
Home » neuropathic pain
Category Archives: neuropathic pain
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Nispomeben
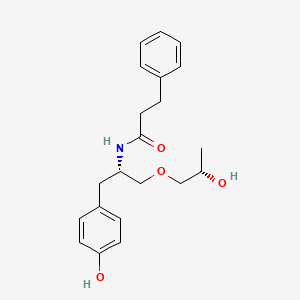
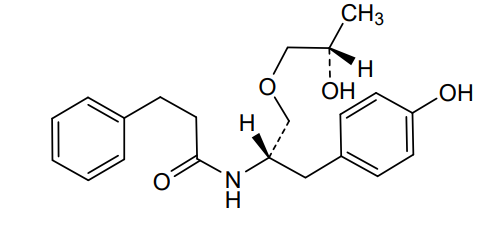
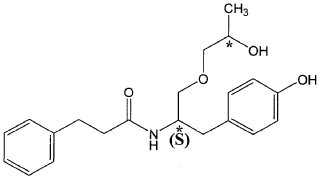
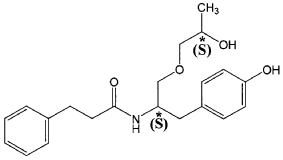
Nispomeben
CAS 1443133-41-2
MF C21H27NO4 MW357.4 g/mol
N-[(2S)-1-(4-hydroxyphenyl)-3-[(2S)-2-hydroxypropoxy]propan-2-yl]-3-phenylpropanamide
N-{(2S)-1-(4-hydroxyphenyl)-3-[(2S)-2-hydroxypropoxy]propan-2-yl}-3-phenylpropanamide
non-opioid analgesic, 470338M5XD, E1, NRD 135S E1, NRD E1, NRD.E1, NRD135S, NRD135S.E1, NRD135SE.1
Nispomeben is a small molecule drug. Nispomeben has a monoisotopic molecular weight of 357.19 Da.
- OriginatorNovaremed
- ClassAlcohols; Amides; Anti-inflammatories; Benzene derivatives; Non-opioid analgesics; Phenols; Small molecules
- Mechanism of ActionLyn protein-tyrosine kinase modulators
- Phase IINeuropathic pain
- 02 Sep 2025Updated adverse events data from a phase II trial in Neuropathic pain released by Novaremed
- 07 May 2025Novaremed completes enrolment in a phase-II clinical trial in Neuropathic pain in USA (PO) (NCT05480228)
- 16 Sep 2022Phase-II clinical trials in Neuropathic pain in USA (PO) (NCT05480228)
PAT
WO 2013/084238
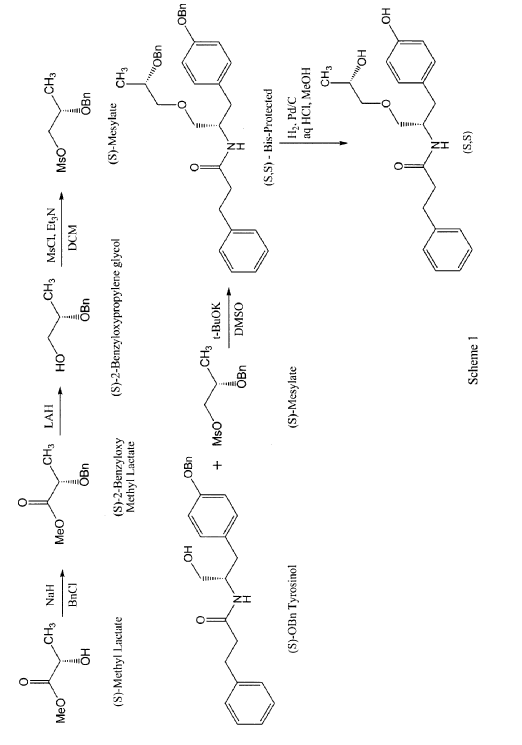
The present invention is based in part on the surprising discovery that the substantially pure enantiomers (S)2-N(3-0-((S)propan 2-ol)-l-propyl-4-hydroxybenzene)-3-phenylpropylamide (also known as the (S,S) enantiomer or El) and (S)2-N(3-0-((R)propan 2-ol)-l -propyl -4-hydroxybenzene)-3-phenylpropyl amide (also known as the (S,R) enantiomer or E2) modulate the activity of specific tyrosine kinases in an opposite manner. It was unexpectedly found that while the (S,S) enantiomer activated protein tyrosine kinases LynA and BLK, the (S,R) enantiomer inhibited their activity. It was further unexpectedly shown that the (S,S) enantiomer was effective as a pain analgesic in animal models of pain, while the (S,R) enantiomer was shown to be ineffective or less effective in these models. Furthermore, the analgesic effect of the (S,S) enantiomer was long acting as it was efficacious for more than 24 hours post administration, in comparison to the commonly used analgesic agent gabapentin which was effective for no longer than 5 hours post administration.
