
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Fluvoxamine

Fluvoxamine
- Molecular FormulaC15H21F3N2O2
- Average mass318.335 Da
- 54739-18-3
(E)-5-Methoxy-1-[4-(trifluoromethyl)phenyl]-1-pentanone O-(2-Aminoethyl)oxime1-Pentanone, 5-methoxy-1-[4-(trifluoromethyl)phenyl]-, O-(2-aminoethyl)oxime, (1E)-2-[({(1E)-5-Methoxy-1-[4-(trifluoromethyl)phenyl]pentylidene}amino)oxy]ethanamine
2-{[(E)-{5-Methoxy-1-[4-(trifluoromethyl)phenyl]pentylidene}amino]oxy}ethanamine1-Pentanone, 5-methoxy-1-(4-(trifluoromethyl)phenyl)-, O-(2-aminoethyl)oxime, (E)-
387954739-18-3[RN]5583954[Beilstein]5-Methoxy-4′-(trifluoromethyl)valerophenone (E)-O-(2-aminoethyl)oximeA selective serotonin reuptake inhibitor that is used in the treatment of DEPRESSION and a variety of ANXIETY DISORDERS.
Fluvoxamine, sold under the brand name Luvox among others, is an antidepressant of the selective serotonin reuptake inhibitor (SSRI) class[5] which is used primarily for the treatment of obsessive–compulsive disorder (OCD).[6] It is also used to treat depression and anxiety disorders, such as panic disorder, social anxiety disorder, and post-traumatic stress disorder.[7][8]
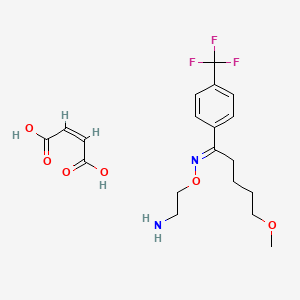
FLUVOXAMINE MALEATE
C19H25F3N2O6, 434.4 g/mol
1-Pentanone, 5-methoxy-1-(4-(trifluoromethyl)phenyl)-, O-(2-aminoethyl)oxime, (E)-, (Z)-2-butenedioate (1:1)
(Z)-but-2-enedioic acid;2-[(E)-[5-methoxy-1-[4-(trifluoromethyl)phenyl]pentylidene]amino]oxyethanamine
Luvox
61718-82-9
CAS 54739-20-7
Fevarin, Luvox CR
Synonyms
- 5-Methoxy-4′-(trifluoromethyl)valerophenone (E)-O-(2-aminoethyl)oxime, maleate (1:1)
- 5-Methoxy-4′-trifluoromethylvalerophenone (E)-O-2-aminoethyloxime monomaleate
- DU23000
- Fevarin
- Fluvoxamine maleate
- Luvox
- Luvox CR
- SME 3110
- UNII-5LGN83G74V
| Originator Company | Solvay SA |
| Active Companies | AbbVie Inc; Abbott Laboratories; Meiji Seika Pharma Co Ltd; Solvay SA |
| Launched (Obsessive compulsive disorder – EU – Dec-1983) |
In the EU, the product is indicated for the treatment of obsessive compulsive disorder (OCD) and for the treatment of major depressive disorder (MDD)
In Japan, Luvox is indicated for the treatment of adult or pediatric OCD, social anxiety disorder (SAD) and MDD
USFDA The drug was approved for the treatment of OCD and SAD in April 2008
CHINA
In 2000, the drug was launched in China for the treatment of OCD and MDD
Patents and Generics
FDA exclusivity expired in the US in June 2000. Generic versions have been on the market since that time. Generic fluvoxamine was still available in the US by May 2007, despite the fact the Solvay/Jazz product had not been relaunched . By October 2004, the drug was also off patent in most European countries .
Medical uses
Fluvoxamine is approved in the United States for OCD,[9][6] and social anxiety disorder.[10] In other countries (e.g., Australia,[11][12] the UK,[13] and Russia[14]) it also has indications for major depressive disorder. In Japan it is currently[when?] approved to treat OCD, SAD and MDD.[15][16] Fluvoxamine is indicated for children and adolescents with OCD.[17] The drug works long-term, and retains its therapeutic efficacy for at least one year.[18] It has also been found to possess some analgesic properties in line with other SSRIs and tricyclic antidepressants.[19][20][21]
There is tentative evidence that fluvoxamine is effective for social phobia in adults.[22] Fluvoxamine is also effective for GAD, SAD, panic disorder and separation anxiety disorder in children and adolescents.[23] There is tentative evidence that fluvoxamine may help some people with negative symptoms of chronic schizophrenia.[24][25]
A double-blind controlled study found that fluvoxamine may prevent clinical deterioration in outpatients with symptomatic COVID-19. The study had important limitations: it was run fully remotely; it had a small sample size (150) and short follow-up duration (15 days).[26] The accompanying editorial noted that, although this study is important enough to choose out of more than 10,000 other COVID-19 related submissions, it “presents only preliminary information” and “the findings should be interpreted as only hypothesis generating; they should not be used as the basis for current treatment decisions.”[27] Similarly, the study authors themselves cautioned that “the trial’s results should not be treated as a measure of fluvoxamine’s effectiveness against COVID-19 but as an encouraging indicator that the drug warrants further testing.”[28] A prospective open-labelled cohort study showed similar results.[29]
Adverse effects
Gastrointestinal side effects are more common in those receiving fluvoxamine than with other SSRIs.[30] Otherwise, fluvoxamine’s side-effect profile is very similar to other SSRIs.[2][9][11][13][31][32]Common (1–10% incidence) adverse effects
- Nausea
- Vomiting
- Weight loss
- Yawning
- Loss of appetite
- Agitation
- Nervousness
- Anxiety
- Insomnia
- Somnolence (drowsiness)
- Tremor
- Restlessness
- Headache
- Dizziness
- Palpitations
- Tachycardia (high heart rate)
- Abdominal pain
- Dyspepsia (indigestion)
- Diarrhea
- Constipation
- Hyperhidrosis (excess sweating)
- Asthenia (weakness)
- Malaise
- Sexual dysfunction (including delayed ejaculation, erectile dysfunction, decreased libido, etc.)
- Xerostomia (dry mouth)
Uncommon (0.1–1% incidence) adverse effects
- Arthralgia
- Hallucination
- Confusional state
- Extrapyramidal side effects (e.g. dystonia, parkinsonism, tremor, etc.)
- Orthostatic hypotension
- Cutaneous hypersensitivity reactions (e.g. oedema [buildup of fluid in the tissues], rash, pruritus)
Rare (0.01–0.1% incidence) adverse effects
- Mania
- Seizures
- Abnormal hepatic (liver) function
- Photosensitivity (being abnormally sensitive to light)
- Galactorrhoea (expulsion of breast milk unrelated to pregnancy or breastfeeding)
Unknown frequency adverse effects
- Hyperprolactinaemia (elevated plasma prolactin levels leading to galactorrhoea, amenorrhoea [cessation of menstrual cycles], etc.)
- Bone fractures
- Glaucoma
- Mydriasis
- Urinary incontinence
- Urinary retention
- Bed-wetting
- Serotonin syndrome — a potentially fatal condition characterised by abrupt onset muscle rigidity, hyperthermia (elevated body temperature), rhabdomyolysis, mental status changes (e.g. coma, hallucinations, agitation), etc.
- Neuroleptic malignant syndrome — practically identical presentation to serotonin syndrome except with a more prolonged onset
- Akathisia — a sense of inner restlessness that presents itself with the inability to stay still
- Paraesthesia
- Dysgeusia
- Haemorrhage
- Withdrawal symptoms
- Weight changes
- Suicidal ideation and behaviour
- Violence towards others[33]
- Hyponatraemia
- Syndrome of inappropriate antidiuretic hormone secretion
- Ecchymoses
Interactions[edit]

Luvox (fluvoxamine) 100 mg film-coated scored tablets
Fluvoxamine inhibits the following cytochrome P450 enzymes:[34][35][36][37][38][39][40][41][42]
- CYP1A2 (strongly) which metabolizes agomelatine, amitriptyline, caffeine, clomipramine, clozapine, duloxetine, haloperidol, imipramine, phenacetin, tacrine, tamoxifen, theophylline, olanzapine, etc.
- CYP3A4 (moderately) which metabolizes alprazolam, aripiprazole, clozapine, haloperidol, quetiapine, pimozide, ziprasidone, etc.[43]
- CYP2D6 (weakly) which metabolizes aripiprazole, chlorpromazine, clozapine, codeine, fluoxetine, haloperidol, olanzapine, oxycodone, paroxetine, perphenazine, pethidine, risperidone, sertraline, thioridazine, zuclopenthixol, etc.[44]
- CYP2C9 (moderately) which metabolizes nonsteroidal anti-inflammatory drugs, phenytoin, sulfonylureas, etc.
- CYP2C19 (strongly) which metabolizes clonazepam, diazepam, phenytoin, etc.
- CYP2B6 (weakly) which metabolizes bupropion, cyclophosphamide, sertraline, tamoxifen, valproate, etc.
By so doing, fluvoxamine can increase serum concentration of the substrates of these enzymes.[34]
The plasma levels of oxidatively metabolized benzodiazepines (e.g., triazolam, midazolam, alprazolam and diazepam) are likely to be increased when co-administered with fluvoxamine. However the clearance of benzodiazepines metabolized by glucuronidation (e.g., lorazepam, oxazepam, temazepam)[45][46] is unlikely to be affected by fluvoxamine.[47] It appears that benzodiazepines metabolized by nitro-reduction (clonazepam, nitrazepam) are unlikely to be affected by fluvoxamine.[48] Using fluvoxamine and alprazolam together can increase alprazolam plasma concentrations.[49] If alprazolam is coadministered with fluvoxamine, the initial alprazolam dose should be reduced to the lowest effective dose.[50][51]
Fluvoxamine and ramelteon coadministration is not indicated.[52][53]
Fluvoxamine has been observed to increase serum concentrations of mirtazapine, which is mainly metabolized by CYP1A2, CYP2D6, and CYP3A4, by 3- to 4-fold in humans.[54] Caution and adjustment of dosage as necessary are warranted when combining fluvoxamine and mirtazapine.[54]
Fluvoxamine seriously affects the pharmacokinetics of tizanidine and increases the intensity and duration of its effects. Because of the potentially hazardous consequences, the concomitant use of tizanidine with fluvoxamine, or other potent inhibitors of CYP1A2, should be avoided.[55]
Fluvoxamine’s interaction with St John’s wort can lead to increased serotonin levels and potentially lead to serotonin syndrome.[citation needed]
Pharmacology
| Site | Ki (nM) |
|---|---|
| SERT | 2.5 |
| NET | 1,427 |
| 5-HT2C | 5,786 |
| α1-adrenergic | 1,288 |
| σ1 | 36 |
Fluvoxamine is a potent selective serotonin reuptake inhibitor with around 100-fold affinity for the serotonin transporter over the norepinephrine transporter.[35] It has negligible affinity for the dopamine transporter or any other site, with the sole exception of the σ1 receptor.[59][60] It behaves as a potent agonist at this receptor and has the highest affinity (36 nM) of any SSRI for doing so.[59] This may contribute to its antidepressant and anxiolytic effects and may also afford it some efficacy in treating the cognitive symptoms of depression.[61] Unlike fluoxetine, fluvoxamine’s metabolites are inactive, without a significant effect on serotonin or norepinephrine uptake.[62]
History
Fluvoxamine was developed by Kali-Duphar,[63] part of Solvay Pharmaceuticals, Belgium, now Abbott Laboratories, and introduced as Floxyfral in Switzerland in 1983.[63] It was approved by the U.S. Food and Drug Administration (FDA) in 1994, and introduced as Luvox in the US.[64] In India, it is available, among several other brands, as Uvox by Abbott.[65] It was one of the first SSRI antidepressants to be launched, and is prescribed in many countries to patients with major depression.[66] It was the first SSRI, a non-TCA drug, approved by the U.S. FDA specifically for the treatment of OCD.[67] At the end of 1995, more than ten million patients worldwide had been treated with fluvoxamine.[68][failed verification] Fluvoxamine was the first SSRI to be registered for the treatment of obsessive compulsive disorder in children by the FDA in 1997.[69] In Japan, fluvoxamine was the first SSRI to be approved for the treatment of depression in 1999[70][71] and was later in 2005 the first drug to be approved for the treatment of social anxiety disorder.[72] Fluvoxamine was the first SSRI approved for clinical use in the United Kingdom.[73]
Society and culture
Manufacturers include BayPharma, Synthon, and Teva, among others.[74]
SYN


SYN
J. Zhejiang Univ. (Medical Sci.) (2003), 32 (5), 441-442

PATENT
WO 2014178064
The present invention relates to an improved and industrially applicable process for the preparation of fluvoxamine maleate of formula I,
Fluvoxamine or (E)-5-methoxy-1 -[4-(trifluoromethyl)phenyl]pentan- 1 -one-O-2-aminoethyl oxime is an antidepressant which functions as a selective serotonin reuptake inhibitor (SSRI). Fluvoxamine is used for the treatment of major depressive disorder (MDD), obsessive compulsive disorder (OCD), and anxiety disorders such as panic disorder and post-traumatic stress disorder (PTSD). Fluvoxamine CR (controlled release) is approved to treat social anxiety disorder.
Fluvoxamine maleate and compounds were first disclosed in US patent 4,085,225. According to said patent, Fluvoxamine maleate prepared by alkylation reaction of 5-methoxy-4′-trifluoromethylvalerophenone oxime, compound of formula III with 2-chloroethylamine hydrochloride in dimethylformamide in the presence of a base such as potassium hydroxide powder for two days at 25°C.
Subsequently the solvent is removed under vacuum then the residue is acidified and extracted with ether to remove the unreacted oxime followed by basification. The obtained fluvoxamine base in ether extract is washed with sodium bicarbonate solution. The fluvoxamine base is then treated with maleic acid in absolute ethanbl and the residue obtained by concentration under vacuum is recrystallized from acetonitrile to obtain fluvoxamine maleate. The process is very much tedious, time consuming as it requires two days for the reaction completion. Operations like removal of dimethylformamide, ether, ethanol makes process cumbersome at plant level. Requirement of
various solvents lead the process to be non-eco-friendly. Moreover the patent is silent about yield and purity of the product.
In an alternate route described in US patent 4,085,225, the oxime of formula III is converted to formula I in a five step process i.e. alkylation of formula III with ethylene oxide. The reaction solvent is ethanol in which lithium is already dissolved. The reaction further involves addition of acetic acid to give the hydroxyethyl compound of formula A as oil. The compound of formula A is purified chromatographically over the silica gel, which is converted to a mesylate compound of formula B by treating with methanesulfonyl chloride and triethylamine at -5 to 0°C, then aminated with ammonia in methanol at 100°C using autoclave for 16 hours followed by removal of methanol and extraction in ether to give fluvoxamine base.
The base is then converted to the maleate salt formula I, which is finally purified by recrystallization from acetonitrile.
There are lots of disadvantages involve like more unit operations, use of various solvents and handling of ethylene oxide which is also known for its carcinogen effect. More unit operations lead to long occupancy of reactors in the plant as well as man power, high energy consumption and require bigger plant. These all parameters make the process commercially unviable as wel l as environmentally non-feasible. Further, purification of the compound of formula A requires cumbersome technique i.e chromatography over silica gel as well as lengthy work-up procedure in U.S. Pat. No. 4,085,225 requires complete removal of organic solvents at various stages.
US patent 6,433,225 discloses the process for preparing fluvoxamine maleate, prepared by alkylating 5-methoxy-4′-trtfluoromethylvalerophenone oxime, compound of formula III with 2-chloroethylamine hydrochloride in toluene and PEG-400 (polyethyleneglycol-400) as facilitator in the presence of a base potassium hydroxide powder at 30-35°C to obtain fluvoxamine base in
toluene layer is then treated with maleic acid in water. The precipitated fluvoxamine maleate is filtered and washed with toluene and dried. The obtained dried cake recrystallized with water to get fluvoxamine maleate. The process disclosed in the patent is silent about actual purity of the product. As per our scientist’s observation alkylation reaction at the temperature of 30-35°C may lead to non completion of reaction and results lower yield. Additional step of purification may further lead to loss of yield.
Thus, present invention fulfills the need of the art and provides an improved and industrially applicable process for preparation of fluvoxamine maleate, which provides fluvoxamine maleate in high purity and overall good yield.
EXAMPLES:
Stage – 1 : Preparation of (1E)-N-hydroxy-5-methoxy-1-(4-trifluoromethyI pheny 1) pentan-1-imine formula III
To a stirred solution of 5-methoxy- 1 -(4-trifluoromethylphenyl) pentan-1 -one ( 150 gm) in methanol (750 ml), sodium carbonate (granule) (72 gm) and hydroxylamine hydrochloride (59.64 gm) were added at temperature 25-30°C. The reaction mass was heated 45-50°C for 10- 15 minutes followed by maintaining the reaction mass at temperature 45-50°C for 8-9 hours under stirring. The reaction mass was cooled to 25-30°C and filtered under vacuum to remove unreacted inorganic matter, then distilled out the methanol completely from the collected filtrate under vacuum at temperature below 50°C. The obtained slurry was cooled to 25-30°C and water (300 ml) was added into the residue followed by the addition of hexane (300×2 ml) and stirred for 30 minutes. The layers were separated. The collected organic layer was stirred for 5- 10 minutes at temperature 25-30°C followed by cooling the mass at temperature -5°C to – 10°C, stirred for 30-40 minutes and filtered at the same temperature. The product was suck dried at -5 to -10°C and further in vacuum at 25-30°C for 2-3 hours to give 138 – 142 gm of title compound. HPLC purity: >98.5%
Stage – 2: Preparation of crude fluvoxamine maleate formula I
To a prepared solution of dimethyl sulphoxide (575 ml), potassium hydroxide flakes ( 1 14.64 gm) and water (69 ml), stage-1 (1 15 gm) was added at temperature 40-45°C. The reaction mixture was stirred to get clear solution followed by adding 2-chloroethylamine hydrochloride (86.36 gm) drop wise into the reaction mixture at temperature 40-45°C and maintained for 1 -2 hour. Water (1 150 ml) was added in to the reaction mixture at temperature 25-30°C and stirred for 20-25 minutes. Then toluene (575 ml x 2) was added and stirred for 30 minutes and preceded for separation of layers followed by washing the toluene layer with water ( 1 1 50 x 5 ml). The solution of maleic acid (48.47 gm) dissolved in water (98 ml) was added into above obtained toluene layer and stirred at temperature 25-30°C for 2-3 hours. The reaction mixture was cooled to 0-5°C and maintained for 30-40 minutes at the same temperature. The obtained material was washed with toluene, filtered and suck dried. The wet cake was then added hexane (600 ml) and stirred for 30 minutes at temperature 25-30°C, filtered, washed with hexane and dried to get 161 gm of title compound. HPLC purity: >98.5%
Stage – 3: Preparation of pure fluvoxamine maleate formula I
In to the reaction assembly, water (600 ml) was added and heated to 40-45°C. Stage -2 ( 1 50 gm) was added into the hot water under stirring. The reaction mixture was stirred for 5- 10 minutes, filtered and cooled to 25°C. Toluene (68 ml) was added into the reaction mixture at temperature 25°C and stirred for 30 minutes. Filtered the solid, washed with 10-15°C chilled water and dried to get the pure 127.5 gm fluvoxamine maleate. HPLC purity: >99.8%
Process for isolation of 5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one formula II
To a solution of cone. HCl (600 ml) and water ( 160 ml), organic residue (250 gm) of ( 1 £)+( 1 Z) of 1 -N-hydroxy-5-methoxy- 1 -[4-(trifluoromethyl) phenyl]pentan-1 -imine and traces of 5-methoxy- 1 -[4-(trifluoromethyl)phenyl]pentan- 1-one (obtained after hexane recovery from stage-1 filtrate) was added at temperature 25-30°C under stirring. The reaction mixture was heated to 67-75°C and maintained for 13-14 hours followed by cool ing the reaction mixture at temperature 25-30°C. Then after hexane (500 x 2 ml) was added into the reaction mixture and stirred for 15 minutes at 25-30°C. The organic layers were separated and sodium bicarbonate solution (25 gm sodium bicarbonate dissolved in 250 ml water) was added into the hexane layer and stirred for 15 minutes. The layers were separated and water (250ml) was added into hexane layer and stirred for 15 minutes at temperature 25-30°C. Further the layers were separated and hexane layer was added activated charcoal ( 12.5 gm) and stirred for 20-30 minutes at temperature 30-35°C. The reaction mixture was filtered and stirred for 5-10 minutes at 25-30°C followed by cooling at 0 to -5°C and stirred for 30-40 minutes at 0 to -5°C. The reaction mixture was filtered and dried to get 150 to l 75 gm of title compound. HPLC purity: >99%.
PATENT
US 20140243544
IN 2013MU01290/WO 2014178064
WO 2014035107
PATENT
https://patents.google.com/patent/US9783492B2/en
Fluvoxamine or (E)-5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one-O-2-aminoethyl oxime is an antidepressant which functions as a selective serotonin reuptake inhibitor (SSRI). Fluvoxamine is used for the treatment of major depressive disorder (MDD), obsessive compulsive disorder (OCD), and anxiety disorders such as panic disorder and post-traumatic stress disorder (PTSD). Fluvoxamine CR (controlled release) is approved to treat social anxiety disorder.
Fluvoxamine maleate and compounds were first disclosed in U.S. Pat. No. 4,085,225. According to said patent, Fluvoxamine maleate prepared by alkylation reaction of 5-methoxy-4′-trifluoromethylvalerophenone oxime, compound of formula III with 2-chloroethylamine hydrochloride in dimethylformamide in the presence of a base such as potassium hydroxide powder for two days at 25° C.

Subsequently the solvent is removed under vacuum then the residue is acidified and extracted with ether to remove the unreacted oxime followed by basification. The obtained fluvoxamine base in ether extract is washed with sodium bicarbonate solution. The fluvoxamine base is then treated with maleic acid in absolute ethanol and the residue obtained by concentration under vacuum is recrystallized from acetonitrile to obtain fluvoxamine maleate. The process is very much tedious, time consuming as it requires two days for the reaction completion. Operations like removal of dimethylformamide, ether, ethanol makes process cumbersome at plant level. Requirement of various solvents lead the process to be non-eco-friendly. Moreover the patent is silent about yield and purity of the product.
In an alternate route described in U.S. Pat. No. 4,085,225, the mine of formula III is converted to formula I in a five step process i.e. alkylation of formula III with ethylene oxide. The reaction solvent is ethanol in which lithium is already dissolved. The reaction further involves addition of acetic acid to give the hydroxyethyl compound of formula A as oil. The compound of formula A is purified chromatographically over the silica gel, which is converted to a mesylate compound of formula B by treating with methanesulfonyl chloride and triethylamine at −5 to 0° C., then aminated with ammonia in methanol at 100° C. using autoclave for 16 hours followed by removal of methanol and extraction in ether to give fluvoxamine base.

The base is then converted to the maleate salt formula I, which is finally purified by recrystallization from acetonitrile.
There are lots of disadvantages in like more unit operations, use of various solvents and handling of ethylene oxide which is also known for its carcinogen effect. More unit operations lead to long occupancy of reactors in the plant as well as man power, high energy consumption and require bigger plant. These all parameters make the process commercially unviable as well as environmentally non-feasible. Further, purification of the compound of formula A requires cumbersome technique i.e chromatography over silica gel as well as lengthy work-up procedure in U.S. Pat. No. 4,085,225 requires complete removal of organic solvents at various stages.
U.S. Pat. No. 6,433,225 discloses the process for preparing fluvoxamine maleate, prepared by alkylating 5-methoxy-4′-trifluoromethylvalerophenone oxime compound of formula III with 2-chloroethylamine hydrochloride in toluene and PEG-400 (polyethyleneglycol-400) as facilitator in the presence of a base potassium hydroxide powder at 30-35°C. to obtain fluvoxamine base in toluene layer is then treated with maleic acid in water. The precipitated fluvoxamine maleate is filtered and washed with toluene and dried. The obtained dried cake recrystallized with water to get fluvoxamine maleate. The process disclosed in the patent is silent about actual purity of the product. As per our scientist’s observation alkylation reaction at the temperature of 30-35° C. may lead to non completion of reaction and results lower yield. Additional step of purification may further lead to loss of yield.
EXAMPLES
Stage-1: Preparation of (1 E)-N-hydroxy-5-methoxy-1-(4-trifluoromethyl phenyl)pentan-1-imine Formula III
To a stirred solution of 5-methoxy-1-(4-trifluoromethylphenyl)pentan-1one (150 gm) in methanol (750 ml), sodium carbonate (granule) (72 gm) and hydroxylamine hydrochloride (59.64 gm) were added at temperature 25-30° C. The reaction mass was heated 45-50° C. for 10-15 minutes followed by maintaining the reaction mass at temperature 45-50° C. for 8-9 hours under stirring. The reaction mass was cooled to 25-30° C. and filtered under vacuum to remove unreacted inorganic matter, then distilled out the methanol completely from the collected filtrate under vacuum at temperature below 50° C. The obtained slurry was cooled to 25-30° C. and water (300 ml) was added into the residue followed by the addition of hexane (300×2 ml) and stirred for 30 minutes. The layers were separated. The collected organic layer was stirred for 5-10 minutes at temperature 25-30° C. followed by cooling the mass at temperature −5° C. to −10° C., stirred for 30-40 minutes and filtered at the same temperature. The product was suck dried at −5 to −10° C. and further in vacuum at 25-30° C. for 2-3 hours to give 138-142 gm of title compound. HPLC purity: >98.5%
Stage-2: Preparation of Crude Fluvoxamine Maleate Formula I
To a prepared solution of dimethyl sulphoxide (575 ml), potassium hydroxide flakes (114.64 gm) and water (69 ml), stage-1 (115 gm) was added at temperature 40-45° C. The reaction mixture was stirred to get clear solution followed by adding 2-chloroethylamine hydrochloride (8636 gm) drop wise into the reaction mixture at temperature 40-45° C. and maintained for 1-2 hour. Water (1150 ml) was added in to the reaction mixture at temperature 25-30° C. and stirred for 20-25 minutes. Then toluene (575 ml×2) was added and stirred for 30 minutes and preceded for separation of layers followed by washing the toluene layer with water (1150×5 ml). The solution of maleic acid (48.47 gm) dissolved in water (98 ml) was added into above obtained toluene layer and stirred at temperature 25-30° C. for 2-3 hours. The reaction mixture was cooled to 0-5° C. and maintained for 30-40 minutes at the same temperature. The obtained material was washed with toluene, filtered and such dried. The wet cake was then added hexane (600 ml) and stirred for 30 minutes at temperature 25-30° C., filtered, washed with hexane and dried to get 161 gm of title compound. HPLC purity: >98.5%
Stage-3: Preparation of Pure Fluvoxamine Maleate Formula I
In to the reaction assembly, water (600 ml) was added and heated to 40-45° C. Stage-2 (150 gm) was added into the hot water under stirring. The reaction mixture was stirred for 5-10 minutes, filtered and cooled to 25° C. Toluene (68 ml) was added into the reaction mixture at temperature 25° C. and stirred for 30 minutes. Filtered the solid, washed with 10-15° C. chilled water and dried to get the pure 127.5 gm fluvoxamine maleate. HPLC purity: >99.8%
Process for isolation of 5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one Formula II
To a solution of conc. HCl (600 ml) and water (160 organic residue (250 gm) of (1 E)+(1 Z) of 1-N-hydroxy-5-methoxy-1-[4trifluoromethyl)phenyl]pentan-1-imine and traces of 5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one (obtained after hexane recovery from stage-1 filtrate) was added at temperature 25-30° C. under stirring. The reaction mixture was heated to 67-75° C. and maintained for 13-14 hours followed by cooling the reaction mixture at temperature 25-30° C. Then after hexane (500×2 ml) was added into the reaction mixture and stirred for 15 minutes at 25-30° C. The organic layers were separated and sodium bicarbonate solution (25 gm sodium bicarbonate dissolved in 250 ml water) was added into the hexane layer and stirred for 15 minutes. The layers were separated and water (250 ml) was added into hexane layer and stirred for 15 minutes at temperature 25-30° C. Further the layers were separated and hexane layer was added activated charcoal (12.5 gm) and stirred for 20-30 minutes at temperature 30-35° C. The reaction mixture was filtered and stirred for 5-10 minutes at 25-30° C. followed by cooling at 0 to −5° C. and stirred for 30-40 minutes at 0 to −5° C. The reaction mixture was filtered and dried to get 150 to 175 gm of title compound. HPLC purity: >99%.
Claims (5)Hide Dependent
We claim:1. An improved process for the preparation of fluvoxamine maleate of formula I,

wherein the improvements comprises the steps of:a). condensing the compound of formula II,

with hydroxylamine hydrochloride in the presence of sodium carbonate granules at temperature 45-50° C. in suitable solvent to form a compound of formula III, wherein the compound of formula III comprises a mixture of (1E)+(1Z) isomers of 1-N-hydroxy-5-methoxy-1-[4(trifluoromethyl)phenyl]pentan-1-imine, and wherein the mixture of (1E)+(1Z) isomers of 1-N-hydroxy-5-methoxy-1-[4(trifluoromethyl)phenyl]pentan-1-imine comprises 98% of E-isomer and 2% of Z-isomer;

b). isolating compound of formula III;c). treating compound of formula III with 2-chloroethylamine hydrochloride in the presence of base in suitable solvent at 40-45° C. to form compound of formula IV;

d). extracting compound of formula IV with suitable solvent to form an organic layer;e). treating organic layer of step d) with maleic acid;f). isolating crude fluvoxamine maleate of formula I; andg). optionally purifying fluvoxamine maleate of formula I.
2. The process according to claim 1, wherein in step a), said suitable solvent is selected from the group consisting of alcohol, ketone, nitrile, and hydrocarbons in any suitable proportion or mixtures thereof;in step c), said base is selected from the group consisting of sodium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, lithium carbonate, sodium bicarbonate, potassium bicarbonate, lithium bicarbonate, triethylamine and diisopropylethyamine;in step c), said solvent is selected from the group consisting of dimethylformamide (DMF), dimethylsulphoxide (DMSO) and hexamethylphosphoramide (HMPA) in any suitable proportion or mixtures thereof; andin step d) said suitable solvent is selected from the group consisting of toluene and xylene.3. A process for the isolation of 5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one of formula II from mixture of (1E)+(1Z) of 1-N -hydroxy-5-methoxy-1-[4-(trifluoromethyl) phenyl]pentan-1-imine of formula III by treating compound of formula III with aqueous hydrochloric acid, wherein the mixture of (1E)+(1Z) of 1-N-hydroxy-5-methoxy-1-[4-(trifluoromethyl) phenyl]pentan-1-imine of formula III comprises 98% of E-isomer and 2% of Z-isomer.4. The process according to claim 3, wherein the reaction is performed at temperature 65-75°C.5. The process according to claim 1, wherein in step a), said suitable solvent is methanol.
Publication numberPriority datePublication dateAssigneeTitleUS4081551A *1975-03-201978-03-28U.S. Philips CorporationOxime ethers having anti-depressive activityUS4085225A1975-03-201978-04-18U.S. Philips CorporationOxime ethers having anti-depressive activityCN1079733A *1993-04-081993-12-22中国科学院成都有机化学研究所The synthetic method of a-benzoin oximeUS6433225B11999-11-122002-08-13Sun Pharamaceutical Industries, Ltd.Process for the preparation of fluvoxazmine maleateCN101654419A *2009-09-122010-02-24西北师范大学Preparation method of fluvoxamine maleate
Syn
US 6433225 SUN
https://patents.google.com/patent/US6433225B1/en
EXAMPLE 1
To a stirred mixture of toluene (1.20 lit.), PEG-400 (0.4 lit) and powdered potassium hydroxide (86.0 g on 100% basis, 1.53 mol.) at ambient temperature is added 5-methoxy-4′-trifluoromethylvalerophenone oxime (100 g, 0.363 mol.), followed by 2-chloroethyl amine hydrochloride (50.56 g, 0.435 mol.). The mixture is stirred at 30-35° C. for 2 hours. Water (1.2 lit.) is then added, stirred for 30 mins. and the aqueous layer is separated out. The organic layer is washed with water (˜3×500 ml) until the washings are neutral. To the washed organic layer is added a solution of maleic acid (14.14 g, 0.363 mol.) in water (65 ml) and the mixture is stirred at 25-30° C. temperature for 2 hours, then cooled to 5-10° C. when the maleate salt crystallizes out. The crystallized fluvoxamine maleate is filtered, washed with toluene (200 ml) and sucked to dryness. The crude fluvoxamine maleate thus obtained is dissolved in water (300 ml) at 50-55° C. to get a clear solution, then gradually cooled to 5-8° C. and then further stirred at this temperature for 2 hours. The recrystallised fluvoxamine maleate is filtered, washed with chilled water (5° C., 100 ml) and sucked dry. The product is finally dried at 50-55° C. to constant weight. The fluvoxamine maleate obtained complies with the specifications of British Pharmacopoeia, 1999.EXAMPLE 2
This process when scaled up in pilot plant on 4.0 kg scale input of 5-methoxy-4′-trifluoromethylvalerophenone oxime gave 4.5 kg (71.2%) of fluvoxamine maleate, complying to the specifications of British Pharmacopoeia, 1999.

SYN
US 4085225
https://patents.google.com/patent/US4085225A/en
EXAMPLE 15-Methoxy-4′-trifluoromethylvalerophenone O-(2-aminoethyl) oxime maleate (1:1).
20.4 Mmol (5.3 g) of 5-methoxy-4′-trifluoromethylvalerophenone (melting point 43°-44° C), 20.5 mmol (3.1 g) of 2-aminooxyethylaminedihydrochloride and 10 ml of pyridine were refluxed for 15 hours in 20 ml of absolute ethanol. After evaporating the pyridine and the ethanol in vacuo, the residue was dissolved in water. This solution was washed with petroleum ether and 10 ml of 50% sodium hydroxide solution were then added. Then three extractions with 40 ml of ether were carried out. The ether extract was washed successively with 20 ml of 5% sodium bicarbonate solution and 20 ml of water. After drying on sodium sulphate, the ether layer was evaporated in vacuo. Toluene was then evaporated another three times (to remove the pyridine) and the oil thus obtained was dissolved in 15 ml of absolute ethanol. An equimolar quantity of maleic acid was added to said solution and the solution was then heated until a clear solution was obtained. The ethanol was then removed in vacuo and the residue was crystallized from 10 ml of acetonitrile at +5° C. After sucking off and washing with cold acetonitrile, it was dried in air. The melting point of the resulting title compound was 120°-121.5° C.

SYN
GB 1535226

References
- ^ Jump up to:a b Use During Pregnancy and Breastfeeding
- ^ Jump up to:a b c d e f “Product Information Luvox”. TGA eBusiness Services. Abbott Australasia Pty Ltd. 15 January 2013. Retrieved 21 October 2013.
- ^ van Harten J (March 1993). “Clinical pharmacokinetics of selective serotonin reuptake inhibitors”. Clinical Pharmacokinetics. 24 (3): 203–20. doi:10.2165/00003088-199324030-00003. PMID 8384945. S2CID 84636672.
- ^ “Luvox”. ChemSpider. Royal Society of Chemistry. Archived from the original on 15 November 2013. Retrieved 21 October 2013.
- ^ “Fluvoxamine Maleate Information”. U.S. Food and Drug Administration(FDA). 15 July 2015. Archived from the original on 29 November 2019. Retrieved 28 November 2019.
- ^ Jump up to:a b McCain JA (July 2009). “Antidepressants and suicide in adolescents and adults: a public health experiment with unintended consequences?”. P T. 34(7): 355–78. PMC 2799109. PMID 20140100.
- ^ Figgitt DP, McClellan KJ (October 2000). “Fluvoxamine. An updated review of its use in the management of adults with anxiety disorders”. Drugs. 60 (4): 925–54. doi:10.2165/00003495-200060040-00006. PMID 11085201.
- ^ Irons J (December 2005). “Fluvoxamine in the treatment of anxiety disorders”. Neuropsychiatric Disease and Treatment. 1 (4): 289–99. PMC 2424117. PMID 18568110.
- ^ Jump up to:a b “Fluvoxamine Maleate tablet, coated prescribing information”. DailyMed. 14 December 2018. Retrieved 28 November 2019.
- ^ “Luvox CR approved for OCD and SAD”. MPR. 29 February 2008. Retrieved 2 March 2019.
- ^ Jump up to:a b Rossi S, ed. (2013). Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790-9-3.
- ^ “Luvox Tablets”. NPS MedicineWise. Retrieved 22 October 2018.
- ^ Jump up to:a b Joint Formulary Committee (2013). British National Formulary (BNF)(65 ed.). London, UK: Pharmaceutical Press. ISBN 978-0-85711-084-8.
- ^ “Summary of Full Prescribing Information: Fluvoxamine”. Drug Registry of Russia (RLS) Drug Compendium (in Russian). Retrieved 21 March 2015.
- ^ “2005 News Releases”. Astellas Pharma. Retrieved 16 September 2018.
- ^ “International Approvals: Ebixa, Depromel/Luvox, M-Vax”. http://www.medscape.com. Retrieved 16 September 2018.
- ^ “US-FDA Fluvoxamine Product Insert”. March 2005.
- ^ Wilde MI, Plosker GL, Benfield P (November 1993). “Fluvoxamine. An updated review of its pharmacology, and therapeutic use in depressive illness”. Drugs. 46(5): 895–924. doi:10.2165/00003495-199346050-00008. PMID 7507038.
- ^ Kwasucki J, Stepień A, Maksymiuk G, Olbrych-Karpińska B (2002). “[Evaluation of analgesic action of fluvoxamine compared with efficacy of imipramine and tramadol for treatment of sciatica–open trial]”. Wiadomosci Lekarskie. 55 (1–2): 42–50. PMID 12043315.
- ^ Schreiber S, Pick CG (August 2006). “From selective to highly selective SSRIs: a comparison of the antinociceptive properties of fluoxetine, fluvoxamine, citalopram and escitalopram”. European Neuropsychopharmacology. 16 (6): 464–8. doi:10.1016/j.euroneuro.2005.11.013. PMID 16413173. S2CID 39278756.
- ^ Coquoz D, Porchet HC, Dayer P (September 1993). “Central analgesic effects of desipramine, fluvoxamine, and moclobemide after single oral dosing: a study in healthy volunteers”. Clinical Pharmacology and Therapeutics. 54 (3): 339–44. doi:10.1038/clpt.1993.156. PMID 8375130. S2CID 8229797.
- ^ Williams T, Hattingh CJ, Kariuki CM, Tromp SA, van Balkom AJ, Ipser JC, Stein DJ (October 2017). “Pharmacotherapy for social anxiety disorder (SAnD)”. The Cochrane Database of Systematic Reviews. 10 (10): CD001206. doi:10.1002/14651858.CD001206.pub3. PMC 6360927. PMID 29048739.
- ^ Cheer SM, Figgitt DP (2002). “Spotlight on fluvoxamine in anxiety disorders in children and adolescents”. CNS Drugs. 16 (2): 139–44. doi:10.2165/00023210-200216020-00006. PMID 11825104. S2CID 26774895.
- ^ Silver H (2001). “Fluvoxamine as an adjunctive agent in schizophrenia”. CNS Drug Reviews. 7 (3): 283–304. doi:10.1111/j.1527-3458.2001.tb00200.x. PMC 6741705. PMID 11607044.
- ^ Polcwiartek C, Nielsen J (March 2016). “The clinical potentials of adjunctive fluvoxamine to clozapine treatment: a systematic review”. Psychopharmacology. 233 (5): 741–50. doi:10.1007/s00213-015-4161-1. PMID 26626327. S2CID 12168939.
- ^ Lenze EJ, Mattar C, Zorumski CF, Stevens A, Schweiger J, Nicol GE, Miller JP, Yang L, Yingling M, Avidan MS, Reiersen AM (December 2020). “Fluvoxamine vs Placebo and Clinical Deterioration in Outpatients With Symptomatic COVID-19: A Randomized Clinical Trial”. JAMA. 324 (22): 2292–2300. doi:10.1001/jama.2020.22760. PMID 33180097.
- ^ Seymour CW, Bauchner H, Golub RM (December 2020). “COVID-19 Infection-Preventing Clinical Deterioration”. JAMA. 324 (22): 2300. doi:10.1001/jama.2020.21720. PMID 33180115.
- ^ [+https://scitechdaily.com/antidepressant-fluvoxamine-may-prevent-covid-19-infections-from-worsening/ “Antidepressant Fluvoxamine May Prevent COVID-19 Infections From Worsening”] Check
|url=value (help). - ^ https://academic.oup.com/ofid/advance-article/doi/10.1093/ofid/ofab050/6124100
- ^ Brayfield A, ed. (13 August 2013). Fluoxetine Hydrochloride. Martindale: The Complete Drug Reference. London, UK: Pharmaceutical Press. Retrieved 24 November 2013.
- ^ Taylor D, Paton C, Shitij K (2012). The Maudsley prescribing guidelines in psychiatry. West Sussex: Wiley-Blackwell. ISBN 978-0-470-97948-8.
- ^ “Faverin 100 mg film-coated tablets – Summary of Product Characteristics (SPC)”. electronic Medicines Compendium. Abbott Healthcare Products Limited. 14 May 2013. Retrieved 21 October 2013.
- ^ “Top Ten Legal Drugs Linked to Violence”. Time. 7 January 2011. Retrieved 10 September 2014.
- ^ Jump up to:a b Ciraulo DA, Shader RI (2011). Ciraulo DA, Shader RI (eds.). Pharmacotherapy of Depression (2nd ed.). Springer. p. 49. doi:10.1007/978-1-60327-435-7. ISBN 978-1-60327-435-7.
- ^ Jump up to:a b Brunton L, Chabner B, Knollman B (2010). Goodman and Gilman’s The Pharmacological Basis of Therapeutics (12th ed.). New York: McGraw-Hill Professional. ISBN 978-0-07-162442-8.
- ^ Baumann P (December 1996). “Pharmacokinetic-pharmacodynamic relationship of the selective serotonin reuptake inhibitors”. Clinical Pharmacokinetics. 31 (6): 444–69. doi:10.2165/00003088-199631060-00004. PMID 8968657. S2CID 31923953.
- ^ DeVane CL, Gill HS (1997). “Clinical pharmacokinetics of fluvoxamine: applications to dosage regimen design”. The Journal of Clinical Psychiatry. 58 Suppl 5 (Suppl 5): 7–14. PMID 9184622.
- ^ DeVane CL (1998). “Translational pharmacokinetics: current issues with newer antidepressants”. Depression and Anxiety. 8 Suppl 1 (Suppl 1): 64–70. doi:10.1002/(SICI)1520-6394(1998)8:1+<64::AID-DA10>3.0.CO;2-S. PMID 9809216.
- ^ Bondy B, Spellmann I (March 2007). “Pharmacogenetics of antipsychotics: useful for the clinician?”. Current Opinion in Psychiatry. 20 (2): 126–30. doi:10.1097/YCO.0b013e328017f69f. PMID 17278909. S2CID 23859992.
- ^ Kroon LA (September 2007). “Drug interactions with smoking”. American Journal of Health-System Pharmacy. 64 (18): 1917–21. doi:10.2146/ajhp060414. PMID 17823102.
- ^ Waknine Y (13 April 2007). “Prescribers Warned of Tizanidine Drug Interactions”. Medscape News. Medscape. Retrieved 1 February 2008.
- ^ “Fluvoxamine (Oral Route) Precautions”. Mayo Clinic. Retrieved 2 November2018.
- ^ Hemeryck A, Belpaire FM (February 2002). “Selective serotonin reuptake inhibitors and cytochrome P-450 mediated drug-drug interactions: an update”. Current Drug Metabolism. 3 (1): 13–37. doi:10.2174/1389200023338017. PMID 11876575.
- ^ “Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers”.
- ^ Raouf M (2016). Fudin J (ed.). “Benzodiazepine Metabolism and Pharmacokinetics” (PDF).
- ^ Peppers MP (1996). “Benzodiazepines for alcohol withdrawal in the elderly and in patients with liver disease”. Pharmacotherapy. 16 (1): 49–57. doi:10.1002/j.1875-9114.1996.tb02915.x. PMID 8700792. S2CID 1389910.
- ^ “fluvoxamine maleate: PRODUCT MONOGRAPH” (PDF). 2016.
- ^ “Luvox Data Sheet” (PDF). Medsafe, New Zealand. 2017.
- ^ Suzuki Y, Shioiri T, Muratake T, Kawashima Y, Sato S, Hagiwara M, Inoue Y, Shimoda K, Someya T (April 2003). “Effects of concomitant fluvoxamine on the metabolism of alprazolam in Japanese psychiatric patients: interaction with CYP2C19 mutated alleles”. European Journal of Clinical Pharmacology. 58 (12): 829–33. doi:10.1007/s00228-003-0563-9. PMID 12698310. S2CID 32559753.
- ^ Gerlach M, Warnke A, Greenhill L (2014). Psychiatric Drugs in Children and Adolescents: Basic Pharmacology and Practical Applications. Springer-Verlag Wien. p. 131. ISBN 978-3-7091-1500-8.
- ^ Fleishaker JC, Hulst LK (1994). “A pharmacokinetic and pharmacodynamic evaluation of the combined administration of alprazolam and fluvoxamine”. European Journal of Clinical Pharmacology. 46 (1): 35–9. doi:10.1007/bf00195913. PMID 8005185. S2CID 2161450.
- ^ Obach RS, Ryder TF (August 2010). “Metabolism of ramelteon in human liver microsomes and correlation with the effect of fluvoxamine on ramelteon pharmacokinetics”. Drug Metabolism and Disposition. 38 (8): 1381–91. doi:10.1124/dmd.110.034009. PMID 20478852. S2CID 8421997.
- ^ Pandi-Perumal SR, Spence DW, Verster JC, Srinivasan V, Brown GM, Cardinali DP, Hardeland R (12 April 2011). “Pharmacotherapy of insomnia with ramelteon: safety, efficacy and clinical applications”. Journal of Central Nervous System Disease. 3: 51–65. doi:10.4137/JCNSD.S1611. PMC 3663615. PMID 23861638.
- ^ Jump up to:a b Anttila AK, Rasanen L, Leinonen EV (October 2001). “Fluvoxamine augmentation increases serum mirtazapine concentrations three- to fourfold”. The Annals of Pharmacotherapy. 35 (10): 1221–3. doi:10.1345/aph.1A014. PMID 11675851. S2CID 44807359.
- ^ Granfors MT, Backman JT, Neuvonen M, Ahonen J, Neuvonen PJ (April 2004). “Fluvoxamine drastically increases concentrations and effects of tizanidine: a potentially hazardous interaction”. Clinical Pharmacology and Therapeutics. 75(4): 331–41. doi:10.1016/j.clpt.2003.12.005. PMID 15060511. S2CID 25781307.
- ^ Ishikawa M, Ishiwata K, Ishii K, Kimura Y, Sakata M, Naganawa M, et al. (October 2007). “High occupancy of sigma-1 receptors in the human brain after single oral administration of fluvoxamine: a positron emission tomography study using [11C]SA4503”. Biological Psychiatry. 62 (8): 878–83. doi:10.1016/j.biopsych.2007.04.001. PMID 17662961. S2CID 728565.
- ^ Schatzberg AF, Nemeroff CB (2009). The American Psychiatric Publishing textbook of psychopharmacology (4th ed.). Arlington, VA: American Psychiatric Pub. p. 354. ISBN 978-1-585-62386-0. OCLC 320111564.
- ^ Yahata M, Chiba K, Watanabe T, Sugiyama Y (September 2017). “Possibility of Predicting Serotonin Transporter Occupancy From the In Vitro Inhibition Constant for Serotonin Transporter, the Clinically Relevant Plasma Concentration of Unbound Drugs, and Their Profiles for Substrates of Transporters”. Journal of Pharmaceutical Sciences. 106 (9): 2345–2356. doi:10.1016/j.xphs.2017.05.007. PMID 28501470.
- ^ Jump up to:a b Hashimoto K (September 2009). “Sigma-1 receptors and selective serotonin reuptake inhibitors: clinical implications of their relationship”. Central Nervous System Agents in Medicinal Chemistry. 9 (3): 197–204. doi:10.2174/1871524910909030197. PMID 20021354.
- ^ Westenberg HG, Sandner C (April 2006). “Tolerability and safety of fluvoxamine and other antidepressants”. International Journal of Clinical Practice. 60 (4): 482–91. doi:10.1111/j.1368-5031.2006.00865.x. PMC 1448696. PMID 16620364.
- ^ Hindmarch I, Hashimoto K (April 2010). “Cognition and depression: the effects of fluvoxamine, a sigma-1 receptor agonist, reconsidered”. Human Psychopharmacology. 25 (3): 193–200. doi:10.1002/hup.1106. PMID 20373470. S2CID 26491662.
- ^ Hrdina PD (July 1991). “Pharmacology of serotonin uptake inhibitors: focus on fluvoxamine”. Journal of Psychiatry & Neuroscience. 16 (2 Suppl 1): 10–8. PMC 1188307. PMID 1931931.
- ^ Jump up to:a b Sittig’s Pharmaceutical Manufacturing Encyclopedia (PDF) (3rd ed.). William Andrew. 2008. p. 1699. ISBN 978-0-8155-1526-5. Retrieved 17 October2013.
- ^ Leslie LK, Newman TB, Chesney PJ, Perrin JM (July 2005). “The Food and Drug Administration’s deliberations on antidepressant use in pediatric patients”. Pediatrics. 116 (1): 195–204. doi:10.1542/peds.2005-0074. PMC 1550709. PMID 15995053.
- ^ “Brand Index―Fluvoxamine India”. Archived from the original on 19 October 2013. Retrieved 18 October 2013.
- ^ Omori IM, Watanabe N, Nakagawa A, Cipriani A, Barbui C, McGuire H, Churchill R, Furukawa TA (March 2010). “Fluvoxamine versus other anti-depressive agents for depression”. The Cochrane Database of Systematic Reviews (3): CD006114. doi:10.1002/14651858.CD006114.pub2. PMC 4171125. PMID 20238342.
- ^ “OCD Medication”. Archived from the original on 14 October 2013. Retrieved 17 October 2013.
- ^ “Fluvoxamine Product Monograph” (PDF). 1999.
- ^ “Luvox Approved For Obsessive Compulsive Disorder in Children and Teens”. Archived from the original on 16 January 2009. Retrieved 8 February 2014.
- ^ Higuchi T, Briley M (February 2007). “Japanese experience with milnacipran, the first serotonin and norepinephrine reuptake inhibitor in Japan”. Neuropsychiatric Disease and Treatment. 3 (1): 41–58. doi:10.2147/nedt.2007.3.1.41. PMC 2654524. PMID 19300537.
- ^ “Human Metabolome Database: Showing metabocard for Fluvoxamine (HMDB0014322)”. http://www.hmdb.ca. Retrieved 15 September 2018.
- ^ “Solvay’s Fluvoxamine maleate is first drug approved for the treatment of social anxiety disorder in Japan”.
- ^ Walker R, Whittlesea C, eds. (2007) [1994]. Clinical Pharmacy and Therapeutics (4th ed.). Edinburgh: Churchill Livingstone Elsevier. ISBN 978-0-7020-4293-5.
- ^ “Fluvoxamine”. http://www.drugbank.ca. Retrieved 22 October 2019.
External links
- “Fluvoxamine”. Drug Information Portal. U.S. National Library of Medicine.
/////////DU23000, Fevarin, Fluvoxamine maleate, Luvox, Luvox CR, SME 3110, UNII-5LGN83G74V, Fluvoxamine, sme 3110, DU 23000
#DU23000, #Fevarin, #Fluvoxamine maleate, #Luvox, #Luvox CR, #SME 3110, #UNII-5LGN83G74V, #Fluvoxamine, #sme 3110, #DU 23000
Evinacumab
(Heavy chain)
EVQLVESGGG VIQPGGSLRL SCAASGFTFD DYAMNWVRQG PGKGLEWVSA ISGDGGSTYY
ADSVKGRFTI SRDNSKNSLY LQMNSLRAED TAFFYCAKDL RNTIFGVVIP DAFDIWGQGT
MVTVSSASTK GPSVFPLAPC SRSTSESTAA LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP
AVLQSSGLYS LSSVVTVPSS SLGTKTYTCN VDHKPSNTKV DKRVESKYGP PCPPCPAPEF
LGGPSVFLFP PKPKDTLMIS RTPEVTCVVV DVSQEDPEVQ FNWYVDGVEV HNAKTKPREE
QFNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKGLPSSIEK TISKAKGQPR EPQVYTLPPS
QEEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT PPVLDSDGSF FLYSRLTVDK
SRWQEGNVFS CSVMHEALHN HYTQKSLSLS LGK
(Light chain)
DIQMTQSPST LSASVGDRVT ITCRASQSIR SWLAWYQQKP GKAPKLLIYK ASSLESGVPS
RFSGSGSGTE FTLTISSLQP DDFATYYCQQ YNSYSYTFGQ GTKLEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Disulfide bridge: H22-H96, H140-L214, H153-H209, H232-H’232, H235-H’235, H267-H327, H373-H431, H’22-H’96, H’140-L’214, H’153-H’209, H’267-H’327, H’373-H’431, L23-L88, L134-L194, L’23-L’88, L’134-L’194)
Evinacumab
エビナクマブ (遺伝子組換え)
Immunoglobulin G4, anti-(human protein ANGPTL3 (angiopoietin-like 3)) (human monoclonal REGN1500 heavy chain), disulfide with human monoclonal REGN1500 light chain, dimer
| Formula | C6480H9992N1716O2042S46 |
|---|---|
| CAS | 1446419-85-7 |
| Mol weight | 146081.9345 |
Protein Sequence
Sequence Length: 1334, 453, 453, 214, 214multichain; modified (modifications unspecified)
FDA APPROVED, 2021/2/11, EVKEEZA
Antihyperlipidemic, Anti-angiopietin like 3
Monoclonal antibody
Treatment of dyslipidemia
- REGN 1500
- REGN-1500
- REGN1500
Sequence:
1EVQLVESGGG VIQPGGSLRL SCAASGFTFD DYAMNWVRQG PGKGLEWVSA51ISGDGGSTYY ADSVKGRFTI SRDNSKNSLY LQMNSLRAED TAFFYCAKDL101RNTIFGVVIP DAFDIWGQGT MVTVSSASTK GPSVFPLAPC SRSTSESTAA151LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP AVLQSSGLYS LSSVVTVPSS201SLGTKTYTCN VDHKPSNTKV DKRVESKYGP PCPPCPAPEF LGGPSVFLFP251PKPKDTLMIS RTPEVTCVVV DVSQEDPEVQ FNWYVDGVEV HNAKTKPREE301QFNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKGLPSSIEK TISKAKGQPR351EPQVYTLPPS QEEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT401PPVLDSDGSF FLYSRLTVDK SRWQEGNVFS CSVMHEALHN HYTQKSLSLS451LGK
Sequence:
1EVQLVESGGG VIQPGGSLRL SCAASGFTFD DYAMNWVRQG PGKGLEWVSA51ISGDGGSTYY ADSVKGRFTI SRDNSKNSLY LQMNSLRAED TAFFYCAKDL101RNTIFGVVIP DAFDIWGQGT MVTVSSASTK GPSVFPLAPC SRSTSESTAA151LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP AVLQSSGLYS LSSVVTVPSS201SLGTKTYTCN VDHKPSNTKV DKRVESKYGP PCPPCPAPEF LGGPSVFLFP251PKPKDTLMIS RTPEVTCVVV DVSQEDPEVQ FNWYVDGVEV HNAKTKPREE301QFNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKGLPSSIEK TISKAKGQPR351EPQVYTLPPS QEEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT401PPVLDSDGSF FLYSRLTVDK SRWQEGNVFS CSVMHEALHN HYTQKSLSLS451LGK
Sequence:
1DIQMTQSPST LSASVGDRVT ITCRASQSIR SWLAWYQQKP GKAPKLLIYK51ASSLESGVPS RFSGSGSGTE FTLTISSLQP DDFATYYCQQ YNSYSYTFGQ101GTKLEIKRTV AAPSVFIFPP SDEQLKSGTA SVVCLLNNFY PREAKVQWKV151DNALQSGNSQ ESVTEQDSKD STYSLSSTLT LSKADYEKHK VYACEVTHQG201LSSPVTKSFN RGEC
Sequence:
1DIQMTQSPST LSASVGDRVT ITCRASQSIR SWLAWYQQKP GKAPKLLIYK51ASSLESGVPS RFSGSGSGTE FTLTISSLQP DDFATYYCQQ YNSYSYTFGQ101GTKLEIKRTV AAPSVFIFPP SDEQLKSGTA SVVCLLNNFY PREAKVQWKV151DNALQSGNSQ ESVTEQDSKD STYSLSSTLT LSKADYEKHK VYACEVTHQG201LSSPVTKSFN RGEC
Sequence Modifications
| Type | Location | Description |
|---|---|---|
| bridge | Cys-22 – Cys-96 | disulfide bridge |
| bridge | Cys-140 – Cys-214” | disulfide bridge |
| bridge | Cys-153 – Cys-209 | disulfide bridge |
| bridge | Cys-232 – Cys-232′ | disulfide bridge |
| bridge | Cys-235 – Cys-235′ | disulfide bridge |
| bridge | Cys-267 – Cys-327 | disulfide bridge |
| bridge | Cys-373 – Cys-431 | disulfide bridge |
| bridge | Cys-22′ – Cys-96′ | disulfide bridge |
| bridge | Cys-140′ – Cys-214”’ | disulfide bridge |
| bridge | Cys-153′ – Cys-209′ | disulfide bridge |
| bridge | Cys-267′ – Cys-327′ | disulfide bridge |
| bridge | Cys-373′ – Cys-431′ | disulfide bridge |
| bridge | Cys-23” – Cys-88” | disulfide bridge |
| bridge | Cys-134” – Cys-194” | disulfide bridge |
| bridge | Cys-23”’ – Cys-88”’ | disulfide bridge |
| bridge | Cys-134”’ – Cys-194”’ | disulfide bridge |
PATENTS
WO 2017024062
US 20170305999
Evinacumab, sold under the brand name Evkeeza, is a monoclonal antibody medication for the treatment of homozygous familial hypercholesterolemia (HoFH).[1][2]
Evinacumab is a recombinant human IgG4 monoclonal antibody targeted against angiopoietin-like protein 3 (ANGPTL3) and the first drug of its kind. The ANGPTL family of proteins serve a number of physiologic functions – including involvement in the regulation of lipid metabolism – which have made them desirable therapeutic targets in recent years.2 Loss-of-function mutations in ANGPTL3 have been noted to result in hypolipidemia and subsequent reductions in cardiovascular risk, whereas increases in function appear to be associated with cardiovascular risk, and it was these observations that provided a rationale for the development of a therapy targeted against ANGPTL3.3
In February 2021, evinacumab became the first-and-only inhibitor of ANGPTL3 to receive FDA approval after it was granted approval for the adjunctive treatment of homozygous familial hypercholesterolemia (HoFH) under the brand name “Evkeeza”.8 Evinacumab is novel in its mechanism of action compared with other lipid-lowering therapies and therefore provides a unique and synergistic therapeutic option in the treatment of HoFH.
Common side effects include nasopharyngitis (cold), influenza-like illness, dizziness, rhinorrhea (runny nose), and nausea. Serious hypersensitivity (allergic) reactions have occurred in the Evkeeza clinical trials.[2]
Evinacumab binds to the angiopoietin-like protein 3 (ANGPTL3).[2] ANGPTL3 slows the function of certain enzymes that break down fats in the body.[2] Evinacumab blocks ANGPTL3, allowing faster break down of fats that lead to high cholesterol.[2] Evinacumab was approved for medical use in the United States in February 2021.[2][3]
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Evkeeza | Injection, solution, concentrate | 150 mg/1mL | Intravenous | Regeneron Pharmaceuticals, Inc. | 2021-02-11 | Not applicable |  | |
| Evkeeza | Injection, solution, concentrate | 150 mg/1mL | Intravenous | Regeneron Pharmaceuticals, Inc. | 2021-02-11 | Not applicable | |
History
The effectiveness and safety of evinacumab were evaluated in a double-blind, randomized, placebo-controlled, 24-week trial enrolling 65 participants with homozygous familial hypercholesterolemia (HoFH).[2] In the trial, 43 participants received 15 mg/kg of evinacumab every four weeks and 22 participants received the placebo.[2] Participants were taking other lipid-lowering therapies as well.[2]
The primary measure of effectiveness was the percent change in low-density lipoprotein (LDL-C) from the beginning of treatment to week 24.[2] At week 24, participants receiving evinacumab had an average 47% decrease in LDL-C while participants on the placebo had an average 2% increase.[2]
The U.S. Food and Drug Administration (FDA) granted the application for evinacumab orphan drug, breakthrough therapy, and priority review designations.[2] The FDA granted approval of Evkeeza to Regeneron Pharmaceuticals, Inc.[2]
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761181s000lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l m n “FDA approves add-on therapy for patients with genetic form of severely”. U.S. Food and Drug Administration (FDA). 11 February 2021. Retrieved 12 February 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “FDA Approves First-in-class Evkeeza (evinacumab-dgnb) for Patients with Ultra-rare Inherited Form of High Cholesterol” (Press release). Regeneron Pharmaceuticals. 11 February 2021. Retrieved 12 February 2021 – via PR Newswire.
Further reading
- Dewey FE, Gusarova V, Dunbar RL, O’Dushlaine C, Schurmann C, Gottesman O, et al. (July 2017). “Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease”. N Engl J Med. 377 (3): 211–221. doi:10.1056/NEJMoa1612790. PMC 5800308. PMID 28538136.
External links
- “Evinacumab”. Drug Information Portal. U.S. National Library of Medicine.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Angiopoietin-like 3 (ANGPTL3) |
| Clinical data | |
| Trade names | Evkeeza |
| Other names | REGN1500, evinacumab-dgnb |
| License data | US DailyMed: Evinacumab |
| Routes of administration | Intravenous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 1446419-85-7 |
| DrugBank | DB15354 |
| ChemSpider | none |
| UNII | T8B2ORP1DW |
| KEGG | D11753 |
| Chemical and physical data | |
| Formula | C6480H9992N1716O2042S46 |
| Molar mass | 146083.95 g·mol−1 |
//////////////
#Evinacumab, #Peptide, #APPROVALS 2021, #FDA 2021, #Monoclonal antibody, #dyslipidemia, #エビナクマブ (遺伝子組換え) , #REGN 1500, #REGN-1500, #REGN1500, #Anthony melvin crasto, #world drug tracker. # new drug approvals, #pharma
Tozinameran, Pfizer–BioNTech COVID‑19 vaccine


