
Home » 0rphan drug status
Category Archives: 0rphan drug status
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Engasertib
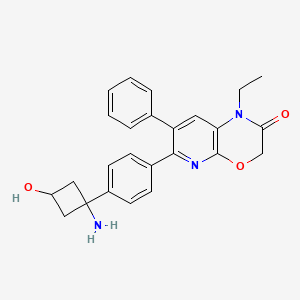
Engasertib
CAS 1313439-71-2
MF C25H25N3O3 MW415.5 g/mol
6-[4-(1-amino-3-hydroxycyclobutyl)phenyl]-1-ethyl-7-phenylpyrido[2,3-b][1,4]oxazin-2-one
6-{4-[(1S,3S)-1-amino-3-hydroxycyclobutyl]phenyl}-1-ethyl-7-phenyl-1H-pyrido[2,3-b][1,4]oxazin-2(3H)-one
serine/threonine kinase inhibitor, ALM-301, VAD-044, ALM 301, VAD 044, Orphan Drug, K2US8HW4TQ
Engasertib is an oral, once-daily AKT inhibitor developed by Vaderis Therapeutics, primarily investigated as a targeted therapy for Hereditary Hemorrhagic Telangiectasia (HHT). Clinical trials show it safely reduces the frequency and duration of bleeding episodes without an FDA-approved equivalent currently available
Core Information
- Mechanism of Action: Engasertib is a highly selective inhibitor of AKT1 and AKT2. In HHT, mutations in the ALK1 pathway lead to abnormal blood vessel growth driven by an excess of the AKT protein. By inhibiting AKT, the drug promotes vascular stability and reduces vessel fragility.
- Target Indication: Hereditary Hemorrhagic Telangiectasia (HHT) — a rare, severe genetic disorder causing vascular abnormalities and frequent, heavy bleeding, particularly nosebleeds (epistaxis)
Clinical Efficacy & Safety
- Proof-of-Concept Trial: A 12-week, placebo-controlled study with 75 HHT patients evaluated daily doses of 30 mg and 40 mg.
- The 40 mg cohort demonstrated a 41% reduction in mean bleeding duration and a 28% reduction in bleeding frequency, compared to 24% and 18% in the placebo group.
- 61% of patients in the 40 mg group rated their clinical condition as “Much Better”.
- Extended Efficacy: In long-term open-label extensions, benefits were sustained and amplified over 12 months, resulting in a 66% reduction in mean bleeding duration and a 55% reduction in bleeding frequency.
- Side Effects: Generally well-tolerated. The most common side effects (reversible and manageable with supportive care) were mild-to-moderate rash and hyperglycemia
- OriginatorAlmac Discovery
- DeveloperVaderis Therapeutics
- ClassAntineoplastics; Small molecules; Vascular disorder therapies
- Mechanism of ActionProto-oncogene protein c-akt inhibitors
- Orphan Drug StatusYes – Hereditary haemorrhagic telangiectasia
- Phase IVascular disorders
- PreclinicalBreast cancer; Prostate cancer
- No development reportedHereditary haemorrhagic telangiectasia
- 28 Dec 2025No recent reports of development identified for phase-I development in Hereditary haemorrhagic telangiectasia in Belgium (PO, Capsule)
- 28 Dec 2025No recent reports of development identified for phase-I development in Hereditary haemorrhagic telangiectasia in France (PO, Capsule)
- 28 Dec 2025No recent reports of development identified for phase-I development in Hereditary haemorrhagic telangiectasia in Italy (PO, Capsule)
SYN
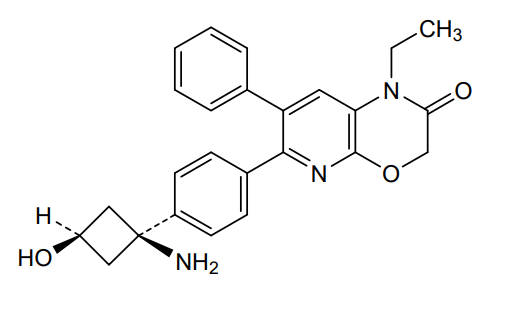
Example 139: 6-(4-((1s.3s)-1-amino-3-hydroxycyclobutyl)phenyl)-1-ethyl-7-phenyl-1H-pyrido[2,3-b][1,4]oxazin-2(3H)-one
Step 1: tert-butyl ((1s.3s)-1-(4-(1-ethyl-2-oxo-7-phenyl-2.3-dihydro-1H-pyrido[2,3-b][1,4]oxazin-6-yl)phenyl)-3-hydroxycyclobutyl)carbamate
In a 15 mL reaction tube was added 6-bromo-1-ethyl-7-phenyl-1H-pyrido[2,3-b][1,4]oxazin-2(3H)-one (50 mg, 0.150 mmol), tert-butyl ((1s,3s)-3-hydroxy-1-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)cyclobutyl)carbamate (49 mg, 0.125 mmol) and cesium carbonate (204 mg, 0.625 mmol) in a mixture of 1,4-dioxane (2.3 ml) and water (0.8 ml) to give a colourless solution. This was degassed by bubbling nitrogen for 15 minutes, followed by the addition of [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II) dichloromethane adduct (20 mg, 0.025 mmol) and degassing for a further 5 minutes. The reaction mixture was heated to 50°C under a nitrogen atmosphere for one hour then allowed to cool to room temperature, diluted with water (5 ml) and extracted into ethyl acetate (3 x 5 ml). The combined organic phases were dried over Na2SO4, filtered and concentrated to dryness under reduced pressure. The residue was purified by Biotage chromatography (cyclohexane:ethyl acetate, gradient elution from 90:10 to 0:100) to give the desired product as an off-white solid (45 mg, 70% yleld). Ή-NMR (500 MHz, CDCl3) δ 7.29-7.35 (5H, m), 7.28 (1H, s), 7.18-7.24 (4H, m), 4.96 (1H, br s), 4.88 (2H, s), 4.05 (1H, br s), 4.01 (2H, q), 2.98 (2H, br s), 2.75 (2H, br s), 1.20-1.51 (9H, br m), 1.32 (3H, t). LCMS (Method D) RT = 1.25 min, M+H+ = 516.20.
Step 2: 6-(4-((1s,3s)-1-amino-3-hydroxycyclobutyl)phenyl)-1-ethyl-7-phenyl-1H-pyrido[2,3-b][1,4]oxazin-2(3H)-one
tert-butyl ((1s,3s)-1-(4-(1-ethyl-2-oxo-7-phenyl-2,3-dihydro-1H-pyrido[2,3-b][1,4]oxazin-6-yl)phenyl)-3-hydroxycyclobutyl)carbamate (45 mg, 0.087 mmol) was dissolved in TFA (1 mL) and stirred for 30 seconds. The solution was immediately concentrated to dryness under reduced pressure. The residue was dissolved in diethyl ether (~3 mL) and concentrated to dryness under reduced pressure three times. The residue was then slurried in diethyl ether (3 mL) and after settling the supernatant solvent removed by pipette. This was repeated three times. The remaining solvent was removed by freeze drylng overnight to give the desired compound as an off-white solid (33 mg, 71% yleld).
1H-NMR (500 MHz, MeOD) δ 7.55 (1H, s), 7.39-7.42 (4H, m), 7.27-7.31 (3H, m), 7.20-7.24 (2H, m), 4.93 (2H, s), 4.01-4.11 (3H, m), 3.03-3.11 (2H, m), 2.42-2.50 (2H, m), 1.28 (3H, t). LCMS (Method D) RT = 0.74 min, M+H+ = 416.20.
SYN
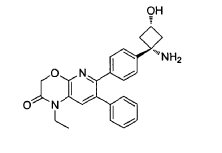
EXAMPLES
Example 1: Synthesis of 6-(4-(l-amino-3-hvdroxycvclobutyl)phenyl)-l-ethyl-7-phenyl-lH-pyrido[2,3-b][l,4]oxazin-2(3H)-one
6-(4-(l-amino-3-hydroxycyclobutyl)phenyl)-l-ethyl-7-phenyl-lH-pyrido[2,3-b][l,4]oxazin-2(3H)-one (referred to herein as “VAD044 free base”) was synthesized in accordance with the protocol as set out in W02011077098 – see in particular Examples 97, 113 and 139 (reproduced below):
Synthesis of 6-(4-((ls,3s)-l-amino-3-hvdroxycvclobutyl)phenyl)-l-ethyl-7-phenyl-lH-pyrido[2,3-bHl,41oxazin-2(3H)-one: from WO2Q11077098 Example 139:
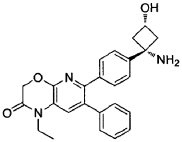
Step 1: tert-butyl((ls,3s)-l-(4-(l-ethyl-2-oxo-7-phenyl-2,3-dihydro-lH-pyrido[2,3-b][l,4]oxazin-6-yl)phenyl)-3-hvdroxycvclobutyl)carbamate
In a 15 mL reaction tube was added 6-bromo-l-ethyl-7-phenyl-lH-pyrido[2,3-b][l,4]oxazin-2(3H)-one* (50 mg, 0.150 mmol), tert-butyl((ls,3s)-3-hydroxy-l-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)cyclobutyl)carbamate** (49 mg, 0.125 mmol) and cesium carbonate (204 mg, 0.625 mmol) in a mixture of 1,4-dioxane (2.3 ml) and water (0.8 ml) to give a colourless solution. This was degassed by bubbling nitrogen for 15 minutes, followed by the addition of [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(ll) dichloromethane adduct (20 mg, 0.025 mmol) and degassing for a further 5 minutes. The reaction mixture was heated to 50°C under a nitrogen atmosphere for one hour then allowed to cool to room temperature, diluted with water (5 ml) and extracted into ethyl acetate (3 x 5 ml). The combined organic phases were dried over Na2SO4, filtered and concentrated to dryness under reduced pressure. The residue was purified by Biotage chromatography (cyclohexane:ethyl acetate, gradient elution from 90:10 to 0:100) to give the desired product as an off- white solid (45 mg, 70% yield). 1H-NMR (500 MHz, CDCI3) 6 7.29-7.35 (5H, m), 7.28 (1H, s), 7.18-7.24 (4H, m), 4.96 (1H, br s), 4.88 (2H, s), 4.05 (1H, br s), 4.01 (2H, q), 2.98 (2H, br s), 2.75 (2H, br s), 1.20-1.51 (9H, br m), 1.32 (3H, t). LCMS (Method D) RT = 1.25 min, M+H+ = 516.20.

tert-butyl((ls,3s)-l-(4-(l-ethyl-2-oxo-7-phenyl-2,3-dihydro-lH-pyrido[2,3-b][l,4]oxazin-6-yl)phenyl)-3- hydroxycyclobutyl)carbamate (45 mg, 0.087 mmol) was dissolved in TFA (1 mL) and stirred for 30 seconds. The solution was immediately concentrated to dryness under reduced pressure. The residue was dissolved in diethyl ether (~3 mL) and concentrated to dryness under reduced pressure three times. The residue was then slurried in diethyl ether (3 mL) and after settling the supernatant solvent removed by pipette. This was repeated three times. The remaining solvent was removed by freeze drying overnight to give the desired compound as an off-white solid (33 mg, 71% yield). 1H-NMR (500 MHz, MeOD) 6 7.55 (1H, s), 7.39- 7.42 (4H, m), 7.27-7.31 (3H, m), 7.20- 7.24 (2H, m), 4.93 (2H, s), 4.01-4.11 (3H, m), 3.03-3.1 1 (2H, m), 2.42-2.50 (2H, m), 1.28 (3H, t). LCMS (Method D) RT = 0.74 min, M+H+ = 416.20.

To a suspension of sodium hydride (5.31 g, 133 mmol) in 1,4-dioxane (250 ml), ethyl glycolate (12.56 ml,
133 mmol) was added drop wise over a period of 30 minutes ensuring that the temperature was maintained below 30°C. The resulting thick suspension was stirred at room temperature for 15 minutes.
In a separate II round- bottomed flask was added 5-bromo-2-chloro-3-nitropyridine (21 g, 88 mmol) in
1,4-dioxane (150 ml) to give a brown solution. The suspension of sodium hydride and ethyl glycolate was added drop wise over a period of 30 minutes at 0°C. The resulting reaction mixture was heated to 80°C overnight.
The reaction mixture was concentrated under reduced pressure and the crude residue was purified by
Biotage silica chromatography (gradient 0% to 10% ethyl acetate in n-hexanes) to give the title compound
(1 ,8g, 44%).1H NMR (500 MHz, CDCI3) 6 8.48 (1H, s), 8.42 (1H, s), 5.07 (2H, s), 4.28-4.24 (2H, q), 1.31-1.28
(3H, t).
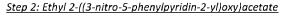
In a II round-bottomed flask was added ethyl 2-(5-bromo-3-nitropyridin-2-yloxy)acetate (18.33 g, 60.1 mmol), phenylboronic acid (10.99 g, 90 mmol), triphenylphosphine (4.73 g, 18.02 mmol) and cesium fluoride (45.6 g, 300 mmol) in 1,2-dimethoxyethane (300 ml) to give a yellow solution. The reaction mixture was degassed by bubbling nitrogen for 30 minutes. Pallad ium (II ) acetate (2.023 g, 9.01 mmol) was added and the mixture was heated to 75°C under a nitrogen atmosphere overnight. The reaction mixture was allowed to cool to room temperature and concentrated to dryness under reduced pressure to give a brown solid. This was re-dissolved in dichloromethane, filtered and concentrated to dryness under reduced pressure to give a brown solid The crude residue was purified via Biotage chromatography (gradient 5% to 60% ethyl acetate in n-hexanes) to give the title compound (6.9g, 38%). 1H NMR (500 MHz, CDCI3) 6 8.58 (1H, s), 8.56 (1H, s), 7.59-7.52 (2H, m), 7.48-7.46 (2H, m), 7.45-7.43 (1H, m), 5.13 (2H, s), 4.30-4.26 (2H, q), 1.33-1.30 (3H, t).
