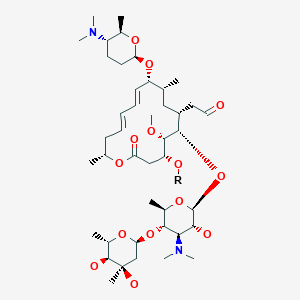
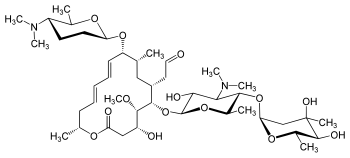
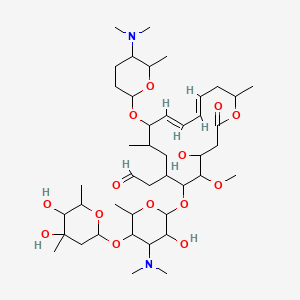
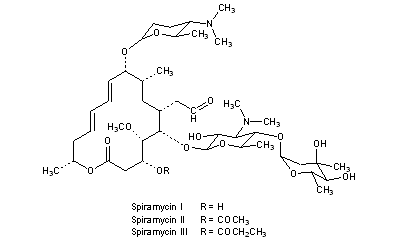
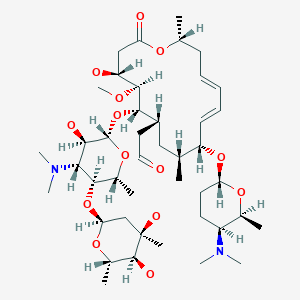
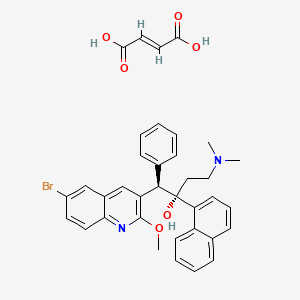
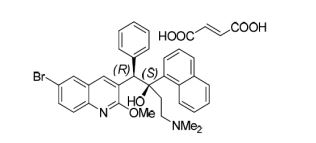
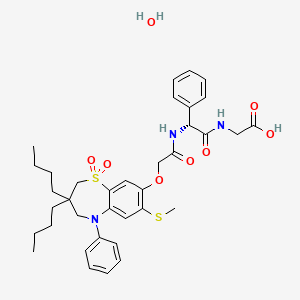
Fidaxomicin (C52H74Cl2O18, Mr = 1058.0 g/mol)
Launched – 2011 MERCK, Clostridium difficile-associated diarrhea
CUBIST ….INNOVATOR
OPT-80
PAR-101
Also tiacumicin B or lipiarmycin A3,
A bacterial RNA polymerase inhibitor as macrocyclic antibiotic used to treat clostridium difficile-associated diarrhea (CDAD).

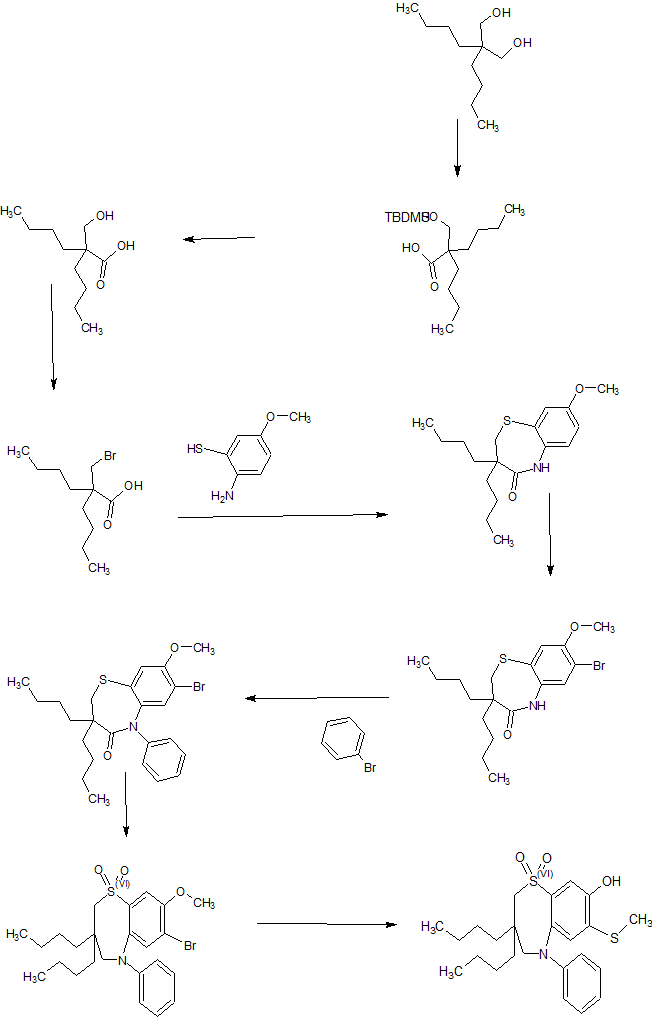
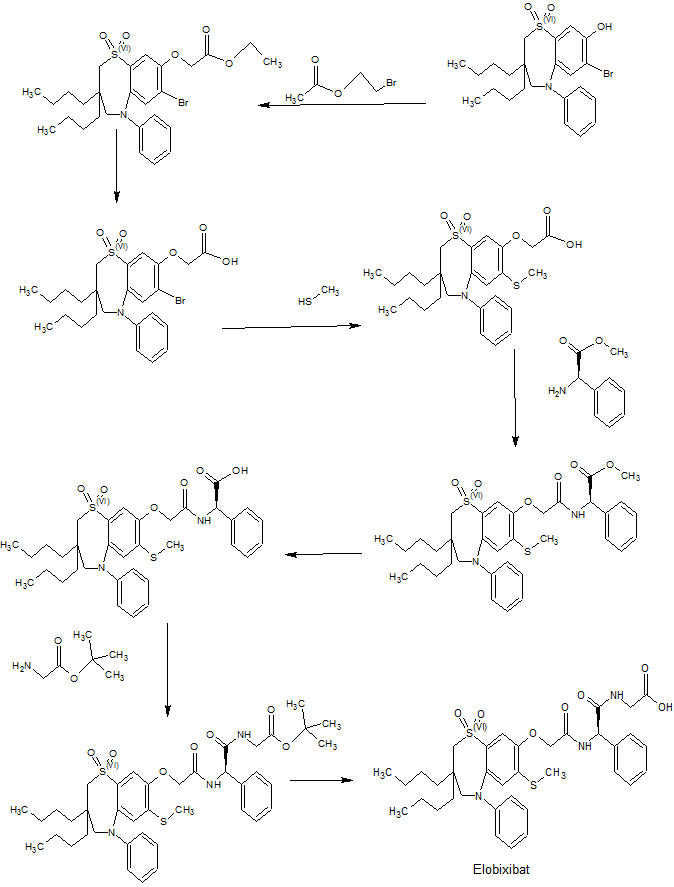
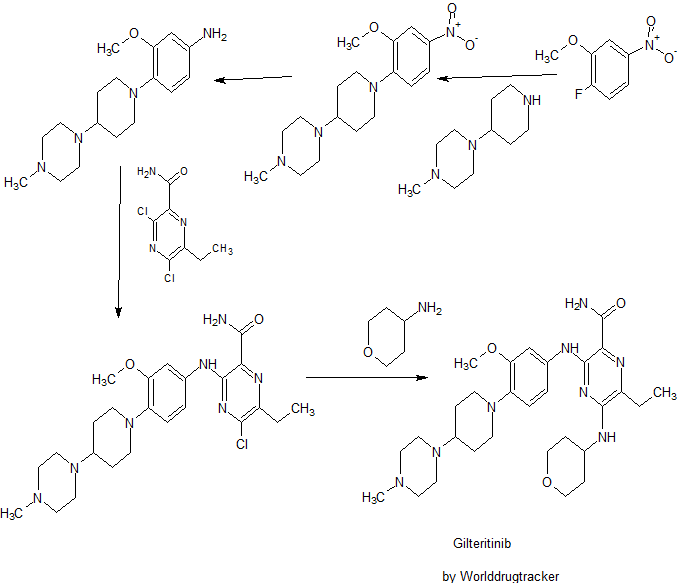
SYNTHESIS

