl)-1,3-thiazol-2-yl]amino}phenyl)benzamide | |
| Clinical data | |
| Trade names | Masivet, Kinavet |
| AHFS/Drugs.com | International Drug Names |
| Identifiers | |
| 790299-79-5 | |
| L01XE22 | |
| PubChem | CID 10074640 |
| ChemSpider | 8250179 |
| ChEMBL | CHEMBL1908391 |
| Chemical data | |
| Formula | C28H30N6OS |
| 498.64 g/mol | |
| Patent | Submitted | Granted |
|---|---|---|
| 2-(3-Aminoaryl)amino-4-aryl-thiazoles for the treatment of diseases [US7423055] | 2004-06-10 | 2008-09-09 |
| 2-(3-aminoaryl)amino-4-aryl-thiazoles and their use as c-kit inhibitors [US2005239852] | 2005-10-27 | |
| Use of C-Kit Inhibitors for Treating Fibrosis [US2007225293] | 2007-09-27 | |
| Use of Mast Cells Inhibitors for Treating Patients Exposed to Chemical or Biological Weapons [US2007249628] | 2007-10-25 | |
| Use of c-kit inhibitors for treating type II diabetes [US2007032521] | 2007-02-08 | |
| Use of tyrosine kinase inhibitors for treating cerebral ischemia [US2007191267] | 2007-08-16 | |
| Use of C-Kit Inhibitors for Treating Plasmodium Related Diseases [US2008004279] | 2008-01-03 | |
| Tailored Treatment Suitable for Different Forms of Mastocytosis [US2008025916] | 2008-01-31 | |
| 2-(3-AMINOARYL) AMINO-4-ARYL-THIAZOLES AND THEIR USE AS C-KIT INHIBITORS [US2008255141] | 2008-10-16 | |
| Use Of C-Kit Inhibitors For Treating Inflammatory Muscle Disorders Including Myositis And Muscular Dystrophy [US2008146585] | 2008-06-19 |
| Patent | Submitted | Granted |
|---|---|---|
| Aminothiazole compounds as kinase inhibitors and methods of using the same [US8940894] | 2013-05-10 | 2015-01-27 |
| Aminothiazole compounds as kinase inhibitors and methods of using the same [US8492545] | 2012-03-08 | 2013-07-23 |
| Patent | Submitted | Granted |
|---|---|---|
| Use of Dual C-Kit/Fgfr3 Inhibitors for Treating Multiple Myeloma [US2008207572] | 2008-08-28 | |
| PROCESS FOR THE SYNTHESIS OF 2-AMINOTHIAZOLE COMPOUNDS AS KINASE INHIBITORS [US8153792] | 2010-05-13 | 2012-04-10 |
| COMBINATION TREATMENT OF SOLID CANCERS WITH ANTIMETABOLITES AND TYROSINE KINASE INHIBITORS [US8227470] | 2010-04-15 | 2012-07-24 |
| Anti-IGF antibodies [US8580254] | 2008-06-19 | 2013-11-12 |
| COMBINATIONS FOR THE TREATMENT OF B-CELL PROLIFERATIVE DISORDERS [US2009047243] | 2008-07-17 | 2009-02-19 |
| TREATMENTS OF B-CELL PROLIFERATIVE DISORDERS [US2009053168] | 2008-07-17 | 2009-02-26 |
| Anti-IGF antibodies [US8318159] | 2009-12-11 | 2012-11-27 |
| SURFACE TOPOGRAPHIES FOR NON-TOXIC BIOADHESION CONTROL [US2010226943] | 2009-08-31 | 2010-09-09 |
| EGFR/NEDD9/TGF-BETA INTERACTOME AND METHODS OF USE THEREOF FOR THE IDENTIFICATION OF AGENTS HAVING EFFICACY IN THE TREATMENT OF HYPERPROLIFERATIVE DISORDERS [US2010239656] | 2010-05-10 | 2010-09-23 |
| ANTI CD37 ANTIBODIES [US2010189722] | 2008-08-08 | 2010-07-29 |
Note: Compound name must be entered under “Substance Identification” and then “Names and Synonyms” selected to view synonyms.
Editors’ Pick
3, Avenue George V
75008 PARIS – FRANCE
Tel. : +33 (0)1 47 20 00 14
Fax. : +33 (0)1 47 20 24 11
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
Flupirtine Revisited

Flupirtine, D 9998
2-amino-6-(4-fluoro-benzylamino)- pyridin-3-yl)-carbamic acid ethyl ester, is unique as a non-opioid, non-NSAID, non-steroidal analgesic with a favorable tolerability. It first became available in Europe in 1984, and was sold mainly under the names Katadolon, Trancolong, Awegal, Efiret, Trancopal Dolo, and Metanor
PHASE 2
MS
- Neuronal potassium channels (7)
- Membrane resting potential (6)
- NMDA receptor channels (indirectly)(14)
- Originally developed by Asta Medica (1) (4)
- Being developed and commercialized to treat fibromyalgia by Synthetic Biologics (1)
Flupirtine

75507-68-5 maleate
56995-20-1 (free base)
56995-20-1 (free base)
| LAUNCHED | 1986 NEUROPATHIC PAIN |
Flupirtine maleate is the INN for 2-amino-3-ethylcarbamato-6- (4-fluoro-benzylamino) maleate, CAS: 75507-68-5, molar mass 420.40 g / mol, molecular formula C1 5 H17FN4O2 • C4H4O4, and corresponds to the structure of formula I.

Flupirtine maleate is used, for example, under the trade name Katadolon® as an analgesic.
56995-20-1
CAS Name: [2-Amino-6-[[(4-fluorophenyl)methyl]amino]-3-pyridinyl]carbamic acid ethyl ester
Additional Names: 2-amino-6-[(p-fluorobenzyl)amino]-3-pyridinecarbamic acid ethyl ester
Trademarks: D-9998
Molecular Formula: C15H17FN4O2
Molecular Weight: 304.32
Percent Composition: C 59.20%, H 5.63%, F 6.24%, N 18.41%, O 10.51%
Properties: Crystals from isopropanol, mp 115-116°. 5% ethanol soln is colorless, turns green on exposure to air for 20 hours. LD50 orally in mice, rats: 617, 1660 mg/kg (Jakovlev).
Melting point: mp 115-116°
Toxicity data: LD50 orally in mice, rats: 617, 1660 mg/kg (Jakovlev)
Derivative Type: Hydrochloride
Molecular Formula: C15H17FN4O2.HCl
Molecular Weight: 340.78
Percent Composition: C 52.87%, H 5.32%, F 5.57%, N 16.44%, O 9.39%, Cl 10.40%
Properties: Crystals from water, mp 214-215°. When prepd industrially contains intensely blue by-product.
Melting point: mp 214-215°
Derivative Type: Maleate
CAS Registry Number: 75507-68-5
Trademarks: Katadolon (AWD)
Molecular Formula: C15H17FN4O2.C4H4O4
Molecular Weight: 420.39
Percent Composition: C 54.28%, H 5.04%, F 4.52%, N 13.33%, O 22.84%
Properties: Colorless crystals from isopropanol, mp 175.5-176°. Formed as mixture of two crystalline forms A and B; mixtures containing 60-90% A are preferred.
Melting point: mp 175.5-176°
Therap-Cat: Analgesic.
TARGET:
Neuronal potassium channels
Membrane resting potential
NMDA receptor channels (indirectly)
STATUS FOR MS:
Phase II
COMMERCIAL:
Originally developed by Asta Medica
Being developed and commercialized to treat fibromyalgia by Synthetic Biologics
Marketed for pain indications in various European countries by Meda
TRADE NAME:
Effirma (US)
Katadolon (Brazil, Germany, Latvia, Estonia, Slovakia, Lithiania, Russian Federation)
SYNONYMS:
EINECS 260-503-8,UNII-MOH3ET196H, Effirma (US), Katadolon (Brazil, Germany, Latvia, Estonia, Slovakia, Lithiania, Russian Federation)
SYSTEMATIC NAME:
Carbamic acid, (2-amino-6-(((4-fluorophenyl)methyl)amino)-3-pyridinyl)-, ethyl ester
PROPERTIES:
Molecular weight: 304
MECHANISMS/EFFECTS
HUMAN:
Stabilizes membrane resting potential by activating neuronal Kv7 potassium channels
Indirectly antagonizes NMDA receptors
Reduces muscle spasticity in humans
Prevents apoptosis and reduced formation of reactive oxygen species by in cultured human retinal pigment epithelial cells

Structures of flupirtine, D13223, and retigabine.
Regulatory and Commercial Status
STATUS FOR MS:
Phase II
HIGHEST STATUS ACHIEVED (FOR ANY CONDITION):
Approved in Europe
ADMINISTRATION:
Oral
COMMERCIAL:
Originally developed by Asta Medica
Being developed and commercialized to treat fibromyalgia by Synthetic Biologics
Marketed for pain indications in various European countries by Meda
Flupirtine is an aminopyridine that functions as a centrally acting non-opioid analgesic. It first became available in Europe in 1984, and is sold mainly under the names Katadolon, Trancolong, Awegal, Efiret, Trancopal Dolo, and Metanor.[5] Flupirtine is sold by Intas Pharma under the brand name Pruf in India. Like nefopam, it is unique among analgesics in that it is a non-opioid, non-NSAID, non-steroidal centrally acting analgesic. In 2010 the chemically related drug (the difference being that the pyridine group in flupirtine is replaced with a phenyl group) retigabine (INN; ezogabine [USAN]) was approved by the FDA as an anticonvulsant for the treatment of refractory partial-onset seizures in treatment-experienced patients.[6] Retigabine also works by opening the neuronal KCNQ/Kv7 potassium channel, just like flupirtine.
History
Flupirtine was originally developed by Asta Medica, with the synthesis of the compound and the development of the drug described in patents from the 1970s to the 2000s.[7][8][9][10][11][12]
It was approved for the treatment of pain in 1984 in Europe. However, it has never been introduced to the United States market for any indication. In 2008, Adeona Pharmaceuticals, Inc. (now called Synthetic Biologics, Inc.) obtained an option to license issued and patent pending applications relating to flupirtine’s use in the treatment of ophthalmic indications, particularly retinitis pigmentosa.[13]
Mechanism of Action
Flupirtine is a selective neuronal potassium channel opener that also has NMDA receptor antagonist and GABAA receptor modulatory properties.[14]
Uses
Flupirtine is used as an analgesic for acute and chronic pain, in moderate-to-severe cases.[15] Its muscle relaxant properties make it popular for back pain and other orthopedic uses, but it is also used for migraines, in oncology, postoperative care, and gynecology.
Flupirtine has been noted for its neuroprotective properties, and it is being investigated for possible use in Creutzfeldt–Jakob disease, Alzheimer’s disease, and multiple sclerosis.[16][17] It has also been proposed as a possible treatment for Batten disease.[18]
Flupirtine underwent a clinical trial as a treatment for multiple sclerosis[19] and fibromyalgia.[20] Flupirtine showed promise for fibromyalgia due to its different action than the three approved by U.S. FDA drugs: Lyrica (pregabalin), Savella (milnacipran), and Cymbalta (duloxetine).[21] Additionally, there are case reports regarding flupirtine as a treatment for fibromyalgia.[22] Adeona Pharmaceuticals (now called Synthetic Biologics) sub-licensed its patents for using flupirtine for fibromyalgia to Meda AB in May 2010.[21]
Side Effects
The most serious side effect is frequent hepatotoxicity which prompted regulatory agencies to issue several warnings and restrictions.[23][24]
Flupirtine is devoid of negative psychological or motor function effects, or effects on reproductive function.[25][26]
Abuse and Dependence
Although some studies have reported flupirtine has no addictive properties,[27][28] there was suggestion that it may possess some abuse potential and liability.[29] There were at least two registered cases of flupirtine abuse.[30] Drug tolerance does not develop in most cases; however, tolerance may develop in single cases.[30]
Flupirtine is 2-amino-3-carbethoxyamino-6-(p-fluorobenzylamino) pyridine; CAS No: 56995-20-1 , an aminopyridine that functions as a centrally acting non-opioid analgesic. It first became available in Europe in 1984, and is sold mainly under the names atadolon, Trancolong, Awegal, Efiret, Trancopal Dolo, and Metanor. It is unique as a non- opioid, non-NSAID, non-steroidal analgesic. Flupirtine is used as an analgesic for acute and chronic pain, in moderate to severe cases. Its muscle relaxant properties make it popular for back pain and other orthopaedic uses, but it is also used for migraines, in oncology, postoperative care, and gynaecology. Flupirtine has been noted for its neuro-protective properties, as well as its possible uses for Creutzfeld- Jakob disease, Alzheimer’s disease, and multiple sclerosis are being investigated. It has also been proposed as a possible treatment for Batten disease. Flupirtine also acts as an antioxidant and prevent free radical- mediated structural damage.
US3481943 (hereinafter referred as ‘943) discloses the process for the preparation of flupirtine hydrochloride of formula (T) wherein p- fluorobenzylamine (formula R) is reacted with 2-amino-3-nitro-6- chloropyridine (Q) in n-propanol using potassium carbonate to prepare 2-amino-3-nitro-6-p-fluorobenzylamino-pyridine of formula (S) which is hydrogenated in dioxane using raney nickel at 50 C under a gauge pressure of 30 atmospheres. Solution is filtered off to remove the catalyst and then reacted with chloroformic acid ethyl ester (ethyl chloroformate) while stirring. The product is filtered off and recrystallized from water to give flupirtine hydrochloride salt of formula (T). The process disclosed therein in ‘943 is depicted as given below

Drawbacks associated with the process disclosed in ‘943 are:
1) The yield of 2-amino-3-nitro-6-p-fluorobenzylamino-pyridine of formula S obtained is around 40% only. ‘943 does not disclose the preparation of maleate salt of flupirtine.
2) During the preparation hydrochloride salt of flupirtine on an industrial scale, intensely blue colored by products are formed which are either difficult to remove or can not be removed completely.
3) Use of n-propanol as reaction solvent is expensive. reaction mass thereby hindering the progress of the reaction. Another most probable reason attributed for getting poor yield of 40% in the said process could be masking of hydrochlorides of both the reactants of formulae (Q’) and (R’) (as both reactants are amino compounds and form hydrochlorides) over potassium carbonate making it unavailable for further reaction posing problem towards the completion of reaction thereby adversely affecting the yield.

DE3133519 (US4481205) discloses the preparation of flupirtine maleate of formula (IA), wherein 2-amino-3-nitro-6-chloro-pyridine of formula (S) is prepared by taking 2,6-dichloro-3-nitropyridine of formula (P) (90%, water wet) in isopropanol at 20°-30°C and purging ammonia gas (or dropping liquid ammonia) into the said reaction mixture and then resulting 2-amino-3-nitro-6-chloro-pyridine of formula (Q) is reacted with p-fluorobenzylamine (R) in isopropanol using triethyl amine as a base ; the reaction mixture is refluxed for 6 hours. Thereupon after addition of a large volume of water the compound 2-amino-3-nitro-6-(p- fluorobenzylamino)-pyridine of formula (S) precipitates.
2-amino-3-nitro-6-(4-fluorobenzylamino) pyridine of formula (S) is then hydrogenated in the presence of raney nickel at 5 bar at 60°C to give 2,3- diamino-6-(4-fluorobenzylamino) pyridine using 2-methoxy ethanol as hydrogenating solvent. This is followed by acylation with ethyl chloroformate using triethylamine as a base under inert gas atmosphere to obtain flupirtine base of formula (I). The catalyst is filtered off and filtrate containing dissolved triethyi amine hydrochloride is directly added to solution of maleic acid in isopropanol resulting into formation of crude flupirtine maleate (IA). It also discloses the importance of the exclusion of atmospheric oxygen by an intensive supply of inert gas and closed reactor system to avoid development of troublesome coloured complexes.
The purification of crude flupirtine maleate is carried out by converting crude flupirtine maleate into crude flupirtine base by contacting with ammonia or sodium hydroxide solution. Then the crude flupirtine base is recrystallized from isopropanol and, after contacting with activated carbon/kieselguh’r, it is reacted with a solution of maleic acid in isopropanol to give flupirtine maleate of formula (IA). The reaction scheme of DE3133519 is depicted herein below.

Drawbacks associated with the process disclosed in DE3133519 (US4481205) are:
1) Use of gaseous ammonia or liquid ammonia for the preparation of 2-amino-3-nitro-6-chloro-pyridine of formula (Q) starting from 2, 6- dichloro-3-nitropyridine of formula (P) contributes towards increased level of impurities of formulae X and Y as the gaseous ammonia and liquid ammonia as sources of ammonia are in concentrated forms and it is not easy to control the purging or addition in appropriate quantities and as a consequence it results in the formation of higher amounts of impurities and poor yield of the desired compound.

