

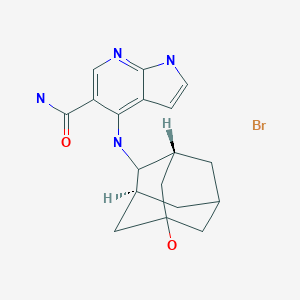

Candidate: TAK-981
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA approves first treatment for pediatric patients with lupus
The U.S. Food and Drug Administration today approved Benlysta (belimumab) intravenous (IV) infusion for treatment of children with systemic lupus erythematosus (SLE) – often referred to as simply “lupus” – a serious chronic disease that causes inflammation and damage to various body tissues and organs. This is the first time that the FDA has approved a treatment for pediatric patients with SLE. Benlysta has been approved for use in adult patients since 2011.
“The agency expedited the review and approval of this application because Benlysta IV fulfils an unmet need for therapies, specifically in pediatric patients with SLE. While there is no cure for lupus, treatment can help our youngest patients control their disease with the hope of …
April 26, 2019
Release
The U.S. Food and Drug Administration today approved Benlysta (belimumab) intravenous (IV) infusion for treatment of children with systemic lupus erythematosus (SLE) – often referred to as simply “lupus” – a serious chronic disease that causes inflammation and damage to various body tissues and organs. This is the first time that the FDA has approved a treatment for pediatric patients with SLE. Benlysta has been approved for use in adult patients since 2011.
“The agency expedited the review and approval of this application because Benlysta IV fulfils an unmet need for therapies, specifically in pediatric patients with SLE. While there is no cure for lupus, treatment can help our youngest patients control their disease with the hope of improving their quality of life and lowering their risk of long-term organ damage and disability,” said Janet Woodcock, M.D., director of the FDA’s Center for Drug Evaluation and Research.
While childhood-onset SLE is rare, when diagnosed, it is generally more active in children and adolescents than adult patients, particularly in how it impacts organs such as the kidneys and central nervous system. As a result of the disease starting early in life, pediatric patients with SLE are at a higher risk for developing increased organ damage and complications from the disease as well as adverse events from the life-long treatments usually required.
The efficacy of Benlysta IV for the treatment of SLE in pediatric patients was studied over 52 weeks in 93 pediatric patients with SLE. The proportion of pediatric patients achieving the composite primary endpoint, the SLE response index (SRI-4), was higher in pediatric patients receiving Benlysta IV plus standard therapy compared to placebo plus standard therapy. Pediatric patients who received Benlysta IV plus standard therapy also had a lower risk of experiencing a severe flare, as well as longer duration of time until a severe flare (160 days versus 82 days). The drug’s safety and pharmacokinetic profiles in pediatric patients were consistent with those in adults with SLE.
Benlysta’s doctor and patient information includes a warning for mortality, serious infections, hypersensitivity and depression, based on data from the clinical studies in adults with SLE. The drug should not be administered with live vaccines. The manufacturer is required to provide a Medication Guide to inform patients of the risks associated with Benlysta.
The most common side effects in patients included nausea, diarrhea and fever. Patients also commonly experienced infusion reactions, so healthcare professionals are advised to pre-treat patients with an antihistamine.
The FDA granted this application a Priority Review designation. The FDA granted the approval of Benlysta to GlaxoSmithKline.
////////////Benlysta, belimumab, fda 2019, Priority Review, GlaxoSmithKline
Ferric carboxymaltose , カルボキシマルトース第二鉄


Ferric carboxymaltose
カルボキシマルトース第二鉄
CAS: 9007-72-1
Molecular Formula, C24H44FeO25–
Molecular Weight, 788.43616 g/mol
(2S,3S,4S,5R)-4-[(2R,3R,4R,5S,6R)-5-[(2R,3R,4R,5S,6R)-3,4-dihydroxy-6-(hydroxymethyl)-5-[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoxan-2-yl]oxy-3,4-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-2,3,5,6-tetrahydroxyhexanoate;iron(3+);oxygen(2-);hydroxide;hydrate
Iron dextri-maltose
Iron(3+) hydroxide oxide poly-(1–4)-alpha-D-glucopyranosyl-(1–4)-D-gluconate hydrate
Polynuclear iron (III)-hydroxide 4(R)-(poly-(1–4)-O-alpha-D-glucopyranosyl)-oxy-2(R),3(S),5(R),6-tetrahydroxy-hexanoate
Poly[D-glucopyranosyl(1–4)]-D-gluconic acid complex of hydrated iron(III) oxide
japan pmda approved, 2019/3/26, Ferinject
Treatment of patients with iron deficiency anemia
Hematinic, Supplement (iron)
LAUNCHED, 2007, Vifor Pharma, Anemia, iron deficiency
1 Injectafer
2. Ferinject
3. Iron Dextri-maltose
4. Unii-6897gxd6oe
5. Vit 45
6. Vit-45
7. Ferric Carboxymaltose [usan:inn:ban]
8. Iron(3+) Hydroxide Oxide Poly-(1?4)-alpha-d-glucopyranosyl-(1?4)-d-gluconate Hydrate
9. 889138-31-2
10. 9007-72-1
11 Z-213
In 2013, Vifor Pharma and Zeria Pharmaceutical signed an exclusive licensing agreement for the product’s development and commercialization in Japan for the treatment of iron deficiency anemia.
Ferric carboxymaltose is an intravenously-administered iron complex which was first launched in Germany following E.U. approval in 2007 for the treatment of iron deficiency anemia (IDA)
PATENT
WO 2011055374
US 20120214986
IN 2011MU03463
IN 2013CH03474
WO 2016181195
IN 2015CH02360
CN 106236707
CN 106977621
EP 3339329
PATENT
Iron deficiency anaemia (IDA) is a common haematological complication with potentially serious clinical consequences that may require intravenous iron therapy.
Ferric carboxymaltose (FCM) is a stable, non-dextran iron formulation administered intravenously in large single doses to treat IDA. It is an iron complex that consists of a ferric hydroxide core stabilized by a carbohydrate shell. It is commercially available in the market under the trade name Ferinject®
Ferric carboxymaltose has been designed to provide high iron utilisation and to have a better benefit to risk profile than iron dextran and iron sucrose therapy. In the case of iron dextran, a key risk is the reaction with anti-dextran antibodies leading to the well known dextran induced anaphylactic reactions. In the case of iron sucrose, the negative characteristics include high pH, high osmolarity, low dosage limits and the long duration of administration.
Ferric carboxymaltose allows for controlled delivery of iron within the cells of the reticuloendothelial system and subsequent delivery to the iron-binding proteins ferritin and transferrin, with minimal risk of release of large amounts of ionic iron in the serum.
U.S. Pat. No. 3,076,798 discloses a process for the preparation of iron(III)-polymaltose complex compounds. The iron(III)-polymaltose complex compound
preferably has a molecular weight in the range from 20,000 to 500,000 daltons, preferably from 30,000 to 80,000 daltons.
U.S. Patent No. 7,612,109 discloses water-soluble iron carbohydrate complexes (ferric carboxymaltose complexes) obtainable from an aqueous solution of an iron (III) salt, preferably iron (III) chloride, and an aqueous solution of the oxidation product of one or more maltodextrins using an aqueous hypochlorite solution.
PCT application No.WO2011/055374, discloses a process for the preparation of iron (III) carboxymaltose complex using ferric hydroxide.
In Netherlands article, starch 41 (1989) Nr .8, S. 303-309 transition metal ions enhance the selectivity of oxidations by H2O2 to produce polysaccharides to polydicarbonates by glycol cleavage of the C2-C3 vicinal diol moiety.
Even though many prior art processes reported methods for the preparation of Iron(III) carboxymaltose, each process has some limitations with respect to yield, purity and scale-up etc.
EXAMPLES
Example- 1: Preparation of trivalent iron carboxymaltose
Step (i)
20grams of anhydrous iron(III)chloride was dissolved in 50ml of purified water at room temperature for 10 minutes stirring. To this 2gm of maltodextrin (13-17 dextrose equivalents) was added and stirred for 10 minutes at room temperature. The obtained brownish-yellow clear solution was cooled to 0-5°C and the pH of the reaction mixture was adjusted to 7.0 by adding 20% aqueous sodium hydroxide solution. A brown colour precipitate obtained was maintained for 1 hour at 0-5°C and collected through filtration (Wet cake wt. ~ 65. Og). The cake was suck dried and used for next step.
Step (ii)
20grams of maltodextrin having a dextrose equivalents of 13-17 were dissolved in 50ml of purified water and the solution was metered in the course of 20 minutes to a stirred mixture of 2.66gm of Starks catalyst (methyl trioctyl ammonium hydrogen sulfate prepared in-situ from 2gm of Aliquat 336 and 0.66gm of NaHSO4.H2O), 0.8gm of sodium tungstate dihydrate and 0.37gm of TEMPO at RT. 31.12gm of hydrogen peroxide solution (50-55% w/v) was then added drop wise over a period of 40 minutes at 25-30°C and raised the temperature to90-95°C and stirred for 3 hours. After cooling to room temperature, a second portion of 15.5gm of H2O2 solution was metered in the course of 15 minutes at 25-30°C and the resulting solution was again refluxed at 90-95°C for 1 hour. After cooling to 35-40°C, wet cake of step (i) (ferric hydroxide maltodextrin complex) was added, with stirring. 14.0ml of 20% aqueous sodium hydroxide solution was added to adjust the reaction mass pH to 10- 10.5 and the slurry was heated to 50°C, stirred for 30 minutes. Then the reaction mixture was acidified to pH 5.5 by adding hydrochloric acid solution and the mixture was maintained at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 14 hours. Then the reaction mixture was cooled to room temperature and filtered through a celite pad. Thereafter, the iron(III)complex was isolated by precipitation by adding ethanol (237. Og) drop wise at room temperature. The obtained brown amorphous solid was dried under vacuum at 50°C for 2-3 hours. Molecular weight = 202 kDa. Iron content = 23.38% w/w
Example-2:
20grams of maltodextrin (13-17 dextrose equivalents) were dissolved in 50ml of purified water and the solution was added to a stirred mixture of 2.66gm of Starks catalyst and 0.2gm of Na2WO4.2H2O at room temperature in the course of 20 minutes. 24grams of H2O2 solution was metered in the course of 45 minutes at 25-30°C and raised the temperature to 90-95°C and stirred for 2 hours and cooled to room temperature.
The solution was added to another portion of a stirred mixture of 1.33gm of Starks catalyst and 0.2gm of Na2WO4.2H2O at room temperature. Thereafter, 12gm of
H2O2solution was added drop wise over a period of 20 minutes at 25-30°C and the resulting reaction mixture was again refluxed at 90-95°C for 2 hours. After cooling to 25-30°C, wet cake of step (i) from example- 1 was added and stirred for 10 minutes. 14ml of 20% NaOH solution was added to adjust the reaction mass pH to 10- 10.5 and the slurry was heated to 50°C, stirred for 30 minutes. Then the mixture was acidified to pH 5.5 by adding hydrochloric acid solution and the solution was maintained at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 13 hours. Then the reaction solution was cooled to room temperature, adjusted pH to 5.5 to 6.0 with 20% NaOH solution and filtered through a celite pad. Then the iron(III)complex was isolated by precipitating with ethanol (331.0g) addition drop wise at room temperature. The obtained brown amorphous solid was dried in vacuum at 50°C for 2-3 hours. Molecular weight = 200 kDa. Iron content = 25.57 % w/w
Example-3:
20grams of maltodextrin (13-17 dextrose equivalents) were dissolved in 100ml of purified water and the solution was added to a stirred mixture of 2.66gm of Starks catalyst, 0.8gm of Na2WO4.2H2O and 0.37gm of TEMPO at room temperature over a period of 15 minutes. 30grams of H2O2solution was added drop wise in the course of 1 hour at 25-30°C and raised the temperature to 90-95°C, stirred for 3 hours and cooled to room temperature.
At 25-30°C, wet cake of step (i) from example- 1 was added and stirred for 10 minutes. A pH of 10-10.5 was established by adding 12ml of 20% NaOH solution and the slurry was heated to 50°C, stirred at this temperature for 30 minutes. Then the reaction mixture was acidified to pH 5.5 with hydrochloric acid addition and the mixture was maintained at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 14 hours. The reaction mixture was allowed to cool to room temperature, adjusted pH to 6.0 to 6.5 with 20% NaOH solution and filtered through a celite pad. Then the iron(III)complex was isolated by precipitating with ethanol (343.0g) addition drop wise at room temperature. The obtained brown
amorphous solid was dried in vacuum at 50°C for 2-3 hours. Molecular weight = 260 kDa. Iron content = 23.67 % w/w
Example-4:
20grams of maltodextrin (13-17 dextrose equivalents) were dissolved in 50ml of purified water and the solution was added to a stirred mixture of 2.66gm of Starks catalyst, 0.8gm of Na2WO4.2H2O and 0.37g of TEMPO at room temperature over a period of 15 minutes. 30grams of H2O2 solution was added drop wise over a period of 1 hour at 55-60°C and the temperature was raised to 90-95 °C, stirred for 3 hours and cooled to room temperature. After cooling to 25-30°C, wet cake of step (i) from example- 1 was added and stirred for 10 minutes. A pH of 10-10.5 was established by adding 12ml of 20% NaOH solution and the slurry was heated to 50°C, stirred at this temperature for 30 minutes. Then the reaction mixture was acidified to pH 5.5 with hydrochloric acid addition and was maintained at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 12 hours. The reaction mixture was allowed to cool to room temperature, adjusted pH to 6.0 to 6.5 with 20% NaOH solution and filtered through a celite pad. Then the iron(III)complex was isolated by precipitating with ethanol (343.0g) addition drop wise at room temperature. The obtained brown amorphous solid was dried under vacuum at 50°C for 2-3 hours. Molecular weight = 261 kDa. Iron content = 22.85 % w/w
Example-5:
Step (i)
16grams of anhydrous iron(III)chloride was dissolved in 50ml of purified water at room temperature for 10 min stirring. The obtained brownish-yellow clear solution was cooled to 0-5°C and the pH was adjusted to 7.0 first by adding aqueous sodium carbonate solution (21gm of Na2CO3dissolved in 102 ml of purified water) and then by adding 20% NaOH solution. A brown colour precipitate obtained was maintained for 1 hour at 0-5°C and collected through filtration (Wet wt. ~54.0g). The cake was suck dried and used for next step.
Step (ii)
20grams of maltodextrin (13-17 dextrose equivalents) were dissolved in 50ml of purified water and the solution was added to a stirred mixture of 2.66gm of Starks catalyst, 0.8gm of Na2WO4.2H2O and 0.37gm of TEMPO at room temperature over a period of 15 minutes. 30gm of H2O2solution was added drop wise over a period of 1 hour at 25-30°C and the temperature was raised to 90-95°C, stirred for 3 hours and cooled to room temperature.
At 25-30°C, wet cake of step (i) added and stirred for 10 minutes. 20% NaOH solution was added drop wise to adjust the reaction mass pH tolO-10.5 and the slurry was heated to 50°C, stirred for 30 minutes. Then the solution was acidified to pH 5.5 with hydrochloric acid addition and the solution was kept at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 12 hours. The reaction mixture was allowed to cool to room temperature, adjusted pH to 6.0 to 6.5 with 20% NaOH solution and filtered through a celite pad. Then the iron(III)complex was isolated by precipitating with ethanol (315.0g) addition drop wise at room temperature. The obtained brown amorphous solid was dried under vacuum at 50°C for 2-3 hours. Molecular weight = 236 kDa. Iron content = 22.35 % w/w
Example-6:
Step (i)
20grams of anhydrous ferric chloride was dissolved in 50ml of purified water at room temperature for 10 min stirring. The obtained brownish-yellow clear solution was cooled to 0-5°C and the pH was adjusted to 7.0 by adding 20% NaOH solution. A brown colour precipitate obtained was stirred for 1 hour at 0-5°C and collected through filtration. The cake was suck dried and used for next step.
Step (ii)
20grams of maltodextrin (13-17 dextrose equivalents) were dissolved in 50ml of purified water and the solution was added to a stirred mixture of 2.66gm of Starks catalyst, 0.8gm of Na2WO4.2H2O and 0.37g of TEMPO at room temperature over a
period of 15 minutes. 36gm of H2O2 solution was metered in the course of 1 hour at 25-30°C and the resulting solution was heated to 90-95°C, stirred for 3 hours and cooled to room temperature.
After cooling to 25-30°C, wet cake of step (i) was added and stirred for 10 min. 12ml of 20% NaOH solution was added drop wise to adjust the reaction mass pH to 10-10.5 and the slurry was heated to 50°C, kept at this temperature for 30 minutes. Then the solution was acidified to pH 5.5 with hydrochloric acid addition and the solution was maintained at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 12 hours. The reaction mixture was allowed to cool to room temperature, adjusted pH to 6.0 to 6.5 with 20% NaOH solution and filtered through a celite pad. Then the iron(III)complex was isolated by precipitating with ethanol (315.0g) addition at room temperature. The obtained brown amorphous solid was dried under vacuum at 50°C for 2-3 hours. Molecular weight = 365 kDa. Iron content = 23.93 % w/w
Example-7:
20grams of maltodextrin (13-17 dextrose equivalents) were dissolved in 50ml of purified water and the solution was added to a stirred mixture of 2.66gm of Starks catalyst, 0.2gm of Na2WO4.2H2O and 0.37gm of TEMPO at room temperature over a period of 15 minutes. 30gm of H2O2 solution was added drop wise in the course of 1 hour at 25-30°C and the temperature was raised to 90-95°C, stirred for 3 hours and cooled to room temperature.
At 25-30°C, wet cake of step (i) from example-6 was added and stirred for 10 minutes. A pH of 10-10.5 was established by adding 12.0ml of 20% NaOH solution and the slurry was heated to 50°C, stirred at this temperature for 30 minutes. Then the solution was acidified to pH 5.5 with hydrochloric acid addition and the solution was kept at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 12 hours. The reaction mixture was allowed to cool to room temperature; pH was adjusted to 6.0 to 6.5 with 20% NaOH solution and filtered through a celite pad. Then the iron(III)complex was isolated by precipitating with ethanol (276.0g) addition drop wise at room temperature. The obtained brown amorphous solid was dried in vacuum at 50°C for 2-3 hours. Molecular weight = 366 kDa. Iron content = 21.2 % w/w
Example-8:
20grams of maltodextrin (13-17 dextrose equivalents) were dissolved in 50ml of purified water and the solution was added to a stirred mixture of 2.66gm of Starks catalyst and 0.8gm of Na2WO4.2H2O at room temperature over a period of 15 minutes. 30grams of H2O2 solution was metered in the course of 1 hour at 25-30°C and the temperature was raised to 90-95°C, stirred for 3 hours and cooled to room temperature.
At 25-30°C, wet cake of step (i) from example-6 was added and stirred for 10 minutes. A pH of 10-10.5 was established by adding 12ml of 20% NaOH solution and the slurry was heated to 50°C, stirred at this temperature for 30 minutes. Then the solution was acidified to pH 5.5 with hydrochloric acid addition and the solution was maintained at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 12 hours. The reaction mixture was allowed to cool to 25-30°C, adjusted pH to 6.0 to 6.5 with 20% NaOH solution and filtered through a celite pad. Then the iron(III)complex was isolated by precipitating with ethanol (315.0g) addition drop wise at room temperature. The obtained brown amorphous solid was dried in vacuum at 50°C for 2-3 hours. Molecular weight = 340 kDa. Iron content = 23.28 % w/w
Example-9:
20grams of maltodextrin (13-17 dextrose equivalents) were dissolved in 50ml of purified water and the solution was added to a stirred mixture of 2.66gm of Starks catalyst, 0.8gm of Na2WO4.2H2O and 0.37gm of TEMPO at room temperature over a period of 15 minutes. 30grams of H2O2 solution was added drop wise over a period of 1 hour at 25-30°C and the resulting solution was heated to 90-95°C, stirred for 3 hours and cooled to room temperature.
At 25-30°C, wet cake of step (i) from example- 1 was added and stirred for 10 minutes. A pH of 10-10.5 was established by adding 12ml of 20% NaOH solution and the slurry was heated to 50°C, stirred at this temperature for 30 minutes. Then the solution was acidified to pH 5.5 with hydrochloric acid addition and the solution was maintained at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 12 hours. The reaction mixture was allowed to cool to room temperature, adjusted pH to 6.0 to 6.5 with 20% NaOH solution and filtered through a celite pad. Then the iron(III)complex was isolated by precipitating with ethanol (304.0g) addition drop wise at room temperature. The obtained brown amorphous solid was dried under vacuum at 50°C for 2-3 hours. Molecular weight = 352 kDa. Iron content = 23.0 % w/w
Example-10:
20grams of maltodextrin (13-17 dextrose equivalents) were dissolved in 50ml of purified water and the solution was added to a stirred mixture of 2.66gm of Starks catalyst, 0.8gm of Na2WO4.2H2O and 0.37gm of TEMPO at room temperature over a period of 15 minutes. 30grams of H2O2 solution was added drop wise in the course of 60 minutes at 25-30°C and the temperature was raised to 90-95°C, stirred for 3 hours and cooled to room temperature.
At 25-30°C, wet cake of step (i) from example- 1 was added and stirred for 10 minutes. 12ml of 20% NaOH solution was added drop wise to adjust the reaction mixture pH to 10-10.5 and the temperature of the slurry was raised to 50°C, stirred at this temperature for 30 minutes. Then the reaction mixture was acidified to pH 5.5 with hydrochloric acid addition and was maintained at 50°C for another 30 minutes. Further temperature was raised to 95-100°C and stirred for 12 hours. The reaction mixture was allowed to cool to room temperature, adjusted pH to 6.0 to 6.5 with 20% NaOH solution and filtered through a celite pad. Then the iron(III)complex was
isolated by precipitating with ethanol (276.0g) addition drop wise at room temperature. The obtained brown amorphous solid was dried in vacuum at 50°C for 2-3 hours. Molecular weight = 348 kDa. Iron content = 24.6 % w/w
/////////Ferric carboxymaltose , カルボキシマルトース第二鉄 ,Injectafer, Ferinject, Iron dextri-maltose, Unii-6897gxd6oe, Vit 45, Vit-45, japan 2019, Z-213
C(C1C(C(C(C(O1)OC2C(OC(C(C2O)O)OC3C(OC(C(C3O)O)OC(C(CO)O)C(C(C(=O)[O-])O)O)CO)CO)O)O)O)O.O.[OH-].[O-2].[Fe+3]
Thiotepa, チオテパ ,тиотепа , ثيوتيبا , 塞替派 ,
Thiotepa, チオテパ
- Use:antineoplastic, alkylating agent
- Chemical name:1,1′,1”-phosphinothioylidynetrisaziridine
- Formula:C6H12N3PS
- MW:189.22 g/mol
- CAS:52-24-4
- EINECS:200-135-7
- LD50:14500 μg/kg (M, i.v.); 38 mg/kg (M, p.o.);
9400 μg/kg (R, i.v.) -
Aziridine, 1,1′,1”-phosphinothioylidynetris-Aziridine, 1-[bis(1-aziridinyl)phosphinothioyl]-N,N’,N”-TriethylenethiophosphoramidePhosphorothioic tri(ethyleneamide)SZ2975000T3NTJ APS&- AT3NTJ&- AT3NTJ [WLN]ThiofozilThiotef1,1′,1”-Phosphorothioyltriaziridine
JAPAN APPROVED, Rethio, PMDA, 2019/3/26
тиотепа [Russian] [INN]
ثيوتيبا [Arabic] [INN]
塞替派 [Chinese] [INN]
Thiotepa (INN,[1] chemical name: N,N′,N′′-triethylenethiophosphoramide) is an alkylating agent used to treat cancer.
Thiotepa is an organophosphorus compound with the formula SP(NC2H4)3.[2] It is an analog of N,N′,N′′-triethylenephosphoramide (TEPA), which contains tetrahedral phosphorus and is structurally akin to phosphate. It is manufactured by heating aziridine with thiophosphoryl chloride.
History
Thiotepa was developed by the American Cyanamid company in the early 1950s and reported to media outlets in 1953.[3] In 1959, thiotepa was registered with the Food and Drug Administration (FDA) as a drug therapy for several solid cancers.[4]
On January 29, 2007, the European Medicines Agency designated thiotepa as an orphan drug. On April 2, 2007, the United States FDA designated thiotepa as a conditioning treatment for use prior to hematopoietic stem cell transplantation.[5] Adienne Pharma & Biotech (Italy), the owner of thiotepa (Tepadina) applied for these designations.
Use
Thiotepa is indicated for use in combination with other chemotherapeutic agents. This can be with or without total body irradiation (TBI), as a conditioning treatment prior to allogeneic or autologous hematopoietic progenitor cell transplantation (HPCT) in hematological diseases in adult and pediatric patients. These diseases include Hodgkin’s disease and leukaemia. Thiotepa is also used with high-dose chemotherapy with HPCT support to treat certain solid tumors in adult and pediatric patients.[6]
Thiotepa is used in the palliation of many neoplastic diseases. The best results are found in the treatment of adenocarcinoma of the breast, adenocarcinoma of the ovary, papillary thyroid cancer and bladder cancer. Thiotepa is used to control intracavitary effusions caused by serosal neoplastic deposits.[6]
Intravesical use
Thiotepa is used as intravesical chemotherapy in bladder cancer.[7]
It may be used prophylactically to prevent seeding of tumor cells at cystoscopic biopsy; as an adjunctive agent at the time of biopsy; or as a therapeutic agent to prevent recurrence after cystoscopic resection of bladder tumor (transurethral resection of bladder tumor, TURBT). For intravesical use, thiotepa is given in 30 mg doses weekly, for four to six weeks. Efficacy in tumor control may reach 55 percent. The main toxicity of this therapy is bone marrow suppression due to systemic absorption of the drug.
Side effects
The main side effect of thiotepa is bone marrow suppression resulting in leukopenia, thrombocytopenia and anemia.[8] Liver and lung toxicity may also occur.
SYN

NMR

Fig 5. 1H NMR spectrum of C6H13N3PS in CDCL3 at 400 MHz.
R.J. Abraham, M. Mobli Modelling 1H NMR Spectra of Organic Compounds: Theory, Applications and NMR Prediction Software, Wiley, Chichester, 2008.

