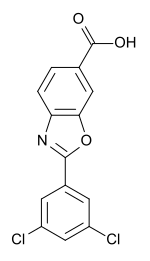
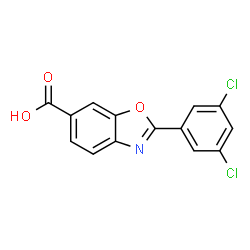
Tafamidis
- Molecular Formula C14H7Cl2NO3
- Average mass 308.116 Da
TAFAMIDIS, Fx-1006A
PF-06291826
2-(3,5-Dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid
6-Benzoxazolecarboxylic acid, 2-(3,5-dichlorophenyl)-
Vyndaqel
Tafamidis meglumine
Familial amyloid polyneuropathy LAUNCHED PFIZER 2011 EU
PHASE 3, at FDA, Amyloidosis, PFIZER
Tafamidis (INN, or Fx-1006A,[1] trade name Vyndaqel) is a drug for the amelioration of transthyretin-related hereditary amyloidosis(also familial amyloid polyneuropathy, or FAP), a rare but deadly neurodegenerative disease.[2][3] The drug was approved by the European Medicines Agency in November 2011 and by the Japanese Pharmaceuticals and Medical Devices Agency in September 2013.[4]
In 2011 and 2012, orphan drug designation was assigned in Japan and the U.S., respectively, for the treatment of transthyretin amyloid polyneuropathy. This designation was assigned in the E.U. in 2012 for the treatment of senile systemic amyloidosis. In 2017, fast drug designation was assigned in the U.S. for the treatment of transthyretin cardiomyopathy.
Tafamidis is a novel specific transthyretin (TTR) stabilizer or dissociation inhibitor. TTR is a tetramer that is responsible in transporting the retinol-binding protein-vitamin A complex and minimally transporting thyroxine in the blood. In TTR-related disorders such as transthyretin familial amyloid polyneuropathy (TTR-FAP), tetramer dissociation is accelerated that results in unregulated amyloidogenesis and amyloid fibril formation. Eventually the failure of autonomic and peripheral nervous system is induced. Tafamidiswas approved by the European Medicines Agency (EMA) in 2011 under the market name Vyndaqel for the treatment of transthyretin familial amyloid polyneuropathy (TTR-FAP) in adult patients with early-stage symptomatic polyneuropathy to delay peripheral neurologic impairment. Tafamidis is an investigational drug under the FDA and in June 2017, Pfizer received FDA Fast Track Designation for tafamidis

The marketed drug, a meglumine salt, has completed an 18 month placebo controlled phase II/III clinical trial,[5][6] and an 12 month extension study[7] which provides evidence that tafamidis slows progression of Familial amyloid polyneuropathy.[8] Tafamidis (20 mg once daily) is used in adult patients with an early stage (stage 1) of familial amyloidotic polyneuropathy.[9][10]
Tafamidis was discovered in the Jeffery W. Kelly Laboratory at The Scripps Research Institute[11] using a structure-based drug design strategy[12] and was developed at FoldRx pharmaceuticals, a biotechnology company Kelly co-founded with Susan Lindquist. FoldRx was led by Richard Labaudiniere when it was acquired by Pfizer in 2010.
