
Home » ANTIBODIES
Category Archives: ANTIBODIES
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Imdevimab
| (Heavy chain) QVQLVESGGG VVQPGRSLRL SCAASGFTFS NYAMYWVRQA PGKGLEWVAV ISYDGSNKYY ADSVKGRFTI SRDNSKNTLY LQMNSLRTED TAVYYCASGS DYGDYLLVYW GQGTLVTVSS ASTKGPSVFP LAPSSKSTSG GTAALGCLVK DYFPEPVTVS WNSGALTSGV HTFPAVLQSS GLYSLSSVVT VPSSSLGTQT YICNVNHKPS NTKVDKKVEP KSCDKTHTCP PCPAPELLGG PSVFLFPPKP KDTLMISRTP EVTCVVVDVS HEDPEVKFNW YVDGVEVHNA KTKPREEQYN STYRVVSVLT VLHQDWLNGK EYKCKVSNKA LPAPIEKTIS KAKGQPREPQ VYTLPPSRDE LTKNQVSLTC LVKGFYPSDI AVEWESNGQP ENNYKTTPPV LDSDGSFFLY SKLTVDKSRW QQGNVFSCSV MHEALHNHYT QKSLSLSPGK (Light chain) QSALTQPASV SGSPGQSITI SCTGTSSDVG GYNYVSWYQQ HPGKAPKLMI YDVSKRPSGV SNRFSGSKSG NTASLTISGL QSEDEADYYC NSLTSISTWV FGGGTKLTVL GQPKAAPSVT LFPPSSEELQ ANKATLVCLI SDFYPGAVTV AWKADSSPVK AGVETTTPSK QSNNKYAASS YLSLTPEQWK SHRSYSCQVT HEGSTVEKTV APTECS (Disulfide bridge: H22-H96, H147-H203, H223-L215, H229-H’229, H264-H324-H370-H428, H’22-H’96, H’147-H’203, H’223-L’215, H’264-H’324, H’370-H’428, L22-L90, L138-L197, L’22-L’90, L’138-L’197) |
イムデビマブ;
- Immunoglobulin G1, anti-(severe acute respiratory syndrome coronavirus 2 spike glycoprotein) (human monoclonal REGN10987 γ1-chain), disulfide with human monoclonal REGN10987 λ-chain, dimer
| Formula | C6396H9882N1694O2018S42 |
|---|---|
| CAS | 2415933-40-1 |
| Mol weight | 144141.7693 |
Monoclonal antibody
Treatment and prophylaxis of SARS-CoV-2 infection
ANTIVIRAL
SARS-CoV-2 spike glycoprotein
- REGN 10987
- RG 6412
Fact Sheet – US Food and Drug Administration
https://www.fda.gov › media › download
PDFBenefit of treatment with casirivimab and imdevimab has not been observed in patients hospitalized due to COVID-19. Monoclonal antibodies, such as casirivimab.
Casirivimab/imdevimab, sold under the brand name REGEN-COV,[1] is an experimental medicine developed by the American biotechnology company Regeneron Pharmaceuticals. It is an artificial “antibody cocktail” designed to produce resistance against the SARS-CoV-2 coronavirus responsible for the COVID-19 pandemic.[3][4] It consists of two monoclonal antibodies, casirivimab (REGN10933) and imdevimab (REGN10987) that must be mixed together.[1][5][6] The combination of two antibodies is intended to prevent mutational escape.[7]
Trials
In a clinical trial of people with COVID-19, casirivimab and imdevimab, administered together, were shown to reduce COVID-19-related hospitalization or emergency room visits in people at high risk for disease progression within 28 days after treatment when compared to placebo.[2] The safety and effectiveness of this investigational therapy for use in the treatment of COVID-19 continues to be evaluated.[2]
The data supporting the emergency use authorization (EUA) for casirivimab and imdevimab are based on a randomized, double-blind, placebo-controlled clinical trial in 799 non-hospitalized adults with mild to moderate COVID-19 symptoms.[2] Of these participants, 266 received a single intravenous infusion of 2,400 milligrams casirivimab and imdevimab (1,200 mg of each), 267 received 8,000 mg casirivimab and imdevimab (4,000 mg of each), and 266 received a placebo, within three days of obtaining a positive SARS-CoV-2 viral test.[2]
The prespecified primary endpoint for the trial was time-weighted average change in viral load from baseline.[2] Viral load reduction in participants treated with casirivimab and imdevimab was larger than in participants treated with placebo at day seven.[2] However, the most important evidence that casirivimab and imdevimab administered together may be effective came from the predefined secondary endpoint of medically attended visits related to COVID-19, particularly hospitalizations and emergency room visits within 28 days after treatment.[2] For participants at high risk for disease progression, hospitalizations and emergency room visits occurred in 3% of casirivimab and imdevimab-treated participants on average compared to 9% in placebo-treated participants.[2] The effects on viral load, reduction in hospitalizations and ER visits were similar in participants receiving either of the two casirivimab and imdevimab doses.[2]
As of September 2020, REGEN-COV is being evaluated as part of the RECOVERY Trial.[8]
On 12 April 2021, Roche and Regeneron announced that the Phase III clinical trial REGN-COV 2069 met both primary and secondary endpoints, reducing risk of infection by 81% for the non-infected patients, and reducing time-to-resolution of symptoms for symptomatic patients to one week vs. three weeks in the placebo group.[9]
Authorization
On 21 November 2020, the U.S. Food and Drug Administration (FDA) issued an emergency use authorization (EUA) for casirivimab and imdevimab to be administered together for the treatment of mild to moderate COVID-19 in people twelve years of age or older weighing at least 40 kilograms (88 lb) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19.[2][10][11] This includes those who are 65 years of age or older or who have certain chronic medical conditions.[2] Casirivimab and imdevimab must be administered together by intravenous (IV) infusion.[2]
Casirivimab and imdevimab are not authorized for people who are hospitalized due to COVID-19 or require oxygen therapy due to COVID-19.[2] A benefit of casirivimab and imdevimab treatment has not been shown in people hospitalized due to COVID-19.[2] Monoclonal antibodies, such as casirivimab and imdevimab, may be associated with worse clinical outcomes when administered to hospitalized people with COVID-19 requiring high flow oxygen or mechanical ventilation.[2]
The EUA was issued to Regeneron Pharmaceuticals Inc.[2][10][12]
On 1 February 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) started a rolling review of data on the REGN‑COV2 antibody combination (casirivimab/imdevimab), which is being co-developed by Regeneron Pharmaceuticals, Inc. and F. Hoffman-La Roche, Ltd (Roche) for the treatment and prevention of COVID‑19.[13][14] In February 2021, the CHMP concluded that the combination, also known as REGN-COV2, can be used for the treatment of confirmed COVID-19 in people who do not require supplemental oxygen and who are at high risk of progressing to severe COVID-19.[15]
The Central Drugs Standards Control Organisation (CDSCO) in India, on 5 May 2021, granted an Emergency Use Authorisation to Roche (Genentech)[16] and Regeneron[17] for use of the casirivimab/imdevimab cocktail in the country. The announcement came in light of the second wave of the COVID-19 pandemic in India. Roche India maintains partnership with Cipla, thereby permitting the latter to market the drug in the country.[18]
Deployment
Although Regeneron is headquartered in Tarrytown, New York (near New York City), REGEN-COV is manufactured at the company’s primary U.S. manufacturing facility in Rensselaer, New York (near the state capital at Albany).[19] In September 2020, to free up manufacturing capacity for REGEN-COV, Regeneron began to shift production of its existing products from Rensselaer to the Irish city of Limerick.[20]
Regeneron has a deal in place with Roche (Genentech)[21]to manufacture and market REGEN-COV outside the United States.[10][22]
On 2 October 2020, Regeneron Pharmaceuticals announced that US President Donald Trump had received “a single 8 gram dose of REGN-COV2” after testing positive for SARS-CoV-2.[23][24] The drug was provided by the company in response to a “compassionate use” (temporary authorization for use) request from the president’s physicians.[23]
References
- ^ Jump up to:a b c “REGEN-COV- casirivimab and imdevimab kit”. DailyMed. Retrieved 18 March 2021.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “Coronavirus (COVID-19) Update: FDA Authorizes Monoclonal Antibodies for Treatment of COVID-19”. U.S. Food and Drug Administration (FDA) (Press release). 21 November 2020. Retrieved 21 November 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Kelland K (14 September 2020). “Regeneron’s antibody drug added to UK Recovery trial of COVID treatments”. Reuters. Retrieved 14 September 2020.
- ^ “Regeneron’s COVID-19 Response Efforts”. Regeneron Pharmaceuticals. Retrieved 14 September 2020.
- ^ Morelle R (14 September 2020). “Antibody treatment to be given to Covid patients”. BBC News Online. Retrieved 14 September2020.
- ^ “Safety, Tolerability, and Efficacy of Anti-Spike (S) SARS-CoV-2 Monoclonal Antibodies for Hospitalized Adult Patients With COVID-19”. ClinicalTrials. 3 September 2020. Retrieved 14 September2020.
- ^ Baum A, Fulton BO, Wloga E, Copin R, Pascal KE, Russo V, et al. (August 2020). “Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies”. Science. 369 (6506): 1014–1018. Bibcode:2020Sci…369.1014B. doi:10.1126/science.abd0831. PMC 7299283. PMID 32540904.
- ^ “RECOVERY COVID-19 phase 3 trial to evaluate Regeneron’s REGN-COV2 investigational antibody cocktail in the UK”. Recovery Trial. Retrieved 14 September 2020.
- ^ “Phase III prevention trial showed subcutaneous administration of investigational antibody cocktail casirivimab and imdevimab reduced risk of symptomatic COVID-19 infections by 81%”. streetinsider.com. Archived from the original on 2021-04-12. Retrieved 2021-04-12.
- ^ Jump up to:a b c “Regeneron Reports Positive Interim Data with REGEN-COV Antibody Cocktail used as Passive Vaccine to Prevent COVID-19”(Press release). Regeneron Pharmaceuticals. 26 January 2021. Retrieved 19 March 2021 – via PR Newswire.
- ^ “Fact Sheet For Health Care Providers Emergency Use Authorization (EUA) Of Casirivimab And Imdevimab” (PDF). U.S. Food and Drug Administration (FDA).
- ^ “Casirivimab and Imdevimab”. Regeneron Pharmaceuticals. Retrieved 19 March 2021.
- ^ “EMA starts rolling review of REGN‑COV2 antibody combination (casirivimab / imdevimab)” (Press release). European Medicines Agency (EMA). 1 February 2021. Retrieved 1 February 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “EMA reviewing data on monoclonal antibody use for COVID-19” (Press release). European Medicines Agency (EMA). 4 February 2021. Retrieved 4 March 2021.
- ^ “EMA issues advice on use of REGN-COV2 antibody combination (casirivimab / imdevimab)” (Press release). European Medicines Agency (EMA). 26 February 2021. Retrieved 5 March 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^https://www.businesswire.com/news/home/20200818005847/en/Genentech-and-Regeneron-Collaborate-to-Significantly-Increase-Global-Supply-of-REGN-COV2-Investigational-Antibody-Combination-for-COVID-19
- ^ https://timesofindia.indiatimes.com/india/india-approves-roche/regeneron-antibody-cocktail-to-treat-covid-19/articleshow/82407551.cms
- ^ “Roche receives Emergency Use Authorisation in India for its investigational Antibody Cocktail (Casirivimab and Imdevimab) used in the treatment of Covid-19 | Cipla”. http://www.cipla.com. Retrieved 2021-05-06.
- ^ Williams, Stephen (3 October 2020). “Experimental drug given to President made locally”. The Daily Gazette.
- ^ Stanton, Dan (11 September 2020). “Manufacturing shift to Ireland frees up US capacity for Regeneron’s COVID antibodies”. BioProcess International.
- ^https://www.businesswire.com/news/home/20200818005847/en/Genentech-and-Regeneron-Collaborate-to-Significantly-Increase-Global-Supply-of-REGN-COV2-Investigational-Antibody-Combination-for-COVID-19
- ^ “Roche and Regeneron link up on a coronavirus antibody cocktail”. CNBC. 19 August 2020. Retrieved 14 September 2020.
- ^ Jump up to:a b Thomas K (2 October 2020). “President Trump Received Experimental Antibody Treatment”. The New York Times. ISSN 0362-4331. Retrieved 2 October 2020.
- ^ Hackett DW (3 October 2020). “8-Gram Dose of COVID-19 Antibody Cocktail Provided to President Trump”. http://www.precisionvaccinations.com. Archived from the original on 3 October 2020.
External links
- “Casirivimab”. Drug Information Portal. U.S. National Library of Medicine.
- “Imdevimab”. Drug Information Portal. U.S. National Library of Medicine.
- “Casirivimab and Imdevimab EUA Letter of Authorization” (PDF). U.S. Food and Drug Administration (FDA).
- “Frequently Asked Questions on the Emergency Use Authorization of Casirivimab + Imdevimab” (PDF). U.S. Food and Drug Administration (FDA).
| REGN10933 (blue) and REGN10987 (orange) bound to SARS-CoV-2 spike protein (pink). From PDB: 6VSB, 6XDG. | |
| Combination of | |
|---|---|
| Casirivimab | Monoclonal antibody against spike protein of SARS-CoV-2 |
| Imdevimab | Monoclonal antibody against spike protein of SARS-CoV-2 |
| Clinical data | |
| Trade names | REGEN-COV |
| Other names | REGN-COV2 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Casirivimab |
| Routes of administration | Intravenous |
| ATC code | None |
| Legal status | |
| Legal status | US: Unapproved (Emergency Use Authorization)[1][2] |
| Identifiers | |
| DrugBank | DB15691 |
| KEGG | D11938D11939 |
////////Imdevimab, ANTI VIRAL, PEPTIDE, CORONA VIRUS, COVID19, APPROVALS 2020, FDA 2020, イムデビマブ, REGN 10987, RG 6412,
NEW DRUG APPROVALS
one time
$10.00
Sacituzumab govitecan-hziy

