

Making small-molecule drugs usually goes something like this: set up a reaction, purify the intermediate, change a solvent, and repeat, repeat, repeat to get the final product. But there’s a lot of waste involved, which is why chemists stress the environmental benefits of an alternate approach: biocatalysis. Engineering enzymes to make reactions happen saves a lot of materials, minimizes chemical and hazardous waste, and even uses less plasticware and glassware. And not having to isolate intermediates saves time.
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TRANILAST
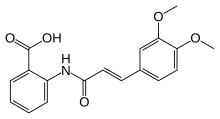
Tranilast
- Molecular FormulaC18H17NO5
- Average mass327.331 Da
2-{[(2E)-3-(3,4-Dimethoxyphenyl)prop-2-enoyl]amino}benzoic acid
3,4-DAA
5070
53902-12-8 [RN]
Tranilast (INN, brand name Rizaben) is an antiallergic drug. It was developed by Kissei Pharmaceuticals and was approved in 1982 for use in Japan and South Korea for bronchial asthma. Indications for keloid and hypertrophic scar were added in the 1980s.
Kissei has developed and launched tranilast in Japan and South Korea for the treatment of allergic rhinitis, asthma and atopic dermatitis. Kissei, in collaboration with GlaxoSmithKline was additionally developing tranilast for the prevention of restenosis following percutaneous transluminal coronary angioplasty.
Medical uses
It is used Japan, South Korea, and China to treat asthma, keloid scars, and hypertrophic scars, and as an ophthalmic solution for allergic pink eye.[1]
It should not be taken women who are or might become pregnant, and it is secreted in breast milk.[1]
Interactions
People who are taking warfarin should not also take tranilast, as they interact.[1] It appears to inhibit UGT1A1 so will interfere with metabolism of drugs that are affected by that enzyme.[1]
Adverse effects
When given systemically, tranilast appears to cause liver damage; in a large well-conducted clinical trial it caused elevated transaminases three times the upper limit of normal in 11 percent of patients, as well as anemia, kidney failure, rash, and problems urinating.[1]
Given systemically it inhibits blood formation, causing leukopenia, thrombocytopenia, and anemia.[1]
Society and culture
As of March 2018 it was marketed in Japan, China, and South Korea under the brand names Ao Te Min, Arenist, Brecrus, Garesirol, Hustigen, Krix, Lumios, Rizaben, Tramelas, Tranilast and it was marketed as a combination drug with salbutamol under the brand name Shun Qi.[2]
In 2016 the FDA proposed that tranilast be excluded from the list of active pharmaceutical ingredients that compounding pharmacies in the US could formulate with a prescription.[1]
Pharmacology
It appears to work by inhibiting the release of histamine from mast cells; it has been found to inhibit proliferation of fibroblasts but its biological target is not known.[3] It has been shown to inhibit the release of many cytokines in various cell types, in in vitro studies.[3] It has also been shown to inhibit NALP3 inflammasome activation and is being studied as a treatment for NALP3-driven inflammatory diseases.[4]
Chemistry
Tranilast is an analog of a metabolite of tryptophan, and its chemical name is 3′,4′-dimethoxycinnamoyl) anthranilic acid (N-5′).[3]
It is almost insoluble in water, easily soluble in dimethylsulfoxide, soluble in dioxane, and very slightly soluble in ether. It is photochemically unstable in solution.[3]
Orally active anti-allergic agent. Prepn: K. Harita et al., DE 2402398; idem, US 3940422 (1974, 1976 both to Kissei).
Y. Kamijo, M. Kobayashi, and A. Ajisawa, Jpn. Kokai, 77/83,428 (1977) via Chem. Abstr.,
88:6,569f (1978).
Research
After promising results in three small clinical trials, tranilast was studied in a major clinical trial (the PRESTO trial) by SmithKline Beecham in partnership with Kissei for prevention of restenosis after percutaneous transluminal coronary revascularization,[5] but was not found effective for that application.[1][6]
As of 2016, Altacor was developing a formulation of tranilast to prevent of scarring following glaucoma surgery and had obtained an orphan designation from the EMA for this use.[7][8]
History
It was developed by Kissei and first approved in Japan and South Korea for asthma in 1982, and approved uses for keloid and hypertrophic scars were added later in the 1980s.[3]
PATENT
tranilast product case US03940422 , expired in all the regional territories.
PATENT
WO2013144916 claiming tranilast complexes and cocrystals with nicotinamide, saccharin, gentisic acid, salicylic acid, urea, 4-aminobenzoic acid and 2,4-dihydroxybenzoic acid
Patent
WO-2020035546
Nuformix Ltd
Novel crystalline forms of tranilast or its salts as histamine H1 receptor antagonist useful for treating allergy, allergic rhinitis and atopic dermatitis.
Tranilast, (2-[[3-(3,4-dimethoxyphenyl)-l-oxo-2-propenyl]amino] benzoic acid, shown below), was originally developed as an anti-allergy drug due to its ability to inhibit the release of inflammatory mediators, such as histamine, from mast cells and basophils (P. Zampini. IntJ Immunopharmacol. 1983;
Tranilast
Tranilast has been marketed in Japan, China and South Korea by Kissei Pharmaceutical Co. Ltd, for allergic conditions such as allergic conjunctivitis, bronchial asthma, allergic rhinitis and atopic dermatitis, under the Rizaben® brand name for more than thirty years. More recently tranilast has also been shown to have anti-proliferative properties. Tranilast was shown to inhibit the proliferation of fibroblasts and suppress collagen synthesis (M. Isaji. Biochem Pharmacol. 1987; 36: 469-474) and also to inhibit the transformation of fibroblasts to myofibroblasts and their subsequent contraction (M. Isaji. Life Sci. 1994; 55: 287-292). This additional behaviour led to tranilast gaining additional approval for the treatment of keloids and hypertrophic scars.
[004] Over recent years many researchers have explored the anti-proliferative effects of tranilast to assess its potential in fibrotic and cancerous conditions. Its anti-proliferative action is believed to be due to its ability to inhibit transforming growth factor beta (TGF-b) (H. Suzawa. Jpn J Pharmacol. 1992 Oct; 60(2): 91-96). Fibrosis is a condition that can affect most organs of the body and fibroblast proliferation, differentiation and collagen synthesis are known to be key factors in the progression of most types of fibrosis. Tranilast has been shown in-vivo to have potential beneficial effects in
numerous fibrotic conditions. Tranilast has been shown in-vivo to have potential in lung fibrosis (M. Kato. Eur RespirJ. 2013; 42(57): 2330), kidney fibrosis (DJ Kelly, J Am Soc Nephrol. 2004; 15(10): 2619-29), cardiac fibrosis (J Martin, Cardiovasc Res. 2005; 65(3): 694-701), ocular fibrosis (M J Moon, BMC Opthalmol. 2016; 16: 166) and liver fibrosis (M Uno, Hepatology. 2008; 48(1): 109-18.
[005] Tranilast’s anti-tumor action has also recently been demonstrated, in-vitro and in-vivo. Tranilast has been shown to inhibit the proliferation, apoptosis and migration of several cell lines including breast cancer (R. Chakrabarti. Anticancer Drugs. 2009 Jun; 20(5): 334-45) and prostate cancer (S. Sato. Prostate. 2010 Feb; 70(3): 229-38) cell lines. In a study of mammary carcinoma in mice tranilast was found to produce a significant reduction in metastasis (R. Chakrabarti. Anticancer Drugs. 2009 Jun; 20(5): 334-45). In a pilot study in humans, tranilast was shown to have the potential to improve the prognosis of patients with advanced castration-resistant prostate cancer (K. Izumi. Anticancer Research. 2010 Jul; 30: 73077-81). In-vitro studies also showed the therapeutic potential of tranilast in glioma (M Platten. IntJ Cancer. 2001; 93:53-61), pancreatic cancer (M Hiroi, J Nippon Med Sch. 2002; 69: 224-234) and gastric carcinoma (M Yashiro, Anticancer Res. 2003; 23: 3899-3904).
[006] Given the wide range of fibrotic conditions and cancers for which tranilast could have a potential therapeutic benefit, as well as the different patient types and specific areas of the body requiring treatment, it is anticipated that patients would benefit from having multiple delivery methods for the administration of tranilast so as to best suit the patient’s needs. The pharmaceutical compositions could include, for example, a solid oral dosage, a liquid oral dosage, an injectable composition, an inhalable composition, a topical composition or a transdermal composition.
[007] Kissei Pharmaceutical Co. Ltd explored the anti-proliferative effect of tranilast in the prevention of restenosis associated with coronary intervention. In a Phase II clinical study Kissei found that the current approved dose of tranilast (300 mg/day) was insufficient to prevent restenosis and that a higher dose of 600 mg/day was needed to achieve a decrease in restenosis rates (H. Tamai, Am Heart J.1999; 138(5): 968-75). However, it was found that a 600 mg daily dosage can result in a ten-fold inter-patient variation in plasma concentrations of the drug (30-300 pmol/L) (H Kusa ma. Atherosclerosis. 1999; 143: 307-313) and in the Phase III study of tranilast for the prevention of restenosis the dose was further increased to 900mg daily (D Holmes, Circulation. 2002; 106(10): 1243-1250).
[008] The marketed oral form of tranilast (Rizaben®) contains tranilast in its pure crystalline form. Crystalline tranilast has extremely low aqueous solubility (solubility of 14.5 pg/ml in water and 0.7 pg/ml in pH 1.2 buffer solution (Society of Japanese Pharmacopoeia. 2002)). Whilst, high energy amorphous forms are often used as a means of improving the solubility of poorly soluble drug
compounds, literature shows that an amorphous form of tranilast is not completely photostable in the solid state and that it undergoes photodegradation on storage when exposed to light (S. Onoue. EurJ Pharm Sci. 2010; 39: 256-262).
[009] It is expected that the very low solubility of tranilast is a limiting factor in the oral bioavailability of the drug. Given the limited time any drug has to firstly dissolve in the
gastrointestinal tract and then be absorbed into the bloodstream, this issue will become even more limiting as the oral dose of tranilast is increased. The poor solubility of tranilast is also possibly a key factor in the high inter-patient variability reported for higher dose tranilast pharmacokinetics. As a BCS class II drug (low solubility/high permeability) it is expected that absorption from the gastrointestinal tract is hampered by the dissolution rate of the drug in gastrointestinal media as well as its overall solubility. For treatment of chronic proliferative diseases such as fibrosis and cancer it is vital for the delivery method of a drug to produce consistent, predictable plasma levels that are maintained above the minimum effective concentration. To achieve efficacious oral delivery of tranilast at higher doses there is a need for new solid forms of the drug with both high solubility and rapid dissolution rates.
[010] Given the severity of conditions involving cancer or fibrosis there is also a need for systemic treatment options by which tranilast can be delivered by healthcare specialists that do not require the patient to swallow solid oral dosage forms. Alternative dosage forms suitable for these needs could include, for example, injectable compositions, liquid oral formulations or nebulized inhaled formulations. These would require a liquid formulation of tranilast suitable for systemic delivery. [Oil] Given the potential of tranilast to treat ocular diseases, such as allergic conjunctivitis, Kissei Pharmaceutical Co. Ltd recognised the need to develop an eye drop formulation of tranilast for localised treatment. However, as well as having very low aqueous solubility, tranilast is also photochemically unstable when stored in solution, resulting in significant degradation (N Hori, Chem. Pharm. Bull. 1999; 47(12): 1713-1716). Therefore, the only way Kissei were able to achieve an eye drop liquid composition of tranilast was to use both solubilising and stabilising agents in the formulation (US Patent 5356620). The resulting 0.5% (w/v) eye drop formulation is currently also marketed under the Rizaben® brand name. However, the focus of this formulation and of the subsequent research that has attempted to produce alternative solution formulations of tranilast has always been solely on external delivery of tranilast using compositions such as eye drops and skin ointments etc. None of the liquid formulations of tranilast previously described have been produced for systemic delivery such as for oral or IV delivery. Excipients used in the previously reported external preparations are not suitable for systemic delivery. Also, despite the successful
development of an eye drop formulation of tranilast, the package insert of the marketed Rizaben® eye drops states that the product should not be stored in a refrigerator as crystals may precipitate.
[012] Thus, there remains a need for aqueous pharmaceutical compositions of tranilast suitable for systemic delivery. Given the potential photochemical degradation issue of long term storage of tranilast in solution and also the disadvantage of the larger storage facilities needed to store bulkier solution based formulations it would also be advantageous to develop a stable highly soluble solid form of tranilast that can be quickly dissolved at the time of treatment by the patient or healthcare provider to produce the required liquid formulation.
[013] Following efforts to make a liquid formulation of tranilast, Kissei made the statement that tranilast and pharmaceutically acceptable salts thereof are too insoluble in water to prepare an aqueous solution (US Patent 5356620). Since that US patent the only crystalline pharmaceutically acceptable salt to have been published is the sodium salt (N Geng, Cryst. Growth Des. 2013; 13: 3546-3553). In line with the findings of Kissei the authors of this paper stated that the apparent solubility of the crystalline tranilast sodium salt is even less than that of pure tranilast. Also, when they performed a dissolution study of tranilast in a sodium containing media they found that as the tranilast dissolved it gradually precipitated out of solution as its sodium salt indicating that the sodium salt has a lower thermodynamic solubility than the pure drug. The authors of this paper also successfully prepared the non-pharmaceutically acceptable crystalline cytosine salt of tranilast. Despite this crystalline cytosine salt showing approximately a two-fold solubility improvement over pure crystalline tranilast, not only would this crystalline cytosine salt not be suitable for systemic delivery to a patient due to cytosine not having FDA acceptability but this improvement in solubility would not be great enough to produce high dose tranilast liquid formulations such as an injectable formulation.
[014] Patent application EP1946753 discloses an attempt to prepare an external preparation of tranilast and claims the preparation of ionic liquid salts of tranilast with organic amines. The inventors claim that blending tranilast with the organic amine results in a liquid form. This application does not disclose the formation of any solid state, crystalline tranilast salts with organic amines. They demonstrate that these ionic liquid forms of tranilast have higher solubility in solvents suitable for external application to the skin and that these preparations have higher photostability than pure tranilast in the same formulation. However, this improved photostability still results in a significant proportion of the tranilast being photo-degraded and would not be suitable for long term storage. Also, the solvents used for preparation of these ionic liquid salt formulations are not suitable for internal delivery of tranilast. Moreover, there is no mention in EP1946753 of improved solubility in aqueous or bio-relevant media.
PATENT
https://patents.google.com/patent/US20150119428
-
Tranilast, (2-[[3-(3,4-dimethoxyphenyl)-1-oxo-2-propenyl]amino]benzoic acid), shown below, is a therapeutic agent that exhibits an anti-allergic effect. It has been shown to inhibit the release of inflammatory mediators, such as histamine, from mast cells and basophils (P. Zampini. Int J Immunopharmacol. 1983; 5(5): 431-5). Tranilast has been used as an anti-allergic treatment, for several years in Japan and South Korea, for conditions such as allergic conjunctivitis, bronchial asthma, allergic rhinitis and atopic dermatitis.
-

- [0004]
Tranilast is currently marketed in Japan and South Korea by Kissei Pharmaceutical Co. Ltd under the Rizaben® brand name. As well as displaying an anti-allergic effect tranilast has been shown to possess anti-proliferative properties. Tranilast was found to inhibit the proliferation of fibroblasts and suppress collagen synthesis (M. Isaji. Biochem Pharmacol. 1987; 36: 469-474) and also to inhibit the transformation of fibroblasts to myofibroblasts and their subsequent contraction (M. Isaji. Life Sci. 1994; 55: 287-292). On the basis of these effects tranilast is now also indicated for the treatment of keloids and hypertrophic scars. Its anti-fibrotic action is believed to be due to its ability to inhibit transforming growth factor beta (TGF-β) (H. Suzawa. Jpn J Pharmacol. 1992 October; 60(2): 91-96). TGF-β induced fibroblast proliferation, differentiation and collagen synthesis are known to be key factors in the progression of idiopathic pulmonary fibrosis and tranilast has been shown in-viva to have potential in the treatment of this chronic lung disease (T. Jiang. Afr J Pharm Pharmaco. 2011; 5(10): 1315-1320). Tranilast has also been shown in-vivo to be have potential beneficial effects in the treatment of airway remodelling associated with chronic asthma (S. C. Kim. J Asthma 2009; 46(9): 884-894.
- [0005]
It has been reported that tranilast also has activity as an angiogenesis inhibitor (M. Isaji. Br. J Pharmacol. 1997; 122(6): 1061-1066). The results of this study suggested that tranilast may be beneficial for the treatment of angiogenic diseases such as diabetic retinopathy and age related macular degeneration. As well as showing inhibitory effects on mast cells and fibroblasts, tranilast has also demonstrated an ability to diminish tumor necrosis factor-alpha (TNF-α) from cultured macrophages (H. O. Pae. Biochem Biophys Res Commun. 371: 361-365) and T-cells (M. Platten. Science. 310: 850-855), and inhibited NF-kB-dependent transcriptional activation in endothelial cells (M. Spieker. Mol Pharmacol. 62: 856-863). Recent studies have revealed that tranilast attenuates inflammation and inhibits bone destruction in collagen induced arthritis in mice suggesting the possible usefulness of tranilast in the treatment of inflammatory conditions such as arthritis (N. Shiota. Br. Pharmacol. 2010; 159 (3): 626-635).
- [0006]
As has recently been demonstrated, in-vitro and in-vivo, tranilast also possesses an anti-tumor action. Tranilast has been shown to inhibit the proliferation, apoptosis and migration of several cell lines including breast cancer (R. Chakrabarti. Anticancer Drugs. 2009 June; 20(5): 334-45) and prostate cancer (S. Sato. Prostate. 2010 February; 70(3): 229-38) cell lines. In a study of mammary carcinoma in mice tranilast was found to produce a significant reduction in metastasis (R. Chakrabarti. Anticancer Drugs. 2009 June; 20(5): 334-45). In a pilot study in humans, tranilast was shown to have the potential to improve the prognosis of patients with advanced castration-resistant prostate cancer (K. Izurni. Anticancer Research. 2010 July; 30: 73077-81).
- [0007]
It has been reported that tranilast has the ability to induce or enhance neurogenesis and, therefore, could be used as an agent to treat neuronal conditions such as cerebral ischernia, glaucoma, multiple sclerosis, amyotrophic lateral sclerosis, Alzheimer’s disease, neurodegenerative trinucleotide repeat disorders, neurodegenerative lyosomal storage diseases, spinal cord injury and trauma, dementia, schizophrenia and peripheral neuropathy (A. Schneider. EP2030617).
- [0008]
Tranilast’s beneficial properties have been reported to have utility in several ocular conditions. Tranilast is currently approved in Japan and Korea far the treatment of allergic conjunctivitis. WO2010137681 claims the use of tranilast as a prophylactic or therapeutic agent for the treatment of retinal diseases. The anti-fibrotic properties of tranilast have been reported to be of benefit in maintaining the filtering blob during glaucoma surgery and this has been demonstrated in a pilot study in humans (E. Chihara.J Glaucoma. 1999; 11(2): 127-133). There have also been several reported cases of the beneficial use of tranilast in the prevention of postoperative recurrence of pterygium (C. Fukui. Jap J Opthalmol. 1999; 12: 547-549). Tsuji recently reported that tranilast may be beneficial not only in the prevention of ptergium recurrence, but also for the inhibition of symblepharon and granuloma formation (A. Tsuji. Tokai J Exp Clin Med. 2011; 36(4): 120-123). Collectively it has been demonstrated that tranilast possesses anti-allergic, anti-fibrotic, anti-inflammatory, anti-tumor, neurogenesis enhancing end angiogenesis inhibitory properties and as such may be useful for the treatment of diseases associated with such properties.
- [0009]
Tranilast occurs as a yellow crystalline powder that is identified by CAS Registry Number: 53902-12-8. As is typical of cinnamic acid derivatives (G. M. J. Schmidt J Chem. Soc. 1964: 2000) tranilast is photochemically unstable when in solution, tranforming into cis-isomer and dimer forms on exposure to light (N. Hori. Cehm Pharm Bull. 1999; 47: 1713-1716). Although pure crystalline tranilast is photochemically stable in the solid state it is practically insoluble in water (14.5 μg/ml) and acidic media (0.7 μg/ml in pH 1.2 buffer solution) (Society of Japanese Pharmacopoeia. 2002). Although tranilast has shown activity in various indications, it is possible that the therapeutic potential of the drug is currently limited by its poor solubility and photostability. High energy amorphous forms are often used as a means of improving the solubility of poorly soluble APIs, however, literature shows that amorphous solid dispersions of tranilast are not completely photostable in the solid state and that they undergo photodegradation on storage when exposed to light (S. Onoue. Eur J Pharm Sci. 2010; 39: 256-262). US20110136835 describes a combination of tranilast and allopurinol and its use in the treatment of hyperuricemia associated with gout and has one mention of a “co-crystal form”, but lacks any further description or characterization.
Patent
Publication numberPriority datePublication dateAssigneeTitle
Family To Family Citations
JP2001072605A *1999-09-032001-03-21Lion CorpTransdermal and transmucosal absorption-promoting agent composition
US6585997B22001-08-162003-07-01Access Pharmaceuticals, Inc.Mucoadhesive erodible drug delivery device for controlled administration of pharmaceuticals and other active compounds
CA2548281C2003-12-092013-11-12Medcrystalforms, LlcMethod of preparation of mixed phase co-crystals with active agents
WO2007046544A1 *2005-10-212007-04-26Medrx Co., Ltd.Preparation for external application comprising salt of mast cell degranulation inhibitor having carboxyl group with organic amine
WO2008078730A1 *2006-12-262008-07-03Translational Research, Ltd.Preparation for transnasal application
EP2030617A12007-08-172009-03-04Sygnis Bioscience GmbH & Co. KGUse of tranilast and derivatives thereof for the therapy of neurological conditions
CN101683330A *2008-09-232010-03-31沈阳三川医药科技有限公司Oral compound pharmaceutic preparation containing tranilast and salbutamol
US20110136835A1 *2009-09-142011-06-09Nuon Therapeutics, Inc.Combination formulations of tranilast and allopurinol and methods related thereto
EP2429495A4 *2009-05-152014-01-22Shin Nippon Biomedical Lab LtdIntranasal pharmaceutical compositions with improved pharmacokinetics
WO2010137681A12009-05-292010-12-02参天製薬株式会社Prophylactic or therapeutic agent for retinal diseases comprising tranilast, method for prevention or treatment of retinal diseases, and tranilast or pharmaceutically acceptable salt thereof and use thereof
JP2011093849A *2009-10-302011-05-12Kissei Pharmaceutical Co LtdEasily dissolvable powder inhalant composed of tranilast
Family To Family Citations
CN106344550A *2016-09-282017-01-25江苏省人民医院Application of tranilast to preparation of medicines for treating pneumoconiosis
CN107286210A *2017-06-192017-10-24昆药集团股份有限公司A kind of Acegastrodine compound and preparation method thereof, preparation and application
References
- ^ Jump up to:a b c d e f g h “FDA Proposed Rules” (PDF). Federal Register. 81 (242): 91071–91082. December 16, 2016. Another version of same published at here
- ^ “International brands for Tranilast”. Drugs.com. Retrieved 10 March 2018.
- ^ Jump up to:a b c d e Darakhshan, S; Pour, AB (January 2015). “Tranilast: a review of its therapeutic applications”. Pharmacological Research. 91: 15–28. doi:10.1016/j.phrs.2014.10.009. PMID 25447595.
- ^ Y. Huang et al, “Tranilast directly targets NLRP3 to treat inflammasome-driven diseases.”, EMBO Mol Med., 10(4), 2018
- ^ “Kissei’s existing business flat but R&D pipeline should lead to growth”. The Pharma Letter. 8 September 2000.
- ^ Holmes, D. R; Savage, M; Lablanche, J. M; Grip, L; Serruys, P. W; Fitzgerald, P; Fischman, D; Goldberg, S; Brinker, J. A; Zeiher, A. M; Shapiro, L. M; Willerson, J; Davis, B. R; Ferguson, J. J; Popma, J; King Sb, 3rd; Lincoff, A. M; Tcheng, J. E; Chan, R; Granett, J. R; Poland, M (2002). “Results of Prevention of REStenosis with Tranilast and its Outcomes (PRESTO) Trial”. Circulation. 106 (10): 1243–50. doi:10.1161/01.CIR.0000028335.31300.DA. PMID 12208800.
- ^ “Tranilast – Altacor: ALT-401”. AdisInsight. Retrieved 10 March 2018.
- ^ “EU/3/10/756 Orphan Designation”. European Medicines Agency. 6 August 2010. Retrieved 10 March 2018.
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
Oral |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| ChemSpider | |
| UNII | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.150.125 |
| Chemical and physical data | |
| Formula | C18H17NO5 |
| Molar mass | 327.336 g·mol−1 |
| 3D model (JSmol) | |
///////////////Tranilast, Rizaben, antiallergic, Kissei Pharmaceuticals, Japan, South Korea, bronchial asthma, keloid, hypertrophic scar
Enavogliflozin, DWP-16001


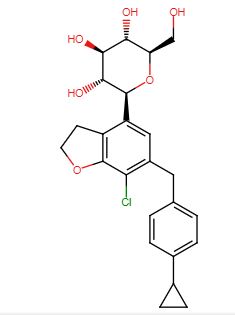

Enavogliflozin, DWP-16001
(2S,3R,4R,5S,6R)-2-(7-chloro-6-(4-cyclopropylbenzyl)-2,3-dihydrobenzofuran-4-yl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
CAS: 1415472-28-4
Chemical Formula: C24H27ClO6
Molecular Weight: 446.92
Elemental Analysis: C, 64.50; H, 6.09; Cl, 7.93; O, 21.48
Green Cross Corp INNOVATOR
Daewoong Pharmaceutical Co Ltd
Enavogliflozin is an antidiabetic (hypoglycemic).
Daewoong is investigating DWJ-304 , a sodium/glucose cotransporter 2 (SGLT-2) inhibitor, for treating type 2 diabetes. By February 2017, preclinical development was underway. Daewoong is developing DWP-16001 , presumed to be enavogliflozin, a SGLT-2 inhibitor, for treating type 2 diabetes. In September 2019, launch was expected in 2023.
Enavogliflozin (DWP16001/GCC5694A) is a selective SLC5A2 and SGLT2 inhibitor developed to treat diabetes and obesity.[1][2][3][4][5] It was developed by GC Pharma[6][7] and Daewoong Pharmaceutical.[7]
Enavogliflozin has been approved for clinical use in South Korea,[7] and Ecuador,[8] and applied for approval in Brazil, Mexico, Peru, and Colombia.[8]
PATENT
WO2012165914
DWP-16001 expire in EU states until June 2032 and US in November 2033.
PATENT
US 2014274918
PATENT
US2019169174
Paragraph 0305; 0340; 0347
H NMR (400 MHz, CD3OD) δ 7.02 (d, J=8.0 Hz, 2H), 6.92 (d, J=8.0 Hz, 2H), 6.81 (s, 1H), 4.59 (t, J=8.8 Hz, 2H), 4.11 (d, J=9.2 Hz, 1H), 3.96 (ABq, ΔvAB=19.0 Hz, JAB=15.2 Hz, 2H), 3.87-3.84 (m, 1H), 3.67-3.63 (m, 1H), 3.47-3.37 (m, 3H), 3.35-3.33 (m, 3H), 1.85-1.79 (m, 1H), 0.91-0.86 (m, 2H), 0.61-0.57 (m, 2H)
PATENT
WO2017217792 , claiming process for preparing diphenylmethane derivative.





1H NMR(400 MHz, CD 3OD) δ 7.02(d, J = 8.0 Hz, 2H), 6.92(d, J = 8.0 Hz, 2H), 6.81(s, 1H), 4.59(t, J = 8.8 Hz, 2H), 4.11(d, J = 9.2 Hz, 1H), 3.96(ABq, Δv AB = 19.0 Hz, J AB = 15.2 Hz, 2H), 3.87-3.84(m, 1H), 3.67-3.63(m, 1H), 3.47-3.37(m, 3H), 3.35-3.33(m, 3H), 1.85-1.79(m, 1H), 0.91-0.86(m, 2H), 0.61-0.57(m, 2H); [M+Na] + 469.
PATENT
WO-2020036382
The present invention relates to a method for producing an intermediate useful for the synthesis of a diphenylmethane derivative that can be used as a SGLT inhibitor. A method for synthesizing a compound of formula 7 according to the present invention has solved the problem of an existing synthesis process which requires an additional process due to the synthesis of Grignard reagent and the management of a related substance. In addition, the process can be simplified by minimizing the formation of the related substance and eliminating the need for reprocessing of reaction products, thereby becoming capable of maximizing a yield of a diphenylmethane derivative.
Process for preparing intermediates of SGLT inhibitor and their use for the synthesis of diphenyl-methane derivative, which can be used as SGLT inhibitors.
Sodium-dependent glucose cotransporters (SGLT) allow the transport of Na + along the concentration gradient simultaneously with the transport of glucose across the concentration gradient. Currently two important SGLT isoforms have been cloned, known as SGLT1 and SGLT2. SGLT1 is located in the intestine, kidney and heart and regulates cardiac glucose transport. SGLT1 is a high affinity low dose transporter and therefore only accounts for a portion of renal glucose reuptake. In contrast, SGLT2 is a low affinity, high dose transporter located primarily in the apica domain of epithelial cells in the early proximal manure tubules. In healthy individuals, over 99% of the plasma glucose filtered out of the renal glomeruli is reabsorbed and less than 1% of the total filtered glucose is excreted in the urine. It is estimated that 90% of renal glucose reuptake is promoted by SGLT2 and the remaining 10% is mediated by SGLT1 in the late proximal canal. Genetic mutations in SGLT2 do not have a particular adverse effect on carbohydrate metabolism but cause increased kidney glucose secretion of about 140 g / day following mutation. Human mutation studies have been the subject of therapeutic studies because SGLT2 is believed to be responsible for most renal glucose resorption.
[3]
Korean Unexamined Patent Publication No. 2017-0142904 discloses a method for producing a diphenylmethane derivative having inhibitory activity against SGLT2. Since the above document prepares diphenylmethane derivatives by a convergent synthesis method in which each group is individually synthesized and then coupled, the synthesis route is more concise and yield is higher than the linear synthesis method disclosed in the prior art. It is disclosed that it can increase and reduce the risks inherent in sequential synthesis pathways.
[4]
However, the preparation method of the diphenylmethane derivative according to Korean Patent Publication No. 2017-0142904 uses a heavy metal such as pyridinium chlorochromate (PCC) to burden safety management, and the Grignard reagent. In addition to the need for a separate manufacturing process, the cost of the additional process is not only incurred, but also the management of the flexible material is necessary because the flexible material from the Grignard reagent manufacturing process is included in the final product. In addition, since the product generated after the reaction between the intermediate and the Grignard reagent includes additional flexible materials, there is a problem that a reprocessing process of such flexible materials is required.
Step 3. 4- bromo- 7- chloro -6- (4- cyclopropylbenzyl ) -2,3- dihydrobenzofuran (Compound 6)
(4-bromo-7-chloro-2,3-dihydrobenzofuran-6-yl) (4-cyclopropylphenyl) methanone (Compound 5) in a mixture of dichloromethane (9.7 mL) and acetonitrile (9.7 mL) at -15 ° C. g, 2.57 mmol) was added Et 3 SiH (1.2 mL, 7.71 mmol) and BF 3 -Et 2 O (0.79 mL, 6.42 mmol) in this order. The reaction mixture was allowed to warm to room temperature and then stirred for 4 hours. After completion of the reaction by TLC, the reaction solution was added with saturated NaHCO 3aqueous solution (40 mL) to terminate the reaction, and extracted with ethyl acetate. The organic layer obtained by extraction was dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. The concentrated residue was purified by silica gel chromatography to give the title compound 6 (0.84 g, 89.9%) as an off-white solid.
[387]
1 H NMR (500 MHz, CDCl 3): δ 7.07 (d, J = 10.0 Hz, 2H), 6.99 (d, J = 10.0 Hz, 2H), 6.80 (s, 1H), 4.70 (t, J = 11.0 Hz , 2H), 3.97 (s, 2H), 3.26 (t, J = 11.0 Hz, 2H), 1.88-1.84 (m, 1H), 0.95-0.90 (m, 2H), 0.68-0.64 (m, 2H); LC-MS: [M + H] & lt; + & gt; 363.

REFERENCES
1: Markiewicz M, Jungnickel C, Stolte S, Białk-Bielińska A, Kumirska J, Mrozik W. Ultimate biodegradability and ecotoxicity of orally administered antidiabetic drugs. J Hazard Mater. 2017 Jul 5;333:154-161. doi: 10.1016/j.jhazmat.2017.03.030. Epub 2017 Mar 16. PubMed PMID: 28349868.
2: Holt RI. Trials of new anti-diabetes agents. Diabet Med. 2017 Feb;34(2):147. doi: 10.1111/dme.13306. PubMed PMID: 28090726.
References
- Hwang, Jun Gi; Lee, SeungHwan; Huh, Wan; Han, Jumi; Oh, Jaeseong; Jang, In-Jin; Yu, Kyung-Sang (September 2022). “Dose-dependent glucosuria of DWP16001, a novel selective sodium–glucose cotransporter-2 inhibitor, in healthy subjects”. British Journal of Clinical Pharmacology. 88 (9): 4100–4110. doi:10.1111/bcp.15348. PMID 35395697.
- Kim, Ju-Hyun; Kim, Dong Kyun; Choi, Won-Gu; Ji, Hye-Young; Choi, Ji-Soo; Song, Im-Sook; Lee, Sangkyu; Lee, Hye Suk (11 September 2020). “In Vitro Metabolism of DWP16001, a Novel Sodium-Glucose Cotransporter 2 Inhibitor, in Human and Animal Hepatocytes”. Pharmaceutics. 12 (9): 865. doi:10.3390/pharmaceutics12090865. PMC 7558535. PMID 32932946.
- Kim, Byungwook; Huh, Ki Young; Hwang, Jun Gi; Nah, JaeJin; Huh, Wan; Cho, Jae Min; Jang, In-Jin; Yu, Kyung-Sang; Kim, Yun; Lee, SeungHwan (April 2023). “Pharmacokinetic and pharmacodynamic interaction between DWP16001, an sodium–glucose cotransporter 2 inhibitor and metformin in healthy subjects”. British Journal of Clinical Pharmacology. 89 (4): 1462–1470. doi:10.1111/bcp.15613. PMID 36422809. S2CID 253838705.
- Rhee, Beomseok; Mahbubur, Rahman Md; Jin, Changfan; Choi, Ji-Soo; Lim, Hyun-Woo; Huh, Wan; Park, Joon Seok; Han, Jumi; Kim, Sokho; Lee, Youngwon; Park, Jinho (December 2022). “Evaluation of safety and anti-obesity effects of DWP16001 in naturally obese dogs”. BMC Veterinary Research. 18 (1): 237. doi:10.1186/s12917-022-03324-2. PMC 9214997. PMID 35733159.
- Yoon, Sukyong; Park, Min Soo; Jin, Byung Hak; Shin, Hyobin; Na, Jaejin; Huh, Wan; Kim, Choon Ok (3 July 2023). “Pharmacokinetic and pharmacodynamic interaction of DWP16001, a sodium-glucose cotransporter-2 inhibitor, with phentermine in healthy subjects”. Expert Opinion on Drug Metabolism & Toxicology. 19 (7): 479–485. doi:10.1080/17425255.2023.2249397. PMID 37593838. S2CID 265846294.
- Kong, Young Kyu; Song, Kwang-Seop; Jung, Myung Eun; Kang, Misuk; Kim, Hyeon Jung; Kim, Min Ju (2022-01-15). “Discovery of GCC5694A: A potent and selective sodium glucose co-transporter 2 inhibitor for the treatment of type 2 diabetes”. Bioorganic & Medicinal Chemistry Letters. 56: 128466. doi:10.1016/j.bmcl.2021.128466. ISSN 0960-894X.
- Kim, Min-Soo; Song, Yoo-Kyung; Choi, Ji-Soo; Ji, Hye Young; Yang, Eunsuk; Park, Joon Seok; Kim, Hyung Sik; Kim, Min-Joo; Cho, In-Kyung; Chung, Suk-Jae; Chae, Yoon-Jee; Lee, Kyeong-Ryoon (2023-03-14). “Physiologically Based Pharmacokinetic Modelling to Predict Pharmacokinetics of Enavogliflozin, a Sodium-Dependent Glucose Transporter 2 Inhibitor, in Humans”. Pharmaceutics. 15 (3): 942. doi:10.3390/pharmaceutics15030942. ISSN 1999-4923. PMC 10058973. PMID 36986803.
- “Daewoong Pharmaceutical’s diabetes drug wins approval in Ecuador – 매일경제 영문뉴스 펄스(Pulse)”. Pulse (in Korean). 2024. Retrieved 2025-04-04.
 |
|
| Clinical data | |
|---|---|
| Other names | DWP16001 |
| ATC code | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C24H27ClO6 |
| Molar mass | 446.92 g·mol−1 |
| 3D model (JSmol) | |
/////// DWJ-304, Daewoong Pharmaceutical, DWP-16001, SGLT-2 inhibitor, type 2 diabetes, KOREA, Enavogliflozin, GCC 5694A
ClC1=C2C(CCO2)=C([C@@H]3O[C@H](CO)[C@@H](O)[C@H](O)[C@H]3O)C=C1CC4=CC=C(C5CC5)C=C4
SYN
Synthesis 2024, 56, 906–943
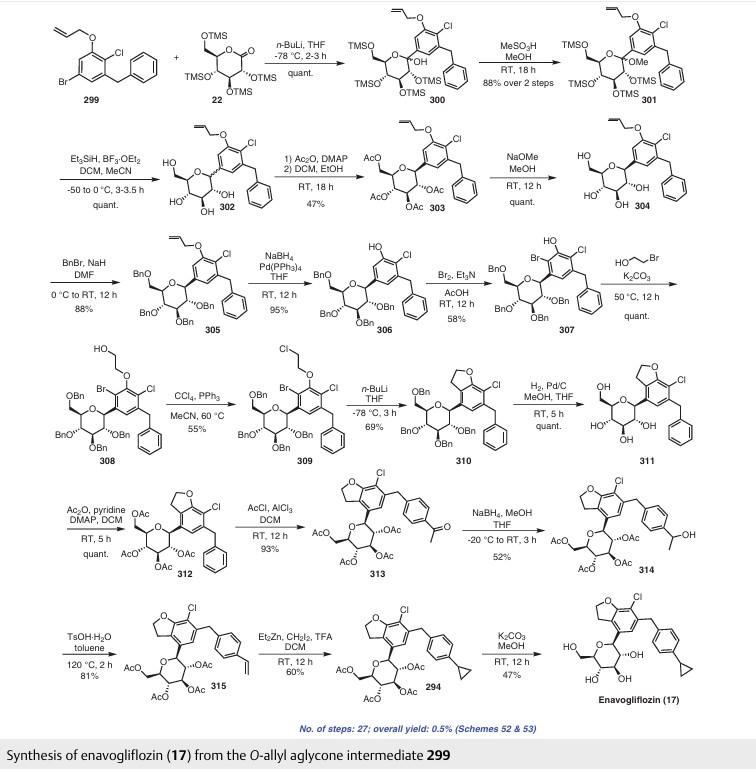
Enavogliflozin (17) (DWP-16001) was developed by Green Cross Corp. and Daewoong Pharmaceuticals Co. Ltd.with the intention of addressing type 2 diabetes. Its chemical name is (2S,3R,4R,5S,6R)-2-(7-chloro-6-(4-cyclopropylbenzyl)-2,3-dihydrobenzofuran-4-yl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol. In clinical trials, this medication exhibited remarkable efficacy as both an antidiabetic agent and an SGLT inhibitor. The initial synthetic pathway for producing envogliflozin (17), in addition to other C-aryl-glycoside-type derivatives, was documented in the United States, specifically through patent application
number US9034921B2.75 Enavogliflozin (17) belongs to the category of C-aryl glycoside derivatives and its synthesis encompasses 19 steps, ultimately achieving an overall yield of 12%.
The process starts with the preparation of aglycone key intermediate 290 (Scheme 50), and involves a series chemical transformations starting from the commercially available 3-methoxy-2-nitrobenzoic acid (275). Following the successful synthesis of the aglycone intermediate 290, the process was advanced by employing n-BuLi for lithium–halogen exchange on 290 and subsequent addition of lithiated 290 to O-silyl-protected compound 22 at –78 °C (Scheme51). This sequence yielded the TMS-protected lactol inter
mediate 291 in quantitative yield. By subjecting this intermediate to treatment with MsOH/MeOH, the desired product 292 was obtained, achieving a 2-step overall yield of 88%. During these reactions, the O-silyl groups of the C-glucoside 292 were cleaved. Furthermore, the reduction of intermediate 292 was executed in the presence of triethylsilane and boron trifluoride–diethyl etherate complex, lead
ing to the formation of desmethoxy intermediate 293 in 100% yield. Subsequent acetylation of the four hydroxy groups was performed using acetic anhydride and a catalytic quantity of DMAP, producing the tetra-acetyl intermediate 294 in a yield of 59%. Ultimately, removal of the acetyl groups was achieved in the presence of NaOH, culminating in the generation of the final product, enovagliflozin (17),
with a 100% yield (Scheme 51). This synthetic procedure is plagued by significant limitations, including an extended route to obtain the aglycone intermediate 290, the application of protection and deprotection chemistry, and the necessity of cryogenic conditions to obtain the lactol intermediate 291. According to the reaction sequences given in Schemes 50 and 51, the overall yield of the final compound is calculated to be 12% via a total of 19 steps. In another approach, enavogliflozin (17) was synthesized in 27 steps with an overall yield of 0.5% (Schemes 52and 53).76 Initially, the synthesis of O-allyl aglycone intermediate 299 was achieved in nine steps starting from 3 methoxy-2-nitrobenzoic acid (275) (see Scheme 51). Methylation of compound 275 using methyl iodide and potassium carbonate in DMF afforded methyl ester 276 in 98% yield. Reduction of the NO2 group of 276 was carried out using Pd/C to afford aryl amine 277 in excellent yield. Next, bromination of 277 was facilitated by using NBS in DMF and
ethyl acetate to afford the brominated compound 278 in 86% yield. Chlorination was then carried out on intermediate 278 under diazotization reaction conditions to afford 279. The ester group of 279 was hydrolyzed under basic conditions to afford the aryl carboxylic acid 295. Subsequently, the preparation of acid chloride 296 from 295 was achieved using oxalyl chloride and a catalytic amount of DMF, which was coupled with benzene under Friedel Crafts acylation conditions to give the aryl benzophenone
intermediate 297. Reduction of the keto group of 297 was achieved by using triethylsilane and TFA/TfOH. Finally, the O-allyl aglycone intermediate 299 was obtained, when in termediate 298 was subjected to O-allylation using allylbromide and potassium carbonate in acetone. Next, the O-allyl aglycone intermediate 299 was subjected to a Br/Li exchange reaction using n-BuLi and addition of the obtained lithiated compound was carried out on gluconolactone 22 to afford the lactol intermediate 300 in quantitative yield (Scheme 53). The hydroxy group of 300 was methylated using methanesulfonic acid in methanol to
give 301 in 88% yield over two steps from 299. Demethoxylation and TMS cleavage was carried out on 301 using triethylsilane and BF3·Et2O to furnish intermediate 302. This hydroxy intermediate was protected using acetic anhydride and DMAP to afford the acetylated compound 303, deprotection of which with sodium methoxide gave product 304 in 100% yield. Benzylation of 304 was carried out using BnBr and NaH to give tetra-O-benzylated compound 305. Next, the O-allyl group of 305 was reduced to give alcohol 306 in 95% yield. Bromination followed by O-alkylation of the intermediate 306 then furnished compound 308. The hydroxy group of 308 was replaced by a chlorine atom under treatment with CCl4 and PPh3 to afford 309. Using n-BuLi, intramolecular cyclization was carried out to give com
pound 310 in 69% yield and subsequent debenzylation by treatment with Pd/C and H2 afforded compound 311. Again, acetylation of the free hydroxy groups of 311 was achieved using acetic anhydride and DMAP to give the O-acetylated intermediate 312. A Friedel–Crafts reaction on the aryl moiety of intermediate 312 gave acylated product 313 in 93% yield. The keto group of 313 was reduced using sodium borohydride in methanol to give the 314, which was further reduced under acidic conditions to give the alkene intermediate 315. The Simmons–Smith cyclopropanation was achieved on the alkene intermediate 315 to give compound 294 in 60% yield. Finally, the acetyl groups were removed
from the sugar moiety of 294 to give enavogliflozin (17) in 47% yield. This synthetic route also contains major disadvantages in terms of the use of protection/deprotection strategies, a lengthy linear process and employs several harmful reagents.
(75) Choi, S.; Song, K. S.; Lee, S. H.; Kim, M. J.; Seo H. J.; Park, E.-J.; Kong, Y.; Park, S. O.; Kang, H.; Jung, M. E.; Lee, K.; Kim, H. J.; Lee, J. S.; Lee, M. W.; Kim, M.-S.; Hong, D. H.; Kang, M. US9034921B2, 2015.
(76) Yoon, H.-K.; Park, S.-H.; Yoon, J.-S.; Choi, S.; Seo, H. J.; Park, E.-J.; Kong, Y.; Song, K.-S.; Kim, M. J.; Park S. O. WO2017217792A1, 2017.
.