Tafamidis functions by kinetic stabilization of the correctly folded tetrameric form of the transthyretin (TTR) protein.[13] In patients with FAP, this protein dissociates in a process that is rate limiting for aggregation including amyloid fibril formation, causing failure of the autonomic nervous system and/or the peripheral nervous system (neurodegeneration) initially and later failure of the heart. Kinetic Stabilization of tetrameric transthyretin in familial amyloid polyneuropathy patients provides the first pharmacologic evidence that the process of amyloid fibril formation causes this disease, as treatment with tafamidis dramatically slows the process of amyloid fibril formation and the degeneration of post-mitotic tissue. Sixty % of the patients enrolled in the initial clinical trial have the same or an improved neurologic impairment score after six years of taking tafamidis, whereas 30% of the patients progress at a rate ≤ 1/5 of that predicted by the natural history. Importantly, all of the V30M FAP patients remain stage 1 patients after 6 years on tafamidis out of four stages of disease progression. [Data presented orally by Professor Coelho in Brazil in 2013][7]
The process of wild type transthyretin amyloidogenesis also appears to cause wild-type transthyretin amyloidosis (WTTA), also known as senile systemic amyloidosis (SSA), leading to cardiomyopathy as the prominent phenotype.[14] Some mutants of transthyretin — including V122I, which is primarily found in individuals of African descent — are destabilizing, enabling heterotetramer dissociation, monomer misfolding, and subsequent misassembly of transthyretin into a variety of aggregate structures [15] including amyloid fibrils[16]leading to familial amyloid cardiomyopathy.[17] While there is clinical evidence from a small number of patients that tafamidis slows the progression of the transthyretin cardiomyopathies,[18] this has yet to be demonstrated in a placebo-controlled clinical trial. Pfizer has enrolled a placebo-controlled clinical trial to evaluate the ability of tafamidis to slow the progression of both familial amyloid cardiomyopathy and senile systemic amyloidosis (ClinicalTrials.gov identifier: NCT01994889).
Regulatory Process
Tafamidis was approved for use in the European Union by the European Medicines Agency in November 2011, specifically for the treatment of early stage transthyretin-related hereditary amyloidosis or familial amyloid polyneuropathy or FAP (all mutations). In September 2013 Tafamidis was approved for use in Japan by the Pharmaceuticals and Medical Devices Agency, specifically for the treatment of transthyretin-related hereditary amyloidosis or familial amyloid polyneuropathy or FAP (all mutations). Tafamidis is also approved for use in Brazil, Argentina, Mexico and Israel by the relevant authorities.[19] It is currently being considered for approval by the United States Food and Drug Administration (FDA) for the treatment of early stage transthyretin-related hereditary amyloidosis or familial amyloid polyneuropathy or FAP.
In June 2012, the FDA Peripheral and Central Nervous System Drugs Advisory Committee voted “yes” (13-4 favorable vote) when asked if the findings of the pivotal clinical study with tafamidis were “sufficiently robust to provide substantial evidence of efficacy for a surrogate endpoint that is reasonably likely to predict a clinical benefit”. The Advisory Committee voted “no” 4-13 to reject the drug–in spite of the fact that both primary endpoints were met in the efficacy evaluable population (n=87) and were just missed in the intent to treat population (n=125), apparently because more patients than expected in the intent to treat population were selected for liver transplantation during the course of the trial, not owing to treatment failure, but because their name rose to the top of the transplant list. However, these patients were classified as treatment failures in the conservative analysis used.
Pfizer (following its acquisition of FoldRx ), under license from Scripps Research Institute , has developed and launched tafamidis, a small-molecule transthyretin stabilizer, useful for treating familial amyloid polyneuropathy.
SYN
European Journal of Medicinal Chemistry, 121, 823-840; 2016

SYN 2

INNOVATORS
THE SCRIPPS RESEARCH INSTITUTE [US/US]; 10550 N Torrey Pines Road, La Jolla, CA 92037 (US)
KELLY, Jeffrey, W.; (US).
SEKIJIMA, Yoshiki; (US)


Dr. Jeffery W. Kelly
Lita Annenberg Hazen Professor of Chemistry
Co-Chairman, Department of Molecular Medicine
Click here to download a concise version of Dr. Jeffery Kelly’s curriculum vitae.

PATENT
WO2004056315
Example 5: Benzoxazoles as Transthyretin Amyloid Fibril Inhibitors
Transthyretin’s two thyroxine binding sites are created by its quaternary structural interface. The tetramer can be stabilized by small molecule binding to these sites, potentially providing a means to treat TTR amyloid disease with small molecule drugs. Many families of compounds have been discovered whose binding stabilizes the tetrameric ground state to a degree proportional to the small molecule dissociation constants Km and Ka2. This also effectively increases the dissociative activation barrier and inhibits amyloidosis by kinetic stabilization. Such inhibitors are typically composed of two aromatic rings, with one ring bearing halogen substituents and the other bearing hydrophilic substituents. Benzoxazoles substituted with a carboxylic acid at C(4)-C(7) and a halogenated phenyl ring at C(2) also appeared to complement the TTR thyroxine binding site. A small library of these compounds was therefore prepared by dehydrocyclization of N-acyl amino-hydroxybenzoic acids as illustrated in Scheme 1.

