
Home » COVID-19
Category Archives: COVID-19
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Olgotrelvir
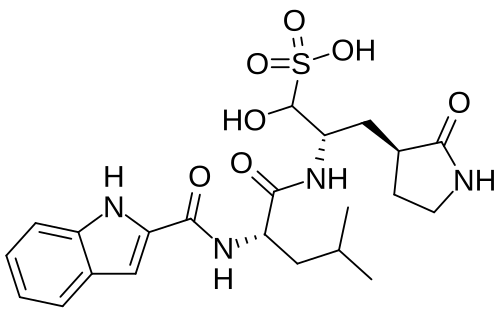
Olgotrelvir
STI-1558, HY-156655, CS-0887294, STI 1558, HY 156655, CS 0887294
Cas 2763596-71-8
494.6 g/mol, C22H30N4O7S, ZP3BDH359D
C22H30N4O7S 3-Pyrrolidinepropaney, α-hydroxy-β-[[(2S)-2-[(1H-indol-2-ylcarbonyl)amino]-4-methyl-1-oxopentyl]amino]-2-oxo-, (βS,3S)-
- (2S)-2-[(S)-2-(1H-Indole-2-carboxamido)-4-methylpentanamido]-1-hydroxy-3-[(S)-2-oxopyrrolidin-3-yl]propane-1-sulfonic acid
- 3-Pyrrolidinepropanesulfonic acid, alpha-hydroxy-beta-[[(2S)-2-[(1H-indol-2-ylcarbonyl)amino]-4-methyl-1-oxopentyl]amino]-2-oxo-, (betaS,3S)-
Olgotrelvir sodium, C22H30N4O7S.Na, CAS 2763596-71-8
3-Pyrrolidinepropanesulfonic acid, α-hydroxy-β-[[(2S)-2-[(1H-indol-2-ylcarbonyl)amino]-4-methyl-1-oxopentyl]amino]-2-oxo-, sodium salt (1:1), (βS,3S)-
Olgotrelvir (STI-1558) is an experimental antiviral medication being studied as a potential treatment for COVID-19. It is believed to work by inhibiting the SARS-CoV-2 main protease (Mpro), a key enzyme that SARS-CoV-2 needs to replicate,[1][2][3][4] and by blocking viral entry.[2][5]
SCHEME
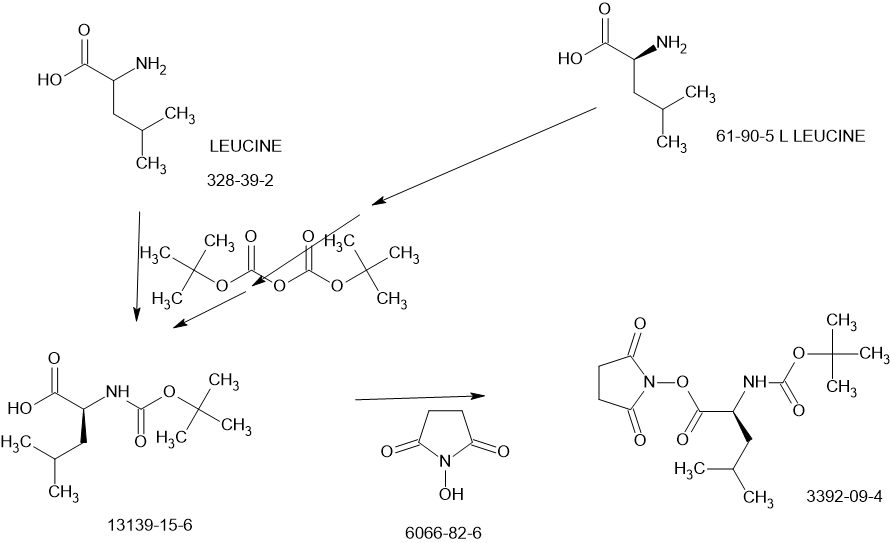
Main
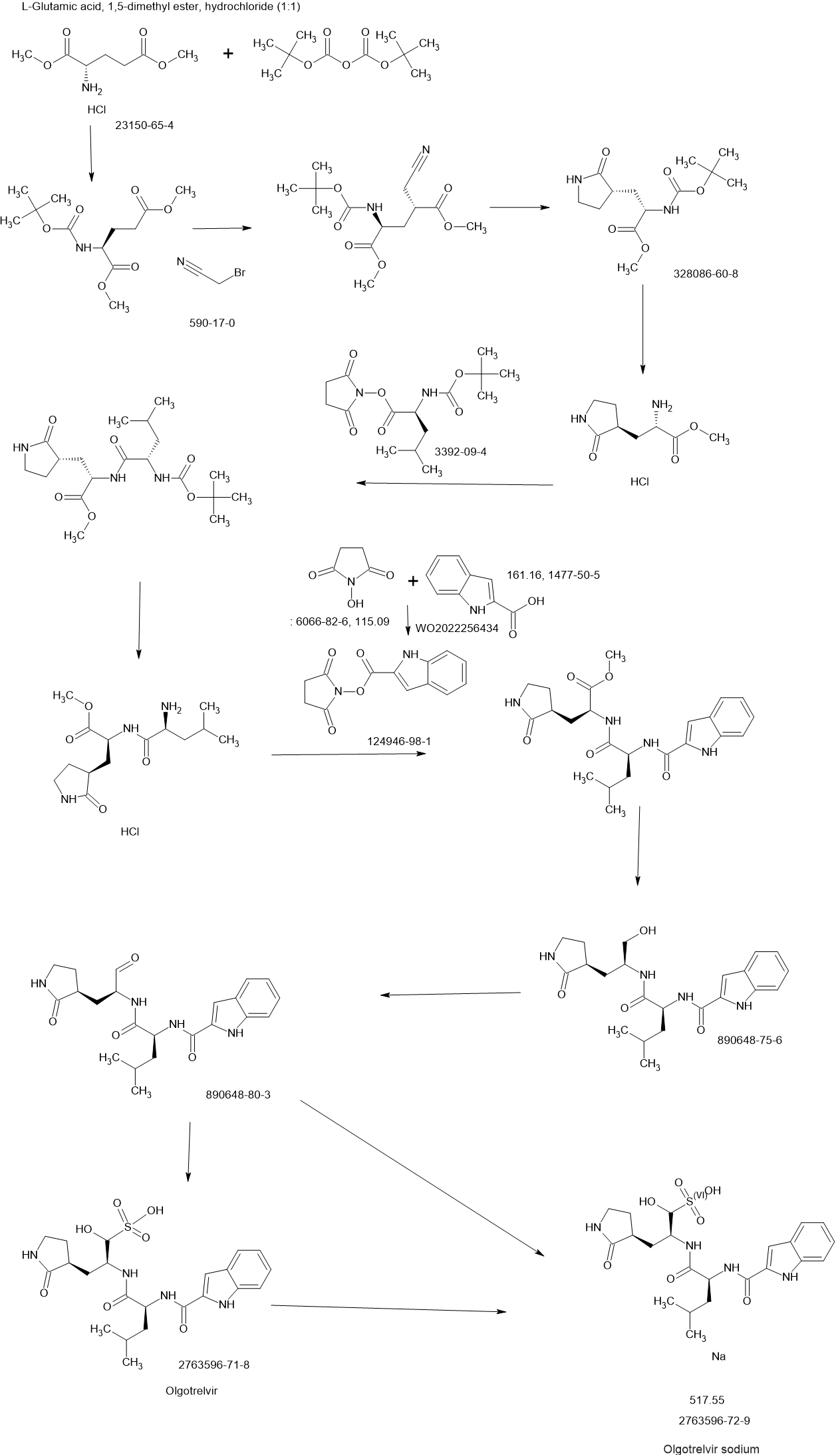
PATENT
US20230322668 – PROTEASE INHIBITORS AS ANTIVIRALS
Example S1: Synthesis of Compounds A-1-a, A-1-b, A-1-c and A-1-d
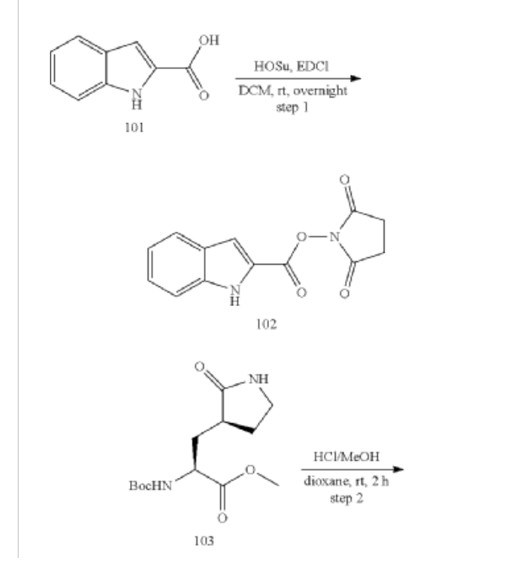




