
Home » AIDS
Category Archives: AIDS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
An Improved Process for the Preparation of Tenofovir Disoproxil Fumarate

Tenofovir Disoproxil Fumarate
For full details see end of page
![]()
PAPER

The current three-step manufacturing route for the preparation of tenofovir disoproxil fumarate (1) was assessed and optimized leading to a higher yielding, simpler, and greener process. Key improvements in the process route include the refinement of the second stage through the replacement of the problematic magnesium tert-butoxide (MTB) with a 1:1 ratio of a Grignard reagent and tert-butanol. The development of a virtually solvent-free approach and the establishment of a workup and purification protocol which allows the isolation of a pure diethyl phosphonate ester (8) was achieved
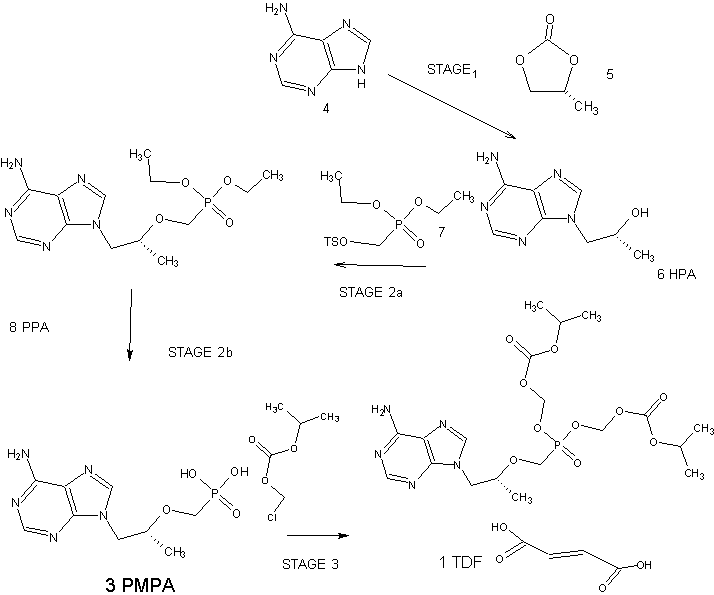
see………….http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00364
An Improved Process for the Preparation of Tenofovir Disoproxil Fumarate
![]()
Department of Chemistry, Natural and Agricultural Sciences, University of Pretoria, 2 Lynnwood Road, Hatfield, 0002, Gauteng, South Africa

///////

Tenofovir Disoproxil Fumarate
5-[[(1R)-2-(6-Amino-9H-purin-9-yl)-1-methylethoxy]methyl]-2,4,6,8-tetraoxa-5-phosphanonanedioic Acid 1,9-Bis(1-methylethyl) Ester 5-Oxide (2E)-2-Butenedioate; GS 4331-05; PMPA Prodrug; Tenofovir DF; Virea; Viread;
GILEAD-4331-300
201341-05-1 – free base, (Tenofovir Disoproxil
Fumarate |
202138-50-9 |
| 113-115°C (dec.) |
| CAS No.: | 202138-50-9 |
|---|---|
| Name: | Tenofovir disoproxil fumarate |
| Molecular Structure: | |
 |
|
| Formula: | C19H30N5O10P.C4H4O4 |
| Molecular Weight: | 635.51 |
| Synonyms: | TDF;PMPA prodrug;Tenofovir Disoproxil Fumarate [USAN];9-((R)-2-((Bis(((isopropoxycarbonyl)oxy)methoxy)phosphinyl)methoxy)propyl)adenine, fumarate;201341-05-1;Bis(NeopentylOC)PMPA;Viread;GS 4331-05 (*1:1 Fumarate salt*);Viread (*1:1 Fumarate salt*);Truvada;Tenofovir DF;[[(2R)-1-(6-aminopurin-9-yl)propan-2-yl]oxymethyl-(propan-2-yloxycarbonyloxymethoxy)phosphoryl]oxymethyl propan-2-yl carbonate; |
|
|
|
|
|
Tenofovir disoproxil is an antiretroviral medication used to prevent and treat HIV/AIDS and to treat chronic hepatitis B.[1] The active substance is tenofovir, while tenofovir disoproxil is a prodrug that is used because of its better absorption in the gut.
The drug is on the World Health Organization’s List of Essential Medicines, the most important medications needed in a basic health system.[2] It is marketed by Gilead Sciences under the trade name Viread (as the fumarate, TDF).[3] As of 2015 the cost for a typical month of medication in the United States is more than 200 USD.[4]
Medical uses
- HIV-1 infection: Tenofovir is indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults and pediatric patients 2 years of age and older.[5] This indication is based on analyses of plasma HIV-1 RNA levels and CD4 cell counts in controlled studies of tenofovir in treatment-naive and treatment-experienced adults.
- Tenofovir is indicated for the treatment of chronic hepatitis B in adults and pediatric patients 12 years of age and older.[5][6]
HIV risk reduction
A Cochrane review examined the use of tenofovir for prevention of HIV before exposure. It found that both tenofovir alone and the tenofovir/emtricitabine combination decreased the risk of contracting HIV.[7]
The U. S. Centers for Disease Control and Prevention (CDC) conducted a study in partnership with the Thailand Ministry of Public Health to ascertain the effectiveness of providing people who inject drugs illicitly with daily doses of the antiretroviral drug tenofovir as a prevention measure. The results of the study were released in mid-June 2013 and revealed a 48.9%-reduced incidence of the virus among the group of subjects who received the drug, in comparison to the control group who received a placebo. The principal investigator of the study stated: “We now know that pre-exposure prophylaxis can be a potentially vital option for HIV prevention in people at very high risk for infection, whether through sexual transmission or injecting drug use.”[8]
Adverse effects
The most common side effects associated with tenofovir include nausea, vomiting, diarrhea, and asthenia. Less frequent side effects include hepatotoxicity, abdominal pain, and flatulence.[9] Tenofovir has also been implicated in causing renal toxicity, particularly at elevated concentrations.[10]
Tenofovir can cause acute renal failure, Fanconi syndrome, proteinuria, or tubular necrosis.[citation needed] These side effects are due to accumulation of the drug in proximal tubules.[citation needed] Tenofovir can interact with didanosine by increasing didanosine’s concentration.[citation needed] It also decreases the concentration of atazanavir sulfate.[citation needed]
Mechanism of action
Tenofovir is a defective adenosine nucleotide that selectively interferes with the action of reverse transcriptase, but only weakly interferes with mammalian DNA polymerases α, β, and mitochondrial DNA polymerase γ.[11] Tenofovir prevents the formation of the 5′ to 3′ phosphodiester linkage essential for DNA chain elongation. A phosphodiester bond cannot be formed because the tenofovir molecule lacks an —OH group on the 3′ carbon of its deoxyribose sugar.[11] Once incorporated into a growing DNA strand, tenofovir causes premature termination of DNA transcription. The drug is classified as a nucleotide analogue reverse transcriptase inhibitor (NRTI), that inhibits reverse transcriptase.[11] Reverse transcriptase is a crucial viral enzyme in retroviruses such as human immunodeficiency virus (HIV) and in hepatitis B virus infections.[5]
History
Tenofovir was initially synthesized by Antonín Holý at the Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic in Prague. The patent[12] filed by Holý in 1984 makes no mention of the potential use of the compound for the treatment of HIV infection, which had only been discovered one year earlier.
In 1985, De Clercq and Holý described the activity of PMPA against HIV in cell culture.[13] Shortly thereafter, a collaboration with the biotechnology company Gilead Sciences led to the investigation of PMPA’s potential as a treatment for HIV infected patients. In 1997 researchers from Gilead and the University of California, San Francisco demonstrated that tenofovir exhibits anti-HIV effects in humans when dosed by subcutaneous injection.[14]
The initial form of tenofovir used in these studies had limited potential for widespread use because it was not absorbed when administered orally. A medicinal chemistry team at Gilead developed a modified version of tenofovir, tenofovir disoproxil.[15] This version of tenofovir is often referred to simply as “tenofovir”. In this version of the drug, the two negative charges of the tenofovir phosphonic acid group are masked, thus enhancing oral absorption.
Tenofovir disoproxil was approved by the U.S. FDA on October 26, 2001, for the treatment of HIV, and on August 11, 2008, for the treatment of chronic hepatitis B.[16][17]
Drug forms
Tenofovir disoproxil is a prodrug form of tenofovir. It is also marketed under the brand name Reviro by Dr. Reddy’s Laboratories. Tenofovir is also available in a fixed-dose combination with emtricitabine in a product with the brand name Truvada for once-a-day dosing. Efavirenz/emtricitabine/tenofovir disoproxil (brand name Atripla) — a fixed-dose triple combination of tenofovir, emtricitabine, and efavirenz, was approved by the FDA on 12 July 2006 and is now available, providing a single daily dose for the treatment of HIV.
Therapeutic drug monitoring
Tenofovir may be measured in plasma by liquid chromatography. Such testing is useful for monitoring therapy and to prevent drug accumulation and toxicity in people with kidney or liver problems.[18][19][20]
PATENT
http://www.google.com/patents/EP2545063A2?cl=en
Tenofovir Disoproxil is chemically known as 9-[-2-(R)-[[bis [[(isopropoxycarbonyl) oxy]methoxy] phosphinoyl]methoxy]propyl]-adenine, having the following structural formula-I.

Formula-I
Tenofovir is a highly potent antiviral agent, particularly for the therapy or prophylaxis of retroviral infections and belongs to a class of drugs called Nucleotide Reverse Transcriptase Inhibitors (NRTI) which blocks reverse transcriptase an enzyme crucial to viral production in HIV-infected people.
Tenofovir Disoproxil and its pharmaceutically acceptable salts were first disclosed in US 5,922,695. This patent discloses the preparation of Tenofovir Disoproxil by the esterification of Tenofovir with chloromethyl isopropyl carbonate using l-methyl-2- pyrrolidinone and triethylamine. In this patent Tenofovir Disoproxil is converted into its Fumarate salt without isolation. PCT Publication WO 2008007392 discloses process for the preparation of Tenofovir Disoproxil fumarate, wherein the isolated crystalline Tenofovir Disoproxil is converted into fumarate salt.
Tenofovir Disoproxil processes in the prior art are similar to process disclosed in product patent US 5,922,695. According to the prior art processes, Tenofovir Disoproxil fumarate obtained is having low yields and also show the presence of impurities such as dimers.
scheme- 1.

Tenofovir disoproxil chloromethyl isopropyl carbonate

Tenofovir disoproxil fumarate
Example 1 : Process for the preparation of Tenofovir Disoproxil fumarate
Toluene (500 ml) was added to the Tenofovir (100 gm) and stirred at room temperature. To this triethylamine (66.31 gm) was added, temperature was raised to 90° C and water was collected by azeotropic distillation at 110°C. Toluene was completely distilled under vacuum at same temperature. The reaction mixture was cooled to room temperature and to this a mixture of N-methyl pyrrolidine (300 gm), triethylamine (66.31 gm), Tetrabutyl ammonium bromide (52.8 gm) and trimethyl silyl chloride (17.8 gm) were added. The above reaction mixture was heated to 50-55 °C and was added slowly chloromethyl. isopropyl carbonate (CMIC) and maintained the reaction mixture at 50-55°C for 5 hrs. (Qualitative HPLC analysis shows about 85% product formation). The above reaction mixture was cooled to room temperature and filtered. The filtrate was added to DM water at 5-10°C and extract with dichloromethane. The combined dichloromethane layer was concentrated under vacuum and the crude was Co-distilled with cyclohexane and this crude was taken into isopropyl alcohol (1000 ml). To this fumaric acid (38 gm) was added and temperature was raised to 50° C. The reaction mixture was filtered and filtrate was cooled to 5-10° C. The obtained solid was filtered and washed with isopropyl alcohol. The compound was dried under vacuum to yield Tenofovir Disoproxil fumarate (140 gm).
Example-2 : Preparation of Tenofovir
N-methyl-2-pyrrolidone (25 gm) was taken along with toluene (150 gm) into a reaction vessel. l-(6-amino-purin-9-yl)-propan-2-ol (100 gm); toluene-4-sulfonic acid diethoxy phosphoryl methyl ester (200 gm) and magnesium ter-butoxide (71.2 gm) were also taken at’ 25-35°C. Temperature was raised to 74-75 °C and maintained for 5-6hrs. After completion of reaction, acetic acid (60 gm) was added and maintained for 1 hr. Later aq.HBr (332 gm) was taken and heated to 90-95 °C. After reaction completion, salts were filtered and filtrate was subjected to washings with water and extracted into methylene dichloride. Later pH was adjusted using CS lye below 10 °C. Tenofovir product was isolated using acetone.
Yield: 110 gm.
Example 3 : Preparation of Tenofovir disoproxil
(R)-9-[2-(phosphonomethoxy)propyl]adenine (25 gm), triethyl amine (25 ml) and cyclohexane (200 ml) were combined and heated to remove water and the solvent was distilled off under vacuum. The reaction mass was cooled to room temperature N-methyl pyrrolidinone (55 ml), triethyl amine (25 ml) and tetra butyl ammonium bromide(54 gms) were added to the reaction mixture. The reaction mass was heated to 50-60°C and chloromethyl isopropyl carbonate (65 gm) was added and maintained for 4-8 hrs at 50- 60°C and then cooled to 0°C. The reaction mass was diluted with chilled water or ice and precipitated solid product was filtered. The mother liquor was extracted with methylene chloride (150 ml). The methylene chloride layer was washed with water (200 ml). The filtered solid and the methylene chloride layer were combined and washed with water and the solvent was distilled under vacuum. Ethyl acetate was charged to the precipitated solid. The reaction mass was then cooled to 0-5 °C and maintained for 6 hrs. The solid was filtered and dried to produce Tenofovir disoproxil (45 gm).
CLIPS
The reaction of chloromethyl chloroformate (I) with isopropyl alcohol (II) by means of pyridine or triethylamine in ether gives the mixed carbonate (III), which is then condensed with (R)-PMPA (IV) by means of diisopropyl ethyl-amine in DMF.
| US 5922695; WO 9804569 |

CLIP 2
1) The protection of isobutyl D-(+)-lactate (I) with dihydropyran (DHP)/HCl in DMF gives the tetrahydropyranyloxy derivative (II), which is reduced with bis(2-methoxyethoxy)aluminum hydride in refluxing ether/ toluene yielding 2(R)-(tetrahydropyranyloxy)-1-propanol (III). The tosylation of (III) with tosyl chloride as usual affords the expected tosylate (VI), which is condensed with adenine (V) by means of Cs2CO3 in hot DMF, affording 9-[2(R)-(tetrahydropyranyloxy)propyl]adenine (VI). The deprotection of (VI) with sulfuric acid affords 9-[2(R)-hydroxypropyl]adenine (VII), which is N-benzoylated with benzoyl chloride/chlorotrimethylsilane in pyridine to give the benzamide (VIII), which is condensed with tosyl-oxymethylphosphonic acid diisopropyl ester (IX) by means of NaH in DMF to yield 9-[2(R)-(diisopropoxyphosphorylmethoxy)propyl]adenine (X). Finally, this compound is hydrolyzed by means of bromotrimethylsilane in acetonotrile.

2) The reaction of the previously described (R)-2-(2-tetrahydropyranyloxy)-1-propanol (III) with benzyl bromide (XI) by means of NaH in DMF, followed by a treatment with Dowex 50X, gives 1-benzyloxy-2(R)-propanol (XII), which is condensed with tosyloxymethylphosphonic acid diisopropyl ester (IX) by means of NaH in THF, yielding 2-benzyloxy-1(R)-methylethoxymethylphosphonic acid diisopropyl ester (XIII). The hydrogenolysis of (XIII) over Pd/C in methanol affords 2-hydroxy-1(R)-methylethoxymethylphosphonic acid diisopropyl ester (XIV), which is tosylated with tosyl chloride/dimethyl-aminopyridine in pyridine to give the expected tosylate (XV). The condensation of (XV) with adenine (VI) by means of Cs2CO3 in hot DMF yields 9-[2(R)-(diisopropoxyphosphorylmethoxy)propyl]adenine (X), which is finally hydrolyzed as before.

3) The catalytic hydrogenation of (S)-glycidol (XVI) over Pd/C gives the (R)-1,2-propanediol (XVII), which is esterified with diethyl carbonate (XVIII)/NaOEt, yielding the cyclic carbonate (XIX). The reaction of (XIX) with adenine (V) by means of NaOH in DMF affords 9-[2(R)-hydroxypropyl]adenine (VII), which is condensed with tosyloxymethylphosphonic acid diethyl ester (XX) by means of lithium tert-butoxide in THF, giving 9-[2(R)-(diethoxyphosphorylmethoxy)propyl]adenine (XXI). Finally, this compound is hydrolyzed with bromotrimethylsilane as before. Compound (XX) is obtained by reaction of diethyl phosphite (XXII) with paraformaldehyde, yielding hydroxy- methylphosphonic acid diethyl ester (XXIII), which is finally tosylated as usual.

References
- 1 “Viread”. The American Society of Health-System Pharmacists. Retrieved 31 July 2015.
- 2
- “WHO Model List of EssentialMedicines” (PDF). World Health Organization. October 2013. Retrieved 22 April 2014.
- 3
- Emau P, Jiang Y, Agy MB, et al. (2006). “Post-exposure prophylaxis for SIV revisited: Animal model for HIV infection”. AIDS Res Ther 3: 29. doi:10.1186/1742-6405-3-29. PMC 1687192. PMID 17132170.
- 4
- Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 66. ISBN 9781284057560.
- 5
- Gilead Sciences, Inc. Prescribing Information. Revised: November 2012.
- 6
- Guidelines for the prevention, care and treatment of persons with chronic hepatitis B infection, WHO, Publication details:Pages: 166 Publication date: March 2015 Languages: English ISBN 978 92 4 154905 9
- 7
- Okwundu CI, Uthman OA, Okoromah CAN (2012). “Antiretroviral pre-exposure prophylaxis (PrEP) for preventing HIV in high-risk individuals”. Cochrane Database Syst Rev 7 (7): CD007189. doi:10.1002/14651858.CD007189.pub3. PMID 22786505.
- 8
- Emma Bourke (14 June 2013). “Preventive drug could reduce HIV transmission among injecting drug users”. The Conversation Australia. The Conversation Media Group. Retrieved 17 June 2013.
- 9
- USPDI. Thompson. 2005. pp. 2741–2.
- 10
- “Viread Prescribing Guidelines” (PDF). U.S. Food and Drug Administration. March 2006. Archived from the original (PDF) on 2007-09-30. Retrieved 2007-02-12.
- 11
- Drugbank: Tenofovir
- 12
- “Patent US4808716 – 9-(phosponylmethoxyalkyl) adenines, the method of preparation and … – Google Patents”.
- 13
- A US 4724233 A, De Clercq, Erik; Antonin Holy & Ivan Rosenberg, “Therapeutical application of phosphonylmethoxyalkyl adenines”, published 1985-04-25
- 14
- Deeks SG, Barditch-Crovo P, Lietman PS, et al. (September 1998). “Safety, pharmacokinetics, and antiretroviral activity of intravenous 9-[2-(R)-(Phosphonomethoxy)propyl]adenine, a novel anti-human immunodeficiency virus (HIV) therapy, in HIV-infected adults”. Antimicrob. Agents Chemother. 42 (9): 2380–4. PMC 105837. PMID 9736567.
- 15
- “Patent US5977089 – Antiviral phosphonomethoxy nucleotide analogs having increased oral … – Google Patents”.
- 16
- FDA letter of approval (regarding treatment of hepatitis B)
- 17
- FDA Clears Viread for Hepatitis B
- 18
- Delahunty T, Bushman L, Robbins B, Fletcher CV (2009). “The simultaneous assay of tenofovir and emtricitabine in plasma using LC/MS/MS and isotopically labeled internal standards”. J. Chrom. B 877 (20–21): 1907–1914. doi:10.1016/j.jchromb.2009.05.029.
- 19
- Kearney BP, Yale K, Shah J, Zhong L, Flaherty JF (2006). “Pharmacokinetics and dosing recommendations of tenofovir disoproxil fumarate in hepatic or renal impairment”. Clin. Pharmacokinet. 45 (11): 1115–24. doi:10.2165/00003088-200645110-00005. PMID 17048975.
- 20
- R. Baselt, Disposition of Toxic Drugs and Chemicals in Man, 8th edition, Biomedical Publications, Foster City, California, 2008, pp. 1490–1492.
External links
| WO2008007392A2 | Jul 11, 2007 | Jan 17, 2008 | Matrix Lab Ltd | Process for the preparation of tenofovir |
| US5922695 | Jul 25, 1997 | Jul 13, 1999 | Gilead Sciences, Inc. | Antiviral phosphonomethyoxy nucleotide analogs having increased oral bioavarilability |
| WO2015051874A1 | Sep 22, 2014 | Apr 16, 2015 | Zentiva, K.S. | An improved process for the preparation of tenofovir disoproxil and pharmaceutically acceptable salts thereof |
| CN103360425A * | Apr 1, 2012 | Oct 23, 2013 | 安徽贝克联合制药有限公司 | Synthesis method of tenofovir disoproxil and fumarate thereof |
| CN103374038A * | Apr 11, 2012 | Oct 30, 2013 | 广州白云山制药股份有限公司广州白云山制药总厂 | Preparation method of antiviral medicine |
| CN103848868A * | Dec 4, 2012 | Jun 11, 2014 | 蚌埠丰原涂山制药有限公司 | Method for preparing tenofovir |
| CN103848869A * | Dec 4, 2012 | Jun 11, 2014 | 上海医药工业研究院 | Method for preparing tenofovir |
| CN103980319A * | Apr 24, 2014 | Aug 13, 2014 | 浙江外国语学院 | Preparation method of tenofovir |
| CN103980319B * | Apr 24, 2014 | Dec 2, 2015 | 浙江外国语学院 | 一种泰诺福韦的制备方法 |
| EP2860185A1 | Oct 9, 2013 | Apr 15, 2015 | Zentiva, k.s. | An improved process for the preparation of Tenofovir disoproxil and pharmaceutically acceptable salts thereof |
The chemical name of tenofovir disoproxil fumarate is 9-[(R)-2[[bis[[(isopropoxycarbonyl)oxy]methoxy]phosphinyl]methoxy]propyl]adenine fumarate (1:1). It has a molecular formula of C19H30N5O10P • C4H4O4 and a molecular weight of 635.52. It has the following structural formula:
|
Tenofovir disoproxil fumarate is a white to off-white crystalline powder with a solubility of 13.4 mg/mL in distilled water at 25 °C. It has an octanol/phosphate buffer (pH 6.5) partition coefficient (log p) of 1.25 at 25 °C.
VIREAD is available as tablets or as an oral powder.
VIREAD tablets are for oral administration in strengths of 150, 200, 250, and 300 mg of tenofovir disoproxil fumarate, which are equivalent to 123, 163, 204 and 245 mg of tenofovir disoproxil, respectively. Each tablet contains the following inactive ingredients: croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, and pregelatinized starch. The 300 mg tablets are coated with Opadry II Y-3010671-A, which contains FD&C blue #2 aluminum lake, hypromellose 2910, lactose monohydrate, titanium dioxide, and triacetin. The 150, 200, and 250 mg tablets are coated with Opadry II 32K-18425, which contains hypromellose 2910, lactose monohydrate, titanium dioxide, and triacetin.
VIREAD oral powder is available for oral administration as white, taste-masked, coated granules containing 40 mg of tenofovir disoproxil fumarate per gram of oral powder, which is equivalent to 33 mg of tenofovir disoproxil. The oral powder contains the following inactive ingredients: mannitol, hydroxypropyl cellulose, ethylcellulose, and silicon dioxide.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
Bis{[(isopropoxycarbonyl)oxy]methyl} ({[(2R)-1-(6-amino-9H-purin-9-yl)-2-propanyl]oxy}methyl)phosphonate
|
|
| Clinical data | |
| Trade names | Viread |
| AHFS/Drugs.com | monograph |
| Pregnancy category |
|
| Routes of administration |
Oral (tablets) |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 25% |
| Identifiers | |
| CAS Number | 201341-05-1 |
| ATC code | J05AF07 (WHO) |
| PubChem | CID 5481350 |
| ChemSpider | 4587262 |
| UNII | F4YU4LON7I |
| ChEBI | CHEBI:63717 |
| NIAID ChemDB | 080741 |
| Chemical data | |
| Formula | C19H30N5O10P |
| Molar mass | 519.443 g/mol |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
({[(2R)-1-(6-amino-9H-purin-9-yl)propan-2-yl]oxy}methyl)phosphonic acid
|
|
| Clinical data | |
| MedlinePlus | a602018 |
| Routes of administration |
In form of prodrugs |
| Pharmacokinetic data | |
| Protein binding | < 1% |
| Biological half-life | 17 hours |
| Excretion | Renal |
| Identifiers | |
| CAS Number | 147127-20-6 |
| ATC code | None |
| PubChem | CID 464205 |
| DrugBank | DB00300 |
| ChemSpider | 408154 |
| UNII | 99YXE507IL |
| KEGG | D06074 |
| ChEBI | CHEBI:63625 |
| ChEMBL | CHEMBL483 |
| Synonyms | 9-(2-Phosphonyl-methoxypropyly)adenine (PMPA) |
| Chemical data | |
| Formula | C9H14N5O4P |
| Molar mass | 287.213 g/mol |
///////
FOSTEMSAVIR ,фостемсавир , فوستيمسافير , 磷坦姆沙韦 ,ホステムサビル;
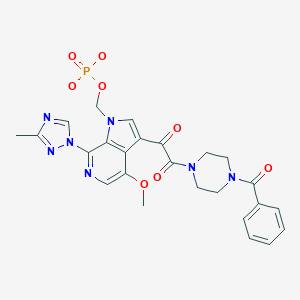

