
Home » organic chemistry
Category Archives: organic chemistry
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Development of an Efficient Manufacturing Process for a Key Intermediate in the Synthesis of Edoxaban

Development of an Efficient Manufacturing Process for a Key Intermediate in the Synthesis of Edoxaban

We report the development of a novel synthetic method to access a key intermediate in the synthesis of edoxaban. The main features of the new synthetic method are an improvement in the approach for the synthesis of a key chiral bromolactone, application of an interesting cyclization reaction utilizing neighboring group participation to construct a differentially protected 1,2-cis-diamine, and implementation of plug-flow reactor technology to enable the reaction of an unstable intermediate on multihundred kilogram scale. The overall yield for the preparation of edoxaban was significantly increased by implementing these changes and led to a more efficient and environmentally friendly manufacturing process.
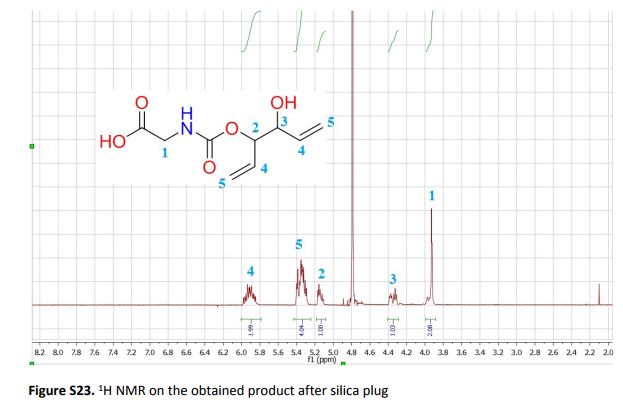
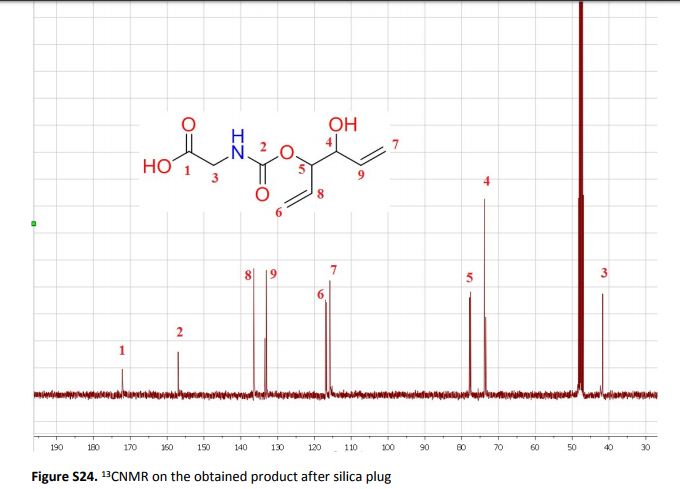
Synthesis of highly functional carbamates through ring-opening of cyclic carbonates with unprotected α-amino acids in water
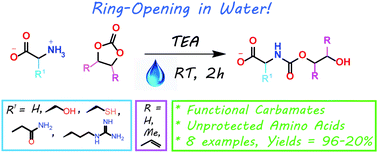
DOI: 10.1039/C7GC02862H, Paper
Ring opening of cyclic carbonates with unprotected amino acids in water – a route to highly functional carbamates.
Synthesis of highly functional carbamates through ring-opening of cyclic carbonates with unprotected α-amino acids in water
Abstract
The present work shows that it is possible to ring-open cyclic carbonates with unprotected amino acids in water. Fine tuning of the reaction parameters made it possible to suppress the degree of hydrolysis in relation to aminolysis. This enabled the synthesis of functionally dense carbamates containing alkenes, carboxylic acids, alcohols and thiols after short reaction times at room temperature. When Glycine was used as the nucleophile in the ring-opening with four different five membered cyclic carbonates, containing a plethora of functional groups, the corresponding carbamates could be obtained in excellent yields (>90%) without the need for any further purification. Furthermore, the orthogonality of the transformation was explored through ring-opening of divinylenecarbonate with unprotected amino acids equipped with nucleophilic side chains, such as serine and cysteine. In these cases the reaction selectively produced the desired carbamate, in 70 and 50% yield respectively. The synthetic design provides an inexpensive and scalable protocol towards highly functionalized building blocks that are envisioned to find applications in both the small and macromolecular arena.