US20230026438 – PROTEASE INHIBITORS AS ANTIVIRALS
WO2022256434 – PROTEASE INHIBITORS AS ANTIVIRALS
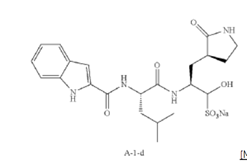
Example S1: Synthesis of Compounds A-1-a, A-1-b, A-1-c and A-1-d
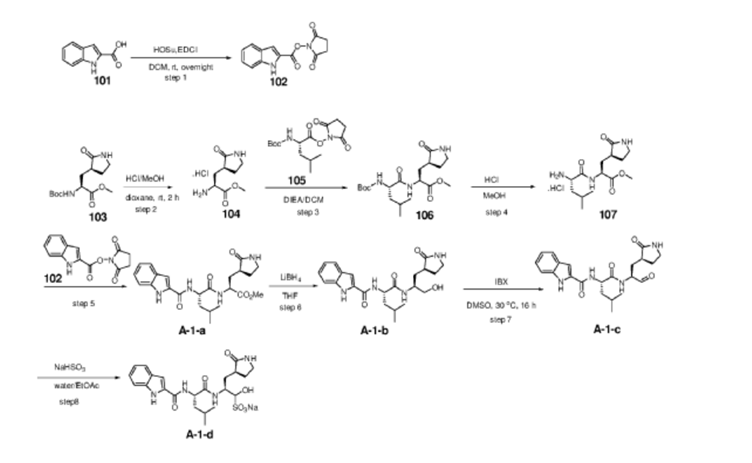
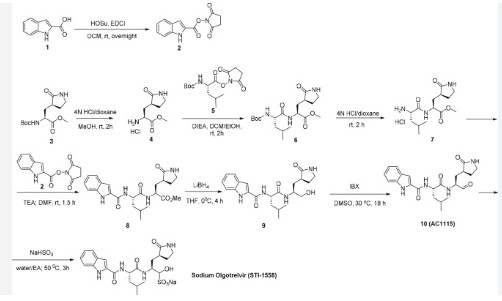
[00224] To a dichloromethane (2.5 L) solution of 1H-indole-2-carboxylic acid (compound 101) (200 g, 1.24 mol) and N-hydroxy succinimide (157.1 g, 1.37 mol) was added EDCI (286 g, 1.49 mmol) at 0℃. After stirring at room temperature overnight, the solvent was removed under reduced pressure. The resulting solid was triturated with deionized water, and the solid was collected and dried under reduced pressure to give the compound 102 as a light-brown solid (310 g, 96%).
1H NMR (400 MHz, CDCl3) δ 9.01 (s, 1H), 7.70 (d, J = 8.2 Hz, 1H), 7.49 – 7.35 (m, 3H), 7.19 (t, J = 7.4 Hz, 1H), 2.92 (s, 4H).
[00225] To a stirred mixture of methyl (2S)-2-{[(tert-butoxy)carbonyl]amino}-3-[(3S)-2- oxopyrrolidin-3-yl]-propanoate (compound 103) (500 g, 1748.24 mmol) in MeOH (200 mL) was
added 4M HCl in 1,4-dioxane (2000 mL) at room temperature. The mixture was stirred at rt for 2 h. LCMS indicated completion of the reaction. The reaction mixture was concentrated under reduced pressure to afford methyl (2S)-2-amino-3-[(3S)-2-oxopyrrolidin-3-yl]propanoate hydrochloride salt (compound 104) (389 g, 1721 mmol, 98%) as a light-yellow solid, which was used for next step without further purification. LCMS= [M+H]+: 187.1.
[00226] To a stirred mixture of methyl (2S)-2-amino-3-[(3S)-2-oxopyrrolidin-3-yl]propanoate hydrochloride (389 g, 1721 mmol) (compound 104) and DIEA (866.162 mL, 5240.94 mmol) in DCM (1800 mL) and EtOH (500 mL) was added 2,5-dioxopyrrolidin-1-yl (2R)-2-{[(tert-butoxy)carbonyl]amino}-4-methyl-pentanoate (compound 105) (573.66 g, 1746.98 mmol) at room temperature. The reaction mixture was stirred at room temperature for 2 h. LCMS indicated completion of the reaction. The reaction mixture was successively washed with water (1.0 L x 2), 0.5 M HCl (1.1 L), sat. NaHCO3 (1 L) and water (1 L). The organic layer was separated, dried with anhydrous Na2SO4, filtered and concentrated under reduced pressure to afford the compound 106 (700 g, 1752.23 mmol, >99%) as a light-yellow solid, which was used for next step without further purification. LCMS = [M+H]+: 400.3.
(400 MHz, DMSO-d6) δ 8.32 (d, J = 8.0 Hz, 1H), 7.62 (s, 1H), 6.88 (d, J = 8.0 Hz, 1H), 4.40 – 4.28 (m, 1H), 3.94 (dd, J = 15.1, 8.1 Hz, 1H), 3.74 – 3.52 (m, 3H), 3.15 (t, J = 8.8 Hz, 1H), 3.06 (dd, J = 16.4, 9.2 Hz, 1H), 2.33 (t, J = 9.2 Hz, 1H), 2.14 – 2.00 (m, 2H), 1.68 – 1.51 (m, 3H), 1.42 – 1.34 (m, 11H), 0.87 (dd, J = 11.4, 6.6 Hz, 6H).
[00227] A mixture of methyl (2S)-2-[(2S)-2-{[(tert-butoxy)carbonyl]amino}-4-methylpentanamido]-3-[(3S)-2-oxopyrrolidin-3-yl]propanoate (compound 106) (590 g, 1476.88 mmol) in HCl/dioxane (3 L) was stirred at room temperature for 2 h. LC-MS indicated completion of the reaction. The reaction mixture was concentrated under reduced pressure to give compound 107 as a yellow solid (490 g, 99%), which was used for next step without further purification.
LCMS = [M+H]+: 300.2.
[00228] To a stirred mixture of methyl (S)-2-((S)-2-amino-4-methylpentanamido)-3-((S)-2-oxopyrrolidin-3-yl)propanoate hydrochloride (compound 107) (418 g, 1235 mmol) and TEA (519.020 mL, 3734.03 mmol) in DMF (2500 mL) at room temperature was added 2,5-dioxopyrrolidin-1-yl 1H-indole-2-carboxylate (compound 102) (353 g, 1369.15 mmol) . The reaction mixture was stirred for 1.5 h. LCMS indicated that the reaction was complete. EtOAc (6 L) was added into the reaction mixture, which was then washed with brine (6 L x 6). The organic layers were combined, dried over anhydrous sodium sulfate, and concentrated down under reduced
pressure. Compound A-1-a was obtained as an off-white solid (414 g. Y: 76%), which was used for next step without further purification. LCMS = [M+H]+: 443.3. 1H NMR (400 MHz, DMSO-d6) δ 11.55 (s, 1H), 8.54 (t, J = 12.2 Hz, 1H), 8.40 (d, J = 8.1 Hz, 1H), 7.62 (d, J = 8.1 Hz, 2H), 7.43 (d, J = 8.2 Hz, 1H), 7.24 (t, J = 10.3 Hz, 1H), 7.18 (t, J = 7.5 Hz, 1H), 7.04 (t, J = 7.5 Hz, 1H), 4.65 – 4.50 (m, 1H), 4.44 – 4.28 (m, 1H), 3.72 – 3.55 (s, 3H), 3.19 – 3.06 (m, 2H), 2.36 (ddd, J = 13.8, 10.3, 4.0 Hz, 1H), 2.16 – 2.03 (m, 2H), 1.79 – 1.49 (m, 5H), 0.92 (dt, J = 14.4, 7.2 Hz, 6H).
[00229] To a stirred solution of methyl (S)-2-((S)-2-(1H-indole-2-carboxamido)-4-methylpentanamido)-3-((S)-2-oxopyrrolidin-3-yl)propanoate (compound A-1-a) (500 g, 1131 mmol) in THF (20 L) LiBH4 (74 g, 3393 mmol) was added portionwise at 0 ℃. The reaction mixture was stirred at 0 ℃ for 4 h. After reaction was completed (monitored by LCMS), the reaction mixture was quenched with sat. aqueous NH4Cl until no more gas formed. The mixture was washed with brine (5 L x 4), organic layer was collected, dried over anhydrous sodium sulfate, filtered, and concentrated down in vacuum. The resulting residue was purified by silica column chromatography (DCM : MeOH = 15 : 1) to give the desired product compound A-1-b (310 g, 66%) as a white solid. LCMS = [M+H]+: 415.2.
NMR (400 MHz, DMSO-d6) δ 11.57 (s, 1H), 8.39 (d, J = 8.2 Hz, 1H), 7.79 (d, J = 9.0 Hz, 1H), 7.61 (d, J = 7.9 Hz, 1H), 7.52 (s, 1H), 7.42 (d, J = 8.3 Hz, 1H), 7.26 (d, J = 1.4 Hz, 1H), 7.17 (t, J = 7.6 Hz, 1H), 7.03 (t, J = 7.5 Hz, 1H), 4.67 (t, J = 5.6 Hz, 1H), 4.50 (td, J = 9.7, 5.0 Hz, 1H), 3.80 (s, 1H), 3.40 – 3.28 (m, 1H), 3.28 – 3.20 (m, 1H), 3.15 – 2.99 (m, 2H), 2.33 – 2.20 (m, 1H), 2.12 (dt, J = 17.8, 9.4 Hz, 1H), 1.86 – 1.75 (m, 1H), 1.75 – 1.64 (m, 2H), 1.56 (ddd, J = 19.3, 9.6, 6.9 Hz, 2H), 1.45 – 1.35 (m, 1H), 0.91 (dd, J = 15.6, 6.3 Hz, 6H).
[00230] To a stirred solution of N-((S)-1-(((S)-1-hydroxy-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-1H-indole-2-carboxamide (compound A-1-b) (8.3 g, 20 mmol) in DMSO (60 mL) was added 2-iodoxybenzoic acid (IBX) (11.2 g, 40 mmol) at room temperature. The reaction mixture was stirred at 30 ℃ for 18 h, and LCMS indicated completion of the reaction. The reaction mixture was diluted with EtOAc (300 mL) and filtered. The filtrate was washed with mixture of brine and sat. aqueous NaHCO3 (1:1 to 5:1, 200 mL x 5). The organic layer was separated, dried over anhydrous sodium sulfate, filtered, and concentrated down at rt to afford crude product. THF (40 mL) was added, and the mixture was stirred overnight at room temperature. The resulting solid was collected and dried under vacuum to yield the desired product N-((S)-4-methyl-1-oxo-1-(((S)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)-propan-2-yl)amino)pentan-2-yl)-1H-indole-2-carboxamide (compound A-1-c) as a white solid (2.5 g, 31%). LCMS = [M+H]+:
413.2. 1H NMR (400 MHz, CDCl3) δ 9.75 (s, 1H), 9.49 (s, 1H), 8.64 (s, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.40 (d, J = 8.4 Hz, 1H), 7.27 (d, J = 8.4 Hz, 1H), 7.14-7.05 (m, 2H), 7.01 (s, 1H), 6.34 (s, 1H), 4.90 (s, 1H), 4.34 (s, 1H), 3.27–3.22 (m, 2H), 2.43 (s, 1H), 2.30 (s, 1H), 2.01-1.96 (m, 1H), 1.94-1.91 (m, 1H) 1.88 – 1.65 (m, 4H), 1.00-0.98 (m, 6H).
[00231] To a stirred solution of N-((S)-4-methyl-1-oxo-1-(((S)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)pentan-2-yl)-1H-indole-2-carboxamide (compound A-1-c) (31 g, 75.25 mmol) in EtOAc (300 mL) at room temperature was added a solution of NaHSO3 (27.56 mg, 72.73 mmol) in water (100 mL). The reaction mixture was heated at 50 ℃ for 3 h. After completion of reaction (monitored by LCMS), the organic layer was separated and removed. The aqueous layer was washed with EtOAc (100 mL x 5), concentrated down to remove remaining EtOAc, and then lyophilized to provide the desired product sodium (2S)-2-((S)-2-(1H-indole-2-carboxamido)-4-methylpentanamido)-1-hydroxy-3-((S)-2-oxopyrrolidin-3-yl)propane-1-sulfonate (compound A-1-d) as off-white solid (32 g, 85%). LCMS = [M-Na+2H]+: 495.2. 1H NMR (400 MHz, DMSO-d6) δ 11.57 (s, 1H), 8.45 (dd, J = 20.7, 8.2 Hz, 1H), 7.72 (dd, J = 48.9, 9.2 Hz, 1H), 7.62 (d, J = 8.1 Hz, 1H), 7.50 – 7.38 (m, 2H), 7.25 (dd, J = 5.1, 1.4 Hz, 1H), 7.18 (t, J = 7.6 Hz, 1H), 7.04 (t, J = 7.5 Hz, 1H), 5.43 (dd, J = 50.7, 5.9 Hz, 1H), 4.57 – 4.41 (m, 1H), 4.33 – 4.03 (m, 1H), 4.01 – 3.82 (m, 1H), 3.19 – 2.92 (m, 2H), 2.29 – 2.08 (m, 2H), 2.06 – 1.90 (m, 1H), 1.83 – 1.51 (m, 5H), 1.00 – 0.83 (m, 6H).
PAPER
https://www.sciencedirect.com/science/article/pii/S2666634023004026
Mechanism of action
Olgotrelvir is a prodrug that first converts to its active form, AC1115.[2][5] AC1115 is believed to work by inhibiting the SARS-CoV-2 main protease (also known as 3C-like protease). This protein is a crucial enzyme responsible for cleaving viral polyproteins into functional subunits essential for viral replication. By binding to the active site of the protease, the drug prevents this cleavage process, effectively halting viral assembly and impeding the virus’s ability to produce future virions.[1][2][3][5]
Olgotrelvir also appears to inhibit cathepsin L (CTSL),[2][5] a protein implicated in facilitating viral entry of SARS-CoV-2 into the host cell.[2][5][6]
Clinical trials
In September 2023, the drug’s developer, Sorrento Therapeutics, announced top-line data that olgotrelvir had met its primary endpoints in a phase III clinical trial that enrolled 1,212 patients with mild or moderate COVID-19. The drug appeared to shorten the recovery time of 11 COVID-19 symptoms in olgotrelvir-treated patients by 2.4 days on average compared to patients in the placebo group. The drug was also shown to reduce the viral load at day 4 in treated patients compared to the placebo group. Side effects were mostly mild and infrequent, with the most common being nausea (1.5% vs. 0.2%) and skin rash (3.3% vs. 0.3%), which occurred more often in the olgotrelvir group.[7][8][9]
References
- ^ Jump up to:a b Tong X, Keung W, Arnold LD, Stevens LJ, Pruijssers AJ, Kook S, et al. (November 2023). “Evaluation of in vitro antiviral activity of SARS-CoV-2 Mpro inhibitor pomotrelvir and cross-resistance to nirmatrelvir resistance substitutions”. Antimicrobial Agents and Chemotherapy. 67 (11): e0084023. doi:10.1128/aac.00840-23. PMC 10649086. PMID 37800975.
Other examples of Mpro inhibitors in late-stage development include STI-1558, currently in the phase 3 clinical trial in adult subjects with mild or moderate COVID-19 (NCT05716425).
- ^ Jump up to:a b c d e f Hackett DW (26 June 2023). “Second Generation Oral Mpro Inhibitor for COVID-19 Treatment Proceeds in Phase 3 Study”. Precision Vaccinations. Retrieved 27 December 2023.
- ^ Jump up to:a b “Coronavirus disease 2019 (COVID-19) emerging treatments”. BMJ Best Practice US. Archived from the original on 27 December 2023. Retrieved 27 December 2023.
- ^ Janin YL (September 2023). “On the origins of SARS-CoV-2 main protease inhibitors”. RSC Medicinal Chemistry. 15 (1): 81–118. doi:10.1039/D3MD00493G. ISSN 2632-8682. PMC 10809347. PMID 38283212. S2CID 264103864.
- ^ Jump up to:a b c d e Mao L, Shaabani N, Zhang X, Jin C, Xu W, Argent C, et al. (January 2024). “Olgotrelvir, a dual inhibitor of SARS-CoV-2 Mpro and cathepsin L, as a standalone antiviral oral intervention candidate for COVID-19”. Med (New York, N.Y.). 5 (1): 42–61.e23. doi:10.1016/j.medj.2023.12.004. PMID 38181791.
- ^ Berdowska I, Matusiewicz M (October 2021). “Cathepsin L, transmembrane peptidase/serine subfamily member 2/4, and other host proteases in COVID-19 pathogenesis – with impact on gastrointestinal tract”. World Journal of Gastroenterology. 27 (39): 6590–6600. doi:10.3748/wjg.v27.i39.6590. PMC 8554394. PMID 34754154.
- ^ Jiang R, Han B, Xu W, Zhang X, Peng C, Dang Q, et al. (June 2024). “Olgotrelvir as a Single-Agent Treatment of Nonhospitalized Patients with Covid-19”. NEJM Evidence. 3 (6): EVIDoa2400026. doi:10.1056/EVIDoa2400026. PMID 38804790.
- ^ Sherman AC, Baden LR (June 2024). “How To Measure Benefit in a Changing Pandemic – Olgotrelvir for SARS-CoV-2”. NEJM Evidence. 3 (6): EVIDe2400144. doi:10.1056/EVIDe2400144. PMID 38804789.
- ^ “Sorrento Announces Phase 3 Trial Met Primary Endpoint and Key Secondary Endpoint in Mild or Moderate COVID-19 Adult Patients Treated with Ovydso (Olgotrelvir), an Oral Mpro Inhibitor as a Standalone Treatment for COVID-19” (Press release). BioSpace. 12 September 2023. Retrieved 27 December 2023.
| Clinical data | |
|---|---|
| Trade names | Ovydso |
| Other names | STI-1558, HY-156655, CS-0887294 |
| Routes of administration | By mouth |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2763596-71-8 |
| PubChem CID | 166157331 |
| UNII | ZP3BDH359D |
| KEGG | D12777 |
| Chemical and physical data | |
| Formula | C22H30N4O7S |
| Molar mass | 494.56 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
//////Olgotrelvir, STI-1558, HY-156655, CS-0887294, STI 1558, HY 156655, CS 0887294, ZP3BDH359D
ENSITRELVIR
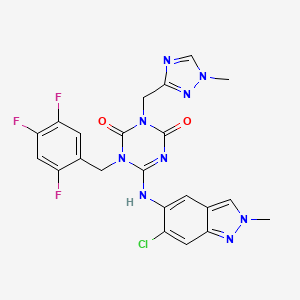
Ensitrelvir
S-217622, S 217622, Xocova, SHIONOGI,
6-[(6-chloro-2-methylindazol-5-yl)amino]-3-[(1-methyl-1,2,4-triazol-3-yl)methyl]-1-[(2,4,5-trifluorophenyl)methyl]-1,3,5-triazine-2,4-dione
CAS 2647530-73-0
| C22H17ClF3N9O2531.9 | |
| Synonyms | BDBM513874bioRxiv20220126.477782, S-217622 |
|---|
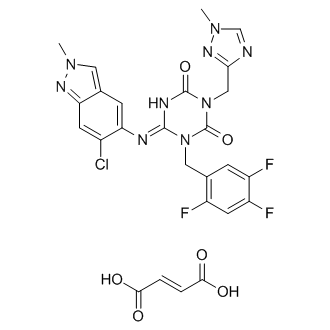
Ensitrelvir fumarate
CAS No. : 2757470-18-9
C22 H17 Cl F3 N9 O2 . C4 H4 O4
1,3,5-Triazine-2,4(1H,3H)-dione, 6-[(6-chloro-2-methyl-2H-indazol-5-yl)imino]dihydro-3-[(1-methyl-1H-1,2,4-triazol-3-yl)methyl]-1-[(2,4,5-trifluorophenyl)methyl]-, (6E)-, (2E)-2-butenedioate (1:1)
| Formula: | C26H21ClF3N9O6 |
|---|---|
| M. Wt. : | 647.95 |
FDA 2026, APPROVALS 2026, 5/29/2026, Xocova
To use as post-exposure prophylaxis of coronavirus disease 2019 (COVID-19) following contact with an individual who has COVID-19
A Phase 1 study of S-217622 in healthy adult participants (jRCT2031210202)
Japan Registry of Clinical Trials Web Site 2021, July 16
PMDA APPROVED 2022/11/22, Xocova
Ensitrelvir[1] (code name S-217622, brand name Xocova)[2] is an antiviral drug developed by Shionogi in partnership with Hokkaido University, which acts as an orally active 3C-like protease inhibitor for the treatment of COVID-19 infection.[3][4] It is taken by mouth, and has been successfully tested against the recently emerged Omicron variant.[5]
About S-217622
S-217622, a therapeutic drug for COVID-19, is a 3CL protease inhibitor created through joint research between Hokkaido University and Shionogi. SARS-CoV-2 has an enzyme called 3CL protease, which is essential for the replication of the virus. S-217622 suppresses the replication of SARS-CoV-2 by selectively inhibiting 3CL protease. Shionogi has already been submitting the non-clinical, manufacturing/CMC data, and clinical trial data obtained so far to the PMDA. Currently the Phase 3 part of a Phase 2/3 clinical trial in patients with mild/moderate symptoms and the Phase 2b/3 part in patients with asymptomatic/only mild symptoms are in progress.
SYN
J.Med.Chem.2024,67,4376−4418
Ensitrelvir fumaric acid (3), also referred to as S-217622, is an oral noncovalent SARS-CoV-2 main protease (Mpro) inhibitordeveloped by Shionogi & Co. that was approved by the japan Pharmaceuticals and Medical Devices Agency (PMDA)for the treatment of disease caused by SARS-CoV-2 (COVID
19) infection. Dosed once daily for 5 days, ensitrelvirsuppresses the replication of SARS-CoV-2 in infected patients as a result of its inhibition of the viral mpro.25,26
Ensitrelvir retains potent inhibitory activity against many of the most common M mutants and exhibits antiviral activity against a wide variety of circulating SARS-CoV-2 variants. 27is the second Mpro
Ensitrelvir inhibitor approved for the treatment of 28 disease caused by COVID-19. Unlike the first approved treatment, Paxlovid, ensitrelvir does not require coadministration with a CYP3A4 inhibitor to attenuate metabolism in vivo.
Furthermore, crystal structures of ensitrelvir in complex with the main proteases of three other human-infecting coronaviruses (MERS-CoV, SARS-CoV, and HCoV-NL63)
A convergent, kilogram-scale synthesis of ensitrelvir suitable for manufacturing has been described in the literature by researchers at Shionogi. 30 The synthetic approach involved the union of two key building blocks indazole 3.7 and 1,3,5triazinone 3.14, each necessitating development of a scale worthy route. The preparation of triazinone 3.14 necessitated construction of a triazolyl methylene chloride subunit which
began with the reduction of triazole ester 3.1 with aluminum hydride 3.2 (a less pyrophoric alternative to LAH yet still required aqueous Rochelle salt quench to chelate excess aluminum) 31to provide alcohol 3.3, which was then convertedto the corresponding chloride and isolated as the triazole HCl
salt 3.4 (Scheme 6). Assembly of indazole intermediate 3.7began with regioselective nitration of benzaldehyde 3.5followed by treatment with hydrazine hydrate in aqueous EtOHtoprovide indazole3.6(Scheme7).Fascinatingly, the Shionogi team isolated a variety of byproducts during the
conversionof3.5to3.6whichsupportedtheirhypothesisforareaction mechanism that likely equilibrated through a dibenzylidenehydrazine intermediateenroute tothedesired
indazole3.6.Ascreenofelectrophilicmethyl sourcesrevealed thatMeerwein’s salt facilitatedthebest conversionof 3.6to the correspondingN2-monomethylated indazole; subsequenthydrogenative nitro reduction furnished the key indazole intermediate 3.7.Construction of the ensitrelvir core started with reaction of carboximidamide 3.8 with t-butyl isocyanate followed by N,N′carbonyldiimidazole (CDI) to secure 1,3,5-triazinone 3.10(Scheme 8). Subsequent N-alkylation with bromide 3.11provided benzyl triazinone 3.12. Substitution of the pyrazolewith m-cresol was accomplished under acidic conditions. The
authors report that m-cresol was identified as a leaving group that facilitated introduction of indazole 3.7 with a minimal number of byproducts in a later step of the synthesis. The TFA-mediated reaction concomitantly removed the N-tertbutyl group providing compound 3.13 in 91% yield. Nalkylation with chloride 3.4 in the presence of a base resulted in intermediate 3.14 which was then treated with building
block 3.7 in the presence of anhydrous acetic acid. Isolation of ensitrelvir fumaric acid was achieved by exposure to fumaric acid in aqueous acetone.
(25) Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Imamura, T.;
Sonoyama, T.; Ichihashi, G.; Sanaki, T.; Tsuge, Y.; Uehara, T.;
Mukae, H. A phase 2/3 study of S-217622 in participants with SARS
CoV-2 infection (Phase 3 part). Medicine 2023, 102, No. e33024.
(26) Mukae, H.; Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Imamura,
T.; Sonoyama, T.; Fukuhara, T.; Ichihashi, G.; Sanaki, T.; Baba, K.;
Takeda, Y.; Tsuge, Y.; Uehara, T. A randomized phase 2/3 study of
ensitrelvir, a novel oral SARS-CoV-2 3C-like protease inhibitor, in
Japanese patients with mild-to-moderate COVID-19 or asymptomatic
SARS-CoV-2 infection: results of the phase 2a part. Antimicrob. Agents
Chemother. 2022, 66, No. 00697.
(27) Kawashima, S.; Matsui, Y.; Adachi, T.; Morikawa, Y.; Inoue, K.;
Takebayashi, S.; Nobori, H.; Rokushima, M.; Tachibana, Y.; Kato, T.
Ensitrelvir is effective against SARS-CoV-2 3CL protease mutants
circulating globally. Biochem. Biophys. Res. Commun. 2023, 645, 132−
136.
(28) Unoh, Y.; Uehara, S.; Nakahara, K.; Nobori, H.; Yamatsu, Y.;
Yamamoto, S.; Maruyama, Y.; Taoda, Y.; Kasamatsu, K.; Suto, T.;
et al. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL
protease inhibitor clinical candidate for treating COVID-19. J. Med.
Chem. 2022, 65, 6499−6512
(29) Lin, C.; Jiang, H.; Li, W.; Zeng, P.; Zhou, X.; Zhang, J.; Li, J.
Structural basis for the inhibition of coronaviral main proteases by
ensitrelvir. Structure 2023, 31, 1016.
(30) Kawajiri, T.; Kijima, A.; Iimuro, A.; Ohashi, E.; Yamakawa, K.;
Agura, K.; Masuda, K.; Kouki, K.; Kasamatsu, K.; Yanagisawa, S.; et al.
Development of a manufacturing process toward the convergent
synthesis of the COVID-19 antiviral Ensitrelvir. ACS Cent. Sci. 2023,
9, 836−843.
(31) Gugelchuk, M.; Silva, III, L. F.; Vasconcelos, R. S.; Quintiliano,
S. A. P. Sodium bis(2-methoxyethoxy)aluminum hydride. In
Encyclopedia of Reagents for Organic Synthesis; Charette, A., Bode, J.,
Rovis, T., Shenvi, R., Eds.; John Wiley & Sons, Ltd., 2007.