The isolated enantiomers according to some embodiments of the invention may be synthesized as a racemate by known in the art methods described for example in US 7,754,771, US 7,642,290, US 7,674,829 or US 2011/0086910. The racemate may be further separated by known in the art methods for the separation of chiral compounds. According to an exemplary embodiment, the enantiomers may be synthesized as a racemate (comprising (S)2-N(3-0-((S)propan 2-ol)-l-propyl-4-hydroxybenzene)-3-phenylpropylamide and (S)2-N(3-0-((R)propan 2-ol)-l-propyl-4-hydroxybenzene)-3-phenylpropylamide and be further separated by a supercritical fluid chromatography (SFC) in combination with chiral stationary phases. Specifically, the (S,S) and (S,R) compounds may be separated on RegisPack™ column a polysaccharide coated chiral column (with a tris-(3,5-dimethylphenyl) carbamoyl cellulose selector) generally used for enantiomeric separations of a wide range of racemate classes (Figure 7A-C).
According to some embodiments, the enantiomers may be synthesized directly using for example, the process described in scheme 1 for the preparation of the (S,S) enantiomer.
PAT
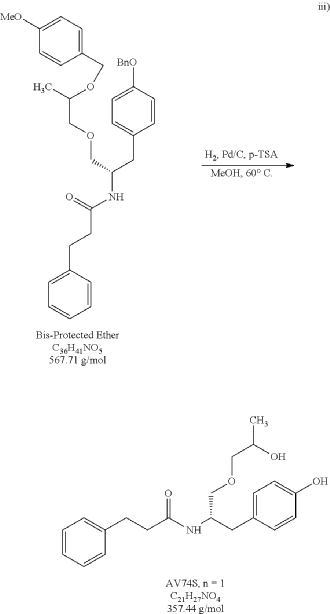
The bis-protected ether (15.7 g) was exposed to one-pot hydrogenation-debenzylation conditions (10% loading of 10% Pd/C and 0.25 eq of p-toluenesulfonic acid) in methanol. After 2 hours at 60° C. under a hydrogen atmosphere, HPLC analysis indicated that the hydrogenation of the benzyl and the debenzylation of PMB ring was complete. The reaction mixture was filtered over Celite and concentrated under reduced pressure. The residue was dissolve in ethyl acetate and a saturated aqueous sodium bicarbonate treatment was conducted to effectively remove p-toluenesulfonic acid, then DURP to provide 12.13 g of an oil (PR030-120-4). Desired product was isolated from an EA/Heptane recrystallization to provide 8.83 g of a white solid (PR030-120-6, 89.4% yield). The purity of PR030-120-6 was 99.3% via HPLC analysis. 1H NMR and Mass spec analysis supported the assigned structure for desired product.
PAT
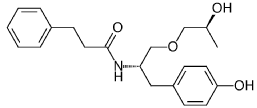
((S,S)-2-N(3-0-(propan-2-ol)-1 -propyl-4-hydroxybenzene)-3-phenylpropylamide), including its enantiomers and diastereomers may be prepared as described in WO 2013/084238,
Example 1 – Preparation of -2-N(3-Q-(propan-2-ol)-1-propyl-4-hvdroxybenzene)-3-
phenylpropylamide
(S,S)-2-N(3-0-(propan-2-ol)-1 -propyl-4-hydroxybenzene)-3-phenylpropylamide was prepared as described in WO 2013/084238 and US 201 1/0086910.
In a first step, 2 g of methyl lactate was reacted with excess of benzyl bromide to get 880 mg of (S)-benzyloxymethyl lactate. The reaction was performed by slurring sodium hydride in THF and cooling down to approximately -15°C. The reaction mixture was then allowed to warm slowly to room temperature and stirred for approximately 1 to 2 hours. The reaction was quenched with saturated ammonium chloride solution and extracted with MTBE twice followed by the removal of solvent on a rotary evaporator to obtain a crude oil. The crude product was purified by column chromatography to yield pure (S)-2-benzyloxymethyl lactate. The (R)-2-benzyloxymethyl lactate isomer was present at 0.93% only. The yield of this step may be increased by avoiding the presence of moisture in the reaction solution.