References
- ^ “International Non-Proprietary Names for Pharmaceutical Preparations. Recommended International Non-Proprietary Names (Rec. I.N.N.): List 4” (PDF). World Health Organization. March 1962. p. 111. Retrieved 27 November 2016.
- ^ Maanen, M. J.; Smeets, C. J.; Beijnen, J. H. (2000). “Chemistry, pharmacology and pharmacokinetics of N,N’,N” -triethylenethiophosphoramide (ThioTEPA)”. Cancer Treatment Reviews. 26 (4): 257–268. doi:10.1053/ctrv.2000.0170. PMID 10913381.
- ^ Sykes, M. P.; Karnofsky, D. A.; Philips, F. S.; Burchenal, J. H. (1953). “Clinical studies on triethylenephosphoramide and diethylenephosphoramide, compounds with nitrogen-mustard-like activity”. Cancer. 6 (1): 142–148. doi:10.1002/1097-0142(195301)6:1<142::AID-CNCR2820060114>3.0.CO;2-W.
- ^ Kim, Kyu-Won; Roh, Jae Kyung; Wee, Hee-Jun; Kim, Chan (2016). Cancer Drug Discovery: Science and History. Springer. p. 82. ISBN 978-94-024-0844-7.
- ^ “EMA Grants Adienne Marketing Rights for Tepadina”. dddmag.com. Drug Discovery & Development. 19 March 2010. Retrieved 25 November 2011.
- ^ Jump up to:a b “Urgent, thioTEPA update” (PDF). Food and Drug Administration. Adienne Pharma & Biotech. 5 April 2011. Retrieved 25 November 2011.
- ^ Droller M. Urothelial Tumors PMPH-USA, 2004. p. 207 ISBN 1550091735
- ^ Agnelli, G.; de Cunto, M.; Gresele, P.; del Favero, A. (1982). “Early onset life-threatening myelosuppression after low dose of intravesical thiotepa”. Postgraduate Medical Journal. 58 (680): 380–381. doi:10.1136/pgmj.58.680.380. PMC 2426344. PMID 6812036.
External links
REF
US 2 670 347 (American Cyanamid; 1954; prior. 1952)
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Consumer Drug Information |
| MedlinePlus | a682821 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
IV, intracavitary, intravesical |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Metabolism | Hepatic (CYP2B, CYP3A) |
| Elimination half-life | 1.5–4.1 hours |
| Excretion | Renal 6 hours for thiotepa 8 hours for TEPA |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.000.124 |
| Chemical and physical data | |
| Formula | C6H12N3PS |
| Molar mass | 189.23 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////////Thiotepa, チオテパ ,тиотепа , ثيوتيبا , 塞替派 , JAPAN 2019
VIXOTRIGINE, NEW PATENT, WO-2019071162, BIOGEN INC
VIXOTRIGINE, NEW PATENT, WO-2019071162, BIOGEN INC
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019071162&redirectedID=true
vixotrigine
Process for preparing α-carboxamide pyrrolidine derivatives (particularly vixotrigine ) and its intermediates are modulators of use-dependent voltage-gated sodium channels
Biogen, following the acquisition of Convergence Pharmaceuticals, that previously acquired clinical assets from GSK is developing vixotrigine a voltage-gated sodium channel 1.7 inhibitor, for the oral treatment of neuropathic pain, primarily trigeminal neuralgia.
| CHEN, Weirong; US |
| COUMING, Vinny; US |
| IRDAM, Erwin; US |
| KIESMAN, William, F.; US |
| KWOK, Daw-long, A.; US |
| MACK, Tamera, L.; US |
| OPALKA, Suzanne, M.; US |
| PATIENCE, Daniel, B.; US |
| WALKER, Donald, G.; US |
| LIANG, Wenli; US |
The invention relates to a novel process for preparing a-carboxamide pyrrolidine derivatives, in particular (2S, 5R)-5-(4-((2-fluorobenzyl)oxy)phenyl)pyrrolidine-2-carboxamide, and to novel intermediates for use in said process along with processes for preparing said intermediates.
(2S, 5R)-5-(4-((2-fluorobenzyl)oxy)phenyl)pyrrolidine-2-carboxamide:
is described in WO 2007/042239 as having utility in the treatment of diseases and conditions mediated by modulation of use-dependent voltage-gated sodium channels. The synthetic preparation of (2S, 5R)-5-(4-((2-fluorobenzyl)oxy)phenyl)pyrrolidine-2-carboxamide is described in both WO 2007/042239 and WO 2011/029762.
Description 1a: Methyl (S)-5-(4-(benzyloxy)phenyl)-2-((iert-butoxycarbonyl)amino)-5-oxopentanoate (D1a) (Batch Process using Grignard Procedure)
A reactor was charged with THF (350 kg) and the solvent was degassed by nitrogen sparging for about 30 min at 20 – 30 °C. To the degassed THF was charged l-(benzyloxy)- 4- bromobenzene (137 kg (1.78 equiv)). The solids were dissolved at 20 – 30 °C with agitation and under an inert atmosphere of nitrogen.
A reactor was charged with Mg (21.3 kg (3.0 equiv)) and THF (131 kg) and the mixture was degassed by nitrogen sparging for about 30 min at 20 – 30 °C. To this mixture was added -5% of the 1-(benzyloxy)-4-bromobenzene – THF solution followed by heating to 50 – 60 °C under an inert atmosphere of nitrogen. With good agitation, DIBAL-H in toluene (1 M; 2.5 kg (0.01 equiv)) was added followed by heating the mixture to 60 – 70 °C and aging for about 1 h. The remaining amount of the 1-(benzyloxy)-4-bromobenzene – THF solution was added followed by a THF rinse (36 kg) of the reactor. The mixture was aged for about 1 h at 60 – 70 °C and was cooled to 20 – 30 °C under an inert atmosphere of nitrogen.
A reactor was charged with THF (382 kg) and the solvent was degassed by nitrogen sparging for about 30 min at 20 – 30 °C. To the degassed THF was charged l-(ferf-butyl) 2-methyl (S)- 5- oxopyrrolidine 1 ,2-dicarboxylate (71 kg (1.0 equiv)), and the resulting solution was cooled to -60 to -70 °C under an inert atmosphere of nitrogen. To this solution was added the Grignard solution while maintaining a reaction temperature of <-60 °C. The reactor that contained the Grignard solution was rinsed with THF (61 kg) and the reaction was aged at -60 to -70 °C for about 1 h. The progress of the reaction was monitored (HPLC).
Upon completion, 2-propanol (56 kg) was added while maintaining a reaction temperature of -60 to -70 °C, and the reaction was aged for about 30 min. Water (296 kg) was added while maintaining a reaction temperature of <10 °C; the contents of the reactor were warmed to 20 – 30 °C following the addition. The pH of the mixture was adjusted to 6 – 7 by addition of 51 wt% acetic acid in water (70 kg). MTBE (220 kg) was added and the mixture was agitated for about 30 min. The layers were separated, the organic layer was clarified by filtration and was concentrated to about 3 – 4V. MTBE (220 kg) was added and the resulting solution was concentrated to about 3 – 4V. MTBE (150 kg) was added and the resulting solution was heated to 35 – 45 °C. n-Heptane (250 kg) was added slowly while maintaining a reaction temperature of 35 – 45 °C, the mixture was aged for 1 – 2 h, cooled to 0 – 5 °C and aged for 3 – 5 h. The solids were isolated by filtration, washed with n-heptane (74 kg) and dried in vacuo at 50 – 60 °C to constant weight to afford 96.7 kg (77.5%) of the title compound.
Description 1 b: Methyl (S)-5-(4-(benzyloxy)phenyl)-2-((iert-butoxycarbonyl)amino)-5-
oxopentanoate (D1 b) (Batch Process using Grignard Procedure) (Alternative
Procedure)
A reactor was charged with degassed THF (1090 kg) and 1-(benzyloxy)-4-bromobenzene (329 kg (1.46 equiv)). The solids were dissolved at 20 – 25 °C with agitation and under an inert atmosphere of nitrogen.
A reactor was charged with Mg turnings (31.9 kg (1.53 equiv)) and degassed THF (389 kg) under an inert atmosphere of nitrogen. To this mixture was added -5% of the l-(benzyloxy)-4-bromobenzene – THF solution (-70 kg) followed by heating to 50 – 60 °C. With good agitation, DIBAL-H in toluene (1.5M; 4.55 kg (0.0093 equiv)) was added followed by addition of toluene (2.16 kg) into the reactor through the charging line. The mixture was heated to 60
– 70 °C and aged for about 1 h. The remaining amount of the 1-(benzyloxy)-4-bromobenzene
– THF solution was added followed by a degassed THF rinse (51 kg) of the reactor. The mixture was aged for about 1 h at 60 – 70 °C and was cooled to 20 – 30 °C under an inert atmosphere of nitrogen.
A reactor was charged with degassed THF (1090 kg) and 1-(te/f-butyl) 2-methyl (S)-5-oxopyrrolidine 1 ,2-dicarboxylate (208 kg (1.0 equiv)), and the resulting solution was cooled to -60 to -70 °C under an inert atmosphere of nitrogen. To this solution was added the Grignard solution while maintaining a reaction temperature of <-50 °C. The reactor that contained the Grignard solution was rinsed with degassed THF (208 kg) and the reaction was aged at -60 to -70 °C for about 1 h. The progress of the reaction was monitored (HPLC).
Upon completion, 2-propanol (164 kg) was added while maintaining a reaction temperature of <-40 °C, and the reaction was aged for 20 – 30 min. Water (100 kg) was added while maintaining a reaction temperature of <-20 °C; the contents of the reactor were warmed to -10 to -20 °C following the addition. The mixture was transferred into another reactor and water (940 kg) was added while maintaining a reaction temperature of <10 °C; the contents of the reactor were warmed to 20 – 30 °C following the addition. The pH of the mixture was adjusted to 6.0 – 7.0 by addition of 50 wt% acetic acid in water (-170 kg). MTBE (647 kg) was added and the mixture was agitated for 20 – 30 min. The layers were separated, and the organic layer was stirred for 20 – 30 min with a brine solution prepared from NaCI (48 kg) and water (390 kg). The layers were separated, the organic layer was clarified by filtration and the filtration apparatus was washed with THF (30 kg). The solution was concentrated to about 5.5 – 6X the input mass of 1-(te/f-butyl) 2-methyl (S)-5-oxopyrrolidine 1 ,2-dicarboxylate at a temperature of 45 – 50 °C. MTBE (647 kg) was added and the resulting solution was concentrated to about 5.5 – 6X the input mass of 1-(te/f-butyl) 2-methyl (S)-5-oxopyrrolidine 1 ,2-dicarboxylate at a temperature of 45 – 50 °C. MTBE (661 kg) was added and the resulting solution was concentrated to about 5.5 – 6X the input mass of 1 -(te/f-butyl) 2-methyl (S)-5-oxopyrrolidine 1 ,2-dicarboxylate at a temperature of 45 – 50 °C. MTBE (77 kg) was added, the solution was sampled and analysed for residual THF content (if the result was >15%, MTBE (661 kg) was added and the solution was concentrated at 45 – 50 °C to about 5.5 – 6X the input mass of 1-(te/f-butyl) 2-methyl (S)-5-oxopyrrolidine 1 ,2-dicarboxylate). The solution was cooled to 35 – 45 °C and n-Heptane (726 kg) was added slowly while maintaining a reaction temperature of 35 – 45 °C. The mixture was aged for 1 – 2 h, cooled to 15 – 25 °C over 2 – 3 h, cooled to 0 – 5 °C and aged for 3 – 5 h. The solids were isolated by centrifugation and washed with n-heptane (214 kg). The wet solids (-328 kg) were dissolved in THF (683 kg) at 40 – 50 °C. The solution was cooled to 35 – 45 °C and n-heptane (564 kg) was added slowly while maintaining a reaction temperature of 35 – 45 °C. The mixture was aged for 1 -2 h, cooled to 15 – 25 °C over 2 – 3 h, cooled to 0 – 5 °C and aged for 3 – 5 h. The solids were isolated by centrifugation, washed with n-heptane (167 kg) and dried in vacuo at 50 – 60 °C to constant weight to afford 252 kg (69%) of the title compound.
Description 1c: Methyl (S)-5-(4-(benzyloxy)phenyl)-2-((fert-butoxycarbonyl)amino)-5-oxopentanoate (D1c) (Batch Process using Magnesium “ate” Procedure)
A reactor was charged with THF (249 kg) and the solvent was degassed by nitrogen sparging for about 30 min at 20 – 30 °C. To the degassed THF was charged l-(ferf-butyl) 2-methyl (S)-5-oxopyrrolidine 1 ,2-dicarboxylate (71 kg (1.0 equiv)), and the resulting solution was stirred at 20 to 30 °C under an inert atmosphere of nitrogen.
A reactor was charged with THF (460 kg) and the mixture was degassed by nitrogen sparging for about 30 min at 20 – 30 °C. To the degassed THF was charged 1-(benzyloxy)-4-bromobenzene (93 kg (1.2 equiv)) and the solution was degassed in triplicate. The solution was cooled to -40 to -50 °C under an inert atmosphere of nitrogen. To this solution was added /‘-PrMgCI – THF solution (51.3 kg, 2M; 0.36 equiv) while maintaining a reaction temperature of <-40 °C. To this solution was added n-BuLi – hexane solution (71.3 kg, 2.5M; 0.90 equiv) while maintaining a reaction temperature of <-40 °C. The contents of the reactor were aged at -40 to -50 °C for 1 – 1.5 h. The solution was cooled to -60 to -70 °C under an inert atmosphere of nitrogen.
The 1-(te/f-butyl) 2-methyl (S)-5-oxopyrrolidine 1 ,2-dicarboxylate – THF solution was added to the reactor containing the organomagnesium “ate” solution while maintaining a reaction temperature of -60 to -70 °C; the contents of the reactor were aged for about 1 h. The progress of the reaction was monitored (HPLC).
Upon completion, 10% NH4CI solution (389 kg) was added while maintaining a reaction temperature of < -40 °C. Following the addition, the contents of the reactor were warmed to 20 – 30 °C. The pH of the mixture was adjusted to 6 – 7 by addition of 50 wt% acetic acid in water (24.4 kg). n-Heptane (97 kg) was added and the mixture was agitated for 20 – 30 min at 20 – 30 °C. The layers were separated and the organic layer was concentrated in vacuo to about 270 L at <50 °C. The contents of the reactor were cooled to 20 – 30 °C and n-heptane (490 kg) was added followed by slurry aging for 2 – 3 h. The slurry was cooled to 0 – 5 °C and aged for 2 – 3 h. The solids were isolated by filtration, washed with a solution composed of n-heptane (58 kg) and THF (25 kg) and were dried in vacuo at 50 – 60 °C to constant weight to afford 102.95 kg (82.5%) of the title compound.
A reactor was charged with the title compound (102.95 kg) and THF (469 kg). The contents of the reactor were warmed to 40 – 50 °C, aged for 1 – 2 h, cooled to 20 – 30 °C and concentrated to a volume of about 250 L. n-Heptane (490 kg) was added and the mixture was agitated for 2 – 3 h at 20 – 30 °C. The mixture was cooled to 0 – 5 °C and aged for 2 – 3 h. The solids were isolated by filtration, washed with n-heptane (213 kg) and dried in vacuo at 50 – 60 °C to constant weight to afford 87.95 kg (70.5%) of the title compound.
Description 1d: Methyl (S)-5-(4-(benzyloxy)phenyl)-2-((fert-butoxycarbonyl)amino)-5-oxopentanoate (Did) (Batch Process using Turbo Grignard Procedure)
A clean 100 mL EasyMax reactor was swept with dry nitrogen, the flow was reduced and /‘-PrMgCI-LiCI complex in THF (41.7g, 1.3M, 1.0 eq) was added to the reactor and the temperature was set to 20 °C. Bis(dimethylamino)ethyl ether (9.13 g, 1.0 eq) was added in a single portion, the mixture was stirred for 5 min, and 4-benzyloxybromobenzene (15.0 g, 1.0 eq) was added in a single portion. The reaction was heated to 40°C under an inert atmosphere of nitrogen and held at this temperature until full conversion was observed (ca. 3.5h).
A clean 100 mL EasyMax reactor was swept with dry nitrogen, the flow was reduced and dry THF (45 mL). 1-(te/f-butyl) 2-methyl (S)-5-oxopyrrolidine 1 ,2-dicarboxylate (5.0 g, 1.0 eq) was charged in a single portion and the solution was cooled to -35 °C under an inert atmosphere of nitrogen. The Grignard solution (26.4 mL, 0.85M, 1.1 eq) was then added at a rate of 0.5 mL/min while maintaining a reaction temperature of <-30°C. The progress of the reaction was monitored (HPLC). Upon completion the reaction was neutralized by the addition of a 14.6 wt% AcOH/water solution (24 mL). The reaction was then warmed to -10 °C, then to 0 °C. A 20% aqueous NH4CI solution (10.3 g) was added followed by a pH adjustment with 1 M HCI (14 mL), then with 6M HCI to an endpoint of pH 1. The reaction mixture was transferred to a separatory funnel with the aid of 25 ml of THF. The phases were separated and the organic layer washed with saturated aqueous NaCI solution (16 g). The organic layer was concentrated under reduced pressure at <50°C to afford a crude product solution (19.4 g).
The crude product solution was transferred to a clean 100 mL EasyMax reactor and was heated to 35 °C. Heptane (20 mL) was then added over about 30 sec. The mixture was cooled to 10°C and held for about 30 min. The solids were filtered, washed twice with 2: 1 heptane/MTBE mixture (14 mL) and dried to constant weight to afford 4.147 g (47%) of the title compound.
Description 1e: Methyl (S)-5-(4-(benzyloxy)phenyl)-2-((fert-butoxycarbonyl)amino)-5-oxopentanoate (Die) (Flow Process using Intermittent Continuous Stirred Tank Reactor)
Reactor 1 was charged with 1-(benzyloxy)-4-bromobenzene (145 g (1.0 eq)) and the reactor was flushed with nitrogen. THF (490 g) was added and solids were dissolved at 20 – 30 °C by agitation; the solution was kept under an inert atmosphere of nitrogen.
Reactor 2 was charged with Mg (13.66 g (1.02 eq relative to reactor 1 charge)) and the reactor was flushed with nitrogen. Iodine (0.14 g (0.001 eq relative to the 1-(benzyloxy)-4-bromobenzene charge)) was charged followed by addition of 5% of the prepared 1-(benzyloxy)-4-bromobenzene – THF solution. The contents of the reactor were warmed to 50 – 65 °C and after color dissipation, the remainder of the prepared 1-(benzyloxy)-4-bromobenzene – THF solution (Reactor 1) was added while maintaining a reaction temperature of 50 – 70 °C. The contents of the reactor were stirred at 60 – 70 °C for about 1 h, cooled to 20 – 30 °C and held under an inert atmosphere of nitrogen.
Grignard Solution Batch 1
Reactor 3 was charged with 1-(benzyloxy)-4-bromobenzene (2.755 kg (1.0 eq)) and the reactor was flushed with nitrogen. THF (9.29 kg) was added and solids were dissolved at 20 – 30 °C by gentle agitation; the solution was kept under an inert atmosphere of nitrogen. Reactor 4 was charged with Mg (259.2 g (1.02 eq relative to the reactor 3 charge)) and the reactor was flushed with nitrogen. The contents of Reactor 2 were charged and the mixture was warmed to 50 – 65 °C. The prepared 1-(benzyloxy)-4-bromobenzene – THF solution in Reactor 3 was added while maintaining a reaction temperature of 50 – 70 °C. The contents of the reactor were stirred at 60 – 70 °C for about 1 h and cooled to 20 – 30 °C. About 95% of this Grignard solution was transferred into Reactor 5 and held under an inert atmosphere of nitrogen. A sample was pulled from Reactor 5 for analysis (residual 1-(benzyloxy)-4-bromobenzene (HPLC); Grignard reagent concentration). The remaining 5% of this Grignard solution was held in Reactor 4 under an inert atmosphere of nitrogen.
Grignard Solution Batch 2
Reactor 3 was charged with 1-(benzyloxy)-4-bromobenzene (2.90 kg (1.0 eq)) and the reactor was flushed with nitrogen. THF (9.78 kg) was added and solids were dissolved at 20 – 30 °C by gentle agitation; the solution was kept under an inert atmosphere of nitrogen.
Reactor 4 was charged with Mg (273.1 g (1.02 eq relative to the reactor 3 charge)) and the mixture was warmed to 50 – 65 °C. The prepared 1-(benzyloxy)-4-bromobenzene – THF solution in Reactor 3 was added while maintaining a reaction temperature of 50 – 70 °C. The contents of the reactor were stirred at 60 – 70 °C for about 1 h and cooled to 20 – 30 °C. About 95% of this Grignard solution was transferred into Reactor 6 and held under an inert atmosphere of nitrogen. A sample was pulled from Reactor 6 for analysis (residual 1-(benzyloxy)-4-bromobenzene (HPLC); Grignard reagent concentration). The remaining 5% of this Grignard solution was held in Reactor 4 under an inert atmosphere of nitrogen.
Grignard Solution Batch 3
Reactor 3 was charged with 1-(benzyloxy)-4-bromobenzene (2.90 kg (1.0 eq)) and the reactor was flushed with nitrogen. THF (9.78 kg) was added and solids were dissolved at 20 – 30 °C by gentle agitation; the solution was kept under an inert atmosphere of nitrogen.
Reactor 4 was charged with Mg (273.2 g (1.02 eq relative to the reactor 3 charge)) and the mixture was warmed to 50 – 65 °C. The prepared 1-(benzyloxy)-4-bromobenzene – THF solution in Reactor 3 was added while maintaining a reaction temperature of 50 – 70 °C. The contents of the reactor were stirred at 60 – 70 °C for about 1 h, cooled to 20 – 30 °C and held under an inert atmosphere of nitrogen. A sample was pulled for analysis (residual 1-(benzyloxy)-4-bromobenzene (HPLC); Grignard reagent concentration).
Reaction of Grignard Reagent with l-(ferf-butyl) 2-methyl (S)-5-oxopyrrolidine 1 ,2-dicarboxylate
The reaction was performed in 12 cycles; a representative cycle is described below. In total, 6.46 kg of 1-(te/f-butyl) 2-methyl (S)-5-oxopyrrolidine 1 ,2-dicarboxylate was processed forward to the title compound.
Reactor 7 was charged with 1-(te/f-butyl) 2-methyl (S)-5-oxopyrrolidine 1 ,2-dicarboxylate (2.21 kg) and THF (5.89 kg) and the solids were dissolved at 20 – 30 °C by gentle agitation under an inert atmosphere of nitrogen.
Reactor 8 was charged with THF (0.98 kg) and the solvent was cooled to about -10 °C under an inert atmosphere of nitrogen. Solutions of the Grignard reagent (3.2 kg) in Reactor 6 and the 1-(te/f-butyl) 2-methyl (S)-5-oxopyrrolidine 1 ,2-dicarboxylate – THF solution (2.0 kg) in Reactor 7 were simultaneously pumped into Reactor 8 over 15 min while maintaining a reaction temperature of <30 °C. The contents of Reactor 8 were stirred for an additional 15 min; the final reaction temperature was 0 – 10 °C. The contents of Reactor 8 were transferred to Reactor 9, cooled to about -5 °C and the reaction was quenched by addition of 1 M aqueous H2SO4 solution (1.20 equiv) while maintaining a reaction temperature of <10 °C. The mixture was stirred for 30 min, was transferred to Reactor 10 and was heated to 25 – 30 °C. The mixture was transferred to Reactor 1 1 , toluene (2.39 kg) was charged and the mixture was agitated. The mixture was transferred to Settler 1 and the organic layer was transferred to Reactor 12 using a metering pump. Water (1.65 kg) wash charged to Reactor 12, the mixture was agitated, transferred to Settler 2 and the organic layer was transferred to a storage container using a metering pump.
Product Isolation
The contents of the storage container (organic streams from 12 reaction cycles) was concentrated in Reactor 13 to an endpoint of 65 °C (pot temperature) at 200 torr. The contents of the reactor were cooled to 30 °C, then to 0 to -10 °C and aged for 0.5 – 2 h. The solids were isolated by filtration, washed with toluene (7.50 kg) and dried in vacuo at 50 °C and < 10 torr to give 8.76 kg (77%) of the title compound.
Description 1f: Methyl (S)-5-(4-(benzyloxy)phenyl)-2-((fert-butoxycarbonyl)amino)-5-oxopentanoate (D1f) (Flow Process using Plug Flow Reactor)
A flow reactor with two reagent inputs, ¾ inch tubing for reagent transfer, and two ½ inch jacketed static mixers connected in series (35 mL volume) was assembled. Gear pumps were used to transfer reagents to the flow reactor. Mass flow meters were used to measure the flow rates of the reagents. Thermocouples were placed to monitor the temperature of the (4-benzyloxy)phenylmagnesium bromide (Grignard) and l-(terf-butyl) 2-methyl (S)-5-oxopyrrolidine 1 ,2-dicarboxylate solutions prior to entering the tube-in-tube mixer T, as well as the out-flowing reaction stream from the static mixers. A fourth thermocouple measured the
temperature of the collection vessel. A peristaltic pump was used to transfer an aqueous acetic acid quench solution to the reaction stream as it exited from the static mixers. A standard T-mixer was used to join these reaction streams. The quenched reaction mixture flowed through a cooled coil into a jacketed collecting vessel. The approximate residence time through the static mixers was calculated to be -4.5 seconds.
Solution A: 0.57M (4-benzyloxy)phenylmagnesium bromide (Grignard) solution in THF (1.3 equiv used).
Solution B: 0.44M l-(ferf-butyl) 2-methyl (S)-5-oxopyrrolidine 1 ,2-dicarboxylate (0.750 kg) in THF (6.5 L)
Solution C: 2.9M glacial acetic acid (517 g) in water (3.013 L) to provide a 2.9 M solution. The quenched reaction mixture flowed into a collecting vessel containing 20% aqueous NH4CI (1.465 kg) at 0 °C.
The pre-cooling loop for Solution B was set to a bath temperature of -20 to -22 °C. The static mixer jacket coolant was set to a temperature of -25 °C. The pre-cooling loop for Solution A was set to a jacket temperature of -5 °C. The continuous quench tube reactor was set to a bath temperature of 0 °C.
After the jacket temperatures and cooling baths were allowed to reach desired temperatures, Solution A was pumped at a rate of -250 mL/min through the outside tube of the tube-in-tube mixer and met the Solution B that was pumped through the inner tube at a rate of 250 mL/min. Simultaneously to the reagent streams, the flow rate of the 2.9M aqueous acetic acid solution was initiated and set to approximately 130 mL/min. Reagent flow rates were measured with mass flow meters and temperatures were measured with thermocouples.
The reaction was run for about 20 min; a total of 5.663 kg of Solution B, 6.237 kg of Solution A and 3.530 kg of 2.9M aqueous acetic acid solution were charged during the reaction. The lines were rinsed with THF (1.252 kg) immediately after the reaction was finished.
The pH of the aqueous layer in the collection vessel was measured at 6.08. The pH was adjusted to 5.05 with 1 N HCI (2.05 kg) followed by the addition of 1V: 1V AcOH/water (162 g). The reactor jacket temperature was set to 10 °C and the contents of the reactor were stirred for 12 h. The pH of the mixture was further adjusted to 2.06 by adding 37% HCI (0.301 kg) and the mixture was stirred at 0 – 10 °C for 15 to 30 min.
The aqueous layer was separated and the organic layer was stirred for 20 min with a 25% brine solution (1.995 kg). The aqueous layer was separated; the organic layer was held at 10 °C overnight. The organic layer was concentrated at 35 – 40 °C (jacket temperature) and 25-30 mm Hg. Upon reaching a volume of about 9.5 L, a well developed slurry was noted. The concentration was continued to a volume of about 4.5 L. The slurry was warmed to 31 °C and heptane (3.145 kg) was added. The slurry was heated to 35 °C, stirred for 30 min, and was cooled to and held at 20 to 22 °C. The slurry was cooled to 10 °C and stirred for at least 2 h. Solids were collected by filtration and washed with 2: 1 heptane/MTBE (2 x 1.5 L). The solids were dried to constant weight in vacuo to yield 990 g (86.8%) of the title compound.
Description 1g: methyl (S)-5-(4-(benzyloxy)phenyl)-2-((tert-butoxycarbonyl)amino)-5-oxopentanoate
A reactor was charged with degassed THF (1 199 kg) and 1-(benzyloxy)-4-bromobenzene (450 kg). The solids were dissolved at 20 – 25 °C with agitation and under an inert atmosphere of nitrogen. The mixture was heated to reflux for 15 min, then cooled to 20 – 30 °C.
A reactor was charged with Mg turnings (43.6 kg) and degassed THF (399 kg) under an inert atmosphere of nitrogen. To this mixture, a solution of DIBAL-H (25% in toluene, 6.2 kg) was added followed by addition of toluene (3.7 L) into the reactor through the charging line. The mixture was heated to reflux for 10 – 15 minutes followed by charging of 5% of the 1-(benzyloxy)-4-bromobenzene – THF solution. The contents of the reactor were held for 1 h under reflux; reaction initiation was confirmed. The remainder of the 1-(benzyloxy)-4-bromobenzene – THF solution was added over 3 – 4 h. Following the charge, the temperature was adjusted to 20 – 30 °C.
A reactor was charged with degassed THF (760 kg) and 1-(te/f-butyl) 2-methyl (S)-5-oxopyrrolidine 1 ,2-dicarboxylate (284.9 kg), and the resulting solution was heated to reflux under an inert atmosphere of nitrogen, maintained at reflux for 10 – 15 min, then cooled to -60 °C to -70 °C. To this solution was added the Grignard solution while maintaining a reaction temperature of <-50 °C. The reactor which contained the 1-(benzyloxy)-4-bromobenzene -THF solution was rinsed with degassed THF (22 kg) and the rinse was charged into the reaction. The contents of the reactor were aged at -60 to -70 °C for about 1 h. The progress of the reaction was monitored for completion (HPLC).
A reactor was charged with 2-propanol (285 L) and THF (253 kg). With good agitation the reaction was quenched into this THF – 2-propanol solution while keeping the temperature between -20 °C and 0 °C. The reactor was rinsed forward with THF (53 kg), and the mixture was stirred vigorously for 5 – 10 min. Water (712 L) was added while maintaining a reaction temperature of <20 °C; the pH of the mixture was adjusted to 6.0 – 7.0 by addition of 50 wt% acetic acid in water (-170 kg) while controlling the temperature below 20 °C. The reaction mixture was warmed to 20 – 30 °C, stirred for 20 – 30 min and the phases were separated. Sodium chloride (42 kg) and water (255 L) were charged, the mixture was stirred for 55 – 65 min, and the phases were separated. THF (125 kg) was charged and the solution was concentrated by distillation under vacuum at a temperature of 40 – 45 °C. The distillation was stopped when the weight of the reaction mixture was between 5.5 – 6. OX the weight of the input mass of 1-(te/f-butyl) 2-methyl (S)-5-oxopyrrolidine 1 ,2-dicarboxylate. The reaction mixture was heated to 35 – 45 °C. Heptane (994 kg) was charged to the reaction mixture, the contents of the reactor were maintained at 35 – 45 °C, aged for 1 – 2 h, cooled to 15 – 25 °C over 2 – 3 h, cooled to 0 – 5 °C and aged for 3 – 5 h. The solids were isolated by centrifugation in three portions; each portion was washed with heptane (97 kg) followed by acetonitrile (59 kg) to give 389 kg of wet product. Based on LOD measurements, 375.3 kg (76.6 %) of the title compound was obtained.
Description 1 h: benzyl (S)-5-(4-(benzyloxy)phenyl)-2-((tert-butoxycarbonyl)amino)-5-oxopentanoate
A reactor was charged with 1-benzyl 2-methyl (S)-5-oxopyrrolidine 1 ,2-dicarboxylate (69.3 g) and anhydrous THF (450 g) and the resulting solution was cooled to about -65 °C under an
inert atmosphere of nitrogen. A solution of 0.8M (4-benzyloxy)phenylmagnesium bromide in THF (1.1 eq) was added over about 2 h, and the progress of the reaction was monitored by HPLC. Upon completion, the reaction was quenched by simultaneous addition of 1 M sulfuric acid (1.1 eq) and toluene (264 g) over about 30 min. The resulting mixture was warmed from -10 °C to ambient temperature and was aged for about 30 min. The phases were separated, and the organic layer was washed with 10 wt% brine (180 g) and water (180 g). The organic solution was concentrated to about 6V at about 50 °C and <170 mbar (distillate: 650 g / 710 ml_). The resulting solution was heated to about 65 °C and a solution of toluene (105 g) and methylcyclohexane (200 g) was added dropwise while maintaining a temperature of about 65 °C. The solution was cooled to 0 – 5 °C and aged for about 1 h. The solids were isolated by filtration, washed with cold (0 – 5 °C) methylcyclohexane (200 g in 6 portions) and dried at 45 °C in vacuo to constant weight to give 76.6 g (66%) of the title compound.
Description 1 i: methyl (S)-2-(((benzyloxy)carbonyl)amino)-5-(4-(benzyloxy)ph oxopentan
Solution A: 0.8M (4-benzyloxy)phenylmagnesium bromide solution in THF
Solution B: 0.88M 1 -benzyl 2-methyl (S)-5-oxopyrrolidine 1 ,2-dicarboxylate (25.0 g) solution in anhydrous THF
Solution C: 1 M aqueous sulfuric acid
Equipment: plug flow reactor with a Y-mixer; 10 ml_ reaction loop
Reaction conditions:
· reagent flow rates:
o solution A: 5.27 ml_ / min (1.3 eq)
o solution B: 4.72 ml_ / min (1.0 eq)
o solution C: 5.75 ml_ / min (1.5 eq)
• residence time: 1 min
· reaction temperature: 25 °C
• collection time: 2 h (theory: 0.36 mol title product)
• the quenched reaction mixture flowed into a collecting vessel
Following collection of the quenched reaction mixture, the phases were separated and the upper organic layer was concentrated to dryness in vacuo. The solids were dissolved in fresh THF (5.5V) at 45 °C. The solution was cooled to -5 °C over about 160 min and was aged overnight. The solids were collected by filtration, washed with heptane (5.5V, total) and dried to constant weight at 55 °C in vacuo to afford 18.61 g (45%) of the title product.
The combined filtrate and wash containing additional solids was transferred to a reactor, cooled to -5 °C over 2 h and aged for an additional 4 h. The solids were collected by filtration, washed with heptane (2 X 2V) and dried to constant weight at 55 °C in vacuo to afford 1 1.37 g (27%) of the title product.
Description 1j: methyl (S)-5-(4-(benzyloxy)phenyl)-2-((tert-butoxycarbonyl)amino)-5-oxopentanoate – flow chemistry procedure
The CSTR flow setup consists of one 1 L stirred tank for reaction, one 1 L settling tank and one 10L Schlenk type collection vessel. The stirred tank was equipped with a solid addition device, a reflux condenser, and a dip-tube (set to a 500 ml_ working volume) with an inner transfer line.
Step 1 : A stirred tank reactor was pre-charged with THF (70 ml), and magnesium (50.8 g, 5 eq), and stirred at room temperature overnight. The solid addition device was filled with magnesium. The reaction was initiated by adding (4-(benzyloxy)phenyl)magnesium bromide 0.77M solution (7.7 g, 5.9 mmol). The jacket temperature was increased to 55 °C. A solution of 1-bromo-4-benzylphenol (0.85 M in THF) was added at a rate of 7.8 ml/min to the stirred reaction vessel. After seven minutes, solid addition of magnesium started at a rate of 0.161 g/min. The total amount of magnesium for the entire run was (175 g, 7.18 mol,
1 equiv) and was calculated to keep 5 eq of magnesium in the stirred tank reactor over the course of the run. When the liquid level in the tank reached the level of the dip tube, a pump activated pulling material to the settling tank at a rate to maintain the 500 mL filling level in the CSTR. The approximate residence time of the solution in the jacketed reactor was 62 minutes. The product was transferred into the settling tank (unstirred), held for another residence time (1 hour), and subsequently transferred to a final collection vessel. The entire process was run for 18 hours.
Step 2: Grignard Addition: The equipment consists of tubular pipe reactor, heat exchanger, and a series of centrifugal phase separators. The tubular reactor accommodates mixing of two reagents for the conversion to methyl (S)-5-(4-(benzyloxy)phenyl)-2-((tert-butoxycarbonyl)amino)-5-oxopentanoate and quenching of the product solution with an acid solution. The centrifugal phase separators separate the product containing organic phase from the waste aqueous phase. The reagent (methyl-N-boc-pyroglutamate, Grignard, and sulfuric acid solutions were transferred continuously at controlled flowrates from their respective storage tank to pass through the tubular pipe reactor, heat exchanger and finally to the centrifugal extractors.
Reaction/Quench/Work-up: The 0.82 M Grignard solution was fed continuously from the storage tank at a flow rate of 32.6 mL/min (1.19 eq), simultaneously a 0.817 M methyl N-boc-pyroglutamate solution stream was fed continuously at 27.4 ml/min through a heat exchanger to pre-cool it to -8°C. The tubular reactor where the reaction between the reagent N-boc-pyroglutamate and Grignard solution occurred was attached to a heat exchange unit with chiller fluid set at 10°C. After passing through the reaction zone, 1.0 M sulfuric acid was introduced at a rate 22.4 ml/min. The residence time of the solution from reagent introduction to acid quench was 8 seconds. From sulfuric acid introduction to phase split the residence time was ca. 80 seconds. The quenched mixture passed through another heat exchanger to increase the temperature to 30°C for phase split. This material was directly fed into a centrifugal extractor to remove the aqueous component. The obtained organic layer was subsequently mixed with a solution of brine and sodium bicarbonate (14.5 ml/min) in a second centrifugal extractor. The final product containing organic layer was collected into a glass bottle. The process was run for 3.7 hours.
Crystallization: The product-containing organic layer above was transferred to a 10 L reactor for solvent switch to a lower water content THF-Heptane solvent system by vacuum distillation. A total of 6867 mL THF (appx. 9.5% v/v) in Heptane was added to the reactor and
subsequently distilled in appx. 2 equal portions maintaining distillation under reduced pressure (appx. 600-700 mbar) at temperature within 60-65°C to replace the original solvent (water-containing THF).3 The final solution obtained (appx. 11.5L) was cooled to 0-5°C with a cooling rate 0.5C/min and the resulting slurry was filtered, washed with Heptane and dried under vacuum at 60°C to obtain 1.765 kg of product.
Description 2a: Methyl (S)-5-(4-(benzyloxy)phenyl)-3,4-dihydro-2H-pyrrole-2-carboxylate
A reactor was charged with methyl (S)-5-(4-(benzyloxy)phenyl)-2-((te/f-butoxycarbonyl)amino)-5-oxopentanoate (180 kg) and ACN (486 kg) and the slurry temperature was adjusted to 10 – 15 °C. A solution of methanesulfonic acid (117.5 kg (2.9 eq)) in ACN (75 kg) was added while maintaining a reaction temperature of <25 °C. The reaction temperature was adjusted to 22 – 26 °C and the contents of the reactor were stirred for 1 – 1.5 h. The progress of the reaction was monitored (HPLC). Upon completion, the contents of the reactor were cooled to 10 – 15 °C and a solution of 4. ON NH4OH (299 kg) was added to a pH of 7 – 8 while maintaining a reaction temperature of <25 °C. The phases were separated and the upper organic layer was heated to 30 – 40 °C. While maintaining a reaction temperature of 30 – 40 °C, 2-propanol (101 kg) and water (430 kg) were added to the reactor. The solution was cooled to 17 – 19 °C and was seeded (1.8 kg). The slurry was stirred for 1 – 2 h at 14 – 19 °C, cooled to 7 – 12 °C, aged for 1 – 2 h and cooled to 2 – 7 °C. Water (890 kg) was added and the slurry was aged for 2 – 3 h at 2 – 7 °C. The solids were isolated by filtration, washed with a solution composed of 2-propanol (61 kg) and water (270 kg) and dried in vacuo at 50 – 60 °C to constant weight to afford 1 19.6 kg (90%) of the title compound.