Scheme 1: General Synthesis of Benzoxazoles
Reagents: (a) ArCOCl, THF, pyridine (Ar = Phenyl, 3,5-Difluorophenyl, 2,6-Difluorophenyl, 3,5-Dichlorophenyl, 2,6-Dichlorophenyl, 2-(Trifluoromethyl)phenyl, and 3-(Trifluoromethyl)phenyl); (b) TsOH*H2O, refluxing xylenes; (c) TMSCHN2, benzene, MeOH; (d) LiOH, THF, MeOH, H2O (8-27% yield over 4 steps).
The benzoxazoles were evaluated using a series of analyses of increasing stringency. WT TTR (3.6 μM) was incubated for 30 min (pH 7, 37 °C) with a test compound (7.2 μM). Since at least one molecule ofthe test compound must bind to each molecule of TTR tetramer to be able to stabilize it, a test compound concentration of 7.2 μM is only twice the minimum effective concentration. The pH was then adjusted to 4.4, the optimal pH for fibrilization. The amount of amyloid formed after 72 h (37 °C) in the presence ofthe test compound was determined by turbidity at 400 nm and is expressed as % fibril formation (ff), 100%) being the amount formed by TTR alone. Ofthe 28 compounds tested, 11 reduced fibril formation to negligible levels (jf< 10%; FIG. 7).
The 11 most active compounds were then evaluated for their ability to bind selectively to TTR over, all other proteins in blood. Human blood plasma (TTR cone. 3.6 -5.4 μM) was incubated for 24 h with the test compound (10.8 μM) at 37 °C. The TTR and any bound inhibitor were immunoprecipitated using a sepharose-bound polyclonal TTR antibody. The TTR with or without inhibitor bound was liberated from the resin at high pH, and the inhibitor: TTR stoichiometry was ascertained by HPLC analysis (FIG. 8). Benzoxazoles with carboxylic acids in the 5- or 6-position, and 2,6-dichlorophenyl (13, 20) or 2-trifluoromethylphenyl (11, 18) substituents at the 2-position displayed the highest binding stoichiometries. In particular, 20 exhibited excellent inhibitory activity and binding selectivity. Hence, its mechanism of action was characterized further.
To confirm that 20 inhibits TTR fibril formation by binding strongly to the tetramer, isothermal titration calorimetry (ITC) and sedimentation velocity experiments were conducted with wt TTR. ITC showed that two equivalents of 20 bind with average dissociation constants of Kdi = Kd2 = 55 (± 10) nM under physiological conditions. These are comparable to the dissociation constants of many other highly efficacious TTR
amyloidogenesis inhibitors. For the sedimentation velocity experiments, TTR (3.6 μM) was incubated with 20 (3.6 μM, 7.2 μM, 36 μM) under optimal fibrilization conditions (72 h, pH 4.4, 37 °C). The tetramer (55 kDa) was the only detectable species in solution with 20 at 7.2 or 36 μM. Some large aggregates formed with 20 at 3.6 μM, but the TTR remaining in solution was tetrameric.
T119M subunit inclusion and small molecule binding both prevent TTR amyloid formation by raising the activation barrier for tetramer dissociation. An inhibitor’s ability to do this is most rigorously tested by measuring its efficacy at slowing tetramer dissociation in 6 M urea, a severe denaturation stress. Thus, the rates of TTR tetramer dissociation in 6 M urea in the presence and absence of 20, 21 or 27 were compared (FIG. 9). TTR (1.8 μM) was completely denatured after 168 h in 6 M urea. In contrast, 20 at 3.6 μM prevented tetramer dissociation for at least 168 h (> 3 the half-life of TTR in human plasma). With an equimolar amount of 20, only 27% of TTR denatured in 168 h. Compound 27 (3.6 μM) was much less able to prevent tetramer dissociation (90% unfolding after 168 h), even though it was active in the fibril formation assay. Compound 21 did not hinder the dissociation of TTR at all. These results show that inhibitor binding to TTR is necessary but not sufficient to kinetically stabilize the TTR tetramer under strongly denaturing conditions; it is also important that the dissociation constants be very low (or that the off rates be very slow). Also, the display of functional groups on 20 is apparently optimal for stabilizing the TTR tetramer; moving the carboxylic acid from C(6) to C(7), as in 27, or removing the chlorines, as in 21, severely diminishes its activity.