In a second step, 880 mg (S)-2-benzyloxymethyl lactate obtained in step 1 were reduced using lithium aluminum hydride to obtain (S)-2-benzyloxypropylene glycol in 83.8% yield with 98.7% purity. A solution of pure (S)-2-benzyloxymethyl lactate in methylene chloride was stirred and a solution of lithium aluminum hydride was slowly added thereto at approximately 5°C. The reaction was monitored by TLC and quenched by USP-PW water very carefully. No racemization occurred in this step.
In a third step, the (S)-2-benzyloxypropylene glycol was then reacted with methane sulfonyl chloride in methylene chloride in the presence of triethyl amine to yield the mesylate in 88% yield. A solution of step 2 was stirred in methylene chloride and methane sulfonyl chloride was added to it dropwise at <5°C. After the addition was complete, the progress of the reaction was monitored by TLC. The reaction was quenched with USP-PW water. After the layers were separated, the aqueous layer was back extracted with methylene chloride. The methylene chloride layers were then combined and washed with USP-PW water 3 times to remove most of the methane sulfonic acid. No racemization occurred in this step.
In a fourth step, the mesylate (of step 3) was coupled with S-O-benzyl tyrosinol to form the bis-protected product in 22.7% yield, with a purity of 97.4%. The reaction was carried out at room temperature using a combination of DMF as the solvent and sodium hydride as the base. The reaction went to completion after stirring for at least 12 hours at room temperature.
In a fifth step, 340 mg of the product of step 4 were reduced by hydrogenation in the presence of 10% palladium on carbon catalyst and hydrochloric acid using methylene chloride as a solvent at 50°C. The reaction went to completion in approximately 4 hours with no racemization to yield the desired product in 84.3% yield and 98.9% purity. More specifically, the catalyst was removed by filtration and the filtrate was then concentrated at 33°C. The resulting mixture of solid and oil was mixed with ethyl acetate. The resulting slurry was filtered and the solids washed with ethyl acetate and dried under vacuum at 40 to 45°C to obtain the desired product.
PAT
Example 1—Preparation of (S,S)-2-N(3-O-(propan-2-ol)-1-propyl-4-hydroxybenzene)-3-phenylpropylamide
| (S,S)-2-N(3-O-(propan-2-ol)-1-propyl-4-hydroxybenzene)-3-phenylpropylamide was prepared as described in WO 2013/084238 and US 2011/0086910. |
PAT
- Method of treating or preventing painPublication Number: US-2016317479-A1Priority Date: 2009-09-09
- Method of treating or preventing painPublication Number: US-8802734-B2Priority Date: 2009-09-09Grant Date: 2014-08-12
- Method of Treating or Preventing PainPublication Number: US-2014350099-A1Priority Date: 2009-09-09
- N-substituted benzenepropanamide or benzenepropenamide for use in the treatment of pain and inflammationPublication Number: WO-2011030205-A1Priority Date: 2009-09-09
- Method of treating or preventing painPublication Number: US-2011086910-A1Priority Date: 2009-09-09
- Isolated stereoisomeric forms of (S)2-N(3-O-(propan 2-Ol)-1-propyl-4-hydroxybenzene)-3-phenylpropylamidePublication Number: US-9381173-B2Priority Date: 2011-12-08Grant Date: 2016-07-05
- Isolated Stereoisomeric Forms Of (S)2-N(3-O-(Propan 2-Ol)-1-Propyl-4-Hydroxybenzene)-3-PhenylpropylamidePublication Number: US-2014275270-A1Priority Date: 2011-12-08
- N-substituted benzenepropanamide and benzenepropenamide for use in the prevention or the treatment of affective disordersPublication Number: US-9133103-B2Priority Date: 2011-09-21Grant Date: 2015-09-15