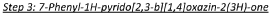
In a 500 ml round-bottomed flask was added ethyl 2-(3-nitro-5-phenylpyridin-2-yloxy)acetate (4.6 g, 15.22 mmol) in hydrochloric acid, 37% (40 ml) to give a yellow suspension. The mixture was cooled to 0-5°C followed by the portion wise addition of tin powder (9.94 g, 84 mmol). The addition proved to be exothermic. Caution should be taken while adding. The mixture was then stirred at room temperature for further 30 minutes until all foaming ceased. The reaction mixture was heated to 80°C under a nitrogen atmosphere for 3 hours. The reaction mixture cooled to room temperature and diluted with water (800ml). The white precipitate was isolated by filtration, washed with water (100 ml) and sucked dry to give a white solid. The solid was azeotroped with toluene (3 x 30 ml) to give a white solid as the title compound (2.6g, 77%). XH NMR (500 MHz, (CD3)2SO) 6 10.41 (1H, s), 8.10 (1H, s), 7.59 (2H, d), 7.49-7.42 (2H, t), 7.39-7-38 (1H, d), 4.83 (2H, s).
Step 4: 6-bromo-7-phenyl-lH-pyrido[2,3-b][l,4]oxazin-2(3H)-one
In a 10ml microwave vial was 7-phenyl-l H-pyrido[2,3-b][l ,4]oxazin-2(3H)-one (50 mg, 0.221 mmol) and N-bromosuccinimide (78.6 mg, 0.441 mmol) in dimethylformamide (1 ml). The reaction mixture was heated to 80°C under microwave irradiation for 30 minutes. The reaction mixture was cooled to room
temperature and diluted with ethyl acetate (10ml). The organic solution was washed with water (2x10ml) and brine (2x10ml). The organic phase was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude residue was purified via Biotage chromatography (gradient 0% to 5% methanol in dichloromethane) to give the title compound as a yellow solid (61 mg, 90%). 1H NMR (500 MHz, CD3OD) 6 7.48-7.32 (5H, m), 7.12 (1 H, s), 4.82 (2H, s).
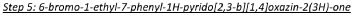
In a 15 mL reaction tube was added 6-bromo-7-phenyl-lH-pyrido[2,3-b][l,4]oxazin-2(3H)-one (300 mg, 0.983 mmol), iodoethane (0.095 mL, 1.180 mmol) and potassium carbonate (408 mg, 2.95 mmol) in anhydrous N,N-dimethylformamide (1 mL) to give a brown suspension. This was stirred at 50 °C under a nitrogen atmosphere for 60 minutes. The reaction mixture was diluted with saturated sodium bicarbonate solution (5 mL) and extracted into ethyl acetate (3 x 5 mL). The combined organic phases were washed with 50:50 water.brine (3 x 5 mL), dried over Na2SO4, filtered and concentrated to dryness under reduced pressure to give a brown solid. This was purified by Biotage chromatography (25g silica cartridge, cyclohexane:ethyl acetate, gradient elution from 90:10 to 20:80) to give the title compound as a beige solid (160 mg, 48.8 % yield). XH NMR (500 MHz, CDCI3) 6 7.58-7.37 (5H, m), 7.21 (1H, s), 4.86 (2H, s), 3.96 (2H, q), 1.27 (3H, t). LCMS (Method D) RT 1.293 min, M+l= 334.

n a 40 mL reaction tube was added tert-butyl(ls,3s)-l-(4-bromophenyl)-3- hydroxycyclobutylcarbamate*** (0.25 g, 0.731 mmol) in anhydrous tetrahydrofuran (14 ml) to give a colourless solution. This was degassed by bubbling nitrogen for 20 minutes, followed by the addition of [l,l’-bis(diphenylphosphino)ferrocene]dichloropalladium(ll) dichloromethane adduct (60 mg, 0.073 mmol). After bubbling nitrogen for a further 15 minutes, potassium acetate (143 mg, 1.461 mmol) and bis(pinacolato)diboron (223 mg, 0.877 mmol) were added. The reaction mixture was heated to reflux overnight then concentrated to dryness under reduced pressure and purified by Biotage chromatography (cyclohexane:ethyl acetate, gradient elution from 88:12 to 0:100) to give the desired product as a colourless oil that solidified upon standing (240 mg, 84% yield). 1H-NMR (500 MHz, CDCI3) 6 7.71 (2H, d), 7.44 (2H, d), 4.15 (1H, br s), 2.87-2.98 (2H, m), 2.27-2.44 (2H, m), 1.22-1.49 (21H, br m).
(*** synthesis described in WO2009148887 and WO2009148916)
ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
- Inhibitors of akt activityPublication Number: EP-2516435-B1Priority Date: 2009-12-23Grant Date: 2014-08-06
- Inhibitors of akt activityPublication Number: WO-2011077098-A1Priority Date: 2009-12-23
- Inhibitors of akt activityPublication Number: EP-2516435-B8Priority Date: 2009-12-23Grant Date: 2014-10-15
- Inhibitors of akt activityPublication Number: EP-2516435-A1Priority Date: 2009-12-23
- Inhibitors of AKT activityPublication Number: US-9221838-B2Priority Date: 2009-12-23Grant Date: 2015-12-29
- Allosteric akt inhibitors for use in the treatment of hereditary hemorrhagic telangiectasiaPublication Number: US-2024092801-A1Priority Date: 2020-09-30
- Allosteric akt inhibitors for use in the treatment of hereditary hemorrhagic telangiectasiaPublication Number: WO-2022069552-A1Priority Date: 2020-09-30
- Allosteric akt inhibitors for use in the treatment of hereditary hemorrhagic telangiectasiaPublication Number: EP-4221713-A1Priority Date: 2020-09-30
- Inhibitors of akt activityPublication Number: US-2013116243-A1Priority Date: 2009-12-23
- Inhibitors of akt activityPublication Number: WO-2011077098-A9Priority Date: 2009-12-23
////////engasertib, anax labs, serine/threonine kinase inhibitor, ALM-301, VAD-044, ALM 301, VAD 044, Orphan Drug, K2US8HW4TQ
Repinatrabit
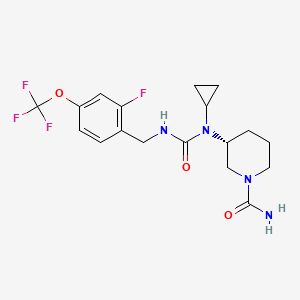
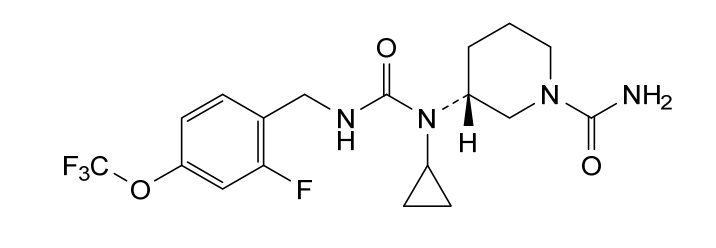
Repinatrabit
CAS 2837993-05-0
MF C18H22F4N4O3 MW 418.4 g/mol
(3R)-3-[cyclopropyl-[[2-fluoro-4-(trifluoromethoxy)phenyl]methylcarbamoyl]amino]piperidine-1-carboxamide
(3R)-3-[cyclopropyl({[2-fluoro-4-(trifluoromethoxy)phenyl]methyl}carbamoyl)amino]piperidine-1-
carboxamide
solute carrier family 6 member 19 (SLC6A19) inhibitor(phenylketonuria), JNT-517, JNT 517, orphan drug, rare pediatric disease designations, Jnana Therapeutics, 5P44NDU6AC, JN 11804, JN-11804
Repinatrabit (JNT-517) is an investigational, oral, small-molecule drug developed by Jnana Therapeutics (now part of Otsuka Pharmaceutical) to treat Phenylketonuria (PKU). It acts as a selective inhibitor of the SLC6A19 transporter, reducing blood phenylalanine (Phe) levels by increasing its urinary excretion.
Key Details About Repinatrabit:
- Mechanism: It targets a novel, cryptic allosteric site to block kidney reabsorption of phenylalanine, aiming to be a first-in-class oral therapy for all PKU patients, regardless of age or genotype.
- Clinical Trials: Otsuka initiated a global Phase 3 study (NCT06971731) in December 2025 to evaluate its safety and efficacy, following positive results from earlier studies.
- Status: The FDA has granted it orphan drug and rare pediatric disease designations.
- A Study to Evaluate the Safety and Efficacy of JNT-517 in Participants With Phenylketonuria (PKU)CTID: NCT06971731Phase: Phase 3Status: RecruitingDate: 2026-02-04
- A Phase 2 Study of JNT-517 in Adolescent Participants With PhenylketonuriaCTID: NCT06637514Phase: Phase 2Status: RecruitingDate: 2025-08-19
- First-in-Human, Multiple Part Clinical Study of JNT-517 in Healthy Participants and in Participants With PhenylketonuriaCTID: NCT05781399Phase: Phase 1/Phase 2Status: Active, not recruitingDate: 2025-07-31
- A Study to Evaluate the Long-Term Safety and Efficacy of JNT-517 in Participants With PhenylketonuriaCTID: NCT06628128Phase: Phase 3Status: Not yet recruitingDate: 2025-06-03
EMA Drug Information,
Type, Orphan designations
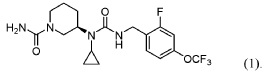
(R)-3-(1-Cyclopropyl-3-(2-fluoro-4-(trifluoromethoxy)benzyl)ureido)piperidine-1-carboxamide
Intended Use, Treatment of hyperphenylalaninaemia, Status of Orphan Designation, Positive
First Published Date, 2024-08-22
PAT
SYN
PAT
PAT
- Crystalline forms of a piperidine inhibitor of slc6a19 functionPublication Number: WO-2024118721-A1Priority Date: 2022-11-30
- Crystalline forms of a piperidine inhibitor of slc6a19 functionPublication Number: EP-4626420-A1Priority Date: 2022-11-30
- Dosing regimen for treatment of PKU with SLC6A19 functional piperidine inhibitorsPublication Number: CN-120091814-APriority Date: 2022-09-14
- Small molecule inhibitors of mammalian slc6a19 functionPublication Number: US-2024208923-A1Priority Date: 2021-03-10
- Small molecule inhibitors of mammalian slc6a19 functionPublication Number: US-2025289797-A1Priority Date: 2021-03-10
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
///////////repinatrabit, ANAX, JNT-517, JNT 517, orphan drug, rare pediatric disease designations, Jnana Therapeutics, 5P44NDU6AC, JN 11804, JN-11804
Nuvisertib
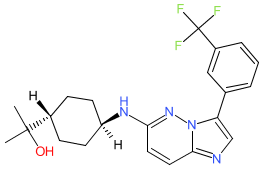
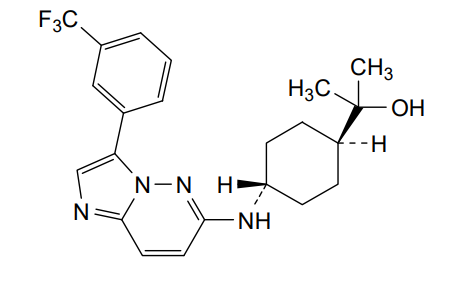

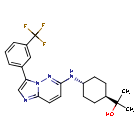
Nuvisertib
CAS 1361951-15-6
MF C22H26ClF3N4O MW418.5 g/mol
2-[(1r,4r)-4-({3-[3-(trifluoromethyl)phenyl]imidazo[1,2-b]pyridazin-6-yl}amino)cyclohexyl]propan-2-ol
serine/ threonine kinase inhibitor, antineoplastic, Orphan Drug, myelofibrosis, SGI-9481, SGI 9481, TP-3654, TP 3654, EOB0N7BOY4
The chemical structure for nuvisertib was obtained from proposed INN list 130 (Feb. 2024), in which the compound is described as a serine/ threonine kinase inhibitor with antineoplastic action. A structure match to clinical lead TP-3654 was made via PubChem. TP-3654 is declared as an orally available, second-generation pan-PIM kinase inhibitor [1-2].
| References |
| 1. Foulks JM, Carpenter KJ, Luo B, Xu Y, Senina A, Nix R, Chan A, Clifford A, Wilkes M, Vollmer D et al.. (2014) A small-molecule inhibitor of PIM kinases as a potential treatment for urothelial carcinomas. Neoplasia, 16 (5): 403-12. [PMID:24953177] |
| 2. Wu CP, Li YQ, Chi YC, Huang YH, Hung TH, Wu YS. (2021) The Second-Generation PIM Kinase Inhibitor TP-3654 Resensitizes ABCG2-Overexpressing Multidrug-Resistant Cancer Cells to Cytotoxic Anticancer Drugs. Int J Mol Sci, 22 (17). [PMID:34502348] |
Nuvisertib is an orally available, second-generation and selective ATP-competitive inhibitor of proviral integration site for Moloney murine leukemia virus (PIM) kinases, with potential antineoplastic activity. Upon oral administration, nuvisertib selectively binds to and prevents the activation of the PIM kinases. This prevents the activation of PIM-mediated signaling pathways and inhibits proliferation in cells that overexpress PIM. PIMs, constitutively active proto-oncogenic serine/threonine kinases, are upregulated in various types of cancers and play key roles in tumor cell proliferation and survival.