Fostemsavir
GSK3684934
CAS 864953-29-7
- Molecular FormulaC25H26N7O8P
- Average mass583.490 Da
- ホステムサビル;
[3-[2-(4-benzoylpiperazin-1-yl)-2-oxoacetyl]-4-methoxy-7-(3-methyl-1,2,4-triazol-1-yl)pyrrolo[2,3-c]pyridin-1-yl]methyl dihydrogen phosphate
- BMS 663068
- BMS663068
- Fostemsavir tromethamine
- UNII-2X513P36U0
Fostemsavir tromethamine [USAN],
CAS 864953-39-9,
MW 704.6303

Fostemsavir (GSK3684934/BMS-663068) is an experimental HIV entry inhibitor and a prodrug of temsavir (BMS-626529). It is under development by [ViiV Healthcare / GlaxoSmithKline]] for use in the treatment of HIV infection. By blocking the gp120 receptor of the virus, it prevents initial viral attachment to the host CD4+ T cell and entry into the host immune cell; its method of action is a first for HIV drugs.[1] Because it targets a different step of the viral lifecycle, it offers promise for individuals with virus that has become highly resistant to other HIV drugs.[2] Since gp120 is a highly conserved area of the virus, the drug is unlikely to promote resistance to itself via generation of CD4-independent virus.[3]



Example 6Preparation of Compound I from Compound D′ (Example 5)

N-Benzoylpiperazine HCl, Compound Db, (11.73 g, 51.74 mmol) was added to a mixture of Compound D′ (14.83 g, 47.03 mmol) (prepared in Example 5) in dry THF (265 mL) and dry DMF (29.5 mL). NaOt-Bu, 30% w/w (52.3 mL, 147 mmol) was added dropwise (30 min.) keeping the temperature at 17-21° C. The resulting yellow slurry was stirred at 17-20° for 1 h more, then cooled to about 5° C. The mixture was slowly poured into cold water (90 mL) and the flask rinsed with additional water (10 mL). The pH of the resulting yellow solution was adjusted to 6-7 with slow addition (˜20 min., 5-12° C.) of 1 N HCl (105 mL). The resulting slurry was warmed and stirred at room temperature for 1.5 h. The slurry was filtered and the cake washed with water (2×60 mL) then dried in vacuo at 65-70° C. for 5 h giving 18.4 g Compound I as a white solid (82.6%), HPLC AP 99.4. 1H NMR (400 MHz, d6-DMSO): δ 2.48 (s, 3H), 3.43 (b, 4H), 3.67 (b, 4H), 3.99 (s, 3H), 7.45 (s, 5H), 7.88 (s, 1H), 8.24 (s, 1H), 9.22 (s, 1H), 12.39 (s, 1H). 13C NMR (100 MHz, d6-DMSO): 13.85, 40.65, 45.22, 56.85, 114.19, 121.02, 122.78, 123.65, 127.06, 128.42, 129.61, 129.70, 135.51, 138.59, 142.18, 149.23, 161.38, 166.25, 169.30, 185.51.
If necessary, the product could be further purified by recrystallization from acetic acid-water-ethanol, ethanol-water, or acetone-water. For example: A mixture of Compound I (25.0 g), glacial acetic acid (260 mL) and DI water (13.8 mL) was heated to 80° C. and held with stirring (overhead) until a solution was obtained (40 min.). The batch was cooled to 70° C. and seeded (0.5 g). With slow agitation (100 rpm), EtOH (300 mL) was added slowly (1 h), keeping the temperature at 70° C. The resulting slurry was kept at 70° C. for 1 h more with very slow stirring. The slurry was cooled to 20° C. over 2 hours and held at 20° C. for over 4 hours. The slurry was filtered, the wet cake washed with EtOH (125 mL), and the solid dried in vacuo at 70° C. (≧16 h), giving 22.6 g Compound I as a white solid (88.4%).

The development of a short and efficient synthesis of a complex 6-azaindole, BMS-663068, is described. Construction of the 6-azaindole core is quickly accomplished starting from a simple pyrrole, via a regioselective Friedel–Crafts acylation, Pictet–Spengler cyclization, and a radical-mediated aromatization. The synthesis leverages an unusual heterocyclic N-oxide α-bromination to functionalize a critical C–H bond, enabling a highly regioselective copper-mediated Ullmann–Goldberg–Buchwald coupling to install a challenging triazole substituent. This strategy resulted in an efficient 11 step linear synthesis of this complex clinical candidate

Attachment inhibitor BMS-663068 is currently in clinical development for the treatment of HIV infection. Key steps in the synthesis depicted are (1) a radical-mediated redox-aromatization to generate the 6-azaindole (B → C) and (2) the regioselective bromination of an N-oxide using PyBroP (D → E).
High regioselectivity was observed in the copper(I)-mediated Ullmann–Goldberg–Buchwald coupling (H → K) using the diamine ligand J (N1/N2 = 22:1), whereas a thermal SNAr reaction gave N1/N2 = 1:1. Alternative conditions for the bromination of the N-oxide D led mainly to deoxygenation.
………………………………….
http://www.google.com/patents/US20050209246
Preparation of Compound IVc

Procedure: To a solution of the acid 6-81 (3.01 g, 10 mmol) and benzoylpiperazine hydrochloride (3.39 g, 15 mmol) in DMF (50 mL) was added triethylamine (10.1 g, 100 mmol, 10 eq.), followed by 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDC; 5.75 g, 30 mmol) under N2 and the mixture stirred at room temperature for 22 h after sonication and at 40° C. for 2 h. The mixture was concentrated in vacuo to remove DMF and TEA, and to the residual solution was added water (200 mL) under stirring and sonication. The precipitates formed were collected, washed with water and dried in vacuo to obtain 2.8 g (5.9 mmol, Y. 59%) of the title compound IVc as off-white solid. The filtrate was extracted with CH2Cl2 (x2). The CH2Cl2 extracts were dried (Na2SO4), filtered and concentrated to gum which was triturated with Et2O to obtain a solid. This solid was suspended and triturated with MeOH to obtain 400 mg of the title compound IVc as off-white solid. Total yield: 3.2 g (6.8 mmol, Y. 68%): MS m/z 474 (MH); HRMS (ESI) m/z calcd for C24H24N7O4 (M+H) 474.1890, found 474.1884 (Δ-1.2 ppm); 1H NMR (DMSO-d6) δ ppm 2.50 (3H, s, overlapped with DMSO peaks), 3.43 (4H, br, CH2N), 3.68 (4H, br, CH2N), 3.99 (3H, s, CH3O), 7.46 (5H, br. s, Ar—Hs), 7.88 (1H, s, indole-H-5), 8.25 (1H, s, indole-H-2), 9.25 (1H, s, triazole-H-5), 12.40 (1H, s, NH); 13C-NMR (DMSO-d6) δ ppm 13.78 ,40.58, 45.11, 56.78, 114.11, 120.95, 122.71, 123.60, 126.98, 128.34, 129.6, 135.43, 138.52, 142.10, 149.15, 161.29, 166.17, 169.22, 185.42; UV (MeOH) λ max 233.6 nm (ε 3.43×104), 314.9 nm (ε 1.73×104); Anal: Calc for C24H24N7O4.1/5H2O; C, 60.42; H, 4.94; N, 20.55, Found; C 60.42, H 5.03, N 20.65; KF (H2O) 0.75%.
This reaction can also be performed by use of HATU and DMAP to provide more consistent yield of the title compound: To a suspension of the acid 6-81 (15.6 mmol) and HATU [O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophos phonate] (8.90 g, 23.4 mmol; 1.5 eq.) in DMF (60 mL) and CH2Cl2 (60 mL) was added a mixture of DMAP (5.72 g, 46.8 mmol, 3 eq.) and benzoylpiperazine hydrochloride (5.30 g, 23.4 mmol; 1.5 eq.) in DMF (60 mL) at room temperature and the mixture was stirred under nitrogen atmosphere for 4 hrs. The mixture was concentrated in vacuo to remove CH2Cl2 and most of DMF, and to the residual solution was added water under stirring and sonication. The precipitates formed were collected, washed with water and dried in vacuo to obtain 5.38 g (11.4 mmol, Y. 72.8%) of the title compound IVc as off-white solid: HPLC >95% (AP, uv at 254 nm)
EXAMPLE 5Preparation of Ica, (Disodium Salt)

General Procedure: A suspension of IVc (0.24 g, 0.5 mmol) in anhydrous THF (4 mL) under nitrogen atmosphere was treated with sodium hydride (60% oil dispersion, 0.08 g, 2.0 mmol), and stirred until gas evolution ceased (approximately 5 minutes). The reaction mixture was treated with iodine (0.13 g, 0.5 mmol) and stirred for 2-3 minutes followed by addition of di-tert-butyl chloromethyl phosphate (1.6 g, 6.0 mmol, crude). A stream of nitrogen was allowed to pass over the reaction to facilitate the removal of much or all of the THF. The reaction mixture was stirred overnight. HPLC analysis of crude indicated starting IVc (ca. 56%) and desired adduct (ca. 32%).
Several crude reaction mixtures (a total of 6.7 mmol based on starting material IVc) were re-dissolved in dichloromethane, combined, concentrated in vacuo to remove any remaining THF. The residue was suspended in dichloromethane and TFA (1:1, approximately 40 mL total volume). The mixture was stirred for 1.5-2 hours and then solvent was removed in vacuo. The residue was suspended in dichloromethane and extracted into water (approximately 60 mL) made weakly basic with solid or aqueous sodium bicarbonate. The aqueous layer was reduced in volume by rotary evaporator if required and the solution was loaded onto a C-18 reverse phase column (approximately 80 g of C-18, YMC ODS-Aq, 50 micron) and eluted with water, followed by water containing 2.5% acetonitrile. Fractions containing pure product were pooled and organic solvent was removed by rotary evaporator. Purified product was recovered after lyophilization to give 1.00 g (1.30 mmol, 19% over 2 steps) of the title compound Ica (disodium salt) as an off-white powder: HPLC purity>99% AP at 254 nm (gradient 0-100% B/A; A 10% CH3CN-90% H2O-0.1% TFA, B 90% CH3CN-10% H2O-0.1 % TFA, gradient time 4 min, column YMC ODS-Aq 4.6×50 mm 3 micron); MS-ESI— m/z 482 (M−H minus 2Na)−; HRMS (ESI) m/z calcd for C25H27N7O8P (M+H minus 2Na)+584.1659, found 584.1651 (Δ-1.3 ppm); 1H NMR (D2O, 500 MHz) δ ppm 2.53, 2.54 (3H, 2s), 3.56 (2H, s, CH2N), 3.72 (2H, br.s, CH2N), 3.78, 3.83 (2H, 2br.s, CH2N), 3.94, 3.96 (2H, 2br.s, CH2N), 4.14 (3H, s, CH3O), 5.38, 5.40 (2H, 2d, J=11 Hz), 7.45-7.59 (5H, m, Ar—Hs), 8.07, 8.09 (1H, 2s, indole-H-5), 8.64, 8.67 (1H, 2s, indole-H-2), 8.87, 8.89 (1H, 2s, triazole-H-5); 13C NMR (125.7 MHz, D2O) δ ppm 15.43 (N-Me), 44.03, 44.47, 44.66, 45.05, 48.20, 48.82, 49.60, 50.23, 59.78 (OMe), 75.81 (NCH2O), 115.6, 126.0, 127.2, 129.6, 131.0, 131.7, 132.1, 133.5, 136.8, 147.6, 150.1, 154.2, 164.8, 170.4, 175.8, 189.2; UV (H2O) λmax 220 nm (ε 3.91×104), 249 nm (ε 2.00×104), 303 nm (ε 1.60×104); Anal: Calc for C25H24N7O8PNa2. 8H2O. 0.2NaHCO3; C, 38.39; H, 5.14; N, 12.44, P, 3.93, Na, 6.42 Found; C, 38.16; H, 4.81; N, 12.43, P, 3.72, Na, 6.05; KF (H2O) 17.3%. A less pure fractions were collected to obtain 0.22 g (0.29 mmol, Y. 4%) of the title compound Ica (disodium salt): HPLC purity>95% (AP at 254 nm).
EXAMPLE 7Preparation of Crystalline Ic (Free Acid Mono-Hydrate)

To a mixture of IVc (600 mg, 1.27 mmol) in anhydrous THF (10 ml) in an oven-dried round bottle flask under nitrogen at r.t. was added NaH (153 mg, 6.38 mmol, dry powder, 95%), and the white suspension stirred until no gas evolution was observed. The mixture was then added I2 (375 mg, 1.48 mmol), and stirred at r.t. for 3 h. To the reaction mixture was added NaH (153 mg, 6.38 mmol, dry powder, 95%), and the mixture stirred for about 5 to 10 min. The crude chloromethyl di-tert-butylphosphate (2.0 g, about 1.6 ml, 7.79 mmol) was added to the mixture, which was then stirred at r.t. for 15 h. LCMS analysis of the reaction showed a >97% conversion of the starting material. After evaporation of the volatiles, the residue was added CH2Cl2 (10 ml), cooled in an ice-water bath, slowly added TFA (10 ml) and stirred at r.t. for 3 h. The reaction mixture was then evaporated, and the residue partitioned between CH2Cl2 (50 ml) and H2O (50 ml). The CH2Cl2 layer was poured into the reaction flask that contained some undissolved brownish solid, and this mixture was extracted with a dilute aqueous NaHCO3 solution (50 ml). The aqueous mixture was purified by reverse phase preparative HPLC (solvent A: 10% MeOH-90% H2O-0.1% TFA; solvent B: 90% MeOH-10% H2O-0.1% TFA; start % B=0, final % B=100; gradient time=6 min; flow rate=45 ml/min; column: phenomenex-Luna 30×50 mm, S5; fraction collected: 3.65 to 4.05 min). The fractions collected were evaporated to dryness, and the residue dried under high vacuum to obtain the acid Ic as a pale yellow solid (356.6 mg); 1H NMR: (500 MHz, CD3OD) δ 9.05 (s, 1H), 8.46 (s, 1H), 8.04 (s, 1H), 7.47 (b s, 5H), 5.93 (d, J=12, 2H), 4.10 (s, 3H), 4.00-3.40 (b s, 8H), 2.53 (s, 3H); 19F NMR analysis showed that the material contained residual TFA, (the percentage was not quantified); Analytical HPLC method: Start % B=0, Final % B=100, Gradient time=2 min, Flow Rate=5 mL/min, Column: Xterra MS C18 7u 3.0×50 mm, LC/MS: (ES+) m/z (M+H)+=584, HPLC Rt=0.983.
172.2 mg of the purified acid Ic was dissolved in 1 ml of H2O and then about 0.3 ml of absolute EtOH (200 proof) was added. The mixture was left standing in a refrigerator (temperature about 3° C.) overnight, after which time, crystalline material was observed. The mixture was then warmed to ambient temperature, diluted with H2O to a volumn of 3 mL, and then 20 mL of MeCN was added slowly. Following the completion of addition, the mixture was stirred at r.t. for 2 h and then filtered. The solid collected (90 mg) was dried in vacuo, and then under high vacuum. This material was shown by powder x-ray studies to be crystalline; Elemental Analysis calculated for C25H26N7O8P.H2O: C 49.92; H 4.69; N 16.30; observed: C 49.66; H 4.62; N 15.99; mp=205° C. (measured by differential scanning calorimetry). The 1H NMR pattern for crystalline material was compared with that from the purified acid and both were consistent with the structure.
EXAMPLE 10Preparation of Icb (mono tromethamine salt): [3-[(4-benzoylpiperazin-1-yl)(oxo)acetyl]-4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-yl)-1H-pyrrolo[2, 3-c]pyridin-1-yl]methyl dihydrogen phosphate, 2-amino-2-(hydroxymethyl)propane-1,3-diol salt (1:1). The sequence of reactions is described in Scheme for Example 10.
Scheme for Example 10


Preparation of di-tert-butyl chloromethyl phosphate

A mixture of tetrabutylammonium di-tert-butyl phosphate (57 g, 0.126 mol, Digital Specialty Chemicals) and chloroiodomethane (221 g, 1.26 mol) was stirred at room temperature for four hours before the volatiles were removed under vacuum. 500 ml of ethyl ether was added to the residue and insoluble solid was filtered away. Concentration of the filtrate in vacuo and removal of remaining volatiles using a vacuum pump provided di-tert-butyl chloromethyl phosphate as a light brown or yellow oil, which was utilized in the next step without further purification.
Preparation of IIc: (3-(2-(4-benzoylpiperazin-1-yl)-2-oxoacetyl)-4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-yl)-1H-pyrrolo[2,3-c]pyridin-1-yl)methyl di-tert-butyl phosphate

NaH (2.6 g, 10.3 mmol, 95% in oil, Seq.) was added slowly into a suspension of IVc (10.0 g, 21.1 mmol) in dry THF (100 ml) and the mixture was allowed to stir for 0.5 hour at room temperature. A solution of iodine (5.27 g, 20.8 mmol) dissolved in dry THF (10 ml) was added slowly into the stirring solution at a rate which prevented foaming or a violent reaction. The resultant mixture was stirred for an additional 3 hours before a second 2.6 g portion of NaH was introduced. After 15 minutes at ambient temperature di-tert-butyl chloromethyl phosphate, the entire batch of di-tert-butyl chloromethyl phosphate, obtained from step one, was added. After stirring for 16 hours, the reaction mixture was poured into iced NH4OAc (30%) (120 ml), followed by extraction with EtOAc (3×300 ml). The combined organic extracts were washed with water (100 ml) and then brine (100 ml), dried over Na2SO4, and concentrated under vacuum to afford a residue, which was purified by silica gel chromatography (elution with EtOAc/Et3N (50/1) and then EtOAc/MeOH (100/1)) to give 8.0 g (˜75% AP, ˜41% yield) of diester IIc as a light yellow solid.
1H NMR (500 MHz, CD3OD) δ8.82 (s, 1H), 8.41 (s, 1H), 8.04 (s, 1H), 7.47 (b, 5H), 6.00 (d, 2H, J=14.5 Hz), 4.10 (s, 3H), 4.00-3.40 (b, 8H), 2.49 (s, 3H), 1.28 (s, 18H); 13C NMR (125 MHz, CD3OD) δ18.6, 176.4, 172.9, 168.0, 162.6, 152.6, 147.5, 144.0, 136.5, 131.5, 130.8, 129.9, 129.1, 128.3, 126.1, 124.0, 116.2, 85.8, 75.4, 61.6, 57.7, 30.1, 22.2, 13.7; HRMS m/z: (M+H)+ calcd for C33H43N7O8P 696.29, found 696.34.
Preparation of Icb (mono L tromethamine salt): [3-[(4-benzoylpiperazin-1-yl)(oxo)acetyl]-4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-yl)-1H-pyrrolo[2,3-c]pyridin-1-yl]methyl dihydrogen phosphate, 2-amino-2-(hydroxymethyl)propane-1,3-diol salt (1:1)

