







Home » Drug discovery
Category Archives: Drug discovery
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Sreeni Labs Private Limited, Hyderabad, India ready to deliver New, Economical, Scalable Routes to your advanced intermediates & API’s in early Clinical Drug Development Stages

Sreeni Labs Private Limited, Hyderabad, India is ready to take up challenging synthesis projects from your preclinical and clinical development and supply from few grams to multi-kilo quantities. Sreeni Labs has proven route scouting ability to design and develop innovative, cost effective, scalable routes by using readily available and inexpensive starting materials. The selected route will be further developed into a robust process and demonstrate on kilo gram scale and produce 100’s of kilos of in a relatively short time.
Accelerate your early development at competitive price by taking your route selection, process development and material supply challenges (gram scale to kilogram scale) to Sreeni Labs…………
INTRODUCTION
Sreeni Labs based in Hyderabad, India is working with various global customers and solving variety of challenging synthesis problems. Their customer base ranges from USA, Canada, India and Europe. Sreeni labs Managing Director, Dr. Sreenivasa Reddy Mundla has worked at Procter & Gamble Pharmaceuticals and Eli Lilly based in USA.
The main strength of Sreeni Labs is in the design, development of innovative and highly economical synthetic routes and development of a selected route into a robust process followed by production of quality product from 100 grams to 100s of kg scale. Sreeni Labs main motto is adding value in everything they do.
They have helped number of customers from virtual biotech, big pharma, specialty chemicals, catalog companies, and academic researchers and drug developers, solar energy researchers at universities and institutions by successfully developing highly economical and simple chemistry routes to number of products that were made either by very lengthy synthetic routes or by using highly dangerous reagents and Suzuki coupling steps. They are able to supply materials from gram scale to multi kilo scale in a relatively short time by developing very short and efficient synthetic routes to a number of advanced intermediates, specialty chemicals, APIs and reference compounds. They also helped customers by drastically reducing number of steps, telescoping few steps into a single pot. For some projects, Sreeni Labs was able to develop simple chemistry and avoided use of palladium & expensive ligands. They always begin the project with end in the mind and design simple chemistry and also use readily available or easy to prepare starting materials in their design of synthetic routes
Over the years, Sreeni labs has successfully made a variety of products ranging from few mg to several kilogram scale. Sreeni labs has plenty of experience in making small select libraries of compounds, carbocyclic compounds like complex terpenoids, retinal derivatives, alkaloids, and heterocyclic compounds like multi substituted beta carbolines, pyridines, quinolines, quinolones, imidazoles, aminoimidazoles, quinoxalines, indoles, benzimidazoles, thiazoles, oxazoles, isoxazoles, carbazoles, benzothiazoles, azapines, benzazpines, natural and unnatural aminoacids, tetrapeptides, substituted oligomers of thiophenes and fused thiophenes, RAFT reagents, isocyanates, variety of ligands, heteroaryl, biaryl, triaryl compounds, process impurities and metabolites.
Sreeni Labs is Looking for any potential opportunities where people need development of cost effective scalable routes followed by quick scale up to produce quality products in the pharmaceutical & specialty chemicals area. They can also take up custom synthesis and scale up of medchem analogues and building blocks. They have flexible business model that will be in sink with customers. One can test their abilities & capabilities by giving couple of PO based (fee for service) projects.
Some of the compounds prepared by Sreeni labs;
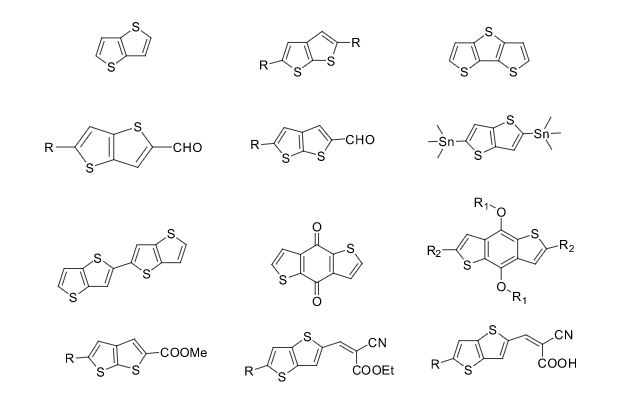
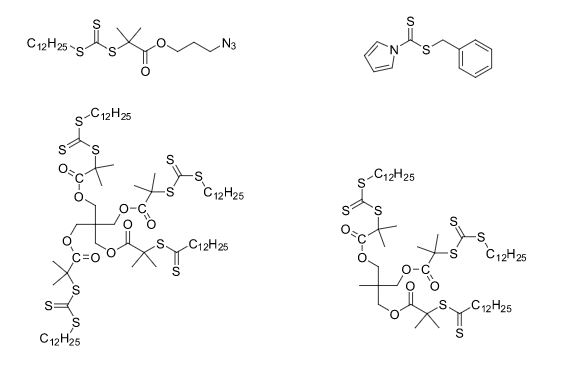



See presentation below
LINK ON SLIDESHARE
Sreeni Labs Profile from Sreenivasa Reddy
Managing Director at Sreeni Labs Private Limited\
Few Case Studies : Source SEEENI LABS
QUOTE………….
One virtual biotech company customer from USA, through a common friend approached Sreeni Labs and told that they are buying a tetrapeptide from Bachem on mg scale at a very high price and requested us to see if we can make 5g. We accepted the challenge and developed solution phase chemistry and delivered 6g and also the process procedures in 10 weeks time. The customer told that they are using same procedures with very minor modifications and produced the tetrapeptide ip to 100kg scale as the molecule is in Phase III.
One East coast customer in our first meeting told that they are working with 4 CROs of which two are in India and two are in China and politely asked why they should work with Sreeni Labs. We told that give us a project where your CROs failed to deliver and we will give a quote and work on it. You pay us only if we deliver and you satisfy with the data. They immediately gave us a project to make 1.5g and we delivered 2g product in 9 weeks. After receiving product and the data, the customer was extremely happy as their previous CRO couldn’t deliver even a milligram in four months with 3 FTEs.
One Midwest biotech company was struggling to remove palladium from final API as they were doing a Suzuki coupling with a very expensive aryl pinacol borane and bromo pyridine derivative with an expensive ligand and relatively large amount of palldium acetate. The cost of final step catalyst, ligand and the palladium scavenging resin were making the project not viable even though the product is generating excellent data in the clinic. At this point we signed an FTE agreement with them and in four months time, we were able to design and develop a non suzuki route based on acid base chemistry and made 15g of API and compared the analytical data and purity with the Suzuki route API. This solved all three problems and the customer was very pleased with the outcome.
One big pharma customer from east coast, wrote a structure of chemical intermediate on a paper napkin in our first meeting and asked us to see if we can make it. We told that we can make it and in less than 3 weeks time we made a gram sample and shared the analytical data. The customer was very pleased and asked us to make 500g. We delivered in 4 weeks and in the next three months we supplied 25kg of the same product.
Through a common friend reference, a European customer from a an academic institute, sent us an email requesting us to quote for 20mg of a compound with compound number mentioned in J. med. chem. paper. It is a polycyclic compound with four contiguous stereogenic centers. We gave a quote and delivered 35 mg of product with full analytical data which was more pure than the published in literature. Later on we made 8g and 6g of the same product.
One West coast customer approached us through a common friend’s reference and told that they need to improve the chemistry of an advanced intermediate for their next campaign. At that time they are planning to make 15kg of that intermediate and purchased 50kg of starting raw material for $250,000. They also put five FTEs at a CRO for 5 months to optimize the remaining 5 steps wherein they are using LAH, Sodium azide, palladium catalyst and a column chromatography. We requested the customer not to purchase the 50kg raw material, and offered that we will make the 15kg for the price of raw material through a new route in less than three months time. You pay us only after we deliver 15 kg material. The customer didn’t want to take a chance with their timeline as they didn’t work with us before but requested us to develop the chemistry. In 7 weeks time, we developed a very simple four step route for their advanced intermediate and made 50g. We used very inexpensive and readily available starting material. Our route gave three solid intermediates and completely eliminated chromatographic purifications.
One of my former colleague introduced an academic group in midwest and brought us a medchem project requiring synthesis of 65 challenging polyene compounds on 100mg scale. We designed synthetic routes and successfully prepared 60 compounds in a 15 month time.
UNQUOTE…………
The man behind Seeni labs is Dr. Sreenivasa Reddy Mundla

Dr. Sreenivasa Reddy Mundla.
Managing Director at Sreeni Labs Private Limited
Sreeni Labs Private Limited
Road No:12, Plot No:24,25,26
- IDA, Nacharam
Hyderabad, 500076
Telangana State, India
Links
LINKEDIN https://in.linkedin.com/in/sreenivasa-reddy-10b5876
FACEBOOK https://www.facebook.com/sreenivasa.mundla
RESEARCHGATE https://www.researchgate.net/profile/Sreenivasa_Mundla/info
EMAIL mundlasr@hotmail.com, Info@sreenilabs.com, Sreeni@sreenilabs.com
Dr. Sreenivasa Reddy Mundla

Dr. M. Sreenivasa Reddy obtained Ph.D from University of Hyderabad under the direction Prof Professor Goverdhan Mehta in 1992. From 1992-1994, he was a post doctoral fellow at University of Wisconsin in Professor Jame Cook’s lab. From 1994 to 2000, worked at Chemical process R&D at Procter & Gamble Pharmaceuticals (P&G). From 2001 to 2007 worked at Global Chemical Process R&D at Eli Lilly and Company in Indianapolis.
In 2007 resigned to his job and founded Sreeni Labs based in Hyderabad, Telangana, India and started working with various global customers and solving various challenging synthesis problems.
The main strength of Sreeni Labs is in the design, development of a novel chemical route and its development into a robust process followed by production of quality product from 100 grams to 100’s of kg scale.
They have helped number of customers by successfully developing highly economical simple chemistry routes to number of products that were made by Suzuki coupling. they are able to shorten the route by drastically reducing number of steps, avoiding use of palladium & expensive ligands. they always use readily available or easy to prepare starting materials in their design of synthetic routes.
Sreeni Labs is Looking for any potential opportunities where people need development of cost effective scalable routes followed by quick scale up to produce quality products in the pharmaceutical & specialty chemicals area. They have flexible business model that will be in sink with customers. One can test their abilities & capabilities by giving PO based projects
Experience
Founder & Managing Director
Sreeni Labs Private Limited
August 2007 – Present (8 years 11 months)
Sreeni Labs Profile
Principal Research Scientist
Eli Lilly and Company
March 2001 – August 2007 (6 years 6 months)
Senior Research Scientist
Procter & Gamble
July 1994 – February 2001 (6 years 8 months)
Education
University of Hyderabad
Doctor of Philosophy (Ph.D.),
1986 – 1992