Nuvisertib, also known as TP-3654, is an oral, investigational, and highly selective PIM1 kinase inhibitor being studied in a Phase 1/2 clinical trial for intermediate- or high-risk myelofibrosis (MF). It is not currently an approved medication.
Key Information
- Mechanism of Action: Nuvisertib targets the PIM1 kinase pathway, which is often overactive in myelofibrosis and can promote cancer cell growth. By inhibiting this pathway, nuvisertib is being investigated for its potential to manage symptoms, reduce spleen size, improve blood counts, and slow the progression of bone marrow fibrosis.
- Current Status: Nuvisertib is in ongoing Phase 1/2 clinical trials (NCT04176198) as a monotherapy and in combination with JAK inhibitors like ruxolitinib and momelotinib.
- Designations: Nuvisertib has received Orphan Drug Designation for myelofibrosis
Study of TP-3654 in Patients With Advanced Solid Tumors
CTID: NCT03715504
Phase: Phase 1
Status: Completed
Date: 2023-11-14
SYN
WO2013013188
Example 31
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US427659372&_cid=P10-MHWTVL-76212-1
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US130491286&_cid=P10-MHWU33-81462-1
31. 4-((3-(3-(Trifluoromethyl)phenyl)imidazo[1,2-b]pyridazin-6-yl)amino)-trans-cyclohexyl)propan-2-ol (EX. 8-31)
| EX. 8-31 was prepared by similar procedures as in EX. 8-1 using 2-(trans-4-aminocyclohexyl)propan-2-ol. |

| 1H-NMR (CD 3OD/400 MHz): δ 8.82 (s, 1H), 8.19 (m, 1H), 7.88 (s, 1H), 7.62 (m, 3H), 6.70 (d, J=9.6 Hz, 1H), 3.71 (m, 1H), 2.26 (m, 2H), 1.95 (m, 2H), 1.36 (m, 1H), 1.27 (m, 4H), 1.21 (s, 6H). MS (ES +, m/z): (M+H) +: 419.6. |
| To a solution of trans-4-((tert-butoxycarbonyl)amino)cyclohexanecarboxylic acid (823 g, 3.38 mol) in EtOAc (4000 mL) was added EA/HCl (2500 mL). The mixture was stirred at 0° C. overnight. The reaction mixture was filtered and dried in vacuo to give a product of hydrochloride salt of trans-4-aminocyclohexanecarboxylic acid as white solid (604 g, 99.42% yield). |
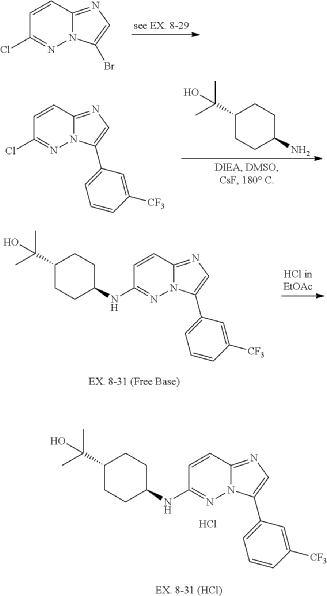
| 6-chloro-3-(3-(trifluoromethyl)phenyl)imidazo[1,2-b]pyridazine was prepared according to procedure in EX. 8-29. |
PAT
- Heterocyclic protein kinase inhibitorsPublication Number: ES-2834093-T3Priority Date: 2011-07-21Grant Date: 2021-06-16
- Substituted imidazo[1,2-b]pyridazines as protein kinase inhibitorsPublication Number: US-2021238183-A1Priority Date: 2011-07-21
- Imidazo[1,2-b]pyridazine and pyrazolo[1,5-a]pyrimidine derivatives and their use as protein kinase inhibitorsPublication Number: US-2012058997-A1Priority Date: 2006-11-06
- Substituted imidazo[1,2-b]pyridazines as protein kinase inhibitorsPublication Number: US-9416132-B2Priority Date: 2011-07-21Grant Date: 2016-08-16
- Heterocyclic protein kinase inhibitorsPublication Number: WO-2013013188-A1Priority Date: 2011-07-21
- Heterocyclic protein kinase inhibitorsPublication Number: EP-3409278-B1Priority Date: 2011-07-21Grant Date: 2020-09-16
- Substituted imidazo[1,2-B]pyridazines as protein kinase inhibitorsPublication Number: US-10875864-B2Priority Date: 2011-07-21Grant Date: 2020-12-29
- Heterocyclic protein kinase inhibitorsPublication Number: EP-3812387-A1Priority Date: 2011-07-21
- Substituted imidazo[1,2-B]pyridazines as protein kinase inhibitorsPublication Number: US-10392392-B2Priority Date: 2011-07-21Grant Date: 2019-08-27
- Heterocyclic protein kinase inhibitorsPublication Number: US-2014329807-A1Priority Date: 2011-07-21
- Substituted imidazo[1,2-b]pyridazines as protein kinase inhibitorsPublication Number: US-2017002014-A1Priority Date: 2011-07-21
- Substituted imidazo[1,2-b]pyridazines as protein kinase inhibitorsPublication Number: US-2019071446-A1Priority Date: 2011-07-21
- Substituted imidazo[1,2-b]pyridazines as protein kinase inhibitorsPublication Number: US-2020102313-A1Priority Date: 2011-07-21
- Heterocyclic protein kinase inhibitorsPublication Number: EP-2734205-B1Priority Date: 2011-07-21Grant Date: 2018-03-21
- Heterocyclic protein kinase inhibitorsPublication Number: EP-3409278-A1Priority Date: 2011-07-21
- Heterocyclic protein kinase inhibitorsPublication Number: JP-2014520898-APriority Date: 2011-07-21
- Heterocyclic protein kinase inhibitorsPublication Number: JP-6105578-B2Priority Date: 2011-07-21Grant Date: 2017-03-29
- Substituted imidazo[1,2-B]pyridazines as protein kinase inhibitorsPublication Number: US-10047093-B2Priority Date: 2011-07-21Grant Date: 2018-08-14

AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
REF
– Nuvisertib (TP-3654), an investigational highly selective oral PIM1 kinase inhibitor, is being evaluated in patients with relapsed or refractory myelofibrosis (MF) –
– Nuvisertib demonstrated symptom and spleen responses correlating with cytokine modulation in the preliminary Phase 1/2 data recently presented at the European Hematology Association (EHA) 2025 Congress –
MARLBOROUGH, Mass., June 12, 2025 /PRNewswire/ — Sumitomo Pharma America, Inc. (SMPA) today announced that the U.S. Food and Drug Administration (FDA) granted Fast Track Designation to nuvisertib (TP-3654) for the treatment of patients with intermediate or high-risk myelofibrosis (MF). The FDA Fast Track Designation is granted to investigational therapies being developed to treat serious or life-threatening conditions that demonstrate the potential to address unmet medical needs. Nuvisertib is an oral, investigational, highly selective inhibitor of PIM1 kinase, which demonstrated clinical activity including symptom and spleen responses correlating with cytokine modulation in the updated preliminary Phase 1/2 data presented at the European Hematology Association (EHA) 2025 Congress in Milan, Italy.
MF, a serious and rare type of blood cancer, is characterized by the buildup of fibrous tissues in the bone marrow which is caused by dysregulation in the Janus-associated kinase (JAK) signaling pathway. The clinical manifestations of MF include an enlarged spleen, debilitating symptoms and reduction in hemoglobin and/or platelets. MF affects 1 in 500,000 people worldwide.1
“This positive momentum for nuvisertib signals strong promise in our pipeline and reflects our dedication to addressing unmet medical needs on behalf of patients with myelofibrosis and their families,” said Tsutomu Nakagawa, Ph.D, President and Chief Executive Officer of SMPA. “Receiving FDA Fast Track Designation for nuvisertib in the treatment of myelofibrosis reinforces our confidence in its potential as a treatment option for patients facing a poor prognosis with limited treatment options. We are committed to working closely with the FDA to progress the clinical development of nuvisertib and bring an alternative treatment option to patients with myelofibrosis.”
Updated data from the ongoing Phase 1/2 study of nuvisertib in patients with relapsed/refractory MF were presented at the EHA Congress on June 12, 2025. Preliminary data showed that nuvisertib monotherapy appears to be well tolerated with no dose-limiting toxicities (DLTs). Evaluable patients showed clinical activity including a ≥25% spleen volume reduction (SVR25) in 22.2% of patients and a ≥50% reduction in total symptom score (TSS50) of 44.4% of patients, as well as improvement of bone marrow fibrosis (42.9% patients), hemoglobin (24% patients) and platelet count (26.7% patients). Data also showed that nuvisertib treatment led to significant cytokine modulation [reduction of pro-inflammatory cytokines (e.g. EN-RAGE, MIP-1β) and increase of anti-inflammatory cytokines (e.g. adiponectin)], which demonstrated significant (p<0.001) correlation with symptom and spleen responses. Preclinical2 and emerging clinical data support the development of nuvisertib in combination with JAK inhibitors for the treatment of patients with MF.
“The data observed to date demonstrate promising clinical activity for nuvisertib and the strong potential for selective PIM1 inhibition to slow the progression of myelofibrosis,” said Jatin Shah, MD, Chief Medical Officer, Oncology. “Patients with myelofibrosis are in need of new therapeutic approaches, including combination treatment options, that can provide increased and durable response rates with limited hematologic adverse events. The FDA Fast Track Designation reinforces the potential of nuvisertib to provide clinical benefits for patients with myelofibrosis, an unmet medical need.”
About Nuvisertib (TP-3654)
Nuvisertib (TP-3654) is an oral investigational selective inhibitor of PIM1 kinase, which has shown potential antitumor and antifibrotic activity through multiple pathways, including induction of apoptosis in preclinical models.2,3 Nuvisertib was observed to inhibit proliferation and increase apoptosis in murine and human hematopoietic cells expressing the clinically relevant JAK2 V617F mutation.3 Nuvisertib alone and in combination with ruxolitinib showed white blood cell and neutrophil count normalization, and also reduced spleen size and bone marrow fibrosis in JAK2 V617F and MPLW515L murine models of myelofibrosis.2 The safety and efficacy of nuvisertib is currently being clinically evaluated in a Phase 1/2 study in patients with intermediate and high-risk myelofibrosis (NCT04176198). The U.S. Food and Drug Administration (FDA) granted Orphan Drug Designation to nuvisertib for the indication of myelofibrosis in May 2022. The Japan Ministry of Health, Labour and Welfare (MHLW) granted Orphan Drug Designation to nuvisertib for the treatment of myelofibrosis in November 2024.
About Sumitomo Pharma
Sumitomo Pharma Co., Ltd., is a global pharmaceutical company based in Japan with key operations in the U.S. (Sumitomo Pharma America, Inc.), Canada (Sumitomo Pharma Canada, Inc.), and Europe (Sumitomo Pharma Switzerland GmbH) focused on addressing patient needs in oncology, urology, women’s health, rare diseases, psychiatry & neurology, and cell & gene therapies. With several marketed products in the U.S., Canada, and Europe, a diverse pipeline of early- to late-stage assets, we aim to accelerate discovery, research, and development to bring novel therapies to patients sooner. For more information on SMPA, visit our website https://www.us.sumitomo-pharma.com or follow us on LinkedIn.
The Sumitomo corporate symbol mark is a trademark of Sumitomo Pharma Co., Ltd., used under license. SUMITOMO PHARMA is a trademark of Sumitomo Pharma Co., Ltd., used under license. SUMITOMO is a registered trademark of Sumitomo Chemical Co., Ltd., used under license. Sumitomo Pharma America, Inc. is a U.S. subsidiary of Sumitomo Pharma Co., Ltd.
©2025 Sumitomo Pharma America, Inc. All rights reserved.
References
- U.S. National Library of Medicine. (n.d.). Primary myelofibrosis: Medlineplus Genetics. MedlinePlus. https://medlineplus.gov/genetics/condition/primary-myelofibrosis/
- Dutta A., Nath D, Yang Y, et al. Genetic ablation of Pim1 or pharmacologic inhibition with TP-3654 ameliorates myelofibrosis in murine models. Leukemia. 2022; 36 (3): 746-759. doi: 10.1038/s41375-021-01464-2.
- Foulks JM, Carpenter KJ, Luo B, et al. A small-molecule inhibitor of PIM kinases as a potential treatment for urothelial carcinomas. Neoplasia. 2014;16(5):403-412.