The role ofthe substituents in 20 is evident from its co-crystal stracture with TTR (FIG. 10). Compound 20 orients its two chlorine atoms near halogen binding pockets 2 and 2′ (so-called because they are occupied by iodines when thyroxine binds to TTR). The 2,6 substitution pattern on the phenyl ring forces the benzoxazole and phenyl rings out of planarity, optimally positioning the carboxylic acid on the benzoxazole to hydrogen bond to the ε-NH3+ groups of Lys 15/15′. Hydrophobic interactions between the aromatic rings of 20 and the side chains of Leu 17, Leu 110, Ser 117, and Val 121 contribute additional binding energy.
PAPER
ChemMedChem (2013), 8(10), 1617-1619.
Nature Reviews Drug Discovery (2012), 11(3), 185-186
PAPER
Design and synthesis of pyrimidinone and pyrimidinedione inhibitors of dipeptidyl peptidase IV
J Med Chem 2011, 54(2): 510
PATENT
WO-2017190682
Novel crystalline forms of tafamidis methylglucamine (designated as Form E), processes for their preparation and compositions comprising them are claimed. Also claimed is their use for treating familial amyloid neuropathy. Represents first PCT filing from Crystal Pharmatech and the inventors on this API.
https://patentscope.wipo.int/search/en/detail.jsf;jsessionid=2C2DC88BD4DC90B179C38EC5283D0941.wapp2nA?docId=WO2017190682&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=FullText
CLIP
http://pubs.rsc.org/en/content/articlelanding/2016/ob/c5ob02496j/unauth#!divAbstract

2-(3, 5-Dichlorophenyl)benzo[d]oxazole-6-carboxylic acid (Tafamidis)
m.p. = 200.4–202.7 °C; Rf = 0.37 (petroleum ether/ethyl acetate/acetic acid = 6:1:0.01).
IR (cm-1 , KBr): 3383, 1685, 1608, 1224, 769;
1H NMR (DMSO-d6, 400 MHz) (ppm) 8.27 (s, 1H), 8.18 (d, J = 6.8 Hz, 1H), 8.04–8.02 (m, 1H), 7.94 (s, 1H), 7.88 (d, J = 1.6 Hz, 1H), 7.67 (dd, J = 6.8 Hz, 5.2 Hz, 1H);
13C NMR (DMSOd6, 100 MHz) (ppm) 167.2, 162.1, 150.1, 145.0, 137.8, 133.7, 131.4, 128.6, 126.8, 124.3, 120.5, 112.6.
Data was consistent with that reported in the literature. [27]Yamamoto, T.; Muto, K.; Komiyama, M.; Canivet, J.; Yamaguchi, J.; Itami, K. Chem. Eur. J. 2011, 17, 10113.
Clip
http://synth.chem.nagoya-u.ac.jp/wordpress/publication/nicatalystscopemechanism?lang=en

CLIP
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3386102/

The transthyretin amyloidoses (ATTR) are invariably fatal diseases characterized by progressive neuropathy and/or cardiomyopathy. ATTR are caused by aggregation of transthyretin (TTR), a natively tetrameric protein involved in the transport of thyroxine and the vitamin A–retinol-binding protein complex. Mutations within TTR that cause autosomal dominant forms of disease facilitate tetramer dissociation, monomer misfolding, and aggregation, although wild-type TTR can also form amyloid fibrils in elderly patients. Because tetramer dissociation is the rate-limiting step in TTR amyloidogenesis, targeted therapies have focused on small molecules that kinetically stabilize the tetramer, inhibiting TTR amyloid fibril formation. One such compound, tafamidis meglumine (Fx-1006A), has recently completed Phase II/III trials for the treatment of Transthyretin Type Familial Amyloid Polyneuropathy (TTR-FAP) and demonstrated a slowing of disease progression in patients heterozygous for the V30M TTR mutation. Herein we describe the molecular and structural basis of TTR tetramer stabilization by tafamidis. Tafamidis binds selectively and with negative cooperativity (Kds ∼2 nM and ∼200 nM) to the two normally unoccupied thyroxine-binding sites of the tetramer, and kinetically stabilizes TTR. Patient-derived amyloidogenic variants of TTR, including kinetically and thermodynamically less stable mutants, are also stabilized by tafamidis binding. The crystal structure of tafamidis-bound TTR suggests that binding stabilizes the weaker dimer-dimer interface against dissociation, the rate-limiting step of amyloidogenesis.