Description 2b: Methyl (S)-5-(4-(benzyloxy)phenyl)-3,4-dihydro-2H-pyrrole-2-carboxylate (D2b) (Alternative Procedure)
A reactor was charged with methyl (S)-5-(4-(benzyloxy)phenyl)-2-((tert-butoxycarbonyl)amino)-5-oxopentanoate (532 kg) and ACN (1670 kg) and the slurry temperature was adjusted to 20 – 25 °C. Methanesulfonic acid (346 kg (2.9 eq)) was added while maintaining a reaction temperature of <26 °C. The contents of the reactor were stirred for 1 h; the progress of the reaction was monitored (HPLC). Upon completion, the contents of the reactor were cooled to <10 °C and a solution of 4.6N NH40H (773 kg) was added until a pH of 7 – 8 was reached while maintaining a reaction temperature of <25 °C. The phases were separated and the upper organic layer was heated to 30 – 35 °C. The organic layer was filtered through a plate filter to remove small particulates. While maintaining a reaction temperature of 30 – 35 °C, 2-propanol (301 kg) and water (1277 kg) were added to the reactor. The solution was cooled to 18 – 22 °C and precipitation occurred. The slurry was stirred for at least 30 minutes at 18 – 22 °C and then cooled to 0 – 10 °C. While maintaining a temperature of 0 – 10 °C, water (2128 kg) was added and the reaction mixture was aged for not less than 2 hours at 0 – 10 °C. The solids were isolated by filtration, washed with a solution composed of 2-propanol (188 kg) and water (798 kg) and dried in vacuo at 50 – 55 °C to constant weight to afford 319 kg (83%) of the title compound.
Description 2c: methyl (S)-5-(4-(benzyloxy)phenyl)-3,4-dihydro-2H-pyrrole-2-carboxylate – flow chemistry procedure with MsOH/ACN
Solution A: 0.79M methanesulfonic acid in anhydrous ACN
Solution B: 0.25M l-(terf-butyl) (S)-5-(4-(benzyloxy)phenyl)-2-((tert-butoxycarbonyl)amino)-5-oxopentanoate solution in anhydrous THF
Solution C: 4.6N NH4OH solution in water
Equipment: plug flow reactor with a Y-mixer; 10 ml_ stainless steel reaction loop
Reaction equivalents:
· solution A: 3.0 (3.764 mL / min)
• solution B: 1.0 (3.946 mL / min)
• solution C: 2.7 (0.579 mL / min)
Residence time: 1.3 min
Reaction temperature: 130 °C
After reaching steady state, the reaction stream was collected for 102 min in a 1 L flask immersed in an ice water bath. The base solution from pump C and the reaction stream
were simultaneously collected with good stirring. Following the run, the pH was adjusted to 7 with by charging additional 4.6N ammonium hydroxide solution (about 15 mL). The phases were split, and the organic layer was concentrated to dryness by rotary evaporation in vacuo. The resulting residue was dissolved in ACN (120 mL) and distilled water (5 mL) at 25 °C and 500 rpm in a 100 mL EZMax reactor. The solution was cooled to 22 °C and water – I PA solution (2/1 (v/v), 80 mL) was added over about 30 min. The solution was further cooled to 18 °C, seeded (5 wt%) and cooled to about 0 °C over 2 h. Water (139 mL) was added to the slurry over about 30 min, and the mixture was aged for about 20 min. The temperature of the slurry was raised to 20 °C, held for about 40 min, re-cooled to about 0 °C over 90 min and aged for an additional 90 min. The solids were collected by filtration and dried to constant weight in vacuo at 55 °C to give 28.9 g (92%, corrected for seed) of the title compound.
Description 2d: methyl (S)-5-(4-(benzyloxy)phenyl)-3,4-dihydro-2H-pyrrole-2-carboxylate – flow chemistry procedure with H2SO4/ACN
Three solutions were prepared for the flow reaction. Solution A: 0.25M l-(terf-butyl) (S)-5-(4-(benzyloxy)phenyl)-2-((tert-butoxycarbonyl)amino)-5-oxopentanoate solution in anhydrous THF; Solution B: 0.75M sulfuric acid in anhydrous ACN;
A plug flow reactor with a Y-mixer and a 10 mL reaction loop was used with 1 reaction equivalent of solution A, and 2 reaction equivalents of solution B; a residence time of 7.5 minutes; a reaction temperature of 95 °C; and a collection time: 73.7 minutes (theory: 22.1 mmol title product).
The collected product stream was neutralized to pH 7 – 8 using 4.6N NH4OH solution in water. HPLC analysis of the organic layer showed it contained 98.0 area% of the desired product. The lower organic layer was removed, and the organic layer was cooled to about 22 °C, aged for about 30 min and cooled to 0 – 5 °C over about 1 h. Water (38 mL) was added over 10 min, and the resulting slurry was filtered, and was washed with a solution composed of IPA (0.45V) and water (1.5V). The solids were dried in vacuo at 55 °C to yield 2.62 g (38%) of the title compound.
Description 2e: methyl (S)-5-(4-(benzyloxy)phenyl)-3,4-dihydro-2H-pyrrole-2-carboxylate – flow chemistry procedure with MsOH/THF-PhMe
Solution A: 1.5M methanesulfonic acid in 1 : 1 (v/v) anhydrous THF – anhydrous PhMe Solution B: 0.25M l-(terf-butyl) (S)-5-(4-(benzyloxy)phenyl)-2-((tert-butoxycarbonyl)amino)-5-oxopentanoate solution in anhydrous THF
Solution C: 4.6N NH4OH solution in water
Equipment: plug flow reactor with a Y-mixer; 10 ml_ PFA coil reactor
Reaction equivalents:
• solution A: 3.0 (1.667 mL / min)
· solution B: 1.0 (3.333 mL / min)
• solution C: 6.0 (1.087 mL / min)
Residence time: 2.0 min
Reaction temperature: 150 °C
After reaching steady state, the reaction stream was collected for 1 17 min in a 1 L flask immersed in an ice water bath. The base solution from pump C and the reaction stream were simultaneously collected with good stirring for the first 60 min; for the remainder of the collection time, only the reaction stream was collected. Following the run, the pH was adjusted to 7 with by charging additional 4.6N ammonium hydroxide solution. The phases were split, and the organic layer was concentrated to dryness by rotary evaporation in vacuo. The resulting residue was transferred to a 400 mL EZMax reactor using ACN (120 mL) and the temperature of the mixture was raised to 35 °C. To the mixture was added water – IPA solution (2/1 (v/v), 78 mL) over about 10 min. The resulting solution was cooled to 18 °C over about 30 min, seeded (208 mg), further cooled to about 0 °C over 2 h and aged overnight. Water (135 mL) was added to the slurry over about 1 h, and the mixture was aged for about 4 h. The temperature of the slurry was raised to 13 °C, re-cooled to about 0 °C over 3 h and aged overnight. The solids were collected by filtration and dried to constant weight in vacuo at 55 °C to give 8.18 g (27%, corrected for seed) of the title compound.
Description 2f: methyl (S)-5-(4-(benzyloxy)phenyl)-3,4-dihydro-2H-pyrrole-2-carboxylate – method A
A reactor was charged with methyl (S)-5-(4-(benzyloxy)phenyl)-2-((te/f-butoxycarbonyl)amino)-5-oxopentanoate (100.0 g) and ACN (400 ml_) and the reaction temperature was adjusted to about 25 °C. Concentrated sulfuric acid (45.3 g) was added over about 10 min while maintaining a reaction temperature of <50 °C. The contents of the reactor were stirred at 40 – 50 °C; the progress of the reaction was monitored for completion (HPLC). Upon completion, the reaction was cooled to about 25 °C. A solution of 4.6N NH4OH (215 ml_) was added with good stirring to a pH of about 7. The phases were separated, and the organic layer was split into two equal portions of about 256 ml_ for product isolation studies.
Portion A
To one portion was added a solution composed of 2-propanol (36.5 ml_) and water (120 ml_) with good stirring at about 22 °C. The resulting slurry was aged briefly at 22 °C, then cooled to 5 °C over about 1 h. Water (100 ml_) was added to the slurry while maintaining a reaction temperature of <10 °C. The solids were filtered, washed with a solution composed of 2-propanol (27.5 ml_) and water (75 ml_) and dried to constant weight in vacuo to give 30.87 g (85%) of the title compound.
Portion B
To one portion was added water (150 ml_) with good stirring at about 22 °C. The resulting slurry was aged briefly at 22 °C, then cooled to 5 °C over about 1 h. Water (100 ml_) was added to the slurry while maintaining a reaction temperature of <10 °C. The solids were filtered, washed with a solution composed of 2-propanol (27.5 ml_) and water (75 ml_) and dried to constant weight in vacuo to give 31.90 g (88%) of the title compound.
Description 2g: methyl (S)-5-(4-(benzyloxy)phenyl)-3,4-dihydro-2H-pyrrole-2-carboxylate – method B
A reactor was charged with methyl (S)-5-(4-(benzyloxy)phenyl)-2-((te/f-butoxycarbonyl)amino)-5-oxopentanoate (776.5 kg) and ACN (1743.5 kg) and the slurry temperature was adjusted to 15 – 25 °C. Methanesulfonic acid (482.1 kg) was added while maintaining a reaction temperature of <26 °C. The contents of the reactor were stirred at 20 – 25 °C for about 1 h. The progress of the reaction was monitored (HPLC); while awaiting results, the contents of the reactor were cooled to 0 – 10 °C. A solution of 4.6N NH4OH (590 kg) was added over about 25 min to a pH of 2 – 3 while maintaining a reaction temperature of <30 °C. Additional 4.6N NH4OH solution (519 kg) was added to a final pH of 7 – 8 while maintaining a reaction temperature of <25 °C. The phases were separated and the upper organic layer was heated to 25 – 30 °C. The organic layer was filtered and the filtrate was cooled to 20 – 25 °C. While maintaining this temperature range, a solution of 2-propanol (362.8 kg) and water (924.3 kg) were added to the reactor. The solution was cooled to 15 -20 °C and was seeded (3.7 kg, 0.5 wt%). The slurry was cooled to 0 – 5 °C over at least 2 h and aged for at least 30 min. Water (2403.1 kg) was added while maintain a reaction temperature of <20 °C. The slurry was cooled to 0 – 5 °C and aged for 30 – 40 min. The slurry was warmed to 15 – 20 °C, aged for 30 – 40 min, cooled to 0 – 5 °C over at least 1 h and aged for at least 2 h. The solids were isolated by filtration, washed with a solution composed of 2-propanol (283.2 kg) and water (1079.5 kg) and dried in vacuo at 50 – 55 °C to constant weight to afford 466.0 kg (87%) of the title compound.
Description 3a: l-(ferf-butyl) 2-methyl (2S, 5 ?)-5-(4-hydroxyphenyl)pyrrolidine-1 ,2-dicarboxylate (D3)
A hydrogenation reactor was charged with methyl (S)-5-(4-(benzyloxy)phenyl)-3,4-dihydro-2H-pyrrole-2-carboxylate (30 kg) and MeOH (120 kg), and the slurry was heated to solution at 30 – 40 °C. The solution was cooled to 15 – 25 °C followed by addition of d -tert-butyldicarbonate (21.8 kg, 1.03 eq) and water wet 20% Pd(OH)2/C (0.9 kg, 3 wt%). The
contents of the reactor were degassed under vacuum followed by pressurization with nitrogen. The contents of the reactor were degassed under vacuum followed by pressurization with hydrogen (3 – 4 bar). After 2 h at 22 – 27 °C, the reactor was vented and re-pressurized with hydrogen (3 – 4 bar). The progress of the reaction was monitored for completion (HPLC). After 4.5 h, the reactor was vented and MeOH (90 kg) was charged. The contents of the reactor were warmed to 32 – 42 °C and held for 20 – 30 min. The catalyst was removed by filtration through a bed of diatomite (13 kg) and the spent filter cake was washed with warm (40 – 45 °C) MeOH (25 kg). The combined filtrate and wash was concentrated in vacuo to 2 volumes at <40 °C and MeOH was charged (56 kg). The slurry was heated to 50 – 56 °C and the solution was aged for about 1.5 h. The solution was cooled to 20 – 30 °C, the slurry was aged for about 1 h, water (60 kg) was added and the slurry was aged for about 2 h. The slurry was cooled to about -5 °C and aged for about 8 h. The solids were isolated by centrifugation, washed with 1 :4 (v/v) MeOH – water (57.5 kg) and dried in vacuo at 50 – 60 °C to constant weight to afford 27.6 kg (88.5%) of the title compound.
Description 3b: 1 -(tert-butyl) 2-methyl (2S, 5R)-5-(4-hydroxyphenyl)pyrrolidine-1,2-dicarboxylate – method A
A hydrogenation reactor was charged with 20% Pd(OH)2/C (water wet; 5.7 kg), methyl (S)-5-(4-(benzyloxy)phenyl)-3,4-dihydro-2H-pyrrole-2-carboxylate (186.4 kg), MeOH (8.85V), water (20 kg) and di-te/f-butyldicarbonate (132 kg). The reactor was pressurized with nitrogen followed by venting (three times). The reactor was pressurized with hydrogen followed by venting (three times). The reactor was pressurized with hydrogen (15 bar). After about 2 h at 25 °C, the reactor was vented and re-pressurized with hydrogen (15 bar). The progress of the reaction was monitored for completion (HPLC). After about 4.25 h, the reactor was vented and its contents were filtered, and the filtrate was concentrated in vacuo to about 4.4 volumes at about 35 °C and at about 240 mbar. The contents of the reactor were reheated to 55 – 60 °C, the solution was cooled to 20 – 30 °C over about 2 h and the slurry was aged for about 1 h. Water (285 kg) was added over about 1 h and the slurry was aged for about 1 h. The slurry was cooled to 3 – 7 °C over about 2 h and aged for about 3 h. The solids were isolated by filtration, washed with 1 :4 (v/v) MeOH – water (359 kg) and dried in vacuo at 50 – 55 °C to constant weight to afford 174.6 kg (90%) of the title compound.
Description 3c: l-(tert-butyl) 2-methyl (2S, 5R)-5-(4-hydroxyphenyl)pyrrolidine-1 ,2-dicarboxylate – method B
A hydrogenation reactor was charged with 20% Pd(OH)2/C (water wet; 3 wt%), methyl (S)-5-(4-(benzyloxy)phenyl)-3,4-dihydro-2H-pyrrole-2-carboxylate (100 g), MeOH (4.5V), water (5 g) and 90 wt% di-te/f-butyldicarbonate in THF (1.00 eq). The reactor was pressurized with nitrogen followed by venting (three times). The reactor was pressurized with hydrogen followed by venting (three times). The reactor was pressurized with hydrogen (15 bar). After 1 h at 25 °C, the reactor was vented and re-pressurized with hydrogen (15 bar). The progress of the reaction was monitored for completion (HPLC). After 5 h, the reactor was vented and its contents were warmed to about 45 °C. The catalyst was removed by filtration through a warmed filter, and the filtrate was re-heated to 45 – 55 °C and held for about 30 minutes. The filtrate was concentrated in vacuo to about 4.4 volumes at 30 – 40 °C. The residue was cooled to 20 – 30 °C over at least 1 h, water (1.5V) was added over about 45 minutes and the slurry was aged for about 1 h. The slurry was cooled to 3 – 7 °C over about 2 h and aged for about 3 h. The solids were isolated by filtration, washed with 1 :4 (v/v) MeOH – water (2V) and dried in vacuo at 50 – 60 °C to constant weight to afford 88.9 g (86%) of the title compound.
Description 3d: 1 -(tert-butyl) 2-methyl (2S, 5R)-5-(4-hydroxyphenyl)pyrrolidine-1,2-dicarboxylate – flow chemistry procedure
reaction 2
The flow direction was from top to bottom (feed solution and hydrogen); and the hydrogen flow rate was 50 ml_ / min (while maintaining desired reaction pressure).
A 25 ml_ tube was packed with glass wool, sand, spherical catalyst beads (3% Pd/0-AI203 (1.0 – 1.2 mm spherical pellets)), sand and glass wool to give a 10 ml_ packed bed volume. 4 wt% methyl (S)-5-(4-(benzyloxy)phenyl)-3,4-dihydro-2H-pyrrole-2-carboxylate and di-ferf- butyldicarbonate (1.2 eq) in MeOH at -5 °C (feed solution 1) was then passed through the flow reactor at 0.08 – 0.10 ml_ / min, at a temperature of 53 – 61 °C and at a pressure of 10 – 15 bar. The collected solution contained a mixture of 1-(te/f-butyl) 2-methyl (2S,5f?)-5-(4- (benzyloxy)phenyl)pyrrolidine-1 ,2-dicarboxylate and 1-(te/f- butyl) 2-methyl (2S, 5f?)-5-(4- hydroxyphenyl)pyrrolidine-1 ,2-dicarboxylate in MeOH (feed solution 2) was passed through the flow reactor at 0.10 mL / min, at a temperature of 78 – 81 °C and at a pressure of 3 bar to produce about 600 g of a methanol solution primarily containing 1-(te/f-butyl) 2-methyl (2S, 5f?)-5-(4-hydroxyphenyl)pyrrolidine-1 ,2-dicarboxylate. This solution was concentrated in vacuo at a temperature of about 40 °C to a net weight of about 3.6X the amount of the input methyl (S)-5-(4-(benzyloxy)phenyl)-3,4-dihydro-2H-pyrrole-2-carboxylate. After stirring the mixture at ambient temperature for 15 – 20 min, water (2V) was added over about 30 min, the resulting mixture was aged for about 30 min, cooled to about 0 °C and aged for about 30 min. Solids were isolated by filtration, washed with ice cold 1 :4 (v/v) MeOH – water (2 X 1V) and dried to constant weight in vacuo at 55 °C to afford 23.51 g (88%) of the title compound.
Description 4a: iert-butyl (2S, 5/?)-2-carbamoyl-5-(4-((2- fluorobenzyl)oxy)phenyl)pyrrolidine-1-carboxylate (D4a) (K2CO3 / ACN procedure using 2-fluorobenzyl bromide)
A reactor was charged with 1-(te/f-butyl) 2-methyl (2S, 5f?)-5-(4-hydroxyphenyl)pyrrolidine- 1 ,2-dicarboxylate (1 10 kg), powdered K2CO3 (71.5 kg (1.5 equiv)) and ACN (429 kg). With good stirring, 2-fluorobenzyl bromide (68.2 kg (1.05 equiv)) and ACN (15 kg) were charged and the mixture was heated to 86 – 94 °C; the progress of the reaction was monitored (HPLC). Upon completion, the slurry was cooled to 40 – 50 °C, filtered and the spent filter cake was washed with fresh ACN (175 kg).
To the ACN filtrate was charged powdered K2CO3 (94.6 kg (2.0 equiv)) and formamide (308 kg (20 equiv)) and the mixture was heated to 86 – 94 °C; the progress of the reaction was monitored (HPLC). Upon completion, the slurry was cooled to 70 – 75 °C and water (1 150 kg) was added while maintaining a reaction temperature of >70 °C. Following the addition the solution was aged for about 30 min, cooled to 65 – 70 °C, seeded (0.55 kg) and aged for 3 – 4 h. The slurry was cooled to 50 – 60 °C, aged 3 – 4 h, cooled to 20 – 30 °C and aged for 3 – 4 h. The solids were isolated by centrifugation, washed twice with water (220 kg) and dried in vacuo at 30 – 40 °C for 4 – 8 h and at 50 – 60 °C for 4 – 8 h to yield 128.75 kg (87.5%) of the title compound.
Description 4b: iert-butyl (2S, 5 ?)-2-carbamoyl-5-(4-((2- fluorobenzyl)oxy)phenyl)pyrrolidine-1-carboxylate (D4b) (NaOMe – MeOH procedure using 2-fluorobenzyl bromide in DMF)
A reactor was charged with 1-(te/f-butyl) 2-methyl (2S, 5f?)-5-(4-hydroxyphenyl)pyrrolidine- 1 ,2-dicarboxylate (1.0 kg), anhydrous DMF (2.9 L), 2-fluorobenzyl bromide (430 mL (1.12 equiv)) and anhydrous DMF (0.1 L). The solution was cooled to about 15 °C. With good stirring, 741 mL (1.05 equiv) 4.4M NaOMe-MeOH solution was added while maintaining a temperature of <20 °C. Following the charge, the contents of the reactor were warmed to about 25 °C, aged for about 1 h and 44 mL (0.06 equiv) 4.4M NaOMe-MeOH solution was added over about 5 min. The progress of the reaction was monitored (HPLC).
Upon completion, formamide (2.5 L) was charged followed by addition of 81 1 mL (1.15 equiv) 4.4M NaOMe-MeOH solution while maintaining a temperature of <25 °C. The contents of the reactor were aged for about 1 h and 516 mL (0.73 equiv) 4.4M NaOMe-MeOH solution was added while maintaining a temperature of <25 °C. The progress of the reaction was monitored (HPLC). Upon completion, a solution of glacial acetic acid (350 mL (2.0 equiv) in water (2.2 L)) was added over about 10 min. The slurry was heated to about 70 °C and aged for about 1 h. Water (1.8 L) was added over about 1 h and the slurry was cooled to about 3 °C over 3 h and aged for about 10 h. The solids were isolated by filtration, washed twice with water (2 L) and dried to constant weight in vacuo at 80 °C to afford 1.21 kg (94%) of the title compound.
Description 4c: iert-butyl (2S, 5 ?)-2-carbamoyl-5-(4-((2- fluorobenzyl)oxy)phenyl)pyrrolidine-1-carboxylate (D4c) (NaOMe – MeOH procedure using 2-fluorobenzyl bromide in DMF) (Alternative Procedure)
A reactor was charged with 1-(te/f-butyl) 2-methyl (2S, 5f?)-5-(4-hydroxyphenyl)pyrrolidine- 1 ,2-dicarboxylate (100 g), anhydrous DMF (290 mL), 2-fluorobenzyl bromide (42.2 mL (1.10 equiv)) and anhydrous DMF (10 mL). The solution was cooled to about 15 °C. With good stirring, 75 mL (1.06 equiv) 4.4M NaOMe-MeOH solution was added over a period of approximately 30 min while maintaining a temperature of <20 °C. Following the charge, the contents of the reactor were warmed to about 25 °C and aged for about 2 h. The progress of the reaction was monitored (HPLC).
Upon completion, formamide (250 mL) was charged followed by addition of 133 mL (1.88 equiv) 4.4M NaOMe-MeOH solution over approximately 45 min while maintaining a temperature of <25 °C. The contents of the reactor were aged for about 4 h. The progress of the reaction was monitored (HPLC). Upon completion, a solution of glacial acetic acid (35 mL (2.0 equiv) in water (100 mL) was added over about 30 min. The slurry was heated to about 60 °C. Water (300 mL) was then charged to the reactor over about 1 h, and the slurry was aged for about 1 h. The slurry was cooled to about 3 °C over 3 h and aged for about 1 h. The solids were isolated by filtration, washed twice with water (200 mL) and dried to constant weight in vacuo at 80 °C to afford 120.0 g (93%) of the title compound.
Description 4d: iert-butyl (2S, 5/?)-2-carbamoyl-5-(4-((2- fluorobenzyl)oxy)phenyl)pyrrolidine-1-carboxylate (D4d) (NaOMe – MeOH procedure using 2-fluorobenzyl chloride in DMF)
A reactor was charged with 1-(te/f- butyl) 2-methyl (2S, 5R)-5-(4-hydroxyphenyl)pyrrolidine- 1 ,2-dicarboxylate (7.50 g), anhydrous DMF (22.5 mL) and 2-fluorobenzyl chloride (3.20 mL (1.15 equiv)). The solution was cooled to about 15 °C. With good stirring, 5.6 mL (1.06 equiv) 4.4M NaOMe-MeOH solution was added while maintaining a temperature of <25 °C. Following the charge, the contents of the reactor were warmed to about 45 °C over 20 min. The progress of the reaction was monitored (HPLC).
Upon completion, the contents of the reactor were cooled to about 25 °C over about 10 min. Formamide (19 mL) was charged followed by addition of 5.8 mL (1.1 equiv) 4.4M NaOMe- MeOH solution while maintaining a temperature of <25 °C. The contents of the reactor were aged for about 1 h and 3.7 mL (0.7 equiv) 4.4M NaOMe-MeOH solution was added while maintaining a temperature of <25 °C. The progress of the reaction was monitored (HPLC). Upon completion, a solution of glacial acetic acid (2.6 mL (2.0 equiv)) in water (7.5 mL) was added over about 25 min. The slurry was heated to about 65 °C and water (22.5 mL) was added to the solution over about 1 h. The slurry was aged for about 30 min, was cooled to 0 – 5 °C over about 3 h and aged for about 30 min. The solids were isolated by filtration, washed twice with water (7.5 mL) and dried to constant weight in vacuo at 80 °C to afford 8.39 g (90%) of the title compound.
Description 4e: iert-butyl (2S, 5/?)-2-carbamoyl-5-(4-((2- fluorobenzyl)oxy)phenyl)pyrrolidine-1-carboxylate (D4e) (NaOMe – MeOH procedure using 2-fluorobenzyl chloride in DMSO)
A reactor was charged with 1-(te/f- butyl) 2-methyl (2S, 5R)-5-(4-hydroxyphenyl)pyrrolidine-1 ,2-dicarboxylate (7.50 g), anhydrous DMSO (22.5 mL) and 2-fluorobenzyl chloride (3.20 mL (1.15 equiv)). The solution was cooled to about 15 °C. With good stirring, 5.5 mL (1.06 equiv) 4.5M NaOMe-MeOH solution was added while maintaining a temperature of <25 °C. Following the charge, the contents of the reactor were warmed to about 25 °C over 5 min. The progress of the reaction was monitored (HPLC).
Upon completion, formamide (19 mL) was charged followed by addition of 9.73 mL (1.88 equiv) 4.5M NaOMe-MeOH solution over about 45 min. The progress of the reaction was monitored (HPLC). Upon completion, a solution of glacial acetic acid (2.6 mL (2.0 equiv)) in water (7.5 mL) was added over about 25 min. The slurry was heated to about 65 °C and water (22.5 mL) was added to the solution over about 1 h. The slurry was aged for about 30 min, was cooled to 0 – 5 °C over about 3 h and aged for about 30 min. The solids were isolated by filtration, washed twice with water (7.5 mL) and dried to constant weight in vacuo at 80 °C to afford 8.72 g (90%) of the title compound.
Description 4f: iert-butyl (2S, 5/?)-2-carbamoyl-5-(4-((2-fluorobenzyl)oxy)phenyl)pyrrolidine-1-carboxylate (D4f) (f-BuOK procedure using 2-fluorobenzyl bromide in ACN – formamide)
f-BuOK
A reactor was charged with 1-(te/f- butyl) 2-methyl (2S, 5R)-5-(4-hydroxyphenyl)pyrrolidine-1 ,2-dicarboxylate (10.0 g), anhydrous ACN (30 mL), 2-fluorobenzyl bromide (4.18 mL (1.05 equiv)) and formamide (10 mL). The solution was cooled to 0 – 5 °C. With good stirring, 3.67 g (1.05 equiv) f-BuOK was added followed by warming the contents of the reactor to about 15 °C. The progress of the reaction was monitored (HPLC).
Upon completion, the contents of the reactor were cooled to 0 – 5 °C and 4.71 g (1.35 equiv) f-BuOK was added followed by warming the contents of the reactor to about 15 °C. The progress of the reaction was monitored (HPLC). Upon completion, the contents of the reactor were warmed to about 65 °C and a solution of glacial acetic acid (4.14 mL (2.3 equiv)) in water (10 mL) was added. Additional water (40 mL) was added over about 30 min. The contents of the reactor were cooled to 0 – 5 °C and filtered. The filter cake was washed twice with water (10 mL) and dried to constant weight in vacuo at 80 °C to afford 10.93 g (85%) of the title compound.
Description 4g: iert-butyl (2S, 5 ?)-2-carbamoyl-5-(4-((2-fluorobenzyl)oxy)phenyl)pyrrolidine-1-carboxylate (D4g) (f-BuONa procedure using 2-fluorobenzyl bromide in ACN – formamide)
f-BuONa-THF
A reactor was charged with 1-(te/f- butyl) 2-methyl (2S, 5R)-5-(4-hydroxyphenyl)pyrrolidine-1 ,2-dicarboxylate (10.0 g), anhydrous ACN (30 mL), 2-fluorobenzyl bromide (4.18 mL (1.05 equiv)) and formamide (1.5 mL). The solution was cooled to 0 – 5 °C. With good stirring, 16.4 mL (1.05 equiv) 2M f-BuONa – THF solution was added followed by warming the contents of the reactor to about 15 °C. The progress of the reaction was monitored (HPLC).
Upon completion, the contents of the reactor were cooled to 0 – 5 °C and formamide (8.5 mL) was added, followed by 21 mL (1.35 equiv) 2M f-BuONa – THF solution, and the contents of the reactor were warmed to about 15 °C. The progress of the reaction was monitored (HPLC). Upon completion, the contents of the reactor were warmed to about 65 °C and a solution of glacial acetic acid (4.14 mL (2.3 equiv)) in water (10 mL) was added. Additional water (40 mL) was added over about 30 min. The contents of the reactor were cooled to 0 – 5 °C and filtered. The filter cake was washed twice with water (10 mL) and dried to constant weight in vacuo at 80 °C to afford 11.06 g (86%) of the title compound.
Description 4h: tert-butyl (2S, 5S)-2-carbamoyl-5-(4-((2-
A reactor was charged with 1-(te/f-butyl) 2-methyl (2S, 5f?)-5-(4-hydroxyphenyl)pyrrolidine-1 ,2-dicarboxylate (264 Kg), anhydrous DMF (748 kg) and 2-fluorobenzyl bromide (171 Kg (1.10 eq)). The solution was cooled to about 15 °C. With good stirring, 157 Kg (1.06 eq) 30% NaOMe-MeOH solution was added over at least 30 min while maintaining a temperature between 20 – 30 °C. Following the charge, the line was rinsed forward with MeOH (18 kg), and the batch was maintained at about 25 °C for at least 1 h. The progress of the reaction was monitored for completion (HPLC).
Upon completion, formamide (749 Kg) was charged followed by a line rinse with MeOH (18 kg). 279 Kg (1.88 eq) 30% NaOMe-MeOH solution was added over at least 45 min while maintaining a temperature of about 25 °C followed by a line rinse with MeOH (18 kg). The contents of the reactor were maintained at about 25 °C with agitation for about 4 h. The progress of the reaction was monitored for completion (HPLC). Upon completion, the batch was transferred to a second reactor and the equipment was rinsed forward with MeOH (155 Kg). Glacial acetic acid (97 Kg) was added to the batch over at least 15 min while maintaining a temperature of 20 – 30 °C followed by the addition of water (264 Kg). The batch was heated to 60 °C and water (792 Kg) was added over at least 2 h with good agitation. The batch was maintained at 60 °C with agitation for at least 1 h. The batch was cooled to about 2 °C over at least 3 h and aged for at least 1 h. The solids were isolated by filtration and washed twice with water (528 Kg per wash). The wet cake was dried to constant weight in vacuo at 67 °C to afford 315.4 kg (93%) of the title compound.
Description 4i: tert-butyl (2S, 5S)-2-carbamoyl-5-(4-((2-
A reactor was charged with 1-(te/f-butyl) 2-methyl (2S, 5f?)-5-(4-hydroxyphenyl)pyrrolidine-1 ,2-dicarboxylate (70 g), anhydrous DMF (198.2 g) and 2-fluorobenzyl bromide (45.3 g (1.10 equiv)). With good agitation, 41.4 g (1.06 equiv) 30% NaOMe-MeOH solution was added over about 60 min while maintaining a temperature of 20 – 30 °C. The addition funnel was rinsed forward into the reactor with MeOH (2.4 g). The batch was maintained at about 25 °C for at least 1 h; the progress of the reaction was monitored for completion (HPLC).
Upon completion, formamide (238.1 g) was charged followed by rinsing forward the charging equipment with MeOH (2.4 g). 30% NaOMe-MeOH solution (66.5 g (1.70 equiv)) was added over 45 min while maintaining temperature at about 25 °C. The addition funnel was rinsed forward into the reactor with MeOH (2.4 g). The batch was stirred for about 4 h at 25 °C; the progress of the reaction was monitored for completion (HPLC). Upon completion, the batch was transferred to a second reactor and the equipment was rinsed forward with MeOH (20.6 g). Glacial acetic acid (25.7 g) was added while maintaining a temperature of 20 – 30 °C. Water (70 g) was added over about 20 min and the batch was heated to 60 °C. Water (280 g) was added over at least 2 h with good agitation. The batch was maintained at 60 °C with agitation for at least 1 h, cooled to 0-3 °C over at least 3 h and aged for at least 1 h. The solids were isolated by filtration, washed with water/MeOH 70:30 v/v (140 ml_) and water (140 g). The wet cake was dried to constant weight in vacuo at 80 °C to afford 83.7 g (93%) of the title compound.
Description 5a: (2S,5 ?)-5-(4-((2-fluorobenzyl)oxy)phenyl)pyrrolidine-2-carboxamide (E1)
A reactor was charged with te/f-butyl (2S, 5f?)-2-carbamoyl-5-(4-((2-fluorobenzyl)oxy)phenyl)pyrrolidine-1-carboxylate (which may be prepared as described in Description 4) (375.1 kg) and ACN (825.6 kg). With good agitation, methanesulfonic acid (1 14.8 kg (1.3 equiv) was added while maintaining a reaction temperature of 20 – 25 °C followed by ACN (50 kg). The contents of the reactor were warmed to 40 – 50 °C and aged for 2 – 3 h. The progress of the reaction was monitored (HPLC). Upon completion, a solution of 1.0N NH4OH (377 kg) was added while maintaining a reaction temperature of 40 – 50 °C. The reaction temperature was raised to 48 – 52 °C and 1.0N NH4OH (1495 kg) was added slowly with good stirring while maintaining the reaction temperature within this range. The slurry was cooled to -3 to 3 °C over 3 – 4 h and was aged for 1 – 2 h. The solids were isolated by centrifugation (3 drops) and each portion was washed twice with water (182 – 189 kg). The solids were dried in vacuo at 30 °C for 4 h, at 50 °C for 4 h and to constant weight at 80 °C (10 h) to afford 256.4 kg (90.5%) of the title compound.
Description 5b: (2S,5 ?)-5-(4-((2-fluorobenzyl)oxy)phenyl)pyrrolidine-2-carboxamide hydrochloride (1 :1 ) (E2)
A reactor was charged with 2-propanol (672 kg) and the solvent was cooled to -10 to 0 °C. With good agitation, HCI (90 kg) was introduced while maintaining a reaction temperature of -10 – 0 °C. A sample of the solution was removed for concentration determination.
A reactor was charged with te/f-butyl (2S, 5f?)-2-carbamoyl-5-(4-((2-fluorobenzyl)oxy)phenyl)pyrrolidine-1-carboxylate (which may be prepared as described in Description 4) (160 kg) and 2-propanol (1280 kg). Wth good agitation, the prepared HCI – 2- propanol solution (5.3 eq) was added while maintaining a reaction temperature of 20 – 30 °C. The contents of the reactor were warmed to 30 – 35 °C and aged for 12 – 16 h. The progress of the reaction was monitored (HPLC). Upon completion, the contents of the reactor were cooled to 0 – 10 °C, concentrated and aged for 2 – 3 h at 0 – 10 °C. The solids were filtered, washed with 2-propanol (105 kg) and dried in vacuo at 60 – 70 °C for 15 – 20 h to afford 132 kg (96%) of the title compound.
Description 5c: (2S,5R)-5-(4-((2-fluorobenzyl)oxy)phenyl)pyrrolidine-2-carboxamide – method A
A reactor was charged with terf-butyl (2S, 5S)-2-carbamoyl-5-(4-((2- fluorobenzyl)oxy)phenyl)pyrrolidine-1-carboxylate (307 Kg) and acetonitrile (612 Kg). With good agitation, methanesulfonic acid (30 Kg (1.28 equiv)) was added over at least 30 min while maintaining a reaction temperature of 20 – 30 °C. The batch was warmed to 30 °C, aged for about 30 min and heated to 45 °C over about 30 min. The batch was maintained at 45°C for 2 h; the progress of the reaction was monitored for completion (HPLC). Upon completion, the batch was transferred to a second reactor, rinsed forward with acetonitrile (108 Kg) and 1.7% aqueous NH4OH solution (304 Kg) was added while maintaining a temperature of about 40 – 50 °C. The reaction temperature was raised to about 46 – 52 °C and 1.7% NH4OH solution (1216 Kg) was added slowly over 2 h with good stirring while maintaining the reaction temperature within this range. The batch was aged at 50 °C for about 1 h, cooled to 0 °C over at least 3 h and aged for about 1 h. The solids were isolated by filtration and washed twice with water (614 Kg per wash). The solids were dried in vacuo at 70 °C to constant weight to afford 218 Kg (94%) of the title compound.
Description 5d: (2S,5R)-5-(4-((2-fluorobenzyl)oxy)phenyl)pyrrolidine-2-carboxamide – meth
A reactor was charged with terf-butyl (2S, 5S)-2-carbamoyl-5-(4-((2- fluorobenzyl)oxy)phenyl)pyrrolidine-1-carboxylate (100 g) and ACN (199.5 g). With good agitation, methanesulfonic acid (29.7 g (1.28 equiv)) was added while maintaining a reaction temperature of 20 – 30 °C. The batch was warmed to 30 °C, aged for at least 30 min and heated to 45 °C over at least 30 min. The batch was maintained at 45 °C for 2 h; the progress of the reaction was monitored for completion (HPLC). Upon completion, the batch was transferred to a second reactor; the first reactor was rinsed forward with ACN (35.4 g). A solution of 1.7% aqueous NH4OH (99.0 g) was added at 40 – 50 °C over at least 15 min. The reaction temperature was raised to 49 °C and 1.7% NH4OH solution (396.0 g) was added slowly over at least 2 h with good stirring while maintaining the reaction temperature at about 49 °C. The slurry was aged for 30 – 90 min, cooled to 0°C over 3 h and aged for at least 1 h. The solids were isolated by filtration and washed with water/acetonitrile 90: 10 v/v (200 mL) and water (200 g). The solids were dried in vacuo at 70 °C to constant weight to afford 71.6 g (94%) of the title compound.
Description 5e: (2S,5R)-5-(4-((2-fluorobenzyl)oxy)phenyl)pyrrolidine-2-carboxamide hydrochloride
A reactor was charged with terf-butyl (2S, 5S)-2-carbamoyl-5-(4-((2-fluorobenzyl)oxy)phenyl)pyrrolidine-1-carboxylate (160 kg) and isopropanol (1280 kg) at 20 -30 °C. A solution of 2.6M HCI in isopropanol (5.3 eq) was added over about 2 h at 20 – 35 °C. The contents of the reactor were warmed to 30 – 35 °C, and the progress of the reaction was monitored for completion (HPLC). The contents of the reactor were cooled to about 10 °C over about 3 h, concentrated in vacuo for about 1 h and aged at 5 – 10 °C for about 2 h under an inert atmosphere of nitrogen. Solids were filtered, washed with isopropanol (125 kg) and dried to constant weight in vacuo at 60 – 70 °C to give 132.05 kg (96%) of the title compound.
Description 6a: l-(tert-butyl) 2-methyl (2S, 5R)-5-(4-hydroxyphenyl)pyrrol
dicarbo
A hydrogenation reactor was charged with 10% Pd(OH)2/C (water wet; 1.06 g), benzyl (S)-5-(4-(benzyloxy)phenyl)-2-((tert-butoxycarbonyl)amino)-5-oxopentanoate (23 g), MeOH (140
ml_) and di-te/f-butyldicarbonate (1 1.3 g, 1.02 eq). The reactor was pressurized with hydrogen (8 bar) and stirred (300 rpm) for 3 h at ambient temperature followed by stirring at 50 °C for an additional 5 h. The contents of the reactor were cooled to ambient temperature and filtered. The filtrate was concentrated to dryness and the residue was reconstituted in warm MeOH (30 ml_). The contents of the flask were cooled to ambient temperature. The solids were isolated by filtration and dried in vacuo at 60 °C to constant weight to afford 9.6 g (60%) of the title compound.
Description 6b: 1 -(tert-butyl) 2-methyl (2S, 5R)-5-(4-hydroxyphenyl)pyrrolidine-1,2-dicarbo
A hydrogenation reactor was charged with 20% Pd(OH)2/C (water wet; 2.25 g), benzyl (S)-5-(4-(benzyloxy)phenyl)-2-((terf-butoxycarbonyl)amino)-5-oxopentanoate (74.59 g (71.00 g activity)), di-terf-butyldicarbonate (35.27 g, 1.01 eq) and MeOH (415 g). Following three vacuum / nitrogen break cycles, the reactor was pressurized with hydrogen (4 bar) and stirred (-2200 rpm) for about 105 min at 25 °C, then heated to 35 °C and held for an additional 1 h. The reactor was vented, additional MeOH (59 g) was charged, and the reduction was continued at 35 °C, 4 bar and -2200 rpm. The progress of the reaction was monitored for completion (HPLC). Celite® (2.5 g) was added, and the mixture was filtered through a pad of Celite® (2.5 g) and the spent pad was washed with warm MeOH (59 g). The filtrate was concentrated at 40 °C and 200 mbar to a net weight of about 179 g. The contents of the flask were warmed to solution at about 55 °C, slowly cooled to ambient temperature and aged for about 30 min. Water (100 g) was added over about 1 h, and the mixture was aged overnight at ambient temperature. The mixture was cooled to 0-5 °C, aged for about 3 h and filtered. The solids were washed with cold 1 :4 (v/v) MeOH – water (2 X 48 g) and dried in vacuo at 55 °C to constant weight to afford 43.98 g (89%) of the title compound.
///////////VIXOTRIGINE, NEW PATENT, WO-2019071162, BIOGEN INC
Peficitinib hydrobromide, ペフィシチニブ臭化水素酸塩