With Sreenivasa Mundla, Narahara sastry, Ram Kishan Rao, Jagadeesh Bharatam, Jagadish Gunjur and Jagadish Bharatham.
PUBLICATIONS
Jianye Zhang · Zhiqian Dong · Sreenivasa Reddy Mundla · X Eric Hu · William Seibel ·Ruben Papoian · Krzysztof Palczewski · Marcin Golczak
Article: ChemInform Abstract: Regioselective Synthesis of 4Halo ortho-Dinitrobenzene Derivative
Aug 2010 · ChemInform
Hong-yu Li · William T. McMillen · Charles R. Heap · Denis J. McCann · Lei Yan · Robert M. Campbell · Sreenivasa R. Mundla · Chi-Hsin R. King · Elizabeth A. Dierks · Bryan D. Anderson · Karen S. Britt · Karen L. Huss
Apr 2008 · Journal of Medicinal Chemistry
Hong-yu Li · Yan Wang · William T. McMillen · Arindam Chatterjee · John E. Toth ·Sreenivasa R. Mundla · Matthew Voss · Robert D. Boyer · J. Scott Sawyer
Feb 2008 · ChemInform
Hong-yu Li · Yan Wang · William T. McMillen · Arindam Chatterjee · John E. Toth ·Sreenivasa R. Mundla · Matthew Voss · Robert D. Boyer · J. Scott Sawyer
Nov 2007 · Tetrahedron
Hong-yu Li · Yan Wang · Charles R Heap · Chi-Hsin R King · Sreenivasa R Mundla · Matthew Voss · David K Clawson · Lei Yan · Robert M Campbell · Bryan D Anderson · Jill R Wagner ·Karen Britt · Ku X Lu · William T McMillen · Jonathan M Yingling
Apr 2006 · Journal of Medicinal Chemistry

Hui Cao · Sreenivasa R. Mundla · James M. Cook
Aug 2003 · Tetrahedron Letters
Article: ChemInform Abstract: A New Method for the Synthesis of 2,6-Dinitro and 2Halo6-nitrostyrenes
Nov 2000 · ChemInform
Article: ChemInform Abstract: A Novel Method for the Efficient Synthesis of 2-Arylamino-2-imidazolines
-
Nov 2000 · ChemInformAug 2000 · Tetrahedron LettersAug 2000 · Tetrahedron Letters


TGF-β inhibitors
The present invention provides 2-(6-methyl-pyridin-2-yl)-3-[6-amido-quinolin-4-yl) -5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole monohydrate, i.e., Formula I.

EXAMPLE 1 Preparation of 2-(6-methyl-pyridin-2-yl)-3-[6-amido-quinolin-4-yl-5,6-dihydro-4H -pyrrolo[1,2-b]pyrazole monohydrate

Galunisertib
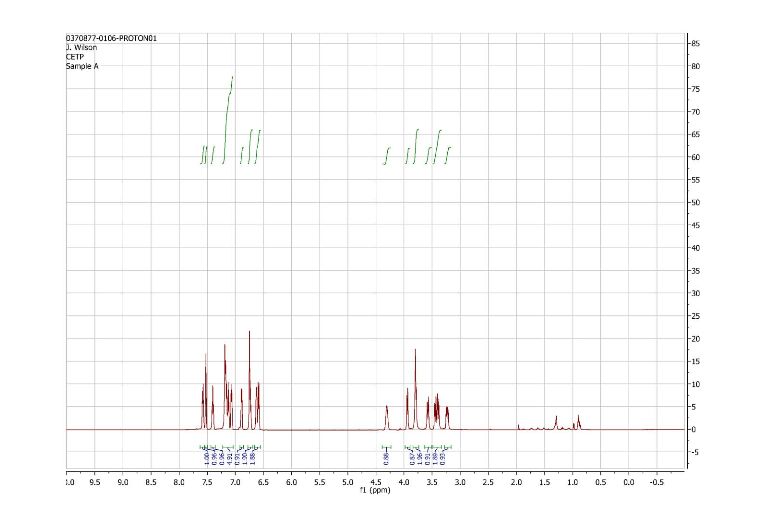
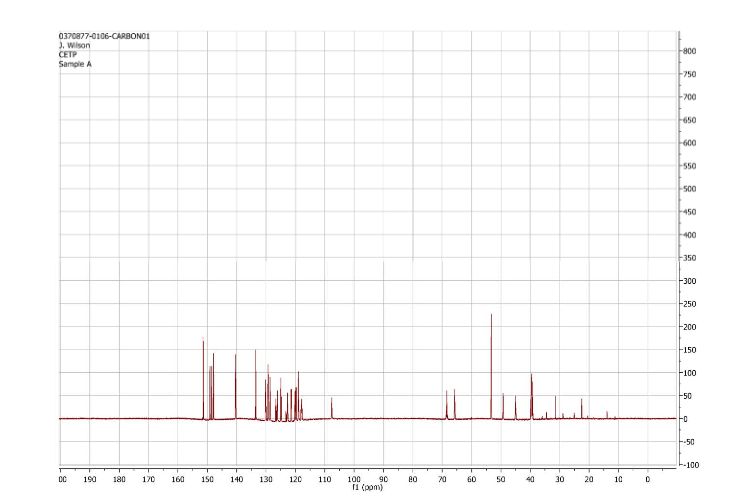
1H NMR (CDCl3): δ=9.0 ppm (d, 4.4 Hz, 1H); 8.23-8.19 ppm (m, 2H); 8.315 ppm (dd, 1.9 Hz, 8.9 Hz, 1H); 7.455 ppm (d, 4.4 Hz, 1H); 7.364 ppm (t, 7.7 Hz, 1H); 7.086 ppm (d, 8.0 Hz, 1H); 6.969 ppm (d, 7.7 Hz, 1H); 6.022 ppm (m, 1H); 5.497 ppm (m, 1H); 4.419 ppm (t, 7.3 Hz, 2H); 2.999 ppm (m, 2H); 2.770 ppm (p, 7.2 Hz, 7.4 Hz, 2H); 2.306 ppm (s, 3H); 1.817 ppm (m, 2H). MS ES+: 370.2; Exact: 369.16

ABOVE MOLECULE IS
https://newdrugapprovals.org/2016/05/04/galunisertib/
Galunisertib
Phase III
A TGF-beta receptor type-1 inhibitor potentially for the treatment of myelodysplastic syndrome (MDS) and solid tumours.
LY-2157299
CAS No.700874-72-2
READ MY PRESENTATION ON
KEYWORDS Sreenivasa Mundla Reddy, Managing Director, Sreeni Labs Private Limited, Hyderabad, Telangana, India, new, economical, scalable routes, early clinical drug development stages, Custom synthesis, custom manufacturing, drug discovery, PHASE 1, PHASE 2, PHASE 3, API, drugs, medicines
Drug Discovery, Hit to Lead
//////
Merck’s Novel Indoline Cholesterol Ester Transfer Protein Inhibitors (CETP)
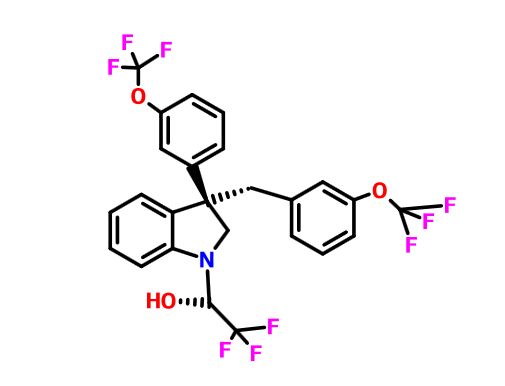
Indoline 7 as in ACS MEDCHEM LETTERS, DOI: 10.1021/acsmedchemlett.5b00404
and
eg 10 as in WO2015054088
(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethoxy)-phenyl)indolin-l-yl)propan-2-ol.
1H-Indole-1-ethanol, 2,3-dihydro-3-[3-(trifluoromethoxy)phenyl]-3-[[3-(trifluoromethoxy)phenyl]methyl]-α-(trifluoromethyl)-, (αR)-
cas 1699732-96-1 R ISOMER
MF C26 H20 F9 N O3, MW 565.43
Merck Sharp & Dohme Corp. INNOVATOR