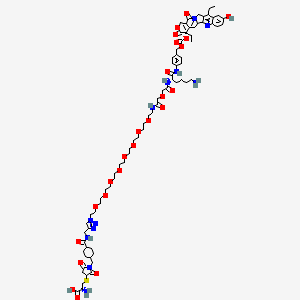

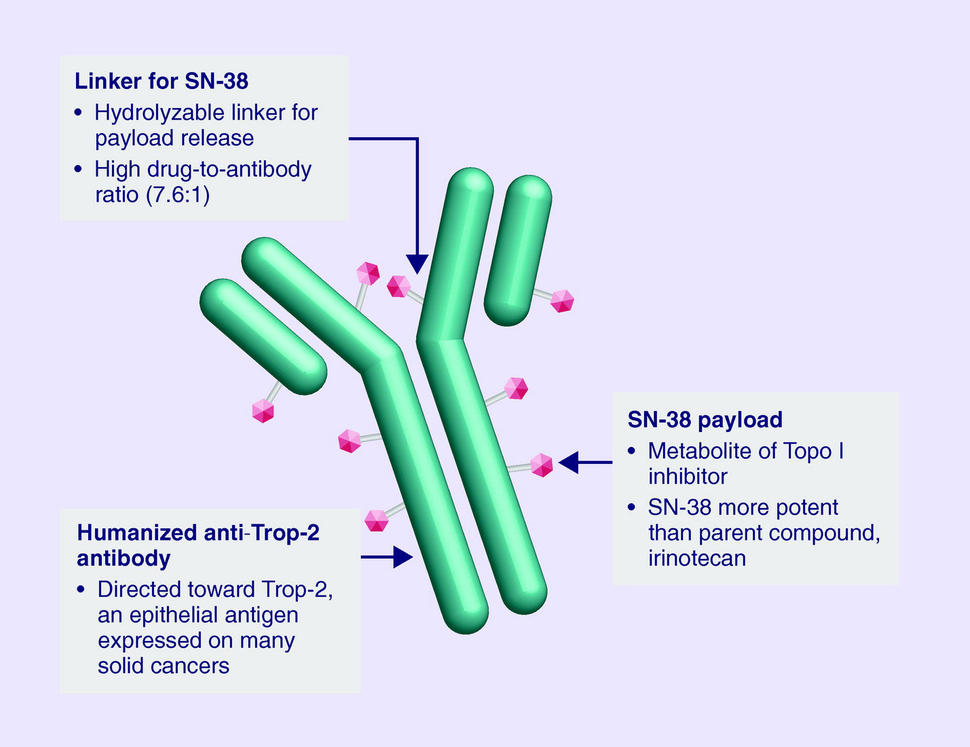
Sacituzumab govitecan-hziy
1601.8 g/mol
(2R)-2-amino-3-[1-[[4-[[1-[2-[2-[2-[2-[2-[2-[2-[2-[2-[[2-[2-[[(2S)-6-amino-1-[4-[[(19S)-10,19-diethyl-7-hydroxy-14,18-dioxo-17-oxa-3,13-diazapentacyclo[11.8.0.02,11.04,9.015,20]henicosa-1(21),2,4(9),5,7,10,15(20)-heptaen-19-yl]oxycarbonyloxymethyl]anilino]-1-oxohexan-2-yl]amino]-2-oxoethoxy]acetyl]amino]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethyl]triazol-4-yl]methylcarbamoyl]cyclohexyl]methyl]-2,5-dioxopyrrolidin-3-yl]sulfanylpropanoic acid
Trodelvy
- hRS 7SN38
- hRS7-SN38
- IMMU 132
- IMMU-132
CAS: 1491917-83-9
UNII-DA64T2C2IO component ULRUOUDIQPERIJ-PQURJYPBSA-N
UNII-SZB83O1W42 component ULRUOUDIQPERIJ-PQURJYPBSA-N
| Efficacy | Antineoplastic, Topoisomerase I inhibitor |
|---|---|
| Disease | Breast cancer (triple negative) |
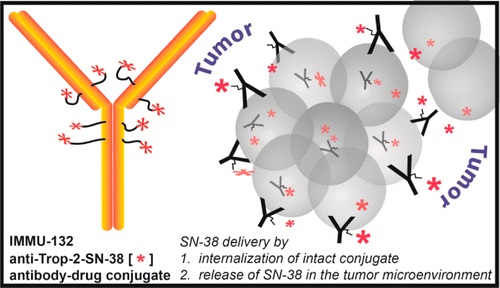