,
LYS 228

LYS228
BOS-228
LYS-228
Molecular Formula, C16-H18-N6-O10-S2
Molecular Weight, 518.4783
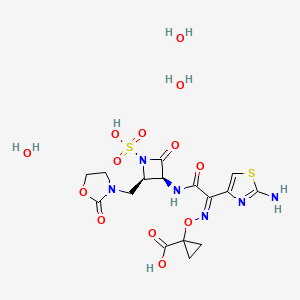
(3S,4R)-3-((Z)-2-(2-Ammoniothiazol-4-yl)-2-((1-carboxycyclopropoxy)imino)acetamido)-2-oxo-4-((2-oxooxazolidin-3-yl)methyl)azetidine-1-sulfonate
RN: 1810051-96-7
UNII: 29H7N9XI1B
UNII-005B24W9YP
005B24W9YP
Lys-228 trihydrate
2091840-43-4
Yclopropanecarboxylic acid, 1-(((Z)-(1-(2-amino-4-thiazolyl)-2-oxo-2-(((3S,4R)-2-oxo-4-((2-oxo-3-oxazolidinyl)methyl)-1-sulfo-3-azetidinyl)amino)ethylidene)amino)oxy)-, hydrate (1:3)
1-[(Z)-[1-(2-amino-1,3-thiazol-4-yl)-2-oxo-2-[[(3S,4R)-2-oxo-4-[(2-oxo-1,3-oxazolidin-3-yl)methyl]-1-sulfoazetidin-3-yl]amino]ethylidene]amino]oxycyclopropane-1-carboxylic acid;trihydrate
BOS-228 (LYS-228) is a monobactam discovered at Novartis and currently in phase II clinical development at Boston Pharmaceuticals for the treatment of complicated urinary tract infection and complicated intraabdominal infections in adult patients.
The compound has been granted fast track and Qualified Infectious Disease Product (QIDP) designation from the FDA.
In October 2018, Novartis licensed to Boston Pharmaceuticals worldwide rights to the product.
Paper
https://pubs.acs.org/doi/10.1021/acs.oprd.9b00330
Patent
US 20150266867
PATENT
WO 2017050218
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017050218&tab=FULLTEXT
Compound X: 1- ( ( (Z) – (1- (2-aminothiazol-4-yl) -2-oxo-2- ( ( (3S, 4R) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) -1-sulfoazetidin-3-yl) amino) ethylidene) amino) oxy) cyclopropanecarboxylic acid.
[0126]
Step 1: Benzhydryl 1- ( ( (Z) – (1- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) -2-oxo-2- ( ( (3S, 4R) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidin-3-yl) amino) ethylidene) amino) oxy) cyclopropanecarboxylate. To a solution of (Z) -2- ( (1- ( (benzhydryloxy) carbonyl) cyclopropoxy) imino) -2- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) acetic acid (854 mg, 1.59 mmol) prepared according to published patent application US2011/0190254, Intermediate B (324 mg, 1.75 mmol) and HATU (785 mg, 2.07 mmol) in DMF (7.9 mL) , DIPEA was added (832 μL, 4.77 mmol) . After 1 h of stirring, it was poured into water and extracted with EtOAc. Brine was added to the aqueous layer, and it was further extracted with ethyl acetate (EtOAc) (3x) . The combined organic layers were dried over Na 2SO 4 and concentrated in vacuo. The crude residue was purified via silica gel chromatography (0-10%MeOH-DCM) to afford the title compound (1.09 g, 97%) as a beige foam. LCMS: R t = 0.97 min, m/z =705.3 (M+1) Method 2m_acidic.
[0127]
Instead of HATU, a variety of other coupling reagents can be used, such as any of the typical carbodiimides, or CDMT (2-chloro-4, 6-dimethoxy-1, 3, 5-triazine) and N-methylmorpholine to form the amide bond generated in Step 1.
[0128]
Step 2: (3S, 4R) -3- ( (Z) -2- ( (1- ( (benzhydryloxy) carbonyl) cyclopropoxy) imino) -2- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) acetamido) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidine-1-sulfonic acid. Benzhydryl 1- ( ( (Z) – (1- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) -2-oxo-2- ( ( (3S, 4R) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidin-3-yl) amino) ethylidene) amino) oxy) cyclopropanecarboxylate (1.00 g, 1.42 mmol) in DMF (7.0 mL) at 0 ℃ was treated with SO 3·DMF (448 mg, 2.84 mmol) . After 2 h of stirring at rt, the solution was poured into ice-cold brine and extracted with EtOAc (3x) . The combined organic layers were dried over Na 2SO 4 and concentrated in vacuo, affording the title compound (assumed quantitative) as a white solid. LCMS: Rt =0.90 min, m/z = 785.2 (M+1) Method 2m_acidic.
[0129]
Step 3: 1- ( ( (Z) – (1- (2-aminothiazol-4-yl) -2-oxo-2- ( ( (3S, 4R) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) -1-sulfoazetidin-3-yl) amino) ethylidene) amino) oxy) cyclopropanecarboxylic acid.
[0130]
[0131]
To a solution of (3S, 4R) -3- ( (Z) -2- ( (1- ( (benzhydryloxy) carbonyl) cyclopropoxy) imino) -2- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) acetamido) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidine-1-sulfonic acid (1.10 g, 1.40 mmol) in DCM (1.5 mL) at 0℃, TFA (5.39 mL, 70.0 mmol) was added, and after 10 minutes, the ice bath was removed. Additional TFA (3.24 mL, 42.0 mmol) was added after 1 hr at rt and the solution was diluted with DCM and concentrated in vacuo after an additional 30 min. Optionally, anisole may be added to the TFA reaction to help reduce by-product formation, which may increase the yield of desired product in this step. The crude residue was purified by reverse phase prep HPLC (XSelect CSH, 30 x 100 mm, 5 μm, C18 column; ACN-water with 0.1%formic acid modifier, 60 mL/min) , affording the title compound (178 mg, 23%) as a white powder. LCMS: R t = 0.30 min, m/z = 518.9 (M+1) Method 2m_acidic; 1H NMR (400 MHz, DMSO-d 6) δ 9.27 (d, J = 9.0 Hz, 1H) 6.92 (s, 1H) 5.23 (dd, J = 9.1, 5.7 Hz, 1H) 4.12-4.23 (m, 3H) 3.72-3.62 (m, 2H assumed; obscured by water) 3.61-3.52 (m, 1H assumed; obscured by water) 3.26 (dd, J = 14.5, 5.9 Hz, 1H) 1.36 (s, 4H) . 1H NMR (400 MHz, D 2O) δ 7.23 (s, 1H) , 5.48 (d, J = 5.8 Hz, 1H) , 4.71-4.65 (m, 1H) , 4.44 (t, J = 8.2 Hz, 2H) , 3.89-3.73 (m, 3H) , 3.54 (dd, J = 14.9, 4.9 Hz, 1H) , 1.65-1.56 (m, 2H) , 1.56-1.46 (m, 2H) . The product of this process is amorphous. Compound X can be crystallized from acetone, ethanol, citrate buffer at pH 3 (50 mM) , or acetate buffer at pH 4.5 (50 mM) , in addition to solvents discussed below.
PAPER
Bioorganic & Medicinal Chemistry Letters (2018), 28(4), 748-755.
https://www.sciencedirect.com/science/article/pii/S0960894X18300064
PATENT
WO 2019026004
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019026004&tab=PCTDESCRIPTION
Over the past several decades, the frequency of antimicrobial resistance and its association with serious infectious diseases have increased at alarming rates. The increasing prevalence of resistance among nosocomial pathogens is particularly disconcerting. Of the over 2 million (hospital-acquired) infections occurring each year in the United States, 50 to 60% are caused by antimicrobial-resistant strains of bacteria. The high rate of resistance to commonly used antibacterial agents increases the morbidity, mortality, and costs associated with nosocomial infections. In the United States, nosocomial infections are thought to contribute to or cause more than 77,000 deaths per year and cost approximately $5 to $10 billion annually.
Important causes of Gram-negative resistance include extended-spectrum 13- lactamases (ESBLs), serine carbapenemases (KPCs) and metallo-13-lactamases (for example NDM-1 ) in Klebsiella pneumoniae, Escherichia coli, and Proteus mirabilis, high-level third-generation cephalosporin (AmpC) 13-lactamase resistance among Enterobacter species and Citrobacter freundii, and multidrug-resistance genes observed in Pseudomonas, Acinetobacter, and Stenotrophomonas. The problem of antibacterial resistance is compounded by the existence of bacterial strains resistant to multiple antibacterials. For example, Klebsiella pneumonia harboring NDM-1 metallo-13- lactamase carries frequently additional serine-13-lactamases on the same plasmid that carries the NDM-1 .
Thus there is a need for new antibacterials, particularly antibacterial compounds that are effective against existing drug-resistant microbes, or are less susceptible to development of new bacterial resistance. Monobactam antibiotic, which is referred to herein as Compound X, is primarily effective against Gram-negative bacteria, including strains that show resistance to other monobactams.
The present invention relates to a process for the preparation of monobactam antibiotic Compound X and intermediates thereof.
More particularly, the present invention relates to a process for the preparation of Compound X
Compound X
also referred to as 1 -(((Z)-(1 -(2-aminothiazol-4-yl)-2-oxo-2-(((3S,4R)-2-oxo-4-((2-oxooxazolidin-3-yl)methyl)-1 -sulfoazetidin-3-yl)amino)ethylidene)amino)oxy)cyclopropanecarboxylic acid, or a salt thereof, or a solvate including hydrate thereof.
Patent application number PCT/US2015/02201 1 describes certain monobactam antibiotics. Compound X may be prepared using the method disclosed in PCT/US2015/02201 1 , in particular example 22, and in PCT/CN2016/099482.
A drawback from these processes is that they exhibit a large number of process steps and intermediate nitrogen protection/deprotection steps, reducing the overall yield and efficiency. Furthermore, these processes require several chromatographic purification steps to be carried out in course of the processes. We have found that the preparation of Compound X, as previously prepared on a manufacturing scale, possesses a number of disadvantages, in particular poor handling characteristics.
It would thus be beneficial to develop alternative or improved processes for the production of Compound X that do not suffer from some or all of these disadvantages.
Compound x Compound x
Scheme 1
Preparation of Compound X from Intermediates 22 and 2A
Scheme 3
Examples
The Following examples are merely illustrative of the present disclosure and they should not be considered as limiting the scope of the disclosure in any way, as these examples and other equivalents thereof will become apparent to those skilled in the art in the light of the present disclosure, and the accompanying claims.
Synthesis of Compound 8 (R = benzyl)
1 .50kg oxazolidin-2-one (7b) was charged into the reactor. 7.50kg THF was charged and the stirring started. The mixture was cooled to 10~20°C. 2.18kg potassium fert-butoxide was charged intol 2.00kg THF and stirred to dissolve.
The potassium fert-butoxide solution was added dropwise into the reactor while maintaining the temperature at 10-20 °C. The reaction was stirred for 1 ~2hrs at 10-20 °C after the addition. The solution of 2.36kg methyl-2-chloroacetate (7a) in 3.00kg of THF was added to the reactor while maintaining the temperature at 10-20 °C. The reaction mixture was stirred for 16-18 h at 20-25 °C. The IPC (in process control) showed completion of the reaction. The mixture was centrifuged and the wet cake was washed with 7.50kg THF. The filtrate was concentrated and the crude 7 was provided as reddish brown liquid, which was used for the next step without further purification,
1H NMR (400 MHz, CHLOROFORM- /) δ ppm 3.65 – 3.71 (m, 2 H) 3.74 (s, 3 H) 4.02 (s, 2 H) 4.34 – 4.45 (m, 2 H).
The dried reactor was exchanged with N2 three times. 3.71 kg LiHMDS solution in THF/Hep (1 M) and 1 .30kg THF were charged under nitrogen protection. The stirring was started and the solution was cooled to -70—60 °C. The solution of 0.71 kg benzyl acetate (6) in 5.20 kg THF was added dropwisely at -70— 60 °C, and the resulted mixture was stirred for 1 -1 .5 h after the addition. The solution of 0.65kg 7 in 3.90kg THF was added dropwise while maintaining the temperature at -70—60 °C, then stirred for 30-40 minutes. The reaction mixture was warmed to 20-25 °C and stirring was continued for 0.5-1 .0 h. IPC showed 6 was less than1 .0% (Otherwise, continue the reaction till IPC passes). The reaction mixture was poured into 13.65 kg aqueous citric acid below 10 °C. The mixture was stirred for 15-20 minutes after the addition. Phases were separated and the organic layer was collected. The aqueous layer was extracted with EA (6.50kg * 2). The organic layer was combined, washed by 6.50 kg 28% NaCI solution and dried with 0.65
kg anhydrous MgSC . The mixture was filtered and the wet cake was washed with 1 .30kg EA. The filtrate was concentrated under vacuum to provide crude 8. The crude 8 was stirred in 2.60 kg MTBE at 20-25 °C for 1 -1 .5 h. The mixture was cooled to 0-10 °C and stirred for 1 .5-2.0 h and filtered. The filter cake was washed with 0.65kg pre-cooled MTBE and dried under vacuum (<-0.096Mpa) at 20-25 °C for 12~16hrs till a constant weight to give 513 g of 8 as a white solid, Yield: 45%, HPLC purity 96.4%,1 H NMR (400 MHz, CHLOROFORM-c δ ppm 3.48 – 3.55 (m, 1 H) 3.56 – 3.63 (m, 2 H) 3.66 – 3.74 (m, 1 H) 4.17 – 4.26 (m, 2 H) 4.31 – 4.44 (m, 2H) 5.12 – 5.24 (m, 2 H) 7.30 – 7.44 (m, 5 H).
Synthesis of Compound 9 (R = benzyl)
The dried reactor was charged with 3.75kg HOAc and 1 .50 kg 8. The stirring was started and the reaction mixture was cooled to 0-5 °C. 3.53kg aqueous NaN02 was added dropwise at 0-10 °C, and the reaction mixture was stirred for 15-30 minutes after the addition. IPC showed 8 was less than 0.2%. The reaction mixture was treated with 7.50kg EA and 7.50 kg water. Phases were separated and the organic layer was collected. The aqueous layer was extracted with EA (7.50kg * 2). The organic layers were combined, washed with 7.50 kg 28% NaCI solution, and concentrated under vacuum to provide crude 9. The crude 9 was slurried with 5.25 kg water at 10-20 °C for 3~4hrs, and filtered. The wet cake was washed with 1 .50kg water. The solid was dried under vacuum (<-0.096 Mpa) at 45-50 °C for 5-6 h till a constant weight to give 1 .44 Kg of 9, yield: 86.9%, HPLC purity 92.9%,1H NMR (400 MHz, CHLOROFORM- /) δ ppm 3.60 – 3.76 (m, 2 H) 4.44 (t, J=8.07 Hz, 2 H) 4.60 (s, 2 H) 5.25 – 5.41 (m, 2 H) 7.30 – 7.43 (m, 5 H) 1 1 .62 (br s, 1 H).
Synthesis of Compound 9a (R = benzyl)
9
The dried reactor was charged with 0.58 kg Zn, 4.72kg (Βο Ο, 6.00 kg water, 1 .20 kg NH4CI and 6.00kg THF. The reaction mixture was stirred and heated to 50-55 °C. The solution of 0.60 kg 9 in 4.20kg THF
was added dropwisely while maintaining the temperature at 50-55 °C. The reaction mixture was stirred for 0.5-1 .Ohrs after the addition. IPC showed 9 was less than 0.1 %. The reaction mixture was treated withl .50 kg ethyl acetate and stirred for 15-20 minutes. Phase was separated and the water layer was extracted by1 .50 kg ethyl acetate. The organic layers were combined, washed with 6.00 kg 28% NaCI solution and concentrated under vacuum to provide crude 9a. The crude 9a was stirred with 3.60kg*2 n-heptane to remove excess (Βο Ο. The residue was purified by silica gel chromatography column eluted with ethyl acetate: Heptane= 1 :1 to provide crude 9a solution. The solution was concentrated under reduced pressure to obtain crude 9a. The crude 9a was slurried with 1 .80 kg MTBE for 2.0-3. Ohrs, filtered, and the wet cake was washed with MTBE. The solid was dried under vacuum (<-0.096 Mpa) at 50-55 °C for 16-18 h till a constant weight to give 392 g of 9a as a white solid, Yield: 51 %, HPLC purity 98.1 %,1H NMR (400 MHz, DMSO-cfe) δ ppm 1.17 – 1 .57 (m, 9 H) 3.39 – 3.61 (m, 2 H) 4.20 – 4.45 (m, 3 H) 5.10 – 5.32 (m, 3 H) 5.75 (s, 1 H) 7.38 (br s, 5 H) 7.75 – 7.99 (m, 1 H).
Synthesis of compound (VII) (R = benzyl, X = CI)
9a VII
The dried reactor was charged with 13.0kg HCI in IPA and the stirring was started. 1 .33 kg 9a was charged in portions at 20-25 °C. The mixture was stirred at 20-25 °C for 3-4 h. IPC showed 9a was less than 0.1 %. The reaction solution was concentrated under vacuum 40-45 °C. The residue was treated with 21 .58kg MTBE at 20-25 °C for 3-4 h. The mixture was filtered and the wet cake was washed with 2.60kg MTBE. The solid was dried under vacuum (<-0.096 Mpa) at 45-50 °C for 5-6 h till a constant weight to give 1 .045 Kg of compound VII (R = benzyl, X = CI) as a yellow solid, Yield: 93.7%, HPLC purity 99.2%,1 H NMR (400 MHz, DMSO-cfe) δ ppm 3.16 – 3.74 (m, 3 H) 4.10 – 4.35 (m, 4 H) 5.09 – 5.39 (m, 2 H) 7.27 – 7.60 (m, 5 H) 8.72 (br s, 2 H).
Synthesis of compound (Vile) (R = benzyl)
VII Vile
To an autoclave (3L) were added VII (R = benzyl, X = CI) (100 g, 304.2 mmol, 1 .0 equiv.), DCM (2650 g, 26.5 equiv., w/w) and (S-BINAP)RuCl2 (2.4 g, 3.04 mmol, 0.01 equiv.), successively. Air in the autoclave was replaced with N2 5 times. N2 in the autoclave was was replaced with H2 5 times. The solution was stirred with 250-260 r/min and H2 (2.1 ±0.1 MPa) at 40±5°C for 24 h. The reaction mixture was filtered, and the filter cake was washed with DCM (400 g, 4.0 equiv., w/w). The filter cake was slurried with IPA (785 g, 7.85 equiv., w/w) and H2O (40 g, 0.4 equiv., w/w) overnight (18-20 h). The mixture was filtered. The filter cake was washed with IPA (200 g, 2.0 equiv., w/w) and dried at 45±5°C overnight (18-20 h). Vile (R = benzyl) was obtained as off-white solid, 80.4 g, 79.9% yield, 95.5% purity, 97.6% de, >99.5% ee. 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.34-3.38 (m, 2 H) 3.50-3.52 (m, 1 H) 3.60-3.62 (m, 1 H) 4.18-4.24 (m, 4 H) 5.23 (s, 2H) 6.16 (s, 1 H) 7.32 (m, 5H) 8.74 (s, 1 H).
Alternative synthesis of compound 9a (R = benzyl)
5b
Mg(OtBu)2
To a flask was added 5a (1 .88 g, 12.93 mmol), THF (40 mL), and CDI (2.20 g, 13.58 mmol) at 25 °C. The mixture was stirred for 3 h. To the reaction mixture was added 5b (2.00 g, 6.47 mmol), and Mg(OfBu)2 (2.21 g, 12.93 mmol). The reaction mixture was stirred at 25 °C for 24 h. The reaction mixture was concentrated under vacuum to remove most of the THF solvent. To the concentrated solution was added MTBE (40 mL), followed by addition of an aqueous solution of HCI (1 M, 60mL) to adjust to pH = 2-3. Two phases were separated, and the water phase was extracted with MTBE (20 mL). The combined organic phase was washed with aqueous NaHCC (5%, 50 mL) and brine (20%, 40 mL). The organic phase was concentrated to a weight of -19 g, and a lot of white solid was obtained in the concentration process. The suspension was cooled to 0 °C, and filtered. The filter cake was washed with cold MTBE (5 mL) and dried under vacuum to obtain product 9a (1 .6g, 63% yield).
Synthesis of compound (Vile) (R = benzyl, PG = Cbz)
Vile Vile
To a flask (5 L) were added Vile (R = benzyl) (140 g, 423.2 mmol, LOequiv.), H20 (1273 g, 9.09 equiv., w/w) and toluene (2206 g, 15.76 equiv., w/w). The solution was stirred and cooled to 0-5 °C with ice bath. Then NaHCOa (78.4 g, 933 mmol, 2.22 equiv.) was added and CbzCI (89.6 g, 527 mmol, 1 .24 equiv.) was dropped into the stirring solution, respectively. The solution was stirred at 30±5 °C overnight (18-20 h). Heptane (3612 g, 25.8 equiv., w/w) was added dropwise to the stirring solution over 1 h at 20-30 °C. The mixture was filtered. The filter cake was washed with heptane (280 g, 2.00 equiv., w/w) and MTBE (377 g, 2.69 equiv., w/w), respectively. The filter cake was dried at 45±5°C overnight (18-20 h). Vile (R = benzyl, PG = Cbz) was obtained as an off-white solid, 169.4 g, 93% yield, 96.7% purity, 98% de, >99.5% ee, 1 H NMR (400 MHz, DMSO-cfe) δ ppm 3.23-3.24 (m, 1 H) 3.30 (m, 1 H) 3.51 -3.55 (m, 2 H) 3.99 (s, 1 H) 4.17-4.21 (m, 3 H) 5.02-5.03 (m, 2H) 5.12 (s, 2H) 5.46-5.48 (d, 1 H) 7.33-7.36 (m, 10H) 7.75-7.73 (d, 1 H).
Synthesis of compound (IV) (PG = Cbz)
Vile IV
Vile (R = benzyl) (220 g, 513.5 mmol, 1 .0 equiv.) was dissolved in THF (1464g, 6.65 equiv., w/w). The solution was filtered. The filter cake was washed with THF (488g, 2.22 equiv., w/w). The filtrate (Vile) was collected. To an autoclave (3L) were added the filtrate (Vile). The reactor was cooled down to -75 – -65 °C with dry-ice/EtOH bath, and bubbled with NH3 for not less than 4 h. Then the solution was stirred at 25±5 °C with NH3 (0.5-0.6 MPa) for 24 h. The autoclave was deflated to release NH3. The reaction solution was concentrated with a rotary evaporator to remove THF until the residue was around 440 g. The residue was slurried with EA (2200 g, 10 equiv., w/w) at 70±2 °C, then cooled to 25±5 °C and stirred for 16-18 h. The mixture was filtered. The filter cake was washed with EA (440 g). The filter cake was slurried with EA (1320 g, 6.00 equiv. w/w), and the temperature was raised to 70±2 °C, then cooled to 25±5 °C and stirred for 16-20 h. The mixture was filtered. The filter cake was washed with EA, and dried at 50±5 °C overnight (18-20 h). IV (PG = Cbz) was obtained as off-white solid, 141 g, 81 .5% yield, 99.1 % purity, >99.5% assay, 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.12 – 3.23 (m, 2 H) 3.31 (br s, 1 H) 3.56 (t, J=8.01 Hz, 2 H) 3.88 (quin, J=6.02 Hz, 1 H) 3.93 – 4.03 (m, 1 H) 4.20 (t, J=8.01 Hz, 2 H) 5.02 (s, 2 H) 5.27 (d, J=5.87 Hz, 1 H) 7.12 (s, 1 H) 7.22 – 7.45 (m, 5 H).
Synthesis of compound (III) (PG = Cbz, LG = S02CH3)
IV III
To a flask was added IV (PG = Cbz) (14.00 g, 41 .50 mmol, 1 .00 equiv), and dry 1 , 2-dimethoxyethane (300 mL) under N2. The mixture was stirred at -5°C ~ 0°C for 1 h to obtain a good suspension. MsCI (7.89 g, 68.89 mmol, 5.33 mL, 1 .66 eq) in 1 , 2-dimethoxyethane (20.00 mL) was added dropwise during 30 min, and Et3N (12.60 g, 124.50 mmol, 17.26 mL, 3.00 eq) in 1 , 2-dimethoxyethane (20.00 mL) was added dropwise during 30 min side to side. The reaction mixture was stirred for additional 5 min at -5°C ~ 0°C, and was quenched with water (6 mL). The reaction mixture was concentrated to remove DME. The solid was slurried in water (250 mL) and MTBE (125 mL) for 1 h. The solid was collected by filtration, and then slurried in water (250 mL) for 1 hr. The solid was collected by filtration, and washed with water (25 mL) to give white solid. The solid was slurried in EA (150 mL) and dried in vacuum at 60°C for 24 h to give III (PG = Cbz, LG = SO2CH3) (15.00 g, 36.1 1 mmol, 87.01 % yield), 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.17 (s, 3 H) 3.26 (br d, J=15.04 Hz, 1 H) 3.47 – 3.57 (m, 1 H) 3.64 (br d, J=6.36 Hz, 2 H) 4.22 (br dd, J=17.79, 8.50 Hz, 2 H) 4.50 (br s, 1 H) 4.95 – 5.17 (m, 3 H) 7.21 – 7.56 (m, 5H) 7.43 (s, 1 H) 7.63 – 7.89 (m, 2 H).
Synthesis of compound II (PG = Cbz, LG = SO2CH3, M+ = NBu4+)
O OMs o CISO3H, 2-picoline – ° O ?yO
HN Bu4NHS04< NHCbz
“Cbz
III II
To a flask was added 2-picoline (1 1 .50 g, 12.23 mL) and DMF (10 mL). The solution was cooled to 5 SC, followed by slow addition of chlorosulfonic acid (7.20 g, 4.14 mL). The temperature was increased to 20 SC. Ill (PG = Cbz, LG = SO2CH3) (5.13 g, 12.35 mmol) was added to the reaction mixture. The reaction mixture was heated to 42 SC for 18h. IPC (in process control) showed complete conversion of starting material. The reaction was cooled to 20 SC and dropwise added to a solution of tetrabutylammonium hydrogen sulfate (4.6 g, 13.6 mmol) in the mixed solvents of dichloromethane (100 mL) and water (100 mL) at 5SC. The phases were separated and the water phase was extracted with dichloromethane (2*50mL). The combined organic phase was washed with water (5*100mL). The organic phase was concentrated to dryness and purified by column chromatography (dichloromethane/methanol = 15/1 v/v) to afford II (PG = Cbz, LG = SO2CH3, M+ = NBii4+) (8.4 g, 92.30%), 1 H NMR (400 MHz, CHLOROFORM-c/) δ ppm 0.99 (t, J=7.34 Hz, 12 H) 1 .36 – 1 .50 (m, 8 H) 1 .54 – 1 .76 (m, 8 H) 3.15 (br d, J=8.31 Hz, 2 H) 3.21 – 3.35 (m, 8 H) 3.47 (br dd, J=14.73, 7.27 Hz, 1 H) 3.54 – 3.65 (m, 1 H) 3.67 – 3.81 (m, 2 H) 4.17 – 4.32 (m, 1 H) 4.39 – 4.62 (m, 1 H) 4.74 (br s, 1 H) 5.1 1 (s, 3 H) 5.32 – 5.50 (m, 1 H) 6.47 (br s, 1 H) 7.29 – 7.47 (m, 5 H) 8.69 – 8.94 (m, 1 H).
Synthesis of compound (IA)
A solution of II (PG = Cbz, LG = SO2CH3, M+ = NBu4+) (4.0 g) in dichloromethane (38 mL) was pumped to tube A at rate of 2.0844 mL/min, and a solution of KHCO3 (3.0 g) in water (100 mL) was pumped to tube B at a rate of 1 .4156 mL/min side to side. These two streams were mixed in a cross-mixer then flowed to a tube coil that was placed in an oil bath at 100 °C. The residence time of the mixed stream in the coil was 2 min. The reaction mixture flowed through a back-pressure regulator that was set at ~ 7 bars, and was collected to a beaker. After completion of the collection, two phases was separated. The organic phase was concentrated to dryness. The residue was slurried in ethyl acetate (5 mL). The solid was filtered and the filter cake was dried to give IA (2.6 g, 75%),
1H NMR (400 MHz, CHLOROFORM-c/) δ ppm 1.00 (t, J=7.27 Hz, 12 H) 1 .42 (sxt, J=7.31 Hz, 8 H) 1 .62 (quin, J=7.83 Hz, 8 H) 3.13 – 3.39 (m, 8 H) 3.54 – 3.69 (m, 2 H) 3.81 (dd, J=14.98, 2.51 Hz, 1 H) 3.96 – 4.13 (m, 1 H) 4.22 – 4.47 (m, 3 H) 4.99 – 5.23 (m, 3 H) 6.42 (br d, J=9.29 Hz, 1 H) 7.26 – 7.44 (m, 5 H).
Synthesis of compound 2A
Step 1
To a stirring solution of compound 16b (2 g, 10.14mmol, 1 .0 eq) in DMF (20 ml_) was added CS2CO3 (5.29g, 16.22 mmol, 1 .6 eq), then the resulting solution was stirred at room temperature for 10mins, then compound 16a (5.27g, 20.28mmol, 2eq) was added dropwise to the mixture for 2 minutes, then the resulting solution was stirred for another 2 hours. TLC showed the starting material was consumed completely. The mixture was added with water (60mL) and extracted with MTBE (20mL*3). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The crude was slurried in heptane to give 1 .65 g 16 as a white solid (Yield: 57%), 1H NMR (400 MHz, DMSO-cfe) δ ppm 7.48-7.28 (m, 10 H), 5.00-4.96 (t, J=6.0 Hz, 1 H), 3.81 (s, 3H), 3.44-3.42 (m, 2H), 2.40-2.37 (m, 2H).
Compound 16 (1 g, 2.66mmol, 1 eq) was dissolved in THF (20mL) under Nitrogen, and cooled to -40 °C. NaHMDS (1 .6mL, 2.0M THF solution, 1 .2 eq) was added dropwise. The reaction was stirred for 1 h at -40 °C. HPLC indicated the reaction was finished. The reaction was quenched with 10% Citric acid, extracted with MTBE (25 ml_ x 2). The combined organic layers were washed with brine (30 ml_), dried with Na2S04, filtered and concentrated to give 17 as a yellow solid, which was used for the next step without purification (assay yield: 65%); 1H NMR (400 MHz, DMSO-cfe) δ ppm 7.27-7.13 (m, 10 H), 3.46 (s, 3H), 1 .21 -1 .17(dd, J=7.2, 10.4 Hz, 2H ); 1 .14-1 .1 1 (dd, J=7.2, 10.4 Hz, 2H).
Step 3
Compound 17 (100 mg) was dissolved in methanol (5 mL) and 2.0 M HCI IPAC solution (5 mL). The solution was heated at 45 °C for 3 days. HPLC indicated the reaction was finished. The reaction was cooled to room temperature and was diluted with 10 mL water. The reaction mixture was washed with MTBE (10 mL x 2), organic layer was discarded and the aqueous layer was concentrated to give compound 2A HCI (32 mg, 62% yield), 1 H NMR (400 MHz, DMSO-cfe) δ ppm 3.80-3.44 (br, 4H), 1 .56 (s, 2H), 1 .38 (s, 2H).
Step 4
To a solution of 2A HCI (0.70 g, 4.57 mmol) in methanol (5 mL) was added triethylamine (1 .26 mL, 9.14 mmol) at room temperature. The solution was stirred for 20 min, and the solvent was removed under vacuum. To the residue was added IPAC (10 mL) leading to precipitation. The solid was filtered, and the filtrate was concentrated to provide 2A (0.50g, 94% yield) containing ca. 6 wt% Et3N-HCI.
Synthesis of Compound X from compound of formula (I), (IA)
Compound x
To a flask was charged 21 (1 .00 g, 68.43 wt%, 2.50 mmol) and DMF (10 mL). The suspension was cooled to -20 °C, to which was added diphenylphosphinic chloride (0.52 mL, 2.75 mmol). The solution was stirred at -20 °C for 30 min, followed by addition of a mixed solution of (IA) (1 .52g, 3.00 mmol) and triethylamine (0.52 mL, 3.76 mmol) in DMF (2mL). The reaction mixture was stirred at 20 °C for 20 h, followed by addition of MTBE (20 mL). The reaction mixture was adjusted to pH = 2-3 using aqueous HCI solution (37%). To the mixture was added isopropanol (100 mL). The resulting mixture was stirred for 4 h to obtain a suspension. The suspension was filtered and the filter cake was dried under vacuum to afford crude 22 (1 .17 g). The crude 22 was slurried in a combined solvent of THF/H2O (= 12 mL / 3mL), and filtered to afford 22 (0.744 g, 75 wt% by Q-NMR, 53.3% yield). 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.47 – 3.55 (m, 2 H) 3.59 – 3.63 (m, 2 H) 4.13 – 4.21 (m, 3 H ) 5.05 (dd, J=8.8, 5.6 Hz, 1 H) 8.22 (s, 1 H) 9.73 (d, J=8.7 Hz, 1 H).
To a suspension of 22 (580 mg, 75 wt%, 1 .037 mmol) in DMAC (1 .5 mL) was added 2A (214.3 mg, 85 wt%, 1 .556 mmol). The reaction was stirred at 25 °C for 3 days, and in process control showed 22, Compound X = 4/96, and Z/E = 91 /9. the mixture was slowly added into 15ml acetone to precipitate yellowish solid. The reaction mixture was filtered to afford Compound X (0.7 g, 34 wt% by QNMR, 44% yield).
Synthesis of compound 3 (R2 = CH(Ph)2)
R2 = CH(Ph)2
2-(2-aminothiazol-4-yl)-2-oxoacetic acid (Y) (10.00 g, 47.93 mmol) and compound W (R2 = CH(Ph)2) (13.31 g, 46.98 mmol) were suspended in DMAC (40 mL), followed by addition of triethylamine (5.01 mL, 35.95 mmol). The reaction mixture was stirred at 20 °C for 5 h. HPLC showed completion of the reaction, and Z/E
= 97/3. To the reaction mixture was added water (120 mL) with stirring. The mixture was stirred for 20 min to obtain a suspension. The suspension was filtered and the filter cake was washed with water (50 mL).
The filter cake was slurried in a combined solvent of THF/ethyl acetate (50 mL / 50 mL) at 60 °C and cooled to 20 °C. The solid was filtered and dried at 50 °C for 3 h to get 3 (R2 = CH(Ph)2) (19.5 g, 88% yield). 1H
NMR (400 MHz, DMSO-cfe) δ ppm 1.37 -1 .42 (m, 2 H) 1 .44 – 1 .49 (m, 2 H) 6.87 (s, 1 H) 6.94 (s, 1 H) 7.22
– 7.30 (m, 6 H) 7.45 – 7.49 (m, 4 H).
Alternative Synthesis of Compound X from compound of formula (I), (IA)
Compound x
IA (40.14 g, 62.63 mmol) was dissolved in methanol (200 ml_), followed by addition of Pd/C (10%, 1 .1 g). The reaction mixture was maintained under hydrogen atmosphere (1 -2 bar) at 20 °C for 24 h. In process control showed completion of the reaction. The reaction mixture was filtered. The filtrate was concentrated to give an oil of IB (M+ = NBu4+) (58.20 g, 55 wt% by Q-NMR, 100% yield). 1 H NMR (400 MHz, DMSO-cfe) δ ppm 0.93 (t, J=7.3 Hz, 12 H) 1 .23 – 1 .36 (m, 8 H) 1 .57 (m, 8 H) 2.99 – 3.28 (m, 8 H) 3.37 (dd, J=14.3, 7.5 Hz, 1 H) 3.65 – 3.70 (m, 3 H) 3.84 – 3.88 (m, 1 H) 4.08 (d, J=5.6 Hz, 1 H) 4.18 – 4.22 (m, 2 H).
3 (R2 = CH(Ph)2) (0.95 g, 2.17 mmol) was dissolved in THF (20 ml_). To the solution was added /V-methyl morpholine (0.77 g, 7.60 mmol) and 2-chloro-4,6-dimethoxy-1 ,3,5-triazine (0.57 g, 3.26 mmol). The reaction mixture was stirred at 20 °C for 1 h followed by addition of IB (M+ = NBu +) (2.70 g, 48.98 wt%, 2.61 mmol). The reaction was stirred at 20 °C for 5 h. In process control showed completion of the reaction. To the reaction mixture was added ethyl acetate (20 ml_). The organic phase was washed with brine (10 ml_). Solvent was removed. Acetone (40ml) was added to dissolve residue. TFA (1 .24 g, 10.86 mmol) dissolved in acetone (3 ml) was added slowly. The white solid was filtered and washed by acetone (10 ml) two times. Dried at 40 °C for 5h to get compound 4 (R2 = CH(Ph)2). 1 H NMR (400 MHz, DMSO-cfe) δ ppm 1 .49 – 1 .55 (m, 4 H) 3.27 (dd, J=14.4, 6.2 Hz, 1 H) 3.49 – 3.65 (m, 2 H) 3.71 (dd, J=14.4, 6.2 Hz, 1 H) 4.04 – 4.10 (m, 1 H) 4.07 (dd, J=16.0, 8.6 Hz, 1 H) 4.17 (dd, J=1 1 .8, 6.0 Hz, 1 H) 5.28 (dd, J=9.0, 5.7 Hz, 1 H) 6.88 (s, 1 H) 7.03 (s, 1 H) 7.18 – 7.32 (m, 6 H) 7.43 (m, 4 H) 9.45 (d, J=9.0 Hz, 1 H).
Crude 4 (R2 = CH(Ph)2) (2.13 g) was dissolved in dichloromethane (20 ml_). The solution was cooled to 0 °C. To the solution was added anisole (0.68 ml_, 6.24 mmol) and trifluoroacetic acid (2.16 ml_, 28.08 mmol). The reaction was warmed to 20 °C, and stirred for 15 h. In process control showed completion of the
reaction. The aqueous phase was separated and added to acetone (40 mL) to obtain a suspension. The suspension was filtered to afford Compound X (0.98 g, 54.5% yield over two steps). 1 H NMR (400 MHz, DMSO-c/e) δ ppm 1.40 (m, 4 H) 3.26 (dd, J=14.4, 6.0 Hz, 1 H) 3.54 – 3.69 (m, 3 H) 4.14 – 4.21 (m, 3 H) 5.25 (dd, J= 8.9, 5.7 Hz, 1 H) 7.02 (s, 1 H) 9.38 (d, J=9.0 Hz, 1 H).
REF
Synthesis and optimization of novel monobactams with activity against carbapenem-resistant Enterobacteriaceae – Identification of LYS228
57th Intersci Conf Antimicrob Agents Chemother (ICAAC) (June 1-5, New Orleans) 2017, Abst SATURDAY-297
//////////////LYS228, LYS 228, BOS-228, LYS-228, monobactam, Novartis, phase II, Boston Pharmaceuticals, complicated urinary tract infection, complicated intraabdominal infections, fast track, Qualified Infectious Disease Product, QIDP,
Nc1nc(cs1)\C(=N\OC2(CC2)C(=O)O)\C(=O)N[C@H]3[C@@H](CN4CCOC4=O)N(C3=O)S(=O)(=O)O
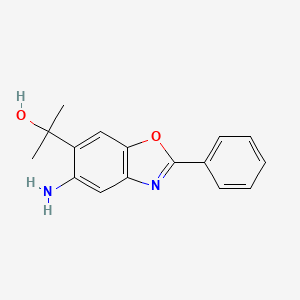
ADX-103
ADX-103
CAS 916056-81-0
Preclinical, Antiinflammatory Ophthalmic Agents, Diabetic Retinopathy,
Agents for Ophthalmic Drugs
MF C16 H16 N2 O2
5-Amino-α,α-dimethyl-2-phenyl-6-benzoxazolemethanol
MW 268.31
6-Benzoxazolemethanol, 5-amino-α,α-dimethyl-2-phenyl-
Aldeyra Therapeutics Inc
ADX-103 , an aldehyde trap being investigated by Aldeyra for the treatment of dry eye syndrome; in May 2018, preclinical data were presented at 2018 ARVO Meeting in Honolulu, HI. Aldeyra, in collaboration with an undisclosed company, is also investigating an anti-inflammatory agent for treating ocular inflammation.
PATENT
WO-2020033344
Novel crystalline forms of a specific benzoxazole and it’s salts, process for their preparation, and compositions comprising them are claimed, useful for treating dry eye, inflammation and diabetes, through action as an aldehyde scavenger.
It has now been found that compounds of the present invention, and compositions thereof, are useful for treating, preventing, and/or reducing a risk of a disease, disorder, or condition in which aldehyde toxicity is implicated in the pathogenesis. In general, salt forms or freebase forms, and pharmaceutically acceptable compositions thereof, are useful for treating or lessening the severity of a variety of diseases or disorders as described in detail herein. Such compounds are represented by the chemical structure below, denoted as compound A:
or a pharmaceutically acceptable salt thereof.
[0008] Compounds of the present invention, and pharmaceutically acceptable compositions thereof, are useful for treating a variety of diseases, disorders or conditions, associated with toxic aldehydes. Such diseases, disorders, or conditions include those described herein.
[0009] Compounds provided by this invention are also useful for the study of certain aldehydes in biology and pathological phenomena.
Scheme 1 – Synthesis of Compound A
Step 1: Synthesis of Compound A2
[00549] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with methanol (10L). Compound A1 (2.0kg) was added, followed by further methanol to rinse (9L). The reaction mixture was warmed to Tjacket=40°C. Once temperature had stabilized, sulfuric acid (220 mL, 0.4eq.) was slowly added. Once addition was complete, agitation was maintained for 30 mins then the vessel was heated to Tjmt=62°C. Reaction progress was
monitored by LC-MS analysis of reaction mixture. The reaction does not go to completion but is deemed complete when no change is apparent in ratio of starting material : product.
[00550] The vessel contents were cooled to Tjmt=24°C and stirred 60 minutes before filtration under vacuum. The filter cake was air dried for 2 hours and the contents then dissolved in ethyl acetate (18L) which was then washed sequentially with saturated sodium bicarbonate (8L), water (8L) and brine (8L) before drying over sodium sulfate, filtration and evaporation in vacuo. Compound A2 (1.5kg, 68.1%) was obtained as a bright orange powder.
Step 2: Synthesis of Compound A3
[00551] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with /V,/V-dimethylformamide (16L). Compound A2 (1.5kg) was added and the brown reaction mixture set to cool to Tint<20oC. Once temperature had stabilized, A-bromosucci ni mi de (l.5kg, 1.1 eq.) was added portion wise, maintaining Tint<27°C. Once addition was complete, the reaction was allowed to stir until starting material content was <1% AUC (250nm) by LCMS analysis.
[00552] A secondary jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with ethyl acetate (16L) and deionized water (22L). The reaction mixture was vacuum transferred into this vessel and held at high agitation for not less than 30 minutes. The aqueous layer was discharged and the organic layer washed with saturated sodium chloride (2 x 8L) then dried over sodium sulfate before evaporation in vacuo to Compound A3 as a deep brown oil (2.lkg, 100.8%), suitable for use in following step without purification.
Step 3: Synthesis of Compound A4
[00553] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with dichloromethane (9L). Compound A3 (2.lkg) was added and the reaction mixture cooled to Tmt<l°C. A solution of Di-/er/-butyl dicarbonate (3.6kg, 2.2 eq.) in dichloromethane (0.5L) was added followed by a solution of A, A-di methyl ami nopyri di ne (92g, 0.1 eq.) in dichloromethane (0.5L). The resultant clear brown solution was stirred for 30 minutes whereupon pyridine (1.3L, 1.7 eq.) was dropwise added, maintaining Tint<5°C. Upon complete addition internal temperature was ramped from Tint=l°C to Tint=20°C over 18 hours.
[00554] The reaction mixture was sequentially washed with saturated sodium chloride (3 x 4.5L), 10 % w/v aqueous citric acid (2 x 4L), saturated sodium bicarbonate (4L), aqueous hydrochloric acid (1M, 4L), saturated sodium bicarbonate (4L) and saturated sodium chloride (4L) then dried over sodium sulfate and evaporated in vacuo with one azeotropic distillation with toluene (2L) to a very dark, heavy tar (3.4kg).
[00555] The isolated tar was mixed with absolute ethanol (3.1L) for 2 days whereupon it was filtered providing light cream colored, granular solids and a black mother liquor. The solids were washed with ice-cold ethanol (3 x 1L) and dried to constant mass. Compound A4 was obtained as off- white granules (1.7 kg, 50.2%).
Step 4: Synthesis of Compound AS
[00556] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with reagent alcohol (6.1 L) and Compound A4 (0.8kg), Tmt<20°C. Iron powder (0.5kg, 5.0 eq.) was added and the suspension stirred vigorously for 30 minutes. Acetic acid (glacial, 1.6L, 15.7 eq.) was added, maintaining Tint<30C.
[00557] Once LCMS confirmed complete consumption of starting material, ethyl acetate (10.2L) and water (10.2L) were added. Sodium bicarbonate (2.3kg, 15.9 eq.) was added portion wise and the layers separated once gas evolution had ceased. The aqueous layer was washed with ethyl acetate until LCMS indicated no further product was being extracted (8 x 2L) and the combined organic layers were sequentially washed with deionized water (6L) then saturated sodium chloride (6L) before drying over magnesium sulfate and evaporation in vacuo. Compound A5 was obtained as a light orange solid (0.7kg, 91.5%).
Step 5: Synthesis of Compound A6
[00558] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with dichloromethane (9L), Compound A5 (0.7kg), and the reaction mixture cooled to Tint 20°C. Benzoyl chloride (0.3L, 1.5 eq.) was added and the reaction stirred 15 minutes. N,N-dimethylaminopyridine (7g, 0.04 eq.) in dichloromethane (0.1L) was added and the reaction stirred 15 minutes. Pyridine (0.5L, 2.5 eq.) was dropwise added, maintaining Tint<20°C. Upon complete addition the reaction was stirred until LCMS indicated consumption of starting material.
[00559] The reaction mixture was washed with deionized water (11L) and the organic layer extracted sequentially with aqueous hydrochloric acid (1M, 3 x 5L), saturated aqueous sodium bicarbonate (11 L), saturated sodium chloride (11 L), dried over magnesium sulfate and evaporated in vacuo. Compound A6 was obtained as a cream colored solid, suitable for use without further purification (0.9kg, 100.7%).
Step 6: Synthesis of Compound A 7
[00560] A 30L jacketed vessel equipped with mechanical agitation, baffle and nitrogen bleed was charged with l,2-dimethoxy ethane (16L) and temperature set to Tint=2l°C. Compound A6 (0.9kg) was added and stirred to dissolution. Copper iodide (0.3kg, 1.0 eq.) was added and the mixture stirred 15 minutes. l, lO-phenanthroline (0.3kg, 1.2 eq.) was added and the mixture stirred 15 minutes. Cesium carbonate (l .5kg, 3.0 eq.) was added and the reaction was stirred for 15 minutes. The reaction temperature was ramped to Tint=80-85oC and maintained for 23 hours whereupon it was cooled to Tmt=20°C.
[00561] The reaction mixture was filtered through a celite pad, washing sequentially with deionized water (8L) and ethyl acetate (8L). The organic layer was extracted sequentially with deionized water (2 x 5L), saturated sodium chloride (4L), dried over sodium sulfate and evaporated in vacuo. Compound A7 was obtained as a brown solid, suitable for use without further purification (0.8kg, 104.1%).
Step 7: Synthesis of Compound A8
[00562] A 12L 3 -neck round bottom flask with nitrogen bleed and mechanical stirring was charged with a solution of Compound A7 (0.8kg) in dichloromethane (3.6L) and cooled to Tmt<5°C in an ice bath. Hydrochloric acid in dioxane (4M, 1 2L, 3.1 eq.) was added dropwise with vigorous stirring, maintaining Tmt<25°C. Once addition was complete, the reaction mixture was allowed to stir for 18 hours at Tint=20-25oC.
[00563] The reaction mixture was filtered and the filter cake washed with dichloromethane (2 x 1L) and dried to constant mass. The hydrochloride salt of Compound A8 was isolated as an off-white solid (0.5kg, 88.7%).
Step 8: Synthesis of Compound A
[00564] A 12L 3 -neck round bottom flask with nitrogen bleed and mechanical stirring was charged with a solution of Compound A8 (0.5kg) in tetrahydrofuran (4.8L) and cooled to Tint<-30°C in a dry-ice / acetone bath. Methylmagnesium bromide (3.4M in 2-methyltetrahydrofuran, 2.4L, 5.0eq.) was added slowly, maintaining Tmt<-lO°C. Once addition was complete, the reaction was allowed to warm to room temperature overnight.
[00565] Saturated aqueous ammonium chloride (2L) and ethyl acetate (2L) were added and the reaction mixture stirred for 30 minutes. The aqueous layer was extracted with further ethyl acetate (2 x 2L) and the combined organic layers washed with saturated sodium chloride (2L), dried over sodium sulfate and evaporated in vacuo to a dark heavy oil. The heavy oil was purified by column chromatography on silica gel, eluting with ethyl acetate : heptane 1 : 19 to 1 : 1. Pure Compound A was obtained after evaporation and drying as a brown powder (99.8 g, 23.0%).
Example 1 – Preparation of Free Base Forms A, B and C of Compound A
Compound A
Primary Polymorph Screen
[00566] Based on solubility screen results, a primary polymorph screen using an initial set of 24 solvents, as shown in Table 18, was performed as follows: A) To 24 x 20 mL vials, approximately 50 mg of the received ADX-103 was added; B) The solids were then slurried in 2 mL of the solvents and left placed in an incubator/shaker to temperature cycle between ambient and 40 °C in 4 hour cycles; C) After 72 hours temperature cycling, the mother liquors were removed from the vials and split evenly between 4 x 2 mL vials. The vials were then split between evaporation, crash cooling to 2 °C and -18 °C and anti-solvent addition; and D) Any solids
recovered were analysed by XRPD, any new patterns identified were also analysed by TG/DTA and PLM.
Table 18. Solvents Selected for Initial Primary Polymorph Screen
PATENT
WO2018039197 , as compound I-8.
PATENT
WO 2006127945
WO 2011072141
WO 2014116593
US 20150344447
WO 2020028820
////////////ADX-103, Preclinical, Antiinflammatory, Ophthalmic Agents, Diabetic Retinopathy, Aldeyra Therapeutics Inc,
CC(C)(O)c1cc2oc(nc2cc1N)c3ccccc3
Islatravir (MK-8591, EFdA)
Islatravir (MK-8591, EFdA)
2′-Deoxy-4′-ethynyl-2-fluoroadenosine
- Molecular FormulaC12H12FN5O3
- Average mass293.254 Da
- 865363-93-5
2′-Deoxy-4′-ethynyl-2-fluoroadenosine
4′-Ethynyl-2-Fluoro-2′-Deoxyadenosine
Adenosine, 2′-deoxy-4′-C-ethynyl-2-fluoro-
(2R,3S,5R)-5-(6-amino-2-fluoro-9H-purin-9-yl)-2-ethynyl-2-(hydroxymethyl)tetrahydrofuran-3-ol
2′-deoxy-4′-c-ethynyl-2-fluoroadenosine
865363-93-5 [RN]
9H-Purin-6-amine, 9-(2-deoxy-4-C-ethynyl-β-D-erythro-pentofuranosyl)-2-fluoro-
E2FDA
(2R,3S,5R)-5-(6-amino-2-fluoropurin-9-yl)-2-ethynyl-2-(hydroxymethyl)oxolan-3-ol
MK-8591
UNII:QPQ082R25D
FDA APPROVED 2026, 4/20/2026, doravirine and islatravir, Idvynso
To treat HIV-1 infection (as a complete regimen) in adults to replace the current antiretroviral regimen in those who are virologically-suppressed on a stable antiretroviral regimen with no history of virologic treatment failure and no known substitutions associated with resistance to doravirine
Islatravir is known to be a nucleoside reverse transcriptase inhibitor, useful for treating HIV-1 and -2 infection and AIDS.
Islatravir (MK-8591, EFdA), useful for the treatment of eg HIV, AIDS and related diseases.
Merck & Co and Idenix , under license from Yamasa Shoyu , are developing islatravir, a nucleoside reverse transcriptase inhibitor, for the oral prevention and treatment of HIV-1 and HIV-2 infection; in July 2019, data from a phase IIb trial in patients with HIV-1 infection were presented.In August 2015, Merck licensed Codexis ‘ CodeEvolver® protein engineering platform technology to develop enzymes for use in the manufacture of the pharmaceutical products such as islatravir.
Islatravir (4′-ethynyl-2-fluoro-2′-deoxyadenosine, EFdA, or MK-8591) is an investigational drug for the treatment of HIV infection.[1]It is classified as a nucleoside reverse transcriptase translocation inhibitor (NRTTI).[2] Merck is developing a subdermal drug-eluting implant to administer islatravir.[3][4]
Biological activity