Peficitinib hydrobromide
ペフィシチニブ臭化水素酸塩
ASP015K,
Rheumatoid Arthritis
1H-Pyrrolo(2,3-b)pyridine-5-carboxamide, 4-((5-hydroxytricyclo(3.3.1.13,7)dec-2-yl)amino)-, hydrobromide (1:1), stereoisomer
4-{[(1R,2s,3S,5r)-5-Hydroxyadamantan-2-yl]amino}-1H-pyrrolo[2,3-b]pyridine-5-carboxamide hydrobromide (1:1)
1H-Pyrrolo[2,3-b]pyridine-5-carboxamide, 4-[[(1R,3S)-5-hydroxytricyclo[3.3.1.13,7]dec-2-yl]amino]-, hydrobromide (1:1)
U55XHZ5X6P
| Formula |
C18H22N4O2. HBr
|
|---|---|
| CAS |
1353219-05-2 HBR
944118-01-8 BASE
|
| Mol weight |
407.3048
|
PMDA, 2019/3/26 JAPAN APPROVED, Smyraf

Peficitinib hydrobromide is used in the treatment of Psoriasis and Rheumatoid Arthritis
Peficitinib (formerly known as ASP015K) is a pyrrolo[2,3-b]pyridine derivative orally administered once-daily JAK inhibitor in development for the treatment of Rheumatoid Arthritis. In preclinical studied Peficitinib inhibited JAK1 and JAK3 with IC50 of 3.9 and 0.7 nM, respectively. Peficitinib also inhibited IL-2-dependent T cell proliferation in vitro and STAT5 phosphorylation in vitro and ex vivo. Furthermore, Peficitinib dose-dependently suppressed bone destruction and paw swelling in an adjuvant-induced arthritis model in rats via prophylactic or therapeutic oral dosing regimens.In clinical trials, Peficitinib treatment prescribed at 50, 100 and 150 mg amounts each showed statistically significantly higher ACR20 response rates compared to the placebo and response rates increased up to the 150 mg dosage. Adverse events included neutropenia, headache, and abdominal pain. The treatment-emergent adverse events occurring more frequently in the Peficitinib group compared with the placebo group included diarrhea, nasopharyngitis, and increased serum creatine phosphokinase activity. No cases of serious infections were reported. Herpes zoster occurred in four patients (two each in the peficitinib 25 and 100 mg cohorts). The authors concluded that treatment with peficitinib as monotherapy for 12 weeks in Japanese patients with moderate to severe RA is efficacious and showed an acceptable safety profile.
SYN

CLIP
Bioorganic & Medicinal Chemistry
Volume 26, Issue 18, 1 October 2018, Pages 4971-4983
Discovery and structural characterization of peficitinib (ASP015K) as a novel and potent JAK inhibitor
Abstract
Janus kinases (JAKs) are considered promising targets for the treatment of autoimmune diseases including rheumatoid arthritis (RA) due to their important role in multiple cytokine receptor signaling pathways. Recently, several JAK inhibitors have been developed for the treatment of RA. Here, we describe the identification of the novel orally bioavailable JAK inhibitor 18, peficitinib (also known as ASP015K), which showed moderate selectivity for JAK3 over JAK1, JAK2, and TYK2 in enzyme assays. Chemical modification at the C4-position of lead compound 5 led to a large increase in JAK inhibitory activity and metabolic stability in liver microsomes. Furthermore, we determined the crystal structures of JAK1, JAK2, JAK3, and TYK2 in a complex with peficitinib, and revealed that the 1H-pyrrolo[2,3–b]pyridine-5-carboxamide scaffold of peficitinib forms triple hydrogen bonds with the hinge region. Interestingly, the binding modes of peficitinib in the ATP-binding pockets differed among JAK1, JAK2, JAK3, and TYK2. WaterMap analysis of the crystal structures suggests that unfavorable water molecules are the likely reason for the difference in orientation of the 1H-pyrrolo[2,3-b]pyridine-5-carboxamide scaffold to the hinge region among JAKs.




PATENT
WO 2011162300
Diseases that have good JAK3 inhibitory activity and are caused by undesired cytokine signaling (eg rejection in living transplantation, rheumatism, psoriasis, autoimmune diseases, asthma, atopic dermatitis, Alzheimer’s disease, atherosclerosis etc. Patent Document 1 discloses fused heterocyclic compounds and salts thereof which are useful as therapeutic agents and / or prophylactic agents for diseases (eg, cancer, leukemia, etc.) caused by abnormal cytokine signaling. Among them, 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo represented by the following formula (I) disclosed in the example compound Ex121 [2,3-b] pyridine-5-carboxamide exhibits good activity, and is particularly a compound expected as a therapeutic agent for suppressing rejection during organ / tissue transplantation, rheumatism, psoriasis and the like.
[Chemical formula 1]