Sacituzumab Govitecan is an antibody drug conjugate containing the humanized monoclonal antibody, hRS7, against tumor-associated calcium signal transducer 2 (TACSTD2 or TROP2) and linked to the active metabolite of irinotecan, 7-ethyl-10-hydroxycamptothecin (SN-38), with potential antineoplastic activity. The antibody moiety of sacituzumab govitecan selectively binds to TROP2. After internalization and proteolytic cleavage, SN-38 selectively stabilizes topoisomerase I-DNA covalent complexes, resulting in DNA breaks that inhibit DNA replication and trigger apoptosis. TROP2, also known as epithelial glycoprotein-1 (EGP-1), is a transmembrane calcium signal transducer that is overexpressed by a variety of human epithelial carcinomas; this antigen is involved in the regulation of cell-cell adhesion and its expression is associated with increased cancer growth, aggressiveness and metastasis.
NEW DRUG APPROVALS
one time
$10.00
CLIP
FDA Approves Trodelvy®, the First Treatment for Metastatic Triple-Negative Breast Cancer Shown to Improve Progression-Free Survival and Overall Survival
– Trodelvy Significantly Reduced the Risk of Death by 49% Compared with Single-Agent Chemotherapy in the Phase 3 ASCENT Study –
– Trodelvy is Under Regulatory Review in the EU and in the United Kingdom, Canada, Switzerland and Australia as Part of Project Orbis –April 07, 2021 07:53 PM Eastern Daylight Time
FOSTER CITY, Calif.–(BUSINESS WIRE)–Gilead Sciences, Inc. (Nasdaq: GILD) today announced that the U.S. Food and Drug Administration (FDA) has granted full approval to Trodelvy® (sacituzumab govitecan-hziy) for adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (TNBC) who have received two or more prior systemic therapies, at least one of them for metastatic disease. The approval is supported by data from the Phase 3 ASCENT study, in which Trodelvy demonstrated a statistically significant and clinically meaningful 57% reduction in the risk of disease worsening or death (progression-free survival (PFS)), extending median PFS to 4.8 months from 1.7 months with chemotherapy (HR: 0.43; 95% CI: 0.35-0.54; p<0.0001). Trodelvy also extended median overall survival (OS) to 11.8 months vs. 6.9 months (HR: 0.51; 95% CI: 0.41-0.62; p<0.0001), representing a 49% reduction in the risk of death.
Trodelvy is directed to the Trop-2 receptor, a protein frequently expressed in multiple types of epithelial tumors, including TNBC, where high expression is associated with poor survival and relapse. Prior to the FDA approval of Trodelvy, patients with previously treated metastatic TNBC had few treatment options in this high unmet-need setting. The FDA granted accelerated approval to Trodelvy in April 2020 based on objective response rate and duration of response results in a Phase 1/2 study. Today’s approval expands the previous Trodelvy indication to include treatment in adult patients with unresectable locally advanced or metastatic TNBC who have received two or more prior systemic therapies, at least one of them for metastatic disease.
“Women with triple-negative breast cancer have historically had very few effective treatment options and faced a poor prognosis,” said Aditya Bardia, MD, MPH, Director of Breast Cancer Research Program, Mass General Cancer Center and Assistant Professor of Medicine at Harvard Medical School, and global principal investigator of the ASCENT study. “Today’s FDA approval reflects the statistically significant survival benefit seen in the landmark ASCENT study and positions sacituzumab govitecan-hziy as a potential standard of care for pre-treated TNBC.”
“A metastatic TNBC diagnosis is frightening. As an aggressive and difficult-to-treat disease, it’s a significant advance to have an FDA-approved treatment option with a proven survival benefit for patients with metastatic disease that continues to progress,” said Ricki Fairley, Founder and CEO of Touch, the Black Breast Cancer Alliance. “For far too long, people with metastatic TNBC had very few treatment options. Today’s news continues the progress of bringing more options to treat this devastating disease.”
Among all patients evaluable for safety in the ASCENT study (n=482), Trodelvy had a safety profile consistent with the previously approved FDA label. The most frequent Grade ≥3 adverse reactions for Trodelvy compared to single-agent chemotherapy were neutropenia (52% vs. 34%), diarrhea (11% vs. 1%), leukopenia (11% vs. 6%) and anemia (9% vs. 6%). Adverse reactions leading to treatment discontinuation occurred in 5% of patients receiving Trodelvy.
“Today’s approval is the culmination of a multi-year development program and validates the clinical benefit of this important treatment in metastatic TNBC,” said Merdad Parsey, MD, PhD, Chief Medical Officer, Gilead Sciences. “Building upon this milestone, we are committed to advancing Trodelvy with worldwide regulatory authorities so that, pending their decision, Trodelvy may become available to many more people around the world who are facing this difficult-to-treat cancer.”
Regulatory submissions for Trodelvy in metastatic TNBC have been filed in the United Kingdom, Canada, Switzerland and Australia as part of Project Orbis, an initiative of the FDA Oncology Center of Excellence (OCE) that provides a framework for concurrent submission and review of oncology products among international partners, as well as in Singapore through our partner Everest Medicines.The European Medicines Agency has also validated a Marketing Authorization Application for Trodelvy in the European Union. All filings are based on data from the Phase 3 ASCENT study.
Trodelvy Boxed Warning
The Trodelvy U.S. Prescribing Information has a BOXED WARNING for severe or life-threatening neutropenia and severe diarrhea; see below for Important Safety Information.
About Trodelvy
Trodelvy (sacituzumab govitecan-hziy) is a first-in-class antibody and topoisomerase inhibitor conjugate directed to the Trop-2 receptor, a protein frequently expressed in multiple types of epithelial tumors, including metastatic triple-negative breast cancer (TNBC), where high expression is associated with poor survival and relapse.
Trodelvy is also being developed as an investigational treatment for metastatic urothelial cancer, hormone receptor-positive/human epidermal growth factor receptor 2-negative (HR+/HER 2-) metastatic breast cancer and metastatic non-small cell lung cancer. Additional evaluation across multiple solid tumors is also underway.
About Triple-Negative Breast Cancer (TNBC)
TNBC is an aggressive type of breast cancer, accounting for approximately 15% of all breast cancers. The disease is diagnosed more frequently in younger and premenopausal women and is more prevalent in African American and Hispanic women. TNBC cells do not have estrogen and progesterone receptors and have limited HER 2. Medicines targeting these receptors therefore are not typically effective in treating TNBC.
About the ASCENT Study
The Phase 3 ASCENT study, an open-label, active-controlled, randomized confirmatory trial, enrolled more than 500 patients with relapsed/refractory metastatic triple-negative breast cancer (TNBC) who had received two or more prior systemic therapies (including a taxane), at least one of them for metastatic disease. Patients were randomized to receive either Trodelvy or a chemotherapy chosen by the patients’ treating physicians. The primary efficacy outcome was progression-free survival (PFS) in patients without brain metastases at baseline, as measured by a blinded, independent, centralized review using RECIST v1.1 criteria. Additional efficacy measures included PFS for the full population (all patients with and without brain metastases) and overall survival (OS). More information about ASCENT is available at http://clinicaltrials.gov/show/NCT02574455.
Important Safety Information for Trodelvy
BOXED WARNING: NEUTROPENIA AND DIARRHEA
- Severe, life-threatening, or fatal neutropenia may occur. Withhold TRODELVY for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment. Consider G-CSF for secondary prophylaxis. Initiate anti-infective treatment in patient with febrile neutropenia without delay.
- Severe diarrhea may occur. Monitor patients with diarrhea and give fluid and electrolytes as needed. Administer atropine, if not contraindicated, for early diarrhea of any severity. At the onset of late diarrhea, evaluate for infectious causes and, if negative, promptly initiate loperamide. If severe diarrhea occurs, withhold TRODELVY until resolved to ≤ Grade 1 and reduce subsequent doses.
CONTRAINDICATIONS
- Severe hypersensitivity to TRODELVY
WARNINGS AND PRECAUTIONS
Neutropenia: Dose modifications may be required due to neutropenia. Neutropenia occurred in 62% of patients treated with TRODELVY, leading to permanent discontinuation in 0.5% of patients. Grade 3-4 neutropenia occurred in 47% of patients. Febrile neutropenia occurred in 6%.
Diarrhea: Diarrhea occurred in 64% of all patients treated with TRODELVY. Grade 3 diarrhea occurred in 12% of patients. Neutropenic colitis occurred in 0.5% of patients. Withhold TRODELVY for Grade 3-4 diarrhea and resume when resolved to ≤ Grade 1. At onset, evaluate for infectious causes and if negative, promptly initiate loperamide, 4 mg initially followed by 2 mg with every episode of diarrhea for a maximum of 16 mg daily. Discontinue loperamide 12 hours after diarrhea resolves. Additional supportive measures (e.g., fluid and electrolyte substitution) may also be employed as clinically indicated. Patients who exhibit an excessive cholinergic response to treatment can receive appropriate premedication (e.g., atropine) for subsequent treatments.
Hypersensitivity and Infusion-Related Reactions: TRODELVY can cause severe and life-threatening hypersensitivity and infusion-related reactions, including anaphylactic reactions. Hypersensitivity reactions within 24 hours of dosing occurred in 37% of patients. Grade 3-4 hypersensitivity occurred in 1% of patients. The incidence of hypersensitivity reactions leading to permanent discontinuation of TRODELVY was 0.4%. Pre-infusion medication is recommended. Observe patients closely for hypersensitivity and infusion-related reactions during each infusion and for at least 30 minutes after completion of each infusion. Medication to treat such reactions, as well as emergency equipment, should be available for immediate use.
Nausea and Vomiting: Nausea occurred in 67% of all patients treated with TRODELVY. Grade 3-4 nausea occurred in 5% of patients. Vomiting occurred in 40% of patients and Grade 3-4 vomiting occurred in 3% of these patients. Premedicate with a two or three drug combination regimen (e.g., dexamethasone with either a 5-HT3 receptor antagonist or an NK-1 receptor antagonist as well as other drugs as indicated) for prevention of chemotherapy-induced nausea and vomiting (CINV). Withhold TRODELVY doses for Grade 3 nausea or Grade 3-4 vomiting and resume with additional supportive measures when resolved to Grade ≤ 1. Additional antiemetics and other supportive measures may also be employed as clinically indicated. All patients should be given take-home medications with clear instructions for prevention and treatment of nausea and vomiting.
Increased Risk of Adverse Reactions in Patients with Reduced UGT1A1 Activity: Individuals who are homozygous for the uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1)*28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia and may be at increased risk for other adverse reactions with TRODELVY. The incidence of Grade 3-4 neutropenia in genotyped patients was 69% in patients homozygous for the UGT1A1*28, 48% in patients heterozygous for the UGT1A1*28 allele and 46% in patients homozygous for the wild-type allele. The incidence of Grade 3-4 anemia in genotyped patients was 24% in patients homozygous for the UGT1A1*28 allele, 8% in patients heterozygous for the UGT1A1*28 allele, and 10% in patients homozygous for the wild-type allele. Closely monitor patients with known reduced UGT1A1 activity for adverse reactions. Withhold or permanently discontinue TRODELVY based on severity of the observed adverse reactions in patients with evidence of acute early-onset or unusually severe adverse reactions, which may indicate reduced UGT1A1 function.
Embryo-Fetal Toxicity: Based on its mechanism of action, TRODELVY can cause teratogenicity and/or embryo-fetal lethality when administered to a pregnant woman. TRODELVY contains a genotoxic component, SN-38, and targets rapidly dividing cells. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TRODELVY and for 6 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRODELVY and for 3 months after the last dose.
ADVERSE REACTIONS
In the ASCENT study (IMMU-132-05), the most common adverse reactions (incidence ≥25%) were nausea, neutropenia, diarrhea, fatigue, alopecia, anemia, vomiting, constipation, rash, decreased appetite, and abdominal pain. The most frequent serious adverse reactions (SAR) (>1%) were neutropenia (7%), diarrhea (4%), and pneumonia (3%). SAR were reported in 27% of patients, and 5% discontinued therapy due to adverse reactions. The most common Grade 3-4 lab abnormalities (incidence ≥25%) in the ASCENT study were reduced hemoglobin, lymphocytes, leukocytes, and neutrophils.
DRUG INTERACTIONS
UGT1A1 Inhibitors: Concomitant administration of TRODELVY with inhibitors of UGT1A1 may increase the incidence of adverse reactions due to potential increase in systemic exposure to SN-38. Avoid administering UGT1A1 inhibitors with TRODELVY.
UGT1A1 Inducers: Exposure to SN-38 may be substantially reduced in patients concomitantly receiving UGT1A1 enzyme inducers. Avoid administering UGT1A1 inducers with TRODELVY
Please see full Prescribing Information, including BOXED WARNING.
About Gilead Sciences
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer. Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California.
Sacituzumab govitecan, sold under the brand name Trodelvy, is a Trop-2-directed antibody and topoisomerase inhibitor drug conjugate indicated for the treatment of metastatic triple-negative breast cancer (mTNBC) in adult patients that have received at least two prior therapies.[1][2]
The most common side effects are nausea, neutropenia, diarrhea, fatigue, anemia, vomiting, alopecia (hair loss), constipation, decreased appetite, rash and abdominal pain.[1][2] Sacituzumab govitecan has a boxed warning about the risk of severe neutropenia (abnormally low levels of white blood cells) and severe diarrhea.[1][2] Sacituzumab govitecan may cause harm to a developing fetus or newborn baby.[1] Women are advised not to breastfeed while on sacituzumab govitecan and 1 month after the last dose is administered.[3]
