
Home » Uncategorized
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Lixosicone
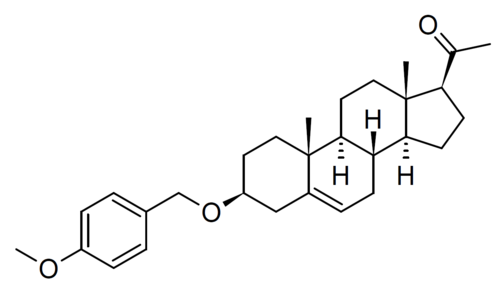
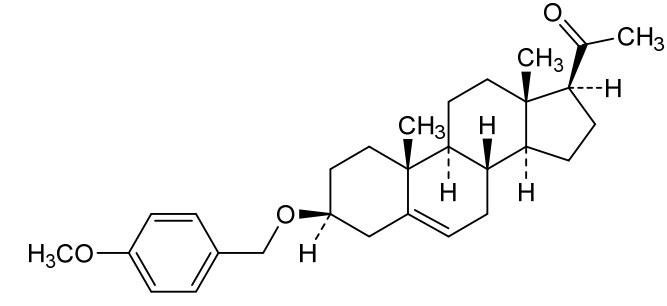
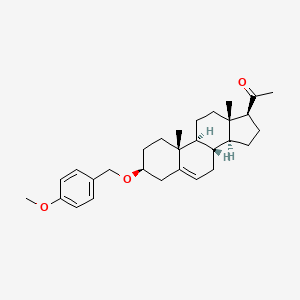
Lixosicone
CAS 1610878-71-1
MF C29H40O3 MW 436.6 g/mol
1-[(3S,8S,9S,10R,13S,14S,17S)-3-[(4-methoxyphenyl)methoxy]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-17-yl]ethanone
- 3beta-[(4-methoxyphenyl)methoxy]pregn-5-en-20-one
- (3beta)-3-[(4-Methoxyphenyl)methoxy]pregn-5-en-20-one
- Pregn-5-en-20-one, 3-[(4-methoxyphenyl)methoxy]-, (3beta)-
- 1-[(3S,8S,9S,10R,13S,14S,17S)-3-[(4-methoxyphenyl)methoxy]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-17-yl]ethanone
- 3?-(4-Methoxybenzyloxy)pregn-5-en-20-one; 1-((3S,8S,9S,10R,13S,14S,17S)-3-((4-Methoxybenzyl)oxy)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)ethan-1-one
3β-[(4-methoxyphenyl)methoxy]pregn-5-en-20-one
cannabinoid CB1 receptor signalling inhibitor, AEF0117, AEF 0117, 9LG9CT78SV
AEF0117 is a small molecule drug. AEF0117 is under investigation in clinical trial NCT05554926 (Study of [4-14C] AEF0117 Following a Single Oral Dose in Healthy Male Subjects).
Lixosicone (AEF0117, 3β-(4-methoxybenzyloxy)pregn-5-en-20-one) is a compound derived from pregnenolone by Aelis Farma, which acts as a biased negative allosteric modulator of the cannabinoid CB1 receptor, representing a new class of compounds referred to as CB1-selective signalling-specific inhibitors (CB1-SSi). It binds to an allosteric site on the CB1 receptor and modifies the downstream signalling produced as a result of CB1 activation, preventing CB1 mediated changes to mitogen-activated protein kinase (MAPK) phosphorylation but without affecting the signalling mediated by cyclic AMP. Unlike pregnenolone, AEF0117 is specific for the CB1-SSi activity and lacks the neurosteroid action typical of many structurally related compounds.[1]
In Phase II human clinical trials in patients diagnosed with cannabis use disorder, AEF0117 was found to partly but not completely block the effects of THC, and reduced cannabis self-administration but without producing an acute withdrawal syndrome and with relatively mild side effects. It is hoped that compounds of this type may be useful either as medications for the treatment of cannabinoid dependence, or could be used alongside medicinal cannabis to reduce unwanted side effects while retaining therapeutic efficacy.[2]
As of March 2026, lixosicone is in phase 2 clinical trials for treatment of substance-related disorders.[3] It is being developed by Aelis Farma.[3]
Clinical Development
The compound has shown promising results in clinical settings:
- Phase 2a results: In clinical trials evaluated by the National Institute on Drug Abuse, lixosicone significantly reduced the positive subjective effects (“the high”) of cannabis by 19% at lower doses and up to 38% at higher doses compared to a placebo.
- Reduced consumption: Testing demonstrated that the drug successfully reduced cannabis self-administration.
- Safety profile: Across early-stage evaluations, the drug was found to be safe, well-tolerated, and did not precipitate adverse behavioral withdrawal symptoms
SYN
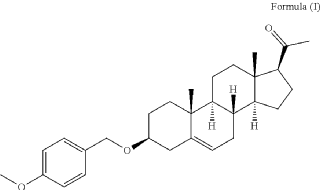
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN309682512&_cid=P10-MQRGHV-67541-1
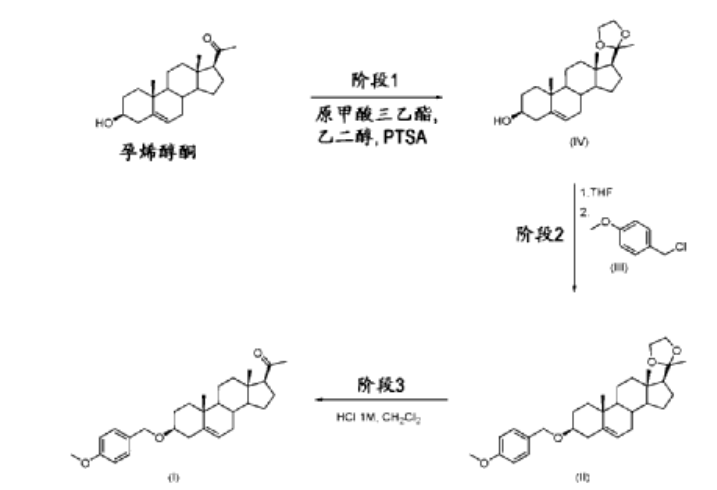
| Stage 1: Compound of formula (IV): (3S,8S,9S,10R,13S,14S,17S)-10,13-dimethyl-17-(2-methyl) (-1,3-dioxane-2-yl)-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecano-1H-cyclopentadiene Preparation of [a]phenanthrene-3-ol |
| Phase 1 was carried out in batches. Ethylene glycol (11.676 kg), pregnenolone (6.992 kg), and p-toluenesulfonic acid (0.840 kg, 4.42 mol, 0.2 equivalents) were charged into the reactor. The reaction mixture was stirred at 15°C–25°C for 25 minutes. Triethyl orthoformate (20.939 kg) was added in three portions, and the mixture was stirred at 15°C–25°C for at least 1 hour. Once complete, the reaction mixture was collected and slowly poured into a sodium bicarbonate solution (2.943 kg in 35.5 L of water) at 0°C–10°C. At the end of the addition, the reaction mixture was stirred at 0°C–10°C for 1 hour, then filtered and washed with water (12 L). The filtrate was also washed with 2-propanol (12 L) and dried under vacuum under a nitrogen stream. The dried solids were collected and charged into the reactor with 2-propanol (35 L). The slurry was heated under reflux for 2 hours. The reaction mixture was cooled to room temperature and stirred at room temperature for 12 hours. The reaction mixture was then cooled to between 0°C and 10°C and stirred for 2 hours. The solid was filtered and washed with 2-propanol (12 l), and then dried under vacuum under a nitrogen stream. Compound (IV) (8.031 kg) was obtained in a yield of 100.8% (uncorrected yield). |
| Phase 2: Compound of formula (II): 2-(3S,8S,9S,10R,13S,14S,17S)-3-((4-methoxybenzyl)oxy (10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecano-1-H-cyclopentadien[a]) Preparation of phenanthrene-17-yl)-2-methyl-1,3-dioxolane |
| Compound (IV) (3.460 kg) and tetrahydrofuran (THF) (69 l) were charged into a reactor. The reaction mixture was stirred at 20°C–25°C for 80 minutes. The reaction mixture was filtered, and the solution of compound (IV) in THF was charged into the reactor. t-BuOK (2.835 kg) was added in portions to the THF solution of compound (IV) at 20°C–25°C. At the end of the addition, p-methoxybenzyl chloride (2.832 kg) and THF (4 l) of formula (III) were added to the reaction mixture via a feeding funnel. The reaction mixture was heated at 38°C–42°C. TBAI (1.555 kg) was added in portions to the reaction mixture at 38°C–42°C. The reaction mixture was heated at 55°C–60°C for 16 hours and 30 minutes. |
| Once complete, the reaction mixture is concentrated under vacuum to distill off 34-36 l of THF. The reaction mixture is then cooled to room temperature. Water (52 l) is added to the reactor, which is then cooled to 0-10 °C. The reaction mixture is carefully poured onto the water while maintaining the temperature at 0-10 °C. At the end of the addition, the reaction mixture is stirred at 0-10 °C for 1 hour and 50 minutes. The reaction mixture is filtered and washed with water (13 l). The filtrate is washed with acetonitrile (13.5 l), and the solids are dried under vacuum for 4 days under a nitrogen stream. |
| The solid was collected and acetonitrile (13 L) was added to the reactor. The mixture was heated under reflux for 4 hours. An additional acetonitrile (11 L) was added to the reactor and heated under reflux until a clear solution was obtained. The reaction mixture was cooled to room temperature and stirred at room temperature for 14 hours. The reaction mixture was cooled to 0 °C–10 °C and stirred at 0 °C–10 °C for 45 minutes, then filtered. Acetonitrile (10.5 L) was added to the reactor, cooled to 0 °C–10 °C, and then added to the filter to wash the filtrate. The solid was dried under vacuum under a nitrogen stream for 21 hours. Compound (II) (2.449 kg) was obtained in a yield of 59.2%. |
| • Stage 3: Preparation of compound (I): 3pMBP |
| Compound (II) (2.448 kg) and dichloromethane (10 L) were charged into a reactor. The solution was stirred for 20 minutes. 1 M hydrochloric acid (4.9 L) was added to the solution at 15 °C–25 °C. The reaction mixture was stirred until complete at 15 °C–25 °C. Dichloromethane (8 L) was added (to completely dissolve any precipitate) and phase separation was allowed. The organic layer was washed twice with water (5 L). The organic layer was collected and 2-propanol (24.5 L) was charged into a reactor at 15 °C–25 °C. The reaction mixture was concentrated under vacuum at a temperature below 40 °C. After completion, the reaction mixture was heated to reflux. 2-propanol (40 L) was added until a clear solution was observed. The reaction mixture was cooled to room temperature and stirred at room temperature for 12 hours. The reaction mixture was cooled to 0 °C–10 °C and stirred at 0 °C–-10 °C for 1 hour. The solid was filtered and washed with 2-propanol (5 l), then dried under vacuum with a nitrogen flow rate while the filter was heated at 35 °C–45 °C for 20 hours. Compound (I) was obtained in 85.8% yield (1.907 kg). |
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019162328&_cid=P10-MQRGHV-67541-1
PAT
- 3β-(4-Methoxybenzyloxy)pregnant-5-en-20-one for the treatment of cannabinoid-related disordersPublication Number: CN-111757740-BPriority Date: 2018-02-20Grant Date: 2023-12-15
- 3beta-(4-methoxybenzyloxy)paragan-5-en-20-one for use in the treatment of cannabinoid-related disordersPublication Number: IL-276697-B2Priority Date: 2018-02-20
- 3-BETA-(4-METHOXYBENZYLOXY)PREGN-5-EN-20-ONE FOR USE IN THE TREATMENT OF CANNABINOID-RELATED DISORDERSPublication Number: PT-3755339-TPriority Date: 2018-02-20
- 3beta-(4-methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of Cannabinoids-Related DisordersPublication Number: US-2023226076-A1Priority Date: 2018-02-20
- 3beta-(4-methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of cannabinoids-related disordersPublication Number: EP-3755339-A1Priority Date: 2018-02-20
- 3beta-(4-methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of Cannabinoids-Related DisordersPublication Number: US-2021030768-A1Priority Date: 2018-02-20
- 3-beta-(4-methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of cannabinoids-related disordersPublication Number: EP-3755339-B1Priority Date: 2018-02-20Grant Date: 2024-01-03
- 3BETA-(4-METHOXYBENZYLOXY)PREGN-5-EN-20-ONE FOR USE IN THE TREATMENT OF CANNABINOIDS RELATED DISORDERSPublication Number: MA-51891-B1Priority Date: 2018-02-20
- 3Beta-(4-Methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of cannabinoid-related disordersPublication Number: MD-3755339-T2Priority Date: 2018-02-20
- 3-BETA-(4-METHOXYBENZYLLOXY)PREGN-5-EN-20-ONE FOR USE IN THE TREATMENT OF CANNABINOID-RELATED DISORDERSPublication Number: HR-P20240276-T1Priority Date: 2018-02-20
- 3beta-(4-methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of cannabinoids-related disordersPublication Number: WO-2019162328-A1Priority Date: 2018-02-20
- 3BETA-(4-METHOXYBENCYLOXY)PREGN-5-EN-20-ONE FOR USE IN THE TREATMENT OF CANNABINOID-RELATED DISORDERS.Publication Number: MX-2020008687-APriority Date: 2018-02-20
- 3β-(4-methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of cannabinoids-related disordersPublication Number: US-11484537-B2Priority Date: 2018-02-20Grant Date: 2022-11-01
- 3beta-(4-methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of cannabinoids-related disordersPublication Number: AU-2019223049-A1Priority Date: 2018-02-20
- 3beta-(4-methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of cannabinoids-related disordersPublication Number: CA-3090975-A1Priority Date: 2018-02-20
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
“AEF 0117”. AdisInsight. 13 March 2026. Retrieved 17 April 2026.