- N-Substituted Benzenepropanamide and Benzenepropenamide For Use in the Prevention or the Treatment of Affective DisordersPublication Number: US-2014275273-A1Priority Date: 2011-09-21
- N-substituted benzenepropanamide and benzenepropenamide for use in the prevention or the treatment of affective disordersPublication Number: EP-2758046-B1Priority Date: 2011-09-21Grant Date: 2015-10-21
- Compounds for treatment or prevention of an infection resulting from a coronavirus and/or a coronavirus-induced diseasePublication Number: EP-3939578-A1Priority Date: 2020-07-13
- Compounds for use in the treatment or prophylaxis of pain, inflammation and/or autoimmunityPublication Number: EP-3860582-B1Priority Date: 2019-01-23Grant Date: 2022-05-04
- Compounds for use in the treatment or prophylaxis of pain, inflammation and/or autoimmunityPublication Number: WO-2020152226-A1Priority Date: 2019-01-23
- Compounds for use in the treatment or prophylaxis of pain, inflammation and/or autoimmunityPublication Number: US-2022002228-A1Priority Date: 2019-01-23
- Pharmaceutical composition comprising stereoisomers of n-(1-(4-hydroxyphenyl)-3-(2-hydroxypropoxy)propan-2-yl)-3-phenylpropanamide for the prevention and treatment of type ii diabetesPublication Number: WO-2015173813-A1Priority Date: 2014-05-14



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

////////nispomeben, non-opioid analgesic, 470338M5XD, E1, NRD 135S E1, NRD E1, NRD.E1, NRD135S, NRD135S.E1, NRD135SE.1, Neuropathic pain
TRK 700
![1-[4-(Dimethylamino)piperidin-1-yl]-3-(1-methylimidazol-2-yl)propan-1-one.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=71738795&t=l)
TRK-700
CAS 1463432-16-7C14 H24 N4 O264.371-Propanone, 1-[4-(dimethylamino)-1-piperidinyl]-3-(1-methyl-1H-imidazol-2-yl)-
1-[4-(dimethylamino)piperidin-1-yl]-3-(1-methylimidazol-2-yl)propan-1-one
- 1-[4-(Dimethylamino)-1-piperidinyl]-3-(1-methyl-1H-imidazol-2-yl)-1-propanone
- OriginatorToray Industries
- ClassAnalgesics
- Mechanism of ActionUndefined mechanism
- Phase IIPostherpetic neuralgia
- PreclinicalPeripheral nervous system diseases
- 12 Sep 2018Pharmacodynamics data from a preclinical trial in Peripheral neuropathy presented at the 17th World Congress on Pain (WCP-2018)
- 01 Jul 2017Toray Industries completes a phase II trial for Postherpetic neuralgia (In adults, In the elderly) in Japan (PO) (NCT02701374)
- 21 May 2017Toray Industries completes a phase I drug-drug interaction trial in Healthy volunteers in Japan (PO) (NCT03043248)
developed by Toray for treating neuropathic pain and investigating for fibromyalgia. In August 2021, this drug was reported to be in phase 1 clinical development.
PATENT
WO 2016136944
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016136944

(Reference Example 22) Synthesis of (E) -methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate:
[Chemical 56]
1-methyl-1H-imidazol-2-carbaldehyde (10.0 g, Methyl (triphenylphosphoranylidene) acetate (33.4 g, 99.9 mmol) was added to a solution of 90.8 mmol) in dichloromethane (240 mL) at room temperature, and the mixture was stirred for 16 hours and then concentrated under reduced pressure. The residue was washed with a mixed solvent of hexane / dichloromethane = 19/1, and the washing liquid was concentrated. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give (E) -methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate as a white solid (11.9 g, 71. 6 mmol, 79%).
1 H-NMR (400 MHz, CDCl 3 ) δ: 3.76 (3H, s), 3.81 (3H, s), 6.82 (1H, d, J = 15.6 Hz), 6.98 (1H, brs), 7.16 (1H, brs), 7.53 (1H, d, J = 15.6Hz).
ESI-MS: m / z = 167 (M + H) + .