- Stockholm, Sweden
- PostDoc Position
Research experience


Dr. Eric Johnston, Ph.D.

Chief Technology Officer
Dr. Eric V. Johnston obtained his Master of Science degree in 2008 at the Department of Organic Chemistry, Stockholm University, Sweden. In the same year, he started his graduate studies under the supervision of Prof. Jan-Erling Bäckvall. During his PhD, he worked on the development of new homogeneous and heterogeneous transition-metal catalysts.
After receiving his PhD in 2012, he joined Prof. Samuel J. Danishefskys research group at Memorial Sloan-Kettering Cancer Center, New York, USA as a postdoctoral fellow supported by The Swedish Research Council. Here he was engaged in the total chemical synthesis of glycolsylated proteins that play important roles in modern cancer treatment.
In 2014 he returned to the Department of Organic Chemistry at Stockholm University to establish his own group. The goal of his research is to contribute new advances to the strategy and methodology for the preparation of synthetic macromolecules such as proteins, glycopeptides, sequence and length-controlled polymers. He is also a Co-Supervisor for Prof. Björn Åkermarks research group, which aims at studying and developing new homogeneous, as well as heterogeneous, water oxidation catalysts.
How flow chemistry can make processes greener…………Supercritical fluids
Safe, small scale access to supercritical fluids
The ability to safely access high temperatures and pressures in flow reactors has implications not only on the rate of chemical reactions, but also on the types of solvents one can use. Many greensolvents such as methanol and acetone have boiling points too low for certain batch applications, whereas performing reactions at high pressure in a flow reactor may allow for their safe use at elevated temperatures.
Supercritical fluids are particularly interesting, since these solvents are entirely inaccessible without high pressure conditions. The use of supercritical fluids in a flow system offers numerous advantages over batch reactors.
Reactions may be performed on a small scale, improving safety and reducing the amount of material required. Depending on the type of reactor, it may be possible to visualize the reaction to evaluate the phase behaviour. Moreover, the reaction can be analyzed and the temperature and pressure subsequently changed without stopping the reaction and cleaning the vessel, as is necessary in a simple autoclave.

Continuous methods for utilizing supercritical fluids for extraction,1 chromatography,2 and as a reaction medium3 have all been commercialized, particularly for supercritical carbon dioxide (scCO2).4 Academic examples using scMeOH, scH2O, and scCO2 for continuous reactions such as hydrogenations, esterifications, oxidations, and Friedel–Crafts reactions have been reported.5
A recent example that illustrates many of the green advantages of performing supercritical fluid chemistry in flow is in the ring opening of phthalic anhydride with methanol by Verboom and co-workers (Scheme 1).6 They designed a microreactor with a volume of just 0.32 μL that can withstand very high pressures.

The exceptionally small channel causes a large build-up of pressure, and supercritical conditions with pressures of up to 110 bar and temperatures up to 100 °C can occur inside the reactor, giving an ‘on-chip’ phase transition. The channel size increases near the outlet, allowing the fluid to expand to atmospheric conditions.
Thus, the total volume of scCO2 under high pressure is exceptionally small, alleviating the major hazards of operating under supercritical conditions. The reaction was thoroughly studied on this small scale, allowing the authors to determine rate constants at several different temperatures and pressures.

 |
||
| Scheme 1 Small scale continuous use of supercritical fluids. | ||
Near- and supercritical water (scH2O) can be an interesting green solvent only obtainable at very high temperature (Tc = 374 °C) and pressure (Pc = 221 bar). It is commonly used for completeoxidation of organic waste materials to CO2; however, it has also been shown to be an effective solvent for selective oxidations.7 Given the harshness of the reaction conditions, it is not surprising that side product formation is common and highly dependent on the reaction time. For fast reactions in a batch reactor, precise control of reaction time is challenging, as the vessel takes time to heat and cool. In contrast, rapid heating, cooling, and quenching can be accomplished in a continuous process, allowing for well defined reaction times.
Fine tuning of the temperature, pressure, and time is also easier in a continuous process, as these variables can be changed without stopping and starting the reaction between samples. Thus, more data points can be obtained with less material and fewer heating and cooling cycles.