US 11484537, Piazza PV, Fabre S, Metna M, Monlezun S, Busquet-Garcia A, Cota D, Marsicano G, Revest JM, Vallée M, “3β-(4-methoxybenzyloxy)pregn-5-en-20-one for use in the treatment of cannabinoids-related disorders.”, issued 1 November 2022, assigned to Universite de Bordeaux.
Haney M, Vallée M, Fabre S, Collins Reed S, Zanese M, Campistron G, et al. (June 2023). “Signaling-specific inhibition of the CB1 receptor for cannabis use disorder: phase 1 and phase 2a randomized trials”. Nature Medicine. 29 (6): 1487–1499. doi:10.1038/s41591-023-02381-w. PMC 10287566. PMID 37291212.
 | |
| Clinical data | |
|---|---|
| Other names | AEF0117; AEF-0117; 3β-(4-Methoxybenzyloxy)pregn-5-en-20-one |
| Drug class | Cannabinoid CB1 receptor negative allosteric modulator |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1610878-71-1 |
| PubChem CID | 139433957 |
| ChemSpider | 129421614 |
| Chemical and physical data | |
| Formula | C29H40O3 |
| Molar mass | 436.636 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////lixosicone, anax labs, cannabinoid CB1 receptor signalling inhibitor, AEF0117, AEF 0117, 9LG9CT78SV
Lirucitinib
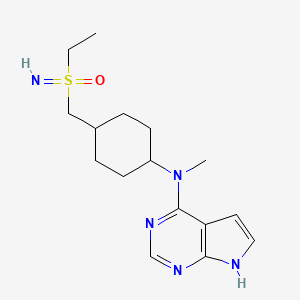
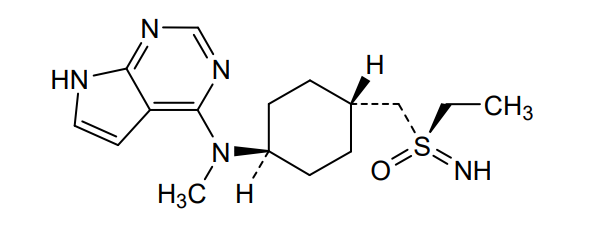
Lirucitinib
CAS 2458115-78-9
MF C16H25N5OS MW335.5 g/mol
N-[4-[(ethylsulfonimidoyl)methyl]cyclohexyl]-N-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine
(R)-ethyl(imino)({trans-4-[methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclohexyl}methyl)-λ6
-sulfanone
Janus tyrosine kinase inhibitor, anti-inflammatory, GGW101, GGW 101
Lirucitinib is a Janus kinase (JAK) inhibitor primarily known as a novel, Class I veterinary drug. It specifically targets the JAK1 enzyme to block itch-inducing (pruritic) and inflammation-causing cytokines in the body.
Core Information
- Primary Use: The drug is developed for veterinary medicine to treat acute and chronic pruritic (severe itch) skin diseases in dogs, which are commonly caused by allergies, parasites, or infections.
- Approval Status: It received a Class I New Veterinary Drug Certificate from the Ministry of Agriculture and Rural Affairs (MARA) in China.
- Human Medicine: As of 2026, there are no clinical indications or indications that Lirucitinib is being tested for use in humans.
- Chemical Profile: It is an orally active small molecule with the chemical formula C₁₆H₂₅N₅OS and acts specifically as the (R)-enantiomer of the compound.
SYN
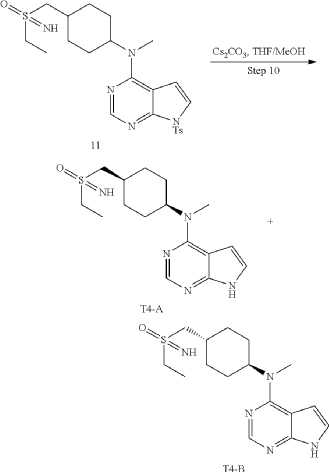
11 (1.0 g, 2.04 mmol) (prepared in step 9), tetrahydrofuran/methanol (10 mL), and cesium carbonate (1.33 g, 4.08 mmol) were added into a 25 mL single-necked flask, refluxed for 12 h, concentrated, and poured into dichloromethane and saturated salt solution, the organic phase was dried with anhydrous sodium sulfate, concentrated, and subjected to a conventional preparation method and a chiral preparation method to obtain product A as a white solid (20 mg, yield: 2.9%), LC-MS: 336 [M+H]+, H 1-NMR: 1H NMR (400 MHz, DMSO) δ 11.61 (s, 1H), 8.09 (s, 1H), 7.13 (s, 1H), 6.54 (s, 1H), 4.67 (s, 1H), 3.90-3.83 (m, 1H), 3.17 (s, 3H), 3.06-2.93 (m, 4H), 2.12-2.01 (m, 3H), 1.73-1.70 (m, 4H), 1.31-1.22 (m, 5H) and product B as a white solid (25 mg, yield: 3.7%), LC-MS: 336 [M+H]+, H 1—NMR: 1H NMR (400 MHz, DMSO) δ 11.59 (s, 1H), 8.09 (s, 1H), 7.12 (dd, J=3.3, 2.6 Hz, 1H), 6.54 (s, 1H), 4.67 (s, 1H), 3.58 (s, 1H), 3.17 (s, 3H), 3.06-2.89 (m, 4H), 2.16-1.93 (m, 3H), 1.74-1.69 (m, 4H), 1.25-1.23 (m, 5H).
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
- Jak inhibitor and preparation method thereforPublication Number: EP-3915989-B1Priority Date: 2019-01-30Grant Date: 2023-06-28
- Jak inhibitor and preparation method thereforPublication Number: US-2022106319-A1Priority Date: 2019-01-30
- Jak inhibitor and preparation method thereforPublication Number: WO-2020155931-A1Priority Date: 2019-01-30
- Jak inhibitor and preparation method thereforPublication Number: EP-3915989-A1Priority Date: 2019-01-30
/////////lirucitinib, anax labs, Janus tyrosine kinase inhibitor, anti-inflammatory, GGW101, GGW 101
Linvemastat
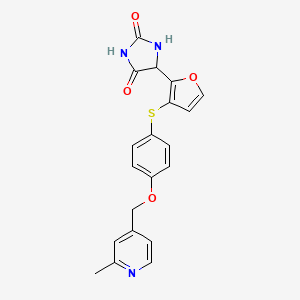
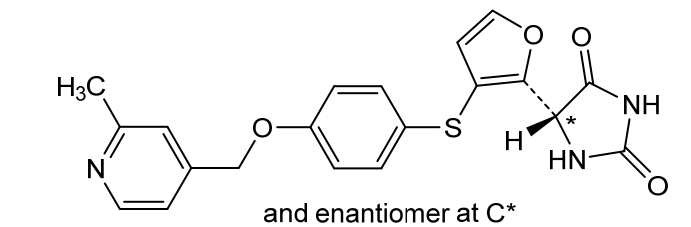
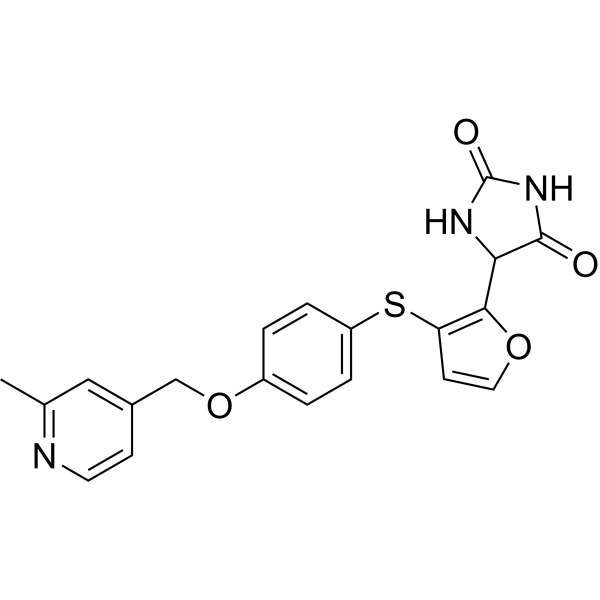
Linvemastat
CAS 2389060-50-6
MF C20H17N3O4S MW395.43
5-[3-[4-[(2-methyl-4-pyridinyl)methoxy]phenyl]sulfanylfuran-2-yl]imidazolidine-2,4-dione
- 2,4-Imidazolidinedione, 5-[3-[[4-[(2-methyl-4-pyridinyl)methoxy]phenyl]thio]-2-furanyl]-
- 5-[3-[[4-[(2-Methyl-4-pyridinyl)methoxy]phenyl]thio]-2-furanyl]-2,4-imidazolidinedione
- rac-(14R)-72-methyl-5-oxa-3-thia-7(4)-pyridina-1(4)-imidazolidina-2(2,3)-furana-4(1,4)-benzenaheptaphane-12,15-dione

matrix metalloproteinase-12 inhibitor, lung diseases, FP 020, FP2AMM4SVF
Linvemastat (also known by its developmental code FP-020) is an investigational, orally active small-molecule drug designed to selectively inhibit matrix metalloproteinase-12 (MMP-12). It is being developed as a potential disease-modifying therapy for chronic inflammatory and fibrotic diseases, primarily targeting respiratory conditions and gastrointestinal disorders.