500 mg (˜p75 AP, 0.54 mmol) of diester IIc was dissolved in a mixture of water (2.5 ml) and acetone (2.5 ml). The resulting mixture was stirred at 40° C. for 16 hours to complete the solvolysis. To this reaction mixture was added 3.0M aqueous TRIS (mono tromethamine) solution to adjust pH to 3.32. Acetone (30 ml) was slowly added to the reaction mixture in 1 hour.* After complete addition of acetone, the solution was stirred overnight to complete the crystallization of Icb. The solid was collected by filtration and rinsed with 20:1 acetone-water (2×5 mL). The white crystalline solid was dried under house vacuum under nitrogen atomosphere at 50° C. for 24 h to afford 290 mg of Icb (>98.5 AP).
*After adding about 15 and 20 ml of acetone, the reaction mixture was seeded with crystalline Icb.
Icb obtained in the above operation: 1H NMR (500 MHz, CD3OD) δ8.83 (s, 1H), 8.52 (s, 1H), 8.02 (s, 1H) 7.49 (b, 5H), 5.469 (d, 2H, J=13 Hz), 4.11 (s, 3H), 4.00-3.40 (m, 8H), 3.66 (s, 6H), 2.50 (s, 3H); 13C NMR (125 MHz, CD3OD) δ185.6, 171.9, 167.4, 161.4, 151.7, 146.9, 143.8, 135.4, 130.3, 129.7, 128.8, 127.2, 124.9, 122.6, 114.3, 73.5, 61.8, 59.9, 56,5, 46.0, 41.7, 12.6. HRMS m/z: (M-trisamine+H)+ calcd for C25H27N7O8P 584.1659, found 584.1664. Anal. Calcd. C, 49.43; H, 5.29; N, 15.90; P, 4.39; found: C, 49.18; H, 5.38; N, 15.59; P, 4.26. Melting Point 203° C.
Obtained via other process (hydrolysis with TFA in methylene chloride), salt Icb is ˜1 molar mono tromethamine salt with 0.47% of water, 0.1% of acetone and 0.05% of methanol. 1H NMR (500 MHz, d6-DMSO, 30° C.) δ8.77 (s, 1H), 8.48 (s, 1H), 8.00 (s, 1H) 7.44 (b, 5H), 5.42 (d, 2H, J=15 Hz), 4.02 (s, 3H), 3.70-3.30 (m, 8H), 3.41 (s, 6H), 2.38 (s, 3H); 13C NMR (125 MHz, CDCl3, 30° C.) δ184.8, 169.0, 165.8, 160.3, 150.4, 146.2, 143.2, 135.4, 129.4, 128.9, 128.2, 127.7, 126.9, 123.2, 122.2, 112.9, 72.3, 60.7, 59.0, 56.7, 13.4. MS m/z: (M-trisamine+H)+ calcd for C25H27N7O8P 584.2, found 584.0. Anal. Calcd. C, 49.11; H, 5.37; N, 15.76; P, 4.32; found: C, 48.88; H, 5.28; N, 15.71; P, 4.16. M.P. 201-205° C.
EXAMPLE 13Alternate preparation of Icb (Pro-drug of IVc)

To a 10 L reactor equipped with an overhead stirrer, thermocouple, distillation apparatus, and nitrogen inlet was charged IVc (200.00 g, 422.39 mmol), Cs2CO3 (344.06 g, 1.06 mol), KI (140.24 g, 844.81 mmol) and NMP (1.00 L, 10.38 mol). The reaction was stirred at room temperature resulting in a light brown heterogeneous suspension. Di-tert-butyl chloromethyl phosphate (273.16 g, 1.06 mol) was added via addition funnel and the reaction mixture was heated to 30° C. for 16-24 hours with stirring after which time the reaction was cooled to 5° C. To the reaction was added DCM (1.5 L) then the reaction was slowly quenched with water (3.5 L) maintaining the reaction temperature under 20° C. resulting in a biphasic mixture. The product rich bottom layer was separated, washed with water (3.5 L×3), then transferred back to the reactor. The solution was concentrated under vacuum to a volume of 1 L keeping the temperature below 25° C. IPA was added (2 L) then the reaction was concentrated under vacuum to a volume of 2 L keeping the temperature below 25° C. The reaction was then seeded with IIc (0.200 g), stirred overnight at room temperature resulting in a slurry. The slurry was filtered and the wet cake was washed with MTBE (1 L), dried in a vacuum oven at 50° C. overnight resulting in a yellow/white powder (207.1 g, 70%). 1H NMR (400 MHz, CDCl3) δ 8.54 (s, 1H), 8.18 (s, 1H), 7.91 (s, 1H), 7.42 (s, 5H), 5.95 (d, J=14.2 Hz, 2H), 4.06 (s, 3H), 3.97-3.36 (m, 8H), 2.50 (s, 3H), 1.27 (s, 18H); 3C NMR (100 MHz, CDCl3) δ 184.64, 170.65, 165.91, 161.60, 150.82, 145.38, 141.89, 134.96, 130.20, 129.59, 128.68, 127.58, 127.10, 124.77, 122.64, 115.22, 83.90, 83.83, 73.69, 73.63, 56.95, 46.04, 41.66, 29.61, 29.56, 13.90; ES+ MS m/z (rel. intensity) 696 (MH+,10), 640 (MH+-isobutylene, 30), 584 (MH+-2 isobutylene, 100).

To a 10 L 4 neck reactor equipped with a thermocouple, overhead stirrer, condenser and nitrogen inlet was added IIc (200.24 g, 287.82 mmol), acetone (800.00 ml, 10.88 mol) and water (800.00 ml, 44.41 mol). The reaction was heated to 40° C. and stirred for 18-24 hours. The reaction was cooled to 20° C. then tromethamine (33.62 g, 277.54 mmol) was added. The reaction was heated to 40° C. then stirred for an additional hour until all solids were dissolved. The reaction was cooled to 20° C. then filtered through a 10 micron cuno filter into a 10 L 4 neck reactor equipped with a thermocouple, overhead stirrer, and nitrogen inlet. Acetone (3 L) was added rapidly, followed by seeding with Icb (0.500 g), then additional acetone (3 L) was added. The reaction was stirred at room temperature overnight resulting in a slurry then filtered. The wet cake was washed with acetone (800 ml) then dried in a vacuum oven at 50° C. overnight resulting in a fluffy white powder (165.91 g, 82%).
Supplementary Information:
Isolation of the Free-Acid Intermediate IC:

In a 250 mL 3 neck reactor equipped with a thermocouple, overhead stirrer, condenser and nitrogen inlet was added IIc (10.0 g, 14.37 mmol), acetone (40.00 ml, 544.15 mmol) and water (40.00 ml, 2.22 mol). The reaction was heated to 40° C. and stirred for 14-24 hours. The reaction was cooled to 20° C. then stirred for three hours, resulting in a slurry. The slurry was filtered, then the wet cake washed with acetone (40.00 ml) then dried in a vacuum oven at 50° C. overnight resulting in a fluffy white powder (7.00 g, 83%). NMR (400 MHz, DMSO-d6) δ 8.84 (s, 1H), 8.47 (s, 1H), 8.06 (s, 1H), 7.45 (s, 5H), 5.81 (d, J=12.3 Hz, 2H), 4.03 (s, 3H), 3.91-3.19 (m, 8H), 2.39 (s, 3H); 13C NMR (500 MHz, DMSO-d6) δ 185.20, 169.32, 165.85, 160.75, 150.51, 146.30, 143.24, 135.53, 129.74, 129.22, 128.46, 127.34, 127.09, 123.67, 122.73, 113.94, 72.90 (d, 2JC-P=5 Hz), 57.01, 45.2 (bs), 40.8 (bs), 13.66. ES+ MS m/z (rel. intensity) 486 (MH+−H3PO4, 100).
References
- ^ HIV Attachment Inhibitor BMS-663068 Looks Good in Early Studies
- ^ HIV attachment inhibitor BMS-663068 shows good safety and efficacy in phase 2b study
- ^ Activity of the HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068, against CD4-independent viruses and HIV-1 envelopes resistant to other entry inhibitors
 |
|
 |
|
| Names | |
|---|---|
| IUPAC name
{3-[(4-Benzoyl-1-piperazinyl)(oxo)acetyl]-4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-yl)-1H-pyrrolo[2,3-c]pyridin-1-yl}methyl dihydrogen phosphate
|
|
| Other names
BMS-663068, GSK3684934
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
| KEGG | |
|
PubChem CID
|
|
| Properties | |
| C25H26N7O8P | |
| Molar mass | 583.498 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
////////////////фостемсавир , فوستيمسافير , 磷坦姆沙韦 ,BMS 663068, Fostemsavir, GSK 3684934, PHASE 3, ホステムサビル;
FDA Approves Tybost (cobicistat) for use in the treatment of HIV-1 Infection

Cobicistat, GS-9350
1004316-88-4
| C 40 H 53 N 7 O 5 S 2 |
N-[1(R)-Benzyl-4(R)-[2(S)-[3-(2-isopropylthiazol-4-ylmethyl)-3-methyl]ureido]-4-(4-morpholinyl)butyramido]-5-phenylpentyl]carbamic acid thiazol-5-ylmethyl ester
(1,3-thiazol-5-yl) methyl (5S, 8R, 11R) -8,11-dibenzyl-2-methyl-5-[2 – (morpholin-4-yl) ethyl] -1 – [2 – (propan-2-yl) -1,3-thiazol-4-yl] -3,6-dioxo-2 ,4,7,12-tetraazatridecan-13-oate
cytochrome P450 3A4 (CYP3A4) inhibitor
FDA Approves Tybost (cobicistat) for use in the treatment of HIV-1 Infection
September 24, 2014 — The U.S. Food and Drug Administration (FDA) has approved Tybost (cobicistat), a CYP3A inhibitor used in combination with atazanavir or darunavir for the treatment of human immunodeficiency virus type 1 (HIV-1) infection
Cobicistat is a pharmacokinetic enhancer that works by inhibiting the enzyme (CYP3A) that metabolizes atazanavir and darunavir. It increases the systemic exposure of these drugs and prolongs their effect. Cobicistat is also one of the ingredients in the combination HIV drug Stribild, which was approved by the FDA in August, 2012.
Tybost comes in 150 mg tablets and is administered once daily in combination with the protease inhibitors atazanavir (Reyataz), or darunavir (Prezista).
Because Tybost inhibits CYP3A, other medications metabolized by CYP3A may result in increased plasma concentrations and potentially severe side effects, which may be life-threatening or even fatal. Extra care should be exercised by healthcare professionals to ensure than other medications are reviewed and their concentrations monitored, especially when initiating new medicines or changing doses.
The approval of Tybost was based on the following clinical trials:
•The data to support the use of atazanavir and Tybost were from a phase 2 and 3 trial in treatment-naïve adults comparing atazanavir/cobicistat 300/150 mg and atazanavir/ritonavir 300/100 mg once daily each in combination with Truvada. The atazanavir/cobicistat based regimen was non-inferior to the atazanavir/ritonavir based regimen.
•The data to support the use of cobicistat with darunavir is from a multiple dose trial in healthy subjects comparing the relative bioavailability of darunavir/cobicistat 800/150 mg to darunavir/ritonavir 800/100 mg.
The most common adverse drug reactions observed with Tybost (in combination with atazanavir) in clinical trials were jaundice, ocular icterus, and nausea.
Tybost is a product of Gilead Sciences, Foster City, CA.

Cobicistat (formerly GS-9350) is a licensed drug for use in the treatment of infection with the human immunodeficiency virus (HIV).
Like ritonavir (Norvir), cobicistat is of interest not for its anti-HIV properties, but rather its ability to inhibit liver enzymes that metabolize other medications used to treat HIV, notablyelvitegravir, an HIV integrase inhibitor currently under investigation itself. By combining cobicistat with elvitegravir, higher concentrations of elvitgravir are achieved in the body with lower dosing, theoretically enhancing elvitgravir’s viral suppression while diminishing its adverse side-effects. In contrast with ritonavir, the only currently approved booster, cobicistat has no anti-HIV activity of its own.[1]
Cobicistat, a cytochrome P450 CYP3A4 inhibitor, was approved in the E.U. in 2013 as a pharmacokinetic enhancer of the HIV-1 protease inhibitors atazanavir and darunavir in adults. First launch took place in 2014 in United Kingdom. In 2012, Gilead filed a New Drug Application in the U.S. for the same indication. In April 2013, the FDA issued a Complete Response Letter from the FDA. In 2014 the FDA accepted Gilead’s resubmission.
Cobicistat is a component of the four-drug, fixed-dose combination HIV treatmentelvitegravir/cobicistat/emtricitabine/tenofovir (known as the “Quad Pill” or Stribild).[1][2] The Quad Pill/Stribild was approved by the FDA in August 2012 for use in the United States and is owned by Gilead Sciences.
Cobicistat is a potent inhibitor of cytochrome P450 3A enzymes, including the importantCYP3A4 subtype. It also inhibits intestinal transport proteins, increasing the overall absorption of several HIV medications, including atazanavir, darunavir and tenofovir alafenamide fumarate.[3]
The drug candidate acts as a pharmaco-enhancer to boost exposure of HIV protease inhibitors. In 2011, cobicistat was licensed to Japan Tobacco by Gilead for development and commercialization in Japan as a stand-alone product for the treatment of HIV infection. In 2012, orphan drug designation was assigned in Japan for the pharmacokinetic enhancement of anti-HIV agent.
Oxidative metabolism by cytochrome P450 enzymes is one of the primary mechanisms of drug metabolism.. It can be difficult to maintain therapeutically effective blood plasma levels of drugs which are rapidly metabolized by cytochrome P450 enzymes. Accordingly, the blood plasma levels of drugs which are susceptible to cytochrome P450 enzyme degradation can be maintained or enhanced by co-administration of cytochrome P450 inhibitors, thereby improving the pharmacokinetics of the drug.
While certain drugs are known to inhibit cytochrome P450 enzymes, more and/or improved inhibitors for cytochrome P450 monooxygenase are desirable. Particularly, it would be desirable to have cytochrome P450 monooxygenase inhibitors which do not have appreciable biological activity other than cytochrome P450 inhibition. Such inhibitors can be useful for minimizing undesirable biological activity, e.g., side effects. In addition, it would be desirable to have P450 monooxygenase inhibitors that lack significant or have a reduced level of protease inhibitor activity. Such inhibitors could be useful for enhancing the effectiveness of antiretroviral drugs, while minimizing the possibility of eliciting viral resistance, especially against protease inhibitors.
…………………………….
Cobicistat (GS-9350): A potent and selective inhibitor of human CYP3A as a novel pharmacoenhancer
ACS Med Chem Lett 2010, 1(5): 209
http://pubs.acs.org/doi/abs/10.1021/ml1000257
http://pubs.acs.org/doi/suppl/10.1021/ml1000257/suppl_file/ml1000257_si_001.pdf

Cobicistat (3, GS-9350) is a newly discovered, potent, and selective inhibitor of human cytochrome P450 3A (CYP3A) enzymes. In contrast to ritonavir, 3 is devoid of anti-HIV activity and is thus more suitable for use in boosting anti-HIV drugs without risking selection of potential drug-resistant HIV variants. Compound 3 shows reduced liability for drug interactions and may have potential improvements in tolerability over ritonavir. In addition, 3 has high aqueous solubility and can be readily coformulated with other agents.
…………………………………
http://www.google.com/patents/CN103694196A?cl=en
CN 103694196
oxidative metabolism by cytochrome P450 enzymes is one of the main mechanisms of drug metabolism, generally by administration of cytochrome P450 inhibitors to maintain or increase the degradation of cytochrome P450 enzymes are sensitive to the drug plasma levels, in order to improve the pharmacokinetics of drugs dynamics, can be used to enhance the effectiveness of anti-retroviral drugs. For example W02008010921 discloses compounds of formula I as a cytochrome P450 monooxygenase specific compounds (Cobicistat):

W02008010921 discloses the synthesis of compounds of formula I with a variety of, as one of the methods of the following routes
Shows:

The reagents used in the method is expensive, and more difficult to remove by-products, long reaction time, high cost, is not conducive to industrial
Production.
W02010115000 on these routes has been improved:

The first step in the route used for the ring-opening reaction reagent trimethylsilyl iodide, trimethylsilyl iodide expensive. W02010115000 reports this step and the subsequent ring-opening reaction of morpholine substitution reaction yield of two steps is not high, only 71%, so that only iodotrimethylsilane a high cost of raw material is not suitable for industrial production.



Preparation of compounds of formula I
Example [0126] Implementation
[0127] I1-a (20g) was dissolved in dichloromethane, was added 50% K0H (5.5g) solution, control the internal temperature does not exceed 25 ° C, TLC analysis ΙΙ-a disappears. Was cooled to O ~ 10 ° C, was added (2R, 5R) -5 – amino-1 ,6 – diphenyl-2 – hexyl-carbamic acid 5 – methyl-thiazole ester hydrochloride (14.8g), stirred for I ~ 2 h, 1 – hydroxybenzotriazole triazole (5.5g), stirred for I h, 1 – ethyl – (3 – dimethylaminopropyl) carbodiimide hydrochloride (15g), and incubated for 5 ~ 10 hours, TLC analysis of the starting material disappeared, the reaction was completed. The reaction was quenched with aqueous acetic acid, methylene chloride layer was separated, washed with saturated aqueous NaHCO3, washed with water, dried and concentrated. By HPLC purity of 99.1%. Adding ethanol, the ethanol was evaporated to give the product compound of part I of a solution in ethanol. Molar yield 88%, LC-MS: M +1 = 777.1 [0128] All publications mentioned in the present invention are incorporated by reference as if each reference was individually incorporated by reference, as cited in the present application. It should also be understood that, after reading the foregoing teachings of the present invention, those skilled in the art that various modifications of the present invention or modifications, and these equivalents falling as defined by the appended claims scope of claims of the present application.
…………………………
US 2014088304
http://www.google.com/patents/US20140088304
International Patent Application Publication Number WO 2008/010921 and International Patent Application Publication Number WO 2008/103949 disclose certain compounds that are reported to be useful to modify the pharmacokinetics of a co-administered drug, e.g. by inhibiting cytochrome P450 monooxygenase. One specific compound identified therein is a compound of the following formula I:

There is currently a need for improved synthetic methods and intermediates that can be used to prepare the compound of formula I and its salts
Schemes 1-4 below.




Preparation of a Compound of Formula IV

Scheme V.
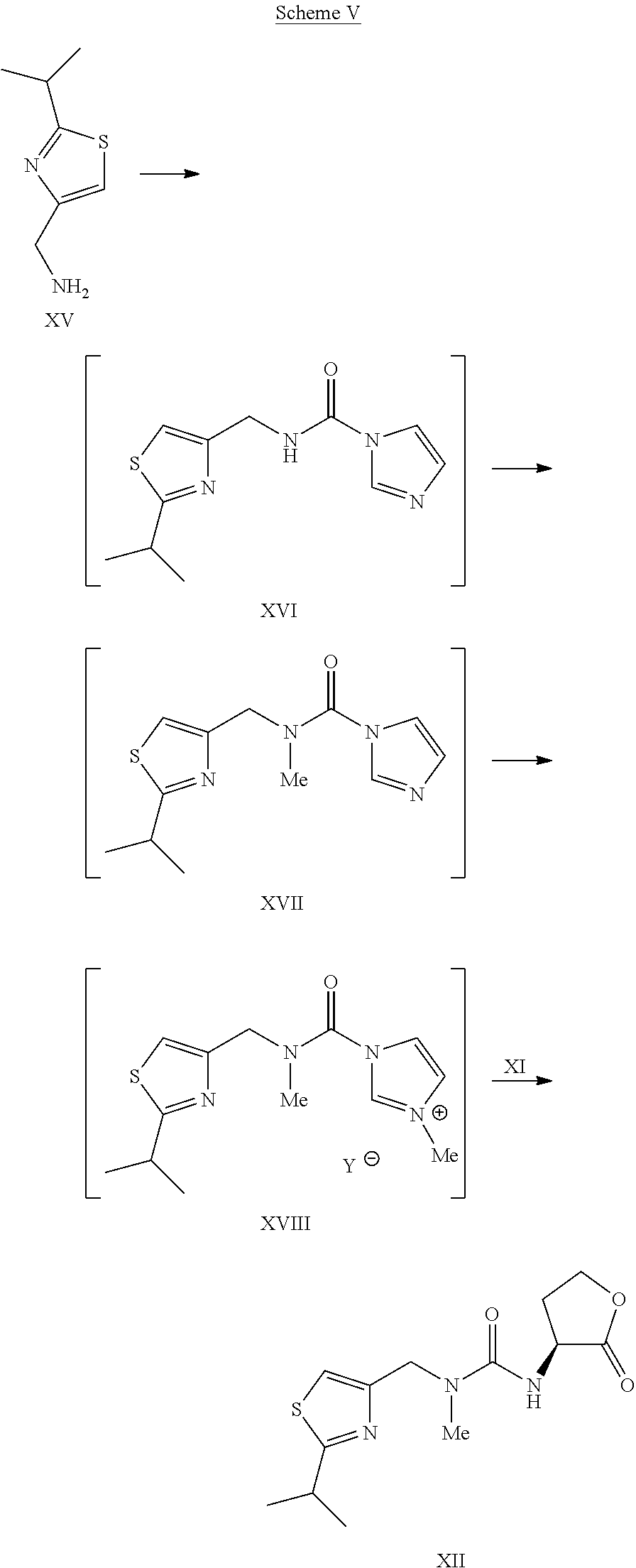
Example 14Preparation of Compound I