Using the collective body of known (CETP) inhibitors as inspiration for design, a structurally novel series of tetrahydroquinoxaline CETP inhibitors were discovered. An exemplar from this series, compound 5, displayed potent in vitro CETP inhibition and was efficacious in a transgenic cynomologus-CETP mouse HDL PD (pharmacodynamic) assay. However, an undesirable metabolic profile and chemical instability hampered further development of the series. A three-dimensional structure of tetrahydroquinoxaline inhibitor 6 was proposed from 1H NMR structural studies, and this model was then used in silico for the design of a new class of compounds based upon an indoline scaffold. This work resulted in the discovery of compound 7, which displayed potent in vitro CETP inhibition, a favorable PK–PD profile relative to tetrahydroquinoxaline 5, and dose-dependent efficacy in the transgenic cynomologus-CETP mouse HDL PD assay.
chemical compounds that inhibit cholesterol ester transfer protein (CETP) and are expected to have utility in raising HDL-C, lowering LDL-C, and in the treatment and prevention of atherosclerosis.
see………….http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.5b00404
http://pubs.acs.org/doi/suppl/10.1021/acsmedchemlett.5b00404/suppl_file/ml5b00404_si_001.pdf
Discovery of Novel Indoline Cholesterol Ester Transfer Protein Inhibitors (CETP) through a Structure-Guided Approach
†Department of Medicinal Chemistry and ‡Department of Structural Chemistry, Merck Research Laboratories, Merck & Co, Inc., P.O. Box 2000, Rahway, New Jersey 07065, United States
§Department of Pharmacology, ∥Department of Drug Metabolism and Pharmacokinetics, and ⊥Department of Biology, Merck Research Laboratories, Merck & Co, Inc., P.O. Box 2000, Kenilworth, New Jersey 07033, United States
ACS Med. Chem. Lett., Article ASAP
DOI: 10.1021/acsmedchemlett.5b00404
Publication Date (Web): January 4, 2016
Copyright © 2016 American Chemical Society
*E-mail: jonathan_wilson@merck.com.
PATENT
Atherosclerosis and its clinical consequences, including coronary heart disease
(CHD), stroke and peripheral vascular disease, represent a truly enormous burden to the health care systems of the industrialized world. In the United States alone, approximately 13 million patients have been diagnosed with CHD, and greater than one half million deaths are attributed to CHD each year. Further, this toll is expected to grow over the next quarter century as an epidemic in obesity and diabetes continues to grow.
It has long been recognized that in mammals, variations in circulating lipoprotein profiles correlate with the risk of atherosclerosis and CHD. The clinical success of HMG-CoA reductase inhibitors, especially the statins, in reducing coronary events is based on the reduction of circulating low density lipoprotein cholesterol (LDL-C), levels of which correlate directly with an increased risk for atherosclerosis. More recently, epidemiologic studies have
demonstrated an inverse relationship between high density lipoprotein cholesterol (HDL-C) levels and atherosclerosis, leading to the conclusion that low serum HDL-C levels are associated with an increased risk for CHD.
Metabolic control of lipoprotein levels is a complex and dynamic process involving many factors. One important metabolic control in man is the cholesteryl ester transfer protein (CETP), a plasma glycoprotein that catalyzes the movement of cholesteryl esters from HDL to the apoB containing lipoproteins, especially VLDL (see Hesler, C.B., et. al. (1987) Purification and characterization of human plasma cholesteryl ester transfer protein. J. Biol. Chem. 262(5), 2275-2282)). Under physiological conditions, the net reaction is a heteroexchange in which CETP carries triglyceride to HDL from the apoB lipoprotein and transports cholesterol ester from HDL to the apoB lipoprotein.
In humans, CETP plays a role in reverse cholesterol transport, the process whereby cholesterol is returned to the liver from peripheral tissues. Intriguingly, many animals do not possess CETP, including animals that have high HDL levels and are known to be resistant to coronary heart disease, such as rodents (see Guyard-Dangremont, V., et. al, (1998)
Phospholipid and cholesteryl ester transfer activities in plasma from 14 vertebrate species. Relation to atherogenesis susceptibility, Comp. Biochem. Physiol. B Biochem. Mol. Biol. 120(3), 517-525). Numerous epidemiologic studies correlating the effects of natural variation in CETP activity with respect to coronary heart disease risk have been performed, including studies on a small number of known human null mutations (see Hirano, K.-L, Yamashita, S. and Matsuzawa, Y. (2000) Pros and cons of inhibiting cholesteryl ester transfer protein, Curr. Opin. Lipidol. 11(6), 589-596). These studies have clearly demonstrated an inverse correlation between plasma HDL-C concentration and CETP activity (see Inazu, A., et. al. (2000) Cholesteryl ester transfer protein and atherosclerosis, Curr. Opin. Lipidol. 11(4), 389-396), leading to the hypothesis that pharmacologic inhibition of CETP lipid transfer activity may be beneficial to humans by increasing levels of HDL-C while lowering LDL-C.
Despite the significant therapeutic advance that statins such as simvastatin and atorvastatin represent, statins only achieve a risk reduction of approximately one-third in the treatment and prevention of atherosclerosis and ensuing atherosclerotic disease events.
Currently, few pharmacologic therapies are available that favorably raise circulating levels of HDL-C. Certain statins and some fibrates offer modest HDL-C gains. Niacin provides an effective therapy for raising HDL-C but suffers from patient compliance issues, due in part to side effects such as flushing. Drugs that inhibit CETP (CETP inhibitors) have been under development with the expectation that they will effectively raise HDL cholesterol levels and also reduce the incidence of atherosclerosis in patients. Torcetrapib was the first drug that was tested in a long-term outcomes clinical trial. The clinical trial of torcetrapib was terminated early due to a higher incidence of mortality in patients to whom torcetrapib and atorvastatin were administered concomitantly compared with patients who were treated with atorvastatin alone. The cause of the increased mortality is not completely understood, but it is not believed to be associated with the CETP inhibiting effects of the drug.
Two other drug candidates, dalcetrapib and anacetrapib, are currently being tested in Phase III clinical trials, including large scale outcomes trials. Data from the recently completed DEFINE Phase III trial of anacetrapib are promising. Patients who were being treated with anacetrapib along with baseline statin therapy showed an increase of HDL-C of 138% and a decrease of LDL-C of 40%> compared with patients who were treated with just a statin. See: N. Engl. J. Med. 2010: 363: 2406-15. The data in the DEFINE trial were sufficient to indicate that an increase in mortality for patients treated with anacetrapib is unlikely. Additional drug candidates are still being sought that may have properties that are advantageous compared with the CETP inhibitors that have so far been studied or are currently being studied. Such properties may include, for example, higher potency, reduced off-target activity, better pharmacodynamics, higher bioavailability, or a reduced food effect compared with many of the highly lipophilic compounds that have so far been studied. “Food effect” refers to the variability in exposure to the active drug that occurs depending on when the patient had last eaten, whether or not the drug is administered with food, and the fat content of the food.
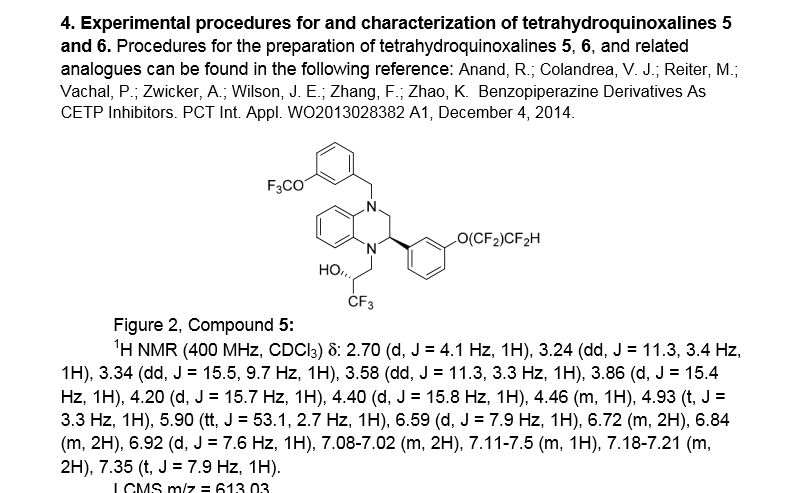
Example 18 as in patent

(R)- 1,1, 1 -trifluoro-3-((R)-4-(3-trifluoromethoxy)benzyl)-2-(3-(l, 1 ,2,2,-tetrafluoroethoxy)phenyl)-3,4- dihydroquinoxalin- 1 (2H)-yl)propan-2-ol
SPA: 15 nM
Example 18 was prepared from 2-bromo-l-(3-(l , 1 ,2,2,-tetrafluoroethoxy)phenyl)ethanone in three steps, using the reactions detailed in Schemes A6, A2 and Al . Spectral data are as follows: 1H NMR (400 MHz, CDC13) £2.70 (bd, J=4.1 Hz, IH), 3.24 (dd, J=l 1.3, 3.4 Hz, IH), 3.34 (dd, J=15.5, 9.7 Hz, IH), 3.58 (dd, J=l 1.3, 3.3 Hz, IH), 3.86 (d, J=15.4 Hz, IH), 4.20 (d, J=15.7 Hz, IH), 4.40 (d, J=15.8 Hz, IH), 4.46 (m, IH), 4.927 (t, J=3.3 Hz, IH), 5.90 (tt, J=53.1 , 2.7 Hz, IH), 6.59 (d, J= 7.9 Hz, IH), 6.72 (m, 2H), 6.84 (m, 2H), 6.92 (d, J=7.6 Hz, IH), 7.20 (m, 2H), 7.35 (t, J=7.9 Hz, IH), MS m/z = 613.03.
Scheme A12

Methyl 3 – { 1 – [(R)-3 ,3 ,3 -trifluoro-2-hy droxypropyl] -4- [3 -(trifluoromethoxy) benzyl]-l,2,3,4-tetrahydroquinoxalin-2-yl}benzoate (700 mg, 1.262 mmol) is made as described in Example 16 but with one stereochemical center unresolved. The compound was dissolved in MeOH (12.6mL), lithium hydroxide monohydrate (530 mg, 12.62 mmol) was added, and the reaction mixture was heated to 60°C for 4 hours. The crude mixture was dissolved in saturated ammonium chloride solution and extracted into EtOAc, the organic phase was dried with anhydrous magnesium sulfate, filtered, concentrated, and purified on a silica gel column with a 0-100% Hex/EtOAc gradient. The major peak was concentrated to afford 3-{l-[(R)-3,3,3-trifluoro-2-hydroxypropyl]-4-[3-(trifluoromethoxy)benzyl]-l,2,3,4-tetra-hydroquinoxalin-2-yl} benzoic acid. MS m/z = 541.09.

1H and 13C NMR spectra for compound 7
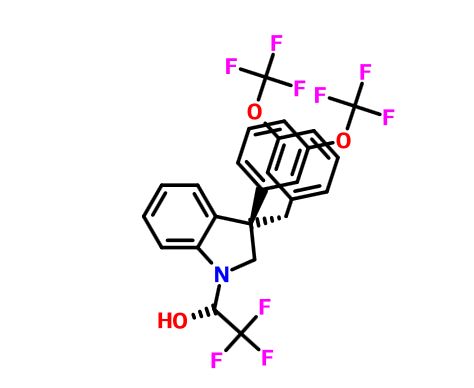
(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethoxy)-phenyl)indolin-l-yl)propan-2-ol.
Patent
WO2015054088
http://google.com/patents/WO2015054088A1?cl=en
Scheme Al

Scheme A2

Scheme A3

R = Ar, NR2l C02R, CN, S02Me
es
es
SEE EXAMPLE ………SIMILAR BUT NOT SAME

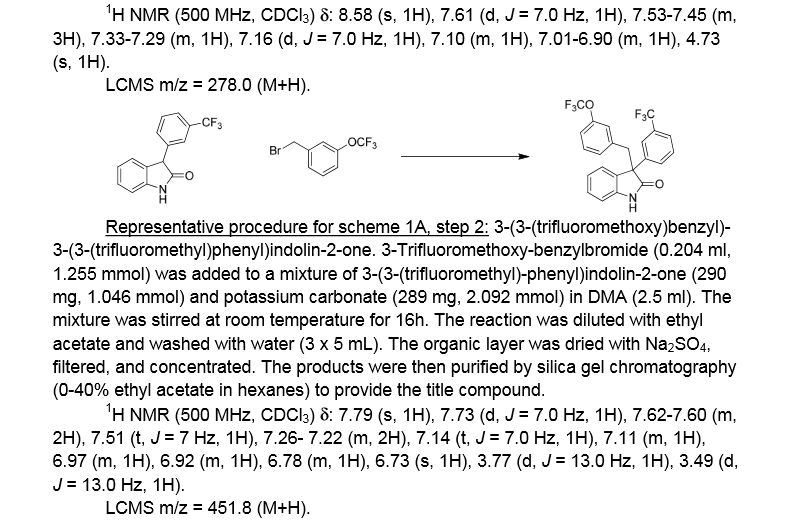
Example 1. (2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethyl)-phenyl)indolin-l-yl)propan-2-ol. This material was prepared according to Scheme Al, as described below.