Syn
Discovery of S-217622, a Non-Covalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19
View ORCID ProfileYuto Unoh, View ORCID ProfileShota Uehara, View ORCID ProfileKenji Nakahara, View ORCID ProfileHaruaki Nobori, Yukiko Yamatsu, View ORCID ProfileShiho Yamamoto, View ORCID ProfileYuki Maruyama, View ORCID ProfileYoshiyuki Taoda, View ORCID ProfileKoji Kasamatsu, View ORCID ProfileTakahiro Suto, Kensuke Kouki, View ORCID ProfileAtsufumi Nakahashi, Sho Kawashima, View ORCID ProfileTakao Sanaki, Shinsuke Toba, Kentaro Uemura, Tohru Mizutare, View ORCID ProfileShigeru Ando, View ORCID ProfileMichihito Sasaki, View ORCID ProfileYasuko Orba, View ORCID ProfileHirofumi Sawa, View ORCID ProfileAkihiko Sato, View ORCID ProfileTakafumi Sato, View ORCID ProfileTeruhisa Kato, View ORCID ProfileYuki Tachibana
doi: https://doi.org/10.1101/2022.01.26.477782
https://www.biorxiv.org/content/10.1101/2022.01.26.477782v1.full
The coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has resulted in millions of deaths and threatens public health and safety. Despite the rapid global spread of COVID-19 vaccines, effective oral antiviral drugs are urgently needed. Here, we describe the discovery of S-217622, the first oral non-covalent, non-peptidic SARS-CoV-2 3CL protease inhibitor clinical candidate. S-217622 was discovered via virtual screening followed by biological screening of an in-house compound library, and optimization of the hit compound using a structure-based drug-design strategy. S-217622 exhibited antiviral activity in vitro against current outbreaking SARS-CoV-2 variants and showed favorable pharmacokinetic profiles in vivo for once-daily oral dosing. Furthermore, S-217622 dose-dependently inhibited intrapulmonary replication of SARS-CoV-2 in mice, indicating that this novel non-covalent inhibitor could be a potential oral agent for treating COVID-19.
Chemistry

The synthetic scheme for compound 1 is described in Scheme 1. Starting from the pyrazole derivative 4, cyclization with Ethyl isocyanatoacetate and CDI was conducted, giving 5 in 90% yield. Then, an alkylation with 5-bromomethyl-1,2,3-trifluorobenzene followed by introduction of a 4-difluoromethoxy-2-methylaniline unit, to give 7 (40% in 2 steps). The ester group in 7 was hydrolyzed and then amidated with methylamine, yielding 1 (58% in 2 steps). Compound 2 was synthesized similarly as shown in Scheme 2.
S-217622 (3) was synthesized as described in Scheme 3. Starting from known compound 9,21 an alkylation with 1-(bromomethyl)-2,4,5-trifluorobenzene gave 10 in 93% yield. Then, the 3-tert-Bu group was removed and the triazole unit was introduced, and the substitution of the SEt moiety with the indazole unit finally gave S-217622 (3).
21 Kai, H.; Kameyama, T.; Horiguchi, T.; Asahi, K.; Endoh, T.; Fujii, Y.; Shintani, T.; Nakamura, K.; Matsumoto, S.; Hasegawa, T.; Oohara, M.; Tada, Y.; Maki, T.; Iida, A. Preparation of triazine derivatives and pharmaceutical compound that contains same and exhibits analgesic activity. WO 2012020749 A1, Feb 16, 2012

Scheme 1.
Reagents and Conditions: (a) ethyl isocyanato-acetate, DBU, CDI, DMA, –10 °C to rt, 90%; (b) 5-bromomethyl-1,2,3-trifluorobenzene, N,N-diisopropylethylamine, DMA, 60 °C; (c) 4-difluoromethoxy-2-methylaniline, tert-butanol, 100 °C, 40% in 2 steps; (d) (i) NaOH aq., THF/MeOH, rt; (ii) methylamine, HATU, N,N-diisopropylethylamine, THF, rt., 58% in 2 steps.

Scheme 2.
Reagents and Conditions: (a) 6-chloro-2-methyl-2H-indazol-5-amine, tert-amyl alcohol, 100 °C, 44% in 2 steps from 5; (b) (i) NaOH aq., THF/MeOH, rt; (ii) methylamine, HATU, N,N-diisopropylethylamine, THF, rt., 29% in 2 steps.

Scheme 3.
Reagents and Conditions: (a) 1-(bromomethyl)-2,4,5-trifluorobenzene, K2CO3, MeCN, 80 °C, 93%; (b) TFA, rt, 97%; (c) 3-(chloromethyl)-1-methyl-1H-1,2,4-triazole hydrochloride, K2CO3, DMF, 60 °C, 45%; (d) 6-chloro-2-methyl-2H-indazol-5-amine, LHMDS, THF, 0 °C to rt., 25%.
(6E)-6-[(6-Chloro-2-methyl-2H-indazol-5-yl)imino]-3-[(1-methyl-1H-1,2,4-triazol-3-yl)methyl]-1-(2,4,5-trifluorobenzyl)-1,3,5-triazinane-2,4-dione (3, S-217622)
To a solution of 12 (300 mg, 0.727 mmol) and 6-chloro-2-methyl-2H-indazol-5-amine (172 mg, 0.946 mmol) in THF (6 mL) was added LHMDS (1M in THF; 1.46 mL, 1.46 mmol) dropwisely at 0 °C. The reaction mixture was stirred at 0 °C for 2.5 h and then at rt for 40 min. The reaction was quenched with aqueous NH4Cl solution, and the aqueous layer was extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CHCl3/MeOH gradient, 0-20% MeOH). The solid was recrystallized from acetone/H2O to afford 3 (S-217622) (95.3 mg, 25%) as a pale brown solid. 1H NMR (400 MHz, DMSO-d6, DCl in D2O) δ 3.90 (3H, s), 4.15 (3H, s), 5.04 (2H, s), 5.26 (2H, s), 7.44 (1H, m), 7.52-7.65 (2H, m), 7.73 (1H, s), 8.40 (1H, s), 9.31 (1H, s). 13C NMR (100 MHz, DMSO-d6, DCl in D2O) δ 37.34, 38.04, 40.06, 40.29, 106.16 (dd, J = 28.2, 21.6 Hz), 116.46-116.70, 116.70, 120.54-120.76, 120.76, 125.93, 129.10, 132.35, 143.84, 145.98, 146.38 (ddd, J = 241.4, 12.5, 3.7 Hz), 146.60, 148.52 (td, J = 247.7, 13.6 Hz), 150.43, 150.50, 155.22 (ddd, J = 244.3, 10.3, 2.2 Hz), 155.58. HRMS-ESI (m/z): [M + H]+ calcd for [C22H18 F3ClN9O2]+ 532.1219; found 532.1221.
Preparation of Compound 3 (S-217622) fumaric acid co-crystal
A mixture of 3 (S-217622) (1.17 g, 2.2 mmol) and fumaric acid (278 mg, 2.4 mmol) in EtOAc (5.9 mL) was stirred at room temperature for 45 min. The suspension was filtrated to afford 3 (S-217622) fumaric acid co-crystal (1.37 g, 95 %) as a white solid. 1H NMR (400 MHz, pyridine-d5) δ 3.64 (s, 3H), 3.99 (s, 3H), 5.56 (s, 2H), 5.61 (s, 2H), 7.16-7.25 (m, 2H), 7.44 (s, 2H), 7.81 (s, 1H), 7.89 (s, 1H), 7.89-7.97 (m, 1H), 8.32 (s, 1H).
Notes
SHIONOGI has applied for a patent covering 1, 2, and 3 (S-217622). Y.U., S.U., K.N., H.N., Y.Y., S.Y., Y.M., Y.T., K.K., T.S., K.K., A.N., S.K., T.S., S.T., K.U., T.M., S.A., A.S., T.S., T.K., and Y.T. are employees of SHIONOGI & Co., Ltd. S.U., K.N., H.N., Y.M., Y.T., K.K., T.S., K.K., S.K., TS, S.T., K.U., T.S., and T.K. are shareholders in SHIONOGI & Co., Ltd. M.S., Y.O., and H.S. are financially supported by the joint research fund from SHIONOGI & Co., Ltd.
- Supporting information[supplements/477782_file02.pdf]
see spectrum at end of page
///////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Oral antiviral medications, in addition to vaccines, are expected to play an important role in treating coronavirus disease 2019 (COVID-19), which is caused by infection with the severe acute respiratory disease coronavirus-2 (SARS-CoV-2).
These drugs must have significant antiviral activity, as well as target specificity, oral bioavailability, and metabolic stability. Although several antiviral compounds have been reported as possible SARS-CoV-2 inhibitors in vitro, only a few of these drugs have been shown to be effective in vivo.
Ensitrelvir, a novel SARS-CoV-2 antiviral
Ensitrelvir (code name S-217622, brand name Xocova), is a new inhibitor of the SARS-CoV-2 major protease (Mpro), also known as 3C-like protease, has been shown to reduce the viral load and help alleviate the severity of SARS-CoV-2 in infected hamsters. In cells, low nanomolar to sub-micromolar doses of S-217622 suppress viral growth. In hamsters, oral treatment of S-217622 showed excellent pharmacokinetic qualities and hastened recovery from acute SARS-CoV-2 infection.
S-217622 also demonstrated antiviral effectiveness against SARS-CoV-2 variants of concern (VOCs), such as the highly pathogenic Delta variant and the newly discovered Omicron variant. Overall, these findings show that S-217622, which is an antiviral drug that is currently being tested in Phase II/III clinical trials, has impressive antiviral efficiency and effectiveness against SARS-CoV-2 and could be a viable oral treatment option for COVID-19.
History
It has reached Phase III clinical trials.[3] The Japanese government is reportedly considering allowing Shionogi permission to apply for approval for medical use before the final steps of trials are completed, potentially speeding up the release for sale. This conditional early approval system has previously been used in Japan to accelerate the progression to market of other antiviral drugs targeting COVID-19, including remdesivir and molnupiravir.[6] In a study of 428 patients, viral load was reduced, but symptoms were not significantly reduced. [7]
It became the first Japanese domestic pill to treat COVID-19, third to be regulatorally approved in Japan; in February 2022.[8]

NEW DRUG APPROVALS
ONE TIME
$10.00
References
- ^ World Health Organization (2021). “International Nonproprietary Names for Pharmaceutical Substances. Proposed INN: List 126” (PDF). WHO Drug Information. 35 (4): 1135.
- ^ Xocova: Powerful New Japanese Pill for Coronavirus Treatment. BioPharma Media, February 2022
- ^ Jump up to:a b Unoh Y, Uehara S, Nakahara K, Nobori H, Yamatsu Y, Yamamoto S, et al. (January 2022). “Discovery of S-217622, a Non-Covalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19”. bioRxiv. doi:10.1101/2022.01.26.477782. S2CID 246367525.
- ^ “Shionogi presents positive Ph II/III results for COVID-19 antiviral S-217622”. thepharmaletter.com. 31 January 2022.
- ^ Shionogi’s new COVID pill appears to ease omicron symptoms. Nikkei Asia, 21 December 2021
- ^ Japan to consider early approval for Shionogi COVID-19 pill. Japan Times, 8 February 2022
- ^ https://www.reuters.com/business/healthcare-pharmaceuticals/japans-shionogi-seeks-approval-oral-covid-19-drug-2022-02-25/[bare URL]
- ^ “Japan’s Shionogi seeks approval for COVID-19 pill”. Reuters. Reuters. 25 February 2022.
| Clinical data | |
|---|---|
| Other names | S-217622 |
| Identifiers | |
| showIUPAC name | |
| PubChem CID | 162533924 |
| Chemical and physical data | |
| Formula | C22H17ClF3N9O2 |
| Molar mass | 531.88 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
Journal reference:
- Sasaki, M., Tabata, K., Kishimoto, M., et al. (2022). Oral administration of S-217622, a SARS-CoV-2 main protease inhibitor, decreases the viral load and accelerates recovery from clinical aspects of COVID-19. bioRxiv. doi:10.1101/2022.02.14.480338. https://www.biorxiv.org/content/10.1101/2022.02.14.480338v1.full.
///////////Ensitrelvir, S-217622, S 217622, Xocova, SHIONOGI, CORONA VIRUS, covid 19


4-Hydroxy-TEMPO, TEMPOL, MBM-02, MTS 01
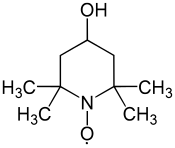
4-Hydroxy-TEMPO, TEMPOL, MBM-02, MTS 01
- Molecular FormulaC9H18NO2
- Average mass172.245 Da
2,2,6,6-Tetramethyl-4-hydroxypiperidinooxy
2,2,6,6-Tetramethyl-4-hydroxypiperidinooxy radical
2,2,6,6-Tetramethyl-4-piperidinol 1-oxyl
CAS 2226-96-2[RN]
4-hydroxy-1-oxyl-2,2,6,6-tetramethylpiperidine
4-Hydroxy-2,2,6,6-tetramethyl-1-piperidin-1-yloxy, free radical
4-Hydroxy-2,2,6,6-tetramethylpiperidine N-oxide
4-Hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl
TEMPOLCAS Registry Number: 2226-96-2
CAS Name: 4-Hydroxy-2,2,6,6-tetramethyl-1-piperidinyloxy
Additional Names: 4-hydroxy-TEMPO; 4-hydroxy-2,2,6,6-tetramethyl piperidine N-oxide; 4-hydroxy-2,2,6,6-tetramethylpiperidinooxy
Molecular Formula: C9H18NO2, Molecular Weight: 172.24
Percent Composition: C 62.76%, H 10.53%, N 8.13%, O 18.58%
Literature References: Stable nitroxyl radical; water-soluble analogue of TEMPO, q.v. Functions as a membrane-permeable radical scavenger. Prepn: E. G. Rozantsev, Bull. Acad. Sci. USSR Div. Chem. Sci.12, 2085 (1964). Energy transfer studies: N. N. Quan, A. V. Guzzo, J. Phys. Chem.85, 140 (1981). IR conformation study: W. A. Bueno, L. Degrève, J. Mol. Struct.74, 291 (1981).Solid state NMR spectra: C. J. Groombridge, M. J. Perkins, J. Chem. Soc. Chem. Commun.1991, 1164. LC/MS/MS determn: I. D. Podmore, J. Chem. Res. Synop.2002, 574. Use as a phase transfer catalyst: X.-Y. Wang et al.,Synth. Commun.29, 157 (1999). Review of effects in animal models for shock, ischemia-reperfusion injury, and inflammation: C. Thiemermann, Crit. Care Med.31, S76-S84 (2003).
Properties: Crystals from ether + hexane, mp 71.5°. uv max (hexane): 240, 450-500 (e ~1800, ~5). uv max (ethanol): 242, 435-455 (e ~3800, ~10). Sol in water.
Melting point: mp 71.5°
Absorption maximum: uv max (hexane): 240, 450-500 (e ~1800, ~5); uv max (ethanol): 242, 435-455 (e ~3800, ~10)
Use: Spin label for EPR studies; phase transfer dehydration catalyst; antioxidant; inhibitor of olefin free radical polymerization.Topical PiperidineNitroxide MTS-01 is a topical gel containing a cell permeable hydrophilic piperidinenitroxide with potential radioprotective and antioxidant activity. As a stable, free radical compound, MTS-01 may be able to protect cells against the damaging effects of reactive oxygen species (ROS), upon exposure to ionizing radiation and oxidative stress. The topically applied MTS-01 may protect normal tissue from radiation-induced toxicity, such as radiation dermatitis, during radiation therapy.
4-Hydroxy-TEMPO is a member of aminoxyls and a member of piperidines. It has a role as a radical scavenger and a catalyst. It derives from a TEMPO.
4-Hydroxy-TEMPO or TEMPOL, formally 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl, is a heterocyclic compound. Like the related TEMPO, it is used as a catalyst and chemical oxidant by virtue of being a stable aminoxyl radical. Its major appeal over TEMPO is that is less expensive, being produced from triacetone amine, which is itself made via the condensation of acetone and ammonia. This makes it economically viable on an industrial scale.[3]

Example synthesis of 4-Hydroxy-TEMPO from phorone, which is itself made from acetone and ammonia
In biochemical research, 4-hydroxy-TEMPO has been investigated as an agent for limiting reactive oxygen species. It catalyzes the disproportionation of superoxide, facilitates hydrogen peroxide metabolism, and inhibits Fenton chemistry.[4] 4-Hydroxy-TEMPO, along with related nitroxides, are being studied for their potential antioxidant properties.[5]
On an industrial-scale 4-hydroxy-TEMPO is often present as a structural element in hindered amine light stabilizers, which are commonly used stabilizers in plastics, it is also used as a polymerisation inhibitor, particularly during the purification of styrene.
It is a promising model substance to inhibit SARS-CoV-2 RNA-dependent RNA polymerase.[6]
SYN