Another disadvantage of using ammonia gas is that it is classified as a hazardous material and is subject to strict regulations and risk management procedures for transport, storage, and handling. These requirements result in additional costs and may generate local community concerns over transporting and storing hazardous materials. While aqueous ammonia used by the inventors requires minimal special handling, social and regulatory requirements.
2) Preparation of 2-amino-3-nitro-6-(p-fluorobenzylamino)-pyridine of formula (S comprises reaction between 2-amino-3-nitro-6-chloro- pyridine of formula (Q) and p-fluorobenzylamine of formula (R) using isopropanol as solvent and triethyl amine as base. To induce separation of 2-amino-3-nitro-6-(p-fluorobenzylamino)-pyridine of formula (S from the reaction mixture in IP A a large volume of water is required which makes reaction mass highly voluminous therefore, not preferred at industrial scale. 3) Basification of crude flupirtine maleate comprising the process of liberating free flupirtine base using ammonia or sodium hydroxide produces an ammonium or sodium salt which pollutes the water.
4) Use of activated charcoal and kieselgulir during the purification of flupirtine base (that contains three amino groups known for their light and colour sensitive nature) takes prolonged time for filtration through filtering bed thereby exposing to environment producing high coloration.
5) The crude flupirtine maleate remains trapped with triethyl amine hydrochloride.
US59591 15A (hereinafter referred as Ί 15) discloses a process for the preparation of flupirtine maleate (IA) as discussed under DE3133519 (US4481205). It also discloses crystalline form “A” of flupirtine maleate by the use of water soluble alcohols (such as ethanol or isopropanol) during synthesis and/or purification. There are three proposed variants in Ί 15 as shown below: process variant:

A: ANFP (S)→hydrogenation→acylation→crude flupirtine base.
B: crude flupirtine base→maleic acid→crude flupirtine maleate
C-E (as shown in scheme-II): not applicable F: crude maleate→pure maleate.
1 s process variant comprises synthesis of oxygen sensitive crude base in situ in process step A and it was converted by a “very rapid” suction filtration process into an aqueous maleic acid solution from which coloured crude flupirtine maleate (IA) is obtained, which is to be purified by recrystallization from isopropanol-water.
2″ process variant:

A: ANFP (S)→hydrogenation→acylation→crude flupirtine base.
B: flupirtine base→maleic acid→crude flupirtine maleate.
C-F (as shown in scheme-II): Not applicable.
G: without isolation of the crude maleate→pure maleate.
As compared to the process step F in 1st variant, process step G in 2nd variant represents substantially shorter alternative process in which the precipitation of crude flupirtine maleate from the flupirtine base formed in situ in isopropanol is effected by Alteration with suction into an aqueous maleic acid solution at 50-60°C and, after that without isolation of the crude maleate, colourless pure material is obtained.
3rd process variant:

A: ANFP (S)→hydrogenation→acylation→cmde flupirtine base (isolated)
B: pure flupirtine base→maleic acid→pure flupirtine maleate.
Herein, after acylation, the flupirtine base (I) is precipitated preferably in ethanol or water and is purified by recrystallization and than treated with maleic acid to prepare pure flupirtine maleate (IA).
Ί 15 disclose hydrogenation of ANFP (S), acylation and precipitation in water-soluble alcohols, such as ethanol or isopropanol.
1) In 1st process variant “very rapid” suction filtration process is a great limitation at plant scale.
2) 2nd process variant also does not produce colorless pure maleate.
3) In 3 process variant, after acylation, the flupirtine base is precipitated preferably in ethanol or water and is purified by recrystallization and than treated with maleic acid to prepare pure flupirtine maleate salt (IA).

It also discloses that although the treatment of final product with activated carbon and recrystallization is known as a reasonably successful procedure to remove impurities. This approach is reluctantly accepted because of the losses in overall yield as it is applied in the last production step of a drug and particularly in the case of flupirtine, it is not a preferred/desirable procedure as it may result into the formation of colored impurities.
US47851 10A discloses a process for the preparation of 2-amino-3-nitro- 6-fluorobenzylamino pyridine of formula (S) comprising reaction of 2- amino-3-nitro-6-methoxypyridine of formula (T) (1 mole) with 4-fluoro- benzylamine of formula R (2-4 mole) optionally as a mineral acid salt in water at a temperature between 70°C and 150°C; preferably between 90° and 120°C. The said condensation is also performed in autoclave as the temperature is above 100°C.It also discloses the necessity of using basic material suitably as an aqueous solution in case when acid addition salts of 4-fluoro-benzylamine of formula (R) is used to liberate the free base for the reaction. It also discloses subsequent reduction of nitro group of 2-amino-3-nitro-6-methoxypyridine by various modes with preference to catalytic hydrogenation optionally in the presence of carriers selected from barium sulphate, calcium sulphate, magnesium sulphate, sodium sulphate etc.

The drawbacks associated with the process described in US47851 10A are: 1) As per the experimental section of the said process of condensation for the preparation of 2-amino-3-nitro-6-fluorobenzylamino pyridine of formula (S) discloses heating at boiling for ten hours. The temperature would be around 100°C as water is used as solvent. However, inventors of the subject invention disclose herein the same process comprising using 6-chlorpyridine instead of 6-methoxy pyridine and water as solvent’, wherein the reaction is carried out at temperature much below boiling point of water and reaction gets completed in 3 hrs compare to 10 hrs at temperature of boiling water as in’ 1 10. Furthermore, the said reaction disclosed herein in the present invention does not require autoclave. There is no teaching or anticipation on this aspect from Ί 10.
2) Excessive use of 2-4 moles of 4-fluoro-benzylamine of formula ( ) for the preparation of 2-amino-3-nitro-6-fluorobenzylamino pyridine of the formula (S) comprising the reaction of 2-amino-3-nitro-6- methoxypyridine of formula (T)with 4-fluoro-benzylamine of formula (R).Unreacted 4-fluoro-benzylamine is then removed by steam distillation which is not only time and energy consuming but also increase in an extra unit operation.
3) In case when acid addition salts of 4-fluoro-benzylamine are used that requires another additional operation of basification to liberate free base to enable 4-fluoro-benzylamine to be available to react further with 2- amino-3-nitro-6-methoxypyridine forming 2-amino-3-nitro-6- fluorobenzylamino pyridine of the formula (S)
DE 31 33 519 describes a process for the preparation of flupirtine maleate as a mixture of polymorphic forms A and B, wherein A is present in a proportion> 60%. The key reaction steps are the hydrogenation of 2-amino-6- (4-fluorobenzylamino) -3-nitropyridine (Formula II) shown in Figure 1, hereinafter also referred to as ANFP, by means of Ra-Nickel for 2,3-diamino- 6- (4-fluoro-benzylamino) -pyridine (Formula III) and subsequent regioselective acylation with chloroformate for free flupirtine base. By precipitation as maleate to blue contaminants that are incurred in the production of HCl salt, are eliminated. Purification of flupirtine maleate is obtained as maleate by releasing the base from the maleate, treatment with activated carbon and reprecipitation. Despite this lengthy and economically expensive purification strategy traces of colored impurities can be difficult to remove.
In WO 98/47872 a process for the preparation of flupirtine maleate is described, in which, in water-soluble alcohols (IPA) is carried out. There are three proposed variants. Option 1 includes an implementation of ANFP to Ra-nickel in the IPA is directly attached to the acylation and the precipitation of a product by Rohmaleat called “very fast” extraction process in an aqueous solution of maleic acid. It falls on a colored Rohmaleat which is to be purified by recrystallization from isopropanol / water. However, the enactment of this variant in the laboratory showed a colored product. In variant 2 should already be colorless an image obtained by aspiration in 50 to 60 0 C warm maleic Rohmaleat. This also could not be confirmed. According to the third variant, the Flupirtinbase formed after acylation is not converted in situ but precipitated in ethanol or water and recrystallized before further reaction with maleic acid. Even with the procedure referred to in this document is a pure white flupirtine maleate is not readily available.
………………….
PATENT
http://www.google.com.tr/patents/WO2010136113A1?cl=en&hl=tr
Example 3 Preparation of flupirtine maleate
50 g of 2-amino-6- (4-fluorobenzylamino) -3-nitropyridine, 2.5 g of palladium on activated carbon and 267 g of isopropanol were hydrogenated with hydrogen at 4.5 bar and 70 0 C. After completion of the reaction was additionally hydrogenated for 8 hours at 70 0 C. Then 20.2 g of ethyl chloroformate, 24.8 g of triethylamine and 4.96 g of ethyl chloroformate at 20 0 C was added. Thereafter, the reaction mixture was stirred for 1.5 h at 55 0 C. It was then filtered at room temperature. The filtrate was then added to a solution of 35.6 g of maleic acid in 1000 g of water at room temperature slowly. The resulting suspension was stirred for 1 h at room temperature. The precipitate was filtered off and washed with water and isopropanol. Dried filter cake (HPLC purity 91.5%) was dissolved in 828 g of isopropanol / water mixture (mass ratio 5.3: 1), and heated to 70 0 C. The resulting clear solution was cooled to room temperature and stirred at room temperature. The precipitate was filtered off and washed with isopropanol / water mixture. The filter cake was dried at 50 0 C. 43 g flupirtine maleate (HPLC purity 97.8%) was obtained as a white-gray solid. The yield was 55%.
………………
PATENT
http://www.google.com/patents/WO2013080215A1?cl=en
The invention relates to an improved process for the preparation of flupirtine of formula (I) and its pharmaceutically acceptable salts, particularly flupirtine maleate of formula (IA) preferably pure crystal modification A of flupirtine maleate.
A process for the preparation of the compound of formula (I)

and pharmaceutically acceptable acid addition salts thereof comprising the steps of:
(a) contacting 2, 6-dichloro-3-nitro pyridine of formula (P) with aqueous ammonia solution in a compatible solvent to produce 2-amino-3-nitro-6- chloro-pyridine of formula (Q);

(b) contacting said compound of formula (Q) with p-fluorobenzylamine taking water as a solvent in presence of a base to produce 2-amino-3- nitro-6-p-fluorobenzylamino-pyridine of formula (S);

(c) reducing nitro group of 2-amino-3-nitro-6-p-fluorobenzylamino- pyridine of formula (S) in a solvent base combination as solvent system in the presence of a catalyst;

(d) contacting 2,3-diamino-6-p-fluorobenzyl amino pyridine produced in step c with an ethyl chloroformate in presence of a base optionally insitu without isolation to produce flupritine base of formula (I);

(e) contacting the said flupritine base of formula (I) with acid solution to produce corresponding acid addition salt.



Scheme (I):


EXAMPLE 1 : Preparation of 2-amino-3-nitro-6-chloro-pyridine.
A solution of 100 gm. 2, 6-dichloro-3-nitro-pyridine in 800 ml isopropyl alcohol is taken in round bottom flask. 300 ml of aqueous ammonia solution (20-25%) is added at 20-25°C. The reaction mass is stirred for 20-24 hours at 20-25°C. After completion of the reaction
The solid is filtered and washed with 100 ml isopropyl alcohol then dried to obtain 70-75 gm 2-amino-3-nitro-6-chloro-pyridine.
EXAMPLE 2: Preparation of 2-amino-3-nitro-6-p-fluorobenzylamino- pyridine.
100 gm of 2-amino-3-nitro-6-chloro-pyridine is taken in 800 ml of water. 90 gm of p-fluorobenzylamine is added dropwise into the reaction mixture at 20-25°C. Then 87 gm triethylamine is also added dropwise into the reaction mixture at 20-25°C. After complete addition, the reaction mass is stirred at 40-45°C for half an hour again the reaction mass is heated to 80-85°C and stirred at this temperature for 3-4 hours. After completion of the reaction, the reaction mass is cooled to 20-25°C and stirred at this temperature for 2-3 hours and then stirred at 15-20°C for 3-4 hours. The solid mass is filtered and then washed with 200 ml of water and 100 ml isopropyl alcohol and then dried in air oven till constant weight to get 140-150 gm. of 2-amino-3-nitro-6-p- fluorobenzylamino-py ridine .
EXAMPLE 3: Preparation of flupirtine maleate.
In an autoclave, 100 gm. 2-amino-3-nitro-6-p-fluorobenzylamino- pyridine is taken in 500 ml. 1, 4-dioxane and 20 ml aqueous ammonia solution. 10 gm of raney nickel is added under nitrogen atmosphere and hydrogenated at 75-80°C for 2-3 hours under 4-5 kg pressure. After completion of the reaction, the reaction mass is cooled and filtered at 40- 45°Cthen in filtrate 45 ml of ethyl chloroformate is added slowly at 5- 10°C. The temperature is raised to 25-30°C and 80 ml triethyl amine is added under nitrogen atmosphere. The reaction mass, is heated at 55- 60°C under stirring for 3-4 hours. After completion of the reaction, the reaction mass is distilled up to 70-80% under vacuum. This concentrated reaction mass is added into aqueous solution of maleic acid (72 gm in 2000 ml DM water at 65-70°C and maintained at 65-70°C for 2 hours under nitrogen to get crude Flupirtine Maleate as a solid. The reaction mass is cooled to 25-30°C in 5-6 hours and maintained at this temperature for next 2-3 hours then filtered. The wet cake is washed with 200 ml water and dried to get 145 gm of flupirtine maleate.
EXAMPLE 4: Preparation of pure flupirtine maleate crystalline modification A.
1 15 gm crude Flupirtine maleate obtained in example 3 is taken in 1 150 ml methanol and 58 ml water. This mixture is heated to reflux and 58 ml water is added slowly to get a clear solution and refluxed for about half an hour. The reaction mixture is cooled slowly to 60°C and seeded with crystals of modification A. Then it is cooled slowly to 20-25°C and maintained at this temperature for 2 hours. The crystalline mass is filtered and washed with 100 ml chilled methanol and dried to give 92 gm. flupirtine maleate crystalline modification A.
………………….
PATENT
http://www.google.com.tr/patents/WO1998047872A1?cl=en
1. Example
Preparation of flupirtine maleate
75 g (0.286 mol) ANFP be in a suspension of 12.5 g of Raney nickel in 400 ml of isopropanol was hydrogenated at 65 ° C and 5 bar hydrogen pressure. After hydrogenation, the solution is then mixed with 26.4 ml of ethyl chloroformate and 50.6 ml of triethylamine. After adding a further 6.3 ml of ethyl chloroformate the reaction solution is stirred at 60 ° C. for 1 hour. Then sucks the hot solution with stirring in a 50 – 60 ° C heated solution of 53.3 g of maleic acid in 1, 5 IH 2 O and washed the catalyst with little isopropanol.
The flupirtine maleate is precipitated in colorless crystal suspension is cooled with further stirring at 20 ° C and maintained at this temperature for 20 minutes. It is suctioned off, washed with 500 ml of water and dried flupirtine maleate in vacuo at 35 ° C.
Yield: 107.55 g (89.6% of theory, based on ANFP.) Example 2
Preparation of flupirtine maleate
18.5 g (0.07 mol) ANFP be analogous to Example 1 in a suspension of 2.0 g of Raney nickel in 140 ml of ethanol 60 – 70 ° C and 5 bar hydrogen pressure After hydrogenation, the further reaction takes place at 40 – 50 ° C with 9.3 g of ethyl chloroformate (0.86 mol) of triethylamine and 9.2 g (0.91 mol) The separated from the catalyst reaction solution is added with stirring to 540 ml of water After 2 hours of stirring at room temperature suctioned the failed base off and washed with water and isopropanol and crystallized in the 3.7-fold amount of isopropanol to yield 18.4 g (86.0% of theory)
The precipitation and modification of pure flupirtine maleate is carried out according to the Examples 7 and 8
………………….
PATENT
CN104086481 (A) – Synthesis method of flupirtine maleate
http://worldwide.espacenet.com/publicationDetails/biblio?CC=CN&NR=104086481A&KC=A&FT=D
The invention provides a synthesis method of flupirtine maleate. Recrystallization by use of methanol is carried out in the refining step of the crude product of the flupirtine maleate so that the product is white in appearance and high in purity, and the crystal form of the product is pure A crystal and same as the crystal form of the commercial products. The optimal reaction solvent, reaction time and reaction temperature are explored and found out by use of a simplified process flow, and a method for preparing the flupirtine maleate in the pure A crystal form, which is high in yield, low in cost and simple to operate, uses easily available raw materials and is applicable to the industrial production is found.
………………
PATENT
http://www.google.com/patents/CN103333103A?cl=en
The preparation of a comprehensive literature about the ratio of maleic acid fluoride Jie Ting to 2_-amino-3-nitro-6-chloro-Jie ratio 唳 as a starting material, by condensation, reduction, acylation, salt and other processes for The most common route, however, due to the reduction, acylation, salt formation method of a three-step operation is different, not only the yield of the synthesis varies widely, and also on the flupirtine maleate product quality. This is mainly because of the intermediate 2,3-diamino-6-fluoro-benzyl amino pyridine and flupirtine multi-aminopyridine derivative, is easy to oxidative deterioration. So the use of continuous operation, not only simple steps, and can improve product quality and yield.
Chinese patent CN102241626 reported to 2,6_ dichloro _3_ nitropyridine as raw material by selective ammonia solution to give 2-amino-3-nitro-6-chloro-approved Li, then with amine fluoride Festival to afford a yellow solid 2-amino-3-nitro-6-p-fluoro-benzylamino-pyridine. After vacuum drying, the use of hydrogenation, and then under nitrogen and ethyl chloroformate acylation catalyst is filtered off and then a salt with maleic acid to give a pale green crude product yield was about 37% (2-amino-3-nitro-6-chloro-pyridin-meter).
Patent No. CN102838534 reported 2-amino-3-nitro-6-chloro-pyridine as starting material, the use of sub-step processing method, in a first reactor, and a condensation-fluorobenzyl amine, and dried in vacuo to give the intermediate 2-amino-3-nitro-6-p-fluorobenzyl-aminopyridine, in a second reactor to Raney nickel as the catalyst, the catalytic hydrogenation of hydrazine hydrate, after filtration the solvent was evaporated to give the intermediate 2,3-solid – diamino-6-p-fluorobenzyl-aminopyridine, in a third reactor with ethyl chloroformate acylated intermediate distillation under reduced pressure to give solid form of flupirtine with an aqueous solution of a salt of maleic acid, after purification, the total Yield 25% ~ 30%.
Patent W02012004391 discloses a method for preparing a high yield of flupirtine maleate method. In 2_-amino-3-nitro-6-chloro-fluoro-section batch Li and amines as raw material for condensation to give 2-amino-3-nitro-6-fluoro-section based on the amino pyridine granted, then using high-pressure hydrogenation the reduction, acylation step in a high pressure hydrogenation reactor concentrated completed, after the catalyst was filtered off and then the salt, the crude yield of greater than 70%. The preparation method using high-pressure hydrogenation apparatus, there are security risks, and takes too long, is not suitable for industrial production.
Patent No. CN102260209 discloses a 2_ amino _3_ _6_ fluorobenzyl nitro-pyridine as starting material, the reduction, acylation and salt-forming step of the continuous operation, the synthetic yield was improved to 58% so, no mention of product purity. Since the acylation step taken ethyl chloroformate, while an organic base is added, so that an increase in a side reaction, the product yield decreases; the same time, 2-amino-3-nitro-6-p-fluoro-benzylamino-pyridine as the raw material, the production cost high.
In the present invention, we consider the key intermediate 2,3-diamino-6-p-fluorobenzyl-aminopyridine and chemical properties of flupirtine, condensation, reduction, acylation, salt formation reaction is concentrated to the same conventional the reactor is completed, each step without intermediate separation, simplifying the process route and operations, improve efficiency, reduce costs, improve the overall yield of the crude by 40 percent following the step by step operation for more than 70% crude purity of more than 99% suitable for industrial scale production.