SOURCE Sumitomo Pharma America
- BLM overexpression as a predictive biomarker for CHK1 inhibitor response in PARP inhibitor–resistant BRCA -mutant ovarian cancerPublication Name: Science Translational MedicinePublication Date: 2023-06-21PMCID: PMC10758289PMID: 37343085DOI: 10.1126/scitranslmed.add7872
- The Second-Generation PIM Kinase Inhibitor TP-3654 Resensitizes ABCG2-Overexpressing Multidrug-Resistant Cancer Cells to Cytotoxic Anticancer DrugsPublication Name: International Journal of Molecular SciencesPublication Date: 2021-08-30PMCID: PMC8431370PMID: 34502348DOI: 10.3390/ijms22179440
- High-Throughput Screening to Identify Inhibitors of the Type I Interferon–Major Histocompatibility Complex Class I Pathway in Skeletal MusclePublication Name: ACS Chemical BiologyPublication Date: 2020-05-27PMCID: PMC7859889PMID: 32459468DOI: 10.1021/acschembio.0c00343
- PIM kinase inhibitors: Structural and pharmacological perspectivesPublication Name: European Journal of Medicinal ChemistryPublication Date: 2019-06-15PMID: 30954777DOI: 10.1016/j.ejmech.2019.03.050
- A Small-Molecule Inhibitor of PIM Kinases as a Potential Treatment for Urothelial CarcinomasPublication Name: Neoplasia (New York, N.Y.)Publication Date: 2014-05PMCID: PMC4198696PMID: 24953177DOI: 10.1016/j.neo.2014.05.004
- BLM overexpression as a predictive biomarker for CHK1 inhibitor response in PARP inhibitor–resistant BRCA -mutant ovarian cancerPublication Name: Science Translational MedicinePublication Date: 2023-06-21PMCID: PMC10758289PMID: 37343085DOI: 10.1126/scitranslmed.add7872
- The Second-Generation PIM Kinase Inhibitor TP-3654 Resensitizes ABCG2-Overexpressing Multidrug-Resistant Cancer Cells to Cytotoxic Anticancer DrugsPublication Name: International Journal of Molecular SciencesPublication Date: 2021-08-30PMCID: PMC8431370PMID: 34502348DOI: 10.3390/ijms22179440
- High-Throughput Screening to Identify Inhibitors of the Type I Interferon–Major Histocompatibility Complex Class I Pathway in Skeletal MusclePublication Name: ACS Chemical BiologyPublication Date: 2020-05-27PMCID: PMC7859889PMID: 32459468DOI: 10.1021/acschembio.0c00343
- PIM kinase inhibitors: Structural and pharmacological perspectivesPublication Name: European Journal of Medicinal ChemistryPublication Date: 2019-06-15PMID: 30954777DOI: 10.1016/j.ejmech.2019.03.050
- A Small-Molecule Inhibitor of PIM Kinases as a Potential Treatment for Urothelial CarcinomasPublication Name: Neoplasia (New York, N.Y.)Publication Date: 2014-05PMCID: PMC4198696PMID: 24953177DOI: 10.1016/j.neo.2014.05.004
///////Nuvisertib, serine/ threonine kinase inhibitor, antineoplastic, Orphan Drug, myelofibrosis, SGI-9481, SGI 9481, TP-3654, TP 3654, EOB0N7BOY4
Iodofalan (131I)
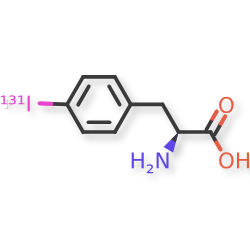
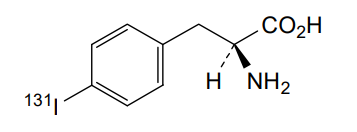
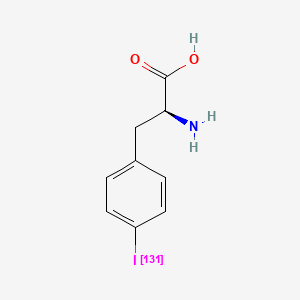
Iodofalan (131I)
CAS 76641-05-9
MFC9H10131INO2
Molecular FormulaC9H10INO2
Molecular Weight295.09
4-(131I)iodo-L-phenylalanine
(2S)-2-amino-3-(4-iodophenyl)propanoic acid
radiopharmaceutical, antineoplastic, Phase 2, Glioblastoma, 606VTF676Y, 131I-TLX-101, ACD 101
- 4-Iodophenylalanine I-131
- 4-(131I)Iodo-L-phenylalanine
- 4-Iodo-L-phenylalanine-131I
- ACD-101
- L-Phenylalanine, 4-(iodo-131I)-
- OriginatorTherapeia
- DeveloperTelix Pharmaceuticals; Therapeia
- ClassAmino acids; Antineoplastics; Radioisotopes; Radiopharmaceutical diagnostics; Radiopharmaceuticals; Small molecules
- Mechanism of ActionApoptosis stimulants; Positron-emission tomography enhancers
- Orphan Drug StatusYes – Glioblastoma
- Phase IIGlioblastoma
- 14 Oct 2025Telix Pharmaceuticals receives IND approval for TLX 101 in Glioblastoma
- 27 Jul 2025Telix Pharmaceuticals plans a phase III IPAX BrIGHT trial for Glioblastoma (Monotherapy, Combination therapy, Recurrent, Second-line therapy or greater) in Australia(IV) (NCT07100730)(EudraCT2025-521785-10) in September 2025
- 16 Apr 2025Telix has submitted for ethics approval a registration-enabling study of TLX101 in recurrent glioblastoma.
Iodofalan (131I) is a radiopharmaceutical that has garnered significant attention in oncological research due to its targeted therapeutic potential. This compound, which includes the radioactive isotope Iodine-131, has been explored for its efficacy in treating certain types of cancers, particularly those associated with the thyroid. Various research institutions worldwide have been studying Iodofalan (131I) to better understand its clinical benefits, optimize its usage, and minimize potential side effects. As a drug type, Iodofalan (131I) is categorized as a targeted radiopharmaceutical therapy, which leverages the properties of radioactive isotopes to destroy cancer cells with precision. Currently, its primary indications include differentiated thyroid cancer and non-resectable metastatic thyroid cancer, among other investigational uses.
Iodofalan (131I) Mechanism of Action
The mechanism of action for Iodofalan (131I) centers on the properties of Iodine-131, a beta-emitting isotope. When administered, Iodofalan (131I) is selectively absorbed by thyroid cells. This selectivity is due to the thyroid gland’s natural ability to uptake iodine, a key element required for the production of thyroid hormones. Cancerous thyroid tissues retain this ability, making them ideal targets for Iodofalan (131I) therapy.
Once absorbed by the thyroid cancer cells, the radioactive decay of Iodine-131 begins. This decay process emits beta particles, which possess sufficient energy to destroy nearby cells. The radiation from these beta particles causes direct DNA damage, leading to cell death. Additionally, the gamma radiation emitted by Iodine-131 can be used diagnostically to track the distribution and uptake of the compound in the body via imaging techniques such as SPECT (Single Photon Emission Computed Tomography).
The dual role of Iodofalan (131I) in both treatment and diagnostic contexts underscores its importance in managing thyroid cancers. By delivering a localized radiation dose to thyroid cancer cells, Iodofalan (131I) minimizes damage to surrounding healthy tissues, which is a significant advantage over traditional external beam radiotherapy.
What is the indication of Iodofalan (131I)?
The primary indication for Iodofalan (131I) is the treatment of differentiated thyroid cancer, a category that includes papillary and follicular thyroid cancers. These subtypes are characterized by their ability to absorb iodine, making them particularly amenable to radioiodine therapy. Iodofalan (131I) is typically used in cases where the thyroid cancer is not amenable to surgical removal or has metastasized to other parts of the body. In such scenarios, the radiopharmaceutical offers a non-invasive therapeutic option that can target and destroy cancer cells even in distant metastatic sites.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US42129729&_cid=P21-MHE8B5-15309-1
EXAMPLE 1
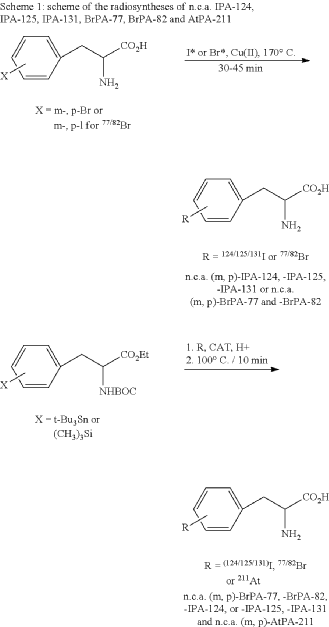
EXAMPLE 2
General synthesis of 3,4-[124I]iodo-L-phenylalanine (m, p-IPA-124), 3,4-[125I]iodo-L-phenylalanine (m,p-IPA-125) and 3,4-[131I]iodo-L-phenylalanine (m,p-IPA-131) by non-isotopic radioiodo-debromination
PAT
- Pharmaceutical combinations and uses thereofPublication Number: US-2024197715-A1Priority Date: 2022-11-18
- Pharmaceutical combinations and uses thereofPublication Number: WO-2024105610-A1Priority Date: 2022-11-18
- Iodine-labeled homoglutamic acid and glutamic acid derivativesPublication Number: US-2013034497-A1Priority Date: 2009-11-17
- MALIGNAS NEOPLASIAS THERAPY.Publication Number: ES-2341575-T3Priority Date: 2005-11-25Grant Date: 2010-06-22
- Therapy of malignant neoplasiasPublication Number: US-2007128108-A1Priority Date: 2005-11-18
- Therapy of malignant neoplasias
- Publication Number: US-9682158-B2
- Priority Date: 2005-11-18
- Grant Date: 2017-06-20
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////Iodofalan (131I), radiopharmaceutical, antineoplastic, Phase 2, Glioblastoma, 606VTF676Y, 131I-TLX-101, ACD 101
Gildeuretinol
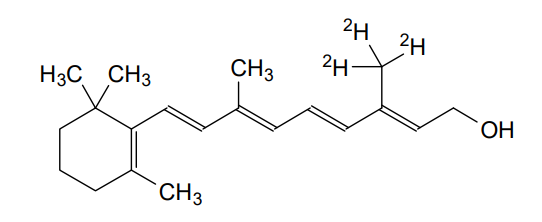
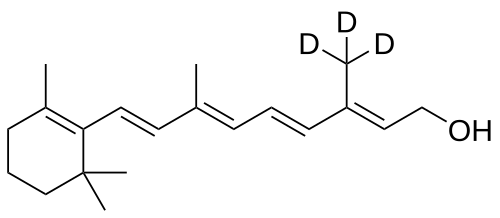
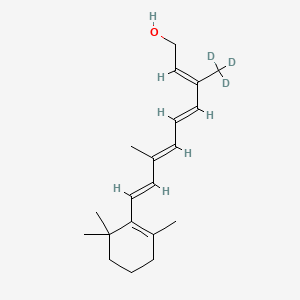
Gildeuretinol
CAS118139-35-8
MF C20H272H3O, MW 289.5 g/mol
(2E,4E,6E,8E)-3-(2H3)methyl-7-methyl-9-(2,6,6-trimethylcyclohex-1-en-1-yl)nona-2,4,6,8-tetraen-1-ol; (20,20,20-2H3)retinol
(2E,4E,6E,8E)-7-methyl-3-(trideuteriomethyl)-9-(2,6,6-trimethylcyclohexen-1-yl)nona-2,4,6,8-tetraen-1-ol
vitamin A analogue, Orphan Drug, Stargardt disease, breakthrough therapy, Pediatric Rare Disease designations, ALK-001, KL-49, ALK 001, KL 49
- OriginatorColumbia University
- DeveloperAlkeus Pharmaceuticals
- ClassEye disorder therapies; Retinoids; Vitamins
- Mechanism of ActionDimerisation inhibitors; Vitamin A replacements
- Orphan Drug StatusYes – Stargardt disease
- Phase II/IIIDry age-related macular degeneration
- Phase IIStargardt disease
- No development reportedRetinal dystrophies
- 08 Sep 2025Gildeuretinol – Alkeus Pharmaceuticals receives Orphan Drug status for Stargardt disease in European Union
- 09 Jan 2025Alkeus Pharmaceuticals announces intention to submit an NDA to US FDA for Stargardt disease in 2025
- 09 Jan 2025Efficacy and adverse event data from phase II trial for Stargardt disease released by Alkeus Pharmaceuticals
Gildeuretinol is an investigational new drug being developed by Alkeus Pharmaceuticals, Inc. for the treatment of retinal diseases, particularly Stargardt disease and geographic atrophy secondary to age-related macular degeneration (AMD). Stargardt disease is caused by a defect in the ABCA4 gene that clears toxic byproducts resulting from the dimerization of vitamin A. Gildeuretinol is new molecular entity designed to reduce the dimerization of vitamin A in the eye without affecting the visual cycle.[1]
Gildeuretinol has received breakthrough therapy, orphan drug and Pediatric Rare Disease designations from the U.S. Food and Drug Administration.[2]

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Zaydon YA, Tsang SH (July 2024). “The ABCs of Stargardt disease: the latest advances in precision medicine”. Cell & Bioscience. 14 (1) 98. doi:10.1186/s13578-024-01272-y. PMC 11282698. PMID 39060921.