SYN
Inorganica Chimica Acta, 370(1), 469-473; 2011
| IR | (KBr)vmax/cm-1: 3413 (m(O-H)) |
| Crystal Structure Data | Empirical formula C25H26NO8F6Cu; Formula weigh 646.02; T (K) 293(2); λ/Å 0.71073; Crystal system monoclinic; Space group P21/c; a (Å) 10.132(2); b (Å) 25.103(5); c (Å) 13.578(5); α (°) 90; β (°) 121.67(2); γ (°) 90; V (Å3) 2939.2(14); Z = 4 |
SYN
Bioorganic & Medicinal Chemistry Letters, 22(2), 920-923; 2012
SYN
https://pubs.acs.org/doi/10.1021/ol0712024
SYN
https://pubs.acs.org/doi/10.1021/es302157j
PAT
CN 113429392
SYN
Journal of the American Chemical Society, 138(29), 9069-9072; 2016
https://pubs.acs.org/doi/10.1021/jacs.6b05421
file:///C:/Users/Inspiron/Downloads/ja6b05421_si_001.pdf
| 1H NMR | (400 MHz, CDCl3) δH 3.89 (1H, tt, J = 11.4, 4.3 Hz, H4), 1.82.-.1.77 (2H, m, H3, H5), 1.43 (2H, t, J = 11.9 Hz, H3, H5), 1.14 (6H, s, 2 ×CH3), 1.07 (6H, s, 2 ×CH3); |
| 13C NMR | (100 MHz, CDCl3) δH 63.1, 47.5, 31.6, 20.6. |
| IR | (thin film, νmax / cm-1) 3407, 1472, 1376, 1174, 1161, 1066; |
| Rf | 0.22 (ethyl acetate / petroleum ether (1:1)); |
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
////////////////////////////////////////
Thursday, June 3, 2021
NIH researchers identify potential new antiviral drug for COVID-19
Compound targets essential viral enzyme and prevents replication in cells.https://www.nih.gov/news-events/news-releases/nih-researchers-identify-potential-new-antiviral-drug-covid-19 small spherical structures in the center of the image are SARS-CoV-2 virus particles. The string-like protrusions extending from the cells are cell projections or pseudopodium. NIAID
small spherical structures in the center of the image are SARS-CoV-2 virus particles. The string-like protrusions extending from the cells are cell projections or pseudopodium. NIAID
The experimental drug TEMPOL may be a promising oral antiviral treatment for COVID-19, suggests a study of cell cultures by researchers at the National Institutes of Health. TEMPOL can limit SARS-CoV-2 infection by impairing the activity of a viral enzyme called RNA replicase. The work was led by researchers at NIH’s Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD). The study appears in Science.
“We urgently need additional effective, accessible treatments for COVID-19,” said Diana W. Bianchi, M.D., NICHD Director. “An oral drug that prevents SARS-CoV-2 from replicating would be an important tool for reducing the severity of the disease.”
The study team was led by Tracey A. Rouault, M.D., head of the NICHD Section on Human Iron Metabolism. It discovered TEMPOL’s effectiveness by evaluating a more basic question on how the virus uses its RNA replicase, an enzyme that allows SARS-CoV-2 to replicate its genome and make copies of itself once inside a cell.
Researchers tested whether the RNA replicase (specifically the enzyme’s nsp12 subunit) requires iron-sulfur clusters for structural support. Their findings indicate that the SARS-CoV-2 RNA replicase requires two iron-sulfur clusters to function optimally. Earlier studies had mistakenly identified these iron-sulfur cluster binding sites for zinc-binding sites, likely because iron-sulfur clusters degrade easily under standard experimental conditions.
Identifying this characteristic of the RNA replicase also enables researchers to exploit a weakness in the virus. TEMPOL can degrade iron-sulfur clusters, and previous research from the Rouault Lab has shown the drug may be effective in other diseases that involve iron-sulfur clusters. In cell culture experiments with live SARS-CoV-2 virus, the study team found that the drug can inhibit viral replication.
Based on previous animal studies of TEMPOL in other diseases, the study authors noted that the TEMPOL doses used in their antiviral experiments could likely be achieved in tissues that are primary targets for the virus, such as the salivary glands and the lungs.
“Given TEMPOL’s safety profile and the dosage considered therapeutic in our study, we are hopeful,” said Dr. Rouault. “However, clinical studies are needed to determine if the drug is effective in patients, particularly early in the disease course when the virus begins to replicate.”
The study team plans on conducting additional animal studies and will seek opportunities to evaluate TEMPOL in a clinical study of COVID-19.
NIH authors on the study include researchers from the National Cancer Institute, the National Institute of Allergy and Infectious Diseases, and the National Institute of Neurological Disorders and Stroke. Authors from the Pennsylvania State University are funded by NIH’s National Institute of General Medical Sciences.
About the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD): NICHD leads research and training to understand human development, improve reproductive health, enhance the lives of children and adolescents, and optimize abilities for all. For more information, visit https://www.nichd.nih.gov.
About the National Institutes of Health (NIH): NIH, the nation’s medical research agency, includes 27 Institutes and Centers and is a component of the U.S. Department of Health and Human Services. NIH is the primary federal agency conducting and supporting basic, clinical, and translational medical research, and is investigating the causes, treatments, and cures for both common and rare diseases. For more information about NIH and its programs, visit www.nih.gov.
NIH…Turning Discovery Into Health®
Article
Maio N, et al. Fe-S cofactors in the SARS-CoV-2 RNA-dependent RNA polymerase are potential antiviral targets. Science DOI: 10.1126/science.abi5224(link is external) (2021)
References
- ^ Zakrzewski, Jerzy; Krawczyk, Maria (1 January 2011). “Reactions of Nitroxides. Part XII [1]. – 2,2,6,6-Tetramethyl-1-oxyl- 4-piperidyl Chloroformate – A New Reactive Nitroxyl Radical. A One-pot Synthesis of 2,2,6,6-Tetramethyl-1-oxyl-4-piperidyl N,N-Dialkyl-carbamates”. Zeitschrift für Naturforschung B. 66 (5). doi:10.1515/znb-2011-0509.
- ^ Jump up to:a b c d Sigma-Aldrich Co., 4-Hydroxy-TEMPO. Retrieved on 2015-08-24.
- ^ Ciriminna, Rosaria; Pagliaro, Mario (15 January 2010). “Industrial Oxidations with Organocatalyst TEMPO and Its Derivatives”. Organic Process Research & Development. 14 (1): 245–251. doi:10.1021/op900059x.
- ^ Wilcox, C. S.; Pearlman, A. (2008). “Chemistry and Antihypertensive Effects of Tempol and Other Nitroxides”. Pharmacological Reviews. 60 (4): 418–69. doi:10.1124/pr.108.000240. PMC 2739999. PMID 19112152.
- ^ Lewandowski, M; Gwozdzinski, K. (2017). “Nitroxides as Antioxidants and Anticancer Drugs”. International Journal of Molecular Sciences. 18 (11): 2490. doi:10.3390/ijms18112490. PMC 5713456.
- ^ Maio, N.; Lafont, B.A.P.; Sil, D.; Li, Y.; Bollinger, M.; Krebs, C. (2021). “Fe-S cofactors in the SARS-CoV-2 RNA-dependent RNA polymerase are potential antiviral targets”. Science. 373 (6551): 236–241. doi:10.1126/science.abi5224.
| Names | |
|---|---|
| Preferred IUPAC name(4-Hydroxy-2,2,6,6-tetramethylpiperidin-1-yl)oxyl | |
| Other namestempol; tanol; TMPN; 4-Oxypiperidol; nitroxyl 2; HyTEMPO | |
| Identifiers | |
| CAS Number | 2226-96-2 |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:180664 |
| ChEMBL | ChEMBL607023 |
| ChemSpider | 121639 |
| ECHA InfoCard | 100.017.056 |
| PubChem CID | 137994 |
| UNII | U78ZX2F65X |
| CompTox Dashboard (EPA) | DTXSID4041280 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C9H18NO2 |
| Molar mass | 172.248 g·mol−1 |
| Appearance | Orange crystals |
| Melting point | 71–73 °C (160–163 °F; 344–346 K)[1] |
| Solubility in water | 629.3 g/l (20 °C) |
| Hazards | |
| GHS labelling: | |
| Pictograms | [2] |
| Signal word | Warning[2] |
| Hazard statements | H302, H315, H319, H335[2] |
| Precautionary statements | P261, P305+P351+P338[2] |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
//////////////////4-Hydroxy-TEMPO, TEMPOL, MBM-02, MTS 01, ZJ 701, CORONA VIRUS, COVID 19
https://www.clinicaltrials.gov/ct2/show/NCT04729595
CC1(CC(CC(N1[O])(C)C)O)C

NEW DRUG APPROVALS
ONE TIME
$10.00
Regdanvimab

| (Heavy chain) QITLKESGPT LVKPTQTLTL TCSFSGFSLS TSGVGVGWIR QPPGKALEWL ALIDWDDNKY HTTSLKTRLT ISKDTSKNQV VLTMTNMDPV DTATYYCARI PGFLRYRNRY YYYGMDVWGQ GTTVTVSSAS TKGPSVFPLA PSSKSTSGGT AALGCLVKDY FPEPVTVSWN SGALTSGVHT FPAVLQSSGL YSLSSVVTVP SSSLGTQTYI CNVNHKPSNT KVDKRVEPKS CDKTHTCPPC PAPELLGGPS VFLFPPKPKD TLMISRTPEV TCVVVDVSHE DPEVKFNWYV DGVEVHNAKT KPREEQYNST YRVVSVLTVL HQDWLNGKEY KCKVSNKALP APIEKTISKA KGQPREPQVY TLPPSRDELT KNQVSLTCLV KGFYPSDIAV EWESNGQPEN NYKTTPPVLD SDGSFFLYSK LTVDKSRWQQ GNVFSCSVMH EALHNHYTQK SLSLSPGK (Light chain) ELVLTQPPSV SAAPGQKVTI SCSGSSSNIG NNYVSWYQQL PGTAPKLLIY DNNKRPSGIP DRFSGSKSGT SATLGITGLQ TGDEADYYCG TWDSSLSAGV FGGGTELTVL GQPKAAPSVT LFPPSSEELQ ANKATLVCLI SDFYPGAVTV AWKADGSPVK AGVETTKPSK QSNNKYAASS YLSLTPEQWK SHRSYSCQVT HEGSTVEKTV APTECS (Disulfide bridge: H22-H97, H155-H211, H231-L215, H237-H’237, H240-H’240, H272-H332, H378-H436, H’22-H’97, H’155-H’211, H’231-L’215, H’272-H’332, H’378-H’436, L22-L89, L138-L197, L’22-L’89, L’138-L’197) |
>Regdanvimab light chain: ELVLTQPPSVSAAPGQKVTISCSGSSSNIGNNYVSWYQQLPGTAPKLLIYDNNKRPSGIP DRFSGSKSGTSATLGITGLQTGDEADYYCGTWDSSLSAGVFGGGTELTVLGQPKAAPSVT LFPPSSEELQANKATLVCLISDFYPGAVTVAWKADGSPVKAGVETTKPSKQSNNKYAASS YLSLTPEQWKSHRSYSCQVTHEGSTVEKTVAPTECS
>Regdanvimab heavy chain: QITLKESGPTLVKPTQTLTLTCSFSGFSLSTSGVGVGWIRQPPGKALEWLALIDWDDNKY HTTSLKTRLTISKDTSKNQVVLTMTNMDPVDTATYYCARIPGFLRYRNRYYYYGMDVWGQ GTTVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHT FPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPC PAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKT KPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVY TLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSK LTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
Regdanvimab
レグダンビマブ;
EMA APPROVED, 2021/11/12, Regkirona
Treatment of adults with coronavirus disease 2019 (COVID-19)
MONOCLONAL ANTIBODY, ANTI VIRAL, PEPTIDE
CAS: 2444308-95-4, CT-P59
Regdanvimab, sold under the brand name Regkirona, is a human monoclonal antibody used for the treatment of COVID-19.[1] The antibody is directed against the spike protein of SARS-CoV-2. It is developed by Celltrion.[2][3] The medicine is given by infusion (drip) into a vein.[1][4]
The most common side effects include infusion-related reactions, including allergic reactions and anaphylaxis.[1]
Regdanvimab was approved for medical use in the European Union in November 2021.[1]
Regdanvimab is a monoclonal antibody targeted against the SARS-CoV-2 spike protein used to treat patients with COVID-19 who are at risk of progressing to severe COVID-19.
Regdanvimab (CT-P59) is a recombinant human IgG1 monoclonal antibody directed at the receptor binding domain (RBD) of the SARS-CoV-2 spike protein.4 It blocks the interaction between viral spike proteins and angiotensin-converting enzyme 2 (ACE2) that allows for viral entry into the cell, thereby inhibiting the virus’ ability to replicate. Trials investigating the use of regdanvimab as a therapeutic candidate for the treatment of COVID-19 began in mid-2020.1,3 It received its first full approval in South Korea in September 2021,3 followed by the EU in November 2021.5
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Synthesis Reference
Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, Jeong JH, Kim M, Kim JI, Kim P, Bae JS, Shim EY, Lee MS, Kim MS, Noh H, Park GS, Park JS, Son D, An Y, Lee JN, Kwon KS, Lee JY, Lee H, Yang JS, Kim KC, Kim SS, Woo HM, Kim JW, Park MS, Yu KM, Kim SM, Kim EH, Park SJ, Jeong ST, Yu CH, Song Y, Gu SH, Oh H, Koo BS, Hong JJ, Ryu CM, Park WB, Oh MD, Choi YK, Lee SY: A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat Commun. 2021 Jan 12;12(1):288. doi: 10.1038/s41467-020-20602-5.
Celltrion’s Monoclonal Antibody Treatment regdanvimab, Approved by the European Commission for the Treatment of COVID-19
- The European Commission (EC) granted marketing authorisation for Celltrion’s regdanvimab following positive opinion by the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) last week (11/11/2021)
- Celltrion continues to discuss supply agreements with regulatory agencies and contractors in more than 30 countries in Europe, Asia and LATAM to accelerate global access to regdanvimab
- The use of regdanvimab across the Republic of Korea is rapidly increasing to address the ongoing outbreaks
November 14, 2021 08:04 PM Eastern Standard Time
INCHEON, South Korea–(BUSINESS WIRE)–Celltrion Group announced today that the European Commission (EC) has approved Regkirona (regdanvimab, CT-P59), one of the first monoclonal antibody treatments granted marketing authorisation from the European Medicines Agency (EMA). The EC granted marketing authorisation for adults with COVID-19 who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID-19. The decision from the EC follows a positive opinion by the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) on November 11th, 2021.1
“Today’s achievement, coupled with CHMP positive opinion for regdanvimab, underscores our ongoing commitment to addressing the world’s greatest health challenges,” said Dr. HoUng Kim, Ph.D., Head of Medical and Marketing Division at Celltrion Healthcare. “Typically, the recommendations from the CHMP are passed on to the EC for rapid legally binding decisions within a month or two, however, given the unprecedented times, we have received the EC approval within a day. As part of our global efforts to accelerate access, we have been communicating with the governments and contractors in 30 countries in Europe, Asia and LATAM. We will continue working with all key stakeholders to ensure COVID-19 patients around the world have access to safe and effective treatments.”
Monoclonal antibodies are proteins designed to attach to a specific target, in this case the spike protein of SARS-CoV-2, which works to block the path the virus uses to enter human cells. The EC approval is based on the global Phase III clinical trial involving more than 1,315 people to evaluate the efficacy and safety of regdanvimab in 13 countries including the U.S., Spain, and Romania. Data showed regdanvimab significantly reduced the risk of COVID-19 related hospitalisation or death by 72% for patients at high-risk of progressing to severe COVID-19.
Emergency use authorisations are currently in place in Indonesia and Brazil, and the monoclonal antibody treatment is fully approved in the Republic of Korea. In the U.S., regdanvimab has not yet been approved by the Food and Drug Administration (FDA), but the company is in discussion with the FDA to submit applications for an Emergency Use Authorisation (EUA).
As of November 12th, 2021, more than 22,587 people have been treated with regdanvimab in 129 hospitals in the Republic of Korea.
Notes to Editors:
About Celltrion Healthcare
Celltrion Healthcare is committed to delivering innovative and affordable medications to promote patients’ access to advanced therapies. Its products are manufactured at state-of-the-art mammalian cell culture facilities, designed and built to comply with the US FDA cGMP and the EU GMP guidelines. Celltrion Healthcare endeavours to offer high-quality cost-effective solutions through an extensive global network that spans more than 110 different countries. For more information please visit: https://www.celltrionhealthcare.com/en-us.
About regdanvimab (CT-P59)
CT-P59 was identified as a potential treatment for COVID-19 through screening of antibody candidates and selecting those that showed the highest potency in neutralising the SARS-CoV-2 virus. In vitro and in vivo pre- clinical studies showed that CT-P59 strongly binds to SARS-CoV-2 RBD and significantly neutralise the wild type and mutant variants of concern. In in vivo models, CT-P59 effectively reduced the viral load of SARS-CoV-2 and inflammation in lung. Results from the global Phase I and Phase II/III clinical trials of CT-P59 demonstrated a promising safety, tolerability, antiviral effect and efficacy profile in patients with mild-to-moderate symptoms of COVID-19.2 Celltrion also has recently commenced the development of a neutralising antibody cocktail with CT-P59 against new emerging variants of SARS-CoV-2.
Medical uses
In the European Union, regdanvimab is indicated for the treatment of adults with COVID-19 who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID-19.[1]
Society and culture
Names
Regdanvimab is the proposed international nonproprietary name (pINN).[5]
Legal status
In March 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) started a rolling review of data on regdanvimab.[6][7] In October 2021, the EMA started evaluating an application for marketing authorization for the monoclonal antibody regdanvimab (Regkirona) to treat adults with COVID-19 who do not require supplemental oxygen therapy and who are at increased risk of progressing to severe COVID 19.[8] The applicant is Celltrion Healthcare Hungary Kft.[8] The European Medicines Agency (EMA) concluded that regdanvimab can be used for the treatment of confirmed COVID-19 in adults who do not require supplemental oxygen therapy and who are at high risk of progressing to severe COVID-19.[4]
In November 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) recommended granting a marketing authorization in the European Union for regdanvimab (Regkirona) for the treatment of COVID-19.[9][10] The company that applied for authorization of Regkirona is Celltrion Healthcare Hungary Kft.[10] Regdanvimab was approved for medical use in the European Union in November 2021.[1]
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Spike protein of SARS-CoV-2 |
| Clinical data | |
| Trade names | Regkirona |
| Other names | CT-P59 |
| License data | EU EMA: by INN |
| Routes of administration | Intravenous infusion |
| ATC code | None |
| Legal status | |
| Legal status | EU: Rx-only [1] |
| Identifiers | |
| CAS Number | 2444308-95-4 |
| DrugBank | DB16405 |
| UNII | I0BGE6P6I6 |
| KEGG | D12241 |
- Tuccori M, Ferraro S, Convertino I, Cappello E, Valdiserra G, Blandizzi C, Maggi F, Focosi D: Anti-SARS-CoV-2 neutralizing monoclonal antibodies: clinical pipeline. MAbs. 2020 Jan-Dec;12(1):1854149. doi: 10.1080/19420862.2020.1854149. [Article]
- Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, Jeong JH, Kim M, Kim JI, Kim P, Bae JS, Shim EY, Lee MS, Kim MS, Noh H, Park GS, Park JS, Son D, An Y, Lee JN, Kwon KS, Lee JY, Lee H, Yang JS, Kim KC, Kim SS, Woo HM, Kim JW, Park MS, Yu KM, Kim SM, Kim EH, Park SJ, Jeong ST, Yu CH, Song Y, Gu SH, Oh H, Koo BS, Hong JJ, Ryu CM, Park WB, Oh MD, Choi YK, Lee SY: A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat Commun. 2021 Jan 12;12(1):288. doi: 10.1038/s41467-020-20602-5. [Article]
- Syed YY: Regdanvimab: First Approval. Drugs. 2021 Nov 1. pii: 10.1007/s40265-021-01626-7. doi: 10.1007/s40265-021-01626-7. [Article]
- EMA Summary of Product Characteristics: Regkirona (regdanvimab) concentrate for solution for intravenous infusion [Link]
- EMA COVID-19 News: EMA recommends authorisation of two monoclonal antibody medicines [Link]
- EMA CHMP Assessment Report: Celltrion use of regdanvimab for the treatment of COVID-19 [Link]
- Protein Data Bank: Crystal Structure of COVID-19 virus spike receptor-binding domain complexed with a neutralizing antibody CT-P59 [Link]
References
- ^ Jump up to:a b c d e f g “Regkirona EPAR”. European Medicines Agency. Retrieved 12 November 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Celltrion Develops Tailored Neutralising Antibody Cocktail Treatment with CT-P59 to Tackle COVID-19 Variant Spread Using Its Antibody Development Platform” (Press release). Celltrion. 11 February 2021. Retrieved 4 March 2021 – via Business Wire.
- ^ “Celltrion Group announces positive top-line efficacy and safety data from global Phase II/III clinical trial of COVID-19 treatment candidate CT-P59” (Press release). Celltrion. 13 January 2021. Retrieved 4 March 2021 – via Business Wire.
- ^ Jump up to:a b “EMA issues advice on use of regdanvimab for treating COVID-19”. European Medicines Agency. 26 March 2021. Retrieved 15 October 2021.
- ^ World Health Organization (2020). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 124 – COVID-19 (special edition)” (PDF). WHO Drug Information. 34 (3): 660–1.
- ^ “EMA starts rolling review of Celltrion antibody regdanvimab for COVID-19” (Press release). European Medicines Agency (EMA). 24 February 2021. Retrieved 4 March 2021.
- ^ “EMA review of regdanvimab for COVID-19 to support national decisions on early use” (Press release). European Medicines Agency (EMA). 2 March 2021. Retrieved 4 March 2021.
- ^ Jump up to:a b “EMA receives application for marketing authorisation Regkirona (regdanvimab) treating patients with COVID-19”. European Medicines Agency. 4 October 2021. Retrieved 15 October 2021.
- ^ “Regkirona: Pending EC decision”. European Medicines Agency. 11 November 2021. Retrieved 11 November 2021.
- ^ Jump up to:a b “COVID-19: EMA recommends authorisation of two monoclonal antibody medicines”. European Medicines Agency (EMA) (Press release). 11 November 2021. Retrieved 11 November 2021.
Further reading
- Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, et al. (January 2021). “A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein”. Nature Communications. 12 (1): 288. doi:10.1038/s41467-020-20602-5. PMC 7803729. PMID 33436577.
External links
- “Regdanvimab”. Drug Information Portal. U.S. National Library of Medicine.
///////////Regdanvimab, Regkirona, MONOCLONAL ANTIBODY, ANTI VIRAL, EU 2021, APPROVALS 2021, EMA 2021, COVID 19, CORONAVIRUS, PEPTIDE, レグダンビマブ , CT-P59, CT P59