3-(3-(trifluoromethyl)phenyl)indolin-2-one. Oxindole (1.598 g, 12 mmol), 3-bromo-a,a,a-trifluoromethyltoluene (2.009 ml, 14.40 mmol), potassium carbonate (3.32 g, 24.00 mmol), Pd2dba3 (0.220 g, 0.240 mmol), and 2-(dicyclohexylphosphino)-2′,4′,6′-triisopropylbiphenyl (0.458 g, 0.960 mmol) were combined in THF (12 ml) and the mixture was degassed with nitrogen. The solution was then heated to 80 °C for 18h. The mixture was cooled to room temperature, filtered through silica eluting with ethyl acetate, and concentrated. The material was then purified by silica gel chromatography (Biotage lOOg SNAP cartridge, 0-50% ethyl acetate in hexanes) to provide 3-(3-(trifluoromethyl)phenyl)indolin-2-one as a white solid.
1H NMR (500 MHz) δ 8.58 (s, 1H), 7.61 (d, J=7 Hz, 1H), 7.53-7.45 (m, 3H), 7.33-7.29 (m, 1H), 7.16 (d, J=7 Hz, 1H), 7.10 (m, 1H), 7.01-6.90 (m, 1H), 4.73 (s, 1H).

3 -(3 -(trifluoromethoxy)benzyl)-3 -(3 -(trifluoromethyl)phenyl)indolin-2-one . 3 -Trifluoromethoxy-benzylbromide (0.204 ml, 1.255 mmol) was added to a mixture of 3-(3-(trifluoromethyl)-phenyl)indolin-2-one (290 mg, 1.046 mmol) and potassium carbonate (289 mg, 2.092 mmol) (sodium carbonate may be used in place of potassium carbonate) in DMA (2.5 ml). The mixture was stirred at r.t. for 16h. The reaction was diluted with ethyl acetate and washed with water (3×5 mL). The organic layer was dried with Na2S04, filtered, and concentrated. The products were then purified by silica gel chromatography (Biotage 50g SNAP cartridge; 0-40%> ethyl acetate in hexanes) to provide 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)-phenyl)indolin-2-one .
1H NMR (500 MHz) δ 7.79 (s, 1H), 7.73 (d, J=7 Hz, 1H), 7.62-7.60 (m, 2H), 7.51 (t, J=7 Hz, 1H), 7.26- 7.22 (m, 2H), 7.14 (t, J=7.0 Hz, 1H), 7.11 (m, 1H), 6.97 (m, 1H), 6.92 (m, 1H), 6.78 (m, 1H), 6.73 (s, 1H), 3.77 (d, J=13 Hz, 1H), 3.49 (d, J=13 Hz, 1H).
LCMS m/z = 451.8 (M+H)

3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indoline. Borane tetrahydrofuran complex (1.673 ml, 1.673 mmol) was added to a solution of 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indolin-2-one (302 mg, 0.669 mmol) in THF (1.5 ml). The mixture was heated to 70 °C for 20h. The reaction was cooled to room temperature and quenched with saturated NH4C1 solution, and this mixture was stirred vigorously for 20 minutes. The product was extracted with ethyl acetate. The extracts were dried over Na2S04, filtered, and concentrated. The product was purified by silica gel chromatography (Biotage 25g SNAP cartridge, 0-50% ethyl acetate in hexanes) to provide 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indoline. This material may also be used without purification in the final step of the sequence, epoxide opening.
1H NMR (500 MHz) δ 7.66 (s, IH), 7.59 (d, J=7 Hz, IH), 7.53 (d, J=7 Hz, IH), 7.45 (t, J=8 Hz, IH), 7.18-7.13 (m, 2H), 7.04 (d, J=8 Hz, IH), 6.98 (d, J=7 Hz, IH), 6.81 (t, J=7.5 Hz, IH), 6.71 (m, 2H), 6.60 (s, IH), 3.83 (m, IH), 3.75-3.73 (m, 2H), 3.46 (d, J=13 Hz, IH), 3.41 (d, J=13 Hz, IH).
= 437.9 (M+H)

(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)-phenyl)indolin-l-yl)propan-2-ol. (S)-2-(trifluoromethyl)oxirane (81 μΐ, 0.933 mmol) was added to a solution of 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indoline (136 mg, 0.311 mmol) in l,l,l,3,3,3-hexafluoro-2-propanol (412 μΐ, 3.91 mmol). The reaction was stirred at room temperature overnight. The solvent was removed and the product was purified by silica gel chromatography (Biotage 25 g SNAP cartridge; 0-25% ethyl acetate in hexanes) to provide (2R)- 1 ,1,1 -trifluoro-3 -(3 -(3 -(trifluoromethoxy)benzyl)-3 -(3 -(trifluoromethyl)phenyl)indolin- 1 -yl)propan-2-ol.
1H NMR (500 MHz) (mixture of diastereomers) δ 7.72 (s, 0.5 H), 7.69 (s, 0.5 H), 7.65 (d, J=6.5 Hz, 0.5 H), 7.61 (d, J=7.5 Hz, 0.5 H), 7.56 (s, 1H), 7.50 (m, 1H), 7.25-7.17 (m, 2H), 7.07 (broad s, 2H), 6.91-6.89 (m, 1H), 6.79-6.75 (m, 1H), 6.53 (m, 2H), 4.00 (broad s, 1H), 3.83 (d, J= 9 Hz, 0.5H), 3.77 (d, J=9 Hz, 0.5H), 3.59-3.55 (m, 1H), 3.45-3.43 (m, 1H), 3.39-3.29 (m, 2H), 3.21-3.15 (m, 1H), 2.32 (m, 0.5H), 2.15 (m, 0.5H).
LCMS m/z = 549.8 (M+H)
Examples 1-25, in the table below, were prepared according to Scheme Al in a
SEE EG 10…….(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethoxy)-phenyl)indolin-l-yl)propan-2-ol.

ABOUT AUTHOR
Jonathan Wilson
Associate Principal Scientist at Merck

https://www.linkedin.com/in/jonathan-wilson-23206523
Experience
Associate Principal Scientist
Merck
October 2013 – Present (2 years 4 months)
Senior scientist
Merck
May 2009 – October 2013 (4 years 6 months)

Postdoctoral researcher
Princeton University
October 2007 – May 2009 (1 year 8 months)
Associate Medicinal Chemist
Merck
2000 – 2002 (2 years)
Education


///////CETP inhibition, cholesterol ester transfer protein, HDL, indoline, tetrahydroquinoxaline, merck, discovery
c21ccccc1N(C[C@@]2(c3cccc(c3)OC(F)(F)F)Cc4cc(ccc4)OC(F)(F)F)C(C(F)(F)F)O
FC(F)(F)Oc1cccc(c1)C3(CN(C[C@@H](O)C(F)(F)F)c2ccccc23)Cc4cccc(OC(F)(F)F)c4
see…………http://worlddrugtracker.blogspot.in/2016/01/mercks-novel-indoline-cholesterol-ester.html
PNQ 370 useful in treating Parkinson’s disease from ADVINUS
2016

PNQ 370
Advinus Therapeutics Ltd
Adenosine A2a receptor antagonist
for treating disease or disorder susceptible to improvement by antagonism of A2A receptor.
Advinus Therapeutics is investigating PNQ-370, presumed to be lead from a series of small molecule therapeutics including PD-2 and PD-3, as adenosine A2a receptor antagonist, for the potential treatment of Parkinson’s disease . In November 2012, this drug was in preclinical development .
![]()
KEEP WATCHING THIS POST AS I ARRIVE AT THE STRUCTURE…………..








ONE OF THE ABOVE OR SIMILAR
INTRODUCTION
The effects of adenosine are mediated through at least four specific cell membrane receptors so far identified and classified as Ai, A2A, A2B and A3 belonging to G protein-coupled receptor family. The Ai and A3 receptors down-regulate cellular cAMP levels through their coupling to G protein, which inhibit adenylate cyclase. In contrast, A2A and A2B receptors couple to G protein that activate adenylate cyclase and increase intracellular levels of cAMP. Through these receptors, adenosine regulates the wide range of physiological functions.
Advances in understanding the role of adenosine and its receptors in physiology and pathophysiology, as well as new developments in medicinal chemistry of these receptors have identified potential therapeutic areas for drug development. With the combination of pharmacological data, using selective ligands and genetically modified mice, important progress has been made toward an understanding of the role of ARs in a variety of diseases, such as inflammatory conditions, sepsis, heart attack, ischemia-reperfusion injury, vascular injury, spinal cord injury, chronic obstructive pulmonary disease (COPD), asthma, diabetes, obesity, inflammatory bowel disease, retinopathy, and Parkinson’s Disease (PD).
Happy new year wishes 2016


Movement disorder constitutes a serious health problem, especially among the elderly. These movement disorders can often be the result of brain lesions. Disorders involving the basal ganglia which result in movement disorders include Parkinson’s disease, Huntington’s chorea and Wilson’s disease. Tremor, rigidity, akinesia and postural changes are four classic symptoms of Parkinson’s disease, it is also associated with depression, dementia and overall cognitive decline. Parkinson’s disease has a prevalence of 1 per 1000 of the total population and increases to 1 per 100 for those aged over 60 years. Degeneration of dopaminergic neurons in the substantia nigra and the subsequent reductions in the interstitial concentrations of dopamine in the striatum are critical to the development of Parkinson’s disease. About 80% of cells from the substantia nigra can be destroyed before the clinical symptoms of Parkinson’s disease become apparent
PD is a progressive, incurable disorder with no definite preventive treatment, although drugs are available to alleviate the symptoms and/or slow down the progress of the disease. Current therapy is based on dopamine replacement therapy, the most common drug treatments being dopaminomimetic agents, including L-DOPA, a dopamine precursor, as well as direct or indirect dopamine receptor agonists. L-DOPA is the mainstay in the treatment of PD, but because of tolerance problems and a wide range of adverse reactions, including involuntary movements and vomiting, a strong demand for new therapies exists. Among the various strategies, A2A AR blockers are considered a potential approach to treatment of the disease. Within the brain A2A ARs are richly expressed in the striatum, nucleus accumbens, and olfactory tubercle. A coexpression of A2A with D2 dopamine receptors has been reported in the GABAergic striatopallidal neurons where adenosine and dopamine agonists exert antagonistic effects in the regulation of locomotor activity. Activation of A2A ARs in striatopallidal neurons decreases the affinity of D2 receptors for dopamine, antagonizing the effects of D2 receptors.
The negative interaction between A2A and D2 receptors is at the basis of the use of A2A antagonists as a novel therapeutic approach in the treatment of PD. (Pharmacol. Ther. 2005, 105, 267). The recent discovery that the A2A can form functional heteromeric receptor complexes with other Gprote in-coupled receptors such as D2 and the mGlu5 receptors has also suggested new opportunities for the potential of A2A antagonists in PD. (J. Mol. Neurosci. 2005, 26, 209).
A2A knockout (KO) mice transient focal ischemia caused less neuronal damage in comparison to their wild-type (WT) littermates (J. Neurosci. 1999, 19, 9192.). Therefore, it seems that tonic activation of A2A ARs may be responsible for dangerous signal during injury, in contrast to the neuroprotective effects induced by endogenous Al activation. Recently, selective inactivation or reconstitution of A2A ARs in bone-marrow cells revealed their contribution to the development of ischemic brain injury (J.F. Nat. Med. 2004, 10, 1081) Blockade of A2A ARs has recently been implicated in the treatment of movement disorders such as Parkinson’s disease (Trends Pharmacol. Sci. 1997, 18, 338-344) and in the treatment of cerebral ischaemia (Life Sci. 1994, 55, 61-65).
The potential utility of A2A AR antagonists in the treatment of Parkinson’s disease has been reviewed (CNS drugs, 1998, 10, 31 1-320). One advantage of A2A AR antagonist therapy is that the underlying neurodegenerative disorder may also be treated ((Ann. N. Y. Acad. Sci. 1997, 825 (Neuroprotective Agents), 3048). In particular, blockade of A2A AR function confers neuroprotection against MPTP-induced neurotoxicity in mice (Neurosci. 2001, 21, RC143).
Alzheimer’s disease (AD) is a neurodegenerative disorder of the central nervous system manifested by cognitive and memory deterioration, a variety of neuropsychiatric symptoms, behavioral disturbances, and progressive impairment of daily life activities. Recent research suggests that adenosine receptors play important roles in the modulation of cognitive function. Epidemiological studies have found an association between coffee (a nonselective adenosine receptor antagonist) consumption and improved cognitive function in AD patients and in the elderly. Long-term administration of caffeine in transgenic animal models showed a reduced amyloid burden in brain with better cognitive performance.