[Chemical formula 1]
The solid stability of a compound which has become a drug development candidate is an important factor both in industrial operation and in maintaining quality. In the stability of the drug substance itself, it is necessary to evaluate the stability of the quality necessary to maintain the efficacy and safety of the drug, and to obtain the information necessary for setting the storage method and the shelf life of the drug. For this reason, the stability test is considered to be one of the most important tests in the manufacture of pharmaceuticals (Heat measurement, 2004, 31 (2), pp. 80-86).
Patent Document 1 discloses the free form of the compound of the formula (I) but does not disclose as a crystal. There is a need for a drug substance which is more suitable for formulation, and is physically and chemically stable from the viewpoint of quality assurance.
Example 1
(Production Method of Hydrobromide Salt Form B 45)
(In the Case of Addition of Seed Crystals )
After nitrogen substitution, the reaction vessel was charged with 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy- 2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide (145.0 kg), water (377 L), EtOH (1508 L), 48% hydrobromic acid (74.9 kg) It charged sequentially at room temperature and started stirring. 48% hydrobromic acid was added, taking care that the pH was in the range of 1.5 to 1.9. The reaction mixture was heated and stirred until the internal temperature reached 70 ° C. or higher. After confirming that the solution was completely dissolved, the solution was stirred for 5 minutes or more, and the solution was subjected to clear filtration at an internal temperature of 70 ° C. or higher, and the pot and line were washed with warm EtOH (290 L). At an internal temperature of about 50 ° C., 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide odor Hydrochloric acid salt seed crystals (B45, 145 g) were added, and the mixture was ripened and stirred overnight at an internal temperature of 40 to 50 ° C. Subsequently, the mixture was cooled to an internal temperature of 20 to 30 ° C. over 1 hour or more, and the mixture was aged and stirred at the same temperature for 1 hour or more. At an internal temperature of 20 to 30 ° C., EtOAc (4350 L) was added dropwise over 1 hour, and the mixture was aged and stirred overnight at the same temperature. The precipitated crystals were filtered. The wet crystals were washed with a solution of EtOH / EtOAc (145 L / 290 L). The wet crystals are dried under reduced pressure at an external temperature of 40 ° C. overnight under reduced pressure to give 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3 -b] Pyridine-5-carboxamide hydrobromide crystal (B45, 161 kg) was obtained.
(Production Method of Hydrobromide Salt Form B 45)
(In the Case of Addition of Seed Crystals )
After nitrogen substitution, the reaction vessel was charged with 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy- 2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide (145.0 kg), water (377 L), EtOH (1508 L), 48% hydrobromic acid (74.9 kg) It charged sequentially at room temperature and started stirring. 48% hydrobromic acid was added, taking care that the pH was in the range of 1.5 to 1.9. The reaction mixture was heated and stirred until the internal temperature reached 70 ° C. or higher. After confirming that the solution was completely dissolved, the solution was stirred for 5 minutes or more, and the solution was subjected to clear filtration at an internal temperature of 70 ° C. or higher, and the pot and line were washed with warm EtOH (290 L). At an internal temperature of about 50 ° C., 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide odor Hydrochloric acid salt seed crystals (B45, 145 g) were added, and the mixture was ripened and stirred overnight at an internal temperature of 40 to 50 ° C. Subsequently, the mixture was cooled to an internal temperature of 20 to 30 ° C. over 1 hour or more, and the mixture was aged and stirred at the same temperature for 1 hour or more. At an internal temperature of 20 to 30 ° C., EtOAc (4350 L) was added dropwise over 1 hour, and the mixture was aged and stirred overnight at the same temperature. The precipitated crystals were filtered. The wet crystals were washed with a solution of EtOH / EtOAc (145 L / 290 L). The wet crystals are dried under reduced pressure at an external temperature of 40 ° C. overnight under reduced pressure to give 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3 -b] Pyridine-5-carboxamide hydrobromide crystal (B45, 161 kg) was obtained.
[0037]
(Another method of producing hydrobromide salt B45 type crystal)
(In the case of no addition of seed crystals) After
sufficiently drying the reaction vessel and replacing with nitrogen, water (585 L) is charged and subsequently 4- {[ (1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide (225 kg), EtOH (2250 L) was charged Stirring was started. The internal temperature was adjusted to 25 ° C., and 48% hydrobromic acid (127.8 kg) was charged at the same temperature, and the vessel and kettle wall were washed with EtOH (90 L). After the completion of the charging, it was confirmed that the reaction solution had been dissolved, the pH was measured, and the pH was confirmed to be in the range of 1.5 to 1.9. When the pH was out of the range, the pH was adjusted to a predetermined pH using 48% hydrobromic acid (48% hydrobromic acid: about 11.6 kg). The temperature was raised until the internal temperature reached 70 ° C., and after confirmation of dissolution, the mixture was stirred for 5 minutes or more. The solution was subjected to clear filtration while maintaining the internal temperature at 60 ° C. or higher, and washed through a filter from a dissolution vessel with warm EtOH (450 L) preheated to 50 ° C. or higher. The clarified filtrate was gradually cooled to an internal temperature of 45 ° C., and filtered EtOAc (6750 L) was added dropwise over 6 hours at an internal temperature of 45 ° C. After the dropping was completed, the mixture was stirred at an internal temperature of 45 ° C. for 10 hours or more. Subsequently, it was cooled to an internal temperature of 25 ° C. using a follow-up temperature control cooler, and stirred at an internal temperature of 25 ° C. for 3 hours. The predetermined supernatant concentration and the crystal form of the precipitated crystals were confirmed and filtered. A mixed solvent of EtOH / EtOAc (225 L / 450 L) was prepared and cake washed using this mixed solvent. The obtained wet crystals are dried under reduced pressure at an external temperature of 40 ° C. for 10 hours or more, and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [ 2,3-b] pyridine-5-carboxamido hydrobromide crystal (B45, 250 kg) was obtained.
(In the case of no addition of seed crystals) After
sufficiently drying the reaction vessel and replacing with nitrogen, water (585 L) is charged and subsequently 4- {[ (1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide (225 kg), EtOH (2250 L) was charged Stirring was started. The internal temperature was adjusted to 25 ° C., and 48% hydrobromic acid (127.8 kg) was charged at the same temperature, and the vessel and kettle wall were washed with EtOH (90 L). After the completion of the charging, it was confirmed that the reaction solution had been dissolved, the pH was measured, and the pH was confirmed to be in the range of 1.5 to 1.9. When the pH was out of the range, the pH was adjusted to a predetermined pH using 48% hydrobromic acid (48% hydrobromic acid: about 11.6 kg). The temperature was raised until the internal temperature reached 70 ° C., and after confirmation of dissolution, the mixture was stirred for 5 minutes or more. The solution was subjected to clear filtration while maintaining the internal temperature at 60 ° C. or higher, and washed through a filter from a dissolution vessel with warm EtOH (450 L) preheated to 50 ° C. or higher. The clarified filtrate was gradually cooled to an internal temperature of 45 ° C., and filtered EtOAc (6750 L) was added dropwise over 6 hours at an internal temperature of 45 ° C. After the dropping was completed, the mixture was stirred at an internal temperature of 45 ° C. for 10 hours or more. Subsequently, it was cooled to an internal temperature of 25 ° C. using a follow-up temperature control cooler, and stirred at an internal temperature of 25 ° C. for 3 hours. The predetermined supernatant concentration and the crystal form of the precipitated crystals were confirmed and filtered. A mixed solvent of EtOH / EtOAc (225 L / 450 L) was prepared and cake washed using this mixed solvent. The obtained wet crystals are dried under reduced pressure at an external temperature of 40 ° C. for 10 hours or more, and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [ 2,3-b] pyridine-5-carboxamido hydrobromide crystal (B45, 250 kg) was obtained.
[0038]
1 H-NMR (600 MHz, d 6 -DMSO) δ: 1.49 (2 H, m), 1. 68 (2 H, m), 1.71 (2 H, m), 1. 80 (2 H, m), 1. 91 (2 H, m), 2.10 (1H, m), 2.20 (2 H, m), 3. 70-4.00 (1 H, brs), 4. 28 (1 H, m), 6. 66 (1 H, m), 7. 39 (1 H, m), 7. 75 (1 H, brs), 8. 38 (1 H, brs), 8.5 5 (1 H, s), 11. 17 (1 H, d, 7.8 Hz), 12.5 (1 H, brs), 14. 17 (1 H, brs)
Elemental analysis: theoretical value: C 53.08%, H 5.69% , N 13.76%, O 7.86% , Br 19.62%;
Found:. C 53.02%, H 5.74 %, N 13.73%, Br 19.42%
molecular composition: C 18 H 22 N 4 O 2 . HBr
MS: 327.0 (M From the result of + H) +
elemental analysis, 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5 The carboxamido hydrobromide was a monohydrobromide.
Elemental analysis: theoretical value: C 53.08%, H 5.69% , N 13.76%, O 7.86% , Br 19.62%;
Found:. C 53.02%, H 5.74 %, N 13.73%, Br 19.42%
molecular composition: C 18 H 22 N 4 O 2 . HBr
MS: 327.0 (M From the result of + H) +
elemental analysis, 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5 The carboxamido hydrobromide was a monohydrobromide.
[0039]
Example 2
(hydrobromide A87 crystal)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] Pyridine-5-carboxamide (6.0 g) was charged in EtOH / water (57.6 mL / 14.4 mL). At 50-60 ° C., 48% hydrobromic acid was added, stirred for 15 minutes more, and washed with EtOH (18 mL). At 45 ° C.-55 ° C. EtOAc (180 mL) was added dropwise over 30 minutes. Crystals were precipitated upon stirring at 15 ° C to 25 ° C. The crystals were collected by filtration and washed with a mixed solvent of EtOH / EtOAc (6 mL / 12 mL). The crystals are dried under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide odor Seed crystals of hydrofluoride (Form A87, 6.11 g) were obtained.
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide (3.0 g), EtOH (24 mL), water (6 mL), and 48% hydrobromic acid (1.55 g) were charged sequentially at room temperature. After charging, the mixture was heated to an internal temperature of 60 ° C. or higher and stirred. After confirming that the solution was completely in solution, the solution was subjected to clear filtration at an internal temperature of 60 ° C. or higher, and washed with warm EtOH (9 mL). EtOH (21 mL) is added dropwise at an internal temperature of 70 ° C. or higher, and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H at an internal temperature of 70 ° C. Seed crystals (A87, 30 mg) of pyrrolo [2,3-b] pyridine-5-carboxamide hydrobromide were added, and the mixture was ripened and stirred overnight at an internal temperature of 65 to 70 ° C. Subsequently, it was cooled to an internal temperature of 20 to 30 ° C., and ripening stirring was carried out at the same temperature overnight. At an internal temperature of 20 to 30 ° C., EtOAc (90 mL) was added dropwise over 1 hour, and the mixture was aged and stirred at the same temperature for 1 hour or more. The precipitated crystals were collected by filtration. The wet crystals were washed with a solution of EtOH / EtOAc (3 mL / 12 mL). The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide crystal (Form A87, 3.09 g) was obtained.
(hydrobromide A87 crystal)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] Pyridine-5-carboxamide (6.0 g) was charged in EtOH / water (57.6 mL / 14.4 mL). At 50-60 ° C., 48% hydrobromic acid was added, stirred for 15 minutes more, and washed with EtOH (18 mL). At 45 ° C.-55 ° C. EtOAc (180 mL) was added dropwise over 30 minutes. Crystals were precipitated upon stirring at 15 ° C to 25 ° C. The crystals were collected by filtration and washed with a mixed solvent of EtOH / EtOAc (6 mL / 12 mL). The crystals are dried under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide odor Seed crystals of hydrofluoride (Form A87, 6.11 g) were obtained.
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide (3.0 g), EtOH (24 mL), water (6 mL), and 48% hydrobromic acid (1.55 g) were charged sequentially at room temperature. After charging, the mixture was heated to an internal temperature of 60 ° C. or higher and stirred. After confirming that the solution was completely in solution, the solution was subjected to clear filtration at an internal temperature of 60 ° C. or higher, and washed with warm EtOH (9 mL). EtOH (21 mL) is added dropwise at an internal temperature of 70 ° C. or higher, and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H at an internal temperature of 70 ° C. Seed crystals (A87, 30 mg) of pyrrolo [2,3-b] pyridine-5-carboxamide hydrobromide were added, and the mixture was ripened and stirred overnight at an internal temperature of 65 to 70 ° C. Subsequently, it was cooled to an internal temperature of 20 to 30 ° C., and ripening stirring was carried out at the same temperature overnight. At an internal temperature of 20 to 30 ° C., EtOAc (90 mL) was added dropwise over 1 hour, and the mixture was aged and stirred at the same temperature for 1 hour or more. The precipitated crystals were collected by filtration. The wet crystals were washed with a solution of EtOH / EtOAc (3 mL / 12 mL). The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide crystal (Form A87, 3.09 g) was obtained.
[0040]
Example 3
(hydrobromide A61 crystal)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] Pyridine-5-carboxamide (5.0 g), EtOH (48 mL), water (12 mL), and 48% hydrobromic acid (2.58 g) were charged sequentially at room temperature. After charging, the mixture was heated to an internal temperature of 70 ° C. and stirred. After confirming complete dissolution, the solution was clarified by filtration at an internal temperature of 70 ° C., and washed with warm EtOH (15 mL). The internal temperature was cooled to 50 to 60 ° C., and EtOAc (150 mL) was added dropwise over 1 hour at the same temperature. After the addition was completed, the solution was gradually cooled to 20 to 30 ° C., and the mixture was aged and stirred at the same temperature for 1 hour or more. The precipitated crystals were collected by filtration. The wet crystals were washed with a solution of EtOH / EtOAc (5 mL / 10 mL). The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide crystal (Form A61, 5.19 g) was obtained.
(hydrobromide A61 crystal)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] Pyridine-5-carboxamide (5.0 g), EtOH (48 mL), water (12 mL), and 48% hydrobromic acid (2.58 g) were charged sequentially at room temperature. After charging, the mixture was heated to an internal temperature of 70 ° C. and stirred. After confirming complete dissolution, the solution was clarified by filtration at an internal temperature of 70 ° C., and washed with warm EtOH (15 mL). The internal temperature was cooled to 50 to 60 ° C., and EtOAc (150 mL) was added dropwise over 1 hour at the same temperature. After the addition was completed, the solution was gradually cooled to 20 to 30 ° C., and the mixture was aged and stirred at the same temperature for 1 hour or more. The precipitated crystals were collected by filtration. The wet crystals were washed with a solution of EtOH / EtOAc (5 mL / 10 mL). The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide crystal (Form A61, 5.19 g) was obtained.
[0041]
Example 4
(hydrobromide A36 type crystal)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] To a suspension of pyridine-5-carboxamide (500 mg) in EtOAc, 48% hydrobromic acid (258 μL) was added, and the mixture was stirred with heating under reflux for 1 hour, and further allowed to cool to room temperature. The precipitated crystals were collected by filtration and washed with EtOAc. The resulting crystals are dried at 60 ° C. under reduced pressure to give 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-]. b] Pyridine-5-carboxamide monohydrobromide crystal (Form A36, 625 mg) was obtained.
(hydrobromide A36 type crystal)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] To a suspension of pyridine-5-carboxamide (500 mg) in EtOAc, 48% hydrobromic acid (258 μL) was added, and the mixture was stirred with heating under reflux for 1 hour, and further allowed to cool to room temperature. The precipitated crystals were collected by filtration and washed with EtOAc. The resulting crystals are dried at 60 ° C. under reduced pressure to give 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-]. b] Pyridine-5-carboxamide monohydrobromide crystal (Form A36, 625 mg) was obtained.
[0042]
Example 5
(B11-type crystal of hydrobromide monohydrate)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2 , 3-b] Pyridine-5-carboxamide (5.0 g), EtOH (48 mL), water (12 mL), 48% hydrobromic acid (2.58 g) were sequentially charged at room temperature. After charging, the mixture was heated to an internal temperature of 70 ° C. or higher and stirred. After confirming that the solution had completely dissolved, the solution was subjected to clear filtration at an internal temperature of 70 ° C. or higher, and washed with warm EtOH (15 mL). 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide odor at an internal temperature of about 35 ° C. Hydrochloric acid salt seed crystals (A87, 49.0 mg) were added, and the mixture was aged with an internal temperature of 30 to 40 ° C. for 4 hours. Subsequently, the mixture was cooled to an internal temperature of 20 to 30 ° C., and aged and stirred overnight at the same temperature. At an internal temperature of 20-25 ° C., EtOAc (150 mL) was added dropwise over 1 hour, and the mixture was aged and stirred at the same temperature for 30 minutes or longer. The precipitated crystals were collected by filtration. The wet crystals were washed with a solution of EtOH / EtOAc (5 mL / 10 mL). The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide monohydrate crystal (Form B11, 5.24 g) was obtained.
(B11-type crystal of hydrobromide monohydrate)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2 , 3-b] Pyridine-5-carboxamide (5.0 g), EtOH (48 mL), water (12 mL), 48% hydrobromic acid (2.58 g) were sequentially charged at room temperature. After charging, the mixture was heated to an internal temperature of 70 ° C. or higher and stirred. After confirming that the solution had completely dissolved, the solution was subjected to clear filtration at an internal temperature of 70 ° C. or higher, and washed with warm EtOH (15 mL). 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide odor at an internal temperature of about 35 ° C. Hydrochloric acid salt seed crystals (A87, 49.0 mg) were added, and the mixture was aged with an internal temperature of 30 to 40 ° C. for 4 hours. Subsequently, the mixture was cooled to an internal temperature of 20 to 30 ° C., and aged and stirred overnight at the same temperature. At an internal temperature of 20-25 ° C., EtOAc (150 mL) was added dropwise over 1 hour, and the mixture was aged and stirred at the same temperature for 30 minutes or longer. The precipitated crystals were collected by filtration. The wet crystals were washed with a solution of EtOH / EtOAc (5 mL / 10 mL). The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide monohydrate crystal (Form B11, 5.24 g) was obtained.
[0043]
Example 6
(B21-type crystal of hydrobromide dihydrate)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2 , 3-b] Pyridine-5-carboxamide (5.0 g), EtOH (18 mL), water (12 mL), 48% hydrobromic acid (2.58 g) were sequentially charged at room temperature. After charging, the mixture was heated to an internal temperature of 60 ° C. or higher and stirred. After confirming that the solution was completely dissolved, the solution was subjected to clear filtration at an internal temperature of 60 ° C. or higher, and washed with warm EtOH (10 mL). The mixture was cooled to an internal temperature of about 45 to 50 ° C. and aged for 2 hours while stirring. Subsequently, the reaction solution is cooled to an internal temperature of 20 to 30 ° C., and aged at the same temperature and stirred overnight. At an internal temperature of 20 to 30 ° C., EtOAc (160 mL) was added dropwise over 1 hour, and the mixture was aged and stirred for 1 hour or more at the same temperature. The precipitated crystals were filtered. The wet crystals were washed with a solution of EtOH / EtOAc (3 mL / 12 mL). The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide dihydrate crystals (Form B21, 6.05 g) were obtained.
(B21-type crystal of hydrobromide dihydrate)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2 , 3-b] Pyridine-5-carboxamide (5.0 g), EtOH (18 mL), water (12 mL), 48% hydrobromic acid (2.58 g) were sequentially charged at room temperature. After charging, the mixture was heated to an internal temperature of 60 ° C. or higher and stirred. After confirming that the solution was completely dissolved, the solution was subjected to clear filtration at an internal temperature of 60 ° C. or higher, and washed with warm EtOH (10 mL). The mixture was cooled to an internal temperature of about 45 to 50 ° C. and aged for 2 hours while stirring. Subsequently, the reaction solution is cooled to an internal temperature of 20 to 30 ° C., and aged at the same temperature and stirred overnight. At an internal temperature of 20 to 30 ° C., EtOAc (160 mL) was added dropwise over 1 hour, and the mixture was aged and stirred for 1 hour or more at the same temperature. The precipitated crystals were filtered. The wet crystals were washed with a solution of EtOH / EtOAc (3 mL / 12 mL). The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide dihydrate crystals (Form B21, 6.05 g) were obtained.
[0044]
Example 7
(Tautomerism of each crystal)
(Tautomerism of each crystal)
[0045]
Example 7-1
(Crystal form conversion; hydrobromide B21 → A61)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H- Pyrrolo [2,3-b] pyridine-5-carboxamide hydrobromide dihydrate (Form B21, 300 mg) and EtOH (3 mL) were sequentially charged at room temperature and suspended overnight. After suspension, the crystals were collected by filtration at room temperature and the wet crystals were washed with EtOH. The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide crystal (Form A61, 258 mg) was obtained.
(Crystal form conversion; hydrobromide B21 → A61)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H- Pyrrolo [2,3-b] pyridine-5-carboxamide hydrobromide dihydrate (Form B21, 300 mg) and EtOH (3 mL) were sequentially charged at room temperature and suspended overnight. After suspension, the crystals were collected by filtration at room temperature and the wet crystals were washed with EtOH. The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide crystal (Form A61, 258 mg) was obtained.
[0046]
Example 7-2
(Crystal form conversion; hydrobromide B11 → B21)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H- Pyrrolo [2,3-b] pyridine-5-carboxamide hydrobromide monohydrate (form B11, 2.0 g), EtOH (7 mL), water (3 mL) were sequentially charged at room temperature and suspended overnight. It became cloudy. After suspension, the crystals were collected by filtration at room temperature and the wet crystals were washed with 70% aqueous EtOH. The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide dihydrate crystals (Form B21, 1.54 g) were obtained.
(Crystal form conversion; hydrobromide B11 → B21)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H- Pyrrolo [2,3-b] pyridine-5-carboxamide hydrobromide monohydrate (form B11, 2.0 g), EtOH (7 mL), water (3 mL) were sequentially charged at room temperature and suspended overnight. It became cloudy. After suspension, the crystals were collected by filtration at room temperature and the wet crystals were washed with 70% aqueous EtOH. The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide dihydrate crystals (Form B21, 1.54 g) were obtained.
[0047]
Example 7-3
(Crystal form conversion; hydrobromide A61 form → B21 form)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H- Pyrrolo [2,3-b] pyridine-5-carboxamide hydrobromide (form A61, 1.0 g), EtOH (3.5 mL) and water (1.5 mL) were sequentially charged at room temperature and suspended overnight. After suspension, the crystals were filtered at room temperature and the wet crystals were washed with 70% aqueous EtOH. The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide dihydrate crystals (Form B21, 827 mg) were obtained.
(Crystal form conversion; hydrobromide A61 form → B21 form)
4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H- Pyrrolo [2,3-b] pyridine-5-carboxamide hydrobromide (form A61, 1.0 g), EtOH (3.5 mL) and water (1.5 mL) were sequentially charged at room temperature and suspended overnight. After suspension, the crystals were filtered at room temperature and the wet crystals were washed with 70% aqueous EtOH. The wet crystals are dried overnight under vacuum and 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2,3-b] pyridine-5-carboxamide Hydrobromide dihydrate crystals (Form B21, 827 mg) were obtained.
[0048]
Reference Example 1
( Example of Preparation of Monohydrate Crystalline Compound (I) Free Form)
4-Chloro-1H-pyrrolo [2,3-b] pyridine-5-carboxamide (44.5 g) under nitrogen atmosphere 1s, 3R, 4s, 5S) -4-aminoadamantan-1-ol (57.0 g) and tributylamine (162.6 mL) were charged in NMP (222.5 mL), and heated and stirred at a bath temperature of 200 ° C. for 2.5 hours. The reaction solution was allowed to cool, and then the reaction solution was added dropwise while stirring in water / Et 2 O (6 L / 0.5 L), followed by stirring for 30 minutes. The obtained solid was collected by filtration, washed twice with water (400 mL), washed twice with Et 2 O (300 mL), and dried. The resulting solid was warmed to dissolve in MeOH (1.8 L) and filtered hot. The resulting mother liquor was concentrated under reduced pressure and MeOH (1.8 L) was added to the residue and heated to dissolve. The resulting solution was allowed to cool and stir, and then stirred at room temperature and aged overnight. The precipitated solid was collected by filtration, washed with EtOH and dried under reduced pressure. The resulting solid was suspended in EtOH (250 mL) and stirred at room temperature for 1 h. The solid was collected by filtration, washed with EtOH and dried under reduced pressure. The obtained solid was suspended in water (900 mL) and stirred at a bath temperature of 70 ° C. for 2 hours. The solid was collected by filtration, washed with water and dried under reduced pressure. Furthermore, the solid was suspended in water (900 mL) and stirred at a bath temperature of 70 ° C. for 2 hours. The solid is collected by filtration, washed with water and then dried under reduced pressure to give 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2 , 3-b] Pyridine-5-carboxamide monohydrate crystal (Form A01, 44 g) was obtained.
( Example of Preparation of Monohydrate Crystalline Compound (I) Free Form)
4-Chloro-1H-pyrrolo [2,3-b] pyridine-5-carboxamide (44.5 g) under nitrogen atmosphere 1s, 3R, 4s, 5S) -4-aminoadamantan-1-ol (57.0 g) and tributylamine (162.6 mL) were charged in NMP (222.5 mL), and heated and stirred at a bath temperature of 200 ° C. for 2.5 hours. The reaction solution was allowed to cool, and then the reaction solution was added dropwise while stirring in water / Et 2 O (6 L / 0.5 L), followed by stirring for 30 minutes. The obtained solid was collected by filtration, washed twice with water (400 mL), washed twice with Et 2 O (300 mL), and dried. The resulting solid was warmed to dissolve in MeOH (1.8 L) and filtered hot. The resulting mother liquor was concentrated under reduced pressure and MeOH (1.8 L) was added to the residue and heated to dissolve. The resulting solution was allowed to cool and stir, and then stirred at room temperature and aged overnight. The precipitated solid was collected by filtration, washed with EtOH and dried under reduced pressure. The resulting solid was suspended in EtOH (250 mL) and stirred at room temperature for 1 h. The solid was collected by filtration, washed with EtOH and dried under reduced pressure. The obtained solid was suspended in water (900 mL) and stirred at a bath temperature of 70 ° C. for 2 hours. The solid was collected by filtration, washed with water and dried under reduced pressure. Furthermore, the solid was suspended in water (900 mL) and stirred at a bath temperature of 70 ° C. for 2 hours. The solid is collected by filtration, washed with water and then dried under reduced pressure to give 4-{[(1R, 2s, 3S, 5s, 7s) -5-hydroxy-2-adamantyl] amino} -1H-pyrrolo [2 , 3-b] Pyridine-5-carboxamide monohydrate crystal (Form A01, 44 g) was obtained.
REFERENCES
1: D’Amico F, Fiorino G, Furfaro F, Allocca M, Danese S. Janus kinase inhibitors for the treatment of inflammatory bowel diseases: developments from phase I and phase II clinical trials. Expert Opin Investig Drugs. 2018 Jul;27(7):595-599. doi: 10.1080/13543784.2018.1492547. Epub 2018 Jul 6. Review. PubMed PMID: 29938545.
2: Sands BE, Sandborn WJ, Feagan BG, Lichtenstein GR, Zhang H, Strauss R, Szapary P, Johanns J, Panes J, Vermeire S, O’Brien CD, Yang Z, Bertelsen K, Marano C; Peficitinib-UC Study Group. Peficitinib, an Oral Janus Kinase Inhibitor, in Moderate-to-Severe Ulcerative Colitis: Results From a Randomized, Phase 2 Study. J Crohns Colitis. 2018 Jun 15. doi: 10.1093/ecco-jcc/jjy085. [Epub ahead of print] PubMed PMID: 29917064.
3: Veale DJ, McGonagle D, McInnes IB, Krueger JG, Ritchlin CT, Elewaut D, Kanik KS, Hendrikx T, Berstein G, Hodge J, Telliez JB. The rationale for Janus kinase inhibitors for the treatment of spondyloarthritis. Rheumatology (Oxford). 2018 Apr 3. doi: 10.1093/rheumatology/key070. [Epub ahead of print] PubMed PMID: 29618084.
4: Cline A, Cardwell LA, Feldman SR. Advances in treating psoriasis in the elderly with small molecule inhibitors. Expert Opin Pharmacother. 2017 Dec;18(18):1965-1973. doi: 10.1080/14656566.2017.1409205. Epub 2017 Nov 27. Review. PubMed PMID: 29171774.
5: Baker KF, Isaacs JD. Novel therapies for immune-mediated inflammatory diseases: What can we learn from their use in rheumatoid arthritis, spondyloarthritis, systemic lupus erythematosus, psoriasis, Crohn’s disease and ulcerative colitis? Ann Rheum Dis. 2018 Feb;77(2):175-187. doi: 10.1136/annrheumdis-2017-211555. Epub 2017 Aug 1. Review. PubMed PMID: 28765121.
6: Zhu T, Howieson C, Wojtkowski T, Garg JP, Han D, Fisniku O, Keirns J. The Effect of Verapamil, a P-Glycoprotein Inhibitor, on the Pharmacokinetics of Peficitinib, an Orally Administered, Once-Daily JAK Inhibitor. Clin Pharmacol Drug Dev. 2017 Nov;6(6):548-555. doi: 10.1002/cpdd.344. Epub 2017 Mar 16. PubMed PMID: 28301084.
7: Genovese MC, Greenwald M, Codding C, Zubrzycka-Sienkiewicz A, Kivitz AJ, Wang A, Shay K, Wang X, Garg JP, Cardiel MH. Peficitinib, a JAK Inhibitor, in Combination With Limited Conventional Synthetic Disease-Modifying Antirheumatic Drugs in the Treatment of Moderate-to-Severe Rheumatoid Arthritis. Arthritis Rheumatol. 2017 May;69(5):932-942. doi: 10.1002/art.40054. PubMed PMID: 28118538.
8: Ito M, Yamazaki S, Yamagami K, Kuno M, Morita Y, Okuma K, Nakamura K, Chida N, Inami M, Inoue T, Shirakami S, Higashi Y. A novel JAK inhibitor, peficitinib, demonstrates potent efficacy in a rat adjuvant-induced arthritis model. J Pharmacol Sci. 2017 Jan;133(1):25-33. doi: 10.1016/j.jphs.2016.12.001. Epub 2016 Dec 23. PubMed PMID: 28117214.
9: Zhu T, Parker B, Wojtkowski T, Nishimura T, Garg JP, Han D, Fisniku O, Keirns J. Drug Interactions Between Peficitinib, an Orally Administered, Once-Daily Janus Kinase Inhibitor, and Rosuvastatin in Healthy Subjects. Clin Pharmacokinet. 2017 Jul;56(7):747-757. doi: 10.1007/s40262-016-0474-4. PubMed PMID: 27878567.
10: Semerano L, Decker P, Clavel G, Boissier MC. Developments with investigational Janus kinase inhibitors for rheumatoid arthritis. Expert Opin Investig Drugs. 2016 Dec;25(12):1355-1359. Epub 2016 Oct 31. PubMed PMID: 27748152.
11: Kivitz AJ, Gutierrez-Ureña SR, Poiley J, Genovese MC, Kristy R, Shay K, Wang X, Garg JP, Zubrzycka-Sienkiewicz A. Peficitinib, a JAK Inhibitor, in the Treatment of Moderate-to-Severe Rheumatoid Arthritis in Patients With an Inadequate Response to Methotrexate. Arthritis Rheumatol. 2017 Apr;69(4):709-719. doi: 10.1002/art.39955. PubMed PMID: 27748083.
12: Lam S. JAK inhibitors: A broadening approach in rheumatoid arthritis. Drugs Today (Barc). 2016 Aug;52(8):467-469. PubMed PMID: 27722215.
13: Roskoski R Jr. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol Res. 2016 Sep;111:784-803. doi: 10.1016/j.phrs.2016.07.038. Epub 2016 Jul 26. Review. PubMed PMID: 27473820.
14: Iwata S, Tanaka Y. Progress in understanding the safety and efficacy of Janus kinase inhibitors for treatment of rheumatoid arthritis. Expert Rev Clin Immunol. 2016 Oct;12(10):1047-57. doi: 10.1080/1744666X.2016.1189826. Epub 2016 Jun 6. Review. PubMed PMID: 27253519.
15: Cao YJ, Sawamoto T, Valluri U, Cho K, Lewand M, Swan S, Lasseter K, Matson M, Holman J Jr, Keirns J, Zhu T. Pharmacokinetics, Pharmacodynamics, and Safety of ASP015K (Peficitinib), a New Janus Kinase Inhibitor, in Healthy Subjects. Clin Pharmacol Drug Dev. 2016 Nov;5(6):435-449. doi: 10.1002/cpdd.273. Epub 2016 Jun 30. PubMed PMID: 27162173.
16: Nielsen OH, Seidelin JB, Ainsworth M, Coskun M. Will novel oral formulations change the management of inflammatory bowel disease? Expert Opin Investig Drugs. 2016 Jun;25(6):709-18. doi: 10.1517/13543784.2016.1165204. Epub 2016 Mar 28. Review. PubMed PMID: 26967267.
17: Yiu ZZ, Warren RB. Novel Oral Therapies for Psoriasis and Psoriatic Arthritis. Am J Clin Dermatol. 2016 Jun;17(3):191-200. doi: 10.1007/s40257-016-0179-3. Review. PubMed PMID: 26923915.
18: Takeuchi T, Tanaka Y, Iwasaki M, Ishikura H, Saeki S, Kaneko Y. Efficacy and safety of the oral Janus kinase inhibitor peficitinib (ASP015K) monotherapy in patients with moderate to severe rheumatoid arthritis in Japan: a 12-week, randomised, double-blind, placebo-controlled phase IIb study. Ann Rheum Dis. 2016 Jun;75(6):1057-64. doi: 10.1136/annrheumdis-2015-208279. Epub 2015 Dec 15. PubMed PMID: 26672064; PubMed Central PMCID: PMC4893099.
19: Oda K, Cao YJ, Sawamoto T, Nakada N, Fisniku O, Nagasaka Y, Sohda KY. Human mass balance, metabolite profile and identification of metabolic enzymes of [¹⁴C]ASP015K, a novel oral janus kinase inhibitor. Xenobiotica. 2015;45(10):887-902. doi: 10.3109/00498254.2015.1026864. Epub 2015 May 19. PubMed PMID: 25986538.
20: Nakada N, Oda K. Identification and characterization of metabolites of ASP015K, a novel oral Janus kinase inhibitor, in rats, chimeric mice with humanized liver, and humans. Xenobiotica. 2015;45(9):757-65. doi: 10.3109/00498254.2015.1019594. Epub 2015 Jun 12. PubMed PMID: 25869242.
/////////////////Peficitinib hydrobromide, Smyraf, JAPAN 2019, ペフィシチニブ臭化水素酸塩 , ASP015K, Rheumatoid Arthritis
O=C(C1=CN=C(NC=C2)C2=C1N[C@@H]3[C@]4([H])C[C@@]5([H])C[C@](C4)(O)C[C@]3([H])C5)N.[H]Br
Cefamandole, セファマンドール ,цефамандол , سيفاماندول , 头孢孟多 ,
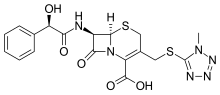

CAS Registry Number: 34444-01-4
CAS Name: (6R,7R)-7-[[(2R)-Hydroxyphenylacetyl]amino]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid
Additional Names: 7-mandelamido-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid; 7-D-mandelamido-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-3-cephem-4-carboxylic acid; 7-D-mandelamido-3-(1-methyl-1,2,3,4-tetrazole-5-thiomethyl)-D3-cephem-4-carboxylic acid; CMT
Manufacturers’ Codes: compd 83405
Molecular Formula: C18H18N6O5S2
Molecular Weight: 462.50
Percent Composition: C 46.74%, H 3.92%, N 18.17%, O 17.30%, S 13.87%
Literature References: Broad-spectrum semi-synthetic cephalosporin antibiotic. Prepn: C. W. Ryan, DE 2018600; idem, US3641021 (1970, 1972 to Lilly); J. M. Greene, DE 2312997; idem, US 3840531 (1973, 1974 to Lilly). Biological properties: W. E. Wick, D. A. Preston, Antimicrob. Agents Chemother. 1, 221 (1972). Antibacterial activity: S. Eykyn et al., ibid. 3, 657 (1973); H. C. Neu, ibid. 6, 177 (1974); A. D. Russell, J. Antimicrob. Chemother. 1, 97 (1975). Pharmacologic studies: B. R. Meyers et al.,Antimicrob. Agents Chemother. 9, 140 (1976); R. S. Griffith et al., ibid. 10, 814 (1976). Comprehensive description: R. H. Bishara, E. C. Rickard, Anal. Profiles Drug Subs. 9, 125-154 (1980).
Derivative Type: Nafate
CAS Registry Number: 42540-40-9
Trademarks: Bergacef (Bergamon); Cedol (Tiber); Cefam (Magis); Cefiran (Poli); Cemado (Francia); Cemandil (SIT); Fado (Errekappa); Kefadol (Lilly); Kefandol (Lilly); Lampomandol (AGIPS); Mandokef (Lilly); Mandol (Lilly); Mandolsan (San Carlo); Neocefal (Metapharma); Pavecef (IBP)
Molecular Formula: C19H17N6NaO6S2
Molecular Weight: 512.49
Percent Composition: C 44.53%, H 3.34%, N 16.40%, Na 4.49%, O 18.73%, S 12.51%
Properties: White, odorless needles, mp 190° (dec). uv max (H2O): 269 nm (e 10800). pKa 2.6-3.0. Sol in water, methanol. Practically insol in ether, chloroform, benzene, cyclohexane.
Melting point: mp 190° (dec)
pKa: pKa 2.6-3.0
Absorption maximum: uv max (H2O): 269 nm (e 10800)
Therap-Cat: Antibacterial.
Keywords: Antibacterial (Antibiotics); ?Lactams; Cephalosporins.
- Use:antibiotic
- Chemical name:[6R-[6α,7β(R*)]]-7-[(hydroxyphenylacetyl)amino]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid
- Formula:C18H18N6O5S2
- MW:462.51 g/mol
- CAS-RN:34444-01-4
- InChI Key:OLVCFLKTBJRLHI-AXAPSJFSSA-N
- InChI:InChI=1S/C18H18N6O5S2/c1-23-18(20-21-22-23)31-8-10-7-30-16-11(15(27)24(16)12(10)17(28)29)19-14(26)13(25)9-5-3-2-4-6-9/h2-6,11,13,16,25H,7-8H2,1H3,(H,19,26)(H,28,29)/t11-,13-,16-/m1/s1
- EINECS:252-030-0
Derivatives
Formate monosodium salt (nafate)
- Formula:C19H17N6NaO6S2
- MW:512.50 g/mol
- CAS-RN:42540-40-9
- EINECS:255-877-4
- LD50:3915 mg/kg (M, i.v.);
2562 mg/kg (R, i.v.)
Cefamandole (INN, also known as cephamandole) is a second-generation broad-spectrumcephalosporinantibiotic. The clinically used form of cefamandole is the formateestercefamandole nafate, a prodrug which is administered parenterally. Cefamandole is no longer available in the United States.
The chemical structure of cefamandole, like that of several other cephalosporins, contains an N-methylthiotetrazole (NMTT or 1-MTT) side chain. As the antibiotic is broken down in the body, it releases free NMTT, which can cause hypoprothrombinemia (likely due to inhibition of the enzymevitamin K epoxide reductase)(vitamin K supplement is recommended during therapy) and a reaction with ethanol similar to that produced by disulfiram (Antabuse), due to inhibition of aldehyde dehydrogenase.
Cefamandole has a broad spectrum of activity and can be used to treat bacterial infections of the skin, bones and joints, urinary tract, and lower respiratory tract. The following represents cefamandole MIC susceptibility data for a few medically significant microorganisms.
- Escherichia coli: 0.12 – 400 μg/ml
- Haemophilus influenzae: 0.06 – >16 μg/ml
- Staphylococcus aureus: 0.1 – 12.5 μg/ml
CO2 is generated during the normal constitution of cefamandole and ceftazidime, potentially resulting in an explosive-like reaction in syringes.[2]
SYNTHESIS
US 3641021
US 3840531 US 3974153 US 3903278 US 2018600 US 2065621 DE 2018600 DE 2065621 DE 2730579
DE 2312997