Example 4:
The 4Kg2_ amino-3-nitro-6-chloropyridine, 4.5Kg triethylamine, 40L of isopropanol into the reactor, stirred and heated to reflux for turn; the 4.4Kg of benzylamine was added to the fluorine reactor, the reaction under reflux conditions for 3 hours. After heating was stopped, the reaction solution was added to 40L of purified water, a lot of yellow solid was precipitated was filtered and the resulting wet product remains in the reaction vessel. To the reaction kettle was added 1.8Kg Raney nickel, 40L of isopropanol, stirred and heated to reflux for open, 7Kg80% hydrazine hydrate was added dropwise, the reaction was refluxed for 3 hours, after completion of the reaction down to room temperature in a nitrogen atmosphere, was added rapidly 3.6Kg chloro carboxylic acid ethyl ester, the reaction at room temperature for 3 hours. 3Kg of triethylamine was added, free 2 hours, filtered and the filtrate was added to 5Kg / 100L of maleic acid in isopropanol, cooling crystallization to give an off-white solid, 50 ° C blast drying, weight 7.8Kg, the yield was 80.5 %, purity 99.6%.
A sub-step treatment process research and data [0034] Comparative Example
The method according to Chinese patent CN102838534 disclosed flupirtine maleate was prepared, and a number of specific steps
………….
PATENT
FIG. 1 is flupirtine maleate 1H NMR.
[0021] FIG. 2 is flupirtine maleate A crystal X-ray diffraction pattern



Example 3
2-Amino-3-nitro-6-chloropyridine 246g, and 254g of triethylamine were added to 800ml of ethanol-necked flask and stirred under heating to reflux, fluorine was slowly added dropwise benzylamine 80g, reaction of 6 hours, the reaction was completed After the dropwise addition of purified water 500ml, cooled slowly with stirring to room temperature, filtered, dried to give 2-amino-3-nitro-6-p-fluoro-benzylamino-pyridine.
[0033] The ferric chloride hexahydrate was dissolved in purified water 41g 200ml, adding activated charcoal 20g, heated to 50 ° C, a saturated solution of sodium hydroxide was added 45g (24g of sodium hydroxide dissolved in 21ml water), 60 ° C with stirring I hours, cooled to room temperature, filtered, and dried to give ferric hydroxide / activated carbon catalyst.
[0034] A mixture of 2-amino-3-nitro-6-p-fluorobenzyl-aminopyridine 104.Sg, ferric hydroxide / activated carbon catalyst was added to 20g 2L reaction flask was added 95% ethanol 1200ml, heated with stirring to 90 ° C. Insulation 60% hydrazine hydrate was added dropwise 250g. Drops Bi insulation response to 3h. Completion of the reaction, the reaction solution is filtered hot with concentrated hydrochloric acid to 240ml and 95% ethanol IOOOml reaction flask. (TlO ° C crystallization I h, filtered, dried to give 2,3-amino-6-fluoro-benzyl-aminopyridine on
Hydrochloride.
[0035] A mixture of 2,3-diamino-6-p-fluoro-benzylamino-pyridine hydrochloride 132g, 800ml of isopropanol was added to a 2L reaction flask, the temperature control to 28 至 30 ° C, was slowly added dropwise acetic acid ester 39g. Stirred for 0.5 hours, was slowly added dropwise triethylamine 70g, after stirring for 0.5 hours, complement ethyl chloroformate 5g, stirred for 15 minutes, additional triethylamine remaining 10g. Continue stirring for I hour. The reaction solution was concentrated under reduced pressure to about 800ml of distillate was distilled out. The remaining reaction solution was poured into an aqueous solution of maleic acid with a good (39g of maleic acid was dissolved in purified water IlOOml), stirred for 30 minutes at room temperature, (T5 ° C was stirred for 5 ~ 8 hours, filtered, dried to give the maleic acid flupirtine crude.
[0036] The crude flupirtine maleate product 100g, 2000ml of ethanol into the reaction flask and heated to 70~80 ° C, was added 5g of activated carbon and dissolved, and incubated I hour, filtered hot, O~5 ° C CRYSTALLIZATION 3 hours, filtered and dried to give crude I. The crude product I 90g, 450ml of ethanol into the reaction flask and heated 20h, and then slowly cooled to room temperature, O~5 ° C for 2 hours, filtered, and dried to give crystal form A of flupirtine maleate product.
…………………..
PATENT
http://www.google.com/patents/CN102838534A?cl=en
flupirtine maleate is a non-opioid analgesic effects on the central nervous system drugs, which is a selective neuronal potassium channel opener (Selective Neuronal Potassium Channel Opener, SNEPCO), has analgesic, muscle relaxant and neuroprotective triple effect. Acute pain treatment is mainly used for various types of moderate, such as surgery, trauma-induced pain and headache / migraine and abdominal spasms.
flupirtine maleate English name: Flupirtine Maleate, chemical name: 2_ amino-6 – [((4-fluorophenyl) methyl) amino] pyridine-3-carboxylic acid ethyl ester maleic salt; Chemical Abstracts (CAS) number = 75507-68-5; formula = C15H17FN4O2 · C4H4O4; molecular weight: 420.39; its structural formula is:

From a structural perspective, flupirtine maleate molecular compounds, the derivatives of benzene and pyridine derivatives synthetically produced flupirtine, flupirtine and then forming an organic salt with maleic acid. Comprehensive literature, synthetic routes flupirtine maleate there are two major, now its main synthetic steps described below.
Route 1 (W0 98 / 47872Α1): The route to 2,6_ dichloro _3_ nitropyridine as raw material substitution, ammoniated, high-pressure hydrogenation, acylation, a process salt, refined and so on. The reaction formula is as follows:

Route 2 (US5959115A) to 2_ amino _3_ nitro _6_ methoxypyrido as the starting material, and on fluorobenzylamine substitution reaction to produce 2-amino-3-nitro–6 – fluorobenzyl amine of pyridine, the yield was 95.2%, and the high-pressure hydrogenation, the catalyst was filtered off, and then the occurrence of an acylation reaction with ethyl chloroformate to give the hydrochloride salt of flupirtine, three-step total yield of 53.3%. The reaction formula is as follows:

Route 1 starting material is different, but relatively speaking, the route I easily controlled reaction conditions, and 2-amino-3-nitro-6-chloro-pyridine is a common chemical raw materials, easy to buy on the market, This can shorten the reaction route. Route 2 two-step reaction process route is short, but the starting 2-amino-3-nitro-6-methoxy-approved Li expensive, hydrogenation, acidification two steps yield only 56.0%.
Chinese Patent Application Publication No. CN102241626A are disclosed and CN102260209A flupirtine maleate preparation method, but the application of these two methods for the preparation of a laboratory scale, for the industrial mass production were not optimized.
The method for purifying of flupirtine maleate in the final product are as follows:
650C ± 5 ° C under the flupirtine maleate crude and ethanol mass ratio of 1: 30-40 mixed, crude completely dissolved, then add 680g of activated carbon and stirred for 15–30 minutes, and hot filtration, the filtrate, stirring down to room temperature, and then cooled to 0 ° C crystallization 5–10 hours, filtered and the filter cake to take the filter cake can be dried.
BELOW AS FREE BASE
| Ethyl N-[2-amino-6-[(4-fluorophenyl)methylamino]pyridin-3-yl]carbamate | |
| CAS No.: | 56995-20-1 |
|---|---|
| Synonyms: |
|
| Formula: | C15H17FN4O2 |
| Exact Mass: | 304.13400 |
1H NMR INTERPRETATIONS/PREDICTIONS
![ethyl N-[2-amino-6-[(4-fluorophenyl)methylamino]pyridin-3-yl]carbamate NMR spectra analysis, Chemical CAS NO. 56995-20-1 NMR spectral analysis, ethyl N-[2-amino-6-[(4-fluorophenyl)methylamino]pyridin-3-yl]carbamate H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-18/000/006/679/56995-20-1-1h.png)
13C NMR INTERPRETATIONS/PREDICTIONS
![ethyl N-[2-amino-6-[(4-fluorophenyl)methylamino]pyridin-3-yl]carbamate NMR spectra analysis, Chemical CAS NO. 56995-20-1 NMR spectral analysis, ethyl N-[2-amino-6-[(4-fluorophenyl)methylamino]pyridin-3-yl]carbamate C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-18/000/006/679/56995-20-1-13c.png)
…….
PAPER
Helvetica Chimica Acta, , vol. 77, # 8 p. 2175 – 2190
 AND
AND  GIVES PRODUCT
GIVES PRODUCT
 ALSO AN INTHelvetica Chimica Acta, , vol. 77, # 8 p. 2175 – 2190
ALSO AN INTHelvetica Chimica Acta, , vol. 77, # 8 p. 2175 – 2190
…………..
HPLC
Instrumentation An HPLC system (Agilent HPLC Model-1200) equipped with a C18 (Agilent BDS, 250 mm x 4.6 mm, 5µ) column, binary pump, rheodyne loop injector with 20 μL, and a photodiode array detector was used. The software used for HPLC data acquisition was EZChrome Elite. A flash chromatograph equipped with silica gel as the column material, and VWD-UV detection (using the software Analogix IF 280 V 5.10) was used for the isolation and purification of degradation products. 1 H-NMR was recorded on the Varian Unity Inova at 400 MHz (using TMS as internal standard and DMSO-d6 as solvent), 13C-NMR (Mercury Plus at (abundance 100 MHz), using DMSO-d6 as solvent), and mass spectral studies were performed on the API 3000 ABPCIES instrument.
Method Development and Optimization of the Chromatographic Conditions In preliminary experiments, the drug was subjected to the reversed-phase mode using a C18 column (Agilent, 250 x 4.6 mm, 5µ) and mobile phases consisting of water (pH 3.0 adjusted with orthophosphoric acid) and methanol by varying the % aqueous phase from 10% to 30%. The drug was retained on the column, but the peak shape was not good. It was noted that increasing the % aqueous phase in the mobile phase composition increases the retention time of flupiritine maleate. Based on the suitable retention time for SIAM, the 20% aqueous phase was optimized. To reduce the tailing effect, 0.2% triethylamine (TEA) was added and the pH was adjusted to 3.0 with orthophosphoric acid and the corresponding retention of FLU was 10.3 ± 0.3 min. Finally, the mobile phase of 0.2% v/v TEA (pH-adjusted to 3.0 with OPA) and methanol in the ratio of 20:80% v/v was optimized. The flow rate was 1.0 mLmin−1. The injection volume was 20 µL and the PDA detection wavelength was at 254 nm. The chromatogram obtained in the optimized condition is shown in Fig. 2. It was observed that eight degradation products were formed with retention times 3.9 ± 0.2 min (D1), 4.8 ± 0.2 min (D2), 6.4 ± 0.1 min (D3), 6.8 ± 0.2 min (D4), 8.2 ± 0.2 min(D5), 12.0 ± 0.2 min (D6), 14.1 ± 0.1 min (D7), and 15.0 ± 0.1 min (D8), respectively. The chromatographic resolution among all of the peaks was more than 2. The % degradation was about 5–30% depending on stress conditions.
………………..
paper
J Pharm Biomed Anal. 2014 Mar;90:27-34. doi: 10.1016/j.jpba.2013.11.015. Epub 2013 Nov 27.
Flupirtine maleate is a centrally acting, non-opioid, nonsteroidal antiinflammatory analgesic. During the manufacturing of flupirtine maleate, two unknown impurities present in the laboratory batches in the range of 0.05-1.0% along with the known impurities in HPLC analysis. These unknown impurities were obtained from the enriched mother liquor by column chromatography. Based on the complete spectral analysis (MS, (1)H, (13)C, 2D NMR and IR) and knowledge of the synthetic scheme of flupirtine maleate, these two new impurities were designated as diethyl 5-((4-fluorobenzyl)amino)-2-oxo-1H-imidazo[4,5-b]pyridine-1,3(2H)-dicarboxylate (impurity-I) and diethyl(6-((4-fluorobenzyl)amino)pyridine-2,3-diyl)dicarbamate (impurity-II). Impurity isolation, identification, structure elucidation and the formation of impurities were also discussed. Preparation and structure elucidation of impurity-III were also first reported in this paper.
…………………
journal of pharmaceutical and biomedical analysis, 90, 2014, 27-34
References: Substituted pyridine with central analgesic properties. Prepn: K. Thiele, W. von Bebenburg, ZA 6902364(1970 to Degussa); W. von Bebenburg et al., Chem. Ztg. 103, 387 (1979); eidem, ibid. 105, 217 (1981).
Prepn of maleate: W. von Bebenburg, S. Pauluhn, BE 890331; eidem, US 4481205 (1980, 1984 both to Degussa).
Comparison of pharmacology with other analgesics: V. Jakovlev et al., Arzneim.-Forsch. 35, 30 (1985).
Pharmacokinetic studies: K. Obermeier et al., ibid. 60.
Effect on driving ability: B. Biehl, ibid. 77.
Clinical trials in treatment of cancer pain: W. Scheef, D. Wolf-Gruber, ibid. 75.
Efficacy in treatment of pain after hysterectomy: R. A. Moore et al., Br. J. Anaesth. 55, 429 (1983).
Symposium on pharmacology and clinical efficacy: Postgrad. Med. J. 63, Suppl. 3, 1-113 (1987).
References
1
- Abrams, SML; L.R.I. Baker, P. Crome, A.S.T. White, A. Johnston, S.I. Ankier, S.J. Warrington, P. Turner, G. Niebch (1988). “Pharmacokinetics of flupirtine in elderly volunteers and in patients with moderate renal impairment”. The Fellowship of Postgraduate Medicine 64 (751): 361–363. doi:10.1136/pgmj.64.751.361. PMC 2428663. PMID 3200777.
- 2
- Narang, PK; Tourville JF; Chatterji DC; Gallelli JF (1984). “Quantitation of flupirtine and its active acetylated metabolite by reversed-phase high-performance liquid chromatography using fluorometric detection”. Journal of Chromatography 305 (1): 135–143. doi:10.1016/S0378-4347(00)83321-6. PMID 6707137.
- 3
- Methling, K; Reszka P; Lalk M; Vrana O; Scheuch E; Siegmund W; Terhaag B; Bednarski PJ (2008). “Investigation of the in Vitro Metabolism of the Analgesic Flupirtine”. Drug Metabolism and Disposition 37: 479–493. doi:10.1124/dmd.108.024364.
- 4
- Blackburn-Munro, G; W. Dalby-Brown; N. R. Mirza; J. D. Mikkelsen; R. E. Blackburn-Munro (2005). “Retigabine: Chemical Synthesis to Clinical Application”. CNS Drug Reviews 11 (1): 1–20. doi:10.1111/j.1527-3458.2005.tb00033.x. PMID 15867950.
- 5
- Flupirtine Drugs.com. Accessed 20 September 2011.
- 6
- “POTIGA® (ezogabine) Tablets, CV. Full Prescribing Information”. GlaxoSmithKline and Valeant Pharmaceuticals. Revised: September, 2013. Initial U.S. Approval: 2011. Retrieved 2 June 2014. Check date values in:
|date=(help) - 7
- http://www.freepatentsonline.com/5721258.html Primary and secondary neuroprotective effect of flupirtine in neurodegenerative diseases The synthesis of flupirtine and its pharmaceutically acceptable salts is described in EP 160 865 and 199 951. EP0199951 December, 1986 Process for the preparation of 2-amino-3-nitro-6-(4-fluorobenzylamino) pyridine and of 2-amino-3-carbethoxyamino-6-(4-fluorobenzylamino) pyridine.
- 8
- http://patent.ipexl.com/EP/EP0199951.html#reference Process for the preparation of 2-amino-3-nitro-6-(4-fluorobenzylamino) pyridine and of EPO Patent EP0199951 1986 German.
- 9
- http://www.patentfish.com/2-amino-3-carbethoxyamino-6-4-fluoro-benzylamino Process for the preparation of 2-amino-3-nitro-6-(4-fluorobenzylamino) pyridine and of 2-amino-3-carbethoxyamino-6-(4-fluorobenzylamino) pyridine EP 0199951 B1 1986. English.
- 10
- http://patent.ipexl.com/US/4481205.html 2-Amino-3-carbethoxyamino-6-(p-fluoro-benzylamino)-pyridine-maleate United States Patent 4481205. 1981
- 11
- http://www.freepatentsonline.com/3998834.html Novel N-(4-piperidinyl)-N-phenylamides and -carbamates having very potent analgesic activity, methods of preparing same and useful intermediates therefor. Patent 3998834. 1976.
- 12
- http://www.faqs.org/patents/app/20090306150 CARBOXYLIC ACID SALTS OF 2-AMINO-3-CARBETHOXYAMINO-6-(4-FLUORO-BENZYLAMINO)-PYRIDINE patent 20090306150. 2009. Flupirtine is commonly used in the form of pharmaceutically acceptable acid addition salts. Commercially, flupirtine is available as its maleate addition salt under the trademark Katadolon®. There are two known polymorphs of flupirtine maleate, designated in the art as flupirtine maleate A and B. European patentEP 0 977 736 discloses pure flupirtine maleate crystalline form A and a process for its preparation. Flupirtine and mixtures of flupirtine maleate polymorphs A and B can be synthesised according to EP 0 199 951.
- 13
- “Adeona Pharmaceuticals and National Neurovision Research Institute (NNRI) Collaborate to Test Flupirtine for Retinitis Pigmentosa”. Ann Arbor, MI and Owings Mills, MD: Synthetic Biologics, Inc. December 2, 2008. Retrieved 2 June 2014.
- 14
- Kornhuber, J.; Bleich, S.; Wiltfang, J.; Maler, M.; Parsons, C. G. (1999). “Flupirtine shows functional NMDA receptor antagonism by enhancing Mg2+ block via activation of voltage independent potassium channels. Rapid communication”. Journal of neural transmission (Vienna, Austria : 1996) 106 (9–10): 857–867. doi:10.1007/s007020050206. PMID 10599868.
- 15
- McMahon, FG; Arndt WF Jr, Newton JJ, Montgomery PA, Perhach JL. (1987). “Clinical experience with flupirtine in the U.S”. Postgraduate Medical Journal 63 (3): 81–85. PMID 3328854.
- 16
- Klawe, C; Maschke, M (2009). “Flupirtine: pharmacology and clinical applications of a nonopioid analgesic and potentially neuroprotective compound”. Expert opinion on pharmacotherapy 10 (9): 1495–500. doi:10.1517/14656560902988528. PMID 19505216.
- 17
- Swedberg MD, Shannon HE, Nickel B, Goldberg SR (September 1988). “Pharmacological mechanisms of action of flupirtine: a novel, centrally acting, nonopioid analgesic evaluated by its discriminative effects in the rat”. J. Pharmacol. Exp. Ther. 246 (3): 1067–74. PMID 2901483.
- 18
- Dhar S, Bitting RL, Rylova SN et al. (April 2002). “Flupirtine blocks apoptosis in batten patient lymphoblasts and in human postmitotic CLN3- and CLN2-deficient neurons”. Ann. Neurol. 51 (4): 448–66. doi:10.1002/ana.10143. PMID 11921051.
- 19
- Flupirtine as Oral Treatment in Multiple Sclerosis (FLORIMS) Clinical Trials.gov Accessed 20 September 2011.
- 20
- Pipex Pharmaceuticals (PPXP)’ Oral Flupirtine Receives IND With FDA for Phase II Clinical Trial for Fibromyalgia 4/21/2008
- 21
- “Partnered Program. Effirma™ for Fibromyalgia”. Synthetic Biologics, Inc. Retrieved 2 June 2014.
- 22
- Stoll AL, Belmont MA. (2000) “Fibromyalgia Symptoms Relieved by Flupirtine: An Open-Label Case Series” Psychosomatics 41:371-372. Accessed 20 September 2011.
- 23
- EMA information about flupirtine
- 24
- article in Deutsches Ärzteblatt
- 25
- Singal, Rikki; Parveen Gupta; Nidhi Jain; Samita Gupta (2012). “Role of Flupirtine in the Treatment of Pain – Chemistry and its Effects”. Mædica — a Journal of Clinical Medicine 7 (2): 163–166. PMID 23401726.
- 26
- “DRUGDEX® Evaluations – Flupirtine”. Retrieved 24 March 2013.
- 27
- Preston, KL; Funderburk, FR; Liebson, IA; Bigelow, GE (Mar 1991). “Evaluation of the Abuse Potential of the Novel Analgesic Flupirtine Maleate”. Drug and Alcohol Dependence 27 (2): 101–113. doi:10.1016/0376-8716(91)90027-v. PMID 2055157.
- 28
- Sofia, RD; Diamantis, W; Gordon, R (1987). “Abuse Potential and Physical Dependence Liability Studies with Flupirtine Maleate in Laboratory Animals”. Postgraduate Medical Journal. 63 Suppl 3: 35–40. PMID 3447127.
- 29
- Gahr, M; Freudenmann, RW; Connemann, BJ; Hiemke, C; Schönfeldt–Lecuona, C (Dec 2013). “Abuse Liability of Flupirtine Revisited: Implications of Spontaneous Reports of Adverse Drug Reactions”. Journal of Clinical Pharmacology 53 (12): 1328–1333. doi:10.1002/jcph.164. PMID 24037995.
- 30
Stoessel, C; Heberlein, A; Hillemacher, T; Bleich, S; Kornhuber, J (Aug 16, 2010). “Positive Reinforcing Effects of Flupirtine—Two Case Reports”. Progress in Neuro-psychopharmacology & Biological Psychiatry 34 (6): 1120–1121. doi:10.1016/j.pnpbp.2010.03.031. PMID 20362025. Retrieved 2 June 2014.
References
2. Flupirtine
Note: Compound name must be entered under “Substance Identification” and then “Names and Synonyms” selected to view synonyms.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| ethyl {2-amino-6-[(4-fluorobenzyl)amino]pyridin-3-yl}carbamate | |
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Pharmacokinetic data | |
| Bioavailability | 90% (oral), 70% (rectal)[1] |
| Metabolism | Hepatic to 2-amino-3-acetylamino-6-(para-fluorobenzylamino) pyridine (which has 20-30% the analgesic potential of its parent compound), para-fluorohippuric acid[2] and a mercapturic acid metabolite, presumably formed from a glutathione adduct[3] |
| Half-life | 6.5 hrs (average), 11.2-16.8 hrs (average 14 hrs) (elderly), 8.7-10.9 hrs (average 9.8 hrs) (in those with moderate-level renal impairment)[1] |
| Excretion | 72% of flupirtine and its metabolites appear in urine and 18% appear in feces[4] |
| Identifiers | |
| 56995-20-1 |
|
| N02BG07 | |
| PubChem | CID 53276 |
| IUPHAR ligand | 2598 |
| ChemSpider | 48119 |
| UNII | MOH3ET196H |
| KEGG | D07978 |
| ChEMBL | CHEMBL255044 |
| Chemical data | |
| Formula | C15H17FN4O2 |
| 304.32 g/mol | |
K 912, NC 6300, Epirubicin nano