To the solution of L-thiazole morpholine ethyl ester oxalate salt XIVa (35.6 kg) in water (66.0 kg) was charged dichloromethane (264 kg), followed by a slow addition of 15 wt % KHCO3 solution (184.8 kg). The resulting mixture was agitated for about 1 hour. The layers were separated and the organic layer was washed with water (132 kg). The organic layer was concentrated under vacuum to dryness. Water (26.5 kg) was charged and the content temperature was adjusted to about 10° C., followed by slow addition of 45% KOH solution (9.8 kg) while maintaining the content temperature at less than or equal to 20° C. The mixture was agitated at less than or equal to 20° C. until the reaction was judged complete by HPLC. The reaction mixture was concentrated under vacuum to dryness and co-evaporated five times with dichloromethane (132 kg each time) under reduced pressure to dryness. Co-evaporation with dichloromethane (132 kg) was continued until the water content was <4% by Karl Fischer titration. Additional dichloromethane (264 kg) was charged and the content temperature was adjusted to −18° C. to −20° C., followed by addition of monocarbamate.HCl salt IXa (26.4 kg). The resulting mixture was agitated at −18° C. to −20° C. for about 1 hour. HOBt (11.4 kg) was charged and the reaction mixture was again agitated at −18° C. to −20° C. for about 1 hour. A pre-cooled solution (−20° C.) of EDC.HCl (21.4 kg) in dichloromethane (396 kg) was added to the reaction mixture while the content temperature was maintained at less than or equal to −20° C. The reaction mixture was agitated at −18° C. to −20° C. until the reaction was judged complete. The content temperature was adjusted to about 3° C. and the reaction mixture quenched with a 10 wt % aqueous citric acid solution (290 kg). The layers were separated and the organic layer was washed once with 15 wt % potassium bicarbonate solution (467 kg) and water (132 kg). The organic layer was concentrated under reduced pressure and then co-evaporated with absolute ethanol.
The product I was isolated as the stock solution in ethanol (35.0 kg product, 76.1% yield).
1H NMR (dDMSO) δ□ 9.05 (s, 1H), 7.85 (s, 1H), 7.52 (d, 1H), 7.25-7.02 (m, 12H), 6.60 (d, 1H), 5.16 (s, 2H), 4.45 (s, 2H), 4.12-4.05 (m, 1H), 3.97-3.85 (m, 1H), 3.68-3.59 (m, 1H), 3.57-3.45 (m, 4H), 3.22 (septets, 1H), 2.88 (s, 3H), 2.70-2.55 (m, 4H), 2.35-2.10 (m, 6H), 1.75 (m, 1H), 1.62 (m, 1H), 1.50-1.30 (m, 4H), 1.32 (d, 6H).
13C NMR (CD3OD) δ 180.54, 174., 160.1, 157.7, 156.9, 153.8, 143.8, 140.1, 140.0, 136.0, 130.53, 130.49, 129.4, 127.4, 127.3, 115.5, 67.7, 58.8, 56.9, 55.9, 54.9, 53.9, 51.6, 49.8, 42.7, 42.0, 35.4, 34.5, 32.4, 32.1, 29.1, 23.7.
Example 13Preparation of L-Thiazole Morpholine Ethyl Ester Oxalate Salt XIVa

To a solution of (L)-thiazole amino lactone XII (33.4 kg) in dichloromethane (89.5 kg) was charged dichloromethane (150 kg) and absolute ethanol (33.4 kg). The content temperature was then adjusted to about 10° C., followed by slow addition of TMSI (78.8 kg) while the content temperature was maintained at less than or equal to 22° C. and agitated until the reaction was judged complete. The content temperature was adjusted to about 10° C., followed by a slow addition of morpholine (49.1 kg) while the content temperature was maintained at less than or equal to 22° C. Once complete, the reaction mixture was filtered to remove morpholine.HI salt and the filter cake was rinsed with two portions of dichloromethane (33.4 kg). The filtrate was washed twice with water (100 kg). The organic layer was concentrated under vacuum to dryness. Acetone (100 kg) was then charged to the concentrate and the solution was concentrated under reduced pressure to dryness. Acetone (233.8 kg) was charged to the concentrate, followed by a slow addition of the solution of oxalic acid (10 kg) in acetone (100 kg). The resulting slurry was refluxed for about 1 hour before cooling down to about 3° C. for isolation. The product XIVa was filtered and rinsed with acetone (66.8 kg) and dried under vacuum at 40° C. to afford a white to off-white solid (40 kg, 71% yield). 1H NMR (CDCl3) δ □7.00 (s, 1H), 6.35 (broad s, 1H), 4.60-4.40 (m, 3H), 4.19 (quartets, 2H), 4.00-3.90 (m, 4H), 3.35-3.10 (m, 7H), 3.00 (s, 3H), 2.40-2.30 (m, 1H), 2.15-2.05 (m, 1H), 1.38 (d, 6H), 1.25 (triplets, 3H).
……………………………………..
W02008010921
http://www.google.co.in/patents/WO2008010921A2?cl=en
Preparation of Example A
Scheme 1

Example A Compound 2
To a solution of Compound 1 (ritonavir) (1.8 g, 2.5 mmol) in 1,2- dichloroethane (15 mL) was added l,l’-thiocarbonyldiimidazole (890 mg, 5.0 mmol). The mixture was heated at 75 SC for 6 hours and cooled to 25 SC. Evaporation under reduced pressure gave a white solid. Purification by flash column chromatography (stationary phase: silica gel; eluent: EtOAc) gave Compound 2 (1.6 g). m/z: 831.1 (M+H)+. Example A
To the refluxing solution of tributyltin hydride (0.78 mL, 2.9 mmol) in toluene (130 mL) was added a solution of Compound 2 (1.6 g, 1.9 mmol) and 2,2′- azobisisobutyronitrile (31 mg, 0.19 mmol) in toluene (30 mL) over 30 minutes. The mixture was heated at 1152C for 6 hours and cooled to 25 BC. Toluene was removed under reduced pressure. Purification by flash column chromatography (stationary phase: silica gel; eluent: hexane/EtOAc = 1/10) gave Example A (560 mg). m/z: 705.2 (M+H)+. 1H-NMR (CDCl3) δ 8.79 (1 H, s), 7.82 (1 H, s), 7.26-7.05 (10 H, m), 6.98 (1 H, s), 6.28 (1 H, m), 6.03 (1 H, m), 5.27 (1 H7 m), 5.23 (2 H, s), 4.45-4.22 (2 H, m), 4.17 (1 H, m), 3.98 (1 H, m), 3.75 (1 H, m), 3.25 (1 H7 m), 2.91 (3 H, s), 2.67 (4 H, m), 2.36 (1 H, m), 1.6-1.2 (10 H, m), 0.85 (6 H, m).
| EP1183026A2 * | 25 May 2000 | 6 Mar 2002 | Abbott Laboratories | Improved pharmaceutical formulations |
| US20060199851 * | 2 Mar 2006 | 7 Sep 2006 | Kempf Dale J | Novel compounds that are useful for improving pharmacokinetics |
| Thiazol-5-ylmethyl N-[1-benzyl-4-[[2-[[(2-isopropylthiazol-4-yl)methyl-methyl-carbamoyl]amino]-4-morpholino-butanoyl]amino]-5-phenyl-pentyl]carbamate | |
| Clinical data | |
|---|---|
| Legal status |
fda approved sept 2014
|
| Identifiers | |
| CAS number | 1004316-88-4 |
| ATC code | V03AX03 |
| PubChem | CID 25151504 |
| ChemSpider | 25084912 |
| UNII | LW2E03M5PG |
| Chemical data | |
| Formula | C40H53N7O5S2 |
| Mol. mass | 776.023 g/mol |
| US7939553 * | Jul 6, 2007 | May 10, 2011 | Gilead Sciences, Inc. | co-administered drug (as HIV protease inhibiting compound, an HIV (non)nucleoside/nucleotide inhibitor of reverse transcriptase, capsid polymerization inhibitor, interferon, ribavirin analog) by inhibiting cytochrome P450 monooxygenase; ureido- or amido-amine derivatives; side effect reduction |
- Highleyman, L.
Elvitegravir “Quad” Single-tablet Regimen Shows Continued HIV Suppression at 48 Weeks
- R Elion, J Gathe, B Rashbaum, and others. The Single-Tablet Regimen of Elvitegravir/Cobicistat/Emtricitabine/Tenofovir Disoproxil Fumarate (EVG/COBI/FTC/TDF; Quad) Maintains a High Rate of Virologic Suppression, and Cobicistat (COBI) is an Effective Pharmacoenhancer Through 48 Weeks. 50th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC 2010). Boston, September 12–15, 2010.
- Lepist, E. -I.; Phan, T. K.; Roy, A.; Tong, L.; MacLennan, K.; Murray, B.; Ray, A. S. (2012). “Cobicistat Boosts the Intestinal Absorption of Transport Substrates, Including HIV Protease Inhibitors and GS-7340, in Vitro”. Antimicrobial Agents and Chemotherapy 56 (10): 5409–5413. doi:10.1128/AAC.01089-12. PMC 3457391. PMID 22850510.
-
Patent No all US
Expiry 5814639 Sep 29, 2015 5814639*PED Mar 29, 2016 5914331 Jul 2, 2017 5914331*PED Jan 2, 2018 5922695 Jul 25, 2017 5922695*PED Jan 25, 2018 5935946 Jul 25, 2017 5935946*PED Jan 25, 2018 5977089 Jul 25, 2017 5977089*PED Jan 25, 2018 6043230 Jul 25, 2017 6043230*PED Jan 25, 2018 6642245 Nov 4, 2020 6642245*PED May 4, 2021 6703396 Mar 9, 2021 6703396*PED Sep 9, 2021 7176220 Nov 20, 2023 7635704 Oct 26, 2026 8148374 Sep 3, 2029
ABACAVIR…….For the treatment of HIV-1 infection, in combination with other antiretroviral agents.
 |
|

Chemical structure of abacavir
|
{(1S,4R)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]cyclopent-2-en-1-yl}methanol
(-)-cis-4-[2-Amino-6-(cyclopropylmethylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol
(1S, 4R)-4-[2-amino-6-(cyclopropylamino)-9H purin-9-yl]-2- cyclopentene-1 -methanol
Abacavir
Abacavir (ABC) is a powerful nucleoside analog reverse transcriptase inhibitor (NRTI) used to treat HIV and AIDS. [Wikipedia] Chemically, it is a synthetic carbocyclic nucleoside and is the enantiomer with 1S, 4R absolute configuration on the cyclopentene ring. In vivo, abacavir sulfate dissociates to its free base, abacavir.
Abacavir (ABC) ![]() i/ʌ.bæk.ʌ.vɪər/ is a nucleoside analog reverse transcriptase inhibitor (NRTI) used to treat HIV and AIDS. It is available under the trade name Ziagen (ViiV Healthcare) and in the combination formulations Trizivir (abacavir, zidovudine andlamivudine) and Kivexa/Epzicom (abacavir and lamivudine). It has been well tolerated: the main side effect is hypersensitivity, which can be severe, and in rare cases, fatal. Genetic testing can indicate whether an individual will be hypersensitive; over 90% of patients can safely take abacavir. However, in a separate study, the risk of heart attack increased by nearly 90%.[1]
i/ʌ.bæk.ʌ.vɪər/ is a nucleoside analog reverse transcriptase inhibitor (NRTI) used to treat HIV and AIDS. It is available under the trade name Ziagen (ViiV Healthcare) and in the combination formulations Trizivir (abacavir, zidovudine andlamivudine) and Kivexa/Epzicom (abacavir and lamivudine). It has been well tolerated: the main side effect is hypersensitivity, which can be severe, and in rare cases, fatal. Genetic testing can indicate whether an individual will be hypersensitive; over 90% of patients can safely take abacavir. However, in a separate study, the risk of heart attack increased by nearly 90%.[1]
Viral strains that are resistant to zidovudine (AZT) or lamivudine (3TC) are generally sensitive to abacavir (ABC), whereas some strains that are resistant to AZT and 3TC are not as sensitive to abacavir.
It is on the World Health Organization’s List of Essential Medicines, a list of the most important medication needed in a basic health system.[2]
Abacavir is a nucleoside reverse transcriptase inhibitor (NRTI) with activity against Human Immunodeficiency Virus Type 1 (HIV-1). Abacavir is phosphorylated to active metabolites that compete for incorporation into viral DNA. They inhibit the HIV reverse transcriptase enzyme competitively and act as a chain terminator of DNA synthesis. The concentration of drug necessary to effect viral replication by 50 percent (EC50) ranged from 3.7 to 5.8 μM (1 μM = 0.28 mcg/mL) and 0.07 to 1.0 μM against HIV-1IIIB and HIV-1BaL, respectively, and was 0.26 ± 0.18 μM against 8 clinical isolates. Abacavir had synergistic activity in cell culture in combination with the nucleoside reverse transcriptase inhibitor (NRTI) zidovudine, the non-nucleoside reverse transcriptase inhibitor (NNRTI) nevirapine, and the protease inhibitor (PI) amprenavir; and additive activity in combination with the NRTIs didanosine, emtricitabine, lamivudine, stavudine, tenofovir, and zalcitabine.
Brief background information
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| – | J05AF06 | C 14 H 18 N 6 O | 286.34 g / mol | 136470-78-5 |
| succinate | J05AF06 | C 14 H 18 N 6 O · C 4 H 6 O | 356.43 g / mol | 168146-84-7 |
| sulfate | J05AF06 | C 14 H 18 N 6 O · 1 / 2H 2 SO 4 | 670.76 g / mol | 188062-50-2 |
| Systematic (IUPAC) name | |
|---|---|
| {(1S,4R)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]cyclopent-2-en-1-yl}methanol | |
| Clinical data | |
| Trade names | Ziagen |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a699012 |
| Pregnancy cat. | B3 (AU) C (US) |
| Legal status | POM (UK) ℞-only (US) |
| Routes | Oral (solution or tablets) |
| Pharmacokinetic data | |
| Bioavailability | 83% |
| Metabolism | Hepatic |
| Half-life | 1.54 ± 0.63 h |
| Excretion | Renal (1.2% abacavir, 30% 5′-carboxylic acid metabolite, 36% 5′-glucuronide metabolite, 15% unidentified minor metabolites). Fecal (16%) |
| Identifiers | |
| CAS number | 136470-78-5 |
| ATC code | J05AF06 |
| PubChem | CID 441300 |
| DrugBank | DB01048 |
| ChemSpider | 390063 |
| UNII | WR2TIP26VS |
| KEGG | D07057 |
| ChEBI | CHEBI:421707 |
| ChEMBL | CHEMBL1380 |
| NIAID ChemDB | 028596 |
| Chemical data | |
| Formula | C14H18N6O |
| Mol. mass | 286.332 g/mol |
Abacavir is a carbocyclic synthetic nucleoside analogue and an antiviral agent. Intracellularly, abacavir is converted by cellular enzymes to the active metabolite carbovir triphosphate, an analogue of deoxyguanosine-5′-triphosphate (dGTP). Carbovir triphosphate inhibits the activity of HIV-1 reverse transcriptase (RT) both by competing with the natural substrate dGTP and by its incorporation into viral DNA. Viral DNA growth is terminated because the incorporated nucleotide lacks a 3′-OH group, which is needed to form the 5′ to 3′ phosphodiester linkage essential for DNA chain elongation.
Application
-
an antiviral agent, is used in the treatment of AIDS
-
ingibitor convertibility transkriptazы
Classes of substances
-
Adenine (6-aminopurines)
-
Aminoalcohols
-
Cyclopentenes and cyclopentadienes
-
Tsyklopropanы
-
-
-
| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| Canada | 2289753 | 2007-01-23 | 2018-05-14 |
| Canada | 1340589 | 1999-06-08 | 2016-06-08 |
| Canada | 2216634 | 2004-07-20 | 2016-03-28 |
| United States | 6641843 | 2000-02-04 | 2020-02-04 |
| United States | 5089500 | 1992-12-26 | 2009-12-26 |
PATENT
US5034394
Synthesis pathway
Abacavir, (-) cis-[4-[2-amino-6-cyclopropylamino)-9H-purin-9-yl]-2-cyclopenten-yl]-1 – methanol, a carbocyclic nucleoside which possesses a 2,3-dehydrocyclopentene ring, is referred to in United States Patent 5,034,394 as a reverse transcriptase inhibitor. Recently, a general synthetic strategy for the preparation of this type of compound and intermediates was reported [Crimmins, et. al., J. Org. Chem., 61 , 4192-4193 (1996) and 65, 8499-8509-4193 (2000)].
-
Abacavir is the International Nonproprietary Name (INN) of {(1S,4R)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol and CAS No. 136470-78-5. Abacavir and therapeutically acceptable salts thereof, in particular the hemisulfate salt, are well-known as potent selective inhibitors of HIV-1 and HIV-2, and can be used in the treatment of human immunodeficiency virus (HIV) infection.
-

-
EP 434450-A discloses certain 9-substituted-2-aminopurines including abacavir and its salts, methods for their preparation, and pharmaceutical compositions using these compounds.
-
Different preparation processes of abacavir are known in the art. In some of them abacavir is obtained starting from an appropriate pyrimidine compound, coupling it with a sugar analogue residue, followed by a cyclisation to form the imidazole ring and a final introduction of the cyclopropylamino group at the 6 position of the purine ring.
-
According to the teachings of EP 434450-A , the abacavir base is finally isolated by trituration using acetonitrile (ACN) or by chromatography, and subsequently it can be transformed to a salt of abacavir by reaction with the corresponding acid. Such isolation methods (trituration and chromatography) usually are limited to laboratory scale because they are not appropriate for industrial use. Furthermore, the isolation of the abacavir base by trituration using acetonitrile gives a gummy solid (Example 7) and the isolation by chromatography (eluted from methanol/ethyl acetate) yields a solid foam (Example 19 or 28).
-
Other documents also describe the isolation of abacavir by trituration or chromatography, but always a gummy solid or solid foam is obtained (cf. WO9921861 and EP741710 ), which would be difficult to operate on industrial scale.
-
WO9852949 describes the preparation of abacavir which is isolated from acetone. According to this document the manufacture of the abacavir free base produces an amorphous solid which traps solvents and is, therefore, unsuitable for large scale purification, or for formulation, without additional purification procedures (cf. page 1 of WO 9852949 ). In this document, it is proposed the use of a salt of abacavir, in particular the hemisulfate salt which shows improved physical properties regarding the abacavir base known in the art. Said properties allow the manufacture of the salt on industrial scale, and in particular its use for the preparation of pharmaceutical formulations.
-
However, the preparation of a salt of abacavir involves an extra processing step of preparing the salt, increasing the cost and the time to manufacture the compound. Generally, the abacavir free base is the precursor compound for the preparation of the salt. Thus, depending on the preparation process used for the preparation of the salt, the isolation step of the abacavir free base must also be done.
The structure of abacavir corresponds to formula (I):

………………………………
http://www.google.co.in/patents/US5034394
EXAMPLE 21(-)-cis-4-[2-Amino-6-(cyclopropylmethylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol
The title compound of Example 7, (2.00 g, 6.50 mmol) was dissolved in 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (Aldrich, 20 mL). Phosphoryl chloride (2.28 mL, 24.0 mmol) was added to the stirred, cooled (-10° C.) solution. After 3 minutes, cold water (80 mL) was added. The solution was extracted with chloroform (3×80 mL). The aqueous layer was diluted with ethanol (400 mL) and the pH adjusted to 6 with saturated aqueous NaOH. The precipitated inorganic salts were filtered off. The filtrate was further diluted with ethanol to a volume of 1 liter and the pH adjusted to 8 with additional NaOH. The resulting precipitate was filtered and dried to give the 5′-monophosphate of (±)-cis-4-[2-amino-6-(cyclopropylmethylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol as white powder (4.0 mmoles, 62% quantitated by UV absorbance); HPLC analysis as in Example 17 shows one peak. This racemic 5′ -monophosphate was dissolved in water (200 mL) and snake venom 5′-nucleotidase (EC 3.1.3.5) from Crotalus atrox (5,000 IU, Sigma) was added. After incubation at 37° C. for 10 days, HPLC analysis as in Example 17 showed that 50% of the starting nucleotide had been dephosphorylated to the nucleoside. These were separated on a 5×14 cm column of DEAE Sephadex A25 (Pharmacia) which had been preequilibrated with 50 mM ammonium bicarbonate. Title compound was eluted with 2 liters of 50 mM ammonium bicarbonate. Evaporation of water gave white powder which was dissolved in methanol, adsorbed on silica gel, and applied to a silica gel column. Title compound was eluted with methanol:chloroform/1:9 as a colorless glass. An acetonitrile solution was evaporated to give white solid foam, dried at 0.3 mm Hg over P2 O5 ; 649 mg (72% from racemate); 1 H-NMR in DMSO-d6 and mass spectrum identical with those of the racemate (title compound of Example 7); [α]20 D -48.0°, [α]20 436 -97.1°, [α]20 365 -149° (c=0.14, methanol).
Anal. Calcd. for C15 H20 N6 O.0.10CH3 CN: C, 59.96; H, 6.72; N, 28.06. Found: C, 59.93; H, 6.76; N, 28.03.
Continued elution of the Sephadex column with 2 liters of 100 mM ammonium bicarbonate and then with 2 liters of 200 mM ammonium bicarbonate gave 5′-monophosphate (see Example 22) which was stable to 5′-nucleotidase.
…………………………………………
| Синтез a) |
|---|
        |
| Синтез b) |
     |
| Preparation c) |
    |
| Synthesis d) |
 |

An enantiopure β-lactam with a suitably disposed electron withdrawing group on nitrogen, participated in a π-allylpalladium mediated reaction with 2,6-dichloropurine tetrabutylammonium salt to afford an advanced cis-1,4-substituted cyclopentenoid with both high regio- and stereoselectivity. This advanced intermediate was successfully manipulated to the total synthesis of (−)-Abacavir.

http://pubs.rsc.org/en/content/articlelanding/2012/ob/c2ob06775g#!divAbstract
………………………………….
http://www.google.com.ar/patents/EP2085397A1?cl=en
Example 1: Preparation of crystalline Form I of abacavir base using methanol as solvent
-
[0026]Abacavir (1.00 g, containing about 17% of dichloromethane) was dissolved in refluxing methanol (2.2 mL). The solution was slowly cooled to – 5 °C and, the resulting suspension, was kept at that temperature overnight under gentle stirring. The mixture was filtered off and dried under vacuum (7-10 mbar) at 40 °C for 4 hours to give a white solid (0.55 g, 66% yield, < 5000 ppm of methanol). The PXRD analysis gave the diffractogram shown in FIG. 1.
……………………………………..
http://www.google.com/patents/WO2008037760A1?cl=en
Abacavir, is the International Nonproprietary Name (INN) of {(1 S,4R)-4-[2- amino-6-(cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol, and CAS No. 136470-78-5. Abacavir sulfate is a potent selective inhibitor of HIV-1 and HIV-2, and can be used in the treatment of human immunodeficiency virus (HIV) infection.
The structure of abacavir hemisulfate salt corresponds to formula (I):