The U.S. Food and Drug Administration (FDA) considers it to be a first-in-class medication.[4]
Mechanism
Sacituzumab govitecan is a conjugate of the humanized anti-Trop-2 monoclonal antibody linked with SN-38, the active metabolite of irinotecan.[5] Each antibody having on average 7.6 molecules of SN-38 attached.[6] SN-38 is too toxic to administer directly to patients, but linkage to an antibody allows the drug to specifically target cells containing Trop-2.
Sacituzumab govitecan is a Trop-2-directed antibody and topoisomerase inhibitor drug conjugate, meaning that the drug targets the Trop-2 receptor that helps the cancer grow, divide and spread, and is linked to topoisomerase inhibitor, which is a chemical compound that is toxic to cancer cells.[1] Approximately two of every ten breast cancer diagnoses worldwide are triple-negative.[1] Triple-negative breast cancer is a type of breast cancer that tests negative for estrogen receptors, progesterone receptors and human epidermal growth factor receptor 2 (HER2) protein.[1] Therefore, triple-negative breast cancer does not respond to hormonal therapy medicines or medicines that target HER2.[1]
Development
Immunomedics announced in 2013, that it had received fast track designation from the US Food and Drug Administration (FDA) for the compound as a potential treatment for non-small cell lung cancer, small cell lung cancer, and metastatic triple-negative breast cancer. Orphan drug status was granted for small cell lung cancer and pancreatic cancer.[7][8] In February 2016, Immunomedics announced that sacituzumab govitecan had received an FDA breakthrough therapy designation (a classification designed to expedite the development and review of drugs that are intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition) for the treatment of patients with triple-negative breast cancer who have failed at least two other prior therapies for metastatic disease.[9][10]
History
Sacituzumab govitecan was added to the proposed INN list in 2015,[11] and to the recommended list in 2016.[12]
Sacituzumab govitecan-hziy was approved for use in the United States in April 2020.[1][13][14][2]
Sacituzumab govitecan-hziy was approved based on the results of IMMU-132-01, a multicenter, single-arm clinical trial (NCT01631552) of 108 subjects with metastatic triple-negative breast cancer who had received at least two prior treatments for metastatic disease.[1][14][2] Of the 108 patients involved within the study, 107 were female and 1 was male.[15] Subjects received sacituzumab govitecan-hziy at a dose of 10 milligrams per kilogram of body weight intravenously on days one and eight every 21 days.[14][15] Treatment with sacituzumab govitecan-hziy was continued until disease progression or unacceptable toxicity.[15] Tumor imaging was obtained every eight weeks.[14][2] The efficacy of sacituzumab govitecan-hziy was based on the overall response rate (ORR) – which reflects the percentage of subjects that had a certain amount of tumor shrinkage.[1][14] The ORR was 33.3% (95% confidence interval [CI], 24.6 to 43.1). [1][14][15] Additionally, with the 33.3% of study participants who achieved a response, 2.8% of patients experienced complete responses.[15] The median time to response in patients was 2.0 months (range, 1.6 to 13.5), the median duration of response was 7.7 months (95% confidence interval [CI], 4.9 to 10.8), the median progression free survival was 5.5 months, and the median overall survival was 13.0 months.[15] Of the subjects that achieved an objective response to sacituzumab govitecan-hziy, 55.6% maintained their response for six or more months and 16.7% maintained their response for twelve or more months.[1][14]
Sacituzumab govitecan-hziy was granted accelerated approval along with priority review, breakthrough therapy, and fast track designations.[1][14] The U.S. Food and Drug Administration (FDA) granted approval of Trodelvy to Immunomedics, Inc.[1]
References
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA Approves New Therapy for Triple Negative Breast Cancer That Has Spread, Not Responded to Other Treatments”. U.S. Food and Drug Administration (FDA). 22 April 2020. Retrieved 22 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f “Drug Trial Snapshot: Trodelvy”. U.S. Food and Drug Administration (FDA). 22 April 2020. Retrieved 29 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ (PDF)https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761115s000lbl.pdf. Missing or empty
|title=(help) - ^ “New Drug Therapy Approvals 2020”. U.S. Food and Drug Administration (FDA). 31 December 2020. Retrieved 17 January2021. This article incorporates text from this source, which is in the public domain.
- ^ Sacituzumab Govitecan (IMMU-132), an Anti-Trop-2/SN-38 Antibody-Drug Conjugate: Characterization and Efficacy in Pancreatic, Gastric, and Other Cancers. 2015
- ^ “Novel Agents are Targeting Drivers of TNBC”. http://www.medpagetoday.com. 28 June 2016.
- ^ “Sacituzumab govitecan Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 22 April 2020.
- ^ “Sacituzumab govitecan Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 22 April 2020.
- ^ “New Therapy Shows Early Promise, Continues to Progress in Triple-Negative Breast Cancer”. Cure Today.
- ^ “U.S. Food and Drug Administration (FDA) Grants Breakthrough Therapy Designation to Immunomedics for Sacituzumab Govitecan for the Treatment of Patients With Triple-Negative Breast Cancer”(Press release). Immunomedics. 5 February 2016. Retrieved 25 April 2020 – via GlobeNewswire.
- ^ World Health Organization (2015). “International nonproprietary names for pharmaceutical substances (INN): proposed INN: list 113”. WHO Drug Information. 29 (2): 260–1. hdl:10665/331080.
- ^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 75”. WHO Drug Information. 30 (1): 151–3. hdl:10665/331046.
- ^ “Trodelvy: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 22 April 2020.
- ^ Jump up to:a b c d e f g h “FDA grants accelerated approval to sacituzumab govitecan-hziy for metastatic triple negative breast cancer”. U.S. Food and Drug Administration (FDA). 22 April 2020. Retrieved 23 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f “Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer”. The New England Journal of Medicine.
Further reading
- Bardia A, Mayer IA, Vahdat LT, et al. (February 2019). “Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer”. N. Engl. J. Med. 380 (8): 741–751. doi:10.1056/NEJMoa1814213. PMID 30786188.
- Weiss J, Glode A, Messersmith WA, et al. (August 2019). “Sacituzumab govitecan: breakthrough targeted therapy for triple-negative breast cancer”. Expert Rev Anticancer Ther. 19 (8): 673–679. doi:10.1080/14737140.2019.1654378. PMID 31398063. S2CID 199518147.
External links
- “Sacituzumab govitecan”. Drug Information Portal. U.S. National Library of Medicine.
- “Sacituzumab govitecan”. ADC Review.
- “Sacituzumab govitecan”. National Cancer Institute.
- Clinical trial number NCT01631552 for “Phase I/II Study of IMMU-132 in Patients With Epithelial Cancers” at ClinicalTrials.gov
- Sacituzumab govitecan at the US National Library of Medicine Medical Subject Headings (MeSH)
| Monoclonal antibody | |
|---|---|
| Type | ? |
| Source | Humanized (from mouse) |
| Target | Trop-2 |
| Clinical data | |
| Trade names | Trodelvy |
| Other names | IMMU-132, hRS7-SN-38, sacituzumab govitecan-hziy |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620034 |
| License data | US DailyMed: Sacituzumab_govitecan |
| Pregnancy category | Contraindicated |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only |
| Identifiers | |
| CAS Number | 1491917-83-9 |
| PubChem CID | 91668186 |
| DrugBank | DB12893 |
| ChemSpider | none |
| UNII | M9BYU8XDQ6 |
| KEGG | D10985 |
| Chemical and physical data | |
| Formula | C76H104N12O24S |
| Molar mass | 1601.79 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| show |
//////////sacituzumab govitecan-hziy, fda 2021, approvals 2021, Trodelvy , hRS 7SN38, hRS7-SN38, IMMU 132, IMMU-132, MONOCLONAL ANTIBODY, Sacituzumab govitecan, sacituzumab govitecan-hziy, CANCER, MONOCLONAL ANTIBODIES
#sacituzumab govitecan-hziy, #fda 2021, #approvals 2021, #Trodelvy , #hRS 7SN38, #hRS7-SN38, #IMMU 132, #IMMU-132, #MONOCLONAL ANTIBODY, #Sacituzumab govitecan, #sacituzumab govitecan-hziy, #CANCER, #MONOCLONAL ANTIBODIES
CCC1=C2CN3C(=CC4=C(C3=O)COC(=O)C4(CC)OC(=O)OCC5=CC=C(C=C5)NC(=O)C(CCCCN)NC(=O)COCC(=O)NCCOCCOCCOCCOCCOCCOCCOCCOCCN6C=C(N=N6)CNC(=O)C7CCC(CC7)CN8C(=O)CC(C8=O)SCC(C(=O)O)N)C2=NC9=C1C=C(C=C9)O
NEW DRUG APPROVALS
ONE TIME
$10.00
Romosozumab, ロモソズマブ (遺伝子組換え)
Romosozumab
ロモソズマブ (遺伝子組換え)
AMG 785
Immunoglobulin G2, anti-(human sclerostin) (human-mouse monoclonal 785A070802 heavy chain), disulfide with human-mouse monoclonal 785A070802 κ-chain, dimer
- Immunoglobulin G2, anti-(human sclerostin) (humanized monoclonal 785A070802 heavy chain), disulfide with humanized monoclonal 785A070802 κ-chain, dimer
| Formula |
C6452H9926N1714O2040S54
|
|---|---|
| CAS |
909395-70-6
|
| Mol weight |
145875.6186
|
Monoclonal antibody
Treatment of osteoporosis
Osteoporosis agent, Sclerostin activity inhibitor
JAPAN APPROVED 2019/1/8, Evenity
Romosozumab (AMG 785) is a humanized monoclonal antibody that targets sclerostin for the treatment of osteoporosis.[1]
Romosozumab was originally discovered by Chiroscience,[2] which was acquired by Celltech (now owned by UCB).[3] Celltech entered in a partnership with Amgen in 2002 for the product’s development.[4]
In 2016 results from 12 months of a clinical study were reported.[5]
Some results from the FRAME[6] and ARCH clinical studies were reported on in 2017.[7]
Japan’s Ministry of Health, Labor and Welfare has granted a marketing authorization for romosozumab (EVENITY) for the treatment of osteoporosis in patients at high risk of fracture. Developed by Amgen and UCB, romosozumab is a humanized IgG2 monoclonal antibody that targets sclerostin. The approval in Japan is based on results from the Phase 3 FRAME and BRIDGE studies, which included 7,180 postmenopausal women with osteoporosis and 245 men with osteoporosis, respectively.
A biologics license application (BLA) for romosozumab as a treatment of osteoporosis in postmenopausal women at high risk for fracture was submitted to the U.S. Food and Drug Administration (FDA) in July 2016, but additional safety and efficacy data was requested in the FDA’s complete response letter, as announced by Amgen and UCB in July 2017. In July 2018, Amgen and UCB announced that the BLA had been resubmitted. In addition to data from early-stage clinical studies, the original BLA included data from the Phase 3 FRAME study. The resubmitted BLA includes results from the more recent Phase 3 ARCH study, an alendronate-active comparator trial including 4,093 postmenopausal women with osteoporosis who experienced a fracture, and the Phase 3 BRIDGE study. The FDA’s Bone, Reproductive and Urologic Drugs Advisory Committee is scheduled to review data supporting the BLA for romosozumab at a meeting on January 16, 2019.
The European Medicines Agency is also currently reviewing a marketing application for romosozumab.
|
Commercial production of cell culture-derived products (for example, protein-based products, such as monoclonal antibodies (mAbs)), requires optimization of cell culture parameters in order for the cells to produce enough product to meet clinical and commercial demands. However, when cell culture parameters are optimized for improving productivity of a protein product, it is also necessary to maintain desired quality specifications of the product such as glycosylation profile, aggregate levels, charge heterogeneity, and amino acid sequence integrity (Li, et al., 2010 , mAbs., 2(5):466-477).
|
References
- ^ “Statement On A Nonproprietary Name Adopted By The USAN Council: Romosozumab” (PDF). American Medical Association.
- ^ Quested, Tony (June 7, 2015). “Cream of life science entrepreneurs’ first venture was selling doughnuts”. Business Week. Cambridge, England: Q Communications. Retrieved December 24, 2018.
- ^ Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003 Dec 1;22(23):6267-76.
- ^ Celltech group Annual Report and Accounts 2002
- ^ Cosman; et al. (2016). “Romosozumab Treatment in Postmenopausal Women with Osteoporosis”. The New England Journal of Medicine. 375: 1532–1543. doi:10.1056/NEJMoa1607948. PMID 27641143.
- ^ Efficacy and Safety of Romosozumab Treatment in Postmenopausal Women With Osteoporosis (FRAME)
- ^ Bone Loss Drug Effective, But is it Safe? Oct 2017
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from mouse) |
| Target | Sclerostin |
| Clinical data | |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| ChemSpider |
|
| KEGG | |
| Chemical and physical data | |
| Formula | C6452H9926N1714O2040S54 |
| Molar mass | 145.9 kg/mol |
///////////Romosozumab, ロモソズマブ (遺伝子組換え) , JAPAN 2019, Monoclonal antibody, Osteoporosis, AMG 785
|
Monoclonals for bone, musculoskeletal, circulatory, and neurologic systems
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Bone (“-os-“, “-s(o)-“) |
|
||||||||
| Musculoskeletal (“-mul-“) |
|
||||||||
| Circulatory (“-c(i[r])-“) |
|
||||||||
| Neurologic (“-ne(u)(r)-“) |
|
||||||||
| Angiogenesis inhibitor (“-anibi-“) |
|
||||||||
| Growth factor (“-gr(o)-“) |
|
||||||||
|
|||||||||
Lanadelumab, ラナデルマブ
(Heavy chain)
EVQLLESGGG LVQPGGSLRL SCAASGFTFS HYIMMWVRQA PGKGLEWVSG IYSSGGITVY
ADSVKGRFTI SRDNSKNTLY LQMNSLRAED TAVYYCAYRR IGVPRRDEFD IWGQGTMVTV
SSASTKGPSV FPLAPSSKST SGGTAALGCL VKDYFPEPVT VSWNSGALTS GVHTFPAVLQ
SSGLYSLSSV VTVPSSSLGT QTYICNVNHK PSNTKVDKRV EPKSCDKTHT CPPCPAPELL
GGPSVFLFPP KPKDTLMISR TPEVTCVVVD VSHEDPEVKF NWYVDGVEVH NAKTKPREEQ
YNSTYRVVSV LTVLHQDWLN GKEYKCKVSN KALPAPIEKT ISKAKGQPRE PQVYTLPPSR
EEMTKNQVSL TCLVKGFYPS DIAVEWESNG QPENNYKTTP PVLDSDGSFF LYSKLTVDKS
RWQQGNVFSC SVMHEALHNH YTQKSLSLSP G
(Light chain)
DIQMTQSPST LSASVGDRVT ITCRASQSIS SWLAWYQQKP GKAPKLLIYK ASTLESGVPS
RFSGSGSGTE FTLTISSLQP DDFATYYCQQ YNTYWTFGQG TKVEIKRTVA APSVFIFPPS
DEQLKSGTAS VVCLLNNFYP REAKVQWKVD NALQSGNSQE SVTEQDSKDS TYSLSSTLTL
SKADYEKHKV YACEVTHQGL SSPVTKSFNR GEC
(dimer; dishulfide bridge: H22-H96, H149-H205, H225-L213, H231-H’231, H234-H’234, H266-H326, H372-H430, H’22-H’96, H’149-H’205, H’225-L’213, H’266-H’326, H’372-H’430, L23-L88, L133-L193, L’23-L’88, L’133-L’193)
Lanadelumab
DX 2930
Fda approved 2018/8/23, Takhzyro
| Formula |
C6468H10016N1728O2012S48
|
|---|---|
| Cas |
1426055-14-2
|
| Mol weight |
145714.225
|
Peptide, Monoclonal antibody
Prevention of angioedema in patients with hereditary angioedema
Immunomodulator, Plasma kallikrein inhibitor
breakthrough therapy, UNII: 2372V1TKXK