SYN
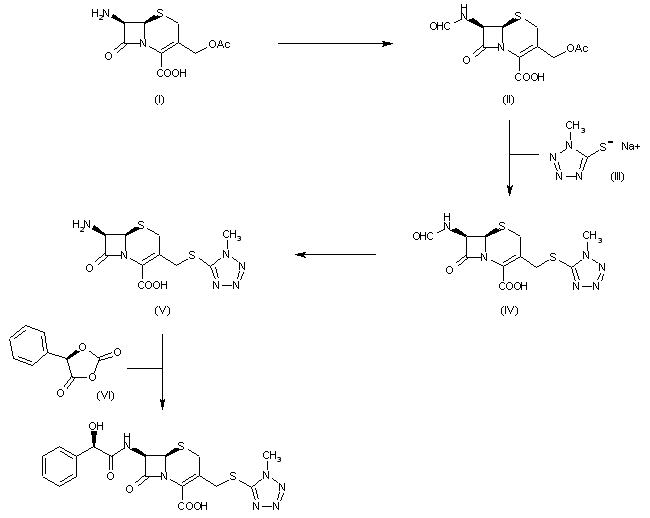
The formylation of 7-aminocephalosporanic acid (I) by the usual techniques produces 7-formamidocephalosporanic acid (II), which is then treated with the sodium salt of 1-methyl-1H-tetrazole-5-thiol (III) to yield 7-formamido-3-(1-methyl-1H-tetrazol-5-ylthio)methyl-3-cephem-4-carboxylic acid (IV). The resulting product (IV) is deformylated affording 7-amino-3-(1-methyl-1H-tetrazol-5-ylthio)methyl-3-cephem-4-carboxylic acid (V), which is finally acylated with anhydro-O-carboxymandelic acid (VI) using the usual techniques.
References
- ^ http://www.toku-e.com/Assets/MIC/Cefamandole%20sodium%20salt.pdf
- ^ Stork CM (2006). “Antibiotics, antifungals, and antivirals”. In Nelson LH, Flomenbaum N, Goldfrank LR, Hoffman RL, Howland MD, Lewin NA. Goldfrank’s toxicologic emergencies. New York: McGraw-Hill. p. 847. ISBN 0-07-143763-0. Retrieved 2009-07-03.
-
- US 3 641 021 (Lilly; 8.2.1972; appl. 18.4.1969).
- DE 2 018 600 (Lilly; appl. 17.4.1970; USA-prior. 18.4.1969).
- DAS 2 065 621 (Lilly; appl. 17.4.1970; USA-prior. 18.4.1969).
- US 3 840 531 (Lilly; 8.10.1974; appl. 21.3.1972).
- US 3 903 278 (Smith Kline Corp.; 2.9.1975; prior. 4.11.1971).
- DOS 2 730 579 (Pierrel S.p.A.; appl. 6.7.1977; GB-prior. 10.7.1976).
-
preparation and/or purification via the trimethylsilyl-derivatives:
- DOS 2 711 095 (Lilly; appl. 14.3.1977; USA-prior. 17.3.1976).
-
purification:
- US 4 115 644 (Lilly; 19.9.1978; appl. 19.9.1978).
- DOS 2 839 670 (Lilly; appl. 12.9.1978; USA-prior. 19.9.1977).
-
crystalline sodium salt:
- US 4 054 738 (Lilly; 18.10.1977; appl. 22.12.1975).
- US 4 168 376 (Lilly; 18.9.1979; appl. 5.6.1978).
-
lithium salt:
- GB 1 546 757 (Lilly; appl. 10.4.1975; valid from 7.4.1976).
-
O-formyl-derivative:
- US 3 928 592 (Lilly; 23.12.1975; appl. 21.2.1974).
- GB 1 493 676 (Lilly; appl. 20.2.1975; USA-prior. 22.2.1974).
- GB 1 546 898 (Lilly; appl. 7.4.1976; USA-prior. 11.4.1975).
- DOS 2 506 622 (Lilly; appl. 17.2.1975; USA-prior. 22.2.1974).
-
crystalline sodium salt of O-formylcefamandole:
- US 4 006 138 (Lilly; 1.2.1977; appl. 11.4.1975).
-
complex of cefamandole sodium with 1,4-dioxane and water:
- US 3 947 414 (Lilly; 30.3.1976; appl. 23.12.1974).
-
complex of cefamandole sodium with ethyl l-(–)-lactate:
- US 3 947 415 (Lilly; 30.3.1976; appl. 23.12.1974).
 |
|
| Clinical data | |
|---|---|
| Trade names | former Mandol |
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| MedlinePlus | a601206 |
| Pregnancy category |
|
| Routes of administration |
Intramuscular, intravenous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | 75% |
| Elimination half-life | 48 minutes |
| Excretion | Mostly renal, as unchanged drug |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.047.285 |
| Chemical and physical data | |
| Formula | C18H18N6O5S2 |
| Molar mass | 462.505 g/mol g·mol−1 |
| 3D model (JSmol) | |
Tegaserod, テガセロド
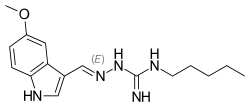
Tegaserod
- Molecular FormulaC16H23N5O
- Average mass301.387 Da
- テガセロド
145158-71-0 cas
HTF 919 / HTF-919 / SDZ HTF 919 / SDZ-HTF-919
N’-[(E)-[(5-methoxy-1H-indol-3-yl)methylidene]amino]-N-pentylguanidine
(2E)-2-[(5-Methoxy-1H-indol-3-yl)methylene]-N-pentylhydrazinecarboximidamide [ACD/IUPAC Name]
(2E)-2-[(5-methoxy-1H-indol-3-yl)methylidene]-N’-pentylhydrazinecarboximidamide
(2E)-2-[(5-methoxy-1H-indol-3-yl)methylidene]-N-pentylhydrazinecarboximidamide
145158-71-0 [RN]
7606
Hydrazinecarboximidamide, 2-[(5-methoxy-1H-indol-3-yl)methylene]-N-pentyl-, (2E)
Sundaram Venkataraman, Srinivasulu Gudipati, Brahmeshwararao Mandava Venkata Naga, Goverdhan Banda, Radhakrishna Singamsetty, “Process for preparing form I of tegaserod maleate.” U.S. Patent US20050272802, issued December 08, 2005.US20050272802
Tegaserod maleate [USAN]
189188-57-6
Tegaserod
CAS Registry Number: 145158-71-0
CAS Name: 2-[(5-Methoxy-1H-indol-3-yl)methylene]-N-pentylhydrazinecarboximidamide
Molecular Formula: C16H23N5O
Molecular Weight: 301.39
Percent Composition: C 63.76%, H 7.69%, N 23.24%, O 5.31%
Literature References: Selective serotonin 5HT4-receptor partial agonist. Prepn: R. K. A. Giger, H. Mattes, EP 505322; eidem, US5510353 (1992, 1996 both to Sandoz); K.-H. Buchheit et al., J. Med. Chem. 38, 2331 (1995). Clinical pharmacology: S. Appel et al., Clin. Pharmacol. Ther. 62, 546 (1997); and pharmacokinetics: idem et al., J. Clin. Pharmacol. 37, 229 (1997). Clinical trial in irritable bowel syndrome: S. A. Müller-Lissner et al., Aliment. Pharmacol. Ther. 15, 1655 (2001); in female patients: J. Novick et al.,ibid. 16, 1877 (2002). Review of clinical efficacy: B. W. Jones et al., J. Clin. Pharm. Ther. 27, 343-352 (2002); of mechanism of action, efficacy and safety: M. Corsetti, J. Tack, Expert Opin. Pharmacother. 3, 1211-1218 (2002).
Properties: mp 155°.
Melting point: mp 155°
Derivative Type: Maleate
CAS Registry Number: 189188-57-6
Manufacturers’ Codes: SDZ-HTF-919
Trademarks: Zelmac (Novartis); Zelnorm (Novartis)
Molecular Formula: C16H23N5O.C4H4O4
Molecular Weight: 417.46
Percent Composition: C 57.54%, H 6.52%, N 16.78%, O 19.16%
Therap-Cat: Gastroprokinetic; in treatment of irritable bowel syndrome.
Keywords: Gastroprokinetic; Serotonin Receptor Agonist.
Tegaserod is a 5-HT4 agonist manufactured by Novartis and sold under the names Zelnorm and Zelmac for the management of irritable bowel syndrome and constipation.[1] Approved by the FDA in 2002, it was subsequently removed from the market in 2007 due to FDA concerns about possible adverse cardiovascular effects. Before then, it was the only drug approved by the United States Food and Drug Administration to help relieve the abdominal discomfort, bloating, and constipation associated with irritable bowel syndrome. Its use was also approved to treat chronic idiopathic constipation.[2]
In 2000, originator Novartis established an alliance with Bristol-Myers Squibb for the codevelopment and copromotion of tegaserod maleate, which is now available in more than 55 countries worldwide for the treatment of IBS with constipation. In 2015, Zelnorm was acquired by Sloan Pharma from Novartis.
Novartis’ brand name Zelnorm (tegaserod) had originally received approval from the US FDA in 2002 for the treatment of irritable bowel syndrome with constipation (IBS-C) [5, 8]. It was, however, voluntarily withdrawn from widespread use in the US market in 2007 after concerns arose over the possibility that tegaserod could potentially cause dangerous cardiovascular events in patients [5,8]. Since then, closer evaluations of the original data suggesting such cardiovascular risk have resulted in the limited reintroduction or ‘re-approval’ of tegaserod for treatment of IBS-C specifically in female patients less than 65 years of age and whom are considered to be at a lower risk of a cardiovascular event than the broader population . Zelnorm (tegaserod) by Sloan Pharma subsequently gained re-approval in April of 2019 [5]. Nevertheless, tegaserod remains un-approved in certain regions [7].
Despite the relative complications involved in its history of regulatory approval, ever since its first introduction in 2002 tegaserod remains the only therapy for IBS-C that possesses the unique mechanism of action of acting on serotonin-4 (5-HT(4)) receptors in smooth muscle cells and in the gastrointestinal wall to facilitate actions like esophageal relaxation, peristaltic gut movement, and natural secretions in the gut, among others
Mechanism of action
The drug functions as a motility stimulant, achieving its desired therapeutic effects through activation of the 5-HT4 receptors of the enteric nervous system in the gastrointestinal tract. It also stimulates gastrointestinal motility and the peristaltic reflex, and allegedly reduces abdominal pain.[3] Additionally, tegaserod is a 5-HT2B receptor antagonist.[4]
Withdrawal from market
On 30 March 2007, the United States Food and Drug Administration requested that Novartis withdraw Zelnorm from shelves.[5] The FDA alleges a relationship between prescriptions of the drug and increased risks of heart attack or stroke. An analysis of data collected on over 18,000 patients demonstrated adverse cardiovascular events in 13 of 11,614 patients treated with Zelnorm (a rate of 0.11%) as compared with 1 of 7,031 patients treated with placebo (a rate of 0.01%). Novartis alleges all of the affected patients had preexisting cardiovascular disease or risk factors for such, and further alleges that no causal relationship between tegaserod use and cardiovascular events has been demonstrated.[6] On the same day as the FDA announcement, Novartis Pharmaceuticals Canada announced that it was suspending marketing and sales of the drug in Canada in response to a request from Health Canada.[7] In a large cohort study based on a US health insurance database, no increase in the risk of cardiovascular events were found under tegaserod treatment.[8] Currently, tegaserod may only be used in emergency situations only with prior authorization from the FDA.[9]
Paper
The serotonin 5-HT4 receptor. 2. Structure-activity studies of the indole carbazimidamide class of agonists
J Med Chem 1995, 38(13): 2331
https://pubs.acs.org/doi/abs/10.1021/jm00013a010
PATENT
US 5510353
WO 2005105740
WO 2007119109
WO 2007126889
CN 103467358
WO 2006116953
Syn
PATENT
https://patents.google.com/patent/US20090306170A1/en

-
In a preferred embodiment of the first aspect of the present invention, the process of preparing tegaserod or a salt thereof comprises the steps of:
- (a) coupling S-methyl-isothiosemicarbazide or a salt thereof and 5-methoxy-indole-3-carboxaldehyde to form 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide:
-

-
and
- (b) reacting the 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide with n-pentyl amine to form tegaserod:
-

- [0013]
The skilled person will appreciate that:
-
- S-methyl-isothiosemicarbazide and salts thereof exist in two tautomeric forms:
-
-

-
- 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide exists in four tautomeric forms:
-
-

-
- tegaserod exists in four tautomeric forms:
-
-

- [0017]
It is to be understood that where tautomeric forms occur, the present invention embraces all tautomeric forms and their mixtures, i.e. although S-methyl-isothio-semicarbazide and 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemi-carbazide are mostly defined for convenience by reference to one isothiosemicarbazide form only, and although tegaserod is mostly defined for convenience by reference to one guanidino form only, the invention is not to be understood as being in any way limited by the particular nomenclature or graphical representation employed.
- [0018]
When an S-methyl-isothiosemicarbazide salt is used in the process of the present invention, this may be an acid addition salt with acids, including but not limited to inorganic acids such as hydrohalogenic acids (for example, hydrofluoric, hydrochloric, hydrobromic or hydroiodic acid) or other inorganic acids (for example, nitric, perchloric, sulfuric or phosphoric acid), or organic acids such as organic carboxylic acids (for example, propionic, butyric, glycolic, lactic, mandelic, citric, acetic, benzoic, salicylic, succinic, malic or hydroxysuccinic, tartaric, fumaric, maleic, hydroxymaleic, mucic or galactaric, gluconic, pantothenic or pamoic acid), organic sulfonic acids (for example, methanesulfonic, trifluoromethanesulfonic, ethanesulfonic, 2-hydroxyethanesulfonic, benzenesulfonic, p-toluenesulfonic, naphthalene-2-sulfonic or camphorsulfonic acid) or amino acids (for example, ornithinic, glutamic or aspartic acid). Preferably the S-methyl-isothiosemicarbazide salt is a hydrohalide (such as the hydrofluoride, hydrochloride, hydrobromide, or hydroiodide) or a sulfonate (such as the methanesulfonate, benzenesulfonate, or p-toluenesulfonate). Preferably the S-methyl-isothiosemicarbazide salt is S-methyl-isothiosemicarbazide hydroiodide.
-
-
The following synthetic scheme demonstrates a preferred process of the present invention.
-
- [0032]
The invention is now demonstrated by the following non-limiting illustrative example.
-
EXAMPLE Step 1: Schiff’s Base Formation of 5-methoxy-indole-3-carboxaldehyde and S-methyl-isothiosemi-carbazide hydroiodide
-
- [0033]
5-Methoxy-indole-3-carboxaldehyde (1.5 g, 1 eq) and S-methyl-isothiosemicarbazide hydroiodide (3.99 g, 2 eq) in methanol (15 ml, 10 vol) were stirred in the presence of triethylamine (3 ml, 2 vol) at 25-30° C. for 2 hours. After completion of the reaction, the methanol was removed by distillation under reduced pressure at 45-50° C. and ethyl acetate (10.5 ml, 7 vol) was added to the residue to precipitate out the product. The product, 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemi-carbazide, was separated by filtration, washed with ethyl acetate (3 ml, 2 vol) and dried under vacuum at 45-50° C. The yield was almost quantitative (˜100%).
- [0033]
Step 2: Conversion of 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide to 1-((5-methoxy-1H-indol-3-yl)methyleneamino)-3-pentyl-guanidine (Tegaserod)
-
- [0034]
A solution of 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide (8.0 g, 1 eq) and n-pentyl amine (2.65 g, 1 eq) was refluxed in methanol (8 ml, 1 vol) at 66° C. for 4 hours. After completion of the reaction, the methanol was removed by distillation under reduced pressure at 45-50° C. to obtain tegaserod free base as a yellowish brown solid. Yield=97%. HPLC purity=95%.
- [0034]
Step 3: Conversion of 1-((5-methoxy-1H-indol-3-yl)methyleneamino)-3-pentyl-guanidine (Tegaserod) to Tegaserod Maleate
- [0035]
1-((5-Methoxy-1H-indol-3-yl)methyleneamino)-3-pentyl-guanidine (55 g, 1 eq) was taken in methanol (357.5 ml, 6.5 vol) and stirred. To this reaction mixture was added at room temperature a solution of maleic acid (74.15 g, 3.5 eq) in water (137.5 ml, 2.5 vol) and the reaction mixture stirred for one hour at room temperature. The solid obtained was then filtered through a Buchner funnel and dried at 700 mmHg and 500° C. Yield=36.8 g, 48.42%. HPLC purity=99.45%.
Polymorphs
WO 2007084697
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007084697
EXAMPLES
PXRD:
EV 320 251 655 US Powder X-ray diffraction (“PXRD”) analysis using a SCINTAG powder X-ray diffϊactometer model X’TRA equipped with a solid-state detector. Copper radiation of λ=1.5418 A was used. The sample was introduced using a round standard aluminum sample holder with round zero background quartz plate in the bottom.
Thermal Gravimetric Analysis TTGA):
TGA/SDTA 85 r, Mettler Toledo , Sample weight 7-15 mg.
Heating rate: 100C/ min., in N2 stream: flow rate: 50 ml/min
Example 1 : Preparation of Tegaserod maleate Form B
To a mixture of 90 g MICHO and 63 g NaOH [47 %] was added a solution of 212 g AGPΗI dissolved in 566 mL of water at room temperature. The resultant reaction mixture was heated to 400C. After 3 hours, 522 mL of ethyl acetate was added and the reaction mixture was stirred for an additional hour. The organic phase was washed with water (3 x 450 mL), and vacuum filtered. After addition of 211 mL ethyl acetate and 870 mL of n-propanol, the mixture was heated to 600C and a solution of maleic acid (86.5 g in 180 mL water), at the same temperature, was added to the reaction mixture and stirred at the same temperature. After 2 hours the reaction mixture was cooled to about 100C and stirred for an additional hour. The resulting solid was filtered off, washed with n-propanol, and dried in a vacuum oven over night to give 195.8 g of tegaserod maleate Form B.
6
EV 320251 655 US
PATENT
Tegaserod maleate is an aminoguanidine indole 5HT4 agonist for the treatment of irritable bowel syndrome (IBS). Tegaserod maleate has the following structure:
According to the prescribing information (Physician’s Desk Reference, 57th Ed., at Page 2339), tegaserod as the maleate salt is a white to off-white crystalline powder and is slightly soluble in ethanol and very slightly soluble in water. Tegaserod maleate is available commercially as ZELNORM®, in which it is present as crystalline form.
Tegaserod maleate is disclosed in US patent No. 5,510,353 and in its equivalent EP 0 505 322 (example 13), and is reported to have a melting point of 1900C (table 1 example 13).
The literature (Buchheit K.H, et al., J.Med.Chem., 1995, 38, 2331) describes a general method for the condensation of amino guanidines with indole-3-carbadehydes in methanol in the presence of HCl (pH 3-4). The product obtained after solvent evaporation maybe converted to its hydrochloride salt by treatment of the methanolic solution with diethylether/HCl followed by recrystallization from
methanol/diethylether. Tegaserod base prepared according to this general method is characterized solely by a melting point of 155 0C (table 3 compound 5b). Additional Tegaserod maleate characterization was done by 1H and 13C-NMR according to the literature (Jing J. et. al., Guangdong Weiliang Yuansu Kexue, 2002, 9/2, 51).
WO 04/085393 discloses four crystalline forms of tegaserod maleate. The search report for WO 04/085393 further identifies WO 00/10526, and Drugs Fut. 1999, 24(1) which provides an overview for tegaserod maleate. Additional crystalline forms of tegaserod maleate are provided in WO 2005/058819, one of which is characterized by an X-ray Diffraction pattern having peaks at 15.7, 16.9, 17.2, 24.1, 24.6 and 25.2±0.2 two theta (designated as Form B in that PCT publication).
The solid state physical properties of tegaserod salt may be influenced by controlling the conditions under which tegaserod salt is obtained in solid Form. Solid state physical properties include, for example, the flowability of the milled solid. Flowability affects the ease with which the material is handled during processing into a pharmaceutical product. When particles of the powdered compound do not flow past each other easily, a formulation specialist must take that fact into account in developing a tablet or capsule formulation, which may necessitate the use of glidants such as colloidal silicon dioxide, talc, starch or tribasic calcium phosphate.
Another important solid state property of a pharmaceutical compound is its rate of dissolution in aqueous fluid. The rate of dissolution of an active ingredient in a patient’s stomach fluid may have therapeutic consequences since it imposes an upper limit on the rate at which an orally- administered active ingredient may reach the patient’s bloodstream. The rate of dissolution is also a consideration in
formulating syrups, elixirs and other liquid medicaments. The solid state Form of a compound may also affect its behavior on compaction and its storage stability.
These practical physical characteristics are influenced by the conformation and orientation of molecules in the unit cell, which defines a particular polymorphic Form of a substance. The polymorphic form may give rise to thermal behavior different from that of the amorphous material or another polymorphic Form. Thermal behavior is measured in the laboratory by such techniques as capillary melting point, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) and may be used to distinguish some polymorphic forms from others. A particular polymorphic Form may also give rise to distinct spectroscopic properties that may be detectable by powder X-ray crystallography, solid state C NMR spectrometry and infrared spectrometry.
The discovery of new polymorphic forms of a pharmaceutically useful compound provides a new opportunity to improve the performance characteristics of a pharmaceutical product. It enlarges the repertoire of materials that a formulation scientist has available for designing, for example, a pharmaceutical dosage form of a drug with a targeted release profile or other desired characteristic.
The polymorphic forms may further help in purification of tegaserod, particularly if they possess high crystallinity. In the event of metastability, a metastable polymorphic form may be used to prepare a more stable polymorph.
Hence, discovery of new polymorphic forms and new processes help in advancing a formulation scientist in preparation of tegaserod as an active pharmaceutical ingredient in a formulation.
The present invention provides an additional polymorphic form of a maleate salt of tegaserod.
Example 1 : Preparation of sesqui-tefiaserod maleate Foπn H2 through tegaserod base
To a mixture of AGPΗI (112.7 g) in 283 mL of water was added 5-MICHO (45 g) followed by NaOH (52.8 g, 47%) and stirred at room temperature. After three hours, 522 mL of ethyl acetate were added and the mixture stirred for an additional four hours. After phase separation at 400C the organic phase was washed with water (3 x 218 ml), and filtrated under vacuum. The resulting solution was heated to 60 0C and a solution of maleic acid (14.4 g) in 45 mL water was dropped during half hour, and the reaction mixture stirred at the same temperature for an additional two hours. The mixture was cooled to 100C during one hour, kept under stirring at the same temperature for 12 hrs and then filtered under vacuum. The wet product was washed twice with 65 ml of ethyl acetate and dried in a vacuum oven at 45°C for 16 hours to give 85% of the product.
Example 2: Preparation of sesqui-tegaserod maleate Form H2
45 gr MICHO were added to a 1 L reactor at RT. A solution of 112.7 gr of AGP HI and 283 ml water was added to the reactor. 52.8 gr of NaOH 47% were added to the mixture while stirring. The mixture was heated to 400C and stirred for 12 hrs. 522 ml of Ethyl Acetate were added and the mixture was stirred for 4 hrs.
After phase separation at 400C the organic phase was washed with water (3 x 218 ml), and filtrated under vacuum.
The mixture was heated to 600C and a mixture o 14.4 gr of Maleic Acid in 45 ml water was dropped during 5 min.
The mixture was stirred at 600C for 2 hrs.
The mixture was cooled to 100C during 1 hour, stirred at 100C for 13 hrs and then filtered under vacuum. The wet product was washed twice with 65 ml of n-Propanol. The wet product was dried in a vacuum oven at 45°C.
Yield: 71.2%
Example 3: Preparation of Tegaserod maleate Form B from Sesqui-tegaserod maleate Form H2
6.9 g of maleic acid were added to a slurry of Sesqui-Tegaserod maleate Form H2 (41.5 g) in 208 ml n-propanol at room temperature. The mixture was stirred for 5 hours at the same temperature, filtered and washed with n-propanol. After drying on vacuum oven at 450C for 15 hours the product was analyzed by XRD and found to be Form B (89% yield).
PATENT
https://patents.google.com/patent/WO2005058819A2/en


PATENT
The formation of hydrazones is catalyzed by both general acids and general bases. General base catalysis of dehydration of the tetrahedral intermediate involves nitrogen deprotonation concerted with elimination of hydroxide ion as shown in the Scheme (Sayer J.M., et al. J. Am. Chem. Soc. 1973, 95, 4277). R fast O I H h° NH2R’ R- -NHR’ R R

In many cases, the equilibrium constant for their formation in aqueous solution is high. The additional stability may be attributed to the participation of the atom adjacent to the nitrogen in delocalized bonding. – + RRC = N – NH2 ~*→- RRC – N = NH2
In order to obtain only the maleic salt, the product when using an acid halide (HA) or other acids has to first be converted into the free base, before the addition of maleic acid (Path a), which results in an additional step to the synthesis. On the other hand, the reaction of the present invention in the presence of organic or inorganic base results in the formation of tegaserod free base which gives only the maleate salt after the addition of maleic acid (Path b).


TGS

TGS-MA
EXAMPLES
HPLC method for detecting the level of the impurities:
Column: Atlantis dcl8(150*4.6),
Mobile phase: A.80% KH2PO4(0.02M) pH=5, 20% acetonitrile(ACN), B.100% ACN. Gradient: time 0= A: 100 B: 0, time 25 min= A:50%, B:50%, time 30 min= A:50%, B:50%, + 10 minutes of equilibration time. Wavelength= 225 nm
Sample concentration: 0.5 mg/mL
Temperature = 25°C
Example 1- Preparation of Tegaserod maleate in water with HCl.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by HCl (37%) until pH 4. The mixture was heated to reflux for 1 hour and then cooled to room temperature. To the resulting slurry was added a solution of NaHCO3 (10%) until pH 9, and heated to 65°C for 20 minutes. After cooling, 100 mL of EtOAc were added, and the organic phase washed with water. A solution of maleic acid (3.48 g, 0.03 mol) in 100 mL EtOAc was added, and the resulting solid was filtered off and washed with EtOAc to give 6.27 g of crude tegaserod maleate with a purity of 99.70% (by HPLC).
Example 2- Preparation of Tegaserod maleate in water with HCl in two steps. a. Preparation of Tegaserod free base.
To a mixture of AGP-HI (163.3 g, 0.6 mol) in 375 mL water was added 5-MICHO (52.5 g, 0.3 mol) followed by HCl (37%) until pH 4. The mixture was heated to reflux for 1 hour and then cooled to room temperature. To the resulting slurry was added a liter of a solution of NaHCO (10%) until pH 9, and heated to 65 °C for one hour. After cooling, 1500 mL of EtOAc were added, and the organic phase washed with water. The remaining organic phase was evaporated to dryness to give tegaserod free base with a purity of 87.42 % (by HPLC). b. Preparation of Tegaserod maleate. To a solution of 2 g of tegaserod free base in MeOH was added a solution of maleic acid (1.28 g, 0.011 mol) in 10 mL MeOH. The resulting solid was filtered off and washed with MeOH to give 1.09 g of crude tegaserod maleate with a purity of 96.81 % (by HPLC).
Example 3- Preparation of Tegaserod maleate in water with TEA.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 100 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by TEA (11.0 mL, 0.08 mol) and stirred at room temperature. After one hour, 25 mL of EtOAc was added, and the organic phase washed with water. A solution of maleic acid (3.48 g, 0.03 mol) in 100 mL EtOAc was added, and the resulting solid was filtered off and washed with EtOAc to give 7.92 g of crude tegaserod maleate with a purity of 94 % (by HPLC).
Example 4- Preparation of Tegaserod maleate in water with NaHCO3. To a mixture of AGP-HI (10.88 g, 0.04 mol) in 100 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by NaHCO3 (6.72 g, 0.08 mol) and heated to reflux for 1 hour. After cooling, 50 mL of EtOAc was added, and the organic phase washed with water. A solution of maleic acid (3.48 g, 0.03mol) in 100 mL EtOAc was added, and the resulting solid was filtered off and washed with EtOAc to give 6.71 g of crude tegaserod maleate with a purity of 98 % (by HPLC) .
Example 5- Preparation of Tegaserod maleate in water with NaHCO3 in two steps. a. Preparation of Tegaserod free base. To a mixture of AGP-HI (32.66 g, 0.12 mol) in 300 mL water was added 5-MICHO (10.51 g, 0.06 mol) followed by NaHCO3(20.16 g, 0.24 mol) and heated to reflux for 1 hour. After cooling, 150 mL of EtOAc was added, and the organic phase washed with water and evaporated to dryness to give 20.4 g of tegaserod free base (91.55%) purity by HPLC). b. Preparation of Tegaserod maleate.
To a solution of 2g of the resulting tegaserod free base in 8 mL MeOH was added a solution of maleic acid (1.28 g, 0.011 mol) in 5 mL MeOH. The resulting solid was filtered off and washed with MeOH to give 2.1 g of crude tegaserod maleate with a purity of 99.63 % (by HPLC).
Example 6- Preparation of Tegaserod maleate in water with Na2CO3. To a mixture of AGP-HI (10.88 g, 0.04 mol) in 100 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by Na2CO3 (4.24 g, 0.04 mol) and heated to reflux for 1 hour. After cooling, 50 mL of EtOAc was added, and the organic phase washed with water. A solution of maleic acid (3.48 g, 0.03 mol) in 100 mL EtOAc was added, and the resulting solid was filtered off and washed with EtOAc to give 6.48 g of crude tegaserod maleate with a purity of 98.2 % (by HPLC).
Example 7- Preparation of Tegaserod maleate in MeOH with TEA in two steps. a. Preparation of tegaserod free base
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL MeOH was added 5-MICHO (3.50 g, 0.02 mol) followed by triethylamine (11.0 mL, 0.08 mol). After 1 h at room temperature the mixture was evaporated to dryness, and washed with water, giving 5.79 g of tegaserod free base (86.90 % purity by HPLC). b. Preparation of tegaserod maleate
To a solution of 2 g of the resulting tegaserod free base in 10 mL MeOH was added a solution of maleic acid (1.16 g, 0.01 mol) in water. The resulting solid was filtrated and washed with water to give 1.45 g of crude tegaserod maleate as a white solid (94.60 % purity by HPLC). Crystallization in MeOH improved the purity to 98.94% by HPLC.
Example 8- Preparation of Tegaserod maleate in IPA with K2CO3.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL IPA was added 5-MICHO (3.50 g, 0.02 mol) followed by K2CO3 (5.53g, 0.04 mol). After 22 h at room temperature the mixture was washed with brine. The organic phase was treated with a solution of maleic acid (3.48 g, 0.03 mol) in IPA. The resulting solid was filtrated and washed with IPA to give 3.26 g of a white solid (98.97% purity by HPLC).
Example 9- Preparation of Tegaserod maleate in TEA.
To a mixture of AGP-HI (10.88 g, 0.04 mol) and 5-MICHO (3.50 g, 0.02 mol) was added 11 mL of TEA (0.08 mol). After 2 h at room temperature 25 mL of EtOAc were added and the mixture was stirred for 1 h. The resulting solid was filtrated and washed with 25 mL EtOAc, to give 5.7 g of crude.
2 g of the residue was dissolved in 13 mL MeOH and treated with 7 mL of a solution of maleic acid (2.7 g, 0.023 mol) in water. The resulting solid was filtered and washed with water to give 1.5 g of tegaserod maleate (99.26 % purity by HPLC). Crystallization of the solid in MeOH improved the purity to 99.89%) by HPLC.
Example 10- Preparation of Tegaserod maleate in toluene/water with NaHCO3. a. Preparation of tegaserod free base To a mixture of AGP-HI (10.88 g, 0.04 mol) in 200 mL of water/toluene 1:1 was added 5-MICHO (3.50 g, 0.02 mol) followed by NaHCO3 (6.72 g, 0.08 mol) and heated to reflux for 1 hour. After cooling, the solid was filtrated out of the mixture and washed with water. After drying 6.25 g of tegaserod free base was obtained (93.8 % purity by HPLC). b. Preparation of tegaserod maleate To a solution of 3 g of the product in 10 mL MeOH was added a solution of maleic acid (2.31 g, 0.02 mol) in 10 mL water. The resulting solid was filtered off and washed with a solution of MeOH / water to give 2.50 g of crude tegaserod maleate with a purity of 96.6 % (by HPLC).
Example 11- Preparation of Tegaserod maleate in water with NaOH. a. Preparation of tegaserod free base
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL of water was added 5-MICHO (3.50 g, 0.02 mol) followed by NaOH (2 g, 0.05 mol) and stirred at room temperature. After 3 hours 50 mL of EtOAc was added, and the organic phase washed with water and evaporated to dryness to give 5.6 g of tegaserod free base (98.80% purity by HPLC). b. Preparation of Tegaserod maleate.
To a solution of 1.6 g of tegaserod free base in 15 mL ethyl acetate was added a solution of maleic acid (0.7 g, 0.006 mol) in 5 mL ethyl acetate. The resulting solid was filtered off and washed with ethyl acetate to give 1.65 g of crude tegaserod maleate, with a purity of 99.87 % (by HPLC)
Example 12- Preparation of Tegaserod maleate in water with maleic acid. To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL of water was added 5-MICHO (3.50 g, 0.02 mol) followed by maleic acid (9.3 g, 0.08 mol) and heated to reflux for 1 hour. After cooling, the solid was filtrated out of the mixture and washed with water. After drying 6.92 g of tegaserod maleate crude was obtained (92.4 % purity by HPLC).
Example 13- Preparation of Tegaserod maleate in methanol with maleic acid.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL of methanol was added 5- MICHO (3.50 g, 0.02 mol) followed by maleic acid (9.29 g, 0.08 mol) and heated to reflux for 2 hours. After cooling, the solid was filtrated out of the mixture and washed with water. After drying 6.51 g of tegaserod maleate crude was obtained (97.4 % purity by HPLC).
Example 14- Preparation of Tegaserod maleate in water with NaOH in one pot. To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL of water was added 5-MICHO (3.50 g, 0.02 mol) followed by NaOH (2 g, 0.05 mol) and stirred at room temperature. After 4 hours a solution of maleic acid (4.35 g, 0.0375 mol) in 25 mL water was added, and the reaction mixture was stirred overnight. The resulting solid was filtered off and washed with water to give 7.87 g of crude tegaserod maleate (99.16% purity by HPLC).
Example 15- Preparation of Tegaserod maleate in water with NaOH in one pot.
To a mixture of AGP-HI (174.2 g, 0.64 mol) in 362 mL of water was added 5-MICHO (56.2 g, 0.32 mol) followed by NaOH (68.1 g, 47%) and stirred at room temperature. After 4.5 hours, 640 mL of EtOAc was added, and the organic phase washed with water, treated with active carbon and filtrated through hyper flow bed. A solution of maleic acid (44.57 g, 0.38 mol) in 415 mL ethyl acetate / water 97:3 was added, and the reaction mixture was heating to 65 °C and stirrer overnight. The resulting solid was filtered off and washed with water and ethyl acetate to give 121.4 g of crude tegaserod maleate (up to 99.88 % purity by HPLC).
Example 16- Preparation of Tegaserod maleate (from Tegaserod acetate).
To a solution of 8.2 g of tegaserod acetate in 15 mL ethyl acetate heated to 65 °C was added a solution of 3.3 g maleic acid in 5 ml ethyl acetate/water 95:5, and the mixture was stirred at the same temperature for an additional 2 hours, followed by cooling to room temperature and stirring overnight. The resulting solid was filtered off and washed with ethyl acetate/water 95:5. After drying on vacuum oven at 45 °C for 15 hours, 9.18 g of tegaserod maleate were obtained. Tegaserod acetate is prepared according to Examples 19, 20 and 21 of U.S. Appl. No. 11/015,875 and PCT/US04/42822.
Example 19 of U.S. Appl. No. 11/015,875 reads as follows: A slurry of tegaserod base amorphous (6 g) in 50 mL ethyl acetate was stirred at 20- 30 °C for 24 hours. The solid was filtrated and washed with 15 mL of same solvent and dried in a vacuum oven at 40 °C for 16 hours.
Example 20 of U.S. Appl. No. 11/015,875 reads as follows:
A slurry of tegaserod base amorphous (6 g) in 50 mL ethyl acetate was stirred at reflux for 24 hours. The solid was filtrated and washed with 15 mL of same solvent and dried in a vacuum oven at 40 °C for 16 hours.
Example 21 of U.S. Appl. No. 11/015,875 reads as follows:
To a slurry of tegaserod maleate Form A (15 g) in EtOAc (210 mL) and water (210 mL) was added 38.4 g of NaOH 47%. The mixture was stirred overnight and the resulting white solid was isolated by filtration and washed with 100 mL of water. Drying in vacuum oven at 40 °C for 16 hours gives 12.38 g (90% yield). Tegaserod acetate was characterized by H and C-NMR.
Example 17: General method for the preparation of Tegaserod maleate Form A from crystallization.
Tegaserod maleate (1 g) was combined with the appropriate solvent (5 mL), and heated to reflux. Then, additional solvent was added until complete dissolution. After the compound was dissolved, the oil bath was removed and the solution was cooled to room temperature. The solid was filtrated and washed with 5 mL of the same solvent and dried in a vacuum oven at 40 C for 16 hours.