NEW DRUG APPROVALS
ONE TIME
$10.00
MVC COVID-19 vaccine, Taiwan’s covid vaccine
Medigen vaccine
MVC COVID-19 vaccine
- MVC-COV1901
track it https://covid19.trackvaccines.org/vaccines/24/
MVC-COV1901 is a vaccine candidate developed and commercialized by Medigen Vaccine Biologics Corporation. The vaccine candidate contains a perfusion form of the SARS-Cov2 recombinant spike protein. Medigen has combined forces with Dynavax, which offers an advanced adjuvant, CpG 1018 (also known as ISS-1018), for use with its vaccine. As of September 2020, the vaccine candidate is in Phase 1 clinical trials to assess its safety and immunogenicity (NCT04487210).
The MVC COVID-19 vaccine, designated MVC-COV1901 and also known as the Medigen COVID-19 vaccine, is a protein subunit COVID-19 vaccine developed by Medigen Vaccine Biologics Corporation [zh] in Taiwan, American company Dynavax Technologies and the U.S. National Institute of Health.[1][2]
This vaccine is made by the recombinant S-2P spike protein adjuvanted with CpG 1018 supplied by Dynavax.[3] Preliminary results from Phase I trials on 77 participants were published in June 2021, indicating what the authors described as “robust” immune system response elicited by the vaccine.[4]
The study authors have assessed the humoral immune response by measuring quantities of binding IgG to S protein, and also the cellular immune response by measuring the quantities of IFN-γ and IL-4 secreting T cells.[4]
Taiwan-based Medigen Vaccine Biologics Corporation (MVC) and Dynavax Technologies Corporation, in the US, have announced the rollout of its COVID-19 vaccine, MVC-COV1901. Approximately 600,000 people are anticipated to receive the Medigen vaccine this week.
Ryan Spencer, Chief Executive Officer of Dynavax commented, “We are pleased that Medigen’s vaccine is now available for the people of Taiwan. We are very excited for this first, of hopefully multiple, EUAs and approvals for COVID-19 vaccines that include CpG 1018 adjuvant. Considering the limitations of current vaccines and the global vaccine shortage, we believe adjuvanted vaccines can contribute significantly to current vaccination efforts.”
In July, MVC received Taiwan Emergency Use Authorization and approval for inclusion in Taiwan’s COVID-19 vaccine immunization program, MVC-COV1901.
MVC COVID-19 vaccine is indicated for adults over 20 years old and is administered in two doses 28 days apart for prevention of COVID-19.
The Advisory Committee recommended that MVC should submit safety monitoring report monthly during the declared EUA period and should submit a vaccine effectiveness report within one year after obtaining EUA approval.
(CNN)Taiwan’s President Tsai Ing-wen received her first shot of the island’s homegrown Covid-19 vaccine on Monday, a public show of support for the new drug which is central to plans for inoculation self sufficiency amid low immunization rates and struggles to obtain vaccines from overseas.Monday’s island-wide rollout of the Medigen Covid-19 vaccine, developed by Taipei-based Medigen Vaccine Biologics Corporation, comes after the drug was approved for emergency use last month by Taiwanese authorities for anyone above 20 years old, with at least 28 days between the two doses.The vaccine has yet to complete phase 3 clinical trials and no efficacy data is available. Paul Torkehagen, Medigen’s director of overseas business development, told CNN in May that the company designed a “very large” phase 2 clinical trial to ensure the vaccine’s safety and effectiveness, with 3,800 participants. Normally, a stage 2 clinical trial only involves several hundred people. Data from the trials showed that 99.8% of participants were able to form antibodies against Covid-19 after taking two doses of the vaccine, Medigen’s CEO Charles Chen said. 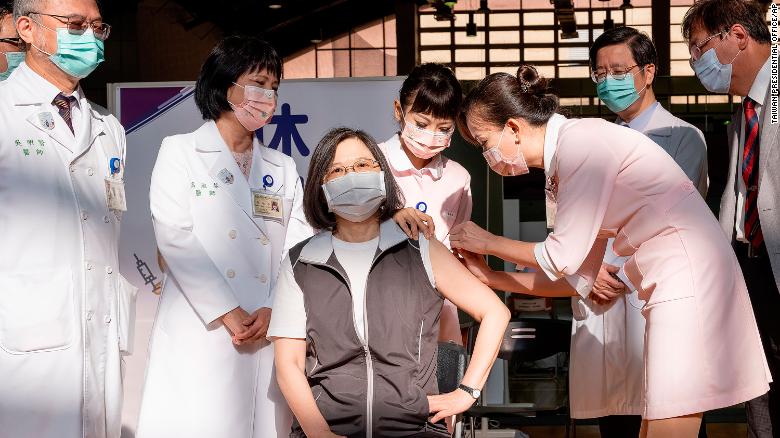
Taiwanese President Tsai Ing-wen, center, receives her first shot of the island’s first domestically developed coronavirus vaccine at the Taiwan University Hospital in Taipei, Taiwan on Monday, August 23.
Taiwan’s Centers for Disease Control said in a July 19 statement that the vaccine posed no serious health effects. Taiwan has ordered 5 million doses of the vaccine from Medigen and more than 700,000 people have already signed up to receive it, according to Reuters.In a Facebook post after receiving the vaccine at a hospital in Taipei, Tsai said she hadn’t suffered from any post-vaccination pain and thanked the health care workers who had administered the shot.”Taking the vaccine can protect yourself, your family, as well as medical staff,” Tsai wrote. “Let’s do our part in boosting Taiwan’s collective defense against the virus!”With its borders sealed to most travelers and strict measures enacted to contain local outbreaks, Taiwan has so far been largely successful in containing Covid-19, reporting fewer than 16,000 total confirmed infections and 828 deaths. But the island has struggled to vaccinate its more than 23 million population, partly due to difficulties obtaining doses from international suppliers.Taiwan’s government has only managed to import around 10 million Covid-19 vaccines, according to Reuters. In July it ordered another 36 million doses of the Moderna shot.Fewer than 5% of Taiwan’s population has received both doses of their Covid-19 vaccine, according to Reuters, as the island delays second dose vaccinations so more people can receive a first shot.On Monday, Taiwan reported four new Covid-19 cases, according to the Central Epidemic Command Center (CECC). Authorities announced on the weekend they would ease virus prevention measures to allow for larger gatherings and the opening of study centers and indoor amusement parks.But Health and Welfare Minister Chen Shih-chung said current Covid-19 restrictions — which include the closure of bars and nightclubs — would remain in place until at least September 6, with the possibility of an extension if the global outbreak continued to grow.Taiwan could become increasingly isolated if it keeps pursuing its “Covid zero” strategy, with both Australia and New Zealand hinting they might abandon the approach once vaccinations reach a certain level.In an opinion piece published on Sunday, Australian Prime Minister Scott Morrison said that while lockdowns to prevent Covid-19 transmission were “sadly necessary for now,” they may not be once vaccination rates increased to the targets of 70% and 80%.”This is what living with Covid is all about. The case numbers will likely rise when we soon begin to open up. That is inevitable,” he said.In neighboring New Zealand, which has also attempted to eliminate the virus within its borders, Covid-19 response minister Chris Hipkins told local media the highly-contagious Delta variant raised “some pretty big questions about what the long-term future of our plans are.”“At some point we will have to start to be more open in the future,” he said.
History
On 16 February 2020, Medigen Vaccine Biologics Corp. (MVC) signed a collaboration agreement with National Institutes of Health (NIH) for COVID-19 vaccine development. The partnership will allow MVC to obtain NIH’s COVID-19 vaccine and related biological materials to conduct animal studies in Taiwan.[5]
On 23 July 2020, Medigen Vaccine Biologics (MVC) announced collaboration with Dynavax Technologies to develop COVID-19 vaccine. The COVID-19 candidate vaccine will have the combination of SARS-CoV2 spike protein created by MVC and Dynavax’s vaccine adjuvant CpG 1018, which was used in a previously FDA-approved adult hepatitis B vaccine.[6][7]
Clinical trials
On 13 October 2020, Medigen Vaccine Biologics received Taiwan’s government subsidies for the initiation of Phase 1 Clinical Trial in Taiwan starting early October. The Phase 1 Clinical Trial was held at National Taiwan University Hospital with 45 participants ranging the age of 20-50.[8][9]
On 25 January 2021, Medigen Vaccine Biologics initiated Phase 2 Clinical Trial for its COVID-19 vaccine candidate MVC-COV1901 with the first participant being dosed. The Phase 2 Clinical Trial for the MVC COVID-19 vaccine was a randomized, double-blinded, and multi-center clinical trial, planned to enroll 3,700 participants of any age 20 above.[3][10][11]
On 10 June 2021, Medigen Vaccine Biologics released its COVID-19 vaccine Phase 2 interim analysis results, which demonstrates good safety profile in participants. The Phase 2 Clinical Trial in the end included 3,800 participants with all participants receiving second dose by 28 April 2021. Medigen Vaccine Biologics announced that it will request Emergency Use Authorization (EUA) with the concluding of the Phase 2 Clinical Trial.[12]
On 20 July 2021, Medigen Vaccine Biologics filed a Phase 3 Clinical Trial IND application with Paraguay’s regulatory authority, which was later approved. The Phase 3 Clinical Trial, however, was different from regular Phase 3 Clinical Trial, which uses immune-bridging trial to compare the performance of MVC COVID-19 vaccine with the Oxford-AstraZeneca COVID-19 vaccine.[13] The decision was a controversial announcement as immune-bridging trials were not fully approved or widely accepted by health authorities. In addition, the accuracy of immune-bridging trials were also been questioned for years.[citation needed]
Adolescents trial
In July 2021, Medigen commenced phase II trials for adolescents aged 12-18.[14]
Authorization
| Full authorization Emergency authorization |
See also: List of COVID-19 vaccine authorizations § Medigen
On July 19, 2021, MVC COVID-19 vaccine obtained Emergency Use Authorization (EUA) approval from the Taiwanese government after fulfilling EUA requirements set by Taiwanese authority.[15] The EUA, however, was met with controversy due to the lack of efficacy data and Phase 3 Clinical Trial. On August 23, 2021, President Tsai Ing-Wen was among the first Taiwanese to receive a dose of the vaccine. [16]
References
- ^ “Dynavax and Medigen Announce Collaboration to Develop a Novel Adjuvanted COVID-19 Vaccine Candidate”. GlobeNewswire. 23 July 2020. Retrieved 7 June 2021.
- ^ 黃驛淵 (10 June 2021). “【獨家】【國產疫苗解盲1】高端實體疫苗針劑首曝光 「每天9萬劑」生產基地直擊” (in Chinese). Mirror Media.
- ^ Jump up to:a b “Medigen Vaccine Biologics COVID-19 Vaccine Adjuvanted with Dynavax’s CpG 1018 Announces First Participant Dosed in Phase 2 Clinical Trial in Taiwan”. http://www.medigenvac.com. Retrieved 7 August 2021.
- ^ Jump up to:a b Hsieh SM, Liu WD, Huang YS, Lin YJ, Hsieh EF, Lian WC, Chen C, Janssen R, Shih SR, Huang CG, Tai IC, Chang SC (25 June 2021). “Safety and immunogenicity of a Recombinant Stabilized Prefusion SARS-CoV-2 Spike Protein Vaccine (MVCCOV1901) Adjuvanted with CpG 1018 and Aluminum Hydroxide in healthy adults: A Phase 1, dose-escalation study”. EClinicalMedicine: 100989. doi:10.1016/j.eclinm.2021.100989. ISSN 2589-5370. PMC 8233066. PMID 34222848.
- ^ “MVC and NIH Collaborate to Develop COVID-19 Vaccine”. http://www.medigenvac.com. Retrieved 7 August 2021.
- ^ “Medigen Collaborates with Dynavax to Develop Novel Adjuvanted COVID-19 Vaccine Candidate”. http://www.medigenvac.com. Retrieved 7 August 2021.
- ^ “MVC Signed an License Agreement with NIH on COVID-19 Vaccine”. Medigen. 5 May 2020. Retrieved 27 July 2021.
- ^ “Medigen’s COVID-19 Vaccine Combined with Dynavax’s CpG 1018 Adjuvant Receives Taiwan Government Subsidy with First Participant Dosed in Early October”. http://www.medigenvac.com. Retrieved 7 August 2021.
- ^ “A Study to Evaluate MVC-COV1901 Vaccine Against COVID-19 in Adult (COVID-19)”. clinicaltrials.gov. United States National Library of Medicine. Retrieved 11 March 2021.
- ^ “A Study to Evaluate the Safety and Immunogenicity of MVC-COV1901 Against COVID-19”. clinicaltrials.gov. United States National Library of Medicine. Retrieved 11 March 2021.
- ^ “A Study to Evaluate MVC-COV1901 Vaccine Against COVID-19 in Elderly Adults”. clinicaltrials.gov. United States National Library of Medicine. 28 March 2021. Retrieved 3 April 2021.
- ^ “MVC Released COVID-19 Vaccine Phase 2 Interim Analysis Result”. http://www.medigenvac.com. Retrieved 7 August 2021.
- ^ “MVC Announces Paraguay Approval of IND Application for Phase 3 Clinical Trial”. http://www.medigenvac.com. Retrieved 7 August 2021.
- ^ “A Study to Evaluate MVC-COV1901 Vaccine Against COVID-19 in Adolescents”. clinicaltrials.gov. United States National Library of Medicine. 6 July 2021. Retrieved 6 July 2021.
- ^ “MVC COVID-19 Vaccine Obtains Taiwan EUA Approval”. http://www.medigenvac.com. Retrieved 7 August 2021.
- ^ Taiwan begins contested rollout of new Medigen domestic vaccine, Nikkei Asia, Erin Hale, August 23, 2021
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Protein subunit |
| Clinical data | |
| Other names | MVC-COV1901 |
| Routes of administration | Intramuscular |
| Legal status | |
| Legal status | Full and Emergency Authorizations: List of MVC COVID-19 vaccine authorizations |
| Identifiers | |
| DrugBank | DB15854 |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| COVID-19 (disease)SARS-CoV-2 (virus)CasesDeaths |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showEconomic impact and recession |
| showImpacts |
| COVID-19 portal |
////////Medigen vaccine, MVC COVID-19 vaccine, SARS-CoV-2, covid 19, corona virus, taiwan, approvals 2021, iss 1018, CpG 1018, MVC-COV1901
NEW DRUG APPROVALS
one time
$10.00
Soberana 02, FINLAY-FR-2