(Reference Example 27) Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one:
[Chemical 61]
(E) )-Methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate (0.180 g, 1.08 mmol) in ethanol (4.0 mL) solution of palladium-carbon (10% wet, 15 mg) at room temperature In a hydrogen atmosphere, the mixture was stirred for 4 hours. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. Methanol (1.0 mL) was added to the obtained residue at room temperature to dissolve it, and the mixture was cooled to 0 ° C. An aqueous sodium hydroxide solution (1.0 N, 1.19 mL, 1.19 mmol) was added to the reaction solution at 0 ° C., the mixture was stirred at room temperature for 2 hours, and then concentrated under reduced pressure. Chloroform (10.0 mL) was added to the obtained residue at room temperature to dissolve it. Add diisopropylethylamine (0.568 mL, 3.25 mmol), HBTU (0.616 g, 1.63 mmol) and 4- (dimethylamino) piperidine (0.125 g, 0.975 mmol) to the reaction solution at room temperature, and add the reaction solution. The mixture was stirred at the same temperature for 16 hours. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was washed with a 10% aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography (NH silica gel, chloroform / methanol) and 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propane. -1-one (0.179 g, 0.68 mmol, 63%) was obtained as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3) δ: 1.29-1.43 (2H, m), 1.80-1.88 (2H, m), 2.27 (6H, s), 2.29-2.38 (1H, m), 2.54-2.63 (1H, m), 2.88-3.04 ( 5H, m), 3.62 (3H, s), 3.98-4.05 (1H, m), 4.57-4.65 (1H, m), 6.79 (1H, d, J = 1.2 Hz), 6.91 (1H, d, J = 1.2 Hz).
ESI-MS: m / z = 265 (M + H) + .
(Comparative Example 1) Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one hydrochloride:
[Chemical 66]
1- (4- (Dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one (1.50 g, 5.67 mmol) diethyl ether (60) A dioxane solution of hydrogen chloride (4.0 M, 3.69 mL, 14.8 mmol) was added to the (0.0 mL) solution at 0 ° C. The reaction mixture was stirred at the same temperature for 1 hour and then at room temperature for 30 minutes. The precipitated white solid was collected by filtration, washed with diethyl ether (100 mL), dried at room temperature for 36 hours, and then 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-). Imidazole-2-yl) propan-1-one hydrochloride (1.41 g, 4.18 mmol, 74%) (hereinafter, the compound of Comparative Example 1) was obtained as a white solid.
1 1 H-NMR (400 MHz, D 2 O) δ: 1.53-1.80 (2H, m), 2.12-2.23 (2H, m), 2.68-2.80 (1H, m), 2.88 (6H, s), 3.01- 3.08 (2H, m), 3.15-3.26 (3H, m), 3.47-3.58 (1H, m), 3.84 (3H, s), 4.08-4.16 (1H, m), 4.50-4.59 (1H, m), 7.29-7.33 (2H, m).
ESI-MS; 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) as propan-1-one : m / z = 265 (M + H) + .
(Comparative Example 2) Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one sulfate monohydrate:
[Chemical 67]
1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one (6.72 g, 25.4 mmol) Concentrated sulfuric acid (2.49 g, 25.4 mmol), water (1.83 g, 102 mmol) and 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl) in a DMSO (100 mL) solution. Seed crystals (50 mg, 0.13 mmol) of -1H-imidazol-2-yl) propan-1-one sulfate monohydrate were added at 80 ° C. The reaction was stirred at the same temperature for 2.5 hours, at 50 ° C. for 2.5 hours and at room temperature for 15 hours. The precipitated white solid was collected by filtration, washed successively with DMSO (20 mL) and methyl ethyl ketone (40 mL), dried at room temperature, and then 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl). -1H-imidazol-2-yl) propan-1-one sulfate monohydrate (8.42 g, 22.1 mmol, 87%) (hereinafter, the compound of Comparative Example 2) was obtained as white crystals.
1 1 H-NMR (400 MHz, DMSO-d 6)) δ: 1.36 (1H, m), 1.58 (1H, m), 1.95 (2H, br), 2.44-2.57 (1H, m), 2.65 (6H, s), 2.74-2.88 (4H, m), 3.00 (1H, t, J = 12.0 Hz), 3.22 (1H, m), 3.61 (3H, s), 4.02 (1H, d, J = 14.0 Hz), 4.47 (1H, d, J = 12.8 Hz), 6.87 (1H, d, J = 1.2 Hz), 7.11 (1H, d, J = 1.2 Hz).
ESI-MS; 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-) As 1H-imidazol-2-yl) propan-1-one: m / z = 265 (M + H) + .
NEW DRUG APPROVALS
ONE TIME
$10.00
PATENT
WO-2021153744
PATENT
WO-2021153743
Novel crystalline polymorphic form of 1-(4-(dimethylamino) piperidin-1-yl)-3-(1-methyl-1H-imidazol-2-yl)propan-1-one, useful as an analgesic in treating neuropathic pain and/or fibromyalgia.Pain is an experience with unpleasant sensations and emotions that occurs when or may cause tissue damage. Pain is mainly classified into nociceptive pain, neuropathic pain or psychogenic pain according to its cause. In addition, fibromyalgia is known as pain of unknown cause.