How Linvemastat Works
- Targeting MMP-12: MMP-12 is an enzyme secreted by macrophages that breaks down extracellular matrix proteins like elastin.
- Controlling Damage: Overexpression of MMP-12 is strongly linked to tissue destruction, chronic inflammation, and fibrosis in the lungs and gut.
- High Selectivity: Unlike older matrix metalloproteinase inhibitors, linvemastat targets MMP-12 with high specificity, avoiding off-target interactions with other vital MMP enzymes.
Primary Therapeutic Indications under Research
- Severe, Uncontrolled Asthma: Evaluated to reduce airway inflammation and improve overall lung function.
- Chronic Obstructive Pulmonary Disease (COPD): Investigated for preventing structural lung damage and progressive emphysema.
- Inflammatory Bowel Disease (IBD): Researched to minimize continuous gut wall inflammation and intestinal fibrosis.
- Other Fibrotic Conditions: Explored in preclinical models for conditions like idiopathic pulmonary fibrosis (IPF) and kidney damage.
Current Clinical Status
Initially developed by Foresee Pharmaceuticals, global development rights for the program were transitioned to Primevera Therapeutics LLC.
- Phase 1 Trial: Completed trials in healthy volunteers demonstrated a favorable safety and pharmacokinetic profile, with only minor, recoverable side effects like mild nausea or headache.
- Phase 2 Trial: Undergoing mid-stage clinical evaluation, such as the global, randomized syMMPonia study. This trial is measuring changes in forced expiratory volume (FEV₁) to assess the drug’s impact on adults with partially controlled, moderate-to-severe asthma.
Effect of Linvemastat in Patients With Partially Controlled Asthma (syMMPonia)
CTID: NCT07191535
Phase: Phase 2
Status: Not yet recruiting
Date: 2025-10-20
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019222157&_cid=P10-MQOLFP-71317-1
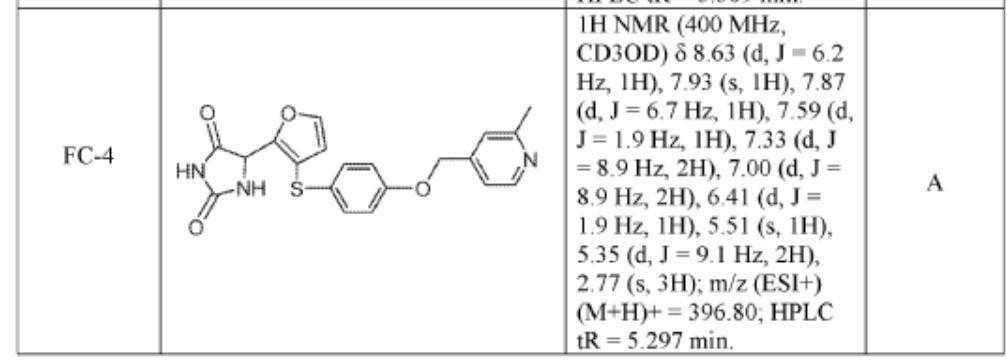
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US276884719&_cid=P10-MQOLBE-66887-1

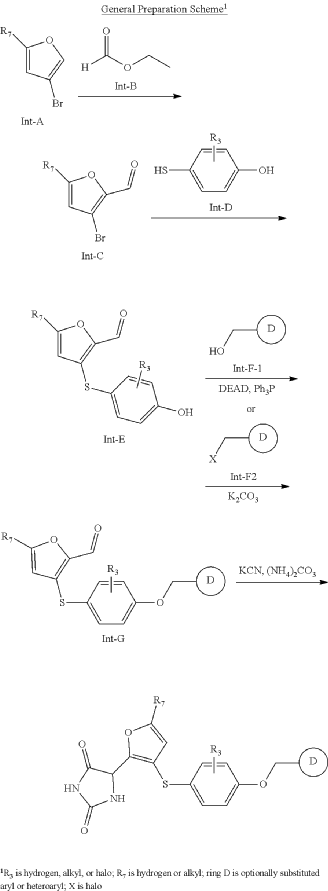
SYN
PAT
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
- Matrix metalloproteinase (MMP) inhibitors and methods of use thereofPublication Number: US-10851089-B2Priority Date: 2018-05-15Grant Date: 2020-12-01
- Matrix metalloproteinase (mmp) inhibitors and methods of use thereofPublication Number: US-2019352287-A1Priority Date: 2018-05-15
//////////////linvemastat, anax labs, matrix metalloproteinase-12 inhibitor, lung diseases, FP 020, FP2AMM4SVF
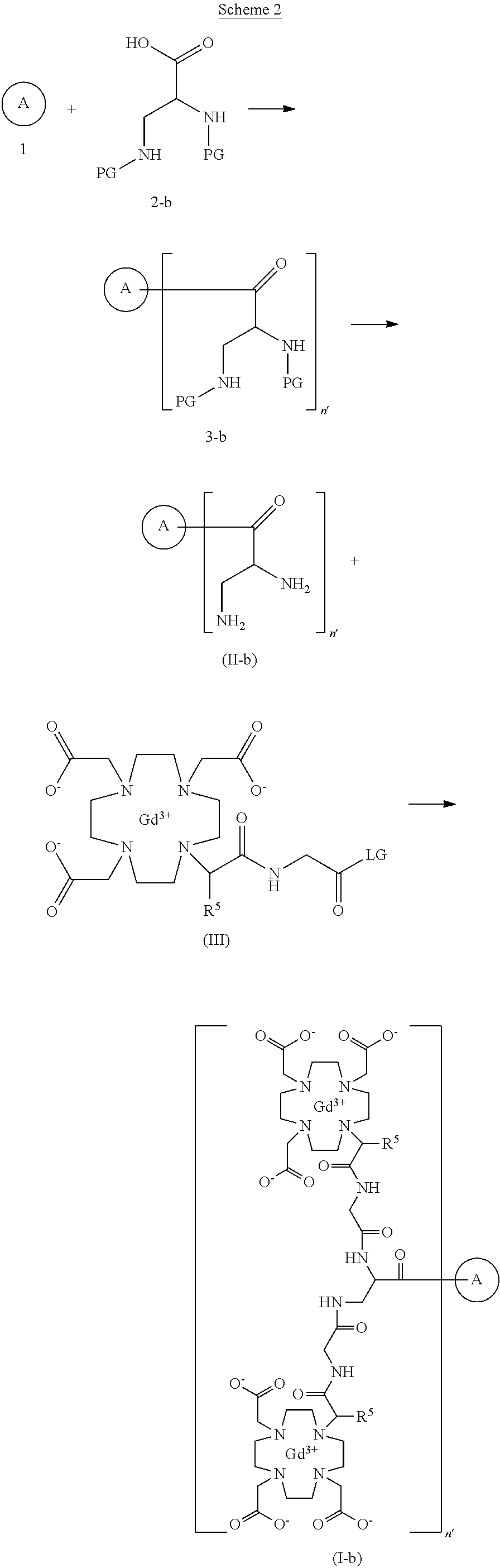
Gadoquatrane
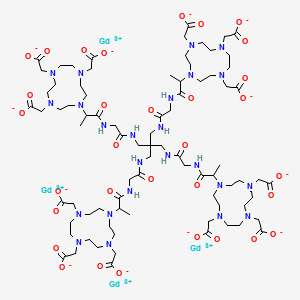
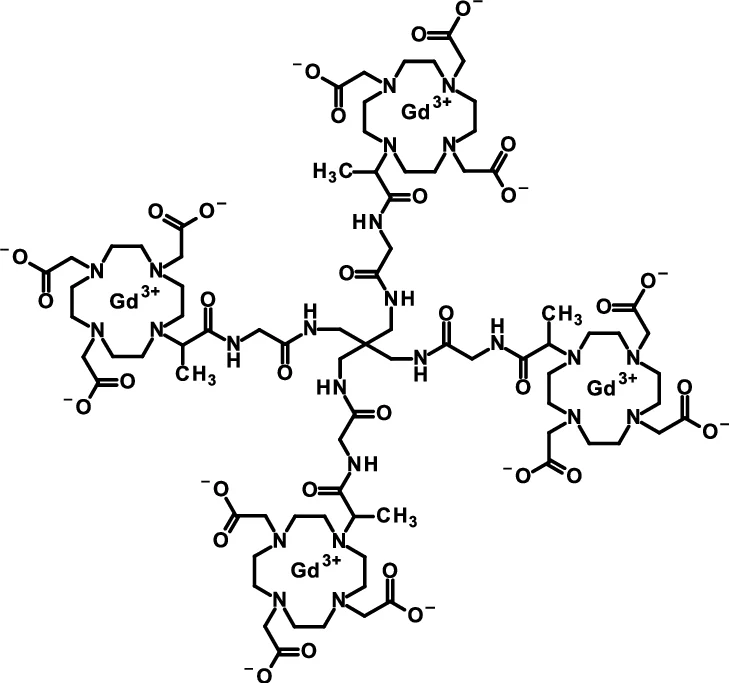
Gadoquatrane
CAS2048221-65-2MW2579.0 g/mol
FDA 2026, APPROVALS 2026, Ambelvist, OZG7J613HK, BAY-1747846, BAY 1747846
2-[4,10-bis(carboxylatomethyl)-7-[1-oxo-1-[[2-oxo-2-[[3-[[2-[2-[4,7,10-tris(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]propanoylamino]acetyl]amino]-2,2-bis[[[2-[2-[4,7,10-tris(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]propanoylamino]acetyl]amino]methyl]propyl]amino]ethyl]amino]propan-2-yl]-1,4,7,10-tetrazacyclododec-1-yl]acetate;tetrakis(gadolinium(3+))
To detect and visualize lesions with abnormal vascularity, in conjunction with MRI
Gadoquatrane (marketed as AMBELVIST®) is a low-dose, macrocyclic gadolinium-based contrast agent (GBCA) developed by Bayer for use in magnetic resonance imaging (MRI). It is designed to enhance the visualization of lesions in the central nervous system (CNS) and other body regions in adult and pediatric patients.