Soberana 02
FINLAY-FR-2
cas 2543416-58-4
A SARS-CoV-2 vaccine comprising a conjugate of the spike protein RBD domain with tetanus toxoid (Finlay Vaccine Institute of Cuba)
Soberana 02, is a conjugate vaccine developed by Instituto Finlay de Vacunas.[517]
Cuba[518]
Iran[517]
517 Zimmer, Carl; Corum, Jonathan; Wee, Sui-Lee. “Coronavirus Vaccine Tracker”. The New York Times. Retrieved 30 June 2021.
518 Sesin, Carmen (14 May 2021). “Cuba begins mass Covid-19 vaccine inoculation before concluding trials”. NBC News. Retrieved 2 July 2021.
Soberana 02, technical name FINLAY-FR-2, is a COVID-19 vaccine produced by the Finlay Institute, a Cuban epidemiological research institute. It is a conjugate vaccine. This candidate followed a previous one called SOBERANA-01 (FINLAY-FR-1).[2] Professor Ihosvany Castellanos Santos said that the antigen is safe because it contains parts instead of the whole live virus, and therefore it does not require extra refrigeration, like other candidates in the world.[3] According to the WHO candidate landscape vaccine document, this vaccine requires two doses, the second one being administered 28 days after the first shot.[4]
The name of the vaccine, Soberana, is a Spanish word that means “sovereign”.[5]

Efficacy
It has shown an efficacy of 62% after only two doses, according to BioCubaFarma, though a pre-print or details of the study have not been released.[6][7][8]
Pharmacology
FINLAY-FR-2 is a conjugate vaccine. It consists of the receptor binding domain of the SARS-CoV-2 spike protein conjugated chemically to tetanus toxoid.[2]
Manufacturing
The spike protein subunit is produced in Chinese hamster ovary cell culture.[2] In a pre-print article scientists from Cuba explain details of the vaccines technology and production.[9][non-primary source needed]
| Production Deliveries Planned Production Potential Production |
Deliveries (0)Effective production (implies deliveries) (1)
Planned production
- Iran
Potential Production
- Ghana
- Argentina
In Cuba
The Cuban government says it is planning to produce 100 million doses of its vaccine to respond to its own demand and that of other countries.[12][13] Cuba has also suggested that, once it’s approved, it will offer the vaccine to tourists visiting the country.[14][15][16]
The production of the first batch of about 100,000 doses will start in April.[17] José Moya, representative of the World Health Organization and the Pan American Health Organization (PAHO) in Cuba, suggested that after the vaccine passes all clinical stages, it could be included as part of PAHO’s Revolving Fund.[18]
The roll-out began with an “Interventional Trial”[19] that consisted of inoculating 150,000 at-risk participants which seems to be defined as health-care workers.[20][21] On April 11, 2021, the Ministry of Public Health of Cuba announced that 75,000 health-care workers were inoculated with their first dose of either of the two Cuba’s Phase III vaccines (the other being Abdala).[22][23]
Outside Cuba
Vietnam, Iran, Venezuela, Argentina,[24][25][26] Pakistan, India, the African Union, Jamaica and Suriname[27] have expressed interest in purchasing the vaccine, although they are waiting on Phase 3 results.[28][29]
Iran has signed an agreement to manufacture the vaccine[30] and Argentina is negotiating one.[24][25][26] Additionally, the Cuban government offered a “transfer of technology” to Ghana and will also supply “active materials” needed to make the vaccine.[31][32][33]
While the price is currently unknown, the commercialization strategy of the vaccine will be a combination of the “impact on health” and the capability of Cuba’s system to financially support “the production of vaccines and drugs for the country”, per the director of the Finlay Institute, Vicente Vérez.[34]
Clinical trials
Phase I
FINLAY-FR-2, which started being developed in October 2020, had 40 volunteers for its Phase I, according to the Cuban Public Registry of Clinical Trials, with an open, sequential and adaptive study to assess safety, reactogenicity and explore immunogenicity of the vaccine.[35]
Phase II
Phase IIa involved 100 Cubans, and phase IIb of the vaccine will have 900 volunteers between 19 and 80 years.[36][37] Vicente Vérez, director general of the Finlay Vaccine Institute, said that the vaccine has shown to give an immune response after 14 days.[38] The second phase has been supervised by Iranian officials from the Pasteur Institute.[5]
Phase III
Phase III commenced at the beginning of March as originally scheduled,[39][15] and “ready to publish” results are expected by June.[40][41][42] The trial volunteers are divided into three groups: some will receive two doses of the vaccine 28 days apart, another group will get two doses plus a third immune booster (Soberana Plus[43][44][45]), and the third a placebo.[39]
Although the trials involve thousands of adult volunteers recruited in Havana,[46] Cuba’s public health officials have said that they will also need to conduct phase III trials abroad because the island doesn’t have an outbreak of sufficient scale to produce meaningful statistics on vaccine protection.[5][14]
On March 13, 2021, the Cuban Biotechnology and Pharmaceutical Industries Business Group (BioCubaFarma) announced on social media that it had sent 100,000 doses of its Soberana 02 coronavirus vaccine candidate to the Pasteur Institute of Iran for clinical testing, “as part of the collaboration with other countries in the development of COVID-19 vaccines.” [47]
On April 26, 2021, it was reported that a Phase III conducted by the Pasteur Institute of Iran was approved to be started in Iran[48][49][50] It was previously reported that the Institute will host Phase 3 but the pre-requisites were “technology transfer and joint production”.[51][5]
Mexico plans to host a phase 3 trial.[52]
Interventional Study
The “Interventional Study” is set both in Havana,[53] Cuba’s capital and Santiago de Cuba, Cuba’s second most populous city [54][55] and in other provinces.[56] On May 6, 2021, the Finlay Institute of Vaccines announced on social media that the following adverse events have been observed: injection site pain (20%), inflammation at the injection site (5%), and general discomfort (5%).[57][58]
Authorizations
| Full authorization Emergency authorization |
See also: List of COVID-19 vaccine authorizations § Soberana 02
References
- ^ “Cuba’s Soberana Plus against Covid-19 is showing good results”. Prensa Latina. Retrieved 10 May 2021.
- ^ Jump up to:a b c Malik JA, Mulla AH, Farooqi T, Pottoo FH, Anwar S, Rengasamy KR (January 2021). “Targets and strategies for vaccine development against SARS-CoV-2”. Biomedicine & Pharmacotherapy. 137: 111254. doi:10.1016/j.biopha.2021.111254. PMC 7843096. PMID 33550049.
- ^ Santos IC (January 2021). “Rapid response to: Covid 19: Hope is being eclipsed by deep frustration”. BMJ. 372: n171. doi:10.1136/bmj.n171.
- ^ “Draft landscape and tracker of COVID-19 candidate vaccines”. http://www.who.int. World Health Organization. Retrieved 2021-02-04.
- ^ Jump up to:a b c d Rasmussen SE, Eqbali A (12 January 2021). “Iran, Cuba, Under U.S. Sanctions, Team Up for Covid-19 Vaccine Trials”. The Wall Street Journal.
- ^ “Cuba’s homegrown Covid vaccine shows promise”. http://www.ft.com. Retrieved 2021-06-20.
- ^ “Cuba encouraged by early efficacy results of homegrown COVID-19 vaccine”. http://www.zawya.com. Retrieved 2021-06-20.
- ^ Acosta, Nelson (2021-06-20). “Cuba encouraged by early results of homegrown COVID-19 vaccine amid worst outbreak”. The Age. Retrieved 2021-06-20.
- ^ Valdes-Balbin, Yury; Santana-Mederos, Darielys; Quintero, Lauren; Fernández, Sonsire; Rodriguez, Laura; Ramirez, Belinda Sanchez; Perez, Rocmira; Acosta, Claudia; Méndez, Yanira; Ricardo, Manuel G.; Hernandez, Tays (2021-02-09). “SARS-CoV-2 RBD-Tetanus toxoid conjugate vaccine induces a strong neutralizing immunity in preclinical studies”. doi:10.1101/2021.02.08.430146.
- ^ Melimopoulos, Elizabeth. “Is Cuba closing in on COVID vaccine sovereignty?”. http://www.aljazeera.com. Retrieved 2021-05-07.
- ^ “Optimism as Cuba set to test its own Covid vaccine”. BBC News. 2021-02-16. Retrieved 2021-05-07.
- ^ “Cuba espera fabricar 100 millones de dosis de su candidato vacunal Soberana 02”. Nodal (in Spanish). 21 January 2021.
- ^ “Vaccino, Cuba pronta a produrre 100 milioni di dosi di ‘Soberana 02′”. Dire (in Italian). 21 January 2021.
- ^ Jump up to:a b Ribeiro G (4 February 2021). “Cuba to offer coronavirus vaccines to tourists”. Brazilian Report.
- ^ Jump up to:a b “Coronavirus: Vacuna cubana Soberana 02 alista fase 3 y ensayos”. Deutsche Welle (in Spanish). 5 February 2021.
- ^ Meredith S (23 February 2021). “‘Sun, sea, sand and Soberana 02’: Cuba open to inoculating tourists with homegrown Covid vaccine”. CNBC.
- ^ “Coronavirus: Vacuna cubana Soberana 02 alista fase 3 y ensayos”. Deutsche Welle (in Spanish). 5 February 2021.
Las expectativas sobre Soberana 02 son tales que el titular del organismo estatal que desarrolló la vacuna, Vicente Vérez, confirmó que mientras se aguarden los resultados de la Fase 3 solo en La Habana, en abril se dará inicio a la producción del primer lote, de alrededor de 100 mil dosis.
- ^ “Cuba anuncia fase 3 de la vacuna Soberana 02”. La Jornada(in Spanish). 7 February 2021.
Una vez que superen las etapas clínicas, la OMS podría contar con el fármaco cubano, afirmó Moya, y “pasar a ser parte del grupo de vacunas que se oferten a través del Fondo Rotatorio”, un mecanismo que desde hace cuatro décadas permite gestionar antígenos e insumos a los países de las Américas.
- ^ “SOBERANA – INTERVENTION | Registro Público Cubano de Ensayos Clínicos”. rpcec.sld.cu. Retrieved 2021-04-11.
- ^ “Cuba says it’s ‘betting it safe’ with its own Covid vaccine”. NBC News. Retrieved 2021-04-11.
- ^ “Cuba begins testing 2nd COVID-19 vaccine on health care workers”. medicalxpress.com. Retrieved 2021-04-11.
- ^ Ministry of Public Health of Cuba (11 April 2021). “[Translated] “The administration of the 1st dose of the Cuban vaccine candidates #Soberana02 and #Abdala to the 75 thousand health workers and Biocubafarma who are part of the intervention study taking place in #LaHabana has concluded.””. Twitter. Retrieved 2021-04-11.
- ^ “Cuban scientists, health workers received first anti-Covid-19 dose”. http://www.plenglish.com/index.php?o=rn&id=66247&SEO=cuban-scientists-health-workers-received-first-anti-covid-19-dose (in Spanish). Retrieved 2021-04-11.
- ^ Jump up to:a b “ILARREGUI (EMBAJADOR EN CUBA): “DURANTE ESTE AÑO PODREMOS TENER VACUNAS CUBANAS EN ARGENTINA””. RadioCut. Retrieved 2021-05-07.
- ^ Jump up to:a b Argentina, Cadena 3. “Argentina comenzó a negociar con Cuba la vacuna Soberana”. Cadena 3 Argentina (in Spanish). Retrieved 2021-05-07.
- ^ Jump up to:a b de 2021, 6 de Mayo. “Sin definiciones sobre cuándo podrían llegar, el Gobierno avanza para conseguir las vacunas Soberana y Abdala de Cuba”. infobae (in Spanish). Retrieved 2021-05-07.
- ^ admin (2021-04-09). “Cuba’s COVID-19 Vaccines Being Sought After by CARICOM Countries”. Caribbean News. Retrieved 2021-05-07.
- ^ Guenot, Marianne (2021-02-15). “Cuba is working on a homegrown COVID-19 vaccine program. It has a history of fighting disease without help from the West”. Business Insider France (in French). Retrieved 2021-05-07.
- ^ Página12 (2021-01-22). “Soberana 02: Cuba prepara cien millones de dosis de la vacuna contra el coronavirus | “No somos una multinacional. Nuestro fin es crear salud”, dijo el director del Instituto Finlay de Vacunas”. PAGINA12. Retrieved 2021-05-07.
- ^ “Cuban coronavirus vaccine to start third clinical trial phase in Iran”. Tehran Times. 2021-04-18. Retrieved 2021-05-07.
- ^ Banini | 0542440286, Awofisoye Richard. “CEO OF FDA DISCUSSES PRODUCTION OF COVID-19 VACCINE WITH CUBAN AMBASSADOR”. http://www.fdaghana.gov.gh. Retrieved 2021-05-05.
- ^ “Cuba To Transfer COVID-19 Vaccine Technology To Ghana”. http://www.gnbcc.net. Retrieved 2021-05-05.
- ^ “Cuban government offers to transfer COVID-19 Soberana 02 vaccine technology to Ghana”. Rio Times Online. 16 February 2021.
- ^ “Coronavirus: Cuba will produce 100 million doses of its Soberana 02 vaccine”. OnCubaNews English. 2021-01-21. Retrieved 2021-05-07.
- ^ “SOBERANA 02 | Registro Público Cubano de Ensayos Clínicos”. Cuban Registry of Clinical Trials (in Spanish). Retrieved 24 January 2021.
- ^ Cuba inicia nova fase de testes com vacina que desenvolve contra covid-19 (in Portuguese), Universo Online, 19 January 2021, Wikidata Q105047566
- ^ “Cuba apuesta por crear primera vacuna de América Latina contra el covid-19”. France 24 (in Spanish). 2021-01-21. Retrieved 24 January 2021.
- ^ “Cuba negotiates with other countries to develop phase 3 of Soberana 02 vaccine”. OnCubaNews English. 2020-12-30. Retrieved 24 January 2021.
- ^ Jump up to:a b “Cuban-developed vaccine enters Phase III trial”. ABS CBN. 5 March 2021.
- ^ Mega, Emiliano Rodríguez (2021-04-29). “Can Cuba beat COVID with its homegrown vaccines?”. Nature. doi:10.1038/d41586-021-01126-4. PMID 33927405.
- ^ “Cuban Vaccine Ready in July. Interview with the Cuban Ambassador to the Czech Republic”. Pressenza. 2021-03-23. Retrieved 2021-04-29.
- ^ Augustin, Ed (2021-05-12). “Cuba deploys unproven homegrown vaccines, hoping to slow an exploding virus outbreak”. The New York Times. ISSN 0362-4331. Retrieved 2021-05-14.
- ^ “L’esempio cubano sui vaccini”. http://www.ilfoglio.it (in Italian). Retrieved 2021-05-07.
- ^ Avances de las vacunas cubanas contra la COVID-19, retrieved 2021-05-07
- ^ Mega, Emiliano Rodríguez (2021-04-29). “Can Cuba beat COVID with its homegrown vaccines?”. Nature. doi:10.1038/d41586-021-01126-4. PMID 33927405.
- ^ Yaffe, Helen. “Cuba’s five COVID-19 vaccines: the full story on Soberana 01/02/Plus, Abdala, and Mambisa”. LSE Latin America and Caribbean blog. Retrieved 2021-03-31.
- ^ “Cuba sends 100,000 doses of the Soberana 02 vaccine candidate to Iran” oncubanews.com. Retrieved 19 March 2021.
- ^ “Iran-Cuba vaccine enters phase three clinical trials”. Tehran Times. 2021-04-26. Retrieved 2021-04-28.
- ^ “Cuban coronavirus vaccine to start third clinical trial phase in Iran”. Tehran Times. 2021-04-18. Retrieved 2021-04-28.
- ^ “América Latina apura una vacuna propia. Cuba, adelante; México avanza. Pero no son los únicos”. http://www.poresto.net (in Spanish). Retrieved 2021-04-28.
- ^ Marsh S (2021-01-09). “Cuba to collaborate with Iran on coronavirus vaccine”. Reuters. Retrieved 2021-01-24.
- ^ “Mexico Hopes to Work With Cuba on Covid Vaccine Phase 3 Trial”. Bloomberg.com. 2021-02-14. Retrieved 2021-05-07.
- ^ Marsh, Sarah (2021-03-24). “Nearly all Havana to receive experimental Cuban COVID-19 vaccines”. Reuters. Retrieved 2021-04-28.
- ^ BioCubaFarma (April 6, 2021). “[Translated] Updating the vaccination process with vaccine candidates #Soberana02 and #Abdala during ongoing clinical trials.#VacunasCubanasCovid19”. Twitter (in Spanish). Retrieved 2021-04-11.
- ^ “Intervention study with Covid-19 vaccine candidate Abdala begins”. Radio Cadena Agramonte. Retrieved 2021-04-28.
- ^ “Cuba administers over 62,000 doses in intervention trials”. http://www.plenglish.com/index.php?o=rn&id=66012&SEO=cuba-administers-over-62000-doses-in-intervention-trials (in Spanish). Retrieved 2021-04-28.
- ^ “[Trnslated] In more than 62 thousand applied doses of #Soberana02 the safety of the vaccine has been demonstrated. Adverse effects have been: 👉 Pain at the injection site (20%). 👉 Redness at the injection site (5%). 👉 Feeling of general malaise (5%)”. Twitter. Retrieved 2021-05-07.
- ^ “[Translated]In more than 62 thousand applied doses of #Soberana02 the safety of the vaccine has been demonstrated. Adverse effects have been: 👉 Pain at the injection site (20%). 👉 Redness at the injection site (5%). 👉 Feeling of general malaise (5%)”. Facebook. Retrieved 2021-05-07.
External links
| Scholia has a profile for SOBERANA 02 (Q105047585). |
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Conjugate |
| Clinical data | |
| Other names | FINLAY-FR-2, SOBERANA PLUS[1] |
| Routes of administration | Intramuscular |
| Legal status | |
| Legal status | Full and Emergency Authorizations: List of Soberana 02 authorizations |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| COVID-19 (disease)SARS-CoV-2 (virus)CasesDeaths |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showEconomic impact and recession |
| showImpacts |
| COVID-19 portal |
/////////////////SARS-CoV-2, covid 19, corona virus, vaccine, iran, cuba, Soberana 02, FINLAY-FR-2
Nature (London, United Kingdom) (2021),