SEQUENCE1
gagaauaaac uaguauucuu cuggucccca cagacucaga gagaacccgc51caccauguuc guguuccugg ugcugcugcc ucuggugucc agccagugug101ugaaccugac caccagaaca cagcugccuc cagccuacac caacagcuuu151accagaggcg uguacuaccc cgacaaggug uucagaucca gcgugcugca201cucuacccag gaccuguucc ugccuuucuu cagcaacgug accugguucc251acgccaucca cguguccggc accaauggca ccaagagauu cgacaacccc301gugcugcccu ucaacgacgg gguguacuuu gccagcaccg agaaguccaa351caucaucaga ggcuggaucu ucggcaccac acuggacagc aagacccaga401gccugcugau cgugaacaac gccaccaacg uggucaucaa agugugcgag451uuccaguucu gcaacgaccc cuuccugggc gucuacuacc acaagaacaa501caagagcugg auggaaagcg aguuccgggu guacagcagc gccaacaacu551gcaccuucga guacgugucc cagccuuucc ugauggaccu ggaaggcaag601cagggcaacu ucaagaaccu gcgcgaguuc guguuuaaga acaucgacgg651cuacuucaag aucuacagca agcacacccc uaucaaccuc gugcgggauc701ugccucaggg cuucucugcu cuggaacccc ugguggaucu gcccaucggc751aucaacauca cccgguuuca gacacugcug gcccugcaca gaagcuaccu801gacaccuggc gauagcagca gcggauggac agcuggugcc gccgcuuacu851augugggcua ccugcagccu agaaccuucc ugcugaagua caacgagaac901ggcaccauca ccgacgccgu ggauugugcu cuggauccuc ugagcgagac951aaagugcacc cugaaguccu ucaccgugga aaagggcauc uaccagacca1001gcaacuuccg ggugcagccc accgaaucca ucgugcgguu ccccaauauc1051accaaucugu gccccuucgg cgagguguuc aaugccacca gauucgccuc1101uguguacgcc uggaaccgga agcggaucag caauugcgug gccgacuacu1151ccgugcugua caacuccgcc agcuucagca ccuucaagug cuacggcgug1201uccccuacca agcugaacga ccugugcuuc acaaacgugu acgccgacag1251cuucgugauc cggggagaug aagugcggca gauugccccu ggacagacag1301gcaagaucgc cgacuacaac uacaagcugc ccgacgacuu caccggcugu1351gugauugccu ggaacagcaa caaccuggac uccaaagucg gcggcaacua1401caauuaccug uaccggcugu uccggaaguc caaucugaag cccuucgagc1451gggacaucuc caccgagauc uaucaggccg gcagcacccc uuguaacggc1501guggaaggcu ucaacugcua cuucccacug caguccuacg gcuuucagcc1551cacaaauggc gugggcuauc agcccuacag agugguggug cugagcuucg1601aacugcugca ugccccugcc acagugugcg gcccuaagaa aagcaccaau1651cucgugaaga acaaaugcgu gaacuucaac uucaacggcc ugaccggcac1701cggcgugcug acagagagca acaagaaguu ccugccauuc cagcaguuug1751gccgggauau cgccgauacc acagacgccg uuagagaucc ccagacacug1801gaaauccugg acaucacccc uugcagcuuc ggcggagugu cugugaucac1851cccuggcacc aacaccagca aucagguggc agugcuguac caggacguga1901acuguaccga agugcccgug gccauucacg ccgaucagcu gacaccuaca1951uggcgggugu acuccaccgg cagcaaugug uuucagacca gagccggcug2001ucugaucgga gccgagcacg ugaacaauag cuacgagugc gacaucccca2051ucggcgcugg aaucugcgcc agcuaccaga cacagacaaa cagcccucgg2101agagccagaa gcguggccag ccagagcauc auugccuaca caaugucucu2151gggcgccgag aacagcgugg ccuacuccaa caacucuauc gcuaucccca2201ccaacuucac caucagcgug accacagaga uccugccugu guccaugacc2251aagaccagcg uggacugcac cauguacauc ugcggcgauu ccaccgagug2301cuccaaccug cugcugcagu acggcagcuu cugcacccag cugaauagag2351cccugacagg gaucgccgug gaacaggaca agaacaccca agagguguuc2401gcccaaguga agcagaucua caagaccccu ccuaucaagg acuucggcgg2451cuucaauuuc agccagauuc ugcccgaucc uagcaagccc agcaagcgga2501gcuucaucga ggaccugcug uucaacaaag ugacacuggc cgacgccggc2551uucaucaagc aguauggcga uugucugggc gacauugccg ccagggaucu2601gauuugcgcc cagaaguuua acggacugac agugcugccu ccucugcuga2651ccgaugagau gaucgcccag uacacaucug cccugcuggc cggcacaauc2701acaagcggcu ggacauuugg agcaggcgcc gcucugcaga uccccuuugc2751uaugcagaug gccuaccggu ucaacggcau cggagugacc cagaaugugc2801uguacgagaa ccagaagcug aucgccaacc aguucaacag cgccaucggc2851aagauccagg acagccugag cagcacagca agcgcccugg gaaagcugca2901ggacgugguc aaccagaaug cccaggcacu gaacacccug gucaagcagc2951uguccuccaa cuucggcgcc aucagcucug ugcugaacga uauccugagc3001agacuggacc cuccugaggc cgaggugcag aucgacagac ugaucacagg3051cagacugcag agccuccaga cauacgugac ccagcagcug aucagagccg3101ccgagauuag agccucugcc aaucuggccg ccaccaagau gucugagugu3151gugcugggcc agagcaagag aguggacuuu ugcggcaagg gcuaccaccu3201gaugagcuuc ccucagucug ccccucacgg cgugguguuu cugcacguga3251cauaugugcc cgcucaagag aagaauuuca ccaccgcucc agccaucugc3301cacgacggca aagcccacuu uccuagagaa ggcguguucg uguccaacgg3351cacccauugg uucgugacac agcggaacuu cuacgagccc cagaucauca3401ccaccgacaa caccuucgug ucuggcaacu gcgacgucgu gaucggcauu3451gugaacaaua ccguguacga cccucugcag cccgagcugg acagcuucaa3501agaggaacug gacaaguacu uuaagaacca cacaagcccc gacguggacc3551ugggcgauau cagcggaauc aaugccagcg ucgugaacau ccagaaagag3601aucgaccggc ugaacgaggu ggccaagaau cugaacgaga gccugaucga3651ccugcaagaa cuggggaagu acgagcagua caucaagugg cccugguaca3701ucuggcuggg cuuuaucgcc ggacugauug ccaucgugau ggucacaauc3751augcuguguu gcaugaccag cugcuguagc ugccugaagg gcuguuguag3801cuguggcagc ugcugcaagu ucgacgagga cgauucugag cccgugcuga3851agggcgugaa acugcacuac acaugaugac ucgagcuggu acugcaugca3901cgcaaugcua gcugccccuu ucccguccug gguaccccga gucucccccg3951accucggguc ccagguaugc ucccaccucc accugcccca cucaccaccu4001cugcuaguuc cagacaccuc ccaagcacgc agcaaugcag cucaaaacgc4051uuagccuagc cacaccccca cgggaaacag cagugauuaa ccuuuagcaa4101uaaacgaaag uuuaacuaag cuauacuaac cccaggguug gucaauuucg4151ugccagccac acccuggagc uagcaaaaaa aaaaaaaaaa aaaaaaaaaa4201aaaagcauau gacuaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa4251aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaa
Sequence Modifications
| Type | Location | Description |
|---|---|---|
| modified base | g-1 | m7g |
| modified base | g-1 | 3′-me |
| modified base | a-2 | am |
| uncommon link | g-1 – a-2 | 5′->5′ triphosphate |
Tozinameran
Pfizer–BioNTech COVID-19 vaccine
トジナメラン (JAN);
コロナウイルス修飾ウリジンRNAワクチン;
RNA (recombinant 5′-[1,2-[(3′-O-methyl)m7G-(5’→5′)-ppp-Am]]-capped all uridine→N1-methylpseudouridine-substituted severe acute respiratory syndrome coronavirus 2 secretory signal peptide contg. spike glycoprotein S1S2-specifying plus 5′- and 3′-untranslated flanking region-contg. poly(A)-tailed messenger BNT162b2), inner salt
Nucleic Acid Sequence
Sequence Length: 42841106 a 1315 c 1062 g 801 umodified
APPROVED JAPAN Comirnaty, 2021/2/14
CAS 2417899-77-3
| Active immunization (SARS-CoV-2) |
Tozinameran is mRNA encoding full length of spike protein analog of SARS-CoV-2
Target Severe acute respiratory syndrome coronavirus 2 spike glycoprotein
Coronavirus disease – COVID-19
| FORM | ROUTE | STRENGTH |
|---|---|---|
| Injection, suspension | Intramuscular | 0.23 mg/1.8mL |
| Suspension | Intramuscular | 30 mcg |
| NAME | INGREDIENTS | DOSAGE | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Pfizer-BioNTech Covid-19 Vaccine | Pfizer-BioNTech Covid-19 Vaccine (0.23 mg/1.8mL) | Injection, suspension | Intramuscular | Pfizer Manufacturing Belgium NV | 2020-12-12 | Not applicable | |
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Comirnaty | 30 mcg | Intramuscular | Bio N Tech Manufacturing Gmb H | 2021-01-06 | Not applicable |  | ||
| Pfizer-BioNTech Covid-19 Vaccine | Suspension | 30 mcg | Intramuscular | Biontech Manufacturing Gmbh | 2020-12-14 | Not applicable |  | |
| Pfizer-BioNTech Covid-19 Vaccine | Injection, suspension | 0.23 mg/1.8mL | Intramuscular | Pfizer Manufacturing Belgium NV | 2020-12-12 | Not applicable | |
The Pfizer–BioNTech COVID‑19 vaccine (pINN: tozinameran), sold under the brand name Comirnaty,[13] is a COVID-19 vaccine developed by the German company BioNTech in cooperation with Pfizer. It is both the first COVID-19 vaccine to be authorized by a stringent regulatory authority for emergency use[14][15] and the first cleared for regular use.[16]
It is given by intramuscular injection. It is an RNA vaccine composed of nucleoside-modified mRNA (modRNA) encoding a mutated form of the spike protein of SARS-CoV-2, which is encapsulated in lipid nanoparticles.[17] The vaccination requires two doses given three weeks apart.[18][19][20] Its ability to prevent severe infection in children, pregnant women, or immunocompromised people is unknown, as is the duration of the immune effect it confers.[20][21][22] As of February 2021, it is one of two RNA vaccines being deployed against COVID‑19, the other being the Moderna COVID‑19 vaccine. A third mRNA-based COVID-19 vaccine, CVnCoV, is in late-stage testing.[23]
Trials began in April 2020; by November, the vaccine had been tested on more than 40,000 people.[24] An interim analysis of study data showed a potential efficacy of over 90% in preventing infection within seven days of a second dose.[19][20] The most common side effects include mild to moderate pain at the injection site, fatigue, and headache.[25][26] As of December 2020, reports of serious side effects, such as allergic reactions, have been very rare,[a] and no long-term complications have been reported.[28] Phase III clinical trials are ongoing: monitoring of the primary outcomes will continue until August 2021, while monitoring of the secondary outcomes will continue until January 2023.[18]
In December 2020, the United Kingdom was the first country to authorize the vaccine on an emergency basis,[28] soon followed by the United States, the European Union and several other countries globally.[29][30][6][31][32]
BioNTech is the initial developer of the vaccine, and partnered with Pfizer for development, clinical research, overseeing the clinical trials, logistics, finances and for manufacturing worldwide with the exception of China.[33] The license to distribute and manufacture in China was purchased by Fosun, alongside its investment in BioNTech.[34][35] Distribution in Germany and Turkey is by BioNTech itself.[36] Pfizer indicated in November 2020, that 50 million doses could be available globally by the end of 2020, with about 1.3 billion doses in 2021.[20]
Pfizer has advanced purchase agreements of about US$3 billion for providing a licensed vaccine in the United States, the European Union, the United Kingdom, Japan, Canada, Peru, Singapore, and Mexico.[37][38] Distribution and storage of the vaccine is a logistics challenge because it needs to be stored at temperatures between −80 and −60 °C (−112 and −76 °F),[39] until five days before vaccination[38][39] when it can be stored at 2 to 8 °C (36 to 46 °F), and up to two hours at temperatures up to 25 °C (77 °F)[40][11] or 30 °C (86 °F).[41][42] In February 2021, Pfizer and BioNTech asked the U.S. Food and Drug Administration (FDA) to update the emergency use authorization (EUA) to permit the vaccine to be stored at between −25 and −15 °C (−13 and 5 °F) for up to two weeks before use.[43]
Development and funding
Before COVID-19 vaccines, a vaccine for an infectious disease had never before been produced in less than several years, and no vaccine existed for preventing a coronavirus infection in humans.[44] After the COVID-19 virus was detected in December 2019,[45] the development of BNT162b2 was initiated on 10 January 2020, when the SARS-CoV-2 genetic sequences were released by the Chinese Center for Disease Control and Prevention via GISAID,[46][47][48] triggering an urgent international response to prepare for an outbreak and hasten development of preventive vaccines.[49][50]
In January 2020, German biotech-company BioNTech started its program ‘Project Lightspeed’ to develop a vaccine against the new COVID‑19 virus based on its already established mRNA-technology.[24] Several variants of the vaccine were created in their laboratories in Mainz, and 20 of those were presented to experts of the Paul-Ehrlich-Institute in Langen.[51] Phase I / II Trials were started in Germany on 23 April 2020, and in the U.S. on 4 May 2020, with four vaccine candidates entering clinical testing. The Initial Pivotal Phase II / III Trial with the lead vaccine candidate ‘BNT162b2’ began in July. The Phase III results indicating a 95% effectiveness of the developed vaccine were published on 18 November 2020.[24]
BioNTech received a US$135 million investment from Fosun in March 2020, in exchange for 1.58 million shares in BioNTech and the future development and marketing rights of BNT162b2 in China,[35] Hong Kong, Macau and Taiwan.[52]
In June 2020, BioNTech received €100 million (US$119 million) in financing from the European Commission and European Investment Bank.[53] In September 2020, the German government granted BioNTech €375 million (US$445 million) for its COVID‑19 vaccine development program.[54]
Pfizer CEO Albert Bourla stated that he decided against taking funding from the US government’s Operation Warp Speed for the development of the vaccine “because I wanted to liberate our scientists [from] any bureaucracy that comes with having to give reports and agree how we are going to spend the money in parallel or together, etc.” Pfizer did enter into an agreement with the US for the eventual distribution of the vaccine, as with other countries.[55]
Clinical trials
See also: COVID-19 vaccine § Clinical trials started in 2020
Preliminary results from Phase I–II clinical trials on BNT162b2, published in October 2020, indicated potential for its efficacy and safety.[17][56] During the same month, the European Medicines Agency (EMA) began a periodic review of BNT162b2.[57]
The study of BNT162b2 is a continuous-phase trial in Phase III as of November 2020.[18] It is a “randomized, placebo-controlled, observer-blind, dose-finding, vaccine candidate-selection, and efficacy study in healthy individuals”.[18] The early-stage research determined the safety and dose level for two vaccine candidates, with the trial expanding during mid-2020 to assess efficacy and safety of BNT162b2 in greater numbers of participants, reaching tens of thousands of people receiving test vaccinations in multiple countries in collaboration with Pfizer and Fosun.[20][35]
The Phase III trial assesses the safety, efficacy, tolerability, and immunogenicity of BNT162b2 at a mid-dose level (two injections separated by 21 days) in three age groups: 12–15 years, 16–55 years or above 55 years.[18] For approval in the EU, an overall vaccine efficacy of 95% was confirmed by the EMA.[58] The EMA clarified that the second dose should be administered three weeks after the first dose.[59]
| Efficacy endpoint | Vaccine efficacy (95% confidence interval) [%] |
|---|---|
| After dose 1 to before dose 2 | 52.4 (29.5, 68.4) |
| ≥10 days after dose 1 to before dose 2 | 86.7 (68.6, 95.4) |
| Dose 2 to 7 days after dose 2 | 90.5 (61.0, 98.9) |
| ≥7 days after dose 2 (subjects without evidence of infection prior to 7 days after dose 2) | |
| Overall | 95.0 (90.0, 97.9) |
| 16–55 years | 95.6 (89.4, 98.6) |
| ≥55 years | 93.7 (80.6, 98.8) |
| ≥65 years | 94.7 (66.7, 99.9) |
The ongoing Phase III trial, which is scheduled to run from 2020 to 2022, is designed to assess the ability of BNT162b2 to prevent severe infection, as well as the duration of immune effect.[20][21][22]
Pfizer and BioNTech started a Phase II/III randomized control trial in healthy pregnant women 18 years of age and older (NCT04754594).[60] The study will evaluate 30 µg of BNT162b2 or placebo administered via intramuscular injection in 2 doses, 21 days apart. The Phase II portion of the study will include approximately 350 pregnant women randomized 1:1 to receive BNT162b2 or placebo at 27 to 34 weeks’ gestation. The Phase III portion of this study will assess the safety, tolerability, and immunogenicity of BNT162b2 or placebo among pregnant women enrolled at 24 to 34 weeks’ gestation. Pfizer and BioNTech announced on 18 February 2021 that the first participants received their first dose in this trial.[61]
Vaccine technology
See also: RNA vaccine and COVID-19 vaccine § Technology platforms
The BioNTech technology for the BNT162b2 vaccine is based on use of nucleoside-modified mRNA (modRNA) which encodes part of the spike protein found on the surface of the SARS-CoV-2 coronavirus (COVID‑19), triggering an immune response against infection by the virus protein.[62]
The vaccine candidate BNT162b2 was chosen as the most promising among three others with similar technology developed by BioNTech.[18][62][56] Prior to choosing BNT162b2, BioNTech and Pfizer had conducted Phase I trials on BNT162b1 in Germany and the United States, while Fosun performed a Phase I trial in China.[17][63] In these Phase I studies, BNT162b2 was shown to have a better safety profile than the other three BioNTech candidates.[63]
Sequence
The modRNA sequence of the vaccine is 4,284 nucleotides long.[64] It consists of a five-prime cap; a five prime untranslated region derived from the sequence of human alpha globin; a signal peptide (bases 55–102) and two proline substitutions (K986P and V987P, designated “2P”) that cause the spike to adopt a prefusion-stabilized conformation reducing the membrane fusion ability, increasing expression and stimulating neutralizing antibodies;[17][65] a codon-optimized gene of the full-length spike protein of SARS-CoV-2 (bases 103–3879); followed by a three prime untranslated region (bases 3880–4174) combined from AES and mtRNR1 selected for increased protein expression and mRNA stability[66] and a poly(A) tail comprising 30 adenosine residues, a 10-nucleotide linker sequence, and 70 other adenosine residues (bases 4175–4284).[64] The sequence contains no uridine residues; they are replaced by 1-methyl-3′-pseudouridylyl.[64]
Composition
In addition to the mRNA molecule, the vaccine contains the following inactive ingredients (excipients):[67][68][8]
- ALC-0315, ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate)
- ALC-0159, 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide
- 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC)
- cholesterol
- dibasic sodium phosphate dihydrate
- monobasic potassium phosphate
- potassium chloride
- sodium chloride
- sucrose
- water for injection
The first four of these are lipids. The lipids and modRNA together form nanoparticles. ALC-0159 is a polyethylene glycol conjugate (that is, a PEGylated lipid).[69]
The vaccine is supplied in a multidose vial as “a white to off-white, sterile, preservative-free, frozen suspension for intramuscular injection“.[11][12] It must be thawed to room temperature and diluted with normal saline before administration.[12]
Authorizations
Expedited
The United Kingdom’s Medicines and Healthcare products Regulatory Agency (MHRA) gave the vaccine “rapid temporary regulatory approval to address significant public health issues such as a pandemic” on 2 December 2020, which it is permitted to do under the Medicines Act 1968.[70] It was the first COVID‑19 vaccine to be approved for national use after undergoing large scale trials,[71] and the first mRNA vaccine to be authorized for use in humans.[14][72] The United Kingdom thus became the first Western country to approve a COVID‑19 vaccine for national use,[73] although the decision to fast-track the vaccine was criticised by some experts.[74]
On 8 December 2020, Margaret “Maggie” Keenan, 90, from Fermanagh, became the first person to receive the vaccine.[75] In a notable example of museums documenting the pandemic, the vial and syringe used for that first dose were saved acquired by The Science Museum in London for its permanent collection.[76] By 20 December, 521,594 UK residents had received the vaccine as part of the national vaccination programme. 70% had been to people aged 80 or over.[77]
After the United Kingdom, the following countries expedited processes to approve the Pfizer–BioNTech COVID‑19 vaccine for use: Argentina,[78] Australia,[79] Bahrain,[80] Canada,[7][81] Chile,[82] Costa Rica,[83] Ecuador,[82] Hong Kong,[84] Iraq,[85] Israel,[86] Jordan,[87] Kuwait,[88] Mexico,[89] Oman,[90] Panama,[91] the Philippines,[92] Qatar,[93] Saudi Arabia,[32][94] Singapore,[95][96] the United Arab Emirates,[97] and the United States.[10]
The World Health Organization (WHO) authorized it for emergency use.[98]
In the United States, an emergency use authorization (EUA) is “a mechanism to facilitate the availability and use of medical countermeasures, including vaccines, during public health emergencies, such as the current COVID‑19 pandemic”, according to the FDA.[99] Following an EUA issuance, BioNTech and Pfizer are expected to continue the Phase III clinical trial to finalize safety and efficacy data, leading to application for licensure (approval) of the vaccine in the United States.[99][100][101] The United States Centers for Disease Control and Prevention (CDC) Advisory Committee on Immunization Practices (ACIP) approved recommendations for vaccination of those aged 16 years or older.[102][103]
Standard
On 19 December 2020, the Swiss Agency for Therapeutic Products (Swissmedic) approved the Pfizer–BioNTech COVID‑19 vaccine for regular use, two months after receiving the application, stating that the vaccine fully complied with the requirements of safety, efficacy and quality. This is the first authorization under a standard procedure.[1][104] On 23 December, a Lucerne resident, a 90-year-old woman, became the first person to receive the vaccine in Switzerland.[105] This marked the beginning of mass vaccination in continental Europe.[106]
On 21 December 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) recommended granting conditional marketing authorization for the Pfizer–BioNTech COVID‑19 vaccine under the brand name Comirnaty.[2][107][108] The recommendation was accepted by the European Commission the same day.[107][109]
On February 23, 2021, the Brazilian Health Regulatory Agency approved the Pfizer–BioNTech COVID-19 vaccine under its standard marketing authorization procedure. It became the first COVID-19 vaccine to receive definitive registration rather than emergency use authorization in the country.[110]
Adverse effects
The adverse effect profile of the Pfizer–BioNTech COVID‑19 vaccine is similar to that of other adult vaccines.[20] During clinical trials, the side effects deemed very common[a] are (in order of frequency): pain and swelling at the injection site, tiredness, headache, muscle aches, chills, joint pain, and fever.[68] Fever is more common after the second dose.[68] These effects are predictable and to be expected, and it is particularly important that people be aware of this to prevent vaccine hesitancy.[111]
Severe allergic reaction has been observed in approximately 11 cases per million doses of vaccine administered.[112][113] According to a report by the US Centers for Disease Control and Prevention 71% of those allergic reactions happened within 15 minutes of vaccination and mostly (81%) among people with a documented history of allergies or allergic reactions.[112] The UK’s Medicines and Healthcare products Regulatory Agency (MHRA) advised on 9 December 2020, that people who have a history of “significant” allergic reaction should not receive the Pfizer–BioNTech COVID‑19 vaccine.[114][115][116] On 12 December, the Canadian regulator followed suit, noting that: “Both individuals in the U.K. had a history of severe allergic reactions and carried adrenaline auto injectors. They both were treated and have recovered.”[67]
On 28 January 2021, the European Union published a COVID-19 vaccine safety update which found that “the benefits of Comirnaty in preventing COVID‑19 continue to outweigh any risks, and there are no recommended changes regarding the use the vaccine.”[113][117] No new side effects were identified.[113]
Manufacturing
_E.jpg)
A doctor holding the Pfizer vaccine
Pfizer and BioNTech are manufacturing the vaccine in their own facilities in the United States and in Europe in a three-stage process. The first stage involves the molecular cloning of DNA plasmids that code for the spike protein by infusing them into Escherichia coli bacteria. In the United States, this stage is conducted at a small pilot plant in Chesterfield, Missouri[118] (near St. Louis). After four days of growth, the bacteria are killed and broken open, and the contents of their cells are purified over a week and a half to recover the desired DNA product. The DNA is stored in tiny bottles and frozen for shipment. Safely and quickly transporting the DNA at this stage is so important that Pfizer has used its company jet and helicopter to assist.[119]
The second stage is being conducted at plants in Andover, Massachusetts[120] in the United States, and in Germany. The DNA is used as a template to build the desired mRNA strands. Once the mRNA has been created and purified, it is frozen in plastic bags about the size of a large shopping bag, of which each can hold up to 5 to 10 million doses. The bags are placed on special racks on trucks which take them to the next plant.[119]
The third stage is being conducted at plants in Portage, Michigan[121] (near Kalamazoo) in the United States, and Puurs in Belgium. This stage involves combining the mRNA with lipid nanoparticles, then filling vials, boxing vials, and freezing them.[119] Croda International subsidiary Avanti Polar Lipids is providing the requisite lipids.[122] As of November 2020, the major bottleneck in the manufacturing process was combining mRNA with lipid nanoparticles.[119]
In February 2021, Pfizer revealed this entire sequence initially took about 110 days on average from start to finish, and that the company was making progress on reducing that number to 60 days.[123] Vaccine manufacturers normally take several years to optimize the process of making a particular vaccine for speed and cost-effectiveness before attempting large-scale production.[123] Due to the urgency presented by the COVID-19 pandemic, Pfizer began production immediately with the process by which the vaccine had been originally formulated in the laboratory, then started to identify ways to safely speed up and scale up that process.[123]
BioNTech announced in September 2020 that it had signed an agreement to acquire from Novartis a manufacturing facility in Marburg, Germany, to expand their vaccine production capacity.[124] Once fully operational, the facility would produce up to 750 million doses per year, or over 60 million doses per month. The site will be the third BioNTech facility in Europe which currently produces the vaccine, while Pfizer operates at least four production sites in the United States and Europe.
Advance orders and logistics
Pfizer indicated in its 9 November press release that 50 million doses could be available by the end of 2020, with about 1.3 billion doses provided globally by 2021.[20] In February 2021, BioNTech announced it would increase production by more than 50% to manufacture two billion doses in 2021.[125]
In July 2020, the vaccine development program Operation Warp Speed placed an advance order of US$1.95 billion with Pfizer to manufacture 100 million doses of a COVID‑19 vaccine for use in the United States if the vaccine was shown to be safe and effective.[34][126][127][128] By mid-December 2020, Pfizer had agreements to supply 300 million doses to the European Union,[129] 120 million doses to Japan,[130] 40 million doses (10 million before 2021) to the United Kingdom,[22] 20 million doses to Canada,[131] an unspecified number of doses to Singapore,[132] and 34.4 million doses to Mexico.[133] Fosun also has agreements to supply 10 million doses to Hong Kong and Macau.[134] The Hong Kong government said it would receive its first batch of one million doses by the first quarter of 2021.[135]
BioNTech and Fosun agreed to supply Mainland China with a batch of 100 million doses in 2021, subject to regulatory approval. The initial supply will be delivered from BioNTech’s production facilities in Germany.[136]
The vaccine is being delivered in vials that, once diluted, contain 2.25 ml of vaccine (0.45 ml frozen plus 1.8ml diluent).[101] According to the vial labels, each vial contains five 0.3 ml doses, however excess vaccine may be used for one, or possibly two, additional doses.[101][137] The use of low dead space syringes to obtain the additional doses is preferable, and partial doses within a vial should be discarded.[101][138] The Italian Medicines Agency officially authorized the use of excess doses remaining within single vials.[139] As of 8 January 2021, each vial contains six doses.[68][140][141][138] In the United States, vials will be counted as five doses when accompanied by regular syringes and as six doses when accompanied by low dead space syringes.[142]
.jpg)
Temperature the Pfizer vaccine must be kept at to ensure effectiveness, roughly between −80 and −60 °C (−112 and −76 °F)
Logistics in developing countries which have preorder agreements with Pfizer—such as Ecuador and Peru—remain unclear.[38] Even high-income countries have limited cold chain capacity for ultracold transport and storage of a vaccine that degrades within five days when thawed, and requires two shots three weeks apart.[38] The vaccine needs to be stored and transported at ultracold temperatures between −80 and −60 °C (−112 and −76 °F),[39][22][38][143][144] much lower than for the similar Moderna vaccine. The head of Indonesia‘s Bio Farma Honesti Basyir stated that purchasing the vaccine is out of the question for the world’s fourth-most populous country, given that it did not have the necessary cold chain capability. Similarly, India’s existing cold chain network can only handle temperatures between 2 and 8 °C (36 and 46 °F), far above the requirements of the vaccine.[145][146]
In January 2021, Pfizer and BioNTech offered to supply 50 million doses of COVID‑19 vaccine for health workers across Africa between March and the end of 2021, at a discounted price of US$10 per dose.[147]
Name
BNT162b2 was the code name during development and testing,[17][148] tozinameran is the proposed international nonproprietary name (pINN),[149] and Comirnaty is the brand name.[1][2] According to BioNTech, the name Comirnaty “represents a combination of the terms COVID‑19, mRNA, community, and immunity.”[150][151]
The vaccine also has the common name “COVID‑19 mRNA vaccine (nucleoside-modified)”[2] and may be distributed in packaging with the name Pfizer–BioNTech COVID‑19 Vaccine.”[152]
How the Pfizer-BioNTech Vaccine Works
By Jonathan Corum and Carl ZimmerUpdated Jan. 21, 2021Leer en español

The German company BioNTech partnered with Pfizer to develop and test a coronavirus vaccine known as BNT162b2, the generic name tozinameran or the brand name Comirnaty. A clinical trial demonstrated that the vaccine has an efficacy rate of 95 percent in preventing Covid-19.
A Piece of the Coronavirus
The SARS-CoV-2 virus is studded with proteins that it uses to enter human cells. These so-called spike proteins make a tempting target for potential vaccines and treatments.

Spikes
Spike
protein
gene
CORONAVIRUS
Like the Moderna vaccine, the Pfizer-BioNTech vaccine is based on the virus’s genetic instructions for building the spike protein.
mRNA Inside an Oily Shell
The vaccine uses messenger RNA, genetic material that our cells read to make proteins. The molecule — called mRNA for short — is fragile and would be chopped to pieces by our natural enzymes if it were injected directly into the body. To protect their vaccine, Pfizer and BioNTech wrap the mRNA in oily bubbles made of lipid nanoparticles.

Lipid nanoparticles
surrounding mRNA
Because of their fragility, the mRNA molecules will quickly fall apart at room temperature. Pfizer is building special containers with dry ice, thermal sensors and GPS trackers to ensure the vaccines can be transported at –94°F (–70°C) to stay viable.
Entering a Cell
After injection, the vaccine particles bump into cells and fuse to them, releasing mRNA. The cell’s molecules read its sequence and build spike proteins. The mRNA from the vaccine is eventually destroyed by the cell, leaving no permanent trace.

VACCINE
PARTICLES
VACCINATED
CELL
Spike
protein
mRNA
Translating mRNA
Three spike
proteins combine
Spike
Cell
nucleus
Spikes
and protein
fragments
Displaying
spike protein
fragments
Protruding
spikes
Some of the spike proteins form spikes that migrate to the surface of the cell and stick out their tips. The vaccinated cells also break up some of the proteins into fragments, which they present on their surface. These protruding spikes and spike protein fragments can then be recognized by the immune system.
Spotting the Intruder
When a vaccinated cell dies, the debris will contain many spike proteins and protein fragments, which can then be taken up by a type of immune cell called an antigen-presenting cell.

Debris from
a dead cell
Engulfing
a spike
ANTIGEN-
PRESENTING
CELL
Digesting
the proteins
Presenting a
spike protein
fragment
HELPER
T CELL
The cell presents fragments of the spike protein on its surface. When other cells called helper T cells detect these fragments, the helper T cells can raise the alarm and help marshal other immune cells to fight the infection.
Making Antibodies
Other immune cells, called B cells, may bump into the coronavirus spikes on the surface of vaccinated cells, or free-floating spike protein fragments. A few of the B cells may be able to lock onto the spike proteins. If these B cells are then activated by helper T cells, they will start to proliferate and pour out antibodies that target the spike protein.

HELPER
T CELL
Activating
the B cell
Matching
surface proteins
VACCINATED
CELL
B CELL
SECRETED
ANTIBODIES
Stopping the Virus
The antibodies can latch onto coronavirus spikes, mark the virus for destruction and prevent infection by blocking the spikes from attaching to other cells.

ANTIBODIES
VIRUS
Killing Infected Cells
The antigen-presenting cells can also activate another type of immune cell called a killer T cell to seek out and destroy any coronavirus-infected cells that display the spike protein fragments on their surfaces.

ANTIGEN-PRESENTING CELL Presenting a spike protein fragment ACTIVATED KILLER T CELL INFECTED CELL Beginning to kill the infected cell
Remembering the Virus
The Pfizer-BioNTech vaccine requires two injections, given 21 days apart, to prime the immune system well enough to fight off the coronavirus. But because the vaccine is so new, researchers don’t know how long its protection might last.

First dose, 0.3ml
Second dose, 21 days later
A preliminary study found that the vaccine seems to offer strong protection about 10 days after the first dose, compared with people taking a placebo:

Cumulative incidence of Covid-19 among clinical trial participants 2.5% 2.0 People taking a placebo
1.5 1.0 Second dose First dose People taking the
Pfizer-BioNTech vaccine
0.5
0
1
2
3
4
8
12
16
Weeks after the first dose
It’s possible that in the months after vaccination, the number of antibodies and killer T cells will drop. But the immune system also contains special cells called memory B cells and memory T cells that might retain information about the coronavirus for years or even decades.
For more about the vaccine, see Pfizer’s Covid Vaccine: 11 Things You Need to Know.
Preparation and Injection
Each vial of the vaccine contains 5 doses of 0.3 milliliters. The vaccine must be thawed before injection and diluted with saline. After dilution the vial must be used within six hours.

A diluted vial of the vaccine at Royal Free Hospital in London.Jack Hill/Agence France-Presse
References
- ^ Jump up to:a b According to the British National Formulary and MedDRA conventions, side effects are “very common” when they occur in more than 1 in 10 instances; “common”, 1 in 100 to 1 in 10; “uncommon”, 1 in 1,000 to 1 in 100; “rare”, 1 in 10,000 to 1 in 1,000; and “very rare” when they occur in less than 1 in 10,000 instances.[27]
- ^ Jump up to:a b c d “Swissmedic grants authorisation for the first COVID-19 vaccine in Switzerland”(Press release). Swiss Agency for Therapeutic Products (Swissmedic). 19 December 2020. Retrieved 19 December 2020.
- ^ Jump up to:a b c d e “Comirnaty EPAR”. European Medicines Agency (EMA). Retrieved 23 December 2020.
- ^ “Comirnaty”. Therapeutic Goods Administration (TGA). Retrieved 25 January 2021.
- ^ “Comirnaty (BNT162b2 [mRNA]) COVID‑19 Vaccine Product Information” (PDF). Therapeutic Goods Administration (TGA). Retrieved 25 January 2021.
- ^ Australian Public Assessment Report for BNT162b2 (mRNA) (PDF) (Report). Therapeutic Goods Administration (TGA). Retrieved 25 January 2021.
- ^ Jump up to:a b “Regulatory Decision Summary – Pfizer-BioNTech COVID-19 Vaccine”. Health Canada. 9 December 2020. Archived from the original on 9 December 2020. Retrieved 9 December 2020.
- ^ Jump up to:a b “Pfizer-BioNTech COVID-19 Vaccine (tozinameran)”. Health Canada. Retrieved 15 December 2020.
- ^ Jump up to:a b “Information for Healthcare Professionals on Pfizer/BioNTech COVID-19 vaccine”. Medicines and Healthcare products Regulatory Agency (MHRA). 10 December 2020. Retrieved 21 December 2020.
- ^ “Conditions of Authorisation for Pfizer/BioNTech COVID-19 vaccine”. Medicines and Healthcare products Regulatory Agency (MHRA). 31 December 2020. Retrieved 8 January2021.
- ^ Jump up to:a b “FDA Takes Key Action in Fight Against COVID-19 By Issuing Emergency Use Authorization for First COVID-19 Vaccine” (Press release). U.S. Food and Drug Administration (FDA). 11 December 2020. Retrieved 11 December 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c “Pfizer-BioNTech COVID-19 Vaccine- rna ingredient bnt-162b2 injection, suspension”. DailyMed. Retrieved 14 December 2020.
- ^ Jump up to:a b c Pfizer-BioNTech COVID-19 Vaccine Emergency Use Authorization Review Memorandum (PDF). U.S. Food and Drug Administration (FDA) (Report). 14 December 2020. Retrieved 14 December 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Comirnaty EPAR”. European Medicines Agency (EMA). Retrieved 23 December 2020.
- ^ Jump up to:a b “UK medicines regulator gives approval for first UK COVID-19 vaccine” (Press release). Medicines and Healthcare products Regulatory Agency (MHRA). 2 December 2020. Retrieved 2 December 2020.
- ^ Boseley S, Halliday J (2 December 2020). “UK approves Pfizer/BioNTech Covid vaccine for rollout next week”. The Guardian. Retrieved 14 December 2020.
- ^ “Swissmedic grants authorisation for the first COVID-19 vaccine in Switzerland” (Press release). Swiss Agency for Therapeutic Products (Swissmedic). 19 December 2020. Retrieved 19 December 2020.
- ^ Jump up to:a b c d e Walsh EE, Frenck RW, Falsey AR, Kitchin N, Absalon J, Gurtman A, et al. (October 2020). “Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates”. The New England Journal of Medicine. 383 (25): 2439–50. doi:10.1056/NEJMoa2027906. PMC 7583697. PMID 33053279.
- ^ Jump up to:a b c d e f Clinical trial number NCT04368728 for “NCT04368728: Study to Describe the Safety, Tolerability, Immunogenicity, and Efficacy of RNA Vaccine Candidates Against COVID-19 in Healthy Individuals” at ClinicalTrials.gov
- ^ Jump up to:a b Palca J (9 November 2020). “Pfizer says experimental COVID-19 vaccine is more than 90% effective”. NPR. Archived from the original on 9 November 2020. Retrieved 9 November 2020.
- ^ Jump up to:a b c d e f g h Herper M (9 November 2020). “Covid-19 vaccine from Pfizer and BioNTech is strongly effective, early data from large trial indicate”. STAT. Archived from the original on 9 November 2020. Retrieved 9 November 2020.
- ^ Jump up to:a b Edwards E (9 November 2020). “Pfizer’s Covid-19 vaccine promising, but many questions remain”. NBC News. Archived from the original on 22 November 2020. Retrieved 12 November 2020.
- ^ Jump up to:a b c d Gallagher J (9 November 2020). “Covid vaccine: First ‘milestone’ vaccine offers 90% protection”. BBC News. Archived from the original on 26 November 2020. Retrieved 9 November 2020.
- ^ “CureVac Initiates Rolling Submission With European Medicines Agency for COVID-19 Vaccine Candidate, CVnCoV”. CureVac (Press release).
- ^ Jump up to:a b c “Update on our COVID-19 vaccine development program with BNT162b2” (PDF)(Press release). BioNTech. 2 December 2020. Retrieved 12 December 2020.
- ^ Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. (December 2020). “Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine”. N Engl J Med. 383 (27): 2603–2615. doi:10.1056/NEJMoa2034577. PMC 7745181. PMID 33301246.
- ^ “Questions and Answers About Pfizer-BioNTech COVID-19 Vaccine”. Pfizer. Retrieved 16 December 2020.
- ^ “Adverse reactions to drugs”. British National Formulary. Retrieved 19 December 2020.
- ^ Jump up to:a b “Coronavirus vaccine”. National Health Service. 7 December 2020. Archived from the original on 7 December 2020. Retrieved 7 December 2020.
- ^ Commissioner, Office of the (3 February 2021). “Pfizer-BioNTech COVID-19 Vaccine”. FDA.
- ^ “EMA recommends first COVID-19 vaccine for authorisation in the EU”. European Medicines Agency.
- ^ “Bahrain becomes second country to approve Pfizer COVID-19 vaccine”. Al Jazeera. Archived from the original on 4 December 2020. Retrieved 5 December 2020.
* “Coronavirus: Saudi Arabia approves Pfizer COVID-19 vaccine for use”. Al Arabiya English. 10 December 2020. Archived from the original on 11 December 2020. Retrieved 10 December 2020.
* Solomon DB, Torres N (11 December 2020). “Mexico approves emergency use of Pfizer’s COVID-19 vaccine”. Reuters. Retrieved 12 December 2020.
* Thomas K (20 November 2020). “F.D.A. Clears Pfizer Vaccine, and Millions of Doses Will Be Shipped Right Away”. The New York Times. Archived from the original on 12 December 2020. Retrieved 11 December 2020.
* “First shipments of Pfizer-BioNTech vaccine in Singapore by end-Dec; enough vaccines for all by Q3 2021”. The Straits Times. 14 December 2020. Retrieved 14 December 2020. - ^ Jump up to:a b Al Mulla Y (13 December 2020). “Kuwait approves emergency use of Pfizer vaccine”. Gulf News. Retrieved 14 December 2020.
- ^ Browne R (11 November 2020). “What you need to know about BioNTech – the European company behind Pfizer’s Covid-19 vaccine”. CNBC. Retrieved 14 January 2021.
- ^ Jump up to:a b Thomas K, Gelles D, Zimmer C (9 November 2020). “Pfizer’s early data shows vaccine is more than 90% effective”. The New York Times. Archived from the original on 23 November 2020. Retrieved 9 November 2020.
- ^ Jump up to:a b c Burger L (15 March 2020). “BioNTech in China alliance with Fosun over coronavirus vaccine candidate”. Reuters. Archived from the original on 14 November 2020. Retrieved 10 November 2020.
- ^ “Pfizer and BioNTech Celebrate Historic First Authorization in the U.S. of Vaccine to Prevent COVID-19”. Pfizer Inc. and BioNTech SE.
- ^ “Securing Singapore’s access to COVID-19 vaccines”. gov.sg. Government of Singapore. 14 December 2020. Retrieved 1 February 2021.
- ^ Jump up to:a b c d e “Deep-freeze hurdle makes Pfizer’s vaccine one for the rich”. Bloomberg. 10 November 2020. Archived from the original on 22 November 2020. Retrieved 12 November 2020.
Vaccine goes bad five days after thawing, requires two shots; Many nations face costly ramp up of cold-chain infrastructure
- ^ Jump up to:a b c “Pfizer-BioNTech COVID-19 Vaccine Vaccination Storage & Dry Ice Safety Handling”. Pfizer. Retrieved 17 December 2020.
- ^ “Information for Healthcare Professionals on Pfizer/BioNTech COVID-19 vaccine”. Government of the United Kingdom. Retrieved 29 January 2021.
- ^ “Recommendation for an Emergency Use Listing of Tozinameran (Covid-19 Mrna Vaccine (Nucleoside Modified)) Submitted by Biontech Manufacturing Gmbh” (PDF). World Health Organization. 26 January 2021.
- ^ “Australian Product Information – Comirnaty (BNT162b2 [mRNA]) COVID-19 Vaccine”(PDF). Therapeutic Goods Administration. Australian Government.
- ^ “Pfizer and BioNTech Submit COVID-19 Vaccine Stability Data at Standard Freezer Temperature to the U.S. FDA”. Pfizer (Press release). 19 February 2021. Retrieved 19 February 2021.
- ^ Gates B (30 April 2020). “The vaccine race explained: What you need to know about the COVID-19 vaccine”. The Gates Notes. Archived from the original on 14 May 2020. Retrieved 2 May 2020.
- ^ “World Health Organization timeline – COVID-19”. World Health Organization. 27 April 2020. Archived from the original on 29 April 2020. Retrieved 2 May 2020.
- ^ Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. (December 2020). “Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine”. The New England Journal of Medicine. 383 (27): 2603–2615. doi:10.1056/NEJMoa2034577. PMC 7745181. PMID 33301246.
development of BNT162b2 was initiated on January 10, 2020, when the SARS-CoV-2 genetic sequence was released by the Chinese Center for Disease Control and Prevention and disseminated globally by the GISAID (Global Initiative on Sharing All Influenza Data) initiative
- ^ Bohn MK, Mancini N, Loh TP, Wang CB, Grimmler M, Gramegna M, et al. (October 2020). “IFCC Interim Guidelines on Molecular Testing of SARS-CoV-2 Infection”. Clinical Chemistry and Laboratory Medicine. 58 (12): 1993–2000. doi:10.1515/cclm-2020-1412. PMID 33027042.
- ^ “CEPI’s collaborative task force to assess COVID-19 vaccines on emerging viral strains”. BioSpectrum – Asia Edition. 23 November 2020.
the first SARS-CoV-2 viral genomes were shared via GISAID on 10 January 2020
- ^ Thanh Le T, Andreadakis Z, Kumar A, Gómez Román R, Tollefsen S, Saville M, Mayhew S (May 2020). “The COVID-19 vaccine development landscape”. Nature Reviews. Drug Discovery. 19 (5): 305–306. doi:10.1038/d41573-020-00073-5. PMID 32273591.
- ^ Fauci AS, Lane HC, Redfield RR (March 2020). “Covid-19 – Navigating the Uncharted”. The New England Journal of Medicine. 382 (13): 1268–1269. doi:10.1056/nejme2002387. PMC 7121221. PMID 32109011.
- ^ Papadopoulos C (14 December 2020). “Chronologie – So entstand der Corona-Impfstoff von Biontech” [Chronology – That’s how the Covid-vaccine of Biontech was being developed] (in German). Südwestrundfunk. Retrieved 20 December 2020.
- ^ 《Fosun Pharma and BioNTech form COVID‑19 vaccine strategic alliance in China》(Fosun Phrama News Content , 15 March 2020) Archived 15 August 2020 at the Wayback Machine
- ^ “Germany: Investment Plan for Europe – EIB to provide BioNTech with up to €100 million in debt financing for COVID-19 vaccine development and manufacturing”. European Investment Bank. 11 June 2020. Archived from the original on 9 November 2020. Retrieved 10 November 2020.
- ^ “BioNTech gets $445 million in German funding for vaccine”. Bloomberg L.P. 15 September 2020. Archived from the original on 9 November 2020. Retrieved 10 November 2020.
- ^ “Pfizer CEO says he would’ve released vaccine data before election if possible”. Axios. 9 November 2020. Archived from the original on 10 November 2020. Retrieved 11 November 2020.
- ^ Jump up to:a b Mulligan MJ, Lyke KE, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. (October 2020). “Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults”. Nature. 586(7830): 589–593. Bibcode:2020Natur.586..589M. doi:10.1038/s41586-020-2639-4. PMID 32785213. S2CID 221126922.
- ^ Hannah B (7 October 2020). “EMA begins rolling review of BNT162b2 COVID-19 vaccine”. European Pharmaceutical Review. Archived from the original on 11 November 2020. Retrieved 11 November 2020.
- ^ Jump up to:a b “EMA Assessment Report” (PDF). Europa (web portal). 21 December 2020. Retrieved 29 December 2020.
- ^ “Clarification of Comirnaty dosage interval”. European Medicines Agency (EMA). 28 January 2021. Retrieved 28 January 2021.
- ^ “Study to Evaluate the Safety, Tolerability, and Immunogenicity of SARS CoV-2 RNA Vaccine Candidate (BNT162b2) Against COVID-19 in Healthy Pregnant Women 18 Years of Age and Older”. ClinicalTrials.gov. Retrieved 21 February 2021.
- ^ “Pfizer and BioNTech Commence Global Clinical Trial to Evaluate COVID-19 Vaccine in Pregnant Women”. pfizer.com (Press release). 18 February 2021. Retrieved 21 February2021.
- ^ Jump up to:a b Gaebler C, Nussenzweig MC (October 2020). “All eyes on a hurdle race for a SARS-CoV-2 vaccine”. Nature. 586 (7830): 501–2. Bibcode:2020Natur.586..501G. doi:10.1038/d41586-020-02926-w. PMID 33077943. S2CID 224808629.
- ^ Jump up to:a b “China’s Fosun to end BioNTech’s COVID-19 vaccine trial, seek approval for another”. Reuters. 3 November 2020. Archived from the original on 12 December 2020. Retrieved 21 November 2020.
- ^ Jump up to:a b c World Health Organization. “Messenger RNA encoding the full-length SARS-CoV-2 spike glycoprotein” (DOC). WHO MedNet. Retrieved 16 December 2020.
- ^ Pallesen J, Wang N, Corbett KS, Wrapp D, Kirchdoerfer RN, Turner HL, et al. (August 2017). “Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen”. Proceedings of the National Academy of Sciences of the United States of America. 114 (35): E7348–E7357. doi:10.1073/pnas.1707304114. PMC 5584442. PMID 28807998.
- ^ Orlandini von Niessen AG, Poleganov MA, Rechner C, Plaschke A, Kranz LM, Fesser S, et al. (April 2019). “Improving mRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3′ UTRs Identified by Cellular Library Screening”. Molecular Therapy. 27 (4): 824–836. doi:10.1016/j.ymthe.2018.12.011. PMC 6453560. PMID 30638957.
- ^ Jump up to:a b “Pfizer-BioNTech COVID-19 vaccine: Health Canada recommendations for people with serious allergies”. Health Canada. 12 December 2020.
- ^ Jump up to:a b c d Comirnaty: Product Information (PDF) (Report). European Medicines Agency(EMA). Retrieved 23 December 2020.
- ^ Public Assessment Report Authorisation for Temporary Supply COVID-19 mRNA Vaccine BNT162b2 (BNT162b2 RNA) concentrate for solution for injection (PDF). Regulation 174(Report). Medicines and Healthcare products Regulatory Agency (MHRA). 15 December 2020.
- ^ “UK medicines regulator gives approval for first UK COVID-19 vaccine”. Medicines and Healthcare products Regulatory Agency (MHRA). 2 December 2020. Archived from the original on 2 December 2020. Retrieved 2 December 2020.
- ^ Neergaard L, Kirka D (2 December 2020). “Britain OKs Pfizer vaccine and will begin shots within days”. Associated Press. Archived from the original on 6 December 2020. Retrieved 6 December 2020.
- ^ Mueller B (2 December 2020). “U.K. Approves Pfizer Coronavirus Vaccine, a First in the West”. The New York Times. Retrieved 2 December 2020.
- ^ Roberts M (2 December 2020). “Covid Pfizer vaccine approved for use next week in UK”. BBC News. Archived from the original on 2 December 2020. Retrieved 2 December 2020.
- ^ Henley J, Connolly, Jones S (3 December 2020). “European and US experts question UK’s fast-track of Covid vaccine”. The Guardian. Archived from the original on 9 December 2020. Retrieved 9 December 2020.
- ^ “First patient receives Pfizer Covid-19 vaccine”. BBC. 8 December 2020. Archivedfrom the original on 8 December 2020. Retrieved 8 December 2020.
- ^ “Vaccine vials and a virtual hug: a history of coronavirus in 15 objects”. The Guardian. 21 February 2021. Retrieved 22 February 2021.
- ^ “COVID-19 Vaccination Statistics –Week ending Sunday 20th December 2020” (PDF). NHS. 24 December 2020.
- ^ “Coronavirus en la Argentina: La ANMAT aprobo el uso de emergencia de la vacuna Pfizer”. La Nación (in Spanish). Retrieved 25 December 2020.
- ^ “TGA provisionally approves Pfizer COVID-19 vaccine”. Therapeutic Goods Administration (Press release). 25 January 2021. Retrieved 26 January 2021.
- ^ “Bahrain becomes second country to approve Pfizer COVID-19 vaccine”. Al Jazeera. Retrieved 5 December 2020.
- ^ “Drug and vaccine authorizations for COVID-19: List of applications received”. Health Canada. 9 December 2020. Retrieved 9 December 2020.
- ^ Jump up to:a b “Chile y Ecuador se adelantan en Sudamérica y autorizan la vacuna de Pfizer”. El Pais. Retrieved 17 December 2020.
- ^ “First Pfizer COVID-19 vaccines set to reach Costa Rica on Wednesday – president”. Reuters. 23 December 2020. Retrieved 24 December 2020.
- ^ “SFH authorises COVID-19 vaccine by Fosun Pharma/BioNTech for emergency use in Hong Kong”. The Government of Hong Kong (Press release). 25 January 2021. Retrieved 26 January 2021.
- ^ “Iraq grants emergency approval for Pfizer COVID-19 vaccine”. MSN. Retrieved 27 December 2020.
- ^ “Israeli Health Minister ‘pleased’ as FDA approves Pfizer COVID-19 vaccine”. The Jerusalem Post. Retrieved 28 December 2020.
- ^ “Jordan approves Pfizer-BioNTech Covid vaccine”. France 24. 15 December 2020. Retrieved 15 December 2020.
- ^ “Kuwait authorizes emergency use of Pfizer-BioNTech COVID-19 vaccine”. Arab News. 13 December 2020. Retrieved 15 December 2020.
- ^ “Mexico Approves Pfizer Vaccine for Emergency Use as Covid Surges”. Bloomberg. 12 December 2020. Retrieved 12 December 2020.
- ^ “Oman issues licence to import Pfizer BioNTech Covid vaccine – TV”. Reuters. 15 December 2020. Retrieved 16 December 2020.
- ^ “Panama approves Pfizer’s COVID-19 vaccine – health ministry”. Yahoo! Finance. Retrieved 16 December 2020.
- ^ “PH authorizes Pfizer’s COVID-19 vaccine for emergency use”. CNN Philippines. 14 January 2021.
- ^ “Qatar, Oman to receive Pfizer-BioNTech COVID-19 vaccine this week”. Reuters. Retrieved 24 December 2020.
- ^ “Saudi Arabia to Launch Its Coronavirus Vaccination Program” (in Spanish). Boomberg. Retrieved 17 December 2020.
- ^ Abdullah Z (14 December 2020). “Pfizer-BioNTech COVID-19 vaccine approved by Singapore, first shipment expected by end-December”. CNA. Retrieved 16 January 2021.
- ^ “Singapore approves use of Pfizer’s COVID-19 vaccine”. AP News. 14 December 2020. Retrieved 15 December 2020.
- ^ “Dubai approves the Pfizer-BioNTech vaccine which will be free of charge”. Emirates Woman. 23 December 2020. Retrieved 28 December 2020.
- ^ “WHO issues its first emergency use validation for a COVID-19 vaccine and emphasizes need for equitable global access”. World Health Organization (WHO) (Press release). 31 December 2020. Retrieved 6 January 2021.
- ^ Jump up to:a b “Emergency Use Authorization for vaccines explained”. U.S. Food and Drug Administration (FDA). 20 November 2020. Archived from the original on 20 November 2020. Retrieved 20 November 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Pfizer-BioNTech COVID-19 Vaccine EUA Letter of Authorization” (PDF). U.S. Food and Drug Administration (FDA). 11 December 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d “Pfizer-BioNTech COVID-19 Vaccine EUA Fact Sheet for Healthcare Providers”(PDF). Pfizer. 11 December 2020.
- ^ Sun LH, Stanley-Becker I. “CDC greenlights advisory group’s decision to recommend Pfizer vaccine for use”. The Washington Post. Retrieved 14 December 2020.
- ^ Oliver SE, Gargano JW, Marin M, Wallace M, Curran KG, Chamberland M, et al. (December 2020). “The Advisory Committee on Immunization Practices’ Interim Recommendation for Use of Pfizer-BioNTech COVID-19 Vaccine — United States, December 2020” (PDF). MMWR. Morbidity and Mortality Weekly Report. 69 (50): 1922–24. doi:10.15585/mmwr.mm6950e2. PMC 7745957. PMID 33332292.
- ^ “COVID-19: Switzerland can start vaccinating vulnerable groups already in December”(Press release). Federal Office of Public Health. 19 December 2020. Retrieved 19 December 2020.
- ^ Erni S (23 December 2020). “90-jährige Luzernerin als erste Person in der Schweiz gegen Corona geimpft”. Neue Luzerner Zeitung. Retrieved 23 December 2020.
- ^ Pralong J (23 December 2020). “La piqûre de l’espoir pratiquée à Lucerne”. Heidi.news. Retrieved 23 December 2020.
- ^ Jump up to:a b “EMA recommends first COVID-19 vaccine for authorisation in the EU”. European Medicines Agency (EMA) (Press release). 21 December 2020. Retrieved 21 December2020.
- ^ “Comirnaty”. Union Register of medicinal products. Retrieved 8 January 2021.
- ^ “Statement by President von der Leyen on the marketing authorisation of the BioNTech-Pfizer vaccine against COVID-19”. European Commission. Retrieved 21 December 2020.
- ^ Cancian, Natália (23 February 2021). “Anvisa aprova registro da vacina da Pfizer contra Covid”. Folha de S. Paulo (in Portuguese). Retrieved 23 February 2021.
- ^ McKenna M (17 December 2020). “Vaccines Are Here. We Have to Talk About Side Effects”. Wired. Retrieved 23 December 2020.
- ^ Jump up to:a b CDC COVID-19 Response Team, Food and Drug Administration (January 2021). “Allergic Reactions Including Anaphylaxis After Receipt of the First Dose of Pfizer-BioNTech COVID-19 Vaccine — United States, December 14–23, 2020” (PDF). MMWR. Morbidity and Mortality Weekly Report. 70 (2): 46–51. doi:10.15585/mmwr.mm7002e1. PMC 7808711. PMID 33444297.
- ^ Jump up to:a b c “COVID-19 vaccine safety update: COMIRNATY” (PDF). European Medicines Agency. 28 January 2021.
- ^ Bostock N (9 December 2020). “MHRA warning after allergic reactions in NHS staff given COVID-19 vaccine”. GP. Archived from the original on 9 December 2020. Retrieved 9 December 2020.
- ^ Booth W, Cunningham E (9 December 2020). “Britain warns against Pfizer vaccine for people with history of ‘significant’ allergic reactions”. The Washington Post. Archivedfrom the original on 9 December 2020. Retrieved 9 December 2020.
- ^ Cabanillas B, Akdis C, Novak N (December 2020). “Allergic reactions to the first COVID-19 vaccine: a potential role of Polyethylene glycol?”. Allergy. doi:10.1111/all.14711. PMID 33320974. S2CID 229284320.
- ^ “First COVID-19 vaccine safety update published”. European Medicines Agency (EMA)(Press release). 28 January 2021. Retrieved 29 January 2021.
- ^ Gray B (23 November 2020). “Pfizer’s Chesterfield workforce playing a key role in coronavirus vaccine development”. St. Louis Post-Dispatch.
- ^ Jump up to:a b c d Johnson CY (17 November 2020). “A vial, a vaccine and hopes for slowing a pandemic — how a shot comes to be”. The Washington Post. Retrieved 21 December2020.
- ^ Hughes M (20 December 2020). “Andover’s piece of the vaccine: Pfizer”. The Eagle-Tribune.
- ^ Shamus KJ (13 December 2020). “Historic journey: Pfizer prepares to deliver 6.4 million doses of COVID-19 vaccines”. Detroit Free Press.
- ^ Mullin R (25 November 2020). “Pfizer, Moderna ready vaccine manufacturing networks”. Chemical & Engineering News. Washington, D.C.: American Chemical Society. Retrieved 21 December 2020.
- ^ Jump up to:a b c Weise, Elizabeth (7 February 2021). “Pfizer expects to cut COVID-19 vaccine production time by close to 50% as production ramps up, efficiencies increase”. USA Today.
- ^ “BioNTech to Acquire GMP Manufacturing Site to Expand COVID-19 Vaccine Production Capacity in First Half 2021 | BioNTech”. investors.biontech.de. Retrieved 5 February2021.
- ^ “Statement on Manufacturing | BioNTech”. investors.biontech.de. Retrieved 5 February2021.
- ^ Erman M, Ankur B (22 July 2020). “U.S. to pay Pfizer, BioNTech $1.95 bln for millions of COVID-19 vaccine doses”. Reuters. Archived from the original on 22 July 2020. Retrieved 22 July 2020.
- ^ “U.S. Government Engages Pfizer to Produce Millions of Doses of COVID-19 Vaccine”. US Department of Health and Human Services. 22 July 2020. Archived from the original on 22 July 2020. Retrieved 23 July 2020.
- ^ Nazaryan A (9 November 2020). “So is Pfizer part of Operation Warp Speed or not? Yes and no”. Yahoo!. Archived from the original on 10 November 2020. Retrieved 9 November 2020.
- ^ Pleitgen F (11 November 2020). “EU agrees to buy 300 million doses of the Pfizer/BioNTech Covid-19 vaccine”. CNN. Archived from the original on 24 November 2020. Retrieved 26 November 2020.
- ^ “Japan and Pfizer reach COVID-19 vaccine deal to treat 60 million people”. The Japan Times. 1 August 2020. Archived from the original on 10 November 2020. Retrieved 21 November 2020.
- ^ Tasker JP (9 November 2020). “Trudeau says promising new Pfizer vaccine could be ‘light at the end of the tunnel'”. CBC News. Archived from the original on 9 November 2020. Retrieved 9 November 2020.
- ^ “Pfizer and BioNTech to Supply Singapore with their BNT162b2 mRNA-based Vaccine Candidate to Combat COVID-19”. pfizer.com.sg. Pfizer Singapore. 14 December 2020. Retrieved 1 February 2021.
- ^ de Salud S. “233. Firma secretario de Salud convenio con Pfizer para fabricación y suministro de vacuna COVID-19”. gob.mx (in Spanish). Retrieved 17 December 2020.
- ^ Ng E (27 August 2020). “Fosun Pharma to supply Covid-19 vaccine to Hong Kong, Macau once approved”. South China Morning Post. Archived from the original on 20 November 2020. Retrieved 21 November 2020.
- ^ Ting V, Lau C, Wong O (11 December 2020). “Hong Kong buys 15 million Covid-19 vaccine doses from Sinovac, Pfizer”. South China Morning Post. Retrieved 18 December2020.
- ^ “BioNTech and Fosun Pharma to Supply China with mRNA-based COVID-19 Vaccine”(Press release). BioNTech. 16 December 2020. Retrieved 16 December 2020.
- ^ “Pfizer-BioNTech COVID-19 Vaccine Frequently Asked Questions”. U.S. Food and Drug Administration. 11 December 2020. Retrieved 29 December 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “Extra dose from vials of Comirnaty COVID-19 vaccine”. European Medicines Agency (EMA). 8 January 2021. Retrieved 8 January 2021.
- ^ “AIFA, possibile ottenere almeno 6 dosi da ogni flaconcino del vaccino BioNTech/Pfizer”. aifa.gov.it (in Italian). Retrieved 29 December 2020.
- ^ “Global information about Comirnaty”. Comirnaty IE. 8 January 2021. Retrieved 16 January 2021.
- ^ “Comirnaty Package Insert” (PDF). BioNTech Manufacturing GmbH.
- ^ Rowland C (22 January 2021). “Biden wants to squeeze an extra shot of vaccine out of every Pfizer vial. It won’t be easy”. The Washington Post. Retrieved 29 January 2021.
- ^ Kollewe J. “Pfizer and BioNTech’s vaccine poses global logistics challenge”. The Guardian. Archived from the original on 10 November 2020. Retrieved 10 November2020.
- ^ Newey S (8 September 2020). “Daunting task of distribution exposed as it emerges some vaccines must be ‘deep frozen’ at −70C”. The Telegraph. Archived from the original on 9 November 2020. Retrieved 10 November 2020.
- ^ “How China’s COVID-19 could fill the gaps left by Pfizer, Moderna, AstraZeneca”. Fortune. 5 December 2020. Archived from the original on 12 December 2020. Retrieved 5 December 2020.
- ^ “Pfizer’s Vaccine Is Out of the Question as Indonesia Lacks Refrigerators: State Pharma Boss”. Jakarta Globe. 22 November 2020. Archived from the original on 7 December 2020. Retrieved 5 December 2020.
- ^ “Pfizer Has Offered South Africa Discounted Covid-19 Vaccines”. Bloomberg. 4 January 2021. Retrieved 5 January 2021.
- ^ Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. (December 2020). “Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine”. N Engl J Med. 383 (27): 2603–2615. doi:10.1056/NEJMoa2034577. PMC 7745181. PMID 33301246.
- ^ World Health Organization (2020). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 124 – COVID-19 (special edition)”(PDF). WHO Drug Information. 34 (3): 666. Archived (PDF) from the original on 27 November 2020. Retrieved 23 November 2020.
- ^ “Pfizer and BioNTech Receive Authorization in the European Union for COVID-19 Vaccine” (Press release). BioNTech. 21 December 2020. Retrieved 26 December 2020 – via GlobeNewswire.
- ^ Bulik BS (23 December 2020). “The inside story behind Pfizer and BioNTech’s new vaccine brand name, Comirnaty”. FiercePharma. Retrieved 25 December 2020.
- ^ “Comirnaty COVID-19 mRNA Vaccine”. Comirnaty Global. Retrieved 31 December2020.
External links
“Tozinameran”. Drug Information Portal. U.S. National Library of Medicine.
- Global Information About Pfizer–BioNTech COVID‑19 Vaccine (also known as BNT162b2) Pfizer
- Comirnaty assessment report European Medicines Agency Committee for Medicinal Products for Human Use
- A Phase 1/2/3 Study to Evaluate the Safety, Tolerability, Immunogenicity, and Efficacy of RNA Vaccine Candidates Against COVID‑19 in Healthy Individuals Pfizer clinical protocol
- Pfizer Vaccince News, updates and tracking of Israel’s vaccinaion campaign
- “How the Pfizer-BioNTech Covid-19 Vaccine Works”. The New York Times.
| A vial of the Pfizer–BioNTech COVID‑19 vaccine | |
| Vaccine description | |
|---|---|
| Target disease | COVID‑19 |
| Type | mRNA |
| Clinical data | |
| Trade names | Comirnaty[1][2] |
| Other names | BNT162b2, COVID-19 mRNA vaccine (nucleoside-modified) |
| License data | EU EMA: by INNUS DailyMed: Pfizer-BioNTech_COVID-19_Vaccine |
| Pregnancy category | AU: B1[3] |
| Routes of administration | Intramuscular |
| ATC code | None |
| Legal status | |
| Legal status | AU: S4 (Prescription only) [4][5]CA: Authorized by interim order [6][7]UK: Conditional and temporary authorization to supply [8][9]US: Unapproved (Emergency Use Authorization)[10][11][12]EU: Conditional marketing authorization granted [2]CH: Rx-only[further explanation needed][1] |
| Identifiers | |
| CAS Number | 2417899-77-3 |
| PubChem SID | 434370509 |
| DrugBank | DB15696 |
| UNII | 5085ZFP6SJ |
| KEGG | D11971 |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| SARS-CoV-2 (virus)COVID-19 (disease) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 Portal |
/////////
#Tozinameran, #APPROVALS 2021, #JAPAN 2021, Comirnaty, #Coronavirus disease, #COVID-19, #BNT162b2 , #BNT162b2, #SARS-CoV-2 Vaccine, #RNA ingredient BNT-162B2, #corona
The Pfizer-BioNTech COVID-19 vaccine (Tozinameran, INN), also known as BNT162b2, is one of four advanced mRNA-based vaccines developed through “Project Lightspeed,” a joint program between Pfizer and BioNTech.2,3 Tozinameran is a nucleoside modified mRNA (modRNA) vaccine encoding an optimized full-length version of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike (S) protein. It is designed to induce immunity against SARS-CoV-2, the virus responsible for causing COVID-19.2 The modRNA is formulated in lipid nanoparticles for administration via intramuscular injection in two doses, three weeks apart.1,3
Tozinameran is undergoing evaluation in clinical trials in both the USA (NCT04368728) and Germany (NCT04380701).4,5 Tozinameran received fast track designation by the U.S. FDA on July 13, 2020.6 On December 11, 2020, the FDA issued an Emergency Use Authorization (EUA) based on 95% efficacy in clinical trials and a similar safety profile to other viral vaccines over a span of approximately 2 months.1 Tozinameran was granted an EUA in the UK on December 2, 2020,8 and in Canada on December 9, 20207 for active immunization against SARS-CoV-2.12
Currently, sufficient data are not available to determine the longevity of protection against COVID-19, nor direct evidence that the vaccine prevents the transmission of the SARS-CoV-2 virus from one individual to another.9 Fact sheets for caregivers, recipients, and healthcare providers are now available.10,11
Tozinameran has not yet been fully approved by any country. In both the UK and Canada, Tozinameran is indicated under an interim authorization for active immunization to prevent COVID-19 caused by SARS-CoV-2 in individuals aged 16 years and older.7,8
On December 11, 2020, the U.S. Food and Drug Administration granted emergency use authorization (EUA) for Tozinameran to prevent COVID-19 caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in patients aged 16 years and above.9 Safety and immune response information for adolescents 12-15 years of age will follow, and studies to further explore the administration of Tozinameran in pregnant women, children under 12 years of age, and those in special risk groups will be evaluated in the future.1
This vaccine should only be administered where appropriate medical treatment for immediate allergic reactions are immediately available in the case of an acute anaphylactic reaction after vaccine administration.12 Tozinameran administration should be postponed in any individual suffering from an acute febrile illness. Its use should be carefully considered in immunocompromised individuals and individuals with a bleeding disorder or on anticoagulant therapy. Appropriate medical treatment should be readily available in case of an anaphylactic reaction following vaccine administration.7,8
Tozinameran contains nucleoside modified mRNA (modRNA) encapsulated in lipid nanoparticles that deliver the modRNA into host cells. The lipid nanoparticle formulation facilitates the delivery of the RNA into human cells.12 Once inside these cells, the modRNA is translated by host machinery to produce the SARS-CoV-2 spike (S) protein antigen, which is subsequently recognized by the host immune system. Tozinameran has been shown to elicit both neutralizing antibody and cellular immune responses to the S protein, which helps protect against subsequent SARS-CoV-2 infection.7,8
Tozinameran is a nucleoside modified mRNA (modRNA) vaccine encoding an optimized full-length version of the SARS-CoV-2 spike (S) protein, translated and expressed in cells in vaccinated individuals to produce the S protein antigen against which an immune response is mounted. As with all vaccines, protection cannot be guaranteed in all recipients, and full protection may not occur until at least seven days following the second dose.7,8
In U.S. clinical trials, the vaccine was 95% effective in preventing COVID-19; eight COVID-19 cases occurred in the vaccine group and 162 cases occurred in the placebo group. Of the total 170 COVID-19 cases, one case in the vaccine group and three cases in the placebo group were considered to be severe infections.1,9
- Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, Perez JL, Perez Marc G, Moreira ED, Zerbini C, Bailey R, Swanson KA, Roychoudhury S, Koury K, Li P, Kalina WV, Cooper D, Frenck RW Jr, Hammitt LL, Tureci O, Nell H, Schaefer A, Unal S, Tresnan DB, Mather S, Dormitzer PR, Sahin U, Jansen KU, Gruber WC: Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med. 2020 Dec 10. doi: 10.1056/NEJMoa2034577. [PubMed:33301246]
- Gen Eng News: BNT162 vaccine candidates [Link]
- BioNTech BNT162 Update [Link]
- Clinical Trial NCT04368728 [Link]
- Clinical Trial NCT04380701 [Link]
- FDA fast track designation: BNT162b1 and BNT162b2 [Link]
- Health Canada Interim Product Monograph: BNT162b2 SARS-CoV-2 Vaccine [Link]
- MHRA Interim Product Monograph: BNT162b2 SARS-CoV-2 Vaccine [Link]
- FDA News Release: FDA Takes Key Action in Fight Against COVID-19 By Issuing Emergency Use Authorization for First COVID-19 Vaccine [Link]
- Pfizer: Fact Sheet for Healthcare Providers Administering Vaccine, Pfizer-BioNtech COVID-19 vaccine [Link]
- Pfizer: Fact Sheet for Recipients and Caregivers, Pfizer BioNTech COVID-19 vaccine [Link]
- FDA Emergency Use Authorization: Full EUA Prescribing information, Pfizer-BioNTech COVID-19 vaccine [Link]
-
PHASESTATUSPURPOSECONDITIONSCOUNT2Active Not RecruitingPreventionCoronavirus Disease 2019 (COVID‑19)12, 3Active Not RecruitingPreventionCoronavirus Disease 2019 (COVID‑19)11, 2Active Not RecruitingPreventionCoronavirus Disease 2019 (COVID‑19)11, 2RecruitingTreatmentCoronavirus Disease 2019 (COVID‑19) / Protection Against COVID-19 and Infections With SARS CoV 2 / Respiratory Tract Infections (RTI) / RNA Virus Infections / Vaccine Adverse Reaction / Viral Infections / Virus Diseases1
NIROGACESTAT