(I)
EP 434450-A discloses certain 9-substituted-2-aminopuhnes including abacavir and its salts, methods for their preparation, and pharmaceutical compositions using these compounds.
Different preparation processes of abacavir are known in the art. In some of them abacavir is obtained starting from an appropriate pyrimidine compound, coupling it with a sugar analogue residue, followed by a cyclisation to form the imidazole ring and a final introduction of the cyclopropylamino group at the 6 position of the purine ring. Pyrimidine compounds which have been identified as being useful as intermediates of said preparation processes include N-2-acylated abacavir intermediates such as N-{6- (cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H-purin- 2-yl}acetamide or N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-
(hydroxymethyl)cyclopent-2-enyl]-9H-purin-2-yl}isobutyramide. The removal of the amino protective group of these compounds using acidic conditions is known in the art. According to Example 28 of EP 434450-A, the amino protective group of the N-{6-(cyclopropylamino)-9-[(1 R,4S)-4- (hydroxymethyl)cyclopent-2-enyl]-9H-purin-2-yl}isobutyramide is removed by stirring with 1 N hydrochloric acid for 2 days at room temperature. The abacavir base, after adjusting the pH to 7.0 and evaporation of the solvent, is finally isolated by trituration and chromatography. Then, it is transformed by reaction with an acid to the corresponding salt of abacavir. The main disadvantages of this method are: (i) the use of a strongly corrosive mineral acid to remove the amino protective group; (ii) the need of a high dilution rate; (iii) a long reaction time to complete the reaction; (iv) the need of isolating the free abacavir; and (v) a complicated chromatographic purification process.
Thus, despite the teaching of this prior art document, the research of new deprotection processes of a N-acylated {(1 S,4R)-4-[2-amino-6- (cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol is still an active field, since the industrial exploitation of the known process is difficult, as it has pointed out above. Thus, the provision of a new process for the removal of the amino protective group of a N-acylated {(1 S,4R)-4-[2-amino-6-
(cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol is desirable.
Example 1 : Preparation of abacavir hemisulfate
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (6.56 g, 18.40 mmol) was slurried in a mixture of isopropanol (32.8 ml) and 10% solution of NaOH (36.1 ml, 92.0 mmol). The mixture was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and tert-butyl methyl ether (32.8 ml) was added. The layers were separated and H2SO4 96% (0.61 ml, 11.03 mmol) was added dropwise to the organic layer. This mixture was cooled to 0-50C and the resulting slurry filtered off.
The solid was dried under vacuum at 40 0C. Abacavir hemisulfate (5.98 g, 97%) was obtained as a white powder.
Example 6: Preparation of abacavir
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (1.0 g, 2.80 mmol) was slurried in a mixture of isopropanol (2 ml) and 10% solution of NaOH (1.1 ml, 2.80 mmol). The mixture was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and tert-butyl methyl ether (2 ml) was added. The aqueous layer was discarded, the organic phase was cooled to 0-5 0C and the resulting slurry filtered off. The solid was dried under vacuum at 400C. Abacavir (0.62 g, 77%) was obtained as a white powder.
Example 7: Preparation of abacavir
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (1.25 g, 3.51 mmol) was slurried in a mixture of isopropanol (2.5 ml) and 10% solution of NaOH (1.37 ml, 3.51 mmol). The mixture was refluxed for 1 h and concentrated to dryness. The residue was crystallized in acetone. Abacavir (0.47 g, 47%) was obtained as a white powder.
Example 8: Preparation of abacavir
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (1.25 g, 3.51 mmol) was slurried in a mixture of isopropanol (2.5 ml) and 10% solution of NaOH (1.37 ml, 3.51 mmol). The mixture was refluxed for 1 h and concentrated to dryness. The residue was crystallized in acetonitrile. Abacavir (0.43 g, 43%) was obtained as a white powder.
Example 9: Preparation of abacavir
A mixture of N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent- 2-enyl]-9H-purin-2-yl}isobutyramide (10 g, 28 mmol), isopropanol (100 ml) and 10% solution of NaOH (16.8 ml, 42 mmol) was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and washed several times with 25% solution of NaOH (10 ml). The wet organic layer was neutralized to pH 7.0-7.5 with 17% hydrochloric acid and it was concentrated to dryness under vacuum. The residue was crystallized in ethyl acetate (150 ml) to afford abacavir (7.2 g, 90%).
Example 10: Preparation of abacavir
A mixture of N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent- 2-enyl]-9H-purin-2-yl}isobutyramide (10 g, 28 mmol), isopropanol (100 ml) and 10% solution of NaOH (16.8 ml, 42 mmol) was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and washed several times with 25% solution of NaOH (10 ml). The wet organic layer was neutralized to pH 7.0-7.5 with 17% hydrochloric acid and it was concentrated to dryness under vacuum. The residue was crystallized in acetone (300 ml) to afford abacavir (7.0 g, 88%).
…………………………………
http://www.google.com/patents/WO2004089952A1?cl=en
Abacavir of formula (1) :

or (1 S,4R)-4-[2-Amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1 – methanol and its salts are nucleoside reverse transcriptase inhibitors. Abacavir sulfate is a nucleoside reverse transcriptase inhibitor and used in the treatment of human immunodeficiency virus infection. Abacavir sulfate and related compounds and their therapeutic uses are disclosed in US 5,034,394.
Crystalline forms of abacavir sulfate have not been reported in the literature. Moreover, the processes described in the literature do not produce abacavir sulfate in a stable, well-defined and reproducible crystalline form. It has now been discovered that abacavir sulfate can be prepared in three stable, well-defined and consistently reproducible crystalline forms.
Example 1
Abacavir free base (3.0 gm, obtained by the process described in example 21 of US 5,034,394) is dissolved in ethyl acetate (15 ml) and cone, sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 3 hours at 20°C and filtered to give 3.0 gm of form I abacavir sulfate. Example 2 Abacavir free base (3.0 gm) is dissolved in acetone (20 ml) and cone, sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 6 hours at 25°C and filtered to give 2.8 gm of form I abacavir sulfate.
Example 3 Abacavir free base (3.0 gm) is dissolved in acetonitrile (15 ml) and sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 2 hours at 25°C and the separated solid is filtered to give 3.0 gm of form II abacavir sulfate.
Example 4 Abacavir free base (3.0 gm) is dissolved in methyl tert-butyl ether (25 ml) and sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 1 hours at 25°C and the separated solid is filtered to give 3.0 gm of form II abacavir sulfate.
Example 5 Abacavir free base (3.0 gm) is dissolved in methanol (15 ml) and sulfuric acid (0.3 ml) is added to the solution. The contents then are cooled to 0°C and diisopropyl ether (15 ml) is added. The reaction mass is stirred for 2 hours at about 25°C and the separated solid is filtered to give 3.0 gm of form III abacavir sulfate
…………………………….
http://www.google.com.ar/patents/WO1999021861A1?cl=en
The present invention relates to a new process for the preparation of the chiral nucleoside analogue (1S, 4R)-4-[2-amino-6-(cyclopropylamino)-9H purin-9-yl]-2- cyclopentene-1 -methanol (compound of Formula (I)).
The compound of formula (I) is described as having potent activity against human immunodeficiency virus (HIV) and hepatitis B virus (HBV) in EPO34450.

Results presented at the 34th Interscience Conference on Antimicrobial Agents and Chemotherapy (October 4-7, 1994) demonstrate that the compound of formula I has significant activity against HIV comparable to, and if not better than, some current anti HIV drugs, such as zidovudine and didanosine.
Currently the compound of Formula (I) is undergoing clinical investigation to determine its safety and efficacy in humans. Therefore, there exists at the present time a need to supply large quantities of this compound for use in clinical trials.
Current routes of synthesising the compound of formula (I) involve multiple steps and are relatively expensive. It will be noted that the compound has two centres of asymmetry and it is essential that any route produces the compound of formula (I) substantially free of the corresponding enantiomer, preferably the compound of formula (I) is greater than 95% w/w free of the corresponding enantiomer.
Processes proposed for the preparation of the compound of formula (I) generally start from a pyrimidine compound, coupling with a 4-amino-2-cyclopentene-1- methanol analogue, cyciisation to form the imidazole ring and then introduction of the cyclopropylamine group into the 6 position of the purine, such routes include those suggested in EPO434450 and WO9521161. Essentially both routes disclosed in the two prior patent applications involve the following steps:-
(i) coupling (1S, 4R)-4-amino-2-cyclopentene-1 -methanol to N-(4,6-dichloro-5- formamido-2-pyrimidinyl) acetamide or a similar analogue thereof, for example N- (2-amino-4,6-dichloro-5-pyrimidinyl) formamide;
(ii) ring closure of the resultant compound to form the intermediate (1 S, 4R)-4- (2-amino-6-chloro-9H-purin-9-yl)-2-cyclopentene-1 -methanol;
(iii) substituting the halo group by a cyclopropylamino group on the 6 position of the purine ring.
The above routes are multi-step processes. By reducing the number of processing steps significant cost savings can be achieved due to the length of time to manufacture the compound being shortened and the waste streams minimised.
An alternative process suggested in the prior art involves the direct coupling of carbocyclic ribose analogues to the N atom on the 9 position of 2-amino-6-chloro purine. For example WO91/15490 discloses a single step process for the formation of the (1S, 4R)- 4-(2-amino-6-chloro-9H-purin-9-yl)-2-cyclopentene-1- methanol intermediate by reacting (1S, 4R)-4-hydroxy-2-cyclopentene-1 -methanol, in which the allylic hydroxyl group has been activated as an ester or carbonate and the other hydroxyl group has a blocking group attached (for example 1 ,4- bis- methylcarbonate) with 2-amino-6-chloropurine.
However we have found that when synthesising (1S, 4R)-4-(2-amino-6-chloro-9H- purin-9-yl)-2-cyclopentene-1- methanol by this route a significant amount of an N- 7 isomer is formed (i.e. coupling has occurred to the nitrogen at the 7- position of the purine ring) compared to the N-9 isomer desired. Further steps are therefore required to convert the N-7 product to the N-9 product, or alternatively removing the N-7 product, adding significantly to the cost. We have found that by using a transition metal catalysed process for the direct coupling of a compound of formula (II) or (III),

Example 1 (1 S. 4R)-4-[2-Amino-6-(cvclopropylamino)-9H purin-9-vπ-2-cvclopentene-1 – methanol
Triphenylphosphine (14mg) was added, under nitrogen, to a mixture of (1S.4R)- 4-hydroxy-2-cyclopentene -1 -methanol bis(methylcarbonate) (91 mg), 2-amino-6- (cyclopropylamino) purine (90mg), tris(dibenzylideneacetone)dipalladium (12mg) and dry DMF (2ml) and the resulting solution stirred at room temperature for 40 min.
The DMF was removed at 60° in vacuo and the residue partitioned between ethyl acetate (25ml.) and 20% sodium chloride solution (10ml.). The ethyl acetate solution was washed with 20% sodium chloride (2x12ml.) and with saturated sodium chloride solution, then dried (MgSO4) and the solvent removed in vacuo.
The residue was dissolved in methanol (10ml.), potassium carbonate (17mg) added and the mixture stirred under nitrogen for 15h.
The solvent was removed in vacuo and the residue chromatographed on silica gel
(Merck 9385), eluting with dichloromethane-methanol [(95:5) increasing to (90:10)] to give the title compound (53mg) as a cream foam.
δ(DMSO-d6): 7.60 (s.1 H); 7.27 (s,1 H); 6.10 (dt,1 H); 5.86 (dt, 1 H); 5.81 (s,2H); 5.39 (m,1H); 4.75 (t,1H); 3.44 (t,2H); 3.03 (m, 1H): 2.86 (m,1H);2.60 (m,1H); 1.58 (dt, 1 H); 0.65 (m, 2H); 0.57 (m,2H).
TLC SiO2/CHCI3-MeOH (4:1 ) Rf 0.38; det. UN., KMnO4
Trade Names
| Page | Trade name | Manufacturer |
|---|---|---|
| Germany | Kiveksa | GlaxoSmithKline |
| Trizivir | -»- | |
| Ziagen | -»- | |
| France | Kiveksa | -»- |
| Trizivir | -»- | |
| Ziagen | -»- | |
| United Kingdom | Kiveksa | -»- |
| Trizivir | -»- | |
| Ziagen | -»- | |
| Italy | Trizivir | -»- |
| Ziagen | -»- | |
| Japan | Épzikom | -»- |
| Ziagen | -»- | |
| USA | Épzikom | -»- |
| Trizivir | -»- | |
| Ziagen | -»- | |
| Ukraine | Virol | Ranbaksi Laboratories Limited, India |
| Ziagen | GlaksoSmitKlyayn Inc.., Canada | |
| Abamun | Tsipla Ltd, India | |
| Abacavir sulfate | Aurobindo Pharma Limited, India |
Formulations
-
Oral solution 20 mg / ml;
-
Tablets of 300 mg (as the sulfate);
-
Trizivir tablets 300 mg – abacavir in fixed combination with 150 mg of lamivudine and 300 mg zidovudine
ZIAGEN is the brand name for abacavir sulfate, a synthetic carbocyclic nucleoside analogue with inhibitory activity against HIV-1. The chemical name of abacavir sulfate is (1S,cis)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol sulfate (salt) (2:1). Abacavir sulfate is the enantiomer with 1S, 4R absolute configuration on the cyclopentene ring. It has a molecular formula of (C14H18N6O)2•H2SO4 and a molecular weight of 670.76 daltons. It has the following structural formula:
 |
Abacavir sulfate is a white to off-white solid with a solubility of approximately 77 mg/mL in distilled water at 25°C. It has an octanol/water (pH 7.1 to 7.3) partition coefficient (log P) of approximately 1.20 at 25°C.
ZIAGEN Tablets are for oral administration. Each tablet contains abacavir sulfate equivalent to 300 mg of abacavir as active ingredient and the following inactive ingredients: colloidal silicon dioxide, magnesium stearate, microcrystalline cellulose, and sodium starch glycolate. The tablets are coated with a film that is made of hypromellose, polysorbate 80, synthetic yellow iron oxide, titanium dioxide, and triacetin.
ZIAGEN Oral Solution is for oral administration. Each milliliter (1 mL) of ZIAGEN Oral Solution contains abacavir sulfate equivalent to 20 mg of abacavir (i.e., 20 mg/mL) as active ingredient and the following inactive ingredients: artificial strawberry and banana flavors, citric acid (anhydrous), methylparaben and propylparaben (added as preservatives), propylene glycol, saccharin sodium, sodium citrate (dihydrate), sorbitol solution, and water.
In vivo, abacavir sulfate dissociates to its free base, abacavir. All dosages for ZIAGEN are expressed in terms of abacavir.
History
Abacavir was approved by the Food and Drug Administration (FDA) on December 18, 1998 and is thus the fifteenth approved antiretroviral drug in the United States. Its patent expired in the United States on 2009-12-26.
Links
-
US 5 089 500 (Burroughs Wellcome; 18.2.1992; GB-prior. 27.6.1988).
-
Synthesis a)
-
EP 434 450 (Wellcome Found .; 26.6.1991; appl. 21.12.1990; prior-USA. 22.12.1989).
-
Crimmins, MT et al .: J. Org. Chem. (JOCEAH) 61 4192 (1996).
-
EP 1 857 458 (Solmag; appl. 5.5.2006).
-
EP 424 064 (Enzymatix; appl. 24.4.1991; GB -prior. 16.10.1989).
-
U.S. 6 340 587 (Beecham SMITHKLINE; 22.1.2002; appl. 20.8.1998; GB -prior. 22.8.1997).
-
-
Синтез b)
-
Olivo, HF et al .: J. Chem. Soc., Perkin Trans. 1 (JCPRB4) 1998, 391.
-
-
Preparation c)
-
U.S. 5 034 394 (Wellcome Found .; 23.7.1991; appl. 22.12.1989; GB -prior. 27.6.1988).
-
-
Synthesis d)
-
WO 9 924 431 (Glaxo; appl. 12.11.1998; WO-prior. 12.11.1997).
-
| WO2008037760A1 * | Sep 27, 2007 | Apr 3, 2008 | Esteve Quimica Sa | Process for the preparation of abacavir |
| EP1905772A1 * | Sep 28, 2006 | Apr 2, 2008 | Esteve Quimica, S.A. | Process for the preparation of abacavir |
| US8183370 | Sep 27, 2007 | May 22, 2012 | Esteve Quimica, Sa | Process for the preparation of abacavir |
| EP0434450A2 | 21 Dec 1990 | 26 Jun 1991 | The Wellcome Foundation Limited | Therapeutic nucleosides |
| EP0741710A1 | 3 Feb 1995 | 13 Nov 1996 | The Wellcome Foundation Limited | Chloropyrimide intermediates |
| WO1998052949A1 | 14 May 1998 | 26 Nov 1998 | Glaxo Group Ltd | Carbocyclic nucleoside hemisulfate and its use in treating viral infections |
| WO1999021861A1 | 24 Oct 1997 | 6 May 1999 | Glaxo Group Ltd | Process for preparing a chiral nucleoside analogue |
| WO1999039691A2 * | 4 Feb 1999 | 12 Aug 1999 | Brooks Nikki Thoennes | Pharmaceutical compositions |
| WO2008037760A1 * | 27 Sep 2007 | 3 Apr 2008 | Esteve Quimica Sa | Process for the preparation of abacavir |
References
- Jump up^ SFGate.com
- Jump up^ “WHO Model List of EssentialMedicines”. World Health Organization. October 2013. Retrieved 22 April 2014.
- Jump up^ https://online.epocrates.com/noFrame/showPage.do?method=drugs&MonographId=2043&ActiveSectionId=5
- Jump up^ Mallal, S., Phillips, E., Carosi, G. et al. (2008). “HLA-B*5701 screening for hypersensitivity to abacavir”. New England Journal of Medicine 358: 568–579.doi:10.1056/nejmoa0706135.
- Jump up^ Rauch, A., Nolan, D., Martin, A. et al. (2006). “Prospective genetic screening decreases the incidence of abacavir hypersensitivity reactions in the Western Australian HIV cohort study”. Clinical Infectious Diseases 43: 99–102. doi:10.1086/504874.
- Jump up^ Heatherington et al. (2002). “Genetic variations in HLA-B region and hypersensitivity reactions to abacavir”. Lancet 359: 1121–1122.
- Jump up^ Mallal et al. (2002). “Association between presence of HLA*B5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir”. Lancet359: 727–732. doi:10.1016/s0140-6736(02)07873-x.
- Jump up^ Rotimi, C.N.; Jorde, L.B. (2010). “Ancestry and disease in the age of genomic medicine”. New England Journal of Medicine 363: 1551–1558.
- Jump up^ Phillips, E., Mallal, S. (2009). “Successful translation of pharmacogenetics into the clinic”. Molecular Diagnosis & Therapy 13: 1–9. doi:10.1007/bf03256308.
- Jump up^ Phillips, E., Mallal S. (2007). “Drug hypersensitivity in HIV”. Current Opinion in Allergy and Clinical Immunology 7: 324–330. doi:10.1097/aci.0b013e32825ea68a.
- Jump up^http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm123927.htmAccessed November 29, 2013.
- Jump up^ http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=ca73b519-015a-436d-aa3c-af53492825a1
- Jump up^ Martin MA, Hoffman JM, Freimuth RR et al. (May 2014). “Clinical Pharmacogenetics Implementation Consortium Guidelines for HLA-B Genotype and Abacavir Dosing: 2014 update”. Clin Pharmacol Ther. 95 (5): 499–500. doi:10.1038/clpt.2014.38.PMC 3994233. PMID 24561393.
- Jump up^ Swen JJ, Nijenhuis M, de Boer A et al. (May 2011). “Pharmacogenetics: from bench to byte–an update of guidelines”. Clin Pharmacol Ther. 89 (5): 662–73.doi:10.1038/clpt.2011.34. PMID 21412232.
- Jump up^ Shear, N.H., Milpied, B., Bruynzeel, D.P. et al. (2008). “A review of drug patch testing and implications for HIV clinicians”. AIDS 22: 999–1007.doi:10.1097/qad.0b013e3282f7cb60.
- Jump up^ http://www.drugs.com/fda/abacavir-ongoing-safety-review-possible-increased-risk-heart-attack-12914.html Accessed November 29, 2013.
- Jump up^ Ding X, Andraca-Carrera E, Cooper C et al. (December 2012). “No association of abacavir use with myocardial infarction: findings of an FDA meta-analysis”. J Acquir Immune Defic Syndr. 61 (4): 441–7. doi:10.1097/QAI.0b013e31826f993c.PMID 22932321.
- Illing PT et al. 2012, Nature, doi:10.1038/nature11147
External links
- Full Prescribing Information
- Abacavir pathway on PharmGKB
- Abacavir dosing guidelines from the Clinical Pharmacogenetics Implementation Consortium (CPIC)
- Abacavir dosing guidelines from the Dutch Pharmacogenetics Working Group (DPWG)
EXTRA INFO
How to obtain carbocyclic nucleosides?
Carbocyclic nucleosides are synthetically the most challenging class of nucleosides, requiring multi-step and often elaborate synthetic pathways to introduce the necessary stereochemistry. There are two main strategies for the preparation of carbocyclic nucleosides. In the linear approach a cyclopentylamine is used as starting material and the heterocycle is built in a stepwise manner (see Scheme 1).
Scheme 1: Linear approach for the synthesis of abacavir.[5]
The more flexible strategy is a convergent approach: a functionalized carbocyclic moiety is condensed with a heterocycle rapidly leading to a variety of carbocyclic nucleosides. Initially, we started our syntheses from cyclopentadiene 1 that is deprotonated and alkylated with benzyloxymethyl chloride to give the diene 2. This material is converted by a hydroboration into cyclopentenol 3 or isomerized into two thermodynamically more stable cyclopentadienes 4a,b. With the protection and another hydroboration step to 5 we gain access to an enantiomerically pure precursor for the synthesis of a variety of carbocyclic 2’-deoxynucleosides e.g.:carba-dT, carba-dA or carba-BVDU.[6] The isomeric dienes 4a,b were hydroborated to the racemic carbocyclic moiety 6.