Core Highlights:
- Lower Gadolinium Exposure: It requires a dose of 0.04 mmol/kg, which results in 60% less gadolinium exposure compared to standard macrocyclic GBCAs.
- Regulatory Approval: The FDA approved it in June 2026 for use in adults and pediatric patients, including term neonates. It was also approved in Japan in March 2026.
- Efficacy & Safety: Phase III clinical trials (the QUANTI studies) showed it effectively detects lesions with abnormal vascularity while maintaining an efficacy and safety profile comparable to other standard macrocyclic agents.
- Structure: Gadoquatrane features a tetrameric, macrocyclic structure that gives it high relaxivity and stability in the body
SYN
https://patents.google.com/patent/US20250114485A1
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
- Gadolinium chelate compounds for use in magnetic resonance imagingPublication Number: US-11491245-B2Priority Date: 2015-06-04Grant Date: 2022-11-08
- Gadolinium chelate compounds for use in magnetic resonance imagingPublication Number: US-2025114485-A1Priority Date: 2015-06-04
- Formulation of contrast media and process of preparation thereofPublication Number: US-2024252690-A1Priority Date: 2018-11-23
- Formulation of contrast media and process of preparation thereofPublication Number: US-12303573-B2Priority Date: 2018-11-23Grant Date: 2025-05-20
- New gadolinium chelate compounds for use in magnetic resonance imagingPublication Number: EP-3611169-A1Priority Date: 2015-06-04
- New gadolinium chelate compounds for use in magnetic resonance imagingPublication Number: US-2023113481-A1Priority Date: 2015-06-04
- New gadolinium chelate compounds for use in magnetic resonance imagingPublication Number: EP-3303307-B1Priority Date: 2015-06-04Grant Date: 2019-09-04
- Generation of artificial contrast-enhanced computed tomography imagesPublication Number: US-2024346718-A1Priority Date: 2023-04-13
- Generation of artificial contrast-enhanced radiological imagesPublication Number: WO-2024100233-A1Priority Date: 2022-11-12
- Generation of artificial contrast-enhanced radiological imagesPublication Number: EP-4616420-A1Priority Date: 2022-11-12
- Automated analysis of radiological imagesPublication Number: WO-2024083466-A1Priority Date: 2022-10-17
- Formulation of contrast media and process of preparation thereofPublication Number: US-11944690-B2Priority Date: 2018-11-23Grant Date: 2024-04-02
/////////gadoquatrane, anax labs, FDA 2026, APPROVALS 2026, Ambelvist, OZG7J613HK, BAY-1747846, BAY 1747846
Lasmotinib
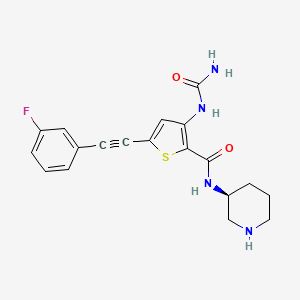
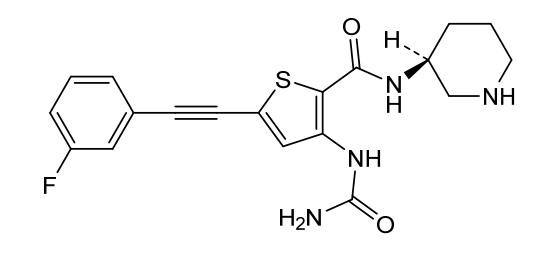
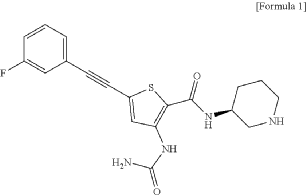
Lasmotinib
CAS 2127107-15-5
MF C19H19FN4O2S MW386.4 g/mol
3-(carbamoylamino)-5-[2-(3-fluorophenyl)ethynyl]-N-[(3S)-piperidin-3-yl]thiophene-2-carboxamide
- N-(2-(N-((3S)(3-Piperidyl))carbamoyl)-5-(2-(3-fluorophenyl)ethynyl)(3-thienyl))aminamide
- 2-Thiophenecarboxamide, 3-((aminocarbonyl)amino)-5-(2-(3-fluorophenyl)ethynyl)-N-(3S)-3-piperidinyl-
- 3-(carbamoylamino)-5-[2-(3-fluorophenyl)ethynyl]-N-[(3S)-piperidin-3-yl]thiophene-2-carboxamide
- 2-Thiophenecarboxamide, 3-[(aminocarbonyl)amino]-5-[2-(3-fluorophenyl)ethynyl]-N-(3S)-3-piperidinyl-
- 3-(carbamoylamino)-5-(2-(3-fluorophenyl)ethynyl)-N-((3S)-piperidin-3-yl)thiophene-2-carboxamide
3-(carbamoylamino)-5-[(3-fluorophenyl)ethynyl]-N-[(3S)-piperidin-3-yl]thiophene-2-carboxamide
tyrosine kinase inhibitor, antineoplastic, PHI-101, PHI 101, U2UY9TBQ8Z
Lasmotinib (also known by its research code PHI-101) is a next-generation, orally bioavailable targeted cancer therapy. It functions as a dual FLT3 and CHK2 inhibitor. It is primarily being investigated to treat Acute Myeloid Leukemia (AML) and ovarian cancer.
How It Works
- FLT3 Inhibition: It targets FMS-like tyrosine kinase 3 (FLT3), an enzyme that is often mutated in AML. Lasmotinib is designed to attack not just single activating mutations (ITD or TKD), but also difficult-to-treat double and triple-resistant mutations.
- CHK2 Inhibition: It also inhibits Checkpoint Kinase 2 (CHK2), preventing cancer cells from repairing DNA damage. This causes the cancer cells to undergo apoptosis (programmed cell death).
Key Clinical Advantages
- High Efficacy: In relapsed or refractory AML patients who have previously failed other FLT3 inhibitors, lasmotinib has demonstrated high rates of composite complete remission.
- Safety Profile: Preclinical and early-stage trials indicate a promising safety profile with a very low or 0% occurrence rate of cardiotoxicity (heart damage), which is a common hurdle for some other FLT3-targeting drugs.
Current Development & Combinations
- Developer: Discovered by Seoul National University Hospital and being developed by Pharos iBio.
- Synergistic Therapies: Lasmotinib is currently moving into global clinical trials as a powerful combination therapy. Research shows it synergizes strongly with existing treatments like Venetoclax or Azacytidine, as well as with emerging Menin inhibitors (such as bleximenib) to achieve deep tumor growth inhibition
Lasmotinib is an orally bioavailable inhibitor of checkpoint kinase 2 (chk2), with potential antineoplastic and chemopotentiating activities. Upon oral administration, lasmotinib binds to and inhibits the activity of chk2, which may prevent the repair of DNA damage caused by DNA-damaging agents. This may result in tumor cell apoptosis and potentiate the antitumor efficacies of various chemotherapeutic agents. Chk2, an ATP-dependent serine–threonine kinase, is a key component in the DNA replication-monitoring checkpoint system and is activated by double-stranded breaks (DSBs); activated chk2 is overexpressed by a variety of cancer cell types.
- Chk2 Inhibitor for Recurrent EpitheliAl periToneal, fallopIan or oVarian cancEr (CREATIVE Phase IA Trial)CTID: NCT04678102Phase: Phase 1Status: Unknown statusDate: 2023-06-26
- Evaluation of the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of PHI 101 for the Treatment of AMLCTID: NCT04842370Phase: Phase 1Status: Unknown statusDate: 2021-04-20
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=JP405710409&_cid=P21-MQIVJB-43702-2
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US465154324&_cid=P21-MQIVJB-43702-2
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024015484&_cid=P21-MQIVJB-43702-2
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2025210599&_cid=P21-MQIVJB-43702-2
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
- Inhibitors of brutons tyrosine kinasePublication Number: US-2021070748-A1Priority Date: 2015-06-02
- New, substituted quinoline compounds as inhibitors of S-nitrosoglutathion reductasePublication Number: HU-E025653-T2Priority Date: 2010-10-08
- New hybrid oligomers. Their preparation process and pharmaceutical compositions containing themPublication Number: AU-2006256439-A1Priority Date: 2005-03-18
- NEW THIOPHENE COMPOUND SUBSTITUTED IN POSITIONS 2,3,5, USED AS A PROTEIN KINASE INHIBITORPublication Number: BR-112018016729-B1Priority Date: 2016-02-16
- 2, 3, 5-substituted thiophene compounds as protein kinase inhibitorsPublication Number: CN-108884066-BPriority Date: 2016-02-16Grant Date: 2021-08-24
- 2,3,5-substituted thiophene compound as protein kinase inhibitorPublication Number: US-10442796-B2Priority Date: 2016-02-16Grant Date: 2019-10-15
- Novel compound of 2,3,5-substituted thiophene as a protein kinase inhibitorPublication Number: RU-2724957-C2Priority Date: 2016-02-16Grant Date: 2020-06-29
- Novel 2,3,5-substituted thiophene compounds as protein kinase inhibitorsPublication Number: KR-101965326-B1Priority Date: 2016-02-16Grant Date: 2019-04-03
- Novel 2,3,5-substituted thiophene compound as protein kinase inhibitorPublication Number: WO-2017142325-A1Priority Date: 2016-02-16
- Novel 2,3,5-substituted thiophene compound as protein kinase inhibitorPublication Number: US-2019047993-A1Priority Date: 2016-02-16
- Novel 2,3,5-substituted thiophene compounds as protein kinase inhibitorsPublication Number: KR-20180136425-APriority Date: 2016-02-16
- Novel 2,3,5-substituted thiophene compound as protein kinase inhibitorPublication Number: EP-3418275-B1Priority Date: 2016-02-16Grant Date: 2021-03-17
- Novel 2,3,5-substituted thiophene compounds that are protein kinase inhibitorsPublication Number: JP-2019504900-APriority Date: 2016-02-16
- Use of 2,3,5-substituted thiophene compound for enhancement of radiotherapyPublication Number: EP-3804719-A1Priority Date: 2018-05-30
- Use of 2,3,5-substituted thiophene compound to prevent, ameliorate, or treat breast cancersPublication Number: EP-3804718-A1Priority Date: 2018-05-30
- Novel 2,3,5-substituted thiophene compound as protein kinase inhibitorPublication Number: EP-3418275-A1Priority Date: 2016-02-16
- New 2,3,5-substituted thiophene compound as a protein kinase inhibitorPublication Number: RU-2018130703-APriority Date: 2016-02-16
- Novel 2,3,5-substituted thiophene compounds as protein kinase inhibitorsPublication Number: KR-20190035671-APriority Date: 2016-02-16
- Radiotherapy-enhancing applications of 2,3,5-substituted thiophene compoundsPublication Number: JP-2021525285-APriority Date: 2018-05-30
- Use of 2,3,5-substituted thiophene compound for enhancement of radiotherapyPublication Number: US-2021205289-A1Priority Date: 2018-05-30
- Use of 2,3,5-Substituted Thiophene Compound for Prevention, Improvement or Treatment of Breast CancerPublication Number: KR-102227117-B1Priority Date: 2018-05-30Grant Date: 2021-03-15
- Use of 2,3,5-Substituted Thiophene Compound for Prevention, Improvement or Treatment of Breast CancerPublication Number: KR-20190136976-APriority Date: 2018-05-30
- Use of 2,3,5-substituted thiophene compound to prevent, ameliorate, or treat breast cancersPublication Number: US-2021205290-A1Priority Date: 2018-05-30
////////lasmotinib, anax labs, tyrosine kinase inhibitor, antineoplastic, PHI-101, PHI 101, U2UY9TBQ8Z
Larubrilstat
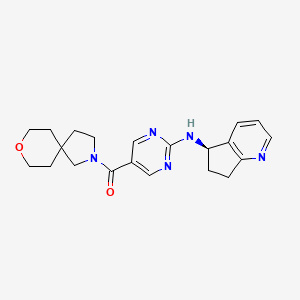
Larubrilstat
CAS 2765226-31-9
MF C21H25N5O2 MW379.5 g/mol
[2-[[(5R)-6,7-dihydro-5H-cyclopenta[b]pyridin-5-yl]amino]pyrimidin-5-yl]-(8-oxa-2-azaspiro[4.5]decan-2-yl)methanone
- [2-[[(5R)-6,7-dihydro-5H-cyclopenta[b]pyridin-5-yl]amino]pyrimidin-5-yl]-(8-oxa-2-azaspiro[4.5]decan-2-yl)methanone
- Methanone, [2-[[(5R)-6,7-dihydro-5H-cyclopenta[b]pyridin-5-yl]amino]-5-pyrimidinyl]-8-oxa-2-azaspiro[4.5]dec-2-yl-
(2-{[(5R)-6,7-dihydro-5H-cyclopenta[b]pyridin-5-yl]amino}pyrimidin-5-yl)(8-oxa-2-azaspiro[4.5]decan-2-
yl)methanone
vascular non-inflammatory molecule-1 (VNN1) inhibitor, AG6K4Y29B4
Larubrilstat is the International Nonproprietary Name (INN) for an experimental, small-molecule vascular non-inflammatory molecule-1 (VNN1) inhibitor. VNN1, also commonly known as Vanin-1 or pantetheinase, is an enzyme involved in tissue response to oxidative stress and inflammation.