4-Amino-3-hydroxybenzoic acid (AHBA) is reacted with HCl (3 to 6 M equivalents) in methanol (8 to 9 L/kg). Methyl t-butyl ether (TBME) (9 to 11 L/kg) is then added to the reaction mixture. The product, methyl 4-amino-3-hydroxybenzoate hydrochloride salt, is isolated by filtration and then reacted with 3,5-dichlorobenzoyl chloride (0.95 to 1.05 M equivalents) in the presence of pyridine (2.0 to 2.5 M equivalents) in dichloromethane (DCM), (8 to 9 L/kg) as a solvent. After the distillation of DCM, acetone and water are added to the reaction mixture, producing methyl 4-(3,5-dichlorobenzoylamino)-3- hydroxy-benzoate. This is recovered by filtration and reacted with p-toluenesulfonic acid monohydrate (0.149 to 0.151 M equivalents) in toluene (12 to 18 L/kg) at reflux with water trap. Treatment with charcoal is then performed. After the distillation of toluene, acetone (4-6 L/kg) is added. The product, methyl 2-(3,5-dichlorophenyl)-benzoxazole-6- carboxylate, is isolated by filtration and then reacted with LiOH (1.25 to 1.29 M equivalents) in the presence of tetrahydrofuran (THF) (7.8 to 8.2 L/kg) and water (7.8 to 8.2 L/kg) at between 40 and 45 °C. The pH of the reaction mixture is adjusted with aqueous HCl to yield 2-(3,5-dichloro-phenyl)-benzoxazole-6-carboxylic acid, the free acid of tafamidis. This is converted to the meglumine salt by reacting with N-methyl-Dglucamine (0.95 to 1.05 M equivalents) in a mixture of water (4.95 to 5.05 L/kg)/isopropyl alcohol (19.75 to 20.25 L/kg) at 65-70 °C. Tafamidis meglumine (dglucitol, 1-deoxy-1-(methylamino)-,2-(3,5-dichlorophenyl)-6-benzoxazole carboxylate) is then isolated by filtration.
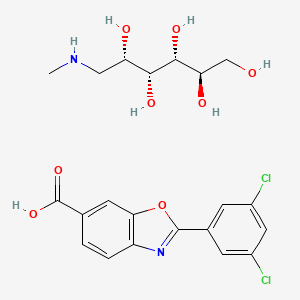
2 The following fragments were identified from electrospray ionization mass spectra acquired in positive-ion mode: meglumine M+ (C7H18NO5+, m/z = 196.13), M (carboxylate form) +2H (C14H6Cl2NO3, m/z = 308.13), M (salt) + H (C21H24Cl2N2O8, m/z = 504.26). 1 H-nuclear magnetic resonance spectra were acquired on a 700 MHz Bruker AVANCE II spectrometer in acetone:D2O (~8:2). Data were reported as chemical shift in ppm (δ), multiplicity (s = singlet, dd = double of doublets, m = multiplet), coupling constant (J Hz), relative integral and assignment: δ = 8.14 (m, JH2-H5 = 0.6 and JH2-H6 = 1.5, 1H, H2), 8.02 (dd, JH9-H11 = 1.9 and JH13-H11 = 1.9, 2H, H9 and H13), 7.97 (dd, JH6-H5 = 8.25, 1H, H6), 7.67 (dd, JH5-H2 = 0.6 and JH5-H6 = 8.25, 1H, H5), 7.58 (m, JH11-H9 = 1.9 and JH11-H13 = 1.9, 1H, H11), 4.08 (m, JH16-H17 = 4.9, 1H, H16), 3.79 (dd, JH17-H18 = 2.2, 1H, H17), 3.73 (dd, JH19-H20 = 3.2, 1H, H20), 3.69 (m, JH19-H20 = 3.2, 1H, H19), 3.61 (m, JH18-H19 = 12.25, 1H, H18), 3.58 (m, JH19-H20′ = 5.8 and JH20-H20′ = 11.7, 1H, H20′ ), 3.19 (m, JH15-H15′ = 12.9 and JH15′-H16 = 9.25 and JH15-H16 = 3.5, 2H, H15).