The Poliakoff group used these advantageous to perform a detailed study on the oxidation of p-xylene to terephthalic acid in scH2O, a reaction carried out on industrial scale in acetic acid (Scheme 2).8 By using a flow reactor, reaction times as low as 9 seconds could be used. The equivalents of oxygen could also be finely varied on a small scale through the controlled thermal decomposition of H2O2.
Studying this aerobic oxidation with such precision in a batch process would prove highly challenging. Under optimal conditions, excellent selectivity for the desired product could be obtained. Further research by the same group identified improved conditions for this transformation.9

 |
||
| Scheme 2 Selective oxidation in supercritical water. | ||

Schematic Diagram of sample Supercritical CO2 system
| Solvent | Molecular weight | Critical temperature | Critical pressure | Critical density |
|---|---|---|---|---|
| g/mol | K | MPa (atm) | g/cm3 | |
| Carbon dioxide (CO2) | 44.01 | 304.1 | 7.38 (72.8) | 0.469 |
| Water (H2O) (acc. IAPWS) | 18.015 | 647.096 | 22.064 (217.755) | 0.322 |
| Methane (CH4) | 16.04 | 190.4 | 4.60 (45.4) | 0.162 |
| Ethane (C2H6) | 30.07 | 305.3 | 4.87 (48.1) | 0.203 |
| Propane (C3H8) | 44.09 | 369.8 | 4.25 (41.9) | 0.217 |
| Ethylene (C2H4) | 28.05 | 282.4 | 5.04 (49.7) | 0.215 |
| Propylene (C3H6) | 42.08 | 364.9 | 4.60 (45.4) | 0.232 |
| Methanol (CH3OH) | 32.04 | 512.6 | 8.09 (79.8) | 0.272 |
| Ethanol (C2H5OH) | 46.07 | 513.9 | 6.14 (60.6) | 0.276 |
| Acetone (C3H6O) | 58.08 | 508.1 | 4.70 (46.4) | 0.278 |
| Nitrous oxide (N2O) | 44.013 | 306.57 | 7.35 (72.5) | 0.452 |
Table 2 shows density, diffusivity and viscosity for typical liquids, gases and supercritical fluids.
| Density (kg/m3) | Viscosity (µPa∙s) | Diffusivity (mm²/s) | |
|---|---|---|---|
| Gases | 1 | 10 | 1–10 |
| Supercritical Fluids | 100–1000 | 50–100 | 0.01–0.1 |
| Liquids | 1000 | 500–1000 | 0.001 |
- F. Sahena, I. S. M. Zaidul, S. Jinap, A. A. Karim, K. A. Abbas, N. A. N. Norulaini and A. K. M. Omar, J. Food Eng., 2009, 95, 240–253
- D. J. Dixon and K. P. Jhonston, in Encyclopedia of Separation Technology, ed. D. M. Ruthven, John Wiley, 1997, 1544–1569
- P. Licence, J. Ke, M. Sokolova, S. K. Ross and M. Poliakoff, Green Chem., 2003, 5, 99–104
- X. Han and M. Poliakoff, Chem. Soc. Rev., 2012, 41, 1428–1436
- S. Marre, Y. Roig and C. Aymonier, J. Supercrit. Fluids, 2012, 66, 251–264
- F. Benito-Lopez, R. M. Tiggelaar, K. Salbut, J. Huskens, R. J. M. Egberink, D. N. Reinhoudt, H. J. G. E. Gardeniers and W. Verboom, Lab Chip, 2007, 7, 1345–1351
- R. Holliday, B. Y. M. Jong and J. W. Kolis, J. Supercrit. Fluids, 1998, 12, 255–260
- P. A. Hamley, T. Ilkenhans, J. M. Webster, E. García-Verdugo, E. Vernardou, M. J. Clarke, R. Auerbach, W. B. Thomas, K. Whiston and M. Poliakoff, Green Chem., 2002, 4, 235–238
- E. Pérez, J. Fraga-Dubreuil, E. García-Verdugo, P. A. Hamley, M. L. Thomas, C. Yan, W. B. Thomas, D. Housley, W. Partenheimer and M. Poliakoff, Green Chem., 2011, 13, 2397–2407