Current Status
- Development Context: Larubrilstat is a designated compound linked to therapeutic exploration in inflammatory pathways. Research and patent filings, such as those cataloged by the IUPHAR/BPS Guide to PHARMACOLOGY, track its evaluation alongside similar Vanin-1 inhibitors
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US425298584&_cid=P20-MQHGA8-93141-1
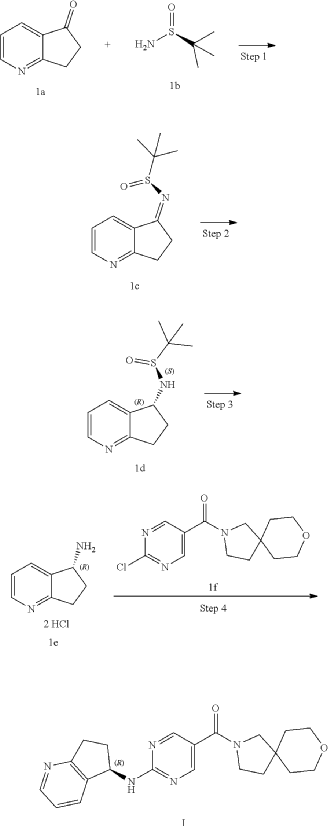
COMP 2-1 IS PRODUCT
Example 2: Synthesis of Compound 2, Compound 2-1 and Compound 2-2
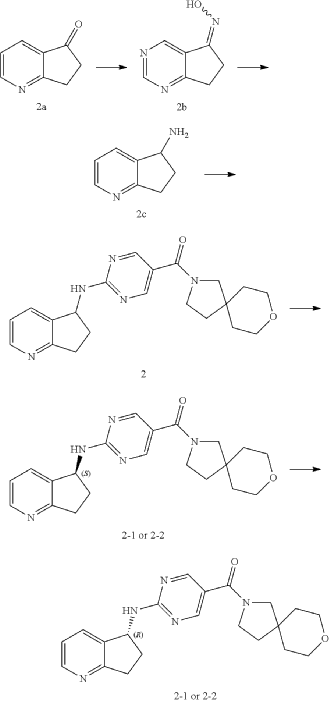
| Step 1 |
Step 2
Step 3
| Compound 2 (66 mg) was obtained from compound if (168 mg) and compound 2c (100 mg) according to the method of Example 1. |
| Two enantiomers 2-1 (retention time: 8.483 min) and 2-2 (retention time: 13.580 min) were obtained by chiral separation of compound 2. |
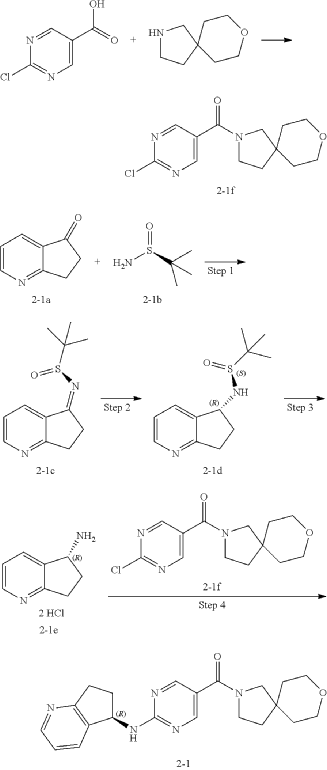
Step 4: Preparation of Compound 2-1
| The absolute stereochemical configuration of compound 2-1 was determined by comparative determination of the above preparation method of the chiral compounds. |
PAT
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
- Pyrimidine carboxamide compound and application thereofPublication Number: US-2024083873-A1Priority Date: 2020-09-25
- Pyrimidine carboxamide compound and application thereofPublication Number: WO-2022063197-A1Priority Date: 2020-09-25
- Pyrimidine carboxamide compound and application thereofPublication Number: WO-2022063333-A1Priority Date: 2020-09-25
////////larubrilstat, ANAX LABS, vascular non-inflammatory molecule-1 (VNN1) inhibitor, AG6K4Y29B4
Laporolimus
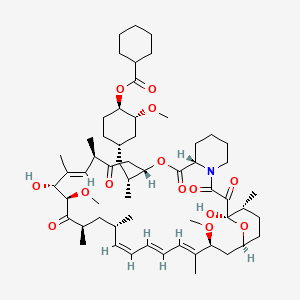
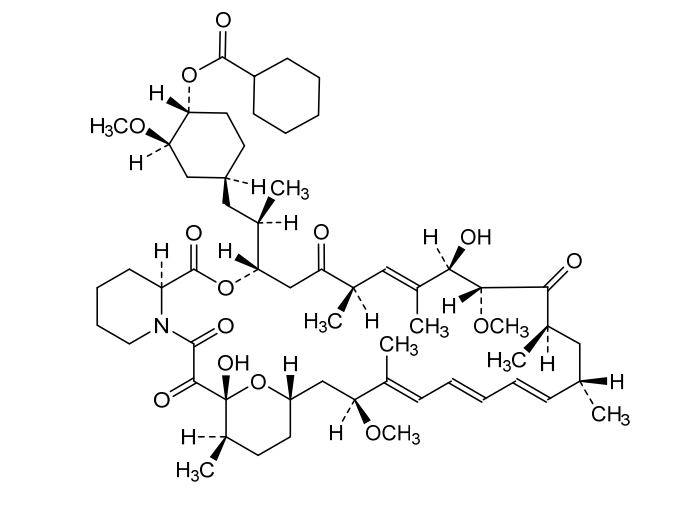
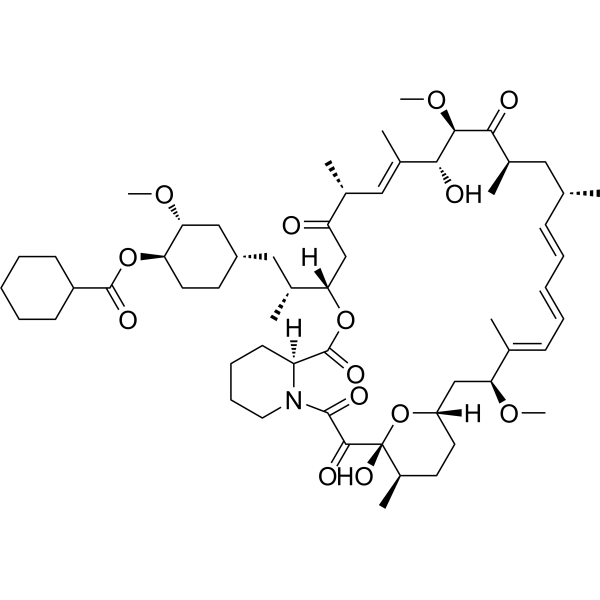
Laporolimus
Rapamycin, 42-cyclohexanecarboxylate
CAS 1504576-27-5
MF C58H89NO14 MW 1024.3 g/mol
[(1R,2R,4S)-4-[(2R)-2-[(1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28E,30S,32S,35R)-1,18-dihydroxy-19,30-dimethoxy-15,17,21,23,29,35-hexamethyl-2,3,10,14,20-pentaoxo-11,36-dioxa-4-azatricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraen-12-yl]propyl]-2-methoxycyclohexyl] cyclohexanecarboxylate
(1R,2R,4S)-4-{(2R)-2-[(3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,27-dihydroxy-10,21-dimethoxy6,8,12,14,20,26-hexamethyl-1,5,11,28,29-pentaoxo1,4,5,6,9,10,11,12,13,14,21,22,23,24,25,26,27,28,29,3
1,32,33,34,34a-tetracosahydro-3H-23,27-epoxypyrido[2,1-c][1,4]oxazacyclohentriacontin-3-yl]propyl}-2-methoxycyclohexyl
cyclohexanecarboxylate
immunosuppressant, CRC-015, CRC 015, F5041W3RVA, Rapamycin, 42-cyclohexanecarboxylate
Laporolimus is an experimental immunosuppressant compound that acts as an mTOR (mechanistic target of rapamycin) pathway inhibitor. It is chemically classified as a macrolide derivative and is also known by its chemical synonym, rapamycin 42-cyclohexanecarboxylate.
Currently, Laporolimus is designated for research use only and has not been approved for clinical medical applications in humans or animals.
Key Technical Details
- Mechanism of Action: It blocks the mTOR signaling pathway, which is responsible for regulating cell growth, proliferation, and immune cell activation.