Neuropathic pain is pathological pain caused by dysfunction of the peripheral or central nervous system itself, and is caused by direct damage or compression of nervous tissue even though nociceptors are not stimulated. It refers to the pain that occurs. As a therapeutic agent for neuropathic pain, an anticonvulsant, an antidepressant, anxiolytic, or an antiepileptic drug such as gabapentin or pregabalin is used.
Fibromyalgia is a disease in which systemic pain is the main symptom and neuropsychiatric symptoms and autonomic nervous system symptoms are secondary symptoms. Pregabalin approved in the United States and Japan, duloxetine and milnacipran approved in the United States are mainly used as therapeutic agents for fibromyalgia, and non-approved agents for fibromyalgia are not approved. It has also been used for steroidal anti-inflammatory agents, opioid compounds, antidepressants, anticonvulsants and antiepileptic drugs. However, the therapeutic effects of non-steroidal anti-inflammatory drugs and opioid compounds are generally considered to be low (Non-Patent Document 1).
On the other hand, Patent Document 1 discloses that certain substituted piperidins have cardiotonic activity, and Patent Document 2 discloses that an imidazole derivative exhibits an FXa inhibitory effect. Patent Document 3 suggests that the substituted piperidins may have a medicinal effect on overweight or obesity, and Patent Documents 4 to 6 and Non-Patent Document 2 indicate that the imidazole derivative has an analgesic effect. It is disclosed.
In addition, the quality of pharmaceutical products needs to be maintained over a long period of time such as distribution and storage, and the compound as an active ingredient is required to have high chemical and physical stability. Therefore, as the active ingredient of a pharmaceutical product, a crystal that can be expected to have higher stability than an amorphous substance is generally adopted. Further, if crystals are obtained, a purification effect due to recrystallization during production can be expected. Further, it is preferable to have low hygroscopicity from the viewpoint of maintaining stability and handling during manufacturing, storage, formulation and analysis of the drug substance. In addition, since a drug needs to be dissolved in the digestive tract in order to exhibit its medicinal effect, it is preferable that the drug has excellent solubility, which is a physical property contrary to stability.
In order to obtain crystals of a compound that is an active ingredient of a pharmaceutical product, it is necessary to study various conditions for precipitating crystals from the solution. It is common to carry out crystallization under the condition of being dissolved in.
Patent documents
Patent Document 1: French Patent Invention No. 2567885
Patent Document 2: Japanese Patent Application Laid-Open No. 2006-0083664
Patent Document 3: International Publication No. 2003/031432
Patent Document 4: International Publication No. 2013/147160
Patent Document 5: International Publication No. 2015/046403
Patent Document 6: International Publication No. 2016/136944
Non-patent literature
Non-Patent Document 1: Okifuji et al., Pain and Therapy, 2013, Volume 2, p. 87-104
Non-Patent Document 2: Takahashi et al., Toxicological Pathology, 2019, Vol. 47. p. 494-503
Compound (I) was synthesized by the method described in the following reference example. For the compounds used in the synthesis of the reference example compounds for which the synthesis method is not described, commercially available compounds were used.
(Reference Example 4) Synthesis of amorphous compound (I):
[Chemical formula 2] 2 of
crude ethyl 3- (1-methyl-1H-imidazol-2-yl) propanol (5.00 g, 27.4 mmol) Aqueous sodium hydroxide solution (1.0N, 30.2 mL, 30.2 mmol) was added to a solution of -propanol (55 mL) at 0 ° C., and the mixture was stirred at room temperature for 12 hours. 2-Propanol (220 mL) was added to the reaction solution at room temperature, and crude 4- (dimethylamino) piperidine (3.17 g, 24.7 mmol) and DMT-MM (8.35 g, 30.2 mmol) were added at room temperature to react. The liquid was stirred at the same temperature for 3 hours. A 10% aqueous sodium chloride solution and a 1.0N aqueous sodium hydroxide solution were added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give compound (I) (6.98 g) as an amorphous substance.