Islatravir has activity against HIV in animal models,[5] and is being studied clinically for HIV treatment and prophylaxis.[6] Islatravir is a nucleoside analog reverse transcriptase translocation inhibitor that unlike other such inhibitors, inhibits HIV through multiple mechanisms,[5] providing rapid suppression of the virus, when tested in macaques and mice.[7] Nevertheless, there are HIV strains resistant to islatravir and research is ongoing.[8]
PATENTS
WO2020014046 ,
PATENT
WO2020014047
PATENT
WO2020014050 (assigned to Codexis ), covering engineered phosphopentomutase (PPM) enzymes, useful in the synthesis of pharmaceutical compounds including islatravir.
PATENT
WO-2020014041
4’-Ethynyl-2’-deoxy nucleoside analogs are known for activity against HIV, AIDS and related diseases.
One example of a 4’-ethynyl nucleoside analog is 4’-ethynyl-2-fluoro-2’-deoxyadenosine (EFdA, also known as MK-8591) which is a nucleoside reverse transcriptase translocation inhibitor that blocks HIV-l and SIV viral replication in vitro (Kawamoto, A., Kodama, E., Sarafianos S. F. et al, Int. J. Biochem. Cell Biol.; 40(l l):24lO-2O [2008]; Ohrui, H., Kohgo, S., Hayakawa, H. et al, Nucleosides, Nucleotides & Nucleic Acids, 26, 1543-1546
[2007]) and in vivo (Hattori, S., Ide, K., Nakata, H. et al. Antimicrobial. Agents and
Chemotherapy, 53, 3887-3893 [2009]). EFdA is claimed in US Patent No. 7,339,053 (referred to in the‘053 patent as 2,-deoxy-4’-C-ethynyl-2-fluoroadenosine). EFdA has the following chemical structure:
EFdA is metabolized in cells to its active triphosphate anabolite which inhibits HIV reverse transcriptase. In contrast to nucleoside reverse transcriptase inhibitors (NsRTIs) and nucleotide reverse transcriptase inhibitors (NtRTIs) currently available for the treatment of HIV infection which lack a 3′-OH group to block incorporation of incoming nucleotide, EFdA retains a 3′ OH group and acts as a chain terminator by preventing translocation of the primer template in the reverse transcriptase (RT) active site and preventing binding of incoming
deoxyribonucleotide triphosphates (dNTPs). In addition, the pucker of the modified ribose ring of EFdA is believed to contribute to inhibition of reverse transcriptase by placing the 3′-OH in a vector in which phosphotransfer from the incoming nucleotide is inefficient. (Michailidis E, et ak, Mechanism of inhibition of HIV-l reverse transcriptase by 4’-ethynyl-2-fluoro-2’-deoxyadenosine triphosphate, J Biol Chem 284:35681-35691 [2009]; Michailidis E, et ak, 4’-Ethynyl-2-fluoro-2’-deoxyadenosine (EFdA) inhibits HIV-l reverse transcriptase with multiple mechanisms, J Biol Chem 289:24533-24548 [2014] ).
In in-vitro HIV replication assays, EFdA is a potent antiretroviral and exhibits comparable antiviral activity against clinical isolates across all subtypes that have been evaluated. It is rapidly anabolized in both lymphoid derived cell lines and in peripheral blood mononuclear cells to the active triphosphate in vitro, and the intracellular half-life of EFdA Triphosphate (EFdA- TP) exceeds 72 hrs. (Stoddart, C. A., Galkina, et ak, Oral Administration of the Nucleoside EFdA (4’-Ethynyl-2-Fluoro-2’-Deoxyadenosine) Provides Rapid Suppression of HIV Viremia in Humanized Mice and Favorable Pharmacokinetic Properties in Mice and the Rhesus Macaque, Antimicrob Agents Chemother, 2015 Jul; 59(7): 4190-4198, Published online 2015 May 4).
EFdA has been shown to have efficacy in animal models of HIV infection including humanized mouse models and an SIV infected rhesus macaque model. Pharmacokinetic studies of orally administered EFdA in mouse and rhesus monkey have demonstrated rapid absorption and high plasma concentrations. A long intracellular half-life was demonstrated by the fact that isolated peripheral blood mononuclear cells from the rhesus macaque were refractory to SIV infection 24 hr after drug administration. (Ibid.)
Previous syntheses of 4’-ethynyl nucleoside analogs including EFdA suffer from modest stereoselectivity in the formation of the C-N bond between the ethynyl-deoxyribose sugar and the 2-fluoroadenine (also referred to as 2-fluoro-9H-purin-6-amine) nucleobase. The previous syntheses also require protecting groups to carry out the glycosylation reaction which reduces the efficiency of the syntheses.
The synthesis described in Kei Fukuyama, et ak, Synthesis of EFdA via a
Diastereoselective Aldol Reaction of a Protected 3-Keto Furanose, Organic Letters 2015, 17(4), pp. 828-831; DOI: 10.102 l/ol5036535) is a l4-step synthesis from D-glucose diacetonide that uses diastereoselective reactions to set the three stereocenters. The stereochemistry of the anomeric center is controlled by having a 2′-acetoxy directing group that is subsequently removed by hydrolysis and deoxygenation. This route requires 4 chromatographic purifications, and the stoichiometric use of a toxic organotin reagent for late-stage deoxygenation.
In another route (see Mark McLaughlin, et al., Enantioselective Synthesis of 4′-Ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) via Enzymatic Desymmetrization, Organic Letters 2017, 19 (4), pp. 926-929), the fully-substituted 4′- carbinol is generated stereoselectively with an enzymatic desymmetrization. The 3 ‘-stereocenter is set with a catalytic asymmetric transfer hydrogenation, and the anomeric 1 ‘-linkage is established in modest stereoselectivity using substrate control, with an upgrade in stereochemical purity achieved by crystallization of an intermediate. This process requires 15 steps, requires the use of several protecting groups and generates the glycosyl linkage between the nucleobase and sugar fragments in low
stereoselectivity (1.8: 1).
A l2-step synthesis for making EFdA from R-glyceraldehyde acetonide is described in Kageyama, M., et al., Concise Synthesis of the Anti-HIV Nucleoside EFdA, Biosci. Biotechnol. Biochem, 2012 , 76, pp. 1219 -1225; and Enantioselective Total Synthesis of the Potent Anti-HIV Nucleoside EFdA, Masayuki Kageyama, et al., Organic Letters 2011 13 (19), pp. 5264-5266 [DOL 10.1021 / ol202116k] . The syntheses use the chiral starting material to set the 3′-stereocenter with moderate diastereoselectivity. After chromatographic separation of stereoisomers, the new stereocenter is used to guide a diastereoselective alkyne addition to set the fully-substituted 4′-stereocenter. The anomeric 1 ‘-position is established with little stereocontrol and requires chromatography to separate the anomers. This route requires chromatographic separation of diastereoisomers at two different stages and starts from an expensive chiral starting material.
Kohgo, S., et al., Design, Efficient Synthesis, and Anti-HIV Activity of 4′-C-Cyano- and 4′-C-Ethynyl-2′-deoxy Purine Nucleosides, Nucleosides, Nucleotides and Nucleic Acids, 2004, 23, pp. 671-690 [ DOL 10.1081/NCN-120037508] describes a synthetic route that starts from an existing nucleoside and modifies both the sugar and nucleobase portions. It is an 18-step synthesis starting from 2-amino-2′-deoxy adenosine with a low 2.5% overall yield.
It is known that enzymes such as purine nucleoside phosphorylase (PNP, EC 2.4.2.1) can form the glycosyl linkage in nucleosides and nucleoside analogs in high stereoselectivity and without the use of protecting groups. See for example the review: New Trends in Nucleoside Biotechnology, Mikhailopulo, I. A., Miroshnikov, A.I,. Acta Naturae 2010, 2, pp. 36-58.
However, the current scope of the sugar fragments capable of undergoing reaction catalyzed by PNP has been limited to the a- 1 -phosphates of natural ribose and deoxyribose along with a small number of analogs with small H, NH2, or F substituents at the C2’ and C3’ positions and replacements of the C5’ OH group. There have been no reports of successful glycosylation catalyzed by PNP using sugars with carbon substituents on the ring or any substitution at the C4’ position.
Access to the ribose and deoxyribose a- 1 -phosphate substrates for the PNP-catalyzed glycosylation has been demonstrated by translocation of the phosphate group from the 5’-hydroxyl to G -hydroxyl position with the enzyme phosphopentomutase (PPM, EC 5.4.2.7) (see Mikhailopulo, I. A., et al. supra). However, the scope of the sugars for which PPM is capable of catalyzing this reaction has been limited to ribose, arabinose, 2-deoxyribose, and 2,3-dideoxyribose. No examples have been reported of successful reaction with sugar phosphates containing any additional substituents.
Deoxyribose phosphate aldolase (DERA, EC 4.1.2.4) enzymes are known to catalyze the aldol addition of acetaldehyde to other short-chain aldehydes (see review: Stephen M. Dean, et al., Recent Advances in Aldolase-Catalyzed Asymmetric Synthesis, Adv. Synth. Catal. 2007, 349, pp. 1308 – 1320; DOI: 10. l002/adsc.200700115). However, no examples have been reported with aldehydes bearing a fully substituted carbon a to the aldehyde.
ETS Patent 7,229, 797 describes the formation of deoxyribonucleosides from the natural unsubstituted deoxyribose 1 -phosphate by use of purine nucleoside phosphorylase (PNP) and additionally using enzymes such as sucrose phosphorylase to remove the inorganic phosphate byproduct and drive the equilibrium. It does not disclose enzyme engineering for the creation of PNP enzymes that can generate nucleosides from the unnatural 4-ethynyl-D-2-deoxyribose 1-phosphate, nor that through engineering of PPM and DERA enzymes to act on unnatural substrates, 4-ethynyl-D-2-deoxyribose 1 -phosphate can be generated.
In view of the difficult and lengthy synthetic options developed to date for producing 4’-ethynyl nucleoside analogs, it would be desirable to develop an improved enzymatic synthesis for 4’-ethynyl nucleoside analogs such as EFdA that reduces the number of process steps, minimizes the use of protecting groups, improves the stereoselectivity of glycosylation and avoids the use of toxic materials.
Surprisingly, it has been found that PPM enzymes have some activity with the 3-atom ethynyl substituent at the 4’ position on ribose and that the PPM enzyme activity could be improved by introducing mutations into the enzymes to successfully develop a reaction for
isomerization of
4-ethynyl-D-2-deoxyribose 5-phosphate (6) to 4-ethynyl-D-2-deoxyribose 1 -phosphate (6.5) catalyzed by PPM to enable a more efficient method for production of 4’-ethynyl-2’-deoxy nucleosides.
Additionally, PNP enzymes have also been found to have some activity with the 3-atom ethynyl substituent at the 4 position on deoxyribose and that the PNP enzyme activity could be improved by introducing mutations into the enzymes to successfully develop a glycosylation reaction catalyzed by PNP to enable a more efficient method for production of 4’ -ethynyl -2’-deoxy nucleosides.
Even further improvement to the overall synthetic method came from the finding that
DERA enzymes, particularly the DERA from Shewanella halifaxensis, have activity for aldol reaction with 2-ethynyl-glyceraldehyde 3-phosphate which has a fully substituted a-carbon. This discovery allowed for the efficient synthesis of 4-ethynyl-D-2-deoxyribose 5-phosphate, a precursor to 4’-ethynyl-2’-deoxy nucleoside analogs, e.g., including EFdA.
SUMMARY OF THE INVENTION
The present invention involves the use of engineered enzymes in a novel enzymatic synthesis of 4’-ethynyl-2’-deoxy nucleoside analogs, including EFdA, that eliminates the use of protecting groups on intermediates, improves the stereoselectivity of glycosylation and greatly reduces the number of process steps needed to make said compounds compared to prior methods, among other process improvements. It further relates to novel intermediates which are an integral part of the enzymatic process.
The overall process is summarized in the following Scheme 1 and Scheme 2; the latter scheme provides an alternative method for making compound 5:
Scheme 1
kinase
p p y
Scheme 1A
kinase galactose oxidase
3 2X+ 9
2
p p y
It has been discovered that 4’-ethynyl-2’-deoxy nucleoside analogs such as EFdA can be synthesized employing a final step one-pot process by combining 4-ethynyl-D-2-deoxyribose 5-phosphate (6) with two enzymes, phosphopentomutase (PPM) [for example but not limited to SEQ ID NO.: 8] and purine nucleoside phosphorylase (PNP) [for example but not limited to SEQ ID NO.: 9, SEQ ID NO.: 15], as shown in Scheme 2.
Scheme 2
Scheme 2A
Several upstream intermediates used in the present process for the synthesis of the final product 4’-ethynyl-2’-deoxy nucleosides and analogs thereof are also made using enzymatic reaction methods as shown in Scheme 3; Scheme 3 A and Scheme 3B
Scheme 3
Scheme 3A
o2
pTsOH
deoxyribose
aldolase
Scheme 3B
Experimental Procedures
Preparation of 2-ethynyl-2-hvdroxypropane-l,3-diyl diacetate 12)
Method A:
To a -35 °C solution of diacetoxyacetone (1) (159 g, 914.0 mmol) in THF (1000 mL) was added 1600 mL of a 0.5 M solution of ethynyl magnesium chloride in THF maintaining the temperature below -20 °C. After the reaction reached completion, acetic acid (78 mL) in 400 mL methyl tert-butyl ether (MTBE) was added dropwise keeping the temperature below -20 °C. MTBE (800 mL) was then added and the mixture was warmed to room temp. Saturated NaCl in water (1000 mL) was added followed by saturated NH4CI solution in water (1050 mL). The organic layer was separated, dried over Na2SC>4 and evaporated to give compound (2) as an oil (160 g, 88%). 1H NMR (CDCI3, 500 MHz): d 4.26 (dd, 4 H), 2.55 (s, 1H), 2.14 (s, 6H).
Preparation of 2-ethynyl-propane-l,2,3-triol 13)
Method B:
To a solution of 2-ethynyl-2-hydroxypropane-l,3-diyl diacetate (2) (70 g, 350 mmol) in ethanol was added a 0.5M solution of sodium methoxylate in methanol (69.9 mL, 35.0 mmol) at room temperature (rt). The reaction was stirred at rt for 2 hours (h) to reach completion. The solvents were evaporated and the residue was re-dissolved in 100 mL water and extracted with 3 x 50 mL MTBE. The aqueous layer was sparged with nitrogen to remove residual solvents to give a 40.9% solution of 2-ethynyl-propane-l,2,3-triol (3) (108 g , 100% yield) as determined by nuclear magnetic resonance (NMR) (maleic acid as internal standard). lH NMR (D2O, 500 MHz): d 3.60 (dd, 4 H), 2.85 (s, 1H).
Alternate Preparations o ethynyl-glvcer aldehyde 14)
Method Cl:
In a stirred reactor, 2-ethynyl-propane-l,2,3-triol (3) (1.1 g, 9.47 mmol) in sodium phosphate buffer (30 mL, 100 mM, pH 7.0) containing antifoam 204 (Sigma A6426, 1 drop ~ 20 pL) was warmed to 30 °C with air sparging at 12.5 seem. Galactose oxidase (GOase, SEQ ID NO.: 1) (250 mg), Horseradish Peroxidase* (Type I, 5 mg) and bovine catalase** (5 mg) dissolved in sodium phosphate buffer (5 mL 100 mM, pH 7.0) were added to the reactor, followed by the addition of CuS04 aq. solution (100 mM, 150 pL). The reaction mixture was stirred at 600 rpm with air sparging for 47h to give (f?)-2-ethynyl-glyceraldehyde (4) in 47% conversion (by NMR) and 72% e.e. . (The product was not isolated). lH NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H).
* Horse Radish Peroxidase: wild type peroxidase from horseradish Type I, commercially available from SIGMA (P8125), isolated from horseradish roots (Amoracia rusticana).
** Bovine catalase: heme-dependent catalase from bovine source, commercially available from Sigma (C1345)
Method C2:
In a stirred 100 L jacketed reactor charged with deionized water (56.2 kg), sodium dihydrogen phosphate (1.212 kg, 10 moles) was added. The pH was adjusted to 7.02 using 10 N sodium hydroxide solution (852.6 g) at 25 °C. The reactor was charged with Antifoam 204 (A6426, 10 mL), followed CuS04*5H20 (6.5 g). Galactose oxidase (451.2 g) (SEQ ID NO.: 10) was added and stirred for 15 min while sparged with air. Horseradish peroxidase* (200.2 g) and catalase** (502.6 g) were added and the reactor was rinsed with water (2.0 kg). Next 2-ethynyl-propane- 1,2, 3 -triol (3) solution in water (9.48%, 30.34 kg, 24.72 mol) was added followed by an additional portion of Antifoam 204 (A6426, 10 mL). The reaction was sparged with air and
stirred overnight to give 94.0 kg of (A)-2-ethynyl-glyceraldehyde (4) in 66% conversion (by NMR) and 84% e.e. Assay yield 60%: 1H NMR (D20, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H).
* Horse Radish Peroxidase: wild type peroxidase from horseradish purified, commercially available from Toyobo (PEO-301), isolated from horseradish roots (Amoracia rusticana).
** Bovine catalase: heme-dependent catalase from bovine source, commercially available from Sigma (C1345).
The above reaction was also performed using the galactose oxidase (SEQ ID NO.: 11) and the product (4) was obtained in 67% conversion (by NMR) and 88% e.e. and assay yield 59%: 1H NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H).
Method C3:
In a 100 mL Easy Max vessel equipped with sparger and flow controller, water (82 mL) and PIPES potassium buffer (5mL, 0.5 M) were charged. The pH was adjusted to 7.5 using 5 M KOH solution at 25 °C. Antifoam 204 (200 pL) was added, followed by evolved galactose oxidase (SEQ ID NO.: 17, 450 mg enzyme powder) and copper(II) sulfate pentahydrate (100 pL, 100 mM). The reaction mixture was sparged with air at 125 standard cubic centimeters per minute (seem) for 15 min. Bovine catalase (Cl 345, Sigma-Aldrich, 150 mg, 2000-5000 U/mg, 0.75 MU) was charged, followed by horseradish peroxidase (HRP, Toyobo PEO-301, 100 mg,
130 U/mg, 1.3 kU) and the aqueous solution of 2-ethynyl-propane-l,2,3-triol (3) (25 wt%, 12 mL, 25.8 mmol). The reaction mixture was stirred at 30 °C with aeration at 125 seem and sampled using EasySampler over 20h to give 70% conversion and form compound (4) ((A)- 2-ethynyl-glyceraldehyde) in 58% assay yield and 99% e.e. lH NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H). The crude reaction stream was carried directly into the subsequent phosphorylation step.
Method C4: Oxidation with immobilized galactose oxidase
Galactose
Oxidase
immobilized
3
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (16 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 160 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution. In a vessel evolved galactose oxidase (SEQ ID NO.: 17, 2.00 g) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 5.00 mL), followed by addition of binding buffer (50 mL) and the resin. The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 160 mL) and potassium PIPES buffer (10 column volumes, 160 mL; 50 mM, pH 7.5) and it was used directly in a reaction. Reaction procedure:
In a 100 mL Easy Max vessel equipped with sparger and flow controller, water (82 mL) and PIPES potassium buffer (5mL, 1 M) were charged. The pH was adjusted to 7.5 using 5 M KOH solution at 25 °C. Antifoam 204 (200 pL) was added, followed by evolved galactose oxidase immobilized on the resin (SEQ ID NO.: 17, 750 mg enzyme powder per 6 mL resin) and copper(II) sulfate pentahydrate (100 pL, 100 mM). The reaction mixture was sparged with air at 125 standard cubic centimeters per minute (seem) for 15 min. Bovine catalase (C1345, Sigma-Aldrich, 210 mg, 2000-5000 U/mg, 1.05 MU) was charged, followed by horseradish peroxidase (HRP, Toyobo PEO-301, 100 mg, 130 U/mg, 1.3 kU) and the aqueous solution of 2-ethynyl-propane- 1,2, 3 -triol (3) (25 wt%, 13 mL, 29.4 mmol). The reaction mixture was stirred at 25 °C with aeration at 125 seem. After 22h the reaction reached 91% conversion to give 200 mM (//)-2-ethynyl-glyceraldehyde (4) solution (100 mL, 68% assay yield, 97% e.e. lH NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H). The crude reaction stream was carried directly into the subsequent phosphorylation step.
Method C5: Optional Isolation of aldehyde via formation of aminal (8)
Step 1: Preparation of (S)-2-( \ .3-dibenzylimidazolidin-2-yl )but-3-yne- l 2-diol
A 100 L jacketed cylindrical vessel equipped with nitrogen bubbler, mechanical stirrer and thermocouple was charged with crude oxidase reaction stream containing (f?)-2-ethynyl-glyceraldehyde ((4), 26.0 kg, 1.85 wt% aldehyde, 3.64 mol) and inerted with N2 atmosphere. The aqueous solution was warmed to 20 °C and Af,A-di methyl dodecan- 1 -ami ne oxide (DDAO) (30 wt% in water, 798 g, 0.96 mol;) was added, followed by MTBE (55.3 kg, 76 L) and N,N -dibenzylethane-l, 2-diamine (1.55 kg, 6.43 mol). The brown, biphasic mixture was stirred overnight at 20 °C under nitrogen atmosphere. After 17 hours the stirring was stopped and the organic phase was removed and discarded. A light brown MTBE solution of fV)-2-( l ,3-dibenzylimidazolidin-2-yl)but-3-yne-l,2-diol (56.5 kg, 2.02 wt% aminal, 3.39 mmol, 93% assay yield) was obtained.
Six similar MTBE solutions were processed together in a single distillation and crystallization step (in total 374.4 kg of solution, containing 7.91 kg aminal).
A 50 L jacketed cylindrical vessel equipped with mechanical stirrer, distillation head (condenser at -20 °C) and thermocouple was charged with aminal solution (45 L). Vacuum was applied to the vessel (65-95 torr) and the jacket was set to 40 °C. Solvent was removed by distillation until a volume of 35 L had been reached. At this point, the internal temperature was 6.1 °C and an off-white solid had begun to crystallize. The remaining MTBE solution was slowly added, maintaining a constant volume of 35-40 L and an internal temperature of 0-10 °C. Once all the MTBE solution had been added the volume was decreased to 25 L. Distillation was halted, the vessel was inerted with nitrogen and the jacket temperature was decreased to 10 °C. The resulting pale yellow suspension was aged at this temperature for 2 hours and the solids were collected by filtration. The filter cake was washed with cold (-2 °C) MTBE (12.7 kg) and then dried under nitrogen flow for 7 hours. (5)-2-(l,3-dibenzylimidazolidin-2-yl)-but-3-yne-l,2-diol was obtained as an off-white crystalline solid (5.75 kg) lff NMR (500 MHz, DMSO-i¾) d 7.42 – 7.35 (m, 4H), 7.32 (td, J= 7.5, 1.6 Hz, 4H), 7.27 – 7.21 (m, 2H), 5.10 (t, J= 5.6 Hz, 1H), 5.03 (s, 1H), 4.28 (d, J= l3.3Hz, 1H), 4.16 (d, J= 13.3 Hz, 1H), 3.76 (s, 1H), 3.70 – 3.58 (m, 4H), 3.21 (d, J= 0.9 Hz, 1H), 2.90 – 2.80 (m, 2H), 2.60 – 2.51 (m, 2H).13C NMR (126 MHz, DMSO-i¾) d 140.0, 140.0, 128.5, 128.3, 128.2, 128.1, 126.8, 126.8, 88.6, 86.9, 75.0, 74.0, 66.4, 60.7, 60.5, 50.4, 50.3, 39.5. HR-MS (ESI) Aminal (M + H+) C21H25N202+ calculated 337.1911; found 337.1922.
Step 2: Prep l (8)
A 4 L jacketed cylindrical vessel equipped with nitrogen bubbler and mechanical stirrer was charged with of TsOH»H20 (12.0 g, 63.1 mmol), water (60 mL), (ri)-2-(l,3-dibenzylimidazolidin-2-yl)but-3-yne-l,2-diol (110 g, 327 mmol) and MTBE (1700 mL). The biphasic mixture was placed under nitrogen and the jacket temperature was set to 15 °C. A solution of TsOH»H20 (114 g, 599.3 mmol) in water (600 mL) was added dropwise over 1.5 hours with overhead stirring (200 rpm). After addition had completed, the jacket temperature was lowered to 5 °C and the resulting slurry was aged for 1 hour. The solids were removed by filtration and washed with cold water (270 mL). The biphasic solution was transferred to a separating funnel and the organic phase was removed and discarded. The aqueous phase was treated with DOWEX™ MARATHON™ A resin (hydroxide form, 11.0 g) and AMBERLYST® 15 resin (hydrogen form, 11.0 g) while sparging with N2 at a rate of 200 seem for 24 hours to remove residual MTBE. The resins were removed by filtration to give a colorless aqueous solution of (f?)-2-hydroxy-2-(hydroxymethyl)but-3-ynal (774 g, 4.6 wt% aldehyde, 82% yield). lH MR (500 MHz, D2O) d 5.01 (s, 1H), 3.77 (d, J= 11.7 Hz, 1H), 3.73 (d, J= 11.7 Hz, 1H), 2.92 (s, 1H). 13C NMR (126 MHz, D2O) d 129.4, 125.4, 90.3, 81.0, 76.0, 73.9, 65.3. HRMS
(ESI) Aldehyde dimer (2M + Na+) CioHi2Na06+ calculated 251.0526; found 251.0530.
Alternate Preparations o ethvnyl-glvceraldehvde 3-phosphate (5):
Method Dl: Acetate kinase: ATP -regeneration system
Pantothenate kinase PanK
ATP
Acetate kinase
4 Acetate phosphate
5
In a stirred reactor, to a solution of adenosine diphosphate disodium salt (40 mg, 0.087 mmol) and magnesium chloride (38 mg, 0.400 mmol) in HEPES buffer (66 mM, pH 7.5, 30 mL) was added (i?)-2-ethynyl-glyceraldehyde (4) (1.9 mL, 210 g/L solution in water, 3.51 mmol), followed by acetate kinase (SEQ ID NO.: 3) (40 mg), and pantothenate kinase (SEQ ID NO.: 2) (120 mg). The reaction mixture was warmed to 25 °C and a solution of acetyl phosphate lithium potassium salt (1.3 g, 7.01 mmol) in HEPES buffer (50 mM, pH 7.5, 10 mL) was added dropwise over 4 hours, with pH maintained at 7.5 using 5M sodium hydroxide. The reaction was stirred for 18 hours to give (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) in 85% conversion (by HPLC) (The product was not isolated). iH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Method D2: Pyruvate oxidase ATP -regeneration system
Pan
Pyruvate oxidase
Pyruvate
Phosphate
02
In a stirred reactor, a solution of sodium pyruvate (3.11 g, 28 mmol) and phosphoric acid (0.523 mL, 7.71 mmol) in 76 mL water pH 7.5 was charged with (i?)-2-ethynyl-glyceraldehyde (4) (3.8 mL, 210 g/L solution in water, 7.01 mmol), adenosine diphosphate disodium salt (80 mg, 0.174 mmol), thiamine pyrophosphate (40 mg, 0.086 mmol), flavin adenine dinucleotide disodium salt hydrate (64 mg, 0.077 mmol), and magnesium chloride (400 pL, 1 M solution in water, 0.4 mmol). The pH was re-adjusted to 7.5 with 5M aq sodium hydroxide and the reaction volume was re-adjusted to 80 mL with water. Acetate kinase (SEQ ID NO.: 3) (80 mg), pyruvate oxidase (SEQ ID NO.: 4) (80 mg, lyophilized cell free extract), pantothenate kinase (SEQ ID NO.: 2) (400 mg), and catalase (800 pL, ammonium sulfate suspension CAT-101, Biocatalytics) were added. The reaction was stirred at 500 rpm and 30 °C with air sparging for 72 hours to give (//)-2-ethynyl-glyceraldehyde 3 -phosphate 5 in 95% conversion (by HPLC) (The product was not isolated). lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
The above reaction was also performed using the pantothenate kinase (SEQ ID NO.: 13) and the product 5 was obtained in 66% conversion. (The product was not isolated). iH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H).
Method D3: Acetate kinase: ATP -regeneration system using immobilized enzymes
Panth
Acetate phosphate
Enzyme immobilization procedure:
NUVIA™ Immobilized Metal-ion Affinity Chromatography (IMAC) nickel-charged resin (168 mL based on settled volume) was added to a filter funnel and washed with binding buffer (1.6 L; 500 mM sodium chloride, 50 mM sodium phosphate, pH 8.0). In a vessel, pantothenate kinase
(8.4 g) (SEQ ID NO.: 12) and acetate kinase (2.8 g) (SEQ ID NO.: 3) were dissolved in binding buffer (500 mL). The washed resin was charged to the vessel and the solution was stirred for 4 hours at 20 °C. The resin was filtered and washed first with binding buffer (1.6 L) followed by piperazine-N,N’-bis(2-ethanesulfonic acid) (PIPES) buffer (840 mL; 50 mM, pH 6.5). The washed resin was used directly in the next step.
Reaction procedure:
To a 1 L reactor, a solution of (f?)-2-ethynyl-glyceraldehyde (4) in water (608.7 g, 4.6 wt%, 212 mmol) was charged and cooled to 5 °C. To the cooled solution piperazine-N,N’-bis(2-ethanesulfonic acid) (PIPES) buffer (32.7 mL, 1 M, pH 6.5, 32.7 mmol), magnesium chloride (9.33 mL, 1 M, 9.33 mmol), acetyl phosphate diammonium salt (51.8 g, 265 mmol), adenosine diphosphate disodium salt hydrate (1.17 g, 2.12 mmol), and water (192 mL) were added. The solution was allowed to stir and pH was adjusted to 6.4 using 5 N KOH. The reaction was warmed to 20 °C and 168 mL of resin with co-immobilized pantothenate kinase (SEQ ID NO.: 12) and acetate kinase (SEQ ID NO.: 3) was added. The reaction was stirred for 10 hours with 5 N KOH used to maintain a pH of 6.4 to give (f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) in
92% conversion (by HPLC) and 91% yield (by 3 lp NMR with tetraphenylphosphonium chloride as internal standard) (the product was not isolated). lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Preparation of 4-ethynyl-D-2-deoxyribose 5-phosphate 16)
Method E:
To a solution of (f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) (5, 20 mL, 5.3 mmol) in water, a solution of acetaldehyde in water (40 wt.%, 2.02 mL, 15.9 mmol) was added at room
temperature, followed by the addition of Deoxyribose-phosphate aldolase (DERA) (SEQ ID NO. : 6), 25 mg solution in triethanolamine hydrochloride buffer (1 mL, 1 M, pH 7.0). The reactor was sealed and the mixture was stirred overnight at 30 °C and 600 rpm to give 4-ethynyl-D-2-deoxyribose 5-phosphate (6) in 99% conv. and 99% e.e., 99% d.e. as a 1 : 1 anomer mixture (The product was not isolated) a-anomer: lH NMR (D2O, 600 MHz) 5 5.31 (t, 1H), 4.13 (t, 1H), 3.81-3.72 (m, 2H), 2.89 (s, 1H), 2.42-2.34 (m, 1H), 1.87-1.79 (m, 1H); 13c NMR (D2O, 151 MHz) 5 97.7 (s), 81.4 (d), 79.4 (s), 78.9 (s), 71.1 (s), 67.7 (d), 39.6 (s). b-anomer: 1H NMR
(D2O, 600 MHz) 5 5.40 (dd, 1H), 4.28 (t, 1H), 3.88-3.80 (m, 2H), 2.87 (s, 1H), 2.13-2.06 (m,
1H), 2.04-1.97 (m, 1H); 13C NMR (D20, 151 MHz) 5 97.3 (s), 82.2 (d), 78.7 (s), 78.5 (s), 71.3 (s), 68.4 (d), 39.6 (s). LC-MS: (ES, m/z): calculated for C7H10O7P (M-H): 237.0; found 237.0
Alternate Preparations of (2 ?,3A,5 ?)-5-(6-amino-2-fluoro-9H-purin-9-yl)-2-ethynyl-2-(hydroxymethyl)tetrahydrofuran-3-ol monohydrate (7) [alternative name 4’-ethynyl-2-fluoro- 2’-deoxvadenosine or EFdAI
Method FI:
Ammonium ((2f?,3ri)-2-ethynyl-3,5-dihydroxytetrahydrofuran-2-yl)m ethyl hydrogen phosphate (1.00 g, 3.91 mmol) was dissolved in 10 mL of pH 7.5 buffer (100 mM triethanolamine ΉO containing 5 mM MnCl2). The solution pH was adjusted to 7.3 with 5 N NaOH. To the solution was added 2-fluoroadenine (0.599 g, 3.91 mmol) and sucrose (2.68 g, 7.82 mmol). The enzyme solution was prepared by dissolving phosphopentomutase (SEQ ID NO. : 8) (100 mg), purine nucleoside phosphorylase (SEQ ID NO.: 9) (50 mg), and sucrose phosphorylase (SEQ ID NO. :
7) (10 mg) in 10 mL of the pH 7.5 buffer. The enzyme solution was added to the reagent mixture and the resulting suspension was shaken at 40 °C. After 20 h, the suspension was cooled to 0 °C and filtered, rinsing with cold water. The solid was suction dried to give the title compound (1.12 g, 92%) as a single isomer.
iH NMR: (300 MHz, DMSO-d6, ppm): d 7.68 (br s, 2H), 7.32 (d, J = 2.0 Hz, 1H), 6.44 (t, J =
5.8 Hz, 1H), 5.52 (d, J = 5.6 Hz, 1H), 5.27 (t, J = 6.0 Hz, 1H), 4.44 (q, J = 6.4 Hz, 1H), 3.60 (q, J = 6.0 Hz, 1H), 3.53 (q, J = 6.4 Hz, 1H), 3.48 (s, 1H), 2.48-2.41 (m, 1H), 2.37-2.30 (m, 1H). 13c NMR (150.92 MHz, DMSO-d6, ppm) d 158.5 (d, JCF = 203.5), 157.6 (d, JCF = 21.2), 150.2 (d, JCF = 20.2), 139.7 (d, JCF = 2.4), 117.4 (d, JCF = 4.0), 85.1, 82.0, 81.4, 78.7, 70.1, 64.2, 38.1. LC-MS: (ES, m/z): calculated for C12H12FN5O3 (M+Na): 316.0822; found 316.0818.
The PPM and PNP enzymes used in this step were each derived from mutations starting from the enzymes from E. coli ( Escherichia coli). The sucrose phosphorylase (SP) used in this step was derived from Alloscardovia omnicolens ; SP derived from other organisms could also be used.
Method F2:
To an aqueous solution of (f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) (950 mL, 157 mmol) containing piperazine-N,N’-bis(2-ethanesulfonic acid) (PIPES) buffer at a pH from about 5.5 to 6.0 was added triethanolamine (7.09 g, 47.5 mmol). The pH of the solution was adjusted from 7.1 to 7.6 using potassium hydroxide (8 mL, 8M). Manganese(II) chloride hydrate (0.592 g, 4.70 mmol) was added followed by sucrose (161 g, 470 mmol), giving a pH of 7.5 To the solution
was added the following enzymes: deoxyribose-phosphate aldolase (SEQ ID NO. : 14) (461 mg), sucrose phosphorylase (SEQ ID NO. : 7) (494 mg), phosphopentomutase (SEQ ID NO.: 8)(2.63 g), and purine nucleoside phosphorylase (SEQ ID NO. : 15) (659 mg). Once the enzymes were dissolved, 2-fluoroadenine (19.80 g, 125 mmol) was added. The reaction was heated to 35 °C and acetaldehyde was added (40 wt% in isopropyl alcohol, 29.8 mL, 235 mmol). After reacting for 2h, the mixture was seeded with EFdA crystalline product (0.96 g, 2 mol%). After reacting over 26 h at 35 °C, the slurry was cooled to 0 °C, and the solids were collected by filtration, washing with water two times (40 mL ea.). The solids were dried under a nitrogen sweep. Yield 43.2 g, 92 wt%, 96.2% corrected. ¾ NMR: (300 MHz, DMSO-d6, ppm): d 7.68 (br s, 2H), 7.32 (d, J = 2.0 Hz, 1H), 6.44 (t, J = 5.8 Hz, 1H), 5.52 (d, J = 5.6 Hz, 1H), 5.27 (t, J = 6.0 Hz, 1H), 4.44 (q, J = 6.4 Hz, 1H), 3.60 (q, J = 6.0 Hz, 1H), 3.53 (q, J = 6.4 Hz, 1H), 3.48 (s, 1H), 2.48-2.41 (m, 1H), 2.37-2.30 (m, 1H). 13C NMR (150.92 MHz, DMSO-d6, ppm) d 158.5 (d, JCF = 203.5), 157.6 (d, JCF = 21.2), 150.2 (d, JCF = 20.2), 139.7 (d, JCF = 2.4), 117.4 (d, JCF = 4.0), 85.1, 82.0, 81.4, 78.7, 70.1, 64.2, 38.1. LC-MS: (ES, m/z): calculated for C12H12FN5O3 (M+Na): 316.0822; found 316.0818.
Alternate Preparations of -2-ethvnyl-propane-l,2,3-triol 1 1-phosphate 19) :
Method Gl: Acetate kinase: ATP-regeneration system using enzymes SEQ. ID No.: 2 and SEQ. ID No.: 3
Panthotenate kinase PanK
ATP
Acetate kinase
Acetate phosphate
A 50 mL reactor was charged with a solution of 2-ethynyl-propane-l,2,3-triol (3) in water (9.29 g, 9.46 wt%, 7.57 mmol) potassium PIPES buffer (1.02 mL, 1 M, pH 6.5, 1.02 mmol), magnesium chloride (292 pL, 1 M, 0.292 mmol), acetyl phosphate diammonium salt (1.851 g, 89 wt%, 9.46 mmol), adenosine diphosphate disodium salt hydrate (ADP, 42 mg, 0.076 mmol, 0.01 eq), and water (28 mL). The pH was adjusted to 6.4 using 5 M KOH, the solution was warmed to 20 °C and evolved pantothenate kinase PanK SEQ. ID No.: 2 (264 mg) and acetate kinase AcK SEQ. ID No. : 3 (88 mg) were added. The reaction was stirred for 16 hours with pH maintained at 6.4 using 5 N KOH. The final reaction contents provided C.V)-2-ethynyl -propane- 1 ,2,3-triol 1-phosphate (9) in >95% e.e. and 99% conversion (by 31P NMR). The product was not isolated. ¾ NMR (D2O, 500 MHz) d 3.89 (m, 2H), 3.72 (d, 7= 11.6 Hz, 1 H), 3.65 (d, J= 11.6 Hz, 1H),
2.93 (s, 1H). 13C NMR (D2O, 126 MHz) d 82.9 (s), 75.1 (s), 71.0 (d, J= 6.9 Hz), 67.0 (d, J= 4.5 Hz), 64.7 (s). 31P NMR (D2O, 202 MHz) d 3.39. HRMS: (ESI, m/z): calculated for [M-l] CsHsOeP: 195.0058; Found 195.0068 [M-H] : 195.0058.
Method G2: Acetate kinase: ATP-regeneration system using enzyme SEQ. ID No.: 20 and enzyme SEQ. ID No.: 21
Panthotenate kinase PanK
– – ATP
Acetate kinase
Acetate phosphate
To a jacketed reactor aqueous solution 2-ethynyl-propane-l,2,3-triol (3) (11.47 kg, 8.7% wt, 8.61 mol) and water (7.5kg) was charged, followed by 1M BIS-TRIS methane buffer pH 6.5 (1L) and magnesium chloride (41.4 g). ATP (48g, 0.086 mol, 0.01 equivalent) and diammonium acetyl phosphate (2.021 kg, 89%, 10.33 mmol) were added, the solution was warmed up to 20 °C and the pH of the solution was re-adjusted to 6.8 using KOH (270.4 g). Evolved pantothenate kinase SEQ. ID No.: 20 (20.4 g) and evolved acetate kinase SEQ. ID No.: 21 (3 g) were then charged as solids. The reaction was stirred for at 20 °C for l6h during which pH dropped to 5.5.
Quantitative conversion of 2-ethynyl-propane-l,2,3-triol (3) was obtained as judged by ‘H and 31P NMR. Such prepared (ri)-2-ethynyl-propane-l,2,3-triol l-phosphate (9) solution (397 mM, 22.5 kg, 98% yield) was used in subsequent oxidation step without any further purification. ‘H NMR (D2O, 500 MHz) d 3.89 (m, 2H), 3.72 (d, 7= 11.6 Hz, 1 H), 3.65 (d, J= 11.6 Hz, 1H),
2.93 (s, 1H).
Method G3: Acetate kinase: ATP-regeneration system using enzyme SEQ. ID No.: 20 and enzyme SEQ. ID No.: 21 with deuterated compound (3) to assign absolute stereochemistry and demonstrate desymmetrizing phosphorylation.
Acetate phosphate
Z-d2, 95:5 er
Evolved pantothenate kinase SEQ. ID No. : 20 (100 pL of 10 g/L solution in water ) and evolved acetate kinase SEQ. ID No. : 21 (100 pL of 2g/L solution in water) were added to a solution containing diammonium acetyl phosphate (41 mg), 2-ethynyl-propane-l, l-72-l,2,3-triol ((A)- 3-d2, 20 mg, 170 pmol), magnesium chloride (10 pL of 1 M solution in water), ADP (10 pL of 100 g/L solution in water), and sodium phosphate buffer (10 pL of 1 M solution in water) in water (800 pL) at pH 6.5. The reaction was incubated for 24h at rt to give deuterated 2-ethynyl-propane-l,2,3-triol l-phosphate analogs (S)-9-(3,3-d2) and (S)-9-(l,l-d2) in 95:5 ratio and 99% overall yield. The ratio of phosphorylated compounds was determined by 31P NMR to be -95:5, confirming stereoselective phosphorylation of the 2-ethynyl-propane-l,2,3-triol (3) at the pro-(S) hydroxyl group (i.e. a desymmetrizing phosphorylation). 1H NMR (D2O, 500 MHz) d 3.89 (m, 2H), 3.72 (d, 7= 11.6 Hz, 1 H), 3.65 (d, J= 11.6 Hz, 1H), 2.93 (s, 1H). 13C NMR (D20, 126 MHz) d 82.9 (s), 75.1 (s), 71.0 (d, J= 6.9 Hz), 67.0 (d, J= 4.5 Hz), 64.7 (s).
Method G4: Acetate kinase: ATP-regeneration system using immobilized enzymes SEQ. ID No. : 20 and enzyme SEQ. ID No. : 21
Panthotenate kinase PanK
– – ATP
Acetate kinase
Acetate phosphate
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (75 mL based on settled volume) was added to a filter funnel and washed with water (9 column volumes, 3 x 225 mL) and binding buffer (1 column volume, 75mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0). In a vessel pantothenate kinase (SEQ ID NO. : 20, 6.0 g) lyophilized powder was resuspended in binding buffer (200 mL) and the washed resin was added. The solution was mixed using rotating mixer at 25 °C for 6h. The resin was filtered and washed with binding buffer (6 column volumes, 6 x 225 mL) and BIS-TRIS buffer (8 column volumes, 600 mL; 50 mM, pH 6.2).
Reaction procedure:
An aqueous solution of 2-ethynyl-propane-l,2,3-triol (3) (574 g, 8.7% wt, 0.430 mol) and water (350 mL) was charged to a jacketed reactor, followed by 1M BIS-TRIS methane buffer pH 6.5 (50 mL) and magnesium chloride (2.033 g, 0.01 mol). ATP (2.37g, 0.0043 mol, 0.01 equivalent) and diammonium acetyl phosphate (101 g, 89%, 0.530 mmol, 1.2 eq) were added, the solution was warmed up to 20 °C and the pH of the solution was re-adjusted to 6.8 using 5 M KOH.
Resin with immobilized pantothenate kinase SEQ. ID No. : 20 (25 mL) and evolved acetate kinase SEQ. ID No. : 21 (0.15 g) were then charged as solids. The reaction was stirred for at 20 °C for l6h during which the pH dropped to 5.5. Quantitative conversion of 2-ethynyl-propane- I,2,3-triol (3) to (ri)-2-ethynyl-propane-l,2,3-triol l-phosphate (9) was obtained as judged by ¾ and 31P NMR. ¾ NMR (D20, 500 MHz) d 3.89 (m, 2H), 3.72 (d, J= 11.6 Hz, 1 H), 3.65 (d, J =
I I .6 Hz, 1H), 2.93 (s, 1H).
Alternate Preparations of (i?V2-ethvnyl-glvceraldehvde 3-phosphate 15):
Method HI: Immobilized galactose oxidases SEP ID No.: 16
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (10 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 100 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give 16 g of washed resin. In a vessel evolved galactose oxidase (SEQ ID NO.: 16, 750 mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 5.00 mL), followed by addition of binding buffer (20 mL) and the washed resin (3.0g). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 100 mL) and BIS-TRIS buffer (10 column volumes, 100 mL; 50 mM, pH 7.5) and it was used directly in the glycosylation reaction.
Reaction procedure:
The resin with immobilized galactose oxidase SEQ ID NO.: 16 (3.0 g) was added to a solution of S)-2-ethynyl-propane-l,2,3-triol l-phosphate (9, 5.4 mmol, 270 mM, 20 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), followed by addition of copper (II) sulphate solution in water (30 pL, 100 mM) and horseradish peroxidase (PEO-301, 18 mg) and bovine catalase (C1345, 120 mg) resuspended in water (600 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 22 °C for 4 days to reach final conversion of 77% and give (f?)-2-ethynyl-glyceraldehyde 3 -phosphate (5) in 95% e.e. The enzyme resin was filtered off and the solution of the(f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) was used
directly in the glycosylation reaction. iH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Method H2: Immobilized galactose oxidases SEP ID No.: 17
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (10 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 100 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give l6g of washed resin. In a vessel, evolved galactose oxidase (SEQ ID NO.: 16, 750 mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 5.00 mL), followed by addition of binding buffer (20 mL) and the washed resin (3.0g). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 100 mL) and BIS-TRIS methane buffer (10 column volumes, 100 mL; 50 mM, pH 7.5) and it was used directly in the reaction.
Reaction procedure:
The resin with immobilized evolved galactose oxidase SEQ ID NO.: 17 (3.0 g) was added to a solution of (ri)-2-ethynyl-propane-l,2,3-triol l-phosphate (9, 5.4 mmol, 270 mM, 20 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), followed by addition of copper (II) sulphate solution in water (30 pL, 100 mM) and horseradish peroxidase (PEO-301, 18 mg) and bovine catalase (C1345, 120 mg) resuspended in water (600 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 22 °C for 4 days to reach final conversion of 77% and give (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) in 95% e.e. The enzyme resin was filtered off and the solution of the (i?)-2-ethynyl-glyceraldehyde 3 -phosphate (5) was used directly in the glycosylation reaction. lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Method H3: Immobilized galactose oxidases SEQ ID No.: 18
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (3 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 30 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give 2.4 g of washed resin. In a vial evolved galactose oxidase (SEQ ID NO.: 18, 75mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 1.00 mL), followed by addition of binding buffer (5 mL) and the washed resin (400 mg). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 4 mL) and BIS-TRIS methane buffer (10 column volumes, 4 mL; 50 mM, pH 7.5) and it was used directly in a reaction.
Reaction procedure:
Immobilized evolved GOase SEQ ID NO.: 18 was added (400 mg) to a solution of (5)-2-ethynyl-propane-l,2,3-triol l-phosphate solution ((9), 5.4 mmol, 270 mM, 1 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), , followed by addition of horseradish peroxidase (PEO-301, 1 mg) and catalase from Corynebacterium glutamicum (Roche, lyophilizate, #11650645103, 3 mg) resuspended in water (100 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 30 °C for 48h. Final conversion after 2 days reached 90% conversion and the (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) >99% e.e. The enzyme resin was filtered off and the solution of the (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) was used directly without further purification. lH NMR (D2O, 400 MHz): d 5.02 (s, 1H),
4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (MΉ): 193.1; found 193.0.
Method H4: Immobilized galactose oxidases SEP ID No.: 19
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (3 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 30 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give 2.4 g of washed resin. In a vial evolved galactose oxidase (SEQ ID NO.: 19, 75mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 1.00 mL), followed by addition of binding buffer (5 mL) and the washed resin (400 mg). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 4 mL) and BIS-TRIS methane buffer (10 column volumes, 4 mL; 50 mM, pH 7.5) and it was used directly in a reaction.
Reaction procedure:
Immobilized evolved GOase SEQ ID NO.: 18 was added (400 mg) to a solution of (5)-2-ethynyl-propane-l,2,3-triol l-phosphate solution (9, 5.4 mmol, 270 mM, 1 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), , followed by addition of horseradish peroxidase (PEO-301, 1 mg) and catalase from Corynebacterium glutamicum (Roche, lyophilizate, #11650645103, 3 mg) resuspended in water (100 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 30 °C for 48h. Final conversion after 2 days reached 100% conversion and (i?)-2-ethynyl-glyceraldehyde 3 -phosphate (5) was obtained in >99% e.e. The enzyme resin was filtered off and the solution of the (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) was used directly without further purification. lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
PATENT
CA 2502109
WO 2017053216
US 20200010834
US 20200010868
PAPER
Organic letters (2017), 19(4), 926-929.
Organic Letters (2017), 19(4), 926-929.
Journal of medicinal chemistry (2018), 61(20), 9218-9228.
Bioscience, Biotechnology, and Biochemistry (2020), 84(2), 217-227.
PAPER
Organic letters (2011), 13(19), 5264-6.
A concise enantioselective total synthesis of 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA), an extremely potent anti-HIV agent, has been accomplished from (R)-glyceraldehyde acetonide in 18% overall yield by a 12-step sequence involving a highly diastereoselective ethynylation of an α-alkoxy ketone intermediate.