Distinguishing Laporolimus from Clinical Alternatives
Because it ends with the suffix -limus, it shares structural and nomenclature similarities with widely used clinical immunosuppressants. However, its legal status and development stage differ significantly:
| Drug Name | Clinical Availability | Primary Mechanism | Primary Uses |
|---|---|---|---|
| Laporolimus | None (Research Only) | mTOR Inhibitor | Laboratory research |
| Sirolimus (Rapamycin) | Approved | mTOR Inhibitor | Transplant rejection, coating coronary stents |
| Tacrolimus | Approved | Calcineurin Inhibitor | Organ transplant prophylaxis, severe eczema |
If you are researching this compound for a laboratory study, you can review its structural data and biochemical properties via the PubChem Laporolimus Compound Page
Laporolimus (CAS 1504576-27-5) is an immunosuppressive agent and mTOR inhibitor structurally derived from rapamycin as a cyclohexanecarboxylate derivative. Its total chemical synthesis is highly complex, typically achieved via semisynthesis starting from natural macrolides produced by Streptomyces fermentation.
Semisynthetic Pathway
Because the core macrocyclic lactone (a 36-membered polyketide ring) is incredibly challenging to build from scratch, researchers and pharmaceutical manufacturers rely on a derivatization approach:
- Fermentation: The baseline macrolide is produced via large-scale fermentation of Streptomyces hygroscopicus (similar to the base rapamycin process).
- Purification: The naturally produced macrocyclic core is isolated and purified from the fermentation broth using column chromatography.
- Esterification: The C-42 hydroxyl group of the macrolide core is selectively protected and subjected to acylation with a cyclohexanecarboxylic acid derivative (or reactive cyclohexanecarbonyl chloride).
- Deprotection & Purification: The C-42 cyclohexanecarboxylate is then deprotected and purified via preparative chromatography to yield pure laporolimus.
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
/////////laporolimus, immunosuppressant, CRC-015, CRC 015, F5041W3RVA, Rapamycin, 42-cyclohexanecarboxylate
Lanoracopan
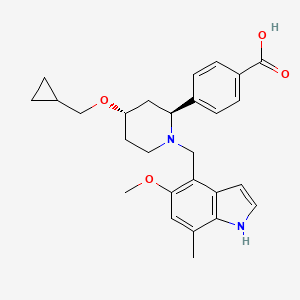
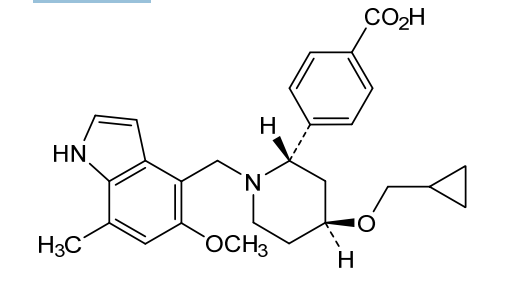
Lanoracopan
CAS 2797066-85-2
MFC27H32N2O4 MW448.6 g/mol
4-[(2S,4S)-4-(cyclopropylmethoxy)-1-[(5-methoxy-7-methyl-1H-indol-4-yl)methyl]piperidin-2-yl]benzoic acid
4-{(2S,4S)-4-(cyclopropylmethoxy)-1-[(5-methoxy-7-methyl-1H-indol-4-yl)methyl]piperidin-2-yl}benzoic acid
complement factor B inhibitor, MY 008211A, Factor B-IN-5, Y5UN7AE8SF
Lanoracopan (also known by its developmental code MY008211A or Factor B-IN-5) is an investigational small-molecule drug that acts as a potent complement factor B (CFB) inhibitor. It is designed to target and regulate the alternative pathway of the complement system, which is a crucial part of the body’s innate immune defense
Clinical Development & Indications
Originally developed by Shanghai Meiyue Biotech Development Co. Ltd., the drug has transitioned from early discovery into active clinical trials. It is primarily being evaluated for:
- Paroxysmal Nocturnal Hemoglobinuria (PNH): Lanoracopan (as MY008211A tablets) is currently undergoing Phase 2 and Phase 2/3 clinical trials. These studies assess its long-term safety, tolerability, and efficacy in patients suffering from PNH who experience active hemolysis (the premature destruction of red blood cells).
- Renal Impairment Studies: Clinical research is also actively evaluating the drug’s safety profile and pharmacokinetics in individuals with varying degrees of kidney function.
Current Status
Lanoracopan is recognized under the World Health Organization’s proposed International Nonproprietary Names (INN) registry. It is not yet approved for public use or commercial medical prescriptions by global regulatory bodies. Currently, it is primarily available to the scientific community as a reference standard for laboratory research use only
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023020566&_cid=P21-MQD5YH-25997-1
[0727]4-((2S,4S)-4-(cyclopropylmethoxy)-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoic acid (compound 4)
[0728]
4-((2S,4S)-4-(cyclopropylmethoxy)-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoic acid
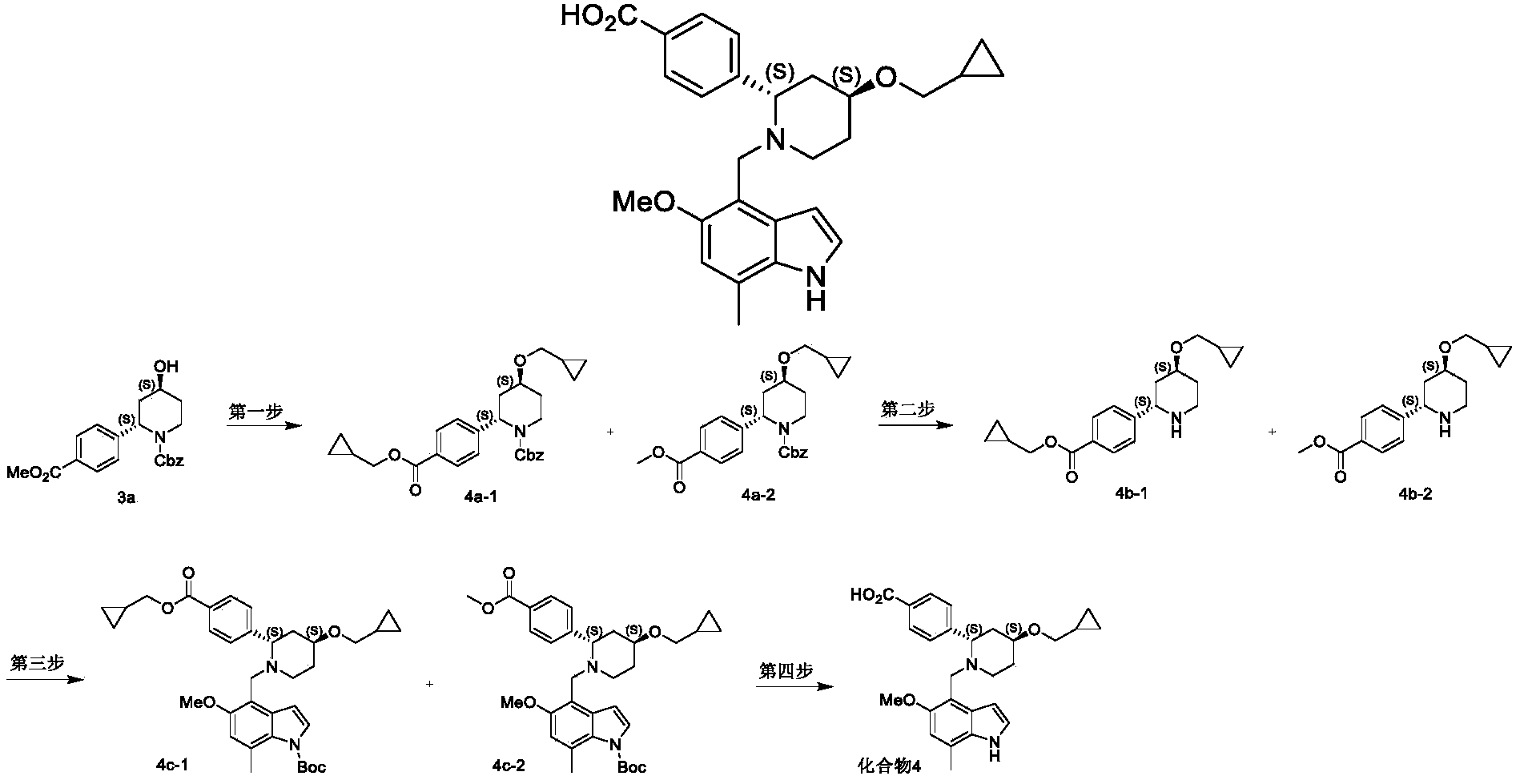
[0755]The mixture (130 mg) of the above-mentioned tert-butyl 4-(((2S,4S)-4-(cyclopropylmethoxy)-2-(4-(((cyclopropylmethoxy)carbonyl)phenyl)piperidin-1-yl)methyl)-5-methoxy-7-methyl-1H-indole-1-carboxylic acid (tert-butyl ester) and 4-(((2S,4S)-4-(cyclopropylmethoxy)-2-(4-(methoxycarbonyl)phenyl)piperidin-1-yl)methyl)-5-methoxy-7-methyl-1H-indole-1-carboxylic acid (tert-butyl ester) (4c-2) was dissolved in 10 mL of methanol, and solid potassium carbonate (149 mg, 1.08 mmol) was added. The mixture was heated to 85 °C and refluxed for 3 h. The reaction solution was cooled to room temperature and concentrated under reduced pressure to obtain the crude product. The crude product was dissolved in a mixed solvent of 10 mL THF, 5 mL methanol, and 2 mL water. Lithium hydroxide monohydrate (181 mg, 4.3 mmol) was added, and the mixture was stirred at room temperature for 16 h. The reaction system was concentrated under reduced pressure, and the crude product was subjected to Pre-HPLC (instrument and preparative column: Glison GX-281 preparative HPLC system; Sunfire C18 column, 5 μm, inner diameter × length = 30 mm × 150 mm). Preparation method: The crude product was dissolved in methanol and dimethyl sulfoxide, and filtered through a 0.45 μm filter membrane to prepare the sample solution. Mobile phase system: acetonitrile/aqueous solution containing 5 mmol/L ammonium acetate. Gradient elution method: Acetonitrile was used to elute 60% of the solution with a 5% gradient (elution time 15 min), and the solution was lyophilized to obtain 4-((2S,4S)-4-(cyclopropylmethoxy)-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoic acid (compound 4) (5 mg).
[0756]
1H NMR(400MHz,CD 3OD)δ8.10(d,2H),7.60(d,2H),7.28(d,1H),6.73(s,1H),6.32(s,1H),4.70–4.40(m,1H),4.32–4.14(m,1H),4.09–3.90(m,1H),3.88–3.79(m,1H),3.75(s, 3H),3.42–3.34(m,2H),3.30–3.14(m,2H),2.49(s,3H),2.26–2.10(m,2H),2.06–1.90(m,2H),1.19–1.04(m,1H),0.64–0.50(m,2H),0.31–0.22(m,2H).