Example 18: Preparation of Tegaserod maleate in water with p-TSOH.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by para-toluenesulfonic acid monohydrate (0.45 g, 0.0024 mol). The mixture was heated to reflux for 4 hour and then cooled to room temperature. The resulting solid was filtered off and washed with water to give 8.32 g of a white solid (84.74 % purity by HPLC).
Example 19: Preparation of Tegaserod maleate from Tegaserod Hemi-maleate hemihydrate
To a solution of 1.72 g of Tegaserod Hemi-maleate hemihydrate in 20 mL ethyl acetate at room temperature was added a solution of 0.134 g maleic acid in 5 ml ethyl acetate/water 95:5, and the mixture was stirred at the same temperature for overnight. The resulting solid was filtered off and washed with ethyl acetate/water 95:5. After drying on vacuum oven at 45°C for 15 hours, 1.68 g of tegaserod maleate were obtained. Tegaserod Hemi-maleate hemihydrate was prepared according to Example 23 of U.S. Appl. No. 11/015,875 and PCT/US04/42822. Example 23 of U.S. Appl. No. 11/015,875 and PCT/US04/42822 reads as follows: A solution of maleic acid (2.32 g in 22 mL ethyl acetate/water 97:3) was added to a mixture of tegaserod base in ethyl acetate, and the reaction mixture was heated to 65 °C and stirrer overnight. The resulting solid was filtered off and washed with water and ethyl acetate. Drying in vacuum oven at 40 °C for 16 hours gives 12.19 g of Tegaserod hemi-maleate hemihydrate. Depending on the base polymorph used a solution or slurry is obtained. When using amorphous tegaserod base, a solution is obtained, while when using any other base polymorph of tegaserod, a slurry is obtained.
PATENT
https://patents.google.com/patent/WO2009063247A1/en
Tegaserod, chemically named 2-[(5-methoxy-liϊ-indol-3-yl)methylene]-IV-pentylhydrazine- carboximidamide, is a selective serotonin 4 (5-HT4) receptor agonist, which can be used to treat gastrointestinal disorders such as heartburn, bloating, postoperative ileus, abdominal pain and discomfort, epigastric pain, nausea, vomiting, regurgitation, intestinal pseudoobstruction, irritable bowel syndrome and gastro-oesophageal reflux. Tegaserod as the maleate salt is marketed for the short-term treatment of irritable bowel syndrome in women whose primary bowel symptom is constipation.
Tegaserod, represented by the formula (I), was first described in US 5 510 353 as well as processes for its preparation. The maleate salt of tegaserod is also disclosed, but interestingly a method of manufacturing tegaserod maleate is not disclosed. The only characterizing data is the melting point which is disclosed as 1900C for the maleate salt and 124°C for the tegaserod base.

WO 2006/116953 describes crystalline forms of the hydrobromide, dihydrogen phosphate and oxalate salts of tegaserod. Also claimed is a process for preparing the hydrochloride, hydrobromide, dihydrogen phosphate, tartrate, citrate, lactate, mesylate, oxalate, succinate, glutarate, adipate, salicylate, sulfate, mandelate, camphor sulfonate and hydrogen sulfate salts of tegaserod from a specific crystalline form of tegaserod base. Another process described is a method of preparing the dihydrogen phosphate, maleate, tartrate, citrate, mesylate, lactate, succinate, oxalate, hydrochloride, salicylate, glutarate, adipate, hydrobromide, sulfate and hydrogen sulfate from a hydrogen halide salt of tegaserod.
There are often major hurdles to overcome before an active pharmaceutical ingredient (API) can be formulated into a composition that can be marketed. For example, the rate of dissolution of an API that has poor aqueous solubility is often problematic. The aqueous solubility is a major influence on the bioavailability of the API such that a poorly soluble API can mean the API is not available to have a pharmaceutical effect on the body. The API can also cause problems during manufacture of a pharmaceutical composition. For example, flowability, compactability and stickiness are all factors affected by the solid state properties of an API.
It has thus always been an aim of the pharmaceutical industry to provide many forms of an API in order to mitigate the problems described above. Different salts, crystalline forms also known as polymorphs, solvates and amorphous forms are all forms of an API that can have different physiochemical and biological characteristics. Indeed, it has been discovered that the tegaserod maleate product on the market, Zelnorm , has been linked to an increase in heart problems in a proportion of individuals. One possible reason is that the maleate moiety reacts with the tegaserod, resulting over time in the production of a toxic impurity.
This impurity could be a contributor to the heart problems seen in some patients.
PATENT
https://patents.google.com/patent/WO2004085393A1/en
Figure 1 is a x-ray powder diffraction pattern of tegaserod maleate Form I. Figure 2 is a x-ray powder diffraction pattern of tegaserod maleate Form II. Figure 3 is a x-ray powder diffraction pattern of tegaserod maleate Form III. Figure 4 is a x-ray powder diffraction pattern of tegaserod maleate Form IV. x-Ray powder diffraction spectrum was measured on a Siemens D5000 x- ray powder diffractometer having a copper-Kα radiation.
The following examples further illustrate the invention.
Example 1 Tegaserod free base (10 gm) is dissolved in acetone (100 ml). Maleic acid (4 gm) is added to the solution and the contents are maintained for 1 hour at 25°C. The separated solid is filtered to give 12.5 gm of tegaserod maleate Form I.
Example 2 Tegaserod maleate Form II (5 gm) and acetone (70 ml) are mixed and refluxed for 1 hour and cooled to 25°C and filtered to give 4.8 gm of tegaserod maleate Form I.
Example 3 Tegaserod maleate Form I (10 gm) is dissolved in methanol (100 ml). Acetonitrile (150 ml) is added to the solution and the contents are heated to reflux. The contents are then cooled to 25°C and maintained for 30 minutes. The separated crystals are collected by filtration to give 9 gm of tegaserod maleate Form II.
Example 4 Tegaserod free base (10 gm) is dissolved in methanol (100 ml) and maleic acid (4 gm) is added to the solution. Then the contents are maintained for 30 minutes at 25°C. Then the separated solid is filtered to give 13 gm of tegaserod maleate Form III.
Example 5
Tegaserod maleate (5 gm) is dissolved in methanol (50 ml) and the solution is maintained at 25°C for 30 minutes. The separated crystals are collected by filtration to give 4.8 gm of tegaserod maleate Form III. Example 6 Tegaserod free base (10 gm) is dissolved in methanol (50 ml), maleic acid (4 gm) is added and the contents are refluxed for 30 minutes and then the resulting solution is cooled to 25°C. Methylene dichloride (200 ml) is added and the contents are maintained for 30 minutes at 25°C. The separated solid is collected by filtration to give 13 gm of tegaserod maleate Form IV.
Example 7 Maleic acid (4 gm) is added to a solution of tegaserod free base (10 gm) in methanol (50 ml). The contents are maintained for 30 minutes at 25°C and isopropyl alcohol (150 ml) is mixed and contents are maintained for 30 minutes at 25°C. The separated solid is collected by filtration to give 12.5 gm of tegaserod maleate Form IV
CLIP
References
- ^ “New Data for Zelnorm”. Archived from the original on December 9, 2007. Retrieved March 30, 2007.
- ^ “FDA approves first treatment for women with irritable-bowel syndrome”. Archived from the original on February 5, 2007. Retrieved March 30, 2007.
- ^ Rossi, S. (2004). Australian Medicines Handbook. Adelaide: Health Communication Network. ISBN 0-9578521-4-2.
- ^ Beattie DT, Smith JA, Marquess D, et al. (November 2004). “The 5-HT4 receptor agonist, tegaserod, is a potent 5-HT2B receptor antagonist in vitro and in vivo”. Br. J. Pharmacol. 143 (5): 549–60. doi:10.1038/sj.bjp.0705929. PMC 1575425. PMID 15466450.
- ^ “FDA Announces Discontinued Marketing of GI Drug, Zelnorm, for Safety Reasons”. FDA Press Release. 30 March 2007.
- ^ “Zelnorm” (PDF). Novartis. Archived from the original (PDF) on 2007-04-10. Retrieved 2007-03-30.
- ^ “Novartis suspends Canadian marketing and sales of Zelnorm in response to request from Health Canada”. Retrieved 2007-03-30.
- ^ Loughlin J, Quinn S, Rivero E, Wong J, Huang J, Kralstein J, Earnest DL, Seeger JD (2010). “Tegaserod and the Risk of Cardiovascular Ischemic Events: An Observational Cohort Study”. J Cardiovasc Pharmacol Ther. 15 (2): 151–7. doi:10.1177/1074248409360357. PMID 20200325.
- ^ http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm103223.htm
- Beattie DT, Smith JA, Marquess D, Vickery RG, Armstrong SR, Pulido-Rios T, McCullough JL, Sandlund C, Richardson C, Mai N, Humphrey PP: The 5-HT4 receptor agonist, tegaserod, is a potent 5-HT2B receptor antagonist in vitro and in vivo. Br J Pharmacol. 2004 Nov;143(5):549-60. Epub 2004 Oct 4. [PubMed:15466450]
- Talley NJ: Irritable bowel syndrome. Intern Med J. 2006 Nov;36(11):724-8. doi: 10.1111/j.1445-5994.2006.01217.x. [PubMed:17040359]
- Borman RA, Tilford NS, Harmer DW, Day N, Ellis ES, Sheldrick RL, Carey J, Coleman RA, Baxter GS: 5-HT(2B) receptors play a key role in mediating the excitatory effects of 5-HT in human colon in vitro. Br J Pharmacol. 2002 Mar;135(5):1144-51. doi: 10.1038/sj.bjp.0704571. [PubMed:11877320]
- Vickers AE, Zollinger M, Dannecker R, Tynes R, Heitz F, Fischer V: In vitro metabolism of tegaserod in human liver and intestine: assessment of drug interactions. Drug Metab Dispos. 2001 Oct;29(10):1269-76. [PubMed:11560869]
- FDA approves the reintroduction of Zelnorm™ (tegaserod) for Irritable Bowel Syndrome with Constipation (IBS-C) in women under 65 [Link]
- Tegaserod 2019 FDA Label [File]
- EMA Refusal Assessment Report for Zelnorm (Tegaserod) [File]
- FDA Joint Meeting of the Gastrointestinal Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee Briefing Document for Zelnorm (tegaserod maleate) [File]
Title
ANDRES: “Labelling of HTF919…” J.LABELLED COMP.RADIOPHARM., vol. 42, 1999, pages 1008-1009, XP002354977 *
BUCHHEIT K H ET AL: “THE SEROTONIN 5-HT4 RECEPTOR. 2. STRUCTURE-ACTIVITY STUDIES OF THE INDOLE CARBAZIMIDAMIDE CLASS OF AGONISTS” JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. WASHINGTON, US, vol. 38, 1995, pages 2331-2338, XP000867864 ISSN: 0022-2623 cited in the application *
GRAUL A ET AL: “TEGASEROD MALEATE” DRUGS OF THE FUTURE, BARCELONA, ES, vol. 24, no. 1, 1999, pages 38-44, XP000874672 ISSN: 0377-8282 *
LALEZARI ET AL.: “Selective synthesis of …” J. HETEROCYCL. CHEM., vol. 8, 1971, pages 689-691, XP002354978 *
WAN ET AL.: “Improved synthesis of tegaserod maleate” CHINESE J. MED. CHEM., vol. 13, no. 1, 2003, pages 40-41, XP009057178 *
WO2004085393A1 *2003-03-252004-10-07Hetero Drugs LimitedNovel crystalline forms of tegaserod maleate
WO2006116953A1 *2005-05-022006-11-09Zentiva, A.S.A method for the preparation of tegaserod and slected salts thereof
WO2007084761A1 *2006-01-182007-07-26Teva Pharmaceutical Industries Ltd.Maleate salt of tegaserod and crystalline forms thereof
WO2007084761A1 *2006-01-182007-07-26Teva Pharmaceutical Industries Ltd.Maleate salt of tegaserod and crystalline forms thereof
WO2007120924A1 *2006-04-172007-10-25Teva Pharmaceutical Industries Ltd.Preparation of tegaserod maleate free of iodide
WO2007126889A1 *2006-03-272007-11-08Teva Pharmaceutical Industries Ltd.Preparation of tegaserod acetate
WO2007146717A3 *2006-06-122008-03-27Joginder S BajwaProcess for making salts of n-hydroxy-3-[4-[[[2-(2-methyl-1h-indol-3-yl)ethyl]amino]methyl]phenyl]-2e-2-propenamide
EP1955998A1 *2007-02-072008-08-13Chemo Ibérica, S.A.New addition salt of N-amino-N’-pentylguanidine, the process for its preparation and use thereof for obtaining tegaserod
CL2008000070A1 *2007-01-172008-07-25Lg Life Sciences LtdMaleic acid mono (3 – [({1 – [(2-amino-9 H -purin-9-yl) methyl] cyclopropyl} oxy) methyl] -8,8-dimethyl-3,7-dioxo-2,4 , 6-trioxa-3 lambda 5 -phosphanon-1-yl pivalate; pharmaceutical composition comprising said mono, and use to treat virus h
US5510353A *1991-03-221996-04-23Sandoz Ltd.Certain aminoguanidine compounds, pharmaceutical compositions containing them and their use in treating gastrointestinal motility disorders and disorders associated with cephalic pain
Family To Family Citations
WO2006116953A1 *2005-05-022006-11-09Zentiva, A.S.A method for the preparation of tegaserod and slected salts thereof
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Zelnorm, Zelmac |
| AHFS/Drugs.com | Monograph |
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 10% |
| Protein binding | 98% |
| Metabolism | Gastric and hepatic |
| Elimination half-life | 11 ± 5 hours |
| Excretion | Fecal and renal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C16H23N5O |
| Molar mass | 301.39 g/mol g·mol−1 |
| 3D model (JSmol) | |
| PATENT NUMBER | PEDIATRIC EXTENSION | APPROVED | EXPIRES (ESTIMATED) | |
|---|---|---|---|---|
| US5510353 | No | 1996-04-23 | 2013-04-26 |  |
References
-
- Buchheit, K.-H. et al.: J. Med. Chem. (JMCMAR) 38, 2331 (1995).
- US 5 510 353 (Novartis; 23.4.1996; GB-prior. 22.3.1991).
- EP 505 322 (Sandoz; GB-prior. 22.3.1991).
-
Preparation of 5-methoxyindole:
- Tsuji, Y. et al.: J. Org. Chem. (JOCEAH) 55 (2), 580 (1990).
- Jones, G.B. et al.: J. Org. Chem. (JOCEAH) 58 (20), 5558 (1993).
- Kondo, Y. et al.: J. Org. Chem. (JOCEAH) 62 (19), 6507 (1997).
- JP 3 024 055 (Kawaken Fine Chemicals; 1.2.1991; J-prior. 21.6.1989).
/////////Tegaserod, HTF 919, HTF-919, SDZ HTF 919, SDZ-HTF-919, テガセロド , Sloan Pharma, Novartis,
CCCCCNC(=N)N\N=C\C1=CNC2=C1C=C(OC)C=C2
BMS 986236
BMS-986236
CAS 2058035-15-5
MW C22 H25 N9 O
MF 431.49
1-(5-(4-(3-Hydroxy-3-methylbutyl)-1H-1,2,3-triazol-1-yl)-4-(isopropylamino)pyridin-2-yl)-1H-pyrazolo[3,4-b]pyridine-5-carbonitrile
1H-Pyrazolo[3,4-b]pyridine-5-carbonitrile, 1-[5-[4-(3-hydroxy-3-methylbutyl)-1H-1,2,3-triazol-1-yl]-4-[(1-methylethyl)amino]-2-pyridinyl]-
1-[5-[4-(3-hydroxy-3-methylbutyl)triazol-1-yl]-4-(propan-2-ylamino)pyridin-2-yl]pyrazolo[3,4-b]pyridine-5-carbonitrile
|
The present invention generally relates to heteroaryl substituted aminopyridine compounds useful as kinase inhibitors, including the modulation of IRAK-4. Provided herein are heteroaryl substituted aminopyridine compounds, compositions comprising such compounds, and methods of their use. The invention further pertains to pharmaceutical compositions containing at least one compound according to the invention that are useful for the treatment of conditions related to kinase modulation and methods of inhibiting the activity of kinases, including IRAK-4 in a mammal.
|
PATENT
US2018186799
https://patentscope.wipo.int/search/en/detail.jsf?docId=US222843237&tab=PCTDESCRIPTION&maxRec=1000
PATENT
Gardner, D. S.; Santella, J. B.; Paidi, V. R.; Wu, H.; Duncia, J. V.; Nair, S. K.; Hynes, J. (BMS, USA). Heteroaryl Substituted Aminopyridine Compounds. PCT Int. Appl. WO/2016/210034 A1, 2016.
https://patents.google.com/patent/WO2016210034A1/en
Clip
https://pubs.acs.org/doi/10.1021/acs.oprd.9b00023
Development of a Scalable Synthesis for the Potent Kinase Inhibitor BMS-986236; 1-(5-(4-(3-Hydroxy-3-methylbutyl)-1H-1,2,3-triazol-1-yl)-4-(isopropylamino)pyridin-2-yl)-1H-pyrazolo[3,4-b]pyridine-5-carbonitrile
Pirama Nayagam Arunachalam†, Prakasam Kuppusamy†, Sivakumar Ganesan†, Suresh Krishnamoorthy†, Roshan Y. Nimje†, Lokesh Babu Jarugu†, Nanjundaswamy Kanikahalli Chikkananjaiah†, China Anki Reddy†, Prakash Anjanappa†, Murali Botlagunta†, Sridhar Vanteru†, Nageswararao Maddala†, Muniyappa Shankar†, Satheesh Nair†, John Hynes, Jr.‡, Joseph B. Santella, III‡, Percy H. Carter‡  , Richard Rampulla‡, Muthalagu Vetrichelvan*† , Anuradha Gupta† , Arun Kumar Gupta†, and Arvind Mathur‡
, Richard Rampulla‡, Muthalagu Vetrichelvan*† , Anuradha Gupta† , Arun Kumar Gupta†, and Arvind Mathur‡
, Richard Rampulla‡, Muthalagu Vetrichelvan*† , Anuradha Gupta† , Arun Kumar Gupta†, and Arvind Mathur‡† Department of Discovery Synthesis, Biocon Bristol-Myers Squibb Research Center, Biocon Park, Bommasandra IV Phase, Jigani Link Road, Bangalore-560 099, India
‡ Discovery Chemistry, Bristol-Myers Squibb, P.O. Box 5400, Princeton, New Jersey 08543-4000, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.9b00023
*E-mail: muthalagu.vetrichelvan@syngeneintl.com.
A scalable route to 1-(5-(4-(3-hydroxy-3-methylbutyl)-1H-1,2,3-triazol-1-yl)-4-(isopropylamino)pyridin-2-yl)-1H-pyrazolo[3,4-b]pyridine-5-carbonitrile (1, BMS-986236) was developed by incorporating an alternate azide intermediate following safety-driven processes. The newly developed process involved mitigating safety hazards and eliminating the column chromatography purification. The issue of trace metal contamination in the final API observed in the first-generation synthesis has been overcome.
1 (92.5 g, 73% yield, 99.5% purity by HPLC) as a cream-colored solid.
1H NMR (400 MHz, DMSO-d6) δ = 9.21–8.86 (m, 2H), 8.66 (s, 1H), 8.45–8.24 (m, 2H), 7.49 (s, 1H), 6.57 (d, J = 7.5 Hz, 1H), 4.33 (s, 1H), 3.83 (d, J = 7.0 Hz, 1H), 2.91–2.72 (m, 2H), 1.97–1.68 (m, 2H), 1.24 (d, J = 6.5 Hz, 12H).
13C NMR (100 MHz, DMSO) δ = 151.7, 150.8, 149.8, 147.9, 147.7, 143.7, 136.8, 136.3, 122.9, 118.9, 117.6, 116.0, 102.8, 99.4, 68.4, 43.6, 42.7, 29.2, 21.7, 20.2.
HRMS [M + H]+ calcd for C22H25N9O 432.2255, found 432.2259.



//////// BMS-986236, BMS 986236
CC(C)(O)CCc1cn(nn1)c2cnc(cc2NC(C)C)n4ncc3cc(cnc34)C#N
TAK-981

TAK-981
C25 H28 Cl N5 O5 S2, 578.103
[(1R,2S,4R)-4-[(5-[4-[(1R)-7-Chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl)amino]-2-hydroxycyclopentyl]methyl sulfamate
[(1R,2S,4R)-4-[[5-[4-[(1R)-7-Chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methyl-thiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxy-cyclopentyl]methyl sulfamate
Sulfamic acid, [(1R,2S,4R)-4-[[5-[[4-[(1R)-7-chloro-1,2,3,4-tetrahydro-1-isoquinolinyl]-5-methyl-2-thienyl]carbonyl]-4-pyrimidinyl]amino]-2-hydroxycyclopentyl]methyl ester
| CAS 1858276-04-6 FREE
CAS 1858279-63-6 HYDRATE |
|
| MW | 578.103 |
- Originator Takeda Oncology
- Class Antineoplastics
- Mechanism of Action Small ubiquitin-related modifier protein inhibitors
- Phase I Lymphoma; Solid tumours
- 01 Oct 2018 Phase-I clinical trials in Solid tumours (Late-stage disease, Metastatic disease) and and Lymphoma (Refractory metastatic disease, Second-line therapy or greater) in USA (IV) (NCT03648372)
- 03 Sep 2018 Takeda Oncology plans a phase I trial for Solid tumours (Late-stage disease, Metastatic disease) and Lymphoma (Refractory metastatic disease, Second-line therapy or greater) in September 2018 (IV) (NCT03648372)
- 03 Sep 2018 Preclinical trials in Lymphoma in USA (IV) prior to September 2018 (NCT03648372)
Takeda is evaluating TAK-981, a SUMO-Activating Enzyme (SAE) inhibitor, in early clinical trials for the treatment of adult patients with advanced or metastatic solid tumors or with relapsed or refractory lymphomas.
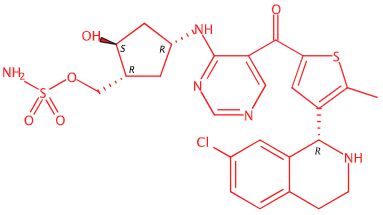
Small ubiquitin-like modifier (SUMO) is a member of the ubiquitin-like protein (Ubl) family that is covalently conjugated to cellular proteins in a manner similar to Ub-conjugation (Kerscher, O., Felberbaum, R., and Hochstrasser, M. 2006. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 22: 159-80). Mammalian cells express three major isoforms: SUMO l , SUM02 and SUM03. SUM02 and SUM03 share -95% amino acid sequence homology but have -45% sequence homology with SUMO l (Kamitani, T., Kito, K., Nguyen, H. P., Fukuda-Kamitani, T., and Yeh, E. T. 1998. Characterization of a second member of the sentrin family of ubiquitin-like proteins. J Biol Chem. 273( 18): 1 1349-53). SUMO proteins can be conjugated to a single lysine residue of a protein (monosumoylation) or to a second SUMO protein that is already conjugated to a protein forming a SUMO chain (polysumoylation). Only SUM02/3 can form such chains because they possess internal consensus SUMO modification sites (Tatham, M. H., Jaffray, E., Vaughan, O. A., Desterro, J. M., Botting, C. H., Naismith, J. H., Hay, R. T. 2001. Polymeric chains of SUMO-2 and SUM 0-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J Biol Chem. 276(38):35368-74). An additional isoform, SUM04, is found in kidney, lymph node and spleen cells, but it is not known whether SUM04 can be conjugated to cellular proteins.
[0003] SUMO l , SUM02 and SUM03 are activated in an ATP-dependent manner by the SUMO-activating enzyme (SAE). SAE is a heterodimer that consists of SAE 1 (SUMO-activating enzyme subunit 1) and SAE2 (UBA2). SAE, like other El activating enzymes, uses ATP to adenylate the C-terminal glycine residue of SUMO. In a second step, a thioester intermediate is then formed between the C-terminal glycine of SUMO and a cysteine residue in SAE2. Next, SUMO is transferred from the El to the cysteine residue of the SUMO conjugating enzyme (E2), UBC9. Unlike the Ub pathway that contains many E2 enzymes, Ubc9 is currently the only known conjugating enzyme for SUMO and functions with SUMOl , SUM02 and SUM03 proteins. SUMO proteins are then conjugated to the target protein, either directly or in conjunction with an E3 ligase, through isopeptide bond formation with the epsilon amino group of a lysine side chain on a target protein. Several SUMO E3 ligases, including PIAS (protein inhibitor of activated signal transducer and activator of transcription protein) proteins and Ran-binding protein 2 (RanBP2), and polycomb 2 (Pc2), have been identified (Johnson, E. S., and Gupta, A. A. 2001. An E3-like factor that promotes SUMO conjugation to the yeast septins. Cell. 106(6):735-44; Pichler, A., Gast, A., Seeler, J. S., Dejean, A.; Melchior, F. 2002. The nucleoporin RanBP2 has SUMOl E3 ligase activity. Cell. 108(1): 109-20; Kagey, M. H., Melhuish, T. A., and Wotton, D. 2003. The polycomb protein Pc2 is a SUMO E3. Cell. 1 13(1): 127- 37). Once attached to cellular targets, SUMO modulates the function, subcellular localization, complex formation and/or stability of substrate proteins (Miiller, S., Hoege, C, Pyrowolakis, G., and Jentsch, S. 2001. SUMO, ubiquitin’s mysterious cousin. Nat Rev Mol Cell Biol. 2(3):202-10). SUMO- conjugation is reversible through the action of de-sumoylating enzymes called SENPs (Hay, R. T. 2007. SUMO-specific proteases: a twist in the tail. Trends Cell Biol. 17(8):370-6) and the SUMO proteins can then participate in additional conjugation cycles.
[0004] SAE-initiated SUMO-conjugation plays a major role in regulating diverse cellular processes, including cell cycle regulation, transcriptional regulation, cellular protein targeting, maintenance of genome integrity, chromosome segregation, and protein stability (Hay, R. T. 2005. SUMO: a history of modification. Mol Cell. 18( 1): 1 -12; Gill, G. 2004. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 18(17):2046-59). For example, SUMO- conjugation causes changes in the subcellular localization of RanGAPl by targeting it to the nuclear pore complex (Mahajan, R., Delphin, C., Guan, T., Gerace, L., and Melchior, F. 1997. A small ubiquitin-related polypeptide involved in targeting RanGAPl to nuclear pore complex protein RanBP2. Cell. 88(1):97- 1070). Sumoylation counteracts ubiquitination and subsequently blocks the degradation of Ι Β, thereby negatively regulating NF-κΒ activation (Desterro, J. M., Rodriguez, M. S., Hay, R. T. 1998. SUMO- 1 modification of IkappaB alpha inhibits NF-kappaB activation. Mol Cell. 2(2):233-9). Sumoylation has been reported to play an important role in transcription exhibiting both repressive and stimulatory effects. Many of the transcriptional nodes that are modulated play important roles in cancer. For example, sumoylation stimulates the transcriptional activities of transcription factors such as p53 and HSF2 (Rodriguez, M. S., Desterro, J. M., Lain, S., Midgley, C. A., Lane, D. P., and Hay, R. T. 1999. SUMO- 1 modification activates the transcriptional response of p53. EMBO J. 18(22):6455-61 ; Goodson, M. L., Hong, Y., Rogers, R., Matunis, M. J., Park-Sarge, O. K., Sarge, K. D. 2001. Sumo- 1 modification regulates the DNA binding activity of heat shock transcription factor 2, a promyelocytic leukemia nuclear body associated transcription factor. J Biol Chem. 276(21 ): 18513-8). In contrast, SUMO-conjugation represses the transcriptional activities of transcription factors such as LEF (Sachdev, S., Bruhn, L., Sieber, H., Pichler, A., Melchior, F., Grosschedl, R. 2001. PIASy, a nuclear matrix-associated SUMO E3 ligase, represses LEF1 activity by sequestration into nuclear bodies. Genes Dev. 15(23):3088- 103) and c-Myb (Bies, J., Markus, J., and Wolff, L. 2002. Covalent attachment of the SUMO- 1 protein to the negative regulatory domain of the c-Myb transcription factor modifies its stability and transactivation capacity. / Biol Chem. 277( 1 1):8999-9009). Thus, SUMO-conjugation controls gene expression and growth control pathways that are important for cancer cell survival.
[0005] Altered expression of SAE pathway components have been noted in a variety of cancer types: (Moschos, S. J., Jukic, D. M., Athanassiou, C., Bhargava, R., Dacic, S., Wang, X., Kuan, S. F., Fayewicz, S. L., Galambos, C., Acquafondata, M., Dhir, R., and Becker, D. 2010. Expression analysis of Ubc9, the single small ubiquitin-like modifier (SUMO) E2 conjugating enzyme, in normal and malignant tissues. Hum Pathol. 41(9): 1286-980); including multiple myeloma (Driscoll, J. J., Pelluru, D., Lefkimmiatis, K., Fulciniti, M., Prabhala, R. H., Greipp, P. R., Barlogie, B., Tai, Y. T., Anderson, K. C, Shaughnessy, J. D. Jr., Annunziata, C. M., and Munshi, N. C. 2010. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood. 1 15(14):2827-34); and breast cancer (Chen, S. F., Gong, C, Luo, M., Yao, H. R., Zeng, Y. J., and Su, F. X. 201 1. Ubc9 expression predicts chemoresistance in breast cancer. Chin J Cancer. 30(9):638-44), In addition, preclinical studies indicate that Myc-driven cancers may be especially sensitive to SAE inhibition (Kessler, J. D., Kahle, K. T., Sun, T., Meerbrey, K. L., Schlabach, M. R., Schmitt, E. M., Skinner, S. O., Xu, Q., Li, M. Z., Hartman, Z. C, Rao, M., Yu, P., Dominguez-Vidana, R., Liang, A. C, Solimini, N. L., Bernardi, R. J., Yu, B., Hsu, T., Golding, I., Luo, J., Osborne, C. K., Creighton, C. J., Hilsenbeck, S. G., Schiff, R., Shaw, C. A., Elledge, S. J., and Westbrook, T. F. 2012. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 335(6066):348-53; Hoellein, A., Fallahi, M., Schoeffmann, S., Steidle, S., Schaub, F. X., Rudelius, M., Laitinen, I., Nilsson, L., Goga, A., Peschel, C, Nilsson, J. A., Cleveland, J. L., and Keller, U. 2014. Myc-induced SUMOylation is a therapeutic vulnerability for B-cell lymphoma. Blood. 124( 13):2081 -90). Since SUMO-conjugation regulates essential cellular functions that contribute to the growth and survival of tumor cells, targeting SAE could represent an approach to treat proliferative disorders such as cancer.
[0006] SAE inhibitors may also be applicable for the treatment of other diseases and conditions outside of oncology. For example, SUMO modifies proteins that play important roles in neurodegenerative diseases (Steffan, J. S., Agrawal, N., Pallos, J., Rockabrand, E., Trotman, L. C, Slepko, N., Hies, K., Lukacsovich, T., Zhu, Y. Z., Cattaneo, E., Pandolfi, P. P., Thompson, L. M., Marsh, J. L. 2004. SUMO modification of Huntington and Huntington’s disease pathology. Science. 304(5667): 100-4); Dorval, V., and Fraser, P. E. 2006. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J Biol Chem. 281 ( 15):9919-24; Ballatore, C, Lee, V. M., and Trojanowski, J. Q. 2007. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 8(9):663-72). Sumoylation also has been reported to play important role in pathogenic viral infection, inflammation and cardiac function (Lee, H. R., Kim, D. J., Lee, J. M., Choi, C. Y., Ahn, B. Y., Hayward, G. S., and Ahn, J. H. 2004. Ability of the human cytomegalovirus ΓΕ1 protein to modulate sumoylation of PML correlates with its functional activities in transcriptional regulation and infectivity in cultured fibroblast cells. / Virol. 78(12):6527-42; Liu, B., and Shuai, K. 2009. Summon SUMO to wrestle with inflammation. Mol Cell. 35(6):731-2; Wang, J., and Schwartz, R. J. 2010. Sumoylation and regulation of cardiac gene expression. Circ Rei. l07( l): 19-29). [0007] It would be beneficial therefore to provide new SAE inhibitors that possess good therapeutic properties, especially for the treatment of proliferative, inflammatory, cardiovascular and neurodegenerative disorders.
PATENT
WO 2016004136

https://patents.google.com/patent/WO2016004136A1/en
Example 133: [(lR,2S,4R)-4-[[5-[4-[(lR)-7-Chloro-l,2,3,4-tetrahydroisoquinolin-l-yl]-5-methyl- thiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxy-cyclopentyl]methyl sulfamate I-263a