REFERENCES
US 4918174
WO 2006085838
J ANTIBIOTICS 1987, 40, PG 567-574 AND 575-588
Idaxomicin(trade names Dificid, Dificlir, and previously OPT-80 and PAR-101) is the first in a new class of narrow spectrum macrocyclic antibiotic drugs.[2] It is a fermentation product obtained from the actinomycete Dactylosporangium aurantiacum subspecies hamdenesis.[3][4] Fidaxomicin is non-systemic, meaning it is minimally absorbed into the bloodstream, it is bactericidal, and it has demonstrated selective eradication of pathogenic Clostridium difficile with minimal disruption to the multiple species of bacteria that make up the normal, healthy intestinal flora. The maintenance of normal physiological conditions in the colon can reduce the probability of Clostridium difficile infection recurrence.[5] [6]
Fidaxomicin is an antibiotic approved and launched in 2011 in the U.S. for the treatment of Clostridium difficile-associated diarrhea (CDAD) in adults 18 years of age and older. In September 2011, the product received a positive opinion in the E.U. and final approval was assigned in December 2011.
First E.U. launch took place in the U.K. in June 2012. Optimer Pharmaceuticals, now part of Cubist (now, Merck & Co.), is conducting phase III clinical trials for the prevention of Clostridium difficile-associated diarrhea in patients undergoing hematopoietic stem cell transplant
In 2014 Astellas initiated in Europe a phase III clinical study for the treatment of Clostridium difficile infection in pediatric patients. Preclinical studies are ongoing for potential use in the prevention of methicillin-resistant Staphylococcus (MRS) infection.


The compound is a novel macrocyclic antibiotic that is produced by fermentation. Its narrow-spectrum activity is highly selective for C. difficile, thus preserving gut microbial ecology, an important consideration for the treatment of CDAD.
It is marketed by Cubist Pharmaceuticals after acquisition of its originating company Optimer Pharmaceuticals. The target use is for treatment of Clostridium difficile infection.
In May 2005, Par Pharmaceutical and Optimer entered into a joint development and collaboration agreement for fidaxomicin. However, rights to the compound were returned to Optimer in 2007. The compound was granted fast track status by the FDA in 2003. In 2010, orphan drug designation was assigned to fidaxomicin in the U.S. by Optimer Pharmaceuticals for the treatment of pediatric Clostridium difficile infection (CDI). In 2011, the compound was licensed by Optimer Pharmaceuticals to Astellas Pharma in Europe and certain countries in the Middle East, Africa, the Commonwealth of Independent States (CIS) and Japan for the treatment of CDAD. In 2011, fidaxomicin was licensed to Cubist by Optimer Pharmaceuticals for comarketing in the U.S. for the treatment of CDAD. In July 2012, the product was licensed by Optimer Pharmaceuticals to Specialised Therapeutics Australia in AU and NZ for the treatment of Clostridium difficile-associated infection. OBI Pharma holds exclusive commercial rights in Taiwan, where the compound was approved for the treatment of CDAD in September 2012, and in December 2012, the product was licensed to AstraZeneca in South America with commercialization rights also for the treatment of CDAD. In October 2013, Optimer Pharmaceuticals was acquired by Cubist.
Fidaxomicin is available in a 200 mg tablet that is administered every 12 hours for a recommended duration of 10 days. Total duration of therapy should be determined by the patient’s clinical status. It is currently one of the most expensive antibiotics approved for use. A standard course costs upwards of £1350.[7]
Fidaxomicin (also known as OPT-80 and PAR-101 ) is a novel antibiotic agent and the first representative of a new class of antibacterials called macrocycles. Fidaxomicin is a member of the tiacumicin family, which are complexes of 18-membered macrocyclic antibiotics naturally produced by a strain of Dactylosporangium aurantiacum isolated from a soil sample collected in Connecticut, USA.
The major component of the tiacumicin complex is tiacumicin B. Optically pure R-tiacumicin B is the most active component of Fidaxomicin. The chiral center at C(19) of tiacumicinB affects biological activity, and R-tiacumicin B has an R-hydroxyl group attached at this position. The isomer displayed significantly higher activity than other tiacumicin B-related compounds and longer post-antibiotic activity.
Tiacumicins are a family of structurally related compounds that contain the 18-membered macrolide ring shown below.
At present, several distinct Tiacumicins have been identified and six of these
(Tiacumicin A-F) are defined by their particular pattern of substituents R1, R2, and R3 (US Patent No. 4,918,174; J. Antibiotics, 1987, 575-588).
The Lipiarmycins are a family of natural products closely related to the Tiacumicins. Two members of the Lipiarmycin family (A3 and B3) are identical to Tiacumicins B and C respectively (J. Antibiotics, 1988, 308-315; J. Chem. Soc. Perkin Trans 1, 1987, 1353-1359).
The Tiacumicins and the Lipiarmycins have been characterized by numerous physical methods. The reported chemical structures of these compounds are based on spectroscopy (UV-vis, IR and !H and 13C NMR), mass spectrometry and elemental analysis (See for example: J. Antibiotics, 1987, 575-588; J. Antibiotics, 1983, 1312-
1322).
Tiacumicins are produced by bacteria, including Dactylosporangium aurantiacum subspecies hamdenensis, which may be obtained from the ARS Patent Collection of the Northern Regional Research Center, United States Department ofAgriculture, 1815 North University Street, Peoria, IL 61604, accession number NRRL
18085. The characteristics of strain AB 718C-41 are given in J. Antibiotics, 1987,567-574 and US Patent No. 4,918,174.
Lipiarmycins are produced by bacteria including Actinoplanes deccanensis (US Patent No. 3,978,211). Taxonomical studies of type strain A/10655, which has been deposited in the ATCC under the number 21983, are discussed in J. Antibiotics,1975, 247-25.
Tiacumicins, specifically Tiacumicin B, show activity against a variety of bacterial pathogens and in particular against Clostridium difficile, a Gram-positive bacterium (Antimicrob. Agents Chemother. 1991, 1108-1111). Clostridium difficile is an anaerobic spore-forming bacterium that causes an infection of the bowel.
As per WIPO publication number 2006085838, Fidaxomicin is an isomeric mixture of the configurationally distinct stereoisomers of tiacumicin B, composed of 70 to 100% of R-tiacumicin B and small quantities of related compounds, such as S-tiacumicin B and lipiarmycin A4. Fidaxomicin was produced by fermentation of the D aurantiacum subspecies hamdenensis (strain 718C-41 ). It has a narrow spectrum antibacterial profile mainly directed against Clostridium difficile and exerts a moderate activity against some other gram-positive species.
Fidaxomicin is bactericidal and acts via inhibition of RNA synthesis by bacterial RNA polymerase at a distinct site from that of rifamycins. The drug product is poorly absorbed and exerts its activity in the gastrointestinal (Gl) tract, which is an advantage when used in the applied indication, treatment of C. difficile infection (CDI) (also known as C. difficile-associated disease or diarrhoea [CDAD]). Fidaxomicin is available as DIFICID oral tablet in US market.
Its CAS chemical name is Oxacyclooctadeca-3,5,9, 13, 15-pentaen-2-one, 3-[[[6-deoxy-4-0-(3,5dichloro-2-ethyl-4,6-dihydroxybenzoyl)-2-0-methyl-P-D-manno pyranosyl]oxy]methyl]-12[[6-deoxy-5-C-methyl-4-0-(2-methyl-1 -oxopropyl)- -D-lyxo-hexo pyranosyl]oxy]-1 1 -ethyl-8-hydroxy-18-[(1 R)-1 -hydroxyethyl] -9,13,15-trimethyl-, (3E.5E, 8S.9E.1 1 S.12R.13E, 15E.18S)-.
Structural formula (I) describes the absolute stereochemistry of fidaxomicin as determined by x-ray.