Google+
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SON
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Total synthesis of the proposed structure of Astakolactin……….Dr Corey Johnson

picture credit…………Dr Corey johnson
Astakolactin is a sesterpene from the Ionian Sea near Greece possessing considerable biological properties. Hence, that’s why the authors decided to synthesize it, and also why the we’re all interested in its structure. In the conclusion of this paper, no biological studies were performed, but the characterization matches that of the natural product, which is a big deal.
read at
http://chemistrycorey.blogspot.in/2014/11/total-synthesis-of-proposed-structure.html
A lovely blog and its great author

- Corey R. Johnson
- Philly native, JCSU alumnus, Brandeis alumnus, Co-author of several scholarly journal articles…
Optimisation of Conditions for O-Benzyl and N-Benzyloxycarbonyl Protecting Group Removal using an Automated Flow Hydrogenator

K.R. Knudsen, J. Holden, S.V. Ley and M. Ladlow, Adv. Syn. Cat. 2007, 349, 535-538.
http://onlinelibrary.wiley.com/doi/10.1002/adsc.200600558/abstract
A versatile, fully automated flow hydrogenator has been developed that is able to perform sequential flow optimisation experiments, flow library hydrogenation, or iterative scale-up hydrogenation. The behaviour of a palladium catalyst in effecting removal of O-benzyl and N-benzyloxycarbonyl protecting groups has been investigated. Significant observations relating to maintaining optimal throughput are reported. A small library of peptidic derivatives has been deprotected in high yield and purity.
System configuration:
The system used was configured from a Gilson liquid handler (233XL), driven with a 10 mL
syringe pump (402). The syringe pump was connected to the sampling needle via a 2-way 6
position switching valve. This single channel liquid handler was used to perform both substrate
manipulation and fraction collection. The liquid handler was connected via a 2-way 6 position
injection valve to a Thales H-CubeTM flow hydrogenator driven with a KnauerTM A120 high
pressure pump. The collection vials were housed in specially designed gas tight blocks (2 x 7)
which were fitted with PTFA seals to enable penetration by the liquid handler needle, and
continuously purged with nitrogen in order to dilute and vent excess hydrogen safely. The
hardware was controlled using a single graphic user interface (HydroMateTM, Figure 2) which
utilised either RS232 or GSIOC connectivity to interface with the Thales and Gilson devices
respectively. Throughout 30 mm, 4 mm id 10% Pd/C catalyst cartridges (CatCartTM) were used
in conjunction with a 5 mL sample injection loop, although larger cartridges are also available.
The control software exploits software ‘wizards’ to assist the user in compiling a sequence of
optimisation experiments, or alternatively permits the implementation of a series of repetitive
experiments for either: (i) catalyst evaluation, (ii) reaction optimisation, (iii) compound library
synthesis, or (iv) as part of an automated, unattended scale up campaign (Figure 1). Experiments
may be devised with variations in scale, temperature, flow rate, and pressure in addition to
periodicity of fraction collection.
Analysis: RP-HPLC was run on a Hewlett Packard 1050 instrument. Column: Supelcosilä
ABZ+
PLUS column, 3.3 cm, 4.6 mm f, 3 mm. Eluent: A: water, 0.1% TFA, B: acetonitrile 95%,
water 5%, TFA 0.05%. Gradient: 10 to 95% B in A (1 mL min-1
) over 8 min. Detection: UV
(diode array detector).
A Microcapillary Flow Disc (MFD) Reactor for Organic Synthesis