1 1 H-NMR (400 MHz, CDCl 3 ) δ: 1.29-1.43 (2H, m), 1.80-1.88 (2H, m), 2.27 (6H, s), 2.29-2.38 (1H, m), 2.54-2.63 (1H, m), 2.88-3.04 (5H, m), 3.62 (3H, s), 3.98-4.05 (1H, m), 4.57-4.65 (1H, m), 6.79 (1H, d, J = 1.2 Hz) ), 6.91 (1H, d, J = 1.2 Hz).
ESI-MS: m / z = 265 (M + H) + .
(Reference Example 5) Synthesis of crude 4- (dimethylamino) piperidine:
[Chemical
formula 3] 1-benzyloxycarbonyl-4- (dimethylamino) piperidine (20.1 g, 77.0 mmol) in methanol (154.0 mL) Palladium-carbon (10% wet, 2.01 g) was added thereto, and the mixture was stirred at room temperature for 19 hours under a hydrogen atmosphere. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to give a crude product of 4- (dimethylamino) piperidine (9.86 g).
(Reference Example 6) Synthesis of crude ethyl 3- (1-methyl-1H-imidazol-2-yl) propanoate:
[Chemical
formula 4] Sodium hydride (55%, 4.36 g, 100 mmol) aqueous solution and tetrahydrofuran (150 mL) To the mixture was added triethylphosphonoacetate (19.1 mL, 95.0 mmol) at 0 ° C. After stirring the reaction solution for 20 minutes, a solution of 1-methyl-1H-imidazol-2-carbaldehyde (10.0 g, 91.0 mmol) in tetrahydrofuran (150 mL) was added at 0 ° C., and then ethanol (30 mL) was added in the same manner. The mixture was added at temperature and stirred at room temperature for 2 hours. A 10% aqueous sodium chloride solution was added to the reaction mixture, and the mixture was extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, chloroform / methanol). After adding methanol (310 mL) to the residue, palladium-carbon (10% wet, 1.40 g) was added, and the mixture was stirred at room temperature for 3 hours under a hydrogen atmosphere. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to obtain a crude product (14.2 g) of ethyl 3- (1-methyl-1H-imidazol-2-yl) propanoate.
(Reference Example 7) Synthesis of 1-benzyloxycarbonyl-4- (dimethylamino) piperidine:
[Chemical
formula 5] dichloromethane (55.7 mL) of 1-benzyloxycarbonyl-4-oxopiperidine (13.0 g, 55.7 mmol) ) Solution of dimethylamine in tetrahydrofuran (2.0 M, 34.8 mL, 69.7 mmol), acetic acid (0.32 mL, 5.6 mmol) and sodium triacetoxyborohydride (4.8 g, 22.6 mmol). Added at ° C. After stirring the reaction solution at the same temperature for 30 minutes, sodium triacetoxyborohydride (4.8 g, 22.6 mmol) was added at 0 ° C. The reaction mixture was stirred at the same temperature for 30 minutes, sodium triacetoxyborohydride (8.1 g, 38.2 mmol) was added at 0 ° C., and the mixture was stirred at room temperature for 12 hours. The reaction solution was cooled to 0 ° C. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, n-hexane / ethyl acetate) and then again by flash chromatography (silica gel, chloroform / methanol) to obtain 1-benzyloxycarbonyl-4- (dimethylamino) piperidine (dimethylamino) piperidine. 13.6 g, 51.8 mmol, 93%) was obtained as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3) δ: 1.34-1.46 (2H, m), 1.78-1.86 (2H, m), 2.28 (6H, s), 2.29-2.34 (1H, m), 2.75-2.85 (2H, m), 4.14-4.28 ( 2H, m), 5.12 (2H, s), 7.29-7.36 (5H, m).
ESI-MS: m / z = 263 (M + H) + .
(Reference Example 8) Synthesis of 1-benzyloxycarbonyl-4-oxopiperidine:
[Chemical
formula 6] Hydrochloride (130 mL) and water (130 mL) of 4-piperidinone hydrochloride monohydrate (10.0 g, 65.1 mmol) Sodium carbonate (13.8 g, 130.2 mmol) and benzyl chloroformate (8.79 mL, 61.8 mmol) were added to the mixed solution with and at 0 ° C., and the mixture was stirred at room temperature for 3 hours. The reaction mixture was extracted with ethyl acetate. The organic layer was washed with 10% aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, n-hexane / ethyl acetate) to give 1-benzyloxycarbonyl-4-oxopiperidine (13.1 g, 56.2 mmol, 86%) as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3 ) δ: 2.42-2.50 (4H, m), 3.78-3.82 (4H, m), 5.18 (2H, s), 7.32-7.38 (5H, m).