CLIP
http://onlinelibrary.wiley.com/store/10.1002/chem.201101091/asset/supinfo/chem_201101091_sm_miscellaneous_information.pdf?v=1&s=7badb204a12057710743c1711a744253eccd636a
Concise Synthesis of Tafamidis (Scheme 8)
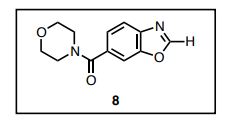
4-(6-Benzoxazoyl)morpholine (8)

A mixture of 4-amino-3-hydroxybenzoic acid (1.53 g, 10 mmol) and trimethyl orthofomate (3 mL) was heated at 100 ºC for 5 h. After cooling to room temperature, trimethyl orthofomate was removed under reduced pressure. To a solution of benzoxazole 6-carboxylic acid in CH2Cl2 (10 mL) were added DMF (0.1 mL) and oxalyl chloride (1.8 mL, 20 mmol) and the resultant mixture was stirred at room temperature for 12 h. After cooling to room temperature, DMF and oxalyl chloride were removed under reduced pressure to yield the corresponding acid chloride as a solid. Thus-generated acid chloride and morpholine (2.2 mL) were stirred at room temperature for 3 h. After removing solvents under reduced pressure, the mixture was treated with saturated aqueous sodium bicarbonate (20 mL) and ethyl acetate (20 mL). The layers were separated, and the aqueous layer was extracted with ethyl acetate (2 × 20 mL). The combined organic layer was washed with brine (20 mL), dried with anhydrous magnesium sulfate, and the solvent removed under reduced pressure. Purification of the resulting oil by flash column chromatography on silica (5% methanol in CHCl3 as eluent) afforded heteroarene 8 (1.30 g, 56%) as a white solid. Rf = 0.47 (MeOH/CHCl3 = 1:20). 1 H NMR (600 MHz, CDCl3) δ 8.23 (s, 1H), 7.83 (d, J = 8.3 Hz, 1H), 7.71 (s, 1H) 7.44 (d, J = 7.6 Hz, 1H), 4.00–3.25 (br, 8H). 13C NMR (150 MHz, CDCl3) δ 169.52, 153.87, 149.67, 141.24, 132.90, 123.79, 120.76, 110.48, 66.81. HRMS (DART) m/z calcd for C12H13N2O3 [MH]+ : 233.0926, found 233.0926.
4-(3,5-Dichlorophenyl 6-benzoxazoyl)morpholine

To a 20-mL glass vessel equipped with J. Young® O-ring tap containing a magnetic stirring bar were added Ni(cod)2 (13.9 mg, 0.05 mmol), 2,2’-bipyridyl (7.8 mg, 0.05 mmol), LiOt-Bu (60 mg, 0.75 mmol), 8 (174.2 mg, 0.5 mmol), 3,5-dichloroiodobenzene (9: 203.9 mg, 0.75 mmol), followed by dry 1,2-dimethoxyethane (2.0 mL). The vessel was sealed with an O-ring tap and then heated at 100 °C in an 8-well reaction block with stirring for 24 h. After cooling the reaction mixture to room temperature, the mixture was passed through a short silica gel pad (EtOAc). The filtrate was concentrated and the residue was subjected to preparative thin-layer chromatography (5% methanol in CHCl3 as eluent) to afford SI-2 (139.6 mg, 74 %) as a white foam. Rf = 0.70 (MeOH/CHCl3 = 1:20). 1 H NMR (600 MHz, CDCl3) δ 8.16 (d, J = 2.0 Hz, 2H), 7.82 (d, J = 7.6 Hz, 1H), 7.70 (s, 1H), 7.55 (d, J = 2.0 Hz, 1H), 7.45 (d, J = 7.6 Hz, 1H), 4.00–3.25 (br, 8H). 13C NMR (150 MHz, CDCl3) δ 169.38, 161.78, 150.40, 142.90, 135.82, 132.95, 131.61, 129.26, 125.91, 124.23, 120.41, 110.26, 66.77. HRMS (DART) m/z calcd for C18H15Cl2N2O3 [MH]+ : 377.0460 found 377.0465.