Cancer Research UK

Irish Cancer Society
Macmillan Cancer Support
NHS Evidence
 Epirubicin – Substance Summary
Epirubicin – Substance Summary
PubChem
MedlinePlus
– See more at: http://www.cancerindex.org/Epirubicin#sthash.BBioCC6K.dpuf
PHASE 1 JAPAN SOLID TUMOURS
DNA/RNA Synthesis Inhibitor
WITH Nano Carrier Co.,Ltdhttp://pdf.irpocket.com/C4571/qnwX/eFou/vG1J.pdf
KOWA COMPANY LTD
CAS FREE FORM. 56420-45-2
Smiles
- O=C2c1c(O)c5c(c(O)c1C(=O)c3cccc(OC)c23)C[C@@](O)(C(=O)CO)C[C@@H]5O[C@@H]4O[C@H]([C@H](O)[C@@H](N)C4)C
- NMR
- http://file.selleckchem.com/downloads/nmr/S122302-Epirubicin-Hydrochloride-NMR-Selleck.pdf
- http://www.medkoo.com/Product-Data/Epirubicin/Epirubicin-QC-TZC20130604web.pdf
-
Epirubicin CAS No.: 56420-45-2 Synonyms: - 4′-Epi-DX;
- Epirubicina;
- WP 697;
- Pidorubicin;
- 4′-epiadriamycin;
- Adriblastin;
- epi-dx;
- Epiadriamycin;
- farmorubicin;
- imi28;
- Farmarubicin;
Formula: C27H29NO11 Exact Mass: 543.17400
NC-6300, an epirubicin-incorporating micelle, extends the antitumor effect and reduces the cardiotoxicity of epirubicin.
Epirubicin is widely used to treat various human tumors. However, it is difficult to achieve a sufficient antitumor effect because of dosage limitation to prevent cardiotoxicity. We hypothesized that epirubicin-incorporating micelle would reduce cardiotoxicity and improve the antitumor effect. NC-6300 comprises epirubicin covalently bound to PEG polyaspartate block copolymer through an acid-labile hydrazone bond. The conjugate forms a micellar structure of 40-80 nm in diameter in an aqueous milieu. NC-6300 (10, 15 mg/kg) and epirubicin (10 mg/kg) were given i.v. three times to mice bearing s.c. or liver xenograft of human hepatocellular carcinoma Hep3B cells. Cardiotoxicity was evaluated by echocardiography in C57BL/6 mice that were given NC-6300 (10 mg/kg) or epirubicin (10 mg/kg) in nine doses over 12 weeks. NC-6300 showed a significantly potent antitumor effect against Hep3B s.c. tumors compared with epirubicin. Moreover, NC-6300 also produced a significantly longer survival rate than epirubicin against the liver orthotopic tumor of Hep3B. With respect to cardiotoxicity, epirubicin-treated mice showed significant deteriorations in fractional shortening and ejection fraction. In contrast, cardiac functions of NC-6300 treated mice were no less well maintained than in control mice. This study warrants a clinical evaluation of NC-6300 in patients with hepatocellular carcinoma or other cancers.

K-912(NC-6300)の概要 K-912(NC-6300)は、世界的に幅広く使用されているアントラサイクリン系の抗が ん剤の一つであるエピルビシンを内包したミセル化ナノ粒子製剤で、その特性により、 エピルビシンの有する心毒性の軽減が期待できます。さらに、pH 応答性システムを採 用することで、腫瘍細胞内でのエピルビシンの放出量を高め、既存のエピルビシンに比 べより強力な抗腫瘍効果が期待できます。
Epirubicin is an anthracycline drug used for chemotherapy. It can be used in combination with other medications to treat breast cancer in patients who have had surgery to remove the tumor. It is marketed by Pfizer under the trade name Ellence in the US andPharmorubicin or Epirubicin Ebewe elsewhere.
Similarly to other anthracyclines, epirubicin acts by intercalating DNA strands. Intercalation results in complex formation which inhibits DNA and RNA synthesis. It also triggers DNA cleavage by topoisomerase II, resulting in mechanisms that lead to cell death. Binding to cell membranes and plasma proteins may be involved in the compound’s cytotoxic effects. Epirubicin also generates free radicalsthat cause cell and DNA damage.
Epirubicin is favoured over doxorubicin, the most popular anthracycline, in some chemotherapy regimens as it appears to cause fewer side-effects. Epirubicin has a different spatial orientation of the hydroxyl group at the 4′ carbon of the sugar – it has the opposite chirality – which may account for its faster elimination and reduced toxicity. Epirubicin is primarily used against breast and ovarian cancer, gastric cancer, lung cancer and lymphomas.
Development history
The first trial of epirubicin in humans was published in 1980.[1] Upjohn applied for approval by the U.S. Food and Drug Administration(FDA) in node-positive breast cancer in 1984, but was turned down because of lack of data.[2] It appears to have been licensed for use in Europe from around this time however.[3] In 1999 Pharmacia (who had by then merged with Upjohn) received FDA approval for the use of epirubicin as a component of adjuvant therapy in node-positive patients.
Patent protection for epirubicin expired in August 2007.
References
- Bonfante, V; Bonadonna, G; Villani, F; Martini, A (1980). “Preliminary clinical experience with 4-epidoxorubicin in advanced human neoplasia”. Recent results in cancer research 74: 192–9. PMID 6934564. PM6934564.
- “On Target”.
- According to the proprietary database iddb.com
External links
- http://www.bccancer.bc.ca/HPI/DrugDatabase/DrugIndexPro/Epirubicin.htm
- http://www.pfizerpro.com/page_not_found?rid=/wyeth_html/home/minisites/oncology/ellence/pi/description.html
1H NMR PREDICT

13C NMR PREDICT

COSY
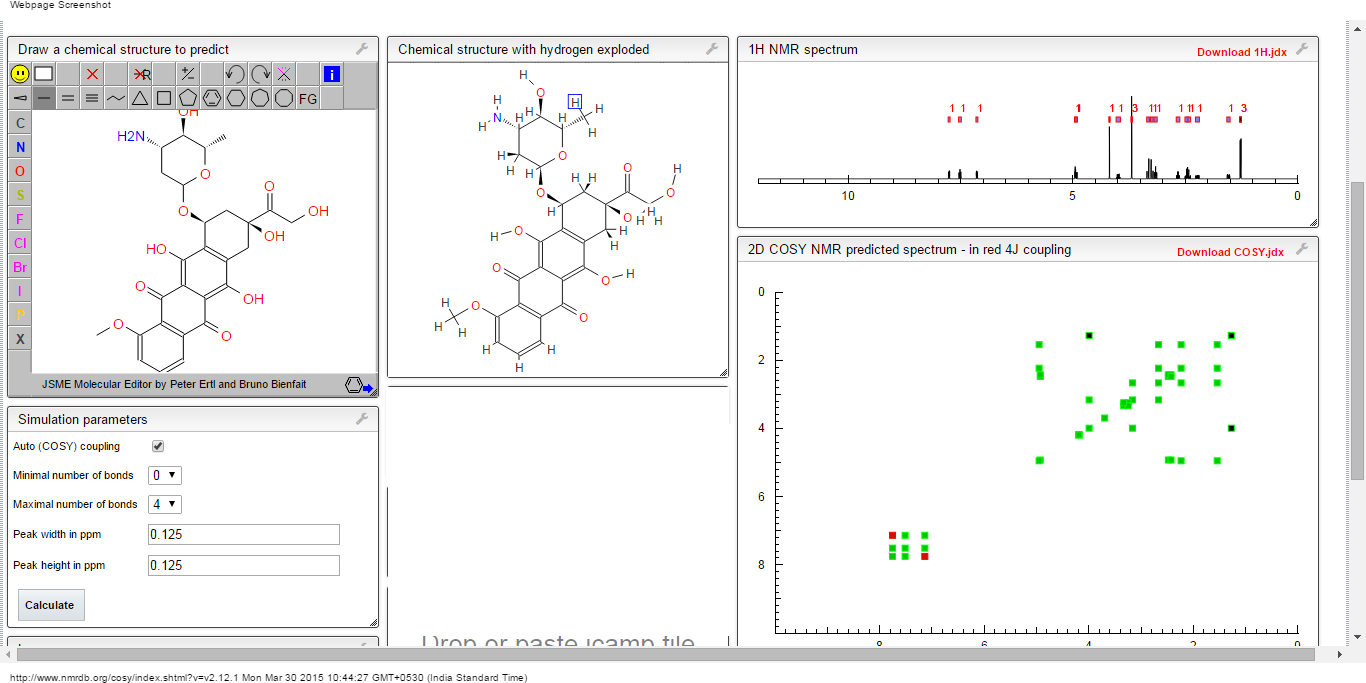
1H NMR

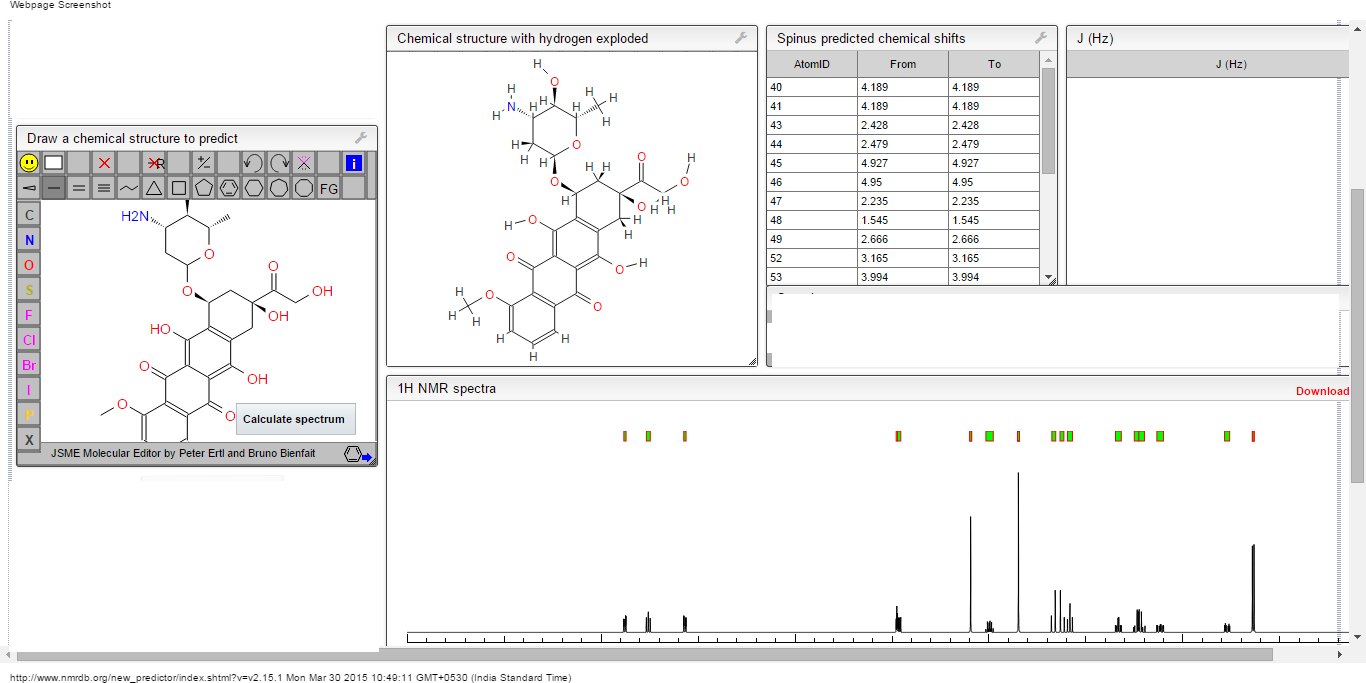
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (8R,10S)-10-((2S,4S,5R,6S)-4-amino-5-hydroxy-6-methyltetrahydro-2H-pyran-2-yl)-6,8,11-trihydroxy-8-(2-hydroxyacetyl)-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione | |
| Clinical data | |
| Trade names | Ellence |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a603003 |
| Intravenous | |
| Pharmacokinetic data | |
| Bioavailability | NA |
| Protein binding | 77% |
| Metabolism | Hepatic glucuronidationand oxidation |
| Excretion | Biliary and renal |
| Identifiers | |
| 56420-45-2 |
|
| L01DB03 | |
| PubChem | CID 41867 |
| DrugBank | DB00445 |
| ChemSpider | 38201 |
| UNII | 3Z8479ZZ5X |
| KEGG | D07901 |
| ChEBI | CHEBI:47898 |
| ChEMBL | CHEMBL417 |
| Chemical data | |
| Formula | C27H29NO11 |
| 543.519 g/mol | |
KOWA COMPANY LTD