NIROGACESTAT
(2S)-2-[[(2S)-6,8-difluoro-1,2,3,4-tetrahydronaphthalen-2-yl]amino]-N-[1-[1-(2,2-dimethylpropylamino)-2-methylpropan-2-yl]imidazol-4-yl]pentanamide
489.6 g/mol, C27H41F2N5O
CAS 1290543-63-3
FDA APPROVED 11/27/2023, To treat adults with progressing desmoid tumors who require systemic treatment, Ogsiveo
PF-03084014, 1290543-63-3, PF-3084014, 865773-15-5QZ62892OFJUNII:QZ62892OFJUNII-QZ62892OFJнирогацестат [Russian] [INN]نيروغاسيستات [Arabic] [INN]尼罗司他 [Chinese] [INN]ニロガセスタット;
orphan drug designation in June 2018 for the treatment of desmoid tumors, and with a fast track designation
Nirogacestat, also known as PF-03084014, is a potent and selective gamma secretase (GS) inhibitor with potential antitumor activity. PF-03084014 binds to GS, blocking proteolytic activation of Notch receptors. Nirogacestat enhances the Antitumor Effect of Docetaxel in Prostate Cancer. Nirogacestat enhances docetaxel-mediated tumor response and provides a rationale to explore GSIs as adjunct therapy in conjunction with docetaxel for men with CRPC (castration-resistant prostate cancer).
Nirogacestat was disclosed to be a gamma-secretase inhibitor, which can inhibit Aβ-peptide production. SpringWorks Therapeutics (a spin-out of Pfizer ) is developing nirogacestat, as hydrobromide salt, a gamma-secretase inhibitor, for treating aggressive fibromatosis. In February 2021, nirogacestat was reported to be in phase 3 clinical development.
Nirogacestat is a selective gamma secretase (GS) inhibitor with potential antitumor activity. Nirogacestat binds to GS, blocking proteolytic activation of Notch receptors; Notch signaling pathway inhibition may follow, which may result in the induction of apoptosis in tumor cells that overexpress Notch. The integral membrane protein GS is a multi-subunit protease complex that cleaves single-pass transmembrane proteins, such as Notch receptors, at residues within their transmembrane domains. Overexpression of the Notch signaling pathway has been correlated with increased tumor cell growth and survival.
Nirogacestat has been used in trials studying the treatment of Breast Cancer, HIV Infection, Desmoid Tumors, Advanced Solid Tumors, and Aggressive Fibromatosis, among others.
Nirogacestat (Gamma Secretase Inhibitor)
Nirogacestat is an oral, selective, small molecule, gamma secretase inhibitor (GSI) in Phase 3 clinical development for patients with desmoid tumors. Gamma secretase is a protease complex that cleaves, or divides, multiple transmembrane protein complexes, including Notch, which, when dysregulated, can play a role in activating pathways that contribute to desmoid tumor growth.
Gamma secretase has also been shown to directly cleave BCMA, a therapeutic target that is highly expressed on multiple myeloma cells. By inhibiting gamma secretase with nirogacestat, membrane-bound BCMA can be preserved, thereby increasing target density while simultaneously reducing levels of soluble BCMA, which may serve as decoy receptors for BCMA-directed therapies. Together, these mechanisms combine to potentially enhance the activity of BCMA therapies and improve outcomes for multiple myeloma patients. SpringWorks is seeking to advance nirogacestat as a cornerstone of multiple myeloma combination therapy in collaboration with industry leaders who are advancing BCMA therapies.
SpringWorks Therapeutics Announces Clinical Collaboration with Pfizer
By Satish October 05, 2020
SpringWorks Therapeutics today announced that the company has entered into a clinical trial collaboration agreement with Pfizer to evaluate SpringWorks Therapeutics’ investigational gamma secretase inhibitor (GSI), nirogacestat, in combination with Pfizer’s anti-B-cell maturation antigen (BCMA) CD3 bispecific antibody, PF‐06863135, in patients with relapsed or refractory multiple myeloma.
Gamma secretase inhibition prevents the cleavage and shedding of BCMA from the surface of myeloma cells. In preclinical models, nirogacestat has been shown to increase the cell surface density of BCMA and reduce levels of soluble BCMA, thereby enhancing the activity of BCMA-targeted therapies, including CD3 bispecific antibodies.
Saqib Islam, Chief Executive Officer of SpringWorks Therapeutics Said: This collaboration is another important step in continuing to advance our goal of developing nirogacestat as a best-in-class BCMA potentiator, and we are pleased to work with Pfizer to study nirogacestat in combination with PF‐06863135, which has recently demonstrated promising monotherapy clinical data, We now have five collaborations with industry-leading BCMA developers to evaluate nirogacestat in combinations across modalities. We look forward to generating clinical data with our collaborators to further evaluate the ability of nirogacestat to improve outcomes for patients with multiple myeloma.

Under the terms of the agreement, Pfizer will sponsor and conduct the Phase 1b/2 study to evaluate the safety, tolerability and preliminary efficacy of the combination, and will assume all costs associated with the study, other than expenses related to the manufacturing of nirogacestat and certain expenses related to intellectual property rights. Pfizer and SpringWorks Therapeutics will also form a joint development committee to manage the clinical study, which is expected to commence in the first half of 2021.
Chris Boshoff, MD, PhD, Chief Development Officer for Pfizer Oncology at Pfizer Said: Entering into this clinical collaboration is a proud milestone in our strong relationship with SpringWorks,We believe that studying nirogacestat in combination with PF-06863135 could hold significant therapeutic promise for patients with relapsed or refractory multiple myeloma, and we look forward to working together to advance this important area of research.
In addition to its ongoing clinical collaborations with BCMA-directed therapies, SpringWorks is also currently conducting a global Phase 3, double-blind, randomized, placebo-controlled clinical trial (the DeFi Trial) to evaluate nirogacestat in adults with progressing desmoid tumors.
About Nirogacestat
Nirogacestat is an investigational, oral, selective, small molecule gamma secretase inhibitor in Phase 3 clinical development for desmoid tumors, which are rare and often debilitating and disfiguring soft-tissue tumors. Gamma secretase cleaves multiple transmembrane protein complexes, including Notch, which is believed to play a role in activating pathways that contribute to desmoid tumor growth.
In addition, gamma secretase has been shown to directly cleave membrane-bound BCMA, resulting in the release of the BCMA extracellular domain, or ECD, from the cell surface. By inhibiting gamma secretase, membrane-bound BCMA can be preserved, increasing target density while reducing levels of soluble BCMA ECD, which may serve as decoy receptors for BCMA-directed therapies. Nirogacestat’s ability to enhance the activity of BCMA-directed therapies has been observed in preclinical models of multiple myeloma. SpringWorks is evaluating nirogacestat as a BCMA potentiator and has five collaborations with industry-leading BCMA developers to evaluate nirogacestat in combinations across modalities, including with an antibody-drug conjugate, two CAR T cell therapies and two bispecific antibodies. In addition, SpringWorks and Fred Hutchinson Cancer Research Center have entered into a sponsored research agreement to further characterize the ability of nirogacestat to modulate BCMA and potentiate BCMA directed therapies using a variety of preclinical and patient-derived multiple myeloma models developed by researchers at Fred Hutch.
Nirogacestat has received Orphan Drug Designation from the U.S. Food and Drug Administration (FDA) for the treatment of desmoid tumors (June 2018) and from the European Commission for the treatment of soft tissue sarcoma (September 2019). The FDA also granted Fast Track and Breakthrough Therapy Designations for the treatment of adult patients with progressive, unresectable, recurrent or refractory desmoid tumors or deep fibromatosis (November 2018 and August 2019).
About PF‐06863135
PF‐06863135 is an anti-B-cell maturation antigen (BCMA) CD3 bispecific antibody being investigated in a Phase 1 clinical study to treat relapsed or refractory multiple myeloma. This bispecific antibody can be administered subcutaneously and has been optimized for binding affinity to both BCMA and CD3, enabling more potent T-cell-mediated tumor cell toxicity.
Source: SpringWorks Therapeutics
FDA Grants Breakthrough Designation to Nirogacestat for Desmoid Tumors
The FDA has granted nirogacestat, an investigational gamma-secretase inhibitor, with a breakthrough therapy designation for the treatment of adult patients with progressive, unresectable, recurrent or refractory desmoid tumors or deep fibromatosis.
The FDA has granted nirogacestat (PF-03084014), an investigational gamma-secretase inhibitor, with a breakthrough therapy designation for the treatment of adult patients with progressive, unresectable, recurrent or refractory desmoid tumors or deep fibromatosis.1
The breakthrough designation was granted as a result of positive findings seen in phase I and II trials of nirogacestat monotherapy in patients with desmoid tumors. A phase III trial has also been initiated investigating nirogacestat in patients with desmoid tumors or aggressive fibromatosis (NCT03785964).
“We are committed to pursuing the rapid development of nirogacestat given the important need for new therapies for patients with desmoid tumors and are pleased to receive this breakthrough therapy designation,” Saqib Islam, CEO of SpringWorks, the company developing the small molecule inhibitor, said in a statement. “We are currently enrolling adult patients in our phase III DeFi trial and will continue to work closely with the FDA with the goal of bringing nirogacestat to patients as quickly as possible.”
The open-label, single-center phase II trial of nirogacestat enrolled 17 patients with desmoid tumors who were not eligible for surgical resection or definitive radiation therapy and who had experienced disease progression after at least 1 prior treatment regimen. Patients received 150 mg twice per day of continuous, oral nirogacestat in 21-day cycles.2
The median age of patients was 34 years (range, 19-69), 82% of the patients were female, and 53% of patients had aCTNNB1T41A somatic missense mutation. The median number of prior therapies was 4 (range, 1-9), which included cytotoxic chemotherapy in 71% and a tyrosine kinase inhibitor in 59%.
Sixteen patients were evaluable for response. After a median follow-up of more than 25 months, 5 patients (29%) achieved a partial response and 11 (65%) had stable disease, for a disease control rate of 100%. Ten patients (59%) remained on treatment with nirogacestat for more than 2 years.
Grade 1/2 adverse events were observed in all patients, with diarrhea (76%) and skin disorders (71%) being the most common toxicities. The only treatment-related grade 3 event was reversible hypophosphatemia, which was reported in 8 patients (47%) and was considered to be a class effect of gamma-secretase inhibitors. Four patients met the criteria for dose reduction.
Findings from the phase I study also showed a disease control rate of 100% with nirogacestat. However, the median progression-free survival was not reached in either study due to a lack of patients progressing on treatment. Only 1 patient discontinued treatment due to an adverse event between the 2 studies.1
The FDA had previously granted nirogacestat with an orphan drug designation in June 2018 for the treatment of desmoid tumors, and with a fast track designation in November 2018 for the treatment of adult patients with progressive, unresectable, recurrent or refractory desmoid tumors or deep fibromatosis.
References
- SpringWorks Therapeutics Receives Breakthrough Therapy Designation for Nirogacestat for the Treatment of Adult Patients with Progressive, Unresectable, Recurrent or Refractory Desmoid Tumors [press release]. Stamford, CT: SpringWorks Therapeutics, Inc; August 29, 2019. https://bit.ly/30IV0Eb. Accessed September 3, 2019.
- Kummar S, O’Sullivan Coyne G, Do KT, et al. Clinical Activity of the γ-Secretase Inhibitor PF-03084014 in Adults With Desmoid Tumors (Aggressive Fibromatosis).J Clin Oncol.2017;35(14):1561-1569. doi: 10.1200/JCO.2016.71.1994.
PAPER

Bioorganic & medicinal chemistry letters (2011), 21(9), 2637-40.
https://www.sciencedirect.com/science/article/abs/pii/S0960894X10018822



PATENT
WO 2016089208
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016089208
PATENT
WO-2021029854
Novel, stable crystalline polymorphic (A to N) and amorphous forms of nirogacestat hydrobromide , useful for treating desmoid tumors such as multiple myeloma, a cancer having a mutation in a Notch pathway gene, adenoid cystic carcinoma and T-cell acute lymphoblastic leukemia.
(S)-2-(((S)-6,8-difluoro-l,2,3,4-tetrahydronaphthalen-2-yl)amino)-N-(l-(2- methyl- l-(neopentylamino) propan-2-yl)-lH-imidazol-4-yl)pentanamide (“Compound 1”) is a gamma-secretase inhibitor which can inhibit Ab-peptide production.
[0003] Not all compounds that are gamma-secretase inhibitors have characteristics affording the best potential to become useful therapeutics. Some of these characteristics include high affinity at the gamma-secretase, duration of gamma-secretase deactivation, oral bioavailability, tissue distribution, and stability (e.g., ability to formulate or crystallize, shelf life). Favorable characteristics can lead to improved safety, tolerability, efficacy, therapeutic index, patient compliance, cost efficiency, manufacturing ease, etc.
[0004] In addition, the isolation and commercial -scale preparation of a solid state form of hydrobromide salts of Compound 1 and corresponding pharmaceutical formulations having acceptable solid state properties (including chemical stability, thermal stability, solubility, hygroscopicity, and/or particle size), compound manufacturability (including yield, impurity rejection during crystallization, filtration properties, drying properties, and milling properties), and formulation feasibility (including stability with respect to pressure or compression forces during tableting) present a number of challenges.
[0005] Accordingly, there is a current need for one or more solid state forms of hydrobromide salts of Compound 1 that have an acceptable balance of these properties and can be used in the preparation of pharmaceutically acceptable solid dosage forms.
Crystalline Form A
[0147] In one aspect, the present disclosure relates to crystalline Form A of a hydrobromide salt of (S)-2-(((S)-6,8-difluoro-l,2,3,4-tetrahydronaphthalen-2-yl)amino)- N-(l -(2 -methyl- l-(neopentylamino) propan-2-yl)-lH-imidazol-4-yl)pentanamide having Formula (I),
[0148] In one embodiment, crystalline Form A is anhydrous.
[0149] In another embodiment, the melting point of crystalline Form A is about 254 °C.
[0150] In another embodiment, Form A is characterized by an XRPD pattern having peaks at 8.8 ± 0.2, 9.8 ± 0.2, and 23.3 ± 0.2 degrees two theta when measured by Cu Ka radiation. In another embodiment, Form A is characterized by an XRPD pattern having peaks at 8.8 ± 0.2, 9.8 ± 0.2, 23.3 ± 0.2, 25.4 ± 0.2, 28.0 ± 0.2, and 29.3 ± 0.2 degrees two theta when measured by Cu Ka radiation. In another embodiment, Form A is characterized by an XRPD pattern having peaks at 8.8 ± 0.2, 9.8 ± 0.2, 20.0 ± 0.2, 23.3 ± 0.2, 25.4 ± 0.2, 28.0 ± 0.2, 29.3 ± 0.2, and 32.5 ± 0.2 degrees two theta when measured by Cu Ka radiation.
Patent
Product case, WO2005092864 ,
hold protection in the EU states until March 2025, and expire in the US in February 2026 with US154 extension.
PATENT
WO2020208572 , co-assigned to GSK and SpringWorks, claiming a combination of nirogacestat with anti-BCMA antibody (eg belantamab mafodotin ), for treating cancer.
PATENT
US10590087 , for a prior filing from Pfizer, claiming crystalline forms of nirogacestat hydrobromide.
////////////NIROGACESTAT, orphan drug designation, esmoid tumors, fast track designation, PF-03084014, PF 03084014, QZ62892OFJ , UNII:QZ62892OFJ ,UNII-QZ62892OFJ, ,нирогацестат , نيروغاسيستات , 尼罗司他 , ニロガセスタット, phase 3
CCCC(C(=O)NC1=CN(C=N1)C(C)(C)CNCC(C)(C)C)NC2CCC3=C(C2)C(=CC(=C3)F)F
Fenfluramine Hydrochloride

Fenfluramine
- DEA No. 1670
- S 768
2020/12/18, FDA APPROVED, Fintepla
Fenfluramine hydrochloride
| Formula | C12H16F3N. HCl |
|---|---|
| CAS | 404-82-0458-24-2 (FREE) |
| Mol weight | 267.7183 |
Antiobesity
| Efficacy | Appetite suppressant |
|---|---|
| Disease | Dravet syndrome |
(+-)-Fenfluramine hydrochloride
Racemic fenfluramine hydrochloride
Fenfluramine hydrochloride [USAN]
1-(m-Trifluoromethylphenyl)-2-(ethylamino)propane hydrochloride
Research Code:ZX-008
MOA:Serotonin agonist
Indication:Dravet syndrome
Company:Zogenix (Originator)
Synonyms of Fenfluramine [INN:BAN]
- (+-)-Fenfluramine
- BRN 4783711
- dl-Fenfluramine
- DL-Fenfluramine
- EINECS 207-276-3
- Fenfluramina
- Fenfluramina [DCIT]
- Fenfluramine
- Fenfluraminum
- Fenfluraminum [INN-Latin]
- HSDB 3080
- Obedrex
- Pesos
- Ponderax PA
- Rotondin
- S 768
- UNII-2DS058H2CF
mp 160-161 °C, ethyl acetate US 3198834
nmr Salsbury, Jonathon S.; Magnetic Resonance in Chemistry 2005, V43(11), P910-917 C
IR BIORAD: Infrared spectral data from the Bio-Rad/Sadtler IR Data Collection was obtained from Bio-Rad Laboratories, Philadelphia, PA (US).FenfluramineCAS Registry Number: 458-24-2CAS Name:N-Ethyl-a-methyl-3-(trifluoromethyl)benzeneethanamineAdditional Names:N-ethyl-a-methyl-m-(trifluoromethyl)phenethylamine; 2-ethylamino-1-(3-trifluoromethylphenyl)propaneManufacturers’ Codes: S-768Molecular Formula: C12H16F3NMolecular Weight: 231.26Percent Composition: C 62.32%, H 6.97%, F 24.65%, N 6.06%Literature References: Prepn: L. G. Beregi et al.,FRM1658; eidem,US3198833 (1963, 1965 both to Sci. Union et Cie Soc. Franc. Recherche Méd.). Prepn of optical isomers: eidem,US3198834 (1965 to Sci. Union et Cie Soc. Franc. Recherche Med.). Pharmacology: Presse Med.71, 181 (1963). Pharmacology and toxicity of isomers and racemate: J. C. Le Douarec et al.,Arch. Int. Pharmacodyn. Ther.161, 206 (1966). Pharmacokinetics: S. Caccia et al.,Eur. J. Clin. Pharmacol.29, 221 (1985). Clinical trial of dextrofenfluramine in refractory obesity: N. Finer et al.,Curr. Ther. Res.38, 847 (1985). Comprehensive review: Pinder et al.,Drugs10, 241-323 (1975).Properties: bp12 108-112°. LD50 i.p. in mice: 144 mg/kg (US3198833).Boiling point: bp12 108-112°Toxicity data: LD50 i.p. in mice: 144 mg/kg
Derivative Type: HydrochlorideCAS Registry Number: 404-82-0Trademarks: Acino (IMA); Adipomin (Streuli); Obedrex (Beta); Pesos (Valeas); Ponderal (Servier); Ponderax (Selpharm); Ponderex (Robins); Pondimin (Robins); Rotondin (Casasco)Molecular Formula: C12H16F3N.HClMolecular Weight: 267.72Percent Composition: C 53.84%, H 6.40%, F 21.29%, N 5.23%, Cl 13.24%Properties: Crystals from ethanol + ether, mp 166°.Melting point: mp 166°
Derivative Type:d-FormCAS Registry Number: 3239-44-9Additional Names: Dexfenfluramine; dextrofenfluramineProperties: [a]D25 +9.5° (c = 8 in ethanol). LD50 orally in rats: 114.6 mg/kg (Le Douarec).Optical Rotation: [a]D25 +9.5° (c = 8 in ethanol)Toxicity data: LD50 orally in rats: 114.6 mg/kg (Le Douarec)
Derivative Type:d-Form hydrochlorideCAS Registry Number: 3239-45-0Trademarks: Adifax (Servier); Glypolix (Stroder); Isomeride (Ardix); Redux (Wyeth-Ayerst)Properties: Crystals from ethyl acetate, mp 160-161°.Melting point: mp 160-161°
Derivative Type:l-FormCAS Registry Number: 37577-24-5Properties: [a]D25 -9.6° (c = 8 in ethanol). LD50 orally in rats: 195 mg/kg (Le Douarec).Optical Rotation: [a]D25 -9.6° (c = 8 in ethanol)Toxicity data: LD50 orally in rats: 195 mg/kg (Le Douarec)
Derivative Type:l-Form hydrochlorideCAS Registry Number: 3616-78-2Properties: Crystals from ethyl acetate, mp 160-161°.Melting point: mp 160-161°
NOTE: This is a controlled substance: 21 CFR, 1308.14.Therap-Cat: Anorexic.Keywords: Anorexic.
A centrally active drug that apparently both blocks serotonin uptake and provokes transport-mediated serotonin release.
Fenfluramine Hydrochloride has been filed an IND application with the FDA in USA to initiate phase III trials by Brabant Pharma (acquired by Zogenix in 2014) for the treatment of dravets syndrome (also known as severe myoclonic epilepsy of infancy, SMEI), this compound has been granted orphan drug designation in Europe and U.S..
Fenfluramine Hydrochloride was launched in 1963 by Servier in France and in 1973 by Wyeth (now a wholly owned subsidiary of Pfizer) in US for the treatment of obesity. However, it was withdrawn from the market in 1997 due to heart disease.
Dravet syndrome is a pediatric encephalopathy that typically manifests within the first year of life following exposure to elevated temperatures. It is characterized by recurrent pharmacoresistant seizures, which increase in frequency and severity with disease progression. Concomitantly with these seizures, patients typically display delayed development and neurocognitive impairment.6,9,10,11 Fenfluramine is a serotonergic phenethylamine originally used as an appetite suppressant until concerns regarding cardiotoxicity in obese patients lead to its withdrawal from the market in 1997.6,12,13 Through its ability to modulate neurotransmission, fenfluramine has reemerged as an effective therapy against pharmacoresistant seizures, such as those involved in Dravet syndrome.3,5,8
Fenfluramine was granted initial FDA approval in 1973 prior to its withdrawal; it was granted a new FDA approval on June 25, 2020, for treatment of Dravet syndrome patients through the restricted FINTEPLA REMS program. It is currently sold under the name FINTEPLA® by Zogenix INC.16
Fenfluramine, sold under the brand name Fintepla, is a medication used for the treatment of seizures associated with Dravet syndrome in people age two and older.[2][3]
The most common adverse reactions include decreased appetite; drowsiness, sedation and lethargy; diarrhea; constipation; abnormal echocardiogram; fatigue or lack of energy; ataxia (lack of coordination), balance disorder, gait disturbance (trouble with walking); increased blood pressure; drooling, salivary hypersecretion (saliva overproduction); pyrexia (fever); upper respiratory tract infection; vomiting; decreased weight; risk of falls; and status epilepticus.[2]
Dravet syndrome is a pediatric encephalopathy that typically manifests within the first year of life following exposure to elevated temperatures. It is characterized by recurrent pharmacoresistant seizures, which increase in frequency and severity with disease progression. Concomitantly with these seizures, patients typically display delayed development and neurocognitive impairment.6,9,10,11 Fenfluramine is a serotonergic phenethylamine originally used as an appetite suppressant until concerns regarding cardiotoxicity in obese patients lead to its withdrawal from the market in 1997.6,12,13 Through its ability to modulate neurotransmission, fenfluramine has reemerged as an effective therapy against pharmacoresistant seizures, such as those involved in Dravet syndrome.3,5,8
Fenfluramine was granted initial FDA approval in 1973 prior to its withdrawal; it was granted a new FDA approval on June 25, 2020, for treatment of Dravet syndrome patients through the restricted FINTEPLA REMS program. It is currently sold under the name FINTEPLA® by Zogenix INC.16
Medical uses
Fenfluramine is indicated for the treatment of seizures associated with Dravet syndrome in people age two and older.[2][3]
Dravet syndrome is a life-threatening, rare and chronic form of epilepsy.[2] It is often characterized by severe and unrelenting seizures despite medical treatment.[2]
Adverse effects
The U.S. Food and Drug Administration (FDA) fenfluramine labeling includes a boxed warning stating the drug is associated with valvular heart disease (VHD) and pulmonary arterial hypertension (PAH).[2] Because of the risks of VHD and PAH, fenfluramine is available only through a restricted drug distribution program, under a risk evaluation and mitigation strategy (REMS).[2] The fenfluramine REMS requires health care professionals who prescribe fenfluramine and pharmacies that dispense fenfluramine to be specially certified in the fenfluramine REMS and that patients be enrolled in the REMS.[2] As part of the REMS requirements, prescribers and patients must adhere to the required cardiac monitoring with echocardiograms to receive fenfluramine.[2]
At higher therapeutic doses, headache, diarrhea, dizziness, dry mouth, erectile dysfunction, anxiety, insomnia, irritability, lethargy, and CNS stimulation have been reported with fenfluramine.[4]
There have been reports associating chronic fenfluramine treatment with emotional instability, cognitive deficits, depression, psychosis, exacerbation of pre-existing psychosis (schizophrenia), and sleep disturbances.[4][5] It has been suggested that some of these effects may be mediated by serotonergic neurotoxicity/depletion of serotonin with chronic administration and/or activation of serotonin 5-HT2A receptors.[5][6][7][8]
Heart valve disease
The distinctive valvular abnormality seen with fenfluramine is a thickening of the leaflet and chordae tendineae. One mechanism used to explain this phenomenon involves heart valve serotonin receptors, which are thought to help regulate growth. Since fenfluramine and its active metabolite norfenfluramine stimulate serotonin receptors, this may have led to the valvular abnormalities found in patients using fenfluramine. In particular norfenfluramine is a potent inhibitor of the re-uptake of 5-HT into nerve terminals.[9] Fenfluramine and its active metabolite norfenfluramine affect the 5-HT2B receptors, which are plentiful in human cardiac valves. The suggested mechanism by which fenfluramine causes damage is through over or inappropriate stimulation of these receptors leading to inappropriate valve cell division. Supporting this idea is the fact that this valve abnormality has also occurred in patients using other drugs that act on 5-HT2B receptors.[10][11]
According to a study of 5,743 former users conducted by a plaintiff’s expert cardiologist, damage to the heart valve continued long after stopping the medication.[12] Of the users tested, 20% of women, and 12% of men were affected. For all ex-users, there was a 7-fold increase of chances of needing surgery for faulty heart valves caused by the drug.[12]
Overdose
In overdose, fenfluramine can cause serotonin syndrome and rapidly result in death.[13][14]
Pharmacology
Pharmacodynamics
Fenfluramine acts primarily as a serotonin releasing agent.[15][16] It increases the level of serotonin, a neurotransmitter that regulates mood, appetite and other functions.[15][16] Fenfluramine causes the release of serotonin by disrupting vesicular storage of the neurotransmitter, and reversing serotonin transporter function.[17] The drug also acts as a norepinephrine releasing agent to a lesser extent, particularly via its active metabolite norfenfluramine.[15][16] At high concentrations, norfenfluramine, though not fenfluramine, also acts as a dopamine releasing agent, and so fenfluramine may do this at very high doses as well.[15][16] In addition to monoamine release, while fenfluramine binds only very weakly to the serotonin 5-HT2 receptors, norfenfluramine binds to and activates the serotonin 5-HT2B and 5-HT2C receptors with high affinity and the serotonin 5-HT2A receptor with moderate affinity.[18][19] The result of the increased serotonergic and noradrenergic neurotransmission is a feeling of fullness and reduced appetite.
The combination of fenfluramine with phentermine, a norepinephrine–dopamine releasing agent acting primarily on norepinephrine, results in a well-balanced serotonin–norepinephrine releasing agent with weaker effects of dopamine release.[15][16]
| Drug | NE | DA | 5-HT | Type | Ref |
|---|---|---|---|---|---|
| Fenfluramine | 739 | >10,000 | 79.3–108 | SRA | [20][15][16] |
| D-Fenfluramine | 302 | >10,000 | 51.7 | SNRA | [20][15] |
| L-Fenfluramine | >10,000 | >10,000 | 147 | SRA | [15][21] |
| Norfenfluramine | 168–170 | 1,900–1,925 | 104 | SNRA | [15][16] |
| Phentermine | 39.4 | 262 | 3,511 | NDRA | [20] |
Pharmacokinetics
The elimination half-life of fenfluramine has been reported as ranging from 13 to 30 hours.[4] The mean elimination half-lives of its enantiomers have been found to be 19 hours for dexfenfluramine and 25 hours for levfenfluramine.[13] Norfenfluramine, the major active metabolite of fenfluramine, has an elimination half-life that is about 1.5 to 2 times as long as that of fenfluramine, with mean values of 34 hours for dexnorfenfluramine and 50 hours for levnorfenfluramine.[13]
Chemistry
Fenfluramine is a substituted amphetamine and is also known as 3-trifluoromethyl-N-ethylamphetamine.[13] It is a racemic mixture of two enantiomers, dexfenfluramine and levofenfluramine.[13] Some analogues of fenfluramine include norfenfluramine, benfluorex, flucetorex, and fludorex.
History
Fenfluramine was developed in the early 1960s and was introduced in France in 1963.[13] Approximately 50 million Europeans were treated with fenfluramine for appetite suppression between 1963 and 1996.[13] Fenfluramine was approved in the United States in 1973.[13] The combination of fenfluramine and phentermine was proposed in 1984.[13] Approximately 5 million people in the United States were given fenfluramine or dexfenfluramine with or without phentermine between 1996 and 1998.[13]
In the early 1990s, French researchers reported an association of fenfluramine with primary pulmonary hypertension and dyspnea in a small sample of patients.[13] Fenfluramine was withdrawn from the U.S. market in 1997 after reports of heart valve disease[22][23] and continued findings of pulmonary hypertension, including a condition known as cardiac fibrosis.[24] It was subsequently withdrawn from other markets around the world. It was banned in India in 1998.[25]
Fenfluramine was an appetite suppressant which was used to treat obesity.[13] It was used both on its own and, in combination with phentermine, as part of the anti-obesity medication Fen-Phen.[13]
In June 2020, fenfluramine was approved for medical use in the United States with an indication to treat Dravet syndrome.[2][26]
The effectiveness of fenfluramine for the treatment of seizures associated with Dravet syndrome was demonstrated in two clinical studies in 202 subjects between ages two and eighteen.[2] The studies measured the change from baseline in the frequency of convulsive seizures.[2] In both studies, subjects treated with fenfluramine had significantly greater reductions in the frequency of convulsive seizures during the trials than subjects who received placebo (inactive treatment).[2] These reductions were seen within 3–4 weeks, and remained generally consistent over the 14- to 15-week treatment periods.[2]
The U.S. Food and Drug Administration (FDA) granted the application for fenfluramine priority review and orphan drug designations.[2][27][28] The FDA granted approval of Fintepla to Zogenix, Inc.[2]
On 15 October 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Fintepla, intended for the treatment of seizures associated with Dravet syndrome.[29] Fenfluramine was approved for medical use in the European Union in December 2020.[3]
Society and culture
Recreational use
Unlike various other amphetamine derivatives, fenfluramine is reported to be dysphoric, “unpleasantly lethargic“, and non-addictive at therapeutic doses.[30] However, it has been reported to be used recreationally at high doses ranging between 80 and 400 mg, which have been described as producing euphoria, amphetamine-like effects, sedation, and hallucinogenic effects, along with anxiety, nausea, diarrhea, and sometimes panic attacks, as well as depressive symptoms once the drug had worn off.[30][31][32] At very high doses (e.g., 240 mg, or between 200–600 mg), fenfluramine induces a psychedelic state resembling that produced by lysergic acid diethylamide (LSD).[32][33] Indirect (via induction of serotonin release) and/or direct activation of the 5-HT2A receptor would be expected to be responsible for the psychedelic effects of the drug at sufficient doses.
Research
Under the development code ZX008, the pharmaceutical company Zogenix is studying fenfluramine’s potential to treat seizures.[34] Clinical trials have studied the use of fenfluramine in patients with Dravet syndrome.[35] Results of a Phase III clinical trial showed a 64% reduction in seizures.[36]Route 1
Reference:1. J. Org. Chem.1979, 44, 3580-3583.Route 2
Reference:1. EP0810195A1.
2. Chem. Ind. Times 2002, 16, 33-34.Route 3
Reference:1. ACS Symp. Ser. 2009, 1003, 165-181.
ref
BE 609630
FR 1658 M 19630218
US 3198834
DE 1593595
US 3769319
NL 7215548
Ger. (East) (1974), DD 108971
EP 3170807
SYN
US20170174613
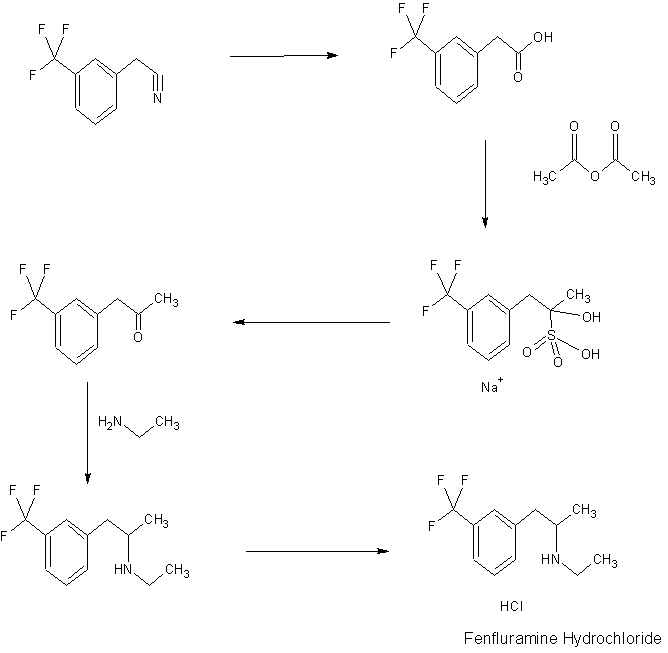
PATENT
US 20170174613
| Step 4.2: Crystallization of Fenfluramine Hydrochloride |
| Procedure: Charge Fenfluramine.HCl (crude) (1.00 wt, 1.0 eq.) and TBME (10.0 vol, 7.4 wt) to the vessel and commence stirring. Heat the suspension to reflux (50 to 58° C.). Charge ethanol (5.0 vol, 3.9 wt) maintaining the temperature at 50 to 58° C. Addition time 20 minutes. Stir at 50 to 58° C. for 5 to 10 minutes and check for dissolution. Stir the solution at 50 to 58° C. for 5 to 10 minutes, targeting 54 to 58° C. Clarify the reaction mixture through a 0.1 μm in-line filter at 54 to 58° C., followed by a line rinse with TBME (1 vol, 0.7 wt). Cool the solution to 48 to 50° C. Charge Fenfluramine.HCl, code FP0188 (0.01 wt). Check for crystallization. Allow the suspension to cool to 15 to 20° C., target 17° C. over 5 to 5.5 hours at an approximately constant rate. Stir the mixture at 15 to 20° C., target 17° C. for 2 to 3 hours. Filter the mixture and wash the filter-cake with clarified TBME (2×3.0 vol, 2×2.2 wt) at 5 to 15° C. Dry the solid at up to 40° C. until the TBME content is <0.5% w/w TBME and the ethanol content is <0.5% w/w EtOH by 1H-NMR analysis. 4 to 8 hours. Determine the w/w assay of the isolated Fenfluramine.HCl by 1H-NMR analysis. |
| Yields and Profiles: The yield for the stage 4 Demonstration batch is summarized in Table 1E below. Input: 750.0 g uncorr. Fenfluramine.HCl crude (1.00 eq, 1.00 wt uncorr.) for input calculation. FIG. 3 shows an exemplary HPLC chromatogram of a crystallized fenfluramine hydrochloride sample (210 nm UV absorbance). |
PATENT
US 20180208543
| Fenfluramine, i.e., 3-trifluoromethyl-N-ethylamphetamine, has the following chemical structure: |
| The marketing of fenfluramine as a pharmaceutical active ingredient in the United States began in 1973 and was used in a therapy in combination with phentermine to prevent and treat obesity. Anyway, in 1997 fenfluramine was withdrawn from the market in the United States and immediately thereafter in other countries, since its use was associated with the onset of cardiac fibrosis and pulmonary hypertension. As a consequence of this event, the pharmaceutical compounds containing this active ingredient were withdrawn from the market. However, fenfluramine, even after its exit from the market, has continued to attract scientific interest, as will become apparent from the discussion presented hereinafter. |
| In the literature, over the years, numerous syntheses or processes have been reported for preparing fenfluramine or its dextrorotatory enantiomer dexfenfluramine or an analog containing a highly electron-attractor group on the aromatic ring as in the fenfluramine molecule (see for example Pentafluorosulfanyl Serotonin Analogs: Synthesis, Characterization, and Biological Activity, John T. Welch and Dongsung Lim Chapter 8, pp 165-181 DOI: 10.1021/bk-2009-1003.ch008). Many of these synthesis paths are long and provide for multiple synthesis steps that can include reagents that are dangerous or scarcely environment-friendly and are therefore scarcely convenient for an industrial synthesis. Hereinafter, any reference to “fenfluramine” is understood to reference the racemic form, i.e., (RS)-N-ethyl-1-[3-(trifluoromethyephenyl]propan-2-amine. |
| To the best of the knowledge of the inventors, the first method for fenfluramine synthesis reported in the literature dates back to 1962 and is referenced in patent BE609630 and in similar patents U.S. Pat. No. 3,198,833 and FR1324220. All the synthesis methods reported in these patents provide for numerous synthesis steps. By way of example, one of the methods provides for the transformation into oxime of a ketone, 1-(3-trifluoromethyephenyl-propan-2-one, as shown here: |
| The oxime is then hydrogenated in the presence of Raney nickel catalyst so as to yield the corresponding primary amine, which is acetylated subsequently with ethanoic anhydride before being converted into fenfluramine by reduction with lithium aluminum hydride. |
| As can be seen, the final step of this chemical process provides for the use of lithium aluminum hydride and the persons skilled in the art will acknowledge that the use of this reagent should be avoided, if possible, on an industrial level, since it is extremely flammable and is the source of accidents. Furthermore, lithium is a potentially neurotoxic metal and therefore its use should be avoided where possible. Furthermore, the Raney nickel catalyst is used in the oxime reduction step and can contaminate the final active ingredient; the use of hydroxylamine also entails problems of toxicity for workers assigned to production. |
| A further disadvantage of this process is, as already mentioned earlier, the number of steps, not only because a large number of synthesis steps entails a reduction of the overall yield of active ingredient, but also because each synthesis step in principle can generate impurities and a larger number of steps can therefore entail a higher number of impurities in the final active ingredient. Many of these impurities, furthermore, due to their structural similarity to fenfluramine, are difficult to eliminate and remove from a fenfluramine preparation. One impurity for example that can be formed in the process described above and is difficult to eliminate is the following: |
| This impurity, which is a primary amine, shares physical-chemical properties that are similar to fenfluramine and therefore, like fenfluramine, it can form a hydrochloride salt by treatment with hydrochloric acid and thus contaminate the active ingredient fenfluramine hydrochloride. Furthermore, this impurity—as a free base—has a boiling point that is similar to that of fenfluramine (73° C. vs. 89° C. at 6 mmHg respectively), and therefore its elimination by distillation also can be problematic. |
| The process described above can in principle generate other impurities, which are listed in FIG. 1. |
| EP 0441160 claims a synthesis in 5 steps of dexfenfluramine, dextrorotatory enantiomer of fenfluramine. This synthesis can be adapted easily to produce fenfluramine instead of its dextrorotatory enantiomer simply by performing the first reduction step with a non-chiral reducing agent. In the first step, in fact: |
(MOL) (CDX) a ketone, 1-(3-trifluoromethyl)phenyl-propan-2-one, is first reduced to the corresponding alcohol in the presence of yeast, D-glucose, ethanol and water. Then the alcohol is converted into the tosylate in the second step:
(MOL) (CDX)
| This reaction occurs in the presence of triethylamine and tosyl chloride in methylene chloride as solvent. After purification, the tosylate is converted to fenfluramine by means of three successive steps: |
| In the first of these three steps, the tosylate is converted into an azide intermediate by reaction with sodium azide in dimethylformamide. The azide intermediate is then hydrogenated in the presence of a catalyst, palladium on carbon. Finally, the resulting primary amine is converted into fenfluramine by reaction with acetaldehyde and sodium borohydride. |
| Persons skilled in the art may see easily that this process is not desirable from an industrial standpoint due to reasons related to environmental risk, safety and costs. For example, the sodium azide used in the process is a notoriously explosive compound and its use at the industrial level is dangerous. Furthermore, palladium is an expensive material and its use in the process entails an increase in the production costs of fenfluramine. Furthermore, palladium can contaminate the finished active ingredient. |
| In another method for the synthesis of dexfenfluramine in 3-4 steps, reported by Goument et al. in Bulletin of the Chemical Society of France (1993), 130, p. 450-458, 3-bromobenzotrifluoride is subjected to a Grignard reaction with enantiopure 1,2-propylene-epoxide to yield 1-[3-(trifluoromethyl)phenyl]propan-2-ol as shown hereafter: |
| If this reaction is performed with racemic 1,2-propylene-epoxide, the synthesis can be adapted to the preparation of fenfluramine. |
| The alcohol thus obtained is first transformed into trifluoromethyl sulfonate by reaction with trifluoromethanesulfonic anhydride and then treated with ethylamine to yield fenfluramine, as shown in the diagram hereinafter: |
| In this article, the authors acknowledge that the main byproducts of the reaction are isomer alkenes having the following chemical structures: |
| The process proposed by Goument et al. is not interesting from the industrial standpoint for a series of reasons. First of all, it is known that the use of Grignard reagents, especially on an industrial scale, is problematic, because these compounds are often pyrophoric and corrosive. Furthermore, 1,2-propylene epoxide is a suspected carcinogenic compound. Finally, the formation of the three isomer alkenes as byproducts listed above is a disadvantage of the process. In the article, Goument presents methods for activation of the intermediate alcohol which are alternative to trifluoromethylsulfonate, for example by converting it to chloride (via thionyl chloride) or to mesylate (via mesyl chloride), but these process variations share the same disadvantages as the main process analyzed above. |
| In addition to the methods with multiple synthesis steps discussed so far in detail, the literature reports other methods or processes for producing fenfluramine or dexfenfluramine. In general, persons skilled in the art acknowledge that the syntheses in the literature for producing dexfenfluramine sometimes can be applied to the preparation of fenfluramine simply by replacing the initial materials and/or enantiopure reagents with the corresponding racemates while maintaining the reaction conditions. For example, patents that present long synthesis methods in multiple steps are the following: |
| DE1593595 and U.S. Pat. No. 3,769,319 |
| NL7215548 |
| EP810195 and EP882700 (dexfenfluramine) |
| EP0301925 (dexfenfluramine) |
| Other examples of preparation of fenfluramine, taken from non-patent literature, are the following: |
| Synthesis, November 1987, p. 1005-1007 |
| J. Org. Chem, 1991, 56, p. 6019 |
| Tetrahedron, 1994, 50(1), p. 171 |
| Bull. Soc. Chim. France, 1993, 130(4), p. 459-466 (dexfenfluramine) |
| Chirality, 2002, 14(4), p. 325-328 (dexfenfluramine) |
| Without analyzing in detail the individual methods described in these patents or articles, it can be stated in summary that all these methods are not attractive and interesting from the industrial standpoint because these are processes with many synthesis steps or because the initial materials described therein are not easily available and therefore have to be prepared separately, with a further expenditure of time and with further costs, or because they provide for the use of reagents that are dangerous/explosive/toxic or because they entail the use of catalysts based on heavy metals that can contaminate the final active ingredient. |
| One should consider that in the literature there are methods for the preparation of fenfluramine that did not provide for long syntheses and multiple steps but are shorter and consist of one or two steps. These processes, which therefore would be more interesting from the industrial standpoint, have other specific disadvantages, as will become apparent in detail hereinafter. For example, in the literature there is a first group of articles or patents that describe the reaction between 1-(3-trifluoromethyl)phenyl-propan-2-one and ethylamine in the presence of hydrogen gas and of a transition metal as catalyst: |
| In particular, in Huagong Shikan, 2002, 16(7), p. 33, the reaction is performed with hydrogen gas (2.9-3.38 atm), at 65-75° C., for 9 hours, in the presence of Raney nickel. Likewise, in patent DD108971 (1973), Raney nickel and hydrogen gas and methanol are used as solvent to perform this reaction. |
| In HU55343, instead, a similar reaction in one step is performed with hydrogen gas in the presence of another transition metal catalyst, such as palladium on carbon. |
| Although these three methods describe short single-step processes, they have the disadvantage of the use of hydrogen gas. As is known to persons skilled in the art, hydrogen gas is a dangerous gas due to the inherent danger of forming explosive mixtures with air and must be used by expert personnel in expensive facilities dedicated to its use and built with special precautions. Despite being used in purpose-built facilities, the use of hydrogen at the industrial level is inherently dangerous and to be avoided if possible. Another danger element that is shared by the processes described above is the fact that the reactions are performed under pressure. The third industrial disadvantage then arises from the use of heavy metal catalysts, which have a high cost and therefore increase the overall cost of the final active ingredient and may then contaminate the active ingredient fenfluramine even after filtration of the catalyst and purification of said active ingredient. |
| Analysis of the background art shows, however, that an attempt has been made to devise a process for the production or synthesis of fenfluramine that is short (one or two steps) and does not entail the use of hydrogen gas or of catalysts based on nickel or palladium or the like. In particular, for example, Synthesis 1987, 11, p. 1005, and then DECHEMA Monographien (1989), 112 (Org. Elektrochem.—Angew. Elektrothermie), 367-74, present a method for the synthesis of fenfluramine which starts from 1-(3-trifluoromethyl)phenyl-propan-2-one, which is made to react with ethylamine in great excess, in an electrochemical process, which uses a mercury cathode in a water/ethanol solution with pH 10-11. One obtains fenfluramine with 87% yield. This process has some drawbacks from an industrial standpoint: it is a process of the electrochemical type and therefore requires special equipment which is scarcely widespread, dedicated cells and reactors, and it is not possible to use the classic multipurpose reactors available in the pharmaceutical industry. Furthermore, the use of mercury at the industrial level poses severe environment safety problems, requiring constant health monitoring on workers who manage the equipment and systems for the management and destruction of wastewater that are particularly onerous; finally, mercury can be transferred from the cathode to the reaction environment and therefore to the active ingredient, and this obviously is to be considered very dangerous due to the accumulation of the metal in human beings; small traces of mercury are very toxic. |
| Another method for fenfluramine synthesis in a single step is the one presented in J. Org. Chem, 1979, 44(20), p. 3580. Here the reaction is described between an alkene derivative and ethylamine in the presence of sodium borohydride and mercury nitrate: |
| Again, this process is not interesting from an industrial standpoint since it has the same problems, if not even greater ones, related to the use of mercury (used here as a water-soluble salt) discussed previously. The complication introduced in this process with the use of mercury nitrate together with sodium borohydride highlights the level of innovation of the synthesis path found here. |
| In the past, therefore, it has not been possible to provide a process for synthesizing fenfluramine in a small number of steps by using modern reducing agents that are commonly and easily used. Indeed, while Gaodeng Xuexiao Huaxue Xuebao, 9(2), 1988, p. 134-139, describes and exemplifies the synthesis of 2-N-ethyl-1-phenyl propane by means of (1) the treatment of the precursor ketone with ethylamine followed by (2) sodium cyanoborohydride as reducing agent, Xuexiao Huaxue Xuebao provides no example for fenfluramine. Moreover, for the latter, Xuexiao Huaxue Xuebao indicates a melting point for the hydrochloride of 161° C., a data item that matches the value indicated in the literature initially (see BE609630); these facts prove thats fenfluramine synthesis with cyanoborohydride was not performed, otherwise one cannot explain why the author did not transcribe, in the document, the example of a product that at the time was very important. It should be noted in fact that 1-phenyl propan-2-one and 1-(3-trifluoromethyl)phenyl-propan-2-one can have different reactivities to reductive amination due to the presence of a highly electron-attractor-trifluoromethyl group, hence the need for an example to demonstrate its feasibility. The use of cyanoborohydride shares some disadvantages with other methods discussed in the preceding paragraphs. The excellent selectivity for reductive aminations of this reagent is highly appreciated, but its application can be less advantageous with respect to other reducing systems in the synthesis of fenfluramine, where the latter is intended for therapeutic application in human beings. The reasons for this are the possible contamination of the finished pharmaceutical active ingredient with cyanide ions, the toxicity of the reagent itself and finally the danger of its use. It is known to persons skilled in the art that sodium cyanoborohydride can release hydrocyanic acid if the pH of the reaction environment is acid enough and it is known that hydrocyanic acid is a powerful poison, since it competes with oxygen for hemoglobin coordination. As a consequence of this, particular care must be taken in its use and in the disposal of the production wastewater, which can be contaminated by cyanides. Not least, one must consider that the cost of sodium cyanoborohydride is considerable. |
| To conclude, it can be seen that more than 50 years after the publication of its first synthesis dated 1962, there are still numerous disadvantages or limitations in the synthesis paths developed in the past decades in the literature for the preparation of fenfluramine. |
| Moreover, recently there has been renewed pharmaceutical interest in the fenfluramine molecule, since the possibility of its therapeutic use in severe disorders of infancy has appeared in the medical literature. For example, mention can be made of Ceulemans et al., Epilepsia, 53(7), pages 1131 to 1139, 2012. |
| According to a certain part of medical literature, fenfluramine might therefore be interesting as a medication in a chronic therapy for the treatment of symptoms of epilepsy and other correlated severe disorders. |
| Based on recent medical developments, therefore, the need exists for a synthesis method that is better than the existing ones and can overcome in particular the disadvantages of the processes that are present in the literature. Particularly important, in view of use in chronic therapies for children such as epilepsy and other severe disorders, it would be fundamentally important to identify a path for synthesis of the active ingredient fenfluramine that does not entail the use of heavy metals and/or transition metals, which in a chronic therapy might accumulate in the body of the patients over the years, with severe consequences on health. |
| More generally, it is desirable to identify a synthesis path that uses reagents from which (or from the transformation products of which) it is then possible to easily purify fenfluramine. |
| It would be equally desirable to identify a synthesis path that comprises a small number of synthesis steps and uses reagents that are widely commercially available and easy to use. |
| At the same time, the new identified synthesis path should avoid if possible the formation of byproducts. |
DESCRIPTION OF THE FIGURES
| FIG. 1: impurities generated theoretically by means of the reagents used in the first fenfluramine synthesis according to BE609630. |
| FIG. 2: DSC of crude fenfluramine hydrochloride, obtained by reduction with sodium cyanoborohydride according to test 12 (table B) of the description that follows. |
| FIG. 3: DSC of fenfluramine hydrochloride recrystallized from 2-butanol as in example 2b (reduction with sodium borohydride). |
SUMMARY OF THE INVENTION
| The inventors of the present application have found surprisingly that the aim and objects indicated above are achieved by a new method for the synthesis of fenfluramine or of a pharmaceutically acceptable salt thereof, comprising the transformation of a ketone having the structure (I): |
(MOL) (CDX) wherein R 1 is CF 3, with ethylamine or with a salt thereof, and with a reducing agent chosen from the group consisting of alkaline cation or ammonium borohydride, alkaline cation or ammonium triacetoxyborohydride and alkaline cation or ammonium cyanoborohydride, in which the alkaline cation is always different from lithium cation and mixtures thereof, to yield fenfluramine, optionally followed by the transformation of the obtained fenfluramine into a pharmaceutically acceptable salt.
| Furthermore, the inventors of the present invention have also discovered a new preparation of fenfluramine, which can be obtained by means of the method described hereinafter, and new pharmaceutical compositions that contain it. |
Example 1
Synthesis of Fenfluramine
| A suspension of sodium hydroxide (34.62 g-0.866 mol, 3.5 eq) in 170 mL of methanol, under mechanical agitation, receives the addition, drop by drop, over the course of 30 minutes, of a solution of ethylamine hydrochloride (70.59 g-0.866 mol, 3.5 eq) in 165 mL of methanol, followed by 1-(3-trifluoromethyl)phenyl-propan-2-one (50 g-0.247 mol). The mixture is left under agitation at 20° C. for 4.5 hours, then cooling to 0° C. is performed and a solution of sodium borohydride (9.36 g-0.247 mol) in 19 mL of sodium hydroxide 1M in water is then added drop by drop, keeping the temperature below 10° C. The reaction is then left under agitation at 20° C. for another 2 hours. Once the reaction is complete, 270 mL of methanol are removed at a reduced pressure at 40° C. and then 200 mL of water are added and the mixture is extracted with heptane (200 mL). The aqueous phase is eliminated and the organic phase is washed with water (200 mL×3). The organic phase is concentrated at 50° C. at reduced pressure to yield free base fenfluramine as colorless oil. Yield: 72%; purity: 77%—as listed in test 3 of table A above. |
Example 2
Purification of Fenfluramine
| Purification of free base fenfluramine can be performed in two ways: |
| distillation of the free base |
| crystallization of the fenfluramine hydrochloride salt |
| Depending on the degree of purity that is desired, both purification processes are performed in sequence (distillation first and then crystallization), or only one of the two purification processes is performed. |
Example 2a
Distillation
| Free base fenfluramine (10 g), prepared as in Example 1, is distilled under reduced pressure with a distillation column of the Vigreux type: the distillation heads are eliminated, the fraction that is distilled at 89-90° C. at 6 mmHg, which is the active ingredient fenfluramine (8.5 g) with a high degree of purity, is collected. |
Example 2b
Conversion into Hydrochloride Salt and Crystallization
| Crude fenfluramine, prepared as in Example 1, or purified fenfluramine as in Example 2a, is dissolved in 125 mL of ethyl acetate, and cooling is performed to 0° Celsius under agitation. 272 mL of a solution of 1M HCl in ethyl acetate are added drop by drop at 0° C. The precipitate that forms is filtered and washed with ethyl acetate (125 mL×2) to yield approximately 55 g of solid fraction. The solid fraction is crystallized by 2-butanol (260 mL), keeping the solid for 22 hours at 3° C. under slow agitation before filtering it. Filtering is performed and washing is performed with cold 2-butanol. The solid fraction, fenfluramine hydrochloride, is dried in a vacuum stove, yielding 51.7 g of product. A DSC of the resulting product is shown in FIG. 3. |
PAPER
Journal of Organic Chemistry (1979), 44(20), 3580-3.J. Org. Chem. 1979, 44, 20, 3580–3583
Publication Date:September 1, 1979
https://doi.org/10.1021/jo01334a031https://pubs.acs.org/doi/abs/10.1021/jo01334a031
PATENT
https://patents.google.com/patent/US20170174613A1/en
- Fenfluramine is an amphetamine drug that was once widely prescribed as an appetite suppressant to treat obesity. Fenfluramine is devoid of the psychomotor stimulant and abuse potential of D-amphetamine and interacts with the 5-hydroxytryptamine (serotonin, 5-HT) receptors to release 5-HT from neurons. Fenfluramine has been investigated as having anticonvulsive activity in the treatment of Dravet Syndrome, or severe myoclonic epilepsy in infancy, a rare and malignant epileptic syndrome. This type of epilepsy has an early onset in previously healthy children.
- [0003]
Anorectic treatment with fenfluramine has been associated with the development of cardiac valvulopathy and pulmonary hypertension, including the condition cardiac fibrosis which led to the withdrawal of fenfluramine from world-wide markets. Interaction of fenfluramine’s major metabolite norfenfluramine with the 5-HT2B receptor is associated with heart valve hypertrophy. In the treatment of epilepsy, the known cardiovascular risks of fenfluramine are weighed against beneficial anticonvulsive activity.