Tafamidis[19 ] Razavi, H.; Palaninathan, S. K.; Powers, E. T.; Wiseman, R. L.; Purkey, H. E.; Mohamedmohaideen, N. N.; Deechongkit, S.; Chiang, K. P.; Dendle, M. T. A.; Sacchettini, J. C.; Kelly, J. W. Angew. Chem. Int. Ed. 2003, 42, 2758.]

HF·pyridine (0.5 mL) was added to a stirred solution of SI-2 (32 mg, 0.09 mmol) in THF (0.5 mL) at 70 ºC for 12 h. After cooling the reaction mixture to room temperature, the mixuture was diluted with EtOAc and washed sequentially with sat.NaHCO3, 2N HCl and brine. The organic layer was concentrated and the residue was subjected to preparative thin-layer chromatography (1% acetic acid, 5% methanol in CHCl3 as eluent) to afford tafamidis (24.7 mg, 94%) as a white foam.
1 H NMR (600 MHz, DMSO-d6) δ 8.23 (s, 1H), 8.08 (d, J = 1.4 Hz, 2H), 8.00 (d, J = 8.3 Hz, 1H), 7.88 (m, 2H).
13C NMR (150 MHz, DMSO-d6) δ 166.6, 162.0, 150.0, 144.6, 135.1, 131.7, 129.1, 128.7, 126.5, 125.8, 120.0, 112.2.
HRMS (DART) m/z calcd for C14H8Cl2NO3 [MH]+ : 307.9881, found 307.9881.


References
- Jump up^ Bulawa, C.E.; Connelly, S.; DeVit, M.; Wang, L. Weigel, C.;Fleming, J. Packman, J.; Powers, E.T.; Wiseman, R.L.; Foss, T.R.; Wilson, I.A.; Kelly, J.W.; Labaudiniere, R. “Tafamidis, A Potent and Selective Transthyretin Kinetic Stabilizer That Inhibits the Amyloid Cascade”. Proc. Natl. Acad. Sci., 2012 109, 9629-9634.
- Jump up^ Ando, Y., and Suhr, O.B. (1998). Autonomic dysfunction in familial amyloidotic polyneuropathy (FAP). Amyloid, 5, 288-300.
- Jump up^ Benson, M.D. (1989). “Familial Amyloidotic polyneuropathy”. Trends in Neurosciences, 12.3, 88-92, PMID 2469222, doi:10.1016/0166-2236(89)90162-8.
- Jump up^ http://www.businesswire.com/news/home/20111117005505/en/Pfizer%E2%80%99s-Vyndaqel%C2%AE-tafamidis-Therapy-Approved-European-Union
- Jump up^ Clinical trial number NCT00409175 for “Safety and Efficacy Study of Fx-1006A in Patients With Familial Amyloidosis” at ClinicalTrials.gov
- Jump up^ Coelho, T.; Maia, L.F.; Martins da Silva, A.; Cruz, M.W.; Planté-Bordeneuve, V.; Lozeron, P.; Suhr, O.B.; Campistol, J.M.; Conceiçao, I.; Schmidt, H.; Trigo, P. Kelly, J.W.; Labaudiniere, R.; Chan, J., Packman, J.; Wilson, A.; Grogan, D.R. “Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial”. Neurology, 2012, 79, 785-792.
- ^ Jump up to:a b Coelho, T.; Maia, L.F.; Martins da Silva, A.; Cruz, M.W.; Planté-Bordeneuve, V.; Suhr, O.B.; Conceiçao, I.; Schmidt, H. H. J.; Trigo, P. Kelly, J.W.; Labaudiniere, R.; Chan, J., Packman, J.; Grogan, D.R. “Long-term Effects of Tafamidis for the Treatment of Transthyretin Familial Amyloid Polyneuropathy”. J. Neurology, 2013 260, 2802-2814.