Nano Carrier Co


P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
S-flurbiprofen (TT-063)
cas 51543-39-6, 244.26,
C15 H13 F O2
[1,1′-Biphenyl]-4-acetic acid, 2-fluoro-α-methyl-, (αS)-
- [1,1′-Biphenyl]-4-acetic acid, 2-fluoro-α-methyl-, (S)-
- (+)-(S)-Flurbiprofen
- (+)-Flurbiprofen
- (2S)-2-(2-Fluoro-1,1′-biphenyl-4-yl)propanoic acid
- (2S)-2-(2-Fluoro-4-biphenyl)propanoic acid
- (S)-Flurbiprofen
- Dexflurbiprofen
- Esflurbiprofen
- S-(+)-Flurbiprofen
- d-Flurbiprofen
On October 20, 2014, Taisho filed for manufacturing and marketing approval for TT-063 from the Ministry of Health, Labour and Welfare as a new drug candidate that will follow the Type 2 diabetes treatment Lusefi®, which was launched in May 2014. TT-063 is a patch formulation that has been co-developed by Taisho and TOKUHON Corporation with the aim of obtaining an indication for osteoarthritis. In Phase 3 clinical trials comparing TT-063 with therapeutic drugs already on the market, TT-063 has been found to be more effective than the control drugs in patients with osteoarthritis of the knee joint (January 16, 2014 announcement ).
Furthermore, Taisho is also preparing to file for approval from the Ministry of Health, Labour and Welfare for CT-064, an oral formulation of the osteoporosis treatment agent Bonviva launched in August 2013. Taisho has confirmed the effectiveness of CT-064 for osteoporosis patients through Phase 3 clinical trials (September 22, 2014 announcement).
In the central nervous system field, TS-091 transitioned from Phase 1 to Phase 2 in Japan in May 2014. Clinical trials of TS-091 have commenced to confirm the effectiveness of this drug in patients with central disorders of hypersomnolence. In addition, Phase 1 clinical trials of TS-091 have commenced overseas. TS-111 and TS-121 are undergoing Phase 1 clinical trials overseas with the aim of obtaining an indication for depression.
Faced with intensifying competition in new drug discovery, we will jointly implement R&D activities with research institutions outside the Taisho Group, and with companies in Japan and overseas, as we work to enhance our drug development pipeline (lineup of drugs in development). Our goal is to discover many more new drugs, primarily in our priority fields.
| Company | Taisho Pharmaceutical Holdings Co. Ltd. |
| Description | Topical anti-inflammatory analgesic patch containing S-flurbiprofen |
| Molecular Target | |
| Mechanism of Action | |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase III |
| Standard Indication | Osteoarthritis |
| Indication Details | Treat osteoarthritis (OA) and scapulohumeral periarthritis |
| Regulatory Designation | |
| Partner |

Scheme 2.
Reagents and conditions: (a) THF, EDC, Et3N; (b) TFA; (c) 0.5 equiv 2,5-dimethoxybenzoquinone, EtOH, 50–80 °C for 3–5 h; (d) 1 equiv naphthoquinone, MeOH, rt, overnight.
http://www.sciencedirect.com/science/article/pii/S0960894X13011773
……………………………………………
http://www.google.com/patents/CN104478703A?cl=en
Preparation of R – (+) _ flurbiprofen:
[0027] The racemic flurbiprofen as a starting material, to obtain an intermediate product of formula I as shown and then the ester prepared as shown in Formula II with 5-isosorbide monobenzyl ether, ester hydrolysis after obtained R – (+) – flurbiprofen;
[0028]

[0029] wherein, in formula I, X is Cl or Br;
[0030] (2) by the R – (+) _ flurbiprofen obtained (RS) – flurbiprofen:
[0031] The R _ (+) _ flurbiprofen 200mg, potassium hydroxide 150mg, 0. 5mL water into IOmL reaction flask and heated to 120 ° C and held for 2h, then water was added 15mL, cooled to room temperature, the resulting stirring the mixed solution with 10% hydrochloric acid to pH = 0. 5, extracted with ethyl acetate, combined several layers, washed with water until neutral, the organic solvent is recovered, the resulting residue was added at 60~90 ° C under an appropriate amount of petroleum ether by recrystallization, obtained (RS) – flurbiprofen 100mg, 50% yield.
[0032] (3) Preparation of (S) -⑴- flurbiprofen:
[0033] In 25mL single-necked flask, followed by adding (RS) – flurbiprofen 123mg, Portugal TOA 29. 8mg, isopropanol lmL, the mixture was stirred at reflux until clear, half the amount of the solvent evaporated under reduced pressure except , set the refrigerator overnight. The precipitate was collected by suction filtration as white crystals, after washing a small amount of isopropanol, which was dissolved in water, washed with 10% aqueous sodium hydroxide (10% NaOH mean mass fraction) adjusted pH = 13, the sheet-like precipitate was filtered off Portuguese octylamine white crystals. The resulting filtrate was added dropwise with stirring 10% hydrochloric acid to pH = 1, extracted with ethyl acetate, the organic layer was washed with water to recover the solvent, the resulting residue was purified by an appropriate amount of petroleum ether and recrystallized at 60~90 ° C. The product was collected by filtration, and dried in vacuo to give a white (S) – (+) _ flurbiprofen needle crystal 45. 3mg, 65% yield, mp 102~103 ° C, [α] = + 44 ° (C = 1, methanol), ee value of 92.6% (ee value measurement method: (S) – (+) – flurbiprofen after chiral amine derivatization reagents, by HPLC analysis).
[0034] wherein in step (3) is a byproduct eleven R _ (+) _ flurbiprofen, its follow step (1) of racemic reused.
[0035] Step (1) of the specific operation is as follows:
[0036] (la) 1:. Synthesis of 2,6-sorbitol dehydration -D- -5- benzyl ether: 4: 3
[0037] 250ml volumetric flask isosorbide 18. 25g (125mmol), lithium hydroxide monohydrate 5. 25g (125mmol) and 60ml of dimethyl sulfoxide (DMSO), heated to 90 ° C, stirred for 30min, constant pressure equalizing dropping funnel was added dropwise benzyl chloride 14. 4ml (125mmol), 90 ° C the reaction 19-20h, reaction mixture was adjusted to pH 1 with 2M hydrochloric acid, extracted with ethyl acetate (50ml * 3), the organic layers combined, washed with water ( 30ml * 2), dried over anhydrous sodium sulfate overnight, filtered and concentrated residue Cheng baby gel column chromatography (petroleum ether: ethyl acetate = 5: 1) to give a cream solid, that is 1: 4: 3: 2,6 Dehydration -D- sorbitol -5- benzyl ether 24. 5g, m.p. 59 ~61 ° C.
[0038] (Ib) · 2- (2- fluoro-4-biphenylyl) propionyl chloride Synthesis
[0039] 50ml vial before racemic flurbiprofen was added 2. 44g (IOmmol), anhydrous toluene 20ml, freshly distilled thionyl chloride was added dropwise 0. 8ml (Ilmmol), N, N- dimethylformamide amide (DMF) 2 dropwise, stirred at room temperature 2h, the solvent was distilled off under reduced pressure to give a pale yellow gum, i.e., 2- (2-fluoro-4-biphenylyl) propionyl chloride, it was used directly in the reaction without isolation.
[0040] (lc). R-2- (2- fluoro-4-biphenylyl) propionic acid 5- isosorbide monobenzyl ether ester synthesis
[0041] The (Ib) resulting acid chloride was dissolved in 20ml of dry toluene was added dropwise at room temperature, dimethyl amine 3. 5ml, solid precipitation, stirred for about Ih, ice salt bath, a bath temperature of minus 10-15Ό, stirred at this temperature IOmin so, and then the constant pressure dropping funnel (Ia) 5 isosorbide monobenzyl ether (2. 83g, 12mmol) in toluene, keeping the reaction temperature, stirring 8h. The ice bath was removed and the reaction mixture under reduced pressure to remove the solvent, the residue was extracted with ethyl acetate. The extract was washed with water, dried over anhydrous sodium sulfate overnight, ethyl acetate was removed under reduced pressure, the residue was a white gel, recrystallized from petroleum ether to give a white solid that R-2- (2- fluoro-4-biphenylyl) propionic acid 5- isosorbide monobenzyl ether ester 3. 65g (7. 88mmol), in order to put the racemic flurbiprofen yield based on 78.8%.
[0042] (ld) R – Synthesis of flurbiprofen – (+)
[0043] Under ice bath (Ic) obtained R-2- (2- fluoro-4-biphenylyl) propionic acid monobenzyl ether isosorbide 5- ester 2. 3Ig (5mmol) was dissolved in 20ml of acetone / water (1/1) was added Iml hydrochloric acid to adjust pH to 3, stirred for 3-4h, the reaction solution was extracted with ethyl acetate (20ml * 2), sash organic layer was washed with ice (10ml * 2), dried over anhydrous sodium sulfate overnight , filtration, and the filtrate was concentrated, the residue was recrystallized from ether to give white crystals, i.e. L-flurbiprofen 1.02g (4 18mmol.), yield 83.5%, optical purity 93% (HPLC method); input-racemic flurbiprofen dollars, the total yield of 78.8% * 83.5% = 65.8%.
[0044] Step (1) reaction of the formula:
[0045]

P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
Roseroot herb shows promise as potential depression treatment option
Rhodiola rosea (R. rosea), or roseroot, may be a beneficial treatment option for major depressive disorder (MDD), according to results of a study in the journal Phytomedicine led by Jun J. Mao, MD, MSCE, associate professor of Family Medicine, Community Health and Epidemiology and colleagues at the Perelman School of Medicine of University of Pennsylvania.
The proof of concept trial study is the first randomized, double-blind, placebo-controlled, comparison trial of oralR. rosea extract versus the conventional antidepressant therapy sertraline for mild to moderate major depressive disorder.
Depression is one of the most common and debilitating psychiatric conditions, afflicting more than 19 million Americans each year, 70 percent of whom do not fully respond to initial therapy. Costs of conventional antidepressants and their sometimes substantial side effects often result in a patient discontinuing use prematurely. Others opt to try natural products or supplements instead.
All of the study’s…
View original post 293 more words
PRI-724, ICG 001, What is correct structure?

PRI 724 AND ICG001 do confuse us, my efforts to unlock this confusion
STRUCTURE 4
4-(((6S,9S,9aS)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-ylmethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazin-6-yl)methyl)phenyl dihydrogen phosphate……………seems most likely PRI 724

STRUCTURE 5
Cas 1422253-37-9
(6S,9S,9aS)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-yImethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazine- 1 -carboxamide.

compd 2 and 1
OR
COMPD 3
 http://www.medkoo.com/Anticancer-trials/PRI-724.htm and similar/Same
http://www.medkoo.com/Anticancer-trials/PRI-724.htm and similar/Same
http://www.nature.com/nrc/journal/v14/n4/fig_tab/nrc3690_T1.html
compd 3.both above str are same
One of compd 1,2, 3, 4, 5 see at the end as an update , CAN BE ICG001, PRI-724,
Beta-catenin (CTNNB1) inhibitor
ICG001, also known as PRI-724, is a potent, specific inhibitor of the canonical Wnt signaling pathway in cancer stem cells with potential antineoplastic activity. Wnt signaling pathway inhibitor PRI-724 specifically inhibits the recruiting of beta-catenin with its coactivator CBP (the binding protein of the cAMP response element-binding protein CREB); together with other transcription factors beta-catenin/CBP binds to WRE (Wnt-responsive element) and activates transcription of a wide range of target genes of Wnt/beta-catenin signaling. Blocking the interaction of CBP and beta-catenin by this agent prevents gene expression of many proteins necessary for growth, thereby potentially suppressing cancer cell growth. The Wnt/beta-catenin signaling pathway regulates cell morphology, motility, and proliferation; aberrant regulation of this pathway leads to neoplastic proliferation.
JAPAN
4-(((6S,9S)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl) octahy- dro-1H-pyrazino[2,1-c][1,2,4]triazine-6-yl)methyl) phenyl dihydrogen phosphate
(6S,9S)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl) octahydro-1H-pyrazino[2,1-c] [I,z,4]triazine-1-carboxamide,
4-(((6S,9S,9aS)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-ylmethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazin-6-yl)methyl)phenyl dihydrogen phosphate
(6S,9S,9aS)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-yImethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazine- 1 -carboxamide.
Compound A as in wo 2014061827……..4-(((6S,9S,9aS)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-ylmethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazin-6-yl)methyl)phenyI dihydrogen phosphate in WO2014061827
4-(((6S,9S)-1-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl)octahydro-1H-pyrazino[2,1-c][1,2,4]triazine-6-yl)methyl)phenyl dihydrogen phosphate (presumed to be PRI-724; first disclosed in WO2009148192), useful for treating cancer, neurodegenerative diseases, glaucoma and idiopathic pulmonary fibrosis.
Eisai, under license from PRISM Pharma, is developing PRI-724, an inhibitor of CREB binding protein or beta-catenin complex formation, for treating cancer (phase 1, as of March 2015) and HCV-induced cirrhosis (preclinical trial).
Follows on from WO2014061827, claiming the use of PRI-724 for treating pulmonary fibrosis.
IS IT

cas 847591-62-2…………http://www.medkoo.com/Anticancer-trials/PRI-724.htm
(6S,9aS)-N-Benzyl-6-(4-hydroxybenzyl)-8-(naphthalen-1-ylmethyl)-4,7-dioxoperhydropyrazino[1,2-a]pyrimidine-1-carboxamide
COMPD 3
OR

COMPD 2
PRI724
1198780-43-6, 578.66, C33 H34 N6 O4
(6S,9S)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl) octahydro-1H-pyrazino[2,1-c] [I,z,4]triazine-1-carboxamide,

COMPD1
PRI 724
4-(((6S,9S)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl) octahy- dro-1H-pyrazino[2,1-c][1,2,4]triazine-6-yl)methyl) phenyl dihydrogen phosphate
COMPD 1
SEE
http://www.google.co.in/patents/WO2009148192A1?cl=en