Processes for preparing islatravir and its analogs comprising the reaction of a substituted tetrahydrofuran compound with purine nucleoside phosphorylase and a nucleobase, followed by stereochemical synthesis, glycosylation, reduction, oxidation and isolation are claimed. Also claimed are novel intermediates of islatravir and processes for their preparation and their use for the preparation of islatravir.
(2R,3S,5R)-5-(6-Amino-2-fluoropurin-9-yl)-2-ethynyl-2-(hydroxymethyl)- tetrahydrofuran-3-ol (1). To a stirred solution of 16 (66.2 mg, 0.115 mmol) in MeOH/CH2Cl2 (2:1, 1.5 mL) was added NH4F (85.1 mg, 2.30 mmol) at room temperature. After 16 h, MeOH (0.5 mL) was added, and the resulting mixture was stirred for an additional 27 h. To the mixture was added 10% NaOH in MeOH (1.5 mL) to adjust the pH of the mixture to ca. 10. After 10 min, Dowex 50W×8 (200– 400 mesh (H)) was added until the pH of the mixture reached ca. 4. To the resulting mixture was added CaCO3 (259 mg, 2.59 mmol), and the mixture was stirred for 30 min. The mixture was filtered through a pad of Celite, and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 10:1) to give 29.3 mg (87%) of 1. Mp: 220.0–221.4 °C (dec.); [α] 25 D +12.4 (c 0.97, MeOH); IR: νmax 3315 (br m), 3179 (br m), 1690 (vs), 1356 (vs); 1 H NMR (600 MHz, DMSO-d6): δ 2.43 (1H, ddd, J = 13.2, 7.3, 7.3 Hz), 2.70 (1H, ddd, J = 13.2, 6.8, 5.1 Hz), 3.52 (1H, s), 3.54 (1H, dd, J = 11.7, 6.4 Hz), 3.65 (1H, dd, J = 11.7, 5.0 Hz), 4.57 (1H, m), 5.32 (1H, m), 5.60 (1H, m), 6.24 (1H, dd, J = 7.2, 5.1 Hz), 7.82 (1H, br s), 7.92 (1H, br s), 8.31 (1H, s); 13C NMR (150 MHz): δ 38.3, 64.4, 70.3, 79.2, 81.7, 82.2, 85.4, 117.6, 140.0, 150.4 (d, JCF = 20.7 Hz), 157.8 (d, JCF = 21.2 Hz), 158.8 (d, JCF = 203.4 Hz); HRMS (FAB): m/z calcd for C12H13FN5 O3, 294.1002; found, 294.1000 ([M+H]+ ).


PAPER
Organic Letters (2011), 13(19), 5264-5266.
PAPER
Bioscience, biotechnology, and biochemistry (2012), 76(6), 1219-25.
EFdA (4′-ethynyl-2-fluoro-2′-deoxyadenosine), a nucleoside reverse transcriptase inhibitor with extremely potent anti-HIV activity, was concisely synthesized from (R)-glyceraldehyde acetonide in an 18% overall yield by a 12-step sequence involving highly diastereoselective ethynylation of an α-alkoxy ketone intermediate. The present synthesis is superior, both in overall yield and in the number of steps, to the previous one which required 18 steps from an expensive starting material and resulted in a modest overall yield of 2.5%.
PAPER
Bioscience, Biotechnology, and Biochemistry (2012), 76(6), 1219-1225.
Organic letters (2015), 17(4), 828-31.
Organic Letters (2015), 17(4), 828-831.
PAPER

Some pharmaceutical companies are investigating biocatalysis at different points in their drug development pipelines, but mostly at one or two steps into the making of a small molecule. Scientists at Merck & Co. have taken this further—they are reporting an entire drug synthesis using a chain of nine enzymes, five of which had been engineered, to produce an experimental HIV drug at high yield in just a few steps (Science 2019, DOI: 10.1126/science.aay8484).
This biocatalytic cascade is turning heads. For the most part, scientists aren’t using biocatalysis to manufacture a compound so much as to develop it, says Princeton University chemist Todd Hyster. The Merck process stitches together nine enzymes to get good yields of the final product, which Hyster says is no small feat.
It literally took my breath away.
Alison Narayan, assistant professor, University of Michigan
“I was blown away,” Hyster says of the first time he saw Merck scientists talk about this work. “It’s something that was very complicated.”
Mark Huffman, a chemist who led the work at Merck with Anna Fryszkowska, says they turned to biocatalysis in order to overcome a couple of key hurdles in synthesizing some molecules. One is stereochemistry. Islatravir is a nucleoside that blocks the HIV enzyme reverse transciptase and traditionally, in medicinal chemistry, it’s been hard to get the stereochemistry of nucleosides right, Huffman says. But this is something enzymes are designed by nature to do. The other is preventing unwanted side reactions. A number of steps in the traditional chemical synthesis of islatravir put the compound’s functional groups at risk of being lopped off, so they must be protected. Huffman says enzymes are specific in the types of reactions they catalyze, so there’s little to no risk of an unwanted side reaction.
Scientists at Merck & Co. use nine enzymes, five of which are engineered, to biocatalytically make an HIV drug called islatravir without having to purify intermediates. Enzymes not engineered by Merck are not pictured.
On top of that, Huffman says, they are doing these reactions at neutral pH, in aqueous solvents, and at room temperature, which cuts down on electricity and the need for multiple bioreactors running under different conditions. Islatravir normally takes between 12 and 18 steps to make. With biocatalysis, the team has cut this down to three.
“You don’t have rigorous equipment requirements,” he says. “You’re usually running [these reactions] under much milder conditions.”
To run the cascade, the team started with 2-ethynylglycerol, and added a mixture of three enzymes to run one group of reactions. They then added more enzymes to drive a second set of reactions. Then, they remove the enzymes from the solution, which are immobilized and easy to filter out, and use four more enzymes to drive the final reactions that lead to islatravir. There are no intermediate purification steps. The overall yield is about 51% using biocatalysis, compared to yields of 7% and 15% using two more traditional syntheses.
To make their biocatalysts, the team surveyed natural enzymes, mostly from microbes, that interacted with the different intermediates in islatravir production. One of the reasons why Huffman says islatravir is an ideal small molecule to produce using biocatalysis is that most organisms have to make and break down nucleosides, so there are several natural enzymes found across multiple species. This gave the team a lot of starting material from which to alter amino acids and build the enzymes they needed to do their syntheses. By making adjustments to active sites and other areas of the enzymes, the team built five of the nine enzymes needed to make islatravir biochemically.
Huffman says that while islatravir is a good molecule to show that scientists can build large biocatalytic cascades, Merck is also looking at biocatalysis to make other small molecules and biologic drugs.
Alison Narayan, a biocatalysis chemist at the University of Michigan, calls Merck “bold” for putting the time, money, and people behind this change in production—it takes a lot of resources to try an entire synthesis via biocatalysis. And, she says, they’ve succeeded spectacularly. “It literally took my breath away,” Narayan says of her first exposure to this project in 2018. “I think it’s a huge accomplishment.”
She says that Merck’s islatravir work shows that industry is starting to appreciate what biocatalysis can do for their drug pipelines and their financial bottom lines. Alongside Merck, companies like GlaxoSmithKline and Pfizer are also exploring biocatalysis at different points in drug development and manufacturing.
“It’s an important proof of concept,” Narayan says. “This is a practical way to build molecules, and this will be the way that people will build molecules when you take into consideration efficiency, green-ness, and constructing an effective synthesis. Biocatalysis has a lot to offer.”
PAPER
Biocatalytic cascades go viral
IN SECTION: PERSPECTIVE | BIOCATALYSIS
An investigational drug targeting the HIV virus is synthesized with nine enzymes
Natural biosynthesis assembles a vast array of complex natural products starting from a limited set of building blocks, under physiological conditions, and in the presence of numerous other biomolecules. Organisms rely on the extraordinary selectivity of enzymes and their ability to operate under similar reaction conditions, meaning that these catalysts are perfectly adapted to mediate cascade reactions. In these multistep processes, the product of one biocatalytic step becomes the substrate for the next transformation (Display footnote number:1-3). On page 1255 of this issue, Huffman et al. (Display footnote number:4) report the development of an impressive nine-enzyme biocatalytic cascade for the synthesis of the investigational drug islatravir for the treatment of human HIV.
This study represents a partnership between scientists from Merck and Codexis. These two companies have a history of successfully collaborating to develop biocatalysts for the synthesis of important pharmaceuticals. Almost a decade ago, they developed a chemoenzymatic route for the synthesis of the type 2 diabetes drug sitagliptin (Januvia), relying on a key enzyme-catalyzed transamination with a highly engineered (R)-selective transaminase (Display footnote number:5). The work was considered a landmark example of directed evolution and functioned to highlight the potential application of biocatalysis to revolutionize industrial chemical processes.
The cascade for synthesizing islatravir was inspired by the bacterial nucleoside salvage pathway, which recycles precious nucleosides by using three key enzymes: a purine nucleoside phosphorylase (PNP), a phosphopentomutase (PPM), and a deoxyribose-5-phosphate aldolase (DERA) (see the figure). However, to achieve the synthesis of the target molecule, Huffman et al. required the natural nucleoside degradative cascade to run in reverse. The reversible nature of enzymes is central to the design of this cascade and is one of the important features that sets biocatalysts apart from the majority of traditional chemical catalysts.
The success of the cascade developed by the team also relied on all three enzymes accepting non-natural substrates bearing a fully substituted carbon at the C-4 position of the 2-deoxyribose ring. The authors reconstructed the reverse nucleoside salvage pathway from a PNP and PPM found in Escherichia coli and a DERA from Shewanella halifaxensis. The native E. coli enzymes required engineering to improve their activity. The DERA displayed existing high activity and stereoselectivity for the formation of the desired sugar phosphate enantiomer, but it required engineering to improve its ability to operate at high substrate concentration.
One of the many advantages of performing biocatalytic cascade reactions is the effective displacement of unfavorable reaction equilibria that can be achieved through product removal. However, despite performing the PNP and PPM steps in tandem, the reaction proceeded with poor conversion, and the inorganic phosphate by-product inhibits the enzymes. An elegant solution to these issues was the inclusion of an auxiliary sucrose phosphorylase, along with its sugar substrate, which removed free phosphate and effectively displaced the reaction equilibrium toward product formation.
Having assembled enzymes for the three key steps in the cascade, Huffman et al. sought to develop a biocatalytic route for the synthesis of the DERA substrate 2-ethynylglyceraldehyde 3-phosphate. Extensive screening of a broad range of kinases resulted in the identification of pantothenate kinase (PanK) from E. coli, which displayed low levels of activity (∼1% conversion) toward the (R)-enantiomer of the target aldehyde. Despite the modest initial activity, directed evolution was successfully used to substantially improve the productivity and stability of this enzyme. Finally, after 12 rounds of evolution, the authors reversed the enantioselectivity and improved the activity, stability, and expression of a galactose oxidase variant for the desymmetrization of the starting substrate, 2-ethynylglycerol.
Opens in modal lightbox
Viewable Image – engineering a biocatalytic cascade
Image Caption
GRAPHIC: A. KITTERMAN/SCIENCE
Advancements in protein engineering, rapid gene sequencing, and the availability of low-cost DNA synthesis have made it possible to alter the properties of enzymes and fine-tune them for biocatalytic applications (Display footnote number:6-8). The work by Huffman et al. is a milestone in cascade design, largely because of the number of biocatalysts operating in tandem and the engineering feat required to optimize five of the nine enzymes involved in the synthesis. It also highlights how biosynthetic or degradative pathways can be a source of inspiration for the design of efficient biocatalytic cascades and how sequences can be reconstituted using enzymes recruited from multiple sources—in this case, of bacterial, fungal, plant, and mammalian origin. The diverse role that biocatalysts can play is also exemplified in this work, where five engineered enzymes are directly involved in the synthesis of the target molecule, and four additional enzymes function to recycle coenzyme, remove inhibitory by-products, and maintain the correct oxidation state of the copper cofactor.
Previous approaches reported for the synthesis of islatravir relied on multistep syntheses and require protecting group manipulations and intermediate purification steps (Display footnote number:9, 10). The incorporation of a key biocatalytic step or steps has the potential to revolutionize synthetic design strategies by making possible transformations that are not accessible using solely chemical approaches (Display footnote number:11, 12). The application of enzymes in industry and the development of chemoenzymatic routes to complex molecules is now well established. However, multistep syntheses exclusively comprising biocatalytic transformations are rare (Display footnote number:13), and this contribution sets a new standard for the synthesis of complex molecules with enzymatic cascades.
School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland. Email: elaine.oreilly@ucd.ie.
REFERENCES AND NOTES
1. S. P. France, L. J. Hepworth, N. J. Turner, S. L. Flitsch, ACS Catal. 7, 710 (2017).
2. S. Gandomkar, A. Żadło-Dobrowolska, W. Kroutil, ChemCatChem 11, 225 (2019).
3. P. Both et al., Angew. Chem. Int. Ed. 55, 1511 (2016).
4. M. A. Huffman et al., Science 366, 1255 (2019).
5. C. K. Savile et al., Science 329, 305 (2010).
6. F. H. Arnold, Angew. Chem. Int. Ed. 57, 4143 (2018).
7. C. Zeymer, D. Hilvert, Annu. Rev. Biochem. 87, 131 (2018).
8. C. A. Denard, H. Ren, H. Zhao, Curr. Opin. Chem. Biol. 25, 55 (2015).
9. M. McLaughlin et al., Org. Lett. 19, 926 (2017).
10. M. Kageyama, T. Nagasawa, M. Yoshida, H. Ohrui, S. Kuwahara, Org. Lett. 13, 5264 (2011).
11. N. J. Turner, E. O’Reilly, Nat. Chem. Biol. 9, 285 (2013).
12. M. Hönig, P. Sondermann, N. J. Turner, E. M. Carreira, Angew. Chem. Int. Ed. 56, 8942 (2017).
13. S. Wu et al., Nat. Commun. 7, 11917 (2016).
ACKNOWLEDGMENTS
J.R. acknowledges the School of Chemistry, University College Dublin, for support.
References
- ^ Kawamoto, A; Kodama, E; Sarafianos, SG; Sakagami, Y; Kohgo, S; Kitano, K; Ashida, N; Iwai, Y; Hayakawa, H; Nakata, H; Mitsuya, H; Arnold, E; Matsuoka, M (2008). “2′-deoxy-4′-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants”. The International Journal of Biochemistry & Cell Biology. 40 (11): 2410–20. doi:10.1016/j.biocel.2008.04.007. PMID 18487070.
- ^ Roy M. Gulick (2018). “Investigational Antiretroviral Drugs: What is Coming Down the Pipeline”. Top Antivir Med. 25 (4): 127–132. PMC 5935216. PMID 29689540.
- ^ “Someday, an Arm Implant May Prevent H.I.V. Infection for a Year”. New York Times. July 23, 2019.
- ^ “Merck Presents Early Evidence on Extended Delivery of Investigational Anti-HIV-1 Agent Islatravir (MK-8591) via Subdermal Implant”(Press release). July 23, 2019.
- ^ Jump up to:a b Michailidis, Eleftherios; Huber, Andrew D.; Ryan, Emily M.; Ong, Yee T.; Leslie, Maxwell D.; Matzek, Kayla B.; Singh, Kamalendra; Marchand, Bruno; Hagedorn, Ariel N.; Kirby, Karen A.; Rohan, Lisa C.; Kodama, Eiichi N.; Mitsuya, Hiroaki; Parniak, Michael A.; Sarafianos, Stefan G. (2014). “4′-Ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) Inhibits HIV-1 Reverse Transcriptase with Multiple Mechanisms”. Journal of Biological Chemistry. 289 (35): 24533–48. doi:10.1074/jbc.M114.562694. PMC 4148878. PMID 24970894.
- ^ Grobler, Jay (February 22–25, 2016). Long-Acting Oral and Parenteral Dosing of MK-8591 for HIV Treatment or Prophylaxis. Boston, Massachusetts. Conference on Retroviruses and Opportunistic Infections. 98.
- ^ Stoddart, Cheryl A.; Galkina, Sofiya A.; Joshi, Pheroze; Kosikova, Galina; Moreno, Mary E.; Rivera, Jose M.; Sloan, Barbara; Reeve, Aaron B.; Sarafianos, Stefan G.; Murphey-Corb, Michael; Parniak, Michael A. (2015). “Oral Administration of the Nucleoside EFdA (4′-Ethynyl-2-Fluoro-2′-Deoxyadenosine) Provides Rapid Suppression of HIV Viremia in Humanized Mice and Favorable Pharmacokinetic Properties in Mice and the Rhesus Macaque”. Antimicrobial Agents and Chemotherapy. 59 (7): 4190–8. doi:10.1128/AAC.05036-14. PMC 4468726. PMID 25941222.
- ^ Bruno Marchand. “The Crystal Structure of EFdA‐Resistant HIV‐1 Reverse Transcriptase Reveals Structural Changes in the Polymerase Active Site” (PDF).
 | |
| Names | |
|---|---|
| IUPAC name
2′-Deoxy-4′-ethynyl-2-fluoroadenosine
| |
| Other names
EFdA; MK-8591
| |
| Identifiers | |
|
3D model (JSmol)
| |
| ChemSpider | |
|
PubChem CID
| |
| UNII | |
| Properties | |
| C12H12FN5O3 | |
| Molar mass | 293.258 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
| |
////////////////Islatravir, MK-8591, EFdA, PHASE 2, HIV-1 , HIV-2, FDA 2026, APPROVALS 2026, Idvynso
C#CC1(C(CC(O1)N2C=NC3=C(N=C(N=C32)F)N)O)CO
LNS-8801 (SRR G-1)
LNS-8801 (SRR G-1)
CAS 925419-53-0
Chemokine receptor-like 2 agonist
C21 H18 Br N O3, 412.28
Ethanone, 1-[(3aS,4R,9bR)-4-(6-bromo-1,3-benzodioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]-
1-((3aS,4R,9bR)-4-(6-bromobenzo[d][1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl)ethan-1-one
- 1-[(3aS,4R,9bR)-4-(6-Bromo-1,3-benzodioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]ethanone
- G 1
- G 1 (estrogen receptor agonist)
Linnaeus Therapeutics Inc
NOTE 1-((3aR,4S,9bR)-4-(6-bromobenzo[d][1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl)ethan-1-one (RSS G-1 or LNS8812)
Novel crystalline forms of an enantiomerically purified LNS-8801 (SRR G-1), or a derivative useful for treating cancer, hearing disorder, depression and myocardial infarction.
Linnaeus Therapeutics is developing LNS-8801, a G-protein coupled estrogen receptor (GPER) agonist, an oral capsule formulation for the treatment of cancer including melanoma, pancreatic ductal adenocarcinoma, non-small cell lung cancer, solid tumor, hematological cancer, advanced cancer and colon carcinoma.
In October 2019, a phase I trial was initiated in patients with locally advanced or metastatic cancer.
PATENT
WO-2020023391
Novel crystalline and amorphous forms of 1-((3aS,4R,9bR)-4-(6-bromobenzo[d][1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl)ethan-1-one (here referred to as SRR-G-1 or LNS-8801) and 1-((3aS,4R,9bR)-4-(6-bromobenzo[d][1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl)ethan-1-one (RSS G-1 or LNS8812) and its derivative (having chiral purity of >90%; substantially free of its opposite enantiomer; designated as Form A-C) and their co-crystal (eg benzenesulfonic acid) and compositions and combinations comprising them are claimed. Also claimed is their use for treating cancers (disorder expresses GPER), depression, myocardial infarction, osteoporosis, rheumatoid arthritis, insomnia, inflammatory bowel disease, Crohn’s disease, thymic atrophy and hearing disorder and further type-2 diabetes. Further claimed are methods for reducing the likelihood of pregnancy after intercourse, increasing skin pigmentation and skin protection and preventing cancer, reoccurrence of cancer and inhibiting the progression of cancer
Embodiments of the present invention relate to an enantiomerically purified agonist of the G-protein coupled estrogen receptor (GPER), pharmaceutical compositions comprising an enantiomerically purified SRR G-l, or a derivative thereof, and methods of treating disease states and conditions in subjects in need thereof, and methods of treating disease states and conditions mediated through GPER receptors.
[0003] Estrogens mediate multiple complex physiological responses throughout the body. The responses are in turn mediated through the binding of estrogen to receptors. The classical receptors bind steroids, such as estrogen, and are soluble cytoplasmic/nuclear proteins that function as transcription factors. These receptors are known as estrogen receptor alpha and beta (two closely related proteins) that mediate transcriptional activity. GPER is a 7-transmembrane G protein-coupled receptor that also binds to estrogen with high affinity (Kd~6 nM) and mediates rapid cellular responses including cyclic adenosine monophosphate signaling, calcium mobilization and phosphatidylinositol 3,4,5 trisphosphate production.
[0004] Diseases whose development, progression, and or response to therapy, may be influenced by endogenous, and/or pharmacologic activation of GPER signaling include cancer (including the prevention of cancer, prevention of the reoccurrence of cancer, and the inhibition of the progression of cancer; and particularly melanoma, pancreatic, lymphomas, uveal melanoma, non-small cell lung cancer, breast, reproductive and other hormone-dependent cancers, leukemia, colon cancer, prostate, bladder cancer), reproductive (genito-urological) including endometritis, prostatitis, polycystic ovarian syndrome, bladder control, hormone-related disorders, hearing disorders, cardiovascular conditions including hot flashes and profuse sweating, hypertension, stroke, obesity, diabetes, osteoporosis, hematologic diseases, vascular diseases or conditions such as venous thrombosis, atherosclerosis, among numerous others and disorders of the central and peripheral nervous system, including depression, insomnia, anxiety, neuropathy, multiple sclerosis, neurodegenerative disorders such as Parkinson’s disease and Alzheimer’s disease, as well as inflammatory bowel disease, Crohn’s disease, coeliac (celiac) disease and related disorders of the intestine.
Embodiments of the present invention encompass compounds comprising enantiomerically purified G-l and methods of use in the treatment of diseases. G-l is a racemic mixture of the enantiomers l-((3aS,4R,9bR)-4-(6-bromobenzo[if][l,3]dioxol-5-yl)-3a.4.5.9b-tetrahydro-3H-cyclopenta|c |quinolin-8-yl)ethan- 1 -one (henceforth referred to as “SRR G-l” or “LNS8801”) and l-((3aR,4S,9bS)-4-(6-bromobenzo[if][l,3]dioxol-5-yl)-3a.4.5.9b-tetrahydro-3H-cyclopenta|c |quinolin-8-yl)ethan- 1 -one (henceforth referred to as “RSS G-l” or“LNS8812”).
Enantiomerically purified G-l has been purified in favor of its l-((3aS,4R,9bR)-4-(6-bromobenzo[ri][l,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl)ethan-l-one enantiomer over the corresponding l-((3aR,4S,9bS)-4-(6-bromobenzo[ri][l,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl)ethan-l-one enantiomer. Unless specifically described, SRR Gl, or a derivative thereof includes, any physical form, including an amorphous form or any crystalline solid forms such as A, B, C or combinations thereof.
Experimental Section
Scheme 1
[0166] A synthesis of G-l is described in Org. Biomol Client., 2010,8, 2252-2259, which is hereby incorporated by reference, and depicted in Scheme 1. A catalytic amount of Sc(OTf)3 (0.492 g, 1.0 mmol) in anhydrous acetonitrile (2.0 cm3) was added to the mixture of 6-bromopiperonal (2.30 g, 10.0 mmol). / aminoacetophenone (1.30 g, 10.0 mmol) and cyclopentadiene (3.30 g, 50.0 mmol) in acetonitrile (25 cm3). The reaction mixture was stirred at ambient temperature (~23 °C) for 2.0 h. The volatiles were removed in vacuo. The residue was purified by preparative silica gel column chromatography using ethyl acetate-hexanes (10 : 90) to provide G-l (4.03 g, 98%, dr. = 94 : 6) as a white solid. The minor diastereomer was substantially removed by recrystallization to yield a racemic mixure of SRR G-l and RSS G-l.
Example 1: Isolation of the SRR G-l and RSS G-l Enantiomers
[0167] Starting with a highly purified sample of G-l, (±)l-(4-(6-bromobenzo[ri][l,3]dioxol-5-yl)-(3aS*,4R*,9bR*)-tetrahydro-3H-cyclopenta[c]quinolin-8-yl)ethan-l-one, (99.4% purity) purchased from Tocris Bioscience, the material was dissolved in 90: l0:0. l(v/v/v) methyl tert-butyl ether / ethanol / diethyl amine and subjected to preparative chromatography using a column packed with Chrialpak 1A resin. Elution was conducted with 90: l0:0. l(v/v/v) methyl tert-butyl ether / ethanol / diethyl amine and the fractions corresponding to each enantiomer were collected and concentered to a solid. The early eluting enantiomer was determined to be the SRR G-l enantiomer by single crystal x-ray structural analysis.
Example 2: SRR G-l Polymorph Screen
[0168] Starting with SRR G-l prepared according to Example 1, a polymorph screening study was conducted analyzing the solids isolated from slurry of the solid, or from fast and slow evaporation and cooling of solutions (Table 1). Two crystal forms were identified, an anhydrous form designated Form A and mono dichloromethane solvate designated Form B. On exposure to elevated temperature the Form B crystal form desolvates to form the Form C crystal form. Amorphous material was generated from purified SRR G-l by two different methods; quick evaporating a diethyl ether solution of SRR G-l or rotary evaporating from a solution of a dichloromethane solution of SRR G-l.
Table 1
PAPER
Anti-Cancer Agents in Medicinal Chemistry (2019), 19(6), 760-771.