(I)
WIPO publication number 2004014295 discloses a process for preparation of Tiacumicins that comprises fermentation of Dactylosporangium aurantiacum NRRL18085 in suitable culture medium. It also provides process for isolation of tiacumicin from fermentation broth using techniques selected from the group consisting of: sieving and removing undesired material by eluting with at least one solvent or a solvent mixture; extraction with at least one solvent or a solvent mixture; Crystallization; chromatographic separation; High-Performance Liquid Chromatography (HPLC); MPLC; trituration; and extraction with saturated brine with at least one solvent or a solvent mixture. The product was isolated from /so-propyl alcohol (IPA) having a melting point of 166-169 °C.
U.S. Patent No. 7378508 B2 discloses polymorphic forms A and B of fidaxomicin, solid dosage forms of the two forms and composition thereof. As per the ‘508 patent form A is obtained from methanol water mixture and Form B is obtained from ethyl acetate.
J. Antibiotics, vol. 40(5), 575-588 (1987) discloses purification of Tiacumicins using suitable solvents wherein tiacumicin B exhibited a melting point of 143-145 °C.
PCT application WO2013170142A1 describes three crystalline forms of Fidaxomicn namely, Form-Z, Form-Z1 and Form-C. IN2650/CHE/2013 describes 6 crystalline polymorphic forms of Fidaxomicin namely, Forms I, Form la, Form II, Form Ha, Form III and Form Ilia).
Mechanism
Fidaxomicin binds to and prevents movement of the “switch regions” of bacterial RNAP polymerase. Switch motion is important for opening and closing of the DNA:RNA clamp, a process that occurs throughout RNA transcription but especially during opening of double standed DNA during transcription initiation.[8] It has minimal systemic absorption and a narrow spectrum of activity; it is active against Gram positive bacteria especially clostridia. The minimal inhibitory concentration (MIC) range for C. difficile (ATCC 700057) is 0.03–0.25 μg/mL.[3]
Clinical trials
Good results were reported by the company in 2009 from a North American phase III trial comparing it with oral vancomycin for the treatment of Clostridium difficile infection (CDI)[9][10] The study met its primary endpoint of clinical cure, showing that fidaxomicin was non-inferior to oral vancomycin (92.1% vs. 89.8%). In addition, the study met its secondary endpoint of recurrence: 13.3% of the subjects had a recurrence with fidaxomicin vs. 24.0% with oral vancomycin. The study also met its exploratory endpoint of global cure (77.7% for fidaxomicin vs. 67.1% for vancomycin).[11] Clinical cure was defined as patients requiring no further CDI therapy two days after completion of study medication. Global cure was defined as patients who were cured at the end of therapy and did not have a recurrence in the next four weeks.[12]
Fidaxomicin was shown to be as good as the current standard-of-care, vancomycin, for treating CDI in a Phase III trial published in February 2011.[13] The authors also reported significantly fewer recurrences of infection, a frequent problem with C. difficile, and similar drug side effects.
Approvals and indications
For the treatment of Clostridium difficile-associated diarrhea (CDAD), the drug won an FDA advisory panel’s unanimous approval on April 5, 2011[14] and full FDA approval on May 27, 2011.[15]
PAPER
Enantioselective synthesis of putative lipiarmycin aglycon related to fidaxomicin/tiacumicin B
Angew Chem Int Ed 2015, 54(6): 1929
Enantioselective Synthesis of Putative Lipiarmycin Aglycon Related to Fidaxomicin/Tiacumicin B (pages 1929–1932)
Dr. William Erb, Dr. Jean-Marie Grassot, Dr. David Linder, Dr. Luc Neuville and Prof. Dr. Jieping Zhu
Article first published online: 24 NOV 2014 | DOI: 10.1002/anie.201409475

Chain gang: In the synthesis of the title compound, the ene-diene ring-closing metathesis was used for the formation of the 18-membered macrolactone and the stereogenic centers of the molecule were installed by Brown’s alkoxyallylboration, allylation, and an Evans aldol reaction, while iterative Horner–Wadsworth–Emmons reactions were used for chain elongation.
http://onlinelibrary.wiley.com/doi/10.1002/anie.201409475/full
http://onlinelibrary.wiley.com/store/10.1002/anie.201409475/asset/supinfo/anie_201409475_sm_miscellaneous_information.pdf?v=1&s=75d40b6f8b214578d5a65518e7f384f03f377c35
PAPER
Total synthesis of the glycosylated macrolide antibiotic fidaxomicin
Org Lett 2015, 17(14): 3514
http://pubs.acs.org/doi/abs/10.1021/acs.orglett.5b01602
http://pubs.acs.org/doi/suppl/10.1021/acs.orglett.5b01602/suppl_file/ol5b01602_si_001.pdf
The first enantioselective total synthesis of fidaxomicin, also known as tiacumicin B or lipiarmycin A3, is reported. This novel glycosylated macrolide antibiotic is used in the clinic for the treatment of Clostridium difficile infections. Key features of the synthesis involve a rapid and high-yielding access to the noviose, rhamnose, and orsellinic acid precursors; the first example of a β-selective noviosylation; an effective Suzuki coupling of highly functionalized substrates; and a ring-closing metathesis reaction of a noviosylated dienoate precursor. Careful selection of protecting groups allowed for a complete deprotection yielding totally synthetic fidaxomicin.