- [0097]
Chemical Abstract Service (CAS) Registry Number (RN): 404-82-0 (HCl Salt), 458-24-2 (Parent Free Base) - [0098]
Chemical Name: N-ethyl-α-methyl-3-(trifluoromethyl)-benzeneethanamine hydrochloride (1:1). Other Names: Fenfluramine HCl, DL-Fenfluramine, (±)-Fenfluramine - [0099]
Structure of Hydrochloride Salt: - [0100]
Stereochemistry: Fenfluramine HCl has one chiral center and is being developed as the racemate and contains dexfenfluramine and levofenfluramine - [0101]
Molecular Formula of hydrochloride salt: C12H16F3N.HCl - [0102]
Molecular Mass/Weight: 267.72 g/mol
2. General Properties
- [0103]
Table 1 summarizes the chemical and physical properties of Fenfluramine HCl. - TABLE 1 General Properties of Fenfluramine HCl Drug Substance Property Result Appearance (color, White to off-white powder physical form) DSC (melting 170° C. (melt/sublimation) point)a TGA Onset 147° C. 0.03% at 150° C. 91% at 220° C. (evaporation) pKa (water) 10.15-10.38 Solubility (mg/mL) Resultant pH 25° C. 37° C. Solubility pH 6.69 (water) 54.13 71.22 (Aqueous) pH 1.73 buffer 25.34 53.68 pH 3.43 buffer 29.50 61.97 pH 6.41 buffer 37.42 95.60 0.9% NaCl (water) 22.98 — Solvent Solubility 25° C. (mg/mL) Solubility (Organic Ethanol 150 Solvents) Dichloromethane 30-35 Ethyl Acetate, 1-5 mg Tetrahydrofuran, Toluene, Acetonitrile UV Absorption Maxima: 210, 265 nm Solution pH (water) 6.69 Hygroscopicity @30% RH: ~0.05% (Dynamic Vapor @60% RH: ~0.07% Sorption (DVS) @90% RH: ~0.20%a) Polymorphism Fenfluramine HCl has been consistently isolated as a single crystalline Form 1 as determined by DSC and x-ray powder diffraction (XRPD) Solvation/Hydration Fenfluramine HCl is isolated as a nonhydrated, nonsolvated solid Solution Stability 8 weeks @ pH 6.7 phosphate buffer medium at 40° C. and 60° C. using concentrations of 0.5, 2.5 and 5.0 mg/ml. All conditions, no new impurities >0.1% by HPLC. Solid Stability 8 weeks @ 40° C., 60° C. and 80° C. 7 days at 150° C. All conditions, no new impurities >0.1% by HPLC.
3. Synthesis of Fenfluramine Drug Substance
- [0104]
Scheme 3.1 shows a 2-step route of synthesis used to manufacture initial clinical supplies of Fenfluramine HCl from ketone (2). The batch size is 4 kg performed in laboratory glassware (kilo lab). No chromatography is required and the process steps are amenable to scale-up. In process 1 there is one isolated intermediate Fenfluramine Free Base (1) starting from commercially supplied 1-(3-(trifluoromethyl)phenyl) acetone (Ketone 2). All steps are conducted under cGMPs starting from Ketone (2). - [0105]
Scheme 3.2 shows a 4-step route of synthesis to Fenfluramine HCl that can be used for commercial supply. Route 2 utilizes the same 2-step process used by Route 1 to convert Ketone (2) to Fenfluramine HCl with the exception that Ketone (2) is synthesized under cGMP conditions starting from 3-(Trifluoromethyl)-phenyl acetic acid (Acid 4). Bisulfate Complex (3) is an isolatable solid and can be purified before decomplexation to Ketone (2). In-situ intermediates which are oils are shown in brackets. Batch sizes of 10 Kg are performed. Commercial batch sizes of 20 kg are performed in fixed pilot plant equipment. Steps 1-2 of Scheme 3.2 to manufacture Ketone (2) have been demonstrated on a 100 g scale to provide high purity ketone (2) of >99.8% (GC & HPLC). Conversion of Ketone (2) to Fenfluramine using either Route 1 or 2 has provided similar purity profiles. - Starting materials are designated by enclosed boxes. Bracketed and non bracketed compounds respectively indicate proposed in-situ and isolated intermediates. NMI=N-Methyl Imidazole.
4.1. Narrative Description (Route 1)
- [0106]
Step 1: Reductive Amination (Preparation of Fenfluramine Free Base 1) - [0107]
A solution of ethylamine, water, methanol, and 1-(3-(trifluoromethyl)phenyl) acetone (Ketone 2) was treated with sodium triacetoxyborohydride and stirred for 16 h at 25° C. at which time HPLC analysis (IPC-1; In Process Control No. 1) showed the reaction to be complete and sodium hydroxide solution was added until pH>10. Toluene was added and the phases separated, and the aqueous phase (IPC-2) and organic phase (IPC-3) are checked for remaining Fenfluramine and Fenfluramine alcohol and the organic phase was reduced. Purified water was added and the pH adjusted to <2 using conc. HCl and the phases were separated. The aqueous phase was washed with toluene and the toluene phase (IPC-4) and the aqueous phase (IPC-5) was checked for Fenfluramine and Fenfluramine alcohol content. The aqueous phase containing product is pH adjusted to >10 using sodium hydroxide solution. The basic aqueous phase was extracted with MTBE until removal of Fenfluramine from the aqueous phase was observed by HPLC (<0.5 mg/ml) (IPC-6). The organic phase was dried over sodium sulfate and filtered. The filtrate was concentrated in vacuo to give the intermediate product Fenfluramine Free Base 1 as a pale yellow oil tested per specifications described herein which showed by NMR the material to contain 2.93% toluene giving an active yield of 88.3% with a purity of 98.23% by HPLC (0.67% Fenfluramine alcohol). - [0108]
Step 2: Salt Formation (Preparation of Fenfluramine HCl) - [0109]
To a flask was charged ethanol and acetyl chloride. The solution was stirred slowly overnight before ethyl acetate was added. The HCl in ethyl acetate solution formed was polish filtered into a clean carboy and retained for later use. To a vessel was added Fenfluramine free base 1 and MTBE. The Fenfluramine solution in MTBE was collected in two carboys before the vessel was cleaned and checked for particulate residue. The Fenfluramine solution was polish filtered into a vessel and cooled and HCl in ethyl acetate solution was added giving a final pH of 6-7. The batch was stirred for 1 h and filtered. The product was dried under vacuum at 40° C. The product (96.52% yield) was tested per IPC-7 had a purity of 99.75% by HPLC and GC headspace analysis showed MTBE (800 ppm) and EtOAc (150 ppm) to be present. The product was then tested per specifications shown herein.
4.2. Narrative Description (Route 2)
- [0110]
Step 1: Preparation of Ketone Bisulfite Adduct - [0111]
Procedure: Charge acetic anhydride, (2.8 vol, 3.0 wt, 5.0 eq.) to a vessel and commence stirring. Cool the solution to −5 to 5° C., targeting −4° C. Charge 1-methylimidazole, (0.2 vol, 0.21 wt, 0.5 eq.) to the mixture at −5 to 5° C. Caution: very exothermic. If necessary, adjust the temperature to 0 to 5° C. Charge ZX008 acid, (1.00 wt, 1.0 eq.) to the mixture at 0 to 5° C. Caution: exothermic. Stir the mixture at 0 to 5° C. until ≦2.1% area ZX008 acid by HPLC analysis, typically 7 to 9 hours. Charge 15% w/w sodium chloride solution (2.0 vol) to the mixture at 0 to 5° C., 60 to 90 minutes. Caution: very exothermic which will be slightly delayed. Warm the mixture to 18 to 23° C. over 45 to 60 minutes and continue stirring for a further 30 to 45 minutes at 18 to 23° C. Charge TBME, (5.0 vol, 3.7 wt) to the mixture and stir for 10 to 15 minutes at 18 to 23° C. Separate the aqueous layer and retain the organic layer. Back-extract the aqueous layer with TBME, (2×3.0 vol, 2×2.2 wt) at 18 to 23° C. retaining each organic layer. Adjust the pH of the combined organic layer to pH 6.5 to 9.0, targeting 7.0 by charging 20% w/w sodium hydroxide solution (5.3 to 8.3 vol) at 18 to 23° C. Caution: exothermic. Separate the aqueous layer and retain the organic layer. Wash the organic layer with 4% w/w sodium hydrogen carbonate solution (2×3.0 vol) at 18 to 23° C. Determine the residual ZX008 acid content in the organic layer by HPLC analysis, pass criterion ≦0.10% area ZX008 acid. Wash the organic layer with purified water, (2×3.0 vol) at 18 to 23° C. Concentrate the organic layer under reduced pressure to ca. 2 vol at 40 to 45° C., targeting 43° C. - [0112]
Determine the w/w assay of ZX008 ketone (WIP) in the mixture by 1H-NMR analysis for information only and calculate the contained yield of ZX008 ketone (WIP) in the mixture. Note: This step can be removed from the process since the process is robust and consistently delivers 80 to 90% th yield. The achieved yield was factored into the charges of the subsequent steps. - [0113]
Charge n-heptane, (4.0 vol, 2.7 wt) to the mixture at 40 to 45° C., targeting 43° C. Concentrate the mixture to ca. 2 vol at 40 to 45° C., targeting 43° C. Determine the TBME content in the mixture by 1H-NMR analysis, (pass criterion ≦5.0% w/w TBME vs. ZX008 ketone). Charge n-heptane, (2.4 vol, 1.6 wt) at 40 to 45° C., targeting 43° C., vessel A. To vessel B, charge sodium metabisulfite, (0.82 wt, 0.88 eq.) at 18 to 23° C. To vessel B, charge a solution of sodium hydrogen carbonate, (0.16 wt, 0.4 eq.) in purified water, code RM0120 (2.0 vol) at 18 to 23° C. followed by a line rinse with purified water, code RM0120 (0.4 vol) at 18 to 23° C. Caution: gas evolution. Heat the contents of vessel B to 40 to 45° C., targeting 43° C. Charge the contents from vessel A to vessel B followed by a line rinse with n-heptane, (0.8 vol, 0.5 wt) at 40 to 45° C., targeting 43° C. Stir the mixture for 1 to 1.5 hours at 40 to 45° C., targeting 43° C. Charge n-heptane, code RM0174 (3.2 vol, 2.2 wt) to the mixture with the temperature being allowed to cool to 18 to 45° C. at the end of the addition. Cool the mixture to 18 to 23° C. at approximately constant rate over 45 to 60 minutes. Stir the mixture at 18 to 23° C. for 1.5 to 2 hours. - [0114]
Sample the mixture to determine the residual ZX008 ketone content by 1H-NMR analysis, (pass criterion ≦10.0% mol, target 5.0% mol ZX008 ketone vs. ZX008 ketone bisulfite adduct). Filter the mixture and slurry wash the filter-cake with n-heptane, (2×2.0 vol, 2×1.4 wt) at 18 to 23° C. Dry the solid at up to 23° C. until the water content is <10.0% w/w water by KF analysis according to AKX reagent. At least 16 hours. Determine the w/w assay of the isolated ZX008 ketone bisulfite adduct by 1H-NMR analysis and calculate the contained yield of ZX008 ketone bisulfite adduct. - [0115]
Yields and Profiles: The yield for the stage 1 Demonstration batch is summarized Table below. Input: 1700.0 g uncorr., acid, 99.50% area (QC, HPLC), 2-isomer not detected, 4-isomer 0.02% area, RRT1.58 (previously not observed) 0.48% area as per the preparative method. The analytical data is summarized in Table 1A below. - TABLE 1A Table for isolated yields for step 1 Demonstration batch Corr. % area Reference Corr. Yield % w/w (HPLC, number Input Output (% th)** (1H-NMR)* QC) Comments Batch A1 1700.0 g 1500.1 g 89.1 45.0 —.— Crude ketone as TBME sol. Batch A2 1500.1 g 1716.1 77.8 76.0 98.15 Bisulfite adduct only 67.3 Overall product
- [0116]
Step 2: Preparation of Ketone - [0117]
Procedure: Charge toluene, (5.0 vol, 4.3 wt), and purified water, (5.0 vol) to the vessel and commence stirring. If necessary, adjust the temperature to 18 to 23° C. and charge ZX008 ketone bisulfite adduct, (1.00 wt corrected for % w/w assay) to the mixture at 18 to 23° C. Charge 20% w/w sodium hydroxide solution to the mixture at 18 to 23° C. adjusting the pH of the mixture to pH 8.0 to 12.0, targeting 9.0 (0.5 to 1.0 vol). - Separate the lower aqueous layer and retain the top organic layer. Wash the organic layer with purified water, (3.0 vol) at 18 to 23° C. Concentrate the organic layer under reduced pressure to ca. 2 vol at 45 to 50° C., targeting 48° C. Charge methanol, (5.0 vol, 4.0 wt) to the mixture at 45 to 50° C., targeting 48° C. Re-concentrate the mixture under reduced pressure to ca. 2 vol at 45 to 50° C., targeting 48° C. Repeat steps 7 and 8 once before continuing with step 9. Cool the mixture to 18 to 23° C. Clarify the mixture into a tared, suitably-sized drum followed by a methanol (1.0 vol, 0.8 wt) line rinse at 18 to 23° C. Determine the w/w assay of ZX008 ketone (WIP) in the mixture by 1H-NMR analysis and calculate the contained yield of ZX008 ketone (WIP) in the mixture. Determine the toluene content in the mixture by 1H-NMR analysis.
- [0118]
Yields and Profiles: The yield for the step 2 Demonstration batch is summarized in Table 1B below. Input: 1200.0 g corr. Ketone bisulfite adduct, 76.0% w/w assay (NMR, using DMB as internal standard in d6-DMSO), (1.00 eq, 1.00 wt corr. for w/w assay) for input calculation. - TABLE 1B Table for isolated yields for step 2 Demonstration batch % w/w % area Corr. Corr. Corr. Yield (1H- (HPLC, Input Output (% th) NMR)* QC) Comments 1200.0 g 858.15 g 108.3 25.5 99.31 Purified ketone
- [0119]
Step 3: Preparation of Fenfluramine HCl Crude - [0120]
Procedure: Charge the ZX008 ketone (corr. for assay, 1.00 wt, 1.00 eq. isolated as solution in MeOH in stage 2) to a vessel. Charge methanol, code RM0036 (5.0 vol, 4.0 wt) to the mixture at 18 to 23° C. Cool the solution to 0 to 5° C. Charge 70 wt % aqueous ethylamine solution (1.3 vol, 1.6 wt, 4.0 eq) to the mixture at 0 to 10° C., over 15 to 30 minutes, followed by a line rinse with methanol (1.0 vol, 0.8 wt). Warm the mixture to 15 to 20° C. and stir the mixture for a further 60 to 70 minutes at 15 to 20° C. Adjust the mixture to 15 to 18° C. if required, targeting 15° C. Charge sodium triacetoxyborohydride (2.4 wt, 2.25 eq.) to the mixture in approximately 10 portions, keeping the mixture at 15 to 20° C., targeting 17° C. Addition time 1.5 to 2 hours. Caution: Exothermic. Stir the mixture at 15 to 20° C. until complete by HPLC analysis, pass criterion ≦3.0% area ZX008 ketone, typically 2 to 3 hours. Adjust the pH of the mixture to pH>12 by charging 20% w/w aqueous sodium hydroxide solution (5.0 to 6.0 vol) to the mixture at 15 to 40° C. Addition time 10 to 30 minutes. Caution: Exothermic. If necessary, adjust the temperature to 18 to 23° C. Extract the mixture with toluene (3×3.0 vol, 3×2.6 wt) at 18 to 23° C., retaining and combining the top organic layer after each extraction. Wash the combined organic layer with purified water, (1.0 vol) at 18 to 23° C. Heat the mixture to 40 to 50° C., targeting 48° C. Concentrate the mixture under reduced pressure at constant volume maintaining ca. 5 vol by charging the organic layer at approximately the same rate as the distillation rate at 40 to 50° C., targeting 48° C. Cool the mixture to 18 to 23° C. Charge purified water (10.0 vol) to the mixture at 18 to 23° C. Adjust the pH of the mixture to 0.1<pH<1.5 at 18 to 23° C. by charging concentrated hydrochloric acid, 0.5 vol. Do not delay from this step until neutralization. - [0121]
Separate the layers at 18 to 23° C. retaining the bottom aqueous layer. Wash the aqueous layer with toluene, (3.0 vol, 2.6 wt) at 18 to 23° C. retaining the aqueous layer. Adjust the pH of the aqueous layer to pH>12 by charging 20% w/w sodium hydroxide solution at 18 to 23° C. 0.8 to 0.9 vol. Caution: Exothermic. Charge TBME, code RM0002 (2.0 vol, 1.5 wt) to the basic aqueous layer. Separate the layers at 18 to 23° C. retaining the top organic layer. Back-extract the aqueous layer with TBME (2×2.0 vol, 2×1.5 wt) at 18 to 23° C. retaining the organic layers. Wash the combined organic layer with purified water, (2×1.0 vol) at 18 to 23° C. Concentrate the combined organic layers under reduced pressure at 40 to 50° C., targeting 48° C. to ca. 3 vol. Determine the residual toluene content of the mixture by 1H-NMR analysis. Sample for determination of residual water content by KF analysis, AKX reagent. Charge TBME (8.7 vol, 6.4 wt) to the mixture at 40 to 50° C. Cool the solution to 0 to 5° C., targeting 2° C. Charge concentrated hydrochloric acid (0.54 vol, 0.46 wt) maintaining the temperature <15° C. Caution: Exothermic. Line rinse with TBME (1.0 vol, 0.7 wt). If necessary, adjust the temperature to 0 to 10° C. and stir the mixture at 0 to 10° C. for a further 2 to 3 hours. Filter the mixture and wash the filter-cake with TBME (2×4.4 vol, 2×3.3 wt) at 0 to 10° C. Dry the solid at up to 40° C. until the TBME content is <0.5% w/w TBME by 1H-NMR analysis. 4 to 8 hours. - [0122]
Yields and Profiles: The yield for the step 3 Demonstration batch is summarized in Table 1C below. Input: 856.8 g corr. Ketone, 44.2% w/w assay (NMR, using TCNB as internal standard in CDCl3), (1.00 eq, 1.00 wt corr. for w/w assay) for input calculation. FIG. 2 and Table 1D shows an exemplary HPLC chromatogram of a crude preparation of fenfluramine hydrochloride (210 nm UV absorbance). - TABLE 1C Table for isolated yields for step 3 Demonstration batch Corr. % area Reference Corr. Corr. Yield % w/w (HPLC, number Input Output (% th) (1H-NMR)* QC) Comments Batch A1 856.8 g 836.31 g 85.3 44.2 99.15 Fenfluramine free base (in situ intermediate) Batch A2 880.7 84.0 based 99.5 100.00 Fenfluramine•HCl on ketone crude (step 3 an bisulfite d 4.1) adduct (77.6 based on purified ketone)
- TABLE 1D Purity of crude fenfluramine hydrochloride by HPLC (see FIG. 2) Processed Channel Descr. DAD AU Ch 1 Sample 210, Bw 4 Peak Results USP USP USP Name RT RelRT Area Height Tailing Resolution Plate Count EP s/n % Area 1 NorFenfluramine 7.46 2 2-Fenfluramine 7.68 3 Fenfluramine 8.67 1.000 3789064 778178 1.7 70796 2549.8 99.15 4 4-Fenfluramine 8.95 5 11 34 1.308 6073 1449 1.2 23.5 215529 3 8 0.16 6 ZX008 acid 12.93 7 Fenfluramine alcohol 14.16 1.633 15266 2972 1.3 24.8 215040 8.7 0.40 8 ZX008 ketone 14.83 9 Fenfluramine acetamide 15.55 10 TOLUENE 15 75 11 15.92 1.836 4110 1122 2.7 0.11 12 16.60 1.915 6861 1630 1.5 451209 4.3 0.18 Sum 3821374 100.00
- [0123]
Step 4.2: Crystallization of Fenfluramine Hydrochloride - [0124]
Procedure: Charge Fenfluramine.HCl (crude) (1.00 wt, 1.0 eq.) and TBME (10.0 vol, 7.4 wt) to the vessel and commence stirring. Heat the suspension to reflux (50 to 58° C.). Charge ethanol (5.0 vol, 3.9 wt) maintaining the temperature at 50 to 58° C. Addition time 20 minutes. Stir at 50 to 58° C. for 5 to 10 minutes and check for dissolution. Stir the solution at 50 to 58° C. for 5 to 10 minutes, targeting 54 to 58° C. Clarify the reaction mixture through a 0.1 μm in-line filter at 54 to 58° C., followed by a line rinse with TBME (1 vol, 0.7 wt). Cool the solution to 48 to 50° C. Charge Fenfluramine HCl, code FP0188 (0.01 wt). Check for crystallization. Allow the suspension to cool to 15 to 20° C., target 17° C. over 5 to 5.5 hours at an approximately constant rate. Stir the mixture at 15 to 20° C., target 17° C. for 2 to 3 hours. Filter the mixture and wash the filter-cake with clarified TBME (2×3.0 vol, 2×2.2 wt) at 5 to 15° C. Dry the solid at up to 40° C. until the TBME content is <0.5% w/w TBME and the ethanol content is <0.5% w/w EtOH by 1H-NMR analysis. 4 to 8 hours. Determine the w/w assay of the isolated Fenfluramine.HCl by 1H-NMR analysis. - [0125]
Yields and Profiles: The yield for the stage 4 Demonstration batch is summarized in Table 1E below. Input: 750.0 g uncorr. Fenfluramine HCl crude (1.00 eq, 1.00 wt uncorr.) for input calculation. FIG. 3 shows an exemplary HPLC chromatogram of a crystallized fenfluramine hydrochloride sample (210 nm UV absorbance). - TABLE 1E Table for isolated yields for stage 4 Demonstration batch Uncorr. Uncorr. Uncorr. Yield HPLC (% area, Input Output (% th) QC) Comments 750.0 g 608.0 81.1 100.00* Fenfluramine•HCl
PATENT
https://patents.google.com/patent/EP3170807A1/en
- Fenfluramine, i.e., 3-trifluoromethyl-N-ethylamphetamine, has the following chemical structure:
- [0003]
The marketing of fenfluramine as a pharmaceutical active ingredient in the United States began in 1973 and was used in a therapy in combination with phentermine to prevent and treat obesity. However, in 1997 fenfluramine was withdrawn from the market in the United States and immediately thereafter in other countries, since its ingestion was associated with the onset of cardiac fibrosis and pulmonary hypertension. As a consequence of this event, the pharmaceutical compounds containing this active ingredient were withdrawn from the market. However, fenfluramine, even after its exit from the market, has continued to attract scientific interest, as will become apparent from the discussion presented hereinafter. - [0004]
In the literature, over the years, numerous syntheses or processes have been reported for preparing fenfluramine or its dextrorotatory enantiomer dexfenfluramine or an analog containing a highly electron-attractor group on the aromatic ring as in the fenfluramine molecule (see for example Pentafluorosulfanyl Serotonin Analogs: Synthesis, Characterization, and Biological Activity, John T. Welch and Dongsung Lim Chapter 8, pp 165-181 DOI: 10.1021/bk-2009-1003.ch008). Many of these synthesis paths are long and foresee multiple stages or synthesis steps that can include reagents that are dangerous or scarcely environment-friendly and are therefore scarcely convenient for an industrial synthesis. Hereinafter, any reference to “fenfluramine” is understood to referto the racemic form, i.e, (RS)-N-ethyl-1-[3-(trifluoromethyl)phenyl]propan-2-amine. - [0005]
To the best of the knowledge of the inventors, the first method for fenfluramine synthesis reported in the literature dates back to 1962 and is referenced in patent BE609630 and in analogous patents US3198833 and FR1324220 . All the synthesis methods reported in these patents provide for numerous synthesis steps. By way of example, one of the methods provides for the transformation into oxime of a ketone, 1-(3-trifluoromethyl)phenyl-propan-2-one, as shown here: - [0006]
The oxime is then hydrogenated in the presence of Raney nickel catalyst so as to yield the corresponding primary amine, which is acetylated subsequently with ethanoic anhydride before being converted into fenfluramine by reduction with lithium aluminum hydride. - [0007]
As can be seen, the final step of this chemical process provides for the use of lithium aluminum hydride and the persons skilled in the art will acknowledge that the use of this reagent should be avoided, if possible, on an industrial level, since it is extremely flammable and is the source of accidents. Furthermore, lithium is a potentially neurotoxic metal and therefore its use should be avoided where possible. Furthermore, the Raney nickel catalyst is used in the oxime reduction step and can contaminate the final active ingredient; the use of hydroxylamine also entails problems of toxicity for workers assigned to production. - [0008]
A further disadvantage of this process is, as already mentioned earlier, the number of steps, not only because a large number of synthesis steps entails a reduction of the overall yield of active ingredient, but also because each synthesis step in principle can generate impurities and a larger number of steps can therefore entail a higher number of impurities in the final active ingredient. Many of these impurities, furthermore, due to their structural similarity to fenfluramine, are difficult to eliminate and remove from a fenfluramine preparation. One impurity for example that can be formed in the process described above and is difficult to eliminate is the following: - [0009]
This impurity, which is a primary amine, shares physical-chemical properties that are similar to fenfluramine and therefore, like fenfluramine, it can form a hydrochloride salt by treatment with hydrochloric acid and thus contaminate the active ingredient fenfluramine hydrochloride. Furthermore, this impurity – as a free base – has a boiling point that is similar to that of fenfluramine (73°C vs. 89°C at 6 mmHg respectively), and therefore its elimination by distillation also can be problematic. - [0010]
The process described above can in principle generate other impurities, which are listed in Figure 1 . - [0011]
EP 0441160 claims a synthesis in 5 steps of dexfenfluramine, dextrorotatory enantiomer of fenfluramine. This synthesis can be adapted easily to produce fenfluramine instead of its dextrorotatory enantiomer simply by performing the first reduction step with a non-chiral reducing agent. In the first step, in fact:a ketone, 1-(3-trifluoromethyl)phenyl-propan-2-one, is first reduced to the corresponding alcohol in the presence of yeast, D-glucose, ethanol and water. Then the alcohol is converted into the tosylate in the second step: - [0012]
This reaction occurs in the presence of triethylamine and tosyl chloride in methylene chloride as solvent. After purification, the tosylate is converted to fenfluramine by means of three successive steps: - [0013]
In the first of these three steps, the tosylate is converted into an azide intermediate by reaction with sodium azide in dimethylformamide. The azide intermediate is then hydrogenated in the presence of a catalyst, palladium on carbon. Finally, the resulting primary amine is converted into fenfluramine by reaction with acetaldehyde and sodium borohydride. - [0014]
Persons skilled in the art may see easily that this process is not desirable from an industrial standpoint due to reasons related to environmental risk, safety and costs. For example, the sodium azide used in the process is a notoriously explosive compound and its use at the industrial level is dangerous. Furthermore, palladium is an expensive material and its use in the process entails an increase in the production costs of fenfluramine. Furthermore, palladium can contaminate the finished active ingredient. - [0015]
In another method for the synthesis of dexfenfluramine in 3-4 steps, reported by Goument et al. in Bulletin of the Chemical Society of France (1993), 130, p. 450-458, 3-bromobenzotrifluoride is subjected to a Grignard reaction with enantiopure 1,2-propylene-epoxide to yield 1-[3-(trifluoromethyl)phenyl]propan-2-ol as shown hereafter: - [0016]
If this reaction is performed with racemic 1,2-propylene-epoxide, the synthesis can be adapted to the preparation of fenfluramine. - [0017]
The alcohol thus obtained is first transformed into trifluoromethyl sulfonate by reaction with trifluoromethanesulfonic anhydride and then treated with ethylamine to yield fenfluramine, as shown in the diagram hereinafter: - [0018]
In this article, the authors acknowledge that the main byproducts of the reaction are isomer alkenes having the following chemical structures: - [0019]
The process proposed by Goument et al. is not interesting from the industrial standpoint for a series of reasons. First of all, it is known that the use of Grignard reagents, especially on an industrial scale, is problematic, because these compounds are often pyrophoric and corrosive. Furthermore, 1,2-propylene epoxide is a suspected carcinogenic compound. Finally, the formation of the three isomer alkenes as byproducts listed above is a disadvantage of the process. In the article, Goument presents methods for activation of the intermediate alcohol which are alternative to trifluoromethylsulfonate, for example by converting it to chloride (via thionyl chloride) or to mesylate (via mesyl chloride), but these process variations share the same disadvantages as the main process analyzed above. - [0020]
In addition to the methods with multiple synthesis steps discussed so far in detail, the literature reports other methods or processes for producing fenfluramine or dexfenfluramine. In general, persons skilled in the art acknowledge that the syntheses in the literature for producing dexfenfluramine sometimes can be applied to the preparation of fenfluramine simply by replacing the initial materials and/or enantiopure reagents with the corresponding racemates while maintaining the reaction conditions. For example, patents that present long synthesis methods in multiple steps are the following: - [0021]
Other examples of preparation of fenfluramine, taken from non-patent literature, are the following:- Synthesis, Nov.1987, p. 1005-1007
- J.Org.Chem, 1991, 56, p. 6019
- Tetrahedron, 1994, 50(1), p. 171
- Bull. Soc. Chim. France, 1993, 130(4), p. 459-466 (dexfenfluramine)
- Chirality, 2002, 14(4), p. 325-328 (dexfenfluramine)
- [0022]
Without analyzing in detail the individual methods described in these patents or articles, it can be stated in summary that all these methods are not attractive and interesting from the industrial standpoint because these are processes with many synthesis steps or because the initial materials described therein are not easily available and therefore have to be prepared separately, with a further expenditure of time and with further costs, or because they provide for the use of reagents that are dangerous/explosive/toxic or because they entail the use of catalysts based on heavy metals that can contaminate the final active ingredient. - [0023]
One should consider that in the literature there are methods for the preparation of fenfluramine that do not provide for long syntheses and multiple steps but are shorter and consist of one or two steps. These processes, which therefore would be more interesting from the industrial standpoint, have other specific disadvantages, as will become apparent in detail hereinafter. For example, in the literature there is a first group of articles or patents that describe the reaction between 1-(3-trifluoromethyl)phenyl-propan-2-one and ethylamine in the presence of hydrogen gas and of a transition metal as catalyst: - [0024]
In particular, in Huagong Shikan, 2002, 16(7), p. 33, the reaction is performed with hydrogen gas (2.9 – 3.38 atm), at 65-75°C, for 9 hours, in the presence of Raney nickel. Likewise, in patent DD108971 (1973), Raney nickel and hydrogen gas and methanol are used as solvent to perform this reaction. - [0025]
In HU55343 , instead, a similar reaction in one step is performed with hydrogen gas in the presence of another transition metal catalyst, such as palladium on carbon. - [0026]
Although these three methods describe short single-step processes, they have the disadvantage of the use of hydrogen gas. As is known to persons skilled in the art, hydrogen gas is a dangerous gas due to the inherent danger of forming explosive mixtures with air and must be used by expert personnel in expensive facilities dedicated to its use and built with special precautions. Despite being used in purpose-built facilities, the use of hydrogen at the industrial level is inherently dangerous and to be avoided if possible. Another danger element that is shared by the processes described above is the fact that the reactions are performed under pressure. The third industrial disadvantage then arises from the use of heavy metal catalysts, which have a high cost and therefore increase the overall cost of the final active ingredient and -on the other hand- may contaminate the active ingredient fenfluramine even after filtration of the catalyst and purification of said active ingredient. - [0027]
Analysis of the background art shows, however, that an attempt has been made to devise a process for the production or synthesis of fenfluramine that is short (one or two steps) and does not entail the use of hydrogen gas or of catalysts based on nickel or palladium or the like. In particular, for example, Synthesis 1987, 11, p. 1005, and then DECHEMA Monographien (1989), 112 (Org. Elektrochem.–Angew. Elektrothermie), 367-74, present a method for the synthesis of fenfluramine which starts from 1-(3-trifluoromethyl)phenyl-propan-2-one, which is made to react with ethylamine in great excess, in an electrochemical process, which uses a mercury cathode in a water/ethanol solution with pH 10-11. One obtains fenfluramine with 87% yield. This process has some drawbacks from an industrial standpoint: it is a process of the electrochemical type and therefore requires special equipment which is scarcely widespread, dedicated cells and reactors, and it is not possible to use the classic multipurpose reactors available in the pharmaceutical industry. Furthermore, the use of mercury at the industrial level poses severe environment safety problems, requiring constant health monitoring on workers who manage the equipment and systems for the management and destruction of wastewater that are particularly onerous; finally, mercury can be transferred from the cathode to the reaction environment and therefore to the active ingredient, and this obviously is to be considered very dangerous due to the accumulation of the metal in human beings; small traces of mercury are very toxic. - [0028]
Another method for fenfluramine synthesis in a single step is the one presented in J.Org.Chem, 1979, 44(20), p. 3580. Here the reaction is described between an alkene derivative and ethylamine in the presence of sodium borohydride and mercury nitrate: - [0029]
Again, this process is not interesting from an industrial standpoint since it has the same problems, if not even greater ones, related to the use of mercury (used here as a water-soluble salt) discussed previously. The complication introduced in this process with the use of mercury nitrate together with sodium borohydride highlights the level of innovation of the synthesis path found here. - [0030]
In past years, therefore, it has not been possible to provide a process for synthesizing fenfluramine in a small number of steps by using modern reducing agents that are commonly and easily used. Indeed, while Gaodeng Xuexiao Huaxue Xuebao, 9(2), 1988, p. 134-139, describes and exemplifies the synthesis of 2-N-ethyl-1-phenyl propane by means of (1) the treatment of the precursor ketone with ethylamine followed by (2) sodium cyanoborohydride as reducing agent, Xuexiao Huaxue Xuebao provides no example for fenfluramine. Moreover, for the latter, Xuexiao Huaxue Xuebao indicates a melting point for the hydrochloride of 161°C, a data item that matches the value indicated in the literature initially (see BE609630 ); these facts prove that fenfluramine synthesis with cyanoborohydride was not performed, otherwise one cannot explain why the author did not transcribe, in the document, the example of a product that at the time was very important. It should be noted in fact that 1-phenyl propan-2-one and 1-(3-trifluoromethyl)phenyl-propan-2-one can have different reactivities to reductive amination due to the presence of a highly electron-attractor -trifluoromethyl group, hence the need for an example to demonstrate its feasibility. The use of cyanoborohydride shares some disadvantages with other methods discussed in the preceding paragraphs. The excellent selectivity for reductive aminations of this reagent is highly appreciated, but its application can be less advantageous with respect to other reducing systems in the synthesis of fenfluramine, where the latter is intended for therapeutic application in human beings. The reasons for this are the possible contamination of the finished pharmaceutical active ingredient with cyanide ions, the toxicity of the reagent itself and finally the danger of its use. It is known to persons skilled in the art that sodium cyanoborohydride can release hydrocyanic acid if the pH of the reaction environment is acid enough and it is known that hydrocyanic acid is a powerful poison, since it competes with oxygen for hemoglobin coordination. As a consequence of this, particular care must be taken in its use and in the disposal of the production wastewater, which can be contaminated by cyanides. Not least, one must consider that the cost of sodium cyanoborohydride is considerable. - [0031]
To conclude, it can be seen that more than 50 years after the publication of its first synthesis dated 1962, there are still numerous disadvantages or limitations in the synthesis paths developed in the past decades in the literature for the preparation of fenfluramine. - [0032]
Moreover, recently there has been renewed pharmaceutical interest in the fenfluramine molecule, since the possibility of its therapeutic use in severe disorders of infancy has appeared in the medical literature. For example, mention can made of Ceulemans et al., Epilepsia, 53(7), pages 1131 to 1139, 2012. - [0033]
According to a certain part of medical literature, fenfluramine might therefore be interesting as a medication in a chronic therapy for the treatment of symptoms of epilepsy and other correlated severe disorders. - [0034]
Based on recent medical developments, therefore, the need exists for a synthesis method that is better than the existing ones and can overcome in particular the disadvantages of the processes that are present in the literature. Particularly important, in view of use in chronic therapies for children such as epilepsy and other severe disorders, it would be fundamentally important to identify a path for synthesis of the active ingredient fenfluramine or of isomers thereof and/or analogs thereof that does not entail the use of heavy metals and/or transition metals, which in a chronic therapy might accumulate in the body of the patients over the years, with severe consequences on health. - [0035]
More generally, it is desirable to identify a synthesis path that uses reagents from which (or from the transformation products of which) it is then possible to easily purify fenfluramine (or isomers and/or analogs thereof). - [0036]
It would be equally desirable to identify a synthesis path that comprises a small number of synthesis steps and uses reagents that are widely commercially available and easy to use. - [0037]
At the same time, the new identified synthesis path should avoid if possible the formation of byproducts.
EXAMPLES
- [0082]
The present invention is exemplified by, but not limited to, the following examples:
Example 1 – Synthesis of fenfluramine
- [0083]
A suspension of sodium hydroxide (34.62 g – 0.866 mol, 3.5 eq) in 170 mL of methanol, under mechanical agitation, receives the addition, drop by drop, over the course of 30 minutes, of a solution of ethylamine hydrochloride (70.59 g – 0.866 mol, 3.5 eq) in 165 mL of methanol, followed by 1-(3-trifluoromethyl)phenyl-propan-2-one (50 g – 0.247 mol). The mixture is left under agitation at 20°C for 4.5 hours, then cooling to 0°C is performed and a solution of sodium borohydride (9.36 g – 0.247 mol) in 19 mL of sodium hydroxide 1M in water is then added drop by drop, keeping the temperature below 10°C. The reaction is then left under agitation at 20°C for another 2 hours. Once the reaction is complete, 270 mL of methanol are removed at a reduced pressure at 40°C and then 200 mL of water are added and the mixture is extracted with heptane (200 mL). The aqueous phase is eliminated and the organic phase is washed with water (200 mL x 3). The organic phase is concentrated at 50°C at reduced pressure to yield free base fenfluramine as colorless oil. Yield: 72%; purity: 77% – as listed in test 3 of table A above.
Example 2 – Purification of fenfluramine
- [0084]
Purification of free base fenfluramine can be performed in two ways: - distillation of the free base
- crystallization of the fenfluramine hydrochloride salt
- [0085]
Depending on the degree of purity that is desired, both purification processes are performed in sequence (distillation first and then crystallization), or only one of the two purification processes is performed.
Example 2a – Distillation:
- [0086]
Free base fenfluramine (10 g), prepared as in Example 1, is distilled under reduced pressure with a distillation column of the Vigreux type: the distillation heads are eliminated, the fraction that is distilled at 89-90°C at 6 mmHg, which is the active ingredient fenfluramine (8.5 g) with a high degree of purity, is collected.
Example 2b – Conversion into hydrochloride salt and crystallization:
- [0087]
Crude fenfluramine, prepared as in Example 1, or purified fenfluramine as in Example 2a, is dissolved in 125 mL of ethyl acetate, and cooling is performed to 0°Celsius under agitation. 272 mL of a solution of 1M HCl in ethyl acetate are added drop by drop at 0°C. The precipitate that forms is filtered and washed with ethyl acetate (125 mL x 2) to yield approximately 55 g of solid fraction. The solid fraction is crystallized by 2-butanol (260 mL), keeping the solid for 22 hours at 3°C under slow agitation before filtering it. Filtering is performed and washing is performed with cold 2-butanol. The solid fraction, fenfluramine hydrochloride, is dried in a vacuum stove, yielding 51.7 g of product. A DSC of the resulting product is shown in Figure 3 .
CLIP
- Synthetic Method of Dexfenfluramine hydrochloride
- (CAS NO.: ), with its systematic name of (S)-N-Ethyl-alpha-methyl-m-(trifluoromethyl)phenethylamine hydrochloride, could be produced through many synthetic methods.Following is one of the synthesis routes:
 The action of d-camphoric acid on (rac)-fenfluramine (I) affords the camphorate of (+)-fenfluramine (II). After purification of this salt by crystallization, sodium hydroxide in methylene chloride is added, forming (+)-fenfluramine (III) after removal of camphoric acid. Finally, the action of hydrogen chloride in methyl cyclohexane on (+)-fenfluramine produces the corresponding salt: (+)-fenfluramine hydrochloride.
The action of d-camphoric acid on (rac)-fenfluramine (I) affords the camphorate of (+)-fenfluramine (II). After purification of this salt by crystallization, sodium hydroxide in methylene chloride is added, forming (+)-fenfluramine (III) after removal of camphoric acid. Finally, the action of hydrogen chloride in methyl cyclohexane on (+)-fenfluramine produces the corresponding salt: (+)-fenfluramine hydrochloride.
PAPER
https://www.designer-drug.com/pte/12.162.180.114/dcd/chemistry/fenfluramine.html
Fenfluramine 1 is the active ingredient of a obesity drug acting on the digestion of carbohydrates, the activity being restricted mainly to the S enantiomer [1, 2], which can be obtained by separation of the diastereoisomers [3] or by preferential crystallisation of derivates, which were identified of being conglomerates [4]. Only two syntheses of optical active fenfluramine have been described until now: one by stereoselective reduction of the imine derived from the ketone 2 and (R) or (S)-alpha-phenylethylamine [5], the other starting from (S)-alanine [6]. Two recent publications [7, 8] about the synthesis of (S)-fenfluramine via the intermediate alcohol (S)-3 (scheme 1) made us publish our previous results [9]. Through yeast reduction of the ketone 2, the authors obtain the alcohol (S)-3, the configuration of which they inverse in three steps. The alcohol (R)-3, via the intermediate tosylate (R)-4a and further the azide (S)-5 leads to the amine (S)-6 after reduction and finally to the (S)-fenfluramine (S)-1 after reductive amination in presence of acetaldehyde:
The (S)-fenfluramine is such obtained in 7 steps starting from the alcohol (S)-3 or in 4 steps from the alcohol (R)-3.
In this article we present a new way of preparing the two enantiomers of the alcohol 3, a new two step synthesis of (S)-fenfluramine starting from the alcohol (R)-3, a one step synthesis of (S)-fenfluramine starting from the azide (S)-5, which doesn’t pass over the intermediate primary amine (S)-6 and finally a much faster process (3 steps) of preparing (S)-fenfluramine starting from the alcohol (S)-3.
Results and discussion
Synthesis of 1-[3-(trifluoromethyl)phenyl]propan-2-ol (R)-3
The racemic alcohol is seldom mentioned. It’s one of the metabolites of fenfluramine in the human body, secreted in urine [10]. It can also be obtained by metabolic transformations of an oxime by different kinds of microorganisms [11]. It was used as an intermediate for the synthesis of a family of anorexics [12] and a family of antispasmodic and psychotherapeutic agents [13]. It was obtained by the reaction of methyloxirane 7 with the magnesium compound 8, with a yield of 50%. This same reaction was described earlier as being little regioselective [14], a fact we observed too [9].
The only synthesis of the optically active alcohol 3 is the reduction of the ketone 2 with yeast as described above. One abtains the S enantiomer, the R enantiomer is obtained by inversion.
On our part we used the condensation of the commercial [15] methyloxirane (R)-7, of which many syntheses are known [16], with the magnesium compound 8 and cuprous chloride [17, 18].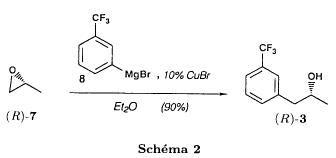
The yield is about 90% and the reaction very selective (purity GC: 93%). The optical purity of the methyloxirane 7 was determined by 1H-NMR in presence of the europium complex Eu(hfc)3 [19]. The optical purity of the alcohol (R)-3 was obtained by 1H-NMR and HPLC over silica of the Mosher derivate [20]. The comparison of these values show that the chiral centre is preserved. This procedure has the advantage of allowing us the preparation of the alcohol (S)-3 with the same reaction, because the methyloxirane (S)-7 is also commercially available and multiple syntheses are known [21].
Two step synthesis of the fenfluramine (S)-1 starting from the alcohol (R)-3
With the goal of obtaining the simplest procedure we have studied at first the transformation of the alcohol (R)-3 into fenfluramine (S)- 1 in two steps via the intermediate of the easily obtained sulfonates (R)-4: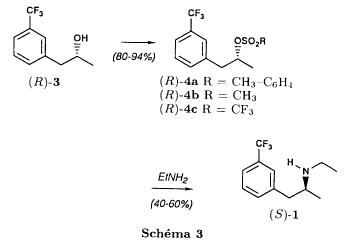
The substitution of the mesitylate (R)-4b and the tosylate (R)-4a with ethylamine was realised with medium yields always between 40 and 50% in spite of the large number of conditions tested: solvents (DMSO, DMF, ethanol, ethylamine), different dilutions (in proportions from 1 to 5) and temparatures from 50 to 160°C (with different times of contact). With the triflate (R)-4c the yield of the substitution is 60% but under non comparable conditions (-20°C in acetonitrile) because of its higher reactivity. In all cases the non aminated, and thus easily separated, byproducts are mainly the alkenes 9, 10Z and 10E (10E >> 10Z > 9).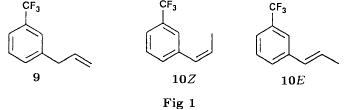
The enantiomeric purity of the amine (S)-1 is analysed by HPLC chromatography through silica of the camphanylated derivate [22] and compared to the previously analysed alcohol (R)-3: we have thus shown that the optical centre is conserved during the nucleophilic substitution. One had indeed to fear that due to the participation of the aromatic ring as neighbour group there could be partial or complete racemisation with an phenonium ion as intermediate. With the results obtained, which match with the literature [23-26], one can suppose that the trifluoromethyl group in meta position is sufficiently deactivating the aromatic ring in order to prevent participation in the substitution. We probably have thus in our case a pure nucleophilic SN2 substitution in competition with an elimination reaction. We believe that this elimination reaction is due to the simultaneous nucleophilic and basic properties of the ethylamine.
Although the yields are medium, this method has the advantage of being relatively fast because it permits to prepare fenfluramine (S)-1 starting from the alcohol (R)-2 in two steps instead of four [7, 8]. As far as we know it was never mentioned in literature.
Synthesis of fenfluramine (S)-1 from 2-azido-1-[3-(trifluoromethyl)phenyl]propane (S)-5
The substitution of the mesylate (R)-4b by sodium azide (scheme 4), an only slightly basic nucleophile compared to ethylamine, forms no elimination side products. One obtains the optically pure azide (S)-5 with a yield of 95%.
The enantiomeric purity couldn’t be directly analysed on the azide (S)-5. Only for analytical purposes did we reduce it into the amine (S)- 6. Among the numerous methods for the reductions of azides to amines mentioned in the literature [27] we chose the catalytic hydrogenation with 5% Pd on calcium carbonate at standard temperature and pressure [27f]. The HPLC analysis through silica column of the champhanyl derivate [22] of the amine (S)-6 such obtained shows that the enantiomeric centre was totally inverted during the substitution when compared to the enantiomeric purity of the alcohol (R)-3.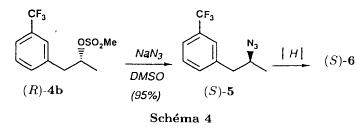
The reductive amination of the amine 6 in presence of acetaldehyde is known for a long time [28]. It was used recently in the works listed in the introduction [7, 8]. On our part, we propose another synthetic route for fenfluramine (S)-1 starting from the azide (S)-5 which does not go via the primary amine (S)-6 (Schema 5).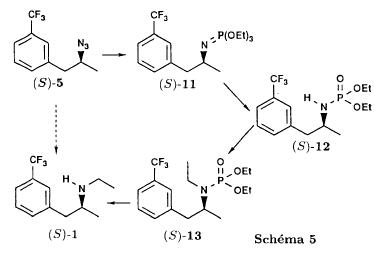
The reaction of Staudinger, reacting a stoichiometric quantity of triethylphosphite on the azide (S)-5 in THF at room temperature [29], gives quantitative yields of the phosphorimide 11 in 48 hours. It’s total conversion into the phosphoramide 13, by reacting with ethyl iodide [30] could not be realised [9]. We always obtained different mixtures of the phosphoramides 12 and 13 (referential compounds prepared from the amines 6 and 1). We also noted that the phosphorimide 11 can’t be isolated. When the solvent is evaporated, a partly transformation into the phosphoramide 12 takes place. This transformation is completed in less then 2h by simple heating to 100°C under argon after evaporation of the solvent. Because the phosphorimides are strongly basic compounds, we believe that an intramolecular arrangement of the phosphorimide, pictured in Schema 6, takes place.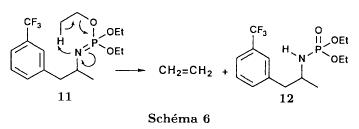
Having the phosphoramide 12, we investigated the alkylation into the phosphoramide 13 in DMF at room temperature [31, 32]: one deprotonates with sodium hydride then alkylates with diethyl sulfate. After treatment with hydrogen bromide [33], one obtains fenfluramine 1 with a yield of 85% and a purity of 97% (GC).
With the goal of simplifying the reaction scheme by avoiding the isolation of the intermediates we have again studied the transformation 5 -> 11 -> 12 in DMF (Schema 7). First, we noted that the reaction of Staudinger can be directly realised in this solvent. Thereafter we pinned down the transformation of the phosphorimide 11 into the phosphoramide 12 by reaction with water [34]. One then proceeds as described above. The transformation is thus performed without isolation of a single intermediate with a yield of 83%.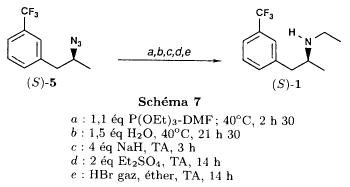
HPLC analysis on silica column of the camphanyl derivate of the amine (S)-1 [22] shows that the optical centre is conserved during the whole transformation.
Synthesis via the intermediate 2-chloro-1-[3-(trifluoromethyl)phenyl]propane 14
The yeast reduction of the ketone 2 gives the alcohol (S)-3, of which the authors have inverted the configuration to get the pharmacological active S enantiomer of fenfluramine [7, 8]. Independent research, using the epoxidation method of Sharpless [9, 35] lead us too to the alcohol (S)- 3 which we tried to convert into fenfluramine (S)-1 using a different method. The reaction scheme we kept uses the chloride 14 and proceeds via two inversions of the optical centre (scheme 8). Not owning enough alcohol (S)-3 during the studies, we tested the principle starting with the alcohol (R)-3, produced earlier, and studied the transformation into the azide (R)-5 (scheme 8), the latter being able to lead to (R)-fenfluramine using different methods, like the one outlined above: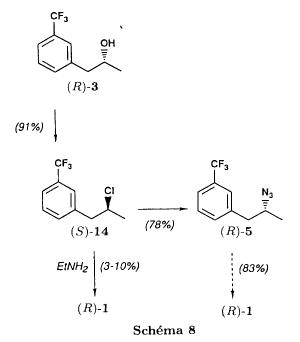
It is well known that the action of thionylchloride on an optically active alcohol gives the corresponding chlorine derivate, with inversion of the configuration in presence of bases and with retention of the configuration in the other case. We have performed the reaction with a catalytical amount of pyridine. One thus obtains the chloride (S)-14 with 91% yield and a purity of 91% (GC): it contains 9% of the elimination products 9, 10Z and 10E which are not separable by chromatography on silica.
The direct substitution of the chloride 14 with ethylamine with similar conditions to those used for the mesylate (R)-4b (EtNH2, DMSO, 110°C, 5h30 or EtNH2 (solvent and reactant), 140°C, 5h), gives mainly the elimination products. The yield of fenfluramine is below 10%.
By action of sodium azide in DMSO, on the other hand, one obtains the azide (R)-5 with a yield of 78%, the elimination products formed here or in the last step can be removed by chromatography on silica. HPLC analysis on silica of the camphanyl derivate of the amine (R)-6 [22] obtained by catalytic reduction of the azide (R)-5 has confirmed the double inversion without racemisation after comparison with the starting alcohol (R)-3. Then the fenfluramine (R)-1 is prepared without racemisation with a 83% yield starting from the azide (R)-5 like detailed above.
This procedure with two inversions allows to transform the alcohol 3 in the azide 5 with the same configuration in two steps with a global yield (non optimised) of 70% and without racemisation. It’s thus preferred over the recently published one [7, 8], which needs 5 steps for a lower global yield (55%) and in addition features an epimerisation of 10% [8]. It’s a promising way to fenfluramine (S)-1 starting from the alcohol (S)- 3.
References
- ^ “Fintepla- fenfluramine solution”. DailyMed. Retrieved 8 January 2021.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “FDA Approves New Therapy for Dravet Syndrome”. U.S. Food and Drug Administration (FDA) (Press release). 25 June 2020. Retrieved 25 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d “Fintepla EPAR”. European Medicines Agency. Retrieved 8 January2021.
- ^ Jump up to:a b c d Richard C. Dart (2004). Medical Toxicology. Lippincott Williams & Wilkins. pp. 874–. ISBN 978-0-7817-2845-4. Archived from the original on 2018-05-09.
- ^ Jump up to:a b James O’Donnell (Pharm. D.); Gopi Doctor Ahuja (2005). Drug Injury: Liability, Analysis, and Prevention. Lawyers & Judges Publishing Company. pp. 276–. ISBN 978-0-913875-27-8.
- ^ Integrating the Neurobiology of Schizophrenia. Academic Press. 27 February 2007. pp. 142–. ISBN 978-0-08-047508-0.
- ^ The Pharmacology of Corticotropin-releasing Factor (CRF). Effects on Sensorimotor Gating in the Rat. 2006. pp. 12–. ISBN 978-0-549-53661-1.
- ^ Christian P. Muller; Barry Jacobs (30 December 2009). Handbook of the Behavioral Neurobiology of Serotonin. Academic Press. pp. 630–. ISBN 978-0-08-087817-1.
- ^ Vickers SP, Dourish CT, Kennett GA (2001). “Evidence that hypophagia induced by d-fenfluramine and d-norfenfluramine in the rat is mediated by 5-HT2C receptors”. Neuropharmacology. 41 (2): 200–9. doi:10.1016/s0028-3908(01)00063-6. PMID 11489456. S2CID 23374227.
- ^ Roth, B. L. (2007). “Drugs and Valvular Heart Disease”. New England Journal of Medicine. 356 (1): 6–9. doi:10.1056/NEJMp068265. PMID 17202450.
- ^ Rothman, R. B.; Baumann, M. H. (2009). “Serotonergic Drugs and Valvular Heart Disease”. Expert Opinion on Drug Safety. 8 (3): 317–329. doi:10.1517/14740330902931524. PMC 2695569. PMID 19505264.
- ^ Jump up to:a b Dahl, C. F.; Allen, M. R.; Urie, P. M.; Hopkins, P. N. (2008). “Valvular Regurgitation and Surgery Associated with Fenfluramine Use: An Analysis of 5743 Individuals”. BMC Medicine. 6: 34. doi:10.1186/1741-7015-6-34. PMC 2585088. PMID 18990200.
- ^ Jump up to:a b c d e f g h i j k l m Donald G. Barceloux (3 February 2012). Medical Toxicology of Drug Abuse: Synthesized Chemicals and Psychoactive Plants. John Wiley & Sons. pp. 255–262. ISBN 978-1-118-10605-1. Archived from the original on 9 May 2018.
- ^ Mann SC, Caroff SN, Keck PE, Lazarus A (20 May 2008). Neuroleptic Malignant Syndrome and Related Conditions. American Psychiatric Pub. pp. 111–. ISBN 978-1-58562-744-8.
- ^ Jump up to:a b c d e f g h i Rothman RB, Clark RD, Partilla JS, Baumann MH (2003). “(+)-Fenfluramine and its major metabolite, (+)-norfenfluramine, are potent substrates for norepinephrine transporters”. J. Pharmacol. Exp. Ther. 305 (3): 1191–9. doi:10.1124/jpet.103.049684. PMID 12649307. S2CID 21164342.
- ^ Jump up to:a b c d e f g Setola V, Hufeisen SJ, Grande-Allen KJ, Vesely I, Glennon RA, Blough B, Rothman RB, Roth BL (2003). “3,4-methylenedioxymethamphetamine (MDMA, “Ecstasy”) induces fenfluramine-like proliferative actions on human cardiac valvular interstitial cells in vitro”. Mol. Pharmacol. 63 (6): 1223–9. doi:10.1124/mol.63.6.1223. PMID 12761331. S2CID 839426.
- ^ Nestler, E. J. (2001). Molecular Neuropharmacology: A Foundation for Clinical Neuroscience. McGraw-Hill.
- ^ Giuseppe Di Giovanni; Vincenzo Di Matteo; Ennio Esposito (2008). Serotonin-dopamine Interaction: Experimental Evidence and Therapeutic Relevance. Elsevier. pp. 393–. ISBN 978-0-444-53235-0.
- ^ Fitzgerald LW, Burn TC, Brown BS, Patterson JP, Corjay MH, Valentine PA, Sun JH, Link JR, Abbaszade I, Hollis JM, Largent BL, Hartig PR, Hollis GF, Meunier PC, Robichaud AJ, Robertson DW (2000). “Possible role of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with fenfluramine”. Mol. Pharmacol. 57 (1): 75–81. PMID 10617681.
- ^ Jump up to:a b c Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI, Partilla JS (January 2001). “Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin”. Synapse. 39 (1): 32–41. doi:10.1002/1098-2396(20010101)39:1<32::AID-SYN5>3.0.CO;2-3. PMID 11071707.
- ^ Rothman RB, Baumann MH (2002). “Therapeutic and adverse actions of serotonin transporter substrates”. Pharmacol. Ther. 95 (1): 73–88. doi:10.1016/s0163-7258(02)00234-6. PMID 12163129.
- ^ Connolly, H. M.; Crary, J. L.; McGoon, M. D.; Hensrud, D. D.; Edwards, B. S.; Edwards, W. D.; Schaff, H. V. (1997). “Valvular Heart Disease Associated with Fenfluramine-Phentermine”. New England Journal of Medicine. 337 (9): 581–588. doi:10.1056/NEJM199708283370901. PMID 9271479.
- ^ Weissman, N. J. (2001). “Appetite Suppressants and Valvular Heart Disease”. The American Journal of the Medical Sciences. 321 (4): 285–291. doi:10.1097/00000441-200104000-00008. PMID 11307869. S2CID 46466276.
- ^ FDA September 15, 1997. FDA Announces Withdrawal Fenfluramine and Dexfenfluramine (Fen-Phen) Archived 2013-07-19 at the Wayback Machine
- ^ “Drugs banned in India”. Central Drugs Standard Control Organization, Dte.GHS, Ministry of Health and Family Welfare, Government of India. Archived from the original on 2015-02-21. Retrieved 2013-09-17.
- ^ “FDA Approves FINTEPLA (fenfluramine) for the Treatment of Seizures Associated with Dravet Syndrome” (Press release). Zogenix Inc. 25 June 2020. Retrieved 25 June 2020 – via GlobeNewswire.
- ^ “Fenfluramine Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 25 June 2020.
- ^ “Fenfluramine Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 25 June 2020.
- ^ “Fintepla: Pending EC decision”. European Medicines Agency (EMA). 16 October 2020. Retrieved 16 October 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b John Calvin M. Brust (2004). Neurological Aspects of Substance Abuse. Butterworth-Heinemann. pp. 117–. ISBN 978-0-7506-7313-6.
- ^ United States. Congress. Senate. Select Committee on Small Business. Subcommittee on Monopoly and Anticompetitive Activities (1976). Competitive problems in the drug industry: hearings before Subcommittee on Monopoly and Anticompetitive Activities of the Select Committee on Small Business, United States Senate, Ninetieth Congress, first session. U.S. Government Printing Office. pp. 2–.
- ^ Jump up to:a b Drug Addiction II: Amphetamine, Psychotogen, and Marihuana Dependence. Springer Science & Business Media. 27 November 2013. pp. 258–. ISBN 978-3-642-66709-1.
- ^ Drug Addiction II: Amphetamine, Psychotogen, and Marihuana Dependence. Springer Science & Business Media. 27 November 2013. pp. 249–. ISBN 978-3-642-66709-1.
- ^ “Zogenix ends enrolment in second Phase lll trial of ZX008”. Drug Development Technology. Kable. 1 February 2018. Archived from the original on 2018-02-04. Retrieved 3 February 2018.
- ^ “A Two-Part Study to Investigate the Dose-Ranging Safety and Pharmacokinetics, Followed by the Efficacy and Safety of ZX008 (Fenfluramine Hydrochloride) Oral Solution as an Adjunctive Therapy in Children ≥2 Years Old and Young Adults With Dravet Syndrome”. ClinicalTrials.gov. Archived from the original on 29 September 2017. Retrieved 9 May 2018.
- ^ “Zogenix drug cuts seizure rate in patients with severe epilepsy”. statnews.com. 29 September 2017. Archived from the original on 30 April 2018. Retrieved 9 May 2018.
Further reading
- Gustafsson BI, Tømmerås K, Nordrum I, Loennechen JP, Brunsvik A, Solligård E, Fossmark R, Bakke I, Syversen U, Waldum H (March 2005). “Long-term serotonin administration induces heart valve disease in rats”. Circulation. 111 (12): 1517–22. doi:10.1161/01.CIR.0000159356.42064.48. PMID 15781732.
- Welch, J. T.; Lim, D. S. (2007). “The Synthesis and Biological Activity of Pentafluorosulfanyl Analogs of Fluoxetine, Fenfluramine, and Norfenfluramine”. Bioorganic & Medicinal Chemistry. 15 (21): 6659–6666. doi:10.1016/j.bmc.2007.08.012. PMID 17765553.
External links
- “Fenfluramine”. Drug Information Portal. U.S. National Library of Medicine.
- “Fenfluramine hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
- Inchem.org – Fenfluramine hydrochloride
| Clinical data | |
|---|---|
| Trade names | Fintepla |
| Other names | ZX008 |
| AHFS/Drugs.com | Professional Drug Facts |
| MedlinePlus | a620045 |
| License data | US DailyMed: Fenfluramine |
| Pregnancy category | AU: B2 |
| Routes of administration | By mouth |
| ATC code | A08AA02 (WHO) N03AX26 (WHO) |
| Legal status | |
| Legal status | US: Schedule IV [1][2]EU: Rx-only [3] |
| Pharmacokinetic data | |
| Elimination half-life | 13–30 hours[4] |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 458-24-2 |
| PubChem CID | 3337 |
| IUPHAR/BPS | 4613 |
| DrugBank | DB00574 |
| ChemSpider | 3220 |
| UNII | 2DS058H2CF |
| KEGG | D07945 C06996 |
| ChEBI | CHEBI:5000 |
| ChEMBL | ChEMBL87493 |
| CompTox Dashboard (EPA) | DTXSID4023044 |
| ECHA InfoCard | 100.006.616 |
| Chemical and physical data | |
| Formula | C12H16F3N |
| Molar mass | 231.262 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Chirality | Racemic mixture |
| SMILES[hide]FC(F)(C1=CC(CC(C)NCC)=CC=C1)F | |
| InChI[hide]InChI=1S/C12H16F3N/c1-3-16-9(2)7-10-5-4-6-11(8-10)12(13,14)15/h4-6,8-9,16H,3,7H2,1-2H3 Key:DBGIVFWFUFKIQN-UHFFFAOYSA-N |
CLIP
http://www.inchem.org/documents/pims/pharm/pim938.htm
////////////Fenfluramine, 塩酸フェンフルラミン , dravet, AHR-3002, ZX-008, Fintepla
CCNC(C)CC1=CC(=CC=C1)C(F)(F)F.Cl
PATENT
https://patents.google.com/patent/US20170174613A1/en
- [0093]
Many general references providing commonly known chemical synthetic schemes and conditions useful for synthesizing the disclosed compounds are available (see, e.g., Smith and March, March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Fifth Edition, Wiley-Interscience, 2001; or Vogel, A Textbook of Practical Organic Chemistry, Including Qualitative Organic Analysis, Fourth Edition, New York: Longman, 1978). - [0094]
Compounds as described herein can be purified by any purification protocol known in the art, including chromatography, such as HPLC, preparative thin layer chromatography, flash column chromatography and ion exchange chromatography. Any suitable stationary phase can be used, including normal and reversed phases as well as ionic resins. In certain embodiments, the disclosed compounds are purified via silica gel and/or alumina chromatography. See, e.g., Introduction to Modern Liquid Chromatography, 2nd Edition, ed. L. R. Snyder and J. J. Kirkland, John Wiley and Sons, 1979; and Thin Layer Chromatography, ed E. Stahl, Springer-Verlag, New York, 1969. - [0095]
During any of the processes for preparation of the subject compounds, it may be necessary and/or desirable to protect sensitive or reactive groups on any of the molecules concerned. This may be achieved by means of conventional protecting groups as described in standard works, such as J. F. W. McOmie, “Protective Groups in Organic Chemistry”, Plenum Press, London and New York 1973, in T. W. Greene and P. G. M. Wuts, “Protective Groups in Organic Synthesis”, Third edition, Wiley, New York 1999, in “The Peptides”; Volume 3 (editors: E. Gross and J. Meienhofer), Academic Press, London and New York 1981, in “Methoden der organischen Chemie”, Houben-Weyl, 4th edition, Vol. 15/1, Georg Thieme Verlag, Stuttgart 1974, in H.-D. Jakubke and H. Jescheit, “Aminosauren, Peptide, Protein”, Verlag Chemie, Weinheim, Deerfield Beach, and Basel 1982, and/or in Jochen Lehmann, “Chemie der Kohlenhydrate: Monosaccharide and Derivate”, Georg Thieme Verlag, Stuttgart 1974. The protecting groups may be removed at a convenient subsequent stage using methods known from the art. - [0096]
The subject compounds can be synthesized via a variety of different synthetic routes using commercially available starting materials and/or starting materials prepared by conventional synthetic methods. A variety of examples of synthetic routes that can be used to synthesize the compounds disclosed herein are described in the schemes below.
Example 11. Fenfluramine Nomenclature & Structure
- [0097]
Chemical Abstract Service (CAS) Registry Number (RN): 404-82-0 (HCl Salt), 458-24-2 (Parent Free Base) - [0098]
Chemical Name: N-ethyl-α-methyl-3-(trifluoromethyl)-benzeneethanamine hydrochloride (1:1). Other Names: Fenfluramine HCl, DL-Fenfluramine, (±)-Fenfluramine - [0099]
Structure of Hydrochloride Salt: - [0100]
Stereochemistry: Fenfluramine HCl has one chiral center and is being developed as the racemate and contains dexfenfluramine and levofenfluramine - [0101]
Molecular Formula of hydrochloride salt: C12H16F3N.HCl - [0102]
Molecular Mass/Weight: 267.72 g/mol
2. General Properties
- [0103]
Table 1 summarizes the chemical and physical properties of Fenfluramine HCl. - TABLE 1 General Properties of Fenfluramine HCl Drug Substance Property Result Appearance (color, White to off-white powder physical form) DSC (melting 170° C. (melt/sublimation) point)a TGA Onset 147° C. 0.03% at 150° C. 91% at 220° C. (evaporation) pKa (water) 10.15-10.38 Solubility (mg/mL) Resultant pH 25° C. 37° C. Solubility pH 6.69 (water) 54.13 71.22 (Aqueous) pH 1.73 buffer 25.34 53.68 pH 3.43 buffer 29.50 61.97 pH 6.41 buffer 37.42 95.60 0.9% NaCl (water) 22.98 — Solvent Solubility 25° C. (mg/mL) Solubility (Organic Ethanol 150 Solvents) Dichloromethane 30-35 Ethyl Acetate, 1-5 mg Tetrahydrofuran, Toluene, Acetonitrile UV Absorption Maxima: 210, 265 nm Solution pH (water) 6.69 Hygroscopicity @30% RH: ~0.05% (Dynamic Vapor @60% RH: ~0.07% Sorption (DVS) @90% RH: ~0.20%a) Polymorphism Fenfluramine HCl has been consistently isolated as a single crystalline Form 1 as determined by DSC and x-ray powder diffraction (XRPD) Solvation/Hydration Fenfluramine HCl is isolated as a nonhydrated, nonsolvated solid Solution Stability 8 weeks @ pH 6.7 phosphate buffer medium at 40° C. and 60° C. using concentrations of 0.5, 2.5 and 5.0 mg/ml. All conditions, no new impurities >0.1% by HPLC. Solid Stability 8 weeks @ 40° C., 60° C. and 80° C. 7 days at 150° C. All conditions, no new impurities >0.1% by HPLC.
3. Synthesis of Fenfluramine Drug Substance
- [0104]
Scheme 3.1 shows a 2-step route of synthesis used to manufacture initial clinical supplies of Fenfluramine HCl from ketone (2). The batch size is 4 kg performed in laboratory glassware (kilo lab). No chromatography is required and the process steps are amenable to scale-up. In process 1 there is one isolated intermediate Fenfluramine Free Base (1) starting from commercially supplied 1-(3-(trifluoromethyl)phenyl) acetone (Ketone 2). All steps are conducted under cGMPs starting from Ketone (2). - [0105]
Scheme 3.2 shows a 4-step route of synthesis to Fenfluramine HCl that can be used for commercial supply. Route 2 utilizes the same 2-step process used by Route 1 to convert Ketone (2) to Fenfluramine HCl with the exception that Ketone (2) is synthesized under cGMP conditions starting from 3-(Trifluoromethyl)-phenyl acetic acid (Acid 4). Bisulfate Complex (3) is an isolatable solid and can be purified before decomplexation to Ketone (2). In-situ intermediates which are oils are shown in brackets. Batch sizes of 10 Kg are performed. Commercial batch sizes of 20 kg are performed in fixed pilot plant equipment. Steps 1-2 of Scheme 3.2 to manufacture Ketone (2) have been demonstrated on a 100 g scale to provide high purity ketone (2) of >99.8% (GC & HPLC). Conversion of Ketone (2) to Fenfluramine using either Route 1 or 2 has provided similar purity profiles. - Starting materials are designated by enclosed boxes. Bracketed and non bracketed compounds respectively indicate proposed in-situ and isolated intermediates. NMI=N-Methyl Imidazole.
4.1. Narrative Description (Route 1)
- [0106]
Step 1: Reductive Amination (Preparation of Fenfluramine Free Base 1) - [0107]
A solution of ethylamine, water, methanol, and 1-(3-(trifluoromethyl)phenyl) acetone (Ketone 2) was treated with sodium triacetoxyborohydride and stirred for 16 h at 25° C. at which time HPLC analysis (IPC-1; In Process Control No. 1) showed the reaction to be complete and sodium hydroxide solution was added until pH>10. Toluene was added and the phases separated, and the aqueous phase (IPC-2) and organic phase (IPC-3) are checked for remaining Fenfluramine and Fenfluramine alcohol and the organic phase was reduced. Purified water was added and the pH adjusted to <2 using conc. HCl and the phases were separated. The aqueous phase was washed with toluene and the toluene phase (IPC-4) and the aqueous phase (IPC-5) was checked for Fenfluramine and Fenfluramine alcohol content. The aqueous phase containing product is pH adjusted to >10 using sodium hydroxide solution. The basic aqueous phase was extracted with MTBE until removal of Fenfluramine from the aqueous phase was observed by HPLC (<0.5 mg/ml) (IPC-6). The organic phase was dried over sodium sulfate and filtered. The filtrate was concentrated in vacuo to give the intermediate product Fenfluramine Free Base 1 as a pale yellow oil tested per specifications described herein which showed by NMR the material to contain 2.93% toluene giving an active yield of 88.3% with a purity of 98.23% by HPLC (0.67% Fenfluramine alcohol). - [0108]
Step 2: Salt Formation (Preparation of Fenfluramine HCl) - [0109]
To a flask was charged ethanol and acetyl chloride. The solution was stirred slowly overnight before ethyl acetate was added. The HCl in ethyl acetate solution formed was polish filtered into a clean carboy and retained for later use. To a vessel was added Fenfluramine free base 1 and MTBE. The Fenfluramine solution in MTBE was collected in two carboys before the vessel was cleaned and checked for particulate residue. The Fenfluramine solution was polish filtered into a vessel and cooled and HCl in ethyl acetate solution was added giving a final pH of 6-7. The batch was stirred for 1 h and filtered. The product was dried under vacuum at 40° C. The product (96.52% yield) was tested per IPC-7 had a purity of 99.75% by HPLC and GC headspace analysis showed MTBE (800 ppm) and EtOAc (150 ppm) to be present. The product was then tested per specifications shown herein.
4.2. Narrative Description (Route 2)
- [0110]
Step 1: Preparation of Ketone Bisulfite Adduct - [0111]
Procedure: Charge acetic anhydride, (2.8 vol, 3.0 wt, 5.0 eq.) to a vessel and commence stirring. Cool the solution to −5 to 5° C., targeting −4° C. Charge 1-methylimidazole, (0.2 vol, 0.21 wt, 0.5 eq.) to the mixture at −5 to 5° C. Caution: very exothermic. If necessary, adjust the temperature to 0 to 5° C. Charge ZX008 acid, (1.00 wt, 1.0 eq.) to the mixture at 0 to 5° C. Caution: exothermic. Stir the mixture at 0 to 5° C. until ≦2.1% area ZX008 acid by HPLC analysis, typically 7 to 9 hours. Charge 15% w/w sodium chloride solution (2.0 vol) to the mixture at 0 to 5° C., 60 to 90 minutes. Caution: very exothermic which will be slightly delayed. Warm the mixture to 18 to 23° C. over 45 to 60 minutes and continue stirring for a further 30 to 45 minutes at 18 to 23° C. Charge TBME, (5.0 vol, 3.7 wt) to the mixture and stir for 10 to 15 minutes at 18 to 23° C. Separate the aqueous layer and retain the organic layer. Back-extract the aqueous layer with TBME, (2×3.0 vol, 2×2.2 wt) at 18 to 23° C. retaining each organic layer. Adjust the pH of the combined organic layer to pH 6.5 to 9.0, targeting 7.0 by charging 20% w/w sodium hydroxide solution (5.3 to 8.3 vol) at 18 to 23° C. Caution: exothermic. Separate the aqueous layer and retain the organic layer. Wash the organic layer with 4% w/w sodium hydrogen carbonate solution (2×3.0 vol) at 18 to 23° C. Determine the residual ZX008 acid content in the organic layer by HPLC analysis, pass criterion ≦0.10% area ZX008 acid. Wash the organic layer with purified water, (2×3.0 vol) at 18 to 23° C. Concentrate the organic layer under reduced pressure to ca. 2 vol at 40 to 45° C., targeting 43° C. - [0112]
Determine the w/w assay of ZX008 ketone (WIP) in the mixture by 1H-NMR analysis for information only and calculate the contained yield of ZX008 ketone (WIP) in the mixture. Note: This step can be removed from the process since the process is robust and consistently delivers 80 to 90% th yield. The achieved yield was factored into the charges of the subsequent steps. - [0113]
Charge n-heptane, (4.0 vol, 2.7 wt) to the mixture at 40 to 45° C., targeting 43° C. Concentrate the mixture to ca. 2 vol at 40 to 45° C., targeting 43° C. Determine the TBME content in the mixture by 1H-NMR analysis, (pass criterion ≦5.0% w/w TBME vs. ZX008 ketone). Charge n-heptane, (2.4 vol, 1.6 wt) at 40 to 45° C., targeting 43° C., vessel A. To vessel B, charge sodium metabisulfite, (0.82 wt, 0.88 eq.) at 18 to 23° C. To vessel B, charge a solution of sodium hydrogen carbonate, (0.16 wt, 0.4 eq.) in purified water, code RM0120 (2.0 vol) at 18 to 23° C. followed by a line rinse with purified water, code RM0120 (0.4 vol) at 18 to 23° C. Caution: gas evolution. Heat the contents of vessel B to 40 to 45° C., targeting 43° C. Charge the contents from vessel A to vessel B followed by a line rinse with n-heptane, (0.8 vol, 0.5 wt) at 40 to 45° C., targeting 43° C. Stir the mixture for 1 to 1.5 hours at 40 to 45° C., targeting 43° C. Charge n-heptane, code RM0174 (3.2 vol, 2.2 wt) to the mixture with the temperature being allowed to cool to 18 to 45° C. at the end of the addition. Cool the mixture to 18 to 23° C. at approximately constant rate over 45 to 60 minutes. Stir the mixture at 18 to 23° C. for 1.5 to 2 hours. - [0114]
Sample the mixture to determine the residual ZX008 ketone content by 1H-NMR analysis, (pass criterion ≦10.0% mol, target 5.0% mol ZX008 ketone vs. ZX008 ketone bisulfite adduct). Filter the mixture and slurry wash the filter-cake with n-heptane, (2×2.0 vol, 2×1.4 wt) at 18 to 23° C. Dry the solid at up to 23° C. until the water content is <10.0% w/w water by KF analysis according to AKX reagent. At least 16 hours. Determine the w/w assay of the isolated ZX008 ketone bisulfite adduct by 1H-NMR analysis and calculate the contained yield of ZX008 ketone bisulfite adduct. - [0115]
Yields and Profiles: The yield for the stage 1 Demonstration batch is summarized Table below. Input: 1700.0 g uncorr., acid, 99.50% area (QC, HPLC), 2-isomer not detected, 4-isomer 0.02% area, RRT1.58 (previously not observed) 0.48% area as per the preparative method. The analytical data is summarized in Table 1A below. - TABLE 1A Table for isolated yields for step 1 Demonstration batch Corr. % area Reference Corr. Yield % w/w (HPLC, number Input Output (% th)** (1H-NMR)* QC) Comments Batch A1 1700.0 g 1500.1 g 89.1 45.0 —.— Crude ketone as TBME sol. Batch A2 1500.1 g 1716.1 77.8 76.0 98.15 Bisulfite adduct only 67.3 Overall product
- [0116]
Step 2: Preparation of Ketone - [0117]
Procedure: Charge toluene, (5.0 vol, 4.3 wt), and purified water, (5.0 vol) to the vessel and commence stirring. If necessary, adjust the temperature to 18 to 23° C. and charge ZX008 ketone bisulfite adduct, (1.00 wt corrected for % w/w assay) to the mixture at 18 to 23° C. Charge 20% w/w sodium hydroxide solution to the mixture at 18 to 23° C. adjusting the pH of the mixture to pH 8.0 to 12.0, targeting 9.0 (0.5 to 1.0 vol). - Separate the lower aqueous layer and retain the top organic layer. Wash the organic layer with purified water, (3.0 vol) at 18 to 23° C. Concentrate the organic layer under reduced pressure to ca. 2 vol at 45 to 50° C., targeting 48° C. Charge methanol, (5.0 vol, 4.0 wt) to the mixture at 45 to 50° C., targeting 48° C. Re-concentrate the mixture under reduced pressure to ca. 2 vol at 45 to 50° C., targeting 48° C. Repeat steps 7 and 8 once before continuing with step 9. Cool the mixture to 18 to 23° C. Clarify the mixture into a tared, suitably-sized drum followed by a methanol (1.0 vol, 0.8 wt) line rinse at 18 to 23° C. Determine the w/w assay of ZX008 ketone (WIP) in the mixture by 1H-NMR analysis and calculate the contained yield of ZX008 ketone (WIP) in the mixture. Determine the toluene content in the mixture by 1H-NMR analysis.
- [0118]
Yields and Profiles: The yield for the step 2 Demonstration batch is summarized in Table 1B below. Input: 1200.0 g corr. Ketone bisulfite adduct, 76.0% w/w assay (NMR, using DMB as internal standard in d6-DMSO), (1.00 eq, 1.00 wt corr. for w/w assay) for input calculation. - TABLE 1B Table for isolated yields for step 2 Demonstration batch % w/w % area Corr. Corr. Corr. Yield (1H- (HPLC, Input Output (% th) NMR)* QC) Comments 1200.0 g 858.15 g 108.3 25.5 99.31 Purified ketone
- [0119]
Step 3: Preparation of Fenfluramine HCl Crude - [0120]
Procedure: Charge the ZX008 ketone (corr. for assay, 1.00 wt, 1.00 eq. isolated as solution in MeOH in stage 2) to a vessel. Charge methanol, code RM0036 (5.0 vol, 4.0 wt) to the mixture at 18 to 23° C. Cool the solution to 0 to 5° C. Charge 70 wt % aqueous ethylamine solution (1.3 vol, 1.6 wt, 4.0 eq) to the mixture at 0 to 10° C., over 15 to 30 minutes, followed by a line rinse with methanol (1.0 vol, 0.8 wt). Warm the mixture to 15 to 20° C. and stir the mixture for a further 60 to 70 minutes at 15 to 20° C. Adjust the mixture to 15 to 18° C. if required, targeting 15° C. Charge sodium triacetoxyborohydride (2.4 wt, 2.25 eq.) to the mixture in approximately 10 portions, keeping the mixture at 15 to 20° C., targeting 17° C. Addition time 1.5 to 2 hours. Caution: Exothermic. Stir the mixture at 15 to 20° C. until complete by HPLC analysis, pass criterion ≦3.0% area ZX008 ketone, typically 2 to 3 hours. Adjust the pH of the mixture to pH>12 by charging 20% w/w aqueous sodium hydroxide solution (5.0 to 6.0 vol) to the mixture at 15 to 40° C. Addition time 10 to 30 minutes. Caution: Exothermic. If necessary, adjust the temperature to 18 to 23° C. Extract the mixture with toluene (3×3.0 vol, 3×2.6 wt) at 18 to 23° C., retaining and combining the top organic layer after each extraction. Wash the combined organic layer with purified water, (1.0 vol) at 18 to 23° C. Heat the mixture to 40 to 50° C., targeting 48° C. Concentrate the mixture under reduced pressure at constant volume maintaining ca. 5 vol by charging the organic layer at approximately the same rate as the distillation rate at 40 to 50° C., targeting 48° C. Cool the mixture to 18 to 23° C. Charge purified water (10.0 vol) to the mixture at 18 to 23° C. Adjust the pH of the mixture to 0.1<pH<1.5 at 18 to 23° C. by charging concentrated hydrochloric acid, 0.5 vol. Do not delay from this step until neutralization. - [0121]
Separate the layers at 18 to 23° C. retaining the bottom aqueous layer. Wash the aqueous layer with toluene, (3.0 vol, 2.6 wt) at 18 to 23° C. retaining the aqueous layer. Adjust the pH of the aqueous layer to pH>12 by charging 20% w/w sodium hydroxide solution at 18 to 23° C. 0.8 to 0.9 vol. Caution: Exothermic. Charge TBME, code RM0002 (2.0 vol, 1.5 wt) to the basic aqueous layer. Separate the layers at 18 to 23° C. retaining the top organic layer. Back-extract the aqueous layer with TBME (2×2.0 vol, 2×1.5 wt) at 18 to 23° C. retaining the organic layers. Wash the combined organic layer with purified water, (2×1.0 vol) at 18 to 23° C. Concentrate the combined organic layers under reduced pressure at 40 to 50° C., targeting 48° C. to ca. 3 vol. Determine the residual toluene content of the mixture by 1H-NMR analysis. Sample for determination of residual water content by KF analysis, AKX reagent. Charge TBME (8.7 vol, 6.4 wt) to the mixture at 40 to 50° C. Cool the solution to 0 to 5° C., targeting 2° C. Charge concentrated hydrochloric acid (0.54 vol, 0.46 wt) maintaining the temperature <15° C. Caution: Exothermic. Line rinse with TBME (1.0 vol, 0.7 wt). If necessary, adjust the temperature to 0 to 10° C. and stir the mixture at 0 to 10° C. for a further 2 to 3 hours. Filter the mixture and wash the filter-cake with TBME (2×4.4 vol, 2×3.3 wt) at 0 to 10° C. Dry the solid at up to 40° C. until the TBME content is <0.5% w/w TBME by 1H-NMR analysis. 4 to 8 hours. - [0122]
Yields and Profiles: The yield for the step 3 Demonstration batch is summarized in Table 1C below. Input: 856.8 g corr. Ketone, 44.2% w/w assay (NMR, using TCNB as internal standard in CDCl3), (1.00 eq, 1.00 wt corr. for w/w assay) for input calculation. FIG. 2 and Table 1D shows an exemplary HPLC chromatogram of a crude preparation of fenfluramine hydrochloride (210 nm UV absorbance). - TABLE 1C Table for isolated yields for step 3 Demonstration batch Corr. % area Reference Corr. Corr. Yield % w/w (HPLC, number Input Output (% th) (1H-NMR)* QC) Comments Batch A1 856.8 g 836.31 g 85.3 44.2 99.15 Fenfluramine free base (in situ intermediate) Batch A2 880.7 84.0 based 99.5 100.00 Fenfluramine•HCl on ketone crude (step 3 an bisulfite d 4.1) adduct (77.6 based on purified ketone)
- TABLE 1D Purity of crude fenfluramine hydrochloride by HPLC (see FIG. 2) Processed Channel Descr. DAD AU Ch 1 Sample 210, Bw 4 Peak Results USP USP USP Name RT RelRT Area Height Tailing Resolution Plate Count EP s/n % Area 1 NorFenfluramine 7.46 2 2-Fenfluramine 7.68 3 Fenfluramine 8.67 1.000 3789064 778178 1.7 70796 2549.8 99.15 4 4-Fenfluramine 8.95 5 11 34 1.308 6073 1449 1.2 23.5 215529 3 8 0.16 6 ZX008 acid 12.93 7 Fenfluramine alcohol 14.16 1.633 15266 2972 1.3 24.8 215040 8.7 0.40 8 ZX008 ketone 14.83 9 Fenfluramine acetamide 15.55 10 TOLUENE 15 75 11 15.92 1.836 4110 1122 2.7 0.11 12 16.60 1.915 6861 1630 1.5 451209 4.3 0.18 Sum 3821374 100.00
- [0123]
Step 4.2: Crystallization of Fenfluramine Hydrochloride - [0124]
Procedure: Charge Fenfluramine.HCl (crude) (1.00 wt, 1.0 eq.) and TBME (10.0 vol, 7.4 wt) to the vessel and commence stirring. Heat the suspension to reflux (50 to 58° C.). Charge ethanol (5.0 vol, 3.9 wt) maintaining the temperature at 50 to 58° C. Addition time 20 minutes. Stir at 50 to 58° C. for 5 to 10 minutes and check for dissolution. Stir the solution at 50 to 58° C. for 5 to 10 minutes, targeting 54 to 58° C. Clarify the reaction mixture through a 0.1 μm in-line filter at 54 to 58° C., followed by a line rinse with TBME (1 vol, 0.7 wt). Cool the solution to 48 to 50° C. Charge Fenfluramine HCl, code FP0188 (0.01 wt). Check for crystallization. Allow the suspension to cool to 15 to 20° C., target 17° C. over 5 to 5.5 hours at an approximately constant rate. Stir the mixture at 15 to 20° C., target 17° C. for 2 to 3 hours. Filter the mixture and wash the filter-cake with clarified TBME (2×3.0 vol, 2×2.2 wt) at 5 to 15° C. Dry the solid at up to 40° C. until the TBME content is <0.5% w/w TBME and the ethanol content is <0.5% w/w EtOH by 1H-NMR analysis. 4 to 8 hours. Determine the w/w assay of the isolated Fenfluramine.HCl by 1H-NMR analysis. - [0125]
Yields and Profiles: The yield for the stage 4 Demonstration batch is summarized in Table 1E below. Input: 750.0 g uncorr. Fenfluramine HCl crude (1.00 eq, 1.00 wt uncorr.) for input calculation. FIG. 3 shows an exemplary HPLC chromatogram of a crystallized fenfluramine hydrochloride sample (210 nm UV absorbance). - TABLE 1E Table for isolated yields for stage 4 Demonstration batch Uncorr. Uncorr. Uncorr. Yield HPLC (% area, Input Output (% th) QC) Comments 750.0 g 608.0 81.1 100.00* Fenfluramine•HCl
5. In-Process Controls
- [0126]
Table 2 summarizes the in-process controls (IPCs) by IPC number as cited in the narrative procedures above used for Process 1. - TABLE 2 In-Process Controls Performed during Process 1 Critical IPC Synthesis Process No. Step Sample Description Method Acceptance Criteria 1 1 Reaction Reaction HPLC NMT 3.0% Ketone (1) Mixture Completion 2 1 Extraction Purity HPLC Report percent Aqueous Fenfluramine Free Base and Phase Fenfluramine Alcohol 3 1 Extraction Purity HPLC Report percent Organic Fenfluramine Free Base and Phase Fenfluramine Alcohol 4 1 Extraction Purity HPLC Report percent Organic Fenfluramine Free Base and Phase Fenfluramine Alcohol 5 1 Extraction Purity HPLC NLT 98.0% Fenfluramine Aqueous HCl Phase LT 1.0% Fenfluramine Alcohol 6 1 Extraction Purity HPLC Report percent result of Aqueous Fenfluramine HCl Phase Fenfluramine Alcohol 7 2 Reaction Purity 1H-NMR Residual Solvents by 1H- Mixture NMR Ethanol NMT 0.50% w/w Ethyl Acetate NMT 0.50% w/w Methanol NMT 0.50% w/w Toluene NMT 0.50% w/w MTBE NMT 0.50% w/w
6. Starting Materials
- [0127]
This section provides information and specification controls for the starting materials used to produce clinical supplies of fenfluramine per the routes shown herein. - TABLE 3 Starting Materials via the Route 1 Chemical Name Code [CAS. No.] Name Structure Source Step 1-(3- (Trifluoromethyl)- phenylacetone [21906-39-8] Ketone (1)Fluorochem 1 Ethyl Amine Ethyl EtNH2 Alfa Aesar 1 (70% in water) Amine [75-04-7]
- TABLE 4 Starting Materials via Route 2 Chemical Name Code [CAS. No.] Name Structure Source Step 3-(Trifluoromethyl)- phenylacetic acid [351-35-9] Acid (1a)To be determined 1 Acetic Anhydride [108-24-7] Acetic AnhydrideVarious 1 Ethyl Amine Ethyl EtNH2 Various 3 (70% in water) Amine [75-04-7]
- [0128]
Table 5 provides a list of the intermediates for the Route 2 synthesis. Both routes share the same intermediate Fenfluramine Free Base (1). Fenfluramine Free Base (1) was treated as an isolated intermediate in the Route 1 process however the Route 2 process uses fixed equipment where both Ketone (2) and Fenfluramine Free Base 1, both non-isolatable oils, are telescoped as a solution and controlled as in-situ intermediates. The Bisulfate Complex (3) is isolated as a solid thus is amenable to treatment as an isolated intermediate and released as such. Crude Fenfluramine HCl can be isolated as an intermediate before recrystallization. - [0129]
A Specification and Testing Strategy for Intermediates is used. Additional tests and acceptance criteria are be added based upon review of data from the primary stability batches and process validation critical parameter studies. Analytical reference standards are used in full characterization of each intermediate. HPLC methods to determine assay and impurities are the same as the drug substance release method and are validated for Accuracy, Precision: Repeatability, Intermediate Precision, Selectivity/Specificity, Detection limit, Quantitation limit, Linearity, Range, and Robustness. - TABLE 5 In-Situ and Isolated Intermediates Chemical Name [CAS No] Code Name Step No. Control Structure Bisulfate Complex of Ketone 1 Bisulfate Complex (3) Step 1 Isolated (Solid)1-(3- (Trifluoromethyl)- phenylacetone [21906-39-8] Ketone (2) Step 2 In-Situ (oil)Fenfluramine Free Base [458-24-2] Fenfluramine Free Base (1) Step 3 In-Situ (oil)Fenfluramine HCl [404-82-0] Crude Fenfluramine HCl Step 4 Isolated (Solid)
7. Characterization
- [0130]
Physiochemical Characteristics of Drug Substance. - [0131]
Fenfluramine HCl is developed as a single polymorph Form 1. A polymorphism and pre-formulation study has been conducted. Under a wide range of solvents and conditions crystalline material is produced of the same polymorph Form 1 based on a well-defined XRPD pattern and a consistent reproducible endotherm by DSC analysis. A summary of the chemophysical properties of Fenfluramine HCl from this study is provided below. Tabulated data includes example diffractograms, DSCs, and micrographs. - [0132]
The input Fenfluramine HCl (from precipitative isolation) was characterized to provide reference data and also to determine if the salt was of the same form as that identified from previous salt formations. The XRPD pattern of the salt reveals a crystalline solid that visually matches the reflection patterns obtained from formal crystallization of Fenfluramine HCl and has been arbitrarily termed Form 1. Comparison of the μATR-FTIR data for the salt from various batches gave profiles that had a 99.95% match. - [0133]
Thermal data analysis matched previous data obtained with only one major endotherm on the DSC thermograph peaking at 172.3° C. that matches the beginning of potential decomposition shown in a TGA thermograph. This also matches the reported melting point quoted for the reference standard. - [0134]
Isolation of the amorphous form has been shown to be difficult, with attempts using three common methods (rapid solvent evaporation, anti-solvent precipitation and lyophilization) all yielding highly crystalline solids that very closely share the same XRPD pattern of the input Form 1. - [0135]
Stability analysis of the salt after one week at 40° C./0% RH, three weeks at 40° C./75% RH, and under photostability conditions revealed that the input Form 1 has been maintained with no new impurities observed at 0.1% threshold. - [0136]
Results from DSC heat cycling analysis of Fenfluramine HCl are comparable to results generated when the material was held at 170° C. No crystallization event was noted and the amorphous was not generated but rather Form 1 was returned. - [0137]
Holding Fenfluramine HCl at approximately 170° C. for several hours causes a melt and evaporation event to take place with recombination and cooling to provide a white solid. Analysis of the white solid by XRPD, DSC and 1H NMR indicates no change in chemical or physical form, purity, or dissociation. - [0138]
Forced degradation studies carried out have proven Fenfluramine HCl to be stable under a range of conditions. Thermal modulation of Fenfluramine HCl repeatedly yielded the input Form 1.
8. Impurities
- [0139]
Impurities in a drug substance can be organic impurities (process impurities or drug substance-related degradants), inorganic impurities (salt residues or metals) and residual solvents; some of these impurities must be evaluated as to whether or not they are genotoxic agents. These impurities are taken into consideration and controlled in Fenfluramine HCl preparation by using either compendia or validated analytical methods per the specifications or by separate “for information only” testing. The following sections address the actual and potential impurities in Fenfluramine HCl. - [0140]
Actual Impurities and the Qualification of Synthesis Batch - [0141]
No impurities reported in cGMP drug substance batches intended for use in humans have exceeded the ICHQ3A qualification thresholds of 0.15% (Table 8). All impurities >0.1% are identified and handled as described in ICH Q3A unless they are genotoxic impurities. - [0142]
Process Impurities - [0143]
Table 6 lists the known potential impurities arising from the route of synthesis. All of these impurities are controlled to below ICHQ3A qualification threshold of 0.15% by either process changes and/or control of starting material input purities. - TABLE 6 Fenfluramine HCl Known Potential Process Impurities (Route 1) Observed Observed in in Development cGMP Name PLC Batches Batches [Cas. No.] Source (RRT) ≧0.10%1) ≧0.10%1) Ketone (2) Starting RRT No No [351-35-9] Material or 0.89 Intermediate Fenfluramine By-product RRT Yes No Alcohol 1.60 [621-45-4] Norfenfluramine By-product RRT Yes Yes [1886-26-6] 1.67 2-Fenfluramine Starting RRT No No [172953-70-7] Material 0.89 (isomer) 4-Fenfluramine Starting RRT Yes Yes [1683-15-4] Material 1.02 (isomer) N-(3- By-product RRT Yes Yes (trifluoromethyl)- 0.53-0.57 benzyl)ethanamine [90754-95-3] 1)ICH Q3A Identification threshold. The Reporting threshold (LOQ) for the HPLC method is 0.05%.
- [0144]
Degradation Impurities - [0145]
No change in impurity profile is observed upon long-term storage based on forced degradation studies under the ICH Q1A(R2) conditions of heat (solid, solution), acid, base, oxidizing, and ICH Q1B photostability conditions (solid, solution). Fenfluramine HCL is stable for 7 days as a solid at 150° C. (99.90 parent area %), as a solution in water-acetonitrile at 70° C. (99.73 parent area %), as a solution in acid, base, or photosensitizing conditions at ambient. Only oxidizing conditions (peroxide conditions) produced degradation of Fenfluramine HCl to 94.42% after 1 day producing several new related substances at −1% each consistent by LC-MS with +16 oxidation by-products - [0146]
Organic Volatiles/Residual Solvents - [0147]
Table 11 in the Batch Analysis section summarizes the solvents used in the process and the resulting amounts found in drug substance. All solvents used in the GMP steps are controlled at ICH Q3A limits using a suitably qualified Head-Space (HS) GC method. - [0148]
Inorganic Impurities - [0149]
Heavy Metals conform to either USP <231> or ICP method USP <233> as well as ICH Q3D. - [0150]
Genotoxic Impurities - [0151]
The ICH guidelines Q3A and Q3B are not sufficient to provide guidance on impurities that are DNA-reactive. The European Medicines Agency (EMA) guideline (2006) “Guideline on the Limits of Genotoxic Impurities” (EMA 2006) and the ICH Guideline M7 (2014) “Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk” (ICH Guideline M7) are taken into consideration in controlling for potential genotoxic impurities. The diazonium route to prepare ketone (2) described in FIG. 5 has a disadvantage due to the potential formation of genotoxic intermediates shown as boxed compounds (e.g., N-hydroxyaryl, N-nitrosamine and Nitro compound). Muller et al. (Regulatory Toxicology and Pharmacology 44 (2006) 198-211) list potential functional alert groups that can be genotoxic. Safety guidances and regulations indicate that analysis of a process and identification of potential genotoxic agents, and control of such impurities at sub 10 parts per million levels is critical for safety. Often removal of such impurities and/or demonstrating their absence is costly and time consuming and sometimes difficult to achieve technically. For these reasons, selecting synthetic routes that circumvent the potential for such toxic intermediates is important. Because of the potential problems with the diazo route discussed above, as well as potential safety issues using diazo (shock-sensitive) intermediates, as well as the lower purity profiles with this route, this route is less preferred than the preferred route to ketone (2) starting from Nitrile (5). This route produces no potential genotoxic agents and leads to high purity Ketone (2) after isolation by distillation or via the bisulfite salt adduct—hydrolysis sequence. - [0152]
Additionally, attempts to remove isomer by-products present in commercial supplies of Aniline were unsuccessful whereas crystallization the Acid (4) resulting from hydrolysis of the nitrile (5) provides crystalline Acid (4) which can be purified to remove isomers early in synthesis. Removing impurities and/or isomers early in a synthesis is preferred if it is known such impurities track to final product, as the need to crystallize a final product at the end of a synthesis is more costly in losses and impacts cost of goods more greatly than removing early in synthesis before raw materials are invested along the process. - TABLE 7 Potential Impurities in Fenfluramine Synthesis Synthesis Route No. Compound Route 1 Route 2 CAS. No. 1No Starting Material [351-35-9] 2Starting Material Intermediate [21906-39-8] 4No Intermediate Not Available 5Potential Impurity Potential Impurity [621-45-4] 6Potential Impurity Potential Impurity [1886-26-6] 7Potential Impurity Potential Impurity [172953-70-7] 8Potential Impurity Potential Impurity [1683-15-4] 9Potential Impurity Potential Impurity [90754-95-3]
- TABLE 8 Batch Analyses of Fenfluramine HCl Drug Substance Test Batch 1 Batch 2 Batch 3 Batch 4 Appearance* White solid White solid White solid White solid Identification: FTIR* a) a Conforms Conforms Identification: 1H-NMR Conforms Conforms Conforms Conforms Identification: 13C-NMR Conforms Conforms Conforms Conforms Identification: MS Conforms Conforms Conforms Conforms Purity (HPLC area %) 99.57 99.77 b) b Assay (w/w %)* 99.49 100.37 100.79 100.13 Anhydrous Basis (HPLC) Impurities 2-Fenfluramine ND ND ND ND (HPLC 4-Fenfluramine) 0.16 0.15 0.11 0.12 area %) Fenfluramine Alcohol ND ND ND ND 1-((3-trifluoromethyl)phenyl)acetone ND ND ND ND Acetamide 0.27 ND ND ND N-(3-(trifluoromethyl)- ND 0.08 0.07 0.13 benzyl)ethanamine (RRT 0.53-0.57) Total 0.43 0.23 0.19 0.25 Residual Solvents Methanol ND ND ND ND (GC): ppm Ethanol ND ND ND ND MTBE 597 844 472 800 Ethyl Acetate 115 164 79 150 Toluene 4 7 ND ND Residue on Ignition (w/w %) 0.01 0.02 0.04 ND Heavy Metals (as Pb) <10 ppm <10 ppm b b Heavy Metals ICP (ppm) As a a <0.1 <0.1 Cd a a 0.1 0.1 Hg a a <0.1 <0.1 Pb a a 0.2 <0.4 Co a a <0.1 0.1 Mo a a <0.1 <0.1 Se a a <0.1 <0.1 V a a <0.1 <0.1 Water Determination* 0.21 0.08 0.02 0.03 (Karl Fischer) Chloride content by titration 13.19 13.09 12.92 12.93 XRPD* Form 1 Form 1 Form 1 Form 1 Differential Scanning Onset 169.42° C. 169.23° C. 169.85° C. 168.70° C. Calorimetry (DSC)* Peak 172.82° C. 171.55° C. 172.22° C. 171.97° C. Particle Size % Volume mean (D) a 11 11 19 Malvern (μm) D10 a 1 1 1 D50 a 5 7 9 D90 a 17 26 32 Microbial Total aerobic a a LT 100 CFU/g LT 100 CFU/g Limits Tests* microbial Count USP <61> Total yeast and a a LT 100 CFU/g LT 100 CFU/g molds count USP <62> Absence of a a Absent Absent Pathogens a)These tests were added to the specifications recently thus only recent lots have been tested using this test. b)These tests have been dropped from the specifications thus only historical lots have been tested using this test.
Example 3Method for Hydrolysis of Nitrile (5) to Acid
- [0153]
- TABLE 9 Step Operation 1. Charge 3-(trifluoromethyl)phenyl acetonitrile (1.0 eq., 1.00 wt) and purified water (5.0 vol) to a vessel and commence stirring. 2. Dissolve sodium hydroxide (1.1 wt, 5.0 eq.) in purified water (4.0 vol) at up to 40° C. in a suitable make-up vessel. Caution very exothermic. 3. Charge the aqueous sodium hydroxide solution to the mixture from step 1 at up to 40° C. followed by a line rinse with purified water, code RM0120 (1.0 vol) at up to 40° C. 4. Heat the mixture to 75 to 85° C., target 80° C. over 1 to 2 hours. 5. Heat the mixture at 80° C. until ≦0.1% area nitrile by HPLC analysis, expected 4 to 6 hours. 6. Cool the mixture to 18 to 23° C. 7. Adjust the pH of the mixture to pH ≦2 by charging 6M HCl (expected 7.0 vol) to the mixture at 18 to 23° C. Caution exothermic. 8. Stir the mixture for 15 to 30 minutes at 18 to 23° C. 9. Filter and wash the filter-cake with purified water (2 × 5.0 vol) at 18 to 23° C. 10. Slurry wash the filter-cake with n-heptane, code RM (2 × vol) at 0 to 5° C. 11. Dry the isolated solid at up to 45° C. until the water content is ≦.0.2% w/w by KF analysis according to MET/AN/0163, AKX-reagent. 12. Crystallization of crude stage 1 acid (1.00 wt for input calculation) 13. Charge the crude stage 1 acid (1.00 wt), ethyl acetate (0.75 vol) and n-heptane (10.5 vol) to a vessel and commence stirring. 14. Heat the mixture to 50° C. to achieve dissolution. 15. Cool the mixture to 5° C. and age at 5° C. for at least 30 mins. 16. Filter and wash the filter-cake with n-heptane (2 × 5.0 vol). 17. Dry the isolated solid at up to 45° C. until the residual solvent content by 1H-NMR analysis is ≦.0% w/w EtOAc and ≦.0% w/w n-heptane. Expected yield: 60 to 90% th uncorr. 68 to 103% w/w Expected purity: 93.00 to 99% area by HPLC
Example 4Evaluation of Minor Components Formed During Dakin-West Reaction in Preparation of Ketone (2)
- [0154]
The impurities formed during the Dakin-West chemistry and their subsequent removal using the distillation or via isolation of the product ketone as the bisulfite salt are described. The two major impurities found are shown below. - [0155]
Table 10 shows a table of analytical data for crude Ketone (2) isolated from Dakin-West reaction before and after bisulfite purification. In entry 1 crude Ketone isolated directly from the Dakin-West step (pre-bisulfite treatment) is 61.66% purity (e.g. about 62%) and contains 1.98% (e.g., about 2%) and 4.64% (e.g., about 5%) respectively of impurities having RRTs 1.20 and 1.34, which are proposed to be the acetate and dimer impurities (e.g., depicted above), respectively. In entry 2 which is post bisulfite treatment these are other impurities are removed leading to an overall purity of 95.55% (e.g., about 96%). Other entries shown in Table 10 provide other examples of this impurity enhancement by bisulfite treatment of crude Dakin-West ketone. The last two entries use pure Fluorchem ketone as input to the salt formation step and re-isolation of ketone thus illustrating that the salt formation and re-isolation does not produce any impurities itself. Additionally use of bicarbonate extraction procedure during reaction workup provides an improvement in purity of the resulting composition as it serves to remove any unreacted acid. Crude Ketone (2) made by the Diazo route showed similar improvements in purity when treated with bisulfite and isolated. - TABLE 10 Analytical purity data for crude Ketone (2) isolated from Dakin-West reaction before and after bisulfite purification. RRT is relative retention time (min) in chromatogram. RRT 0.93 1.00 1.009 1.06 Entry 0.85 Aniline 0.95 0.99 Ketone 1.004 Nitrile 1.02 Acid 1.10 1.15 1.34 1.38 1 1.38 1.76 0.04 0.49 61.66 nd 0.29 0.29 0.26 1.98 0.66 4.64 0.14 2 0.82 nd nd nd 95.53 0.31 0.14 nd 0.23 0.01 0.10 0.43 0.27 3 nd nd nd nd 77.82 nd nd nd nd 3.12 0.01 7.76 6.16 4 nd nd nd nd 98.82 nd 0.63 nd nd nd 0.02 0.30 0.22 5 0.08 nd nd 0.05 72.02 nd 0.02 nd nd 7.11 0.04 3.58 10.33 6 nd nd nd nd 99.49 nd 0.02 nd nd nd 0.02 0.11 0.24 7 0.15 0.23 nd nd 98.35 nd nd nd nd nd nd nd 0.24 8 nd nd nd nd 99.84 nd nd nd nd nd nd nd nd Entry 1 (Crude ketone from Route 1); Entry 2 (Ketone Route 1 post bisulfite release); Entry 3 (Crude ketone using crude acid); Entry 4 (Ketone using crude acid Post bisulfite); Entry 5 (Crude ketone using cryst. acid); Entry 6 (Crude ketone using cryst. acid post bisulfite); Entry 7 (Crude ketone using cryst. acid); Entry 8 (Fluorochem ketone); Entry 9 (Fluorochem ketone post bisulfite).
Example 5
- [0156]
- Additional Method for Preparation of 1-(3-trifluoromethyl)phenyl-propan-2-one
- [0157]
35 mL of water and 45 g of 37% (w/w) aqueous hydrochloric acid are put in a flask equipped with stirrer and dropping funnel. 24.25 Grams (0.151 moles) of m-trifluoromethylaniline are added after having cooled to 10 degree C. with an ice bath and then, at 5 degree C., an aqueous solution containing 12.43 g (0.180 moles) of sodium nitrite in 150 ml of water is slowly added. The reaction mixture is stirred for 30 minutes and then is poured during 30 minutes into a mixture made by 90 ml of water, 1.35 g (0.014 moles) of cuprous chloride, 2.30 g (0.013 moles) of cupric chloride dihydrate, 50 ml of acetone, 40.8 g (0.300 moles) of sodium acetate trihydrate and 23 g (0.230 moles) of isopropenyl acetate while keeping the reaction temperature at 30 degree C. After further 30 minutes of stirring, the reaction mixture is brought to 20 degree C., 50 ml of methylene chloride are added and the two layers are separated. - The aqueous layer is discarded while the organic layer is concentrated under vacuum until an oil is obtained which is treated with 35 g of sodium metabisulfite, 70 ml of water and 150 ml of heptane under stirring at room temperature for 12 hours. The suspension is filtered, the bisulfite complex is washed on the filter with 50 ml of heptane and then suspended in a two-phase mixture made by 100 ml of methylene chloride and 150 ml of a 10% (w/v) aqueous solution of sodium hydroxide. The layers are separated after one hour of stirring at room temperature, the aqueous phase is discarded while the organic layer is washed with water and evaporated under vacuum to give pure ketone.
Umbralisib