Advinus’ Pharma Development Bangalore operation, located on a 8-acre campus with 220,000 sq ft of modern facilities, offers end-to-end pre-clinical to early clinical development platform for pharma product development
Antagonists of adenosine A2A receptors mimic these beneficial effects of caffeine on cognitive function. Neuronal cell cultures with amyloid beta in the presence of an A2A receptor antagonist completely prevented amyloid beta-induced neurotoxicity. These findings suggest that the adenosinergic system constitutes a new therapeutic target for AD, and caffeine and A2A receptor antagonists may have promise to manage cognitive dysfunction in AD (Curr Neuropharmacol. 2009 September; 7(3): 207-216).
High expression of A2A ARs has been found in platelets, leukocytes, vascular smooth muscle, and endothelial cells with important implications in the regulation of inflammatory responses. It is now well established that stimulation of the A2A AR in immune cells induces anti-inflammatory effects, mostly due to its ability to increase cAMP levels, which has strong immunosuppressive effects (Trends Immunol. 2005, 26, 299). Stimulation of A2A ARs inhibits neutrophil adherence to the endothelium, degranulation of activated neutrophils and monocytes, plus superoxide anion generation. A2A ARs have been recently defined as sensors and terminators of proinflammatory activities. The strongest evidence for the key role of A2A in inflammation is derived by the elegant study using mice deficient in A2A ARs (Nature 2001, 414, 916).

The state-of-the-art facility in Pune, Advinus Drug Discovery, develops its own drug candidates to out-license them at preclinical or clinical stages
In this model the lack of A2A subtype leads to increased tissue inflammation and damage, thus suggesting a negative and nonredundant regulatory role for the A2A AR. This model permits one to appreciate that adenosinergic regulation of immune cells is fundamental in normal physiological control of inflammation in vivo in spite of the fact that other Gs-protein-coupled receptors and cAMP elevating ligands are present, such as cathecolamines, prostaglandins, dopamine, and histamine (Trends Immunol. 2005, 26, 299). Interestingly, the A2A AR has been demonstrated to be involved in promotion of wound healing and angiogenesis in healing wounds (Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R283).
Moreover, it plays an active role in the pathogenesis of dermal fibrosis, suggesting a role for antagonists as novel therapeutic approach in the treatment and prevention of dermal fibrosis in diseases such as scleroderma (Arthritis Rheum. 2006, 54, 2632) as well as hepatic fibrosis (Br. J. Pharmacol. 2006 Aug; 148(8): 1 144-55). Studies also suggest that A2A receptor antagonists may be beneficial for social memory impairment and hypertension (Behav Brain Res. 2005 Apr 30;159(2):197-205), sepsis (J Immunol. 2006 May 1 ; 176(9): 5616-26), spinal cord injury and neuroprotection (J Neuroinflammation. 201 1 Apr 12;8:31), retinopathy (IVOS, Jan. 2000, vol. 41 (1), 230-243, depression (Neurology. 2003 Dec 9;61(1 1 Suppl 6):S82-7), narcolepsy and other sleep related disorders (Prog Neurobiol. 2007 Dec;83(5):332-47), attention-deficit hyperactivity disorder (ADHD) (Behav Pharmacol. 2009 Mar;20(2): 134-45; Clinical Genetics (2000), 58(1), 31-40 and references therein),

Dr Rashmi Barbhaiya, CEO & Managing Director

… Dr Rashmi Barbhaiya, CEO & Managing Director and Dr Kasim Mookthiar, Chief Scientific Officer and SVP, Drug Discovery, Advinus Therapeutics …
Antagonists of the A2A receptor are potentially useful therapies for the treatment of addiction. Major drugs of abuse (opiates, cocaine, ethanol, and the like) either directly or indirectly modulate dopamine signaling in neurons particularly those found in the nucleus accumbens, which contain high levels OfA2A adenosine receptors. Dependence has been shown to be augmented by the adenosine signaling pathway, and it has been shown that administration of an A2A receptor antagonist redues the craving for addictive substances (“The Critical Role of Adenosine A2A Receptors and Gi βγ Subunits in Alcoholism and Addiction: From Cell Biology to Behavior”, by Ivan Diamond and Lina Yao, (The Cell Biology of Addiction, 2006, pp 291-316) and “Adaptations in Adenosine Signaling in Drug Dependence: Therapeutic Implications”, by Stephen P. Hack and Macdonald J. Christie, Critical Review in Neurobiology, Vol. 15, 235-274 (2003)). See also Alcoholism: Clinical and Experimental Research (2007), 31(8), 1302-1307.
A2A receptors may be beneficial for the treatment or prevention of disorders such as a movement disorder, for example, Parkinson’s disease or progressive supernuclear palsy, Restless leg syndrome, nocturnal myoclonus, cerebral ischaemia, Huntington’s disease, multiple system atrophy, corticobasal degeneration, Wilson’s disease or other disorders of basal ganglia which results in dyskinesias, post traumatic stress disorder. See for example WO200013682, WO200012409, WO2009156737, WO20091 1442, WO2008121748, WO2001092264, WO2007038284, WO2008002596, WO20091 1 1449, WO20091 1 1442, WO2008121748, WO2009156737, WO2003022283, WO2005044245, WO2008077557, WO20091 1 1449, WO2009705138, WO20091 1 1442, WO2007035542, WO20080870661, WO2008070529, WO20051 16026, WO2009055548, WO2007133983, WO2010045006, WO2010045015, WO2010045008 WO2009015236.
![]()