Lanadelumab (INN) (alternative identifier DX-2930[1]) is a human monoclonal antibody (class IgG1 kappa)[2] that targets plasma kallikrein (pKal)[1] in order to promote prevention of angioedema in patients with hereditary angioedema.[3][4] In phase 1 clinical trialsLanadelumab was well tolerated and was reported to reduce cleavage of kininogen in the plasma of patients with hereditary angioedeman and decrease the number of patients experiencing attacks of angioedema.[1][5][6][7] As of 2017 ongoing trials for Lanadelumab include two phase 3 studies focused on investigating the utility of Lanadelumab in preventing of acute angioedema attacks in hereditary angioedema patients[8][9]
This drug was produced by Dyax Corp and currently under development by Shire.[10] Lanadelumab has been designated by the U.S. Food and Drug Administration (FDA) as a breakthrough therapy.[11]

References
- ^ Jump up to:a b c Banerji, Aleena; Busse, Paula; Shennak, Mustafa; Lumry, William; Davis-Lorton, Mark; Wedner, Henry J.; Jacobs, Joshua; Baker, James; Bernstein, Jonathan A. (2017-02-23). “Inhibiting Plasma Kallikrein for Hereditary Angioedema Prophylaxis”. The New England Journal of Medicine. 376 (8): 717–728. doi:10.1056/NEJMoa1605767. ISSN 1533-4406. PMID 28225674.
- Jump up^ Kenniston, Jon A.; Faucette, Ryan R.; Martik, Diana; Comeau, Stephen R.; Lindberg, Allison P.; Kopacz, Kris J.; Conley, Gregory P.; Chen, Jie; Viswanathan, Malini (2014-08-22). “Inhibition of Plasma Kallikrein by a Highly Specific Active Site Blocking Antibody”. The Journal of Biological Chemistry. 289 (34): 23596. doi:10.1074/jbc.M114.569061. PMC 4156074
 . PMID 24970892.
. PMID 24970892. - Jump up^ Statement On A Nonproprietary Name Adopted By The USAN Council – Lanadelumab, American Medical Association.
- Jump up^ World Health Organization (2015). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 114”(PDF). WHO Drug Information. 29 (4).
- Jump up^ Chyung, Yung; Vince, Bradley; Iarrobino, Ryan; Sexton, Dan; Kenniston, Jon; Faucette, Ryan; TenHoor, Chris; Stolz, Leslie E.; Stevens, Chris (2014-10-01). “A phase 1 study investigating DX-2930 in healthy subjects”. Annals of Allergy, Asthma & Immunology. 113 (4): 460–466.e2. doi:10.1016/j.anai.2014.05.028. ISSN 1534-4436. PMID 24980392.
- Jump up^ “A Single Increasing Dose Study to Assess Safety and Tolerability of DX-2930 in Healthy Subjects – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2017-03-24.
- Jump up^ “Double-Blind, Multiple Ascending Dose Study to Assess Safety, Tolerability and Pharmacokinetics of DX-2930 in Hereditary Angioedema (HAE) Subjects – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2017-03-24.
- Jump up^ “Efficacy and Safety Study of DX-2930 to Prevent Acute Angioedema Attacks in Patients With Type I and Type II HAE – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2017-03-24.
- Jump up^ “Long-term Safety and Efficacy Study of DX-2930 to Prevent Acute Angioedema Attacks in Patients With Type I and Type II HAE – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2017-03-24.
- Jump up^ “Lanadelumab – AdisInsight”. adisinsight.springer.com. Retrieved 2017-03-24.
- Jump up^ “Dyax Corp. Receives FDA Breakthrough Therapy Designation for DX-2930 for Prevention of Attacks of Hereditary Angioedema”. http://www.businesswire.com. Retrieved 2017-03-24.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | kallikrein |
| Clinical data | |
| Synonyms | DX-2930 |
| ATC code |
|
| Identifiers | |
| CAS Number | |
| ChemSpider |
|
| UNII | |
| Chemical and physical data | |
| Formula | C6468H10016N1728O2012S47 |
| Molar mass | 145.7 kDa |
///////////Lanadelumab, Peptide, Monoclonal antibody, FDA 2018, ラナデルマブ ,Immunomodulator, Plasma kallikrein inhibitor, DX 2930, breakthrough therapy, Takhzyro
“DRUG APPROVALS INTERNATIONAL” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
FDA approves first biosimilar to Neulasta, Fulphila (pegfilgrastim) to help reduce the risk of infection during cancer treatment
The U.S. Food and Drug Administration today approved Fulphila (pegfilgrastim-jmdb) as the first biosimilar to Neulasta (pegfilgrastim) to decrease the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells), in patients with non-myeloid (non-bone marrow) cancer who are receiving myelosuppressive chemotherapy that has a clinically significant incidence of febrile neutropenia.
June 4, 2018
Release
The U.S. Food and Drug Administration today approved Fulphila (pegfilgrastim-jmdb) as the first biosimilar to Neulasta (pegfilgrastim) to decrease the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells), in patients with non-myeloid (non-bone marrow) cancer who are receiving myelosuppressive chemotherapy that has a clinically significant incidence of febrile neutropenia.
“Bringing new biosimilars to patients is a top priority for the FDA, and a key part of our efforts to help promote competition that can reduce drug costs and promote access,” said FDA Commissioner Scott Gottlieb, M.D. “We’ll continue to prioritize reviews of these products to help ensure that biosimilar medications are brought to the market efficiently and through a process that makes certain that these new medicines meet the FDA’s rigorous standard for approval. This summer, we’ll release a comprehensive new plan to advance new policy efforts that promote biosimilar product development. Biologics represent some of the most clinically important, but also costliest products that patients use to promote their health. We want to make sure that the pathway for developing biosimilar versions of approved biologics is efficient and effective, so that patients benefit from competition to existing biologics once lawful intellectual property has lapsed on these products.”
Biological products are generally derived from a living organism and can come from many sources, such as humans, animals, microorganisms or yeast. A biosimilar is a biological product that is approved based on data showing that it is highly similar to a biological product already approved by the FDA (reference product) and has no clinically meaningful differences in terms of safety, purity and potency (i.e., safety and effectiveness) from the reference product, in addition to meeting other criteria specified by law.
The FDA’s approval of Fulphila is based on review of evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamic data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrates Fulphila is biosimilar to Neulasta. Fulphila has been approved as a biosimilar, not as an interchangeable product.
The most common side effects of Fulphila are bone pain and pain in extremities. Patients with a history of serious allergic reactions to human granulocyte colony-stimulating factors such as pegfilgrastim or filgrastim products should not take Fulphila.
Serious side effects from treatment with Fulphila include rupture of the spleen, acute respiratory distress syndrome, serious allergic reactions including anaphylaxis, acute inflammation of the kidney (glomerulonephritis), an abnormally high level of white blood cells (leukocytosis), capillary leak syndrome and the potential for tumor growth. Fatal sickle cell crises have occurred.
The FDA granted approval of Fulphila to Mylan GmbH.

//////////// pegfilgrastim, fda 2018, Fulphila, Neulasta, Mylan GmbH, biosimilars, MONOCLONAL ANTIBODY,
Tildrakizumab-asmn
| Heavy chain: | |
| QVQLVQSGAEVKKPGASVKVSCKASGYIFITYWMTWVRQAPGQGL | |
| EWMGQIFPASGSADYNEKFEGRVTMTTDTSTSTAYMELRSLRSDD | |
| TAVYYCARGGGGFAYWGQGTLVTVSSASTKGPSVFPLAPSSKSTS | |
| GGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYS | |
| LSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTC | |
| PPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDP | |
| EVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNG | |
| KEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKN | |
| QVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFL | |
| YSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK | |
| Light chain: | |
| DIQMTQSPSSLSASVGDRVTITCRTSENIYSYLAWYQQKPGKAPK | |
| LLIYNAKTLAEGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQH | |
| HYGIPFTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCL | |
| LNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT | |
| LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC |
Tildrakizumab-asmn
Immunoglobulin G1, anti-(human interleukin 23) (human-Mus musculus monoclonal heavy chain), disulfide with human-Mus musculus monoclonal light chain, dimer
CAS 1326244-10-3, BLA 761067
Tildrakizumab (SCH 900222/MK-3222)
ILUMYA; MK-3222; SCH-900222; SUNPG 1622; SUNPG 1622 I; SUNPG 1623 I; SUNPG 1623 II; SUNPG 1623 III; SUNPG 1623 IV; SUNPG1623; Tildrakizumab-asmn
DRUG BANK https://www.drugbank.ca/drugs/DB14004
Company Sun Pharmaceuticals
Approval Status FDA Approved March 2018 FOR Psoriasis, plaque
Treatments plaque psoriasis
Protein chemical formulaC6426H9918N1698O2000S46
Protein average weight144400.0 DaSequences
>Tildrakizumab Sequence MLGSRAVMLLLLLPWTAQGRAVPGGSSPAWTQCQQLSQKLCTLAWSAHPLVGHMDLREEG DEETTNDVPHIQCGDGCDPQGLRDNSQFCLQRIHQGLIFYEKLLGSDIFTGEPSLLPDSP VGQLHASLLGLSQLLQPEGHHWETQQIPSLSPSQPWQRLLLRFKILRSLQAFVAVAARVF AHGAATLSP
| Monoclonal antibody | |
|---|---|
| Type | ? |
| Source | Humanized (from mouse) |
| Target | IL23 |
| Clinical data | |
| Trade names | Ilumya |
| Synonyms | Tildrakizumab-asmn |
| Routes of administration |
Subcutaneous injection |
| ATC code |
|
| Identifiers | |
| CAS Number | |
| ChemSpider |
|
| KEGG | |
| Chemical and physical data | |
| Formula | C6426H9918N1698O2000S46 |
| Molar mass | 144.4 kg/mol |
- Originator Schering-Plough
- Developer Almirall S.A.; Merck & Co; Schering-Plough; Sun Pharmaceutical Industries
- Class Antipsoriatics; Monoclonal antibodies
- Mechanism of Action Interleukin 23 inhibitors
- Orphan Drug StatusNo
- New Molecular EntityYes
Highest Development Phases
- Registered Plaque psoriasis
- Phase II Ankylosing spondylitis; Psoriatic arthritis
- Discontinued Autoimmune disorders
Most Recent Events
- 21 Mar 2018 Registered for Plaque psoriasis in USA (SC) – First global approval
- 16 Feb 2018 Adverse events data from two phase III trials (reSURFACE 1 and 2) in chronic Plaque psoriasis presented at the 76th Annual Meeting of the American Academy of Dermatology (AAD-2018)
- 16 Feb 2018 Pharmacokinetics data from population PK model in healthy volunteers and patients with psoriasis presented at the 76th Annual Meeting of the American Academy of Dermatology (AAD-2018)
Ilumya (tildrakizumab-asmn) is an interleukin-23 antagonist.
Humanized monoclonal IgG1-kappa antibody against IL-23p19; produced in CHO cells
Immunoglobulin G1, anti-(human interleukin 23) (human-Mus musculus monoclonal heavy chain), disulfide with human-Mus musculus monoclonal light chain, dimer
Ilumya is specifically indicated for the treatment of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
Ilumya is supplied as a solution for subcutaneous injection. The recommended dose is 100 mg at Weeks 0, 4, and every twelve weeks thereafter.

Tildrakizumab (Ilumya) is a monoclonal antibody designed for the treatment of immunologically mediated inflammatory disorders.[1] In the United States, it is approved for the treatment of moderate-to-severe plaque psoriasis.[2]
Tildrakizumab was designed to block interleukin-23, a cytokine that plays an important role in managing the immune system and autoimmune disease. Originally developed by Schering-Plough, this drug is now part of Merck‘s clinical program, following that company’s acquisition of Schering-Plough.
Sun Pharmaceutical acquired worldwide rights to tildrakizumab for use in all human indications from Merck in exchange for an upfront payment of U.S. $80 million. Upon product approval, Sun Pharmaceutical will be responsible for regulatory activities, including subsequent submissions, pharmacovigilance, post approval studies, manufacturing and commercialization of the approved product. [3]

As of March 2014, the drug was in phase III clinical trials for plaque psoriasis. The two trials enrolled nearly 2000 patients. [4][5]
In 2016, tildrakizumab became the first IL-23p19 inhibitor to demonstrate positive results in Phase-3 clinical trials for the treatment of moderate-to-severe plaque psoriasis, further validating the importance of the role of IL-23 in psoriasis. Sun Pharma signed a licensing pact with Spain’s Almirall for marketing tildrakizumab in Europe [6]
In March 2018, it was approved by the Food and Drug Administration for the treatment of moderate-to-severe plaque psoriasis as an injection for subcutaneous use in the United States.[2]
In 2014, Sun Pharma acquired worldwide rights to tildrakizumab from Merck; upon product approval, Sun Pharma is responsible for regulatory activities, including subsequent submissions, pharmacovigilance, post approval studies, manufacturing and commercialization of the product. In 2016, Almirall sublicensed the product for the development and marketing in Europe for the treatment of psoriasis.
See also
- Ustekinumab, a monoclonal antibody targeting both IL-12 and IL-23 and used to treat plaque psoriasis, launched in the United States under the brand name Stelara
- Guselkumab, another experimental, IL-23-specific monoclonal antibody. (FDA approved in 2017)
- Risankizumab, another experimental, IL-23-specific monoclonal antibody. (In Phase 3 clinical trials for plaque psoriasis as of 2017)
References
- Jump up^ Statement On A Nonproprietary Name Adopted By The USAN Council – Tildrakizumab, American Medical Association.
- ^ Jump up to:a b “FDA approves Ilumya for plaque psoriasis”. National Psoriasis Foundation. March 22, 2018.
- Jump up^ http://www.merck.com/licensing/our-partnership/sunpharma_partnership.html
- Jump up^ http://clinicaltrials.gov/ct2/show/NCT01729754?term=SCH-900222&phase=2&fund=2&rank=1
- Jump up^ http://clinicaltrials.gov/ct2/show/NCT01722331?term=SCH-900222&phase=2&fund=2&rank=2
- Jump up^ http://www.business-standard.com/content/b2b-pharma/sun-pharma-signs-licensing-pact-with-spain-s-almirall-for-tildrakizumab-in-europe-116072800225_1.html
Mechanism of Action
Tildrakizumab is a humanized IgG1/k monoclonal antibody that selectively binds to the p19 subunit of IL-23 and inhibits its interaction with the IL-23 receptor. IL-23 is a naturally occurring cytokine that is involved in inflammatory and immune responses. Tildrakizumab inhibits the release of proinflammatory cytokines and chemokines.
FDA APPROVAL DATA
BLA 761067
https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2018/761067Orig1s000REPLACEMENT_ltr.pdf
Please refer to your Biologics License Application (BLA) dated and received March 23, 2017 and your amendments, submitted under section 351(a) of the Public Health Service Act for ILUMYA (tildrakizumab-asmn) injection. We also refer to our approval letter dated March 20, 2018 which contained the following error: the Final Report Submission date was incorrectly listed for postmarketing requirement 3357-3. This replacement approval letter incorporates the correction of the error. The effective approval date will remain March 20, 2018, the date of the original approval letter.
LICENSING We have approved your BLA for ILUMYA (tildrakizumab-asmn) effective this date. You are hereby authorized to introduce or deliver for introduction into interstate commerce, ILUMYA under your existing Department of Health and Human Services U.S. License No. 0002. ILUMYA is indicated for the treatment of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
MANUFACTURING LOCATIONS Under this license, you are approved to manufacture ILUMYA drug substance at . The final formulated drug product will be manufactured, filled, labeled, and packaged at MSD Ireland, Carlow, Ireland. You may label your product with the proprietary name, ILUMYA, and market it in 100 mg/1 mL single-dose prefilled syringe
DATING PERIOD The dating period for ILUMYA drug product shall be 36 months from the date of manufacture when stored at 2-8°C. The date of manufacture shall be defined as the date of final sterile filtration of the formulated drug product. The dating period for your drug substance shall be months from the date of manufacture when stored at We have approved the stability protocols in your license application for the purpose of extending the expiration dating period of your drug substance and drug product under 21 CFR 601.12.
PATENTS
WO 2014109927
PAPER
https://www.tandfonline.com/doi/full/10.4161/19420862.2015.988944
Tildrakizumab (SCH 900222/MK-3222) targets the p19 subunit of IL-23. The mAb was developed by Schering-Plough, which was acquired by Merck & Co. in 2009, and it was then licensed by Merck to Sun Pharmaceutical Industries Ltd in September 2014. Clinical development and regulatory activities will be conducted by Merck, but funded by Sun Pharma. As of October 2014, the safety and efficacy of tildrakizumab are being evaluated in 2 Phase 3 studies that are ongoing but not recruiting patients. Both studies include patients with moderate-to-severe chronic plaque psoriasis and subcutaneously administered drug. The 52-week Phase 3 NCT01729754 study has 4 arms (200 mg tildrakizumab; 100 mg tildrakizumab; 50 mg etanercept; and placebo only), and includes an optional long-term safety extension study. The estimated enrollment is 1050, and the estimated primary completion date is October 2019. The 64-week Phase 3 NCT01722331 study is evaluating the effects of either 200 mg or 100 mg tildrakizumab to placebo; it includes an optional long-term safety extension study. The estimated enrollment is 885, and the estimated primary completion date is June 2015.