/////////RSS G-1, LNS8812, phase I, Locally advanced, metastatic cancer, LNS-8801, SRR G-1, Linnaeus Therapeutics
CC(=O)c1cc4c(cc1)N[C@@H](c2cc3OCOc3cc2Br)[C@H]5CC=C[C@@H]45
CC-90010
CC-90010
C21 H21 N O4 S, 383.46
CAS 1706738-98-8
1(2H)-Isoquinolinone, 4-[2-(cyclopropylmethoxy)-5-(methylsulfonyl)phenyl]-2-methyl-
- 4-[2-(Cyclopropylmethoxy)-5-(methylsulfonyl)phenyl]-2-methyl-1(2H)-isoquinolinone
- 4-[2-(Cyclopropylmethoxy)-5-(methanesulfonyl)phenyl]-2-methylisoquinolin-1(2H)-one
- 4-[2-(Cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-1-one
Quanticel Pharmaceuticals Inc, Michael John BennettJuan Manuel BetancortAmogh BoloorStephen W. KaldorJeffrey Alan StaffordJames Marvin Veal
![]()
Celgene (now a wholly owned subsidiary of Bristol-Myers Squibb ) , following its acquisition of Quanticel , is developing CC-90010, an oral inhibitor of BET (bromodomain and extraterminal) proteins, for the potential treatment of solid tumors and non-Hodgkin’s lymphoma. In August 2019, a phase I trial for diffuse astrocytoma, grade III anaplastic astrocytoma and recurrent glioblastoma was planned
PATENT
WO2018075796 claiming solid composition comprising a bromodomain inhibitor, preferably 4-[2-(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-1-one in crystalline form A.
PATENT
WO2015058160 (compound 89, page 103).

Example 89: 4-[2-(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-l-one
Step 1 : 2-methyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)isoquinolin-l-one
[00344] A suspension of 4-bromo-2-methylisoquinolin-l-one (100 mg, 0.42 mmol), bis(pinacolato)diboron (214 mg, 0.84 mmol), Pd(dppf)Cl2 (31 mg, 0.04 mmol) and potassium acetate (104 mg, 1.05 mmol) in dioxane (2 mL) under nitrogen was warmed up to 90 °C for 135 minutes. It was then cooled down to room temperature and diluted with ethyl acetate (8 mL). The mixture was washed with aqueous saturated solution of NaHC03 (8 mL) and brine (8 mL). The organic phase was separated, dried over Na2S04, filtered and concentrated under reduced pressure. The residue was purifed by normal phase column chromatography (10-90% EtOAc/Hexanes) to give the title compound (44 mg, 37%). 1H NMR (CDC13, 400 MHz) δ 8.43 (d, J = 7.9 Hz, 1 H), 8.40 (dd, J = 8.2 Hz, 0.9 Hz, 1 H), 7.68 (s, 1 H), 7.65 (ddd, J = 8.2, 8.2, 1.1 Hz, 1 H), 7.46 (t, J = 7.5 Hz, 1 H), 3.63 (s, 3H), 1.38 (s, 12H). LCMS (M+H)+ 286. Step 2: 4-[2-(cyclopropylmethox -5-methylsulfonylphenyl]-2-methylisoquinolin-l-one
[00345] The title compound was prepared in a manner similar to Example 18, step 3, substituting 2-bromo-l-(cyclopropylmethoxy)-4-methylsulfonylbenzene for 4-bromo-2-methylisoquinolin-l(2H)-one and 2-methyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)isoquinolin-l-one for N-benzyl-2-methoxy-5-(tetramethyl-l,3,2-dioxaborolan-2-yl)benzamide. 1H NMR (DMSO-d6, 400 MHz) δ 0.09 (m, 2 H), 0.29 (m, 1H), 0.35 (m, 1H),
0.94 (m, 1H), 3.22 (s, 3H), 3.57 (s, 3H), 3.95 (m, 2H), 7.16 (d, J = 7.9 Hz, 1H), 7.37 (d, J =
8.8 Hz, 1H), 7.53 (m, 2H), 7.65 (t, J = 7.6 Hz, 1H), 7.81 (d, J = 2.4 Hz, 1H), 7.97 (dd, J = 8.8,
2.4 Hz, 1H), 8.30 (d, J = 8.1 Hz, 1H). LCMS (M+H)+ 384.
[00346] Alternatively, 4-[2-(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-l-one can be prepared as described below.
Step 1 : 2-methyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)isoquinolin-l-one
[00347] A mixture of 4-bromo-2-methylisoquinolin-l-one (8.0 g, 33.6 mmol),
bis(pinacolato)diboron (17.1 g, 67.2 mmol), KOAc (6.6 g, 67.2 mmol), Pd2(dba)3 (3.1 g, 3.36 mmol) and X-Phos (1.6 g, 3.36 mmol) in anhydrous dioxane (200 mL) was stirred at 60 °C for 12 h. The reaction mixture was concentrated and the residue was purified by column chromatography on silica gel (PE : EA = 15 : 1) to give the title compound (6.0 g, 62 %) as a solid.
Step 2: 4-[2-(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-l-one
[00348] The title compound from Step 1 (5.0 g, 17.5 mmol), 2-bromo-l-(cyclopropylmethoxy)-4-methylsulfonylbenzene (6.4 g, 21 mmol), K3PO4 (9.3 g, 43.9 mmol) and Pd(dppf)Cl2 (1.4 g, 1.75 mmol) in a dioxane/water (100 mL / 10 mL) mixture were stirred at 60 °C for 12 hrs. The reaction mixture was concentrated under reduced pressure and the residue was purified by column chromatography on silica gel (EA : DCM = 1 : 4).
Appropriate fractions were combined and concentrated under reduce pressure. The resultant solid was recrystallized from DCM / MTBE (1 : 1, 50 mL) to give the title compound (4.0 g, 60 %) as a white solid. 1H NMR: (CDC13, 400 MHz) δ 8.51 (dd, Ji = 8.0 Hz, J2 = 0.8 Hz, 1 H), 7.98 (dd, Ji = 8.4 Hz, J2 = 2.4 Hz, 1 H), 7.86 (d, J = 2.4 Hz, 1 H), 7.53 (m, 2 H), 7.16 (d, J = 7.6 Hz, 1 H), 7.10 (m, 2 H), 3.88 (m, 2 H), 3.66 (s, 3 H), 3.09 (s, 3 H), 1.02-0.98 (m, 1 H), 0.44-0.38 (m, 2 H), 0.11-0.09 (m, 2 H). LCMS: 384.1 (M+H)+
Patent
WO-2020023438
A process for preparing bromodomain inhibitor, particularly 4-[2(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-1-one (having HPLC purity of 99%; compound 1; (hereafter referred to as C-90010)) and its hydrates, solvates, prodrugs and salts comprising the reaction of a substituted 4-(methylsulfonyl)phenol compound with a quinoline derivative, followed by purification is claimed. Also claimed are novel intermediates of CC-90010 and their processes for preparation. Further claimed are novel crystalline form of CC-90010. CC-90010 is known and disclosed to be a bromodomain containing protein inhibitor, useful for treating cancer.
Scheme 10: Synthesis of Compound 1
[0090] Acetonitrile (1.6L) was charged to a mixture of Compound 2 (156.7g, 460 mmol), Compound 3 (lOOg, 420 mmol) and potassium phosphate tribasic (223g, l.OSmol). Agitation
was begun and water (400mL) charged to the batch. The system was vacuum purged three times with nitrogen and charged with Pd(PPh3)2Cl2 (2.9g, 4 mmol) and the system vacuum purged three times with nitrogen. The batch was heated to about 65 to about 75 °C (or any temperature in between and including these two values) and contents stirred for at least about 16 hours until reaction was complete by HPLC analysis. The batch was cooled to about 60 to about 70 °C (or any temperature in between and including these two values), agitation halted and the mixture allowed to settle. The bottom aqueous layer was removed. Water (150mL) and acetonitrile (700mL) were charged at about 60 to about 70°C (or any temperature in between and including these two values). Ecosorb C-941 (15g) and Celite (lOg) were charged to the reaction vessel at about 60 to about 70°C (or any temperature in between and including these two values). After lh, the mixture was filtered to remove solids. The solids were washed twice each with 18% water in acetonitrile (500 mL) at about 60 to about 70°C (or any temperature in between and including these two values). The filtrates were combined and concentrated under atmospheric pressure to a final volume of 1.5L. The batch was cooled to about 60 to about 65°C (or any temperature in between and including these two values) and seeded with Compound 1 (1 g). After lh, water (500 mL) was charged over at least 1 hour at about 60 to about 65°C (or any temperature in between and including these two values). The slurry was cooled to about 15 to about 25°C (or any temperature in between and including these two values) over 4 hours. The product was collected by suction filtration. The wet cake was washed with 45% water in acetonitrile (500mL) twice. The product was dried under vacuum at about 40°C with nitrogen purge. Yield: 139g of 1.
[0091] The above procedure for coupling Compound 3 and Compound 2 to produce
Compound 1 may be modified in any of the ways that follow. Reaction solvents: Different reaction solvents from acetonitrile can be used, including tetrahydrofuran, 2-methyl tetrahydrofuran, toluene, and isopropanol. Boronic ester: Different boronic esters from Compound 2 can be used, including pinacolato ester compound 7, and the free boronic acid of Compound 2. Examples of boronic esters can be found in Lennox et al., Chem. Soc. Rev., 43: 412 (2014). Carbon treatment: Different carbon treatments from Ecosorb C-941 could be used. Different amounts of carbon, from 0.01 to 0.5X weight can be used. The carbon can be eliminated. Different amounts of Celite, from 0.01 to 0.5X weight can be used.
Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used.
The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying. Catalysts: Different metal and ligand combination could be used. Examples of metal/ligand combinations can be found in Maluenda, Irene; Navarro, Oscar, Molecules, 2015, 20, 7528. Various catalysts can be including: XPhos-3G (cas# 1445085-55-1); cataCXium® A Pd 3G (CAS# 1651823-59-4); PdCk(DtBPF) (CAS# 95408-45-0); SPhos 3G (Cas# 1445085-82-4); AmPhos 3G (Cas# 1820817-64-8); PCy3 3G (Cas# 1445086-12-3); Pd PEPPSI IPent Cas#l 158652-41-5);
Pd(PPh3)2Cb (Cas# 13965-03-2). Examples of catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
VI. Purification of Compound 1 fCC-900101 bv crystallization from formic acid and water
[0092] Described herein are methods of purifying Compound 1 by crystallization from formic acid and water. Also described are methods for obtaining three different polymorphs of Compound 1, including the most stable form, Form 1 and two metastable forms, Form 4
The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying. Catalysts: Different metal and ligand combination could be used. Examples of metal/ligand combinations can be found in Maluenda, Irene; Navarro, Oscar, Molecules, 2015, 20, 7528. Various catalysts can be including: XPhos-3G (cas# 1445085-55-1); cataCXium® A Pd 3G (CAS# 1651823-59-4); PdCh(DtBPF) (CAS# 95408-45-0); SPhos 3G (Cas# 1445085-82-4); AmPhos 3G (Cas# 1820817-64-8); PCy3 3G (Cas# 1445086-12-3); Pd PEPPSI IPent Cas#l 158652-41-5);
Pd(PPh3)2Cl2 (Cas# 13965-03-2). Examples of catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
VI. Purification of Compound 1 (CC-90010! bv crystallization from formic acid and water
[0092] Described herein are methods of purifying Compound 1 by crystallization from formic acid and water. Also described are methods for obtaining three different polymorphs of Compound 1, including the most stable form, Form 1 and two metastable forms, Form 4
33 -a
and Form 5. Supporting data (XRPD, DSC, photomicroscopy) for all three forms is provided in the examples below.
[0093] The stmcture of Compound 1 (CC-90010) is shown below:
Example 1: Synthesis of Compound 1
[0217] Synthesis of compound 1 was accomplished according to Scheme 1 below. Referring to Scheme 1, synthesis commenced with bromination of starting material 4-(methylsulfonyl)phenol 4, to produce compound 5. Compound 5 was O-alkylated with (bromomethyl)cyclopropane to produce compound 6. Boronate Compound 2 was then formed by borylation of Compound 6 with Pd catalyst and bis(pinacolato)diboron to produce transient Compound 7, which was subsequenctly treated with diethanolamine (DBA) to afford cross-coupling partner Compound 2. Cross-coupling partner Compound 3 was formed in one pot starting from commercially available Compound 8. Compound 8 was N-methylated and brominated to afford Compound 3. Compounds 2 and 3 were cross-coupled (Norio, M. and Suzuki, A., Chem. Rev., 95(7), 2457-2483 (1995)) to afford the target compound 1.
Scheme 1: Synthesis of compound 1
1.1: Bromination of 4
[0218] The bromination of Compound 4 to produce Compound 5 itself is simple, however stopping at the mono-brominated Compound 5 was challenging. The bis-brominated Compound 5-a (see Scheme 2 below) is a particularly pernicious impurity as it couples downstream to form a di ffi cult-to-purge impurity.
Scheme 2: Bromination of Compound 4
[0219] The key to high purity with reasonable yield was to exploit the solubility differences of the starting material Compound 4 (46 mg/ml at 20 °C) and the product Compound 5 (8 mg/ml) in CH2CI2. These solubility differences are summarized in Table 3 below.
[0220] This solubility difference is exploited by performing the reaction at a high
concentration to drive Compound 5 out of solution once formed, thereby minimizing its ability to react further with the brominating reagent to form Compound 5-a diBr. The reaction is seeded with Compound 5 to initiate its crystallization.
[0221] In Fig. 22 (Conversion of Compound 4 to Compound 5: Effect of Sulfuric Acid) it can be seen that in the absence of acid the initial reaction to Compound 5 is rapid, however the conversion plateaus at about 30% Compound 5. The main side product was found to be the impurity Compound 5-a diBr (see Fig. 23: Conversion of Compound 5 and Compound 5-a diBr: No H2SO4). Addition of increasing amounts of sulfuric acid leads to a higher conversion to desired Compound 5.
[0222] Fig. 24 (Compound 4 to Compound 5 Reaction Profile: Portion-wise Addition of NBS, Seeding) depicts further reaction control. The portion-wise addition ofNBS after addition of catalytic sulfuric acid minimizes the temperature rise, and the addition of Compound 5 after an initial NBS charge promotes the reactive crystallization of Compound 5. After about 6 to 7 hours of reaction it can be seen that the major product is Compound 5, with only a small (<5%) of the di-brominated impurity formed. In contrast, in a reaction where Compound 4 and all of the NBS were charged followed by the addition of 4 volumes of methylene chloride, a rapid exotherm resulted and undesired Compound 5-a diBr was found to be the major product.
[0223] Thus, the reaction was run under a high concentration in CH2CI2 with a portion-wise solid addition of NBS (to control both availability of the electrophile and the exotherm). An end of reaction slurry sample typically showed not more than 5% of the starting material Compound 4 remaining. After filtration the crude cake was washed with cold CH2CI2 and the OkCk-washed filter cake contained not more than 0.5% by weight dibrominated Compound 5-a. It also contained a large amount of HPLC-silent succinimide.
[0224] The following procedure was carried out: Compound 4 (25g, 145mmol) followed by CH2CI2 (lOOmL) were added to a reaction vessel and agitated. The batch was adjusted to 17 °C to 23 °C. Sulfuric acid was charged (2.7mL, Slmmol) to the batch maintaining 17 °C to 23°C. The batch was stirred at 17 °C to 23 °C for 10 minutes to 20 minutes. The first portion of A-bromosuccimide (NBS) was charged (6.5g, 36.5 mmol) to the batch at 17 °C to 23°C and stirred for at least 30 min. The second portion of NBS was charged (6.5g, 36.5 mmol) to the batch at 17 °C to 23°C and stirred for at least 30 min. The batch was seeded with
Compound 5 (0.02wt) and stirred for ca. 30 min at 17 °C to 23 °C to induce crystallization.
[0225] The third portion of NBS was charged (6.5g, 36.5 mmol) to the batch at 17 °C to 23 °C and stirred for at least 30 min. NBS (6.5g, 36.5 mmol) was charged to the batch at 17 °C to 23 °C and stirred for at least 30 min. Additional CH2CI2 was charged (50mL) to the batch while maintaining 17 °C to 23 °C to aid in agitation and transfer for filtration. The batch was stirred at 17 °C to 23 °C until complete by HPLC analysis (~20 – 40 h). The product was collected by suction filtration. The filter cake was slurry washed with CH2CI2 (3 x 50mL) at 17 °C to 23 °C (target 20 °C). The filter cake was slurry washed with purified water (3.0vol) at 65 °C to 75 °C for 2 to 3 hours. Then, the filter cake was slurry washed with purified water (3 x 1.0 vol, 3 x 1.0 wt) at 17 °C to 23°C. The wet cake was dried under vacuum with nitrogen bleed at 60 °C. Yield: 27g 5 (74% molar) >97% by weight. ¾ NMR (500 MHz, de-DMSO) 8.01 (1H, d, 4J = 2.1 Hz, RO-Ar meta- H ), 7.76 (1H, dd, J = 8.6 and 4J = 2.1 Hz, RO-Ar meta-H ), 7.14 (1H, d, J = 8.6 Hz, RO-Ar ortho- H), 3.38 (1H, br s, OH), 3.20 (3H, s,
CHJ); MS (ES-) calc. 249/251; found 249/251. Melting point (MP): (DSC) 188 °C.
[0226] The above procedure allowed for the following modifications. Solvents: Alternative solvents could be used. Examples include chlorinated solvents, such as chloroform or 1,2 dichloroethane, and non-chlorinated solvents such as acetonitrile, tetrahydrofuran, or 2- methyltetrahydrofuran. Reaction concentration: The reaction concentration can be varied from about 2X vol to about 20 X vol (with respect to Compound 4). Brominating agents: Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination reagent stoichiometry: Different amounts of the brominating reagent can be used, from about 0.8 equiv to about 1.9 equiv. Bromination reagent addition: The brominating reagent can be added all at once, portion wise in about 2 to about 20 portions, or continuously. The addition times can vary from about 0 to about 72 hours. Temperature: Reaction temperatures from about 0 °C to about 40 °C could be used. Acids: Different acids can be envisioned, including benzenesulfonic acid, para-toluenesulfonic acid, triflic acid, hydrobromic acid, and trifluoroacetic acid. Isolation: Instead of directly filtering the product and washing with methylene chloride and water, at the end of reaction an organic solvent capable of dissolving Compound 5 could be charged, followed by an aqueous workup to remove succinimide, and addition of an antisolvent or solvent exchange to an appropriate solvent to crystallize Compound 4. Drying: A temperature range of about 10 to about 60 °C could be used for drying.
[0227] An alternative process to Compound 5 has also been developed. This process is advantageous in that it does not use a chlorinated solvent, and provides additional controls over the formation of the Compound 5-a dibromo impurity. See Oberhauser, T. J Org. Chem 1997, 62, 4504-4506. The process is as follows. Compound 4 (10 g, 58 mmol) and acetonitrile (100 ml) were charged to the reactor and agitated. The batch was cooled to -20 °C. Triflic acid (CF3SO3H or TfOH, 5.5 mL, 62 mmol) was charged while maintaining a batch temperature of -10 to -25 °C. N-bromosuccinimide was charged (NBS, 11.4 g, 64 mmol), stirred at -10 to -25 °C for 30 minutes, then warmed to 20 °C over 3 to 4 hours. Agitation was continued at 15 °C to 25 °C until reaction completion. If the reaction conversion plateaued before completion, the reaction was cooled to -5 to -15 °C, and additional NBS was added, the amount based off of unreacted starting material, followed by warming to 15 °C to 25 °C and reacting until complete.
[0228] After reaction completion, the batch was warmed to 40 °C to 50 °C and concentrated under reduced pressure to 40 mL. The batch was cooled to -5 °C to -15 °C and the resulting product solids were filtered off. The solids were slurry washed three times, each with 20 mL water, for at least 15 minutes. The final cake was dried at 50 °C to 60 °C under reduced pressure to furnish 10 g of 5 containing less than 0.1% MeCN, 0.07% water, and 0.1% triflic acid (TfOH) by weight.
[0229] Alternatives to the above procedure employing MeCN and TfOH are as follows. Brominating agents: Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination Reagent Stoichiometry: Different amounts of the brominating reagent can be used, from about 0.8 equiv to about 2 equiv. Drying: A temperature range of about 10 °C to about 60 °C could be used for drying.
[0230] The impurity 5-a is was prepared and characterized as follows. 10 g of Compound 4 and sulfuric acid (35 mol%) were dissolved in MeOH (10 vol). The mixture was set to stir at 20 °C to 25 °C for 5-10 min and 2.0 equivalents of NBS were charged in one portion. The resulting yellow mixture was stirred for three days at 20-25 °C. The batch was concentrated under reduced pressure and the resulting solid was slurried in water at 95-100 °C for 3 hours. After a second overnight slurry in CH2CI2 at room temperature, the batch was filtered and dried to give a white solid 5-a (15.0 g, 78%). ¾ NMR (500 MHz, de-DMSO), 8.05 (2H, s, ArH), 3.40 (1H, br s, HO-Ar), 3.28 (3H, s, CH3); MS (ES‘) calc. 327/329/331; found
327/329/331; MP (DSC): 226 °C (onset 221 °C, 102 J/g); lit. 224-226 °C.
1.2: O-alkylation of 5 to produce 6
[0231] Compound 6 was prepared according to Scheme 7 below.
Scheme 7: O-alkylation of 5 to produce 6
[0232] Compound 5 (100 g, 398 mmol) and methyl ethyl ketone (MEK, 700 mL) were charged to the reaction vessel and agitated. Potassium carbonate (K2CO3, 325 mesh 82.56 g, 597 mmol) was then charged to the stirred reaction vessel at 15 °C to 25 °C.
Bromomethylcyclopropane (64.4 mL, 664 mmol) was charged to the reaction vessel over at least 1 hour, maintaining the temperature between 15 °C to 25 °C. MEK (200 mL) was added into the reactor and the reactor heated to 65 to 75 °C. The contents of the reaction vessel were stirred at 65 to 75°C for approximately 10 hours until reaction was complete by HPLC analysis. Water (3.0 vol, 3.0wt) was charged to the vessel maintaining the temperature at 65 to 75 °C. The batch was stirred at 65 to 75 °C. The phases were allowed to separate at 65°C to 75 °C and the lower aqueous phase was removed. Water (300 mL) was charged to the vessel maintaining the temperature at 65 °C to 75 °C. The batch was agitated for at least 10 minutes at 65 to 75 °C. The phases were allowed to separate at 65 °C to 75 °C and the lower aqueous phase was removed. The water wash was repeated once. The temperature was adjusted to 40 to 50°C. The mixture was concentrated to car. 500 mL under reduced pressure. The mixture was distilled under reduced pressure at up to 50 °C with MEK until the water content was <1.0% w/w. n-heptane (500mL) was charged to the vessel maintaining the temperature at 40 to 50 °C. The mixture was continuously distilled under vacuum with n-heptane (300mL), maintaining a 1L volume in the reaction vessel. Compound 6 seeds (0.0 lwt) were added at 40 to 50 °C. The mixture was continuously distilled under reduced pressure at up to 50 °C with n-heptane (300mL) while maintaining 1L volume in the reactor. The batch was cooled to 15 to 25 °C and aged for 2 hours. The product was collected by suction filtration. The filter cake was washed with a solution of 10% MEK in n-heptane (5vol) at 15 to 25°C. The filter cake was dried under reduced pressure at up to 40 °C under vacuum with nitrogen flow to afford 95g of 6. 1H NMR (500 MHz, de-DMSO) 8.07 (1H, d, 4J = 2.2 Hz, ArH), 7.86 (1H, d, J = 8.7 Hz, meta-ArH), 7.29 (1H, d, J = 8.8 Hz, ortho-AiK),
4.04 (2H, d, J = 6.9 Hz, OCH2CH), 3.21 (3H, s, CH3), 1.31-1.24 (1H, m, OCH), 0.62- 0.58 (2H, m, 2 x CHCHaHb), 0.40-0.37 (2H, m, 2 x CHC¾Hb); MS (ES+) calc. 305/307; found 305/307; MP: (DSC) 93 °C.
[0233] The following modifications of the above reaction, synthesis of 6 from 5, may be employed as well. Solvent: Different solvents could be used, for example acetone, methyl isobutyl ketone, ethyl acetate, isopropyl acetate, acetonitrile, or 2-methyl tetrahydrofuran. Reaction volume: Reaction volumes of 3 to 30 volumes with respect to 3 could be used. Base: Different inorganic bases, such as cesium carbonate or phosphate bases (sodium, potassium, or cesium) could be used. Also, organic bases, such as trimethylamine or diisopropyldiimide could be used. Base particle size: Different particle sizes of potassium carbonate from 325 mesh could be used. Reaction temperature: A lower temperature, such
as 50 °C could be used. A higher temperature, such as about 100 °C could be used. Any temperature above the boiling point of the solvent could be run in a pressure vessel.
Isolation: Different solvent ratios of MEK to n-heptane could be used. Different amounts of residual water can be left. Different amounts of seeds, from 0 to 50% could be used.
Seeding could take place later in the process and/or at a lower temperature. An un-seeded crystallization can be employed. A different isolation temperature, from 0 °C to 50 °C could be used. A different wash could be used, for example a different ratio of MEK to n-heptane. A different antisolvent from n-heptane could be used, such as hexane, pentane, or methyl tert-butyl ether. Alternatively, the batch could be solvent exchanged into a solvent where Compound 3 has a solubility of less than 100 mg/ml and isolated from this system. Drying: A temperature range of 10 to 60 °C could be used for drying.
[0234] Compound 10, shown below may also be formed as a result of O-alkylation of unreacted 4 present in product 5, or alternatively from or via a palladium mediated proteodesbromination or proteodesborylation in subsequent chemistry discussed in Example 1.3 below.
[0235] Preparation of methylsulfonylphenyl(cyclopropylmethyl) ether 10: Compound 4 (0.86 g, 5.0 mmol) and K2CO3 (1.04 g, 7.5 mmol) were slurried in acetone (17 mL, 20 vols). Cyclopropylmethyl bromide (0.73 mL, 7.5 mmol) was added in several small portions over ~1 minute and the reaction mixture heated to 50 °C for 48 hours, then cooled to 25 °C. Water (5.0 mL) was added with stirring and the acetone was evaporated on a rotary evaporator from which a fine white solid formed which was filtered off and returned to a vessel as a damp paste. A 1 : 1 mixture of MeOH/ water (8 mL) was added and heated to 40 °C with stirring. After 1 hour, the white solid was filtered off. Some residual solid was washed out with fresh water that was also rinsed through the cake, which was then isolated and left to air dry over the two days to give a dense white solid 10 (1.00 g, 88%). ¾ NMR (500 MHz, CDCb) 7.85
(2H, d, J = 8.8 Hz, RO-Ar ortho-H), 7.00 (2H, d, J = 8.8 Hz, RO-Ar meta- H), 3.87 (2H, d, J = 7.0 Hz, OCH2CH), 3.02 (3H, s, CHs), 1.34-1.23 (1H, m, OCH2CH), 0.72-0.60 (2H, m, 2 x CHCHflHb), 0.42-0.31 (2H, m, 2 x CHCH^.
1.3: Synthesis and Isolation Coupling Partner Boronic Ester 2
[0236] The final bond forming step to Compound 1 is a Suzuki-Miyaura coupling between Compounds 2 and 3, as shown in Scheme 3 below (Norio, M. and Suzuki, A., Chem. Rev., 95(7), 2457-2483 (1995)). Early studies demonstrated that the boronic ester of the isoquinolinone Compound 3-a had poor physical attributes and solid phase stability (Kaila, N. et al., J. Med Chem., 57: 1299-1322 (2014)). The pinacolatoboronate of the O-alkyl phenol, Compound 7, had acceptable solid phase stability and could be isolated via crystallization.
Scheme 3: Suzuki-Miyaura coupling between 2 and 3
[0237] Process robustness studies for the isolation of Compound 7, however, indicated that Compound 7 has poor solution stability, decomposing primarily to the proteodeborylated compound 10, as shown in Scheme 4 below. This was particularly problematic as the isolation process involved a solvent exchange from 2-MeTHF (2-methyl tetrahydrofuran) to iPrOAc (isopropyl acetate), which is not a fast unit operation on scale.
Scheme 4: Modification of 7
[0238] A search for a more stable boronic ester was undertaken. Early attempts targeted making N-methyliminodiacetic acid (MID A) boronate Compound 2-a (E. Gilis and M. Burke,“Multi step Synthesis of Complex Boronic Acids from Simple MIDA Boronates,” J Am. Chem. Soc., 750(43): 14084-14085 (2008)), however, all attempts resulted in product decomposition. Applicant then turned to a relatively obscure boronate formed by the addition of diethanolamine to Compound 7 (Bonin et al., Tetrahedron Lett., 52: 1132-1135 (2011)). Addition of diethanolamine to a solution of Compound 7 led to rapid ester formation and concomitant crystallization of Compound 2.
[0239] The discovery of boronic ester Compound 2 allowed for a simple, fast, high-yielding, high-purity process comprising the following procedure. Tetrahydrofuran (THF, 1500mL) was charged to a flask containing Compound 6 (100g, 328 mmol), bis(pinacolato)diboron (90.7g, 357 mmol) and cesium acetate (CsOAc, 158g, 822 mmol). The system was vacuum purged three times with nitrogen. Pd(PPh3)2Cl2 (13.8g, 20 mmol) was charged to the reaction and the system was vacuum purged three times with nitrogen. The reaction was then heated to 55 to 65°C.
[0240] The batch was stirred for approximately 8 hours until reaction was complete by HPLC analysis. The batch was cooled to 15 to 25 °C (target 20 °C ) and charged with silica gel (20g) and Ecosorb C-941 (20g). After lh, the mixture was filtered to remove solid. The residual solids were washed twice, each with THF (300mL). The filtrate and washes were combined. In a separate vessel, diethanolamine (34.5mL, 360 mmol) was dissolved in THF (250 mL). The diethanolamine solution in THF (25mL) was then charged to the batch. After 10 minutes, the batch was seeded with 2 (1 g) and aged for 1 to 2 hours. The remaining of the diethanolamine solution in THF was charged to the batch over at least 2 hours and the slurry was stirred for at least 2 hours. The product 2 was collected by suction filtration. The wet cake was washed thrice with THF (200mL). The material was dried under vacuum at 40 °C with nitrogen purge yielding 94.6g of 2.
[0241] The reaction to synthesize Compound 2 from Compound 6 described above may be modified as follows. Solvent: Different solvents from THF could be used, such as 1,4 dioxane or 2-methyltetrahydrofuran. Reaction volume: The reaction volume can be varied from 4 to 50 volumes with respect to compound 2. Catalyst and base: Different palladium catalyst and bases can be used for the borylation. Examples can be found in Chow et al., RSC Adv., 3 : 12518-12539 (2013). Borylation reaction temperature: Reaction temperatures from room temperature (20 °C) to solvent reflux can be used. Carbon/ Silica treatment:
The treatment can be performed without silica gel. The process can be performed without a carbon treatment. Different carbon sources from Ecosorb C-941 can be used. Different amounts of silica, from 0.01X to IX weight equivalents, can be used. Different amounts of Ecosorb C-941, from 0.01X to IX weight equivalents, can be used. Crystallization: A different addition rate of diethanolamine can be used. Different amounts of diethanolamine, from 1.0 to 3.0 molar equivalents can be used. A different cake wash with more or less THF can be used. Different amount of seeds from 0.0001X wt to 50X wt can be used.
Alternatively, the process can be unseeded. Drying: A temperature range of 10 °C to 60 °C could be used for drying.
[0242] The subsequent Suzuki-Miyaura coupling between Compounds 2 and 3 also proceeded well, providing over 20 kg of crude compound 1 with an average molar yield of 80% and LCAP of 99.7%.
1.4: Synthesis of Coupling Partner 3
[0243] Cross-coupling partner 3 was prepared by two different processes corresponding to Schemes 8 and 9 shown below.
Scheme 8: Process A for preparation of 3
[0244] According to Process A, Compound 9 (100g, 628 mmol) was dissolved in acetonitrile (450 mL) at room temperature. In a separate vessel, N-bromosuccinimide (NBS, 112g, 628 mmol) was suspended in acetonitrile (1 L). Compound 9 in acetonitrile was charged to the NBS slurry over at least 45 minutes. The contents of the reaction vessel were warmed to 45 °C to 55 °C and the batch stirred until the reaction was complete by HPLC analysis. The batch was cooled to 35 °C to 45 °C and ensured dissolution. Norit SX plus carbon (lOg) was charged to the mixture and the reaction mixture adjusted to 55 °C to 60 °C. The mixture was stirred at 55 °C to 60 °C for about lh and the mixture filtered at 55 °C to 60 °C to remove solids. The solids were washed with acetonitrile (500mL) at 55 °C to 60 °C. The volume of the combined filtrate was reduced to 900 mL by distilling off acetonitrile under reduced pressure. The batch with Compound 3 (lg) and stirred at 35 °C to 45 °C for at least 60 minutes. The contents of the reaction vessel were cooled to 15 °C to 25 °C over at least 1 hour. Water (2000 mL) was charged to the reaction vessel over at least 90 minutes and the slurry aged for at least 60 minutes. The product was collected by suction filtration. The cake was washed with a premixed 5% solution of acetonitrile in water (300mL). The wet cake was dried under vacuum at 40 °C with nitrogen purge. Yield: 120g of 3.
[0245] The above procedure, Process A for this synthesis of 3, may be practiced with alternative reagents and conditions as follows. Solvents: Alternative solvents could be used. Examples include chlorinated solvents, such as methylene chloride, chloroform or 1,2 dichloroethane, and non-chlorinated solvents such as tetrahydrofuran, or 2-methyltetrahydrofuran. Reaction concentration: The reaction concentration can be varied from 2X vol to 40 X vol (with respect to Compound 9). Brominating agents: Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination reagent Stoichiometry: Different amounts of the brominating reagent can be used, from 0.8 equiv to 2 equiv. Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used. The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 °C to 60 °C could be used for drying.
Scheme 9: Process B for preparation of 3
[0246] According to Process B, Compound 3 can be formed starting from 8 via non-isolated compound 9 as follows. Compound 8 (80 g, 55 mmol), cesium carbonate (CS2CO3, 215 g, 66 mmol), and acetonitrile (800 mL) were charged to the reactor. The temperature was adjusted from 15 to 25 °C and iodomethane charged to the reactor (Mel, 86 g, 0.61 mol) while maintaining a batch temperature below 25 °C. The batch was heated to 40 °C and agitated for 10 hours to form Compound 9. The batch was cooled to 25 °C, filtered into a fresh reactor to remove solids, and the solids washed twice with acetonitrile. The combined organic layers were concentrated via atmospheric distillation to about 320 mL.
[0247] In a separate reactor N-bromosuccinimide (NBS, 98.1 g, 0.55 mol) was charged to acetonitrile (800 mL) and agitated. The batch containing Compound 9 was transferred to the NBS solution while maintaining a batch temperature of 15 to 25 °C. The batch was heated to 45 to 55 °C and agitated for at least 4 hours to allow for reaction completion to Compound 3. Upon reaction completion, Norit SX Plus activated carbon (8 g) was charged, and agitated at 45 to 55 °C for one hour. The batch was filtered into a fresh vessel, the Norit SX plus cake was washed with 400 ml of 45 to 55 °C acetonitrile. The acetonitrile layers were combined, cooled to 35 to 45 °C, and distilled under reduced pressure to 720 mL. The batch was adjusted to a temperature of 40 °C, charged with Compound 3 seeds (0.8 g), agitated for one hour, cooled to 15 to 25 °C over at least on hour, then charged with water (1600 mL) over at least two hours. The mixture was agitated for an additional one to two hours, filtered, the cake washed with a premixed 5% solution of acetonitrile in water (240 mL). The wet cake was dried under vacuum at 40°C with nitrogen purge. Yield: 52 g of 3.
[0248] Process B to synthesize Compound 3, described above, may be modified as follows. Solvents: Alternative solvents could be used. Examples include chlorinated solvents, such as methylene chloride, chloroform or 1,2 dichloroethane, and non-chlorinated solvents such as tetrahydrofuran, or 2-methyltetrahydrofuran. Reaction concentration: The reaction concentration can be varied from 2X vol to 40 X vol (with respect to Compound 8).
Alkylating reagent: Alternative methylating reagents to methyl iodide can be used such as dimethylsulfate. Alkylating reagent stoichiometry: 1 to 10 molar equivalents of methyl iodide may be used. Base: Different inorganic bases, such as potassium carbonate or phosphate bases (sodium, potassium, or cesium) could be used. Brominating agents:
Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination reagent stoichiometry: Different amounts of the brominating reagent can be used, from 0.8 equiv to 2 equiv. Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used. Seeding levels from 0.0001% to 50% can be used. The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying.
1.5: Cross-coupling of 2 and 3 to Produce Target Compound 1
[0249] 1 is synthesized by Suzuki cross-coupling of 3 and 2 according to Scheme 10 and as described below.
Scheme 10: Synthesis of 1
[0250] Acetonitrile (1.6L) was charged to a mixture of Compound 2 (156.7g, 460 mmol), Compovmd 3 (lOOg, 420 mmol) and potassium phosphate tribasic (223 g, l.OSmol). Agitation was begun and water (400mL) charged to the batch. The system was vacuum purged three times with nitrogen and charged with Pd(PPh3)2Cl2 (2.9g, 4 mmol) and the system vacuum
purged three times with nitrogen. The batch was heated to 65 to 75°C and contents stirred for at least 16 hours until reaction was complete by HPLC analysis. The batch was cooled to 60 to 70°C, agitation halted and the mixture allowed to settle. The bottom aqueous layer was removed. Water (150mL) and acetonitrile (700mL) were charged at 60 to 70°C. Ecosorb C-941 (15g) and Celite (lOg) were charged to the reaction vessel at 60 to 70°C. After lh, the mixture was filtered to remove solids. The solids were washed twice each with 18% water in acetonitrile (500 mL) at 60 to 70°C. The filtrates were combined and concentrated under atmospheric pressure to a final volume of 1.5L. The batch was cooled to 60 to 65°C and seeded with Compound 1 (1 g). After lh, water (500 mL) was charged over at least 1 hour at 60 to 65°C. The slurry was cooled to 15 to 25°C over 4 hours. The product was collected by suction filtration. The wet cake was washed with 45% water in acetonitrile (500mL) twice. The product was dried under vacuum at 40°C with nitrogen purge. Yield: 139g of 1.
[0251] The above procedure for coupling Compound 3 and Compound 2 to produce
Compound 1 may be modified in any of the ways that follow. Reaction solvents: Different reaction solvents from acetonitrile can be used, including tetrahydrofuran, 2-methyl tetrahydrofuran, toluene, and isopropanol. Boronic ester: Different boronic esters from Compound 2 can be used, including pinacolato ester compound 7, and the free boronic acid of Compound 2. Examples of boronic esters can be found in Lennox, Alister, J.J., Lloyd-Jones, Guy C. Chem. Soc. Rev., 2014, 43, 412. Carbon treatment: Different carbon treatments from Ecosorb C-941 could be used. Different amounts of carbon, from 0.01 to 0.5X weight can be used. The carbon can be eliminated. Different amounts of Celite, from 0.01 to 0.5X weight can be used. Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used. The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying. Catalysts: Different metal and ligand combination could be used. Examples of metal/ligand combinations can be found in Maluenda, Irene; Navarro, Oscar, Molecules, 2015, 20, 7528. Various catalysts can be including: XPhos-3G (cas# 1445085-55-1);
cataCXium® A Pd 3G (CAS# 1651823-59-4); PdCk(DtBPF) (CAS# 95408-45-0); SPhos 3G (Cas# 1445085-82-4); AmPhos 3G (Cas# 1820817-64-8); PCy3 3G (Cas# 1445086-12-3); Pd PEPPSI IPent Cas#l 158652-41-5); Pd(PPh3)2Cl2 (Cas# 13965-03-2). Examples of
catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
1.6: Crystallization of 1
[0252] The final isolation of Compound 1 requires a polish filtration. For this, the batch must be completely soluble. Unfortunately, Compound 1 has low solubility in almost all
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Class 3 and common Class 2 (e.g. THF, MeCN) solvents (ICH
Harmonized Guideline“Impurities: Guideline for Residual Solvents Q3C(R6)” October 20, 2016). A reasonable solubility was obtained in a warm MeCN-water mix, but this is not an optimal system (requires a heated filtration, MeCN has a residual solvent limit of only 410 ppm). Additional solvents with reasonable solubility (>50 mg/ml) include N-methyl-2- pyrrolidone (NMP) and dimethylacetamide (DMAc); but the development of isolations from these solvents required large volumes and raised residual solvent limit concerns (530 ppm or less for NMT and 1090 ppm or less for DMAc).
catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
1.6: Crystallization of 1
[0252] The final isolation of Compoxmd 1 requires a polish filtration. For this, the batch must be completely soluble. Unfortunately, Compound 1 has low solubility in almost all
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Class 3 and common Class 2 (e.g. THF, MeCN) solvents (ICH
Harmonized Guideline“Impurities: Guideline for Residual Solvents Q3C(R6)” October 20, 2016). A reasonable solubility was obtained in a warm MeCN-water mix, but this is not an optimal system (requires a heated filtration, MeCN has a residual solvent limit of only 410 ppm). Additional solvents with reasonable solubility (>50 mg/ml) include N-methyl-2- pyrrolidone (NMP) and dimethylacetamide (DMAc); but the development of isolations from these solvents required large volumes and raised residual solvent limit concerns (530 ppm or less for NMT and 1090 ppm or less for DMAc).
[0253] Formic acid is one ICH Class 3 solvent in which Compound 1 is highly soluble, having a solubility greater than 250 mg/ml at 20 °C. The solubility curve of Compound 1 in formic acid-Water is quite steep (see Figure 7), which enables a volumetrically efficient process.
[0254] Initial attempts to recrystallize crude Compound 1 involved dissolving in formic acid, polish filtering, and charging polish filtered water to about 20% supersaturation, followed by seeding with the thermodynamically most stable form (Form 1), followed by slow addition of water to the final solvent ratio, filtration, washing, and drying. Applicant observed that during the initial water charge, if the batch self-seeded it formed a thick slurry. X-ray diffraction (XRD), differential scanning calorimetry (DSC), and photomicroscopy demonstrated that a metastable form was produced. Once seeded with Form 1, the batch converted to the desired form (Form 1) prior to the addition of the remaining water. This process worked well during multiple lab runs, consistently delivering the desired form and purity with about 85% yield.
[0255] Unfortunately, upon scale-up, the batch did not convert to Form 1 after seeding. Additional water was charged and the batch began to convert to the desired form (mix of Form 1 and the metastable form by X-ray powder diffraction (XRPD)). When additional water was charged, the XRPD indicated only the metastable form. After a few hours with no change, Applicant continued the water charge to the final solvent ratio, during which time the batch eventually converted to Form 1. This process is summarized in Figure 8.
[0256] It was subsequently found by closer analysis of the plant and laboratory retains that a new metastable form was formed during scale up, with a similar, but different XRPD pattern. This form (metastable B) could be reproduced in the laboratory, but only when the batch has a high formic acid:water ratio and is seeded with Form 1. Without Form 1 seeds, metastable A is the kinetic form. Both metastable forms converted to Form 1 with additional water and/or upon drying, leading Applicant to believe that the metastable forms are formic acid solvates. These findings are summarized in Figure 9.
[0257] While there is little risk in not being able to control the final form, there is a risk of forming a difficult-to-stir slurry which can lead to processing issues. The crystallization procedure was therefore modified to keep a constant formic acid-water ratio. This was performed by charging 2.4X wt. formic acid and 1.75X wt. water (final solvent composition)
to the crystallizer with 0.03X wt. Form 1 seeds, and performing a simultaneous addition of Compound 1 in 6. IX wt. formic acid and 4.4X wt. water. The batch filtered easily and was washed with formic acid/water, then water, and dried under reduced pressure to yield 8.9 kg of Compound 1 (92% yield) with 99.85% LCAP and N.D. formic acid.
Example 2: Exemplary high throughput experimentation reaction
[0258] The following procedure is an exemplary high throughput experimentation reaction.
[0259] An overview of the reaction is shown below in Scheme 5:
Scheme 5: Reaction conditions tested for cross-coupling reaction of 2 and 3
[0260] Pd catalysts were dosed into the 24-well reactor vial as solutions (100 pL of 0.01 M solution in tetrahydrofuran (THF) or dichloroethane (DCE) depending upon the solubility of the ligand). Plates of these ligands are typically dosed in advance of the reaction, the solvent is removed by evacuation in an evaporative centrifuge and plates are stored in the glovebox. The catalysts screened in the coupling are the following: XPhos, SPhos, CataCXium A, APhos, P(Cy)3, PEPPSI-IPent. For the first five ligands, these were initially screened as the Buchwald Pd G2/G3 precatalysts.
[0261] To the plates was then added a stock solution of Compound 3 (10 pmol) and Compound 2 (12 pmol) dissolved in the following solvents: dimethylformamide (DMF),
tetrahydrofuran (THF), butanol (/r-BuOH), and toluene. The base was then added as a stock solution (30 mmol) in 20 mL of water.
[0262] A heatmap summarizing catalyst performance is shown in Figs. 10A and 10B. High performance liquid chromatography (HPLC) yields for this screening span from <5% up to -85%. Larger circles indicate higher yield. Lighter circles indicate higher cleanliness.
[0263] A similarly designed screening of base and solvent also indicate that a range of alcoholic solvents (methanol, ethanol, propanol, 2-butanol, 2-propanol, and /-amyl alcohol) are also all viable in this coupling chemistry. Bases such as potassium phosphate, potassium carbonate, potassium acetate, and potassium hydroxide were all successful in achieving the coupling. Fig. 10B shows a heatmap with HPLC yields ranging from -50 – 95%. Larger, darker circles indicate higher yield.
[0264] This chemistry from microvial screening has been scaled to a laboratory process. To a 3 -necked jacketed 250 mL flask equipped with overhead stirring, nitrogen inlet, and thermocouple was added Compound 3 (1.0 eq, 4.00 grams), Compound 2 (1.2 eq, 1.71 x wt), potassium carbonate (3.0 eq, 1.74 x wt). The reactor was inerted three times and then degassed 2-propanol (24 x vol.) followed by degassed water (6 x vol) was then added.
Stirring was then initiated at 300 rpms. The reactor was then stirred and blanketed with nitrogen for 1 hour. The catalyst was then added (0.01 eq, 0.028 x wt) and stirring continued (300 rpms) and the reactor was heated into the Tj = 65 °C.
[0265] After 2 hours, with full conversion confirmed analytically, trioctylphosphine (0.1 eq, 0.16 x wt) dosed, and reaction mixture allowed to cool slowly to room temperature hours.
The reaction mixture was then filtered, washed with 2-propanol (4 x vol), 2-propanol: water (4: 1, 4 x vol), and then with water (4 x vol). Note: If 2 is dimer present in cake, an additional ethyl acetate (EtOAc) wash (4 x vol) can be added for purging. The cake was then transferred to a vacuum oven to dry overnight at 40 °C, -40 cm Hg, under nitrogen flow. After transfer to a bottle, 6.03 grams of 1 were isolated, 98.6% assay, 91% overall yield.
Scheme 6: Alternative reagents and solvents for cross-coupling
[0266] Based on the previously delineated results, it was expected that a variety of monodentate (PPI13 [triphenylphosphine], PBu3 [tributylphosphine], etc) and bidentate phosphines (dppf [1,1 ‘-bis(diphenylphosphino)ferrocene], BINAP [2,2 -bis(diphenylphosphino)- 1 , 1 -binaphthyl], Xantphos [4,5-bis(diphenylphosphino)-9,9-dimethylxanthene], dppe [l,2-bis(diphenylphosphino)ethane], etc) ligated to any number of Pd sources (Pd halides, Pd(H) precatalyts, Pd(0) sources) could reasonably be employed to arrive at the Compound 1 crude material. A range of organic solvents ranging from non-polar (heptane, benzene), protic (alcohols), polar aprotic (dimethylsulfoxide, dimethylformamide, dimethylacetamide, acetonitrile) as well as a variety of esters and ketones (acetone, 2-butanone, ethylacetate) should also serve as effective solvents for this reactivity. Finally, inorganic bases of varying strength (phosphates, carbonates, acetates, etc) along with organic variants such as triethylamine, l,8-diazabicyclo(5.4.0)undec-7-ene, and others in a wide pKa range are viable as stoichiometric basic additives.
Example 3: Exemplary Compound 5 process
[0267] The purpose of this example was to describe an exemplary process for making Compound 5.
[0268] Charge 4 (lOg, 58mmol) and acetonitrile (lOOmL) to a reaction vessel and start the stirrer. Adjust the batch to -18 °C to -22 °C (target -20 °C). Charge triflic acid (5.5mL, 62mmol) to the batch maintaining -10 °C to -25 °C (target -20 °C). Stir the batch at -10 °C to -25 °C (target -20 °C) for 10 to 20 minutes. Charge NBS (11.38g, 64mmol) to the batch at -10 °C to -25 °C (target -20 °C) and stir for ca. 30 min at -10 °C to -25 °C (target -20 °C). Warm the batch to 20 °C over 3-4 hours (reaction will occur when internal temp is between 5 °C and 15 °C). Stir the batch at 15 °C to 25 °C (target 20 °C) for approximately 1 hour and sample for reaction completion.
[0269] If Compound 4 relative to Compound 5 is more than 5%:
[0270] Cool the bath to -5 °C to -15 °C (target -10 °C) (cooling below 0 °C to ensure selectivity). Charge NBS to the batch according to the follow formula: Mass of NBS = (% Compound 4 x lOg). Warm the batch to 20 °C over 1-2 hours. Stir the batch at 15 °C to 25 °C (target 20 °C) for approximately 1 hour and check reaction for completion. Proceed to next line.
[0271] If Compound 4 relative to Compound 5 is less than 5%:
[0272] Warm the batch to 40 °C to 50 °C (target 48 °C). Concentrate the batch under reduced pressure to a final volume of ~40mL. Cool the batch to -15 °C to -5 °C (target -10 °C) and stir for ca. lh. Filter the batch by suction filtration. Slurry wash the filter cake with purified water (3 x 20mL) at 15 °C to 25 °C (target 20 °C) for 10 to 15 minutes each wash. Remove a sample of the filter cake for analysis by ¾ NMR. Continue washing cake until the residual succimide is below 1.0%mol% relative to 5. Dry the filter cake at up to 60°C under vacuum and nitrogen purge. Analyse the 5 by HPLC analysis (97%w/w to 99%w/w). Expected yield: 60-85% theory (90-110% w/w).
Example 4: Purification of Compound 1 (CC-90010) by crystallization from formic acid and water.
[0273] This example describes a method for the purification of Compound 1 by
crystallization from formic acid and water. Also detailed are methods for obtaining three different polymorphs of Compound 1, including the most stable form, Form 1.
[0274] Figure 11 shows XH NMR of Compound 1 (CC-90010). Solvent: d6DMSO; and Figure 12 shows microscopy of Compound 1 (CC-90010) Form I. Figure 13 shows XRPD of Compound 1 (CC-90010) Form I, with peak information detailed in Table 6:
PATENT
US 20190008852
WO 2018081475
US 20180042914
WO 2016172618
WO 2015058160
/////////CC-90010, solid tumors , non-Hodgkin’s lymphoma, PHASE 1, CANCER, QUANTICEL
CS(=O)(=O)c4cc(C1=CN(C)C(=O)c2ccccc12)c(OCC3CC3)cc4
Delgocitinib