- Fitch J (22 November 2024). “Gildeuretinol for Stargardt disease receives Rare Pediatric Disease, Fast Track Designations”. Contemporary Pediatrics.
| Clinical data | |
|---|---|
| Other names | ALK-001, KL-49 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 118139-35-8 |
| PubChem CID | 169490774 |
| UNII | PSZ7W5NR24 |
| KEGG | D12713 |
| ChEMBL | ChEMBL5314606 |
| Chemical and physical data | |
| Formula | C20H30D3O |
| Molar mass | 292.500 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////Gildeuretinol, vitamin A analogue, Orphan Drug, Stargardt disease, breakthrough therapy, Pediatric Rare Disease designations, ALK-001, KL-49, ALK 001, KL 49, PSZ7W5NR24
Enzomenib
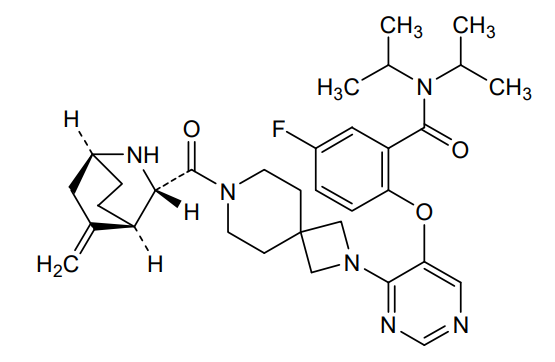
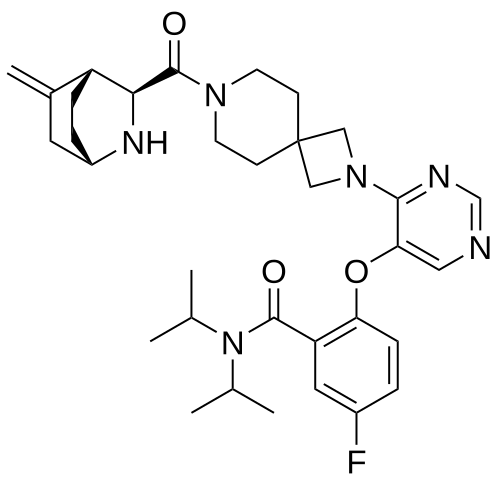
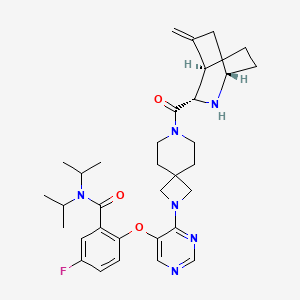
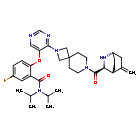
Enzomenib
CAS 2412555-70-3
MF C33H43FN6O3 MW 590.7 g/mol
5-fluoro-2-[4-[7-[(1S,3S,4R)-5-methylidene-2-azabicyclo[2.2.2]octane-3-carbonyl]-2,7-diazaspiro[3.5]nonan-2-yl]pyrimidin-5-yl]oxy-N,N-di(propan-2-yl)benzamide
5-fluoro-2-[(4-{7-[(1S,3S,4R)-5-methylidene-2-azabicyclo[2.2.2]octane-3-carbonyl]-2,7-
diazaspiro[3.5]nonan-2-yl}pyrimidin-5-yl)oxy]-N,Ndi(propan-2-yl)benzamide
menin-MLL (mixed-lineage leukemia) protein, interaction inhibitor, antineoplastic, DSP-5336, Fast Track, Orphan Drug designations
Enzomenib is an investigational new drug that is being evaluated for the treatment of acute leukemia.[1] It is a small molecule inhibitor that targets the interaction between menin and mixed-lineage leukemia (MLL) proteins.[2] Enzomenib particularly in patients with KMT2A (MLL) rearrangements or NPM1 mutations.[3]
The U.S. Food and Drug Administration (FDA) has granted both Fast Track and Orphan Drug designations to Enzomenib.[4]
Enzomenib is an orally bioavailable, small molecule inhibitor of menin, with potential antineoplastic activity. Upon oral administration, enzomenib targets and binds to the nuclear protein menin, thereby preventing the interaction between the two proteins menin and menin-mixed lineage leukemia (MLL; myeloid/lymphoid leukemia; KMT2A) and the formation of the menin-MLL complex. This reduces the expression of downstream target genes and results in an inhibition of the proliferation of MLL-rearranged leukemic cells. The menin-MLL complex plays a key role in the survival, growth, transformation and proliferation of certain kinds of leukemia cells.
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US295244745&_cid=P21-MGISYZ-31333-1
Example 3 to 19
| The following compounds of Examples 3 to 19 were prepared according to a similar method to Example 1 by using each corresponding starting compound. |

PAT
Optically active azabicyclo derivatives
Publication Number: JP-7614262-B2
Priority Date: 2018-08-27
Grant Date: 2025-01-15
- Optically active azabicyclo derivativesPublication Number: CN-112585140-BPriority Date: 2018-08-27Grant Date: 2023-07-04
- Optically active azabicyclo ring derivativePublication Number: JP-2023134729-APriority Date: 2018-08-27
- Chiral azabicyclyl compound derivativePublication Number: TW-I815954-BPriority Date: 2018-08-27Grant Date: 2023-09-21
- Optically active azabicyclo ring derivativePublication Number: US-11911381-B2Priority Date: 2018-08-27Grant Date: 2024-02-27
- Optically active azabicyclo ring derivativePublication Number: US-2024148727-A1Priority Date: 2018-08-27
- Optically active azabicyclic derivativePublication Number: AU-2019327006-A1Priority Date: 2018-08-27
- Optically active azabicyclic derivativePublication Number: EP-3845533-A1Priority Date: 2018-08-27
- Optically active azabicyclo ring derivativePublication Number: US-2021338668-A1Priority Date: 2018-08-27
- Optically active azabicyclo ring derivativePublication Number: US-11369605-B2Priority Date: 2018-08-27Grant Date: 2022-06-28
- Optically active azabicyclo ring derivativePublication Number: US-2022288072-A1Priority Date: 2018-08-27
- Optically active azabicyclo ring derivativePublication Number: US-2020157114-A1Priority Date: 2018-08-27
- Optically active azabicyclic derivativePublication Number: WO-2020045334-A1Priority Date: 2018-08-27
- Optically active azabicyclo ring derivativesPublication Number: JP-2020105191-APriority Date: 2018-08-27
- Chiral azabicyclyl compound derivativePublication Number: TW-202024082-APriority Date: 2018-08-27
- Optically active azabicyclo ring derivativePublication Number: US-10815241-B2Priority Date: 2018-08-27Grant Date: 2020-10-27
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Enzomenib – Sumitomo Pharma”. AdisInsight. Springer Nature Switzerland AG.
- Dempke WC, Desole M, Chiusolo P, Sica S, Schmidt-Hieber M (September 2023). “Targeting the undruggable: menin inhibitors ante portas”. Journal of Cancer Research and Clinical Oncology. 149 (11): 9451–9459. doi:10.1007/s00432-023-04752-9. PMC 11798168. PMID 37103568.
- “Sumitomo Pharma Presents New Clinical Data on DSP-5336 at the European Hematology Association 2024 Congress”. Sumitomo Pharma Co., Ltd. 14 June 2024.
- Flaherty C (15 July 2024). “FDA Grants Fast Track Designation to DSP-5336 in KMT2A/NMP1+ AML”. OncLive.
| Clinical data | |
|---|---|
| Other names | DSP-5336 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2412555-70-3 |
| PubChem CID | 146430058 |
| DrugBank | DB18514 |
| ChemSpider | 129534736 |
| UNII | VW83Y2JLZ5 |
| ChEMBL | ChEMBL5314915 |
| Chemical and physical data | |
| Formula | C33H43FN6O3 |
| Molar mass | 590.744 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////enzomenib, Interaction inhibitor, antineoplastic, DSP 5336, Fast Track, Orphan Drug designations
Claziprotamide
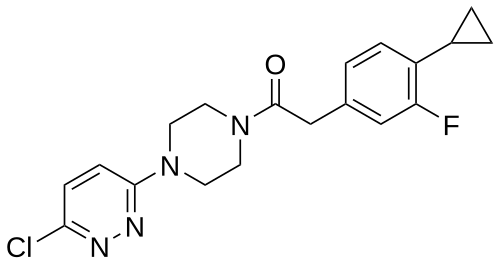
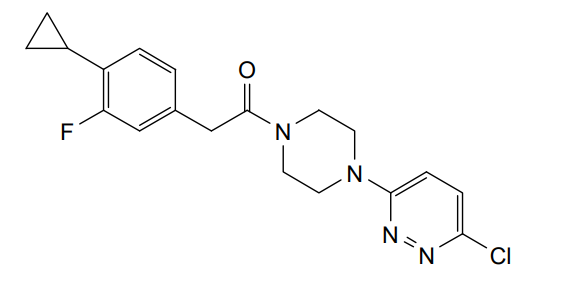
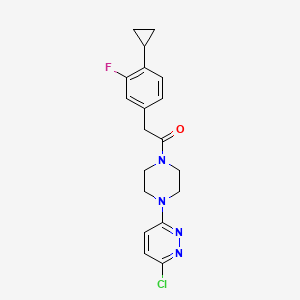
Claziprotamide
CAS 2361124-03-8, BBP 671
MF C19H20ClFN4O MW374.8 g/mol
1-[4-(6-chloropyridazin-3-yl)piperazin-1-yl]-2-(4-cyclopropyl-3-fluorophenyl)ethan-1-one
1-[4-(6-chloropyridazin-3-yl)piperazin-1-yl]-2-(4-cyclopropyl-3-fluorophenyl)ethan-1-one
pantothenate kinases 1 and 3 (PanK1 and PanK3) positive allosteric modulator
Claziprotamide is an investigational new drug that is being evaluated for the treatment of rare metabolic disorders such as pantothenate kinase-associated neurodegeneration (PKAN) and neurodegeneration with brain iron accumulation (NBIA). It acts as a positive allosteric modulator (PAM) of pantothenate kinases 1 and 3 (PANK1 and PANK2) which are critical for coenzyme A biosynthesis and cellular metabolism.[1][2]
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US319558593&_cid=P22-MG32VL-67777-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019133635&_cid=P22-MG32PO-63930-1
SCHEME 4B.
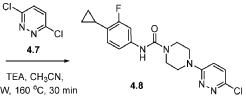
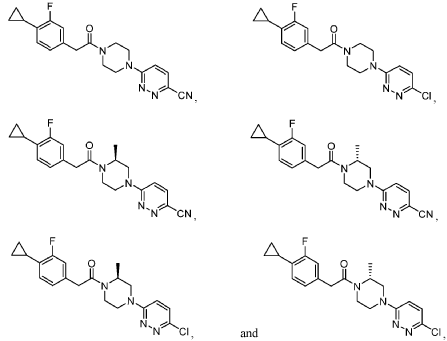
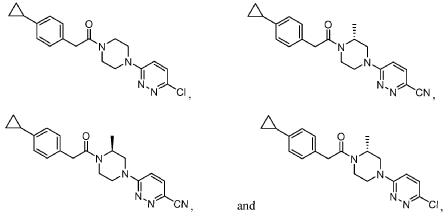
[00184] In one aspect, compounds of type 4.8, and similar compounds, can be prepared according to reaction Scheme 4B above. Thus, compounds of type 4.6 can be prepared by a urea bond formation reaction between an appropriate amine, e.g., 4.2 as shown above, and an appropriate isocyanate, e.g., 4.5 as shown above. Appropriate amines and appropriate isocyanates are commercially available or prepared by methods known to one skilled in the art. The nucleophilic substitution is carried out in the presence of an appropriate solvent, e.g., diethyl ether, for an appropriate period of time, e.g., 3 hours. The nucleophilic substitution is followed by a deprotection reaction. The deprotection reaction is carried out in the presence of an appropriate deprotecting agent, e.g., trifluoroacetic acid, in an appropriate solvent, e.g., dichloromethane, for an appropriate period of time, e.g., 1 hour. Compounds of type 4.8 can be prepared by an arylation reaction of appropriate amine, e.g., 4.6 as shown above, and an appropriate aryl halide, e.g., 4.7 as shown above. Appropriate aryl halides are commercially available or prepared by methods known to one skilled in the art. The arylation reaction is carried out in the presence of an appropriate base, e.g., triethylamine, in an appropriate solvent, e.g., acetonitrile, at an appropriate temperature, e.g, 160 °C, for an appropriate period of time, e.g., 30 minutes using microwave irradiations. As can be appreciated by one skilled in the art, the above reaction provides an example of a generalized approach wherein compounds similar in structure to the specific reactants above (compounds similar to compounds of type 3.6, 4.1, 4.2, and 4.3), can be substituted in the reaction to provide 4-aryl-N-phenylpiperazine-l -carboxamide derivatives similar to Formula 4.4.
PAT
- Methods of treating disorders associated with castorPublication Number: US-2023321092-A1Priority Date: 2017-12-27
- Small molecule modulators of pantothenate kinasesPublication Number: US-11891378-B2Priority Date: 2017-12-27Grant Date: 2024-02-06
- Small molecule modulators of pantothenate kinasesPublication Number: US-2024287039-A1Priority Date: 2017-12-27
- Small molecule modulator of pantothenate kinasePublication Number: KR-102728619-B1Priority Date: 2017-12-27Grant Date: 2024-11-08
- Small molecule modulators of pantothenate kinasesPublication Number: US-2021061788-A1Priority Date: 2017-12-27
- Methods of treating disorders associated with castorPublication Number: US-11547709-B2Priority Date: 2017-12-27Grant Date: 2023-01-10
- Small molecule modulators of pantothenate kinasesPublication Number: AU-2018395222-B2Priority Date: 2017-12-27Grant Date: 2023-06-08
- Small molecule modulators of pantothenate kinasePublication Number: CN-111818922-BPriority Date: 2017-12-27Grant Date: 2023-06-13
- Small molecule modulators of pantothenate kinasePublication Number: JP-7352565-B2Priority Date: 2017-12-27Grant Date: 2023-09-28
- Small molecule modulators of pantothenate kinasesPublication Number: EP-3731844-A1Priority Date: 2017-12-27
- Small molecule modulators of pantothenate kinasePublication Number: KR-20200130242-APriority Date: 2017-12-27
- MODULATORS OF SMALL MOLECULES OF PANTOTENATE KINASESPublication Number: BR-112020012875-A2Priority Date: 2017-12-27
- Small molecule modulator of pantothenate kinasePublication Number: JP-2021508739-APriority Date: 2017-12-27
- Methods of treating disorders associated with castorPublication Number: US-2021023081-A1Priority Date: 2017-12-27
- Treatment of organic acidemias or pantothenate kinase associated neurodegeneration with modulators of pantothenate kinasesPublication Number: WO-2023230560-A1Priority Date: 2022-05-26
- Methods of treating disorders associated with castorPublication Number: WO-2022133034-A1Priority Date: 2020-12-16
- Small molecule modulators of pantothenate kinasesPublication Number: WO-2019133635-A1Priority Date: 2017-12-27
- Small molecule modulators of pantothenate kinasesPublication Number: AU-2018395222-A1Priority Date: 2017-12-27
- Small molecule modulators of pantothenate kinasePublication Number: CN-111818922-APriority Date: 2017-12-27
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | BBP-671 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2361124-03-8 |
| PubChem CID | 142616838 |
| ChemSpider | 129431674 |
| UNII | 74N47PKZ3K |
| PDB ligand | Y92 (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C19H20ClFN4O |
| Molar mass | 374.84 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Tangallapally R, Subramanian C, Yun MK, Edwards A, Sharma LK, Yang L, et al. (August 2024). “Development of Brain Penetrant Pyridazine Pantothenate Kinase Activators”. Journal of Medicinal Chemistry. 67 (16): 14432–14442. doi:10.1021/acs.jmedchem.4c01211. PMC 11345825. PMID 39136313.