About PRI-724
PRI-724 is an antiproliferative small molecule that selectively inhibits the CBP/beta-catenin complex, which modulates the beta-catenin dependent pathway of Wnt signaling. Activation of the Wnt/beta-catenin signaling pathway is observed in various tumor cells and results in proliferation and metastasis. PRI-724 exhibits a selective antiproliferative effect, inhibiting various cancer cell lines in vitroand substantially inhibiting tumor growth in animal studies. PRI-724 is currently in clinical trials in oncology indications, partnered with Eisai Co., Ltd. PRI-724 also has potential to provide therapeutic benefit in non-oncology areas such as fibrosis and clinical trials in that indication are targeted to start in the second half of 2013.
About PRISM Pharma Co., Ltd.
PRISM Pharma Co., Ltd. has developed its platform technology to modulate inter-cellular protein-protein interactions using peptide mimetic small molecules and found various hit compounds including PRI-724.
SEE
Eisai Research Institute; PRISM Pharma Co Ltd
出願人:エ_ ザイ■ ア_ ル■ アンド■ ディ_ ■
マネジメン卜株式会社(EISAI R&D MANAGEMENT
CO., LTD.) [JP /JP ];亍1128088 東京都文京区
小石川四丁目6 番1 O 号Tokyo (JP).株式会社P
R I S M P h a r m a (PRISM PHARMA CO.,
LTD.) [JP /JP ];亍2268510神奈川県横浜市緑区長津
田町 4 2 5 9 — 3 Kanagawa (JP)
(IO) 国際公開番号
2 0 1 5 ^ ® S 3 .2 0 1 5 )
WO 2015/037587 Al
This method of producing 4-(((6S,9S)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl) octahy- dro-1H-pyrazino[2,1-c][1,2,4]triazine-6-yl)methyl) phenyl dihydrogen phosphate involves a step for adding a reaction solution (I) comprising (6S,9S)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl) octahydro-1H-pyrazino[2,1-c] [I,z,4]triazine-1-carboxamide, triethylamine and a solvent to a reaction solution (2) comprising a phosphorylating agent and a solvent.
1
1H-NMR (600MHz, METHAN0L-d4) δ (ppm):1.15 (d, J=6 Hz, 3H), 2.65 (s, 3H), 3.12 (d, J=18 Hz, 1H), 3.35 (d, J=7 Hz, 2H), 3.48 (d, J=18 Hz,1H), 4.15 (m,1H), 4.32 (d, J=15 Hz, 1H), 4.40 (d, J=15 Hz, 1H), 5.33(d, J=16 Hz, 1H), 5.41(d, J=16 Hz, 1H), 5.44 (d, J=7 Hz, 1H), 5.64 (d, J=10 Hz, 1H), 7.07 (dd, J=9,1 Hz, 2H), 7.15 (d, J=9 Hz, 2H), 7.24 (t, J=7 Hz, 1H), 7.27 (d, J=7 Hz, 2H), 7.34 (t, J=8 Hz, 2H), 7.55 (d d, J=8, 4 Hz, 1H), 7.60 (brd, J=6 Hz, 1H), 7.62 (dd, J=8, 7 Hz, 1H), 7.88 (dd, J=8,1 Hz, 1H), 8.38 (dd, J=8, 2 Hz, 1H), 8.90 (dd, J =4, 2 Hz, 1H).
…………………………………………………………………….
SEE
http://www.google.co.in/patents/WO2009148192A1?cl=en
SYNTHESIS OF COMPD 2
PART A
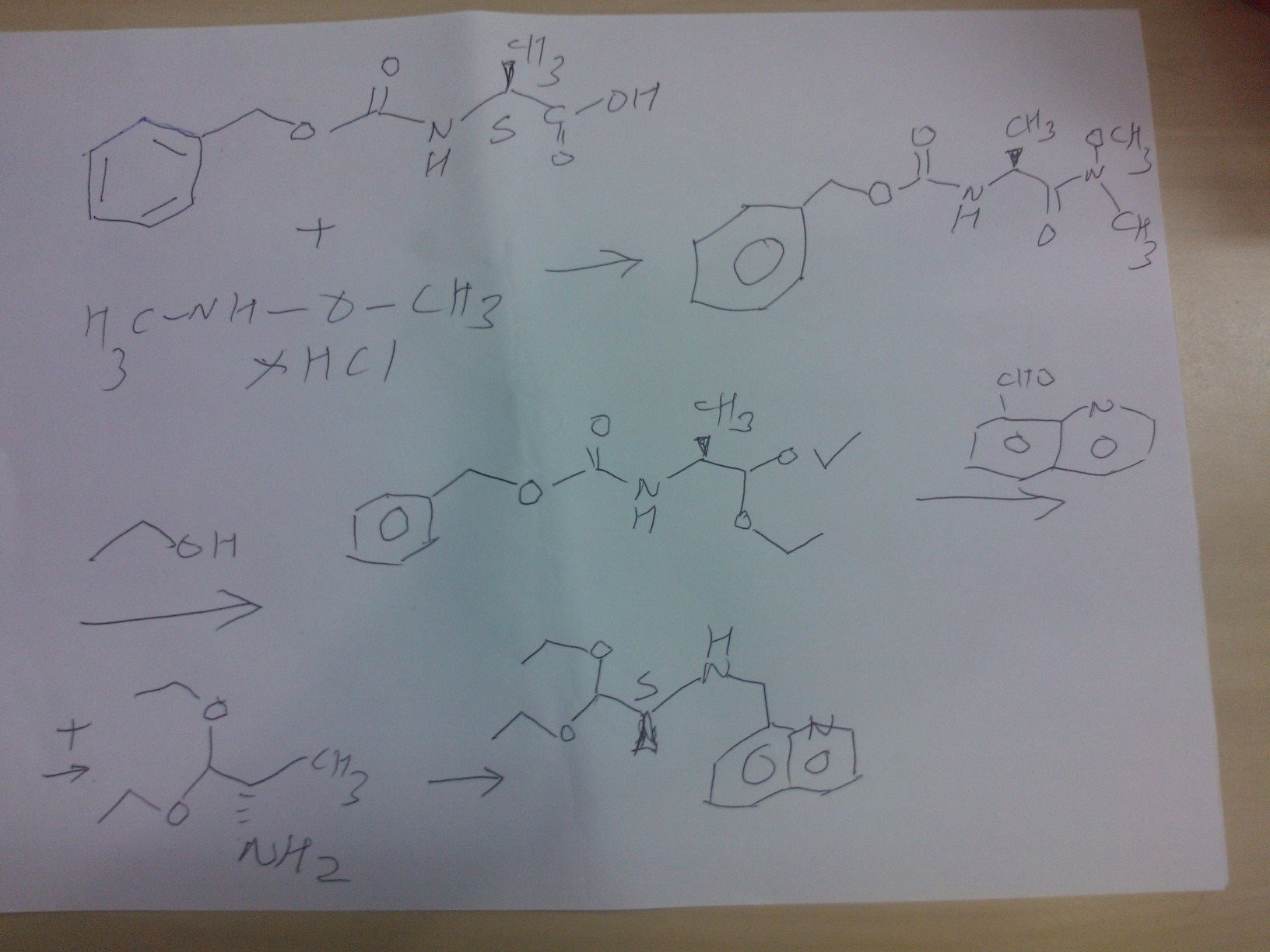
Synthesis Part A
step A
(S)-benzyl 1-(methoxy(methyl)amino)-1-oxopropan-2-ylcarbamate
Reaction of the foll……………….N-methoxy-N-methylamine hydrochloride, 1N sodium hydroxide , (S)-2-(benzyloxycarbonylamino)propanoic acidand 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride to obtain
(S)-benzyl 1-(methoxy(methyl)amino)-1-oxopropan-2-ylcarbamate.
STEP B
(S)-benzyl 1,1-diethoxypropan-2-ylcarbamate
Reaction of the foll……………….(S)-benzyl 1-(methoxy(methyl)amino)-1-oxopropan-2-ylcarbamate, 2M lithium aluminium hydride in tetrahydrofuran solution to obtain (S)-benzyl 1,1-diethoxypropan-2-ylcarbamate
STEP C
(S)-1,1-diethoxypropan-2-amine
Reaction of the foll……………….(S)-benzyl 1,1-diethoxypropan-2-ylcarbamate, 5% palladium on carbon title compound . (S)-1,1-diethoxypropan-2-amine,
STEP D
(S)-1,1-diethoxy-N-(quinolin-8-ylmethyl)propan-2-amine,
Reaction of the foll……………….(S)-1,1-diethoxypropan-2-amine,was reacted with 8-Quinolinecarboaldehyde to obtain the title
compound (S)-1,1-diethoxy-N-(quinolin-8-ylmethyl)propan-2-amine
PART B
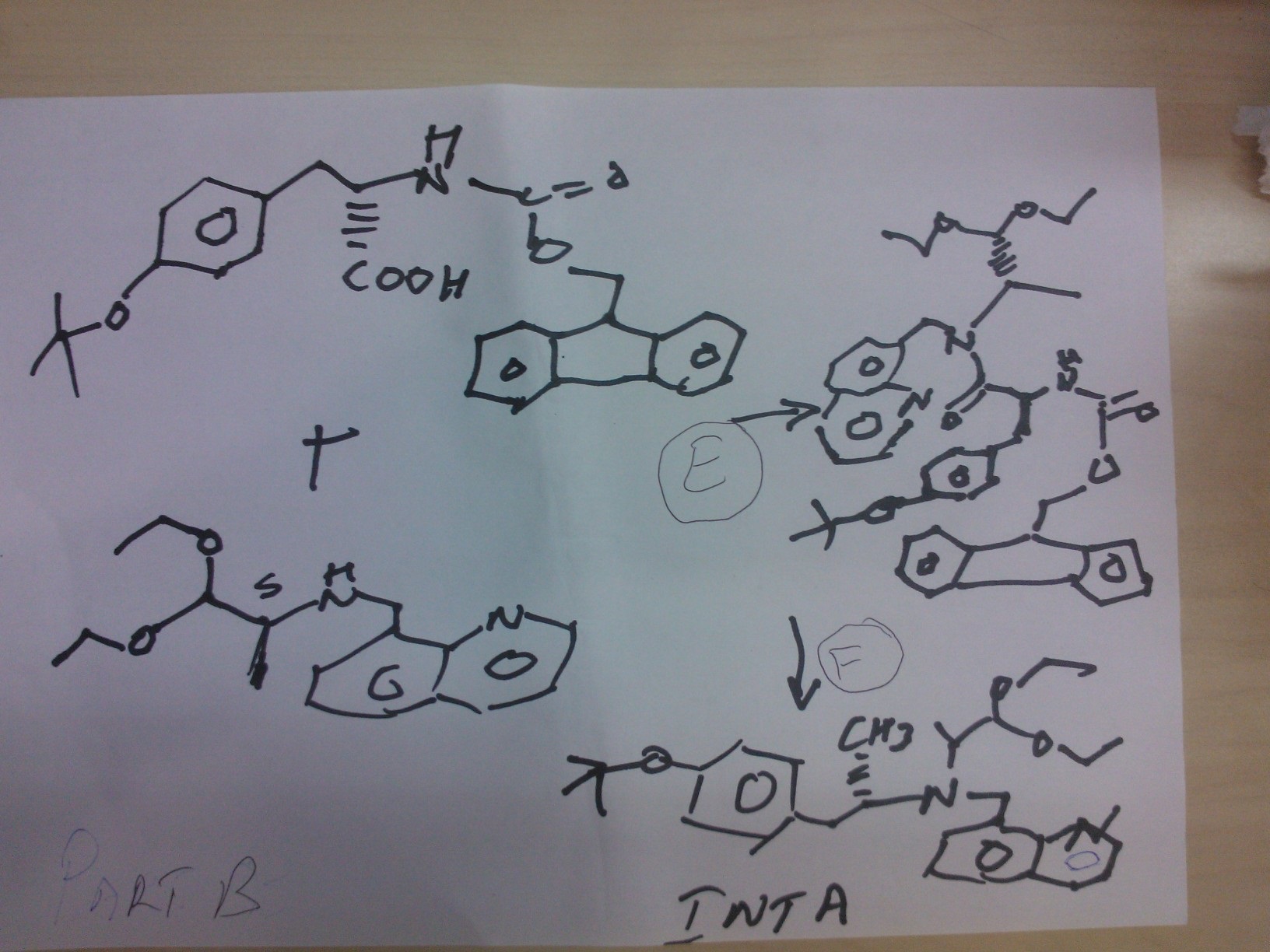
STEP E
(9H-fluoren-9-yl)methyl (S)-3-(4-tert-butoxyphenyl)-1-(((S)-1,1-diethoxypropan-2-yl)(quinolin-8-ylmethyl)amino)-1-oxopropan-2-ylcarbamate
Reaction of the foll………………. (S)-1,1-diethoxy-N-(quinolin-8-ylmethyl)propan-2-amine, (S)-2-(((9H-fluoren-9-yl)methoxy)carbonylamino)-3-(4-tertbutoxyphenyl)propanoic acid to obtain the title compound (9H-fluoren-9-yl)methyl (S)-3-(4-tert-butoxyphenyl)-1-(((S)-1,1-diethoxypropan-2-yl)(quinolin-8-ylmethyl)amino)-1-oxopropan-2-ylcarbamate
STEP f
(S)-2-amino-3-(4-tertbutoxyphenyl)-N-((S)-1,1-diethoxypropan-2-yl)-N-(quinolin-8-ylmethyl)propanamide INT A
Reaction of the foll………………. (9H-fluoren-9-yl)methyl (S)-3-(4-tert-butoxyphenyl)-1-(((S)-1,1-diethoxypropan-2-yl)(quinolin-8-ylmethyl)amino)-1-oxopropan-2-ylcarbamate and piperidine to
obtain the title compound (S)-2-amino-3-(4-tertbutoxyphenyl)-N-((S)-1,1-diethoxypropan-2-yl)-N-(quinolin-8-ylmethyl)propanamide INT A
PART C

STEP g
ethyl 2-(1-methylhydrazinyl)acetate
Reaction of the foll……………….methylhydrazine 7 was reacted with ethyl 2-bromoacetate 1to obtain the title compound
STEP h
ethyl 2-(1-Methyl-2-(benzylcarbamoyl)hydrazinyl)acetate
Reaction of the foll………………. ethyl 2-(1-methylhydrazinyl)acetateand benzyl isocyanate to obtain the title
compound ethyl 2-(1-Methyl-2-(benzylcarbamoyl)hydrazinyl)acetate
STEP i
2-(2-(benzylcarbamoyl)-1-methylhydrazinyl)acetic acid
Reaction of the foll………………. ethyl 2-(1-allyl-2-
(benzylcarbamoyl)hydrazinyl)acetate and lithium hydroxide monohydrate to obtain the title compound 2-(2-(benzylcarbamoyl)-1-methylhydrazinyl)acetic acid
STEP j
N-benzyl-2-(2-((S)-3-(4-tert-butoxyphenyl)-1-(((S)-1,1-
diethoxypropan-2-yl)(quinolin-8-ylmethyl)amino)-1-oxopropan-2-ylamino)-2-oxoethyl)-2-
methylhydrazinecarboxamide……… precursor
Reaction of the foll………………. 2-(2-(benzylcarbamoyl)-1-methylhydrazinyl)acetic acid and (S)-2-amino-3-(4-tert-butoxyphenyl)-N-((S)-1,1-diethoxypropan-2-yl)-N-(quinolin-8-ylmethyl)propanamide ( INT A )yielded the title compound ie the precursor
PART D
THIS PRECURSOR GIVES FINAL PRODUCT
Synthesis of (6S,9S)-N-benzyl-6-(4-hydroxybenzyl)-2,9-
dimethyl-8-(naphthalen-1-ylmethyl)-4,7-dioxooctahydro-1H-pyrazino[2,1-c][1,2,4]triazine-1-
carboxamide ……….final
fOLL reactants……….. N-benzyl-2-(2-((S)-3-(4-tert-butoxyphenyl)-1-(((S)-1,1-diethoxypropan-2-yl)(naphthalen-1-ylmethyl)amino)-1-oxopropan-2-ylamino)-2-oxoethyl)-2-methylhydrazinecarboxamide, ie the precursor and 10%-water/HCOOH gave (6S,9S)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-ylmethyl)octahydro-1Hpyrazino[2,1-c][1,2,4]triazine-1-carboxamide
RT 4.22; Mass 578.9
COMPD 3

(6S,9aS)-N-Benzyl-6-(4-hydroxybenzyl)-8-(naphthalen-1-ylmethyl)-4,7-dioxoperhydropyrazino[1,2-a]pyrimidine-1-carboxamide
SEE
US 6762185
……………………………..
SEE
http://www.google.com/patents/WO2012141038A1?cl=en

novel compounds, agent for inducing differentiation into hepatocytes of mesenchymal stem cells, Wnt / β- catenin signaling pathway inhibitor, method for producing hepatocytes with them on hepatocytes such as by their production.
Liver disease is said to be Japan’s national disease, a large number of patients suffering from liver disease. In addition, the annual number of deaths from hepatocellular carcinoma amounts to about 30 004 thousand people. Recently, hepatocellular cancer outcome is improved by advances in treatment, but the increase of advanced cancer, with hepatic dysfunction cirrhosis to merge, so-called hepatic failure death has increased. Liver failure therapy, although liver transplantation is ideal, it is difficult in Japan to obtain sufficient donors, it is necessary to develop a liver regeneration therapy with stem cells.
As stem cells that have the potential to differentiate into liver cells, bone marrow cells, tissue stem cells, such as umbilical cord blood cells can be expected.Therefore, a number of research institutions, for the realization of by regenerative medicine liver cell transplantation treatment of chronic liver failure patient, to differentiate human tissue stem cells into functional hepatocytes, truly clinically applicable efficient differentiation induction technology you are conducting research and development with the goal of developing a.
For example, in the laboratory of Shioda Professor of Tottori University Graduate School of Medicine, reported that the Wnt / β- catenin signaling pathway were differentiated into hepatocytes showed that suppressed by RNA interference at the time of induction of differentiation from human mesenchymal stem cells into hepatocytes you are (Non-Patent Document 1 and Non-Patent Documents 3-5).Furthermore, studies to induce differentiation of hepatocytes in other institutions have been conducted (Non-Patent Document 2, Patent Documents 1 and 2).
On the other hand, recently, from 4,000 or more screening of large compound libraries, Wnt / β- catenin signaling pathway inhibitory low molecular compound 5 types have been identified (Non-Patent Documents 6-9).
Kohyo 2009-535035 JP Patent Publication No. 2010-75631
Atsushi Yanagitani et al., ” retinoic Acid Receptor Dominant Level Negative Form Causes steatohepatitis and Liver Tumors in Transgenic Mice “, Hepatology, Vol. 40, No. 2, 2004, P. 366-375 Seoyoung Park et al.,”Hexachlorophene Inhibits Wnt / beta-catenin Pathway by Promoting Siah-Mediated beta-catenin Degradation “, Mol Pharmacol Vol. 70, No. 3, 960-966, 2006 Yoko Yoshida et al.,” A role of Wnt / beta-catenin Signals in hepatic fate Specification of human umbilical cord blood-derived mesenchymal stem cells “, Am J Physiol Gastrointest Liver Physiol 293:. G1089-G1098, 2007 Shimomura T et al,” Hepatic differentiation of human bone marrow-derived UE7T-13 cells: Effects of cytokines and CCN family Gene expression “, Hepatol Res., 37, 1068-79, 2007 Ishii K et al.,” Hepatic differentiation of human bone marrow-derived mesenchymal stem cells by tetracycline-regulated Hepatocyte Nuclear factor 3Beta “Hepatology, 48, 597- 606, 2008 Maina Lepourcelet et al., ” Small-molecule Antagonists of the oncogenic Tcf / beta-catenin protein complex “, CANCER CELL, JANUARY 2004, VOL. 5, 91-102 Emami KH et al.,” A Small molecule inhibitor of beta-catenin / CREB-binding protein Transcription “, Proc Natl Acad Sci US A. 2004 Aug 24; 101 (34):.. 12682-7 Jufang Shan et al,”Identification of a Specific Inhibitor of the Dishevelled PDZ Domain ” , Biochemistry 2005 Nov 29; 44 (47):.. 15495-503 Trosset JY et al, ” Inhibition of protein-protein Interactions: the discovery of beta-catenin Druglike Inhibitors by combining virtual and Biophysical Screening . “, Proteins 2006 Jul 1 ; 64 (1): 60-7
However, the conventional techniques described above literature, had a room for improvement in the following points.
Patent Documents 1 and 2, it has been described for proteins to induce stem cells from Hikimomiki cells, due to the use of the protein formulation as a differentiation inducing agent, a room for further improvement in terms of stability and safety and there was.
Non-Patent Document 1 and Non-Patent Document 3 to 5, and have reported that induced differentiated hepatocytes from human mesenchymal stem cells, the use of siRNA as a differentiation inducing agent, such as stability and safety there is room for further improvement in the surface. Non-Patent Document 2, 6 to 9, is not described with respect to method of inducing differentiation into hepatocytes.
The present invention has been made in view of the above circumstances, and an object thereof is to provide an effective low-molecular compounds that induce differentiation into hepatocytes from mesenchymal stem cells. Or, it is intended that the low-molecular compound was used to provide a secure differentiation inducing method is excellent from the mesenchymal stem cell differentiation efficiency of liver cells.
According to the present invention, there is provided formula (1) and one or more compounds selected from the group of compounds represented by the formula (2), a salt thereof or a solvate thereof.