Delgocitinib
デルゴシチニブ
3-[(3S,4R)-3-methyl-7-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1,7-diazaspiro[3.4]octan-1-yl]-3-oxopropanenitrile
1,6-Diazaspiro(3.4)octane-1-propanenitrile, 3-methyl-beta-oxo-6-(7H-pyrrolo(2,3-d)pyrimidin-4-yl)-, (3S,4R)-
3-((3S,4R)-3-methyl-6-(7H-pyrrolo(2,3-d)pyrimidin-4-yl)-1,6-diazaspiro(3.4)octan-1-yl)-3-oxopropanenitrile
| Formula |
C16H18N6O
|
|---|---|
| CAS |
1263774-59-9
|
| Mol weight |
310.3537
|
Approved, Japan 2020, Corectim, 2020/1/23, atopic dermatitis, Japan Tobacco (JT)
Torii
7/23/2025 fda approved, Anzupgo
| To treat moderate-to-severe chronic hand eczema when topical corticosteroids are not advisable or produce an inadequate response |
UNII-9L0Q8KK220, JTE-052, LP-0133, ROH-201, 9L0Q8KK220, LEO 124249A, LEO 124249, HY-109053
CS-0031558, D11046, GTPL9619, JTE-052A, JTE052
Delgocitinib, also known as LEO-124249 and JTE052, is a potent and selective JAK inhibitor. JTE-052 reduces skin inflammation and ameliorates chronic dermatitis in rodent models: Comparison with conventional therapeutic agents. JTE-052 regulates contact hypersensitivity by downmodulating T cell activation and differentiation.
Delgocitinib is a JAK inhibitor first approved in Japan for the treatment of atopic dermatitis in patients 16 years of age or older. Japan Tobacco is conducting phase III clinical trials for the treatment of atopic dermatitis in pediatric patients. Leo is developing the drug in phase II clinical trials for the treatment of inflammatory skin diseases, such as atopic dermatitis, and chronic hand eczema and for the treatment of discoid lupus erythematosus. Rohto is evaluating the product in early clinical development for ophthalmologic indications.
In 2014, the drug was licensed to Leo by Japan Tobacco for the development, registration and marketing worldwide excluding Japan for treatment of inflammatory skin conditions. In 2016, Japan Tobacco licensed the rights of co-development and commercialization in Japan to Torii. In 2018, Japan Tobacco licensed the Japanese rights of development and commercialization to Rohto for the treatment of ophthalmologic diseases.
Delgocitinib, sold under the brand name Corectim among others, is a medication used for the treatment of autoimmune disorders and hypersensitivity, including inflammatory skin conditions.[3] Delgocitinib was developed by Japan Tobacco and approved in Japan for the treatment of atopic dermatitis.[3] In the United States, delgocitinib is in Phase III clinical trials and the Food and Drug Administration has granted delgocitinib fast track designation for topical treatment of adults with moderate to severe chronic hand eczema.[4]
Delgocitinib works by blocking activation of the JAK-STAT signaling pathway which contributes to the pathogenesis of chronic inflammatory skin diseases.[5]
PATENTS
WO 2018117151
IN 201917029002
IN 201917029003
IN 201917029000
PATENTS
WO 2011013785
https://patents.google.com/patent/WO2011013785A1/en
[Production Example 6]: Synthesis of Compound 6

(1) Optically active substance of 2-benzylaminopropan-1-ol

To a solution of (S)-(+)-2-aminopropan-1-ol (50.0 g) and benzaldehyde (74 ml) in ethanol (500 ml) was added 5% palladium carbon (5.0 g) at room temperature and normal pressure. Hydrogenated for 8 hours. The reaction mixture was filtered through celite and concentrated under reduced pressure to give the title compound (111.2 g).
1 H-NMR (DMSO-D 6 ) δ: 7.34-7.27 (4H, m), 7.23-7.18 (1H, m), 4.53-4.47 (1H, m), 3.76 (1H, d, J = 13.5 Hz) , 3.66 (1H, d, J = 13.5 Hz), 3.29-3.24 (2H, m), 2.65-2.55 (1H, m), 1.99 (1H, br s), 0.93 (3H, d, J = 6.4 Hz) .
(2) Optically active substance of [benzyl- (2-hydroxy-1-methylethyl) -amino] acetic acid tert-butyl ester

To a mixture of optically active 2-benzylaminopropan-1-ol (111.2 g), potassium carbonate (111.6 g) and N, N-dimethylformamide (556 ml) cooled to 0 ° C., tert-butyl bromoacetate was added. Ester (109 ml) was added dropwise over 20 minutes and stirred at room temperature for 19.5 hours. The mixture was acidified to pH 2 by adding 2M aqueous hydrochloric acid and 6M aqueous hydrochloric acid, and washed with toluene (1000 ml). The separated organic layer was extracted with 0.1 M aqueous hydrochloric acid (300 ml). The combined aqueous layer was adjusted to pH 10 with 4M aqueous sodium hydroxide solution and extracted with ethyl acetate (700 ml). The organic layer was washed successively with water (900 ml) and saturated aqueous sodium chloride solution (500 ml). The separated aqueous layer was extracted again with ethyl acetate (400 ml). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure to give the title compound (160.0 g).
1 H-NMR (DMSO-D 6 ) δ: 7.37-7.26 (4H, m), 7.24-7.19 (1H, m), 4.26 (1H, dd, J = 6.9, 3.9 Hz), 3.76 (1H, d, J = 14.1 Hz), 3.68 (1H, d, J = 13.9 Hz), 3.45-3.39 (1H, m), 3.29-3.20 (1H, m), 3.24 (1H, d, J = 17.2 Hz), 3.13 ( 1H, d, J = 17.0 Hz), 2.84-2.74 (1H, m), 1.37 (9H, s), 0.96 (3H, d, J = 6.8 Hz).
(3) Optically active substance of [benzyl- (2-chloropropyl) -amino] acetic acid tert-butyl ester

(3)-(1) Optically active form of [benzyl- (2-chloro-1-methylethyl) -amino] acetic acid tert-butyl ester

To a solution of [benzyl- (2-hydroxy-1-methylethyl) -amino] acetic acid tert-butyl ester optically active substance (160.0 g) cooled to 0 ° C. in chloroform (640 ml) was added thionyl chloride (50.0 ml). Was added dropwise and stirred at 60 ° C. for 2 hours. The reaction mixture was cooled to 0 ° C., saturated aqueous sodium hydrogen carbonate solution (1000 ml) and chloroform (100 ml) were added and stirred. The separated organic layer was washed with a saturated aqueous sodium chloride solution (500 ml), and the aqueous layer was extracted again with chloroform (450 ml). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain the title compound (172.9 g).
1 H-NMR (CDCl 3 ) δ: 7.40-7.22 (5H, m), 4.05-3.97 (0.4H, m), 3.93-3.81 (2H, m), 3.70-3.65 (0.6H, m), 3.44- 3.38 (0.6H, m), 3.29 (0.8H, s), 3.27 (1.2H, d, J = 2.4 Hz), 3.24-3.15 (0.6H, m), 3.05-2.99 (0.4H, m), 2.94 -2.88 (0.4H, m), 1.50 (1.2H, d, J = 6.4 Hz), 1.48 (3.6H, s), 1.45 (5.4H, s), 1.23 (1.8H, d, J = 6.8 Hz) .
(3)-(2) Optically active form of [benzyl- (2-chloropropyl) -amino] acetic acid tert-butyl ester

[Benzyl- (2-chloro-1-methylethyl) -amino] acetic acid tert-butyl ester optically active substance (172.9 g) was dissolved in N, N-dimethylformamide (520 ml) and stirred at 80 ° C. for 140 minutes. did. The reaction mixture was cooled to 0 ° C., water (1200 ml) was added, and the mixture was extracted with n-hexane / ethyl acetate (2/1, 1000 ml). The organic layer was washed successively with water (700 ml) and saturated aqueous sodium chloride solution (400 ml), and the separated aqueous layer was extracted again with n-hexane / ethyl acetate (2/1, 600 ml). The combined organic layers were concentrated under reduced pressure, and the obtained residue was purified by silica gel column chromatography (eluent: n-hexane / ethyl acetate = 50/1 to 40/1) to give the title compound (127.0 g )
1 H-NMR (CDCl 3 ) δ: 7.37-7.29 (4H, m), 7.28-7.23 (1H, m), 4.05-3.97 (1H, m), 3.91 (1H, d, J = 13.5 Hz), 3.86 (1H, d, J = 13.7 Hz), 3.29 (2H, s), 3.03 (1H, dd, J = 13.9, 6.6 Hz), 2.91 (1H, dd, J = 13.9, 6.8 Hz), 1.50 (3H, d, J = 6.4 Hz), 1.48 (9H, s).
(4) Optically active substance of 1-benzyl-3-methylazetidine-2-carboxylic acid tert-butyl ester

To a solution of [benzyl- (2-chloropropyl) -amino] acetic acid tert-butyl ester optically active substance (60.0 g) cooled to −72 ° C. and hexamethylphosphoramide (36.0 ml) in tetrahydrofuran (360 ml), Lithium hexamethyldisilazide (1.0 M tetrahydrofuran solution, 242 ml) was added dropwise over 18 minutes, and the temperature was raised to 0 ° C. over 80 minutes. A saturated aqueous ammonium chloride solution (300 ml) and water (400 ml) were sequentially added to the reaction mixture, and the mixture was extracted with ethyl acetate (500 ml). The organic layer was washed successively with water (700 ml) and saturated aqueous sodium chloride solution (500 ml), and the separated aqueous layer was extracted again with ethyl acetate (300 ml). The combined organic layers were dried over anhydrous sodium sulfate, concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography (developing solvent: n-hexane / ethyl acetate = 50/1 to 4/1). To give the title compound (50.9 g).
1 H-NMR (CDCl 3 ) δ: 7.34-7.21 (5H, m), 3.75 (1H, d, J = 12.6 Hz), 3.70-3.67 (1H, m), 3.58 (1H, d, J = 12.6 Hz ), 3.05-3.01 (1H, m), 2.99-2.95 (1H, m), 2.70-2.59 (1H, m), 1.41 (9H, s), 1.24 (3H, d, J = 7.1 Hz).
(5) Optically active substance of 3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester

1-Benzyl-3-methylazetidine-2-carboxylic acid tert-butyl ester optically active substance (43.5 g) and di-tert-butyl dicarbonate (38.2 g) in tetrahydrofuran / methanol (130 ml / 130 ml) solution 20% Palladium hydroxide carbon (3.5 g) was added thereto, and hydrogenated at 4 atm for 2 hours. The mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to give the title compound (48.0 g).
1 H-NMR (DMSO-D 6 ) δ: 4.44 (1H, d, J = 8.8 Hz), 3.99-3.77 (1H, m), 3.45-3.37 (1H, m), 3.00-2.88 (1H, m) , 1.45 (9H, s), 1.40-1.30 (9H, m), 1.02 (3H, d, J = 7.2 Hz).
(6) Optically active substance of 3-methyl-2- (3-methyl-but-2-enyl) -azetidine-1,2-dicarboxylic acid di-tert-butyl ester

Optically active substance (48.0 g) of 3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester cooled to -69 ° C. and 1-bromo-3-methyl-2-butene (25.4 ml) Lithium hexamethyldisilazide (1.0 M tetrahydrofuran solution, 200 ml) was added to a tetrahydrofuran solution (380 ml). The reaction mixture was warmed to −20 ° C. in 40 minutes and further stirred at the same temperature for 20 minutes. A saturated aqueous ammonium chloride solution (200 ml) and water (300 ml) were successively added to the reaction mixture, and the mixture was extracted with n-hexane / ethyl acetate (1 / 1,500 ml). The separated organic layer was washed successively with water (200 ml) and saturated aqueous sodium chloride solution (200 ml), dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (eluent: n-hexane / ethyl acetate = 15/1 to 8/1) to give the titled compound (44.5 g).
1 H-NMR (CDCl 3 ) δ: 5.29-5.21 (1H, m), 3.77-3.72 (1H, m), 3.49-3.44 (1H, m), 2.73-2.52 (3H, m), 1.76-1.74 ( 3H, m), 1.66-1.65 (3H, m), 1.51 (9H, s), 1.43 (9H, s), 1.05 (3H, d, J = 7.3 Hz).
(7) Optically active substance of 3-methyl-2- (2-oxoethyl) azetidine-1,2-dicarboxylic acid di-tert-butyl ester

3-methyl-2- (3-methyl-but-2-enyl) -azetidine-1,2-dicarboxylic acid di-tert-butyl ester optically active substance (44.5 g) in chloroform / cooled to −70 ° C. An ozone stream was passed through the methanol solution (310 ml / 310 ml) for 1 hour. To this reaction mixture, a solution of triphenylphosphine (44.7 g) in chloroform (45 ml) was added little by little, and then the mixture was warmed to room temperature. To this mixture were added saturated aqueous sodium thiosulfate solution (200 ml) and water (300 ml), and the mixture was extracted with chloroform (500 ml). The separated organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to obtain the title compound (95.0 g). This product was subjected to the next step without further purification.
1 H-NMR (DMSO-D 6 ) δ: 9.65 (1H, t, J = 2.6 Hz), 3.79-3.74 (1H, m), 3.45-3.40 (1H, m), 2.99-2.80 (3H, m) , 1.46 (9H, s), 1.34 (9H, s), 1.06 (3H, d, J = 7.2 Hz).
(8) Optically active substance of 2- (2-benzylaminoethyl) -3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester

To a solution of the residue (95.0 g) obtained in (7) in tetrahydrofuran (300 ml) was added benzylamine (34 ml) at room temperature, and the mixture was stirred for 2 hours. The mixture was cooled to 0 ° C., sodium triacetoxyborohydride (83.3 g) was added, and the mixture was stirred at room temperature for 1.5 hours. Water (300 ml) was added to the reaction mixture, and the mixture was extracted with n-hexane / ethyl acetate (1/3, 600 ml). The separated organic layer was washed with water (300 ml) and saturated aqueous sodium chloride solution (200 ml), and then extracted twice with 5% aqueous citric acid solution (300 ml, 200 ml) and three times with 10% aqueous citric acid solution (250 ml × 3). . The combined aqueous layers were basified to pH 10 with 4M aqueous sodium hydroxide solution and extracted with chloroform (300 ml). The organic layer was washed with a saturated aqueous sodium chloride solution (200 ml), dried over anhydrous magnesium sulfate and concentrated under reduced pressure to obtain the title compound (46.9 g).
1 H-NMR (DMSO-D 6 ) δ: 7.34-7.26 (4H, m), 7.22-7.17 (1H, m), 3.74-3.65 (2H, m), 3.61 (1H, t, J = 7.8 Hz) , 3.28 (1H, t, J = 7.5 Hz), 2.76-2.66 (2H, m), 2.57-2.45 (1H, m), 2.15 (1H, br s), 2.05-1.89 (2H, m), 1.42 ( 9H, s), 1.27 (9H, s), 0.96 (3H, d, J = 7.1 Hz).
(9) Optically active substance of 2- (2-benzylaminoethyl) -3-methylazetidine-2-dicarboxylic acid dihydrochloride

2- (2-Benzylaminoethyl) -3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester optically active substance (46.5 g), 4M hydrochloric acid 1,4-dioxane (230 ml) and water (4.1 ml) was mixed and stirred at 80 ° C. for 2 hours. The mixture was concentrated under reduced pressure, azeotroped with toluene, and then slurry washed with n-hexane / ethyl acetate (1/1, 440 ml) to give the title compound (30.1 g).
1 H-NMR (DMSO-D 6 ) δ: 10.24 (1H, br s), 9.64 (2H, br s), 8.90 (1H, br s), 7.58-7.53 (2H, m), 7.47-7.41 (3H , m), 4.21-4.10 (2H, m), 4.02-3.94 (1H, m), 3.46-3.37 (1H, m), 3.20-3.10 (1H, m), 2.99-2.85 (2H, m), 2.69 -2.54 (2H, m), 1.10 (3H, d, J = 7.2 Hz).
(10) Optically active substance of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octan-5-one

To a solution of 2- (2-benzylaminoethyl) -3-methylazetidine-2-dicarboxylic acid dihydrochloride optically active substance (29.1 g) and N, N-diisopropylethylamine (65 ml) in chloroform (290 ml), At room temperature, O- (7-azabenzotriazol-1-yl) -N, N, N ′, N′-tetramethyluronium hexafluorophosphate (41.3 g) was added and stirred for 4 hours. To this reaction mixture were added saturated aqueous sodium hydrogen carbonate solution (200 ml) and water (100 ml), and the mixture was extracted with chloroform (200 ml). The organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent: chloroform / methanol = 20/1 to 10/1) to give the titled compound (21.3 g).
1 H-NMR (DMSO-D 6 ) δ: 7.38-7.31 (2H, m), 7.30-7.22 (3H, m), 4.52 (1H, d, J = 14.8 Hz), 4.29 (1H, d, J = 14.8 Hz), 3.35-3.27 (2H, m), 3.22-3.17 (1H, m), 3.05 (2H, dd, J = 9.5, 4.0 Hz), 2.77-2.66 (1H, m), 2.16-2.10 (1H , m), 1.96-1.87 (1H, m), 0.94 (3H, d, J = 7.1 Hz).
(11) Optically active substance of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester

Concentrated sulfuric acid (4.8 ml) was slowly added dropwise to a suspension of lithium aluminum hydride (6.8 g) in tetrahydrofuran (300 ml) under ice cooling, and the mixture was stirred for 30 minutes. To this mixture was added dropwise a solution of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octan-5-one optically active substance (21.3 g) in tetrahydrofuran (100 ml) at the same temperature. Stir for 45 minutes. Water (7.0 ml), 4M aqueous sodium hydroxide solution (7.0 ml) and water (14.0 ml) were sequentially added to the reaction mixture, and the mixture was stirred as it was for 30 minutes. To this mixture was added anhydrous magnesium sulfate and ethyl acetate (100 ml), and the mixture was stirred and filtered through celite. Di-tert-butyl dicarbonate (23.4 g) was added to the filtrate at room temperature and stirred for 3 hours. The mixture was concentrated under reduced pressure to a half volume and washed twice with a saturated aqueous ammonium chloride solution (200 ml × 2). N-Hexane (200 ml) was added to the separated organic layer, and the mixture was extracted 5 times with a 10% aqueous citric acid solution. The separated aqueous layer was basified with 4M aqueous sodium hydroxide solution and extracted with chloroform. The organic layer was washed with a saturated aqueous sodium chloride solution (200 ml), dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (eluent: chloroform / methanol = 40/1 to 20/1) to give the titled compound (15.6 g).
1 H-NMR (DMSO-D 6 ) δ: 7.34-7.27 (4H, m), 7.26-7.21 (1H, m), 3.84-3.69 (1H, m), 3.62-3.47 (2H, m), 3.19- 3.05 (1H, m), 3.02-2.92 (1H, m), 2.76-2.69 (1H, m), 2.47-2.24 (4H, m), 1.95-1.77 (1H, m), 1.36 (9H, s), 1.03 (3H, d, J = 7.0 Hz).
(12) Optically active substance of 3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester

20% of optically active form of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester (10.0 g) in tetrahydrofuran / methanol (50 ml / 50 ml) solution Palladium hydroxide on carbon (2.0 g) was added and hydrogenated at 4 atm for 24 hours. The mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to give the title compound (7.3 g).
1 H-NMR (DMSO-D 6 ) δ: 3.88-3.71 (1H, m), 3.44-3.06 (2H, m), 3.02-2.64 (4H, m), 2.55-2.38 (1H, m), 2.31- 2.15 (1H, m), 1.81-1.72 (1H, m), 1.37 (9H, s), 1.07 (3H, d, J = 7.0 Hz).
(13) Optical activity of 3-methyl-6- (7H-pyrrolo [2,3-d] pyrimidin-4-yl) -1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester body

The optically active substance (6.9 g) of 3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester was converted into 4-chloro-7H-pyrrolo [2,3-d] pyrimidine ( 4.3 g), potassium carbonate (7.7 g) and water (65 ml) and stirred for 4 hours at reflux. The mixture was cooled to room temperature, water (60 ml) was added, and the mixture was extracted with chloroform / methanol (10/1, 120 ml). The organic layer was washed successively with water, saturated aqueous ammonium chloride solution and saturated aqueous sodium chloride solution, and dried over anhydrous sodium sulfate. To this mixture, silica gel (4 g) was added, stirred for 10 minutes, filtered through celite, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent: chloroform / ethyl acetate = 1/1, then chloroform / methanol = 50/1 to 20/1) to give the title compound (10.0 g). Obtained.
1 H-NMR (DMSO-D 6 ) δ: 11.59 (1H, br s), 8.09 (1H, s), 7.12-7.09 (1H, m), 6.64-6.59 (1H, m), 4.09-3.66 (5H , m), 3.39-3.21 (1H, m), 2.64-2.44 (2H, m), 2.27-2.06 (1H, m), 1.36 (3H, s), 1.21 (6H, s), 1.11 (3H, d , J = 6.5 Hz).
(14) Optically active form of 4- (3-methyl-1,6-diazaspiro [3.4] oct-6-yl) -7H-pyrrolo [2,3-d] pyrimidine dihydrochloride

Optically active form of 3-methyl-6- (7H-pyrrolo [2,3-d] pyrimidin-4-yl) -1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester (9 0.5 g), 4M hydrochloric acid 1,4-dioxane (50 ml), chloroform (50 ml) and methanol (100 ml) were mixed and stirred at 60 ° C. for 30 minutes. The mixture was concentrated under reduced pressure and azeotroped with toluene to give the title compound (9.3 g).
1 H-NMR (DMSO-D 6 ) δ: 12.91 (1H, br s), 9.97-9.64 (2H, m), 8.45-8.35 (1H, m), 7.58-7.47 (1H, m), 7.04-6.92 (1H, m), 4.99-4.65 (1H, m), 4.32-3.21 (7H, m), 3.04-2.90 (1H, m), 2.46-2.31 (1H, m), 1.27 (3H, d, J = 6.0 Hz).
(15) 3- [3-Methyl-6- (7H-pyrrolo [2,3-d] pyrimidin-4-yl) -1,6-diazaspiro [3.4] oct-1-yl] -3-oxo Optically active form of propionitrile

4- (3-Methyl-1,6-diazaspiro [3.4] oct-6-yl) -7H-pyrrolo [2,3-d] pyrimidine dihydrochloride optically active substance (8.8 g) was converted to 1- The mixture was mixed with cyanoacetyl-3,5-dimethylpyrazole (6.8 g), N, N-diisopropylethylamine (20 ml) and 1,4-dioxane (100 ml) and stirred at 100 ° C. for 1 hour. The mixture was cooled to room temperature, saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform / methanol (10/1). The separated organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent: chloroform / methanol = 30/1 to 9/1). The residue obtained by concentration under reduced pressure was slurry washed with n-heptane / ethanol (2/1, 90 ml) to obtain a solid (7.3 g). The solid was slurried again with n-heptane / ethanol (5/1, 90 ml) to give the title compound as crystals 1 (6.1 g).
1 H-NMR (DMSO-D 6 ) δ: 11.60 (1H, br s), 8.08 (1H, s), 7.11 (1H, dd, J = 3.5, 2.4 Hz), 6.58 (1H, dd, J = 3.4 , 1.9 Hz), 4.18-4.14 (1H, m), 4.09-3.93 (3H, m), 3.84-3.73 (1H, m), 3.71 (1H, d, J = 19.0 Hz), 3.66 (1H, d, J = 18.7 Hz), 3.58 (1H, dd, J = 8.2, 6.0 Hz), 2.70-2.58 (2H, m), 2.24-2.12 (1H, m), 1.12 (3H, d, J = 7.1 Hz).
[Α] D = + 47.09 ° (25 ° C., c = 0.55, methanol)
1-Butanol (39 ml) was added to the obtained crystal 1 (2.6 g), and the mixture was heated and stirred at 100 ° C. After complete dissolution, the solution was cooled to room temperature by 10 ° C. every 30 minutes and further stirred at room temperature overnight. The produced crystals were collected by filtration, washed with 1-butanol (6.2 ml), and dried under reduced pressure to give crystals 2 (2.1 g) of the title compound.
PATENTS
WO 2017006968
WO 2018117152
WO 2018117151
PATENT
WO 2018117153
https://patentscope.wipo.int/search/zh/detail.jsf?docId=WO2018117153&tab=FULLTEXT
Janus kinase (JAK) inhibitors are of current interest for the treatment of various diseases including autoimmune diseases, inflammatory diseases, and cancer. To date, two JAK inhibitors have been approved by the U.S. Food & Drug Administration (FDA). Ruxolitinib has been approved for the treatment of primary myelofibrosis and polycythemia vera (PV), and tofacitinib has been approved for the treatment of rheumatoid arthritis. Other JAK inhibitors are in the literature. The compound 3-((3S,4R)-3-methyl-6-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1,6-diazaspiro[3.4]octan-1-yl)-3-oxopropanenitrile (Compound A) (see structure below) is an example of a spirocyclic JAK inhibitor reported in U.S. Pat. Pub. Nos. 2011/0136778 and International Pat. Pub. No. PCT/JP2016/070046.
[Chem. 1]
Step A. Preparation of S-MABB-HC (Compound [5])
[Chem. 2]