Umbralisib tosylate
| Formula | C31H24F3N5O3. C7H8O3S |
|---|---|
| Cas | 1532533-72-4 FREE 1532533-67-7 |
| Mol weight | 743.7508 |
FDA APPR 2021/2/5
ウムブラリシブトシル酸塩;
| Treatment of cancer and B-cell related disorders |
Antineoplastic
RP-5152; RP-5237; PI3K delta inhibitors (cancer), Rhizen/Incozen; PI3K delta inhibitors (B-cell lymphoma/hematological cancers), Incozen/Rhizen; TGR-1202; TG-1202; RV-1001; umbralisib tosylate; umbralisib; RP-5264; RP-5307; dual PI3Kdelta/CK1 inhibitor (cancer), TG Therapeutics; Ukoniq
Umbralisib (TGR-1202) is an orally available PI3K delta inhibitor, targeting the delta isoform with nanomolar potency and several fold selectivity over the alpha, beta, and gamma isoforms of PI3K. The delta isoform of PI3K is strongly expressed in cells of hematopoietic origin and is believed to be important in the proliferation and survival of B-cell lymphocytes. Inhibition of PI3K delta signaling with umbralisib has demonstrated robust activity in numerous pre-clinical models and primary cells from patients with hematologic malignancies. Umbralisib is currently in Phase 3 clinical development in combination with Ublituximab for patients with hematologic malignancies.
Umbralisib, sold under the brand name Ukoniq, is a medication for the treatment of marginal zone lymphoma (MZL) and follicular lymphoma (FL).[2] It is taken by mouth.[2]
The most common side effects include increased creatinine, diarrhea-colitis, fatigue, nausea, neutropenia, transaminase elevation, musculoskeletal pain, anemia, thrombocytopenia, upper respiratory tract infection, vomiting, abdominal pain, decreased appetite, and rash.[2]
Umbralisib is a kinase inhibitor including PI3K-delta and casein kinase CK1-epsilon.[2][3][4] Umbralisib was approved for medical use in the United States in February 2021.[2][5]
In April 2019, the FDA granted umbralisib Orphan drug designations for the treatment of nodal MZL, extranodal MZL, and splenic MZL. In January 2019, the FDA granted Breakthrough Therapy Designation for the treatment of MZL in patients who had received at least one prior anti-CD20 regimen, based on the interim data from the MZL umbralisib monotherapy cohort in the UNITY-NHL study. In March 2020, the drug was granted Orphan status for treatment of FL By June 2019, the confirmation of registration path to submit umbralisib for accelerated approval was obtained from the MZL cohort of the UNITY-NHL Phase IIb trial .
In August 2020, the FDA accepted the NDA for review; the MZL indication (patients with previously treated MZL who have received at least one prior anti-CD20 based regimen) was accepted for Priority Review with a PDUFA date of February 15, 2021, while the FL indication (patients with previously treated FL who have received at least two prior systemic therapies) was accepted for standard review with a PDUFA date of June 15, 2021.
In February 2021, the drug was granted accelerated approval by the FDA for second-line MZL and for fourth-line FL, based on results of UNITY-NHL. At that time, commercial launch was expected in the coming days
Medical uses
Umbralisib is indicated for adults with relapsed or refractory marginal zone lymphoma (MZL) who have received at least one prior anti-CD20-based regimen; and adults with relapsed or refractory follicular lymphoma (FL) who have received at least three prior lines of systemic therapy.[2][1]
Umbralisib is a kinase inhibitor. The active pharmaceutical ingredient is umbralisib tosylate with the molecular formula C38H32F3N5O6S and a molecular weight of 743.75 g/mol. The chemical name for umbralisib tosylate is (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo [3, 4-d] pyrimidin-1-yl)-ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4- methylbenzenesulfonate and has the following structure:
 |
Umbralisib tosylate is white to light brown powder that is freely soluble in dimethyl sulfoxide, soluble in methanol, and practically insoluble in water. The ionization constant (pKa) of umbralisib tosylate is 2.71.
UKONIQ tablets are for oral administration. Each tablet contains 200 mg of umbralisib free base equivalent to 260.2 mg of umbralisib tosylate. The tablets also contain inactive ingredients: croscarmellose sodium, hydroxypropyl betadex, hydroxypropyl cellulose, magnesium stearate and microcrystalline cellulose.
The tablet coating film consists of FD&C Blue No. 1, FD&C Yellow No. 5, ferric oxide yellow, hypromellose 2910, polydextrose, polyethylene glycol 8000, titanium dioxide and triacetin.
Indications & Dosage
INDICATIONS
Marginal Zone Lymphoma
UKONIQ is indicated for the treatment of adult patients with relapsed or refractory marginal zone lymphoma (MZL) who have received at least one prior anti-CD20-based regimen.
This indication is approved under accelerated approval based on overall response rate [see Clinical Studies]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).

Follicular Lymphoma
UKONIQ is indicated for the treatment of adult patients with relapsed or refractory follicular lymphoma (FL) who have received at least three prior lines of systemic therapy.
This indication is approved under accelerated approval based on overall response rate [see Clinical Studies]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
Adverse effects
The prescribing information provides warnings and precautions for adverse reactions including infections, neutropenia, diarrhea and non-infectious colitis, hepatotoxicity, and severe cutaneous reactions.[2]
History
It has undergone clinical studies for chronic lymphocytic leukemia (CLL).[6][7] Three year data (including follicular lymphoma and DLBCL) was announced June 2016.[8] It is in combination trials for various leukemias and lymphomas, such as mantle cell lymphoma (MCL)[9][10] and other lymphomas.[11]
Umbralisib was granted breakthrough therapy desgination by the U.S. Food and Drug Administration (FDA) for use in people with marginal zone lymphoma (MZL), a type of cancer with no specifically approved therapies.[12]
FDA approval was based on two single-arm cohorts of an open-label, multi-center, multi-cohort trial, UTX-TGR-205 (NCT02793583), in 69 participants with marginal zone lymphoma (MZL) who received at least one prior therapy, including an anti-CD20 containing regimen, and in 117 participants with follicular lymphoma (FL) after at least two prior systemic therapies.[2] The application for umbralisib was granted priority review for the marginal zone lymphoma (MZL) indication and orphan drug designation for the treatment of MZL and follicular lymphoma (FL).[2][13][14][15][16]
SYN
WO 2014071125