<Example 1> synthetic ICG-001 of synthesis (1) ICG-001 of the IC-2 is an oligopeptide having two rings of β- turn mimic structure in central skeleton, and transcription by β-catenin / Tcf complex can function as a potent antagonist for activation has been reported (Drug Discov. Today 2005, 10, 1467-1474). Synthesis of ICG-001 in accordance with the literature (Tetrahedron 2007, 63, 12912-12916), was subjected to examination.

(1-1) of Compound 1 Synthesis 1-naphtaldehyde (Wako Pure Chemical) (1.56 g, 10 mmol) and 2,2-diethoxyethanamine (Tokyo Kasei Kogyo) (1.33 g, 10 mmol) were mixed 100 I was stirred 20 min at o C. After cooling to room temperature, diluted with EtOH (20 mL), was added portionwise NaBH 4 (0.38 g, 10 mmol), at room temperature, and stirred for 16 h. After completion of the reaction, was distilled off by concentration under reduced pressure EtOH, the product was extracted with AcOEt. The resulting product was purified by silica gel column chromatography (hexane / AcOEt = 5/1) to give the to give compound 1 (2.29 g, 8.5 mmol, 85%).

(1-2) Synthesis of Compound 3 Fmoc-L-Tyr (t-Bu) -OH (0.87 g, 1.9 mmol) in DMF (7 mL) solution of a condensing agent HATU (0.76 g, 2.0 mmol) and diisopropylethylamine (DIEA) (0.35 mL, 2.0 mmol) was added and after stirring for 20 min, compound 1 (0.54 g, a 2.0 mmol) was added, at room temperature, 16 h the mixture was stirred. After the reaction, DMF was distilled off by concentration under reduced pressure, and the resulting product was purified by column chromatography (hexane / AcOEt = 10/1), compound 2 was obtained (1.33 g, 1.9 mmol, 93%). The resulting compound 2 (1.33 g, 1.9 mmol) was dissolved in CH 2 Cl 2 (20 mL), was added diethylamine (DEA) (10 ml, excess), at room temperature, was 2 h stirring.After confirming the completion of the reaction by TLC, vacuum was distilled off CH 2 Cl 2 by concentration, the resulting product was purified by silica gel column chromatography (AcOEt), to give compound 3 (0.92 g, 1. 8 mmol, 92%).

(1-3) Synthesis Fmoc-β-Ala-OH (0.53 g, 1.7 mmol) of compound 5 in DMF (8 mL) solution of a condensing agent HATU (0.70 g, 1.8 mmol) and diisopropylethylamine (DIEA) (0.32 mL, 1.8 mmol) was added and after stirring for 20 min, compound 3 (0.92 g, 1.8 mmol) was added, at room temperature, and stirred for 14 h. After the reaction, DMF was distilled off by concentration under reduced pressure, the resulting product was purified by column chromatography (hexane / AcOEt = 1/1), compound 4 was obtained (1.2 g, 1.5 mmol, 82%). Obtained compound 4 (1.2 g, 1.5 mmol) was dissolved in CH 2 Cl 2 (20 mL), was added diethylamine (DEA) (9 mL, excess), at room temperature, and stirred for 1 h. After confirming the completion of the reaction by TLC, was distilled off CH 2 Cl 2 by concentration under reduced pressure, and the resulting product was purified by silica gel column chromatography (AcOEt / EtOH = 1/1), to give compound 5 (0 .66 g, 1.2 mmol, 80%).

(1-4) synthetic compounds 5 (0.66 g, 1.2 mmol) of compound 7 CH 2 Cl 2 of solution (8 mL) to benzylisocyanate (0.16 g, 1.2 mmol) of CH 2 Cl 2 solution (8 mL) was added, at room temperature, and stirred for 12 h. After confirming the completion of the reaction by TLC, was distilled off CH 2 Cl 2 by concentration under reduced pressure, and the resulting product was purified by column chromatography (AcOEt / EtOH = 1/1), to give compound 6 (0. 59 g, 0.85 mmol, 73%). The obtained compound 6 (0.59 g, 0.85 mmol) at room temperature in the formic acid (9 ml), I was stirred 20 h. Was evaporated formic acid by concentration under reduced pressure, the resulting product was purified by column chromatography (AcOEt), Compound 7a to (ICG-001) was obtained as a white solid (0.26 g, 0.48 mmol, 57 %).
The resulting product, MS spectra and were identified from the 1 H NMR spectrum (with the literature value) (Fig. 1).

STRUCTURE 4
4-(((6S,9S,9aS)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-ylmethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazin-6-yl)methyl)phenyl dihydrogen phosphate
STRUCTURE 5
(6S,9S,9aS)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-yImethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazine- 1 -carboxamide.
Cas 1422253-37-9
2H-Pyrazino[2,1-c][1,2,4]triazine-1(6H)-carboxamide, hexahydro-6-[(4-hydroxyphenyl)methyl]-2,9-dimethyl-4,7-dioxo-N-(phenylmethyl)-8-(8-quinolinylmethyl)-, (6S,9S,9aS)-
Structure can represented as

P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

FDA approves new treatment for diabetic retinopathy in patients with diabetic macular edema

03/25/2015
The U.S. Food and Drug Administration today expanded the approved use for Eylea (aflibercept) injection to treat diabetic retinopathy in patients with diabetic macular edema.
March 25, 2015
Release
The U.S. Food and Drug Administration today expanded the approved use for Eylea (aflibercept) injection to treat diabetic retinopathy in patients with diabetic macular edema.
Diabetic retinopathy (DR) is the most common diabetic eye disease and is a leading cause of blindness in adults in the United States. According to the Centers for Disease Control and Prevention, diabetes (type 1 and type 2) affects more than 29 million people in the United States and is the leading cause of new blindness among people ages 20 to 74 years. In 2008, 33 percent of adults with diabetes aged 40 years or older had some form of DR. In some cases of DR with diabetic macular edema (DME), abnormal new blood vessels grow on the surface of the retina. Severe vision loss or blindness can occur if the new blood vessels break.
“Diabetes is a serious public health crisis, affecting more patients every year,” said Edward Cox, M.D., M.P.H, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval gives patients with diabetic retinopathy and diabetic macular edema another therapy to treat this vision-impairing complication.”
In February, the FDA approved Lucentis (ranibizumab injection) 0.3 mg to treat DR in patients with DME.
Eylea is administered by a physician as an injection into the eye once a month for the first five injections and then once every two months. It is intended to be used along with appropriate interventions to control blood sugar, blood pressure and cholesterol.
The safety and efficacy of Eylea to treat DR in patients with DME were evaluated in 679 participants in two clinical studies where participants were randomly assigned to receive Eylea or macular laser photocoagulation, a laser-based treatment used to burn small areas of the retina. At week 100, participants being treated with Eylea showed significant improvement in the severity of their DR, compared to patients who did not receive Eylea.
The most common side effects associated with Eylea include bleeding of the conjunctiva (the tissue that lines the inside of the eyelids and covers the white part of the eye); eye pain; cataracts; floaters; increased pressure inside the eye (increased intraocular pressure); and separation of the interior jelly of the eye from the retina (vitreous detachment). Serious adverse reactions include infection within the eye (endophthalmitis) and retinal detachments.
The FDA granted breakthrough therapy designation to Eylea for the treatment of DR with DME. The FDA can designate a drug a breakthrough therapy at the request of the sponsor if preliminary clinical evidence indicates the drug may demonstrate a substantial improvement over available therapies for patients with serious or life-threatening conditions. The FDA also reviewed the new use for Eylea under the agency’s priority review program, which provides for an expedited review of drugs that demonstrate the potential to be a significant improvement in safety or effectiveness in the treatment of a serious condition.
The FDA previously approved Eylea to treat wet (neovascular) age-related macular degeneration, a condition in which abnormal blood vessels grow and leak fluid into the macula. Eylea is also approved to treat DME and macular edema secondary to retinal vein occlusions, both of which cause fluid to leak into the macula resulting in blurred vision.
Eylea is marketed by Tarrytown, N.Y.-based Regeneron Pharmaceuticals Inc. Lucentis is marketed by South San Francisco, California-based Genentech, a subsidiary of Roche Pharmaceuticals.
GSK 923295, a CENP-E Inhibitor

GSK-923295A
1088965-37-0
Synonym: GSK-923295; GSK 923295; GSK923295.
CENP-E Inhibitor
IUPAC/Chemical name:
3-Chloro-N-{(1S)-2-[(N,N-dimethylglycyl)amino]-1-[(4-{8-[(1S)-1-hydroxyethyl]imidazo[1,2-a]pyridin-2-yl}phenyl)methyl]ethyl}-4-[(1-methylethyl)oxy]benzamide
3-chloro-N-[(1S)-2-[[2-(dimethylamino)acetyl]amino]-1-[[4-[8-[(1S)-1-hydroxyethyl]imidazo[1,2-a]pyridin-2-yl]phenyl]methyl]ethyl]-4-(1-methylethoxy)- Benzamide,
3-Chloro-N-{(1S)-2-[(N,N-dimethylglycyl)amino]-1-[(4-{8-[(1S)-1-hydroxyethyl]imidazo[1,2-a]pyridin-2-yl}phenyl)methyl]ethyl}-4-[(1-methylethyl)oxy]benzamide
3-Chloro-N-[(1S)-2-[(N,N-dimethylglycyl)amino]-1-({4-[8-(1-hydroxyethyl)imidazo[1,2-a]pyridin-2-yl]phenyl}methyl)ethyl]-4-[(1-methylethyl)oxy]benzamide
3-Chloro-N-[1-(N,N-dimethylglycinamido)-3-[4-[8-[1(S)-hydroxyethyl]imidazo[1,2-a]pyridin-2-yl]phenyl]propan-2(S)-yl]-4-isopropoxybenzamide
C32H38ClN5O4
Exact Mass: 591.26123
Molecular Weight: 592.12822
Elemental Analysis: C, 64.91; H, 6.47; Cl, 5.99; N, 11.83; O, 10.81
Kinesin-like protein KIF11 inhibitor; Centromere protein E inhibitor
GSK-923295 is a novel antimitotic inhibitor of centromere-associated protein E (CENP-E) with potential anticancer activity. GSK923295A demonstrated significant antitumor activity against solid tumor models, inducing CRs in Ewing sarcoma, rhabdoid, and rhabdomyosarcoma xenografts.
GSK-923295, a small-molecule inhibitor of centromere associated protein (CENP), is in early clinical development at Cytokinetics for the treatment of refractory cancer. No recent development has been reported for early clinical research which had been ongoing at GlaxoSmithKline.
Clinical study showed that GSK923295 had dose-proportional pharmacokinetics and a low number of grade 3 or 4 adverse events. The observed incidence of myelosuppression and neuropathy was low. Further investigations may provide a more complete understanding of the potential for GSK923295 as an antiproliferative agent.
GSK923295 is a first-in-class, specific allosteric inhibitor of CENP-E kinesin motor ATPase with Ki of 3.2 nM, and less potent to mutant I182 and T183. Phase 1.
The compound potently inhibits CENP-E ATPase activity and exerts broad-spectrum antiproliferative activity against cancer cells and xenografts. GSK-923295 has demonstrated a broad spectrum of activity against a range of human tumor xenografts grown in nude mice, including models of colon, breast, ovarian, lung and other tumors.
Cytokinetics was developing GSK-923295, the lead from a series of small-molecule mitotic kinesin spindle protein inhibitors, for treating cancer including advanced solid tumors. However, since October 2014, the program was no longer listed on the Cytokinetics’ website
In 2001, a strategic alliance was established between Cytokinetics and GlaxoSmithKline to discover, develop and commercialize novel small-molecule therapeutics targeting mitotic kinesins for applications in the treatment of cancer and other diseases.

…………………….
PATENT
US8772507
http://www.google.com/patents/US8772507


1,1-Dimethylethyl [(1S)-2-(4-bromophenyl)-1-(hydroxymethyl)ethyl]carbamate

To a solution of 4-bromo-N-{[(1,1-dimethylethyl)oxy]carbonyl}-L-phenylalanine (72.6 mmol), in anhydrous diethyl ether (550 mL) at 0° C. was added slowly lithium aluminum hydride, 95% (108.9 mmol). The resulting solution was stirred for an additional 2 h at 0° C. The reaction was then carefully quenched with a saturated aqueous solution of sodium bicarbonate (73 mL) which stirred at RT for half an hour. Lithium aluminium salts crashed out of solution and were removed by filtration. The filtrate was concentrated and vacuum pumped for 24 h to afford the title product as a white solid (97%). ESMS [M+H]+: 331.2.
1,1-Dimethylethyl {(1S)-2-(4-bromophenyl)-1-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]ethyl}carbamate

To a solution of 1,1-dimethylethyl [(1S)-2-(4-bromophenyl)-1-(hydroxymethyl)ethyl]carbamate (70.6 mmol), tripheylphosphine (84.7 mmol), and phthalimide (84.7 mmol) in anhydrous tetrahydrofuran (550 mL) at 0° C. was added dropwise diisopropyl azodicarboxylate (84.7 mmol) over 10 minutes. The reaction continued to stir allowing to warm to RT over 5 h. The reaction was then concentrated in vacuo and product was triturated out of solution using ethyl acetate (500 mL). The precipitate was filtered, washed with ethyl acetate (3×100 mL), and dried to afford the title product as a white solid (57%). ESMS [M+H]+: 460.4.
1,1-Dimethylethyl {(1S)-2-[4-(bromoacetyl)phenyl]-1-[(1,3-d oxo-1,3-dihydro-21′-isoindol-2-yl)methyl]ethyl}carbamate

A solution of 1,1-dimethylethyl {(1S)-2-(4-bromophenyl)-1-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]ethyl}carbamate (21.7 mmol), 1-ethoxyvinyltri-n-butylin (43.5 mmol), and trans-dichlorobis(triphenylphosphine)palladium(II) (5 mol %) were stirred in anhydrous dioxane (300 mL) at 100° C. for 3 h. The reaction was then concentrated in vacuo and redissolved in a solution of tetrahydrofuran and water (3:1, 400 mL). The mixture was treated with N-bromosuccinimide (108.8 mmol) and stirred at RT for half an hour. The reaction solution was then concentrated to dryness and redissolved in ethyl acetate (150 mL). Precipate formed upon addition of hexanes (350 mL) and was filtered and dried to afford the title product as yellow solid (71%). ESMS [M+H]+: 502.4.
1,1-Dimethylethyl [(1S)-2-(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)-1-({4-[8-(1-hydroxyethyl)imidazo[1,2-a]pyridin-2-yl]phenyl}methyl)ethyl]carbamate

A mixture of 1,1-dimethylethyl{(1S)-2-{4-(bromoacetyl)phenyl]-1-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]ethyl}carbamate (1.90 g, 3.79 mmol), 1-(2-amino-3-pyridinyl)ethanol (0.523 g, 3.79 mmol), and solid sodium bicarbonate (0.398 g, 4.72 mmol) in isopropanol (24 mL) was refluxed for 3.0 h. The mixture was concentrated in vacuo and the residue dissolved in ethyl acetate, washed with water and saturated sodium chloride, dried (Na2SO4), and concentrated to give the title compound (1.79 g, 87%) as a light pink solid. MS (ES+) m/e 541 [M+H]+.
3-Chloro-N-[(1S)-2-(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)-1-({4-[8-(1-hydroxyethyl)imidazo[1,2-a]pyridin-2-yl]phenyl}methyl)ethyl]-4-[(1-methylethyl)oxy]benzamide

A mixture of 1,1-dimethylethyl [(1S)-2-(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)-1-({4-[8-(1-hydroxyethyl)imidazo[1,2-a]pyridin-2-yl]phenyl}methyl)ethyl]carbamate (1.79 g, 3.31 mmol) and 4 M HCl in 1,4-dioxane (20 mL, 80 mmol) was stirred at room temperature for 45 minutes. The reaction was concentrated to dryness and redissolved in DMF (30 mL). To this solution was added N,N-diisopropylethylamine (2.14 g, 16.55 mmol) and pentafluorophenyl 3-chloro-4 [(1-methylethyl)oxy]benzoate (1.36 g, 3.31 mmol). The mixture was stirred overnight at room temperature, diluted with water, and extracted into ethyl acetate. The extracts were washed with water, dried (Na2SO4), and concentrated in vacuo to give the title compound (2.10 g, 100%) as a tan solid. MS (ES+) m/e 637 [M+H]+.
N-[(1S)-2-Amino-1-({4-[8-(1-hydroxyethyl)imidazo[1,2-a]pyridin-2-yl]phenyl}methyl)ethyl]-3-chloro-4-[(1-methylethyl)oxy]benzamide