Step 1
[Chem. 3]
 S-BAPO [1] (35.0 g, 212 mmol) was added to water (175 mL) at room temperature under nitrogen atmosphere. To the resulting suspension were added toluene (53 mL) and potassium carbonate (32.2 g, 233 mmol) at room temperature. To the resulting solution was added dropwise TBBA (434.4 g, 223 mmol) at room temperature, and then the used dropping funnel was washed with toluene (17 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 65°C for 21 hours, and then cooled to room temperature. After toluene (105 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The organic layer was washed with water (175 mL), aqueous layer was removed, and then the solvent was removed out of the organic layer in vacuo. Toluene (105 mL) was added to the residue and the toluene solution was concentrated. The operation was repeated two more times to give a toluene solution of S-BBMO [2] (74.0 g, 212 mmol in theory). The given toluene solution of S-BBMO was used in the next step, assuming that the yield was 100 %.
S-BAPO [1] (35.0 g, 212 mmol) was added to water (175 mL) at room temperature under nitrogen atmosphere. To the resulting suspension were added toluene (53 mL) and potassium carbonate (32.2 g, 233 mmol) at room temperature. To the resulting solution was added dropwise TBBA (434.4 g, 223 mmol) at room temperature, and then the used dropping funnel was washed with toluene (17 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 65°C for 21 hours, and then cooled to room temperature. After toluene (105 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The organic layer was washed with water (175 mL), aqueous layer was removed, and then the solvent was removed out of the organic layer in vacuo. Toluene (105 mL) was added to the residue and the toluene solution was concentrated. The operation was repeated two more times to give a toluene solution of S-BBMO [2] (74.0 g, 212 mmol in theory). The given toluene solution of S-BBMO was used in the next step, assuming that the yield was 100 %.
A crude product of S-BBMO which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.36-7.13 (5H, m), 4.26 (1H, dd, J = 6.8, 3.9 Hz), 3.72 (2H, dd, J = 14.2, 6.8 Hz), 3.47-3.38 (1H, m), 3.30-3.08 (3H, m), 2.79 (1H, sext, J = 6.8 Hz), 1.35 (9H, s), 0.96 (3H, d, J = 6.8 Hz).
MS: m/z = 280 [M+H] +
[Chem. 2]
Step 1
[Chem. 3]
A crude product of S-BBMO which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.36-7.13 (5H, m), 4.26 (1H, dd, J = 6.8, 3.9 Hz), 3.72 (2H, dd, J = 14.2, 6.8 Hz), 3.47-3.38 (1H, m), 3.30-3.08 (3H, m), 2.79 (1H, sext, J = 6.8 Hz), 1.35 (9H, s), 0.96 (3H, d, J = 6.8 Hz).
MS: m/z = 280 [M+H] +
[0134]
Step 2
[Chem. 4]
 To the toluene solution of S-BBMO [2] (74.0 g, 212 mmol) were added toluene (200 mL), tetrahydrofuran (35 mL), and then triethylamine (25.7 g, 254 mmol) at room temperature under nitrogen atmosphere. To the mixture was added dropwise methanesulfonyl chloride (26.7 g, 233 mmol) at 0°C, and then the used dropping funnel was washed with toluene (10 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at room temperature for 2 hours and further at 65°C for 22 hours, and then cooled to room temperature. After sodium bicarbonate water (105 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The organic layer was washed with water (105 mL), aqueous layer was removed, and then the solvent was removed out of the organic layer in vacuo. Toluene (105 mL) was added to the residue, and the toluene solution was concentrated. The operation was repeated two more times to give a toluene solution of R-BCAB [3] (75.3 g, 212 mmol in theory). The given toluene solution of R-BCAB was used in the next step, assuming that the yield was 100 %.
To the toluene solution of S-BBMO [2] (74.0 g, 212 mmol) were added toluene (200 mL), tetrahydrofuran (35 mL), and then triethylamine (25.7 g, 254 mmol) at room temperature under nitrogen atmosphere. To the mixture was added dropwise methanesulfonyl chloride (26.7 g, 233 mmol) at 0°C, and then the used dropping funnel was washed with toluene (10 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at room temperature for 2 hours and further at 65°C for 22 hours, and then cooled to room temperature. After sodium bicarbonate water (105 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The organic layer was washed with water (105 mL), aqueous layer was removed, and then the solvent was removed out of the organic layer in vacuo. Toluene (105 mL) was added to the residue, and the toluene solution was concentrated. The operation was repeated two more times to give a toluene solution of R-BCAB [3] (75.3 g, 212 mmol in theory). The given toluene solution of R-BCAB was used in the next step, assuming that the yield was 100 %.
A crude product of R-BCAB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.28-7.11 (5H, m), 4.24-4.11 (1H, m), 3.80 (2H, d, J = 3.6 Hz), 3.24 (2H, d, J = 3.6 Hz), 2.98-2.78 (2H, m), 1.46-1.37 (12H, m).
MS: m/z = 298 [M+H] +
[Chem. 4]
A crude product of R-BCAB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.28-7.11 (5H, m), 4.24-4.11 (1H, m), 3.80 (2H, d, J = 3.6 Hz), 3.24 (2H, d, J = 3.6 Hz), 2.98-2.78 (2H, m), 1.46-1.37 (12H, m).
MS: m/z = 298 [M+H] +
[0135]
Step 3
[Chem. 5]
 To the toluene solution of R-BCAB [3] (75.3 g, 212 mmol) were added tetrahydrofuran (88.0 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (42.0 mL) at room temperature under nitrogen atmosphere. To the resulting solution was added dropwise a solution of lithium bis(trimethylsilyl)amide /tetrahydrofuran (195 mL, 233 mmol) at 0°C, and then the used dropping funnel was washed with tetrahydrofuran (17.0 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 0°C for 1 hour, and then warmed to room temperature. After water (175 mL) and toluene (175 mL) were added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with aqueous ammonium chloride (175 mL) and then water (175 mL), and the solvent was removed out of the organic layer in vacuo. Ethyl acetate (175 mL) was added to the residue and the ethyl acetate solution was concentrated. The operation was repeated two more times to give an ethyl acetate solution of S-MABB [4] (66.5 g, 212 mmol in theory). The given ethyl acetate solution of S-MABB was used in the next step, assuming that the yield was 100 %.
To the toluene solution of R-BCAB [3] (75.3 g, 212 mmol) were added tetrahydrofuran (88.0 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (42.0 mL) at room temperature under nitrogen atmosphere. To the resulting solution was added dropwise a solution of lithium bis(trimethylsilyl)amide /tetrahydrofuran (195 mL, 233 mmol) at 0°C, and then the used dropping funnel was washed with tetrahydrofuran (17.0 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 0°C for 1 hour, and then warmed to room temperature. After water (175 mL) and toluene (175 mL) were added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with aqueous ammonium chloride (175 mL) and then water (175 mL), and the solvent was removed out of the organic layer in vacuo. Ethyl acetate (175 mL) was added to the residue and the ethyl acetate solution was concentrated. The operation was repeated two more times to give an ethyl acetate solution of S-MABB [4] (66.5 g, 212 mmol in theory). The given ethyl acetate solution of S-MABB was used in the next step, assuming that the yield was 100 %.
A crude product of S-MABB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.28-7.25 (10H, m), 3.75 (1H, d, J = 12.7 Hz), 3.68 (1H, d, J = 1.4 Hz), 3.66 (1H, d, J = 6.7 Hz), 3.46 (2H, d, J = 12.7 Hz), 3.30-3.17 (2H, m), 2.95 (1H, dd, J = 6.2, 1.2 Hz), 2.77 (1H, dd, J = 6.1, 2.2 Hz), 2.65-2.55 (1H, m), 2.48-2.40 (2H, m), 1.35 (9H, s), 1.35 (9H, s), 1.12 (3H, d, J = 7.2 Hz), 1.09 (3H, d, J = 6.2 Hz).
MS: m/z = 262 [M+H] +
[Chem. 5]
A crude product of S-MABB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.28-7.25 (10H, m), 3.75 (1H, d, J = 12.7 Hz), 3.68 (1H, d, J = 1.4 Hz), 3.66 (1H, d, J = 6.7 Hz), 3.46 (2H, d, J = 12.7 Hz), 3.30-3.17 (2H, m), 2.95 (1H, dd, J = 6.2, 1.2 Hz), 2.77 (1H, dd, J = 6.1, 2.2 Hz), 2.65-2.55 (1H, m), 2.48-2.40 (2H, m), 1.35 (9H, s), 1.35 (9H, s), 1.12 (3H, d, J = 7.2 Hz), 1.09 (3H, d, J = 6.2 Hz).
MS: m/z = 262 [M+H] +
[0136]
Step 4
[Chem. 6]
 To the ethyl acetate solution of S-MABB [4] (66.5 g, 212 mmol in theory) were added ethyl acetate (175 mL) and active carbon (3.5 g) under nitrogen atmosphere, and then the mixture was stirred at room temperature for 2 hours. The active carbon was removed by filtration, and the residue on the filter was washed with ethyl acetate (175 mL). The washings were added to the filtrate. To the solution was added S-MABB-HC crystal (17.5 mg) that was prepared according to the method described herein at 0°C, and then 4 M hydrogen chloride/ethyl acetate (53.0 mL, 212 mmol) was dropped thereto at 0°C. The reaction mixture was stirred at 0°C for 17 hours, and then the precipitated solid was collected on a filter, and washed with ethyl acetate (70 mL). The resulting wet solid was dried in vacuo to give S-MABB-HC [5] (48.3 g, 162 mmol, yield: 76.4 %).
To the ethyl acetate solution of S-MABB [4] (66.5 g, 212 mmol in theory) were added ethyl acetate (175 mL) and active carbon (3.5 g) under nitrogen atmosphere, and then the mixture was stirred at room temperature for 2 hours. The active carbon was removed by filtration, and the residue on the filter was washed with ethyl acetate (175 mL). The washings were added to the filtrate. To the solution was added S-MABB-HC crystal (17.5 mg) that was prepared according to the method described herein at 0°C, and then 4 M hydrogen chloride/ethyl acetate (53.0 mL, 212 mmol) was dropped thereto at 0°C. The reaction mixture was stirred at 0°C for 17 hours, and then the precipitated solid was collected on a filter, and washed with ethyl acetate (70 mL). The resulting wet solid was dried in vacuo to give S-MABB-HC [5] (48.3 g, 162 mmol, yield: 76.4 %).
S-MABB-HC which was prepared by the same process was measured about NMR, MS, and Cl-content.
1H-NMR (DMSO-d 6) δ: 11.08 (1H, br s), 10.94 (1H, br s), 7.52-7.42 (10H, m), 5.34 (1H, t, J = 8.4 Hz), 4.90 (1H, br s), 4.45-4.10 (5H, m), 3.92-3.49 (3H, br m), 3.10-2.73 (2H, br m), 1.35 (9H, s), 1.29 (9H, s), 1.24 (3H, d, J = 6.7 Hz), 1.17 (3H, d, J = 7.4 Hz).
MS: m/z = 262 [M+H-HCl] +
Cl content (ion chromatography): 11.9 % (in theory: 11.9 %).
[Chem. 6]
S-MABB-HC which was prepared by the same process was measured about NMR, MS, and Cl-content.
1H-NMR (DMSO-d 6) δ: 11.08 (1H, br s), 10.94 (1H, br s), 7.52-7.42 (10H, m), 5.34 (1H, t, J = 8.4 Hz), 4.90 (1H, br s), 4.45-4.10 (5H, m), 3.92-3.49 (3H, br m), 3.10-2.73 (2H, br m), 1.35 (9H, s), 1.29 (9H, s), 1.24 (3H, d, J = 6.7 Hz), 1.17 (3H, d, J = 7.4 Hz).
MS: m/z = 262 [M+H-HCl] +
Cl content (ion chromatography): 11.9 % (in theory: 11.9 %).
[0137]
Step B. Preparation of S-MACB-HC (Compound [6])
[Chem. 7]
 To a solution of S-MABB-HC [5] (5.0 g, 16.8 mmol) in methanol (15.0 mL) was added 5 % palladium carbon (made by Kawaken Fine Chemicals Co., Ltd., PH type, 54.1 % water-content 1.0 g) at room temperature under nitrogen atmosphere. The reaction vessel was filled with hydrogen, the reaction mixture was stirred at hydrogen pressure of 0.4 MPa at room temperature for 12 hours, the hydrogen in the reaction vessel was replaced with nitrogen, and then the 5 % palladium carbon was removed by filtration. The reaction vessel and the 5 % palladium carbon were washed with methanol (10 mL). The washings were added to the filtrate to give a methanol solution of S-MACB-HC [6] (24.8 g, 16.8 mmol in theory). The given methanol solution of S-MACB-HC was used in the next step, assuming that the yield was 100 %.
To a solution of S-MABB-HC [5] (5.0 g, 16.8 mmol) in methanol (15.0 mL) was added 5 % palladium carbon (made by Kawaken Fine Chemicals Co., Ltd., PH type, 54.1 % water-content 1.0 g) at room temperature under nitrogen atmosphere. The reaction vessel was filled with hydrogen, the reaction mixture was stirred at hydrogen pressure of 0.4 MPa at room temperature for 12 hours, the hydrogen in the reaction vessel was replaced with nitrogen, and then the 5 % palladium carbon was removed by filtration. The reaction vessel and the 5 % palladium carbon were washed with methanol (10 mL). The washings were added to the filtrate to give a methanol solution of S-MACB-HC [6] (24.8 g, 16.8 mmol in theory). The given methanol solution of S-MACB-HC was used in the next step, assuming that the yield was 100 %.
A crude product of S-MACB-HC which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 9.60 (br s, 1H), 4.97 (d, 1H, J = 9.2 Hz), 4.61 (d, 1H, J = 8.4 Hz), 4.01 (dd, 1H, J = 10.0, 8.4 Hz), 3.78-3.74 (m, 1H), 3.54 (dd, 1H, J = 9.6, 8.4 Hz), 3.35 (dd, 1H, J = 10.0, 6.0 Hz), 3.15-3.03 (m, 1H), 3.00-2.88 (m, 1H), 1.49 (s, 9H), 1.47 (s, 9H), 1.22 (d, 3H, J = 6.8 Hz), 1.14 (d, 3H, J = 7.2 Hz).
MS: m/z = 172 [M+H] + (free form)
[Chem. 7]
A crude product of S-MACB-HC which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 9.60 (br s, 1H), 4.97 (d, 1H, J = 9.2 Hz), 4.61 (d, 1H, J = 8.4 Hz), 4.01 (dd, 1H, J = 10.0, 8.4 Hz), 3.78-3.74 (m, 1H), 3.54 (dd, 1H, J = 9.6, 8.4 Hz), 3.35 (dd, 1H, J = 10.0, 6.0 Hz), 3.15-3.03 (m, 1H), 3.00-2.88 (m, 1H), 1.49 (s, 9H), 1.47 (s, 9H), 1.22 (d, 3H, J = 6.8 Hz), 1.14 (d, 3H, J = 7.2 Hz).
MS: m/z = 172 [M+H] + (free form)
[0138]
Step C. Preparation of S-ZMAB (Compound [7])
[Chem. 8]
 To the methanol solution of S-MACB-HC [6] (24.8 g, 16.8 mmol in theory) was added dropwise N,N-diisopropylethylamine (4.8 g, 36.9 mmol) at room temperature under nitrogen atmosphere, and then the used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. To the resulting reaction mixture was added dropwise benzyl chloroformate (3.0 g, 17.6 mmol) at 0°C, and then the used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 0°C for 1 hour, and then the solvent was removed in vacuo. After toluene (25.0 mL) and an aqueous solution of citric acid (25.0 mL) was added to the residue and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with sodium bicarbonate water (25.0 mL) and then water (25.0 mL), and the solvent in the organic layer was removed out of the organic layer in vacuo. Toluene (15.0 mL) was added to the residue and the toluene solution was concentrated. The operation was repeated one more time to give a toluene solution of S-ZMAB [7] (6.9 g, 16.8 mmol in theory). The given toluene solution of S-ZMAB was used in the next step, assuming that the yield was 100 %.
To the methanol solution of S-MACB-HC [6] (24.8 g, 16.8 mmol in theory) was added dropwise N,N-diisopropylethylamine (4.8 g, 36.9 mmol) at room temperature under nitrogen atmosphere, and then the used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. To the resulting reaction mixture was added dropwise benzyl chloroformate (3.0 g, 17.6 mmol) at 0°C, and then the used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 0°C for 1 hour, and then the solvent was removed in vacuo. After toluene (25.0 mL) and an aqueous solution of citric acid (25.0 mL) was added to the residue and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with sodium bicarbonate water (25.0 mL) and then water (25.0 mL), and the solvent in the organic layer was removed out of the organic layer in vacuo. Toluene (15.0 mL) was added to the residue and the toluene solution was concentrated. The operation was repeated one more time to give a toluene solution of S-ZMAB [7] (6.9 g, 16.8 mmol in theory). The given toluene solution of S-ZMAB was used in the next step, assuming that the yield was 100 %.
A crude product of S-ZMAB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (CDCl 3) δ: 7.38-7.28 (m, 10H), 5.16-5.04 (m, 4H), 4.60 (d, 1H, J = 9.2 Hz), 4.18-4.12 (m, 2H), 4.04 (t, 1H, J = 8.6 Hz), 3.66 (dd, 1H, J = 7.6, 7.2 Hz), 3.50 (dd, 1H, J = 8.0, 5.2 Hz), 3.05-2.94 (m, 1H), 2.60-2.50 (m, 1H), 1.43 (br s, 18H), 1.33 (d, 3H, J = 6.5 Hz), 1.15 (d, 3H, J = 7.2 Hz).
MS: m/z = 328 [M+Na] +.
[Chem. 8]
A crude product of S-ZMAB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (CDCl 3) δ: 7.38-7.28 (m, 10H), 5.16-5.04 (m, 4H), 4.60 (d, 1H, J = 9.2 Hz), 4.18-4.12 (m, 2H), 4.04 (t, 1H, J = 8.6 Hz), 3.66 (dd, 1H, J = 7.6, 7.2 Hz), 3.50 (dd, 1H, J = 8.0, 5.2 Hz), 3.05-2.94 (m, 1H), 2.60-2.50 (m, 1H), 1.43 (br s, 18H), 1.33 (d, 3H, J = 6.5 Hz), 1.15 (d, 3H, J = 7.2 Hz).
MS: m/z = 328 [M+Na] +.
[0139]
Step D. Preparation of RS-ZMBB (Compound [8])
[Chem. 9]
 To the toluene solution of S-ZMAB [7] (6.9 g, 16.8 mmol) was added tetrahydrofuran (15.0 mL) at room temperature under nitrogen atmosphere. A solution of lithium bis(trimethylsilyl)amide/tetrahydrofuran (14.7 mL, 17.6 mmol) was added dropwise to the toluene solution at -70°C. The used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at -70°C for 6 hours, and then a solution of TBBA (3.4 g, 17.6 mmol) in tetrahydrofuran (2.5 mL) was added dropwise to the reaction mixture at -70°C. The used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at -70°C for 1 hour, and then warmed to room temperature. To the reaction mixture were added an aqueous ammonium chloride (25 mL) and toluene (25 mL) and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with an aqueous solution of citric acid (25 mL, x 2), sodium bicarbonate water (25 mL), and then water (25 mL), and then the solvent was removed out of the organic layer in vacuo. Acetonitrile (15 mL) was added to the residue and the acetonitrile solution was concentrated. The operation was repeated two more times. Acetonitrile (15 mL) and active carbon (0.25 g) were added to the residue, the mixture was stirred at room temperature for 2 hours. The active carbon was removed by filtration, and the reaction vessel and the residue on the filter was washed with acetonitrile (10 mL). The washings were added to the filtration, and then the filtration was concentrated in vacuo to give an acetonitrile solution of RS-ZMBB [8] (13.2 g, 16.8 mmol in theory). The given acetonitrile solution of RS-ZMBB was used in the next step, assuming that the yield was 100 %.
To the toluene solution of S-ZMAB [7] (6.9 g, 16.8 mmol) was added tetrahydrofuran (15.0 mL) at room temperature under nitrogen atmosphere. A solution of lithium bis(trimethylsilyl)amide/tetrahydrofuran (14.7 mL, 17.6 mmol) was added dropwise to the toluene solution at -70°C. The used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at -70°C for 6 hours, and then a solution of TBBA (3.4 g, 17.6 mmol) in tetrahydrofuran (2.5 mL) was added dropwise to the reaction mixture at -70°C. The used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at -70°C for 1 hour, and then warmed to room temperature. To the reaction mixture were added an aqueous ammonium chloride (25 mL) and toluene (25 mL) and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with an aqueous solution of citric acid (25 mL, x 2), sodium bicarbonate water (25 mL), and then water (25 mL), and then the solvent was removed out of the organic layer in vacuo. Acetonitrile (15 mL) was added to the residue and the acetonitrile solution was concentrated. The operation was repeated two more times. Acetonitrile (15 mL) and active carbon (0.25 g) were added to the residue, the mixture was stirred at room temperature for 2 hours. The active carbon was removed by filtration, and the reaction vessel and the residue on the filter was washed with acetonitrile (10 mL). The washings were added to the filtration, and then the filtration was concentrated in vacuo to give an acetonitrile solution of RS-ZMBB [8] (13.2 g, 16.8 mmol in theory). The given acetonitrile solution of RS-ZMBB was used in the next step, assuming that the yield was 100 %.
A crude product of RS-ZMBB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.38-7.29 (m, 5H), 5.09-4.96 (m, 2H), 3.91 (t, 0.4H, J = 8.0 Hz), 3.79 (t, 0.6H, J = 8.0 Hz), 3.55 (t, 0.4H, J = 7.2 Hz), 3.46 (t, 0.6H, J = 7.5 Hz), 3.14-3.04 (m, 1H), 2.83-2.72 (m, 2H), 1.38 (br s, 9H), 1.37 (br s, 3.6H), 1.34 (br s, 5.4H), 1.12-1.09 (m, 3H).
MS: m/z = 420 [M+H] +.
[Chem. 9]
A crude product of RS-ZMBB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.38-7.29 (m, 5H), 5.09-4.96 (m, 2H), 3.91 (t, 0.4H, J = 8.0 Hz), 3.79 (t, 0.6H, J = 8.0 Hz), 3.55 (t, 0.4H, J = 7.2 Hz), 3.46 (t, 0.6H, J = 7.5 Hz), 3.14-3.04 (m, 1H), 2.83-2.72 (m, 2H), 1.38 (br s, 9H), 1.37 (br s, 3.6H), 1.34 (br s, 5.4H), 1.12-1.09 (m, 3H).
MS: m/z = 420 [M+H] +.
[0140]
Step E. Preparation of RS-ZMAA-DN . 2H 2 O (Compound [9])
[Chem. 10]
 To the acetonitrile solution of RS-ZMBB [8] (13.2 g, 16.8 mmol in theory) was added acetonitrile (15 mL) at room temperature under nitrogen atmosphere. p-Toluenesulfonic acid mono-hydrate (6.4 g, 33.6 mmol) was added to the solution at room temperature. The reaction mixture was stirred at 50°C for 12 hours, and then cooled to room temperature, and water (7.5 mL) was added dropwise to the reaction mixture. The reaction mixture was cooled to 0°C, and then 4 mol/L aqueous sodium hydroxide (17.6 mL, 70.5 mmol) was added dropwise thereto. After stirring the reaction mixture at room temperature for 1 hour, acetonitrile (75 mL) was added dropwise thereto at room temperature, and the reaction mixture was stirred for 3 hours. The precipitated solid was collected on a filter, and washed with a mixture of acetonitrile : water = 4 : 1 (10 mL) and then acetonitrile (10 mL). The resulting wet solid was dried in vacuo to give RS-ZMAA-DN .2H 2O [9] (5.2 g, 13.4 mmol, yield: 85.4 %).
To the acetonitrile solution of RS-ZMBB [8] (13.2 g, 16.8 mmol in theory) was added acetonitrile (15 mL) at room temperature under nitrogen atmosphere. p-Toluenesulfonic acid mono-hydrate (6.4 g, 33.6 mmol) was added to the solution at room temperature. The reaction mixture was stirred at 50°C for 12 hours, and then cooled to room temperature, and water (7.5 mL) was added dropwise to the reaction mixture. The reaction mixture was cooled to 0°C, and then 4 mol/L aqueous sodium hydroxide (17.6 mL, 70.5 mmol) was added dropwise thereto. After stirring the reaction mixture at room temperature for 1 hour, acetonitrile (75 mL) was added dropwise thereto at room temperature, and the reaction mixture was stirred for 3 hours. The precipitated solid was collected on a filter, and washed with a mixture of acetonitrile : water = 4 : 1 (10 mL) and then acetonitrile (10 mL). The resulting wet solid was dried in vacuo to give RS-ZMAA-DN .2H 2O [9] (5.2 g, 13.4 mmol, yield: 85.4 %).
RS-ZMAA-DN .2H 2O which was prepared by the same process was measured about NMR, MS, Na-content, and water-content.
1H-NMR (DMSO-d 6) δ: 7.32-7.22 (m, 5H), 4.97 (d, 1H, J = 12.7 Hz), 4.84 (d, 1H, J = 12.7 Hz), 3.79 (t, 1H, J = 8.0 Hz), 3.29 (d, 1H, J = 14.8 Hz), 3.16-3.12 (m, 1H), 2.17-2.09 (m, 2H), 1.07 (d, 3H, J = 6.9 Hz).
MS: m/z = 352 [M+H] + (anhydrate)
Na content (ion chromatography): 13.3 % (after correction of water content)(13.1 % in theory)
Water content (Karl Fischer’s method): 9.8 % (9.3 % in theory)
[Chem. 10]
RS-ZMAA-DN .2H 2O which was prepared by the same process was measured about NMR, MS, Na-content, and water-content.
1H-NMR (DMSO-d 6) δ: 7.32-7.22 (m, 5H), 4.97 (d, 1H, J = 12.7 Hz), 4.84 (d, 1H, J = 12.7 Hz), 3.79 (t, 1H, J = 8.0 Hz), 3.29 (d, 1H, J = 14.8 Hz), 3.16-3.12 (m, 1H), 2.17-2.09 (m, 2H), 1.07 (d, 3H, J = 6.9 Hz).
MS: m/z = 352 [M+H] + (anhydrate)
Na content (ion chromatography): 13.3 % (after correction of water content)(13.1 % in theory)
Water content (Karl Fischer’s method): 9.8 % (9.3 % in theory)
[0141]
Step F. Preparation of RS-ZMAA (Compound [10])
[Chem. 11]
 To 1 mol/L hydrochloric acid (180 mL) were added RS-ZMAA-DN .2H 2O [9] (30 g, 77.5 mmol) and acetonitrile (60 mL), and the mixture was stirred at room temperature for about 15 minutes. After ethyl acetate (240 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The organic layer was washed with 10 % brine (60 mL x 2). The organic layer was stirred with magnesium sulfate (6 g), the magnesium sulfate was removed by filtration, and the residue on the filter was washed with ethyl acetate (60 mL). The filtrate and the washings are combined, and the solvent was removed out in vacuo. Tetrahydrofuran (240 mL) was added to the residue and the tetrahydrofuran solution was concentrated. The operation was repeated two more times. Tetrahydrofuran (60 mL) was added to the residue to give a tetrahydrofuran solution of RS-ZMAA [10]. The given tetrahydrofuran solution of RS-ZMAA was used in the next step, assuming that the yield was 100 %.
To 1 mol/L hydrochloric acid (180 mL) were added RS-ZMAA-DN .2H 2O [9] (30 g, 77.5 mmol) and acetonitrile (60 mL), and the mixture was stirred at room temperature for about 15 minutes. After ethyl acetate (240 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The organic layer was washed with 10 % brine (60 mL x 2). The organic layer was stirred with magnesium sulfate (6 g), the magnesium sulfate was removed by filtration, and the residue on the filter was washed with ethyl acetate (60 mL). The filtrate and the washings are combined, and the solvent was removed out in vacuo. Tetrahydrofuran (240 mL) was added to the residue and the tetrahydrofuran solution was concentrated. The operation was repeated two more times. Tetrahydrofuran (60 mL) was added to the residue to give a tetrahydrofuran solution of RS-ZMAA [10]. The given tetrahydrofuran solution of RS-ZMAA was used in the next step, assuming that the yield was 100 %.
RS-ZMAA which was prepared by the same process was measured about NMR and MS.
1H-NMR (DMSO-D 6) δ: 7.35-7.28 (m, 5H), 5.06-4.94 (m, 2H), 3.86 (dt, 1H, J = 48.4, 7.9 Hz), 3.50 (dt, 1H, J = 37.9, 7.4 Hz), 3.16-3.02 (br m, 1H), 2.91-2.77 (br m, 2H), 1.08 (d, 3H, J = 6.9 Hz)
MS: m/z = 308 [M+H] +.
[Chem. 11]
RS-ZMAA which was prepared by the same process was measured about NMR and MS.
1H-NMR (DMSO-D 6) δ: 7.35-7.28 (m, 5H), 5.06-4.94 (m, 2H), 3.86 (dt, 1H, J = 48.4, 7.9 Hz), 3.50 (dt, 1H, J = 37.9, 7.4 Hz), 3.16-3.02 (br m, 1H), 2.91-2.77 (br m, 2H), 1.08 (d, 3H, J = 6.9 Hz)
MS: m/z = 308 [M+H] +.
[0142]
Step G. Preparation of RS-ZMOO (Compound [11])
[Chem. 12]
 To the tetrahydrofuran solution of RS-ZMAA [10] (25.8 mmol in theory) was added tetrahydrofuran (50 mL) under nitrogen atmosphere. Boron trifluoride etherate complex (4.40 g) was added dropwise thereto at 0°C to 5°C. The used dropping funnel was washed with tetrahydrofuran (5 mL) and the washings were added to the reaction mixture. To the reaction mixture was added dropwise 1.2 mol/L borane-tetrahydrofuran complex (43.0 mL) at 0°C to 5°C, and the reaction mixture was stirred at 0°C to 5°C for about 30 minutes, and then further stirred at room temperature overnight. To the reaction mixture was added dropwise 1.2 mol/L borane-tetrahydrofuran complex (21.1 mL) at 0°C to 5°C, and then the reaction mixture was stirred at room temperature overnight. After stirring, water (40 mL) was added dropwise to the reaction mixture at 0°C to 15°C. To the reaction mixture was added sodium bicarbonate (5.42 g) at 0°C to 15°C. The sodium bicarbonate left in the vessel was washed with water (10 mL), and the washings were added to the reaction mixture. The reaction mixture was stirred at room temperature for 2 hours, and then toluene (50 mL) was added thereto and the reaction mixture was further stirred. The organic layer was separated out. The resulting organic layer was washed with 10 % brine (20 mL x 1), a mixture (x 3) of 5 % sodium bicarbonate water (20 mL) and 10 % brine (20 mL), a mixture (x 1) of 5 % aqueous potassium hydrogensulfate (10 mL) and 10 % brine (10 mL), and then 10 % brine (20 mL x 2). The organic layer was stirred with magnesium sulfate (8.9 g), the magnesium sulfate was removed by filtration, and the residue on the filter was washed with toluene (20 mL). The washings were added to the filtration, and then the filtrate was concentrated in vacuo. To the concentrated residue was added toluene (80 mL). The solution was concentrated in vacuo, and toluene (15 mL) was added thereto to give a toluene solution of RS-ZMOO [11]. The given toluene solution of RS-ZMOO was used in the next step, assuming that the yield was 100 %.
To the tetrahydrofuran solution of RS-ZMAA [10] (25.8 mmol in theory) was added tetrahydrofuran (50 mL) under nitrogen atmosphere. Boron trifluoride etherate complex (4.40 g) was added dropwise thereto at 0°C to 5°C. The used dropping funnel was washed with tetrahydrofuran (5 mL) and the washings were added to the reaction mixture. To the reaction mixture was added dropwise 1.2 mol/L borane-tetrahydrofuran complex (43.0 mL) at 0°C to 5°C, and the reaction mixture was stirred at 0°C to 5°C for about 30 minutes, and then further stirred at room temperature overnight. To the reaction mixture was added dropwise 1.2 mol/L borane-tetrahydrofuran complex (21.1 mL) at 0°C to 5°C, and then the reaction mixture was stirred at room temperature overnight. After stirring, water (40 mL) was added dropwise to the reaction mixture at 0°C to 15°C. To the reaction mixture was added sodium bicarbonate (5.42 g) at 0°C to 15°C. The sodium bicarbonate left in the vessel was washed with water (10 mL), and the washings were added to the reaction mixture. The reaction mixture was stirred at room temperature for 2 hours, and then toluene (50 mL) was added thereto and the reaction mixture was further stirred. The organic layer was separated out. The resulting organic layer was washed with 10 % brine (20 mL x 1), a mixture (x 3) of 5 % sodium bicarbonate water (20 mL) and 10 % brine (20 mL), a mixture (x 1) of 5 % aqueous potassium hydrogensulfate (10 mL) and 10 % brine (10 mL), and then 10 % brine (20 mL x 2). The organic layer was stirred with magnesium sulfate (8.9 g), the magnesium sulfate was removed by filtration, and the residue on the filter was washed with toluene (20 mL). The washings were added to the filtration, and then the filtrate was concentrated in vacuo. To the concentrated residue was added toluene (80 mL). The solution was concentrated in vacuo, and toluene (15 mL) was added thereto to give a toluene solution of RS-ZMOO [11]. The given toluene solution of RS-ZMOO was used in the next step, assuming that the yield was 100 %.
RS-ZMOO which was prepared by the same process was measured about NMR and MS.
1H-NMR (CDCl 3) δ: 7.39-7.30 (m, 5H), 5.10 (s, 2H), 4.15-4.01 (br m, 2H), 3.83-3.73 (br m, 3H), 3.48 (dd, 1H, J = 8.3, 6.4 Hz), 2.59-2.50 (br m, 1H), 2.46-2.40 (br m, 1H), 2.07-1.99 (m, 1H), 1.14 (d, 3H, J = 7.2 Hz)
MS: m/z = 280 [M+H]+.
[Chem. 12]
RS-ZMOO which was prepared by the same process was measured about NMR and MS.
1H-NMR (CDCl 3) δ: 7.39-7.30 (m, 5H), 5.10 (s, 2H), 4.15-4.01 (br m, 2H), 3.83-3.73 (br m, 3H), 3.48 (dd, 1H, J = 8.3, 6.4 Hz), 2.59-2.50 (br m, 1H), 2.46-2.40 (br m, 1H), 2.07-1.99 (m, 1H), 1.14 (d, 3H, J = 7.2 Hz)
MS: m/z = 280 [M+H]+.
[0143]
Step H. Preparation of RS-ZMSS (Compound [12])
[Chem. 13]
 To the toluene solution of RS-ZMOO [11] (23.7 mmol in theory) was added toluene (55 mL) under nitrogen atmosphere. And, triethylamine (5.27 g) was added dropwise thereto at -10°C to 10°C, and the used dropping funnel was washed with toluene (1.8 mL) and the washings were added to the reaction mixture. To this reaction mixture was added dropwise methanesulfonyl chloride (5.69 g) at -10°C to 10°C, and then the used dropping funnel was washed with toluene (1.8 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 0°C to 10°C for about 2 hours, and then water (28 mL) was added dropwise thereto at 0°C to 20°C. The reaction mixture was stirred at 0°C to 20°C for about 30 minutes, and then, the organic layer was separated out. The resulting organic layer was washed twice with 10 % brine (18 mL). The organic layer was stirred with magnesium sulfate (2.75 g), the magnesium sulfate was removed by filtration, and the residue on the filter was washed with toluene (18 mL). The washings were added to the filtrate, and then the solvent was removed from the filtrate in vacuo. To the concentrated residue was added toluene up to 18 mL to give a toluene solution of RS-ZMSS [12]. The given toluene solution of RS-ZMSS was used in the next step, assuming that the yield was 100 %.
To the toluene solution of RS-ZMOO [11] (23.7 mmol in theory) was added toluene (55 mL) under nitrogen atmosphere. And, triethylamine (5.27 g) was added dropwise thereto at -10°C to 10°C, and the used dropping funnel was washed with toluene (1.8 mL) and the washings were added to the reaction mixture. To this reaction mixture was added dropwise methanesulfonyl chloride (5.69 g) at -10°C to 10°C, and then the used dropping funnel was washed with toluene (1.8 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 0°C to 10°C for about 2 hours, and then water (28 mL) was added dropwise thereto at 0°C to 20°C. The reaction mixture was stirred at 0°C to 20°C for about 30 minutes, and then, the organic layer was separated out. The resulting organic layer was washed twice with 10 % brine (18 mL). The organic layer was stirred with magnesium sulfate (2.75 g), the magnesium sulfate was removed by filtration, and the residue on the filter was washed with toluene (18 mL). The washings were added to the filtrate, and then the solvent was removed from the filtrate in vacuo. To the concentrated residue was added toluene up to 18 mL to give a toluene solution of RS-ZMSS [12]. The given toluene solution of RS-ZMSS was used in the next step, assuming that the yield was 100 %.
RS-ZMSS which was prepared by the same process was measured by NMR and MS.
1H-NMR (DMSO-D 6) δ: 7.37-7.27 (br m, 5H), 5.10-4.98 (m, 2H), 4.58-4.22 (br m, 4H), 3.84 (dt, 1H, J = 45.6, 8.1 Hz), 3.48-3.33 (br m, 1H), 3.17-3.10 (m, 6H), 2.81-2.74 (br m, 1H), 2.22-2.12 (m, 2H)
MS: m/z = 436 [M+H] +.
[Chem. 13]
RS-ZMSS which was prepared by the same process was measured by NMR and MS.
1H-NMR (DMSO-D 6) δ: 7.37-7.27 (br m, 5H), 5.10-4.98 (m, 2H), 4.58-4.22 (br m, 4H), 3.84 (dt, 1H, J = 45.6, 8.1 Hz), 3.48-3.33 (br m, 1H), 3.17-3.10 (m, 6H), 2.81-2.74 (br m, 1H), 2.22-2.12 (m, 2H)
MS: m/z = 436 [M+H] +.
[0144]
Step I. Preparation of SR-ZMDB (Compound [13])
[Chem. 14]
 To a toluene solution of RS-ZMSS [12] (23.7 mmol in theory) was added toluene (55 mL) under nitrogen atmosphere. And, benzylamine (17.8 g) was added dropwise thereto at room temperature, and the used dropping funnel was washed with toluene (9.2 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at room temperature for about 1 hour, at 55°C to 65°C for about 3 hours, and then at 70°C to 80°C for 6 hours. After the reaction mixture was cooled to room temperature, 10 % NaCl (28 mL) was added dropwise thereto, and the reaction mixture was stirred at room temperature for about 30 minutes. After toluene (37 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with a mixture (x 2) of 10 % brine (18 mL) and acetic acid (2.84 g), and then 10 % brine (11 mL, x 1). The solvent of the organic layer was removed in vacuo to a half volume, and acetic anhydride (1.45 g) was added to the concentrated residue at room temperature. The mixture was stirred for about 3 hours. To the reaction mixture were added dropwise a solution of potassium hydrogensulfate (3.87 g) and water (92 mL) at room temperature. The reaction mixture was stirred, and then the aqueous layer was separated out. The resulting aqueous layer was washed with toluene (18 mL), and toluene (73 mL) and then sodium bicarbonate (6.56 g) were added to the aqueous layer at room temperature, and the mixture was stirred. The organic layer was separated out, and washed with 10 % brine (11 mL). The organic layer was stirred with magnesium sulfate (2.75 g), the magnesium sulfate was removed by filtration. The residue on the filter was washed with toluene (18 mL), and the washings were added to the filtrate, and then the filtrate was concentrated in vacuo. Toluene (44 mL) was added to the concentrated residue to give a toluene solution of SR-ZMDB [13]. The given toluene solution of SR-ZMDB was used in the next step, assuming that the yield was 100 %.
To a toluene solution of RS-ZMSS [12] (23.7 mmol in theory) was added toluene (55 mL) under nitrogen atmosphere. And, benzylamine (17.8 g) was added dropwise thereto at room temperature, and the used dropping funnel was washed with toluene (9.2 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at room temperature for about 1 hour, at 55°C to 65°C for about 3 hours, and then at 70°C to 80°C for 6 hours. After the reaction mixture was cooled to room temperature, 10 % NaCl (28 mL) was added dropwise thereto, and the reaction mixture was stirred at room temperature for about 30 minutes. After toluene (37 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with a mixture (x 2) of 10 % brine (18 mL) and acetic acid (2.84 g), and then 10 % brine (11 mL, x 1). The solvent of the organic layer was removed in vacuo to a half volume, and acetic anhydride (1.45 g) was added to the concentrated residue at room temperature. The mixture was stirred for about 3 hours. To the reaction mixture were added dropwise a solution of potassium hydrogensulfate (3.87 g) and water (92 mL) at room temperature. The reaction mixture was stirred, and then the aqueous layer was separated out. The resulting aqueous layer was washed with toluene (18 mL), and toluene (73 mL) and then sodium bicarbonate (6.56 g) were added to the aqueous layer at room temperature, and the mixture was stirred. The organic layer was separated out, and washed with 10 % brine (11 mL). The organic layer was stirred with magnesium sulfate (2.75 g), the magnesium sulfate was removed by filtration. The residue on the filter was washed with toluene (18 mL), and the washings were added to the filtrate, and then the filtrate was concentrated in vacuo. Toluene (44 mL) was added to the concentrated residue to give a toluene solution of SR-ZMDB [13]. The given toluene solution of SR-ZMDB was used in the next step, assuming that the yield was 100 %.
1H-NMR (CDCl 3) δ: 7.35-7.20 (m, 10H), 5.08 (d, 2H, J = 23.6 Hz), 3.94 (q, 1H, J = 7.9 Hz), 3.73-3.42 (br m, 2H), 3.30-3.23 (m, 1H), 3.05 (dd, 1H, J = 19.7, 9.5 Hz), 2.79 (dt, 1H, J = 69.6, 6.1 Hz), 2.57-2.32 (br m, 4H), 1.96-1.89 (m, 1H), 1.09 (d, 3H, J = 6.9 Hz)
MS: m/z = 351 [M+H] +.
[Chem. 14]
1H-NMR (CDCl 3) δ: 7.35-7.20 (m, 10H), 5.08 (d, 2H, J = 23.6 Hz), 3.94 (q, 1H, J = 7.9 Hz), 3.73-3.42 (br m, 2H), 3.30-3.23 (m, 1H), 3.05 (dd, 1H, J = 19.7, 9.5 Hz), 2.79 (dt, 1H, J = 69.6, 6.1 Hz), 2.57-2.32 (br m, 4H), 1.96-1.89 (m, 1H), 1.09 (d, 3H, J = 6.9 Hz)
MS: m/z = 351 [M+H] +.
[0145]
Step J. Preparation of SR-MDOZ (Compound [14])
[Chem. 15]
 To a solution of 1-chloroethyl chloroformate (3.72 g) in toluene (28 mL) was added dropwise the toluene solution of SR-ZMDB [13] (23.7 mmol in theory) at 0°C to 10°C under nitrogen atmosphere, and then the used dropping funnel was washed with toluene (4.6 mL) and the washings were added to the reaction mixture. To the reaction mixture was added triethylamine (718 mg) at 0°C to 10°C, and the reaction mixture was stirred at 15°C to 25°C for about 2 hours. Then, methyl alcohol (46 mL) was added to the reaction mixture, and the mixture was stirred at 50°C to 60°C for additional about 2 hours. The solvent of the reaction mixture was removed in vacuo to a volume of about less than 37 mL. To the concentrated residue was added dropwise 2 mol/L hydrochloric acid (46 mL) at 15°C to 20°C, and the mixture was stirred, and the aqueous layer was separated out. The resulting aqueous layer was washed with toluene (28 mL, x 2). To the aqueous layer were added 20 % brine (46 mL) and tetrahydrofuran (92 mL), and then 8 mol/L aqueous sodium hydroxide (18 mL) was added dropwise thereto at 0°C to 10°C. The organic layer was separated out from the reaction mixture, washed with 20 % brine (18 mL, x 2), and then the solvent of the organic layer was removed in vacuo. To the concentrated residue was added tetrahydrofuran (92 mL), and the solution was concentrated in vacuo. The operation was repeated one more time. The concentrated residue was dissolved in tetrahydrofuran (92 mL). The solution was stirred with magnesium sulfate (2.75 g), and the magnesium sulfate was removed by filtration. The residue on the filter was washed with tetrahydrofuran (28 mL), the washings were added to the filtrate, and the filtrate was concentrated in vacuo. The volume of the concentrated residue was adjusted to about 20 mL with tetrahydrofuran to give a tetrahydrofuran solution of SR-MDOZ [14] (net weight: 4.01 g, 15.4 mol, yield: 65.0 %).
To a solution of 1-chloroethyl chloroformate (3.72 g) in toluene (28 mL) was added dropwise the toluene solution of SR-ZMDB [13] (23.7 mmol in theory) at 0°C to 10°C under nitrogen atmosphere, and then the used dropping funnel was washed with toluene (4.6 mL) and the washings were added to the reaction mixture. To the reaction mixture was added triethylamine (718 mg) at 0°C to 10°C, and the reaction mixture was stirred at 15°C to 25°C for about 2 hours. Then, methyl alcohol (46 mL) was added to the reaction mixture, and the mixture was stirred at 50°C to 60°C for additional about 2 hours. The solvent of the reaction mixture was removed in vacuo to a volume of about less than 37 mL. To the concentrated residue was added dropwise 2 mol/L hydrochloric acid (46 mL) at 15°C to 20°C, and the mixture was stirred, and the aqueous layer was separated out. The resulting aqueous layer was washed with toluene (28 mL, x 2). To the aqueous layer were added 20 % brine (46 mL) and tetrahydrofuran (92 mL), and then 8 mol/L aqueous sodium hydroxide (18 mL) was added dropwise thereto at 0°C to 10°C. The organic layer was separated out from the reaction mixture, washed with 20 % brine (18 mL, x 2), and then the solvent of the organic layer was removed in vacuo. To the concentrated residue was added tetrahydrofuran (92 mL), and the solution was concentrated in vacuo. The operation was repeated one more time. The concentrated residue was dissolved in tetrahydrofuran (92 mL). The solution was stirred with magnesium sulfate (2.75 g), and the magnesium sulfate was removed by filtration. The residue on the filter was washed with tetrahydrofuran (28 mL), the washings were added to the filtrate, and the filtrate was concentrated in vacuo. The volume of the concentrated residue was adjusted to about 20 mL with tetrahydrofuran to give a tetrahydrofuran solution of SR-MDOZ [14] (net weight: 4.01 g, 15.4 mol, yield: 65.0 %).
SR-MDOZ which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (CDCl 3) δ: 7.37-7.28 (m, 5H), 5.08 (dd, 2H, J = 16.8, 12.8 Hz), 4.00 (dd, 1H, J = 17.1, 8.3 Hz), 3.40-3.31 (m, 1H), 3.24 (d, 1H, J = 12.7 Hz), 3.00 (dd, 1H, J = 54.9, 12.4 Hz), 2.87-2.57 (m, 3H), 2.47-2.27 (m, 1H), 1.91-1.80 (m, 1H), 1.14 (d, 3H, J = 7.2 Hz)
MS: m/z = 261 [M+H] +.
[Chem. 15]
SR-MDOZ which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (CDCl 3) δ: 7.37-7.28 (m, 5H), 5.08 (dd, 2H, J = 16.8, 12.8 Hz), 4.00 (dd, 1H, J = 17.1, 8.3 Hz), 3.40-3.31 (m, 1H), 3.24 (d, 1H, J = 12.7 Hz), 3.00 (dd, 1H, J = 54.9, 12.4 Hz), 2.87-2.57 (m, 3H), 2.47-2.27 (m, 1H), 1.91-1.80 (m, 1H), 1.14 (d, 3H, J = 7.2 Hz)
MS: m/z = 261 [M+H] +.
[0146]
Step K. Preparation of SR-MDOZ-OX (Compound [15])
[Chem. 16]
 Under nitrogen atmosphere, oxalic acid (761 mg) was dissolved in tetrahydrofuran (40 mL), and the tetrahydrofuran solution of SR-MDOZ [14] (3.84 mmol in theory) was added dropwise to the solution of oxalic acid at room temperature. To the solution was added SR-MDOZ-OX crystal (1 mg) that was prepared according to the method described herein at room temperature, and the mixture was stirred at room temperature for about 3.5 hours to precipitate the crystal. To the slurry solution was added dropwise the tetrahydrofuran solution of SR-MDOZ (3.84 mmol) at room temperature, and the mixture was stirred at room temperature for about 1 hour. The slurry solution was heated, and stirred at 50°C to 60°C for about 2 hours, and then stirred at room temperature overnight. The slurry solution was filtrated, and the wet crystal on the filter was washed with tetrahydrofuran (10 mL), dried in vacuo to give SR-MDOZ-OX [15] (2.32 g, 6.62 mol, yield: 86.2 %).
Under nitrogen atmosphere, oxalic acid (761 mg) was dissolved in tetrahydrofuran (40 mL), and the tetrahydrofuran solution of SR-MDOZ [14] (3.84 mmol in theory) was added dropwise to the solution of oxalic acid at room temperature. To the solution was added SR-MDOZ-OX crystal (1 mg) that was prepared according to the method described herein at room temperature, and the mixture was stirred at room temperature for about 3.5 hours to precipitate the crystal. To the slurry solution was added dropwise the tetrahydrofuran solution of SR-MDOZ (3.84 mmol) at room temperature, and the mixture was stirred at room temperature for about 1 hour. The slurry solution was heated, and stirred at 50°C to 60°C for about 2 hours, and then stirred at room temperature overnight. The slurry solution was filtrated, and the wet crystal on the filter was washed with tetrahydrofuran (10 mL), dried in vacuo to give SR-MDOZ-OX [15] (2.32 g, 6.62 mol, yield: 86.2 %).
SR-MDOZ-OX which was prepared by the same process was measured about NMR, MS, and elementary analysis.
1H-NMR (DMSO-D 6) δ: 7.37-7.30 (m, 5H), 5.15-5.01 (m, 2H), 3.92 (dt, 1H, J = 43.5, 8.4 Hz), 3.48-3.12 (br m, 5H), 2.67-2.56 (m, 1H), 2.46-2.35 (m, 1H), 2.12-2.05 (m, 1H), 1.13 (d, 3H, J = 6.9 Hz)
MS: m/z = 261 [M+H] +
elementary analysis: C 58.4wt % , H 6.4wt % , N 7.9 % wt % (theoretically, C 58.3wt % , H 6.3wt % , N 8.0wt % )
[Chem. 16]
SR-MDOZ-OX which was prepared by the same process was measured about NMR, MS, and elementary analysis.
1H-NMR (DMSO-D 6) δ: 7.37-7.30 (m, 5H), 5.15-5.01 (m, 2H), 3.92 (dt, 1H, J = 43.5, 8.4 Hz), 3.48-3.12 (br m, 5H), 2.67-2.56 (m, 1H), 2.46-2.35 (m, 1H), 2.12-2.05 (m, 1H), 1.13 (d, 3H, J = 6.9 Hz)
MS: m/z = 261 [M+H] +
elementary analysis: C 58.4wt % , H 6.4wt % , N 7.9 % wt % (theoretically, C 58.3wt % , H 6.3wt % , N 8.0wt % )
[0147]
Step L. Preparation of SR-MDPZ (Compound [16])
[Chem. 17]
 To SR-MDOZ-OX [15] (12.0 g, 34.2 mmol) were added ethanol (36 mL), water (72 mL), CPPY [20] (5.36 g, 34.9 mmol), and then K 3PO 4 (21.8 g, 103 mmol) under nitrogen atmosphere. The reaction mixture was stirred at 80°C for 5 hours, and then cooled to 40°C. Toluene (120 mL) was added thereto at 40°C, and the organic layer was separated out. The resulting organic layer was washed with 20 % aqueous potassium carbonate (48 mL), followed by washing twice with water (48 mL). The solvent of the organic layer was then removed in vacuo. tert-butanol (60 mL) was added to the residue and the tert-butanol solution was concentrated. The operation was repeated two more times. tert-Butanol (36 mL) was added to the concentrated residue to give a solution of SR-MDPZ [16] in tert-butanol (61.1 g, 34.2 mmol in theory). The given tert-butanol solution of SR-MDPZ was used in the next step, assuming that the yield was 100 %.
To SR-MDOZ-OX [15] (12.0 g, 34.2 mmol) were added ethanol (36 mL), water (72 mL), CPPY [20] (5.36 g, 34.9 mmol), and then K 3PO 4 (21.8 g, 103 mmol) under nitrogen atmosphere. The reaction mixture was stirred at 80°C for 5 hours, and then cooled to 40°C. Toluene (120 mL) was added thereto at 40°C, and the organic layer was separated out. The resulting organic layer was washed with 20 % aqueous potassium carbonate (48 mL), followed by washing twice with water (48 mL). The solvent of the organic layer was then removed in vacuo. tert-butanol (60 mL) was added to the residue and the tert-butanol solution was concentrated. The operation was repeated two more times. tert-Butanol (36 mL) was added to the concentrated residue to give a solution of SR-MDPZ [16] in tert-butanol (61.1 g, 34.2 mmol in theory). The given tert-butanol solution of SR-MDPZ was used in the next step, assuming that the yield was 100 %.
SR-MDPZ which was prepared by the same process was isolated as a solid from a mixture of ethyl acetate and n-heptane, and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.41-7.26 (br m, 3H), 7.22-7.08 (br m, 3H), 6.64-6.51 (br m, 1H), 5.07-4.91 (br m, 2H), 4.09-3.67 (br m, 5H), 3.47-3.32 (br m, 1H), 2.67-2.55 (br m, 2H), 2.21-2.15 (br m, 1H), 1.11 (d, 3H, J = 6.9 Hz).
MS: m/z = 378 [M+H] +
[Chem. 17]
SR-MDPZ which was prepared by the same process was isolated as a solid from a mixture of ethyl acetate and n-heptane, and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.41-7.26 (br m, 3H), 7.22-7.08 (br m, 3H), 6.64-6.51 (br m, 1H), 5.07-4.91 (br m, 2H), 4.09-3.67 (br m, 5H), 3.47-3.32 (br m, 1H), 2.67-2.55 (br m, 2H), 2.21-2.15 (br m, 1H), 1.11 (d, 3H, J = 6.9 Hz).
MS: m/z = 378 [M+H] +
[0148]
Step M. Preparation of SR-MDOP (Compound [17])
[Chem. 18]
 To the solution of SR-MDPZ [16] in tert-butanol (34.2 mmol in theory) were added ammonium formate (10.8 g, 171 mmol), water (60 mL), and 10 % palladium carbon (made by Kawaken Fine Chemicals Co., Ltd., M type, 52.6 % water-content, 1.20 g) under nitrogen atmosphere. The reaction mixture was stirred at 40°C for 13 hours, and then cooled to room temperature, and the resulting precipitate was removed by filtration. The reaction vessel and the residue on the filter were washed with tert-butanol (24 mL), the washings was added to the filtrate, and 8 M aqueous sodium hydroxide (25.7 mL, 205 mmol) and sodium chloride (13.2 g) were added to the filtrate. The reaction mixture was stirred at 50°C for 2 hours, and then toluene (84 mL) was added thereto at room temperature, and the organic layer was separated out. The resulting organic layer was washed with 20 % brine (60 mL), stirred with anhydrous sodium sulfate, and then the sodium sulfate was removed by filtration. The residue on the filter was washed with a mixture of toluene : tert-butanol = 1 : 1 (48 mL), the washings was added to the filtrate, and the filtrate was concentrated in vacuo. To the concentrated residue was added toluene (60 mL), and the solution was stirred at 50°C for 2 hours, and then the solvent was removed in vacuo. To the concentrated residue was added toluene (60 mL) again, and the solution was concentrated. To the concentrated residue was added toluene (48 mL), and the solution was stirred at room temperature for 1 hour, and then at ice temperature for 1 hour. The precipitated solid was collected on a filter, and washed with toluene (24 mL). The resulting wet solid was dried in vacuo to give SR-MDOP [17] (7.07 g, 29.1 mmol, yield: 84.8 %).
To the solution of SR-MDPZ [16] in tert-butanol (34.2 mmol in theory) were added ammonium formate (10.8 g, 171 mmol), water (60 mL), and 10 % palladium carbon (made by Kawaken Fine Chemicals Co., Ltd., M type, 52.6 % water-content, 1.20 g) under nitrogen atmosphere. The reaction mixture was stirred at 40°C for 13 hours, and then cooled to room temperature, and the resulting precipitate was removed by filtration. The reaction vessel and the residue on the filter were washed with tert-butanol (24 mL), the washings was added to the filtrate, and 8 M aqueous sodium hydroxide (25.7 mL, 205 mmol) and sodium chloride (13.2 g) were added to the filtrate. The reaction mixture was stirred at 50°C for 2 hours, and then toluene (84 mL) was added thereto at room temperature, and the organic layer was separated out. The resulting organic layer was washed with 20 % brine (60 mL), stirred with anhydrous sodium sulfate, and then the sodium sulfate was removed by filtration. The residue on the filter was washed with a mixture of toluene : tert-butanol = 1 : 1 (48 mL), the washings was added to the filtrate, and the filtrate was concentrated in vacuo. To the concentrated residue was added toluene (60 mL), and the solution was stirred at 50°C for 2 hours, and then the solvent was removed in vacuo. To the concentrated residue was added toluene (60 mL) again, and the solution was concentrated. To the concentrated residue was added toluene (48 mL), and the solution was stirred at room temperature for 1 hour, and then at ice temperature for 1 hour. The precipitated solid was collected on a filter, and washed with toluene (24 mL). The resulting wet solid was dried in vacuo to give SR-MDOP [17] (7.07 g, 29.1 mmol, yield: 84.8 %).
SR-MDOP which was prepared by the same process was measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.57 (br s, 1H), 8.07 (s, 1H), 7.10 (d, 1H, J = 3.2 Hz), 6.58 (d, 1H, J = 3.2 Hz), 3.92-3.59 (br m, 4H), 3.49 (dd, 1H, J = 8.3, 7.2 Hz), 2.93 (dd, 1H, J = 7.2, 6.1 Hz), 2.61-2.53 (m, 2H), 2.12-2.01 (br m, 2H), 1.10 (d, 3H, J = 6.9 Hz).
MS: m/z = 244 [M+H] +.
[Chem. 18]
SR-MDOP which was prepared by the same process was measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.57 (br s, 1H), 8.07 (s, 1H), 7.10 (d, 1H, J = 3.2 Hz), 6.58 (d, 1H, J = 3.2 Hz), 3.92-3.59 (br m, 4H), 3.49 (dd, 1H, J = 8.3, 7.2 Hz), 2.93 (dd, 1H, J = 7.2, 6.1 Hz), 2.61-2.53 (m, 2H), 2.12-2.01 (br m, 2H), 1.10 (d, 3H, J = 6.9 Hz).
MS: m/z = 244 [M+H] +.
[0149]
Step N. Preparation of Compound A mono-ethanolate (Compound [18])
[Chem. 19]
 Under nitrogen atmosphere, acetonitrile (60 mL) and triethylamine (416 mg, 4.11 mmol) were added to SR-MDOP [17] (5.00 g, 20.5 mmol), and to the solution was added dropwise a solution of DPCN [21] (3.69 g, 22.6 mmol) in acetonitrile (35 mL) at 45°C, and then the used dropping funnel was washed with acetonitrile (5.0 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 45°C for 3 hours, and then cooled to room temperature. After 5 % sodium bicarbonate water (25 mL), 10 % brine (25 mL), and ethyl acetate (50 mL) were added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The solvent of the organic layer was removed out in vacuo. Tetrahydrofuran (50 mL) was added to the residue and the tetrahydrofuran solution was concentrated. The operation was repeated three more times. To the concentrated residue was added tetrahydrofuran (50 mL), and water was added the solution to adjust the water content to 5.5 %. The resulting precipitate was removed by filtration. The reaction vessel and the residue on the filter were washed with tetrahydrofuran (15 mL), the washings were added to the filtrate, and the solvent was removed out of the filtrate in vacuo. To the concentrated residue were added ethanol (50 mL) and Compound A crystal (5.1 mg) that was prepared according to the method described in the following Example 15. The mixture was stirred at room temperature for 1 hour, and concentrated in vacuo. To the residue was ethanol (50 mL), and the solution was concentrated again. To the concentrated residue was added ethanol (15 mL), and the solution was stirred at room temperature for 1 hour. The precipitated solid was collected on the filter, and washed with ethanol (20 mL). The resulting wet solid was dried in vacuo to give Compound A mono-ethanolate [18] (6.26 g, 17.6 mmol, yield: 85.5 %).
Under nitrogen atmosphere, acetonitrile (60 mL) and triethylamine (416 mg, 4.11 mmol) were added to SR-MDOP [17] (5.00 g, 20.5 mmol), and to the solution was added dropwise a solution of DPCN [21] (3.69 g, 22.6 mmol) in acetonitrile (35 mL) at 45°C, and then the used dropping funnel was washed with acetonitrile (5.0 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 45°C for 3 hours, and then cooled to room temperature. After 5 % sodium bicarbonate water (25 mL), 10 % brine (25 mL), and ethyl acetate (50 mL) were added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The solvent of the organic layer was removed out in vacuo. Tetrahydrofuran (50 mL) was added to the residue and the tetrahydrofuran solution was concentrated. The operation was repeated three more times. To the concentrated residue was added tetrahydrofuran (50 mL), and water was added the solution to adjust the water content to 5.5 %. The resulting precipitate was removed by filtration. The reaction vessel and the residue on the filter were washed with tetrahydrofuran (15 mL), the washings were added to the filtrate, and the solvent was removed out of the filtrate in vacuo. To the concentrated residue were added ethanol (50 mL) and Compound A crystal (5.1 mg) that was prepared according to the method described in the following Example 15. The mixture was stirred at room temperature for 1 hour, and concentrated in vacuo. To the residue was ethanol (50 mL), and the solution was concentrated again. To the concentrated residue was added ethanol (15 mL), and the solution was stirred at room temperature for 1 hour. The precipitated solid was collected on the filter, and washed with ethanol (20 mL). The resulting wet solid was dried in vacuo to give Compound A mono-ethanolate [18] (6.26 g, 17.6 mmol, yield: 85.5 %).
Compound A mono-ethanolate which was prepared by the same process was measured by NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.11 (dd, 1H, J = 3.5, 2.3 Hz), 6.58 (dd, 1H, J = 3.5, 1.8 Hz), 4.34 (t, 1H, J = 5.1 Hz), 4.16 (t, 1H, J = 8.3 Hz), 4.09-3.92 (m, 3H), 3.84-3.73 (m, 1H), 3.71 (d, 1H, J = 19.0 Hz), 3.65 (d, 1H, J = 19.0 Hz), 3.58 (dd, 1H, J = 8.2, 5.9 Hz), 3.44 (dq, 2H, J = 6.7, 5.1 Hz), 2.69-2.60 (m, 2H), 2.23-2.13 (br m, 1H), 1.12 (d, 3H, J = 7.1 Hz), 1.06 (t, 3H, J = 6.7 Hz).
MS: m/z = 311 [M+H] +
[Chem. 19]
Compound A mono-ethanolate which was prepared by the same process was measured by NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.11 (dd, 1H, J = 3.5, 2.3 Hz), 6.58 (dd, 1H, J = 3.5, 1.8 Hz), 4.34 (t, 1H, J = 5.1 Hz), 4.16 (t, 1H, J = 8.3 Hz), 4.09-3.92 (m, 3H), 3.84-3.73 (m, 1H), 3.71 (d, 1H, J = 19.0 Hz), 3.65 (d, 1H, J = 19.0 Hz), 3.58 (dd, 1H, J = 8.2, 5.9 Hz), 3.44 (dq, 2H, J = 6.7, 5.1 Hz), 2.69-2.60 (m, 2H), 2.23-2.13 (br m, 1H), 1.12 (d, 3H, J = 7.1 Hz), 1.06 (t, 3H, J = 6.7 Hz).
MS: m/z = 311 [M+H] +
[0150]
Step O. Purification of Compound A (Compound [19])
[Chem. 20]
 Compound A mono-ethanolate [18] (4.00 g, 11.2 mmol) and n-butanol (32 mL) were mixed under nitrogen atmosphere, and the mixture was dissolved at 110°C. The mixture was cooled to 85°C, and Compound A crystal (4.0 mg) that was prepared according to the method described herein was added thereto, and the mixture was stirred at 85°C for 2 hours, at 75°C for 1 hour, and then at room temperature for 16 hours. The precipitated solid was collected on a filter, and washed with n-butanol (8.0 mL) and then ethyl acetate (8.0 mL). The resulting wet solid was dried in vacuo to give Compound A [19] (3.18 g, 10.2 mmol, yield: 91.3 %).
Compound A mono-ethanolate [18] (4.00 g, 11.2 mmol) and n-butanol (32 mL) were mixed under nitrogen atmosphere, and the mixture was dissolved at 110°C. The mixture was cooled to 85°C, and Compound A crystal (4.0 mg) that was prepared according to the method described herein was added thereto, and the mixture was stirred at 85°C for 2 hours, at 75°C for 1 hour, and then at room temperature for 16 hours. The precipitated solid was collected on a filter, and washed with n-butanol (8.0 mL) and then ethyl acetate (8.0 mL). The resulting wet solid was dried in vacuo to give Compound A [19] (3.18 g, 10.2 mmol, yield: 91.3 %).
Compound A which was prepared by the same process was measured by NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.11 (dd, 1H, J = 3.5, 2.5 Hz), 6.58 (dd, 1H, J = 3.5, 1.8 Hz), 4.16 (t, 1H, J = 8.3 Hz), 4.09-3.93 (m, 3H), 3.84-3.73 (m, 1H), 3.71 (d, 1H, J = 19.0 Hz), 3.65 (d, 1H, J = 19.0 Hz), 3.58 (dd, 1H, J = 8.2, 5.9 Hz), 2.69-2.59 (m, 2H), 2.23-2.13 (m, 1H), 1.12 (d, 3H, J = 7.2 Hz).
MS: m/z = 311 [M+H] +
[Chem. 20]
Compound A which was prepared by the same process was measured by NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.11 (dd, 1H, J = 3.5, 2.5 Hz), 6.58 (dd, 1H, J = 3.5, 1.8 Hz), 4.16 (t, 1H, J = 8.3 Hz), 4.09-3.93 (m, 3H), 3.84-3.73 (m, 1H), 3.71 (d, 1H, J = 19.0 Hz), 3.65 (d, 1H, J = 19.0 Hz), 3.58 (dd, 1H, J = 8.2, 5.9 Hz), 2.69-2.59 (m, 2H), 2.23-2.13 (m, 1H), 1.12 (d, 3H, J = 7.2 Hz).
MS: m/z = 311 [M+H] +
[0151]
Using Compound A, which was prepared by the same method, the single crystal X-ray analysis was carried out.
(1) Preparation of Single crystal
To 10 mg of Compound A in a LaPha ROBO Vial(R) 2.0 mL wide-mouthed vial was added 0.5 mL of chloroform. The vial was covered with a cap, in which Compound A was completely dissolved. In order to evaporate the solvent slowly, a hole was made on the septum attached in the cap with a needle of a TERUMO(R) syringe, and the vial was still stood at room temperature. The resulting single crystal was used in the structural analysis.
(2) Measuring instrument
Beam line: SPring-8 BL32B2
Detector: Rigaku R-AXIS V diffractometer
(3) Measuring method
The radiant light of 0.71068Å was irradiated to the single crystal to measure X-ray diffraction data.
(4) Assay method
Using the X-ray anomalous scattering effect of the chlorine atom in the resulting Compound A chloroform-solvate, the absolute configuration of Compound A was identified as (3S,4R). Based on the obtained absolute configuration of Compound A, the absolute configurations of each process intermediate were identified.
(1) Preparation of Single crystal
To 10 mg of Compound A in a LaPha ROBO Vial(R) 2.0 mL wide-mouthed vial was added 0.5 mL of chloroform. The vial was covered with a cap, in which Compound A was completely dissolved. In order to evaporate the solvent slowly, a hole was made on the septum attached in the cap with a needle of a TERUMO(R) syringe, and the vial was still stood at room temperature. The resulting single crystal was used in the structural analysis.
(2) Measuring instrument
Beam line: SPring-8 BL32B2
Detector: Rigaku R-AXIS V diffractometer
(3) Measuring method
The radiant light of 0.71068Å was irradiated to the single crystal to measure X-ray diffraction data.
(4) Assay method
Using the X-ray anomalous scattering effect of the chlorine atom in the resulting Compound A chloroform-solvate, the absolute configuration of Compound A was identified as (3S,4R). Based on the obtained absolute configuration of Compound A, the absolute configurations of each process intermediate were identified.
REFERENCES
1: Nakagawa H, Nemoto O, Yamada H, Nagata T, Ninomiya N. Phase 1 studies to assess the safety, tolerability and pharmacokinetics of JTE-052 (a novel Janus kinase inhibitor) ointment in Japanese healthy volunteers and patients with atopic dermatitis. J Dermatol. 2018 Jun;45(6):701-709. doi: 10.1111/1346-8138.14322. Epub 2018 Apr 17. PubMed PMID: 29665062; PubMed Central PMCID: PMC6001687.
2: Nakagawa H, Nemoto O, Igarashi A, Nagata T. Efficacy and safety of topical JTE-052, a Janus kinase inhibitor, in Japanese adult patients with moderate-to-severe atopic dermatitis: a phase II, multicentre, randomized, vehicle-controlled clinical study. Br J Dermatol. 2018 Feb;178(2):424-432. doi: 10.1111/bjd.16014. Epub 2018 Jan 15. PubMed PMID: 28960254.
3: Tanimoto A, Shinozaki Y, Yamamoto Y, Katsuda Y, Taniai-Riya E, Toyoda K, Kakimoto K, Kimoto Y, Amano W, Konishi N, Hayashi M. A novel JAK inhibitor JTE-052 reduces skin inflammation and ameliorates chronic dermatitis in rodent models: Comparison with conventional therapeutic agents. Exp Dermatol. 2018 Jan;27(1):22-29. doi: 10.1111/exd.13370. Epub 2017 Jul 3. PubMed PMID: 28423239.
4: Nomura T, Kabashima K. Advances in atopic dermatitis in 2015. J Allergy Clin Immunol. 2016 Dec;138(6):1548-1555. doi: 10.1016/j.jaci.2016.10.004. Review. PubMed PMID: 27931536.
5: Amano W, Nakajima S, Yamamoto Y, Tanimoto A, Matsushita M, Miyachi Y, Kabashima K. JAK inhibitor JTE-052 regulates contact hypersensitivity by downmodulating T cell activation and differentiation. J Dermatol Sci. 2016 Dec;84(3):258-265. doi: 10.1016/j.jdermsci.2016.09.007. Epub 2016 Sep 13. PubMed PMID: 27665390.
6: Tanimoto A, Shinozaki Y, Nozawa K, Kimoto Y, Amano W, Matsuo A, Yamaguchi T, Matsushita M. Improvement of spontaneous locomotor activity with JAK inhibition by JTE-052 in rat adjuvant-induced arthritis. BMC Musculoskelet Disord. 2015 Nov 6;16:339. doi: 10.1186/s12891-015-0802-0. PubMed PMID: 26546348; PubMed Central PMCID: PMC4636776.
7: Amano W, Nakajima S, Kunugi H, Numata Y, Kitoh A, Egawa G, Dainichi T, Honda T, Otsuka A, Kimoto Y, Yamamoto Y, Tanimoto A, Matsushita M, Miyachi Y, Kabashima K. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol. 2015 Sep;136(3):667-677.e7. doi: 10.1016/j.jaci.2015.03.051. Epub 2015 Jun 24. PubMed PMID: 26115905.
8: Tanimoto A, Ogawa Y, Oki C, Kimoto Y, Nozawa K, Amano W, Noji S, Shiozaki M, Matsuo A, Shinozaki Y, Matsushita M. Pharmacological properties of JTE-052: a novel potent JAK inhibitor that suppresses various inflammatory responses in vitro and in vivo. Inflamm Res. 2015 Jan;64(1):41-51. doi: 10.1007/s00011-014-0782-9. Epub 2014 Nov 12. PubMed PMID: 25387665; PubMed Central PMCID: PMC4286029.
References
- “Anzupgo EPAR”. European Medicines Agency. 25 July 2024. Retrieved 25 July 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- “Anzupgo PI”. Union Register of medicinal products. 23 September 2024. Retrieved 27 September 2024.
- Dhillon S (April 2020). “Delgocitinib: First Approval”. Drugs. 80 (6): 609–615. doi:10.1007/s40265-020-01291-2. PMID 32166597. S2CID 212681247.
- Park B (5 August 2020). “Delgocitinib Cream Gets Fast Track Status for Chronic Hand Eczema”. empr.com.
- Szalus K, Trzeciak M, Nowicki RJ (November 2020). “JAK-STAT Inhibitors in Atopic Dermatitis from Pathogenesis to Clinical Trials Results”. Microorganisms. 8 (11): 1743. doi:10.3390/microorganisms8111743. PMC 7694787. PMID 33172122.
- “Meeting highlights from the Committee for Medicinal Products for Human Use (CHMP) 22-25 July 2024”. European Medicines Agency (Press release). 25 July 2024. Retrieved 29 July 2024.
/////////Delgocitinib, デルゴシチニブ , JAPAN 2020, 2020 APPROVALS, Corectim, UNII-9L0Q8KK220, JTE-052, 9L0Q8KK220, LEO 124249A, LEO 124249, HY-109053, CS-0031558, D11046, GTPL9619, JTE-052A, JTE052, LP-0133 , ROH-201, atopic dermatitis
CC1CN(C12CCN(C2)C3=NC=NC4=C3C=CN4)C(=O)CC#N