- “Claziprotamide”. PatSnap.
/////////Claziprotamide, BBP 671, ORPHAN DRUG
Atumelnant
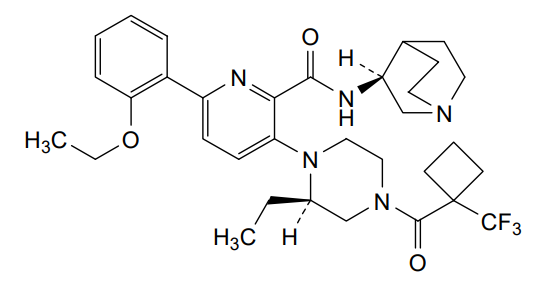
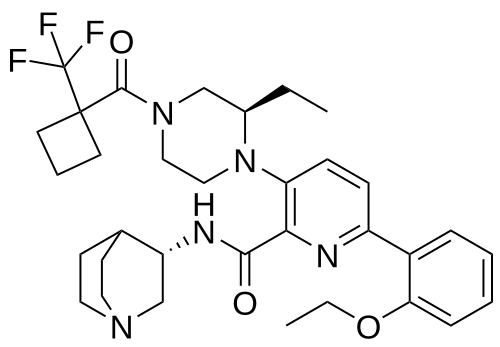
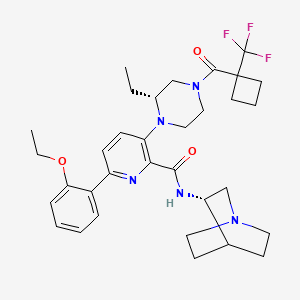
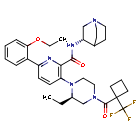
Atumelnant
CAS 2392970-97-5
MF C33H42F3N5O3 MW 613.7 g/mol
CRN04894, NR57FH6U1N
CRINETICS PHARMA, Orphan Drug Status, Congenital adrenal hyperplasia
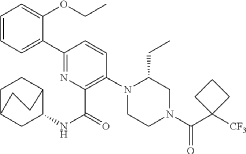
N-[(3S)-1-azabicyclo[2.2.2]octan-3-yl]-6-(2-ethoxyphenyl)-3-[(2R)-2-ethyl-4-[1-(trifluoromethyl)cyclobutanecarbonyl]piperazin-1-yl]pyridine-2-carboxamide
N-[(3S)-1-azabicyclo[2.2.2]octan-3-yl]-6-(2-ethoxyphenyl)-3-{(2R)-2-ethyl-4-[1-(trifluoromethyl) cyclobutane-1-carbonyl]piperazin-1-yl}pyridine-2-carboxamide
Adrenocorticotropic hormone receptor antagonist
- OriginatorCrinetics Pharmaceuticals
- ClassAmides; Antineoplastics; Antisecretories; Benzene derivatives; Cyclobutanes; Ethers; Fluorocarbons; Ketones; Piperazines; Pyridines; Quinuclidines; Small molecules
- Mechanism of ActionMelanocortin type 2 receptor antagonists
- Orphan Drug StatusYes – Congenital adrenal hyperplasia
- Phase IICongenital adrenal hyperplasia; Cushing syndrome
- No development reportedEctopic ACTH syndrome
- 21 Aug 2025Atumelnant receives Orphan Drug status for Congenital adrenal hyperplasia in the US
- 07 Aug 2025Crinetics pharmaceuticals plans phase II/III clinical trial for Cushing’s disease in 1H 2026
- 08 May 2025Crinetics Pharmaceuticals plans the phase III CALM-CAH trial for Congenital adrenal hyperplasia (In adults) (PO), in the second half of 2025
Atumelnant (INNTooltip International Nonproprietary Name; developmental code name CRN04894) is an investigational new drug developed by Crinetics Pharmaceuticals for the treatment of adrenocorticotropic hormone (ACTH)-dependent endocrine disorders.[1] It is a selective antagonist of the melanocortin type 2 receptor (MC2R), also known as the ACTH receptor, which is primarily expressed in the adrenal glands.[1][2] The drug is orally active.[1] Atumelnant is being evaluated to treat conditions such as congenital adrenal hyperplasia (CAH) and ACTH-dependent Cushing’s syndrome caused for example by pituitary adenomas.[3]
Atumelnant is an orally bioavailable nonpeptide antagonist of the adrenocorticotropic hormone (ACTH) receptor (ACTHR; melanocortin receptor 2; MC2R), with potential steroid hormone production inhibitory activity. Upon oral administration, atumelnant competes with ACTH for receptor binding to MC2R in the adrenal cortex and inhibits ACTH signaling. This may inhibit the synthesis and secretion of steroid hormones. MC2R, a member of the melanocortin receptor subfamily of type 1 G protein-coupled receptors, plays a key role in adrenal steroidogenesis.
PAPER
Discovery of CRN04894: A Novel Potent Selective MC2R Antagonist
Publication Name: ACS Medicinal Chemistry Letters
Publication Date: 2024-03-19, PMCID: PMC11017392, PMID: 38628803
DOI: 10.1021/acsmedchemlett.3c00514
PATENTS
- Melanocortin subtype-2 receptor antagonists and uses thereofPublication Number: IL-279152-B2Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2024300920-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor antagonists and uses thereofPublication Number: IL-279152-B1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: JP-2024009837-APriority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: KR-102695210-B1Priority Date: 2018-06-05Grant Date: 2024-08-13
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2024109866-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: CN-112533904-BPriority Date: 2018-06-05Grant Date: 2024-10-29
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-10981894-B2Priority Date: 2018-06-05Grant Date: 2021-04-20
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2021002254-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2021238164-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-11566015-B2Priority Date: 2018-06-05Grant Date: 2023-01-31
- Melanocortin subtype-2 receptor (MC2R) antagonists and their usesPublication Number: JP-7359783-B2Priority Date: 2018-06-05Grant Date: 2023-10-11
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2020216415-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-10766877-B2Priority Date: 2018-06-05Grant Date: 2020-09-08
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: CN-112533904-APriority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: EP-3802500-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: KR-20210005995-APriority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists for the treatment of diseasePublication Number: CN-117043146-APriority Date: 2021-03-19
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-10562884-B2Priority Date: 2018-06-05Grant Date: 2020-02-18
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-10604507-B2Priority Date: 2018-06-05Grant Date: 2020-03-31
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2019367481-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2020010452-A1Priority Date: 2018-06-05
Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: US-2022313691-A1Priority Date: 2021-03-19 - Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: WO-2022197798-A1Priority Date: 2021-03-19
- Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: TW-202302108-APriority Date: 2021-03-19
- Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: AU-2022240609-A1Priority Date: 2021-03-19
- Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: EP-4308553-A1Priority Date: 2021-03-19
- Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of acth-dependent cushing’s syndromePublication Number: WO-2024211343-A1Priority Date: 2023-04-05
- Crystalline melanocortin subtype-2 receptor (mc2r) antagonistPublication Number: TW-202430167-APriority Date: 2022-12-16
- Crystalline melanocortin subtype-2 receptor (mc2r) antagonistPublication Number: US-2024208963-A1Priority Date: 2022-12-16
- Crystalline melanocortin subtype-2 receptor (mc2r) antagonistPublication Number: WO-2024130091-A1Priority Date: 2022-12-16
- Treatment of congenital adrenal hyperplasia and polycystic ovary syndromePublication Number: WO-2023163945-A1Priority Date: 2022-02-2
SYN
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US278278493&_cid=P22-MFXDN2-76849-1
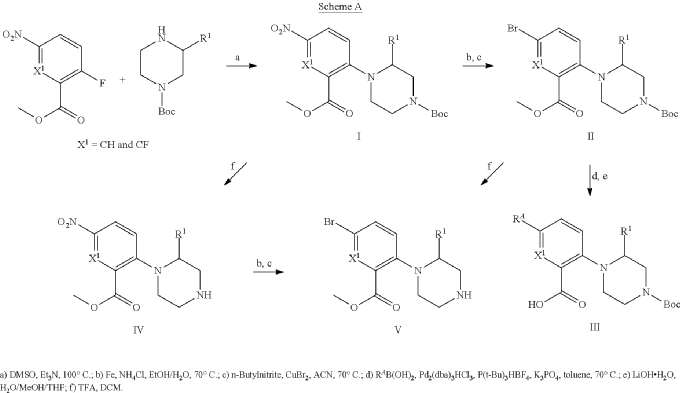
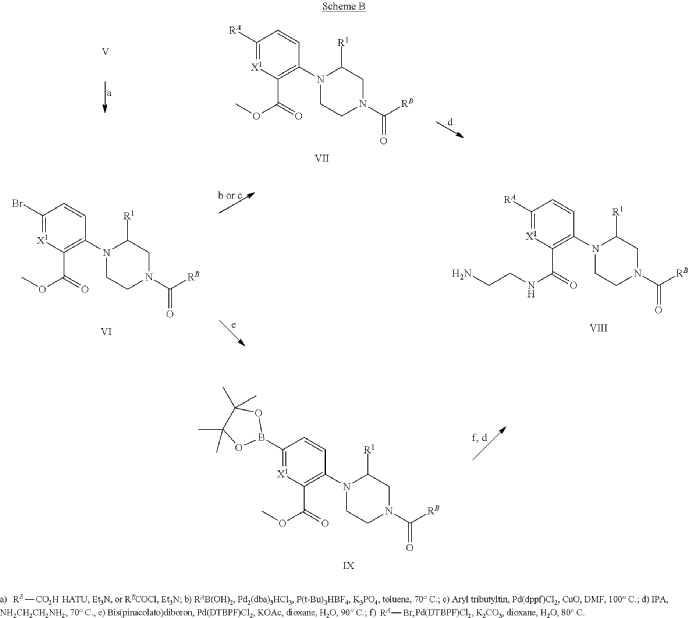
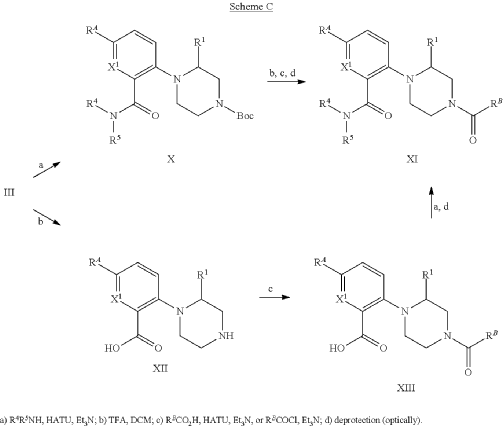
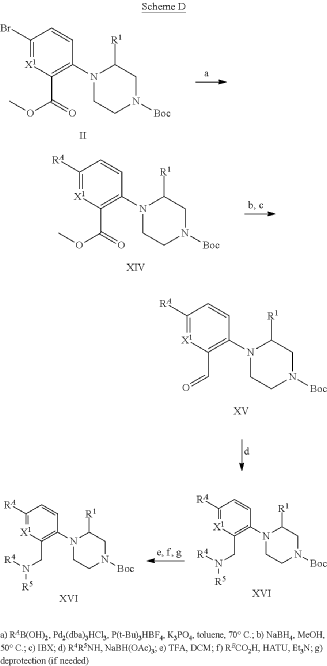
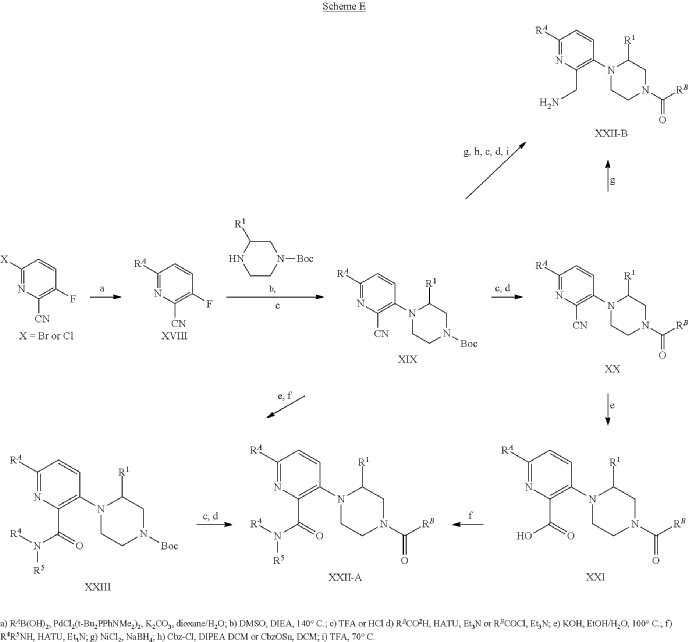
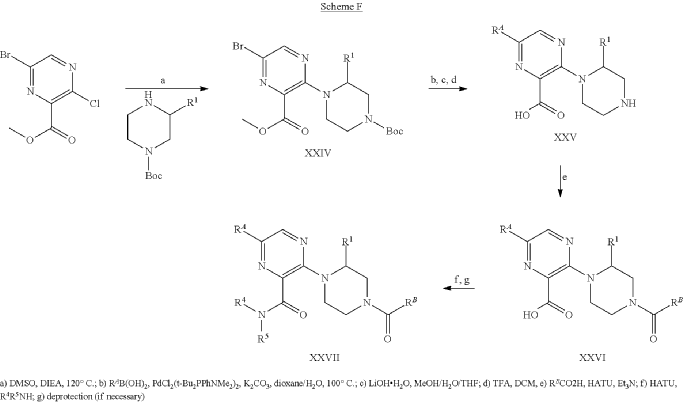
Example 31: N-[(3S)-1-azabicyclo[2.2.2]octan-3-yl]-6-(2-ethoxyphenyl)-3-[(2R)-2-ethyl-4-[1-(trifluoromethyl)cyclobutanecarbonyl]piperazin-1-yl]pyridine-2-carboxamide (Compound 1-410)
Step 31-1, Preparation of 6-(2-ethoxyphenyl)-3-[(2R)-2-ethyl-4-[1-(trifluoromethyl)cyclobutanecarbonyl]piperazin-1-yl]pyridine-2-carboxylic acid
Step 31-2, Preparation of N-[(3S)-1-azabicyclo[2.2.2]octan-3-yl]-6-(2-ethoxyphenyl)-3-[(2R)-2-ethyl-4-[1-(trifluoromethyl)cyclobutanecarbonyl]piperazin-1-yl]pyridine-2-carboxamide
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Crinetics Pharmaceuticals”. AdisInsight. 21 January 2025. Retrieved 25 February 2025.