clip
First new chemical entity discovered by Indian scientists gets USFDA approval
Rhizen has retained commercialisation rights for India while also being the manufacturing and supply partner for Umbralisib. Alembic owns 50 per cent stake in Rhizen
Umbralisib, a novel cancer drug discovered and out-licensed by India’s Alembic Pharmaceuticals and its associate drug discovery company Rhizen Pharmaceuticals, has received the drug regulatory approval for sales in the US market. The drug is touted to be the first new chemical entity (NCE) discovered by Indian scientists to secure a US Food and Drug Administration (FDA) approval.
Switzerland based Rhizen had discovered the molecule in 2012 and two years later was licensed to US based TG Therapeutics, which has worldwide sales rights. Rhizen has retained commercialisation rights for India while also being the manufacturing and supply partner for Umbralisib. Alembic owns 50 per cent stake in Rhizen.
Umbralisib is a novel, next generation, oral, once daily drug for adult patients with relapsed or refractory lymphoma and relapsed or refractory marginal zone lymphoma (MZL) that resists treatments and drugs. Such cancers affect over 3-4 lakh patients in the US every year. The drug is estimated to have a global market worth US$ 1-1.5 billion.
“We are extremely proud of this historic milestone for Rhizen, and of the fact that Umbralisib is the first NCE discovered by Indian scientists to secure a US FDA approval,” said Pranav Amin, Chairman, Rhizen Pharmaceuticals & Managing Director of Alembic Pharmaceuticals.
“We are keen to bring Umbralisib to Indian patients and we plan to initiate activities towards registration and approval there soon,” said Swaroop Vakkalanka, President & CEO of Rhizen Pharmaceuticals.
Ahmedabad-based Zydus Cadila had a few months ago got ‘Fast Track Designation’ by the US Food and Drug Administration (USFDA) for Saroglitazar in the treatment of patients with Primary Biliary Cholangitis (PBC), a liver disorder due to progressive destruction of the bile ducts.
PATENT
WO 2021009509
Umbralisib, having the chemical designation (S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one, is an orally available PI3K delta inhibitor. Umbralisib has the following structure:
Inhibition of PI3K delta signaling with umbralisib has demonstrated activity in several pre-clinical models and primary cells from patients with hematologic malignancies. In a Phase 2 trial, umbralisib provided effective PI3K-delta inhibition and appeared well-tolerated among patients with relapsed/refractory marginal zone lymphoma. Umbralisib is currently in Phase 3 clinical development in combination with ublituximab for patients with hematologic malignancies. Hematologic malignancies are forms of cancer that begin in the cells of blood-forming tissue, such as the bone marrow, or in the cells of the immune system. Examples of hematologic cancer are acute and chronic leukemias, lymphomas, multiple myeloma and myelodysplastic syndromes. Lymphomas can include follicular lymphoma (FL), small lymphocytic lymphoma (SLL), non-Hodgkin lymphoma (NHL), and diffuse large B-cell lymphoma (DLBCL), among others. Leukemia can include chronic lymphocytic leukemia (CLL), among others. The U.S. Food and Drug Administration (FDA) has granted orphan drug designation to umbralisib for the treatment of patients with follicular lymphoma and for the treatment of patients with nodal, extranodal, and splenic marginal zone lymphoma.
U.S. Patent No. 9,150,579 discloses umbralisib and pharmaceutically acceptable salts thereof, such as 4-methylbenzenesulfonate (also known as tosylate), sulphate, hydrochloride, benzenesulfonate, maleate, and camphor sulfonate salts. U.S. Patent Nos. 9,969,740 and 10,414,773 and U.S. Patent Application Publication No. 2019/0382411 disclose solid state forms of a p-toluenesulfonic acid salt (PTSA) of umbralisib. None of these references disclose an amorphous form of umbralisib monotosylate.
An amorphous form of a compound is considered to be a solid state form that lacks long-range order relative to crystalline solid state forms of the compound. The amorphous form is chemically identical to other crystalline solid state forms but can exhibit different physical properties such as intrinsic solubility, rate of dissolution, density, mechanical property, chemical and physical stability, hygroscopicity, and morphology. The differences in intrinsic solubility also may lead to a difference in the rate of absorption, thus impacting bioavailability. Generally, amorphous compounds have a higher solubility than crystalline compounds.
EXAMPLES
Examples 1-3, which follow herein, provide embodiments of the preparation of amorphous umbralisib monotosylate.
Example 1
Preparation of Amorphous Umbralisib Monotosylate by Dry Grinding of Crystalline Umbralisib Tosylate Salt
Form I of umbralisib tosylate salt is dried under vacuum at about 40 °C in an oven for at least about 3 days to remove any residual ethyl acetate. About 30 mg of the dried umbralisib tosylate salt is ground manually using a mortar (about 6 cm in diameter) and pestle for about 3 minutes. The ground umbralisib tosylate salt is identified as being amorphous by XRPD. FIG. 1 is a representative XPRD pattern for amorphous umbralisib monotosylate prepared according to Example 1.
The amorphous umbralisib monotosylate prepared according to Example 1 is characterized by a Tg of about 51 °C, as depicted in the mDSC thermogram contained in FIG. 2.
A DVS of amorphous umbralisib monotosylate prepared according to Example 1 indicates the sample is hygroscopic, with about a 4% weight change between about 0-90% relative humidity, as depicted in FIG. 3, and less than about a 1% weight change in the sample over three cycles, as depicted in FIG. 4.
An XRPD pattern of the sample after DVS indicates that the sample is still amorphous, as depicted in FIG. 5.
Example 2
Preparation of Amorphous Umbralisib Monotosylate by Dissolution of
Crystalline Umbralisib Tosylate Salt in Methanol and Its Evaporation Therefrom
About 470 mg of Form I of umbralisib tosylate salt is dissolved in about 20 mL of methanol at about 50 °C. A solid umbralisib tosylate salt is obtained by evaporation of the solution under vacuum at about 40 °C in an oven overnight. The isolated product is identified as being amorphous umbralisib monotosylate by XRPD. FIG. 6 is a representative XPRD pattern for amorphous umbralisib monotosylate prepared according to Example 2.
The amorphous umbralisib monotosylate prepared according to Example 2 is characterized by a Tg of about 75 °C, as depicted in the mDSC thermogram contained in FIG. 7.
A TGA of amorphous umbralisib monotosylate prepared according to Example 2 shows about a 0.9% weight loss up to about 120 °C, as depicted in FIG. 8.
A DVS of amorphous umbralisib monotosylate prepared according to Example 2 indicates that the sample is hygroscopic, with about a 4% weight change between about 0-90% relative humidity, as depicted in FIG. 9, with about a 0.5% weight change in the sample over three cycles, as depicted in FIG. 10.
An XRPD pattern of the sample after DVS indicates that the sample is still amorphous, as depicted in FIG. 11.
‘ H NMR is carried out on a sample of amorphous umbralisib monotosylate prepared according to Example 2 in DMSO-d6 which indicates an umbralisib tosylate salt with a 1 :0.9 ratio of free base to acid, as depicted in FIG. 12. The peak at 8.25 ppm is representative of a single proton in the free base and the peaks at 2.30 ppm are the three protons from p-toluenesulfonic acid. A trace amount (about 0.07%) of methanol is observed at 3.16 ppm.
FTIR spectra is collected on amorphous umbralisib monotosylate prepared according to Example 2, as depicted in FIG. 13(a) and on starting crystalline umbralisib tosylate salt, as depicted in FIG. 13(b).
XRPD of amorphous umbralisib monotosylate prepared according to Example 2 after storage at about 40 °C under vacuum conditions for about two weeks indicates that the sample is still amorphous, as depicted in FIG. 14. Further, mDSC of amorphous umbralisib monotosylate after storage at about 40 °C under vacuum conditions for about two weeks indicates that the Tg is increased to about 83 °C, as depicted in FIG. 15.
Example 3
Solution Preparation of Amorphous Umbralisib Monotosylate from Umbralisib
Free Base and p-Toluenesulfonic Acid
Umbralisib free base and p-toluenesulfonic acid are each separately dissolved in MeOH. Specifically, about 72 mg of umbralisib free base is dissolved in about 3mL of MeOH at about 50 °C and about 24 mg of p-toluenesulfonic acid is dissolved in about 0.25 mL of MeOH at about 50 °C. The two solutions are mixed and stirred at room temperature for about 1 hr and then at about 4 °C overnight. The solution is transferred to a vacuum oven at about 40 °C overnight to evaporate the MeOH. Amorphous umbralisib monotosylate, identified by XRPD, is obtained. FIG. 16 is a representative XPRD pattern for amorphous umbralisib monotosylate prepared according to Example 3.
PATENT
WO 2015181728
TGR-1202, chemically known as (S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-3-(3-fluorophenyl)-4H-chromen-4-one, has the following chemical structure:
[04] The preparation of TGR-1202 and its salts is described in International Publication No. WO 2014/006572 and U.S. Patent Publication No. 2014/0011819, each of which is incorporated herein by reference in its entirety for all purposes. TGR-1202 is an investigational drug currently undergoing multiple clinical trials in the area of haematological malignancies.
[05] WO 2014/006572 and US 2014/0011819 describe the synthesis of TGR-1202 (Example B l) and also disclose the therapeutic activity of this molecule to inhibit, regulate and/or modulate the signal transduction of PI3K.
Example 1: Preparation of the PTSA Salt of TGR-1202 (Form A)
[103] 7100 g of TGR-1202 was charged in a reactor containing 56.8 litres of acetone and stirred at ambient temperature. 4680 g of p-toluene sulphonic acid was added and the reaction mixture was heated at a temperature of 60-65° C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 142 litres of diethyl ether was then added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass. The solid mass was re-suspended in diethyl ether, stirred for 6 hours, and then filtered to yield a solid mass which was subsequently dissolved in 56.8 litres of acetone, filtered through a HiFlow bed, and concentrated under reduced pressure. The resulting residue mass was stirred with water overnight, then filtered and vacuum dried to yield 6600 g of the PTSA salt of TGR-1202. HPLC: 99.21% and chiral purity of 99.64:0.36 (S:R).
Example 2: Preparation of the PTSA Salt of TGR-1202 (Form B)
1000 g of TGR-1202 was charged in a reactor containing 8 litres of acetone and stirred at ambient temperature. 666 g of p-toluene sulphonic acid was then added and the reaction mixture was heated at a temperature of 60-65 °C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 20 litres of diethyl ether was added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass which was then vacuum dried to yield 1150 g of the PTSA salt of TGR-1202. HPLC: 99.33% and chiral purity: 99.61:0.39 (S:R).
PATENT
WO 2014006572
Intermediate 1
[104] Intermediate 1: 6-fluoro-3-(3-fluorophenyl)-2-(l-hydroxyethyl)-4H-chromen-4-one: To a solution of 2-(l-bromoethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (15.0 g,
40.84 mmol) in DMSO (150 ml), n-butanol (7.5 ml) was added and heated to 120°C for 3h. The reaction mixture was cooled to RT, quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (7.90 g, 64%). H-NMR (δ ppm, CDC13, 400 MHz): 7.85 (dd, J = 8.1, 3 Hz, 1H), 7.54 (dd, J = 9.2, 4.2 Hz, 1H), 7.47-7.37 (m, 2H), 7.15-6.98 (m, 3H), 4.74 (quintet, J = 6.8 Hz, 1H), 2.23 (d, J = 7.4 Hz, 1H), 1.54 (d, J = 6.6 Hz, 3H).
Intermediate 2
[105] Intermediate 2: 2-acetyl-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one: DMSO (5.60 ml, 79.14 mmol) was added to dichloromethane (40 ml) cooled to -78°C, followed by oxalyl chloride (3.40 ml, 39.57 mmol). After 10 min. intermediate 1 (6.00 g, 19.78 mmol) in dichloromethane (54 ml) was added dropwise and stirred for 20 min. Triethylamine (12 ml) was added and stirred for lh. The reaction mixture was quenched with water and extracted with dichloromethane. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (4.2 g, 71%) which was used as such in the next step.
Intermediate 3
OH
[106] Intermediate 3: (S)-6-fluoro-3-(3-fluorophenyl)-2-(l-hydroxyethyl)-4H-chromen-4-one: To intermediate 2 (2.00 g, 6.66 mmol), R-Alpine borane (0.5M in THF, 20 ml) was added and heated to 60°C for 20h. The reaction mixture quenched with aq. 2N HC1, and
extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.51 g, 75%). Enantiomeric excess: 94.2%, enriched in the fast eluting isomer (retention time: 8.78 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 4
[107] Intermediate 4: (R)-l-(6-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl 4-chlorobenzoate: To a solution of intermediate 3 (1.45 g, 4.78 mmol) in THF (15 ml), 4-chlorobenzoic acid (0.748 g, 4.78 mmol) and triphenylphosphine (1.88 g, 7.17 mmol) were added and heated to 45 C followed by diisopropylazodicarboxylate (1.4ml, 7.17 mmol). After lh, the reaction mixture was concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.81 g, 86%) which was used without purification in the next step.
Intermediate 5
Method A
[108] Intermediate 5: (R)-6-fluoro-3-(3-fluorophenyl)-2-(l-hydroxyethyl)-4H-chromen-4-one: To intermediate 4 (1.75 g, 3.96 mmol) in methanol (17 ml) cooled to 10°C, potassium carbonate (0.273 g, 1.98 mmol) was added and stirred for 30 min. The reaction mixture was concentrated, acidified with 2N HC1 solution, extracted with ethyl acetate, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (1.05 g, 87%). Enantiomeric excess: 93.6%, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Method B:
[109] Step-1 : (R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one : To l-(5-fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone (11.00 g, 44.31 mmol ) in dichloromethane, HATU (33.7 g, 88.63 mmol) and R-(+)2-benzyloxypropionic acid (9.58 g, 53.17 mmol) were added and stirred for 10 min. Triethylamine (66.7 ml, 0.47 mol) was added dropwise and stirred at RT for 24h. The reaction mixture was quenched with water, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (10.5 g, 60%). ‘H-NMR (δ ppm, CDC13, 400 MHz): 7.85 (dd, J = 8.1,3 Hz, 1H), 7.58 (dd, J = 9.1, 4.1 Hz, 1H), 7.47-7.39 (m, 1H), 7.39-7.34 (m, 1H), 7.28-7.20 (m, 3H), 7.20-7.14 (m, 2H), 7.16-7.07 (m, 1H), 6.99-6.89 (m, 2H), 4.50-4.31 (m, 3H), 1.56 (d, J = 6.4 Hz, 3H).
[110] Step-2 : (R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (10.5 g, 26.69 mmol) in dichloromethane (110 ml) cooled to 0°C, aluminium chloride (5.35 g, 40.03 mmol) was added portionwise and stirred at RT for 6h. The reaction mixture was quenched with 2N HC1 solution, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the desired intermediate as a yellow solid (6.1 g, 76%). Enantiomeric excess: 97.7%, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 13
[121] Intermediate 13: 3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-amine: To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF 110 ml), ethanol (55 ml) and water (55 ml), intermediate 12 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min. Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h, the reaction mixture was filtered though celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
Example Bl
(S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
[127] To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate ( 0.15 ml, 0.749 mmol) was added heated to 45°C. After 2h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 %). MP: 139-142°C. Mass : 571.7 (M H-NMR (δ ppm, CDC13, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J = 8.2,3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J = 8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J = 8.4 Hz, 1H), 6.11 (q, J = 7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J = 6.1 Hz, 1H), 2.00 (d, J = 7.1Hz, 3H), 1.42 (d, J = 6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC
on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64min.).
PATENT
US 2014/0011819 describe the synthesis of TGR-1202 (Example B l)
PATENT
US 20150290317
US 20150174263
WO 2014071125
WO 2014006572
WO 2013188763*
References
- ^ Jump up to:a b c d e f “Ukoniq (umbralisib) tablets, for oral use” (PDF). TG Therapeutics.
- ^ Jump up to:a b c d e f g h i j “FDA grants accelerated approval to umbralisib for marginal zone lymphoma and follicular lymphoma”. U.S. Food and Drug Administration (FDA). 5 February 2021. Retrieved 5 February 2021. This article incorporates text from this source, which is in the public domain.
- ^ Lunning M, Vose J, Nastoupil L, Fowler N, Burger JA, Wierda WG, et al. (November 2019). “Ublituximab and umbralisib in relapsed/refractory B-cell non-Hodgkin lymphoma and chronic lymphocytic leukemia”. Blood. 134 (21): 1811–20. doi:10.1182/blood.2019002118. PMC 7042665. PMID 31558467.
- ^ Burris HA, Flinn IW, Patel MR, Fenske TS, Deng C, Brander DM, et al. (April 2018). “Umbralisib, a novel PI3Kδ and casein kinase-1ε inhibitor, in relapsed or refractory chronic lymphocytic leukaemia and lymphoma: an open-label, phase 1, dose-escalation, first-in-human study”. Lancet Oncology. 19 (4): 486–96. doi:10.1016/S1470-2045(18)30082-2. PMID 29475723.
- ^ “TG Therapeutics Announces FDA Accelerated Approval of Ukoniq (umbralisib)” (Press release). TG Therapeutics. 5 February 2021. Retrieved 5 February 2021 – via GlobeNewswire.
- ^ Inman S (19 March 2016). “Novel BTK, PI3K Inhibitors on Horizon for Relapsed CLL”. OncLive. Archived from the original on 1 May 2016.
- ^ “Therapy Focus –- TG Could Benefit From Zydelig Setback”. Seeking Alpha. 29 March 2016.
- ^ “TG Therapeutics, Inc. Announces First Patient Enrolled in the Registration-Directed UNITY-DLBCL Phase 2b Trial”. TG Therapeutics Inc. June 2016.
- ^ Clinical trial number NCT02268851 for “A Phase I/Ib Safety and Efficacy Study of the PI3K-delta Inhibitor TGR-1202 and Ibrutinib in Patients With CLL or MCL” at ClinicalTrials.gov
- ^ “Follow-Up Data for Combination of TGR-1202 (umbralisib) plus Ibrutinib in Patients with Relapsed or Refractory CLL and MCL”(Press release). TG Therapeutics. 14 June 2017 – via Globenewswire.
- ^ Clinical trial number NCT02793583 for “Study to Assess the Efficacy and Safety of Ublituximab + TGR-1202 With or Without Bendamustine and TGR-1202 Alone in Patients With Previously Treated Non-Hodgkin’s Lymphoma (UNITY-NHL)” at ClinicalTrials.gov
- ^ Columbus G (22 January 2019). “FDA Grants Umbralisib Breakthrough Designation for Marginal Zone Lymphoma”. OncLive. Archived from the original on 23 January 2019.
- ^ “Orphan Treatment of extranodal marginal zone lymphoma”. U.S. Food and Drug Administration (FDA). 11 April 2019. Retrieved 5 February 2021.
- ^ “Orphan Treatment of splenic marginal zone lymphoma”. U.S. Food and Drug Administration (FDA). 11 April 2019. Retrieved 5 February 2021.
- ^ “Orphan Treatment of Follicular Lymphoma”. U.S. Food and Drug Administration (FDA). 11 April 2019. Retrieved 5 February2021.
- ^ “Orphan Treatment of nodal marginal zone lymphoma”. U.S. Food and Drug Administration (FDA). 11 April 2019. Retrieved 5 February 2021.
External links
- “Umbralisib”. Drug Information Portal. U.S. National Library of Medicine.
- “Umbralisib”. NCI Drug Dictionary. National Cancer Institute.
| Clinical data | |
|---|---|
| Trade names | Ukoniq |
| Other names | RP5264; TGR-1202 |
| License data | US DailyMed: Umbralisib |
| Pregnancy category | Not recommended[1] |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Pharmacokinetic data | |
| Metabolism | CYP2C9, CYP3A4, and CYP1A2[1] |
| Elimination half-life | 91 h[1] |
| Excretion | Feces, urine[1] |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 1532533-67-7 |
| PubChem CID | 72950888 |
| DrugBank | DB14989 |
| ChemSpider | 34979945 |
| UNII | 38073MQB2A |
| ChEMBL | ChEMBL3948730 |
| Chemical and physical data | |
| Formula | C31H24F3N5O3 |
| Molar mass | 571.560 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]CC(C)OC1=C(C=C(C=C1)C2=NN(C3=NC=NC(=C23)N)C(C)C4=C(C(=O)C5=C(O4)C=CC(=C5)F)C6=CC(=CC=C6)F)F |
Feb. 9, 2021 04:45 UTC Rhizen Pharmaceuticals AG Announces That Its Partnered Asset, Umbralisib (UKONIQ™), Has Received US FDA Accelerated Approval for Adult Patients With Relapsed or Refractory MZL & FL
Umbralisib (UKONIQ™) granted accelerated approval by US FDA for the treatment of adult patients with relapsed or refractory marginal zone lymphoma (MZL), follicular lymphoma (FL).
Umbralisib, a novel next generation inhibitor of PI3K delta & CK1 epsilon, was discovered by Rhizen Pharmaceuticals and subsequently licensed to TG Therapeutics, who led the asset’s clinical development.
Rhizen and its affiliate Alembic Pharma to support TG Therapeutics towards UKONIQ’s commercialization as its manufacturing & supply partner; Rhizen plans to register and commercialize Umbralisib in India.
BASEL, Switzerland–(BUSINESS WIRE)–Rhizen Pharmaceuticals, a clinical-stage oncology-focused biopharmaceutical company, today announced that its novel next generation PI3K-delta inhibitor, Umbralisib, which was licensed to TG Therapeutics (NASDAQ:TGTX), has secured US FDA accelerated approval for the treatment of:
adult patients with relapsed or refractory marginal zone lymphoma (MZL) who have received at least one prior anti-CD20 based regimen, and
adult patients with relapsed or refractory follicular lymphoma (FL) who have received at least three prior lines of systemic therapy.
Accelerated approval was granted for these indications, under a priority review (MZL), based on the results of the Phase 2 UNITY-NHL Trial (NCT02793583); in MZL, an ORR of 49% with 16% complete responses and in FL an ORR of 43% with 3% complete responses were achieved, respectively. Umbralisib was earlier granted Breakthrough Therapy Designation (BTD) for the treatment of MZL and orphan drug designation (ODD) for the treatment of MZL and FL.
Umbralisib is a novel, next generation, oral, once daily, inhibitor of phosphoinositide 3 kinase (PI3K) delta and casein kinase 1 (CK1) epsilon and was discovered by Rhizen Pharma and subsequently licensed to TG Therapeutics (NASDAQ:TGTX) at an IND stage (TGR 1202) in 2012. In 2014, both parties entered into a licensing agreement as a part of which TGTX obtained worldwide rights and Rhizen has retained commercialization rights for India while also being the manufacturing and supply partner for Umbralisib.
Swaroop Vakkalanka, President & CEO of Rhizen Pharmaceuticals said: “Umbralisib’s approval offers MZL & FL patients a new treatment option and is a huge validation of Rhizen’s drug discovery & development capabilities. This is a momentous occasion in Rhizen’s journey as a successful biotech that speaks of the true ability of our team to discover & develop safe and effective therapies that can last the rigors of drug development. Further, we are keen to bring Umbralisib to Indian patients and we plan to initiate activities towards registration and approval there soon.”
Pranav Amin, Chairman, Rhizen Pharmaceuticals & Managing Director of Alembic Pharmaceuticals Ltd said: “We are extremely proud of this historic milestone for Rhizen, and of the fact that Umbralisib is the first NCE discovered by Indian scientists to secure a US FDA approval. We are committed to working together with TG Therapeutics and Rhizen Pharma to ensure uninterrupted supply of UKONIQ™. Umbralisib is the first discovery asset to come out of Rhizen’s R&D efforts and this approval heralds the promise of the rest of Rhizen’s deep pipeline and continuing efforts.”
About Umbralisib:
Umbralisib is the first and only oral inhibitor of phosphoinositide 3 kinase (PI3K) delta and casein kinase 1 (CK1) epsilon. PI3K-delta is known to play an important role in supporting cell proliferation and survival, cell differentiation, intercellular trafficking and immunity and is expressed in both normal and malignant B-cells. CK1-epsilon is a regulator of oncoprotein translation and has been implicated in the pathogenesis of cancer cells, including lymphoid malignancies. Umbralisib is indicated for the treatment of adult patients with relapsed or refractory marginal zone lymphoma (MZL) who have received at least one prior anti-CD20-based regimen and for the treatment of adult patients with relapsed or refractory follicular lymphoma (FL) who have received at least three prior lines of systemic therapy. These indications are approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial. More information on Umbralisib or UKONIQ™ can be found at https://www.tgtherapeutics.com/prescribing-information/uspi-ukon.pdf.
About Alembic Pharmaceuticals Ltd:
Alembic Pharmaceuticals Limited, a vertically integrated research and development pharmaceutical company, has been at the forefront of healthcare since 1907. Headquartered in India, Alembic is a publicly listed company that manufactures and markets generic pharmaceutical products all over the world. Alembic’s state of the art research and manufacturing facilities are approved by regulatory authorities of many developed countries including the USFDA. Alembic is one of the leaders in branded generics in India. Alembic’s products that are marketed through a marketing team of over 5000 are well recognized by doctors and patients.
Information about Alembic can be found at http://www.alembicpharmaceuticals.com/.
(Reuters: ALEM.NS) (Bloomberg: ALPM) (NSE: APLL TD) (BSE: 533573)
About Rhizen Pharmaceuticals A.G.:
Rhizen Pharmaceuticals is an innovative, clinical-stage biopharmaceutical company focused on the discovery and development of novel onco-therapeutics. Since its establishment in 2008, Rhizen has created a diverse pipeline of proprietary drug candidates targeting several cancers and immune associated cellular pathways. Rhizen is headquartered in Basel, Switzerland. For additional information, please visit http://www.rhizen.com.
View source version on businesswire.com: https://www.businesswire.com/news/home/20210208005742/en/ Contacts
////////////ウムブラリシブトシル酸塩 , Umbralisib, fda 2021, 2021 approvals, TGR 1202, TGR-1202-101, RP 5264, Umbralisib tosylate, RP-5307 , TGR-1202, TGR-1202 PTSA, FU8XW5V3FS , RP-5264, AK173784,
old post pasted

TGR 1202, TGR-1202-101, RP 5264, UmbralisibAK173784;(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one(S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-3-(3-fluorophenyl)-4H-chromen-4-one,2-[(1S)-1-[4-amino-3-(3-fluoro-4-propan-2-yloxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]ethyl]-6-fluoro-3-(3-fluorophenyl)chromen-4-one CAS TOSYLATE 1532533-72-4 Umbralisib tosylateCAS 1532533-67-7, 1514919-95-9
| Molecular Formula: | C31H24F3N5O3 |
|---|---|
| Molecular Weight: | 571.54917 g/mol |
RP-5307
TGR-1202
TGR-1202 PTSA
FU8XW5V3FS (UNII code)
RP-5264 (free base)
A PI3K inhibitor potentially for treatment of chronic lymphocytic leukemia, leukemia,lymphoma,B-cell
TGR‐1202, a next generation PI3K-δ delta inhibitor. TGR-1202 (RP-5264) is a highly specific, orally available, PI3K delta inhibitor, targeting the delta isoform with nanomolar potency and several fold selectivity over the alpha, beta, and gamma isoforms of PI3K.
TG Therapeutics, under license from Rhizen Pharmaceuticals, is developing TGR-1202 (structure shown; formerly RP-5264), a lead from a program of PI3K delta inhibitors, for the potential oral treatment of hematological cancers including Hodgkin lymphoma, non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), B-cell lymphoma and mantle cell lymphoma (MCL)
Incozen Therapeutics Pvt Ltd
TG Therapeutics
TGR-1202 potential to perform as the best PI3K inhibitor in its class and the possible superiority of TG-1101 over Rituxan®.
| Rhizen Pharmaceuticals S.A. | |
| Description | Phosphoinositide 3-kinase (PI3K) delta inhibitor |
Leukemia, chronic lymphocytic PHASE 3, TG Therapeutics
Orphan Drug
Umbralisib is a novel phosphatidylinositol 3-kinase delta (PI3Kdelta) inhibitor under development at TG Therapeutics in phase III clinical trials, in combination with ublituximab, for the treatment of chronic lymphocytic leukemia (CLL) and for the treatment of diffuse large B-cell lymphoma (DLBCL). The company refers to the combination regimen of ublituximab and TGR-1202 as TG-1303. The drug is also in phase II clinical development for the oral treatment of hematologic malignancies, as a single agent or in combination therapy. Phase I clinical trials are ongoing in patients with select relapsed or refractory solid tumors, such as adenocarcinoma of the pancreas, adenocarcinoma of the colon, rectum, gastric and GE junction cancer, and GI Stromal Tumor (GIST).
In 2016, orphan drug designation was assigned to the compound in the U.S. for the treatment of CLL. In 2017, additional orphan drug designation was granted in the U.S. for the treatment of CLL and DLBCL, in combination with ublituximab.
Originated by Rhizen Pharmaceuticals, the product was jointly developed by Rhizen Pharmaceuticals and TG Therapeutics since 2012. In 2014, exclusive global development and commercialization rights (excluding India) were licensed to TG Therapeutics.
CLINICAL TRIALS……….https://clinicaltrials.gov/search/intervention=TGR-1202
B-cell lymphoma; Chronic lymphocytic leukemia; Hematological neoplasm; Hodgkins disease; Mantle cell lymphoma; Non-Hodgkin lymphoma
Phosphoinositide-3 kinase delta inhibitor
SYNTHESIS


Rhizen Pharmaceuticals Announces Out-licensing Agreement for TGR-1202, a Novel Next Generation PI3K-delta Inhibitor
Rhizen to receive upfront payment of $8.0 million — Rhizen to retain global manufacturing and supply rights — Rhizen to retain development and commercialization for India
Rhizen to retain development and commercialization for India
September 23, 2014 09:00 ET | Source: Rhizen Pharmaceuticals SA
La Chaux-de-Fonds, Switzerland, Sept. 23, 2014 (GLOBE NEWSWIRE) — Rhizen Pharmaceuticals S.A. today announced an out-licensing agreement for TGR-1202, a novel next generation PI3K-delta inhibitor. TG Therapeutics exercised its option for early conversion to a licensing agreement from a 50:50 joint venture partnership.
In exchange for this licensing agreement, TG Therapeutics will pay Rhizen an upfront payment of $8.0 million ($4.0 million in cash and $4.0 million in TG Therapeutics common stock). In addition to the upfront payment, Rhizen will be eligible to receive regulatory filing, approval and sales based milestones in the aggregate of approximately $240 million, and tiered royalties based on net sales.
Swaroop Vakkalanka, Ph.D. and President of Rhizen stated, “We are extremely happy and take pride in discovering a novel, next generation, once-daily PI3K-delta inhibitor under active development led by TG Therapeutics. We are encouraged by the progress of TRG-1202 to date, and the speed at which TG Therapeutics is developing the asset in various hematological malignancies. We look forward to the day this novel drug reaches cancer patients in need of new and safe therapies.”
About Rhizen Pharmaceuticals S.A.:
Rhizen Pharmaceuticals is an innovative, clinical-stage biopharmaceutical company focused on the discovery and development of novel therapeutics for the treatment of cancer, immune and metabolic disorders. Since its establishment in 2008, Rhizen has created a diverse pipeline of proprietary drug candidates targeting several cancers and immune associated cellular pathways. Rhizen is headquartered in La-Chaux-de-Fonds, Switzerland. For additional information, please visit Rhizen’s website, www.rhizen.com.

TGR-1202.with Idelalisib and IPI-145 (left to right) for comparison.
IPI 145
PATENTS
WO 2011055215
http://www.google.com/patents/WO2011055215A2?cl=en

PATENT
WO 2015181728
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015181728
TGR-1202, chemically known as (S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-3-(3-fluorophenyl)-4H-chromen-4-one, has the following chemical structure:

Example 1: Preparation of the PTSA Salt of TGR-1202 (Form A)

7100 g of TGR-1202 was charged in a reactor containing 56.8 litres of acetone and stirred at ambient temperature. 4680 g of p-toluene sulphonic acid was added and the reaction mixture was heated at a temperature of 60-65° C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 142 litres of diethyl ether was then added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass. The solid mass was re-suspended in diethyl ether, stirred for 6 hours, and then filtered to yield a solid mass which was subsequently dissolved in 56.8 litres of acetone, filtered through a HiFlow bed, and concentrated under reduced pressure. The resulting residue mass was stirred with water overnight, then filtered and vacuum dried to yield 6600 g of the PTSA salt of TGR-1202. HPLC: 99.21% and chiral purity of 99.64:0.36 (S:R).
Example 2: Preparation of the PTSA Salt of TGR-1202 (Form B)

1000 g of TGR-1202 was charged in a reactor containing 8 litres of acetone and stirred at ambient temperature. 666 g of p-toluene sulphonic acid was then added and the reaction mixture was heated at a temperature of 60-65 °C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 20 litres of diethyl ether was added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass which was then vacuum dried to yield 1150 g of the PTSA salt of TGR-1202. HPLC: 99.33% and chiral purity: 99.61:0.39 (S:R).
Table 1 lists the XRPD pattern peaks and relative peak intensities for the products of Examples 1 and 2.
TABLE 1

The tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 2 exhibited a Cmax about 2.5 fold and an area under the curve (AUC) about 1.9 fold greater than that of the tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 1. The results are provided in Table 8 below.
TABLE 8

PATENT
WO 2014071125
http://www.google.com/patents/WO2014071125A1?cl=en
formula (A) that is a ΡΒΚδ selective inhibitor,

(A)
Synthesis of Compound of Formula A
Unless otherwise stated, purification implies column chromatography using silica gel as the stationary phase and a mixture of petroleum ether (boiling at 60-80°C) and ethyl acetate or dichloromethane and methanol of suitable polarity as the mobile phases. The term “RT” refers to ambient temperature (25-28°C).
Intermediate 1 : 2-( l-bromoethyl)-6-fluoro-3-f3-fluorophenyl)-4H-chromen-4-one
Step-1 [l-(5-Fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone]: 3- Fluorophenylacetic acid (7.33 g, 47.56 mmoles) was dissolved in 25 ml dichloromethane. To this mixture, oxalylchloride (7.54 g, 59.46 mmoles) and DMF (3 drops) were added at 0°C and stirred for 30 min. The solvent was evaporated and dissolved in 25 ml dichloromethane. To this mixture, 4-fluoroanisole (5.00 g, 39.64 mmoles) was added and cooled to 0°C. At 0°C A1C13 (7.95 g, 59.46 mmoles) was added and the reaction mixture was warmed to RT and stirred for 12 hours. The reaction mixture was quenched by the addition of 2N HC1, extracted with ethyl acetate, dried over sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound as colorless solid (4.5 g, 45% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 11.34 (s, 1H), 7.75 (dd, J=9.4, 3.1 Hz, 1H), 7.42 (m, 2H), 7.12 (m, 3H), 7.05 (dd, J=9.0, 4.5 Hz, 1H), 4.47 (s, 2H).
Step-2 [2-Ethyl-6-fiuoro-3-(3-fluorophenyl)-4H-chromen-4-one]: l-(5-Fluoro-2- hydroxyphenyl)-2-(3-fluorophenyl)ethanone obtained from Step-1 (3.00 g, 12.08 mmoles) was placed in a round bottom flask and to this triethylamine (25 ml) and propionic anhydride (4.92 g, 37.82 mmoles) were added, and the mixture was refluxed for 24 hours. After cooling to RT, the reaction mixture was acidified by the addition of IN HC1 solution, extracted with ethyl acetate, washed with sodium bicarbonate solution, dried with sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound as off-yellow solid (1.80 g, 52% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 7.80 (m, 1H), 7.76 (m, 2H), 7.51 (dd, J=8.0, 6.4 Hz), 7.22 (m, 1H), 7.18 (m, 2H), 2.56 (q, J=7.6 Hz, 2H), 1.20 (t, J=7.6 Hz, 3H).
Step-3: To a solution of 2-Ethyl-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one obtained from Step-2 (1.80 g, 6.28 mmoles) in carbon tetrachloride (20 ml), N- bromosuccinimide (1.11 g, 6.28 mmoles) was added and heated to 80°C. Azobisisobutyronitrile (10 mg) was added to the reaction mixture at 80°C. After 12 hours, the reaction mixture was cooled to RT, diluted with dichloromethane and washed with water. The organic layer was dried over sodium sulphate and concentrated under reduced pressure to afford the crude title compound as yellow solid (1.25 g, 55% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 7.91 (dd, J=9.2, 4.3 Hz, 1H), 7.81 (dt, j=8.2, 2.8 Hz, 1H), 7.74 (dd, J=8.3, 3.1 Hz, 1H), 7.57 (m, 1H), 7.32 (dt, J=8.5, 2.4 Hz, 1H), 7.19 (m, 2H), 5.00 (q, J=6.8 Hz, 1H), 1.97 (d, J=6.8 Hz, 3H).
Intermediate 2: 6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

To a solution of Intermediate 1 (15.0 g, 40.84 mmol) in DMSO (150 ml), n-butanol (7.5 ml) was added and heated to 120°C for 3 hours. The reaction mixture was cooled to RT, quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (7.90 g, 64%). 1H-NMR (δ ppm, CDC13, 400 MHz): 7.85 (dd, J = 8.1, 3 Hz, 1H), 7.54 (dd, J = 9.2, 4.2 Hz, 1H), 7.47-7.37 (m, 2H), 7.15-6.98 (m, 3H), 4.74 (quintet, J= 6.8 Hz, 1H), 2.23 (d, J = 7.4 Hz, 1H), 1.54 (d, J = 6.6 Hz, 3H).
Intermediate 3 : 2-acetyl-6-fluoro-3-( 3-fluorophenyl)-4H-chromen-4-one

DMSO (5.60 ml, 79.14 mmol) was added to dichloromethane (40 ml), and cooled to – 78°C, followed by oxalyl chloride (3.40 ml, 39.57 mmol). After 10 min., intermediate 2 (6.00 g, 19.78 mmol) in dichloromethane (54 ml) was added dropwise and stirred for 20 min.
Triethylamine (12 ml) was added and stirred for 1 hour. The reaction mixture was quenched with water and extracted with dichloromethane. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (4.2 g, 71%) which was used as such in the next step.
Intermediate 4: fS)-6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

To intermediate 3 (2.00 g, 6.66 mmol), R-Alpine borane (0.5 M in THF, 20 ml) was added and heated to 60°C for 20 hours. The reaction mixture quenched with 2N HC1, and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.51 g, 75%).
Enantiomeric excess: 94.2%, enriched in the fast eluting isomer (retention time: 8.78 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 5: fR)-l-f6-fluoro-3-f3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl 4- chlorobenzoate

To a solution of intermediate 4 (1.45 g, 4.78 mmol) in THF (15 ml), 4-chlorobenzoic acid (0.748 g, 4.78 mmol) and triphenylphosphine (1.88 g, 7.17 mmol) were added and heated to 45°C followed by diisopropylazodicarboxylate (1.4 ml, 7.17 mmol). After 1 hour, the reaction mixture was concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.81 g, 86%) which was used without purification in the next step. Intermediate 6: fR)-6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

Method A
Intermediate 5 (1.75 g, 3.96 mmol) in methanol (17 ml) was cooled to 10°C, potassium carbonate (0.273 g, 1.98 mmol) was added and stirred for 30 min. The reaction mixture was concentrated, acidified with 2N HCl solution, extracted with ethyl acetate, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (1.05 g, 87% yield). Enantiomeric excess: 93.6%>, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Method B
Step-1 [(R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one]: To l-(5-fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone (11.00 g, 44.31 mmol) in dichloromethane, HATU (33.7 g, 88.63 mmol) and R-(+)2-benzyloxypropionic acid (9.58 g, 53.17 mmol) were added and stirred for 10 min. Triethylamine (66.7 ml, 0.47 mol) was added dropwise and stirred at RT for 24 hours. The reaction mixture was quenched with water, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate:
petroleum ether to afford the title compound as a yellow solid (10.5 g, 60%> yield). 1H-NMR (δ ppm, CDCls, 400 MHz): 7.85 (dd, J = 8.1,3 Hz, 1H), 7.58 (dd, J = 9.1, 4.1 Hz, 1H), 7.47-7.39 (m, 1H), 7.39-7.34 (m, 1H), 7.28-7.20 (m, 3H), 7.20-7.14 (m, 2H), 7.16-7.07 (m, 1H), 6.99-6.89 (m, 2H), 4.50-4.31 (m, 3H), 1.56 (d, J = 6.4 Hz, 3H).
Step-2: (R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one obtained in Step-1 (10.5 g, 26.69 mmol) in dichloromethane (110 ml) was cooled to 0°C, aluminium chloride (5.35 g, 40.03 mmol) was added portionwise and stirred at RT for 6 hours. The reaction mixture was quenched with 2N HCl solution, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford intermediate 6 a yellow solid (6.1 g, 76% yield). Enantiomeric excess: 97.7%, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 7: 4-bromo-2-fluoro-l-isopropoxybenzene

To a solution of 4-bromo-3-fluorophenol (10 g, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8 ml, 62.62 mmol) and triphenylphosphine (20.6 g, 78.52 mmol) were added and heated to 45°C followed by diisopropylazodicarboxylate (15.4 ml, 78.52 mmol). The mixture was refluxed for 1 hour, concentrated and the residue was purified by column
chromatography with ethyl acetate: petroleum ether to afford the title compound as a colorless liquid (13.1 g, 99% yield), which was used without purification in the next step.
Intermediate 8: 2-f3-fluoro-4-isopropoxyphenyl)-4,4,5.,5-tetramethyl-l,3i2-dioxaborolane

Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15 g, 58.96 mmol) were added to a solution of intermediate 7 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [l, -Bis(diphenylphosphino)ferrocene]dichloro palladium(II) CH2CI2 (4.4 g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12 hours, the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 9: 3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-dlpyrimidin-4-amine

To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF (110 ml), ethanol (55 ml) and water (55 ml), intermediate 8 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min.
Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12 hours, the reaction mixture was filtered through celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
(RS)- 2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
To a solution of intermediate 9 (0.080 g, 0.293 mmol) in DMF (2 ml), potassium carbonate (0.081 g, 0.587 mmol) was added and stirred at RT for 10 min. To this mixture intermediate 1 (0.215 g, 0.587 mmol) was added and stirred for 12 hours. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as a pale yellow solid (0.045 g). MP: 175-177°C. 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 8.20 (s, 1H), 7.85 (dd, J = 81, 3.0 Hz, 1H), 7.48-7.33 (m, 5H), 7.14 (t, J= 8.3 Hz, 1H), 7.02 (m, 2H), 6.90 (m, 1H), 6.10 (q, J = 7.1 Hz, 1H), 5.42 (s, 2H), 4.64 (quintet, J = 6.0 Hz, 1H), 1.99 (d, J = 7.1 Hz, 3H), 1.42 (d, J= 6.1 Hz, 6H).
fS)-2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (“S-isomer”)
To a solution of intermediate 9 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 6 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.15 ml, 0.749 mmol) was added heated to 45°C. After 2 hours, the reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 % yield). MP: 139-142°C. Mass: 571.7 (M+). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64 min.). fR)-2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-ehromen-4-one
To a solution of intermediate 8 (0.284 g, 0.989 mmol) in THF (5.0 ml), intermediate 4 (0.250 g, 0.824 mmol) and tris(4-methoxy)phenylphosphine (0.435 g, 1.23 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.25 ml, 1.23 mmol) was added stirred at RT. After 12 hours, the reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate :
petroleum ether to afford the title compound as an off-white solid (0.105 g, 22 % yield). MP: 145-148°C. Mass: 571.7 (M+). Enantiomeric excess: 95.4% as determined by HPLC on a chiralpak AD-H column, enriched in the late eluting isomer (retention time = 14.83 min.).
PATENT
WO 2014006572
http://www.google.com/patents/WO2014006572A1?cl=en
 B1 IS DESIRED
B1 IS DESIRED
(S)-2- (l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-6- fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (compound-B l)
Intermediate 11
[119] Intermediate 11: 4-bromo-2-fluoro-l-isopropoxybenzene:To a solution of 4-bromo-2- fluorophenol (lOg, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8ml, 62.62 mmol) and triphenylphosphine (20.6g, 78.52 mmol) were added and heated to 45 C followed by diisopropylazodicarboxylate (15.4ml, 78 52 mmol). The mixture was refluxed for lh, concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a colourless liquid (13. lg, 99%) which was used without purification in the next step. Intermediate 12
[120] Intermediate 12: 2-(3-fluoro-4-isopropoxyphenyl)-4,4,5,5-tetramethyl- 1,3,2- dioxaborolane: Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15g, 58.96 mmol) were added to a solution of intermediate 11 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [1,1 ‘- Bis(diphenylphosphino)ferrocene]dichloro palladium(II).CH2Cl2 (4.4g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 13
[121] Intermediate 13: 3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- amine: To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF 110 ml), ethanol (55 ml) and water (55 ml), intermediate 12 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min. Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h, the reaction mixture was filtered though celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
Example Bl
(S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
[127] To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate ( 0.15 ml, 0.749 mmol) was added heated to 45°C. After 2h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 %). MP: 139- 142°C. Mass : 571.7 (M H-NMR (δ ppm, CDC13, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J = 8.2,3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J = 8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J = 8.4 Hz, 1H), 6.11 (q, J = 7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J = 6.1 Hz, 1H), 2.00 (d, J = 7.1Hz, 3H), 1.42 (d, J = 6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64min.).

NEW DRUG APPROVALS
one time
$10.00
PATENT
US 2014/0011819 describe the synthesis of TGR-1202 (Example B l)
http://www.google.co.in/patents/US20140011819
Example B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
- To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.15 ml, 0.749 mmol) was added heated to 45° C. After 2 h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate:petroleum ether to afford the title compound as an off-white solid (0.049 g, 20%). MP: 139-142° C. Mass: 571.7 (M+).1H-NMR (δ ppm, CDCl3, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J=8.2, 3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J=8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J=8.4 Hz, 1H), 6.11 (q, J=7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J=6.1 Hz, 1H), 2.00 (d, J=7.1 Hz, 3H), 1.42 (d, J=6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=10.64 min)
4-Methylbenzenesulfonate Salt of Compound B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4-methylbenzenesulfonate
- (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4-methylbenzenesulfonate: To (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (22.7 g, 39.69 mmol) in isopropanol (600 ml), p-toluenesulphonic acid (8.30 g, 43.66 mmol) was added and refluxed for 1 h. The reaction mixture was concentrated, co-distilled with petroleum ether and dried. To the residue water (300 ml) was added and stirred for 30 min. The solid was filtered, washed with petroleum ether and dried under vacuum to afford the title compound as off-white solid (28.2 g, 95%). MP: 138-141° C. 1H-NMR (δ ppm, CDCl3, 400 MHz): 8.11 (s, 1H), 7.85 (dd, J=8.0, 3.0 Hz, 1H), 7.80 (d, J=8.2 Hz, 2H), 7.51 (dd, J=9.3, 4.3 Hz, 1H), 7.45 (dd, J=7.5, 3.1 Hz, 1H), 7.42-7.31 (m, 3H), 7.29 (m, 2H), 7.22 (d, J=8.0 Hz, 2H), 7.16 (t, J=8.3 Hz, 1H), 7.08 (dt, J=8.5, 2.5 Hz, 1H), 6.97 (br s, 1H), 6.88 (br s, 1H), 6.11 (q, J=7.2 Hz, 1H), 4.67 (quintet, J=6.0 Hz, 1H), 2.36 (s, 3H), 2.03 (d, J=7.1 Hz, 3H), 1.43 (d, J=6.0 Hz, 6H). Mass: 572.4 (M++1-PTSA). Enantiomeric excess: 93.4% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=12.35 min.)
Sulphate Salt of Compound B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one sulfate
- (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one sulphate: To (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (15.0 g, 26.24 mmol) in isopropanol (600 ml) was cooled to 0° C. To this Sulphuric acid (2.83 g, 28.86 mmol) was added and stirred at room temperature for 24 h. The reaction mass was filtered and washed with petroleum ether and dried under vacuum. To the solid, water (150 ml) was added and stirred for 30 min. The solid was filtered, washed with petroleum ether and dried under vacuum to afford the title compound as off-white solid (13.5 g, 76%). MP: 125-127° C. 1H-NMR (δ ppm, CDCl3, 400 MHz): 8.11 (s, 1H), 7.85 (dd, J=8.0, 3.0 Hz, 1H), 7.51 (dd, J=9.2, 4.2 Hz, 1H), 7.45-7.31 (m, 3H), 7.29 (m, 1H), 7.15 (t, J=8.3 Hz, 1H), 7.08 (dt, J=8.5, 2.4 Hz, 1H), 6.96 (br s, 1H), 6.88 (br s, 1H), 6.09 (q, J=7.1 Hz, 1H), 4.676 (quintet, J=6.1 Hz, 1H), 2.01 (d, J=7.1 Hz, 3H), 1.42 (d, J=6.1 Hz, 6H). Mass: 572.2 (M++1-H2SO4). Enantiomeric excess: 89.6% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=12.08 min.)
- Various other acid addition salts of compound B1 were prepared as provided in Table 1.
- TABLE 1 Melting PointAcidMethod of preparation(° C.) Hydro-Compound B1 (1 eq.) dissolved in THF,130-132chloricexcess HCl/Et2O was added, the clearacidsolution obtained was evaporated completely. The residue obtained was washed with water.p-Compound B1 (1 eq.) dissolved in138-141° C.Toluene-isopropyl alcohol (IPA), refluxed forsulfonic30 min., acid (1.1 eq.) in IPA was added,acidthe clear solution obtained was evaporated completely. The residue obtained was washed with water.Benzene-Compound B1 (1 eq.) dissolved in IPA,170-172sulphonicrefluxed for 30 min., acid(1.1 eq.) in IPAacidwas added, the clear solution not obtained, the residue was evaporated completely and was washed with water.MaleicCompound B1 (1 eq.) dissolved in IPA,107-109acidrefluxed for 30 min., acid (1.1 eq.) in IPA was added, the clear solution not obtained, the residue was evaporated completely and was washed with water.CamphorCompound B1 (1 eq.) dissolved in IPA,120-121sulfonicrefluxed for 30 min., acid (1.1 eq.) in IPAacidwas added, the clear solution not obtained, the residue was evaporated completely and was washed with water.SulphuricCompound B1 (1 eq.) dissolved in IPA,125-127acidrefluxed for 30 min., acid(1.1 eq.) in IPA was added, the clear solution obtained was evaporated completely. The residue obtained was washed with water.
REFERENCES
WO 2014/006572 and U.S. Patent Publication No. 2014/0011819,
http://www.tgtherapeutics.com/O’ConnorTGR202Single%20AgentEHA&Lugano2015.pdf
- TGR-1202: Phase I/II started 09/28/2015Week in Review, Clinical Status Rhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) …
- Ublituximab: Phase I/II started 09/28/2015Week in Review, Clinical Status LFB S.A., Les Ulis, France TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: Ublituximab (TGTX-1101, TG-1101, LFB-R603) Business: Cancer Molecular target: CD20 Description: Glycoengineered mAb against CD20 …
- COMPANY NEWS: TG rises on SPA for combination CLL therapy 09/17/2015The Daily Extra, Company News TG Therapeutics Inc. (NASDAQ:TGTX) rose $2.65 (23%) to $14.37 after the company said it received an SPA from FDA for the Phase III UNITY-CLL trial of ublituximab (TG-1101) in combination with TGR-1202 to treat chronic …
- Targets & Mechanisms: The battle for IRAK 04/23/2015
Nimbus, Aurigene and TG Therapeutics are chasing IRAK4 inhibitors for cancerBC Innovations, Targets & Mechanisms Now that Nimbus has put IRAK4 on the map for B cell lymphoma, several companies are closing in with their own inhibitors, and they’re all on track for IND-enabling studies this year. - TGR-1202: Additional Phase I/II data 01/26/2015Week in Review, Clinical Results Rhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) …
- Ublituximab: Additional Phase I/II data 01/26/2015Week in Review, Clinical Results LFB S.A., Les Ulis, France TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Ildong Pharmaceutical Co. Ltd. (KSE:000230), Seoul, South Korea Product: Ublituximab (TGTX-1101, TG-1101, LFB-R603) Business: Cancer …
- TGR-1202: Phase I started 12/15/2014Week in Review, Clinical Status Rhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) …
- Rhizen, TG Therapeutics deal 12/08/2014Week in Review, Deals Rhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Business: Cancer TG Therapeutics exercised an option under a 2012 deal to license exclusive, worldwide …
| Patent | Submitted | Granted |
|---|---|---|
| NOVEL SELECTIVE PI3K DELTA INHIBITORS [US2014011819] | 2013-07-02 | 2014-01-09 |
| Treatment Of Cancers Using PI3 Kinase Isoform Modulators [US2014377258] | 2014-05-30 | 2014-12-25 |
////////Umbralisib
CC(C)OC1=C(C=C(C=C1)C2=NN(C3=C2C(=NC=N3)N)C(C)C4=C(C(=O)C5=C(O4)C=CC(=C5)F)C6=CC(=CC=C6)F)F
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021164789&_cid=P12-KSZQ3G-94695-1Phosphatidylinositol 3-kinase (phosphatidylinositol-3-kinase, PI3K) is composed of the regulatory subunit p85 or p101, and the catalytic subunit p110 (subdivided into four subtypes: p110a, p110b, p110g, and p110d) Lipid kinase catalyzes the phosphorylation of the inositol ring 3′-OH of phosphatidylinositol 4,5-bisphosphate (phosphatidylinositol 4,5-bisphosphate, PIP2) to phosphatidylinositol 3,4,5-triphosphate (phosphatidylinositol 4,5-bisphosphate, PIP2). 3,4,5-trisphosphate, PIP3) and activate downstream Akt, which plays a key role in cell proliferation, survival and metabolism. In tumor cells, PI3K is overexpressed, which leads to rapid proliferation and growth of tumor cells.
The tumor suppressor gene PTEN (phosphatase, tension homolog deleted on chromosome ten) dephosphorylates PIP3 to generate PIP2, which leads to negative feedback regulation of the PI3K signaling pathway, inhibits cell proliferation and promotes cell apoptosis. PI3K gene mutation and amplification frequently occur in cancer, and PTEN gene deletion in cancer, etc., suggest that PI3K overexpression is closely related to tumorigenesis.
TGR-1202 is a second-generation PI3Kδ inhibitor developed by TG Therapeutic. Compared with the first-generation PI3Kδ inhibitor, it can significantly reduce the toxicity of liver and gastrointestinal tract in clinical trials, and patients with large B-cell lymphoma are also exposed to TGR. -1202 There is a partial response. Patent WO2014006572 discloses the structure of TGR-1202. ACP-196 is a second-generation BTK inhibitor that has been approved for marketing by the FDA. It has been reported in the literature (PLoS ONE 12(2):e0171221.). The combination of a PI3Kδ inhibitor and a BTK inhibitor can inhibit BCR signaling in two ways. Access, thereby playing a synergistic effect.Example 1: Preparation of the compound of formula (I)
Step 1: Synthesis of compound BB-1-3
To a solution of BB-1-1 (23g, 205.17mmol, 1eq) in polyphosphoric acid (23g, 17.84mmol) was added BB-1-2 (43.90g, 266.71mmol, 1.3eq). The reaction solution was stirred at 125°C for 5 hours under the protection of nitrogen. After the completion of the reaction, water (300 mL) was added to the reaction solution to quench the reaction, and a solid precipitated out, which was directly filtered to obtain a filter cake. The filter cake was washed with water (100 mL) once, and then purified by column chromatography (PE:EA=1:1) to obtain the target compound BB-1-3. 1 H NMR (400MHz, CDCl 3 ) δ 8.94 (br s, 1H), 7.68 (br d, J=5.3 Hz, 2H), 6.65 (s, 1H), 4.51 (s, 2H).
Step 2: Synthesis of compound BB-1-4
To a solution of BB-1-3 (21.02g, 98.87mmol, 1eq) in glacial acetic acid (210mL) was added NBS (19.36g, 108.75mmol, 1.1eq). The reaction solution was stirred at 25°C for 1 hour under the protection of nitrogen. After the completion of the reaction, water (200 mL) was added to the reaction solution to quench the reaction, and a solid was formed, which was filtered to obtain a filter cake. After washing three times with water (30mL*3), the filter cake was dissolved in dichloromethane (100mL), dried over anhydrous sodium sulfate, concentrated, and then beaten with methyl tert-butyl ether (50mL) once. The filter cake was collected by filtration to obtain the target Compound BB-1-4 batch one. The aqueous phase was extracted with dichloromethane (100mL*3) and combined with the mother liquor obtained by washing with methyl tert-butyl ether, and then subjected to column chromatography (petroleum ether: ethyl acetate = 1:1, target product Rf = 0.43) ) Purification to obtain the target compound BB-1-4 batch two. The two batches were dissolved and combined with dichloromethane and spin-dried to obtain the target compound BB-1-4. 1 H NMR (400 MHz, CDCl 3 ) δ 8.93 (dd, J = 1.3, 3.1 Hz, 1H), 7.80-7.69 (m, 2H), 4.74 (s, 2H).
Step 3: Synthesis of compound BB-1-5
To a solution of BB-1-4 (3g, 10.29mmol, 1eq) in N,N-dimethylformamide (30mL), potassium acetate (1.52g, 15.44mmol, 1.5eq) was added. The reaction solution was stirred at 40°C for 3.5 hours under the protection of nitrogen. After the reaction was completed, water (60 mL) was added to the reaction solution to quench the reaction, and a large amount of solid was formed, which was filtered to obtain a filter cake. The filter cake was dissolved in dichloromethane (100 mL), dried over anhydrous sodium sulfate, and concentrated to obtain the target compound BB-1-5 batch one. The aqueous phase was extracted with methyl tert-butyl ether (100 mL*3) to obtain the organic phase, dried over anhydrous sodium sulfate, and concentrated to obtain BB-1-5 batch two, which was obtained by combining the two batches. Used directly in the next reaction. 1 H NMR (400MHz, CDCl 3 ) δ 9.07-8.88 (m, 1H), 7.71 (dd, J=1.7, 5.8 Hz, 2H), 5.31-5.26 (m, 2H), 2.22 (s, 3H).
Step 4: Synthesis of compound BB-1-6
To the dioxane (37 mL) solution of BB-1-5 (3.77 g, 11.96 mmol, 1 eq), hydrochloric acid (12M, 3.49 mL, 3.5 eq) was added. The reaction solution was stirred at 40°C for 3.5 hours under the protection of nitrogen. After the completion of the reaction, the reaction solution was concentrated, water (2 mL) was added, the pH was adjusted to 9 with ammonia water, and the filter cake was collected by filtration. After dissolving with dichloromethane (100 mL), drying with anhydrous sodium sulfate, and concentration, the target compound BB-1-6 was obtained. Used directly in the next reaction. 1 H NMR(400MHz,DMSO-d 6 )δ8.95(dd,J=2.9,4.6Hz,1H), 8.15(dd,J=2.6,7.0,9.6Hz,1H), 7.86(dd,J=5.3 , 9.6 Hz, 1H), 5.35 (t, J = 5.9 Hz, 1H), 4.58 (d, J = 6.1 Hz, 2H).
Step 5: Synthesis of compound BB-1-7
To BB-1-6 (2.6g, 9.52mmol, 1eq) and 3-fluorophenylboronic acid (2.66g, 19.04mmol, 2eq) in acetonitrile/water (12.5mL, volume ratio: 3/1), add carbonic acid Sodium ( 5.05g, 47.61mmol, 5eq ) and Pd(PPh 3 ) 4 (550.15mg, 476.09μmol, 0.05eq). The reaction solution was stirred at 85°C for 4 hours under the protection of nitrogen. After the reaction was completed, dichloromethane (50 mL) was added to the reaction solution, and then water (5 mL) was slowly added to quench the reaction, and then extracted with dichloromethane (50 mL*3). The organic phases were combined, dried over anhydrous sodium sulfate, concentrated, and purified by column chromatography (petroleum ether: ethyl acetate=0:1) to obtain BB-1-7. 1 H NMR(400MHz,DMSO-d 6 )δ8.94(dd,J=3.1,4.8Hz,1H), 8.12(ddd,J=2.6,7.1,10.0Hz,1H), 7.84(dd,J=5.3 , 10.1 Hz, 1H), 7.62 (s, 1H), 7.27-7.15 (m, 3H), 5.25 (t, J = 5.9 Hz, 1H), 4.28 (d, J = 5.7 Hz, 2H).
Step 6: Synthesis of compound BB-1-8
In a three-neck flask, at -78°C, to a solution of oxalyl chloride (1.85g, 14.57mmol, 1.28mL, 3eq) in dichloromethane (20mL) was added DMSO (2.28g, 29.14mmol, 2.28mL, 6eq). The reaction solution was stirred at -78°C for 1 hour under the protection of nitrogen. A solution of BB-1-7 (1.4g, 4.86mmol, 1eq) in dichloromethane (20mL) was added, and the mixture was stirred at -78°C for 1 hour. Triethylamine (4.91g, 48.57mmol, 6.76mL, 10eq) was added, and after stirring at -78°C for 1 hour, the reaction solution was raised to 25°C and stirred for 1 hour. After the reaction was completed, dichloromethane (20 mL) was added to the reaction solution at 0° C., water (10 mL) was added slowly to quench the reaction, and it was extracted with dichloromethane (30 mL*3). The organic phases were combined, dried over anhydrous sodium sulfate, and concentrated to obtain the target compound BB-1-8. LCMS, m/z=287.0 [M+1].
Step 7: Synthesis of compound BB-1
In a three-neck flask, at 0°C, to a tetrahydrofuran (50 mL) solution of BB-1-8 (1.9 g, 6.64 mmol, 1 eq) was added methyl magnesium bromide (3M, 5.53 mL, 2.5 eq). The reaction solution was stirred at 25°C for 5 hours under the protection of nitrogen. After the reaction was completed, at 0°C, water (10 mL) was slowly added to the reaction solution to quench the reaction, and then extracted with dichloromethane (10 mL*3). The organic phases were combined, dried with anhydrous sodium sulfate, concentrated, and subjected to preparative high performance liquid chromatography (column: Phenomenex Luna C18 200*40mm*10μm; mobile phase: [water (0.1%TFA)-acetonitrile]; B%: 15%- 35%, 10min) Purification (B is acetonitrile) to obtain BB-1.
Step 8: Synthesis of compound WX001-2
To the N,N-dimethylformamide (20mL) solution of WX001-1 (2g, 10.47mmol, 1eq) and potassium carbonate (4.34g, 31.41mmol, 3eq), add 2-iodopropane (3.56g, 20.94 mmol, 2.09mL, 2eq). The reaction solution was stirred at 90°C for 12 hours under the protection of nitrogen. After the reaction was completed, water (20 mL) was added to the reaction solution to quench the reaction, and then extracted with methyl tert-butyl ether (10 mL*3). The organic phases were combined, dried over anhydrous sodium sulfate, concentrated, and purified by column chromatography (petroleum ether: ethyl acetate = 5:1) to obtain the target compound WX001-2. 1 H NMR(400MHz, CDCl 3 )δ7.23(dd,J=2.4,10.6Hz,1H), 7.16(td,J=1.9,8.8Hz,1H), 6.85(t,J=8.7Hz,1H) , 4.49 (spt, J = 6.1 Hz, 1H), 1.35 (d, J = 6.1 Hz, 6H).
Step 9: Synthesis of compound WX001-3
To the dioxane (20mL) solution of WX001-2 (2g, 8.58mmol, 1eq), add double pinacol borate (2.40g, 9.44mmol, 1.1eq), potassium acetate (1.68g, 17.16 mmol, 2eq) and Pd(dppf)Cl 2 (627.87mg, 858.09μmol, 0.1eq). The reaction solution was stirred at 90°C for 3 hours under the protection of nitrogen. After the reaction was completed, water (20 mL) was added to the reaction solution to quench the reaction, and then extracted with dichloromethane (30 mL*3). The organic phases were combined, dried over anhydrous sodium sulfate, concentrated, and the crude product was purified by column chromatography (petroleum ether: ethyl acetate = 5:1) to obtain the target compound WX001-3. 1 H NMR (400MHz, CDCl 3 ) δ7.54-7.44 (m, 2H), 6.95 (t, J = 8.1 Hz, 1H), 4.60 (spt, J = 6.1 Hz, 1H), 1.37 (s, 3H) ,1.36(s,3H),1.33(s,12H).
Step 10: Synthesis of compound WX001-5
To WX001-3 (2.65g, 9.46mmol, 1eq) and WX001-4 (2.47g, 9.46mmol, 1eq) N,N-dimethylformamide/ethanol/water (265mL, volume ratio: 2/1/ 1) In the solution, add Pd(PPh 3 ) 4 (546.55 mg, 472.97 μmol, 0.05 eq) and sodium carbonate (3.01 g, 28.38 mmol, 3 eq). The reaction solution was stirred at 80°C for 12 hours under the protection of nitrogen. After the completion of the reaction, the reaction solution was filtered while hot (80℃) to obtain the mother liquor. After the mother liquor was spin-dried, dichloromethane (30mL) and water (30mL) were added. A large amount of insoluble matter was formed. After filtration, it was subjected to preparative high performance liquid chromatography. Purified to obtain the target compound WX001-5. LCMS, m/z=288.1 [M+1].
Step 11: Synthesis of compound WX001-6
To BB-1 (230mg, 760.90μmol, 1eq), WX001-5 (218.60mg, 760.90μmol, 1eq) and PPh 3 (299.36mg, 1.14mmol, 1.5eq) in tetrahydrofuran (50mL) solution at 25℃ , Add diisopropyl azodicarboxylate (230.79 mg, 1.14 mmol, 221.91 μL, 1.5 eq). The reaction solution was stirred at 45°C for 5 hours under the protection of nitrogen. After the completion of the reaction, the reaction solution was directly concentrated, and purified by a thin-layer chromatography plate (dichloromethane:methanol=15:1) to obtain an isomer mixture WX001-6.
Step 12: Synthesis of the compound of formula (I)
WX001-6 was purified by supercritical fluid chromatography (column: DAICEL CHIRALPAK AD-H (250mm*30mm, 5μm); mobile phase: [0.1% ammonia in ethanol]; B%: 22%-22%, 8min) (B It is 0.1% ammonia in ethanol) to obtain the compound of formula (I) (retention time is 2.29 min), and the structure of the compound of formula (I) is confirmed by a single crystal to be correct. 1 H NMR (400MHz, CD 3 OD) δ 8.95 (br s, 1H), 8.03 (s, 1H), 8.00-7.92 (m, 1H), 7.91-7.82 (m, 1H), 7.42-7.29 (m ,2H),7.27-7.09(m,1H),7.22(br t,J=8.6Hz,1H),6.96-6.71(m,2H),6.20(q,J=6.6Hz,1H),4.68(td , J=6.1, 11.9 Hz, 1H), 1.91 (d, J=7.0 Hz, 3H), 1.36 (d, J=5.7 Hz, 6H); LCMS, m/z=572.2 [M+1].
////////////TGR 1202
TROFINETIDE

Trofinetide
- Molecular FormulaC13H21N3O6
- Average mass315.322 Da
Tofinetide , NNZ-256610076853400-76-7[RN]
glycyl-2-methyl-L-prolyl-L-glutamic acid
H-Gly-PMe-Glu-OHL-Glutamic acid, glycyl-2-methyl-L-prolyl-UNII-Z2ME8F52QLZ2ME8F52QLтрофинетид [Russian] [INN]تروفينيتيد [Arabic] [INN]曲非奈肽 [Chinese] [INN]
| IUPAC Condensed | H-Gly-aMePro-Glu-OH |
|---|---|
| Sequence | GXE |
| HELM | PEPTIDE1{G.[*C(=O)[C@@]1(CCCN1*)C |$_R2;;;;;;;;_R1;$|].E}$$$$ |
| IUPAC | glycyl-alpha-methyl-L-prolyl-L-glutamic acid |
An (1-3) IGF-1 analog with neuroprotective activity.
OPTICAL ROT; -52.4 ° Conc: 0.19 g/100mL; water ; 589.3 nm; Temp: 20 °C; Len: 1.0 dm…Tetrahedron 2005, V61(42), P10018-10035
EU Customs Code CN, 29339980
Harmonized Tariff Code, 293399
- L-Glutamic acid, glycyl-2-methyl-L-prolyl-
- glycyl-2-methyl-L-prolyl-L-glutamic acid
- Glycyl-L-2-methylprolyl-L-glutamic acid
FDA APPROVED 2023/3/10, Daybue
853400-76-7 CAS
トロフィネチド;
Trofinetide (NNZ-2566) is a drug developed by Neuren Pharmaceuticals that acts as an analogue of the neuropeptide (1-3) IGF-1, which is a simple tripeptide with sequence Gly–Pro–Glu formed by enzymatic cleavage of the growth factor IGF-1 within the brain. Trofinetide has anti-inflammatory properties and was originally developed as a potential treatment for stroke,[1][2] but has subsequently been developed for other applications and is now in Phase II clinical trials against Fragile X syndrome and Rett syndrome.[3][4][5]
Trofinetide (NNZ-2566), a neuroprotective analogue of glypromate, is a novel molecule that has a profile suitable for both intravenous infusion and chronic oral delivery. It is currently in development to treat traumatic brain injury.
In February 2021, Neuren is developing trofinetide (NNZ-2566, phase 2 clinical ), a small-molecule analog of the naturally occurring neuroprotectant and N-terminus IGF-1 tripeptide Glypromate (glycine-proline-glutamate), for intravenous infusion treatment of various neurological conditions, including moderate to severe traumatic brain injury (TBI), stroke, chronic neurodegenerative disorders and peripheral neuropathies. At the same time, Neuren is also investigating an oral formulation of trofinetide (phase 3 clinical) for similar neurological indications, including mild TBI.
Autism Spectrum Disorders and neurodevelopment disorders (NDDs) are becoming increasingly diagnosed. According to the fourth edition of the American Psychiatric Association’s (APA) Diagnostic and Statistical Manual oƒ Mental Disorders (DSM-4), Autism spectrum disorders (ASD) are a collection of linked developmental disorders, characterized by abnormalities in social interaction and communication, restricted interests and repetitive behaviours. Current classification of ASD according to the DSM-4 recognises five distinct forms: classical autism or Autistic Disorder, Asperger syndrome, Rett syndrome, childhood disintegrative disorder and pervasive developmental disorder not otherwise specified (PDD-NOS). A sixth syndrome, pathological demand avoidance (PDA), is a further specific pervasive developmental disorder.
More recently, the fifth edition of the American Psychiatric Association’s (APA) Diagnostic and Statistical Manual oƒ Mental Disorders (DSM-5) recognizes recognises Asperger syndrome, childhood disintegrative disorder, and pervasive developmental disorder not otherwise specified (PDD-NOS) as ASDs.
This invention applies to treatment of disorders, regardless of their classification as either DSM-4 or DSM-5.
Neurodevelopment Disorders (NDDs) include Fragile X Syndrome (FXS), Angelman Syndrome, Tuberous Sclerosis Complex, Phelan McDermid Syndrome, Rett Syndrome, CDKL5 mutations (which also are associated with Rett Syndrome and X-Linked Infantile Spasm Disorder) and others. Many but not all NDDs are caused by genetic mutations and, as such, are sometimes referred to as monogenic disorders. Some patients with NDDs exhibit behaviors and symptoms of autism.
As an example of a NDD, Fragile X Syndrome is an X-linked genetic disorder in which affected individuals are intellectually handicapped to varying degrees and display a variety of associated psychiatric symptoms. Clinically, Fragile X Syndrome is characterized by intellectual handicap, hyperactivity and attentional problems, autism spectrum symptoms, emotional lability and epilepsy (Hagerman, 1997a). The epilepsy seen in Fragile X Syndrome is most commonly present in childhood, but then gradually remits towards adulthood. Hyperactivity is present in approximately 80 percent of affected males (Hagerman, 1997b). Physical features such as prominent ears and jaw and hyper-extensibility of joints are frequently present but are not diagnostic. Intellectual handicap is the most common feature defining the phenotype. Generally, males are more severely affected than females. Early impressions that females are unaffected have been replaced by an understanding of the presence of specific learning difficulties and other neuropsychiatric features in females. The learning disability present in males becomes more defined with age, although this longitudinal effect is more likely a reflection of a flattening of developmental trajectories rather than an explicit neurodegenerative process.
The compromise of brain function seen in Fragile X Syndrome is paralleled by changes in brain structure in humans. MRI scanning studies reveal that Fragile X Syndrome is associated with larger brain volumes than would be expected in matched controls and that this change correlates with trinucleotide expansion in the FMRP promoter region (Jakala et al, 1997). At the microscopic level, humans with Fragile X Syndrome show abnormalities of neuronal dendritic structure, in particular, an abnormally high number of immature dendritic spines (Irwin et al, , 2000).
Currently available treatments for NDDs are symptomatic – focusing on the management of symptoms – and supportive, requiring a multidisciplinary approach. Educational and social skills training and therapies are implemented early to address core issues of learning delay and social impairments. Special academic, social, vocational, and support services are often required. Medication, psychotherapy or behavioral therapy may be used for management of co-occurring anxiety, ADHD, depression, maladaptive behaviors (such as aggression) and sleep issues, Antiepileptic drugs may be used to control seizures.
Patent
WO 2014085480,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014085480