A mixture of 3-chloro-N-[(1S)-2-(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)-1-({4-[8-(1-hydroxyethyl)imidazo[1,2-a]pyridin-2-yl]phenyl}methyl)ethyl]-4-[(1-methylethyl)oxy]benzamide (2.10 g, 3.30 mmol) and hydrazine monohydrate (0.83 g, 16.5 mmol) in ethanol (30 mL) was heated at 57° C. overnight. The reaction was cooled, diluted with ethanol, filtered, and concentrated to give the title compound (1.67 g, 100%) as a pale yellow powder. MS (ES+) m/e 507 [M+H]+.
3-Chloro-N-[(1S)-2-[(N,N-dimethylglycyl)amino]-1-({4-[8-(1-hydroxyethyl)imidazo[1,2-a]pyridin-2-yl]phenyl}methyl)ethyl]-4-[(1-methylethyl)oxy]benzamide

A mixture of N-[(1S)-2-amino-1-({4-[8-(1-hydroxyethyl)imidazo[1,2-a]pyridin-2-yl]phenyl}methyl)ethyl]-3-chloro-4-[(1-methylethyl)oxy]benzamide (0.912 g, 1.80 mmol), EDCI (0.69 g, 3.6 mmol), N,N-diisopropylethylamine (0.466 g, 3.6 mmol), and N,N-dimethylglycine (0.372 g, 3.6 mmol) in methylene chloride (17 mL) was stirred overnight at room temperature. The reaction was diluted with water, washed with brine, dried (Na2SO4), and concentrated. The residue was purified by flash chromatography on silica gel (8%-10% MeOH:CH2Cl2) to give the title compound (0.515 g, 48%) as a pale yellow solid. MS (ES+) ink 592 [M+H]+.
………………….
WO2005107762
https://www.google.im/patents/WO2005107762A2
Example 1

cheme E:
ide


NaHCOj, IPA 100 ‘C


1 , 1 -Dimethylethyl [( 1 S)-2-(4-bromophenyl)- 1 -(hydroxymethyl)ethyl]carbamate:

To a solution of 4-bromo-N-{[(l ,1 -dimethylethyl)oxy] carbonyl }-L- phenylalanine (72.6 mmol), in anhydrous diethyl ether (550 mL) at 0 °C was added slowly lithium aluminum hydride, 95% (108.9 mmol). The resulting solution was stiπed for an additional 2 h at 0 °C, The reaction was then carefully quenched with a saturated aqueous solution of sodium bicarbonate (73 mL) which stiπed at RT for half an hour. Lithium aluminium salts crashed out of solution which were removed by filtration. The filtrate was concentrated and vacuum pumped for 24 h to afford the title product as a white solid (97%).
ESMS [M+H]+: 331.2.
1,1 -Dimethylethyl {(lS)-2-(4-bromophenyl)-l-[(l,3-dioxo-l,3-dihydro-2H-isoindol-2- yl)methyl]ethyl}carbamate:

To a solution of 1 ,1 -dimethylethyl [(lS)-2-(4-bromophenyl)-l –
(hydroxymethyl)ethyl]carbamate (70.6 mmol), tripheylphosphine (84.7 mmol), and phthalimide (84.7 mmol) in anhydrous tetrahydrofuran (550 mL) at 0 °C was added dropwise diisopropyl azodi carboxyl ate (84.7 mmol) over 10 minutes. The reaction continued to stir allowing to wai to RT over 5h, The reaction was then concentrated in vacuo and product was tritarated out of solution usingl acetate (500 mL). The precipitate was filtered, washed with ethyl acetate (3 x 100 mL), and dried to afford the title product as a white solid (57%).
ESMS [M+H]+: 460.4.
1 ,1 -Dimethylethyl {(15)-2-[4-(bromoacetyl)phenyl]-l -[(l,3-dioxo-l ,3-dihydro-2H-isoindol- 2-yl)methyl]ethyl}carbamate:

A solution of 1,1 -dimethyl ethyl {(lS)-2-(4-bromophenyl)-l-[(l,3-dioxo-l,3- dihydro-2H-isoindol-2-yl)methyl]ethyl}carbamate (21.7 mmol), 1-ethoxyvinyltri-n-butylin (43.5 mmol), and /ra/?s–dichlorobis(triphenylphospine)palladιum(II) (5 mol%) were stiπed in anhydrous dioxane (300 mL) at 100 °C for 3h. The reaction was then concentrated in vacuo and redissolved in a solution of tetrahydrofuran and water (3:1, 400mL) and treated with N- bromosuccinimide (108.8 mmol) and stined at RT for half an hour. The reaction solution was then concentrated to dryness and redissolved in ethyl acetate (150 mL) and precipate formed upon addition of hexanes (350 mL). The precipitate was filtered and dried to afford the title product as yellow solid (71%). ESMS [M+Η]+: 502.4. l,l-Dimethylethyl [(lS)-2-(l ,3-dioxo-l,3-dihydro-2H-isoindol-2-yl)-l-({4-[8-(l- hydroxyethyl)imidazo[l,2-β]pyridin-2-yl]phenyl}methyl)ethyl]carbamate:

A mixture of l!l-dimethylethyl{(lS)-2-{4-(biOinoacetyl)phenyl]-l-[(l,3- dioxo-l ,3-dihydro-2H-isoindol-2-yl)methyl]ethyl}carbamate (1.90 g, 3.79 mmol), l-(2- amino-3-pyτidinyl)ethanol (0.523 g, 3.79 mmol), and solid sodium bicarbonate (0.398 g, 4,72 mmol) in isopropanol (24 mL) was refluxed for 3.0 h. and concentrated in vacuo. The residue was dissolved in ethyl acetate, washed with water and saturated sodium chloride, dried (Na2S04), and concentrated to give the title compound (1.79 g, S7%) as a light pink solid. MS(ES+) m/e 541 [M+Η]+.
3-Chloro-N-[(lS)-2-(l,3-dioxo-l ,3-dihydro-2H-isoindol-2-yl)-l-({4-[8-(l- hydroxyethyl)imidazo[l,2-Λ]pyridin-2-yl]phenyl}methyl)ethyl]-4-[(l – methylethyl)oxy]benzamide:

A mixture of 1,1 -dimethylethyl [(15)-2-(l,3-dioxo-l,3-dihydro-2H-isoindol-2- yl)-l-({4-[8-(l-hydroxyethyl)imidazo[l,2-fl]pyridin-2-yl]phenyl}methyl)ethyl]carbamate (1.79 g, 3.31 mmol) and 4M ΗC1 in 1,4-dioxane (20 mL, 80 mmol) was stirred at room temperature for 45 minutes. The reaction was concentrated to dryness ,redissolved in DMF (30 mL), and to this solution was added N,N-diisopropylethylamine (2.14 g, 16,55 mmol) and pentafluorophenyl 3-chloro-4 [(l-methylethyl)oxy]benzoate (1.36 g, 3.31 mmol). The mixture was stirred overnight at room temperature, diluted with water, and extracted into ethyl acetate. The extracts were washed with water, dried (Na SO ), and concentrated in vacuo to give the title compound (2.10 g, 100%) as a tan solid. MS(ES+) m/e 637 [M+H]+.
N-[(lS)-2-Amino-l-({4-[8-(l-hydroxyethyl)imidazo[l,2-α]p>tidin-2- yl]phenyl}methyl)eth)’l]-3-chloro-4-[(l-methylethyl)oxy]benzamide:

A mixture of 3-chloro-N-[(lS)-2-(l,3-dioxo-l ,3-dihydro-2N-isoindol-2-yl)-l-
({4-[8-(l -hydiOxyethyl)imidazo[l,2-β]pyridin-2-yl]phenyl}methyl)ethyl]-4-[(l- methylethyl)oxy]benzamide (2.10 g, 3.30 mmol) and hydrazine monohydrate (0.83 g, 16.5 mmol) in ethanol (30 mL) was heated at 57°C ovemight. The reaction was cooled, diluted with ethanol, filtered, and concentrated to give the title compound(1.67 g, 100%) as a pale yellow powder. MS(ES+) m/e 507 [M+H]+.
3-Chloro-N-[(15)-2-[(7VN-dimethylglycyl)amino]-l-({4-[8-(l-hydroxyethyl)imidazo[l ,2- «]pyitdin-2-yl]phenyl}methyl)ethyl]-4-[(l-methylethyl)oxy]benzamide:

A mixture ofN-[(lS)-2-amino-l-({4-[S-(l-hydroxyethyl)imidazo[l,2- α]pyridin-2-yl]phenyl)methyl)ethyl]-3-chloro-4-[(l-methylethyl)oxy]benzamide (0.912 g, 1 ,80 mmol), EDCI (0.69 g, 3,6 mmol), NN-diisopropylethylamine (0.466 g, 3,6 mmol), and N,N-dimethylglycine (0.372 g, 3.6 mmol) in methylene chloride (17 mL) was stirred overnight at room temperature. The reaction was diluted with water, washed with brine, dried (Νa2S0 ), and concentrated. The residue was purified by flash chromatography on silica gel (8%-10% MeOH:CH2Cl2) to give the title compound ( 0.515 g, 48%) as a pale yellow solid. MS(ES+) m/e 592 [M+H]+.
SEE
WO2008 / 138561
………………..
Organic Process Research & Development (2010), 14(5), 1254-1263
Org. Process Res. Dev., 2010, 14 (5), pp 1254–1263
DOI: 10.1021/op100186c
http://pubs.acs.org/doi/abs/10.1021/op100186c

The discovery and development of an efficient manufacturing route to the CENP-E inhibitor 3-chloro-N-{(1S)-2-[(N,N-dimethylglycyl)amino]-1-[(4-{8-[(1S)-1-hydroxyethyl]imidazo[1,2-a]pyridin-2-yl}phenyl)methyl]ethyl}−4-[(1-methylethyl)oxy]benzamide (GSK923295A) is described. The existing route to GSK923295A was expensive, nonrobust, used nonideal reagents, and consistently struggled to deliver the API needed for clinical studies. The new synthesis commences from the readily available l-phenylalaninol, which is smoothly converted through to GSK923295A using key Friedel−Crafts acylation as well as selective acylation chemistries. Downstream chemistry to GSK923295A is both high yielding and robust, and the resulting process has been demonstrated first on the kilo scale and subsequently in the pilot plant where 55 kg was successfully prepared. The resulting process is simple, uses cheaper raw materials, is greener in that it avoids using aluminum, tin, and bromination chemistries, and obviates the need for chromatographic purification. Also discussed are the route derived impurities, how they were unambiguously prepared to confirm structure and processing amendments to control their formation, and enhancements to the new process to facilitate future processing.
1H NMR (400 MHz, CD3OD) δH 1.34 (6H, d, J = 6.0, (CH3)2), 1.59 (3H, d, J = 7.0, CH3CH), 2.21 (6H, s, N(CH3)2), 2.87−3.01 (4H, m, CH2Ph and CH2N(CH3)2), 3.49 (2H, m, CH2NPhthal), 4.50 (1H, m, CHNH), 4.70 (1H, m, (CH3)2CHO)), 5.49 (1H, q, J = 7.0, CHOH), 6.88 (1H, t, J = 7.0, H-j), 7.08 (1H, d, J = 7.5, H-b), 7.33−7.37 (3H, m, H-k and H-d), 7.63 (1H, dd, J = 7.5 and 2.0, H-c), 7.78 (1H, s, H-a), 7.83 (2H, d, J = 7.0, H-e), 8.09 (1H, m, H-h), 8.27 (1H, d, J = 8.0, H-i);
13C NMR (100 MHz, CD3OD) δC 22.2, 24.1, 39.3, 43.8, 46.1, 53.0, 63.7, 66.2, 73.0, 110.4, 113.8, 115.3, 121.2, 124.5, 126.1, 127.5, 128.4, 128.5, 130.6, 130.7, 133.3, 136.0, 139.4, 145.1, 146.1, 157.6, 168.5 and 173.6;
HRMS (ESI+) m/z calculated for [M+H]+ C32H39N5O4Cl 592.2691, found 592.2684.
…………………….
PREDICTIONS
http://orgspectroscopyint.blogspot.in/2015/03/gsk-923295.html
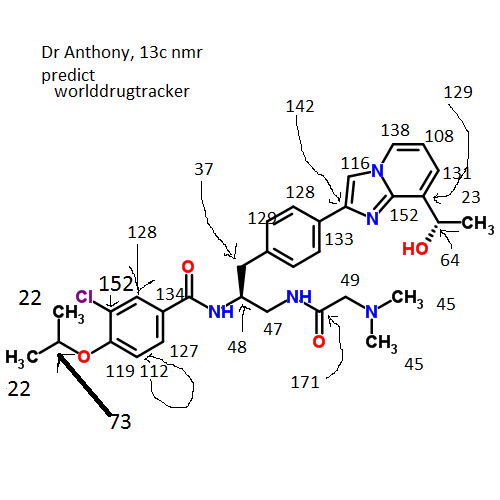

1H NMR PREDICT

see http://orgspectroscopyint.blogspot.in/2015/03/gsk-923295.html
ACS Medicinal Chemistry Letters (2010), 1(1), 30-34
http://pubs.acs.org/doi/abs/10.1021/ml900018m

Inhibition of mitotic kinesins represents a novel approach for the discovery of a new generation of anti-mitotic cancer chemotherapeutics. We report here the discovery of the first potent and selective inhibitor of centromere-associated protein E (CENP-E) 3-chloro-N-{(1S)-2-[(N,N-dimethylglycyl)amino]-1-[(4-{8-[(1S)-1-hydroxyethyl]imidazo[1,2-a]pyridin-2-yl}phenyl)methyl]ethyl}-4-[(1-methylethyl)oxy]benzamide (GSK923295; 1), starting from a high-throughput screening hit, 3-chloro-4-isopropoxybenzoic acid 2. Compound 1 has demonstrated broad antitumor activity in vivo and is currently in human clinical trials.
SEE
WO-2015037460
Method for producing optically active 3-(biphenyl-4-yl)-2-[(t-butoxycarbonyl)amino]propan-1-ol
Process for preparing optically active 3-(biphenyl-4-yl)-2-[(t-butoxycarbonyl)amino]propan-1-ol, useful as an intermediate in the synthesis of pharmaceuticals described in WO2005107762 and WO2008138561 (such as GSK-923295 and tubulysin derivatives respectively). Appears to be a new area of interest to the assignee.
…………..
WO2010118207
|
References |
1: Mayes PA, Degenhardt YY, Wood A, Toporovskya Y, Diskin SJ, Haglund E, Moy C, Wooster R, Maris JM. Mitogen-activated protein kinase (MEK/ERK) inhibition sensitizes cancer cells to centromere-associated protein E inhibition. Int J Cancer. 2013 Feb 1;132(3):E149-57. doi: 10.1002/ijc.27781. Epub 2012 Sep 28. PubMed PMID: 22948716.
2: Chung V, Heath EI, Schelman WR, Johnson BM, Kirby LC, Lynch KM, Botbyl JD, Lampkin TA, Holen KD. First-time-in-human study of GSK923295, a novel antimitotic inhibitor of centromere-associated protein E (CENP-E), in patients with refractory cancer. Cancer Chemother Pharmacol. 2012 Mar;69(3):733-41. doi: 10.1007/s00280-011-1756-z. Epub 2011 Oct 22. PubMed PMID: 22020315.
3: Lock RB, Carol H, Morton CL, Keir ST, Reynolds CP, Kang MH, Maris JM, Wozniak AW, Gorlick R, Kolb EA, Houghton PJ, Smith MA. Initial testing of the CENP-E inhibitor GSK923295A by the pediatric preclinical testing program. Pediatr Blood Cancer. 2012 Jun;58(6):916-23. doi: 10.1002/pbc.23176. Epub 2011 May 16. PubMed PMID: 21584937; PubMed Central PMCID: PMC3163687.
4: Balamuth NJ, Wood A, Wang Q, Jagannathan J, Mayes P, Zhang Z, Chen Z, Rappaport E, Courtright J, Pawel B, Weber B, Wooster R, Sekyere EO, Marshall GM, Maris JM. Serial transcriptome analysis and cross-species integration identifies centromere-associated protein E as a novel neuroblastoma target. Cancer Res. 2010 Apr 1;70(7):2749-58. doi: 10.1158/0008-5472.CAN-09-3844. Epub 2010 Mar 16. PubMed PMID: 20233875; PubMed Central PMCID: PMC2848992.
5: Wood KW, Lad L, Luo L, Qian X, Knight SD, Nevins N, Brejc K, Sutton D, Gilmartin AG, Chua PR, Desai R, Schauer SP, McNulty DE, Annan RS, Belmont LD, Garcia C, Lee Y, Diamond MA, Faucette LF, Giardiniere M, Zhang S, Sun CM, Vidal JD, Lichtsteiner S, Cornwell WD, Greshock JD, Wooster RF, Finer JT, Copeland RA, Huang PS, Morgans DJ Jr, Dhanak D, Bergnes G, Sakowicz R, Jackson JR. Antitumor activity of an allosteric inhibitor of centromere-associated protein-E. Proc Natl Acad Sci U S A. 2010 Mar 30;107(13):5839-44. doi: 10.1073/pnas.0915068107. Epub 2010 Feb 18. PubMed PMID: 20167803; PubMed Central PMCID: PMC2851928.