Step 1: 7-Chloro-l-[5-(l,3-dioxolan-2-yl)-2-methyl-3-thienyl]-l,2,3,4-tetrahydroisoquinoline
[00714] An oven-dried 2-neck 250 mL round bottom flask under nitrogen was charged with THF (40 mL) and cooled to -74 °C . Added 2.50 M ra-BuLi in hexane (6.92 mL, 17.3 mmol). Added a solution of Int-1 (4.00 g, 16.0 mmol) in THF (60 mL) slowly keeping the internal temperature less than -70 °C . Stirred with cooling 5 min. A second oven-dried 250 mL round bottom flask under nitrogen was charged with THF (60 mL) and Int-50 (2.04 g, 12.4 mmol) and the resulting solution was cooled to 0 °C . Added boron trifluoride diethyl ether complex ( 1.71 mL, 13.6 mmol) slowly and cooled to -30 °C . The contents of the first flask were transferred via cannula to the second flask. Reaction was quenched with saturated aqueous NaHC03 and warmed to rt. Water was added, and the mixture was extracted three times with EtOAc. Combined organic portions were washed with brine, dried over anhydrous Na2S04, filtered, and concentrated in vacuo. Residue was purified via flash column chromatography eluting with a hexane / EtOAc gradient (0 to 100% EtOAc) to afford the title compound as a white solid ( 1.88g, 45%). Ή NMR (400 MHz, Chloroform-d) δ 7.17 – 7.01 (m, 2H), 6.83 – 6.61 (m, 2H), 5.92 (s, 1H), 5.09 (s, 1H), 4.17 – 4.04 (m, 2H), 4.03 – 3.92 (m, 2H), 3.37 – 3.25 (m, 1H), 3.13 – 2.91 (m, 2H), 2.82 – 2.69 (m, 1H), 2.46 (s, 3H). LCMS: (AA) M+l 336.1
Step 2: ieri-Butyl 7-chIoro-l-[5-(l,3-dioxolan-2-yl)-2-methyl-3-thienyl]-3,4-dihydroisoquinoIine -2(lH)-carboxyIate [00715] A 50 mL round bottom flask under nitrogen was charged with 7-chloro-l -[5-(l ,3-dioxolan-2- yl)-2-methyl-3-thienyl]- l ,2,3,4-tetrahydroisoquinoline (5.67 g, 16.9 mmol) and DCM ( 100 mL), to which was added triethylamine (4.71 mL, 33.8 mmol), di-ieri-butyldicarbonate (4.61 g, 21.1 mmol), and N,N-dimethylaminopyridine (23 mg, 0.18 mmol). Reaction was stirred for 1 h at rt and then poured into saturated NaHC03 solution. Mixture was extracted three times with DCM, and the combined organic portions were washed with brine, dried over Na2S04, filtered, and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford 6.96g (95%) of the title compound. LCMS: (AA) M+ l 436.1
Step 3: tert-Butyl 7-chloro-l-(5-formyl-2-methyl-3-thienyl)-3,4-dihydroisoquinoline -2(1H)- carboxylate
[00716] A 1 L round bottom flask was charged with ferf-butyl 7-chloro-
1 -[5-( 1 ,3-dioxolan-2-yl)-2-methyl-3-thienyl]-3 ,4-dihydroisoquinoline-2( 1 H)-carboxylate (7.30 g, 16.7 mmol), methanol (200 mL), and water (20 mL), to which was added a solution of 12M HC1 (4.00 mL, 130 mmol) in methanol (200 mL), and the reaction was stirred at rt for 1 h. Reaction was quenched via addition of 50mL of saturated NaHC03 and stirred for 5 min. Methanol was removed in vacuo, and the resulting aqueous mixture was extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford the title compound (4.55g, 70%). Ή NMR (400 MHz, Chloroform-d) δ 9.67 (s, 1 H), 7.27 – 7.15 (m, 2H), 7.12 (s, 1 H), 6.98 – 6.94 (m, 1 H), 6.34 (m, l H), 4.15 (s, 1 H), 3.18 – 3.06 (m, 1 H), 3.05 – 2.93 (m, 1H), 2.82 – 2.73 (m, 1 H), 2.69 (s, 3H), 1.50 (s, 9H). LCMS: (AA) M+Na 414.2
Step 4: tert-Butyl 7-chIoro-l-{5-[(4-chloropyrimidin-5-yl)(hydroxy)methyI]-2-methyl-3-thienyl}- 3,4-dihydroisoquinoline-2(lH)-carboxylate
[00717] An oven-dried 500 mL 3-neck round bottom flask under nitrogen was charged with 4-chloro- 5-iodopyrimidine (4.08 g, 17.0 mmol) and 2-methyltetrahydrofuran ( 150 mL). An addition funnel containing a solution of rert-butyl 7-chloro- l -(5-formyl-2-methyl-3-thienyl)-3,4- dihydroisoquinoline-2(l H)-carboxylate (4.75 g, 12.1 mmol) in 2-methyltetrahydrofuran (50 mL) was attached, and the contents of the reaction flask were cooled to -75 °C . 2.50 M n-BuLi in hexane ( 14.1 mL, 35.2 mmol) was added in small portions keeping the internal temperature less than -70 °C , at which point the contents of addtion funnel were added in a single portion. Upon completion of addition, the reaction was quenched by adding 20 mL of saturated NaHC03 in small portions and warmed to rt. The aqueous mixture was extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford the title compound (4.85g, 79%). LCMS: (AA) M+Na 528.1
Step 5: tert-Butyl 7-chloro-l-{5-[(4-chloropyrimidin-5-yl)(hydroxy)methyl]-2-methyl-3-thienyl}- 3,4- dihydroisoquinoline-2(lH)-carboxylate
[00718] A 1 L round bottom flask was charged with fe/Y-butyl 7-chloro- l – { 5-[(4-chloropyrimidin-5- yl)(hydroxy)methyl]-2-methyl-3-thienyl}-3,4-dihydroisoquinoline-2(l H)-carboxylate (4.85 g, 9.58 mmol) and DCM (300 mL). Manganese (IV) oxide (14.2 g, 163 mmol) was added and the reaction was stirred at rt for 18 h. Mixture was filtered through Celite, and the filter cake was rinsed with hot EtOAc. Filtrate was concentrated in vacuo to afford the title compound (4.47g , 93%). Ή NMR (400 MHz, Chloroform-d) δ 9.09 (s, 1 H), 8.70 (s, 1 H), 7.24 – 7.16 (m, 1 H), 7.16
– 7.07 (m, 1 H), 7.00 – 6.90 (m, 2H), 6.32 (s, 1 H), 4.28 – 3.97 (m, 1H), 3.14 – 2.89 (m, 2H), 2.78
– 2.65 (m, 4H), 1 .53 – 1.43 (m, 9H).
Step 6: tert-Butyl (lR)-7-chloro-l-[5-[4-[[(lR,3R,4S)-3-(hydroxymethyl)-4-triisopropylsiIyloxy- cyclopentyl]amino]pyrimidine-5-carbonyl]-2-methyl-3-thienyl]-3,4-dihydro-lH-isoquinoline-2- carboxylate
[00719] A 1 L round bottom flask under nitrogen was charged with iert-butyl 7-chloro- l – { 5-[(4- chloropyrimidin-5-yl)carbonyI]-2-methyl-3-thienyl }-3,4-dihydroisoquinoline-2( l H)-carboxylate (4.47 g, 8.86 mmol), DMF (20.0 mL, 258 mmol), Int-259 (3.06 g, 10.6 mmol), and triethylamine (3.09 mL, 22.2 mmol) and the mixture was stirred at rt for 18 h. Reaction mixture was poured into water and saturated NaHC03, and then extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a 70/30 to 60/40 hexane/EtOAc gradient to afford 0.56g of first-eluting diastereomer 1 (not pictured), 4.3 l g of a mixture of diastereomers, and 1.1 lg ( 17%) of second-eluting diastereomer 2 (the title compound). The mixture of diastereomers thus obtained was resubjected to the described chromatography conditions two additional times to afford a total of 2.62 g of the desired diastereomer. Ή NMR (400 MHz, Methanol-d4) δ 8.54 – 8.46 (m, 2H), 7.27 – 7.19 (m, 2H), 7.09 – 6.99 (m, 2H), 6.37 (s, 1H), 4.87 – 4.75 (m, 1H), 4.38 – 4.29 (m, 1H), 4.20 – 4.09 (m, 1H), 3.66 – 3.52 (m, 2H), 3.28- 3.14 (m, 2H), 3.02 – 2.89 (m, 1 H), 2.89 – 2.78 (m, 1 H), 2.68 (s, 3H), 2.54 – 2.41 (m, 1 H), 2.22 – 2.09 (m, 2H), 1.86 – 1.73 (m, 1H), 1.50 (s, 8H), 1.39 – 1.23 (m, 2H), 1.15 – 1.04 (m, 20H).
LCMS: (AA) M+ 1 755.3
Step 7: tert-Butyl (lR)-7-chloro-l-[2-methyl-5-[4-[[(lR,3R,4S)-3-(sulfamoyloxymethyl)-4- triisopropylsilyloxy-cyclopentyl]amino]pyrimidine-5-carbonyl]-3-thienyl]-3,4-dihydro-lH- isoquinoline-2-carboxylate [00720] A solution of ie/t-butyl (lR)-7-chloro-l-[5-[4-[[( lR,3R,4S)-3-(hydroxymethyl)-4- triisopropylsilyloxy-cyclopentyl]amino]pyrimidine-5-carbonyl]-2-methyl-3-thienyl]-3,4-dih lH-isoquinoline-2-carboxylate (2.46 g, 3.26 mmol) in 2-methyltetrahydrofuran (25 mL), and DMF (25 mL) was cooled to 0 °C. Triethylamine ( 1.82 mL, 13.0 mmol) and chlorosulfonamide (1.50 g, 13.0 mmol) were added and the reaction was stirred for 10 min. Added methanol (0.53 mL, 13.0 mmol) and stirred for 15 min. Reaction mixture was poured into saturated NaHC03, extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford the title compound (2.41g, 89%). Ή NMR (400 MHz, Methanol-d4) δ 8.58 – 8.45 (m, 2H), 7.29 – 7.17 (m, 2H), 7.1 1 – 6.98 (m, 2H), 6.36 (s, 1 H), 4.84 – 4.73 (m, 1H), 4.44 – 4.33 (m, 1H), 4.21 – 4.08 (m, 4H), 3.27- 3.17 (m, 1 H),3.02 – 2.89 (m, 1 H), 2.88 – 2.78 (m, 1 H), 2.67 (s, 3H), 2.57 – 2.47 (m, 1 H), 2.41 – 2.30 (m, 1 H), 2.23 – 2.13 (m, 1 H), 1.87- 1.78 (m, 1 H), 1.50 (s, 9H), 1.43 – 1 .33 (m, 1 H), 1 .17 – 1.04 (m, 20H). LCMS: (AA) M+l 834.3
Step 8: [(lR,2S,4R)-4-[[5-[4-[(lR)-7-Chloro-l,2,3,4-tetrahydroisoquinolin-l-yl]-5-methyl- thiophene-2-carbonyl]pyrimidin-4-yI]aniino]-2-hydroxy-cyclopentyl]methyl sulfamate
[00721] A solution of f«?r/-butyl ( l R)-7-chloro- l -[2-methyl-5-[4-[[( l R,3R,4S)-3-
(sulfamoyloxymethyl)-4-triisopropylsilyloxy-cyclopentyl]amino]pyrimidine-5-carbonyl]-3- thienyl]-3,4-dihydro- l H-isoquinoline-2-carboxylate (2.41 g, 2.89 mmol) in CH3CN ( 10 mL) was cooled in an ice bath to + 1 °C . Phosphoric acid ( 10 mL, 200 mmol) was added dropwise and the reaction was stirred with ice bath cooling for 60 min. The mixture was warmed to rt and stirred for an additional 3 h. Reaction was poured into a stirring mixture of 50 mL water and 50 mL EtOAc, and the the pH was adjusted to ~9 by slowly adding 200 mL of saturated NaHC03 with stirring. Resulting aqueous mixture was extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a gradient that began with 100% DCM and increased in polarity to 80% DCM / 20% methanol / 2% ammonium hydroxide gradient to afford the title compound (1.50 g, 90%). Ή NMR (400 MHz, Methanol-d4) δ 8.61 (s, 1H), 8.52 (s, 1 H), 7.27 (s, 1 H), 7.18 – 7.13 (m, 2H), 6.73 – 6.68 (m, 1 H), 5.23 (s, 1H), 4.81 – 4.70 (m, 1 H), 4.26 – 4.10 (m, 3H), 3.29 – 3.23 (m, 2H), 3.1 1 – 2.96 (m, 2H), 2.87 – 2.76 (m, 1H), 2.60 (s, 3H), 2.55 – 2.42 (m, 1 H), 2.33 – 2.19 (m, 1H), 2.18 – 2.07 (m, 1H), 1.95 – 1.81 (m, 1H), 1.47 – 1.35 (m, 1 H). LCMS: (AA) M+l 580.0
CLIP

Credit: Tien Nguyen/C&EN
Presenter: Steven Paul Langston, associate director at Takeda Pharmaceuticals International
Target: Sumo activating enzyme
Disease: Solid tumors
Reporter’s notes: Langston gave the last talk of the morning session, placing him in the “precarious position of being between you and lunch,” he said. Takeda acquired this drug development program, falling under the umbrella of immuno-oncology, along with Millenium Pharmaceuticals in 2008. The team targeted a pathway known as SUMOylation, a protein post translation modification that is implicated in a number of cellular processes including immune response. In SUMOylation, enzymes attach a small protein to another protein. They found that inhibiting this pathway activates a type I interferon response in immune cells. How the molecule, TAK-981, inhibits this pathway is quite complicated, Langston said. TAK-981 forms an adduct with a small ubiquitin like modifier (SUMO) to inhibit a SUMO activating enzyme that catalyzes SUMOylation. While the synthesis of TAK-981 is fairly short, it requires a nonideal chiral chromatography separation after the first step. TAK-981 is in Phase I clinical trials as an intravenous infusion for patients with metastatic solid tumors or lymphomas.
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2018311239 | HETEROARYL COMPOUNDS USEFUL AS INHIBITORS OF SUMO ACTIVATING ENZYME | 2018-03-16 | |
| US9962386 | HETEROARYL COMPOUNDS USEFUL AS INHIBITORS OF SUMO ACTIVATING ENZYME | 2017-04-17 | |
| US9683003 | HETEROARYL COMPOUNDS USEFUL AS INHIBITORS OF SUMO ACTIVATING ENZYME | 2015-06-30 | 2016-01-14 |
//////////TAK-981, TAK 981, Phase I, Lymphoma, Solid tumours, TAKEDA,
Cc3sc(cc3[C@@H]1NCCc2ccc(Cl)cc12)C(=O)c5cncnc5N[C@@H]4C[C@H](COS(N)(=O)=O)[C@@H](O)C4
BIIB-095

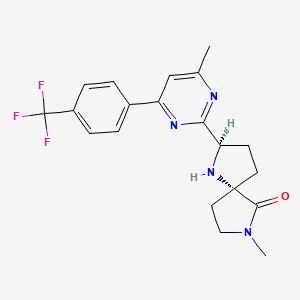
BIIB-095
ROTATION (+)
1493790-64-9 CAS free form,
1493772-48-7 cas Hcl salt
cas 1493790-65-0, 1496563-32-6 ,SULPHATE ???
cas 1496563-31-5 SULFATE 1;1
cas 1496563-32-6 SULFATE HYDRATE 1;1;1
(2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one
1,7-Diazaspiro[4.4]nonan-6-one, 7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)phenyl]-2-pyrimidinyl]-, (2R,5S)-
C20 H21 F3 N4 O, 390.40
- Originator Biogen
- Class Analgesics
- Mechanism of Action Nav1.7 voltage-gated sodium channel inhibitors
- Phase I Neuropathic pain
- 29 Mar 2018 Phase-I clinical trials in Neuropathic pain (In volunteers) in United Kingdom (PO) (NCT03454126)
- 05 Mar 2018 Biogen plans a phase I trial for Pain, including Neuropathic pain (In volunteers) in USA (PO) (NCT03454126)
- 05 Mar 2018 Preclinical trials in Neuropathic Pain in USA (PO), before March 2018
In March 2018, a randomized, double blind, placebo controlled, single and multiple-ascending dose, dose-escalation phase I study ( NCT03454126; 255HV101; 2017-003982-90) was initiated in the UK in healthy subjects (expected n = 80) to evaluate the safety, tolerability and pharmacokinetics of BIIB-095. At that time, the trial was expected to complete in December 2018
Biogen is developing BIIB-095, a voltage-gated sodium channel 1.7 inhibitor, for the potential oral treatment of neuropathic pain [2027279], [2027426]. In March 2018, a phase I trial was initiated in healthy subjects
Biogen is developing oral agent BIIB-095 for the treatment of chronic pain, including neuropathic pain. A phase I clinical trial is under way in healthy volunteers.
The compound was first claimed in WO2013175205 , for treating schizophrenia, assigned to subsidiary Convergence Pharmaceuticals Limited , naming some of the inventors. This might present the structure of BIIB-095 , a voltage-gated sodium channel 1.7 inhibitor, being developed by Biogen for the oral treatment of neuropathic pain; in March 2018, a phase I trial was initiated in healthy subjects.
PATENT
WO2013175205

CONTD………………

INTERMEDIATE

WO 2013175206
US 20150119404
https://patents.google.com/patent/US20150119404
Patent
WO-2019067961
Novel salts (citrate, mesylate, hydrosulfate, saccharinate and oxalate) forms of 7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1,7-diazaspiro[4.4]nonan-6-one, processes for their preparation and compositions comprising them are claimed. Also claimed are their use for treating diseases and conditions mediated by modulation of voltage-gated sodium channels.
Voltage-gated sodium channels are responsible for the initial phase of the action potential, which is a wave of electrical depolarisation usually initiated at the soma of the neuron and propagated along the axon to the terminals. At the terminals, the action potential triggers the influx of calcium and the release of neurotransmitter. Drugs, such as lidocaine, that block voltage-gated sodium channels are used as local anaesthetics. Other sodium channel blockers, such as lamotrigine and carbamazepine are used to treat epilepsy. In the latter case, partial inhibition of voltage-gated sodium channels reduces neuronal excitability and reduces seizure propagation. In the case of local anaesthetics, regional block of sodium channels on sensory neurons prevents the conduction of painful stimuli. A key feature of these drugs is their state-dependent mechanism of action. The drugs are thought to stabilise an inactivated conformation of the channel that is adopted rapidly after the channel opens. This inactivated state provides a refractory period before the channel returns to its resting (closed) state ready to be reactivated. As a result, state-dependent sodium channel blockers inhibit the firing of neurons at high frequency, for example in response to painful stimuli, and will help to prevent repetitive firing during periods of prolonged neuronal depolarisation that might occur, for example, during a seizure. Action potentials triggered at lower frequencies, for example in the heart, will not be significantly affected by these drugs, although the safety margin differs in each case, since at high enough concentrations each of these drugs is capable of blocking the resting or open states of the channels.
The voltage-gated sodium channel family is made up of 9 subtypes, four of which are found in the brain, NaV1.1 , 1.2, 1.3 and 1.6. Of the other subtypes, NaV1.4 is found only in skeletal muscle, NaV1.5 is specific to cardiac muscle, and NaV1.7, 1.8, and 1.9 are found
predominantly in sensory neurons. The hypothesised binding site for state-dependent sodium channel blockers is the local anaesthetic (LA) binding site in the inner vestibule of the pore on transmembrane S6 of domain IV. Critical residues are located in a highly conserved region among the different subtypes, thus presenting a challenge for the design of new subtype selective drugs. Drugs such as lidocaine, lamotrigine and carbamazepine do not distinguish between the subtypes. However, selectivity can be achieved, and can be further enhanced functionally, as a result of the different frequencies at which the channels operate.
Drugs that block voltage-gated sodium channels in a state-dependent manner are also used in the treatment of bipolar disorder, either to reduce symptoms of mania or depression, or as mood stabilisers to prevent the emergence of mood episodes. Clinical and preclinical evidence also suggests that state-dependent sodium channel blockers may help to reduce the symptoms of schizophrenia. For example, lamotrigine has been shown to reduce symptoms of psychosis induced by ketamine in healthy human volunteers, and furthermore, studies in patients suggest that the drug can augment the antipsychotic efficacy of some atypical antipsychotic drugs, such as clozapine or olanzapine. It is hypothesised that efficacy in these psychiatric disorders may result in part from a reduction of excessive glutamate release. The reduction in glutamate release is thought to be a consequence of sodium channel inhibition in key brain areas, such as the frontal cortex. However, interaction with voltage-gated calcium channels may also contribute to the efficacy of these drugs.
WO 2013/175205 (Convergence Pharmaceuticals Limited) describes (2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one hydrochloride, sulfuric acid salt and sulfuric acid salt hydrate which are claimed to be modulators of voltage-gated sodium channels. The object of the invention is to identify alternative salts of said compound which have advantageous properties.
Example 1
(2R,5S)-7-Methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1,7-diazaspiro[4.4]nonan-6-
To a solution of (2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one (which may be prepared in accordance with the procedure described in Example 1 of WO 2013/175205) (4.45g, 0.0114 mol) dissolved in absolute ethanol (66.82 ml, 15 vol) at 45 °C was added a solution of citric acid in ethanol (1 M, 1.05 equiv. 12 ml) over a period of 2-3 minutes. The solution was aged at 45 °C for a period of 1 hour. After 30 minutes a seed of citrate salt (0.1 wt%) was added and the mixture allowed to cool over approximately 2 hours and mature for 18 hours at ambient temperature (approximately 10-15 °C). Following maturation the salt was noted to be a very thick suspension (white) that required mobilisation with 20 ml additional ethanol and a further maturation period of 2 hours at ambient temperature. Filtration was carried out under vacuum and the vessel and cake rinsed with 15 ml ethanol. The de-liquored cake was dried further in a vacuum oven at 50 °C to provide 6.0 g of crystalline white solid (91 % yield).
H NMR (400MHz, DMSO-D6): δΗ 1.90-2.05 (2H, m), 2.10-2.20 (2H, m,), 2.20-2.30 (1 H, m), -2.50 (1 H, m, partially masked by solvent)), 2.55-2.68 (4H, m), 2.56 (3H, s), 2.79 (3H, s),
3.28-3.40 (2H, m), 4.79 (1 H, t, J= 8.0 Hz), 7.92 (2H, d, J = 8.4 Hz), 8.03 (1 H, s), 8.45 (2H, d, J= 8.8Hz) ppm, (exchangeables not reported)
Characterisation of Example 1
The XRPD of Example 1 is presented in FIG. 1 and the DSC/TGA of Example 1 is presented in FIG. 2. The citrate salt of Example 1 displayed the following characteristics:
1 endotherm onset: 171.82°C
peak maximum: 174.55°C
There was an endotherm post the main endotherm.
There was no weight reduction until ca 168°C had been reached. The weight reduction commenced with the start of the main endotherm and coincided with the endotherm post the main endotherm which indicated that this thermal event was the onset of compound decomposition and loss of citric acid. Thermal events >220°C were due to compound decomposition.
The XPRD data in FIG. 1 demonstrated that under different extremes of humidity indicate a stable crystalline form of the citrate salt of Example 1 with no tendency to form hydrates. This is supported by DSC/TGA data in FIG. 2 which show clear transitions and no evidence of solvates.
Aqueous solubility of the citrate salt (Example 1) = 22mg/ml (25°C).
Example 2
(2R,5S)-7-Methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1,7-diazaspiro[4.4]nonan-6-one ) salt (E2)
To a solution of (2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one (which may be prepared in accordance with the procedure described in Example 1 of WO 2013/175205) (4.45g, 0.0114 mol) dissolved in absolute ethanol (66.82 ml, 15 vol) at 45 °C was added a solution of methanesulfonic acid in ethanol (1 M, 1.05 equiv. 12 ml) over a period of 2-3 minutes. The solution was aged at 45 °C for a period of 1 hour. After 10 minutes nucleation and gradual crystallisation was noted to afford a thick mixture. Additional ethanol was added (10 ml) to mobilise the suspension that was then allowed to cool over approximately 2 hours and mature for 18 hours at ambient temperature (approximately 10-15 °C). Following maturation the salt was noted to be a thin, mobile suspension (white) that was filtered under vacuum and the vessel and cake rinsed with 15 ml ethanol. The de-liquored cake was dried further in a vacuum oven at 50 °C to provide 4.0 g of crystalline white solid (72% yield).
H NMR (400MHz, DMSO-D6): δΗ 2.1-2.45 (4H, m), 2.27 (3H, s), 2.50-2.75 (2H, m), 2.61 (3H, s), 2.86 (3H, s), 3.35-3.50 (2H, m), 5.20 (1 H, t, J = 8 Hz), 7.96 (2H, d, J = 8.8 Hz), 8.17 (1 H, s), 8.51 (2H, d, J = 8.4Hz), 9.45 (1 H, br), 10.16 (1 H, br) ppm.
Characterisation of Example 2
The XRPD of Example 2 is presented in FIG. 3 and the DSC/TGA of Example 2 is presented in FIG. 4. The DSC thermograph of the methanesulfonate (mesylate) (Example 2) displayed the following characteristics:
One distinct endotherm onset: 247.34°C
peak maximum: 250.34°C
The TGA thermograph showed no weight reduction until ca 250°C had been reached. The weight reduction commenced with the start of the main endotherm and indicated that this thermal event was the onset of compound decomposition. There is no evidence of entrapped solvents or water.
The XPRD data in FIG. 3 demonstrated that under different extremes of humidity indicate a stable crystalline form of the mesylate salt of Example 2 with no tendency to form hydrates. This is supported by DSC/TGA data in FIG. 4 which show clear transitions and no evidence of solvates.
Aqueous solubility of the mesylate salt (Example 2) = 65mg/ml (25°C).
Example 3
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one hydrosulfate single crystals: 25.0 mg of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluorome
one hydrosulfate was added to 4 mL vial. 1.000 mL of anhydrous EtOH was added, and the sample was filtered. Anhydrous hexanes were added dropwise until the solution neared the precipitation point. The vial was sealed and left undisturbed for 24 hr, after which time a crop of single crystals was evident. The sample was sent for single crystal analysis and confirmed as the anhydrous (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one hydrosulfate form (FIGs. 5A-5B).
Example 4
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one freebase: 8.00 g of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one hydrosulfate (JM Lot R-2017-4323 D 301) was added to a 1 L Erlenmeyer flask and suspended and stirred vigorously in 400 mL of THF. 20% K2C03 (250 mL) was added and dissolved. The mixture was transferred to 1 L sep. funnel. 100 mL EtOAc was added and the aqueous and organic layers were separated. The aqueous layer was re-extracted with 50 mL of EtOAc and the combined organics were back-extracted with brine (100 mL) and water (100 mL). Due to fairly poor separation, a significant quantity of MgSCU was required to dry the solution. The solution was reduced via Rotavap (45 °C) to -50 mL, transferred to a 100 mL RB flask, reduced down to -10 mL, transferred to 20 mL scintillation vial and continued to be reduced to a thick oil. The oil was left on the Rotavap for another hour and a “wet” solid was obtained. Loosened solids on the bottom of the vial were left on the Rotavap for 1 hr with no heat applied to obtain a chunky solid. The contents was transferred to a mortar and pestle, ground to powder and fine granules, placed back in a 20 mL scintillation vial and left on a Rotavap overnight to obtain a dry solid (5.1 g). The XRPD pattern of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one freebase is shown in FIG. 6.
Example 5
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one saccharinate: 199.7 mg of (2R,5S)-7-Methyl-2-(4-
methyl-6-(4-(trifluoromethyl)phenyl)pyrirnidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one free base (0.5115 mmol) was dissolved in 4.2 mL of 2-Me-THF. 98.1 mg of saccharin (0.5106 mmol) was dissolved in 4.2 mL of 2-Me-THF. Saccharin was added to the freebase, and after 15 seconds the mixture began to precipitate and solidify. 10 mL of 2-Me-THF was added and stirred at max rpm as to provide a thick white suspension in 10 min. The suspension was filtered, air dried under vacuum for 10 min on frit, then dried under a stream of nitrogen for 30 min resulting in 215 mg of white solid product. The XRPD pattern for (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one saccharinate is shown in FIG. 7.
Example 6
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one oxalate: 403 mg of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one freebase was dissolved in 4.03 mL EtOH. 1.000 mL of this solution was added to a 4 mL vial. 23.8 mg of oxalic acid was dissolved in 1.000 mL of EtOH and added dropwise to the stirring (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one freebase solution. After 5 min, a white precipitate was evident and 2.000 mL of EtOH was added to the slurry to aid stirring. The resulting suspension was stirred overnight. The following day the suspension was filtered and dried on a frit under vacuum for 10 min yielding 106 mg of white solid. The XRPD pattern for (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one oxalate is shown in FIG. 8.
Example 7
The single crystal structural information and refinement parameters for (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one hydrosulfate are shown in Table 1.
Table 1.
Largest peak, hole / e A-3 0.363, -0.264
The most prominent XRPD diffraction peaks were (2Θ): 7.8±0.2°, 8.1±0.2°, 12.6±0.2°, 14.3±0.2°, 16.5±0.2°, 18.5±0.2°, 19.6±0.2°, 24.8±0.2° and 25.3±0.2°.
PATENTS
US2018360833NOVEL PYRIMIDINYL-DIAZOSPIRO COMPOUNDS2018-06-27
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2017304303 | Novel Pyrimidinyl-DiazoSpiro Compounds | 2017-07-11 | |
| US9737536 | Novel Pyrimidinyl-DiazoSpiro Compounds | 2016-05-25 | 2016-09-15 |
| US2016184306 | Novel Pyrimidinyl-DiazoSpiro Compounds | 2016-02-15 | 2016-06-30 |
| US9309254 | NOVEL COMPOUNDS | 2013-05-22 | 2015-04-30 |
| US9376445 | NOVEL COMPOUNDS | 2013-05-22 | 2015-06-18 |
////////////////BIIB-095, BIIB095, BIIB 095, PHASE 1
CC1=NC(=NC(=C1)C2=CC=C(C=C2)C(F)(F)F)C3CCC4(N3)CCN(C4=O)C