The identity of the synthetic compound to an authentic sample of fidaxomicin (1) was confirmed by coinjection on RP-HPLC and an equimolar mixed NMR-sample with an authentic sample. Rƒ = 0.44 (MeOH/CH2Cl2 1/10).
HRMS ESI calcd. for [C52H74Cl2NaO18] + [M+Na]+ : 1079.4144; found:1079.4151.
1H NMR (600 MHz, Methanol-d4 , containing HCOO- ) δ 7.23 (d, J = 11.5 Hz, 1H), 6.60 (dd, J = 14.9, 11.8 Hz 1H), 5.95 (ddd, J = 14.7, 9.5, 4.8 Hz, 1H), 5.83 (s, 1H), 5.57 (ap t, J = 8.2 Hz, 1H), 5.14 (ap d, J = 10.7, 1H), 5.13 (dd, J = 9.7 Hz, 1H), 5.02 (d, J = 10.2 Hz, 1H), 4.74-4.70 (m, 1H), 4.71 (s, 1H), 4.64 (s, 1H), 4.61 (d, J = 11.6 Hz, 1H), 4.44 (d, J = 11.6 Hz, 1H), 4.22 (ap s, 1H), 4.02 (p, J = 6.3 Hz, 1H), 3.92 (dd, J = 3.2, 1.2 Hz, 1H), 3.75 (ddd, J = 13.9, 10.2, 3.3 Hz, 1H) 3.71 (d, J = 9.7 Hz 1H), 3.58-3.52 (m, 2H) 3.54 (s, 3H), 3.15-3.06 (m, 1H), 3.04-2.95 (m, 1H), 2.76-2.66 (m, 3H), 2.60 (hept, J= 7.0 Hz, 1H), 2.49 (ddd, J = 14.9, 9.5, 4.4 Hz, 1H), 2.43 (ddd, J = 13.8, 8.8, 4.5 Hz, 1H), 2.05-1.98 (m, 1H), 1.82 (d, J = 1.3 Hz, 3H), 1.76 (ap s, 3H), 1.66 (ap s, 3H), 1.32-1.27 (m, 4H), 1.22-1.15 (m, 12H), 1.15 (s, 3H), 1.13 (s, 3H), 0.88 (t, J = 7.4 Hz, 3H).
RP-HPLC tR = 14.87 min (A: H2O+0.1% HCOOH; Solvent B: MeCN+0.1% HCOOH; 1 mL/min; T = 20°C; B[%] (tR [min])= 10 (0 to 3); 100 (15).
PATENT
WO 2004014295
http://www.google.co.in/patents/WO2004014295A2?cl=en
The term “Tiacumicin B” refers to molecule having the structure shown below:
Example 1
Dactylosporangium aurantiacum subsp. hamdenensis AB 718C-41 NRRL 18085 (-20 °C stock), was maintained on 1 mL of Medium No. 104 (Table 1). After standard sterilization conditions (30 min., 121 °C, 1.05 kg/cm2) the seed flask (250 mL) containing Medium No. 104 (50 mL) was inoculated with AB 718C-41 NRRL 18085 on a shaker (set @ 250 rpm) at 30 °C for 72 hr. Five percent vegetative inoculum from the first passage seed flask was then transferred aseptically to a fermentation flask containing the same ingredients as in Table 1.
Table 1: Ingredients of Medium No. 104

Fermentation flasks were incubated on a rotary shaker at 30 °C for 3 to 12 days. Samples of the whole culture fermentation broth were filtered. The filter cake was washed with MeOH and solvents were removed under reduced pressure. The residue was re-constituted in methanol to the same volume of the original fermentation broth. Analysis was performed using a Waters BREEZE HPLC system coupling with Waters 2487 2-channel UV/Vis detector. Tiacumincins were assayed on a 50 x 4.6 μm I.D., 5 μm YMC ODS-A column (YMC catalog # CCA AS05- 0546WT) with a mobile phase consisting of 45% acetonitrile in water containing 0.1% phosphoric acid at a flow rate of 1.5 mL/minute. Tiacumicins were detected at 266 nm. An HPLC chromatogram of a crude product (Tiacumicin B retention time @ 12.6 minutes) is shown in Fig. 1. In this example the crude yield of Tiacumicin B was about 250 mg/L after 7 days. After purification by HPLC, the yield of Tiacumicin B was about 100 mg/L.
Example 2
After standard sterilization conditions (30 min, 121 °C, 1.05 kg/cm2) the seed flask (250 mL) containing Medium No. 104 (50 mL) was inoculated with AB 718C- 41 NRRL 18085 and incubated on a shaker (set @ 250 rpm) at 30° C for 72 hr. Five percent vegetative inoculum from the first passage seed flask was transferred aseptically to a seed flask containing the same ingredients as in Table 1 and was incubated on a rotary shaker at 30 °C for 72 hr. Five percent inoculum from the second passage seed flasks was then used to inoculate with AB 718C-41 NRRL 18085 in a 5-liter fermenter containing Medium No. 104 (2.5 L). Excessive foam formation was controlled by the addition of an antifoaming agent (Sigma A-6426). This product is a mixture of non-silicone organic defoamers in a polyol dispersion.
Glucose consumption was monitored as a growth parameter and its level was controlled by the addition of the feeding medium. Feeding medium and conditions in Example 2 were as follows:
Feeding medium:
Fermenter Medium: No. 104
Fermenter Volume: 5 liters
Sterilization: 40 minutes, 121° C, 1.05 kg/cm2
Incubation Temperature: 30 °C.
Aeration rate: 0.5-1.5 volumes of air per culture volume and minute
Fermenter Agitation: 300-500 rpm
The fermentation was carried out for 8 days and the XAD-16 resin was separated from the culture broth by sieving. After washing with water the XAD-16 resin was eluted with methanol (5-10 x volume of XAD-16). Methanol was evaporated and the oily residue was extracted three times with ethyl acetate. The extracts were combined and concentrated under reduced pressure to an oily residue. The oily residue was dried and washed with hexane to give the crude product as a pale brown powder and its HPLC chromatogram (Tiacumincin B rete tion time @ 11.8 minutes) is shown in Figure 2. This was purified by silica gel column (mixture of ethyl acetate and hexane as eluent) and the resultant material was further purified by RP-HPLC (reverse phase HPLC) to give Tiacumicin B as a white solid. The purity was determined to be >95% by HPLC chromatography and the chromatogram (Tiacumincin B retention time @ 12.0 minutes) is shown in Figure 3. Analysis of the isolated Tiacumincin B gave identical !H and 13C NMR data to those reported in J. Antibiotics, 1987, 575-588, and these are summarized below. Tiacumicin B: mp 129-140 °C (white powder from RP-HPLC); mp 166-169 °C (white needles from isopropanol); [α]D 20-6.9 (c 2.0, MeOH); MS m/z (ESI) 1079.7(M + Na)+; H NMR (400 MHz, CD3OD) δ 7.21 (d, IH), 6.59 (dd, IH), 5.95 (ddd, IH), 5.83 (br s, IH), 5.57 (t, IH), 5.13 (br d, IH), 5.09 (t, IH), 5.02 (d, IH), 4.71 (m, IH), 4.71 (br s, IH), 4.64 (br s, IH), 4.61 (d, IH), 4.42 (d, IH), 4.23 (m, IH), 4.02 (pentet, IH), 3.92 (dd, IH), 3.73 (m, 2H), 3.70 (d, IH), 3.56 (s, 3H), 3.52-3.56 (m, 2H), 2.92 (m, 2H), 2.64-2.76 (m, 3H), 2.59 (heptet, IH), 2.49 (ddd, IH), 2.42 (ddd, IH), 2.01 (dq, IH), 1.81 (s, 3H), 1.76 (s, 3H), 1.65 (s, 3H), 1.35 (d, 3H), 1.29 (m, IH), 1.20 (t, 3H), 1.19 (d, 3 H), 1.17 (d, 3H), 1.16 (d, 3H), 1.14 (s, 3H), 1.12 (s, 3H), 0.87 (t, 3H); 13C NMR (100 MHz, CD3OD) δ 178.4, 169.7, 169.1, 154.6, 153.9, 146.2, 143.7, 141.9, 137.1, 137.0, 136.4, 134.6, 128.5, 126.9, 125.6, 124.6, 114.8, 112.8, 108.8, 102.3, 97.2, 94.3, 82.5, 78.6, 76.9, 75.9, 74.5, 73.5, 73.2, 72.8, 71.6, 70.5, 68.3, 63.9, 62.2, 42.5, 37.3, 35.4, 28.7, 28.3, 26.9, 26.4, 20.3, 19.6, 19.2, 18.7, 18.2, 17.6, 15.5, 14.6, 14.0, 11.4.
PATENT
http://www.google.com/patents/US7378508
macrolide of Formula I:

Structure of R-Tiacumicin B
The structure of the R-Tiacumicin B (the major most active component) is shown below in Formula I. The X-ray crystal structure of the R-Tiacumicin B was obtained as a colorless, parallelepiped-shaped crystal (0.08×0.14×0.22 mm) grown in aqueous methanol. This x-ray structure confirms the structure shown below. The official chemical name is 3-[[[6-Deoxy-4-O-(3,5-dichloro-2-ethyl-4,6-dihydroxybenzoyl)-2-O-methyl-β-D-mannopyranosyl]oxy]-methyl]-12(R)-[[6-deoxy-5-C-methyl-4-O-(2-methyl-1-oxopropyl)-β-D-lyxo-hexopyranosyl]oxy]-11(S)-ethyl-8(S)-hydroxy-18(S)-(1(R)-hydroxyethyl)-9,13,15-trimethyloxacyclooctadeca-3,5,9,13,15-pentaene-2-one.
7.2.1 Analytical Data of R-Tiacumicin B
The analytical data of R-Tiacumicin B (which is almost entirely (i.e., >90%) R-Tiacumicin).
mp 166-169° C. (white needle from isopropanol);
[α]D 20-6.9 (c 2.0, MeOH);
MS m/z (ESI) 1079.7(M+Na)+;
1H NMR (400 MHz, CD3OD) δ 7.21 (d, 1H), 6.59 (dd, 1H), 5.95 (ddd, 1H), 5.83 (br s, 1H), 5.57 (t, 1H), 5.13 (br d, 1H), 5.09 (t, 1H), 5.02 (d, 1H), 4.71 (m, 1H), 4.71 (br s, 1H), 4.64 (br s, 1H), 4.61 (d, 1H), 4.42 (d, 1H), 4.23 (m, 1H), 4.02 (pentet, 1H), 3.92 (dd, 1H), 3.73 (m, 2H), 3.70 (d, 1H), 3.56 (s, 3H), 3.52-3.56 (m, 2H), 2.92 (m, 2H), 2.64-2.76 (m, 3H), 2.59 (heptet, 1H), 2.49 (ddd, 1H), 2.42 (ddd, 1H), 2.01 (dq, 1H), 1.81 (s, 3H), 1.76 (s, 3H), 1.65 (s, 3H), 1.35 (d, 3H), 1.29 (m, 1H), 1.20 (t, 3H), 1.19 (d, 3H), 1.17 (d, 3H), 1.16 (d, 3 H), 1.14 (s, 3H), 1.12 (s, 3H), 0.87 (t, 3H);
13C NMR (100 MHz, CD3OD) δ 178.4, 169.7, 169.1, 154.6, 153.9, 146.2, 143.7, 141.9, 137.1, 137.0, 136.4, 134.6, 128.5, 126.9, 125.6, 124.6, 114.8, 112.8, 108.8, 102.3, 97.2, 94.3, 82.5, 78.6, 76.9, 75.9, 74.5, 73.5, 73.2, 72.8, 71.6, 70.5, 68.3, 63.9, 62.2, 42.5, 37.3, 35.4, 28.7, 28.3, 26.9, 26.4, 20.3, 19.6, 19.2, 18.7, 18.2, 17.6, 15.5, 14.6, 14.0, 11.4.
PATENT
WO2013170142
EXAMPLES
Example 1; General procedure for the preparation of crude Fidaxomycin
Fidaxomycin was prepared by:
i) culturing a microorganism in a nutrient medium to accumulate Fidaxomycin in the nutrient medium;
ii) isolating crude Fidaxomycin from the nutrient medium by methods known from the art;
iii) purifying Fidaxomycin by reversed phase chromatography using a mixture of acetonitrile, water and acetic acid as eluent; and iv) isolating the purified Fidaxomycin from the fractions.
Actionplanes deccanenesis was used during the cultivation. The nutrient medium comprises the following combination based on weight: from about 0% to about 5% Sucrose; from about 0% to about 3% Starch; from about 0.1% to about 1.0 % Soy peptone; from about 2% to about 5% Cotton seed meal; from about 0.01% to about 0.1% Potassium-dihydrogen Phosphate; from about 0.05% to about 0.5% Dipotassium-hydrogen Phosphate; from about 0.05% to about 0.5% Antifoam agent; from about 0% to about 2% Amberlite XAD-16N resin. The preferred temperature of the cultivation is from 28 to 32°C, and the pH is between 6.0 and 8.0. During the cultivation C-source is continuously fed.
The Fidaxomycin fermentation production can also be done by the following procedure:
The Fidaxomycin fermentation production can include a step of inoculation followed by fermentation as follows:
Inoculation: Actinoplanes deccanenesis strain is inoculated into the seed medium. The inoculation parameters are adjusted and maintained until the inoculum transferred to the main fermentation. The inoculum medium comprises: from about 0 to about 5% glucose, from about 0 to about 1% yeast extract, from about 0 to about 1% soy peptone, from about 0 to about 0.5% CaCo3, from about 0 to about 0.2% MgS0 -7H20, from about 0 to about 0.2% K2HP04, from about 0 to about 0.2% KC1, from about 0 to about 0.3% Polypropylene glycol. The pH is adjusted by adding Hydrochloric acid and/or Sodium/potassium hydroxide.
Inoculation parameters :
Inoculation time: 40-48 ± 24 hours.
At the end of the inoculation, the inoculum (or a part of it) is transferred into the sterile fermentation medium at a ratio of 8-15 ± 5 %.
Fermentation: the fermentation medium comprises: from about 0 to aboutl0% Sucrose/Hydrolyzed Starch, from about 0 to about 1% Soy peptone, from about 0 to about 5% Cotton seed meal, from about 0 to about 0.3% K2HP04, from about 0 to about 0.2% KH2P04, from about 0 to aboutl% KC1, from about 0 to about 0.5% Polypropylene glycol (PPG). The pH is adjusted by adding Hydrochloric acid and/or Sodium/potassium hydroxide.
The sterile fermentation medium is seeded with the inoculum.
Feeding:
C-source is fed during the fermentation, For C-source feeding sucrose or hydrolyzed-starch can be applied. Total amount of fed C-source is 0 – 15% related to the initial volume.
Fermentation parameters :
In case of foaming, sterile antifoaming agent should be added.
Fermentation time: 168-192 ± 24 hours.
The inoculation/fermentation medium may also include from about 0% to about 2% Amberlite XAD-16N resin.
Upon completion of fermentation, the Fidaxomycin is extracted from the fermented broth with an organic solvent such as, for example, ethyl acetate, isobutyl acetate or isobutanol. The organic phase is concentrated and the Fidaxomycin is precipitated by addition of an antisolvent such as, for example, n-hexane. Optionally the precipitate can be suspended in a second antisolvent. After filtration and drying, crude Fidaxomycin is obtained.
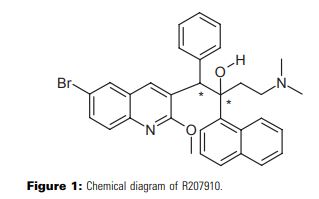
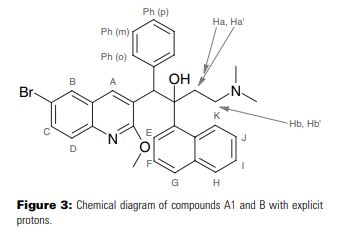
DIFICID (fidaxomicin) is a macrolide antibacterial drug for oral administration. Its CAS chemical name is Oxacyclooctadeca-3,5,9,13,15-pentaen-2-one, 3-[[[6-deoxy-4-O-(3,5-dichloro-2-ethyl-4,6-dihydroxybenzoyl)-2-Omethyl- β-D- mannopyranosyl]oxy]methyl]-12-[[6-deoxy-5-C-methyl-4-O-(2-methyl-1-oxopropyl)-β-D-lyxohexopyranosyl] oxy]-11-ethyl-8 -hydroxy-18-[(1R)-1-hydroxyethyl]-9,13,15-trimethyl-,(3E,5E,8S,9E,11S,12R,13E,15E,18S)-. The structural formula of fidaxomicin is shown in Figure 1.
Figure 1: Structural Formula of Fidaxomicin