Scheme 2: Convergent approach for the synthesis of carba-dT.
The asymmetric synthesis route and the racemic route above are short and efficient ways to diverse carbocyclic D- or L-nucleosides (Scheme 2). Different heterocycles can be condensed to these precursors leading to carbocyclic purine- and pyrimidine-nucleosides. Beside α- and β-nucleosides, carbocyclic epi– andiso-nucleosides in the 2’-deoxyxylose form were accessable.[7]
What else is possible? The racemic cyclopentenol 6 can be coupled by a modified Mitsunobu-reaction.Moreover, this strategy offers the possibility of synthesizing new carbocyclic nucleosides by functionalizing the double bond before or after introduction of the nucleobase (scheme 3).[8]

Scheme 3: Functionalized carbocyclic nucleosides based on cyclopentenol 6.
Other interesting carbocyclic precursors like cyclopentenol 7 can be used to synthesize several classes of carbocyclic nucleoside analogues, e.g.: 2’,3’-dideoxy-2’,3’-didehydro nucleosides (d4-nucleosides), 2’,3’-dideoxynucleosides (ddNs), ribonucleosides, bicyclic nucleosides or even 2’-fluoro-nucleosides.

Scheme 4: Functionalized carbocyclic thymidine analogues based on cyclopentenol 7.
[1] V. E. Marquez, T. Ben-Kasus, J. J. Barchi, K. M. Green, M .C. Nicklaus, R. Agbaria, J. Am. Chem. Soc.2004,126, 543.
[2] A. D. Borthwick, K. Biggadike, Tetrahedron 1992, 48, 571.
[3] H. Bricaud, P. Herdewijn, E. De Clercq, Biochem. Pharmacol. 1983, 3583.
[4] P. L. Boyer, B. C. Vu, Z. Ambrose, J. G. Julias, S. Warnecke, C. Liao, C. Meier, V. E. Marquez, S. H. Hughes, J. Med. Chem. 2009, 52, 5356.
[5] S. M. Daluge, M. T. Martin, B. R. Sickles, D. A. Livingston, Nucleosides, Nucleotides Nucleic Acids 2000,19, 297.
[6] O. R. Ludek, C. Meier, Synthesis 2003, 2101.
[7] O. R. Ludek, T. Kraemer, J. Balzarini, C. Meier, Synthesis 2006, 1313.
[8] M. Mahler, B. Reichardt, P. Hartjen, J. van Lunzen, C. Meier, Chem. Eur. J. 2012, 18, 11046-11062.
BI 224436 an investigational new drug under development for the treatment of HIV infection


(2S)-2-tert-butoxy-2-(4-(2,3-dihydropyrano[4,3,2-de]quinolin-7-yl)-2- methylquinolin-3-yl)acetic acid
BI 224436
1155419-89-8 cas no
mw
| 442.51 |
3-Quinolineacetic acid, 4-(2,3-dihydropyrano[4,3,2-de]quinolin-7-yl)-α-(1,1-dimethylethoxy)-2-methyl-, (αS,4R)-
hemi-succinate of (2S)-2-tert-butoxy-2-(4-(2,3-dihydropyrano[4,3,2-de]quinolin-7-yl)-2-methylquinolin-3-yl)acetic acid)
BI 224436 is an investigational new drug under development for the treatment of HIV infection. BI 224436 is the first non-catalytic site integrase inhibitor (NCINI). It inhibits HIV replication via binding to a conserved allosteric pocket of the HIV integrase enzyme. This makes the drug distinct in mechanism of action compared to raltegravir and elvitegravir, which bind at the catalytic site.[2] In October 2011, Gilead Sciences purchased exclusive rights to develop BI 224436 and several related compounds under investigation in Boehringer Ingelheim’s noncatalytic site integrase inhibitor program.[3][4]
Novel hemi-succinate salt form of (2S)-2-tert-butoxy-2-(4-(2,3-dihydropyrano[4,3,2-de]quinolin-7-yl)-2-methylquinolin-3-yl)acetic acid (presumed to be BI-224436) and its crystalline forms is desc in WO-2014055618.
Gilead, under license from BI, was developing BI-224436 for the oral treatment of HIV infection. In September 2011, this drug had entered phase 1 trials. Picks up from WO2012138670, claiming a process for the preparation of the same drug. Also see the concurrently published WO2014055603. This compound is claimed specifically in WO2009062285 and generically in WO2007131350.
BI 224436 has antiviral EC50 values ranging between 4 and 15 nM against different HIV-1 laboratory strains and CC50 values >90 μM in different cells, including peripheral blood mononuclear cells. BI 224436 also has a low, 2.2-fold shift in antiviral potency in the presence of 50% human serum and by virtue of a steep dose-response curve slope, BI 224436 exhibits serum-shifted EC95 values ranging between 22 and 75 nM. Drug combination studies performed in cell-based antiviral assays have shown that BI 224436 displays, at the least, an additive effect in combination with any of the marketed antiviral classes including INSTIs. BI 224436 has drug-like ADME properties including a Caco-2 cell permeability of 14 .10-6 cm/sec, solubility > 24 mg/ml in the pH range 2.0-6.8 and low cytochrome P450 inhibition. Moreover BI 224436 shows excellent PK profiles in rat (CL=0.7% QH; F= 54%), monkey (CL= 23% QH; F= 82%) and dog (CL= 8%QH; F= 81%).

http://www.natap.org/2011/ICAAC/ICAAC_32.htm
……………………
Discovery of BI 224436, a Noncatalytic Site Integrase Inhibitor (NCINI) of HIV-1
ACS Med. Chem. Lett., 2014, 5 (4), pp 422–427
DOI: 10.1021/ml500002n
http://pubs.acs.org/doi/abs/10.1021/ml500002n

1H NMR: 12.4 (br, 1H), 8.52 (d, 1H, J = 4.4Hz), 7.94 (d, 1H, J = 7.9 Hz),7.65-7.61 (m, 1H), 7.45 (d,
1H, J = 8.2 Hz), 7.31-7.24 (m, 2H), 7.12 (d, 1H, J = 7.9 Hz), 6.94-6.92 (m, 1H), 4.99 (s, 1H), 4.57-4.47
(m, 2H), 3.37-3.30 (m, 2H), 2.86 (s, 3H), 0.82 (s, 9H).
13C NMR: 172.2, 158.4, 153.1, 150.1, 146.6,
146.1, 145.0, 141.0, 130.8 (br), 130.6 (br), 128.9, 128.0, 127.2, 127.1 (br) 126.4, 125.6, 118.0, 116.7,
109.1, 75.2, 70.8, 65.6, 27.7, 27.5, 24.9.
HRMS: m/z calc. for C27H26N2O4 + H+: 443.1965, m/z found:
443.1951 (-3.2 ppm).
UPLC-MS: rt = 0.68 min, m/z 443.3 [M + H]+, purity: >99.9% @ 254 nm.
http://pubs.acs.org/doi/suppl/10.1021/ml500002n/suppl_file/ml500002n_si_001.pdf
………………………….
http://www.google.com/patents/WO2012138670A1?cl=en
General Scheme IA:

G1 1001
wherein Y is I, Br or CI;
General Scheme 11 A:

wherein:
Example 1

1 a 1 b
1a (600 g, 4.1 mol) was charged into a dry reactor under nitrogen followed by addition of Ac20 (1257.5 g, 12.3 mol, 3 eq.). The resulting mixture was heated at 40 °C at least for 2 hours. The batch was then cooled to 30 °C over 30 minutes. A suspension of 1b in toluene was added to seed the batch if no solid was observed. After toluene (600 ml_) was added over 30 minutes, the batch was cooled to -5— 10 °C and was held at this temperature for at least 30 minutes. The solid was collected by filtration under nitrogen and rinsed with heptanes (1200 ml_). After being dried under vacuum at room temperature, the solid was stored under nitrogen at least below 20 °C. The product 1 b was obtained with 77% yield. 1H NMR (500 MHz, CDCI3): δ = 6.36 (s, 1 H), 3.68 (s, 2H), 2.30 (s, 3H). Example 2

2a 2b
2a (100g, 531 mmol) and 1b (95 g, 558 mmol) were charged into a clean and dry reactor under nitrogen followed by addition of fluorobenzene (1000 mL). After being heated at 35-37 °C for 4 hours, the batch was cooled to 23 °C. Concentrated H2S04 (260.82 g, 2659.3 mmol, 5 eq.) was added while maintaining the batch temperature below 35 °C. The batch was first heated at 30-35 °C for 30 minutes and then at 40- 45 °C for 2 hours. 4-Methyl morpholine (215.19 g, 2127 mmol, 4 eq.) was added to the batch while maintaining the temperature below 50 °C. Then the batch was agitated for 30 minutes at 40-50 °C. eOH (100 mL) was then added while maintaining the temperature below 55 °C. After the batch was held at 50-55 °C for 2 hours, another portion of MeOH (100 mL) was added. The batch was agitated for another 2 hours at 50-55 °C. After fluorobenzene was distilled to a minimum amount, water (1000 mL) was added. Further distillation was performed to remove any remaining fluorobenzene. After the batch was cooled to 30 °C, the solid was collected by filtration with cloth and rinsed with water (400 mL) and heptane (200 mL). The solid was dried under vacuum below 50 °C to reach KF < 0.1%. Typically, the product 2b was obtained in 90% yield with 98 wt%. 1H NMR (500 MHz, DMSO- d6): δ = 10.83 (s, 1 H), 9.85 (s, bs, 1 H), 7.6 (d, 1 H, J
Hz), 6.40 (s, 1 H), 4.00 (s, 2 H), 3.61 (s, 3 H). Example 3

2b 3a
2b (20 g, 64 mmol) was charged into a clean and dry reactor followed by addition of THF (140 mL). After the resulting mixture was cooled to 0 °C, Vitride® (Red-AI, 47.84 g, 65 wt%, 154 mmol) in toluene was added while maintaining an internal temperature at 0-5 °C. After the batch was agitated at 5-10 °C for 4 hours, IPA (9.24 g, 153.8 mmol) was added while maintaining the temperature below 10 °C. Then the batch was agitated at least for 30 minutes below 25 °C. A solution of HCI in IPA (84.73 g, 5.5 M, 512 mmol) was added into the reactor while maintaining the temperature below 40 °C. After about 160 mL of the solvent was distilled under vacuum below 40 °C, the batch was cooled to 20-25 °C and then aqueous 6M HCI (60 mL) was added while maintaining the temperature below 40 °C. The batch was cooled to 25 °C and agitated for at least 30 minutes. The solid was collected by filtration, washed with 40 mL of IPA and water (1V/1V), 40 mL of water and 40 mL of heptanes. The solid was dried below 60 °C in a vacuum oven to reach KF < 0.5%. Typically, the product 3a was obtained in 90-95% yield with 95 wt%. 1H NMR (400 MHz, DMSO-d6): δ = 10.7 (s, 1 H), 9.68 (s, 1 H), 7.59 (d, 1 H, J = 8.7 Hz), 6.64 (, 1 H, J = 8.7 Hz), 6.27 (s, 1 H), 4.62 (bs, 1 H), 3.69 (t, 2H, J = 6.3 Hz), 3.21 (t, 2H, J = 6.3 Hz).
Example 4

3a (50 g, 174.756 mmol) and acetonitrile (200 mL) were charged into a dry and clean reactor. After the resulting mixture was heated to 65 °C, POCI3 (107.18 g, 699 mmol, 4 eq.) was added while maintaining the internal temperature below 75 °C. The batch was then heated at 70-75 °C for 5-6 hours. The batch was cooled to 20 °C. Water (400 mL) was added at least over 30 minutes while maintaining the internal temperature below 50 °C. After the batch was cooled to 20-25 °C over 30 minutes, the solid was collected by filtration and washed with water (100 mL). The wet cake was charged back into the reactor followed by addition of 1 M NaOH (150 mL). After the batch was agitated at least for 30 minutes at 25-35 °C, it was verified that the pH was greater than 12. Otherwise, more 6M NaOH was needed to adjust the pH >12. After the batch was agitated for 30 minutes at 25-35 °C, the solid was collected by filtration, washed with water (200 mL) and heptanes (200 mL). The solid was dried in a vacuum oven below 50 °C to reach KF < 2%. Typically, the product 4a was obtained at about 75-80% yield. H NMR (400 MHz, CDCI3): δ = 7.90 (d, 1 H, J = 8.4 Hz), 7.16 (s, 1 H), 6.89 (d, 1 H, J = 8.4 Hz), 4.44 (t, 2 H, J = 5.9 Hz), 3.23 (t, 2 H, J = 5.9 Hz). 13C NMR (100 MHz, CDCI3): δ = 152.9, 151.9, 144.9, 144.1 , 134.6, 1 19.1 , 1 17.0, 1 13.3, 1 1 1.9, 65.6, 28.3.
Example 5

4a 5a
Zn powder (54 g, 825 mmol, 2.5 eq.) and TFA (100 mL) were charged into a dry and clean reactor. The resulting mixture was heated to 60-65 °C. A suspension of 4a (100 g, 330 mmol) in 150 mL of TFA was added to the reactor while maintaining the temperature below 70 °C. The charge line was rinsed with TFA (50 mL) into the reactor. After 1 hour at 65±5 °C, the batch was cooled to 25-30 °C. Zn powder was filtered off by passing the batch through a Celite pad and washing with methanol (200 mL). About 400 mL of solvent was distilled off under vacuum. After the batch was cooled to 20-25 °C, 20% NaOAc (ca. 300 mL) was added at least over 30 minutes to reach pH 5-6. The solid was collected by filtration, washed with water (200 mL) and heptane (200 mL), and dried under vacuum below 45 °C to reach KF ≤ 2%. The solid was charged into a dry reactor followed by addition of loose carbon (10 wt%) and toluene (1000 mL). The batch was heated at least for 30 minutes at 45-50 °C. The carbon was filtered off above 35 °C and rinsed with toluene (200 mL). The filtrate was charged into a clean and dry reactor. After about 1000 mL of toluene was distilled off under vacuum below 50 °C, 1000 mL of heptane was added over 30 minutes at 40-50 °C. Then the batch was cooled to 0±5 °C over 30 minutes. After 30 minutes, the solid was collected and rinsed with 200 mL of heptane. The solid was dried under vacuum below 45 °C to reach KF≤ 500 ppm. Typically, the product 5a was obtained in about 90-95 % yield. 1H NMR (400 MHz, CDCI3): δ = 8.93 (m, 1 H), 7.91 (dd, 1 H, J = 1.5, 8 Hz), 7.17 (m 1 H), 6.90 (dd, 1 H, J = 1 .6, 8.0 Hz), 4.46-4.43 (m, 2 H), 3.28-3.23 (m, 2 H). 13C NMR (100 MHz, CDCI3): δ = 152.8, 151 .2, 145.1 , 141.0, 133.3, 1 18.5, 1 18.2, 1 14.5, 1 1 1.1 , 65.8, 28.4.
Example 6

5a 6a
5a (1.04 kg, 4.16 mol) and toluene (8 L) were charged into the reactor. The batch was agitated and cooled to -50 to -55 °C. BuLi solution (2.5 M in hexanes, 1.69 L, 4.23 mol) was charged slowly while maintaining the internal temperature between – 45 to -50 °C. The batch was agitated at -45 °C for 1 hour after addition. A solution of triisopropyl borate (0.85 kg, 4.5 mol) in MTBE (1 .48 kg) was charged. The batch was warmed to 10 °C over 30 minutes. A solution of 5 N HCI in I PA (1 .54 L) was charged slowly at 10 °C, and the batch was warmed to 20 °C and stirred for 30 minutes. It was seeded with 6a crystal (10 g). A solution of aqueous concentrated HCI (0.16 L) in IPA (0.16 L) was charged slowly at 20 °C in three portions at 20 minute intervals, and the batch was agitated for 1 hour at 20 °C. The solid was collected by filtration, rinsed with MTBE (1 kg), and dried to provide 6a (943 g, 88.7 % purity, 80% yield). 1H NMR (400 MHz, D20): δ 8.84 (d, 1 H, J = 4 Hz)
1 H), 7.68 (d, 1 H, J = 6 Hz), 7.09 (m, 1 H), 4.52 (m, 2H), 3.47 (m, 2H).
Example 7

Iodine stock solution was prepared by mixing iodine (57.4 g, 0.23 mol) and sodium iodide (73.4 g, 0.49 mol) in water (270 mL). Sodium hydroxide (28.6 g, 0.715 mol) was charged into 220 mL of water. 4-Hydroxy-2 methylquinoline 7a (30 g, 0.19 mol) was charged, followed by acetonitrile (250 mL). The mixture was cooled to 10 °C with agitation. The above iodine stock solution was charged slowly over 30 minutes. The reaction was quenched by addition of sodium bisulfite (6.0 g) in water (60 mL). Acetic acid (23 mL) was charged over a period of 1 hour to adjust the pH of the reaction mixture between 6 and 7. The product was collected by filtration, washed with water and acetonitrile, and dried to give 7b (53 g, 98%). MS 286 [M + 1].
Example 8

7b 8a
4-Hydroxy-3-iodo-2-methylquinoline 7b (25 g, 0.09 mol) was charged to a 1-L reactor. Ethyl acetate (250 mL) was charged, followed by triethylamine (2.45 mL, 0.02 mol) and phosphorus oxychloride (12 mL, 0.13 mol). The reaction mixture was heated to reflux until complete conversion (~1 hour), then the mixture was cooled to 22 °C. A solution of sodium carbonate (3 .6 g, 0.3 mol) in water (500 mL) was charged. The mixture was stirred for 20 minutes. The aqueous layer was extracted with ethyl acetate (120 mL). The organic layers were combined and concentrated under vacuum to dryness. Acetone (50 mL) was charged. The solution was heated to 60 °C. Water (100 mL) was charged, and the mixture was cooled to 22 °C. The product was collected by filtration and dried to give 8a (25 g, 97.3 % pure, 91.4 % yield). MS 304 [M + 1].
(Note: 8a is a known compound with CAS # 1033931-93-9. See references: (a) J. Org Chem. 2008, 73, 4644-4649. (b) Molecules 2010, 15, 3171 -3178. (c) Indian J. Chem. Sec B: Org. Chem. Including Med Chem. 2009, 488(5), 692-696.)
Example 9

8a 9a
8a (100 g, 0.33 mol) was charged to the reactor, followed by copper (I) bromide dimethyl sulfide complex (3.4 g, 0.017 mol) and dry THF (450 mL). The batch was cooled to -15 to -12 °C. i-PrMgCI (2.0 M in THF, 173 mL, 0.346 mol) was charged into the reactor at the rate which maintained the batch temperature < -10 °C. In a 2nd reactor, methyl chlorooxoacetate (33 mL, 0.36 mol) and dry THF (150 mL) were charged. The solution was cooled to -15 to -10 °C. The content of the 1 st reactor (Grignard/cuprate) was charged into the 2nd reactor at the rate which maintained the batch temperature < -10 °C. The batch was agitated for 30 minutes at -10 °C. Aqueous ammonium chloride solution ( 0%, 300 mL) was charged. The batch was agitated at 20 – 25 °C for 20 minutes and allowed to settle for 20 minutes. The aqueous layer was separated. Aqueous ammonium chloride solution (10%, 90 mL) and sodium carbonate solution (10%, 135 mL) were charged to the reactor. The batch was agitated at 20 – 25 °C for 20 minutes and allowed to settle for 20 minutes. The aqueous layer was separated. Brine (10%, 240 mL) was charged to the reactor. The batch was agitated at 20 – 25 °C for 20 minutes. The aqueous layer was separated. The batch was concentrated under vacuum to -1/4 of the volume (about 80 mL left). 2-Propanol was charged (300 mL). The batch was concentrated under vacuum to -1/3 of the volume (about 140 mL left), and heated to 50 °C.
Water (70 mL) was charged. The batch was cooled to 20 – 25 °C, stirred for 2 hours, cooled to – 0 °C and stirred for another 2 hours. The solid was collected by filtration, washed with cold 2-propanol and water to provide 58.9 g of 9a obtained after drying (67.8 % yield). 1H NMR (400 MHz, CDCI3): δ 8.08 (d, 1 H, J = 12 Hz), 7.97 (d, 1 H, J = 12 Hz), 7.13 (t, 1 H, J = 8 Hz), 7.55 (t, 1 H, J= 8 Hz), 3.92 (s, 3H), 2.63 (s, 3H). 13C NMR (100 MHz, CDCI3): δ 186.6, 161.1 , 155.3, 148.2, 140.9, 132.0, 129.0, 128.8, 127.8, 123.8, 123.7, 53.7, 23.6.