A Microcapillary Flow Disc (MFD) Reactor for Organic Synthesis,
C.H. Hornung, M.R. Mackley, I.R. Baxendale and S.V. Ley and, Org. Proc. Res. Dev., 2007, 11, 399-405.
http://pubs.acs.org/doi/abs/10.1021/op700015f
This paper reports proof of concept, development, and trials for a novel plastic microcapillary flow disc (MFD) reactor. The MFD was constructed from a flexible, plastic microcapillary film (MCF), comprising parallel capillary channels with diameters in the range of 80−250 μm. MCFs were wound into spirals and heat treated to form solid discs, which were then capable of carrying out continuous flow reactions at elevated temperatures and pressures and with a controlled residence time. Three reaction schemes were conducted in the system, namely the synthesis of oxazoles, the formation of an allyl-ether, and a Diels−Alder reaction. Reaction scales of up to four kilograms per day could be achieved. The potential benefits of the MFD technology are compared against those of other reactor geometries including both conventional lab-scale and other microscale devices.
Continuous Flow Ligand-Free Heck Reactions Using Monolithic Pd[0] Nanoclusters

Continuous Flow Ligand-Free Heck Reactions Using Monolithic Pd[0] Nanoclusters
N. Nikbin, M. Ladlow, S.V. Ley, Org. Proc. Res. Dev., 2007, 11, 458-462.
http://pubs.acs.org/doi/abs/10.1021/op7000436

Flow-through reactor setup.

An automated reactor has been developed for performing ligand-free Heck reactions in continuous flow mode. The reactor utilises a monolithic reactor cartridge derivatised with Pd(0) nanoparticles in-line with a scavenging cartridge containing Quadrapure-TU to efficiently capture palladium residues and thereby afford Heck products directly in high purity.
Piecing together the puzzle: understanding a mild, metal free reduction method for large scale synthesis of hydrazines

D.L. Browne,* I.R. Baxendale, S.V. Ley, Tetrahedron2011, 67, 10296-10303.
http://www.sciencedirect.com/science/article/pii/S0040402011015304

A key intermediate for the synthesis of hydrazines via a mild, metal free reduction of diazonium salts has been isolated and characterized by X-ray analysis. The presence of this intermediate is general, as demonstrated by the preparation of a number of analogues. A discussion of the mechanism and potential benefits of such a process are also described.
FLOW SYNTHESIS IN ACTION

I.R. Baxendale, S.C. Schou, J. Sedelmeier, S.V. Ley, Chem. Eur. J. 2010, 16, 89-94.
READ
http://onlinelibrary.wiley.com/doi/10.1002/chem.200902906/abstract
Multi-step in flow: The palladium-catalysed acylation of terminal alkynes for the synthesis of yne![[BOND]](https://i0.wp.com/onlinelibrarystatic.wiley.com/undisplayable_characters/00f8ff.gif) ones as well as their further transformation to various heterocycles in a continuous-flow mode is presented. Furthermore, an extension of the simple flow configuration that allows for easy batch splitting and the generation of a heterocyclic library is described (see scheme).
ones as well as their further transformation to various heterocycles in a continuous-flow mode is presented. Furthermore, an extension of the simple flow configuration that allows for easy batch splitting and the generation of a heterocyclic library is described (see scheme).
Advanced Intermediate Flow Studies: Nevirapine
After looking through a number of flow articles that describe and illustrate processes toward the production of drug final products and advanced intermediates, I thought an article from Florida State — Tyler McQuade (open source Beilstein JOC 2013) was informative and storytelling. He was able to show some of the challenges that go into designing a flow methodology around process that have already been worked out in batch mode, and had been looked at in a number of labs already.
Before talking about the chemistry, Professor McQuade talks about a number of concerns in transferring technology from batch to flow: DOE, solvent exchange (precipitation and moving from one reaction to another), Cost of Goods Analysis – reaction concentrations, solvent costs, process time, by-product formation and purification. There certainly is a lot that goes into the strategy. To give you the framework: this group was looking to make a continuous process…
View original post 247 more words