Patent
WO 2016024243, New patent, Dr Reddy’s Laboratories Ltd, Fidaxomicin
WO2016024243, FIDAXOMICIN POLYMORPHS AND PROCESSES FOR THEIR PREPARATION
DR. REDDY’S LABORATORIES LIMITED [IN/IN]; 8-2-337, Road No. 3, Banjara Hills, Telangana State, India Hyderabad 500034 (IN)
CHENNURU, Ramanaiah; (IN).
PEDDY, Vishweshwar; (IN).
RAMAKRISHNAN, Srividya; (IN)
Aspects of the present application relate to crystalline forms of Fidaxomicin IV, V & VI and processes for their preparation. Further aspects relate to pharmaceutical compositions comprising these polymorphic forms of fidaxomicin

The occurrence of different crystal forms, i.e., polymorphism, is a property of some compounds. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physico-chemical properties.
Polymorphs are different solid materials having the same molecular structure but different molecular arrangement in the crystal lattice, yet having distinct physico-chemical properties when compared to other polymorphs of the same molecular structure. The discovery of new polymorphs and solvates of a pharmaceutical active compound provides an opportunity to improve the performance of a drug product in terms of its bioavailability or release profile in vivo, or it may have improved stability or advantageous handling properties. Polymorphism is an unpredictable property of any given compound. This subject has been reviewed in recent articles, including A. Goho, “Tricky Business,” Science News, August 21 , 2004. In general, one cannot predict whether there will be more than one form for a compound, how many forms will eventually be discovered, or how to prepare any previously unidentified form.
There remains a need for additional polymorphic forms of fidaxomicin and for processes to prepare polymorphic forms in an environmentally-friendly, cost-effective, and industrially applicable manner.

G.V. Prasad, chairman, Dr Reddy’s Laboratories
EXAMPLES
Example 1 : Preparation of fidaxomicin Form IV:
Fidaxomicin (0.5 g) and a mixture of 1 ,4-Dioxane (10 mL), THF (10 ml) and water (20mL) were charged in Easy max reactor (Mettler Toledo). The reactor was set to temperature cycle with following parameters:
Starting temperature: 25 °C;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 25 °C over a period of 2 hours;
Temperature maintained at 25 °C for 6 hours.
After completion of temperature cycling process, the slurry was filtered under suction, followed by drying in air tray dryer (ATD) at 40°C to a constant weight to produce crystalline fidaxomicin form-IV.
Example 2: Preparation of fidaxomicin Form V:
Fidaxomicin (1 g) and a mixture of propylene glycol (10 mL) and water (20mL) were charged in Easy max reactor (Mettler Toledo). The reactor was set to temperature cycle with following parameters:
Starting temperature is 25 °C;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 25 °C over a period of 2 hours;
Temperature maintained at 25 °C for 6 hours.
After completion of temperature cycling process, the slurry was filtered under suction, followed by drying in air tray dryer (ATD) at 40°C to a constant weight to produce crystalline fidaxomicin form-V.
Example 3: Preparation of fidaxomicin Form VI:
Fidaxomicin (0.5 mg) and MIBK (10 mL) were charged in Easy max reactor (Mettler Toledo) and the mixture was heated to 80°C. n-heptane (20 mL) was added to the solution at the same temperature. The mixture was stirred for 1 hour. The reaction mass was then cooled to 25°C. Solid formed was filtered at 25°C and dried at 40°C in air tray dryer (ATD) to a constant weight to produce crystalline fidaxomicin form VI.
Example 4: Preparation of fidaxomicin Form V:
Fidaxomicin (500 mg) and a mixture of R-propylene glycol (5 mL) and water (15 mL) were charged in Easy max reactor (Mettler Toledo). The reactor was set to temperature cycle with following parameters:
Starting temperature is 25 °C;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 25 °C over a period of 2 hours;
Temperature maintained at 25 °C for 2 hours.
After completion of temperature cycling process, the slurry was filtered and dried at 25°C to produce crystalline fidaxomicin form-V.
Example 5: Preparation of fidaxomicin Form V:
Fidaxomicin (1 g) and a mixture of S-propylene glycol (3 ml_) and water (30 mL) were charged in Easy max reactor (Mettler Toledo). The reactor was set to temperature cycle with following parameters:
Starting temperature is 25 °C;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 25 °C over a period of 2 hours;
Temperature maintained at 25 °C for 2 hours.
After completion of temperature cycling process, the slurry was filtered and dried at 25°C to produce crystalline fidaxomicin form-V.
Example 6: Preparation of fidaxomicin Form V:
Fidaxomicin (40 g) and a mixture of propylene glycol (400 mL) and water (1600 mL) were charged in Chem glass reactor. The reactor was set to temperature cycle with following parameters:
Starting temperature is 25 °C;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 25 °C over a period of 2 hours;
Temperature maintained at 25 °C for 6 hours.
After completion of temperature cycling process, the slurry was filtered under suction, followed by drying in air tray dryer (ATD) at 40°C to a constant weight to produce crystalline fidaxomicin form-V.

The 10-member board at pharmaceutical major Dr Reddy’s thrives on diversity. Liberally sprinkled with gray hairs, who are never quite impressed with powerpoint presentations, “they want information to be pre-loaded so that the following discussions (at the board level) are fruitful,” says Satish Reddy, Chairman, Dr Reddy’s. That said, the company has now equipped its board members with a customized application (that runs on their tablets) to manage board agenda and related processes.
see at
http://articles.economictimes.indiatimes.com/2014-10-31/news/55631761_1_board-members-board-agenda-dr-reddy-s