Catalyst preparation: To a suitable sized, clean and dry reactor was charged dichloro(pentamethylcyclopentadienyl)rhodium (III) dimer (800 ppm relative to 9a, 188.5 mg) and the ligand (2000 ppm relative to 9a, 306.1 mg). The system was purged with nitrogen and then 3 ml. of acetonitrile and 0.3 ml_ of triethylamine was charged to the system. The resulting solution was agitated at room temperature for not less than 45 minutes and not more than 6 hours. Reaction: To a suitable sized, clean and dry reactor was charged 9a (1.00 equiv, 100.0 g (99.5 wt%), 377.4 mmol). The reaction was purged with nitrogen. To the reactor was charged acetonitrile (ACS grade, 4 L/Kg of 9a, 400 mL) and
triethylamine (2.50 equiv, 132.8 mL, 943 mmol). Agitation was initiated. The 9a solution was cooled to Tint= -5 to 0 °C and then formic acid (3.00 equiv, 45.2 mL, 1 132 mmol) was charged to the solution at a rate to maintain Tint not more than 20 °C. The batch temperature was then adjusted to Tint= -5 to -0 °C. Nitrogen was bubbled through the batch through a porous gas dispersion unit (Wiimad-LabGlass No. LG-8680-1 0, VWR catalog number 14202-962) until a fine stream of bubbles was obtained. To the stirring solution at Tint= -5 to 0 °C was charged the prepared catalyst solution from the catalyst preparation above. The solution was agitated at Tint= -5 to 0 °C with the bubbling of nitrogen through the batch until HPLC analysis of the batch indicated no less than 98 A% conversion (as recorded at 220 nm, 10-14 h). To the reactor was charged isopropylacetate (6.7 L/Kg of 9a, 670 ml_). The batch temperature was adjusted to Tint= 18 to 23 °C. To the solution was charged water (10 L/Kg of 9a, 1000 mL) and the batch was agitated at Tint= 18 to 23 °C for no less than 20 minutes. The agitation was decreased and or stopped and the layers were allowed to separate. The lighter colored aqueous layer was cut. To the solution was charged water (7.5 L/Kg of 9a, 750 mL) and the batch was agitated at Tint= 18 to 23 °C for no less than 20 minutes. The agitation was decreased and or stopped and the layers were allowed to separate. The lighter colored aqueous layer was cut. The batch was then reduced to 300 mL (3 L/Kg of 9a) via distillation while maintaining Text no more than 65 °C. The batch was cooled to Tint= 35 to 45 °C and the batch was seeded (10 mg). To the batch at Tint= 35 to 45 °C was charged heptane (16.7 L/Kg of 9a, 1670 mL) over no less than 1.5 hours. The batch temperature was adjusted to Tint= -2 to 3 °C over no less than 1 hour, and the batch was agitated at Tint= -2 to 3 °C for no less than 1 hour. The solids were collected by filtration. The filtrate was used to rinse the reactor (Filtrate is cooled to Tint= -2 to 3 °C before filtration) and the solids were suction dried for no less than 2 hours. The solids were dried until the LOD is no more than 4 % to obtain 82.7 g of 10a (99.6- 100 wt%, 98.5% ee, 82.5% yield). 1H-NMR (CDCI3, 400 MHz) δ: 8.20 (d, J= 8.4 Hz, 1 H), 8.01 (d, J= 8.4 Hz, 1 H), 7.73 (t, J= 7.4 Hz, 1 H), 7.59 (t, J= 7.7 Hz, H), 6.03 (s, 1 H), 3.93 (s, 1 H), 3.79 (s, 3H), 2.77 (s, 3H). 13C-NMR (CDCI3, 100 MHz) δ: 173.5, 158.3, 147.5, 142.9, 130.7, 128.8, 127.7, 127.1 , 125.1 , 124.6, 69.2, 53.4, 24.0.
Example 11

10a 6a
10a (2.45 kg, 96.8% purity, 8.9 mol), 6a (2.5 kg, 88.7% purity, 8.82 mol), tris(dibenzylideneacetone)dipalladium(0) (Pd2dba3, 40 g, 0.044 mol), (S)-3-ieri-butyl- 4-(2,6-dimethoxypheny1 )-2,3-dihydrobenzo[d][1 ,3]oxaphosphole (32 g, 0.01 1 mol), sodium carbonate (1.12 kg, 10.58 mol), 1-pentanol (16.69 L), and water (8.35 L) were charged to the reactor. The mixture was de-gassed by sparging with argon for 10-15 minutes, was heated to 60-63 °C, and was agitated until HPLC analysis of the reaction shows <1 A% (220 nm) of the 6a relative to the combined two atropisomer products (-15 hours). The batch was cooled to 18-23 °C. Water (5 L) and heptane (21 L) were charged. The slurry was agitated for 3 – 5 hours. The solids were collected by filtration, washed with water (4 L) and heptane/toluene mixed solvent (2.5 L toluene/5 L heptane), and dried. The solids were dissolved in methanol (25 L) and the resulting solution was heated to 50 °C and circulated through a CUNO carbon stack filter. The solution was distilled under vacuum to ~ 5 L. Toluene (12 L) was charged. The mixture was distilled under vacuum to ~ 5 L and cooled to 22 °C. Heptane (13 L) was charged to the contents over 1 hour and the resulting slurry was agitated at 20-25 °C for 3 – 4 hours. The solids were collected by filtration and washed with heptanes to provide 2.58 kg of 11a obtained after drying (73% yield). 1H NMR (400 MHz, CDCI3): δ 8.63 (d, 1 H, J = 8 Hz), 8.03 (d, 1 H, J = 12 Hz), 7.56 (t, 1 H, J = 8 Hz), 7.41 (d, 1 H, J = 8 Hz), 7.19 (t, 1 H, J = 8 Hz), 7.09 (m, 2H), 7.04 (d, 1 H, J = 8 Hz), 5.38 (d, 1 H, J = 8 Hz), 5.14 (d, 1 H, J = 8 Hz), 4.50 (t, 2H, J = 4 Hz), 3.40 (s, 3H), 3.25 (t, 2H, J = 4 Hz), 2.91 (s, 3H). 13C NMR (100 MHz, CDCI3): δ 173.6, 158.2, 154.0, 150.9, 147.3, 147.2, 145.7, 141.3, 132.9, 123.0, 129.4, 128.6, 127.8, 126.7, 126.4, 125.8, 1 18.1 , 1 17.3, 109.9, 70.3, 65.8, 52.3, 28.5, 24.0.
Example 12

11a 12a
To a suitable clean and dry reactor under a nitrogen atmosphere was charged 11a (5.47 Kg, 93.4 wt%, 1 .00 equiv, 12.8 mol) and fluorobenzene (10 vols, 51.1 kg) following by trifluoromethanesulfonimide (4 mol%, 143 g, 0.51 mol) as a 0.5 M solution in DCM (1.0 Kg). The batch temperature was adjusted to 35-41 °C and agitated to form a fine slurry. To the mixture was slowly charged i-butyl-2,2,2- trichloroacetimidate 12b as a 50 wt% solution (26.0 Kg of f-butyl-2,2,2- trichloroacetimidate (1 19.0 mol, 9.3 equiv), the reagent was -48-51 wt% with the remainder 52-49 wt% of the solution being – 1.8:1 wt:wt heptane: fluorobenzene) over no less than 4 hours at Tint= 35-41 °C. The batch was agitated at Tint= 35-41 °C until HPLC conversion (308 nm) was >96 A%, then cooled to Tint= 20-25 °C and then triethylamine (0.14 equiv, 181 g, 1 .79 mol) was charged followed by heptane (12.9 Kg) over no less than 30 minutes. The batch was agitated at Tint= 20-25 °C for no less than 1 hour. The solids were collected by filtration. The reactor was rinsed with the filtrate to collect all solids. The collected solids in the filter were rinsed with heptane (1 1 .7 Kg). The solids were charged into the reactor along with 54.1 Kg of DM Ac and the batch temperature adjusted to Tint= 70-75 °C. Water ( .2 Kg) was charged over no less than 30 minutes while the batch temperature was maintained at Tint= 65-75 °C. 12a seed crystals (34 g) in water (680 g) was charged to the batch at Tlnt= 65-75 °C. Additional water (46.0 Kg) was charged over no less than 2 hours while maintaining the batch temperature at Tint= 65-75 °C. The batch temperature was adjusted to Tint= 18-25 °C over no less than 2 hours and agitated for no less than 1 hour. The solids were collected by filtration and the filtrate used to rinse the reactor. The solids were washed with water (30 Kg) and dried under vacuum at no more than 45 °C until the LOD < 4% to obtain 12a (5.275 Kg, 99.9 A% at 220 nm, 99.9 wt% via HPLC wt% assay, 90.5% yield). 1H-NMR (CDCI3, 400
MHz) δ: 8.66-8.65 (m, 1 H), 8.05 (d, J= 8.3 Hz, 1 H), 7.59 (t, J= 7.3 Hz, 1 H), 7.45 (d, J= 7.8 Hz, 1 H), 7.21 (t, J= 7.6 Hz, 1 H), 7.13-7.08 (m, 3H), 5.05 (s, 1 H), 4.63-4.52 (m, 2H), 3.49 (s, 3H), 3.41 -3.27 (m, 2H), 3.00 (s, 3H), 0.97 (s, 9H). 13C-NMR (CDCI3, 100 MHz) δ: 172.1 , 159.5, 153.5, 150.2, 147.4, 146.9, 145.4, 140.2, 131.1 , 130.1 , 128.9, 128,6, 128.0, 127.3, 126.7, 125.4, 117.7, 117.2, 109.4, 76.1 , 71.6, 65.8, 51 .9, 28.6, 28.0, 25.4. Example 13

To a suitable clean and dry reactor under a nitrogen atmosphere was charged 12a (9.69 Kg, 21.2 mol) and ethanol (23.0 Kg). The mixture was agitated and the batch temperature was maintained at Τίηί= 20 to 25 °C. 2 M sodium hydroxide (17.2 Kg) was charged at Tint= 20 to 25 °C and the batch temperature was adjusted to Tint= 60- 65°C over no less than 30 minutes. The batch was agitated at Tint= 60-65°C for 2-3 hours until HPLC conversion was >99.5% area (12a is <0.5 area%). The batch temperature was adjuted to Tlnt= 50 to 55°C and 2M aqueous HCI (14.54 Kg) was charged. The pH of the batch was adjusted to pH 5.0 to 5.5 (target pH 5.2 to 5.3) via the slow charge of 2M aqueous HCI (0.46 Kg) at Tint= 50 to 55°C. Acetonitrile was charged to the batch (4.46 Kg) at Tint= 50 to 55°C. A slurry of seed crystals (1001 , 20 g in 155 g of acetonitrile) was charged to the batch at Tint= 50 to 55°C. The batch was agitated at Tint= 50 to 55°C for no less than 1 hour (1-2 hours). The contents were vacuum distilled to -3.4 vol (32 L) while maintaining the internal temperature at 45-55°C. A sample of the batch was removed and the ethanol content was determined by GC analysis; the criterion was no more than 10 wt% ethanol. If the ethanol wt% was over 10%, an additional 10% of the original volume was distilled and sampled for ethanol wt%. The batch temperature was adjusted to Tint= 18-22°C over no less than 1 hour. The pH of the batch was verified to be pH= 5 – 5.5 and the pH was adjusted, if necessary, with the slow addition of 2 M HCI or 2 M NaOH aqueous solutions. The batch was agitated at Tint= 18-22°C for no less than 6 hours and the solids were collected by filtration. The filtrate/mother liquid was used to remove all solids from reactor. The cake with was washed with water (19.4 Kg) (water temperature was no more than 20 °C). The cake was dried under vacuum at no more than 60 °C for 12 hours or until the LOD was no more than 4% to obtain 1001 (9.52 Kg, 99.6 A% 220 nm, 97.6 wt% as determined by HPLC wt% assay, 99.0% yield).
…………………
compd 1144
http://www.google.com/patents/WO2009062285A1?cl=en


……………………
http://www.google.com/patents/WO2012138669A1?cl=en
Compound (I), (2S)-2-tert-butoxy-2-(4-(2,3-dihydropyrano[4,3,2-de]quinolin-7-yl)-2- methylquinolin-3-yl)acetic acid, is an HIV non-catalytic site integrase inhibitor.

Compound (I) falls within the scope of the HIV inhibitors disclosed in WO
2007/131350. Compound (I) is disclosed specifically as compound no. 1144 in WO 2009/062285. Compound (I) can be prepared according to the general procedures found in WO 2007/13 350 and WO 2009/062285, which are hereby incorporated by reference.
Example 1

1 a 1b
1a (600 g, 4.1 mol) was charged into a dry reactor under nitrogen followed by addition of Ac20 (1257.5 g, 12.3 mol, 3 eq.). The resulting mixture was heated at 40 °C at least for 2 hours. The batch was then cooled to 30 °C over 30 minutes. A suspension of 1b in toluene was added to seed the batch if no solid was observed. After toluene (600 mL) was added over 30 minutes, the batch was cooled to -5 ~ -10 °C and was held at this temperature for at least 30 minutes. The solid was collected by filtration under nitrogen and rinsed with heptanes (1200 mL). After being dried under vacuum at room temperature, the solid was stored under nitrogen at least below 20 °C. The product 1b was obtained with 77% yield. 1H NMR (500 MHz, CDCI3): δ = 6.36 (s, 1 H), 3.68 (s, 2H), 2.30 (s, 3H).
Example 2

2a (100 g, 531 mmol) and 1 b (95 g, 558 mmol) were charged into a clean and dry reactor under nitrogen followed by addition of fluorobenzene ( 000 mL). After being heated at 35-37 °C for 4 hours, the batch was cooled to 23 °C. Concentrated H2S04 (260.82 g, 2659.3 mmol, 5 eq.) was added while maintaining the batch temperature below 35 °C. The batch was first heated at 30-35 °C for 30 minutes and then at 40- 45 °C for 2 hours. 4-Methyl morpholine (215.19 g, 2127 mmol, 4 eq.) was added to the batch while maintaining the temperature below 50 °C. Then the batch was agitated for 30 minutes at 40-50 °C. MeOH ( 00 mL) was then added while maintaining the temperature below 55 °C. After the batch was held at 50-55 °Cfor 2 hours, another portion of MeOH (100 mL) was added. The batch was agitated for another 2 hours at 50-55 °C. After fluorobenzene was distilled to a minimum amount, water (1000 mL) was added. Further distillation was performed to remove any remaining fluorobenzene. After the batch was cooled to 30 °C, the solid was collected by filtration with cloth and rinsed with water (400 mL) and heptane (200 mL). The solid was dried under vacuum below 50 °C to reach KF < 0.1 %. Typically, the product 2b was obtained in 90% yield with 98 wt%. 1H NMR (500 MHz, DMSO- cfe): δ = 10.83 (s, 1 H), 9.85 (s, bs, 1 H), 7.6 (d, 1 H, J = 8.7 Hz), 6.55 (d, 1 H, J = 8.7 Hz), 6.40 (s, 1 H), 4.00 (s, 2 H), 3.61 (s, 3 H).
Example 3

2b 3a
2b (20 g, 64 mmol) was charged into a clean and dry reactor followed by addition of THF (140 mL). After the resulting mixture was cooled to 0 °C, Vitride® (Red-AI, 47.84 g, 65 wt%, 154 mmol) in toluene was added while maintaining an internal temperature at 0-5 °C. After the batch was agitated at 5-10 °C for 4 hours, IPA (9.24 g, 153.8 mmol) was added while maintaining the temperature below 10 °C. Then the batch was agitated at least for 30 minutes below 25 °C. A solution of HCI in IPA (84.73 g, 5.5 M, 512 mmol) was added into the reactor while maintaining the temperature below 40 °C. After about 160 mL of the solvent was distilled under vacuum below 40 °C, the batch was cooled to 20-25 °C and then aqueous 6M HCI (60 mL) was added while maintaining the temperature below 40 °C. The batch was cooled to 25 °C and agitated for at least 30 minutes. The solid was collected by filtration, washed with 40 mL of IPA and water (1 V/1 V), 40 mL of water and 40 mL of heptanes. The solid was dried below 60 °C in a vacuum oven to reach KF < 0.5%. Typically, the product 3a was obtained in 90-95% yield with 95 wt%. 1H NMR (400 MHz, DMSO-c/e): 5 = 10.7 (s, 1 H), 9.68 (s, 1 H), 7.59 (d, 1 H, J = 8.7 Hz), 6.64 (, 1 H, J = 8.7 Hz), 6.27 (s, 1 H), 4.62 (bs, 1 H), 3.69 (t, 2H, J = 6.3 Hz), 3.21 (t, 2H, J = 6.3 Hz).
Example 4

3a 4a
3a (50 g, 174.756 mmol) and acetonitrile (200 mL) were charged into a dry and clean reactor. After the resulting mixture was heated to 65 °C, POC13 (107.18 g, 699 mmol, 4 eq.) was added while maintaining the internal temperature below 75 °C. The batch was then heated at 70-75 °C for 5-6 h. The batch was cooled to 20 °C. Water (400 mL) was added at least over 30 minutes while maintaining the internal temperature below 50 °C. After the batch was cooled to 20-25 °C over 30 minutes, the solid was collected by filtration and washed with water (100 mL). The wet cake was charged back into the reactor followed by addition of 1 M NaOH (150 mL). After the batch was agitated at least for 30 minutes at 25-35 °C, verify that the pH was greater than 12. Otherwise, more 6M NaOH was needed to adjust the pH >12. After the batch was agitated for 30 minutes at 25-35 °C, the solid was collected by filtration, washed with water (200 mL) and heptanes (200 mL). The solid was dried in a vacuum oven below 50 °C to reach KF < 2%. Typically, the product 4a was obtained at about 75-80% yield. 1H NMR (400 MHz, CDCI3): δ = 7.90 (d, 1 H, J = 8.4 Hz), 7.16 (s, 1 H), 6.89 (d, 1 H, J = 8.4 Hz), 4.44 (t, 2 H, J = 5.9 Hz), 3.23 (t, 2 H, J = 5.9 Hz). 13C NMR (100 MHz, CDCI3): δ = 152.9, 151.9, 144.9, 144.1 , 134.6, 119.1 , 1 17.0, 1 13.3, 1 1 1.9, 65.6, 28.3.
Example 5

4a 5a
Zn powder (54 g, 825 mmol, 2.5 eq.) and TFA (100 mL) were charged into a dry and clean reactor. The resulting mixture was heated to 60-65 °C. A suspension of 4a (100 g, 330 mmol) in 150 mL of TFA was added to the reactor while maintaining the temperature below 70 °C. The charge line was rinsed with TFA (50 mL) into the reactor. After 1 hour at 65±5 °C, the batch was cooled to 25-30 °C. Zn powder was filtered off by passing the batch through a Celite pad and washing with methanol (200 mL). About 400 mL of solvent was distilled off under vacuum. After the batch was cooled to 20-25 °C, 20% NaOAc (ca. 300 mL) was added at least over 30 minutes to reach pH 5-6. The solid was collected by filtration, washed with water (200 mL) and heptane (200 mL), and dried under vacuum below 45 °C to reach KF ≤ 2%. The solid was charged into a dry reactor followed by addition of loose carbon (10 wt%) and toluene (1000 mL). The batch was heated at least for 30 minutes at 45-50 °C. The carbon was filtered off above 35 °C and rinsed with toluene (200 mL). The filtrate was charged into a clean and dry reactor. After about 1000 mL of toluene was distilled off under vacuum below 50 °C, 1000 mL of heptane was added over 30 minutes at 40-50 °C. Then the batch was cooled to 0±5 °C over 30 minutes. After 30 minutes, the solid was collected and rinsed with 200 mL of heptane. The solid was dried under vacuum below 45 °C to reach KF≤ 500 ppm. Typically, the product 5a was obtained in about 90-95 % yield. 1H NMR (400 MHz, CDCI3): δ = 8.93 (m, 1 H), 7.91 (dd, 1 H, J = 1.5, 8 Hz), 7.17 (m 1 H), 6.90 (dd, 1 H, J = 1.6, 8.0 Hz), 4.46-4.43 (m, 2 H), 3.28-3.23 (m, 2 H). 13C NMR (100 MHz, CDCI3): δ = 152.8, 151 .2, 145.1 , 141.0, 133.3, 1 18.5, 1 18.2, 1 14.5, 1 1 1 .1 , 65.8, 28.4.
Example 6

5a (1.04 kg, 4.16 mol) and toluene (8 L) were charged into the reactor. The batch was agitated and cooled to -50 to -55 °C. BuLi solution (2.5 M in hexanes, 1.69 L, 4.23 mol) was charged slowly while maintaining the internal temperature between – 45 to -50 °C. The batch was agitated at -45 °C for 1 hour after addition. A solution of triisopropyl borate (0.85 kg, 4.5 mol) in MTBE (1.48 kg) was charged. The batch was warmed to 10 °C over 30 minutes. A solution of 5 N HCI in IPA (1.54 L) was charged slowly at 10 °C, and the batch was warmed to 20 °C and stirred for 30 minutes. It was seeded with 6a crystal (10 g). A solution of aqueous concentrated HCI (0.16 L) in IPA (0.16 L) was charged slowly at 20 °C in three portions at 20 minute intervals, and the batch was agitated for 1 hour at 20 °C. The solid was collected by filtration, rinsed with MTBE (1 kg), and dried to provide 6a (943 g, 88.7 % purity, 80% yield). 1H NMR (400 MHz, D20): δ 8.84 (d, 1 H, J = 4 Hz), 8.10 (m, 1 H), 7.68 (d, 1 H, J = 6 Hz), 7.09 (m, 1 H), 4.52 (m, 2H), 3.47 (m, 2H).
Example 7

7a 7b
Iodine stock solution was prepared by mixing iodine (57.4 g, 0.23 mol) and sodium iodide (73.4 g, 0.49 mol) in water (270 mL). Sodium hydroxide (28.6 g, 0.715 mol) was charged into 220 mL of water. 4-Hydroxy-2 methylquinoline 7a (30 g, 0.19 mol) was charged, followed by acetonitrile (250 mL). The mixture was cooled to 10 °C with agitation. The above iodine stock solution was charged slowly over 30 minutes. The reaction was quenched by addition of sodium bisulfite (6.0 g) in water (60 mL). Acetic acid (23 mL) was charged over a period of 1 hour to adjust the pH of the reaction mixture between 6 and 7. The product was collected by filtration, washed with water and acetonitrile, and dried to give 7b (53 g, 98%). MS 286 [M + 1].

7b 8a
4-Hydroxy-3-iodo-2-methylquinoline 7b (25 g, 0.09 mol) was charged to a 1 -L reactor. Ethyl acetate (250 mL) was charged, followed by triethylamine (2.45 mL, 0.02 mol) and phosphorus oxychloride (12 mL, 0.13 mol). The reaction mixture was heated to reflux until complete conversion (~1 hour), then the mixture was cooled to 22 °C. A solution of sodium carbonate (31.6 g, 0.3 mol) in water (500 mL) was charged. The mixture was stirred for 20 minutes. The aqueous layer was extracted with ethyl acetate (120 mL). The organic layers were combined and concentrated under vacuum to dryness. Acetone (50 mL) was charged. The solution was heated to 60 °C. Water (100 mL) was charged, and the mixture was cooled to 22 °C. The product was collected by filtration and dried to give 8a (25 g, 97.3 % pure, 91.4 % yield). MS 304 [M + 1].
(Note: 8a is a known compound with CAS # 1033931-93-9. See references: (a) J. Org Chem. 2008, 73, 4644-4649. (b) Molcules 2010, 15, 3171-3178. (c) Indian J. Chem. Sec B: Org. Chem. Including Med Chem. 2009, 48B(5), 692-696.)