- “Atumelnant (CRN04894)”. crinetics.com. 14 August 2020.
- Varlamov EV, Gheorghiu ML, Fleseriu M (December 2024). “Pharmacological management of pituitary adenomas – what is new on the horizon?”. Expert Opinion on Pharmacotherapy. 26 (2): 119–125. doi:10.1080/14656566.2024.2446625. PMID 39718553.
| Clinical data | |
|---|---|
| Other names | CRN04894 |
| Routes of administration | Oral[1] |
| Drug class | Melanocortin MC2 receptor antagonist[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2392970-97-5 |
| PubChem CID | 146361282 |
| IUPHAR/BPS | 13339 |
| ChemSpider | 129750231 |
| UNII | NR57FH6U1N |
| KEGG | D13102 |
| Chemical and physical data | |
| Formula | C33H42F3N5O3 |
| Molar mass | 613.726 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////Atumelnant, CRN04894, CRN 04894, NR57FH6U1N, CRINETICS PHARMA, Orphan Drug Status, Congenital adrenal hyperplasia, PHASE 3
Asengeprast
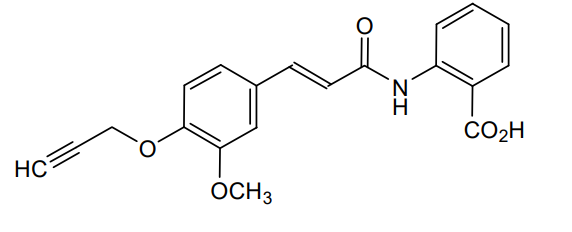
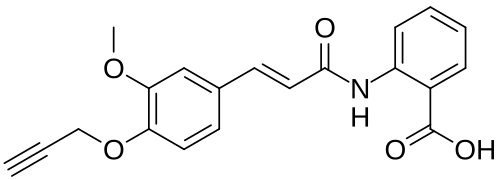
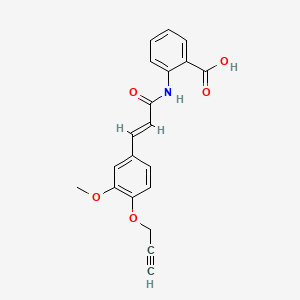
Asengeprast
CAS 1001288-58-9
FT011, FT 011, orphan drug status, systemic sclerosis, SHP-627, SHP 627,
Fast Track
2-[[(E)-3-(3-methoxy-4-prop-2-ynoxyphenyl)prop-2-enoyl]amino]benzoic acid
2-[(2E)-3-{3-methoxy-4-[(prop-2-yn-1-yl)oxy]phenyl}prop-2-enamido]benzoic acid G protein-coupled receptor 68 (GPR68) antagonist,
anti-inflammatory
MF C20H17NO5 MW 351.4 g/mol. C6V7ZU2NPR
Asengeprast (development code FT011) is an experimental scleroderma drug candidate.[1] It is a small molecule inhibitor of the G-protein coupled receptor GPR68 with antifibrotic activity.[2] It is being developed by Certa Therapeutics.
The European Medicines Agency (EMA) and the U.S. Food and Drug Administration (FDA) has granted orphan drug status to FT011, for systemic sclerosis (SSc).[3]
Asengeprast has been reported to attenuate fibrosis and chronic heart failure in experimental diabetic cardiomyopathy.[4] Asengeprast can also inhibit kidney fibrosis and prevent kidney failure.[5] It was developed by structure-activity optimization of the antifibrotic activity of cinnamoyl anthranilates, by assessment of their ability to prevent TGF-beta-stimulated production of collagen.[6]
Effects of FT011 in Systemic Sclerosis, CTID: NCT04647890
Phase: Phase 2, Status: Completed, Date: 2023-12-20
SYN
Evaluation and optimization of antifibrotic activity of cinnamoyl anthranilates
Publication Name: Bioorganic & Medicinal Chemistry Letters
Publication Date: 2009-12-15
PMID: 19879136
DOI: 10.1016/j.bmcl.2009.09.120
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018144620&_cid=P21-MFTHV7-45829-1
PAT
Publication Number: WO-2008003141-A1
Priority Date: 2006-07-05
- Tranilast analogues (substituted cinnamoyl anthranilate compounds) for treatment of conditions associated with firbrosisPublication Number: NZ-574028-APriority Date: 2006-07-05
- Therapeutic CompoundsPublication Number: US-2010130497-A1Priority Date: 2006-07-05
- Therapeutic compoundsPublication Number: US-2014357628-A1Priority Date: 2006-07-05
- Therapeutic compoundsPublication Number: US-8765812-B2Priority Date: 2006-07-05Grant Date: 2014-07-01
- Therapeutic compoundsPublication Number: US-9561201-B2Priority Date: 2006-07-05Grant Date: 2017-02-07
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Asengeprast Ligand page”. IUPHAR/BPS Guide to PHARMACOLOGY.
- “Certa Therapeutics website”.
- Inácio P (23 July 2024). “Certa’s FT011 granted orphan drug status in Europe for SSc”. Scleroderma News.
- Zhang Y, Edgley AJ, Cox AJ, Powell AK, Wang B, Kompa AR, et al. (May 2012). “FT011, a new anti-fibrotic drug, attenuates fibrosis and chronic heart failure in experimental diabetic cardiomyopathy”. European Journal of Heart Failure. 14 (5): 549–562. doi:10.1093/eurjhf/hfs011. PMID 22417655.
- Gilbert RE, Zhang Y, Williams SJ, Zammit SC, Stapleton DI, Cox AJ, et al. (2012). “A purpose-synthesised anti-fibrotic agent attenuates experimental kidney diseases in the rat”. PLOS ONE. 7 (10): e47160. Bibcode:2012PLoSO…747160G. doi:10.1371/journal.pone.0047160. PMC 3468513. PMID 23071743.
- Zammit SC, Cox AJ, Gow RM, Zhang Y, Gilbert RE, Krum H, et al. (December 2009). “Evaluation and optimization of antifibrotic activity of cinnamoyl anthranilates”. Bioorganic & Medicinal Chemistry Letters. 19 (24): 7003–7006. doi:10.1016/j.bmcl.2009.09.120. PMID 19879136.
| Chemical structure of asengeprast (FT011) | |
| Clinical data | |
|---|---|
| Other names | FT011 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1001288-58-9 |
| PubChem CID | 23648966 |
| ChemSpider | 24664633 |
| UNII | C6V7ZU2NPR |
| ChEMBL | ChEMBL1075834 |
| Chemical and physical data | |
| Formula | C20H17NO5 |
| Molar mass | 351.358 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- FT011, a Novel Cardiorenal Protective Drug, Reduces Inflammation, Gliosis and Vascular Injury in Rats with Diabetic RetinopathyPublication Name: PLOS ONEPublication Date: 2015-07-29PMCID: PMC4519240PMID: 26222724DOI: 10.1371/journal.pone.0134392
- A new anti-fibrotic drug attenuates cardiac remodeling and systolic dysfunction following experimental myocardial infarctionPublication Name: International Journal of CardiologyPublication Date: 2013-09-30PMID: 23219315DOI: 10.1016/j.ijcard.2012.11.067
- Attenuation of Armanni–Ebstein lesions in a rat model of diabetes by a new anti-fibrotic, anti-inflammatory agent, FT011Publication Name: DiabetologiaPublication Date: 2012-12-16PMID: 23242170DOI: 10.1007/s00125-012-2805-9
- A Purpose-Synthesised Anti-Fibrotic Agent Attenuates Experimental Kidney Diseases in the RatPublication Name: PLoS ONEPublication Date: 2012-10-10PMCID: PMC3468513PMID: 23071743DOI: 10.1371/journal.pone.0047160
- FT011, a new anti‐fibrotic drug, attenuates fibrosis and chronic heart failure in experimental diabetic cardiomyopathyPublication Name: European Journal of Heart FailurePublication Date: 2012-05PMID: 22417655DOI: 10.1093/eurjhf/hfs011
///////////Asengeprast, FT011, FT 011, orphan drug status, systemic sclerosis, SHP-627, SHP 627, C6V7ZU2NPR, Fast Track
Tebapivat
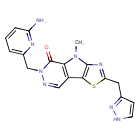
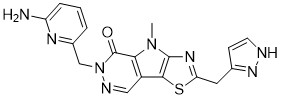
Tebapivat
CAS 2283422-04-6
WeightAverage: 392.44
Monoisotopic: 392.116778341
Chemical FormulaC18H16N8OS
10-[(6-aminopyridin-2-yl)methyl]-7-methyl-4-(1H-pyrazol-5-ylmethyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one
6-[(6-aminopyridin-2-yl)methyl]-4-methyl-2-[(1H-pyrazol-3-yl)methyl]-4,6-dihydro-5H-[1,3]thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5-one
6-[(6-aminopyridin-2-yl)methyl]-4-methyl-2-[(1H-pyrazol-3-yl)methyl]-4,6-dihydro-5H-[1,3]thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5-one
- AG946
- CS-0115951
- HY-135884
- ORG4KGP5ZS
- OriginatorAgios Pharmaceuticals
- ClassAntianaemics; Small molecules
- Mechanism of ActionPyruvate kinase stimulants
- Orphan Drug StatusYes – Myelodysplastic syndromes
- Phase IIAnaemia; Sickle cell anaemia
- 01 May 2025Phase-II clinical trials in Sickle cell anaemia in USA (PO) (NCT06924970)
- 01 May 2025Agios plans to initiate a phase II clinical trial for Sickle cell disease(PO) in mid-2025.
- 21 Feb 2025Agios Pharmaceuticals completes a phase I bioavailability trial (In volunteers) in USA (PO, capsule) (NCT06745271)
Tebapivat is under investigation in clinical trial NCT05490446 (A Study of Tebapivat (AG-946) in Participants With Anemia Due to Lower-risk Myelodysplastic Syndromes (LR-MDS)).
Tebapivat is an orally available activator of the red cell isoform of pyruvate kinase (PK-R; PKR), with potential to improve hemolytic anemia and related-symptoms in patients with pyruvate kinase deficiency (PKD). Upon oral administration, tebapivat binds to and activates PKR, thereby enhancing glycolytic pathway activity in red blood cells (RBCs), improving adenosine triphosphate (ATP) levels and reducing 2,3-diphosphoglycerate (2,3-DPG) levels. This may result in increased oxygen affinity, improved RBC deformability, decreased sickle RBC hemolysis, increased hemoglobin (Hb) levels and improved RBC membrane function. Mutations in PKR cause deficiency in PKR which prevents adequate RBC glycolysis, leading to a build-up of the upstream glycolytic intermediate 2,3-DPG and deficiency in the PKR product ATP.