Dr. Reddy’s Laboratories Managing Director and Chief Operating Officer Satish Reddy addressing
References
- 1 “DIFICID” (PDF). TGA eBusiness Services. Specialised Therapeutics Australia Pty Ltd. 23 April 2013. Retrieved 31 March 2014.
- 2 Revill, P.; Serradell, N.; Bolós, J. (2006). “Tiacumicin B”. Drugs of the Future 31 (6): 494. doi:10.1358/dof.2006.031.06.1000709.
- 3″Dificid, Full Prescribing Information” (PDF). Optimer Pharmaceuticals. 2013.
- 4 “Fidaxomicin”. Drugs in R&D 10: 37. 2012. doi:10.2165/11537730-000000000-00000.
- 5Louie, T. J.; Emery, J.; Krulicki, W.; Byrne, B.; Mah, M. (2008). “OPT-80 Eliminates Clostridium difficile and is Sparing of Bacteroides Species during Treatment of C. Difficile Infection”. Antimicrobial Agents and Chemotherapy 53 (1): 261–3. doi:10.1128/AAC.01443-07. PMC 2612159. PMID 18955523.
- 6Johnson, Stuart (2009). “Recurrent Clostridium difficile infection: A review of risk factors, treatments, and outcomes”. Journal of Infection 58 (6): 403–10. doi:10.1016/j.jinf.2009.03.010. PMID 19394704.
- 7http://www.medicinescomplete.com/mc/bnf/current/PHP18388-dificlir.htm#PHP18388-dificlir
- 8Srivastava, Aashish; Talaue, Meliza; Liu, Shuang; Degen, David; Ebright, Richard Y; Sineva, Elena; Chakraborty, Anirban; Druzhinin, Sergey Y; Chatterjee, Sujoy; Mukhopadhyay, Jayanta; Ebright, Yon W; Zozula, Alex; Shen, Juan; Sengupta, Sonali; Niedfeldt, Rui Rong; Xin, Cai; Kaneko, Takushi; Irschik, Herbert; Jansen, Rolf; Donadio, Stefano; Connell, Nancy; Ebright, Richard H (2011). “New target for inhibition of bacterial RNA polymerase: ‘switch region'”. Current Opinion in Microbiology 14 (5): 532–43. doi:10.1016/j.mib.2011.07.030. PMC 3196380. PMID 21862392.
- 9″Optimer’s North American phase 3 Fidaxomicin study results presented at the 49th ICAAC” (Press release). Optimer Pharmaceuticals. September 16, 2009. Retrieved May 7, 2013.
- 10″Optimer Pharmaceuticals Presents Results From Fidaxomicin Phase 3 Study for the Treatment” (Press release). Optimer Pharmaceuticals. May 17, 2009. Retrieved May 7, 2013.
- 11Golan Y, Mullane KM, Miller MA (September 12–15, 2009). Low recurrence rate among patients with C. difficile infection treated with fidaxomicin. 49th interscience conference on antimicrobial agents and chemotherapy. San Francisco.
- 12Gorbach S, Weiss K, Sears P; et al. (September 12–15, 2009). Safety of fidaxomicin versus vancomycin in treatment of Clostridium difficile infection. 49th interscience conference on antimicrobial agents and chemotherapy. San Francisco.
- 13Louie, Thomas J.; Miller, Mark A.; Mullane, Kathleen M.; Weiss, Karl; Lentnek, Arnold; Golan, Yoav; Gorbach, Sherwood; Sears, Pamela; Shue, Youe-Kong; Opt-80-003 Clinical Study, Group (2011). “Fidaxomicin versus vancomycin for Clostridium difficile infection”. New England Journal of Medicine 364 (5): 422–31. doi:10.1056/NEJMoa0910812. PMID 21288078.
- 14Peterson, Molly (Apr 5, 2011). “Optimer wins FDA panel’s backing for antibiotic fidaxomicin”. Bloomberg.
- 15Nordqvist, Christian (27 May 2011). “Dificid (fidaxomicin) approved for Clostridium difficile-associated diarrhea”. Medical News Today.
ARNONE A ET AL: “STRUCTURE ELUCIDATION OF THE MACROCYCLIC ANTIBIOTIC LIPIARMYCIN“, JOURNAL OF THE CHEMICAL SOCIETY, PERKIN TRANSACTIONS 1, CHEMICAL SOCIETY, LETCHWORTH; GB, 1 January 1987 (1987-01-01), pages 1353-1359, XP000578201, ISSN: 0300-922X, DOI: 10.1039/P19870001353
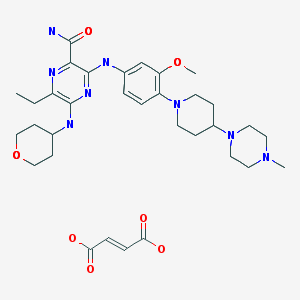
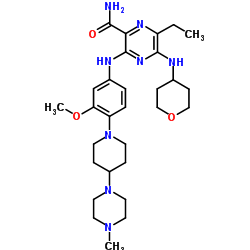
Fidaxomicin
 |
| Systematic (IUPAC) name |
|
3-(((6-Deoxy-4-O-(3,5-dichloro-2-ethyl-4,6-dihydroxybenzoyl)-2-O-methyl-β-D-mannopyranosyl)oxy)-methyl)-12(R)-[(6-deoxy-5-C-methyl-4-O-(2-methyl-1-oxopropyl)-β-D-lyxo-hexopyranosyl)oxy]-11(S)-ethyl-8(S)-hydroxy-18(S)-(1(R)-hydroxyethyl)-9,13,15-trimethyloxacyclooctadeca-3,5,9,13,15-pentaene-2-one
|
| Clinical data |
| Trade names |
Dificid, Dificlir |
| Licence data |
US FDA:link |
Pregnancy
category |
- AU: B1
- US: B (No risk in non-human studies)
|
| Legal status |
|
Routes of
administration |
Oral |
| Pharmacokinetic data |
| Bioavailability |
Minimal systemic absorption[1] |
| Biological half-life |
11.7 ± 4.80 hours[1] |
| Excretion |
Urine (<1%), faeces (92%)[1] |
| Identifiers |
| CAS Number |
873857-62-6  |
| ATC code |
A07AA12 |
| PubChem |
CID 11528171 |
| ChemSpider |
8209640  |
| UNII |
Z5N076G8YQ |
| KEGG |
D09394 |
| ChEBI |
CHEBI:68590 |
| ChEMBL |
CHEMBL1255800 |
| Synonyms |
Clostomicin B1, lipiarmicin, lipiarmycin, lipiarmycin A3, OPT 80, PAR 01, PAR 101, tiacumicin B |
| Chemical data |
| Formula |
C52H74Cl2O18 |
| Molar mass |
1058.04 g/mol |
| US4918174 |
26 Sep 1986 |
17 Apr 1990 |
Abbott Laboratories |
Tiacumicin compounds |
| WO2009025439A1 * |
6 May 2008 |
26 Feb 2009 |
Genotech Co Ltd |
Method of extraction and yield-up of tricyclo compounds by adding a solid adsorbent resin as their carrier in fermentation medium |
| WO2014023616A1 * |
30 Jul 2013 |
13 Feb 2014 |
Olon Spa |
Procedure for the production of tiacumicin b |
| WO2014111254A1 |
14 Jan 2014 |
24 Jul 2014 |
Astellas Pharma Europe Ltd |
Composition of tiacumicin compounds |
| WO2015091851A1 |
18 Dec 2014 |
25 Jun 2015 |
Xellia Pharmaceuticals Aps |
Process for the preparation of tiacumicin |
| WO2015169451A1 |
11 May 2015 |
12 Nov 2015 |
Astellas Pharma Europe Ltd |
Treatment regimen tiacumicin compound |
| CN101128114B |
31 Jan 2005 |
28 Mar 2012 |
浩鼎生技公司 |
18-membered macrocycles and analogs thereof |
| CN102614207B * |
31 Jan 2005 |
13 Jan 2016 |
默克夏普&多梅有限公司 |
18元环大环化合物及其类似物 |
| EP1848273A1 * |
31 Jan 2005 |
31 Oct 2007 |
Optimer Pharmaceuticals, Inc. |
18-membered macrocycles and analogs thereof |
| EP2070530A1 |
13 May 2005 |
17 Jun 2009 |
Optimer Pharmaceuticals, Inc. |
Treatment of diseases associated with the use of antibiotics |
| EP2125850A1 † |
22 Jan 2008 |
2 Dec 2009 |
Optimer Pharmaceuticals, Inc. |
Macrocyclic polymorphs, compositions comprising such polymorphs, and methods of use and manufacture thereof |
| EP2305244A1 |
13 May 2005 |
6 Apr 2011 |
Optimer Pharmaceuticals, Inc. |
Treatment of diseases associated with the use of antibiotics |
| EP2305245A1 |
13 May 2005 |
6 Apr 2011 |
Optimer Pharmaceuticals, Inc. |
Treatment of diseases associated with the use of antibiotics |
| EP2468761A1 |
22 Jan 2008 |
27 Jun 2012 |
Optimer Pharmaceuticals, Inc. |
Macrocyclic polymorphs, compositions comprising such polymorphs, and methods of use and manufacture thereof |
| US7378508 |
31 Jul 2007 |
27 May 2008 |
Optimer Pharmaceuticals, Inc. |
Polymorphic crystalline forms of tiacumicin B |
| US7863249 |
11 Apr 2008 |
4 Jan 2011 |
Optimer Pharmaceuticals, Inc. |
Macrolide polymorphs, compositions comprising such polymorphs, and methods of use and manufacture thereof |
| US7906489 |
31 Jul 2007 |
15 Mar 2011 |
Optimer Pharmaceuticals, Inc. |
18-membered macrocycles and analogs thereof |
| US8044030 |
28 Nov 2008 |
25 Oct 2011 |
Optimer Pharmaceuticals, Inc. |
Antibiotic macrocycle compounds and methods of manufacture and use thereof |
| US8586551 |
31 Aug 2009 |
19 Nov 2013 |
Optimer Pharmaceuticals, Inc. |
18-membered macrocycles and analogs thereof |
| US8859510 |
22 Jan 2008 |
14 Oct 2014 |
Optimer Pharmaceuticals, Inc. |
Macrocyclic polymorphs, compositions comprising such polymorphs, and methods of use and manufacture thereof |
| US8883986 |
4 Mar 2009 |
11 Nov 2014 |
Optimer Pharmaceuticals, Inc. |
Macrolide polymorphs, compositions comprising such polymorphs, and methods of use and manufacture thereof |
| US8916527 |
15 Mar 2013 |
23 Dec 2014 |
Optimer Pharmaceuticals, Inc. |
Antibiotic macrocycle compounds and methods of manufacture and use thereof |
| US20110166090 * |
|
7 Jul 2011 |
Youe-Kong Shue |
18-Membered Macrocycles and Analogs Thereof |
| US20140107054 * |
21 Dec 2012 |
17 Apr 2014 |
Optimer Pharmaceuticals, Inc. |
Method of treating clostridium difficile-associated diarrhea |
| US3978211 * |
Oct 31, 1974 |
Aug 31, 1976 |
Gruppo Lepetit S.P.A. |
Lipiarmycin and its preparation |
| US4918174 |
Sep 26, 1986 |
Apr 17, 1990 |
Abbott Laboratories |
Tiacumicin compounds |
| US5583115 |
May 9, 1995 |
Dec 10, 1996 |
Abbott Laboratories |
Dialkyltiacumicin compounds |
| US5767096 |
Jul 12, 1996 |
Jun 16, 1998 |
Abbott Laboratories |
Bromotiacumicin compounds |
| US20060257981 * |
Jul 15, 2003 |
Nov 16, 2006 |
Optimer Pharmaceuticals, Inc. |
Tiacumicin production |
| US20070173462 * |
May 13, 2005 |
Jul 26, 2007 |
Optimer Pharmaceuticals, Inc. |
Treatment of diseases associated with the use of antibiotics |
| WO2004014295A2 |
Jul 15, 2003 |
Feb 19, 2004 |
Optimer Pharmaceuticals Inc |
Tiacumicin production |
| WO2005112990A2 |
May 13, 2005 |
Dec 1, 2005 |
Optimer Pharmaceuticals Inc |
Treatment of diseases associated with the use of antibiotics |
| WO2006085838A1 * |
Jan 31, 2005 |
Aug 17, 2006 |
Optimer Pharmaceuticals Inc |
18-membered macrocycles and analogs thereof |
| DE2455230A1 * |
Nov 21, 1974 |
May 28, 1975 |
Lepetit Spa |
Lipiarmycin, verfahren zu seiner herstellung, mikroorganismus zur durchfuehrung des verfahrens und arzneimittel |
| EP2125850A1 |
Jan 22, 2008 |
Dec 2, 2009 |
Optimer Pharmaceuticals, Inc. |
Macrocyclic polymorphs, compositions comprising such polymorphs, and methods of use and manufacture thereof |
| US7378508 |
Jul 31, 2007 |
May 27, 2008 |
Optimer Pharmaceuticals, Inc. |
Polymorphic crystalline forms of tiacumicin B |
| Braga et al., “Making crystals from crystals: a green route tocrystal engineering and polymorphism” Chemical Communications (2005) pp. 3635-3645. |
| 2 |
* |
Chemical Abstracts registry entry 56645-60-4, Tiacumicin B, Copyright 2007, American Chemical Society, p. 1-2. |
| 3 |
* |
Dean, J., Analytical Chemistry Handbook, Published bt McGraw-Hill, Inc., pp. 10.23-10.26. |
| 4 |
|
J.E. Hochlowski et al., Tiacumicins, A Novel Complex of 18-Membered Macrolides, J. Antibiotics, vol. XL, No. 5, pp. 575-588 (May 1987). |
| 5 |
* |
Jain et al., “Polymorphism in Pharmacy” Indian Drugs (1986) vol. 23, No. 6, pp. 315-329. |
| 6 |
* |
Pharmaceutical Dosage Forms: Tablets, vol. 2, Published by Marcel Dekker, Inc., ed. by Lieberman, Lachman, and Schwartz, pp. 462-472. |
| 7 |
* |
Polymorphism in Pharmaceutical Solids, published 1999 by Marcel Dekker Inc, ed. by Harry G. Brittain, pp. 1-2. |
| 8 |
|
Robert N. Swanson et al., In Vitro and In Vivo Evaluation of Tiacumicins B and C against Clostridium difficile, Antimicrob. Agents Chemother., Jun. 1991, pp. 1108-1111. |
| 9 |
* |
The Condensed Chemical Dictionary, Tenth Edition, published 1981 by the Van Nostrand Reinhold Company, revised by Gessner G. Hawley, p. 35 and 835. |