EP 0 366 638 discloses GPE (a tri-peptide consisting of the amino acids Gly-Pro-Glu) and its di-peptide derivatives Gly-Pro and Pro-Glu. EP 0 366 638 discloses that GPE is effective as a neuromodulator and is able to affect the electrical properties of neurons.
WO95/172904 discloses that GPE has neuroprotective properties and that administration of GPE can reduce damage to the central nervous system (CNS) by the prevention or inhibition of neuronal and glial cell death.
WO 98/14202 discloses that administration of GPE can increase the effective amount of choline acetyltransferase (ChAT), glutamic acid decarboxylase (GAD), and nitric oxide synthase (NOS) in the central nervous system (CNS).
WO99/65509 discloses that increasing the effective amount of GPE in the CNS, such as by administration of GPE, can increase the effective amount of tyrosine hydroxylase (TH) in the CNS to increase TH-mediated dopamine production in the treatment of diseases such as Parkinson’s disease.
WO02/16408 discloses certain GPE analogs having amino acid substitutions and certain other modification that are capable of inducing a physiological effect equivalent to GPE within a patient. The applications of the GPE analogs include the treatment of acute brain injury and neurodegenerative diseases, including injury or disease in the CNS.
EXAMPLES
The following examples are intended to illustrate embodiments of this invention, and are not intended to limit the scope to these specific examples. Persons of ordinary skill in the art can apply the disclosures and teachings presented herein to develop other embodiments without undue experimentation and with a likelihood of success. All such embodiments are considered part of this invention.
Example 1: Synthesis of N,N-Dimethylglycyl-L-prolyl)-L-glutamic acid
The following non-limiting example illustrates the synthesis of a compound of the invention, N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
All starting materials and other reagents were purchased from Aldrich; BOC=tert-butoxycarbonyl; Bn=benzyl.
BOC-L-proline-(P-benzyl)-L-glutamic acid benzyl ester
To a solution of BOC-proline [Anderson GW and McGregor AC: J. Amer. Chem. Soc: 79, 6810, 1994] (10 mmol) in dichloromethane (50 mi), cooled to 0°C, was added triethylamine (1 .39 ml, 10 mmol) and ethyl chloroformate (0.96 ml, 10 mmol). The resultant mixture was stirred at 0 °C for 30 minutes. A solution of dibenzyl-L-glutamate (10 mmol) was then added and the mixture stirred at 0° C for 2 hours then warmed to room temperature and stirred overnight. The reaction mixture was washed with aqueous sodium bicarbonate and citric acid (2 mol 1-1) then dried (MgSO4) and concentrated at reduced pressure to give BOC-L-proline-L-glutamic acid dibenzyl ester (5.0 g, 95%).
L-proline-L-glutamic acid dibenzyl ester
A solution of BOC-L-glutamyl-L-proline dibenzyl ester (3.4 g, 10 mmol), cooled to 0 °C, was treated with trifluoroacetic acid (25 ml) for 2 h. at room temperature. After removal of the volatiles at reduced pressure the residue was triturated with ether to give L-proline-L-glutamic acid dibenzyl ester.
N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
A solution of dicyclohexylcarbodiimide (10.3 mmol) in dichloromethane (10 ml) was added to a stirred and cooled (0 °C) solution of L-proline-L-glutamic acid dibenzyl ester (10 mmol), N,N-dimethylglycine (10 mmol) and triethylamine ( 10.3 mmol) in dichloromethane (30 ml). The mixture was stirred at 0°C overnight and then at room temperature for 3 h. After filtration, the filtrate was evaporated at reduced pressure. The resulting crude dibenzyl ester was dissolved in a mixture of ethyl acetate (30 ml) and methanol (30 ml) containing 10% palladium on charcoal (0.5 g) then hydrogenated at room temperature and pressure until the uptake of hydrogen ceased. The filtered solution was evaporated and the residue recrystallised from ethyl acetate to yield the tripeptide derivative.
It can be appreciated that following the method of the Examples, and using alternative amino acids or their amides or esters, will yield other compounds of Formula 1.
Eample 2: Synthesis of Glycyl-L-2-Methyl-L-Prolyl-L-Glutamate
L-2-Methylproline and L-glutamic acid dibenzyl ester p-toluenesulphonate were purchased from Bachem, N-benzyloxycarbonyl-glycine from Acros Organics and bis(2-oxo-3-oxazolidinyl)phosphinic chloride (BoPCl, 97%) from Aldrich Chem. Co.
Methyl L-2-methylprolinate hydrochloride 2
Thionyl chloride (5.84 cm3, 80.1 mmol) was cautiously added dropwise to a stirred solution of (L)-2-methylproline 1 (0.43 g, 3.33 mmol) in anhydrous methanol (30 cm3) at -5 °C under an atmosphere of nitrogen. The reaction mixture was heated under reflux for 24 h, and the resultant pale yellow-coloured solution was. concentrated to dryness in vacuo. The residue was dissolved in a 1 : 1 mixture of methanol and toluene (30 cm3) then concentrated to dryness to remove residual thionyl chloride. This procedure was repeated twice more, yielding hydrochloride 2 (0.62 g, 104%) as an hygroscopic, spectroscopically pure, off-white solid: mp 127- 131 °C; [α]D -59.8 (c 0.24 in CH2Cl2); vmax (film)/cm-1 3579, 3398 br, 2885, 2717, 2681 , 2623, 2507, 1743, 1584, 1447, 1432, 1374, 1317, 1294, 1237, 1212, 1172, 1123, 981 , 894, 861 and 764; δH (300 MHz; CDCl3; Me4Si) 1.88 (3H, s, Proα-CH3), 1 .70-2.30 (3H, br m, Proβ-HAΗΒ and Proγ-H2), 2.30-2.60 (1H, br m, Proβ-HAΗΒ), 3.40-3.84 (2H, br m, Proδ-H2), 3.87 (3H, s, CO2CH3), 9.43 (1H, br s, NH) and 10.49 ( 1H, br s, HCl); δC (75 MHz; CDCl3) 21.1 (CH3, Proα-CH3), 22.4 (CH2, Proγ-C), 35.6 (CH2, Proβ-C), 45.2 (CH2, Proδ-C), 53.7 (CH3, CO2CH3), 68.4 (quat., Proα-C) and 170.7 (quat, CO); m/z (FAB+) 323.1745 [M2.H35Cl.H+: (C7H13NO2)2. H35Cl.H requires 323.1738] and 325.1718 [M2.H37Cl.H+: (C7H13NOz)2. H37Cl.H requires 325.1708],
N-Benxyloxycarbonyl-glycyl-L-2-methylproline 5
Anhydrous triethylamine (0.45 cm3, 3.23 mmol) was added dropwise to a mixture of methyl L-2-methylprolinate hydrochloride 2 (0.42 g, 2.34 mmol) and N-benzyloxycarbonyl-glycine (98.5%) 3 (0.52 g, 2.45 mmol) in methylene chloride (16 cm3), at 0 °C, under an atmosphere of nitrogen. The resultant solution was stirred for 20 min and a solution of 1 ,3-dicyclohexylcarbodiimide (0.56 g, 2.71 mmol) in methylene chloride (8 cm3) at 0 °C was added dropwise and the reaction mixture was warmed to room temperature and stirred for a further 20 h. The resultant white mixture was filtered through a Celite™ pad to partially remove 1 ,3-dicyclohexylurea, and the pad was washed with methylene chloride (50 cm3). The filtrate was washed successively with 10% aqueous hydrochloric acid (50 cm3) and saturated aqueous sodium hydrogen carbonate (50 cm3), dried (MgSO4), filtered, and concentrated to dryness in vacuo. Further purification of the residue by flash column chromatography (35 g SiO2; 30-70% ethyl acetate – hexane; gradient elution) afforded tentatively methyl N-benzyloxycarbonyl-glycyl-L-2-methylprolinate 4 (0.56 g), containing 1 ,3-dicyclohexylurea, as a white semi-solid: Rf 0.65 (EtOAc); m/z (ΕI+) 334.1534 (M+. C17H22N2O5 requires 334.1529) and 224 ( 1 ,3-dicyclohexylurea).
To a solution of impure prolinate 4 (0.56 g, ca. 1.67 mmol) in 1,4-dioxane (33 cm3) was added dropwise 1 M aqueous sodium hydroxide (10 cm3, 10 mmol) and the mixture was stirred for 19 h at room temperature. Methylene chloride ( 100 cm3) was then added and the organic layer extracted with saturated aqueous sodium hydrogen carbonate (2 x 100 cm3). The combined aqueous layers were carefully acidified with hydrochloric acid (32%), extracted with methylene chloride (2 x 100 cm3), and the combined organic layers dried (MgSO4), filtered, and
concentrated to dryness in vacuo. Purification of the ensuing residue (0.47 g) by flash column chromatography ( 17 g SiO2; 50% ethyl acetate – hexane to 30% methanol – dichloromethane; gradient elution) gave N-protected dipeptide 5 (0.45 g, 60%) as a white foam in two steps from hydrochloride 2. Dipeptide 5 was shown to be exclusively the frafw-orientated conformer by NMR analysis: Rf 0.50 (20% MeOH – CH2Cl2); [α]D -62.3 (c 0.20 in CH2Cl2); vmax (film)/cm-1 3583, 3324 br, 2980, 2942, 1722, 1649, 1529, 1454, 1432, 1373, 1337, 1251 , 1219, 1179, 1053, 1027, 965, 912, 735 and 698; δH (300 MHz; CDCl3; Me4Si) 1.59 (3H, s, Proα-CH3), 1 .89 (1H, 6 lines, J 18.8, 6.2 and 6.2, Proβ-HAHB), 2.01 (2H, dtt, J 18.7, 6.2 and 6.2, Proγ-H2), 2.25-2.40 (1H, m, Proβ-HAΗΒ), 3.54 (2H, t, J 6.6, Proδ-H2), 3.89 (1H, dd, J 17.1 and 3.9, Glyα-HAHB), 4.04 (1H, dd, J 17.2 and 5.3, Glyα-HAΗΒ), 5.11 (2H, s, OCH2Ph), 5.84 (I H, br t, J 4.2, N-H), 7.22-7.43 (5H, m, Ph) and 7.89 (1 H, br s, -COOH); δC (75 MHz; CDCl3) 21.3 (CH3, Proα-CH3), 23.8 (CH2, Proγ-C), 38.2 (CH2, Proβ-C), 43.6 (CH2, Glyα-C), 47.2 (CH2, Proδ-C), 66.7 (quat, Proα-C), 66.8 (CH2, OCH2Ph), 127.9 (CH, Ph), 127.9 (CH, Ph), 128.4, (CH, Ph), 136.4 (quat., Ph), 156.4 (quat., NCO2), 167.5 (quat., Gly-CON) and 176.7 (quat., CO); m/z (EI+) 320.1368 (M+. C16Η20Ν2Ο5 requires 320.1372).
Dibenzyl N-benzyloxycarbonyl-glycyl-L-2-methylprolyl-L-glutamate 7
Triethylamine (0.50 cm3, 3.59 mmol) was added dropwise to a solution of dipeptide 5 (0.36 g, 1.12 mmol) and L-glutamic acid dibenzyl ester /Moluenesulphonate 6 (0.73 g, 1.46 mmol) in methylene chloride (60 cm3) under nitrogen at room temperature, and the reaction mixture stirred for 10 min. Bis(2-oxo-3-oxazoIidinyl)phosphinic chloride (BoPCl, 97%) (0.37 g, 1.41 mmol) was added and the colourless solution stirred for 17 h. The methylene chloride solution was washed successively with 10% aqueous hydrochloric acid (50 cm3) and saturated aqueous sodium hydrogen carbonate (50 cm3), dried (MgSO4), filtered, and evaporated to dryness in vacuo. Purification of the resultant residue by repeated (2x) flash column chromatography (24 g SiO2; 30-70% ethyl acetate – hexane; gradient elution) yielded ƒully protected tripeptide 7 (0.63 g, 89%) as a colourless oil. Tripeptide 7 was shown to be exclusively the trans-orientated conformer by NMR analysis: Rf 0.55 (EtOAc); [α]D -41.9 (c 0.29 in CH2Cl2); vmax (film)/cm-1 3583, 3353 br, 2950, 1734, 1660, 1521, 1499, 1454, 1429, 1257, 1214, 1188, 1166, 1051, 911, 737 and 697; δH (400 MHz; CDCl3; Me4Si) 1.64 (3H, s, Proot-CH3), 1.72 (1H, dt, J 12.8, 7.6 and 7.6, Proβ-HAHB), 1.92 (2H, 5 lines, J 6.7, Proγ-H2), 2.04 (1H, 6 lines, J 7.3 Gluβ-HAHB), 2.17-2.27 (1H, m, Gluβ-HAΗΒ), 2.35-2.51 (3H, m, Proβ-HAΗΒ and Gluγ-H2), 3.37-3.57 (2H, m, Proδ-H2), 3.90 (1 H, dd, J 17.0 and 3.6, Glyα-HAHB), 4.00 (1H, dd, J 17.1 and 5.1, Glyα-HAΗΒ), 4.56 (1H, td, J 7.7 and 4.9, Glyα-H), 5.05-5.20 (6H, m, 3 x OCH2Ph), 5.66-5.72 (1H, br m, Gly-NH), 7.26-7.37 (15H, m, 3 x Ph) and 7.44 (1H, d, J 7.2, Glu-NH); δC (100 MHz; CDCl3) 21.9 (CH3, Proα-CH3), 23.4 (CH2, Proγ-C), 26.6 (CH2, Gluβ-C), 30.1 (CH2, Gluγ-C), 38.3 (CH2, Proβ-C),
43.9 (CH2, Glyα-C), 47.6 (CH2, Proδ-C), 52.2 (CH, Glua-C), 66.4 (CH2, OCH2Ph), 66.8 (CH2, OCH2Ph), 67.1 (CH2, OCH2Ph), 68.2 (quat, Proα-C), 127.9 (CH, Ph), 128.0 (CH, Ph), 128.1, (CH, Ph), 128.2, (CH, Ph), 128.2, (CH, Ph), 128.3, (CH, Ph), 128.4, (CH, Ph), 128.5, (CH, Ph), 128.5, (CH, Ph), 135.2 (quat., Ph), 135.7 (quat., Ph), 136.4 (quat, Ph), 156.1 (quat, NCO2), 167.3 (quat., Gly-CO), 171.4 (quat., CO), 172.9 (quat., CO) and 173.4 (quat., CO); m/z (FAB+) 630.2809 (MH+. C35H40N3O8 requires 630.2815).
Glycyl-L-2-methylprolyl-L-glutamic acid (G-2-MePE)
A mixture of the protected tripeptide 7 (0.63 g, 1.00 mmol) and 10 wt % palladium on activated carbon (0.32 g, 0.30 mmol) in 91 :9 methanol – water (22 cm3) was stirred under an atmosphere of hydrogen at room temperature, protected from light, for 23 h. The reaction mixture was filtered through a Celite™ pad and the pad washed with 75 :25 methanol – water (200 cm3). The filtrate was concentrated to dryness under reduced pressure and the residue triturated with anhydrous diethyl ether to afford a 38: 1 mixture of G-2-MePE and tentatively methylamine 8 (0.27 g, 86%) as an extremely hygroscopic white solid. Analytical reverse-phase HPLC studies on the mixture [Altech Econosphere C 18 Si column, 150 x 4.6 mm, 5 ☐m; 5 min flush with H2O (0.05% TFA) then steady gradient over 25 min to MeCN as eluent at flow rate of 1 ml/min; detection using diode array] indicated it was a 38: 1 mixture of two eluting peaks with retention times of 13.64 and 14.44 min at 207 and 197 nm, respectively. G-2-MePE was shown to be a 73 :27 trans:cis mixture of conformers by 1H NMR analysis (the ratio was estimated from the relative intensities of the double doublet and triplet at δ 4.18 and 3.71 , assigned to the Gluα-H protons of the major and minor conformers, respectively):
mp 144 °Cɸ;
[ α]D -52.4 (c 0.19 in H2O);
δα (300 MHz; D2O; internal MeOH) 1.52 (3H, s, Proα-CH3), 1.81-2.21 (6H, m, Proβ-H2, Proγ-H, and Gluβ-H2), 2.34 (1.46H, t, J 7.2, Gluy-H2), 2.42* (0.54H, t, 77.3, Gluγ-H2), 3.50-3.66 (2H, m, Pro6-H2), 3.71 * (0.27H, t, J 6.2, Gluoc-H), 3.85 (1H, d, J 16.6, Glyα-HAHB), 3.92 (1H, d, J 16.6, Glyα-HAΗΒ) and 4.18 (0.73H, dd, J 8.4 and 4.7, Glua-H);
δC (75 MHz; D2O; internal MeOH) 21.8 (CH3, Proα-CH3), 25.0 (CH2, Proγ-C), 27.8* (CH2: Gluβ-C), 28.8 (CH2, Gluβ-C), 32.9 (CH2, Gluγ-C), 40.8 (CH2, Proβ-C), 42.7 (CH2, Glyα-C), 49.5 (CH2, Proδ-C), 56.0* (CH, Gluα-C), 56.4 (CH, Gluα-C), 69.8 (quat, Proα-C), 166.5 (quat., Gly-CO), 177.3 (quat., Pro-CON), 179.2 (quat., Gluα-CO), 180.2* (quat., Gluγ-CO) and 180.6 (quat., Gluγ-CO);
m/z (FAB+) 3 16.1508 (MH+. C13H22N3O6 requires 316.1509).
PATENT
WO02094856
Example
The following non-limiting example illustrates the synthesis of a compound of the invention, NN-dimethylglycyl-L-prolyl-L-glutamic acid.
All starting materials and other reagents were purchased from Aldrich;
BOC = tert-butoxycarbonyl; Bn = benzyl.
BOC-(γ-benzyl)-L-prolyl-L-glutamic acid benzyl ester
To a solution of BOC-proline [Anderson GW and McGregor AC: J. Amer. Chem.
Soc: 79, 6180, 1957] (10 mmol) in dichloromethane (50 ml), cooled to 0 °C, was added triethylamine (1.39 ml, 10 mmol) and ethyl chloroformate (0.96 ml, 10 mmol). The resultant mixture was stirred at 0 °C for 30 minutes. A solution of dibenzyl L-glutamate (10 mmol) was then added and the mixture stirred at 0 °C for 2 hours then warmed to room temperature and stirred overnight. The reaction mixture was washed with aqueous sodium bicarbonate and citric acid (2 mol l“1) then dried (MgS04) and concentrated at reduced pressure to give BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (5.0 g, 95%).
(7-Benzyl)-L-prolyl-L-glutamic acid dibenzyl ester
A solution of BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (3.4 g, 10 mmol), cooled to 0 °C, was treated with trifluoroacetic acid (25 ml) for 2 hr at room temperature. After removal of the volatiles at reduced pressure the residue was triturated with ether to give (γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (I).
N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
A solution of dicyclohexylcarbodiimide (10.3 mmol) in dichloromethane (10 ml) was added to a stirred and cooled (0 °C) solution of (7-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (10 mmol), TVN-dimethylglycine (10 mmol) and triethylamine
(10.3 mmol) in dichloromethane (30 ml). The mixture was stirred at 0 °C overnight and then at room temperature for 3 h. After filtration, the filtrate was evaporated at reduced pressure. The resulting crude dibenzyl ester was dissolved in a mixture of ethyl acetate (30 ml) and methanol (30 ml) containing 10% palladium on charcoal (0.5 g) then hydrogenated at room temperature and pressure until the uptake of hydrogen ceased. The filtered solution was evaporated and the residue recrystallized from ethyl acetate to yield the tri-peptide derivative.
It will be evident that following the method of the Example, and using alternative amino acids or their amides or esters, will yield other compounds of Formula 1.
Testing; Material and Methods
The following experimental protocol followed guidelines approved by the
University of Auckland animal ethics committee.
Preparation of cortical astrocyte cultures for harvest of metabolised cell culture supernatant
One cortical hemisphere from a postnatal day 1 rat was used and collected into
4ml of DMEM. Trituration was done with a 5ml glass pipette and subsequently through an 18 gauge needle. Afterwards, the cell suspension was sieved through a lOOμm cell strainer and washed in 50ml DMEM (centrifugation for 5min at 250g). The sediment was resuspended into 20ml DMEM+10% fetal calf serum. 10 Milliliters of suspension was added into each of two 25cm3 flasks and cultivated at 37°C in the presence of 10% C02, with a medium change twice weekly. After cells reached confluence, they were washed three times with PBS and adjusted to Neurobasal/B27 and incubated for another 3 days. This supernatant was frozen for transient storage until usage at -80°C.
Preparation of striatal and cortical tissue from rat E18/E19 embryos
A dam was sacrificed by C02-treatment in a chamber for up to 4 minutes and was prepared then for cesarean section. After surgery, the embryos were removed from their amniotic sacs, decapitated and the heads put on ice in DMEM/F12 medium for striatum and PBS + 0.65% D(+)-glucose for cortex.
Striatal tissue extraction procedure and preparation of cells
Whole brain was removed from the skull with the ventral side facing upside in DMEM/F12 medium. The striatum was dissected out from both hemispheres under a stereomicroscope and the striatal tissue was placed into the Falcon tube on ice.
The collected striatal tissue was triturated by using a PI 000 pipettor in 1ml of volume. The tissue was triturated by gently pipetting the solution up and down into the pipette tip about 15 times, using shearing force on alternate outflows. The tissue pieces settled to the bottom of the Falcon tube within 30 seconds, subsequently the supernatant was transferred to a new sterile Falcon tube on ice. The supernatant contained a suspension of dissociated single cells. The tissue pieces underwent a second trituration to avoid excessively damaging cells already dissociated by over triturating them. 1 Milliliter of ice-cold DMEM/F12 medium was added to the tissue pieces in the first tube and triturated as before. The tissue pieces were allowed to settle and the supernatant was removed to a new sterile Falcon tube on ice. The cells were centrifuged at 250g for 5 minutes at 4°C. The resuspended cell pellet was ready for cell counting.
Plating and cultivation of striatal cells
Striatal cells were plated into Poly-L-Lysine (O.lmg/ml) coated 96-well plates (the inner 60 wells only) at a density of 200,000 cells /cm2 in Neurobasal/B27 medium (Invitrogen). The cells were cultivated in the presence of 5% C02 at 37°C under 100% humidity. Complete medium was changed on days 1, 3 and 6.
Cortical tissue extraction procedure and preparation of cells
The two cortical hemispheres were carefully removed by a spatula from the whole brain with the ventral side facing upside into a PBS +0.65% D(+)-glucose containing petri dish. Forcips were put into the rostral part (near B. olfactorius) of the cortex for fixing the tissue and two lateral – sagittal oriented cuttings were done to remove the paraform and entorhinal cortices. The next cut involved a frontal oriented cut at the posterior end to remove the hippocampal formation. A final frontal cut was done a few millimeters away from the last cut in order to get hold of area 17/18 of the visual cortex.
The collected cortices on ice in PBS+0.65% D(+)-glucose were centrifuged at 350g for 5min. The supernatant was removed and trypsin/EDTA (0.05%/0.53mM) was added for 8min at 37°C. The reaction was stopped by adding an equal amount of DMEM+10%) fetal calf serum. The supernatant was removed by centrifugation followed by two subsequent washes in Neurobasal/B27 medium.
The cells were triturated once with a glass Pasteur pipette in 1 ml of
Neurobasal/B27 medium and subsequently twice by using a 1ml insulin syringe with a 22 gauge needle. The cell suspension was passed through a lOOμm cell strainer and subsequently rinsed by 1ml of Neurobasal B27 medium. Cells were counted and adjusted to 50,000 cells per 60μl.
Plating and cultivation of cortical cells
96-well plates were coated with 0.2mg/ml Poly-L-Lysine and subsequently coated with 2μg/ml laminin in PBS, after which 60μl of cortical astrocyte-conditioned medium was added to each well. Subsequently, 60μl of cortical cell suspension was added. The cells were cultivated in the presence of 10% C02 at 37°C under 100%) humidity. At day 1, there was a complete medium change (1:1- Neurobasal/B27 and astrocyte-conditioned medium) with addition of lμM cytosine-β-D-arabino-furanoside (mitosis inhibitor). On the second day, 2/3 of medium was changed. On day 5, 2/3 of the medium was changed again.
Cerebellar microexplants from P8 animals: preparation, cultivation and fixation
The laminated cerebellar cortices of the two hemispheres were explanted from a P8 rat, cut into small pieces in PBS + 0.65% D(+)glucose solution and triturated by a 23gauge needle and subsequently pressed through a 125 μm pore size sieve. The microexplants that were obtained were centrifuged (60 g) twice (media exchange) into serum-free BSA-supplemented START V-medium (Biochrom). Finally, the
microexplants were reconstituted in 1500 μl STARTV-medium (Biochrom). For cultivation, 40μl of cell suspension was adhered for 3 hours on a Poly-D-Lysine
(O.lmg/ml) coated cover slip placed in 35mm sized 6-well plates in the presence of 5% C02 under 100% humidity at 34°C. Subsequently, 1ml of STARTV-medium was added together with the toxins and drugs. The cultures were monitored (evaluated) after 2-3 days of cultivation in the presence of 5% C02 under 100% humidity. For cell counting analysis, the cultures were fixed in rising concentrations of paraformaldehyde (0.4%, 1.2%, 3% and 4% for 3min each) followed by a wash in PBS.
Toxin and drug administration for cerebellar, cortical and striatal cells: analysis
All toxin and drug administration experiments were designed that 1/100 parts of okadaic acid (30nM and lOOnM concentration and 0.5mM 3-nitropropionic acid for cerebellar microexplants only), GPE (InM -ImM) and G-2Methyl-PE (InM-lmM) were used respectively at 8DIV for cortical cultures and 9DIV for striatal cultures. The incubation time was 24hrs. The survival rate was determined by a colorimetric end-point MTT-assay at 595nm in a multi-well plate reader. For the cerebellar microexplants four windows (field of 0.65 mm2) with highest cell density were chosen and cells displaying neurite outgrowth were counted.
Results
The GPE analogue G-2Methyl-PE exhibited comparable neuroprotective capabilities within all three tested in vitro systems (Figures 12-15).
The cortical cultures responded to higher concentrations of GPE (Figure 12) /or
G-2Methyl-PE (lOμM, Figure 13) with 64% and 59% neuroprotection, respectively.
Whereas the other 2 types of cultures demonstrated neuroprotection at lower doses of G-2Methyl-PE (Figures 14 and 15). The striatal cells demonstrated
neuroprotection within the range of InM to ImM of G-2Methyl-PE (Figure 15) while the postnatal cerebellar microexplants demonstrated neuroprotection with G-2Methyl-PE in the dose range between InM and lOOnM (Figure 14).
While this invention has been described in terms of certain preferred embodiments, it will be apparent to a person of ordinary skill in the art having regard to that knowledge and this disclosure that equivalents of the compounds of this invention may be prepared and administered for the conditions described in this application, and all such equivalents are intended to be included within the claims of this application.
PATENT
WO-2021026066
Composition and kits comprising trofinetide and other related substances. Also claims a process for preparing trofinetide and the dosage form comprising the same. Disclosed to be useful in treating neurodegenerative conditions, autism spectrum disorders and neurodevelopmental disorders.
Trofinetide is a synthetic compound, having a similar core structure to Glycyl-Prolyl-Glutamic acid (or “GPE”). Trofinetide has been found to be useful in treating neurodegenerative conditions and recently has been found to be effective in treating Autism Spectrum disorders and Neurodevelopmental disorders.
Formula (Ila),
Example 1: Trofinetide Manufacturing Process
In general, trofinetide and related compounds can be manufactured from a precursor peptide or amino acid reacted with a silylating or persilylating agent at one or more steps. In the present invention, one can use silylating agents, such as N-trialkylsilyl amines or N-trialkylsilyl amides, not containing a cyano group.
Examples of such silylating reagents include N,O-bis(trimethylsilyl)acetamide (BSA), N,O-bis(trimethylsilyl)trifluoroacetamide, hexamethyldisilazane, N-methyl-N-(trimethylsilyl)acetamide (TMA), N-methyl-N-(trimethylsilyl)trifluoroacetamide, N-(trimethylsilyl)acetamide, N-(trimethylsilyl)diethylamine, N-(trimethylsilyl)dimethylamine, 1-(trimethylsilyl)imidazole, 3-(trimethylsilyl)-2-oxazolidone.
Step 1: Preparation of Z-Gly-OSu
Several alternative procedures can be used for this step.
Procedure 1A
One (1) eq of Z-Gly-OH and 1.1 eq of Suc-OH were solubilized in 27 eq of iPrOH and 4 eq of CH2Cl2 at 21 °C. The mixture was cooled and when the temperature reached -4 °C, 1.1 eq of EDC.HCl was added gradually, keeping the temperature below 10 °C. During the reaction a dense solid appeared. After addition of EDC.HCl, the mixture was allowed to warm to 20 °C. The suspension was cooled to 11 °C and filtered. The cake was washed with 4.9 eq of cold iPrOH and 11 eq of IPE before drying at 34 °C (Z-Gly-OSu dried product -Purity: 99.5%; NMR assay: 96%; Yield: 84%).
Procedure 1B
This Procedure is for a variant of Procedure 1A, and differs by replacing iPrOH with ACN. One (1) eq of Z-Gly-OH and 1.1 eq of Suc-OH were solubilized in 22 eq of ACN at 35 °C. The mixture was cooled in an ice bath. When the temperature reached 1 °C, 0.9 eq of DCC in 5.5 eq of ACN was added gradually to keep the temperature below 5 °C. The coupling reaction took about 20 hrs. During the reaction, DCU precipitated and was removed by filtration at the end of the coupling. After filtration, DCU was washed with ACN to recover the product. The mixture of Z-Gly-OSu was then concentrated to reach 60% by weight. iPrOH (17 eq) was added to initiate the crystallization. Quickly after iPrOH addition a dense solid appeared. An additional 17 eq of iPrOH was needed to liquify the suspension. The suspension was cooled in an ice bath and filtered. The solid was washed with 9 eq of iPrOH before drying at 45 °C (Z-Gly-OSu dried product – Purity: 99.2%; HPLC assay: 99.6%; Yield: 71%).
Step 2: Preparation of Z-Gly-MePro-OH
Several alternative procedures can be used for this step.
Procedure 2A
One (1) eq of MePro.HCl was partially solubilized in 29 eq of CH2Cl2 at 35 °C with 1.04 eq of TEA and 1.6 eq of TMA. The mixture was heated at 35 °C for 2 hrs to perform the silylation. Then 1.02 eq of Z-Gly-OSu was added to the mixture. The mixture was kept at 35 °C for 3 hrs and then 0.075 eq of butylamine was added to quench the reaction. The mixture was allowed to return to room temperature and mixed for at least 15 min. The Z-Gly-MePro-OH was extracted once with 5% w/w NaHCO3 in 186 eq of water, then three times successively with 5% w/w NaHCO3 in 62 eq of water. The aqueous layers were pooled and the pH was brought to 2.2 by addition of 34 eq of HCl as 12N HCl at room temperature. At this pH, Z-Gly-MePro-OH formed a sticky solid that was solubilized at 45 °C with approximately 33 eq of EtOAc and 2.3 eq of iButOH. Z-Gly-MePro-OH was extracted into the organic layer and washed with 62 eq of demineralized water. The organic layer was then dried by azeotropic distillation with 11.5 eq of EtOAc until the peptide began to precipitate. Cyclohexane (12 eq) was added to the mixture to complete the precipitation. The suspension was cooled at 5 °C for 2 hrs and filtered. The solid was washed with 10 eq of cyclohexane before drying at 45 °C (Z-Gly-MePro-OH dried product – Purity: 100%; HPLC assay: 100%; Yield 79%).
Procedure 2B
This Procedure is for a variant of Procedure 2A. One (1) eq of MePro.HCl was partially solubilized in 36.6 eq of CH2Cl2 at 34 °C with 1.01 eq of TEA and 0.1 eq of TMA. Then 1.05 eq of Z-Gly-OSu was added to the mixture, followed by 1.0 eq of TEA. The mixture was maintained at 35 °C for approximately 1 hr, cooled to 25 to 30 °C and 0.075 eq of DMAPA was added to stop the reaction. One hundred (100) eq of water, 8.6 eq of HCl as 12N HCl and 0.3 eq of KHSO4 were added to the mixture (no precipitation was observed, pH=1.7). Z-Gly-MePro-OH was extracted into the organic layer and washed twice with 97 eq of demineralized water with 0.3 eq of KHSO4, then 100 eq of demineralized water, respectively. EtOAc (23 eq) was added to the mixture and CH2Cl2 was removed by distillation until the peptide began to precipitate. Cyclohexane (25 eq) was added to the mixture to complete the precipitation. The suspension was cooled at -2 °C overnight and filtered. The solid was washed with 21 eq of cyclohexane before drying at 39 °C (Z-Gly-MePro-OH dried product – Purity: 98.7%; NMR assay: 98%; Yield 86%).
Procedure 2C
In reactor 1, MePro.HCl (1 eq) was suspended in EtOAc (about 7 eq). DIPEA (1 eq) and TMA (2 eq) were added, and the mixture heated to dissolve solids. After dissolution, the solution was cooled to 0 °C. In reactor 2, Z-Gly-OH (1 eq) was suspended in EtOAc (about 15 eq). DIPEA (1 eq), and pyridine (1 eq) were added. After mixing, a solution was obtained, and cooled to -5 °C. Piv-Cl (1 eq) was added to reactor 2, and the contents of reactor 1 added to reactor 2. Upon completed addition, the contents of reactor 2 were taken to room temperature. The conversion from Z-Gly-OH to Z-Gly-MePro-OH was monitored by HPLC. When the reaction was complete, the reaction mixture was quenched with DMAPA (0.1 eq), and washed with an aqueous solution comprised of KHSO4, (about 2.5 wt%), NaCl (about 4 wt%), and conc. HCl (about 6 wt%) in 100 eq H2O. The aqueous layer was re-extracted with EtOAc, and the combined organic layers washed with an aqueous solution comprised of KHSO4 (about 2.5 wt%) and NaCl (about 2.5 wt%) in 100 eq H2O, and then with water (100 eq). Residual water was removed from the organic solution of Z-Gly-MePro-OH by vacuum distillation with EtOAc. The resulting suspension was diluted with heptane (about 15 eq) and cooled to 0 °C. The product was isolated by filtration, washed with cold heptane (about 7 eq), and dried under vacuum at 45 °C. Z-Gly-MePro-OH (85% yield) was obtained.
Step 3: Preparation of Z-Gly-MePro-Glu-OH
Several alternative procedures can be used in this step.
Procedure 3A
H-Glu-OH (1.05 eq) was silylated in 2 eq of CH2Cl2 with 3.5 eq of TMA at 65 °C. Silylation was completed after 2 hrs. While the silylation was ongoing, 1.0 eq of Z-Gly-MePro-OH and 1.0 eq of Oxyma Pure were solubilized in 24 eq of CH2Cl2 and 1.0 eq of DMA at room temperature in another reactor. EDC.HCl (1.0 eq.) was added. The activation rate reached 97% after 15 min. The activated Oxyma Pure solution, was then added to silylated H-Glu-OH at 40 °C and cooled at room temperature. Coupling duration was approximately 15 min, with a coupling rate of 97%. Addition of 8.2% w/w NaHCO3 in 156 eq of water to the mixture at room temperature (with the emission of CO2) was performed to reach pH 8. Z-Gly-MePro-Glu-OH was extracted in water. The aqueous layer was washed twice with 29 eq of CH2Cl2. Residual CH2Cl2 was removed by concentration. The pH was brought to 2.5 with 2.5N HCl, followed by 1.4 eq of solid KHSO4 to precipitate Z-Gly-MePro-Glu-OH. The mixture was filtered and the solid was washed with 3 x 52 eq of water. The filtered solid was added to 311 eq of demineralized water and heated to 55-60 °C. iPrOH (29 eq) was added gradually until total solubilization of the product. The mixture was slowly cooled to 10 °C under moderate mixing during 40 min to initiate the crystallization. The peptide was filtered and washed with 2 x 52 eq of water before drying at 45 °C (Z-Gly-MePro-Glu-OH dried product – Purity: 99.5%; NMR assay: 96%; Yield 74%).
Procedure 3B
One (1) eq of Z-Gly-MePro-OH and 1.05 eq of Suc-OH were solubilized in 40 eq of ACN and 30 eq of CH2Cl2 at room temperature. The mixture was cooled in an ice bath, and when the temperature was near 0 °C, 1.05 eq of DCC dissolved in 8 eq of ACN was added gradually, keeping the temperature below 5 °C. After addition of DCC, the mixture was progressively heated from 0 °C to 5 °C over 1 hr, then to 20 °C between 1 to 2 hrs and then to 45 °C between 2 to 5 hrs. After 5 hrs, the mixture was cooled to 5 °C and maintained overnight. The activation rate reached 98% after approximately 24 hrs. DCU was removed by filtration and washed with 13.5 eq of ACN. During the activation step, 1.1 eq of H-Glu-OH was silylated in 30 eq of ACN with 2.64 eq of TMA at 65 °C. Silylation was completed after 2 hrs. Z-Gly-MePro-OSu was then added gradually to the silylated H-Glu-OH at room temperature, with 0.4 eq of TMA added to maintain the solubility of the H-Glu-OH. The mixture was heated to 45 °C and 0.7 eq of TMA was added if precipitation occurred. The coupling duration was about 24 hrs to achieve a coupling rate of approximately 91%. The reaction was quenched by addition of 0.15 eq of butylamine and 2.0 eq of TEA. Water (233 eq) was added and the mixture concentrated until gelation occurred. Z-Gly-MePro-Glu-OH was extracted in water by addition of 5% w/w NaHCO3 in 233 eq of water and 132 eq of CH2Cl2. The aqueous layer was washed twice with 44 eq of CH2Cl2. Residual CH2Cl2 was removed by distillation. The pH was brought to 2.0 with 24 eq of HCl as 12N HCl followed by 75 eq of HCl as 4N HCl. At this pH, Z-Gly-MePro-Glu-OH precipitated. The mixture was cooled in an ice bath over 1 hr and filtered. The solid was washed with 186 eq of cold water before drying at 45 °C (Z-Gly-MePro-Glu-OH dried product – HPLC Purity: 98.4%; NMR assay: 100%; Yield 55%).
Procedure 3C
This Procedure is for a variant of Procedure 3A. H-Glu-OH (1.05 eq) was silylated in 3.7 eq of CH2Cl2 with 3.5 eq of TMA at 62 °C. Silylation was completed after approximately 1.5 to 2 hrs, as evidenced by solubilization. During the silylation step, 1.0 eq of Z-Gly-MePro-OH and 1.0 eq of Oxyma Pure were solubilized in 31.5 eq of CH2Cl2 at 22 °C. One (1.06) eq of EDC.HCl was added to complete the activation. The silylated H-Glu-OH was then added to the activated Oxyma Pure solution. The temperature was controlled during the addition to stay below 45 °C. Desilylation was performed by addition of a mixture of 2.5% w/w KHSO4 in 153 eq of water and 9 eq of iPrOH to reach a pH of 1.65. Residual CH2Cl2 was removed by concentration. The mixture was cooled to 12 °C to precipitate the Z-Gly-MePro-Glu-OH. The mixture was filtered and the solid was washed with 90 eq of water before drying at 36 °C.
Procedure 3D
This Procedure is for a variant of Procedure 3A. H-Glu-OH (1.05 eq.) was silylated in 3.9 eq of CH2Cl2 with 3.5 eq of TMA at 62 °C. Silylation was completed after 2 hrs, as evidenced by Solubilization. During the silylation step, 1 eq of Z-Gly-MePro-OH and 1 eq of Oxyma Pure were solubilized in 25 eq of CH2Cl2 at 23 °C. One (1) eq of EDC.HCl was added. To complete the activation, an additional 0.07 eq of EDC. HCl was added. Silylated H-Glu-OH was then added to the activated Oxyma Pure solution. Temperature was controlled during the addition to stay below 45 °C. Desilylation was performed by addition of a mixture of 2.5% w/w KHSO4 in 160 eq of water and 9.6 eq of iPrOH to reach pH 1.63.
Residual CH2Cl2 was removed by concentration. The mixture was cooled to 20 °C to precipitate the Z-Gly-MePro-Glu-OH. The mixture was filtered and the solid was washed with 192 eq of water before drying at about 25 °C for 2.5 days. The solid was then solubilized at 64 °C by addition of 55 eq of water and 31 eq of iPrOH. After solubilization, the mixture was diluted with 275 eq of water and cooled to 10 °C for crystallization. The mixture was filtered and the solid was washed with 60 eq of water before drying at 27 °C (Z-Gly-MePro-Glu-OH dried product – Purity: 99.6%; NMR assay: 98%; Yield 74%).
Procedure 3E
In reactor 1, H-Glu-OH (1.05 eq) was suspended in ACN (about 2.2 eq). TMA (about 3.5 eq) added, and the mixture was heated to dissolve solids. After dissolution, the solution was cooled to room temperature. In reactor 2, Z-Gly-MePro-OH (1 eq) was suspended in ACN (14 eq). Oxyma Pure (1 eq) and EDC.HCl (1 eq) were added. The mixture was stirred at room temperature until the solids dissolved. The contents of reactor 2 were added to reactor 1. The conversion from Z-Gly-MePro-OH to Z-Gly-MePro-Glu-OH was monitored by HPLC. Upon completion the reaction mixture was added to an aqueous solution comprised of KHSO4 (about 2.5 wt%) dissolved in about 100 eq H2O. ACN was removed from the aqueous suspension of Z-Gly-MePro-Glu-OH by vacuum distillation with H2O. After stirring at room temperature, the product in the resulting suspension was isolated by filtration and washed with water. The solid obtained was dissolved in an aqueous solution comprised of NaHCO3 (about 5 wt%) in 110 eq H2O, and recrystallized by addition of an aqueous solution comprised of KHSO4 (about 10 wt%) in 90 eq H2O. The product was isolated by filtration, washed with water, and dried under vacuum at 45 °C. Z-Gly-MePro-Glu-OH (75% yield) was obtained.
Step 4: Deprotection and Isolation of Trofinetide
Several alternative procedures can be used in this step.
Procedure 4A
Z-Gly-MePro-Glu-OH (1 eq) was suspended in water (about 25 eq) and EtOAc (about 15 eq). Pd/C (0.025 eq by weight and containing 10% Pd by weight) was added, and the reaction mixture hydrogenated by bubbling hydrogen through the reaction mixture at room temperature. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC, and upon reaction completion the catalyst was removed by filtration, and the layers separated. Residual EtOAc was removed from the aqueous solution containing trofinetide by sparging with nitrogen or washing with heptane. The aqueous solution was spray-dried to isolate the product. Trofinetide (90% yield) was obtained. Alternatively, deprotection can be accomplished using MeOH only, or a combination of iPrOH and MeOH, or by use of ethyl acetate in water.
Procedure 4B
This Procedure is for a variant of Procedure 4A, excluding EtOAc. Z-Gly-MePro-Glu-OH (1 eq) was suspended in water (about 50 eq). Pd/C (0.05 eq, 5% Pd by weight) was added, and the reaction mixture hydrogenated at room temperature with a pressure of 5 bar. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC. Upon
reaction completion the catalyst was removed by filtration, and the aqueous layer washed with EtOAc (about 5 eq). Residual EtOAc was removed from the aqueous solution containing trofinetide by sparging with nitrogen or washing with heptane. The aqueous solution was spray-dried to isolate the product. Trofinetide (90% yield) was obtained.
Procedure 4C
This Procedure is for a variant of Procedure 4A, replacing EtOAc with MeOH. Z-Gly-MePro-Glu-OH (1 eq) was suspended in MeOH (100 eq) and water (12 eq). Pd/Si (0.02 eq by weight) was added and the mixture was heated at 23 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC, and upon reaction completion the catalyst was removed by filtration and the layers were washed with MeOH and iPrOH. The solvents were concentrated under vacuum at 45 °C, and trofinetide precipitated. The precipitate was filtered and dried at 45 °C to provide trofinetide.
Procedure 4D
This Procedure is for a variant of Procedure 4A, replacing Pd/C with Pd/Si. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 105 eq of MeOH and 12 eq of water. Pd/Si (0.02 eq by weight) was added and the mixture was heated at 23 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (conversion rate approximately 99% after 1 hr), the catalyst was filtered off and washed with 20-30 eq of MeOH. iPrOH (93 eq) was added and MeOH was replaced by iPrOH by concentration at 45 °C under vacuum. The peptide was concentrated until it began to precipitate. The peptide was filtered and dried at 45 °C (H-Gly-MePro-Glu-OH dried product: Purity: 98.1%; NMR assay: 90%; Yield 81%).
Procedure 4E
This Procedure is for a variant of Procedure 4A, removing H2O and replacing Pd/C with Pd/Si. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 44 eq of MeOH. Pd/Si type 340 (0.02 eq by weight) was added and the mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (conversion rate about 99.9%, after 3-3.5 hrs), the catalyst was filtered off and washed with 8 eq of MeOH. Deprotected peptide was then precipitated in 56 eq of iPrOH. After 30 min at 5 °C, the peptide was filtered and washed with three times with 11 eq of iPrOH before drying at 25 °C (H-Gly-MePro-Glu-OH dried product: Purity: 99.4%; HPLC assay: ~98%; Yield: 81%).
Procedure 4F
This Procedure is for a variant of Procedure 4A. One (1) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 14 eq of EtOAc and 25 eq of water. Pd/C (0.01 eq by weight) was added and the mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (conversion rate about 100%, after about 3.5 hrs), the catalyst was filtered off and washed with a mixture of 3.5 eq of EtOAc and 6 eq of water. The aqueous layer was then ready for spray-drying (Aqueous H-Gly-MePro-Glu-OH peptide solution: Purity: 98.6%; Yield: ~95%).
Procedure 4G
This Procedure is for a variant of Procedure 4A, replacing Pd/C with Pd/Si, EtOAc with MeOH, and removing H2O. Pd/Si type 340 (0.02 eq by weight) was added to 2.9 vols of MeOH for pre-reduction during 30 min. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 34 eq of MeOH. The reduced palladium was then transferred to the peptide mixture. The mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. Pd/C type 39 (0.007 eq by weight) was added to the mixture to increase reaction kinetics. At the end of the deprotection, the catalyst was filtered off and washed with 13.6 eq of MeOH. The deprotected peptide was then precipitated in 71 eq of iPrOH. After about 40 min, the peptide was filtered and washed with 35 eq of iPrOH. The peptide was dried below 20 °C and was then ready for solubilization in water and spray-drying.
Procedure 4H
This Procedure is for a variant of Procedure 4A. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 24.8 eq of water and 13.6 eq of EtOAc. Pd/C type 39 (0.025 eq by weight) was added to the peptide mixture. The mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (19 hrs), the catalyst was removed by filtration and washed with 5.3 eq of water and 2.9 eq of EtOAc. The biphasic mixture was then decanted to remove the upper organic layer. The aqueous layer was diluted with water to reach an H-Gly-MePro-Glu-OH concentration suitable for spray-drying the solution.
Example 2: Alternative Trofinetide Manufacturing Process
An alternative method for synthesis of Trofinetide is based on U.S. Patent No.
8,546,530 adapted for a tripeptide as follows.
The persilylated compounds used to synthesis Formula (Ia) (trofinetide) are obtained by silylating a corresponding peptide or amino acid by reaction with a silylating agent, optionally in an organic solvent. The persilylated peptide or amino acid can be isolated and purified if desired. One can use the persilylated peptide or amino acid in situ, e.g. by combining a solution containing persilylated peptide or amino acid with a solution containing, optionally activated, peptide or amino acid.
In step 2, the persilylated compound of an amino acid is obtained by silylating a corresponding amino acid (for example, H-MePro-OH) by reaction with a silylating agent, optionally in an organic solvent. The persilylated amino acid can be isolated and purified if desired. One can use the persilylated amino acid in situ, e.g. by combining a solution containing the persilylated amino acid with a solution containing, optionally activated, amino acid (for example, Z-Gly-OH).
In step 3, the persilylated compound of an amino acid is obtained by silylating a corresponding amino acid (for example, H-Glu-OH) by reaction with a silylating agent, optionally in an organic solvent. The persilylated amino acid or peptide can be isolated and purified if desired. It is however useful to use the persilylated amino acid or peptide in situ, e.g. by combining a solution containing the persilylated amino acid with a solution containing, optionally activated (for example, by using EDC.HCl and Oxyma Pure), peptide (for example, Z-Gly-MePro-OH).
In the present invention, it is useful to use silylating agents, such as N-trialkylsilyl amines or N-trialkylsilyl amides, not containing a cyano group. Examples of such silylating reagents include N,O-bis(trimethylsilyl)acetamide (BSA), N,O-bis(trimethylsilyl)trifluoroacetamide, hexamethyldisilazane, N-methyl-N-(trimethylsilyl)acetamide (TMA), N-methyl-N-(trimethylsilyl)trifluoroacetamide, N-(trimethylsilyl)acetamide, N-(trimethylsilyl)diethylamine, N-(trimethylsilyl)dimethylamine, 1-(trimethylsilyl)imidazole, 3-(trimethylsilyl)-2-oxazolidone.
The reaction of step 2 is generally carried out at a temperature from 0 °C to 100 °C, optionally from 10 °C to 40 °C, and optionally from 15 °C to 30 °C.
The reaction of step 3 is generally carried out at a temperature from 0 °C to 100 °C, optionally from 10 °C to 60 °C, optionally from 15 °C to 50 °C.
In the reaction of step 2, generally 0.5 to 5 equivalents, optionally 1 to 3 equivalents, optionally about 1.5 to 2.5 equivalents of silylating agent are used relative to the molar amount of functional groups to be silylated. Use of 2 to 4 equivalents of silylating agent relative to the molar amount of functional groups to be silylated is also possible. “Functional groups to be silylated” means particular groups having an active hydrogen atom that can react with the silylating agent such as amino, hydroxyl, mercapto or carboxyl groups.
In the reaction of step 3, generally 0.5 to 5 equivalents, optionally 2 to 4.5 equivalents, optionally about 3 to 4 equivalents of silylating agent are used relative to the molar amount of functional groups to be silylated. Use of 2.5 to 4.5 equivalents of silylating agent relative to the molar amount of functional groups to be silylated is also possible.
It is understood that “persilylated” means an amino acid or peptide or amino acid analogue or peptide analogue in which the groups having an active hydrogen atom that can react with the silylating agent are sufficiently silylated to ensure that a homogeneous reaction medium for a coupling step is obtained.
In the process according to the invention, the reaction between the amino acid or peptide and the persilylated amino acid or peptide is often carried out in the presence of a carboxyl group activating agent. In that case the carboxylic activating reagent is suitably selected from carbodiimides, acyl halides, phosphonium salts and uronium or guanidinium salts. More optionally, the carboxylic activating agent is an acyl halide, such as isobutyl chloroformate or pivaloyl chloride or a carbodiimide, such as EDC.HC1 or DCC.
Good results are often obtained when using additional carboxylic activating reagents which reduce side reactions and/or increase reaction efficiency. For example, phosphonium and uronium salts can, in the presence of a tertiary base, for example, N,N-diisopropylethylamine (DIPEA) and triethylamine (TEA), convert protected amino acids into activated species. Other reagents help prevent racemization by providing a protecting reagent. These reagents include carbodiimides (for example, DCC) with an added auxiliary nucleophile (for example, 1-hydroxy-benzo triazole (HOBt), 1-hydroxy-azabenzotriazole (HOAt), or Suc-OH) or derivatives thereof. Another reagent that can be utilized is TBTU. The mixed anhydride method, using isobutyl chloroformate, with or without an added auxiliary nucleophile, is also used, as is the azide method, due to the low racemization associated with it. These types of compounds can also increase the rate of carbodiimide-mediated couplings. Typical additional reagents include also bases such as N,N-diisopropylethylamine (DIPEA), triethylamine (TEA) or N-methylmorpholine (NMM).
When the silylation is carried out in the presence of a solvent, said solvent is optionally a polar organic solvent, more optionally a polar aprotic organic solvent. An amide type solvent such as N,N-dimethylformamide (DMF) or N,N-dimethylacetamide (DMAC)
can be used. In the present invention for step 2, one can use an alkyl acetate solvent, in particular ethyl acetate is more particularly optional.
In the present invention for step 3, one can use a chlorinated hydrocarbon solvent or alkyl cyanide solvent, in particular dichloromethane or acetonitrile are more particularly optional.
In another embodiment, silylation is carried out in a liquid silylation medium consisting essentially of silylating agent and amino acid or peptide.
In the present invention, amino acid or peptide is understood to denote in particular an amino acid or peptide or amino acid analogue or peptide analogue which is bonded at its N-terminus or optionally another position, to a carboxylic group of an amino protected amino acid or peptide.
Example 3: Specifications for Compositions Containing Compounds of Formula (I)
1 ICH guideline Q3C on impurities: guideline for residual solvents
Example 4: Alternative Manufacturing of Trofinetide Example 1, Step 4, Procedure 4B
This Procedure is for a variant of Step 4, Procedure 4B. Z-Gly-MePro-Glu-OH (1 eq) was added in portions to Pd/C (0.027 eq by weight and containing 5% Pd by weight) in about 50 eq of water. The reaction mixture was hydrogenated at 20 °C at a pressure of 5 bar for at least 4 cycles of 4 hrs each. Pd/C (0.0027 eq by weight) was charged between cycles, as needed, to speed up the reaction. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC. Upon reaction completion the catalyst was removed by filtration, washed with water (12.5 eq) and the aqueous layer washed with EtOAc (about 14 eq). After phase separation, residual EtOAc was removed from the aqueous solution containing
trofinetide by sparging with nitrogen under vacuum at 20 °C for about 3 hrs. The aqueous solution was filtered. The final concentration of trofinetide was about 25 wt% and the solution was then ready for spray-drying to isolate the product.
Example 5: Alternative Composition of Trofinetide
A composition comprising a compound of Formula (I)
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (II):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, and/or a compound of Formula (III):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, wherein R1, R2, R3 and R4 independently are selected from the group consisting of hydrogen and C1-4 alkyl, provided that least one of R1, R2, R3 and R4 is C1-4 alkyl, and wherein the composition comprises at least 90 wt%, such as 91 wt%, 92 wt%, 93 wt%, 94 wt%, 95 wt%, 96 wt%, or 97 wt% of the compound of Formula (I) on an anhydrous basis.
Example 6: Alternative Composition of Trofinetide
A composition comprising a compound of Formula (Ia)
or a hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (II):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, and/or a compound of Formula (III):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, wherein R1, R2, R3 and R4 independently are selected from the group consisting of hydrogen and C1-4 alkyl, provided that least one of R1, R2, R3 and R4 is C1-4 alkyl, and wherein the composition comprises at least 90 wt%, such as 91 wt%, 92 wt%, 93 wt%, 94 wt%, 95 wt%, 96 wt%, or 97 wt% of the compound of Formula (Ia) on an anhydrous basis.
Example 7: A Product of Trofinetide
A product, including a kit containing a dosage form with instructions for use, comprising a compound of Formula (Ia)
or a hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (IIa)
or a hydrate, or pharmaceutically acceptable salt thereof, wherein the product comprises between 95 wt% and 105 wt%, such as 96 wt%, 97 wt%, 98 wt%, 99 wt%, 100 wt%, 101
wt%, 102 wt%, 103 wt%, or 104 wt% of the specified amount of the compound of Formula (Ia) in the product.
Example 8: A Product of Trofinetide
A product, including a kit containing a dosage form with instructions for use, comprising a compound of Formula (Ia)
or a hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (IIa)
or a hydrate, or pharmaceutically acceptable salt thereof, and additionally comprising one or more compounds selected from the group consisting of Formula (III), Formula (IIIa), Formula (IV), Formula (V), Formula (VI), Formula (VII), Formula (VIII), and Formula (IX), wherein the composition comprises between 95 wt% and 105 wt%, such as 96 wt%, 97 wt%, 98 wt%, 99 wt%, 100 wt%, 101 wt%, 102 wt%, 103 wt%, or 104 wt% of the specified amount of the compound of Formula (Ia) in the product.
Example 9: Analysis of Products and Compositions
The products and compositions disclosed herein may be analyzed by liquid chromatography, a suitable chromatographic method using UPLC, e.g. using materials and conditions such as Waters Acquity CSH C18, 1.7 µm, 150 x 2.1 mm column, water with 0.1 % TFA (mobile phase A), and water/ACN 70/30 + 0.1 % TFA (mobile phase B), ranging from (4% phase A/6% phase B to 100% phase B and flushed with 4% phase A/6% phase B).
Flow rate: 0.35 ml/min, Column temperature: 40 °C, autosampler temperature: 4 °C, injection volume: 4 ml (e.g. prepared by weighing about 10 mg of powder in a 10 ml volumetric flask and diluted to volume with water). Examples of detectors are UV (ultraviolet, UV 220 nm) and MS (mass spectrometry).
INDUSTRIAL APPLICABILITY
This invention finds use in the pharmaceutical, medical, and other health care fields.
PATENT
WO2014085480 ,
claiming use of trofinetide for treating autism spectrum disorders including autism, Fragile X Syndrome or Rett Syndrome.
EP 0 366 638 discloses GPE (a tri-peptide consisting of the amino acids Gly-Pro- Glu) and its di-peptide derivatives Gly-Pro and Pro-Glu. EP 0 366 638 discloses that GPE is effective as a neuromodulator and is able to affect the electrical properties of neurons.
W095/172904 discloses that GPE has neuroprotective properties and that administration of GPE can reduce damage to the central nervous system (CNS) by the prevention or inhibition of neuronal and glial cell death.
WO 98/14202 discloses that administration of GPE can increase the effective amount of choline acetyltransferase (ChAT), glutamic acid decarboxylase (GAD), and nitric oxide synthase (NOS) in the central nervous system (CNS).
WO99/65509 discloses that increasing the effective amount of GPE in the CNS, such as by administration of GPE, can increase the effective amount of tyrosine hydroxylase (TH) in the CNS for increasing TH-mediated dopamine production in the treatment of diseases such as Parkinson’s disease.
WO02/16408 discloses GPE analogs capable of inducing a physiological effect equivalent to GPE within a patient. The applications of the GPE analogs include the treatment of acute brain injury and neurodegenerative diseases, including but not limited to, injury or disease in the CNS.
Example
The following non-limiting example illustrates the synthesis of a compound of the invention, NN-dimethylglycyl-L-prolyl-L-glutamic acid.
All starting materials and other reagents were purchased from Aldrich;
BOC = tert-butoxycarbonyl; Bn = benzyl.
BOC-(γ-benzyl)-L-prolyl-L-glutamic acid benzyl ester
To a solution of BOC-proline [Anderson GW and McGregor AC: J. Amer. Chem.
Soc: 79, 6180, 1957] (10 mmol) in dichloromethane (50 ml), cooled to 0 °C, was added triethylamine (1.39 ml, 10 mmol) and ethyl chloroformate (0.96 ml, 10 mmol). The resultant mixture was stirred at 0 °C for 30 minutes. A solution of dibenzyl L-glutamate (10 mmol) was then added and the mixture stirred at 0 °C for 2 hours then warmed to room temperature and stirred overnight. The reaction mixture was washed with aqueous sodium bicarbonate and citric acid (2 mol l“1) then dried (MgS04) and concentrated at reduced pressure to give BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (5.0 g, 95%).
(7-Benzyl)-L-prolyl-L-glutamic acid dibenzyl ester
A solution of BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (3.4 g, 10 mmol), cooled to 0 °C, was treated with trifluoroacetic acid (25 ml) for 2 hr at room temperature. After removal of the volatiles at reduced pressure the residue was triturated with ether to give (γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (I).
N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
A solution of dicyclohexylcarbodiimide (10.3 mmol) in dichloromethane (10 ml) was added to a stirred and cooled (0 °C) solution of (7-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (10 mmol), TVN-dimethylglycine (10 mmol) and triethylamine
(10.3 mmol) in dichloromethane (30 ml). The mixture was stirred at 0 °C overnight and then at room temperature for 3 h. After filtration, the filtrate was evaporated at reduced pressure. The resulting crude dibenzyl ester was dissolved in a mixture of ethyl acetate (30 ml) and methanol (30 ml) containing 10% palladium on charcoal (0.5 g) then hydrogenated at room temperature and pressure until the uptake of hydrogen ceased. The filtered solution was evaporated and the residue recrystallized from ethyl acetate to yield the tri-peptide derivative.
It will be evident that following the method of the Example, and using alternative amino acids or their amides or esters, will yield other compounds of Formula 1.
PAPER
Tetrahedron (2005), 61(42), 10018-10035. (CLICK HERE)
The synthesis of ten proline-modified analogues of the neuroprotective tripeptide GPE is described. Five of the analogues incorporate a proline residue with a hydrophobic group at C-2 and two further analogues have this side chain locked into a spirolactam ring system. The pyrrolidine ring was also modified by replacing the γ-CH2 group with sulfur and/or incorporation of two methyl groups at C-5.
Graphical Abstract

PAPER
Bioorganic & Medicinal Chemistry Letters (2005), 15(9), 2279-2283
A series of GPE analogues, including modifications at the Pro and/or Glu residues, was prepared and evaluated for their NMDA binding and neuroprotective effects. Main results suggest that the pyrrolidine ring puckering of the Pro residue plays a key role in the biological responses, while the preference for cis or trans rotamers around the Gly-Pro peptide bond is not important.
Graphical abstract
A series of Pro and/or Glu modified GPE analogues is described. Compounds incorporating PMe and dmP showed higher affinity for glutamate receptors than GPE and neuroprotective effects similar to those of this endogenous tripeptide in culture hippocampal neurons exposed to NMDA.