centre: Mr Ratan Tata, Chairman, Tata Sons, flanked by Dr Rashmi Barbhaiya (left), Managing Director and CEO, Advinus, and Mr R. Gopalakrishnan, …
ONE EXAMPLE………..
COMPD A1
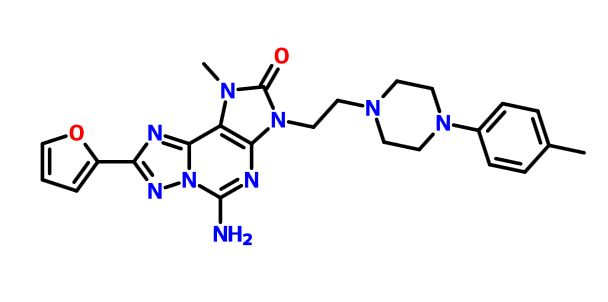
MF C26 H31 N9 O4
2H-[1,2,4]Triazolo[5,1-i]purin-2-one, 5-amino-8-(2-furanyl)-1,3-dihydro-3-[2-[4-[4-(2-methoxyethoxy)phenyl]-1-piperazinyl]ethyl]-1-methyl-
mw 533.58
cas 1367365-26-1
| Molecular Formula: | C26H31N9O4 |
|---|---|
| Molecular Weight: | 533.58224 g/mol |
 A1
A15-amino-8-(furan-2-yl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-1-yl]ethyl]-1-methyl-[1,2,4]triazolo[5,1-f]purin-2-one
Example Al :
5-amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin- 1 -yl]ethyl]- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one
5-Amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-l-
5-amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin- 1 -yl]ethyl]- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one
Step-1 : 2-[(2,5-Diamino-6-chloro-pyrimidin-4-yI)amino]ethanol
A mixture of 4,6-dichloropyrimidine-2,5-diamine (28g, 156mmol), ethanolamine (18ml, 312mmol) and ethanol (250ml) were heated at 100-1 10 °C for 16 hours. The mixture was cooled and solvent was removed. To the residue methanol (100ml) was added and stirred for 20 minutes. The solid was filtered off to obtain 2-[(2,5-diamino-6-chloro-pyrimidin-4-yl)amino]ethanol (22.0g, 70%).
‘H MR(400MHz, DMSO d6): δ 3.36-3.40 (m, 2H); 3.50-3.54 (m, 2H); 3.88 (bs, 2H); 4.74 (t, J=5.6Hz, 1H); 5.63 (bs, 2H); 6.51 (t, J=5.6Hz, 1H)
Step-2: 2-Amino-6-chloro-9-(2-hydroxyethyl)-7H-purin-8-one
A mixture of 2-[(2,5-diamino-6-chloro-pyrimidin-4-yl)amino]ethanol obtained in step 1 (l O.Og, 49.26mmol) in acetonitrile (400ml) were cooled to 0 °C. To this reaction mixture K2C03 (20.39gm, 147.7mmol) and 4-nitrophenyl chloroformate (19.8g, 98.52mmol)was added and stirred at 25-27 °C for 24 hours. This reaction mixture was filtered and washed with acetonitrile (300ml) and diethyl ether (300ml) respectively. Solid obtained was dried to obtain crude 2-amino-6-chloro-9-(2-hydroxyethyl)-7H-purin-8-one as a yellow solid. Small amount of crude material was purified by column chromatography to obtain pure product. ‘HNMR(400MHz, DMSO d6): δ 3.61-3.66 (m, 2H); 3.72-3.75 (m, 2H); 4.85 (t, J=6Hz, 1H); 6.60 (s, 2H); 1 1.21 (s, 1 H)
Step-3: 2-Amino-6-chloro-9-(2-hydroxyethyl)-7-methyl-purin-8-one
A mixture of 2-amino-6-chloro-9-(2-hydroxyethyl)-7H-purin-8-one obtained in step 2 (13g, 56.7mmol) , K2C03 (1 1.5g, 84mmol), methyl iodide (12g, 85.15mmol) and DMF (130ml) were stirred at 25-30 °C for 16 hours. The reaction mixture was concentrated and purified by column chromatography using 60-120 silica gel and 4% methanol in DCM as an eluent to obtain 2-amino-6-chloro-9-(2-hydroxyethyl)-7-methyl-purin-8-one (8g, 58%) as an off white solid.
‘HNMR(400MHz, DMSO d6): δ 3.42 (s, 3H); 3.65 (t, J=5.6Hz, 2H); 3.78 (t, J=5.6Hz, 2H); 4.85 (t, J=5.6Hz, 1H); 6.69 (bs, 2H).
Step-4: 2-Amino-6-hydrazino-9-(2-hydroxyethyl)-7-methyI-purin-8-one
A mixture of 2-amino-6-chloro-9-(2-hydroxyethyl)-7-methyl-purin-8-one obtained in step 3 (8g, 32.9mmol) , Hydrazine hydrate (16ml ,32.9mmol) and ethanol (300ml) were heated at 100-1 10 °C for 16 hours. The reaction mixture was concentrated and solid obtained was filtered off and dried to obtain 2-amino-6-hydrazino-9-(2-hydroxyethyl)-7-methyl-purin-8-one (7g, 89 %) as an off white solid.
‘HNMR(400MHz, DMSO d6): δ 3.37 (s, 3H); 3.58-3.61 (m, 2H); 3.71 (t, J=6Hz, 2H); 4.29 (bs, 2H); 4.87 (t, J=5.6Hz, 1H), 6.00 (bs, 2H); 7.63 (s, 1H).
Step-5: N’-[2-Amino-9-(2-hydroxyethyl)-7-methyl-8-oxo-purin-6-yl]furan-2-carbohydrazide
2-amino-6-hydrazino-9-(2-hydroxyethyl)-7-methyl-purin-8-one (4.5g, 18.18mmol) obtained in step 4, 2-furoic acid (2.53g, 22.5mmol), HOBT (2.53g, 18.8 mmol) and N-methylmorpholine were taken in dimethylformamide (40ml). l-Ethyl-3(3′-dimethylaminopropryl)carbodiimide hydrochloride (EDCI.HCl) (5.4g, 28.2mmol) was added to the reaction mixture and stirred at 25-27 °C for 14 hours. The reaction mixture was evaporated and residue was purified by column chromatography to obtain N’-[2-amino-9-(2-hydroxyethyl)-7-methyl-8-oxo-purin-6-yl]furan-2-carbohydrazide (5.3g, 84%) as an off white solid.
‘HNMR (400MHZ, DMSO d6): δ 3.43 (s, 3H); 3.59-3.63 (m, 2H); 3.74 (t, J=6Hz, 2H); 4.88 (t, J=5.6Hz, 1H); 5.98 (bs, 2H); 6.67 (bs, 1H); 7.25 (d, J=3.2Hz, 1H); 7.90 (s, 1H); 8.35 (s, 1H); 10.28 (s, lH).
Step-6: 5-Amino-8-(2-furyl)-3-(2-hydroxyethyl)-l-methyl-[l^,4]triazolo[5,l-flpurin-2-one
A mixture of N’-[2-amino-9-(2-hydroxyethyl)-7-methyl-8-oxo-purin-6-yl]furan-2-carbohydrazide obtained in step 5 (5.3g, 15.9mmol), Ν,Ο-bistrimethylsilylacetamide (27ml, 1 1 1.4mmol) and hexamethyldisilazane (83ml, 397mmol) were heated at 1 10-120 °C for 16 hours. The reaction mixture was quenched with methanol (100ml) and water (100ml) and organic volatiles were evaporated. The solid obtained was filtered off and washed with water (30ml) followed by diethyl ether (100ml) to obtain 5-amino-8-(2-furyl)-3-(2-hydroxyethyl)-l-methyl-[l,2,4]triazolo[5,l-f]purin-2-one (3.50g, 71%) as an off white solid.
‘HNMR (400MHZ, DMSO d6): δ 3.56 (s, 3H); 3.67-3.70 (m, 2H); 3.84-3.87 (m, 2H); 4.88 (t, J=5.6Hz, 1H); 6.73 (bs, 1H); 7.20 (bs, 1H); 7.79 (bs, 2H); 7.94 (bs, 1H).
Step-7: 2-[5-Amino-8-(2-furyl)-l-methyl-2-oxo-[l,2,4]triazolo[5,l-fJpurin-3-yl]ethyl 4-methylbenzenesulfonate
A mixture of 5-amino-8-(2-furyl)-3-(2-hydroxyethyl)-l -methyl-[l,2,4]triazolo[5, l-fJpurin-2-one obtained in step 6 (3.5g, l lmmol), p-toluene sulphonylchloride (5.2 g, 27mmol) were taken in pyridine (30ml)and stirred at 25-27 °C for 16 hours. To the reaction mixture hexane (100ml) was added and solid obtained was filtered off and washed with water (100ml) followed by hexane (100ml) to obtain 2-[5-amino-8-(2-furyl)-l-methyl-2-oxo-[l,2,4]triazolo[5, l-f]purin-3-yl]ethyl 4-methylbenzenesulfonate (4.1g, 78%) as a brown solid. ‘HNMR (400MHz, DMSO d6): δ 2.02 (s, 3H); 3.49 (s, 3H); 3.99 (t, J=4.8Hz, 2H); 4.71 (t, J=4.8Hz, 2H); 6.73-6.75 (m, 1H); 7.01 (d, J=8Hz, 2H); 7.23 (d, J=3.2Hz, 1H); 7.41 (d, J=8.4Hz, 2H); 7.78 (bs, 2H); 7.96 (d, J=1.2Hz, 1H).
Step-8: : 5-Amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-l-yl]ethyl]-l-methyl-[l,2,4]triazolo[5,l-f)purin-2-one
A mixture of 2-[5-amino-8-(2-furyl)-l-methyl-2-oxo-[l ,2,4]triazolo[5, l-f]purin-3-yl]ethyl 4-methylbenzenesulfonate obtained in step 7 (0.25g, 0.533mmol), l-[4-(2-Methoxy-ethoxy)-phenyl]-piperazine (0.188g, 0.799mmol) and DIPEA (0.27ml, 1.599mmol) were taken in DMF (5ml) and stirred at 80 °C for 16 hours. To the reaction mixture water (100ml) was added and solid obtained was filtered off. The crude product was purified by column chromatography to obtain 5-amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin- 1 -yl]ethyl]- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one (0.135g, 47%) as an off white solid
‘HNMR (400MHz, DMSO d6): δ 2.60 (bs, 4H); 2.68 (t, J=6.4Hz, 2H); 2.96 (bs, 4H); 3.29 (s, 3H); 3.56 (s, 3H); 3.59-3.62 (m, 2H); 3.94-4.00 (m, 4H); 6.71 -6.73 (m, 1H); 6.79-6.86 (m, 4H); 7.19 (dd, J=3.2Hz, 1.2Hz, 1H); 7.80 (bs, 2H); 7.94 (bs, 1H).
ANOTHER……..
Example Gl: 5-Amino-l-ethyl-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-l-yl]ethyl]-[l,2,4]triazolo[5,l-i]purin-2-one

Step-1 : 2-Amino-6-chloro-7-ethyl-9-(2-hydroxyethyl)purin-8-one
(Procedure is same as step-3 in example Al)
‘HNMR (400MHz, DMSO d6): δ 1.21 (t, J=7.2Hz, 3H); 3.64 (s, 2H); 3.78 (t, J=6Hz, 2H);
3.92 (q, J=7.2Hz, 2H); 4.92 (bs, I H); 6.7 (bs, 2H).
Step-2 : 2-Amino-7-ethyl-6-hydrazino-9-(2-hydroxyethyl)purin-8-one
(Procedure is same as step-4 in example Al)
‘ HNMR (400MHz, DMSO d6): δ 1.07 (t, J=6.8Hz, 3H); 3.59 (q, J=6Hz, 2H); 3.72 (t, J=6Hz,
2H); 3.91 (q, J=6.8Hz, 2H); 4.32 (bs, 2H); 4.86 (t, J=5.6Hz, IH); 5.99 (bs, 2H), 7.55 (bs, IH).
Step-3: N’-[2-Amino-7-ethyl-9-(2-hydroxyethyl)-8-oxo-purin-6-yl]furan- 2carbohydrazide (Procedure is same as step-5 in example Al)
Crude product was used in next step
Step-4: 5-Amino-l-ethyI-8-(2-furyl)-3-(2-hydroxyethyl)-[l,2,4]triazolo[5,l-flpurin-2-one
(Procedure is same as step-6 in example Al)
‘H MR (400MHZ, DMSO d6): δ 1.34 (t, J=7.2Hz, 3H); 3.67 (q, J=5.6Hz, 2H); 3.84 (t, J=5.6Hz, 2H); 4.01 (q, J=7.2Hz, 2H); 4.87 (t, J=6Hz, IH); 6.70 (bs, IH); 7.17 (d, J=2.8Hz, I H); 7.18 (bs, 2H); 7.92 (bs, IH).
Step-5: 2-[5-Amino-l-ethyl-8-(2-furyl)-2-oxo-[l,2,4]triazoIo[5,l-f|purin-3-yl]ethyl 4- methylbenzenesulfonate (procedure is same as step-7 in example Al)
lHNMR (400MHz, DMSO d6): δ 1.35 (t, J=7.2Hz, 3H); 2.00 (s, 3H); 3.95-4.00 (m, 4H); 4.47 (bs, 2H); 6.74 (s, IH); 7.00 (d, J=7.6Hz, 2H); 7.22 (s, IH); 7.42 (d, J=7.6Hz, 2H); 7.78 (bs, 2H); 7.97 (bs, IH).
Step-6: 5-Amino-l-ethyl-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyi]piperazin-l- yl]ethyl]-[l,2,4]triazolo[5,l-f]purin-2-one (procedure is same as step-8 in example Al)
HNMR(400MHz, DMSO d6): δ 1.35 (t, J=7.2Hz, 3H); 2.60 (bs, 4H); 2.68 (t, J=6.8Hz, 2H); 2.95 (bs, 4H); 3.28(s, 3H);3.61 (t, J=4.4Hz, 2H); 3.94-4.04 (m, 6H); 6.72 (dd, J=2Hz, 3.6Hz, I H); 6.78-6.85 (m, 4H); 7.19 (d, J=3.2Hz, IH); 7.81(bs, 2H); 7.94 (s, IH).
Representative compounds of the present disclosure were tested and had micromolar to nanomolar activity.