///////////Fidaxomicin, OPT-80, PAR-101, japan 2018
CC[C@H]1/C=C(/[C@H](C/C=C/C=C(/C(=O)O[C@@H](C/C=C(/C=C(/[C@@H]1O[C@H]2[C@H]([C@H]([C@@H](C(O2)(C)C)OC(=O)C(C)C)O)O)\C)\C)[C@@H](C)O)\CO[C@H]3[C@H]([C@H]([C@@H]([C@H](O3)C)OC(=O)C4=C(C(=C(C(=C4O)Cl)O)Cl)CC)O)OC)O)\C
Fidaxomicin
-
- Synonyms:OPT-80; PAR-101; Tiacumicin B
- ATC:A07AA12
- Use:macrocyclic, antibotic, RNA polymerase inhibitor
- Chemical name:3-(((6-Deoxy-4-O-(3,5-dichloro-2-ethyl-4,6-dihydroxybenzoyl)-2-O-methyl-β-D-mannopyranosyl)oxy)-methyl)-12(R)-[(6-deoxy-5-C-methyl-4-O-(2-methyl-1-oxopropyl)-β-D-lyxo-hexopyranosyl)oxy]-11(S)-ethyl-8(S)-hydroxy-18(S)-(1(R)-hydroxyethyl)-9,13,15-trimethyloxacyclooctadeca-3,5,9,13,15-pentaene-2-one
- Formula:C52H74Cl2O8
- MW:1058.0 g/mol
- CAS-RN:873857-62-6
Synthesis Path

Trade Names
| Country |
Trade Name |
Vendor |
Annotation |
| USA |
Dificid |
Optimer Pharmceuticals, 2011 |
|
References
-
- WO 2004 014295 (Optimer Pharmaceuticals; 19.2.2004; USA-prior. 29.7.2002).
- US 7 507 564 (Optimer Pharmaceuticals; 24.3.2009; USA-prior. 29.7.2002).
- US 7 378 508 (Optimer Pharmaceuticals; 27.5.2008; USA-prior. 22.7.2007).
- US 3 978 211 (Gruppo Lepetit; 31.10.1974; GB-prior. 22.11.1973).
- US 4 918 174 (Abbott Laboratories; 17.10.1990; USA-prior. 26.9.1986).
- EP 923 594 (Abbott Laboratories; 2.10.2002; USA-prior. 12.7.1996).

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....