SCHEME
COUPLER
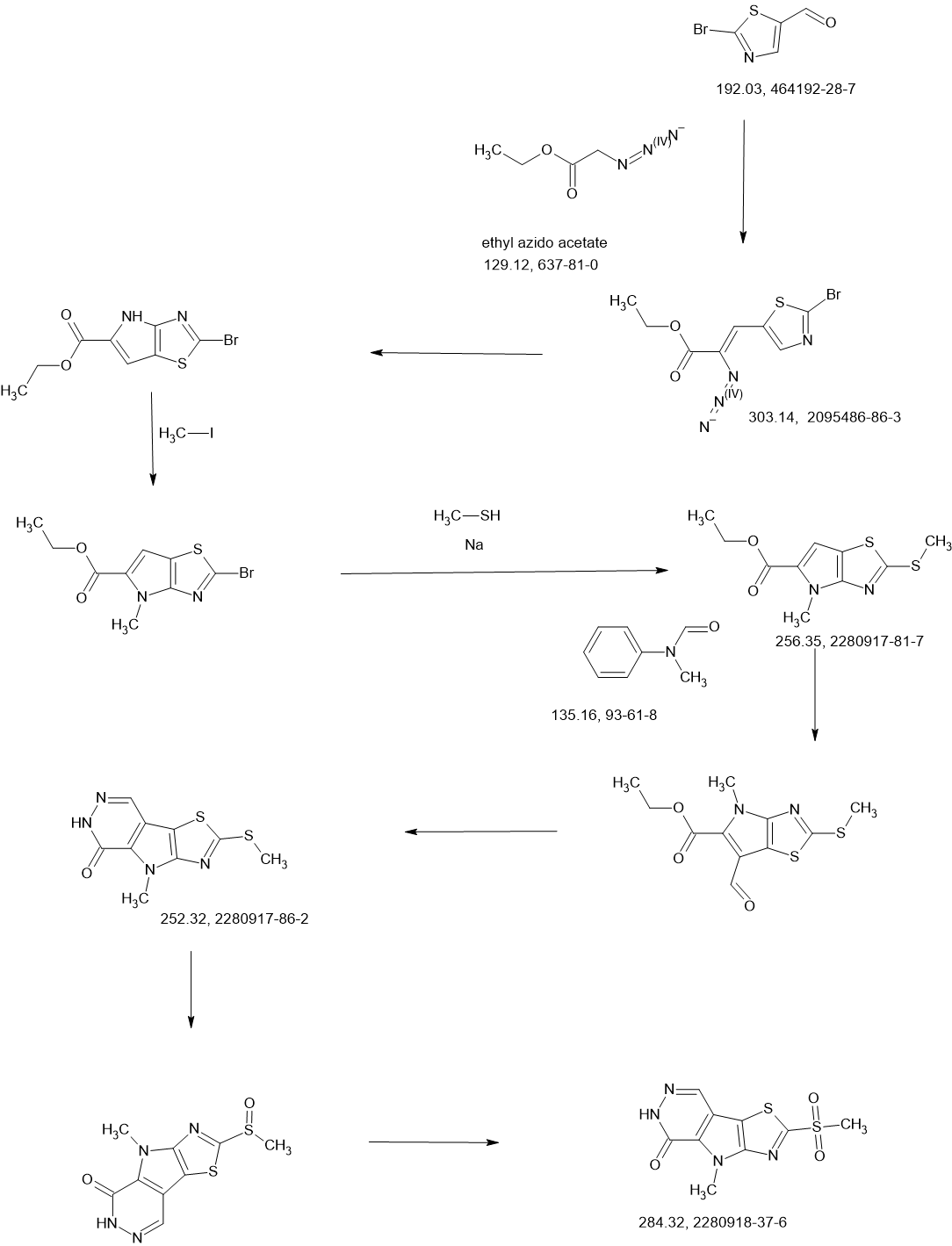
COUPLER
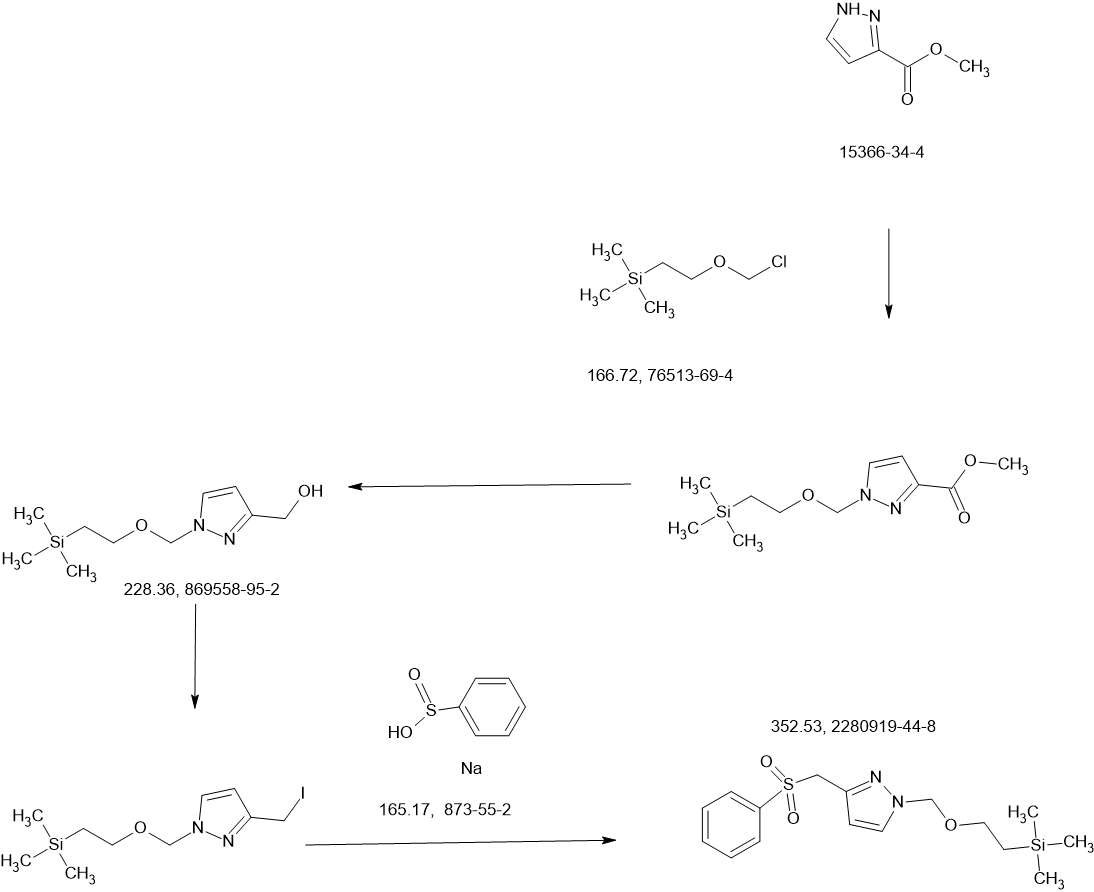
MAIN
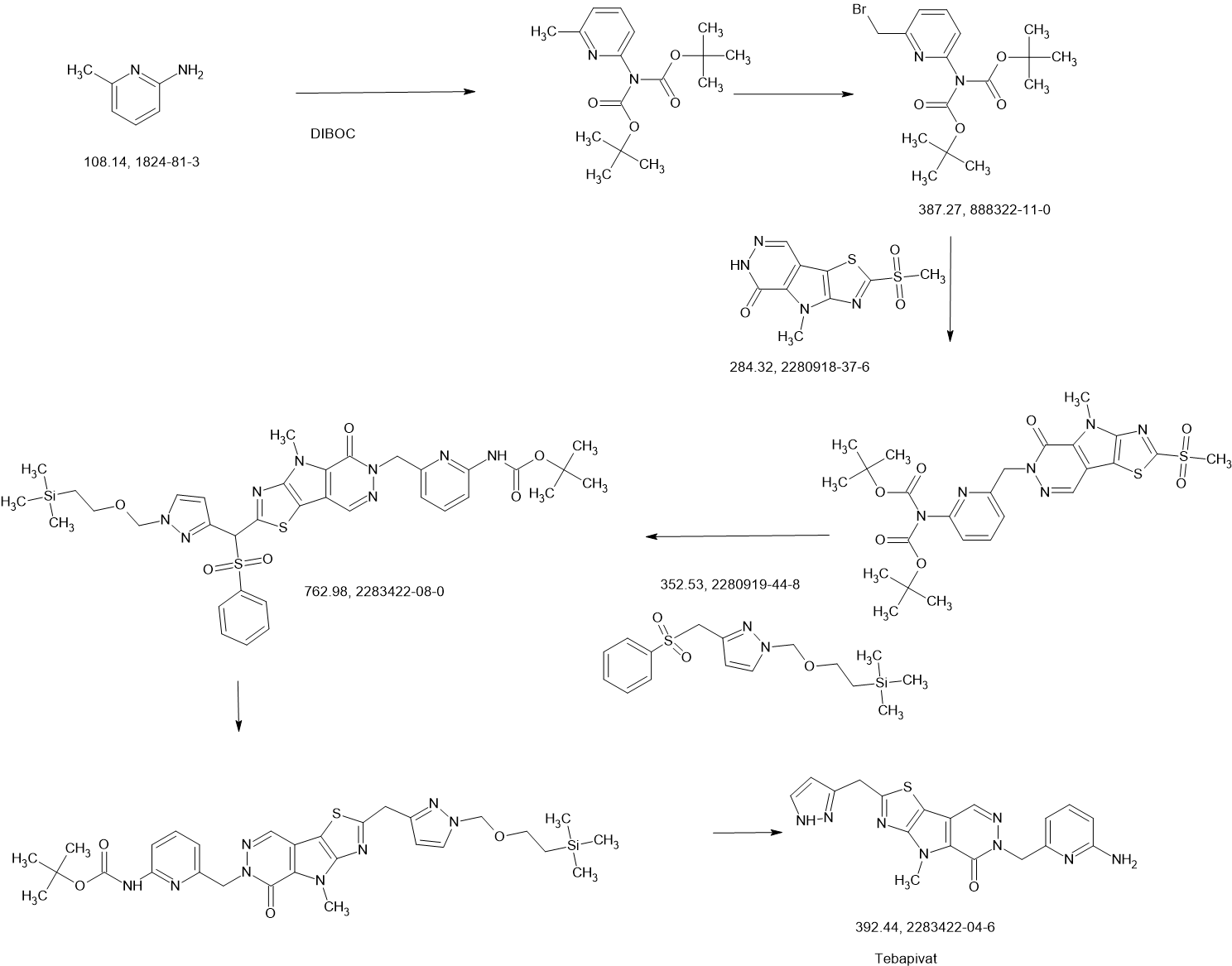
PATENT
Agios Pharmaceuticals, Inc.
WO2019035864
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019035864&_cid=P22-MDGSEF-03229-1
Example 8A. Synthesis of 2-((1H-pyrazol-3-yl)methyl)-6-((6-aminopyridin-2-yl)methyl)- 4-methyl-4H-thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5(6H)-one and 6-((6- aminopyridin-2-yl)methyl)-4-methyl-2-(1H-pyrazole-3-carbonyl)-4H- thiazolo[5′,4′:4,5]pyrroIo[2,3-d]pyridazin-5(6H)-one
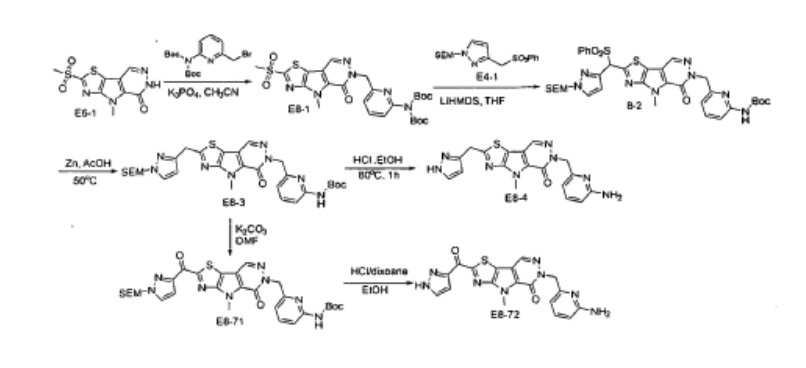


Step F. Synthesis of 6-((6-aminopyridin-2-yl)methyl)-4-methyl-2-(1H-pyrazole-3- carbonyl)-4H-thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5(6H)-one To a solution of tert- butyl (6-((4-methyl-5-oxo-2-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-3-carbonyl)- 4H-thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-6(5H)-yl)methyl)pyridin-2-yl)carbamate (20 mg, 0.03 mmol) in EtOH (1 mL) was added HCl (1 mL, 4 mol/L in dioxane). The mixture was stirred at 80 °C for lhr and cooled down. The precipitate was collected by filtration and neutralized with sat. NaHCO3, washed with water and dried to afford 5 mg of 6-((6- aminopyridin-2-yl)methyl)-4-methyl-2-(1H-pyrazole-3-carbonyl)-4H- thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5(6H)-one. LC-MS (ESI): m/z 407 (M+H)+. 1H NMR (400 MHz, DMSO-d6) δ: 8.75 (s, 1H), 7.96 (s, 1H), 7.50 (s, 1H), 7.31-7.22 (m, 1H), 6.31 (d, 1H), 6.14 (d, 1H), 5.91 (s, 2H), 5.23 (s, 2H), 4.38 (s, 3H).
PATENT
WO2023091414
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023091414&_cid=P22-MDGSRV-15431-1
PATENT
WO2019035863
WO2019035865
WO2019035864
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////Tebapivat, 2283422-04-6, AG946, CS-0115951, HY-135884, AG 946, CS 0115951, HY 135884, ORG4KGP5ZS, AGIOS, Orphan Drug, PHASE 2,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}