NEW DRUG APPROVALS
one time
$10.00
CVnCoV, zorecimeran, CureVac COVID-19 vaccine
CVnCoV
cas 2541470-90-8
An optimized, non-chemical modified mRNA encoding the prefusion-stabilized full-length spike protein of SARS-CoV-2 virus (Curevac)
zorecimeran, CureVac COVID-19 vaccine
CureVac/Bayer
GSK
NCT04674189 NCT04449276 NCT04515147 NCT04652102
EudraCT-2020-004066-19
mRNA-based vaccine
PHASE 3
| CVnCoV | Humoral and cellular responses | CD4+ T-cells, CD8+ T-cells | N/A | N/A | Rhesus macaque | [124] |
124. Rauch S, Gooch K, Hall Y, Salguero FJ, Dennis MJ, Gleeson FV. et al. mRNA vaccine CVnCoV protects non-human primates from SARS-CoV-2 challenge infection. bioRxiv. 2020. 2020 12.23.424138
The CureVac COVID-19 vaccine is a COVID-19 vaccine candidate developed by CureVac N.V. and the Coalition for Epidemic Preparedness Innovations (CEPI).[1] The vaccine showed inadequate results in its Phase III trials with only 47% efficacy.[2] The European Medicines Agency stated that: “(…) medicine developers should design studies to demonstrate a rate of efficacy of at least 50%.”[3].
The CVnCov Vaccine (or CV07050101) is in development by CureVac AG. The vaccine uses mRNA technology to create a protein associated with SARS-CoV2, and upon administration and replication, to initiate subsequent immune responses in the body. As of June 2020, the company received regulatory approval from German and Belgian Authorities to commence Phase 1 clinical trials of this vaccine (NCT04449276).
Efficacy
On 16 June 2021,[4] CureVac said its vaccine showed 47% efficacy from its Phase III trial. This was based on interim analysis of 134 COVID cases in its Phase III study conducted in Europe and Latin America. The final analysis for the trials requires a minimum of 80 additional cases.[2]
Pharmacology
CVnCoV is an mRNA vaccine that encodes the full-length, pre-fusion stabilized coronavirus spike protein, and activates the immune system against it.[5][6][7] CVnCoV technology does not interact with the human genome.[6] CVnCoV uses unmodified RNA,[8] unlike the Pfizer–BioNTech COVID-19 vaccine and Moderna COVID-19 vaccine, which both use nucleoside-modified RNA.[9]
Manufacturing
Manufacturing of mRNA vaccines can be performed rapidly in high volume,[10] including use of portable, automated printers (“RNA microfactories”) for which CureVac has a joint development partnership with Tesla.[11]
mRNA vaccines require stringent cold chain refrigeration throughout manufacturing, distribution and storage.[12][13] The CureVac technology for CVnCoV uses a non-modified, more natural mRNA less affected by hydrolysis, enabling storage at 5 °C (41 °F) and relatively simplified cold chain requirements that facilitate up to three months of storage and distribution to world regions that do not have specialized ultracold equipment.[6][10]
CureVac has a European-based network to accelerate manufacturing of CVnCoV, if proven safe and effective, for production of up to 300 million doses in 2021 and 600 million doses in 2022.[10][14] An estimated 405 million doses will be provided to EU states.[14]
Clinical trials
In November 2020, CureVac reported results of a Phase I-II clinical trial that CVnCoV (active ingredient zorecimeran) was well-tolerated, safe, and produced a robust immune response.[15][16]
In December 2020, CureVac began a Phase III clinical trial of CVnCoV with 36,500 participants.[17][18] Bayer will provide clinical trial support and international logistics for the Phase III trial, and may be involved in eventual manufacturing should the vaccine prove to be safe and effective.[19][20] In February 2021, the EU’s CHMP started a rolling review of CVnCoV.[21][22] In April 2021, the same procedure began in Switzerland.[23]
Brand names
The manufacturer currently markets the vaccine under the name CVnCoV.[24] Zorecimeran is the proposed international nonproprietary name (pINN).[25]
References
- ^ “CureVac focuses on the development of mRNA-based coronavirus vaccine to protect people worldwide”. CureVac(Press release). 15 March 2020. Retrieved 17 February 2021.
- ^ Jump up to:a b Burger, Ludwig (16 June 2021). “CureVac fails in pivotal COVID-19 vaccine trial with 47% efficacy”. Reuters. Retrieved 17 June 2021.
- ^ https://www.ema.europa.eu/en/human-regulatory/overview/public-health-threats/coronavirus-disease-covid-19/treatments-vaccines/vaccines-covid-19/covid-19-vaccines-studies-approval#what-is-the-level-of-efficacy-that-can-be-accepted-for-approval?-section
- ^ “CureVac Provides Update on Phase 2b/3 Trial of First-Generation COVID-19 Vaccine Candidate, CVnCoV”. 16 June 2021.
- ^ https://www.curevac.com/wp-content/uploads/2020/10/20201023-CureVac-Manuscript-draft-preclinical-data.pdf
- ^ Jump up to:a b c Schlake T, Thess A, Fotin-Mleczek M, Kallen KJ (November 2012). “Developing mRNA-vaccine technologies”. RNA Biology. 9(11): 1319–30. doi:10.4161/rna.22269. PMC 3597572. PMID 23064118.
- ^ “Understanding mRNA COVID-19 vaccines”. US Centers for Disease Control and Prevention. 18 December 2020. Retrieved 5 January 2021.
- ^ “COVID-19”. CureVac. Retrieved 21 December 2020.
- ^ Dolgin, Elie (25 November 2020). “COVID-19 vaccines poised for launch, but impact on pandemic unclear”. Nature Biotechnology: d41587–020–00022-y. doi:10.1038/d41587-020-00022-y. PMID 33239758. S2CID 227176634.
- ^ Jump up to:a b c Nawrat A (3 December 2020). “Q&A with CureVac: resolving the ultra-cold chain logistics of Covid-19 mRNA vaccines”. Pharmaceutical Technology. Retrieved 5 January 2021.
- ^ “Tesla to make molecule printers for German COVID-19 vaccine developer CureVac”. Reuters. 2 July 2020. Retrieved 19 December 2020.
- ^ Kartoglu U, Milstien J (July 2014). “Tools and approaches to ensure quality of vaccines throughout the cold chain”. Expert Review of Vaccines. 13 (7): 843–54. doi:10.1586/14760584.2014.923761. PMC 4743593. PMID 24865112.
- ^ Hanson CM, George AM, Sawadogo A, Schreiber B (April 2017). “Is freezing in the vaccine cold chain an ongoing issue? A literature review”. Vaccine. 35 (17): 2127–2133. doi:10.1016/j.vaccine.2016.09.070. PMID 28364920.
- ^ Jump up to:a b Kansteiner F (17 November 2020). “CureVac, armed with COVID-19 vaccine deal, plots ‘pandemic-scale’ Euro manufacturing expansion”. FiercePharma, Questex LLC. Retrieved 5 January2021.
- ^ “CureVac’s Covid-19 vaccine induces immune response in study”. Clinical Trials Arena. 3 November 2020. Retrieved 5 January 2021.
- ^ “CureVac’s COVID-19 vaccine triggers immune response in Phase I trial”. Reuters. 2 November 2020. Retrieved 5 January2021.
- ^ “Multicenter Clinical Study Evaluating the Efficacy and Safety of Investigational SARS-CoV-2 mRNA Vaccine CVnCoV in Adults 18 Years of Age and Older”. EU Clinical Trials Register. 19 November 2020. Retrieved 5 January 2021.
Proposed INN: zorecimeran
- ^ “A Study to Determine the Safety and Efficacy of SARS-CoV-2 mRNA Vaccine CVnCoV in Adults”. ClinicalTrials.gov. 8 December 2020. NCT04652102. Retrieved 19 December 2020.
- ^ Burger L (7 January 2021). “CureVac strikes COVID-19 vaccine alliance with Bayer”. Reuters. Retrieved 17 February 2021.
- ^ “CureVac and Bayer join forces on COVID-19 vaccine candidate CVnCoV”. CureVac (Press release). 7 January 2021. Retrieved 17 February 2021.
- ^ “EMA starts rolling review of CureVac’s COVID-19 vaccine (CVnCoV)”. European Medicines Agency (EMA) (Press release). 11 February 2021. Retrieved 12 February 2021.
- ^ “CureVac Initiates Rolling Submission With European Medicines Agency for COVID-19 Vaccine Candidate, CVnCoV”. CureVac(Press release).
- ^ “CureVac starts review process in Switzerland for COVID-19 vaccine hopeful”. Reuters. 19 April 2021. Retrieved 19 April 2021.
- ^ “Celonic and CureVac Announce Agreement to Manufacture over 100 Million Doses of CureVac’s COVID-19 Vaccine Candidate, CVnCoV”. CureVac (Press release). 30 March 2021. Retrieved 14 April 2021.
- ^ World Health Organization (October 2020). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 124 – COVID-19 (special edition)” (PDF). WHO Drug Information. 34 (3): 668–69. Archived (PDF) from the original on 27 November 2020.
External links
| Scholia has a profile for zorecimeran (Q97154239). |
- “Zorecimeran”. Drug Information Portal. U.S. National Library of Medicine.
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | mRNA |
| Clinical data | |
| Other names | CVnCoV, CV07050101 |
| Routes of administration | Intramuscular |
| ATC code | None |
| Identifiers | |
| DrugBank | DB15844 |
| UNII | 5TP24STD1S |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| COVID-19 (disease)SARS-CoV-2 (virus) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 portal |
- Rego GNA, Nucci MP, Alves AH, Oliveira FA, Marti LC, Nucci LP, Mamani JB, Gamarra LF: Current Clinical Trials Protocols and the Global Effort for Immunization against SARS-CoV-2. Vaccines (Basel). 2020 Aug 25;8(3). pii: vaccines8030474. doi: 10.3390/vaccines8030474. [Article]
- Speiser DE, Bachmann MF: COVID-19: Mechanisms of Vaccination and Immunity. Vaccines (Basel). 2020 Jul 22;8(3). pii: vaccines8030404. doi: 10.3390/vaccines8030404. [Article]
- CureVac & Covid-19 [Link]
- Smart Patients [Link]
- Regulatory News [Link]
////////////zorecimeran, CVnCoV, CV07050101, CORONA VACCINE, COVID 19, VACCINE, CUREVAC, SARS-CoV-2, CV07050101, SARS-CoV-2 mRNA vaccine
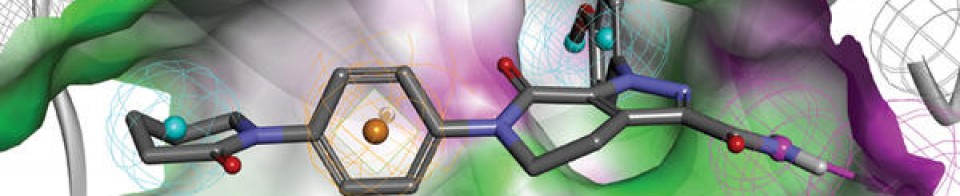
NEW DRUG APPROVALS
one time
$10.00
COVAX-19
Vaxine Pty Ltd company logo