(Example 1) Production of A-type crystal of
compound (I): Amorphous compound (6.98 g) of compound (I) prepared in Reference Example 4 is purified and concentrated with chloroform / methanol by silica gel column chromatography. After that, the wall surface of the flask was rubbed with a spartel and mechanical stimulation was applied to obtain A-type crystals of compound (I) as a powder. For the obtained crystals, measurement of powder X-ray diffraction using a powder X-ray diffractometer (Rigaku Co., Ltd .; 2200 / RINT ultima + PC) and TG-DTA using a TG-DTA device (Rigaku Co., Ltd .; TG8120) Was done. The results of these measurements are shown in FIGS. 1 and 2.
Diffraction angle 2θ: 5.9, 16.5, 17.7, 20.8, 26.7 °
Endothermic peak: 55 ° C
PATENT
WO2013147160
Example 1 Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one:
[Chemical 27]
(E) )-Methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate (0.180 g, 1.08 mmol) in ethanol (4.0 mL) solution of palladium-carbon (10% wet, 15 mg) at room temperature In a hydrogen atmosphere, the mixture was stirred for 4 hours. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. Methanol (1.0 mL) was added to the obtained residue at room temperature to dissolve it, and the mixture was cooled to 0 ° C. An aqueous sodium hydroxide solution (1.0 N, 1.19 mL, 1.19 mmol) was added to the reaction solution at 0 ° C., the mixture was stirred at room temperature for 2 hours, and then concentrated under reduced pressure. Chloroform (10.0 mL) was added to the obtained residue at room temperature to dissolve it. Add diisopropylethylamine (0.568 mL, 3.25 mmol), HBTU (0.616 g, 1.63 mmol) and 4- (dimethylamino) piperidine (0.125 g, 0.975 mmol) to the reaction solution at room temperature, and add the reaction solution. The mixture was stirred at the same temperature for 16 hours. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was washed with a 10% aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (NH silica gel, chloroform / methanol) and 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan- 1-one (0.179 g, 0.68 mmol, 63%) (hereinafter, the compound of Example 1) was obtained as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3) δ: 1.29-1.43 (2H, m), 1.80-1.88 (2H, m), 2.27 (6H, s), 2.29-2.38 (1H, m), 2.54-2.63 (1H, m), 2.88-3.04 ( 5H, m), 3.62 (3H, s), 3.98-4.05 (1H, m), 4.57-4.65 (1H, m), 6.79 (1H, d, J = 1.2 Hz), 6.91 (1H, d, J = 1.2 Hz).
ESI-MS: m / z = 265 (M + H) + .
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2016136944-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| JP-WO2013147160-A1 | Cyclic amine derivatives and their pharmaceutical use | 2012-03-29 | |
| TW-201350119-A | Cyclic amine derivatives and their medical uses | 2012-03-29 | |
| WO-2013147160-A1 | Cyclic amine derivative and use thereof for medical purposes | 2012-03-29 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| RU-2667062-C1 | Dynamic cyclic amine and pharmaceutical application thereof | 2015-02-27 | 2018-09-14 |
| TW-201639826-A | Cyclic amine derivatives and their medical uses | 2015-02-27 | |
| TW-I682927-B | Cyclic amine derivatives and their medical uses | 2015-02-27 | 2020-01-21 |
| US-10173999-B2 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2019-01-08 |
| US-2018065950-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| EP-3263565-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| EP-3263565-B1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2019-06-26 |
| ES-2744785-T3 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2020-02-26 |
| JP-6569671-B2 | Cyclic amine derivatives and their pharmaceutical use | 2015-02-27 | 2019-09-04 |
| JP-WO2016136944-A1 | Cyclic amine derivatives and their pharmaceutical use | 2015-02-27 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2019189781-A1 | Agent for inhibiting rise in intraneuronal calcium concentration | 2018-03-30 | |
| AU-2016224420-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| AU-2016224420-B2 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2019-08-22 |
| CA-2977614-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| CN-107250128-B | Cyclic amine derivatives and its medical usage | 2015-02-27 | 2019-07-26 |
//////////TRK-700, phase 1, neuropathic pain, fibromyalgia, toray
O=C(CCc1nccn1C)N1CCC(CC1)N(C)C