[0757]
LCMS m/z=449.2[M+1] +
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
- Benzo nitrogen-containing heteroaromatic ring derivative and use thereof in medicinePublication Number: WO-2023020566-A1Priority Date: 2021-08-18
- Benzo nitrogen-containing heteroaromatic ring derivative and use thereof in medicinePublication Number: US-2024383917-A1Priority Date: 2021-08-18
- A benzazaaromatic ring derivative and its application in medicinePublication Number: CN-118043314-APriority Date: 2021-08-18
- Benzo nitrogen-containing heteroaromatic ring derivative and use thereof in medicinePublication Number: EP-4389742-A1Priority Date: 2021-08-18
- Complement factor b inhibitor, and pharmaceutical composition thereof, preparation method therefor and use thereofPublication Number: EP-4194451-A1Priority Date: 2020-08-07
- Complement factor b inhibitor, and pharmaceutical composition thereof, preparation method therefor and use thereofPublication Number: EP-4282486-A2Priority Date: 2020-08-07
- Complement factor b inhibitor, and pharmaceutical composition, preparation method and use thereofPublication Number: US-2023286947-A1Priority Date: 2020-08-07
/////////lanoracopan, anax labs, complement factor B inhibitor, MY 008211A, Factor B-IN-5, Y5UN7AE8SF
Lanisidenib
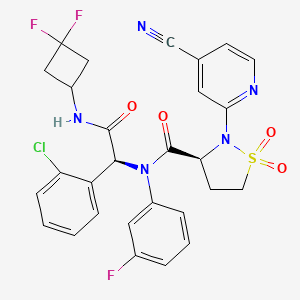
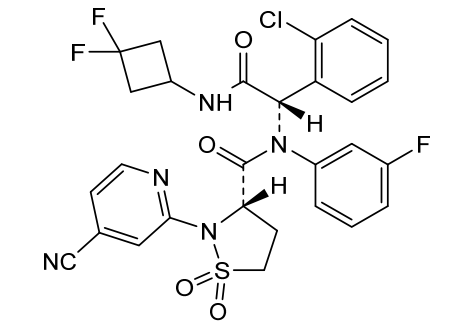
Lanisidenib
Cas 2135537-20-9
MF C28H23ClF3N5O4S MW618.03 g/mol
(3S)-N-[(1S)-1-(2-chlorophenyl)-2-[(3,3-difluorocyclobutyl)amino]-2-oxoethyl]-2-(4-cyano-2-pyridinyl)-N-(3-fluorophenyl)-1,1-dioxo-1,2-thiazolidine-3-carboxamide
IUPAC Name: (3S)-N-{(1S)-1-(2-chlorophenyl)-2-[(3, 3-difluorocyclobutyl)amino]-2-oxoethyl}-2-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-1,1-dioxo-1λ⁶,2-thiazolidine-3-carboxamide
(3S)-N-{(1S)-1-(2-chlorophenyl)-2-[(3,3-difluorocyclobutyl)amino]-2-oxoethyl}-2-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-1,1-dioxo1λ6,2-thiazolidine-3-carboxamide
isocitrate dehydrogenase inhibitor, antineoplastic, G5J396CG5J
Lanisidenib is a potent, selective isocitrate dehydrogenase (IDH) inhibitor that exhibits antineoplastic (anti-cancer) activity. It works by targeting abnormal IDH enzymes, which are frequently mutated in various malignancies, such as certain myeloid leukemias and solid tumours. By blocking these mutant enzymes, it halts the production of oncometabolites that drive cancer progression
Research and Availability
The compound is primarily utilized in biochemical research and preclinical drug screening platforms. Specialty chemical suppliers, such as MedChemExpress and AdooQ BioScience, distribute it exclusively for laboratory research
SYN
Inhibitors of Mutant Isocitrate Dehydrogenases 1 and 2 (mIDH1/2): An Update and Perspective
Publication Name: Journal of Medicinal Chemistry
Publication Date: 2018-05-31
PMID: 29847930
DOI: 10.1021/acs.jmedchem.8b00159
PAT
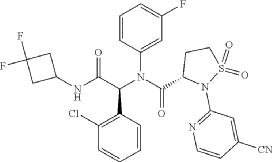
| Step F: (S)—N—((S)-1-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-2-(4-cyanopyridin-2-yl-N-(3-fluorophenyl)-isothiazolidine-3-carboxamide 1,1-dioxide |
| At room temperature, 3-amino-5-Fluorouridine (57 mg, 0.508 mmol) and o-chlorobenzaldehyde (72 mg, 0.512 mmol) were dissolved in methanol, and stirred for 30 min. (S)-2-(4-cyanopyridin-2-yl)isothiazolidine-3-carboxylic acid 1,1-dioxide (136 mg, 0.508 mmol) was then added into the mixed solution, stirred for 10 min, then added with 1,1-difluoro-3-isocyanocyclobutane (prepared according to the method described in patent CN103097340, 60 mg, 0.508 mmol), and stirred overnight. The solvent was removed and the residue was separated by thin layer chromatography, to give the title compound (S)—N—((S)-1-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-2-(4-cyanopyridin-2-yl-N-(3-fluorophenyl)-isothiazolidine-3-carboxamide 1,1-dioxide (the compound of formula I). |
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019057142&_cid=P20-MQBQHW-03190-1
A sulfonamide compound with the structure shown in Formula I has the chemical name: (S)-N-((S)-1-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-2-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-isothiazolidin-3-carboxamide 1,1-dioxide.
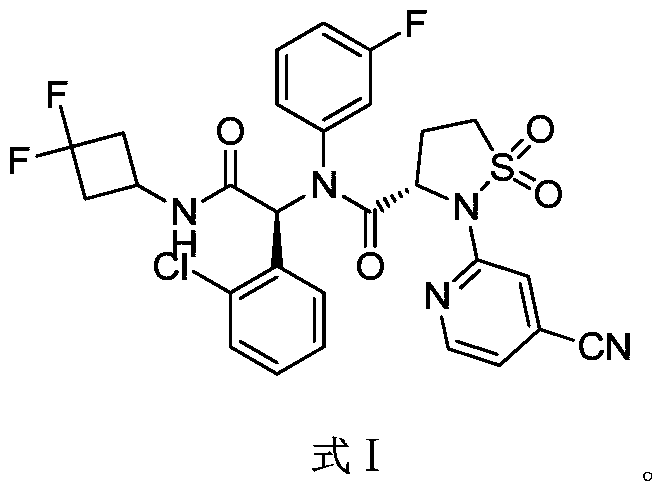
Step F: (S)-N-((S)-1-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-2-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-isothiazolidin-3-carboxamide 1,1-dioxide
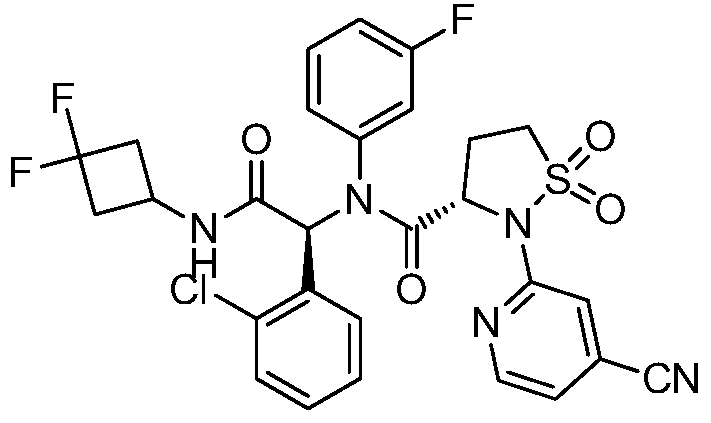
At room temperature, 3-amino-5-fluoropyridine (57 mg, 0.508 mmol) and o-chlorobenzaldehyde (72 mg, 0.512 mmol) were dissolved in methanol and stirred for 30 minutes. Then, (S)-2-(4-cyanopyridin-2-yl)isothiazolidin-3-carboxylic acid 1,1-dioxide (136 mg, 0.508 mmol) was added to the mixture and stirred for 10 minutes. Finally, 1,1-difluoro-3-isocyanocyclobutane (refer to the patent) was added. Prepared by the method described in CN103097340, 60 mg (0.508 mmol), stirred overnight, solvent removed, and separated by thin-layer chromatography to obtain the title compound (S)-N-((S)-1-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-2-(4-cyanopyridin-2-yl)-N-(3-fluorophenyl)-isothiazolidin-3-carboxamide 1,1-dioxide (compound of formula I).
[0134]
1H-NMR(400MHz,CDCl 3):δ=8.46(m,1H),7.67(d,J=8.8Hz,1H),7.63(s,1H),7.22-6.84(m,8H),6.47(d,J=3.6,1H),6.08(s,1H),4.82(d,J=6.1Hz,1H),4.33(m,1H),3.68-3.60(m,1H),3.40-3.28(m,1H),3.10-2.98(m,2H),2.68-2.38(m,4H)。
[0135]
m/z=618[M+H] +。
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PAT
- Sultam compound and application method thereofPublication Number: US-11111240-B2Priority Date: 2016-03-22Grant Date: 2021-09-07
- Sultam Compound And Application Method ThereofPublication Number: US-2021047314-A1Priority Date: 2016-03-22
- Lactam compounds and methods of using the samePublication Number: CN-113666922-APriority Date: 2016-03-22
- Endosulfonamide compound and method of use thereofPublication Number: TW-201736354-APriority Date: 2016-03-22
- Sultam compound and application method thereofPublication Number: EP-3434671-B1Priority Date: 2016-03-22Grant Date: 2020-10-21
- Internal sulfonamide compounds and methods of use thereofPublication Number: CN-109071471-BPriority Date: 2016-03-22Grant Date: 2021-05-07
- Sultam compounds and methods of use thereofPublication Number: KR-102389985-B1Priority Date: 2016-03-22Grant Date: 2022-04-22
- Endosulfonamide compounds and methods of usePublication Number: TW-I729094-BPriority Date: 2016-03-22Grant Date: 2021-06-01
- Crystalline sulfamide compoundsPublication Number: KR-102707847-B1Priority Date: 2017-09-22Grant Date: 2024-09-23
- Crystalline sulfamide compoundPublication Number: KR-20200057049-APriority Date: 2017-09-22
- Crystalline sulfamide compoundPublication Number: CA-3076405-A1Priority Date: 2017-09-22
- Crystalline sulfamide compoundPublication Number: US-2020291012-A1Priority Date: 2017-09-22
- Crystalline sulfamide compoundPublication Number: US-11254665-B2Priority Date: 2017-09-22Grant Date: 2022-02-22
- Preparation method of lactam compoundPublication Number: CN-118580235-APriority Date: 2023-03-03
- A kind of internal sulfonamide compound crystalPublication Number: CN-111065630-APriority Date: 2017-09-22
- Crystalline sulfamide compoundPublication Number: EP-3686191-A1Priority Date: 2017-09-22
- A kind of internal sulfonamide compound crystallizationPublication Number: CN-111065630-BPriority Date: 2017-09-22Grant Date: 2022-12-30
- Crystalline sulfamide compoundPublication Number: EP-3686191-B1Priority Date: 2017-09-22Grant Date: 2022-12-14
///////////lanisidenib, anax labs, isocitrate dehydrogenase inhibitor, antineoplastic, G5J396CG5J
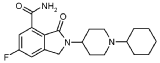
Itareparib
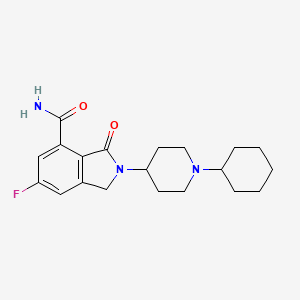
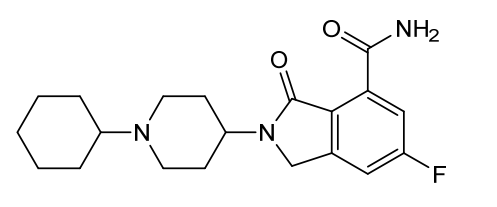
Itareparib
CAS 1606995-47-4
MF C20H26FN3O2 MW359.4 g/mol
2-(1-Cyclohexyl-4-piperidinyl)-6-fluoro-2,3-dihydro-3-oxo-1H-isoindole-4-carboxamide
1H-ISOINDOLE-4-CARBOXAMIDE, 2-(1-CYCLOHEXYL-4-PIPERIDINYL)-6-FLUORO-2,3-DIHYDRO-3-OXO-
2-(1-cyclohexylpiperidin-4-yl)-6-fluoro-3-oxo-2,3-dihydro-1H-isoindole4-carboxamide
poly (ADP-ribose) polymerase (PARP) inhibitor, antineoplastic, NMS-03305293, NMS-293, NMS 03305293, NMS 293, KFI1190L8L, NV 578,
Itareparib is the inhibitor for PARP and exhibits antineoplastic activity.