8a (100 g, 0.33 mol) was charged to the reactor, followed by copper (I) bromide dimethyl sulfide complex (3.4 g, 0.017 mol) and dry THF (450 mL). The batch was cooled to – 5 to – 2 °C. i-PrMgCI (2.0 M in THF, 173 mL, 0.346 mol) was charged into the reactor at the rate which maintains the batch temperature < -10 °C.
In a 2nd reactor, methyl chlorooxoacetate (33 mL, 0.36 mol) and dry THF (150 mL) was charged. The solution was cooled to -15 to -10 °C. The content of the 1 st reactor (Grignard/cuprate) was charged into the 2nd reactor at the rate which maintained the batch temperature < -10 °C. The batch was agitated for 30 minutes at -10 °C. Aqueous ammonium chloride solution (10%, 300 mL) was charged. The batch was agitated at 20 – 25 °C for 20 minutes and allowed to settle for 20 minutes. The aqueous layer was separated. Aqueous ammonium chloride solution (10%, 90 mL) and sodium carbonate solution (10%, 135 mL) were charged to the reactor. The batch was agitated at 20 – 25 °C for 20 minutes and allowed to settle for 20 minutes. The aqueous layer was separated. Brine (10%, 240 mL) was charged to the reactor. The batch was agitated at 20 – 25 °C for 20 minutes. The aqueous layer was separated. The batch was concentrated under vacuum to -1/4 of the volume (about 80 mL left). 2-Propanol was charged (300 mL). The batch was concentrated under vacuum to -1/3 of the volume (about 140 mL left), and heated to 50 °C. Water (70 mL) was charged. The batch was cooled to 20 – 25 °C, stirred for 2 hours, cooled to -10 °C and stirred for another 2 hours. The solid was collected by filtration, washed with cold 2-propanol and water to provide 58.9 g of 9a obtained after drying (67.8 % yield). 1H NMR (400 MHz, CDCI3): δ 8.08 (d, 1 H, J = 12 Hz), 7.97 (d, 1 H, J = 12 Hz), 7.13 (t, 1 H, J = 8 Hz), 7.55 (t, 1 H, J = 8 Hz), 3.92 (s, 3H), 2.63 (s, 3H). 13C NMR (100 MHz, CDCI3): δ 186.6, 161.1 , 155.3, 148.2, 140.9, 132.0, 129.0, 128.8, 127.8, 123.8, 123.7, 53.7, 23.6.
Example 10

Catalyst preparation: To a suitable sized, clean and dry reactor was charged dichloro(pentamethylcyclopentadienyl)rhodium(lll) dimer (800 ppm relative to 9a, 188.5 mg) and the ligand (2000 ppm relative to 9a, 306.1 mg). The system was purged with nitrogen and then 3 ml_ of acetonitrile and 0.3 ml_ of triethylamine was charged to the system. The resulting solution was agitated at RT for not less than 45 minutes and not more than 6 hours.
Reaction: To a suitable sized, clean and dry reactor was charged 9a (1.00 equiv, 100.0 g (99.5 wt%), 377.4 mmol). The reaction was purged with nitrogen. To the reactor was charged acetonitrile (ACS grade, 4 L/Kg of 9a, 400 ml_) and
triethylamine (2.50 equiv, 132.8 ml_, 943 mmol). Agitation was initiated. The 9a solution was cooled to Tint= -5 to 0 °C and then formic acid (3.00 equiv, 45.2 ml_, 1 132 mmol) was charged to the solution at a rate to maintain Tint not more than 20 °C. The batch temperature was then adjusted to Tlnt= -5 to -0 °C. Nitrogen was bubbled through the batch through a porous gas dispersion unit (Wilmad-LabGlass No. LG-8680-1 10, VWR catalog number 14202-962) until a fine stream of bubbles was obtained. To the stirring solution at Jml= -5 to 0 °C was charged the prepared catalyst solution from the catalyst preparation above. The solution was agitated at Tint= -5 to 0 °C with the bubbling of nitrogen through the batch until HPLC analysis of the batch indicated no less than 98 A% conversion (as recorded at 220 nm, 10-14 h). To the reactor was charged isopropylacetate (6.7 L/Kg of 9a, 670 mL). The batch temperature was adjusted to Tint= 18 to 23 °C. To the solution was charged water (10 L/Kg of 9a, 1000 mL) and the batch was agitated at Tint= 18 to 23 °C for no less than 20 minutes. The agitation was decreased and or stopped and the layers were allowed to separate. The lighter colored aqueous layer was cut. To the solution was charged water (7.5 L/Kg of 9a, 750 mL) and the batch was agitated at Tint= 18 to 23 °C for no less than 20 minutes. The agitation was decreased and or stopped and the layers were allowed to separate. The lighter colored aqueous layer was cut. The batch was then reduced to 300 mL (3 L/Kg of 9a) via distillation while maintaining Text no more than 65 °C. The batch was cooled to Tint= 35 to 45 °C and the batch was seeded ( 0 mg). To the batch at Tint= 35 to 45 °C charged heptane (16.7 L/Kg of 9a, 1670 mL) over no less than 1.5 hours. Adjusted the batch temperature to Tint= -2 to 3 °C over no less than 1 hour, and agitated the batch at Tint= -2 to 3 °C for no less than 1 hour. Collected the solids by filtration. Used the filtrate to rinse the reactor (Filtrate is cooled to
-2 to 3 °C before filtration) and the solids were suction dried for no less than 2 hours. The solids were dried until the LOD was no more than 4 % to obtain 82.7 g of 10a (99.6-100 wt%, 98.5% ee, 82.5% yield). 1H- NMR (CDCI3, 400 MHz) δ: 8.20 (d, J= 8.4 Hz, 1 H), 8.01 (d, J= 8.4 Hz, 1 H), 7.73 (t, J= 7.4 Hz, 1 H), 7.59 (t, J= 7.7 Hz, 1 H), 6.03 (s, 1 H), 3.93 (s, 1 H), 3.79 (s, 3H), 2.77 (s, 3H). 13C-NMR (CDCI3, 100 MHz) δ: 173.5, 158.3, 147.5, 142.9, 130.7, 128.8, 127.7, 127.1 , 125.1 , 124.6, 69.2, 53.4, 24.0.
Example 11

10a 6a 11a
10a (2.45 kg, 96.8% purity, 8.9 mol), 6a (2.5 kg, 88.7% purity, 8.82 mol), tris(dibenzylideneacetone)dipalladium(0) (Pd2dba3, 40 g, 0.044 mol), (S)-3-iert-butyl-4-(2,6-dimethoxyphenyl)-2,3-dihydrobenzo[d][1 ,3]oxaphosphole (32 g, 0.01 1 mol), sodium carbonate (1.12 kg, 10.58 mol), 1 -pentanol (16.69 L), and water (8.35 L) were charged to the reactor. The mixture was de-gassed by sparging with argon for 10-15 minutes, was heated to 60-63 °C, and was agitated until HPLC analysis of the reaction shows <1 A% (220 nm) of the 6a relative to the combined two atropisomer products (-15 hours). The batch was cooled to 8-23 °C. Water (5 L) and heptane (21 L) were charged. The slurry was agitated for 3 – 5 hours. The solids were collected by filtration, washed with water (4 L) and heptane/toluene mixed solvent (2.5 L toluene/5 L heptane), and dried. The solids were dissolved in methanol (25 L) and the resulting solution was heated to 50 °C and circulated through a CUNO carbon stack filter. The solution was distilled under vacuum to ~ 5 L. Toluene (12 L) was charged. The mixture was distilled under vacuum to – 5 L and cooled to 22 °C. Heptane (13 L) was charged to the contents over 1 hour and the resulting slurry was agitated at 20-25 °C for 3 – 4 hours. The solids were collected by filtration and washed with heptanes to provide 2.58 kg of 11a obtained after drying (73% yield). 1H NMR (400 MHz, CDCI3): δ 8.63 (d, 1 H, J = 8 Hz), 8.03 (d, 1 H, J = 12 Hz), 7.56 (t, 1 H, J = 8 Hz), 7.41 (d, 1 H, J = 8 Hz), 7.19 (t, 1 H, J = 8 Hz), 7.09 (m, 2H), 7.04 (d, 1 H, J = 8 Hz), 5.38 (d, 1 H, J = 8 Hz), 5.14 (d, 1 H, J = 8 Hz), 4.50 (t, 2H, J = 4 Hz), 3.40 (s, 3H), 3.25 (t, 2H, J = 4 Hz), 2.91 (s, 3H). 13C NMR (100 MHz, CDCI3): δ 173.6, 158.2, 154.0, 150.9, 147.3, 147.2, 145.7, 141.3, 132.9, 123.0, 129.4, 128.6, 127.8, 126.7, 126.4, 125.8, 1 18.1 , 1 17.3, 109.9, 70.3, 65.8, 52.3, 28.5, 24.0.

To a suitable clean and dry reactor under a nitrogen atmosphere was charged 1a (5.47 Kg, 93.4 wt%, 1 .00 equiv, 12.8 mol) and fluorobenzene (10 vols, 51.1 kg) following by trifluoromethanesulfonimide (4 mol%, 143 g, 0.51 mol) as a 0.5 M solution in DCM (1.0 Kg). The batch temperature was adjusted to 35-41 °C and agitated to form a fine slurry. To the mixture was slowly charged i-butyt-2,2,2- trichloroacetimidate 12b as a 50 wt% solution (26.0 Kg of f-butyl-2,2,2- trichloroacetimidate (119.0 mol, 9.3 equiv), the reagent was -48-51 wt% with the remainder 52-49 wt% of the solution being ~ 1.8:1 wt:wt heptane: fluorobenzene) over no less than 4 hours at Tint= 35-41 °C. The batch was agitated at Tint= 35-41 °C until HPLC conversion (308 nm) was >96 A%, then cooled to Tlnt= 20-25 °C and then triethylamine (0.14 equiv, 181 g, 1.79 mol) was charged followed by heptane (12.9 Kg) over no less than 30 minutes. The batch was agitated at Tint= 20-25 °C for no less than 1 hour. The solids were collected by filtration. The reactor was rinsed with the filtrate to collect all solids. The collected solids in the filter were rinsed with heptane (1 1.7 Kg). The solids were charged into the reactor along with 54.1 Kg of DM Ac and the batch temperature adjusted to Tint= 70-75 °C. Water (1 1.2 Kg) was charged over no less than 30 minutes while the batch temperature was maintained at Tint= 65-75 °C. 12a seed crystals (34 g) in water (680 g) was charged to the batch at Tint= 65-75 °C. Additional water (46.0 Kg) was charged over no less than 2 hours while maintaining the batch temperature at Tint= 65-75 °C. The batch temperature was adjusted to Tint= 18-25 °C over no less than 2 hours and agitated for no less than 1 hour. The solids were collected by filtration and the filtrate used to rinse the reactor. The solids were washed with water (30 Kg) and dried under vacuum at no more than 45 °C until the LOD < 4% to obtain 12a (5.275 Kg, 99.9 A% at 220 nm, 99.9 wt% via HPLC wt% assay, 90.5% yield). H-NMR (CDCI3l 400 MHz) δ: 8.66-8.65 (m, 1 H), 8.05 (d, J= 8.3 Hz, 1 H), 7.59 (t, J= 7.3 Hz, 1 H), 7.45 (d, J= 7.8 Hz, 1 H), 7.21 (t, J= 7.6 Hz, 1 H), 7.13-7.08 (m, 3H), 5.05 (s, H), 4.63-4.52 (m, 2H), 3.49 (s, 3H), 3.41 -3.27 (m, 2H), 3.00 (s, 3H), 0.97 (s, 9H). 13C-NMR (CDCI3, 100 MHz) δ: 172.1 , 159.5, 153.5, 150.2, 147.4, 146.9, 145.4, 140.2, 131.1 , 130.1 , 128.9, 128.6, 128.0, 127.3, 126.7, 125.4, 1 17.7, 1 17.2, 109.4, 76.1 , 71.6, 65.8, 51.9, 28.6, 28.0, 25.4.
Example 13

To a suitable clean and dry reactor under a nitrogen atmosphere was charged 12a (9.69 Kg, 21.2 mol) and ethanol (23.0 Kg). The mixture was agitated and the batch temperature was maintained at Tjnt= 20 to 25 °C. 2 M sodium hydroxide (17.2 Kg) was charged at Tint= 20 to 25 °C and the batch temperature was adjusted to Tlnt= 60- 65°C over no less than 30 minutes. The batch was agitated at Tint= 60-65°C for 2-3 hours until HPLC conversion was >99.5% area (12a is <0.5 area%). The batch temperature was adjuted to Tint= 50 to 55°C and 2M aqueous HCI (14.54 Kg) was charged. The pH of the batch was adjusted to pH 5.0 to 5.5 (target pH 5.2 to 5.3) via the slow charge of 2M aqueous HCI (0.46 Kg) at Tint= 50 to 55°C. Acetonitrile was charged to the batch (4.46 Kg) at Τ,ηί= 50 to 55°C. A slurry of seed crystals (1001 , 20 g in 155 g of acetonitrile) was charged to the batch at Tint= 50 to 55°C. The batch was agitated at Tint= 50 to 55°C for no less than 1 hour (1-2 hours). The contents were vacuum distilled to -3.4 vol (32 L) while maintaining the internal temperature at 45-55°C. A sample of the batch was removed and the ethanol content was determined by GC analysis; the criterion was no more than 10 wt% ethanol. If the ethanol wt% was over 10%, an additional 10% of the original volume was distilled and sampled for ethanol wt%. The batch temperature was adjusted to Tint= 8-22°C over no less than 1 hour. The pH of the batch was verified to be pH= 5 – 5.5 and the pH was adjusted, if necessary, with the slow addition of 2 M HCI or 2 M NaOH aqueous solutions. The batch was agitated at Tint= 18-22°C for no less than 6 hours and the solids were collected by filtration. The filtrate/mother liquid was used to remove all solids from reactor. The cake with was washed with water (19.4 Kg) (water temperature was no more than 20 °C). The cake was dried under vacuum at no more than 60 °C for 12 hours or until the LOD was no more than 4% to obtain 1001 (9.52 Kg, 99.6 A% 220 nm, 97.6 wt% as determined by HPLC wt% assay, 99.0% yield). Example 14
Hydrochloride salt of Compound (I), Type A
Compound (I) (263 mg) was added to a vial of ethanol (1.5 ml_), and then 36.5% HCL aqueous solution (59 mg) was added. The mixture was heated to 70 °C; and stirred at this temperature until solid material was obtained. The mixture was cooled to 20 °C over a period of 10 hours. After cooling, isopropanol (400 μΙ_) was added over a period of 3 hours. The resulting solids were collected and characterized as the hydrochloride salt of Compound (I), Type A.
The hydrochloride salt of Compound (I), Type A was prepared analogously to the aforementioned procedure using methyl ethyl ketone, tetrahydrofuran, acetonitrile, ethyl acetate, dichloroethane and methyl-t-buyl ether instead of ethanol.

References
- Pharmacodynamics of BI 224436 for HIV-1 in an in vitro hollow fiber infection model system
- Levin, Jules. BI 224436, a Non-Catalytic Site Integrase Inhibitor, is a potent inhibitor of the replication of treatment-naïve and raltegravir-resistant clinical isolates of HIV-1. Conference Reports for NATAP. ICAAC Chicago Sept 17-20 2011.
- Gilead Negotiates Worldwide License to BI’s Early Clinical Stage HIV Program. Genetic Engineering and Biotechnology News. 6 Oct 2011.
- Highleyman, Liz. ICAAC: New Integrase Inhibitor BI 224436 Active against Raltegravir-Resistant HIV. HIVandHepatitis.com. 7 Oct 2011.
- WO-2014055618

Janssen signs licensing agreement with PATH for development of HIV-1 drug

rilpivirine.
Janssen R&D Ireland has signed a licensing agreement with PATH for the early development of a long-acting depot formulation of the human immunodeficiency virus type 1 (HIV-1) drug rilpivirine.

Rilpivirine, a non-nucleoside reverse transcriptase inhibitor (NNRTI), is being developed as potential pre-exposure prophylaxis against HIV infection
read all at
Rilpivirine (TMC278, trade name Edurant) is a pharmaceutical drug, developed by Tibotec, for the treatment of HIVinfection.[1][2] It is a second-generation non-nucleoside reverse transcriptase inhibitor (NNRTI) with higher potency, longer half-lifeand reduced side-effect profile compared with older NNRTIs, such as efavirenz.[3][4]
Rilpivirine entered phase III clinical trials in April 2008,[5][6] and was approved for use in the United States in May 2011.[7] A fixed-dose drug combining rilpivirine with emtricitabine and tenofovir, was approved by the U.S. Food and Drug Administration in August 2011 under the brand name Complera.[8]

Like etravirine, a second-generation NNRTI approved in 2008, rilpivirine is a diarylpyrimidine (DAPY). Rilpivirine in combination with emtricitabine and tenofovir has been shown to have higher rates of virologic failure than Atripla in patients with baseline HIV viral loads greater than 100,000 copies.
- Rilpivirine bound to proteins in the PDB

- “TMC278 – A new NNRTI”. Tibotec. Retrieved 2010-03-07.
- Stellbrink HJ (2007). “Antiviral drugs in the treatment of AIDS: what is in the pipeline ?”. Eur. J. Med. Res. 12 (9): 483–95.PMID 17933730.
- Goebel F, Yakovlev A, Pozniak AL, Vinogradova E, Boogaerts G, Hoetelmans R, de Béthune MP, Peeters M, Woodfall B (2006).“Short-term antiviral activity of TMC278–a novel NNRTI–in treatment-naive HIV-1-infected subjects”. AIDS 20 (13): 1721–6.doi:10.1097/01.aids.0000242818.65215.bd. PMID 16931936.
- Pozniak A, Morales-Ramirez J, Mohap L et al. 48-Week Primary Analysis of Trial TMC278-C204: TMC278 Demonstrates Potent and Sustained Efficacy in ART-naïve Patients. Oral abstract 144LB.
- ClinicalTrials.gov A Clinical Trial in Treatment naïve HIV-1 Patients Comparing TMC278 to Efavirenz in Combination With Tenofovir + Emtricitabine
- ClinicalTrials.gov A Clinical Trial in Treatment naïve HIV-Subjects Patients Comparing TMC278 to Efavirenz in Combination With 2 Nucleoside/Nucleotide Reverse Transcriptase Inhibitors
- “FDA approves new HIV treatment”. FDA. Retrieved 2011-05-20.
- “Approval of Complera: emtricitabine/rilpivirine/tenofovir DF fixed dose combination”. FDA. August 10, 2011.
-
Rilpivirine hydrochloride, 4-[[4-[[4-(2-Cyanoethenyl)-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile monohydrochloride, is a non-nucleoside reverse transcriptase inhibitor (NNRTI) of human immunodeficiency virus type 1 (HIV-1) and indicated for the treatment of HIV-1 infection in treatment-naïve adult patients in combination with other antiretroviral agents. The product received marketing approval in the US (brand name Edurant) and is represented by the following general formula (I):

-
[0003]EP1419152 B1 claims amongst others Rilpivirine base and Rilpivirinehydrochloride per se as well as pharmaceutical compositions comprising the same. However, only concrete examples for preparingRilpivirine base are given in said patent but no concrete examples describing the production of the hydrochloride salt are provided.
-
[0004]EP1632232 B1 claims amongst others a solid pharmaceutical composition comprising crystalline forms A, B, C or D of Rilpivirinehydrochloride. In addition said patent claims a process for the production of Rilpivirine hydrochloride by reacting Rilpivirine base with hydrochloric acid in the presence of a suitable acid, such as acetic acid.
-
[0005]Polymorphism is a phenomenon relating to the occurrence of different crystal forms for one molecule. There may be several different crystalline forms for the same molecule with distinct crystal structures and varying in physical properties like melting point, XRPD pattern and FTIR spectrum. These polymorphs are thus distinct solid forms which share the molecular formula of the compound from which the crystals are made up, however they may have distinct advantageous physical properties such as e.g. chemical stability, physical stability, hygroscopicity, solubility, dissolution rate, bioavailability, etc.
-
[0006]The bioavailability of a compound intended to be administered orally, is dependent on the compounds solubility in aqueous systems as well as the compounds permeability as mentioned in EP1632232 B1 . Hydrates are known to be less soluble in aqueous systems than anhydrous forms of the same compound. Hence anhydrous forms of Rilpivirinehydrochloride are preferred over hydrated forms. Rilpivirinehydrochloride form D of EP1632232 B1 is a hydrate and thus no suitable candidate for the preparation of an orally administered medicament, whereas form E of the present invention is an anhydrate.
-
[0007]The novel polymorph E of Rilpivirine hydrochloride of the present invention shows high solubility in aqueous systems e.g. a higher solubility than forms A and C of EP1632232 B1 and is thus especially suitable for the preparation of an orally administered medicament.
-
[0008]In addition the crystalline forms A and C of EP1632232 B1 are difficult to make in a reliable manner as these forms are obtained from the same solvent system. As the polymorphs A and C of Rilpivirinehydrochloride are obtainable from the same solvent system acetic acid/water, the production processes are especially critical and sensitive because the single crystalline forms are only obtainable in pure form in a quite narrow range of temperature as described in the concrete examples A.a) and A.c) of EP1632232 B1 . In contrast form E of the present invention is reliably obtained by crystallization from ethanol as form E is the only polymorph of Rilpivirine hydrochloride obtained from this solvent system.
-
[0009]According to example A.b) of EP1632232 B1 form B is obtained by recrystallizing Rilpivirine hydrochloride from propanone using an initial Rilpivirine hydrochloride concentration of 0.3 g/L. However, this concentration is not suitable for up-scaling as larger amounts of Rilpivirine hydrochloride would ask for tremendous solvent volumina and hence the usage of tremendously large reaction vessels. In contrast form E of the present invention is also obtained by applying higher initial Rilpivirine hydrochloride concentrations such as e.g. ≥10 g/L and is thus suitable for large scale production.
-
[0010]Hence, aim of the present invention is to circumvent the drawbacks of the known forms A, B, C and D ofEP1632232 B1 by providing an anhydrous polymorph of Rilpivirine hydrochloride, which is obtainable in an easy and reliable manner also in large scale. In addition the novel polymorph shows high solubility in aqueous systems making it especially suitable for the preparation of an orally administered medicament.


CFDA Approves Clinical Trials for Novel China AIDS Treatment, Azi Fu (Azvudine)
A novel treatment for AIDS, developed in the Zhengzhou University labs of Junbiao Chang, PhD, has been approved by the CFDA for human trials. The molecule, which has been in development for ten years, is a reverse transcriptase (RT) inhibito, called Azi Fu (Azvudine). According to Dr. Chang, Azi Fu is more effective at blocking the mutated forms of the virus than currently available treatments. He also believes the molecule has the potential to lower treatment costs
read all at

http://www.chinabiotoday.com/articles/20130523
![]() read at
read at