- Jump up^ Ando, Y.; Sekijima, Y.; Obayashi, K.; Yamashita, T.; Ueda, M.; Misumi, Y.; Morita, H.; Machii, K; Ohta, M.; Takata, A; Ikeda, S-I. “Effects of tafamidis treatment on transthyretin (TTR) stabilization, efficacy, and safety in Japanese patients with familial amyloid polyneuropathy (TTR-FAP) with Val30Met and non-Varl30Met: A phase III, open-label study”. J. Neur. Sci., 2016 362, 266-271, doi:10.1016/j.jns.2016.01.046.
- Jump up^ Andrade, C. (1952). “A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves”. Brain: a Journal of Neurology, 75, 408-427.
- Jump up^ Coelho, T. (1996). “Familial amyloid polyneuropathy: new developments in genetics and treatment”. Current Opinion in Neurology, 9, 355-359.
- Jump up^ Razavi, H.; Palaninathan, S.K. Powers, E.T.; Wiseman, R.L.; Purkey, H.E.; Mohamadmohaideen, N.N.; Deechongkit, S.; Chiang, K.P.; Dendle, M.T.A.; Sacchettini, J.C.; Kelly, J.W. “Benzoxazoles as Transthyretin Amyloid Fibril Inhibitors: Synthesis, Evaluation and Mechanism of Action”. Angew. Chem. Int. Ed., 2003, 42, 2758-2761.
- Jump up^ Connelly, S., Choi, S., Johnson, S.M., Kelly, J.W., and Wilson, I.A. (2010). “Structure-based design of kinetic stabilizers that ameliorate the transthyretin amyloidoses”. Current Opinion in Structural Biology, 20, 54-62.
- Jump up^ Hammarstrom, P.; Wiseman, R. L.; Powers, E.T.; Kelly, J.W. “Prevention of Transthyretin Amyloid Disease by Changing Protein Misfolding Energetics”. Science, 2003, 299, 713-716
- Jump up^ Westermark, P., Sletten, K., Johansson, B., and Cornwell, G.G., 3rd (1990). “Fibril in senile systemic amyloidosis is derived from normal transthyretin”. Proc Natl Acad Sci U S A, 87, 2843-2845.
- Jump up^ Sousa, M.M., Cardoso, I., Fernandes, R., Guimaraes, A., and Saraiva, M.J. (2001). “Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy: evidence for toxicity of nonfibrillar aggregates”. The American Journal of Pathology, 159, 1993-2000.
- Jump up^ Colon, W., and Kelly, J.W. (1992). “Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro”. Biochemistry 31, 8654-8660.
- Jump up^ Jacobson, D.R., Pastore, R.D., Yaghoubian, R., Kane, I., Gallo, G., Buck, F.S., and Buxbaum, J.N. (1997). “Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans”. The New England Journal of Medicine, 336, 466-473.
- Jump up^ Maurer, M.S.; Grogan, D.R.; Judge, D.P.; Mundayat, R.; Lombardo, I.; Quyyumi, A.A.; Aarts, J.; Falk, R.H. “Tafamidis in transthyretin amyloid cardiomyopathy: effects on transthyretin stabilization and clinical outcomes.” Circ. Heart. Fail. 2015 8, 519-526.
- Jump up^http://www.pfizer.com/sites/default/files/news/Brazil%20Approval%20Press%20Statement%2011-7-16_0.pdf
//////////////TTAFAMIDIS, Fx-1006A, PF-06291826, Orphan Drug, SCRIPP, PFIZER
C1=CC2=C(C=C1C(=O)O)OC(=N2)C3=CC(=CC(=C3)Cl)Cl
CNC[C@@H]([C@H]([C@@H]([C@@H](CO)O)O)O)O.c1cc2c(cc1C(=O)O)oc(n2)c3cc(cc(c3)Cl)Cl
“NEW DRUG APPROVALS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....


















 LESINURAD
LESINURAD