Itareparib (development code NMS-03305293 or NMS-293) is an experimental, next-generation PARP1-selective oral inhibitor being developed by the biopharmaceutical company Nerviano Medical Sciences for the treatment of various advanced solid tumors and brain cancers
Key Characteristics & Mechanism
Unlike first-generation poly(ADP-ribose) polymerase (PARP) inhibitors, itareparib features a highly specialized mechanism designed to improve clinical safety and versatility:
- Non-Trapping Profile: Traditional PARP inhibitors trap the PARP enzyme onto DNA, forming PARP-DNA complexes. This trapping causes significant bone marrow toxicity (myelosuppression), leading to severe side effects like anemia, neutropenia, and thrombocytopenia. Itareparib is engineered to be “non-trapping,” avoiding these complexes to protect healthy blood cells.
- High Brain Penetrance: It crosses the blood-brain barrier effectively, making it uniquely suitable for treating primary and secondary central nervous system (CNS) malignancies.
- Ideal Combinability: Because it does not cause overlapping bone marrow toxicity, it can be safely paired with other DNA-damaging therapies like traditional chemotherapies and antibody-drug conjugates (ADCs).
Clinical Development & Target Indications
Itareparib is currently advancing through Phase I and Phase II clinical trials. It is being investigated across several oncology settings:
- Glioblastoma (GBM): Evaluated in Phase II clinical studies for relapsed, IDH wild-type glioblastoma in combination with the chemotherapy drug temozolomide (TMZ).
- Ovarian Cancer: Evaluated in Phase Ia/Ib trials (such as trial NCT06930755) in combination with topotecan for patients with recurrent, platinum-resistant ovarian, fallopian tube, or peritoneal cancers. [1]
- Small Cell Lung Cancer (SCLC) & Astrocytoma: Explored in ongoing combination trials targeting highly aggressive tumors where conventional PARP inhibitors are limited by overlapping toxicity.
- Study of NMS-03305293 in Adult Patients With Relapsed Ovarian CancerCTID: NCT06930755Phase: Phase 1Status: RecruitingDate: 2026-05-28
- Study of NMS-03305293 in Adult Patient With Relapsed Small Cell Lung CancerCTID: NCT06931626Phase: Phase 1Status: RecruitingDate: 2025-11-12
- Ph I/II Study of NMS-03305293+TMZ in Adult Patients With Recurrent GlioblastomaCTID: NCT04910022Phase: Phase 1/Phase 2Status: Active, not recruitingDate: 2025-08-19
- Study of NMS-03305293 in Pts with Selected Advanced/Metastatic Solid TumorsCTID: NCT04182516Phase: Phase 1Status: TerminatedDate: 2024-09-19
A Phase I/II Combination Study of NMS-03305293 and Temozolomide in Adult Patients with Recurrent Glioblastoma
EudraCT: 2020-003417-35
Phase: Phase 1, Phase 2
Status: Trial now transitioned
Date: 2021-11-10
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US275481284&_cid=P10-MQA9O8-42416-1
2-(1-Cyclohexyl-piperidin-4-yl)-6-fluoro-3-oz-2,3-dihydro-1H-isoindole-4-carboxylic Acid Amide (I), cpd 29 [R═F; n=m=0; R1=piperidin-4-yl; R2=1-cyclohexyl]
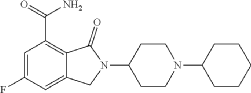
| To a stirred solution of 2-(1-cyclohexyl-piperidin-4-yl)-6-fluoro-3-oxo-2,3-dihydro-1H-isoindole-4-carbonitrile (IV) (100 mg, 0.3 mmol) in acetic acid (5 mL), concentrated sulfuric acid (2.7 mL) was added dropwise during 30 min. The reaction was then warmed at 80° C. for 9 h, cooled at room temperature and poured into cold water (10 mL). The aqueous phase was then made basic by adding concentrated aqueous ammonia and extracted with dichloromethane (3×10 mL). The combined organic phases were washed with 2N aqueous sodium hydroxide (2×12 mL) and brine, dried over Na 2SO 4 and evaporated to dryness in vacuo. The title compound was obtained as a white solid (43 mg, 40%) after purification through column chromatography ((dichloromethane/methanol/ammonia solution, 7N in methanol:97/2/1). |
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014064149&_cid=P10-MQA9K8-39764-1
2-(1-Cyclohexyl-piperidin-4-yl)-6-fluoro-3-oxo-2,3-dihydro-1 H-isoindole-4-carboxylic acid amide (I), cpd 29
[R = F; n = m = 0; R1 = piperidin-4-yl; R2 = 1-cyclohexyl]
To a stirred solution of 2-(1-cyclohexyl-piperidin-4-yl)-6-fluoro-3-oxo-2,3-dihydro-1 H-isoindole-4-carbonitrile (IV) (100 mg, 0.3 mmol) in acetic acid (5 mL), concentrated sulfuric acid (2.7 mL) was added dropwise during 30 min. The reaction was then warmed at 80 °C for 9 h, cooled at room temperature and poured into cold water (10 mL). The aqueous phase was then made basic by adding concentrated aqueous ammonia and extracted with dichloromethane (3 x 10 mL). The combined organic phases were washed with 2N aqueous sodium hydroxide (2 X 12 mL) and brine, dried over Na2S04 and evaporated to dryness in vacuo. The title compound was obtained as a white solid (43 mg, 40%) after purification through column chromatography ((dichloromethane/methanol/ammonia solution, 7N in methanol: 97/2/1).
1H NMR (400.5 MHz, DMSO- cfe) δ ppm 1.00 – 1.14 (m, 1 H), 1.14 – 1.28 (m, 4 H), 1.53 – 1.61 (m, 1 H), 1.67 – 1.80 (m, 6 H), 2.25 – 2.36 (m, 3 H), 2.88 – 2.95 (m, 2 H), 3.94 – 4.03 (m, 1 H), 4.55 (s, 2 H), 7.66 (dd, JHF = 7.7, JHH = 2.6 Hz, 1 H), 7.85 (br. s., 1 H), 7.89 (dd, JHF = 10.9, JHH = 2.6 Hz, 1 H), 10.78 (br. s., 1 H).
HRMS (ESI+): calcd. for C20H27FN3O2 [M + H]+ 360.2082; found 360.2098
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: US-2020407314-A1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective PARP-1 inhibitorsPublication Number: US-11773064-B2Priority Date: 2012-10-26Grant Date: 2023-10-03
- 4-Carboxamide-isoindolinone derivatives as selective PARP-1 inhibitorsPublication Number: JP-6314147-B2Priority Date: 2012-10-26Grant Date: 2018-04-18
- 4-carboxamido-isoindolinone derivatives as selective PARP-1 inhibitorsPublication Number: ES-2813530-T3Priority Date: 2012-10-26Grant Date: 2021-03-24
- 4-carboxamido-isoindolinone derivatives as selective PARP-1 inhibitorsPublication Number: US-10385018-B2Priority Date: 2012-10-26Grant Date: 2019-08-20
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: US-2015274662-A1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: WO-2014064149-A1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: US-11420940-B2Priority Date: 2012-10-26Grant Date: 2022-08-23
- DERIVATIVES OF 4-CARBOXAMIDO-ISOINDOLINONA AS SELECTIVE INHIBITORS OF PARP-1.Publication Number: MX-2015005245-APriority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: US-2019330151-A1Priority Date: 2012-10-26
- COMPOUNDS DERIVED FROM 4-CARBOXAMIDO-ISOINDOLINONE, PROCESS OF PREPARATION OF THESE, IN VITRO METHOD TO SELECTIVELY INHIBIT PARP-1 PROTEIN ACTIVITY, PHARMACEUTICAL COMPOSITION AND USE OF THE REFERRED COMPOUNDSPublication Number: BR-112015009130-B1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: US-2022363636-A1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: CA-2889581-A1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective PARP-1 inhibitorsPublication Number: US-10800739-B2Priority Date: 2012-10-26Grant Date: 2020-10-13
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: EP-2912032-A1Priority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: EP-2912032-B1Priority Date: 2012-10-26Grant Date: 2020-05-27
- DERIVATIVES 4-CARBOXAMIDO-ISOINDOLINONE AS PARP-1 SELECTIVE INHIBITORS, METHOD FOR THEIR PRODUCTION AND APPLICATIONPublication Number: EA-028506-B1Priority Date: 2012-10-26
- 4-Formylamino-isoindolinone derivatives as selective PARP-1 inhibitorsPublication Number: CN-104768948-APriority Date: 2012-10-26
- 4-carboxamido-isoindolinone derivatives as selective parp-1 inhibitorsPublication Number: CA-2889581-CPriority Date: 2012-10-26Grant Date: 2021-06-29
////////itareparib, ANAX LABS, poly (ADP-ribose) polymerase (PARP) inhibitor, antineoplastic, NMS-03305293, NMS-293, NMS 03305293, NMS 293, KFI1190L8L, NV 578,

{kind=link}