Mar 21, 2018, 09:04 ET
MUMBAI, India and PRINCETON, N.J., March 21, 2018 /PRNewswire/ — Sun Pharmaceutical Industries Ltd. (Reuters: SUN.BO, Bloomberg: SUNP IN, NSE: SUNPHARMA, BSE: 524715, “Sun Pharma” and includes its subsidiaries and/or associate companies) today announced that the U.S. Food and Drug Administration (FDA) has approved ILUMYA™ (tildrakizumab-asmn) for the treatment of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy. ILUMYA selectively binds to the p19 subunit of IL-23 and inhibits its interaction with the IL-23 receptor leading to inhibition of the release of pro-inflammatory cytokines and chemokines. ILUMYA is administered at a dose of 100 mg by subcutaneous injection every 12 weeks, after the completion of initial doses at weeks 0 and 4. ILUMYA is contraindicated in patients with a previous serious hypersensitivity reaction to tildrakizumab or to any of the excipients.
“With the approval of ILUMYA and our long-standing commitment in dermatology, we are focused on making a difference for people living with moderate-to-severe plaque psoriasis,” said Abhay Gandhi, President and Chief Executive Officer, North America, Sun Pharma. “We are committed to working with all relevant stakeholders to make ILUMYA available to appropriate people with plaque psoriasis.”
The FDA approval of ILUMYA for the treatment of adults with moderate-to-severe plaque psoriasis was supported by data from the pivotal Phase-3 reSURFACE clinical development program. In the two multicenter, randomized, double-blind, placebo-controlled trials (reSURFACE 1 and reSURFACE 2), 926 adult patients were treated with ILUMYA (N=616) or placebo (N=310). Results from these studies were published in The Lancet in July 2017, with primary endpoints presented at the 25th European Academy of Dermatology and Venereology (EADV) Congress.
Both Phase-3 studies met the primary efficacy endpoints, demonstrating significant clinical improvement with ILUMYA 100 mg compared to placebo when measured by at least 75 percent of skin clearance (Psoriasis Area Sensitivity Index or PASI 75) and Physician’s Global Assessment (PGA) score of “clear” or “minimal” at week 12 after two doses.
|
Efficacy Primary Endpoint at Week 12 in Adults with Plaque Psoriasis (NRI*) |
||||
|
reSURFACE 1 Study (NCT01722331) |
reSURFACE 2 Study (NCT01729754) |
|||
|
ILUMYA 100 mg n=309 |
Placebo n=154 |
ILUMYA 100 mg n=307 |
Placebo n=156 |
|
|
PGA of “clear” (0) or “minimal” (1)† |
179 (58%) |
11 (7%) |
168 (55%) |
7 (4%) |
|
PASI 75† |
197 (64%) |
9 (6%) |
188 (61%) |
9 (6%) |
|
PASI 90 |
107 (35%) |
4 (3%) |
119 (39%) |
2 (1%) |
|
PASI 100 |
43 (14%) |
2 (1%) |
38 (12%) |
0 (0%) |
* NRI = Non-Responder Imputation † Co-Primary Endpoints
Of the patients in the reSURFACE 1 study 74 percent (229 patients) achieved 75 percent skin clearance at week 28 after three doses, and 84 percent of patients who continued receiving ILUMYA 100 mg maintained PASI 75 at week 64 compared to 22 percent of patients who were re-randomized to placebo. In addition, 69 percent of the patients receiving ILUMYA 100 mg who had a PGA score of “clear” or “minimal” at week 28 maintained this response at week 64 compared to 14 percent of patients who were re-randomized to placebo.
Full Prescribing Information and Medication Guide for ILUMYA are attached:
PDF: https://mma.prnewswire.com/media/656994/Sun_Pharma_ILUMYA_US_Prescribing_Information.pdf
PDF: https://mma.prnewswire.com/media/656995/Sun_Pharma_ILUMYA_US_Medication_Guide.pdf
IMPORTANT SAFETY INFORMATION (continued)
Cases of angioedema and urticaria occurred in ILUMYA treated subjects in clinical trial. If a serious hypersensitivity reaction occurs, discontinue ILUMYA immediately and initiate appropriate therapy.
ILUMYA may increase the risk of infection. Treatment with ILUMYA should not be initiated in patients with a clinically important active infection until the infection resolves or is adequately treated. Consider the risks and benefits of treatment prior to prescribing ILUMYA in patients with a chronic infection or a history of recurrent infection. Instruct patients receiving ILUMYA to seek medical help if signs or symptoms of clinically important chronic or acute infection occur. If a patient develops a clinically important or serious infection, or is not responding to standard therapy, closely monitor and discontinue ILUMYA until the infection resolves.
Evaluate patients for TB infection prior to initiating treatment with ILUMYA. Initiate treatment of latent TB prior to administering ILUMYA. Monitor patients for signs and symptoms of active TB during and after ILUMYA treatment. Do not administer ILUMYA to patients with active TB infection.
Prior to initiating ILUMYA, consider completion of all age-appropriate immunizations according to current immunization guidelines. Avoid use of live vaccines in patients treated with ILUMYA.
The most common (≥1%) adverse reactions associated with ILUMYA include upper respiratory infections, injection site reactions, and diarrhea. Adverse reactions that occurred at rates less than 1% but greater than 0.1% in the ILUMYA group and at a higher rate than in the placebo group included dizziness and pain in extremity.
About the Phase-3 reSURFACE Trials
The Phase-3 studies (reSURFACE 1 and reSURFACE 2) were randomized, placebo-controlled, multicenter, three-part studies designed to demonstrate efficacy of ILUMYA in moderate-to-severe plaque psoriasis compared to placebo and comparative drug and to assess safety and tolerability. Part one of the studies randomized patients into three or four treatment arms, including ILUMYA 100 mg, ILUMYA 200 mg, placebo and etanercept (reSURFACE 2 only). After Week 12, patients on placebo were then re-randomized into ILUMYA 100 mg and 200 mg treatment arms to proceed into part two of the studies. Finally, in part three of the reSURFACE 1 study, responders (PASI ≥75) and partial responders (PASI ≥50 and PASI <75) to ILUMYA were re-randomized after Week 28 to continue the same treatment, a different dose of ILUMYA or placebo. Partial and non-responders to etanercept were treated with ILUMYA 200 mg in part three of the reSURFACE 2 study. Patients with guttate, erythrodermic, or pustular psoriasis were excluded.
About Psoriasis
Psoriasis is a chronic immune disease that appears on the skin. It is a non-contagious disorder that speeds the growth cycle of skin cells1 and results in thick scaly areas of skin2. The most common form, affecting about 80 to 90 percent of people living with psoriasis, is called plaque psoriasis3. It appears as red, raised areas of skin covered with flaky white scales, which may be itchy and painful and can crack and bleed2. Many people with plaque psoriasis continue to struggle with the ongoing, persistent nature of this chronic disease.
About Sun Dermatology
Sun Dermatology (the branded dermatology division of a wholly owned subsidiary of Sun Pharma) is committed to expanding its dermatology portfolio to bring healthcare providers and patients around the world more treatment options and ongoing support for conditions like moderate-to-severe plaque psoriasis. Sun Pharma, along with its subsidiaries, is ranked fourth in dermatology prescription volume within the U.S. per IMS and is fifth largest specialty generic pharmaceutical company globally. In addition to ILUMYA, Sun Dermatology is comprised of several branded products indicated for the treatment of acne and actinic keratosis with a focus on other dermatologic conditions.
About Sun Pharma, Merck & Co., Inc., Kenilworth, NJ, USA, Agreement
Sun Pharmaceutical Industries Ltd.’s wholly owned subsidiary licensed worldwide rights to ILUMYA from a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, in 2014. Funded by a Sun Pharma subsidiary, Merck & Co., Inc., Kenilworth, NJ, USA was responsible for the completion of Phase-3 trials and submission of a Biologics License Application to the United States Food and Drug Administration (FDA), as well as manufacturing finished goods to support Sun Pharma’s initial product launch. Sun Pharma will be responsible for all post-approval regulatory activities, including subsequent submissions, pharmacovigilance, post approval studies, manufacturing and commercialization of the approved product. Sun Pharma will also be responsible for all regulatory, pharmacovigilance, post approval studies, manufacturing and commercialization of approved products for all non-U.S. markets. Merck & Co., Inc., Kenilworth, NJ, USA is eligible to receive milestone payments and royalties on sales of ILUMYA.
About Sun Pharma, Almirall S.A, Europe, Agreement
Sun Pharma and its wholly owned subsidiary and Almirall (Spanish Stock Exchange ticker: ALM) closed on July 2016 a licensing agreement on the development and commercialization of tildrakizumab-asmn for psoriasis in Europe. Under the terms of the licensing agreement, Almirall is able to lead European studies, and participate in larger Global clinical studies for plaque psoriasis indication subject to the terms of the Sun Pharma – Merck & Co., Inc., Kenilworth, NJ, USA agreements, as well as certain cost sharing agreements. Sun Pharma will be eligible to receive development and regulatory milestone payments and, additionally, sales milestone payments and royalties on net sales. Sun Pharma will continue to lead development of tildrakizumab-asmn for other indications, where Almirall will have right of first negotiation for certain indications in Europe. The agreement between Sun Pharma and Almirall remains subject to the exclusive licensing agreement between Sun Pharma and Merck & Co., Inc., Kenilworth, NJ, USA.
About Sun Pharmaceutical Industries Ltd. (CIN – L24230GJ1993PLC019050)
Sun Pharma is the world’s fifth largest specialty generic pharmaceutical company and India’s top pharmaceutical company. A vertically integrated business, economies of scale and an extremely skilled team enable us to deliver quality products in a timely manner at affordable prices. It provides high-quality, affordable medicines trusted by customers and patients in over 150 countries across the world. Sun Pharma’s global presence is supported by 41 manufacturing facilities spread across 6 continents, R&D centres across the globe and a multi-cultural workforce comprising over 50 nationalities. In India, the company enjoys leadership across 11 different classes of doctors with 30 brands featuring amongst top 300 pharmaceutical brands in India. Its footprint across emerging markets covers over 100 markets and 6 markets in Western Europe. Its Global Consumer Healthcare business is ranked amongst Top 10 across 3 global markets. Its API business footprint is strengthened through 14 world class API manufacturing facilities across the globe. Sun Pharma fosters excellence through innovation supported by strong R&D capabilities comprising about 2,000 scientists and R&D investments of approximately 8% of annual revenues. For further information, please visit www.sunpharma.com & follow us on Twitter @SunPharma_Live.
References
1. National Psoriasis Foundation. Facts about psoriasis. www.psoriasis.org/sites/default/files/for-media/MediaKit.pdf. Accessed on February 22, 2018.
2. National Psoriasis Foundation. About Psoriasis. www.psoriasis.org/about-psoriasis. Accessed on February 22, 2018.
3. Menter A, Gottlieb A, Feldman SR, Van Voorhees AS et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol 2008 May; 58(5):826-50.
////////////////tildrakizumab-asmn, FDA 2018, MERCK, Schering-Plough, MONOCLONAL ANTIBODY, SCH 900222, MK-3222, Psoriasis, plaque, BLA 761067, SCH-900222, SUNPG 1622, SUNPG 1622 I, SUNPG 1623 I, SUNPG 1623 II, SUNPG 1623 III, SUNPG 1623 IV, SUNPG1623,
FDA approves Mylotarg (gemtuzumab ozogamicin) for treatment of acute myeloid leukemia
The U.S. Food and Drug Administration today approved Mylotarg (gemtuzumab ozogamicin) for the treatment of adults with newly diagnosed acute myeloid leukemia whose tumors express the CD33 antigen (CD33-positive AML). The FDA also approved Mylotarg for the treatment of patients aged 2 years and older with CD33-positive AML who have experienced a relapse or who have not responded to initial treatment (refractory).
Mylotarg originally received accelerated approval in May 2000 as a stand-alone treatment for older patients with CD33-positive AML who had experienced a relapse. Mylotarg was voluntarily withdrawn from the market after subsequent confirmatory trials failed to verify clinical benefit and demonstrated safety concerns, including a high number of early deaths. Today’s approval includes a lower recommended dose, a different schedule in combination with chemotherapy or on its own, and a new patient population.
“We are approving Mylotarg after a careful review of the new dosing regimen, which has shown that the benefits of this treatment outweigh the risk,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Mylotarg’s history underscores the importance of examining alternative dosing, scheduling, and administration of therapies for patients with cancer, especially in those who may be most vulnerable to the side effects of treatment.”
AML is a rapidly progressing cancer that forms in the bone marrow and results in an increased number of white blood cells in the bloodstream. The National Cancer Institute of the National Institutes of Health estimates that approximately 21,380 people will be diagnosed with AML this year and that 10,590 patients with AML will die of the disease.
Mylotarg is a targeted therapy that consists of an antibody connected to an anti-tumor agent that is toxic to cells. It is thought to work by taking the anti-tumor agent to the AML cells that express the CD33 antigen, blocking the growth of cancerous cells and causing cell death.
The safety and efficacy of Mylotarg in combination with chemotherapy for adults were studied in a trial of 271 patients with newly diagnosed CD33-positive AML who were randomized to receive Mylotarg in combination with daunorubicin and cytarabine or to receive daunorubicin and cytarabine without Mylotarg. The trial measured “event-free survival,” or how long patients went without certain complications, including failure to respond to treatment, disease relapse or death, from the date they started the trial. Patients who received Mylotarg in combination with chemotherapy went longer without complications than those who received chemotherapy alone (median, event-free survival 17.3 months vs. 9.5 months).
The safety and efficacy of Mylotarg as a stand-alone treatment were studied in two, separate trials. The first trial included 237 patients with newly diagnosed AML who could not tolerate or chose not to receive intensive chemotherapy. Patients were randomized to receive treatment with Mylotarg or best supportive care. The trial measured “overall survival,” or how long patients survived from the date they started the trial. Patients who received Mylotarg survived longer than those who received only best supportive care (median overall survival 4.9 months vs. 3.6 months). The second trial was a single-arm study that included 57 patients with CD33-positive AML who had experienced one relapse of disease. Patients received a single course of Mylotarg. The trial measured how many patients achieved a complete remission. Following treatment with Mylotarg, 26 percent of patients achieved a complete remission that lasted a median 11.6 months.
Common side effects of Mylotarg include fever (pyrexia), nausea, infection, vomiting, bleeding, low levels of platelets in the blood (thrombocytopenia), swelling and sores in the mouth (stomatitis), constipation, rash, headache, elevated liver function tests, and low levels of certain white blood cells (neutropenia). Severe side effects of Mylotarg include low blood counts, infections, liver damage, blockage of the veins in the liver (hepatic veno-occlusive disease), infusion-related reactions, and severe bleeding (hemorrhage). Women who are pregnant or breastfeeding should not take Mylotarg, because it may cause harm to a developing fetus or a newborn baby. Patients with hypersensitivity to Mylotarg or any component of its formulation should not use Mylotarg.
The prescribing information for Mylotarg includes a boxed warning that severe or fatal liver damage (hepatotoxicity), including blockage of veins in the liver (veno-occlusive disease or sinusoidal obstruction syndrome), occurred in some patients who took Mylotarg.
Mylotarg received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Mylotarg to Pfizer Inc.



| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from mouse) |
| Target | CD33 |
| Clinical data | |
| Trade names | Mylotarg |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a607075 |
| Pregnancy category |
|
| Routes of administration |
Intravenous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| DrugBank | |
| ChemSpider |
|
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Molar mass | 151–153 g/mol |
Gemtuzumab ozogamicin (marketed by Wyeth as Mylotarg) is a drug-linked monoclonal antibody (an antibody-drug conjugate) that was used to treat acute myelogenous leukemia from 2000 to 2010. It was withdrawn from market in June 2010 when a clinical trial showed the drug increased patient death and added no benefit over conventional cancer therapies.
Mechanism and side effects
Gemtuzumab is a monoclonal antibody to CD33 linked to a cytotoxic agent from the class of calicheamicins. CD33 is expressed in most leukemic blast cells but also in normal hematopoietic cells, the intensity diminishing with maturation of stem cells.
Common side effects of administration included shivering, fever, nausea and vomiting. Serious side effects included severe myelosuppression (suppressed activity of bone marrow, which is involved in formation of various blood cells [found in 98% of patients]), disorder of the respiratory system, tumor lysis syndrome, Type III hypersensitivity, venous occlusion, and death.
History
Gemtuzumab ozogamicin was created in a collaboration between Celltech and Wyeth that began in 1991.[1][2] The same collaboration later produced inotuzumab ozogamicin.[3] Celltech was acquired by UCB in 2004[4] and Wyeth was acquired by Pfizer in 2009.[5]
In the United States, it was approved under an accelerated-approval process by the FDA in 2000 for use in patients over the age of 60 with relapsed acute myelogenous leukemia (AML); or those who are not considered candidates for standard chemotherapy.[6] The accelerated approval was based on the surrogate endpoint of response rate.[7] It was the first antibody-drug conjugate to be approved.[8]
Within the first year after approval, the FDA required a black box warning be added to Gemtuzumab packaging. The drug was noted to increase the risk of veno-occlusive disease in the absence of bone marrow transplantation.[9] Later the onset of VOD was shown to occur at increased frequency in Gemtuzumab patients even following bone marrow transplantation.[10] The drug was discussed in a 2008 JAMA article, which criticized the inadequacy of postmarketing surveillance of biologic agents.[11]
A randomized phase 3 comparative controlled trial (SWOG S0106) was initiated in 2004 by Wyeth in accordance with the FDA accelerated-approval process. The study was stopped[when?] prior to completion due to worrisome outcomes. Among the patients evaluated for early toxicity, fatal toxicity rate was significantly higher in the gemtuzumab combination therapy group vs the standard therapy group. Mortality was 5.7% with gemtuzumab and 1.4% without the agent (16/283 = 5.7% vs 4/281 = 1.4%; P = .01).[7]
In June 2010, Pfizer withdrew Mylotarg from the market at the request of the US FDA.[12][13] However, some other regulatory authorities did not agree with the FDA decision, with Japan’s Pharmaceuticals and Medical Devices Agency stating in 2011 that the “risk-benefit balance of gemtuzumab ozogamicin has not changed from its state at the time of approval”.[14]
In early 2017 Pfizer reapplied for US and EU approval, based on a meta-analysis of prior trials and results of the ALFA-0701 clinical trial, an open-label Phase III trial in 280 older people with AML. [8]
References
- Jump up^ “Mylotarg”. Informa Biomedtracker. Retrieved 19 August 2017.
- Jump up^ Niculescu-Duvaz, I (December 2000). “Technology evaluation: gemtuzumab ozogamicin, Celltech Group.”. Current opinion in molecular therapeutics. 2 (6): 691–6. PMID 11249747.
- Jump up^ Damle, NK; Frost, P (August 2003). “Antibody-targeted chemotherapy with immunoconjugates of calicheamicin.”. Current opinion in pharmacology. 3 (4): 386–90. PMID 12901947. doi:10.1016/S1471-4892(03)00083-3.
- Jump up^ “Celltech sold to Belgian firm in £1.5bn deal”. The Guardian. 18 May 2004.
- Jump up^ Sorkin, Andrew Ross; Wilson, Duff (25 January 2009). “Pfizer Agrees to Pay $68 Billion for Rival Drug Maker Wyeth”. The New York Times.
- Jump up^ Bross PF, Beitz J, Chewn G, Chen XH, Duffy E, Kieffer L, Roy S, Sridhara R, Rahman A, Williams G, Pazdur R (2001). “Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia.”. Clin Cancer Res. 7 (6): 1490–6. PMID 11410481.
- ^ Jump up to:a b Gemtuzumab Voluntarily Withdrawn From US Market. June 2010
- ^ Jump up to:a b Stanton, Dan (February 1, 2017). “Pfizer resubmits US and EU application for withdrawn ADC Mylotarg”. BioPharma Reporter.
- Jump up^ Giles FJ, Kantarjian HM, Kornblau SM, Thomas DA, Garcia-Manero G, Waddelow TA, David CL, Phan AT, Colburn DE, Rashid A, Estey EH (2001). “Mylotarg (gemtuzumab ozogamicin) therapy is associated with hepatic venoocclusive disease in patients who have not received stem cell transplantation.”. Cancer. 92 (2): 406–13. PMID 11466696. doi:10.1002/1097-0142(20010715)92:2<406::AID-CNCR1336>3.0.CO;2-U.
- Jump up^ Wadleigh M, Richardson PG, Zahrieh D, Lee SJ, Cutler C, Ho V, Alyea EP, Antin JH, Stone RM, Soiffer RJ, DeAngelo DJ (2003). “Prior gemtuzumab ozogamicin exposure significantly increases the risk of veno-occlusive disease in patients who undergo myeloablative allogeneic stem cell transplantation.”. Blood. 102 (5): 1578–82. PMID 12738663. doi:10.1182/blood-2003-01-0255.
- Jump up^ The Research on Adverse Drug Events and Reports (RADAR) Project, JAMA
- Jump up^ Mylotarg (gemtuzumab ozogamicin): Market Withdrawal, US FDA
- Jump up^ Pfizer pulls leukemia drug from U.S. market, Reuters
- Jump up^ Pharmaceuticals and Medical Devices Safety Information, No. 277, February 2011 (PDF) (Technical report). Pharmaceuticals and Medical Devices Agency of Japan. 2011.
FDA approval brings first gene therapy to the United States