PATENT
US 20060251649
WO 2006127702
US 20070004641
US 20080145335
WO 2012102832
WO 2014085480
US 20140147491
References
- ^ Bickerdike MJ, Thomas GB, Batchelor DC, Sirimanne ES, Leong W, Lin H, et al. (March 2009). “NNZ-2566: a Gly-Pro-Glu analogue with neuroprotective efficacy in a rat model of acute focal stroke”. Journal of the Neurological Sciences. 278 (1–2): 85–90. doi:10.1016/j.jns.2008.12.003. PMID 19157421. S2CID 7789415.
- ^ Cartagena CM, Phillips KL, Williams GL, Konopko M, Tortella FC, Dave JR, Schmid KE (September 2013). “Mechanism of action for NNZ-2566 anti-inflammatory effects following PBBI involves upregulation of immunomodulator ATF3”. Neuromolecular Medicine. 15 (3): 504–14. doi:10.1007/s12017-013-8236-z. PMID 23765588. S2CID 12522580.
- ^ Deacon RM, Glass L, Snape M, Hurley MJ, Altimiras FJ, Biekofsky RR, Cogram P (March 2015). “NNZ-2566, a novel analog of (1-3) IGF-1, as a potential therapeutic agent for fragile X syndrome”. Neuromolecular Medicine. 17 (1): 71–82. doi:10.1007/s12017-015-8341-2. PMID 25613838. S2CID 11964380.
- ^ Study Details – Rett Syndrome Study
- ^ Neuren’s trofinetide successful in Phase 2 clinical trial in Fragile X
| PHASE | STATUS | PURPOSE | CONDITIONS | COUNT |
|---|---|---|---|---|
| 3 | Enrolling by Invitation | Treatment | Rett’s Syndrome | 1 |
| 3 | Recruiting | Treatment | Rett’s Syndrome | 1 |
| 2 | Completed | Supportive Care | Injuries, Brain | 1 |
| 2 | Completed | Treatment | Fragile X Syndrome (FXS) | 1 |
| 2 | Completed | Treatment | Injuries, Brain | 1 |
| 2 | Completed | Treatment | Rett’s Syndrome | 2 |
| 2 | Terminated | Treatment | Concussions | 1 |
| 1 | Completed | Treatment | Brain Injuries,Traumatic | 2 |
| Legal status | |
|---|---|
| Legal status | US: Investigational New Drug |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 853400-76-7 |
| PubChem CID | 11318905 |
| ChemSpider | 9493869 |
| UNII | Z2ME8F52QL |
| Chemical and physical data | |
| Formula | C13H21N3O6 |
| Molar mass | 315.322 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]C[C@]1(CCCN1C(=O)CN)C(=O)N[C@@H](CCC(=O)O)C(=O)O | |
| InChI[hide]InChI=1S/C13H21N3O6/c1-13(5-2-6-16(13)9(17)7-14)12(22)15-8(11(20)21)3-4-10(18)19/h8H,2-7,14H2,1H3,(H,15,22)(H,18,19)(H,20,21)/t8-,13-/m0/s1Key:BUSXWGRAOZQTEY-SDBXPKJASA-N |
////////////Tofinetide , NNZ 2566, PHASE 2, PHASE 3. NEUREN, Amino Acids, Peptides, Proteins,
CC1(CCCN1C(=O)CN)C(=O)NC(CCC(=O)O)C(=O)O
J. Med. Chem. 2025, 68, 2147−2182
Trofinetide (Daybue). Trofinetide (8) was developed by Neuren Pharmaceuticals and Acadia Pharmaceuticals for the treatment of rare childhood neurodevelopmental disorders and
was approved by the USFDA in March 2023 for adults and pediatric patients two years of age or older with Rett syndrome. 59 In most cases, Rett syndrome is caused by loss of-function mutations in the X-linked gene that encodes methyl CpG-binding protein 2 (MeCP2). 60,61 MeCP2 is a critical transcriptional regulator required for normal neurological development. In the past, treatment of Rett syndrome has
been limited to symptom management based on knowledge from treating other conditions, 62
but new research has focused on targeting the underlying genetic cause and finding agents to
restore MeCP2 function. Trofinetide is an orally available synthetic analog of glycine-proline-glutamate (GPE), the Nterminal tripeptide metabolite of insulin like growth factor-1 (IGF-1). GPE has been shown to partially reverse Rett-like symptoms in MeCP2 deficient mouse models and trofenitide was developed to have an improved pharmakokinetic profile to GPE. 64 Its use has shown significant improvement over placebo in clinical trials.
Anumberofaccountsrelated to the preparation of trofinetide have been reported with various protecting group strategies, but they are moreamenabletosmall-scaleproductionduetoreagent selection and challenging isolations requiring column chromatography.65−67 A commercially viable synthesis of the drug has been described by researchers at Neuren Pharmaceuticals and is depicted in Scheme 12.68
This synthesis takes advantage of in situ silyl protection/deprotection during its amidation steps to
avoid lengthy protecting group manipulations. Activation of Cbz protected glycine 8.1 with hydroxysuccinimide 8.2 using EDCI provided 8.3 in 84% yield as a direct drop crystallization
(isolated by crystallization directly from the reaction mixture). This activated ester was then coupled with commercially available methyl proline 8.4 69 by first silylation of the carboxylic acid of the proline analog in situ with 8.5, then adding 8.3 to couple with the amine. Deprotection of the silyl ester during the
workup provided amide 8.6 in 79% yield. In a similar sequence, 8.6 was activated with Oxyma Pure and EDCI while in a second vessel 8.7 was silyl protected using 8.5. Subsequent combination of these streams followed by workup and crystallization gave amide 8.8 in good yield. Finally, Cbz deprotection was
accomplished using Pd/C and hydrogen. Trofinetide (8) was extracted into the aqueous layer and isolated by spray drying in 90% yield.
(60) Kyle, S. M.; Vashi, N.; Justice, M. J. Rett syndrome: a
neurological disorder with metabolic components. Open Biol. 2018,
8, No. 170216.
(61) Collins, B. E.; Neul, J. L. Rett syndrome and MECP2 duplication
syndrome: disorders of MeCP2 dosage. Neuropsychiatr. Dis. Treat.
2022, 18, 2813−2835.
(62) Fu, C.; Armstrong, D.; Marsh, E.; Lieberman, D.; Motil, K.; Witt,
R.; Standridge, S.; Nues, P.; Lane, J.; Dinkel, T.; et al. Consensus
guidelines on managing Rett syndrome across the lifespan. BMJ
Paediatr. Open 2020, 4, No. e000717.
(63) Neul, J. L.; Percy, A. K.; Benke, T. A.; Berry-Kravis, E. M.; Glaze,
D.G.;Marsh,E.D.;Lin,T.;Stankovic,S.;Bishop,K. M.;Youakim,J.M.
Trofinetide for the treatment of Rett syndrome: a randomized phase 3
study. Nat. Med. 2023, 29, 1468−1475.
(64) Neul, J. L.; Percy, A. K.; Benke, T. A.; Berry-Kravis, E. M.; Glaze,
D. G.; Peters, S. U.; Jones, N. E.; Youakim, J. M. Design and outcome
measures of LAVENDER, a phase 3 study of trofinetide for Rett
syndrome. Contemp. Clin. Trials 2022, 114, No. 106704.
(65) Glass, L.; Bickerdike, M. J.; Snape, M. F. Treatment of autism
spectrum disorders using glycyl-l-2-methylprolyl-l-glutamic acid. US
20140147491, 2014.
(66) Glass, L.; Bickerdike, M. J.; Snape, M. F. Treatment of autism
spectrum disorders using glycyl L2012102832, 2012.-2-methylprolyl
L-glutamic acid. WO 2012102832, 2012
(67) Brimble, M.A.;Harris, P. W.R.;Sieg, F. Preparation of analogsof
glycyl-prolyl-glutamate as neuroprotective agents. US 20080145335,
2008
(68) Blower, C.; Peterson, M.; Shaw, J. M.; Bonnar, J. A.; Moniotte, E.
D. F. P.; Bousmanne, M. B. C.; Betti, C.; Decroos, K. W. L.; Ayoub, M.
Compositions of trofinetide. WO 2021026066, 2021.
(69) Beck, A. K.; Blank, S.; Job, K.; Seebach, D.; Sommerfeld, T.
Synthesis of (S)-2-methylproline: a general method for the preparation
of α-branched amino acids (L.-proline, 2-methyl-). Org. Synth. 1995, 72, 62−73

.
IMRECOXIB
Imrecoxib (Hengyang)
CHINA 2012 osteoarthritis2H-Pyrrol-2-one, 1,5-dihydro-3-(4-methylphenyl)-4-[4-(methylsulfonyl)phenyl]-1-propyl-
3-(4-Methylphenyl)-4-[4-(methylsulfonyl)phenyl]-1-propyl-1,5-dihydro-2H-pyrrol-2-one395683-14-4[RN]
Imrecoxib was approved by China Food and Drug Administration (CFDA) on May 20, 2011. It was developed and marketed as 恒扬® by HengRui Pharmaceuticals.
Imrecoxib is a selective COX-2 inhibitor indicated for treatment of osteoarthritis.
恒扬® is available as tablet for oral use, containing 100 mg of free Imrecoxib, and the recommend dose is 100 mg twice daily.
Common name: Imrecoxib; BAP-909; BAP 909; BAP909
Trademarks: Hengyang
Molecular Formula: C21H23NO3S
CAS Registry Number: 395683-14-4
IUPAC Name: 4-(4-methane-sulfonyl-phenyl)-1-propyl-3-p-tolyl-1,5-dihydropyrrol-2-one
Molecular Weight: 369.48
SMILES: O=C1N(CCC)CC(C2=CC=C(S(=O)(C)=O)C=C2)=C1C3=CC=C(C)C=C3
Mechanism: COX-2 Inhibitor; Cyclooxygenase-2 Inhibitor
Activity: Treatment of Osteoarthritis; Analgesic; Antipyritic; Antiinflammatory Drug
Status: Launched 2011 (China)
Originator: HengRuiDrug Name:ImrecoxibResearch Code:BAP-909Trade Name:恒扬®MOA:Selective cyclooxygenase-2 (COX-2) inhibitorIndication:Osteoarthritis (OA)Status:ApprovedCompany:HengRui (Originator)Sales:ATC Code:Approved Countries or Area
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2011-05-20 | Marketing approval | 恒扬 | Osteoarthritis (OA) | Tablet, Film coated | 100 mg | HengRui |
Reference:1. US7112605B2.Route 2
Reference:1. CN102206178A.
2. Chinese Chem. Lett. 2001, 12, 775-778.Route 3
Reference:1. CN104193664A.
Imrecoxib | NSAID | Treatment of Osteoarthritis | COX-2 Inhibitor
Imrecoxib [4-(4-methane-sulfonyl-phenyl)-1-propyl-3-p-tolyl-1,5-dihydropyrrol-2-one] is a novel and moderately selective cyclooxygenase-2 (COX-2) inhibitor that possesses anti-inflammatory effect by inhibition of COX-2 mRNA expression. It belongs to the family of non-steroid anti-inflammtory drugs (NSAIDs). Imrecoxib was found to inhibit COX-1 and COX-2 with IC50 value of 115 ± 28 nM and 18 ± 4 nM, respectively [1].
| Imrecoxib: 2D and 3D Structure |
Imrecoxib effectively inhibited carrageenan-induced acute inflammation at the doses of 5, 10, and 20 mg-kg-1 ig and adjuvant-induced chronic inflammation at the doses of 10 and 20 mg-kg -1·d-1 ig.
NSAIDs and Imrecoxib:
Non-steroidal anti-inflammatory drugs (NSAIDs) are used extensively for the treatment of inflammatory conditions, including pain-releasing, anti-pyretic and rheumatoid arthritis. These functions are believed to inhibit the enzyme cyclooxygenase (COX) that is involved in the biosynthesis of prostaglandins G and H from arachidonic acid. So far two isozymes of COX are known: COX-1 and COX-2. COX-1 is constitutively produced in a variety of tissues and appears to be important to the maintenance of normal physiological functions, including gastric and renal cytoprotection. The COX-2 is an inducible isozyme, which is produced in cells under the stimulation of endotoxins, cytokines, and hormones and catalyzes the production of prostaglandins which cause inflammation.
The currently therapeutic use of NSAIDs has been associated with the inhibition of both COX-1 and COX-2 and causes well-known side effects at the gastrointestinal and renal level. Therefore, the selective COX-2 inhibitors could provide anti-inflammatory agents devoid of the undesirable effects associated with classical, nonselective NSAIDs. In addition, COX-2 is over-expressed in colon cancer tissue. COX-2 inhibitors possess potential prophylactic and therapeutic application to colon cancer.
Imrecoxib is designed in a manner such that it has “moderate selectivity” for COX-2 over COX-1. This balanced inhibition to both COX-1 and COX-2 was pursued to maintain the homeostasis of the two enzymes in the body,which is presumably critical to normal functions of the cardiovascular system.
Imrecoxib was launched in China with the trade name of Hengyang for the treatment of osteoarthritis in May 2011. Hengyang is available as tablet for oral use, containing 100 mg of free Imrecoxib, and the recommend dose is 100 mg twice daily.
SYN
Imrecoxib Synthesis
Chin Chem Lett 2001, 12, 775-778 (also Ref 2. This route is quoted as industrial method in various texts)
CN104193664A (an improvement here as Br is replaced with Cl)

US7112605B2 (primary reference for synthesis routes)

Identification:

| 1H NMR (Estimated) for Imrecoxib |
Experimental: 1H-NMR (CDCl3, TMS, 400MHz) 1.008 (3H, t, J = 7.2Hz), 1.701-1.756 (2H, m), 2.376 (3H, s), 3.078 (3H, s), 3.575 (2H, t, J = 7.2Hz), 4.317 (2H, s), 7. 175 (2H, d, J = 8.0Hz), 7.294 (2H, d, J = 8.0Hz), 7.505 (2H, t, J = 6.8Hz), 7.870 (2H, t, J = 6.8Hz)
Sideeffects:
Being a mild COX-2 inhibitor, it is expected not to cause any serious cardiovascular risks. Similarly, it should not have any serious gastrointestinal problems too, as it not a good inhibitor of COX-1. None of the reports though have listed any serious adverse event reported by patients in the clinical trials.
References:
- Cheng, G. F.;et. al. Imrecoxib: A novel and selective cyclooxygenase 2 inhibitor with anti-inflammatory effect. Acta Pharmacol Sin 2004, 25(7), 927-931.
- Zhang, F.;et. al.Method for preparing imrecoxib. CN102206178A
- Chao, W.;et. al. Synthesis method of imrecoxib. CN104193664A
- Bai, A. P.;et. al. Synthesis and in vitro Evaluation of a New Class of Novel Cyclooxygenase-2 Inhibitors: 3, 4-diaryl-3-pyrrolin-2 ones.Chin Chem Lett 2001, 12, 775-7785.
- Guo, Z. Discovery of imrecoxib. Chin J New Drugs2012, 21, 223.
- Guo, Z.;et. al. Sulfonyl-containing 3,4-diaryl-3-pyrrolin-2-ones, preparation method, and medical use thereof. US7112605B2
![MS 2 spectrum of the [M þ H] þ ion (m/z 370) of imrecoxib (inset, full-scan mass spectrum).](https://www.researchgate.net/profile/Dafang_Zhong/publication/6930659/figure/fig4/AS:394580018122760@1471086616006/MS-2-spectrum-of-the-M-th-H-th-ion-m-z-370-of-imrecoxib-inset-full-scan-mass.png)
SYN
Imrecoxib (Hengyang)
Imrecoxib, a new non-steroid anti-inflammtory drug (NSAID), was launched in China with the trade name of Hengyang for the treatment of osteoarthritis in 2012. It was originally designed and synthesized by Guo and co-workers at the Institute of Materia Medica (IMM) of the Chinese Academy of Medical Sciences in collaboration with Hengrui Pharmaceuticals.88 Imrecoxib, which is a moderately selective COX-2 inhibitor (with IC50 values against COX-1 and COX-2 being 115 ± 28 and 18 ± 4 nM, respectively),89 is the subject of twwo synthetic routes reported across several publications.90–93
The most likely process-scale route to this drug is described in Scheme 15, 93 which began with 2-bromo-40 -(methylsulfonyl)-acetophenone (84) and p-tolylacetic acid (85) as starting materials. In the presence of base, a-bromoketone 84 was treated with acid 85 which resulted in lactone 86 in 72% yield across the two-step sequence. Exposure of lactone 86 with propylamine triggered a ring-opening-ring closing reaction, which resulted in imrecoxib (XIII) directly in 85% yield.93

88. Guo, Z. R. Chin. J. New Drugs 2012, 21, 223.
89. Chen, X. H.; Bai, J. Y.; Shen, F.; Bai, A. P.; Guo, Z. R.; Cheng, G. F. Acta Pharmacol. Sin. 2004, 25, 927.
90. Bai, A. P.; Guo, Z. R.; Hu, W. H.; Shen, F.; Cheng, G. F. Chin. Chem. Lett. 2001, 12, 775.
91. Guo, Z.; Cheng, G.; Chu, F.; Yang, G.; Xu, B. CN Patent 1134413 C, 2001.
92. Guo, Z.; Cheng, G.; Chu, F. US Patent 2004/0029951 A1, 2004.
93. Zhang, F. Y.; Shen, X. M.; Sun, P. Y. CN Patent 102206178 A, 2011
Patent
CN 111747879
PATENT
CN 111747874
CN 111747873
CN 110386891
CN 109553564
CN 109553563
CN 108997188
CN 108947884
CN 108912030
CN 108864003
CN 108707100
CN 107586268
CN 104193664
CN 102206178
CN 101774958
US 20040029951
PATENT
CN 109678775
https://patents.google.com/patent/CN102206178A/en
Ai Rui former times cloth (N-n-propyl-3-p-methylphenyl-4-is to methylsulfonyl phenyl-3-pyrrolidin-2-one) is the nonsteroidal anti-inflammatory drug that a kind of appropriateness suppresses COX-2; put down in writing the synthetic method of Ai Rui former times cloth in the prior art (US20040029951), may further comprise the steps:
1) is raw material with the methylsulfonyl methyl phenyl ketone, makes alpha-brominated methylsulfonyl methyl phenyl ketone through bromo;
2) sodium borohydride reduction of alpha-brominated methylsulfonyl methyl phenyl ketone obtains the Styrene oxide 98min. derivative;
3) reaction of Styrene oxide 98min. derivative and Tri N-Propyl Amine generates N-n-propyl-capable biology of beta-hydroxyphenyl ethamine;
4) tolyl-acetic acid and the reaction of excessive thionyl chloride are generated the methylbenzene Acetyl Chloride 98Min.;
5) methylbenzene Acetyl Chloride 98Min. and the capable biological respinse of N-n-propyl-beta-hydroxyphenyl ethamine are generated N-n-propyl-N-[2-hydroxyl-2-to the methylsulfonyl styroyl] phenylacetamide;
6) Jone ‘ s reagent or pyridine chromium trioxide oxidation N-n-propyl-N-[2-hydroxyl-2-are to the methylsulfonyl styroyl] phenylacetamide obtains the capable biology of oxo phenylacetamide;
7) the above-mentioned oxo phenylacetamide of condensation makes end product Ai Rui former times cloth under the alkaline medium effect.
Because existing preparation method’s route is longer, and relate to reduction, oxidation, steps such as acid amides coupling, solvent load is big, the cost height, particularly to use oxygenants such as Jone ‘ s reagent or pyridine chromium trioxide in the oxidation step, low and the product of this oxidation step productive rate is difficult for separation and purification, and is difficult to control because chromium metal residual quantity control criterion in bulk drug is extremely strict, thereby makes this preparation method be difficult to be applicable to scale operation.
Synthetic route 1
Step 1), preparation alpha-brominated methylsulfonyl methyl phenyl ketone (III)
51.0g 4-methylsulfonyl methyl phenyl ketone and 760mL acetic acid is added to has magnetic agitation, in three mouthfuls of glass flask of the 1000mL of thermometer and constant pressure funnel.Be heated to 40 ℃, beginning slowly drips 41.1g liquid bromine, after dripping, continues to stir 30 minutes at 40 ℃.Reaction solution 50 ℃ concentrate after, add entry, stir, filter, washing, oven dry obtains the thick product of 70.5g, adds ethyl acetate/normal hexane mixed solvent, reflux 1 hour, slowly be cooled to 25 ℃, filtering drying gets the alpha-brominated methylsulfonyl methyl phenyl ketone of 56.5g off-white color solid (III), yield 80.0%.
1H-NMR(CDCl 3,TMS,400MHz):3.120(3H,s),4.485(2H,s),8.101(2H,dd,J=2.0Hz),8.191(2H,dd,J=2.0Hz)
MS(M+1):279.05
Step 2), prepare 4-(4-methylsulfonyl phenyl)-3-(4-aminomethyl phenyl)-2,5-dihydrofuran-2-ketone (II)
Experiment condition A
With the alpha-brominated methylsulfonyl methyl phenyl ketone of 44.3g (III), 24.0g 4-methylphenyl acetic acid and 600mL acetonitrile are added to and have magnetic agitation, in the 500mL there-necked flask of thermometer and constant pressure funnel.Be added dropwise to the 24.0mL triethylamine by constant pressure funnel, temperature is controlled at 25 ℃, after adding, continues to stir 1 hour.Add the 36.0mL triethylamine again, reaction solution is heated to 75 ℃, stirring reaction 18 hours.Cool to 25 ℃, concentrate, add ethyl acetate, washing, organic phase concentrates the back and adds ethyl acetate and ethanol, stirs, and filters and obtains 28.0g light yellow solid compound (II), yield 53.4%.
1H-NMR(CDCl 3,TMS)2.398(3H,s),3.091(3H,s),5.192(2H,s),7.216(2H,d,J=8.0Hz),7.292(2H,d,J=8.0Hz),7.543(2H,d,J=8.0Hz),7.933(2H,d,J=8.0Hz)
MS(M+1):329.02
Similarly, compound (II) can prepare under experiment condition B, C, D.
Experiment condition B
With the alpha-brominated methylsulfonyl methyl phenyl ketone of 5.0g (III), 2.7g 4-methylphenyl acetic acid and 70mL acetonitrile are added to and have magnetic agitation, in the 100mL there-necked flask of thermometer and constant pressure funnel.Be added dropwise to the 2.3mL tetramethyl guanidine by constant pressure funnel, temperature is controlled at 20 ℃, after adding, continues to stir 1.5 hours.Add the 4.6mL tetramethyl guanidine again, 20 ℃ of stirring reactions 2 hours.Concentrate, add ethyl acetate, washing, organic phase concentrates the back and adds ethyl acetate and ethanol, stirs, and filters and obtains 2.5g light yellow solid compound (II), yield 42.0%.
Experiment condition C
With the alpha-brominated methylsulfonyl methyl phenyl ketone of 1.85g (III), 1.0g 4-methylphenyl acetic acid and 20mL ethanol are added to and have magnetic agitation, in the 50mL there-necked flask of thermometer and constant pressure funnel.Be added dropwise to the 1.0mL triethylamine by constant pressure funnel, temperature is controlled at 25 ℃, after adding, continues to stir 3 hours.Add the 2.0mL triethylamine again, 80 ℃ of stirring reactions 18 hours.Concentrate, add ethyl acetate, washing, organic phase concentrates the back and adds ethyl acetate and ethanol, stirs, and filters and obtains 0.83g light yellow solid compound (II), yield 38.1%.
Experiment condition D
With the alpha-brominated methylsulfonyl methyl phenyl ketone of 1.0g (III), 0.54g 4-methylphenyl acetic acid and 12mL acetonitrile are added to and have magnetic agitation, in the 50mL there-necked flask of thermometer and constant pressure funnel.Add 1.0g salt of wormwood, 25 ℃ were reacted 2 hours.50 ℃ of stirring reactions are 5 hours then.Concentrate, add ethyl acetate, washing, organic phase concentrates the back and adds ethyl acetate and ethanol, stirs, and filters and obtains 0.13g light yellow solid compound (II), yield 11%.
Step 3), preparation N-n-propyl-3-p-methylphenyl-4-are to methylsulfonyl phenyl-3-pyrrolidin-2-one (Ai Rui former times cloth (I))
Experiment condition A
With the 25.0mL Tri N-Propyl Amine, be added drop-wise in the 17.5mL acetic acid at 10 ℃, add the back and stir, in the Tri N-Propyl Amine acetate that generates, add 10.0g compound (II).Under the nitrogen protection, be heated to 160 ℃, stirring reaction 8 hours.Cool to 40 ℃, add methylene dichloride and water, standing demix.Organic phase concentrates in the residue of back and adds ethanol, and reflux cools to 25 ℃, filters, and oven dry obtains 8.2g white solid product compound (I), yield 72.8%.
1H-NMR(CDCl 3,TMS,400MHz)1.008(3H,t,J=7.2Hz),1.701-1.756(2H,m),2.376(3H,s),3.078(3H,s),3.575(2H,t,J=7.2Hz),4.317(2H,s),7.175(2H,d,J=8.0Hz),7.294(2H,d,J=8.0Hz),7.505(2H,t,J=6.8Hz),7.870(2H,t,J=6.8Hz)
MS(M+1):370.17
Similarly, compound (I) can prepare under experiment condition B, C, D.
Experiment condition B
2.9g Tri N-Propyl Amine hydrochloride and 1.0g compound (II) are mixed, under the nitrogen protection, be heated to 170 ℃, stirring reaction 2 hours.Cool to 40 ℃, add methylene dichloride and water, standing demix.Organic phase concentrates in the residue of back and adds ethanol, and reflux cools to 25 ℃, filters, and oven dry obtains 0.9g white solid product compound (I), yield 80.0%.
Experiment condition C
Digest compound (II) with 2.0,3 milliliters of Tri N-Propyl Amines, 1.75 gram Tri N-Propyl Amine hydrochlorides add in the tube sealing of nitrogen protections, are heated to 140 ℃, react 20 hours.Be cooled to room temperature, add methylene dichloride and water, standing demix.Organic phase concentrates in the residue of back and adds ethanol, and reflux cools to 25 ℃, filters, and oven dry obtains 1.8g white solid product compound (I), yield 80.0%.
Experiment condition D
With the 0.5mL Tri N-Propyl Amine, be added drop-wise in the 0.35mL acetic acid at 10 ℃, add the back and stir, in the Tri N-Propyl Amine acetate that generates, add 0.5g compound (II).Under the nitrogen protection, be heated to 120 ℃, stirring reaction 4 hours.Cool to 40 ℃, add methylene dichloride and water, standing demix.Obtain 0.14g compound (I) after organic phase is concentrated and purified, yield 24.2%.Publication numberPriority datePublication dateAssigneeTitleCN104072467A *2014-07-072014-10-01太仓博亿化工有限公司Synthesis method of 5-chloro-2-benzofuranyl-p-chlorophenyl-oneCN104193664A *2014-08-222014-12-10山东铂源药业有限公司Synthesis method of imrecoxibCN107586268A *2016-07-072018-01-16江苏恒瑞医药股份有限公司A kind of preparation method of imrecoxib and its intermediateCN108864003A *2018-06-152018-11-23江苏美迪克化学品有限公司A kind of preparation method of imrecoxib intermediate and imrecoxibCN108947884A *2018-06-292018-12-07江苏美迪克化学品有限公司A kind of Preparation Method And Their Intermediate of imrecoxibCN109553564A *2017-09-252019-04-02江苏恒瑞医药股份有限公司A kind of purification process of imrecoxibCN109678775A *2017-10-182019-04-26江苏恒瑞医药股份有限公司A kind of crystal form and preparation method thereof of COX-2 selective depressantCN107586268B *2016-07-072021-01-19江苏恒瑞医药股份有限公司Preparation method of dapoxib and intermediate thereofPublication numberPriority datePublication dateAssigneeTitleUS5489693A *1992-04-281996-02-06Linz; GuenterCyclic imino derivatives, pharmaceutical compositions containing these compounds and processes preparing themCN101386590A *2007-09-132009-03-18中国医学科学院药物研究所Pyrrolidone containing hydroxymethyl and carboxyl, preparation method and medicament composition and use thereofCN101497580A *2009-01-092009-08-05华南理工大学HIV-1 inhibitor 2-pyrrolidinone derivative, as well as synthesizing method and use thereof
PAPER
Chinese Chemical Letters (2001), 12(9), 775-778.
PATENT
CN 110386891,

CLIP
For Chinese drugmaker Hengrui, R&D plans pan out
Ambitious program to launch innovative drugs starts to pay off for generics producerby Jean-François TremblayJULY 17, 2017 | APPEARED IN VOLUME 95, ISSUE 29

Credit: Jean-François Tremblay/C&ENHengrui recently invested in a custom-made phage-display library screening system for its Shanghai lab.
Launching their own innovative pharmaceuticals is a common goal for managers of generic drug firms. But it remains a dream for many. Jiangsu Hengrui Medicine, one of China’s largest generic drug makers, has advanced further than most. It has already launched two of its own drugs in China and licensed rights to another to a U.S. firm.
JIANGSU HENGRUI MEDICINE AT A GLANCE
▸ Headquarters: Lianyungang, Jiangsu, China
▸ 2016 sales: $1.6 billion
▸ 2016 profits: $390 million
▸ Employees: More than 13,000, 2,000 of whom work for a Shanghai-based unit developing and commercializing innovative drugs
▸ Innovative drug R&D staff: 800
Obtaining these results required substantial resources, though. Back in 2004, Hengrui built a large R&D lab in Shanghai, hired world-class researchers to lead it, equipped the facility with the latest instruments, and staffed it with hundreds of scientists.
Initially, the project looked like a money pit. In Chinese industry circles, many doubted that it would amount to anything. But revenues from the company’s innovative drugs are starting to pour in, and R&D at Hengrui is well on its way to financial sustainability.
Over the past 10 years, China has made great strides in growing an innovative drug industry. For all the talk, cynics say, China has yet to foster a blockbuster with $1 billion or more in annual sales. But as Hengrui and other Chinese firms launch their own drugs at home and license the foreign rights to others, it is becoming clear that an innovative drug industry is taking root.
“Producing generic drugs funds our R&D,” says Weikang Tao, a Hengrui vice president who doubles as chief executive officer of Shanghai Hengrui, the company’s innovative drug subsidiary.
Overall, Hengrui invests more than 10% of its sales in R&D, “which is big by Chinese standards,” Tao says. The drug giant Pfizer by comparison spent about 15% of its sales on R&D in 2016. With sales of $1.6 billion last year, Hengrui does most of its business in China. But it also exports finished drugs to the U.S., making it one of the few Chinese firms to have the U.S. Food & Drug Administration’s okay to do so.
Hengrui was formed in 1970 as a state-owned company. It began investing in its own R&D in 2004 and has since cultivated an innovative drug subsidiary that employs 2,000 people, including more than 800 at a Shanghai lab and about 20 at a subsidiary in Princeton, N.J. Other staffers work in the usual functions found in an innovative drug firm: clinical trial management, regulatory affairs, marketing and sales, and so on.
The Shanghai subsidiary recruits in China and internationally. Tao, who joined Hengrui in 2014, is a Chinese-trained physician who earned a Ph.D. in molecular and cell biology at the University of Medicine & Dentistry of New Jersey. He focused on tumor cell biology during a postdoc at Princeton University and worked in research at Merck & Co. for 10 years. Hengrui is constantly hiring, he notes.
Hengrui’s research facilities appear to be well equipped. Earlier this year the firm opened a biologics drug lab and a pilot plant for process development in Shanghai. “We spent nearly $7 million just on equipment for the biologics lab,” Tao says.

The lab is equipped with a custom-made automated phage-display library screening system that speeds up the process of discovering antibody drugs. “The machine can do automatically in a few hours what would otherwise take days for several scientists,” says Jiakang Sun, group leader of in vivo pharmacology at Shanghai Hengrui Pharmaceutical. With the phage-display system, Sun adds, a library displaying millions of human antibodies can be screened in vitro to find antibodies that bind to a specific antigen.
However well-staffed and well-equipped, Hengrui’s labs are still smaller than those of Merck or other major drug firms. But Hengrui has made notable strides recently. In 2015, it became the first Chinese firm to license a drug candidate to a U.S. firm. Incyte agreed to pay $25 million up front, and several hundred million dollars more once certain milestones are met, for the rights outside China and Taiwan to camrelizumab, a cancer treatment in Phase III human clinical trials in China.

In China, Hengrui’s priority market, the firm launched the osteoarthritis treatment imrecoxib in 2011 and the gastric cancer drug apatinib in 2014. The two will eventually achieve combined annual sales of $160 million, Hengrui expects.
Together with the licensing deal with Incyte, this will allow the firm to nearly recoup its R&D investment. Launching a few more compounds, particularly in the U.S., would make innovative R&D at Hengrui solidly profitable. The company is making good progress in that direction. A neutropenia treatment awaits final market approval in China, and five others have reached Phase III trials. Hengrui also has drugs in Phase I clinical trials in the U.S.
“I wouldn’t say that our lab is more productive than a lab operated by a multinational drug firm,” Tao says. Merck and other major players operate excellent facilities staffed by top people, he says. “But I would say that our researchers work very hard, and our decision-making at the top is very quick.”
Unlike biotech start-ups that tend to be built around groundbreaking technology, promising drug leads, or star researchers, Hengrui at first approached innovative drug development with a conservative strategy designed to reduce the risk of failure.
Relying on developmental compounds licensed from other organizations, the company initially aimed to develop drugs with the same mechanisms of action as others already on the market. Imrecoxib, for example, is part of the well-known family of COX-2 inhibitor anti-inflammatory drugs.
Later, it sought to invent compounds offering slight improvements over existing ones. Today, Hengrui is aiming to launch pharmaceuticals that are clearly superior to the competition. The company’s ultimate goal, Tao says, is to develop groundbreaking pharmaceuticals.
“We went from me-too to me-better to now best in class, and then we will do first in class,” he says.
And as Hengrui’s research strategy has become more ambitious, its scientists have broadened the range of diseases and drugs that they work on. Six or seven years ago, Tao says, Hengrui limited itself to the development of small-molecule drugs that treat cancer. Today the company is looking at small molecules, peptides, antibodies, antibody-drug conjugates, and other drug types to treat diseases as diverse as psoriasis and diabetes. “We have expanded our focus,” Tao says.
Most research is conducted in-house, Tao says. This includes medicinal chemistry, process chemistry, biology, drug metabolism, and pharmacokinetics. But the company leans on contract research firms for certain specific tasks, such as developing animal models. “They help accelerate our R&D,” Tao says.
Although Hengrui is a pioneer in launching new drugs in China, several other Chinese firms have made progress in advancing their own drug development. For instance, in the southern city of Dongguan, the generic drug producer HEC Pharma is conducting Phase II trials of the hepatitis C drug yimitasvir.
In Beijing, the biotech firm BeiGene just sold the U.S. company Celgene rights in much of the world to one of its immuno-oncology compounds for $263 million. Celgene also agreed to inject $150 million into BeiGene.
Chinese companies increasingly have the resources required to sustain innovative drug discovery and development, China watchers say. “Hengrui has the financial resources and the commitment to become a world-class innovative drugmaker,” says George Baeder, a former pharmaceutical industry executive who is now a director of China Global Insight, a California-based think tank.
But developing drugs and selling them are two different things, Baeder warns. “It is easy to underestimate the complexity a firm faces when moving into the arena of innovative medicines,” he says. “Chinese companies typically lack the capabilities in medical affairs, marketing, and sales needed to build a successful franchise.”
For the time being, Hengrui’s innovative drug subsidiary will stay focused on developing new drugs and not worry about the fine points of marketing them. Tao expects the former will keep his firm busy. “Don’t be surprised if several of our drugs begin clinical trials in the U.S., Europe, and Australia in the next year or two,” he says.
Rich pipeline
Hengrui boasts a diverse portfolio of drugs in late-stage development.
| China approval stage | Name | Application | Mechanism or target |
|---|---|---|---|
| Phase II clinical trials | Hetrombopaga | Idiopathic thrombocytopenia | Thrombopoietin receptor agonist |
| HR7056 | Anesthesia | na | |
| Pyrotiniba | Non-small cell lung cancer | EGFR/HER2 | |
| SHR3680b | Prostate cancer | Androgen receptor | |
| Phase III | Apatinib | Liver and non-small cell lung cancer | VEGFR-2 |
| Camrelizumabc | Cancer | PD-1 blocker | |
| Pyrotinib | HER2-positive breast cancer | EGFR/HER2 | |
| Retagliptin | Type 2 diabetes | Dipeptidyl peptidase 4 | |
| SHR3824 | Type 2 diabetes | SGLT2 inhibitor | |
| New drug application (China) | Mecapegfilgrastim | Neutropenia | PEG G-CSF |
| Launched | Apatinib | Gastric cancer | VEGFR-2 |
| Imrecoxib | Osteoarthritis | COX-2 inhibitor |
a In Phase I in U.S. b In Phase I in Australia. c The U.S. firm Incyte has acquired the rights to this drug outside China. na = not available. Source: Hengrui
////////////////Imrecoxib, Hengyang, CHINA 2012, osteoarthritis
Telacebec

Telacebec
- Molecular FormulaC29H28ClF3N4O2
- Average mass557.006 Da
Telacebec, IAP6, CAS No. 1334719-95-7телацебек [Russian] [INN]تيلاسيبيك [Arabic] [INN]特雷贝克105731334719-95-7[RN]55G92WGH3X
6-Chloro-2-ethyl-N-(4-{4-[4-(trifluoromethoxy)phenyl]-1-piperidinyl}benzyl)imidazo[1,2-a]pyridine-3-carboxamide
Imidazo[1,2-a]pyridine-3-carboxamide, 6-chloro-2-ethyl-N-[[4-[4-[4-(trifluoromethoxy)phenyl]-1-piperidinyl]phenyl]methyl]-Q203Q-203T56 AN DNJ C2 HG BVM1R D- AT6NTJ DR DOXFFF
Qurient Therapeutics and Russia licensee Infectex are developing telacebec, an oral formulation which targets QcrB subunit of the cytochrome bc1 complex, for treating multi drug resistant or extensively drug resistant Mycobacterium tuberculosis infection. Qurient is also investigating telacebec for treating buruli ulcer (an infection caused by Mycobacterium ulcerans ). In January 2021, a global phase II trial was expected to begin by December 2021 for the treatment of buruli ulcer.
syn
Angewandte Chemie, International Edition, 57(4), 1108-1111; 2018

PATENT
WO-2021018387
Novel crystalline forms of telacebec , processes for their preparation and compositions comprising them are claimed. Also claimed is their use for treating bacterial infection.
Different forms of 6-chloro-2-ethyl-AT-(4-(4-(4- (trifluoromethoxy)phenvDpiperidine-i-vDbenzvDimidazolT.2-alpyridine- 3-carboxamide
The present invention relates to different forms of the compound 6-chloro-2-ethyl-lV-(4-(4-(4-(trifhioromethoxy)phenyl)piperidine-i-yl)benzyl)imidazo[i,2-a]pyridine-3-carboxamide and to methods of making such forms/compounds. The present invention furthermore relates to mono-acid addition salts thereof, to methods of making such mono-acid addition salts and to pharmaceutical compositions comprising any of the aforementioned compounds. Furthermore, the present invention relates to uses of any of these compounds.
Tuberculosis as a disease continues to result in millions of deaths each year. Inadequate use of chemotherapy has led to an increasing number of drug resistant cases. This situation is likely to worsen with the emergence of extremely resistant strains to all currently known drugs. Current chemotherapy consists of compounds that directly target Mycobacterium tuberculosis, either by neutralizing general information pathways and critical processes such as RNA polymerization and protein synthesis inhibition or by interfering with mycobacterial specific cell envelop synthesis. The most widely used dedicated anti-tubercular drugs isoniazid, ethionamide, and pyriazin amide are pro-drugs that first require activation. They are administered to a patient for a course of several months. Patients infected with multi-drug resistant strains of M. tuberculosis may have to undergo combination therapies for extended periods of time.
WO 2011/113606 describes various anti-tubercular compounds and their use in the treatment of bacterial infections, including compound“Q203” which chemically is 6-chloro-2-ethyl-!V-(4-(4-(4-(trifluoromethoxy)phenyl)piperidine-i-yl)benzyl)imidazo[i,2-a]pyridine-3-carboxamide. In a publication by Pethe et al. (Nature Medicine, 19, 1157-1160 (2013), this compound is reported to be active against tuberculosis by interfering with the bacterial energy metabolism, inhibiting cytochrome bci activity which is an essential component of the electron transport chain required for synthesis of ATP.
Whilst the compound shows promise for future therapy of tuberculosis and related infections, there continues to be a need for forms thereof that are particularly suitable for pharmaceutical administration. In particular there is a need to provide forms that are showing an improved solubility in comparison to the free base of this compound. Furthermore, there is a need in the art to provide for forms that show an improved stability.
In a first aspect the present invention relates to a compound 6-chloro-2-ethyl-N-(4-(4-(4-(trifluoromethoxy)phenyl)piperidine-i-yl)benzyl)imidazo[i,2-a]pyridine-3-carboxamide ditosylate having the structure
PATENT
WO2011113606 .
WO 2017049321
WO 2012143796
PAPER
Scientific reports (2019), 9(1), 8608.
Angewandte Chemie, International Edition (2018), 57(4), 1108-1111.
European journal of medicinal chemistry (2017), 136, 420-427.
European Journal of Medicinal Chemistry (2017), 136, 420-427.
European journal of medicinal chemistry (2017), 125, 807-815.
Nature communications (2016), 7, 12393.
Nature medicine (2013), 19(9), 1157-60
PAPER
Journal of Medicinal Chemistry (2014), 57(12), 5293-5305.
https://pubs.acs.org/doi/10.1021/jm5003606J. Med. Chem. 2014, 57, 12, 5293–5305
Publication Date:May 28, 2014
https://doi.org/10.1021/jm5003606

A critical unmet clinical need to combat the global tuberculosis epidemic is the development of potent agents capable of reducing the time of multi-drug-resistant (MDR) and extensively-drug-resistant (XDR) tuberculosis therapy. In this paper, we report on the optimization of imidazo[1,2-a]pyridine amide (IPA) lead compound 1, which led to the design and synthesis of Q203 (50). We found that the amide linker with IPA core is very important for activity against Mycobacterium tuberculosis H37Rv. Linearity and lipophilicity of the amine part in the IPA series play a critical role in improving in vitro and in vivo efficacy and pharmacokinetic profile. The optimized IPAs 49 and 50 showed not only excellent oral bioavailability (80.2% and 90.7%, respectively) with high exposure of the area under curve (AUC) but also displayed significant colony-forming unit (CFU) reduction (1.52 and 3.13 log10 reduction at 10 mg/kg dosing level, respectively) in mouse lung.
6-Chloro-2-ethyl-N-(4-{4-[4-(trifluoromethoxy)phenyl]piperidin-1-yl}benzyl)imidazo[1,2-a]pyridine-3-carboxamide (50)
Mp = 164.0 °C; 1H NMR (400 MHz, CDCl3) δ 1.37 (t, J = 7.6 Hz, 3H), 1.82–1.97 (m, 4H), 2.64–2.70 (m, 1H), 2.80–2.87 (m, 2H), 2.93 (q, J = 7.6 Hz, 2H), 3.80–3.83 (m, 2H), 4.61 (d, J = 5.2 Hz, 2H), 6.00 (br t, J = 5.2 Hz, 1H), 6.96–6.99 (m, 2H), 7.15 (d, J = 8.0 Hz, 2H), 7.24–7.30 (m, 5H), 7.52 (dd, J = 9.6, 0.8 Hz, 1H), 9.53 (dd, J = 2.0, 0.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 13.3, 23.6, 33.4, 42.0, 43.3, 50.4, 115.4, 117.0, 121.2, 121.6, 121.9, 126.3, 128.2, 128.3, 128.7, 128.9, 144.5, 144.7, 147.7, 151.4, 151.5, 161.2; 19F NMR (376 MHz, CDCl3) δ 58.31 (s, 3F); LC/MS (ESI) m/z 557 [M + H]+; HRESIMS calcd for C29H29ClF3N4O2 [M + H]+ 557.1926, found 557.1918.



19F NMR (376 MHz, CDCl3) δ 58.31 (s, 3F);

13C NMR (100 MHz, CDCl3) δ 13.3, 23.6, 33.4, 42.0, 43.3, 50.4, 115.4, 117.0, 121.2, 121.6, 121.9, 126.3, 128.2, 128.3, 128.7, 128.9, 144.5, 144.7, 147.7, 151.4, 151.5, 161.2;

1H NMR (400 MHz, CDCl3) δ 1.37 (t, J = 7.6 Hz, 3H), 1.82–1.97 (m, 4H), 2.64–2.70 (m, 1H), 2.80–2.87 (m, 2H), 2.93 (q, J = 7.6 Hz, 2H), 3.80–3.83 (m, 2H), 4.61 (d, J = 5.2 Hz, 2H), 6.00 (br t, J = 5.2 Hz, 1H), 6.96–6.99 (m, 2H), 7.15 (d, J = 8.0 Hz, 2H), 7.24–7.30 (m, 5H), 7.52 (dd, J = 9.6, 0.8 Hz, 1H), 9.53 (dd, J = 2.0, 0.8 Hz, 1H);
CLIP
June 3, 2019. Qurient press release:
SEONGNAM-SI, South Korea–(BUSINESS WIRE)– Qurient Co. Ltd. today announced positive results from the Phase 2a EBA (early bactericidal activity) clinical trial for telacebec (Q203), a first-in-class, orally-available antibiotic for the treatment of tuberculosis (TB). Telacebec is a selective inhibitor with high specificity for the cytochrome bc1 complex of Mycobacterium tuberculosis. This complex is a critical component of the electron transport chain, and inhibition disrupts the bacterium’s ability to generate energy.
The EBA trial assessed the pharmacokinetics, safety, and activity of telacebec in three dose strength (100 mg, 200 mg and 300 mg) in the treatment of adult patients with pulmonary TB. Telacebec met the primary objective of rate of change in the time to positivity (TTP) in sputum over days 0 to 14. Telacebec was safe and well tolerated throughout the different dose strengths. Full results from EBA trial are expected to be presented at future scientific meetings.
Phase 2. EBA began July 2018 in South Africa. As of March 2019, study is active, not enrolling.
June 2018. Q203 has a non-proprietary name assigned: telacebec. USAN: -cebec Cytochrome bc1 complex inhibitors in Mycobacterium tuberculosis.
Phase 1. Description from clinicaltrials.gov: Randomized, double-blind, placebo-controlled, dose-escalation study in healthy male and female volunteers. Subjects randomly assigned to 1 of 7 treatment cohorts (Cohorts 1 – 7) of 8 subjects each, receiving either Q203 or placebo (6 active treatment : 2 placebo) in a fasting state. Dose escalation to the next cohort may be considered when at least 6 out of 8 subjects, in a cohort, completes all procedures and none of the subjects has a clinically significant adverse event (AE) that is being followed, or at the discretion of the PI if no drug-related serious adverse events (SAEs) have occurred. A food effect cohort will be enrolled to test administration of Q203 in a fed state, at 100 mg dose level (this dose level may change based on PK analysis results). Subjects who received 100mg dose in a fasting state will return and receive the second dose, with food. Subjects will be followed up for AEs, SAE or pregnancy for 30 days postdrug administration.
Related Links
Qurient Press Release. June 2019.Kalia NP et al. 2017. Exploiting the synthetic lethality between terminal respiratory oxidases to kill M. tuberculosis and clear host infection.. PNAS.114.7426
Related Links
- Qurient website
- 2013 Pethe K–Nature Medicine–Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis
- 2014 Kang S — JMC – Lead Optimization of a Novel Series of Imidazo[1,2-a]pyridine Amides Leading to a Clinical Candidate (Q203) as a Multi- and Extensively-Drug-Resistant Anti-tuberculosis Agent
//////////////Telacebec, IAP6, 1334719-95-7, PHASE 2, QURIENT, TUBERCULOSIS, телацебек , تيلاسيبيك , 特雷贝克 , Q 203
Iguratimod
Iguratimod
- Molecular FormulaC17H14N2O6S
- Average mass374.368 Da
- UNII-4IHY34Y2NVигуратимодإيغوراتيمود艾拉莫德
123663-49-0[RN]
3-Formylamino-7-methylsulfonylamino-6-phenoxy-4H-1-benzopyran-4-one
4IHY34Y2NV8176IGU
Methanesulfonamide, N-[3-(formylamino)-4-oxo-6-phenoxy-4H-1-benzopyran-7-yl]-
N-(3-Formamido-4-oxo-6-phenoxy-4H-chromen-7-yl)methanesulfonamide
product patent US4954518
Research Code:T-614
Trade Name:Iremod® / Kolbet® / Careram®
MOA:Nuclear factor NF-κB activation inhibitor
Indication:Rheumatoid arthritis
Status:Approved
Company:Simcere (Originator) , Taisho Toyama,EisaiSales:ATC Code:
Approved Countries or Area
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2012-06-29 | Marketing approval | Careram | Rheumatoid arthritis | Tablet, Film coated | 25 mg | Eisai | |
| 2012-06-29 | Marketing approval | Kolbet | Rheumatoid arthritis | Tablet, Film coated | 25 mg | Toyama Chemical, Taisho Toyama |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2011-08-15 | Marketing approval | 艾得辛/Iremod | Rheumatoid arthritis | Tablet, Film coated | 25 mg | Simcere |
Iguratimod was first approved by China Food and Drug Administration (CFDA) on August 15, 2011, then approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on June 29, 2012. It was developed by Simcere and marketed as 艾得辛®/Iremod® by Simcere and as Kolbet® by Taisho Toyama and by Eisai in Japan.
Iguratimod is a nuclear factor NF-κB activation inhibitor used in the treatment of rheumatoid arthritis.
Iremod® is available as tablet for oral use, containing 25 mg of free Iguratimod, and the recommended dose is 25 mg once daily or 25 mg at a time, twice daily.
Iguratimod: (Iremod)-First approval: 2009
Iguratimod is a disease-modifying antirheumatic drug (DMARD) that was approved for use in rheumatoid arthritis (RA) patients in China and Japan in 2009
Toyama Chemical , Taisho Toyama Pharmaceutical , Eisai , Simcere and Tianjin Institute of Pharmaceutical Research have codeveloped and launched iguratimod, an inflammatory cytokine and IL-6 gene inhibiting compound that also inhibits immunoglobulin production in B cells. Iguratimod is indicated for the oral treatment of rheumatoid arthritis.
Iguratimod is an anti-inflammatory small molecule drug used for the treatment of rheumatoid arthritis, together with methotrexate in Japan and China.[1] As of 2015 the biological target was not known, but it prevents NF-κB activation and subsequently selectively inhibits COX-2 and several inflammatory cytokines.[1]
Adverse effects include elevated transaminases, nausea, vomiting, stomach pain; rashes, and itchiness.[1]
It is a derivative of 7-methanesulfonylamino-6-phenoxychromones and is a chromone with two amide groups; it was first published in 2000.[1][2] It was submitted for regulatory approval in Japan in 2003; the application was withdrawn in 2009, and it was resubmitted with additional data in 2011 and approved for marketing in Japan in 2012.[1] Eisai and Toyama Chemical market it in Japan.[3] Approval was obtained in China in 2011 by Simcere, independently of the Japanese originators.[1][4]
During discovery and development it was called T-614 and it is marketed under the names Careram and Kolbet.[5]
Syn
Indian Pat. Appl., 2014MU01507

SYN


Reference:1. US4954518.Route 2
Reference:1. Chem. Pharm. Bull. 2000, 48, 131-139.
2. Chin. J. New. Drugs. 2006, 15, 2042-2044.
3. Shanghai Chem. Ind. 2007, 32, 22-24.Route 3
Reference:1. CN1462748A.
2. Chem. Pharm. Bull. 2000, 48, 131-139.
SYN
AU 8823489; CH 679397; FR 2621585; GB 2210879; JP 1995267943; US 4954518
The reduction of 3-nitro-4-phenoxyphenol (I) with Fe, aqueous HCl gives 3-amino-4-phenoxyphenol (II), which is acylated with methanesulfonyl chloride by means of pyridine in dichloromethane affording 3-(methylsulfonamido)-4-phenoxyphenol (III). The condensation of (III) with 3-chloropropionic acid (IV) by means of NaOH in water gives 3-[3-(methylsulfonamido)-4-phenoxyphenoxy]propionic acid (V), which is cyclized by means of polyphosphoric acid at 65-70 C yielding 7-(methylsulfonamido)-6-phenoxy-3,4-dihydro-2H-1-benzopyran-4-one (VI). The bromination of (VI) with Br2 in CHCl3 affords 3-bromo-7-(methylsulfonamido)-6-phenoxy-3,4-dihydro-2H-1-benzopyran-4-one (VII), which is treated with sodium azide in DMF at 70-75 C giving 3-amino-7-(methylsulfonamido)-6-phenoxy-2H-1-benzopyran-4-one (VIII). Finally, this compound is formylated with formic acid in acetic anhydride.
SYN
Chem Pharm Bull 2000,48(1),131
A preparative-scale synthetic route for T-614 has been reported: The reaction of 4-chloro-3-nitroanisole (I) with potassium phenolate in hot DMF gives 4-phenoxy-3-nitroanisole (II), which is reduced to the corresponding 3-amino compound (III) by treatment with Fe and HCl. The reaction of (III) with mesyl chloride in pyridine affords the sulfonamide (IV), which is acylated with 2-aminoacetonitrile (V) and AlCl3 in nitrobenzene/HCl giving the 2-aminoacetophenone (VI). Formylation of (VI) at the NH2 with acetic formic anhydride yields the formamide (VII), which is demethylated with AlCl3 and NaI in acetonitrile affording the phenol (VIII). Finally, this compound is cyclized with dimethylformamide dimethylacetal in DMF.
SYN
https://www.sciencedirect.com/science/article/abs/pii/S0968089614001230
Synthetic approaches to the 2012 new drugs
Hong X. Ding, … Christopher J. O’Donnell, in Bioorganic & Medicinal Chemistry, 2014

13 Iguratimod (Careram®, Iremod®)
Iguratimod, which was discovered by Toyama Pharmaceuticals and jointly co-developed with Eisai in Japan, was approved by the PMDA (Pharmaceuticals and Medical Devices Agency) of Japan on June 29, 2012 for the treatment of rheumatoid arthritis.83 This drug was also independently developed by Simcere Pharmaceutical Group and is marketed as Iremod® in China. The drug exhibited inhibitory effects on granuloma inflammation, and was shown to be efficacious for the prevention of joint destruction in adjuvant arthritis.84,85 While several synthesis of iguratimod have been published,86 the most likely scale synthesis, which does not require chromatographic purification, is described in Scheme 14.87
The synthesis began with commercially available 3-nitro-4-chloro anisole (78) which was reacted with potassium phenoxide (generated from phenol and potassium t-butoxide at 110 °C) to provide the corresponding nitrophenyl ether which was subsequently reduced and sulfonylated to furnish sulfonamide 79. Next, this diphenyl ether was submitted to a Friedel–Crafts reaction with aminoacetonitrile hydrochloride which gave rise to aminomethylacetophenone 80 in 90% yield. This aminoketone was then formylated with formic trimethylacetic anhydride 81 at room temperature to afford formamide 82 in 91% yield, and this material was immediately subjected to O-demethylation conditions with aluminum trichloride and sodium iodide in acetonitrile to give the phenol 83 in 95% yield. Finally, treatment of the aminomethyl acetophenone phenol 83 with N,N-dimethylformamide dimethylacetal in DMF at low temperatures furnished iguratimod (XII) in 87% yield
83. Eisai and Toyama Chemical Receive Approval to Market Anti-rheumatic Agent Iguratimod in Japan, 2012, http://www.eisai.com/news/news201239.html, [Access Date: 2012-July-29].
84. Tanaka, K.; Shimotori, T.; Makino, S.; Aikawa, Y.; Inaba, T.; Yoshida, C.; Takano, S. Arzneim.-Forsch. 1992, 42, 935.
85. Tanaka, K.; Makino, S.; Shimotori, T.; Aikawa, Y.; Inaba, T.; Yoshida, C. Arzneim.-Forsch. 1992, 42, 945.
86. Takano, S.; Yoshida, C.; Inaba, T.; Tanaka, K.; Takeno, R.; Nagaki, H.; Shimotori, T.; Makino, S. US Patent 4954518 A, 1990.
87. Inaba, T.; Tanaka, K.; Takeno, R.; Nagaki, H.; Yoshida, C.; Takano, S. Chem. Pharm. Bull. 2000, 48, 131.
SYN

https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/slct.202003553A Convenient Synthesis of Iguratimod‐Amine Precursor via NHC‐Catalyzed Aldehyde‐Nitrile Cross Coupling ReactionNithya MurugeshProf. Ramasamy KarvembuDr. Seenuvasan VedachalamFirst published: 24 November 2020https://doi.org/10.1002/slct.202003553
A protocol for the synthesis of iguratimod‐amine precursor has been developed using N‐heterocyclic carbene (NHC)‐catalyzed aldehyde‐nitrile cross coupling reaction with overall atom efficiency of 71 %. The first step involves a nucleophilic aromatic substitution (SNAr) of 1‐chloro‐4‐methoxy‐2‐nitrobenzene (1) with phenol to produce 4‐methoxy‐2‐nitro‐1‐phenoxybenzene (2) which further undergoes nitro reduction followed by mesylation to produce N‐(5‐methoxy‐2‐phenoxyphenyl)methanesulfonamide (4). Furthermore, it was subjected to Vilsmeier‐Haack formylation and demethylation (using BBr3) to produce N‐(4‐formyl‐5‐hydroxy‐2‐phenoxyphenyl)methanesulfonamide (6). Subsequently, O‐alkylation followed by NHC‐catalyzed aldehyde‐nitrile cross coupling yields the amine precursor of iguratimod (8).
N-(3-Amino-4-oxo-6-phenoxy-4H-chromen-7-yl)methanesulfonamide (8):4=4. S. Vedachalam, J. Zeng, B. K. Gorityala, M. Antonio, X.-W. Liu, Org. Lett. 2010, 12, 352–355.
Compound 7 (290 mg, 0.83 mmol) and triazolium carbene catalyst (34 mg, 0.1245 mmol) were dissolved in dry CH2Cl2 under N2 atmosphere. To this, DBU (24.7 µL, 0.16 mmol) was added at room temperature, and the mixture was stirred for 12 h. After the completion of reaction, the reaction mixture was dried, and the residue was purified by column chromatography to yield compound 8. Yield: 200 mg, 70 %; m. p. 162 ℃; 1H NMR (500 MHz, DMSO-d6): δ 8.25 (s, 1H), 7.96 (s, 1H), 7.78 (s, 1H), 7.45 (t, J = 8.0 Hz, 2H), 7.25–7.21 (m, 3H), 7.16 (s, 1H), 4.91 (s, 2H), 3.23 (s, 3H); 13C NMR (125 MHz, DMSO-d6): δ 171.2, 155.0, 152.1, 150.4, 137.9, 133.0, 132.8, 131.0 (2C), 125.9, 122.4, 121.1 (2C), 117.2, 110.0, 39.0; FTIR (KBr): v 3423, 3347, 1620, 1592, 1487, 1342, 1210, 1155, 970, 757 cm-1 ; HR-MS (ESI): m/z calcd. for C16H15N2O5S 347.0702, found 347.0714 [M+H]+ …..https://chemistry-europe.onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fslct.202003553&file=slct202003553-sup-0001-misc_information.pdf
Patent
WO 2021020481
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021020481&tab=FULLTEXT&_cid=P11-KKXGLE-06477-1The following formula (1)
[Chemical 1]
Iguratimod (chemical name: N- [3- (formylamino) -4-oxo-6-phenoxy-4H-chromen-7-yl] methanesulfonamide), which is indicated by, has excellent anti-inflammatory and antipyretic and analgesic effects. It is a very useful compound as a therapeutic agent exhibiting anti-arthritis and anti-allergic effects (see Patent Document 1).
A plurality of synthetic routes are known for the method for producing iguratimod and its derivatives (hereinafter, may be referred to as iguratimod derivatives as a term meaning both iguratimod and its derivatives), and all of them use intermediates of iguratimod derivatives. This is a stepwise manufacturing method. The present inventors have studied the stepwise production of intermediates of iguratimod derivatives of formulas (II) to (XI) by the following synthetic route to synthesize the iguratimod derivatives represented by formula (XII). doing.
[Chemical formula 2]R 1 = hydroxyl protecting group, R 2 = amino protecting group, Ar = aromatic ring group which may have a substituent, X 2 = halogen atom.[Chemical 3]
Ms = methylsulfonyl group[Chemical 4]
Production Example 1
(Production of igratimodo: Patent Documents 3 and 4)
N, N-dimethylformamide 150 mL, N, N-dimethylformamide dimethyl acetal 40.9 g (N, N-dimethylformamide dimethyl acetal ) in a 1000 mL four-necked flask equipped with two stirring blades having a diameter of 10 cm ( 343 mmol) was added, and the mixture was cooled to 10 ° C. with stirring. 8.2 g (137 mmol) of glacial acetic acid and 50.0 g (137 mmol) of formylaminomethyl (2-hydroxy-4-methylsulfonylamino-5-phenoxyphenyl) ketone were sequentially added thereto, and the temperature was raised to 20 ° C. The reaction was carried out at the same temperature for 5 hours. 250 mL of methylene chloride was added to the reaction suspension, and 500 mL of water was added dropwise to the obtained solution. After adjusting the pH to 5 with a 10% aqueous hydrochloric acid solution, the mixture was stirred at 20 ° C. for 1 hour. The obtained precipitated crystals were separated, washed successively with 50 mL of methylene chloride, 50 mL of water and 50 mL of ethanol, and then dried at 50 ° C. for 12 hours. Next, the obtained crystals were dissolved in a mixed solvent of 7.7 g (137 mmol) of potassium hydroxide, 750 mL of water and 750 mL of acetone, and then neutralized with 2N hydrochloric acid water, and the obtained precipitated crystals were separated. Then, after washing with 50 mL of water, it was dried at 50 ° C. for 12 hours to obtain 42.8 g of iguratimod (iguratimod purity: 99.72%, N-methyl compound: 0.23%).
References
- ^ Jump up to:a b c d e f Tanaka K, Yamaguchi T, Hara M (May 2015). “Iguratimod for the treatment of rheumatoid arthritis in Japan”. Expert Review of Clinical Immunology. 11 (5): 565–73. doi:10.1586/1744666X.2015.1027151. PMID 25797025. S2CID 25134255.
- ^ Inaba T, Tanaka K, Takeno R, Nagaki H, Yoshida C, Takano S (January 2000). “Synthesis and antiinflammatory activity of 7-methanesulfonylamino-6-phenoxychromones. Antiarthritic effect of the 3-formylamino compound (T-614) in chronic inflammatory disease models”. Chemical & Pharmaceutical Bulletin. 48 (1): 131–9. doi:10.1248/cpb.48.131. PMID 10705489.
- ^ Bronson J, Dhar M, Ewing W, Lonberg N (2012). “Chapter Thirty-One – To Market, To Market—2011”. Annual Reports in Medicinal Chemistry. 47: 499–569. doi:10.1016/B978-0-12-396492-2.00031-X.
- ^ “Iguratimod – Simcere”. AdisInsight. Retrieved 27 May 2018.
- ^ “Iguratimod – Toyama Chemical”. AdisInsight. Retrieved 27 May 2018.
| Clinical data | |
|---|---|
| Trade names | Careram; Kolbet |
| Other names | T-614 |
| ATC code | None |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 123663-49-0 |
| PubChem CID | 124246 |
| ChemSpider | 110694 |
| UNII | 4IHY34Y2NV |
| ChEMBL | ChEMBL2107455 |
| CompTox Dashboard (EPA) | DTXSID0048971 |
| ECHA InfoCard | 100.236.037 |
| Chemical and physical data | |
| Formula | C17H14N2O6S |
| Molar mass | 374.37 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]O=S(=O)(Nc3c(Oc1ccccc1)cc2c(O/C=C(\C2=O)NC=O)c3)C | |
| InChI[hide]InChI=1S/C17H14N2O6S/c1-26(22,23)19-13-8-15-12(17(21)14(9-24-15)18-10-20)7-16(13)25-11-5-3-2-4-6-11/h2-10,19H,1H3,(H,18,20)Key:ANMATWQYLIFGOK-UHFFFAOYSA-N |
////////////IGURATIMOD, UNII-4IHY34Y2NV , игуратимод , إيغوراتيمود , 艾拉莫德 , T-614, T 614, Kolbet, Careram, Rheumatoid arthritis, JAPAN 2012, CHINA 2011
CS(=O)(=O)NC1=C(C=C2C(=C1)OC=C(C2=O)NC=O)OC3=CC=CC=C3

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
.svg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
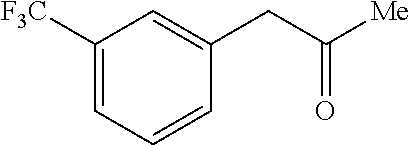{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
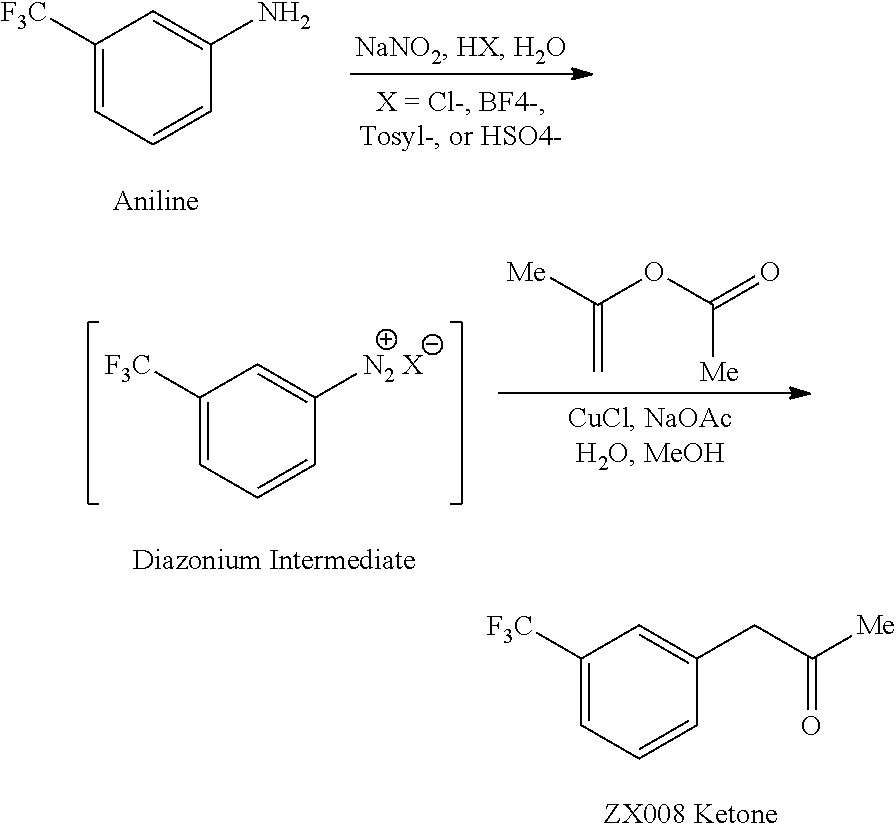{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}