 A1 ABOVE
A1 ABOVE
A7 ABOVE
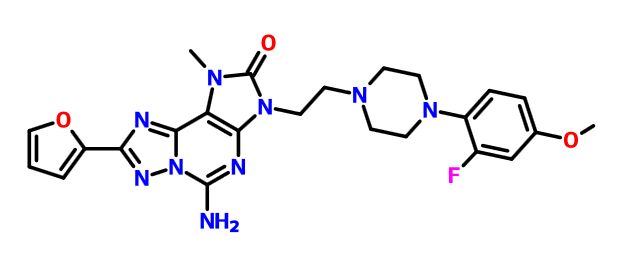
A9 ABOVE
A13 ABOVE
A31 ‘HNMR (400MHz, DMSO d6): δ 2.62 (bs,4H); 2.68 (t, J=6.8Hz, 2H); 2.85 (bs, 4H); 3.28 (s, 3H); 3.57 (s, 3H); 3.59-3.62 (m, 2H); o 3.95 (t, J=6.8Hz, 2H); 4.01-4.04 (m, 2H);
5-Amino-3-[2-[4-[2-fluoro-4-(2- 6.66-6.68 (m, 1H); 6.72 (dd, J=2 Hz,3.6Hz, methoxyethoxy)phenyl]piperazin-l-yl]ethyl]-8- 1H); 6.79 (dd, J=2.8Hz, 14Hz, 1H); 6.92 (t, (2-furyl)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f|purin-2- J=9.6Hz, 1H); 7.19 (d, J=3.2Hz, 1 H); 7.93 one (bs, 2H); 7.93-7.94 (m, 1H).
A31 ABOVE
A32 HNM (400MHz, DMSO d6): δ 2.59 (bs,
4H); 2.68(t, J=6.4Hz, 2H); 3.27(t, J=4.8Hz, 4H); 3.56 (s, 3H); 3.96 (t, J=6.4Hz, 2H);
0 6.72(dd, J=2Hz, 3.6Hz, 1H); 6.99 (d, J=8.8Hz,
4-[4-[2-[5-Amino-8-(2-furyl)-l-methyl-2-oxo- 2H); 7.19 (d, J=3.6Hz, 1H);7.56 (d, J=8.8Hz, [ 1 ,2,4]triazolo[5, 1 -f]purin-3-yl]ethyl]piperazin- 2H); 7.80 (bs, 2H); 7.93 (bs, lH).
l-yl]benzonitrile
A32 ABOVE
A36 ‘HNMR(400MHz, CDCI3): δ θ.09 (d,
J=4.4Hz, 2H); 0.50 (d, J=6.8Hz, 2H); 0.82- 0.89 (m, 1H); 2.24 (d, J=6.0Hz, 2H): 2.52- 2.72 (m, 8H); 2.80 (t, J=6.4Hz, 2H); 3.76 (s,
5-Amino-3-[2-[4-(cyclopropylmethyl)piperazin- 3H); 4.07 (t, J=6.8Hz, 2H); 5.89 (bs, 2H); l -yl]ethyl]-8-(2-furyl)-l-methyl- 6.61 (bs, 1H); 7.22 (d, J=2.4Hz, 1H); 7.64 (s, [ 1 ,2,4]triazolo[5, 1 -f]purin-2-one 1H).
A36 ABOVE
A38 ‘HNMR(400MHz, CDCI3): δ 2.62 . (t,
J=4.4Hz, 4H); 2.79 (t, J=6.4Hz, 2H); 2.81 (s, 6H); 3.22 (t, J=4.4Hz, 4H): 3.77 (s, 3H); 4.06 (t, J=6.8Hz, 2H); 5.74 (bs, 2H); 6.60 (dd,
4-[2-[5-Amino-8-(2-fiiryl)- 1 -methyl-2-oxo- J=2.0Hz, 3.2Hz, 1H); 7.24 (d, J=3.6Hz, 1H);
[ 1 ,2,4]triazolo[5, 1 -f]purin-3-yl]ethyl]-N,N- 7.65 (s, 1H).
dimethy l-piperazine- 1 -sulfonamide
A38 ABOVE
A39 ‘HNMR(400MHZ, DMSO d6): δ 1.89-1.94
im, 1H); 2.09-2.18 .(m, 1 H); 2.60 (bs, 4H); 2.67 (t, J=6.4Hz, 2H); 2.96 (bs, 4H); 3.56 (s, 3H); 3.69-3.85 (m, 4H); 3.95 (t, J=6.4Hz,
2H); 4.89 (bs, 1H); 6.72 (dd, J=2.0, 3.2Hz,
5-Amino-8-(2-furyl)-l -methyl-3-[2-[4-(4- 1H); 6.78 (d, J=9.2Hz, 2H); 6.85 (d, J=9.2Hz, tetrahydrofuran-3-yloxyphenyl)piperazin- 1 – 2H): 7.20 (d, J=3.2Hz, 1 H); 7.80 (bs, 2H); yl]ethyl]-[l ,2,4]triazolo[5,l-f]purin-2-one
7.93 (s, 1H).
A39 ABOVE
A42 ‘HNMR(400MHz, CDCI3): δ
2.26 (s,3H); 2.94-2.97 (m, 6H); 3.72 (s, 2H); 3.75 (s, 3H); 4.17 (t, J=6.4Hz, 2H); 5.74 (bs, 2H); 6.59 (dd, J=1.6Hz, 3.6Hz, 1H);7.13 (s, J=3.6Hz, IH); 7.21-7.24 (m, IH); 7.63 (s,
5-Amino-8-(2-furyl)-l-methyl-3-[2-(3-methyl- IH); 8.20 (bs, IH),
7,8-dihydro-5H- 1 ,6-naphthyridin-6-yl)ethyl]- [ 1 ,2,4]triazolo[5, 1 -f]purin-2-one
A42 ABOVE
A57 HNMR(400MHz, DMSO d6): δ 2.95 (t,
J=8Hz, 2H); 3.52 (s, 3H); 3.69 (s, 3H ), 3.97 (t, J=8Hz, 2H); 6.71 (dd, J=2Hz, 3.6Hz, I H );
5-Amino-8-(2-furyl)-3-[2-(4- 6.80 (dd, J=2Hz, 6.8Hz, 2H); 7.10 (d, methoxyphenyl)ethyl]- 1 -methyl- J=8.8Hz, 2H); 7.18 (dd, J=0.8Hz, 3.2Hz, I H );
[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one 7.80 (bs, 2H), 7.94 (dd, J=lHz, 2Hz, I H ).
A57 ABOVE
A58 HNMR(400MHz, DMSO d6): δ 2.61 (bs,
4H); 2.68 (bs, 2H); 3.05(bs, 4H); 3.57 (s, 3H ), 3.96 (bs, 2H); 6.72 (bs, IH); 6.92 (d, J=8Hz, 2H); 7.01 (d, J=10Hz, 2H );7.03(d, J=148Hz, IH); 7.19 (bs , 1 H); 7.80 (bs, 2H); 7.94 (s,
5-amino-3-[2-[4-[4- IH).
(difluoromethoxy)phenyl]piperazin-l-yl]ethyl]- 8-(2-furyl)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -fjpurin- 2-one
A58 ABOVE
A62 O ‘HNMR (400MHz, DMSO d6): δ 0.66-0.70
(m, 4H); 1.90-1.94 (m, lH); 2.41 (bs, 4H); 2.65 (t, J=6Hz, 2H); 3.38 (bs, 2H); 3.56 (bs, 5H); 3.93 (t, J=6.4 Hz, 2H); 6.71 (bs, 1H );
5-Amino-3-[2-[4- 7.19 (d, J=2.4Hz, 1H); 7.79 (bs, 2H); 7.93 (bs,
(cyclopropanecarbonyl)piperazin- 1 -yl]ethyl]-8- 1H).
(2-furyl)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -fjpurin-2- one
A62 ABOVE
A63 ‘HNMR (400MHz, DMSO d6): δ 0.07-0.10
(m, 2H); 0.40-0.44 (m, 2H); 0.88-0.94 (m,lH); 2.21 (d, J=6.4Hz, 2H); 2.41-2.45 (m, 4H); 2.64 (t, J=6.4Hz, 2H); 3.38 (bs,4H); 3.56
5-Amino-3-[2-[4-(2- (s, 3H); 3.93 (t, J=6.4Hz, 2H); 6.72 (dd, cyclopropylacetyl)piperazin-l -yl]ethyl]-8-(2- J=2Hz,3.6 Hz, 1H); 7.19-7.20 (m, 1H); 7.80 fury 1)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -fJpurin-2- (bs, 2H); 7.93 (d, J=0.8 Hz, 1H).
one
A63 ABOVE
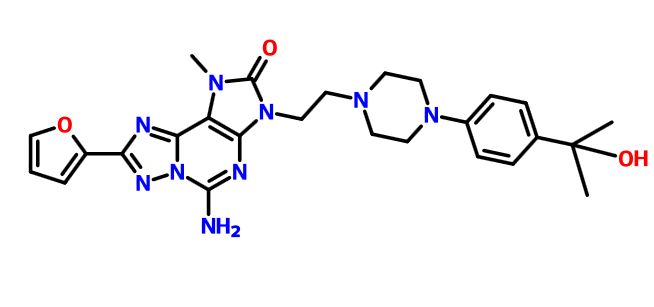
C1 ABOVE

E1 ABOVE

D3 ABOVE
G1 ABOVE
G2
H2
M1
M2
M3
M6
ETC AS IN TABLE……………..