 |
|
| Clinical data | |
|---|---|
| Trade names | Corectim, others |
| Other names | JTE-052; JTE-052A |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C16H18N6O |
| Molar mass | 310.361 g·mol−1 |
| 3D model (JSmol) | |
https://pubs.acs.org/doi/10.1021/acs.oprd.1c00031
https://www.chemicalbook.com/article/synthesis-of-delgocitinib.htm
Synthesis of Delgocitinib
Delgocitinib is synthesised using bromolactone as raw material by chemical reaction. The specific synthesis steps are as follows:

Synthesis of Delgocitinib
Dec 26,2023
Synthesis of Delgocitinib
Delgocitinib is synthesised using bromolactone as raw material by chemical reaction. The specific synthesis steps are as follows:

A stereocontrolled kilogram scale synthesis of delgocitinib has been disclosed, beginning with an SN2 reaction involving bromolactone 128 and benzyl amine to provide α-amino lactone 129, which was isolated as the HCl salt after precipitation from hydrochloric acid in ethyl acetate. Amine 129 was then acylated with enantiomerically pure acid chloride 131 (prepared by thionyl chloride treatment of commercial acid 130) to furnish lactone 132. In the crucial spirocyclic ring ringforming sequence of the synthesis, lactone 132 was treated with LHMDS to form an enolate that underwent SN2 displacement of the chloride, forming the spirolactone 133 and establishing both stereocenters with 98:2 dr and 96% ee.
The lactone ring of 133 was then opened by an attack of potassium phthalimide on the γ- carbon, and the resulting carboxylic acid was converted to the ethyl ester by treatment with ethyl iodide. Finally, treatment with diethylenetriamine released phthalimide, providing a free amine for subsequent cyclization to spirolactam 134 via the corresponding ethyl ester intermediate. This sequence took place in 80% yield over four steps and provided the spirolactam in >99% de after recrystallization.
The carbonyl groups within spirolactam 134 were then reduced with lithium aluminum hydride and aluminum chloride in THF, and the resulting diamine 135 was crystallized as a succinic acid salt in 86% yield. The SNAr reaction of 135 with chloropyrrolopyrimidine 136 followed by hydrogenative removal of the benzyl protecting group provided amine 137 in 92% yield over 2 steps. Finally, amine 137 was acylated with cyanoacetyl pyrazole 138 and recrystallized from n-butanol with 3 wt % BHT to provide delgocitinib in 86% yield, >99% ee, and >99% de.
Dotinurad ドチヌラド
Dotinurad
ドチヌラド
(3,5-dichloro-4-hydroxyphenyl)-(1,1-dioxo-2H-1,3-benzothiazol-3-yl)methanone
| Formula |
C14H9Cl2NO4S
|
|---|---|
| CAS |
1285572-51-1
|
| Mol weight |
358.1966
|
PMDA, Urece, APROVED JAPAN 2020/1/23, Antihyperuricemic
305EB53128UNII-305EB53128
1285572-51-1,
Dotinurad is a urate transporter inhibitor.
Patents
WO 2011040449

https://patents.google.com/patent/WO2011040449A1/en
Uric acid is produced by metabolizing a purine produced by the degradation of a nucleic acid in the body and adenosine triphosphate (ATP), which is an energy source of the living body, to xanthine, and further undergoes oxidation by xanthine oxidase or xanthine dehydrogenase. In humans, uric acid (dissociation constant pKa = 5.75) is the final metabolite of purines and exists in the body as free forms or salts.
Uric acid is normally excreted in the urine, but when uric acid production exceeds excretion and blood uric acid increases, hyperuricemia occurs. If a state in which the blood level of uric acid exceeds the upper limit of solubility (about 7 mg / dL) continues for a long period of time, crystals of urate (usually sodium salt) precipitate.
In the blood, the precipitated crystals deposit on cartilage tissue and joints, form precipitates and become gouty nodules, causing acute gouty arthritis, and then transition to chronic gouty arthritis.
When uric acid crystals are precipitated in urine, renal disorders such as interstitial nephritis (gouty kidney), urinary calculi, and the like are caused. After the seizures of acute gouty arthritis have subsided, drug therapy is given along with lifestyle improvement guidance to correct hyperuricemia.
Correcting hyperuricemia and appropriately managing uric acid levels are also important in preventing acute gouty arthritis, gouty kidneys, urinary tract stones, and the like.
Hyperuricemia is considered to be associated with a high rate of lifestyle-related diseases such as obesity, hyperlipidemia, impaired glucose tolerance, and hypertension (see Non-Patent Document 1 (pp7-9)). Increased serum uric acid levels are positively related to cardiovascular mortality, and higher serum uric acid levels increase mortality due to ischemic heart disease. It has been suggested that it is associated with the risk of death from disease (see Non-Patent Document 2).
Furthermore, serum uric acid levels have also been shown to be a powerful risk factor for myocardial infarction and stroke (see Non-Patent Document 3). To date, hyperuricemia is obesity, hyperlipidemia, dyslipidemia, impaired glucose tolerance, diabetes, metabolic syndrome, kidney disease (eg, renal failure, urine protein, end-stage renal disease (ESRD), etc.), heart It is known to be associated with vascular diseases (for example, hypertension, coronary artery disease, carotid artery disease, vascular endothelial disorder, arteriosclerosis, cardiac hypertrophy, cerebrovascular disease, etc.) or risk factors of these diseases (Non-Patent Documents 2 to 11) reference). In cerebrovascular dementia, it has also been reported that the concentration of uric acid in the cerebrospinal cord is increased (see Non-Patent Document 12).
Under such circumstances, it has been suggested that the treatment for lowering the blood uric acid level may delay the progression of kidney disease and reduce the risk of cardiovascular disease (Non-Patent Documents 5, 8, 13, 14), it has been reported that it should also be applied to asymptomatic hyperuricemia (see Non-Patent Document 14).
Therefore, reducing the blood uric acid level in the above-mentioned diseases is effective for the treatment or prevention of these diseases, and is considered to be important in terms of preventing recurrence of cardiovascular accidents and maintaining renal function.
The main factors that increase blood uric acid levels include excessive uric acid production and decreased uric acid excretion. Therefore, as a method for lowering blood uric acid level, it is conceivable to suppress the production of uric acid or promote the excretion of uric acid, and allopurinol is a drug having the former mechanism of action (uric acid production inhibitor). Benzbromarone, probenecid, JP-A 2006-176505 (Patent Document 1) and the like are known as drugs having the latter mechanism of action (uric acid excretion promoters).
According to the Japanese guidelines for treatment of hyperuricemia and gout, in principle, uric acid excretion-promoting agents are applied to hyperuricemia-reducing types and uric acid production-inhibiting agents are applied to excessive uric acid production types, respectively. (See Non-Patent Document 1 (pp31-32)).
In Japan, it is said that about 60% of hyperuricemia patients have a reduced uric acid excretion type, and about 25% are a mixed type of reduced uric acid excretion type and excessive uric acid production type (Non-patent Document 15). About 85% of the patients showed a decrease in uric acid excretion, and the average value of uric acid clearance was significantly lower than that of healthy individuals even in patients with excessive uric acid production, and the decrease in uric acid excretion was fundamental in all gout patients. Is also reported (Non-Patent Document 16).
Therefore, in hyperuricemia (especially gout), treatment for patients with reduced uric acid excretion is considered to be important, and the existence significance of uric acid excretion promoters is extremely large.
Among the major uric acid excretion promoters, probenecid is weakly used and is rarely used because of its gastrointestinal tract disorders and interactions with other drugs. On the other hand, severe liver damage has been reported for benzbromarone, which has a strong uric acid excretion promoting action and is widely used in Japan as a uric acid excretion promoting drug (see Non-Patent Document 17).
Benzbromarone or its analogs inhibit mitochondrial respiratory chain enzyme complex activity, uncoupling action, respiration inhibition, fatty acid β oxidation inhibition, mitochondrial membrane potential reduction, apoptosis, generation of reactive oxygen species, etc. Has been suggested to be involved in the development of liver damage (see Non-Patent Documents 18 and 19). Hexahydrate, which is the active body of benzbromarone, is also toxic to mitochondria.
Furthermore, benzbromarone has an inhibitory action on cytochrome P450 (CYP), which is a drug metabolizing enzyme. In particular, the inhibition against CYP2C9 is very strong, suggesting the possibility of causing a pharmacokinetic drug interaction (non-) (See Patent Documents 20 and 21).
Furthermore, although a nitrogen-containing fused ring compound having a URAT1 inhibitory action, which is a kind of uric acid transporter, and having a structure similar to that of the compound of the present invention is described in JP-A-2006-176505 (Patent Document 1), the effect is sufficient. In addition, no practical uric acid excretion promoter has been developed yet.
Recently, it has been found that the uric acid excretion promoting action depends on the urinary concentration of a drug having the same action, that is, the uric acid excretion promoting drug is excreted in the urine and exhibits a medicinal effect (Patent Document 2). Non-Patent Documents 22 and 23).
Therefore, a stronger pharmacological effect is expected if it is a uric acid excretion promoter that is excreted more in the urine, but the above existing uric acid excretion promoters have a very low concentration in urine, and a satisfactory activity can be obtained sufficiently. I can’t say that.
Regarding the urinary excretion of drugs, it is assumed that the administered drug is excreted as it is as an unchanged form or converted into an active metabolite and excreted. In the latter case, the active metabolite is produced. There is a risk that the individual difference in the amount becomes large, and in order to obtain stable drug efficacy and safety, a drug excreted as an unchanged substance is more desirable.
As described above, there is a demand for the development of a highly safe pharmaceutical having a high unchanged body urine concentration and a remarkable uric acid excretion promoting action as compared with existing uric acid excretion promoting drugs.
JP 2006-176505 A WO2005 / 121112
Treatment Guidelines for Hyperuricemia and Gout (1st Edition) pp7-9 and pp31-32, Gout and Nucleic Acid Metabolism, Volume 26, Supplement 1, 2002 Japan Gout and Nucleic Acid Metabolism Society JAMA 283: 2404-2410 (2000) Stroke 37: 1503-1507 (2006) Nephrology 9: 394-399 (2004) Semin. Nephrol. 25: 43-49 (2005)J. Clin. Hypertens. 8: 510-518 (2006) J. Hypertens. 17: 869-872 (1999) Curr. Med. Res. Opin. 20: 369-379 (2004) Curr. Pharm. Des. 11: 4139-4143 (2005)Hypertension 45: 991-996 (2005) Arch. Intern. Med. 169: 342-350 (2009) J. Neural. Transm. Park Dis. Dement. Sect. 6: 119-126 (1993) Am. J. Kidney Dis. 47: 51-59 (2006) Hyperuricemia and gout 9: 61-65 (2001) Japanese clinical trials 54: 3230-3236 (1996) Japanese clinical trial 54: 3248-3255 (1996) J. Hepatol. 20: 376-379 (1994) J. Hepatol. 35: 628-636 (2001) Hepatology 41: 925-935 (2005) Saitama Medical University Journal (J. Saitama. Med. School) 30: 187-194 (2004) Drug Metab. Dispos. 31: 967-971 (2003) 42nd Annual Meeting of the Japanese Gout and Nucleic Acid Metabolism General Assembly Program / Abstracts, p59 (2009) ACR 2008 Annual Scientific Meeting, No. 28





PATENT
JP 2011074017
PATENT
WO 2018199277
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018199277
//////////Dotinurad, Antihyperuricemic, JAPAN 2020, 2020 APPROVALS , ドチヌラド , VOFLAIHEELWYGO-UHFFFAOYSA-N, HY-109031, CS-0030545
C1N(C2=CC=CC=C2S1(=O)=O)C(=O)C3=CC(=C(C(=C3)Cl)O)Cl