Vaxine’s promising new COVID-19 vaccine candidate
A new multivalent COVID-19 vaccine developed by Australian company Vaxine to tackle the new virus variants could be game-changer in the fight against COVID-19
The world desperately needs a vaccine that blocks virus transmission and protects against all the variants. Covax-19 vaccine may soon change history”— Sharen Pringle, Vaxine Business Mananager
ADELAIDE, SA, AUSTRALIA, May 16, 2021 /EINPresswire.com/ — Professor Petrovsky, who is the Chairman and Research Director of Australian-based Vaxine Pty Ltd, explains that the two biggest challenges to tackling the COVID-19 pandemic are to develop a vaccine that completely prevents virus transmission something other COVID-19 have not been completely successful in achieving, and the second being to find a vaccine that protects equally against all the evolving immune-escape variants.
Professor Petrovsky has been researching coronavirus vaccines for the last 17 years, having previously published scientific papers on vaccines against both the SARS and MERS coronaviruses, which were highly protective in relevant animal models. He also recently published data from a collaboration with the US Army on development of a promising Ebola vaccine that protected mice against this most lethal disease after just a single vaccine dose. He has now successfully taken the same approach to design a protein-based vaccine against COVID-19.
Studies in a broad range of animal models including mice, hamsters, ferrets and monkeys, have recently revealed the high potential of this vaccine that is currently known as Covax-19(TM), but which likely will be soon rebranded as in its latest iteration it moves into late stage human trials in a number of countries.
Recent breakthrough data generated by Vaxine’s partner, Professor Kaissar Tabynov who leads the International Center for Vaccinology at the Kazakh National Agrarian University has shown that Vaxine’s unique spike protein antigen which is produced using insect cells in culture, was unique in that it not only totally protected hamsters from infection themselves but also prevented them from transmitting the virus to unvaccinated animals that were placed in the same cage two days after the vaccinated animals had been challenged with virus. Protection against transmission was not seen in hamsters given other vaccines making this finding unique to Vaxine’s spike protein antigen.
This hamster data reinforced findings in hamster, ferret and monkey challenge study performed by collaborating US Universities, who showed that two doses of Vaxine’s Covax-19 vaccine provided complete clearance of recoverable virus from the lungs and nose of animals when sampled just days after an infectious challenge.
“COVAX-19 vaccine has now been shown to be highly protective against the original Wuhan strain of the virus in hamster, ferret and monkey infection models performed by independent academic institutions in multiple countries, attesting to the strength of our protein-based vaccine approach”, says Prof. Petrovsky.
“A key element in the success of Covax-19 vaccine is the inclusion of Vaxine’s Advax adjuvant technology which acts as a turbocharger to drive an optimal immune response against the virus” explains Prof. Petrovsky who has been working on this promising vaccine adjuvant technology for the last 20 years with funding support from the US National Institutes of Health.
“We have now shown that our COVAX-19 vaccine can provide effective immunity including an ability to block nasal virus replication and this in turn successfully prevents transmission of the virus to vaccine-naïve animals,” he explains.
Follow on studies to confirm and expand upon these initial findings are currently underway at several US universities as well as Kazakh National Agrarian University, with a manuscript describing some of the initial animal data currently under review at a leading vaccine journal.
In another major breakthrough the team has now developed the vaccine into a multivariant format designed to protect against all the recently described variant strains of COVID-19, with work also underway on the most recently described Indian strains.
While the data is still preliminary says Prof. Petrovsky, the immune responses to the multivalent vaccine in mice are generating equally strong antibody binding activity against all the major virus variants. “This is extremely exciting as the world desperately needs vaccines able to protect against all the new strains of the virus including the UK, South African and Brazilian strains. By contrast , the currently available vaccines are clearly not as strong against some of these variants as they are against the original Wuhan strain” he explains.
Already there have been multiple confirmed cases of vaccine breakthrough where otherwise healthy individuals who have received mRNA, adenovirus or inactivated whole virus vaccines have become infected generally with either the South African or Brazilian variants.
This problem of immune-escape will only get worse over time as more complex variants emerge which is why Vaxine has been putting all its energy into finding a robust solution to this issue before proceeding with Phase 3 clinical trials of its Covax-19 vaccine.
Dr. Petrovsky went on to conclude “Now we have a multivalent formulation of Covax-19 vaccine that is showing high promise in animal studies, we plan to work as fast as we can to advance this new vaccine formulation in human trials, while expanding manufacturing capacity to ensure we are able to produce enough vaccine to meet the enormous global demand that will be attracted by such a successful vaccine.”
“To help us in this task Vaxine is looking to assemble a global network of partner organisations in countries around the world to assist Vaxine with vaccine development, clinical trials, manufacturing, distribution and sales. This is going to be a mammoth effort as we go to war against this insidious virus that continues to wreak havoc around the globe, with WHO recently predicting that the second year of the pandemic is likely to be much worse even than the first, an ominous warning for many countries that still remain poorly prepared and lacking in local vaccine manufacturing capability.
Vaxine wishes to help developing countries to establish their own local state-of-the-art vaccine manufacturing facilities, providing advice on appropriate facility design and undertaking technology transfer of its state of the art protein production technology to such facilities.
Countries in the developing world can no longer afford to sit and wait for outside organisations like COVAX to solve their vaccine supply problems, instead Vaxine proposes to help such countries find their own local solutions to the vaccine supply bottleneck for this.
Sharen Pringle
Vaxine Pty Ltd
437 033 400
email us here……..https://www.einnews.com/pr_news/541113168/covid-19-vaccine-breakthrough
Currently, the Australian influenza vaccine and adjuvant specialist and the Polish protein drug maker have just inked a memorandum of understanding, so the terms of a future contract remain to be defined. However, the technology behind is interesting.
The partners intent to utilize an insect cell-based recombinant spike protein of SARS-CoV–2 in combination with Vaxine’s proprietary Advax™ adjuvant and have already started Phase I testing in Australia with first result expected later this month. The company announced it will use artificial intelligence to evalutate clinical data in real time and announced the ambition to complete Phase II and III trials at the end of this year. “Supported by Microsoft technology, we aim to collect and analyse the COVAX-19™ trial data in real time, rather than waiting until the end of the trial before seeing if the vaccine is working, which is the traditional process,” said Vaxine’s Research Director Professor Nikolai Petrovsky from Flinders University in Adelaide.
Preclinically, Vaxine Pty Ltd’s syntetic spike protein with the company’s non-inflammatory Advax™ adjuvant, induced antibody and T-cell immune responses against the co-administered antigen. In various animal models, Covax-19 vaccination provided robust protection against an infection with the novel coronavirus.
The Phase I of Vaxine Pty Ltd in running since July in 40 healthy volunteers. If results are positive, the Australian vaccine maker is to expand studies and manufacturing to Europe. Under a future agreement Mabion SA would lead clinical development, manufacturing, regulatory negotiations and could exclusively market the vaccine in the EU and – optionally – in the US……..https://european-biotechnology.com/up-to-date/latest-news/news/mabion-to-licence-covid-19-jab-from-vaxine-pty-ltd.html
////////////////COVAX-19, corona virus, covid 19, Vaxine, australia, vaccine
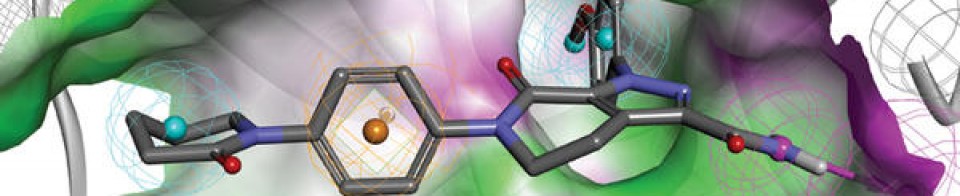
NEW DRUG APPROVALS
ONE TIME
$10.00
ARCT-021 (LUNAR-COV19)
ARCT-021 (LUNAR-COV19)
cas 2541451-24-3
A lipid-enabled and UnlockedNucleomonomer Agent modified RNA (LUNAR) of self-replicating RNA for vaccination against spike protein of SARS-CoV-2 (Arcturus)
Self-replicating RNA vaccine
| Arcturus Therapeutics and Duke-NUS Medical School, Singapore |
- OriginatorArcturus Therapeutics
- ClassCOVID-19 vaccines; RNA vaccines; Viral vaccines
- Mechanism of ActionImmunostimulants
- Orphan Drug StatusNo
- New Molecular EntityNo
- Available For LicensingYes – COVID 2019 infections
- Phase IICOVID 2019 infections
- 01 Mar 2021Arcturus Therapeutics has patent pending for STARR platform in USA
- 01 Mar 2021Immunogenicity data from a preclinical studies in COVID-2019 infections released by Arcturus Therapeutics
- 01 Mar 2021Arcturus Therapeutics completes a phase I/II trial in COVID-2019 infection in the Singapore
ref International Journal of Biological Sciences (2021), 17(6), 1446-1460. https://www.ijbs.com/v17p1446.htm
| LUNAR-COV19 | T7 | m7GpppNmN | Yes | VEEV-FL-S | N1-methyl pseudouridine | Silicon column | |
| protein | [54] |
ARCT-021: Currently undergoing phase 1/2 clinical trials, it combines two technologies, i.e., saRNA STARR™ and LUNAR® lipid-mediated delivery method. It was designed to enhance and extend antigen expression, enabling vaccination at lower doses [87]. In addition, LUNAR® lipids are pH-sensitive and biodegradable, causing minimal lipid accumulation in cells after multiple dosing [87]The Arcturus COVID-19 vaccine, commonly known as ARCT-021 and LUNAR-COV19, is a COVID-19 vaccine candidate developed by Arcturus Therapeutics.
| LUNAR- COV19 | 1 | Day 0 | 0.2 μg and 10 μg (Preclinical) | IM | Arcturus Therapeutics | N/A | Phase 2 | NCT04668339 NCT04480957 | [54] |
54. de Alwis R, Gan ES, Chen S, Leong YS, Tan HC, Zhang SL. et al. A Single Dose of Self-Transcribing and Replicating RNA Based SARS-CoV-2 Vaccine Produces Protective Adaptive Immunity In Mice. bioRxiv. 2020. 2020 09.03.280446
Development
Arcturus Therapeutics partnered with Singapore’s Duke–NUS Medical School to develop a COVID-19 vaccine.[1] The company also partnered with Catalent, a contract development and manufacturing organization, to manufacture multiple batches of Arcturus’ COVID-19 mRNA vaccine candidate.[2]
Clinical research
Phase I-II
LUNAR-COV19 clinical trials in humans began in July 2020.[3] On 4 January 2021, Arcturus Therapeutics started phase-2 clinical trials.[4]
Deployment
Arcturus has entered into development and supply agreements with the Economic Development Board of Singapore and supply agreements with the Israel Ministry of Health for LUNAR-COV19.[5][6]
Arcturus Therapeutics Receives FDA Allowance to Proceed with Phase 2 Study of ARCT-021 (LUNAR-COV19) Vaccine Candidate in the United States
Phase 2 study to be conducted in the U.S. and Singapore, and will evaluate both single dose and two dose priming regimens of ARCT-021 in up to 600 participants
Anticipate interim Phase 2 data in early 2021; targeting global Phase 3 study start in Q2 2021 which could allow application for emergency use authorization/conditional approval in H2 2021January 04, 2021 07:01 AM Eastern Standard Time
SAN DIEGO–(BUSINESS WIRE)–Arcturus Therapeutics Holdings Inc. (the “Company”, “Arcturus”, Nasdaq: ARCT), a leading clinical-stage messenger RNA medicines company focused on the development of infectious disease vaccines and significant opportunities within liver and respiratory rare diseases, today announced that the Company has received allowance of the Investigational New Drug (IND) application from the U.S. Food and Drug Administration (FDA) for the Phase 2 clinical study of its vaccine candidate ARCT-021 following review of data from the Phase 1/2 study.
Arcturus Therapeutics Receives FDA Allowance to Proceed with Phase 2 Study of ARCT-021 (LUNAR-COV19) Vaccine Candidate in the United States
Arcturus previously announced that the ARCT-021 Phase 2 study had been approved to proceed by the Singapore Health Sciences Authority (HSA), who reviewed the same data as reviewed by the FDA. These Phase 1/2 study results demonstrated favorable tolerability and both humoral and cellular immunogenicity following administration of ARCT-021.
The Phase 2 study will enroll 600 participants, with 450 receiving ARCT-021 and 150 receiving placebo. Both older and younger adult participants will be included. Early interim analyses of safety and immunogenicity will be performed to inform dose selection for a Phase 3 study, which is targeted to start in Q2 2021, if the Phase 2 study is successful.
“Allowance of the IND for our ARCT-021 Phase 2 clinical study represents an important milestone for the program and we look forward to starting to screen study participants at U.S. and Singapore clinical sites very soon,” said Steve Hughes, M.D., Chief Medical Officer of Arcturus. “We have advanced ARCT-021 to Phase 2 based on promising interim results from our Phase 1/2 study and extensive preclinical data. Our prior clinical results show that ARCT-021 administration results in humoral and cellular immunogenicity, and we are encouraged by an increasing body of evidence highlighting the potential importance of T cells in providing protection against SARS-CoV-2 infection and COVID-19. We believe that ARCT-021 holds promise to be a highly effective vaccine with a differentiated clinical profile, including the potential to only require a single dose for protection.”
About Arcturus Therapeutics
Founded in 2013 and based in San Diego, California, Arcturus Therapeutics Holdings Inc. (Nasdaq: ARCT) is a clinical-stage mRNA medicines and vaccines company with enabling technologies: (i) LUNAR® lipid-mediated delivery, (ii) STARR™ mRNA Technology and (iii) mRNA drug substance along with drug product manufacturing expertise. Arcturus’ diverse pipeline of RNA therapeutic and vaccine candidates includes self-replicating mRNA vaccine programs for SARS-CoV-2 (COVID-19) and Influenza, and other programs to potentially treat Ornithine Transcarbamylase (OTC) Deficiency, Cystic Fibrosis, and Cardiovascular Disease along with partnered programs including Glycogen Storage Disease Type 3, Hepatitis B Virus, and non-alcoholic steatohepatitis (NASH). Arcturus’ versatile RNA therapeutics platforms can be applied toward multiple types of nucleic acid medicines including messenger RNA, small interfering RNA, replicon RNA, antisense RNA, microRNA, DNA, and gene editing therapeutics. Arcturus’ technologies are covered by its extensive patent portfolio (205 patents and patent applications, issued in the U.S., Europe, Japan, China and other countries). Arcturus’ commitment to the development of novel RNA therapeutics has led to collaborations with Janssen Pharmaceuticals, Inc., part of the Janssen Pharmaceutical Companies of Johnson & Johnson, Ultragenyx Pharmaceutical, Inc., Takeda Pharmaceutical Company Limited, CureVac AG, Synthetic Genomics Inc., Duke-NUS Medical School, and the Cystic Fibrosis Foundation. For more information visit www.ArcturusRx.com. In addition, please connect with us on Twitter and LinkedIn.
References
- ^ Teo J (15 April 2020). “Coronavirus: Clinical trials for Singapore’s vaccine project could start in August”. The Straits Times. Retrieved 27 April 2020.
- ^ Stanton D (6 May 2020). “With Arcturus, Catalent bags another COVID project”. Bioprocess Insider. Retrieved 8 May 2020.
- ^ Clinical trial number NCT04480957 for “Phase 1/2 Ascending Dose Study of Investigational SARS-CoV-2 Vaccine ARCT-021 in Healthy Adult Subjects” at ClinicalTrials.gov
- ^ “Arcturus Therapeutics Receives FDA Allowance to Proceed with Phase 2 Study of ARCT-021 (LUNAR-COV19) Vaccine Candidate in the”. Bloomberg. 4 January 2021. Retrieved 17 January 2021.
- ^ Anwar N (26 November 2020). “Singapore’s co-developed vaccine candidate is in ‘good shape’ for delivery in 2021”. CNBC. Retrieved 18 March 2021.
- ^ Cheok M, Mookerjee I (5 August 2020). “Singapore Will Get First Claim to Any Successful Arcturus Vaccine”. Bloomberg. Retrieved 18 March 2021.
External links
| Scholia has a profile for Lunar-COV19 (Q98713328). |
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | mRNA |
| Clinical data | |
| Other names | ARCT-021, LUNAR-COV19 |
| Routes of administration | Intramuscular |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| COVID-19 (disease)SARS-CoV-2 (virus) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 portal |
/////////COVID-19, SARS-CoV-2, corona virus, singapore, ARCT 021, LUNAR-COV19
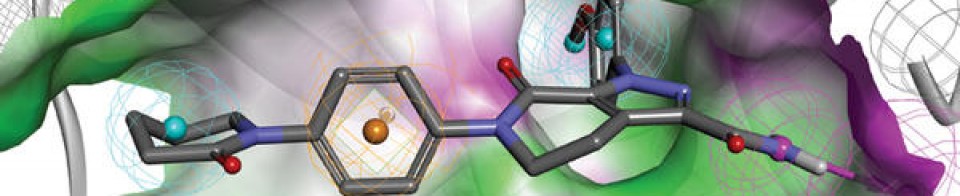
NEW DRUG APPROVALS
one time
$10.00
IIBR-100

IIBR-100
Brilife
Recombinant vesicular stomatitis virus (rVSV) vaccine
Israel Institute for Biological Research
Hadassah Medical Center; Sheba Medical Center Hospital
The SARS-CoV-2 virus is responsible for the COVID-19 pandemic. The pandemic emerged from Wuhan Province in China in December 2019 and was declared by the WHO Director-General a Public Health Emergency of International Concern on 30 January 2020.
In this study, a vaccine developed by IIBR for SARS-CoV-2 virus will be assessed for its safety and potential efficacy in volunteers. The study is comprised of two phases, a dose-escalation phase (phase I) during which subjects (18-55 years old) will be randomly allocated to receive a single administration of IIBR-100 100 at low, mid or high dose or saline or two administrations of IIBR-100 at low dose, or saline, 28 days apart.
Based on results obtained during phase I, and cumulative phase I data review, the expansion phase (phase II) has begun, during which larger cohorts as well as elderly age subjects will be randomly allocated to receive a single administration of IIBR-100 at low, mid or high dose or saline, or two administrations of IIBR-100 at low, mid or high dose (prime-boost) or saline, 28 days apart. Additional top-dose (prime-boost) may be implemented when immunogenicity of any prime-boost arm is considered insufficient.
Based on immunogenicity preliminary data and DSMB recommendations, the two administrations of mid, high and top dose (prime-boost) or saline will continue.
The subjects will be followed for a period of up to 12 months post last vaccine administration to assess the safety and efficacy of the vaccine.
https://clinicaltrials.gov/ct2/show/NCT04608305
IIBR-100 also known as Brilife is a COVID-19 vaccine candidate developed by The Israel Institute for Biological Research.[1][2]
References
- ^ Clinical trial number NCT04608305 for “Phase I/II Randomized, Multi-Center, Placebo-Controlled, Dose-Escalation Study to Evaluate the Safety, Immunogenicity and Potential Efficacy of an rVSV-SARS-CoV-2-S Vaccine (IIBR-100) in Adults” at ClinicalTrials.gov
- ^ Jeffay N (29 December 2020). “As Israel goes vaccine-wild, will the homegrown version lose its shot?”. The Times of Israel. Retrieved 1 January 2021.
candidate developed by The Israel Institute for Biological Research.[1][2]
Israeli institute’s COVID vaccine candidate said very effective in animal trials
Secretive Israeli research center’s shot shows near 100% efficacy in non-human trials, is on par with US company Moderna’s candidate, TV report says
Israeli researchers at a top secret research center have made progress on a coronavirus vaccine that shows a high level of effectiveness in animals, according to a Friday TV report.
However, there is no guarantee that the vaccine under development will be effective in humans, or will be available soon.
The Israel Institute for Biological Research (IIBR), a secretive unit that works under the Prime Minister’s Office, developed a vaccine that shows close to 100 percent protection against the virus in lab animals, the Channel 12 report said, citing “a security source.”
The vaccine under development is on par in effectiveness with a vaccine being developed by US biotechnology company Moderna, the report said.
Unlike vaccines developed abroad, the domestic vaccine will first be delivered to Israeli citizens, it added. If successful, it was expected to provide protection against the disease with a single dose.
The institute has not started human trials but was preparing to manufacture 10 to 15 million doses, report said.
Hebrew media have reported on potential breakthroughs at the shadowy institute several times before, starting in mid-March, with the Defense Ministry pushing back on some of the claims to tamper expectations.

Magen David Adom medical workers test Israelis for the coronavirus at a drive-through site in Lod, on July 10, 2020. (Yossi Aloni/Flash90)
IIBR said last month that it had completed successful coronavirus vaccine trials on rodents, paving the way for further testing on other animals and then possibly human trials.
In a paper published on the website of bioRxiv, an online repository for papers that haven’t yet been peer-reviewed, the institute, which is based in Ness Ziona, said it hopes to have a finished vaccine in a year, or possibly even earlier.
In the abstract of the report, the researchers say their vaccine, which they tested on hamsters, “results in rapid and potent induction of neutralizing antibodies against SARS-CoV-2,” the virus that causes COVID-19.
Earlier this month a vaccine adviser to the government cautioned that there was no guarantee that the shots being developed will prove widely effective.
In May, the institute confirmed that it had isolated an antibody it believed could be used to develop treatments against the virus. The development would not be useful in the creation of a vaccine, but would rather be a move toward a drug treatment for those who have already contracted the disease.
Tal Zaks, Moderna’s Israeli chief medical officer, described to Channel 12 on Friday the company’s push into Phase 3 testing of its vaccine candidate, which was developed with the National Institutes of Health, and began its first injections Monday.
The trial, the world’s largest vaccine study, plans to test the vaccine on 30,000 volunteers.
There’s still no guarantee that the experimental vaccine, developed by the National Institutes of Health and Moderna Inc., will really offer protection.
“The first time we saw the first model, that the vaccine, even if it’s just in mice, successfully stimulated the immune system to identify the virus and neutralize it, I knew that we hadn’t missed anything, that we had the correct vaccine,” he said.
“And of course the second ‘ah-ha’ moment was when we saw the first clinical results, when it was clear that in humans we weren’t just getting to antibody levels we were seeing in sick people, which is what we aspired to, but we were getting to even higher levels,” Zaks said.
A Nurse gives a volunteer an injection, as the world’s biggest study of a possible COVID-19 vaccine, developed by the US National Institutes of Health and Moderna Inc., gets underway on July 27, 2020, in Binghamton, NY. (AP Photo/Hans Pennink)
Last month Israel signed a deal with Moderna for the potential purchase of its coronavirus vaccine if it ends up proving effective.
Moderna said the vaccination was administered in Savannah, Georgia, the first site to get underway among more than seven dozen trial sites scattered around the country.
Several other vaccines made by China and by Britain’s Oxford University earlier this month began smaller final-stage tests in Brazil and other hard-hit countries.
The massive studies aren’t just to test if the shots work — they’re needed to check each potential vaccine’s safety. And following the same study rules will let scientists eventually compare all the shots.
It normally takes years to create a new vaccine from scratch, but scientists are setting speed records this time around, spurred by knowledge that vaccination is the world’s best hope against the pandemic.
If everything goes right with the final studies, it still will take months for the first data to trickle in from the Moderna test, followed by the Oxford one.
Governments around the world are trying to stockpile millions of doses of those leading candidates so if and when regulators approve one or more vaccines, immunizations can begin immediately. But the first available doses will be rationed, presumably reserved for people at highest risk from the virus.
Coronavirus cases in Israel rose by 1,791 in 24 hours on Friday and the national death toll hit 512, according to the latest Health Ministry figures.
The total case count stood at 70,970, with 320 patients in serious condition, including 98 on ventilators. The number of recovered patients reached 43,850.
Israel has the fifth-highest number of new coronavirus infections per capita in the world, overtaking the United States, according to data compiled by a scientific publication based at Oxford University.
And while Israel has seen the number of new coronavirus cases rocket to more than 2,000 a day in recent weeks, a new Hebrew University report published on Thursday asserted that Israel has managed to gain control of the second wave of the coronavirus, thanks to a recent stabilization in the number of seriously and moderately ill patients.
The curve for seriously and moderately ill patients began to spike in late June before stabilizing in recent days, the researchers reported. They credited the restrictions imposed by the government in recent weeks to limit crowding for helping to flatten the curve.
According to the report, the death toll will climb by roughly 200 in the coming three weeks as a result of the high infection rate over the past month.
Experts have blamed a too-speedy reopening and the lack of an effective contact-tracing program as main factors in the virus resurgence, which has come as new daily coronavirus cases around the world have also reached record highs.
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Viral vector |
| Clinical data | |
| Other names | Brilife |
| Routes of administration | Intramuscular |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| COVID-19 (disease)SARS-CoV-2 virus (variants) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 portal |
//////IIBR-100, Brilife, COVID-19, vaccine, israel, corona virus, covid 19, SARS-CoV-2
NEW DRUG APPROVALS
one time
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
.svg){kind=link}
{kind=link}