The U.S. Food and Drug Administration issued a historic action today making the first gene therapy available in the United States, ushering in a new approach to the treatment of cancer and other serious and life-threatening diseases.
The FDA approved Kymriah (tisagenlecleucel) for certain pediatric and young adult patients with a form of acute lymphoblastic leukemia (ALL).
“We’re entering a new frontier in medical innovation with the ability to reprogram a patient’s own cells to attack a deadly cancer,” said FDA Commissioner Scott Gottlieb, M.D. “New technologies such as gene and cell therapies hold out the potential to transform medicine and create an inflection point in our ability to treat and even cure many intractable illnesses. At the FDA, we’re committed to helping expedite the development and review of groundbreaking treatments that have the potential to be life-saving.”
Kymriah, a cell-based gene therapy, is approved in the United States for the treatment of patients up to 25 years of age with B-cell precursor ALL that is refractory or in second or later relapse.
Kymriah is a genetically-modified autologous T-cell immunotherapy. Each dose of Kymriah is a customized treatment created using an individual patient’s own T-cells, a type of white blood cell known as a lymphocyte. The patient’s T-cells are collected and sent to a manufacturing center where they are genetically modified to include a new gene that contains a specific protein (a chimeric antigen receptor or CAR) that directs the T-cells to target and kill leukemia cells that have a specific antigen (CD19) on the surface. Once the cells are modified, they are infused back into the patient to kill the cancer cells.
ALL is a cancer of the bone marrow and blood, in which the body makes abnormal lymphocytes. The disease progresses quickly and is the most common childhood cancer in the U.S. The National Cancer Institute estimates that approximately 3,100 patients aged 20 and younger are diagnosed with ALL each year. ALL can be of either T- or B-cell origin, with B-cell the most common. Kymriah is approved for use in pediatric and young adult patients with B-cell ALL and is intended for patients whose cancer has not responded to or has returned after initial treatment, which occurs in an estimated 15-20 percent of patients.
“Kymriah is a first-of-its-kind treatment approach that fills an important unmet need for children and young adults with this serious disease,” said Peter Marks, M.D., Ph.D., director of the FDA’s Center for Biologics Evaluation and Research (CBER). “Not only does Kymriah provide these patients with a new treatment option where very limited options existed, but a treatment option that has shown promising remission and survival rates in clinical trials.”
The safety and efficacy of Kymriah were demonstrated in one multicenter clinical trial of 63 pediatric and young adult patients with relapsed or refractory B-cell precursor ALL. The overall remission rate within three months of treatment was 83 percent.
Treatment with Kymriah has the potential to cause severe side effects. It carries a boxed warning for cytokine release syndrome (CRS), which is a systemic response to the activation and proliferation of CAR T-cells causing high fever and flu-like symptoms, and for neurological events. Both CRS and neurological events can be life-threatening. Other severe side effects of Kymriah include serious infections, low blood pressure (hypotension), acute kidney injury, fever, and decreased oxygen (hypoxia). Most symptoms appear within one to 22 days following infusion of Kymriah. Since the CD19 antigen is also present on normal B-cells, and Kymriah will also destroy those normal B cells that produce antibodies, there may be an increased risk of infections for a prolonged period of time.
The FDA today also expanded the approval of Actemra (tocilizumab) to treat CAR T-cell-induced severe or life-threatening CRS in patients 2 years of age or older. In clinical trials in patients treated with CAR-T cells, 69 percent of patients had complete resolution of CRS within two weeks following one or two doses of Actemra.
Because of the risk of CRS and neurological events, Kymriah is being approved with a risk evaluation and mitigation strategy (REMS), which includes elements to assure safe use (ETASU). The FDA is requiring that hospitals and their associated clinics that dispense Kymriah be specially certified. As part of that certification, staff involved in the prescribing, dispensing, or administering of Kymriah are required to be trained to recognize and manage CRS and neurological events. Additionally, the certified health care settings are required to have protocols in place to ensure that Kymriah is only given to patients after verifying that tocilizumab is available for immediate administration. The REMS program specifies that patients be informed of the signs and symptoms of CRS and neurological toxicities following infusion – and of the importance of promptly returning to the treatment site if they develop fever or other adverse reactions after receiving treatment with Kymriah.
To further evaluate the long-term safety, Novartis is also required to conduct a post-marketing observational study involving patients treated with Kymriah.
The FDA granted Kymriah Priority Review and Breakthrough Therapy designations. The Kymriah application was reviewed using a coordinated, cross-agency approach. The clinical review was coordinated by the FDA’s Oncology Center of Excellence, while CBER conducted all other aspects of review and made the final product approval determination.
The FDA granted approval of Kymriah to Novartis Pharmaceuticals Corp. The FDA granted the expanded approval of Actemra to Genentech Inc.
/////////////Kymriah, Novartis Pharmaceuticals Corp, Actemra, Genentech Inc., gene therapy, fda 2017
FDA approves Amjevita, a biosimilar to Humira

FDA approves Amjevita, a biosimilar to Humira
The U.S. Food and Drug Administration today approved Amjevita (adalimumab-atto) as a biosimilar toHumira (adalimumab) for multiple inflammatory diseases.
FDA approves Amjevita, a biosimilar to Humira
For Immediate Release
September 23, 2016
Release
The U.S. Food and Drug Administration today approved Amjevita (adalimumab-atto) as a biosimilar to Humira (adalimumab) for multiple inflammatory diseases.
Amjevita is approved for the following indications in adult patients:
- moderately to severely active rheumatoid arthritis;
- active psoriatic arthritis;
- active ankylosing spondylitis (an arthritis that affects the spine);
- moderately to severely active Crohn’s disease;
- moderately to severely active ulcerative colitis; and
- moderate to severe plaque psoriasis.
Amjevita is also indicated for moderately to severely active polyarticular juvenile idiopathic arthritis in patients four years of age and older.
Health care professionals should review the prescribing information in the labeling for detailed information about the approved uses.
“This is the fourth FDA-approved biosimilar. The biosimilar pathway is still a new frontier and one that we expect will enhance access to treatment for patients with serious medical conditions,” said Janet Woodcock, M.D., director of the FDA’s Center for Drug Evaluation and Research.
Biological products are generally derived from a living organism and can come from many sources, including humans, animals, microorganisms or yeast. A biosimilar is a biological product that is approved based on a showing that it is highly similar to an already-approved biological product and has no clinically meaningful differences in terms of safety, purity and potency (i.e., safety and effectiveness) from the reference product, in addition to meeting other criteria specified by law.
The FDA’s approval of Amjevita is based on review of evidence that included structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamics data, clinical immunogenicity data and other clinical safety and effectiveness data that demonstrates Amjevita is biosimilar to Humira. It has been approved as a biosimilar, not as an interchangeableproduct.
The most serious known side effects with Amjevita are infections and malignancies. The most common expected adverse reactions with Amjevita are infections and injection site reactions.
Like Humira, the labeling for Amjevita contains a Boxed Warning to alert health care professionals and patients about an increased risk of serious infections leading to hospitalization or death. The Boxed Warning also notes that lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with tumor necrosis factor blockers, including adalimumab products. The drug must be dispensed with a patient Medication Guide that describes important information about its uses and risks.
Amjevita is manufactured by Amgen, Inc., of Thousand Oaks, California. Humira was approved in December 2002 and is manufactured by AbbVie Inc. of North Chicago, Illinois.
| Adalimumab | ||
 |
||
| Farmaceutische gegevens | ||
| t1/2 | 10–20 dagen | |
| Databanken | ||
| CAS-nummer | 331731-18-1 | |
| ATC-code | L04AB04 | |
| DrugBank | BTD00049 | |
| Farmacotherapeutisch Kompas | Adalimumab | |
| Chemische gegevens | ||
| Molaire massa | 144190.3 g/mol | |
///////FDA, Amjevita, biosimilar, Humira, FDA 2016
Perspectives on Anti-Glycan Antibodies Gleaned from Development of a Community Resource Database

Antibodies are used extensively for a wide range of basic research and clinical applications. While an abundant and diverse collection of antibodies to protein antigens have been developed, good monoclonal antibodies to carbohydrates are much less common. Moreover, it can be difficult to determine if a particular antibody has the appropriate specificity, which antibody is best suited for a given application, and where to obtain that antibody. Herein, we provide an overview of the current state of the field, discuss challenges for selecting and using antiglycan antibodies, and summarize deficiencies in the existing repertoire of antiglycan antibodies. This perspective was enabled by collecting information from publications, databases, and commercial entities and assembling it into a single database, referred to as the Database of Anti-Glycan Reagents (DAGR). DAGR is a publicly available, comprehensive resource for anticarbohydrate antibodies, their applications, availability, and quality
Monoclonal antibodies have transformed biomedical research and clinical care. In basic research, these proteins are used widely for a myriad of applications, such as monitoring/detecting expression of biomolecules in tissue samples, activating or antagonizing various biological pathways, and purifying antigens. To illustrate the magnitude and importance of the antibody reagent market, one commercial supplier sells over 50 000 unique monoclonal antibody clones. In a clinical setting, antibodies are used frequently as therapeutic agents and for diagnostic applications. As a result, monoclonal antibodies are a multibillion dollar industry, with antibody therapeutics estimated at greater than $40 billion annually, diagnostics at roughly $8 billion annually, and antibody reagents at $2 billion annually as of 2012
Perspectives on Anti-Glycan Antibodies Gleaned from Development of a Community Resource Database
ACS Editors’ Choice – This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes.
http://pubs.acs.org/doi/full/10.1021/acschembio.6b00244

Jeffrey C. Gildersleeve, Ph.D.
The Gildersleeve group works at the interface of chemistry, glycobiology, and immunology. We use chemical approaches to 1) aid the design and development of cancer and HIV vaccines, 2) identify clinically useful biomarkers, and 3) better understand the roles of carbohydrates in cancer and HIV immunology. To facilitate these studies, we have developed a glycan microarray that allows high-throughput profiling of serum anti-glycan antibody populations.
Link to additional information about Dr. Gildersleeve’s research.
Areas of Expertise
Contact Info
Center for Cancer Research
National Cancer Institute
Building 376, Room 208
Frederick, MD 21702-1201
Ph: 301-846-5699
gildersj@mail.nih.gov (link sends e-mail)
The Gildersleeve group works at the interface of chemistry, glycobiology, and immunology. We use chemical approaches to 1) aid the design and development of cancer and HIV vaccines, 2) identify clinically useful biomarkers, and 3) better understand the roles of carbohydrates in cancer and HIV immunology. To facilitate these studies, we have developed a glycan microarray that allows high-throughput profiling of serum anti-glycan antibody populations. A number of other groups have also developed glycan arrays; our array is unique in that we use multivalent neoglycoproteins as our array components. This format allows us to readily translate array results to other applications and affords novel approaches to vary glycan presentation.
The main focus of our current and future research is to study the roles of anti-glycan antibodies in the development, progression, and treatment of cancer. These projects are shedding new light on how cancer vaccines work and are uncovering new biomarkers for the early detection, diagnosis, and prognosis of cancer. In particular, we are studying immune responses induced by PROSTVAC-VF, a cancer vaccine in Phase III clinical trials for the treatment of advanced prostate cancer. In addition, we are identifying biomarkers for the early detection and prognosis of ovarian and lung cancer. These projects are highly collaborative in nature and are focused on translating basic research from the bench to the clinic. We rely heavily on glycan array technology to study immune responses to carbohydrates, and we continually strive to improve this technology. First, carbohydrate-protein interactions often involve formation of multivalent complexes. Therefore, presentation is a key feature of recognition. We have developed several new approaches to vary carbohydrate presentation on the surface of the array, including methods to vary glycan density and neoglycoprotein density. Second, we use synthetic organic chemistry to obtain a diverse set of tumor-associated carbohydrates and glycopeptides to populate our array.
Collaborations and Carbohydrate Microarray Screening. We are frequently asked to screen lectins, antibodies, and other entities on our array. Although we are not a core facility and do not provide screening services per se, we are happy to collaborate on many projects. Please contact Jeff Gildersleeve for more details.
Scientific Focus Areas:

a small clip
Dr. Eric Sterner, a postdoctoral CRTA Fellow in the Gilderlseeve Lab was presented with a FARE award for his abstract entitled, “Profiling Mutational Significance in Germline-to-Affinity Mature 3F8 Variants” in the NIH-wide FARE 2016 competition. This award is given to abstracts that are deemed outstanding based on scientific merit, originality, experimental design and overall quality and presentation. FARE 2016 is sponsored by the NIH Scientific Directors, the Office of Intramural Training & Education and FelCom. The FARE 2016 Award is a $1000 travel grant to attend and present this work at a scientific meeting within the United States.

Natalie Flanagan
Postbaccalaureate Fellow – Cancer Research Training Award (CRTA) at National Cancer Institute (NCI)
https://www.linkedin.com/in/natalie-flanagan-602a98109
Experience

Postbaccalaureate Fellow – Cancer Research Training Award (CRTA)
National Cancer Institute (NCI)
June 2015 – Present (1 year 1 month)Frederick, Maryland

Organic Chemistry Lab TA
University of Maryland
September 2014 – May 2015 (9 months)College Park, Maryland
– Ran on section of the Organic Chemistry I laboratory course for two semesters
– Worked with students in a laboratory setting and office hours to help them understand course materials and experimental procedures
– Worked with professors and other TAs to help develop and grade examinations

Summer Intern
Pfizer
June 2013 – August 2013 (3 months)Groton, Connecticut
– Used protein crystallization to research ligand binding in a protein kinase system
– Learned a variety of laboratory techniques, including: expression and purification of proteins, and various protein crystallization techniques
– Gained a basic knowledge for how to interpret electron density maps used in three-dimensional protein structure determination
– Presented my research project at an internal poster presentation
Education

University of Maryland College Park
Bachelor’s Degree, Cell Biology and Genetics
2011 – 2015
Activities and Societies: Organic Chemistry Tutor – Primannum Honor Society, Chemistry TA, Phi Beta Kappa, Clinical Volunteer – HSC Pediatric Center
Ledyard High School
High School, General Studies
2007 – 2011
//////////Anti-Glycan Antibodies, Gleaned, Community Resource Database

{kind=link}
{kind=link}