Dr Kasim Mookthiar, CSO & Executive VP (Drug Discovery),

Dr Nimish Vachharajani, Senior VP & Head (Pharmaceuticals & Agrochemical Development),
/////////
n21c(nc4c(c1nc(n2)c3occc3)N(C(N4CCN5CCN(CC5)c6ccc(cc6)OCCOC)=O)C)N
CN1C2=C(N=C(N3C2=NC(=N3)C4=CC=CO4)N)N(C1=O)CCN5CCN(CC5)C6=CC=C(C=C6)OCCOC
Lead-oriented synthesis: a new concept to aid drug-discovery process

Figure 2. a. Fragment-based screening: Small and structurally diverse molecules (circles represent functional groups) are screened for a biological target, and they are combined and modified to generate drug-like compounds. b. Diversity-oriented synthesis: Large collections of structurally diverse and complex molecules are made using a short number of reactions. The resulting compounds are optimized to produce the drug-like compounds. | Credit: P. J. Hajduk,W. R. J. D. Galloway & D. R. Spring Nature, 2011, 470, 42–43. DOI: 10.1038/470042a
The discovery and development of new drugs is a long and expensive process, and despite of it, essential to face present and new diseases. For small molecules, which account for the majority of the marketed drugs, the discovery process generally involves finding a starting point termed hit or lead compound. These molecules have biological activity but need to be optimized to enhance their potency and selectivity (i.e. minimize the toxicity) and improve pharmacokinetic parameters making them suitable to go to the next stage, the pre-clinical tests……….http://mappingignorance.org/2014/07/04/lead-oriented-synthesis-new-concept-aid-drug-discovery-process/
Author
pablo ortiz
PhD at Rijksuniversiteit Groningen
Experience

PhD Student, Rijksuniversiteit Groningen
October 2013 – Present
Asymmetric organometallic catalysis focused on tertiary alcohols and amines

Harutyunyan research group
Master Thesis Project
University of the Basque Country
February 2013 – September 2013 (8 months)Vitoria-Gasteiz Area, Spain
Estereoselective synthesis of quaternary alpha-aminophosphonic acid derivatives (organocatalysis)
Locum pharmacist
Community pharmacy
August 2012 – August 2012 (1 month)La Rioja, Spain
Pre-registration pharmacist
NHS Trust
January 2012 – June 2012 (6 months)Southport, England
In-patient and out-patient dispensing
Clinical pharmacy
Medicines information
Anticoagulant management
Aseptic preparation of medicines
Clinical audit of antimicrobial use
Publications
Tertiary α-diarylmethylamines derived from diarylketimines and organomagnesium reagents(Link)
Chem. Commun. 2015, 51, 703-706.
November 13, 2014
Organomagnesium reagents enable swift and versatile derivatisation of diarylimines to the corresponding α-substituted diarylmethylamines in excellent yields, through fast and clean reactions. Where it occurs, 1,2-reduction can be circumvented using readily accessible dialkylmagnesium reagents.
Asymmetric Synthesis of Functionalized Tetrasubstituted α-Aminophosphonates through Enantioselective Aza-Henry Reaction of Phosphorylated Ketimines(Link)
J. Org. Chem., 2015, 80, 156–164
November 2014
Bifunctional Cinchona alkaloid thioureas efficiently catalyze asymmetric nucleophilic addition of nitromethane to ketimines derived from α-aminophosphonic acids to afford tetrasubstituted α-amino-β-nitro-phosphonates.
Catalytic Asymmetric Alkylation of Aryl Heteroaryl Ketones(Link)
Eur. J. Org. Chem., 2015, 72–76.
November 2014
Tertiary diarylmethanols are highly bioactive structural motifs. A new strategy to access chiral tertiary diarylmethanols through copper-catalyzed direct alkylation of (di)(hetero)aryl ketones by using Grignard reagents was developed. The low reactivity and the similarity of the enantiotopic faces of bis-aromatic ketones were partially overcome, which resulted in moderate to good yields and…more
Synthesis of Tetrasubstituted α-Aminophosphonic Acid Derivatives from Trisubstituted α-Aminophosphonates(Link)
Eur. J. Org. Chem., 2013: 7095–7100
September 2013
Education

Universidad del País Vasco/Euskal Herriko Unibertsitatea
Master’s degree, Synthetic and Industrial Chemistry, 9.2
2012 – 2013
- (Open)1 course
Universidad del País Vasco/Euskal Herriko Unibertsitatea
Bachelor’s degree, Pharmacy, Extraordinary Degree Award, 9.06
2007 – 2012
- (Open)1 honor or award
- (Open)2 courses
Courses
Universidad del País Vasco/Euskal Herriko Unibertsitatea
- How to write and publish a research article
- X Pharmaceutical Chemistry Sessions: New strategies for the design and synthesis of drugs
Universidad del País Vasco/Euskal Herriko Unibertsitatea
- II Organic Chemistry Synthesis and Catalysis Workshop: Methods and strategies in synthesis
GRONINGEN, NETHERLANDS




stadtmitte groningen niederlande stadtwanderung
Piramal Drops Drug Discovery,…………. Pharmaceuticals: Risks and regulations convince the Indian company to reallocate resources

A Piramal scientist at work in Mumbai last month.
Credit: Danish Siddiqui/Reuters/Newscom
In a move that raises questions about the future of drug research in India, Piramal Enterprises will end its drug discovery activities. The decision—which involves possible job losses—will affect several hundred scientists, many of whom were recruited internationally to work in Mumbai in one of India’s most sophisticated pharmaceutical labs.
The company has been considered an Indian leader in drug research since opening its discovery labs in 2004. Within the firm, drug discovery was championed by the vice chairman, Swati A. Piramal, a medical doctor who also holds a master’s degree from the Harvard School of Public Health.
“After reevaluating the risk-benefits of new chemical entity research, the company decided to focus resources on our other areas of R&D with shorter development timelines and different risk profiles,” Piramal tells C&EN.
read all at
http://cen.acs.org/articles/92/i37/Piramal-Drops-Drug-Discovery.html

Piramal Enterprises, which sold off its domestic formulations business to Abbott in a multi-billion dollar deal a few years ago, is now shutting down its Mumbai-based R&D unit which would in effect bring to an end its early stage drug discovery business.
Separate media reports, citing Swati Piramal, part of the promoter group of the diversified firm and wife of group chief Ajay Piramal, said, the decision to move away from the drug discovery business was taken given the costs of basic research.
The company would now focus on molecules at an advanced stage of development; resources would be redeployed from basic research to the clinical unit.
Its other research facilities are located in Chennai, Hyderabad, Ahmedabad and Indore, which would continue to be functional.
Although Piramal Enterprises retains its exposure to healthcare as a sector, after selling the key pharma business, it is now more associated with financial services, including investments in infrastructure and real estate sectors.
In an unrelated development, the firm is forming a joint venture with Navin Fluorine International Limited, an Arvind Mafatlal Group company, to develop, manufacture and sell specialty fluorochemicals with a focus on applications in healthcare, according to a company release.
As per the agreement, Piramal Enterprises will hold 51 per cent of the equity share capital of the proposed joint venture company, whereas the remaining 49 per cent will be held by Navin.
In the first phase of development, the JV is expected to invest around Rs 120 crore in India for this project.
Mumbai-based Navin Fluorine has a turnover of around $100 million. It specialises in specialty fluorine. It had acquired UK-based Manchester Organics, a specialty fluorochemicals research company in 2011.

Piramal Enterprises vice-chairperson Swati Piramal
Read more at: http://www.livemint.com/Companies/7weGbimcrdp7lrKH0YaSnL/Piramal-to-exit-drug-discovery-business.html?utm_source=copy
THE GROWING IMPACT OF CLICK CHEMISTRY ON DRUG DISCOVERY
The growing impact of click chemistry on drug discovery
HC Kolb, KB Sharpless – Drug discovery today, 2003 – Elsevier
Click chemistry is a modular approach that uses only the most practical and reliable
chemical transformations. Its applications are increasingly found in all aspects of drug
discovery, ranging from lead finding through combinatorial chemistry and target-templated …
chemical transformations. Its applications are increasingly found in all aspects of drug
discovery, ranging from lead finding through combinatorial chemistry and target-templated …
Click chemistry is a modular approach that uses only the most practical
and reliable chemical transformations. Its applications are increasingly
found in all aspects of drug discovery, ranging from lead finding through
combinatorial chemistry and target-templated in situchemistry, to proteomics
and DNA research, using bioconjugation reactions. The copper-(I)-catalyzed
1,2,3-triazole formation from azides and terminal acetylenes is a particularly
powerful linking reaction, due to its high degree of dependability, complete
specificity, and the bio-compatibility of the reactants. The triazole products
are more than just passive linkers; they readily associate with biological
targets, through hydrogen bonding and dipole interactions.
and reliable chemical transformations. Its applications are increasingly
found in all aspects of drug discovery, ranging from lead finding through
combinatorial chemistry and target-templated in situchemistry, to proteomics
and DNA research, using bioconjugation reactions. The copper-(I)-catalyzed
1,2,3-triazole formation from azides and terminal acetylenes is a particularly
powerful linking reaction, due to its high degree of dependability, complete
specificity, and the bio-compatibility of the reactants. The triazole products
are more than just passive linkers; they readily associate with biological
targets, through hydrogen bonding and dipole interactions.
GET YOUR PDF AT
http://image.sciencenet.cn/olddata/kexue.com.cn/blog/admin//images/upfiles/2007101622257911935.pdf


Macrocycles in new drug discovery

Macrocycles in new drug discovery
Summary | Full Text | PDF (3354 KB) | PDF Plus (3440 KB) | Add to Favorites | Related
http://www.future-science.com/doi/full/10.4155/fmc.12.93?src=recsys
The use of drug-like macrocycles is emerging as an exciting area of medicinal chemistry, with several recent examples highlighting the favorable changes in biological and physicochemical properties that macrocyclization can afford. Natural product macrocycles and their synthetic derivatives have long been clinically useful and attention is now being focused on the wider use of macrocyclic scaffolds in medicinal chemistry in the search for new drugs for increasingly challenging targets. With the increasing awareness of concepts of drug-likeness and the dangers of ‘molecular obesity’, functionalized macrocyclic scaffolds could provide a way to generate ligand-efficient molecules with enhanced properties. In this review we will separately discuss the effects of macrocyclization upon potency, selectivity and physicochemical properties, concentrating on recent case histories in oncology drug discovery. Additionally, we will highlight selected advances in the synthesis of macrocycles and provide an outlook on the future use of macrocyclic scaffolds in medicinal chemistry.
Drug discovery: a view through the looking glass

Liposome microarray technology is a new solution to the growing costs of drug discovery [1]. As numerous blockbuster drugs’ patents expire and regulatory requirements for new drugs tighten, the future for drug discovery begins to look rather uncertain. For example, through 2015, biologic drugs worth more than US$80 billion in global sales will lose patent protection
Drug discovery: a view through the looking glass
Steven Lenhert, Franklin G Vellanti
Citation | Full Text | PDF (1063 KB) | PDF Plus (1071 KB) |
http://www.future-science.com/doi/full/10.4155/fmc.12.125?src=recsys
Drug discovery: Past and present

The field of biotechnology has revolutionized the drug discovery process. Recombinant DNA-driven drug discovery process is beginning to add new avenues for some old drugs. In its infancy, genetic engineering was considered useful only for the production of therapeutic proteins. Insulin, for example, previously prepared by isolation of pancreatic tissue of bovine or porcine species, can now be prepared identical to human insulin by biotechnology. Companies like Genentech and Biogen were founded solely with this objective. However, proteins do not make ideal drugs, being difficult to administer, rapidly cleared, and potentially immunogenic. Despite these disadvantages, a rapidly increasing number of “biopharmaceuticals” including recombinant proteins, therapeutic monoclonal antibodies, and even antisense oligonucleosides have been approved for indications ranging from metastatic breast cancer (Herceptin) to rheumatoid arthritis (Remicade, Enbrel).
READ AT
| Giridhar R. Drug discovery: Past and present. J Adv Pharm Technol Res 2012;3:2 |
| Giridhar R. Drug discovery: Past and present. J Adv Pharm Technol Res [serial online] 2012 [cited 2014 Aug 15];3:2. Available from: http://www.japtr.org/text.asp?2012/3/1/2/93554 |
